comparative genomics of dairy lactic acid bacteria
TRANSCRIPT

PDF hosted at the Radboud Repository of the Radboud University
Nijmegen
The following full text is a publisher's version.
For additional information about this publication click this link.
http://hdl.handle.net/2066/77580
Please be advised that this information was generated on 2022-07-18 and may be subject to
change.

Comparative genomics of dairy lactic acid bacteria: Proto-cooperation, proteolysis and flavour formation
Mengjin Liu

To my parents
献给我的父亲母亲
ISBN: 978-90-9025309-1
© 2010 M. LiuAll rights reserved.
This research was supported by grant CSI4017 of the Casimir program of the Ministry of Economic Affairs, the Netherlands. The research was carried out at FrieslandCampina Research, Deventer, and the Center for Molecular and Biomolecular Informatics, Radboud University Nijmegen
Cover Design: Mengjin Liu
Print by Ipskamp Drukkers

Comparative genomics of dairy lactic acid bacteria: Proto-cooperation, proteolysis and flavour formation
Een wetenschappelijke proeve op het gebied van de Medische WetenschappenProefschrift
ter verkrijging van de graad van doctoraan de Radboud Universiteit Nijmegen
op gezag van de rector magnificus prof. mr. S.C.J.J. Kortmann,volgens besluit van het College van Decanen
in het openbaar te verdedigen op donderdag 10 Juni 2010om 10.30 uur precies
door
Mengjin Liu
geboren op 22 Juli 1983te Jiangxi, China

Promotor: Prof. dr. R.J. Siezen
Co-promotor: Dr. A. Nauta (FrieslandCampina)
Manuscriptcommissie:Prof. dr. M. Huynen (voorzitter)Prof. dr. G. Smit (Wageningen Universiteit)Prof. dr. J. Hugenholtz (Universiteit van Amsterdam)

Comparative genomics of dairy lactic acid bacteria: Proto-cooperation, proteolysis and flavour formation
An academic essay in Medical ScienceDoctoral Thesis
to obtain the degree of doctorfrom Radboud University Nijmegen
on the authority of the Rector Magnificus prof. dr. S.C.J.J. Kortmann,according to the decision of the Council of Deans
to be defended in public on Thursday, 10 June 2010at precisely 10.30 hours
by
Mengjin Liu
Born on July 22, 1983in Jiangxi, China

Supervisor: Prof. R.J. Siezen
Co- supervisor: Dr. A. Nauta (FrieslandCampina)
Doctoral thesis committee:Prof. M. Huynen (Chairman)Prof. G. Smit (Wageningen University)Prof. J. Hugenholtz (University of Amsterdam)

Table of Contents
Chapter 1 General Introduction 9
Chapter 2 The proteolytic system of lactic acid bacteria revisited: a genomic comparison
21
Chapter 3 Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria
39
Chapter 4 Regulation of cysteine and methionine metabolism in lactic acid bacteria 55
Chapter 5 Combining chemoinformatics with bioinformatics: In silico prediction of flavor-forming pathways by a chemical systems biology approach “Reverse Pathway Engineering”
69
Chapter 6 In silico prediction of horizontal gene transfer events in Lactobacillus delbrueckii ssp. bulgaricus and Streptococcus thermophilus reveals proto-cooperation in yoghurt manufacturing
85
Chapter 7 Summarizing discussion 101
Samenvatting 107
总结概述 111
Appendix: Colour Figures 113
Acknowledgements 123
List of publications 125
Curriculum Vitae 127
Supplemental materials can be found:http://www.cmbi.ru.nl/~mliu/


GENERAL INTRODUCTION
9
CH
APT
ER 1
Chapter 1
GENERAL INTRODUCTION

CHAPTER 1
10
Lactic Acid Bacteria GenomesLactic acid bacteria (LAB) are a heterogeneous family of mainly low G+C Gram-positive,
anaerobic, non-sporulating and acid-tolerant bacteria. They can ferment various nutrients in a homofermentative or heterofermentative fashion into primarily lactic acid, but also into by-products such as acetic acid, formic acid, ethanol and carbon dioxide. They contribute to rapid acidification of food products, but also to flavour, texture and nutrition1.
LAB are naturally found in dairy, plant, meat and cereal fermentation environments, and have a long tradition of use as starter or adjunct cultures in industrial food and feed fermentations. Moreover, some LAB, as well as bifidobacteria, which belong to the high G+C Gram-positive bacteria, are found to be natural inhabitants of the gastro-intestinal tract in humans or other mammals. Some of them have been widely used as probiotics in various functional foods and claimed to stimulate the hosts’ health.
Over decades, LAB used for fermenting cheese, yoghurts, and other fermented dairy products have been extensively studied, also regarding to their contributions to the flavour and texture of the product. However, the mechanisms of flavour-formation by LAB on the genomic and molecular level are still poorly understood. Research of genetics and physiology of LAB may give a better fundamental understanding of industrial starter cultures with predictable and stable characteristics.
0.1
Bacillus subtilis(outgroup)
S. thermophilus
L. lactis cremoris
L. lactis lactis
L. bulgaricusL. acidophilus
L. helveticus
L. johnsoniiL. gasseri
L. casei
L. sakei
Oenococcus oeni
Leuconostoc mesenteroides L. salivarius L. reuteri
L. fermentum
Pediococcus pentosaceus
L. plantarum
L. brevis
Figure 1.1. Phylogenetic tree of LAB based on alignment of concatenated ribosomal proteins sequences. Bacillus subtilis was used as an outgroup. The tree was visualized by TreeView61.
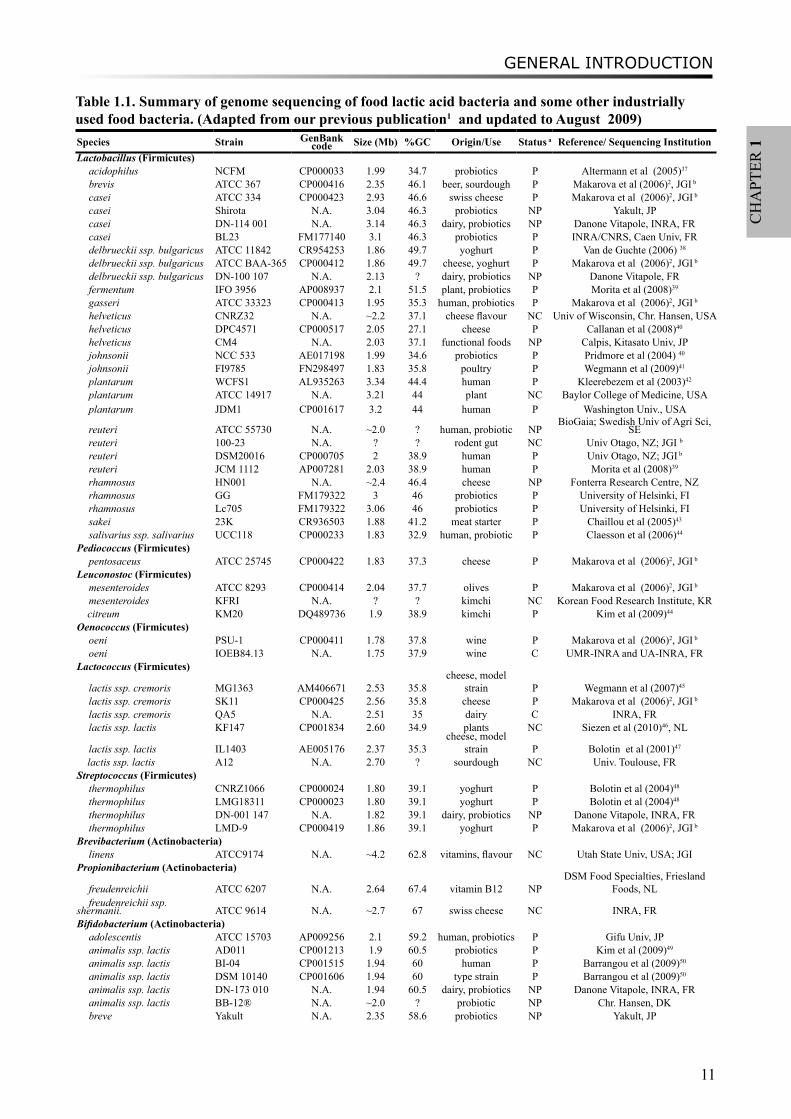
In recent years, genome sequencing of food and health- related LAB has been booming1,2. Around forty LAB genomes have been fully sequenced, including the genera Lactobacilli, Lactococci, Streptococci, Pediococcus, Oenococcus and Leuconotoc (Table 1.1, Figure 1.1). In particular, the Joint Genome Institute has sequenced and published nine LAB genomes in 2006, which contributed significantly to the phylogenetic and functional studies of LAB2. The fully sequenced genomes bring numerous opportunities and challenges to bioinformaticians in the interpretation of encoded functions. For instance, the genome information will allow developing metabolic models and understanding regulation mechanisms. Eventually, revealing the genetic and metabolic potentials of LAB will provide leads for the improvement of industrial products, e.g. control of flavours.
GENERAL INTRODUCTION
11
CH
APT
ER 1
Table 1.1. Summary of genome sequencing of food lactic acid bacteria and some other industrially used food bacteria. (Adapted from our previous publication1 and updated to August 2009)Species Strain GenBank
code Size (Mb) %GC Origin/Use Status a Reference/ Sequencing InstitutionLactobacillus (Firmicutes)
acidophilus NCFM CP000033 1.99 34.7 probiotics P Altermann et al (2005)37
brevis ATCC 367 CP000416 2.35 46.1 beer, sourdough P Makarova et al (2006)2, JGI b
casei ATCC 334 CP000423 2.93 46.6 swiss cheese P Makarova et al (2006)2, JGI b
casei Shirota N.A. 3.04 46.3 probiotics NP Yakult, JPcasei DN-114 001 N.A. 3.14 46.3 dairy, probiotics NP Danone Vitapole, INRA, FRcasei BL23 FM177140 3.1 46.3 probiotics P INRA/CNRS, Caen Univ, FRdelbrueckii ssp. bulgaricus ATCC 11842 CR954253 1.86 49.7 yoghurt P Van de Guchte (2006) 38
delbrueckii ssp. bulgaricus ATCC BAA-365 CP000412 1.86 49.7 cheese, yoghurt P Makarova et al (2006)2, JGI b
delbrueckii ssp. bulgaricus DN-100 107 N.A. 2.13 ? dairy, probiotics NP Danone Vitapole, FRfermentum IFO 3956 AP008937 2.1 51.5 plant, probiotics P Morita et al (2008)39
gasseri ATCC 33323 CP000413 1.95 35.3 human, probiotics P Makarova et al (2006)2, JGI b
helveticus CNRZ32 N.A. ~2.2 37.1 cheese flavour NC Univ of Wisconsin, Chr. Hansen, USAhelveticus DPC4571 CP000517 2.05 27.1 cheese P Callanan et al (2008)40
helveticus CM4 N.A. 2.03 37.1 functional foods NP Calpis, Kitasato Univ, JPjohnsonii NCC 533 AE017198 1.99 34.6 probiotics P Pridmore et al (2004) 40
johnsonii FI9785 FN298497 1.83 35.8 poultry P Wegmann et al (2009)41
plantarum WCFS1 AL935263 3.34 44.4 human P Kleerebezem et al (2003)42
plantarum ATCC 14917 N.A. 3.21 44 plant NC Baylor College of Medicine, USAplantarum JDM1 CP001617 3.2 44 human P Washington Univ., USA
reuteri ATCC 55730 N.A. ~2.0 ? human, probiotic NPBioGaia; Swedish Univ of Agri Sci,
SEreuteri 100-23 N.A. ? ? rodent gut NC Univ Otago, NZ; JGI b
reuteri DSM20016 CP000705 2 38.9 human P Univ Otago, NZ; JGI b
reuteri JCM 1112 AP007281 2.03 38.9 human P Morita et al (2008)39
rhamnosus HN001 N.A. ~2.4 46.4 cheese NP Fonterra Research Centre, NZrhamnosus GG FM179322 3 46 probiotics P University of Helsinki, FIrhamnosus Lc705 FM179322 3.06 46 probiotics P University of Helsinki, FIsakei 23K CR936503 1.88 41.2 meat starter P Chaillou et al (2005)43
salivarius ssp. salivarius UCC118 CP000233 1.83 32.9 human, probiotic P Claesson et al (2006)44
Pediococcus (Firmicutes)pentosaceus ATCC 25745 CP000422 1.83 37.3 cheese P Makarova et al (2006)2, JGI b
Leuconostoc (Firmicutes)mesenteroides ATCC 8293 CP000414 2.04 37.7 olives P Makarova et al (2006)2, JGI b
mesenteroides KFRI N.A. ? ? kimchi NC Korean Food Research Institute, KR citreum KM20 DQ489736 1.9 38.9 kimchi P Kim et al (2009)44
Oenococcus (Firmicutes)oeni PSU-1 CP000411 1.78 37.8 wine P Makarova et al (2006)2, JGI b
oeni IOEB84.13 N.A. 1.75 37.9 wine C UMR-INRA and UA-INRA, FRLactococcus (Firmicutes)
lactis ssp. cremoris MG1363 AM406671 2.53 35.8cheese, model
strain P Wegmann et al (2007)45
lactis ssp. cremoris SK11 CP000425 2.56 35.8 cheese P Makarova et al (2006)2, JGI b
lactis ssp. cremoris QA5 N.A. 2.51 35 dairy C INRA, FRlactis ssp. lactis KF147 CP001834 2.60 34.9 plants NC Siezen et al (2010)46, NL
lactis ssp. lactis IL1403 AE005176 2.37 35.3cheese, model
strain P Bolotin et al (2001)47
lactis ssp. lactis A12 N.A. 2.70 ? sourdough NC Univ. Toulouse, FRStreptococcus (Firmicutes)
thermophilus CNRZ1066 CP000024 1.80 39.1 yoghurt P Bolotin et al (2004)48
thermophilus LMG18311 CP000023 1.80 39.1 yoghurt P Bolotin et al (2004)48
thermophilus DN-001 147 N.A. 1.82 39.1 dairy, probiotics NP Danone Vitapole, INRA, FRthermophilus LMD-9 CP000419 1.86 39.1 yoghurt P Makarova et al (2006)2, JGI b
Brevibacterium (Actinobacteria)linens ATCC9174 N.A. ~4.2 62.8 vitamins, flavour NC Utah State Univ, USA; JGI
Propionibacterium (Actinobacteria)
freudenreichii ATCC 6207 N.A. 2.64 67.4 vitamin B12 NPDSM Food Specialties, Friesland
Foods, NLfreudenreichii ssp.
shermanii. ATCC 9614 N.A. ~2.7 67 swiss cheese NC INRA, FRBifidobacterium (Actinobacteria)
adolescentis ATCC 15703 AP009256 2.1 59.2 human, probiotics P Gifu Univ, JPanimalis ssp. lactis AD011 CP001213 1.9 60.5 probiotics P Kim et al (2009)49
animalis ssp. lactis BI-04 CP001515 1.94 60 human P Barrangou et al (2009)50
animalis ssp. lactis DSM 10140 CP001606 1.94 60 type strain P Barrangou et al (2009)50
animalis ssp. lactis DN-173 010 N.A. 1.94 60.5 dairy, probiotics NP Danone Vitapole, INRA, FRanimalis ssp. lactis BB-12® N.A. ~2.0 ? probiotic NP Chr. Hansen, DKbreve Yakult N.A. 2.35 58.6 probiotics NP Yakult, JP

CHAPTER 1
12
breve M-16V N.A. ~2.3 58.6 human, probiotics NP Morinaga Milk Industry, JPbreve UCC2003 N.A. 2.42 58.7 human, probiotics C Univ College Cork, IElongum NCC2705 AE014295 2.26 60.1 human, probiotics P Schell et al (2005)51
longum BB536 N.A. ~2.4 59.4 human, probiotics NP Morinaga Milk Industry, JPlongum DJO10A CP000605 2.38 60.1 probiotics P Lee et al (2008)52, JGI b
longum ssp. infantis ATCC 15697 CP001095 2.8 59.9 human P Sela et al (2008)53, JGI b
longum biotype infantis M-63 N.A. ~2.9 59.4 human, probiotics NP Morinaga Milk Industry, JPa P, Public; C, Complete (to be published); NC, Not Complete; NP, Not Public; b JGI, Joint Genome Institute, US Department of Energy
Flavour-forming pathways in LABFlavour compounds produced by LAB are derived from various (bio)chemical pathways. Key
flavour components of a food product can be identified by a combination of separation of compounds by gas chromatography (GC) and analytical identification by the human nose (olfactometry)3. In cheese, the precursors of the key flavour compounds include milk fats, milk proteins (caseins), and sugars (lactose) (Table 1.2). In particular, degradation of casein is the most important biochemical pathway leading to formation of a large number of flavours, e.g. alcohols, aldehydes, carboxylic acids etc. The conversion of casein to flavours comprises of two steps: proteolysis and amino acid degradation (Figure 1.2).
Amino acidspeptidases
extracellular
intracellular
Peptides Amino acids
Caseins
proteinases
transport transport
Flavour compounds(alcohols, aldehydes, carboxylic acids, esters, etc)
Amino acidsDegradation
Proteolysis
Figure 1.2. General protein conversion pathway of LAB leading to flavour formation, consisting of two parts, i.e. proteolysis and amino acids degradation.
ProteolysisProteolysis of casein by LAB is initiated by a cell-wall bound proteinase (PrtP) which degrades
the protein into oligopeptides4,5. The oligopeptides are subsequently taken up by peptide transport systems, and further degraded into di/tri-peptides or amino acids by a set of intracellular peptidases6,7. The cell-wall bound proteinases have been characterized from several LAB, such as Lactococcus lactis, L. helveticus, L. rhamnosus, L. bulgaricus, and S. thermophilus7. In lactococci, they are usually encoded on a plasmid and in lactobacilli they are mostly genome-encoded. The prtP gene is sometimes adjacent to a gene encoding a membrane-bound lipoprotein (PrtM) that is responsible for the maturation of PrtP. However, it is reported that PrtM is not required for the maturation of PrtP in L. bulgaricus, where it is not found to be encoded in the flanking region of the prtP gene8. Peptides generated by the actions of PrtP extracellularly are imported into the cell by several transport systems of LAB. Oligopeptides are transported by the Opp system, whereas di/tri/tetra-peptides are transported by a proton motive force-driven transporter DtpT or an ATP-driven Dpp system9. After peptides are taken up into the cell, they are degraded into amino acids by concerted actions of peptidases including aminopeptidases that cleave amino acids from the N-terminus of the peptides, endopeptidases that act on the internal peptide bonds, di- and tri-peptidases that degrade di- and tri-peptides respectively, as well as proline-specific peptidases which hydrolyze peptides carrying proline residues10. Multiple copies of peptidases can be encoded in one bacterial genome, for instance L. helveticus encodes three PepO paralogs which are important for the bacteria’s ability to reduce bitterness in cheese by degrading bitter peptides11.

GENERAL INTRODUCTION
13
CH
APT
ER 1
Table 1.2. Overview of cheese key flavour components derived from amino acid metabolism. a
Flavour compoundsb Descriptionc Possible precursor Type of cheese
Alcohols3-Methylbutanol Fresh cheese, breathtaking, alcoholic Leucine Gouda2-Phenylethanol Flowery Phenylalanine GoudaOctenol (1-octen-3-ol) Mushroom Fatty acids Camembert, Mozzarella2-Heptanol Mushroom NA Gorgonzola, Grana Padano2-Butanol Wine NA Camembert
Aldehydes3-Methylbutanal Malty, powerful, cheese Leucine Gouda, Cheddar, Camembert,
Emmental, Swiss (Maasdam)2-Methylpropanal Banana, malty, chocolate like Valine Parmigiano ReggianoAcetaldehyde Pungent, ethereal Combined pathways Swiss, Cantalet, Manchego, FetaPhenylacetaldehyde (phenylethanal) Hawthorne, honey, sweet Phenylalanine Camembert, GruyereNonanal Fat, citrus, green NA Grana Padano, Mozzarella
Ketones2-Heptanone Fruity, spicy Fatty acids Emmental, Gorgonzola2-Nonanone Fruity, floral Fatty acids RagusanoAcetoin (3-hydroxy-2-butanone) Buttery Pyruvate Cheddar1-Octen-3-one (octenone) Mushroom Fatty acids? Cheddar, Camembert, Emmental,
RagusanoPhenethyl acetate (2-phenylethyl acetate) Rose, honey, tobacco Combined pathways Camembert, Swiss (Maasdam)2-Undecanone Orange, fresh, green Fat? CamembertDiacetyl (2,3-Butanedione) Buttery, strong Sugar, Aspartic acid Gouda, Cheddar, Camembert,
Emmental, Swiss (Maasdam)δ-Dodecalactone Fatty, peach Fatty acids? Gouda, Cheddar γ-Dodecalactone Fatty, peach Fatty acids? Goudaδ-Decalactone Coconut NA Gouda, Camembert, Emmentalγ-Decalactone Peach, fat NA Camembert4-Hydroxy-2,5-dimethyl-3(2H)-furanone (furaneol™)
Cotton candy, caramel Amino acids+ carbonylcompounds
Gouda, Cheddar, Emmental
5-Ethyl-2-methyl-4-hydroxy-3(2H)-furanone (methylfuranone, homofuranone)
Cotton candy, caramel Amino acids+ carbonylcompounds
Gouda, Cheddar, Emmental
Fatty acids3-Methylbutanoic acid (Isovaleric acid) Sweet, cheese Leucine Gouda, Cheddar, Camembert,
Goat cheeseAcetic acid Sour Pyruvate Gouda, CheddarButanoic acid (Butyric acid) Rancid, cheese sweaty, butter Fatty acids Gouda, Cheddar, CamembertPentanoic acid Sweaty Fatty acids Gouda, Cheddar (aged)2-Methylbutanoic acid Sweaty Isoleucine Gouda2-Methylpropanoic acid (isobutyric acid) Sour, sweet Valine GoudaPhenylacetic acid Bee wax honey Phenylalanine GoudaPropionic acid (Propanoic acid) pungent, rancid, soy Sugar Cheddar, Emmental, Swiss
(Maasdam)
EstersEthylbutanoate (ethyl-3-methylbutanoate, ethyl butyrate)
Fruity Combined pathways(Leucine+ethanol)
Gouda, Cheddar, Emmental, Swiss (Maasdam)
Ethyl caproate (ethyl hexanoate) Fruity Combined pathways Cheddar, Swiss (Maasdam), Grana Padano, Emmental
Ethyl-3-methylbutanoate (ethyl isovalerate) Fruity Combined pathways Swiss (Maasdam)
Sulfur compoundsDMDS (dimethyldisulphide) Onion Methionine CheddarDMTS (dimethyltrisulphide) Garlic note, cabbage Methionine Cheddar, Camembert, GoudaDMS (dimethylsulphide) Cabbage Methionine Camembert, Cheddar, Swiss,
Gouda3-Methyl(thio)propionaldehyde (Methional) Boiled potato Methionine Gouda, Cheddar, Camembert,
Emmental, Swiss (Maasdam)Methanethiol Gasoline, garlic Methionine Cheddar, Gouda, CamembertSkatole (3-methyl-1H-indole) Mothball, faecal Tryptophan Swiss (Maasdam), Emmental
a. Data shown in this table were derived from the following references 3,54-59, N.A., not known.b. Alternative compound names are in bracketsc. Additional information on flavour description (aroma notes) of compounds was obtained from “Gas chromatography - olfactometry (GCO)
of natural products” 60

CHAPTER 1
14
Amino Acid DegradationAmino acids released by proteolysis or taken up directly from the environment are necessary
for bacterial growth, and meanwhile their catabolism produces various flavour compounds (Figure 1.2). In particular, branched-chain amino acids (i.e. Val, Leu, Ile), aromatic amino acids (i.e. Phe, Tyr, Trp) and sulfuric amino acids (i.e. Cys, Met) are the main sources of flavours derived from milk protein. The first step of amino acid catabolism leading to flavour formation could be transamination or elimination, by LAB encoding transaminases or lyases, respectively. α-Keto acids formed by transamination reactions can be further converted by keto acid decarboxylase, dehydrolases, or via oxidative decarboxylation into flavour components such as aldehydes, carboxylic acids, alcohols3. Hydroxy acids are also formed by this route, but do not contribute to the sensory profile. Elimination is the second flavour-forming route for synthesizing sulfuric flavours from methionine. In LAB, the elimination route is initiated by C-S lyases such as cystathionine β- and γ-lyases which directly release methanethiol from methionine12. Methanethiol is further converted to volatile sulfur compounds or to thioesters by reacting with carboxylic acids. Furthermore, chemical conversions also play an important role in flavour formation, although most of the known flavour-forming reactions are enzymatic. For example, the flavour compound phenylpyruvic acid (α-keto acid of Phe) can be chemically converted to benzaldehyde13. Similarly, two analogous chemical reactions, conversion of α-keto-γ-methylthiobutyrate (α-keto acid of Met) to methylsulfanyl-acetaldehyde and conversion of α-ketoisocaproate (α-keto acid of Leu) to 2-methylpropanal have been characterized by Bonnarme et al.14 and Smit et al.15, respectively.
To conclude, in fermented dairy products such as cheese, the breakdown of casein is one of the main pathways contributing to flavour formation by LAB. A study into the diversity of these pathways can provide important insights for predicting flavour-forming potentials of LAB.
Computational genomics analyses
Comparative genomics analysesOne of the fundamental evolutionary relationships of life is based on conserved genetic elements
(DNA) which underlie functional similarities between species. It allows the use of comparative genomics analyses (comparison of genomes from different species or strains) to predict the function of unknown genes by transferring the function from characterized ones, a process called functional “annotation”. Those functional equivalents from different genomes usually belong to a single ortholog group. Orthologs are genes which diverge via speciation events during evolution, in contrast to the concept of paralogs which evolve via duplication events. Both orthologs and paralogs are homologs, that is evolutionarily related genes (Figure 1.3). Orthologs share the same function whereas paralogs often present distinct related functionalities, except for the case of in-paralogs which are genes arisen by duplication after the speciation event (Figure 1.3).
The most commonly used method for annotation is searching for best homologs in the sequence database using the BLAST tools16. However, BLAST often retrieves false positives especially for those orthologs groups within one large protein superfamily which share different levels of sequence similarity. Orthologous groups can also be identified by bi-directional best hits methods, such as the tool InParanoid17. The best method however is to explore the evolution of protein families, for example by phylogenetic tree analysis18,19. Moreover, the conservation information of gene context can be used to assist in the prediction of orthologs18.
Comparative genomics analyses can also be applied for the identification of transcriptional factor binding motifs. When orthologs share the same gene context, they are more likely to be functionally equivalent and share equivalent regulatory sites19. Thus, orthologous groups can be

GENERAL INTRODUCTION
15
CH
APT
ER 1
divided into subgroups, each of which contains a conserved gene context. Then, searches for conserved regulatory motifs can be preformed by comparing upstream regions of gene members within each subgroup. Motif detection methods include manual identification via multiple sequence alignments or using probabilistic models such as MEME20.
Horizontal gene transfer analysesHorizontal gene transfer (HGT, also called lateral gene transfer) is defined as the exchange
of genetic elements between phylogenetically unrelated organisms. It is considered to play a major role in bacterial evolution during their adaptation to different environments21. Because HGT events have occurred over a long time period, the history of HGT is difficult to be discovered by wet-lab experiments. Instead, various computational methods have been applied to infer the HGT events as molecular footprints on the genomes.
The most common approaches to identify HGT are phylogenetic and compositional methods. By analyzing the genes in multiple taxa, HGT can be inferred by comparing the phylogeny of the protein family and the phylogeny of the species evolutionary tree (e.g. 16s RNA tree) (Figures 1.1 and 1.3). If genes from phylogenetically distantly-related species are clustered together in a gene family tree, it suggests that one of the genes might be acquired by HGT. Compositional methods examine sequence characteristics such as GC content, amino acid usage, the codon usage, and dinucleotide usage. Other approaches include identification of genomic islands by comparing closely related strains22 or identification of HGT mechanisms e.g. detection of mobile elements23. Each method, however, may only detect a subset of horizontally transferred genes or brings in some false positives if performed individually. Combining different computational approaches to complement each other may provide better predictions of the HGT events.
Lc_geneA
St_geneA
Lb_geneA
Lp_geneA
La_geneA
Lj_geneA
Lb_geneB
Lp_geneB
La_geneB
Lj_geneB
St_geneB
Lc_geneB1
Lc_geneB2
orthologs
paralogs
inparalogs
HGT
homologs
Figure 1.3. Example of homolog, ortholog, paralog and inparalog relationships. St. for Streptococcus thermophilus; Lb, La, Lj, Lp for lactobacillus strains: L. bulgaricu, L. acidophilus, L. johnsonii, L. plantarum, respectively; Lc. for Lactococcus lactis. All the genes shown in the tree are homologs. Gene A from those bacteria are shown as examples of ortholog relationships. Gene A and gene B from Lp are out-paralogs. Gene B1 is inparalogs relative to gene B2 from Lc (L. lactis). As St (S. thermophilus) is evolutionally more closely related to Lc (L. lactis), a horizontal gene transfer (HGT) event can be identified between gene A from the two bacteria. The HGT event is indicated by a dashed arrow. Squares represent duplication events, while circles represent speciation events.

CHAPTER 1
16
Metabolic pathway reconstructionGenome-scale reconstruction of metabolic pathways begins with gene annotations of a
fully sequenced genome. Enzymes encoded by the genes are then assigned to the reactions in the metabolic networks by utilizing various databases such as BRENDA24 and KEGG25, and thereby a draft biochemical network can be constructed. Such reconstructed networks should be curated by various experimental data, e.g. genetic data (mutation studies), biochemical data (measured enzyme activity), or physiological data (substrate dependency), etc.26 Once the stoichiometric networks have been reconstructed and curated (in LAB, models of L. plantarum, L. lactis, and S. thermophilus are available27-29), they can serve as a predictive model and may be applied to answer specific questions by various computational analyses. Pathway structure analyses include constraint-based flux-balance analyses of a genome-scale model, and elementary mode analysis using tools such as MetaTool30. The analyses can be used for various applications e.g. phenotype prediction, experimental data interpretation and metabolic engineering31.
However, sometimes those genome-scale models are incomplete, especially regarding to gaps in the non-essential pathways. Only a few of the gaps can be filled by computational methods or manual curation32. Completion of the gaps in these incomplete networks requires prediction of novel reactions or unknown pathways. Several algorithms have been developed to propose new pathways which are missing from the model. For example, UM-PPS (University of Minnesota Pathway Prediction System, http://umbbd.msi.umn.edu/predict/) system33 predicts plausible pathways for microbial degradation of chemical compounds. The predictions use biotransformation rules, based on reactions found in the UM-BBD (University of Minnesota Biocatalysis/Biodegradation Database) database34 or in the scientific literature. Other retrosynthesis tools include Route Designer35, which predicts chemical reactions using organic chemistry transformation rules, and ReBiT (Retro-Biosynthesis Tool)36, a database for identification of enzymes in the plausible pathways.
Outline of thesisThis thesis studies and compares the genome content of a group of gram-positive micro-
organisms, the lactic acid bacteria (LAB), which are important for the food industry, in particular the dairy industry. In order to improve the control of the product characteristics, especially the flavour and taste of dairy products, the metabolic potential of LAB was predicted with various bioinformatics and/or chemoinformatics methods. Flavour-forming pathways from protein are studied in chapters 2, 3 and 4, with regards to gene diversity and transcriptional regulation. A novel approach has been introduced in chapter 5 to reconstruct the metabolic routes starting from the flavour compounds by reverse pathway engineering. From a different angle, chapter 6 explores the proto-cooperation of two yoghurt bacteria on a genetic level.
Chapter 2 describes a comprehensive comparative genomics study of the proteolytic system in lactic acid bacteria (LAB), which initiates flavour formation from milk protein. Protein 3D structures have been compared and superimposed to improve functional annotation of a protein superfamily which contains peptidases PepI/PepR/PepL and esterases EstA. A diverse distribution of proteolytic system components in strains of Lactococcus lactis has been identified based on PanGenome comparative genome hybridization (CGH) analysis.
Following the proteolysis of LAB, amino acids are further converted to flavour compounds. In chapter 3, we perform a comparative genomics analysis on genes which encode enzymes involved in amino acid degradation pathways, especially in branched-chain amino acids, aromatic amino acids and sulfur-containing amino acids metabolism. Annotations of these genes have been improved by combining analyses of phylogeny, gene context and literature evidence. The distribution of the enzymes are found to vary considerably between species and sometimes between strains, and in

GENERAL INTRODUCTION
17
CH
APT
ER 1
general agreed with different flavour-forming capacities of these LAB. The predicted potential of flavour-formation can provide an excellent framework to guide future experimental validation.
Chapter 4 describes the comparative analysis of the transcriptional mechanisms regulating the sulfur-containing amino acid metabolism in LAB. Various regulatory mechanisms have been found for different enzymes in different microorganisms, including regulatory proteins and RNA riboswitches. Binding motifs for CmbR and MetR/MtaR were refined in lactococci and streptococci. Novel putative regulatory motifs were found from upstream regions of several genes in some LAB strains.
Chapter 5 describes a novel approach “Reverse Pathway Engineering (RPE)” which combines chemoinformatics and bioinformatics analyses to predict the missing links between target compounds and their possible metabolic precursors. Using the RPE approach, plausible chemical or enzymatic reactions can be proposed by restrosynthesis methods, and putative enzymes for catalyzing the enzymatic reactions can be predicted by bioinformatics analyses. Several flavour-forming pathways have been explored using the approach. As a positive control, the previously characterized degradation routes of leucine to flavour components were successfully predicted. Some novel enzymatic reactions and the putative enzymes in LAB were identified, such as the conversion of α-hydroxyisocaproate to 3-methylbutanoic acid in the leucine degradation pathway and the formation of dimethylsulfide from methanethiol in the methionine degradation pathway.
Moreover, a study on proto-cooperation of yoghurt starter Lactobacillus delbrueckii ssp. bulgaricus (L. bulgaricus) and Streptococcus thermophilus, is presented in chapter 6. The new insights into proto-cooperation on the genetic level can be revealed by horizontal gene transfer (HGT) events between the bacteria. We performed an in silico analysis, combining gene composition evidence, i.e. GC content and dinucleotide composition, and gene transfer mechanism associated evidence, in order to predict horizontally transferred genes in both bacterial genomes. Several exopolysaccharide (EPS) biosynthesis genes are proposed to be transferred from S. thermophilus to L. bulgaricus, and the gene cluster cbs-cblB/cglB-cysE for metabolism of sulfur-containing amino acids was identified to be transferred from L. bulgaricus or L. helveticus to S. thermophilus.
Chapter 7 summarizes the main conclusions of this thesis and provides perspectives on further studies and future applications.

CHAPTER 1
18
References
1. Liu, M., van Enckevort, F.H. & Siezen, R.J. Genome update: lactic acid bacteria genome sequencing is booming. Microbiology 151, 3811-4 (2005).
2. Makarova, K. et al. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci U S A 103, 15611-6 (2006).3. Smit, G., Smit, B.A. & Engels, W.J.M. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese
products. FEMS Microbiology Reviews 29, 591-610 (2005).4. Pritchard, G.G. & Coolbear, T. The physiology and biochemistry of the proteolytic system in lactic acid bacteria. FEMS
Microbiol Rev 12, 179-206 (1993).5. Siezen, R.J. Multi-domain, cell-envelope proteinases of lactic acid bacteria. Antonie Van Leeuwenhoek 76, 139-55 (1999).6. Kunji, E.R., Mierau, I., Hagting, A., Poolman, B. & Konings, W.N. The proteolytic systems of lactic acid bacteria. Antonie
Van Leeuwenhoek 70, 187-221 (1996).7. Savijoki, K., Ingmer, H. & Varmanen, P. Proteolytic systems of lactic acid bacteria. Appl Microbiol Biotechnol 71, 394-406
(2006).8. Gilbert, C. et al. A new cell surface proteinase: sequencing and analysis of the prtB gene from Lactobacillus delbruekii subsp.
bulgaricus. J Bacteriol 178, 3059-65 (1996).9. Doeven, M.K., Kok, J. & Poolman, B. Specificity and selectivity determinants of peptide transport in Lactococcus lactis and
other microorganisms. Mol Microbiol 57, 640-9 (2005).10. Christensen, J.E., Dudley, E.G., Pederson, J.A. & Steele, J.L. Peptidases and amino acid catabolism in lactic acid bacteria.
Antonie Van Leeuwenhoek 76, 217-46 (1999).11. Sridhar, V.R., Hughes, J.E., Welker, D.L., Broadbent, J.R. & Steele, J.L. Identification of endopeptidase genes from the
genomic sequence of Lactobacillus helveticus CNRZ32 and the role of these genes in hydrolysis of model bitter peptides. Appl Environ Microbiol 71, 3025-32 (2005).
12. Weimer, B., Seefeldt, K. & Dias, B. Sulfur metabolism in bacteria associated with cheese. Antonie Van Leeuwenhoek 76, 247-61 (1999).
13. Nierop Groot, M.N. & de Bont, J.A.M. Conversion of phenylalanine to benzaldehyde initiated by an aminotransferase in lactobacillus plantarum. Appl Environ Microbiol 64, 3009-13 (1998).
14. Bonnarme, P. et al. Methylthioacetaldehyde, a possible intermediate metabolite for the production of volatile sulphur compounds from L-methionine by Lactococcus lactis. FEMS Microbiol Lett 236, 85-90 (2004).
15. Smit, B.A. et al. Chemical conversion of alpha-keto acids in relation to flavor formation in fermented foods. J Agric Food Chem 52, 1263-8 (2004).
16. Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J Mol Biol 215, 403-10 (1990).
17. O’Brien, K.P., Remm, M. & Sonnhammer, E.L. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 33, D476-80 (2005).
18. Huynen, M., Snel, B., Lathe, W., 3rd & Bork, P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res 10, 1204-10 (2000).
19. Francke, C., Kerkhoven, R., Wels, M. & Siezen, R.J. A generic approach to identify Transcription Factor-specific operator motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics 9, 145 (2008).
20. Bailey, T.L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2, 28-36 (1994).
21. Jain, R., Rivera, M.C., Moore, J.E. & Lake, J.A. Horizontal gene transfer in microbial genome evolution. Theor. Popul. Biol. 61, 489-95 (2002).
22. Juhas, M. et al. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33, 376-93 (2009).
23. Zaneveld, J.R., Nemergut, D.R. & Knight, R. Are all horizontal gene transfers created equal? Prospects for mechanism-based studies of HGT patterns. Microbiology 154, 1-15 (2008).
24. Chang, A., Scheer, M., Grote, A., Schomburg, I. & Schomburg, D. BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acids Res 37, D588-92 (2009).
25. Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res 32, D277-80 (2004).
26. Feist, A.M., Herrgard, M.J., Thiele, I., Reed, J.L. & Palsson, B.O. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 7, 129-43 (2009).
27. Oliveira, A.P., Nielsen, J. & Forster, J. Modeling Lactococcus lactis using a genome-scale flux model. BMC Microbiol 5, 39 (2005).
28. Pastink, M.I. et al. Genome-scale model of Streptococcus thermophilus LMG18311 for metabolic comparison of lactic acid bacteria. Appl Environ Microbiol 75, 3627-33 (2009).
29. Teusink, B. et al. Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J Biol Chem 281, 40041-8 (2006).
30. Pfeiffer, T., Sanchez-Valdenebro, I., Nuno, J.C., Montero, F. & Schuster, S. METATOOL: for studying metabolic networks. Bioinformatics 15, 251-7 (1999).
31. Durot, M., Bourguignon, P.Y. & Schachter, V. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol Rev 33, 164-90 (2009).

GENERAL INTRODUCTION
19
CH
APT
ER 1
32. Breitling, R., Vitkup, D. & Barrett, M.P. New surveyor tools for charting microbial metabolic maps. Nat Rev Microbiol 6, 156-61 (2008).
33. Ellis, L.B., Gao, J., Fenner, K. & Wackett, L.P. The University of Minnesota pathway prediction system: predicting metabolic logic. Nucleic Acids Res 36, W427-32 (2008).
34. Ellis, L.B., Roe, D. & Wackett, L.P. The University of Minnesota Biocatalysis/Biodegradation Database: the first decade. Nucleic Acids Res 34, D517-21 (2006).
35. Law, J. et al. Route Designer: a retrosynthetic analysis tool utilizing automated retrosynthetic rule generation. J Chem Inf Model 49, 593-602 (2009).
36. Prather, K.L. & Martin, C.H. De novo biosynthetic pathways: rational design of microbial chemical factories. Curr Opin Biotechnol 19, 468-74 (2008).
37. Altermann, E. et al. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc Natl Acad Sci U S A 102, 3906-12 (2005).
38. van de Guchte, M. et al. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc Natl Acad Sci U S A 103, 9274-9 (2006).
39. Morita, H. et al. Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res 15, 151-61 (2008).
40. Callanan, M. et al. Genome sequence of Lactobacillus helveticus, an organism distinguished by selective gene loss and insertion sequence element expansion. J Bacteriol 190, 727-35 (2008).
41. Wegmann, U. et al. Complete genome sequence of Lactobacillus johnsonii FI9785, a competitive exclusion agent against pathogens in poultry. J Bacteriol 191, 7142-3 (2009).
42. Kleerebezem, M. et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci U S A 100, 1990-5 (2003).
43. Chaillou, S. et al. The complete genome sequence of the meat-borne lactic acid bacterium Lactobacillus sakei 23K. Nat Biotechnol 23, 1527-33 (2005).
44. Kim, J.F. et al. Complete genome sequence of Leuconostoc citreum KM20. J Bacteriol 190, 3093-4 (2008).45. Wegmann, U. et al. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris
MG1363. J Bacteriol 189, 3256-70 (2007).46. Siezen, R.J. et al. Complete genome sequence of Lactococcus lactis subsp. lactis KF147, a plant-associated lactic acid
bacterium. J Bacteriol Submitted(2010).47. Bolotin, A. et al. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome
Res 11, 731-53 (2001).48. Bolotin, A. et al. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus.
Nat Biotechnol 22, 1554-8 (2004).49. Kim, J.F. et al. Genome sequence of the probiotic bacterium Bifidobacterium animalis subsp. lactis AD011. J Bacteriol 191,
678-9 (2009).50. Barrangou, R. et al. Comparison of the complete genome sequences of Bifidobacterium animalis subsp. lactis DSM 10140
and Bl-04. J Bacteriol 191, 4144-51 (2009).51. Schell, M.A. et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract.
Proc Natl Acad Sci U S A 99, 14422-7 (2002).52. Lee, J.H. et al. Comparative genomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to
deletion during pure culture growth. BMC Genomics 9, 247 (2008).53. Sela, D.A. et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization
within the infant microbiome. Proc Natl Acad Sci U S A 105, 18964-9 (2008).54. Thage, B.V. et al. Aroma development in semi-hard reduced-fat cheese inoculated with Lactobacillus paracasei strains with
different aminotransferase profiles. International Dairy Journal 15, 795-805 (2005).55. Broadbent, J.R. et al. Overexpression of Lactobacillus casei D-hydroxyisocaproic acid dehydrogenase in cheddar cheese.
Applied And Environmental Microbiology 70, 4814-4820 (2004).56. Marilley, L. & Casey, M.G. Flavours of cheese products: metabolic pathways, analytical tools and identification of producing
strains. International Journal Of Food Microbiology 90, 139-159 (2004).57. Yvon, M. & Rijnen, L. Cheese flavour formation by amino acid catabolism. International Dairy Journal 11, 185-201
(2001).58. Curioni, P.M.G. & Bosset, J.O. Key odorants in various cheese types as determined by gas chromatography-olfactometry.
International Dairy Journal 12, 959-984 (2002).59. Thierry, A. & Maillard, M.B. Production of cheese flavour compounds derived from amino acid catabolism by
Propionibacterium freudenreichii. Lait 82, 17-32 (2002).60. Acree, T.E. & Arn, H. Flavornet and human odor space. Vol. 2006 (Cornell University 2004).61. Page, R.D. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12, 357-8
(1996).

CHAPTER 1
20
知之者不如好之者,好之者不如乐之者。 - 论语
They who know the truth are not equal to those who love it, and they who love it are not equal to those who delight in it.
- Confucian Analects

PROTEOLYTIC SYSTEM OF LAB REVISITED
21
CH
APT
ER 2Chapter 2
THE PROTEOLYTIC SYSTEM OF LACTIC ACID BACTERIA REVISITED: A GENOMIC COMPARISON
Mengjin Liu, Jumamurat R. Bayjanov, Bernadet Renckens, Arjen Nauta, Roland J. Siezen
BMC Genomics 2010. 11(1):36.

CHAPTER 2
22
Abstract
BackgroundLactic acid bacteria (LAB) are a group of gram-positive, lactic acid producing Firmicutes.
They have been extensively used in food fermentations, including the production of various dairy products. The proteolytic system of LAB converts proteins to peptides and then to amino acids, which is essential for bacterial growth and also contributes significantly to flavor compounds as end-products. Recent developments in high-throughput genome sequencing and comparative genomics hybridization arrays provide us with opportunities to explore the diversity of the proteolytic system in various LAB strains.
ResultsWe performed a genome-wide comparative genomics analysis of proteolytic system
components, including cell-wall bound proteinase, peptide transporters and peptidases, in 22 sequenced LAB strains. The peptidase families PepP/PepQ/PepM, PepD and PepI/PepR/PepL are described as examples of our in silico approach to refine the distinction of subfamilies with different enzymatic activities. Comparison of protein 3D structures of proline peptidases PepI/PepR/PepL and esterase A allowed identification of a conserved core structure, which was then used to improve phylogenetic analysis and functional annotation within this protein superfamily.
The diversity of proteolytic system components in 39 Lactococcus lactis strains was explored using pangenome comparative genome hybridization analysis. Variations were observed in the proteinase PrtP and its maturation protein PrtM, in one of the Opp transport systems and in several peptidases between strains from different Lactococcus subspecies or from different origin.
ConclusionsThe improved functional annotation of the proteolytic system components provides an excellent
framework for future experimental validations of predicted enzymatic activities. The genome sequence data can be coupled to other “omics” data e.g. transcriptomics and metabolomics for prediction of proteolytic and flavor-forming potential of LAB strains. Such an integrated approach can be used to tune the strain selection process in food fermentations.
BackgroundLactic acid bacteria (LAB) have been used for centuries as starter or adjunct cultures in dairy
fermentations. The breakdown of milk proteins (proteolysis) by LAB plays an important role in generating peptides and amino acids for bacterial growth and in the formation of metabolites that contribute to flavor formation of fermented products. The proteolytic system of LAB comprises three major components: (i) cell-wall bound proteinase that initiates the degradation of extracellular casein (milk protein) into oligopeptides, (ii) peptide transporters that take up the peptides into the cell, and (iii) various intracellular peptidases that degrade the peptides into shorter peptides and amino acids. In particular, as caseins are rich in proline, LAB have numerous proline peptidases for degrading proline-rich peptides [1-3]. Amino acids can be further converted into various flavor compounds, such as aldehydes, alcohols and esters [4].
Several reviews have described the proteolytic system of LAB with respect to their biochemical and genetic aspects [1, 5-8]. In the past ten years, however, many LAB genomes have been sequenced, which allows a thorough comparative analysis of their proteolytic systems at a genome scale. In a preliminary study, we described a comparative analysis of cell-wall-bound proteinase and various peptidases from 13 fully or incompletely sequenced LAB which were publicly available in May 2006

PROTEOLYTIC SYSTEM OF LAB REVISITED
23
CH
APT
ER 2
[9]. More recently, over ten additional LAB genomes have become publicly available. These include 8 LAB strains from the Joint Genome Institute and the LAB Genome Consortium [10], the model laboratory strain Lactococcus lactis subsp. cremoris MG1363 [11], a Lactobacillus helveticus strain [12] which is known for its proteolytic capacity as an adjunct culture in cheese, and the probiotic strain Lactobacillus rhamnosus GG [13]. Furthermore, a recent comparative genome hybridization (CGH) analysis of 39 L. lactis strains [14] provides opportunities to explore the diversity of the proteolytic system within the same species.
In this study, we systematically explored the diversity of the cell-wall bound proteinase, the peptidases and the peptide transporters in twenty-two completely sequenced LAB strains. The distinctions between subgroups in large peptidase families such as the PepP/PepQ/PepM family, the PepD family and the PepI/PepR/PepL family are described in detail as examples. The PepI/PepR/PepL family was compared with the EstA family of esterases, the key enzyme for synthesizing various ester flavors [4, 15], since the members of these two families share sequence and structure homology. Furthermore, the results from comparative genomics analysis were used to explore the diversity of members of the proteolytic system in 39 Lactococcus lactis strains by pangenome CGH analysis [14].
Methods
Comparative genome analyses and orthologous groups identificationComplete genome sequences of LAB were obtained from the NCBI microbial genome database
(http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi). The genomes include: Lactobacillus acidophilus NCFM (abbreviation LAC, accession code CP000033), Lactobacillus johnsonii NCC 533 (LJO, AE017198), Lactobacillus gasseri ATCC 33323 (LGA, CP000413), Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842 (LDB, CR954253), Lactobacillus delbrueckii subsp. bulgaricus ATCC BAA365 (LBU, CP000412), Lactobacillus plantarum WCFS1 (LPL, AL935263), Lactobacillus brevis ATCC 367 (LBE, CP000416), Lactobacillus sakei 23K (LSK, CR936503), Lactobacillus salivarius UCC118 (LSL, CP000233), Oenococcus oeni PSU1 (OOE, CP000411), Pediococcus pentosaceus ATCC 25745 (PPE, CP000422), Leuconostoc mesenteroides ATCC 8293 (LME, CP000414), Lactobacillus casei ATCC 334 (LCA, CP000423), Lactococcus lactis subsp. lactis IL1403 (LLX, AE005176), Lactococcus lactis subsp. cremoris MG1363 (LLM, AM406671), Lactococcus lactis subsp. cremoris SK11 (LLA, CP000425), Streptococcus thermophilus CNRZ1066 (STH, CP000024), Streptococcus thermophilus LMG18311 (STU, CP000023), Streptococcus thermophilus LMD9 (STM, CP000419), Lactobacillus reuteri F275 (LRF, CP000705), Lactobacillus helveticus DPC 4571 (LHE, CP000517) and Lactobacillus rhamnosus GG (LRH, FM179322). Incomplete genome sequences of Lactococcus lactis subsp. lactis strains KF147 and KF282 [16] were additionally used for analysis of L. lactis strain diversity by pangenome CGH analysis [14].
Protein sequences of experimentally verified proteolytic system members, i.e. cell-wall bound proteinase, various peptidases and peptide transporters, were derived from the non-redundant protein database Uniprot (http://www.uniprot.org/) [17]. These sequences were used to perform a BLASTP [18] search against all LAB genomes. The corresponding Hidden Markov Models (HMMs) of each protein family were obtained from the Pfam database [19] and utilized to search for homologous genes using the HMMER 2.3.2 package (http://hmmer.janelia.org/). The homologous sequences of each proteinase, peptidase and peptide transporter were collected on basis of the BLAST and HMM search results and redundancies were removed. Orthologous groups (subfamilies) were identified by an in-house developed method [4, 20]. Multiple sequence alignments (MSA) were generated for each homologous group using MUSCLE [21]. Bootstrapped (n=1000) neighbor-joining family trees were constructed with ClustalW [22]. The trees were visualized in LOFT [23] and orthologous groups were

CHAPTER 2
24
identified. The gene contexts were analyzed using the ERGO Bioinformatics Suite [24] to improve ortholog prediction when necessary.
3D structure alignmentPeptidases PepI/PepR/PepL and esterase EstA belong to the same protein superfamily, but they
possess different functionalities. In order to identify substrate specificity of each protein subfamily, a comparison of known protein 3D structures was carried out. As described above, protein sequences of experimentally characterized peptidases PepI, PepR, and PepL, together with EstA esterases were used to search against all the sequenced LAB genomes and other prokaryote genomes in the NCBI database by BLASTP [18]. Moreover, the HMM of the protein α/β hydrolase fold PF00561 from the Pfam database [19], to which PepI/PepR/PepL and EstA belong, was used to search against LAB genomes. Homologs of both PepI/PepR/PepL and EstA families were collected. Similarly, the protein sequences of experimentally verified PepI/PepR/PepL and EstA members were used for BLAST searches against the PDB database (http://www.rcsb.org/pdb/) [25]. The protein sequences, as well as the 3D structures of the best BLAST hits were collected. Other proteins with similar structures were retrieved by the Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/) using the protein structures of the BLAST hits as input.
The retrieved 3D structures of the proteins used as templates in this study are: the tricorn interacting factor F1 with proline iminopeptidase (PIP) activity from Thermoplasma acidophilum (PDB ID: 1MTZ), proline iminopeptidases from Xanthomonas campestris pv. citri (PDB ID: 1AZW) and Serratia marcescens (PDB ID: 1WM1) as members of PepI/R/L subfamilies, and the esterase (PDB ID: 2UZ0) from Streptococcus pneumoniae which belongs to the EstA subfamily. These 3D structures were superimposed and visualized by the YASARA program (version 6.813, http://www.yasara.org/). Conserved superimposable regions (core regions) of the catalytic domain were identified based on the 3D-structure alignment, and these consisted of 4 discontinuous sequence segments that are connected by loops of variable structure.
The amino acid sequences of the four core region segments were aligned with MUSCLE or ClustalW as described [26]. The alignments were manually curated for ambiguously aligned sequences compared to the 3D-structure alignment. Sequences with more than 90% identity were removed. Finally, a MSA was constructed based on concatenated alignments of all the curated local alignments of the core regions [see Additional File 1]. A bootstrapped (n=1000) neighbor-joining tree on basis of the MSA was constructed and orthologous groups, so-called subfamilies, were identified automatically by LOFT.
Pangenome CGH diversity analysisComparative genome hybridization (CGH) data of 39 L. lactis strains was acquired from
pangenome arrays [14]. The pangenome array was constructed on basis of publicly available complete genome sequences of L. lactis subsp. lactis IL1403, L. lactis subsp. cremoris SK11, and incomplete genome sequences of L. lactis strains KF147 and KF282, as described by Bayjanov et al. [14]. The CGH data used in this study can be found under the accession number GSE12638 in the NCBI GEO (NCBI Gene Expression Omnibus) database (http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE12638).
The genes encoding predicted proteolytic system components of the three sequenced L. lactis strains were used to query the database containing pangenome CGH data. We obtained a statistical score of the hybridization signal for each gene from the reference strains against 39 L. lactis strains. A cut-off value 5.5 was used to assign presence or absence of every gene from the proteolytic system in query strains, as described by Bayjanov et al. [14]. In most cases, a gene is regarded present in a specific strain if it has a maximum score higher than 5.5 [14].

