centre de recherches nucleaires ... - osti.gov
TRANSCRIPT

r7,M
piirrr^/rj»."."?^?, ' i-j
THASUOUHG
CRN 95-40I N° d'ordre 1979
i ^ ^
THESE
présentée
pour obtenir le grade de
DOCTEUR DE L'UNIVERSITE LOUIS PASTEUR DESTRASBOURG
par
Sylvie Yona WAKSMAN
Les céramiques byzantines des fouilles de Pergame.1 Caracterisation des productions locales et importées| par analyse élémentaire par les méthodes PIXE et INAA
V et par pétrographie
CENTRE DE RECHERCHES NUCLEAIRES
STRASBOURG
IN2P3
CNRS
UNIVERSITE
LOUIS PASTEUR
2 / ii 1 4

Gestion INIS,Doc, enreg. leN° TRN :D e s t i n a t i o n : l , I + D,D

THESE
présentée
pour obtenir le grade de
DOCTEUR DE L'UNIVERSITE LOUIS PASTEUR DESTRASBOURG
par
Sylvie Yona WAKSMAN
Les céramiques byzantines des fouilles de Pergame.Caractérisation des productions locales et importées
par analyse élémentaire par les méthodes PIXE et INAAet par pétrographie
soutenue le 12 Janvier 1995, devant la commission d'examen
Mrs SENS Jean - Claude rapporteur interne
BARRANDON Jean - Noël rapporteur externe
PICON Maurice rapporteur externe
SCHVOERER Max examinateur
SPIESER Jean - Michel directeur de thèse
HEITZ Charles directeur de thèse
HONNOREZ José membre invité

Remerciements
Ce travail est le fruit d'une collaboration entre le Centre de Recherches Nucléaires de
Strasbourg (CRN) et l'Institut d'Art et Archéologie de Byzance, Université de Strasbourg.
Il a pu être réalisé grâce à une allocation du Ministère de la Recherche et de la
Technologie. Je suis très reconnaissante à Raymond Seltz, ancien directeur du CRN et actuel
correspondant du CNRS à Bonn, pour son accueil enthousiaste. Je remercie la direction
actuelle du CRN de m'avoir donné la possibilité de travailler dans de bonnes conditions et de
faire connaître mes travaux. Mr Monsonego, délégué régional du Ministère de la Recherche et
de la Technologie, a manifesté de l'intérêt pour ces recherches et je l'en remercie vivement.
Maurice Picon, Directeur du Laboratoire de Céramologie de Lyon, m'a fait le plaisir
d'accepter de faire partie de mon jury de thèse et d'en être l'un des rapporteurs. J'ai eu plusieurs
fois recours à ses conseils au cours de mon travail et le remercie vivement de l'attention
bienveillante qu'il m'a accordée. J'ai beaucoup appris de sa façon de travailler, notamment lors
de mes visites à son laboratoire.
Je suis très reconnaissante à Jean-Noel Barrandon, Directeur du Centre Ernest Babelon
d'Orléans, d'avoir bien voulu être rapporteur de cette thèse et de m'avoir permis par ses
remarques de rendre ce travail plus rigoureux.
C'est grâce à Max Schvoerer, Directeur du Centre de Recherches Interdisciplinaire
d'Archéologie Analytique de Bordeaux, que je me suis formée et ai pris goût à l'archéométrie.
Je lui en suis très reconnaissante et le remercie d'avoir examiné ce travail.
Je remercie Jean-Claude Sens, professeur à l'Université de Strasbourg et directeur du
Réacteur Universitaire de Strasbourg, de m'avoir donné accès aux services de ce dernier et
d'avoir accepté d'être rapporteur interne.
Professeur à l'Institut d'Art et Archéologie de Byzance et chargé de la publication des
céramiques byzantines de Pergame, Jean-Michel Spieser a bien voulu me confier l'étude
archéométrique de ces céramiques et assurer l'encadrement archéologique de mon travail.
J'espère sincèrement que ce dernier aura pu être utile à ses recherches. Je le remercie de sa
confiance et de l'ouverture sur le monde byzantin qu'il m'a apportée.
Je remercie mon directeur de thèse Charles Heitz de m'avoir accueilli au CRN dans son
groupe d'Analyses par Techniques Nucléaires. Son savoir-faire d'expérimentateur m'a été très
précieux. Je lui suis très reconnaissante de sa totale disponibilité et de très grande confiance

