charge transfer between tryptophan and tyrosine in casein: a pulse radiolysis study
TRANSCRIPT

150 Biochimica et Biophysica A cta, 705 (1982) 150- i 62 Elsevier Biomedical Press
BBA31233
CHARGE TRANSFER BETWEEN TRYPTOPHAN AND TYROSINE IN PROTEINS
JOHN BUTLER a, EDWARD J. LAND a WALTER A. PROTZ b and A. JOHN SWALLOW a
Paterson Laboratories, Christie Hospital and Holt Radium Institute, Manchester M20 9BX (U.K.) and b Institut fiir Biophysik und Strahlenbiologie, Universitiit Freibur& D- 7800 Freibur 8 (F. R. G.)
(Received December 11th, 1981)
Key words: Charge transfer; Tryptophan; Tyrosine; Pulse radiolysis
With numerous proteins, most of which are not involved in oxidation reduction reactions, azide radicals have been found to react with tryptophan units to give Trp radicals. The Trp" radicals commonly transfer their electron deficiencies to tyrosine with rate constants in the region of 102-104 s - t , woducing TyrO'. For ~-lactogiobulin, which has been studied in must detail, the rate of transfer is independent of protein concentration, so the reaction must he intramolecular. Sodium perchiorate and SDS affect the rate, pmhably because of alterations in protein conformation. The activation energy for the process is 45 kJ.mo1-1, inconsistent with any mechanism involving hydrogen bounds or charge conduction through the chain, but consistent with tunnelling. With other proteins there is evidence of intermolecular transfer as well as intramolecnlar transfer, and there may he several different intramolecular steps corresponding to different tryptophan-tyrosine pairs. Rates are not always the same as those initiated by photoionization of tryptophan, probably because different tryptophan units are involved. The possibility of transformation means that with enzymes the initial site of attack by free radicals is not necessarily the site responsible for any consequent loss in activity. The oxidation-reduction potentials involved in the transfer are such that the process may he important in the mode of action of enzymes such as high-potential copper proteins and peroxidases, and perhaps also in the mode of action of other proteins if the environment is suitable.
Introduction
Many reactions of proteins involve charge transfer, including those initiated by light or ioniz- ing radiation. One possibility is that proteins might act as semiconductors when electrons are removed by the action of electron acceptors [1].
The removal of single electrons from proteins can be conveniently carried out by means of radia- tion. Since OH" radicals tend to add to substrates, as well as to abstract electrons, it is better to use the radiation to generate the radical anion Br 2-
Abbreviations: Trp', TrpH semi-oxidized tryptophan radical, tryptophan; TyrO', TyrOH, semi-oxidized tyrosine radical, tyrosine; SDS, sodium dodecyl sulphate.
0167-4838/82/0000-0000/$02.75 © 1982 Elsevier Biomedical Press
[2,3] or, as more recently adopted, uncharged N~ [4]. These species react most efficiently with tryp- tophan (TrpH), although, at alkaline pH, other amino acids such as tyrosine (TyrOH), cysteine and methionine also show high reactivities. N~ radicals are particularly useful, since their lack of absorption above 300 nm facilitates observations of product species at longer wavelengths and, being uncharged, they may be able to react more easily than the radical anions in hydrophobic environ- ments of proteins.
When azide radicals are generated in a mixture of TrpH and TyrOH in free aqueous solution, intermolecular transfer does not readily occur be- tween Trp', which is the principal initial species, and TyrOH [5,6]. However, when N~ radicals react

151
with dipeptides of tryptophan and tyrosine, one- electron oxidation of the TrpH residue is followed by an efficient, rapid (/Ls) intramolecular process, ( k ~ 6 . 1 0 4 s - I ) which could be regarded as hy- drogen atom transfer from TyrOH to Trp" or, resulting in the same overall effect, transfer of a proton and either an electron or, in the opposite direction, a positive charge [7]. The insertion of one or two glycines between TrpH and TyrOH progressively lowers the intramolecular transfer rate to about 2-104 s -I, but not its efficiency. Insertion of a further glycine, or of an c-amino- caproic acid group, between TrpH and TyrOH leads to increased transfer rates again (approx. 3-104 s-I), showing the importance of peptide flexibility in determining the transfer rate [6]. Pro- teins tend to possess more rigid structures than peptides, and earlier studies of alcohol dehydro- genase [4] yielded evidence of a sl0w (ms) transfor- mation indicating charge transfer between TyrOH and semi-oxidized TrpH. The present paper shows that charge transfer between tryptophan and tyro- sine units can be induced in a variety of proteins and proposes that it could be a fundamental pro- cess in the normal biological action of oxidition/ reduction proteins. A brief report of these studies has recently appeared [8].
Materials and Methods
Charge-transfer reactions in proteins were gen- erally induced in N20-saturated aqueous solutions containing the protein and 20-50 mM NaN 3 by the delivery of single 10-20 ns electron pulses. Azide radicals (N3) are thereby formed within about 0.1 #s after the pulse [9],
H20~e.~ + ' O H + H', etc.
e~q + N 2 0 + H 2 0 - " O H + O H - +N2
"OH+Nf --*OH- +N;
leading to subsequent oxidation of the protein.
N~ + protein --, transients.
Some proteins were also oxidized with Br 2- radi- cals as described previously [2,3]. Transient changes were detected by kinetic absorption spectroscopy,
using a pulse radiolysis apparatus developed from that described [10]. The optical pathlength of the cells was 2.5 cm. Dosimetry was by the thiocyanate method as in previous experiments [6]. Unless otherwise specified, doses (D) were in the region of 3-15 Gy per pulse. Initial radical yields were taken to be G(N~)=G('OH)+G(e~q)~5.6 (100 eV)-i, corresponding to a molar concentration of G(N~) × D × 1.035 × 10 -7 M. Rate constants were reproducible to approx. -+ 1095.
Source and handling of proteins Proteins of the following origins were obtained
from either Boehringer (Mannheim), Serva (Heidelberg) or Sigma (St. Louis): aldolase from rabbit muscle as suspension in (NH4)2SO 4 solu- tion (Boehringer); D-amino-acid oxidase from porcine kidney as suspension in (NH4)zSO 4 solu- tion (Sigma); L-amino-acid oxidase from Crotalus atrox as powder with glycine buffer (Sigma, Type III); carbonic anhydrase B from human erythro- cytes and carbonic anhydrase from bovine erythrocytes (both Sigma); a-chymotrypsin and a-chymotrypsinogen A both from porcine pan- creas (Sigma, Type II); concanavalin A from Canaoalia ensiformis (Sigma, Type IV); cyto- chrome c from horse heart (Sigma, Type III) was used to prepare apocytochrome c according to the method previously described [11]; bovine y-globu- lins (Sigma, Cohn fraction II); a-lactalbumin from bovine milk (Sigma); p-lactoglobulins A + B and A alone from milk (Sigma); lysozyme from hen egg (Serva or Boehringer); papain from papaya latex as powder with balance salts (Sigma, Type IV); pepsin from porcine stomach (Serva, reinst); RNAase from bovine pancreas (Sigma, Type II-A); trypsin from bovine pancreas (Serva, p.A); trypsin inhibitor from soybean (Sigma, Type I-S). Solu- tions of aldolase, D- and L-amino-acid oxidase, apocytochrome c and papain were exhaustively dialyzed against the final solvent before the experi- ment in order to remove salts or buffers. Other proteins or chemicals such as NaN 3 (Merck), 2H 2 ° (Aldrich), SDS (Serva) and dithiothreitol (Sigma) were used as received. The solutions were prepared shortly before each experiment using redistilled water. They were then saturated by gentle bub- bling for 30 rain with N20 and passed through a flow system into the optical cell.