PROTEOLYTIC SYSTEM OF LAB REVISITED
25
CH
APT
ER 2
peptid
ase
F
amily
S
ub
stra
te/
An
no
tatio
n LA
C d
LJO
L
GA
L
DB
LB
U
LH
E
LP
L
LBE
LR
F
LSK
LS
L
LC
A
LRH
P
PE
O
OE
L
ME
LL
X
LLA
LLM
S
TH
S
TU
S
TM
P
rote
inas
e
Cell-
wall
bound
pro
tein
ase
P
rtP
S
8-A
1
1
0
1
1
0
0
0
0
0
0
2 (
1p)
2
0
0
0
0
1 b
0
0
0
1
P
rtM
mat
ura
tion
pro
tein
for
P
rtP
(a
dja
cent
P
rtP
) 0
1
0
0
0
0
0
0
0
0
0
1
1
0
0
0
0
1 b
0
0
0
0
Pep
tid
es
tran
spo
rter
s
OppA
Olig
opeptid
e-
bin
din
g p
rote
in
3
4
4
2
2
1
0
1
0
0
0
1
1
0
1
0
1
2 b
2
4
(2p)
3
(1p)
2
OppB
perm
eas
e
pro
tein
1
1
1
1
1
1
0
1
0
0
0
1
1
0
1
0
1
2 b
2
1
1
1
OppC
perm
eas
e
pro
tein
1
1
1
1
1(p
) 1
0
1
0
0
0
1
1
0
1
0
1
2 b
2
1
1
1
OppD
AT
P-b
ind
ing
pro
tein
1
1
1
1
1
1
0
1
0
0
0
1
1
0
1
0
1
2 b
1
1
1
1
Olig
opeptid
es
AB
C tr
ansp
ort
sy
stem
O
ppF
AT
P-b
indin
g pro
tein
1
1
1
1
1
1
0
1
0
0
0
1
1
0
1
0
1
2 b
1
1
1
1
DppA
/P
di/t
rip
ep
tide-
olig
opeptid
e-
bin
din
g p
rote
in
3
0
0
6
5
1
4
3
0
1
1
3
2
2
3
2
3
3
2
1(p
) 1(p
) 1(p
)
DppB
perm
eas
e
pro
tein
1
0
0
1
1
1
1
1
0
1
1
1
1
1
1
1
1
1
1
1(p
) 1(p
) 1(p
)
DppC
perm
eas
e
pro
tein
1
0
0
1
1
1
1
1
0
1
1
1
1
1
1
1
1
1
1
2(p
) 2(p
) 1(p
)
DppD
AT
P-b
ind
ing
pro
tein
1
0
0
1
1
1
1
1
0
1
1
1
1
1
1
1
1
1
1
1(p
) 1(p
) 1(p
) D
i/tri
pe
ptid
es
AB
C tr
ansp
ort
sy
stem
D
ppF
AT
P-b
indin
g pro
tein
1
0
0
1
1
1
1
1
0
1
1
1
1
1
1
1
1
1
1
1
1
1
di/t
ripeptid
es io
n-lin
ked tra
nsp
ort
er
Dtp
T
P
TR
fam
ily
1
1
1
0
0
0
1
1
1
1
1
1
1
1
0
0
1
1
1
1
1
1
Pep
tid
ases
Pe
pC
C
1-B
X
|(X
)n
1
1(2
)a 1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
Am
inopeptid
ase
P
ep
N
M1
X|(
X)n
1(1
)a 1
1
1
1
1(1
)a 1
1
1
1
1 b
1
1
1
1
1
1
1
1
1
1
1
Pe
pM
M
24-A
M
et|(
X)n
1
1
1
1
1
1
1
2
1
1
1
1
1
1
1
1
1
1
1
1
1
1
Pe
pA
M
1 G
lu/A
sp
|(X
)n
1
1
1
1
1
1
0
0
0
0
0
0
1
0
0
1
1
1
1
1
1
1
(uniq
ue
am
inopeptid
ase
s)
P
cp
C15
pyr
oG
lu|
(X)n
1
1
1
1
1
1
0
1
0
0
0
1
1
0
0
0
0
1 b
1
0
0
0
Pe
pE
/Pep
G
C1-
B
(X)m
|(X
)n
3
3
3
2
2
3
1
1
1
1(p
) 0
1
1
0
1
0
0
0
0
0
0
0
Pe
pO
M
13
(X)m
|(X
)n
2
3
3
1
1
3
1
1
1
1
1
2
2
1
1
1
1
2 b
2
1
1
1
endopeptid
ase
Pe
pF
M
3-B
(X
)m|(
X)n
1
1
1
1
1
1
2
2
0
2
2
2
3
1
3
2
1
2 b
1
1
1
1
Pe
pD
C
69
X|X
5 (
1p)
6
4
3
(1p)
3
(1p)
5
(1p)
4
5
5
5
2
4
3
4
1
0
2
2 (
1p)
2
1(p
) 1(p
) 1
(p)
dip
eptid
ase
Pe
pV
M
20-A
X
|X
1
1
2
1
1
1
1
2
2
2
1
2
2
1
2
2
1
1
1
1
1
1
trip
eptid
ase
P
ep
T
M20-B
X
|X-X
2
2
2
2
2
2
1
1
1
1
1
0
1
1
1
1
1
1
1
1
1
1
Pe
pX
S
15
X-P
ro|(
X)n
1
1
1
1
1
1
1
1
1
1
1
1
1
1(1
) a
1
1
1(1
)a 1
1
1
1
1
Pe
pI
S33
Pro
|X-(
X)n
1
0
0
1
1
1
1
1
0
0
0
0
1
0
0
0
0
0
0
0
0
0
Pe
pR
S
33
Pro
|X
1
1
1
1
1
1
1
1
1
1
0
1
1
1
0
0
0
0
0
0
0
0
Pe
pL
c
S33
Leu|(
X)n
1
0
1
0
0
0
1
0
0
0
0
1
1
0
3
0
0
0
0
0
0
0
Pe
pP
M
24-B
X
|Pro
-(X
)n
1
1
1
1
1
1
1
1
1
0
1
1
1(1
) a
0
1
1(1
)a 1
1
1
1
1
1
pro
line p
eptid
ase
Pe
pQ
M
24-B
X
|Pro
1
1
1
2
2
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
a. P
aral
og
s: T
he
nu
mb
er in
bra
cket
s in
dic
ates
th
e n
um
ber
of e
xtra
par
alo
gs
pre
sen
t, w
hic
h d
o n
ot
bel
on
g t
o t
he
sam
e o
rth
olo
g g
rou
p a
s al
l oth
er p
rote
in fa
mily
mem
ber
s (s
ee p
hyl
og
enet
ic t
rees
). b.
Pla
smid
-en
cod
ed p
rote
ins:
Prt
P, Pr
tM, P
cp a
nd
on
e o
f th
e Pe
pF,
Pep
O, O
pp
tra
nsp
ort
sys
tem
en
cod
ing
gen
es o
f LLA
, as
wel
l as
Pep
N fr
om
LSL
. c.
In
clu
din
g t
he
L. p
lant
arum
an
d L
. cas
ei p
rote
ins
in t
he
inte
rmed
iate
gro
up
in t
he
Pep
L/Pe
pR
fam
ily w
hic
h a
re n
ot
ort
ho
log
s o
f th
e Pe
pR
fam
ily.
d. S
trai
n n
ames
are
: LA
C, L
. aci
doph
ilus
NC
FM; L
JO,
L. jo
hnso
nii N
CC
533
; LG
A, L
. gas
seri
ATC
C 3
3323
; LD
B, L
. bul
gari
cus
ATC
C 1
1842
; LB
U, L
. bul
gari
cus
ATC
C B
AA
365;
LH
E, L
. hel
veti
cus
DPC
457
1; L
PL,
L. p
lant
arum
WC
FS1;
LB
E, L
. bre
vis
ATC
C 3
67;
L
RF,
L. r
eute
ri F
275;
LSK
, L.
sak
ei 2
3K; L
SL, L
. sal
ivar
ius
UC
C11
8; L
CA
, L. c
asei
ATC
C 3
34; L
RH
, L. r
ham
nosu
s G
G; P
PE,
P. p
ento
sace
us A
TCC
257
45; O
OE,
O. o
eni P
SU1;
LM
E, L
. mes
ente
roid
es A
TCC
829
3; L
LX, L
. lac
tis
sub
sp. l
acti
s IL
1403
; LLA
,
L. l
acti
s su
bsp
. cr
emor
is S
K11
; LLM
, L. l
acti
s su
bsp
. cre
mor
is M
G13
63; S
TH, S
. the
rmop
hilu
s C
NRZ
1066
; STU
, S. t
herm
ophi
lus
LMG
1831
1; S
TM, S
. the
rmop
hilu
s LM
D9.
p
. pse
ud
og
enes
(e.g
. wit
h t
run
cati
on
s o
r fra
me-
shift
s)
Figu
re 2
.1. D
istr
ibut
ion
of p
rote
inas
e, p
eptid
e tr
ansp
orte
rs a
nd p
eptid
ases
of t
he p
rote
olyt
ic sy
stem
in L
AB
. The
num
ber o
f ide
ntifi
ed g
enes
is in
dica
ted.
MER
OPS
fam
ilies
are
indi
cate
d fo
r pro
tein
-as
e an
d pe
ptid
ases
. Sha
ding
show
s abs
ence
of a
gen
e (w
hite
), a
sing
le g
ene
or m
ultip
le g
enes
(gre
y). T
he G
I cod
es o
f the
gen
es c
an b
e fo
und
in A
dditi
onal
file
2.

CHAPTER 2
26
Results
The distribution of proteolytic system components in sequenced LAB genomes An overview of the distribution of components of the proteolytic system identified in 22
completely sequenced LAB is given in Figure 2.1. A detailed list of genes with GI codes can be found in Additional File 2. The number of genes encoding putative members of each proteinase, peptide transporter and peptidase subfamily are shown.
The LAB genomes in the L. acidophilus group [4], including L. acidophilus, L. johnsonii, L. gasseri, L. bulgaricus, and L. helveticus strains, encode a relatively higher number and variety of proteolytic system components. Some enzymes are only found in a few LAB strains, such as the cell-wall bound proteinase (PrtP). PrtP was only found on the chromosome of L. acidophilus, L. johnsonii, L. bulgaricus, L. casei, L. rhamnosus and S. thermophilus strain LMD9, as well as on the plasmid of L. lactis subsp. cremoris SK11 [27]. Members of both the PepE/PepG (endopeptidases) and PepI/PepR/PepL (proline peptidases) superfamilies are absent in lactococci and streptococci. On the other hand, many of the peptidases seem to be essential for bacterial growth or survival as they are encoded in all LAB genomes. For instance, aminopeptidases PepC, PepN, and PepM, and proline peptidases PepX and PepQ are present in all genomes, usually with one gene per genome. Some LAB genomes have two peptidase homologs, possibly with the same function (shown in brackets in Figure 2.1), e.g. two PepC homologs (GI codes: 42518641 and 42518638) in L. johnsonii. Other essential peptidases (found in all LAB genomes) such as endopeptidase PepO and dipeptidase PepV are encoded by multiple paralogous genes.
L. acidophilus, L. brevis, L. casei, L. rhamnosus and L. lactis strains possess all three known LAB peptide transport systems, i.e. the di/tripeptide Dpp and DtpT systems and the oligopeptide Opp system [5]. In contrast, L. reuteri strain only has one functional peptide transport system, the DtpT system. Several peptide transporters or peptidases fall into larger protein superfamilies. Examples are (i) the oligopeptide-binding protein OppA and di/tripeptide-binding proteins DppA/DppP in the same peptide-binding protein family, (ii) aminopeptidase PepC together with endopeptidases PepE and PepG belonging to MEROPS peptidase family C1-B, (iii) proline peptidases PepI, PepR and PepL belonging to MEROPS family S33, and (iv) aminopeptidase PepM together with proline peptidases PepP and PepQ belonging to MEROPS family M24 (Figure 2.1). Protein members in those large superfamilies share high sequence similarity, and cannot always be distinguished by simple BLAST sequence homology searches. Using a comparative genomics approach, the large protein families can be divided into subfamilies with putatively different substrate specificities. For example, the aminopeptidase PepC subfamily can be clearly distinguished from the endopeptidase PepE/PepG subfamily as they are separated into distinct groups in a superfamily tree [9]. In other cases, such as the endopeptidase PepF family, several distinct subgroups can be distinguished but the difference in specificity between the subgroups is still unclear [see Additional File 3].
Three large peptidase families (PepP/PepQ/PepM, PepD and PepI/PepR/PepL) will be discussed in detail in the following sections.
Subfamilies of peptidase family PepP/PepQ/PepMPepP, PepQ and PepM belong to the MEROPS peptidase family M24 which requires metal
ions for catalytic activity. PepM is a methionyl aminopeptidase cleaving N-terminal methionine from proteins. PepP is a member of the proline peptidases which cleave off any N-terminal amino acid linked to proline in an oligopeptide. PepQ is also a proline peptidase, however specific for Xaa-Pro dipeptides, where Xaa represents any amino acid (Figure 2.1)
Our phylogenetic analysis shows that PepP, PepQ and PepM are separated into three distinct subgroups in accordance with the known different substrate specificities of each peptidase (Figure

PROTEOLYTIC SYSTEM OF LAB REVISITED
27
CH
APT
ER 2
2.2). PepP and PepQ seem to be more closely related than PepM on the basis of the family tree, which is in agreement with the differences in their catalytic activities. Bacterial PepM is an aminopeptidase belonging to subfamily M24A which usually requires cobalt ions for catalysis, while PepP and PepQ as proline peptidases belong to the subfamily M24B which prefers manganese [1].
[1000]
[990]
[629]
[524]
[777]
[506]
[667]
[949] LRF_148530818PPE_116492999
[848] LBE_116333814 LPL_28378852
OOE_116490638 [632] LSL_90961396
[991] LSK_81428028[1000] LRH_258507787
LCA_116494352
[1000]
[1000]
[726] LDE_PepQ LBU_116514499 LDB_104774401
[851]
[1000] LGA_116629106 LJO_42518586
[1000] LAC_58336767 [1000] LHV_pepQ
LHE_161507001 LME_116617659
[1000]
[1000]
[987] LLA_116512511 LLM_125623626 LLX_15673631
[1000] STM_116627514 [1000]STU_55820708 STH_55822599
[1000] LBU_116514595 LDB_104774485
[970] LRH_258507347
[944]
[1000]
[877]
[1000] LGA_116629404 LJO_42519426
[1000] LHE_161507742 LAC_58337609
[1000] LBU_116514378 LDB_104774293
[758]
[930]
[407]
[981]
[689] LBE_116333619 LPL_28378296 LRF_148531446
LSL_90962059[998] LME_116618421
OOE_116490991
[794]
[1000] LRH_258508682 LCA_116495125
[1000]
[1000]
[778] STM_116628437 STU_55821714 STH_55823634
[1000] LLX_15672673 [1000] LLA_116511487
[1000]LLA_PepP LLM_125624666
[908]
[1000] CBO_153939733 LME_116618966
[1000] ECO_pepM
[1000]
[1000] LBE_116334569 LPL_28377183
[525]
[1000]
[986]
[1000] LHE_161507146 LAC_58336948
[1000] LBU_116513668 LDB_104773672
[1000] LGA_116629358 LJO_42519469
[320]
[1000]
[1000]
[861] LLM_125623440 LLX_15672594 LLA_116511405
[1000] STM_116628246 [538] STU_55821533 STH_55823461
[462]
[832] LME_116618532 OOE_116491667
[699]
[940]
[690] LRF_148531635 LSL_90962243
PPE_116492178
[899]
[982] LBE_116334483 LSK_81429128
[1000] LRH_258508051 LCA_116494589
PepQ
PepP
PepM
Figure 2.2. Superfamily tree of PepP/PepQ/PepM members in LAB. Genome abbreviations can be found in “Methods”. For each gene, the organism abbreviations are followed by GI codes. Homologs from two non-LAB strains are also included, CBO for Clostridium botulinum F str. Langeland and ECO for E. coli. Experimentally characterized genes are highlighted by the dots following the gene names. Small circles represent the speciation events, and squares represent duplication events.
In the PepP subgroup, one gene is found in each LAB genome except in L. sakei and Pediococcus pentosaceus. The absence of the pepP genes in both genomes is very likely due to a gene loss event. The family tree also includes an experimentally verified pepP gene from L. lactis whose protein product has been purified and characterized [28]. Moreover, LAB-derived pepP genes are always flanked on the chromosome by a gene encoding an elongation factor for protein translation. The conserved gene context of pepP among LAB genomes is consistent with the putative important physiological role of PepP in protein maturation, as suggested by Matos et al. [28].

CHAPTER 2
28
Genes from the PepQ cluster are distributed equally in all LAB genomes, generally as one copy per genome. However, the L. delbrueckii bulgaricus strains have two pepQ paralogs. One paralog is clustered with the other orthologs of LAB, whereas the second paralog is located in a separate cluster (LBU_116514595 and LDB_104774485). This might be the result of an ancient duplication (Figure 2.2) or horizontal gene transfer (HGT) event. Rantanen et al. suggested that the second paralogous pepQ of L. bulgaricus is a cryptic gene [29]. Experimentally characterized pepQ genes from L. delbrueckii bulgaricus [30] and L. helveticus (GI: 3282339) are added and highlighted in the tree, supporting the annotation of the subgroups.
In the aminopeptidase PepM subgroup, L. brevis has an extra paralogous gene, which clusters together with the L. plantarum pepM gene. Gene context analysis suggests that pepM genes in all Lactobacillus strains share the same neighbor genes, except the pepM gene from L. plantarum and both the paralogs from L. brevis. One of the L. brevis pepM genes (LBE_116334483) is located in the same operon as a transposase. Based on the protein family tree, we hypothesize that an extra pepM gene was acquired first in the ancestor of L. brevis and L. plantarum, after which one gene was lost from L. plantarum. The L. plantarum pepM gene (LPL_28377183) is flanked by a methionine metabolism related operon (cysK_cblB/cglB_cysE). Therefore, the pepM gene in L. plantarum may have a broader function, probably utilizing proteins and peptides as methionine pool, in addition to the classic PepM function for N-terminal maturation of proteins.
One gene from Leuconostoc mesenteroides (LME_116618966) is located as an intermediate between the PepP/PepQ and PepM subfamilies. It shares higher sequence homology with a putative pepP gene from Clostridium botulinum (Figure 2.2) and has a phage-related gene in its neighborhood. This suggests that the pepP gene from Leuconostoc mesenteroides might be acquired from clostridia.
Subfamilies of peptidase family PepD The PepD dipeptidase family has a broad specificity toward various dipeptides [1]. PepD
has been purified and characterized from L. helveticus by Vesanto et al. [31]. The pepD genes are distributed heterogeneously in LAB genomes, varying from 0 to 6 paralogs. The pepD gene is absent in Leuconostoc mesenteroides and truncated in S. thermophilus strains, while multiple genes are mainly observed in Lactobacillus genomes (Figure 2.1). Recently, Smeianov et al. reported the expression level of four pepD genes from L. helveticus CNRZ32 by a microarray analysis [32]. Five major PepD subfamilies can be clearly distinguished based on the multiple sequence alignment (Figure 2.3). PepD1-4 are assigned with the names according to the four pepD genes from L. helveticus [32]. Due to the lack of experimental evidence, it is still unclear whether the substrate specificities vary between those subfamilies. Microarray analysis of L. helveticus has shown that pepD1, pepD2 and pepD4 were up-regulated in MRS medium compared to growth in milk, while pepD3 was not differentially expressed in both media [31]. It suggests that differences between subgroups of pepD1/pepD2/pepD4 and pepD3 could also be on the level of transcription regulation. Moreover, several genes are located as intermediate between the major PepD subgroups in the superfamily tree. Most of those genes have unclear origins and functions. The protein sequences of LCA_116493607 from L. casei, LRH_258507036 from L. rhamnosus, LJO_42518640 from L. johnsonii, and LBU_116514855 from L. bulgaricus have best BLASTP hits to several recently sequenced lactobacilli, such as L. hilgardii and L. buchneri, suggesting a possible duplication of the gene in a specific Lactobacillus group.
3D-structure comparison to distinguish PepI/PepR/PepL peptidases from EstA family esterases
The proline iminopeptidase PepI possesses aminopeptidase activity toward N-terminal proline peptides, preferably tri-peptides, while prolinase PepR has a broad specificity for dipeptides

PROTEOLYTIC SYSTEM OF LAB REVISITED
29
CH
APT
ER 2
including Pro-Xaa dipeptides [1]. The only characterized PepL is from L. delbrueckii subsp. lactis DSM7290 and it displays high specificity for di-/tri- peptides with N-terminal leucine residues [33]. Interestingly, the PepI/PepR/PepL family and the esterase EstA family belong to the same α/β hydrolase superfamily, since the BLASTP analysis of PepI/PepR/PepL members against the non-redundant protein database also retrieves homologs from the EstA family. Multiple sequence alignment (MSA) of the whole protein sequences of the homologs from those two protein families is not reliable, as large insertions and deletions are present in these sequences, and several regions of the proteins share very low sequence similarity. Therefore, we first compared the 3D structures of four representative proteins by superposition, including proline iminopeptidases from Thermoplasma acidophilum (PDB ID: 1MTZ) [34], Xanthomonas campestris pv. citri (PDB ID: 1AZW) [35], and Serratia marcescens (PDB ID: 1WM1) [36] as members from the PepI/R/L family, and an esterase A (PDB ID: 2UZ0) from Streptococcus pneumoniae [37] as a member from the EstA subfamily (Figure 2.4). The superimposed 3D structures share a highly similar catalytic domain, which displays a typical canonical α/β hydrolase topology consisting of an eight-stranded β-sheet, and have a non-conserved cap domain. Four conserved structural regions in the catalytic domain, separated by variable loops, were identified based on the structure alignment. A detailed comparison of the residues of the catalytic
[560]
[845]
[999]
[727]
[966]
[810]
[984]
[621]
[694]
[804]
[942]
[1000]
[968] PPE_116493193 LSK_81427789
[990] LBE_116334325 LPL_28377780
[1000] LLX_15673540[1000] LLA_116512355
LLM_125623784 [1000] LLM_125623134
LLX_15672234 [1000] LRH_258508967
LCA_116495449
[1000]
[1000] LBU_116513626 LDB_104773634
[1000]
[1000] LGA_116628929 LJO_42518345
[1000] LHV_pepD LHE_161506829
LSK_81428199 LRF_148530839
[1000] LHE_5772891 LAC_58336386
[1000] BLO_pepD OOE_116491743
[1000] LRH_258507036 LCA_116493607
[1000]
[1000] LBE_116332933 LPL_28377159
[1000] LRF_148532067
[1000]
[1000] LGA_116630204 LJO_42519672
[1000] LAC_58337904 [1000] LHV_pepD3
LHE_161507962 [538] LJO_42518640
LBU_116514855
[878]
[1000]
[1000]
[999]
[968]
[885]
[1000] LHV_pepD4 LHE_161508060 LAC_58338085
[1000] LGA_116630307 LJO_42519780
[1000] LBU_116514839 LDB_104774712
LJO_42518668 [577] LRF_148531326
[398]
[306] PPE_116492678 LSL_90961981
[157] LSK_81428507 [436] LPL_28378398
LBE_116333540 LBE_116334103
[999]
[1000]
[672]
[1000] PPE_116491984 LCA_116495641
LSK_81427935 LRF_148530979
[994] LRF_148531929
[510]
[955]
[963] PPE_116493475 LBE_116334140
[556] LPL_28377254 LSL_90961161
[945]
[903]
[1000] LRH_258508153 LCA_116494640
LSK_81427806
[1000]
[1000] LGA_116629440 LJO_42519390
[1000] LAC_58337570 [1000]] LHV_pepD2
LHE_161507712
[[
PepD1
PepD3
PepD4
PepD5
PepD2
Figure 2.3. Superfamily tree of PepD members in LAB. PepD that is experimentally characterized from Bifidobacterium longum NCC2705 (BLO) [52] and pepD genes from L. helveticus CNRZ32 (LHV) analyzed by microarray [32] are indicated by the dots following the gene names. Small circles represent the speciation events, while squares represent duplication events.

CHAPTER 2
30
site and substrate-binding region can be found in Additional File 4. In contrast, the cap domain shows a large structural variation, and the esterase EstA has a much smaller cap domain than the peptidases (Figure 2.4). The cap regions of peptidases cover and close the substrate-binding region, allowing only the N-terminal proline of a peptide to fit into the substrate-binding pocket.
CAP
CORE
CAP
GluHis
AspSer
A. B.
Figure 2.4. Superposition of 3D structures of proline iminopeptidases 1WM1 (yellow) and 1MTZ (green), and esterase 2UZ0 (purple). (A colour version can be found in Appendix) The structure of 1AZW is highly similar to 1WM1 and is not shown. A) The 4 conserved structural core segments are shown as thick tubes, and the variable segments as thin sticks connecting C-alpha atoms. The variable large cap regions of the peptidases, which do not superpose, are at the bottom half of the figure. Note that the esterase has a much shorter connecting segment in this cap region. The red frame indicates the position of the active site, which is shown as the zoomed-in view in Panel B. B) The catalytic site is shown with catalytic residues Ser, His and Asp. The active site is enlarged and rotated by about 180 degrees relative to Panel A. A short stretch of the cap region in both peptidases is shown, bearing the Glu residues that interact with the positive charge of the peptide substrate N-terminus. Note that the side chains of the two Glu residues superpose very well, despite coming from different (non-superposable) parts of the cap region.
A MSA of the concatenated sequences of the four conserved structural regions of the PepI/PepR/PepL and EstA superfamily members from various microorganisms was constructed and manually curated [See Additional File 1]. On basis of the curated MSA, a much improved superfamily tree was constructed for the PepI/PepR/PepL and EstA families, including LAB and other bacteria, as well as the reference proteins with known 3D structures (Figure 2.5). In this 3D alignment tree, the homologs of the superfamily can be clearly separated into four subclusters (Figure 2.5). The first cluster PepIa contains the proline iminopeptidases from Proteobacteria and non-LAB Firmicutes, including the ones from the known structures 1AZW and 1WM1. The second cluster contains the esterase members from LAB, including the representative structure 2UZ0 from S. pneumoniae. The third cluster PepIb contains proline iminopeptidases from Proteobacteria and Actinobacteria, and PepI from Firmicutes (including the ones from LAB), as well as the known structure 1MTZ from Thermoplasma acidophilum. The last cluster PepR/L consists of putative PepL proteins from LAB and the subgroup of prolinase PepR. Experimentally verified proteins PepR from L. helveticus CNRZ32 [38, 39], PepI from L. delbrueckii subsp. bulgaricus CNRZ 397 [40, 41], PepL from L. delbrueckii subsp. lactis DSM7290 [33] and EstA from L. lactis [42] and L. casei BL23 [43] also support this subdivision within the protein superfamily (Figure 2.5). Moreover, PepI from L. helveticus strain 53/7 has also been experimentally characterized [44].
Sequenced lactococcal, streptococcal, leuconostoc and L. salivarius strains lack the genes encoding proline peptidases PepI, PepR or PepL. This agrees with the observation from gene deletion

PROTEOLYTIC SYSTEM OF LAB REVISITED
31
CH
APT
ER 2
experiments in strains harboring those peptidase genes that the physiological role of PepI, PepR or PepL is not essential for cell growth [39, 45, 46]. However, in L. helveticus, the growth rate in milk was slower for a PepI-deletion mutant as compared to the wild type [45]. Similarly, the activity of cell extract of L. helveticus and L. rhamnosus toward several proline dipeptides was significantly reduced in a PepR-deletion mutant [39, 46]. Those observations suggest that PepI/PepR/PepL may contribute specifically to the proteolytic capacity on proline-containing peptides of Lactobacillus strains.
[923]
[886]
[744]
[1000]
[445]
[977]
[1000] YPI_108812503 Yersinia pestis1WM1_Serratia marcescens
MLO_13476474 Mesorhizobium loti[475] MX_108757707
[1000] 1AZW_ Xanthomonas smithiiXFB_170730035 Xylella fastidiosa
[1000] PA_15600273 Pseudomonas aeruginosa PF_70733915 Pseudomonas fluorescens
[1000] BCR_42783975 Bacillus cereus[980] BS_16078154 Bacillus subtilis
CAC_15895946 Clostridium acetobutylicum [1000] LCA_est
[1000]
[954] 2UZ0STU_55821195
[609]
[1000] LLC_estALLA_estA
[727] OOE_116491533[841] OOE_116490775
LME_116618577[589] 1MTZ_Thermoplasma acidophilum
[770]
[461]
[998]
[1000] REM_190894576XCP_66767623 Xanthomonas campestris
RSTS00294 Salmonella typhimurium[1000] MAI_118464592
RMTG03682 Mycobacterium tuberculosis
[1000]
[246]
[1000] LDE_pepILBU_116514711
CBG_170761364 Clostridium botulinum[394]
[998] LPL_28377046LBE_116334786
[1000] LAC_58336439LHE_1615067129]
OOE_116490346LGA_116630389
[930]
[999] OOE_116490640OOE_116491087
[1000]
[1000] LDE_pepLLAC_58338197
[774] LPL_28379370[528] LCA_116494294
[1000]
[384]
[254]
[584] LSK_81427743LBE_116334352
[847] LPL_28377695LCA_116495454
PPE_116492182[997] LBU_116514722
[987] LRF_148531889[607] LAC_58337916
[631]LHE_pepR[677]LJO_42518140
LGA_116628733
EstA
PepIb
PepR
PepIa Proteobacteria
non LABFirmicutes
Archaea
Proteobacteria
Actinobacteria
Firmicutes(including LAB)
PepR/L
LAB
LAB
D E
L. caseiS. pneumoniae
S. thermophilusLc. LactisLc. Lactis
O. oeniO. oeniLeuconostoc
Myxococcus xanthus
Mycobacterium avium
Rhizobium etli
L. bulgaricusL. bulgaricus
L. plantarumL. brevis
L. acidophilusL. helveticusO. oeniL. gasseriO. oeni
O. oeniL. bulgaricus
L. acidophilusL. plantarumL. casei
L. sakeiL. brevis
L. plantarumL. caseiPediococcus
L. bulgaricusL. reuteri
L. acidophilus
L. johnsoniiL. helveticus
L. gasseri
PepLPutative
Figure 2.5. Superfamily tree of PepI/PepR/PepL and EstA members. (A colour version can be found in Appendix) Based on MSA of the concatenated sequences of the four structural core regions identified by the protein 3D structure alignment. The PepR and the PepL subgroup from LAB are shadowed. The bacterial phyla are indicated. Dots indicate the experimentally characterized genes and triangles indicate the protein 3D structures used for the analyses. The event of the substitution of catalytic residue aspartate by glutamate in the PepR subgroup is indicated in the tree.
Diversity of the proteolytic system in L. lactis strains The distribution of proteolytic system components in various L. lactis strains was studied by
comparative genome hybridization (CGH) analysis. PanGenome arrays were made based on ORFs

CHAPTER 2
32
found in four sequenced L. lactis strains, and subsequently used to determine the presence or absence of orthologs in 39 L. lactis strains [14]. Table 2.1 summarizes only the proteolytic system genes with variable absence/presence patterns in the 39 L. lactis strains. All other components described in Figure 2.1 but not shown in Table 2.1, such as PepC, PepN, PepM, PepA, PepD, PepV, PepT, PepP, PepQ, DtpT and most members of the Dpp system, are present in almost all strains. PepE/PepG and PepI/PepR/PepL family members are absent in all L. lactis strains. Those genes are excluded from the table, as well as all genes of strains P7304 and P7266 (see explanation in Table legend). Some plant-derived L. lactis strains such as KF24, NIZOB2244W, LMG9446 and KW10 have the largest set of proteolytic system genes.
Variations are found for proteinase PrtP and its maturation protein PrtM, for peptidases Pcp, PepO2, PepF2 and PepX2, and for genes from peptide transport systems Opp and Dpp (Table 2.1). Most of these genes are known to be present on plasmids [27]: in strain SK11 the prtP, prtM and pcp genes are located on one large plasmid, while the pepO2, pepF2 and oppABCDF2 are co-localized on a different plasmid. The co-presence or co-absence of these genes in other L. lactis strains (Table 2.1), is largely consistent with their coupling in SK11, and suggests that variability is mainly due to the presence or absence of the plasmids. Cell-wall bound proteinase PrtP together with PrtM are mainly present in L. lactis subsp. cremoris, although several L. lactis subsp. lactis strains also harbor these genes (including dairy strains e.g. UC317, ML8, and ATCC19435T).
PepX2 is a PepX homolog of L. lactis subsp. lactis IL1403. It is mainly found in L. lactis subsp. lactis strains from dairy origin. This putative pepX2 gene was originally annotated as a hypothetical protein named YmgC. It contains both a C-terminal domain of X-prolyl dipeptidyl aminopeptidase and a Peptidase_S15 catalytic domain which are usually found in PepX, whereas the PepX N-terminal domain is missing in PepX2. No experimental evidence for the enzyme activity of PepX2 is known. The family tree of PepX shows that this putative pepX2 gene is not clustered in the same orthologous group as its paralogous gene from L. lactis subsp. lactis IL1403 [Additional File 5]. The only members of the PepX2 (YmgC) group in sequenced LAB genomes are from L. lactis subsp. lactis IL1403 and Pediococcus. Their best BLAST hits against the non-redundant protein database are from Listeria monocytogenes, suggesting a HGT event [See Additional File 5].
Discussion
In this study, we performed a systematic genome-wide analysis of all the proteins involved in proteolysis, including cell-wall bound proteinase, peptide transporters, and peptidases, from twenty-two fully sequenced LAB genomes, including Lactobacillus, Lactococcus, Streptococcus, Pediococcus, Oenococcus, and Leuconostoc strains. The comparative genomics analysis was shown to distinguish various subgroups within a protein superfamily, allowing a highly improved annotation of genes and clarification caused by inconsistent annotation.
This information on the distribution of the proteolytic system genes can be used to predict the proteolytic potential of various LAB strains. For instance, L. bulgaricus and L. helveticus have a very extensive set of proteolytic enzymes, which is consistent with previous knowledge that L. bulgaricus serves as the proteolytic organism in yoghurt rather than S. thermophilus [47]. L. helveticus is a proteolytic cheese adjunct culture that has been used to degrade bitter peptides in cheese [48]. Interestingly, L. bulgaricus encodes the Dpp system with preference for uptake of hydrophobic di/tripeptides, complementing S. thermophilus which encodes the general di/tripeptide transporter DtpT in its genome, suggesting that more peptides can be utilized by both bacteria when grown together. LAB species of plant origin, such as L. plantarum, O. oeni, and Leuconostoc mesenteroides, encode less proteolytic enzymes in their genomes, which agrees with their ecological niche that is fiber-rich but contains less proteins.
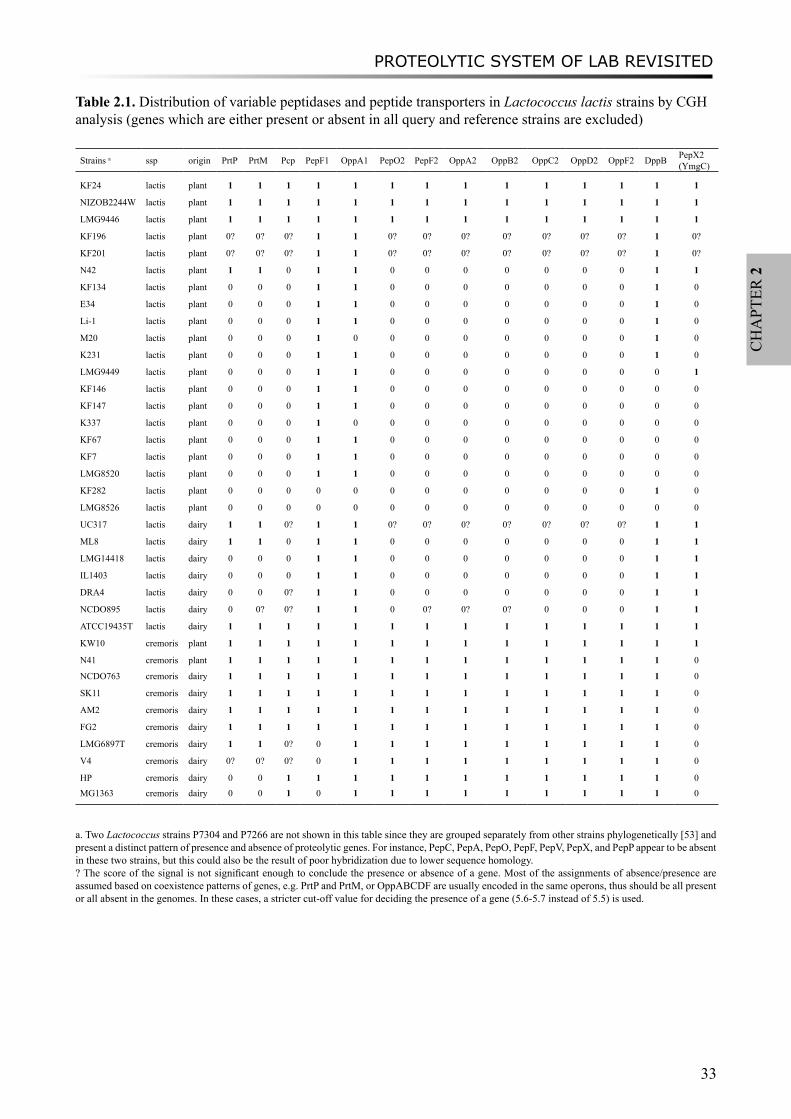
PROTEOLYTIC SYSTEM OF LAB REVISITED
33
CH
APT
ER 2
Table 2.1. Distribution of variable peptidases and peptide transporters in Lactococcus lactis strains by CGH analysis (genes which are either present or absent in all query and reference strains are excluded)
Strains a ssp origin PrtP PrtM Pcp PepF1 OppA1 PepO2 PepF2 OppA2 OppB2 OppC2 OppD2 OppF2 DppB PepX2(YmgC)
KF24 lactis plant 1 1 1 1 1 1 1 1 1 1 1 1 1 1
NIZOB2244W lactis plant 1 1 1 1 1 1 1 1 1 1 1 1 1 1
LMG9446 lactis plant 1 1 1 1 1 1 1 1 1 1 1 1 1 1
KF196 lactis plant 0? 0? 0? 1 1 0? 0? 0? 0? 0? 0? 0? 1 0?
KF201 lactis plant 0? 0? 0? 1 1 0? 0? 0? 0? 0? 0? 0? 1 0?
N42 lactis plant 1 1 0 1 1 0 0 0 0 0 0 0 1 1
KF134 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 1 0
E34 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 1 0
Li-1 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 1 0
M20 lactis plant 0 0 0 1 0 0 0 0 0 0 0 0 1 0
K231 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 1 0
LMG9449 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 0 1
KF146 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 0 0
KF147 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 0 0
K337 lactis plant 0 0 0 1 0 0 0 0 0 0 0 0 0 0
KF67 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 0 0
KF7 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 0 0
LMG8520 lactis plant 0 0 0 1 1 0 0 0 0 0 0 0 0 0
KF282 lactis plant 0 0 0 0 0 0 0 0 0 0 0 0 1 0
LMG8526 lactis plant 0 0 0 0 0 0 0 0 0 0 0 0 0 0
UC317 lactis dairy 1 1 0? 1 1 0? 0? 0? 0? 0? 0? 0? 1 1
ML8 lactis dairy 1 1 0 1 1 0 0 0 0 0 0 0 1 1
LMG14418 lactis dairy 0 0 0 1 1 0 0 0 0 0 0 0 1 1
IL1403 lactis dairy 0 0 0 1 1 0 0 0 0 0 0 0 1 1
DRA4 lactis dairy 0 0 0? 1 1 0 0 0 0 0 0 0 1 1
NCDO895 lactis dairy 0 0? 0? 1 1 0 0? 0? 0? 0 0 0 1 1
ATCC19435T lactis dairy 1 1 1 1 1 1 1 1 1 1 1 1 1 1
KW10 cremoris plant 1 1 1 1 1 1 1 1 1 1 1 1 1 1
N41 cremoris plant 1 1 1 1 1 1 1 1 1 1 1 1 1 0
NCDO763 cremoris dairy 1 1 1 1 1 1 1 1 1 1 1 1 1 0
SK11 cremoris dairy 1 1 1 1 1 1 1 1 1 1 1 1 1 0
AM2 cremoris dairy 1 1 1 1 1 1 1 1 1 1 1 1 1 0
FG2 cremoris dairy 1 1 1 1 1 1 1 1 1 1 1 1 1 0
LMG6897T cremoris dairy 1 1 0? 0 1 1 1 1 1 1 1 1 1 0
V4 cremoris dairy 0? 0? 0? 0 1 1 1 1 1 1 1 1 1 0
HP cremoris dairy 0 0 1 1 1 1 1 1 1 1 1 1 1 0
MG1363 cremoris dairy 0 0 1 0 1 1 1 1 1 1 1 1 1 0
a. Two Lactococcus strains P7304 and P7266 are not shown in this table since they are grouped separately from other strains phylogenetically [53] and present a distinct pattern of presence and absence of proteolytic genes. For instance, PepC, PepA, PepO, PepF, PepV, PepX, and PepP appear to be absent in these two strains, but this could also be the result of poor hybridization due to lower sequence homology. ? The score of the signal is not significant enough to conclude the presence or absence of a gene. Most of the assignments of absence/presence are assumed based on coexistence patterns of genes, e.g. PrtP and PrtM, or OppABCDF are usually encoded in the same operons, thus should be all present or all absent in the genomes. In these cases, a stricter cut-off value for deciding the presence of a gene (5.6-5.7 instead of 5.5) is used.

CHAPTER 2
34
Several examples have been provided for the division of large superfamilies into subfamilies. Clear separation of major subgroups can be observed from the family trees. By including the experimentally characterized genes, different substrate specificities can be assigned to various subfamilies. The PepP/PepQ/PepM and PepI/PepR/PepL superfamilies include subfamilies with distinct substrate specificities. The general dipeptidase superfamily PepD consists of several distinct orthologous groups of which the substrate specificities are still unknown. In most cases, the prediction of orthologous groups and the evolutionary events leading to the variation of substrate specificities are straight-forward using the phylogenetic analysis. However, some orphan genes are present as intermediate groups between the subfamilies with unknown functions and some of them may originate from HGT events.
Peptidases PepI/R/L and the esterase EstA, which is also involved in flavor-formation by LAB, belong to the same α/β hydrolase superfamily. We performed a comparative analysis of 3D structures of representative proteins from each subfamily in order to identify the core regions of the enzymes and to improve the multiple sequence alignment of the superfamily. Orthologs could then be identified more clearly in the protein family tree as constructed on basis of the curated MSA of the core regions. The classic catalytic triad Ser-His-Asp of the α/β hydrolase family is conserved in most of the members of the PepI/R/L and EstA superfamily. However, in the PepR subfamily of LAB (Figure 2.5), the catalytic Asp residues are substituted by Glu residues. Aspartate and glutamate residues are chemically equivalent and differ only in length of the side chain. The substitution of Asp by Glu has been observed in prokaryotic subtilases [49], as well as in an acetylcholinesterase of Torpedo californica and a lipase of Geotrichum candidum [50, 51]. Moreover, two additional peptidases from L. plantarum and L. casei (LPL_28379307 and LCA_116494294) which are not grouped into the PepR subfamily (Figure 2.5) also have glutamate catalytic residues instead of aspartate residues. It suggests that the substitution of Asp to Glu may have happened in the common ancestor of these two proteins and the PepR family. Since the glutamate residue at the catalytic triad is only found to be conserved in the PepR subfamily, it can be used as an extra indication for determining whether a peptidase with unclear function belongs to the PepR subfamily.
One of the applications of our comparative analysis is to explore the diversity of proteolytic system genes in various strains of L. lactis by combining the results from comparative genomics analysis and the hybridization data from pangenome CGH analysis. Distinct patterns were found in the presence and absence of proteolytic enzymes in the two L. lactis subspecies, i.e. subsp. lactis and subsp. cremoris, confirming the proteolytic diversity between the subspecies, and now providing a genetic basis for this diversity. Several strains show corresponding distributions of some proteolytic genes in their genomes, presumably resulting from the presence or absence of plasmids encoding proteolytic system components.
ConclusionsWe performed a genome-wide comparative study on the proteolytic system of LAB, and
demonstrated that the functional annotation of proteolytic system genes can be improved by combining phylogeny, synteny and literature. Examples of the PepP/PepQ/PepM family, the PepD family and the PepI/PepR/PepL family elucidated that protein subfamilies with distinct substrate specificities can be identified. In the case of the PepI/PepR/PepL family, protein 3D-structure alignment allowed us to more clearly distinguish the peptidase subfamilies and an esterase family EstA. Moreover, the complete distribution of proteolytic system components in various sequenced LAB strains was obtained.
The diversity of proteolytic system genes from 39 Lactococcus strains was explored using

PROTEOLYTIC SYSTEM OF LAB REVISITED
35
CH
APT
ER 2
CGH analysis. Several components including proteinase, oligopeptide transport system and peptidases were shown to be distributed unevenly among the Lactococcus strains. The presence or absence of those proteolytic system components are probably the result of the presence or absence of plasmids that encode them.
Knowledge of the variations in proteolytic system components may allow the prediction of proteolytic and flavor-forming potential of bacterial strains, and could direct future experimental tests into the phenotypes of various LAB. Ultimately, this knowledge could be used to improve the sensory characteristics of dairy and other fermented food products by supporting the strain selection process.
List of abbreviations LAB, Lactic Acid Bacteria; HMMs, Hidden Markov Models; MSA, Multiple Sequence Alignments; CGH, Comparative Genome Hybridization; HGT, Horizontal Gene Transfer
Authors’ contributionsML conceived and designed the study, performed the analyses, drafted and revised the manuscript; JRB carried out the pangenome CGH analysis and the diversity analysis of Lactococcus lactis; BR carried out the protein 3D structure alignment; AN coordinated the study and helped revising the manuscript; RJS conceived, designed and coordinated the study, helped drafting and revised the manuscript. All authors read and approved the final manuscript.
AcknowledgementsThis work was supported by grant CSI4017 of the Casimir program of the Ministry of Economic Affairs, the Netherlands.
Additional FilesAdditional file 1: Multiple sequence alignment of core regions of proteins from both the PepI/R/L and EstA families. A manually curated multiple sequence alignment of the concatenated sequences of the four core regions of the PepI/PepR/PepL and EstA superfamily members identified by the protein structure superposition. On basis of this MSA, a family tree was constructed, and is shown in Figure 2.5.Additional file 2: Proteolytic components in LAB. The file contains a table with GI codes of all the genes listed in Figure 2.1.Additional file 3: Superfamily tree of PepF members. The bootstrapped (n=1000) neighbor-joining tree for PepF members in LABAdditional file 4: Comparison of important residues of the conserved core regions and the cap region. The file contains a table describing the four structurally conserved regions and the cap regions. The identified residues within those regions, which are functionally important and/or conserved in PepI/R/L or EstA families, are highlightedAdditional file 5: PepX superfamily tree. The file contains a bootstrapped (n=1000) NJ tree for PepX family homologs of LAB

CHAPTER 2
36
References1. Christensen JE, Dudley EG, Pederson JA, Steele JL: Peptidases and amino acid catabolism in lactic acid bacteria. Antonie
Van Leeuwenhoek 1999, 76:217-246.2. Kim YK, Yaguchi M, Rose D: Isolation and Amino Acid Composition of Para-Kappa-Casein. J Dairy Sci 1969, 52:316-
320.3. Stewart AF, Bonsing J, Beattie CW, Shah F, Willis IM, Mackinlay AG: Complete nucleotide sequences of bovine alpha
S2- and beta-casein cDNAs: comparisons with related sequences in other species. Mol Biol Evol 1987, 4:231-241.4. Liu M, Nauta A, Francke C, Siezen RJ: Comparative genomics of enzymes in flavor-forming pathways from amino acids
in lactic acid bacteria. Appl Environ Microbiol 2008, 74:4590-4600.5. Kunji ER, Mierau I, Hagting A, Poolman B, Konings WN: The proteolytic systems of lactic acid bacteria. Antonie Van
Leeuwenhoek 1996, 70:187-221.6. Savijoki K, Ingmer H, Varmanen P: Proteolytic systems of lactic acid bacteria. Appl Microbiol Biotechnol 2006, 71:394-
406.7. Sousa MJ, Ardö Y, McSweeney PLH: Advances in the study of proteolysis during cheese ripening. International Dairy
Journal 2001, 11:327-345.8. Doeven MK, Kok J, Poolman B: Specificity and selectivity determinants of peptide transport in Lactococcus lactis and
other microorganisms. Mol Microbiol 2005, 57:640-649.9. Liu M, Siezen R: Comparative genomics of flavour-forming pathways in lactic acid bacteria. Australian Journal of
Dairy Technology 2006, 61:61-68.10. Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N et al:
Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci USA 2006, 103:15611-15616.11. Wegmann U, O’Connell-Motherway M, Zomer A, Buist G, Shearman C, Canchaya C, Ventura M, Goesmann A, Gasson MJ,
Kuipers OP et al: Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J Bacteriol 2007, 189:3256-3270.
12. Callanan M, Kaleta P, O’Callaghan J, O’Sullivan O, Jordan K, McAuliffe O, Sangrador-Vegas A, Slattery L, Fitzgerald GF, Beresford T et al: Genome sequence of Lactobacillus helveticus, an organism distinguished by selective gene loss and insertion sequence element expansion. J Bacteriol 2008, 190:727-735.
13. Kankainen M, Paulin L, Tynkkynen S, von Ossowski I, Reunanen J, Partanen P, Satokari R, Vesterlund S, Hendrickx AP, Lebeer S et al: Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human-mucus binding protein. Proc Natl Acad Sci U S A 2009, 106:17193-17198.
14. Bayjanov JR, Wels M, Starrenburg M, van Hylckama Vlieg JE, Siezen RJ, Molenaar D: PanCGH: a genotype-calling algorithm for pangenome CGH data. Bioinformatics 2009, 25:309-314.
15. Smit G, Smit BA, Engels WJM: Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiology Reviews 2005, 29:591-610.
16. Siezen RJ, Starrenburg MJ, Boekhorst J, Renckens B, Molenaar D, van Hylckama Vlieg JE: Genome-scale genotype-phenotype matching of two Lactococcus lactis isolates from plants identifies mechanisms of adaptation to the plant niche. Appl Environ Microbiol 2008, 74:424-436.
17. UniProt Consortium: The universal protein resource (UniProt). Nucleic Acids Res 2008, 36:D190-195.18. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215:403-410.19. Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL
et al: The Pfam protein families database. Nucleic Acids Res 2004, 32:D138-D141.20. Francke C, Kerkhoven R, Wels M, Siezen RJ: A generic approach to identify Transcription Factor-specific operator
motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics 2008, 9:145.
21. Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32:1792-1797.
22. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al: Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23:2947-2948.
23. van der Heijden RT, Snel B, van Noort V, Huynen MA: Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinformatics 2007, 8:83.
24. Overbeek R, Larsen N, Walunas T, D’Souza M, Pusch G, Selkov E, Liolios K, Joukov V, Kaznadzey D, Anderson I et al: The ERGO (TM) genome analysis and discovery system. Nucleic Acids Res 2003, 31:164-171.
25. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Res 2000, 28:235-242.
26. Folkertsma S, van Noort P, Van Durme J, Joosten HJ, Bettler E, Fleuren W, Oliveira L, Horn F, de Vlieg J, Vriend G: A family-based approach reveals the function of residues in the nuclear receptor ligand-binding domain. J Mol Biol 2004, 341:321-335.
27. Siezen RJ, Renckens B, van Swam I, Peters S, van Kranenburg R, Kleerebezem M, de Vos WM: Complete sequences of four plasmids of Lactococcus lactis subsp. cremoris SK11 reveal extensive adaptation to the dairy environment. Appl Environ Microbiol 2005, 71:8371-8382.
28. Matos J, Nardi M, Kumura H, Monnet V: Genetic characterization of pepP, which encodes an aminopeptidase P whose deficiency does not affect Lactococcus lactis growth in milk, unlike deficiency of the X-prolyl dipeptidyl aminopeptidase. Appl Environ Microbiol 1998, 64:4591-4595.
29. Rantanen T, Palva A: Lactobacilli carry cryptic genes encoding peptidase-related proteins: characterization of a

PROTEOLYTIC SYSTEM OF LAB REVISITED
37
CH
APT
ER 2
prolidase gene (pepQ) and a related cryptic gene (orfZ) from Lactobacillus delbrueckii subsp. bulgaricus. Microbiology 1997, 143 ( Pt 12):3899-3905.
30. Morel F, Frot-Coutaz J, Aubel D, Portalier R, Atlan D: Characterization of a prolidase from Lactobacillus delbrueckii subsp. bulgaricus CNRZ 397 with an unusual regulation of biosynthesis. Microbiology 1999, 145 ( Pt 2):437-446.
31. Vesanto E, Peltoniemi K, Purtsi T, Steele JL, Palva A: Molecular characterization, over-expression and purification of a novel dipeptidase from Lactobacillus helveticus. Appl Microbiol Biotechnol 1996, 45:638-645.
32. Smeianov VV, Wechter P, Broadbent JR, Hughes JE, Rodriguez BT, Christensen TK, Ardo Y, Steele JL: Comparative high-density microarray analysis of gene expression during growth of Lactobacillus helveticus in milk versus rich culture medium. Appl Environ Microbiol 2007, 73:2661-2672.
33. Klein JR, Dick A, Schick J, Matern HT, Henrich B, Plapp R: Molecular cloning and DNA sequence analysis of pepL, a leucyl aminopeptidase gene from Lactobacillus delbrueckii subsp. lactis DSM7290. Eur J Biochem 1995, 228:570-578.
34. Goettig P, Groll M, Kim JS, Huber R, Brandstetter H: Structures of the tricorn-interacting aminopeptidase F1 with different ligands explain its catalytic mechanism. EMBO J 2002, 21:5343-5352.
35. Medrano FJ, Alonso J, Garcia JL, Romero A, Bode W, Gomis-Ruth FX: Structure of proline iminopeptidase from Xanthomonas campestris pv. citri: a prototype for the prolyl oligopeptidase family. EMBO J 1998, 17:1-9.
36. Inoue T, Ito K, Tozaka T, Hatakeyama S, Tanaka N, Nakamura KT, Yoshimoto T: Novel inhibitor for prolyl aminopeptidase from Serratia marcescens and studies on the mechanism of substrate recognition of the enzyme using the inhibitor. Arch Biochem Biophys 2003, 416:147-154.
37. Kim MH, Kang BS, Kim S, Kim KJ, Lee CH, Oh BC, Park SC, Oh TK: The crystal structure of the estA protein, a virulence factor from Streptococcus pneumoniae. Proteins 2008, 70:578-583.
38. Dudley EG, Steele JL: Nucleotide sequence and distribution of the pepPN gene from Lactobacillus helveticus CNRZ32. FEMS Microbiol Lett 1994, 119:41-45.
39. Shao W, Yuksel GU, Dudley EG, Parkin KL, Steele JL: Biochemical and molecular characterization of PepR, a dipeptidase, from Lactobacillus helveticus CNRZ32. Appl Environ Microbiol 1997, 63:3438-3443.
40. Atlan D, Gilbert C, Blanc B, Portalier R: Cloning, sequencing and characterization of the pepIP gene encoding a proline iminopeptidase from Lactobacillus delbrueckii subsp. bulgaricus CNRZ 397. Microbiology 1994, 140 ( Pt 3):527-535.
41. Morel F, Gilbert C, Geourjon C, Frot-Coutaz J, Portalier R, Atlan D: The prolyl aminopeptidase from Lactobacillus delbrueckii subsp. bulgaricus belongs to the alpha/beta hydrolase fold family. Biochim Biophys Acta 1999, 1429:501-505.
42. Fernandez L, Beerthuyzen MM, Brown J, Siezen RJ, Coolbear T, Holland R, Kuipers OP: Cloning, characterization, controlled overexpression, and inactivation of the major tributyrin esterase gene of Lactococcus lactis. Appl Environ Microbiol 2000, 66:1360-1368.
43. Yebra MJ, Viana R, Monedero V, Deutscher J, Perez-Martinez G: An esterase gene from Lactobacillus casei cotranscribed with genes encoding a phosphoenolpyruvate:sugar phosphotransferase system and regulated by a LevR-like activator and sigma54 factor. J Mol Microbiol Biotechnol 2004, 8:117-128.
44. Varmanen P, Rantanen T, Palva A: An operon from Lactobacillus helveticus composed of a proline iminopeptidase gene (pepI) and two genes coding for putative members of the ABC transporter family of proteins. Microbiology 1996, 142 ( Pt 12):3459-3468.
45. Yuksel GU, Steele JL: DNA sequence analysis, expression, distribution, and physiological role of the Xaa-prolyldipeptidyl aminopeptidase gene from Lactobacillus helveticus CNRZ32. Appl Microbiol Biotechnol 1996, 44:766-773.
46. Varmanen P, Rantanen T, Palva A, Tynkkynen S: Cloning and characterization of a prolinase gene (pepR) from Lactobacillus rhamnosus. Appl Environ Microbiol 1998, 64:1831-1836.
47. Sieuwerts S, de Bok FA, Hugenholtz J, van Hylckama Vlieg JE: Unraveling microbial interactions in food fermentations: from classical to genomics approaches. Appl Environ Microbiol 2008, 74:4997-5007.
48. Sridhar VR, Hughes JE, Welker DL, Broadbent JR, Steele JL: Identification of endopeptidase genes from the genomic sequence of Lactobacillus helveticus CNRZ32 and the role of these genes in hydrolysis of model bitter peptides. Appl Environ Microbiol 2005, 71:3025-3032.
49. Siezen RJ, Renckens B, Boekhorst J: Evolution of prokaryotic subtilases: Genome-wide analysis reveals novel subfamilies with different catalytic residues. Proteins: Structure, Function and Bioinformatics 2007, 67:681-694.
50. Polgar L: The catalytic triad of serine peptidases. Cell Mol Life Sci 2005, 62:2161-2172.51. Dodson G, Wlodawer A: Catalytic triads and their relatives. Trends Biochem Sci 1998, 23:347-352.52. Seo JM, Ji GE, Cho SH, Park MS, Lee HJ: Characterization of a Bifidobacterium longum BORI dipeptidase belonging
to the U34 family. Appl Environ Microbiol 2007, 73:5598-5606.53. Rademaker JL, Herbet H, Starrenburg MJ, Naser SM, Gevers D, Kelly WJ, Hugenholtz J, Swings J, van Hylckama Vlieg JE:
Diversity analysis of dairy and nondairy Lactococcus lactis isolates, using a novel multilocus sequence analysis scheme and (GTG)5-PCR fingerprinting. Appl Environ Microbiol 2007, 73:7128-7137.

