disease severity and thin filament regulation in m9r tpm3
TRANSCRIPT

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
ORIGINAL ARTICLE
Disease Severity and Thin Filament Regulation in M9R TPM3Nemaline Myopathy
Biljana Ilkovski, PhD, Nancy Mokbel, BSc, MappSc, Raymond A. Lewis, PhD, Kendall Walker, PhD,Kristen J. Nowak, PhD, Ana Domazetovska, PhD, Nigel G. Laing, PhD, Velia M. Fowler, PhD,
Kathryn N. North, MD, and Sandra T. Cooper, PhD
AbstractThe mechanism of muscle weakness was investigated in an Aus-
tralian family with an M9R mutation in TPM3 (>-tropomyosinslow).Detailed protein analyses of 5 muscle samples from 2 patientsshowed that nemaline bodies are restricted to atrophied Type 1(slow) fibers in which the TPM3 gene is expressed. Developmentalexpression studies showed that >-tropomyosinslow is not expressedat significant levels until after birth, thereby likely explaining thechildhood (rather than congenital) disease onset in TPM3 nemalinemyopathy. Isoelectric focusing demonstrated that >-tropomyosinslowdimers, composed of equal ratios of wild-type and M9R->-tropomyosinslow, are the dominant tropomyosin species in 3 separatemuscle groups from an affected patient. These findings suggest thatmyopathy-related slow fiber predominance likely contributes to theseverity of weakness in TPM3 nemaline myopathy because ofincreased proportions of fibers that express the mutant protein.Using recombinant proteins and far Western blot, we demonstrated ahigher affinity of tropomodulin for >-tropomyosinslow comparedwith A-tropomyosin; the M9R substitution within >-tropomyosinslowgreatly reduced this interaction. Finally, transfection of the M9Rmutated and wild-type >-tropomyosinslow into myoblasts revealedreduced incorporation into stress fibers and disruption of thefilamentous actin network by the mutant protein. Collectively, theseresults provide insights into the clinical features and pathogenesis ofM9R-TPM3 nemaline myopathy.
Key Words: Disease severity, Nemaline myopathy, Skeletalmuscle, Tropomodulin, Tropomyosin.
INTRODUCTIONNemaline myopathy (NM) is an inherited neuromus-
cular disease characterized by general muscle weakness androd-shaped structures in skeletal muscle. The disorder rangesin severity from severe (lethal) congenital to childhood- andadult-onset forms. Nemaline myopathy results from mutationsin genes encoding thin filament proteins, including >-skeletalactin, nebulin, A-tropomyosin, troponin T, >-tropomyosinslow(1), and the actin regulator protein cofilin 2 (2). The firstdisease causing mutation (M9R) was identified more than adecade ago in the >-tropomyosinslow gene (TPM3) in a largeAustralian autosomal dominant family with childhood-onsetNM (3). Two additional mutations in TPM3 with recessiveinheritance have been identified in patients with severecongenital (4) and intermediate (5) forms of NM. AnR167H mutation in TPM3 has been identified in a sporadic(6) case and in a large family with NM (7). Every NM patientwho harbors a TPM3 mutation contains rods only withinType 1 muscle fibers (4Y8). In addition, dominant mutationsin TPM3 have recently been identified in 11 patients from 6families with congenital fiber-type disproportion in whom themuscle pathology is characterized by small Type 1 fibers butwith absence of nemaline bodies (9).
The identification of the M9R mutation in TPM3 pavedthe way for research into understanding the molecularmechanisms of NM. A transgenic mouse model of the M9R>-tropomyosinslow mutation has a late onset of muscleweakness, nemaline bodies within its skeletal muscle fibers,a predominance of atrophied oxidative (Type 1) fibers, andhypertrophied glycolytic (Type 2) fibers (10), features thatare also observed in patients with the M9R mutation in TPM3(8). Isolated muscles from M9R transgenic mice do not showany overt differences in the maximal force generated whenmuscle fibers are stimulated at optimum sarcomere lengths,but they exhibit impaired force generation at shorter (i.e.below optimum) sarcomere lengths (11).
Cell biology studies have highlighted functional deficitsof the M9R substitution within TPM3 that may contribute tothe hypotonia and sarcomeric disruption in NM patients pos-sessing the M9R mutation. Studies with recombinant proteinshave shown that introduction of the M9R substitution in
867J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008
J Neuropathol Exp NeurolCopyright � 2008 by the American Association of Neuropathologists, Inc.
Vol. 67, No. 9September 2008
pp. 867Y877
From the Institute for Neuromuscular Research, The Children’s Hospital atWestmead (BI, NM, AD, KNN, STC); Discipline of Paediatrics andChild Health, Faculty of Medicine, University of Sydney, Sydney, NewSouth Wales (NM, AD, KNN, STC); Centre for Medical Research,University of Western Australia, West Australian Institute for MedicalResearch, QEII Medical Centre, Nedlands, Western Australia, Australia(KW, KJN, NGL); and Department of Cell Biology, The ScrippsResearch Institute (RAL, VMF), La Jolla, California.
Send correspondence and reprint requests to: Sandra Cooper, PhD, Neuro-genetics Research Unit, The Children’s Hospital at Westmead, LockedBag 4001, Sydney, New South Wales, Australia; E-mail: [email protected]
Sandra T. Cooper was supported by a USA MDA Development grant and byGrant 301946 from the Australian National Health and Medical ResearchCouncil. Biljana Ilkovski, Kristen J. Nowak, and Kathryn N. North weresupported by Grant 40394 from the Australian National Health andMedical Research Council. Velia M. Fowler and Raymond A. Lewiswere supported by Grant HL083464 from the National Institutes ofHealth. Nigel G. Laing was supported by Fellowship 403904 from theAustralian National Health and Medical Research Council.
A supplementary table is available online at http://www.jneuropath.com.
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
TPM3 reduces the affinity of recombinant M9R >-tropomyosinfor F-actin by as much as 100-fold (12, 13). Circular di-chroism studies showed that the binding of tropomodulin to anN-terminal tropomyosin fragment containing the M9R sub-stitution was abolished (14). Furthermore, substitution of M9Rwithin the human >-tropomyosinfast gene resulted in reducedsensitivity of isometric force production to activating calciumin adenovirally transduced rat cardiac myocytes (15).
Previous analysis of skeletal muscle from 2 relatedpatients bearing the M9R mutation revealed a highly specificpathologic finding of atrophied Type 1 fibers containingnemaline rods and hypertrophied Type 2 fibers without rods(8). We have since obtained 3 muscle samples from 1 ofthese individuals with M9R TPM3 NM and have shown thatthis pathology is not consistent in all 4 muscle samples,thereby providing valuable information with respect to thediagnosis of primary TPM3 mutations in patients with NM.In the present study, we performed a thorough analysis of 4muscle samples fromasinglepatient bearing theM9Rmutation,determining the mutant protein load in each muscle and theeffect on endogenous tropomyosin isoform regulation. Weshow that slow muscle fibers in TPM3 NM muscle express >-tropomyosinslow dimers as the dominant sarcomeric tropo-myosin species, with roughly equal levels of wild-type (WT)and mutant M9R proteins detected within the myofibrillarfraction. We also demonstrate that altered tropomyosin dimerpopulations may affect fiber-type-specific troponin expres-sion, and we examine the localization and binding oftropomodulin within patient muscle expressing a roughlyequal ratio of WT and mutant M9R >-tropomyosinslow.
