characterization of two conditional early mutants of rous sarcoma virus
TRANSCRIPT

VIROLOGY 63, 258-273 (1973)
Characterization of Two Conditional Early Mutants of
Rous Sarcoma Virus’
MAXINE LINIAL’ AND WILLIAM S. MASON3
Department of Microbiology, University of Southern California School of Medicine, 2025 Zonal Avenue, Los Angeles, California 90043
Accepted February 16, 1973
Some of the biological and physical properties of two independently isolated mu- tants of Rous sarcoma virus have been investigated. Both ts 335 and ts 337 are unable either to transform or replicate at 41”. The lesions in the mutants occur very early after infection (prior to 2 hr); however, the lesions appear to be expressed after ad- sorption to and penetration into the host cells. Both mutants contain temperature sensitive structural components, and ts 337 is also antigenically different from the wild type. The virions contain a polymerase activity which appears to be tempera- ture sensitive in both the endogenous reaction and with poly rA:d(pT)io as the ex- ogenous t,emplate. Upon coinfection of chick fibroblasts with ts 335 or Is 337 and Rous- associated virus (RAV), genetically stable wild-t,ype sarcoma virus is produced at a high frequency. Therefore, the information for the function(s) which is mutant in these viruses is also present in RAV. All the parameters measured are consistent with a mutation affecting the function of the avian tumor virus RNA-dependent DNA polymerase.
INTRODUCTION
There have been numerous reports of the isolation of temperature-sensitive mutants of avian sarcoma viruses (Toyoshima and Vogt, 1969a, b; lIartin, 1970; Biquard and Vigier, 1972; Bader and Brown, 1971; Temin, 1971; Rawai and Hanafusa, 1971; Vogt et al., 1972; Wyke, in preparation). The majority of these mutants are deficient only in the ability to induce ccl1 transforma- tion; they are able to replicate normally at the nonpermissive temperature. In order to study the events occurring early after infec-
1 Supported by U. S. Public Health Service Research Grant No. CA 13213 from the National Cancer Institute and Contract No. NCI-NIH 72.2032 under the Special Virus Cancer Program.
2 Fellow of the Jane Coffin Childs Memorial Fund for Medical Research. Present address: Department of Genetics, University of Washing- ton, Seattle, Washington 98105.
3 Fellow of the Leukemia Society of America, Inc.
tion which arc central to both viral replica- tion and cell transformation, coordinate mutants (containing a single mutation af- fecting both replication and transformation) are desirable. The only well studied coordi- nate mutants previously reported are ts 334(75) and ts 336(149), formerly referred to as ts 75 and ts 149 (Friis et al., 1971; Katz and Vogt, 1971). Ts 336(149) is dcfi- cicnt in an unknown factor which is required fairly early after infection. Ts 334(75) is deficient in a function acting late in the replication cycle, possibly a structural poly- peptide of the vii-ion. In neither case can the biological properties of the mutants be ex- nlained in terms of known defects.
In the present study, two coordinate mu- tants, ts 335 and ts 337, have been investi- gated. The function defective in these vi- ruses is necessary for the initiation, but not the maintenance, of both viral replication and cell transformation.
258 Copyright 0 1973 by Academic Press, Inc. 911 rights of reproduction in any form reserved.

EARLY RSV MUTANTS 259
MATERIALS AND METHODS
Viruses and cells. The wild-type virus used in these studies was Prague strain Rous sar- coma virus of subgroup C (PR RSV-C or PR C). Stocks of PR C treated with 5-aza- cytidine (Toyoshima and Vogt, 1969a) were the source of mutants ts 335 (Wyke, in preparation) and ts 337. Permissive and nonpermissive temperatures were 35” and 41”, respectively. Infectivity of all virus is expressed in focus-forming units (FFU) per ml. Infectivity of virus-cell complexes formed in suspension is expressed as focus- forming centers (FFC). While free virus may contribute to focus formation by FFC, the low multiplicity of infection (m.o.i.) of 0.01 or less used in these experiments and rapid virus adsorption (Steck and Rubin, 1966) make such a possibility unlikely.
The source, cultivation, and charact,er- ization of chicken cells have been described previously (Vogt and Ishizaki, 1965; Duff and Vogt, 1969). Unless otherwise noted, all cells used (except in focus assays) were negat,ive in the complement fixation test for avian tumor virus group specific antigen(s) (gs-) (Vogt’ and Friis, 1971) and also nega- tive in the infective cent’er test assay for cell helper factor (chf-) as measured on pri- mary cultures (Hanafusa et al., 1970; Weiss et al. 1973, in press). Growth medium used was either Ml99 or Ham’s FlO, both sup- plemented with 10% tryptose phosphate broth and 5 % calf serum. All infections and assays were done in the presence of 2 &g/ml Polybrene (PB) to enhance adsorption (To- yoshima and Vogt, 1969a). Medium used to make virus stocks, or test viral replication, contained, in addition, 1% dimethyl sulf- oxide (DMSO) which was added one day after infection (Vogt et al., 1970).
V+M assay. Focus assays were performed as previously described (Rubin, 1960; Vogt, 1969). Cells were infected within 3 hr of seeding. If temperature shifts were per- formed up to 2 hr after infection, the focus assay was modified as follows: cells sus- pended in medium with PB were infected at a multiplicity of infection of less than 0.1 FFU/cell. Tubes were incubated in water bat’hs at 35’ or 41’ and shaken period- ically to prevent cell settling. Shifts were
performed by diluting aliquots into medium at the other temperature and continuing incubation, so that all samples remained in suspension for the same length of time. At the end of the incubation, sampIes were plated on 35-mm dishes containing con- fluent lawns of feeder cells. Plates were over- laid one day later, as in the usual focus assay.
Colonies were grown in agar by a modifi- cation of the met-hod of Friis et al. (1971), as described by Linial and Wyke (in prepa- rat,ion). In these experiments 5 X lo5 quail cells were suspended directly into 2 ml of bottom agar (0.6 %) which was then layered in a 60-mm Falcon bacteriological dish and allowed to harden. The virus being tested was used to infect a suspension of cells in growth medium at 35’ or 41’. Dilutions of the infected cells were added to 3 ml of top agar (0.36 %) and poured onto t.he bot,tom agar, allowed to harden, and incubated.
Antiserum. Neutralization tests were car- ried out using chicken anti-B77 (subgroup C sarcoma virus) serum obtained by I<. Toyoshima from birds with regressed tu- mors.
Treatment with lozu pH. The method em- ployed is that of Steck and Rubin (1966). Chick cells were seeded (5 X 105/35 mm plat,e) and ea. 3 hr later virus was adsorbed for 2.5 hr at 35’. After removal of the me- dium, cells were washed 3 times with 1.5 ml portions of growth medium, then twice in ca. 2 min with 1 ml portions of 0.05 M glycine. HCl buffer (pH 2.2). The cult,ures were then washed 3 times with 1.5ml aliquots of growth medium; 2 ml of growth medium was added and incubation was continued at 35” or 41”. Cultures were overlaid with nutrient agar at ea. 22 hr post-infection.
Preparation of virus for polymerase assay. Supernatants were harvested at daily inter- vals from confluently infected fibroblast cultures at 35” and stored at -74”. Ts 335 virus stocks were prepared from C/B cells which were gs- but had not been tested for chf. Virus was purified as described by Katz and Vogt (1971) with the following modifica- t.ions. Equilibrium sedimentation was per- formed by layering virus on a linear 30-55 % (w/v) sucrose gradient in st’andard buffer