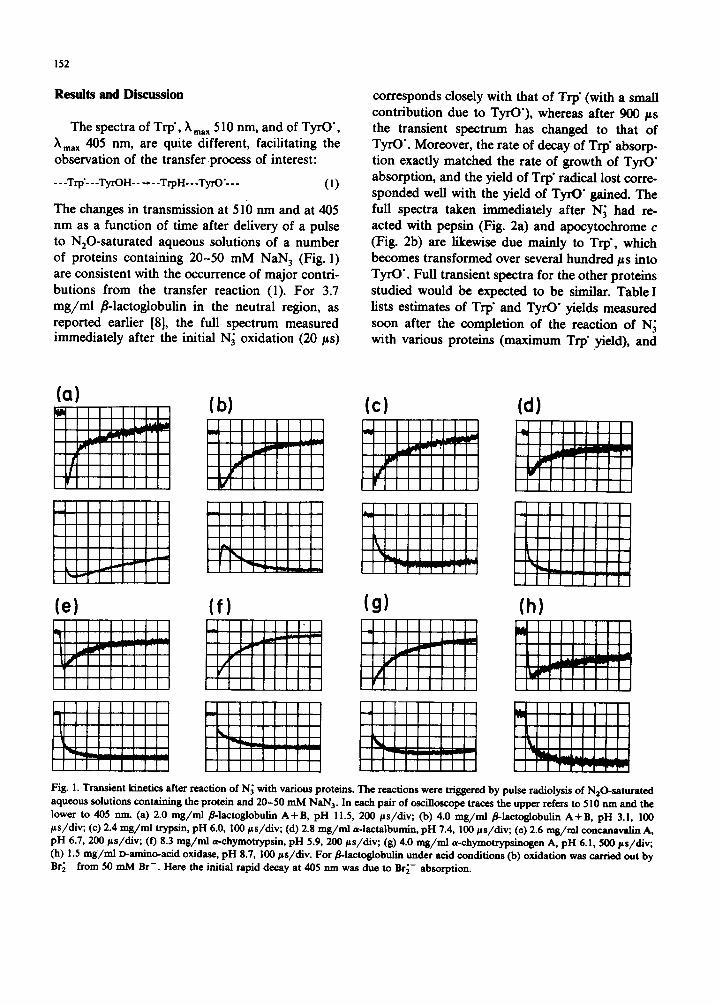
152
Results and Discussion
The spectra of Trp', ~max 510 nm, and of TyrO', ~max 405 nm, are quite different, facilitating the observation of the transfer process of interest:
-- -Trp'---TyrOH ..... T~H-- -TyrO'--- ( 1 )
The changes in transmission at 510 nm and at 405 nm as a function of time after delivery of a pulse to NHO-Saturated aqueous solutions of a number of proteins containing 20-50 mM NaN 3 (Fig. l) are consistent with the occurrence of major contri- butions from the transfer reaction (1). For 3.7 mg/ml ~8-1actoglobulin in the neutral region, as reported earlier [8], the full spectrum measured immediately after the initial N~ oxidation (20 ps)
corresponds closely with that of Trp" (with a small contribution due to TyrO'), whereas after 900/~s the transient spectrum has changed to that of TyrO'. Moreover, the rate of decay of Trp" absorp- tion exactly matched the rate of growth of TyrO" absorption, and the yield of Trp" radical lost corre- sponded well with the yield of TyrO" gained. The full spectra taken immediately after N~ had re- acted with pepsin (Fig. 2a) and apocytochrome c (Fig. 2b) are likewise due mainly to Trp', which becomes transformed over several hundred ps into TyrO'. Full transient spectra for the other proteins studied would be expected to be similar. Table I lists estimates of Trp" and TyrO" yields measured soon after the completion of the reaction of N~ with various proteins (maximum Trp" yield), and
(a) .Ad
,[I
(b) (c) (d)
. , ~ ~' j l l l l l Imil l l l l l Im
~ r j W
Wf l
d&d
!J
|
(e) (f) (g) (h) I
ql =
" 7 ~ - ' ~
Fig. l. Transient kinetics after reaction of N~ with various proteins. The reactions were trisgered by pulse radiolysis of N20-saturated aqueous solutions containing the protein and 20-50 mM NaN 3. In each pair of osciDoscope traces the upper refers to 510 nm and the lower to 405 nm. (a) 2.0 mg/ml/~-lactoglobulin A+B, pH 11.5, 200 ps/div; (b) 4.0 mg/ml ~-lactoglobulin A+B, pH 3.1, 100 ps/div; (c) 2.4 mg/ml trypsin, pH 6.0, 100 ps/div; (d) 2.8 mg/ml a-lactalbumin, pH 7.4, 100 ~s/div; (e) 2.6 ms/m1 concanavalin A, pH 6.7, 200 ps/div, (f) 8.3 ms/m1 a-chymotrypsin, pH 5.9, 200 ps/div; (g) 4.0 ms/rid a-chymotrypsinosen A, pH 6.1, 500/~s/div; (h) 1.5 m$/ml D-amino-acid oxidase, pH 8.7, 100 ps/div. For #-lactoslobulin under acid conditions (b) oxidation was carried out by Br~- from 50 mM Br-. Here the initial rapid decay at 405 nm was due to Br i - absorption.
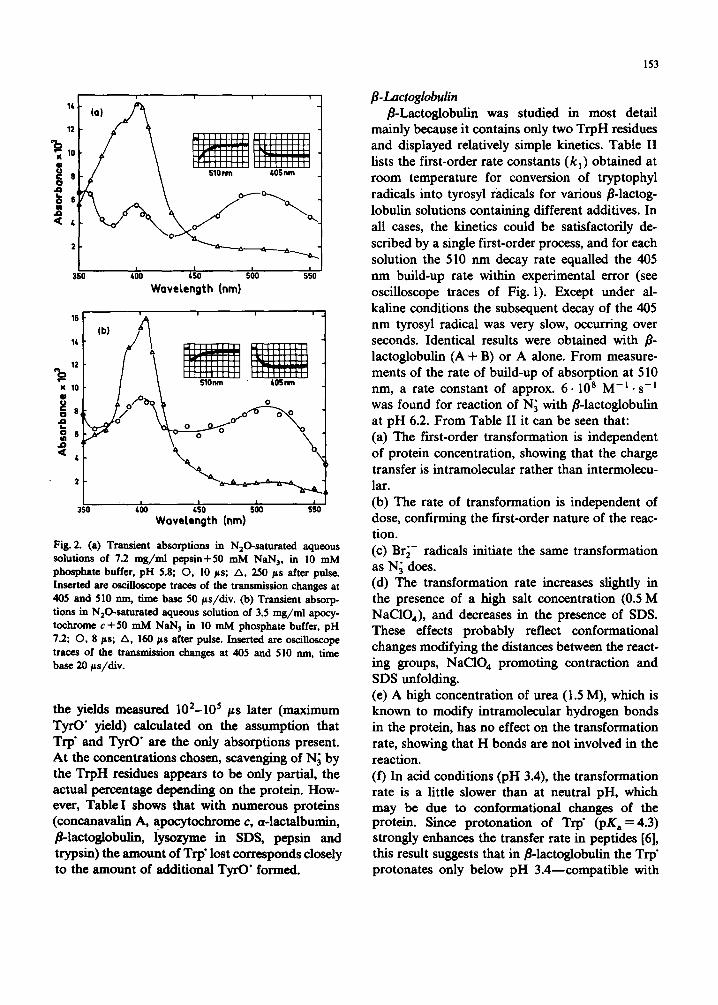
153
i t
12
g '< t
2
350
i i i i
~ 510 nm 406nm
I ,0o ,60 s~0 6~0 Wovetength (rim)
16
b I t
12
x 10 0
2
360
, i i i
f i / ~ Ill II IIIIII H ' ~
0.1 °
,h0 & 6~0 6~o Wovetength (nm)
Fig. 2. (a) Transient absorptions in N20-saturated aqueous solutions of 7.2 mg/ral pepsin+50 mM NAN3, in 10 mM phosphate buffer, pH 5.8; O, 10 ps; A, 250/~s after pulse. Inserted are oscilloscope traces of the transmission changes at 405 and 510 nm, time base 50 ps/div. (b) Transient absorp- tions in N20-saturated aqueous solution of 3.5 mg/ml apocy- tochrome c+50 mM NaN 3 in 10 mM phosphate buffer, pH 7.2; O, 8 Fs; A, 160/is after pulse. Inserted are oscilloscope traces of the transmission changes at 405 and 510 nm, time base 20 es/div.
the yields measured 102-105 ps later (maximum TyrO" yield) calculated on the assumption that Trp" and TyrO" are the only absorptions present. At the concentrations chosen, scavenging of N 3 by the TrpH residues appears to be only partial, the actual percentage depending on the protein. How- ever, Table I shows that with numerous proteins (concanavalin A, apocytochrome c, 0t-lactalbumin, fl-laetoglobulin, lysozyme in SDS, pepsin and trypsin) the amount of Trp" lost corresponds closely to the amount of additional TyrO" formed.