MATERIALS AND METHODS
Skeletal Muscle SamplesWe studied archival frozen human fetal skeletal muscle
biopsies from fetuses delivered at 14, 16, 19, and 22 weeks’gestation. All specimens were deidentified in accordance withthe ethical guidelines of the Children’s Hospital at West-mead. We also studied skeletal muscle biopsies fromindividuals of the following ages: 25 and 36 weeks’gestation, 8 days, 5 months, 5 years, and 28 years. All ofthe samples were from patients with no known form ofneuromuscular disease and showed normal muscle histology.All muscle samples from Patient 1 were obtained at autopsy(at age 46 years); the control (28 and 38 years) and Patient 2muscle samples (22 years) were obtained at biopsy.
Immunohistochemistry AnalysisIndirect immunofluorescence was performed as previ-
ously described (16) with the following exceptions: 1) fortroponin Islow, muscle cryosections were fixed with 3% para-formaldehyde for 3 minutes, washed 3 times in phosphate-buffered saline (PBS), then extracted with j20-C methanolfor 5 minutes; 2) for tropomodulin, immunohistochemistrywas performed as previously described (17). Antibodysources and dilutions are listed in the Supplementary Table.Images were captured as previously described (16). Musclemorphometry was performed on at least 200 fibers usingImageQuant software.
Protein PurificationRecombinant human Tmod1 protein was expressed in
Escherichia coli and purified as described (18). Tropomyo-sins were purified as in Reference 13.
Far Western BlotsIncreasing amounts of purified Tmod1 or tropomyosin
(TPM) isoforms were separated by 12% or 15% sodiumdodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Parallel gels were stained with Coomassie brilliantblue or transferred onto nitrocellulose, and Tmod1 or TPMbinding was assessed by a blot overlay technique (19). Thetransferred proteins were bound to the membrane, followedby two 5-minute washes at 4-C in OverWash Buffer(20 mmol/L of HEPES, pH 7.3, 80 mmol/L of KCl,2 mmol/L of MgCl2, 1 mmol/L of EGTA, 0.2% TritonX-100). The blots were overlayed with 3 Kg of purifiedprotein incubated for 12 to 18 hours at 4-C, and excessprotein was removed by five 5-minute washes with Over-Wash. Primary antibodies (anti-Tmod1 monoclonal mAb95[20] or anti-TPM polyclonal antibodies [17]) were addedand incubated for 12 to 18 hours at 4-C. The blots wererinsed with 2 consecutive OverWash rinses, followed by five10-minute shaking washes. Peroxidase-conjugated secondaryantibody was added for 1 hour in Blotto (50 mmol/L of Tris,pH 7.5, 185 mmol/L of NaCl, 0.05% Tween-20, 3%powdered skim milk). The blots were subsequently washedtwice in Blotto and twice in Otto (Blotto minus milk),followed by enhanced chemiluminescence detection. Westernblots were performed as previously discussed without theprotein overlay step. Gels and blots were scanned and quan-titated using ImageJ (National Institutes of Health). Thepercentage of Tmod1 binding to each TPM is the averageof the Far Western blot signal divided by the correspondingCoomassie signal for each isoform set with the Tmod1-WT>-tropomyosinslow interaction set to 100%. The Far West-ern analysis was repeated multiple times with representativeblots shown.
Western Blot Analysis of Tropomyosin andTropomodulin
Samples were extracted into myofibrillar (insoluble)and soluble protein pools as previously described (16).Tropomyosin isoforms were separated as outlined in Refer-ence 21 and immunoblotted as described in Reference 22using anti-tropomyosin antibody (TM311; 1:10000 dilution;Sigma, St Louis, MO). Separation of tropomodulin wasperformed using 4% to 12% Bis-Tris NuPAGE precast gels(Invitrogen, Carlsbad, CA). Immunoblotting of tropomodulinwas performed as previously described (17).
Isoelectric FocusingIsoelectric focusing was performed using a Multiphor
II system (GE Healthcare, Little Chalfont, Buckinghamshire,UK) according to methods described in Reference 16 using18-cm immobiline dry strips (pH 4.5Y5.5; GE Healthcare)and focused for a total of 60,000 V/h. The isoelectric point(pI) for proteins was calculated using the pI analysis toolavailable at the Web site http://www.expasy.org/.
Ilkovski et al J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008
� 2008 American Association of Neuropathologists, Inc.868
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
Denaturation of Muscle Thin Filament With UreaAn insoluble myofibrillar fraction was obtained from
frozen muscle sections after 2 washes in 1 ml of PBScontaining 0.5% Triton X-100 and protease inhibitors (1:500;Sigma). The pellet was resuspended in 250 Kl of PBScontaining protease inhibitors, and aliquots were incubatedwith increasing concentrations of urea (prepared in PBS). Thesamples were incubated for exactly 15 minutes on ice thencentrifuged at 18,000 � g for 10 minutes at 4-C. Thesupernatant (40 Kl) was removed and solubilized in 2� SDSloading buffer (4% SDS, 40% glycerol, 100 mmol/L of Tris,pH 6.8, 100 mmol/L of dithiothreitol), heat-inactivated at94-C for 3 minutes, and analyzed by SDS-PAGE.
Expression ConstructsThe WT TPM3 cDNA sequence was amplified from
reverse-transcribed human skeletal muscle RNA (AppliedBiosystems, Foster City, CA) using Pfu Turbo DNApolymerase (Stratagene, La Jolla, CA), and the followingprimers (forward, 5¶-cgtgtacggtgggaggtct-3¶; reverse, 5¶-atcctggtcgagctggacg-3¶; SigmaGenosys, Sydney, Australia),which introduced XhoI and EcoR1 restriction sites, respec-tively. The resultant polymerase chain reaction product wascloned into the TOPO (blunt) plasmid (Invitrogen), andclones were sequenced to confirm that no polymerase chainreactionYinduced errors were present. The M9R mutation wasintroduced into an error-free WT-TPM3TOPO clone throughsite-directed mutagenesis (QuickChange site-directed muta-genesis kit; Stratagene, La Jolla, CA). Both the WT-TPM3and M9R-TPM3 cDNA sequences were released from theTOPO (blunt) vector using XhoI and EcoRI (New EnglandBiolabs, Ipswich, MA), gel-purified (Qiagen, Hilden,Germany), and cloned into a similarly digested enhancedgreen fluorescent protein (EGFP)YN1 vector (Clontech,Mountain View, CA). Clones were sequenced to confirm
FIGURE 1. Rods are localized to atrophied type 1 fibers inpatients with the M9R mutation in TPM3. (A, C, E, G) Frozenmuscle sections stained using the modified Gomori trichromemethod (31) in control (A; quadriceps muscle biopsy from a35-year-old woman with no known neuromuscular disease),Patient 1 (bicep [C] and quadriceps [E]; 46 years of age) andPatient 2 (quadriceps [G] 22 years of age). (B, D, F, H) Panelsshow immunolabeling with anti-myosin heavy chain slow(anti-MHCslow) in control (B), Patient 1 (bicep [D] andquadriceps [F]), and Patient 2 (quadriceps [H]). (I) Panelshows immunolabeling with anti-MHCfast in Patient 2. Rodsare dark blue in all fibers of the quadriceps (E) muscle inPatient 1 (arrow). They are evident in atrophied fibers in thebiceps muscle from Patient 1 (C; arrows) and in Patient 2 (G;arrows). Immunolabeling with anti-MHCslow shows that therods are restricted to slow fibers (D, F, H). Staining of controlmuscle for anti-MHCslow shows labeling of approximately 50%of fibers (B). Patient 2 exhibits a complete Type 2 fiberpredominance (I; anti-MHCfast), with approximately 50% ofthese fibers coexpressing anti-MHCslow (H). All images wereoriginally taken at 20� magnification except for inset of (G),which is 100� magnification.