CHAPTER 2
38
There is no such a thing as inaccuracy in a photograph (scientific finding). All photographs (scientific findings) are accurate. None of them is the truth.
- Richard Avedon (commented by the author )

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
39
CH
APT
ER 3
Chapter 3
COMPARATIVE GENOMICS OF ENZYMES IN FLAVOR-FORMING PATHWAYS FROM AMINO ACIDS
IN LACTIC ACID BACTERIA
Mengjin Liu, Arjen Nauta, Christof Francke, Roland J. Siezen
Appl Environ Microbiol. 2008; 74(15): 4590-4600

CHAPTER 3
40
Lactic acid bacteria (LAB) have been widely used as starter or non-starter cultures in the dairy industry for over a thousand years. They also play an essential role in flavor formation during the fermentation of dairy products. Several metabolic routes can lead to the formation of flavor compounds when LAB are growing in milk. One of the main precursors for flavor compounds in milk is casein, although they can also be derived from fatty acids and sugars. The proteolytic system of LAB degrades casein into its constituent amino acids, which can be converted to flavor compounds. Although amino acid catabolism by LAB has been well researched (5, 31, 65, 78), many flavor-forming routes are yet to be discovered.
Over twenty LAB genomes have been fully sequenced (1, 11, 12, 16, 19, 41, 44, 45, 53, 68, 71). The available genomic information provides us with new opportunities to study the flavor-forming potential of LAB. However, one of the main problems that one encounters while reconstructing flavor-forming routes based on the genomic information stored in the public databases is the inconsistency in the functional annotation for many of the relevant genes. These genes are mostly members of larger protein families. Moreover, even when the functional annotation in databases is appropriate, it sometimes reflects only part of the protein’s full functional potential, as broad substrate specificities are often not taken into consideration in the annotation.
We have now improved the functional annotation of the key enzymes in the formation of flavor compounds from amino acids by applying comparative genomics approaches that have been developed within our group to specify the annotation of homologous proteins, by combining phylogeny, gene context and experimental evidence (32). We focused especially on the enzymes involved in the metabolism of sulfur-containing amino acids since these are known to be precursors of many flavor compounds in dairy fermentations. Comparative analysis of the various sequenced LAB species and strains resulted in an overall view of differences in their flavor-forming capacities.
In silico analyses of LAB genomesComplete LAB genome sequences were obtained from the NCBI microbial genome database
(http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi) on Nov. 30, 2007: Lactobacillus acidophilus NCFM (abbreviation LAC, accession code CP000033), Lactobacillus johnsonii NCC 533 (LJO, AE017198), Lactobacillus gasseri ATCC 33323 (LGA, CP000413), Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842 (LDB, CR954253), Lactobacillus delbrueckii subsp. bulgaricus ATCC BAA365 (LBU, CP000412), Lactobacillus plantarum WCFS1 (LPL, AL935263), Lactobacillus brevis ATCC 367 (LBE, CP000416), Lactobacillus sakei 23K (LSK, CR936503), Lactobacillus salivarius UCC118 (LSL, CP000233), Oenococcus oeni PSU1 (OOE, CP000411), Pediococcus pentosaceus ATCC 25745 (PPE, CP000422), Leuconostoc mesenteroides ATCC 8293 (LME, CP000414), Lactobacillus casei ATCC 334 (LCA, CP000423), Lactococcus lactis subsp. lactis IL1403 (LLX, AE005176), Lactococcus lactis subsp. cremoris MG1363 (LLM, AM406671), Lactococcus lactis subsp. cremoris SK11 (LLA, CP000425), Streptococcus thermophilus CNRZ1066 (STH, CP000024), Streptococcus thermophilus LMG18311 (STU, CP000023), Streptococcus thermophilus LMD9 (STM, CP000419), and Lactobacillus reuteri F275 (LRF, CP000705). The complete genome sequence of Lactobacillus reuteri JCM1112 (LRE) was obtained from the ERGO database (50). The phylogeny of these LAB is shown in Figure 3.1.
Protein sequences of experimentally verified enzymes involved in flavor formation were obtained from the Uniprot database (9). These sequences were used to perform BLAST searches against all LAB genomes (3). In addition, Hidden Markov Models (HMMs) of the related protein families were obtained from the Pfam database (10) and used to identify homologs with the HMMER 2.3.2 package (24). Homologous sequences from each enzyme family derived from both BLAST and HMM searches were collected and the redundant sequences were removed. Orthologous relationships between the various homologs were determined on basis of phylogeny and synteny. For

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
41
CH
APT
ER 3
each protein family, a multiple sequence alignment was created with MUSCLE (25). The alignments included representative sequences from literature for which experimental evidence is available. ClustalW was used to generate bootstrapped (n=1000) neighbor-joining trees on basis of the multiple sequence alignments (67). The trees were visualized in LOFT (69), a tool that automatically identifies orthologous groups. When possible, gene context was used to distinguish between functional variants. Gene context was studied using the ERGO Bioinformatics Suite (50). The complete procedure is depicted in Figure S1 in the supplemental material.
Lc. lactis cremoris
Oenococcus oeniLeuconostoc mesenteroides
Pediococcus pentosaceusLb. plantarum
Lb. brevisLb. salivarius
Lb. reuteri
Lb. caseiLb. sakei
Lb. delbrueckii bulgaricusLb. acidophilus
Lb. johnsoniiLb. gasseri
0.05
Lb. acidophilus group
Lc. lactis lactis Streptococcus thermophilus
Figure 3.1. Phylogenetic tree of lactic acid bacteria constructed on the basis of concatenated alignments of four subunits (alpha, beta, beta’, and delta) of the DNA-dependent RNA polymerase. (A colour version can be found in Appendix) Different colors represent the various origins or usages of LAB: red for dairy fermentation, blue for other fermentation such as beer, wine, plants or meat, green for GI-tract bacteria. The Lb. acidophilus group is framed. Lb. for Lactobacillus, Lc. for Lactococcus
Flavor-forming pathways from amino acidsFree amino acids produced by proteolysis are converted to various flavor compounds through
amino acid catabolism. The branched-chain amino acids (valine, leucine, and isoleucine), the aromatic amino acids (tyrosine, tryptophan and phenylalanine) and the sulfur-containing amino acids (methionine, cysteine) are the main amino acid sources for flavor compounds. The conversion of these amino acids into flavors proceeds via two distinct routes: i) transamination and ii) elimination (Figure 3.2 and Figure 3.3) (5, 65, 78). The transamination route is initiated by aminotransferases that convert amino acids into their corresponding α-keto acids. The α-keto acids are then further converted into aldehydes, alcohols and esters, which are important aroma compounds. It was shown that branched-chain amino acids, aromatic amino acids and methionine are catabolized via the transamination route. The elimination route has been described for methionine where activity by carbon-sulfur lyases results in the release of methanethiol.
Enzymes involved in the transamination routeAminotransferases
Transamination of branched-chain amino acids, aromatic amino acid and methionine can be catalyzed by different aminotransferases (Figure 3.2). In lactococcal strains, branched-chain aminotransferase (BcAT) displays an activity towards both the branched-chain amino acids and methionine (56), while in these strains aromatic aminotransferase (ArAT) is active against aromatic amino acids, leucine and methionine (55, 56). In Brevibacterium linens, aspartate aminotransferase (AspAT) is responsible for aspartate transamination, but is also active on the aromatic amino acids (78). In cheese, flavor compounds such as 2-methylbutanoate (sweaty odor) and isobutyric acid (sour and sweet odor) are believed to be derived from the transamination of Ile or Val by BcAT (6, 77, 78).

CHAPTER 3
42
Table 3.1 indicates that a BcAT ortholog is present in all lactococcal and streptococcal strains while it is lacking in a number of lactobacilli such as Lb. johnsonii, Lb. sakei, Lb. reuteri. Therefore the latter strains may be unable to produce 2-methylbutanoate and isobutyric acid.
Genes encoding ArAT and AspAT have been experimentally characterized in Lc. lactis, e.g. the araT gene from Lc. lactis NCDO763 (55), and the aspAT gene from Lc. lactis LM0230 (23). They are found to be homologs, as both belong to the aminotransferase I family (40) (see Figure S2 in the supplemental material). Putative araT genes were found in all LAB genomes except Lb. sakei and Lb. brevis, while the aspAT gene was absent in LAB species of the Lb. acidophilus group (Figure 3.1).
Glutamate dehydrogenase
In LAB, the transaminase reaction is commonly linked to the deamination of glutamate to oxoglutaric acid (α-ketoglutarate), catalyzed by glutamate dehydrogenase (GDH) (Figure 3.2). Yvon et al. proposed that the amount of α-ketoglutarate is the limiting factor for flavor formation from amino acids in cheeses (66, 76). Table 3.1 shows that gdh genes are only found in the genomes of Lb. plantarum, Lb. salivarius and S. thermophilus strains, which agrees with the strain-dependency of the presence of GDH and the lack of GDH activity in most LAB strains (66). However, GDH activity was more commonly found in natural strains used for cheese manufacture (66).
α-Ketoacid conversion enzymesα-Ketoacids can be further converted in three different routes (Figure 3.2). The α-ketoacid
decarboxylase gene (kdcA), for conversion to the corresponding aldehydes, was characterized from Lc. lactis B1157 by Smit et al. (64). No orthologs were found in the sequenced LAB genomes except a gene with partial homology in Lc. lactis subsp. lactis IL1403 (Table 3.1). This is consistent with the observations that KdcA activity was only found in non-dairy lactococcal strains and only small amounts of amino-acid derived aldehydes were produced by most LAB (62-65). However, alcohols and carboxylic acids that can be derived from those aldehydes were detected in many LAB, suggesting alternative pathways to produce these flavor compounds.
Figure 3.2. Generic amino acid (branched-chain amino acids, aromatic amino acids, and methionine) degradation pathway initiated by transamination in LAB. Enzymes names are: BcAT, branched-chain aminotransferase; ArAT, aromatic aminotransferase; AspAT, aspartate aminotransferase; GDH, glutamate dehydrogenase; HycDH, hydroxyacid dehydrogenase; KdcA, alpha-ketoacid decarboxylase; AlcDH, alcohol dehydrogenase; AldDH, aldehyde dehydrogenase; EstA, esterase A. Enzymes specific for branched-chain amino acids degradation (dotted box): KaDH, alpha-ketoacid dehydrogenase complex; PTA, phosphotransacylase; ACK, acyl kinase.
Amino acid (BcAA, ArAA, Met)
Glutamate
Oxoglutaric acidNAD(P)H
NAD(P)+
Keto acid
BcATArATAspATGDH
KdcA
Aldehyde
CO2
NAD(P)H
NAD(P)+
AlcDH
Alcohol
NAD(P)H
NAD(P)+
Carboxylic cid
NAD(P)H
NAD(P)+ HycDH
AldDH
Hydroxyacid
NAD+ NADHCO2
CoA
Pi
CoASH
P
AD
P
AT
P
KaDH
CoA
Elimination route
lyases
Transamination route
EstA
Ester
alcohol
a
PTA
ACK
Acyl-
Acyl-
BcAA
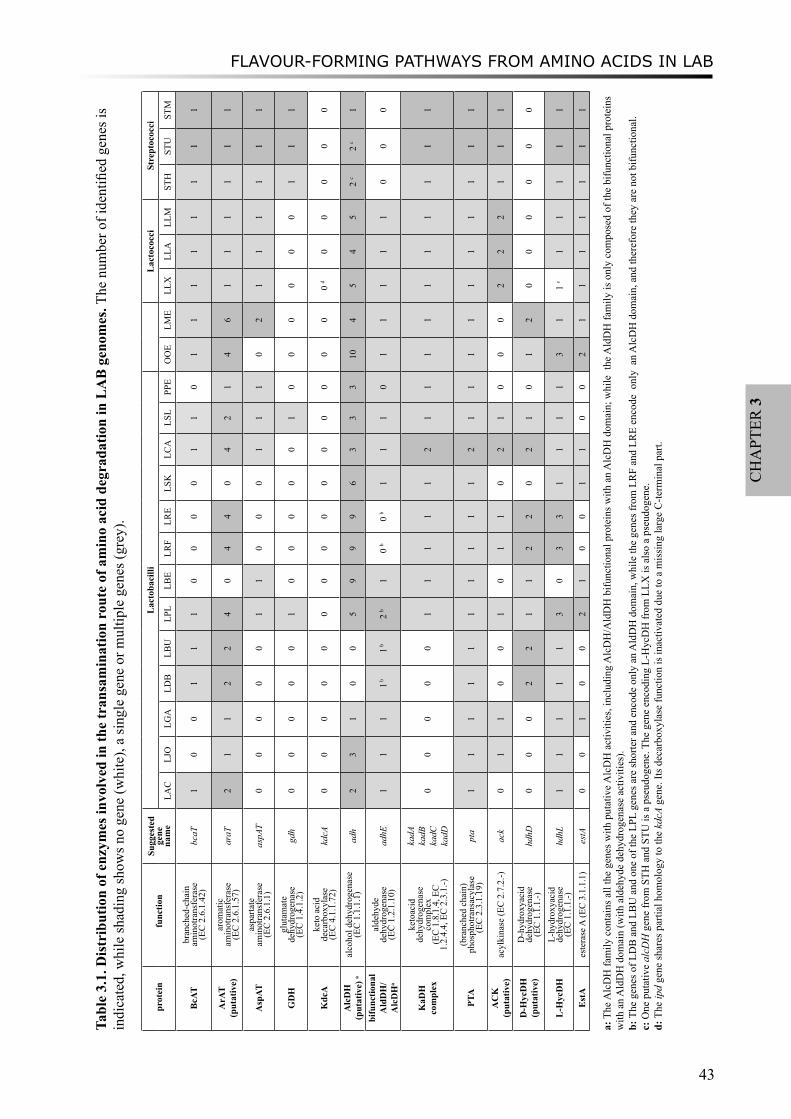
FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
43
CH
APT
ER 3
Tabl
e 3.
1. D
istr
ibut
ion
of e
nzym
es in
volv
ed in
the
tran
sam
inat
ion
rout
e of
am
ino
acid
deg
rada
tion
in L
AB
gen
omes
. The
num
ber o
f ide
ntifi
ed g
enes
is
indi
cate
d, w
hile
shad
ing
show
s no
gene
(whi
te),
a si
ngle
gen
e or
mul
tiple
gen
es (g
rey)
.
pro
tein
func
tion
Sugg
este
d ge
ne
nam
e
Lac
toba
cilli
Lac
toco
cci
Stre
ptoc
occi
LAC
LJ
OLG
ALD
BLB
ULP
LLB
ELR
FLR
ELS
KLC
ALS
LPP
EO
OE
LME
LLX
LLA
LLM
STH
STU
STM
BcA
Tbr
anch
ed-c
hain
am
inot
rans
fera
se
(E
C 2
.6.1
.42)
bcaT
10
01
11
00
00
11
01
11
11
11
1
ArA
T (p
utat
ive)
arom
atic
am
inot
rans
fera
se
(E
C 2
.6.1
.57)
araT
21
12
24
04
40
42
14
61
11
11
1
Asp
ATas
parta
te
amin
otra
nsfe
rase
(EC
2.6
.1.1
)as
pAT
00
00
01
10
00
11
10
21
11
11
1
GD
Hgl
utam
ate
dehy
drog
enas
e (E
C 1
.4.1
.2)
gdh
00
00
01
00
00
01
00
00
00
11
1
Kdc
Ake
to a
cid
deca
rbox
ylas
e
(E
C 4
.1.1
.72)
kdcA
00
00
00
00
00
00
00
00
d0
00
00
Alc
DH
(p
utat
ive)
aal
coho
l deh
ydro
gena
se(E
C 1
.1.1
.1)
adh
23
10
05
99
96
33
310
45
45
2 c
2 c
1
bifu
nctio
nal
Ald
DH
/A
lcD
Ha
alde
hyde
de
hydr
ogen
ase
(EC
1.2
.1.1
0)ad
hE1
11
1 b1 b
2 b
10
b0
b1
11
01
11
11
00
0
KaD
H
com
plex
keto
acid
de
hydr
ogen
ase
com
plex
(EC
1.8
.1.4
, EC
1.
2.4.
4, E
C 2
.3.1
.-)
kadA
kadB
kadC
kadD
00
00
01
11
11
21
11
11
11
11
1
PTA
(bra
nche
d ch
ain)
ph
osph
otra
nsac
ylas
e
(EC
2.3
.1.1
9)pt
a1
11
11
11
11
12
11
11
11
11
11
AC
K
(put
ativ
e)ac
ylki
nase
(EC
2.7
.2.-)
ack
01
10
01
01
10
21
00
02
22
11
1
D-H
ycD
H
(put
ativ
e)D
-hyd
roxy
acid
de
hydr
ogen
ase
(EC
1.1
.1.-)
hdhD
00
02
21
12
20
21
01
20
00
00
0
L-H
ycD
HL-
hydr
oxya
cid
dehy
drog
enas
e
(E
C 1
.1.1
.-)hd
hL1
11
11
30
33
11
11
31
1 c
11
11
1
Est
Aes
tera
se A
(EC
3.1
.1.1
)es
tA0
01
00
21
00
11
00
21
11
11
11
a: T
he A
lcD
H fa
mily
con
tain
s all
the
gene
s with
put
ativ
e Alc
DH
act
iviti
es, i
nclu
ding
Alc
DH
/Ald
DH
bifu
nctio
nal p
rote
ins w
ith a
n A
lcD
H d
omai
n; w
hile
the
Ald
DH
fam
ily is
onl
y co
mpo
sed
of th
e bi
func
tiona
l pro
tein
s w
ith a
n A
ldD
H d
omai
n (w
ith a
ldeh
yde
dehy
drog
enas
e ac
tiviti
es).
b: T
he g
enes
of L
DB
and
LB
U a
nd o
ne o
f the
LPL
gen
es a
re sh
orte
r and
enc
ode
only
an
Ald
DH
dom
ain,
whi
le th
e ge
nes f
rom
LR
F an
d LR
E en
code
onl
y a
n A
lcD
H d
omai
n, a
nd th
eref
ore
they
are
not
bifu
nctio
nal.
c: O
ne p
utat
ive
alcD
H g
ene
from
STH
and
STU
is a
pse
udog
ene.
The
gen
e en
codi
ng L
-Hyc
DH
from
LLX
is a
lso
a ps
eudo
gene
.d:
The
ipd
gene
shar
es p
artia
l hom
olog
y to
the
kdcA
gen
e. It
s dec
arbo
xyla
se fu
nctio
n is
inac
tivat
ed d
ue to
a m
issi
ng la
rge
C-te
rmin
al p
art.

CHAPTER 3
44
α-Ketoacids can be directly converted to carboxylic acids via oxidative decarboxylation. The first step is the conversion of an α-ketoacid to its corresponding acyl-CoA by α-ketoacid dehydrogenase (KaDH), an enzyme complex composed of the four subunits E1α, E1β, E2, and E3. Acyl-CoA is further converted to the corresponding carboxylic acid by a phosphotransacylase (PTA) and acyl kinase (ACK) (Figure 3.2). The oxidative decarboxylation pathway was characterized in Enterococcus faecalis (70), and PTA, ACK and a KaDH complex with specific activity toward their corresponding substrates derived from branched-chain amino acids, were found encoded in one operon (ptb-buk-bkdDABC). Only the Lb. casei genome contained a similar orthologous operon. Nevertheless, homologs of the ptb gene, buk gene, and bkdDABC genes were found encoded separately in different positions of the chromosome in various other LAB, for instance in S. thermophilus. This finding agrees well with the experiments of Helinck et al. (37) who showed these enzyme activities in S. thermophilus strains. Caution is required however, since the best homologs of KaDH in many LAB are annotated as either pyruvate or acetoin dehydrogenase complex, and it is not clear whether these complexes have overlapping substrate specificity.
α-Ketoacids can also be reduced to hydroxyacids by hydroxyacid dehydrogenase (HycDH). Two stereospecific enzymes D-HycDH and L-HycDH are distinguished that belong to the larger D-LDH (D-lactate dehydrogenase) and L-LDH (L-lactate dehydrogenase) protein families, respectively. L-HycDH from Weisella confuse (formerly Lb. confuses) has been characterized while a D-HycDH encoding gene has been cloned from Lb. casei (42, 49). Our analysis showed that HycDH enzymes can be clearly distinguished from their closely related LDH homologs (see Figure S3 in the supplemental material). Table 3.1 indicates that most LAB possess one or more L-HycDH while only some have a D-HycDH. Although we could not find any literature evidence that hydroxyacids can directly lead to flavor formation, the shared precursors of hydroxyacids and some flavor compounds imply that the activity of HycDH could have a negative effect on flavor formation by shunting flavor precursors into non-flavor products.
Alcohol and aldehyde dehydrogenasesAlcohol dehydrogenase (AlcDH) and aldehyde dehydrogenase (AldDH) catalyze the
conversion of aldehydes to alcohols and carboxylic acids, respectively. The NJ trees of both the AlcDH and AldDH family appear to be large and complicated (51, 54). As the experimental evidence of AlcDH and AldDH from LAB is lacking, only several putative AlcDH subfamilies and a bifunctional AlcDH/AldDH subfamily can be distinguished (data not shown). Table 3.1 shows the genes encoding putative AlcDH and bifunctional AlcDH/AldDH proteins with aldehyde dehydrogenase catalytic domains. Most LAB genomes encode multiple AlcDH members, but only a single AlcDH/AldDH ortholog.
EsterasesEsters with short-chain fatty acids as the precursors, such as ethyl butanoate and ethyl
isovalerate, can be formed via amino acid degradation pathways (Figure 3.2). They are important for development of the characteristic “fruity” flavor notes in cheeses. A gene named estA encoding an esterase which can catalyze the biosynthesis of esters derived from short-chain fatty acids was cloned and characterized in lactococcal strains (28), as well as in Lb. helveticus (26) and Lb. casei (74). These estA genes shared high sequence similarity, and could be easily distinguished from other esterases (17, 26) (data not shown). Many LAB genomes, like those of the lactoccoci and streptococci, have one estA gene (Table 3.1). A study of an esterase-negative mutant of Lc. lactis confirmed that the estA encodes the only enzyme for synthesis of short-chain fatty acid esters in this species (48). Therefore, the absence of the estA gene in Lb. acidophilus, Lb. johnsonni, Lb. salivarius and others (Table 3.1), could imply that these strains do not synthesize short-chain fatty acid esters.

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
45
CH
APT
ER 3
Enzymes involved in flavor formation from methionine/cysteine metabolismMethanethiol and other key sulfuric aroma compounds such as dimethyl sulfide (DMS),
dimethyl disulfide (DMDS) and dimethyl trisulfide (DMTS) have a significant impact on cheese sensory profiles (72). They are normally derived from methionine and cysteine. As methionine and cysteine are usually present in only limited amounts in the milk environment, either as free amino acids or in milk proteins (52, 58), the formation of sulfur-containing flavor compounds will depend on both biosynthesis and catabolic pathways of methionine and cysteine. Figure 3.3 gives an overview of the metabolic pathways involved. Hydrogen sulfide can be released from cysteine or homocysteine by the activities of several C-S lyases or cysteine synthase (CysK) (Figure 3.3), which leads to increased H2S levels during cheese ripening (7, 34, 46). Several chemical reactions can also contribute to the sulfuric flavor compounds formation, e.g. converting cystathionine to DMS or liberating methanethiol from methionine in the presence of pyridoxal phosphate (PLP) (72, 73).
Cysteine
L-serine
O-acetyl-L-serine
CysE
CysK
CGS
Cystathionine HomocysteineCGL CBS
Methionine
MetHMetE
Homo-serine
Aspartate 4-semialdehyde
HSDH
NAD(P)H
NAD(P)+
Aspartate
O-phospho-homoserine O-acetyl-homoserineO-succinyl-homoserineHSK
Threonine
HSAT or HSSTATP
ADP
acetyl-CoA
suc-CoA
HSST
AHSHH2S
succinatePi
H2S
H2SCBL CysK MGL
chemical/enzymatic
DMS
MethanethiolMGLCBL CGL
Chemicalwith PLP
H2S, DMS, DMDS, DMTS
Elimination routeglutamate
keto-glutarateAT
KMBA
HMBA
HycDH
Transamination
Methional
alcohol carboxylic acid
CoA
CoA
CBLacetate
CGLCBLCGL
KdcA
route
acetate
acetate
acetyl-CoACoA
Figure 3.3. Cysteine and methionine metabolism in LAB. Both biosynthesis and degradation pathways of cysteine and methionine are included in this map, while the catabolic reactions are framed. Biosynthesis of both amino acids comprises a route from serine, a route from aspartate and the inter-conversion between cysteine and methionine. Degradation of methionine involves the transamination pathway that is initiated by aminotransferases and the elimination pathway initiated by C-S lyases. Enzymes involved in the pathways are: CysE, serine acetyltransferase; CysK; CBL; CGL; MGL; CBS; CGS; HSDH; HSK; HSST; HSAT; AHSH; MetH; MetE; AT, aminotransferases; HycDH; and KdcA. Abbreviations of the metabolites produced are: KMBA, 4-methylthio-2-oxobutanoate; HMBA, 2-hydroxy-4-methylsulfanyl-butyric acid; DMS, dimethyl sulfide; DMDS, dimethyl disulfide; and DMTS, dimethyl trisulfide. Key sulfur-containing flavor compounds are in italic.

CHAPTER 3
46
Tabl
e 3.
2. D
istr
ibut
ion
of g
enes
enc
odin
g en
zym
es in
volv
ed in
Met
hion
ine
and
Cys
tein
e m
etab
olis
m in
LA
B g
enom
es. T
he n
umbe
r of i
dent
ified
gen
es
is in
dica
ted,
whi
le sh
adin
g sh
ows n
o ge
ne (w
hite
), a
sing
le g
ene
or m
ultip
le g
enes
(gre
y).
prot
ein
func
tion
Sugg
este
d ge
ne
nam
e
Lac
toba
cilli
Lac
toco
cci
Stre
ptoc
occi
LAC
LJO
LGA
LDB
LBU
LPL
LBE
LRF
LRE
LSK
LCA
LSL
PPE
OO
ELM
ELL
XLL
ALL
MST
HST
UST
M
Cys
Ese
rine
acet
yltra
nsfe
rase
(E
C 2
.3.1
.30)
cysE
00
01
p1
p1
00
00
00
01
21
11
22
2
Cys
KO
-ace
tyls
erin
e su
lfhyd
ryla
se (E
C
2.5.
1.47
)cy
sK1
00
11
00
11
01
10
00
22
21
11
CB
L/C
GL
(lyas
e gr
oup)
a
cyst
athi
onin
e be
ta
lyas
e (E
C 4
.4.1
.8)
/cys
tath
ioni
ne
gam
ma
lyas
e (E
C4.
4.1.
1)
cblB
/cgl
B1
p0
01
11
01
10
01
02
21
11
11
1
CB
L/C
GL
(AT
grou
p) a
cyst
athi
onin
e be
ta
lyas
e (E
C 4
.4.1
.8)
/cys
tath
ioni
ne
gam
ma
lyas
e (E
C4.
4.1.
1)
cblA
/cgl
A1
10
11
31
11
01
20
00
11
1 p
11
1
MG
Lm
ethi
onin
e ga
mm
a ly
ase
(EC
4.4
.1.1
1)m
gl0
00
0 0
00
00
00
00
00
00
00
00
CB
Scy
stat
hion
ine
beta
synt
hase
(EC
4.
2.1.
22)
cbs
1 p0
01
11
01
10
01
01
10
00
11
1
CG
S b
cyst
athi
onin
e ga
mm
a sy
ntha
se
(EC
2.5
.1.4
8)cg
s0
00
00
10
00
01
00
01
11
p1
11
1
Met
Hho
moc
yste
ine
S-m
ethy
ltran
sfer
ase
(
EC 2
.1.1
.10)
met
H1
00
11
10
11
00
10
00
00
01
11
Met
Eho
moc
yste
ine
met
hyltr
ansf
eras
e
(EC
2.1
.1.1
4)m
etE
00
00
11
01
10
10
00
11
11
11
1
HSD
Hho
mos
erin
e de
hydr
ogen
ase
(EC
1.
1.1.
3)ho
m1
01
11
20
00
01
10
11
11
11
11
HSK
hom
oser
ine
kina
se
(EC
2.7
.1.3
9 )
thrB
10
11
11
10
00
11
01
p1
11
11
11
HSA
Tho
mos
erin
e O
-ace
tyl
trans
fera
se
(E
C 2
.3.1
.31
)m
etX
00
00
00
00
00
00
00
00
00
00
0
HSS
T c
hom
oser
ine
O-s
ucc
inyl
trans
fera
se
(E
C 2
.3.1
.46
)m
etA
10
02
p2
10
11
01
10
11
11
p1
11
1
AH
SHO
-ac
etyl
hom
oser
ine
sulfh
ydry
lase
(EC
2.5
.1.4
9)cy
sD0
00
00
10
00
00
00
11
11
11
11
a: c
bl/c
gl g
enes
fall
into
two
prot
ein
fam
ilies
: the
lyas
e fa
mily
and
the
amin
otra
nsfe
rase
fam
ily. N
ames
of t
he fa
mili
es a
re in
dica
ted
betw
een
brac
kets
. b:
Enz
ymes
in th
e C
GS
subf
amily
may
pos
sess
bot
h cy
stat
hion
ine
gam
ma
synt
hase
and
O-a
cety
lhom
oser
ine
sulfh
ydry
lase
(AH
SH) a
ctiv
ityc:
Enz
ymes
in th
e H
SST
fam
ily m
ay h
ave
hom
oser
ine
O-s
ucci
nyltr
ansf
eras
e ac
tivity
or h
omos
erin
e O
-ace
tyltr
ansf
eras
e ac
tivity
or b
oth
activ
ities
. p:
pse
udog
ene

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
47
CH
APT
ER 3
The metabolism of sulfur-containing amino acids is complex, especially considering the existence of multiple alternative pathways/enzymes for methionine biosynthesis (36). Our genome-wide analysis of LAB showed large differences in the distribution of the related enzymes, indicating a high variability in the presence of the different routes (Table 3.2). Compared to Lb. plantarum and S. thermophilus strains, in which most of these genes seem to be present, the other LAB genomes lack many of these enzymes. Most S. thermophilus strains exhibit no absolute amino acids requirements for growth on minimal medium including sulfur-containing amino acids (43), which agrees with our prediction that all biosynthesis enzymes are present. The distribution of these enzymes can also vary between different strains of the same LAB species. For instance, in the Lb. delbrueckii subsp. bulgaricus ATCC11842 genome, a metE gene is absent and two HSST-encoding genes (metA) are inactivated due to point mutations while those genes are present in Lb. delbrueckii subsp. bulgaricus ATCC BAA365. Similarly, one of the cbl/cgl genes is truncated in Lc. lactis subsp. cremoris MG1363 and cgs and metA appear to be pseudogenes in Lc. lactis subsp. cremoris SK11. It should be noted that those three genes are located in the same operon in Lactococcus genomes, indicating putative frequent mutation events in this region leading to gene inactivation (see Figure S4 in the supplemental material).
Homoserine activation enzymes Methionine biosynthesis from homoserine can be initiated by one of three reactions, a
homoserine kinase reaction, a homoserine trans-succinylase reaction and a homoserine trans-acetylase reaction, catalyzed by HSK, HSST and HSAT, respectively (Figure 3.3). No homolog of HSAT from Bacillus cereus was found using BLAST in any of the sequenced LAB genomes, while HSK and HSST are distributed relatively broadly in LAB. Although HSST and HSAT belong to different protein families and share little sequence similarity, previous studies revealed that they share overlapping substrate specificity (36). In Bacillus subtilis, HSST is likely to possess homoserine trans-acetylase activity (14, 57). Moreover, the orthologous gene in Lb. plantarum is located in the same operon as the genes encoding AHSH and HSDH, as in Bacillus (Figure S4 in the supplemental material). The genetic organization reflects the functional correlation between HSDH, HSST and AHSH in Lb. plantarum, a bacteria that is known to lack a TCA-cycle to make succinyl-CoA and therefore is restricted to using acetyl-CoA to synthesize methionine (33). Thus, we hypothesize that HSAT activities can be complemented by HSST in some LAB, and that HSST together with AHSH can form homocysteine, the precursor of methionine.
Homocysteine methylation enzymes Methylation of homocysteine, the final step of methionine biosynthesis, can be catalyzed by
either of two non-homologous enzymes, i.e. MetH, a cobalamin-dependent methionine synthase, or MetE, a cobalamin-independent methionine synthase. Both MetH and MetE are present in Lb. reuteri strains, S. thermophilus strains and Lb. delbrueckii subsp. bulgaricus strain ATCC BAA365, whereas in some other LAB genomes only one enzyme is found (Table 3.2).
Lyases and synthases: the CysK/CBS familyCysK (cysteine synthase or O-acetylserine-thiol-lyase) and CBS (cystathionine beta-synthase)
belong to the same protein family, the so-called pyridoxal-phosphate dependent β-family (18, 38, 47). Five major subfamilies could be distinguished in the phylogenetic tree of the CysK/CBS protein family with relatively good bootstrap support (Figure 3.4). This protein family includes the tryptophan synthase β-subunit, threonine dehydratase, and threonine synthase subfamilies. The CysK and CBS subfamilies are more closely related to each other than to the other three subfamilies. As a result, the attribution of substrate specificities of these enzymes was less straightforward. In addition, the

CHAPTER 3
48
only characterized cysK gene in LAB is from Lc. lactis subsp. cremoris MG1363 (30), while none of cbs gene products have been studied in any LAB. Actually, all the putative enzymes belonging to this CysK/CBS cluster were originally annotated as cysteine synthase (CysK) in LAB genomes and given genes names cysK or cysM (Table S1 in the supplemental material). A recent study on the enzymes converting methionine to cysteine in Bacillus subtilis (38) has found that both CysK and CBS (formerly named YrhA in B. subtilis) have an O-acetylserine-thiol-lyase (cysteine synthase) activity, though CysK represented 95% of this activity. In addition, an atypical cystathionine β-synthase activity was observed for CBS but not for CysK in B. subtilis.
[780]
[1000][926]
[940]
[433]
[654]
[788][1000]
[1000][1000][1.1.1] LLM_125624565
[1.1.1] LLM_CysK[1.1.1] LLX_15672764[1.1.1] LLA_116511591
[1000] [1.1.2] LLX_15672522[1000] [1.1.2] LLM_125623375
[1.1.2] LLA_116511337[1000] [1.1] STM_116627279
[1000][1.1] STH_55822343[1.1] STU_55820459
[1000][595]
[1000] [1.1] LRF_148532032[1.1] LRE00008
[1.1] LCA_116494039[535] [1.1] LAC_58337517
[999] [1.1] LSL_90962687[1000] [1.1] LBU_116514403
[1.1] LDB_104774314[1.1] BSU_CysK
[1] FN_CysK[1000] [2] BSU_yrhA_CBS
[981][550] [2] LSL_90961016
[2] LAC_58337377
[617][859]
[1000] [2] LRF_148531797[2] LRE00702
[2] OOE_116491721[947] [2] LME_116618386
[657] [2] LPL_28377182[1000]
[1000][2] LBU_116514295[2] LDB_104774207
[1000][2] STM_116627697[2] STH_55822810[2] STU_55820893
[[
]]
CysK
CBS
Tryptophan synthase Threonine dehydratase
Threonine synthase 0.1
Figure 3.4. Phylogenetic tree of the CysK/CBS family. (A colour version can be found in Appendix) Squares represent duplication events, while circles represent speciation events. Bootstrap values are reported for a total of 1,000 replicates. Dots indicate the experimentally verified genes from Lc. lactis subsp. cremoris MG1363 (LLM_CysK), Bacillus subtilis (BSU_CysK and BSU_yrhA_CBS) and Fusobacterium nucleatum (FN_CysK). The subfamilies related to the tryptophan synthase β-subunit, threonine dehydratase and threonine synthase could be distinguished via analysis of the gene context, as most of the corresponding genes are associated with specific amino acid biosynthesis gene clusters (data not shown). The GI code of each protein in the family is preceded by its corresponding genome abbreviation.
The information on cysK and cbs obtained from B. subtilis and on cysK obtained from Fusobacterium nucleatum was added to Figure 3.4 to increase the specificity of the functional annotation of the CysK/CBS cluster (34). The branches with the CysK sequences from B. subtilis, Lc. lactis and Fb. nucleatum contain orthologous sequences from all lactococcal and streptococcal strains, as well as from several Lactobacillus strains, mainly from the Lb. acidophilus group. As Figure 3.4 shows, a relatively recent duplication in the CysK subfamily has occurred in lactococcal genomes. In Lc. lactis subsp. cremoris MG1363, one of the paralogs was found to be co-transcribed with the gene encoding cystathionine beta/gamma lyase (belongs to CBL/CGL lyase group, which will be described later) (Figure S5 in the supplemental material), and to be under the regulation of the LysR-type regulator CmbR (29, 35). In contrast to CysK, a protein orthologous to B. subtilis CBS (YrhA) seems to be absent in lactococcal genomes. This could cause a slower growth of lactococci utilizing homocysteine as the only sulfur source, as this phenotype is observed for a cbs (yrhA) mutant from B. subtilis (38). Our subdivision into CysK and CBS protein subfamilies was strengthened by the observation that almost all the putative cbs orthologs have the same gene context, being located in the same operon together with a cbl/cgl gene (belongs to CBL/CGL lyase group, which will be described later). In fact, the genetic organization is similar to that of the metC-cysK operon in lactococci (Figure S5 in the supplemental material).

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
49
CH
APT
ER 3
Lyases and synthases: the CBL/CGL familiesMethionine elimination is a major pathway of methionine degradation in some cheese micro-
organisms (78). This pathway is initiated by the dethiomethylation reaction, in which methionine is converted to methanethiol by C-S lyases, including methionine γ-lyase (MGL), cystathionine β-lyase (CBL), and cystathionine γ-lyase (CGL). MGL catalyses α, γ-elimination of methionine, and this activity is observed mainly in brevibacteria and corynebacteria (27). The mgl gene from Brevibacterium linens was sequenced, and gene disruption has shown its essential role for flavor formation (4). However, none of the LAB genomes seem to have the mgl gene, which is in agreement with the near absence of the related enzyme activity observed in LAB (21).
CBL and CGL catalyze an α, β-elimination and an α, γ-elimination reaction respectively, converting cystathionine to homocysteine or cysteine (Figure 3.3). Moreover, it was shown that the enzymes are capable of converting other sulfur-containing substrates via α, γ-elimination. For instance, methanethiol is formed from methionine, although the enzyme activity toward methionine was around 10-100 fold lower than toward cystathionine (72, 78). CBL has been purified from various lactic acid bacteria and its gene was previously identified from Lc. lactis (called metC) and Lb. debrueckii subsp. bulgaricus (called patC) (2, 7, 29). CGL enzyme activity has been detected in Lb. reuteri (20), Lb. fermentum (61), and Lc. lactis subsp. cremoris SK11 (13) while the gene from Lc. lactis subsp. cremoris MG1363 was characterized experimentally (22). Analysis of substrate specificities suggests an overlapping function between CBL and CGL. For example, it has been reported that the gene encoding CBL from Lc. lactis subsp. cremoris B78 or MG1363 is also capable of catalyzing the α, γ-elimination reaction like CGL (2, 22).
[514]
[548]
[825]
[997]
[1000]
[654]
[1000]
[948][1.1] STH_55822811[1.1] STU_55820894[1.1] STM_116627698
[978][1.1] LBU_116514296[1.1] LDB_104774208
[1.1] LPL_28377181[1.1] LME_116618387
[823] [1.1] OOE_116491720[995] [1.1] LSL_90961017
[856] [1.1] LAC_58337380[1000] [1.1] LRE01167
[1.1] LRF_148530564[543] [1.1] BSU_yrhB_CGL
[1000]
[1000] [1.1.1] LLC2_MetC[1.1.1] LLA_116511590
[1000][1.1.2] LLC1_MetC[1.1.2] LLM_125624566[1.1.2] LLX_15672763
[948] [1.2] BSU_yjcJ_CBL[1000] [1.2] LME_116618893
[1.2] OOE_116490814[1000] [1.3] LCA_MetB
[1.4] LCA_116494152[944] [2] BSU_yjcI_CGS
[782]
[981] [2] LPL_28379151[2] LME_116618894
[1000]
[1000] [2] LLM_125624948[2] LLX_15673899
[616] [2] SAN_CGS[1000] [2] STM_116627266
[2] STH_55822329[2] STU_55820445
[632] [3] LME_116618337[1000] [4] NAI_CysD
[768]
[1000]
[1000][4] STH_55822940[4] STU_55821020[4] STM_116627784
[830]
[985]
[995] [4] LME_116618888[4] OOE_116490344
[4] LPL_28379077[1000] [4] LLX_15672055
[935][4] LLM_125622970[4] LLA_116510901
CBL/CGL
CBL
CGS/AHSH
AHSH
0.1
Figure 3.5. Neighbor-joining tree of the CBL/CGL lyase family. (A colour version can be found in Appendix) Squares represent duplication events, while circles represent speciation events. Bootstrap values were calculated for a total of 1000 replicates. Experimentally characterized proteins are indicated by dots, including CGL, CBL and CGS from B. subtilis (BSU_yrhB_CGL, BSU_yjcJ_CBL and BSU_yjcI_CGS respectively), the CBL/CGL from Lc. lactis subsp. cremoris MG1363 and NIZO B78 (LLC1_MetC and LLC2_MetC respectively), the MetB from Lb. casei (LCA_MetB), the CGS from Streptococcus anginosus (SAN_CGS), as well as the AHSH (cysD gene) from Emericella nidulans (NAI_CysD). The GI codes of the other LAB proteins in this family are shown after the genome abbreviation.

CHAPTER 3
50
Sequence analysis of the functional domains of experimentally verified cbl and cgl gene products revealed that they fall into two distinct families which share little sequence similarity. The first family is the aminotransferase I protein family which mainly consists of various aminotransferases including the previously mentioned ArAT and AspAT (Figure S2 in the supplemental material). We propose to use cblA/cglA as names for the genes encoding CBL/CGL in this aminotransferase I family. Three experimentally characterized genes encoding CBL or CGL were found to belong to this family, i.e the patC gene of Lb. debrueckii subsp. bulgaricus (7), the ytjE gene from Lc. lactis subsp. lactis IL1403 (46) and the malY gene from Lb. casei (39). All of these enzymes exhibit α, β-elimination activity toward cystathionine, however the one from Lc. lactis can also catalyze α, γ-elimination of methionine. Orthologs of the cblA/cglA genes from the aminotransferase family are found in many LAB genomes (Table 3.2).
The second family to which CBL/CGL enzymes belong has been designated as the lyase family, and includes various methionine or cysteine metabolism-related enzymes: CBL, CGL, and CGS (cystathionine γ-synthase), as well as AHSH (O-acetyl homoserine sulfhydrylase). The NJ tree of this family clearly shows three major subfamilies, the CBL/CGL subfamily, the CGS/AHSH subfamily, and the AHSH subfamily (Figure 3.5). The first major subfamily can be divided into two functional clusters named “CBL/CGL” and “CBL” respectively, with proposed gene names cblB/cglB. They can be distinguished by including experimentally characterized CBL/CGL proteins, encoded by the metC genes from Lc. lactis subsp. cremoris MG1363 and B78 (22, 30), the yrhB gene (renamed cgl in Figure 3.5) (38) and the metC gene (formally yjcJ, renamed cbl in Figure 3.5) from B. subtilis (8). The CGL and CBL from B. subtilis could be separated into two sub-clusters, representing different substrate specificities and physiological roles. Experimentally, the B. subtilis CGL enzyme was found to possess cystathionine γ-lyase activity, but was not able to degrade methionine (38). On the other hand, cystathionine β-lyase activity was detected from the CBL enzyme in B. subtilis, but its activity toward methionine was not determined (8). The metC genes from Lc. lactis subsp. cremoris grouped together with CGL from B. subtilis. However, unlike the B. subtilis CGL, the enzymes encoded by the lactococcal metC genes were found to carry out both α, β-elimination and α, γ-elimination towards different substrates (22). Therefore, the subgroup “CBL/CGL” seems to display a mixture of cystathionine beta- and gamma-lyases activites. It implies that LAB enzymes in this sub-cluster could have either solo CGL activity or a CBL/CGL dual activity.
The second major subfamily in the lyase family contains the yjcI gene (renamed cgs in Figure 3.5) from B. subtilis and the cgs gene from Streptococcus anginosus. Both genes encode bifunctional enzymes which have both cystathionine γ-synthase (CGS) and O-acetyl homoserine sulfhydrylase activity (AHSH) (8, 75). Thus we define this protein subfamily as the (bifunctional) “CGS/AHSH” subfamily, and assumed that all the members may exhibit both CGS and AHSH activities.
The third major subfamily contains unifunctional AHSH enzymes, as suggested by the experimentally characterized cysD gene from the fungus Emericella nidulans and other micro-organisms (15, 59, 60). Notably, almost all the LAB species/strains that contain a member of the AHSH subfamily also seem to contain a CGS/AHSH subfamily member, except for Oenococcus oeni. Unfortunately, to our knowledge, no experimental evidence on the characterization of CGS or AHSH from LAB is available.
Improved annotation and nomenclatureIn this study, we focused on the enzymes involved in flavor-forming pathways, especially
the ones which catalyze the metabolism of sulfur-containing amino acids leading to sulfuric flavor compounds. By combining phylogenetic analysis, experimental evidence and gene context analysis, we could improve the original annotations of many of the genes encoding these enzymes in LAB.

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
51
CH
APT
ER 3
Some of the previous annotations were sometimes inconsistent and caused much confusion. A more consistent nomenclature for genes and proteins is proposed (Table 3.1, Table 3.2, and supplemental material Table S1). Various enzymes belong to large protein families. For instance aminotransferases ArAT, AspAT and cystathionine lyases CBL/CGL fall into the same aminotransferase family I. Subfamilies with different substrate specificities could be distinguished by our method. Some of the enzymes exhibit overlapping substrate specificities, e.g. ArAT, AspAT and BcAT, although they are still expected to have a preference towards certain substrates. The cbs genes from lactic acid bacteria, some of which were previously annotated as cysteine synthase (CysK), are now for first time proposed to be cystathionine beta-synthase instead. Finally, some enzymes are predicted to be able to catalyze different types of reactions, for example proteins from the CBL/CGL subfamily may carry out both α, β-elimination and α, γ-elimination reactions and proteins from CGS/AHSH subfamily might display both cystathionine γ-synthase and O-acetyl homoserine sulfhydrylase activity.
Flavor-forming potentialThe improved gene annotation leads to a better prediction of the flavor-forming potential. S.
thermophilus and Lactococcus strains as well as some other milk-associated LAB e.g. Lb. casei, seem to possess relatively more abundant genes encoding flavor-related enzymes, whereas many of these enzymes are lacking in Lb. gasseri and Lb. johnsonii, which are typical bacteria living in the GI-tract. The Lb. plantarum genome also encodes a large set of enzymes involved in sulfur-containing amino acid metabolism, which might reflect its flexibility to grow under different conditions (41). It is important to keep in mind that only the LAB type strains which have been completely sequenced were analyzed in this study. It has been shown that in some cases the presence of flavor-forming enzymes can vary between strains from the same species. Therefore, in order to explore the flavor-forming ability of other LAB strains which have not been sequenced, especially the ones for industrial use, experimental techniques such as CGH-microarrays (comparative genomic hybridization with microarray) could be applied. On the other hand, high-throughput genome sequencing is now becoming so fast and affordable that numerous industrial and environmental LAB strains can soon be sequenced. The importance of our in silico analysis is in guiding future genomics sequencing and experimentation and thereby validating our predictions.
In conclusion, the identification of key enzymes in flavor forming pathways and the prediction on flavor-forming capacity of various LAB should provide an excellent starting point to direct the selection of potential species and/or strains used for the industrial production of flavored products.
Acknowledgements
This work was supported by grant CSI4017 from the Casimir program of the Ministry of Economic Affairs, the Netherlands. C. Francke is supported by the Kluyver Center for Genomics of Industrial Fermentation. We thank Dr. Johan van Hylckama Vlieg for critically reading the manuscript.

CHAPTER 3
52
References1. Altermann, E., W. M. Russell, M. A. zcarate-Peril, R. Barrangou, B. L. Buck, O. McAuliffe, N. Souther, A. Dobson, T.
Duong, M. Callanan, S. Lick, A. Hamrick, R. Cano, and T. R. Klaenhammer. 2005. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc. Natl. Acad. Sci. U. S. A. 102:3906-3912.
2. Alting, A. C., W. Engels, S. van Schalkwijk, and F. A. Exterkate. 1995. Purification and characterization of cystathionine beta-Lyase from Lactococcus lactis subsp. cremoris B78 and its possible role in flavor development in cheese. Appl. Environ. Microbiol. 61:4037-4042.
3. Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
4. Amarita, F., M. Yvon, M. Nardi, E. Chambellon, J. Delettre, and P. Bonnarme. 2004. Identification and functional analysis of the gene encoding methionine gamma-lyase in Brevibacterium linens. Appl. Environ. Microbiol. 70:7348-54.
5. Ardo, Y. 2006. Flavour formation by amino acid catabolism. Biotechnology Advances 24:238-242.6. Atiles, M. W., E. G. Dudley, and J. L. Steele. 2000. Gene cloning, sequencing, and inactivation of the branched-chain
aminotransferase of Lactococcus lactis LM0230. Appl. Environ. Microbiol. 66:2325-9.7. Aubel, D., J. E. Germond, C. Gilbert, and D. Atlan. 2002. Isolation of the patC gene encoding the cystathionine beta-lyase
of Lactobacillus delbrueckii subsp. bulgaricus and molecular analysis of inter-strain variability in enzyme biosynthesis. Microbiology 148:2029-36.
8. Auger, S., W. H. Yuen, A. Danchin, and I. Martin-Verstraete. 2002. The metIC operon involved in methionine biosynthesis in Bacillus subtilis is controlled by transcription antitermination. Microbiology 148:507-18.
9. Bairoch, A., R. Apweiler, C. H. Wu, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H. Z. Huang, R. Lopez, M. Magrane, M. J. Martin, D. A. Natale, C. O’Donovan, N. Redaschi, and L. S. L. Yeh. 2005. The universal protein resource (UniProt). Nucleic Acids Res. 33:D154-D159.
10. Bateman, A., L. Coin, R. Durbin, R. D. Finn, V. Hollich, S. Griffiths-Jones, A. Khanna, M. Marshall, S. Moxon, E. L. L. Sonnhammer, D. J. Studholme, C. Yeats, and S. R. Eddy. 2004. The Pfam protein families database. Nucleic Acids Res. 32:D138-D141.
11. Bolotin, A., B. Quinquis, P. Renault, A. Sorokin, S. D. Ehrlich, S. Kulakauskas, A. Lapidus, E. Goltsman, M. Mazur, G. D. Pusch, M. Fonstein, R. Overbeek, N. Kyprides, B. Purnelle, D. Prozzi, K. Ngui, D. Masuy, F. Hancy, S. Burteau, M. Boutry, J. Delcour, A. Goffeau, and P. Hols. 2004. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat. Biotechnol. 22:1554-1558.
12. Bolotin, A., P. Wincker, S. Mauger, O. Jaillon, K. Malarme, J. Weissenbach, S. D. Ehrlich, and A. Sorokin. 2001. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp lactis IL1403. Genome Res. 11:731-753.
13. Bruinenberg, P. G., G. De Roo, and G. Limsowtin. 1997. Purification and characterization of cystathionine gamma-Lyase from Lactococcus lactis subsp. cremoris SK11: possible role in flavor compound formation during cheese maturation. Appl. Environ. Microbiol. 63:561-566.
14. Brush, A., and H. Paulus. 1971. The enzymic formation of O-acetylhomoserine in Bacillus subtilis and its regulation by methionine and S-adenosylmethionine. Biochem. Biophys. Res. Commun. 45:735-41.
15. Brzywczy, J., M. Sienko, A. Kucharska, and A. Paszewski. 2002. Sulphur amino acid synthesis in Schizosaccharomyces pombe represents a specific variant of sulphur metabolism in fungi. Yeast 19:29-35.
16. Chaillou, S., M. C. Champomier-Verges, M. Cornet, A. M. Crutz-Le Coq, A. M. Dudez, V. Martin, S. Beaufils, E. rbon-Rongere, R. Bossy, V. Loux, and M. Zagorec. 2005. The complete genome sequence of the meat-borne lactic acid bacterium Lactobacillus sakei 23K. Nat. Biotechnol. 23:1527-1533.
17. Choi, Y. J., C. B. Miguez, and B. H. Lee. 2004. Characterization and heterologous gene expression of a novel esterase from Lactobacillus casei CL96. Appl. Environ. Microbiol. 70:3213-21.
18. Christen, P., and P. K. Mehta. 2001. From cofactor to enzymes. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Chem Rec 1:436-47.
19. Claesson, M. J., Y. Li, S. Leahy, C. Canchaya, J. P. van Pijkeren, A. M. Cerdeno-Tarraga, J. Parkhill, S. Flynn, G. C. O’Sullivan, J. K. Collins, D. Higgins, F. Shanahan, G. F. Fitzgerald, D. van Sinderen, and P. W. O’Toole. 2006. Multireplicon genome architecture of Lactobacillus salivarius. Proc. Natl. Acad. Sci. U. S. A. 103:6718-23.
20. De Angelis, M., A. C. Curtin, P. L. McSweeney, M. Faccia, and M. Gobbetti. 2002. Lactobacillus reuteri DSM 20016: purification and characterization of a cystathionine gamma-lyase and use as adjunct starter in cheesemaking. J. Dairy Res. 69:255-67.
21. Dias, B., and B. Weimer. 1998. Conversion of methionine to thiols by lactococci, lactobacilli, and brevibacteria. Appl. Environ. Microbiol. 64:3320-6.
22. Dobric, N., G. K. Limsowtin, A. J. Hillier, N. P. Dudman, and B. E. Davidson. 2000. Identification and characterization of a cystathionine beta/gamma-lyase from Lactococcus lactis ssp. cremoris MG1363. FEMS Microbiol. Lett. 182:249-54.
23. Dudley, E., and J. Steele. 2001. Lactococcus lactis LM0230 contains a single aminotransferase involved in aspartate biosynthesis, which is essential for growth in milk. Microbiology 147:215-24.
24. Durbin, R., S. Eddy, A. Krogh, and G. and Mitchison. 1998. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press.
25. Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797.
26. Fenster, K. M., K. L. Parkin, and J. L. Steele. 2000. Characterization of an arylesterase from Lactobacillus helveticus CNRZ32. J. Appl. Microbiol. 88:572-83.

