development of oxidative methodologies and application toward
TRANSCRIPT

DEVELOPMENT OF OXIDATIVE METHODOLOGIES AND APPLICATION TOWARD
TETRODOTOXIN CORE
by
Brian Alan Mendelsohn
B.Sc., University of Washington, 2000
A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE
DEGREE OF
DOCTOR OF PHILOSOPHY
in
THE FACULTY OF GRADUATE STUDIES
(Chemistry)
THE UNIVERSITY OF BRITISH COLUMBIA
(Vancouver)
December 2010
© BRIAN ALAN MENDELSOHN, 2010

ii
Abstract
This thesis covers a novel approach to tetrodotoxin that relies on the oxidative amidation of a
phenol and intramolecular nitrile oxide cycloaddition to install a -hydroxynitrile unit among the key
steps. These transformations, and others contained herein, effectively set the tetrasubstituted C-8a
stereocenter, as well C-4a formyl equivalent and C-5, C-7 and C-8 hydroxyl groups. Novel reaction types
were developed in the course of this work, including a new method for the oxidation of oximes to nitrile
oxides using hypervalent iodine reagents. Additionally, I identified a tandem reaction sequence,
involving the dearomatization of a phenol, followed by [3+2]-dipolarcycloaddition, the first of its kind.
This tandem sequence proved a powerful tool for the rapid construction of multicyclic compounds from
structurally simpler starting materials. These studies resulted in advanced intermediates which contained
much of the structure of the tetrodotoxin core.

iii
Preface
The work presented in this thesis was in part a collaborative effort. However, primarily I, in
conjunction with my supervisor Professor Marco A. Ciufolini, developed the ideas and design of the
research projects presented herein.
The introductory section (Chapter 1) of this thesis covers a review of previous work by other
scientists. Chapter 2 of this thesis covers the work I performed during the course of my Ph.D. studies. I
performed the vast majority of the work presented in this thesis, and exceptions are noted in this Preface
section. The data described in Tables 2.10, 2.11, 2.12 and 2.13 was generated in conjunction with Mr.
Tim Jen, a talented undergraduate student who worked under my direction over two summers.
Compounds 2.113 and 2.115 were synthesized by another Ciufolini group member Mr. Florian Tessier.
Dr. Brian Patrick of the Department of Chemistry at UBC performed all crystal structure data
collection and analysis. Mr. David Wong and Mr. Marshall Lapawa of the Department of Chemistry at
UBC performed all high-resolution mass spectrometry experiments and all elemental analyses.

iv
Table of contents
Abstract ........................................................................................................................................................ ii
Preface ......................................................................................................................................................... iii
Table of contents ......................................................................................................................................... iv
List of tables ................................................................................................................................................ xi
List of figures ............................................................................................................................................. xii
List of schemes .......................................................................................................................................... xiv
List of abbreviations .................................................................................................................................. xvi
Acknowledgements ................................................................................................................................... xxi
1 Introduction ..................................................................................................................................1
1.1 Tetrodotoxin ..................................................................................................................................1
1.1.1 Isolation, characterization and natural occurrence ...................................................................2
1.1.2 Tetrodotoxin biosynthesis ........................................................................................................4
1.1.3 Voltage-gated sodium channels................................................................................................5
1.1.4 TTX and naturally occurring voltage-gated sodium channel inhibitors ...................................6
1.2 Synthetic studies ............................................................................................................................7
1.2.1 Kishi’s total synthesis ...............................................................................................................7
1.2.2 Isobe’s total synthesis and related studies ..............................................................................11
1.2.3 Du Bois’ total synthesis ..........................................................................................................23
1.2.4 Sato’s total syntheses .............................................................................................................27
1.2.5 Funabashi ...............................................................................................................................34
1.2.6 Keana ......................................................................................................................................36
1.2.7 Fraser-Reid .............................................................................................................................37
1.2.8 Alonso ....................................................................................................................................40
1.2.9 Taber ......................................................................................................................................41
1.2.10 Fukuyama ...............................................................................................................................43
1.2.11 Ohfune ....................................................................................................................................46
1.2.12 Summary ................................................................................................................................48
2 The oxidative amidation strategy ...............................................................................................50
2.1 General strategy ...........................................................................................................................50
2.2 Oxidative amidation ....................................................................................................................53
2.2.1 Oxidative amidation in total synthesis ...................................................................................54
2.3 Bimolecular oxidative amidation.................................................................................................57

v
2.3.1 Optimization of scalable bimolecular oxidative amidation conditions ..................................57
2.4 Nitrile oxide [3+2] cycloaddition ................................................................................................61
2.5 Kemp-type keto-isooxazoline fragmentation ..............................................................................73
2.5.1 Access to a suitable dihydroxylation substrate .......................................................................76
2.6 Osmylation of substituted cyclohexene derivative ......................................................................79
2.7 Iodine(III)-mediated oxime oxidation to nitrile oxides ...............................................................82
2.7.1 Oximes as nitrile oxide precusors ..........................................................................................82
2.7.2 Optimization of DIB as a reagent for oxime to nitrile oxide oxidations ................................83
2.7.3 Oxidation of -oxo-ketoximes and ’-dioxo-ketoximes ....................................................89
2.7.4 Intramolecular nitrile oxide cycloaddition .............................................................................93
2.8 Tandem oxidative dearomatization/nitrile oxide [3+2] cycloaddition ........................................95
2.8.1 Sorensen’s use of tandem dearomitization/nitrile oxide [3+2] cycloaddition in Cortistatin
core synthesis .........................................................................................................................97
2.9 Diastereoselective tandem oxidative amidation—INOC.............................................................99
2.10 Summary ...................................................................................................................................103
References .................................................................................................................................................104
Appendices ................................................................................................................................................115
A. Experimental protocols .............................................................................................................116
A.1 Preparation of methyl 2-(1-acetamido-4-oxocyclohexa-2,5-dienyl)acetate (2.32) ...................116
A.2 Preparation of methyl 2-((1r,4r)-1-acetamido-4-(tert-butyldiphenylsilyloxy)cyclohexa-2,5-
dienyl)acetate (2.37) .................................................................................................................118
A.3 Preparation of 2-((1r,4r)-1-acetamido-4-(tert-butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetic
acid (2.38) .................................................................................................................................119
A.4 Preparation of N-((1r,4r)-4-(tert-butyldiphenylsilyloxy)-1-(3-nitro-2-oxopropyl)cyclohexa-2,5-
dienyl)acetamide (2.39) ............................................................................................................120
A.5 Preparation of N-((3aS,7aS)-2-oxo-2,3,3a,7a-tetrahydrobenzofuran-3a-yl)acetamide (2.41) ...121
A.6 Preparation of N-((2aR,2a1S,3S,5aS)-3-(tert-butyldiphenylsilyloxy)-7-oxo-2a,2a1,3,5a,6,7-
hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.43) .......................................................122
A.7 Preparation of N-((3aS,7aS,E)-2-(nitromethylene)-2,3,3a,7a-tetrahydrobenzofuran-3a-
yl)acetamide (2.44) ...................................................................................................................123
A.8 Preparation of N-((1r,4r)-1-(2-(tert-butyldimethylsilyloxy)-3-nitropropyl)-4-(tert-
butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetamide (2.46) diastereomers .........................124
A.9 Preparation of compounds 2.49, 2.50, 2.51 and 2.52 ................................................................125
A.9.1 Compound 2.49 ....................................................................................................................126
A.9.2 Compound 2.50 ....................................................................................................................127
A.9.3 Compound 2.51/2.52 ............................................................................................................128

vi
A.10 Preparation of trans-9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-3-nitro-1-
azaspiro[5.5]undeca-2,7,10-trien-4-one (2.55) .........................................................................129
A.11 Preparation of methyl 2-((1S,4S,5R,6S)-1-acetamido-4-(tert-butyldiphenylsilyloxy)-6-cyano-5-
hydroxycyclohex-2-enyl)acetate (2.59) ....................................................................................130
A.12 Preparation of N-((2aR,2a1S,3S,5aS)-3-hydroxy-7-oxo-2a,2a1,3,5a,6,7-hexahydroindeno[1,7-
cd]isoxazol-5a-yl)acetamide (2.60) ..........................................................................................131
A.13 Preparation of methyl 2-((1S,5R,6S)-1-acetamido-6-cyano-5-hydroxy-4-oxocyclohex-2-
enyl)acetate (2.62) ....................................................................................................................132
A.14 Preparation of N-((2aR,2a1S,3R,5aS)-3-(tert-butyldiphenylsilyloxy)-7-oxo-2a,2a1,3,5a,6,7-
hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.66) .......................................................133
A.15 Preparation of methyl 2-((1S,4R,5R,6S)-1-acetamido-4-(tert-butyldiphenylsilyloxy)-6-cyano-5-
hydroxycyclohex-2-enyl)acetate (2.67) ....................................................................................135
A.16 Preparation of compounds 2.76-2.88 ............................................................................................136
A.16.1 Preparation of 3-(4-methoxyphenyl)-5-phenyl-4,5-dihydroisoxazole (2.76) .......................136
A.16.2 Preparation of 3,5-diphenyl-4,5-dihydroisoxazole (2.77) ....................................................137
A.16.3 Preparation of 3-(3-nitrophenyl)-5-phenyl-4,5-dihydroisoxazole (2.78) .............................138
A.16.4 Preparation of 3-pentyl-5-phenyl-4,5-dihydroisoxazole (2.79) ............................................139
A.16.5 Preparation of 3-phenethyl-5-phenyl-4,5-dihydroisoxazole (2.80) ......................................140
A.16.6 Preparation of 3a,4,5,6,7,7a-hexahydro-3-phenyl-4,7-methano-1,2-benzisoxazole (2.81) ..141
A.16.7 Preparation of 3a,4,5,6,7,7a-hexahydro-3-(3-nitrophenyl)-4,7-methano-1,2-benzisoxazole
(2.82) ....................................................................................................................................142
A.16.8 Preparation of 3a,4,5,6,7,7a-hexahydro-3-pentyl-4,7-methano-1,2-benzisoxazole (2.83) ...143
A.16.9 Preparation of 3a,4,5,6,7,7a-hexahydro-3-(2-phenylethyl)-4,7-methano-1,2-benzisoxazole
(2.84) ....................................................................................................................................144
A.16.10 Preparation of 3-(1,1-dimethylethyl)-3a,4,5,6,7,7a-hexahydro4,7-methano-1,2-
benzisoxazole (2.85) .............................................................................................................145
A.16.11 Preparation of 3a,4,5,6,7,7a-hexahydro-3-(4-methoxyphenyl)-4,7-methano-1,2-
benzisoxazole (2.86) .............................................................................................................146
A.16.12 Preparation of 5-(3-bromopropyl)-4,5-dihydro-3-phenyl-isoxazole (2.87) .......................147
A.16.13 Preparation of 3,5-diphenylisoxazole (2.88) ......................................................................148
A.17 Preparation of 1-(3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-benzisoxazol-3-yl)-ethanone (2.90)
..................................................................................................................................................149
A.18 Preparation of 1-(4,5-dihydro-5-phenyl-3-isoxazolyl)-ethanone (2.91) ....................................150
A.19 Preparation of 3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-benzisoxazole-3-carboxylic acid ethyl
ester (2.93) ................................................................................................................................151
A.20 Preparation of 4,5-dihydro-5-phenyl-3-isoxazolecarboxylic acid ethyl ester (2.94) .................153
A.21 Preparation of 2-isonitrosocyclopentanone (2.100) ...................................................................155
A.22 Preparation of compound 2.101 ................................................................................................156

vii
A.23 Preparation of methyl 4-(5-phenyl-4,5-dihydroisoxazol-3-yl)butanoate (2.102) ......................157
A.24 Preparation of 2-isonitrosocyclohexanone (2.103) ....................................................................158
A.25 Preparation of compound 2.104 ................................................................................................159
A.26 Preparation of methyl 5-(5-phenyl-4,5-dihydroisoxazol-3-yl)pentanoate (2.105) ....................160
A.27 Preparation of 3-(hydroxyimino)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one (2.106) .............161
A.28 Preparation of compounds 2.107/2.108 .....................................................................................162
A.29 Preparation of (1S,3R)-methyl 1,2,2-trimethyl-3-(5-phenyl-4,5-dihydroisoxazol-3-yl)
cyclopentanecarboxylate (2.109/2.110) ....................................................................................163
A.30 Preparation of 3,7-dimethyl-6-octenoxime (2.111) ...................................................................164
A.31 Preparation of (6S)-3,3a,4,5,6,7-hexahydro-3,3,6-trimethyl-2,1-benzisoxazole (2.112) ..........165
A.32 Preparation of 4-hydroxy-benzenepropanal oxime (2.123) .......................................................166
A.33 Preparation of N-[(4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-
acetamide (2.124) .....................................................................................................................167
A.34 Preparation of 4,4a,7a,7b-tetrahydro-4a-methoxy-indeno[1,7-cd]isoxazol-7(3H)-one (2.125) 168
A.35 Preparation of N-benzyl, N-tosyl tyrosine (2.130) ....................................................................169
A.36 Preparation of N-[2-(hydroxyimino)-1-[(4-hydroxyphenyl)methyl]ethyl]-4-methyl-N-
(phenylmethyl)-benzenesulfonamide (2.132) ...........................................................................171
A.37 Preparation of N-[(3R,4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-3-[[(4-methylphenyl)sulfonyl]
(phenylmethyl)amino]-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-acetamide (2.134) ..............173
B. Experimental section ................................................................................................................174
B.1 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-(1-acetamido-4-oxocyclohexa-2,5-
dienyl)acetate (2.32) .................................................................................................................174
B.2 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1r,4r)-1-acetamido-4-(tert-
butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetate (2.37) .....................................................175
B.3 1H-NMR spectrum and
13C-NMR spectrum for: 2-((1r,4r)-1-acetamido-4-(tert-
butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetic acid (2.38) ...............................................176
B.4 1H-NMR spectrum and
13C-NMR spectrum for: N-((1r,4r)-4-(tert-butyldiphenylsilyloxy)-1-(3-
nitro-2-oxopropyl)cyclohexa-2,5-dienyl)acetamide (2.39) ......................................................177
B.5 1H-NMR spectrum and
13C-NMR spectrum for: N-((3aS,7aS)-2-oxo-2,3,3a,7a-
tetrahydrobenzofuran-3a-yl)acetamide (2.41) ..........................................................................178
B.6 1H-NMR spectrum and
13C-NMR spectrum for: N-((2aR,2a1S,3S,5aS)-3-(tert-
butyldiphenylsilyloxy)-7-oxo-2a,2a1,3,5a,6,7-hexahydroindeno[1,7-cd]isoxazol-5a-
yl)acetamide (2.43) ...................................................................................................................179
B.7 1H-NMR spectrum and
13C-NMR spectrum for: N-((3aS,7aS,E)-2-(nitromethylene)-2,3,3a,7a-
tetrahydrobenzofuran-3a-yl)acetamide (2.44) ..........................................................................180
B.8 1H-NMR spectrum and
13C-NMR spectrum for: N-((1r,4r)-1-(2-(tert-butyldimethylsilyloxy)-3-
nitropropyl)-4-(tert-butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetamide (2.46) ................181
B.9 1H-NMR spectrum for: compound 2.49 ....................................................................................182

viii
B.10 1H-NMR spectrum and
13C-NMR spectrum for: compound 2.50 .............................................183
B.11 1H-NMR spectrum for: compound 2.51/2.52 ............................................................................184
B.12 1H-NMR spectrum and
13C-NMR spectrum for: trans-9-[[(1,1-
dimethylethyl)diphenylsilyl]oxy]-2-methyl-3-nitro-1-azaspiro[5.5]undeca-2,7,10-trien-4-one
(2.55) ........................................................................................................................................185
B.13 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1S,4S,5R,6S)-1-acetamido-4-(tert-
butyldiphenylsilyloxy)-6-cyano-5-hydroxycyclohex-2-enyl)acetate (2.59) .............................186
B.14 1H-NMR spectrum and
13C-NMR spectrum for: N-((2aR,2a1S,3S,5aS)-3-hydroxy-7-oxo-
2a,2a1,3,5a,6,7-hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.60) .............................187
B.15 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1S,5R,6S)-1-acetamido-6-cyano-5-
hydroxy-4-oxocyclohex-2-enyl)acetate (2.62) .........................................................................188
B.16 1H-NMR spectrum for: N-((2aR,2a1S,3R,5aS)-3-(tert-butyldiphenylsilyloxy)-7-oxo-
2a,2a1,3,5a,6,7-hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.66) .............................189
B.17 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1S,4R,5R,6S)-1-acetamido-4-(tert-
butyldiphenylsilyloxy)-6-cyano-5-hydroxycyclohex-2-enyl)acetate (2.67) .............................190
B.18 1H-NMR spectrum and
13C-NMR spectrum for: 3-(4-methoxyphenyl)-5-phenyl-4,5-
dihydroisoxazole (2.76) ............................................................................................................191
B.19 1H-NMR spectrum and
13C-NMR spectrum for: 3,5-diphenyl-4,5-dihydroisoxazole (2.77) ....192
B.20 1H-NMR spectrum and
13C-NMR spectrum for: 3-(3-nitrophenyl)-5-phenyl-4,5-
dihydroisoxazole (2.78) ............................................................................................................193
B.21 1H-NMR spectrum and
13C-NMR spectrum for: 3-pentyl-5-phenyl-4,5-dihydroisoxazole (2.79)
..................................................................................................................................................194
B.22 1H-NMR spectrum and
13C-NMR spectrum for: 3-phenethyl-5-phenyl-4,5-dihydroisoxazole
(2.80) ........................................................................................................................................195
B.23 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-phenyl-4,7-
methano-1,2-benzisoxazole (2.81) ...........................................................................................196
B.24 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-(3-nitrophenyl)-4,7-
methano-1,2-benzisoxazole (2.82) ...........................................................................................197
B.25 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-pentyl-4,7-methano-
1,2-benzisoxazole (2.83) ..........................................................................................................198
B.26 1H-NMR spe ctrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-(2-phenylethyl)-4,7-
methano-1,2-benzisoxazole (2.84) ...........................................................................................199
B.27 1H-NMR spectrum and
13C-NMR spectrum for: 3-(1,1-dimethylethyl)-3a,4,5,6,7,7a-
hexahydro4,7-methano-1,2-benzisoxazole (2.85) ....................................................................200
B.27 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-(4-methoxyphenyl)-
4,7-methano-1,2-benzisoxazole (2.86) .....................................................................................201
B.28 1H-NMR spectrum and
13C-NMR spectrum for: 5-(3-bromopropyl)-4,5-dihydro-3-phenyl-
isoxazole (2.87) ........................................................................................................................202
B.29 1H-NMR spectrum and
13C-NMR spectrum for: 3,5-diphenylisoxazole (2.88) ........................203

ix
B.30 1H-NMR spectrum and
13C-NMR spectrum for: 1-(3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-
benzisoxazol-3-yl)-ethanone (2.90) ..........................................................................................204
B.31 1H-NMR spectrum and
13C-NMR spectrum for: 1-(4,5-dihydro-5-phenyl-3-isoxazolyl)-
ethanone (2.91) .........................................................................................................................205
B.32 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-
benzisoxazole-3-carboxylic acid ethyl ester (2.93) ..................................................................206
B.33 1H-NMR spectrum and
13C-NMR spectrum for: 4,5-dihydro-5-phenyl-3-isoxazolecarboxylic
acid ethyl ester (2.94) ...............................................................................................................207
B.34 1H-NMR spectrum and
13C-NMR spectrum for: 2-isonitrosocyclopentanone (2.100) .............208
B.35 1H-NMR spectrum and
13C-NMR spectrum for: compound 2.101 ...........................................209
B.36 1H-NMR spectrum and
13C-NMR spectrum for: methyl 4-(5-phenyl-4,5-dihydroisoxazol-3-
yl)butanoate (2.102) .................................................................................................................210
B.37 1H-NMR spectrum and
13C-NMR spectrum for: 2-isonitrosocyclohexanone (2.103)...............211
B.38 1H-NMR spectrum and
13C-NMR spectrum for: compound 2.104 ...........................................212
B.39 1H-NMR spectrum and
13C-NMR spectrum for: methyl 5-(5-phenyl-4,5-dihydroisoxazol-3-
yl)pentanoate (2.105) ................................................................................................................213
B.40 1H-NMR spectrum and
13C-NMR spectrum for: compounds 2.107/2.108 ................................214
B.41 1H-NMR spectrum and
13C-NMR spectrum for: (1S,3R)-methyl 1,2,2-trimethyl-3-(5-phenyl-
4,5-dihydroisoxazol-3-yl) cyclopentanecarboxylate (2.109/2.110) ..........................................215
B.42 1H-NMR spectrum and
13C-NMR spectrum for: diastereomers (6S)-3,3a,4,5,6,7-hexahydro-
3,3,6-trimethyl-2,1-benzisoxazole (2.112) ...............................................................................216
B.43 1H-NMR spectrum and
13C-NMR spectrum for: major diastereomer (6S)-3,3a,4,5,6,7-
hexahydro-3,3,6-trimethyl-2,1-benzisoxazole (2.112) .............................................................217
B.44 1H-NMR spectrum and
13C-NMR spectrum for: N-[(4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-7-
oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-acetamide (2.124) .......................................................218
B.45 1H-NMR spectrum and
13C-NMR spectrum for: 4,4a,7a,7b-tetrahydro-4a-methoxy-indeno[1,7-
cd]isoxazol-7(3H)-one (2.125) .................................................................................................219
B.46 1H-NMR spectrum and
13C-NMR spectrum for: N-benzyl, N-tosyl tyrosine (2.130) ...............220
B.47 1H-NMR spectrum and
13C-NMR spectrum for: N-[2-(hydroxyimino)-1-[(4-
hydroxyphenyl)methyl]ethyl]-4-methyl-N-(phenylmethyl)-benzenesulfonamide (2.132) ......221
B.48 1H-NMR spectrum and
13C-NMR spectrum for: N-[(3R,4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-3-
[[(4-methylphenyl)sulfonyl](phenylmethyl)amino]-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-
acetamide (2.134) .....................................................................................................................222
C. X-ray crystallography data .......................................................................................................223
C.1 X-ray data of methyl 2-(1-acetamido-4-oxocyclohexa-2,5-dienyl)acetate (2.32) .....................223
C.2 X-ray data of N-((3aS,7aS,E)-2-(nitromethylene)-2,3,3a,7a-tetrahydrobenzofuran-3a-
yl)acetamide (2.44) ...................................................................................................................229
C.3 X-ray data of N-((2aR,2a1S,3S,5aS,7R)-3,7-dihydroxy-2a,2a1,3,5a,6,7-hexahydroindeno[1,7-
cd]isoxazol-5a-yl)acetamide (2.53) ..........................................................................................235

x
C.4 X-ray data of trans-9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-3-nitro-1-
azaspiro[5.5]undeca-2,7,10-trien-4-one (2.55) .........................................................................243
C.5 X-ray data of methyl 2-((1S,4R,5R,6S)-1-acetamido-4-(tert-butyldiphenylsilyloxy)-6-cyano-5-
hydroxycyclohex-2-enyl)acetate (2.67) ....................................................................................254
C.6 X-ray data of N-[(4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-
acetamide (2.124) .....................................................................................................................265

xi
List of tables
Table 2.1. Optimization of scalable conditions for bimolecular oxidative amidation. ..............................59
Table 2.2. Larger-scale reproducible conditions for oxidative amidation of phenol 2.31 with acetonitrile.
.....................................................................................................................................................................60
Table 2.3. Reduction of dienone 2.32. .......................................................................................................62
Table 2.4. Initial attempts to dehydrate nitroketone 2.39. .........................................................................65
Table 2.5. Nitroketone 2.39 dehydration optimization. .............................................................................69
Table 2.6. Refinements to [3+2] cycloaddition conditions. .......................................................................70
Table 2.7. Optimization of tricycle 2.43 fragmentation. ............................................................................74
Table 2.8. DIB-mediated bimolecular [3+2] dipolar cycloaddition: optimization studies. .......................84
Table 2.9. DIB-mediated bimolecular [3+2] dipolar cycloaddition: substrate scope. ...............................86
Table 2.10. DIB-mediated bimolecular [3+2] dipolar cycloaddition: optimization studies. .....................87
Table 2.11. DIB-mediated oxidation of -oxo-aldoximes 2.89 and 2.92. .................................................88
Table 2.13. DIB-mediated oxidation of -oxo-ketoximes. .......................................................................92

xii
List of figures
Figure 1.1. The structure of (−)-tetrodotoxin (TTX). ..................................................................................1
Figure 1.2. Orthoester-lactone equilibrium. ................................................................................................3
Figure 1.3. Method of extracting tetrodotoxin.21
.........................................................................................3
Figure 1.4. Some tetrodotoxin derivatives found in nature36
. ......................................................................4
Figure 1.5. Possible TTX biosynthetic pathway. .........................................................................................5
Figure 1.6. Sodium channel inhibitors: tetrodotoxin, saxitoxin and gonyautoxin 3. ...................................6
Figure 1.7. Kishi’s retrosynthetic analysis. .................................................................................................7
Figure 1.7. Isobe’s deoxy tetrodotoxin analogs. ........................................................................................11
Figure 1.8. Isobe’s (−)-tetrodotoxin retrosynthetic rationale from 2-acetoxy-tri-O-acetyl-D-glucal. .......12
Figure 1.9. Isobe’s (−)-tetrodotoxin retrosynthetic rationale from levoglucosenone and isoprene. ..........17
Figure 1.10. Isobe’s updated C-11 hydroxylation and comparison to 1.43. ..............................................22
Figure 1.11. Du Bois’ tetrodotoxin retrosynthetic analysis. ......................................................................23
Figure 1.12. Sato’s retrosynthetic analysis for (±)-tetrodotoxin from myo-inositol. .................................27
Figure 1.13. Sato’s (−)-tetrodotoxin retrosynthetic analysis. ....................................................................30
Figure 1.14. Sato’s updated retrosynthetic analysis for (−)-tetrodotoxin intermediate 1.84. ....................32
Figure 1.15. Funabashi’s retrosynthetic approach to (−)-tetrodotoxin. .....................................................34
Figure 1.16. Fraser-Reid’s retrosynthetic considerations. .........................................................................37
Figure 1.17. Fukuyama’s TTX-core synthon 1.147. .................................................................................43
Figure 1.18. Ohfune’s approach to (−)-tetrodotoxin. ................................................................................46
Figure 1.19. Comparison of retrosynthetic intermediates between completed total syntheses to date......49
Figure 2.1. Retrosynthetic hypothesis for tetrodotoxin. ............................................................................50
Figure 2.2. Generalized strategy for the elaboration of the tetrodotoxin core. ..........................................51
Figure 2.3. Our elaborated tetrodotoxin retrosynthetic analysis. ...............................................................52
Figure 2.4. Oxidative amidation of para-phenols. ....................................................................................53
Figure 2.5. Oxidative amidation of para-phenols. ....................................................................................53
Figure 2.6. Kikugawa-Glover-type reactions. ...........................................................................................54
Figure 2.7. Structures of FR901483 and TAN1251C and Ciufolini’s retrosynthetic logic for the
construction of their ring systems. ...............................................................................................................55
Figure 2.8. Sorensen’s144,145
and Honda’s146-148
oxidative cyclization of phenolic secondary amines. .....55
Figure 2.9. Ciufolini modes of oxidative amidation of phenols. ...............................................................56
Figure 2.10. Possible DIB-mediated bimolecular oxidative amidation mechanism. .................................57
Figure 2.11. Effect of phenol concentration on reaction outcome. ............................................................58
Figure 2.12. Desymmetrization of dienone 2.8. ........................................................................................61

xiii
Figure 2.13. Theoretical dehydration of nitroketone 2.39. ........................................................................64
Figure 2.14. Nitrile oxide [3+2] dipolar cycloaddition..............................................................................64
Figure 2.15. Torssell cyclization: silyl nitronates as 1,3-dipoles...............................................................67
Figure 2.16. Probable enolization of nitroketone 2.39 can inhibit [3+2] cyclization. ...............................72
Figure 2.17. All cis-relationship of compound 2.43. .................................................................................72
Figure 2.18. Comparison of prepared enone 2.62 to proposed tetrodotoxin retron 2.5. ............................75
Figure 2.19. Rationale for expected (but not observed) rate difference between nitroketones 2.39/2.65. 77
Figure 2.20. X-ray image of crystalline 2.67 and rationalization of expected facial selectivity. ..............78
Figure 2.21. Comparison of 1H-NMR: olefin osmylation. ........................................................................80
Figure 2.22. 1H-NMR couplings for 2.68. .................................................................................................81
Figure 2.23. Comparison of advanced intermediate 2.68 with TTX retron 1.170. ....................................81
Figure 2.24. Dimerization of nitrile oxides. ..............................................................................................82
Figure 2.25. Conversion of oximes to nitrile oxides and subsequent trapping. .........................................83
Figure 2.26. Predicted course of the DIB oxidation of -oxo-ketoximes. ................................................89
Figure 2.27. Hypothetical tandem oxidative amidation-intramolecular nitrile oxide cycloaddition. ........95
Figure 2.28. Predicted course of the INOC reaction. ................................................................................99
Figure 2.28. NOe NMR spectral expansion of 2.134. .............................................................................102

xiv
List of schemes
Scheme 1.1. Kishi’s Diels-Alder and Beckmann transformations. ..............................................................8
Scheme 1.2. Kishi’s installation of C-11 and C-6 oxygen atoms. ...............................................................8
Scheme 1.3. Installation of C-9 -acetoxy moiety. .....................................................................................9
Scheme 1.4. Baeyer-Villiger oxidation and intramolecular epoxide-opening cyclization. ..........................9
Scheme 1.5. Kishi’s (±)-tetrodotoxin end game strategy. ............................................................................9
Scheme 1.6. Isobe’s cyclohexane skeleton synthesis: Sonogashira coupling and Claisen rearrangement.
.....................................................................................................................................................................13
Scheme 1.7. Isobe’s cyclohexane skeleton synthesis: C-5 and C-11 hydroxyl installation. ......................14
Scheme 1.8. Isobe’s cyclohexane skeleton synthesis: cyclohexanone and exo-olefin installation. ...........14
Scheme 1.9. Isobe’s introduction of nitrogen through intramolecular conjugate addition. .......................15
Scheme 1.10. Isobe’s stereoselective lactone formation. ...........................................................................16
Scheme 1.11. Isobe’s introduction of the guanidine moiety and completion of the total synthesis. .........17
Scheme 1.12. Isobe’s carbocyclic core formation. ....................................................................................18
Scheme 1.13. Isobe’s use of the Overman rearrangement. ........................................................................18
Scheme 1.14. Isobe’s C-8 oxygenation sequence. .....................................................................................18
Scheme 1.15. Isobe’s inversion of C-8 and oxygenation at C-7. ...............................................................19
Scheme 1.16. Isobe’s C-11/C-6 oxygenation sequence and addition of the C-10 acetylide group. ..........19
Scheme 1.17. Isobe’s epoxide-opening cyclization. ..................................................................................20
Scheme 1.18. Isobe’s ortho lactonization and end-game strategy. ............................................................21
Scheme 1.19. Du Bois’ Rh-carbenoid C-H insertion. ................................................................................24
Scheme 1.20. Du Bois’ methylenation. ......................................................................................................24
Scheme 1.21. Du Bois’ allylic oxidation and establishment of C-4a and C-5 configurations. ..................25
Scheme 1.22. Du Bois’ Rh-catalyzed nitrene C-H insertion and guanidine formation. ............................26
Scheme 1.23. Sato’s setup for spiro -chloroepoxide formation. ..............................................................28
Scheme 1.24. Sato’s -chloroepoxide formation and azide ion-mediated ring-opening. ..........................28
Scheme 1.25. Sato’s end game strategy: lactone/orthoester formation, guanidine formation. ..................29
Scheme 1.26. Sato’s synthesis of nitro cyclitol 1.93 employing an intramolecular Henry reaction. .........31
Scheme 1.27. Sato’s McMurry-Nef transformation to common intermediate 1.90. ..................................31
Scheme 1.28. Sato’s Ferrier(II) sequence to common intermediate 1.90. .................................................33
Scheme 1.29. Funabashi’s approach to (−)-tetrodotoxin. ..........................................................................34
Scheme 1.30. Sato’s early independent work on (−)-tetrodotoxin. ............................................................35
Scheme 1.31. Keana’s most advanced tetrodotoxin intermediate. .............................................................36

xv
Scheme 1.32. Fraser-Reid’s synthesis of dioxadamantane core 1.129 via D-mannosan. ..........................38
Scheme 1.33. Fraser-Reid’s synthesis of advanced intermediate 1.134. ...................................................39
Scheme 1.34. Alonso’s radical-cyclization approach. ...............................................................................40
Scheme 1.35. Taber’s C-H insertion strategy. ...........................................................................................42
Scheme 1.36. Fukuyama’s route to diiodo 1.152 enroute to 1.147. ...........................................................43
Scheme 1.37. Fukuyama’s racemic route to TTX core synthon 1.147. .....................................................45
Scheme 1.38. Ohfune’s approach to C-5, 6, 7, 11 tetraol system in (−)-tetrodotoxin. ..............................47
Scheme 2.1. Initial conditions for bimolecular oxidative amidation.153
.....................................................57
Scheme 2.2. Synthesis of nitroketone 2.39. ...............................................................................................63
Scheme 2.3. Undesired cyclization reaction of intermediate 2.40/2.38. ....................................................63
Scheme 2.4. An undesired reaction of nitroketone intermediate 2.39. ......................................................65
Scheme 2.5. Reduction/protection sequence of nitroketone 2.39. .............................................................66
Scheme 2.6. [3+2]-dipolar cycloaddition of 2.46. .....................................................................................67
Scheme 2.7. Confirmation of structural geometry: X-ray crystallographic analysis of 2.53. ....................68
Scheme 2.8. Unusual Knoevenagel-type condensation of nitroketone 2.39. .............................................69
Scheme 2.9. Optimized conditions for dehydration of nitroketone 2.39. ..................................................71
Scheme 2.10. Ring-fragmentation and comparison to Kemp-elimination188-190
products. .........................73
Scheme 2.11. Kemp-type fragmentation: methanolysis of tricycle 2.43. ..................................................74
Scheme 2.12. Fragmentation sequence: conversion of 2.43 to desymmetrized enone 2.62. .....................75
Scheme 2.13. Synthesis of nitroketones 2.39 and 2.65. .............................................................................76
Scheme 2.14. Synthesis of tricycles 2.43 and 2.66. ...................................................................................77
Scheme 2.15. Methanolysis of tricycle 2.66. .............................................................................................78
Scheme 2.16. Osmylation sequence. ..........................................................................................................79
Scheme 2.17. Dimerization of oxime 2.73. ................................................................................................83
Scheme 2.18. The first intramolecular variant. ..........................................................................................93
Scheme 2.19. Other intramolecular variants from Ciufolini group. ...........................................................94
Scheme 2.20. The synthesis of oxime 2.123. .............................................................................................96
Scheme 2.21. Tandem oxidative amidation—INOC. ................................................................................96
Scheme 2.22. Tandem oxidative methoxylation—INOC. .........................................................................97
Scheme 2.23. Sorensen’s use of tandem oxidative dearomatization—INOC towards the cortistatin
pentacyclic core. ..........................................................................................................................................98
Scheme 2.24. Synthesis of oxime 2.132. ..................................................................................................100
Scheme 2.25. Diastereoselective tandem oxidative amidation—INOC. .................................................101

xvi
List of abbreviations
1D one-dimensional
2D two-dimensional
[]20
D specific rotation at 20 °C and wavelength of sodium D line
[O] oxidation
(S)-CBS (S)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2c][1,3,2]oxazaborole
°C degrees centigrade
AB AB system
ABq AB quartet
Ac acetyl
acac acetylacetonate
AcOH acetic acid
AIBN azobisisobutyronitrile
Alloc allyloxycarbonyl
aq aqueous
B.C.E. before the common era
Bn benzyl
BRSM/brsm based on recovered starting material
Boc tert-butyloxycarbonyl
Boc2O di-tert-butyl-pyrocarbonate
BOM benzyloxymethyl
Bz benzoyl
c concentration
calcd calculated
cat. catalytic
cf. confer
CDI carbonyldiimidazole
cm-1
inverse centimeter/wavenumber
d deuterio

xvii
d / dd / ddd doublet / doublet of doublets / doublet of doublet of doublets
chemical shift
DBU 1,8-diazabicycloundec-7-ene
dd doublet of doublets
DIB diacetoxy iodobenzene (iodobenzene diacetate)
DIBAL diisobutyl aluminum
DMAP 4-(N,N-dimethylamino)-pyridine
DMF dimethylformamide
DMSO dimethyl sulfoxide
CAN cerium ammonium nitrate
CSA camphor sulfonic acid
EDC 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
Et ethyl
etc. et cetera
eq equivalents
g gram
G a generic group (see associated text)
GTX gonyautoxin
h hour(s)
HFIP 1,1,1,3,3,3-hexafluoroisopropanol
HRMS high-resolution mass spectrometry
Hz hertz
IBX 2-iodoxybenzoic acid
IC50 half maximal inhibitory concentration
IDCP iodonium dicollidine perchlorate
INOC intramolecular nitrile oxide-olefin cycloaddition
ISOC intramolecular siloxynitronate-olefin cycloaddition
iPr isopropyl
J coupling constant
Jaa axial-axial coupling constant

xviii
Jae axial-equatorial coupling constant
kg kilogram
L liter
L generic sterically demanding group (see text)
LD50 oral median lethal dose
LDA lithium diisopropylamine
LRMS low-resolution mass spectrometry
M molar
m multiplet
mCPBA meta-chloroperoxybenzoic acid
Me methyl
mg milligram
MHz megahertz
min minute
mL milliliter
MMTr 4-monomethoxytrityl
mol mole
mmol millimole
MOM methoxymethyl
MP melting point
Ms mesyl (methane sulfonyl)
n an integer (0, 1, 2, etc.)
nBu normal butyl (linear butyl)
NBS N-bromosuccinimide
NCS N-chlorosuccinimide
NIS N-iodosuccinimide
NMO N-methyl morpholine oxide
NMR nuclear magnetic resonance
NOe/nOe nuclear Overhauser effect
NOESY nuclear Overhauser effect spectroscopy

xix
Nu generic nucleophile (see text)
o ortho
ON overnight (12-16 h)
P generic protecting group (see text)
p para
PCC pyridinium chlorochromate
Ph phenyl
Pht phthalate
PIFA phenyliodine bis(trifluoroacetate)
Piv pivaloyl
PMB para-methoxy benzyl
ppm parts per million
PPTS pyridinium toluene-4-sulphonate
psi pounds per square inch
PTSA/p-TsOH para-toluene sulfonic acid
PyBOP benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
q quartet
R generic functional group (defined in text)
rt room temperature
s singlet
s seconds
S-H generic solvent
SM reaction starting material
SN2’ bimolecular nucleophilic substitution
STX saxitoxin
t triplet
TBAF tetrabutylammonium fluoride
TBHT di-tert-butyl hyponitrite
TBS tert-butyldimethylsilyl
TBDPS tert-butyldiphenylsilyl

xx
tBu tert-butyl
TEA triethylamine
TEMPO 2,2,6,6-tetramethylpiperidine-1-oxyl
TES triethylsilyl
Tf triflyl/triflate
TFA trifluoroacetic acid
TFAA trifluoroacetic acid anhydride
TFE trifluoroethanol
THF tetrahydrofuran
TLC thin-layer chromatography
TMS trimethylsilyl
TPAP tetrapropylammonium perruthenate
Ts tosyl
TTX tetrodotoxin
g microgram
mol micromole
L microliter
v/v volume per volume
w/ with
w/o without

xxi
Acknowledgements
There is no doubt that this dissertation could not have been possible without the guidance of my
advisor, Professor Marco A. Ciufolini. I owe him many thanks for allowing me to join his laboratory and
giving me a great deal of intellectual freedom on my research endeavors. While many days were
difficult, these times taught me patience and I am grateful for his guidance.
I would also like to thank the members of my advisory committee: Professor Jennifer A. Love,
Professor Michael D. Fryzuk and most especially my second reader Professor Gregory R. Dake. Thank
you for your sagacious advice and discussions over the past five years. I would also like to acknowledge
Graduate Advisor Professor Chris Orvig for his support. The UBC Chemistry Department NMR staff
also was indispensable due to their help and advice in setting up NMR experiments, and thanks go out to
Dr. Nick Burlinson, Dr. Maria B. Ezhova, and Zorana Danilovic. Many thanks also go to Brian
Ditchburn for the many last-minute glassware repairs and many hours spent introducing me to (the art of)
glassblowing.
I have had the pleasure to work in Ciufolini group with some exceptional undergraduate students,
graduate students and post-doctoral researchers. I would like to thank especially Tim Jen, Simon J. Kim,
Srini Masuna, Bhaskar Reddy, Virender S. Aulakh, Bryan Chan, Dylan Turner, Jaclyn Chau, Kam Ho
and Catherine Diering for their friendship and camaraderie. I would also like to acknowledge Dr. Josh
Zaifman and Dr. Mathew Smith for their careful proof-reading of my thesis manuscript.
My Ph.D. work was not possible without the love and support of my Mom and Dad. Thank you
for always listening to me, even when you had no idea what I was talking about.
Finally, I would like to thank my wife Lynne. You are my best friend and my staunchest
supporter. With you, life and all things are much more meaningful.

1
1 Introduction
1.1 Tetrodotoxin
Tetrodotoxin (TTX, Figure 1.1) is recognized as one of the most poisonous non-protein
molecules known.1,2
With a long history traced back to ancient times, tetrodotoxin has left its mark on
Egyptian, Chinese and Haitian civilizations among others. Ancient Egyptian hieroglyphics of several
Fifth Dynasty (ca. 2700 B.C.E.) tombs clearly depict the poisonous tetrodotoxin-containing puffer
Tetraodon stallatus.3,4
An early Chinese record from the second century B.C.E., the Pen-T’so Chin (The
Herbal), lists the eggs of tetraodon (four tooth) fish among its drugs.5
Figure 1.1. The structure of (−)-tetrodotoxin (TTX).
Interestingly, and perhaps horrifyingly, tetrodotoxin poisoning renders the body in a low
metabolic state, yet the brain remains unaffected. Near lethal doses can leave a victim of tetrodotoxin
poisoning in a near-death state for days, while remaining conscious for the duration. Victims of
tetrodotoxin poisoning typically undergo feelings of numbness and weakness, followed by paralysis of
the limbs and chest muscles which typically leads to death by asphyxiation. Currently, there is no known
antidote; however, in vivo studies in mice have indicated possible treatment of tetrodotoxin poisoning
with a monoclonal antibody.6 Patients suffering tetrodotoxin poisoning are typically kept alive on
ventilators until they either recover, or do not. The oral median lethal dose (LD50) of tetrodotoxin in mice
is 334 g per kg.7 The lethal dose by injection is 8 g per kg, or about 0.5 mg for a 75 kg human
assuming the lethal dose is similar for humans and mice.8

2
Ethnobotanist Wade Davis alleged in the 1980s that tetrodotoxin-containing tissue from puffer
fish were an ingredient of Haitian voodooism.9 Davis claimed that the partial limb paralysis and other
symptoms of lethal and non-lethal doses of tetrodotoxin matched depictions of Haitian voodoo zombies,
especially recounts involving the burying of apparently dead victims followed later by exhumation and
revivification. Other scientific studies of modern-day Haitian zombie powders have indicated the
presence of tetrodotoxin.10
More recent analyses generally claim that folklore and stories of zombies
created in this manner differ from victims of tetrodotoxin poisoning11
and question the amount of
tetrodotoxin that can be administered transdermally in the form of a zombie powder. In modern-day
Japan, high-end sushi restaurants still offer carefully prepared puffer fish for adventurous patrons. When
prepared properly, consumption of puffer fish sushi results in tingling sensations of the lips and inner
mouth surfaces. When prepared incorrectly puffer fish consumption can result in death.
A century after its discovery, tetrodotoxin remains an ambitious and worthwhile target that
necessarily expands the envelope of current synthetic chemical technology, as evidenced by the large
amount of synthetic studies (Chapter 1.2). Efficient syntheses could lead to TTX analogues with varying
activities to further the understanding of voltage-gated sodium channels. The unique architecture of
tetrodotoxin has provided synthetic chemists with many opportunities to explore new reaction types and
procedures to piece together tetrodotoxin and analogues. The work described in this thesis details one
such path, as our approach to the tetrodotoxin core yielded new reaction types hitherto unexplored.
1.1.1 Isolation, characterization and natural occurrence
Tetrodotoxin remains a daring target for synthetic chemistry, not only due to its unprecedented
highly hydroxylated dioxa-adamantane structure (Figure 1.1), but also due to its extreme toxicity. It is
this toxicity that initially drove researchers to attempt isolation studies. In 1910, Japanese scientist
Yoshizumi Tahara published and patented complete isolation procedures and characterization
analyses.12,13
Though Tahara’s original extracts are now known to contain only trace quantities of
tetrodotoxin,4 it is remarkable that he was able to contribute as much knowledge as he did given the crude
equipment available during that era. It was not until 1950, well after the isolation of many plant alkaloids
(atropine, morphine and strychnine, etc.) and most vitamins (pantothenic acid and biotin, etc.) when
tetrodotoxin was isolated in pure form from the ovaries of the puffer fish.14
The abundantly available
pool of tetrodotoxin-containing fish spurred wide interest in its isolation and structure elucidation.
Complicating the structure determination of tetrodotoxin is the fact that the compound is zwitterionic,
highly polar (only soluble in aqueous acid) and exists in equilibrium between orthoester form 1.1 and
lactone form 1.2 (Figure 1.2).15

3
Figure 1.2. Orthoester-lactone equilibrium.
The structure of tetrodotoxin was determined through chemical degradations as well as x-ray
crystallographic studies of tetrodotoxin derivatives.1,15-17
In 1964, groups led by R. B. Woodward, H. S.
Mosher, K. Tsuda and T. Goto all reported the correct structure of tetrodotoxin in Kyoto at the Natural
Products Symposium of the International Union of Pure and Applied Chemistry. The absolute
configuration was elucidated via crystallographic studies a few years later.18
With pure crystalline
tetrodotoxin in hand, researchers began to isolate other tetrodotoxin-derivatives from various species of
Tetraodon fish.5,19,20
Recently, a United States patent was issued describing a new process for extracting
tetrodotoxin from the ovaries of the puffer fish.21
This invention doubles the yield of pure crystalline
tetrodotoxin (Figure 1.3).
Figure 1.3. Method of extracting tetrodotoxin.21

4
Tetrodotoxin is found in the tissues of several different animals throughout the world, including
species of puffer fish, trigger fish, gobies,22,23
parrotfish, blue-ringed octopodes (Hapalochlaena), moon
snails,24
sea stars,25
polyclad flatworms,26
nemerteans, xanthid crabs, frogs,27
western newts (Taricha)1
and other species.28
1.1.2 Tetrodotoxin biosynthesis
Puffer fish species grown in captivity do not contain detectable amounts of tetrodotoxin until they
are fed tissues from tetrodotoxin-containing fish.29-31
When tetrodotoxin-containing fish were fed
radiolabeled metabolic precursors (14
C-labeled acetate, arginine, citrulline and glucose), the fish failed to
produce any labeled tetrodotoxin, yet did produce common metabolites (cholesterol, amines, amino acids,
etc.) with 14
C incorporation.32
The elucidation of the biosynthesis of tetrodotoxin is an actively
researched area; current understanding is based upon analogy to other tetrodotoxin derivatives isolated
from various organisms (Figure 1.4).33-35
O
OH
O
OH
HO
NHHN
NH
HO
CH3
OH
11-deoxyTTX
O
OH
O
HO
NHHN
NH
HO
CH3
5,6,11-trideoxyTTX
O
OH
O
OH
HO
NHHN
NH
HO
OH
6-epi TTX
OH
O
OH
O
OH
HO
NHHN
NH
HO
CH3
6,11-dideoxyTTX
5-deoxyTTX
O
OH
O
HO
NHHN
NH
HOOH
OH
O
OH
O
OH
HO
NHHN
NH
HOOH
OH
TTX
11
654 4a 8
8a
7
10
9
Figure 1.4. Some tetrodotoxin derivatives found in nature36
.
Since tetrodotoxin is found in such a variety of different organisms, most of which are unrelated
phylogenically, and since studies of puffer fish and Taricha newts showed a lack of endogenous
tetrodotoxin production in these animals, it has been considered that tetrodotoxin found in these animals
comes from an exogenous source, perhaps through the food chain or the environment.37
Various studies38
have determined that tetrodotoxin is actually biosynthesized by various types of bacteria species
(Pseudoalteromonas tetraodonis28
certain species of Pseudomonas and Vibrio28
Serratia marcescens39
).

5
Little is known about the actual biosynthesis of TTX, but studies have indicated that TTX
precursors may come from the amino acid arginine,40
a known precursor for guanidinium functional
groups in natural products and a C5 isoprene unit (most probably isopentenyl diphosphate33
).41
The
currently accepted biosynthetic pathway is shown in Figure 1.5, yet this proposal has not been confirmed
conclusively.
arginine isopentenyl diphosphate
CH3
OP
OO
OH
P
O
OHOHHN
NH2
HN
H2NCO2H
HNNH2
HN
H2N CO2HCH3
OP
OO
OH
P
O
OHOH
+
TTX
Figure 1.5. Possible TTX biosynthetic pathway.
1.1.3 Voltage-gated sodium channels
The ability for rapid cell-to-cell communication is an important feature of living organisms.
Excitable cells communicate rapidly with one another through short-lived electrical potentials, known as
action potentials, across their membranes. Every neuron has a separation of charges across its membrane,
and at rest this potential is called the resting membrane potential. Electrical signals and transmissions all
involve a temporary disruption of this resting potential.42
The resting membrane potential is determined
by resting ion channels.
Neuronal cells generate action potentials via voltage gated ion channels embedded in the cell’s
plasma membrane.43
These ion channel proteins are closed when the voltage potential across the
membrane is near the resting potential of the cell, but they quickly open when the voltage potential is
raised above a threshold value. When the ion channels open, a rapid flux of ions across the membrane
occurs, resulting in a decrease in the charge separation (voltage) across the membrane (depolarization),
and the electrical signal propagates the length of the membrane. During depolarization, a rapid influx of
sodium ions causes the polarity of the membrane to reverse relative to the resting state, and this closes the
voltage-gated sodium channels. The lipid bilayer of a cell membrane is nearly impervious to the
movement of sodium ions, and thus the cell actively pumps these ions back across the lipid bilayer to
reset the cell for a future action potential. As sodium ions are pumped out of the cell, potassium channels
open to allow the influx of potassium ions into the cell, thus returning the cell to its resting potential.

6
1.1.4 TTX and naturally occurring voltage-gated sodium channel inhibitors
Tetrodotoxin, along with saxitoxin (STX), members of the gonyautoxin (GTX) family of
compounds (Figure 1.6) and the neurotoxic conotoxin peptides, binds tightly to the extracellular pore
opening of the voltage-gated sodium channels.2 Studies suggest that the guanidinium group acts as a
bioisostere for sodium ions, and binds like a plug partway into the sodium channel.44
Studies of TTX
derivatives (Figure 1.4) have indicated that important H-bonding interactions between the hydroxyl
groups and residues on the outside of the pore opening of the sodium channel exist, and are substantially
responsible for the binding to the sodium channel.45
Blockage of the sodium channel effectively
suppresses action potentials by stopping the influx of sodium ions. Sodium channels are transmembrane
proteins, and as such are difficult to isolate and characterize. Tetrodotoxin is an important
pharmacological tool for probing the structure and function of sodium channels.46
By isolating sodium
channel proteins, or protein fragments, structural information can be gleaned by checking for TTX
binding.47
tetrodotoxin
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
saxitoxin gonyautoxin 3
N NH
NH2
HNNH
H2N
HO
HO
O3SOO
O NH2
N NH
NH2
HNNH
H2N
HO
HO O
O NH2
Figure 1.6. Sodium channel inhibitors: tetrodotoxin, saxitoxin and gonyautoxin 3.
In recent years, it has become apparent that small molecules capable of selective, partial blocking
of ion channel function may be useful therapeutic resources to combat a number of human diseases,48
such as Parkinson’s disease, and perhaps chronic pain management in terminally ill patients.49

7
1.2 Synthetic studies
1.2.1 Kishi’s total synthesis
The first total synthesis of racemic tetrodotoxin by Kishi in 1972 remains to this day a historic
conquest in synthetic chemistry, especially when considering the technologies available to his group at
the time.50-53
Kishi’s retrosynthetic analysis (Figure 1.7) features a number of well designed
transformations, including Baeyer-Villiger oxidation, intramolecular epoxide-opening cyclization, Lewis
acid mediated Diels-Alder chemistry and a Beckmann rearrangement. Central to this synthesis is Kishi’s
utilization of the bowl-like shape of the cis-decalin system to direct the stereochemistry of the subsequent
oxidation/reduction steps to complete the racemic synthesis.
H
AcHN
CH3
O
OCH3
O
ON
H3C
HO
(±)-tetrodotoxin
Baeyer-Villigeroxidation
intramolecularepoxide-opening
cyclization
Diels-Alder
Beckmann rearrangement
guanidineformation
orthoacidformation
4
4
4 4a4a
4a
4aNO
H
OO
OHOH
N
HOO
HH2N
HO
H
O
HO
HO
H
H
OAcN
OAc
OAcO
O
AcHN
AcHN OAc
4
4a
O
O NH
H
AcO OAc
O
OAc
Ac
ONH
H
AcO OAc
O
OAc
Ac
O
NH
H
AcO OAc
O
OAc
Ac
OO
O
4
4a
4
4a 6
7
6
76
7
8a
8a
8a
1.8 1.7 1.5
1.4
1.3
1.6
Figure 1.7. Kishi’s retrosynthetic analysis.
Kishi’s retrosynthetic disconnections, and those of nearly all of the other synthetic work in the
area, begin with the release of the guanidine functionality and formation of the orthoester. Woodward’s
pioneering elucidation work15
demonstrated the spontaneous ortholactonization of bridging lactones such
as 1.3. Kishi establishes both the trans-relationship between the C-6 and C-7 centers and the cis-
relationship between C-8a and C-7 via a trans-lactonization/intramolecular epoxide-opening cyclization
operation from 1.4. Seven-membered lactone 1.4 was generated through a Baeyer-Villiger oxidation
from the corresponding ketone 1.5 which derives from 1.6. The synthesis begins from cis-decalin 1.7,
which was produced from oxime 1.8 by Diels-Alder cycloaddition and Beckmann rearrangement.
8
O
OH3C
NHAc
H
NHAc
H3C
O
OH3C
O
O N
CH3
OH
SnCl4H
H3C
O
O
H3C N
OH
1) MsCl, Et3N 2) H2O, heat
4a
8a4a
8a
1.8 1.9 1.7
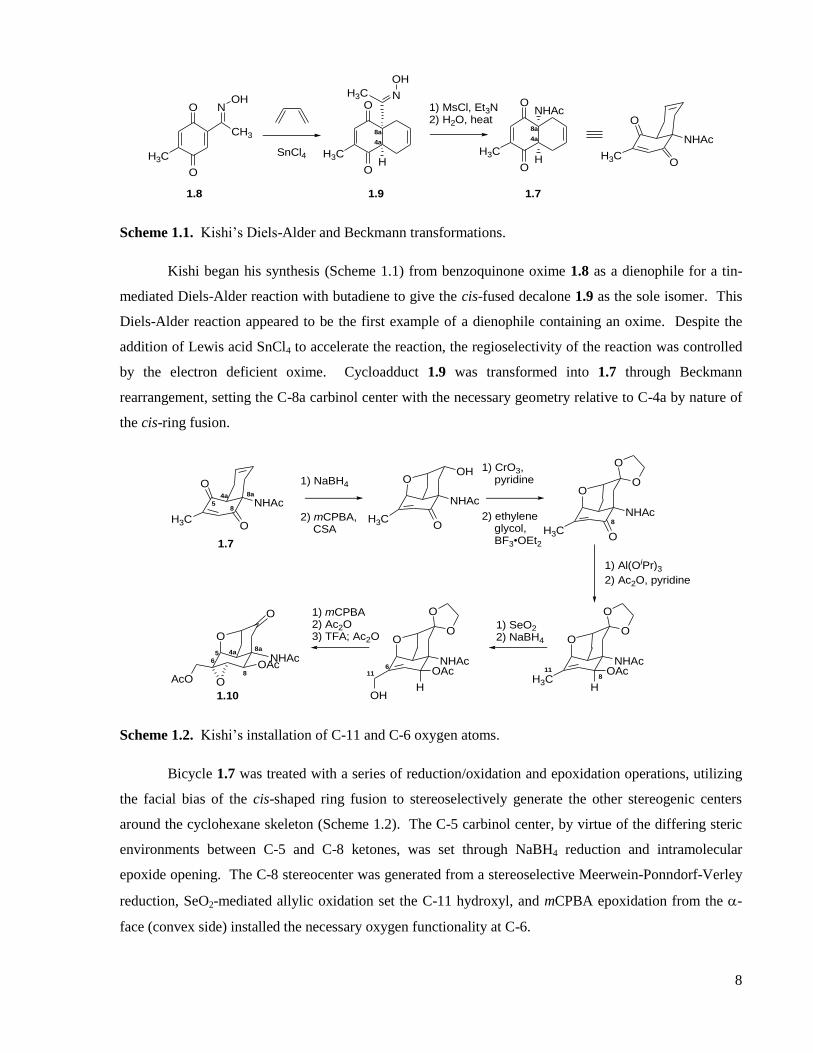
Scheme 1.1. Kishi’s Diels-Alder and Beckmann transformations.
Kishi began his synthesis (Scheme 1.1) from benzoquinone oxime 1.8 as a dienophile for a tin-
mediated Diels-Alder reaction with butadiene to give the cis-fused decalone 1.9 as the sole isomer. This
Diels-Alder reaction appeared to be the first example of a dienophile containing an oxime. Despite the
addition of Lewis acid SnCl4 to accelerate the reaction, the regioselectivity of the reaction was controlled
by the electron deficient oxime. Cycloadduct 1.9 was transformed into 1.7 through Beckmann
rearrangement, setting the C-8a carbinol center with the necessary geometry relative to C-4a by nature of
the cis-ring fusion.
O
OH3C
NHAc
1) NaBH4
2) mCPBA, CSA
NHAc
H3C
OOH
O
1) CrO3, pyridine
2) ethylene glycol, BF3•OEt2
NHAc
H3C
O
O
O
O
1) Al(OiPr)3
2) Ac2O, pyridine
NHAc
H3C
OO
O
OAc
H
1) mCPBA2) Ac2O3) TFA; Ac2O
NHAc
OO
O
OAc
H
1) SeO2 2) NaBH4
O
OAcNHAc
AcO
O
O
8a4a
58
8
8a5 4a
11
6
OH
8
8
6
1.7
1.10
11
Scheme 1.2. Kishi’s installation of C-11 and C-6 oxygen atoms.
Bicycle 1.7 was treated with a series of reduction/oxidation and epoxidation operations, utilizing
the facial bias of the cis-shaped ring fusion to stereoselectively generate the other stereogenic centers
around the cyclohexane skeleton (Scheme 1.2). The C-5 carbinol center, by virtue of the differing steric
environments between C-5 and C-8 ketones, was set through NaBH4 reduction and intramolecular
epoxide opening. The C-8 stereocenter was generated from a stereoselective Meerwein-Ponndorf-Verley
reduction, SeO2-mediated allylic oxidation set the C-11 hydroxyl, and mCPBA epoxidation from the -
face (convex side) installed the necessary oxygen functionality at C-6.

9
O
OAcNHAc
AcO
O
O1) (EtO)3CH, CSA; Ac2O
2) o-Cl2C6H4, heat O
OAcNHAc
AcO
O
OEt
1) mCPBA, K2CO3
2) AcOH, H2OO
OAcNHAc
AcO
O
O
OAc
1.10 1.11 1.5
9 9
Scheme 1.3. Installation of C-9 -acetoxy moiety.
Kishi then turned his attention to the installation of the -acetoxy moiety at C-9 (Scheme 1.3).
Treatment of 1.10 with CSA and triethyl orthoformate gave the corresponding diethyl ketal, which
eliminated after heating in dichlorobenzene to give enol ether 1.11. Another -directed stereoselective
mCPBA-mediated epoxidation generated the C-9 -acetoxy unit after opening the epoxide with aqueous
acetic acid. With 1.5 in hand, Baeyer-Villiger oxidation with mCPBA gave ring-expanded lactone 1.4
(Scheme 1.4). Lactone-ring opening of 1.4 with potassium acetate caused the resulting free-carboxylate
at C-10 to undergo an intramolecular epoxide-ring opening at C-7 giving 1.12. Acetylation of the C-6
hydroxyl moiety and thermal elimination gave 1.13 with the dihydrofuran serving as precursor to a C-4
aldehyde.
Scheme 1.4. Baeyer-Villiger oxidation and intramolecular epoxide-opening cyclization.
4
NO
H
OO
OHOH
N
HOO
HH2N
HO
HO
O
O AcO
NHAcOAc
OAcAcO
Et3OBF4, Na2CO3 O
O
O AcO
NH2OAc
OAcAcO
EtS SEt
NAc1)
2) AcNH2
3) NH3
OO
O AcO
NOAc
OAcAcO
NH2
NAc
1) OsO4 2) NaIO4 3) NH4OH
(±)-tetrodotoxin1.13 1.14 1.15
4 H
Scheme 1.5. Kishi’s (±)-tetrodotoxin end game strategy.
Scheme 1.5 outlines Kishi’s end game strategy. Treatment of 1.13 with Et3OBF4/Na2CO3
effected removal of the acetamide group afforded 1.14, and the monoacetylguanidine moiety was
installed in a three-step operation. Kishi completed the racemic tetrodotoxin synthesis in three additional
steps from 1.15. Dihydroxylation of the dihydrofuran with OsO4 followed by sodium periodate cleavage

10
of the resulting 1,2-diol unmasked the C-4 aldehyde. A final aminolysis of the acetyl groups with
NH4OH caused the formation of the orthoacid and guanidine and yielded racemic tetrodotoxin.
Kishi’s effective work in the construction of the tetrodotoxin core, and full elaboration to the
racemic natural product set a standard for three decades. Kishi’s racemic total synthesis was
accomplished in 32 linear steps and in 0.52% overall yield. Several elegant transformations were
employed, including an unusual Diels-Alder reaction with a dienophile containing an oxime. The
synthesis possesses a high degree of substrate control in the creation of the required stereocenters, all
stemming originally from cis-decalin 1.7. Kishi expertly used substrate-controlled hydride reductions,
stereospecific substrate-controlled epoxidations, carboxylate attack onto an epoxide and stereospecific
substrate-controlled epoxidation of enol ether. Kishi also developed a then-novel procedure for the
creation of the guanidine functionality that was used in several later syntheses of tetrodotoxin.

11
1.2.2 Isobe’s total synthesis and related studies
Before Isobe published his two asymmetric tetrodotoxin syntheses,54,55
his group worked on and
published the synthesis of several deoxy-analogues of tetrodotoxin (Figure 1.7).56-62
In 2003, Isobe
published the first asymmetric total synthesis of (−)-tetrodotoxin. His retrosynthetic analysis is described
in Figure 1.8 and differs significantly from not only his previous work on the deoxy-series of TTX
analogues, but also from his second asymmetric total synthesis of TTX,55
which was based upon his work
in the deoxy-series.
O
OH
O
OH
HO
NHHN
NH
HO
CH3
OH
11-deoxyTTX
O
OH
HO
NHHN
NH
HO
CH3
8,11-trideoxyTTX
O
OH
O
HO
NHHN
NH
HO
CH3
5,11-dideoxyTTX
OH
O
OH
Figure 1.7. Isobe’s deoxy tetrodotoxin analogs.
Isobe’s late stage guanidine formation and closing of the ortho acid closely mirror Kishi’s
approach (Chapter 1.2.1). Installation of the C-8 nitrogen functionality was planned to come from a
diastereoselective Overman rearrangement and the creation of the carbocycle through aldol chemistry.
12
(-)-tetrodotoxin
conjugateaddition
aldolcondensation
guanidineformation
Claisenrearrangement
Sonogashira
NO
H
OO
OHOH
N
HOO
HH2N
HO
H
O
O
O
CH3
CH3
H HN
OBOM
COCCl3AcO
BzO
O
O
CH3
CH3
H
OBOM
BzO
OH
TBDPSO
O
O
TBDPSO
BzO
OCH3
OTBS
H
H3C
O
O
OH
HO
OiPr
OTBS
H
O
O OiPr
OTBS
O
I
O OiPr
OTBS
O
O
OAc
OAc
AcO
OAc
CH3
orthoesterformation
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
O
HO H
1.171.16
1.181.191.20
1.21 1.22 1.23
TMS
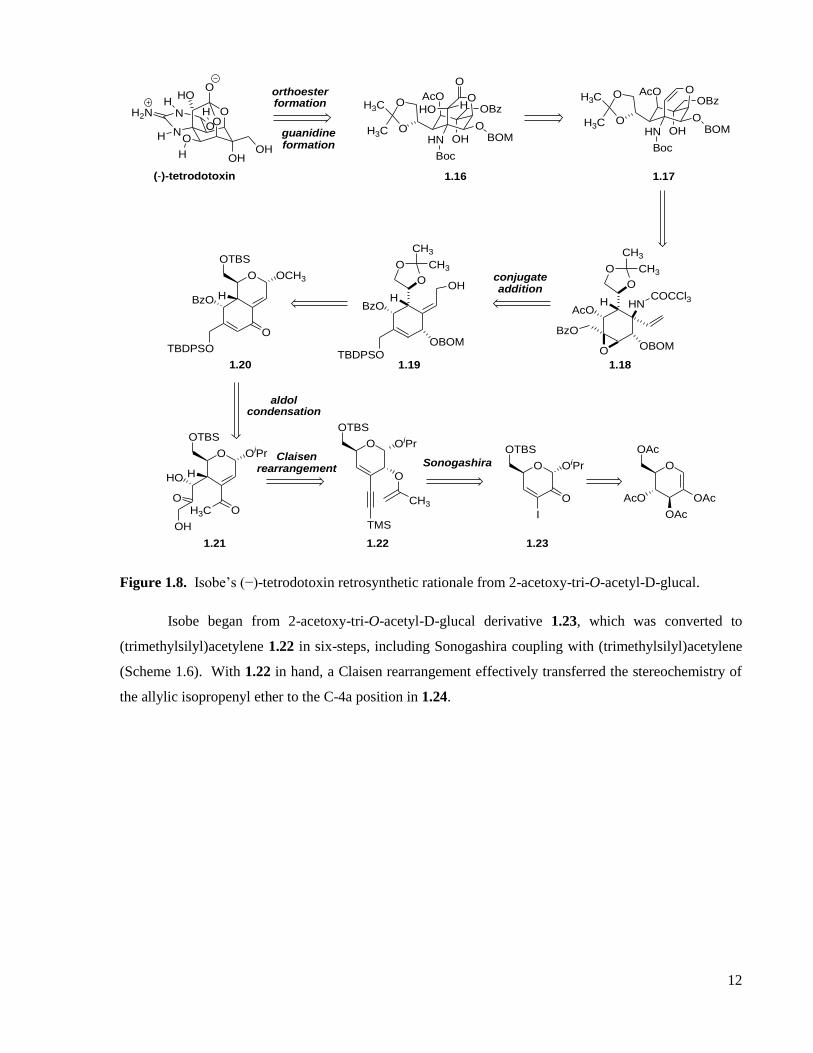
Figure 1.8. Isobe’s (−)-tetrodotoxin retrosynthetic rationale from 2-acetoxy-tri-O-acetyl-D-glucal.
Isobe began from 2-acetoxy-tri-O-acetyl-D-glucal derivative 1.23, which was converted to
(trimethylsilyl)acetylene 1.22 in six-steps, including Sonogashira coupling with (trimethylsilyl)acetylene
(Scheme 1.6). With 1.22 in hand, a Claisen rearrangement effectively transferred the stereochemistry of
the allylic isopropenyl ether to the C-4a position in 1.24.

13
O O
OH
HO iPr
1) TBSCl2) SO3•pyridine, Et3N, DMSO
3) I24) NaBH4, CeCl3
O O
OH
TBSO iPr
I
1) Pd(OAc)2
2) PPTS
H3C OCH3
Si(CH3)3O O
O
TBSO iPr
Si(CH3)3
CH3
K2CO3
o-Cl2C6H4
150 °C
O OTBSO iPr
Si(CH3)3
OH3C
1.22
1.24
4
4
4a
4a
1.23
Scheme 1.6. Isobe’s cyclohexane skeleton synthesis: Sonogashira coupling and Claisen rearrangement.
Isobe next installed the C-5 and C-11 hydroxyl groups through a series of regioselective
enolizations (Scheme 1.7). Compound 1.24 was trapped as the terminal silyl enol ether, and then treated
with lead tetraacetate before deprotections of the C-11 acetate and the trimethylsilyl group gave 1.25.
Enolization of the ketone toward C-5 proved difficult; thus, oxidation of the C-11 hydroxyl allowed for
MOM-trapping of the corresponding (and readily enolizable) -keto aldehyde, which was then reduced
with NaBH4/CeCl3 to 1.26. Epoxidation of 1.26 and treatment of the resulting products with acidic
Amberlyst 15 ion-exchange resin gave dihydroxylacetone 1.21 in a 7:1 ratio of diastereomers at C-5.
Oxymercuration and protecting group adjustments gave compound 1.27 as a precursor to intramolecular
aldol ring closing.
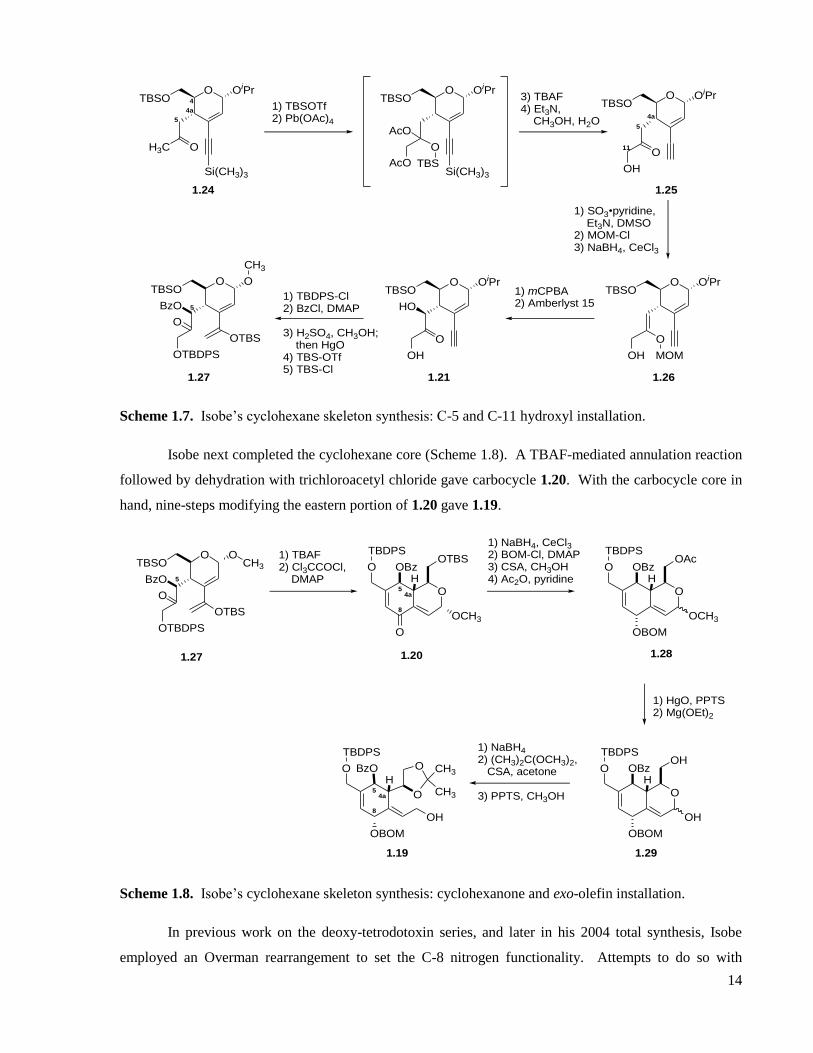
14
O OiPrTBSO
Si(CH3)3
OH3C
1) TBSOTf2) Pb(OAc)4
O OiPrTBSO
Si(CH3)3
O
TBS
AcO
AcO
3) TBAF4) Et3N, CH3OH, H2O
O OiPrTBSO
O
OH
O OiPrTBSO
O
OH
1) SO3•pyridine, Et3N, DMSO2) MOM-Cl3) NaBH4, CeCl3
MOM
1) mCPBA2) Amberlyst 15
O OiPrTBSO
O
OH
1) TBDPS-Cl2) BzCl, DMAP
3) H2SO4, CH3OH; then HgO4) TBS-OTf5) TBS-Cl
HO
4
4a
1.24 1.25
5
4a
5
1.261.211.27
O OTBSO
CH3
O
OTBDPS
BzO
OTBS
5
11
Scheme 1.7. Isobe’s cyclohexane skeleton synthesis: C-5 and C-11 hydroxyl installation.
Isobe next completed the cyclohexane core (Scheme 1.8). A TBAF-mediated annulation reaction
followed by dehydration with trichloroacetyl chloride gave carbocycle 1.20. With the carbocycle core in
hand, nine-steps modifying the eastern portion of 1.20 gave 1.19.
O OTBSO CH3
O
OTBDPS
BzO
OTBS
1) TBAF2) Cl3CCOCl, DMAP
O
HOBz
OCH3
O
O
TBDPSOTBS
1) NaBH4, CeCl32) BOM-Cl, DMAP3) CSA, CH3OH4) Ac2O, pyridine
OH
HBzO
OBOM
O
TBDPS
O
O CH3
CH3
O
HOBz
OCH3
OBOM
O
TBDPSOAc
O
HOBz
OH
OBOM
O
TBDPSOH
1) HgO, PPTS2) Mg(OEt)2
1) NaBH4
2) (CH3)2C(OCH3)2, CSA, acetone
3) PPTS, CH3OH
5
1.27 1.20 1.28
1.291.19
54a
8
54a
8
Scheme 1.8. Isobe’s cyclohexane skeleton synthesis: cyclohexanone and exo-olefin installation.
In previous work on the deoxy-tetrodotoxin series, and later in his 2004 total synthesis, Isobe
employed an Overman rearrangement to set the C-8 nitrogen functionality. Attempts to do so with

15
compound 1.19 failed repeatedly. To overcome this, Isobe introduced the nitrogen functionality through
an intramolecular conjugate addition reaction (Scheme 1.9). Primary alcohol 1.19 was converted to -
unsaturated methyl ester 1.30 in four steps. The C-5 hydroxyl group was then converted to the
corresponding carbamate in two steps with trichloroacetyl isocyanate, and treatment with KOtBu gave
bicycle 1.31. The C-5 stereocenter effectively controls the geometry at C-8a when nitrogen is installed.
The cyclic carbamate of 1.32 was hydrolyzed to give cyclohexene 1.33.
1) DIBAL-H2) TEMPO, NCS
3) NaClO2, NaH2PO4, (CH3)2C=CHCH3
4) (CH3)3Si-CHN2
1) Cl3CCONCO
2) Et3N, CH3OH
3) KOtBuOBOM
NH
CO2CH3
O O
H3C CH3
TBDPSO
O
H
O
H
1) LiBH4
2) MMTr-Cl
OBOM
NH
O O
H3C CH3
TBDPSO
O
H
O
H
OMMTr
1) Boc2O, Et3N, DMAP2) LiOH, CH3OHHO
HOH
NHBoc
BOMO
OMMTr
O
OCH3
CH3
1.19
1.33 1.32
1.30 1.31
5
8a
5
8a
5
8a
8a
5
OH
HBzO
OBOM
O
TBDPS
O
O CH3
CH35
4a
8CO2CH3
HHO
OBOM
O
TBDPS
O
O CH3
CH3
Scheme 1.9. Isobe’s introduction of nitrogen through intramolecular conjugate addition.
Stereoselective lactone formation and inversion at C-5 were then addressed (Scheme 1.10).
Seven steps were taken to convert 1.33 to 1.34 in anticipation of the 6-exo-tet epoxide ring-opening.
When compound 1.34 was treated with DBU in dichlorobenzene at 130 °C, the cyclic vinyl ether 1.17
was generated under stereoelectronic control, presumably though intermediate Z-enolate 1.35. Successive
oxidations with OsO4 and IBX gave -ketolactone 1.37. Reduction of 1.37 with NaBH4 provided 1.16 as
the sole diastereomer; the axial C-5 acetoxy group provided severe steric hindrance, forcing hydride
reduction from the front face. Bicyclic cyclohexane 1.33 contained the necessary functional features and
proper configurations for (−)-tetrodotoxin, and Isobe at this point focused on the introduction of the
guanidine and the completion of the total synthesis.
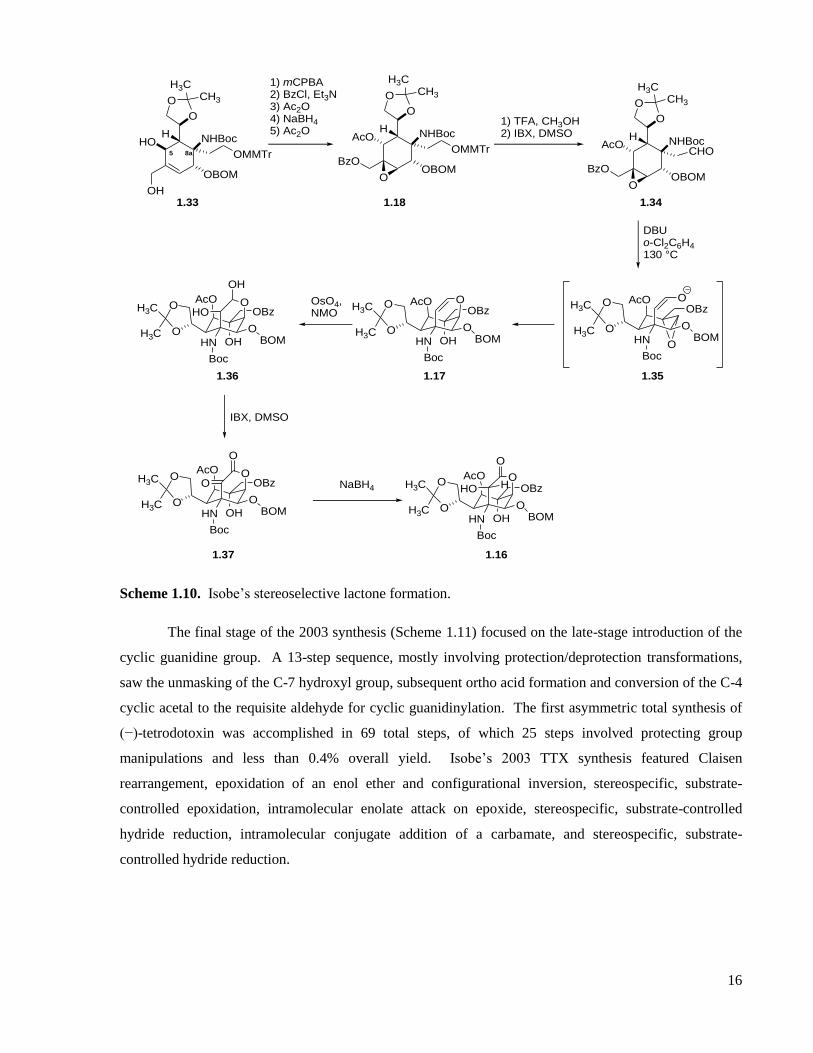
16
1) mCPBA2) BzCl, Et3N3) Ac2O4) NaBH4
5) Ac2O
OH
HHO NHBoc
OBOM
OMMTr
O
O
H3CCH3
BzO
HAcO NHBoc
OBOM
OMMTr
O
O
H3CCH3
O
1) TFA, CH3OH2) IBX, DMSO
BzO
HAcO NHBoc
OBOM
CHO
O
O
H3CCH3
O
DBUo-Cl2C6H4
130 °C
AcO
OHN
Boc
OOBz
O
OH3C
H3C
OBOM
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
OsO4, NMO
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
OH
HO
IBX, DMSO
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
O
O NaBH4
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
O
HO H
1.33 1.18 1.34
1.17
1.161.37
1.36 1.35
5 8a
Scheme 1.10. Isobe’s stereoselective lactone formation.
The final stage of the 2003 synthesis (Scheme 1.11) focused on the late-stage introduction of the
cyclic guanidine group. A 13-step sequence, mostly involving protection/deprotection transformations,
saw the unmasking of the C-7 hydroxyl group, subsequent ortho acid formation and conversion of the C-4
cyclic acetal to the requisite aldehyde for cyclic guanidinylation. The first asymmetric total synthesis of
(−)-tetrodotoxin was accomplished in 69 total steps, of which 25 steps involved protecting group
manipulations and less than 0.4% overall yield. Isobe’s 2003 TTX synthesis featured Claisen
rearrangement, epoxidation of an enol ether and configurational inversion, stereospecific, substrate-
controlled epoxidation, intramolecular enolate attack on epoxide, stereospecific, substrate-controlled
hydride reduction, intramolecular conjugate addition of a carbamate, and stereospecific, substrate-
controlled hydride reduction.
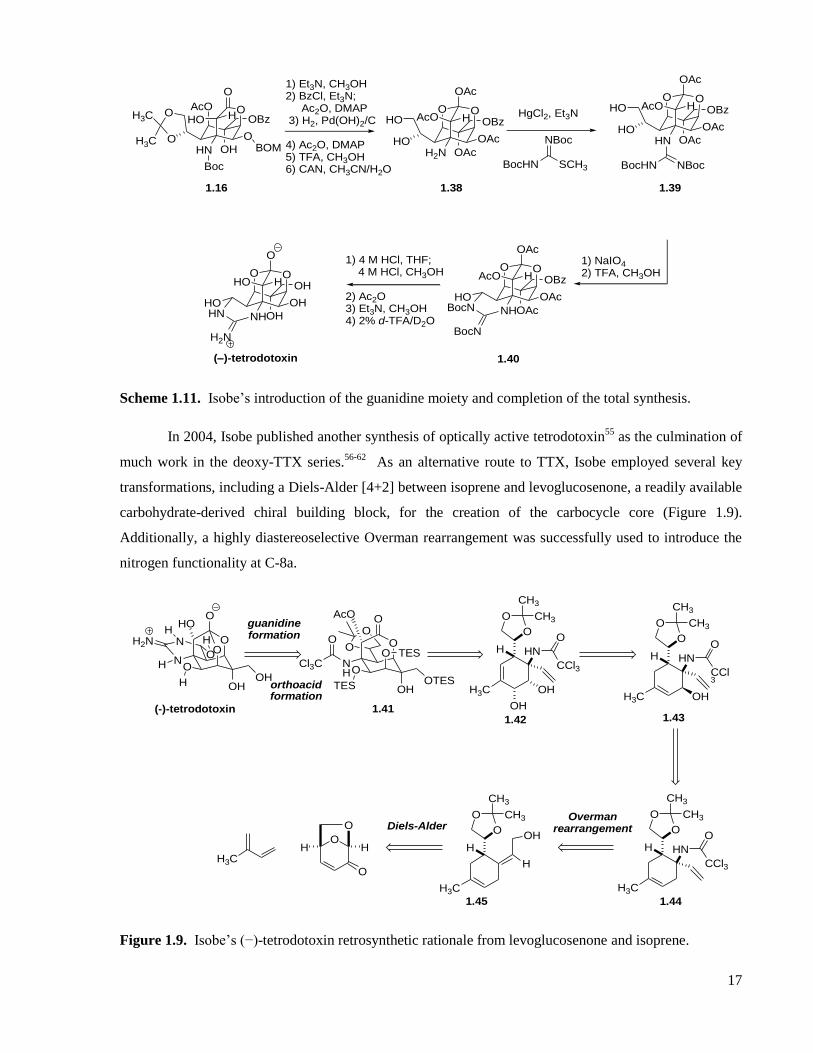
17
1) Et3N, CH3OH 2) BzCl, Et3N; Ac2O, DMAP 3) H2, Pd(OH)2/C 4) Ac2O, DMAP 5) TFA, CH3OH 6) CAN, CH3CN/H2O
AcO
OHHN
Boc
OOBz
O
OH3C
H3C
OBOM
O
HO HO
OAcH2N
OOBzHO
HO OAc
OAc
AcO H
BocHN
NBoc
SCH3
HgCl2, Et3N
O
OAcHN
OOBzHO
HO OAc
OAc
AcO H
NBocBocHN
1) NaIO4
2) TFA, CH3OHO
OAcNH
OOBz
OAc
OAc
AcO H
BocN
BocNHO
1) 4 M HCl, THF; 4 M HCl, CH3OH
2) Ac2O3) Et3N, CH3OH4) 2% d-TFA/D2O
O
OHNH
OOH
OH
O
HO H
H2N
HNHO
()-tetrodotoxin
1.16 1.38
1.40
1.39
Scheme 1.11. Isobe’s introduction of the guanidine moiety and completion of the total synthesis.
In 2004, Isobe published another synthesis of optically active tetrodotoxin55
as the culmination of
much work in the deoxy-TTX series.56-62
As an alternative route to TTX, Isobe employed several key
transformations, including a Diels-Alder [4+2] between isoprene and levoglucosenone, a readily available
carbohydrate-derived chiral building block, for the creation of the carbocycle core (Figure 1.9).
Additionally, a highly diastereoselective Overman rearrangement was successfully used to introduce the
nitrogen functionality at C-8a.
Overmanrearrangement
H3C
O
O
CH3
CH3
H
H
OHO
O
O
H H
Diels-Alder
H3C
H3C
O
O
CH3
CH3
H HN
O
CCl3
H3C
O
O
CH3
CH3
H HN
O
CCl3
OHH3C
O
O
CH3
CH3
H HN
O
CCl3
OH
OH(-)-tetrodotoxin
NO
H
OO
OHOH
N
HOO
HH2N
HO
H
guanidineformation
orthoacidformation
1.411.42 1.43
1.441.45
NHO
TES
Cl3C
O O
O
O TES
OHOTES
O
O
AcO
Figure 1.9. Isobe’s (−)-tetrodotoxin retrosynthetic rationale from levoglucosenone and isoprene.
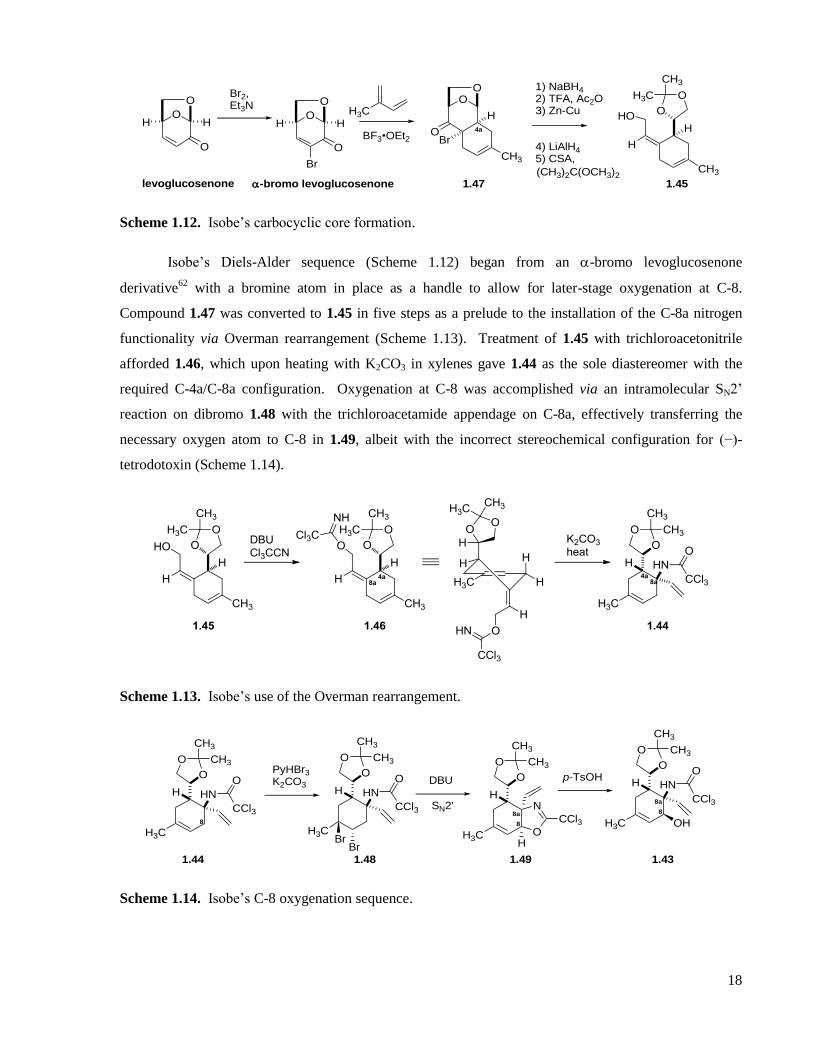
18
O
O
O
H HO
O
O
H H
Br
H3C
BF3•OEt2
O
CH3
O
O
Br
H
CH3
O
O
H3C
CH3
H
H
HO
1) NaBH4
2) TFA, Ac2O3) Zn-Cu
4) LiAlH4
5) CSA,
Br2,Et3N
levoglucosenone -bromo levoglucosenone 1.47 1.45
4a
(CH3)2C(OCH3)2
Scheme 1.12. Isobe’s carbocyclic core formation.
Isobe’s Diels-Alder sequence (Scheme 1.12) began from an -bromo levoglucosenone
derivative62
with a bromine atom in place as a handle to allow for later-stage oxygenation at C-8.
Compound 1.47 was converted to 1.45 in five steps as a prelude to the installation of the C-8a nitrogen
functionality via Overman rearrangement (Scheme 1.13). Treatment of 1.45 with trichloroacetonitrile
afforded 1.46, which upon heating with K2CO3 in xylenes gave 1.44 as the sole diastereomer with the
required C-4a/C-8a configuration. Oxygenation at C-8 was accomplished via an intramolecular SN2’
reaction on dibromo 1.48 with the trichloroacetamide appendage on C-8a, effectively transferring the
necessary oxygen atom to C-8 in 1.49, albeit with the incorrect stereochemical configuration for (−)-
tetrodotoxin (Scheme 1.14).
Scheme 1.13. Isobe’s use of the Overman rearrangement.
H3C
O
O
CH3
CH3
H HN
O
CCl3
PyHBr3
K2CO3
H3C
O
O
CH3
CH3
H HN
O
CCl3
BrBr
DBU
SN2'
H3C
O
O
CH3
CH3
H
O
N
CCl3
H
p-TsOH
H3C
O
O
CH3
CH3
H HN
O
CCl3
OH8 8
8
1.44 1.48 1.49 1.43
8a
8a
Scheme 1.14. Isobe’s C-8 oxygenation sequence.
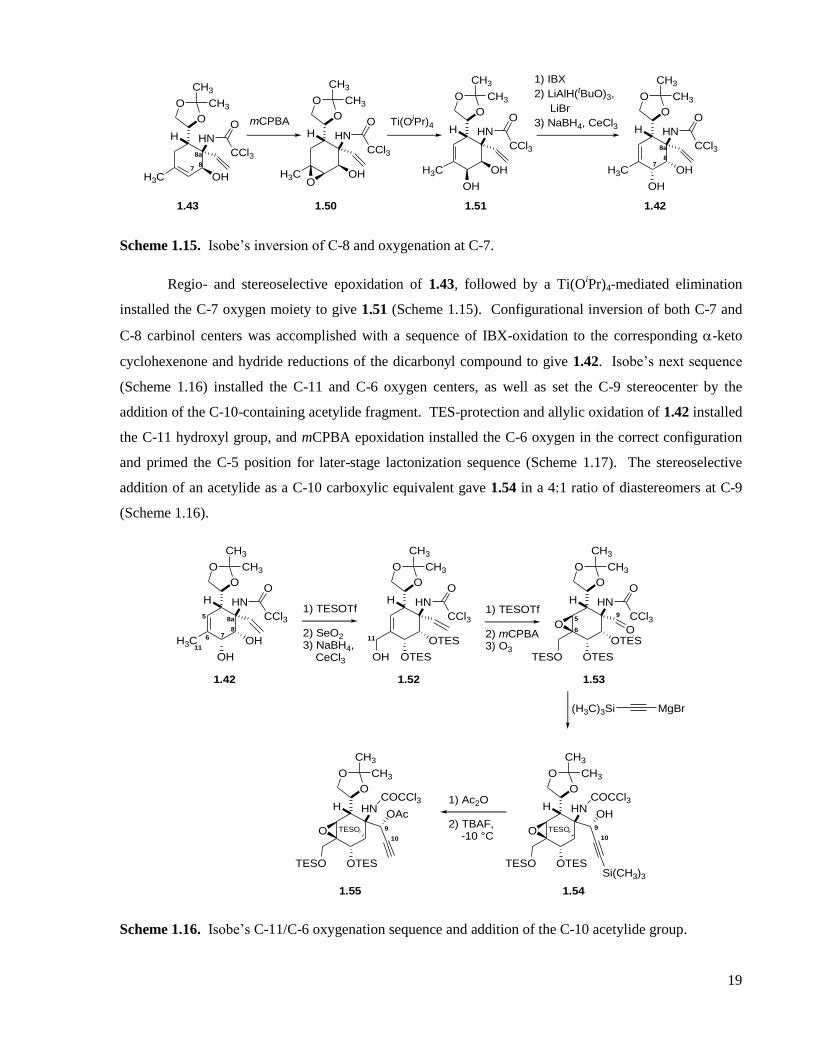
19
H3C
O
O
CH3
CH3
H HN
O
CCl3
OH
mCPBA
H3C
O
O
CH3
CH3
H HN
O
CCl3
OHO
H3C
O
O
CH3
CH3
H HN
O
CCl3
OH
OH
Ti(OiPr)4
H3C
O
O
CH3
CH3
H HN
O
CCl3
OH
OH
1) IBX
2) LiAlH(tBuO)3,
LiBr
3) NaBH4, CeCl3
78
1.43 1.50 1.51 1.42
8a8
8a
7
Scheme 1.15. Isobe’s inversion of C-8 and oxygenation at C-7.
Regio- and stereoselective epoxidation of 1.43, followed by a Ti(OiPr)4-mediated elimination
installed the C-7 oxygen moiety to give 1.51 (Scheme 1.15). Configurational inversion of both C-7 and
C-8 carbinol centers was accomplished with a sequence of IBX-oxidation to the corresponding -keto
cyclohexenone and hydride reductions of the dicarbonyl compound to give 1.42. Isobe’s next sequence
(Scheme 1.16) installed the C-11 and C-6 oxygen centers, as well as set the C-9 stereocenter by the
addition of the C-10-containing acetylide fragment. TES-protection and allylic oxidation of 1.42 installed
the C-11 hydroxyl group, and mCPBA epoxidation installed the C-6 oxygen in the correct configuration
and primed the C-5 position for later-stage lactonization sequence (Scheme 1.17). The stereoselective
addition of an acetylide as a C-10 carboxylic equivalent gave 1.54 in a 4:1 ratio of diastereomers at C-9
(Scheme 1.16).
H3C
O
O
CH3
CH3
H HN
O
CCl3
OH
OH
1) TESOTf
2) SeO2
3) NaBH4, CeCl3
O
O
CH3
CH3
H HN
O
CCl3
OTES
OTESOH
1) TESOTf
2) mCPBA3) O3
O
O
O
CH3
CH3
H HN
O
CCl3
OTES
OTESTESO
O
O
O
CH3
CH3
H HN
TESO
OTESTESO
O
(H3C)3Si MgBr
Si(CH3)3
OH
COCCl31) Ac2O
2) TBAF, -10 °C
O
O
CH3
CH3
H HN
TESO
OTESTESO
O
OAc
COCCl3
8
8a
7
11
5
6 11
5
6
9
9
10
9
10
1.42 1.52 1.53
1.541.55
Scheme 1.16. Isobe’s C-11/C-6 oxygenation sequence and addition of the C-10 acetylide group.