J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008 Molecular Mechanisms of Nemaline Myopathy
� 2008 American Association of Neuropathologists, Inc. 869
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022
Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
that the WT-TPM3EGFP sequence was still error-free and thatthe M9R-TPM3EGFP had the appropriate mutation.
Cell CultureC2C12 cell culture and transfection were performed as
previously described (16).
RESULTS
Clinical Patient DataThis study examines 2 patients (Patients 1 and 2) from
a large autosomal dominant Australian family with NM andan M9R mutation in TPM3 (3). Interestingly, Patients 1 and 2presented with predominantly distal weakness and foot drop,reminiscent of a peripheral neuropathy. Patient 1 wasconfined to a wheelchair in her late 30s and died at age46 years from adult respiratory distress syndrome. Hercousin, Patient 2, presented in midchildhood with foot dropand predominantly distal muscle weakness. She has had arelatively nonprogressive disease course and is ambulant andactive in her mid-20s.
Patients With an M9R Mutation in TPM3 ShowVariable Fiber-Type Distribution With NemalineBodies Detected Only in Type 1 Fibers
Our previous characterization of pathologic findings inNM due to an M9R mutation within TPM3 was performedon single-muscle samples from Patients 1 (biceps) and 2(quadriceps) (8) (Fig. 1). Histologic analysis highlighted astriking pathology associated with this mutation. The musclesamples consisted of a mixed population of rod-containingatrophied Type 1 fibers and hypertrophied Type 2 fibers thatdo not contain rods (Figs. 1C, G). We have since obtained 3additional muscle samples from Patient 1 (quadriceps,deltoid, and adductor pollicis). In contrast to our findings inthe biceps, the quadriceps, deltoid, and adductor pollicisexhibit almost complete Type 1 myofiber predominance, withrods in almost all fibers (Figs. 1E, F). Closer examination offiber size reveals these rod-containing slow fibers are atrophic(quadriceps mean diameter, 32.0 Km T 5.4; n = 200 fibers)compared with the mean fiber diameter from a healthy adultfemale quadriceps muscle (51.3 T 7.9 Km [23]). Thus,
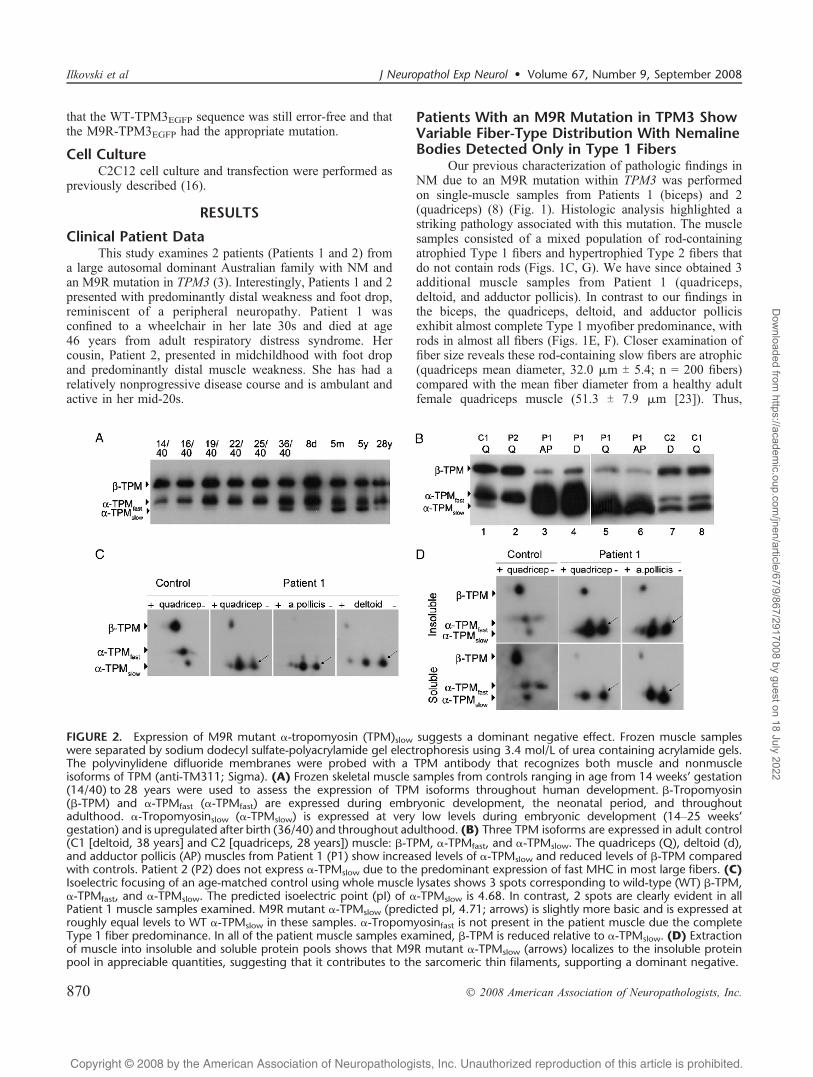
FIGURE 2. Expression of M9R mutant >-tropomyosin (TPM)slow suggests a dominant negative effect. Frozen muscle sampleswere separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 3.4 mol/L of urea containing acrylamide gels.The polyvinylidene difluoride membranes were probed with a TPM antibody that recognizes both muscle and nonmuscleisoforms of TPM (anti-TM311; Sigma). (A) Frozen skeletal muscle samples from controls ranging in age from 14 weeks’ gestation(14/40) to 28 years were used to assess the expression of TPM isoforms throughout human development. A-Tropomyosin(A-TPM) and >-TPMfast (>-TPMfast) are expressed during embryonic development, the neonatal period, and throughoutadulthood. >-Tropomyosinslow (>-TPMslow) is expressed at very low levels during embryonic development (14Y25 weeks’gestation) and is upregulated after birth (36/40) and throughout adulthood. (B) Three TPM isoforms are expressed in adult control(C1 [deltoid, 38 years] and C2 [quadriceps, 28 years]) muscle: A-TPM, >-TPMfast, and >-TPMslow. The quadriceps (Q), deltoid (d),and adductor pollicis (AP) muscles from Patient 1 (P1) show increased levels of >-TPMslow and reduced levels of A-TPM comparedwith controls. Patient 2 (P2) does not express >-TPMslow due to the predominant expression of fast MHC in most large fibers. (C)Isoelectric focusing of an age-matched control using whole muscle lysates shows 3 spots corresponding to wild-type (WT) A-TPM,>-TPMfast, and >-TPMslow. The predicted isoelectric point (pI) of >-TPMslow is 4.68. In contrast, 2 spots are clearly evident in allPatient 1 muscle samples examined. M9R mutant >-TPMslow (predicted pI, 4.71; arrows) is slightly more basic and is expressed atroughly equal levels to WT >-TPMslow in these samples. >-Tropomyosinfast is not present in the patient muscle due the completeType 1 fiber predominance. In all of the patient muscle samples examined, A-TPM is reduced relative to >-TPMslow. (D) Extractionof muscle into insoluble and soluble protein pools shows that M9R mutant >-TPMslow (arrows) localizes to the insoluble proteinpool in appreciable quantities, suggesting that it contributes to the sarcomeric thin filaments, supporting a dominant negative.
Ilkovski et al J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008
� 2008 American Association of Neuropathologists, Inc.870
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
different muscle groups from a single individual affectedwith M9R TPM3 NM respond differently to expression ofthe mutant M9R >-tropomyosinslow isoform with respect toslow-fiber predominance. Importantly, in all muscles exam-ined from both patients bearing this mutation, the abnormal-ities remain restricted to slow fibers in which the TPM3 geneis expressed. Thus, slow-fiber predominance likely contrib-utes to the severity of weakness, and slow-fiber conversionmay contribute to disease progression.