FLAVOUR-FORMING PATHWAYS FROM AMINO ACIDS IN LAB
53
CH
APT
ER 3
27. Ferchichi, M., D. Hemme, M. Nardi, and N. Pamboukdjian. 1985. Production of methanethiol from methionine by Brevibacterium linens CNRZ 918. J. Gen. Microbiol. 131:715-23.
28. Fernandez, L., M. M. Beerthuyzen, J. Brown, R. J. Siezen, T. Coolbear, R. Holland, and O. P. Kuipers. 2000. Cloning, characterization, controlled overexpression, and inactivation of the major tributyrin esterase gene of Lactococcus lactis. Appl. Environ. Microbiol. 66:1360-8.
29. Fernandez, M., M. Kleerebezem, O. P. Kuipers, R. J. Siezen, and R. van Kranenburg. 2002. Regulation of the metC-cysK operon, involved in sulfur metabolism in Lactococcus lactis. J. Bacteriol. 184:82-90.
30. Fernandez, M., W. van Doesburg, G. A. Rutten, J. D. Marugg, A. C. Alting, R. van Kranenburg, and O. P. Kuipers. 2000. Molecular and functional analyses of the metC gene of Lactococcus lactis, encoding cystathionine beta-lyase. Appl. Environ. Microbiol. 66:42-8.
31. Fernandez, M., and M. Zuniga. 2006. Amino acid catabolic pathways of lactic acid bacteria. Crit. Rev. Microbiol. 32:155-83.
32. Francke, C., R. Kerkhoven, M. Wels, and R. J. Siezen. 2008. A generic approach to identify Transcription Factor-specific operator motifs; inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics 9:145.
33. Francke, C., R. J. Siezen, and B. Teusink. 2005. Reconstructing the metabolic network of a bacterium from its genome. Trends Microbiol. 13:550-8.
34. Fukamachi, H., Y. Nakano, M. Yoshimura, and T. Koga. 2002. Cloning and characterization of the L-cysteine desulfhydrase gene of Fusobacterium nucleatum. FEMS Microbiol. Lett. 215:75-80.
35. Golic, N., M. Schliekelmann, M. Fernandez, M. Kleerebezem, and R. van Kranenburg. 2005. Molecular characterization of the CmbR activator-binding site in the metC-cysK promoter region in Lactococcus lactis. Microbiology 151:439-46.
36. Gophna, U., E. Bapteste, W. F. Doolittle, D. Biran, and E. Z. Ron. 2005. Evolutionary plasticity of methionine biosynthesis. Gene 355:48-57.
37. Helinck, S., D. Le Bars, D. Moreau, and M. Yvon. 2004. Ability of thermophilic lactic acid bacteria to produce aroma compounds from amino acids. Appl. Environ. Microbiol. 70:3855-61.
38. Hullo, M. F., S. Auger, O. Soutourina, O. Barzu, M. Yvon, A. Danchin, and I. Martin-Verstraete. 2007. Conversion of methionine to cysteine in Bacillus subtilis and its regulation. J. Bacteriol. 189:187-97.
39. Irmler, S., S. Raboud, B. Beisert, D. Rauhut, and H. Berthoud. 2007. Cloning and characterization of two genes encoding a C-S lyase from Lactobacillus casei. Appl. Environ. Microbiol. 9:9.
40. Jensen, R. A., and W. Gu. 1996. Evolutionary recruitment of biochemically specialized subdivisions of Family I within the protein superfamily of aminotransferases. J. Bacteriol. 178:2161-71.
41. Kleerebezem, M., J. Boekhorst, R. van Kranenburg, D. Molenaar, O. P. Kuipers, R. Leer, R. Tarchini, S. A. Peters, H. M. Sandbrink, M. Fiers, W. Stiekema, R. M. K. Lankhorst, P. A. Bron, S. M. Hoffer, M. N. N. Groot, R. Kerkhoven, M. de Vries, B. Ursing, W. M. de Vos, and R. J. Siezen. 2003. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. U. S. A. 100:1990-1995.
42. Lerch, H. P., H. Blocker, H. Kallwass, J. Hoppe, H. Tsai, and J. Collins. 1989. Cloning, sequencing and expression in Escherichia coli of the D-2-hydroxyisocaproate dehydrogenase gene of Lactobacillus casei. Gene 78:47-57.
43. Letort, C., and V. Juillard. 2001. Development of a minimal chemically-defined medium for the exponential growth of Streptococcus thermophilus. J. Appl. Microbiol. 91:1023-9.
44. Liu, M. J., F. H. J. van Enckevort, and R. J. Siezen. 2005. Genome update: lactic acid bacteria genome sequencing is booming. Microbiology 151:3811-3814.
45. Makarova, K., A. Slesarev, Y. Wolf, A. Sorokin, B. Mirkin, E. Koonin, A. Pavlov, N. Pavlova, V. Karamychev, N. Polouchine, V. Shakhova, I. Grigoriev, Y. Lou, D. Rohksar, S. Lucas, K. Huang, D. M. Goodstein, T. Hawkins, V. Plengvidhya, D. Welker, J. Hughes, Y. Goh, A. Benson, K. Baldwin, J. H. Lee, I. Diaz-Muniz, B. Dosti, V. Smeianov, W. Wechter, R. Barabote, G. Lorca, E. Altermann, R. Barrangou, B. Ganesan, Y. Xie, H. Rawsthorne, D. Tamir, C. Parker, F. Breidt, J. Broadbent, R. Hutkins, D. O’Sullivan, J. Steele, G. Unlu, M. Saier, T. Klaenhammer, P. Richardson, S. Kozyavkin, B. Weimer, and D. Mills. 2006. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:15611-6.
46. Martinez-Cuesta, M. C., C. Pelaez, J. Eagles, M. J. Gasson, T. Requena, and S. B. Hanniffy. 2006. YtjE from Lactococcus lactis IL1403 is a C-S lyase with alpha, gamma-elimination activity toward methionine. Appl. Environ. Microbiol. 72:4878-84.
47. Mehta, P. K., and P. Christen. 2000. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 74:129-84.
48. Nardi, M., C. Fiez-Vandal, P. Tailliez, and V. Monnet. 2002. The EstA esterase is responsible for the main capacity of Lactococcus lactis to synthesize short chain fatty acid esters in vitro. J. Appl. Microbiol. 93:994-1002.
49. Niefind, K., H. J. Hecht, and D. Schomburg. 1995. Crystal structure of L-2-hydroxyisocaproate dehydrogenase from Lactobacillus confusus at 2.2 A resolution. An example of strong asymmetry between subunits. J. Mol. Biol. 251:256-81.
50. Overbeek, R., N. Larsen, T. Walunas, M. D’Souza, G. Pusch, E. Selkov, K. Liolios, V. Joukov, D. Kaznadzey, I. Anderson, A. Bhattacharyya, H. Burd, W. Gardner, P. Hanke, V. Kapatral, N. Mikhailova, O. Vasieva, A. Osterman, V. Vonstein, M. Fonstein, N. Ivanova, and N. Kyrpides. 2003. The ERGO (TM) genome analysis and discovery system. Nucleic Acids Res. 31:164-171.
51. Perozich, J., H. Nicholas, B. C. Wang, R. Lindahl, and J. Hempel. 1999. Relationships within the aldehyde dehydrogenase extended family. Protein Sci. 8:137-46.
52. Pieniazek, D., Z. Grabarek, and M. Rakowska. 1975. Quantitative determination of the content of available methionine

CHAPTER 3
54
and cysteine in food proteins. Nutr. Metab. 18:16-22.53. Pridmore, R. D., B. Berger, F. Desiere, D. Vilanova, C. Barretto, A. C. Pittet, M. C. Zwahlen, M. Rouvet, E. Altermann,
R. Barrangou, B. Mollet, A. Mercenier, T. Klaenhammer, F. Arigoni, and M. A. Schell. 2004. The genome sequence of the probiotic intestinal bacterium Lactobacillus johnsonii NCC 533. Proc. Natl. Acad. Sci. U. S. A. 101:2512-2517.
54. Radianingtyas, H., and P. C. Wright. 2003. Alcohol dehydrogenases from thermophilic and hyperthermophilic archaea and bacteria. FEMS Microbiol. Rev. 27:593-616.
55. Rijnen, L., S. Bonneau, and M. Yvon. 1999. Genetic characterization of the major lactococcal aromatic aminotransferase and its involvement in conversion of amino acids to aroma compounds. Appl. Environ. Microbiol. 65:4873-4880.
56. Rijnen, L., M. Yvon, R. van Kranenburg, P. Courtin, A. Verheul, E. Chambellon, and G. Smit. 2003. Lactococcal aminotransferases AraT and BcaT are key enzymes for the formation of aroma compounds from amino acids in cheese. International Dairy Journal 13:805-812.
57. Rodionov, D. A., A. G. Vitreschak, A. A. Mironov, and M. S. Gelfand. 2004. Comparative genomics of the methionine metabolism in Gram-positive bacteria: a variety of regulatory systems. Nucleic Acids Res 32:3340-53.
58. Rutherfurd, S. M., and P. J. Moughan. 1998. The digestible amino acid composition of several milk proteins: application of a new bioassay. J. Dairy Sci. 81:909-17.
59. Shimizu, H., S. Yamagata, R. Masui, Y. Inoue, T. Shibata, S. Yokoyama, S. Kuramitsu, and T. Iwama. 2001. Cloning and overexpression of the oah1 gene encoding O-acetyl-L-homoserine sulfhydrylase of Thermus thermophilus HB8 and characterization of the gene product. Biochim. Biophys. Acta 1549:61-72.
60. Sie’nko, M., J. Topczewski, and A. Paszewski. 1998. Structure and regulation of cysD, the homocysteine synthase gene of Aspergillus nidulans. Curr. Genet. 33:136-44.
61. Smacchi, E., and M. Gobbetti. 1998. Purification and characterization of cystathionine gamma-lyase from Lactobacillus fermentum DT41. FEMS Microbiol. Lett. 166:197-202.
62. Smit, B. A., W. J. M. Engels, J. Bruinsma, J. Vlieg, J. T. M. Wouters, and G. Smit. 2004. Development of a high throughput screening method to test flavour-forming capabilities of anaerobic micro-organisms. J. Appl. Microbiol. 97:306-313.
63. Smit, B. A., W. J. M. Engels, J. T. M. Wouters, and G. Smit. 2004. Diversity of L-leucine catabolism in various microorganisms involved in dairy fermentations, and identification of the rate-controlling step in the formation of the potent flavour component 3-methylbutanal. Appl. Microbiol. Biotechnol. 64:396-402.
64. Smit, B. A., J. Vlieg, W. J. M. Engels, L. Meijer, J. T. M. Wouters, and G. Smit. 2005. Identification, cloning, and characterization of a Lactococcus lactis branched-chain alpha-keto acid decarboxylase involved in flavor formation. Appl. Environ. Microbiol. 71:303-311.
65. Smit, G., B. A. Smit, and W. J. M. Engels. 2005. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol. Rev. 29:591-610.
66. Tanous, C., A. Kieronczyk, S. Helinck, E. Chambellon, and M. Yvon. 2002. Glutamate dehydrogenase activity: a major criterion for the selection of flavour-producing lactic acid bacteria strains. Antonie Van Leeuwenhoek 82:271-8.
67. Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882.
68. van de Guchte, M., S. Penaud, C. Grimaldi, V. Barbe, K. Bryson, P. Nicolas, C. Robert, S. Oztas, S. Mangenot, A. Couloux, V. Loux, R. Dervyn, R. Bossy, A. Bolotin, J. M. Batto, T. Walunas, J. F. Gibrat, P. Bessieres, J. Weissenbach, S. D. Ehrlich, and E. Maguin. 2006. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc. Natl. Acad. Sci. U. S. A. 103:9274-9.
69. van der Heijden, R. T., B. Snel, V. van Noort, and M. A. Huynen. 2007. Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinformatics 8:83.
70. Ward, D. E., R. P. Ross, C. C. van der Weijden, J. L. Snoep, and A. Claiborne. 1999. Catabolism of branched-chain alpha-keto acids in Enterococcus faecalis: the bkd gene cluster, enzymes, and metabolic route. J. Bacteriol. 181:5433-42.
71. Wegmann, U., M. O’Connell-Motherway, A. Zomer, G. Buist, C. Shearman, C. Canchaya, M. Ventura, A. Goesmann, M. J. Gasson, O. P. Kuipers, D. van Sinderen, and J. Kok. 2007. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 189:3256-70.
72. Weimer, B., K. Seefeldt, and B. Dias. 1999. Sulfur metabolism in bacteria associated with cheese. Antonie Van Leeuwenhoek 76:247-61.
73. Wolle, D. D., D. S. Banavara, and S. A. Rankin. 2006. Short communication: empirical and mechanistic evidence for the role of pyridoxal-5’-phosphate in the generation of methanethiol from methionine. J. Dairy Sci. 89:4545-50.
74. Yebra, M. J., R. Viana, V. Monedero, J. Deutscher, and G. Perez-Martinez. 2004. An esterase gene from Lactobacillus casei cotranscribed with genes encoding a phosphoenolpyruvate:sugar phosphotransferase system and regulated by a LevR-like activator and sigma54 factor. J Mol Microbiol Biotechnol 8:117-28.
75. Yoshida, Y., M. Negishi, and Y. Nakano. 2003. Homocysteine biosynthesis pathways of Streptococcus anginosus. FEMS Microbiol. Lett. 221:277-84.
76. Yvon, M. 2006. Key enzymes for flavour formation by lactic acid bacteria. Aust. J. Dairy Technol. 61:88-96.77. Yvon, M., E. Chambellon, A. Bolotin, and F. Roudot-Algaron. 2000. Characterization and role of the branched-chain
aminotransferase (BcaT) isolated from Lactococcus lactis subsp cremoris NCDO 763. Appl. Environ. Microbiol. 66:571-577.
78. Yvon, M., and L. Rijnen. 2001. Cheese flavour formation by amino acid catabolism. International Dairy Journal 11:185-201.

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
55
CH
APT
ER 4
Chapter 4
REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LACTIC ACID BACTERIA
Mengjin Liu, Celine Prakash, Arjen Nauta, Roland J. Siezen, Christof Francke
Manuscript in preparation

CHAPTER 4
56
AbstractSulphuric volatile compounds provide many dairy products a characteristic odour and taste. The volatile compounds mainly originate during the catabolism of the sulphur-containing amino acids cysteine and methionine by lactic acid bacteria (LAB). By in silico comparative analysis of whole genome sequences, we have systematically studied the transcriptional regulatory mechanisms controlling twelve of the key enzymes of cysteine and methionine metabolism in twenty LAB strains. Potential riboswitches were identified using literature-based Hidden Markov Models. Seven new Met T-boxes were found, whereas the absence of S-boxes in LAB was confirmed. Using a comprehensive footprinting method, we identified many putative transcription factor binding sites that have not been reported before. These include cis-regulatory motifs associated with the cbs-cblB/cglB operon in various lactobacilli, O. oeni, Leuconostoc and S. thermophilus strains, as well as the thrB gene in L. acidophilus and L. gasseri. Moreover, it appeared necessary to redefine the binding motifs related to the transcriptional regulators CmbR and MetR/MtaR reported previously in literature. The redefined motifs obey the dyad symmetry AT-N11-AT that is typical for the LysR family of regulators and correspond well with experimental data. Finally, we have reconstructed the transcriptional regulatory networks of cysteine and methionine metabolism. This knowledge can be used to better control the sulphuric volatile compound formation by different LAB.
IntroductionMany of the characteristic odours in cheese are the result of metabolic reactions involving the
sulphur-containing amino acids. Degradation of cysteine and methionine, leads to the production of flavour components such as methanethiol, dimethyl sulphide (DMS), dimethyl disulphide (DMDS) and dimethyl trisulphide (DMTS). Knowledge of the regulation of the metabolic routes that lead to the formation of these flavour compounds and their precursors will therefore be of great importance to rationally control the flavour profiles of dairy products.
Several studies describing part of the regulation of sulphur-containing amino acid metabolism in lactic acid bacteria (LAB) and other closely related gram-positive bacteria have been published in past years. For instance, Hullo et al. [1] report on the regulatory mechanisms related to methionine and cysteine conversions in Bacillus subtilis, whereas Sperandio et al. describe these relations for Lactococcus lactis [2] and Streptocococus mutans [3]. Furthermore, Kovaleva and Gelfand [4] and Rodionov et al. [5] performed comparative in silico studies for the transcriptional regulators CmbR and MtaR within selected gram-positive bacteria. However, a systematic comparative analysis of the transcriptional control of all key enzymes involved in cysteine and methionine metabolism in LAB is still lacking.
Previously, we have improved the annotation of the key enzymes involved in the metabolism of cysteine and methionine in LAB using a genome-wide comparative analysis [6]. The related reactions and species-specific pathways are displayed in Figure 4.1A. In this study, an in-depth in silico analysis of the associated transcriptional regulatory mechanisms has been carried out. Transcription regulatory motifs preceding the genes/operons encoding the twelve key enzymes in twenty different LAB were identified and compared. Binding motifs for CmbR and MetR/MtaR in lactococci and streptococci were highly improved from the previous reports of distinct binding sequences. New Met T-boxes riboswitches were identified in strains such as L. bulgaricus, L. reuteri and L. casei, and previously found Met T-boxes, such as in L. plantarum, were confirmed.

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
57
CH
APT
ER 4
Materials and Methods
Genomic information and ToolsGenomic information was retrieved from the ERGO resource [7] and from the NCBI microbial
genome database (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi). Multiple sequence alignments and neighbour-joining trees (corrected for multiple substitutions) were generated using ClustalX [8]. BioEdit was used to manipulate the alignments and to toggle between translated protein and nucleotide sequence (version 7.0.9, http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Hidden Markov Models (HMMs) were made and genome-wide HMM searches were performed using the HMMER package (http://hmmer.janelia.org/)[9]
The identification of riboswitches Hidden Markov Models were constructed for the T-box motif and for the S-box (SAM-I) motif
on basis of the available literature [5, 10]. Both HMMs were used to scan the LAB genomes (cut-off e-value 1 [10]) and the locations of putative motifs were retrieved. The amino acid specificity of the detected T-boxes was identified on basis of the specifier codon as described by Wels et al. [10].
The identification of putative regulatory elementsCis-regulatory elements were identified according to the specific footprinting method described
by Francke et al. [11] and de Been et al.[12]. The method relies on the definition of Groups Of Orthologous Functional Equivalents (GOOFEs) on basis of synteny. The orthologous relationships of the genes encoding the enzymes involved in the metabolism of sulphur-containing amino acids in LAB [6] were used to define the GOOFEs. The comparative linear genome maps generated by the Microbial Genome Viewer [13] were used to determine synteny. For every GOOFE, the upstream regions (normally ~200 nucleotides) were collected and conserved sequence elements were searched using MEME (Multiple EM for Motif Elicitation) [14] and/or manually from a multiple sequence alignment. The resulting putative regulatory elements were compared and when necessary redefined. Finally, the regulatory elements were related to all genes present in the downstream operon. Operons were defined as genes on the same strand with intergenic regions less than 250 nucleotides and not interrupted by a termination signal.
Results and DiscussionThe transcription regulation of the genes encoding cysteine and methionine metabolism in
LAB involves both regulator-binding and RNA riboswitch mechanisms. Two specific transcriptional regulators MtaR and CmbR have been shown to be involved in activation as well as repression of genes such as metC-cysK, cysD, metEF and metA in Lactococcus lactis [2, 15], and in streptococci [3, 4]. In addition, two types of riboswitches for the regulation of methionine and cysteine metabolism have been reported in prokaryotes: i.e. the T-box and S-box [5, 10, 16]. A riboswitch is a structural sequence element at the 5’ untranslated region of an mRNA that can switch conformation depending on the binding of an effector molecule and therewith terminates transcription (no binding) or allows read through (upon binding) [17, 18]. We have searched for T-boxes and S-boxes associated with cysteine and methionine metabolism in LAB using literature-based Hidden Markov Models. In addition, we have searched the same upstream regions for other regulatory elements, including those related to MtaR and CmbR, using our comprehensive footprinting procedure [11, 12].
MtaR and CmbR both belong to the LysR family of transcription regulators (LTTRs). A neighbour-joining tree of homologous LysR family proteins in LAB was constructed on basis of a comprehensive BLAST search of homologs (cut-off e-value: 1*e-5) in all sequenced LAB genomes,
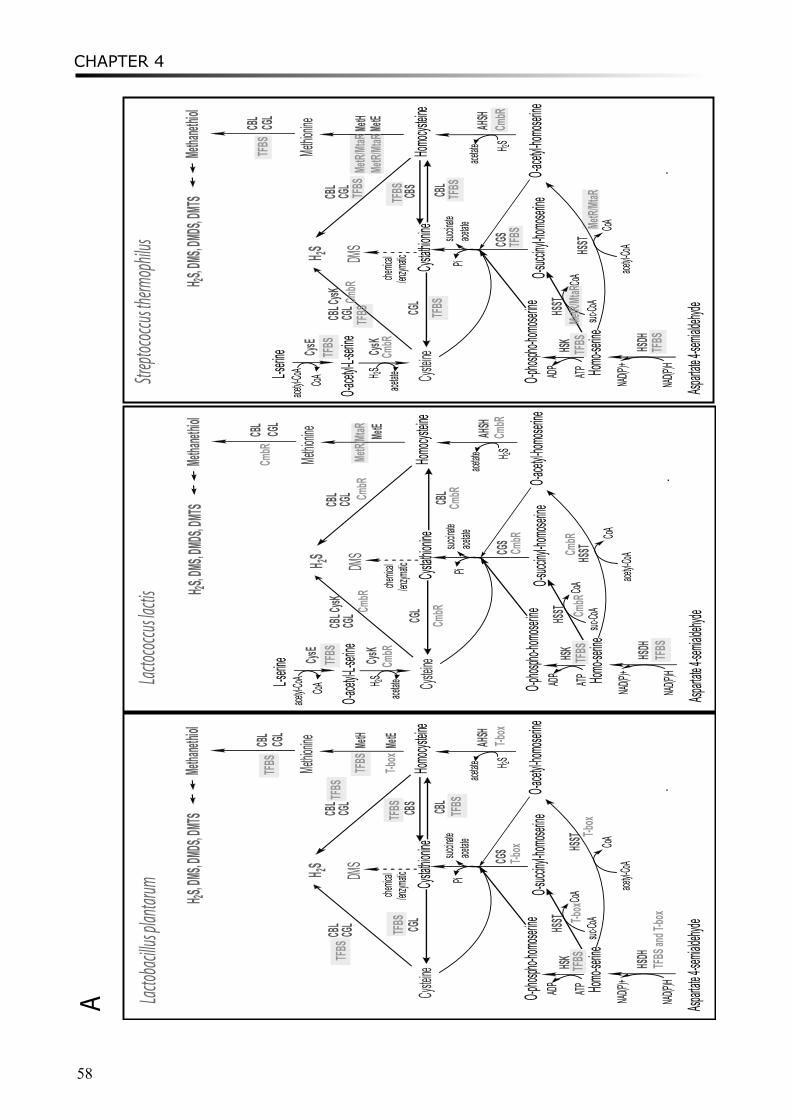
CHAPTER 4
58
Cystein
e
CGS
Cystath
ionine
Homocy
steine
CGL
CBS
Methion
ine MetH
MetE
Homo-se
rine
Asparta
te4-sem
ialdehy
de
HSDH
NAD(P)H
NAD(P) +O-phos
pho-hom
oserine
O-acety
l-homos
erine
O-succ
inyl-hom
oserine
HSKHSS
TATPAD P
acetyl-C
oA
suc-Co
A
HSST
AHSH
H 2S
succina
tePiH 2S
CBL
chemic
al/enz
ymaticDM
S
Methan
ethiol
CBL CGL
H 2S,DMS,
DMDS,
DMTS
CoA
CoA
CBLace
tate
CGLCB
L CGL
acetate
Lactob
acillu
s plan
tarum
Cystein
e
L-serine
O-acety
l-L-serin
e
CysE
CysK
CGS
Cystath
ionine
Homocy
steine
CGL
CB S
Methion
ine MetH
MetE
Homo-se
rine
Asparta
te4-sem
ialdehy
de
HSDH
NAD(P)H
NAD(P)+O-phos
pho-hom
oserine
O-acety
l-homos
erine
O-succ
inyl-hom
oserine
HSKHSS
TATPAD P
acetyl-C
oA
suc-Co
A
HSST
AHSH
H 2S
succina
tePi
H 2S
H 2SCB
LCysK
chemic
al/enz
ymaticDM
S
Methan
ethiol
CBL CGL
H 2S,DMS,
DMDS,
DMTS
CoA
Co A
CBLace
tate
CGLCB
L CGL
acetate
acetate
acetyl-C
oACoA
Cystein
e
L-serine
O-acety
l-L-serin
e
CysE
CysK
CGS
Cystath
ionine
Homocy
steine
CGL
Methion
ine MetE
Homo-se
rine
Asparta
te4-sem
ialdehy
de
HSDH
NAD(P)H
NAD(P) +O-phos
pho-hom
oserine
O-acety
l-homos
erine
O-succ
inyl-hom
oserin e
HSKHSS
TATPAD P
acetyl-C
oA
suc-Co
A
HSST
AHSH
H 2S
succina
tePi
H 2S
H 2SCB
LCysK
chemic
al/enz
ymaticDM
S
Methan
ethiol
CBL CGL
H 2S,DMS,
DMDS,
DMTS
CoA
CoA
CBLace
tate
CGLCB
L CGL
acetate
acetate
acetyl-C
oACo A
Lacto
coccus
lactis
Strep
tococc
us the
rmop
hilu s
TFBS
TFBS
TFBS
TFBS
TFBS
T-box
TFBS
TFBS
T-box
TFBS a
nd T-b
ox
TFBS
T-box
T-box
T-box
TFBS
CmbR
CmbR
CmbR
CmbR
CmbR
CmbR
MetR/
MtaR
CmbR
CmbR
CmbR
TFBS
TFBS
TFBS
CmbR
TFBSCm
bR
TFBS
TFBS
TFBS
TFBS
MetR/
MtaR
TFBS
TFBS
MetR/
MtaR
TFBS
CmbR
TFBS
CmbR
MetR/
MtaR
MetR/
MtaR
A

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
59
CH
APT
ER 4
Lc. lac
tisS.
therm
ophilu
s
Regu
latory
moti
f: cblB
/cglB,
cbs
01
2
sti b 5?
1
CGA
2
ACT
3
TA
4
CA 5
TA
6
TG
7
CG 8
AG
9C 01
AG 11
AC 21
ATG
31
GA
41
GA 51
GAT
61
CTA
71
ATC
81
AT
9102
ACT
12
TGA
22
AG
32A 42T 52T 62T 72
TC
8292
TAG
03
GA
13
GTA
23
GAT
33
CT
43
CT 53
GC
63G 73
TC
83G 93
TC
04
TC 14
CT24T 34T 44T 3
‘
LPL,
LAC,
LSL,
OOE,
LME,
L. bu
lgariu
s,S.
therm
ophilu
s
Regu
latory
moti
f: metH
Regu
latory
moti
f: cblA
/cglA
LPL,
LAC,
LJO,
LB
E, LR
F/LRE
LPL,
LSL
Regu
latory
moti
f: hom
2 (HS
DH), t
hrB (H
SK)
AATGGG
CTGLP
L, LB
E
Regu
latory
moti
f: cys
E
Lc.
lactis
S. the
rmop
hilus
(cysE
1) Cm
bR m
otif: c
blB/cg
lB, cy
sK1
012 bits 5‘
1A 2T 3A 4A 5A 6A 7A 8A 9
TA
10
AG
11T 12T 13A 14A 15
GT
16T 17C 18T 19G 20
AC
21
CT
22A 23T 24A 25
GA
26
CA
27A 28
TA
29
TA
30
TA
31T 32C 33T 34T 35A 36T 3‘
Lc. lac
tis
CmbR
moti
f: cblB
/cglB,
cysM
(CysK
2)
012 bits 5‘
1A 2T 3A 4A 5A 6A 7A 8T 9T 10T 11T 12C 13T 14G 15
GA
16
AC
17G 18A 19T 20T 2122T 23G 24T 25A 26A 27A 28G 3
‘
Lc. lac
tis
CmbR
moti
f: cblA
/cglA,
cgs, m
etA (H
SST)
ATAATAC
GAGGCT
ATLc.
lactis
MetR/
MtaR
moti
f: metE
012 bits 5‘
1A 2T 3A 4G 5T 6T 7
TA
8A 9A 10A 11A 12C 13T 14A 15T 16A 17G 18G 19T 20C 21A 22T 23C 24A 25T 26A 27T 28
TC
29A 30A 31A 32A 33A 34
TG
35G 36T 37A 38T 39T 40A 41T 3‘Lc.
lactis
LBU
Regu
latory
moti
f: hom
(HSD
H) th
rB (H
SK)
Lc.
lactis,
LAC,
LGA,
LSL,
LME
CmbR
moti
f: cysD
(AHS
H)
ATAAAT
TTTTAG
TATLc.
lactis
Regu
latory
moti
f: cb
lA/cgl
A, cgs
CmbR
moti
f: cysK
S. the
rmop
hilus
TCTTTT
cAGAAA
AS.
therm
ophilu
s
MetR/
MtaR
moti
f: metH
S. the
rmop
hilus
MetR/
MtaR
moti
f: metE
012 bits 5‘
1A 2T 3A 4G 5T 6T 7
TA
8A 9A 10A 11A 12C 13T 14A 15T 16A 17G 18G 19T 20C 21A 22T 23C 24A 25T 26A 27T 28
TC
29A 30A 31A 32A 33A 34
TG
35G 36T 37A 38T 39T 40A 41T 3‘
S. the
rmop
hilus
CmbR
moti
f: cysD
(AHS
H)
S. the
rmop
hilus
MetR/
MtaR
moti
f: metA
(HSS
T)
S. the
rmop
hilus
Regu
latory
moti
f: hom
(HSD
H) th
rB (H
SK)
S. the
rmop
hilus
LAC,
LGA
5‘
1A 2T 3A 4G 5T 6T 7T 8A 9A 10A 11G 12T 13T 14A 15T 16A 17G 18C 19T 20A 21A 22C 23T 24A 25G 26A 27G 3‘
012 bits 5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T 10
TC
11T 12C 13T 14A 15T 16C 17T 18C 19T 20T 21T 22G 23A 24T 25A 26G 27T 28T 29T 30T 31T 32T 33T 34A 35T 36T 37G 38T 39G 40A 41T 42A 43A 44G 45C 46T 3‘
B
5‘
1A 2T 3A 4A 5G 6C 7C 8C 9T 10C 11C 12C 13T 14A 15T 16C 17A 18C 19A 20T 21T 22G 23A 24T 25A 26A 27A 28A 29A 30A 31T 32T 33T 34C 35T 36A 37T 3‘
′5
‘
1A 2T 3A 4G 5T 6C 7A 8A 9A 10C 11A 12T 13T 14A 15T 16C 17A 18C 19A 20G 21T 22G 23A 24T 25A 26A 27A 28G 29A 30A 31G 32A 33T 34G 35T 36A 37T 3‘
Figu
re 4
.1. T
rans
crip
tiona
l reg
ulat
ion
of c
yste
ine
and
met
hion
ine
met
abol
ism
in L
. pla
ntar
um, L
c. la
ctis
and
S. t
herm
ophi
lus.(
A c
olou
r ve
rsio
n ca
n be
foun
d in
App
endi
x) (A
) In
the
met
abol
ic m
ap, e
nzym
es
are
give
n in
red
and
the
trans
crip
tiona
l mec
hani
sms
indi
cate
d in
gre
en: T
FBS,
for t
rans
crip
tion
fact
or b
indi
ng m
otif;
T-b
ox fo
r a T
-box
ribo
switc
h. T
he n
ewly
iden
tified
tran
scrip
tiona
l mec
hani
sms
in th
is s
tudy
are
hi
ghlig
hted
usi
ng b
oxes
. (B
) The
iden
tified
put
ativ
e bi
ndin
g se
quen
ces f
or th
e ge
nes i
n di
ffere
nt L
AB
stra
ins a
re sh
own.
Som
e of
the
actu
al b
indi
ng si
tes a
re h
ighl
ight
ed in
yel
low.
Gen
es a
re g
roup
ed b
y th
eir f
unct
ions
. St
rain
abbr
evia
tions
: Lac
toba
cillu
s aci
doph
ilus N
CFM
(abb
revi
atio
n LA
C),
Lact
obac
illus
john
soni
i NC
C 5
33 (L
JO),
Lact
obac
illus
gas
seri
ATC
C 3
3323
(LG
A),
Lact
obac
illus
del
brue
ckii
subs
p. b
ulga
ricu
s ATC
C 1
1842
(L
DB
), L.
del
brue
ckii
subs
p. b
ulga
ricu
s ATC
C B
AA
365
(LB
U),
Lact
obac
illus
pla
ntar
um W
CFS
1 (L
PL),
Lact
obac
illus
bre
vis A
TCC
367
(LB
E), L
acto
baci
llus s
akei
23K
(LSK
), La
ctob
acill
us sa
livar
ius U
CC
118
(LSL
), O
enoc
occu
s oe
ni P
SU1
(OO
E), L
euco
nost
oc m
esen
tero
ides
ATC
C 8
293
(LM
E), L
acto
baci
llus
case
i ATC
C 3
34 (
LCA
), La
ctoc
occu
s la
ctis
sub
sp. l
actis
IL14
03 (
LLX
), La
ctoc
occu
s la
ctis
sub
sp. c
rem
oris
MG
1363
(L
LM),
L. la
ctis
subs
p. c
rem
oris
SK
11 (L
LA),
Stre
ptoc
occu
s the
rmop
hilu
s CN
RZ1
066
(STH
), S.
ther
mop
hilu
s LM
G18
311
(STU
), S.
ther
mop
hilu
s LM
D9
(STM
), an
d La
ctob
acill
us re
uter
i F27
5 (L
RF)
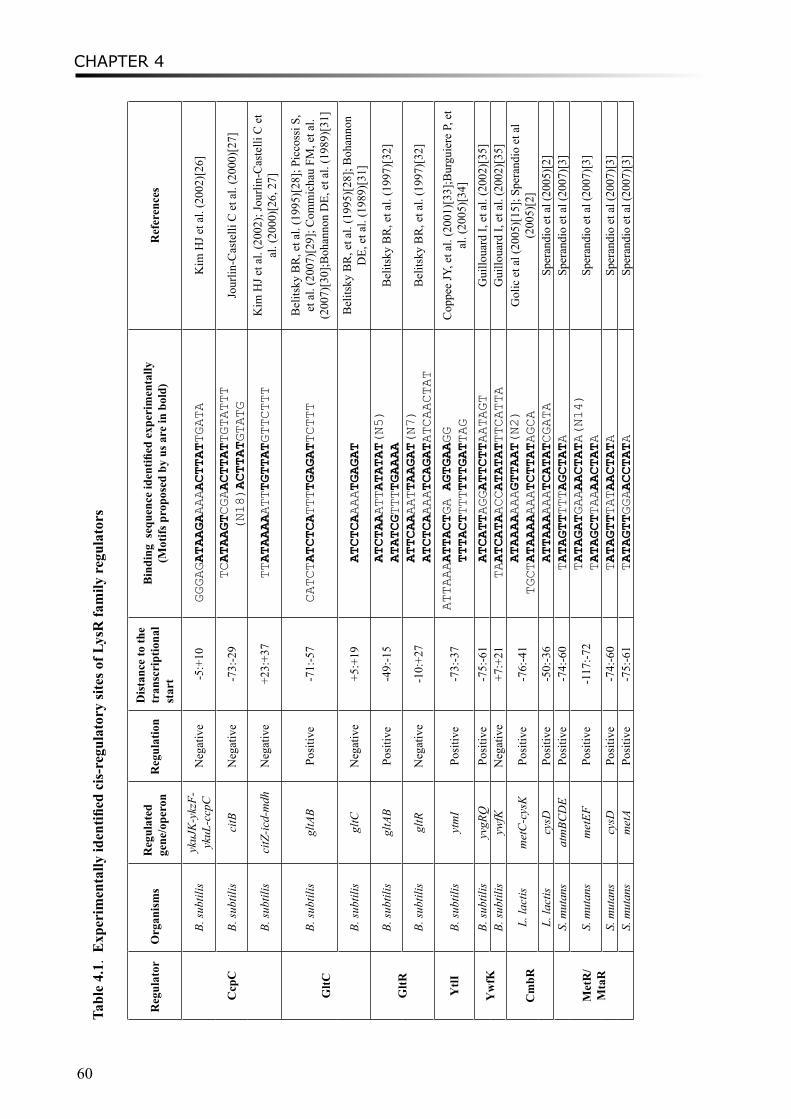
CHAPTER 4
60
Tabl
e 4.
1. E
xper
imen
tally
iden
tified
cis
-reg
ulat
ory
site
s of L
ysR
fam
ily r
egul
ator
s
Reg
ulat
orO
rgan
ism
sR
egul
ated
ge
ne/o
pero
nR
egul
atio
nD
ista
nce
to th
e tr
ansc
ript
iona
l st
art
Bin
ding
seq
uenc
e id
entifi
ed e
xper
imen
tally
(M
otifs
pro
pose
d by
us a
re in
bol
d)R
efer
ence
s
Ccp
C
B. su
btili
syk
uJK
-ykz
F-yk
uL-c
cpC
Neg
ativ
e-5
:+10
GGGAGATAAGAAAAACTTATTGATA
Kim
HJ e
t al.
(200
2)[2
6]
B. su
btili
sci
tBN
egat
ive
-73:
-29
TCATAAGTCGAACTTATTGTATTT
(N18)ACTTATGTATG
Jour
lin-C
aste
lli C
et a
l. (2
000)
[27]
B. su
btili
sci
tZ-ic
d-m
dhN
egat
ive
+23:
+37
TTATAAAAATTTGTTATGTTCTTT
Kim
HJ e
t al.
(200
2); J
ourli
n-C
aste
lli C
et
al. (
2000
)[26
, 27]
GltC
B. su
btili
sgl
tAB
Posi
tive
-71:
-57
CATCTATCTCATTTTGAGATTCTTT
Bel
itsky
BR
, et a
l. (1
995)
[28]
; Pic
coss
i S,
et a
l. (2
007)
[29]
; Com
mic
hau
FM, e
t al.
(200
7)[3
0];B
ohan
non
DE,
et a
l. (1
989)
[31]
B. su
btili
sgl
tCN
egat
ive
+5:+
19 ATCTCAAAATGAGAT
Bel
itsky
BR
, et a
l. (1
995)
[28]
; Boh
anno
n D
E, e
t al.
(198
9)[3
1]
GltR
B. su
btili
sgl
tAB
Posi
tive
-49:
-15
ATCTAAATTATATAT(N5)
ATATCGTTTTGAAAA
Bel
itsky
BR
, et a
l. (1
997)
[32]
B. su
btili
sgl
tRN
egat
ive
-10:
+27
ATTCAAAATTAAGAT(N7)
ATCTCAAAATCAGATATCAACTAT
Bel
itsky
BR
, et a
l. (1
997)
[32]
YtlI
B. su
btili
syt
mI
Posi
tive
-73:
-37
ATTAAAATTACTGA AGTGAAGG
TTTACTTTTTTTGATTAG
Cop
pee
JY, e
t al.
(200
1)[3
3];B
urgu
iere
P, e
t al
. (20
05)[
34]
Yw
fKB.
subt
ilis
yvgR
QPo
sitiv
e-7
5:-6
1 ATCATTAGGATTCTTAATAGT
Gui
lloua
rd I,
et a
l. (2
002)
[35]
B. su
btili
syw
fKN
egat
ive
+7:+
21 TAATCATAACCATATATTTCATTA
Gui
lloua
rd I,
et a
l. (2
002)
[35]
Cm
bRL.
lact
ism
etC
-cys
KPo
sitiv
e-7
6:-4
1 ATAAAAAAAGTTAAT(N2)
TGCTATAAAAAAATCTTATAGCA
Gol
ic e
t al (
2005
)[15
]; Sp
eran
dio
et a
l (2
005)
[2]
L. la
ctis
cysD
Posi
tive
-50:
-36
ATTAAAAAATCATATCGATA
Sper
andi
o et
al (
2005
)[2]
Met
R/
Mta
R
S. m
utan
sat
mBC
DE
Posi
tive
-74:
-60
TATAGTTTTTAGCTATA
Sper
andi
o et
al (
2007
)[3]
S. m
utan
sm
etEF
Posi
tive
-117
:-72
TATAGATGAAAACTATA(N14)
TATAGCTTAAAACTATA
Sper
andi
o et
al (
2007
)[3]
S. m
utan
scy
sDPo
sitiv
e-7
4:-6
0 TATAGTTTATAACTATA
Sper
andi
o et
al (
2007
)[3]
S. m
utan
sm
etA
Posi
tive
-75:
-61
TATAGTTGGAACCTATA
Sper
andi
o et
al (
2007
)[3]

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
61
CH
APT
ER 4
using the sequences of MetR from S. mutans, MtaR from S. agalactiae and CmbR from L. lactis as seeds. The clustering of protein sequences within the tree (Figure 4.2) clearly indicates that MetR of S. mutans and MtaR of S. agalactiae are orthologous and that CmbR is their closest relative.
CmbR
CmbR
[8][
[754]
[1000] OOE_116491070 LME_116617365
[681] LME_116618338
[952]
[656]6]56
[1000]
[1000]
[1000]
[632] STU_55820601 STH_55822491STM_116627404
[524] STA_146318438[1000] SPG_157150504
SPF01218[1000] LLX_15672313
[878] LLA_116511188[988] LLM_125623220 LLC_cmbR
[1000] STU_55820535[563] STM_116627344
STH_55822426
[1000]
[1000]
[989]
[1000] LLA_116512352 LLM_125623787 LLX01592
LLX02384 (pseudogene)
[739]
[1000] LBU_116513310 LDB_104773332
[463] LPL_28378514[1000] SAG_MtaR
[621] STA_146318666
[899]
[1000] SPG_157150437 SPF00013
[999] SMU_MetR[1000] STM_116627706
[1000] STH_55822834 STU_55820914
MetR/MtaR
LysR family
[
Other members from LysR family
Figure 4.2. Bootstrapped (n=1000) partial neighbor-joining tree of LysR family transcription regulators in LAB. Only the branches of the CmbR and MetR/MtaR subfamilies are shown. The CmbR subfamily and MetR/MtaR subfamily are highlighted. If known, GI codes follow the strain abbreviations (see legend to Figure 4.1). SAG for S. agalactiae, SMU for S. mutans, SPG for S. gordonii, SPF for S. pneumoniae, STA for S. suis. Experimentally validated regulatory proteins are indicated by dots following the names.
The LysR-family represents a common type of transcriptional regulators in prokaryotes, as the member proteins consist of a signal molecule binding domain and a helix-turn-helix (HTH) DNA-binding domain. The family contains a number of well-studied proteins, including AlsR, CcpC, CitR, GltR, YwfK and CmbR. Table 4.1 summarizes the experimentally characterized LysR-family members and the sequence of their DNA-binding site. A straightforward comparison of the reported binding sites clearly reveals a common motif structure of the cis-elements, namely ATNNNN---NNNNAT. The motif displays a dyad symmetry and a conserved spacing of three nucleotides. This motif agrees well with the observation made by Schell et al. [19] in 1993 that LysR-family members generally recognize a box with a conserved sequence T-N11-A, located 50-80 bp upstream of the transcriptional start sites. Surprisingly, the motifs that have been reported in later literature in some cases deviate strongly from this clear consensus.
In the following, we will describe the comparative analysis of the riboswitches and cis-regulatory elements in more detail. The findings are compared to earlier reports in literature and the implications of the observed discrepancies will be discussed. The resulting reconstruction of regulatory connections is summarized in Figure 4.1 for the various LAB, where the new findings are highlighted.
RiboswitchesRecently, comprehensive studies have appeared that describe the occurrence and evolution
of T-boxes among prokaryotes [5, 10, 20]. We have used the T-box HMMs of [10] to search newly sequenced LAB genomes and we identified twenty-three T-boxes associated with genes/operons involved in Met/Cys metabolism (supplemental Table S1). As a control we also scanned the LAB genomes included in the previous studies of Wels et al. [10] and Rodionov et al. [5, 10].

CHAPTER 4
62
The conservation of the ATG specifier codon in the multiple sequence alignment of all recovered T-boxes (see supplementary material File 3) implies that they all could respond to the absence of the same signal molecule, methionine. We found that genes cgs, metE, hom1, metA and cysD from L. plantarum and operons metA-cgs-cblB/cglB and metE-metF from Leuconostoc are regulated by a T-box like, in agreement with Wels et al. [10] and Rodionov et al. [5]. Also cysE, encoding a serine acetyltransferase was found to be preceded by a T-box in B. subtilis, as reported by [21]. New Met T-boxes were identified upstream of the genes cysD in Leuconostoc, metE and metA in the two sequenced L. bulgaricus strains, metH and metE in the two sequenced L. reuteri strains, as well as cgs and metE in L. casei (see supplemental Table S1).
In S-box transcriptional regulation, the folding of the leader region of the regulated gene is dependent on the effector molecule S-adenosylmethionine (SAM)[22, 23]. SAM-I riboswitches are extensively found upstream of genes involved in sulphur metabolism and transport in several Firmicutes, especially in Bacillales and Clostridia [5, 24], but they have not been reported in LAB. Correspondingly, no SAM-I riboswitches were detected by us upstream of genes involved in methionine and cysteine metabolism in LAB.
Transcriptional Regulator MetR/MtaR By previous in silico approaches, a 17-bp palindromic sequence TATAGTTtnaAACTATA,
named the MET box, was identified upstream of genes such as metY, metA, metQ, metI and the metEF operon in streptococci and upstream of the metEF operon in L. lactis [4, 5]. However, the regulatory protein which binds to the MET-box was yet to be identified. Rodionov et al. [5] first proposed that the transcriptional regulator MtaR, known to be involved in methionine uptake in S. agalactiae, fulfilled this role. A more recent study showed that MetR, which has 81% identity to MtaR, is the regulator protein that binds to the MET-box in S. mutans [3]. Indeed, MetR and MtaR can be inferred to fulfil the same role as they are orthologs (Figure 4.2). The MET-box motif was identified in the promoter regions of atmB, metE, cysD, metA, and smu.1487 in S. mutans and binding of MetR to these MET-boxes was confirmed with gel mobility shift assays and base substitutions in the MET-boxes [3] (Table 4.1).
We combined the experimentally identified binding sites and our in silico analyses to redefine the binding motif for MetR/MtaR in both lactococcal and streptococcal strains (Figure 4.3). The putative binding sites of MetR/MtaR show a palindromic structure, ATA-N9-TAT, which is typical for the LysR-family (see Table 4.1). The most conserved element ATAGTT is located upstream, whereas the downstream element N3-XXCTAT shows somewhat less conservation. As expected, the complete MetR/MtaR binding motif we thus define, ‘ATAGTT-N3-XXCTAT’, agrees almost completely with the previously defined MET-box, but it is two nucleotides shorter, in agreement with the LysR-family characteristic. As the recovery of putative binding sites in genome-wide searches is very sensitive to the precise composition of the search-motif, we prefer to use the newly defined motif.
A genome-wide search with the redefined MetR/MtaR binding motif recovered the operator binding sites from the upstream region of metH, metE, and metA in the various S. thermophilus genomes and, with the exception of metA, in the L. lactis genomes (Figure 4.1). The MetR/MtaR subfamily is also found in various other LAB including L. plantarum, L. bulgaricus, and Streptococcus species such as S. agalactiae, S. mutans, S. gordonii, S. pneumoniae, and S. suis. Indeed a MetR/MtaR-like motif (ATAGTT-N3-GAAATT) was found upstream of metE from L. bulgarius ATCC BAA365 (supplemental Table S2, Figure 4.1). However, in L. plantarum the MetR/MtaR motif has not been identified upstream of the genes associated in Met/Cys metabolism.
The MetR/MtaR binding sites are found in the upstream region of metH and metE genes in L. lactis and S. thermophilus strains (Figure 4.3), with a putative direct-repeat structure [3]. The first site is mostly around 60-70bp from the transcriptional start and usually displays an activation role when
MetH from StreptococciDirect Repeat of MetR/MtaR motif
MetE from Streptococci
0
1
2
bits
5‘
1A 2T 3A 4G 5T 6T 7TA
8A 9A
10
A
11
A
12
C
13
T
14
A
15
T
16
A
17
G
18
G
19
T
20
C
21
A
22
T
23
C
24
A
25
T
26
A
27
T
28
TC
29
A
30
A
31
A
32
A
33
A
34
TG
35
G
36
T
37
A
38
T
39
T
40
A
41
T3‘
MetE from L. lactisPutative Direct Repeat of MetR/MtaR motif
Direct Repeat of MetR/MtaR motif
HSST(metA) from Streptococci
0
1
2
bits
5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T 10TC
11T 12
C
13T 14A 15T 16
C
17T 18
C
19T 20T 21T 22
G
23A 24T 25A 26
G
27T 28T 29T 30T 31T 32T 33T 34A 35T 36T 37
G
38T 39
G
40A 41T 42A 43A 44
G
45
C
46T3‘
Streptococcus thermophilus
Streptococcus agalactiae
5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T
10
C
11
T
12
T
13
T
14
A
15
T
16
C
17
A
18
T
19
A
20
T
21
A
22
T
23
G
24
A
25
T
26
A
27
G
28
C
29
T
30
A
31
C
32
T
33
A
34
G
35
A
36
T
37
T
38
G
39
T
40
G
41
G
42
T
43
A
44
G
45
G
46
A
47
T3‘
Streptococcus suis
5‘
1A 2T 3A 4G 5T 6T 7T 8G 9A
10
A
11
G
12
C
13
T
14
A
15
T
16
A
17
A
18
C
19
A
20
C
21
C
22
T
23
G
24
G
25
A
26
T
27
T
28
G
29
A
30
A
31
C
32
C
33
A
34
T
35
G
36
G
37
T
38
A
39
T
40
G
41
A
42
T
43
A
44
G
45
A
46
A
47
T3‘
5‘
1 2T 3A 4G 5T 6T 7T 8A 9A
10
A
11
A
12
C
13
T
14
A
15
T
16
A
17
T
18
A
19
T
20
A
21
T
22
T
23
G
24
A
25
A
26
A
27
A
28
C
29
T
30
G
31
A
32
T
33
A
34
A
35
C
36
A
37
A
38
T
39
T
40
A
41
T3‘
A
5‘
1A 2T 3A 4G 5T 6T 7T 8C 9A
10
A
11
A
12
C
13
T
14
A
15
T
16
A
17
T
18
C
19
A
20
A
21
A
22
C
23
C
24
C
25
T
26
A
27
A
28
T
29
T
30
T
31
T
32
T
33
T
34
A
35
C
36
A
37
A
38
T
39
T
40
A
41
T3‘
Streptococcus thermophilus
Streptococcus pneumoniae
5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T
10
G
11
G
12
C
13
T
14
A
15
T
16
A
17
T
18
C
19
A
20
A
21
A
22
A
23
T
24
T
25
A
26
G
27
G
28
G
29
A
30
A
31
A
32
T
33
T
34
C
35
C
36
A
37
A
38
T
39
T
40
A
41
T3‘
Streptococcus gordonii
5‘
1
A
2
T
3
A
4
G
5
T
6
T
7
T
8
A
9
A
10
A
11
G
12
T
13
T
14
A
15
T
16
A
17
G
18
C
19
T
20
A
21
A
22
C
23
T
24
A
25
G
26
A
27
G3‘
Streptococcus thermophilus
5‘
1
A
2
T
3
A
4
G
5
T
6
T
7
T
8
C
9
A
10
G
11
G
12
C
13
T
14
A
15
T
16
A
17
C
18
C
19
T
20
A
21
G
22
G
23
T
24
A
25
A
26
A
27
G3‘
Streptococcus gordonii

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
63
CH
APT
ER 4
binding to the regulator [3]. The second putative binding site, which is closer to the transcriptional start, is far less conserved. Therefore, it is reasonable to assume that the second site has less affinity for the regulator compared to the upstream site. Considering the location of this downstream second site with respect to the promoter and its overlapping with the TATA box, binding of the transcription factor at this site probably results in repression of transcription. This organization has clear implications for the dynamics of MetR/MtaR-mediated transcriptional regulation of these genes. We hypothesize that MetR/MtaR activates the transcription when bound to the high-affinity upstream site, whereas when the concentration of the regulator in the cell is high enough, it will also bind to the second site and repress the transcription.
MetH from StreptococciDirect Repeat of MetR/MtaR motif
MetE from Streptococci
0
1
2
bits
5‘
1A 2T 3A 4G 5T 6T 7TA
8A 9A
10
A
11
A
12
C
13
T
14
A
15
T
16
A
17
G
18
G
19
T
20
C
21
A
22
T
23
C
24
A
25
T
26
A
27
T
28
TC
29
A
30
A
31
A
32
A
33
A
34
TG
35
G
36
T
37
A
38
T
39
T
40
A
41
T3‘
MetE from L. lactisPutative Direct Repeat of MetR/MtaR motif
Direct Repeat of MetR/MtaR motif
HSST(metA) from Streptococci
0
1
2
bits
5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T 10TC
11T 12
C
13T 14A 15T 16
C
17T 18
C
19T 20T 21T 22
G
23A 24T 25A 26
G27T 28T 29T 30T 31T 32T 33T 34A 35T 36T 37
G
38T 39
G
40A 41T 42A 43A 44
G
45
C
46T3‘
Streptococcus thermophilus
Streptococcus agalactiae
5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T
10
C
11
T
12
T
13
T
14
A
15
T
16
C
17
A
18
T
19
A
20
T
21
A
22
T
23
G
24
A
25
T
26
A
27
G
28
C
29
T
30
A31
C32
T33
A34
G35
A36
T37
T38
G39
T40
G41
G
42
T
43
A
44
G
45
G
46
A
47
T3‘
Streptococcus suis
5‘
1A 2T 3A 4G 5T 6T 7T 8G 9A
10
A
11
G
12
C
13
T
14
A
15
T
16
A
17
A
18
C
19
A
20
C
21
C
22
T
23
G
24
G
25
A
26
T
27
T
28
G
29
A
30
A
31
C
32
C
33
A
34
T
35
G
36
G37
T38
A39
T40
G41
A42
T43
A44
G45
A46
A47
T3‘
5‘
1 2T 3A 4G 5T 6T 7T 8A 9A
10
A
11
A
12
C
13
T
14
A
15
T
16
A
17
T
18
A
19
T
20
A
21
T
22
T
23
G
24
A
25
A
26
A
27
A
28
C
29
T
30
G
31
A
32
T
33
A
34
A
35
C
36
A
37
A
38
T
39
T
40
A
41
T3‘
A
5‘
1A 2T 3A 4G 5T 6T 7T 8C 9A
10
A
11
A
12
C
13
T
14
A
15
T
16
A
17
T
18
C
19
A
20
A
21
A
22
C
23
C
24
C
25
T
26
A
27
A
28
T
29
T
30
T
31
T
32
T
33
T
34
A
35
C
36
A
37
A
38
T
39
T
40
A
41
T3‘
Streptococcus thermophilus
Streptococcus pneumoniae
5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T
10
G
11
G
12
C
13
T
14
A
15
T
16
A
17
T
18
C
19
A
20
A
21
A
22
A
23
T
24
T
25
A
26
G
27
G
28
G
29
A
30
A
31
A
32
T
33
T
34
C
35
C
36
A
37
A
38
T
39
T
40
A
41
T3‘
Streptococcus gordonii
5‘
1
A
2
T
3
A
4
G
5
T
6
T
7
T
8
A
9
A
10
A
11
G
12
T
13
T
14
A
15
T
16
A
17
G
18
C
19
T
20
A
21
A
22
C
23
T
24
A
25
G
26
A
27
G3‘
Streptococcus thermophilus
5‘
1
A
2
T
3
A
4
G
5
T
6
T
7
T
8
C
9
A
10
G
11
G
12
C
13
T
14
A
15
T
16
A
17
C
18
C
19
T
20
A
21
G
22
G
23
T
24
A
25
A
26
A
27
G3‘
Streptococcus gordonii
Figure 4.3. MetR/MtaR binding sites in Lactococcus and Streptococcus. The sites are located upstream of metH and metA in the Streptococci as well as upstream of metE in both the Streptococcus and Lactococcus strains (The displayed sequences represent sequences at the bacterial species level). The actual putative binding sites are indicated in grey.
Transcriptional regulator CmbR A second member of the LysR-family, CmbR, was found to positively regulate the metC-cysK
operon in L. lactis. In fact, eighteen genes of L. lactis in the pathways for cysteine and methionine metabolism are controlled by this regulator [2]. These genes, which are clustered in several transcriptional units, include cysD, cysM (cysK homolog), and the operons metB2(cblB/cglB)-cysK and metA-metB1(cgs)-ytjE(cblA/cglA) [2]. It was shown that binding of CmbR to the promoters in L. lactis is stimulated by O-acetyl-L-serine and low cysteine and methionine concentrations [15, 25].

