crystal structures of an oligopeptide-binding protein from the biosynthetic pathway of the...
TRANSCRIPT

Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/40035035
CrystalStructuresofanOligopeptide-BindingProteinfromtheBiosyntheticPathwayoftheβ-LactamaseInhibitorClavulanicAcid
ARTICLEinJOURNALOFMOLECULARBIOLOGY·NOVEMBER2009
ImpactFactor:4.33·DOI:10.1016/j.jmb.2009.11.045·Source:PubMed
CITATIONS
6
READS
54
8AUTHORS,INCLUDING:
AlasdairKennethMackenzie
NorwegianUniversityofLifeSciences(NMBU)
22PUBLICATIONS374CITATIONS
SEEPROFILE
KarinValegård
UppsalaUniversity
37PUBLICATIONS2,069CITATIONS
SEEPROFILE
AmanIqbal
UniversityofToronto
6PUBLICATIONS30CITATIONS
SEEPROFILE
NadiaJeanKershaw
TheWalterandElizaHallInstituteofMedic…
35PUBLICATIONS785CITATIONS
SEEPROFILE
Availablefrom:NadiaJeanKershaw
Retrievedon:04February2016

doi:10.1016/j.jmb.2009.11.045 J. Mol. Biol. (2010) 396, 332–344
Available online at www.sciencedirect.com
Crystal Structures of an Oligopeptide-Binding Proteinfrom the Biosynthetic Pathway of the β-LactamaseInhibitor Clavulanic Acid
Alasdair K. Mackenzie1†, Karin Valegård1†, Aman Iqbal2,Matthew E. C. Caines2, Nadia J. Kershaw2, Susan E. Jensen3,Christopher J. Schofield2⁎ and Inger Andersson1⁎
1Department of MolecularBiology, Swedish University ofAgricultural Sciences, Box 590,S-751 24 Uppsala, Sweden2Department of Chemistry andthe Oxford Centre forIntegrative Biology, Universityof Oxford, The ChemistryResearch Laboratory, MansfieldRoad and the Oxford Centre forIntegrative System Biology,Oxford, OX1 3TA, UK3Department of BiologicalSciences, University of Alberta,Edmonton, Alberta, CanadaReceived 27 July 2009;received in revised form17 November 2009;accepted 18 November 2009Available online24 November 2009
*Corresponding authors. E-mail [email protected]@xray.bmc.uu.se.† A.K.M. and K.V. contributed eqAbbreviations used: CA, clavulan
reading frame; OppA1 and OppA2oligopeptide-binding proteins 1 andoligopeptide-binding protein; SBP,protein; AppA, another oligopeptidglutathione S-transferase; PDB, ProSeMet, selenomethionine; PEG, polyTris-buffered saline.
0022-2836/$ - see front matter © 2009 E
Clavulanic acid (CA) is a clinically important β-lactamase inhibitor that isproduced by fermentation of Streptomyces clavuligerus. The CA biosynthesispathway starts from arginine and glyceraldehyde-3-phosphate andproceeds via (3S,5S)-clavaminic acid, which is converted to (3R,5R)-clavaldehyde, the immediate precursor of (3R,5R)-CA. Open readingframes 7 (orf7) and 15 (orf15) of the CA biosynthesis cluster encodeoligopeptide-binding proteins (OppA1 and OppA2), which are essential forCA biosynthesis. OppA1/2 are proposed to be involved in the bindingand/or transport of peptides across the S. clavuligerus cell membrane.Peptide binding assays reveal that recombinant OppA1 and OppA2 binddi-/tripeptides containing arginine and certain nonapeptides includingbradykinin. Crystal structures of OppA2 in its apo form and in complexwith arginine or bradykinin were solved to 1.45, 1.7, and 1.7 Å resolution,respectively. The overall fold of OppA2 consists of two lobes with a deepcavity in the center, as observed for other oligopeptide-binding proteins.The large cavity creates a peptide/arginine binding cleft. The crystalstructures of OppA2 in complex with arginine or bradykinin reveal that theC-terminal arginine of bradykinin binds similarly to arginine. The resultsare discussed in terms of the possible roles of OppA1/2 in CA biosynthesis.
© 2009 Elsevier Ltd. All rights reserved.
Keywords: clavulanic acid; β-lactam; β-lactamase inhibitor; oligopeptide-binding proteins; solute-binding proteins
Edited by G. SchulzIntroduction
Clavulanic acid (CA) is a clinically importantinhibitor of the class A β-lactamases and is produced
resses:;
ually to this work.ic acid; ORF, open, clavulanate2; OPP,
substrate-bindinge permease A; GST,tein Data Bank;ethylene glycol; TBS,
lsevier Ltd. All rights reserve
by fermentation of the actinomycete Streptomycesclavuligerus. In addition to CA, S. clavuligerus alsoproduces other β-lactam metabolites including‘clavams’ (e.g., alanylclavam) and antibiotics fromthe penicillin and cephalosporin subfamilies of β-lactam antibiotics. Although a β-lactam, CA isinsufficiently active as an antibiotic to be used as asole therapeutic agent and, hence, is used incombinationwith a penicillin antibiotic, for example,amoxicillin (for reviews, see Refs. 1–4).The biosynthesis of CA1–3 (Fig. 1) proceeds via
multiple steps from the precursors D-glyceraldehydeand L-arginine. CA and its immediate biosyntheticprecursor, clavaldehyde, possess the (3R,5R) stereo-chemistry,5 whereas all other isolated clavams havethe (3S,5S) stereochemistry. The enzymes respon-sible for catalyzing the conversion of D-glyceralde-
d.

Fig. 1. Outline of the CA biosynthetic pathway in S. clavuligerus, showing identified intermediates and enzymesinvolved. The proposed branchpoint intermediate, clavaminic acid, is shown in blue and CA is shown in red [CEAS(orf2):carboxyethyl arginine synthase, BLS(orf3): β-lactam synthase, CAS(orf5): clavaminate synthase, PAH(orf4): proclavami-nate synthase, OAT2(orf6): ornithine acetyl transferase, CAD(orf9): clavulanate dehydrogease].3
333OPP Structures from the CA Biosynthetic Pathway
hyde and L-arginine to clavaminic acid [encoded byopen reading frames 2–6 (orf2–orf6) of the CAcluster] have been isolated and crystal structuresfor them have been solved,6–10 along with (3R,5R)-clavaldehyde dehydrogenase (encoded by orf9).11
The product of orf610 is an ornithine acetyltransferase proposed to ‘feed’ arginine into thepathway. Studies on the CA pathway are compli-cated by the presence of three gene clusters in S.clavuligerus with apparently overlapping functions.One gene cluster includes all the genes involveddirectly in CA biosynthesis, self-resistance to CA,and regulation. The second and third gene clustersencode proteins involved in (3S,5S)-clavam bio-synthesis. The three gene clusters appear to havesome functional overlap, since all three encode atleast some of the enzymes catalyzing steps up toclavaminic acid biosynthesis activity, which actsas a branch point between CA and clavambiosynthesis.12–15
The process by which (3S,5S)-clavaminic acid orN-glycyl-clavaminic acid undergoes the requisiteoxidation and epimerization steps to yield (3R,5R)-clavaldehyde is unknown but likely involves one ormore of the products of orf10–orf17 of the CA genecluster. Gene disruption studies on the orfs ofunassigned functions (orf10–orf17) have indicated
that all of the products of these orfs are involved inthe biosynthesis of CA.16 Two of the CA biosynthe-sis cluster genes (orf7 and orf15) are closely related insequence (∼48% identity) but have no functionalcross complementation.17Gene disruption studies coupled to growth in
different media and bioinformatics analyses haveprovided evidence that the products of orf7 andorf15 (named OppA1/OppA2 for CA biosynthesisoligopeptide-binding proteins 1/2) are members ofthe oligopeptide-binding protein (OPP) or substrate-binding protein (SBP) family,17 involved in theimport of peptides in S. clavuligerus. In contrast tothe orf7 disruption, S. clavuligerus disrupted in orf15was unable to grow on a medium containing thepeptide bradykinin (RPPGFSPFR) as a nitrogensource, suggesting a difference in selectivity in theproducts of orf7 and orf15.17 The presence ofoligopeptide transporters in the CA biosynthesiscluster is notable because their involvement inantibiotic biosynthesis has not been previouslyreported; in the light of the OppA1/2 work, itappears that oligopeptide transporters may beinvolved in other pathways, including the calichea-micin biosynthesis clusters.17,18
Sequence analysis of OppA2 revealed similarity tomembers of the periplasmic binding protein-like II

334 OPP Structures from the CA Biosynthetic Pathway
subfamily of OPPs.16,17,19 These OPPs (or, moregenerally, SBPs) form part of the ATP-bindingcassette transport system and are responsible forthe import of nutrients into bacterial cells. OppA2shows the highest sequence similarity with mem-bers of the ‘Sub Family Five’ SBPs,20 which is thelargest of the SBP families with specificity forpeptides. A sequence alignment of OppA2 withOppA1, AppA (another oligopeptide permease A)from Frankia alni, and Dpp from Escherichia coli isshown in Supplementary Fig. S1.Most SBPs are thought to transport target peptides
with a low degree of specificity. However, thegenetic studies on CA biosynthesis suggest thattheir role in CA biosynthesis may be more specific;16
it has been proposed that they might be involved insensing signaling peptides that ‘trigger’ CA bio-synthesis.17 Here, we report structural studies aimedat defining how OppA1/2 bind peptides and theirstructural relationship with SBPs that are notinvolved in antibiotic biosynthesis.