Late Developmental Expression of>-Tropomyosinslow Explains theChildhood-Onset Form of PatientsWith an M9R Mutation in TPM3
All patients with the M9R mutation in TPM3 have thechildhood-onset form of NM. >-Tropomyosinslow and >-tropomyosinfast are expressed in slow and fast skeletal musclefibers, respectively. During human skeletal muscle develop-ment, fast fibers predominate, and complete fiber typingprofile is not established until approximately 9 months ofage, as determined using myosin heavy chain (MHC) fibertype-specific antibodies (24). The developmental expressionof tropomyosin isoforms in human skeletal muscle has notpreviously been defined at the protein level. By examiningthe expression of tropomyosin isoforms during human skel-etal muscle development, we determined that A-tropomyosinand >-tropomyosinfast are the predominant isoforms duringdevelopment (Fig. 2A). In contrast, >-tropomyosinslow isexpressed at extremely low levels during development and isnot expressed at significant levels until around birth. The lackof >-tropomyosinslow expression during development likelyaccounts for the childhood (rather than congenital) onset of
clinical disease in patients with NM due to an M9R mutationin TPM3.
2-Dimensional PAGE Demonstrates Expressionof Mutant M9R >-Tropomyosinslow in PatientMuscle, Suggesting a Dominant NegativeMode of Pathogenesis
Western blot analysis of tropomyosin isoform expres-sion in Patient 1 shows upregulation of >-tropomyosinslowprotein and reduced levels of A-tropomyosin in the quad-riceps muscle (Fig. 2B; lanes 3Y6), confirming findings prev-iously described for the deltoid and adductor pollicis (16, 25).>-Tropomyosinfast was markedly reduced or absent in thequadriceps and adductor pollicis muscle from Patient 1,consistent with complete Type 1 fiber predominance asindicated by positive staining of all fibers with MHCslow andabsent labeling with MHCfast (shown for adductor pollicismuscle; Fig. 3). Normal levels of A-tropomyosin and >-tropomyosinfast were, however, observed in Patient 2, withabsent >-tropomyosinslow (Fig. 2B; lane 2). These resultslikely reflect tropomyosin expression within healthy Type2 fibers in this patient due to extreme atrophy of the Type 1rod-containing fibers and their low relative contribution to thewhole muscle lysate (Fig. 1G).
We exploited the net charge change resulting from theM9R substitution to examine the levels of mutant and WT>-tropomyosinslow in the 3 muscle samples from Patient 1that exhibited complete Type 1 fiber predominance. In wholemuscle lysates from control muscle, 3 spots corresponding toA-tropomyosin, >-tropomyosinfast, and >-tropomyosinslowwere detected using the anti-TM311 antibody (Fig. 2C). Inall three muscles from Patient 1, we confirmed the marked
FIGURE 3. Inappropriate expression of troponin I fast is specific to a patient with an M9R mutation in TPM3. Immuno-histochemistry was performed on an age-matched control (deltoid, 38 years) and Patient 1 (M9R mutation, adductor pollicis)samples. Sequential sections were used to track individual fibers in multiple stains (asterisks). Appropriate expression of fiber typesis evident in control muscle using myosin heavy chain (MHC)- and troponin (Tn)-specific antibodies (AYE). In contrast, the patientwith the M9R mutation in TPM3 (FYJ) expresses Troponin I fast (J) in a subset of fibers despite complete Type 1 fiberpredominance in this muscle, as shown by MHCslow (F). All images were taken at 20� magnification.
J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008 Molecular Mechanisms of Nemaline Myopathy
� 2008 American Association of Neuropathologists, Inc. 871
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
downregulation of A-tropomyosin, absent >-tropomyosinfast,and 2 distinct spots for >-tropomyosinslow. Mutant M9R >-tropomyosinslow protein is more basic (predicted pI, 4.71)compared with WT >-tropomyosinslow (predicted pI, 4.68)and is expressed in roughly equal proportions, with slightvariations in the ratio of mutant to WT >-tropomyosinslow.
To confirm that mutant M9R >-tropomyosinslow waspresent within the insoluble myofibrillar fraction, we firstextracted muscle samples into insoluble and soluble fractionswith 0.5% Triton X-100 and repeated 2-dimensional PAGEanalysis. In both the quadriceps and adductor pollicis(insufficient material was available for the deltoid), mutantM9R Type >-tropomyosinslow was detected in the insolublemyofibrillar protein fraction, strongly supporting its local-ization within the thin filament (Fig. 2D). Densitometricanalysis showed a slight predominance of the relative levelsof myofibrillar WT tropomyosin (quadriceps, 57%; adductorpollicis, 58%) compared with M9R tropomyosin (quadriceps,43%; adductor pollicis, 42%), these experiments could beperformed only once. Thus, mutant M9R and WT >-tropomyosinslow proteins are equally represented within theinsoluble protein fraction that would include proteins of the
thin filament and potentially also proteins sequestered withininsoluble nemaline bodies.
In summary, Figure 2 demonstrates 3 separate musclegroups from Patient 1, each consisting of only Type 1 fibers,which show marked reduction in levels of A-tropomyosinand dominant expression of >-tropomyosinslow, composed ofroughly equal amounts of M9R and WT >-tropomyosinslowproteins. We have previously shown that recombinant M9R->-tropomyosinslow can effectively dimerize with itself, WT->-tropomyosinslow, and with A-tropomyosin, although itshows a preference for > /> dimers, rather than the usual>A heterodimer pairing (25). Thus, our observation that bothM9R and WT >-tropomyosinslow are equally representedwithin the insoluble myofibrillar fraction, together withprevious evidence that M9R can effectively form dimers invitro (25), collectively supports the notion that M9Rtropomyosin is incorporated into the skeletal muscle thinfilament and exerts a dominant negative effect on skeletalmuscle function. In addition, the marked skewing in thetropomyosin dimer population, from >/A heterodimers to >/>homodimers (some containing the mutant M9R protein), mayalso induce a detrimental effect on contractile function.
M9R TPM3 Patient Muscle With CompleteType 1 Fiber Predominance Shows AbnormalExpression of Troponin Ifast in Type 1 FibersExpressing Only Slow Myosin
Because of the altered ratio of tropomyosin isoformexpression observed in Patient 1, we queried whether altered
FIGURE 4. Tropomodulin is normally expressed and boundto the thin filament in a patient with an M9R mutation inTPM3. (A) Immunohistochemistry with tropomodulin shows astriated pattern in control (i; quadriceps muscle) and in Patient1 (iv; adductor pollicis muscle). Labeling with >-actinin 2 alsoshows a striated pattern indicating its localization to the z line(ii and v). Tropomodulin does not colocalize with >-actinin 2(iii and vi) due to its localization at the pointed end of the thinfilament. The images were taken on a Leica SP2 confocal mi-croscope at 60� magnification and 2.5� zoom. (B) Frozenmuscle sections from Patient 1 P1ap, adductor pollicis; P1d,deltoid) and an age-matched control (C, quadriceps) wereextracted into insoluble and soluble protein pools using 0.5%Triton X-100 and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Tropomodulinis expressed in the control and in both of the patient musclesamples in the insoluble pool. Troponin Ifast is detected at highlevels in the control, with only low levels in the patient sam-ples. The polyvinylidene difluoride membrane stained withCoomassie blue myosin is shown to illustrate equal proteinloading for each sample. (C) Myofibrillar proteins were ex-tracted from frozen muscle sections from Patient 1 (adductorpollicis muscle) and an age-matched control using 0.5%Triton X-100 and denatured using varying concentrations ofurea (0.5Y3 mol/L). The samples were solubilized, separatedby SDS-PAGE, and immunoblotted using anti-tropomodulin(Tmod no. 1749). In both the patient and the control samples,most of the tropomodulin (È80%) is denatured at 1.5 mol/Lof urea, suggesting that there is no difference in the bindingdynamics of tropomodulin to the thin filament.