20
The cleavage of the acetylenic unit was accomplished in a two-step procedure starting with
treatment of 1.55 with KMnO4 and sodium periodate (Scheme 1.17). To affect an epoxide-opening
cyclization, -keto acid 1.56 was cleaved with alkaline hydrogen peroxide to furnish 1.57, with the
required C-5 oxygen moiety in place as part of the lactone bridge. Re-protection of 1.57 lead to 1.41.
This epoxide opening sequence was quite reminiscent of Isobe’s 2003 total synthesis54
(Scheme 1.10) in
that the C-10 unit attack at C-5 closed the lactone while setting C-5 or C-7.
O
O
CH3
CH3
H HN
TESO
OTESTESO
O
OAc
COCCl3KMnO4, NaIO4
O
O
CH3
CH3
H HN
TESO
OTESTESO
O
OAc
COCCl3
COOHO
O
O
CH3
CH3
H HN
TESO
OTESTESO
O
OAc
COCCl3
OO
H2O2, NaHCO3
1) TESOTf -40 °C2) Ac2O
NHO
TES
Cl3C
O O
O
O TES
OHOH
O
O
HO
NHO
TES
Cl3C
O O
O
O TES
OHOTES
O
O
AcO
5
1.55 1.56
1.41 1.57
10
5
10
10
5
Scheme 1.17. Isobe’s epoxide-opening cyclization.
Isobe constructed the ortholactone (Scheme 1.18) by TBAF removal of the silyl groups of 1.41
followed by spontaneous ortho acid formation with the newly released C-7 oxygen. Global protection of
the hydroxyl groups with acetic anhydride lead to 1.59, and HIO4-mediated cleavage of the acetonide
yielded the C-4a aldehyde which was protected as the dimethylacetal 1.60. Mixed acetal 1.61 was
generated in a 6:1 ratio of diastereomers at C-8a following selective deprotection of C-9 and C-10 acetate
groups and silyl protection of the ortho acid. Reduction of the trichloroacetamide unit allowed for facile
introduction of the guanidine moiety in a similar manner to both Kishi’s 1972 racemic synthesis (Scheme
1.5) and Isobe’s 2003 synthesis (Scheme 1.11).
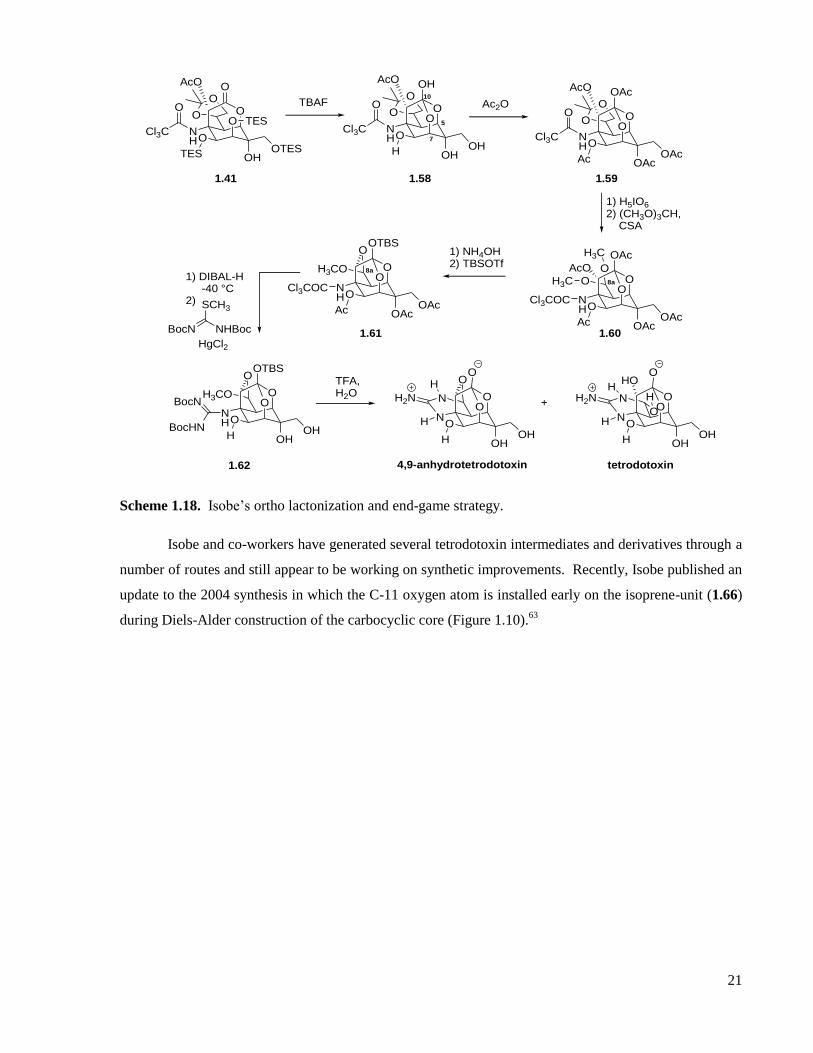
21
NHO
TES
Cl3C
O O
O
O TES
OHOTES
O
O
AcO
TBAF
NHO
H
Cl3C
O O
OH
O
OHOH
O
O
AcO
Ac2O
NHO
Ac
Cl3C
O O
OAc
O
OAcOAc
O
O
AcO
1) H5IO6
2) (CH3O)3CH, CSA
NHO
Ac
Cl3COC
OO
OAcOAc
OOAcO
OAc1) NH4OH2) TBSOTf
NHO
Ac
Cl3COC
OO
OAcOAc
H3CO
OOTBS
10
H3C
7
5
8a
8a
1) DIBAL-H -40 °C2)
BocN
SCH3
NHBoc
TFA, H2O
NHO
H
OO
OHOH
H3CO
OOTBS
BocHN
BocN
NO
H
OO
OHOH
N
OO
H
H2N
H
4,9-anhydrotetrodotoxin
+
NO
H
OO
OHOH
N
HOO
HH2N
H
tetrodotoxin
O
H
HgCl2
1.41 1.58 1.59
1.601.61
1.62
H3C
Scheme 1.18. Isobe’s ortho lactonization and end-game strategy.
Isobe and co-workers have generated several tetrodotoxin intermediates and derivatives through a
number of routes and still appear to be working on synthetic improvements. Recently, Isobe published an
update to the 2004 synthesis in which the C-11 oxygen atom is installed early on the isoprene-unit (1.66)
during Diels-Alder construction of the carbocyclic core (Figure 1.10).63

22
H3C
O
O
CH3
CH3
H HN
O
CCl3
OH7
8
1.43
8a
O
O
CH3
CH3
H HN
O
CCl3
OH7
8
1.63
8a
HO
1111
O
O
CH3
CH3
H HN
O
CCl3
78
1.64
8a
HO
11
O
O
CH3
CH3
H
78
1.65
8a
HO
11
OH
OR
OO
Br
O
+
Diels-Alder
Overman rearrangement
C-8hydroxylation
-bromo levoglucosenone1.66
Figure 1.10. Isobe’s updated C-11 hydroxylation and comparison to 1.43.

23
1.2.3 Du Bois’ total synthesis
Justin Du Bois published an impressive stereoselective synthesis of (−)-tetrodotoxin64
shortly
after Isobe. This route to (−)-TTX (Figure 1.11) followed a similar late-stage guanidine and orthoacid
formation as did Kishi (Figure 1.7) and Isobe (Figures 1.8, 1.9), but ingeniously utilized two C-H
activation steps to install the two tetrasubstituted centers C-6 and C-8a. The installation of the nitrogen
functionality at C-8a through a nitrene insertion reaction into the C-8a C-H bond is unique. An
intramolecular rhodium carbenoid C-H insertion reaction at C-6 was used in the construction of the
carbocycle, a general strategy also employed by Taber in his work (Chapter 1.2.9) on the tetrodotoxin
core.
O
OHNH
OOH
OH
O
HO H
H2N
HNHO
()-tetrodotoxin
O
OH
OO
O
O
O H
CH3
CH3
CH3
CH3
stereospecificC-H amination
H2N
O
Cl
O
OO
O
CH2
H
CH3
H3C
O(H3C)2N
PivO
H3CCH3
stereospecificRh-catalyzedC-H insertion
methylenation
O
OPivO
TBSO
O
N2
O
O
H3C
CH3
HBnO
O
O O
OBn
TBSOO
O
CH3
CH3
HOHC+
orthoacid formation
guanidine formation
6
5
8a
4a
9
8a
6
58a
9
9
4a8
8a49
10
10
6
8
10
9
4
4a
6
aldol
1.72
1.71 1.70 1.69
1.67 1.68
OO
O
H3C CH3O
O
CH3H3C
O
HO
H2N
Figure 1.11. Du Bois’ tetrodotoxin retrosynthetic analysis.
Du Bois began his synthesis from chiral aldehyde 1.71, which is a readily available derivative of
D-isoascorbic acid (Scheme 1.19). A sodium acetate-mediated aldol reaction with dibenzyloxalacetate
1.72 and chiral aldehyde 1.71 provided 1.73, forming the new C-8 to C-8a carbon-carbon bond.
Compound 1.73 was converted into diazoketone 1.70 to test the rhodium-catalyzed C-6 carbene C-H
insertion. Reaction conditions were screened, and reaction with 1.70 and 1.5 mol % Rh2(NHCOCPh3)4
resulted in the production of cyclic ketone 1.75 as the only detected product. The configuration of the C-
9 and C-4a stereocenters were set by exploiting the shape of bicyclic 1.75. Reduction of the C-4a
carbonyl occurred from the less sterically crowded convex face, as did hydrogenation of the C-8a/C-9
olefin. Acetonide-protection gave compound 1.76, which was oxidatively opened at the lactone, and then
methylenated in anticipation of C-5 oxygenation (Scheme 1.20).
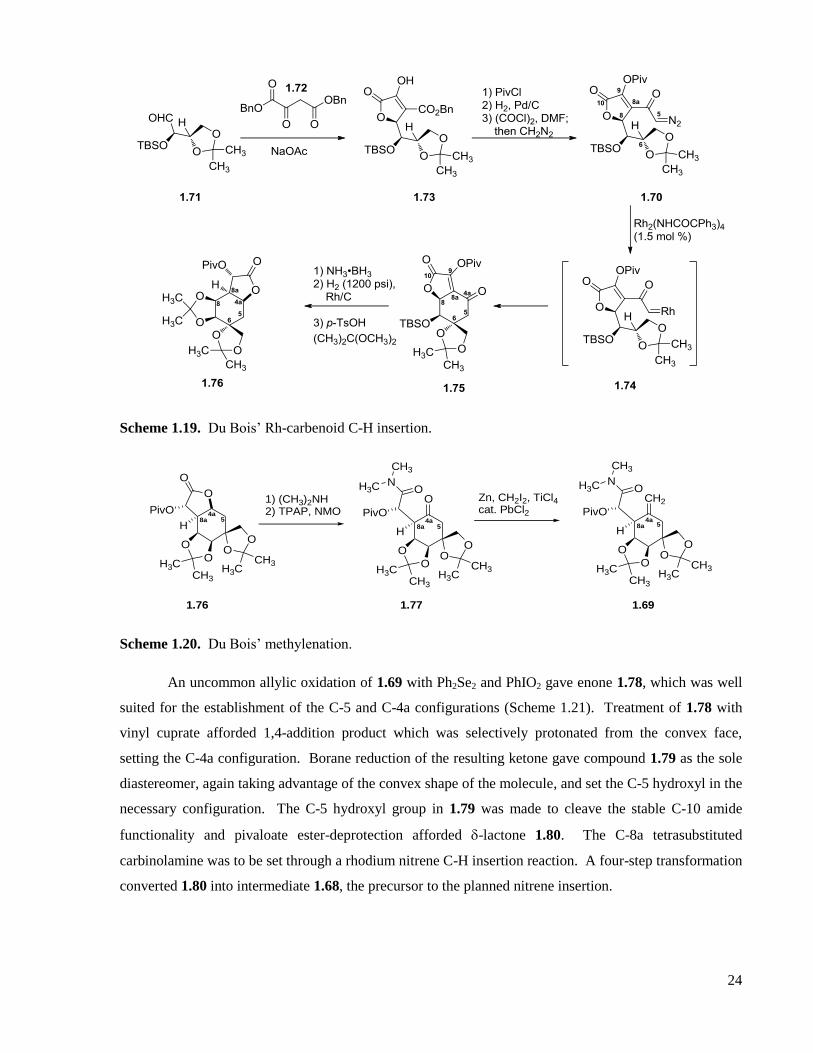
24
Scheme 1.19. Du Bois’ Rh-carbenoid C-H insertion.
1) (CH3)2NH2) TPAP, NMO
Zn, CH2I2, TiCl4cat. PbCl2
OO
O
O
O
PivO
CH3H3C
CH3
H3C
H
CH2
NH3C
CH3
O
OO
O
O
O
PivO
CH3H3C
CH3
H3C
H
1.76
8a 54a
1.77 1.69
8a4a
OO
O
O
O
PivO
CH3H3C
CH3
H3C
H
O
NH3C
CH3
8a4a
55
Scheme 1.20. Du Bois’ methylenation.
An uncommon allylic oxidation of 1.69 with Ph2Se2 and PhIO2 gave enone 1.78, which was well
suited for the establishment of the C-5 and C-4a configurations (Scheme 1.21). Treatment of 1.78 with
vinyl cuprate afforded 1,4-addition product which was selectively protonated from the convex face,
setting the C-4a configuration. Borane reduction of the resulting ketone gave compound 1.79 as the sole
diastereomer, again taking advantage of the convex shape of the molecule, and set the C-5 hydroxyl in the
necessary configuration. The C-5 hydroxyl group in 1.79 was made to cleave the stable C-10 amide
functionality and pivaloate ester-deprotection afforded -lactone 1.80. The C-8a tetrasubstituted
carbinolamine was to be set through a rhodium nitrene C-H insertion reaction. A four-step transformation
converted 1.80 into intermediate 1.68, the precursor to the planned nitrene insertion.
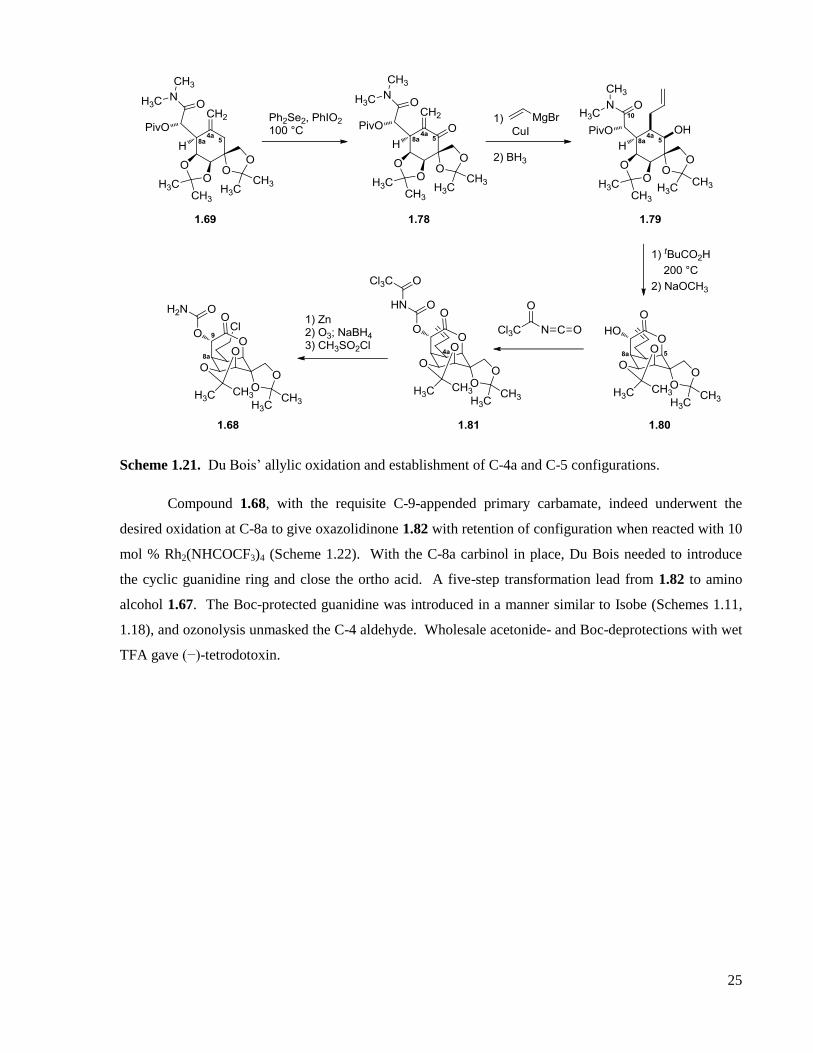
25
Scheme 1.21. Du Bois’ allylic oxidation and establishment of C-4a and C-5 configurations.
Compound 1.68, with the requisite C-9-appended primary carbamate, indeed underwent the
desired oxidation at C-8a to give oxazolidinone 1.82 with retention of configuration when reacted with 10
mol % Rh2(NHCOCF3)4 (Scheme 1.22). With the C-8a carbinol in place, Du Bois needed to introduce
the cyclic guanidine ring and close the ortho acid. A five-step transformation lead from 1.82 to amino
alcohol 1.67. The Boc-protected guanidine was introduced in a manner similar to Isobe (Schemes 1.11,
1.18), and ozonolysis unmasked the C-4 aldehyde. Wholesale acetonide- and Boc-deprotections with wet
TFA gave (−)-tetrodotoxin.

26
Rh2(NHCOCF3)4
(10 mol%),PhI(OAc)2, MgO O
O
O
H3C CH3O
O
CH3H3C
O
OCl
N
O
H
1) NaSePh2) mCPBA3) Boc2O, DMAP4) K2CO3, CH3OH5) H2O, 110 °C
OO
O
H3C CH3O
O
CH3H3C
O
HO
H2N
BocHN
NBoc
SCH3
HgCl2, Et3N
OO
O
H3C CH3O
O
CH3H3C
O
HO
NBocN
NHBoc
H
O3; (CH3)2Sthen aq. TFAO
O
HO
OHOH
OHO
ON
H2NHN
H
H
()-tetrodotoxin
OO
O
H3C CH3O
O
CH3H3C
O
O
OH2N
Cl
8a
9
1.68 1.82 1.67
1.83
Scheme 1.22. Du Bois’ Rh-catalyzed nitrene C-H insertion and guanidine formation.
Du Bois completed his synthesis of (−)-tetrodotoxin in 28 linear steps from known aldehyde 1.71
with 0.96% overall yield. This work demonstrated an alternative approach to the Diels-Alder chemistry
used to build the six-member carbon framework. Du Bois utilized C-H functionalization as a central
feature of his approach, leading to a short and concise synthesis relative to other tetrodotoxin syntheses.
The mild conditions for the C-H functionalization reactions, along with other transformations including
diastereoselective aldol addition, substrate-controlled hydrogenation, and substrate-controlled hydride
reduction, as well as the relative lack of protection/deprotection transformations clearly mark this
synthesis as an impressive achievement.

27
1.2.4 Sato’s total syntheses
In 2005, Ken-Ichi Sato published a racemic synthesis of tetrodotoxin starting from myo-inositol.65
His novel and stereocontrolled synthesis involved the typical orthoesterification/guanidine formation as
the end-game strategy and the central feature of his route was the formation of a spiro -chloroepoxide
and subsequent azide ion-mediated ring-opening (Figure 1.12).
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
(±)-tetrodotoxin
NH2
OO O
OO
O
OH
H3CCH3
C
N
orthoacidformation
OH
OH
HO
HOHO
OH
HOOH
OH
OH
OH OH
OO O
CHO
OO
H3CCH3
CH3H3C
N3
OTBDPS
guanidine formation
azide ring opening
spiro -chloroepoxide
formation
OO
CH3H3C
OO
O
H3C CH3
OTBDPS
O
Cl
myo-inositol
NH
OO OH
OO
O
OMOM
H3CCH3
H3CH3C O
NBoc
NHBoc
1.84 1.85
MOM
TBDPS
1.861.87
MOMMOM
Figure 1.12. Sato’s retrosynthetic analysis for (±)-tetrodotoxin from myo-inositol.
To begin, Sato orthogonally protected the secondary hydroxyl groups of myo-inositol in a seven-
step transformation to give compound 1.88 (Scheme 1.23). Another 10-step sequence converted 1.88 to
compound 1.89, with the C-4a ketone in place to introduce the C-4 carbon fragment. A Peterson
olefination/hydroboration operation and subsequent TBDPS-protection/C-8a oxidation afforded 1.90 with
the requisite C-8a ketone in place to introduce the C-9 carbon unit and set up for the key spiro -
chloroepoxide formation. An approach to advanced tetrodotoxin intermediates using the spiro -
chloroepoxide as a means of installing the C-8a nitrogen and the C-9 carbon center was worked on by
Sato in the early 1990’s66
(Scheme 1.30).
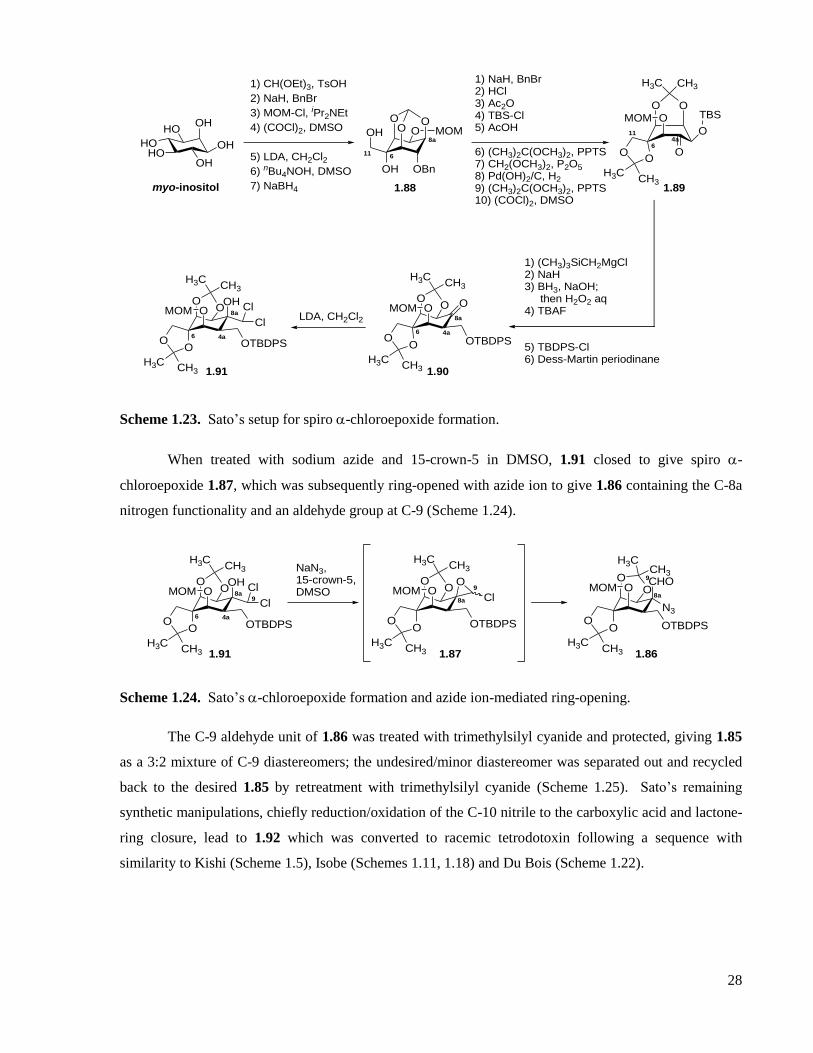
28
OH
OH
HO
HOHO
OH
OO O
O
OH OBn
MOMOH
1) CH(OEt)3, TsOH
2) NaH, BnBr
3) MOM-Cl, iPr2NEt
4) (COCl)2, DMSO
5) LDA, CH2Cl26) nBu4NOH, DMSO
7) NaBH4
1) NaH, BnBr2) HCl3) Ac2O4) TBS-Cl5) AcOH
6) (CH3)2C(OCH3)2, PPTS7) CH2(OCH3)2, P2O5
8) Pd(OH)2/C, H2
9) (CH3)2C(OCH3)2, PPTS10) (COCl)2, DMSO
OOO
H3CCH3
O O
H3C CH3
OMOM
O
TBS
1) (CH3)3SiCH2MgCl2) NaH3) BH3, NaOH; then H2O2 aq4) TBAF
5) TBDPS-Cl6) Dess-Martin periodinane
OO
CH3H3C
OO
O
H3CCH3
OTBDPS
OH
Cl
OO
CH3H3C
OO
O
H3CCH3
OTBDPS
OMOMLDA, CH2Cl2
MOM Cl
myo-inositol
11
11
68a
8a
4a6
6
8a
4a6
4a
1.88 1.89
1.901.91
Scheme 1.23. Sato’s setup for spiro -chloroepoxide formation.
When treated with sodium azide and 15-crown-5 in DMSO, 1.91 closed to give spiro -
chloroepoxide 1.87, which was subsequently ring-opened with azide ion to give 1.86 containing the C-8a
nitrogen functionality and an aldehyde group at C-9 (Scheme 1.24).
OO
CH3H3C
OO
O
H3CCH3
OTBDPS
O
ClMOM
NaN3,15-crown-5,DMSO
OO O
CHO
OO
CH3H3C
CH3
H3C
N3
OTBDPS
MOM
OO
CH3H3C
OO
O
H3CCH3
OTBDPS
OH
ClMOM Cl
1.91
8a
4a6
1.87 1.86
8a8a
9
9
9
Scheme 1.24. Sato’s -chloroepoxide formation and azide ion-mediated ring-opening.
The C-9 aldehyde unit of 1.86 was treated with trimethylsilyl cyanide and protected, giving 1.85
as a 3:2 mixture of C-9 diastereomers; the undesired/minor diastereomer was separated out and recycled
back to the desired 1.85 by retreatment with trimethylsilyl cyanide (Scheme 1.25). Sato’s remaining
synthetic manipulations, chiefly reduction/oxidation of the C-10 nitrile to the carboxylic acid and lactone-
ring closure, lead to 1.92 which was converted to racemic tetrodotoxin following a sequence with
similarity to Kishi (Scheme 1.5), Isobe (Schemes 1.11, 1.18) and Du Bois (Scheme 1.22).
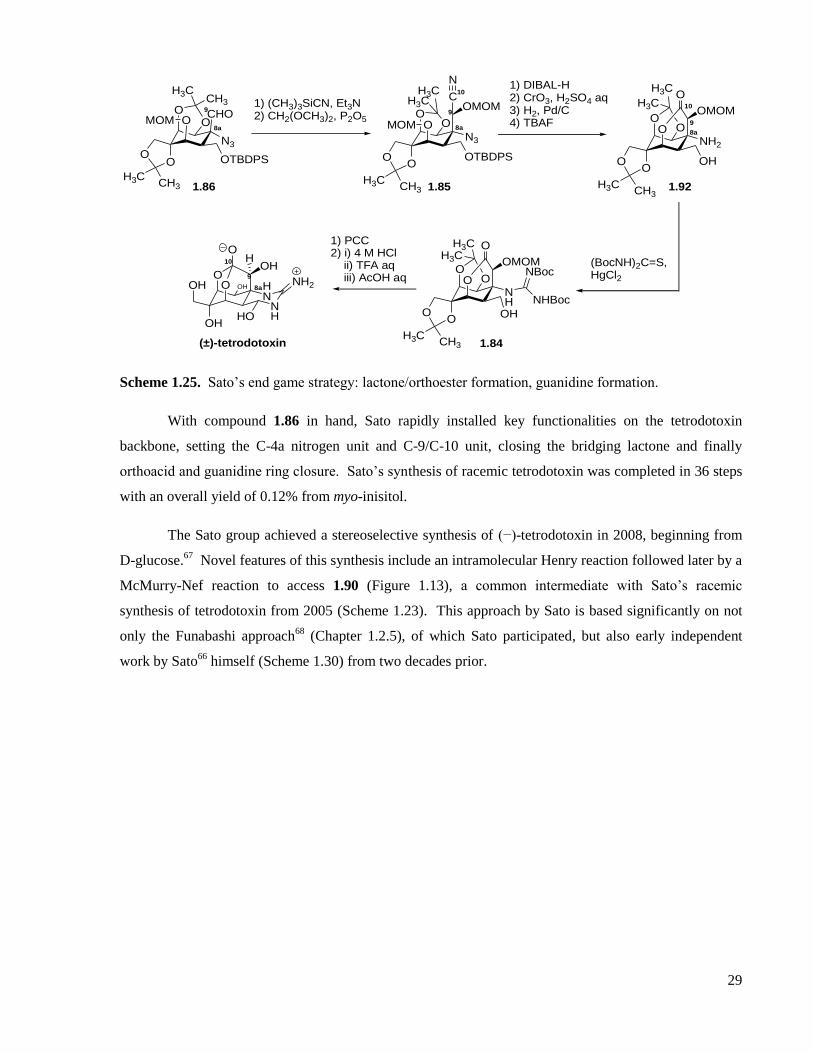
29
1) (CH3)3SiCN, Et3N2) CH2(OCH3)2, P2O5
N3
OO OTBDPS
OO
O
OMOM
H3CCH3
H3C C
N
MOM
NH2
OO OH
OO
O
OMOM
H3CCH3
H3C
H3CO
1) DIBAL-H2) CrO3, H2SO4 aq3) H2, Pd/C4) TBAF
(BocNH)2C=S, HgCl2
NH
OO OH
OO
O
OMOM
H3CCH3
H3CH3C O
NBoc
NHBoc
1) PCC2) i) 4 M HCl ii) TFA aq iii) AcOH aq
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
(±)-tetrodotoxin
OO O
CHO
OO
H3CCH3
CH3
H3C
N3
OTBDPS
MOM
1.86
8a
9
1.85 1.92
1.84
H3C
8a
9
10
10
9
8a
10
9
8a
Scheme 1.25. Sato’s end game strategy: lactone/orthoester formation, guanidine formation.
With compound 1.86 in hand, Sato rapidly installed key functionalities on the tetrodotoxin
backbone, setting the C-4a nitrogen unit and C-9/C-10 unit, closing the bridging lactone and finally
orthoacid and guanidine ring closure. Sato’s synthesis of racemic tetrodotoxin was completed in 36 steps
with an overall yield of 0.12% from myo-inisitol.
The Sato group achieved a stereoselective synthesis of (−)-tetrodotoxin in 2008, beginning from
D-glucose.67
Novel features of this synthesis include an intramolecular Henry reaction followed later by a
McMurry-Nef reaction to access 1.90 (Figure 1.13), a common intermediate with Sato’s racemic
synthesis of tetrodotoxin from 2005 (Scheme 1.23). This approach by Sato is based significantly on not
only the Funabashi approach68
(Chapter 1.2.5), of which Sato participated, but also early independent
work by Sato66
himself (Scheme 1.30) from two decades prior.
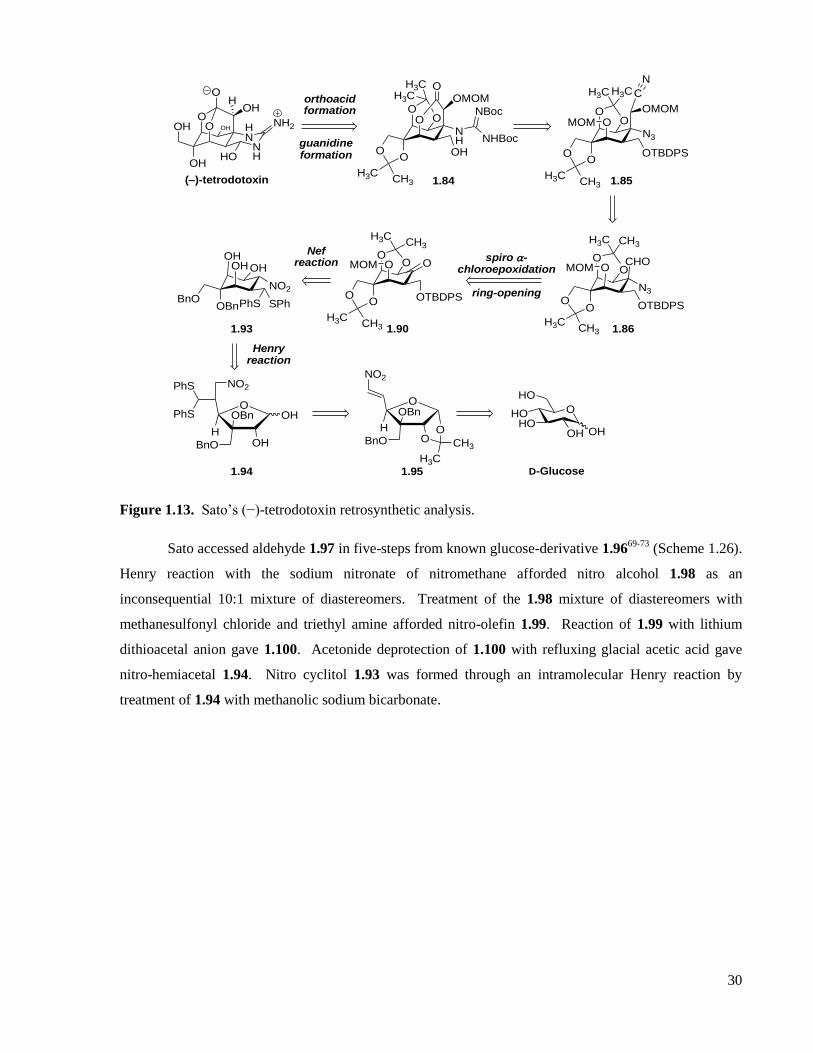
30
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
()-tetrodotoxin
orthoacidformation
O
OH
HOHO
OH
OHOHOH
OBnBnO
NO2
SPh
guanidine formation
D-Glucose
HO
OHO
OBnO
H3C
CH3BnO
NO2
OH
HOH
OBnO
BnO
PhS
PhS
NO2
PhS
Henryreaction
O
OO OTBDPS
OO
O
H3CCH3
H3CCH3
MOMspiro -
chloroepoxidation
ring-opening
Nefreaction
N3
OO OTBDPS
OO
O
OMOM
H3CCH3
H3C C
N
MOMNH
OO OH
OO
O
OMOM
H3CCH3
H3CH3C O
NBoc
NHBoc
OO O
CHO
OO
H3CCH3
CH3H3C
N3
OTBDPS
MOM
H3C
1.86
1.851.84
1.90
1.951.94
1.93
Figure 1.13. Sato’s (−)-tetrodotoxin retrosynthetic analysis.
Sato accessed aldehyde 1.97 in five-steps from known glucose-derivative 1.9669-73
(Scheme 1.26).
Henry reaction with the sodium nitronate of nitromethane afforded nitro alcohol 1.98 as an
inconsequential 10:1 mixture of diastereomers. Treatment of the 1.98 mixture of diastereomers with
methanesulfonyl chloride and triethyl amine afforded nitro-olefin 1.99. Reaction of 1.99 with lithium
dithioacetal anion gave 1.100. Acetonide deprotection of 1.100 with refluxing glacial acetic acid gave
nitro-hemiacetal 1.94. Nitro cyclitol 1.93 was formed through an intramolecular Henry reaction by
treatment of 1.94 with methanolic sodium bicarbonate.
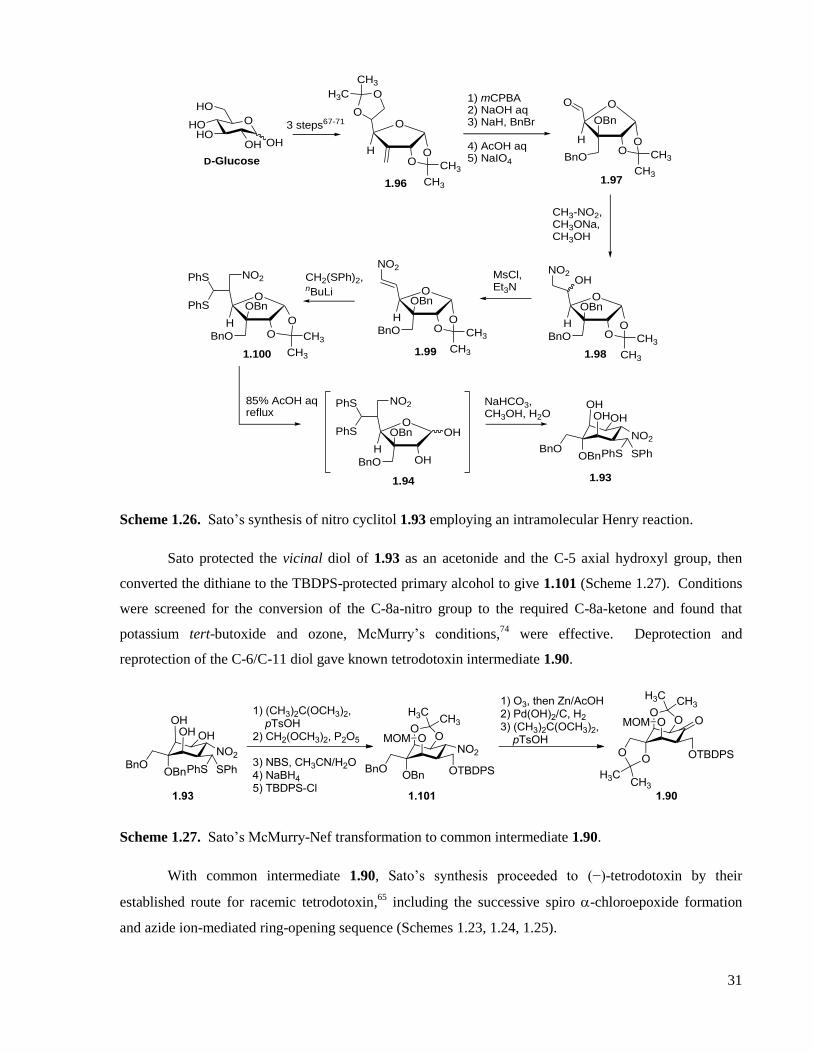
31
3 steps67-71
1) mCPBA2) NaOH aq3) NaH, BnBr
4) AcOH aq5) NaIO4
O
OH
HOHO
OH
D-Glucose
HO
OHO
O
CH3
CH3
O
O
CH3
H3C
OHO
O
CH3
CH3
O
BnO
OBn
CH3-NO2,CH3ONa, CH3OH
OHO
OBnO
CH3
CH3BnO
NO2
OHO
OBnO
CH3
CH3BnO
NO2OH
MsCl,Et3N
CH2(SPh)2,nBuLi
OHO
OBnO
BnO
PhS
PhS
NO2
CH3
CH3
85% AcOH aqreflux
OH
HOH
OBnO
BnO
PhS
PhS
NO2 NaHCO3,CH3OH, H2O
OHOHOH
OBnBnO
NO2
SPhPhS
1.96 1.97
1.98
1.93
1.991.100
1.94
Scheme 1.26. Sato’s synthesis of nitro cyclitol 1.93 employing an intramolecular Henry reaction.
Sato protected the vicinal diol of 1.93 as an acetonide and the C-5 axial hydroxyl group, then
converted the dithiane to the TBDPS-protected primary alcohol to give 1.101 (Scheme 1.27). Conditions
were screened for the conversion of the C-8a-nitro group to the required C-8a-ketone and found that
potassium tert-butoxide and ozone, McMurry’s conditions,74
were effective. Deprotection and
reprotection of the C-6/C-11 diol gave known tetrodotoxin intermediate 1.90.
Scheme 1.27. Sato’s McMurry-Nef transformation to common intermediate 1.90.
With common intermediate 1.90, Sato’s synthesis proceeded to (−)-tetrodotoxin by their
established route for racemic tetrodotoxin,65
including the successive spiro -chloroepoxide formation
and azide ion-mediated ring-opening sequence (Schemes 1.23, 1.24, 1.25).
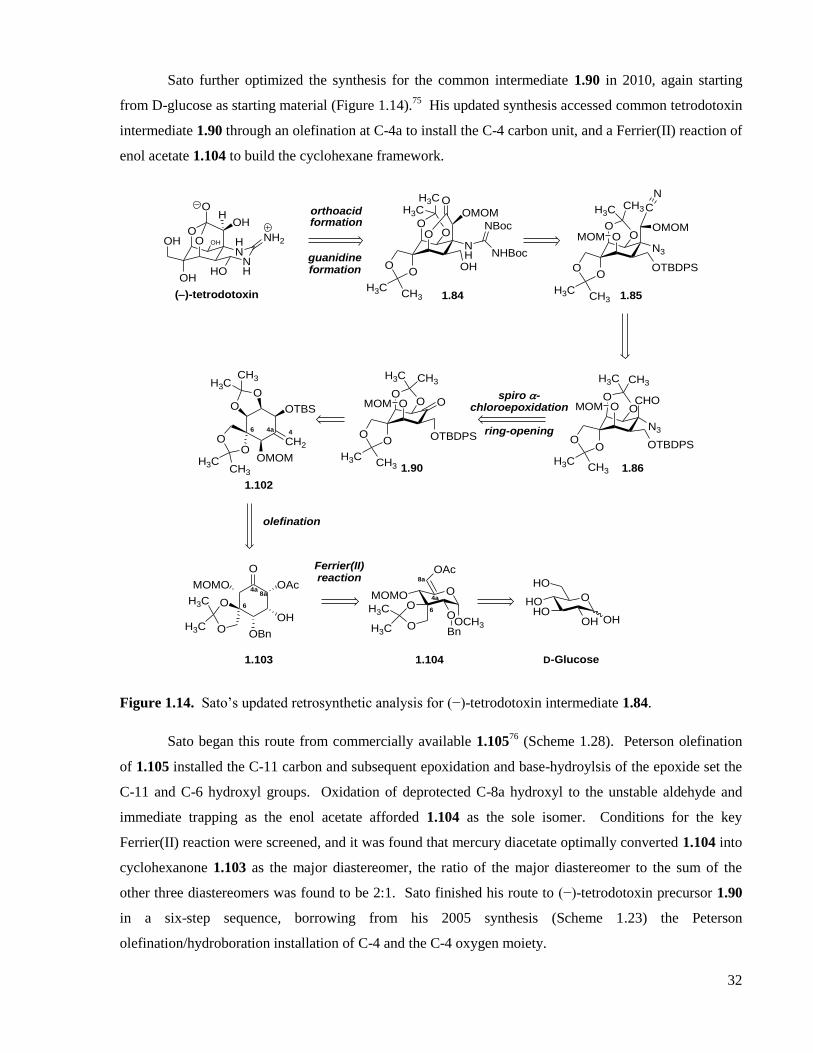
32
Sato further optimized the synthesis for the common intermediate 1.90 in 2010, again starting
from D-glucose as starting material (Figure 1.14).75
His updated synthesis accessed common tetrodotoxin
intermediate 1.90 through an olefination at C-4a to install the C-4 carbon unit, and a Ferrier(II) reaction of
enol acetate 1.104 to build the cyclohexane framework.
O
OCH3
MOMOO
O
OAc
BnO
H3C
H3C
MOMO
O
OAc
OH
OBn
O
O
H3C
H3C
O
O
OO
OMOM
CH2
OTBS
H3CCH3
H3CCH3
Ferrier(II)reaction
olefination
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
()-tetrodotoxin
orthoacidformation
O
OH
HOHO
OH
guanidine formation
D-Glucose
HO
O
OO OTBDPS
OO
O
H3CCH3
H3C CH3
MOMspiro -
chloroepoxidation
ring-opening
N3
OO OTBDPS
OO
OOMOM
H3CCH3
H3C C
N
MOMNH
OO OH
OO
O
OMOM
H3CCH3
H3CH3C O
NBoc
NHBoc
OO O
CHO
OO
H3CCH3
CH3H3C
N3
OTBDPS
MOM
CH3
4a6
4a
4
6
8a
8a4a
6
1.86
1.851.84
1.90
1.1041.103
1.102
Figure 1.14. Sato’s updated retrosynthetic analysis for (−)-tetrodotoxin intermediate 1.84.
Sato began this route from commercially available 1.10576
(Scheme 1.28). Peterson olefination
of 1.105 installed the C-11 carbon and subsequent epoxidation and base-hydroylsis of the epoxide set the
C-11 and C-6 hydroxyl groups. Oxidation of deprotected C-8a hydroxyl to the unstable aldehyde and
immediate trapping as the enol acetate afforded 1.104 as the sole isomer. Conditions for the key
Ferrier(II) reaction were screened, and it was found that mercury diacetate optimally converted 1.104 into
cyclohexanone 1.103 as the major diastereomer, the ratio of the major diastereomer to the sum of the
other three diastereomers was found to be 2:1. Sato finished his route to (−)-tetrodotoxin precursor 1.90
in a six-step sequence, borrowing from his 2005 synthesis (Scheme 1.23) the Peterson
olefination/hydroboration installation of C-4 and the C-4 oxygen moiety.
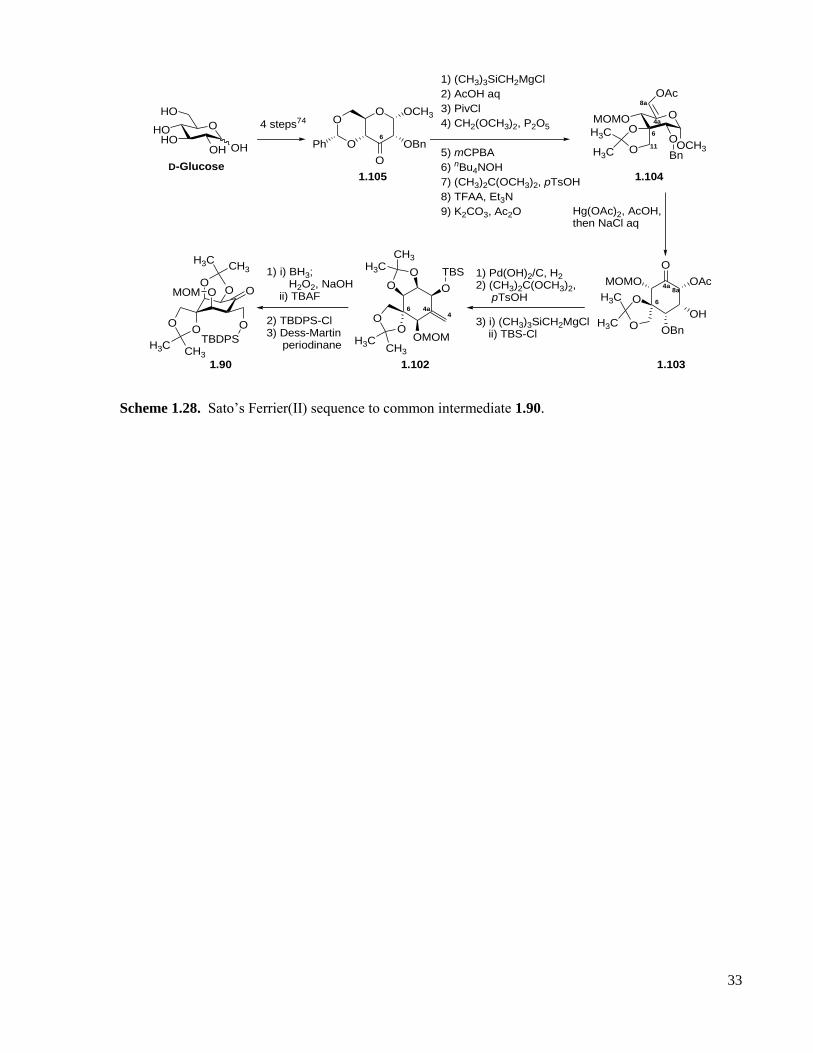
33
4 steps74O
OH
HOHO
OH
D-Glucose
HO O OCH3
OBn
O
O
OPh
1) (CH3)3SiCH2MgCl
2) AcOH aq
3) PivCl
4) CH2(OCH3)2, P2O5
5) mCPBA
6) nBu4NOH
7) (CH3)2C(OCH3)2, pTsOH
8) TFAA, Et3N
9) K2CO3, Ac2O Hg(OAc)2, AcOH,then NaCl aq
1) Pd(OH)2/C, H2
2) (CH3)2C(OCH3)2, pTsOH
3) i) (CH3)3SiCH2MgCl ii) TBS-Cl
1) i) BH3; H2O2, NaOH ii) TBAF
2) TBDPS-Cl3) Dess-Martin periodinane
O
OCH3
MOMOO
O
OAc
BnO
H3C
H3C
MOMO
O
OAc
OH
OBn
O
O
H3C
H3C
O
O
OO
OMOM
O
H3CCH3
H3CCH3
O
OO O
OO
O
H3CCH3
H3CCH3
MOM
4a6
4a
4
6
8a
8a
4a
6
TBDPS
TBS
6
11
1.90
1.104
1.1031.102
1.105
Scheme 1.28. Sato’s Ferrier(II) sequence to common intermediate 1.90.
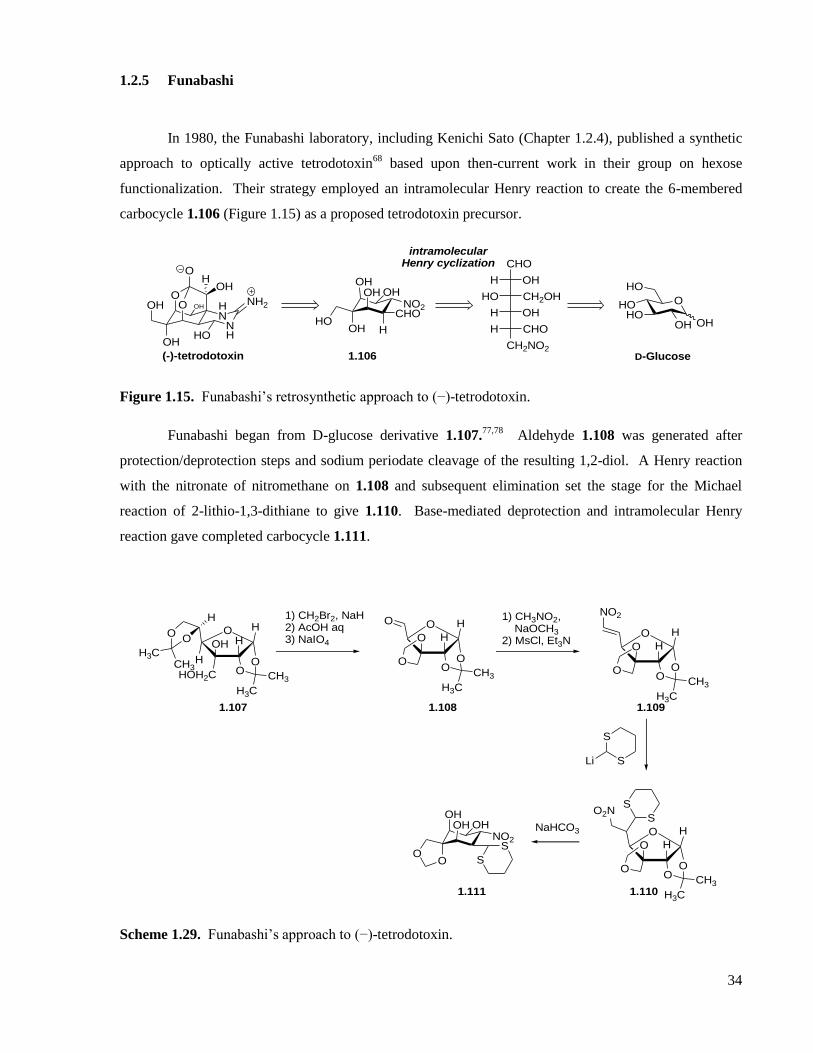
34
1.2.5 Funabashi
In 1980, the Funabashi laboratory, including Kenichi Sato (Chapter 1.2.4), published a synthetic
approach to optically active tetrodotoxin68
based upon then-current work in their group on hexose
functionalization. Their strategy employed an intramolecular Henry reaction to create the 6-membered
carbocycle 1.106 (Figure 1.15) as a proposed tetrodotoxin precursor.
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
(-)-tetrodotoxin
O
OH
HOHO
OH
D-Glucose
HOOHOH OH
OH
CHOHO
NO2
H
CHO
OHH
CH2OHHO
OHH
CHOH
CH2NO2
intramolecularHenry cyclization
1.106
Figure 1.15. Funabashi’s retrosynthetic approach to (−)-tetrodotoxin.
Funabashi began from D-glucose derivative 1.107.77,78
Aldehyde 1.108 was generated after
protection/deprotection steps and sodium periodate cleavage of the resulting 1,2-diol. A Henry reaction
with the nitronate of nitromethane on 1.108 and subsequent elimination set the stage for the Michael
reaction of 2-lithio-1,3-dithiane to give 1.110. Base-mediated deprotection and intramolecular Henry
reaction gave completed carbocycle 1.111.
O
H
H
HOH2C O
OH HO
H
OO
H3C
CH3
CH3
H3C
1.107
O
H
O
O H
O
H3C
CH3
1.109
NO2
O
1) CH2Br2, NaH2) AcOH aq3) NaIO4
S
SLi
O
H
O
O H
O
H3C
CH31.110
O2N
O
S
S
NaHCO3
OHOH OH
OO
NO2
S
S
1.111
1) CH3NO2, NaOCH3
2) MsCl, Et3N
O
H
O
O H
OO
H3C
CH3
O
1.108
Scheme 1.29. Funabashi’s approach to (−)-tetrodotoxin.

35
Intermediate 1.111 appeared to be the most advanced TTX-intermediate assembled by Funabashi,
but his former student Sato pursued this route independently,66
using the intramolecular Henry cyclization
as the key step in assembling the carbocyclic framework as Funabashi. Sato further elaborated the
approach by employing an azide-mediated opening of a spiro -chloro-epoxide derivative (Scheme 1.30)
to create a more advanced (−)-tetrodotoxin precursor 1.115. This early work set the stage for his eventual
conquest of optically active tetrodotoxin65,67
(Chapter 1.2.4) roughly a decade and a half later.
OO
O
OROR
OCH3
O
ClR
OO
O
OROR
OCH3
O
ClR
Cl
NaN3,15-crown-5,DMSO
ORO
OR CHO
OO
N3
OCH3
R
OHOH OH
OO
NO2
S
Ph
Ph
S
1) CH2(OCH3)2, P2O5
2) HgO, HgCl2, BF3•OEt2, (CH3O)3CH
OROR OR
OO
O
OCH3
OCH3
R = MOM
OCH3
OCH3
LDA, CH2Cl2
Li
OCH3
1) NaBH4
2) PMB-Cl, NaH3) H2, Pd/C; then BrCN4) NH4OH
ORO
O
OO
N
H3CO
R
OCH3
OPMB
NH
NH2
H
1.112 1.113
1.1141.115
R
Scheme 1.30. Sato’s early independent work on (−)-tetrodotoxin.

36
1.2.6 Keana
Over nearly two decades, Keana worked on the synthesis of tetrodotoxin and published several
papers in the 1970’s and 1980’s reporting their progress.79-81
Generally, Keana approached the synthesis
of tetrodotoxin by the construction of the carbocyclic framework via Diels-Alder chemistry (Scheme
1.31) while simultaneously, and uniquely forming the cyclic guanidine at an early stage. Diels-Alder
reaction between dienophile pyrimidone 1.116 and 1-acetoxy-3-methyl-butadiene afforded racemic
hydroquinazoline 1.117. Stoichiometric osmium tetraoxide-mediated dihydroxylation of 1.117 gave
compound 1.118 as the major diastereomer. Advanced intermediate 1.118 appeared to be the most
advanced proposed tetrodotoxin precursor reported by Keana, containing several key structural
components.
THF165 °C
OAc
H3C
N
HN
H3C
O
NHAc
O
NH
HN NAc
O
H3C O
H3C
AcO
+OsO4 then H2S
NH
HN NAc
O
H3C O
H3C
AcO
HO
HO
H H
1.117 1.1181.116
Scheme 1.31. Keana’s most advanced tetrodotoxin intermediate.

37
1.2.7 Fraser-Reid
In the 1990s, Fraser-Reid’s laboratory, including Ricardo Alonso (Chapter 1.2.8), published
several reports on progress toward the formal synthesis of tetrodotoxin.82-84
Fraser-Reid reasoned that
then-recent free-radical methods had been shown to be tolerant of a variety of functional groups85,86
whereas previous methods proved incompatible with sugar-derived substrates. Fraser-Reid proposed a
possible avenue to a formal synthesis of tetrodotoxin (Figure 1.16) beginning from a carbohydrate-
derived source.
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
tetrodotoxin
NR'
HO O
OR
OOH
OR
ORH
H
OOR
OR
NHR'OR
CHOOHHO
O
O
OH
NHR'OH
O
O
O
O
OH
OH
OH
D-Mannosan(1,6-anhydro--D-mannopyranose)
OO
OAc
NHAcOAc
OHO OAc
AcO
Kishi-Gotointermediate
1.121 1.120
1.119
1.12
R and R' were suitable protection moieties
Figure 1.16. Fraser-Reid’s retrosynthetic considerations.
Initially, Fraser-Reid began his sugar-based approach to tetrodotoxin from 1,6-anhydro--D-
mannopyranose, exploiting the different reactivity of the sugar’s various hydroxyl groups to achieve
suitably functionalized advanced intermediate 1.123 (Scheme 1.32). Olefination-product 1.124 was
deprotected with HF and reacted with trichloroacetonitrile to give oxazoline 1.125 which was opened
with HCl and protected as the triacetate 1.126. Fraser-Reid was able to directly convert 1.126 to 1.127 by
reaction with tert-butylhyponitrite (TBHT) in refluxing tert-butyl alcohol. After protecting group
manipulation, dioxadamantane 1.129 was afforded from reaction of 1.128 with TES-OTf/Ac2O.

38
O
O
OH
OH
OH
D-Mannosan
O
O
O
OTBDPS
O SnBu2steps841) Br2
2) Ac2OO
O
OAc
TBDPSO O
O
O
OAc
TBDPSO CHCN
CN(EtO)2P
O
O
O
OAc
O CHCN
HF;then Cl3CCN, DBU
N
Cl3C
O
O
OAc
ON
CCl3
NC1) HCl2) Ac2O
O
O
OAc
OAcNHAc
NCO
O
OAc
NHAcOAc
O
1.127 1.126
1.1231.122 1.124
1.125
O
O
OBn
NHAcOBn
O
Ac2O, TES-OTf
O
OAc
OBn
NHAcOBn
O
1.128
O
AcO
OBn
NHAcOBn
O
OAc
1.129
TBHT
TBHT = O N N O
steps84
Scheme 1.32. Fraser-Reid’s synthesis of dioxadamantane core 1.129 via D-mannosan.
Several years later, Fraser-Reid reported the synthesis of an advanced intermediate of similarity
to the Kishi-Goto intermediate 1.12 (Scheme 1.33). Compound 1.128 was allylated under radical
conditions to afford compound 1.130 as the major diastereomer. A reduction/iodination sequence,
followed by ozonolysis under Schreiber conditions87
gave the desired methyl ester 1.131. Reductive
elimination with Zn in refluxing ethanol, PMB-protection of the hemi-acetal and basic hydrolysis gave
compound 1.132 which was ready for an iodolactonization procedure. Iodolactonization of 1.132,
promoted by iodonium dicollidine perchlorate (I(collidine)2ClO4, IDCP), lead to 1.133. Oxygenation was
affected under radical conditions with tributyltin hydride and molecular oxygen to give 1.134, which
correlates to the structurally similar Kishi-Goto intermediate 1.12. Conceivably, this approach could be
exploited to facilitate a synthesis of tetrodotoxin, but further studies by Fraser-Reid have not appeared in
the chemical literature.
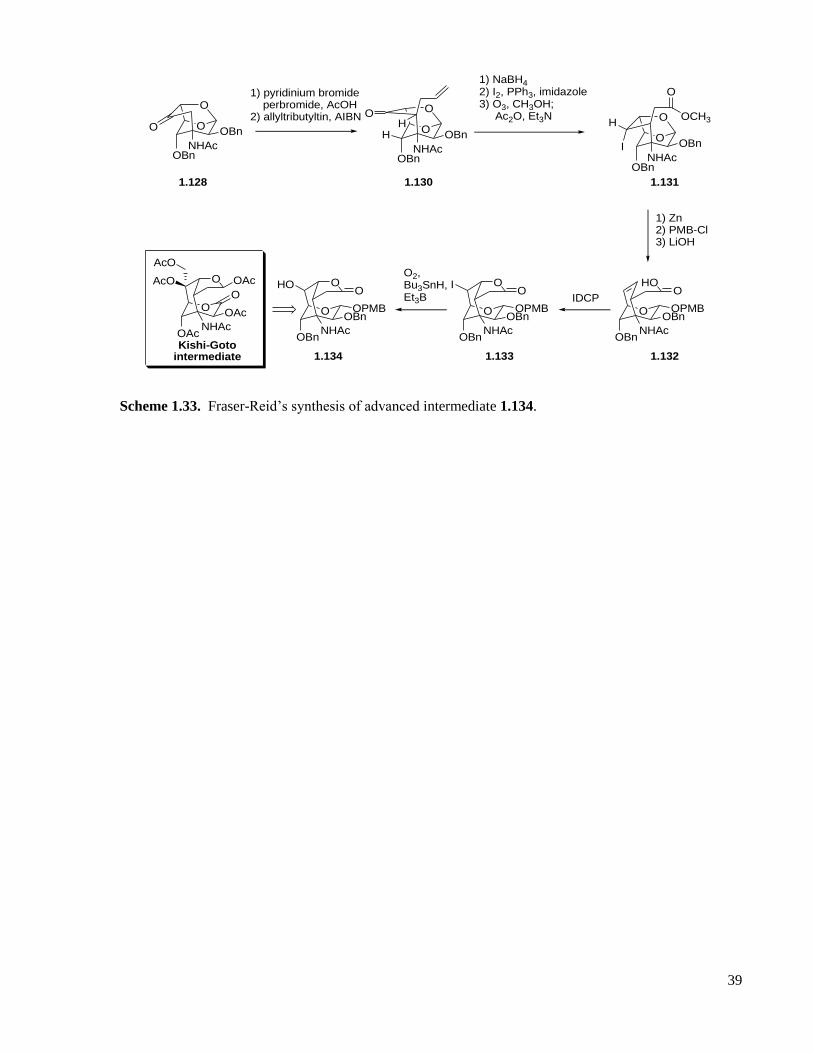
39
OO
OAc
NHAcOAc
OAcO OAc
AcO
Kishi-Gotointermediate
O
O
OBn
NHAcOBn
O
1.128
1) pyridinium bromide perbromide, AcOH2) allyltributyltin, AIBN
O
O
OBn
NHAcOBn
O
1.130
1) NaBH4
2) I2, PPh3, imidazole3) O3, CH3OH; Ac2O, Et3N
O
O
OBn
NHAcOBn
I
1.131
O
HOCH3
1) Zn2) PMB-Cl3) LiOH
IDCP
O2, Bu3SnH, Et3B
OOBn
NHAcOBn
OHOO
OPMB OOBn
NHAcOBn
OIO
OPMB OOBn
NHAcOBn
HOO
OPMB
1.1321.1331.134
HH
Scheme 1.33. Fraser-Reid’s synthesis of advanced intermediate 1.134.

40
1.2.8 Alonso
Following his experience with Fraser-Reid, Ricardo Alonso also independently pursued a
mannose-based radical cyclization approach to the synthesis of tetrodotoxin. His approach (Scheme 1.34)
used a combination of iodoacetal 1.136 as a radical cyclization precursor containing an aldoxime ether as
a radical trap.88,89
The cis ring fusion, and thus the C-8a carbinolamine configuration, were set in a
radical annulation reaction. Upon treatment with AIBN and Ph3SnH, iodoacetal 1.136 underwent an
efficient intramolecular 5-exo-trig cyclization, forcing the approach of the tethered radical chain to the
concave face of the bicyclic structure and setting the C-8a tertiary center’s configuration in compound
1.137. After protection as an oxazolidinone, Jones oxidation and installation of the exomethylene,
compound 1.140 was created in a stereocontrolled manner.
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
tetrodotoxin
O
O
O
OH
O
1,6-anhydro-2,3-isopropylidene--D-
mannopyranose
CH3
CH3
1) TBDPS-Cl
2) HOAc aq
3) nBu2SnO; Br2
4) H2NOCH3
O
O
OH
TBDPSO NOCH3
1) NIS,
2) TBAF
O O
O
O
OHNOCH3
I
OCH2CH3
AIBN, Ph3SnH
O
O
O
OHN CH2
OCH2CH3
O
O
OH
ONHOCH3
OCH2CH3
triphosgeneO
OO
ONOCH3
OCH2CH3
O
CrO3,H2SO4
O
OO
ONOCH3
O
ONaH,
(H2CO)n
O
OO
ONOCH3
O
O
8a
8a
8a
1.135 1.136
1.1371.138
1.139 1.140
OCH3
Scheme 1.34. Alonso’s radical-cyclization approach.

41
1.2.9 Taber
In the early 2000s, Taber proposed an approach to optically active tetrodotoxin based on the
functionalization of a cyclohexenone created by an intramolecular alkylidene carbene cyclization
(Scheme 1.35).90
Taber synthesized an advanced intermediate towards (−)-tetrodotoxin using
established91
carbene-based C-H insertion technology to set the C-6 tertiary alcohol stereocenter. Readily
available 1,2;5,6-di-O-isopropylidene-D-mannitol was used to test the feasibility of Taber’s proposed C-
H insertion strategy. A Sharpless asymmetric epoxidation92
of the corresponding allylic alcohol gave
compound 1.141, which was ring-opened with CuBr/isopropenylmagnesium bromide93,94
and the resulting
diol protected as the cyclic ketal 1.142. Ozonolysis of the terminal olefin gave 1.143 as a precursor to
attempt the intramolecular alkylidene C-H insertion reaction on the C-6 methine. Treatment of 1.143
with trimethylsilyl diazomethane and nbutyl lithium gave compound 1.144 after warming. Taber used the
same approach to set the C-6 tertiary stereocenter was employed by Du Bois (Chapter 1.2.3) in his
synthesis of optically active tetrodotoxin.64
Ozonolysis, annulation and redox adjustment of 1.144,
followed by epimerization of the C-4a center, gave cyclohexenone 1.146, containing stereochemical
features substantially contained in the core of (−)-tetrodotoxin.
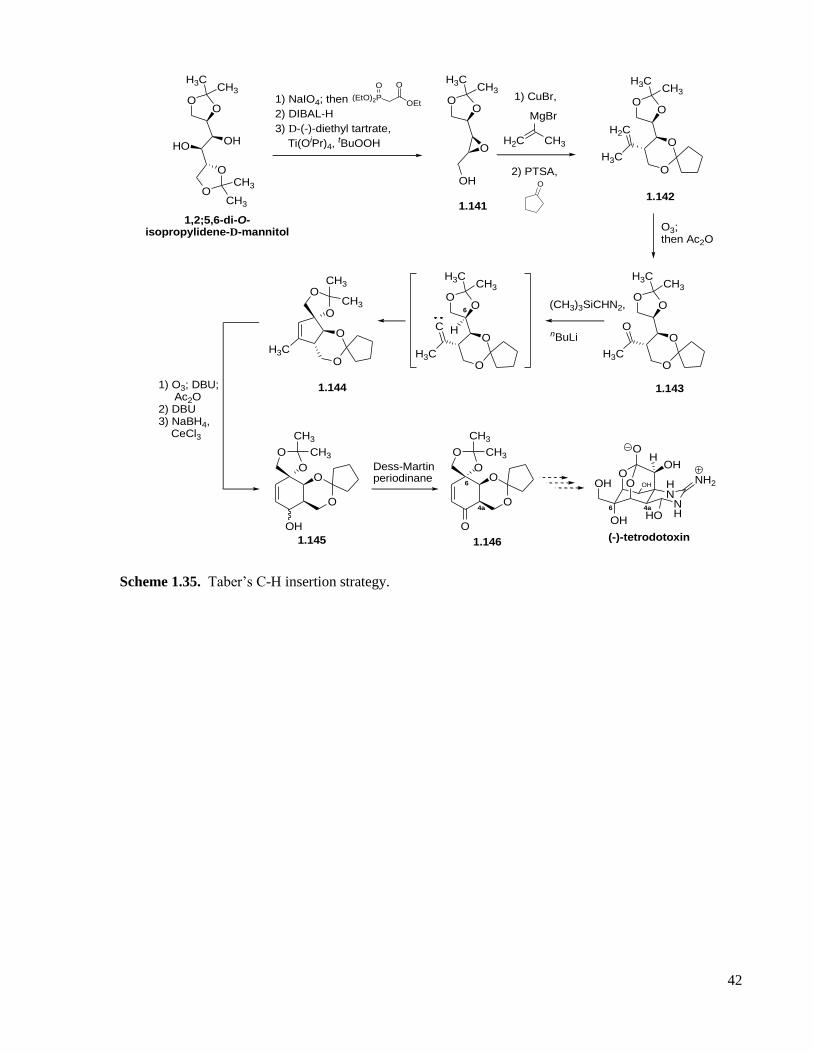
42
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
(-)-tetrodotoxin
OO
OHHO
O
O
H3CCH3
CH3
CH3
1,2;5,6-di-O-isopropylidene-D-mannitol
1) NaIO4; then
2) DIBAL-H
3) D-(-)-diethyl tartrate,
Ti(OiPr)4, tBuOOH
OO
O
OH
H3CCH3
(EtO)2P
O O
OEt
H2C
MgBr
CH3
1) CuBr,O
O
O
O
H3CCH3
H3C
H2C
2) PTSA,O
O3; then Ac2O
OO
O
O
H3CCH3
H3C
O
(CH3)3SiCHN2,O
O
O
O
H3CCH3
H3C
C H
H3CO
O
O
OCH3
CH3
O
O
O
O
O
1) O3; DBU; Ac2O2) DBU3) NaBH4, CeCl3
6
6
Dess-Martin periodinane
OH
O
O
O
O
6
4a
CH3
CH3
CH3
CH3
4a
1.1411.142
1.143
nBuLi
1.144
1.1461.145
Scheme 1.35. Taber’s C-H insertion strategy.
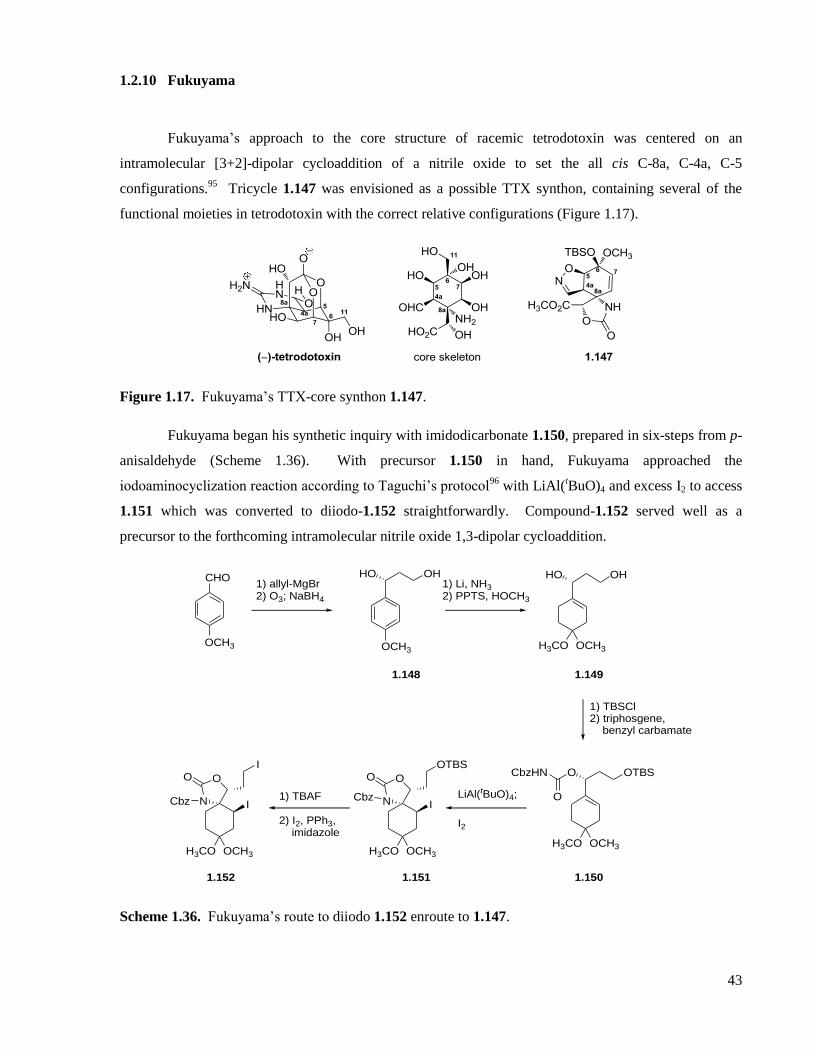
43
1.2.10 Fukuyama
Fukuyama’s approach to the core structure of racemic tetrodotoxin was centered on an
intramolecular [3+2]-dipolar cycloaddition of a nitrile oxide to set the all cis C-8a, C-4a, C-5
configurations.95
Tricycle 1.147 was envisioned as a possible TTX synthon, containing several of the
functional moieties in tetrodotoxin with the correct relative configurations (Figure 1.17).
Figure 1.17. Fukuyama’s TTX-core synthon 1.147.
Fukuyama began his synthetic inquiry with imidodicarbonate 1.150, prepared in six-steps from p-
anisaldehyde (Scheme 1.36). With precursor 1.150 in hand, Fukuyama approached the
iodoaminocyclization reaction according to Taguchi’s protocol96
with LiAl(tBuO)4 and excess I2 to access
1.151 which was converted to diiodo-1.152 straightforwardly. Compound-1.152 served well as a
precursor to the forthcoming intramolecular nitrile oxide 1,3-dipolar cycloaddition.
CHO
OCH3
1) allyl-MgBr2) O3; NaBH4
OCH3
HO OH1) Li, NH3
2) PPTS, HOCH3
OCH3
HO OH
H3CO
OCH3
O OTBS
H3CO
1) TBSCl2) triphosgene, benzyl carbamate
CbzHN
O
OCH3H3CO
I
O
N
OTBSO
Cbz
1.148 1.149
1.1501.151
LiAl(tBuO)4;
I2
OCH3H3CO
I
O
N
IO
Cbz
1.152
1) TBAF
2) I2, PPh3, imidazole
Scheme 1.36. Fukuyama’s route to diiodo 1.152 enroute to 1.147.