Fig. 2. Intensities of di- and tripeptides upon (a) OppAglutathione to GST. The x-axis shows the peptides screened,shown as a percentage. O in peptide ROG is amino acid ornithsubstantial binding to the di-/tripeptides, except with glutatripeptides (data not shown). The error bars are shown as stan
Results
Cloning, expression, and purification of OppA2
The orf7 and orf15 genes were inserted into thepET28a vector retaining the complete N-terminalregion. The resultant constructs were expressed inE. coli BL21 DE3 to enable production of solubleOppA1 and OppA2. A three-step purificationprotocol, involving sequential anion-exchange col-umns (Q-Sepharose and Mono-Q) and gel-filtrationchromatography, was established, and large-scaleproduction of OppA2 was then carried out.Purified OppA1 and OppA2 migrated at about60 kDa by SDS-PAGE and were N95% pure (bySDS-PAGE analysis, see Materials and Methods fordetails). The orf7 (OppA1) and orf15 (OppA2) geneswere also inserted into the pGEX6P1 vector to giveconstructs to produce N-terminally glutathione S-transferase (GST)-tagged proteins (GST-OppA1 and
2 and (b) OppA1 binding relative to that observed forand the y-axis corresponds to the relative spot intensitiesine. When used as a control, GST protein did not show anythione. GST-ORF9 also did not bind to any of the di-/dard error of mean.

335OPP Structures from the CA Biosynthetic Pathway
GST-OppA2) that were used in peptide bindingassays.
Peptide binding assays
Because the work of Lorenzana et al.17 hadindicated that OppA2 is involved in the transportof the arginine-containing peptide bradykinin(RPPGFSPFR) and arginine is involved in the firststep of CA biosynthesis, we initially exploredwhether OppA2 binds arginine-containing peptides.A set of di- and tripeptides, of the form R-X or R-X-G(Fig. S2), were synthesized on a membrane using thepeptide array synthesis method.21,22 In addition toprobing the nature of the amino acid in position 2 (X),we also wanted to investigate the influence of thelength of the peptide. To avoid imposing sequencespecificity on the third ligand, we used glycine atposition 3. The binding of the membrane-boundpeptides to OppA1/2 was investigated using GST-
Fig. 3. Views from the OppA2 crystal structure showing seOppA2 is bilobal and composed of three domains, with domainforming lobe 2. (b) The topological arrangement of the 16 α-hethe three domains, colored according to (a).
tagged OppA1 and OppA2. Glutathione (γ-L-gluta-myl-L-cysteinylglycine) was synthesized on themembrane as a control for the GST protein bindingand to enable the quantification of the binding ofGST-OppA1/2 to the synthesized peptides. A non-apeptide, VDSKNTSSW,23 known to bind to an OPPfrom Bacillus subtilis (AppA), was used as a control.The results revealed that GST-OppA2 bound morestrongly to di- and tripeptides with amino acidspossessing cyclic/aromatic side chains [phenylala-nine (F), histidine (H), tyrosine (Y), and proline (P)] atposition 2 (Fig. 2a). Other amino acids at position 2did not display binding, within the limits ofdetection. Bradykinin (RPPGFSPFR) apparentlybound tightly to GST-OppA2, whereas theVDSKNTSSW peptide bound less strongly toOppA2. Similar binding patterns and spot intensitieswere observed for GST-OppA1 (Fig. 2b), with theonly difference that the VDSKNTSSW peptide wasnot observed to bind to GST-OppA1. Bradykinin
condary and tertiary structures. (a) The overall structure ofs I (blue) and II (green) forming lobe 1 and domain III (red)lices (labeled A–P) and 13 β-strands (labeled 1–13) creating

336 OPP Structures from the CA Biosynthetic Pathway
was also found to bind toGST-OppA1.Overall, theseresults indicate that OppA1 and OppA2 indeed bindto arginine-containing peptides, but with slightlydifferent specificities; that is, in this sense, the resultsare supportive of the reported genetic analyses.17
However, both OppA1 andOppA2 bind bradykinin,in contrast to the genetic analyses indicating thatonly OppA2 is involved in bradykinin transport.Thus, our results imply that biochemical selectivity isnot the only factor in determining the functionalselectivity of OppA1/2.
Crystallization of OppA2
We initiated crystallographic studies to investi-gate the structural nature of peptide binding to
Fig. 4. Structural similarity of OppA2 to the dipeptide-binaligned to domain III of OppA2 to show the relative differenceOppA2 (blue) and DppA (yellow) in an open conformation (closed conformation (PDB code 1DPP).
OppA2. Wild-type OppA2 crystals were obtainedby microseeding of needle cluster crystals into dropsprepared under the same conditions. This proceduregave rod-shaped crystals that diffracted to 1.45 Åresolution (for further details, see Materials andMethods). To investigate the binding of ligands toOppA2, we prepared complexes by soaking crystalsof OppA2 with L-arginine, other amino acids, andpeptides or cocrystallizing OppA2 with bradykinin(see below).