CHAPTER 4
64
As CmbR belongs to the LysR-family of transcription regulators, one would expect its binding site to have a common LysR-family motif. However, each of the studies into CmbR binding to date has resulted in a distinctly different CmbR-binding motif prediction. In the first study, a deletion analysis showed that a direct repeat of ATAAAAAAA is required for metC activation by CmbR [15]. In the second study, the upstream regions of seven transcriptional units found to be regulated by CmbR in L. lactis were analyzed. A first consensus binding sequence TWAAAAATTNNTA was proposed, centered 46 to 53 bp upstream of the transcriptional start, with a second consensus TWAAAWANNTNNA, located 8 to 10 bp upstream [2]. The proposed boxes for CmbR binding of the metB2 and cysD gene promoter regions were confirmed by gel-shift experiments [2, 15]. The recent publication by Kovaleva et al. [4] describes an in silico analysis of upstream regions of cysteine biosynthesis genes in streptococcal species and defines the CmbR-binding motif as TGATA-N9-TATCA-N2-4-TGATA.
[433]
[654]
[788]
[1000]
[1000][1.1.1] LLM_125624565
[1.1.1] LLX_15672764[1.1.1] LLA_116511591
[1000][1.1.2] LLX_15672522
[1000] [1.1.2] LLM_125623375[1.1.2] LLA_116511337
[1000][1.1] STM_116627279
[1000] [1.1] STH_55822343[1.1] STU_55820459
CysK family CBL/CGL CysK1
1,766,725 1,746,725
(lyase group)
782,352 802,352
765,214 785,214
Other LAB
536,730
510,448
515,104
365,216
356,672
364,138
516,730
490,448
495,104
345,216
336,672
344,138
CysK2(CysM)
CysM1
cblB/cglB-cysK1
cysK2(cysM)
′
cysM1
Lactococcus lactis
Lactococcus lactis
Streptococcus species
0
1
2
bits
5‘
1A 2T 3A 4A 5A 6A 7A 8T 9T
10
T
11
T
12
C
13
T
14
G
15
GA
16
AC
17
G
18
A
19
T
20
T
21 22
T
23
G
24
T
25
A
26
A
27
A
28
G3‘
0
1
2
bits
5‘
1A 2T 3A 4A 5A 6A 7A 8A 9TA
10AG
11T 12T 13A 14A 15GT
16T 17
C
18T 19
G
20AC
21CT
22A 23T 24A 25GA
26CA
27A 28TA
29TA
30TA
31T 32
C33T 34T 35A 36T
3‘
5‘
1A 2T 3A 4G 5T 6C 7A 8A 9A 10
C
11A 12T 13T 14A 15T 16
C
17A 18
C
19A 20
G
21T 22
G
23A 24T 25A 26A 27A 28
G
29A 30A 31
G
32A 33T 34
G
35T 36A 37T3‘
Streptococcus thermophilus
5‘
1A 2T 3A 4A 5A 6T 7A 8A 9T
10
A
11
G
12
C
13
T
14
A
15
T
16
C
17
A
18
C
19
T
20
T
21
G
22
A
23
T
24
A
25
A
26
A
27
G
28
A
29
A
30
T
31
T
32
G
33
A
34
T
35
A
36
T3 ‘
Streptococcus agalactiae
5‘
1A 2T 3A 4A 5A 6A 7A 8A 9A
10
A
11
G
12
C
13
T
14
A
15
T
16
G
17
A
18
T
19
A
20
G
21
C
22
C
23
A
24
T
25
A
26
G
27
C
28
T
29
T
30
T
31
T
32
T
33
T
34
A
35
T
36
A
37
T3‘
Streptococcus gordonii
Figure 4.4. Identification of putative CmbR binding sites in the upstream region of cysK and paralogous/orthologous genes involved in sulphur amino acids metabolism in Lactococcus and Streptococcus. A partial bootstrapped (n=1000) neighbour-joining tree of the CysK family is shown together with the corresponding gene context. Putative binding sites were identified in the conserved upstream regions of each sub-cluster and are indicated in grey.
To resolve the discrepancies resulting from the previous analyses, we re-analyzed the reported CmbR-binding sites in L. lactis and Streptococcus species. We identified consensus binding sequences in the upstream region of the CmbR-regulated cysK gene and its paralogs/orthologs from lactococcal and streptococcal genomes (Figure 4.4). Since three sub-clusters of cysK can be distinguished by phylogeny and gene context, a putative CmbR-binding site was identified for each sub-cluster (Figure 4.4). In case of the cysM1 cluster, CmbR-binding sites from other streptococci such as S. agalactiae and S. gordonii were also identified. As Figure 4.4 clearly shows, all the putative CmbR-binding sites in both Lactococcus and Streptococcus can be summarized by a motif of dyad symmetry ATA-N9-TAT, with a preference for a long stretch of adenines following the starting “ATA” (Figure 4.4). This putative CmbR binding motif has the general LysR family signature and, as importantly, all the previous experimental work on CmbR binding fits well into this assignment (Table 4.1).

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
65
CH
APT
ER 4
In the phylogenetic tree, the CmbR subfamily is most closely related to the MetR/MtaR subfamily (Figure 4.2). Therefore, one might expect that their binding sites resemble each other. Although both putative binding motifs have the LysR family signature, the conserved ATA followed by adenines that constitutes the first part of the inverted repeat binding site for CmbR (Figure 4.4) is remarkably different from the highly conserved ATAGTT in the first part of the inverted repeat binding site for MetR/MtaR (Figure 4.3).
On basis of this consensus CmbR-binding site, we have identified additional putative CmbR-binding sites in the region upstream of cgs, metA, and cysD from Lactococcus strains, and cysD from Streptococcus thermophilus strains (Figure 4.1). However, CmbR binding sites were not detected in Lactobacillus genomes, which agrees completely with the absence of a CmbR ortholog in their genomes (Figure 4.2).
Novel cis-regulatory motifs upstream of cbs and thrB genesThe comparative analysis of the upstream regions also revealed several new putative binding
sites, previously not reported (see Figure 4.1 and supplemental Table S2). For example, the cbs-cblB/cglB operon in various lactobacilli strains such as L. salivarius, L. plantarum, L. bulgaricus, L. acidophilus, as well as O. oeni, Leuconostoc and S. thermophilus strains are preceded by clear conserved binding sites. The cbs gene encoding cystathionine beta-synthase gene (CBS) from LAB has a relatively conserved gene context. Gene cbs together with cblB/cglB, and sometimes also with cysE are located in the same operon in L. acidophilus, L. bulgaricus, L. plantarum, Oenococcus oeni, Leuconostoc mesenteroides and S. thermophilus. The upstream regions of the operon from these organisms were used for motif analysis (Figure 4.5A). A novel putative regulatory binding site was identified, which contains a clear palindrome AAAGGGCGCGAA-N(11-18)-TTCGCGCCtTTT.
In the multiple sequence alignment of the upstream regions of thrB encoding homoserine kinase (HSK) from L. acidophilus and L. gasseri, another new conserved motif was observed (Figure 4.5B). The putative motif consists of inverted repeats separated by 15 bp (ATTGTAAC-N15-GTTACAAT). Interestingly, each part of the inverted repeat complements itself (e.g. in the first part, ATTG is followed immediately by its complement sequence TAAC). Remarkably, the 15-bp spacer causes a separation of 21-22 bp between the centre of the first element and the centre of the second element. This means a separation of exactly two full helical turns as illustrated in Figure 4.5B, implying that a possible dimer structure of the regulator could bind to the DNA sequences in the same direction.
ConclusionsWe have performed a genome-wide in silico analysis on the transcription regulation mechanisms
which control cysteine and methionine metabolism. We found clear diversity in the transcriptional regulation of the genes that encode the enzymes involved in methionine/cysteine metabolism in different LAB species. The associated regulators include known regulatory proteins such as CmbR and MetR/MtaR, as well as unknown regulators, but also RNA riboswitches. A complete overview of the findings is given in Figure 4.1. Three model LAB strains, i.e. L. plantarum, L. lactis and S. thermophilus are shown as examples. In L. plantarum, the biosynthesis of methionine, as well as its precursors i.e. homocysteine and cystathionine are mainly regulated by post-transcription initiation by T-box riboswitches, whereas the synthesis of cysteine from homocysteine and degradation of both cysteine and methionine are regulated by as yet unknown transcription factors (Figure 4.1). In L. lactis, most of the genes encoding enzymes in these pathways are regulated by CmbR and some unknown regulators, except for MetE, the enzyme catalyzing the last step of the methionine biosynthesis route, which is regulated by the transcriptional regulator MetR/MtaR. The Met/Cys metabolic pathway in S. thermophilus is regulated by MetR/MtaR and CmbR, as well as by some unknown transcription

CHAPTER 4
66
factors. In lactococci and streptococci, the production of flavour compounds, e.g. H2S, methanethiol, DMS, DMDS, DMTS, catalyzed by cystathionine beta/gamma lyase (CBL/CGL) or cysteine synthase (CysK), are mainly regulated by CmbR (Figure 4.1).
The specific binding motifs of CmbR and MetR/MtaR in lactococci and streptococci were redefined with respect to previous literature. Both motifs follow the characteristic LysR family signature (ATA-N9-TAT) and agree well with experimental observations (Table 4.1).
This insight into the regulatory mechanisms of sulphur-containing amino acids could contribute to the construction of a dynamic bacterial metabolic network, and ultimately lead to development of new strategies for controlling the sulphuric flavour formation in various LAB-fermented food products, for example by modulating the expression of the relevant regulators.
AcknowledgementsThis work was supported by grant CSI4017 of the Casimir program of the Ministry of Economic Affairs, The Netherlands.
LSLCBS CBL/CGL (lyase group)
22,279 42,279
OOE 1,690,039 1,670,039
LME 1,267,171 1,287,171
LPL 241,255 221,255
Lb. bulgaricus 1,118,341 1,138,341
S. thermophilus 774,693 794,693
LAC 1,057,074 1,077,074
CysE
0
1
2
stib
5′
1CGA
2ACT
3TA
4CA
5TA
6TG
7CG
8AG
9C 0 1
AG
11
AC
21
A
TG
31
GA
41
GA
51
G
AT
61
CTA
71
A
TC
81
AT
91 0 2
ACT
12
T
GA
22
AG
32
A 4 2
T 52
T 6 2
T 72
TC
82 92
TAG
03
GA
1 3
GTA
23GAT
33CT
4 3
CT
53
GC
63
G 73
TC
83
G 93
TC
04
TC
14
CT
24
T 3 4
T 4 4
T3′
inverted repeat
cbs-cblB/cglB Common motif
ATAC ATCA0
1
2
bits
5‘
1CT
2CT
3A 4T 5T 6G 7T 8A 9GA
10
C
11
TC
12
T
13
AG
14
AT
15
T
16
T
17
AT
18
TA
19
GC
20 21
AC
22
TC
23
CT
24
AG
25
T
26
G
27
AT
28 29 30 31 32 33 34 35 3‘
TAACATTG CAATGTTA
ATTGTAAC GTTACAAT 22nt
thrB motif
A
B
Figure 4.5. (A) A novel binding motif identified upstream of the cbs-cblB/cglB-(cysE) operon in LAB. Gene context of the operon in various LAB is shown. The cluster cbs-cblB/cglB-cysE of S. thermophilus is probably horizontally transferred from L. bulgaricus [36].The inverted repeat structure within the identified motif is indicated by arrows. (B) A novel binding motif identified upstream of thrB in LAB. The motif contains a palindrome (indicated by red arrows) and each part of the palindrome is a direct repeat of compliment sequences (indicated by blue arrows beneath numbers). The putative location of the identified motif in the DNA double helix structure for binding the regulatory protein is shown in the lower panel of the figure.

REGULATION OF CYSTEINE AND METHIONINE METABOLISM IN LAB
67
CH
APT
ER 4
References
1. Hullo MF, Auger S, Soutourina O, Barzu O, Yvon M, Danchin A, Martin-Verstraete I: Conversion of methionine to cysteine in Bacillus subtilis and its regulation. J Bacteriol 2007, 189:187-197.
2. Sperandio B, Polard P, Ehrlich DS, Renault P, Guedon E: Sulfur amino acid metabolism and its control in Lactococcus lactis IL1403. J Bacteriol 2005, 187:3762-3778.
3. Sperandio B, Gautier C, McGovern S, Ehrlich DS, Renault P, Martin-Verstraete I, Guedon E: Control of methionine synthesis and uptake by MetR and homocysteine in Streptococcus mutans. J Bacteriol 2007, 189:7032-7044.
4. Kovaleva GY, Gelfand MS: Transcriptional regulation of the methionine and cysteine transport and metabolism in streptococci. FEMS Microbiol Lett 2007, 276:207-215.
5. Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS: Comparative genomics of the methionine metabolism in Gram-positive bacteria: a variety of regulatory systems. Nucleic Acids Res 2004, 32:3340-3353.
6. Liu M, Nauta A, Francke C, Siezen RJ: Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria. Appl Environ Microbiol 2008, 74:4590-4600.
7. Overbeek R, Larsen N, Walunas T, D’Souza M, Pusch G, Selkov E, Liolios K, Joukov V, Kaznadzey D, Anderson I et al: The ERGO (TM) genome analysis and discovery system. Nucleic Acids Res 2003, 31:164-171.
8. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al: Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23:2947-2948.
9. Durbin R, Eddy S, Krogh A, and Mitchison G: Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids: Cambridge University Press; 1998.
10. Wels M, Groot Kormelink T, Kleerebezem M, Siezen RJ, Francke C: An in silico analysis of T-box regulated genes and T-box evolution in prokaryotes, with emphasis on prediction of substrate specificity of transporters. BMC Genomics 2008, 9:330.
11. Francke C, Kerkhoven R, Wels M, Siezen RJ: A generic approach to identify Transcription Factor-specific operator motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics 2008, 9:145.
12. de Been M, Bart MJ, Abee T, Siezen RJ, Francke C: The identification of response regulator-specific binding sites reveals new roles of two-component systems in Bacillus cereus and closely related low-GC Gram-positives. Environ Microbiol 2008, 10:2796-2809.
13. Kerkhoven R, van Enckevort FH, Boekhorst J, Molenaar D, Siezen RJ: Visualization for genomics: the Microbial Genome Viewer. Bioinformatics 2004, 20:1812-1814.
14. Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS: MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 2009, 37:W202-208.
15. Golic N, Schliekelmann M, Fernandez M, Kleerebezem M, van Kranenburg R: Molecular characterization of the CmbR activator-binding site in the metC-cysK promoter region in Lactococcus lactis. Microbiology 2005, 151:439-446.
16. Epshtein V, Mironov AS, Nudler E: The riboswitch-mediated control of sulfur metabolism in bacteria. Proc Natl Acad Sci U S A 2003, 100:5052-5056.
17. Tucker BJ, Breaker RR: Riboswitches as versatile gene control elements. Curr Opin Struct Biol 2005, 15:342-348.18. Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS: Riboswitches: the oldest mechanism for the regulation of gene
expression? Trends Genet 2004, 20:44-50.19. Schell MA: Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol 1993, 47:597-626.20. Gutierrez-Preciado A, Henkin TM, Grundy FJ, Yanofsky C, Merino E: Biochemical features and functional implications
of the RNA-based T-box regulatory mechanism. Microbiol Mol Biol Rev 2009, 73:36-61.21. Pelchat M, Lapointe J: In vivo and in vitro processing of the Bacillus subtilis transcript coding for glutamyl-tRNA
synthetase, serine acetyltransferase, and cysteinyl-tRNA synthetase. RNA 1999, 5:281-289.22. Grundy FJ, Henkin TM: The S box regulon: a new global transcription termination control system for methionine and
cysteine biosynthesis genes in gram-positive bacteria. Mol Microbiol 1998, 30:737-749.23. McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM: Transcription termination control of the S box system: direct
measurement of S-adenosylmethionine by the leader RNA. Proc Natl Acad Sci U S A 2003, 100:3083-3088.24. Auger S, Yuen WH, Danchin A, Martin-Verstraete I: The metIC operon involved in methionine biosynthesis in Bacillus
subtilis is controlled by transcription antitermination. Microbiology 2002, 148:507-518.25. Fernandez M, Kleerebezem M, Kuipers OP, Siezen RJ, van Kranenburg R: Regulation of the metC-cysK operon, involved
in sulfur metabolism in Lactococcus lactis. J Bacteriol 2002, 184:82-90.26. Kim HJ, Jourlin-Castelli C, Kim SI, Sonenshein AL: Regulation of the bacillus subtilis ccpC gene by ccpA and ccpC. Mol
Microbiol 2002, 43:399-410.27. Jourlin-Castelli C, Mani N, Nakano MM, Sonenshein AL: CcpC, a novel regulator of the LysR family required for
glucose repression of the citB gene in Bacillus subtilis. J Mol Biol 2000, 295:865-878.28. Belitsky BR, Janssen PJ, Sonenshein AL: Sites required for GltC-dependent regulation of Bacillus subtilis glutamate
synthase expression. J Bacteriol 1995, 177:5686-5695.29. Picossi S, Belitsky BR, Sonenshein AL: Molecular mechanism of the regulation of Bacillus subtilis gltAB expression by
GltC. J Mol Biol 2007, 365:1298-1313.30. Commichau FM, Herzberg C, Tripal P, Valerius O, Stulke J: A regulatory protein-protein interaction governs glutamate
biosynthesis in Bacillus subtilis: the glutamate dehydrogenase RocG moonlights in controlling the transcription factor

CHAPTER 4
68
GltC. Mol Microbiol 2007, 65:642-654.31. Bohannon DE, Sonenshein AL: Positive regulation of glutamate biosynthesis in Bacillus subtilis. J Bacteriol 1989,
171:4718-4727.32. Belitsky BR, Sonenshein AL: Altered transcription activation specificity of a mutant form of Bacillus subtilis GltR, a
LysR family member. J Bacteriol 1997, 179:1035-1043.33. Coppee JY, Auger S, Turlin E, Sekowska A, Le Caer JP, Labas V, Vagner V, Danchin A, Martin-Verstraete I: Sulfur-
limitation-regulated proteins in Bacillus subtilis: a two-dimensional gel electrophoresis study. Microbiology 2001, 147:1631-1640.
34. Burguiere P, Fert J, Guillouard I, Auger S, Danchin A, Martin-Verstraete I: Regulation of the Bacillus subtilis ytmI operon, involved in sulfur metabolism. J Bacteriol 2005, 187:6019-6030.
35. Guillouard I, Auger S, Hullo MF, Chetouani F, Danchin A, Martin-Verstraete I: Identification of Bacillus subtilis CysL, a regulator of the cysJI operon, which encodes sulfite reductase. J Bacteriol 2002, 184:4681-4689.
36. Liu M, Siezen RJ, Nauta A: In silico prediction of horizontal gene transfer events in Lactobacillus bulgaricus and Streptococcus thermophilus reveals protocooperation in yogurt manufacturing. Appl Environ Microbiol 2009, 75:4120-4129.
Supplemental FilesSupplemental Table S1. T-box regulated genes that are involved in cysteine and methionine metabolism in LAB.Supplemental Table S2. Diversity of transcriptional regulatory mechanisms of the genes encoding enzymes involved in cysteine and methionine metabolism in LAB. The table includes the identified binding motifs for both known and unknown transcription factors, as well as for T-box riboswitches.Supplemental File 3. Multiple sequence alignment of Met T-box sequences from LAB and other Firmicutes. The specifier codons are highlighted, indicating that those T-boxes are methionine-specific.

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
69
CH
APT
ER 5
Chapter 5
COMBINING CHEMOINFORMATICS WITH BIOINFORMATICS: IN SILICO PREDICTION OF FLAVOR-FORMING PATHWAYS
BY A CHEMICAL SYSTEMS BIOLOGY APPROACH “REVERSE PATHWAY ENGINEERING”
Mengjin Liu , Bruno Bienfait, Oliver Sacher, Johann Gasteiger, Arjen Nauta, Roland J. Siezen, Jan M.W. Geurts
Manuscript submitted

CHAPTER 5
70
AbstractThe incompleteness of genome-scale metabolic models is a major bottleneck for systems biology calculations. High-throughput technologies, e.g. metabolomics, can identify and quantify huge numbers of metabolites. Many of those metabolites, such as flavor compounds, are considered as non-essential, and their synthesis pathways are relatively less studied and only partially known. In this study, we describe a novel approach, named Reverse Pathway Engineering (RPE)1, which combines chemoinformatics and bioinformatics analyses, to predict the missing links between target compounds and their possible metabolic precursors by providing plausible chemical or enzymatic reactions. We demonstrate the usability of the approach by exploring flavor-forming pathways in lactic acid bacteria. As validation of the approach, known metabolic routes leading to the formation of flavor compounds from leucine were successfully proposed. Unknown flavor-forming reactions, i.e. the conversion of α-hydroxy-isocaproate to 3-methylbutanoic acid and the synthesis of dimethyl sulfide, as well as the corresponding enzymes were identified. The new insights into the flavor-formation mechanisms in LAB will have significant impact on improving the control of aroma formation in fermented food products. Since the input reaction databases and compounds are highly flexible, the RPE approach can be easily extended to a broader spectrum of applications, ranging from health or disease biomarker discovery to synthetic biology.
IntroductionChemical systems biology, a new interdisciplinary field between chemical biology and systems
biology, is currently drawing more and more attention (1). One direction that has emerged is the development of novel strategies in which chemoinformatic and bioinformatic tools are integrated to interpret large high-throughput datasets.
A classical systems biology approach for analyzing cellular processes of microorganisms starts with the reconstruction of biochemical networks based on annotated genomes(2,3). However, those genome-scale metabolic models contain numerous gaps, only a small portion of which can be filled by computational methods or manual curation on the basis of known genetic/biochemical data(4). High-throughput analytical technologies for metabolomics, such as mass spectrometry (MS) and nuclear magnetic resonance spectroscopy (NMR), provide large data sets of chemical compounds from various biological systems(5). Many of these compounds, particularly the products that are non-essential in metabolism (e.g. flavor compounds), cannot be mapped in the metabolic models due to the lack of biochemical and/or genetic information. In many cases, these secondary metabolites or their derivatives can be of great economical value and/or possess potential health benefits(6). Therefore, insight into their precursors and synthetic pathways could have a great impact on the control of their production.
Flavor components in various fermented foods can be produced by lactic acid bacteria (LAB), which are present in starter or adjunct cultures. Several reviews have summarized the flavor-forming pathways of LAB, especially the pathways from amino acids (7-10). However, the flavor-forming network is presumed to be more complex, and previous studies have only revealed small parts of this network. The known flavor-forming pathways still do not meet the requirements to establish a comprehensive, curated predictive model for flavor formation. It happens quite often that the measured aroma compounds, for instance by gas chromatography-mass spectrometry (GC-MS), cannot be mapped to any known flavor-forming pathway. One of the reasons for this is that chemical reactions play an important role in flavor formation. Chemical reactions are usually not included in the genome-scale metabolic models (2,11).
In order to solve the above mentioned problems, we developed a novel approach named Reverse

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
71
CH
APT
ER 5
Pathway Engineering (RPE), which uses small molecules i.e. measured flavor compounds as the input, and suggests the enzymatic or chemical reactions connecting them to known metabolic precursors, thereby predicting the production routes that synthesize the target compounds.
In this paper, we present a use case of the RPE approach for predicting flavor-forming reactions/ pathways, including those from leucine and methionine catabolism. Since the branched-chain amino acid degradation pathways, in particular the leucine degradation pathway, are relatively well studied (12-15), some routes from this pathway were used for validating the method. Novel reactions or alternative routes are proposed, such as the conversion of α-hydroxy-isocaproate to 3-methylbutanoic acid and the chemical reaction to form 2-methylpropanal from α-ketoisocaproate. In addition, an enzymatic reaction to synthesize dimethyl sulfide from methanethiol in the methionine degradation pathway is proposed. Using bioinformatics analyses, plausible enzymes encoded in the genomes of LAB have been identified for catalyzing the predicted enzymatic reactions.
Materials and Methods
Flavor compounds The flavor compounds used as inputs for the RPE approach were selected based on descriptions
from literature or from measurements by GC-MS(7-10,16). In this paper, the studied flavor compounds include 3-methylbutanol, 3-methylbutanoic acid, 2-methylbutyric acid, 2-methylpropanal, and dimethyl sulfide. The structures of the target compounds were obtained from the NCBI PubChem database (http://pubchem.ncbi.nlm.nih.gov/).
Reaction database The BioPath.Database (17) (Molecular Networks GmbH) is a manually curated biochemical
reaction database, where the reactions are stoichiometrically balanced, and where the reaction centres, as well as the bonds broken and formed during a reaction, have been marked (http://www.molecular-networks.com/databases/biopath.html). It contains reactions originally derived from the Roche Applied Science “Biochemical Pathways” wall chart and from a more extensive monograph (18). In particular, for the examples of predicting flavor-forming pathways described in this paper, an extended version of BioPath.Database has been utilized, with 438 additional reactions extracted from a Lactobacillus plantarum genome-scale metabolic model(19). Moreover, 24 reactions were added from the flavor-forming pathways from sulfur-containing amino acids, including both enzymatic and chemical reactions(20). After removing the redundancy, the database contains 3,516 reactions in total, as the resource of reference reactions.
Retrieval of flavor-forming reactions/pathways In order to predict the unknown reactions or pathways leading to flavor formation, a software
package called THERESA (THE REtroSynthetic Analyser) for designing organic syntheses was used (http://www.molecular-networks.com/software/theresa/index.html). THERESA derives a large part of its knowledge from a reaction database. The extended BioPath.Database described above was utilized as the reaction pool. The combined system is called BioPath.Design because it allows the design of biotechnological processes. For each reaction of the BioPath.Database, BioPath.Design extracted automatically the reaction centers plus the transformation steps and then used them for RPE analyses. After the target structure was submitted, BioPath.Design searched its database of biotransformation rules for all reactions that could be applied to the target structure in the reverse direction (retrosynthesis). If a substructure match was identified, BioPath.Design could build a new complete synthesis reaction by adding the missing co-product(s), copying the atom-atom mapping numbers of the reaction centers to the target compound and generating the structure of the reactants.

CHAPTER 5
72
The predicted reactions were ranked based on the presence of reactants and predicted reactions in the database, as well as the simplicity of structures. The predicted reactions were manually inspected and a candidate reaction was selected based on the information of the corresponding reference reactions found in the database or on prior knowledge. When one of the plausible synthesis reactions was selected, the reactant could be used as the input compound to search for its precursor. This process was repeated iteratively until a known metabolic precursor was found and a complete synthetic route was retrieved. A detailed description of THERESA, with examples of RPE predictions, can be found in the Supplemental Methods.
Identification of putative enzymes catalyzing the plausible reactions in LAB by comparative genomics
After a plausible reaction was proposed by BioPath.Design, a list of candidate enzymes which might catalyze the reaction was prepared on the basis of the reference reaction in the BioPath.Database or by inferring from the transformation rule of the predicted reaction(21). Detailed enzyme information, including literature references and sequence information when available, was obtained from the BRENDA database (22) by searching with the EC number or the enzyme name.
We used our previously described comparative genomics approach to identify putative enzymes for reactions (20). In brief, sequences of the target enzymes, which have been characterized experimentally, are obtained from the UniProt/Swiss-Prot database (http://www.uniprot.org/). The genomes of the sequenced LAB and other microorganisms used in this study were obtained from the non-redundant genome databases, the NCBI microbial genome database (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi) and/or the ERGO database(23). The retrieved protein sequence was used as a seed to perform a BLAST search against the microbial genomes to obtain their homologs. Multiple sequence alignments (MSAs) were created by MUSCLE (24), based on the homologous sequences of the target enzymes, as well as the sequences of the experimentally characterized enzymes from literature. Bootstrapped neighbor-joining phylogenetic trees were constructed within ClustalW(25) based on the MSAs and visualized by LOFT(26). Orthologs of the putative enzymes in LAB genomes were identified by phylogenetic analysis. The resulting enzymatic information was integrated into the retrosynthesized reactions and pathways for further analyses or applications. The outcome of a comparative genomics analysis was also used as an indication for verifying the predicted reaction. For example, if no ortholog is found in the target organism which had shown to possess certain enzyme activities, the selected reaction is less likely to be correct.
Results
Reverse pathway engineering approachTo close the gaps between the target chemical compounds and their metabolic precursors, we
developed a novel approach named Reverse Pathway Engineering. The pipeline of the RPE approach, combining the scientific disciplines chemo- and bioinformatics, is shown in Figure 5.1.
The chemoinformatics analyses are carried out by a software system named BioPath.Design, which comprises a reaction database and a retrosynthesis tool called THERESA. Since the unrevealed reactions in metabolic pathways are usually not deposited in any biochemical or chemical database, a search will not retrieve the desired reaction. However, known enzymatic and chemical reactions can be used for generating a pool of transformation rules. BioPath.Design suggests synthesis reactions, including novel ones, by using the biotransformation rules based on those stored in a manually curated reaction database. Moreover, the reaction database of BioPath.Design can be adjusted according to specific tasks, in our case an extended biochemical reaction database named BioPath.Database (upper panel in Figure 5.1).
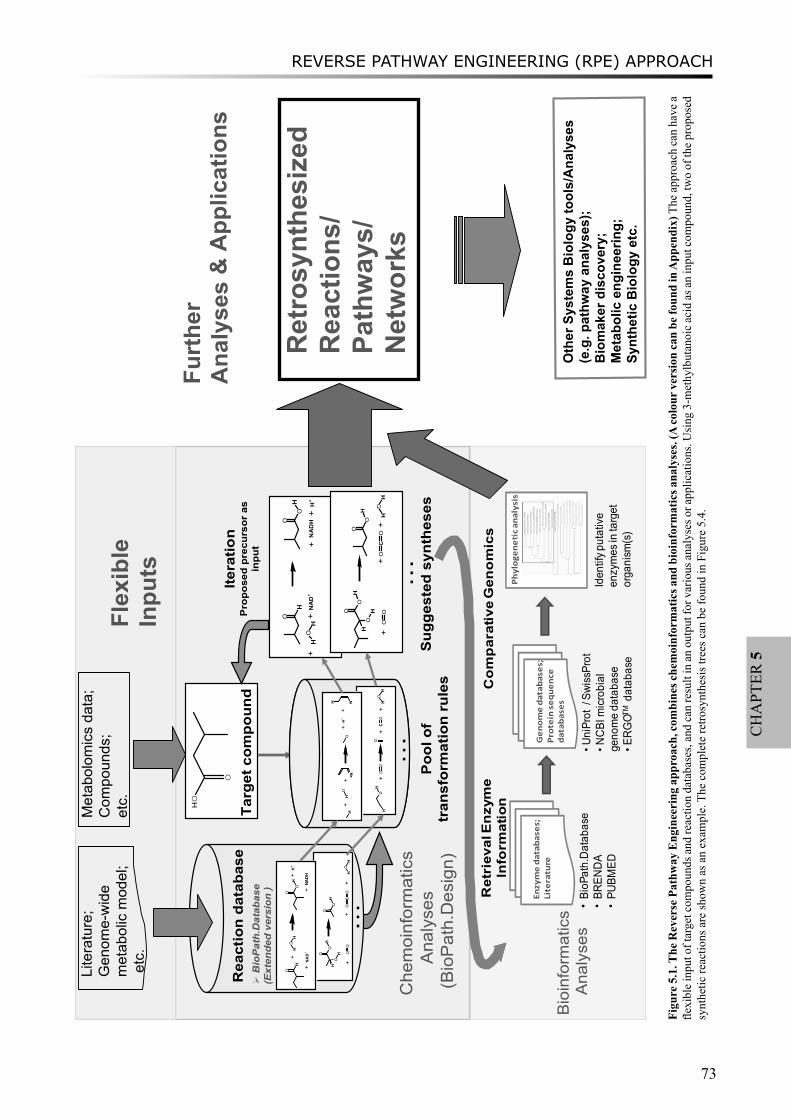
REVERSE PATHWAY ENGINEERING (RPE) APPROACH
73
CH
APT
ER 5
Ret
riev
alE
nzy
me
Info
rmat
ion
•BioP
ath.Da
taba
se•BR
ENDA
•PU
BMED
Co
mp
arat
ive
Gen
om
ics
Enzymedatabases;
Literature
Genomedatabases;
Proteinsequenc e
database s
•UniProt
/SwissProt
•NCB
Imicrob
ial
geno
meda
taba
se•E
RGOT
Mda
taba
se
Phylogene
canalysis
Iden
tifyp
utative
enzymes
intarget
orga
nism
(s)
Bio
info
rmat
ics
A
naly
ses
…
Rea
ctio
n da
taba
se
Sug
gest
ed s
ynth
eses
Targ
et c
ompo
und
�B
ioP
ath.
Dat
abas
e (E
xten
ded
vers
ion
)
……
HO
O
Poo
l of
tran
sfor
mat
ion
rule
s
Itera
tion
Pro
pose
d pr
ecur
sor
as
inpu
t
…
H
O
HO
HN
AD
+
O
OH
++
+H
++
NA
DH
O
OH
OH
H
OO
O
OH
OC
OHOH
++
+
Che
moi
nfor
mat
ics
A
naly
ses
(Bio
Pat
h.D
esig
n)
H
OHOH
NAD+
O
OH
+
+
+H+
+NADH
HO
++
HNH
NO
+H+
+
O
OH
OH
H
OO
O
OH
OC
OH
OH
++
+
OH
HO
OO
CO
HO
H+
++
Lite
ratu
re;
Gen
ome-
wid
e m
etab
olic
mod
el;
etc
.Fl
exib
leIn
puts
Met
abol
omic
s da
ta;
Com
poun
ds;
etc.
Furt
her
Ana
lyse
s&
App
licat
ions
Ret
rosy
nthe
size
d R
eact
ions
/Pa
thw
ays/
Net
wor
ks
Oth
er S
yste
ms
Bio
logy
tool
s/A
naly
ses
(e.g
. pat
hway
ana
lyse
s);
Bio
mak
er d
isco
very
;M
etab
olic
eng
inee
ring;
Sy
nthe
tic B
iolo
gy e
tc.
+
Figu
re 5
.1. T
he R
ever
se P
athw
ay E
ngin
eeri
ng a
ppro
ach,
com
bine
s che
moi
nfor
mat
ics a
nd b
ioin
form
atic
s ana
lyse
s. (A
colo
ur v
ersi
on ca
n be
foun
d in
App
endi
x) T
he a
ppro
ach
can
have
a
flexi
ble
inpu
t of t
arge
t com
poun
ds a
nd re
actio
n da
taba
ses,
and
can
resu
lt in
an
outp
ut fo
r var
ious
ana
lyse
s or a
pplic
atio
ns. U
sing
3-m
ethy
lbut
anoi
c ac
id a
s an
inpu
t com
poun
d, tw
o of
the
prop
osed
sy
nthe
tic re
actio
ns a
re sh
own
as a
n ex
ampl
e. T
he c
ompl
ete
retro
synt
hesi
s tre
es c
an b
e fo
und
in F
igur
e 5.
4.

CHAPTER 5
74
The second step of the RPE approach is to apply bioinformatics analyses to identify the putative enzymes that can catalyze the suggested retrosynthesis reactions (lower panel in Figure 5.1). A list of candidate enzymes is proposed, based on either the reference reactions in BioPath.Database or by inferring them from the biotransformation rule of the predicted reaction. Since incorrect annotation of genes encoding enzymes in metabolic pathways is one of the causes of gaps in the reconstructed networks, a comparative genomics analysis is carried out to improve the annotations. Homologs in specific organisms are obtained from genome sequence databases by using the experimentally characterized protein sequences as the seed, after which orthologous genes are identified by phylogenetic analysis. The results of the bioinformatics analyses are used as an indication to verify the suggested reactions and, later, to be integrated into the retrosynthesis routes.
To exemplify the application of the RPE approach, synthesis routes of flavor compounds in LAB were explored.
HO
N
O
Leucine
HON
OOH
O
Glutamate
HOO
OHO
O
oxoglutaric acid
NAD(P)H
NAD(P)+
HO
O
O
alpha-keto isocaproate
BcAT GDH
KdcA
O
3-methylbutanal
CO2
NAD(P)H
NAD(P)+
AlcDH
HO
3-methylbutanol
NAD(P)H
NAD(P)+
HO
O
3-methylbutanoic acid
NAD(P)H
NAD(P)+ HycDH
AldDH
HO
O
OH
alpha-hydroxy-isocaproate
CoANAD+
NADCO2
S
OCoA
Isovaleryl-CoA
Pi
CoASH
P
O
Isovaleryl-P
ADP
ATPACK
PTA
KaDH
2-methylpropanal
O
Val catabolism
Chemical
S
OCoA
3-methylcrotonyl-CoA
S
OCoA
OH
O
3-methylglutaconyl-CoA
ATP, HCO3
ADP
S
OCoA
OH
OOH
HMG-CoA
OH
O
2-methylbutanoic acid
Ile catabolism
lactate monooxygenase
EstA ethanol
O
O
3-methylbutanoate ethyl ester
(KICA)
(HICA)
Figure 5.2. Leucine catabolism network in lactic acid bacteria, including inter-conversion pathways between leucine degradation and valine catabolism (framed by blue box), and between leucine degradation and isoleucine catabolism (framed by green box). (A colour version can be found in Appendix) Three branches of the further degradation of α-keto isocaporate (KICA) are: i) conversion to the corresponding aldehyde, alcohol or carboxylic acid via α-keto acid decarboxylation, depicted in black, or ii) via oxidative decarboxylation, depicted in blue, or iii) alternatively resulting in α-hydroxy-isocaproate (HICA), depicted in gold. The names of the flavor compounds used as input for RPE approach are shown in italics. The novel predicted reactions are indicated by red dashed arrows. Enzymes names are: BcAT, branched-chain aminotransferase; GDH, glutamate dehydrogenase; HycDH, hydroxyacid dehydrogenase; KdcA, α-ketoacid decarboxylase; AlcDH, alcohol dehydrogenase; AldDH, aldehyde dehydrogenase; EstA, esterase A; KaDH, α-ketoacid dehydrogenase complex; PTA, phosphotransacylase; ACK, acyl kinase.

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
75
CH
APT
ER 5
Validating the RPE approach: mapping flavor molecules to the leucine degradation pathway Leucine catabolism. A leucine catabolism network was reconstructed on the basis of several reported studies (9,13)(Figure 5.2). Some routes in this network were then used to validate the RPE prediction method, using selected flavor compounds from this network.
Leucine catabolism can be divided into three major parts, as shown in Figure 5.2. The first part is the main degradation pathway of leucine, initiated by an aminotransferase reaction converting leucine to α-keto-isocaproate (KICA). The second major part is the degradation of leucine to 2-methylbutanoic acid, as described previously by Ganesan et al. (13). This pathway inter-connects the degradation of leucine and isoleucine since 2-methylbutyric acid is the corresponding carboxylic acid derived from isoleucine. The last major part of leucine catabolism represents the inter-conversion pathway between leucine and valine. The main branches of leucine degradation pathway and the leucine and isoleucine inter-conversion route were used as positive controls for the validation of the RPE approach. The reactions highlighted in red are novel reactions predicted by the RPE approach and will be described in detail in the following sections (Figure 5.2).
Main branches of the flavor-formation pathway from leucine. The main flavor products of leucine degradation are 3-methylbutanal, 3-methylbutanol, and 3-methylbutanoic acid, which provide cheesy, malty and sweaty odors, respectively (10). Since 3-methylbutanal is one of the intermediates in the synthetic routes of the other two compounds, only 3-methylbutanol and 3-methylbutanoic acid were utilized as the input for the retrosynthesis analysis.
One of the retrieved synthetic routes of 3-methylbutanol is shown in Figure 5.3. First, leucine is converted to KICA by a transamination reaction, then further converted to 3-methylbutanal by a decarboxylation reaction. An alcohol dehydrogenase finally catalyzes the conversion of 3-methylbutanal to 3-methylbutanol with NADH as the cofactor. This 3-methylbutanol biosynthetic route predicted by RPE is the same as the one described in literature (Figure 5.2). It should however be noted, that although at the transamination step (step1 in Figure 5.3) pyruvate was predicted to be the acceptor of the amino group based on its simpler structure, oxoglutarate was more often listed as the cofactor in the reference reactions. Thus, according to the frequency of the occurrences of the co-products in the reference reactions, oxoglutarate should be the cofactor for the predicted reaction, which is in line with experimental observation (28).
3-methylbutanol
3-methylbutanal
alpha-keto isocaproate
LeucinePyruvate
NADH H+
H2O
O H
Step 3
Step 2
Step 1
OH
O OH
O
O OH
OH
O
OH
NH
HH
OH
Figure 5.3. Proposed leucine to 3-methylbutanol route by the RPE approach. The retrosynthesis tree was obtained from BioPath.Design when 3-methylbutanol was used as input.

CHAPTER 5
76
When 3-methylbutanoic acid was used as input, three routes were predicted by the RPE approach (Figure 5.4). The first route is via oxidation of 3-methylbutanal (Figure 5.4a), similar to the 3-methylbutanol biosynthesis described above. In the second route, 3-methylbutanoic acid is formed from KICA via isovaleryl-CoA and isovaleryl-phosphate catalyzed by a keto-acid dehydrogenase complex (Figure 5.4b). These proposed routes by RPE are the same as the ones described in literature (29,30) (Figure 5.2). The third one is a novel route (Figure 5.4c), which will be described in detail below (section “suggested novel reactions I: α-hydroxy-isocaproate to 3-methylbutanoic acid”).
Leucine-isoleucine inter-conversion pathway. The inter-conversion route between leucine and isoleucine degradation was also successfully predicted by the RPE approach. This inter-conversion route, in which isovaleryl-CoA is converted to 2-methylbutyric acid, has been proposed in Lactococcus lactis during starvation and was based on gene expression profiling, NMR, and Pathcomp analysis in KEGG (13)(Figure 5.2). By using 2-methylbutanoic acid as input and the intermediate compounds measured by NMR as selecting criteria, we could predict the synthetic route of hydroxymethylglutaryl-CoA from isovaleryl-CoA exactly the same as suggested by Ganesan et al. (13), as well as the route of HMG-CoA to 2-methylbutanoic acid which is very similar as in the study of Ganesan et al. (13) (supplemental material Figure S1).
3-methylbutanoic acid
3-methylbutanoic acid
3-methylbutanoic acid
3-methylbutanal
Isovaleryl-PADP
Isovaleryl-CoA
alpha-keto isocaproate
alpha-hydroxy-isocaproate
alpha-keto isocaproate
a b
c
H+
H+
Phosphate
NADCoA-SH
NADH H+
NADP
H2O
O2
Step 3
Step 2
Step 1
O
OH
O
O
PO O
-
O-
PO-
O-O
O- S
O
CoA
O OH
O
Step 1
O
OH
OH
HO
H
Step 2
Step 1
O
OH
O OH
O
HH
O O
O OH
O
Figure 5.4. Proposed 3-methylbutanoic acid synthesis routes from α-ketoisocaproate by the RPE approach. The retrosynthesis trees correspond to the pathways shown in Figure 5.2. (a) Proposed route for 3-methylbutanal to 3-methylbutanoic acid. (b) Proposed oxidative decarboxylation route converting α-keto isocaproate (KICA) into 3-methylbutanoic acid. (c) Proposed route converting KICA into 3-methylbutanoic acid via α-hydroxy-isocaproate (HICA). A novel reaction converting HICA into 3-methylbutanoic acid by lactate 2-monooxygenase was proposed (step 2).

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
77
CH
APT
ER 5
Predicted novel reactionsSuggested novel reactions I: α-hydroxy-isocaproate to 3-methylbutanoic acid in leucine degradation. Besides the two main biosynthesis routes of 3-methylbutanoic acid (i.e. via α-keto acid decarboxylation and oxidative decarboxylation) described above, a third route of forming 3-methylbutanoic acid from KICA via α-hydroxy-isocaproate (HICA) was also proposed by RPE (Figure 5.2 and Figure 5.4c). Step 1 of this route was predicted as a reduction reaction of KICA to HICA by hydroxy-acid dehydrogenase. This reaction has been described previously and a gene encoding the enzyme responsible for this conversion in L. lactis has been characterized recently (31).
The second step of the predicted route suggested that 3-methylbutanoic acid can be formed from HICA (Figure 5.4c and Figure 5.5). The lactate oxidation reaction catalyzed by lactate 2-monooxygenase is one of the reference reactions (Figure 5.5). Since some enzymes have broader substrate specificities, they may catalyze analogous reactions. Therefore, we hypothesize that lactate 2-monooxygenase could also catalyze the oxidation reaction of HICA.
In order to further explore this hypothesis we performed bioinformatics studies. Lactate 2-monooxygenase activity was identified in Aerococcus viridans, Mycobacterium smegmatis and Geotrichun candidum by previous studies (32-35). It was found that this enzyme can utilize branched longer chain hydroxy acids as the substrate in Mycobacterium smegmatis (36,37). Using the protein sequence of the lactate 2-monooxygenase from Mycobacterium smegmatis, we identified its homologs in lactic acid bacteria by BLASTP. By performing comparative genomics analyses, we identified the orthologs in LAB which are currently annotated as L-lactate oxidase (LOX)(Figure 5.6). Interestingly, Yorita et al. (37) found a mutant form of LOX from Aerococcus viridians, in which alanine 95 was replaced by glycine, yielding an enzyme isoform that has the same oxidase activity on L-lactate as the wild-type LOX, but with an additional enhanced oxidase activity towards longer-chain hydroxy acids. A multiple sequence alignment of LOXs shows that LOX of LAB also has an alanine conserved at the same position as Ala95 in LOX from A. viridans (supplemental material Figure S2). This suggests that LOXs from LAB may only utilize lactate as a substrate, similar to the wild-type LOX from A. viridans. However, broader substrate specificity towards long-chain hydroxy acids may be obtained if the conserved alanine residue in LOX from LAB is mutated to glycine.
+
+
+
L-Lactate
+
Oxygen
Acetate
+
Carbon dioxide
+
Water
Biopath reaction ID RXN00173
Enzyme name lactate 2-monooxygenase
EC number 1.13.12.4 , Search BioPath.Explore
Number of reactions catalysed by this enzyme 4
Reactant name(s) L-Lactate Oxygen
Product name(s) Acetate Carbon_dioxide Water
Pathway {Alcoholic fermentation of pyruvate} {Reactions of Pyruvate and Phosphoenolpyruvate}
Reversible? irreversible
Direction catabolic
Occurrence {in prokarya}
Predicted Reaction:
Related Reaction Found in the Database (Reference Reactions):
Search BioPath.Explore
3-Methylbutanoic acid Carbon dioxide Wateralpha-Hydroxyisocaproate Oxygen
Figure 5.5. The suggested reaction converting α-hydroxy-isocaproate (HICA) to 3-methylbutanoic acid (adapted from BioPath.Design). The suggested reaction is shown in the upper part. One of the reference reactions is shown with the information of the enzyme which catalyzes it.

CHAPTER 5
78
Suggested novel reactions II: chemical reaction converting α-ketoisocaproate to 2-methylpropanal in leucine degradation. In addition to enzymatic reactions, chemical reactions also play an important role in the process of flavor formation. For example, Bonnarme et al. (38) discovered that α-keto-γ-methylthiobutyrate (KMBA) can be chemically converted to methylsulfanyl-acetaldehyde (MTAC) and oxalic acid in the presence of Mn(II), alkaline pH and oxygen. MTAC is the precursor of various volatile sulfur compounds, such as methanethiol. This chemical reaction belongs to the reconstructed flavor-forming pathway from methionine that was included in the extended BioPath.Database in this study.
When 2-methylpropanal was used as the target compound for RPE, one of the reactions predicted by BioPath.Design is the decarboxylation of α-ketoisovaleric acid derived from valine, analogous to the decarboxylation reaction converting KICA to 3-methylbutanal described above. An exact match of this reaction was found by scanning the BioPath.Database, suggesting it has been stored in the database (see Fig. M2, the example from Supplementary Methods).
A novel chemical reaction converting KICA to 2-methylpropanal was also predicted, which inter-connects leucine and valine catabolism (Figure 5.2). The chemical conversion of KMBA to MTAC from methionine catabolism was proposed as the reference reaction (Figure 5.7). In the predicted reaction, KICA reacts with oxygen, resulting in 2-methylpropanal and oxalate. A literature survey confirmed that this plausible chemical reaction has been experimentally characterized by Smit et al. (39). Interestingly, their study described that the chemical reaction can be modulated by Mn (II) concentration and oxygen as well as pH, a similar condition as for the reference reaction.
Suggested novel reactions III: enzymatic reaction converting methanethiol to DMS in the methionine degradation pathway. Methionine catabolism is known as one of the main flavor-forming pathways, resulting in various volatile sulfur compounds such as H2S, methanethiol, dimethyl sulfide (DMS), dimethyl disulfide (DMDS), and dimethyl trisulfide (DMTS) (20,40).
[1000]
[861]
[759]
[858]
[1000]
[441]
[1000] Lactobacillus casei ATCC334 (LSE116495791)
Lactobacillus casei BL23 (LSD191639282)
Lactobacillus plantarum WCFS1 (LPL28379893)
Pediococcus pentosaceus ATCC25745 (PEP116492687)[1000] Lactococcus lactis lactis IL 1403 (LLX15673234)
[1000] Lactobacillus gasseri ATCC33323 (LGA116630404)
Lactobacillus johnsonii NCC533 (LJO42519875)
[879] Lactobacillus sakei 23K (LSK81429009)
Aerococcus viridans (AVI_lox, 849022)
Lactobacillus casei BL23 (LSD191637600)
Lactobacillus acidophilus NCFM (LAC58337859)
[818]
[1000]
[527]
[913] Lactobacillus sakei 23K (LSK81428516)
Lactobacillus plantarum WCFS1 (LPL28378413)
Lactobacillus fermentum IFO3956 (LFF184155671)
Lactobacillus salivarius UCC118 (LSL90961659)
[507]
[1000] Lactobacillus helveticus DPC 4571 (LHL161506798)
Lactobacillus acidophilus NCFM (LAC58336540)
[1000] Lactobacillus casei ATCC334 (LSE116495575)
Lactobacillus casei BL23 (LSD191639057)
Figure 5.6. Bootstrapped (n=1000) neighbor-joining tree of the LOX homologs from LAB. The functional equivalents (orthologs) of LOX from Aerococcus viridans are highlighted by the frame. Genome abbreviations and GI codes of the homologs are in parentheses. Diverse annotations have been assigned to the protein members in the lower groups, such as lactate dehydrogenase, isopentenyl pyrophosphate isomerase, and inosine-5-monophosphate dehydrogenase. Squares indicate duplication events, and circles represent speciation events.

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
79
CH
APT
ER 5
Commonly, methanethiol, a compound derived from elimination of methionine catalyzed by a C-S lyase, is regarded as the precursor of DMS whose odor is described as “boiled cabbage, sulfurous” (41). Two different hypotheses on the reaction mechanisms of DMS formation from methanethiol have been proposed. The first one is the chemical conversion of methanethiol to DMS (40), by either a methylation reaction or disproportionation reaction (42). However, this chemical reaction requires an extremely high temperature (350°C) and acid-base type catalysts (43), which is impossible during fermentation with LAB. The other hypothesis suggests that an enzymatic reaction converts methanethiol to DMS, utilizing S-adenosyl-L-methionine (S-AdoMet) as the methyl-group donor (44).
In order to identify plausible synthesis reactions, DMS was used as input for RPE. The reaction of methanethiol accepting methyl group from S-AdoMet to form DMS and S-adenosyl-L-homocysteine (S-AdoHcy) was predicted by BioPath.Design, in line with the second hypothesis (supplemental material Figure S3). The predicted reaction was based on a reference reaction which converts L-homocysteine into L-methionine using S-AdoMet as methyl-donor. Several enzymes were proposed by the bioinformatics analyses as candidates for catalyzing this reaction, including homocysteine S-methyltransferase (EC 2.1.1.10), methionine synthase (EC 2.1.1.13) and thiol S-methyltransferase (TMT, EC 2.1.1.9). The first two enzymes are known for catalyzing the final step of methionine biosynthesis and are distributed widely among LAB(20), but their activity toward methanethiol is still unclear. TMT is mainly found in animals and plants, whereas it has never been reported in LAB (45). The genetically characterized TMT from Brassica oleracea (cabbage)(46) was used as seed to perform the comparative genomics analyses. TMT orthologs were identified in several Streptococcus thermophilus genomes and in the genome of Propionibacterium freudenreichii ATCC 6207 (supplemental material Figure S4). The plausible TMT ortholog is in agreement with the observation that DMS is formed by P. freudenreichii in Swiss-type cheese (44,47).
DiscussionIn the last decades, high-throughput analytical techniques, such as metabolomics, have developed
rapidly (5,16,48). This increased analytical power leads to challenges in data storage, analysis and interpretation. One example is flavor formation by LAB. Currently, many of the measured flavor compounds cannot be connected to their metabolic precursors. In this study, we describe a novel approach “Reverse Pathway Engineering”, to retrieve the missing links in the incomplete flavor-
+ +
alpha-Keto-gamma-methiolbutyrate
+
Oxygen Methylsulfanyl-acetaldehyde
+
Oxalate
Biopath reaction ID RXN07012
Number of reactions catalysed by this enzyme 0
Reactant name(s) alpha-Keto-gamma-methiolbutyrate Oxygen
Product name(s) Methylsulfanyl-acetaldehyde Oxalate
Occurrence {in prokarya}
Predicted Reaction:
Related Reaction Found in the Database (Reference Reactions):
(chemical reaction)
alpha-Keto-isocarproate Oxygen 2-Methylpropanal Oxalate
Figure 5.7. The predicted chemical reaction converts α-keto-isocaproate (KICA) to 2-methylpropanal, connecting the leucine degradation and the valine degradation pathways. The reference reaction converting α-keto-γ-methylthiobutyrate to methylsulfanyl-acetaldehyde was derived from the additional part of the BioPath.Database containing the reactions of the flavor-forming pathways from sulfur-containing amino acid degradation.