260 LINIAL AND MASON
(0.1 M NaCl, 0.01 M Tris, 0.001 M EDTA, pH 7.4). Gradient fractions containing virus (as determined from parallel centrifugation of 14C-uridine labeled virus) were pooled, diluted in standard buffer, and resedimented (90 min at 40,000 rpm in a SW 41 rotor). Pellets were resuspended by sonication. With the exception of sonication, all operations were carried out at 4”.
Polymerase assay. Protein was assayed using the procedure of Lowry et al. (1951). Bovine plasma albumin (BSA) was used as a calibration standard. Protein concentra- tions are thus expressed as milligram equiva- lents of BSA.
Endogenous polymerase activity of virus particles was determined using the assay system of Green et al. (1970). Reactants were mixed at 4” and immediately placed at 35” or 41”. Final concentrations of reactants are indicated with each experiment.
Polymerase activity directed by an ex- ogenous template was measured following the addition of 10 pg/ml of poly (rA) : d(pT)lo (abbreviated rA:dT) to the standard assay.
Incorporation of radioactivity into macro- molecules is report’ed as counts per minute incorporat’ed into t’richloroacetic acid (TCA)-precipitable materials. Samples were spotted onto Whatman 3RIM filters (2.3 cm diameter) and immediately placed in 10% (w/v) TCA at 4°C for at least 20 min. The filters were subsequently washed three times in 5 % (w/v) TCA (4” C, at least 20 min per wash), twice in ethanol : ether (1: 1)) and once in ether. The filters were allowed to air. dry, placed in scintillation fluid (42 ml of New England Nuclear Liquifluor per liter of toluene) and counted in a Packard Tri-Carb Liquid Scintillation Spect’ropho- t’ometer.
Materials. Thymidine-methyl-H3 5’-tri- phosphate, tetrasodium salt (18 Ci/mmole, 3H-TTP) was obtained from New England Nuclear (Boston, Masschusetts) ; Xaz dATP, dGTP, and dCTP from Sigma Chem- ical Company (St. Louis, ;1Iissouri); Nonidet P-40 (NP-40) from Shell Chemical Company (New York); poly (rA) : d(pT),,, double strand, sodium salt from P-L Biochemicals (Milwaukee, Wisconsin) ; bovine plasma al- bumin (crystalline) from Armour Pharma-
ceutical Company (Kankakee, Illinois) ; tri- chloroacet’ic acid (U.S.P.) from Mallinckrodt (St. Louis, Jlissouri). All other chemicals were of reagent grade. Virus radioactively labeled with 14C-uridine was generously sup- plied by Dr. Robert Friis.
RESULTS
Focus Formation and Viral Replication at
The first experiments were performed to determine whether ts 335 and ts 337 were transformation or coordinate mutants and to determine the genetic stability of the mutants. Individual clones of mutants 2s 33.5 and ts 337 were obtained by picking foci formed at 35”, adding them to susceptible feeder cells, and passaging cultures several times until the entire monolayer was trans- formed. Virus from clones was tested for focus-forming ability at the permissive and nonpermissive temperatures (Table 1). Ts 335 plates with an efficiency of 6 to 1s X 10U4 at 41’. This efficiency is much higher t’han those reported for transformation mu- tants of R.ous sarcoma virus (Martin, 1970; Biquard and Vigier, 1972; Kawai and Hana- fusa, 1971; Wyke, in preparation), but it is comparable t,o the efliciencies for t,he coordi- nate mutant’s ts 334(75) and ts 336(149) (Friis et al., 1971). Ts 337 is much “leakier.” The best clones still plate at the high effi- ciency of 1 X lop3 at 41’. Repeated clonings do not decrease the ability to plate at 41”. However, as reported by Friis et al. (1971) for ts 334(75) and ts 336(149), foci which are formed at 41” cont’ain virus of the trm- peraturc sensitive genotype and not true revertants (Table 2). This is also true for the small percent of colonies formed in agar by the mutants at 41”. The true reversion frequencies of ts 335 and ts 337 have not’ been determined.
Mutants ts 335 and ts 337 are of the coor- dinate type (Table 3). Virus replication in cultures infected and maintained at 41” is markedly reduced. At 4 days after infection, PR C tit’ers are about 3 times higher at 41” than at 35”; at 3 days, the ratio for a given multiplicity of infection is even higher (data not shown). Yields of ts 335 are depressed about 40-fold at 41”; at higher multiplicities