Overall structure of OppA2
The overall structure of OppA2 comprises 16 α-helices (αA–αP) and 13 β-strands (β1–β13) that formthree β-sheets. The secondary-structure elements are
ding protein DppA from E. coli. The structures have beens between the open and closed conformations of DppA. (a)PDB code 1DPE). (b) OppA2 (blue) and DppA (red) in a

337OPP Structures from the CA Biosynthetic Pathway
linked by a series of meandering loops that accountfor ∼60% of the overall structure. The overall foldresults in two lobes and gives rise to a pear-shapedprotein of dimensions ∼72 Å×60 Å×40 Å (Fig. 3a).A large cleft formed between the two lobes runs theentire length of the molecule.The two lobes are formed from three domains of
mixed α/β secondary structure (Fig. 3b). Domain Istarts at β1 and αA and is formed by threepolypeptide segments arising from separate regionsof the sequence (residues 7–65, 214–297, and 529–560). After helix αA, the backbone extends intodomain II (residues 65–213), which is composed oftwo β-hairpins (β2/β3 and β4/β5) and four α-helices (αB–αE). From domain II, the polypeptidebackbone returns to domain I and forms threeadditional antiparallel strands (β6–β8) of a four-stranded mixed β-sheet and three α-helices(αF–αH). Together, domains I and II form the largerlobe; the smaller lobe is formed by domain III(residues 303–531). Domain III is composed of amixed, five-stranded β-sheet (β9–β13) surroundedby eight α-helices (αI–αP). Domain III is connectedto domain I by two inter-domain linker regions(residues 297–302 and 530–532), of which the secondlinker region returns to complete domain I.DALI24 and DEJAVU25 searches of the structural
database supported the proposal that OppA2 isstructurally most closely related to Sub Family Fiveof peptide-binding SBPs. This subfamily includesthe periplasmic peptide-binding proteins DppA,26
AppA,23 and OppA27 [Protein Data Bank (PDB)codes 1DPE, 1XOC, and 1OLA, respectively] and anickel binding protein, NikA28 (PDB code 1UIU).There is also similarity between OppA2 and otherputative SBPs, including Ylib from E. coli, theperiplasmic OPP (TM1223) from Thermotoga mar-itima, and the lipoprotein LpqW29 (PDB codes1UQW, 1VR5, and 2GRV, respectively) for whichthere is structural information but not a clearfunction. An alignment of Cα atoms from SBPand putative SBP structures, including OppA2,reveals a similar arrangement of the three domains,although the architecture and folding of the main-chain backbone in the respective domains differbetween the proteins. A characteristic sequencemotif of Family Five SBPs (GPGEVSDGGRTW-TYRLRRGLRY, identical residues are underlined)20(PROSITE, PS01040), which is located in domain II(residues Gly88-Tyr109),17,30 is also present inOppA2.
Peptide/arginine substrate binding cleft
SBPs or OPPs capture their respective ligands in thecleft formed between the two lobes. Ligand binding isassociated with domain movements, where lobes 1and 2 close around the substrate in a mechanism thathas been likened to a ‘Venus flytrap’.31 As implied bycrystallographic analyses, the movements can belarge; for example, lobe 1 of the DppA undergoes a55° rigid-body rotation when moving between theopen26 and closed32 conformations. Although some
OPPs/SBPs can co-purify with ligands bound, thiswas not the case for recombinant OppA2. However,in both the native and selenomethionine (SeMet)OppA2 forms, cryo-protectant molecules [glycerol,polyethylene glycol (PEG) 400] were observed in thecleft between the two lobes, which is the knownsubstrate binding site for OPPs.27
Structures were obtained for OppA2 in complexwith arginine (a precursor of CA) and bradykinin,both to 1.7 Å resolution. Only small changes wereobserved in the overall OppA2 structures with andwithout ligands (r.m.s.d.'s, 0.13–0.24 Å), and noconformational movements were detected betweenthe two lobes. This observation contrasts with theSeMet structure, which displays a different crystalpacking with two molecules in the asymmetric unitinstead of one. An alignment of the two molecules inthe asymmetric unit of the SeMet structure revealeda small rigid-body difference due to an ∼8° hingemovement between the two lobes and an r.m.s.d.between the twomolecules in the asymmetric unit of1.1 Å. An alignment of the native and SeMetstructures also revealed small rigid-body differences,∼9° to molecule A and ∼14° to molecule B, of theSeMet structure (Fig. S3). Small differences of similarmagnitude were also observed when the SeMetstructure is compared to the structures of thearginine and bradykinin complexes. It is likely thatthese differences result, at least in part, from crystalpacking forces. Overall, it appears that the OppA2structure more closely resembles the open ratherthan the closed conformation compared to thereported OPP/SBP structures.33 A comparison ofthe OppA2 structure with the open and closedstructures of DppA from E. coli is shown in Fig. 4.In the OppA2:arginine complex, the arginine is
bound in the cleft between the two lobes, in closeproximity to the intersection of the three domains(Fig. 5a). The binding of arginine is stabilized by amixture of direct and solvent-mediated hydrogenbonds. The arginine carboxylate is positioned toform a hydrogen bond with the main-chain nitrogenof Gly454 and to a nearby solvent acetate ion, whichalso interacts with the other oxygen of the argininecarboxyl group. The α-amino nitrogen of arginineinteracts with residues in domain III includingAsp456, a binding mode observed in otherOPPs.23,27 Additional binding interactions occurbetween the amino acid arginine and the main-chain carbonyl and amide groups of Gly454. Theindole rings of Trp57 and Trp453 are also positionedclose to the amino acid binding site. The guanidinogroup of arginine points towards residues fromdomains I and II, implying that all three domains areinvolved in arginine binding.In order for arginine to bind in the cleft, the side
chains of Trp57 (domain I) and Gln189 (domain II)are shifted relative to the uncomplexed structures toprovide space for the guanidine group. In addition,one glycerol molecule in the uncomplexed structureis displaced upon binding of arginine. The Nη2
nitrogen of the guanidine group is positioned toform a hydrogen bond with the carbonyl oxygen of
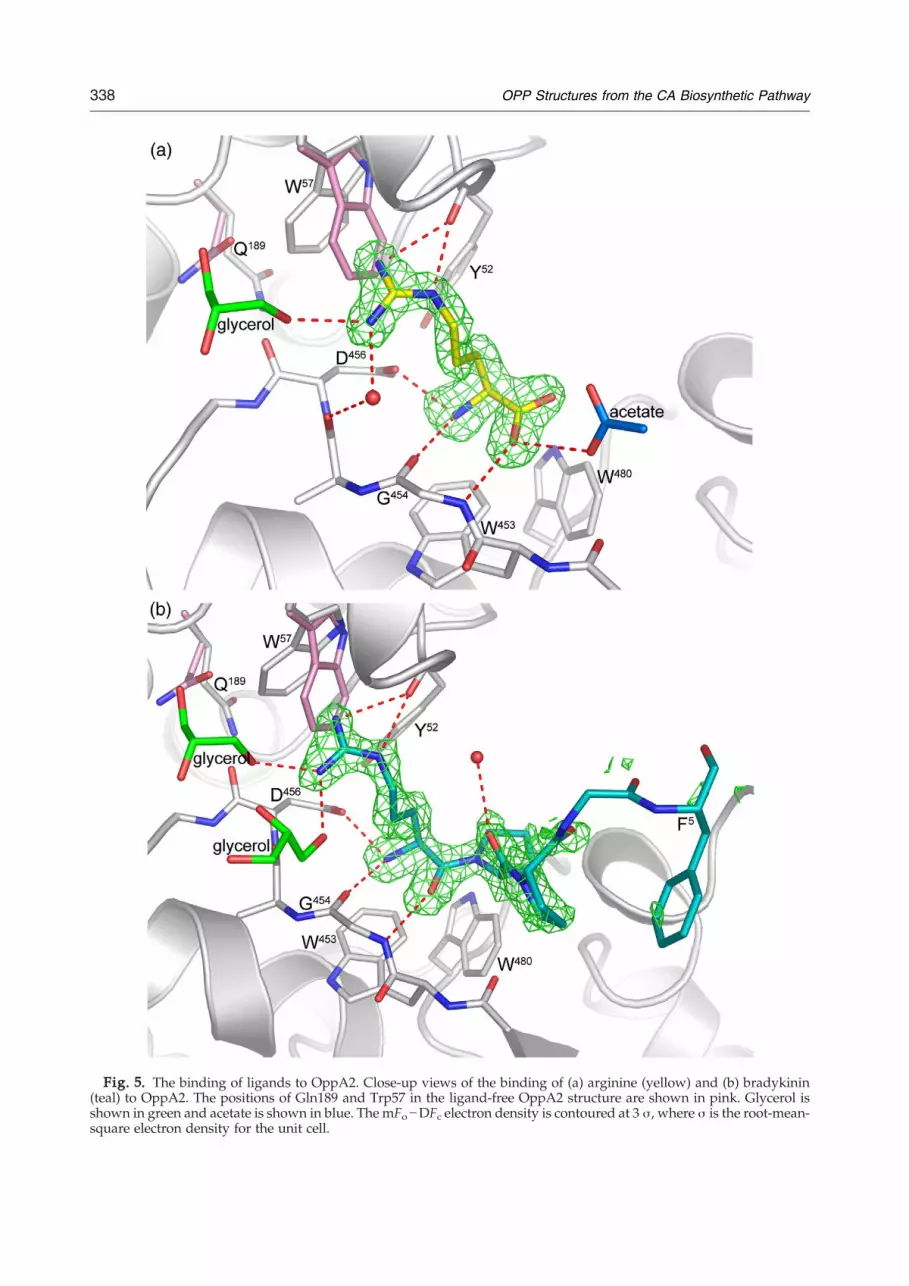
Fig. 5. The binding of ligands to OppA2. Close-up views of the binding of (a) arginine (yellow) and (b) bradykinin(teal) to OppA2. The positions of Gln189 and Trp57 in the ligand-free OppA2 structure are shown in pink. Glycerol isshown in green and acetate is shown in blue. The mFo−DFc electron density is contoured at 3 σ, where σ is the root-mean-square electron density for the unit cell.