CHAPTER 5
80
forming pathways of LAB, by combining chemoinformatics and bioinformatics analyses. To validate the RPE approach, the leucine degradation pathway was explored as it is an important and thoroughly studied flavor-forming pathway in LAB (9,13), although many of the described reactions from this pathway are lacking in biochemical databases. We successfully predicted the main branches of leucine degradation pathway and the inter-conversion route between leucine and isoleucine as described in literature. Some novel enzymatic reactions were identified, such as the conversion of α-hydroxy-isocaproate to 3-methylbutanoic acid in the leucine degradation pathway and the formation of DMS from methanethiol in the methionine degradation pathway.
Since there are no golden standards in the field of pathway prediction at this moment. we compared our approach to one of the previously published tools, Pathcomp (49), which has been used for the prediction of the inter-conversion pathway between leucine and isoleucine (13). Using the same dataset, BioPath.Design provided a broader spectrum of predicted reactions. For instance, RPE predicted that 2-methylacetoacetyl-CoA could be synthesized from either acetoacetyl-CoA or acetyl-CoA, rather than only from acetyl-CoA as reported by Ganesan et al. (13) (supplemental material Figure S1). This is probably due to the fact that different reaction databases were used for the pathway predictions in the two studies. Pathcomp utilizes the KEGG database for predictions while in our study the BioPath.Database was used. As Pathcomp is only able to retrieve known reactions stored in KEGG (49), unknown reactions cannot be predicted. In contrast, BioPath.Design is based on transformation rules, and can propose both known reactions and unknown ones that were not originally stored in the database.
For the novel predicted enzymatic reactions described in this study, the putative enzymes, as well as the candidate genes in LAB were identified by bioinformatics analyses. The conflicting hypotheses on the synthesis of DMS from previous studies were elucidated in this study. In support of the hypothesis that DMS is formed enzymatically, our analyses suggested both a plausible enzymatic reaction and possible enzymes to catalyze such a reaction. Those candidate reactions and enzymes can be used for directing future experiments to validate the hypothesis.
The predicted novel reactions not only fill in the gaps in the cellular models, but also provide leads for metabolic engineering. We discovered an enzymatic reaction which converts a non-flavor compound HICA to the flavor compound 3-methylbutanoic acid. The homologs of the enzyme catalyzing this reaction were found in LAB genomes. One previous study suggested that the homologous enzymes could gain the activity toward HICA by a site-directed mutation. It is well accepted that the synthesis of hydroxy acids such as HICA may have a negative effect on the flavor formation since it shares the same precursors as the other flavor compounds such as 3-methylbutanol and 3-methylbutanoic acid (9,20). The results of the RPE approach provide novel means for increasing the production of the flavor compound 3-methylbutanoic acid by utilizing HICA as a precursor. The novel enzymatic activities of LOX in LAB towards long-chain hydroxy acids can be introduced via protein engineering, similar to what has been described for the biosynthesis of shikimate pathway products by directed evolution of 2-keto-3-deoxy-6-phosphogalactonate aldolase in E. coli (50), or by adaptive evolution e.g. by stressing the bacterial cells with excess amounts of hydroxy acids.
The predicted chemical reaction for forming 2-methylpropanal from KICA takes place under the same environmental conditions as the reference reaction which chemically converts KMBA to MTAC (38,39). This suggests that the experimental conditions of the corresponding reference reaction can be applied to the novel predicted chemical reaction. The RPE approach is not limited to predicting enzymatic reactions, as chemical reactions can be predicted as well, suggesting a powerful role when this approach is applied to complex biological systems which contain both enzymatic and chemical conversions.
Furthermore, the reference reaction of the predicted chemical reaction originally belongs to the reconstructed flavor-forming pathways in the extended BioPath.Database. This indicates the

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
81
CH
APT
ER 5
importance of choosing a comprehensive and suitable reaction database for the RPE approach. For this reason, here an extended BioPath.Database was constructed with extra reactions from the genome-scale metabolic model of L. plantarum and from the reconstructed sulfur flavor-forming pathways. However, this reaction database is still limited and not yet optimized. More enzymatic reactions from LAB metabolism and chemical reactions occurring during milk fermentation, which have been described in literature, can be added for database improvement. It should be noted, however, that including a large number of reactions in the reaction database may introduce more false positives during analyses. Moreover, the most time- and labour-consuming step of this approach is to manually select the most likely reactions from a list of proposed reactions, especially when the list is long. Good selection and ranking criteria are helpful for reducing the manual work. At this moment, our ranking criteria are based on combining the presence of reactants and predicted reactions in the database, as well as the simplicity of structures of the reactants. However, these criteria do not fit all needs, and sometimes can cause errors, e.g. the prediction of a wrong cofactor as described in the previous section. New criteria are being explored, also for different application cases, such as the degree of structural similarity between the compounds in the predicted reactions and the reference reactions, or the reaction conditions used for predicting chemical reactions.
The RPE approach uniquely connects chemical data all the way back to genomic data. This distinguishes it from other retrobiosynthesis methods such as ReBiT (Retro-Biosynthesis Tool)(51), which only covers part of the whole process, i.e. identifying enzymes in designed pathways. Another retrosynthesis tool, Route Designer, uses a set of retrosynthesis rules specific to organic chemistry(27), making it less suitable for biochemical questions. Our approach brings together chemistry and biology, thereby elucidating molecular mechanisms of biochemical systems. This approach can be easily extended to other fields, thanks to the flexibility of using various reaction databases and target compounds. A possible future application could be in the field of biomarker discovery. Recently, a metabolomics study described the correlation between human metabolic phenotypes and specific dietary preferences concerning chocolate consumption(52). In the study, it was demonstrated that several metabolic biomarkers could be connected to human host and gut microbial co-metabolism. Other studies have also identified such metabolites as biomarkers for evaluation of disease risks (53,54). Our approach will be particularly helpful in tackling questions regarding the interplay of host-microbial co-metabolism by predicting the synthesis routes of measured metabolites, proposing plausible enzymes and assigning enzymatic reactions to mammalian cells or gut microbes. Leads for other application areas can also be provided, for instance synthetic biology, helping in silico design of biosynthesis pathways to construct microbial chemical factories(55).
AcknowledgementsThis work was supported by grant CSI4017 of the Casimir program of the Ministry of Economic Affairs, The Netherlands.
AbbreviationsRPE, Reverse Pathway Engineering; GC-MS, gas chromatography mass spectrometry; NMR, nuclear magnetic resonance spectroscopy; LAB, lactic acid bacteria; MSA, multiple sequence alignment; THERESA, THE REtroSynthesis Analyser; KICA, α-keto-isocaproate; HICA, α-hydroxy-isocaproate; LOX, L-lactate oxidase; KMBA, α-keto-γ-methyl thiobutyrate; MTAC, methylsulfanyl-acetaldehyde; DMS, dimethyl sulfide; S-AdoMet, S-adenosyl-L-methionine; S-AdoHcy, S-adenosyl-L-homo cysteine; TMT, thiol S-methyltransferase

CHAPTER 5
82
References
1. Simon, G. M., and Cravatt, B. F. (2008) Nat Chem Biol 4, 639-6422. Feist, A. M., Herrgard, M. J., Thiele, I., Reed, J. L., and Palsson, B. O. (2009) Nat Rev Microbiol 7, 129-1433. Kastenmuller, G., Schenk, M. E., Gasteiger, J., and Mewes, H. W. (2009) Genome Biol 10, R284. Breitling, R., Vitkup, D., and Barrett, M. P. (2008) Nat Rev Microbiol 6, 156-1615. Mapelli, V., Olsson, L., and Nielsen, J. (2008) Trends Biotechnol 26, 490-4976. Pettit, R. K. (2009) Appl Microbiol Biotechnol 83, 19-257. Ardo, Y. (2006) Biotechnol Adv 24, 238-2428. Fernandez, M., and Zuniga, M. (2006) Crit Rev Microbiol 32, 155-1839. Smit, G., Smit, B. A., and Engels, W. J. (2005) FEMS Microbiol Rev 29, 591-61010. Yvon, M. (2006) Austral. Journal of Dairy Technology 61, 88-9611. Durot, M., Bourguignon, P. Y., and Schachter, V. (2009) FEMS Microbiol Rev 33, 164-19012. de la Plaza, M., Fernandez de Palencia, P., Pelaez, C., and Requena, T. (2004) FEMS Microbiol Lett 238, 367-37413. Ganesan, B., Dobrowolski, P., and Weimer, B. C. (2006) Appl Environ Microbiol 72, 4264-427314. Smit, B. A., Engels, W. J., Wouters, J. T., and Smit, G. (2004) Appl Microbiol Biotechnol 64, 396-40215. Smit, B. A., Engels, W. J., and Smit, G. (2009) Appl Microbiol Biotechnol 81, 987-99916. Pastink, M. I., Sieuwerts, S., de Bok, F. A. M., Janssen, P. W. M., Teusink, B., van Hylckama Vlieg, J. E. T., and Hugenholtz,
J. (2008) International Dairy Journal 18, 781-78917. Reitz, M., Sacher, O., Tarkhov, A., Trumbach, D., and Gasteiger, J. (2004) Org Biomol Chem 2, 3226-323718. Michal, G. (1999) Biochemical Pathways: An Atlas of Biochemistry and Molecular Biology, Wiley, New York19. Teusink, B., Wiersma, A., Molenaar, D., Francke, C., de Vos, W. M., Siezen, R. J., and Smid, E. J. (2006) J Biol Chem 281,
40041-4004820. Liu, M., Nauta, A., Francke, C., and Siezen, R. J. (2008) Appl Environ Microbiol 74, 4590-460021. Sacher, O., Reitz, M., and Gasteiger, J. (2009) J Chem Inf Model 49, 1525-153422. Chang, A., Scheer, M., Grote, A., Schomburg, I., and Schomburg, D. (2009) Nucleic Acids Res 37, D588-59223. Overbeek, R., Larsen, N., Walunas, T., D‘Souza, M., Pusch, G., Selkov, E., Jr., Liolios, K., Joukov, V., Kaznadzey, D.,
Anderson, I., Bhattacharyya, A., Burd, H., Gardner, W., Hanke, P., Kapatral, V., Mikhailova, N., Vasieva, O., Osterman, A., Vonstein, V., Fonstein, M., Ivanova, N., and Kyrpides, N. (2003) Nucleic Acids Res 31, 164-171
24. Edgar, R. C. (2004) BMC Bioinformatics 525. Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M.,
Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007) Bioinformatics 23, 2947-294826. van der Heijden, R. T., Snel, B., van, N. V., and Huynen, M. A. (2007) BMC Bioinformatics 8, 8327. Law, J., Zsoldos, Z., Simon, A., Reid, D., Liu, Y., Khew, S. Y., Johnson, A. P., Major, S., Wade, R. A., and Ando, H. Y. (2009)
J Chem Inf Model 49, 593-60228. Yvon, M., Chambellon, E., Bolotin, A., and Roudot-Algaron, F. (2000) Appl Environ Microbiol 66, 571-57729. Helinck, S., Le Bars, D., Moreau, D., and Yvon, M. (2004) Appl Environ Microbiol 70, 3855-386130. Ward, D. E., Ross, R. P., van der Weijden, C. C., Snoep, J. L., and Claiborne, A. (1999) J Bacteriol 181, 5433-544231. Chambellon, E., Rijnen, L., Lorquet, F., Gitton, C., van Hylckama Vlieg, J. E., Wouters, J. A., and Yvon, M. (2009) J
Bacteriol 191, 873-88132. Morimoto, Y., Yorita, K., Aki, K., Misaki, H., and Massey, V. (1998) Biochimie 80, 309-31233. Xu, P., Yano, T., Yamamoto, K., Suzuki, H., and Kumagai, H. (1996) Appl Biochem Biotechnol 56, 277-28834. Sztajer, H., Wang, W., Lunsdorf, H., Stocker, A., and Schmid, R. D. (1996) Appl Microbiol Biotechnol 45, 600-60635. Giegel, D. A., Williams, C. H., Jr., and Massey, V. (1990) J Biol Chem 265, 6626-663236. Takemori, S., and Katagiri, M. (1975) Methods Enzymol 41, 329-33337. Yorita, K., Aki, K., Ohkuma-Soyejima, T., Kokubo, T., Misaki, H., and Massey, V. (1996) J Biol Chem 271, 28300-2830538. Bonnarme, P., Amarita, F., Chambellon, E., Semon, E., Spinnler, H. E., and Yvon, M. (2004) FEMS Microbiol Lett 236, 85-
9039. Smit, B. A., Engels, W. J., Alewijn, M., Lommerse, G. T., Kippersluijs, E. A., Wouters, J. T., and Smit, G. (2004) J Agric Food
Chem 52, 1263-126840. Weimer, B., Seefeldt, K., and Dias, B. (1999) Antonie Van Leeuwenhoek 76, 247-26141. Landaud, S., Helinck, S., and Bonnarme, P. (2008) Appl Microbiol Biotechnol 77, 1191-120542. Schulz, S., and Dickschat, J. S. (2007) Nat Prod Rep 24, 814-84243. Mashkina, A. (2000) Kinetics and Catalysis 41, 216-22144. Bentley, R., and Chasteen, T. G. (2004) Chemosphere 55, 291-31745. Taylor, B. F., and Kiene, R. P. (1989) Microbial metabolism of dimethyl sulfide. in Biogenic Sulfur in the Environment.
(Saltzman, E., and Cooper, W. J. eds.), American Chemical Society, Washington, DC46. Attieh, J., Djiana, R., Koonjul, P., Etienne, C., Sparace, S. A., and Saini, H. S. (2002) Plant Mol Biol 50, 511-52147. Kadota, H., and Ishida, Y. (1972) Annual Review of Microbiology 26, 127-13848. Dunn, W. B., Bailey, N. J., and Johnson, H. E. (2005) Analyst 130, 606-62549. Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004) Nucleic Acids Res 32, D277-28050. Ran, N., and Frost, J. W. (2007) J Am Chem Soc 129, 6130-613951. Martin, C. H., Nielsen, D. R., Solomon, K. V., and Prather, K. L. (2009) Chem Biol 16, 277-286

REVERSE PATHWAY ENGINEERING (RPE) APPROACH
83
CH
APT
ER 5
52. Rezzi, S., Ramadan, Z., Martin, F. P., Fay, L. B., van Bladeren, P., Lindon, J. C., Nicholson, J. K., and Kochhar, S. (2007) J Proteome Res 6, 4469-4477
53. Holmes, E., Loo, R. L., Stamler, J., Bictash, M., Yap, I. K., Chan, Q., Ebbels, T., De Iorio, M., Brown, I. J., Veselkov, K. A., Daviglus, M. L., Kesteloot, H., Ueshima, H., Zhao, L., Nicholson, J. K., and Elliott, P. (2008) Nature 453, 396-400
54. Kinross, J. M., von Roon, A. C., Holmes, E., Darzi, A., and Nicholson, J. K. (2008) Curr Gastroenterol Rep 10, 396-40355. Prather, K. L., and Martin, C. H. (2008) Curr Opin Biotechnol 19, 468-474

CHAPTER 5
84
吾生也有涯,而知也无涯。以有涯随无涯,殆已;已而为知者,殆而已矣。
- 庄子·养生主
There is a limit to our life, but to knowledge there is no limit. With what is limited to pursue after what is
unlimited is a perilous thing; and when, knowing this, we still seek the increase of our knowledge, the peril
cannot be averted.
- Zhuangzi, Nourishing the Lord of Life

HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
85
CH
APT
ER 6
Chapter 6
IN SILICO PREDICTION OF HORIZONTAL GENE TRANSFER EVENTS IN LACTOBACILLUS DELBRUECKII SSP. BULGARICUS
AND STREPTOCOCCUS THERMOPHILUS REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
Mengjin Liu, Roland J. Siezen, Arjen Nauta
Appl Environ Microbiol. 2009; 75(12):4120-9.

CHAPTER 6
86
AbstractLactobacillus delbrueckii ssp. bulgaricus (L. bulgaricus) and Streptococcus thermophilus, used as yoghurt starter cultures, are well-known for their stable co-existence and proto-cooperation during their co-existence in milk. In this study, we show that a close interaction between both species also takes place at the genetic level. We performed an in silico analysis, combining gene composition and gene transfer mechanism associated features, and predicted horizontally transferred genes in both L. bulgaricus and S. thermophilus. Putative horizontal gene transfer (HGT) events between the two bacterial species include EPS biosynthesis genes, transferred from S. thermophilus to L. bulgaricus, and the gene cluster cbs-cblB/cglB-cysE for metabolism of sulfur-containing amino acids which was transferred from L. bulgaricus or L. helveticus to S. thermophilus. The HGT event of the cbs-cblB/cglB-cysE gene cluster was analyzed in detail with respect to both evolutionary and functional aspects. It can be concluded that during co-existence of both yoghurt starter species in a milk environment, agonistic co-evolution at the genetic level has probably been involved in the optimization of their combined growth and interactions.
Introduction Lactobacillus delbrueckii ssp. bulgaricus (L. bulgaricus) and Streptococcus thermophilus
have been used as the starter culture for yoghurt manufacture for thousands of years. Both species are known to stably co-exist in a milk environment and interact beneficially. This so called proto-cooperation, previously defined as biochemical mutualism, involves the exchange of metabolites and/or stimulatory factors (38). Examples of biochemical proto-cooperation between L. bulgaricus and S. thermophilus include the action of cell-wall bound proteases, as produced by L. bulgaricus strains, and formate, required for growth of L. bulgaricus and supplied by S. thermophilus (6, 7). An overview of the interactions between the two yoghurt bacteria, including exchange of CO2, pyruvate, folate etc, can be found in a recently published review by Sieuwerts et al. (43). Putative genetic mechanisms underlying proto-cooperation, however, so far have not been studied in detail. The genomes of two L. bulgaricus strains and three S. thermophilus strains, all used in yoghurt manufacturing, have been fully sequenced (3, 31, 33, 34, 39, 44, 46). The available genomic information could provide new insights into the genetic aspects of proto-cooperation between L. bulgaricus and S. thermophilus through the identification of putative horizontal gene transfer (HGT) events at the genome scale. HGT can be defined as the exchange of genetic material between phylogenetically unrelated organisms (23). It is considered as a major factor in the process of environmental adaptation, for both individual species and entire microbial populations. Especially HGT events between two species existing in the same niche can reflect their inter-related activities and dependencies (13, 17). Nicolas et al. (36) predicted HGT events between Lactobacillus acidophilus and Lactobacillus johnsonii by analyzing 401 phylogenetic trees, also including genes of L. bulgaricus. Several HGT events have been predicted in the S. thermophilus strains CNRZ1066 and LMG18311 (3, 10, 18) as well as in L. bulgaricus ATCC 11842 (46). Moreover, a core genome of S. thermophilus and possibly acquired genes were identified by a comparative genome hybridization study of 47 strains (40).
In this study we describe an in-depth bioinformatics analysis in which we combined gene composition (GC content and dinucleotide composition) and gene transfer mechanism associated features. Thus, we predicted horizontally transferred genes and gene clusters in the five sequenced L. bulgaricus and S. thermophilus genomes, with focus on niche-specific genes and genes required for bacterial growth. Identification of HGT events led to a list of putative transferred genes, some of which could be important for bacterial proto-cooperation and the adaptation to their environment. The evolution and function of the transferred gene cluster cbs-cblB/cglB-cysE (originally called cysM2-metB2-cysE2 in S. thermophilus), involved in metabolism of sulfur-containing amino acids, were analyzed in detail. On the basis of our analysis, it can be concluded that both species probably
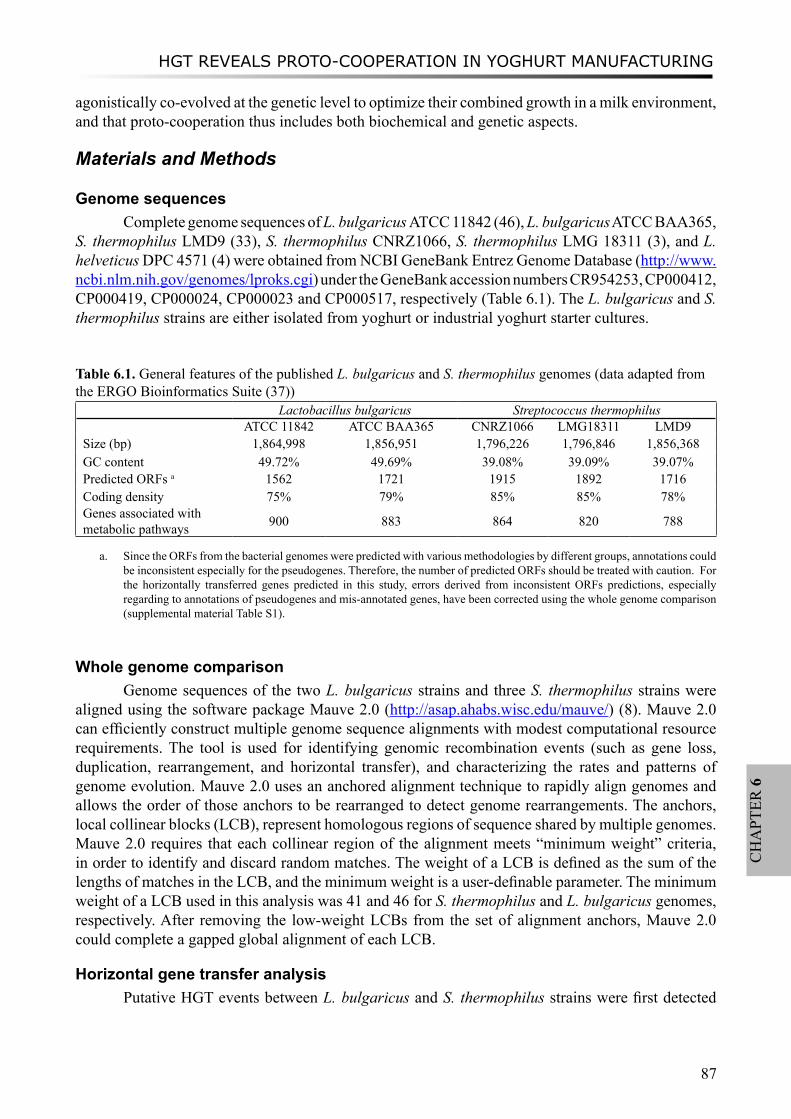
HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
87
CH
APT
ER 6
agonistically co-evolved at the genetic level to optimize their combined growth in a milk environment, and that proto-cooperation thus includes both biochemical and genetic aspects.
Materials and Methods
Genome sequencesComplete genome sequences of L. bulgaricus ATCC 11842 (46), L. bulgaricus ATCC BAA365,
S. thermophilus LMD9 (33), S. thermophilus CNRZ1066, S. thermophilus LMG 18311 (3), and L. helveticus DPC 4571 (4) were obtained from NCBI GeneBank Entrez Genome Database (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi) under the GeneBank accession numbers CR954253, CP000412, CP000419, CP000024, CP000023 and CP000517, respectively (Table 6.1). The L. bulgaricus and S. thermophilus strains are either isolated from yoghurt or industrial yoghurt starter cultures.
Table 6.1. General features of the published L. bulgaricus and S. thermophilus genomes (data adapted from the ERGO Bioinformatics Suite (37))
Lactobacillus bulgaricus Streptococcus thermophilusATCC 11842 ATCC BAA365 CNRZ1066 LMG18311 LMD9
Size (bp) 1,864,998 1,856,951 1,796,226 1,796,846 1,856,368GC content 49.72% 49.69% 39.08% 39.09% 39.07%Predicted ORFs a 1562 1721 1915 1892 1716Coding density 75% 79% 85% 85% 78%Genes associated with metabolic pathways 900 883 864 820 788
a. Since the ORFs from the bacterial genomes were predicted with various methodologies by different groups, annotations could be inconsistent especially for the pseudogenes. Therefore, the number of predicted ORFs should be treated with caution. For the horizontally transferred genes predicted in this study, errors derived from inconsistent ORFs predictions, especially regarding to annotations of pseudogenes and mis-annotated genes, have been corrected using the whole genome comparison (supplemental material Table S1).
Whole genome comparisonGenome sequences of the two L. bulgaricus strains and three S. thermophilus strains were
aligned using the software package Mauve 2.0 (http://asap.ahabs.wisc.edu/mauve/) (8). Mauve 2.0 can efficiently construct multiple genome sequence alignments with modest computational resource requirements. The tool is used for identifying genomic recombination events (such as gene loss, duplication, rearrangement, and horizontal transfer), and characterizing the rates and patterns of genome evolution. Mauve 2.0 uses an anchored alignment technique to rapidly align genomes and allows the order of those anchors to be rearranged to detect genome rearrangements. The anchors, local collinear blocks (LCB), represent homologous regions of sequence shared by multiple genomes. Mauve 2.0 requires that each collinear region of the alignment meets “minimum weight” criteria, in order to identify and discard random matches. The weight of a LCB is defined as the sum of the lengths of matches in the LCB, and the minimum weight is a user-definable parameter. The minimum weight of a LCB used in this analysis was 41 and 46 for S. thermophilus and L. bulgaricus genomes, respectively. After removing the low-weight LCBs from the set of alignment anchors, Mauve 2.0 could complete a gapped global alignment of each LCB.
Horizontal gene transfer analysisPutative HGT events between L. bulgaricus and S. thermophilus strains were first detected

CHAPTER 6
88
by the whole genome comparison using Mauve 2.0. The whole genome alignments were manually inspected to identify putative horizontally transferred genes. Sequence composition analysis was carried out, including calculation of GC composition (of 600 bp fragments) and dinucleotide dissimilarity value δ (of 1000 bp fragments) along the whole genome, using the δρ-Web tool (48) (see supplemental material Figure S2 and Figure S3). Identification of HGT events by using composition differences is based on previous observations by Karlin et al. (24, 25) that each genome has a typical dinucleotide frequency and that related species have a similar genome signature. A high genomic dissimilarity between an input sequence and a representative genome sequence of the species from which the sequence was isolated suggests a heterologous origin of the input sequence. In other words, horizontally acquired genes can have a very different sequence dinucleotide composition, compared to the genome in which they presently reside and the difference can be expressed by the δ value. DNA fragments with significantly different GC composition and/or dinucleotide composition (average ±2SD) as compared to the whole genomes were predicted to be HGT regions.
The predicted horizontally transferred genes and gene clusters were checked for HGT mechanism-associated features such as neighboring mobile elements or tRNA genes using Artemis (41) and Mauve 2.0. Homologs of the genes that were predicted to be transferred between S. thermophilus and L. bulgaricus, were collected by performing BLASTP searches (1) against all the available lactic acid bacteria (LAB) genomes or the non-redundant NCBI protein database. Homologous sequences were aligned with MUSCLE (12), and phylogenetic trees were constructed using the neighborhood joining method implemented in ClustalW (32). The phylogenetic trees were visualized in LOFT (47). The positions of orthologs from L. bulgaricus and S. thermophilus in the phylogenetic trees were checked to confirm whether the predicted genes are horizontally transferred genes between the two genomes.
Results and DiscussionThrough HGT, a genome can be rearranged by the integration and/or deletion of genetic
elements, one of the driving forces in the evolution of organisms (50). HGT events can be detected using phylogenetic and compositional approaches. Information on gene transfer mechanisms, for instance transposases or bacteriophage-related genes found in the neighborhood of the target genes, can improve the prediction of HGT events (50). In order to reveal proto-cooperation between the two co-existing yoghurt species on the genetic level and to understand the rationale of their co-evolution, putative HGT events were predicted and analyzed. HGT events, as detected by combining composition analysis or phylogenetic analysis and gene transfer mechanism associated features, are described for both S. thermophilus and L. bulgaricus strains.
Putative HGT from foreign origin to S. thermophilus Previously, Hols et al. (18) identified putative HGT events in the genomes of S. thermophilus
CNRZ1066 and LMG 18311. In this study, we predicted the horizontally transferred genes in the S. thermophilus LMD9 genome, and also thoroughly reanalyzed the genomes of S. thermophilus CNRZ1066 and LMG 18311 based on both gene composition and gene transfer mechanism associated features. In order to identify strain-specific regions and indications for gene transfer or loci rearrangement, genome alignments of the strains from each species were performed (supplementary material Figure S1). The alignment of the three S. thermophilus genomes showed that strain LMD9 has undergone more genome rearrangements as compared to both other strains. Strain CNRZ1066 and strain LMG 18311 share a more conserved genome context (supplementary material Figure S1A).
Gene composition analysis including GC-content (for details see supplementary material Figure S2) and dinucleotide composition (supplementary material Figure S3) analyses revealed a list of putative horizontally transferred genes in the three S. thermophilus strains (Table 6.2a,

HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
89
CH
APT
ER 6
supplementary material Table S1). In total 197 genes were predicted as potentially acquired in the three S. thermophilus genomes, of which 118 genes are located in 28 gene clusters (Table 6.2a). Over 60% of those genes were found to be associated with gene transfer mechanism associated features, such as transposase, bacteriophage, and tRNA genes (supplementary material Table S1). Compared with the core gene set of 47 S. thermophilus strains, as identified by a recent comparative genomic hybridization study (40), 30 of the core genes overlapped with our HGT prediction. Since most of these core genes are not found to be associated with any mobile element or located in a predicted HGT gene cluster, they may be incorrectly predicted to be horizontally transferred by the gene composition analysis (supplemental material Table S1). The three genomes have several putative horizontally transferred gene clusters in common, including clusters S1, S5, S23, S24, S25, and S26. Cluster S1 encodes several lantibiotic/bacteriocin biosynthesis proteins and an exporter i.e. labB, labC, labT. These genes in S. thermophilus CNRZ1066 and LMG 18311 have been described by Hols et al. (18) as horizontally transferred genes. An ABC-type transporter in cluster S5 (stu0324, str0324, STER_1694) was found to have the best homolog (40% identity) in L. helveticus DPC4571. Cluster S26 encodes enzymes involved in metabolism of sulfur-containing amino acids, as will be described in detail below.
We also found examples of strain-specific HGT events such as EPS (exopolysaccharides) biosynthesis genes. According to the distinct groups of polysaccharides defined by Delcour et al. (9), EPS in this study refer to extracellular polysaccharides which are released into the medium or attached to the cell surface (capsular polysaccharides). EPS are important for the rheology, texture and ‘mouthfeel’ of yoghurt. In addition, they are believed to contribute to probiotic effects (35). Previous studies revealed that EPS biosynthesis genes can vary enormously in different LAB strains (49). The EPS biosynthesis genes are either obtained by HGT or may have evolved much faster than other genes. The clusters S10, S12 and S13 of S. thermophilus LMD9, CNRZ1066 and LMG 18311, respectively, have different gene compositions and contexts and share limited sequence similarities (Table 6.3), suggesting that their origin differs.
Cluster S21 of S. thermophilus LMD9 contains STER_1698, encoding a phage-resistance protein with a Pfam Abi_2 domain. Homologs of this protein were found to be involved in bacteriophage resistance, mediated through abortive infection in Lactococcus species (2). They belong to the AbiD, AbiD1 or AbiF family of proteins encoded by lactococcal plasmids and are mainly active against bacteriophage groups 936 and C2. The unique presence of this phage-resistance gene in strain LMD9 might indicate a strain-dependent anti-phage characteristic. Moreover, the genes present in cluster S19 of the same strain encode a novel phage-resistance system CRISPR3 (clustered regularly interspaced short palindromic repeats) with a low GC content (31%) indicative of a foreign origin. It agrees with the observation of Horvath et al. that CRISPR loci were possibly horizontally transferred and they may play an important role in adaptation to a specific environment (19, 20).
Putative HGT from foreign origin to L. bulgaricus A similar analysis of the genomes of L. bulgaricus ATCC 11842 and ATCC BAA365 also
revealed several putative HGT events (Table 6.2b, supplemental material Table S1, Figure S2 and Figure S3). The chromosomal regions with significantly lower GC content could be the result of horizontal gene transfer from either S. thermophilus or other low-GC organisms. The high δ values, showing higher dissimilarity on dinucleotide composition, and the features of the flanking regions related to mobile elements are indicative for the HGT event (Table 6.2b and supplemental material Table S1). In total 149 genes (including pseudogenes) were identified as putatively horizontally transferred, 120 of which were found to be associated with gene transfer elements. The large majority (130 of the 149 genes) were distributed in 24 gene clusters.
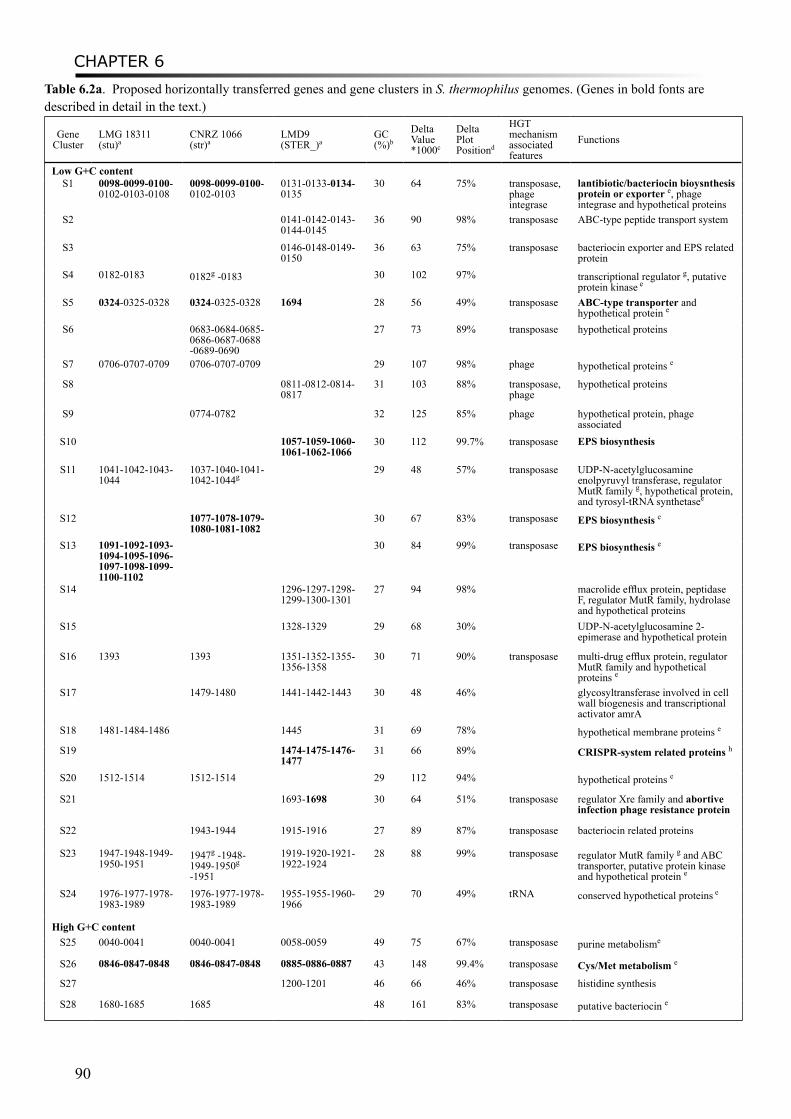
CHAPTER 6
90
Table 6.2a. Proposed horizontally transferred genes and gene clusters in S. thermophilus genomes. (Genes in bold fonts are described in detail in the text.)
Gene Cluster
LMG 18311 (stu)a
CNRZ 1066 (str)a
LMD9 (STER_)a
GC (%)b
DeltaValue*1000c
DeltaPlotPositiond
HGT mechanism associated features
Functions
Low G+C contentS1 0098-0099-0100-
0102-0103-01080098-0099-0100-0102-0103
0131-0133-0134-0135
30 64 75% transposase, phage integrase
lantibiotic/bacteriocin bioysnthesis protein or exporter e, phage integrase and hypothetical proteins
S2 0141-0142-0143-0144-0145
36 90 98% transposase ABC-type peptide transport system
S3 0146-0148-0149-0150
36 63 75% transposase bacteriocin exporter and EPS related protein
S4 0182-0183 0182g -0183 30 102 97% transcriptional regulator g, putative protein kinase e
S5 0324-0325-0328 0324-0325-0328 1694 28 56 49% transposase ABC-type transporter and hypothetical protein e
S6 0683-0684-0685-0686-0687-0688-0689-0690
27 73 89% transposase hypothetical proteins
S7 0706-0707-0709 0706-0707-0709 29 107 98% phage hypothetical proteins e
S8 0811-0812-0814-0817
31 103 88% transposase, phage
hypothetical proteins
S9 0774-0782 32 125 85% phage hypothetical protein, phage associated
S10 1057-1059-1060-1061-1062-1066
30 112 99.7% transposase EPS biosynthesis
S11 1041-1042-1043-1044
1037-1040-1041-1042-1044g
29 48 57% transposase UDP-N-acetylglucosamine enolpyruvyl transferase, regulator MutR family g, hypothetical protein, and tyrosyl-tRNA synthetasee
S12 1077-1078-1079-1080-1081-1082
30 67 83% transposase EPS biosynthesis e
S13 1091-1092-1093-1094-1095-1096-1097-1098-1099-1100-1102
30 84 99% transposase EPS biosynthesis e
S14 1296-1297-1298-1299-1300-1301
27 94 98% macrolide efflux protein, peptidase F, regulator MutR family, hydrolase and hypothetical proteins
S15 1328-1329 29 68 30% UDP-N-acetylglucosamine 2-epimerase and hypothetical protein
S16 1393 1393 1351-1352-1355-1356-1358
30 71 90% transposase multi-drug efflux protein, regulator MutR family and hypothetical proteins e
S17 1479-1480 1441-1442-1443 30 48 46% glycosyltransferase involved in cell wall biogenesis and transcriptional activator amrA
S18 1481-1484-1486 1445 31 69 78% hypothetical membrane proteins e
S19 1474-1475-1476-1477
31 66 89% CRISPR-system related proteins h
S20 1512-1514 1512-1514 29 112 94% hypothetical proteins e
S21 1693-1698 30 64 51% transposase regulator Xre family and abortive infection phage resistance protein
S22 1943-1944 1915-1916 27 89 87% transposase bacteriocin related proteins
S23 1947-1948-1949-1950-1951
1947g -1948-1949-1950g -1951
1919-1920-1921-1922-1924
28 88 99% transposase regulator MutR family g and ABC transporter, putative protein kinase and hypothetical protein e
S24 1976-1977-1978-1983-1989
1976-1977-1978-1983-1989
1955-1955-1960-1966
29 70 49% tRNA conserved hypothetical proteins e
High G+C contentS25 0040-0041 0040-0041 0058-0059 49 75 67% transposase purine metabolisme
S26 0846-0847-0848 0846-0847-0848 0885-0886-0887 43 148 99.4% transposase Cys/Met metabolism e
S27 1200-1201 46 66 46% transposase histidine synthesis
S28 1680-1685 1685 48 161 83% transposase putative bacteriocin e
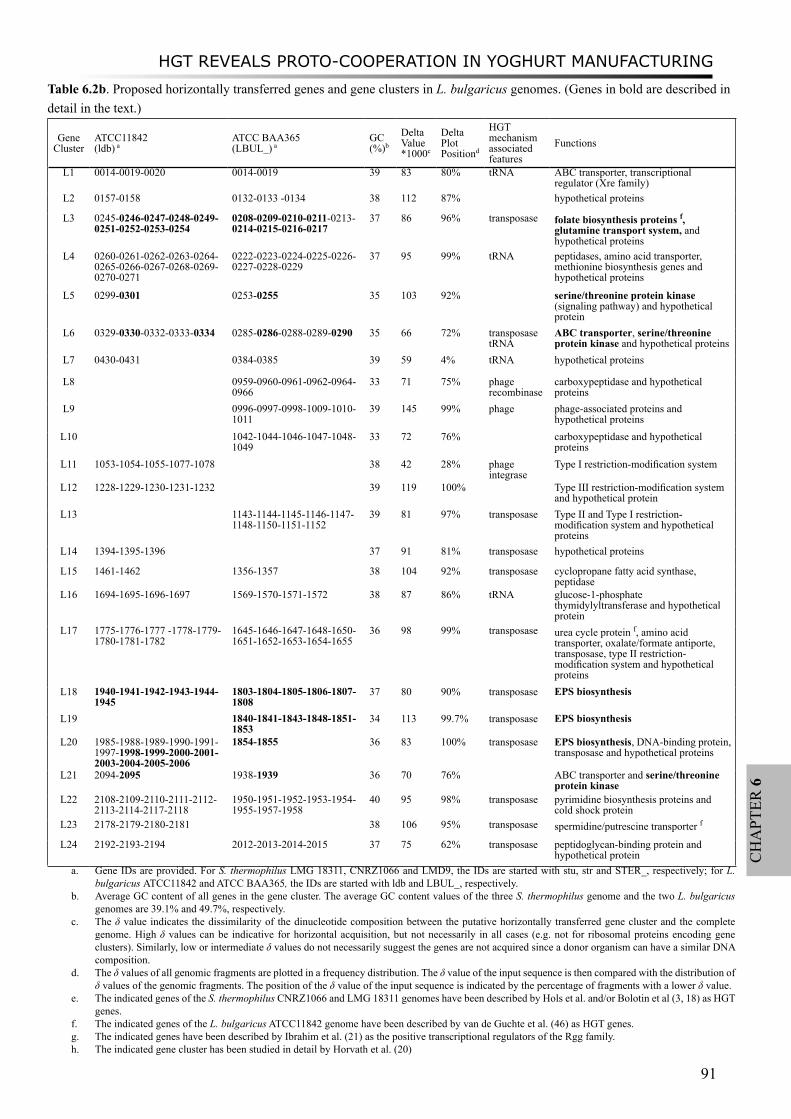
HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
91
CH
APT
ER 6
Table 6.2b. Proposed horizontally transferred genes and gene clusters in L. bulgaricus genomes. (Genes in bold are described in detail in the text.)
Gene Cluster
ATCC11842 (ldb) a
ATCC BAA365 (LBUL_) a
GC (%)b
DeltaValue*1000c
DeltaPlotPositiond
HGT mechanism associated features
Functions
L1 0014-0019-0020 0014-0019 39 83 80% tRNA ABC transporter, transcriptional regulator (Xre family)
L2 0157-0158 0132-0133 -0134 38 112 87% hypothetical proteins
L3 0245-0246-0247-0248-0249-0251-0252-0253-0254
0208-0209-0210-0211-0213-0214-0215-0216-0217
37 86 96% transposase folate biosynthesis proteins f, glutamine transport system, and hypothetical proteins
L4 0260-0261-0262-0263-0264-0265-0266-0267-0268-0269-0270-0271
0222-0223-0224-0225-0226-0227-0228-0229
37 95 99% tRNA peptidases, amino acid transporter, methionine biosynthesis genes and hypothetical proteins
L5 0299-0301 0253-0255 35 103 92% serine/threonine protein kinase (signaling pathway) and hypothetical protein
L6 0329-0330-0332-0333-0334 0285-0286-0288-0289-0290 35 66 72% transposasetRNA
ABC transporter, serine/threonine protein kinase and hypothetical proteins
L7 0430-0431 0384-0385 39 59 4% tRNA hypothetical proteins
L8 0959-0960-0961-0962-0964-0966
33 71 75% phage recombinase
carboxypeptidase and hypothetical proteins
L9 0996-0997-0998-1009-1010-1011
39 145 99% phage phage-associated proteins and hypothetical proteins
L10 1042-1044-1046-1047-1048-1049
33 72 76% carboxypeptidase and hypothetical proteins
L11 1053-1054-1055-1077-1078 38 42 28% phage integrase
Type I restriction-modification system
L12 1228-1229-1230-1231-1232 39 119 100% Type III restriction-modification system and hypothetical protein
L13 1143-1144-1145-1146-1147-1148-1150-1151-1152
39 81 97% transposase Type II and Type I restriction-modification system and hypothetical proteins
L14 1394-1395-1396 37 91 81% transposase hypothetical proteins
L15 1461-1462 1356-1357 38 104 92% transposase cyclopropane fatty acid synthase, peptidase
L16 1694-1695-1696-1697 1569-1570-1571-1572 38 87 86% tRNA glucose-1-phosphate thymidylyltransferase and hypothetical protein
L17 1775-1776-1777 -1778-1779-1780-1781-1782
1645-1646-1647-1648-1650-1651-1652-1653-1654-1655
36 98 99% transposase urea cycle protein f, amino acid transporter, oxalate/formate antiporte, transposase, type II restriction-modification system and hypothetical proteins
L18 1940-1941-1942-1943-1944-1945
1803-1804-1805-1806-1807-1808
37 80 90% transposase EPS biosynthesis
L19 1840-1841-1843-1848-1851-1853
34 113 99.7% transposase EPS biosynthesis
L20 1985-1988-1989-1990-1991-1997-1998-1999-2000-2001-2003-2004-2005-2006
1854-1855 36 83 100% transposase EPS biosynthesis, DNA-binding protein, transposase and hypothetical proteins
L21 2094-2095 1938-1939 36 70 76% ABC transporter and serine/threonine protein kinase
L22 2108-2109-2110-2111-2112-2113-2114-2117-2118
1950-1951-1952-1953-1954-1955-1957-1958
40 95 98% transposase pyrimidine biosynthesis proteins and cold shock protein
L23 2178-2179-2180-2181 38 106 95% transposase spermidine/putrescine transporter f
L24 2192-2193-2194 2012-2013-2014-2015 37 75 62% transposase peptidoglycan-binding protein and hypothetical protein
a. Gene IDs are provided. For S. thermophilus LMG 18311, CNRZ1066 and LMD9, the IDs are started with stu, str and STER_, respectively; for L. bulgaricus ATCC11842 and ATCC BAA365, the IDs are started with ldb and LBUL_, respectively.
b. Average GC content of all genes in the gene cluster. The average GC content values of the three S. thermophilus genome and the two L. bulgaricus genomes are 39.1% and 49.7%, respectively.
c. The δ value indicates the dissimilarity of the dinucleotide composition between the putative horizontally transferred gene cluster and the complete genome. High δ values can be indicative for horizontal acquisition, but not necessarily in all cases (e.g. not for ribosomal proteins encoding gene clusters). Similarly, low or intermediate δ values do not necessarily suggest the genes are not acquired since a donor organism can have a similar DNA composition.
d. The δ values of all genomic fragments are plotted in a frequency distribution. The δ value of the input sequence is then compared with the distribution of δ values of the genomic fragments. The position of the δ value of the input sequence is indicated by the percentage of fragments with a lower δ value.
e. The indicated genes of the S. thermophilus CNRZ1066 and LMG 18311 genomes have been described by Hols et al. and/or Bolotin et al (3, 18) as HGT genes.
f. The indicated genes of the L. bulgaricus ATCC11842 genome have been described by van de Guchte et al. (46) as HGT genes.g. The indicated genes have been described by Ibrahim et al. (21) as the positive transcriptional regulators of the Rgg family.h. The indicated gene cluster has been studied in detail by Horvath et al. (20)

CHAPTER 6
92
Among the predicted HGT events in L. bulgaricus, genes encoding transporter systems, restriction-modification systems and EPS biosynthesis clusters are highly represented (Table 6.2b). Genes present in the EPS biosynthesis clusters L18, L19 and L20 (Table 6.3), have low-GC content values (from 34% to 37%) and relatively high δ values (between 0.08 and 0.11) which are higher than those of about 90% of the genome fragments. This indicates that most EPS biosynthesis genes in L. bulgaricus are possibly derived from phylogenetically unrelated low-GC organisms.
Of the predicted horizontally transferred gene clusters, 15 out of 24 are found in both L. bulgaricus genomes (Table 6.2b). For example, Cluster L6 harboring genes encoding an ABC-type transporter has the best blast hit with Streptococcus pneumoniae TIGR4. Another example is the folate biosynthesis operon folB-folKE-folC-folP (cluster L3) which has been described previously by van Guchte et al.(46). L. bulgaricus strains are not able to produce folate, one of the essential components in human diets (45). This could be due to the absence of the genes involved in the biosynthesis of p-aminobenzoic acid (PABA), a precursor of folate. The PABA biosynthesis genes are present in the genomes of S. thermophilus. During the co-growth of both organisms, the S. thermophilus cells may provide PABA to L. bulgaricus cells, thereby enabling them to produce folate (43, 46). A glutamine transporter system (LBUL_0214-0217 or ldb0251-0254) present in the same cluster L3 could also be acquired by HGT. All the putative horizontally transferred genes indicate that the region harboring gene cluster L3 could be an active region with respect to genomic recombination. The ISL5-like (29) transposase gene (LBUL_0221) in this region provides additional suggestions for the predicted HGT events.
Another interesting finding is the identification of three eukaryotic-type serine/threonine protein kinase genes (Ldb0301, ldb0334 and Ldb2095 or LBUL_0255, LBUL_0290 and LBUL_1939) in gene clusters L5, L6 and L21, respectively. Their protein sequences share about 30% identity with S. thermophilus homologs. Notably, BLAST analysis found the homologs of these genes only present in genomes of S. thermophilus, L. bulgaricus and one Lactococcus strain, but absent in other sequenced LAB genomes. The nucleotide sequence compositions of these protein kinase genes were not very similar to the S. thermophilus or L. bulgaricus genomes, suggesting a putative horizontal transfer event of the kinase genes from a foreign origin to S. thermophilus and L. bulgaricus genomes. Serine/threonine protein kinases are commonly found in eukaryotes, and they have been reported to play a role in bacterial growth and development in many bacterial species (28, 51). The function of the serine/threonine protein kinases in L. bulgaricus and S. thermophilus remains to be discovered.
HGT between L. bulgaricus and S. thermophilus: cbs-cblB/cglB-cysE as an exampleIn addition to the HGT events in L. bulgaricus and S. thermophilus genomes from foreign
origins, we also identified putative HGT events between L. bulgaricus and S. thermophilus genomes. The EPS biosynthesis genes epsI-ML (ldb1998-2000) in cluster L20 of L. bulgaricus were possibly acquired from S. thermophilus, since they share high sequence homology with the eps15 gene (stu1092) of S. thermophilus LMG18311 and eps3I of another S. thermophilus strain, with 46% and 49% identity at the amino acid level, respectively (Table 6.3). EPSI-L (ldb1999-2000) is truncated due to an in-frame stop codon. A phylogenetic analysis using the homologs of EPSI-M and EPSI-L supported the hypothesis that the genes were transferred from S. thermophilus to L. bulgaricus (data not shown). The transfer of EPS biosynthesis genes between S. thermophilus and L. bulgaricus could play a role in (the optimization of) the physical interaction between the species in mixed yoghurt cultures and, thus, in the exchange of metabolites and/or stimulatory factors. This hypothesis is supported by the observation that EPS production is also influenced by co-cultivation of both species (personal communication S. Sieuwerts, C. Ingham and J. van Hylckama Vlieg). For kefir, similar findings have been obtained for L. kefiranofaciens and S. cerevisiae (5). The physical contact between both species enhanced the capsular kefiran production of L. kefiranofaciens.
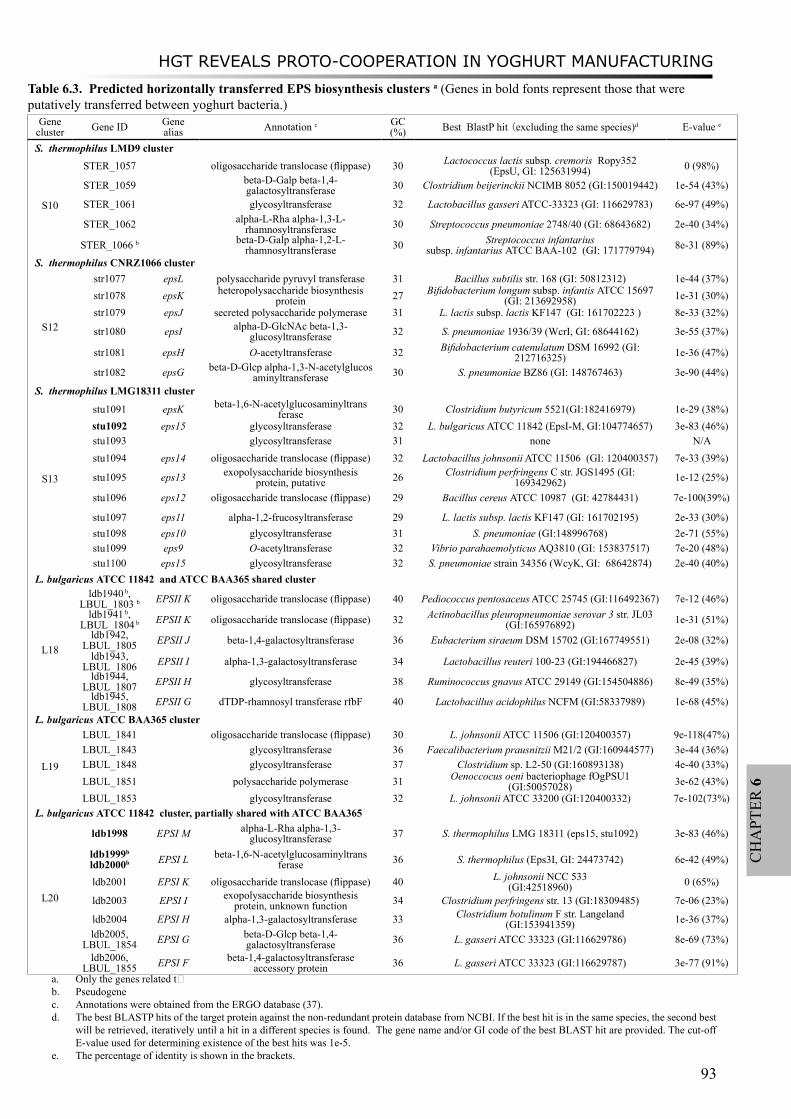
HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
93
CH
APT
ER 6
Table 6.3. Predicted horizontally transferred EPS biosynthesis clusters a (Genes in bold fonts represent those that were putatively transferred between yoghurt bacteria.)
Gene cluster Gene ID Gene
alias Annotation c GC (%) Best BlastP hit (excluding the same species)d E-value e
S. thermophilus LMD9 cluster
S10
STER_1057 oligosaccharide translocase (flippase) 30 Lactococcus lactis subsp. cremoris Ropy352 (EpsU, GI: 125631994) 0 (98%)
STER_1059 beta-D-Galp beta-1,4-galactosyltransferase 30 Clostridium beijerinckii NCIMB 8052 (GI:150019442) 1e-54 (43%)
STER_1061 glycosyltransferase 32 Lactobacillus gasseri ATCC-33323 (GI: 116629783) 6e-97 (49%)
STER_1062 alpha-L-Rha alpha-1,3-L-rhamnosyltransferase 30 Streptococcus pneumoniae 2748/40 (GI: 68643682) 2e-40 (34%)
STER_1066 b beta-D-Galp alpha-1,2-L-rhamnosyltransferase 30 Streptococcus infantarius
subsp. infantarius ATCC BAA-102 (GI: 171779794) 8e-31 (89%)
S. thermophilus CNRZ1066 cluster
S12
str1077 epsL polysaccharide pyruvyl transferase 31 Bacillus subtilis str. 168 (GI: 50812312) 1e-44 (37%)str1078 epsK heteropolysaccharide biosynthesis
protein 27 Bifidobacterium longum subsp. infantis ATCC 15697 (GI: 213692958) 1e-31 (30%)
str1079 epsJ secreted polysaccharide polymerase 31 L. lactis subsp. lactis KF147 (GI: 161702223 ) 8e-33 (32%)
str1080 epsI alpha-D-GlcNAc beta-1,3-glucosyltransferase 32 S. pneumoniae 1936/39 (WcrI, GI: 68644162) 3e-55 (37%)
str1081 epsH O-acetyltransferase 32 Bifidobacterium catenulatum DSM 16992 (GI: 212716325) 1e-36 (47%)
str1082 epsG beta-D-Glcp alpha-1,3-N-acetylglucosaminyltransferase 30 S. pneumoniae BZ86 (GI: 148767463) 3e-90 (44%)
S. thermophilus LMG18311 cluster
S13
stu1091 epsK beta-1,6-N-acetylglucosaminyltransferase 30 Clostridium butyricum 5521(GI:182416979) 1e-29 (38%)
stu1092 eps15 glycosyltransferase 32 L. bulgaricus ATCC 11842 (EpsI-M, GI:104774657) 3e-83 (46%)stu1093 glycosyltransferase 31 none N/Astu1094 eps14 oligosaccharide translocase (flippase) 32 Lactobacillus johnsonii ATCC 11506 (GI: 120400357) 7e-33 (39%)
stu1095 eps13 exopolysaccharide biosynthesis protein, putative 26 Clostridium perfringens C str. JGS1495 (GI:
169342962) 1e-12 (25%)
stu1096 eps12 oligosaccharide translocase (flippase) 29 Bacillus cereus ATCC 10987 (GI: 42784431) 7e-100(39%)
stu1097 eps11 alpha-1,2-frucosyltransferase 29 L. lactis subsp. lactis KF147 (GI: 161702195) 2e-33 (30%)stu1098 eps10 glycosyltransferase 31 S. pneumoniae (GI:148996768) 2e-71 (55%)stu1099 eps9 O-acetyltransferase 32 Vibrio parahaemolyticus AQ3810 (GI: 153837517) 7e-20 (48%)stu1100 eps15 glycosyltransferase 32 S. pneumoniae strain 34356 (WcyK, GI: 68642874) 2e-40 (40%)
L. bulgaricus ATCC 11842 and ATCC BAA365 shared cluster
L18
ldb1940 b, LBUL_1803 b EPSII K oligosaccharide translocase (flippase) 40 Pediococcus pentosaceus ATCC 25745 (GI:116492367) 7e-12 (46%)
ldb1941 b, LBUL_1804 b EPSII K oligosaccharide translocase (flippase) 32 Actinobacillus pleuropneumoniae serovar 3 str. JL03
(GI:165976892) 1e-31 (51%)ldb1942,
LBUL_1805 EPSII J beta-1,4-galactosyltransferase 36 Eubacterium siraeum DSM 15702 (GI:167749551) 2e-08 (32%)ldb1943,
LBUL_1806 EPSII I alpha-1,3-galactosyltransferase 34 Lactobacillus reuteri 100-23 (GI:194466827) 2e-45 (39%)ldb1944,
LBUL_1807 EPSII H glycosyltransferase 38 Ruminococcus gnavus ATCC 29149 (GI:154504886) 8e-49 (35%)ldb1945,
LBUL_1808 EPSII G dTDP-rhamnosyl transferase rfbF 40 Lactobacillus acidophilus NCFM (GI:58337989) 1e-68 (45%)
L. bulgaricus ATCC BAA365 cluster
L19
LBUL_1841 oligosaccharide translocase (flippase) 30 L. johnsonii ATCC 11506 (GI:120400357) 9e-118(47%)LBUL_1843 glycosyltransferase 36 Faecalibacterium prausnitzii M21/2 (GI:160944577) 3e-44 (36%)LBUL_1848 glycosyltransferase 37 Clostridium sp. L2-50 (GI:160893138) 4e-40 (33%)LBUL_1851 polysaccharide polymerase 31 Oenoccocus oeni bacteriophage fOgPSU1
(GI:50057028) 3e-62 (43%)LBUL_1853 glycosyltransferase 32 L. johnsonii ATCC 33200 (GI:120400332) 7e-102(73%)
L. bulgaricus ATCC 11842 cluster, partially shared with ATCC BAA365
L20
ldb1998 EPSI M alpha-L-Rha alpha-1,3-glucosyltransferase 37 S. thermophilus LMG 18311 (eps15, stu1092) 3e-83 (46%)
ldb1999b ldb2000b EPSI L beta-1,6-N-acetylglucosaminyltrans
ferase 36 S. thermophilus (Eps3I, GI: 24473742) 6e-42 (49%)
ldb2001 EPSI K oligosaccharide translocase (flippase) 40 L. johnsonii NCC 533(GI:42518960) 0 (65%)
ldb2003 EPSI I exopolysaccharide biosynthesis protein, unknown function 34 Clostridium perfringens str. 13 (GI:18309485) 7e-06 (23%)
ldb2004 EPSI H alpha-1,3-galactosyltransferase 33 Clostridium botulinum F str. Langeland (GI:153941359) 1e-36 (37%)
ldb2005, LBUL_1854 EPSI G beta-D-Glcp beta-1,4-
galactosyltransferase 36 L. gasseri ATCC 33323 (GI:116629786) 8e-69 (73%)
ldb2006, LBUL_1855 EPSI F beta-1,4-galactosyltransferase
accessory protein 36 L. gasseri ATCC 33323 (GI:116629787) 3e-77 (91%)a. Only the genes related t�b. Pseudogenec. Annotations were obtained from the ERGO database (37).d. The best BLASTP hits of the target protein against the non-redundant protein database from NCBI. If the best hit is in the same species, the second best
will be retrieved, iteratively until a hit in a different species is found. The gene name and/or GI code of the best BLAST hit are provided. The cut-off E-value used for determining existence of the best hits was 1e-5.
e. The percentage of identity is shown in the brackets.