~-Lactoglobulin #-Lactoglobulin was studied in most detail
mainly because it contains only two TrpH residues and displayed relatively simple kinetics. Table II lists the first-order rate constants (kl) obtained at room temperature for conversion of tryptophyl radicals into tyrosyl radicals for various ~-lactog- lobulin solutions containing different additives. In all cases, the kinetics could be satisfactorily de- scribed by a single first-order process, and for each solution the 510 nm decay rate equalled the 405 nm build-up rate within experimental error (see oscilloscope traces of Fig. 1). Except under al- kaline conditions the subsequent decay of the 405 nm tyrosyl radical was very slow, occurring over seconds. Identical results were obtained with fl- lactoglobulin (A + B) or A alone. From measure- ments of the rate of build-up of absorption at 510 nm, a rate constant of approx. 6.108 M - t . s-I was found for reaction of N~ with fl-lactoglobulin at pH 6.2. From Table II it can be seen that: (a) The first-order transformation is independent of protein concentration, showing that the charge transfer is intrarnolecular rather than intermoleeu- lar. (b) The rate of transformation is independent of dose, confirming the first-order nature of the reac- tion. (c) Br~- radicals initiate the same transformation as N 3 does. (d) The transformation rate increases slightly in the presence of a high salt concentration (0.5 M NaC104), and decreases in the presence of SDS. These effects probably reflect conformational changes modifying the distances between the react- ing groups, NaC104 promoting contraction and SDS unfolding. (e) A high concentration of urea (1.5 M), which is known to modify intramolecular hydrogen bonds in the protein, has no effect on the transformation rate, showing that H bonds are not involved in the reaction. (f) In acid conditions (pH 3.4), the transformation rate is a little slower than at neutral pH, which may be due to conformational changes of the protein. Since protonation of Trp" (pKa=4.3) strongly enhances the transfer rate in peptides [6], this result suggests that in #-lactoglobulin the Trp" protonates only below pH 3.4--compatible with
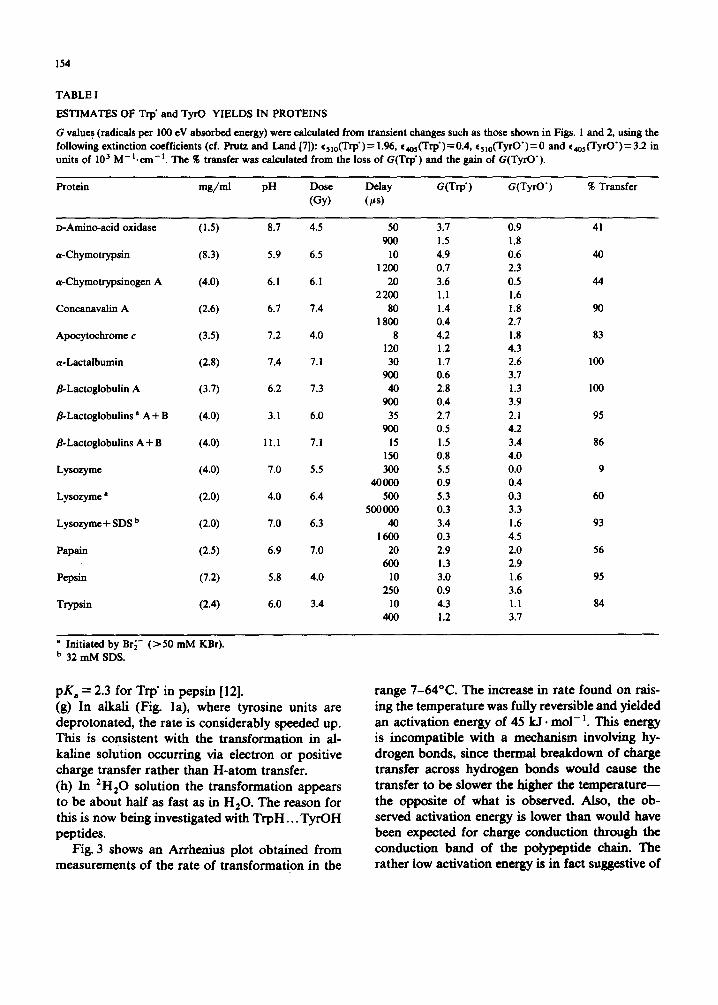
154
TABLE I
ESTIMATES OF Trp' and TyrO YIELDS IN PROTEINS
G values (radicals per 100 eV absorbed energy) were calculated from transient changes such as those shown in Figs. i and 2, using the following extinction coefficients (cf. Prutz and Land [7]): %lo(Trp')= 1.96, e4os(Trp')=0.4, %10(TyrO')=0 and ¢4o5(TyrO')=3.2 in units of 103 M - L c m - t . The % transfer was calculated from the loss of G(Trp') and the gain of G(TyrO').
Protein mg/ml pH Dose Delay G(Trp') G(TyrO') % Transfer (Gy) (~as)
D-Amino-acid oxidase (1.S) 8.7 4.5 50 3.7 0.9 41 900 1.5 1.8
a-Chymotrypsin (8.3) 5.9 6.5 10 4.9 0.6 40 1200 0.7 2.3
a-Chymotrypsinogen A (4.0) 6. I 6.1 20 3.6 0.5 44 2200 1.1 1.6
Concanavalin A (2.6) 6.7 7.4 80 1.4 1.8 90 1 800 0.4 2.7
Apocytochrome c (3.5) 7.2 4.0 8 4.2 1.8 83 120 1.2 4.3
a-Lactalbumin (2.8) 7.4 7.1 30 1.7 2.6 100 900 0.6 3.7
/]-Lactoglobulin A (3.7) 6.2 7.3 40 2.8 1.3 100 900 0.4 3.9
fl-Lactoglobulins a A + B (4.0) 3.1 6.0 35 2.7 2. i 95 900 0.5 4.2
fl-Lactoglobulins A + B (4.0) I l,l 7.1 15 1.5 3.4 86 150 0.8 4.0
Lysozyme (4.0) 7.0 5.5 300 5.5 0.0 9 40000 0.9 0.4
Lysozyme a (2.0) 4.0 6.4 500 5.3 0.3 60 500000 0.3 3.3
Lysozyme + SDS b (2.0) 7.0 6.3 40 3.4 1.6 93 I 600 0.3 4.5
Papain (2.5) 6.9 7.0 20 2.9 2.0 56 600 1.3 2.9
Pepsin (7.2) 5.8 4.0 10 3.0 1.6 95 250 0.9 3.6
Trypsin (2.4) 6.0 3.4 10 4.3 1.1 84 400 1.2 3.7
a Initiated by Br6- ( > 50 mM KBr). b 32 mM SDS.
pK a = 2.3 for Trp" in pepsin [12]. (g) In alkali (Fig. la), where tyrosine units are deprotonated, the rate is considerably speeded up. This is consistent with the transformation in al- kaline solution occurring via electron or positive charge transfer rather than H-atom transfer. (h) In 2H20 solution the transformation appears to be about half as fast as in H20. The reason for this is now being investigated with TrpH... TyrOH peptides.