EARLY RSV MUTANTS 261
TABLE 1
FOCUS-FORMING EFFICIENCIES OF DIFFERENT
TABLE 3
VIRAL ItEPLIC.4TION .4T 41’ AND 35’ CLONES OF MUTANTS
ncuba ,- ti0n
Temp (“Cl
41° 35” 41° 35” 41° 35” 41” 35O 41° 35*
41”/35” :noculum :FFU/pl) Titer (FFU/ml)
Virus I
I( Virus -
_
-
41”
1 x 103 7 x 102
2.8 x 103 9 x 102 4 x 103 1 x 103 3 x 103 6 X 1W
8.4 x 105 8.0 x 105
35”
1.8 x 106 1.2 x 106 1.6 X IO6 1.2 x 106
1 x 106 1 x 106 1 x 106 1 x 105
1.0 x 106 7.0 x 105
41”/35”
5.5 x 10-4 IS 335
5.8 x 10-4 1.8 x 10-a
Is 335
8 X 1OW 4 x IO-3
Is 337
1 X 10-a 3 x lo-3
PR C
0 x 10-a 8.4 x 10-r
PR C
11.4 x 10-r
102
104
5 x 105
5 x 105
5 x 10’
5 x 102 a 2 x 104 a 2 x 104a 3 x 1osa 6 x lo3 b 6 X 10’ *
4.5 x 106b 1.5 x 105 IJ
7 x 104 Q 3 x 104 Q
1s 335
ts 337
PR C
2.5 X 1OP
6.7 x lcF‘r2
1 x 10-i
3 x 100
2.3 X 10”
n Assayed at 3 days post infection (p.i.) b Assayed at 4 days p.i.
TABLE 2
PHENOTYPE OF VIRUS TRANSFORMING AT 41” TABLE 4
41” Foci 41” Agar EFFECT OF TEMPERATURE SHIFT ON Focus colon&
ts 337 20/20 is” 5/5 1s 1s 335 20/20 1s 5/5 1s Virus
FORMATION'"
Time of shift to 41” (hr)
a Foci or agar colonies from several independ- ent clones of ts 337 or ts 335 were picked from plates at 41” and tested for focus formation at
1s 335
35” and 41’. Is 337 PR C
3 9 10 74 2 14
120 112 128
Time of shift to 35” (hr)
0
there is more “leakiness,” and higher titers are obtained at the nonpermissive tempera- ture. However, the virus obtained from 41’ cultures has the same low plating efficiency at 41” as the virus from 35” cultures. With ts 335 ts 337, as with ts 335, the efficiency of virus ts 337 yield at 41” is depressed as the multiplicity PR C
I of infection is lowered (AI. Linial, unpub- y
2
1OW 48 100” 12 100 104
4 8 24 50
26 6 1.2 0.3 9 4 <l
80 84 88 /
lished data).
E$ect of Temperature Shift on Focus Forma- tion
In order to determine whether the two mutants were deficient in initiation and/or maintenance of t,ransformation at 41°, the effect of reciprocal temperature shifts on focus formation was studied (Table 4). If plates infected with ts 335 were exposed to 35” for as little as 6 hr, 10% of the control foci appeared on subsequent incubation at, 41”. Two hours at 35” was enough to in-
u expressed as percent surviving FFU of the control initiated and maintained at 35’.
h Infected and maintained at 41”. Foci were counted at 6 days after infect,ion.
c Infected and maintained at 35”. Foci were counted at 8 days after infect,ion.
crease the number of foci at least 30-fold over the 41” control. Ts 337 appears to require a longer time at 3.5” to yield sub- stantial transformation at 41”. However, a 20-fold increase in focus formation was ob- served aft,er 8 hr at 35”. Therefore both ts 335 and ts-337 require the permissive tem-
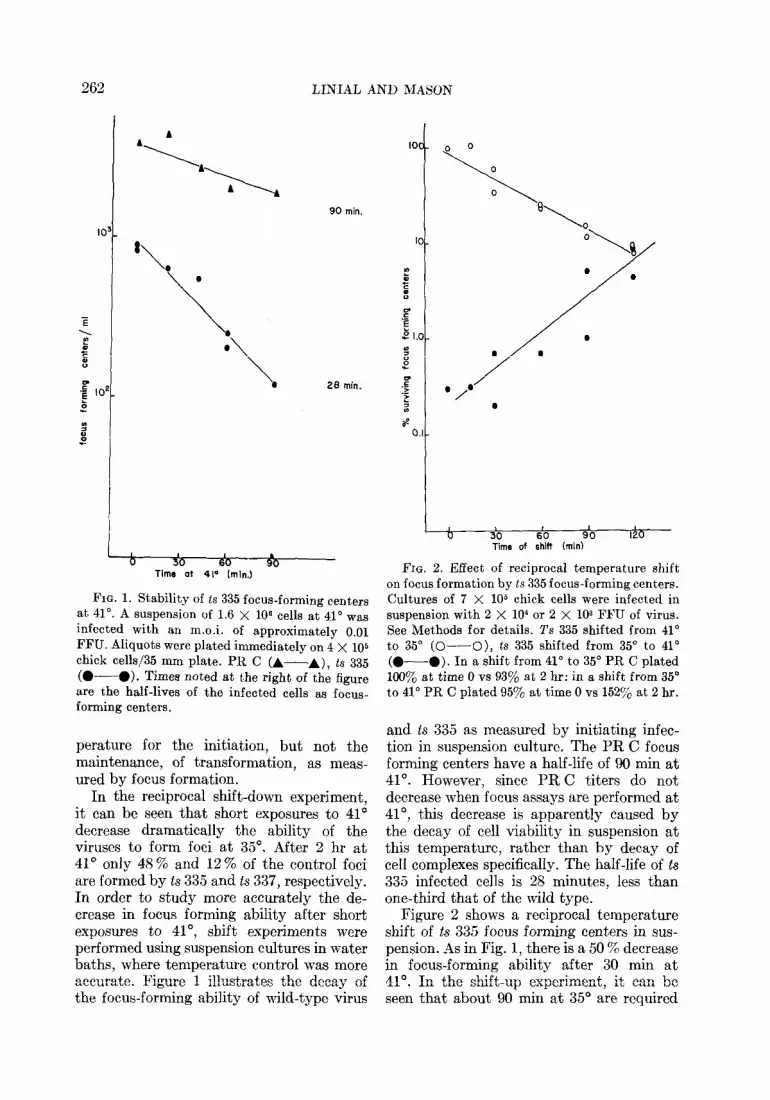
262 LINIAL AND MASON
a \ a
l
\ 0
0
\ l
90 min.
26 min.
IO
t
L
, I I 0
s 30 60 90
Time at 4P [min.)
FIG. 1. Stability of ts 335 focus-forming centers at 41”. A suspension of 1.6 X 106 cells at 41” was infected with an m.o.i. of approximately 0.01 FFU. Aliquots were plated immediately on 4 X 105 chick cells/35 mm plate. PR C (A-A), ts 335 (O---O). Times noted at the right of the figure are the half-lives of the infected cells as focus- forming centers.
perature for the initiation, but not the maintenance, of transformation, as meas- ured by focus formation.
In the reciprocal shift-down experiment, it can be seen that short exposures to 41” decrease dramatically the ability of the viruses to form foci at 35”. After 2 hr at 41’ only 48 % and 12 % of the control foci are formed by ts 335 and ts 337, respectively. In order to study more accurately the de- crease in focus forming ability after short exposures to 41”, shift experiments were performed using suspension cultures in water baths, where temperature control was more accurate. Figure 1 illustrates the decay of the focus-forming ability of wild-type virus
, I I I
0 30 60 90 120 Time of shift (mid
FIG. 2. Effect of reciprocal temperature shift on focus formation by ts 335 focus-forming centers. Cultures of 7 X lo5 chick cells were infected in suspension with 2 X lo4 or 2 X 103 FFU of virus. See Methods for details. Ts 335 shifted from 41’ to 35” (O-O), ts 335 shifted from 35’ to 41’ (o-0). In a shift from 41” to 35’ PR C plated 100% at time 0 vs 93% at 2 hr: in a shift from 35” to 41” PR C plated 95v0 at time 0 vs 152% at 2 hr.
and ts 335 as measured by initiating infec- tion in suspension culture. The PR C focus forming centers have a half-life of 90 min at 41“. However, since PR C titers do not decrease when focus assays are performed at 41°, this decrease is apparently caused by the decay of cell viability in suspension at this temperature, rather than by decay of cell compIexes specifically. The half-life of ts 335 infected cells is 28 minutes, less than one-third that of the wild type.
Figure 2 shows a reciprocal temperature shift of ts 335 focus forming centers in sus- pension. ils in Fig. 1, there is a 50 % decrease in focus-forming ability after 30 min at 41”. In the shift-up experiment, it can be seen that about 90 min at 35’ are required

EARLY RSV MUTANTS 263
TABLE 5
EFFECT OF TEMPERATURE SHIFT ON VIRAL ~<KPLIC!hTI@N"
Virus
PR C wild
type ts 335
Time of
shift (hr)
T Titer (FFU/ml) after shiftb
41” to 3.5” 35” to 41” 3.5” to 41”/ 35”
con. trol
2.3 X lo4 = 1.4 X 103d 0.06 8.5 x lo” 2.4 X lo* 1.0 5.5 x 103 1.2 x 10” 0.5 6.3 x 10” 2.0 x 104 0.9 4.5 x 103 5.2 X lo4 2.2
14.5
- 0 35 mm dishes seeded with 5 X 105 chick cells
were infected with 5 X 10’ FFU of PR C or ts 335 and incubated at 35” or 41’.
b Assayed at 53 hr after infection. c Initiated and maintained at 35’ (35’ control). d Initiated and maintained at 41”.
for a lo-fold increase in focus formation after shift to 41’. Both Fig. 2 and Table 4 show that there is an exponential increase in focus yield with increasing incubation at 35”, rather than a short period of time at 35” at which the mutant function is ex- pressed. It would therefore appear that the mutant funct’ion involved is first expressed very early after infection (prior to 2 hr), but there is a continual increase in expression up to 1S hr.
E$ect of Temperature Shift on Viral Repli- cation
Table 5 shows the effect of reciprocal temperature shift on virus production by ts 335. At 2 days after infection, the titer of PR C wild type was 14.5fold higher at 41’ than at 35”. The titer of ts 335 main- tained throughout at 41” was only 6% that of the 35” control. A IO-fold increase in ts 335 viral titer occurred after only 2 hr at 35’ followed by incubation at 41’. In the shift-down experiment, there was no great decrease in viral titer up to 22 hr at 41”, but this is possibly due to the large viral inoculum (Table 3). The shift-up data indi-
cate that the t’ime course of expression of the mutant function is similar for both viral replication and focus formation.
Growth of Mutants in Agar Suspension
It would appear that there is no good cor- relation between the ability of ts mutants to grow in agar and their ability to form foci at the nonpermissive temperature. The ma- jority of mutants defective in focus forma- tion are unable to grow in agar at 41”. How- ever, ts 334(75) is clearly able to form agar colonies at 41” although it is unable to form foci at that temperature (Friis et al., 1971). Moreover ts 336(149) and ts 338 can also form agar colonies under conditions in which they are no longer able to form foci (Linial and Wylie, in preparation). It was therefore of interest to see the effect of an early co- ordinate mutation on growth in agar (Fig. 3). In a shift-up experiment (Fig. 3a), at least 6 hr at 35’ are required for a lo-fold increase in agar colony formation over the 41“ control. A much shorter time at 35” was required for a comparable increase in focus formation (Fig. 2). This may merely reflect the difference in the physiological state of the cells suspended in agar, rather than a true difference in viral function. In a shift- down experiment (Fig. 3b), the half-life of the ability to form agar colonies is less than 1 hr, and no significant numbers of colonies are detected after about 8 hr. Thus, although agar colony formation and focus formation may involve different viral func- tions, the mutant function in 2s 335 and ts 337 is required for both these parameters of transformation. In addition, this experiment strongly suggests that the lesion in ts 335 and ts 337 is expressed after the formation of virus-cell complexes, since dilution and plating into agar might, be expected to effectively eliminate further infections.
Production of gs Antigen at the Nonpermissive Temperature
The COFAL best’ (Sarma et al., 1964) was used to determine whether avian tumor virus group specific gs antigen could be detected in t’he mutant infected cells a,t 41’. Cells were seeded at 1 X 106/60 mm plate and infected with an m.o.i. of 3. Three days later