qu'il m'a témoignée. Son enthousiasme et son esprit critique m'ont encouragée et stimulée tout
au long de ce travail.
Arthur Pape a participé à ce travail et m'a apporté soutien et conseils. J'ai beaucoup
appris de son sens de la rigueur et le remercie de son aide.
Merci à Isabelle Rossini pour sa collaboration et ses conseils amicaux. Son expérience
dans les analyses par activation neutronique a été très appréciée.
Je dois beaucoup à l'équipe du Réacteur Universitaire de Strasbourg, et tiens à
remercier en particulier Mrs Stampfler et Meyer. Leur contribution à un programme d'analyse
de plus de 2 ans a été tout à fait considérable.
Merci à Jean-Paul Resch, opérateur de l'accélérateur 4 MV du CRN, pour ses
interventions, notamment celles du week-end!
De nombreuses personnes ont contribué à ce travail au sein du CRN. Grand merci à
Albert Walter, qui m'a rendu agréable la fastidieuse tâche de l'échantillonnage; Gaby Bontemps,
pour ses programmes fortran en activation neutronique; David Balouka, pour son aide en
informatique; les équipes réseau et informatique, que je remercie de leur assistance; Andrée
Meens, pour la réalisation des cibles minces monoélémentaires. Merci à mes "équipes
d'adoption": groupe XYZt, groupe de chimie nucléaire et à l'équipe DELPHI (en particulier
Marcos Dracos) de m'avoir fait bénéficier de leurs équipements. Remerciements à Serge Liess,
CNRS, qui a réalisé avec grand soin les photographies des céramiques.
Mes séjours au Laboratoire de Recherches des Musées de France (LRMF) m'ont
permis de bénéficier du concours de plusieurs spécialistes. Je tiens à remercier la direction du
LRMF, et en particulier Jean-Michel Dupouy, directeur adjoint, de m'avoir donné cette
possibilité.
Merci à Joseph Salomon et à Thomas Calligaro, qui ont été nos partenaires pour les
expériences PIXE auprès de l'accélérateur AGLAE - auxquelles je regrette de ne pouvoir
laisser plus de place dans ce mémoire. Je remercie tout particulièrement Thomas de m'avoir
initiée à PIXAN et pour les discussions que nous avons eues à ce sujet.
Ces expériences ont été en partie réalisées dans le cadre du stage de DEA de Thomas
Keutgen, étudiant en physique à l'Université de Louvain la Neuve, que je remercie pour sa
participation enthousiaste au programme d'analyses de glaçures. Bruce Velde, ENS Paris, a été
l'initiateur de ce programme et je le remercie de ses conseils.
Je suis très reconnaissante à Alain Duval qui a été l'artisan des analyses au MEB et à la
microsonde protonique. Sa patience et sa disponibilité ont été très appréciées.
J'ai bénéficié des conseils amicaux de Guirrec Querré et d'Anne Bouquillon dans
l'approche géologique des céramiques. Merci à Guirrec pour son aide dans les examens pétro-
graphiques et à Alain Leclaire pour ses explications auprès du microscope polarisant.
De nombreuses autres personnes m'ont apporté leur concours, en particulier Myriam
Eveno dans les analyses au MEB et Dominique Bagault qui a réalisé les photographies auprès