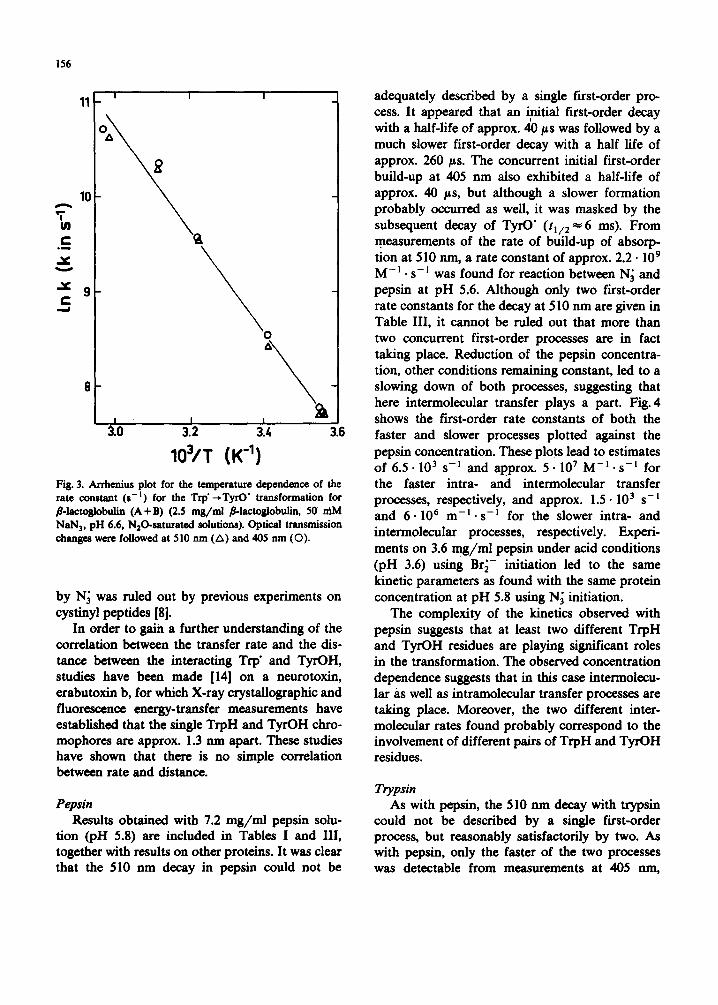
Fig. 3 shows an Arrhenius plot obtained from measurements of the rate of transformation in the
range 7-64°C. The increase in rate found on rais- ing the temperature was fully reversible and yieMed an activation energy of 45 kJ. mol-~. This energy is incompatible with a mechanism involving hy- drogen bonds, since thermal breakdown of charge transfer across hydrogen bonds would cause the transfer to be slower the higher the temperature-- the opposite of what is observed. Also, the ob- served activation energy is lower than would have been expected for charge conduction through the conduction band of the polypeptide chain. The rather low activation energy is in fact suggestive of
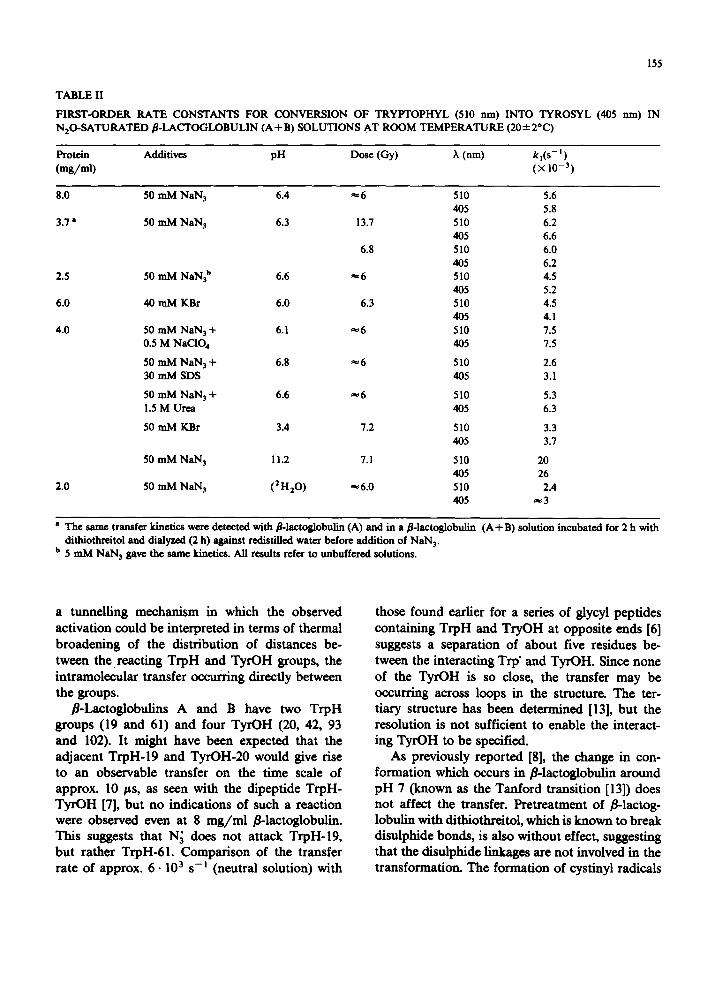
155
TABLE II
FIRST-ORDER RATE CONSTANTS FOR CONVERSION OF TRYPTOPHYL (510 nm) INTO TYROSYL (405 nm) IN N20-SATURATED fl-LACTOGLOBULIN (A+ B) SOLUTIONS AT ROOM TEMPERATURE (20 .4- 2°C)
Protein Additives pH Dose (Gy) 7, (nm) k,(s- I) (mg/ml) ( )< i 0 -- 3)
8.0 50 mM NaN 3 6.4 ~ 6 510 5.6 405 5.8
3.7 a 50 mM NaN 3 6.3 13.7 510 6.2 405 6.6
6.8 510 6.0 405 6.2
2.5 50 mM NaN3 b 6.6 ~ 6 510 4.5 405 5.2
6.0 40 mM KBr 6.0 6.3 510 4.5 405 4.1
4.0 50 mM NaN 3 + 6.1 ~ 6 510 7.5 0.5 M NaCIO 4 405 7.5
50 mM NaN 3 + 6.8 ~ 6 510 2.6 30 mM SDS 405 3.1
50 mM NaN 3 + 6.6 ~ 6 510 5.3 1.5 M Urea 405 6.3
50 mM KBr 3.4 7.2 510 3.3 405 3.7
50 mM NaN 3 11.2 7.1 510 20 405 26
2.0 50 mM NaN 3 (2H20) ~6.0 510 2.4 405 ~ 3
• The same transfer kinetics were detected with 8-1actoglobulin (A) and in a 8-1actoglobulin (A + B) solution incubated for 2 h with dithiothreitol and dialyzed (2 h) against redistilled water before addition of NaN 3.
b 5 mM NaN 3 gave the same kinetics. All results refer to unbuffered solutions.
a tunnelling mechanism in which the observed activation could be interpreted in terms of thermal broadening of the distribution of distances be- tween thereact ing TrpH and TyrOH groups, the intramolecular transfer occurring directly between the groups.
//-Lactoglobulins A and B have two TrpH groups (19 and 61) and four TyrOH (20, 42, 93 and 102). It might have been expected that the adjacent TrpH-19 and TyrOH-20 would give rise to an observable transfer on the time scale of approx. 10 ps, as seen with the dipeptide TrpH- TyrOH [7], hut no indications of such a reaction were observed even at 8 rag/m1 8-1actoglobulin. This suggests that N~ does not attack TrpH-19, but rather TrpH-61. Comparison of the transfer rate of approx. 6.103 s-~ (neutral solution) with
those found earlier for a series of giycyl peptides containing TrpH and TryOH at opposite ends [6] suggests a separation of about five residues be- tween the interacting Trp" and TyrOH. Since none of the TyrOH is so close, the transfer may be occurring across loops in the structure. The ter- tiary structure has been determined [13], but the resolution is not sufficient to enable the interact- ing TyrOH to be specified.