264 LINIAL AND MASON
FIG. 3. Effect of reciprocal temperature shifts on colonies growing in agar. Cell suspensions at a den- sityof 2 x 106cells/ml were infected with an m.o.i. of 0.02 to 0.1, at 35” and 41”. After 1 hr, aliquots were diluted and plated (see Methods). Agar suspension cultures were shifted as indicated, 0 hr being the time of initial infection. Wild tvDe PR C (O-a), ts 335 (X-X), ts 337 (O-O). (a) Shifted from 35” to 41”. (b) Shifted from 4”l” to 35”. .-
infected cells were washed with phosphat’e- buffered saline, harvested, and assayed for gs antigen as described by Vogt and Friis (1971). The wild-type, PR C control had a gs titer 4-fold higher at 41’ than at 35’. There was no detectable gs antigen in either ts 335 or ts 337 infected cells at 41”. Control cultures of ts 335 or ts 337 at 35” exhibited amounts of gs antigen comparable to PR C at 35”. When cultures of ts 335 or ts 337 were shifted from 35” to 41’ one day after infec- tion, the gs antigen titer was increased 2- to 4-fold, as in the case with PR C.
Adsorption and Penetration of ts 335 and ts SW at the Permissive Temperature: E$ect on the Shift to the Nonpermissive Temperature
Because of the extremely early manifest’a- tion of the mutant lesions in ts 335 and ts 337, it was of interest to do a direct deter- mination of whether the mutants were a,ble to adsorb to and penetrate into cells at the nonpermissive temperature. Steck and Rubin (1966) h,ave defined two measurable sequen- tial steps of virus entry in avian tumor virus infection. Soon after virus adsorbs to SUS- ceptible cells, the virus becomes insensitive
to antibody inactivation. This may either reflect penetration beyond the cell surface, or binding to specific receptors on the cell sur- face. Virus at this time is still susceptible to inactivation by brief exposure to low pH. Later, virus becomes insensitive to low pH treatment as well. This possibly represents actual penetration of virus into the cells.
If the temperature sensitive lesions(s) in ts 335 and ts 337 are expressed before penetra- tion, then viruses that have penetrated into the host cells will have passed the step at which focus formation is sensitive to a temperature shift from 35” to 41’. However, if temperature sensitivity of focus forma- tion is expressed after penetration, then focus formation by viruses which have pene- trated at 35” will be transiently sensitive to a 35” to 41’ shift. In the experiment presented in Table 6, chick fibroblasts were infected for 2.Ti hr at 35”, washed with pH 2.2 buffer, and then either shifted to 41” or maintained at 35’. The results indicate t’hat viruses which have. penetrated beyond t’he step defined by sensitivity to pH 2.2 buffer are st,ill sensitive (for focus formation) to a temperature shift from 35” to 41”.
The results presented in this section sug-

EARLY RSV MUTANTS 26.5
TABLE 6 of virus grown in fibroblasts derived from EFFECT OF pH 2.2 ON Focus FORMATION” separate embryos were tested to eliminate
the possibility of an aberrant host com- Virus Treatment ponent. Both ts 335 and ts 337 consistently
had half-lives at 41’ of from one-half to Untreated Growth media pH 2.2 Wash
wash one-fourth t,hat of the wild-type virus.
I 1 I i 35” 35” to 35”* 1 35” to41”c
41” ~___- _~ PR C 1.0 0.5 0.3 0.1 0.05 0.015 ts 335 1.0 0.002 0.3 ca. 0.0007 0.06 0.0001 ts 337 1.0 0.001 0.3 ca. 0.0007 0.12 0.0001 -
0 Fibroblasts were infected with PR C, ts 335, or ts 337 at 35” and washed with pH 2.2 gly- cine.HCl buffer at. 2.5 hr post-infection as de- scribed in Methods. Incubation was then con- tinued at 35’ or 41’. One set of control plates was washed 3 times with growth media at 2.5 hours post.-infection. A second set of control plates was left untreated. Results are expressed as the ratio of foci obtained after the acid wash or growth media wash to foci obtained on untreated plates incubated only at, 35’. The acid treatment had no effect on focus formation at 35’ when applied to cells at 20 hr post-infection. Dilution of virus 20.fold into glycine-HCl for 2 min reduced in- fectivity ca. 30. to lOO-fold. All manipulat.ions were performed at room temperature.
Q Fibroblast,s were infected for 2.5 hr at 35”, washed, and focus development was recorded aft,er incubation at 35’.
c Fibroblasts were infected for 2.5 hr at 35”, washed, and focus development was recorded after incubation at 41”.
gest, that the temperature sensitivity of in- fection exhibited bv ts 335 and ts 337 is not due t.o a defect in either adsorpt,ion or penetration. Moreover, the data in the pre- ceding section suggested that the defect was expressed post-adsorption, and not at the level of free virus. Experiments are presented in subsequent sections which sug- gest a ts lesion in the reverse-transcriptase function of ts 335 and ts 337.
Temperature Sensitivity of Virus Infectivity
Heat inactivation kinetics of wild-type and mutant viruses were performed to de- termine whether ts 335 and ts 337 contain temperature sensitive structural components of t.he virion (Fig. 4). Many different clones
Antigen& Di$erence of ts 337
An anti-B77 antiserum which was found to be very effective in neutralizing wild-type PR C was tested for its neutralizing prop- erties with the two mutants (Table 7). As a control, it was found that, over a period of 15 min, t’here was no difference in the heat stabilit,ies of the three viruses at 3Fj” (84, 84, and SO% of the focus-forming activity re- mained after 15 min with PR C, ts 335, and ts 337, respectively). Ts 335 was neutralized to the same extent as PR C. However, ts 337 was consistently neutralized between three and four times as rapidly as PR C. As with the temperature sensitivity of the virus infect’ivit’y, these results were verified with many different clones of virus grown in fibroblasts derived from separate embryos. The results may reflect structural differrnce bct.ween the two viruses.
Vision DNA Polymerase Activity
The preceding experiment’s demonstrate that, ts 335 and ts 337 are unable to initiate infect’ion at 41’. Initiation of infection may require the proper functioning of the reverse transcriptase system of the virion to tran- scribe viral RNA into DIVA. In order to determine whether the mutants possess a defective virion-associated DNA polymerase act.ivity, this a&ivity has been measured at 35” and 41’. The results presented in Fig. 5 show that the mild-type PR C produces similar incorporation of 3H-TTP when as- sayed either at 35” or 41’ (in the presence of 0.01% (V/V) NP-40 and without added template). In comparison, all four ts 335 clones tested showed negligible incorpora- tion at 41”. However, a decrease of incorpora- tion of only 2-3-fold would appear as neg- ligible incorporation because of the low incorporation at 35”. in a similar experiment with ts 337, no significant incorporation of 3H-TTP has so far been detected at either temperature, though possibly this activity