Ilkovski et al J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008
� 2008 American Association of Neuropathologists, Inc.872
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
tropomyosin dimers affected troponin isoform regulation andrecruitment to the sarcomere (Fig. 3). Troponin Islow andtroponin Tslow labeled all fibers of the quadriceps, adductorpollicis, and deltoid muscles, consistent with complete Type1 fiber predominance (Figs. 3G, H). Interestingly, troponinIfast was detected in Type 1 fibers positive only for MHCslow
(negative for MHCfast, actinin 3, and developmental myosin[data not shown]) in all 3 muscles examined (Fig. 3J; shownfor adductor pollicus). Despite positive labeling of troponinIfast in a subset of muscle fibers by immunohistochemistry,only a very faint band was detected for troponin Ifast by
Western blotting (Fig. 4B). This suggests that immunohis-tochemistry is a more sensitive tool than Western blotting,and that the level of expression of troponin Ifast is below thelevel of detection by Western analysis. Dysregulation oftroponin Ifast in slow fibers was not observed in 4 other NMpatients with Type 1 fiber predominance (3 patients with adefined ACTA1 mutation, V163M, and I136M, and 1 patientwith unknown genetic cause in whom mutations in ACTA1and TPM3 have been excluded [data not shown]). Thissuggests that altered regulation of troponin Ifast is not acommon feature observed in NM and may be specific toPatient 1 (M9R TPM3), perhaps as a consequence of alteredtropomyosin isoform regulation.
The M9R Mutation Abolishes the Interaction ofTmod1 with >-Tropomyosinslow but Does NotDisrupt Tmod1 Localization to Thin FilamentPointed Ends
In vitro binding studies using circular dichroism hadshown that binding of a tropomodulin peptide to an N-terminal fragment of >-tropomyosinfast containing the M9Rsubstitution is abolished (14). Therefore, we explored theexpression, localization, and binding of tropomodulin to thethin filament in muscle from the patient with a M9Rmutation. Colabeling of tropomodulin (Fig. 4A; i and iv)with >-actinin 2 to mark the Z-line (Fig. 4A; ii and v)showed a clear striated staining pattern in the quadriceps andadductor pollicis muscles from Patient 1 identical to that ofthe control (Fig. 4A; iii and vi). Partial overlay was observedafter colabeling for tropomodulin and phalloidin (not shown),consistent with normal localization of tropomodulin at thepointed end of the thin filament (Fig. 4A).
To examine the strength of the association of tropo-modulin to the thin filament further, we used partialdenaturation with increasing concentrations of urea tocompare dissociation of tropomodulin from a muscle myo-fibrillar fraction prepared from Patient 1 and an age-matchedcontrol. Figure 4B demonstrates that tropomodulin localizesexclusively to the insoluble myofibrillar fraction after crudeextraction in 0.5% Triton X-100 in both Patient 1 and controlmuscle. Denaturation of this myofibrillar preparation withincreasing concentrations of urea shows an identical profileof tropomodulin dissociation in both patient and control(Fig. 4C). Thus, our data suggests that tropomodulin is nor-mally localized to the pointed end of the thin filament inpatient muscle containing a roughly equal ratio of M9R/WT>-tropomyosinslow as the dominant tropomyosin species; inaddition, there is no major difference in the tenacity of thisbinding as determined by denaturation of myofibrillar proteinwith urea.
Tmod Far Western AnalysisPurified recombinant tropomyosin proteins (A, >slow,
and M9R>slow) were separated by SDS-PAGE, transferredto polyvinylidene difluoride membranes, and incubated withrecombinant Tmod1 in the overlay solution. Figure 5 demon-strates that Tmod1 binds to both WT >-tropomyosinslow
FIGURE 5. Tmod1 has a decreased interaction with mutantM9R >-tropomyosinslow mutant. (A) Increasing amounts ofTPM isoforms were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the inter-action with Tmod1 (upper panel) was monitored by FarWestern blotting for Tmod1. Parallel gels were Coomassiestained (middle panel) to demonstrate equivalent loading ofthe gel and Western blotted with an anti-TPM polyclonalantibody (bottom panel) to demonstrate that the anti-TPMantibody used in (B) recognizes all of the purified TPMs. (B)Increasing amounts of Tmod1 were separated by SDS-PAGEon parallel gels, and the interaction with wild-type (WT) >-TPMslow (upper panel), M9R >-TPMslow mutant (upper middlepanel), and WT A-TPM (bottom middle panel) was monitoredby Far Western blotting for TPM. A parallel gel was run andCoomassie stained (bottom panel) to monitor loading of thegel. (C) The N-terminal Tmod1-binding domains of thehuman TPM genes are identical except for an aspartic acid[D] to glutamic acid [E] substitution (asterisks representconserved residues). The position of the M9R mutation in >-TPMslow is highlighted and marked by an arrow. The relativeinteraction affinity between Tmod1 and each TPM isoform(determined from A) is indicated on the right, where theTmod1-WT >-TPMslow interaction is set to 100%. >-Tropo-myosinfast binding to Tmod1 was not tested; the Tmod1-binding quotient was deduced to be similar to A-TPM becausetheir Tmod1 interaction domains at the N-terminus areidentical.
J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008 Molecular Mechanisms of Nemaline Myopathy
� 2008 American Association of Neuropathologists, Inc. 873
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022
Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
and A-tropomyosin, but shows a greatly decreased interactionwith the mutant M9R >-tropomyosinslow. Tmod1 alsoshows a distinct preference for binding to >-tropomyosinslowover A-tropomyosin, as indicated by an increase in Tmod1binding (Fig. 5A). Quantification of the relative binding ofTmod1 to the TPM isoforms demonstrated that the inter-action of Tmod1 with the mutant M9R->-tropomyosinslowwas approximately 10% of the level of binding to WT>-tropomyosinslow, whereas binding of Tmod1 to A-tropomyosin was approximately 50% of the level of bindingto WT >-tropomyosinslow (Fig. 5C). Next, we comparedTmod1-TPM interactions using the TPMs in the overlaysolution and Tmod1 on the blots (Fig. 5B). This approach
also showed that binding of M9R->-tropomyosinslow toTmod1 could not be detected (in this approach, binding of>-tropomyosinslow to Tmod1 cannot be compared directlywith binding of A-tropomyosin to Tmod1 because exposuresfor the 2 blots were different).
Comparison of the Tmod1 binding N-terminal sequencesin exon 1a from the TPM1, TPM2, and TPM3 genes codingfor >-tropomyosinfast, A-tropomyosin, and >-tropomyosinslow,respectively, demonstrates that the >fast and the A sequencesare identical, whereas the >slow sequence differs only by aconserved glutamic acid [E] for aspartic acid [D] substitution(Fig. 5C). However, this amino acid change leads toapproximately a 2-fold increase in Tmod1 binding to the >-tropomyosinslow compared with A-tropomyosin (equivalent to>-tropomyosinfast). Nevertheless, the M9R substitution in the>-tropomyosinslow leads to a 10-fold reduction in binding ofTmod1 (our data), similar to the effect of the M9R substitutionin the >-tropomyosinfast sequence (14). Thus, small changeswithin the Tmod1 binding site of TPMs can drastically alterthe interaction between these proteins (Fig. 5C). In addition,our data suggest that the compensatory upregulation of thestronger Tmod1-binding >-tropomyosinslow expression(Fig. 2) is responsible for localizing normal levels of Tmod1to pointed ends in the patients’ skeletal muscles.