As previously reported [8], the change in con- formation which occurs in/~-lactoglobulin around pH 7 (known as the Tanford transition [13]) does not affect the transfer. Pretreatment of fl-lactog- lobulln with dithiothreitol, which is known to break disulphide bonds, is also without effect, suggesting that the disulphide linkages are not involved in the transformation. The formation of cystinyl radicals
156
A "T in c
° ~ ,v, ,,e e-
11
10
9
! I
I I 3.0 3.2
, _J
~ o
I 3.4 3.6
103/T (K -1) Fig. 3. Arrhenius plot for the temperature dependence of the rate constant (s -I) for the Trp" -. TyrO" transformation for fl-lactoglobulin (A+B) (2.5 mg/ml fl-lactoglobulin, 50 mM NaN 3, pH 6.6, N20-saturated solutions). Optical transmission changes were followed at 510 nm (A) and 405 nm (O).
by N 3 was ruled out by previous experiments on cystinyl peptides [8].
In order to gain a further understanding of the correlation between the transfer rate and the dis- tance between the interacting Trp" and TyrOH, studies have been made [14] on a neurotoxin, erabutoxin b, for which X-ray crystallographic and fluorescence energy-transfer measurements have established that the single TrpH and TyrOH chro- mophores are approx. 1.3 nm apart. These studies have shown that there is no simple correlation between rate and distance.
Pepsin Results obtained with 7.2 m g / m l pepsin solu-
tion (pH 5.8) are included in Tables I and III, together with results on other proteins. It was clear that the 510 nm decay in pepsin could not be
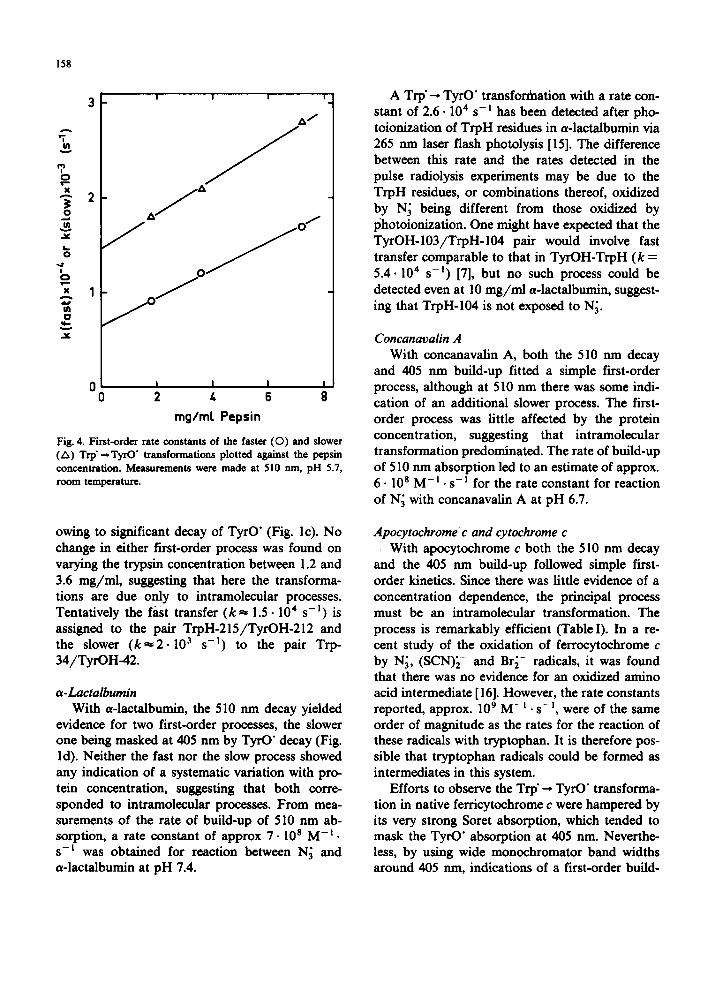
adequately described by a single first-order pro- cess. It appeared that an initial first-order decay with a half-life of approx. 40/~s was followed by a much slower first-order decay with a half life of approx. 260/~s. The concurrent initial first-order build-up at 405 nm also exhibited a half-life of approx. 40 /zs, but although a slower formation probably occurred as well, it was masked by the subsequent decay of TyrO" ( t ] / 2 ~ 6 ms). From measurements of the rate of build-up of absorp- tion at 510 nm, a rate constant of approx. 2.2.10 9 M - i . s - E was found for reaction between N; and pepsin at pH 5.6. Although only two first-order rate constants for the decay at 510 nm are given in Table III, it cannot be ruled out that more than two concurrent first-order processes are in fact taking place. Reduction of the pepsin concentra- tion, other conditions remaining constant, led to a slowing down of both processes, suggesting that here intermolecular transfer plays a part. Fig. 4 shows the first-order rate constants of both the faster and slower processes plotted against the pepsin concentration. These plots lead to estimates of 6.5 • 10 3 s-! and approx. 5.107 M - ] • s - I for the faster intra- and intermolecular transfer processes, respectively, and approx. 1.5.10 3 s -I and 6 .10 6 m - I . s -m for the slower intra- and intermolecular processes, respectively. Experi- ments on 3.6 m g / m l pepsin under acid conditions (pH 3.6) using Br~- initiation led to the same kinetic parameters as found with the same protein concentration at pH 5.8 using N; initiation.
The complexity of the kinetics observed with pepsin suggests that at least two different TrpH and TyrOH residues are playing significant roles in the transformation. The observed concentration dependence suggests that in this case intermolecu- lar as well as intramolecular transfer processes are taking place. Moreover, the two different inter- molecular rates found probably correspond to the involvement of different pairs of TrpH and TyrOH residues.