338 OPP Structures from the CA Biosynthetic Pathway

339OPP Structures from the CA Biosynthetic Pathway
Tyr52 and also to interact with the indole electronsof Trp57. The Nη1 nitrogen is positioned to interactwith the carbonyl oxygen of Ala455 (domain III) viaa bridging water molecule and also interacts with anearby glycerol molecule (Fig. 5a). The Nɛ nitrogeninteracts with the carbonyl of Tyr52. The presence ofthree tryptophan residues in the binding cleft ofOppA2 bears some resemblance to the mechanismby which ProX binds its glycine and proline-betainesubstrates.34 However, in OppA2, the tryptophanresidues Trp57, Trp453, and Trp480 are morespatially separated compared to those in ProX. Ofthese, Trp57 is conserved in some (including OppA1from S. clavuligerus, see Fig. S1) but not all OPPs/SBPs, whereas Trp543 appears to be well conserved(see further below). The observation that OppA2 canbind arginine is consistent with a role for OppA2,either in transport of arginine or of its derivatives inCA biosynthesis.The overall structure of the protein in the OppA2:
bradykinin complex was very similar to the
Fig. 6. Binding of bradykinin in the cavity of (a) OppA2 fr(OppA) from L. lactis (PDB code 3DRG), which is in a ‘closed’orientation with the ligand-binding cavity facing the viewer. Tcolored magenta. (c) Close-up view of bradykinin binding in O(light gray). The structures were aligned to overlap residuesmagenta, whereas bradykinin in the OppA model is colored d
structures of the non-liganded protein or thearginine complex. Bradykinin binds in the OppA2binding cleft via its N-terminal arginine residue(Figs. 5b and 6a). There is an apparent change of theconformation of a loop (residues 424–431) relative tothe apo structure in order to accommodate bradyki-nin, including a small shift in the side-chainconformation of Phe431 to accommodate Pro2 ofbradykinin. Apart from the bradykinin N-terminalarginine, electron density was observed for thefollowing four bradykinin residues: Pro2, Pro3,Gly4, and Phe5. These residues interact predomi-nantly by hydrophobic contacts: Trp480 of OppA2 toPro2 of bradykinin, Phe431 to Pro2 and Pro3, Arg276to Gly4, and Phe402 to Phe5 of bradykinin. The N-terminal arginine of bradykinin is apparently boundin a very similar way to free arginine. Whereas nodensity was observed for residues 6–8 of bradykinin,electron density was present, which could bemodeled as the C-terminal arginine residue. Thisresidue makes crystal contacts with a neighboring
om S. clavuligerus and (b) oligo peptide-binding protein Aform. The structures in (a) and (b) are shown in a similarhe protein is depicted as a gray surface and bradykinin isppA2 from S. clavuligerus (pink) and OppA from L. lactisin domain III. Bradykinin bound to OppA2 is shown inark gray.

340 OPP Structures from the CA Biosynthetic Pathway
molecule. The absence of any conformational changein the protein as well as in the N-terminal residue ofthe ligand when compared to the OppA2:argininecomplex is notable. It is possible that crystal latticeconstraints prevent the active site from closingcompletely on the ligand and that a closed complex(see below) would feature additional protein–ligandinteractions not observed here. The crystal structureof the OPP (OppA) from Lactococcus lactis in complexwith bradykinin has been reported.35 In contrast tothe OppA2 structure (Fig. 6a), the L. lactis OppAbinds bradykinin in a closed conformation (Fig. 6b)with the bound peptide completely buried in thecavity between the two domains. A superposition ofthe bradykinin structures by least-squares fittingwasmade such that theα-carbon atoms of domain III(the smaller lobe, named domain II in Ref. 35) werealigned. This positions bradykinin of OppA2 suchthat residues 1–3 overlap with residues 4–6 ofbradykinin when complexed with the L. lactisOppA (Fig. 6c). As a consequence, bradykininbinds deeper within the closed structure of L. lactisOppA than in OppA2. The binding of bradykinin inthe S. clavuligerus OppA2 structure is achievedthrough small local conformational changes, forexample, a different conformation of the loopbearing residue Asp456. The corresponding residuein the L. lactisOppA is Ala476; the less bulkyAla sidechain apparently favors a deeper binding ofbradykinin.35 The bulky side chain of Trp453 alsointeracts with bradykinin in OppA2. This residue isconserved in a number of OPP/SBPs (Fig. S1). In theL. lactis OppA, the corresponding residue is Trp473,but the side chain of this residue adopts a differentconformation with its indole ring being almostperpendicular relative to the indole ring of Trp453in S. clavuligerus OppA2 (Fig. 6c). Trp480 is notconserved in L. lactis OppA;35 the correspondingresidue is Tyr491. Crystal structures of the open formof L. lactis OppA in complex with various peptideshave been reported.35 Compared to the S. clavuligerusOppA2 structure presented herein, the two lobes aremore separated from each other in the open OppAstructures; the ligands bind to the smaller lobe onlyand are partly disordered, consistent with the broadspecificity of L. lactis OppA for different peptides.OppA2 crystals were soaked with Gly4 and Gly6
and with the amino acids phenylalanine andproline. However, no density could be observedfor these compounds in the corresponding electrondensity maps. CA and a potential mimic of NAG-clavam (N-acetyl-ornithine-glycine) were alsosoaked into crystals, but no electron density wasobserved for either compound. Furthermore, noconformational changes in the side chains of Trp57or Gln189 were observed in the above structures,while the cryo-protectant glycerol could clearly beseen in the electron density maps (data not shown).The positions of the glycerol molecules were similarto those observed in the apo structure. These resultsare in support of the peptide binding assayssuggesting that OppA2 has a preference for bindingarginine and arginine-containing peptides.