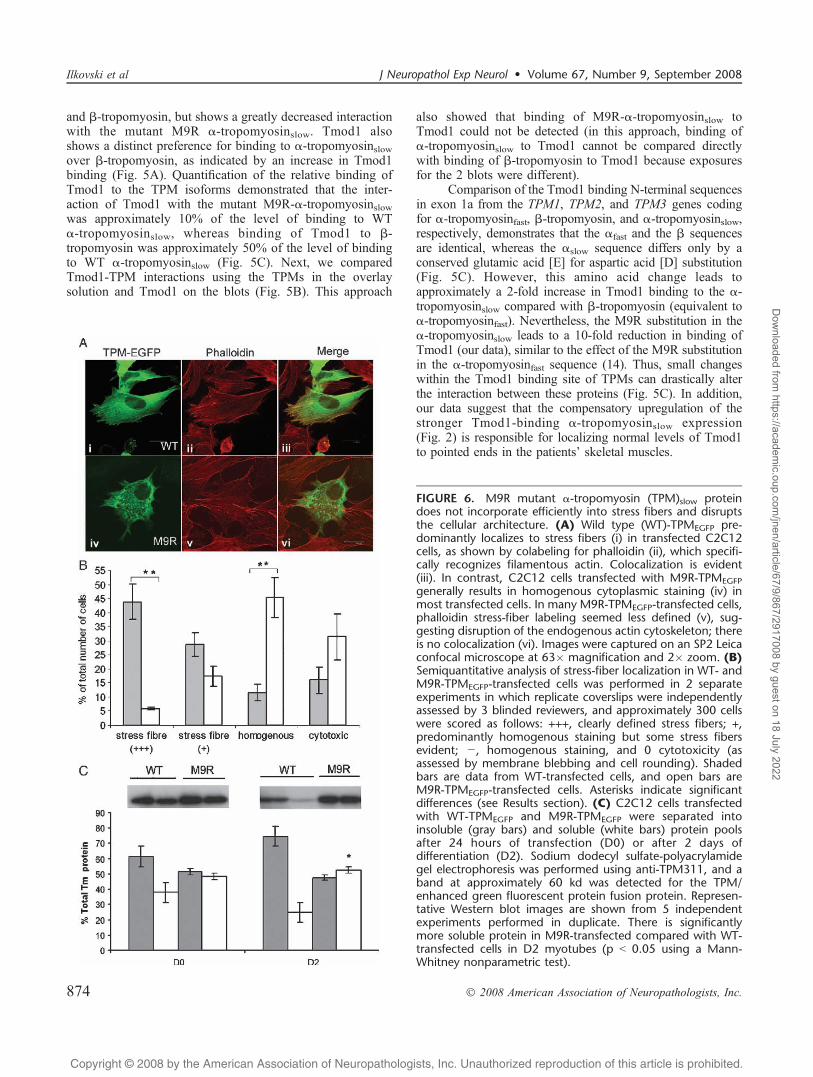
FIGURE 6. M9R mutant >-tropomyosin (TPM)slow proteindoes not incorporate efficiently into stress fibers and disruptsthe cellular architecture. (A) Wild type (WT)-TPMEGFP pre-dominantly localizes to stress fibers (i) in transfected C2C12cells, as shown by colabeling for phalloidin (ii), which specifi-cally recognizes filamentous actin. Colocalization is evident(iii). In contrast, C2C12 cells transfected with M9R-TPMEGFPgenerally results in homogenous cytoplasmic staining (iv) inmost transfected cells. In many M9R-TPMEGFP-transfected cells,phalloidin stress-fiber labeling seemed less defined (v), sug-gesting disruption of the endogenous actin cytoskeleton; thereis no colocalization (vi). Images were captured on an SP2 Leicaconfocal microscope at 63� magnification and 2� zoom. (B)Semiquantitative analysis of stress-fiber localization in WT- andM9R-TPMEGFP-transfected cells was performed in 2 separateexperiments in which replicate coverslips were independentlyassessed by 3 blinded reviewers, and approximately 300 cellswere scored as follows: +++, clearly defined stress fibers; +,predominantly homogenous staining but some stress fibersevident; j, homogenous staining, and 0 cytotoxicity (asassessed by membrane blebbing and cell rounding). Shadedbars are data from WT-transfected cells, and open bars areM9R-TPMEGFP-transfected cells. Asterisks indicate significantdifferences (see Results section). (C) C2C12 cells transfectedwith WT-TPMEGFP and M9R-TPMEGFP were separated intoinsoluble (gray bars) and soluble (white bars) protein poolsafter 24 hours of transfection (D0) or after 2 days ofdifferentiation (D2). Sodium dodecyl sulfate-polyacrylamidegel electrophoresis was performed using anti-TPM311, and aband at approximately 60 kd was detected for the TPM/enhanced green fluorescent protein fusion protein. Represen-tative Western blot images are shown from 5 independentexperiments performed in duplicate. There is significantlymore soluble protein in M9R-transfected compared with WT-transfected cells in D2 myotubes (p G 0.05 using a Mann-Whitney nonparametric test).
Ilkovski et al J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008
� 2008 American Association of Neuropathologists, Inc.874
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
The M9R Mutant Tropomyosin Disrupts theCytoarchitecture and Morphology ofC2C12 Myoblasts
To examine the localization of the M9R mutant tropo-myosin inC2C12myoblasts, we generated aC-terminal-taggedtropomyosin-EGFP fusion construct. Transfection with WT-tropomyosinslow-EGFP (WT-TPMEGFP) into C2C12 myo-blasts resulted in stress fiber staining in most cells (Fig. 6A;iYiii), diffuse cytoplasmic staining in a subset of cells, andevidence of cytotoxicity in a few cells. In contrast, C2C12myoblasts transfected with M9R-TPMEGFP did not typicallylocalize to stress fibers but predominantly showed a homo-genous distribution of staining (Fig. 6A; ivYvi), with manycells appearing cytopathic, as assessed by membrane bleb-bing and rounding (Fig. 6A; iv). In many, but not all, cases,C2C12 myoblasts transfected with M9R-TPMEGFP alsoshowed reduced labeling of phalloidin to stress fibers(Fig. 6A, v), suggesting that expression of the mutant TPMisoform disrupts endogenous actin filaments. By semiquan-titative assessment, M9R-TPMEGFP-transfected cells (+++,6% T 1%) showed a significant reduction in the ability toincorporate into clearly defined stress fibers compared withWT-TPMEGFP-transfected cells (+++, 44% T 12%, p = 0.008,Student t-test; see Fig. 6 legend for details of morphologicscoring). In addition, M9R-TPMEGFP-transfected cells pre-dominantly showed homogenous staining (45% T 14%) com-pared with WT-TPMEGFP-transfected cells (11% T 6%, p =0.011, Student t-test). The proportion of cytotoxic cells inWT-TPMEGFP-transfected cells was 16% T 9% comparedwith 31% T 16% in M9R-TPMEGFP-transfected cells, but thiswas not statistically significant ( p = 0.16, Student t-test).These immunocytochemical observations are supported bycentrifugation studies from transfected C2C12 cells in whichM9R-TPMEGFP showed a significant impairment in its abilityto contribute to insoluble filaments isolated from transfectedC2C12 myoblasts (Fig. 6C, Day 0: insoluble fraction, WT =61% T 5%; M9R = 52% T 4%) and myotubes (Fig. 6C, Day2: insoluble fraction, WT = 75% T 7%, M9R = 47% T 3%,p G 0.05).