d'AGLAE.
Enfin, j'ai beaucoup apprécié les conseils des chercheurs invités Gôran Lôvestam,
Arpad Kiss, Duncan Mac Arthur que j'ai eu l'occasion de rencontrer au LRMF.
Yves Besnus, GSTS Strasbourg, m'a incité à me former en géologie et je lui en suis très
reconnaissante. Je le remercie pour l'aide qu'il m'a apporté dans les observations pétro-
graphiques. Nos discussions à propos des traitements statistiques multivariés et de leur
interprétation géochimique m'ont également été d'un grand secours. La partie de ce mémoire
qui s'y rapporte lui doit beaucoup.
Je remercie Hubert Whitechurch, Philippe Duringer, Florence Beck et José Honnorez,
enseignants en géologie à l'Université de Strasbourg, de m'avoir permis d'assister à leurs cours
et de m'avoir conseillée. C'est également grâce à José Honnorez que la partie géologique de ce
travail a pu être réalisée, et que j'ai pu bénéficier de la collaboration d'Anthony Leruyet, qui a
mené les analyses cristallographiques et participé aux examens pétrographiques dans le cadre
de son stage de maîtrise de géologie. Je suis reconnaissante à Raymond Montigny pour ses
indications en matière de géochimie des magmas. Merci à tous les chercheurs et techniciens de
l'Institut de Géologie et de l'EOPG de Strasbourg que j'ai eu l'occasion de solliciter, en
particulier à Mrs Larqué, Cailleux, Samuel et Rouault. Merci également à Gaby Ehret et à
Mireille Del Nero pour leurs conseils.
J'ai beaucoup apprécié le concours d'Ursula Robert, chercheur au Laboratoire de
pétrologie de l'Université Paris VI et géologue de terrain spécialiste de l'Ouest de la Turquie.
Je la remercie vivement de son accueil chaleureux et de l'aide qu'elle m'a apportée. Merci à
Tuncay Ercan, MTA Ankara, et à Ismail Karamandesi, MTA Izmir, pour leur coopération lors
de mon séjour à Ankara.
Je tiens à remercier Mr Lafon et Mme Adam, enseignants d'histoire de l'art et
archéologie à l'Université de Strasbourg, de m'avoir permis d'assister à leur cours. Cet aperçu
des méthodes de l'archéologie m'aura été très utile. Merci également aux byzantinistes Jean-
Pierre Sodini, Université Paris I, et Véronique François, Université de Strasbourg, pour leur
éclairage sur l'apport de l'archéométrie dans leur discipline.
Un grand merci à mes collègues archéomètres qui ont bien voulu me communiquer
leurs données et m'ont apporté de nombreux échanges fructueux, en particulier à Richard
Jones, Université de Glasgow, pour son standard "Lefkandi brick"; Michael Jones, British
Museum, Londres, pour les données élémentaires de Sardes et d'Ephèse; I. KulefF, Université
de Sofia, qui m'a très aimablement communiqué ses résultats d'analyse; Max Peisach et Carlos
Pineda, Faure, Afrique du Sud, pour nos discussions sur les techniques de classification;
R. Sauer, Vienne, Autriche.

Je remercie les personnes qui m'ont permis d'utiliser leurs matériaux standards pour les
calibrations: Mr Govindaraju, CRPG, Nancy; Mr Heimburger, Institut de Chimie, Strasbourg;
Mr Abbé, Subatech, Nantes.
Je tiens à remercier Mr Pinol, Université de Strasbourg, pour ses conseils dans
l'utilisation de SPAD-N. J'ai également bénéficié de l'assistance de l'ancienne équipe statistique
du Centre de Calcul de Strasbourg, en particulier Mme Grange et Michel Ringenbach.
Ce travail n'aurait pu être réalisé sans le concours des bibliothécaires et documentalistes
qui m'ont facilité l'accès à des fonds spécialisés: Centre de documentation du LRMF,
Bibliothèque d'Art et Archéologie (fondation Doucet), Bibliothèque du Musée de l'Homme (en
particulier Mme Fonton), Bibliothèque de l'Institut de Géologie de Strasbourg, Bibliothèques
d'Archéologie de l'Université de Strasbourg, Bibliothèque Byzantine, Bibliothèque du service
d'archéologie de la DRAC Alsace, Bibliothèque du Musée d'Archéologie de Strasbourg
(remerciements à Mlle Schnitzler), Bibliothèque de l'IFROA (Mme Coros).
Les cartes et la finition des figures ont été réalisées par Alain Saccatti, dont la confiance
tout au long de ce travail a été le meilleur des soutiens.
Grand merci à tous ceux et celles qui m'ont encouragée et ont manifesté leur intérêt
pour cette étude. A mes collègues, et en particulier à Slavica qui a partagé mes nuits de travail,
merci de m'avoir témoigné leur amitié et de m'avoir accompagnée jour après jour.
Enfin, ce travail n'aurait pu aboutir sans l'appui de ma famille. Je lui dédie cette thèse.