266 LINIAL AND MASON
b
FIG. 4. Heat inactivation of wild-type PR C, Is 335, and ts 337. Virus stocks were diluted into pre- warmed growth medium (containing 5oj0 calf serum) and incubated in tubes at 41’. Some change in pH occurred during the course of the experiment. (a) Two independent clones of PR C and ts 337 grown in different chick embryo cultures were tested. PR C (.&--A, A-A), ts 337 (O--O, O---O). (b) PR C (A---A), ts 335 (0-O). Numbers at the right of the figure refer t,o the half-lives of the virus at 41”.
will become apparent when larger amounts of ts 337 virion protein are utilized. PR C and ts 335 have also been assayed at 35” and 41’ in the presence of 0.1% (v/v) NP-40, rather than 0.01% (v/v) NP-40, with results qualitatively similar to those in Fig. 5.
These results could reflect either a de- ficiency in the polymerase system, or in the endogenous template. Therefore, a polym- erase activity was measured at 35’ and 41’ in the presence of rA:dT added as templat’e (Fig. 6). PR C again exhibits similar activity at 35” and 41”. Ts 335 and ts 337 both in- corporate 3H-TTP at 41”, but at a 2- to ‘S-fold lower rate than at 35”. When the concentra- tion of NP-40 was increased from 0.01% (v/v) to 0.1% (v/v), the rate of rA:dT stimulated incorporation of 3H-TTP by PR C increased approximately lo-fold. The two mutants were therefore compared with PR C in the presence of 0.1% (v/3) XP-40. PR C exhibited similar activity at 35” and 41*. However, although the two mutants had approximately the same specific ac- tivity as the wild type at 35”, they incor- porated 3H-TTP at a 2- to 4-fold lower rate
TABLE 7 ANTISERUM INKTIVITION OF ts 3370
Virus Clan@ FFU/ml at % FFU remain-
0 Min 15 Min ing at 15 min
_____ PRC 1 1.7 x 10’ 1.0 x 103 6
2 4.7 x 104 4.7 x 103 10 1s 337 1 1.1 x 104 2.0 x 102 1.8
2 4.4 x 103 1.0 x 102 2.3 ts 335 5.5 x 103 5.0 x 102 9
Q Anti-B77 used at 1:lO dilution in FlO me- dium. Incubation was at 35”.
b Independently cloned foci of PR C and ts 337 grown in different chick embryo cultures.
at 41”; that is, the relative rates of incor- poration of 3H-TTP exhibited by ts 335 and ts 337 at 35” and 41’ were approximately t’he same in 0.01% (v/v) or 0.1% (v/v) NP- 40. These results suggest a deficiency in the polymerase act,ivity of ts 335 and ts 337.
A lesion in the reverse transcriptase ac- tivity of ts 335 and ts 337 might, result in a DNA polymerase activity which is more

EARLY RSV MUTANTS 267
1000 PR-C f
750 /’
500
250 ‘J’1 p”
,/’
P’ 0’
300
5! 200
i5 100
FIG. 6. The rA:dT directed DNA polymerase activity of ts 335 and ts 337. Except as noted, assays were as described for Fig. 5. Virus was added to reaction mixtures to 0.05 mg/ml, rA:dT to 0.01 mg/ml. The concentration of EDTA added with the virus was reduced by diluting the virus at least 25-fold with 1.25X standard buffer lacking EDTA, pelleting the virus by high speed cen- trifugation, and resuspending in the same buffer. Samples of 0.01 were assayed for TCA-precipitable cpm. Results have been expressed as cpm/O.Ol ml, after subtracting the 0 min cpm (approxi- mately 200-300 cpm/O.Ol ml). 35” (0-O); 41” (O- - - -0).
shown in Fig. 7. The inactivations were per- formed in the presence of 0.77% (v/v) NP-40. The DNA polymerase activity of ts 335 and ts 337 shows a similar time course of inactivation at 41”, and the PR C associated activity is clearly less heat sensi- tive than that of either mutant.
The heat sensitivity of the polymerase
FIG. 5. Endogenous DNA Polymerase activity of ts 335 at 35” and 41’. Complete reaction systems at 4” were divided, and equal volumes of 0.2 ml were incubated at 35” or 41”. Samples (0.005 ml) were removed at intervals and assayed for TCA precipitable radioactivity. Final concentrations of reactants were: 0.045 M Tris.HCl, pH 8.1; 2.5 mM MgClz ; 50 mM NaCl; 5 mM dithioerythrit,ol (DTT); 0.1 mM dATP; 0.1 mM dGTP; 0.1 mM dCTP; 100 rCi/ml 3H-TTP; 0.01% (v/v) NP-40; 0.1 mg/ml viral protein; 0.5 mM EDTA (added with virus). The results have been expressed as cpm/O.Ol ml, after subtraction of cpm at 0 min (approximately 250/0.01 ml). Data have been presented for wild-type PR C as well as for four separate clones of ts 335. 35” (@---a); 41” co- - - -0).
rapidly inactivated than the wild type by exposure to high temperatures. The inac- tivation of the rA:dT stimulated DNA polymerase activity by incubation of the wild-type and mutant virions at 41’ is

268 LINIAL AND MASON
4 PR-C.
I.1 1 1 1 1 0 30 60 120 160
Time at 41° C (mid FIG. 7. Inactivation of 11NA polymerase activity by incubation at 41”: PR C; IS 335; ts 337. Inacti-
vations were carried out at 41” in thefollowing: 0.067 M Tris.HCl, pH 8.1; 3.8 mM MgCh; 0.058 M NaCl; 0.0077 M 1)TT; 0.154 mM dATP; 0.154 mM dCTP; 0.154 mM dGTP; 0.77% (v/v) NP-40; virion protein at 0.077 mg/ml. At various times, 0.065.ml aliquots were removed; 3H-TTP, buffer components (Tris. HCl and NaCl) and rA:dT were added to a total volume of 0.1 ml; the mixture was immediately placed at 35” and the rate of incorporation of TCA-precipitable radioactivity measured. In addition, an inacti- vation mixture was incubated at 41” without virus; 0.065.ml aliquots were withdrawn at various times, untreated virus (either PR C, ts 335, or ts 337), 3H-TTP, rA:dT, and buffer components were added to a total volume of 0.1 ml, and the DNA polymerase activity measured at 35”, as above. The final concen- trations at 35” were as described in Fig. 6, with virion protein at 0.005 mg/ml, 0.5% (v/v) NP-40, and 0.01 mg/ml rA:dT. Polymerase activities at 35” were determined from the linear portion of incorpora- tion vs time plots. The surviving polymerase activity has been presented as the ratio of activity at any time (t) to the activity at time zero. (In the absence of rA:dT, the DNA polymerase activity of untreated virus was less than 1% of the rA:dT directed activity of the same virus.) With neither PR C, ts 335, nor ts 337 was a drop in incorporation rate greater than approximately 20% observed as a result of incu- bating the inactivation mixture (minus virus) at 41”. The specific activity of the PR C rA:dT stimu- lated DNA polymerase was approximately twice that of either mutant.
activity of ts 33Fi and ts 337 might be the result of a factor(s) affecting components of the assay other than the reverse-transcript- ase. Howcvcr, the rA:dT stimulated ac- tivit’y of ts 335 was unaffected by addition of an equal amount of heat inactivated virus (ts 33.5; Fig. 8). In addition, the factor(s) responsible for the greater heat sensitivity of the ts 335 associated activity were un- able to inactivate any excess amount of polymerase activity. When equal activities of PR C and ts 335 were mixed and incubated at 4I”, the resulting inactivation approached that of PR C by itself (Fig. 8). These results
suggest that ts 33ri, and possibly ts 337, possess lesions specific to the reverse tran- scriptase system.
Interactions of Mutants and Avian Leukosis Virus
Both avian sarcoma viruses (Temin and Mizutani, 1970) and avian leukosis viruses (Spiegelman et al., 1970) have been shown to possess a RNA-dependent DNA polymerase activity. Since ts 335 and ts 337 appear to have a temperature-sensitive mutation affect- ing this activity, experiments were performed to ascertain whether Rous-associated virus