Cellular localization experiments
Antibodies were raised against the purified pro-teins to investigate the cellular localization ofOppA1/2. Western blot analyses were performedon the insoluble and soluble fractions of S. clavuli-gerus. The Western blotting studies implied thepresence of OppA2 predominantly in the solublefraction of cells grown under optimal CA producingconditions (soymedium; Figs. S4 and S5).NoOppA2was detected in culture supernatants at any sampletime. However, when the analyses were carried outon cells grown in seed medium (TSB+1% starch),conditions where little CA is produced, OppA2 wasapparently distributed between both the soluble andthe insoluble fractions (Fig. S5). Preliminary studiesto determine whether soluble OppA2 arose from thecytoplasmic or periplasmic cell compartment werehampered by extensive lysis of protoplasts duringformation but nonetheless suggested that at leastsome OppA2 was present in the cytoplasm.
Discussion
Overall structure and peptide binding assays
Together, the biochemical and crystallographicanalyses confirm that OppA2 and, likely, OppA1 arevery closely related to known OPPs. The structuralstudies demonstrate that OppA2 possesses the stereo-typical structural features of OPPs, including threedomains that form two lobes with an intervening‘substrate-binding’ cleft and have revealed howOppA2 may bind arginine and bradykinin. Althoughwe have not yet obtained a structure for OppA1, thepeptide binding assays and a model of OppA1 basedon the OppA2 structure are consistent with themhaving similar but different selectivities in support ofthe genetic studies.17 The observed binding ofarginine and bradykinin supports the proposal thatOppA1 and OppA2 act to import peptides, andpossibly arginine, into S. clavuligerus to act as afeedstock for CA and/or clavam biosynthesis.However, the subcellular localization studiesrevealed that, at least under some growth conditions,OppA2 is present in both the soluble and theinsoluble fractions, suggesting that it may haveroles other than as a ‘simple’ peptide transporter.Alternatively, OppA2 may be solely envelope-asso-ciated, but by so labile an interaction that even mildcell breakage conditions can result in solubilization.Only small structural changes were observed upon
bindingof ligands toOppA2.This contrastswithworkon other OPPs/SBPs, where crystallographic studieshave revealed that ligand binding is associated withsignificant domain movements, wherein lobe 1 andlobe 2 close in a Venus flytrap mechanism.26,27,31–33 Itis possible that the lack of conformational changesobserved for ligand binding by OppA2 is due tolimitations imposed by the crystal lattice, but it cannotbe ruled out that it has functional significance,

341OPP Structures from the CA Biosynthetic Pathway
especially if OppA2 has roles in CA biosynthesis otherthan peptide/amino acid transport.The observation that genetic disruption of either
orf7 or orf15 ablates CA production also suggeststhat provision of nutrients for CA biosynthesis maynot be the only role for OppA1/2. There is precedentfor OPPs having roles other than as simple nutrienttransporters and it is possible that OppA1/2 areinvolved in sensing signaling peptides that triggerCA biosynthesis.17 Sanchez and Brana36 haveobserved that peptides accumulating in culturebroths are capable of stimulating CA production.There is evidence thatmorphological development
and β-lactam antibiotic production in S. clavuligerusmay be stimulated by related signaling peptides. Theability of Streptomyces spp. to erect aerial hyphae andspores is regulated by the bld genes.37 The bldKoperon in Streptomyces coelicolor includes five genes(A–E) with homology to the ATP-binding cassettefamily of transmembrane transporters.38 One of thegenes, bldKB, encodes a putative OPP, proposed tobind a signaling peptide that stimulates the forma-tion of aerial mycelia and spores. Notably, both a‘bald’ phenotype and biosynthesis of CA, 5S-clavams, and cephamycin C in S. clavuligerus areblocked by mutations to certain bld genes althoughthe bldK operon has not yet been examined in thisspecies.39 OppA1/2 display ∼36% sequence simi-larity with BldKB and structural similarity to AppA.OrfB encoded by the paralogue cluster of S.
clavuligerus has been implicated to act in thesequestration of unstable intermediates in the alanyl-clavam biosynthetic pathway.14 The reported inter-mediates in CA biosynthesis are structurally relatedto arginine (Fig. 1), suggesting that they could beaccommodated in the OppA1/2 binding sites.Overall, the results support the proposals based
on genetic work that OppA1/2 are involved inpeptide and/or amino acid binding/transport andprovide a structural basis for further detailed studieson their other roles in CA biosynthesis.
Materials and Methods
Cloning, expression, and purification of OppA2
Orf15 was amplified by PCR in the presence of 10%dimethyl sulfoxide from the pCEC001 plasmid.16 Theprimers used were as follows: forward primer, 5′-GGTGGT CAT ATG CCC ACC GCC GCG-3′; reverse primer,5′-GGT GGT GGA TCC CTA GTC TTC TTC GGG-3′.Orf15 was subsequently cloned into pET28a vector(Novagen) using NdeI/BamHI restriction sites. Theorf15/pET28a construct was transformed into E. coliBL21 (DE3) cells. Cells were grown at 37 °C in 2YTmedia containing kanamycin at 50 μg/ml. When the cellsreached anOD600 (optical density at 600 nm) of 0.6–0.8, thetemperature was lowered to 15 °C and the expression ofnative protein was induced by the addition of 1 mMisopropyl thio-β-D-galactoside. Following overnight incu-bation, the cells were harvested by centrifugation at 7000gfor 20 min. The cells were resuspended in lysis bufferconsisting of 0.05 M sodium phosphate, pH 7.5, 0.5 M
NaCl, 2 mM MgCl2, Benzonase Nuclease (Novagen,Denmark), and Complete EDTA (ethylenediaminetetraa-cetic acid)-free Protease Inhibitor tablet (Roche Diagnos-tics GmbH,Mannheim, Germany) and lysed using either aOne Shot cell disruptor (Constant Cell Disruptor Systems,Daventry, UK) or a Vibra Cell sonicator. The lysate wascleared by centrifugation at 20,000g, and the supernatantwas applied onto a HiTrap Chelating HP column (GEHealthcare Bio Sciences AB, Uppsala, Sweden) loadedwith Ni2+. The columnwas washedwith a 0.005- to 0.05-Mstepwise imidazole gradient, before elution of the recom-binant OppA2 using a gradient of 0.05–0.5 M imidazole,50 mM sodium phosphate buffer, pH 7.5, and 0.5 M NaCl.The peak fractions from the Ni-affinity chromatographywere pooled and loaded on a High prep 26/10 desaltingcolumn (GE Healthcare Bio Sciences AB), pre-equilibratedin 0.05 M Tris–HCl, pH 7.5. Protein was concentrated andloaded onto a HiLoad 26/60 Superdex 200 gel-filtrationcolumn (GE Healthcare Bio Sciences AB) to yield OppA2of N95% purity by SDS-PAGE analysis. Pure proteinfractions were pooled and concentrated to 10–15 mg/mlusing a VivaSpin 10,000 molecular weight cutoff concen-trator (Vivascience). SeMet-substituted OppA2 was pre-pared by transforming the oppA2/pETX construct into E.coli 834 (DE3) cells. Cells were grown and proteinexpressed as described above, except the use of SeMetmedium (Molecular Dimensions Ltd, UK) containing L-SeMet. β-Mercaptoethanol was included in all buffers forthe SeMet protein purification.For the production of GST-tagged OppA1 and OppA2,
orf7 and orf15 genes were subcloned from orf7/pET28a andorf15/pET28a vectors into pGEX6P1 vector to give orf7/pGEX6P1 and orf15/pGEX6P1 constructs. The small-scaleexpression of these constructs in E. coli resulted in solubleexpression of N-terminally GST-tagged OppA1/2. Large-scale expression was followed by purification of the GST-OppA1/2 proteins.