44
Deprotection of the dimethyl ketal and concomitant dehydroiodination of the secondary iodide
with hot aqueous acetic acid afforded enone 1.153, which was converted to the silyl enol ether and then
the primary iodide was substituted with sodium nitrite to 1.154 (Scheme 1.37). Fukuyama treated 1.154
with di-tert-butyl dicarbonate and DMAP97
to dehydrate the primary nitro group to the corresponding
nitrile oxide and induce the subsequent intramolecular 1,3-dipolar cycloaddition with the C-5/C-4a olefin
to give tetracycle 1.155 as a single diastereomer. The C-7/C-8 olefin was installed by treatment of silyl
enol ether 1.155 with phenylselenium chloride in methanol and mCPBA oxidation to give unsaturated
mixed acetal 1.156. A DBU-mediated oxazolidinone-opening gave tricycle 1.157 that was primed for
attempts to oxidize the cyclopentene unit. Fukuyama found that dihydroxylation of 1.157 proceeded from
the convex face of the bowl-shaped tricycle, and reductive workup with Na2SO3 gave the corresponding
oxazolidinone. Oxazolidinone formation suitably protected the C-9 hydroxyl group, allowing
manipulation of the C-10 hydroxyl group to proceed specifically after reduction of the isooxazoline C=N
bond and Alloc protection. Oxidation at C-10 with Dess-Martin periodinane and Baeyer-Villiger
oxidation afforded ring-expanded lactone 1.159. Methanolysis of lactone 1.160 and palladium-mediated
deprotection of the Alloc group afforded isooxazoline 1.147 containing the oxidation level at C-4 found in
TTX.
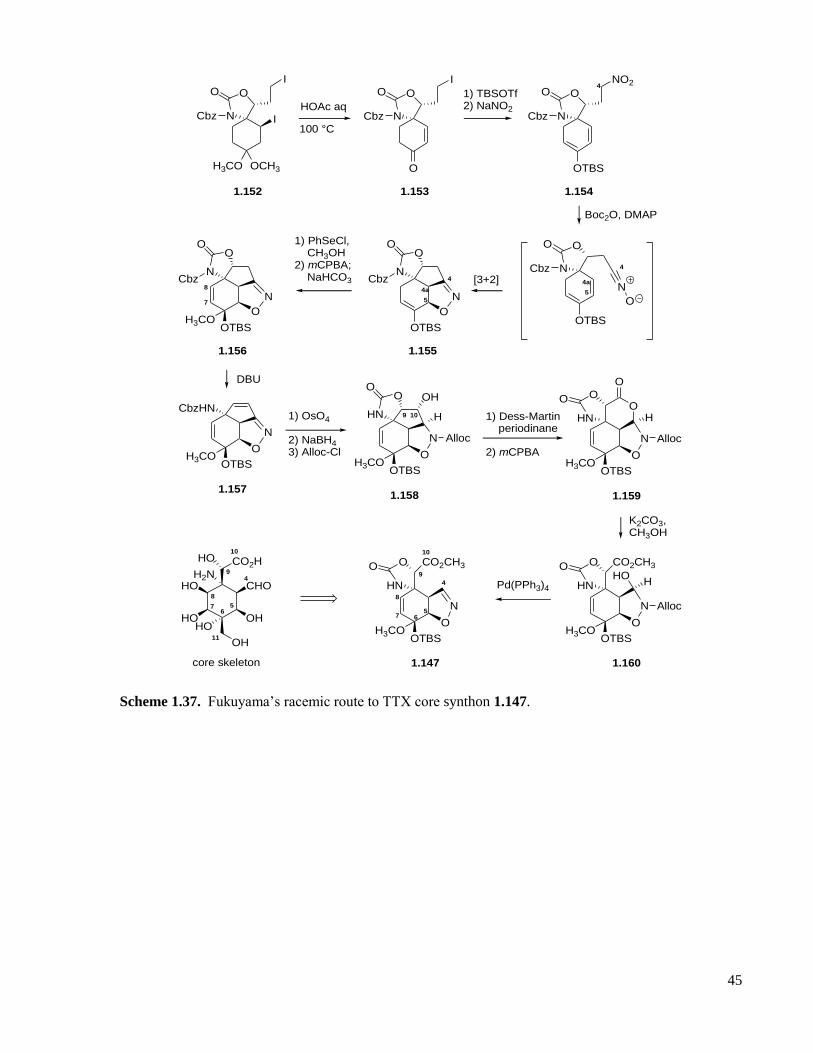
45
H3CO
OH
OCH3H3CO
I
O
N
IO
Cbz
1.152
O
O
N
IO
Cbz
1.153
OTBS
O
N
NO2
O
Cbz
1.154
O
N
O
N
OTBS
O
1.155
OTBS
O
N
N
O
Cbz
O
[3+2]
HOAc aq
1) TBSOTf2) NaNO2
Boc2O, DMAP
O
N
O
N
OTBS
O
Cbz
1.156
H3COO
N
CbzHN
OTBS
1.157
H3COO
N
O
HN
OTBS
O
1.158
Alloc
H
H3COO
N
OTBS
1.159
Alloc
O
O
O
HN
O
H
H3COO
N
OTBS
1.160
Alloc
CO2CH3O
HN
OHO
H
H3COO
N
OTBS
1.147
CO2CH3O
HN
O
11
57
HO
HO
OH
CHO
HO
H2N
CO2HHO
OH
6
core skeleton
57 6
1) PhSeCl, CH3OH2) mCPBA; NaHCO3
DBU
1) OsO4
2) NaBH4
3) Alloc-Cl
1) Dess-Martin periodinane
2) mCPBA
K2CO3, CH3OH
Pd(PPh3)4
4a
58
7
8 8
Cbz
109
9 9
10 10
44
100 °C
4
4
4
4a
5
Scheme 1.37. Fukuyama’s racemic route to TTX core synthon 1.147.

46
1.2.11 Ohfune
In early 2003, the Ohfune group communicated work in their laboratory on the stereoselective
construction of a tetraol system corresponding to the C-5, 6, 7, 11 hydroxyl groups in (−)-tetrodotoxin.98
The consecutive hydroxyl groups in (−)-tetrodotoxin have proved to be one of the challenges in the
construction of the unique core structure. Ohfune approached the tetraol part of tetrodotoxin (Figure
1.18) through a series of repetitive stereoselective epoxidation/base-induced ring-fragmentation reactions
beginning from (−)-quinic acid derivative 1.161 which already contained the correct C-6 geometry.
Figure 1.18. Ohfune’s approach to (−)-tetrodotoxin.
Ohfune generated allyl sulfone 1.162 as the sole regioisomer via the dehydration of diol 1.161
with (PhS)2/Bu3P, as developed in their laboratory (Scheme 1.38).99
Epoxidation occurring from the less-
sterically demanding -face of the allyl sulfone and subsequent base-mediated ring-opening/olefin
isomerization gave 1.164 with the correct configuration of the C-5 hydroxy group. The same mCPBA
epoxidation/base-mediated ring-opening sequence was repeated on 1.164 to generate 1,3-diaxial diol
1.166. Having set the diaxial C-5, C-7 stereochemistry, Ohfune tackled the installation of the C-6, 11
diol. Protection of 1.166 as the cyclic acetal, removal of the phenyl sulfone group and deprotection of the
-OTBS group afforded 1.168. Inversion of the C-4 stereocenter was accomplished through an
oxidation/reduction sequence, and osmium tetraoxide-based dihydroxylation of the exo olefin lead to
1.169 containing the C-5, 6, 7, 11 tetraol system in (−)-tetrodotoxin. Ohfune’s stereoselective route to the
tetraol system by successive epoxidation and allyl sulfone ring-opening operations effectively addressed a
method of accessing the 1,2,3-triaxial hydroxyl groups of (−)-tetrodotoxin on a simpler (−)-quinic acid
derivative.

47
1.161
OO
OCH3
H3C
OCH3
H3C
TBSO OHOH
(PhS)2/Bu3P
1.162
OO
OCH3
H3C
OCH3
H3C
OTBSSPh
mCPBA
1.163
OO
OCH3
H3C
OCH3
H3C
OTBSSO2Ph
O
KOtBu
1.164
OO
OCH3
H3C
OCH3
H3C
OTBSSO2Ph
OHmCPBA
1.165
OO
OCH3
H3C
OCH3
H3C
OTBSSO2Ph
OH
OKOtBu
1.166
OO
OCH3
H3C
OCH3
H3C
OTBSSO2Ph
OH
HO
1.167
OO
OCH3
H3C
OCH3
H3C
OTBSSO2Ph
O
O
NaH, CH2Br2
1) nBuMgCl,
Pd(acac)2
2) TBAF
1.168
OO
OCH3
H3C
OCH3
H3COH CH2
O
O 1) Dess-Martin periodinane
2) NaBH(OCH3)33) TBSOTf4) OsO4 1.169
OO
OCH3
H3C
OCH3
H3C
TBSOO
O
OH
OH
5
7
5
6
11
Scheme 1.38. Ohfune’s approach to C-5, 6, 7, 11 tetraol system in (−)-tetrodotoxin.

48
1.2.12 Summary
Several groups have pursued tetrodotoxin as a synthetic target. The history of the synthesis of
tetrodotoxin and of the tetrodotoxin core is marked with various themes which have evolved over time.
In comparing the completed tetrodotoxin syntheses to date, it is easy to recognize similar finishing
strategies due to tetrodotoxin’s polar guanidine and ortho acid functionalities. Other similarities between
synthetic approaches are abundant. Funabashi created synthetic tetrodotoxin intermediates via
intramolecular Henry addition, a theme used later by Sato in his initial synthesis of optically active (−)-
tetrodotoxin from D-glucose. The Taber group achieved a synthesis of an advanced intermediate for
tetrodotoxin by using an intramolecular carbene insertion to install the C-6 center, a similar method which
was used by Du Bois in his remarkable conquest of (−)-tetrodotoxin where he utilized two impressive C-
H-activation transformations to build (−)-tetrodotoxin, using synthetic methodology published only a
couple of years before. Keana approached the carbocyclic framework synthesis via Diels-Alder
chemistry, as did Kishi and Isobe, but with a twist to simultaneously, and thus far uniquely, install the
cyclic guanidine functionality early-stage. Fraser-Reid completed advanced tetrodotoxin intermediates
via radical cyclization chemistry, an approach further explored by Alonso. Ohfune used repetitive
epoxidation/ring-opening sequences in construction of the tetraol core, similar to work by Isobe. Both
Kishi and Isobe approached the hexasubstituted cyclohexane ring through intramolecular epoxide ring-
opening events, which also set the requisite stereocenters. Fukuyama’s use of a nitrile oxide in an
intramolecular 1,3-dipolar cycloaddition to set the all-cis C-8a/C-4a/C-7 configurations lead to work
described in Chapter 2 of this thesis as an approach to the core of tetrodotoxin.
Despite the major advances in the synthesis of tetrodotoxin and its core, the structural complexity
of tetrodotoxin ensures that it will continue to be a synthetic target for years to come. All current total
syntheses (summarized in Figure 1.19) to date are long (28-40 linear steps), and the densely
functionalized carbocyclic core makes convergent approaches to shorten the synthesis difficult, as evident
by the lack of shorter syntheses almost 40 years after the structural elucidation of tetrodotoxin.
Tetrodotoxin serves as not only a template upon which to test new reactions and transformations, but also
as a source for the very creation and discovery of new reaction types.
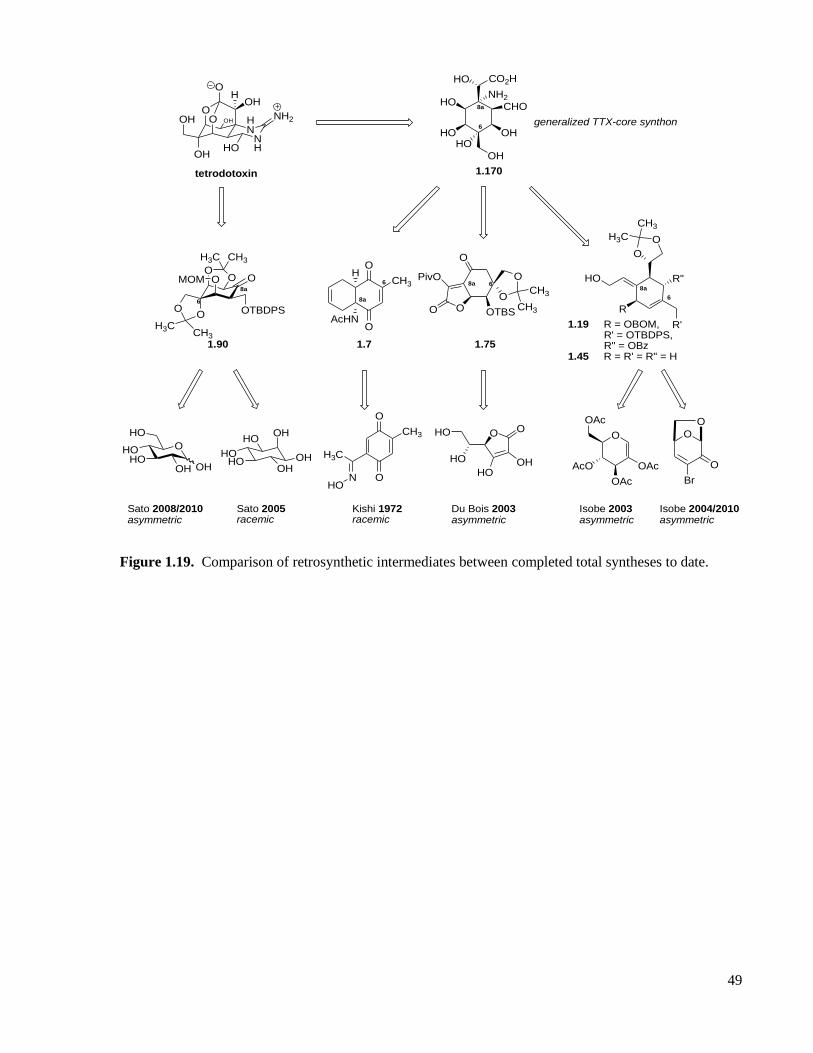
49
6
8a
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
CHO
OHHO
HO
OH
HO
NH2
CO2HHO
O
O
CH3
H
AcHNR
HO
O
O
H3C
CH3
R"
R'
O
PivO
OO OTBS
O
O
CH3
CH3
HOOH
OHOH
HOHO
tetrodotoxin
O
O
CH3
H3C
NHO
O
OAc
OAc
OAcAcO
OHO
HO
O
OHHO
Sato 2005racemic
Kishi 1972racemic
Isobe 2003asymmetric
Du Bois 2003asymmetric
O
OH
HOHO
OH
HO
O
OO OTBDPS
OO
O
H3CCH3
H3C CH3
MOM
Sato 2008/2010asymmetric
O
Br
O
O
Isobe 2004/2010asymmetric
1.170
1.7 1.751.90
8a
6 8a
6 8a 68a
6
generalized TTX-core synthon
1.19 R = OBOM, R' = OTBDPS, R" = OBz1.45 R = R' = R" = H
Figure 1.19. Comparison of retrosynthetic intermediates between completed total syntheses to date.

50
2 The oxidative amidation strategy
2.1 General strategy
It is in the greater context of the afore-mentioned works that we examined tetrodotoxin as a target
for total synthesis. The dense and intricate structure of tetrodotoxin provided an opportunity for the
exploration of new and unique synthetic strategies. Bearing this in mind, we recognized the possibility
described in Figure 2.1. Compound 2.2, which is a generalized product of oxidative amidation of a
phenol of type 2.1 where G = an oxygen functionality, or appropriate precursor thereof, may be mapped
nicely onto compound 1.170. Compound 1.170 emerges upon release of the guanidine and orthoacid
functionalities of tetrodotoxin and is a common retrosynthetic intermediate as per Kishi, Du Bois and
Isobe.
tetrodotoxin
NH
N
OH
OH NH2
HO
OHO
O
O
OHH
H
OH
OH
COOH
HO
NH2
HO
HO
CHO
OH
COOR
O
NHAcG
COOR
HO
G
1.170
2.1 2.2
4a
6
8a
8a
6
4a
4
5
5
Figure 2.1. Retrosynthetic hypothesis for tetrodotoxin.
The conversion of compound 2.2 into 1.170 requires, among other things, the stereocontrolled
introduction of the C-5 hydroxyl group and C-4 aldehyde (or equivalent moiety). Fukuyama’s work95
(Chapter 1.2.10) indicated that the stereocontrolled introduction of these elements across the C-4a/C-5
olefin could be accomplished via an intramolecular nitrile oxide-olefin cycloaddition (INOC)
reaction.100,101
We further envisioned that fragmentation of isooxazoline 2.4 could lead to the revealing of
both the C-5 hydroxyl group and also the C-4 formyl unit masked as a cyano group (Figure 2.2).

51
O
AcHN
COOR
G
O
AcNG
O
N
O
OO
O NH
H
AcN G
Nu
[?]
O
CO
AcHNG
Nu
OH
CNH
H
[?]
[?]
H2C
CO
NG
Nu
O
CNH
H
P
P
[?]
OH
HO
NH2HO
HO
OH
CHO
COOH
OH
2.2
1.170 2.6 2.5
2.3 2.4
H H
H
4
5
Figure 2.2. Generalized strategy for the elaboration of the tetrodotoxin core.
Our elaborated retrosynthetic analysis is depicted in Figure 2.3 and features a common end-game
strategy involving ortholactone and guanidine formation. A sequence involving methylenation to install
the C-11 carbon unit, common to the work of Sato (Schemes 1.26, 1.28), as well as osmium tetraoxide
dihydroxylations to install the four hydroxyl units across C-6, C-11, C-7 and C-8, elaborates the
carbocycle 1.170. Functionalized enone 2.5 is a product of an intramolecular 1,3-dipolar cycloaddition
reaction and subsequent isooxazoline fragmentation, with the pendant C-9 arm delivering the dipole to the
C-4a/C-5 olefin in a stereocontrolled manner. Dienone 2.2 is rapidly and uniquely assembled from an
oxidative amidation reaction of a phenol of type 2.1.
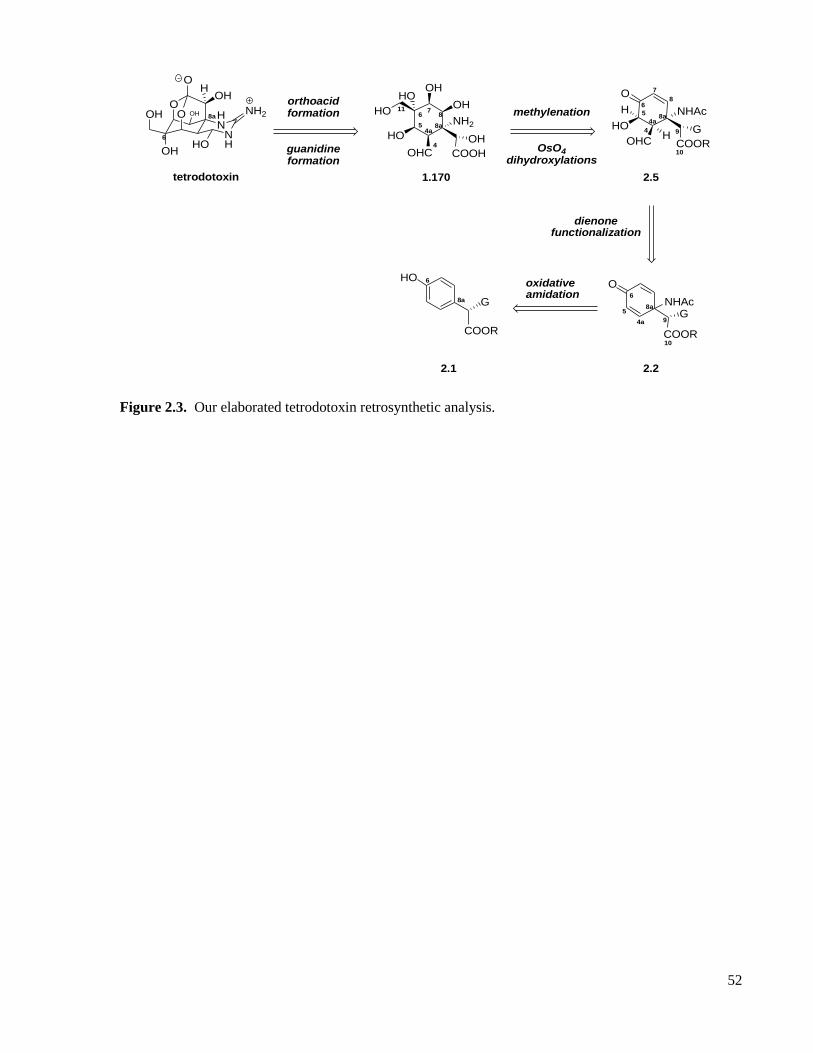
52
orthoacidformation
guanidineformation
methylenation
OsO4 dihydroxylations
COOR
O
H
H
NHAc
HO
OHCG
dienonefunctionalization
oxidativeamidation
OH
OH
COOH
HO
NH2
HO
HO
OHC
OH
1.170
4a
6
8a
4
5
tetrodotoxin
NH
N
OH
OH NH2
HO
OH
OO
O
OHH
H
COOR
O
NHAcG
2.2
8a
6
4a
5
COOR
HO
G
2.1
2.5
4
8a4a
5
6
8a
6
9
9
10
10
7
7
8
8
11
6
8a
Figure 2.3. Our elaborated tetrodotoxin retrosynthetic analysis.

53
2.2 Oxidative amidation
The oxidative amidation of phenols102,103
entails the creation of aza-substituted dienones of type
2.8 from phenolic substrates of type 2.7 (or ortho-substituted derivatives of 2.7). Group N is a suitable
nucleophilic nitrogen moiety in 2.7 and is an amide functionality in 2.8 (Figure 2.4). The dashed-curve
indicates that the N-group may be either covalently tethered to the phenolic ring (intramolecular) or may
be an independent molecule (bimolecular). Hypervalent iodine (III) reagents,104-110
especially PhI(OAc)2
(diacetoxy iodobenzene, DIB) and PhI(OCOCF3)2 (phenyliodine bis(trifluoroacetate), PIFA), are uniquely
competent in effecting oxidative amidation reactions of phenols, a special case of oxidative
dearomatization of phenols.111
HO
oxidation
O
NN
2.82.7
Figure 2.4. Oxidative amidation of para-phenols.
Oxidative amidation reactions of phenols proceed through an electrophilic intermediate such as
2.9 which is trapped with the nucleophilic N-group (Figure 2.5). The chemical literature is rife with
examples of the oxidation of a phenol112
to a transient electrophilic species followed by interception with
suitable nucleophiles. Barton113-115
published initial discoveries in this area in the 1950’s as well as
others.116,117
As synthetic organic technologies evolved, newer and more effective oxidants were used in
phenol oxidation reactions,118
however the development and widespread use of hypervalent iodine
reagents permitted easy access to reactions of this type. Contributions to the chemical literature over the
past 25 years by Kita,119-121
Pelter,122
Barret,123
and Wipf124,125
testify to the transcendence of oxidative
dearomatization of phenols reactions from a novelty-class of reaction to a widely useful paradigm for the
creation of synthetically useful products.
Figure 2.5. Oxidative amidation of para-phenols.

54
Oxidative dearomatization reactions, as described in Figures 2.4 and 2.5, are mechanistically
distinct from related transformations (Figure 2.6) such as Kikugawa-Glover-type reactions.126-140
These
involve the oxidation of an amide unit of type 2.10 to an electrophilic nitrogen-containing intermediate of
type 2.11. The nucleophilic electron-rich aromatic ring attacks the electrophilic nitrogen, resulting in
products of type 2.12 which formally resemble products of oxidative amidation (2.8) as in Figure 2.5.
Kikugawa-Glover-type reactions
RO
2.10
NH
OZ
Z = CH3O, PhtN
RO
2.11
N
OZ
O
2.12
N
OZ
[O]
Figure 2.6. Kikugawa-Glover-type reactions.
2.2.1 Oxidative amidation in total synthesis
The recognition that oxidative amidation technology could allow the rapid assembly of various
nitrogen-containing substances led the development and subsequent refinements of this methodology.
Oxidative amidation reactions of the type described in Figure 2.4 were developed in the Ciufolini
laboratory to approach synthetic challenges in natural product synthesis programs such as FR901483141
and TAN1251C142
(Figure 2.7). The investigation of different modes of oxidative amidation of phenols
can be categorized into three distinct generations (Figure 2.9), all of which evolved to solve specific
synthetic challenges in natural product synthesis. All three generations of oxidative amidation reaction
are promoted by DIB.143
The first generation of oxidative amidation reaction involved the cyclization of
phenolic oxazolines, and was applied to the synthesis of FR901483 and TAN1251C (Figure 2.7).
Independently, Sorensen144,145
and Honda146-148
developed oxidative dearomatization-cyclization reactions
of phenolic secondary amines 2.13 and 2.15 in their syntheses of FR901483 and TAN1251C (Figure 2.8).
The second generation of oxidative amidation involved the intramolecular oxidative cyclization of ortho-
and para-phenolic sulfonamides and was applied to the synthesis of (−)-cylindricine C149
and towards the
himandrine core.150
The third generation oxidative amidation reaction type is the bimolecular reaction of
2- and 4-substituted phenols with nitriles and has been applied towards histrionicotoxins151
and
tetrodotoxin.152
The development of the third generation of oxidative amidation as a reliable large-scale
(50-100g) reaction was significantly advanced as a result of the synthetic ventures described in Chapter
2.3.1 of this thesis.
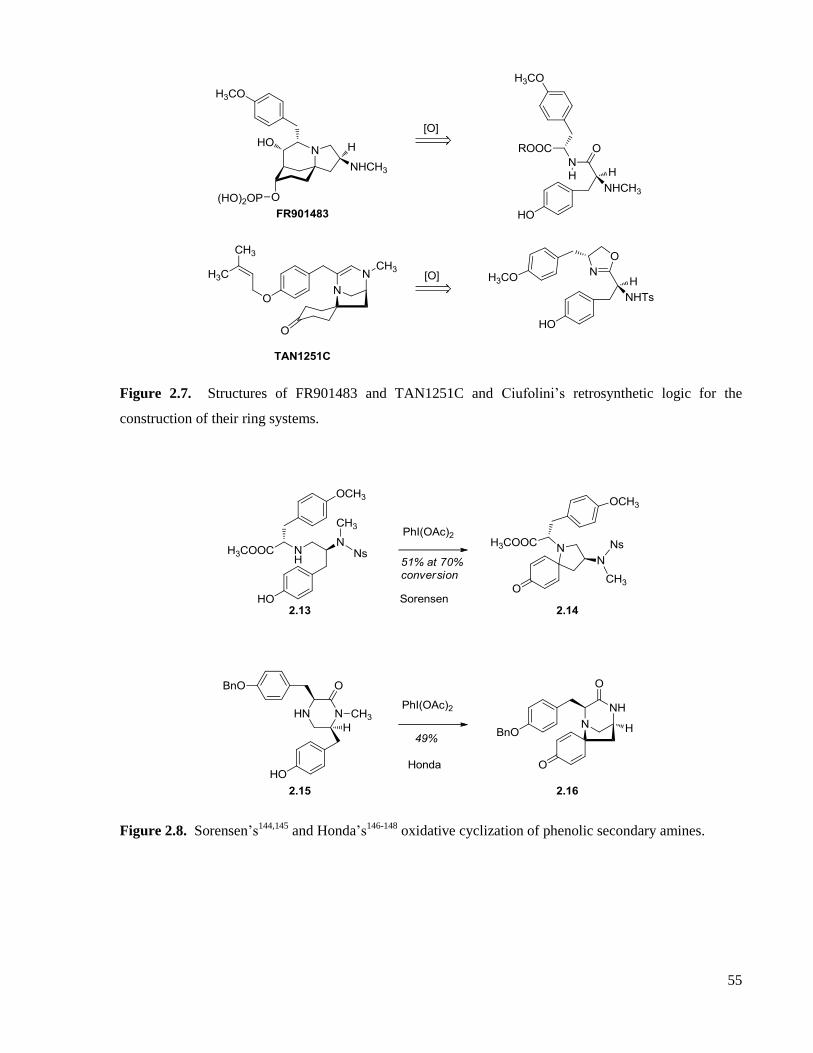
55
Figure 2.7. Structures of FR901483 and TAN1251C and Ciufolini’s retrosynthetic logic for the
construction of their ring systems.
Figure 2.8. Sorensen’s144,145
and Honda’s146-148
oxidative cyclization of phenolic secondary amines.
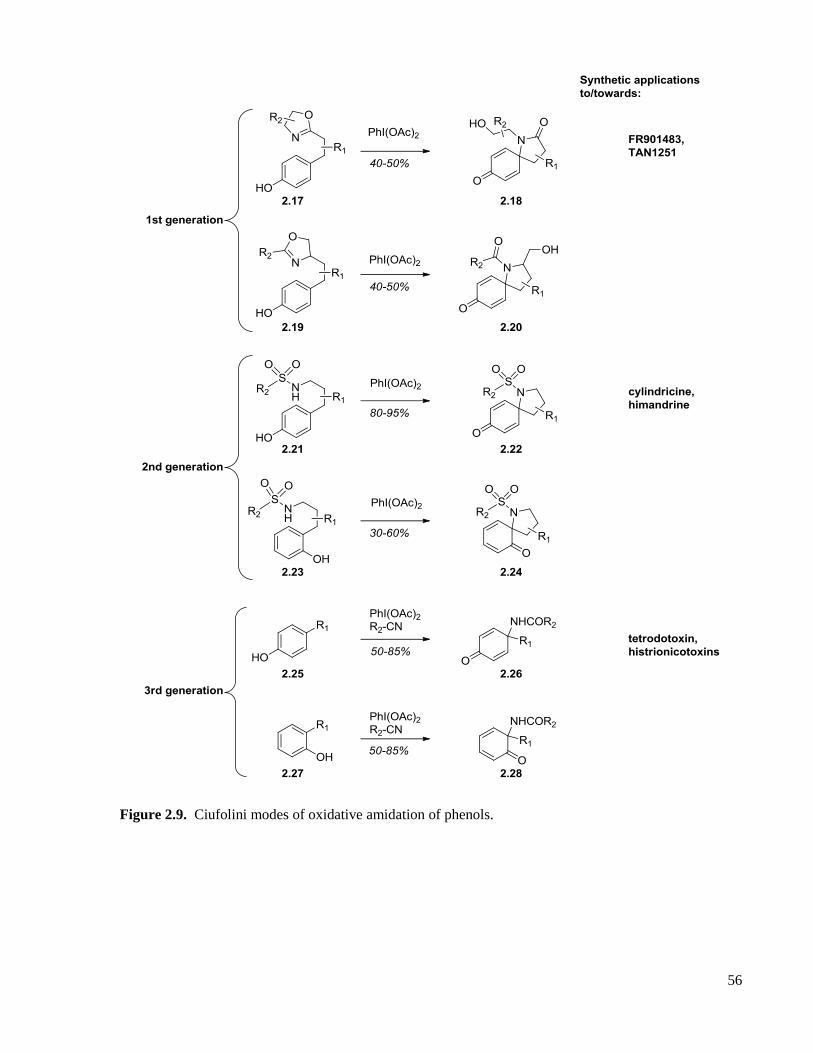
56
Figure 2.9. Ciufolini modes of oxidative amidation of phenols.

57
2.3 Bimolecular oxidative amidation
The initial conditions (Scheme 2.1) for the bimolecular mode of oxidative amidation in the
presence of nitriles153,154
suffered from two main drawbacks: variable yields and costly solvent
(1,1,1,3,3,3-hexafluoroisopropanol, HFIP or 2,2,2-trifluoroethanol, TFE). These fluorinated alcohols
were initially selected as solvents based on work by Kita,119-121
which indicated that phenol oxidations
promoted by DIB proceed best in these solvents, presumably due to their acidic and non-nucleophilic
nature. The development of a procedure which reliably allows for efficient large-scale synthesis of
compounds of type 2.30 and also avoids costly solvents emerged as an important goal.
HO
RPhI(OAc)2,CH3CN, HFIP
O
RN
H
O
CH3
2.302.29
50-85%
Scheme 2.1. Initial conditions for bimolecular oxidative amidation.153
2.3.1 Optimization of scalable bimolecular oxidative amidation conditions
Figure 2.10. Possible DIB-mediated bimolecular oxidative amidation mechanism.

58
Bimolecular oxidative amidation reactions in which the nitrile nucleophile serves as co-solvent
suffer when run at higher concentrations of phenol (Figure 2.11). Phenols are better nucleophiles than
nitriles such as acetonitrile, and thus tar-like oligomers are obtained from oxidative amidation reactions,
which ceteris paribus, are run at higher concentration of phenol starting material. Reactions run at large
scale under the initial conditions for oxidative amidation153
yield products contaminated with oligomeric
matter which complicates the isolation of the desired dienone products (2.30) by requiring lengthy
chromatographic purifications. Fluorinated alcohol solvents such as HFIP and TFE are highly costly, and
their avoidance in these oxidative amidation reactions is merited.
Figure 2.11. Effect of phenol concentration on reaction outcome.
Phenol 2.31 was selected as the starting material to begin optimization studies, since the product
of its oxidative amidation, compound 2.32, was required for work in assembling the tetrodotoxin core.
Our initial optimization studies focused on small-scale optimization (1g of phenol 2.31) based upon the
initial 2005 conditions153
(Table 2.1). This reaction worked best when the phenol was added to a solution
of DIB in acetonitrile/co-solvent in order to keep the concentration of phenol minimized (see Figure 2.11)
and avoid undesired side reaction. A reaction (Table 2.1, entry d) was observed in which PIFA was used
without the presence of any fluoro-solvents in the bimolecular oxidative amidation reaction. This
observation is in accord with work by Wood,155
and suggested that perhaps small amounts of TFA were
sufficient replacement for HFIP or TFE in these types of reactions. Entry e in Table 2.1 further indicated
that DIB could be used in oxidative amidation reactions of this type, and a more thorough optimization of
the bimolecular oxidative amidation reaction was carried out based upon this.154
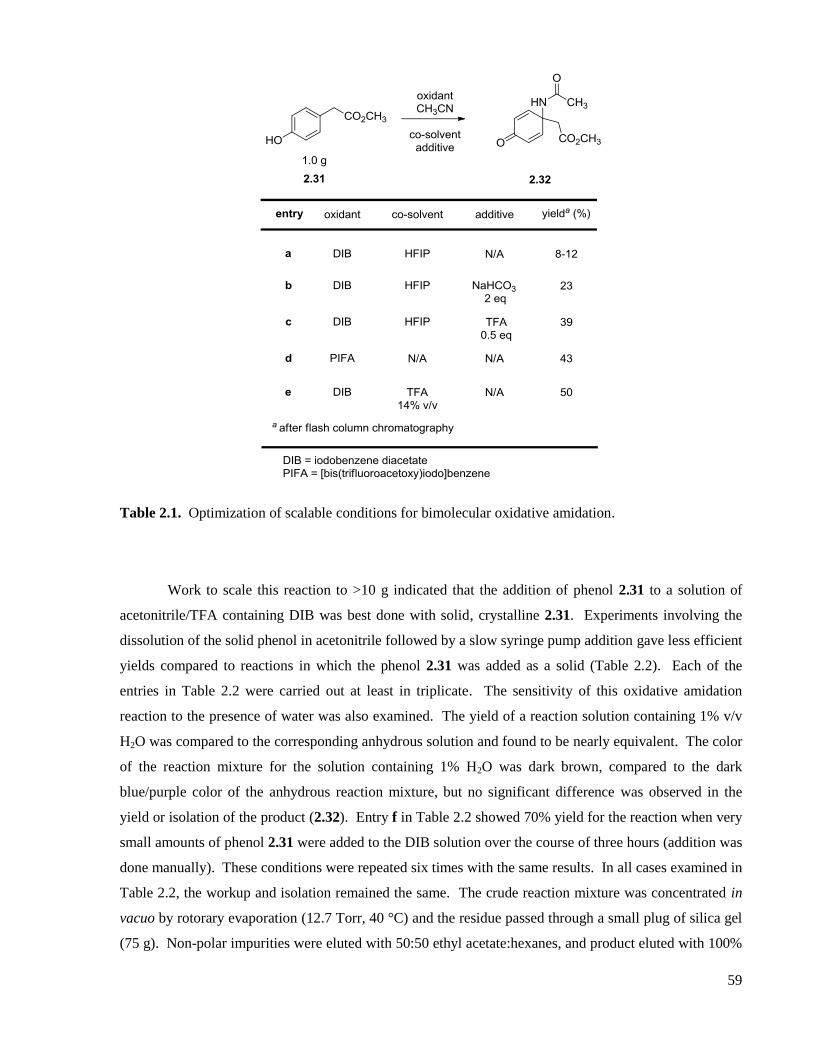
59
Table 2.1. Optimization of scalable conditions for bimolecular oxidative amidation.
Work to scale this reaction to >10 g indicated that the addition of phenol 2.31 to a solution of
acetonitrile/TFA containing DIB was best done with solid, crystalline 2.31. Experiments involving the
dissolution of the solid phenol in acetonitrile followed by a slow syringe pump addition gave less efficient
yields compared to reactions in which the phenol 2.31 was added as a solid (Table 2.2). Each of the
entries in Table 2.2 were carried out at least in triplicate. The sensitivity of this oxidative amidation
reaction to the presence of water was also examined. The yield of a reaction solution containing 1% v/v
H2O was compared to the corresponding anhydrous solution and found to be nearly equivalent. The color
of the reaction mixture for the solution containing 1% H2O was dark brown, compared to the dark
blue/purple color of the anhydrous reaction mixture, but no significant difference was observed in the
yield or isolation of the product (2.32). Entry f in Table 2.2 showed 70% yield for the reaction when very
small amounts of phenol 2.31 were added to the DIB solution over the course of three hours (addition was
done manually). These conditions were repeated six times with the same results. In all cases examined in
Table 2.2, the workup and isolation remained the same. The crude reaction mixture was concentrated in
vacuo by rotorary evaporation (12.7 Torr, 40 °C) and the residue passed through a small plug of silica gel
(75 g). Non-polar impurities were eluted with 50:50 ethyl acetate:hexanes, and product eluted with 100%

60
ethyl acetate. The enriched product 2.32 crystallized upon standing, and was recrystallized from a hot
solution of acetone:diethyl ether (1:1). Very large crystals of compound 2.32 were obtained from
recrystallization (2cm X 2cm X 1cm) which allowed for X-ray crystallographic analysis.156
40-50
HO
CO2CH3
O
HN CH3
O
a after flash column chromatographyb additional product present in supernatant following recrystallization
CO2CH3
DIBCH3CN/TFA
additive
entry yielda (%)
a N/A 25-30
b N/A 20-30
c N/A 40-50
d
70b+
2.31 2.32
additive
H2O1 % v/v
f N/A
DIB = iodobenzene diacetate
addition
syringe pump(3 h)
solid addition(all at once)
solid addition(500 mg/30 min)
solid addition(500 mg/30 min)
solid addition(constant manualaddition over 3 h)
10 g
45-55e N/Asolid addition(250 mg/15 min)
Table 2.2. Larger-scale reproducible conditions for oxidative amidation of phenol 2.31 with acetonitrile.

61
2.4 Nitrile oxide [3+2] cycloaddition
The next objective, once the bimolecular oxidative amidation reaction to create dienone 2.32 was
optimized, was to convert 2.32 into a functionalized enone of type 2.5 as Figure 2.3. The
desymmetrization of dienones of type 2.8 enables stereoselective access to the tetrasubstituted nitrogen-
bearing carbon in enantiomers 2.33 and 2.34 (Figure 2.12). Structures of type 2.33/2.34 would arise from
the selective addition of a generic reagent X-Y across either the pro-(R) or pro-(S) -bond of 2.8. 1,3-
Dipolar cycloaddition reactions offered a rapid route for the construction of a wide variety of five-
membered heterocycles,157,158
and are well-documented methods for the synthesis of isooxazolines.159-162
Isooxazolines are established precursors to amino ketones, oxo alcohols and other functional groups as
well as a variety of natural products from reduction of the N-O bond or other ring-fragmentations.157-162
It
was in this mode that we approached the desymmetrization as described generally in Figure 2.2: a nitrile
oxide 1,3-dipolar cycloaddition reaction with the C-9 arm delivering the dipole to the dienone to install
the functional units at C-4a/C-5.
Figure 2.12. Desymmetrization of dienone 2.8.
In anticipation for the advancement of the ester functionality of 2.32 to an -nitroketone163
as a
precursor to reactive nitrile oxide intermediate 2.3 (Figure 2.2), the reduction and protection of dienone
2.32 was desired. Dienone 2.32 proved to be an excellent Michael-acceptor, and the reduction/protection
sequence suppressed the possibility of unwanted later-stage intramolecular Michael additions. Various
reduction conditions were screened (Table 2.3). Initial conditions involved NaBH4 and
NaBH4/CeCl3164,165
and resulted in roughly a 1:1 ratio of diastereomers 2.35 and 2.36. A DIBAL-H
reduction of 2.32 at –78 °C gave, in good yield, the -diastereomer 2.35. The diastereoselectivity of this

62
reduction can be reversed with a (S)-CBS/BH3 reduction166,167
using 1 mol % of CBS reagent at room
temperature.168
Both diastereomers 2.35 and 2.36 have further downstream utility in this approach to the
tetrodotoxin core. The relative configurations of the doubly allylic alcohols 2.35 and 2.36 was
determined on the basis of X-ray crystallographic studies of later intermediates (vide infra).
Table 2.3. Reduction of dienone 2.32.

63
Compound 2.35, the -diastereomer from the DIBAL-H-mediated reduction of dienone 2.32, was
protected as the TBDPS-ether using standard conditions, and the methyl ester cleaved with aqueous
sodium hydroxide to afford carboxylic acid 2.38 (Scheme 2.2). Acidification of the reaction mixture after
saponification of the methyl ester with aqueous sodium hydroxide needed to be done carefully, ensuring
that the apparent pH of the solution stayed >2 to avoid lactonization to 2.41 as per Scheme 2.3.169
Activation of acid 2.38 with carbonyldiimidazole (CDI)170,171
and condensation of the resultant acid
imidazolide with the nitronate of nitromethane (generated in situ from nitromethane and potassium tert-
butoxide) generated nitroketone 2.39 in high yield after flash column chromatography.172
Scheme 2.2. Synthesis of nitroketone 2.39.
ONHAc
CO2HTBDPS
-OTBDPS = 2.40-OTBDPS = 2.38
pH < 2
O
O
H
NHAc
2.41
quantitative
Scheme 2.3. Undesired cyclization reaction of intermediate 2.40/2.38.
The conversion of nitroketone 2.39 to the reactive intermediate nitrile oxide 2.42 formally
entailed a dehydration reaction. A generic reagent X-Y could be reacted across the nitro group as
indicated in Figure 2.13, affecting the dehydration of nitroketone 2.39 to -keto-nitrile oxide173-175
2.42.

64
O NHAc
H
TBDPS
ON
OO
X-Y
HH
O NHAc
H
TBDPS
ON
OO
H
XY
O NHAc
H
TBDPS
ONO
2.422.39
Figure 2.13. Theoretical dehydration of nitroketone 2.39.
The dehydration of nitroketone 2.39 proved quite troublesome (Table 2.4). Initially this was
examined using various reagents known for their use in nitro-dehydration reactions. A technique for
converting nitroalkanes to their corresponding nitrile oxides was Mukaiyama’s aromatic isocyanate
method176
which used such reagents as 4-chlorophenyl isocyanate.177,178
Reactions with nitroketone 2.39
and 4-chlorophenyl isocyanate gave complex mixtures of products. Other known methods for converting
alkyl nitro compounds into their nitrile oxide derivatives were attempted as well. Shimizu’s method179
using ethyl chloroformate resulted in the slow conversion of nitroketone 2.39 to adduct 2.43. Hassner’s
method,97
used by Fukuyama in his work on the TTX core (Chapter 1.2.10), using 4-(N,N-
dimethylamino)-pyridine (DMAP) and di-tert-butyl-pyrocarbonate (Boc2O) was ineffective as well.
These methods and reagents, and several others, failed to efficiently produce the desired intramolecular
nitrile oxide cycloaddition product 2.43 (Figure 2.14). Common side product 2.44, whose structure was
ascertained by X-ray crystallography, arose formally from the intramolecular O-alkylation of the
nitroketone enolate via allylic displacement of the OTBDPS unit, and could be generated in high yield by
treating the nitroketone 2.39 with triethylamine (Scheme 2.4). The initial data from the studies described
in Table 2.4 were tantalizing: trace amount of the desired tricycle 2.43 appeared to be present according
to 1H-NMR studies on crude reaction materials. This indicated that the transformation indeed must be
possible, but that efficient reaction conditions remained elusive.
O NHAc
H
TBDPS
OO2N
O
O
NHAc
H
TBDPS
NO
O NHAc
H
TBDPS
ONO
[3+2] dipolar cycloaddition
2.432.422.39
Figure 2.14. Nitrile oxide [3+2] dipolar cycloaddition.
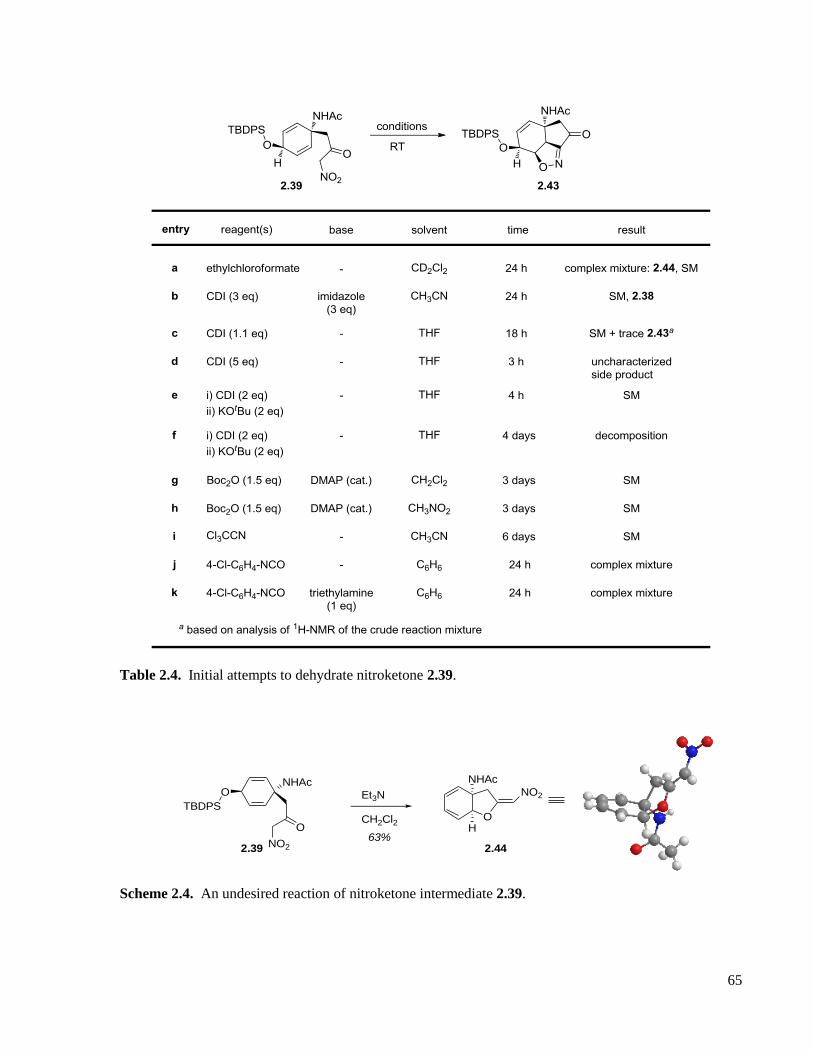
65
Table 2.4. Initial attempts to dehydrate nitroketone 2.39.
ONHAc
TBDPS
O
NO2
Et3N
CH2Cl2 OH
NHAc
2.44
NO2
63%2.39
Scheme 2.4. An undesired reaction of nitroketone intermediate 2.39.

66
Sensing that the -keto-group of nitroketone 2.39 could be responsible for the undesired
reactivity seen thus far, we sought to alleviate this potential issue through a reduction/protection sequence
(Scheme 2.5). Thus, NaBH4 treatment of nitroketone 2.39 provided unstable nitro alcohol 2.45, a
sensitive material that was prone to undergo a retro-Henry fragmentation event, especially upon
manipulations or attempted purification. Accordingly, crude nitro alcohol 2.45 was immediately O-
silylated with TBSCl/imidazole.
O
NO2
TBDPSO
NHAc
2.39
NaBH4 (5 eq)CH3OH
OH
NO2
TBDPSO
NHAc
2.45
TBSCl (1.5 eq)imidazole
OTBS
NO2
TBDPSO
NHAc
2.46
Scheme 2.5. Reduction/protection sequence of nitroketone 2.39.
A careful analysis of 1H-NMR data from the crude reaction mixture to synthesize compound 2.46
indicated the presence of small quantities of tricyclic materials of the type 2.49/2.50/2.51/2.52. The O-
silylation event indicated in Scheme 2.6 proceeded with concomitant Torssell-type cyclization162
(Figure
2.15). Thus purified TBS-ether 2.46 was treated with TBSCl (4 eq) and imidazole (4 eq) and resulted in a
1:1 mixture of tricyclic isooxazolines 2.49/2.50 (Scheme 2.6). The lack of diastereoselectivity signaled
that the steric demand of the OTBS group was insufficient to exert stereocontrol during the cycloaddition
step (see Figure 2.12). Notably, minor amounts of a mixture of diastereomeric nitroso acetals180
2.51 and
2.52 were isolated following flash column chromatography of the desired isooxazolines 2.49/2.50. Upon
prolonged standing, both 2.51 and 2.52 spontaneously converted to the desired 2.49 and 2.50. A sample
of enriched diastereomer 2.51/2.52 (6% yield) was isolated by flash column chromatography and
characterized. These materials (2.51/2.52) converted to compound 2.49/2.50 upon standing. The
presence of nitroso acetals 2.51 and 2.52 implicated the intermediacy of siloxy nitronate162,180-184
2.48 en
route to desired isooxazolines 2.49 and 2.50. The desired product formed from an intramolecular
siloxynitronate-olefin cycloaddition (ISOC) process. It was unclear if the ISOC pathway occurs
simultaneously with the expected INOC pathway or exclusively.
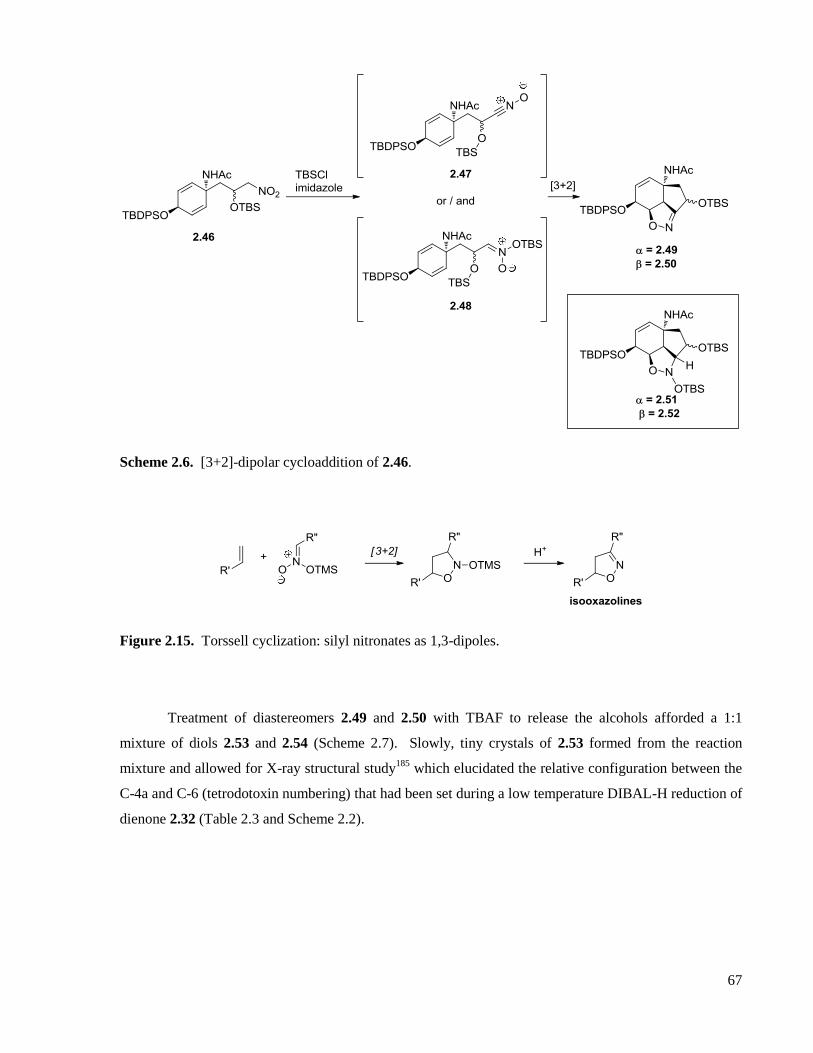
67
Scheme 2.6. [3+2]-dipolar cycloaddition of 2.46.
Figure 2.15. Torssell cyclization: silyl nitronates as 1,3-dipoles.
Treatment of diastereomers 2.49 and 2.50 with TBAF to release the alcohols afforded a 1:1
mixture of diols 2.53 and 2.54 (Scheme 2.7). Slowly, tiny crystals of 2.53 formed from the reaction
mixture and allowed for X-ray structural study185
which elucidated the relative configuration between the
C-4a and C-6 (tetrodotoxin numbering) that had been set during a low temperature DIBAL-H reduction of
dienone 2.32 (Table 2.3 and Scheme 2.2).

68
8a
6
TBDPSO
O N
OTBS
NHAc
TBAF
HO
O N
OH
NHAc
2.53
HO
O N
OH
NHAc
2.54
+
= 2.49 = 2.50
Scheme 2.7. Confirmation of structural geometry: X-ray crystallographic analysis of 2.53.
The success with the TBSCl-mediated conversion of 2.46 to cyclized products 2.49/2.50
prompted attempts to try similar conditions for the originally-planned nitroketone dehydration. Treatment
of nitroketone 2.39 under various conditions based around TBSCl (Table 2.5) were attempted. This
reaction appeared to be quite slow, with conversions to product taking place over approximately a week.
Attempts to accelerate the reaction by the addition of 10 equivalents of TBSCl and imidazole each
resulted in the unexpected formation of the noteworthy dihydropyridone 2.55 as the major product (65%,
Scheme 2.8) with concomitant decrease in the yield of the desired isooxazoline 2.43 (5% yield after flash
column chromatography). The architecturally unique structure of 2.55 was elucidated by X-ray
crystallographic analysis and there appeared to be no other reports of similar molecules in the literature.
The compound formally evolved from an unusual Knoevenagel-type186,187
intramolecular condensation of
the nitroketone arm of 2.39 with the attached acetamide; it was presumed to proceed through the O-silyl
imino ether derivative of the C-8a acetamide unit. Other reactive silylating reagents such as TMS-OTf
and TBS-OTf resulted in the rapid and complete degradation of starting nitroketone 2.39.
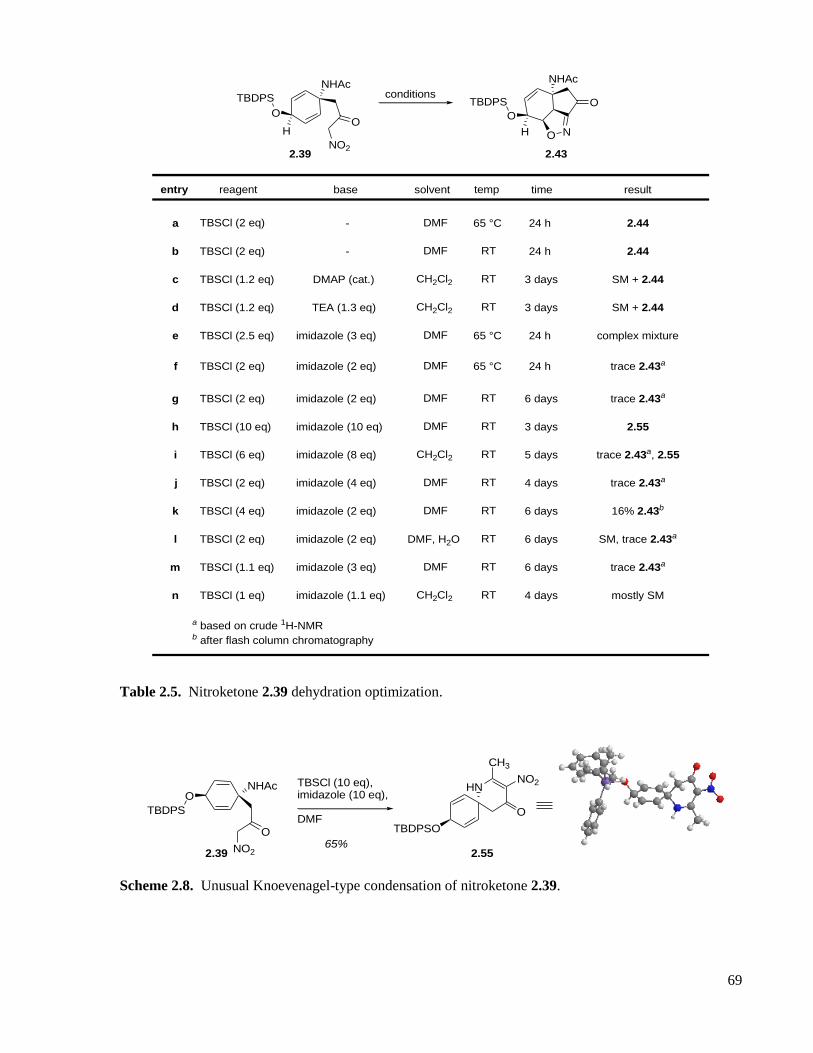
69
2.39 2.43
conditions
O
NHAc
H
TBDPS
O
NO2
O
O
NHAc
H
TBDPS
NO
entry reagent base solvent temp time result
a
b
c
d
e
f
g
h
i
j
k
TBSCl (2 eq) - DMF 65 °C 24 h 2.44
TBSCl (2 eq) - DMF RT 24 h 2.44
TBSCl (1.2 eq) DMAP (cat.) CH2Cl2 RT 3 days SM + 2.44
TBSCl (1.2 eq) TEA (1.3 eq) CH2Cl2 RT 3 days SM + 2.44
TBSCl (2.5 eq) imidazole (3 eq) 65 °CDMF 24 h complex mixture
TBSCl (2 eq) imidazole (2 eq) DMF 65 °C 24 h trace 2.43a
TBSCl (2 eq) imidazole (2 eq) DMF RT 6 days trace 2.43a
TBSCl (10 eq) imidazole (10 eq) DMF RT 3 days 2.55
TBSCl (6 eq) imidazole (8 eq) CH2Cl2 RT 5 days trace 2.43a, 2.55
TBSCl (2 eq) imidazole (4 eq) DMF RT 4 days trace 2.43a
TBSCl (4 eq) imidazole (2 eq) DMF RT 6 days 16% 2.43b
l TBSCl (2 eq) imidazole (2 eq) DMF, H2O RT 6 days SM, trace 2.43a
m TBSCl (1.1 eq) imidazole (3 eq) DMF RT 6 days trace 2.43a
n TBSCl (1 eq) imidazole (1.1 eq) CH2Cl2 RT 4 days mostly SM
a based on crude 1H-NMRb after flash column chromatography
Table 2.5. Nitroketone 2.39 dehydration optimization.
ONHAc
TBDPS
O
NO2 2.55
TBSCl (10 eq),imidazole (10 eq),
DMF
65%
HN
CH3
NO2
O
TBDPSO
2.39
Scheme 2.8. Unusual Knoevenagel-type condensation of nitroketone 2.39.

70
Further attempts to refine conditions for the desired conversion of 2.39 to tricycle 2.43 (Table
2.6) narrowed the search to two-equivalents each of TBSCl and imidazole in dichloromethane at room
temperature. These reactions were run at small scale (25-50 mg each). At this time, it was still not
possible to resolve the question of whether 2.43 is formed via an INOC or an ISOC mechanism or
possibly both. Entry k in Table 2.6 showed that a moderate amount of desired product 2.43 could be
obtained after treatment of nitroketone 2.39 with TBSCl/imidazole (2 equivalents each), after a week-long
reaction time at room temperature.
Table 2.6. Refinements to [3+2] cycloaddition conditions.
The small scale conditions (Table 2.6, entry k) were transferred to a larger scale (1-5 g) and the
same yield was observed (Scheme 2.9). It was noted that the product began to decompose slowly over
time in the reaction mixture, so the reaction was typically stopped after a week and the crude material
purified by silica gel chromatography. The starting material (2.39) was recovered (35%) in addition to

71
the desired product, thus bringing the yield of 2.43 to a moderate 58% based on recovered starting
material (BRSM).
Scheme 2.9. Optimized conditions for dehydration of nitroketone 2.39.
We speculated that the relatively poor reactivity of the -nitro ketone 2.39 could have been due to
the facile enolization of 2.39 (Figure 2.16). When enolate 2.57 was exposed to TBSCl/imidazole, we
speculated that the formation of both 2.56 and 2.58 were possible. Reaction on the oxygen atom which
was formerly the -ketone would lead to synthetic dead-end 2.58 which was unable to undergo either of
the desired INOC or ISOC pathways. Conversely, reaction of the silyl halide on an oxygen atom of the
nitro group could lead to intermediate 2.56 which we postulate undergoes the desired [3+2] cycloaddition
in an ISOC and/or INOC mode. The slow conversion of nitroketone 2.39 to tricycle 2.43 could also have
been due to a possible interconversion of silyl intermediates 2.58 and 2.56.
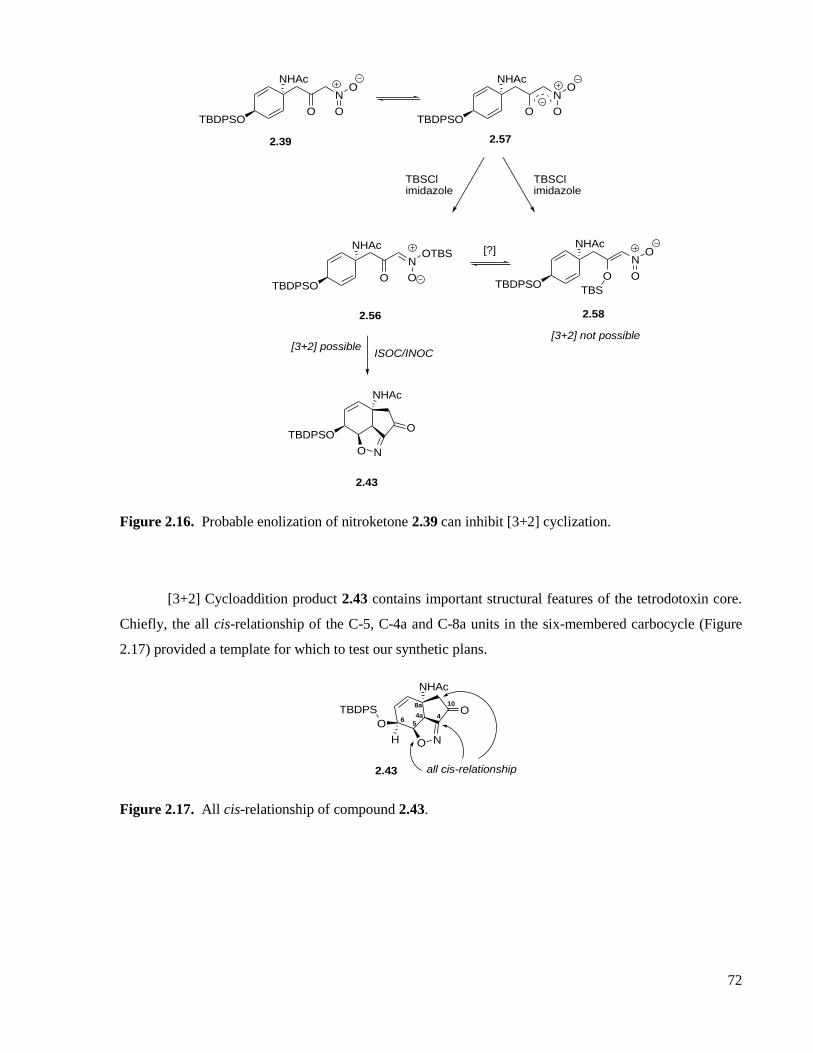
72
OTBDPSO
NHAc
2.39
O
N
TBDPSO
NHAc
2.57
OTBDPSO
NHAc
N
O
O
TBS
2.58
[3+2] not possible
TBSClimidazole
TBSClimidazole
OTBDPSO
NHAc
N
O
OTBS
2.56
[3+2] possible
TBDPSO
O N
O
NHAc
2.43
ISOC/INOC
O
ON
O
O
[?]
Figure 2.16. Probable enolization of nitroketone 2.39 can inhibit [3+2] cyclization.
[3+2] Cycloaddition product 2.43 contains important structural features of the tetrodotoxin core.
Chiefly, the all cis-relationship of the C-5, C-4a and C-8a units in the six-membered carbocycle (Figure
2.17) provided a template for which to test our synthetic plans.
4a6
8a
45
10
O
O
NHAc
H
TBDPS
NO
all cis-relationship2.43
Figure 2.17. All cis-relationship of compound 2.43.

73
2.5 Kemp-type keto-isooxazoline fragmentation
At this juncture, attention turned to focus on the development of a procedure for the
fragmentation of keto-isooxazoline 2.43. The base-mediated ring-opening of benzisooxazoles to give -
cyano phenols is known as the Kemp elimination188-190
(Scheme 2.10). The resulting Kemp-elimination
fragment contained the requisite hydroxyl group (for C-5) and a formyl-equivalent nitrile (for C-4a) if this
concept were applied towards keto-isooxazolines such as 2.43. The tricyclic ring geometry of compounds
like 2.43 suffered from strain, and if a nuclophile such as methoxide were to add to the carbonyl, we
hypothesized that a ring fragmentation event would follow, yielding an -cyano alcohol formally
resembling Kemp-elimination products. As with the previous section, the implementation of an efficient
method to achieve the desired outcome required a good deal of experimentation.
Scheme 2.10. Ring-fragmentation and comparison to Kemp-elimination188-190
products.
Initial attempts to fragment tricyclic isooxazoline structures as per Figure 2.2 were centered on
intermediate 2.43 (Table 2.7). Treatment of 2.43 with sodium methoxide in methanol or potassium
carbonate in alcohols (methanol, ethanol, benzyl alcohol) failed to induce the desired conversion.
Catalytic amounts of lithium carbonate in methanol resulted in some conversion to the desired 2.59,
however reaction times were long and complex mixtures of cyclohexene 2.59 were obtained. Imidazole
in methanol as well as DMAP did not have any effect on isooxazoline 2.59. Finally, it transpired that
efficient recovery (68% isolated yield) of the desired 2.59 was achieved by ring opening with catalytic
amounts of lithium carbonate and imidazole in methanol (Table 2.7, entry g).
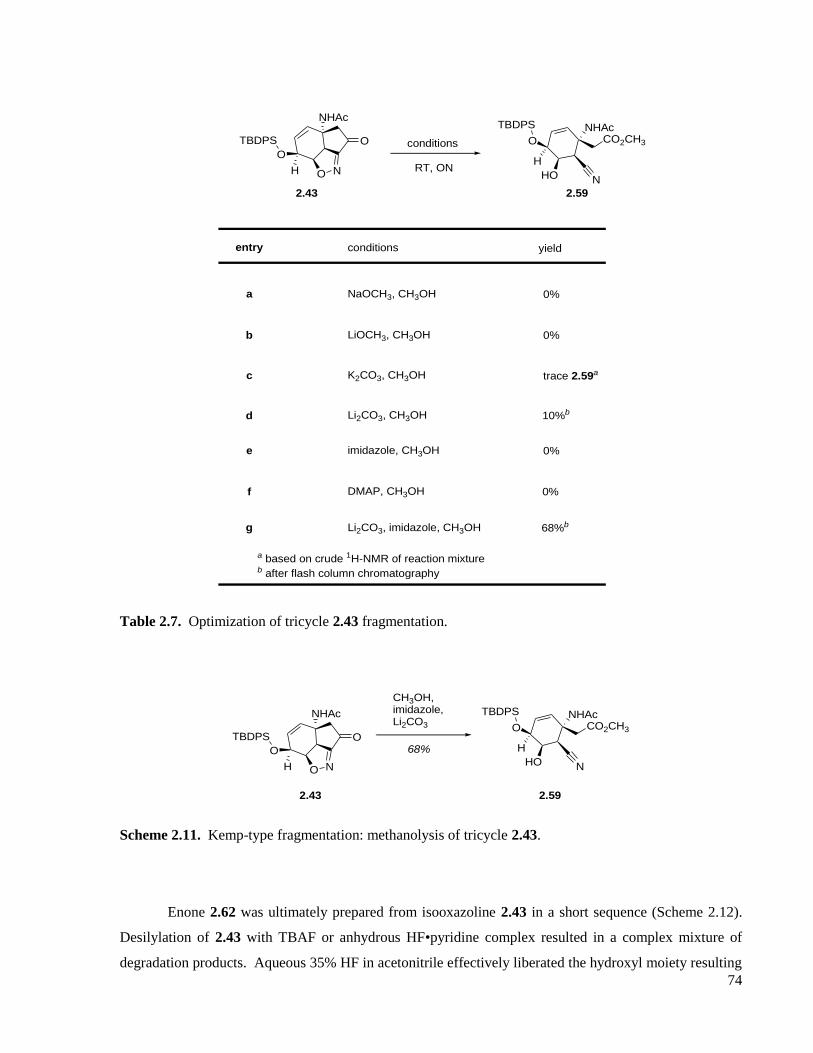
74
conditions
conditions yield
K2CO3, CH3OH trace 2.59a
imidazole, CH3OH 0%
2.43 2.59
NaOCH3, CH3OH 0%
LiOCH3, CH3OH 0%
O
O
NHAc
H
TBDPS
NO
O CO2CH3
H
TBDPS
HO N
NHAc
RT, ON
Li2CO3, imidazole, CH3OH 68%b
DMAP, CH3OH 0%
Li2CO3, CH3OH 10%b
a based on crude 1H-NMR of reaction mixtureb after flash column chromatography
entry
a
b
c
d
e
f
g
Table 2.7. Optimization of tricycle 2.43 fragmentation.
O CO2CH3
H
TBDPS
HO N
NHAc
68%O
O
NHAc
H
TBDPS
NO
CH3OH, imidazole,Li2CO3
2.43 2.59
Scheme 2.11. Kemp-type fragmentation: methanolysis of tricycle 2.43.
Enone 2.62 was ultimately prepared from isooxazoline 2.43 in a short sequence (Scheme 2.12).
Desilylation of 2.43 with TBAF or anhydrous HF•pyridine complex resulted in a complex mixture of
degradation products. Aqueous 35% HF in acetonitrile effectively liberated the hydroxyl moiety resulting

75
in a 70% recovered yield of compound 2.60. A Dess-Martin191,192
oxidation of enol 2.60 provided
diketone 2.61 which degraded upon prolonged chemical manipulations. For this reason, crude diketone
2.61 was treated with catalytic lithium carbonate and imidazole in methanol to afford the target (2.62) in
38% isolated yield over the two chemical transformations.
a) aq HF, THF; b) Dess-Martin periodinane, CH2Cl2; c) CH3OH, imidazole, Li2CO3.
O
CO2CH3
HO N
NHAc
O
O
NHAc
H
TBDPS
NO
c38%over2-steps
a
70%HO
O
NHAc
H NO
b
O
O
NHAc
NO
2.43 2.60 2.61
2.62
Scheme 2.12. Fragmentation sequence: conversion of 2.43 to desymmetrized enone 2.62.
My work in this thesis described thus far had demonstrated the feasibility of the planned approach
to the tetrodotoxin core (Figure 2.3) with the C-8 nitrogen installed through the oxidative amidation of a
phenol and the successful introduction of the C-5 hydroxyl and the C-4a formate-equivalent nitrile.
Planned TTX retron 2.5 (cf. Figure 2.3) possessed similarity to isolated product 2.62; formally a C-9
oxidation and a partial reduction of the C-4a cyano group were necessary to elaborate our TTX-core
intermediate. Other key features of the tetrodotoxin core molecule not yet addressed at this point
included the installation of the C-11 hydroxymethyl unit and the cis-dihydroxylation across the C-7/C-8
olefin.
O
CO2CH3
OHN
NHAc
2.62
H
HO
OHO
NHAc
2.5
H
H CO3CH3
OP8a
6 4a5
7
8
oxidation
reduction
Figure 2.18. Comparison of prepared enone 2.62 to proposed tetrodotoxin retron 2.5.

76
2.5.1 Access to a suitable dihydroxylation substrate
With the synthesis of enone 2.62 accomplished, attention turned to the selective C-7/C-8
dihydroxylation. Advanced intermediates of the type 2.5/2.62 (Figure 2.18) were not suitable substrates
for the exploration of the required selective dihydroxylation due to the lack of steric encumbrance on the
-face of the carbocycle. Cyclohexene-derivative 2.59 was likewise not a suitable substrate as the bulky
-OTBDPS group would likely have directed an osmium tetraoxide-mediated dihydroxylation from the
incorrect face of the molecule. However, if the C-6 epimer of compound 2.59 were obtained (see
compound 2.67), we envisioned that a large -OTBDPS group could provide enough steric demand as to
block the bottom face and appropriately direct the dihydroxylation. With this in mind, we revisited the
reduction of dienone 2.32 (cf. Table 2.3). A CBS-reduction of dienone 2.32 provided an inseparable
mixture of doubly-allylic alcohols 2.35 and 2.36 in approximately 1:2 ratio. This mixture of
diastereomers was treated with the same reaction sequence as in Scheme 2.2 to provide a mixture of
nitroketones 2.39 and 2.65 (Scheme 2.13). In this sequence, none of the diastereomeric product mixtures
were rigorously purified and all compounds were carried forward without purification.
HONHAc
CO2CH3HO
NHAc
CO2CH3+
1 : 2
TBDPS-ClimidazoleCH2Cl2
ONHAc
CO2CH3O
NHAc
CO2CH3+
1 : 2
TBDPS TBDPS
NaOHH2O/THF
ONHAc
CO2HO
NHAc
CO2H+
1 : 2
TBDPS TBDPS
1) CDI, THF2) KOtBu, CH3NO2
ONHAc
ONHAc
+
1 : 2
TBDPS TBDPS
O O
NO2NO2
2.35 2.36 2.37 2.63
2.642.382.652.39
Scheme 2.13. Synthesis of nitroketones 2.39 and 2.65.
Additionally, it warranted mention that we expected the conversion of nitroketone 2.65 to tricycle
2.66 (Scheme 2.14) to proceed at a faster rate relative to the conversion of nitroketone 2.39 to tricycle
2.43. This expectation was based on the hypothesis that the nitrile oxide-containing arm, as it approached
the olefin, could encounter the bulky -OTBDPS unit (Figure 2.19). We expected that this steric clash,
which was not possible for nitroketone 2.65 with its -OTBDPS unit orientated away from the emerging
1,3-dipole, would result in a difference in reaction rate and could enhance the efficiency of the dipolar
cycloaddition of nitroketone 2.65 relative to nitroketone 2.39.

77
Figure 2.19. Rationale for expected (but not observed) rate difference between nitroketones 2.39/2.65.
It was not possible to efficiently separate nitroketone intermediates 2.39 and 2.65, thus the
mixture was treated with the conditions optimized for [3+2] cycloaddition (Chapter 2.4). Similar yields
were obtained using the mixture of diasteromers 2.39 and 2.65 as were obtained from the single
diasteromer 2.39. Recovered nitroketones 2.39 and 2.65 were recycled following flash column
chromatography, rendering tricyclic products 2.43 and 2.66 in 20% and 47% yield respectively based on
the recovered starting materials. The results indicated in Scheme 2.14 showed that our expectation that
nitroketone 2.65 would react faster than nitroketone 2.39 was incorrect.
TBS-Cl,imidazole,
CH2Cl2
+
1 : 2
O N
NHAc
OO
TBDPS
+
O N
NHAc
OO
TBDPS
12%20% BRSM
28%47% BRSM
ONHAc
ONHAc
+TBDPS TBDPS
O O
NO2NO2
40% recovery of starting material
2.39 2.65
2.39 2.652.43 2.66
+
Scheme 2.14. Synthesis of tricycles 2.43 and 2.66.
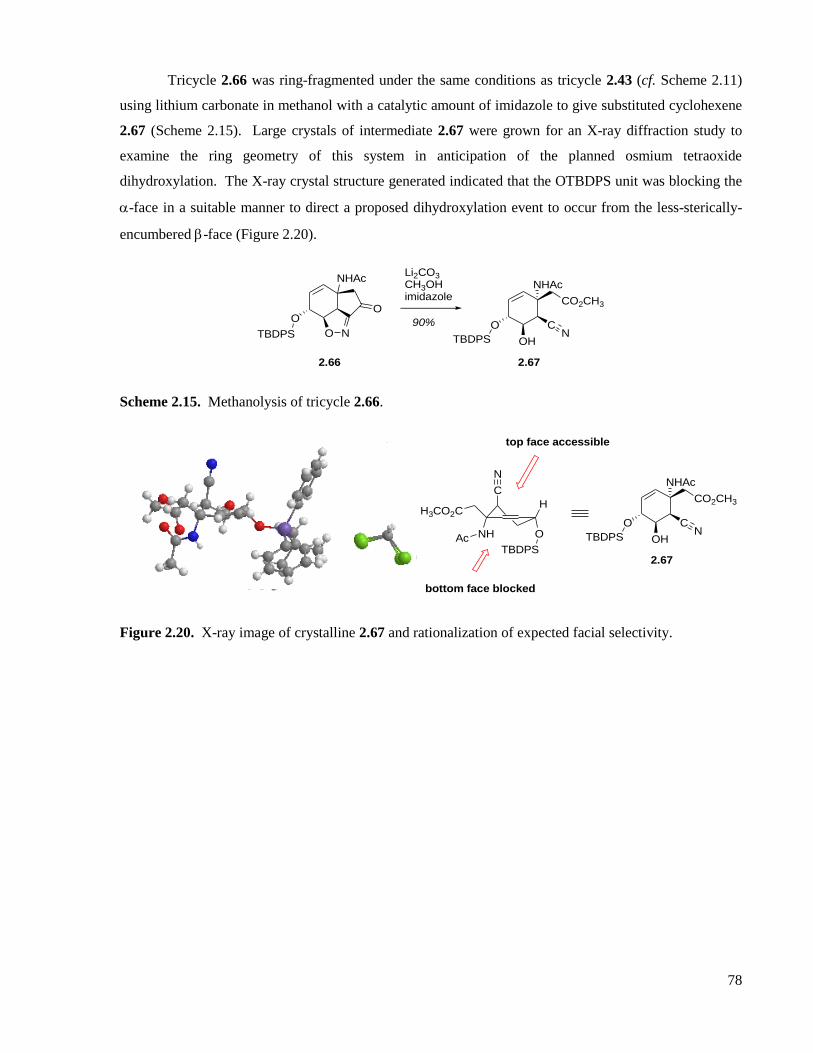
78
Tricycle 2.66 was ring-fragmented under the same conditions as tricycle 2.43 (cf. Scheme 2.11)
using lithium carbonate in methanol with a catalytic amount of imidazole to give substituted cyclohexene
2.67 (Scheme 2.15). Large crystals of intermediate 2.67 were grown for an X-ray diffraction study to
examine the ring geometry of this system in anticipation of the planned osmium tetraoxide
dihydroxylation. The X-ray crystal structure generated indicated that the OTBDPS unit was blocking the
-face in a suitable manner to direct a proposed dihydroxylation event to occur from the less-sterically-
encumbered -face (Figure 2.20).
O N
NHAc
OO
TBDPS
Li2CO3
CH3OH imidazole
OH
O CN
CO2CH3
NHAc
90%
2.66 2.67
TBDPS
Scheme 2.15. Methanolysis of tricycle 2.66.
OH
O CN
CO2CH3
NHAc
2.67
TBDPS
C
N
NH
H3CO2C
Ac O
H
TBDPS
bottom face blocked
top face accessible
Figure 2.20. X-ray image of crystalline 2.67 and rationalization of expected facial selectivity.