EARLY RSV MUTANTS 269
0 50 60 120 180 0 LO 60 120 I80 rime 01 4,oc hi”1 Time 0, 41’C (ml1
FIG. 8. Absence of an inhibitor in the inactivation of DNA polymerase activity at 41”: PR C and ts 335 (virus preparations described in Fig. 7). Inactivation reactions were performed as described in Fig. 7 and contained the following amounts of virus: reaction (1) 0.038 mg/ml of PR C; (2) 0.077 mg/ml of ts 335; (3) 0.077 mg/ml of PR C; (4) 0.038 mg/ml of PR C and 0.077 mg/ml of ts 335; (5) (5) none. Renc- tion (5) was tested at 35” by addition of untreated PR C or ts 335. (With neither virus was a drop in incorporation greater than approximately 25% observed as a result of incubating the inactivation mix- ture (minus virus) at 41”.) In addition, reaction (1) was tested after various times at 41” by addition of untreated PR C (0.0025 mg) to the 35” assays and reaction (2) by t,he addition of ts 335 (0.005 mg). (a) Inactivabion of PR C polymerase activity at 41”: (0) reaction (1); (0) reaction (3); (A) reaction (1) wit,h untreated PR C added to the 35” assays. (b) Inactivation of ts 335 polymerase activity at 41”: (0) reaction (2); (0) reaction (4); (A) reaction (2) with untreated ts 335 added to the 35” assays.
(RAV) could supply the biological function temperature sensitive in the mutants.
Cowqlenmatation
Direct complementation tests were per- formed in two different ways using RAV-2 (subgroup B) and RAV-50 (subgroup D). (1) Fibroblasts were infected in suspension with the mutants at an m.o.i. of 5 and with RAV at an m.o.i. of 50 at 35” or 41”. After 1 hr for adsorption the infected cells were diluted and plated on feeder layers at the same temperature. Focus formation was re- corded after further incubation. Simulta- neous infection with RAV did not enhance focus formation by the mutants at 41’. 2. Fibroblasts were infected with RAV-2 or RAV-50 and passaged several times to yield a high level of infection. These RAV in- fected cells were then used for a focus assay of the mutants. Pre-infection with RAV did not enhance focus formation by the mutants at 41’.
Genetic Reassortment or Recombination
It has been shown by Vogt (1971) that if cells are mixcdly infected with avian sarcoma viruses and avian leukosis viruses of dif-
fering subgroup, genetically stable trans- forming viruses are produced with the sub- group specificity of the parental leukosis virus. Similar reassortment has been demon- strated between the subgroup specificity marker of a leukosis virus and the temprra- ture-sensitive transformation marker of cer- tain mutants (Kawai and Hanafusa, 1972; Wyke and Linial, unpublished results). Tcm- perature sensitive virus of the leukosis sub- group is recovered, but no wild-type sarcoma virus is detected. When such experiments were performed with ts 335 and ts 337, and RAV-2 and RAV-50 (details to be published elsewhere), a large proportion of the progeny virus was no longer temperature sensitive and retained the C subgroup marker. Such virus was genetically stable for several pas- sages. Rarely, sarcoma virus was found with the RAV B or D host range. These were also of wild-type transformation phenotype.
Since recombination with RAV yielded wild-type sarcoma viruses, RAV must possess the gene function(s) which is defective in ts 335 and ts 337. The absence of complementa- tion in the experiments described above was therefore surprising. It will bc interesting to see whcthcr conditions can be defined for

270 LINIAL AND MASON
which complementation can occur between RAV and the mutants.
Ts 335 and ts 337 are early coordinate mutants of l’rague strain Rous sarcoma virus. Although they were isolated inde- pendently (ts 335 was selected, ts 337 was picked at random), their biological behavior is indistinguishable. Both mutants require a short t.ime at 35”, immediately after infec- tion, to initiate both viral replication and cell transformation. Once initiation has oc- curred, replication and transformation can be maintained after a shift to the nonpermis- sive t.emperaturo (41”). In this respect they are, thus far, unique avian tumor virus mutants. The only other early coordinate mutant which has been reported, ts 336(149) (Friis et al., 1971), cannot maintain some parameters of transformation at 41’ (Linial and Wyke, manuscript in preparation).
Ts 335 and ts 337 are also unique in that they contain a structural component which is temperature sensitive, although the mu- tants are only 2- to 4-fold more sensitive t,o inactivation at 41” than the wild type. Cells infected with either mutant, and maintained at 41°, show a dramatic decrease in focus- forming ability. Moreover, a short incuba- tion at 35“ immediately following infection, followed by a shift to 41” leads to a large increase in focus formation (lo- t’o 30-fold increase over controls incubated only at 41”). Since the virion is temperature sensi- tive, this observation could be explained if cell penetration were more eflicient at 35” and the virus was subsequently protected from heat inactivation at 41”. This does not appear to be the case, however, since penetra- tion at 35” (as defined by acid resistance, Table 6) does not protect the ts virus-cell complex from effects of a shift to 41”. The most likely explanation for the relationship between the temperature sensitivity of some structural component of the virion and the biological behavior of the virus is that the ts structural component’ is also necessary int,racellularly for the initiation of viral replication and ccl1 transformation.
In preliminary cxperimcnts, ts 335 appears to possess type specific antigenicity similar to t,he parental wild-type virus. TS 337,
however, does exhibit a difference from both wild-type and ts 33.5, in neutralization of focus formation by antiserum produced against subgroup C sarcoma virus. This anti- genie difference suggests that the mutants may have temperature-sensitive lesions in different gent functions. Alternatively, ts 337 may contain a secondary lesion in those genes specifying type specific antigenicity.
Genetic information contained in avian sarcoma and leukosis viruses reassorts with high frequency for transformation and host range markers (Vogt, 1971; Iiawai and Hanafusa, 1972). Some early coordinate mu- tants like ts 336(149) (Luual, unpublished results) reassort with leukosis virus to pro- duce temperature-sensitive sarcoma virus of the leukosis virus subgroup. The phcnom- enon of reassortment may be a result of the segmentation of the viral genome (Duesberg, 1968; Montagnier et al., 1969; Duesberg and Vogt, 1970; Martin and Duesberg, 1972). There is some physical evidence for more than one type of RlSA subunit (Duesberg and Vogt, 1970). Ts 335 and ts 337 rcassort with a high frequency (ea. 50 70) with leukosis virus to product wild-type sarcoma virus (Wyke and Linial, manuscript in prepara- tion). The wild-type virus is predominantly of the C (parental sarcoma virus) subgroup. With ts 337, in addition, wild-type and t.em- perature-sensitive focus-forming virus of the leukosis virus subgroup can be isolated at low frequency. These experiments suggest that t,he lcukosis virus contains genetic information for the function or functions dcficicnt in ts 335 and ts 337. Since direct complcmentation tests have failed to yield any progeny virus which can form foci at 41°, it would appear that the function cannot be supplied by infection with another virus in a single infective cycle, but must be present in the virions at the time of infec- tion. The data suggest that leukosis viruses do contain some gene& information neces- sary for the initiation of transformation. It also suggests that the lesion in ts 337 is not closely linked with either host range or the bulk of the functions required for focus formation.
Preliminary results presented here suggest that the virion associated DNA polymerase activity of ts 335 and ts 337 is temperature