Table des matières
Table des matières
Introduction.
Chapitre 1
1. Analyse élémentaire par PIXE et par activation neutronique: principes et mise en
oeuvre 3
1.1. Analyse élémentaire par PIXE 4
1.1.1. Principe 4
1.1.1.1. Processus physiques 4
Emission des rayons X 4
Transmission du rayonnement électromagnétique 6
Interaction de particules chargées avec la matière 8
Conclusions pratiques 13
1.1.1.2. Analyse quantitative 14
Détermination absolue, cibles minces, cibles épaisses 14
Standard externe 15
Standard interne 16
1.1.2. Mise en oeuvre pour l'analyse sous vide de pâtes céramiques 17
Echantillonnage 17
Echantillonnage d'une céramique archéologique 17
Préparation des cibles 19
Contamination lors du prélèvement 19
Dispositif et protocole expérimentaux 22
Dispositif expérimental au CRN 22
Protocole d'acquisition, paramètres expérimentaux 24
Mesure du courant 25
Spectrométrie X, dépouillement de spectres par le programme
PIXAN (subroutine BATTY) 27

Table des matières
Détermination expérimentale de l'efficacité du détecteur
Si(Li) 27
Paramètres pour la description théorique du détecteur
Si(Li) 28
Composantes spectrales, modélisation de raies 30
Dépouillement de spectres, incertitudes statistiques de
comptage, limites de détection 31
Détermination des concentrations élémentaires 36
Calcul des rendements par PIXAN (subroutine THICK) 36
Calcul des concentrations par une méthode "mixte",
précision, justesse 36
1.2. Analyse élémentaire par activation neutronique 42
1.2.1. Principe 42
Production de radioéléments par capture neutronique,
schémas de décroissance 42
Equations fondamentales 43
Analyse quantitative en standard externe, sources d'erreurs 45
1.2.2. Mise en oeuvre 47
Dispositif et protocoles expérimentaux 47
Dispositif expérimental 47
Protocoles expérimentaux 47
Spectrométrie gamma 49
Précision, justesse .....51
1.3. Bilan analytique et intercomparaisons 55
1.3.1. Bilan analytique et intercomparaison PIXE - INAA 54
1.3.2. Intercomparaison entre laboratoires 59
Laboratoire de céramologie, Lyon 59
Standard Lefkandi brick 61

Table des matières iii
Chapitre 2
2. Caractérisation des pâtes céramiques pour l'étude de la production et de la diffusion
des céramiques archéologiques. Application au cas des céramiques byzantines de
Pergame 63
2.1. Les études de provenance en céramologie: généralités 63
Le matériau céramique 63
Méthodologie en étude de provenance 65
2.2. La céramique byzantine médiévale:
catégories archéologiques et jalons analytiques 68
2.3. Les céramiques des fouilles de Pergame 89
2.3.1. Contexte historique et archéologique 89
2.3.2. Description des échantillons et classification archéologique 932.3.3. Problématique 111
2.4. Classification et caractérisation des céramiques de Pergame 114
2.4.1. Traitement statistique des résultats d'analyse élémentaire et
classification des échantillons à partir de leur composition 114
2.4.1.1. Statistiques univariées 114
2.4.1.2. Méthodes multivariées 122
Principe des méthodes factorielles 123
Principes des méthodes de classification 125
Classification ascendante hiérarchique (CAH) 125
Classification non hiérarchique 127
Mise en oeuvre 128
Classification 128
Constitution des groupes et confrontation avec la
classification archéologique 136