79
2.6 Osmylation of substituted cyclohexene derivative
Compound 2.67 was treated with N-methyl morpholine oxide and catalytic osmium tetraoxide193
but no reaction was observed after more than a week reaction time (this reaction was monitored
periodically for 60 days). Not dissuaded by the failure of the commonly used Upjohn-procedure, we
attempted to use stoichiometric osmium tetraoxide to cause the osmylation of the C-7/C-8 olefin. In an
NMR-scale experiment (Scheme 2.16), compound 2.67 was dissolved in d5-pyridine and to this solution
was added solid crystals of osmium tetraoxide. The reaction was monitored by 1H-NMR and the addition
of solid OsO4 continued until the starting material 2.67 was fully consumed.
Scheme 2.16. Osmylation sequence.
Osmate ester 2.68 proved to be too fragile for rigorous purification/isolation sequence. Attempts
to isolate pure 2.68 resulted in the decomposition of the product. Additionally, the purported osmate 2.68
appeared to be resistant to (unoptimized) attempts at removing the osmium moiety. The structure of
compound 2.68 was proposed to be that indicated in Scheme 2.16 based on enriched samples of 2.68 and
careful analysis of 1D and 2D NMR data (cf. Figures 2.21 and 2.22).
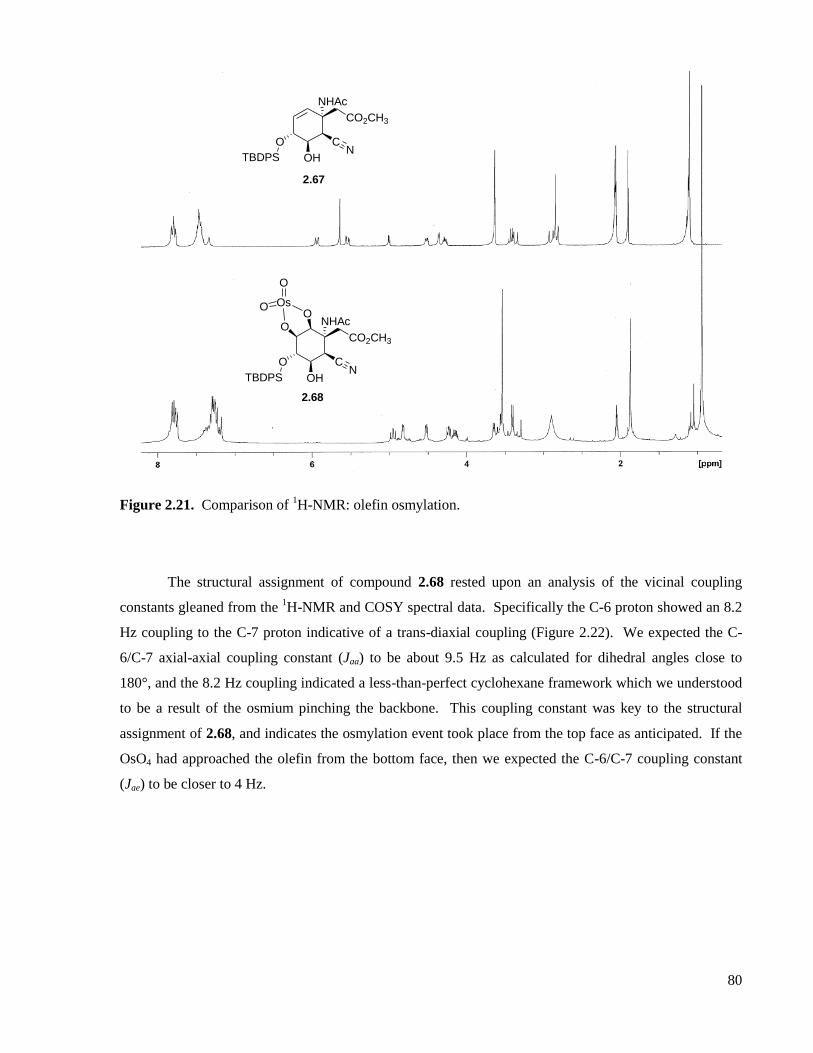
80
Figure 2.21. Comparison of 1H-NMR: olefin osmylation.
The structural assignment of compound 2.68 rested upon an analysis of the vicinal coupling
constants gleaned from the 1H-NMR and COSY spectral data. Specifically the C-6 proton showed an 8.2
Hz coupling to the C-7 proton indicative of a trans-diaxial coupling (Figure 2.22). We expected the C-
6/C-7 axial-axial coupling constant (Jaa) to be about 9.5 Hz as calculated for dihedral angles close to
180°, and the 8.2 Hz coupling indicated a less-than-perfect cyclohexane framework which we understood
to be a result of the osmium pinching the backbone. This coupling constant was key to the structural
assignment of 2.68, and indicates the osmylation event took place from the top face as anticipated. If the
OsO4 had approached the olefin from the bottom face, then we expected the C-6/C-7 coupling constant
(Jae) to be closer to 4 Hz.
OH
O CN
CO2CH3
NHAc
2.67
TBDPS
OH
O CN
CO2CH3
NHAcO
Os
O
O
O
2.68
TBDPS

81
Figure 2.22. 1H-NMR couplings for 2.68.
Compound 2.68 contains key structural features compared to TTX retron 1.170. The lack of C-9
hydroxyl group and the C-11 hydroxymethyl group in compound 2.68 were the major differences
between these structures.
HO
O
NC CO2CH3
NHAc
O
O
2.68
TBDPS OH
OH
COOH
HO
NH2
HO
HO
OHC
OH
1.170
4a
6
8a
4
5
78
10
6
4a8a5
78
4
10
OsO2
Figure 2.23. Comparison of advanced intermediate 2.68 with TTX retron 1.170.

82
2.7 Iodine(III)-mediated oxime oxidation to nitrile oxides
The apparent sluggish reaction times for the conversion of nitroketones 2.39 and 2.65 to their
respective tricyclic isooxazoline products (Scheme 2.14) led us to investigate alternative nitrile oxide
precursors.
2.7.1 Oximes as nitrile oxide precusors
Oximes are established nitrile oxide precursors.100,101
Oxidation of oximes with various reagents
have generated nitrile oxide intermediates which can be trapped in a [3+2]-mode with a variety of
dipolarophiles (Figures 2.24 and 2.25). Nitrile oxides (generated from oximes or otherwise) were also
known to dimerize194
to give furoxans195,196
of the type 2.71 (Figure 2.24); these products were often used
as evidence for the formation of highly reactive nitrile oxide intermediates. Reagents known to oxidize
oximes to nitrile oxide species included lead tetraacetate,197
chloramine-T,198
mercuric acetate,199
1-
chlorobenzotriazole,200
manganese dioxide,201
halogens,202,203
dimethyldioxirane,194
and alkali
hypohalite204
such as tert-butyl hypochlorite205
and NaClO.206,207
Ceric ammonium nitrate has been used
to oxidize aromatic aldoximes to nitrile oxides with modest efficiency.208
Potassium ferricyanide-
mediated oxime oxidations were known to proceed only in aqueous solvents.209
Treatment of oximes
with N-chlorosuccinimide (NCS) generated the corresponding oximoyl chlorides, and subsequent addition
of base can cause the generation of a nitrile oxide as used by Ray and co-workers in their synthesis of
pyrimidoazepine-based derivatives.210,211
H
NOH
2.702.69
NN
O
RR C N OR
2.71
O O
CH3H3C
R
O
Figure 2.24. Dimerization of nitrile oxides.
Oximes can also be converted to nitrile oxides using iodine(III) reagents such as iodosylbenzene
(PhIO)194
and PhICl2212
(Figure 2.25). These iodine(III) reagents were known to require an alkaline
workup after the oxidation event, limiting the use of these reagents to base-insensitive substrates.212,213
Hypervalent iodine reagents have also been known to induce oxidative deoximation214-221
in lieu of nitrile

83
oxide formation. Despite the variety of methods that were available for the oxidation of oximes to nitrile
oxides, finding a new oxidizing agent would contribute to the chemistry of nitrile oxides.
H
NOH
2.702.69
N O
R3
R1R1Ph I
O
CHCl3
C N OR1
R2 R3
R2
2.72
Figure 2.25. Conversion of oximes to nitrile oxides and subsequent trapping.
2.7.2 Optimization of DIB as a reagent for oxime to nitrile oxide oxidations
There was no precedent in the chemical literature for the use of hypervalent iodine reagents
commonly employed for oxidative amidation reactions (DIB or PIFA) to oxidize oximes to nitrile oxides.
Our initial attempts (not optimized) to use DIB as a reagent to oxidize oximes centered on commercially
available oxime 2.73. When treated with DIB in methanol, oxime 2.73 readily formed known furoxan
2.74 which crystallized from the reaction mixture. Furoxan 2.74 was compared to literature data194
and
found to be identical, in addition to an unambiguous X-ray crystallographic derived structure (Scheme
2.17).
H
NOH
2.73
NN
O
OPhI(OAc)2
CH3OH
35-40%unoptimized
2.74
Scheme 2.17. Dimerization of oxime 2.73.
A test of the feasibility of a DIB-mediated oxime oxidation and subsequent capture of the newly
formed nitrile oxide with a dipolarophile began with commercially available oxime 2.75. The test
oxidation of 2.75 with DIB in a variety of solvents with varying equivalents of styrene as the
dipolarophile trap was summarized in Table 2.8. The typical procedure was dissolving oxime 2.75 in the
appropriate solvent followed by the slow addition of the oxime solution to a room temperature solution of
DIB and styrene in the same solvent. After the given time, the reaction mixture was concentrated in
vacuo and the crude reaction residue was purified by flash column chromatography. Excess of styrene
trap had a minor effect on recovered yields. Additionally, methanol was found to be an adequate
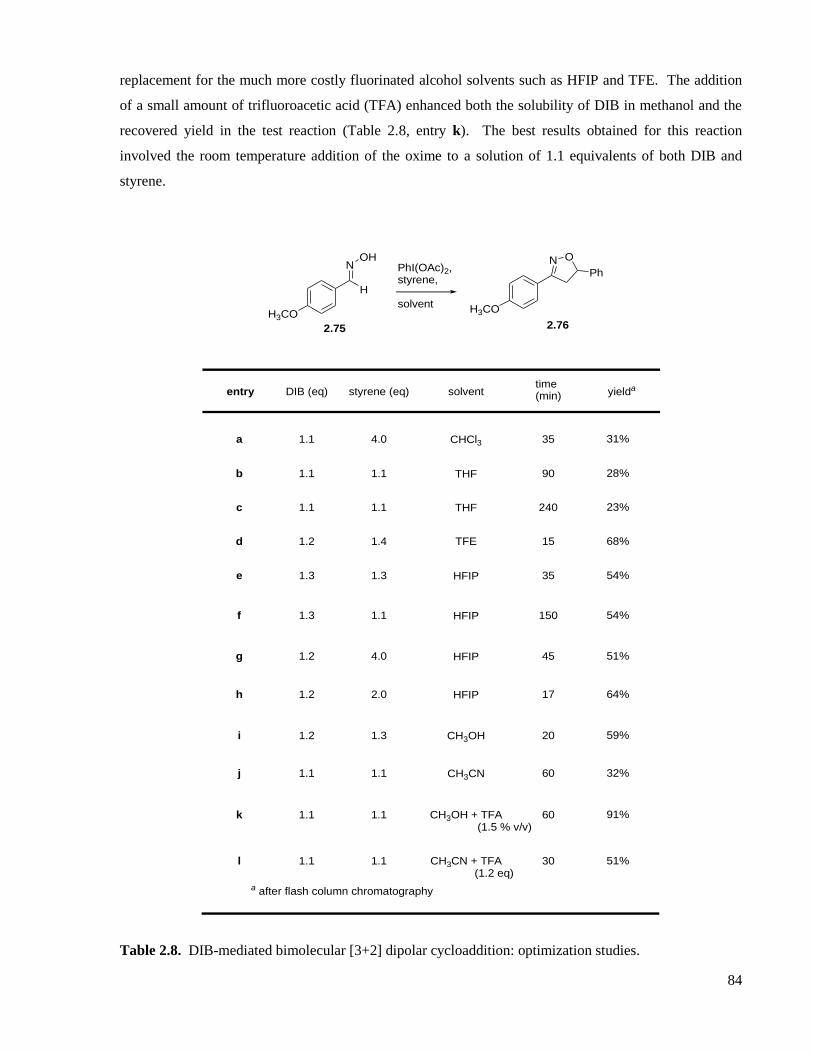
84
replacement for the much more costly fluorinated alcohol solvents such as HFIP and TFE. The addition
of a small amount of trifluoroacetic acid (TFA) enhanced both the solubility of DIB in methanol and the
recovered yield in the test reaction (Table 2.8, entry k). The best results obtained for this reaction
involved the room temperature addition of the oxime to a solution of 1.1 equivalents of both DIB and
styrene.
H
NOH
2.762.75
PhI(OAc)2,styrene,
solvent
entry styrene (eq) solvent yielda
a 31%
b 28%
c 23%
d 68%
e 54%
f 54%
g 51%
h 64%
i 59%
j 32%
k 91%
l 51%
H3CO
DIB (eq)time(min)
N O
Ph
H3CO
1.1 4.0 CHCl3 35
1.1 1.1 THF 90
1.1 1.1 THF 240
1.2 1.4 TFE 15
1.3 1.3 HFIP 35
1.3 1.1 HFIP 150
1.2 4.0 HFIP 45
1.2 2.0 HFIP 17
1.2 1.3 CH3OH 20
1.1 1.1 CH3CN 60
1.1 1.1 CH3OH + TFA (1.5 % v/v)
60
1.1 1.1 CH3CN + TFA (1.2 eq)
30
a after flash column chromatography
Table 2.8. DIB-mediated bimolecular [3+2] dipolar cycloaddition: optimization studies.

85
The optimized conditions from Table 2.8, entry k were applied to a variety of different oximes
and dipolarophiles (Table 2.9). Aromatic oximes with activating and deactivating groups were reacted in
good to excellent recovered yields. The aliphatic oximes tested worked with similar efficiency.
Replacement of the olefin trap with a terminal alkyne resulted in the fully aromatic isoxazole 2.88, but
with diminished efficiency (Table 2.9, entry l). Variable amounts of 3,5-diphenyl-1,2,4-oxadiazole 4-
oxide, the dimer of benzonitrile oxide, were also recovered from the reaction of oxime 2.73 with
phenylacetylide (Table 2.9, entry l). The reaction mixtures for these reactions became unusually dark
colored, perhaps resulting from a competing in situ formation of alkynyliodonium species.107,221-224
The
subsequent reaction of any alkynyliodonium species with methanol (or other nucleophile) could have
reduced the amount of dipolarophile available for reaction with a nitrile oxide. Other terminal alkyne
traps such as 1-hexyne provided isoxazole products in even lower recovered yields.
Similar to aromatic and aliphatic aldoximes, -oxo-aldoximes were oxidized to -oxo-nitrile
oxides in the presence of DIB. The nitrile oxides formed in such a manner were trapped in good to
excellent recovered yields (Table 2.10). A screen of reaction conditions using -oximinoacetone 2.89
and norbornylene as the dipolarophile trap was embarked on and summarized in Table 2.10. Similar to
before225
(Table 2.8), the best conditions involved room temperature reaction of -oximinoacetone 2.89
and 1.2 equivalents of both DIB and norbornylene in methanol with TFA (0.1 – 1% v/v) (Table 2.10,
entries d and e). In this case, we observed a good recovered yield from the oxidation of -
oximinoacetone 2.89 in methanol with no added trifluoroacetic acid (Table 2.10, entry c), though this
appeared to be an exceptional case.

86
Table 2.9. DIB-mediated bimolecular [3+2] dipolar cycloaddition: substrate scope.
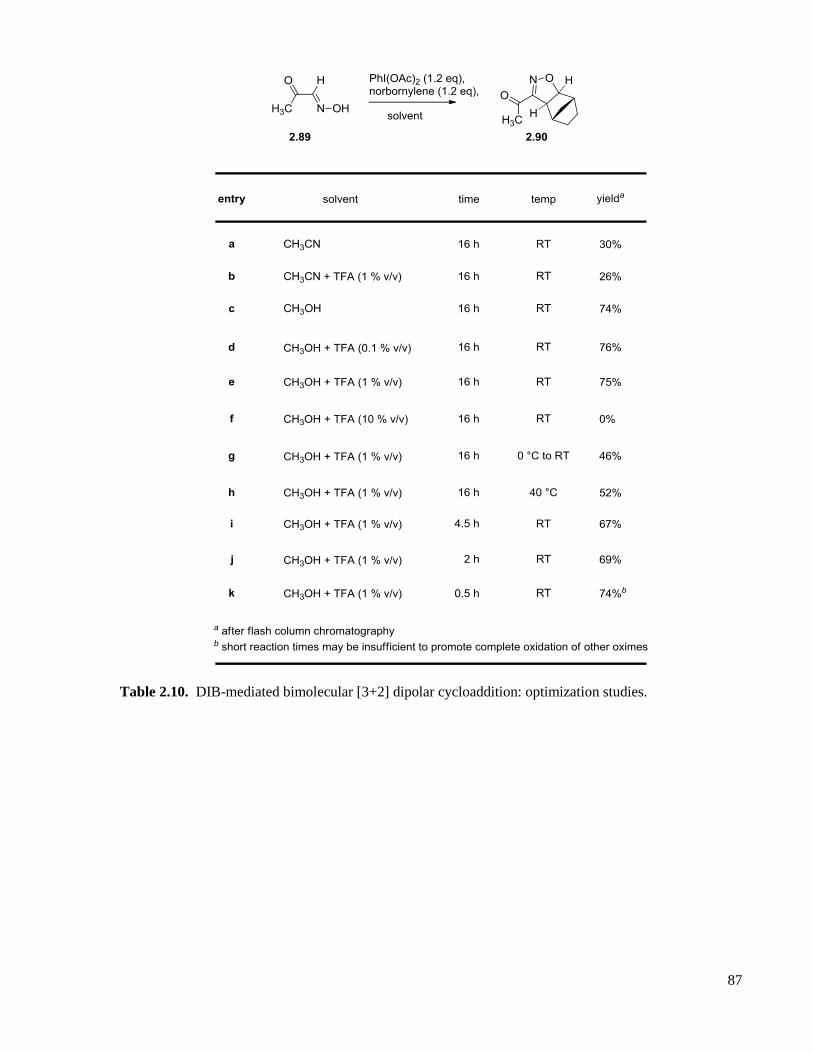
87
Table 2.10. DIB-mediated bimolecular [3+2] dipolar cycloaddition: optimization studies.

88
The DIB-mediated oxidation, and subsequent trapping with norbornylene or styrene, of
commercially available -oxo-aldoximes 2.89 and 2.92, using conditions from Table 2.10, entry k,
proceeded to produce isooxazoline products 2.90/2.91 and 2.93/2.94 with high efficiency (Table 2.11).
Good to excellent recovered yields were obtained. Note that entries d and e in Table 2.10 and entry a in
Table 2.11 were equivalent. This method permitted access to carbethoxy-formonitrile oxide
(CEFNO)174,226-228
and related CEFNO cycloadducts 2.93 and 2.94.
Table 2.11. DIB-mediated oxidation of -oxo-aldoximes 2.89 and 2.92.

89
2.7.3 Oxidation of -oxo-ketoximes and ’-dioxo-ketoximes
The DIB-mediated oxidation of oximes to their corresponding nitrile oxides was not only applied
to aromatic, aliphatic and -oxo-aldoximes (Chapter 2.7.2), but also to -oxo-ketoximes and ’dioxo-
ketoximes. We predicted that -oxo-ketoximes such as compound 2.95 would react with DIB to give
reactive intermediates of the type 2.96, which in turn could react with appropriate nucleophilic solvents
leading to the formation of nitrile oxides of the type 2.97 which could be trapped with various
dipolarophiles (Figure 2.26). Our hope in this endeavor was to broaden the synthetic utility of the DIB-
mediated oxime oxidation reactions to allow for wider range of applications and synthetically useful
products of the type 2.98 and others.
Figure 2.26. Predicted course of the DIB oxidation of -oxo-ketoximes.
We predicted that nitrile oxides would form from -oxo-ketoximes via the oxidative cleavage of
the carbonyl-imino bond (Figure 2.26). As a test of this hypothesis, we reacted ’dioxo-ketoxime
2.99 with DIB and norbornylene under the conditions which worked well for aromatic, aliphatic, and -
oxo-aldoximes (Table 2.12, entry a). The isolated yield from the reaction was a moderate 52%, with
nearly a third of the unreacted oxime recovered from the reaction mixture. Despite the moderate isolated
yields, attempts were made to screen for better conditions by modifying DIB and norbornylene
stoichiometry and solvent conditions (Table 2.12). Unfortunately, all other conditions employed only
served to decrease the recovered yield for this system, and therefore rigorous optimization studies were
left for a future time.
The typical procedure for the reactions listed in Table 2.12 was as follows: a solution of ketoxime
2.99 (1.5 mmol) in an appropriate solvent (3 mL) was added very slowly at room temperature to a stirred
solution of DIB and norbornylene in the same solvent (2 mL). Appropriate amounts of TFA (or HFIP)
were added only to the solution with DIB and norbornylene. Another portion of solvent (0.5 mL) was
used to wash any remaining ketoxime 2.99 into the reaction mixture. After the stated time, the mixture
was evaporated in vacuo and the residue was purified by flash chromatography.
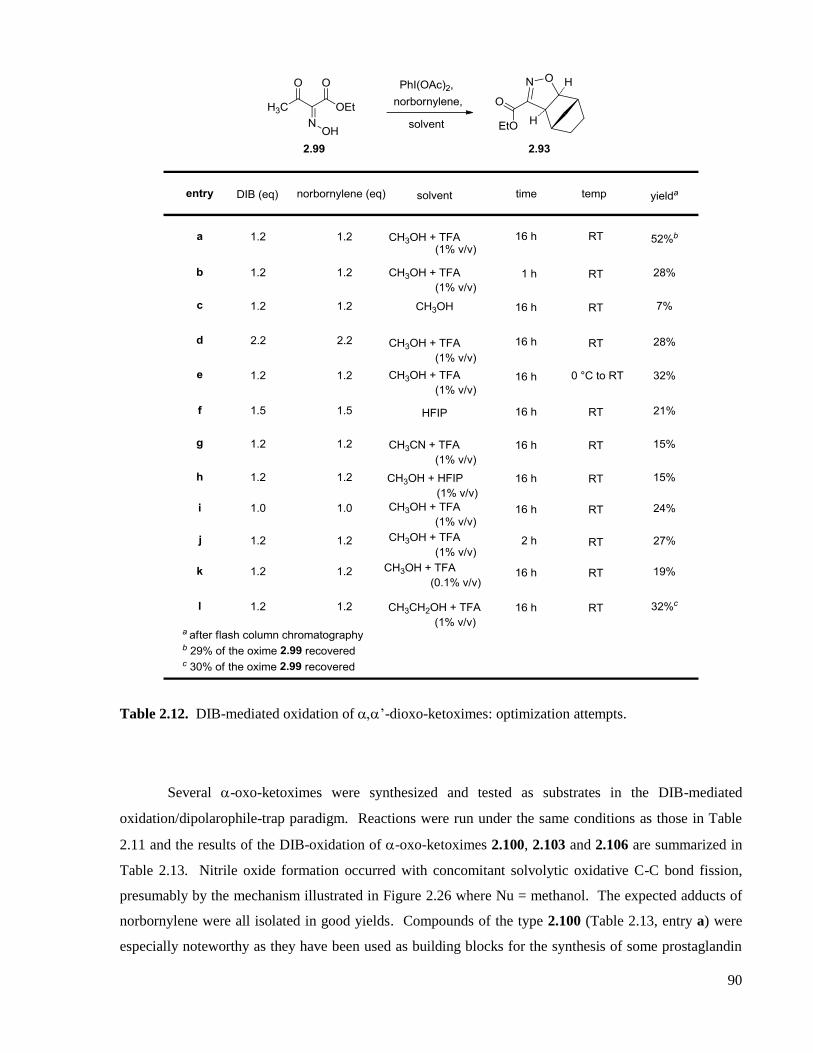
90
Table 2.12. DIB-mediated oxidation of ’-dioxo-ketoximes: optimization attempts.
Several -oxo-ketoximes were synthesized and tested as substrates in the DIB-mediated
oxidation/dipolarophile-trap paradigm. Reactions were run under the same conditions as those in Table
2.11 and the results of the DIB-oxidation of -oxo-ketoximes 2.100, 2.103 and 2.106 are summarized in
Table 2.13. Nitrile oxide formation occurred with concomitant solvolytic oxidative C-C bond fission,
presumably by the mechanism illustrated in Figure 2.26 where Nu = methanol. The expected adducts of
norbornylene were all isolated in good yields. Compounds of the type 2.100 (Table 2.13, entry a) were
especially noteworthy as they have been used as building blocks for the synthesis of some prostaglandin

91
analogs.229
The conversion of (D)-camphor-derived -oxo-ketoxime 2.106 into a 1:1 mixture of exo-
cyclic adducts 2.107/2.108 proceeded in high conversion and recovery. For unknown reasons, the
conversion of this chiral-ketoxime 2.106 with styrene as the dipolarophile trap consistently proceeded
with diminished efficiency relative to the norbornylene adducts (Table 2.13, entry f).
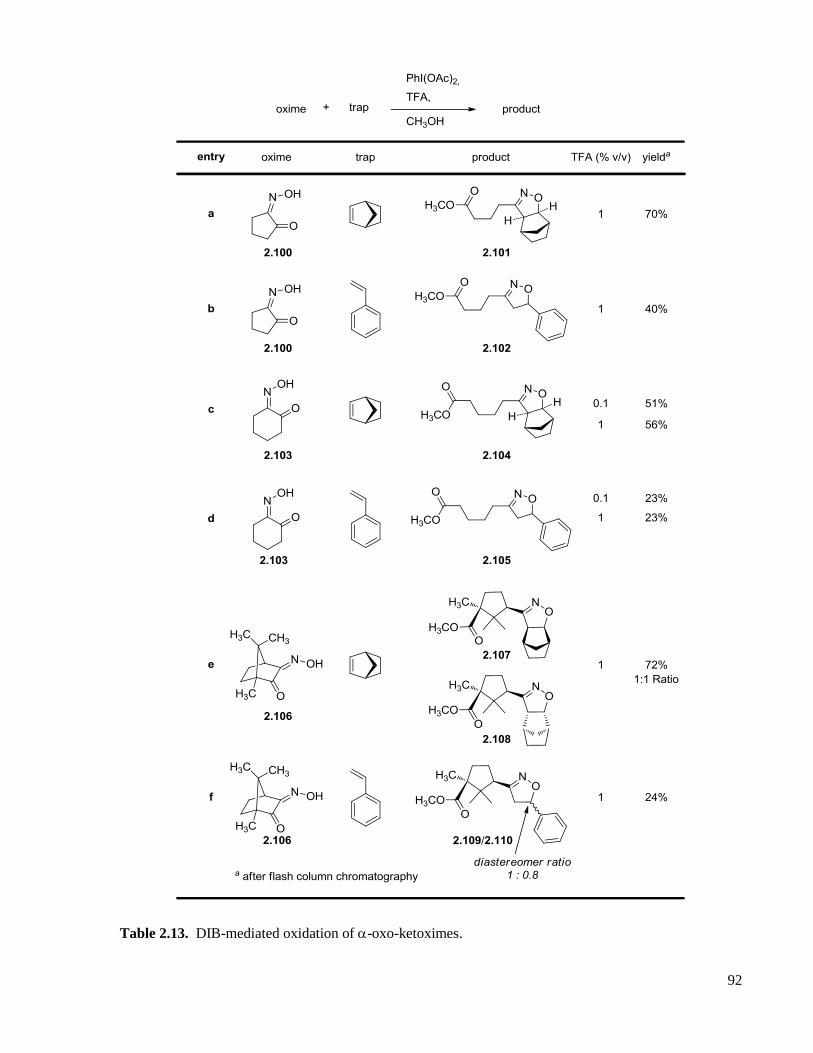
92
Table 2.13. DIB-mediated oxidation of -oxo-ketoximes.

93
2.7.4 Intramolecular nitrile oxide cycloaddition
Several intramolecular versions of the DIB-mediated oxime-oxidation reaction were examined
(Schemes 2.18 and 2.19). The first intramolecular variant of this reaction involved the oxime 2.111, a
derivative of citronellal, which cyclized in 85% conversion upon exposure to conditions optimized from
the bimolecular studies. The citronellal-derived isooxazoline 2.112 was obtained as a mixture of
diastereomers in a ratio of 3.8:1 as determined from 1H-NMR peak integration. Terpene 2.112 was quite
volatile and pleasant smelling, and material was repeatedly lost upon vacuum concentration and during
purification. Thus, the reaction yield was calculated by the addition of a 1H-NMR standard to the crude
reaction mixture. A known amount of 1,3,5-trimethoxybenzene was added to the crude reaction mixture
of 2.112, 1H-NMR data was obtained (the D1 NMR parameter was increased to 20 seconds for
quantitative integration) and the reaction conversion was calculated to be 85% based on the comparison
of the 2.112 methyl 1H-NMR signals with those of the methoxy
1H-NMR signals from the 1,3,5-
trimethoxybenzene. A portion of the crude material was purified fully for characterization purposes, and
had an []20
D = –87.0 (CH2Cl2, c = 0.01). The configuration of this material was not determined due to
the nature of the molecule not lending itself to accurate determination of coupling constants: the
resonances of the axial hydrogen on the methylene adjacent to the oximino group of 2.112 (about 1.8
ppm) and of the methyl-bearing methine of 2.112 (about 1.50 ppm) overlap with those of other ring
hydrogen atoms, precluding the accurate determination of the coupling constants, and thus preventing the
determination of the configuration.
Scheme 2.18. The first intramolecular variant.
Two other intramolecular examples were completed in conjunction with another student in the
Ciufolini group (Scheme 2.19). Florian Tessier synthesized225
oximes 2.113 and 2.115 and we observed
60-69% recovered yields of the tricyclic isooxazolines 2.114 and 2.116 using the optimized conditions.

94
Scheme 2.19. Other intramolecular variants from Ciufolini group.
Shortly after our initial publication on the DIB-mediated INOC reaction,225
another research
group used DIB (and also Koser’s reagent: [hydroxyl(tosyloxy)iodo]benzene) to convert aldoximes to N-
acetoxy or N-hydroxy amides through a nitrile oxide intermediate.230

95
2.8 Tandem oxidative dearomatization/nitrile oxide [3+2] cycloaddition
The use of DIB in both phenol dearomatizations and also in oxime oxidations, under similar
reaction conditions (polar/acidic solvents, room temperature) seemed a convenient twist of fate; I had
wondered if there was some way to tie these two reactions together into one-pot. Extensive
experimentation with the DIB-mediated INOC reaction, both intermolecularly and intramolecularly,
indicated a rate of oxidation of aldoximes to nitrile oxides to be on the order of an hour for complete
conversion for the reactions under typical concentrations of 0.5-1.0 M. Comparatively, the rate of
reaction for DIB-mediated oxidative dearomatizations at similar reaction concentrations was, practically
speaking, instantaneous. This apparent difference in reaction rates boded well for the feasibility of a
tandem oxidative dearomatization of phenols/intramolecular nitrile oxide cycloaddition sequence, with
both oxidations mediated by DIB sequentially (Figure 2.27). Dienones arising from oxidative amidation,
or other oxidative dearomatization type, would be captured by an in situ generated nitrile oxide (2.118) to
afford structures of the type 2.119. Dienone products of oxidative dearomatization were already known
to be able to participate in tandem reactions, especially 1,4-addition processes, and products of these
tandem sequences were densely functionalized, synthetically valuable intermediates. In fact, an oxidative
amidation/conjugate addition procedure was a central step in the synthesis of (–)-cylindricine C.149
Other
examples include a tandem sequence involving an oxidative hydroxylation/Michael cyclization118,124
of
tyrosine derivatives which was used as a key transformation in the synthesis of Stemona alkaloids125
and
of hydroxylated amino acids related to parkacine, aeruginosine and castanospermine.231
However, no
examples existed in the literature involving a tandem oxidative dearomatization followed by a second
oxidation event.
Figure 2.27. Hypothetical tandem oxidative amidation-intramolecular nitrile oxide cycloaddition.
Oxime 2.123 served to explore the feasibility of the hypothesized tandem oxidative amidation-
INOC sequence. Aldoxime 2.123 was synthesized in a straightforward manner from phenol 2.120
(Scheme 2.20). Conversion of compound 2.120 to the corresponding Weinreb-amide232
2.121 occurred
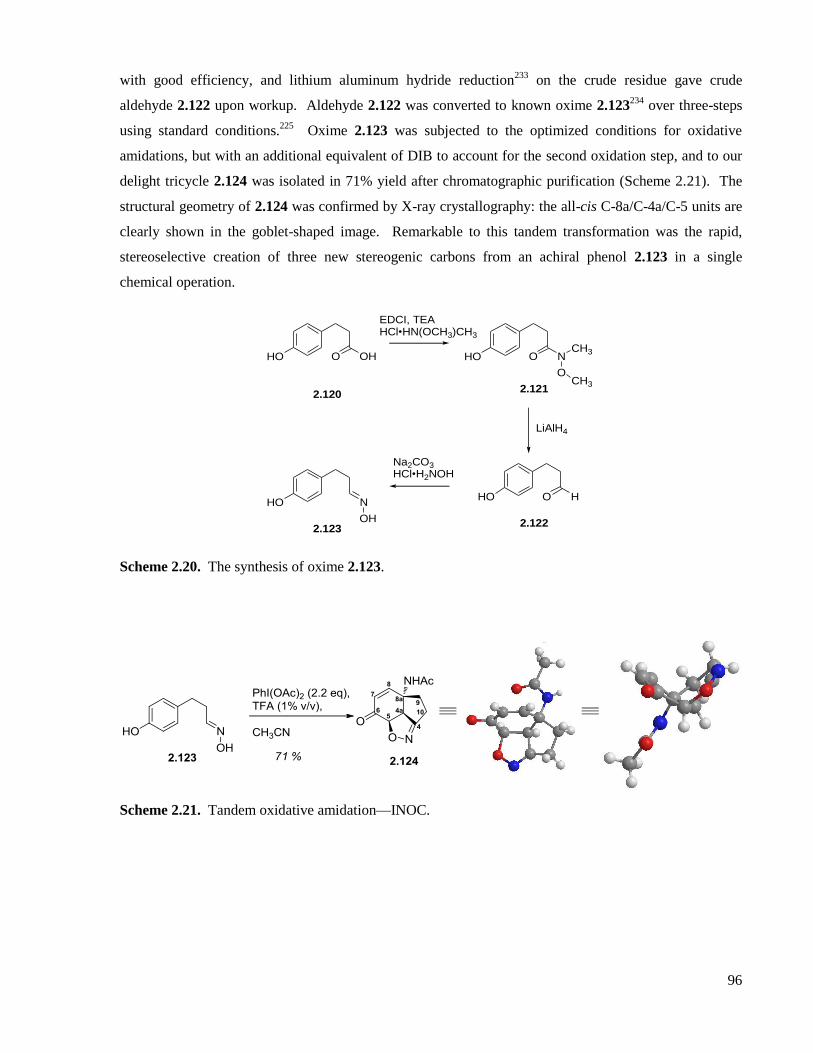
96
with good efficiency, and lithium aluminum hydride reduction233
on the crude residue gave crude
aldehyde 2.122 upon workup. Aldehyde 2.122 was converted to known oxime 2.123234
over three-steps
using standard conditions.225
Oxime 2.123 was subjected to the optimized conditions for oxidative
amidations, but with an additional equivalent of DIB to account for the second oxidation step, and to our
delight tricycle 2.124 was isolated in 71% yield after chromatographic purification (Scheme 2.21). The
structural geometry of 2.124 was confirmed by X-ray crystallography: the all-cis C-8a/C-4a/C-5 units are
clearly shown in the goblet-shaped image. Remarkable to this tandem transformation was the rapid,
stereoselective creation of three new stereogenic carbons from an achiral phenol 2.123 in a single
chemical operation.
HO N
OH2.123
HO N
O
2.121
OCH3
CH3
HO OH
2.120
O
HO H
2.122
O
EDCI, TEAHCl•HN(OCH3)CH3
LiAlH4
Na2CO3
HCl•H2NOH
Scheme 2.20. The synthesis of oxime 2.123.
Scheme 2.21. Tandem oxidative amidation—INOC.

97
2.8.1 Sorensen’s use of tandem dearomitization/nitrile oxide [3+2] cycloaddition in Cortistatin
core synthesis
We also subjected oxime 2.123 to similar reaction conditions optimized for the DIB-mediated
oxidation of oximes (cf. Table 2.9): chiefly acetonitrile was replaced with methanol which changed the
reaction type to a tandem oxidative methoxylation/INOC sequence. Compound 2.125 was isolated in
51% purified yield following reaction of oxime 2.123 with DIB in methanol/TFA. Isooxazoline 2.125
was highly polar and seemed to irreversibly adsorb onto silica gel during purification. Thus, a crude
reaction conversion was determined to be 65% using the same technique and 1H-NMR standard as for
terpene 2.112. This reaction was the first tandem oxidative alkoxylation/INOC sequence to appear in the
chemical literature.
Scheme 2.22. Tandem oxidative methoxylation—INOC.
A few months after this tandem oxidative methoxylation-intramolecular nitrile oxide
cycloaddition was published,225
it was adopted by Erik Sorensen and co-workers as the solution to the
construction of the pentacyclic core structure of the cortistatins235
(Scheme 2.23). Their approach utilized
a phenol as a latent A-ring featuring a tandem intramolecular oxidative cyclodearomatization/dipolar
cycloaddition event. The use of our tandem dearomatization/dipolar cycloaddition sequence in this work
rendered the rapid construction of the corresponding rings.

98
Scheme 2.23. Sorensen’s use of tandem oxidative dearomatization—INOC towards the cortistatin
pentacyclic core.
According to Professor Erik Sorensen, the use of our tandem sequence allowed for his completion
of the pentacyclic Cortistatin core; without this sequence, alternative routes toward the construction of the
pentacytclic core structure would have been less-efficient and contrived.236
The use of this technology by
another group shortly after its initial publication stands as a testament of its potential utility for the
generation of synthetically useful molecules.

99
2.9 Diastereoselective tandem oxidative amidation—INOC
The second oxidation in the tandem sequence (the INOC step) caused desymmetrization of the
dienone intermediate. Specifically, the intramolecular nitrile oxide cycloaddition caused the acetamide-
bearing carbon (C-8a) to become stereogenic (see Figure 2.12). Selectivity for the pro-(S) double bond
could be controlled through utilization of the principle illustrated in Figure 2.28. If the group L were a
sterically demanding group, preferably something suitable for later-stage isooxazoline-ring fragmentation,
then the INOC step of reactive intermediate 2.127 could proceed through either transition states 2.128 or
2.129. For transition state 2.129, the bulky L group is forced into the developing concavity of the
tricyclic framework and would thus suffer from significant steric congestion. In the case of transition
state 2.128, the bulky L group would point to the convexity of the emerging bowl-shaped cycloadduct,
and since the external orientation of the group L would suffer from less unfavorable steric interactions the
reaction is predicted to proceed in this manner.
Figure 2.28. Predicted course of the INOC reaction.
We already had attempted an INOC sequence with compound 2.46, and observed a 1:1 ratio of
the possible diastereomeric products (cf. Scheme 2.6). This result indicated that with regard to compound
2.126/2.127, an OTBS group was not of sufficient bulk to have a diastereoselective effect on the INOC
step as predicted in Figure 2.28. We then considered other entities which could satisfy the requirements
for compounds of the type 2.127. Compound 2.127 could be derived from tyrosine if the L group were a

100
protected nitrogen atom. Oxime 2.132 was synthesized from racemic tyrosine in an eight-step sequence
(Scheme 2.24). We hoped that the N-benzyl tosylamido group (–N(Ts)Bn) would be large enough to
exert the desired diastereoselectivity. Earlier compounds (derivatives of compound 2.132) which we
synthesized that contained a monoprotected nitrogen unit, an N-tosylamido group (–NH-Ts) underwent
oxidative amidation followed by conjugate addition instead of the desired INOC; conjugate addition
products of this type were published in the chemical literature shortly after our finding by Wipf in related
systems.231
Our plan was for the N-benzyl tosylamido group of compound 2.132 to allow for the
alleviation of nonbonding interactions (steric avoidance), avoiding transition state 2.129 and favoring
2.128, while also disfavoring any possible Michael addition pathways.
()-2.132
HO
CO2H
NH2
()-tyrosine
1) SOCl2, CH3OH2) PhCHO3) NaBH4, CH3OH
4) TsCl5) NaOH, H2O/THF
HO
CO2H
NTs
Ph
HN(OCH3)CH3,PyBOP, Et3N
HON
Ts
PhHO
NTs
Ph
N OHO
NO
CH3
CH3
()-2.131
()-2.130
1) LiAlH4
2) H2NOH, Na2CO3
Et2O, H2O
64% over five-steps
11% over three-steps(unoptimized)
Scheme 2.24. Synthesis of oxime 2.132.
The exposure of oxime 2.132 to the action of DIB in acetonitrile/TFA resulted in the formation of
2.134 as a single diastereomer (Scheme 2.25). The observed stereochemical outcome was explained by
considering that the presumed nitrile oxide (2.133) arising from the DIB-mediated oxidation of oxime
2.132, following the dearomatization of the phenol moiety, indeed observed the principle outlined in
Figure 2.28. The transition 2.129 was disfavored due to unfavorable steric compression from the bulky
N-benzyl tosylamido group, represented by group L in Figure 2.28, orientated in the concavity of the
molecule. Transition state 2.128 conveniently avoided these higher-energy interactions with the bulky N-
benzyl tosylamido group residing on the convex face of the incipient product. Thus tricyclic isooxazoline
2.134 was synthesized diastereoselectively in 44% isolated yield. The majority of the remaining mass
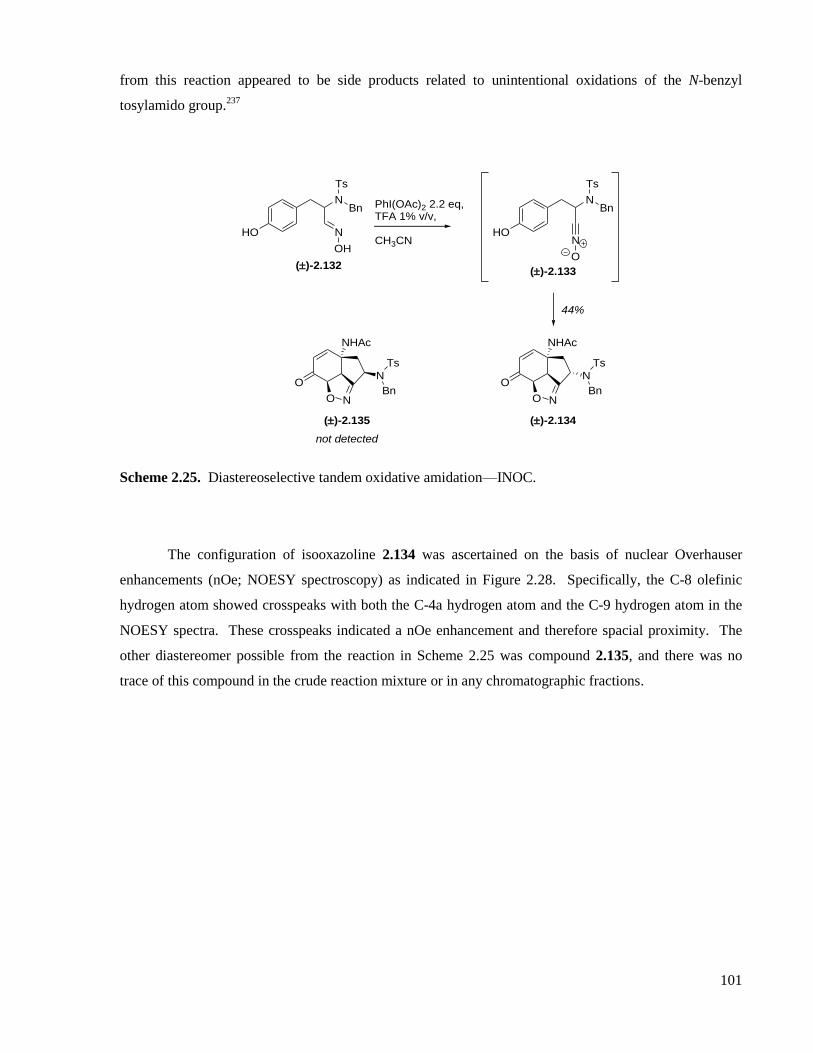
101
from this reaction appeared to be side products related to unintentional oxidations of the N-benzyl
tosylamido group.237
HO
N
N
OH
Bn
Ts
()-2.132
PhI(OAc)2 2.2 eq,TFA 1% v/v,
CH3CN
O
O N
NHAc
N
Bn
Ts
()-2.134
44%
O
O N
NHAc
N
Bn
Ts
()-2.135
not detected
HO
N
N
O
Bn
Ts
()-2.133
Scheme 2.25. Diastereoselective tandem oxidative amidation—INOC.
The configuration of isooxazoline 2.134 was ascertained on the basis of nuclear Overhauser
enhancements (nOe; NOESY spectroscopy) as indicated in Figure 2.28. Specifically, the C-8 olefinic
hydrogen atom showed crosspeaks with both the C-4a hydrogen atom and the C-9 hydrogen atom in the
NOESY spectra. These crosspeaks indicated a nOe enhancement and therefore spacial proximity. The
other diastereomer possible from the reaction in Scheme 2.25 was compound 2.135, and there was no
trace of this compound in the crude reaction mixture or in any chromatographic fractions.
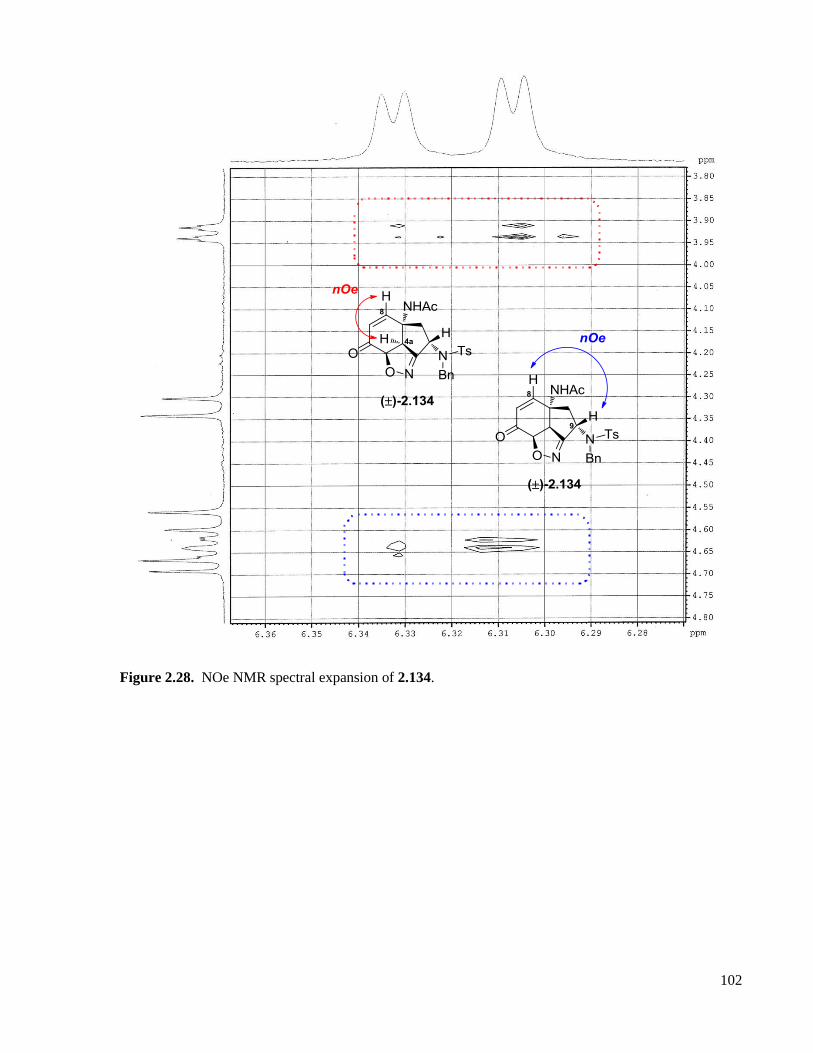
102
Figure 2.28. NOe NMR spectral expansion of 2.134.

103
2.10 Summary
The advances accompanying this route towards the tetrodotoxin core fell into two major
categories. First, we have further optimized a scalable, less-costly and more-reproducible method for the
IIII
-mediated oxidative amidation of 4-hydroxyphenyl acetate (2.31). The challenges associated with this
primarily included developing a method for the slow addition of the phenol to the reaction mixture,
without causing the formation of oligiomeric side products, and also an efficient method for purification
of the dienone products. Second, we have explored new methods for the creation of nitrile oxides from
oximes. Oximes are established precursors to nitrile oxides, but until now there was not a mild and
reliable method for this conversion mediated by hypervalent iodine reagents. Aldoximes and ketoximes
of different types have now been shown to undergo DIB-mediated oxidative transformations to nitrile
oxides, and these have been used to generate a number of synthetically useful isooxazoline and isooxazole
products.
The combination of the two main elements contained in this thesis allowed us to create a tandem
sequence where the oxidative dearomatization step is immediately, and in the same pot, followed by an
oxidative event converting an oxime into the corresponding nitrile oxide and the subsequent
intramolecular [3+2] cycloaddition. This was the first example of a tandem hypervalent iodine—induced
oxidative dearomatization—nitrile oxide cycloaddition, the development of which relied heavily on the
extensive advancement of the individual steps: chiefly the optimization of bimolecular oxidative
amidation of 4-substituted phenols and the oxidation of oximes to nitrile oxides under similar conditions.
This technology was immediately adopted by other synthetic chemists working on related systems.
We have created and detailed new reactivity and have showcased the ability of our tandem
sequence to stereoselectively generate complex tetrodotoxin core intermediates. With access to
functionalizable core structures for TTX, we hope that analogues could be produced with altered and
potentially useful biological activities. The creation of our advanced tetrodotoxin core intermediates
should serve as a solid foundation for future studies to our approach to tetrodotoxin.

104
References
(1) Mosher, H. S.; Fuhrman, F. A.; Buchwald, H. D.; Fischer, H. G. Science 1964, 144, 1100.
(2) Fuhrman, F. A. Tetrodotoxin, Saxitoxin, and the Molecular Biology of the Sodium Channel; New
York Academy of Sciences: New York, 1986; Vol. 479.
(3) Gaillard, C. Mem. Inst. Francais Arch. Orient. 1923, 51, 97.
(4) Halstead, B. W. In Poisonous and Venomous Marine Animals of the World 1988.
(5) Kao, C. Y. Pharmacol. Rev. 1966, 18, 997.
(6) Rivera, V. R.; Poli, M. A.; Bignami, G. S. Toxicon 1995, 33, 1231.
(7) Material Safety Data Sheet Tetrodotoxin ACC# 01139.
https://fscimage.fishersci.com/msds/01139.htm
(8) Material Safety Data Sheet Tetrodotoxin. Sigma-Aldrich Version 1.6 updated 10 March 2007.
(9) Davis, W. The Serpent and the Rainbow; Simon and Schuster: New York, 1985.
(10) Benedek, C., Rivier, L. Toxicon 1989, 27, 473.
(11) Hines, T. Skeptical Inquirer 2008, 32, 60.
(12) Tahara, Y. Biochem. Z. 1911, 10, 255.
(13) Tahara, Y. U.S.A., 1913.
(14) Yokoo, A. J. Chem. Soc. Jpn. 1950, 71, 590.
(15) Woodward, R. B. Pure Appl. Chem. 1964, 9, 49.
(16) Goto, T.; Kishi, Y.; Takahashi, S.; Hirata, Y. Tetrahedron 1965, 21, 2059.
(17) Tsuda, K.; Ikuma, S.; Kawamura, M.; Tachikawa, R.; Sakai, K.; Tamura, C.; Amakasu, O. Chem.
Pharm. Bull. 1964, 12, 1357.
(18) Furusaki, A.; Tomie, Y.; Nitta, I. Bull. Chem. Soc. Jpn. 1970, 43, 3325.
(19) Nakamura, M.; Yasumoto, T. Toxicon 1985, 23, 331.
(20) Shiomi, K.; Inaoka, H.; Yamanaka, H.; Kikuchi, T. Toxicon 1985, 22, 381.
(21) Zhou, M.; Shum, F. H. K.; States, U., Ed.; Wex Medical Instrumentation Co., Ltd., Hong Kong:
United States, 2003; Vol. US 6,552,191 B1.
(22) Hashimoto, Y.; Noguchi, T. Toxicon 1971, 9, 79.
(23) Noguchi, T.; Hashimoto, Y. Toxicon 1973, 11, 305.
(24) Hwang, D. F.; Tai, K. P.; Chueh, C. H.; Lin, L. C.; Jeng, S. S. Toxicon 1991, 8, 1019.

105
(25) Noguchi, T.; Narita, H.; Maruyama, J.; Hashimoto, K. Nippon Suisan Gakkaishi 1982, 48, 1173.
(26) Ritson-Williams, R.; Yotsu-Yamashita, M.; Paul, V. J. Proc. Natl. Acad. Sci. 2006, 103, 3176.
(27) Kim, Y. H.; Brown, G. B.; Mosher, H. S.; Fuhrman, F. A. Science 1975, 189, 151.
(28) Miyazawa, K.; Noguchi, T. J. of Toxicology: Toxin Reviews 2001, 11.
(29) Anderson, P. A. V. J. Exp. Biol. 1987, 231.
(30) Anderson, P. A. V. Evolution of the First Nervous Systems; Plenum Press: New York, 1990.
(31) Anderson, P. A. V.; Holman, M. A. Proc. Natl. Acad. Sci. 1993, 7419.
(32) Shimizu, S.; Kobayashi, M. Chem. Pharm. Bull. 1983, 31, 3625.
(33) Kotaki, Y.; Shimizu, Y. J. Am. Chem. Soc. 1993, 115, 827.
(34) Yotsu-Yamashita, M.; Yamagashi, Y.; Yasumoto, T. Tetrahedron Lett. 1995, 36, 9329.
(35) Jang, J.-H.; Yotsu-Yamashita, M. Toxicon 2007, 50, 947.
(36) Note: the numbering schemes for all chemical structures in this thesis are based on the
corresponding tetrodotoxin (TTX) numbering.
(37) Hwang, D. F.; Lu, Y. H. Recent Advances in Marine Biotechnology 2002, 7, 53.
(38) Matsumura, K. Nippon Juishikai Zasshi 1998, 51, 231.
(39) Yan, Q.; Yu, P. H.-F.; Li, H.-Z. World J. Microbiol. Biotechnol. 2005, 21, 1255.
(40) Dewich, P. Med. Nat. Prod. 2001, 2, 396.
(41) Shimizu, Y.; Norte, M.; Hori, A.; Genenah, A.; Kobayshi, M. J. Am. Chem. Soc. 1984, 106, 6433.
(42) Kandel, E. R.; Schwartz, J. H.; Jessell, T. M. Principles of Neural Science; 4th ed.; New York,
McGraw-Hill, 2000.
(43) Hille, B. Ion Channels of Excitable Membranes; 3rd ed.; Sunderland, MA: Sinauer Associates,
Inc., 2001.
(44) Satin, J.; Limberis, J. T.; Kyle, J. W.; Rogart, R. B.; Fozzard, H. A. Biophys. J. 1994, 67, 1007.
(45) Yotsu-Yamashita, M.; Sugimoto, A.; Takai, A.; Yasumoto, T. J. Pharm. Exp. Therap. 1999, 289,
1688.
(46) Hucho, F. Angew. Chem., Int. Ed. 1995, 34, 39.
(47) Denac, H.; Mevissen, M.; Scholtysik, G. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000, 362,
453.
(48) Slaughter, R. S.; Garcia, M. L.; Kaczorowski, G. J. Current Pharmaceutical Design 1996, 2, 610.

106
(49) Gilman, V. Chem. Eng. News 2004.
(50) Kishi, Y.; Fukuyama, T.; Aratani, M.; Nakatsubo, F.; Goto, T.; Inoue, S.; Tanino, H.; Sugiura, S.;
Kakoi, H. J. Am. Chem. Soc. 1972, 94, 9219.
(51) Kishi, Y.; Aratani, M.; Fukuyama, T.; Nakatsubo, F.; Goto, T.; Inoue, S.; Tanino, H.; Sugiura, S.;
Kakoi, H. J. Am. Chem. Soc. 1972, 94, 9217.
(52) Kishi, Y.; Nakatsubo, F.; Aratani, M.; Goto, T.; Inoue, S.; Kakoi, H. Tetrahedron Lett. 1970, 59,
5129.
(53) Kishi, Y.; Nakatsubo, F.; Aratani, M.; Goto, T.; Inoue, S.; Kakoi, H.; Sagiura, S. Tetrahedron
Lett. 1970, 59, 5127.
(54) Ohyabu, N.; Nishikawa, T.; Isobe, M. J. Am. Chem. Soc. 2003, 125, 8798.
(55) Nishikawa, T.; Urabe, D.; Isobe, M. Angew. Chem., Int. Ed. 2004, 43, 4782.
(56) Nishikawa, T.; Urabe, D.; Yoshida, K.; Iwabuchi, T.; Asai, M.; Isobe, M. Chem. Eur. J. 2004, 10,
452.
(57) Nishikawa, T.; Urabe, D.; Yoshida, K.; Iwabuchi, T.; Asai, M.; Isobe, M. Pure Appl. Chem. 2003,
75, 251.
(58) Nishikawa, T.; Urabe, D.; Yoshida, K.; Iwabuchi, T.; Asai, M.; Isobe, M. Org. Lett. 2002, 4,
2679.
(59) Nishikawa, T.; Asai, M.; Isobe, M. J. Am. Chem. Soc. 2002, 124, 7847.
(60) Asai, M.; Nishikawa, T.; Ohyabu, N.; Yamamoto, N.; Isobe, M. Tetrahedron 2001, 57, 4543.
(61) Nishikawa, T.; Asai, M.; Ohyabu, N.; Yamamoto, N.; Isobe, M. Angew. Chem. Int. Ed. 1999, 38,
3081.
(62) Nishikawa, T.; Asai, M.; Ohyabu, N.; Yamamoto, N.; Fukuda, Y.; Isobe, M. Tetrahedron 2001,
57, 3875.
(63) Satake, Y.; Nishikawa, T.; Hiramatsu, T.; Araki, H.; Isobe, M. Synthesis 2010, 12, 1992.
(64) Hinman, A.; Du Bois, J. J. Am. Chem. Soc. 2003, 125, 11510.
(65) Sato, K.; Akai, S.; Sugita, N.; Ohsawa, T.; Kogure, T.; Shoji, H.; Yoshimura, J. J. Org. Chem.
2005, 70, 7496.
(66) Sato, K.; Kajihara, Y.; Nakamura, Y.; Yoshimura, J. Chem. Lett. 1991, 1559.
(67) Sato, K. I.; Akai, S.; Shoji, H.; Sugita, N.; Yoshida, S.; Nagai, Y.; Suzuki, K.; Nakamura, Y.;
Kajihara, Y.; Funabashi, M.; Yoshimura, J. J. Org. Chem. 2008, 73, 1234.
(68) Funabashi, M.; Wakai, H.; Sato, K.; Yoshimura, J. J. Chem. Soc., Perkin Trans 1 1980, 14.

107
(69) Kinoshita, M.; Taniguchi, M.; Morioka, M.; Takami, H.; Mizusawa, Y. Bull. Chem. Soc. Jpn.
1988, 61, 2147.
(70) Kartha, K. P. R. Tetrahedron Lett. 1986, 27, 3415.
(71) Yoshimura, J.; Sato, K.; Hashimoto, K. Chem. Lett. 1977, 1327.
(72) Szarek, W. A.; Jewell, J. S.; Szczerek, I.; Jones, J. K. N. Can. J. Chem. 1969, 47, 4473.
(73) Rosenthal, A.; Sprinzl, M. Can. J. Chem. 1969, 47, 4477.
(74) McMurry, J. E.; Melton, J.; Padgett, H. J. Org. Chem. 1974, 39, 259.
(75) Akai, S.; Seki, H.; Sugita, N.; Kogure, T.; Nishizawa, N.; Suzuki, K.; Nakamura, Y.; Kajihara,
Y.; Yoshimura, J.; Sato, K. Bull. Chem. Soc. Jpn. 2010, 83, 279.
(76) Fairweather, J. K.; McDonough, M. J.; Stick, R. V.; Tilbrook, D. M. G. Aust. J. Chem. 2004, 57,
197.
(77) Yoshimura, J.; Kobayashi, K.; Sato, K.; Funabashi, M. Bull. Chem. Soc. Jpn. 1972, 45, 1806.
(78) Funabashi, M.; Sato, K.; Yoshimura, J. Bull. Chem. Soc. Jpn. 1976, 49, 788.
(79) Keana, J. F. W.; Bland, J. S.; Boyle, P. J.; Erion, M.; Hartling, R.; Husman, J. R.; Roman, R. B.;
Ferguson, G.; Parvez, M. J. Org. Chem. 1983, 48, 3627.
(80) Keana, J. F. W.; Boyle, P. J.; Erion, M.; Hartling, R.; Husman, J. R.; Richman, J. E.; Roman, R.
B.; Wah, R. M. J. Org. Chem. 1983, 48, 3621.
(81) Keana, J. F. W.; Kim, C. U. J. Org. Chem. 1971, 36, 118.
(82) Fraser-Reid, B.; Burgey, C. S.; Vollerthun, R. Pure Appl. Chem. 1998, 70, 285.
(83) Burgey, C. S.; Vollerthun, R.; Fraser-Reid, B. J. Org. Chem. 1996, 61, 1609.
(84) Alonso, R. A.; Burgey, C. S.; Venkateswara R. B.; Vite, G. D.; Vollerthun, R.; Zottola, M. A.;
Fraser-Reid, B. J. Am. Chem. Soc. 1993, 115, 6666.
(85) Motherwell, W. B.; Crich, D. Free Radical Chain Reactions in Organic Synthesis; Academic
Press, 1992.
(86) Giese, B. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds; Pergamon Press,
Oxford, 1986.
(87) Schreiber, S. L.; Claus, R. E.; Reagan, J. Tetrahedron Lett. 1982, 23, 3867.
(88) Noya, B.; Paredes, M. D.; Ozores, L.; Alonso, R. J. Org. Chem. 2000, 65, 5960.
(89) Noya, B.; Alonso, R. Tetrahedron Lett. 1997, 38, 2745.
(90) Taber, D. F.; Storck, P. H. J. Org. Chem. 2003, 68, 7768.
(91) Lee, E.; Choi, I.; Song, S. Y. J. Chem. Soc., Chem. Commun. 1995, 3, 321.

108
(92) Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974.
(93) Tius, M. A.; Fauq, A. H. J. Org. Chem. 1983, 48, 4131.
(94) Roush, W. R.; Adam, M. A.; Walts, A. E.; Harris, D. J. J. Am. Chem. Soc. 1986, 108, 3422.
(95) Itoh, T.; Watanabe, M.; Fukuyama, T. Synlett 2002, 1323.
(96) Fujita, M.; Kitagawa, O.; Suzuki, T.; Taguchi, T. J. Org. Chem. 1997, 62, 7330.
(97) Basel, Y.; Hassner, A. Synthesis 1997, 309.
(98) Ohtani, Y.; Shinada, T.; Ohfune, Y. Synlett 2003, 619.
(99) Shinada, T., Yoshida, S., Yamamura, S. Tetrahedron Lett. 1998, 39, 6027.
(100) Mulzer, J.; Altenbach, H. J.; Braun, M.; Krohn, K.; Reissig, H. U. Organic Synthesis Highlights;
VCH: Weinheim, 1990; Vol. 1.
(101) Nair, V.; Suja, T. D. Tetrahedron 2007, 63, 12247.
(102) Liang, H.; Ciufolini, M. A. Tetrahedron 2010, 66, 5884.
(103) Ciufolini, M. A.; Braun, N. A.; Canesi, S.; Ousmer, M.; Chang, J.; Chai, D. Synthesis 2007, 3759.
(104) Moriarty, R. M. J. Org. Chem. 2005, 70, 2893.
(105) Wirth, T. Angew. Chem., Int. Ed. 2005, 44, 3656.
(106) Hamamoto, H.; Anilkumar, G.; Tohma, H.; Kita, Y. Chem. Eur. J. 2002, 8, 5377.
(107) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523.
(108) Moriarty, R. M. Org. React. 2001, 57, 327.
(109) Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: London, 1997.
(110) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 1996, 96, 1123.
(111) Pouysegu, L.; Deffieux, D.; Quideau, S. Tetrahedron 2010, 66, 2235.
(112) Yamamura, S. In The Chemistry of Phenols; Rappoport, Z., Ed.; John Wiley & Sons: Chichester,
UK, 2003; Vol. 2, p 1153.
(113) Barton, D. H. R. Pure Appl. Chem. 1964, 9, 35.
(114) Barton, D. H. R.; Deflorin, A. M.; Edwards, O. E. J. Chem. Soc. 1956, 530.
(115) Barton, D. H. R.; Kirby, G. W. J. Chem. Soc. 1962, 806.
(116) Kametani, T.; Fukumoto, K. Yakugaku Kenkyu 1963, 35, 426.

109
(117) Scott, A. I.; McCapra, F.; Buchanan, R. L.; Day, A. C.; Young, D. W. Tetrahedron 1965, 21,
3605.
(118) Rodriguez, S.; Wipf, P. Synthesis 2004, 2767.
(119) Kita, Y.; Fujioka, H. Pure Appl. Chem. 2007, 79, 701.
(120) Kita, Y.; Tohma, H.; Kikuchi, K.; Inagaki, M.; Yakura, T. J. Org. Chem. 1991, 56, 435.
(121) Tamura, Y.; Yakura, T.; Haruta, J. I.; Kita, Y. J. Org. Chem. 1987, 52, 3927.
(122) Pelter, A.; Elgendy, S. Tetrahedron Lett. 1988, 29, 677.
(123) Barret, R.; Daudon, M. Tetrahedron Lett. 1991, 32, 2133.
(124) Wipf, P.; Jung, J. K. Chem. Rev. 1999, 99, 1469.
(125) Wipf, P.; Spencer, S. R. J. Am. Chem. Soc. 2005, 127, 225.
(126) Kawase, M.; Kitamura, T.; Kikugawa, Y. J. Org. Chem. 1989, 54, 3394.
(127) Kikugawa, Y.; Kawase, M. J. Am. Chem. Soc. 1984, 106, 5728.
(128) Kikugawa, Y.; Nagashima, A.; Sakamoto, T.; Miyazawa, E.; Shiiya, M. J. Org. Chem. 2003, 68,
6739.
(129) Kikugawa, Y.; Shimada, M.; Matsumoto, K. Heterocycles 1994, 37, 293.
(130) Kikugawa, Y. K., M. Chem. Lett. 1990, 581.
(131) Glover, S. A.; Goosen, A.; McCleland, C. W.; Schoonraad, J. L. J. Chem. Soc., Perkin Trans. 1
1984, 2255.
(132) Glover, S. A.; Scott, A. P. Tetrahedron 1989, 45, 1763.
(133) Clemente, D. T. V.; Lobo, A. M.; Prabhakar, S.; Marcelo-Curto, M. J. Tetrahedron Lett. 1994,
35, 2043.
(134) Prata, J. V.; Clemente, D. T. S.; Prabhakar, S.; Lobo, A. M.; Mourato, I.; Branco, P. S. J. Chem.
Soc., Perkin Trans. 1 2002, 513.
(135) Wardrop, D. J.; Basak, A. Org. Lett. 2001, 3, 1053.
(136) Wardrop, D. J.; Burge, M. S. Chem. Commun. 2004, 10, 1230.
(137) Wardrop, D. J.; Burge, M. S. J. Org. Chem. 2005, 70, 10271.
(138) Wardrop, D. J.; Burge, M. S.; Zhang, W.; Ortiz, J. A. Tetrahedron Lett. 2003, 44, 2587.
(139) Wardrop, D. J.; Landrie, C. L.; Ortiz, J. A. Synlett 2003, 1352.
(140) Wardrop, D. J.; Zhang, W.; Landrie, C. L. Tetrahedron Lett. 2004, 45, 4229.

110
(141) Ousmer, M.; Braun, N. A.; Ciufolini, M. A. Org. Lett. 2001, 3, 765.
(142) Ousmer, M.; Braun, N. A.; Bavoux, C.; Perrin, M.; Ciufolini, M. A. J. Am. Chem. Soc. 2001, 123,
7534.
(143) Note: whereas PIFA has been used as a reagent for these oxidations, it was not as efficient and we
generally preferred to use DIB.
(144) Scheffler, G.; Seike, H.; Sorensen, E. J. Angew. Chem., Int. Ed. 2000, 39, 4593.
(145) Seike, H.; Sorensen, E. J. Synlett 2008, 695.
(146) Mizutani, H.; Takayama, J.; Soeda, Y.; Honda, T. Tetrahedron Lett. 2002, 43, 2411.
(147) Mizutani, H.; Takayama, J.; Soeda, Y.; Honda, T. Heterocycles 2004, 62, 343.
(148) Mizutani, H.; Takayama, J.; Honda, T. Synlett 2005, 328.
(149) Canesi, S.; Bouchu, D.; Ciufolini, M. A. Angew. Chem., Int. Ed. 2004, 43, 4336.
(150) Liang, H.; Ciufolini, M. A. Org. Lett. 2010, 12, 1760.
(151) Braun, N. A.; Ousmer, M.; Bray, J. D.; Bouchu, D.; Peters, K.; Peters, E. M.; Ciufolini, M. A. J.
Org. Chem. 2000, 65, 4397.
(152) Mendelsohn, B. A.; Ciufolini, M. A. Org. Lett. 2009, 11, 4736.
(153) Canesi, S.; Bouchu, D.; Ciufolini, M. A. Org. Lett. 2005, 7, 175.
(154) Liang, H.; Ciufolini, M. A. J. Org. Chem. 2008, 73, 4299.
(155) Drutu, I.; Njardarson, J. T.; Wood, J. L. Org. Lett. 2002, 4, 493.
(156) Patrick, B. O.; Mendelsohn, B. A.; Ciufolini, M. A. Z. Kristallogr.-New Cryst. Struct. 2008, 223,
205.
(157) Gothelf, K. V.; Jorgensen, K. A. Chem. Rev. 1998, 98, 863.
(158) In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984;
Vol. 1 and 2.
(159) Kanemasa, S.; Tsuge, O. Heterocycles 1990, 30, 719.
(160) Kozikowski, A. P. Acc. Chem. Res. 1984, 17, 410.
(161) Curran, D. P. In Advances in Cycloaddition; Curran, D. P., Ed.; JAI Press: Greenwich, 1988; Vol.
1, p 129.
(162) In Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; Torssell, K. B. G., Ed.; VCH:
New York, 1988.
(163) Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A. Tetrahedron 2005, 61, 8971.

111
(164) Luche, J. L. J. Am. Chem. Soc. 1978, 100, 2226.
(165) Gemal, A. L.; Luche, J. L. J. Am. Chem. Soc. 1981, 103, 5454.
(166) Itsuno, S.; Ito, K.; Hirao, A.; Nakahama, S. J. Chem. Soc., Chem. Commun. 1983, 8, 469.
(167) Corey, E. J.; Shibata, S.; Bakshi, R. K. J. Org. Chem. 1988, 53, 2861.
(168) Huan Liang performed initial/unoptimized CBS-reduction studies on dienone systems such as
2.32.
(169) Cyril Benhaim first observed this type of reaction with a related system.
(170) Staab, H. A. Angew. Chem., Int. Ed. Engl. 1962, 1, 351.
(171) Armstrong, A.; Li, W. N,N'-Carbonyldiimidazole. In e-EROS Encyclopedia of Reagents for
Organic Synthesis; Wiley, 2006.
(172) Cyril Benhaim worked out conditions to prepare a TBS-version of nitroketone 2.39.
(173) Paul, R.; Tchelitcheff, S. Bull. Soc. Chim. Fr. 1962, 2215.
(174) Kozikowski, A. P.; Adamcz, M. J. Org. Chem. 1983, 48, 366.
(175) Martin, S. F.; Dappen, M. S.; Dupre, B.; Murphy, C. J.; Colapret, J. A. J. Org. Chem. 1989, 54,
2209.
(176) Mukaiyama, T.; Hoshino, T. J. Am. Chem. Soc. 1960, 82, 5339.
(177) Mukaiyama, T.; Hoshino, T. J. Am. Chem. Soc. 1960, 82, 5339.
(178) Baranski, A.; Kelarev, V. I. Chem. Nat. Compd. 1992, 28, 253.
(179) Shimizu, T.; Hayashi, T.; Shibafuchi, H.; Teramura, K. Bull. Chem. Soc. Jpn. 1986, 59, 2827.
(180) Denmark, S. E.; Baiazitov, R. Y. J. Org. Chem. 2006, 71, 593.
(181) Andersen, S. H.; Das, N. B.; Joergensen, R. D.; Kjeldsen, G.; Knudsen, J. S.; Sharma, S. C.;
Torssell, K. B. G. Acta. Chem. Scand. B 1982, 36, 1.
(182) Klebe, J. F. J. Am. Chem. Soc. 1964, 86, 3399.
(183) Namboothiri, I. N. N.; Hassner, A.; Gottlieb, H. E. J. Org. Chem. 1997, 62, 485.
(184) Shitkin, V. M.; Ioffe, S. L.; Kashutina, M. V.; Tartakovskii, V. A. Izv. Akad. Nauk SSSR, Ser.
Khim. 1977, 2266.
(185) Patrick, B. O.; Mendelsohn, B. A.; Ciufolini, M. A. Z. Kristallogr.-New Cryst. Struct. 2008, 223,
203.
(186) Knoevenagel, E. Berichte der deutschen chemischen Gesellschaft 1898, 31, 2596.

112
(187) March, J. Advanved Organic Chemistry: Reactions, Mechanisms, and Structure; 3rd ed.; Wiley:
New York, 1985.
(188) Kemp, D. S.; Paul, K. J. Am. Chem. Soc. 1970, 92, 2553.
(189) Casey, M. L.; Kemp, D. S.; Paul, K. G.; Cox, D. D. J. Org. Chem. 1973, 38, 2294.
(190) Kemp, D. S.; Cox, D. D.; Paul, K. G. J. Am. Chem. Soc. 1975, 97, 7312.
(191) Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155.
(192) Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277.
(193) VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973.
(194) Tanaka, S.; Ito, M.; Kishikawa, K.; Kohmoto, S.; Yamamoto, M. Nippon Kagaku Kaishi 2002, 3,
471.
(195) Vigalok, I.; Moisak, I. E.; Svetakov, N. V. Chem. Heterocycl. Compd. 1969, 5, 133.
(196) Duranleau, R. G.; Larkin, J. M.; Newman, S. R. US Patent, 1978, 4089867.
(197) Just, G.; Dahl, L. Tetrahedron 1968, 24, 5251.
(198) Hassner, A.; Rai, K. M. L. Synthesis 1989, 57.
(199) Rai, K. M. L.; Linganna, N.; Hassner, A.; Murthy, C. A. Org. Prep. Proc. Int. 1992, 24, 91.
(200) Kim, J. N.; Rui, E. K. Synth. Commun. 1990, 20, 1373.
(201) Kiegiel, J.; Poplawska, M.; Jozwik, J.; Kosior, M.; Jurczak, J. Tetrahedron Lett. 1999, 40, 5605.
(202) Eibler, E.; Kasbauer, J.; Pohl, H.; Sauer, J. Tetrahedron Lett. 1987, 28, 1097.
(203) Corbett, D. F. J. Chem. Soc., Perkin Trans. 1 1986, 421.
(204) Grundaman, C.; Dean, J. M. J. Org. Chem. 1965, 30, 2809.
(205) Tsuji, M.; Ukaji, Y.; Inomata, K. Chem. Lett. 2002, 1112.
(206) Larsen, K. E.; Torssell, K. B. G. Tetrahedron 1984, 40, 2985.
(207) de la Cruz, P.; Espildora, E.; Garcia, J. J.; de la Hoz, A.; Langa, F.; Martin, N.; Sanchez, L.
Tetrahedron Lett. 1999, 40, 4889.
(208) Giurg, M.; Mlochowski, M. Pol. J. Chem. 1997, 71, 1093.
(209) Gagneux, A. R.; Meier, R. Helv. Chim. Acta. 1970, 53, 1883.
(210) Miller, M. L.; Ray, P. S. Tetrahedron 1996, 52, 5739.
(211) Miller, M. L.; Ray, P. S. Heterocycles 1997, 45, 501.

113
(212) Radhakrishna, A. S.; Sivaprakash, K.; Singh, B. B. Synth. Commun. 1991, 21, 1625.
(213) Varvoglis, A. Synthesis 1984, 709.
(214) De, S. K.; Mallik, A. K. Tetrahedron Lett. 1998, 39, 2389.
(215) Corsaro, A.; Chiacchio, U.; Librando, V.; Pistara, V.; Rescifina, A. Synthesis 2000, 1469.
(216) Chaudhari, S. S.; Akamanchi, K. G. Synthesis 1999, 760.
(217) Bose, D. S.; Narsaiah, A. V. Synth. Commun. 1999, 29, 937.
(218) Chaudhari, S. S.; Akamanchi, K. G. Tetrahedron Lett. 1998, 39, 3209.
(219) Bose, D. S.; Srinivas, P. Synlett 1998, 977.
(220) Stang, P. J. Chem. Rev. 2002, 102, 2523.
(221) Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123.
(222) Lodaya, J. S.; Koser, G. F. J. Org. Chem. 1990, 55, 1513.
(223) Rebrovic, L.; Koser, G. F. J. Org. Chem. 1984, 49, 4700.
(224) Margida, A. J.; Koser, G. F. J. Org. Chem. 1984, 49, 4703.
(225) Mendelsohn, B. A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V. S.; Ciufolini, M. A. Org. Lett.
2009, 11, 1539.
(226) Skinner, G. S. J. Am. Chem. Soc. 1924, 46, 731.
(227) Panizzi, L. Gazz. Chim. Ital. 1939, 69, 332.
(228) Vaughan, W. R.; Spencer, J. L. J. Org. Chem. 1960, 25, 1160.
(229) Bondar, N. F.; Isaenya, L. P.; Skupskaya, R. V.; Lakhvich, F. A. Russ. J. Org. Chem. 2003, 39,
1089.
(230) Ghosh, H.; Patel, B. K. Org. Biomol. Chem. 2010, 8, 384.
(231) Pierce, J. G.; Kasi, D.; Fushimi, M.; Cuzzuppe, A.; Wipf, P. J. Org. Chem. 2008, 73, 7807.
(232) Nahm, S.; Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815.
(233) Goel, O. P.; Krolls, U.; Stier, M.; Kesten, S. Organic Syntheses 1989, 67, 69.
(234) Kusama, H.; Yamashita, Y.; Uchiyama, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1997, 70, 965.
(235) Frie, J. L.; Jeffrey, C. S.; Sorensen, E. J. Org. Lett. 2009, 11, 5394.
(236) Sorensen, E. J., Personal communication.

114
(237) Simon Kim performed some preliminary unpublished work in the area of tandem oxidative
amidation-INOC sequences.

115
Appendices
Unless otherwise indicated, 1H (300 MHz) and
13C (75 MHz) NMR spectra were recorded at
room temperature from either CDCl3 or d6-acetone solutions as indicated. Chemical shifts are reported as
ppm on the d scale and coupling constants, J, are in Hz. Multiplicities are described as s (singlet), d / dd /
ddd (doublet / doublet of doublets / doublet of doublet of doublets), t, (triplet), q (quartet), p (pentet), sept
(septuplet), m (multiplet), ABq (AB quartet), and further qualified as app (apparent), b (broad), c
(complex). All 2D NMR spectra were recorded at 300 MHz (1H) or 75 MHz (
13C) unless otherwise
stated.
Infared (IR) spectra (cm-1
) were recorded on a Perkin-Elmer model 1710 Fourier transform
spectrometer from films deposited on NaCl plates or on a Perkin-Elmer Spectrum 100 Fourier transform
spectrometer from samples deposited on a Universal ATR Sampling Accessory. Optical rotations were
measured on a Jasco P-1010 polarimeter at the sodium D line (589 nm). Unless otherwise stated, low
resolution mass spectra (m/z) were obtained in the electrospray (ESI) mode or the atmospheric pressure
chemical ionization (APCI) mode on a Waters Micromass ZQ mass spectrometer. High-resolution mass
spectra (m/z) were recorded in the ESI or APCI mode on a Micromass LCT mass spectrometer by the
David Wong and Marshall Lapawa of the UBC Mass Spectrometry laboratory. Melting points
(uncorrected) were measured on a Mel-Temp apparatus. All X-ray single crystal measurements were
made by Dr. Brian Patrick on a Bruker X8 APEX II diffractometer or a Bruker APEX DUO
diffractometer (as indicated) and the refinements were performed using the SHELXTL crystallographic
software package of Bruker-AXS.
All reagents and solvents were commercial products and used without further purification except
for tetrahydrofuran (THF) which was freshly distilled from Na/benzophenone under argon atmosphere
and methylene chloride (CH2Cl2) which was freshly distilled from CaH2 under argon atmosphere. Flash
column chromatography was performed with Silicycle 230–400 mesh silica gel. All reactions were
performed in flame-dried or oven-dried glassware equipped with TeflonTM
magnetic stirbars. All flasks
were fitted with rubber septa for the introduction of substrates, reagents and solvents via syrings.

116
A. Experimental protocols
A.1 Preparation of methyl 2-(1-acetamido-4-oxocyclohexa-2,5-dienyl)acetate (2.32)
O
NHAc
CO2CH3
2.32
Chemical Formula: C11H13NO4
Exact Mass: 223.08
Molecular Weight: 223.23
Procedure A
To a 1000-mL round-bottomed flask equipped with magnetic stirring bar was added 21.3 g (67.30 mmol)
of iodosobenzene diacetate. The flask was fitted with rubber septa through which a large-gauge needle
was passed to flush the system with dry argon. After the vessel had been thoroughly purged, 500 mL
acetonitrile and 5 mL TFA was added by syringe and stirred at room temperature for 30 min. To this
suspension was added portions of crystalline methyl 4-hydroxy-phenylacetate 2.31 (250 mg every 15
minutes) until a total of 10.0 g (60.18 mmol) methyl 4-hydroxy-phenylacetate had been added. The
reaction mixture was allowed to stir at room temperature for 16 h. The clear reddish-colored solution was
concentrated to a dark-red colored oil in vacuo on a rotary evaporator (12.7 torr, 40 °C) and passed
through a short plug of silica gel (75 g), eluting non-polar impurities with 50% ethyl acetate in hexanes
and enriched product with 100% ethyl acetate. The enriched product fractions were combined and
concentrated to an orange-colored solid (9.5 g – 11.2 g). The enriched product is dissolved in hot acetone
(50 mL) and diethyl ether (50 mL) and crystallized at –20 °C overnight. Product crystals were collected
by vacuum filtration and washed with minimal amounts of diethyl ether to give 6.95 g – 7.35 g (50 –
55%) of the desired dienone 2.32 as off-white crystals.

117
Procedure B
To a 1000-mL round-bottomed flask equipped with magnetic stirring bar was added 21.3 g (67.30 mmol)
of iodosobenzene diacetate. The flask was fitted with rubber septa through which a large-gauge needle
was passed to flush the system with dry argon. After the vessel had been thoroughly purged, 500 mL
acetonitrile and 5 mL TFA was added by syringe and stirred at room temperature for 30 min. To this
suspension was added manually portions of crystalline methyl 4-hydroxy-phenylacetate 2.31 (continuous
addition of the solid phenol 2.31 over 3 h) until a total of 10.0 g (60.18 mmol) methyl 4-hydroxy-
phenylacetate had been added. The reaction mixture was allowed to stir at room temperature for 16 h.
The clear reddish-colored solution was concentrated to a dark-red colored oil in vacuo on a rotary
evaporator (12.7 torr, 40 °C) and passed through a column of silica gel (~200 g), eluting non-polar
impurities with 50% ethyl acetate in hexanes and enriched product with 100% ethyl acetate. The enriched
product fractions were combined and concentrated to an orange-colored solid which was recrystallized
from hot acetone (~100 mL). Product crystals were collected by vacuum filtration and washed with
minimal amounts of diethyl ether to give 9.4 g (42.13 mmol, 70%) of the desired dienone 2.32 as off-
white crystals. Note: repurification/recrystallization of the supernatant could provide additional 2.32.
1H (d6-acetone): 7.56 (s, 1H), 7.18 (d, 2H, J = 10.23), 6.15 (d, 2H, J = 10.20), 3.61 (s, 3H), 3.02 (s, 2H),
1.88 (s, 3H).
13C (d6-acetone): 184.14, 169.34, 168.81, 148.56, 127.90, 53.09, 50.93, 41.40, 22.24.
MP: 104–105 °C.
IR: 1735, 1675, 1662.
ESI-MS: 246.2 [M + Na]+.
HRMS: calcd for C11H13NO4 [M + Na]+ = 246.0742, found 246.0736.
EA: calcd C 59.19%, H 5.87%, N 6.28%; found C 59.27%, H 5.84%, N 6.24%.

118
A.2 Preparation of methyl 2-((1r,4r)-1-acetamido-4-(tert-butyldiphenylsilyloxy)cyclohexa-2,5-
dienyl)acetate (2.37)
TBDPSO
NHAc
CO2CH3H
2.37
Chemical Formula: C27H33NO4Si
Exact Mass: 463.22
Molecular Weight: 463.64
Commercial DIBAL-H solution (1M in hexanes, 36.8 mL, 36.8 mmol) was added to a cold (–78 °C), well
stirred solution of 2.32 (5.5 g, 24.5 mmol) in THF (250 mL), under argon atmosphere. The mixture was
stirred at –78 C for 4 h, and then it was quenched by the sequential addition of water (4 mL), 10% aq
NaOH (4 mL), and again water (12 mL). The volatile organics were removed in vacuo, and the crude
product was suspended in acetone (200 mL) and filtered through methanol-washed celite. The filtrate was
evaporated in vacuo and the residue (5.1 g, 22.6 mmol, 92% yield) was advanced to the next step without
further purification. Thus, a portion of this material (3.0 g, 13.3 mmol) in CH2Cl2 (30 mL) was treated
with TBDPS-Cl (5.1 mL, 20 mmol) and imidazole (1.4 g, 20 mmol), and the mixture was stirred at rt
under an argon atmosphere for 18 h. The mixture was then treated with 0.05M HCl (30 mL) and
extracted with ethyl acetate. The combined extracts were dried over anhydrous MgSO4, filtered and
concentrated in vacuo. The crude product was purified by flash column chromatography (step gradient of
20-40-100% ethyl acetate/hexanes). Pure fractions were concentrated to give 2.37 (5.3 g, 11.4 mol, 87%)
as a clear viscous oil.
1H (d6-acetone): 7.73 (m, 4H), 7.45 (m, 6H), 6.99 (brs, 1H), 6.14 (d, 2H, J = 10.1), 5.80 (dd, 2H, J2 =
10.3; J1 = 2.8), 4.60 (m, 1H), 3.62 (s, 3H), 2.96 (s, 2H) 1.75 (s, 3H), 1.07 (s, 9H).
13C (d6-acetone): 170.7, 169.7, 136.5, 134.6, 131.0, 130.7, 130.0, 128.6, 64.5, 52.2, 51.6, 43.3, 27.3,
23.5, 19.7.
IR: 1739, 1653.
ESI-MS: 486.2 [M + Na]+.
HRMS: calcd for C27H33NO4Si•Na+ [M + Na]
+ = 486.2077, found 486.2059.
EA: calcd C 69.94%, H 7.17%, N 3.02%; found C 69.71%, H 7.45%, N 3.02%.