DISCUSSIONSince the discovery of the M9R mutation in TPM3, a
number of different model systems have been establishedto improve understanding of the etiology of muscle weak-ness in NM and to determine the functional consequencesof this mutation. Here, we examine 5 different musclesamples obtained from 2 related patients with the M9R mu-tation in TPM3 to investigate proposed mechanisms ofdisease pathogenesis. Analysis of the developmental expres-sion of the sarcomeric tropomyosin isoforms revealed that >-tropomyosinslow is not expressed at robust levels until afterbirth, thereby likely explaining the childhood, rather thancongenital, onset in patients with TPM3-related disease.
Nemaline bodies were evident only in Type 1 fibers;therefore, the pathologic phenotype is chiefly observed infibers that express mutant >-tropomyosinslow protein. M9RPatient 1 showed a complete Type 1 fiber predominance in 3of the 4 muscles examined, resulting in expression of mutant
protein in all fibers. Type 1 myofiber predominance is acommon feature in many inherited myopathies, and it hasbeen postulated to be a compensatory mechanism forconserving energy because of the more efficient oxidativenature of slow muscle. In this case, predominance of Type 1fibers (due to fiber conversion or, perhaps, altered matura-tion) results in whole muscle groups in which all fibersexpress mutant tropomyosin and thereby likely contributingto the severity of the phenotype. Differences in fibercomposition may explain the more severe phenotype ofPatient 1 (wheelchair confinement, death at age 46 due torespiratory distress) compared with that of Patient 2 (moder-ate weakness, remaining ambulatory in her 20s). In the singlequadriceps biopsy examined from Patient 2, rod-containingType 1 fibers are extremely atrophied, and the bulk of musclecross-sectional area consists of mainly Type 2 fibers, ofwhich some are hybrid fibers that coexpress fast and slowMHC.
Fiber atrophy of rod-containing Type 1 fibers is a con-sistent pathologic feature of all muscles examined from bothpatients possessing the M9R mutation in TPM3. Fiber atrophyis a common feature in NM (8) and has been observed inother NM patients with mutations in ACTA1 (26). Fiberatrophy may be a primary manifestation of the disease pro-cess due to expression of the mutant protein; in both Patients1 and 2, it was most pronounced in Type 1 fibers thatcontained rods. This concept is supported by the recentidentification of mutations in TPM3 as a cause of congenitalfiber-type disproportion (9) in which the major pathology issmall Type 1 fibers without rods. Alternatively, atrophy ofslow fibers might be a compensatory mechanism and/or beexacerbated by muscle disuse. Patient 1 became wheelchair-bound in her late 30s after a period of prolonged immobili-zation due to a broken leg. Thus, fiber atrophy observed inthe quadriceps muscle might be due in part to disuse in thatcase. Furthermore, anecdotal evidence from this and otherpatients with NM suggests that immobilization results inaccelerated muscle atrophy and acute chronic deterioration inmuscle strength (27). Interestingly, the biceps muscle ofPatient 1 was the only 1 of 4 muscles examined to contain asubset of hypertrophied Type 2 fibers. This patient likelywould have maintained higher levels of activity in her upperlimbs, including pushing her wheelchair, and this might inpart explain the variation in fiber size in this muscle.
Analysis of tropomyosin isoform expression in 3different muscles from Patient 1 using 2-dimensional gelelectrophoresis shows that >-tropomyosinslow speciesdominate, with roughly equal proportions of mutant andWT >-tropomyosinslow, and marked downregulation of A-tropomyosin. Because these 3 muscle groups showedcomplete Type 1 fiber predominance, we can conclude thatType 1 fibers in Patient 1 express >-tropomyosin dimers asthe dominant tropomyosin species. We also observed ab-normal expression of troponin Ifast in slow fibers (positivefor slow MHC in the absence of fast myosin expression)in Patient 1. This may be due either to dysregulation oftroponin isoform expression secondary to altered expressionof tropomyosin isoforms and/or residual troponin Ifast ex-pression after fast-slow fiber conversion. However, altered
J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008 Molecular Mechanisms of Nemaline Myopathy
� 2008 American Association of Neuropathologists, Inc. 875
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
fiber-specific expression of troponin Ifast has not, however,been observed in skeletal muscle from patients with otherforms of NM with complete Type 1 fiber predominance.
The M9R mutant >-tropomyosinslow isoform localizedto the insoluble protein pool, which consists of the insolublecytoskeleton and the sarcomeric apparatus. This suggests thatthe mutant >-tropomyosin protein is incorporated into thethin filament and has a dominant negative (poisoning) effecton sarcomere thin filament function. We cannot excludethe possibility that the insoluble fraction of M9R mutant >-tropomyosinslow is present in the rods, however. A dominant-negative mode of disease pathogenesis has also beenshown for 4 patients with NM due to mutations in ACTA1(16, 28, 29).
Here, we provide evidence that tropomodulin localizesnormally to the pointed end of the thin filament in M9Rpatient muscle. These results contrast with in vitro bindingresults reported by Greenfield and Fowler (14), whichshowed loss of binding of synthetic M9R mutant peptide totropomodulin. This discrepancy may reflect the differenttropomyosin isoform (>-tropomyosinfast, which is notexpressed at high levels in affected muscles [Fig. 2]) usedin their studies (14), Indeed, we show that Tmod1 binds morestrongly to >-tropomyosinslow than to A-tropomyosin, whichhas a Tmod1-binding site equivalent to >-tropomyosinfast.Thus, the upregulation of >-tropomyosinslow, to whichTmod1 binds better than A-tropomyosin, may account forthe WT localization of Tmod1 in the presence of M9Rmutant protein in patient muscle. Tmod1 may bind toheterodimers of the M9R mutant and WT >-tropomyosinslowat sarcomeric pointed ends, but this has not been tested.Alternatively, WT >-tropomyosinslow homodimers may belocated at most sarcomeric thin filament pointed ends,whereas M9R mutant >-tropomyosinslow could localize alongthe remainder of the thin filament or be sequestered intonemaline rods. In this scenario, Tmod1 would be able tointeract with WT >-tropomyosinslow at or near the sarcomericpointed ends. Additionally, in human muscle, there are likelyto be other proteins that link tropomyosin and tropomodulindirectly or indirectly to sarcomeric pointed ends to maintainand regulate thin filament length. Nebulin is known to binddirectly to tropomodulin (30) and plays a supportive role inpromoting the thin filament stability and length, and thus maycontribute to the normal expression, localization, and bindingof tropomodulin observed in the patient muscle.
Transfection studies in C2C12 myoblasts showed thatM9R mutant >-tropomyosinslow incorporates less efficientlyinto filamentous structures such as stress fibers, results incellular toxicity, and may disrupt or destabilize the endoge-nous actin cytoskeleton of transfected cells. In patientmuscle, myofiber formation is completed prior to expressionof significant levels of the mutant M9R-tropomyosin (i.e.around birth). Thus, defective myotube formation is unlikelyto contribute to disease pathogenesis in M9R TPM3 NM, buta detrimental effect of mutant M9R-tropomyosin on actinfilament stability or formation can influence muscle fibergrowth and may well contribute to the fiber atrophy/hypotrophy consistently observed in slow fibers expressingmutant protein in M9R TPM3 NM.