Crystallization
Crystallization experiments were performed at 20 °Cusing the sitting-drop vapor diffusion method. Nativecrystals were obtained by microseeding from needlecluster crystals grown in 10–15 mg/ml protein, 20–25%(w/v) PEG 6000 in 0.1 M Tris–HCl, pH 8.0, and 0.1 MNaCl, into drops prepared under the same conditions.Crystals were transferred to a cryo-solution containing25% (w/v) PEG 6000, 20% glycerol, 0.1 MNaCl, and 0.1 MTris–HCl, pH 8.0, and immediately flash-cooled in liquidnitrogen. Crystals of the SeMet derivative were obtainedby seeding from small crystals into a drop consisting of9 mg/ml protein, 14% (w/v) PEG 6000, 0.05 M Tris–HCl,pH 8.0, 0.32 M sodium acetate, and 2.5 mM MgCl2.Needle-shaped SeMet crystals grew after ca 3 to 5 days.The OppA2:arginine complex was prepared by soakingnative crystals in a mother liquor solution containing15 mM L-arginine and cryo-solution for 5 min prior toflash-freezing in liquid nitrogen. The complex of OppA2:bradykinin was obtained by co-crystallization of OppA2with 5 mM bradykinin acetate (Sigma-Aldrich).
Data collection and structure determination
Native data to 1.45 Å resolution were collected at ID14-1[European Synchrotron Radiation Facility (ESRF), Greno-ble, France] at 100 K. The data were processed usingMOSFLM40,41 and SCALA.40 The crystal belongs to spacegroup P21, with unit cell dimensions a=53.6 Å, b=84.4 Å,

342 OPP Structures from the CA Biosynthetic Pathway
c=62.4 Å, and β=91.7° (Table 1). Attempts to solve thestructure using molecular replacement were unsuccessful.A single-wavelength anomalous diffraction (SAD) data setto 1.7 Å resolution was collected on a crystal of a SeMetderivative at ID14-3 (ESRF, Grenoble, France) at 100 K(Table 1). The data were processed using DENZO/SCALEPACK,43 MOSFLM,40,41 and SCALA.40 The crystalsbelong to space group P21, with unit cell dimensionsa=53.3 Å, b=120.8 Å, c=80.0 Å, with a β-angle of 94.1°(Table 1). Assuming the presence of two molecules in theasymmetric unit gave a VM of 2.1 Å3 Da−1 with 40.1%solvent.44 Data for the arginine complex were collected atID14-4 (ESRF, Grenoble, France) at 100 K on a MAR CCDdetector (Table 1), with unit cell dimensions isomorphousto the native data. Data for the bradykinin complex werecollected at ID14-3 (ESRF, Grenoble, France) at 100 K on anADSC Q4R detector (Table 1).Ten selenium sites were located using the SHELX suite45
and refined using AUTO-SHARP.46 Solvent flattening anddensity modification in SOLOMON and the use of ARP/wARP47 (implemented in AUTO-SHARP) resulted in thetracing of the main-chain residues from Arg7 to Glu560 forboth molecules. The protein sequence could be easilydocked by themanual insertion of residues into the electrondensity maps from AUTO-SHARP using O,48 except for asmall loop (Gly340-Gly345) with extremely poor electrondensity. Despite the presence of two molecules within theasymmetric unit, electron density averaging could not beperformed due to an imperfect 2-fold symmetry betweenthe two molecules, caused by small domain movements.The model was initially refined in CNS49 using simulatedannealing. Difference maps clearly showed the presence ofadditional density in the binding cleft of both molecules,which were attributed to the cryo-protectant PEG 400.Refinement continued in REFMAC5,50 with TLS refine-ment51 used in the final stages with each domain as a TLSgroup. Solvent molecules were added using ARP/wARP52and were manually inspected in O.48
The native ‘apo’ structure was solved by molecularreplacement (MolRep53), using the SeMet structure as a
Table 1. Data collection and refinement statistics
OppA2 SeMet-SAD OppA2 native
X-ray source ID14:3, ESRF, Grenoble ID14:1, ESRResolution (Å) 60.4–1.7
(1.74–1.70)a62.4–(1.53–
Space group P21 P2Unit cell dimensionsa, b, c (Å) 53.3, 120.8, 80.0 53.6, 84β (°) 94.1 91Molecules/asymmetric unit 2 1No. of observations 587,218 369,No. of unique reflections 85,165 97,9Completeness (%) 81.1 (35.3) 99.9 (Mean I/σ(I) 18.1 (2.7) 12.3Multiplicity 6.5 (3.7) 3.8 (Rmeas
b 0.089 (0.516) 0.062 (No. of Se sites 10Figure of merit 0.39Phasing power 1.27Rcryst 0.169 0.1Rfree 0.198 0.2Wilson B-factor (Å2) 14No. of solvent molecules 883 39r.m.s.d. from ideal geometryBond lengths (Å) 0.009 0.0Bond angles (°) 1.193 1.0a Numbers in parentheses indicate values for the outer resolution sb Rmeas as defined by Diederichs and Karplus.42
search model. The small loop, Gly340-Gly345, was clearlyvisible in the electron density and has been used to modelthe position of this loop in both molecules of the SeMetstructure. Additional density belonging to several glycerolmolecules could also be seen in the binding cleft. Thestructure of the native protein was used as a search modelfor the arginine and bradykinin complexes. The modelswere refined as described for the SeMet structure; therefinement statistics is shown in Table 1.
SPOT synthesis of peptides
The L-arginine-containing di- and tripeptide library wassynthesized on a Multipep™ instrument from Intavis AG(Germany). Fmoc-protected amino acids were obtainedfrom Novabiochem. Pre-activated cellulose membranescoupled with PEG 500 were obtained from Intavis AG.Anti-GST-horseradish peroxidase conjugate antibody(Sigma Aldrich) and the ECL Plus™ Western blottingdetection kit (GE Healthcare) were used for the SPOTanalysis.22 The membrane was immersed in 5 ml ofblocking reagent [5% nonfat dried milk in Tris-bufferedsaline (TBS)–Tween, 8 g NaCl, 0.2 g KCl, and 20 mM Tris–HCl, pH 7.5] and incubated at room temperature for 1 hwhile shaking. After 1 h, the membrane was rinsed withthe TBS–Tween wash buffer twice. GST protein (100 μl)was diluted into 2.5 ml in TBS–Tween buffer to a finalconcentration of 10 μM. The protein solution was added tothe membrane and incubated for 1 h at room temperaturewhile shaking. The membrane was then gently washed for30 min 2× at room temperature. This was followed by theincubation of the membrane in the antibody solution(100 μl of the antibody diluted in 10 ml of wash buffer,1:1000 dilution of antibody). The antibody solution wasfreshly prepared every time from the stock. The mem-brane was washed for 10 min 3× and then a final wash of15 min was required for complete removal of nonspecificantibody binding. The wash buffer was changed afterevery wash. For the peptide-binding detection, solutions
(non-liganded) OppA2:arginine OppA2:bradykinin
F, Grenoble ID14:4, ESRF, Grenoble ID14:3, ESRF, Grenoble1.451.45)
62.5–1.7(1.76–1.70)
62.1–1.7(1.74–1.70)
1 P21 P21
.4, 62.4 53.6, 85.2, 62.5 53.5, 83.3, 62.4
.7 91.7 95.51 1
011 208,239 221,68040 61,692 59,88599.3) 92.2 (60.0) 99.7 (99.8)(3.2) 17.7 (2.6) 18.0 (2.6)3.5) 3.7 (3.1) 3.7 (3.7)0.450) 0.043 (0.131) 0.097 (0.441)
88 0.165 0.18501 0.186 0.223.3 16.4 19.25 521 380
06 0.011 0.01438 1.289 1.464
hell.