119
A.3 Preparation of 2-((1r,4r)-1-acetamido-4-(tert-butyldiphenylsilyloxy)cyclohexa-2,5-
dienyl)acetic acid (2.38)
TBDPSO
NHAc
CO2HH
2.38
Chemical Formula: C26H31NO4Si
Exact Mass: 449.2
Molecular Weight: 449.61
A solution of 2.37 (2.2 g, 4.7 mmol) in THF (70 mL) was treated with aqueous 1M NaOH (15 mL) and
the mixture was stirred for 18 h at rt. The volatile organics were removed in vacuo, and the residue was
partitioned between 50% AcOH/H2O (60 mL) and ethyl acetate (100 mL). The organic layer was dried
over MgSO4, filtered and concentrated to give pure 2.38 (2.1 g, 4.7 mmol, 99% yield) as a glassy-solid.
1H (600 MHz, d6-acetone): 7.73 (m, 4H), 7.46 (m, 6H), 6.96 (brs, 1H), 6.14 (d, 2H, J = 8.7), 5.80 (dd,
2H, J2 = 10.3; J1 = 3.0), 4.62 (m, 1H), 2.88 (s, 2H), 1.74 (s, 3H), 1.06 (s, 9H).
13C (150 MHz, d6-acetone): 172.0, 170.1, 137.1, 135.2, 131.8, 131.3, 130.6, 130.2, 129.2, 65.3, 52.5,
43.6, 27.8, 24.1, 20.3.
ESI-MS: 472.2 [M + Na]+.
HRMS: calcd for C26H31NO4Si•Na+ [M + Na]
+ = 472.1920, found 472.1912.

120
A.4 Preparation of N-((1r,4r)-4-(tert-butyldiphenylsilyloxy)-1-(3-nitro-2-oxopropyl)cyclohexa-
2,5-dienyl)acetamide (2.39)
TBDPSO
NHAc
H
2.39
ONO2
Chemical Formula: C27H32N2O5Si
Exact Mass: 492.21
Molecular Weight: 492.64
Carbonyldiimidazole (874 mg, 5.4 mmol) was added to a solution of acid 2.38 (2.0 g, 4.5 mmol) in THF
(10 mL) and the mixture was stirred at rt for 1 h. Nitromethane (1.5 mL, 26.9 mmol) followed by KOtBu
(2.0 g, 18.0 mmol) were then added and after stirring for an additional 2 h, the volatiles were removed in
vacuo. The residue was partitioned between 50% AcOH/H2O (100 mL) and CH2Cl2 (150 mL). The
organic layer was dried (MgSO4) filtered and concentrated to give crude nitroketone 2.39, which was
purified by flash column chromatography using a step gradient from 30% ethyl acetate/hexanes to 100%
ethyl acetate. This afforded 1.8 g (3.7 mmol, 82% yield) of pure 2.39 as a light-yellow colored amorphous
solid.
1H (d6-acetone): 7.74 (m, 4H), 7.47 (m, 6H), 6.18 (dd, 2H, J2 = 10.2; J1 = 1.7), 5.86 (dd, 2H, J2 = 10.3; J1
= 3.2), 5.67 (s, 2H), 4.59 (m, 1H), 3.30 (s, 2H), 1.78 (s, 3H), 1.07 (s, 9H).
13C (d6-acetone): 195.5, 170.4, 136.6, 134.5, 130.9, 130.8, 130.2, 128.7, 85.2, 64.2, 52.2, 48.7, 27.3,
23.5, 19.7.
IR: 1736, 1700, 1656, 1558.
ESI-MS: 515.2 [M + Na]+.
HRMS: calcd for C27H32N2O5Si•Na+ [M + Na]
+ = 515.1978, found 515.1962.

121
A.5 Preparation of N-((3aS,7aS)-2-oxo-2,3,3a,7a-tetrahydrobenzofuran-3a-yl)acetamide (2.41)
O
O
NHAc
H
2.41
Chemical Formula: C10H11NO3
Exact Mass: 193.07
Molecular Weight: 193.2
Carboxylic acid 2.38 (also 2.40 and mixtures of the two) readily converts to 2.41 upon treatment with
acidic media such as 1M aq HCl. Lactone 2.41 was commonly seen as a side product during the workup
and extraction of carboxylic acid 2.38. Lactone 2.41 was intentionally prepared by exposing compound
2.38 to any amount of HCl for a prolonged period of time.
1H (d6-acetone): 6.12 (m, 1H), 6.00 (m, 2H, 1 olefinic H + NH signals), 5.88 (m, 2H), 5.45 (br d, 1H, J =
3.8), 3.28 (d, 1H, B part of AB, J = 17.5), 2.81 (d, 1H, A part of AB, J = 17.5), 2.01 (s, 3H).
13C (d6-acetone): 174.2, 170.6, 129.4, 125.9, 123.3, 123.0, 80.8, 57.6, 39.7, 23.9.
ESI-MS: 216.3 [M + Na]+.
HRMS: calcd for C10H11NO3 •Na+ [M + Na]
+ = 216.0637, found 216.0634.

122
A.6 Preparation of N-((2aR,2a1S,3S,5aS)-3-(tert-butyldiphenylsilyloxy)-7-oxo-2a,2a1,3,5a,6,7-
hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.43)
O
O
NHAc
H
TBDPS
NO
2.43
Chemical Formula: C27H30N2O4Si
Exact Mass: 474.2
Molecular Weight: 474.62
A solution of 2.39 (27 mg, 55 mol), TBSCl (17 mg, 110 mol) and imidazole (8 mg, 110 mol) in dry
CH2Cl2 (0.5 mL) was stirred at rt for 120 h, then it was concentrated in vacuo. Purification of the crude
2.43 residue by flash column chromatography (50% ethyl acetate/hexanes to 100% ethyl acetate) gave
pure 2.43 (10 mg, 38% yield) as a foamy colorless solid.
1H (d6-acetone): 7.79 (d, 2H, J = 6.8), 7.73 (d, 2H, J = 7.7), 7.64 (brs, 1H), 7.47 (m, 6H), 6.09 (ddd, 1H,
J3 = 10.3; J1 = J2 = 1.6), 5.70 (ddd, 1H, J3 = 10.3; J2 = 3.0; J1 = 1.2), 5.01 (dd, 1H, J3 = 11.1; J2 = 5.0; J1 =
1.4), 4.81 (ddd, 1H, J3 = 4.9; J2 = 2.9; J1 = 1.8), 4.70 (dd, 1H, J2 = 11.1; J1 = 1.0), 3.36 (d, 1H, B part of
AB, J = 18.0), 3.04 (d, 1H, A part of AB, J = 18.0), 1.71 (s, 3H), 1.11 (s, 9H).
13C (d6-acetone): 192.0, 170.3, 163.1, 137.9, 136.6, 136.5, 134.5, 134.3, 130.9, 130.8, 130.4, 128.7,
128.7, 85.4, 67.3, 57.3, 56.0, 52.3, 27.3, 23.0, 19.8.
MP: 92–96 °C.
IR: 1747, 1660, 1599, 1538.
ESI-MS: 497.2 [M + Na]+.
HRMS: calcd for C27H30N2O4Si•Na+ [M + Na]
+ = 497.1873, found 497.1866.

123
A.7 Preparation of N-((3aS,7aS,E)-2-(nitromethylene)-2,3,3a,7a-tetrahydrobenzofuran-3a-
yl)acetamide (2.44)
OH
NHAc
2.44
NO2
Chemical Formula: C11H12N2O4
Exact Mass: 236.08
Molecular Weight: 236.22
A solution of nitroketone 2.39 (33 mg, 67 mol) and triethylamine (50 L) in anhydrous THF (300 L)
was stirred for 24 h at rt, then it was concentrated in vacuo. Flash column chromatography of the residue
(50% ethyl acetate/hexanes to 75% ethyl acetate) afforded 2.44 (10 mg, 42 mol, 63%) as a crystalline
solid (crystallized from ethyl acetate).
1H (d6-acetone): 7.72 (br s, 1H), 7.19 (s, 1H), 6.17 (dd, 1H, J2 = 9.7; J1 = 5.3), 6.10 (d, 1H, J = 9.5), 5.99
(dd, 1H, J2 = 9.7; J1 = 5.4), 5.88 (dd, 1H, J2 = 9.7; J1 = 3.6), 5.67 (d, 1H, J = 3.6), 4.17 (d, 1H, B part of
AB, J = 19.0), 3.40 (d, 1H, A part of AB, J = 19.0), 1.90 (s, 3H).
13C (d6-acetone): 172.9, 169.7, 131.5, 125.8, 122.6, 121.0, 118.1, 87.0, 58.3, 45.5, 22.3.
MP: 154–156 °C.
ESI-MS: 237.2 [M + H]+; 259.1 [M + Na]
+.
HRMS: calcd for C11H12N2O4•Na+ [M + Na]
+ = 259.0695, found 259.0688.

124
A.8 Preparation of N-((1r,4r)-1-(2-(tert-butyldimethylsilyloxy)-3-nitropropyl)-4-(tert-
butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetamide (2.46) diastereomers
OTBS
NO2
TBDPSO
NHAc
2.46
Chemical Formula: C33H48N2O5Si2Exact Mass: 608.31
Molecular Weight: 608.92
Nitro alcohol intermediate:
ESI-MS: 517.1 [M + Na]+.
HRMS: calcd for C27H34N2O5 [M + Na]+ = 517.2135, found 517.2152.
Product 2.46 diastereomers:
Note: NMR data shows overlapping signals which appeared difficult to deconvolute.
1H (d6-acetone, 400 MHz): 7.74 (m, 4H), 7.46 (m, 6H), 6.91 (br s, 1H), 6.15 (c br m, 1H), 5.94 (c m,
2H), 5.85 (br c m, 1H), 4.81 (c m, 1H), 4.56 (br s, 1H), 4.47 (c m, 1H) 4.40 (c m, 1H), 2.47 (c br m, 1H),
2.29 (c br m, 1H), 1.75 (br s, 3H), 1.07 (br s, 9H), 0.86 (br s, 9H), 0.17 (br s, 3H), 0.08 (br s, 3H).
13C (d6-acetone, 100 MHz): 169.59, 136.77, 136.75, 136.42, 134.65, 132.11, 131.34, 130.94, 130.79,
130.65, 128.81, 128.80, 136.77, 136.75, 136.42, 134.65, 132.11, 131.34, 130.94, 130.79, 130.65, 128.81,
128.80, 82.37, 68.63, 64.49, 52.94, 43.88, 27.43, 26.43, 26.18, 23.83, 19.78, 18.57, -3.81, -4.79.
ESI-MS: 631.3 [M + Na]+.
HRMS: calcd for C33H48N2O5 [M + Na]+ = 631.2999, found 631.2994.
EA: calcd C 65.09%, H 7.95%, N 4.6%; found C 65.17%, H 8.00%, N 4.82%.

125
A.9 Preparation of compounds 2.49, 2.50, 2.51 and 2.52
TBDPSO
O N
OTBS
NHAc
= 2.49 = 2.50
Chemical Formula: C33H46N2O4Si2Exact Mass: 590.3
Molecular Weight: 590.9
TBDPSO
O N
OTBS
OTBS
NHAc
H
= 2.51 = 2.52
Chemical Formula: C39H62N2O5Si3Exact Mass: 722.4
Molecular Weight: 723.18
Solid NaBH4 (1.4 g, 36.7 mmol) was added to a cold (0 °C), well stirred solution of nitroketone 2.39
(3.61 g, 7.33 mmol) in methanol (50 mL), and stirring was continued for 2 h. The mixture was carefully
poured into 50% AcOH/H2O (100 mL; CAUTION: release of flammable H2 gas) and the acetic acid
solution was extracted with ethyl acetate (3 x 150 mL). The combined extracts were dried over MgSO4,
filtered and concentrated in vacuo. The alcohol intermediate was found to be extremely sensitive and
readily decomposed in a retro-Henry mode. Thus, the crude reaction residue was immediately dissolved
in CH2Cl2, and to this solution was added imidazole (2.0 g, 29.4 mmol) and TBSCl (4.4 g, 29.3 mmol).
The reaction mixture was stirred for 168 h and then it was concentrated in vacuo. Purification of the
residue by flash column chromatography (step gradient 5, 10, 15, 20, 25, 30, 40, 50 and then 100% ethyl
acetate/hexanes) yielded the following compounds: ester 2.37 (200 mg, 432 mol, 6% yield), 2.49 (725
mg, 1.2 mmol, 17% yield), 2.50 (775 mg, 1.3 mmol, 18% yield), and a mixture of compounds 2.51 and
2.52 (300 mg, 420 mol, 6% yield, note: the spontaneous conversion of 2.51 and 2.52 to the
isooxazolines 2.49 and 2.50 occurred slowly over time).

126
A.9.1 Compound 2.49
TBDPSO
O N
OTBS
NHAc
2.49
Chemical Formula: C33H46N2O4Si2Exact Mass: 590.3
Molecular Weight: 590.9
Colorless oil.
1H (d6-acetone): 7.76 (m, 4H), 7.44 (m, 6H), 5.85 (B-part of AB-type system, app br dt, 1H, J = 10.2),
5.59 (A-part of AB-type system, br d, 1H, J = 10.2), 4.78 (m, 1H), 4.75 (m, 1H), 4.61 (m, 1H), 4.28 (br d,
1H, J = 10.2), 2.90 (B-part of AB-type system, dd, 1H, J2 = 14.0; J1 = 6.9), 2.38 (A-part of AB-type
system, dd, 1H, J2 = 14.0; J1 = 4.0), 1.68 (s, 3H), 1.10 (s, 9H), 0.89 (s, 9H), 0.12 (s, 3H), 0.10 (s, 3H).
ESI-MS: 589.6 [M – H]-; 613.4 [M + Na]
+.
HRMS: calcd for C33H46N2O4Si2•Na+ [M + Na]
+ = 613.2894, found 613.2891.

127
A.9.2 Compound 2.50
TBDPSO
O N
OTBS
NHAc
2.50
Chemical Formula: C33H46N2O4Si2Exact Mass: 590.3
Molecular Weight: 590.9
Colorless oil.
1H (d6-acetone): 7.76 (m, 4H), 7.44 (m, 6H), 7.31 (br s, 1H), 5.88 (B-part of AB-type system, app dt,
1H, J2 = 10.2; J2 = 1.6), 5.69 (A-part of AB-type system, br d, 1H, J = 10.2), 5.11 (dd, 1H, J2 = 9.6; J1 =
4.5 ), 4.66 (m, 1H), 4.63 (m, 1H), 4.28 (br d, 1H, J = 10.2), 2.90 (B-part of AB-type system, dd, 1H, J2 =
13.8; J1 = 9.7), 2.19 (A-part of AB-type system, dd, 1H, J2 = 13.8; J1 = 4.6), 1.67 (s, 3H), 1.08 (s, 9H),
0.90 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H).
13C (d6-acetone): 169.0, 166.0, 135.8, 135.7, 133.8, 133.6, 132.2, 129.8, 127.7, 127.6, 80.2, 66.8, 65.7,
57.8, 51.9, 51.6, 26.4, 25.2, 22.2, 18.9, 17.9, -5.6, -5.9.
ESI-MS: 589.5 [M – H]-; 613.4 [M + Na]
+.
HRMS: calcd for C33H46N2O4Si2•Na+ [M + Na]
+ = 613.2894, found 613.2883.

128
A.9.3 Compound 2.51/2.52
TBDPSO
O N
OTBS
OTBS
NHAc
H
= 2.51 = 2.52
Chemical Formula: C39H62N2O5Si3Exact Mass: 722.4
Molecular Weight: 723.18
Colorless oil.
1H (d6-acetone): 7.71 (m, 4H), 7.44 (m, 6H), 7.02 (brs, 1H), 6.01 (d, 1H, J = 10.1), 5.61 (ddd, 1H, J3 =
10.1; J1 = J2 = 4.3), 4.76 (dd, 1H, J2 = 8.4; J1 = 2.2 ), 4.24 (m, 1H), 4.01 (dd, 1H, J2 = 8.4; J1 = 4.8), 3.80
(app q, 1H, J = 6.3), 3.63 (app t, 1H, J = 8.4), 2.91 (dd, 1H, B part of AB-type system, J = 13.9, 6.3), 2.38
(dd, 1H, A part of AB-type system, J = 13.9, 4.2), 1.88 (s, 3H), 1.08 (s, 9H), 0.91 (s, 9H), 0.89 (s, 9H),
0.13 (s, 6H), 0.07 (s, 3H), 0.05 (s, 3H).
ESI-MS: 729.6 [M + Na]+.
HRMS: calcd for C39H62N2O4Si3•Na+ [M + Na]
+ = 729.3908, found 729.3915.

129
A.10 Preparation of trans-9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-3-nitro-1-
azaspiro[5.5]undeca-2,7,10-trien-4-one (2.55)
2.55
HN
CH3
NO2
O
TBDPSO
Chemical Formula: C27H30N2O4Si
Exact Mass: 474.2
Molecular Weight: 474.62
A solution of nitroketone 2.39 (1.7 g, 3.4 mmol) TBSCl (5.1 g, 34.0 mmol) and imidazole (2.3 g, 34.0
mmol) in anhydrous CH2Cl2 (20 mL) was stirred for 48 h at rt, then the reaction was concentrated in
vacuo. Silica gel flash column chromatography (50% ethyl acetate/hexanes to 100% ethyl acetate) of the
reaction residue afforded 1.1 g (2.2 mmol, 65%) of 2.55 as a crystalline solid, (recrystallized from ethyl
acetate), and 83 mg (180 mol) of isoxazoline 2.43.
1H (d6-acetone): 7.72 (m, 4H), 7.45 (m, 6H), 5.99 (s, 4H), 4.58 (m, 1H), 2.59 (s, 2H), 2.28 (s, 3H), 1.06
(s, 9H).
13C (d6-DMSO): 178.9, 161.9, 135.3, 133.0, 131.2, 130.1, 128.0, 127.5, 125.2, 63.0, 52.5, 45.6, 26.7,
19.7, 18.7.
MP: 212–213 °C
ESI-MS: 475.2 [M +H]+; 497.2 [M + Na]
+.
HRMS: calcd for C27H29N2O4Si [M – H]– = 473.1897, found 473.1913.
EA: calcd C 68.33%, H 6.37%, N 5.90%; found C 68.19%, H 6.39%, N 5.89%.

130
A.11 Preparation of methyl 2-((1S,4S,5R,6S)-1-acetamido-4-(tert-butyldiphenylsilyloxy)-6-cyano-
5-hydroxycyclohex-2-enyl)acetate (2.59)
OH
O CN
CO2CH3
NHAc
2.59
TBDPS
Chemical Formula: C28H34N2O5Si
Exact Mass: 506.22
Molecular Weight: 506.67
Isooxazoline 2.43 (15 mg, 0.0317 mmol) was dissolved in methanol (0.5 mL) at ambient temperature. To
this solution was added imidazole (1 mg, 0.0147 mmol) and lithium carbonate (1 mg, 0.0135 mmol). The
solution was allowed to stir for 1 h, before the reaction mixture was concentrated in vacuo. The crude
residue was passed through a small plug of silica gel (150 mg) using ethyl acetate/hexanes (30-75% ethyl
acetate/hexanes). Compound 2.59 was obtained in 68% yield (11 mg, 0.0217 mmol) as a glassy solid.
1H (d6-acetone): 7.78 (m, 4H), 7.45 (m, 6H), 7.31 (br s, 1H), 5.86 (ddd, 1H, J3 = 10.4; J1 = J2 = 1.8),
5.52 (ddd, 1H, J3 = 10.5; J1 = J2 = 2.9), 4.41 (m, 1H), 4.33 (3, 1H), 4.26 (m, 1H), 3.67 (s, 3H), 3.33 (d,
1H, B part of AB, J = 15.8), 3.17 (d, 1H, A part of AB, J = 15.8), 1.82 (s, 3H), 1.12 (s, 9H).
13C (d6-acetone): 170.9, 170.3, 136.8, 136.7, 134.4, 133.9, 130.9, 130.9, 130.0, 128.7, 128.6, 119.2, 69.0,
68.9, 68.8, 54.8, 51.9, 41.5, 38.3, 27.3, 23.5, 19.9.
IR: 2243, 1737, 1661.
ESI-MS: 529.2 [M + Na]+.
HRMS: calcd for C28H34N2O5Si•Na+ [M + Na]
+ = 529.2135, found 529.2123.

131
A.12 Preparation of N-((2aR,2a1S,3S,5aS)-3-hydroxy-7-oxo-2a,2a1,3,5a,6,7-
hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.60)
HO
O
NHAc
H NO
2.60
Chemical Formula: C11H12N2O4
Exact Mass: 236.08
Molecular Weight: 236.22
To a solution of isooxazoline 2.43 (40 mg, 0.084 mmol) in dry CD3CN (1 mL) was added drop-wise
HF/H2O (35% HF v/v, 3 drops). This reaction was allowed to stir for 96 h, after which volatile organics
were removed in vacuo. The crude product was purified via flash column chromatography (50%
acetone/hexanes, then 75% acetone/hexanes, then 100% acetone) to give pure 2.60 (14 mg, 0.059 mmol,
70% yield) as a glassy solid. Unreacted compound 2.43 was also recovered (3 mg, 0.006 mmol, 7%).
1H (d6-acetone): 7.73 (brs, 1H), 6.05 (ddd, 1H, J3 = 10.3; J1 = J2 = 1.6 ), 5.70 (ddd, 1H, J = 10.3, 3.0,
1.2), 5.22 (ddd, 1H, J3 = 11.1, 5.2, 1.4), 4.85 (dd, 1H, J = 11.1, 0.9), 4.54 (br m, 1H), 4.22 (br d, 1H, J =
8.2), 3.45 (d, 1H, B part of AB, J = 18.0), 3.04 (d, 1H, A part of AB, J = 18.0), 1.85 (s, 3H).
13C (d6-acetone): 192.1, 170.5, 163.5, 138.8, 129.9, 85.5, 65.1, 57.7, 56.0, 52.3, 23.2.
ESI-MS: 259.2 [M + Na]+.
HRMS: calcd for C11H12N2O4•Na+ [M + Na]
+ = 259.0695, found 259.0695.

132
A.13 Preparation of methyl 2-((1S,5R,6S)-1-acetamido-6-cyano-5-hydroxy-4-oxocyclohex-2-
enyl)acetate (2.62)
O
CO2CH3
HO N
NHAc
2.62
Chemical Formula: C12H14N2O5
Exact Mass: 266.09
Molecular Weight: 266.25
To a solution of 2.60 (14 mg, 0.0593 mmol) in CD3CN (1.5 mL) was added Dess-Martin periodinane (30
mg, 71 mol). This solution was monitored by 1H-NMR until the reaction had completed (12 h). The
crude reaction was filtered through celite and concentrated in vacuo. Crude N-[(4aR,7aS,7bR)-3,4,7a,7b-
tetrahydro-3,7-dioxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-acetamide (2.61) was thus obtained.
1H (d6-acetone): 8.16 (br s, 1H), 6.55 (dd, 1H, J = 10.4, 2.0), 6.29
(dd, 1H, J = 10.4, 0.6), 5.19 (dd, 1H, J = 10.7, 0.7), 4.78 (dd, 1H, J =
10.7, 1.8), 3.45 (d, 1H, B part of AB, J = 18.0), 3.23 (d, 1H, A part of
AB, J = 18.0), 1.89 (s, 3H).
ESI-MS: 257.1 [M + Na]+.
HRMS: calcd for C11H10N2O4•Na+ [M + Na]
+ = 257.0538, found
257.0541.
Diketone 2.61 was not purified nor thoroughly characterized due to significant reactivity. Instead, it was
dissolved in dry methanol (700 L) and to this was added Li2CO3 (1 mg, 14 mol), and the mixture was
stirred at rt for 8 h. The suspension was filtered through celite and concentrated in vacuo. The reaction
residue was purified by flash column chromatography (30% ethyl acetate/hexanes, then 75% ethyl
acetate/hexanes, then 100% ethyl acetate) to give pure 2.62 (6 mg, 23 mol, 38% yield) as a glassy solid.
1H (CD3CN): 7.25 (dd, 1H, J = 10.3, 1.5), 6.10 (d, 1H, J = 10.3), 4.74 (d, 1H, J = 5.0), 4.42 (dd, 1H, J2 =
5.0; J1 = 1.4), 3.68 (s, 3H), 3.18 (app. d, 2H, actually compressed AB system, J = 15.7), 1.87 (s, 3H).
13C (CD3CN, 100 MHz): 196.46, 171.67, 170.34, 149.24, 127.54, 117.79, 70.16, 56.20, 52.61, 43.74,
41.19, 23.31.
IR: 2251, 1732, 1699, 1655.
ESI-MS: 289.1 [M + Na]+.
HRMS: calcd for C12H14N2O5•Na+ [M + Na]
+ = 289.0800, found 289.0794.
O
O
NHAc
NO
2.61
Chemical Formula: C11H10N2O4
Exact Mass: 234.06Molecular Weight: 234.21

133
A.14 Preparation of N-((2aR,2a1S,3R,5aS)-3-(tert-butyldiphenylsilyloxy)-7-oxo-2a,2a1,3,5a,6,7-
hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.66)
O
O
NHAc
H
TBDPS
NO
2.66
Chemical Formula: C27H30N2O4Si
Exact Mass: 474.2
Molecular Weight: 474.62
Dienone 2.32 (15.0 g, 67.3 mmol) was dissolved in THF (200 mL) at room temperature under argon
atmosphere. To this solution was added (S)-(–)2-methyl-CBS-oxazaborolidine (1.83 g, 6.73 mmol)
followed by the addition of BH3-DMS complex (6.38 mL, 67.3 mmol). This reaction was allowed to stir
overnight. The crude reaction residue (mostly 2.35 and 2.36) was thoroughly concentrated in vacuo. The
crude reaction residue was dissolved in CH2Cl2 (500 mL) and to this was added imidazole (5.04 g, 74.0
mmol) and TBDPS-Cl (20.3 g, 74.0 mmol). The reaction was allowed to stir overnight at ambient
temperature under an argon atmosphere. The crude reaction mixture was poured into a solution of 0.05M
aqueous HCl (400 mL) and the aqueous phase was extracted with CH2Cl2 (2 x 400 mL). The combined
organic phase was dried over anhydrous MgSO4, filtered through a short plug of silica gel and
concentrated in vacuo. The crude mixture of TBDPS-alcohols 2.37/2.63 was dissolved in THF (500 mL)
and to this was added a solution of 1M aqueous NaOH (270 mL). The biphasic mixture was vigorously
stirred overnight. The volatile organics were removed in vacuo, and the residue was partitioned between
50% AcOH/H2O (500 mL) and ethyl acetate (400 mL). The aqueous layer was extracted a second time
with ethyl acetate (250 mL), and the combined organic layer was dried over anhydrous MgSO4, filtered
and concentrated in vacuo. The crude mixture of carboxylic acids 2.38/2.64 was dissolved in THF (250
mL) and to this was added carbonyldiimidazole (13.1 g, 80.8 mmol) with stirring at room temperature.
The reaction mixture was stirred for 1 h at room temperature, and then nitromethane (100 mL) was added,
followed by the addition of solid potassium tert-butoxide (30.2 g, 269.2 mmol). The reaction mixture
was stirred at room temperature for 2 h and then the volatiles were removed in vacuo. The residue was
partitioned between 50% AcOH/H2O (500 mL) and CH2Cl2 (400 mL). The organic phase was separated
and the aqueous phase was washed with additional CH2Cl2 (2 x 400 mL). The combined organic phase
was dried over anhydrous MgSO4, filtered through a short silica gel plug and concentrated to give

134
enriched nitroketones 2.39/2.65. The mixture of crude nitroketones was dissolved in CH2Cl2 (300 mL)
and to this was added imidazole (9.12 g, 134 mmol) and TBS-Cl (20.20 g, 134 mmol). The reaction
mixture was allowed to stir at room temperature under an argon atmosphere for 5 days. The reaction
mixture was concentrated in vacuo and purified by flash column chromatography using (50% ethyl
acetate/hexanes to 100% ethyl acetate) to give 2.43 (2.7 g, 8% yield over 5 steps) and 2.66 (6.3 g, 20%
yield over 5 steps). Significant amount of nitroketones 2.39 and 2.65 were recovered (~28%) and
recycled.
1H (d6-acetone): 7.73 (m, 4H),7.47 (m, 6H), 6.15 (ddd, 1H, J3 = 10.3; J1 = J2 = 1.1 ), 6.01 (ddd, 1H, J =
10.0, 5.3, 0.8), 5.17 (ddd, 1H, J3 = 10.9, 2.0, 0.8), 5.05 (app d, 1H, J = 5.4), 4.16 (dd, 1H, J = 5.4, 2.0),
3.55 (d, 1H, B part of AB, J = 18.3), 3.12 (d, 1H, A part of AB, J = 18.3), 1.93 (s, 3H), 1.09 (s, 9H).
ESI-MS: 473.5 [M – H]–, 497.4 [M + H]
+, 497.4 [M + Na]
+.
HRMS: calcd for C27H31N2O4•Na+ [M + Na]
+ = 475.2053, found 475.2059.

135
A.15 Preparation of methyl 2-((1S,4R,5R,6S)-1-acetamido-4-(tert-butyldiphenylsilyloxy)-6-cyano-
5-hydroxycyclohex-2-enyl)acetate (2.67)
OH
O CN
CO2CH3
NHAc
2.67
TBDPS
Chemical Formula: C28H34N2O5Si
Exact Mass: 506.22
Molecular Weight: 506.67
Isooxazoline 2.66 (900 mg, 1.90 mmol) was dissolved in methanol (10 mL) at ambient temperature. To
this solution was added imidazole (10 mg) and lithium carbonate (50 mg). The solution was allowed to
stir for 1 h, before the reaction mixture was concentrated in vacuo. The crude residue was passed through
a small plug of silica gel (1 g) using ethyl acetate/hexanes (30-75% ethyl acetate/hexanes). Compound
2.67 was obtained in 90% yield (864 mg, 1.71 mmol) as a glassy solid.
1H (d6-acetone): 7.78 (m, 4H), 7.45 (m, 6H), 7.33 (brs, 1H), 5.92 (ddd, 1H, J3 = 10.3; J2 = 1.3 J1 = 0.4),
5.52 (ddd, 1H, J3 = 10.3, J2 = J1 =2.6), 5.00 (d, 1H, J = 4.7), 4.50 (c m, 1H), 4.34 (dd, 1H, J2 = 4.0 J1 =
1.0), 4.26 (m, 1H), 3.62 (s, 3H), 3.35 (d, 1H, A part of ABq, J = 15.5), 2.88 (d, 1H, B part of ABq, J =
15.5), 1.89 (s, 3H), 1.09 (s, 9H).
13C (d6-acetone): 169.70, 169.69, 135.91, 135.84, 133.97, 133.30, 131.26, 129.84, 129.83, 128.10,
127.69, 127.64, 117.91, 71.61, 69.50, 55.04, 50.94, 39.98, 39.52, 26.46, 22.67, 19.04.
IR: 2247 cm-1
, 1719 cm-1
, 1639 cm-1
.
ESI-MS: 529.5 [M + Na]+.
HRMS: calcd for C28H34N2O5•Na+ [M + Na]
+ = 529.2135, found 529.2122.

136
A.16 Preparation of compounds 2.76-2.88
Representative procedure for the results listed in Tables 2.8 and 2.9:
A solution of oxime (1 mmol) in methanol (1 mL) was added slowly (syringe pump, 1 h) at room
temperature to a stirred solution of DIB (1.1 eq) and olefin (1.1 eq) in methanol (2 mL) containing TFA
(15 L). A white precipitate formed immediately and then slowly redissolved as the reaction progressed.
Upon consumption of starting oxime (TLC, about 1 h), the mixture was concentrated in vacuo and the
residue was purified by flash column chromatography using a step gradient: 5%-10%-20% ethyl
acetate/hexanes.
A.16.1 Preparation of 3-(4-methoxyphenyl)-5-phenyl-4,5-dihydroisoxazole (2.76)
H3CO
ON
Chemical Formula: C16H15NO2
Exact Mass: 253.11
Molecular Weight: 253.3
2.76
Representative procedure is located in Appendix section A.16.
colorless crystals.
91% yield.
1H (CDCl3): 7.63 (d, 2H, J = 8.9), 7.39 (m, 5H), 6.93 (d, 2H, J = 8.9), 5.71 (dd, 1H, J = 10.9, 8.2), 3.84
(s, 3H), 3.76 (dd, 1H, J = 16.5, 10.9), 3.32 (dd, 1H, J = 16.5, 8.2).
13C (CDCl3): 161.24, 155.81, 141.24, 128.88, 128.43, 128.30, 126.03, 122.19, 114.30, 82.44, 55.51,
43.60.
MP: 100-101 °C.
ESI-MS: 276.3 [M + Na]+.
HRMS: calcd for C16H15NO2Na [M + Na]+ = 276.1000, found 276.1001.

137
A.16.2 Preparation of 3,5-diphenyl-4,5-dihydroisoxazole (2.77)
ON
Chemical Formula: C16H15NO2
Exact Mass: 253.11
Molecular Weight: 253.3
2.77
Representative procedure is located in Appendix section A.16.
colorless crystals.
71% yield.
1H (CDCl3): 7.70 (m, 2H), 7.40 (m, 8H), 5.75 (dd, 1H, J = 11.2, 8.4), 3.79 (dd, 1H, J = 16.4, 11.2), 3.35
(dd, 1H, J = 16.4, 8.2).
13C (CDCl3): 156.23, 141.06, 130.27, 129.61, 128.89, 128.87, 128.35, 126.88, 126.00, 82.70, 43.30.
MP: 72-73 °C.
ESI-MS: 224.3 [M + H]+; 246.3 [M + Na]
+.
HRMS: calcd for C15H13NONa [M + Na]+ = 246.0895, found 246.0894.

138
A.16.3 Preparation of 3-(3-nitrophenyl)-5-phenyl-4,5-dihydroisoxazole (2.78)
ON
Chemical Formula: C16H15NO2
Exact Mass: 253.11
Molecular Weight: 253.3
2.78
O2N
Representative procedure is located in Appendix section A.16.
yellow oil.
91% yield.
1H (CDCl3): 8.44 (t, 1H, J = 1.9), 8.27 (ddd, 1H, J = 8.2, 2.4, 1.0), 8.12 (dt, H, J = 7.9, 1.2), 7.61 (t, 1H,
J = 8.0), 7.39 (m, 5H), 5.84 (dd, 1H, J = 8.3, 11.4), 3.84 (dd, 1H, J = 11.4, 16.5), 3.39 (dd, 1H, J = 16.5,
8.3).
13C (CDCl3): 154.62, 148.62, 140.37, 132.37, 131.49, 130.00, 129.05, 128.67, 125.93, 124.74, 121.72,
83.51.
ESI-MS: 269.2 [M + H]+; 291.2 [M + Na]
+.
HRMS: calcd for C15H12N2O3Na [M + Na]+ = 291.0746, found 291.0736.

139
A.16.4 Preparation of 3-pentyl-5-phenyl-4,5-dihydroisoxazole (2.79)
ON
Chemical Formula: C14H19NO
Exact Mass: 217.15
Molecular Weight: 217.31
2.79
Representative procedure is located in Appendix section A.16.
colorless oil.
74% yield.
1H (CDCl3): 7.34 (m, 5H), 5.54 (dd, 1H, J = 10.8, 8.0), 3.36 (dd, 1H, J = 16.7, 10.8), 2.89 (dd, 1H, J =
16.7, 8.0), 2.38, (t, 2H, J = 7.7), 1.59 (m, 2H), 1.32 (m, 4H), 0.89 (m, 3H).
13C (CDCl3): 158.69, 141.51, 128.75, 128.05, 125.80, 81.28, 45.45, 31.46, 27.73, 26.16, 22.40, 14.03.
ESI-MS: 218.4 [M + H]+.
HRMS: calcd for C14H19NONa [M + Na]+ = 240.1364, found 240.1366.

140
A.16.5 Preparation of 3-phenethyl-5-phenyl-4,5-dihydroisoxazole (2.80)
ON
Chemical Formula: C17H17NO
Exact Mass: 251.13
Molecular Weight: 251.32
2.80
Representative procedure is located in Appendix section A.16.
colorless oil.
63% yield.
1H (CDCl3): 7.38-7.19 (m, 10H), 5.53 (dd, 1H, J = 1.0, 8.0), 3.31 (dd, 1H, J = 16.8, 11.0), 2.94 (m, 2H),
2.90 (dd, 1H, J = 16.8, 8.0), 2.71 (m, 2H).
13C (CDCl3): 157.90, 141.37, 140.55, 128.80, 128.73, 128.44, 128.15, 126.52, 125.87, 81.49, 45.72,
32.82, 29.54.
ESI-MS: 274.3 [M + Na]+.
HRMS: calcd for C17H17NONa [M + Na]+ = 274.1208, found 274.1201.

141
A.16.6 Preparation of 3a,4,5,6,7,7a-hexahydro-3-phenyl-4,7-methano-1,2-benzisoxazole (2.81)
ON
Chemical Formula: C14H15NO
Exact Mass: 213.12
Molecular Weight: 213.28
2.81
Representative procedure is located in Appendix section A.16.
colorless crystals.
95% yield.
1H (CDCl3): 7.71 (m, 2H), 7.39 (m, 3H), 4.64 (d, 1H, J = 8.4), 3.50 (d, 1H, J = 8.4), 2.57 (d, 2H, J =
30.4), 1.56 (m, 3H), 1.35 (m, 1H), 1.18 (m, 2H).
13C (CDCl3): 156.98, 129.78, 129.50, 128.77, 126.92, 87.97, 57.15, 43.09, 39.36, 32.41, 27.52, 22.81.
MP: 98-99 °C.
ESI-MS: 214.3 [M + H]+.
HRMS: calcd for C14H15NONa [M + Na]+ = 236.1051, found 236.1049.

142
A.16.7 Preparation of 3a,4,5,6,7,7a-hexahydro-3-(3-nitrophenyl)-4,7-methano-1,2-benzisoxazole
(2.82)
ON
Chemical Formula: C14H14N2O3
Exact Mass: 258.1
Molecular Weight: 258.27
2.82
O2N
Representative procedure is located in Appendix section A.16.
colorless crystals.
77% yield.
1H (CDCl3): 8.47 (t, 1H, J = 1.8), 8.23 (ddd, 1H, J = 8.4, 2.4, 0.9), 8.09 (dt, 1H, J = 7.9, 0.9), 7.58 (t, 1H,
J = 8.0), 4.73 (d, 1H, J = 8.6), 3.52 (d, 1H, J = 8.6), 2.67 (app br s, 1H), 2.52 (app br s, 1H), 1.72-1.32 (m,
4H), 1.23 (m, 2H).
13C (CDCl3): 155.55, 148.67, 132.55, 131.52, 129.90, 124.32, 121.57, 89.00, 56.61, 43.12, 39.26, 32.52,
27.55, 22.76.
MP: 76-77 °C.
ESI-MS: 259.3 [M + H]+.
HRMS: calcd for C14H14N2O3Na [M + Na]+ = 281.0902, found 281.0906.

143
A.16.8 Preparation of 3a,4,5,6,7,7a-hexahydro-3-pentyl-4,7-methano-1,2-benzisoxazole (2.83)
Representative procedure is located in Appendix section A.16.
colorless oil.
91% yield.
1H (CDCl3): 4.39 (d, 1H, J = 8.2), 2.99 (d, 1H, J = 8.2), 2.51 (brd, 1H, J = 3.2), 2.32 (m, 2H), 2.16 (ddd,
1H, J = 15.3, 8.7, 6.0), 1.57 (m, 4H), 1.43 (m, 1H), 1.33, (m, 4H), 1.14 (m, 3H), 0.89 (m, 3H).
13C (CDCl3): 158.97, 86.04, 59.47, 42.96, 38.29, 32.24, 31.64, 27.38, 26.72, 26.08, 22.82, 22.45, 14.04.
ESI-MS: 208.4 [M + H]+; 230.4 [M + Na]
+.
HRMS: calcd for C13H21NONa [M + Na]+ = 230.1521, found 230.1523.

144
A.16.9 Preparation of 3a,4,5,6,7,7a-hexahydro-3-(2-phenylethyl)-4,7-methano-1,2-benzisoxazole
(2.84)
Representative procedure is located in Appendix section A.16.
colorless oil.
79% yield.
1H (CDCl3): 7.33-7.18 (m, 5H), 4.41 (d, 1H, J = 8.2), 2.93 (m, 3H), 2.65 (ddd, 1H, J = 15.8, 8.7, 6.5),
2.48 (m, 2H), 2.30 (brs, 1H), 1.51 (m, 2H), 1.40 (m, 1H), 1.12 (m, 3H).
13C (CDCl3): 158.34, 141.07, 128.63, 128.40, 126.36, 86.26, 59.68, 42.96, 38.32, 32.61, 32.27, 28.70,
27.36, 22.82.
ESI-MS: 264.3 [M + Na]+.
HRMS: calcd for C16H19NONa [M + Na]+ = 264.1364, found 264.1369.

145
A.16.10 Preparation of 3-(1,1-dimethylethyl)-3a,4,5,6,7,7a-hexahydro4,7-methano-1,2-
benzisoxazole (2.85)
Representative procedure is located in Appendix section A.16.
colorless oil.
75% yield.
1H (CDCl3): 4.40 (d, 1H, J = 8.3), 3.05 (dd, 1H, J = 8.3, 1.3), 2.53 (brs, 2H), 1.50 (m, 3H), 1.23 (s, 9H),
1.15 (m, 3H).
13C (CDCl3): 165.27, 87.07, 58.35, 42.79, 39.81, 33.30, 32.18, 29.41, 27.67, 22.85.
ESI-MS: 194.3 [M + H]+; 216.3 [M + Na]
+.
HRMS: calcd for C12H19NONa [M + Na]+ = 216.1364, found 216.1366.

146
A.16.11 Preparation of 3a,4,5,6,7,7a-hexahydro-3-(4-methoxyphenyl)-4,7-methano-1,2-
benzisoxazole (2.86)
ON
Chemical Formula: C15H17NO2
Exact Mass: 243.13
Molecular Weight: 243.3
2.86
H3CO
Representative procedure is located in Appendix section A.16.
colorless crystals.
90% yield.
1H (CDCl3): 7.65 (d, 2H, J = 8.9), 6.91 (d, 2H, J = 8.9), 4.60 (d, 1H, J = 8.3), 3.83 (s, 3H), 3.46 (d, 1H, J
= 8.3), 2.55 (dd, 2H, J = 30.2, 2.9), 1.57 (m, 3H), 1.35 (m, 1H), 1.17 (m, 2H).
13C (CDCl3): 160.86, 156.56, 128.46, 122.07, 114.22, 87.68, 57.49, 55.47, 43.11, 39.41, 32.43, 27.56,
22.83.
MP: 95-96 °C.
ESI-MS: 244.3 [M + H]+; 266.3 [M + Na]
+.
HRMS: calcd for C15H17NO2Na [M + Na]+ = 266.1157, found 266.1152.

147
A.16.12 Preparation of 5-(3-bromopropyl)-4,5-dihydro-3-phenyl-isoxazole (2.87)
Representative procedure is located in Appendix section A.16.
colorless crystals.
83% yield.
1H (CDCl3): 7.65 (m, 2H), 7.40 (m, 3H), 4.76 (m, 1H), 3.50 (m, 2H), 3.43 (dd, 1H, J = 16.4, 10.4), 3.00
(dd, 1H, J = 16.4, 7.7), 2.06 (m, 2H), 1.84 (m, 2H).
13C (CDCl3): 156.50, 130.11, 129.64, 128.76, 126.66, 80.39, 40.16, 33.98, 33.52, 28.80.
MP: 53-54 °C.
ESI-MS: 268.2 and 270.2 [M + H]+.
HRMS: calcd for C12H14NO79
BrNa [M + Na]+ = 290.0156, found 290.0163.

148
A.16.13 Preparation of 3,5-diphenylisoxazole (2.88)
Representative procedure is located in Appendix section A.16.
colorless crystals.
50% yield.
1H (CDCl3): 7.87 (m, 2H), 7.47 (m, 8H), 6.84, (s, 1H).
13C (CDCl3): 170.41, 162.98, 130.22, 130.01, 129.14, 129.01, 128.93, 127.47, 126.82, 125.84, 97.47.
MP: 138-140 °C.
ESI-MS: 222.3 [M + H]+; 244.3 [M + Na]
+.
HRMS: calcd for C15H11NONa [M + Na]+ = 244.0738, found 244.0733.

149
A.17 Preparation of 1-(3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-benzisoxazol-3-yl)-ethanone
(2.90)
A solution of oxime 2.89 (120 mg, 1.4 mmol) in methanol (3 mL) was added dropwise at room
temperature to a solution of norbornylene (158 mg, 1.68 mmol, 1.2 eq), PhI(OAc)2 (541 mg, 1.68 mmol,
1.2 eq), TFA (0.1% v/v, 5 L) and methanol (2 mL) with rapid stirring. The reaction mixture was stirred
overnight. The crude product 2.90 was purified by flash column chromatography using a step-gradient:
5%, 10%, 15%, and 20% ethyl acetate/hexanes. Pure fractions of 2.90 were combined and concentrated
in vacuo, yielding compound 2.90 as clear viscous oil (0.188 g, 76%).
1H (CDCl3): 4.65 (d, 1H, J = 8.4), 3.25 (d, 1H, J = 8.4), 2.61 (br s, 1H), 2.51 (br s, 1H), 2.45 (s, 3H),
1.62-1.46 (m, 2H), 1.38-1.02 (m, 4H).
13C (CDCl3): 193.49, 158.98, 91.01, 54.47, 43.16, 39.20, 32.41, 27.38, 27.16, 22.73.
IR: 1685 cm-1
, 1567 cm-1
.
ESI-MS: 202.3 [M + Na]+.
HRMS: calcd for C10H13NO2Na = 202.0844, found 202.0841.
Literature characterization is also available in Cecchi, L.; De Sarlo, F.; Machetti, F. Eur. J. Org. Chem.
2006, 21, 4852-4860.
Alternative method of synthesis is available in Cecchi, L.; De Sarlo, F.; Machetti, F. Tetrahedron Lett.
2005, 46, 7877-7879.

150
A.18 Preparation of 1-(4,5-dihydro-5-phenyl-3-isoxazolyl)-ethanone (2.91)
A solution of oxime 2.89 (138 mg, 1.6 mmol) in methanol (3 mL) was added dropwise at room
temperature to a solution of styrene (200 mg, 1.92 mmol, 1.2 eq), PhI(OAc)2 (618 mg, 1.92 mmol, 1.2
eq), TFA (0.1% v/v, 5 L) and methanol (2 mL) with rapid stirring. The reaction mixture was stirred
overnight and then concentrated in vacuo. The pale-yellow crude product 2.91 was purified by flash
column chromatography using a step-gradient: 5%, 10%, 15%, and 20% ethyl acetate/hexane. Pure
fractions of 2.91 were combined and concentrated in vacuo, yielding 2.91 as clear viscous oil (0.224 g,
75%).
1H (CDCl3): 7.43-7.27 (m, 5H), 5.77 (dd, 1H, J2 = 11.6, J1 = 8.9), 3.55 (dd, 1H, J2 = 17.9, J1 = 11.6),
3.14 (dd, 1H, J2 = 17.9, J1 = 8.9), 2.55 (s, 3H).
13C (CDCl3): 193.18, 158.02, 139.68, 129.04, 128.83, 126.01, 85.69, 39.89, 26.90.
IR: 1686 cm-1
, 1577 cm-1
.
ESI-MS: 190.5 [M + H]+; 212.3 [M + Na]
+.
HRMS: calcd for C11H11NO2Na = 212.0687, found 212.0686.
Literature characterization is also available in Cecchi, L.; De Sarlo, F.; Machetti, F. Eur. J. Org. Chem.
2006, 21, 4852-4860.
Alternative method of synthesis is available in Cecchi, L.; De Sarlo, F.; Machetti, F. Tetrahedron Lett.
2005, 46, 7877-7879.

151
A.19 Preparation of 3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-benzisoxazole-3-carboxylic acid
ethyl ester (2.93)
Procedure A
A solution of 2.99 (263 mg, 1.65 mmol) in methanol (3 mL) was added dropwise at room temperature to
a solution of norbornylene (186 mg, 1.98 mmol, 1.2 eq), PhI(OAc)2 (638 mg, 1.98 mmol, 1.2 eq), TFA
(1% v/v, 55 L) and methanol (2 mL) with rapid stirring. The reaction mixture was stirred overnight and
then concentrated in vacuo. The crude product was purified by flash column chromatography using a
step-gradient: 5%, 10%, 15%, and 20% ethyl acetate/hexane. Pure fractions of product 2.93 were
combined and concentrated in vacuo, yielding 2.93 as clear viscous oil (0.178 g, 52%). Oxime 2.99 (76
mg, 29%, 52% brsm) was also recovered from the purification of the crude 2.93 indicating that the
reaction required longer reaction time.

152
Procedure B
A solution of 2.92 (158 mg, 1.04 mmol) in methanol (2 mL) was added dropwise at room temperature to
a solution of norbornylene (117 mg, 1.25 mmol, 1.2 eq), PhI(OAc)2 (402 mg, 1.25 mmol, 1.2 eq), TFA
(1% v/v, 70 L) and methanol (5 mL) with rapid stirring. The reaction appeared complete in 30 min by
TLC. The crude product 2.93 was purified by flash column chromatography using a step-gradient: 5%,
10%, 15%, and 20% ethyl acetate/hexane. Pure fractions of 2.93 were collected and concentrated in
vacuo, yielding 2.93 as clear viscous oil (0.194 g, 90%).
1H (CDCl3): 4.65 (d, 1H, J = 8.5), 4.39-4.22 (m, 2H, J = 3.6), 3.27 (d, 1H, J = 8.5), 2.57 (br d, 2H, J =
10.0), 1.62-1.46 (m, 2H), 1.44-1.37 (m, 1H), 1.34 (t, 3H, J = 7.1), 1.29-1.16 (m, 2H), 1.16-1.06 (m, 1H).
13C (CDCl3): 160.91, 152.35, 90.31, 61.86, 55.74, 42.98, 39.43, 32.34, 27.23, 22.70, 14.18.
IR: 1715 cm-1
, 1580 cm-1
.
ESI-MS: 232.3 [M + Na]+.
HRMS: calcd for C11H15NO3Na = 232.0950, found 232.0948.
Literature characterization is also available in Cecchi, L.; De Sarlo, F.; Machetti, F. Eur. J. Org. Chem.
2006, 21, 4852-4860.
Alternative method of synthesis is available in Cecchi, L.; De Sarlo, F.; Machetti, F. Tetrahedron Lett.
2005, 46, 7877-7879.

153
A.20 Preparation of 4,5-dihydro-5-phenyl-3-isoxazolecarboxylic acid ethyl ester (2.94)
Procedure A
A solution of 2.99 (251 mg, 1.58 mmol) in methanol (3 mL) was added dropwise at room temperature to
a solution of styrene (197 mg, 1.90 mmol, 1.2 eq), PhI(OAc)2 (611 mg, 1.90 mmol, 1.2 eq), TFA (1% v/v,
55 L) and methanol (2 mL) with rapid stirring. The reaction mixture was stirred overnight and then
concentrated in vacuo. The crude product was purified by flash column chromatography using a step-
gradient: 5%, 10%, 15%, and 20% ethyl acetate/hexane. Pure fractions of product 2.94 were combined
and concentrated in vacuo, yielding 2.94 as clear viscous oil (0.205 g, 59%). Oxime 2.99 (98 mg, 39 %)
was also recovered from the purification of the crude 2.94 indicating that the reaction required longer
reaction time.

154
Procedure B
A solution of 2.92 (152 mg, 1.02 mmol) in methanol (2 mL) was added dropwise at room temperature to
a solution of styrene (115 mg, 1.22 mmol, 1.2 eq), PhI(OAc)2 (402 mg, 1.22 mmol, 1.2 eq), TFA (1% v/v,
70 L) and methanol (5 mL) with rapid stirring. The reaction appeared complete in 30 min by TLC. The
crude product 2.94 was purified by flash column chromatography using a step-gradient: 5%, 10%, 15%,
and 20% ethyl acetate/hexane. Pure fractions of 2.94 were collected and concentrated in vacuo, yielding
2.94 as clear viscous oil (0.183 g, 82%).
1H (CDCl3): 7.43-7.28 (m, 5H), 5.77 (dd, 1H, J2 = 11.7, J1 = 8.9), 4.36 (q, 2H, J = 7.1), 3.63 (dd, 1H, J2
= 17.9, J1 = 11.7), 3.21 (dd, 1H, J2 = 17.9, J1 = 8.9), 1.37 (t, 3H, J = 7.1).
13C (CDCl3): 160.67, 151.23, 139.63, 128.97, 128.74, 125.96, 85.04, 62.23, 41.55, 14.21.
IR: 1716 cm-1
, 1589 cm-1
.
ESI-MS: 220.3 [M + H]+; 242.3 [M + Na]
+.
HRMS: calcd for C12H13NO3Na = 242.0793, found 242.0797.
Literature characterization is also available in Cecchi, L.; De Sarlo, F.; Machetti, F. Eur. J. Org. Chem.
2006, 21, 4852-4860.
Alternative method of synthesis is available in Cecchi, L.; De Sarlo, F.; Machetti, F. Tetrahedron Lett.
2005, 46, 7877-7879.

155
A.21 Preparation of 2-isonitrosocyclopentanone (2.100)
Oxime 2.100 was synthesized from 3.12 g of α-carbethoxycyclopentanone as detailed by:
Cope, A. C.; Estes, L. L. Jr.; Emery, J. R.; Haven, A. C. Jr. J. Amer. Chem. Soc. 1951, 73, 1199.
1H (CDCl3): 9.92 (br s, 1H), 2.83 (t, 2H, J = 7.6), 2.49 (t, 2H, J = 7.8), 2.07 (quin, 2H, J = 7.7).
13C (CDCl3): 203.91, 156.32, 38.47, 25.33, 17.57.
IR: 1713 cm-1
, 1634 cm-1
.
ESI-MS: 136.2 [M + Na]+.
HRMS: calcd for C5H7NO2Na = 136.0374, found 136.0379.

156
A.22 Preparation of compound 2.101
A solution of 2.100 (50 mg, 0.44 mmol) in methanol (3.5 mL) was added dropwise at room temperature
to a solution of norbornylene (50 mg, 0.53 mmol, 1.2 eq), PhI(OAc)2 (170 mg, 0.53 mmol, 1.2 eq), TFA
(1% v/v, 55 L) and methanol (2 mL) with rapid stirring. The reaction appeared complete in 30 min by
TLC. The crude product 2.101 was purified by flash column chromatography using a step-gradient: 5%,
10%, 15%, and 20% ethyl acetate/hexane. Pure fractions of 2.101 were combined and concentrated in
vacuo, yielding compound 2.101 as clear viscous oil (0.074 g, 70%).
1H (CDCl3): 4.34 (d, 1H, J = 8.2), 3.61 (s, 3H), 2.95 (d, 1H, J = 8.2), 2.44 (br s, 1H), 2.40-2.12 (m, 5H),
2.00-1.76 (m, 2H), 1.56-1.29 (m, 3H), 1.22-0.96 (m, 3H).
13C (CDCl3): 173.50, 157.88, 86.09, 59.33, 51.54, 42.84, 38.18, 33.25, 32.12, 27.21, 26.01, 22.67, 21.40.
IR: 2953 cm-1
, 1732 cm-1
.
ESI-MS: 260.4 [M + Na]+.
HRMS: calcd for C13H19NO3Na = 260.1263, found 260.1265.

157
A.23 Preparation of methyl 4-(5-phenyl-4,5-dihydroisoxazol-3-yl)butanoate (2.102)
A solution of 2.100 (38 mg, 0.34 mmol) in methanol (3.5 mL) was added dropwise at room temperature
to a solution of styrene (43 mg, 0.41 mmol, 1.2 eq), PhI(OAc)2 (131 mg, 0.41 mmol, 1.2 eq), TFA (1%
v/v, 55 L) and methanol (2 mL) with rapid stirring. The reaction was stirred overnight. The crude
product 2.102 was purified by flash column chromatography using a step-gradient: 5%, 10%, 15%, and
20% ethyl acetate/hexane. Pure fractions were combined and concentrated in vacuo, yielding 2.102 as
clear viscous oil (0.034 g, 40%).
1H (CDCl3): 7.47-7.28 (m, 5H), 5.55 (dd, 1H, J2 = 11.4, J1 = 8.2), 3.68 (s, 3H), 3.37 (dd, 1H, J2 = 17.0,
J1 = 10.9), 2.91 (dd, 1H, J2 = 17.0, J1 = 8.2), 2.50-2.36 (m, 4H), 1.94 (quin, 2H, J = 7.3).
13C (CDCl3): 173.50, 157.65, 141.24, 128.77, 128.12, 125.79, 81.46, 51.72, 45.42, 33.26, 27.22, 21.56.
IR: 2951 cm-1
, 1731 cm-1
.
ESI-MS: 270.4 [M + Na]+.
HRMS: calcd for C14H17NO3Na = 270.1106, found 270.1111.
Compound 2.102 was known previously in the literature: Ignatovich, Zh. V.; Chernikhova, T. V.;
Skupskaya, R. V.; Bondar', N. F.; Koroleva, E. V.; Lakhvich, F. A. Chem. Heterocycl. Compd. 1999,
35(2), 248-249.

158
A.24 Preparation of 2-isonitrosocyclohexanone (2.103)
A solution of 1,2-cyclohexanedione (0.500 g, 4.459 mmol) in diethyl ether (5 mL) was combined with a
solution of NaOH (0.178 g, 1.0 eq, 4.459 mmol) and HCl.H2NOH (0.310 g, 1.0 eq, 4.459 mmol) in 5 mL
of water and was stirred vigorously overnight. The biphasic reaction mixture was -extracted with diethyl
ether (5 times) and dried over anhydrous MgSO4. The crude product 2.103 was filtered and conventrated
and purified by flash column chromatography using a step-gradient: 5%, 10%, 15%, 20%, and 30% ethyl
acetate/hexane. Pure fractions of 2.103 were combined and concentrated in vacuo, yielding oxime 2.103
(0.139 g, 25%) as viscous oil.
1H (CDCl3): 9.56 (s, 1H), 2.79 (t, 2H, J = 6.5), 2.58 (t, 2H, J = 6.5), 1.97-1.74 (m, 4H).
13C (CDCl3): 196.61, 154.19, 41.03, 25.19, 22.66, 21.78.
IR: 1701 cm-1
.
ESI-MS: 150.2 [M + Na]+.
HRMS: calcd for C6H9NO2Na = 150.0531, found 150.0533.
A similar preparation is known:
Wu, S.; Fluxe, A.; Janusz, J. M.; Sheffer, J. B.; Browning, G.; Blass, B.; Cobum, K.; Hedges, R.;
Murawsky, M.; Fang, B.; Fadayel, G. M.; Hare, M.; Djandjighian, L. Bioorg. Med. Chem. Lett. 2006, 16,
5859-5863.

159
A.25 Preparation of compound 2.104
A solution of oxime 2.103 (69 mg, 0.54 mmol) in methanol (1.5 mL) was added dropwise at room
temperature to a solution of norbornylene (61 mg, 0.65 mmol, 1.2 eq), PhI(OAc)2 (209 mg, 0.65 mmol,
1.2 eq), TFA (1% v/v, 25 L) and methanol (1 mL) with stirring. The reaction mixture was stirred
overnight. The crude product 2.104 was purified by flash column chromatography using a step-gradient:
5%, 10%, 15%, and 20% ethyl acetate/hexane. Pure fractions of 2.104 were combined and concentrated
in vacuo, yielding 2.104 as clear viscous oil (0.076 g, 56%).
1H (CDCl3): 4.35 (d, 1H, J = 8.2), 3.61 (s, 3H), 2.94 (d, 1H, J = 8.2), 2.46 (s, 1H), 2.37-2.07 (c m, 5H),
1.73-1.30 (c m, 7H), 1.22-0.97 (c m, 3H).
13C (CDCl3): 173.78, 158.31, 86.06, 59.33, 51.50, 42.86, 38.21, 33.63, 32.15, 27.26, 26.38, 25.67, 24.56,
22.70.
IR: 2952 cm-1
, 1733 cm-1
.
ESI-MS: 252.4 [M + H]+; 274.4 [M + Na]
+.
HRMS: calcd for C14H22NO3 = 252.1600, found 252.1595.
Compound 2.104 was previously known in the literature:
Bondar, N. F.; Isaenya, L. P.; Skupskaya, R. V.; Lakhvich, F. A. Russ. J. Org. Chem. 2003, 39(8), 1089-
1094.

160
A.26 Preparation of methyl 5-(5-phenyl-4,5-dihydroisoxazol-3-yl)pentanoate (2.105)
A solution of 2.103 (75 mg, 0.59 mmol) in methanol (1.5 mL) was added dropwise at room temperature
to a solution of styrene (74 mg, 0.71 mmol, 1.2 eq), PhI(OAc)2 (228 mg, 0.71 mmol, 1.2 eq), TFA (0.1%
v/v, 3 L) and methanol (1 mL) with rapid stirring. The reaction mixture was stirred overnight. The
crude product 2.105 was purified by flash column chromatography using a step-gradient: 5%, 10%, 15%,
and 20% ethyl acetate/hexane. Pure fractions of 2.105 were collected and concentrated in vacuo, yielding
2.105 as clear viscous oil (0.036 g, 23%).
1H (CDCl3): 7.41-7.27 (m, 5H), 5.55 (dd, 1H, J2 = 10.7, J1 = 8.2), 3.67 (s, 3H), 3.36 (dd, 1H, J2 = 16.8,
J1 = 10.7), 2.90 (dd, 1H, J2 = 16.8, J1 = 8.2), 2.46-2.30 (m, 4H), 1.80-1.59 (m, 4H).
13C (CDCl3): 173.88, 158.11, 141.41, 128.83, 128.16, 125.86, 81.46, 51.69, 45.47, 33.70, 27.59, 25.89,
24.56.
IR: 2950 cm-1
, 1732 cm-1
.
ESI-MS: 262.5 [M + H]+; 284.4 [M + Na]
+.
HRMS: calcd for C15H19NO3Na = 284.1263, found 284.1267.

161
A.27 Preparation of 3-(hydroxyimino)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one (2.106)
Compound 2.106 was synthesized as detailed by:
Chen, Y. K.; Jeon, S.-J.; Walsh, P. J.; Nugent, W. A. Org. Synth. 2005, 82, 87.

162
A.28 Preparation of compounds 2.107/2.108
A solution of 2.106 (500 mg, 2.76 mmol) in methanol (12.5 mL) was added dropwise at room
temperature to a solution of norbornylene (312 mg, 3.31 mmol, 1.2 eq), PhI(OAc)2 (1067 mg, 3.31 mmol,
1.2 eq), TFA (1% v/v, 225 L) and methanol (10 mL) with rapid stirring. The reaction apeared complete
in 30 min by TLC. The crude products 2.107/2.108 were purified by flash column chromatography using
a step-gradient: 5%, 10%, and 15% ethyl acetate/hexane. Pure fractions were combined and concentrated
in vacuo, yielding an inseparatable mixture of 2.107/2.108 (0.609 g, 72%) as clear viscous oil. The
diastereoselectivity of the product mixture was determined from 1H-NMR integration to be 1 : 1.
Diastereomer 1:
1H (CDCl3): 4.42 (d, 1H, J = 8.3), 3.67 (s, 3H), 3.05 (d, 1H, J = 8.3), 2.86-2.17 (m, 5H), 1.92-1.74 (m,
1H), 1.61-1.36 (m, 4H), 1.29-1.03 (m, 9H), 0.77 (s, 3H).
13C (CDCl3): 176.50, 158.91, 86.28, 60.27, 56.46, 51.66, 46.45, 46.16, 43.16, 38.44, 32.28, 27.46, 24.39,
23.48, 22.88, 22.38, 21.15.
Diastereomer 2:
1H (CDCl3): 4.36 (d, 1H, J = 8.3), 3.69 (s, 3H), 2.94 (d, 1H, J = 8.3), 2.86-2.17 (m, 5H), 2.07-1.92 (m,
1H), 1.61-1.36 (m, 4H), 1.29-1.03 (m, 9H), 0.86 (s, 3H).
13C (CDCl3): 176.68, 158.82, 85.64, 77.36, 60.78, 55.95, 47.20, 45.67, 42.75, 39.43, 32.44, 27.66, 25.81,
22.88, 22.14, 20.48.
IR: 2960 cm-1
, 1723 cm-1
.
ESI-MS: 306.4 [M + H]+; 328.4 [M + Na]
+.
HRMS: calcd for C18H28NO3 = 306.2069, found 306.2062.

163
A.29 Preparation of (1S,3R)-methyl 1,2,2-trimethyl-3-(5-phenyl-4,5-dihydroisoxazol-3-yl)
cyclopentanecarboxylate (2.109/2.110)
A solution of oxime 2.106 (500 mg, 2.76 mmol) in methanol (12.5 mL) was added dropwise at room
temperature to a solution of styrene (345 mg, 3.31 mmol, 1.2 eq), PhI(OAc)2 (1067 mg, 3.31 mmol, 1.2
eq), TFA (1% v/v, 225 L) and methanol (10 mL) with rapid stirring. The reaction appeared complete in
30 min by TLC. The crude products 2.109/2.110 were purified by flash column chromatography using a
step-gradient: 5%, 10%, and 15% ethyl acetate/hexane. Pure fractions were collected and concentrated in
vacuo, yielding a mixture of 2.109/2.110 (0.206 g, 24%) as clear viscous oil. The diastereoselectivity of
the product mixture was determined from 1H-NMR integration to be 1 : 0.8.
Diastereomer 1:
1H (CDCl3): 7.41-7.27 (m, 5H), 5.62-5.47 (m, 1H), 3.66 (s, 3H), 3.44 (dd, 1H, J2 = 16.5, J1 = 10.6), 3.04-
2.80 (m, 2H), 2.73-2.54 (m, 1H), 2.16-1.82 (m, 2H), 1.62-1.46 (m, 1H), 1.28-1.13 (m, 6H), 0.68 (s, 3H).
Diastereomer 2:
1H (CDCl3): 7.41-7.27 (m, 5H), 5.62-5.47 (m, 1H), 3.68 (s, 3H), 3.29 (dd, 1H, J2 = 16.5, J1 = 10.6), 3.04-
2.80 (m, 2H), 2.73-2.54 (m, 1H), 2.16-1.82 (m, 2H), 1.62-1.46 (m, 1H), 1.28-1.13 (m, 6H), 0.79 (s, 3H).
Diastereomers 1 and 2:
13C (CDCl3): 176.45, 176.43, 159.08, 158.75, 141.46, 141.08, 128.83, 128.80, 128.20, 128.08, 125.91,
125.60, 81.58, 81.16, 77.37, 56.15, 56.11, 51.72, 47.17, 47.04, 46.94, 46.76, 46.46, 46.26, 32.58, 24.16,
24.09, 22.85, 22.81, 22.32, 21.21, 21.14.
IR: 2964 cm-1
, 1722 cm-1
.
ESI-MS: 316.4 [M + H]+; 338.4 [M + Na]
+.
HRMS: calcd for C18H28NO3 = 316.1913, found 316.1909.

164
A.30 Preparation of 3,7-dimethyl-6-octenoxime (2.111)
The first report of the preparation of 2.111 appeared to be:
Semmler, Ber., 1893, 26, 2255.
Oxime 2.111 was synthesized according to the procedure outlined in:
Caldwell, A. G.; Jones, E. R. H. J. Chem. Soc. 1946, 599-601.

165
A.31 Preparation of (6S)-3,3a,4,5,6,7-hexahydro-3,3,6-trimethyl-2,1-benzisoxazole (2.112)
A solution of the oxime 2.111 (99 mg, 0.44 mmol) in CH3OH (2 mL) was slowly added to a solution of
DIB (461 mg, 1.43 mmol) and TFA (15 L) in CH3OH (3 mL) over 15 min. The mixture was stirred at
room temperature for 45 min, and then it was concentrated in vacuo. A 1H-NMR spectrum of the crude
product thus obtained revealed the presence of two products in a ratio of 3.8:1. These products proved to
be quite volatile, especially under reduced pressure. This complicated the calculation of a reaction yield.
Accordingly, a crude reaction mixture obtained as detailed above was treated with a known amount (1
mmol) of 1,3,5-trimethoxy benzene as an internal standard, and a 1H-NMR spectrum of the mixture was
recorded using a delay of 20 seconds between pulses. Integration of the signals of the internal standard
and of the products indicated a chemical yield of 85% for the 3.8:1 mixture of products. Flash column
chromatography (33% diethyl ether/pentane) afforded a pure sample of the major diastereomer (30 mg,
18 mmol, 41%) for characterization.
1H (CDCl3): major: 2.72 (ddd, 1H, J = 13.8, 4.1, 1.4), 2.60 (dd, 1H, J = 12.0, 5.4), 1.87-1.68 (m, 2H),
1.55 (m, 1H), 1.4-1.3 (m, 2H), 1.38 (s, 3H), 1.22 (s, 3H), 1.11 (m, 1H), 1.02 (d, 3H, J = 6.5).
13C (CDCl3): major: 160.63, 84.24, 56.22, 33.91, 33.35, 33.31, 28.47, 26.61, 22.25, 22.07.
ESI-MS: 168.3 [M + H]+.
HRMS: calcd for C10H19NO [M + H]+ = 168.1388, found 168.1384.
[]D20
= –87.0° (0.01 g/mL, CH2Cl2).

166
A.32 Preparation of 4-hydroxy-benzenepropanal oxime (2.123)
Known compound 2.123 (Kusama, H.; Yamashita, Y.; Uchiyama, K.; Narasaka, K.; Bull. Chem. Soc.
Jpn., 70, 1997, 965-975) was synthesized from commercially available 3-(4-hydroxyphenyl)propanal
(2.122) according to Oresmaa, L.; Kotikoski, H.; Haukka, M.; Salminen, J.; Oksala, O.; Pohjala, E.;
Moilanen, E.; Vapaatalo, H.; Vainiotalo, P.; Aulaskari, P., J. Med. Chem. 2005, 48, 4231-4236.
ESI-MS: 166.1 [M + H]+; 188.4 [M + Na]
+.
HRMS: calcd for C9H10NO2 [M – H]– = 164.0712, found 164.0708.

167
A.33 Preparation of N-[(4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-
yl]-acetamide (2.124)
A solution of 2.123 (62 mg, 0.375 mmol) in acetonitrile (5 mL) was slowly added to a solution of DIB
(226 mg, 0.825 mmol) and TFA (15 L) in acetonitrile (20 mL). This reaction mixture was stirred at
room temperature for 1 h, and then the volatile organics were removed in vacuo. The crude product 2.124
was purified by flash column chromatography using a step gradient: 25%-50%-100% ethyl
acetate/hexanes, to afford 58 mg of compound 2.124 as colorless crystals (0.266 mmol, 71%).
1H (d6-acetone): 7.84 (brs, 1H), 6.41 (dd, 1H, J = 10.3, 2.0), 6.15 (dd, 1H, J = 10.3, 0.5), 4.72 (dd, 1H, J
= 9.7, 0.5), 4.28 (ddd, 1H, J = 9.7, 2.0, 1.6), 2.69 (m, 1H), 2.64 (m, 2H), 2.37 (m, 1H), 1.84 (s, 1H).
13C (d6-acetone): 191.12, 170.45, 169.26, 146.53, 132.66, 79.80, 63.57, 53.49, 42.18, 42.12, 23.07,
19.34.
MP: 162.163 °C.
ESI-MS: 243.3 [M + Na]+.
HRMS: calcd for C11H12N2O3Na [M + Na]+ = 243.0746, found 243.0752.

168
A.34 Preparation of 4,4a,7a,7b-tetrahydro-4a-methoxy-indeno[1,7-cd]isoxazol-7(3H)-one (2.125)
A solution of 2.123 (38 mg, 0.23 mmol) in methanol (1.3 mL) was slowly added to a solution of DIB
(162 mg, 0.50 mmol) and TFA (15 L) in methanol (2 mL). This reaction mixture was stirred at room
temperature for 1 h, and then the volatile organics were removed in vacuo. The crude product was
purified by flash column chromatography (step gradient 50%-100% ethyl acetate/hexanes) to afford 29
mg of compound 2.125 (0.15 mmol, 51%).
1H (d6-acetone): 6.66 (dd, 1H, J = 10.5, 1.9), 6.36 (dd, 1H, J = 10.5, 0.4), 4.84 (d, 1H, J = 10.1), 4.33
(dd, 1H, J = 10.1, 1.8), 3.20 (s, 3H), 2.69 (m, 2H), 2.49 (m, 2H).
13C (d6-acetone): 192.28, 169.20, 148.93, 134.26, 77.77, 77.23, 60.57, 53.17, 43.32, 20.46.
ESI-MS: 216.3 [M + Na]+.
HRMS: calcd for C10H12NO3 [M + H]+ = 216.0637, found 216.0635.

169
A.35 Preparation of N-benzyl, N-tosyl tyrosine (2.130)
To a solution of SOCl2 (10 mL) in methanol (100 mL) was added tyrosine (10.0 g, 45.9 mmol). This
solution was allowed to stir at room temperature over night. Volatile organics were removed in vacuo by
rotary evaporation. This crude residue was suspended in methanol (10 mL), and added dropwise to a
stirring solution of diethyl ether (300 mL). The white precipitate was collected by filtration and the
supernatant discarded. The methyl ester intermediate was dissolved in acetonitrile (100 mL) and
triethylamine (6.40 mL, 45.9 mmol). Benzaldehyde (5.13 mL, 50.5 mmol) was added via syringe, and
the reaction was allowed to stir at room temperature. The reaction finished (by TLC) after 2 h, and was
concentrated in vacuo. This crude residue was taken up in methanol (200 mL) and cooled in a 0 °C ice
bath for 15 min. To this was added solid NaBH4 (2.08 g, 55.1 mmol), and the reaction mixture was
allowed to warm to room temperature overnight. Methanol and volatile organics were removed in vacuo,
and the crude residue was suspended in ethyl acetate (250 mL) and extracted successively with saturated
aqueous NaHCO3, and saturated aqueous NaCl, dried over anhydrous MgSO4, filtered and concentrated in
vacuo. Without further purification, the residue was suspended in pyridine (25 mL) followed by the
addition of solid tosyl chloride (21.9 g, 115 mmol), and this reaction mixture was stirred over night at
room temperature. Pyridine was removed in vacuo, and the crude residue suspended in ethyl acetate (200
mL) and washed with 0.1 M aqueous HCl (200 mL) and H2O (200 mL). The organic phase was dried
(MgSO4), filtered through a short silica gel plug (10 g) and concentrated in vacuo. This viscous bis-tosyl-
tyrosine ester residue was dissolved in THF (100 mL), and to this was added a solution of NaOH (7.34 g,
183.6 mmol) in H2O (100 mL). (Upon addition of aqueous NaOH, a white cloudy precipitate formed
which slowly disappeared over the course of the reaction.) The reaction was heated to 75 °C and allowed
to stir overnight. The reaction was concentrated in vacuo, and re-suspended in ethyl acetate (250 mL).
The organic solution was washed successively with 1.0 M aqueous HCl (100 mL) and H2O (100 mL),
dried over anhydrous MgSO4, filtered and concentrated in vacuo. Pure 2.130 (12.5 g, 29.4 mmol, 64%)
was obtained as colorless crystals from crystallization in refluxing CH2Cl2.

170
1H (d6-acetone): 8.20 (brs, 1H), 7.73 (d, 2H, J = 8.3), 7.35 (d, 2H, J = 8.3), 7.35-7.23 (m, 5H), 6.90 (d,
2H, J = 8.5), 6.69 (d, 2H, J = 8.5), 4.65 (d, 1H, J = 16.1), 4.64 (m, 1H), 4.43 (d, 1H, J = 16.1), 3.07 (dd,
1H, J = 13.9, 8.3), 2.72 (dd, 1H, J = 13.9, 6.5), 2.41 (s, 3H).
13C (d6-acetone): 171.43, 156.93, 144.23, 138.66, 138.46, 131.03, 130.29, 129.21, 128.92, 128.85,
128.46, 128.09, 115.92, 62.36, 50.20, 36.99, 21.40.
MP: 154-156 °C.
ESI-MS: 426.4 [M + H]+; 448.1 [M + Na]
+; 424.3 [M – H]
-.
HRMS: calcd for C23H23NO5SNa [M + Na]+ = 448.1195, found 448.1209.

171
A.36 Preparation of N-[2-(hydroxyimino)-1-[(4-hydroxyphenyl)methyl]ethyl]-4-methyl-N-
(phenylmethyl)-benzenesulfonamide (2.132)
Carboxylic acid 2.130 (5.00 g, 11.8 mmol) was dissolved in THF (20 mL) at room temperature. To this
solution was added HN(CH3)OCH3•HCl (1.73 g, 17.7 mmol), triethylamine (4.9 mL, 35.4 mmol) and
PyBOP (9.21 g, 17.7 mmol). The reaction was stirred for 12 hours, and then the solvent was removed in
vacuo. The crude residue was diluted with ethyl acetate (200 mL) and washed successively with 100 mL
portions of 0.1 M aqueous HCl and H2O. The organic solution was dried over anhydrous MgSO4, filtered
through a silica gel plug (10 g) and concentrated in vacuo. The crude Weinreb amide product was
dissolved in THF (100 mL), and this solution was cooled in a 0 °C ice-bath. A suspension of LiAlH4
(0.67 g, 17.7 mmol) in THF (50 mL) was added with vigorous stirring. This heterogeneous reaction
mixture was stirred at 0 °C for 4 h. The reaction was quenched at 0 °C by the addition of saturated
aqueous NaHSO4 (3 mL) and stirred for 0.5 h, followed by the addition of aqueous 1 M aqueous HCl (5
mL) and then diluted with H2O (100 mL) and extracted with ethyl acetate (100 mL). The organic phase
was dried over anhydrous MgSO4, filtered through a silica gel plug (10 g) and concentrated in vacuo. The
crude aldehyde was immediately dissolved in diethyl ether (50 mL), and to this was added sequentially a
solution of H2NOH•HCl (3.44 g, 35.4 mmol) in H2O (50 mL) and a solution of Na2CO3 (6.25 g, 59 mmol)
in H2O (50 mL). This reaction was stirred at room temperature over night and then diluted with diethyl
ether (50 mL) and the organic phase was separated, dried over anhydrous MgSO4, filtered and
concentrated in vacuo. Pure 2.132 (0.55 g, 1.30 mmol, 11%) was obtained following flash column
chromatography with 30% ethyl acetate/hexanes. Note: this reaction sequence was not optimized for
higher recovered yield.

172
1H (d6-acetone): 9.97 (s, 1H), 8.14 (s, 1H), 7.72 (d, 2H, J = 8.3), 7.36 (m, 5H), 7.28 (d, 2H, J = 8.3), 7.15
(d, 2H, J = 6.0), 6.82 (d, 2H, J = 8.7), 6.69 (d, 2H, J = 8.3), 4.61 (ddd, 1H, J1 = 9.1, J2 = J3 = 6.0), 4.46
(ABq, 2H, J = 29.4, 16.0), 3.0 (dd, 1H, J = 13.6, 9.1), 2.87 (s, 3H), 2.71 (dd, 1H, J = 13.6, 6.0), 2.43 (s,
3H).
13C (d6-acetone): 156.82, 148.49, 144.24, 138.98, 138.93, 131.09, 130.51, 129.28, 129.16, 129.12,
128.26 (two overlapping signals), 115.90, 60.41, 49.67, 37.74, 21.42.
ESI-MS: 425.2 [M + H]+, 447.2 [M + Na]
+.
HRMS: calcd for C23H24N2O5SNa [M + Na]+ = 447.1354, found 447.1362.