Trypsin As with pepsin, the 510 nm decay with trypsin
could not be described by a single first-order process, but reasonably satisfactorily by two. As with pepsin, only the faster of the two processes was detectable from measurements at 405 rim,
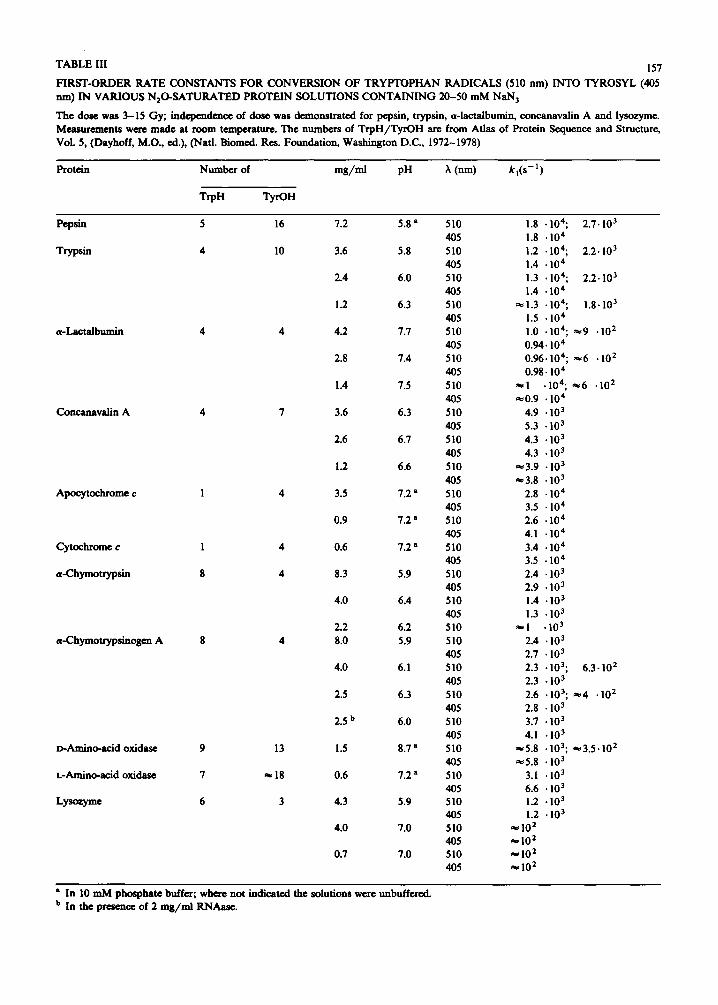
TABLE III 157
F IRST-ORDER RATE CONSTANTS F OR CONVERSION OF T R Y P T O P H A N RADICALS (510 nm) INTO TYROSYL (405 nm) IN VARIOUS N 2 0 - S A T U R A T E D PROTEIN SOLUTIONS C O N T A I N I N G 20-50 m M NaN 3
The dose was 3-15 Gy; independence of dose was demonstrated for pepsin, trypsin, a-lactalbumin, concanavalin A and lysozyme. Measurements were made at room temperature. The numbers of T r p H / T y r O H are from Atlas of Protein Sequence and Structure, VoL 5, (Dayhoff, M.O., ed.), (Natl. Biomed. Res. Foundation, Washington D.C., 1972-1978)
Protein Number of m g / m l pH ?~ (nm) k , ( s - 1 )
TrpH TyrOH
Pepsin 5
Trypsin 4
a-Lacta lbumin 4
Concanavalin A 4
Apocytochrome c 1
Cytochrome c 1
a-Chymotryps in 8
a-Chymotrypsinogen A 8
D-Amino-acid oxidase 9
L-Amino-acid oxidase 7
Lyso~yme 6
16 7.2 5.8 a 510 1.8 • 104; 2,7.103
405 1.8 • 104
I0 3.6 5.8 510 1.2 . 104; 2.2.103
405 1.4 • 10 4
2.4 6.0 510 1.3 .104; 2.2.103
405 1.4 • 10 4
1.2 6.3 5 I0 ~ 1.3 • 104; 1.8.103
405 1.5 • 10 4
4 4.2 7.7 510 1.0 • I04; ~9 .102
405 0.94.104
2.8 7,4 510 0.96.104; ~6 • 10 2
405 0.98.10 4
1.4 7.5 510 ~I .104; ~e6 .10 2
405 ~0.9 • 104 7 3.6 6.3 510 4.9 • 103
405 5.3 • 103
2.6 6.7 510 4.3 • 103
405 4.3 • 103
1.2 6.6 510 ~3.9 • 103
405 ~3.8 • 103
4 3.5 7.2 a 510 2.8 "104
405 3.5 • 104
0.9 7,2 a 510 2.6 "10 4
405 4.1 • 10 4
4 0.6 7.2 a 510 3.4 • 10 4
405 3.5 • 10 4
4 8.3 5.9 510 2.4 • 103
405 2.9 • 103 4.0 6.4 510 1.4 • 103
405 1.3 • 103 2.2 6.2 510 ~ l "10 3
4 8.0 5.9 510 2.4 • 103
405 2.7 • 103 4.0 6.1 510 2.3 • 103; 6.3-102
405 2.3 • 103
2.5 6.3 510 2.6 • 103; #e4 • 10 2
405 2.8 • 103 2.5 b 6.0 510 3.7 • 103
405 4.1 -103 13 1.5 8.7 a 510 ~5 .8 .103; ~3 .5 -10 2
405 ~5 .8 • 103 ~ 1 8 0.6 7.2 a 510 3.1 .103
405 6.6 • 103 3 4.3 5.9 510 1.2 • 103
405 1.2 • 103 4.0 7.0 5 I0 ~ I0 2
405 ~ 102 0.7 7.0 510 ae I0 2
405 ~ 102
• In I0 m M phosphate buffer; where not indicated the solutions were unbuffered. b In the presence of 2 m g / m l RNAase.
158
3
2
1
0 0 2 4 6 8
rag/mr Pepsin
Fig. 4. First-order rate constants of the faster ( 0 ) and slower (A) Trp" --- TyrO" transformations plotted a~ainst the pepsin concentration. Measurements were made at 510 rim, pH 5.7, room temperature.
owing to significant decay of TyrO" (Fig. lc). No change in either first-order process was found on varying the trypsin concentration between 1.2 and 3.6 mg/mi, suggesting that here the transforma- tions are due only to intramolecular processes. Tentatively the fast transfer ( k ~ 1.5. l04 s -I) is assigned to the pair TrpH-215/TyrOH-212 and the slower ( k ~ 2 . l03 s -n) to the pair Trp- 34/TyrOH-42.
a -Lactalbumin With a-lactalbumin, the 510 nm decay yielded
evidence for two first-order processes, the slower one being masked at 405 nm by TyrO" decay (Fig. l d). Neither the fast nor the slow process showed any indication of a systematic variation with pro- tein concentration, suggesting that both corre- sponded to intramolecular processes. From mea- surements of the rate of build-up of 510 nm ab- sorption, a rate constant of approx 7. l0 s M -x. s-L was obtained for reaction between N~ and a-lactalbumin at pH 7.4.
A Trp" -, TyrO" transfordaation with a rate con- stant of 2.6.104 s-n has been detected after pho- toionization of TrpH residues in a-lactalbumin via 265 nm laser flash photolysis [15]. The difference between this rate and the rates detected in the pulse radiolysis experiments may be due to the TrpH residues, or combinations thereof, oxidized by N~ being different from those oxidized by photoionization. One might have expected that the TyrOH-103/TrpH-104 pair would involve fast transfer comparable to that in TyrOH-TrpH (k = 5.4.104 s -m) [7], but no such process could be detected even at 10 mg/ml a-lactalbumin, suggest- ing that TrpH-104 is not exposed to N~.
Concanavalin A With concanavalin A, both the 510 nm decay
and 405 nm build-up fitted a simple first-order process, although at 510 nm there was some indi- cation of an additional slower process. The first- order process was little affected by the protein concentration, suggesting that intramolecular transformation predominated. The rate of build-up of 510 nm absorption led to an estimate of approx. 6.108 M-m. s-n for the rate constant for reaction of N~ with concanavalin A at pH 6.7.
Apocytochrome c and cytochrome c With apocytochrome c both the 510 nm decay
and the 405 nm build-up followed simple first- order kinetics. Since there was little evidence of a concentration dependence, the principal process must be an intramolecular transformation. The process is remarkably efficient (Table I). In a re- cent study of the oxidation of ferrocytochrome c by N~, (SCN)[- and Br~- radicals, it was found that there was no evidence for an oxidized amino acid intermediate [ 16]. However, the rate constants reported, approx. 109 M- m. s- n, were of the same order of magnitude as the rates for the reaction of these radicals with tryptophan. It is therefore pos- sible that tryptophan radicals could be formed as intermediates in this system.
Efforts to observe the Trp" --, TyrO" transforma- tion in native ferricytochrome c were hampered by its very strong Sorer absorption, which tended to mask the TyrO" absorption at 405 nm. Neverthe- less, by using wide monochromator band widths around 405 nm, indications of a first-order build-

159
up at this wavelength, corresponding to an equiva- lent decay at 510 nm, were obtained, although the yield of Trp" appeared to be only about 10~ of that found with apocytochrome c. The first-order transformation rates obtained with apocytochrome c and cytochrome c were, within error, the same. It is known that the tertiary structures of apo- and native cytochrome c are different [ 11]. However, as there is only one tryptophan unit in horse heart cytochrome c and the same very fast reaction is observed in both forms, this single tryptophan-59 must in the two cases be very near a tyrosine, the nearest from the crystal structure being tyrosine-74. These observations do provide direct evidence for the feasibility of a mechanism similar to that pro- posed by Winfield [17] for some reactions of cyto- chrome c.
a-Chymotrypsin and a-chymotrypsinogen A a-Chymotrypsin exhibited a concentration de-
pendence in the transformation rate, indicating the presence of intermolecular as well as intramolecu- lar processes. A solution of a single concentration (10 mg/ml) of a-chymotrypsin in water yielded evidence for a Trp" -~ TyrO" transformation with a rate constant of approx. 4.103 s -~ after photo- ionization of TrpH residues in the protein by 265 nm laser flash photolysis [15]. This rate is some- what faster than the rate expected for this con- centration from the present pulse radiolysis experi- ments. As with a-lactalbumin~ this again suggests that the distribution of TrpH residues oxidized by photoionization differs from that produced by N~ attack, a-Chymotrypsinogen A, on the other hand, showed little evidence for a concentration depen- dence, consistent with predominance of an in- tramolecular ~ansformation. From measurements of the rate of build-up of 510 nm absorption, a rate constant of approx. 6.108 M -1 .s -] was obtained for reaction of N~ with a a-chymotryp- sinogen A. Addition of RNAase to a-chymotryp- sinogen A led to an increase in the rate of decay of Trp" and build-up of TyrO', indicating intermolec- ular electron transfer from TyrOH in RNAase (which contains no TrpH) to Trp" in the a- chymotrypsinogen.