Table des matières iy_
2.4.2. Caractérisation des groupes de composition par les données
élémentaires, minéralogiques et pétrographiques 141
Groupes de composition rattachés à la production locale 146
Groupe byzantin local (byzl) 146
Groupe hellénistique local (hell) 150
Groupe de céramiques non glaçurées (g) 150
Groupe byzantin local altéré (byzp) 153
Groupe byzantin local indéterminé (byzi) 154
Groupes de composition de provenance indéterminée 160
Groupes de céramiques à pâte grasse (f) et de
céramiques à pâte fine (hi) 160
* Différenciation entre f, hi et byzi 160
* Comparaison des groupes f et hi avec
des groupes dereferences 165
Groupe de sgraffito fin (sgraf) 172
Groupe composite (lnop) 175
Conclusion 178
Références 181
Annexes:
1. Paramètres expérimentaux pour l'analyse élémentaire par la méthode PIXE
2. Petit glossaire de termes techniques en céramologie
3. Procédures expérimentales de laboratoires impliqués dans des programmes d'analyse de
céramiques byzantines ou dont les travaux sont cités au Chapitre 2 (2.4.2.)
4. Compositions élémentaires des pâtes des 168 céramiques analysées
5 A. Procédure de sélection des échantillons pour une ACP. Enchaînement ACP-CAH
B. Exemple de partitions obtenues par troncature d'un dendrogramme résultant d'une CAH
6 A. Principes des analyses minéralogiques, pétrographiques et de la microscopie électronique à
balayage
B. Classification proposée par A. Leruyet à partir des analyses minéralogiques et pétrographi-
ques de 32 échantillons
7 A. Mise en oeuvre de l'analyse par PIXE en faisceau extrait: vers un protocole d'analyse non
destructive de glaçures plombifères par PIXE et PIGE
B. Mise en oeuvre de la microsonde protonique pour l'analyse par la méthode PIXE:
application à l'étude de l'interface argile - glaçure et à l'analyse d'inclusions de verres
volcaniques dans les pâtes céramiques
8. Bibliographie, index alphabétique

îniiuuuuiion
Introduction
A la croisée des mondes antique et médiéval, oriental et occidental, la céramique
byzantine est longtemps restée méconnue. Depuis quelques années, elle suscite un intérêt accru
auprès des archéologues ce qui conduit à une meilleure définition de ses caractères spécifiques.
L'identification des centres de productions de types de céramiques comme le "sgraffito fin" [1],
la "Zeuxippus ware" [2], la "St Symeon ware" [3], 1"'Aegean ware" [4], la connaissance de
leurs circuits de diffusion et des influences technologiques et stylistiques qu'ils ont assimilées et
inspirées sont l'une des préoccupations majeures des archéologues et des historiens de l'art de
Byzance.
Si la mise en évidence du caractère producteur d'un site est subordonnée aux indices
donnés par la fouille, les analyses de laboratoire sont susceptibles de discerner des céramiques
de même provenance et de les attribuer à leur centre de production si celui-ci est inventorié et
caractérisé. Or à l'heure actuelle, en dehors de quelques rares exceptions comme Corinthe et
Thessalonique, les données analytiques concernant les ateliers de fabrication de la céramique
byzantine sont quasiment inexistantes.
Dans ce travail, nous nous proposons d'établir un nouveau référentiel pour les études de
provenance au sein du monde byzantin.
Les fouilles de Pergame ont mis au jour un important matériel céramique daté des
12cnic_i4cmc siècles. La découverte de trépieds, de ratCi. de cuisson et de pièces inachevées
dans les fouilles récentes de la ville moyenne atteste d'une production locale. Un
échantillonnage de 160 tessons, représentatif des céramiques trouvées à Pergame [5] et
incluant certaines de ces pièces dont la provenance locale ne peut être mise en doute, a été
sélectionné pour l'analyse. Une dizaine de tessons issus d'un atelier hellénistique de Pergame
ont également été considérés afin de confronter des productions pergaméniennes d'époques
différentes.
La distinction des productions locales et importées et leur caractérisation ont été l'objet
de notre étude. A cette fin, deux approches possibles en étude de provenance ont été mises en
oeuvre de manière complémentaire:
D'une part, l'analyse élémentaire des pâtes céramiques alliée à l'utilisation de traitements
statistiques multivariés; elle permet la classification des céramiques en groupes de
compositions similaires, correspondant aux produits d'un même atelier. Deux méthodes
d'analyse élémentaires parmi les plus performantes à l'heure actuelle, la fluorescence X induite