EARLY RSV MUTANTS 271
sensitive. This temperature sensitivity is apparent in both the endogenous reaction and the reaction stimulated by the addition of poly rA:d(pT)lo as template. It is also apparent in the instability of the rA:d(pT)lo st,imulated activit.y to incubation at 41”.
By utilizing a number of purification pro- cedures, a DXA polymerase activity which appears to exist as a single molecular entity has been isolated from avian myeloblastosis virus (Kacian et al., 1971; Hurwitz and Leis, 1972). A similar enzyme has been isolated from the Schmidt-Ruppin strain of ROUS sarcoma virus (Faras et al., 1972). A tem- perature-sensitive lesion in this enzyme could explain our results wit,h ts 335 and ts 337. However, many other enzyme activit,ies including ribonucleases and endo- and exo- dcoxyribonucleases have been found asso- ciated with avian tumor virus preparations (Mizutani ef al., 1970; Quintrell et al., 1971; Rlizutani and Temin, 1971; Rosenbergova and PristaSov&, 1972). In addition, Molling et al. (1971) have found an activity closely associated with the reverse transcriptase which degrades RNA from RlSA-DNA hy- brids. Leis and Hurwitz (1972) have ob- tained an activity from avian myeloblastosis virus which stimulates DNA synthesis from viral RNA by purified viral polymerase. These or other enzymes may be encoded in the genetic information of the virus. A lesion in any of these, or in controlling ele- ments affecting the enzymes, might affect the transcription of RNA or RNA-DNA hybrid molecules into DNA. Further, a mutation altering nonenzymatic components of the virion, such as t’he nucleic acids, might lead to the results obtained. This pos- sibility is suggested by the failure to find measurable endogenous DNA polymerase activity in ts 337 clones at either temperature.
Experiments are now in progress to purify the polymerase activity from ts 335 and ts 337 and wild-type PR C grown at the permis- sivc temperature. It is hoped that these ex- periments mill clarify the nature of the lesion in these mutants. Different clones of all three of these viruses, and wild-type revertants of the mutants, are being tested to eliminate any aberrant results caused by clonal variation in the parental PR C wild- type stocks.
There have been two reports of avian RNA tumor viruses which are deficient in the RNA-dependent DNA polymerase ac- tivity. Hanafusa and Hanafusa (1971) have shown that a noninfectious virus, RSV a(O), present in some stocks of Bryan st,rain Rous sarcoma virus is lacking all DNA polym- erase activity. In addition, Hanafusa et al. (1972) have found that. RSV a(O) does not appear to possess any protein cross-reacting with antibodies prepared against purified reverse-transcriptase. Robinson and Robin- son (1971) have examined another, pre- sumably independently isolated, clone of RSV ~(0) and have found that it is lacking only one of the two DNA polymerase ac- tivities associated wit’h RNA tumor viruses. This noninfectious virus appears to contain the activity that synthesizes double-stranded DKA (activity 2), but only very low amounts of t.he activity 1 which yields a single stranded DNA product. Because the rela- tionship between the two noninfectious viruses is unknown, it is difficult to explain the differences in their enzyme activit’ies. Since the RSV ~~(0)‘s are nonconditional variants, it is difficult to prove that their lesion is in a viral-coded enzyme. It is possible that they are merely deficient in their ability to package or utilize a host polymerase activity. The existence of tem- perature-sensit#ive (or other conditional mu- tants) in the reverse transcriptase activity would be much stronger evidence for its viral coded nature.
There is a good correlation between the data reported here, and that reported for the RSV a(0) stocks, in regard to the role of DNA polymerase act’ivity in cell infection. Apparently RSV a(O) transformed cells are indistinguishable from those transformed by infectious Bryan RSV. This indicates that the activity (or activities) missing in RSV a(0) is not required for maintenance of transformation. However, work with the defective viruses demands utilization of pseudotypes in order to initiate infection; therefore, the role of the missing activity in initiation of transformation cannot be di- rectly studied. With temperature sensitive mutants, this difficulty is overcome. Results presented here suggest that the lack of a DNA polymerase activity at the nonpermis-