173
A.37 Preparation of N-[(3R,4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-3-[[(4-methylphenyl)sulfonyl]
(phenylmethyl)amino]-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-acetamide (2.134)
A solution of oxime 2.132 (20 mg, 0.047 mmol) in acetonitrile (3 mL) was slowly added to a solution of
DIB (36 mg, 0.113 mmol) and TFA (15 L) in acetonitrile (3 mL) over 10 min. The mixture was stirred
at room temperature for 1 h, and then it was diluted with heptanes (0.4 mL) and concentrated in vacuo by
rotary evaporation. The reaction residue was immediately purified by flash column chromatography
using a step gradient: 50%-100% ethyl acetate/hexanes, to afford 10 mg of compound 2.134 (0.021 mmol,
44%) as a glassy solid.
1H (d6-acetone): 7.82 (d, 2H, J = 8.5), 7.76 (brs, 1H), 7.43 (d, 2H, J = 8.5), 7.36 (m, 5H), 6.32 (dd, 1H, J
= 10.2, 2.0), 6.08 (dd, 1H, J = 10.2, 0.4), 4.68 (dd, 1H, J = 9.8, 0.4), 4.64 (m, 1H), 4.58 (d, 1H, J = 15.9),
4.32 (d, 1H, J = 15.9), 3.93 (dt, 1H, J = 9.8, 2.0), 2.87 (dd, 1H, J = 13.6, 8.1), 2.63 (dd, 1H, J = 13.6, 9.2),
2.44 (s, 3H), 1.77 (s, 3H).
13C (d6-acetone): 189.86, 170.41, 170.33, 166.65, 145.55, 144.85, 137.80, 137.55, 132.59, 130.71,
129.43, 129.31, 128.69, 128.39, 80.95, 62.09, 51.74, 51.66, 51.53, 49.98, 49.92, 22.95, 22.91, 21.44.
ESI-MS: 502.2 [M + Na]+; 478.3 [M – H]
–.
HRMS: calcd for C25H24N3O5S [M - H]- = 478.1437, found 478.1446.
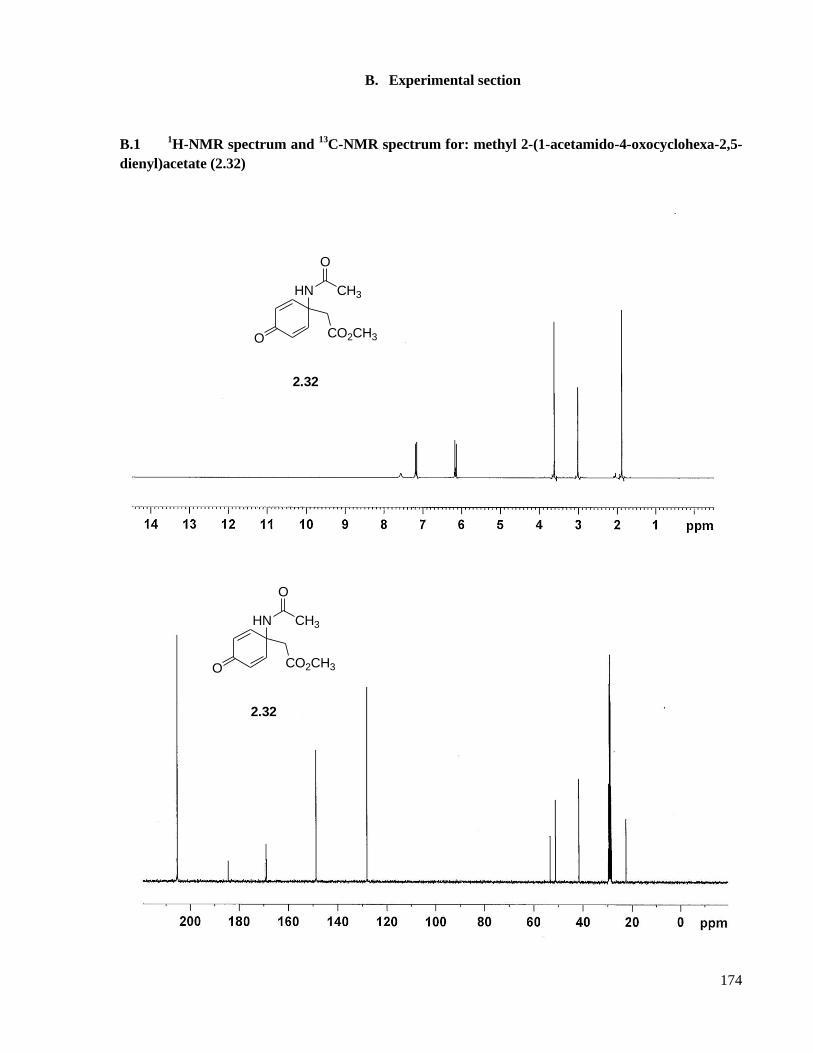
174
B. Experimental section
B.1 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-(1-acetamido-4-oxocyclohexa-2,5-
dienyl)acetate (2.32)
O
HN CH3
O
CO2CH3
2.32
O
HN CH3
O
CO2CH3
2.32
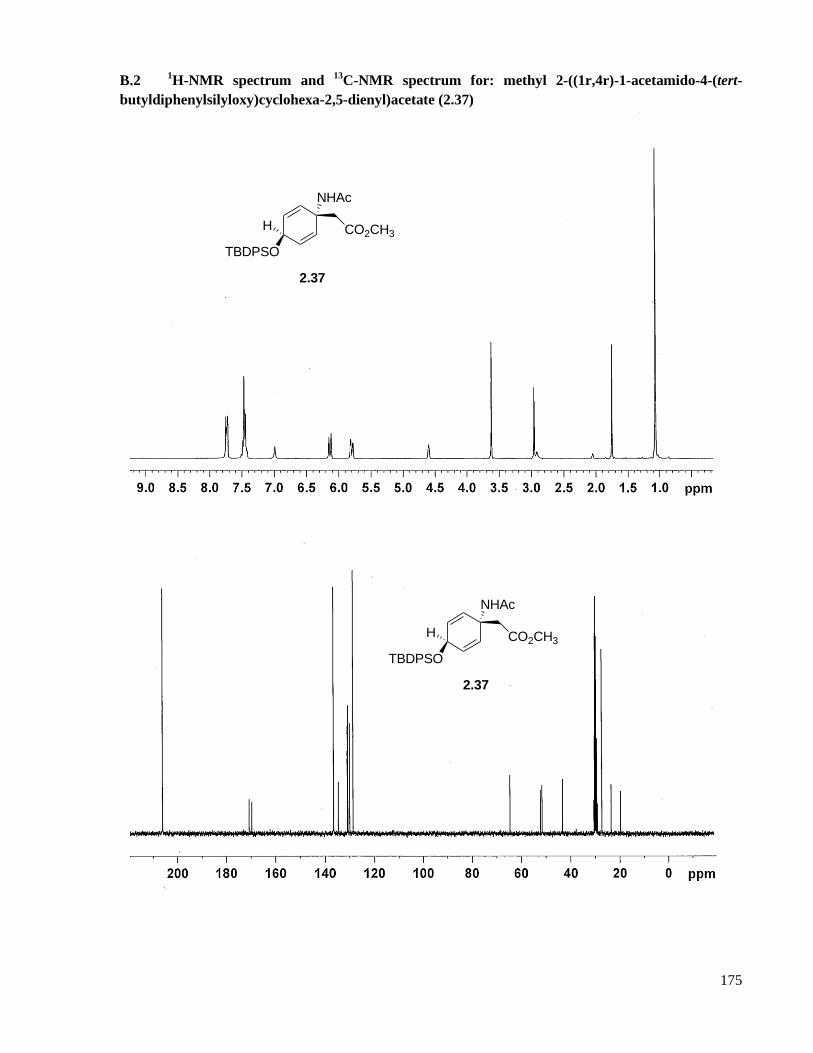
175
B.2 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1r,4r)-1-acetamido-4-(tert-
butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetate (2.37)
TBDPSO
NHAc
CO2CH3H
2.37
TBDPSO
NHAc
CO2CH3H
2.37
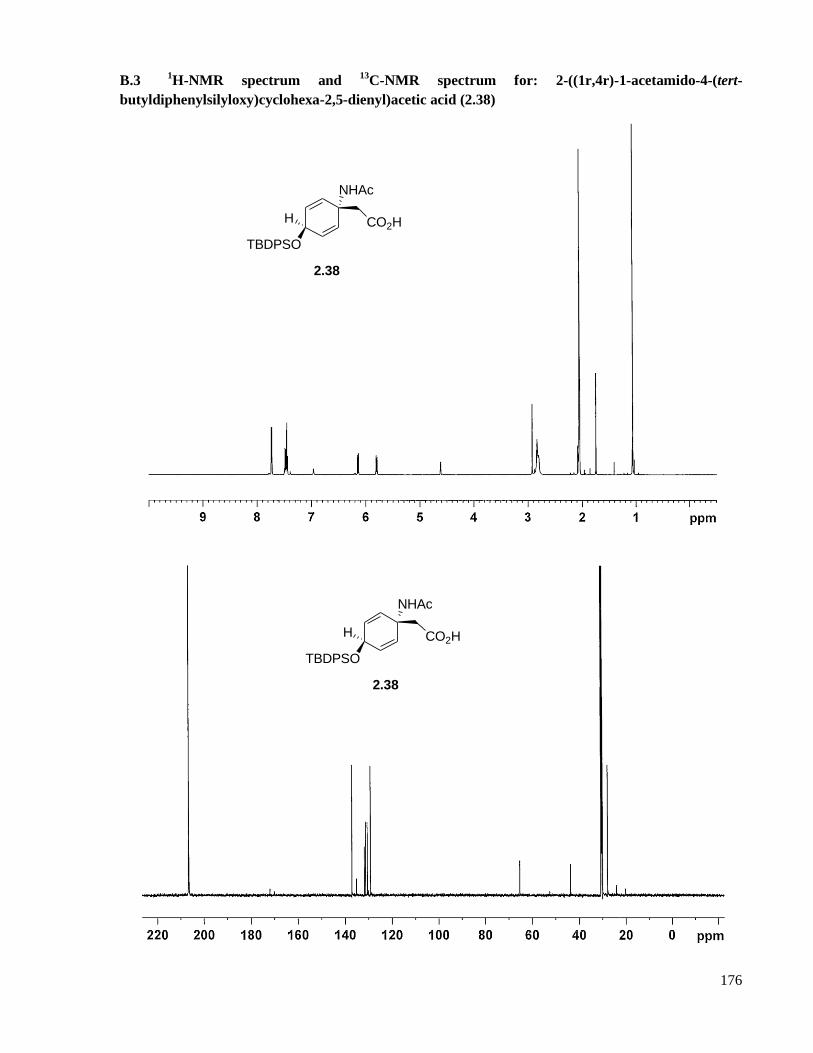
176
B.3 1H-NMR spectrum and
13C-NMR spectrum for: 2-((1r,4r)-1-acetamido-4-(tert-
butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetic acid (2.38)
TBDPSO
NHAc
CO2HH
2.38
TBDPSO
NHAc
CO2HH
2.38
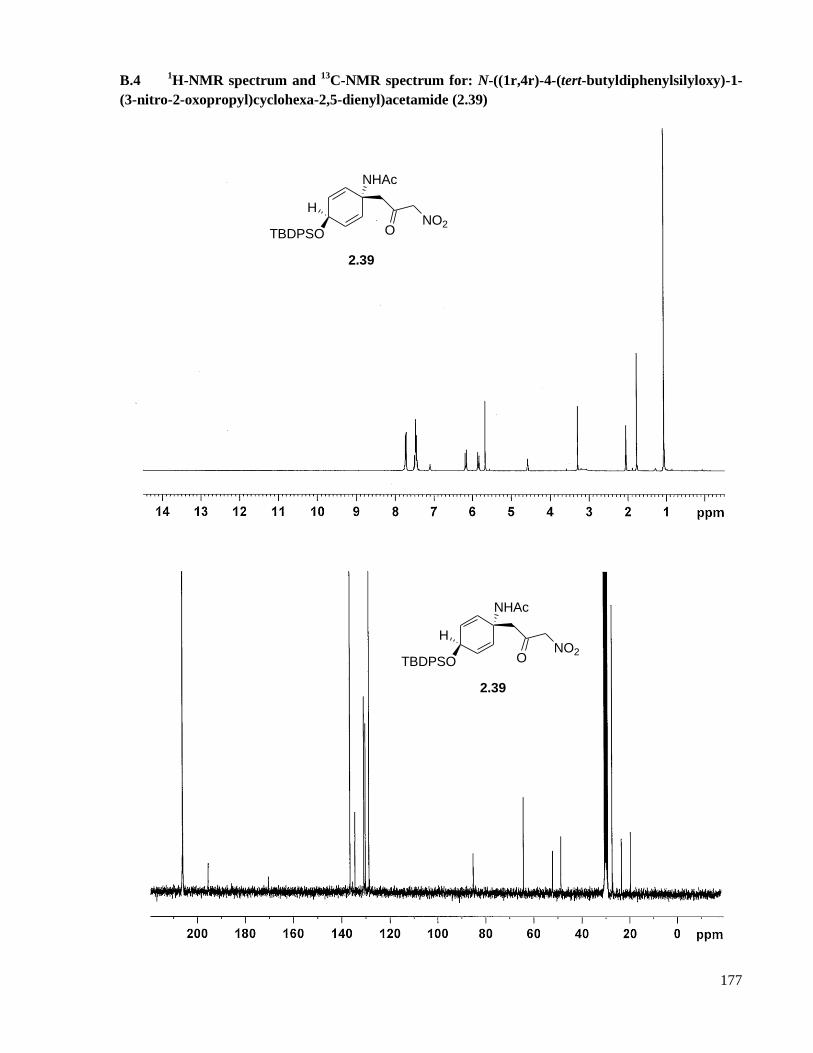
177
B.4 1H-NMR spectrum and
13C-NMR spectrum for: N-((1r,4r)-4-(tert-butyldiphenylsilyloxy)-1-
(3-nitro-2-oxopropyl)cyclohexa-2,5-dienyl)acetamide (2.39)
TBDPSO
NHAc
H
2.39
ONO2
TBDPSO
NHAc
H
2.39
ONO2
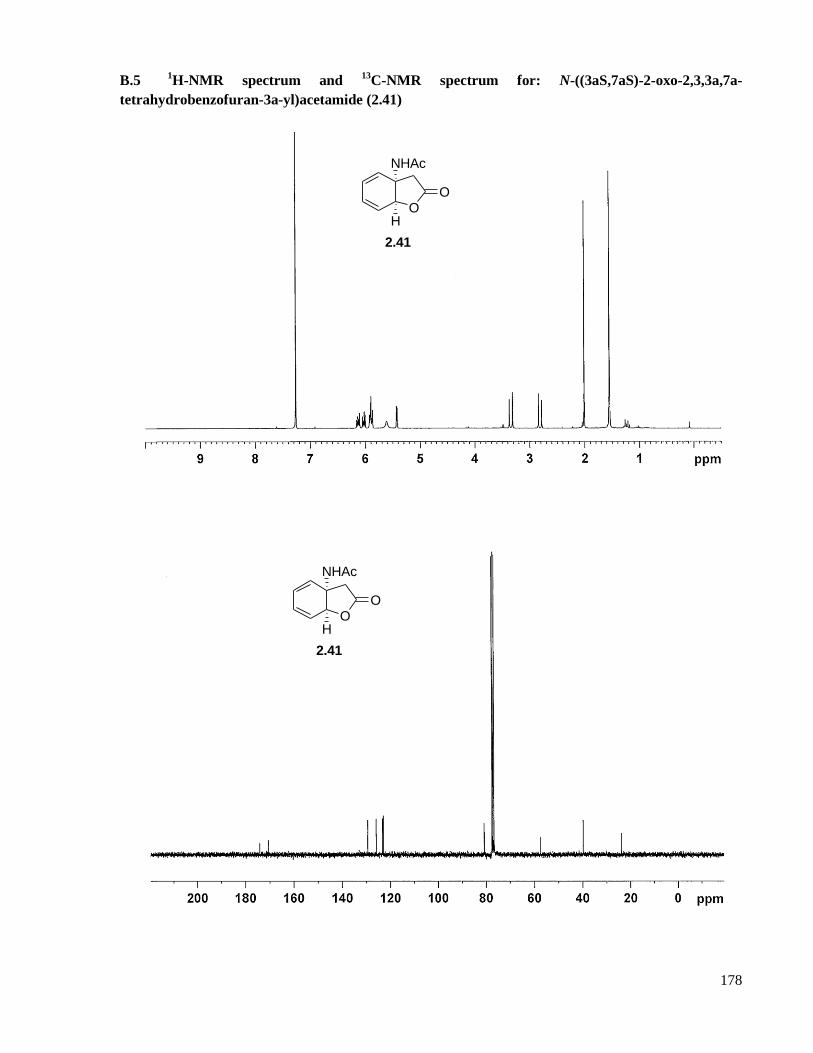
178
B.5 1H-NMR spectrum and
13C-NMR spectrum for: N-((3aS,7aS)-2-oxo-2,3,3a,7a-
tetrahydrobenzofuran-3a-yl)acetamide (2.41)
OO
NHAc
H
2.41
OO
NHAc
H
2.41
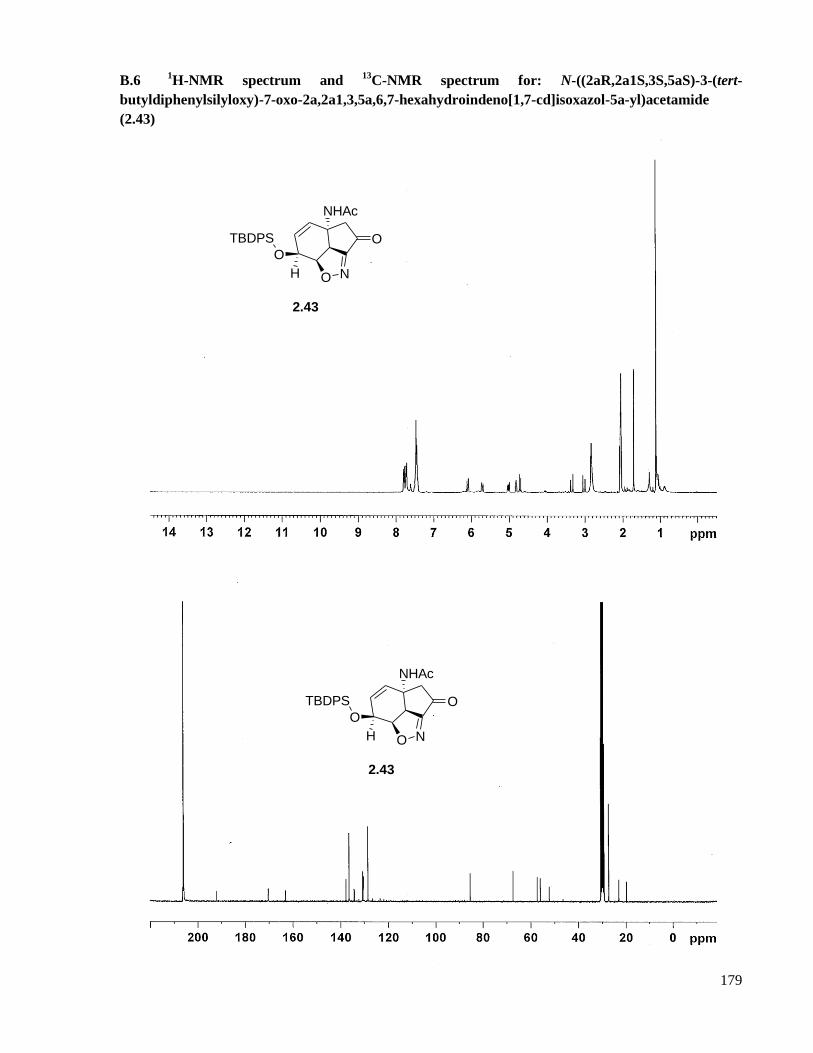
179
B.6 1H-NMR spectrum and
13C-NMR spectrum for: N-((2aR,2a1S,3S,5aS)-3-(tert-
butyldiphenylsilyloxy)-7-oxo-2a,2a1,3,5a,6,7-hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide
(2.43)
O
O
NHAc
H
TBDPS
NO
2.43
O
O
NHAc
H
TBDPS
NO
2.43
30
O
O N
TBDPSO
NHAc
H
H
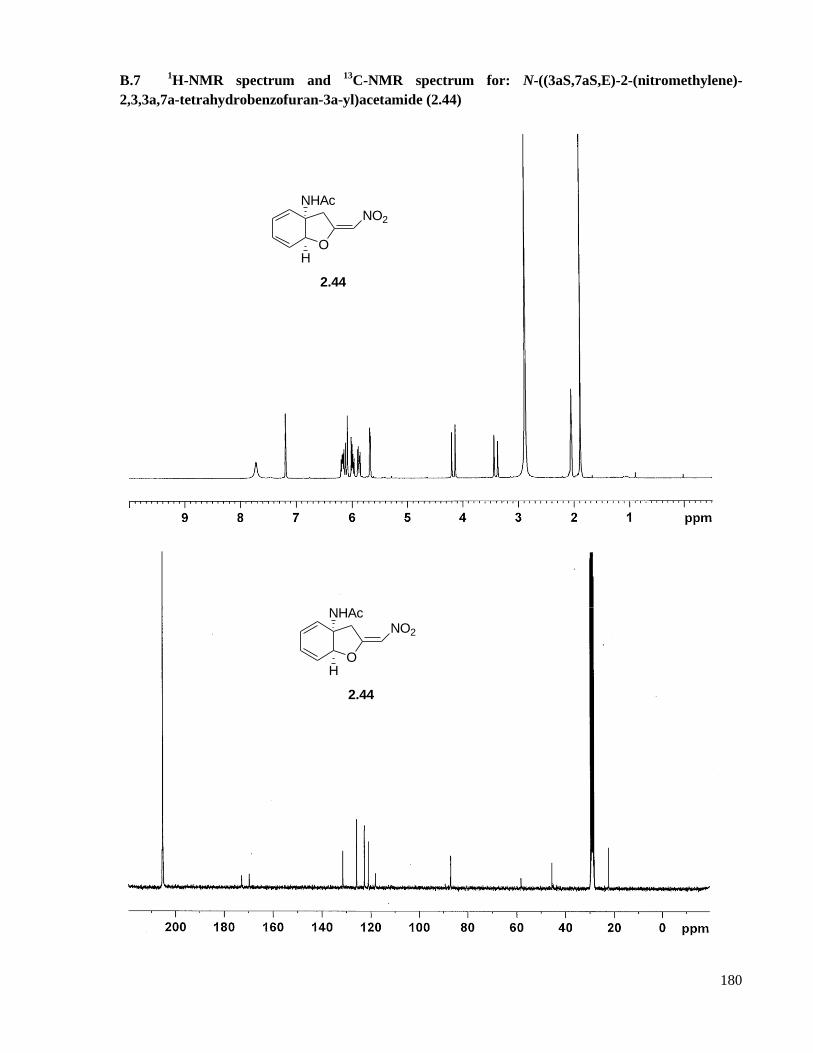
180
B.7 1H-NMR spectrum and
13C-NMR spectrum for: N-((3aS,7aS,E)-2-(nitromethylene)-
2,3,3a,7a-tetrahydrobenzofuran-3a-yl)acetamide (2.44)
OH
NHAc
2.44
NO2
OH
NHAc
2.44
NO2
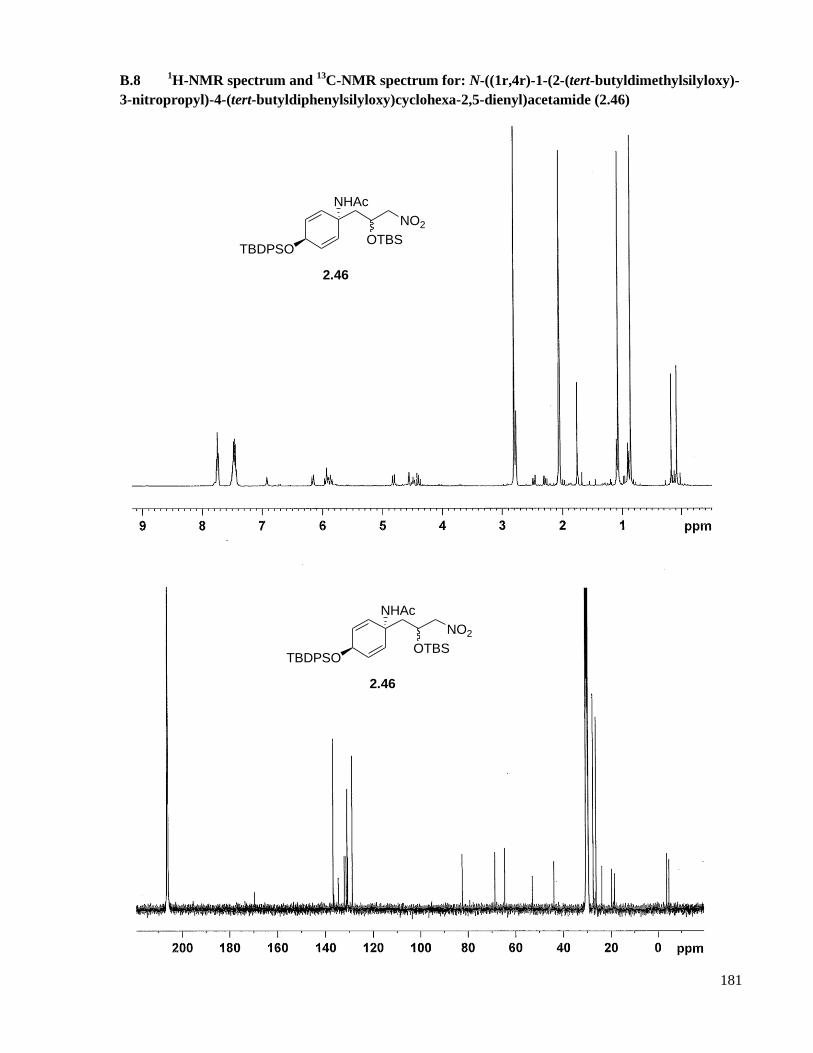
181
B.8 1H-NMR spectrum and
13C-NMR spectrum for: N-((1r,4r)-1-(2-(tert-butyldimethylsilyloxy)-
3-nitropropyl)-4-(tert-butyldiphenylsilyloxy)cyclohexa-2,5-dienyl)acetamide (2.46)
OTBS
NO2
TBDPSO
NHAc
2.46
OTBS
NO2
TBDPSO
NHAc
2.46
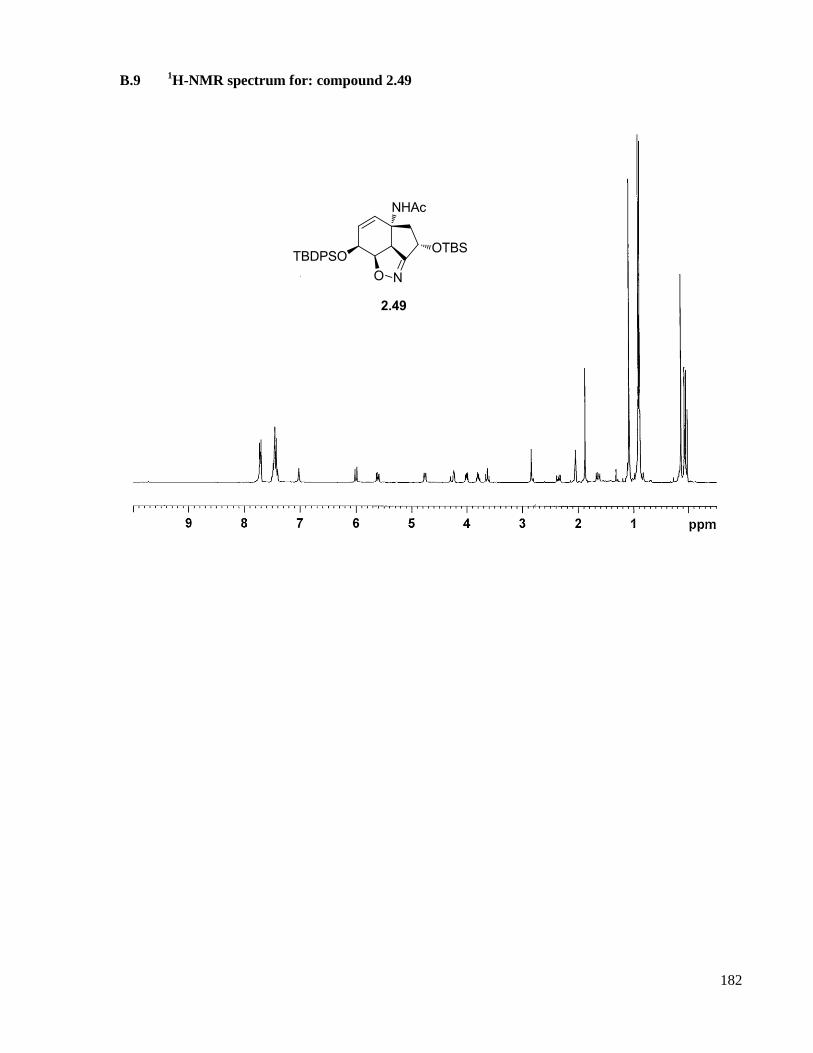
182
B.9 1H-NMR spectrum for: compound 2.49
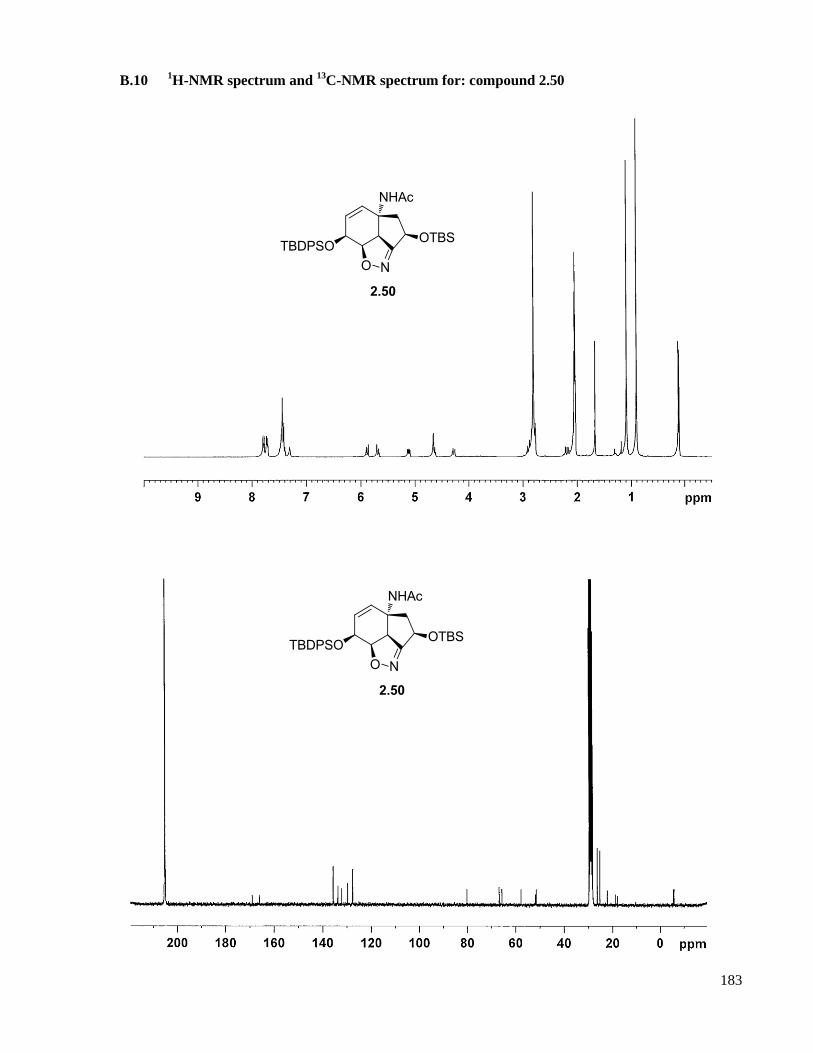
183
B.10 1H-NMR spectrum and
13C-NMR spectrum for: compound 2.50
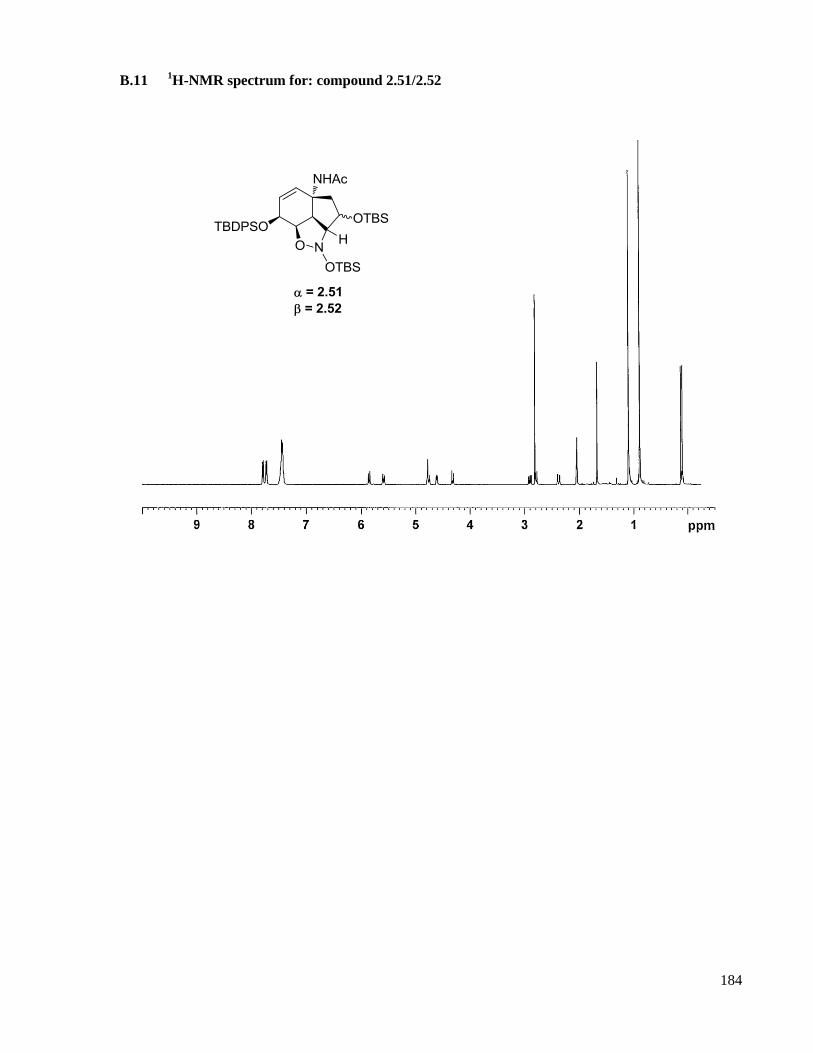
184
B.11 1H-NMR spectrum for: compound 2.51/2.52
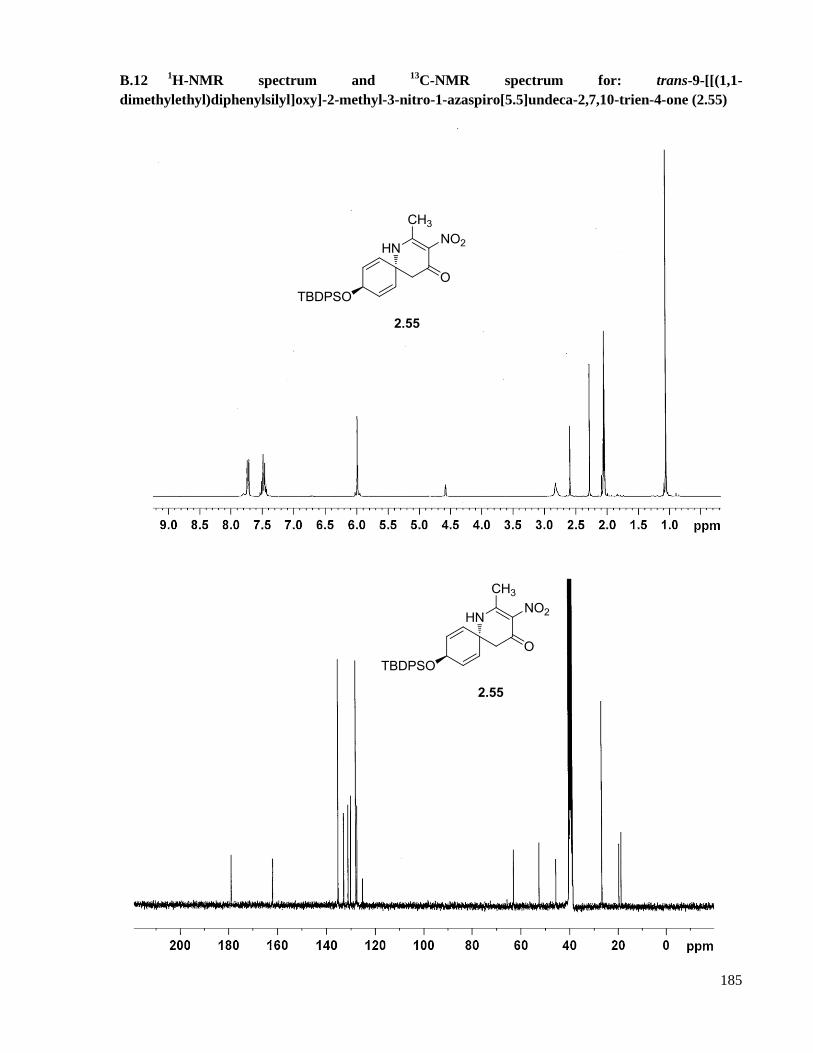
185
B.12 1H-NMR spectrum and
13C-NMR spectrum for: trans-9-[[(1,1-
dimethylethyl)diphenylsilyl]oxy]-2-methyl-3-nitro-1-azaspiro[5.5]undeca-2,7,10-trien-4-one (2.55)
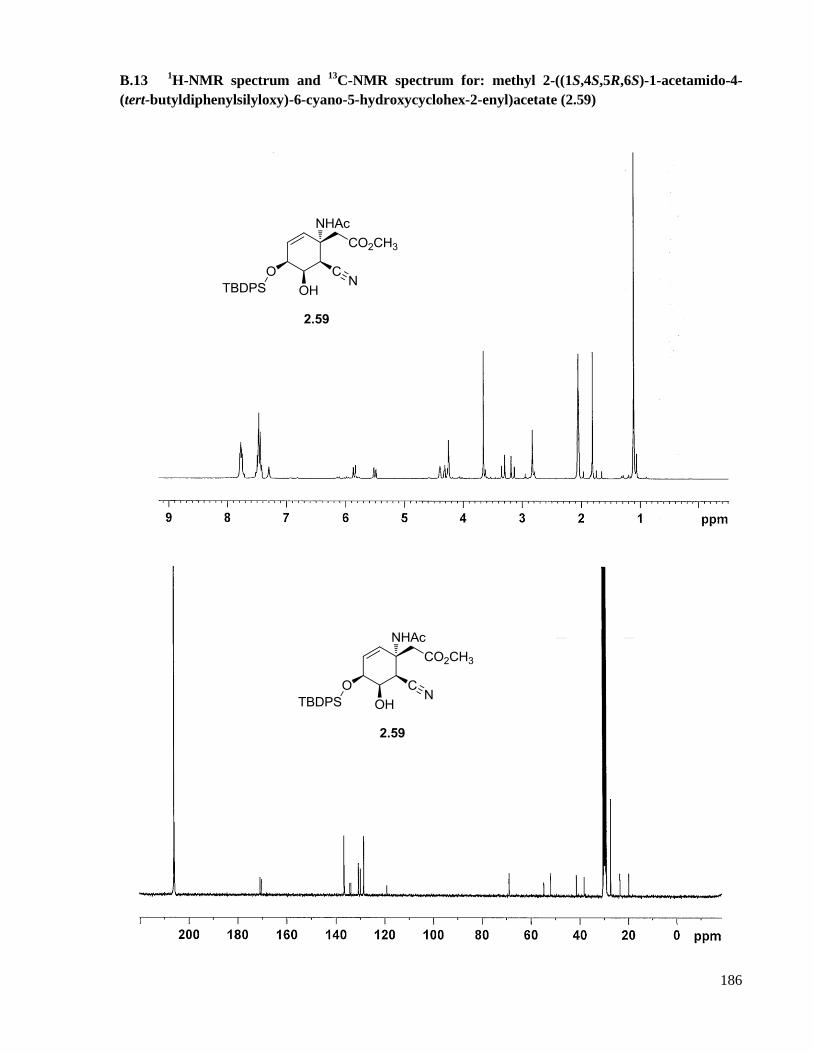
186
B.13 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1S,4S,5R,6S)-1-acetamido-4-
(tert-butyldiphenylsilyloxy)-6-cyano-5-hydroxycyclohex-2-enyl)acetate (2.59)
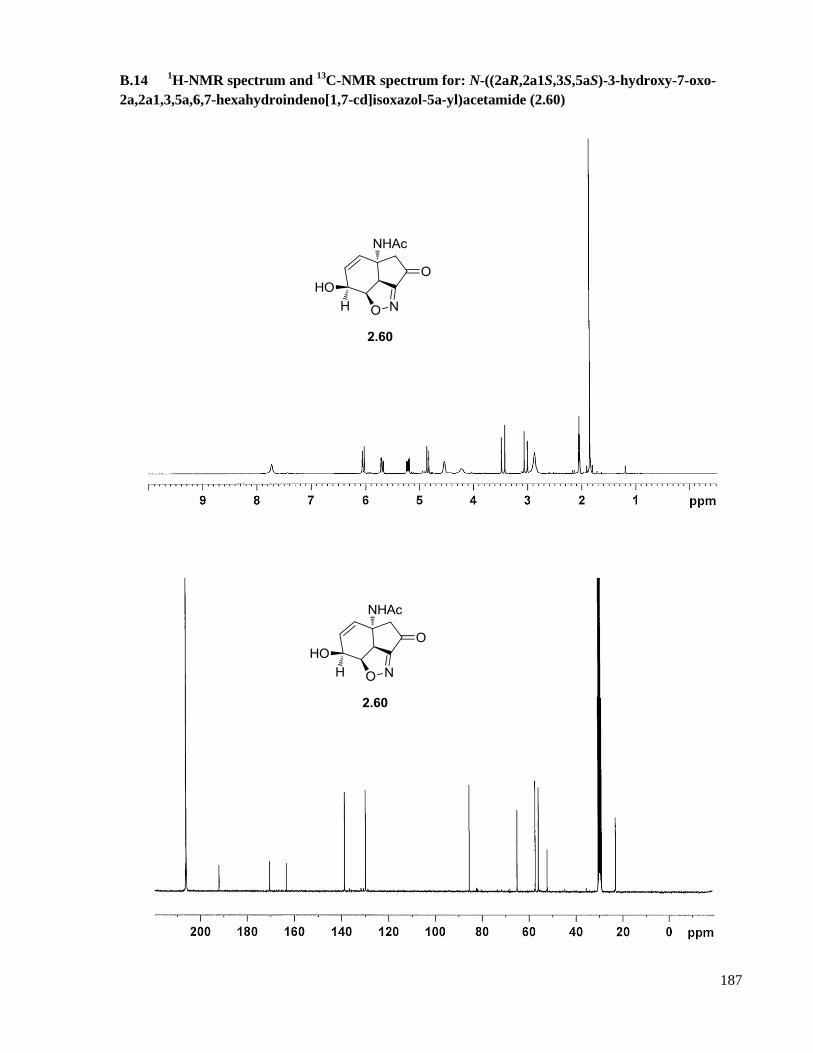
187
B.14 1H-NMR spectrum and
13C-NMR spectrum for: N-((2aR,2a1S,3S,5aS)-3-hydroxy-7-oxo-
2a,2a1,3,5a,6,7-hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.60)
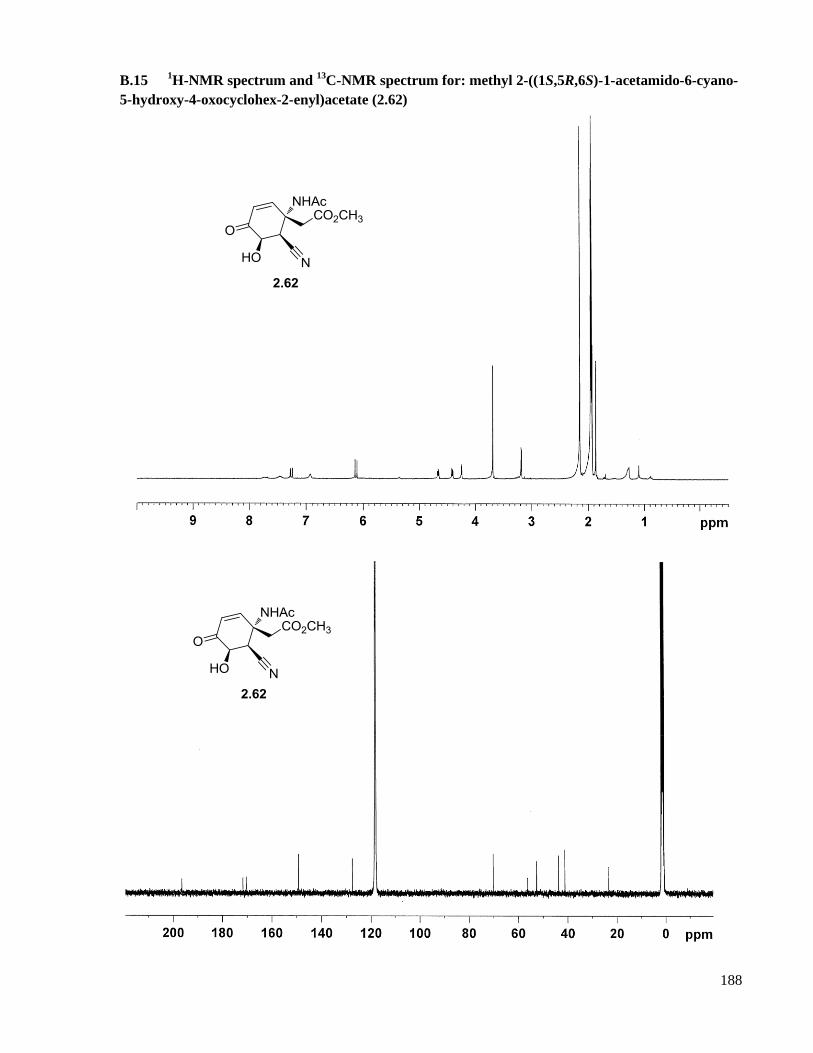
188
B.15 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1S,5R,6S)-1-acetamido-6-cyano-
5-hydroxy-4-oxocyclohex-2-enyl)acetate (2.62)
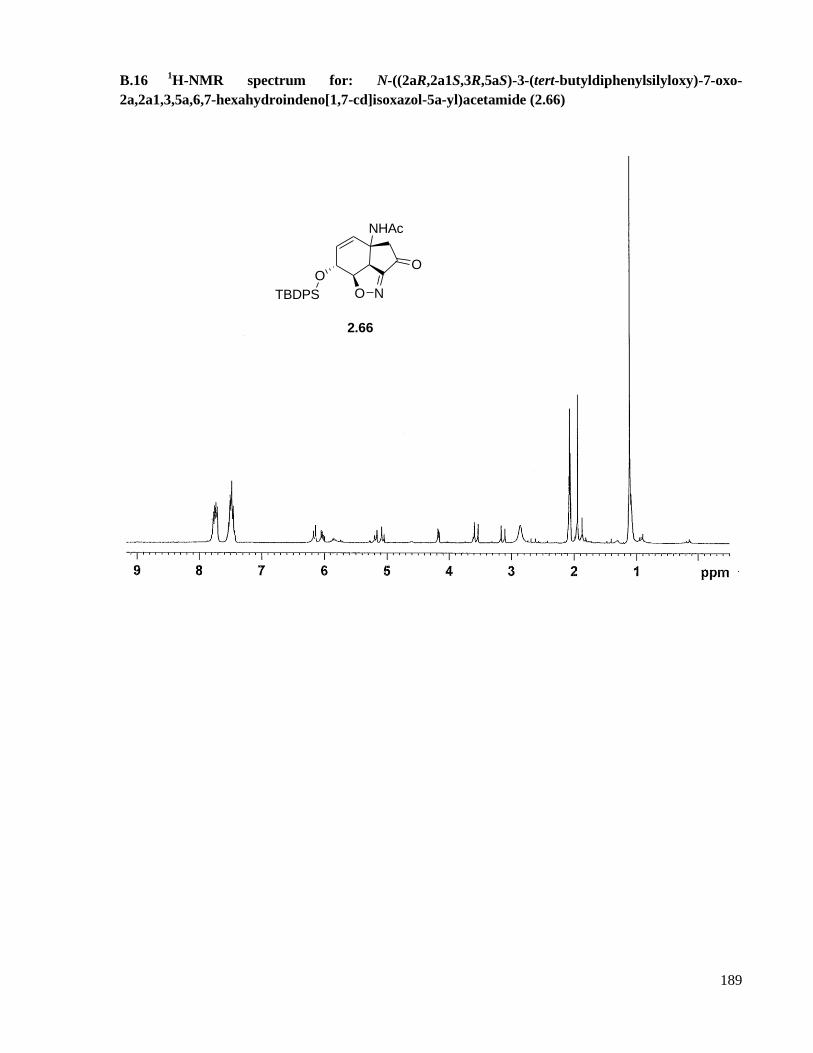
189
B.16 1H-NMR spectrum for: N-((2aR,2a1S,3R,5aS)-3-(tert-butyldiphenylsilyloxy)-7-oxo-
2a,2a1,3,5a,6,7-hexahydroindeno[1,7-cd]isoxazol-5a-yl)acetamide (2.66)
O N
NHAc
OO
TBDPS
2.66

190
B.17 1H-NMR spectrum and
13C-NMR spectrum for: methyl 2-((1S,4R,5R,6S)-1-acetamido-4-
(tert-butyldiphenylsilyloxy)-6-cyano-5-hydroxycyclohex-2-enyl)acetate (2.67)
OH
O CN
CO2CH3
NHAc
2.67
TBDPS
OH
O CN
CO2CH3
NHAc
2.67
TBDPS
191
B.18 1H-NMR spectrum and
13C-NMR spectrum for: 3-(4-methoxyphenyl)-5-phenyl-4,5-
dihydroisoxazole (2.76)
192
B.19 1H-NMR spectrum and
13C-NMR spectrum for: 3,5-diphenyl-4,5-dihydroisoxazole (2.77)
193
B.20 1H-NMR spectrum and
13C-NMR spectrum for: 3-(3-nitrophenyl)-5-phenyl-4,5-
dihydroisoxazole (2.78)
194
B.21 1H-NMR spectrum and
13C-NMR spectrum for: 3-pentyl-5-phenyl-4,5-dihydroisoxazole
(2.79)
N O
2.79
N O
2.79
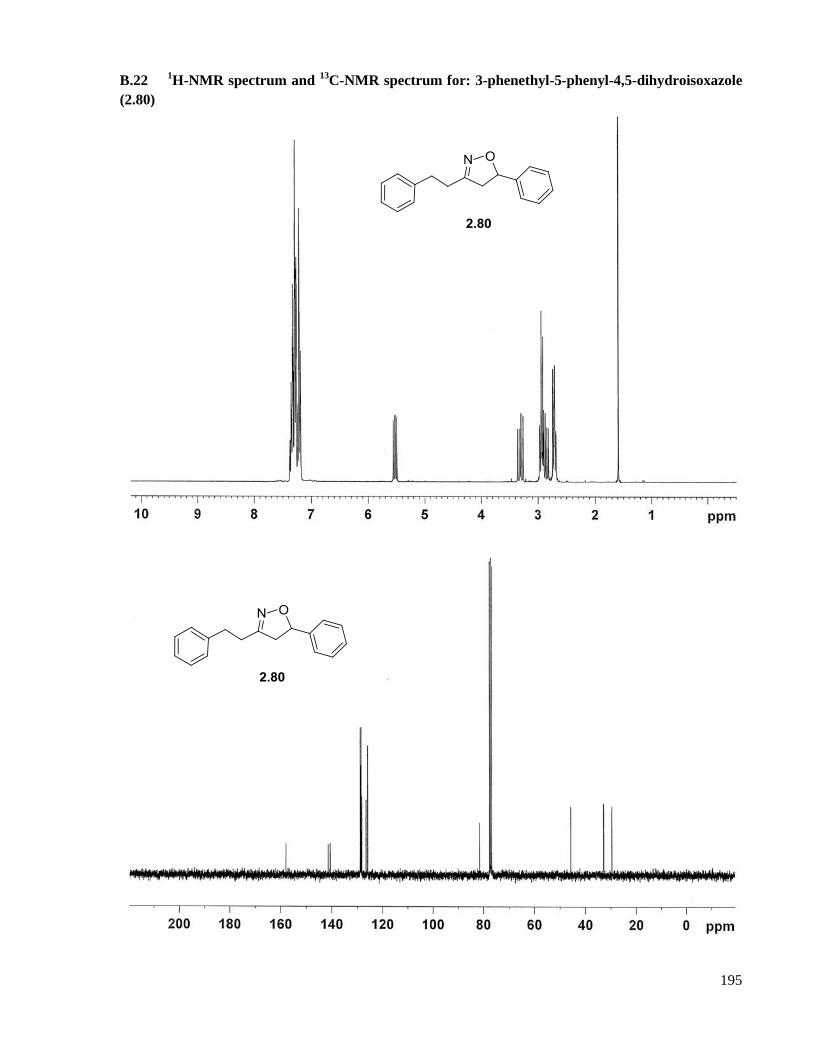
195
B.22 1H-NMR spectrum and
13C-NMR spectrum for: 3-phenethyl-5-phenyl-4,5-dihydroisoxazole
(2.80)
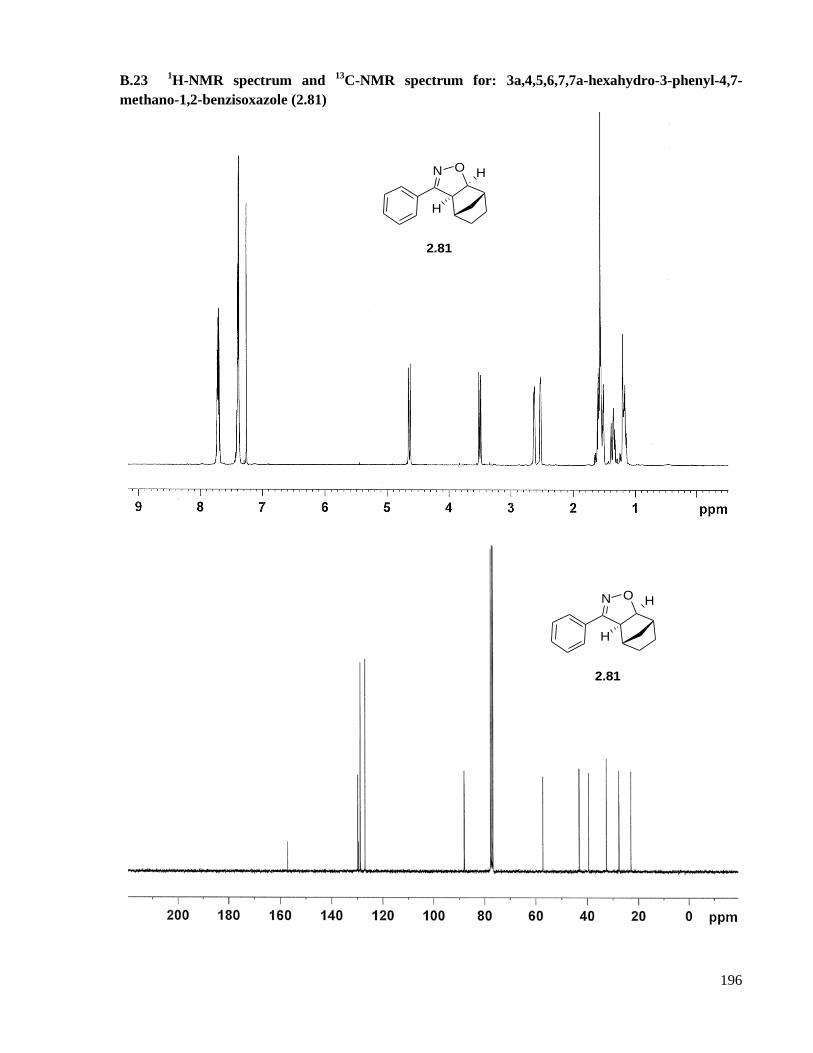
196
B.23 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-phenyl-4,7-
methano-1,2-benzisoxazole (2.81)
N O H
H
2.81
N O H
H
2.81
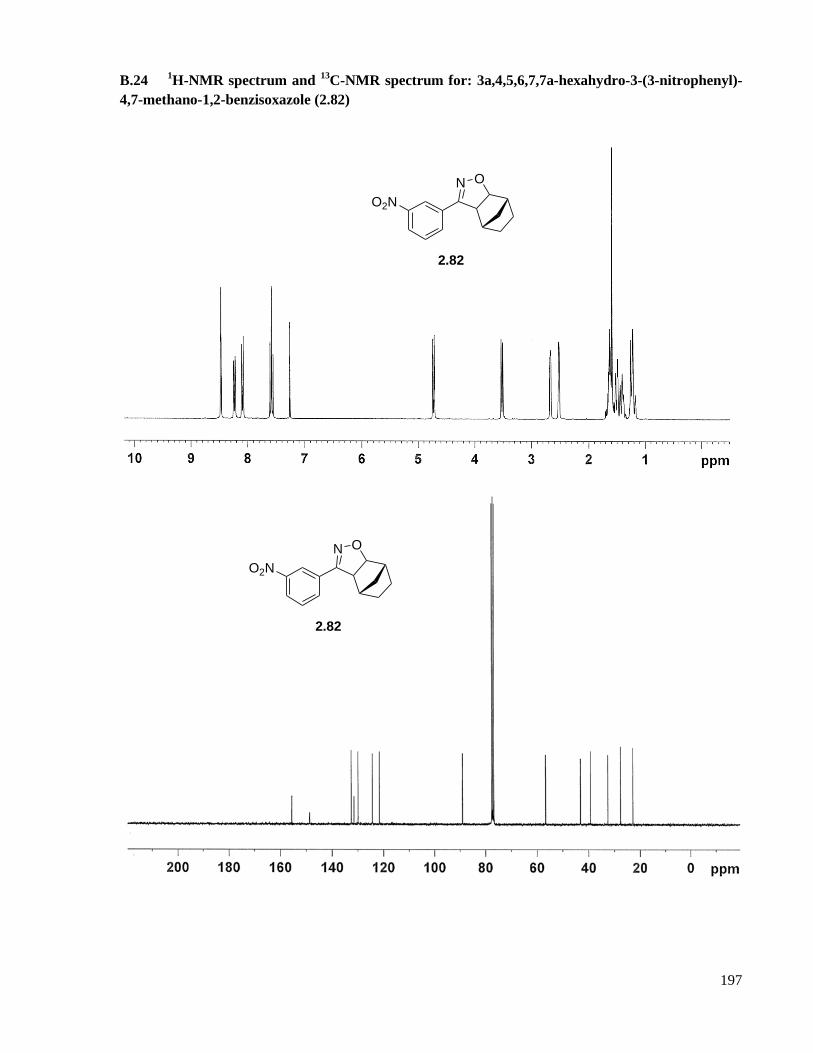
197
B.24 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-(3-nitrophenyl)-
4,7-methano-1,2-benzisoxazole (2.82)
ON
2.82
O2N
ON
2.82
O2N
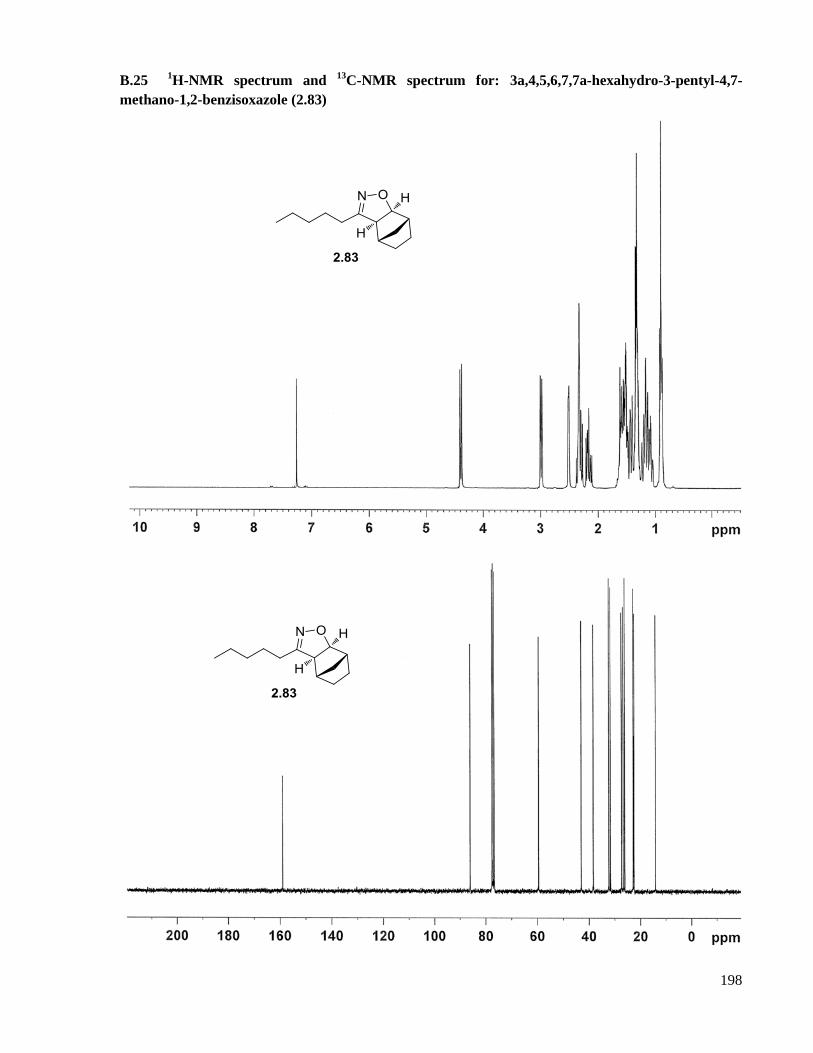
198
B.25 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-pentyl-4,7-
methano-1,2-benzisoxazole (2.83)

199
B.26 1H-NMR spe ctrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-(2-phenylethyl)-
4,7-methano-1,2-benzisoxazole (2.84)
N O H
H
2.84
N O H
H
2.84
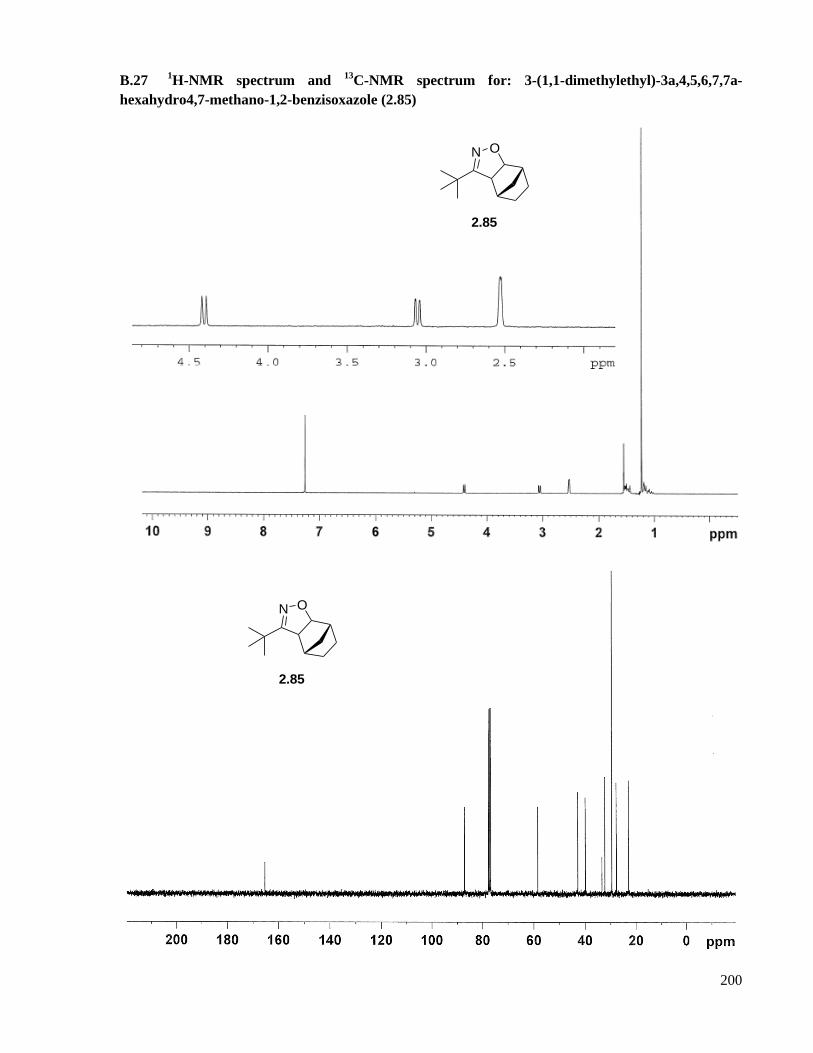
200
B.27 1H-NMR spectrum and
13C-NMR spectrum for: 3-(1,1-dimethylethyl)-3a,4,5,6,7,7a-
hexahydro4,7-methano-1,2-benzisoxazole (2.85)
ON
2.85
ON
2.85
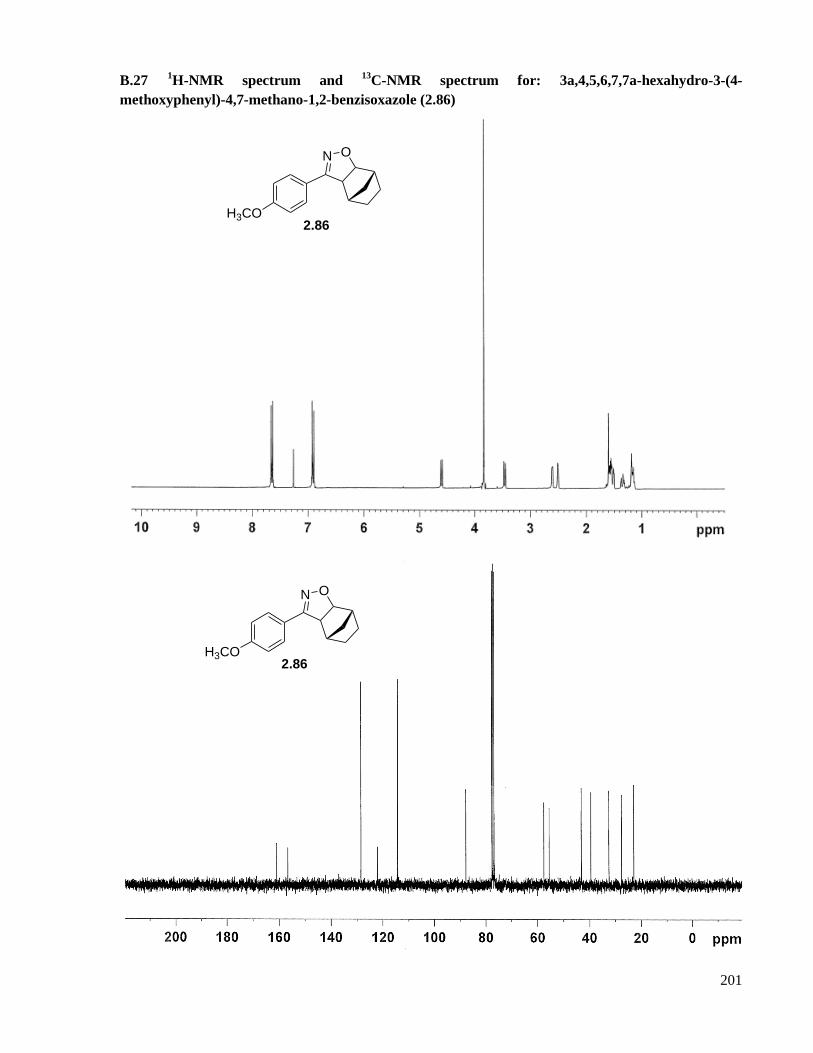
201
B.27 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-3-(4-
methoxyphenyl)-4,7-methano-1,2-benzisoxazole (2.86)
ON
2.86H3CO
ON
2.86H3CO

202
B.28 1H-NMR spectrum and
13C-NMR spectrum for: 5-(3-bromopropyl)-4,5-dihydro-3-phenyl-
isoxazole (2.87)
N O
Br
2.87
N O
Br
2.87
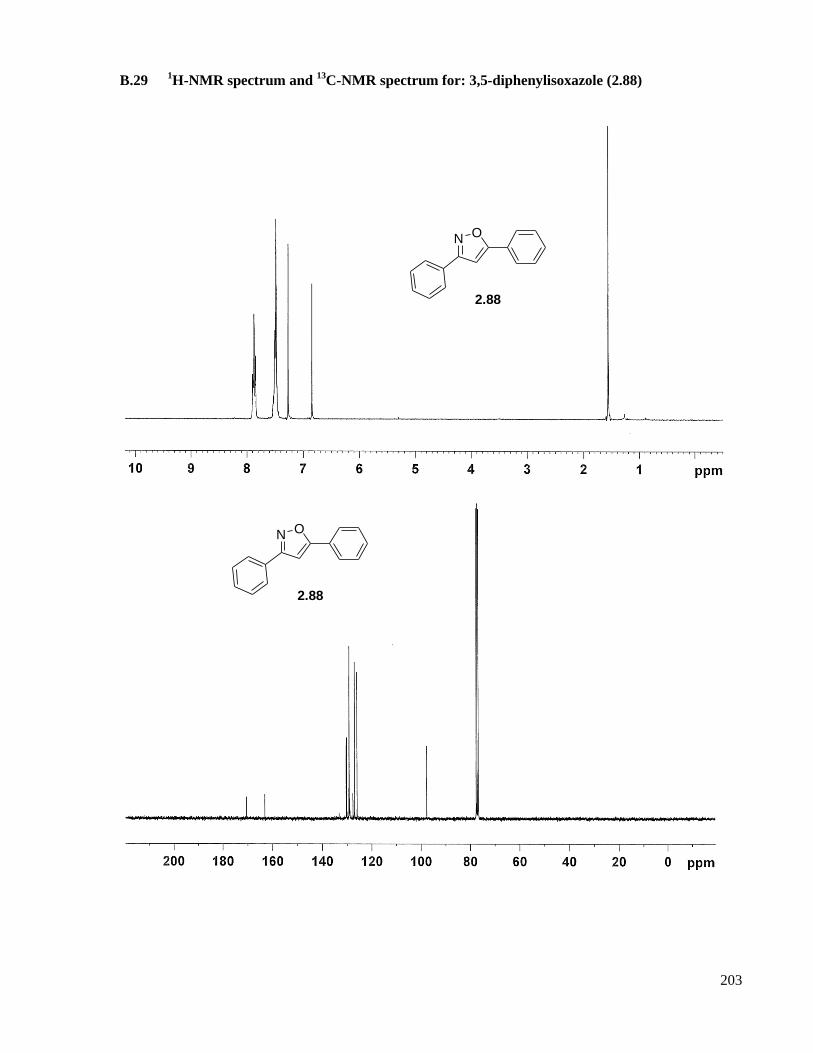
203
B.29 1H-NMR spectrum and
13C-NMR spectrum for: 3,5-diphenylisoxazole (2.88)
N O
2.88
N O
2.88
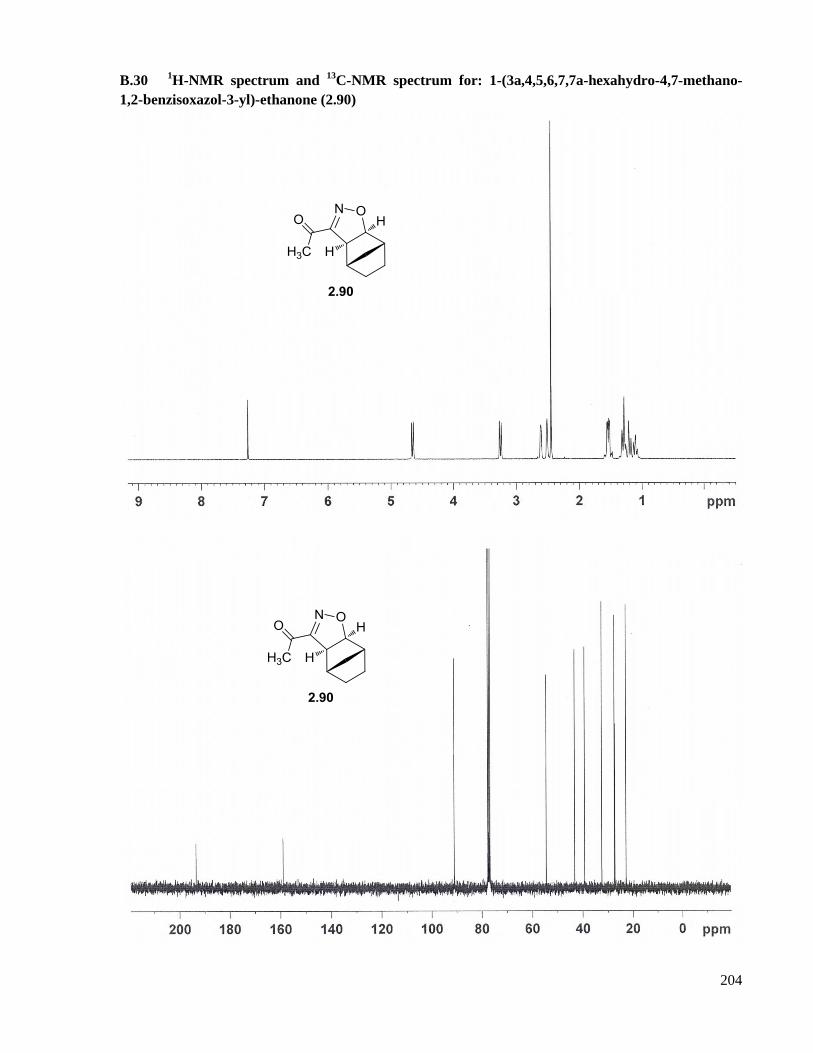
204
B.30 1H-NMR spectrum and
13C-NMR spectrum for: 1-(3a,4,5,6,7,7a-hexahydro-4,7-methano-
1,2-benzisoxazol-3-yl)-ethanone (2.90)
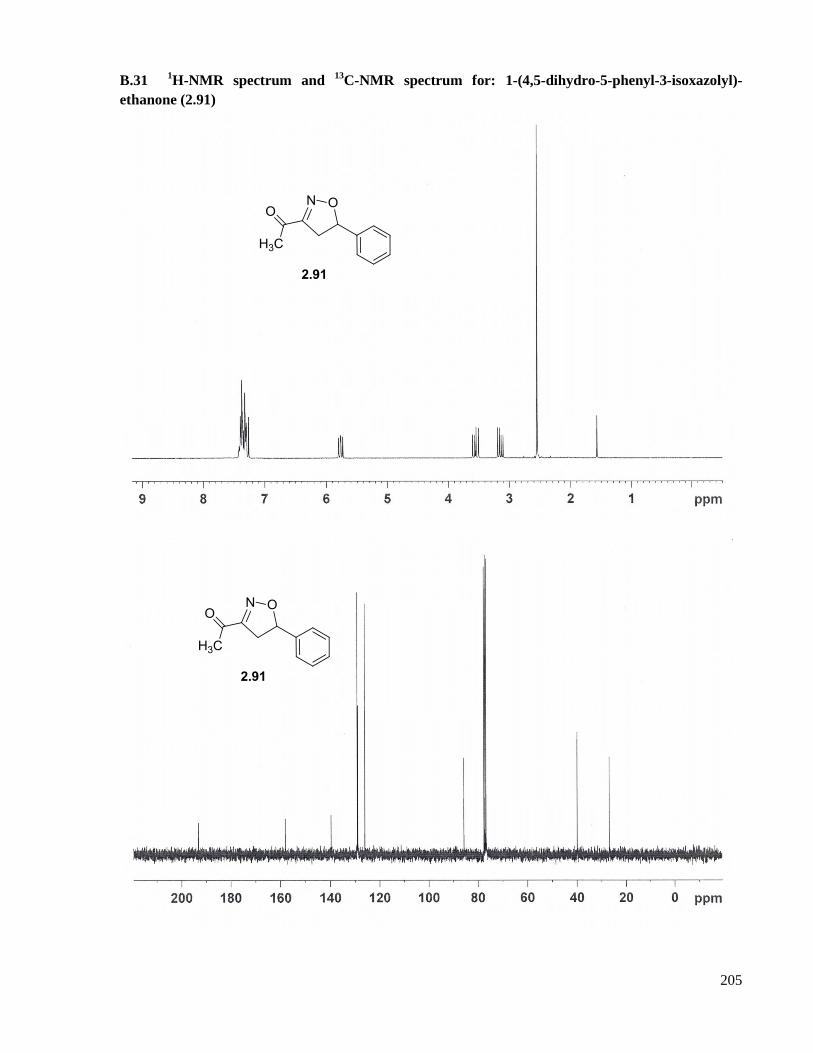
205
B.31 1H-NMR spectrum and
13C-NMR spectrum for: 1-(4,5-dihydro-5-phenyl-3-isoxazolyl)-
ethanone (2.91)
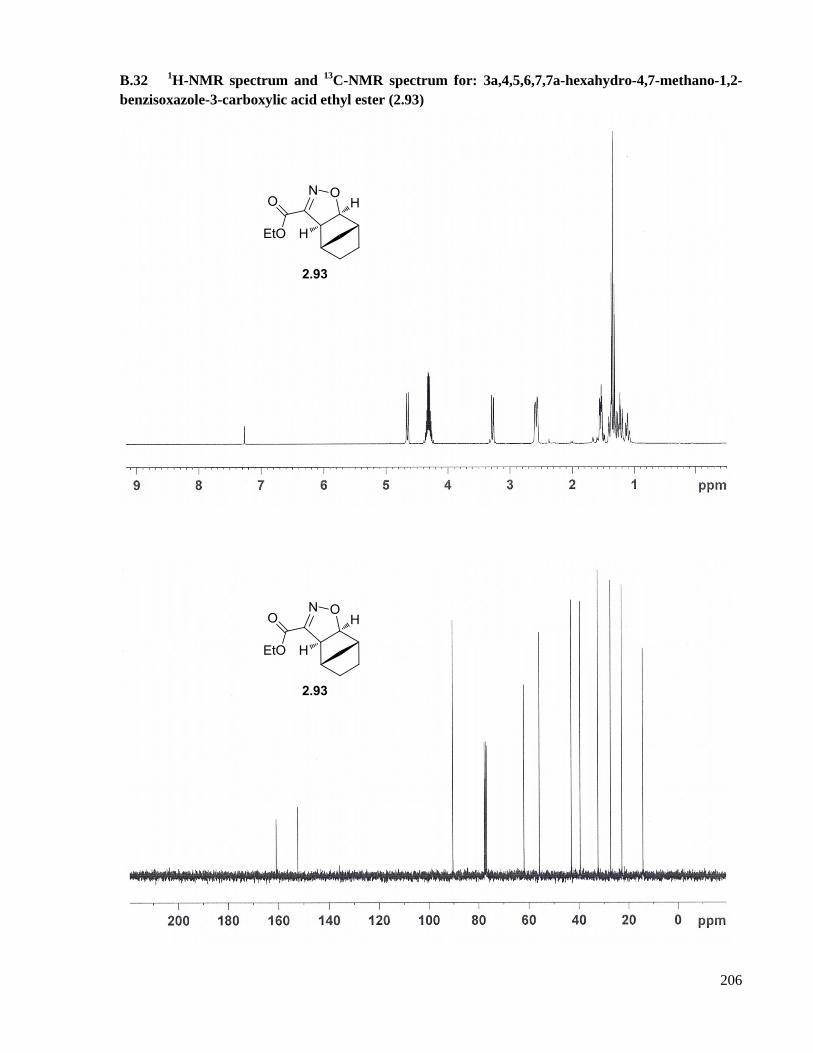
206
B.32 1H-NMR spectrum and
13C-NMR spectrum for: 3a,4,5,6,7,7a-hexahydro-4,7-methano-1,2-
benzisoxazole-3-carboxylic acid ethyl ester (2.93)
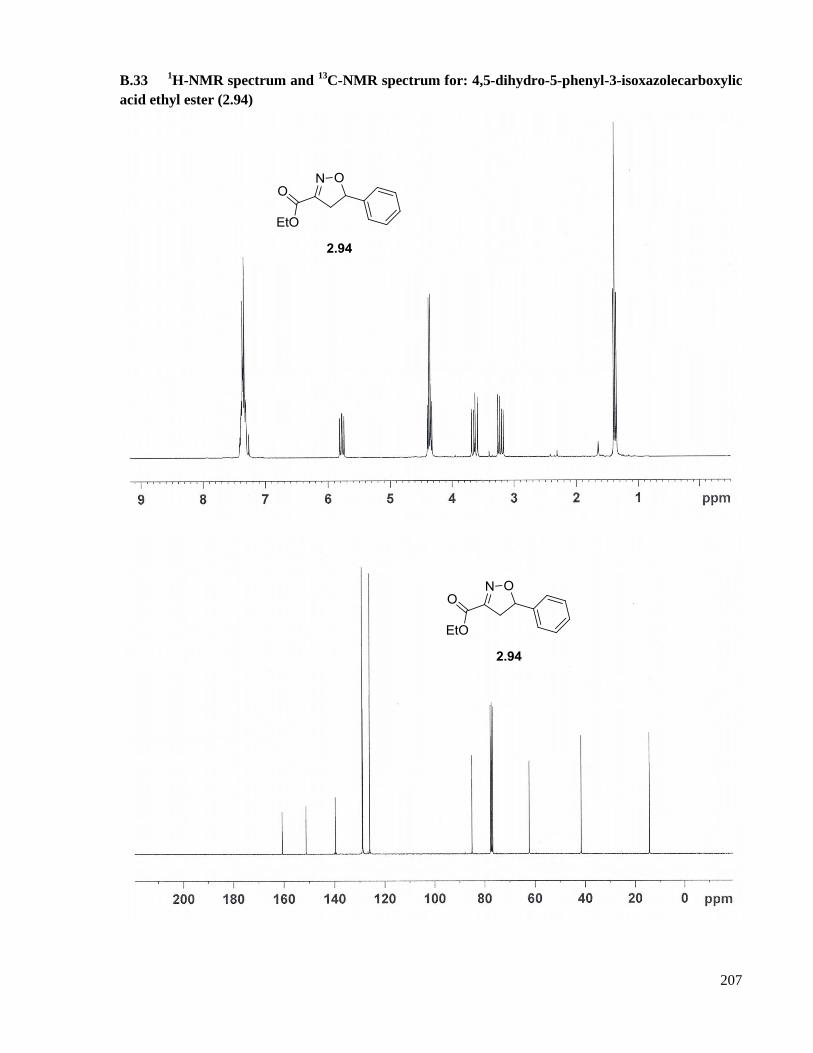
207
B.33 1H-NMR spectrum and
13C-NMR spectrum for: 4,5-dihydro-5-phenyl-3-isoxazolecarboxylic
acid ethyl ester (2.94)
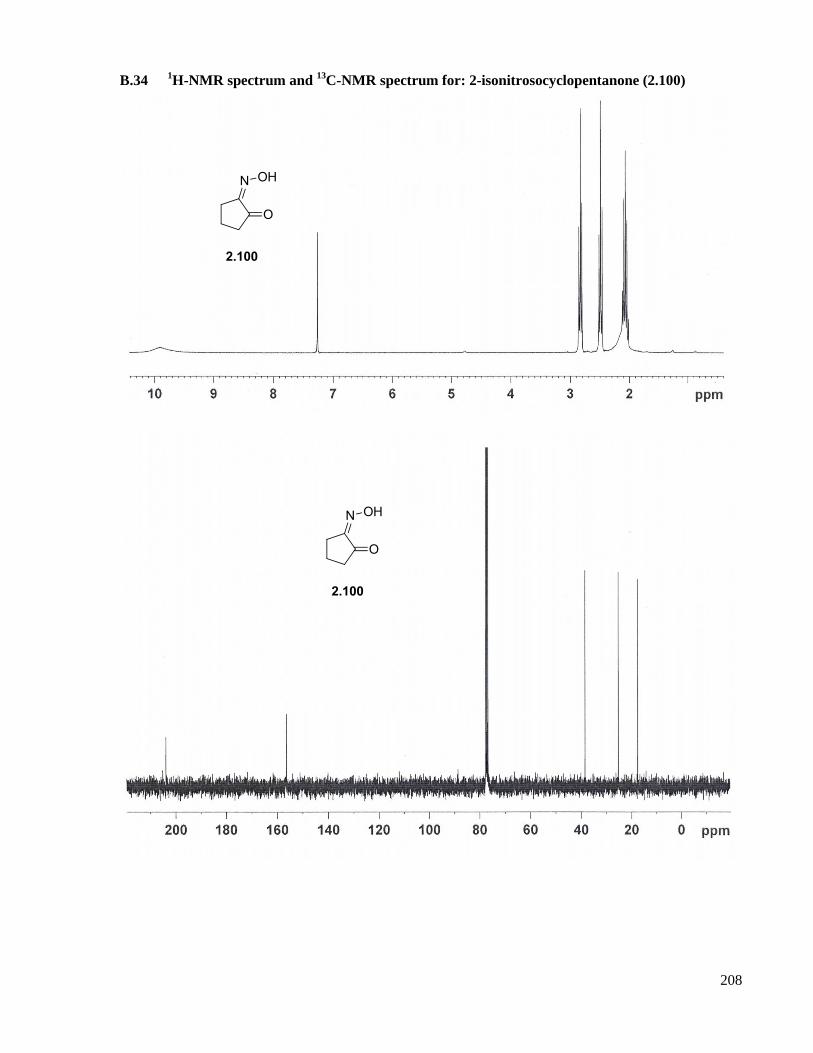
208
B.34 1H-NMR spectrum and
13C-NMR spectrum for: 2-isonitrosocyclopentanone (2.100)