D- and L-amino-acid oxidases Both D- and L-amino-acid oxidases gave evi-
dence for the Trp'-~ TyrO" transformation, but, since each was only studied at one concentration, the relative importance of intra- or intermolecular processes is not known. At the protein concentra- tions studied, the final yield of TyrO" was approx. 30 and 25~ of the initial yield of N~ radicals for D- and L-amino-acid oxidases, respectively.
Lysozyme For lysozyme at pH 5.9, both the decay at 510
nm and the build -up at 405 nm followed first-order kinetics (Table III). At pH 7 the rates were much slower, but were the same for 4.0 and 0.7 mg/ml lysozyme, indicating a mainly intramolecular transformation. However, only a minor fraction of Trp" appears to be transformed into TyrO" at this pH (Table I). The rates were so slow that fluctua- tions in the intensity of the analyzing lamp pre- vented accurate estimates of the corresponding rate constants being obtained. From measure- ments of the rate of build-up of 510 nm absorp- tion, a rate constant of approx. 2.10 9 M -1. s -I was obtained for reaction of N~ with lysozyme. SDS was found to affect dramatically both the kinetics and yields of the transformation at pH 7 (see Table I). The effects of pH and SDS must relate to the structural changes in the protein, which suggests that the present method could be generally useful in the study of the modification of protein structure by environment. Further work on lysozyme is now in progress.
Other proteins Neutral solutions of y- globulins (1.0 mg/ml),
papain (2.5 mg/ml) and bovine and human carbonic anhydrase (2.0 and 1.0 mg/ml) were also studied at 510 and 405 nm, but, although in each case significantly differing kinetics were obtained at these two wavelengths after N~ attack, con- sistent with a contribution from Trp'~TyrO" transformations, in no case did the 510 nm decay match growth at 405 nm. Carbonic anhydrase (2 mg/ml) dissolved in 14 mM SDS, on the other hand, revealed matching decay at 510 nm and build-up at 405 nm with k ~ 10 4 s - l . Aldolase A (2.7 mg/ml) and trypsin inhibitor (1-4 mg/ml) gave little evidence for transfer reactions after N~ attack in neutral solutions.

160
Relevance to the reactions of other oxidizing radicals with enzymes
As mentioned earlier, certain oxidizing radicals possess a considerable degree of selectivity in their reactions with amino acid residues [3]. This selec- tivity, and its variation with pH, has been used to probe active sites of enzymes by comparing the enzyme inactivation by these radicals with the site of their primary attack, often as identified by the characteristic absorption spectra of certain tran- sient amino acid radicals formed in such oxidative reactions (for reviews see Refs. 18 and 19). How- ever, in the probe technique, transient spectra were usually measured 2-300/~s after the pulse, after which time Trp'--, TyrO" transitions of the type described in the present paper will have had time to occur at least partially. Consequently, the damage caused by oxidizing radicals may eventu- ally appear at a position different from the initial site of attack. Such remote positions could be essential to the activity of the enzyme, so that selective radical reactions do not necessarily probe sites which control enzyme activity. Moreover, the present study deals only with migrations of damage from tryptophan to tyrosine, which are compara- tively easy to detect, because of the sharp and distinct absorption spectra of Trp" and TyrO'. Other types of radical transformation are also possible in principle, but could be less easy to observe.
It is often found in free radical probe studies that in going from neutral to alkaline solutions, the amount of TyrO" observed increases. Examples are trypsin [20], carboxypeptidase A [21], papain [22], subtilisins Carlsberg and Nova [23], a- chymotrypsin [24], carbonic anhydrase [25] and D-amino-acid oxidase [26]. The enhanced TyrO" is usually attributed to increased exposure of tyro- sine residues in alkaline conditions, and the in- creased reactivity of TyrO- as compared with TyrOH (pK of TyrOH ~ 10), although it has been suggested [25] that in carbonic anhydrase some of the initial damage to TrpH is transferred to TyrOH and histidine. The present studies demonstrate that an important additional factor may be intra- and/or intermolecular charge transfer between Trp" and TyrOH, which, if other enzymes behave like fl-lactoglobulin, is likely to be strongly base- catalyzed. With trypsin [20], it was noted that
tyrosine residues appeared to be more reactive towards Br 2- and (SCN)2 ~- than expected from experiments with separate amino acids, even at neutral pH. In view of the present studies, it seems likely that in this case the transient absorption spectra caused by radical anion attack were mea- sured after subsequent intramolecular Trp" -, TyrO" transformations had occurred. Such transforma- tions may also be taking place in the free radical probe studies of pepsin [12] and of lactate dehy- drogenase [27].
On the feasibility of tryptophan or tyrosine radicals being biologically generated in a protein
Several experiments (unpublished) using free radicals to generate the reduced tryptophan and tyrosine radicals have shown that this process is energetically very unfavourable. Only the hydrated electron, E ° = - 2 . 9 V [28] was able to produce these radicals at any appreciable rate. The reac- tions of CO~- (E °= -2 .0 V), CH2OH ( E ° = -0 .97 V), CHaCHOH ( E ° = - 1 . 1 V) and (CH3)2(~OH (E ° = - 1.8 V) radicals [29] with the free amino acids were not detected (k < 106 M - i . s-I) . This would indicate that the potential of the amino acid/amino acid anion radical couple is certainly more negative than - 1 V and hence, even allowing for the environment in a protein, it is unlikely that reduced amino acid radicals can be produced in the normal biological reactions of proteins.
The reduction potential of the oxidized tryp- tophan radicals can be evaluated from the equi- librium:
TrpI:I + +SCN- ~TrpH+SCN" (2)
The equilibrium constant K 2 has been evaluated as 6.3.10 -s [30].
If the standard reduction potential of the cou- ple, TrpI2I+/TrpH is E~ and of the couple SCN ' /SCN- is E~ then,
E~' -- E [ =0.059 logK 2
= -0 .425 V
The SCN" radical is in equilibrium with the (SCN)~-:
SCN" + S C N - =(SCN);- (3)

K 3 - - 2 . 1 0 5 M -I [31]. Thus if E~ is the potential of the (SCN)2-/2 SCN- couple:
E~ - E~ =0.059 log K 3
=0.313 V
Thus E~--E~= -0.112V. E~' can be calculated to be 1.25 -+ 0.22 (Butler, J., based on the dissocia- tion constant of ISClq- [32] and the reduction potential of I ' / I - [33]), so that E~' = 1.14-+ 0.2.