343OPP Structures from the CA Biosynthetic Pathway
A and B (provided with theWestern blotting detection kit)were mixed in a ratio of 40:1, to a final volume of 2 ml. Theresultant solution was then added to the membrane andincubated at room temperature for 5 min while coveringthe tube with aluminum foil.The membrane was wrapped cleanly in a cling film and
sandwiched in an X-ray cassette with a film on top. After5 s, the filmwaswashedwith the developing solutions anddried. Binding was quantified by measuring the fluores-cence emission of the spots using a Victor3 multilabel platereader (Perkin Elmer). The peptide array was excited at355 nm and the emission was recorded at 460 nm. Therelative fluorescence intensities of every spot werenormalized by dividing the intensity of the peptide spotby the control glutathione (see Supplementary Table 1).Experiments were carried out in duplicates, and the errorswere calculated as standard deviations (Fig. 2).
Accession numbers
Coordinates and structure factors for OppA2 have beendeposited in the PDB with accession codes 2WOL for theapo structure, 2WOP for the arginine complex, and 2WOKfor the bradykinin complex.
Acknowledgements
We thank ESRF and the European MolecularBiology Laboratory Outstation in Grenoble forproviding beam time and data collection facilitiesand the staff of the ESRF for outstanding support atthe synchrotron. This work was supported by grantsfrom the Swedish Natural Science Research Council(VR), the European Union (No. QLK3-CT-2000-00513), and the Biotechnology and BiologicalSciences Research Council, UK. A.K.M. acknowl-edges the support from the Lawski Foundation.
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2009.11.045
References
1. Baggaley, K. H., Brown, A. G. & Schofield, C. J. (1997).Chemistry and biosynthesis of clavulanic acid andother clavams. Nat. Prod. Rep. 14, 309–333.
2. Townsend, C. A. (1997). Structural studies of naturalproduct biosynthetic proteins. Chem. Biol. 10, 721–730.
3. Kershaw, N. J., Caines, M. E. C., Sleeman, M. C. &Schofield, C. J. (2005). The enzymology of clavam andcarbapenembiosynthesis.Chem.Commun. 34, 4251–4263.
4. Baldwin, J. E. & Schofield, C. J. (1992). The biosynthe-sis of beta-lactams. In The Chemistry of β-Lactams (Page,M. I., ed.), pp. 1–78, Blackie, London, UK.
5. Nicholson, N. H., Baggaley, K. H., Cassells, R.,Davison, M., Elson, S. W., Fulston, M. et al. (1994).Evidence that the immediate biosynthetic precursor ofclavulanic acid is its N-aldehyde analog. J. Chem. Soc.,Chem. Commun. 11, 1281–1282.
6. Zhang, Z., Ren, J., Stammers, D. K., Baldwin, J. E.,Harlos, K. & Schofield, C. J. (2000). Structural origins ofthe selectivity of the trifunctional oxygenase clavami-nic acid synthase. Nat. Struct. Mol. Biol. 7, 127–133.
7. Caines, M. E., Elkins, J. M., Hewitson, K. S. &Schofield, C. J. (2004). Crystal structure and mecha-nistic implications of N2-(2-carboxyethyl)argininesynthase, the first enzyme in the clavulanic acidbiosynthesis pathway. J. Biol. Chem. 279, 5685–5692.
8. Elkins, J. M., Clifton, I. J., Hernandez, H., Doan,L. X., Robinson, C. V., Schofield, C. J. & Hewitson,K. S. (2002). Oligomeric structure of proclavaminicacid amidino hydrolase: evolution of a hydrolyticenzyme in clavulanic acid biosynthesis. Biochem. J.366, 423–434.
9. Miller, M. T., Bachmann, B. O., Townsend, C. A. &Rosenzweig, A. C. (2001). Structure of beta-lactamsynthetase reveals how to synthesize antibioticsinstead of arginine. Nat. Struct. Mol. Biol. 8, 684–689.
10. Elkins, J. M., Kershaw, N. J. & Schofield, C. J. (2005). X-ray crystal structure of ornithine acetyltransferase fromclavulanic acid biosynthesis gene cluster. Biochem. J.385, 565–573.
11. MacKenzie, A. K., Kershaw, N. J., Hernandez, H.,Robinson, C. V., Schofield, C. J. & Andersson, I. (2007).Clavulanic acid dehydrogenase: structural and bio-chemical analysis of the final steps in the biosynthe-sis of the beta-lactamase inhibitor clavulanic acid.Biochemistry, 46, 1523–1533.
12. Egan, L. A., Busby, R. W., Iwata-Reuyl, D. & Town-send, C. A. (1997). Probable role of clavaminic acid asthe terminal intermediate in the common pathway toclavulanic acid and the antipodal clavam metabolites.J. Am. Chem. Soc. 119, 2348–2355.
13. Tahlan, K., Park, H. U., Wong, A., Beatty, P. H. &Jensen, S. E. (2007). Two sets of paralogous genesencode the enzymes involved in the early stages ofclavulanic acid and clavam metabolite biosynthesis inStreptomyces clavuligerus. Antimicrob. Agents Chemother.48, 930–939.
14. Zelyas, N. J., Cai, H., Kwong, T. & Jensen, S. E.(2008). Alanylclavam biosynthetic genes are clusteredtogether with one group of clavulanic acid biosyn-thetic genes in Streptomyces clavuligerus. J. Bacteriol.190, 7957–7965.
15. Jensen, S. E. & Paradkar, A. S. (1999). Biosynthesis andmolecular genetics of clavulanic acid. Antonie vanLeeuwenhoek, 75, 125–133.
16. Jensen, S. E., Paradkar, A. S., Mosher, R. H., Anders,C., Beatty, P. H., Brumlik, M. J. et al. (2004). Fiveadditional genes are involved in clavulanic acidbiosynthesis in Streptomyces clavuligerus. Antimicrob.Agents Chemother. 48, 192–202.
17. Lorenzana, L. M., Perez-Redondo, R., Santamarta, I.,Martin, J. F. & Liras, P. (2004). Two oligopeptide-permease-encoding genes in the clavulanic acidcluster of Streptomyces clavuligerus are essential forproduction of the beta-lactamase inhibitor. J. Bacteriol.186, 3431–3438.
18. Thorson, J. S., Sievers, E. L., Ahlert, J., Shepard, E.,Whitwam, R. E., Onwueme, K. C. & Ruppen, M.(2000). Understanding and exploiting nature's chem-ical arsenal: the past, present and future of calichea-micin research. Curr. Pharm. Des. 6, 1841–1879.
19. Dwyer, M. A. & Hellinga, H. W. (2004). Periplasmiicbinding proteins: a versatile superfamily for proteinengineering. Curr. Opin. Struct. Biol. 14, 495–504.
20. Tam, R. & Saier, M. H., Jr (1993). Structural, functional,and evolutionary relationships among extracellular

344 OPP Structures from the CA Biosynthetic Pathway
solute-binding receptors of bacteria.Microbiol. Rev. 57,320–346.