In summary, we present a detailed pathologic andbiochemical analysis of NM associated with a M9R mutationin TPM3 and provide insights into the disease mechanism.Patients affected with TPM3-NM may have mixed-fibermuscle groups or complete slow-fiber predominance. In allcases, slow fibers are atrophied, and nemaline bodies arelocalized to Type 1 fibers in which the gene is expressed.Atrophy of Type 1 myofibers is likely a primary consequenceof expression of the mutant M9R >-tropomyosinslow proteinand of the resulting dysfunction this mutant protein imparts,although slow-fiber atrophy may also be exacerbated bymuscle disuse. The M9R mutation alters the expression ofother tropomyosin isoforms and results in a switch fromnormal >/A tropomyosin heterodimers to a predominance of>/> dimers, of which some likely contain the mutant M9Rprotein. This alteration in TPM dimer populations may alsodirectly contribute to muscle dysfunction.
REFERENCES1. Sanoudou D, Beggs AH. Clinical and genetic heterogeneity in nemaline
myopathyVa disease of skeletal muscle thin filaments. Trends Mol Med2001;7:362Y68
2. Agrawal PB,Greenleaf RS, TomczakKK, et al. Nemaline myopathy withminicores caused by mutation of the CFL2 gene encoding the skeletalmuscle actin-binding protein, cofilin-2. Am J Hum Gen 2007;80:162Y67
3. Laing NG, Wilton SD, Akkari PA, et al. A mutation in the alphatropomyosin gene TPM3 associated with autosomal dominant nemalinemyopathy NEM1. Nat Genet 1995;10:249
4. Tan P, Briner J, Boltshauser E, et al. Homozygosity for a nonsensemutation in the alpha-tropomyosin slow gene TPM3 in a patient withsevere infantile nemaline myopathy. Neuromuscul Disord 1999;9:573Y79
5. Wattanasirichaigoon D, Swoboda KJ, Takada F, et al. Mutations of theslow muscle alpha-tropomyosin gene, TPM3, are a rare cause ofnemaline myopathy. Neurology 2002;59:613Y17
6. Durling HJ, Reilich P, Muller-Hocker J, et al. De novo missense mutationin a constitutively expressed exon of the slow alpha-tropomyosin geneTPM3 associated with an atypical, sporadic case of nemaline myopathy.Neuromuscul Disord 2002;12:947Y51
7. Penisson-Besnier I, Monnier N, Toutain A, et al. A second pedigree withautosomal dominant nemaline myopathy caused by TPM3 mutation: Aclinical and pathological study. Neuromuscul Disord 2007;17:330Y37
8. Ryan MM, Ilkovski B, Strickland CD, et al. Clinical course correlatespoorly with muscle pathology in nemaline myopathy. Neurology 2003;60:665Y73
9. Clarke FN, Kolski H, Dye ED, et al. Mutations in TPM3 are a commoncause of congenital fibre type disproportion. Ann Neurol 2008;63:329Y37
10. Corbett MA, Robinson CS, Dunglison GF, et al. A mutation inalpha-tropomyosin(slow) affects muscle strength, maturation and hyper-trophy in a mouse model for nemaline myopathy. Hum Mol Genet 2001;10:317Y28
11. de Haan A, van der Vliet MR, Gommans IM, et al. Skeletal muscle ofmice with a mutation in slow alpha-tropomyosin is weaker at lowerlengths. Neuromuscul Disord 2002;12:952Y57
12. Moraczewska J, Greenfield NJ, Liu Y, et al. Alteration of tropomyosinfunction and folding by a nemaline myopathy-causing mutation.Biophys J 2000;79:3217Y25
13. Akkari PA, Song Y, Hitchcock-DeGregori S, et al. Expression andbiological activity of Baculovirus generated wild-type human slow alphatropomyosin and the Met9Arg mutant responsible for a dominantform of nemaline myopathy. Biochem Biophys Res Commun 2002;296:300Y4
14. Greenfield NJ, Fowler VM. Tropomyosin requires an intact N-terminalcoiled coil to interact with tropomodulin. Biophys J 2002;82:2580Y91
15. Michele DE, Albayya FP, Metzger JM. A nemaline myopathy mutationin alpha-tropomyosin causes defective regulation of striated muscleforce production. J Clin Invest 1999;104:1575Y81
Ilkovski et al J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008
� 2008 American Association of Neuropathologists, Inc.876
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022

Copyright @ 200 by the American Association of Neuropathologists, Inc. Unauthorized reproduction of this article is prohibited.8
16. Ilkovski B, Nowak KJ, Domazetovska A, et al. Evidence for adominant-negative effect in ACTA1 nemaline myopathy caused byabnormal folding, aggregation and altered polymerization of mutantactin isoforms. Hum Mol Genet 2004;13:1727Y43
17. Ursitti JA, Fowler VM. Immunolocalization of tropomodulin, tropo-myosin and actin in spread human erythrocyte skeletons. J Cell Sci1994;107:1633Y39
18. Fowler VM, Greenfield NJ, Moyer J. Tropomodulin contains two actinfilament pointed end-capping domains. J Biol Chem 2003;278:40000Y9
19. Fowler VM. Identification and purification of a novel Mr 43,000tropomyosin-binding protein from human erythrocyte membranes. J BiolChem 1987;262:12792Y800
20. Almenar-Queralt A, Lee A, Conley CA, et al. Identification of a noveltropomodulin isoform, skeletal tropomodulin, that caps actin filamentpointed ends in fast skeletal muscle. J Biol Chem 1999;274:28466Y75
21. Pieples K, Wieczorek DF. Tropomyosin 3 increases striated muscleisoform diversity. Biochemistry 2000;39:8291Y97
22. Cooper ST, Lo HP, North KN. Single section Western blot: Improvingthe molecular diagnosis of the muscular dystrophies. Neurology 2003;61:93Y97
23. Doriguzzi C, Mongini T, Palmucci L, et al. Quantitative analysis ofquadriceps muscle biopsy. Results in 30 healthy females. J Neurol Sci1984;66:319Y26
24. Compton AG, Cooper ST, Hill PM, et al. The syntrophin-dystrobrevinsubcomplex in human neuromuscular disorders. J Neuropathol ExpNeurol 2005;64:350Y61
25. Corbett MA, Akkari PA, Domazetovska A, et al. An alphaTropomyosinmutation alters dimer preference in nemaline myopathy. Ann Neurol2005;57:42Y49
26. Ilkovski B, Cooper ST, Nowak K, et al. Nemaline myopathy caused bymutations in the muscle alpha-skeletal-actin gene. Am J Hum Genet2001;68:1333Y43
27. RyanMM,Schnell C, StricklandCD, et al. Nemaline myopathy: A clinicalstudy of 143 cases. Ann Neurol 2001;50:312Y20
28. Wallefeld W, Krause S, Nowak KJ, et al. Severe nemaline myopathycaused by mutations of the stop codon of the skeletal muscle alpha actingene (ACTA1). Neuromuscul Disord 2006;16:541Y47
29. D’Amico A, Graziano C, Pacileo G, et al. Fatal hypertrophic cardiomyop-athy and nemaline myopathy associated with ACTA1 K336E mutation.Neuromuscul Disord 2006;16:548Y52
30. McElhinny AS, Kolmerer B, Fowler VM, et al. The N-terminal end ofnebulin interacts with tropomodulin at the pointed ends of the thinfilaments. J Biol Chem 2001;276:583Y92
31. Engel WK, Cunningham GG. Rapid examination of muscle tissue: Animproved trichrome method for fresh-frozen biopsy sections. Neurology1963;13:919Y23
J Neuropathol Exp Neurol � Volume 67, Number 9, September 2008 Molecular Mechanisms of Nemaline Myopathy
� 2008 American Association of Neuropathologists, Inc. 877
Dow
nloaded from https://academ
ic.oup.com/jnen/article/67/9/867/2917008 by guest on 18 July 2022