CHAPTER 6
94
Another example of an HGT event between S. thermophilus and L. bulgaricus is the cbs-cblB/cglB-cysE gene cluster encoding enzymes involved in sulfur-containing amino acid metabolism. The gene cluster was originally named cysM2-metB2-cysE2 (3), but renamed in our previous study on basis of a consistent functional annotation in LAB (30). Since this HGT event could give insight in the co-evolution in milk and proto-cooperation between the two bacterial species during yoghurt fermentation, we performed an in-depth comparative analysis on the evolution of this gene cluster, in the light of phylogeny, gene context and sequence features.
The cbs-cblB/cglB-cysE gene cluster is located in a 3.6 kb region (including a 600 bp upstream non-coding region) in S. thermophilus cluster S26. The corresponding genes in L. bulgaricus and S. thermophilus share more than 94% nucleotide sequence identity. Using the δρ-Web tool, we analyzed the sequence similarity between this DNA fragment and the whole genomes of S. thermophilus respectively: the cbs-cblB/cglB-cysE fragment has a very high δ value compared to the rest of the genome, indicating a lower sequence composition similarity between the DNA fragment and the S. thermophilus genomes. The δ values of the cbs-cblB/cglB-cysE cluster against S. thermophilus genomes are much higher than the δ values against L. bulgaricus genomes. Moreover, phylogenetic analysis also supported the prediction of this HGT event, and provided more insight in the evolutionary events leading to transfer of this cluster (Figure 6.1). The cbs-cblB/cglB gene clusters of S. thermophilus are closely linked to those of L. bulgaricus and L. helveticus (lactobacilli branch), instead of L. lactis in the classic phylogenetic tree, suggesting a HGT event from L. bulgaricus or L. helveticus to S. thermophilus. Although the sequence composition of this gene cluster is more similar to L. bulgaricus genomes than to the L. helveticus genome (lower δ values and more similar GC content), the difference is not significant enough to conclude that the cluster originates from L. bulgaricus.
[1000]
[1000]
[776]
[1000]
[786]
[635]
[982]
[1000]
[642]
[872]
[997]
[692]
[1000]
[1000]
Lb. helveticus CNRZ 32
Lb. helveticus DPC 4571
Lb. delbrueckii bulgaricus ATCC 11842
Lb. delbrueckii bulgaricus ATCC BAA365 St. thermophilus LMD9
St. thermophilus CNRZ1066
HGT
St. thermophilus LMG18311
Lb. plantarum WCFS1
Leuconostoc mesenteroides ATCC8293 Oenococcus oeni PSU1
Lb. salivarius UCC118Lb. acidophilus NCFM
Lb. reuteri F275
Lc. lactis subsp. cremoris SK11
Lc. lactis subsp. lactis IL1403
Lc. lactis subsp. cremoris MG1363Bacillus subtilis 168
0.1
Figure 6.1. Phylogenetic tree of the cbs-cblB/cglB-cysE gene cluster in LAB genomes. The tree is constructed on the basis of the alignment of concatenated sequences of the cbs and cblB/cglB genes. Since the gene cysE is a pseudogene in a few genomes, it is not taken into account in the phylogenetic analysis. Bootstrap values are reported for a total of 1,000 replicates. Truncated cbs and cblB/cglB genes from Lactobacillus acidophilus NCFM are also included in the tree. Genes from Bacillus subtilis 168 are used as the out-group. The HGT event is indicated by an arrow. Lb., Lactobacillus; St., Streptococus; Lc., for Lactococcus.

HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
95
CH
APT
ER 6
Gene context analysis indicates that L. bulgaricus, L. helveticus and S. thermophilus have the same gene organization in this region (Figure 6.2). Moreover, the locus is flanked by transposase genes, supporting recombination events to occur in this region. Multiple DNA sequence alignment of the cbs-cblB/cglB-cysE cluster revealed a 179 bp DNA fragment with a low GC content (34%) inserted into the open reading frame of the cysE gene in the L. bulgaricus genomes (supplemental material Figure S4). This sequence is responsible for the truncation of the cysE gene in L. bulgaricus. An inverted repeat and a direct repeat are found in close proximity to the boundaries of the insertion sequence (supplemental material Figure S4). The 179bp DNA sequence shares 98% identity with a transposase gene located in the vicinity of an EPS biosynthesis cluster in L. bulgaricus ATCC BAA365 (cluster L18). We found that this insertion element frequently occurs in non-functional genes (pseudogenes) of L. bulgaricus genomes (Table S2). For instance, in L. bulgaricus ATCC BAA365 it was found to be responsible for the truncation of the sucrose-6-phosphate hydrolase gene, a gene important for sucrose metabolism (22).
The cbs-cblB/cglB-cysE cluster in L. helveticus shares over 90% nucleotide sequence identity to the counterparts in L. bulgaricus. Interestingly, the cysE gene in L. helveticus is intact. This suggests that S. thermophilus could have acquired this gene cluster from L. helveticus. Both species are used in conjunction in the manufacture of Bulgarian type yoghurt (11).
Role of the horizontally transferred cbs-cblB/cglB-cysE cluster in proto-cooperationThe enzymes encoded by the cbs-cblB/cglB-cysE gene cluster are involved in the metabolism
of sulfur-containing amino acids. The metabolic pathway of the inter-conversion between cysteine and methionine containing these enzymes is shown in Figure 6.3. Serine acetyltransferase, encoded by
Figure 6.2. Gene context of cbs-cblB/cglB-cysE in L. bulgaricus, L. helveticus and S. thermophilus. (A colour version can be found in Appendix). The clusters have the same gene context, while cysE in L. bulgaricus strains are truncated due to the presence of an insertion sequence (see supplemental material Figure S4). PSG, pseudogene. Dotted arrows also represent the pseudogenes. Predicted terminators are indicated by mushroom symbols. Transposase genes are shadowed by grey diagonal stripes. Modified from a image created with the Microbial Genome Viewer application (26).
816k 819k 822k
1146k 1149k
1125k 1128k 1131kk
783k 786k
777k 780k
cblB/cglB
cblB/cglB
cblB/cglB
cblB/cglB
cbs
cbs
cbs
cbs
cbs
Streptococcusthermophilus LMD9
Lactobacillushelveticus DPC 4571
delbrueckii ssp. bulgaricus ATCC BAA-365
Lactobacillusdelbrueckii ssp. bulgaricus ATCC 11842
Streptococcusthermophilus CNRZ1066
Streptococcusthermophilus LMG 18311
cblB/cglB
CysE PSG
cysE
cysE
cysE
Lactobacillus
CysE PSG
1872k1875k1878kcbs cblB/cglB cysE
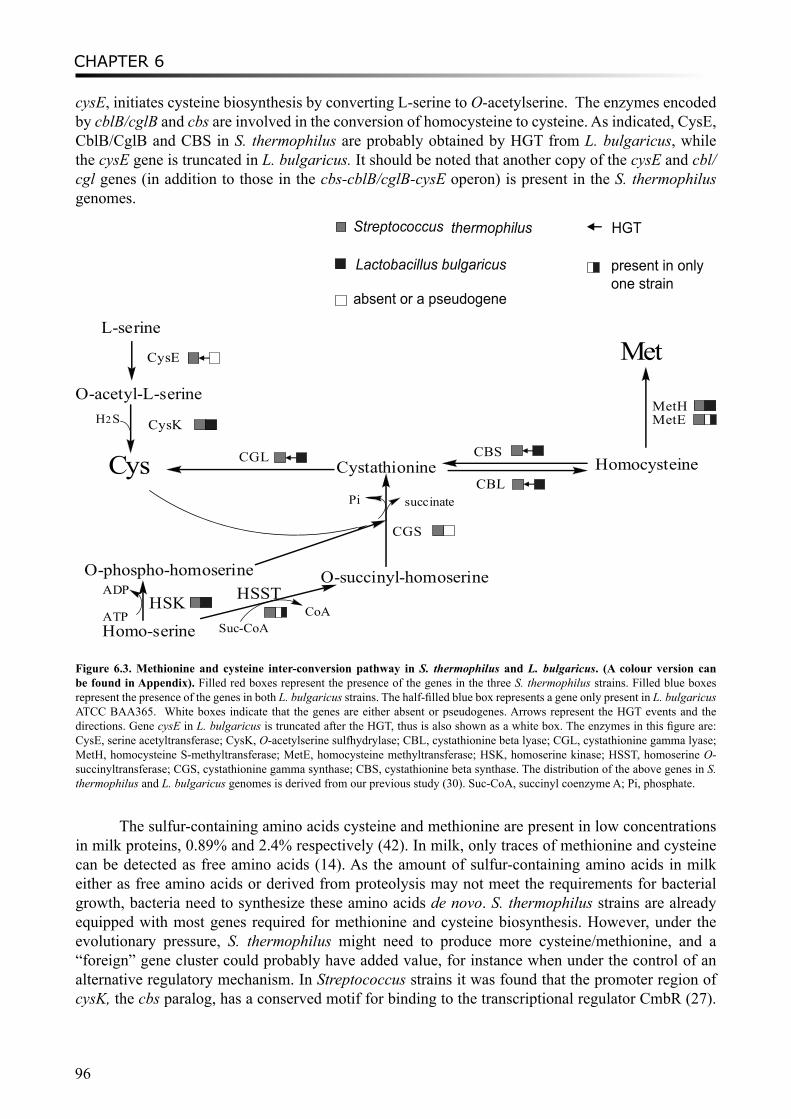
CHAPTER 6
96
cysE, initiates cysteine biosynthesis by converting L-serine to O-acetylserine. The enzymes encoded by cblB/cglB and cbs are involved in the conversion of homocysteine to cysteine. As indicated, CysE, CblB/CglB and CBS in S. thermophilus are probably obtained by HGT from L. bulgaricus, while the cysE gene is truncated in L. bulgaricus. It should be noted that another copy of the cysE and cbl/cgl genes (in addition to those in the cbs-cblB/cglB-cysE operon) is present in the S. thermophilus genomes.
Streptococcus thermophilus
Lactobacillus bulgaricus
Cys
L-serine
O-acetyl-L-serine
CysE
CysK
CGS
Cystathionine HomocysteineCBL
CGL CBS
Met
MetHMetE
Homo-serine
O-phospho-homoserine O-succinyl-homoserineHSK
ATP
ADP
CoASuc-CoA
HSST
succinatePi
H2S
HGT
absent or a pseudogene
present in onlyone strain
Figure 6.3. Methionine and cysteine inter-conversion pathway in S. thermophilus and L. bulgaricus. (A colour version can be found in Appendix). Filled red boxes represent the presence of the genes in the three S. thermophilus strains. Filled blue boxes represent the presence of the genes in both L. bulgaricus strains. The half-filled blue box represents a gene only present in L. bulgaricus ATCC BAA365. White boxes indicate that the genes are either absent or pseudogenes. Arrows represent the HGT events and the directions. Gene cysE in L. bulgaricus is truncated after the HGT, thus is also shown as a white box. The enzymes in this figure are: CysE, serine acetyltransferase; CysK, O-acetylserine sulfhydrylase; CBL, cystathionine beta lyase; CGL, cystathionine gamma lyase; MetH, homocysteine S-methyltransferase; MetE, homocysteine methyltransferase; HSK, homoserine kinase; HSST, homoserine O-succinyltransferase; CGS, cystathionine gamma synthase; CBS, cystathionine beta synthase. The distribution of the above genes in S. thermophilus and L. bulgaricus genomes is derived from our previous study (30). Suc-CoA, succinyl coenzyme A; Pi, phosphate.
The sulfur-containing amino acids cysteine and methionine are present in low concentrations in milk proteins, 0.89% and 2.4% respectively (42). In milk, only traces of methionine and cysteine can be detected as free amino acids (14). As the amount of sulfur-containing amino acids in milk either as free amino acids or derived from proteolysis may not meet the requirements for bacterial growth, bacteria need to synthesize these amino acids de novo. S. thermophilus strains are already equipped with most genes required for methionine and cysteine biosynthesis. However, under the evolutionary pressure, S. thermophilus might need to produce more cysteine/methionine, and a “foreign” gene cluster could probably have added value, for instance when under the control of an alternative regulatory mechanism. In Streptococcus strains it was found that the promoter region of cysK, the cbs paralog, has a conserved motif for binding to the transcriptional regulator CmbR (27).

HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
97
CH
APT
ER 6
This activator regulates the metC-cysK operon encoding a cystathionine lyase and cysteine synthase in Lactococcus lactis (15). This regulatory motif is not found upstream of the cbs-cblB/cglB-cysE gene cluster. However, we did find a GC-rich motif to be conserved in the upstream region of the cbs gene in S. thermophilus, L. bulgaricus, as well as L. plantarum, Oenococcus and Leuconostoc genomes, which may be a binding site for an alternative transcriptional regulator (supplemental material Figure S5). After transferring the intact gene cluster to S. thermophilus, the L. bulgaricus strains may have inactivated their cysE gene thereby losing the ability to synthesize cysteine. In contrast to S. thermophilus, L. bulgaricus lacks an active serine biosynthesis pathway, it is unnecessary to maintain a functional gene involved in this pathway for synthesizing cysteine from serine. A recent proteomics study on S. thermophilus LMG18311 revealed the up-regulation of sulfur-containing amino acids biosynthesis genes, including cbs (cysM2) and cblB/cglB (metB2), when grown in co-culture with L. bulgaricus ATCC11842 (16). The stimulatory effect of L. bulgaricus on this biosynthetic pathway in S. thermophilus suggests that the enzymes are indeed of importance for the proto-cooperation in yoghurt manufacturing.
ConclusionsThe proto-cooperation between L. bulgaricus and S. thermophilus in yoghurt manufacturing
has been previously described, but mainly with respect to their dependency of growth factors and metabolic interactions. In this study, we identified HGT events between the yoghurt bacteria and revealed proto-cooperation on basis of exchanged and/or acquired genetic elements during evolution. The genome-wide analysis generated in a list of genes and gene clusters, most probably obtained by HGT. The EPS biosynthesis proteins EPSI-ML in L. bulgaricus genomes were likely to be acquired from S. thermophilus. A gene cluster encoding the enzymes involved in sulfur-containing amino acids metabolism (cbs-cblB/cglB-cysE) in S. thermophilus was probably transferred from L. bulgaricus strains. The predicted HGT events in bacteria used in yoghurt manufacturing, S. thermophilus and L. bulgaricus, provide important information on their co-evolution and their proto-cooperation strategies for adaptation to the milk environment. The new insights could be used advantageously to improve the control of co-growth of both species in yoghurt manufacturing and, accordingly, to improve product characteristics.
AcknowledgementsThis work was supported by grant CSI4017 of the Casimir program of the Ministry of Economic Affairs, the Netherlands. We thank Dr. Ellen Looijesteijn for critically reading the manuscript and Celine Prakash for the analysis of the conserved regulatory motifs.

CHAPTER 6
98
References
1. Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-10.
2. Anba, J., E. Bidnenko, A. Hillier, D. Ehrlich, and M. C. Chopin. 1995. Characterization of the lactococcal abiD1 gene coding for phage abortive infection. J. Bacteriol. 177:3818-3823.
3. Bolotin, A., B. Quinquis, P. Renault, A. Sorokin, S. D. Ehrlich, S. Kulakauskas, A. Lapidus, E. Goltsman, M. Mazur, G. D. Pusch, M. Fonstein, R. Overbeek, N. Kyprides, B. Purnelle, D. Prozzi, K. Ngui, D. Masuy, F. Hancy, S. Burteau, M. Boutry, J. Delcour, A. Goffeau, and P. Hols. 2004. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat. Biotechnol. 22:1554-1558.
4. Callanan, M., P. Kaleta, J. O’Callaghan, O. O’Sullivan, K. Jordan, O. McAuliffe, A. Sangrador-Vegas, L. Slattery, G. F. Fitzgerald, T. Beresford, and R. P. Ross. 2008. Genome sequence of Lactobacillus helveticus, an organism distinguished by selective gene loss and insertion sequence element expansion. J. Bacteriol. 190:727-35.
5. Cheirsilp, B., H. Shoji, H. Shimizu, and S. Shioya. 2003. Interactions between Lactobacillus kefiranofaciens and Saccharomyces cerevisiae in mixed culture for kefiran production. J. Biosci. Bioeng. 96:279-84.
6. Courtin, P., V. Monnet, and F. Rul. 2002. Cell-wall proteinases PrtS and PrtB have a different role in Streptococcus thermophilus/Lactobacillus bulgaricus mixed cultures in milk. Microbiology 148:3413-3421.
7. Courtin, P., and F. Rul. 2004. Interactions between microorganisms in a simple ecosystem: yogurt bacteria as a study model. Lait 84:125-134.
8. Darling, A. C., B. Mau, F. R. Blattner, and N. T. Perna. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14:1394-1403.
9. Delcour, J., T. Ferain, M. Deghorain, E. Palumbo, and P. Hols. 1999. The biosynthesis and functionality of the cell-wall of lactic acid bacteria. Antonie Van Leeuwenhoek 76:159-84.
10. Delorme, C. 2008. Safety assessment of dairy microorganisms: Streptococcus thermophilus. Int. J. Food Microbiol. 126:274-277.
11. Dimitrov, Z., M. Michaylova, and S. Mincova. 2005. Characterization of Lactobacillus helveticus strains isolated from Bulgarian yoghurt, cheese, plants and human faecal samples by sodium dodecilsulfate polyacrylamide gel electrophoresis of cell-wall proteins, ribotyping and pulsed field gel fingerprinting. Int. Dairy J. 15:998-1005.
12. Edgar, R. C. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113.
13. Feder, M. E. 2007. Evolvability of physiological and biochemical traits: evolutionary mechanisms including and beyond single-nucleotide mutation. J. Exp. Biol. 210:1653-1660.
14. Ghadimi, H., and P. Pecora. 1963. Free amino acids of different kinds of milk. Am. J. Clin. Nutr. 13:75-81.15. Golic, N., M. Schliekelmann, M. Fernandez, M. Kleerebezem, and R. van Kranenburg. 2005. Molecular characterization
of the CmbR activator-binding site in the metC-cysK promoter region in Lactococcus lactis. Microbiology 151:439-446.16. Herve-Jimenez, L., I. Guillouard, E. Guedon, C. Gautier, S. Boudebbouze, P. Hols, V. Monnet, F. Rul, and E. Maguin.
2008. Physiology of Streptococcus thermophilus during the late stage of milk fermentation with special regard to sulfur amino-acid metabolism. Proteomics 8:4273-4286.
17. Hoffmeister, M., and W. Martin. 2003. Interspecific evolution: microbial symbiosis, endosymbiosis and gene transfer. Environmental Microbiology 5:641-649.
18. Hols, P., F. Hancy, L. Fontaine, B. Grossiord, D. Prozzi, N. Leblond-Bourget, B. Decaris, A. Bolotin, C. Delorme, S. Dusko Ehrlich, E. Guedon, V. Monnet, P. Renault, and M. Kleerebezem. 2005. New insights in the molecular biology and physiology of Streptococcus thermophilus revealed by comparative genomics. FEMS Microbiol. Rev. 29:435-463.
19. Horvath, P., A. C. Coute-Monvoisin, D. A. Romero, P. Boyaval, C. Fremaux, and R. Barrangou. 15 Jul 2008. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol. doi:10.1016/j.ijfoodmicro.2008.05.030.
20. Horvath, P., D. A. Romero, A. C. Coute-Monvoisin, M. Richards, H. Deveau, S. Moineau, P. Boyaval, C. Fremaux, and R. Barrangou. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190:1401-1412.
21. Ibrahim, M., P. Nicolas, P. Bessieres, A. Bolotin, V. Monnet, and R. Gardan. 2007. A genome-wide survey of short coding sequences in streptococci. Microbiology 153:3631-44.
22. Iyer, R., and A. Camilli. 2007. Sucrose metabolism contributes to in vivo fitness of Streptococcus pneumoniae. Mol. Microbiol. 66:1-13.
23. Jain, R., M. C. Rivera, J. E. Moore, and J. A. Lake. 2002. Horizontal gene transfer in microbial genome evolution. Theor. Popul. Biol. 61:489-95.
24. Karlin, S. 2001. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 9:335-343.
25. Karlin, S., I. Ladunga, and B. E. Blaisdell. 1994. Heterogeneity of genomes: measures and values. Proc. Natl. Acad. Sci. U.S.A. 91:12837-12841.
26. Kerkhoven, R., F. H. van Enckevort, J. Boekhorst, D. Molenaar, and R. J. Siezen. 2004. Visualization for genomics: the Microbial Genome Viewer. Bioinformatics 20:1812-4.
27. Kovaleva, G. Y., and M. S. Gelfand. 2007. Transcriptional regulation of the methionine and cysteine transport and metabolism in streptococci. FEMS Microbiol. Lett. 276:207-215.
28. Krupa, A., and N. Srinivasan. 2005. Diversity in domain architectures of Ser/Thr kinases and their homologues in

HGT REVEALS PROTO-COOPERATION IN YOGHURT MANUFACTURING
99
CH
APT
ER 6
prokaryotes. BMC Genomics 6:129.29. Lapierre, L., B. Mollet, and J. E. Germond. 2002. Regulation and adaptive evolution of lactose operon expression in
Lactobacillus delbrueckii. J. Bacteriol. 184:928-935.30. Liu, M., A. Nauta, C. Francke, and R. J. Siezen. 2008. Comparative genomics of enzymes in flavor-forming pathways
from amino acids in lactic acid bacteria. Appl. Environ. Microbiol. 74:4590-4600.31. Liu, M., F. H. van Enckevort, and R. J. Siezen. 2005. Genome update: lactic acid bacteria genome sequencing is booming.
Microbiology 151:3811-3814.32. Larkin, M., G. Blackshileds, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWill, F. Valentin, I. M. Wallace, A.
Wilm, R. Lopez, J. D. Tompson,T. J. Gibson, and D. G. Higgins. 2007. ClustalW and ClustalX version 2.0. Bioinformatics 23:2947-8.
33. Makarova, K., A. Slesarev, Y. Wolf, A. Sorokin, B. Mirkin, E. Koonin, A. Pavlov, N. Pavlova, V. Karamychev, N. Polouchine, V. Shakhova, I. Grigoriev, Y. Lou, D. Rohksar, S. Lucas, K. Huang, D. M. Goodstein, T. Hawkins, V. Plengvidhya, D. Welker, J. Hughes, Y. Goh, A. Benson, K. Baldwin, J. H. Lee, I. Diaz-Muniz, B. Dosti, V. Smeianov, W. Wechter, R. Barabote, G. Lorca, E. Altermann, R. Barrangou, B. Ganesan, Y. Xie, H. Rawsthorne, D. Tamir, C. Parker, F. Breidt, J. Broadbent, R. Hutkins, D. O’Sullivan, J. Steele, G. Unlu, M. Saier, T. Klaenhammer, P. Richardson, S. Kozyavkin, B. Weimer, and D. Mills. 2006. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U.S.A. 103:15611-15616.
34. Makarova, K. S., and E. V. Koonin. 2007. Evolutionary genomics of lactic acid bacteria. J Bacteriol 189:1199-208.35. Makino, S., S. Ikegami, H. Kano, T. Sashihara, H. Sugano, H. Horiuchi, T. Saito, and M. Oda. 2006. Immunomodulatory
effects of polysaccharides produced by Lactobacillus delbrueckii ssp. bulgaricus OLL1073R-1. J. Dairy Sci. 89:2873-81.36. Nicolas, P., P. Bessieres, S. D. Ehrlich, E. Maguin, and M. van de Guchte. 2007. Extensive horizontal transfer of core
genome genes between two Lactobacillus species found in the gastrointestinal tract. BMC Evol. Biol. 7:141.37. Overbeek, R., N. Larsen, T. Walunas, M. D’Souza, G. Pusch, E. Selkov, K. Liolios, V. Joukov, D. Kaznadzey, I.
Anderson, A. Bhattacharyya, H. Burd, W. Gardner, P. Hanke, V. Kapatral, N. Mikhailova, O. Vasieva, A. Osterman, V. Vonstein, M. Fonstein, N. Ivanova, and N. Kyrpides. 2003. The ERGO (TM) genome analysis and discovery system. Nucleic Acids Res. 31:164-171.
38. Pette, J. W., and H. Lolkema. 1950. Yoghurt. I: Symbiosis and antibiosis of mixed cultures of Lactobacillus bulgaricus and Streptococcus thermophilus. Neth. Milk Dairy J. 4:197-208.
39. Pfeiler, E. A., and T. R. Klaenhammer. 2007. The genomics of lactic acid bacteria. Trends Microbiol. 15:546-53.40. Rasmussen, T. B., M. Danielsen, O. Valina, C. Garrigues, E. Johansen, and M. B. Pedersen. 2008. Streptococcus
thermophilus core genome: comparative genome hybridization study of 47 strains. Appl. Environ. Microbiol. 74:4703-4710.
41. Rutherford, K., J. Parkhill, J. Crook, T. Horsnell, P. Rice, M. A. Rajandream, and B. Barrell. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944-5.
42. Rutherfurd, S. M., and P. J. Moughan. 1998. The digestible amino acid composition of several milk proteins: application of a new bioassay. J. Dairy Sci. 81:909-917.
43. Sieuwerts, S., F. A. de Bok, J. Hugenholtz, and J. E. van Hylckama Vlieg. 2008. Unraveling microbial interactions in food fermentations: from classical to genomics approaches. Appl. Environ. Microbiol. 74:4997-5007.
44. Siezen, R. J., and H. Bachmann. 2008. Genomics of dairy fermentations. Microbial Biotechnology 1:435-442.45. Sybesma, W., M. Starrenburg, L. Tijsseling, M. H. Hoefnagel, and J. Hugenholtz. 2003. Effects of cultivation conditions
on folate production by lactic acid bacteria. Appl. Environ. Microbiol. 69:4542-4548.46. van de Guchte, M., S. Penaud, C. Grimaldi, V. Barbe, K. Bryson, P. Nicolas, C. Robert, S. Oztas, S. Mangenot, A.
Couloux, V. Loux, R. Dervyn, R. Bossy, A. Bolotin, J. M. Batto, T. Walunas, J. F. Gibrat, P. Bessieres, J. Weissenbach, S. D. Ehrlich, and E. Maguin. 2006. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc. Natl. Acad. Sci. U.S.A. 103:9274-9279.
47. van der Heijden, R. T., B. Snel, V. van Noort, and M. A. Huynen. 2007. Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinformatics 8:83.
48. van Passel, M. W., A. C. Luyf, A. H. van Kampen, A. Bart, and A. van der Ende. 2005. Deltarho-web, an online tool to assess composition similarity of individual nucleic acid sequences. Bioinformatics 21:3053-3055.
49. Welman, A. D., and I. S. Maddox. 2003. Exopolysaccharides from lactic acid bacteria: perspectives and challenges. Trends Biotechnol. 21:269-274.
50. Zaneveld, J. R., D. R. Nemergut, and R. Knight. 2008. Are all horizontal gene transfers created equal? Prospects for mechanism-based studies of HGT patterns. Microbiology 154:1-15.
51. Zhang, C. C. 1996. Bacterial signalling involving eukaryotic-type protein kinases. Mol. Microbiol. 20:9-15.

CHAPTER 6
100
读万卷书,行万里路
- 古训
In order to attain wisdom, it is not enough merely to read books, you must be well travelled as well.
- unknown author

SUMMARIZING DISCUSSION
101
CH
APT
ER 7
Chapter 7
SUMMARIZING DISCUSSION
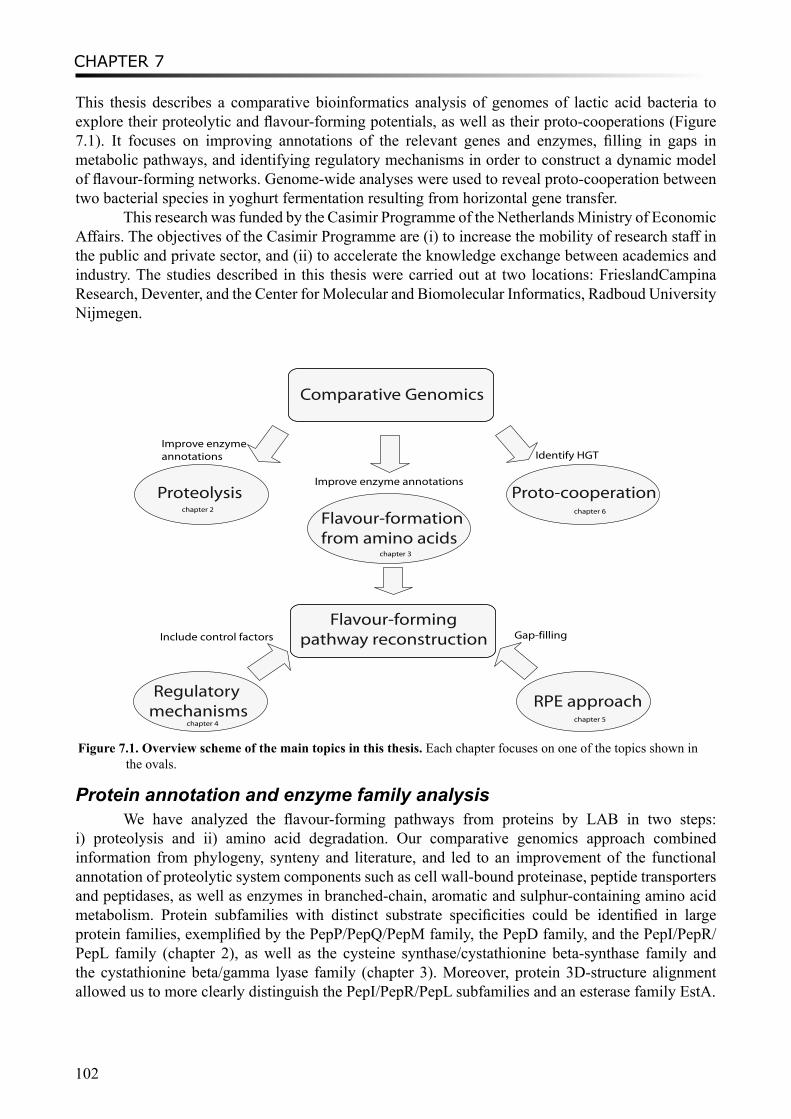
CHAPTER 7
102
This thesis describes a comparative bioinformatics analysis of genomes of lactic acid bacteria to explore their proteolytic and flavour-forming potentials, as well as their proto-cooperations (Figure 7.1). It focuses on improving annotations of the relevant genes and enzymes, filling in gaps in metabolic pathways, and identifying regulatory mechanisms in order to construct a dynamic model of flavour-forming networks. Genome-wide analyses were used to reveal proto-cooperation between two bacterial species in yoghurt fermentation resulting from horizontal gene transfer.
This research was funded by the Casimir Programme of the Netherlands Ministry of Economic Affairs. The objectives of the Casimir Programme are (i) to increase the mobility of research staff in the public and private sector, and (ii) to accelerate the knowledge exchange between academics and industry. The studies described in this thesis were carried out at two locations: FrieslandCampina Research, Deventer, and the Center for Molecular and Biomolecular Informatics, Radboud University Nijmegen.
Comparative Genomics
Proteolysis
Flavour-formationfrom amino acids
Proto-cooperation
Improve enzyme annotations
Improve enzyme annotations
Identify HGT
Flavour-formingpathway reconstruction
Regulatory mechanisms
RPE approach
Gap-fillingInclude control factors
chapter 2
chapter 3
chapter 4chapter 5
chapter 6
Figure 7.1. Overview scheme of the main topics in this thesis. Each chapter focuses on one of the topics shown in the ovals.
Protein annotation and enzyme family analysisWe have analyzed the flavour-forming pathways from proteins by LAB in two steps:
i) proteolysis and ii) amino acid degradation. Our comparative genomics approach combined information from phylogeny, synteny and literature, and led to an improvement of the functional annotation of proteolytic system components such as cell wall-bound proteinase, peptide transporters and peptidases, as well as enzymes in branched-chain, aromatic and sulphur-containing amino acid metabolism. Protein subfamilies with distinct substrate specificities could be identified in large protein families, exemplified by the PepP/PepQ/PepM family, the PepD family, and the PepI/PepR/PepL family (chapter 2), as well as the cysteine synthase/cystathionine beta-synthase family and the cystathionine beta/gamma lyase family (chapter 3). Moreover, protein 3D-structure alignment allowed us to more clearly distinguish the PepI/PepR/PepL subfamilies and an esterase family EstA.

SUMMARIZING DISCUSSION
103
CH
APT
ER 7
Proteolytic and flavour-forming potentialThe improved functional annotations were used to predict the proteolytic capacities in various
species or strains of LAB. For example, the LAB genomes in the L. acidophilus group, including L. acidophilus, L. johnsonii, L. gasseri, L. bulgaricus, and L. helveticus strains, encode a relatively higher number and variety of proteolytic system components. The cell-wall bound proteinase (PrtP) was only found on the chromosome of L. acidophilus, L. johnsonii, L. bulgaricus, L. casei, L. rhamnosus and S. thermophilus strain LMD9, as well as on the plasmid of L. lactis subsp. cremoris SK11. LAB species of plant origin, such as L. plantarum, O. oeni, and Leuconostoc mesenteroides, encode less proteolytic enzymes in their genomes, which agrees with their ecological niche that is fiber-rich but contains less proteins. In addition, proteolytic components in 39 Lactococcus strains, including proteinase, oligopeptide transport system and peptidases were shown to be distributed unevenly, probably due to plasmids which encode them, and reflect the environment from which they were isolated. Similarly, flavour-forming potential from amino acids by various LAB was also predicted. S. thermophilus and Lactococcus strains as well as some other milk-associated LAB e.g. Lb. casei, seem to possess relatively more abundant flavour-forming enzymes, whereas many of them are lacking in typical GI-tract bacteria e.g. L. gasseri and L. johnsonii.
Metabolic pathway reconstructionReconstructions of metabolic networks rely heavily on the correct annotation of genes and
proteins from the genome sequence of the organism. However, many genes and proteins in public databases have wrong annotations or are annotated as unknown function, leading to an incompleteness of the genome-scale metabolic model. Many of the metabolites that are identified by high-throughput techniques cannot be linked to the metabolic networks due to the limited genetic or biochemical knowledge, especially for those which are non-essential in central metabolism. We developed an in silico approach, named Reverse Pathway Engineering (RPE), to improve these models. This approach combines chemoinformatics and bioinformatics analyses, and predicts the missing links between target compounds and their possible metabolic precursors by providing plausible chemical or enzymatic reactions. Based on the transformation rules of a reaction database, which can be adjusted to the specific user case, the RPE approach can propose both chemical and enzymatic reactions, including ones that have never been described in databases or literature. We have validated this approach with the known metabolic routes leading to the formation of flavour compounds from leucine. We identified unknown flavour-forming reactions, i.e. the conversion of α-hydroxy-isocaproate to 3-methylbutanoic acid and the synthesis of dimethyl sulfide, as well as the corresponding enzymes. Those new findings filled in gaps of the incomplete flavour-forming pathways and provided leads to improve the flavour profiles by metabolic engineering.
RegulationRegulatory mechanisms of genes encoding twelve key enzymes associated with cysteine
and methionine metabolism were identified, and included both transcription factor binding sites and RNA riboswitches. Binding motifs of the known transcriptional regulators CmbR and MetR/MtaR in lactococci and streptococci were refined on basis of sequence consensus. Many putative transcription factor binding sites that have not been reported before were proposed using a comprehensive footprinting method. Inclusion of the regulation mechanisms of flavour-forming pathways contributes to the construction of a dynamic metabolic network where the control factors are integrated.

CHAPTER 7
104
Proto-cooperationComparative analyses of genomes can also generate insight into proto-cooperation of bacteria
which evolve in the same niche, e.g. via horizontal gene transfer events. In yoghurt manufacturing, the proto-cooperation between L. bulgaricus and S. thermophilus has been previously described, but mainly with respect to their dependency of growth factors and metabolic interactions. Our analysis of horizontal gene transfer events revealed proto-cooperation on basis of exchanged and/or acquired genetic elements during evolution. The EPS biosynthesis proteins EpsI-ML in L. bulgaricus genomes were predicted to be acquired from S. thermophilus. A gene cluster encoding the enzymes involved in sulfur-containing amino acid metabolism (cbs-cblB/cglB-cysE) in S. thermophilus was probably transferred from L. bulgaricus strains. The HGT events are possibly important for adaptation of both bacteria to the milk environment and optimization of their combined growth and interactions.
General discussion and future perspectivesIn this thesis, we have improved gene annotations by a comprehensive comparative genomics
approach, which combines sequence similarity, phylogeny, gene context and literature evidence. The improved annotations of enzyme activities can be used for predictions of metabolic phenotypes of LAB, with respect to their proteolytic capacity and flavour-forming potential. It should be noted that only the LAB strains which have been sequenced to date were analyzed by our comparative genomics approach, and these are often type strains or model laboratory strains. To explore other strains which have not been sequenced, especially industrial strains, experimental techniques such as comparative genomic hybridization with microarrays could be applied, as exemplified by our CGH pangenome analysis of proteolytic system components in lactococci (chapter 2). In fact, the improvement of the functional annotation of the enzymes involved in target pathways is a starting point for predicting bacterial phenotypical characteristics on the strain level. Furthermore, the comparative genomics analyses could direct molecular or biochemical tests in order to validate the predicted enzyme activities.
Genome-wide metabolic models are generally constructed on basis of the sequence information and gene function assignments. However, for the non-essential metabolic routes, little information is available on the genetic or biochemical level, which therefore leads to the incompleteness of the reconstructed pathways. Although improving problematic gene annotations via comparative genomics can provide a better foundation for metabolic reconstruction, gaps still exist in the metabolic pathways. In these cases, metabolomics techniques such as NMR in combination with isotope-labeled precursors will be helpful by measuring the intermediate metabolites. Pathways can then be reconstructed and gaps can be filled using the retrosynthesis tool within the Reverse Pathway Engineering (RPE) approach, combined with the information of identified intermediates.
When genome-wide metabolic models with curated flavour-forming pathways are obtained, various pathway analyses can be applied to the stoichiometric model, to investigate the metabolic properties and ultimately to identify opportunities for steering specific end-products via metabolic engineering. For instance, high-throughput GC-MS data of flavour profiles measured under different conditions can be projected on metabolic networks. Pathways or reactions that are significantly changed under certain conditions can be identified and condition-dependent cell phenotypes could be determined. Such studies are currently underway at Top Institute (TI) Food and Nutrition, the Netherlands. Another example is to apply flux-balance analysis e.g. to compare flux distributions using the genome-scale models of bacteria with different metabolic capacities to produce target compounds. Key enzymes/genes can be identified as genetic engineering targets, and model-guided metabolic engineering may increase production of the desired compounds.

SUMMARIZING DISCUSSION
105
CH
APT
ER 7
The newly developed strategy (RPE approach), used for forging the gaps between metabolomics and genomics data, can also provide leads for metabolic engineering by predicting alternative reactions/pathways. A hypothesis was generated to produce a key cheese flavour component from a non-flavour compound via a site-directed mutagenesis of a proposed enzyme (described in detail in chapter 5) and is being tested experimentally under cheese fermentation conditions. In addition, the mechanism of biosynthesis of off-flavour compounds, observed in some dairy products, can be investigated by the combination of cheminformatics and bioinformatics analyses in this approach. Candidate spoilage microorganisms that are responsible for the formation of off-flavours in the product could be proposed
Cysteine
L-serine
O-acetyl-L-serine
CysE
CysK
CGS
Cystathionine HomocysteineCGL CBS
Methionine
MetHMetE
Homo-serine
Aspartate 4-semialdehyde
HSDH
NAD(P)H
NAD(P)+
O-phospho-homoserine O-acetyl-homoserineO-succinyl-homoserineHSK
HSSTATP
ADP
acetyl-CoA
suc-CoA
HSST
AHSHH2S
succinatePi
H2S
H2SCBL CysK
chemical/enzymatic
DMS
Methanethiol
CBLCGL
H2S, DMS, DMDS, DMTS
CoA
CoA
CBLacetate
CGLCBLCGL
acetate
acetate
acetyl-CoACoA
Streptococcus thermophilus
TFBS
CmbR
TFBSCmbR
TFBSTFBS
TFBS
TFBSMetR/MtaR
TFBS
TFBSMetR/MtaR
TFBSCmbR
TFBS
MetR/MtaR
MetR/MtaR
Figure 7.2. Methionine and cysteine regulated by various metabolism in S. thermophilus transcriptional mechanisms. (A full version can be found in Chapter 4, Figure 4.1; a colour version can be found in Appendix). TFBS, transcription factor bind site has been identified; however the transcription factor is unknown.
Coupling the transcriptional mechanisms to the metabolic model may provide additional opportunities, since regulatory genes are often responsible for controlling the flux distribution, and may serve as important targets for metabolic engineering. For instance, Figure 7.2 shows that the methionine and cysteine metabolic pathways in S. thermophilus are regulated by several transcription regulators, e.g. CmbR and MetR/MtaR. Different expression levels of these regulators could lead to different flux distributions. Therefore, fluxes could be increased and the production of target products could be steered by modifying the gene expression of regulators. However, some transcription factors which regulate enzymatic reactions in the biosynthesis of sulphuric flavour compounds are unknown as yet, although we have identified conserved binding sites for those regulators. Both computational and experimental methods can be used to search for the candidate regulators. The in silico approach includes identification of the regulon members of the unknown regulator by genome-wide scanning for the identified binding motifs. If the transcription factor is self-regulated, it can be found as a member of the regulon.

CHAPTER 7
106
and corresponding trouble-shooting methods can be applied. Since the reaction database used in the approach is quite flexible, the application field could be easily extended to biomarker discovery i.e. looking for potential metabolite biomarkers that are indicative of specific phenotypes, and to synthetic biology for in silico design of biosynthesis pathways to construct microbial chemical factories. Moreover, the new insights into proto-cooperation of yoghurt bacteria could be used advantageously to improve the control of co-growth of both species in yoghurt manufacturing and, accordingly, to improve product characteristics.

SAMENVATTING
107
Samenvatting

DUTCH SUMMARY
108
Dit proefschrift beschrijft de resultaten van een bio-informatica aanpak waarin, vergelijkenderwijs, het erfelijk materiaal van verschillende melkzuurbacteriën onderling bestudeerd is. Er is op zoek gegaan naar de verschillen en overeenkomsten in eiwitafbrekende en aromavormende eigenschappen. Ook de mogelijke samenwerking van verschillende melkzuurbacteriën, de zogenaamde proto-coöperatie (Figuur 7.1), is aan bod gekomen. Het in dit proefschrift beschreven werk heeft zich met name gericht op het bouwen van een dynamisch model voor aromavormende stofwisselingsroutes. Om dit te kunnen bereiken is gewerkt aan het verbeteren van de annotatie van relevante genen en enzymen, zijn hiaten in stofwisselingsroutes ingevuld en regulerende mechanismen in kaart gebracht. Genoomwijde analyses zijn gebruikt om samenwerking aan te tonen tussen twee bacteriële soorten die gebruikt worden in yoghurt fermentatie. Deze samenwerking is gebaseerd op horizontale genoverdracht.
Het hier beschreven onderzoek is uitgevoerd in het kader van het Casimir programma van het Nederlands Ministerie van Economische Zaken. De doelstellingen van het Casimir programma zijn tweeledig: aan de ene kant het stimuleren van de uitwisseling van onderzoekers tussen de publieke en private sector en aan de andere kant het versnellen van kennisuitwisseling. De beschreven studies zijn uitgevoerd op twee locaties: FrieslandCampina Research in Deventer en het Centrum voor Moleculaire en Biomoleculaire Informatica aan de Radboud Universiteit Nijmegen.
Eiwit annotatie en enzym familie analyseAromavormende stofwisselingsroutes vanuit eiwitafbraak door melkzuurbacteriën zijn in
twee stappen in kaart gebracht: analyse van eiwitafbraak routes en analyse van aminozuurafbraak routes. Door gebruik te maken van fylogenetische informatie, syntenie gegevens en literatuur, werd de functionele annotatie verbeterd van allerlei onderdelen van het eiwitafbraak systeem zoals celwandgebonden proteïnases, peptide transporters en peptidases maar ook enzymen die betrokken zijn bij de stofwisseling van vertakte keten aminozuren, aromatische aminozuren en zwavelhoudende aminozuren.
Eiwit subfamilies met duidelijk afgebakende substraat specificaties konden worden herkend in grotere eiwit families. Voorbeelden hiervan zijn de PepP/PepQ/PepM familie, de PepD familie en ook de PepI/PepR/PepL familie (hoofdstuk 2), maar ook de cysteïne synthase/cystathion beta-synthase familie en de cystathion beta/gamma lyase familie (hoofdstuk 3). Daarnaast was het mogelijk om met 3D eiwitstructuurvergelijking te komen tot een betere (sub)familie classificatie van de PepI/PepR/PepL subfamilie en de esterase familie EstA.
Eiwitafbraak en aromavormend potentieelVerbeterde functionele annotatie is gebruikt om de eiwitafbrekende eigenschappen van
verschillende soorten melkzuurbacteriën nader in kaart te brengen. Hieruit is gebleken dat de genomen van de L. acidophilus groep, die bestaat uit L. acidophilus, L. johnsonii, L. gasseri, L. bulgaricus, en L. helveticus, een groter en meer divers aantal componenten uit het eiwitafbraak systeem in zich dragen dan andere melkzuurbacteriën. Het celwandgebonden proteinase (PrtP) werd alleen aangetroffen op het chromosomale DNA van L. acidophilus, L. johnsonii, L. bulgaricus, L. casei, L. rhamnosus en S. thermophilus strain LMD9, en op het plasmide van L. lactis subsp. cremoris SK11. Melkzuurbacteriën van plantaardige oorsprong, zoals L. plantarum, O. Oeni en Leuconostoc mesenteroides, bevatten minder eiwitafbrekende enzymen in hun genomen. Dit is in overeenstemming met de ecologische niche van deze organismen waarin met name vezels aanwezig zijn en minder eiwitten. Analyse van 39 Lactococcus stammen toonde duidelijk aan dat verschillende componenten van het eiwitafbraak

SAMENVATTING
109
systeem (o.a. proteïnases, oligopeptide transporters en peptidases) onevenredig verdeeld zijn. Dit wordt waarschijnlijk veroorzaakt doordat sommige van deze componenten genetisch gecodeerd liggen op (mobiele) plasmiden en is een weerspiegeling van het milieu waaruit deze organismen afkomstig zijn.
Niet alleen de eiwitafbrekende eigenschappen van verschillende melkzuurbacteriën zijn in kaart gebracht, maar ook de aromavormende. S. Thermophilus, verschillende Lactococcus stammen en een aantal andere melkgerelateerde stammen (b.v. Lb. casei) bevatten vooral veelvoorkomende aromavormende enzymen. Bacteriën uit de darm, zoals L. gasseri en L. johnsonii, lijken deze enzymen te missen.
Reconstructie van stofwisselingsroutesReconstructie van stofwisselingsroutes is alleen mogelijk als de genen en eiwitten van
een organisme correct geannoteerd zijn op basis van hun genoomsequentie. In veel gevallen zijn de annotatie gegevens van genen en eiwitten in publiekelijk beschikbare databases incompleet of foutief, waardoor er geen goede stofwisselingsmodellen gemaakt kunnen worden. Veel producten van stofwisselingsprocessen, die gemeten worden met behulp van moderne technieken met zeer hoge verwerkingscapaciteit, kunnen niet goed geplaatst worden in dit soort modellen doordat er onvoldoende genetische en/of biochemische kennis voorhanden is. Dit is vooral het geval voor de stofwisselingsproducten die niet essentieel zijn voor de centrale stofwisseling. Om het reconstructieproces van stofwisselingsroutes verder te verbeteren is er een in silico methode ontwikkeld genaamd “Reverse Pathway Engineering (RPE)”. Deze nieuwe methode combineert chemo-informatica met bio-informatica en voorspelt de ontbrekende moleculen tussen gemeten functionele moleculen en hun mogelijke cellulaire voorlopers uit stofwisselingsroutes. RPE levert hiervoor mogelijke chemische of enzymatische reacties aan. Op basis van zogenaamde omzettingsregels uit een reactiedatabase genereert de RPE methode een aantal plausibele chemische en enzymatische reacties, waaronder nieuwe reacties die nog niet beschreven zijn in literatuur of publiekelijk beschikbare databases. De omzettingsregels die gebruikt worden kunnen worden aangepast aan de specifieke vraag. Deze nieuwe methode is gevalideerd aan de hand van de al eerder beschreven aromavormende stofwisselingsroutes uitgaande van leucine. Het is interessant om te vermelden dat ook onbekende aromavormende reacties voorspeld werden zoals de conversie van α-hydroxy-isocaproaat naar isovaleriaanzuur en de productie van dimethyl sulfide, maar ook de noodzakelijke enzymen hiervoor. Door het vermogen om dit soort nieuwe routes te voorspellen, kunnen bestaande gaten in incomplete aromavormende stofwisselingsroutes gedicht worden en kan er richting gegeven worden aan het proces van “metabolic engineering” om aromaprofielen te verbeteren.
ReguleringsmechanismenVan twaalf enzymen die een sleutelrol vervullen in de cysteïne en methionine stofwisseling
zijn de activiteitsregulerende mechanismen opgehelderd op gen niveau. Zowel transcriptiefactor bindingsplaatsen als RNA “riboswitches” maken hier deel van uit. In dit proces zijn de bindingsmotieven verder verfijnd van de al bekende transcriptie factoren CmbR en MetR/MtaR, die voorkomen in Lactococci en Streptococci, op grond van overeenstemming in DNA sequenties. Er konden veel nieuwe potentiële transcriptiefactor bindingsplaatsen voorspeld worden door gebruik te maken van een uitgebreide “footprinting” methode. Door dit soort regulerende mechanismen als onderdeel mee te nemen in modellen van aromavormende stofwisselingsroutes wordt een duidelijke bijdrage geleverd aan het verder verbeteren van dit soort modellen. Uiteindelijk zal dit leiden tot allesomvattende, dynamische stofwisselingsmodellen, waarin ook de verschillende factoren die controle uitoefenen op

DUTCH SUMMARY
110
de kinetiek van de processen geïntegreerd zijn.
Samenwerking (proto-coöperatie)Het vergelijken van de genoominformatie van melkzuurbacteriën geeft inzicht in de mogelijke
onderlinge samenwerking tussen die organismen. Doordat deze organismen zich gezamenlijk ontwikkeld hebben in een zelfde ecologische niche kunnen samenwerkingsverbanden tot stand zijn gekomen door horizontale genoverdracht. Het is bekend dat tijdens yoghurt productie L. bulgaricus en S. thermophilus samenwerken waarbij er in het verleden vooral gekeken is naar welke groei- en stofwisselingsfactoren hiervoor noodzakelijk zijn. Analyse van horizontale genoverdracht laat zien dat samenwerking ook plaatsvindt op basis van uitwisseling van genetische elementen gedurende de evolutie. Uit deze analyses kwam naar voren dat de genen van de EPS biosynthese eiwitten EpsI-ML uit het genoom van L. bulgaricus oorspronkelijk afkomstig zijn uit het genoom van S. thermophilus. Verder is het waarschijnlijk dat een gencluster dat codeert voor de enzymen die betrokken zijn bij zwavelbevattende aminozuur stofwisselingsroutes (cbs-cblB/cglB-cysE) in S. thermophilus afkomstig zijn van L. bulgaricus. Horizontale genoverdracht lijkt dus een belangrijk mechanisme waarmee bacteriën zich uiteindelijk hebben aangepast aan de melk matrix en waardoor er optimale groei en samenwerking mogelijk is.