272 LINIAL AND MASON
sive temperature prevents initiation of cell transformation and viral replication, but once initiation occurs, replication and trans- formation can be maintained.
ACKNOWLEDGMENTS
We are grateful to Drs. Robert Friis, Robin Weiss, and John Wyke for many helpful discus- sions. The latter also provided invaluable assist- ance in preparation of this manuscript. Ellis Rucker, Victor Wong, Gene Wade, Jack Zakowski, Phillip Harris, Christine McMillin, and Hsiao- Ching Pang provided helpful technical assistance. Finally, we are indebted to Dr. Peter K. Vogt for his continued support and encouragement of this work.
REFERENCES
BADER, J. P., and BROWN, N. R. (1971). Induc- tion of mutations in an RNA tumour virus by an analogue of a DNA precursor. Nature (Lon- don) New BioE. 23‘4,11-1‘2.
BALTIMORE, D. (1970). RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature (London) 227, 1026-1038.
BALTIMORE, D., and S~o~ea, D. (1971). Primer requirement and template speciftcity of the DNA polymerase of RNA tumor viruses. Proc. Nat. dead. Sci. U.S.A. 68, 1507-1511.
BIQUARD, J.-M., and VIGIER, P. (1972). Charac- teristics of a conditional mutant of Rous sar- coma virus defect,ive in ability to transform cells at high temperature. Virology 47, 444-455.
DUESBERG, P. (1968). Physical properties of Rous sarcoma virus RNA. Proc. Nat. Acacl. Sci. U.S.A. 60, 1511-1518.
DUESBERG, P. H., and VOGT, P. K. (1970). Dif- ferences between the ribonucleic acids of trans- forming and nontransforming avian tumor viruses. Proc. -vat. Acad. Sci. U.S.A. 67, 1673- 1680.
DUFF, R. G., and VOGT, P. K. (1969). Charac- teristics of two new avian tumor virus sub- groups. liirologv 40, 1022-1029.
FARAS, A. J., TAYLOR, J. M., MCDONNELL, J. P., LEVINSON, W. E., and BISHOP, J. M. (1972). Purification and characterization of the deoxy- ribonucleic acid polymerase associated with R.ons sarcoma virus. Biochemistry 11, 2334- 2342.
FRIIS, R. R., TOYOSHII~A, K., and VOGT, P. K. (1971). Conditional lethal mutants of avian sarcoma viruses. 1. Physiology of Is 75 and ts 149. Virology 43, 375-389.
GREEN, M., ROKUTINDI, M., FUJIN~G.~, K., RAY, R. K., ROKUT~ND.~, H., and GURGO, C. (1970). Mechanism of carcinogenesis by RNA
tumor viruses. 1. An RNA-dependent DNA polymerase in murine sarcoma viruses. Proc. Nat. Acad. Sci. U.S.A. 67, 385-393.
HANAFUSA, H., and HANAFUSA, T. (1971). Non- infectious RSV deficient in DNA polymerase. Virology 43, 313-316.
HANAFUSA, H., MIYAMOTO, T., and HANAFUSA, T. (1970). A cell-associated factor essential for for- mation of an infectious form of Rous sarcoma virus. Proc. Nat. Acad. Sci. U.S.A. 66, 314-321.
HANAFUSA, H., BALTIMORE D., SMOLER D., WATSON, K. F., YANIV, A., and SPIEGELMAN, S. (1972). Absence of polymerase proteins in virions of alpha-type Rous sarcoma virus. Science 177, 1188 1191.
HURWITZ, J., and LEIS, J. P. (1972) RNA- dependent DNA polymerase activity of RNA tumor viruses. 1. Directing influence of DNA in the reaction. J. Viral. 9, 116-129.
K.~cI~N, D. L., WATSON, K. F., BURNY, A., and SPIEGELMAN, S. (1971). Purification of the DNA polymerase of avian myeloblastosis virus. Bio- chim. Biophys. Acta 246, 365-383.
KATZ, E., and VOGT, P. K. (1971). Conditional lethal mutants of avian sarcoma viruses. II. Analysis of the t,emperature sensitive lesion in 1s 75. Virology 46, 745-753.
K.4w.41, S., and HANAFUSA, H. (1971). The effects of reciprocal changes in temperature on the transformed state of cells infected with a Rous sarcoma virus mutant. Virology 46, 470-479.
Kaw.u, S., and HANAFUSA, H. (1972). Genetic recombination with avian tumor virus. Virology 49, 37-44.
KAWAI, S., METROKA, C. E., and H.~NAFus.~, H. (1972). Complementation of functions required for cell transformation by double infection wit,h RSV mutants. Virology 49, 302-304.
Lms, J. P., and HURIVITZ, J. (1972). Isolation and characterization of a protein that stimulates DNA synthesis from avian myeloblastosis virus. Proc. Nat. Acad. Sci. U.S.A. 69, 2331-2335.
LOPTRY, 0. H., ROSEBROUGH, N. J., FARR, A. L., and R.IND.~LL, lt. J. (1951). Protein measure- ment wit,h the Folin phenol reagent. J. Biol.
Chem. 193,265-275. MARTIN, 6. S. (1970). Rous sarcoma virus: A
function required for the maintenance of the transformed state. Nature (London) 227, 1021- 1023.
MARTIN, G. S., and DUICSBXWG, P. H. (1972). The 01 subunit on the RNA of transforming avian tumor viruses: I. Occurrence in different strains. II. Spontaneous loss resulting in nontransform- ing variants. Virology 47, 494-497.
MIZUTANI, S., and TEMIN, H. M. (1971). Enzymes and nucleotides in virions of Rous sarcoma virus. J. Viral. 8, 409-416.

EARLY RSV MUTANTS 273
MIZUTANI, S., BOETTIGEH, D., and TEMIN, H. M. (1970). A DNA dependent DNA polymerase and a DNA endonuclease in virions of Rous sar- coma virus. Suture (London) 228,424-427.
MULLING, K., BOLOGNESI, D. P., B.LUEIZ, H., B&EN, W., PLASSMANN, H. W., and HAUSEN, P. (1971). Association of viral reverse tran- scriptase with an enzyme degrading the RNA moiety of RNA-DNA hybrids. Nature (Lo&on) Sew Biol. 234, 240-243.
MONTAGNIER, L., GOLDS, A., and VIGIER, P. (1969). A possible subunit structure of Rous sarcoma virus RNA. J. Gen. Viral. 4, 449-452.
QUINTRELL, N., FANSHIER, L., EVANS, B., LEVIN-
SON, W., and BISHOP, J. M. (1971). Deoxyribo- nucleic acid polymerase(s) of Rous sarcoma virus: effect of virion-associated endonuclease on the enzymatic product. J. Viral. 8, 17-27.
I~OBINSON, W. S., and ROBINSON, H. L. (1971). J)NA polymerase in defective Rous sarcoma virus. Virology 44, 456362.
ROSISNBERGDV~~, M., and PRIST.GOV~, S. (1972). Nuclease activity of large RNA viruses. Acta Viral. (Prague) 16, l-8.
RUBIN, H. (1960). A virus in chick embryos which induces resistance NIL vitro to infection with Rous sarcoma virus. Proc. Nat. Acad. Sri. C7.S.A. 46, 1105-1119.
S.IRM.I, P. S., TURNER, H. C., and HUEBNER,
1:. J. (1964). An avian leukosis group-specific complement fixation reaction. Application for the detection and assay on noncytopathogenic leukosis viruses. Virology 23, 313-321.
SPIEGISLMAN, S., BURNY, A., D.~s, M. R., KEYDAR,
J., SCHLOM, J., TRIVNICEK, M., and WATSON,
K. (1970): Characterization of the products of 11NA-directed DNA polymerases in oncogenic RNA viruses. Nature (Londor~) 227, 563-567.
STIXK, F. T., and RUBIN, H. (1966). The mech- anism of interference between an avian leukosis virus and Rous sarcoma virus. II. Early steps of infection by RSV of cells under conditions of interference. Virology 29,642~653.
TEMIN, H. M. (1971). The role of DNA provirus in carcinogenesis by RNA tumor viruses. In “The Biology of Oncogenic Viruses” (L. G. Silvestri, ed.), pp. 176-187. North-Holland Publ., Amsterdam.
TEMIN, H. M., and MIZUTANI, S. (1970). RNA- dependent DNA polymerase in virions of Rous sarcoma virus. Natzcre (Londo,l) 226, 1211-1213.
TOYOSHIMA, K., and VOGT, P. K. (1969a). En- hancement and inhibition of avian sarcoma viruses by polycations and polyanions. Virology 38, 414-426.
TOYOSHIMA, K., and VOGT, P. K. (196913). Tem- perature sensitive mutants of an avian sarcoma virus. Virology 39, 930-931.
VOGT, P. K. (1969). Focus assay of Rous sarcoma virus. 111 “Fundamental Techniques in Virol- ogy” (K. Habel and N. P. Salzman, eds.), pp. 198-211. Academic Press, New York.
VOGT, P. K. (1971). Genetically stable reassort- ment of markers during mixed infection wit,h avian tumor viruses. Virology 46, 947-952.
VOGT, P. K., and FRIIS, 1:. R. (1971). An avian leukosis virus related to RSV(0): properties and evidence for helper activity. Virology 43, 223-234.
VOGT, P. K., and ISHIZAKI, R. (1965). Reciprocal patt,erns of genetic resistance to avian tumor viruses in two lines of chickens. Virology 26, 664-672.
VOGT, P. K., TOYOSHIM.I, K., and YOSHII, S. (1970). Factors promoting avian tumor virus infections. 1,~ “Defectiveness, Rescue and Stimulation of Oncogenic Viruses” Znt. Symp. Tumor Viruses, 2nd, Royaumont, pp. 229-238.
VOGT, P. K., WYKE, J. A., WEISS, R. A., FRIIS, KATZ, E., and LINIAL, M. (1972). Avian RNA tumor viruses: mutant markers and genetic mixing. Symp. Fundam. Cancer Res. in press.
WEISS, R. A., MASON, W. S., and VOGT, P. K. (1973). Genetic recombination between endog- enous and exogenous avian RNA tumor viruses. Virology 53, 535-552.