This value refers to the standard reduction potential of the protonated tryptophan radical. The corresponding value for the unprotonated radical is given by E~'-0.059 logK4, where K4 is the dissociation constant of the protonated radical TrpH + (pK 4 = 4.3). Thus E ° (Trp', H+/TrpH) = 1.39-+0.2. The reduction potential is dependent on pH such that E~, the basic potential of the couple, will be 0.83 V less than that above [34], and ET, the potential at pH 7, is 0.413 less, i.e., 0.98 -+ 0.2. An independent estimate (see Ref. 35) has indicated that this value could be lower, i.e., E 7 ~ 0.6. Such potentials for this couple are also consistent with the value at pH 7 being more positive than that of ferricyanide ( E ° = 0.4 V) and more positive at pH 3 than that of the Fe(H20)63+/Fe(H20)62+ couple (E ° = 0.77 V) [36]. There are as yet no reliable data on the reduction potential of the oxidized tyrosine radical, but E~3.5 for the phenoxyl radial itself has been given as over 0.5 V [37]. Allowing for the pK of the phenol group (10.0), E 7 for phenoxyl would be 0.68 V or more. The reduction potential of TyrO" is expected to be less, due to the electron-donating effect on the side chain. It is certainly known that tyrosine is oxidized by ferricyanide in alkaline solution [38], so the potential must be less than 0.4 V. The fact that the tryptophan radical efficiently oxidizes tyrosine in peptides [7] over the whole pH range means that its reduction potential, at least in peptides and proteins, must be less than that of tryptophan. The potentials at pH 7 are still quite positive, i.e., approx. 0.5-1.0 V, and it should be stated that, if the potential of the tryptophyl cou- ple in the protein is the same as that in solution, the radical will not be formed in normal biochemi- cal reactions of, for example, the cytochromes (E 7 ~- -0 .3 to +0.4V). However, they could oc- cur as intermediates in the reactions of the high-
161
potential copper proteins such as certain laccases and azurins [39]. They could be formed in systems in which hydrogen peroxide (ET(H202, H + / ' O H ) = 0.85 V) or oxygen (E~(O z, H+/H202) = 0.82 V) are involved (Koppenol and Singh, unpublished results), as, for example, the reaction of cy- tochrome c peroxidase with hydrogen peroxide. Free radicals have been unequivocally detected in the yeast peroxidase system and it has been pro- posed that tryptophan radicals are involved [40]. It should also be considered that the interior en- vironment of a protein may have some dramatic effect on the oxidation-reduction reactions of aromatic amino acid radicals. Effects such as low proton availability, a non-aqueous environment or any possible interaction between the aromatic re- sidues could effectively lower the reduction poten- rials of the radicals, making them more easily formed. Further work is required to discover whether such processes are involved in oxidation/reduction proteins.
Acknowledgements
This work was supported in part by grants from the Cancer Research Campaign and the Medical Research Council. Financial support to W.A. Prutz was given by the Deutsche Forschungsgemein- schaft (Grant Pr 178/1). We thank F.A.P. Rush- ton and R. SwindeU for assistance in preparing the figures and S. Vogel for assistance'in the evalua- tion of data given in Tables II and III.
References
1 Wolstenholme, G.E.W., Fitzsimons, D.W. and Whelan, J. (eds.) (1979) Submolocular Biology and Cancer, Excerpta Medica, Amsterdam
2 Land, E.J. and Swallow, A.J. (1971) Biochim. Biophys. Acta 234, 34-42
3 Adams, G.E., Aldrich, J.E., Bisby, R.H., Cundall, R.B., Redpath, J.L. and Willson, R.L. (1972) Radiat. Res. 49, 278-289
4 Land, E.J. and Prutz, W.A. (1979) Int. J. Radiat. Biol. 36, 75-83
5 Swallow, A.J. (1977) Discuss. Faraday Soc. 63, 289-290 6 Prutz, W.A., Land, E.J. and Sloper, R.W. (1981) J. Chem.
Soc., Faraday Trans. 1, 77, 281-292 7 Prutz, W.A. and Land, E.J. (1979) Int. J. Radiat. Biol. 36,
513-520 8 Prutz, W.A., Butler, J., Land, E.J. and Swallow, A.J. (1980)
Biochem. Biophys. Res. Commun. 96, 408-414

162
9 Hayon, E. and Simic, M. (1970) J. Am. Chem. Soc. 92, 7486-7487
10 Keene, J.P. (1964) J. Sci. Instrum. 41. 493-496 II Fisher, W.R., Taniuchi, H. and Anfinsen, C.B. (1973) J.
Biol. Chem. 248, 3188-3195 12 Adams, G.E., Posener, M.L., Bisby, R.H., Cundall, R.B.
and Key, J.R. (1979) Int. J. Radiat. Biol. 35, 497-507 13 Green, D.W., Aschaffenburg, R., Camerman, A., Coppola,
J.C., DunniU, P., Simmons, R.M., Komorowski, E.S., Sawyer, L., Turner, E.M.C. and Woods, K.F. (1979) J. Mol. Biol. 131,375-397
14 Prutz, W.A., Siebert, F., Butler, J., Land, EJ., Menez, A. and Montenay-Garestier, T. (1982) Biochim. Biophys. Acta 705, 139-149
15 Sloper, R.W. and Land, E.J. (1980) Photochem. Photobiol. 32, 687-689
16 Seki, H. and Imamura, M. (1981) Biochim. Biophys. Acta 635, 81-89
17 Winfield, M.E. (1965)J. Mol. Biol. 12, 600-611 18 Adams, G.E. (1972) Adv. Radiat. Chem. 3, 125-208 19 Bisby, R.H., Cundall, R.B. and Davies, A.K. (1978) Photo-
chem. Photobiol. 28, 825-837 20 Adams, G.E., Redpath, J.L., Bisby, R.H. and CundaU, R.B.
(1973) J. Chem. Sot., Faraday Trans. 1, 69, 1608-1617 21 Roberts, P.B. (1973) Int. J. Radiat. Biol. 24, 143-152 22 Adams, G.E. and Redpath, J.L. (1974) Int. J. Radiat. Biol.
25, 129-138 23 Bisby, R.H., Cundall, R.B., Adams, G.E. and Redpath, J.L.
(1974) J. Chem. Soc., Faraday Trans. 1, 70, 2210-2218 24 Baverstock, K., Cundall, R.B., Adams, G.E. and Redpath,
J.L. (1974) Int. J. Radiat. Biol. 26, 39-46
25 Redpath, J.L., Santus, R., Ovadia, J. and Grossweiner, L.I. (1975) Int. J. Radiat. Biol. 28', 243-253
26 Anderson, R.F., Patel, K.B. and Adams, G.E. (1977) Int. J. Radiat. Biol. 32, 523-531
27 Buchanan, J.D., Armstrong, D.A., Greenstock, C.L. and Ruddock, G.W. (1977) Int. J. Radiat. Biol. 32, 247-257
28 Swallow, A.J. (1973) Radiation Chemistry, Longman, London
29 Breitenkamp, M., Henglein, A. and Lifie, J. (1976) Ber. Bunsenges. Phys. Chem. 80, 973-979
30 Posener, M.L., Adams, G.E., Wardman, P. and Cundall, R.B. (1976) J. Chem. Soc., Faraday Trans. !, 72, 2231-2239
31 Baxendale, J.H., Bevan, P.L.T. and Stott, D.A. (1968) Trans. Faraday Soc. 64, 2389-2397
32 Schoneshofer, M. and Henglein, A. (1970) Bet. Bunsenges. Phys. Chem. 74, 393-398
33 Henglein, A. (1980) Radiat. Phys. Chem. 15, 151-158 34 Latimer, W.M. (1962) Oxidation Potentials, Prentice-Hall,
N.J. 35 Grossweiner, L.I. (1976) Curr. Top. Radiat. Res. Q. 11,
141-199 36 Bums, P.S., Harrod, J.F., Williams, RJ.P. and Wright, P.E.
(1976) Biochim. Biophys. Acta 428, 261-268 37 Steenken, S. and Neta, P. (1979) J. Phys. Chem. 83, i 134-
!137 38 Moore, F.R. and Williams, RJ.P. (1976) Coord. Chem.
Rev. 18, 125-197 39 Beinert, H. (1980) Coord. Chem. Rev_33, 55-85 40 Poulos, T.L. and Kraut, J. (1980) J. Biol. Chem. 255,
8199-8205