21. Hilpert, K., Winkler, D. F. & Handock, R. E. (2007).Peptide arrays on cellulose support: SPOT synthesis, atime and cost efficient method for synthesis of largenumbers of peptides in a parallel and addressablefashion. Nat. Protoc. 2, 1333–1349.
22. Espanel, X. & van Huijsduijnen, R. H. (2005).Applying the SPOT peptide synthesis procedure tothe study of protein tyrosine phosphatase substratespecificity: probing for the heavenly match in vivo.Methods, 35, 64–72.
23. Levdikov, V. M., Blagova, E. V., Brannigan, J. A.,Wright, L., Vagin, A. A. & Wilkinson, A. J. (2005). Thestructure of the oligopeptide-binding protein, AppA,from Bacillus subtilis in complex with a nonapeptide.J. Mol. Biol. 345, 879–892.
24. Holm, L. & Sander, C. (1995). Dali: a network tool forprotein structure comparison. Trends Biochem. Sci. 20,478–480.
25. Kleywegt, G. J. & Jones, T. A. (1997). Detecting foldingmotifs and similarities in protein structures. MethodsEnzymol. 277, 525–545.
26. Nickitenko, A. V., Trakhanov, S. & Quiocho, F. A.(1995). 2 Å resolution structure of DppA, a peri-plasmic dipeptide transport/chemosensory receptor.Biochemistry, 34, 16585–16595.
27. Tame, J. R.,Murshudov,G.N., Dodson, E. J.,Neil, T. K.,Dodson, G. G., Higgins, C. F. & Wilkinson, A. J. (1994).The structural basis of sequence-independent peptidebinding by OppA protein. Science, 264, 1578–1581.
28. Heddle, J., Scott, D. J., Unzai, S., Park, S.-Y. & Tame,J. R. H. (2003). Crystal structures of the liganded andunliganded nickel-binding protein NikA from Escher-ichia coli. J. Biol. Chem. 278, 50322–50329.
29. Marland, Z., Beddoe, T., Zaker-Tabrizi, L., Lucet, I. S.,Brammananth, R., Whisstock, J. C. et al. (2006).Hijacking of a substrate-binding protein scaffold foruse in mycobacterial cell wall biosynthesis. J. Mol. Biol.359, 983–997.
30. Mellado, E., Lorenzana, L. M., Rodriguez-Salz, M.,Diez, B., Liras, P. & Barredo, J. L. (2002). The clavulanicacid biosynthetic cluster of Streptomyces clavuligerus:genetic organization of the region upstream of the cargene. Microbiology, 148, 1427–1438.
31. Quiocho, F. A. & Ledvina, P. S. (1996). Atomic struc-ture and specificity of bacterial periplasmic receptorsfor active transport and chemotaxis: variation ofcommon themes. Mol. Microbiol. 20, 17–25.
32. Dunten, P. & Mowbray, S. L. (1995). Crystal structureof the dipeptide binding protein from Escherichia coliinvolved in active transport and chemotaxis. ProteinSci. 4, 2327–2334.
33. Sleigh, S. H., Tame, J. R., Dodson, E. J. & Wilkinson,A. J. (1997). Peptide binding in OppA, the crystalstructures of the periplasmic oligopeptide bindingprotein in the unliganded form and in complex withlysyllysine. Biochemistry, 36, 9747–9758.
34. Schiefner, A., Breed, J., Bosser, L., Kneip, S., Gade, J.,Holtmann, G. et al. (2004). Cation–pi iterations asdeterminants for binding of the compatible solutesglycine betaine and proline betaine by the periplasmicligand-binding protein ProX from Escherichia coli. J.Biol. Chem. 279, 5588–5596.
35. Berntsson, R. P.-A., Doeven, M. K., Fusetti, F.,Duurkens, R. H., Sengupta, D., Marrink, S.-J. et al.(2009). The structural basis for peptide selection by thetransport receptor OppA. EMBO J. 28, 1332–1340.
36. Sanchez, L. & Brana, A. F. (1996). Cell density
influences antibiotic synthesis in Streptomyces clavuli-gerus. Microbiology, 142, 1209–1220.
37. Nodwell, J. R. & Losick, R. (1998). Purification of anextracellular signalingmolecule involved in productionof aerial mycelium by Streptomyces coelicolor. J. Bacteriol.180, 1334–1337.
38. Nodwell, J. R., McGovern, K. & Losick, R. (1996). Anoligopeptide permease responsible for the import of anextracellular signal governing aerial mycelium forma-tion in Streptomyces coelicolor.Mol.Microbiol. 22, 881–893.
39. Bignell, D. R., Tahlan, K., Colvin, K. R., Jensen, S. E. &Leskiw, B. K. (2005). Expression of ccaR, encoding thepositive activator of cephamycin C and clavulanic acidproduction in Streptomyces clavuligerus, is dependenton bldG. Antimicrob. Agents Chemother. 49, 1529–1541.
40. CollaborativeComputational Project,Number 4. (1994).The CCP4 suite: programs for protein crystallography.Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763.
41. Leslie, A. G. W. (1992). Recent changes to theMOSFLM package for processing film and imageplate data. Joint CCP4+ESF-EAMCB Newsletter onProtein Crystallography, 26.
42. Diederichs, K. & Karplus, P. A. (1997). ImprovedR-factors for diffraction data analysis in macromolec-ular crystallography. Nat. Struct. Mol. Biol. 4, 269–275.
43. Otwinowski, Z. & Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode.Methods Enzymol. 276, 307–326.
44. Matthews, B. W. (1968). Solvent content of proteincrystals. J. Mol. Biol. 33, 491–497.
45. Schneider, T. R. & Sheldrick, G. M. (2002). Substruc-ture solution with SHELXD. Acta Crystallogr., Sect. D:Biol. Crystallogr. 58, 1772–1779.
46. La Fortelle, E. & Bricogne, G. (1997). Maximum-likelihood heavy-atom parameter refinement for themultiple isomorphous replacement and multiwave-length anomalous diffraction methods. Methods Enzy-mol. 276, 472–492.
47. Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Auto-mated protein model building combined with iterativestructure refinement. Nat. Struct. Mol. Biol. 6, 458–463.
48. Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building proteinmodels in electron density maps and the location oferrors in these models.Acta Crystallogr., Sect. A: Found.Crystallogr. 47, 110–119.
49. Brunger,A.T.,Adams,P.D.,Clore,G.M.,DeLano,W.L.,Gros, P., Grosse-Kunstleve, R. W. et al. (1998). Crystal-lography andNMR system: a new system software suitefor macromolecular structure determination. Acta Crys-tallogr., Sect. D: Biol. Crystallogr. 54, 905–921.
50. Murshudov, G. N., Vagin, A. A. & Dodson, E. J.(1997). Refinement of macromolecular structures bythe maximum-likelihood method. Acta Crystallogr.,Sect. D: Biol. Crystallogr. 53, 240–255.
51. Winn, M. D., Isupov, M. N. & Murshudov, G. N.(2001). Use of TLS parameters to model anisotropicdisplacements in macromolecular refinement. ActaCrystallogr., Sect. D: Biol. Crystallogr. 57, 122–133.
52. Lamzin, V. S., Perrakis, A. & Wilson, K. S. (2001). TheARP/wARP suite for automated construction andrefinement of protein models. In International Tables forCrystallography. Vol. F, Crystallography of BiologicalMacromolecules (Rossman, M. G. & Arnold, E., eds),pp. 720–722, Kluwer Academic Publishers, Dordrecht,The Netherlands.
53. Vagin, A. A. & Teplyakov, A. (1997). MOLREP: anautomated program for molecular replacement. J.Appl. Crystallogr. 30, 1022–1025.