209
B.35 1H-NMR spectrum and
13C-NMR spectrum for: compound 2.101
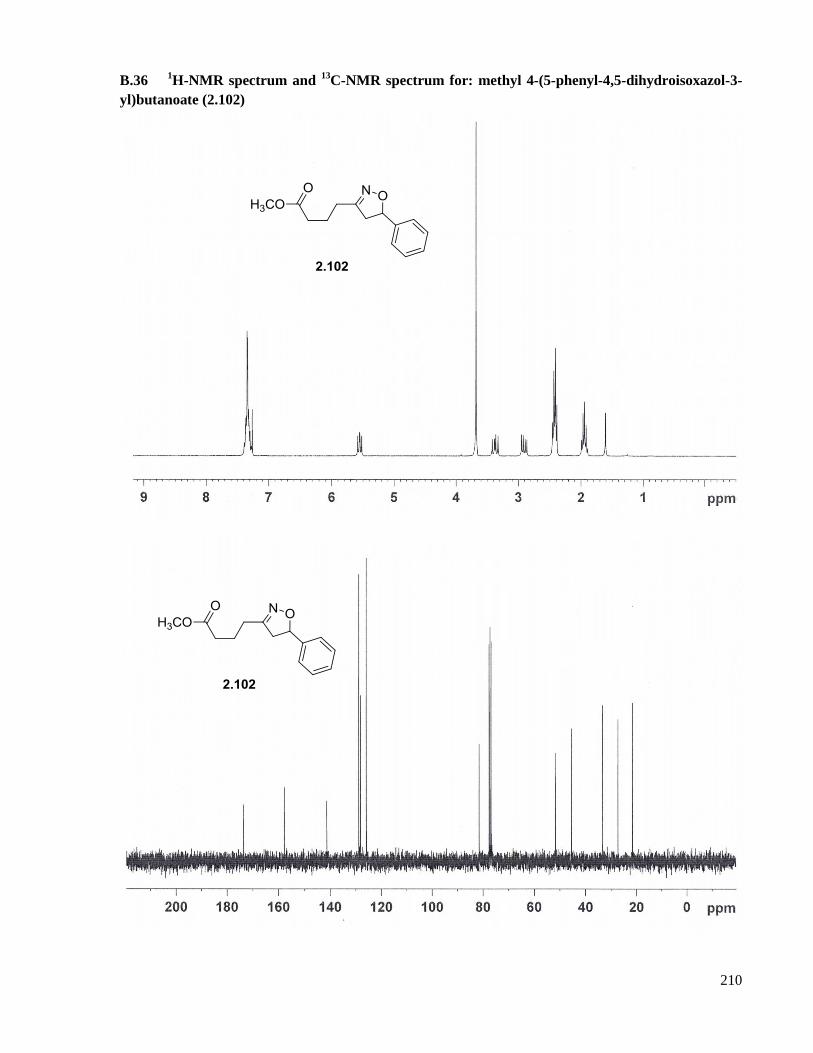
210
B.36 1H-NMR spectrum and
13C-NMR spectrum for: methyl 4-(5-phenyl-4,5-dihydroisoxazol-3-
yl)butanoate (2.102)
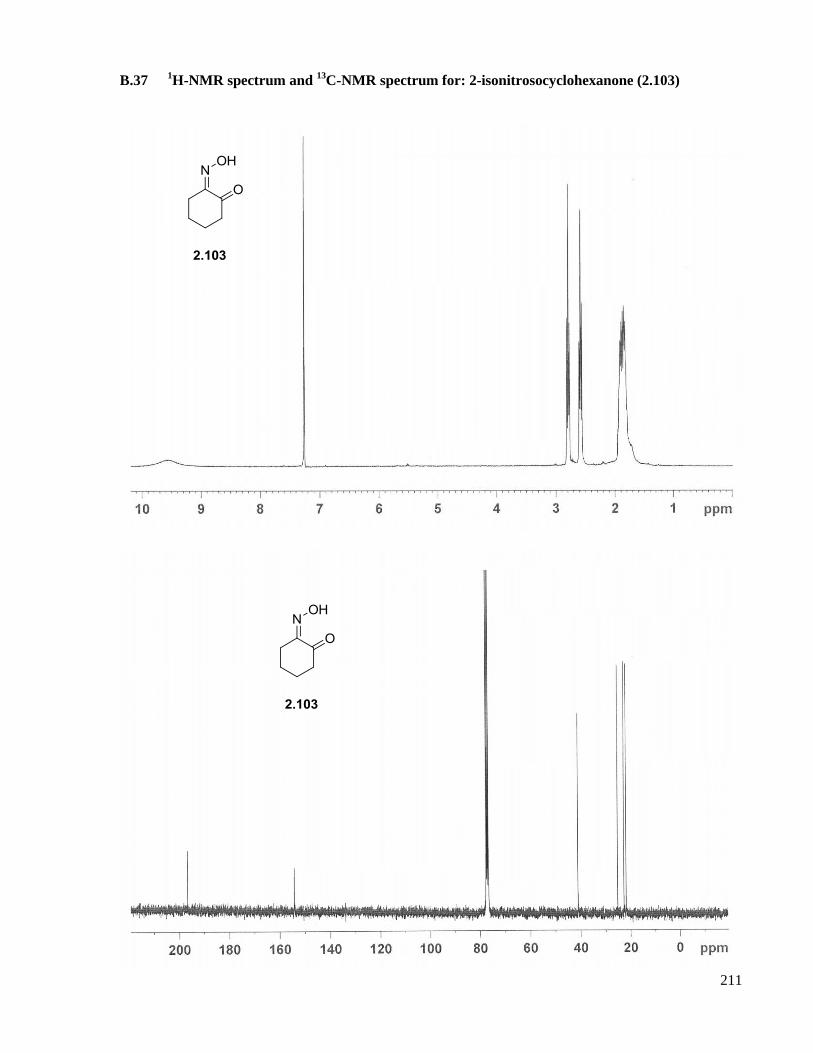
211
B.37 1H-NMR spectrum and
13C-NMR spectrum for: 2-isonitrosocyclohexanone (2.103)
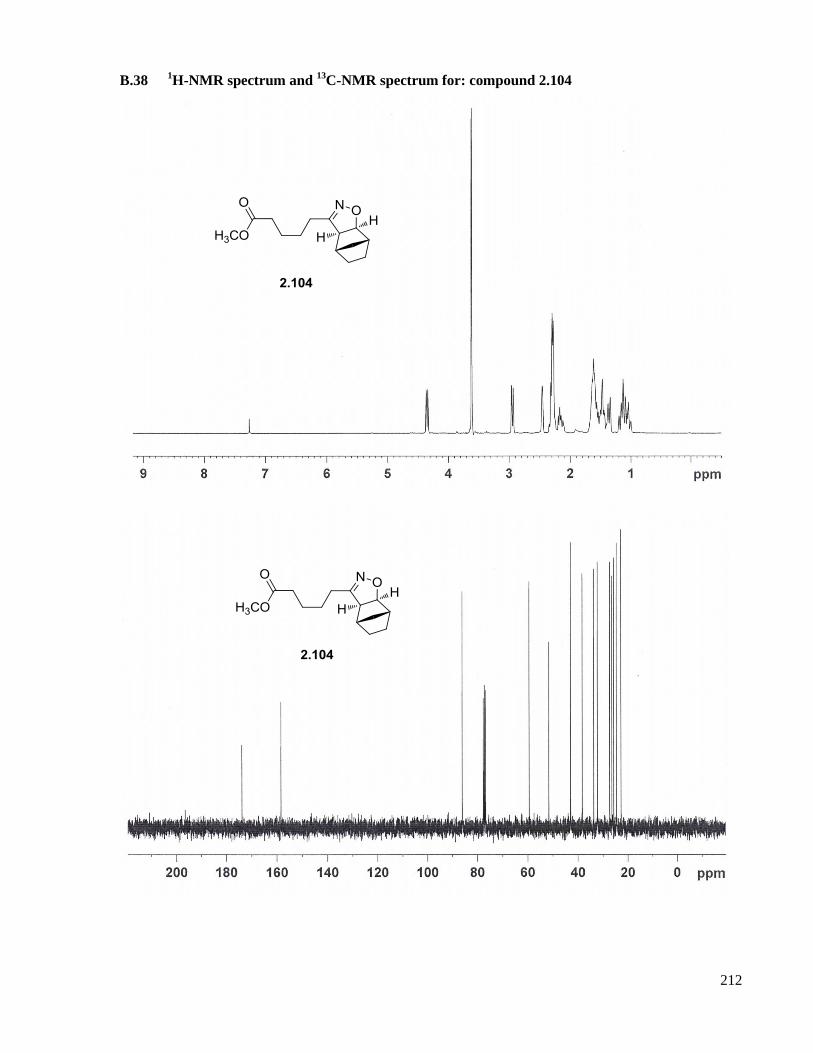
212
B.38 1H-NMR spectrum and
13C-NMR spectrum for: compound 2.104
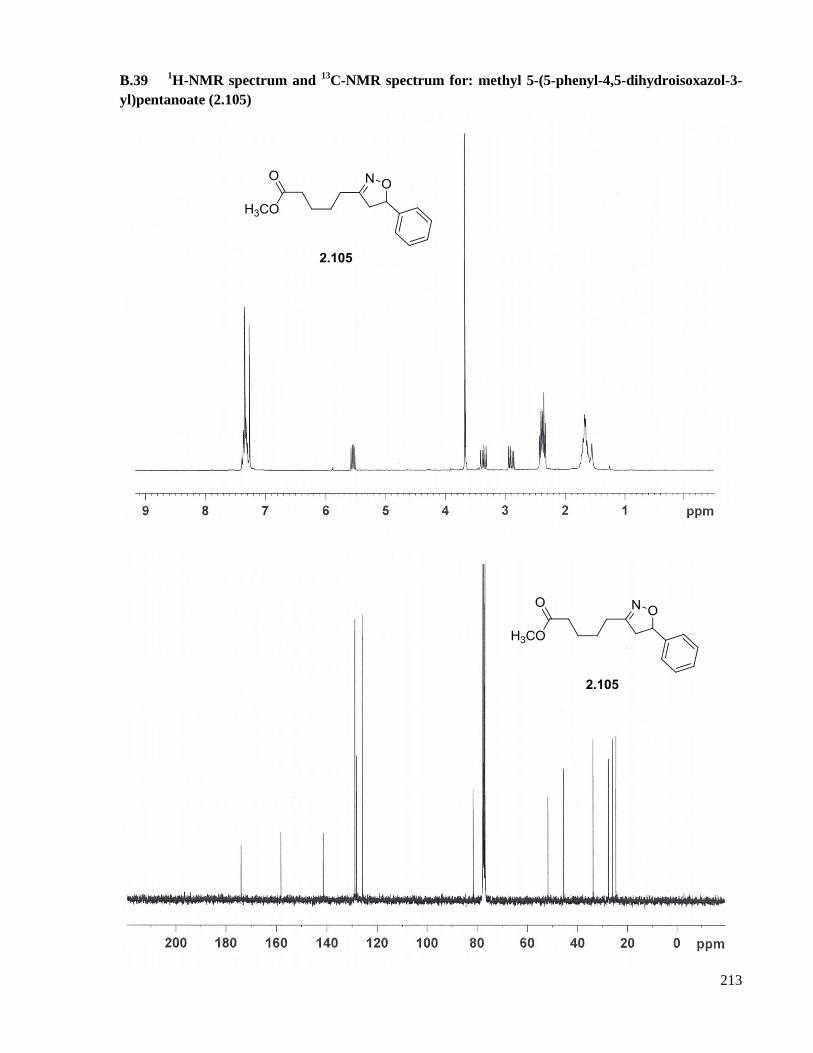
213
B.39 1H-NMR spectrum and
13C-NMR spectrum for: methyl 5-(5-phenyl-4,5-dihydroisoxazol-3-
yl)pentanoate (2.105)
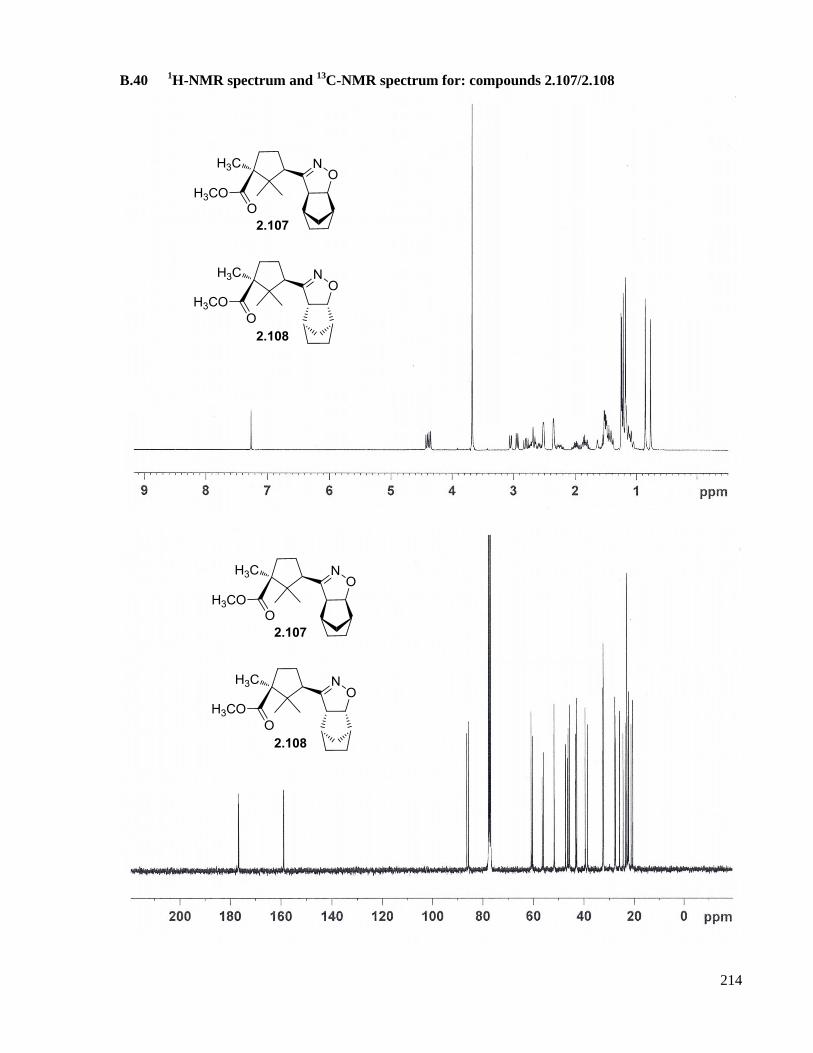
214
B.40 1H-NMR spectrum and
13C-NMR spectrum for: compounds 2.107/2.108
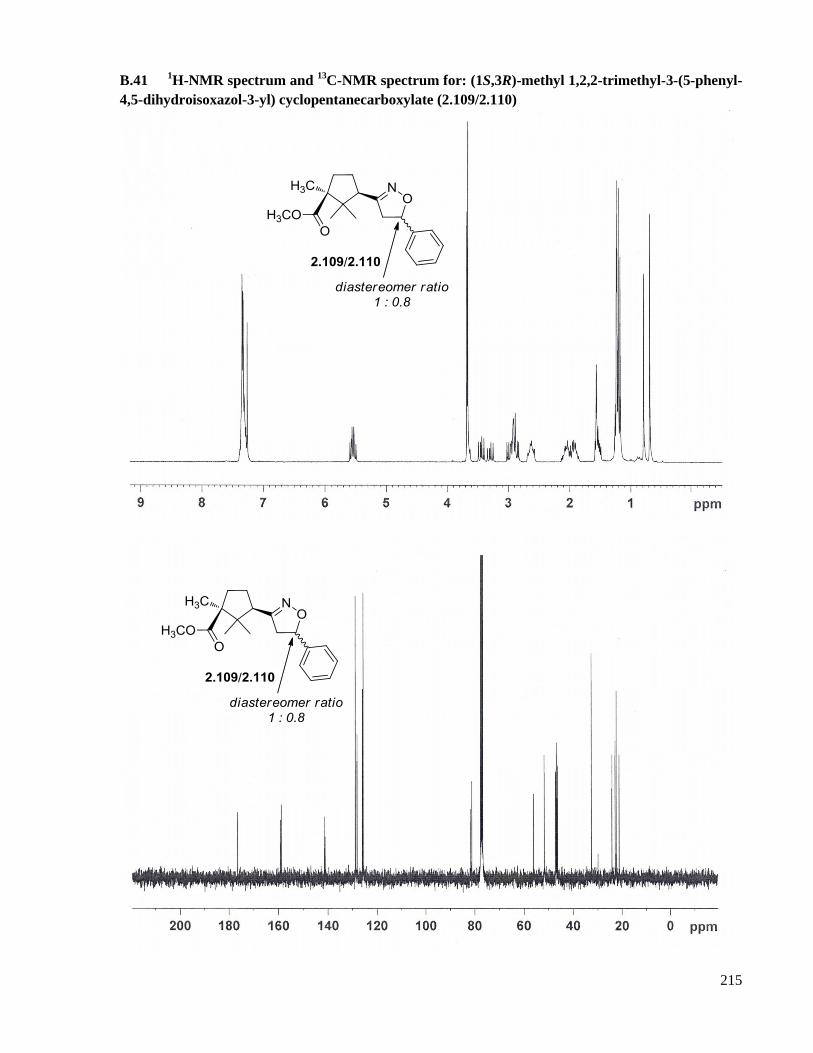
215
B.41 1H-NMR spectrum and
13C-NMR spectrum for: (1S,3R)-methyl 1,2,2-trimethyl-3-(5-phenyl-
4,5-dihydroisoxazol-3-yl) cyclopentanecarboxylate (2.109/2.110)
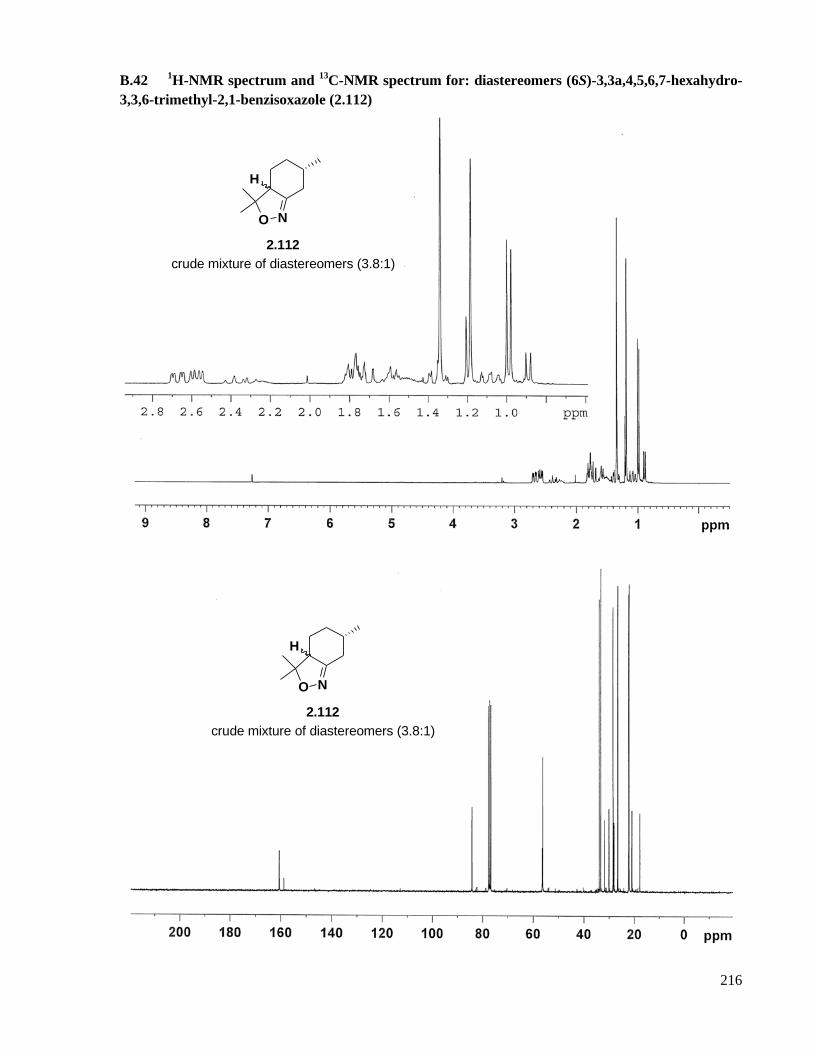
216
B.42 1H-NMR spectrum and
13C-NMR spectrum for: diastereomers (6S)-3,3a,4,5,6,7-hexahydro-
3,3,6-trimethyl-2,1-benzisoxazole (2.112)
NO
H
2.112
crude mixture of diastereomers (3.8:1)
NO
H
2.112
crude mixture of diastereomers (3.8:1)
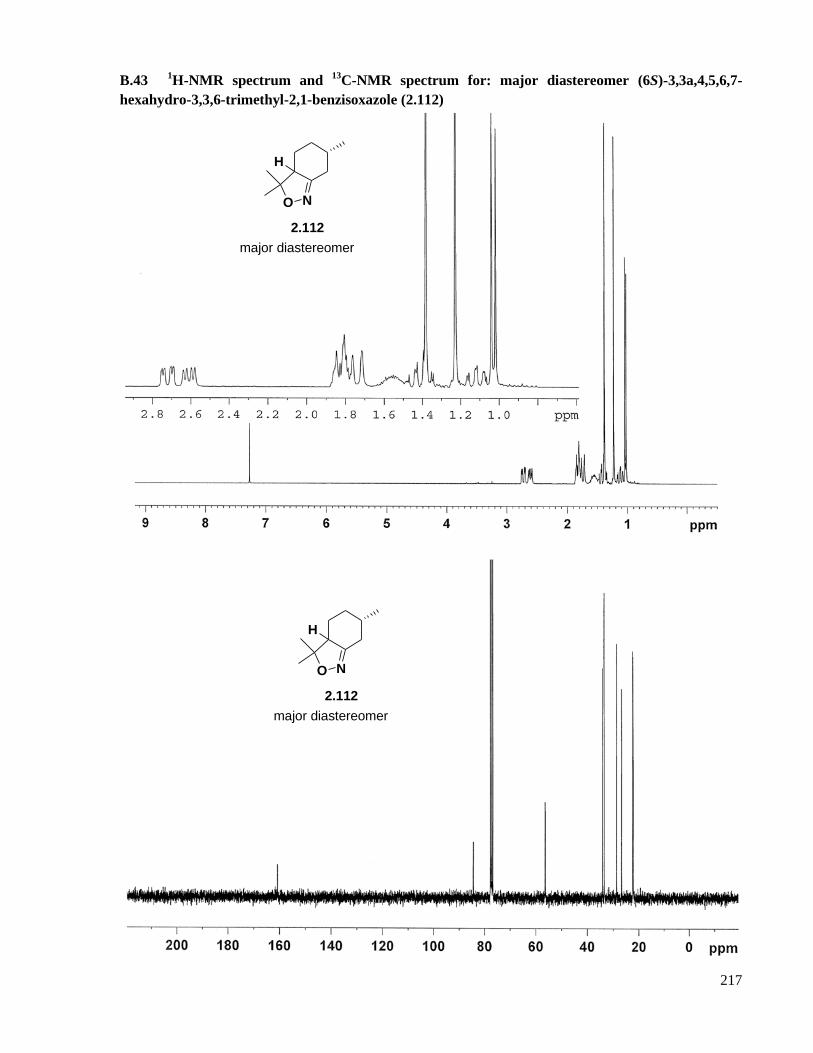
217
B.43 1H-NMR spectrum and
13C-NMR spectrum for: major diastereomer (6S)-3,3a,4,5,6,7-
hexahydro-3,3,6-trimethyl-2,1-benzisoxazole (2.112)
NO
H
2.112
major diastereomer
NO
H
2.112
major diastereomer

218
B.44 1H-NMR spectrum and
13C-NMR spectrum for: N-[(4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-7-
oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-acetamide (2.124)
O
O
NHAc
N
2.124
O
O
NHAc
N
2.124

219
B.45 1H-NMR spectrum and
13C-NMR spectrum for: 4,4a,7a,7b-tetrahydro-4a-methoxy-
indeno[1,7-cd]isoxazol-7(3H)-one (2.125)
O
O
OCH3
N
2.125
O
O
OCH3
N
2.125
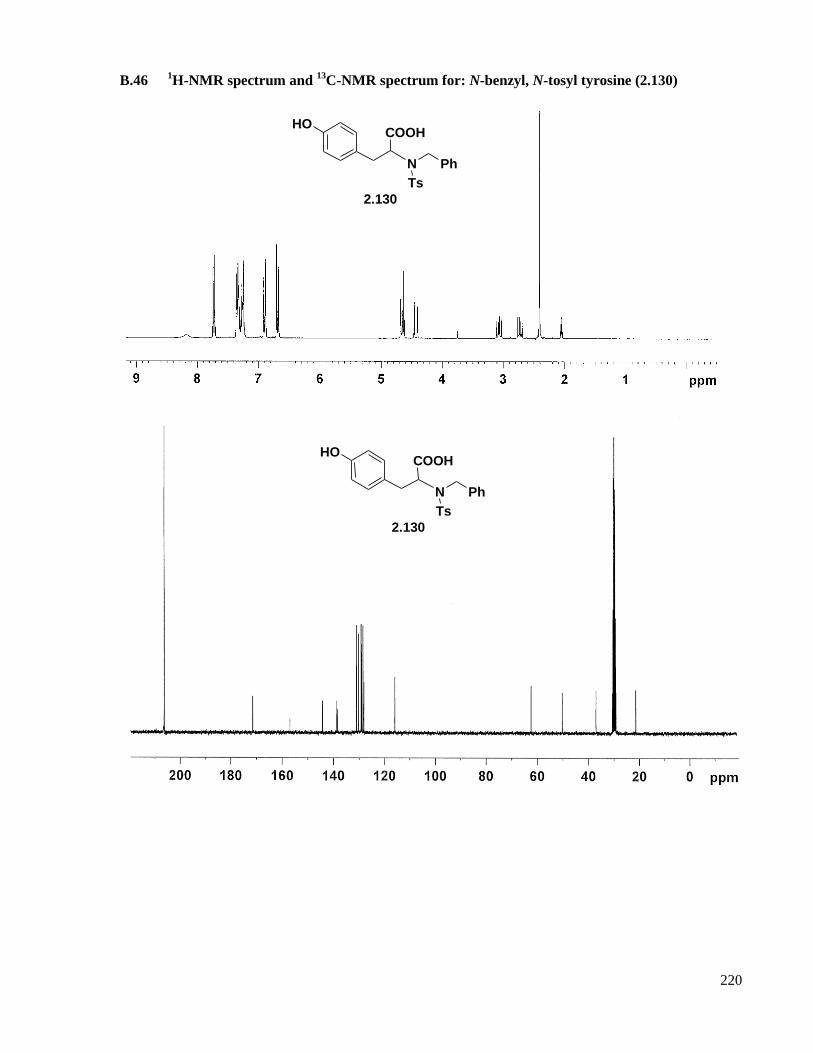
220
B.46 1H-NMR spectrum and
13C-NMR spectrum for: N-benzyl, N-tosyl tyrosine (2.130)
HOCOOH
N
Ts
Ph
2.130
HOCOOH
N
Ts
Ph
2.130
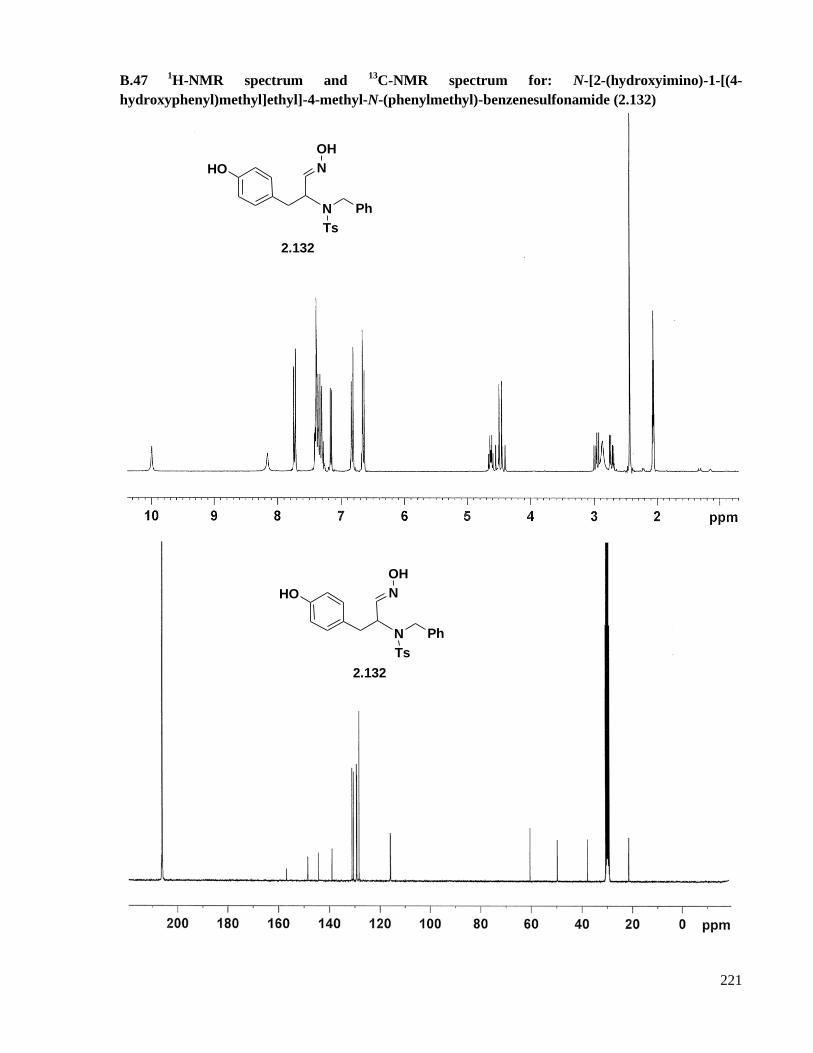
221
B.47 1H-NMR spectrum and
13C-NMR spectrum for: N-[2-(hydroxyimino)-1-[(4-
hydroxyphenyl)methyl]ethyl]-4-methyl-N-(phenylmethyl)-benzenesulfonamide (2.132)
HO N
N Ph
OH
Ts
2.132
HO N
N Ph
OH
Ts
2.132
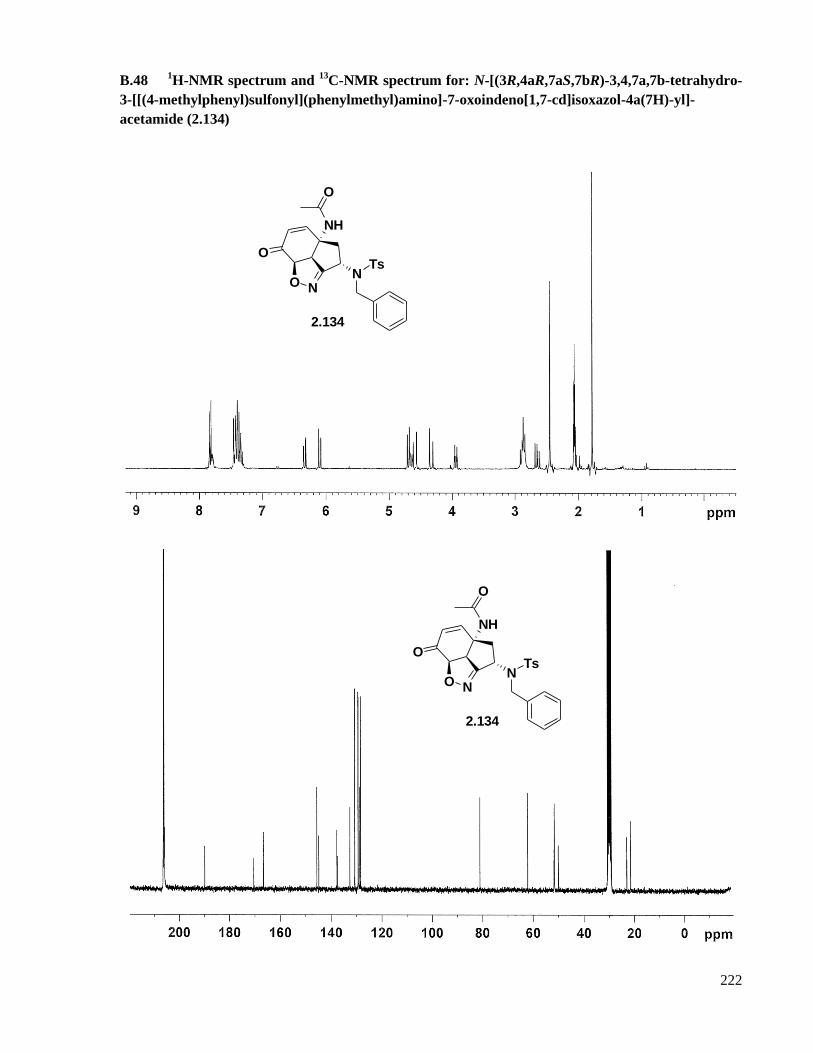
222
B.48 1H-NMR spectrum and
13C-NMR spectrum for: N-[(3R,4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-
3-[[(4-methylphenyl)sulfonyl](phenylmethyl)amino]-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-yl]-
acetamide (2.134)
O
NH
O N
O
NTs
2.134
O
NH
O N
O
NTs
2.134
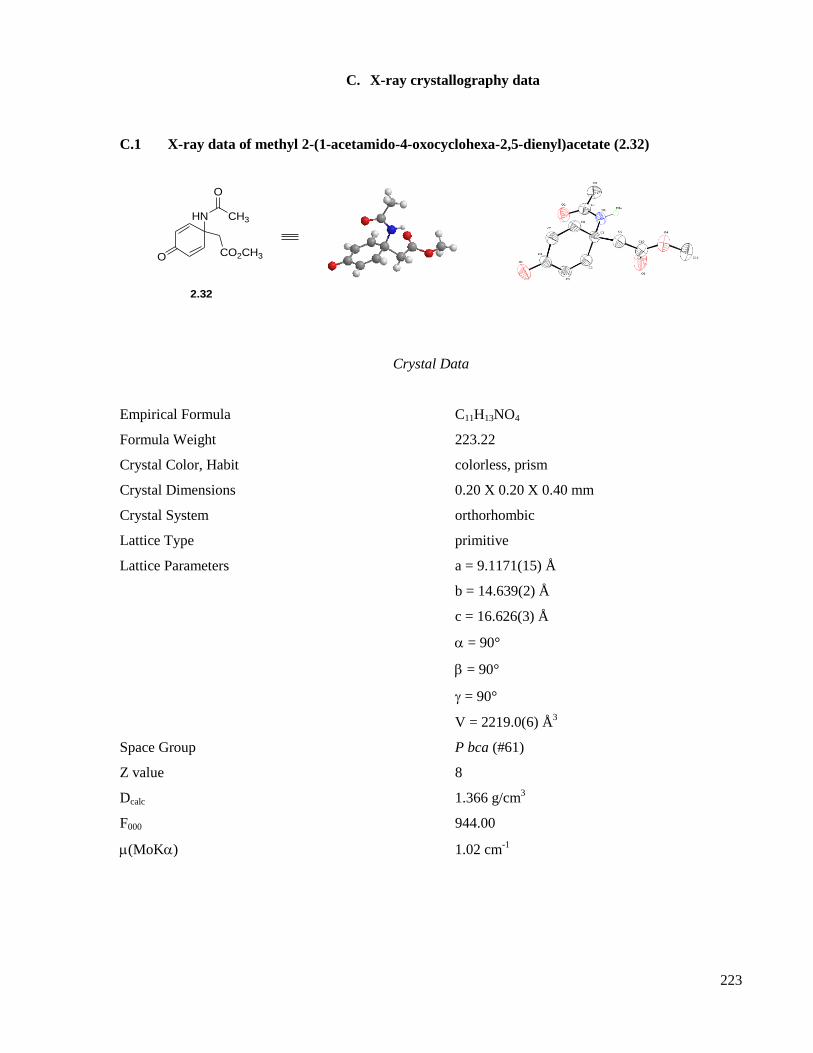
223
C. X-ray crystallography data
C.1 X-ray data of methyl 2-(1-acetamido-4-oxocyclohexa-2,5-dienyl)acetate (2.32)
O
HN CH3
O
CO2CH3
2.32
Crystal Data
Empirical Formula C11H13NO4
Formula Weight 223.22
Crystal Color, Habit colorless, prism
Crystal Dimensions 0.20 X 0.20 X 0.40 mm
Crystal System orthorhombic
Lattice Type primitive
Lattice Parameters a = 9.1171(15) Å
b = 14.639(2) Å
c = 16.626(3) Å
= 90°
= 90°
= 90°
V = 2219.0(6) Å3
Space Group P bca (#61)
Z value 8
Dcalc 1.366 g/cm3
F000 944.00
(MoK) 1.02 cm-1

224
Intensity Measurements
Diffractometer Bruker X8 APEX II
Radiation MoK ( = 0.71073 Å)
graphite monochromated
Data Images 1235 exposures @ 20.0 seconds
Detector Position 36.00 mm
2max 56.0°
No. of Reflections Measured Total: 24567
Unique: 2658 (Rint = 0.024)
Corrections Absorption (Tmin = 0.843, Tmax = 0.980)
Lorentz-polarization
Structure Solution and Refinement
Structure Solution Direct Methods (SIR97)
Refinement Full-matrix least-squares on F2
Function Minimized w (Fo2 – Fc
2)
2
Least Squares Weights w = 1/(2(Fo
2) + (0.0545P)
2 + 0.7113P)
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>0.00(I)) 2658
No. Variables 151
Reflection/Parameter Ratio 17.60
Residuals (refined on F2, all data): R1; wR2 0.059; 0.124
Goodness of Fit Indicator 1.05
No. Observations (I>2.00(I)) 2035
Residuals (refined on F): R1; wR2 0.042; 0.109
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.26 e-/Å
3
Minimum peak in Final Diff. Map -0.23 e-/Å
3

225
Atomic coordinates (x104) and equivalent isotropic displacement parameters
(A2 x 103) for mc010.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
C(1) 8932(2) 4498(1) 8594(1) 43(1)
C(2) 9535(2) 3680(1) 8566(1) 45(1)
C(3) 8715(2) 2875(1) 8315(1) 43(1)
C(4) 7157(2) 3002(1) 8149(1) 44(1)
C(5) 6543(2) 3819(1) 8178(1) 40(1)
C(6) 7334(2) 4672(1) 8428(1) 37(1)
C(7) 6540(2) 5006(1) 9192(1) 44(1)
C(8) 7074(2) 5890(1) 9551(1) 48(1)
C(9) 6339(3) 7110(1) 10383(1) 69(1)
C(10) 7579(2) 5285(1) 7050(1) 43(1)
C(11) 7281(2) 6091(1) 6516(1) 58(1)
N(1) 7151(1) 5385(1) 7820(1) 41(1)
O(1) 9316(1) 2131(1) 8228(1) 60(1)
O(2) 8274(2) 6205(1) 9501(1) 78(1)
O(3) 5988(1) 6284(1) 9952(1) 57(1)
O(4) 8171(1) 4594(1) 6804(1) 59(1)
________________________________________________________________
Bond lengths [A] and angles [deg] for mc010.
_____________________________________________________________
C(1)-C(2) 1.319(2)
C(1)-C(6) 1.5047(19)
C(1)-H(1) 0.9300
C(2)-C(3) 1.456(2)
C(2)-H(2) 0.9300
C(3)-O(1) 1.2275(17)
C(3)-C(4) 1.459(2)
C(4)-C(5) 1.3213(19)
C(4)-H(4) 0.9300
C(5)-C(6) 1.5011(18)
C(5)-H(5) 0.9300
C(6)-N(1) 1.4634(18)
C(6)-C(7) 1.541(2)
C(7)-C(8) 1.506(2)
C(7)-H(7A) 0.9700
C(7)-H(7B) 0.9700
C(8)-O(2) 1.190(2)
C(8)-O(3) 1.326(2)
C(9)-O(3) 1.441(2)
C(9)-H(9A) 0.9600
C(9)-H(9B) 0.9600
C(9)-H(9C) 0.9600
C(10)-O(4) 1.2168(18)
C(10)-N(1) 1.3457(19)
C(10)-C(11) 1.502(2)
C(11)-H(11A) 0.9600
C(11)-H(11B) 0.9600
C(11)-H(11C) 0.9600
N(1)-H(1N) 0.870(18)

226
C(2)-C(1)-C(6) 123.44(13)
C(2)-C(1)-H(1) 118.3
C(6)-C(1)-H(1) 118.3
C(1)-C(2)-C(3) 122.05(13)
C(1)-C(2)-H(2) 119.0
C(3)-C(2)-H(2) 119.0
O(1)-C(3)-C(2) 121.47(14)
O(1)-C(3)-C(4) 121.68(14)
C(2)-C(3)-C(4) 116.83(12)
C(5)-C(4)-C(3) 121.37(13)
C(5)-C(4)-H(4) 119.3
C(3)-C(4)-H(4) 119.3
C(4)-C(5)-C(6) 124.05(13)
C(4)-C(5)-H(5) 118.0
C(6)-C(5)-H(5) 118.0
N(1)-C(6)-C(5) 110.29(12)
N(1)-C(6)-C(1) 110.95(11)
C(5)-C(6)-C(1) 112.01(11)
N(1)-C(6)-C(7) 106.87(11)
C(5)-C(6)-C(7) 105.52(11)
C(1)-C(6)-C(7) 110.94(12)
C(8)-C(7)-C(6) 116.59(12)
C(8)-C(7)-H(7A) 108.1
C(6)-C(7)-H(7A) 108.1
C(8)-C(7)-H(7B) 108.1
C(6)-C(7)-H(7B) 108.1
H(7A)-C(7)-H(7B) 107.3
O(2)-C(8)-O(3) 123.58(15)
O(2)-C(8)-C(7) 127.05(15)
O(3)-C(8)-C(7) 109.36(13)
O(3)-C(9)-H(9A) 109.5
O(3)-C(9)-H(9B) 109.5
H(9A)-C(9)-H(9B) 109.5
O(3)-C(9)-H(9C) 109.5
H(9A)-C(9)-H(9C) 109.5
H(9B)-C(9)-H(9C) 109.5
O(4)-C(10)-N(1) 122.54(14)
O(4)-C(10)-C(11) 122.37(15)
N(1)-C(10)-C(11) 115.08(14)
C(10)-C(11)-H(11A) 109.5
C(10)-C(11)-H(11B) 109.5
H(11A)-C(11)-H(11B) 109.5
C(10)-C(11)-H(11C) 109.5
H(11A)-C(11)-H(11C) 109.5
H(11B)-C(11)-H(11C) 109.5
C(10)-N(1)-C(6) 123.18(12)
C(10)-N(1)-H(1N) 117.2(11)
C(6)-N(1)-H(1N) 119.6(11)
C(8)-O(3)-C(9) 116.70(15)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
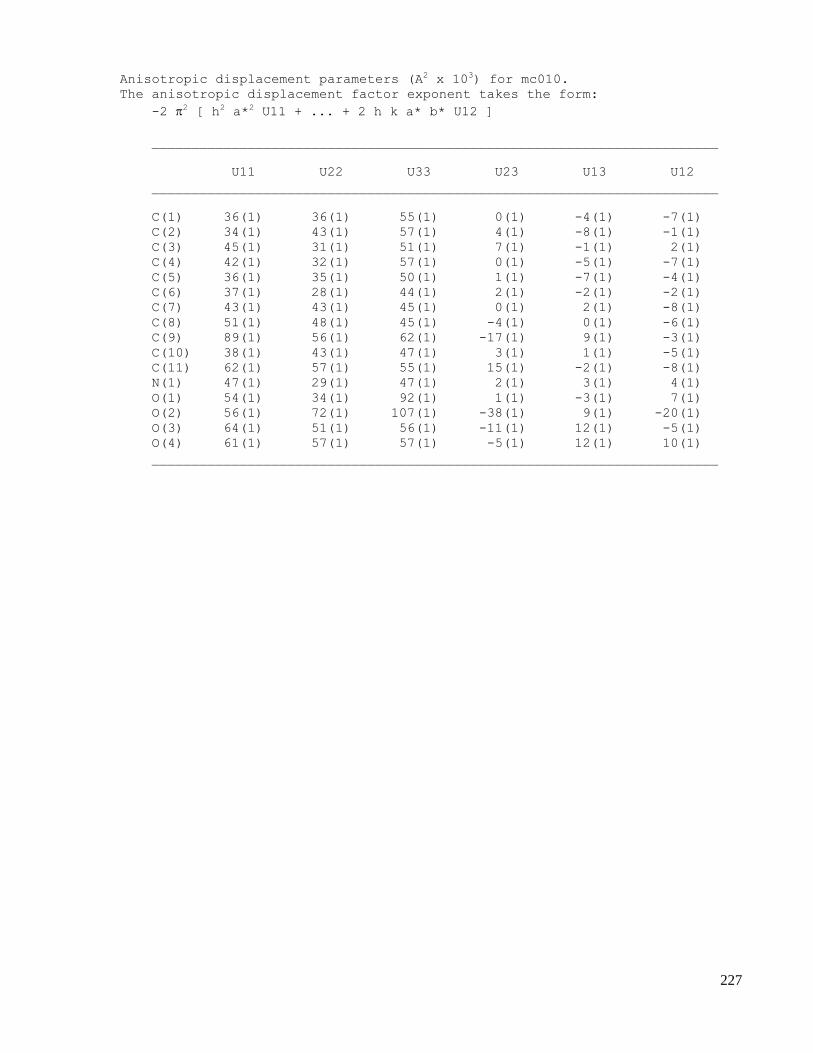
227
Anisotropic displacement parameters (A2 x 103) for mc010.
The anisotropic displacement factor exponent takes the form:
-2 2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
C(1) 36(1) 36(1) 55(1) 0(1) -4(1) -7(1)
C(2) 34(1) 43(1) 57(1) 4(1) -8(1) -1(1)
C(3) 45(1) 31(1) 51(1) 7(1) -1(1) 2(1)
C(4) 42(1) 32(1) 57(1) 0(1) -5(1) -7(1)
C(5) 36(1) 35(1) 50(1) 1(1) -7(1) -4(1)
C(6) 37(1) 28(1) 44(1) 2(1) -2(1) -2(1)
C(7) 43(1) 43(1) 45(1) 0(1) 2(1) -8(1)
C(8) 51(1) 48(1) 45(1) -4(1) 0(1) -6(1)
C(9) 89(1) 56(1) 62(1) -17(1) 9(1) -3(1)
C(10) 38(1) 43(1) 47(1) 3(1) 1(1) -5(1)
C(11) 62(1) 57(1) 55(1) 15(1) -2(1) -8(1)
N(1) 47(1) 29(1) 47(1) 2(1) 3(1) 4(1)
O(1) 54(1) 34(1) 92(1) 1(1) -3(1) 7(1)
O(2) 56(1) 72(1) 107(1) -38(1) 9(1) -20(1)
O(3) 64(1) 51(1) 56(1) -11(1) 12(1) -5(1)
O(4) 61(1) 57(1) 57(1) -5(1) 12(1) 10(1)
_______________________________________________________________________
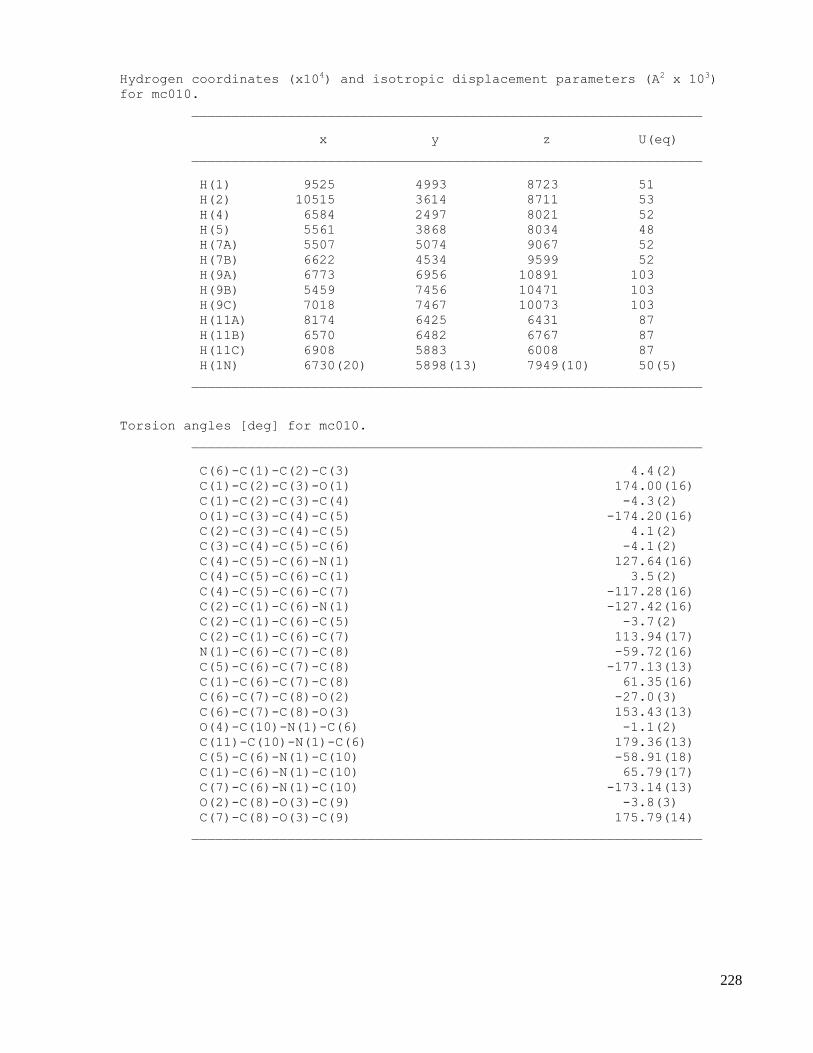
228
Hydrogen coordinates (x104) and isotropic displacement parameters (A2 x 103)
for mc010.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(1) 9525 4993 8723 51
H(2) 10515 3614 8711 53
H(4) 6584 2497 8021 52
H(5) 5561 3868 8034 48
H(7A) 5507 5074 9067 52
H(7B) 6622 4534 9599 52
H(9A) 6773 6956 10891 103
H(9B) 5459 7456 10471 103
H(9C) 7018 7467 10073 103
H(11A) 8174 6425 6431 87
H(11B) 6570 6482 6767 87
H(11C) 6908 5883 6008 87
H(1N) 6730(20) 5898(13) 7949(10) 50(5)
________________________________________________________________
Torsion angles [deg] for mc010.
________________________________________________________________
C(6)-C(1)-C(2)-C(3) 4.4(2)
C(1)-C(2)-C(3)-O(1) 174.00(16)
C(1)-C(2)-C(3)-C(4) -4.3(2)
O(1)-C(3)-C(4)-C(5) -174.20(16)
C(2)-C(3)-C(4)-C(5) 4.1(2)
C(3)-C(4)-C(5)-C(6) -4.1(2)
C(4)-C(5)-C(6)-N(1) 127.64(16)
C(4)-C(5)-C(6)-C(1) 3.5(2)
C(4)-C(5)-C(6)-C(7) -117.28(16)
C(2)-C(1)-C(6)-N(1) -127.42(16)
C(2)-C(1)-C(6)-C(5) -3.7(2)
C(2)-C(1)-C(6)-C(7) 113.94(17)
N(1)-C(6)-C(7)-C(8) -59.72(16)
C(5)-C(6)-C(7)-C(8) -177.13(13)
C(1)-C(6)-C(7)-C(8) 61.35(16)
C(6)-C(7)-C(8)-O(2) -27.0(3)
C(6)-C(7)-C(8)-O(3) 153.43(13)
O(4)-C(10)-N(1)-C(6) -1.1(2)
C(11)-C(10)-N(1)-C(6) 179.36(13)
C(5)-C(6)-N(1)-C(10) -58.91(18)
C(1)-C(6)-N(1)-C(10) 65.79(17)
C(7)-C(6)-N(1)-C(10) -173.14(13)
O(2)-C(8)-O(3)-C(9) -3.8(3)
C(7)-C(8)-O(3)-C(9) 175.79(14)
________________________________________________________________

229
C.2 X-ray data of N-((3aS,7aS,E)-2-(nitromethylene)-2,3,3a,7a-tetrahydrobenzofuran-3a-
yl)acetamide (2.44)
OH
NHAc
2.44
NO2
Crystal Data
Empirical Formula C11H12N2O4
Formula Weight 236.23
Crystal Color, Habit colorless, needle
Crystal Dimensions 0.06 X 0.08 X 0.35 mm
Crystal System monoclinic
Lattice Type primitive
Lattice Parameters a = 8.6012(3) Å
b = 9.6959(4) Å
c = 13.3029(5) Å
= 90.0°
= 96.914(2)°
= 90.0°
V = 1101.35(7) Å3
Space Group P 21/c (#14)
Z value 4
Dcalc 1.425 g/cm3
F000 496.00
(MoK) 1.10 cm-1

230
Intensity Measurements
Diffractometer Bruker X8 APEX II
Radiation MoK ( = 0.71073 Å)
graphite monochromated
Data Images 739 exposures @ 30.0 seconds
Detector Position 36.00 mm
2max 50.0°
No. of Reflections Measured Total: 7510
Unique: 1936 (Rint = 0.033)
Corrections Absorption (Tmin = 0.916, Tmax = 0.993)
Lorentz-polarization
Structure Solution and Refinement
Structure Solution Direct Methods (SIR97)
Refinement Full-matrix least-squares on F2
Function Minimized w (Fo2 – Fc
2)
2
Least Squares Weights w = 1/(2(Fo
2) + (0.0486P)
2 + 0.3430P)
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>0.00(I)) 1936
No. Variables 159
Reflection/Parameter Ratio 12.18
Residuals (refined on F2, all data): R1; wR2 0.056; 0.101
Goodness of Fit Indicator 1.02
No. Observations (I>2.00(I)) 1479
Residuals (refined on F): R1; wR2 0.038; 0.93
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.19 e-/Å
3
Minimum peak in Final Diff. Map -0.15 e-/Å
3

231
Atomic coordinates (x104) and equivalent isotropic displacement parameters
(A2 x 103) for mc013.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
C(1) 8669(2) 1754(2) 9456(1) 35(1)
C(2) 7705(3) 649(2) 9859(2) 47(1)
C(3) 6566(3) 21(2) 9273(2) 54(1)
C(4) 6156(2) 394(2) 8226(2) 51(1)
C(5) 6837(2) 1449(2) 7816(2) 40(1)
C(6) 7981(2) 2369(2) 8440(1) 29(1)
C(7) 7095(2) 3656(2) 8768(1) 31(1)
C(8) 7871(2) 3970(2) 9800(1) 30(1)
C(9) 7764(2) 5075(2) 10401(1) 34(1)
C(10) 10253(2) 1914(2) 7539(1) 29(1)
C(11) 11505(2) 2509(2) 6980(2) 39(1)
N(1) 9248(2) 2811(2) 7876(1) 30(1)
N(2) 6732(2) 6166(2) 10131(1) 35(1)
O(1) 6758(2) 7153(2) 10727(1) 57(1)
O(2) 5814(2) 6126(1) 9346(1) 40(1)
O(3) 8813(2) 2941(1) 10157(1) 42(1)
O(4) 10154(2) 661(1) 7692(1) 36(1)
________________________________________________________________
Bond lengths [A] and angles [deg] for mc013.
_____________________________________________________________
C(1)-O(3) 1.478(2)
C(1)-C(2) 1.493(3)
C(1)-C(6) 1.530(3)
C(1)-H(1) 1.0000
C(2)-C(3) 1.324(3)
C(2)-H(2) 0.9500
C(3)-C(4) 1.441(4)
C(3)-H(3) 0.9500
C(4)-C(5) 1.328(3)
C(4)-H(4) 0.9500
C(5)-C(6) 1.502(3)
C(5)-H(5) 0.9500
C(6)-N(1) 1.460(2)
C(6)-C(7) 1.551(2)
C(7)-C(8) 1.484(3)
C(7)-H(7A) 0.9900
C(7)-H(7B) 0.9900
C(8)-O(3) 1.336(2)
C(8)-C(9) 1.347(3)
C(9)-N(2) 1.401(2)
C(9)-H(9) 0.9500
C(10)-O(4) 1.237(2)
C(10)-N(1) 1.341(2)
C(10)-C(11) 1.496(3)
C(11)-H(11A) 0.9800
C(11)-H(11B) 0.9800
C(11)-H(11C) 0.9800

232
N(1)-H(1N) 0.80(2)
N(2)-O(2) 1.232(2)
N(2)-O(1) 1.241(2)
O(3)-C(1)-C(2) 109.90(16)
O(3)-C(1)-C(6) 104.35(14)
C(2)-C(1)-C(6) 115.07(17)
O(3)-C(1)-H(1) 109.1
C(2)-C(1)-H(1) 109.1
C(6)-C(1)-H(1) 109.1
C(3)-C(2)-C(1) 121.5(2)
C(3)-C(2)-H(2) 119.2
C(1)-C(2)-H(2) 119.2
C(2)-C(3)-C(4) 122.1(2)
C(2)-C(3)-H(3) 119.0
C(4)-C(3)-H(3) 119.0
C(5)-C(4)-C(3) 121.4(2)
C(5)-C(4)-H(4) 119.3
C(3)-C(4)-H(4) 119.3
C(4)-C(5)-C(6) 121.5(2)
C(4)-C(5)-H(5) 119.3
C(6)-C(5)-H(5) 119.3
N(1)-C(6)-C(5) 111.75(15)
N(1)-C(6)-C(1) 109.55(15)
C(5)-C(6)-C(1) 114.50(16)
N(1)-C(6)-C(7) 109.28(14)
C(5)-C(6)-C(7) 108.78(14)
C(1)-C(6)-C(7) 102.51(14)
C(8)-C(7)-C(6) 104.11(14)
C(8)-C(7)-H(7A) 110.9
C(6)-C(7)-H(7A) 110.9
C(8)-C(7)-H(7B) 110.9
C(6)-C(7)-H(7B) 110.9
H(7A)-C(7)-H(7B) 109.0
O(3)-C(8)-C(9) 117.63(17)
O(3)-C(8)-C(7) 111.22(15)
C(9)-C(8)-C(7) 131.15(17)
C(8)-C(9)-N(2) 122.34(18)
C(8)-C(9)-H(9) 118.8
N(2)-C(9)-H(9) 118.8
O(4)-C(10)-N(1) 121.50(17)
O(4)-C(10)-C(11) 121.89(16)
N(1)-C(10)-C(11) 116.61(16)
C(10)-C(11)-H(11A) 109.5
C(10)-C(11)-H(11B) 109.5
H(11A)-C(11)-H(11B) 109.5
C(10)-C(11)-H(11C) 109.5
H(11A)-C(11)-H(11C) 109.5
H(11B)-C(11)-H(11C) 109.5
C(10)-N(1)-C(6) 122.18(16)
C(10)-N(1)-H(1N) 119.9(15)
C(6)-N(1)-H(1) 117.8(15)
O(2)-N(2)-O(1) 121.80(17)
O(2)-N(2)-C(9) 121.03(16)
O(1)-N(2)-C(9) 117.15(17)
C(8)-O(3)-C(1) 110.85(14)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
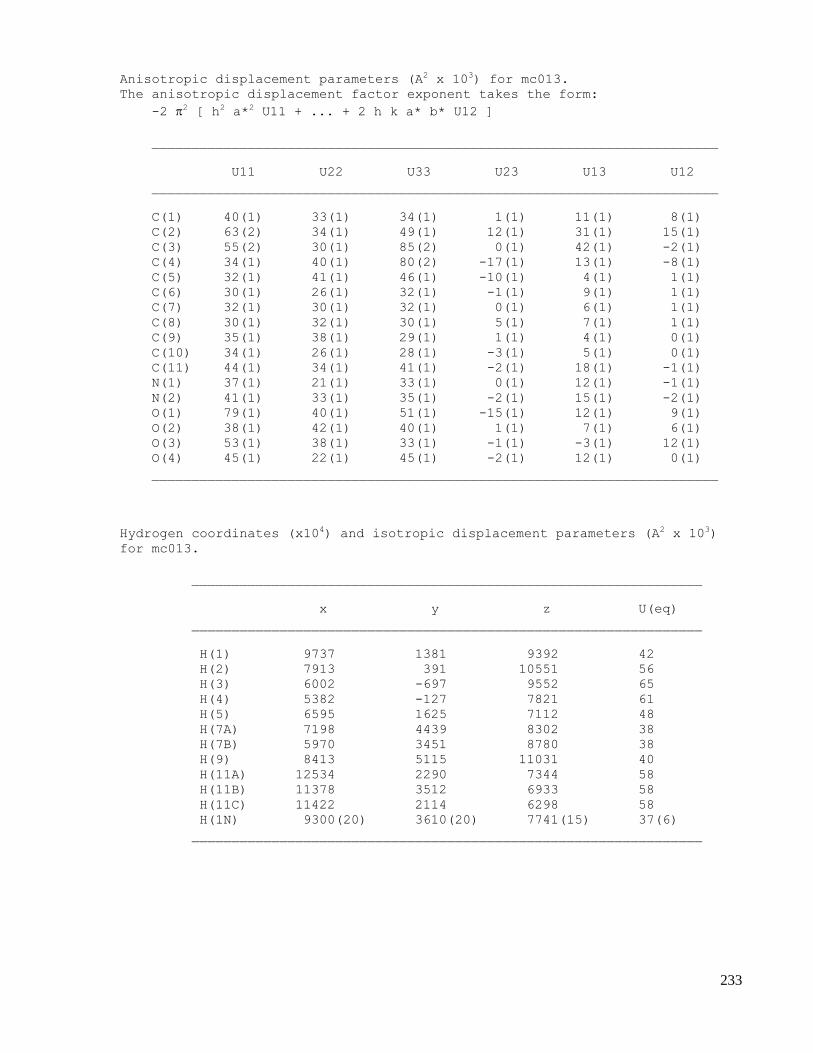
233
Anisotropic displacement parameters (A2 x 103) for mc013.
The anisotropic displacement factor exponent takes the form:
-2 2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
C(1) 40(1) 33(1) 34(1) 1(1) 11(1) 8(1)
C(2) 63(2) 34(1) 49(1) 12(1) 31(1) 15(1)
C(3) 55(2) 30(1) 85(2) 0(1) 42(1) -2(1)
C(4) 34(1) 40(1) 80(2) -17(1) 13(1) -8(1)
C(5) 32(1) 41(1) 46(1) -10(1) 4(1) 1(1)
C(6) 30(1) 26(1) 32(1) -1(1) 9(1) 1(1)
C(7) 32(1) 30(1) 32(1) 0(1) 6(1) 1(1)
C(8) 30(1) 32(1) 30(1) 5(1) 7(1) 1(1)
C(9) 35(1) 38(1) 29(1) 1(1) 4(1) 0(1)
C(10) 34(1) 26(1) 28(1) -3(1) 5(1) 0(1)
C(11) 44(1) 34(1) 41(1) -2(1) 18(1) -1(1)
N(1) 37(1) 21(1) 33(1) 0(1) 12(1) -1(1)
N(2) 41(1) 33(1) 35(1) -2(1) 15(1) -2(1)
O(1) 79(1) 40(1) 51(1) -15(1) 12(1) 9(1)
O(2) 38(1) 42(1) 40(1) 1(1) 7(1) 6(1)
O(3) 53(1) 38(1) 33(1) -1(1) -3(1) 12(1)
O(4) 45(1) 22(1) 45(1) -2(1) 12(1) 0(1)
_______________________________________________________________________
Hydrogen coordinates (x104) and isotropic displacement parameters (A2 x 103)
for mc013.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(1) 9737 1381 9392 42
H(2) 7913 391 10551 56
H(3) 6002 -697 9552 65
H(4) 5382 -127 7821 61
H(5) 6595 1625 7112 48
H(7A) 7198 4439 8302 38
H(7B) 5970 3451 8780 38
H(9) 8413 5115 11031 40
H(11A) 12534 2290 7344 58
H(11B) 11378 3512 6933 58
H(11C) 11422 2114 6298 58
H(1N) 9300(20) 3610(20) 7741(15) 37(6)
________________________________________________________________

234
Torsion angles [deg] for mc013.
________________________________________________________________
O(3)-C(1)-C(2)-C(3) 133.5(2)
C(6)-C(1)-C(2)-C(3) 16.1(3)
C(1)-C(2)-C(3)-C(4) -2.1(3)
C(2)-C(3)-C(4)-C(5) -4.4(3)
C(3)-C(4)-C(5)-C(6) -4.5(3)
C(4)-C(5)-C(6)-N(1) 143.55(19)
C(4)-C(5)-C(6)-C(1) 18.3(3)
C(4)-C(5)-C(6)-C(7) -95.7(2)
O(3)-C(1)-C(6)-N(1) 90.11(17)
C(2)-C(1)-C(6)-N(1) -149.40(16)
O(3)-C(1)-C(6)-C(5) -143.46(15)
C(2)-C(1)-C(6)-C(5) -23.0(2)
O(3)-C(1)-C(6)-C(7) -25.84(17)
C(2)-C(1)-C(6)-C(7) 94.66(17)
N(1)-C(6)-C(7)-C(8) -93.40(17)
C(5)-C(6)-C(7)-C(8) 144.36(16)
C(1)-C(6)-C(7)-C(8) 22.75(17)
C(6)-C(7)-C(8)-O(3) -11.50(19)
C(6)-C(7)-C(8)-C(9) 169.02(19)
O(3)-C(8)-C(9)-N(2) -175.46(16)
C(7)-C(8)-C(9)-N(2) 4.0(3)
O(4)-C(10)-N(1)-C(6) -0.1(3)
C(11)-C(10)-N(1)-C(6) -179.44(16)
C(5)-C(6)-N(1)-C(10) -62.6(2)
C(1)-C(6)-N(1)-C(10) 65.4(2)
C(7)-C(6)-N(1)-C(10) 176.99(16)
C(8)-C(9)-N(2)-O(2) 3.7(3)
C(8)-C(9)-N(2)-O(1) -178.00(18)
C(9)-C(8)-O(3)-C(1) 173.94(16)
C(7)-C(8)-O(3)-C(1) -5.6(2)
C(2)-C(1)-O(3)-C(8) -103.34(18)
C(6)-C(1)-O(3)-C(8) 20.56(19)
________________________________________________________________
Symmetry transformations used to generate equivalent atoms:
Hydrogen Bonds
Donor --- H....Acceptor [ ARU ] D - H H...A D...A D -
H...A
-----------------------------------------------------------------------------
N1 --H1N ..O4 [ 2756.01] 0.807(19) 2.130(19) 2.926(2)
169.5(18)
Translation of ARU-code to Equivalent Position Code
===================================================
[ 2756. ] = 2-x,1/2+y,3/2-z
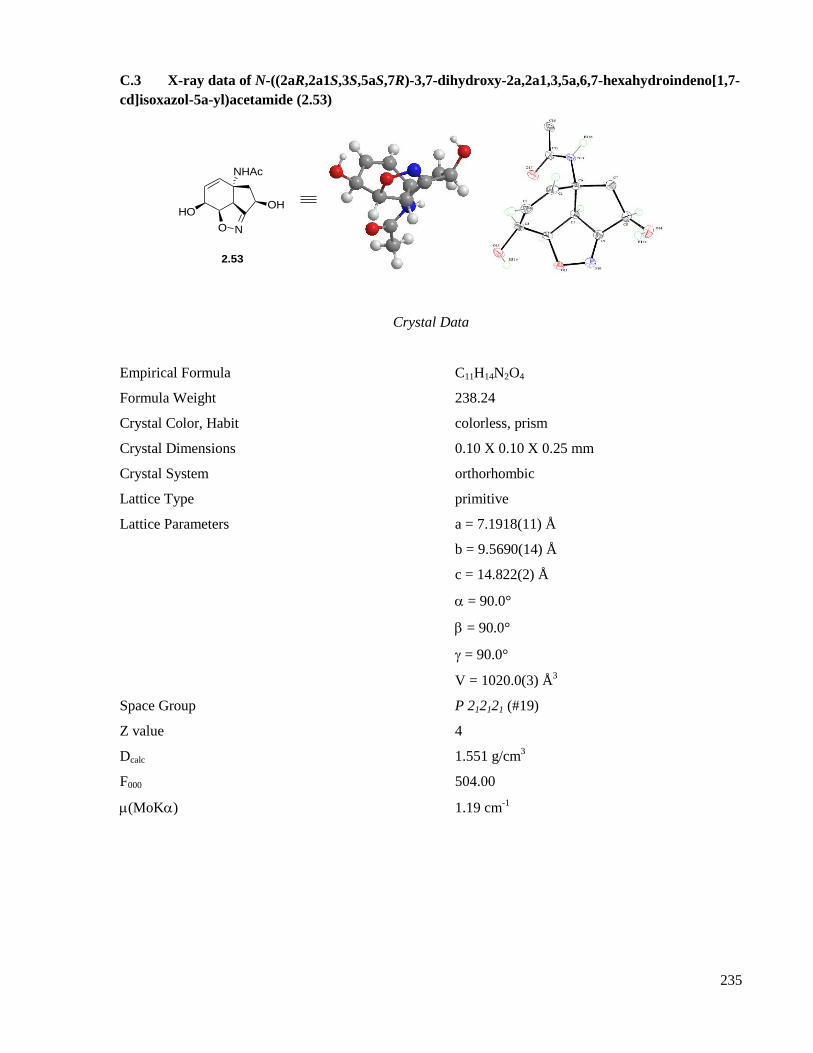
235
C.3 X-ray data of N-((2aR,2a1S,3S,5aS,7R)-3,7-dihydroxy-2a,2a1,3,5a,6,7-hexahydroindeno[1,7-
cd]isoxazol-5a-yl)acetamide (2.53)
HO
O N
OH
NHAc
2.53
Crystal Data
Empirical Formula C11H14N2O4
Formula Weight 238.24
Crystal Color, Habit colorless, prism
Crystal Dimensions 0.10 X 0.10 X 0.25 mm
Crystal System orthorhombic
Lattice Type primitive
Lattice Parameters a = 7.1918(11) Å
b = 9.5690(14) Å
c = 14.822(2) Å
= 90.0°
= 90.0°
= 90.0°
V = 1020.0(3) Å3
Space Group P 212121 (#19)
Z value 4
Dcalc 1.551 g/cm3
F000 504.00
(MoK) 1.19 cm-1

236
Intensity Measurements
Diffractometer Bruker X8 APEX II
Radiation MoK ( = 0.71073 Å)
graphite monochromated
Data Images 1107 exposures @ 10.0 seconds
Detector Position 36.00 mm
2max 56.2°
No. of Reflections Measured Total: 10660
Unique: 2485 (Rint = 0.036)
Corrections Absorption (Tmin = 0.790, Tmax = 0.988)
Lorentz-polarization
Structure Solution and Refinement
Structure Solution Direct Methods (SIR97)
Refinement Full-matrix least-squares on F2
Function Minimized w (Fo2 – Fc
2)
2
Least Squares Weights w = 1/(2(Fo
2) + (0.0367P)
2 + 0.2803P)
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>0.00(I)) 2485
No. Variables 167
Reflection/Parameter Ratio 14.88
Residuals (refined on F2, all data): R1; wR2 0.049; 0.083
Goodness of Fit Indicator 1.03
No. Observations (I>2.00(I)) 2108
Residuals (refined on F): R1; wR2 0.037; 0.078
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.25 e-/Å
3
Minimum peak in Final Diff. Map -0.19 e-/Å
3

237
Atomic coordinates (x104) and equivalent isotropic displacement parameters
(A2 x 103) for mc009.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
C(1) 4914(3) 637(2) 6160(1) 20(1)
C(2) 4089(3) 298(2) 6920(1) 21(1)
C(3) 4957(3) -590(2) 7627(1) 19(1)
C(4) 7042(2) -397(2) 7680(1) 18(1)
C(5) 8007(2) -70(2) 6791(1) 16(1)
C(6) 6865(2) 230(2) 5923(1) 17(1)
C(7) 7885(3) 1478(2) 5473(1) 24(1)
C(8) 9314(3) 2080(2) 6140(1) 20(1)
C(9) 8748(2) 1342(2) 6981(1) 18(1)
C(15) 6673(2) -2263(2) 5523(1) 18(1)
C(16) 6826(3) -3320(2) 4785(1) 25(1)
N(10) 8404(2) 1848(2) 7746(1) 21(1)
N(13) 6891(2) -939(2) 5286(1) 19(1)
O(11) 7435(2) 821(1) 8249(1) 22(1)
O(12) 4112(2) -378(1) 8485(1) 23(1)
O(14) 9386(2) 3530(2) 6173(1) 25(1)
O(17) 6397(2) -2624(1) 6303(1) 25(1)
________________________________________________________________

238
Bond lengths [A] and angles [deg] for mc009.
_____________________________________________________________
C(1)-C(2) 1.314(3)
C(1)-C(6) 1.497(3)
C(1)-H(1) 0.9500
C(2)-C(3) 1.486(2)
C(2)-H(2) 0.9500
C(3)-O(12) 1.425(2)
C(3)-C(4) 1.513(3)
C(3)-H(3) 1.0000
C(4)-O(11) 1.465(2)
C(4)-C(5) 1.522(2)
C(4)-H(4) 1.0000
C(5)-C(9) 1.479(2)
C(5)-C(6) 1.553(2)
C(5)-H(5) 1.0000
C(6)-N(13) 1.464(2)
C(6)-C(7) 1.553(2)
C(7)-C(8) 1.538(2)
C(7)-H(7A) 0.9900
C(7)-H(7B) 0.9900
C(8)-O(14) 1.389(2)
C(8)-C(9) 1.489(2)
C(8)-H(8) 1.0000
C(9)-N(10) 1.259(2)
C(15)-O(17) 1.223(2)
C(15)-N(13) 1.324(2)
C(15)-C(16) 1.494(2)
C(16)-H(16A) 0.9800
C(16)-H(16B) 0.9800
C(16)-H(16C) 0.9800
N(10)-O(11) 1.416(2)
N(13)-H(13N) 0.84(2)
O(12)-H(12O) 0.87(3)
O(14)-H(14O) 0.80(3)
C(2)-C(1)-C(6) 124.04(16)
C(2)-C(1)-H(1) 118.0
C(6)-C(1)-H(1) 118.0
C(1)-C(2)-C(3) 123.73(17)
C(1)-C(2)-H(2) 118.1
C(3)-C(2)-H(2) 118.1
O(12)-C(3)-C(2) 111.63(15)
O(12)-C(3)-C(4) 111.03(14)
C(2)-C(3)-C(4) 112.61(15)
O(12)-C(3)-H(3) 107.1
C(2)-C(3)-H(3) 107.1
C(4)-C(3)-H(3) 107.1
O(11)-C(4)-C(3) 108.54(14)
O(11)-C(4)-C(5) 104.28(14)
C(3)-C(4)-C(5) 115.53(14)
O(11)-C(4)-H(4) 109.4
C(3)-C(4)-H(4) 109.4
C(5)-C(4)-H(4) 109.4
C(9)-C(5)-C(4) 100.79(14)
C(9)-C(5)-C(6) 100.33(13)
C(4)-C(5)-C(6) 120.94(14)
C(9)-C(5)-H(5) 111.1

239
C(4)-C(5)-H(5) 111.1
C(6)-C(5)-H(5) 111.1
N(13)-C(6)-C(1) 111.24(14)
N(13)-C(6)-C(5) 112.68(14)
C(1)-C(6)-C(5) 110.46(13)
N(13)-C(6)-C(7) 107.73(14)
C(1)-C(6)-C(7) 110.08(15)
C(5)-C(6)-C(7) 104.38(14)
C(8)-C(7)-C(6) 109.11(14)
C(8)-C(7)-H(7A) 109.9
C(6)-C(7)-H(7A) 109.9
C(8)-C(7)-H(7B) 109.9
C(6)-C(7)-H(7B) 109.9
H(7A)-C(7)-H(7B) 108.3
O(14)-C(8)-C(9) 117.01(15)
O(14)-C(8)-C(7) 114.94(16)
C(9)-C(8)-C(7) 100.26(14)
O(14)-C(8)-H(8) 108.0
C(9)-C(8)-H(8) 108.0
C(7)-C(8)-H(8) 108.0
N(10)-C(9)-C(5) 116.89(16)
N(10)-C(9)-C(8) 128.73(17)
C(5)-C(9)-C(8) 111.87(14)
O(17)-C(15)-N(13) 122.65(16)
O(17)-C(15)-C(16) 120.88(17)
N(13)-C(15)-C(16) 116.46(16)
C(15)-C(16)-H(16A) 109.5
C(15)-C(16)-H(16B) 109.5
H(16A)-C(16)-H(16B) 109.5
C(15)-C(16)-H(16C) 109.5
H(16A)-C(16)-H(16C) 109.5
H(16B)-C(16)-H(16C) 109.5
C(9)-N(10)-O(11) 107.66(14)
C(15)-N(13)-C(6) 124.01(15)
C(15)-N(13)-H(13N) 121.6(15)
C(6)-N(13)-H(13N) 114.4(15)
N(10)-O(11)-C(4) 110.17(12)
C(3)-O(12)-H(12O) 106.5(16)
C(8)-O(14)-H(14O) 106.0(19)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
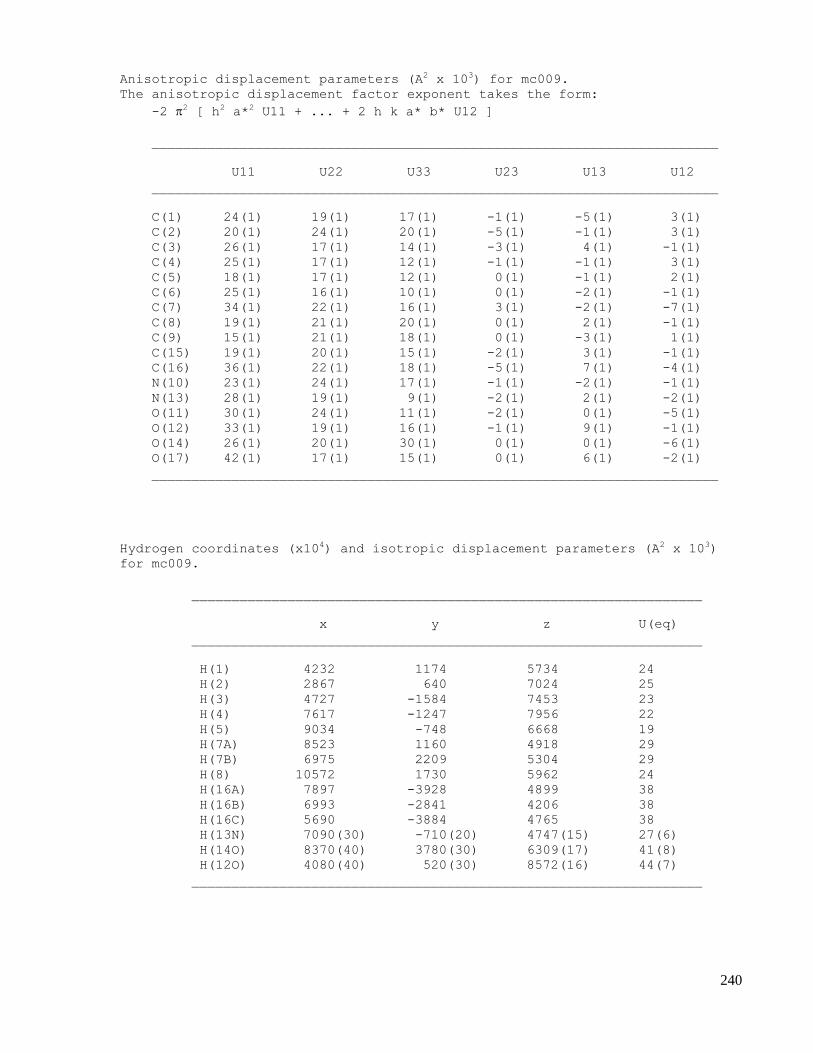
240
Anisotropic displacement parameters (A2 x 103) for mc009.
The anisotropic displacement factor exponent takes the form:
-2 2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
C(1) 24(1) 19(1) 17(1) -1(1) -5(1) 3(1)
C(2) 20(1) 24(1) 20(1) -5(1) -1(1) 3(1)
C(3) 26(1) 17(1) 14(1) -3(1) 4(1) -1(1)
C(4) 25(1) 17(1) 12(1) -1(1) -1(1) 3(1)
C(5) 18(1) 17(1) 12(1) 0(1) -1(1) 2(1)
C(6) 25(1) 16(1) 10(1) 0(1) -2(1) -1(1)
C(7) 34(1) 22(1) 16(1) 3(1) -2(1) -7(1)
C(8) 19(1) 21(1) 20(1) 0(1) 2(1) -1(1)
C(9) 15(1) 21(1) 18(1) 0(1) -3(1) 1(1)
C(15) 19(1) 20(1) 15(1) -2(1) 3(1) -1(1)
C(16) 36(1) 22(1) 18(1) -5(1) 7(1) -4(1)
N(10) 23(1) 24(1) 17(1) -1(1) -2(1) -1(1)
N(13) 28(1) 19(1) 9(1) -2(1) 2(1) -2(1)
O(11) 30(1) 24(1) 11(1) -2(1) 0(1) -5(1)
O(12) 33(1) 19(1) 16(1) -1(1) 9(1) -1(1)
O(14) 26(1) 20(1) 30(1) 0(1) 0(1) -6(1)
O(17) 42(1) 17(1) 15(1) 0(1) 6(1) -2(1)
_______________________________________________________________________
Hydrogen coordinates (x104) and isotropic displacement parameters (A2 x 103)
for mc009.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(1) 4232 1174 5734 24
H(2) 2867 640 7024 25
H(3) 4727 -1584 7453 23
H(4) 7617 -1247 7956 22
H(5) 9034 -748 6668 19
H(7A) 8523 1160 4918 29
H(7B) 6975 2209 5304 29
H(8) 10572 1730 5962 24
H(16A) 7897 -3928 4899 38
H(16B) 6993 -2841 4206 38
H(16C) 5690 -3884 4765 38
H(13N) 7090(30) -710(20) 4747(15) 27(6)
H(14O) 8370(40) 3780(30) 6309(17) 41(8)
H(12O) 4080(40) 520(30) 8572(16) 44(7)
________________________________________________________________

241
Torsion angles [deg] for mc009.
________________________________________________________________
C(6)-C(1)-C(2)-C(3) -2.9(3)
C(1)-C(2)-C(3)-O(12) 158.39(17)
C(1)-C(2)-C(3)-C(4) 32.7(2)
O(12)-C(3)-C(4)-O(11) -41.97(19)
C(2)-C(3)-C(4)-O(11) 84.04(17)
O(12)-C(3)-C(4)-C(5) -158.61(14)
C(2)-C(3)-C(4)-C(5) -32.6(2)
O(11)-C(4)-C(5)-C(9) -2.36(16)
C(3)-C(4)-C(5)-C(9) 116.66(16)
O(11)-C(4)-C(5)-C(6) -111.44(16)
C(3)-C(4)-C(5)-C(6) 7.6(2)
C(2)-C(1)-C(6)-N(13) 102.5(2)
C(2)-C(1)-C(6)-C(5) -23.4(2)
C(2)-C(1)-C(6)-C(7) -138.15(18)
C(9)-C(5)-C(6)-N(13) 145.34(14)
C(4)-C(5)-C(6)-N(13) -105.33(18)
C(9)-C(5)-C(6)-C(1) -89.55(16)
C(4)-C(5)-C(6)-C(1) 19.8(2)
C(9)-C(5)-C(6)-C(7) 28.73(16)
C(4)-C(5)-C(6)-C(7) 138.06(16)
N(13)-C(6)-C(7)-C(8) -131.14(16)
C(1)-C(6)-C(7)-C(8) 107.41(17)
C(5)-C(6)-C(7)-C(8) -11.14(19)
C(6)-C(7)-C(8)-O(14) -137.60(16)
C(6)-C(7)-C(8)-C(9) -11.22(19)
C(4)-C(5)-C(9)-N(10) -0.6(2)
C(6)-C(5)-C(9)-N(10) 123.96(17)
C(4)-C(5)-C(9)-C(8) -164.16(14)
C(6)-C(5)-C(9)-C(8) -39.64(17)
O(14)-C(8)-C(9)-N(10) -3.8(3)
C(7)-C(8)-C(9)-N(10) -128.8(2)
O(14)-C(8)-C(9)-C(5) 157.36(16)
C(7)-C(8)-C(9)-C(5) 32.38(18)
C(5)-C(9)-N(10)-O(11) 3.4(2)
C(8)-C(9)-N(10)-O(11) 163.74(17)
O(17)-C(15)-N(13)-C(6) 1.2(3)
C(16)-C(15)-N(13)-C(6) -177.35(16)
C(1)-C(6)-N(13)-C(15) -80.6(2)
C(5)-C(6)-N(13)-C(15) 44.1(2)
C(7)-C(6)-N(13)-C(15) 158.70(18)
C(9)-N(10)-O(11)-C(4) -4.89(18)
C(3)-C(4)-O(11)-N(10) -119.25(14)
C(5)-C(4)-O(11)-N(10) 4.42(17)
________________________________________________________________

242
Hydrogen Bonds
Donor --- H....Acceptor [ ARU ] D - H H...A D...A D -
H...A
-----------------------------------------------------------------------------
O(12) --H(12O) ..O(17) [ 4656.01] 0.87(3) 1.82(3) 2.6792(19)
170(3)
N(13) --H(13N) ..O(11) [ 2654.01] 0.84(2) 2.25(2) 3.061(2)
162.2(18)
O(14) --H(14O) ..O(12) [ 4656.01] 0.80(3) 1.98(3) 2.771(2)
172(3)
Translation of ARU-code to Equivalent Position Code
===================================================
[ 4656. ] = 1-x,1/2+y,3/2-z
[ 2654. ] = 3/2-x,-y,-1/2+z