总结概述
111
总结概述
背景
乳酸菌(lactic acid bacteria, LAB)是一大类糖代谢主要以乳酸为终产物的革兰氏阳性非芽孢细菌。乳酸菌广泛存在于乳制品,肉类,植物等发酵环境中。他们在食品和饮料的发酵生产过程中起着非常重要的作用,例如,促进产品风味,口感和营养物质的形成。某些乳酸菌,譬如双歧杆菌,长期寄居在人类和其他哺乳动物的肠道,他们作为益生菌被添加至不同的功能食品中。
乳酸菌有着用于制作乳酪,酸奶和其他发酵乳制品悠久历史,可追溯到几个世纪之前。随着现代生活方式对发酵乳制品需求量增长,分子层面对乳酸菌的作用机制和特性的了解将革命性的促进大规模工业生产。但是,乳酸菌的很多特性,比如它们如何影响食品中的风味物质形成却依旧不清楚。因此,对其遗传和生理机制的基础研究将有效地改善发酵食品的质量和功能。
在最近的几年里,随着基因组测序的技术迅速发展,及其成本的降低,效率的提高,许多与食品健康相关的乳酸菌基因组序列相继被测定,包括大约40个乳酸菌染色体基因组,如嗜热链球菌、乳酸乳球菌、嗜酸乳杆菌、约氏乳杆菌、植物乳杆菌、唾液乳杆菌唾液亚种和长双歧杆菌等等。这些完整的基因组序列带来了大量前所未有的可供研究的遗传信息,但同时也带来了不可低估的挑战,比如如何使用生物信息学翻译和理解这些遗传编码中隐藏的生物意义。基因组信息可以用于建立细菌的代谢模型,在电脑中模拟其生长过程,并且理解它们的调控机制,最终,揭示这些乳酸菌的遗传和代谢趋势。了解乳酸菌的基因序列将极大地帮助人们了解它们在食品工业中的作用,实现最优化的生产设计。
本书系统阐述了运用比较基因组学的方法研究不同已被测序乳酸菌的蛋白水解力和形成风味物质的能力,以及酸奶乳酸菌的共生关系。我们重点研究了如何改进相关基因的功能注释,如何填补代谢途径中空白的方法,并通过相应调控机制架构一个风味物质形成路径的动态模型。
蛋白水解,氨基酸降解和风味物质的形成
乳酸菌产出的风味物质来自不同的生化代谢路径。食品产品中的重要风味物质可以结合气相色谱-质谱联用分析仪(GC-MS)和专业嗅觉检测(olfactometry)而确定。在乳酪中,风味物质的代谢前体包括脂肪,酪蛋白(casein),及各种糖类。其中,酪蛋白的降解是最为重要的风味物质形成路径,它产出大量具有香味的醇类,醛类,酯类及含硫化合物。从酪蛋白到风味物质的过程包括两个基本步骤,蛋白质水解和氨基酸降解。
在乳酸菌中,酪蛋白水解是由细胞壁蛋白酶主导的催化反应将蛋白质降解到多肽开始的。这些多肽接着被肽转运系统送入细胞内,然后被细胞内的一系列肽水解酶进一步降解到短链肽或者氨基酸。应用比较基因组学工具,我们具体研究了乳酸菌的蛋白水解酶系统,包括以上描述的蛋白酶和肽水解酶,以及多肽转运系统(第二章)。作为例子,肽水解酶家族PepP/PepQ/PepM, PepD and PepI/PepR/PepL的分类说明了使用生物信息工具可区分有着不同底物特异性的次家族。通过比较脯氨酸多肽水解酶(the proline peptidases PepI/PepR/PepL)和酯酶(esterase A)的蛋白质3D结构,改进了它们所属的超级蛋白家族的功能注释。另外,使用pangenome比较基因组杂交技术(comparative genome hybridization analysis),我们比较了乳酸乳球菌39个菌株的蛋白水解酶系统。 我们发现细胞壁蛋白酶PrtP,多肽转运系统Opp,以及多种多肽水解酶不均匀分布于乳酸乳球菌的不同亚种和有着不同的来源的菌株中(例如用于植物或乳制品发酵的菌株)。

中文概述
112
从酪蛋白到风味物质的第二步是将蛋白质水解得到的氨基酸进一步降解。同样地,应用生物信息方法和比较基因组学手段,我们研究了参与侧链氨基酸,芳香氨基酸和含硫氨基酸降解路径的酶,尤其是直接影响到风味物质形成的酶(第三章)。种系发生学研究表明用演化树可以从超级蛋白质家族中区分出功能分族,不同的酶分族具有不同的底物特异性。通过具体的功能分族,我们大大改进了这些酶蛋白的基因注释。在不同的菌种间,甚至在不同的菌株间,风味物质形成酶分布呈现出巨大差异。这些差异大致与不同乳酸菌的风味物质产出能力相吻合。此研究成果可以更加直接地用于引导未来试验设计,基因组的测序,以及应用于工业快速筛选改进产品风味的乳酸菌菌种。
调控机制
我们同时研究了参与风味物质形成路径的各类酶的调控机制,尤其注重于对调控半胱氨酸和蛋氨酸的代谢途径的研究。通过确定直系同源基因(orthology),基因环境(gene context)比较,转录因子结合序列研究,我们发现并比较了12种酶的编码基因在20种不同的乳酸菌中的调控机制,包括已知的转录因子CmbR和MetR/MtaR,及其可结合未知转录因子的新序列。应用隐马尔可夫模型(Hidden Markov Model)搜索乳酸菌完整基因组,我们确定了另一种RNA层次的调控机制riboswitch, 包括T-box和S-box,同样可以控制某些半胱氨酸和蛋氨酸代谢酶的转录。
代谢路径的重新架构
重新架构生物体的代谢网络往往取决于它基因组的基因或者蛋白的功能注释。很多情况下,已发布的基因组序列里存在着很多错误信息,这些错误信息直接导致了重组代谢网络的不完整。很多高通量方法观测到的代谢产物,尤其是次级代谢产物,不能整合到代谢网络中。为了解决上述问题,本书阐述了一种新的研究方法(Reverse Pathway Engineering),首次结合了化学信息学和生物信息学来预测未知的生化反应,以联系目标代谢产物和它们可能的代谢前物。使用此方法,我们成功证实了已知的风味物质形成路径,并且预测了在风味物质形成路径上的未知酶催化反应。这些新反应的发现填补了风味物质代谢路径中的空白,并为进一步的代谢工程学研究提供了线索。此新方法还可以广泛应用于系统生物学,合成生物学,以及蛋白质工程领域。
共生关系
最后,我们研究了用于酸奶发酵的德氏乳杆菌保加利亚亚种和嗜热链球菌间的共生互利关系,追溯了它们共同进化过程中可能发生的水平基因转移事件。使用电脑模拟(in silico), 结合基因结构和基因转移机制的相关特性研究,我们估测出德氏乳杆菌保加利亚亚种和嗜热链球菌基因组中的水平基因转移事件,包括一系列细胞外多聚糖(EPS)合成基因和一个参与含硫氨基酸代谢的操作子。在此研究中,我们发现它们在遗传因子层面也存在着紧密的联系。这种紧密联系可能是两种乳酸菌于奶制品里长期共存过程中优化的结果。

COLOUR FIGURES
113
Appendix
COLOUR FIGURES

Appendix
114
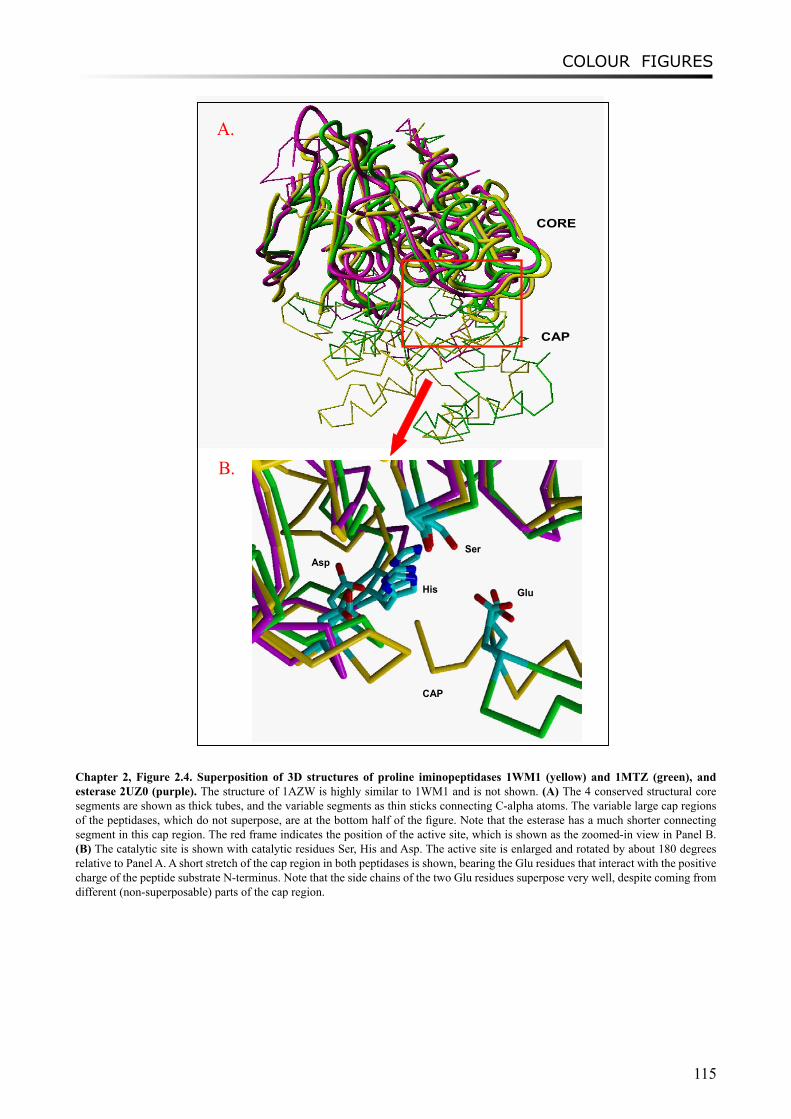
COLOUR FIGURES
115
CAP
CORE
CAP
GluHis
AspSer
A.
B.
Chapter 2, Figure 2.4. Superposition of 3D structures of proline iminopeptidases 1WM1 (yellow) and 1MTZ (green), and esterase 2UZ0 (purple). The structure of 1AZW is highly similar to 1WM1 and is not shown. (A) The 4 conserved structural core segments are shown as thick tubes, and the variable segments as thin sticks connecting C-alpha atoms. The variable large cap regions of the peptidases, which do not superpose, are at the bottom half of the figure. Note that the esterase has a much shorter connecting segment in this cap region. The red frame indicates the position of the active site, which is shown as the zoomed-in view in Panel B. (B) The catalytic site is shown with catalytic residues Ser, His and Asp. The active site is enlarged and rotated by about 180 degrees relative to Panel A. A short stretch of the cap region in both peptidases is shown, bearing the Glu residues that interact with the positive charge of the peptide substrate N-terminus. Note that the side chains of the two Glu residues superpose very well, despite coming from different (non-superposable) parts of the cap region.
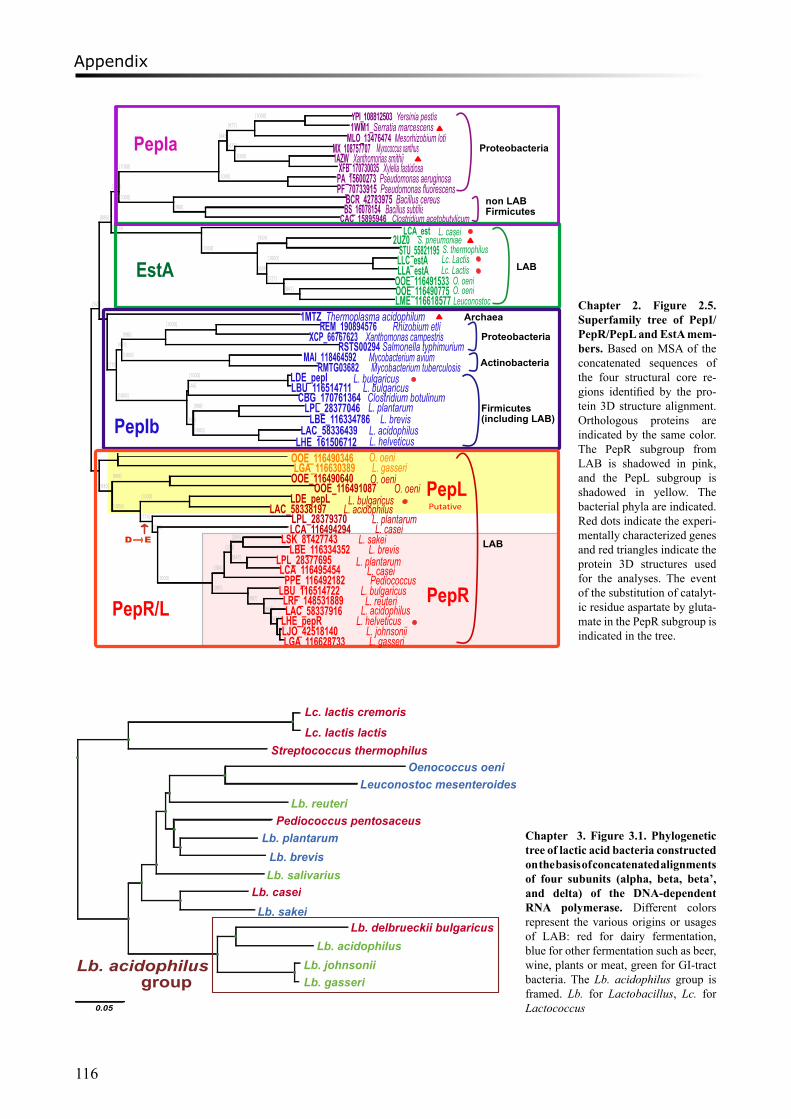
Appendix
116
Lc. lactis cremoris
Oenococcus oeni Leuconostoc mesenteroides
Pediococcus pentosaceus Lb. plantarum
Lb. brevisLb. salivarius
Lb. reuteri
Lb. caseiLb. sakei
Lb. delbrueckii bulgaricus Lb. acidophilus
Lb. johnsonii Lb. gasseri
0.05
Lb. acidophilus group
Lc. lactis lactis Streptococcus thermophilus
Chapter 3. Figure 3.1. Phylogenetic tree of lactic acid bacteria constructed on the basis of concatenated alignments of four subunits (alpha, beta, beta’, and delta) of the DNA-dependent RNA polymerase. Different colors represent the various origins or usages of LAB: red for dairy fermentation, blue for other fermentation such as beer, wine, plants or meat, green for GI-tract bacteria. The Lb. acidophilus group is framed. Lb. for Lactobacillus, Lc. for Lactococcus
[923]
[886]
[744]
[1000]
[445]
[977]
[1000] YPI_108812503 Yersinia pestis 1WM1_Serratia marcescens
MLO_13476474 Mesorhizobium loti[475] MX_108757707
[1000] 1AZW_ Xanthomonas smithiiXFB_170730035 Xylella fastidiosa
[1000] PA_15600273 Pseudomonas aeruginosa PF_70733915 Pseudomonas fluorescens
[1000] BCR_42783975 Bacillus cereus[980] BS_16078154 Bacillus subtilis
CAC_15895946 Clostridium acetobutylicum [1000] LCA_est
[1000]
[954] 2UZ0STU_55821195
[609]
[1000] LLC_estALLA_estA
[727] OOE_116491533[841] OOE_116490775
LME_116618577[589] 1MTZ_Thermoplasma acidophilum
[770]
[461]
[998]
[1000] REM_190894576XCP_66767623 Xanthomonas campestris
RSTS00294 Salmonella typhimurium[1000] MAI_118464592
RMTG03682 Mycobacterium tuberculosis
[1000]
[246]
[1000] LDE_pepILBU_116514711CBG_170761364 Clostridium botulinum
[394]
[998] LPL_28377046LBE_116334786
[1000] LAC_58336439LHE_1615067129]
OOE_116490346LGA_116630389
[930]
[999] OOE_116490640OOE_116491087
[1000]
[1000] LDE_pepLLAC_58338197
[774] LPL_28379370[528] LCA_116494294
[1000]
[384]
[254]
[584] LSK_81427743LBE_116334352
[847] LPL_28377695LCA_116495454
PPE_116492182[997] LBU_116514722
[987] LRF_148531889[607] LAC_58337916
[631]LHE_pepR[677]LJO_42518140
LGA_116628733
EstA
PepIb
PepR
PepIa Proteobacteria
non LABFirmicutes
Archaea
Proteobacteria
Actinobacteria
Firmicutes(including LAB)
PepR/L
LAB
LAB
D E
L. caseiS. pneumoniae
S. thermophilusLc. LactisLc. Lactis
O. oeniO. oeniLeuconostoc
Myxococcus xanthus
Mycobacterium avium
Rhizobium etli
L. bulgaricusL. bulgaricus
L. plantarumL. brevis
L. acidophilusL. helveticusO. oeniL. gasseriO. oeni
O. oeniL. bulgaricus
L. acidophilusL. plantarumL. casei
L. sakeiL. brevis
L. plantarumL. caseiPediococcus
L. bulgaricusL. reuteri
L. acidophilus
L. johnsoniiL. helveticus
L. gasseri
PepLPutative
Chapter 2. Figure 2.5. Superfamily tree of PepI/PepR/PepL and EstA mem-bers. Based on MSA of the concatenated sequences of the four structural core re-gions identified by the pro-tein 3D structure alignment. Orthologous proteins are indicated by the same color. The PepR subgroup from LAB is shadowed in pink, and the PepL subgroup is shadowed in yellow. The bacterial phyla are indicated. Red dots indicate the experi-mentally characterized genes and red triangles indicate the protein 3D structures used for the analyses. The event of the substitution of catalyt-ic residue aspartate by gluta-mate in the PepR subgroup is indicated in the tree.
COLOUR FIGURES
117
[780]
[1000][926]
[940]
[433]
[654]
[788][1000]
[1000][1000][1.1.1] LLM_125624565
[1.1.1] LLM_CysK[1.1.1] LLX_15672764[1.1.1] LLA_116511591
[1000] [1.1.2] LLX_15672522[1000] [1.1.2] LLM_125623375
[1.1.2] LLA_116511337[1000] [1.1] STM_116627279
[1000][1.1] STH_55822343[1.1] STU_55820459
[1000][595]
[1000] [1.1] LRF_148532032[1.1] LRE00008
[1.1] LCA_116494039[535] [1.1] LAC_58337517
[999] [1.1] LSL_90962687[1000] [1.1] LBU_116514403
[1.1] LDB_104774314[1.1] BSU_CysK
[1] FN_CysK[1000] [2] BSU_yrhA_CBS
[981][550] [2] LSL_90961016
[2] LAC_58337377
[617][859]
[1000] [2] LRF_148531797[2] LRE00702
[2] OOE_116491721[947] [2] LME_116618386
[657] [2] LPL_28377182[1000]
[1000][2] LBU_116514295[2] LDB_104774207
[1000][2] STM_116627697[2] STH_55822810[2] STU_55820893
[[
]
CysK
CBS
Tryptophan synthase Threonine dehydratase
Threonine synthase 0.1
[514]
[548]
[825]
[997]
[1000]
[654]
[1000]
[948][1.1] STH_55822811[1.1] STU_55820894[1.1] STM_116627698
[978][1.1] LBU_116514296[1.1] LDB_104774208
[1.1] LPL_28377181[1.1] LME_116618387
[823] [1.1] OOE_116491720[995] [1.1] LSL_90961017
[856] [1.1] LAC_58337380[1000] [1.1] LRE01167
[1.1] LRF_148530564[543] [1.1] BSU_yrhB_CGL
[1000]
[1000] [1.1.1] LLC2_MetC[1.1.1] LLA_116511590
[1000][1.1.2] LLC1_MetC[1.1.2] LLM_125624566[1.1.2] LLX_15672763
[948] [1.2] BSU_yjcJ_CBL[1000] [1.2] LME_116618893
[1.2] OOE_116490814[1000] [1.3] LCA_MetB
[1.4] LCA_116494152[944] [2] BSU_yjcI_CGS
[782]
[981] [2] LPL_28379151[2] LME_116618894
[1000]
[1000] [2] LLM_125624948[2] LLX_15673899
[616] [2] SAN_CGS[1000] [2] STM_116627266
[2] STH_55822329[2] STU_55820445
[632] [3] LME_116618337[1000] [4] NAI_CysD
[768]
[1000]
[1000][4] STH_55822940[4] STU_55821020[4] STM_116627784
[830]
[985]
[995] [4] LME_116618888[4] OOE_116490344
[4] LPL_28379077[1000] [4] LLX_15672055
[935][4] LLM_125622970[4] LLA_116510901
CBL/CGL
CBL
CGS/AHSH
AHSH
0.1
Chapter 3. Figure 3.4. Phylogenetic tree of the CysK/CBS family. Different colors represent orthologous groups. Red Squares represe� -cates. Red dots indicate the experimentally verified genes from Lc. lactis subsp. cremoris MG1363 (LLM_CysK), Bacillus subtilis (BSU_CysK and BSU_yrhA_CBS) and Fusobacterium nucleatum (FN_CysK). The subfamilies related to the tryptophan synthase β-s� -responding genes are associated with specific amino acid biosynthesis gene clusters (data not shown). The GI code of each protein in the family is preceded by its corresponding genome abbreviation.
Chapter 3. Figure 3.5. Neighbor-joining tree of the CBL/CGL lyase family. Different colors represent orthologous groups. Red Squares represent duplication events, while green circles represent speciation events. Bootstrap values were calcu-lated for a total of 1000 replicates. Experimentally characterized pro-teins are indicated by red dots, in-cluding CGL, CBL and CGS from B. subtilis (BSU_yrhB_CGL, BSU_yjcJ_CBL and BSU_yjcI_CGS respectively), the CBL/CGL from Lc. lactis subsp. cremoris MG1363 and NIZO B78 (LLC1_MetC and LLC2_MetC respectively), the MetB from Lb. casei (LCA_MetB), the CGS from Streptococcus angi-nosus (SAN_CGS), as well as the AHSH (cysD gene) from Emeri-cella nidulans (NAI_CysD). The GI codes of the other LAB proteins in this family are shown after the ge-nome abbreviation.
Appendix
118
Cystein
e
CGS
Cystath
ionine
Homocy
steine
CGL
CBS
Methion
ine MetH
MetE
Homo-se
rine
Asparta
te4-sem
ialdehy
de
HSDH
NAD(P)H
NAD(P) +O-phos
pho-ho
moserin
eO-a
cetyl-ho
moserin
eO-s
uccinyl
-homos
erine
HSKHSS
TATPAD P
acetyl-C
oA
suc-Co
A
HSST
AHSH
H 2S
succina
tePiH 2S
CBL
chemic
al/enz
ymaticDM
S
Methan
ethiol
CBL CGL
H 2S,DM
S,DMD
S,DMTS
CoA
CoA
CBLace
tate
CGL
CBL
CGL
acetate
Lacto
bacill
us pla
ntarum
Cystein
e
L-serine
O-acety
l-L-seri
ne
CysE
CysK
CGS
Cystath
ionine
Homocy
steine
CGL
CBS
Methion
ine MetH
MetE
Homo-se
rine
Asparta
te4-sem
ialdehy
de
HSDH
NAD(P)H
NAD(P) +O-phos
pho-ho
moserin
eO-a
cetyl-ho
moserin
eO-s
uccinyl
-homos
erine
HSKHSS
TATPAD P
acetyl-C
oA
suc-Co
A
HSST
AHSH
H 2S
succina
tePi
H 2S
H 2SCB
LCysK
chemic
al/enz
ymaticDM
S
Methan
ethiol
CBL CGL
H 2S,DM
S,DMD
S,DMTS
CoA
CoA
CBLace
tate
CGL
CBL
CGL
acetate
acetate
acetyl-C
oACoA
Cystein
e
L-serine
O-acety
l-L-seri
ne
CysE
CysK
CGS
Cystath
ionine
Homocy
steine
CGL
Methion
ine MetE
Homo-se
rine
Asparta
te4-sem
ialdehy
de
HSDH
NAD(P)H
NAD(P)+O-phos
pho-ho
moserin
eO-a
cetyl-ho
moserin
eO-s
uccinyl
-homos
erine
HSKHSS
TATPADP
acetyl -C
oA
suc-Co
A
HSST
AHSH
H 2S
succina
t ePi
H 2S
H 2SCB
LCysK
chemic
al/enz
ymaticDM
S
Methan
ethiol
CBL CGL
H 2S,DM
S,DMD
S,DMTS
CoA
CoA
CBLace
tate
CGL
CBL
CGL
acetate
acetate
acetyl-C
oACo A
Lacto
coccu
s lacti
sStr
eptoc
occus
therm
ophil
u s
TFBS
TFBS
TFBS
TFBS
TFBS
T-box
TFBS
TFBS
T-box
TFBS
and T
-box
TFBS
T-box
T- box
T-box
TFBS
CmbR
CmbR
CmbR
CmbR
CmbR
CmbR
MetR/
MtaR
CmbR
CmbR
CmbR
TFBS TFBS
TFBS
CmbR
TFBS
CmbR
TFBS
TFBS
TFBSTF
BSMe
tR/Mta
R
TFBS
TFBS
MetR/
MtaR
TFBS
CmbR
TFBS
CmbR
MetR/
MtaR
MetR/
MtaR
A
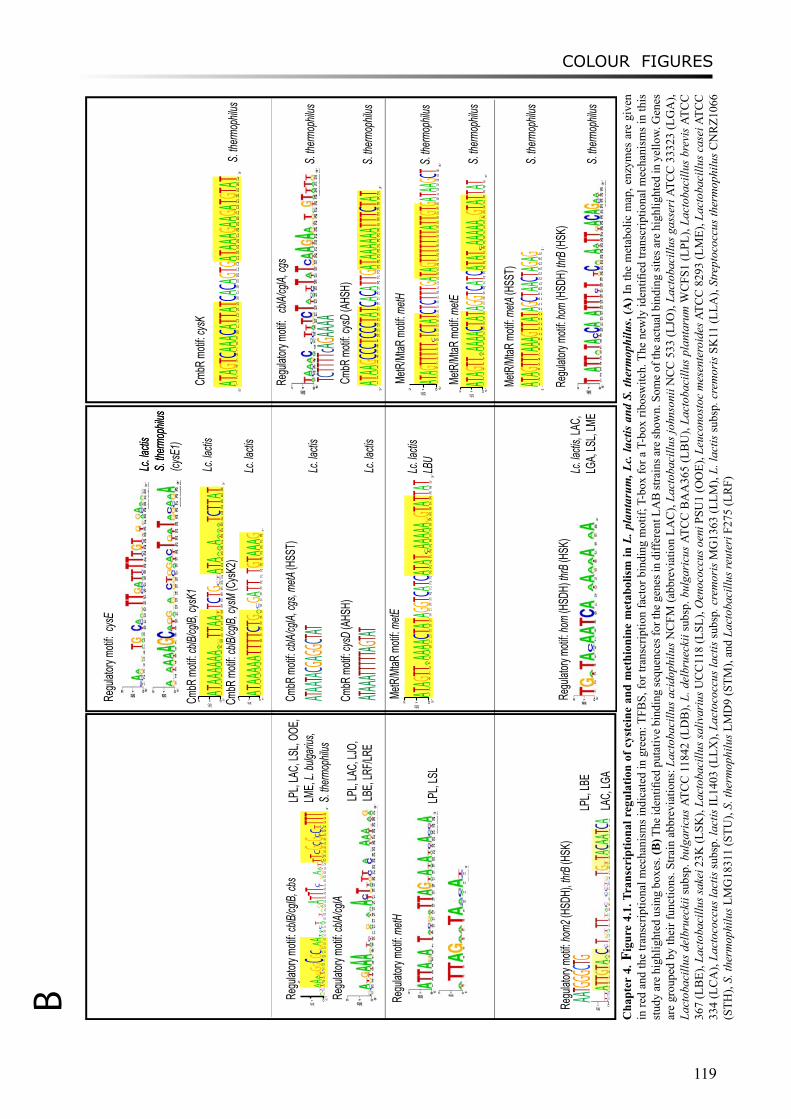
COLOUR FIGURES
119
Lc. la
ctisS.
therm
ophilu
s
Regu
latory
moti
f: cblB
/cglB,
cbs
01
2
sti b 5?
1
CGA
2
ACT
3
TA
4
CA 5
TA
6
TG
7
CG 8
AG
9C 01
AG 11
AC 21
ATG
31
GA
41
GA 51
G
AT
61
CTA
71
ATC
81
AT
9102
ACT
12
TGA
22
AG
32A 42T 52T 62T 72
TC
8292
TAG
03
GA
13
GTA
23
GAT
33
CT
43
CT 53
GC
63G 73
TC
83G 93
TC
04
TC 14
CT
24T 34T 44T 3‘
LPL,
LAC,
LSL,
OOE,
LME,
L. bu
lgariu
s,S.
therm
ophilu
s
Regu
latory
moti
f: metH
Regu
latory
moti
f: cblA
/cglA
LPL,
LAC,
LJO,
LB
E, LR
F/LRE
LPL,
LSL
Regu
latory
moti
f: hom
2 (HS
DH), t
hrB (H
SK)
AATGGG
CTGLP
L, LB
E
Regu
latory
moti
f: cys
E
Lc
. lactis
S. the
rmop
hilus
(cysE
1) Cm
bR m
otif: c
blB/cg
lB, cy
sK1
012 bits 5‘
1A
2T 3A 4A 5A 6A
7A
8A
9
TA
10
AG
11T 12T 13A 14A 15
GT
16T 17C
18T 19G
20
AC
21
CT
22A 23T 24A
25
GA
26
CA
27A
28
TA
29
TA
30
TA
31T 32C
33T 34T 35A
36T 3‘
Lc. la
ctis
CmbR
moti
f: cblB
/cglB,
cysM
(Cys
K2)
012 bits 5‘
1A
2T 3A
4A
5A
6A
7A
8T 9T 10T 11T 12C
13T 14G
15
GA
16
AC
17G
18A
19T 20T 2122T 23G
24T 25A
26A
27A
28G 3‘
Lc. la
ctis
CmbR
moti
f: cblA
/cglA,
cgs,
metA
(HSS
T)
ATAATA
CGAGGC
TATLc
. lactis
MetR
/MtaR
moti
f: metE
012 bits 5‘
1A 2T 3A 4G 5T 6T 7
TA
8A 9A 10A 11A 12C 13T 14A 15T 16A 17G 18G 19T 20C 21A 22T 23C 24A 25T 26A 27T 28
TC
29A 30A 31A 32A 33A 34
TG
35G 36T 37A 38T 39T 40A 41T 3‘
Lc. la
ctisLB
U
Regu
latory
moti
f: hom
(HSD
H) th
rB (H
SK)
Lc
. lactis
, LAC
, LG
A, LS
L, LM
E
CmbR
moti
f: cysD
(AHS
H)
ATAAAT
TTTTAG
TATLc
. lactis
Regu
latory
moti
f: cb
lA/cg
lA, cg
s
CmbR
moti
f: cysK
S. the
rmop
hilus
TCTTTT
cAGAAA
AS.
therm
ophilu
s
MetR
/MtaR
moti
f: metH
S. the
rmop
hilus
MetR
/MtaR
moti
f: metE
012 bits 5‘
1A 2T 3A 4G 5T 6T 7
TA
8A 9A 10A 11A 12C 13T 14A 15T 16A 17G 18G 19T 20C 21A 22T 23C 24A 25T 26A 27T 28
TC
29A 30A 31A 32A 33A 34
TG
35G 36T 37A 38T 39T 40A 41T 3‘
S. the
rmop
hilus
CmbR
moti
f: cysD
(AHS
H)
S. the
rmop
hilus
MetR
/MtaR
moti
f: metA
(HSS
T)
S. the
rmop
hilus
Regu
latory
moti
f: hom
(HSD
H) th
rB (H
SK)
S. the
rmop
hilus
LAC,
LGA
5‘
1A 2T 3A 4G 5T 6T 7T 8A 9A 10A 11G 12T 13T 14A 15T 16A 17G 18C 19T 20A 21A 22C 23T 24A 25G 26A 27G 3‘
012 bits 5‘
1A 2T 3A 4G 5T 6T 7T 8T 9T 10
TC
11T 12C 13T 14A 15T 16C 17T 18C 19T 20T 21T 22G 23A 24T 25A 26G 27T 28T 29T 30T 31T 32T 33T 34A 35T 36T 37G 38T 39G 40A 41T 42A 43A 44G 45C 46T 3‘
B
5‘
1A 2T 3A 4A 5G 6C 7C 8C 9T 10C 11C 12C 13T 14A 15T 16C 17A 18C 19A 20T 21T 22G 23A 24T 25A 26A 27A 28A 29A 30A 31T 32T 33T 34C 35T 36A 37T 3‘
′5
‘
1A 2T 3A 4G 5T 6C 7A 8A 9A 10C 11A 12T 13T 14A 15T 16C 17A 18C 19A 20G 21T 22G 23A 24T 25A 26A 27A 28G 29A 30A 31G 32A 33T 34G 35T 36A 37T 3‘
Cha
pter
4. F
igur
e 4.
1. T
rans
crip
tiona
l reg
ulat
ion
of c
yste
ine
and
met
hion
ine
met
abol
ism
in L
. pla
ntar
um, L
c. la
ctis
and
S. t
herm
ophi
lus.
(A) I
n th
e m
etab
olic
map
, enz
ymes
are
giv
en
in re
d an
d th
e tra
nscr
iptio
nal m
echa
nism
s ind
icat
ed in
gre
en: T
FBS,
for t
rans
crip
tion
fact
or b
indi
ng m
otif;
T-b
ox fo
r a T
-box
ribo
switc
h. T
he n
ewly
iden
tified
tran
scrip
tiona
l mec
hani
sms i
n th
is
stud
y ar
e hi
ghlig
hted
usi
ng b
oxes
. (B
) The
iden
tified
put
ativ
e bi
ndin
g se
quen
ces f
or th
e ge
nes i
n di
ffere
nt L
AB
stra
ins a
re sh
own.
Som
e of
the
actu
al b
indi
ng si
tes a
re h
ighl
ight
ed in
yel
low.
Gen
es
are
grou
ped
by th
eir f
unct
ions
. Stra
in a
bbre
viat
ions
: Lac
toba
cillu
s aci
doph
ilus N
CFM
(abb
revi
atio
n LA
C),
Lact
obac
illus
john
soni
i NC
C 5
33 (L
JO),
Lact
obac
illus
gas
seri
ATC
C 3
3323
(LG
A),
Lact
obac
illus
del
brue
ckii
subs
p. b
ulga
ricu
s AT
CC
118
42 (L
DB
), L.
del
brue
ckii
subs
p. b
ulga
ricu
s AT
CC
BA
A36
5 (L
BU
), La
ctob
acill
us p
lant
arum
WC
FS1
(LPL
), La
ctob
acill
us b
revi
s AT
CC
36
7 (L
BE)
, Lac
toba
cillu
s sak
ei 2
3K (L
SK),
Lact
obac
illus
saliv
ariu
s UC
C11
8 (L
SL),
Oen
ococ
cus o
eni P
SU1
(OO
E), L
euco
nost
oc m
esen
tero
ides
ATC
C 8
293
(LM
E), L
acto
baci
llus c
asei
ATC
C
334
(LC
A),
Lact
ococ
cus l
actis
subs
p. la
ctis
IL14
03 (L
LX),
Lact
ococ
cus l
actis
subs
p. c
rem
oris
MG
1363
(LLM
), L.
lact
is su
bsp.
cre
mor
is S
K11
(LLA
), St
rept
ococ
cus t
herm
ophi
lus C
NR
Z106
6 (S
TH),
S. th
erm
ophi
lus L
MG
1831
1 (S
TU),
S. th
erm
ophi
lus L
MD
9 (S
TM),
and
Lact
obac
illus
reut
eri F
275
(LR
F)
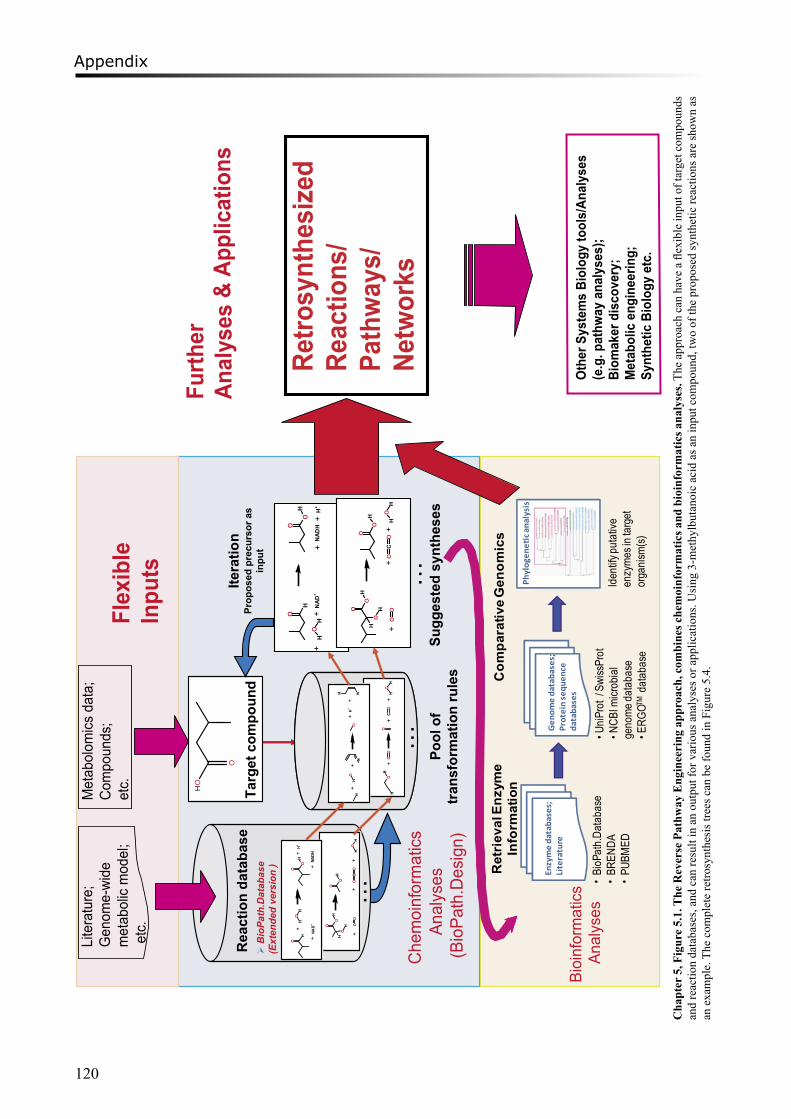
Appendix
120
Ret
riev
alEn
zym
eIn
form
atio
n
•BioP
ath.Da
taba
se•BR
ENDA
•PU
BMED
Com
para
tive
Gen
omic
s
Enzymedatabases;
Literature
Gen
omedatabases;
Proteinsequ
ence
databases
•UniP
rot/
Swiss
Prot
•NCB
Imicrob
ialgeno
meda
taba
se•E
RGOT
Mda
taba
se
Phylogen
eca
nalysis
Iden
tifypu
tative
enzyme
sintarget
orga
nism(
s)
Bioi
nfor
mat
ics
A
naly
ses
…
Rea
ctio
n da
taba
se
Sugg
este
d sy
nthe
ses
Targ
et c
ompo
und
�B
ioPa
th.D
atab
ase
(Ext
ende
d ve
rsio
n )
… …
HO
O
Pool
of
tran
sfor
mat
ion
rule
s
Itera
tion
Prop
osed
pre
curs
or a
s in
put
…
H
O
HO
HN
AD+
O
OH
++
+H
++
NAD
H
O
OH
OH
H
OO
O
OH
OC
OHOH
++
+
Che
moi
nfor
mat
ics
A
naly
ses
(Bio
Path
.Des
ign)
H
OHOH
NAD+
O
OH
+
+
+H+
+NADH
HO
++
HNH
NO
+H+
+
O
OH
OH
H
OO
O
OH
OC
OH
OH
++
+
OH
HO
OO
CO
HO
H+
++
Lite
ratu
re;
Gen
ome-
wide
m
etab
olic
mod
el;
etc
.Fl
exib
leIn
puts
Met
abol
omics
dat
a;Co
mpo
unds
; et
c.
Furth
erAn
alys
es&
Appl
icat
ions
Retro
synt
hesi
zed
Reac
tions
/Pa
thw
ays/
Netw
orks
Oth
er S
yste
ms
Biol
ogy
tool
s/An
alys
es(e
.g. p
athw
ay a
naly
ses)
;Bi
omak
er d
isco
very
;M
etab
olic
eng
inee
ring;
Sy
nthe
tic B
iolo
gy e
tc.
+
Cha
pter
5, F
igur
e 5.
1. T
he R
ever
se P
athw
ay E
ngin
eeri
ng a
ppro
ach,
com
bine
s che
moi
nfor
mat
ics a
nd b
ioin
form
atic
s ana
lyse
s. Th
e ap
proa
ch c
an h
ave
a fle
xibl
e in
put o
f tar
get c
ompo
unds
an
d re
actio
n da
taba
ses,
and
can
resu
lt in
an
outp
ut fo
r var
ious
ana
lyse
s or a
pplic
atio
ns. U
sing
3-m
ethy
lbut
anoi
c ac
id a
s an
inpu
t com
poun
d, tw
o of
the
prop
osed
synt
hetic
reac
tions
are
show
n as
an
exa
mpl
e. T
he c
ompl
ete
retro
synt
hesi
s tre
es c
an b
e fo
und
in F
igur
e 5.
4.
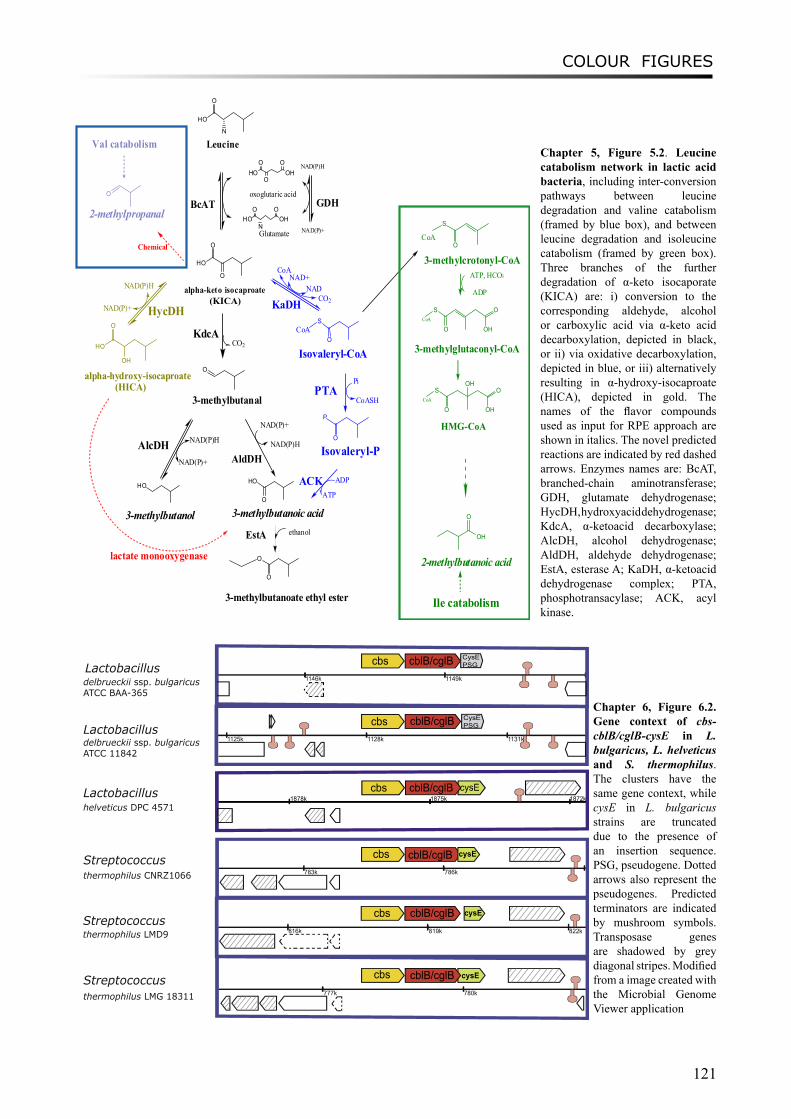
COLOUR FIGURES
121
HO
N
O
Leucine
HON
OOH
O
Glutamate
HOO
OHO
O
oxoglutaric acid
NAD(P)H
NAD(P)+
HO
O
O
alpha-keto isocaproate
BcAT GDH
KdcA
O
3-methylbutanal
CO2
NAD(P)H
NAD(P)+
AlcDH
HO
3-methylbutanol
NAD(P)H
NAD(P)+
HO
O
3-methylbutanoic acid
NAD(P)H
NAD(P)+ HycDH
AldDH
HO
O
OH
alpha-hydroxy-isocaproate
CoANAD+
NADCO2
S
OCoA
Isovaleryl-CoA
Pi
CoASH
P
O
Isovaleryl-P
ADP
ATPACK
PTA
KaDH
2-methylpropanal
O
Val catabolism
Chemical
S
OCoA
3-methylcrotonyl-CoA
S
OCoA
OH
O
3-methylglutaconyl-CoA
ATP, HCO3
ADP
S
OCoA
OH
OOH
HMG-CoA
OH
O
2-methylbutanoic acid
Ile catabolism
lactate monooxygenase
EstA ethanol
O
O
3-methylbutanoate ethyl ester
(KICA)
(HICA)
Chapter 5, Figure 5.2. Leucine catabolism network in lactic acid bacteria, including inter-conversion pathways between leucine degradation and valine catabolism (framed by blue box), and between leucine degradation and isoleucine catabolism (framed by green box). Three branches of the further degradation of α-keto isocaporate (KICA) are: i) conversion to the corresponding aldehyde, alcohol or carboxylic acid via α-keto acid decarboxylation, depicted in black, or ii) via oxidative decarboxylation, depicted in blue, or iii) alternatively resulting in α-hydroxy-isocaproate (HICA), depicted in gold. The names of the flavor compounds used as input for RPE approach are shown in italics. The novel predicted reactions are indicated by red dashed arrows. Enzymes names are: BcAT, branched-chain aminotransferase; GDH, glutamate dehydrogenase; HycDH, hydroxyacid dehydrogenase; KdcA, α-ketoacid decarboxylase; AlcDH, alcohol dehydrogenase; AldDH, aldehyde dehydrogenase; EstA, esterase A; KaDH, α-ketoacid dehydrogenase complex; PTA, phosphotransacylase; ACK, acyl kinase.
816k 819k 822k
1146k 1149k
1125k 1128k 1131kk
783k 786k
777k 780k
cblB/cglB
cblB/cglB
cblB/cglB
cblB/cglB
cbs
cbs
cbs
cbs
cbs
Streptococcusthermophilus LMD9
Lactobacillushelveticus DPC 4571
delbrueckii ssp. bulgaricus ATCC BAA-365
Lactobacillusdelbrueckii ssp. bulgaricus ATCC 11842
Streptococcusthermophilus CNRZ1066
Streptococcusthermophilus LMG 18311
cblB/cglB
CysE PSG
cysE
cysE
cysE
Lactobacillus
CysE PSG
1872k1875k1878kcbs cblB/cglB cysE
Chapter 6, Figure 6.2. Gene context of cbs-cblB/cglB-cysE in L. bulgaricus, L. helveticus and S. thermophilus. The clusters have the same gene context, while cysE in L. bulgaricus strains are truncated due to the presence of an insertion sequence. PSG, pseudogene. Dotted arrows also represent the pseudogenes. Predicted terminators are indicated by mushroom symbols. Transposase genes are shadowed by grey diagonal stripes. Modified from a image created with the Microbial Genome Viewer application
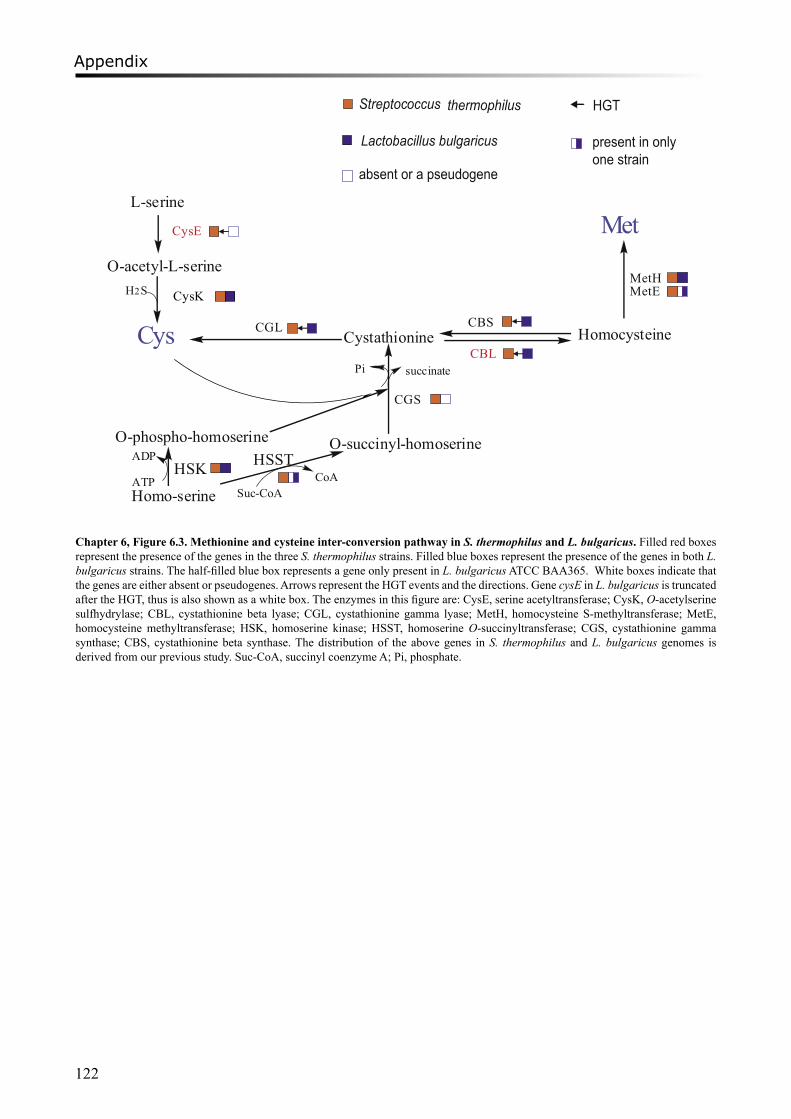
Appendix
122
Streptococcus thermophilus
Lactobacillus bulgaricus
Cys
L-serine
O-acetyl-L-serine
CysE
CysK
CGS
Cystathionine HomocysteineCBL
CGL CBS
Met
MetHMetE
Homo-serine
O-phospho-homoserine O-succinyl-homoserineHSK
ATP
ADP
CoASuc-CoA
HSST
succinatePi
H2S
HGT
absent or a pseudogene
present in onlyone strain
Chapter 6, Figure 6.3. Methionine and cysteine inter-conversion pathway in S. thermophilus and L. bulgaricus. Filled red boxes represent the presence of the genes in the three S. thermophilus strains. Filled blue boxes represent the presence of the genes in both L. bulgaricus strains. The half-filled blue box represents a gene only present in L. bulgaricus ATCC BAA365. White boxes indicate that the genes are either absent or pseudogenes. Arrows represent the HGT events and the directions. Gene cysE in L. bulgaricus is truncated after the HGT, thus is also shown as a white box. The enzymes in this figure are: CysE, serine acetyltransferase; CysK, O-acetylserine sulfhydrylase; CBL, cystathionine beta lyase; CGL, cystathionine gamma lyase; MetH, homocysteine S-methyltransferase; MetE, homocysteine methyltransferase; HSK, homoserine kinase; HSST, homoserine O-succinyltransferase; CGS, cystathionine gamma synthase; CBS, cystathionine beta synthase. The distribution of the above genes in S. thermophilus and L. bulgaricus genomes is derived from our previous study. Suc-CoA, succinyl coenzyme A; Pi, phosphate.

ACKNOWLEDGEMENTS
123
AcknowledgementsThere is a metaphor for life. It is like a road trip. One will come cross many forks in the road
and needs to decide which one of the two paths to take. When struggling with a rough path, some people would regret their choices and think what if they had taken the other one, things would have been much easier. Fortunately, I have never regretted those big decisions. My life path would have been totally different, though, if I had not left my hometown at age 16 to study biology in Beijing University, if I had not chosen to come to the Netherlands, or if I had not decided to pursue the PhD degree. However, for sure, I would have missed those eye-opening opportunities, the life-long benefiting experiences, the amazing trips, and most importantly, the people who I am so grateful to.
First of all, I would like to thank my supervisor, Roland, who gave me the opportunity to work in his group and guided me into the world of bioinformatics. He taught me how to perform research independently, and helped me to improve my English writing skills. His continuous support during the whole period is one of the most essential factors leading to this thesis.
I wish to thank my co-supervisor Arjen. He started to be involved in the project in 2007, when I experienced a very difficult time to progress it further. I am grateful for his coordination of the project, his ideas on how to link the fundamental studies to applications and his contributions to this thesis.
My gratitude also goes to my coach at FrieslandCampina, Jan Geurts. I have learned so much from you, especially about how to become a researcher in industry. Thank you for your advice on improving my personal skills and developing my competencies, for your enlightened ideas and inspiring discussions. Your trust and enthusiasm on my work have been great encouragements. I also want to thank your family for the kindness, especially Mariken and you for the Dutch translation of the summary.
Furthermore, I owe a debt of thanks to: Christof, for the helpful discussions and contributions to several chapters in this thesis; Frank van Enckevort, for his kind supervision at the beginning of my PhD; Bernadet for her assistances on handling the protein structures; my German collaborators, Prof. Gasteiger (Molecular Networks /Uni. Erlangen-Nürnberg) and his group members Dr. Oliver Sacher and Dr. Bruno Bienfait, for their help to design and perform the study described in chapter 5 and comments on the manuscript; and finally my student Celine Prakash for her good job which was done within a limited time frame.
I am grateful for the financial support from the Casimir Programme of the Netherlands Ministry of Economic Affairs, which provided me a great opportunity to perform a relatively applied research in the framework of both academia and industry.
It is a pleasure to thank all the members of our department CMBI, whose names will not be listed
here but can be found at the CMBI website. In particular, I thank all the present and past members of BAMICS group, Sacha, Juma, Barzan, Victor, Daniel, Tilman, Bas, Richard and Greer. Of course my (ex)roomies: Anita, Jos, Lex, Michiel, Mark, Tom and the students. I felt so lucky to work with you guys!
My special thanks will be given to Miaomiao, the only one at work I can chat with in Chinese. I enjoyed our weekly/monthly “dates” at “Chinese restaurants” after work and our many trips together. Thank you and Tim for the invitations to your home. Plus thanks for being my paranimf!
Thank you, all my colleagues from FrieslandCampina Research, Deventer. To my group manager
Rolf Bos, for giving me the freedom to perform research. To members of Life Science group for the great working atmosphere, Alfred, Renske, Anouk, Ellen van den Heuvel, Cordula, Joost, Stefanie, Ruud, Gertjan, Yvonne, Ynte, Janine, Helmie, Anneke, Roy, Wim, Talitha, Elise, especially Ellen

ACKNOWLEDGEMENTS
124
Looijesteijn for the advice and critically reading the manuscripts. To members from sensory group, Christine, Marty, Carina, et al., for the suggestions and fruitful discussions. To all the others whose names have not been mentioned here, who have been so supportive. I am especially thankful for all the help from Herman, Peter and other foreigners who were seated in the old innovation lab. I had a lot of fun there, as well as at the dinners and parties.
Many friends I have made during all these years in the Netherlands, Dongmei, Henry, JiaoYang, Huang Lei, Xing Yutao, Li Xiaoqiang, Ji Wei, Cao Fei, Lih King, Katia, Suzette, Anne Marieke, Deborah, Jonna, Myrna, and all my neighbors from Magnificent Seven. It was a great pleasure to meet you all, and I have had a great time hanging out with you!
Hu Di, thanks for the encouragements and cheering-ups, no matter where you were. I really loved our trips in Europe and China. Your optimism has greatly strengthened me.
Jiang Jing, for our fun moment jumping from the airplane. That was one of the craziest moments and also one of the biggest achievements in my life.
Roy and Anita, thank you for inviting me to your house and letting me play with your cute children. I had such a great time at the dinners, parties, and the Christmas Eve!
Tang Ji, it is great to have you as a friend in the Netherlands. Our trips to Germany and Austria were so much fun. Our addiction to spicy foods brought us together into many food adventures. And thank you for the inspiration for designing the cover.
Irene and Anne, for all the years you have been so nice and supportive to me. Irene, you are my dearest Spanish sister. Thank you for those great moments, travelling, eating, drinking strawberry margarita, and encouraging each other. Also many thanks for inviting me to Spain for the two unforgettable New Years. I was so impressed by the hospitability of your family and enjoyed so much the foods and “siesta” there. Moreover, I really appreciate that you can be my paranimf.
Again, my sincere thanks will be dedicated to those who helped me to get through the hardest period in my life. When I had my leg broken, it was my first time feeling useless and helpless. Luckily, I had got all the help I needed, which I will always remember. Thank you to those who drove me to work and brought me home. Ji and Jing, thank you for giving me a hand with my daily life, for helping me to cook and doing grocery shopping. Irene and Anne, thanks for taking care of me, and painting the new house. Dongmei, thanks for helping me to pack and unpack the boxes. Jan, Roy, Herman and Jan Ebbekink, thank you for helping me with the moving, which I know was not an easy job.
Last but not least, my biggest thank goes to my entire family, especially my parents. As their only child, I have been away from home for most of the time during the past ten years. However, I have never felt the distance between us. Their support is the endless source of my courage.
最后,我要感谢我所有的家人,谢谢你们的支持!亲爱的爸爸妈妈,你们是我今生最大的财富!原谅我这么多年离家在外,让你们一直担
心。这些年来,你们的理解和支持就是我勇气的源泉。没有你们,没有你们的爱,就不会有今天的我,就不会有这篇论文的完成。
我是如此的幸运。谢谢! 2010年于荷兰Mengjin Liu, Deventer, March 2010

125
Publications
Liu M, Bayjanov JR, Renckens B, Nauta A, Siezen RJ (2010) The proteolytic system of lactic acid bacteria revisited: a genomic comparison. BMC Genomics. 11(1):36.
Liu M, Siezen RJ, Nauta A (2009) In silico prediction of horizontal gene transfer events in Lactobacillus delbrueckii ssp. bulgaricus and Streptococcus thermophilus reveals proto-cooperation in yoghurt manufacturing. Appl Environ Microbiol. 75(12):4120-9.
Liu M, Nauta A, Francke C, Siezen RJ (2008) Comparative Genomics of Enzymes in Flavor-Forming Pathways from Amino Acids in Lactic Acid Bacteria. Appl Environ Microbiol. 74(15): 4590-4600
Deng DM, Liu MJ, ten Cate JM, Crielaard W (2007) The VicRK system of Streptococcus mutans responds to oxidative stress. J Dent Res. 86(7):606-10.
Liu M, Siezen RJ (2006) Comparative genomics of flavour-forming pathways in lactic acid bacteria. The Australian Journal of Dairy Technology 61(2): 61-68
Liu M, van Enckevort FHJ, Siezen RJ (2005) Genome update: lactic acid bacteria genome sequencing is booming. Microbiology. 151(Pt 12):3811-3814

126

127
Curriculum Vitae
Mengjin Liu was born on July 22nd 1983, in DeXing, Jiangxi Province, China. From 1999 to 2003, she studied Biology at College of Life Sciences, Peking University in China, and obtained Bachelor Degree of Science majoring in Biotechnology. In 2003, she came to the Netherlands to begin her Master study in the Biomolecular Sciences programme at University of Amsterdam (UvA). In 2005, she completed her master thesis on the two-component system of an oral pathogen Streptococcus mutans under the supervision of Professor Wim Crielaard at Academic Centre for Dentistry in Amsterdam (ACTA) and Swammerdam Institute for Life Science (SILS), UvA. From 2005, she worked as a junior researcher at FrieslandCampina Research (formerly FrieslandFoods Corporate Research), Deventer, and as a PhD student in Bacterial Genomics group headed by Prof. Roland Siezen at the Center for Molecular and Biomolecular Informatics (CMBI) of Radboud University Medical Center Nijmegen. The research project was funded by the Casimir Programme of the Netherlands Ministry of Economic Affairs, to accelerate the knowledge exchange between academics and industry. She is now working as a researcher and project leader in the Life Science group at FrieslandCampina Research.

128
After all... tomorrow is another day...- Margaret Mitchell