243
C.4 X-ray data of trans-9-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-3-nitro-1-
azaspiro[5.5]undeca-2,7,10-trien-4-one (2.55)
2.55
HN
CH3
NO2
O
TBDPSO
Crystal Data
Empirical Formula C27H30N2O4Si
Formula Weight 474.62
Crystal Color, Habit colorless, prism
Crystal Dimensions 0.24 X 0.30 X 0.50 mm
Crystal System monoclinic
Lattice Type primitive
Lattice Parameters a = 14.9015(14) Å
b = 13.8571(13) Å
c = 12.6366(14) Å
= 90°
= 106.644(5)°
= 90°
V = 2500.0(4) Å3
Space Group P 21/c (#14)
Z value 4
Dcalc 1.261 g/cm3
F000 1008.00
(MoK) 1.29 cm-1

244
Intensity Measurements
Diffractometer Bruker X8 APEX II
Radiation MoK ( = 0.71073 Å)
graphite monochromated
Data Images 851 exposures @ 10.0 seconds
Detector Position 36.00 mm
2max 56.6°
No. of Reflections Measured Total: 24445
Unique: 6127 (Rint = 0.032)
Corrections Absorption (Tmin = 0.899, Tmax = 0.970)
Lorentz-polarization
Structure Solution and Refinement
Structure Solution Direct Methods (SIR97)
Refinement Full-matrix least-squares on F2
Function Minimized w (Fo2 – Fc
2)
2
Least Squares Weights w = 1/(2(Fo
2) + (0.0528P)
2 + 0.6852P)
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>0.00(I)) 6127
No. Variables 316
Reflection/Parameter Ratio 19.39
Residuals (refined on F2, all data): R1; wR2 0.058; 0.112
Goodness of Fit Indicator 1.03
No. Observations (I>2.00(I)) 4713
Residuals (refined on F): R1; wR2 0.042; 0.103
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.38 e-/Å
3
Minimum peak in Final Diff. Map -0.25 e-/Å
3
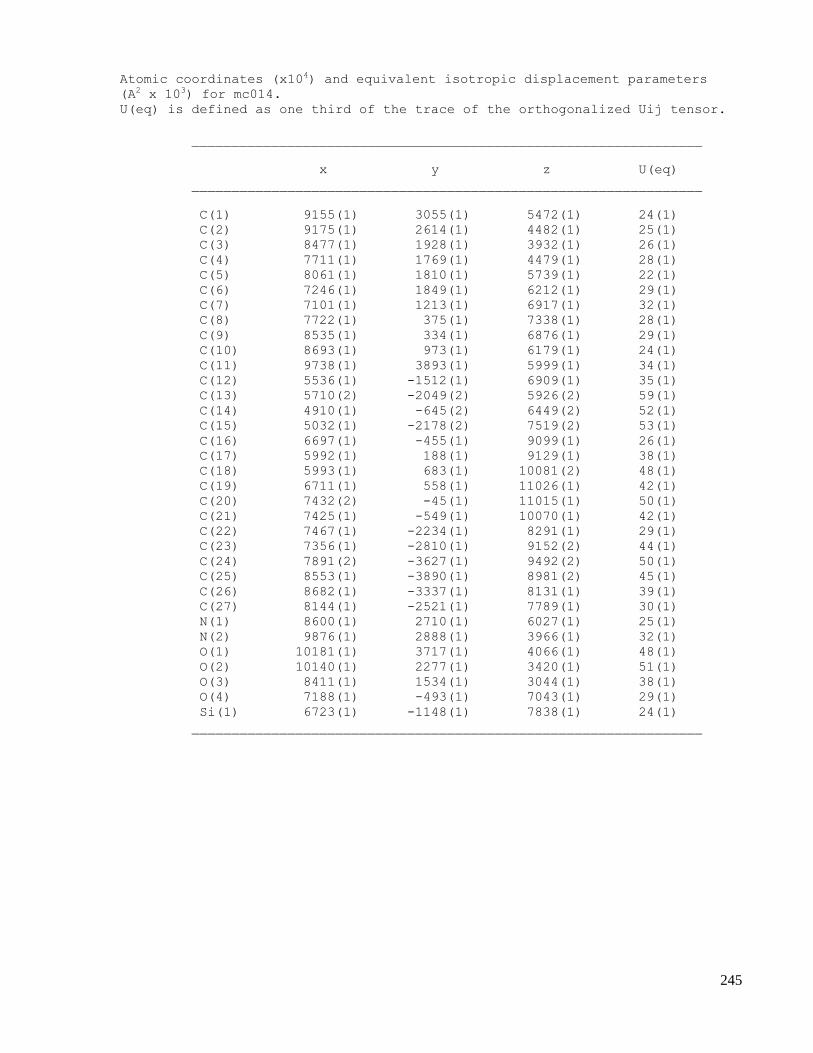
245
Atomic coordinates (x104) and equivalent isotropic displacement parameters
(A2 x 103) for mc014.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
C(1) 9155(1) 3055(1) 5472(1) 24(1)
C(2) 9175(1) 2614(1) 4482(1) 25(1)
C(3) 8477(1) 1928(1) 3932(1) 26(1)
C(4) 7711(1) 1769(1) 4479(1) 28(1)
C(5) 8061(1) 1810(1) 5739(1) 22(1)
C(6) 7246(1) 1849(1) 6212(1) 29(1)
C(7) 7101(1) 1213(1) 6917(1) 32(1)
C(8) 7722(1) 375(1) 7338(1) 28(1)
C(9) 8535(1) 334(1) 6876(1) 29(1)
C(10) 8693(1) 973(1) 6179(1) 24(1)
C(11) 9738(1) 3893(1) 5999(1) 34(1)
C(12) 5536(1) -1512(1) 6909(1) 35(1)
C(13) 5710(2) -2049(2) 5926(2) 59(1)
C(14) 4910(1) -645(2) 6449(2) 52(1)
C(15) 5032(1) -2178(2) 7519(2) 53(1)
C(16) 6697(1) -455(1) 9099(1) 26(1)
C(17) 5992(1) 188(1) 9129(1) 38(1)
C(18) 5993(1) 683(1) 10081(2) 48(1)
C(19) 6711(1) 558(1) 11026(1) 42(1)
C(20) 7432(2) -45(1) 11015(1) 50(1)
C(21) 7425(1) -549(1) 10070(1) 42(1)
C(22) 7467(1) -2234(1) 8291(1) 29(1)
C(23) 7356(1) -2810(1) 9152(2) 44(1)
C(24) 7891(2) -3627(1) 9492(2) 50(1)
C(25) 8553(1) -3890(1) 8981(2) 45(1)
C(26) 8682(1) -3337(1) 8131(1) 39(1)
C(27) 8144(1) -2521(1) 7789(1) 30(1)
N(1) 8600(1) 2710(1) 6027(1) 25(1)
N(2) 9876(1) 2888(1) 3966(1) 32(1)
O(1) 10181(1) 3717(1) 4066(1) 48(1)
O(2) 10140(1) 2277(1) 3420(1) 51(1)
O(3) 8411(1) 1534(1) 3044(1) 38(1)
O(4) 7188(1) -493(1) 7043(1) 29(1)
Si(1) 6723(1) -1148(1) 7838(1) 24(1)
________________________________________________________________

246
Bond lengths [A] and angles [deg] for mc014.
_____________________________________________________________
C(1)-N(1) 1.3182(17)
C(1)-C(2) 1.4001(19)
C(1)-C(11) 1.4886(19)
C(2)-N(2) 1.4309(18)
C(2)-C(3) 1.434(2)
C(3)-O(3) 1.2263(16)
C(3)-C(4) 1.510(2)
C(4)-C(5) 1.5268(18)
C(4)-H(4A) 0.9900
C(4)-H(4B) 0.9900
C(5)-N(1) 1.4722(17)
C(5)-C(10) 1.4980(19)
C(5)-C(6) 1.5000(19)
C(6)-C(7) 1.315(2)
C(6)-H(6) 0.9500
C(7)-C(8) 1.485(2)
C(7)-H(7) 0.9500
C(8)-O(4) 1.4312(17)
C(8)-C(9) 1.489(2)
C(8)-H(8) 1.0000
C(9)-C(10) 1.316(2)
C(9)-H(9) 0.9500
C(10)-H(10) 0.9500
C(11)-H(11A) 0.9800
C(11)-H(11B) 0.9800
C(11)-H(11C) 0.9800
C(12)-C(14) 1.529(3)
C(12)-C(15) 1.530(2)
C(12)-C(13) 1.532(2)
C(12)-Si(1) 1.8894(16)
C(13)-H(13A) 0.9800
C(13)-H(13B) 0.9800
C(13)-H(13C) 0.9800
C(14)-H(14A) 0.9800
C(14)-H(14B) 0.9800
C(14)-H(14C) 0.9800
C(15)-H(15A) 0.9800
C(15)-H(15B) 0.9800
C(15)-H(15C) 0.9800
C(16)-C(17) 1.387(2)
C(16)-C(21) 1.391(2)
C(16)-Si(1) 1.8699(14)
C(17)-C(18) 1.384(2)
C(17)-H(17) 0.9500
C(18)-C(19) 1.367(3)
C(18)-H(18) 0.9500
C(19)-C(20) 1.364(3)
C(19)-H(19) 0.9500
C(20)-C(21) 1.381(2)
C(20)-H(20) 0.9500
C(21)-H(21) 0.9500
C(22)-C(27) 1.394(2)
C(22)-C(23) 1.397(2)
C(22)-Si(1) 1.8606(16)
C(23)-C(24) 1.380(3)
C(23)-H(23) 0.9500

247
C(24)-C(25) 1.375(3)
C(24)-H(24) 0.9500
C(25)-C(26) 1.376(2)
C(25)-H(25) 0.9500
C(26)-C(27) 1.382(2)
C(26)-H(26) 0.9500
C(27)-H(27) 0.9500
N(1)-H(1N) 0.87(2)
N(2)-O(2) 1.2263(17)
N(2)-O(1) 1.2294(17)
O(4)-Si(1) 1.6466(10)
N(1)-C(1)-C(2) 119.64(13)
N(1)-C(1)-C(11) 114.83(12)
C(2)-C(1)-C(11) 125.51(13)
C(1)-C(2)-N(2) 119.75(12)
C(1)-C(2)-C(3) 121.63(12)
N(2)-C(2)-C(3) 118.53(12)
O(3)-C(3)-C(2) 126.56(13)
O(3)-C(3)-C(4) 118.76(13)
C(2)-C(3)-C(4) 114.42(12)
C(3)-C(4)-C(5) 113.26(11)
C(3)-C(4)-H(4A) 108.9
C(5)-C(4)-H(4A) 108.9
C(3)-C(4)-H(4B) 108.9
C(5)-C(4)-H(4B) 108.9
H(4A)-C(4)-H(4B) 107.7
N(1)-C(5)-C(10) 108.85(11)
N(1)-C(5)-C(6) 108.48(11)
C(10)-C(5)-C(6) 112.02(11)
N(1)-C(5)-C(4) 106.81(11)
C(10)-C(5)-C(4) 110.43(12)
C(6)-C(5)-C(4) 110.08(12)
C(7)-C(6)-C(5) 123.71(14)
C(7)-C(6)-H(6) 118.1
C(5)-C(6)-H(6) 118.1
C(6)-C(7)-C(8) 124.08(14)
C(6)-C(7)-H(7) 118.0
C(8)-C(7)-H(7) 118.0
O(4)-C(8)-C(7) 108.79(12)
O(4)-C(8)-C(9) 108.65(12)
C(7)-C(8)-C(9) 112.51(12)
O(4)-C(8)-H(8) 108.9
C(7)-C(8)-H(8) 108.9
C(9)-C(8)-H(8) 108.9
C(10)-C(9)-C(8) 123.93(14)
C(10)-C(9)-H(9) 118.0
C(8)-C(9)-H(9) 118.0
C(9)-C(10)-C(5) 123.72(13)
C(9)-C(10)-H(10) 118.1
C(5)-C(10)-H(10) 118.1
C(1)-C(11)-H(11A) 109.5
C(1)-C(11)-H(11B) 109.5
H(11A)-C(11)-H(11B) 109.5
C(1)-C(11)-H(11C) 109.5
H(11A)-C(11)-H(11C) 109.5
H(11B)-C(11)-H(11C) 109.5
C(14)-C(12)-C(15) 109.31(15)
C(14)-C(12)-C(13) 107.44(16)

248
C(15)-C(12)-C(13) 109.70(16)
C(14)-C(12)-Si(1) 112.75(12)
C(15)-C(12)-Si(1) 110.76(12)
C(13)-C(12)-Si(1) 106.76(12)
C(12)-C(13)-H(13A) 109.5
C(12)-C(13)-H(13B) 109.5
H(13A)-C(13)-H(13B) 109.5
C(12)-C(13)-H(13C) 109.5
H(13A)-C(13)-H(13C) 109.5
H(13B)-C(13)-H(13C) 109.5
C(12)-C(14)-H(14A) 109.5
C(12)-C(14)-H(14B) 109.5
H(14A)-C(14)-H(14B) 109.5
C(12)-C(14)-H(14C) 109.5
H(14A)-C(14)-H(14C) 109.5
H(14B)-C(14)-H(14C) 109.5
C(12)-C(15)-H(15A) 109.5
C(12)-C(15)-H(15B) 109.5
H(15A)-C(15)-H(15B) 109.5
C(12)-C(15)-H(15C) 109.5
H(15A)-C(15)-H(15C) 109.5
H(15B)-C(15)-H(15C) 109.5
C(17)-C(16)-C(21) 116.19(14)
C(17)-C(16)-Si(1) 123.60(11)
C(21)-C(16)-Si(1) 120.19(12)
C(18)-C(17)-C(16) 121.97(16)
C(18)-C(17)-H(17) 119.0
C(16)-C(17)-H(17) 119.0
C(19)-C(18)-C(17) 120.21(17)
C(19)-C(18)-H(18) 119.9
C(17)-C(18)-H(18) 119.9
C(20)-C(19)-C(18) 119.32(16)
C(20)-C(19)-H(19) 120.3
C(18)-C(19)-H(19) 120.3
C(19)-C(20)-C(21) 120.52(17)
C(19)-C(20)-H(20) 119.7
C(21)-C(20)-H(20) 119.7
C(20)-C(21)-C(16) 121.75(16)
C(20)-C(21)-H(21) 119.1
C(16)-C(21)-H(21) 119.1
C(27)-C(22)-C(23) 117.02(14)
C(27)-C(22)-Si(1) 122.43(11)
C(23)-C(22)-Si(1) 120.55(12)
C(24)-C(23)-C(22) 121.62(16)
C(24)-C(23)-H(23) 119.2
C(22)-C(23)-H(23) 119.2
C(25)-C(24)-C(23) 119.96(16)
C(25)-C(24)-H(24) 120.0
C(23)-C(24)-H(24) 120.0
C(24)-C(25)-C(26) 119.86(16)
C(24)-C(25)-H(25) 120.1
C(26)-C(25)-H(25) 120.1
C(25)-C(26)-C(27) 120.14(16)
C(25)-C(26)-H(26) 119.9
C(27)-C(26)-H(26) 119.9
C(26)-C(27)-C(22) 121.39(14)
C(26)-C(27)-H(27) 119.3
C(22)-C(27)-H(27) 119.3
C(1)-N(1)-C(5) 123.81(12)

249
C(1)-N(1)-H(1N) 118.4(12)
C(5)-N(1)-H(1N) 117.1(12)
O(2)-N(2)-O(1) 122.22(13)
O(2)-N(2)-C(2) 117.89(13)
O(1)-N(2)-C(2) 119.88(13)
C(8)-O(4)-Si(1) 127.16(9)
O(4)-Si(1)-C(22) 108.34(6)
O(4)-Si(1)-C(16) 110.63(6)
C(22)-Si(1)-C(16) 108.00(7)
O(4)-Si(1)-C(12) 104.53(6)
C(22)-Si(1)-C(12) 110.14(7)
C(16)-Si(1)-C(12) 115.02(7)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
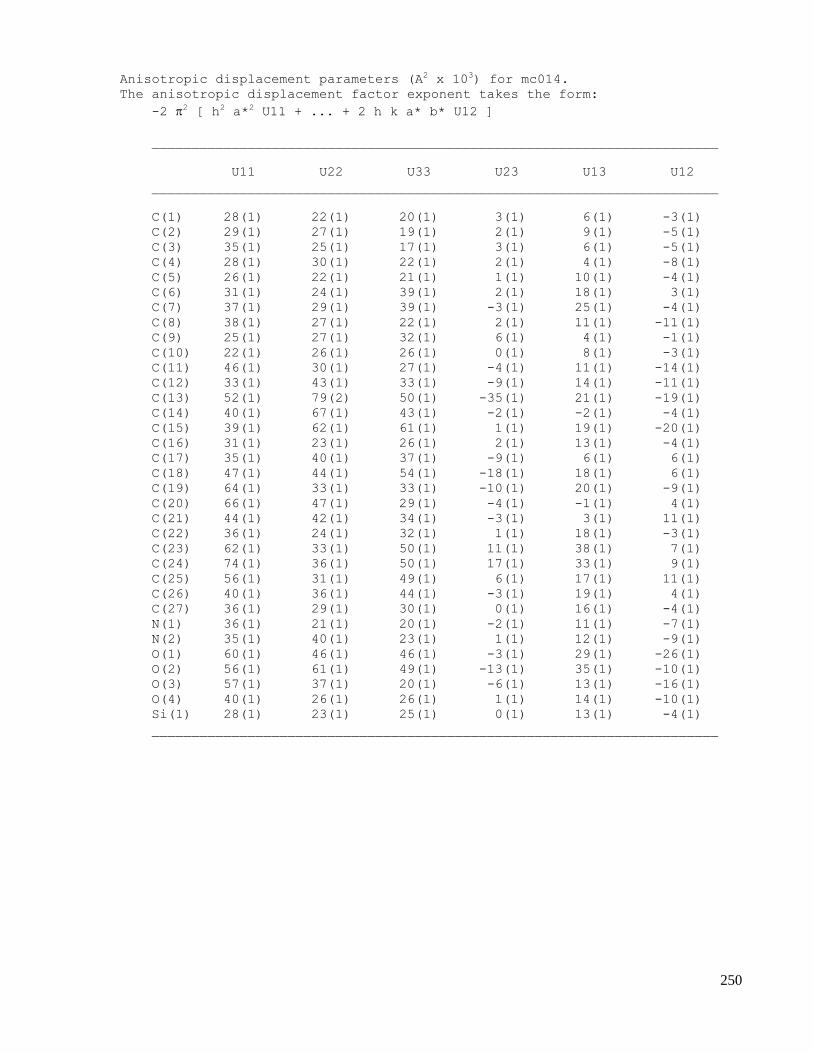
250
Anisotropic displacement parameters (A2 x 103) for mc014.
The anisotropic displacement factor exponent takes the form:
-2 2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
C(1) 28(1) 22(1) 20(1) 3(1) 6(1) -3(1)
C(2) 29(1) 27(1) 19(1) 2(1) 9(1) -5(1)
C(3) 35(1) 25(1) 17(1) 3(1) 6(1) -5(1)
C(4) 28(1) 30(1) 22(1) 2(1) 4(1) -8(1)
C(5) 26(1) 22(1) 21(1) 1(1) 10(1) -4(1)
C(6) 31(1) 24(1) 39(1) 2(1) 18(1) 3(1)
C(7) 37(1) 29(1) 39(1) -3(1) 25(1) -4(1)
C(8) 38(1) 27(1) 22(1) 2(1) 11(1) -11(1)
C(9) 25(1) 27(1) 32(1) 6(1) 4(1) -1(1)
C(10) 22(1) 26(1) 26(1) 0(1) 8(1) -3(1)
C(11) 46(1) 30(1) 27(1) -4(1) 11(1) -14(1)
C(12) 33(1) 43(1) 33(1) -9(1) 14(1) -11(1)
C(13) 52(1) 79(2) 50(1) -35(1) 21(1) -19(1)
C(14) 40(1) 67(1) 43(1) -2(1) -2(1) -4(1)
C(15) 39(1) 62(1) 61(1) 1(1) 19(1) -20(1)
C(16) 31(1) 23(1) 26(1) 2(1) 13(1) -4(1)
C(17) 35(1) 40(1) 37(1) -9(1) 6(1) 6(1)
C(18) 47(1) 44(1) 54(1) -18(1) 18(1) 6(1)
C(19) 64(1) 33(1) 33(1) -10(1) 20(1) -9(1)
C(20) 66(1) 47(1) 29(1) -4(1) -1(1) 4(1)
C(21) 44(1) 42(1) 34(1) -3(1) 3(1) 11(1)
C(22) 36(1) 24(1) 32(1) 1(1) 18(1) -3(1)
C(23) 62(1) 33(1) 50(1) 11(1) 38(1) 7(1)
C(24) 74(1) 36(1) 50(1) 17(1) 33(1) 9(1)
C(25) 56(1) 31(1) 49(1) 6(1) 17(1) 11(1)
C(26) 40(1) 36(1) 44(1) -3(1) 19(1) 4(1)
C(27) 36(1) 29(1) 30(1) 0(1) 16(1) -4(1)
N(1) 36(1) 21(1) 20(1) -2(1) 11(1) -7(1)
N(2) 35(1) 40(1) 23(1) 1(1) 12(1) -9(1)
O(1) 60(1) 46(1) 46(1) -3(1) 29(1) -26(1)
O(2) 56(1) 61(1) 49(1) -13(1) 35(1) -10(1)
O(3) 57(1) 37(1) 20(1) -6(1) 13(1) -16(1)
O(4) 40(1) 26(1) 26(1) 1(1) 14(1) -10(1)
Si(1) 28(1) 23(1) 25(1) 0(1) 13(1) -4(1)
_______________________________________________________________________
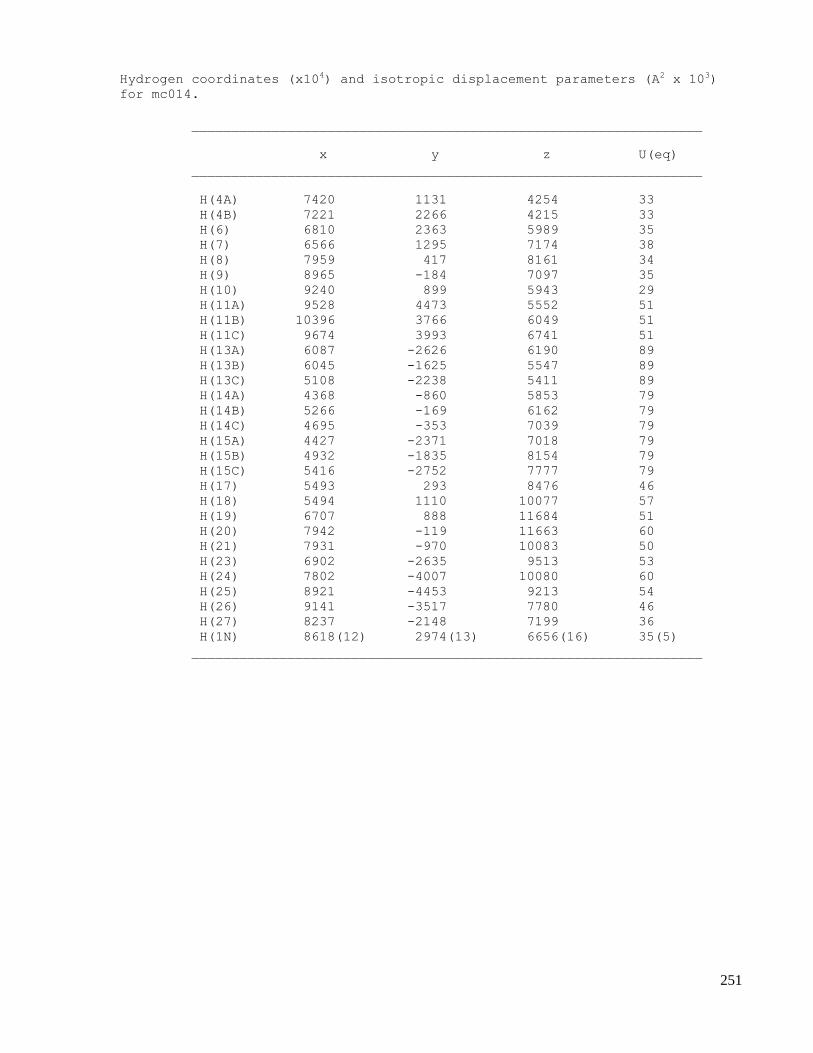
251
Hydrogen coordinates (x104) and isotropic displacement parameters (A2 x 103)
for mc014.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(4A) 7420 1131 4254 33
H(4B) 7221 2266 4215 33
H(6) 6810 2363 5989 35
H(7) 6566 1295 7174 38
H(8) 7959 417 8161 34
H(9) 8965 -184 7097 35
H(10) 9240 899 5943 29
H(11A) 9528 4473 5552 51
H(11B) 10396 3766 6049 51
H(11C) 9674 3993 6741 51
H(13A) 6087 -2626 6190 89
H(13B) 6045 -1625 5547 89
H(13C) 5108 -2238 5411 89
H(14A) 4368 -860 5853 79
H(14B) 5266 -169 6162 79
H(14C) 4695 -353 7039 79
H(15A) 4427 -2371 7018 79
H(15B) 4932 -1835 8154 79
H(15C) 5416 -2752 7777 79
H(17) 5493 293 8476 46
H(18) 5494 1110 10077 57
H(19) 6707 888 11684 51
H(20) 7942 -119 11663 60
H(21) 7931 -970 10083 50
H(23) 6902 -2635 9513 53
H(24) 7802 -4007 10080 60
H(25) 8921 -4453 9213 54
H(26) 9141 -3517 7780 46
H(27) 8237 -2148 7199 36
H(1N) 8618(12) 2974(13) 6656(16) 35(5)
________________________________________________________________

252
Torsion angles [deg] for mc014.
________________________________________________________________
N(1)-C(1)-C(2)-N(2) -170.33(13)
C(11)-C(1)-C(2)-N(2) 8.0(2)
N(1)-C(1)-C(2)-C(3) 13.1(2)
C(11)-C(1)-C(2)-C(3) -168.52(14)
C(1)-C(2)-C(3)-O(3) 176.88(14)
N(2)-C(2)-C(3)-O(3) 0.3(2)
C(1)-C(2)-C(3)-C(4) 2.9(2)
N(2)-C(2)-C(3)-C(4) -173.73(12)
O(3)-C(3)-C(4)-C(5) 149.48(13)
C(2)-C(3)-C(4)-C(5) -36.00(18)
C(3)-C(4)-C(5)-N(1) 50.88(16)
C(3)-C(4)-C(5)-C(10) -67.32(15)
C(3)-C(4)-C(5)-C(6) 168.47(12)
N(1)-C(5)-C(6)-C(7) -121.14(16)
C(10)-C(5)-C(6)-C(7) -1.0(2)
C(4)-C(5)-C(6)-C(7) 122.32(16)
C(5)-C(6)-C(7)-C(8) 0.0(2)
C(6)-C(7)-C(8)-O(4) -120.14(16)
C(6)-C(7)-C(8)-C(9) 0.3(2)
O(4)-C(8)-C(9)-C(10) 121.07(15)
C(7)-C(8)-C(9)-C(10) 0.5(2)
C(8)-C(9)-C(10)-C(5) -1.7(2)
N(1)-C(5)-C(10)-C(9) 121.77(15)
C(6)-C(5)-C(10)-C(9) 1.81(19)
C(4)-C(5)-C(10)-C(9) -121.28(15)
C(21)-C(16)-C(17)-C(18) -2.3(3)
Si(1)-C(16)-C(17)-C(18) 179.49(14)
C(16)-C(17)-C(18)-C(19) 1.1(3)
C(17)-C(18)-C(19)-C(20) 1.1(3)
C(18)-C(19)-C(20)-C(21) -2.0(3)
C(19)-C(20)-C(21)-C(16) 0.7(3)
C(17)-C(16)-C(21)-C(20) 1.5(3)
Si(1)-C(16)-C(21)-C(20) 179.69(15)
C(27)-C(22)-C(23)-C(24) -0.1(3)
Si(1)-C(22)-C(23)-C(24) 179.08(16)
C(22)-C(23)-C(24)-C(25) 0.1(3)
C(23)-C(24)-C(25)-C(26) 0.1(3)
C(24)-C(25)-C(26)-C(27) -0.3(3)
C(25)-C(26)-C(27)-C(22) 0.3(3)
C(23)-C(22)-C(27)-C(26) -0.2(2)
Si(1)-C(22)-C(27)-C(26) -179.27(12)
C(2)-C(1)-N(1)-C(5) 7.0(2)
C(11)-C(1)-N(1)-C(5) -171.48(13)
C(10)-C(5)-N(1)-C(1) 80.84(16)
C(6)-C(5)-N(1)-C(1) -157.03(13)
C(4)-C(5)-N(1)-C(1) -38.40(18)
C(1)-C(2)-N(2)-O(2) 149.99(14)
C(3)-C(2)-N(2)-O(2) -33.4(2)
C(1)-C(2)-N(2)-O(1) -31.1(2)
C(3)-C(2)-N(2)-O(1) 145.53(14)
C(7)-C(8)-O(4)-Si(1) -99.62(13)
C(9)-C(8)-O(4)-Si(1) 137.58(11)
C(8)-O(4)-Si(1)-C(22) -103.19(12)
C(8)-O(4)-Si(1)-C(16) 15.02(14)
C(8)-O(4)-Si(1)-C(12) 139.38(12)
C(27)-C(22)-Si(1)-O(4) -14.59(15)

253
C(23)-C(22)-Si(1)-O(4) 166.32(13)
C(27)-C(22)-Si(1)-C(16) -134.45(13)
C(23)-C(22)-Si(1)-C(16) 46.46(16)
C(27)-C(22)-Si(1)-C(12) 99.18(14)
C(23)-C(22)-Si(1)-C(12) -79.91(15)
C(17)-C(16)-Si(1)-O(4) 84.30(14)
C(21)-C(16)-Si(1)-O(4) -93.80(14)
C(17)-C(16)-Si(1)-C(22) -157.30(13)
C(21)-C(16)-Si(1)-C(22) 24.61(15)
C(17)-C(16)-Si(1)-C(12) -33.84(16)
C(21)-C(16)-Si(1)-C(12) 148.06(13)
C(14)-C(12)-Si(1)-O(4) -60.25(13)
C(15)-C(12)-Si(1)-O(4) 176.89(12)
C(13)-C(12)-Si(1)-O(4) 57.50(14)
C(14)-C(12)-Si(1)-C(22) -176.44(12)
C(15)-C(12)-Si(1)-C(22) 60.71(14)
C(13)-C(12)-Si(1)-C(22) -58.68(15)
C(14)-C(12)-Si(1)-C(16) 61.25(14)
C(15)-C(12)-Si(1)-C(16) -61.60(15)
C(13)-C(12)-Si(1)-C(16) 179.01(13)
________________________________________________________________
Symmetry transformations used to generate equivalent atoms:
Hydrogen Bonds
Donor --- H....Acceptor [ ARU ] D - H H...A D...A D -
H...A
-----------------------------------------------------------------------------
N(1) --H(1N) ..O(3) [ 4555.01] 0.868(19) 1.987(19) 2.8434(16)
168.8(18)
Translation of ARU-code to Equivalent Position Code
===================================================
[ 4555. ] = x,1/2-y,1/2+z
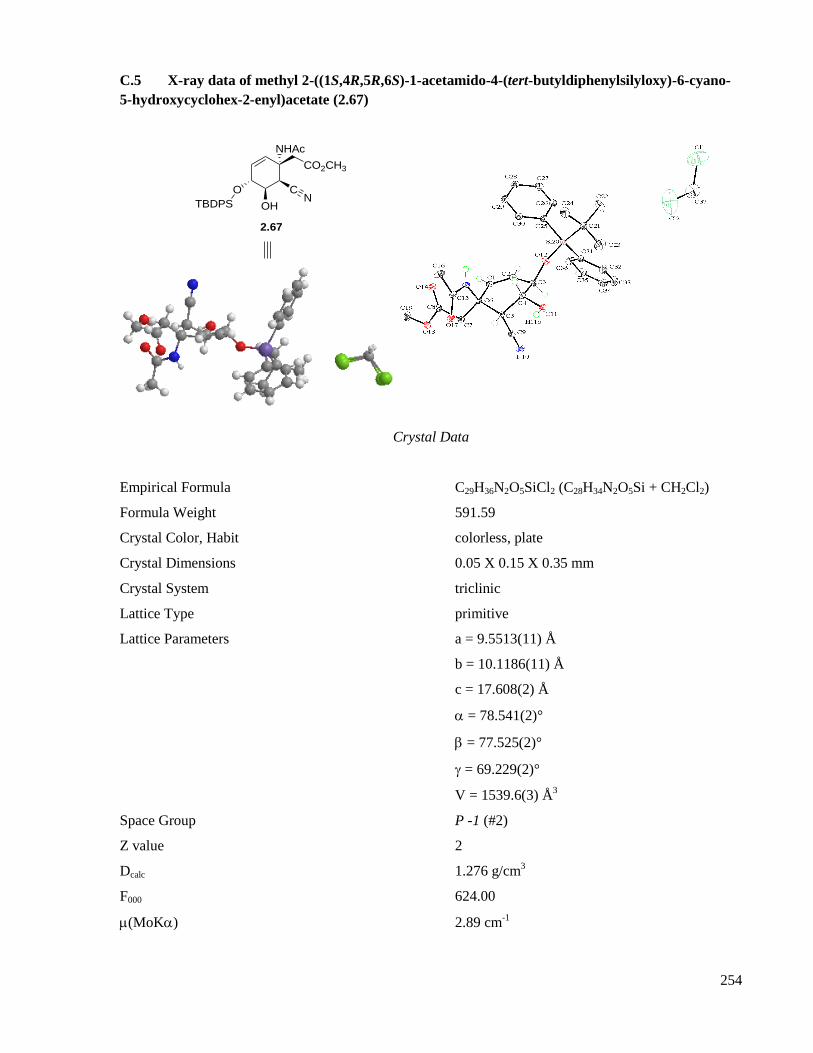
254
C.5 X-ray data of methyl 2-((1S,4R,5R,6S)-1-acetamido-4-(tert-butyldiphenylsilyloxy)-6-cyano-
5-hydroxycyclohex-2-enyl)acetate (2.67)
OH
O CN
CO2CH3
NHAc
2.67
TBDPS
Crystal Data
Empirical Formula C29H36N2O5SiCl2 (C28H34N2O5Si + CH2Cl2)
Formula Weight 591.59
Crystal Color, Habit colorless, plate
Crystal Dimensions 0.05 X 0.15 X 0.35 mm
Crystal System triclinic
Lattice Type primitive
Lattice Parameters a = 9.5513(11) Å
b = 10.1186(11) Å
c = 17.608(2) Å
= 78.541(2)°
= 77.525(2)°
= 69.229(2)°
V = 1539.6(3) Å3
Space Group P -1 (#2)
Z value 2
Dcalc 1.276 g/cm3
F000 624.00
(MoK) 2.89 cm-1

255
Intensity Measurements
Diffractometer Bruker APEX DUO
Radiation MoK ( = 0.71073 Å)
graphite monochromated
Data Images 2229 exposures @ 10.0 seconds
Detector Position 40.00 mm
2max 61.0°
No. of Reflections Measured Total: 34341
Unique: 9366 (Rint = 0.028)
Corrections Absorption (Tmin = 0.872, Tmax = 0.986)
Lorentz-polarization
Structure Solution and Refinement
Structure Solution Direct Methods (SIR97)
Refinement Full-matrix least-squares on F2
Function Minimized w (Fo2 – Fc
2)
2
Least Squares Weights w = 1/(2(Fo
2) + (0.0524P)
2 + 0.5909P)
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>0.00(I)) 9366
No. Variables 393
Reflection/Parameter Ratio 23.83
Residuals (refined on F2, all data): R1; wR2 0.052; 0.106
Goodness of Fit Indicator 1.03
No. Observations (I>2.00(I)) 7568
Residuals (refined on F): R1; wR2 0.039; 0.098
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.55 e-/Å
3
Minimum peak in Final Diff. Map -0.49 e-/Å
3

256
Atomic coordinates (x104) and equivalent isotropic displacement parameters
(A2 x 103) for mc044.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq) occ
________________________________________________________________
C(1) 7750(1) 5449(1) 9308(1) 13(1)
C(2) 8310(1) 5712(1) 8555(1) 13(1)
C(3) 8193(1) 7172(1) 8105(1) 12(1)
C(4) 7168(1) 8344(1) 8591(1) 12(1)
C(5) 7421(1) 7950(1) 9456(1) 11(1)
C(6) 6989(1) 6595(1) 9843(1) 11(1)
C(7) 7512(1) 6041(1) 10650(1) 13(1)
C(8) 7042(1) 4790(1) 11099(1) 13(1)
C(9) 9003(1) 7760(1) 9497(1) 14(1)
C(15) 4331(1) 7951(1) 10366(1) 13(1)
C(16) 2682(1) 8197(1) 10369(1) 22(1)
C(19) 7449(2) 3016(1) 12197(1) 22(1)
C(21) 7092(1) 8049(1) 5858(1) 16(1)
C(22) 7436(2) 7522(2) 5059(1) 31(1)
C(23) 7394(2) 9472(2) 5759(1) 31(1)
C(24) 5398(2) 8319(2) 6185(1) 27(1)
C(25) 7652(1) 4997(1) 6732(1) 14(1)
C(26) 8214(1) 4036(1) 6175(1) 17(1)
C(27) 7773(2) 2831(1) 6274(1) 21(1)
C(28) 6755(2) 2558(1) 6931(1) 20(1)
C(29) 6175(1) 3501(1) 7485(1) 19(1)
C(30) 6616(1) 4703(1) 7388(1) 16(1)
C(31) 10320(1) 6123(1) 6346(1) 15(1)
C(32) 11101(2) 7029(2) 5902(1) 26(1)
C(33) 12685(2) 6578(2) 5743(1) 32(1)
C(34) 13525(2) 5230(2) 6036(1) 27(1)
C(35) 12786(2) 4310(2) 6486(1) 27(1)
C(36) 11207(2) 4749(1) 6632(1) 22(1)
N(10) 10240(1) 7620(1) 9521(1) 20(1)
N(13) 5327(1) 6998(1) 9911(1) 12(1)
O(11) 7457(1) 9632(1) 8254(1) 15(1)
O(12) 7553(1) 7384(1) 7412(1) 15(1)
O(14) 6170(1) 4308(1) 10930(1) 16(1)
O(17) 4738(1) 8584(1) 10769(1) 15(1)
O(18) 7721(1) 4279(1) 11734(1) 16(1)
Si(20) 8194(1) 6654(1) 6593(1) 12(1)
C(37) 9750(14) 9010(30) 2305(7) 53(5) 0.70(2)
Cl(1) 8649(10) 8810(11) 1685(5) 88(2) 0.70(2)
Cl(2) 8626(9) 9554(8) 3186(5) 102(3) 0.70(2)
Cl(2B) 8504(2) 9465(2) 3126(1) 38(1) 0.30(2)
Cl(1B) 8885(3) 8491(3) 1609(1) 44(1) 0.30(2)
C(37B) 9765(4) 8882(9) 2278(2) 25(1) 0.30(2)
________________________________________________________________

257
Bond lengths [A] and angles [deg] for mc044.
_____________________________________________________________
C(1)-C(2) 1.3300(16)
C(1)-C(6) 1.5171(15)
C(1)-H(1) 0.9500
C(2)-C(3) 1.5094(16)
C(2)-H(2) 0.9500
C(3)-O(12) 1.4267(13)
C(3)-C(4) 1.5214(15)
C(3)-H(3) 1.0000
C(4)-O(11) 1.4153(14)
C(4)-C(5) 1.5444(15)
C(4)-H(4) 1.0000
C(5)-C(9) 1.4708(15)
C(5)-C(6) 1.5564(15)
C(5)-H(5) 1.0000
C(6)-N(13) 1.4761(14)
C(6)-C(7) 1.5415(15)
C(7)-C(8) 1.5099(16)
C(7)-H(7A) 0.9900
C(7)-H(7B) 0.9900
C(8)-O(14) 1.2132(14)
C(8)-O(18) 1.3354(14)
C(9)-N(10) 1.1477(16)
C(15)-O(17) 1.2421(14)
C(15)-N(13) 1.3433(15)
C(15)-C(16) 1.5036(16)
C(16)-H(16A) 0.9800
C(16)-H(16B) 0.9800
C(16)-H(16C) 0.9800
C(19)-O(18) 1.4477(15)
C(19)-H(19A) 0.9800
C(19)-H(19B) 0.9800
C(19)-H(19C) 0.9800
C(21)-C(22) 1.5306(18)
C(21)-C(23) 1.5363(19)
C(21)-C(24) 1.5393(18)
C(21)-Si(20) 1.8941(12)
C(22)-H(22A) 0.9800
C(22)-H(22B) 0.9800
C(22)-H(22C) 0.9800
C(23)-H(23A) 0.9800
C(23)-H(23B) 0.9800
C(23)-H(23C) 0.9800
C(24)-H(24A) 0.9800
C(24)-H(24B) 0.9800
C(24)-H(24C) 0.9800
C(25)-C(26) 1.4050(16)
C(25)-C(30) 1.4072(17)
C(25)-Si(20) 1.8814(12)
C(26)-C(27) 1.3936(17)
C(26)-H(26) 0.9500
C(27)-C(28) 1.3925(19)
C(27)-H(27) 0.9500
C(28)-C(29) 1.3895(18)
C(28)-H(28) 0.9500
C(29)-C(30) 1.3917(17)
C(29)-H(29) 0.9500

258
C(30)-H(30) 0.9500
C(31)-C(32) 1.3994(18)
C(31)-C(36) 1.4048(18)
C(31)-Si(20) 1.8812(12)
C(32)-C(33) 1.397(2)
C(32)-H(32) 0.9500
C(33)-C(34) 1.376(2)
C(33)-H(33) 0.9500
C(34)-C(35) 1.387(2)
C(34)-H(34) 0.9500
C(35)-C(36) 1.3935(18)
C(35)-H(35) 0.9500
C(36)-H(36) 0.9500
N(13)-H(13N) 0.842(17)
O(11)-H(11O) 0.85(2)
O(12)-Si(20) 1.6485(9)
C(37)-Cl(2) 1.750(7)
C(37)-Cl(1) 1.753(7)
C(37)-H(37A) 0.9900
C(37)-H(37B) 0.9900
Cl(2B)-C(37B) 1.760(3)
Cl(1B)-C(37B) 1.761(3)
C(37B)-H(37C) 0.9900
C(37B)-H(37D) 0.9900
C(2)-C(1)-C(6) 123.59(10)
C(2)-C(1)-H(1) 118.2
C(6)-C(1)-H(1) 118.2
C(1)-C(2)-C(3) 125.38(10)
C(1)-C(2)-H(2) 117.3
C(3)-C(2)-H(2) 117.3
O(12)-C(3)-C(2) 110.77(9)
O(12)-C(3)-C(4) 106.85(9)
C(2)-C(3)-C(4) 111.34(9)
O(12)-C(3)-H(3) 109.3
C(2)-C(3)-H(3) 109.3
C(4)-C(3)-H(3) 109.3
O(11)-C(4)-C(3) 108.65(9)
O(11)-C(4)-C(5) 111.01(9)
C(3)-C(4)-C(5) 111.12(9)
O(11)-C(4)-H(4) 108.7
C(3)-C(4)-H(4) 108.7
C(5)-C(4)-H(4) 108.7
C(9)-C(5)-C(4) 109.53(9)
C(9)-C(5)-C(6) 111.72(9)
C(4)-C(5)-C(6) 110.88(9)
C(9)-C(5)-H(5) 108.2
C(4)-C(5)-H(5) 108.2
C(6)-C(5)-H(5) 108.2
N(13)-C(6)-C(1) 109.40(9)
N(13)-C(6)-C(7) 111.56(9)
C(1)-C(6)-C(7) 109.42(9)
N(13)-C(6)-C(5) 106.89(8)
C(1)-C(6)-C(5) 108.65(9)
C(7)-C(6)-C(5) 110.84(9)
C(8)-C(7)-C(6) 114.78(9)
C(8)-C(7)-H(7A) 108.6
C(6)-C(7)-H(7A) 108.6
C(8)-C(7)-H(7B) 108.6

259
C(6)-C(7)-H(7B) 108.6
H(7A)-C(7)-H(7B) 107.5
O(14)-C(8)-O(18) 123.73(11)
O(14)-C(8)-C(7) 126.60(10)
O(18)-C(8)-C(7) 109.66(9)
N(10)-C(9)-C(5) 179.20(14)
O(17)-C(15)-N(13) 122.36(10)
O(17)-C(15)-C(16) 121.59(10)
N(13)-C(15)-C(16) 116.04(10)
C(15)-C(16)-H(16A) 109.5
C(15)-C(16)-H(16B) 109.5
H(16A)-C(16)-H(16B) 109.5
C(15)-C(16)-H(16C) 109.5
H(16A)-C(16)-H(16C) 109.5
H(16B)-C(16)-H(16C) 109.5
O(18)-C(19)-H(19A) 109.5
O(18)-C(19)-H(19B) 109.5
H(19A)-C(19)-H(19B) 109.5
O(18)-C(19)-H(19C) 109.5
H(19A)-C(19)-H(19C) 109.5
H(19B)-C(19)-H(19C) 109.5
C(22)-C(21)-C(23) 109.83(12)
C(22)-C(21)-C(24) 108.71(11)
C(23)-C(21)-C(24) 107.74(11)
C(22)-C(21)-Si(20) 111.37(9)
C(23)-C(21)-Si(20) 112.24(9)
C(24)-C(21)-Si(20) 106.79(8)
C(21)-C(22)-H(22A) 109.5
C(21)-C(22)-H(22B) 109.5
H(22A)-C(22)-H(22B) 109.5
C(21)-C(22)-H(22C) 109.5
H(22A)-C(22)-H(22C) 109.5
H(22B)-C(22)-H(22C) 109.5
C(21)-C(23)-H(23A) 109.5
C(21)-C(23)-H(23B) 109.5
H(23A)-C(23)-H(23B) 109.5
C(21)-C(23)-H(23C) 109.5
H(23A)-C(23)-H(23C) 109.5
H(23B)-C(23)-H(23C) 109.5
C(21)-C(24)-H(24A) 109.5
C(21)-C(24)-H(24B) 109.5
H(24A)-C(24)-H(24B) 109.5
C(21)-C(24)-H(24C) 109.5
H(24A)-C(24)-H(24C) 109.5
H(24B)-C(24)-H(24C) 109.5
C(26)-C(25)-C(30) 117.43(11)
C(26)-C(25)-Si(20) 121.76(9)
C(30)-C(25)-Si(20) 120.77(9)
C(27)-C(26)-C(25) 121.27(12)
C(27)-C(26)-H(26) 119.4
C(25)-C(26)-H(26) 119.4
C(28)-C(27)-C(26) 120.20(12)
C(28)-C(27)-H(27) 119.9
C(26)-C(27)-H(27) 119.9
C(29)-C(28)-C(27) 119.51(12)
C(29)-C(28)-H(28) 120.2
C(27)-C(28)-H(28) 120.2
C(28)-C(29)-C(30) 120.29(12)
C(28)-C(29)-H(29) 119.9

260
C(30)-C(29)-H(29) 119.9
C(29)-C(30)-C(25) 121.29(11)
C(29)-C(30)-H(30) 119.4
C(25)-C(30)-H(30) 119.4
C(32)-C(31)-C(36) 116.70(12)
C(32)-C(31)-Si(20) 123.98(10)
C(36)-C(31)-Si(20) 119.30(9)
C(33)-C(32)-C(31) 121.43(13)
C(33)-C(32)-H(32) 119.3
C(31)-C(32)-H(32) 119.3
C(34)-C(33)-C(32) 120.62(14)
C(34)-C(33)-H(33) 119.7
C(32)-C(33)-H(33) 119.7
C(33)-C(34)-C(35) 119.40(13)
C(33)-C(34)-H(34) 120.3
C(35)-C(34)-H(34) 120.3
C(34)-C(35)-C(36) 120.00(14)
C(34)-C(35)-H(35) 120.0
C(36)-C(35)-H(35) 120.0
C(35)-C(36)-C(31) 121.83(13)
C(35)-C(36)-H(36) 119.1
C(31)-C(36)-H(36) 119.1
C(15)-N(13)-C(6) 123.96(9)
C(15)-N(13)-H(13N) 118.8(11)
C(6)-N(13)-H(13N) 117.0(11)
C(4)-O(11)-H(11O) 107.2(13)
C(3)-O(12)-Si(20) 132.22(7)
C(8)-O(18)-C(19) 115.81(10)
O(12)-Si(20)-C(31) 111.71(5)
O(12)-Si(20)-C(25) 108.42(5)
C(31)-Si(20)-C(25) 107.94(5)
O(12)-Si(20)-C(21) 103.04(5)
C(31)-Si(20)-C(21) 116.13(6)
C(25)-Si(20)-C(21) 109.34(5)
Cl(2)-C(37)-Cl(1) 110.7(7)
Cl(2)-C(37)-H(37A) 109.5
Cl(1)-C(37)-H(37A) 109.5
Cl(2)-C(37)-H(37B) 109.5
Cl(1)-C(37)-H(37B) 109.5
H(37A)-C(37)-H(37B) 108.1
Cl(2B)-C(37B)-Cl(1B) 112.94(19)
Cl(2B)-C(37B)-H(37C) 109.0
Cl(1B)-C(37B)-H(37C) 109.0
Cl(2B)-C(37B)-H(37D) 109.0
Cl(1B)-C(37B)-H(37D) 109.0
H(37C)-C(37B)-H(37D) 107.8
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
261
Anisotropic displacement parameters (A2 x 103) for mc044.
The anisotropic displacement factor exponent takes the form:
-2 2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
C(1) 12(1) 10(1) 16(1) -3(1) -4(1) -2(1)
C(2) 12(1) 12(1) 16(1) -5(1) -3(1) -1(1)
C(3) 11(1) 13(1) 12(1) -3(1) -2(1) -3(1)
C(4) 11(1) 11(1) 12(1) -2(1) -2(1) -3(1)
C(5) 10(1) 11(1) 13(1) -2(1) -2(1) -4(1)
C(6) 9(1) 11(1) 13(1) -2(1) -3(1) -3(1)
C(7) 12(1) 13(1) 14(1) -2(1) -4(1) -4(1)
C(8) 11(1) 13(1) 12(1) -3(1) -1(1) -2(1)
C(9) 14(1) 13(1) 14(1) -3(1) -2(1) -4(1)
C(15) 11(1) 11(1) 16(1) -1(1) -2(1) -4(1)
C(16) 11(1) 21(1) 38(1) -13(1) -3(1) -4(1)
C(19) 21(1) 22(1) 19(1) 7(1) -4(1) -8(1)
C(21) 16(1) 15(1) 15(1) -1(1) -3(1) -4(1)
C(22) 38(1) 31(1) 15(1) -4(1) -9(1) 2(1)
C(23) 33(1) 17(1) 45(1) 6(1) -17(1) -9(1)
C(24) 16(1) 31(1) 26(1) 5(1) -6(1) -3(1)
C(25) 12(1) 14(1) 16(1) -2(1) -4(1) -3(1)
C(26) 17(1) 16(1) 18(1) -4(1) 0(1) -5(1)
C(27) 23(1) 16(1) 24(1) -6(1) -4(1) -6(1)
C(28) 22(1) 16(1) 26(1) -1(1) -8(1) -9(1)
C(29) 16(1) 20(1) 19(1) 1(1) -3(1) -8(1)
C(30) 13(1) 17(1) 16(1) -3(1) -3(1) -3(1)
C(31) 14(1) 20(1) 14(1) -4(1) -2(1) -6(1)
C(32) 19(1) 32(1) 26(1) 8(1) -8(1) -12(1)
C(33) 20(1) 50(1) 26(1) 8(1) -5(1) -19(1)
C(34) 14(1) 46(1) 24(1) -12(1) -2(1) -10(1)
C(35) 15(1) 26(1) 40(1) -11(1) -7(1) -1(1)
C(36) 16(1) 18(1) 32(1) -5(1) -4(1) -5(1)
N(10) 15(1) 24(1) 23(1) -5(1) -4(1) -7(1)
N(13) 10(1) 12(1) 15(1) -4(1) -3(1) -4(1)
O(11) 17(1) 11(1) 16(1) -2(1) 1(1) -4(1)
O(12) 15(1) 16(1) 12(1) -4(1) -4(1) -2(1)
O(14) 14(1) 16(1) 18(1) -2(1) -4(1) -6(1)
O(17) 13(1) 15(1) 18(1) -6(1) -1(1) -5(1)
O(18) 18(1) 19(1) 14(1) 2(1) -6(1) -7(1)
Si(20) 12(1) 12(1) 11(1) -2(1) -2(1) -4(1)
C(37) 46(9) 37(7) 70(10) -6(6) -15(7) -5(6)
Cl(1) 64(2) 66(3) 143(5) -23(3) -42(2) -13(2)
Cl(2) 63(3) 81(4) 96(4) 20(2) 10(2) 27(2)
Cl(2B) 38(1) 27(1) 28(1) 3(1) 14(1) 0(1)
Cl(1B) 34(1) 63(1) 42(1) -3(1) -13(1) -23(1)
C(37B) 15(2) 28(2) 29(2) -4(2) 5(2) -9(2)
_______________________________________________________________________
262
Hydrogen coordinates (x104) and isotropic displacement parameters (A2 x 103)
for mc044.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(1) 7831 4491 9522 15
H(2) 8826 4915 8277 16
H(3) 9228 7267 7957 15
H(4) 6088 8473 8572 14
H(5) 6747 8760 9747 13
H(7A) 8631 5760 10573 15
H(7B) 7097 6832 10973 15
H(16A) 2266 7721 10860 33
H(16B) 2579 7807 9925 33
H(16C) 2126 9223 10323 33
H(19A) 6364 3232 12400 32
H(19B) 8020 2710 12636 32
H(19C) 7779 2251 11868 32
H(22A) 6796 8236 4704 46
H(22B) 7228 6622 5125 46
H(22C) 8506 7372 4838 46
H(23A) 8404 9381 5454 47
H(23B) 7342 9722 6276 47
H(23C) 6627 10222 5483 47
H(24A) 4784 9026 5817 40
H(24B) 5159 8677 6692 40
H(24C) 5174 7425 6256 40
H(26) 8908 4210 5724 21
H(27) 8169 2193 5891 25
H(28) 6458 1734 7000 24
H(29) 5473 3324 7933 22
H(30) 6210 5338 7772 19
H(32) 10543 7972 5704 31
H(33) 13186 7208 5429 38
H(34) 14602 4933 5931 33
H(35) 13357 3381 6696 32
H(36) 10716 4102 6933 26
H(13N) 4997(18) 6547(17) 9677(10) 17(4)
H(11O) 6840(20) 10280(20) 8517(12) 36(5)
H(37A) 10555 8090 2418 63
H(37B) 10245 9725 2039 63
H(37C) 10599 8018 2434 30
H(37D) 10215 9631 2014 30
________________________________________________________________

263
Torsion angles [deg] for mc044.
________________________________________________________________
C(6)-C(1)-C(2)-C(3) -4.13(18)
C(1)-C(2)-C(3)-O(12) -126.21(12)
C(1)-C(2)-C(3)-C(4) -7.46(16)
O(12)-C(3)-C(4)-O(11) -76.58(11)
C(2)-C(3)-C(4)-O(11) 162.34(9)
O(12)-C(3)-C(4)-C(5) 161.01(9)
C(2)-C(3)-C(4)-C(5) 39.93(12)
O(11)-C(4)-C(5)-C(9) -60.39(12)
C(3)-C(4)-C(5)-C(9) 60.64(12)
O(11)-C(4)-C(5)-C(6) 175.86(9)
C(3)-C(4)-C(5)-C(6) -63.11(11)
C(2)-C(1)-C(6)-N(13) 99.09(13)
C(2)-C(1)-C(6)-C(7) -138.41(11)
C(2)-C(1)-C(6)-C(5) -17.27(14)
C(9)-C(5)-C(6)-N(13) 168.87(9)
C(4)-C(5)-C(6)-N(13) -68.64(11)
C(9)-C(5)-C(6)-C(1) -73.16(11)
C(4)-C(5)-C(6)-C(1) 49.32(11)
C(9)-C(5)-C(6)-C(7) 47.10(12)
C(4)-C(5)-C(6)-C(7) 169.58(9)
N(13)-C(6)-C(7)-C(8) 56.74(12)
C(1)-C(6)-C(7)-C(8) -64.46(12)
C(5)-C(6)-C(7)-C(8) 175.74(9)
C(6)-C(7)-C(8)-O(14) -9.25(16)
C(6)-C(7)-C(8)-O(18) 171.70(9)
C(4)-C(5)-C(9)-N(10) 19(10)
C(6)-C(5)-C(9)-N(10) 142(10)
C(30)-C(25)-C(26)-C(27) -0.57(18)
Si(20)-C(25)-C(26)-C(27) -178.30(10)
C(25)-C(26)-C(27)-C(28) 0.15(19)
C(26)-C(27)-C(28)-C(29) 0.4(2)
C(27)-C(28)-C(29)-C(30) -0.46(19)
C(28)-C(29)-C(30)-C(25) 0.03(18)
C(26)-C(25)-C(30)-C(29) 0.48(17)
Si(20)-C(25)-C(30)-C(29) 178.23(9)
C(36)-C(31)-C(32)-C(33) 0.9(2)
Si(20)-C(31)-C(32)-C(33) 179.44(12)
C(31)-C(32)-C(33)-C(34) -1.5(2)
C(32)-C(33)-C(34)-C(35) 0.7(2)
C(33)-C(34)-C(35)-C(36) 0.6(2)
C(34)-C(35)-C(36)-C(31) -1.2(2)
C(32)-C(31)-C(36)-C(35) 0.4(2)
Si(20)-C(31)-C(36)-C(35) -178.18(11)
O(17)-C(15)-N(13)-C(6) 0.46(18)
C(16)-C(15)-N(13)-C(6) -178.84(11)
C(1)-C(6)-N(13)-C(15) 177.47(10)
C(7)-C(6)-N(13)-C(15) 56.26(14)
C(5)-C(6)-N(13)-C(15) -65.06(13)
C(2)-C(3)-O(12)-Si(20) -68.26(13)
C(4)-C(3)-O(12)-Si(20) 170.30(8)
O(14)-C(8)-O(18)-C(19) 4.61(16)
C(7)-C(8)-O(18)-C(19) -176.31(10)
C(3)-O(12)-Si(20)-C(31) -30.74(12)
C(3)-O(12)-Si(20)-C(25) 88.08(11)
C(3)-O(12)-Si(20)-C(21) -156.10(10)
C(32)-C(31)-Si(20)-O(12) -88.41(12)

264
C(36)-C(31)-Si(20)-O(12) 90.09(11)
C(32)-C(31)-Si(20)-C(25) 152.49(11)
C(36)-C(31)-Si(20)-C(25) -29.01(11)
C(32)-C(31)-Si(20)-C(21) 29.35(13)
C(36)-C(31)-Si(20)-C(21) -152.15(10)
C(26)-C(25)-Si(20)-O(12) -171.17(9)
C(30)-C(25)-Si(20)-O(12) 11.17(11)
C(26)-C(25)-Si(20)-C(31) -50.00(11)
C(30)-C(25)-Si(20)-C(31) 132.34(9)
C(26)-C(25)-Si(20)-C(21) 77.18(11)
C(30)-C(25)-Si(20)-C(21) -100.48(10)
C(22)-C(21)-Si(20)-O(12) -175.57(10)
C(23)-C(21)-Si(20)-O(12) 60.82(11)
C(24)-C(21)-Si(20)-O(12) -57.03(10)
C(22)-C(21)-Si(20)-C(31) 61.98(11)
C(23)-C(21)-Si(20)-C(31) -61.63(11)
C(24)-C(21)-Si(20)-C(31) -179.47(9)
C(22)-C(21)-Si(20)-C(25) -60.42(11)
C(23)-C(21)-Si(20)-C(25) 175.97(10)
C(24)-C(21)-Si(20)-C(25) 58.12(10)
________________________________________________________________
Hydrogen Bonds
Donor --- H....Acceptor [ ARU ] D - H H...A D...A D -
H...A
-----------------------------------------------------------------------------
O(11) --H(11O) ..O(17) [ 2677.01] 0.85(2) 1.92(2) 2.7578(13)
167(2)
N(13) --H(13N) ..O(14) [ 2667.01] 0.842(17) 2.168(18) 3.0024(15)
170.8(16)
Translation of ARU-code to Equivalent Position Code
===================================================
[ 2677. ] = 1-x,2-y,2-z
[ 2667. ] = 1-x,1-y,2-z
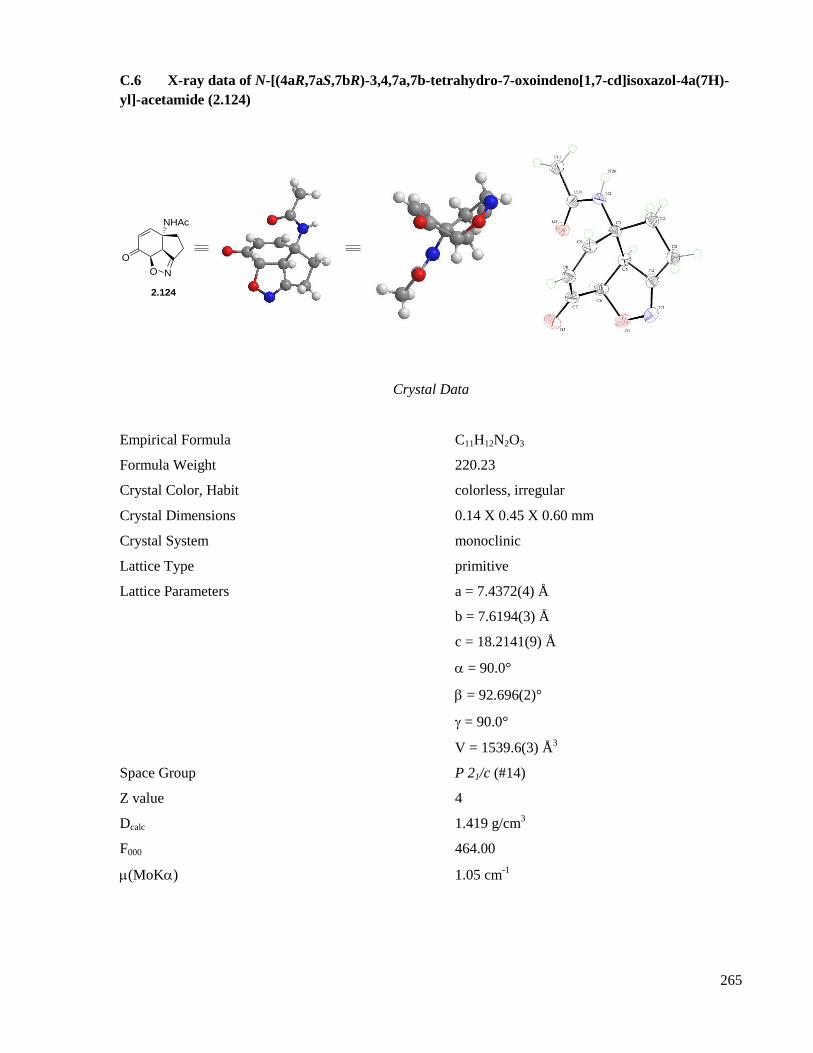
265
C.6 X-ray data of N-[(4aR,7aS,7bR)-3,4,7a,7b-tetrahydro-7-oxoindeno[1,7-cd]isoxazol-4a(7H)-
yl]-acetamide (2.124)
O
O N
NHAc
2.124
Crystal Data
Empirical Formula C11H12N2O3
Formula Weight 220.23
Crystal Color, Habit colorless, irregular
Crystal Dimensions 0.14 X 0.45 X 0.60 mm
Crystal System monoclinic
Lattice Type primitive
Lattice Parameters a = 7.4372(4) Å
b = 7.6194(3) Å
c = 18.2141(9) Å
= 90.0°
= 92.696(2)°
= 90.0°
V = 1539.6(3) Å3
Space Group P 21/c (#14)
Z value 4
Dcalc 1.419 g/cm3
F000 464.00
(MoK) 1.05 cm-1

266
Intensity Measurements
Diffractometer Bruker X8 APEX II
Radiation MoK ( = 0.71073 Å)
graphite monochromated
Data Images 1321 exposures @ 7.0 seconds
Detector Position 36.00 mm
2max 56.0°
No. of Reflections Measured Total: 12787
Unique: 2466 (Rint = 0.030)
Corrections Absorption (Tmin = 0.840, Tmax = 0.985)
Lorentz-polarization
Structure Solution and Refinement
Structure Solution Direct Methods (SIR97)
Refinement Full-matrix least-squares on F2
Function Minimized w (Fo2 – Fc
2)
2
Least Squares Weights w = 1/(2(Fo
2) + (0.0455P)
2 + 0.3215P)
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>0.00(I)) 2466
No. Variables 150
Reflection/Parameter Ratio 16.44
Residuals (refined on F2, all data): R1; wR2 0.043; 0.100
Goodness of Fit Indicator 1.04
No. Observations (I>2.00(I)) 2132
Residuals (refined on F): R1; wR2 0.037; 0.095
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.26 e-/Å
3
Minimum peak in Final Diff. Map -0.21 e-/Å
3
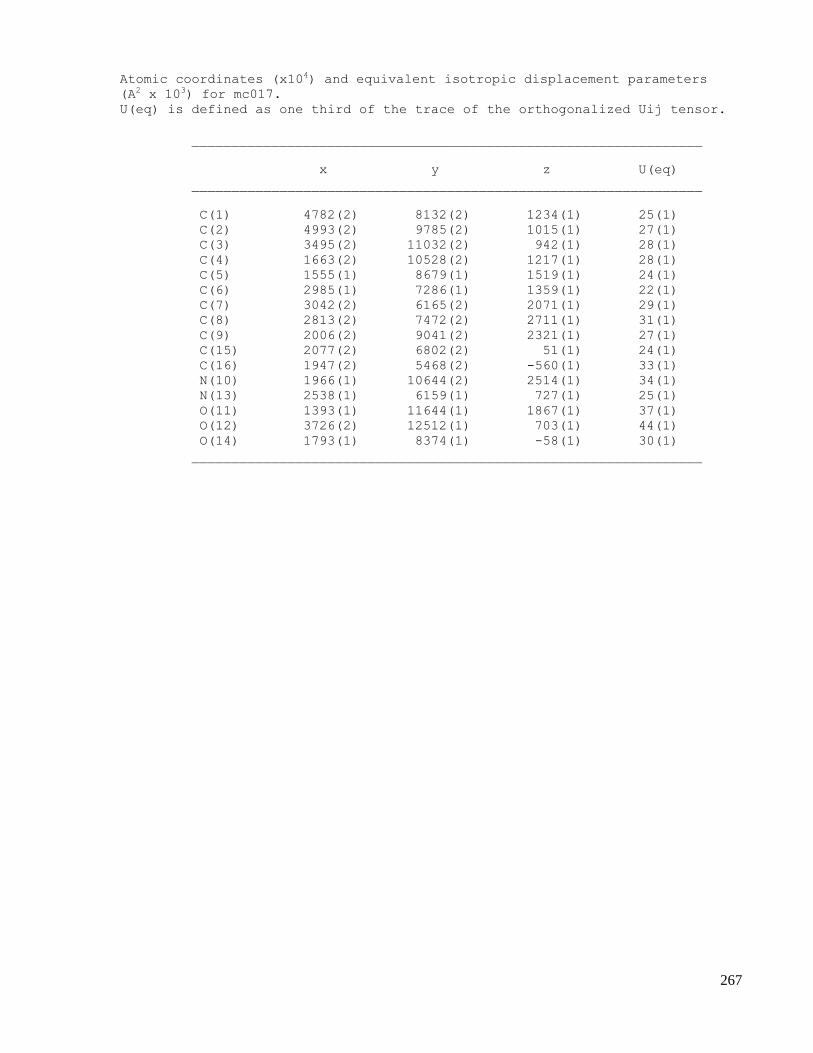
267
Atomic coordinates (x104) and equivalent isotropic displacement parameters
(A2 x 103) for mc017.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
C(1) 4782(2) 8132(2) 1234(1) 25(1)
C(2) 4993(2) 9785(2) 1015(1) 27(1)
C(3) 3495(2) 11032(2) 942(1) 28(1)
C(4) 1663(2) 10528(2) 1217(1) 28(1)
C(5) 1555(1) 8679(1) 1519(1) 24(1)
C(6) 2985(1) 7286(1) 1359(1) 22(1)
C(7) 3042(2) 6165(2) 2071(1) 29(1)
C(8) 2813(2) 7472(2) 2711(1) 31(1)
C(9) 2006(2) 9041(2) 2321(1) 27(1)
C(15) 2077(2) 6802(2) 51(1) 24(1)
C(16) 1947(2) 5468(2) -560(1) 33(1)
N(10) 1966(1) 10644(2) 2514(1) 34(1)
N(13) 2538(1) 6159(1) 727(1) 25(1)
O(11) 1393(1) 11644(1) 1867(1) 37(1)
O(12) 3726(2) 12512(1) 703(1) 44(1)
O(14) 1793(1) 8374(1) -58(1) 30(1)
________________________________________________________________

268
Bond lengths [A] and angles [deg] for mc017.
_____________________________________________________________
C(1)-C(2) 1.3324(16)
C(1)-C(6) 1.5105(15)
C(1)-H(1) 0.9500
C(2)-C(3) 1.4656(16)
C(2)-H(2) 0.9500
C(3)-O(12) 1.2245(14)
C(3)-C(4) 1.5228(17)
C(4)-O(11) 1.4787(14)
C(4)-C(5) 1.5160(16)
C(4)-H(4) 1.0000
C(5)-C(9) 1.5079(16)
C(5)-C(6) 1.5406(15)
C(5)-H(5) 1.0000
C(6)-N(13) 1.4629(14)
C(6)-C(7) 1.5521(15)
C(7)-C(8) 1.5479(17)
C(7)-H(7A) 0.9900
C(7)-H(7B) 0.9900
C(8)-C(9) 1.5011(17)
C(8)-H(8A) 0.9900
C(8)-H(8B) 0.9900
C(9)-N(10) 1.2715(16)
C(15)-O(14) 1.2308(14)
C(15)-N(13) 1.3532(15)
C(15)-C(16) 1.5063(16)
C(16)-H(16A) 0.9800
C(16)-H(16B) 0.9800
C(16)-H(16C) 0.9800
N(10)-O(11) 1.4509(14)
N(13)-H(13N) 0.854(17)
C(2)-C(1)-C(6) 124.47(10)
C(2)-C(1)-H(1) 117.8
C(6)-C(1)-H(1) 117.8
C(1)-C(2)-C(3) 122.69(11)
C(1)-C(2)-H(2) 118.7
C(3)-C(2)-H(2) 118.7
O(12)-C(3)-C(2) 120.69(12)
O(12)-C(3)-C(4) 119.49(11)
C(2)-C(3)-C(4) 119.67(10)
O(11)-C(4)-C(5) 103.44(9)
O(11)-C(4)-C(3) 106.01(9)
C(5)-C(4)-C(3) 114.64(9)
O(11)-C(4)-H(4) 110.8
C(5)-C(4)-H(4) 110.8
C(3)-C(4)-H(4) 110.8
C(9)-C(5)-C(4) 99.69(9)
C(9)-C(5)-C(6) 100.66(9)
C(4)-C(5)-C(6) 121.52(9)
C(9)-C(5)-H(5) 111.2
C(4)-C(5)-H(5) 111.2
C(6)-C(5)-H(5) 111.2
N(13)-C(6)-C(1) 107.54(9)
N(13)-C(6)-C(5) 114.86(9)
C(1)-C(6)-C(5) 111.01(9)
N(13)-C(6)-C(7) 109.35(9)

269
C(1)-C(6)-C(7) 111.82(9)
C(5)-C(6)-C(7) 102.28(9)
C(8)-C(7)-C(6) 105.98(9)
C(8)-C(7)-H(7A) 110.5
C(6)-C(7)-H(7A) 110.5
C(8)-C(7)-H(7B) 110.5
C(6)-C(7)-H(7B) 110.5
H(7A)-C(7)-H(7B) 108.7
C(9)-C(8)-C(7) 102.23(9)
C(9)-C(8)-H(8A) 111.3
C(7)-C(8)-H(8A) 111.3
C(9)-C(8)-H(8B) 111.3
C(7)-C(8)-H(8B) 111.3
H(8A)-C(8)-H(8B) 109.2
N(10)-C(9)-C(8) 130.38(11)
N(10)-C(9)-C(5) 115.88(11)
C(8)-C(9)-C(5) 112.23(10)
O(14)-C(15)-N(13) 122.25(10)
O(14)-C(15)-C(16) 122.18(11)
N(13)-C(15)-C(16) 115.57(10)
C(15)-C(16)-H(16A) 109.5
C(15)-C(16)-H(16B) 109.5
H(16A)-C(16)-H(16B) 109.5
C(15)-C(16)-H(16C) 109.5
H(16A)-C(16)-H(16C) 109.5
H(16B)-C(16)-H(16C) 109.5
C(9)-N(10)-O(11) 106.84(10)
C(15)-N(13)-C(6) 122.79(9)
C(15)-N(13)-H(13N) 118.0(11)
C(6)-N(13)-H(13N) 118.0(11)
N(10)-O(11)-C(4) 107.58(8)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
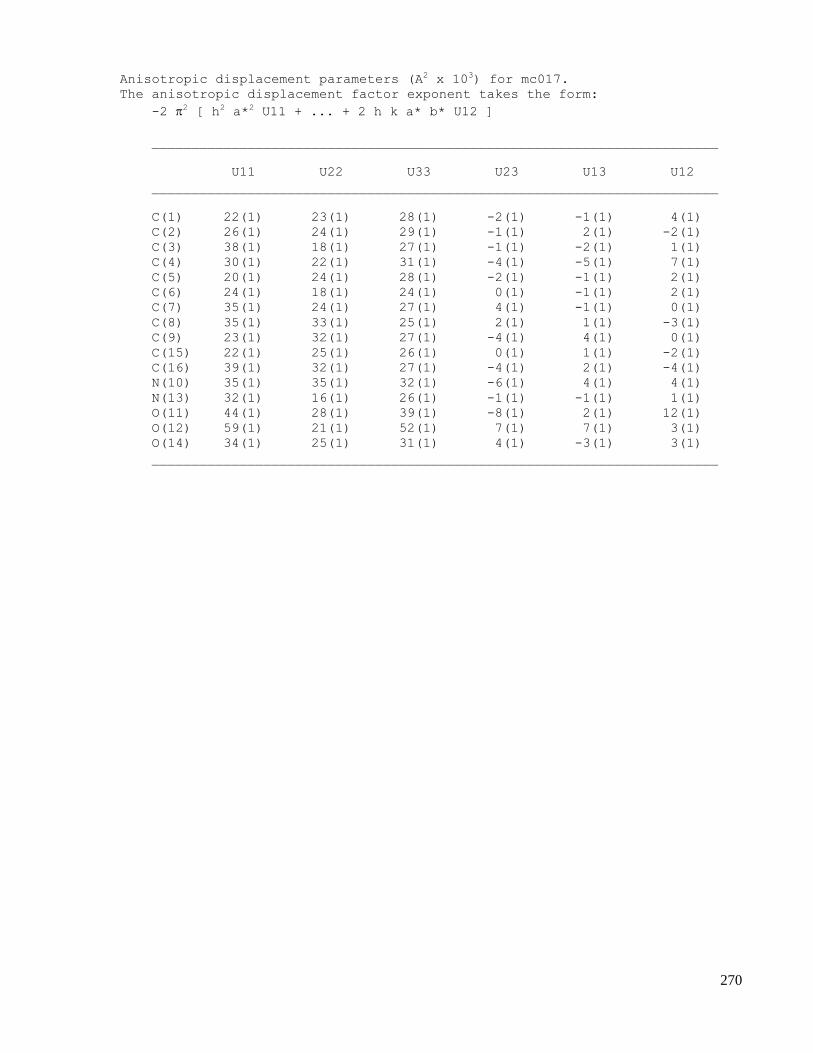
270
Anisotropic displacement parameters (A2 x 103) for mc017.
The anisotropic displacement factor exponent takes the form:
-2 2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
_______________________________________________________________________
U11 U22 U33 U23 U13 U12
_______________________________________________________________________
C(1) 22(1) 23(1) 28(1) -2(1) -1(1) 4(1)
C(2) 26(1) 24(1) 29(1) -1(1) 2(1) -2(1)
C(3) 38(1) 18(1) 27(1) -1(1) -2(1) 1(1)
C(4) 30(1) 22(1) 31(1) -4(1) -5(1) 7(1)
C(5) 20(1) 24(1) 28(1) -2(1) -1(1) 2(1)
C(6) 24(1) 18(1) 24(1) 0(1) -1(1) 2(1)
C(7) 35(1) 24(1) 27(1) 4(1) -1(1) 0(1)
C(8) 35(1) 33(1) 25(1) 2(1) 1(1) -3(1)
C(9) 23(1) 32(1) 27(1) -4(1) 4(1) 0(1)
C(15) 22(1) 25(1) 26(1) 0(1) 1(1) -2(1)
C(16) 39(1) 32(1) 27(1) -4(1) 2(1) -4(1)
N(10) 35(1) 35(1) 32(1) -6(1) 4(1) 4(1)
N(13) 32(1) 16(1) 26(1) -1(1) -1(1) 1(1)
O(11) 44(1) 28(1) 39(1) -8(1) 2(1) 12(1)
O(12) 59(1) 21(1) 52(1) 7(1) 7(1) 3(1)
O(14) 34(1) 25(1) 31(1) 4(1) -3(1) 3(1)
_______________________________________________________________________
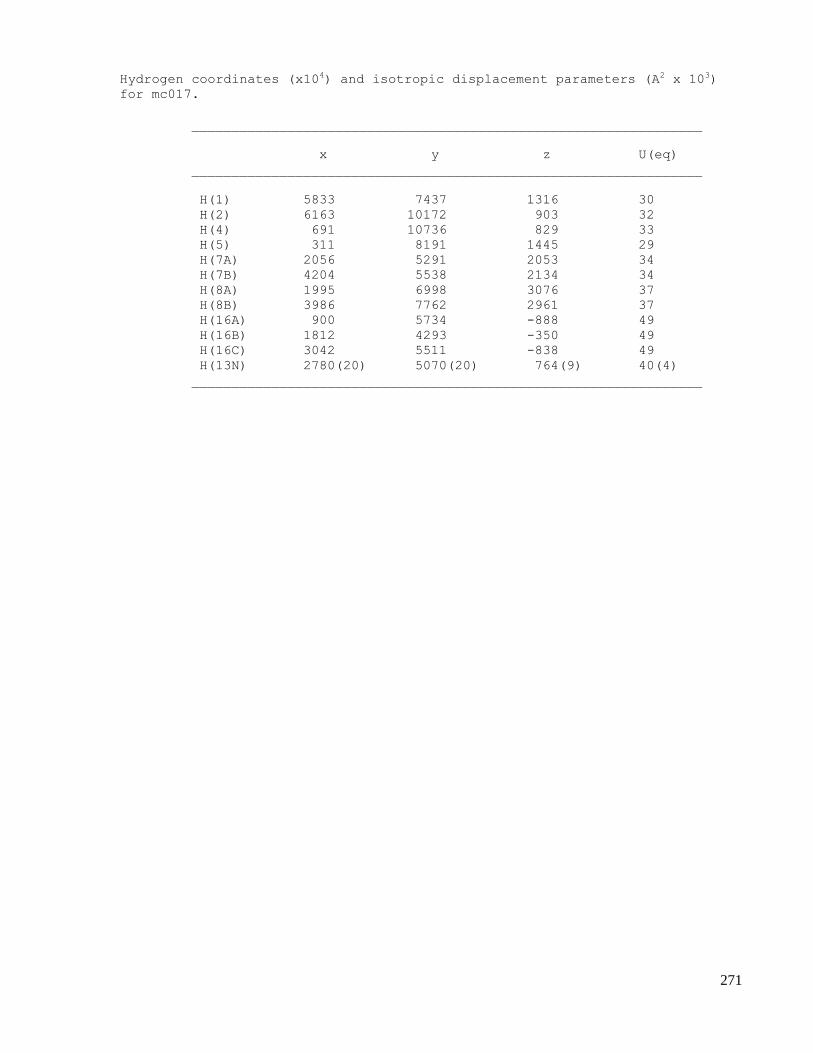
271
Hydrogen coordinates (x104) and isotropic displacement parameters (A2 x 103)
for mc017.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(1) 5833 7437 1316 30
H(2) 6163 10172 903 32
H(4) 691 10736 829 33
H(5) 311 8191 1445 29
H(7A) 2056 5291 2053 34
H(7B) 4204 5538 2134 34
H(8A) 1995 6998 3076 37
H(8B) 3986 7762 2961 37
H(16A) 900 5734 -888 49
H(16B) 1812 4293 -350 49
H(16C) 3042 5511 -838 49
H(13N) 2780(20) 5070(20) 764(9) 40(4)
________________________________________________________________

272
Torsion angles [deg] for mc017.
________________________________________________________________
C(6)-C(1)-C(2)-C(3) 6.23(18)
C(1)-C(2)-C(3)-O(12) -176.07(12)
C(1)-C(2)-C(3)-C(4) 8.35(17)
O(12)-C(3)-C(4)-O(11) -64.99(14)
C(2)-C(3)-C(4)-O(11) 110.64(11)
O(12)-C(3)-C(4)-C(5) -178.42(11)
C(2)-C(3)-C(4)-C(5) -2.79(15)
O(11)-C(4)-C(5)-C(9) -22.36(10)
C(3)-C(4)-C(5)-C(9) 92.58(11)
O(11)-C(4)-C(5)-C(6) -131.31(10)
C(3)-C(4)-C(5)-C(6) -16.37(15)
C(2)-C(1)-C(6)-N(13) 102.91(12)
C(2)-C(1)-C(6)-C(5) -23.50(15)
C(2)-C(1)-C(6)-C(7) -137.03(11)
C(9)-C(5)-C(6)-N(13) 157.55(9)
C(4)-C(5)-C(6)-N(13) -94.01(12)
C(9)-C(5)-C(6)-C(1) -80.20(10)
C(4)-C(5)-C(6)-C(1) 28.24(14)
C(9)-C(5)-C(6)-C(7) 39.22(10)
C(4)-C(5)-C(6)-C(7) 147.65(10)
N(13)-C(6)-C(7)-C(8) -159.61(9)
C(1)-C(6)-C(7)-C(8) 81.40(11)
C(5)-C(6)-C(7)-C(8) -37.44(11)
C(6)-C(7)-C(8)-C(9) 19.32(12)
C(7)-C(8)-C(9)-N(10) -158.72(13)
C(7)-C(8)-C(9)-C(5) 6.46(13)
C(4)-C(5)-C(9)-N(10) 13.30(13)
C(6)-C(5)-C(9)-N(10) 138.18(11)
C(4)-C(5)-C(9)-C(8) -154.18(10)
C(6)-C(5)-C(9)-C(8) -29.31(12)
C(8)-C(9)-N(10)-O(11) 167.17(12)
C(5)-C(9)-N(10)-O(11) 2.43(14)
O(14)-C(15)-N(13)-C(6) -8.92(17)
C(16)-C(15)-N(13)-C(6) 170.69(10)
C(1)-C(6)-N(13)-C(15) -70.52(13)
C(5)-C(6)-N(13)-C(15) 53.59(14)
C(7)-C(6)-N(13)-C(15) 167.86(10)
C(9)-N(10)-O(11)-C(4) -18.10(12)
C(5)-C(4)-O(11)-N(10) 25.77(11)
C(3)-C(4)-O(11)-N(10) -95.20(10)
________________________________________________________________

273
Hydrogen Bonds
Donor --- H....Acceptor [ ARU ] D - H H...A D...A D -
H...A --------------------------------------------------------------------
------------
N(13) --H(13N) ..O(12) [ 1545.01] 0.851(15) 2.077(15) 2.9166(13)
169.0(15)
Translation of ARU-code to Equivalent Position Code
===================================================
[ 1545. ] = x,-1+y,z