Introduction 2_
par faisceau d'ions, ou PIXE, et ('activation neutronique ont été utilisées. Leurs principes, leurs
pratiques et leurs performances sont exposées dans la première partie de cette thèse.
D'autre part, la caractérisation du matériau par des analyses pétrographiques et
minéralogiques; elle prend en compte la nature géologique de l'argile constituant la céramique.
Dans des cas favorables, elle peut mettre cette dernière en relation avec le contexte géologique
du gisement argileux exploité par le potier.
La deuxième partie de la thèse est consacrée à l'étude du cas des céramiques de
Pergame selon ces différentes approches. Le matériel analysé est présenté dans le contexte des
travaux antérieurs sur la céramique byzantine. La classification des tessons en groupes de
composition et la distinction entre groupes de céramiques locales et importées est ensuite
abordée. Enfin, l'utilisation conjointe des données géochimiques, pétrographiques et
minéralogiques permet de caractériser la production pergaménienne et de tester des hypothèses
de provenance pour les céramiques importées.

Chapitre 1.
Analyse élémentaire par PIXE et par activationneutronique: principes et mise en oeuvre

Chapitre 1
1.1. Analyse élémentaire par PIXE
PIXE est l'acronyme de "Particle Induced X-ray Emission": la fluorescence X d'un
matériau "cible" est provoquée par le bombardement avec des particules chargées. Ce mode
d'excitation l'inscrit dans les méthodes d'analyse par faisceau d'ions [6], Le fait que le PIXE soit
souvent considéré, par abus de langage, comme une méthode nucléaire est dû à l'utilisation
d'accélérateurs - équipements associés à la physique nucléaire - pour produire ces faisceaux.
Bien que divers types de projectiles puissent être utilisés en PIXE (particules a, ions lourds),
l'usage courant a privilégié le proton par rapport aux autres particules. Par la suite, le terme
PIXE désignera implicitement "Proton Induced X-ray Emission".
On s'accorde généralement à situer la première utilisation du PIXE en tant que méthode
d'analyse au début des années 1970 [7]. Elle a connu depuis de nombreux développements
techniques (faisceau extrait, microsonde protonique) et trouvé des champs d'applications très
variés, parmi lesquels on peut citer les études environnementales, la géologie, la biologie et la
médecine, ..., sans oublier l'histoire de l'art et l'archéologie [8]. Nous reviendrons bien
évidemment sur ce dernier point au chapitre 2.
1.1.1. Principe
1.1.1.1. Processus physiques
Nous passerons rapidement en revue les processus physiques en jeu, en commençant
par les aspects communs à tous les types d'excitation de la fluorescence X (rayons X primaires,
électrons, rayons gammas, particules, ...), puis en insistant plus particulièrement sur les
spécificités de l'utilisation de protons.
Emission des rayons X
Suite à un processus d'ionisation, une lacune électronique créée dans une couche
électronique interne (K, L, M) de l'atome est comblée par un électron d'une couche plus
externe. La relaxation s'accompagne:
• soit de l'émission d'un rayon X caractéristique d'énergie égale à la différence des
énergies de liaison des deux niveaux concernés: la transition est dite radiative et constitue le
phénomène te fluorescence X\
• soit de l'émission d'un électron du cortège électronique auquel cette énergie a été
directement transférée: la transition est non radiative et donne lieu à l'émission d'un électron
Auger.

Chapitre 1 1
La nomenclature de Siegbahn, usuelle en fluorescence X, est illustrée par la figure 1 :
par exemple, les raies dites Ka correspondent à une lacune initiale dans la couche K comblée
par un électron de la couche L. Toutes les transitions ne sont pas permises: dans ce cas, seules
les transitions L2-K ou L3-K sont autorisées par les règles de sélection de la mécanique
quantique, qui s'expriment en général par:
An> 1
Al = ± l
Aj = ± 1 ou 0
où n, 1 et j sont respectivement le nombre quantique principal et les nombres quantiques
associés aux moments angulaires orbital et total.
Le catalogue exhaustif des énergies de rayons X établi par Bearden [9] a été utilisé au
cours de ce travail.
VI -*-V —
N iv —
M m
L il
^ !
i
||l
> i l 1
a,
P.
!i r ' (1 r t
P:P
Ili
Yi
1
t
P.
I
Pi Pi
ilX l'ies I l ines
Figure 1: Diagramme partiel des niveaux d'énergie montrant les principales raies K et L
(nomenclature de Siegbahn, d'après [10])
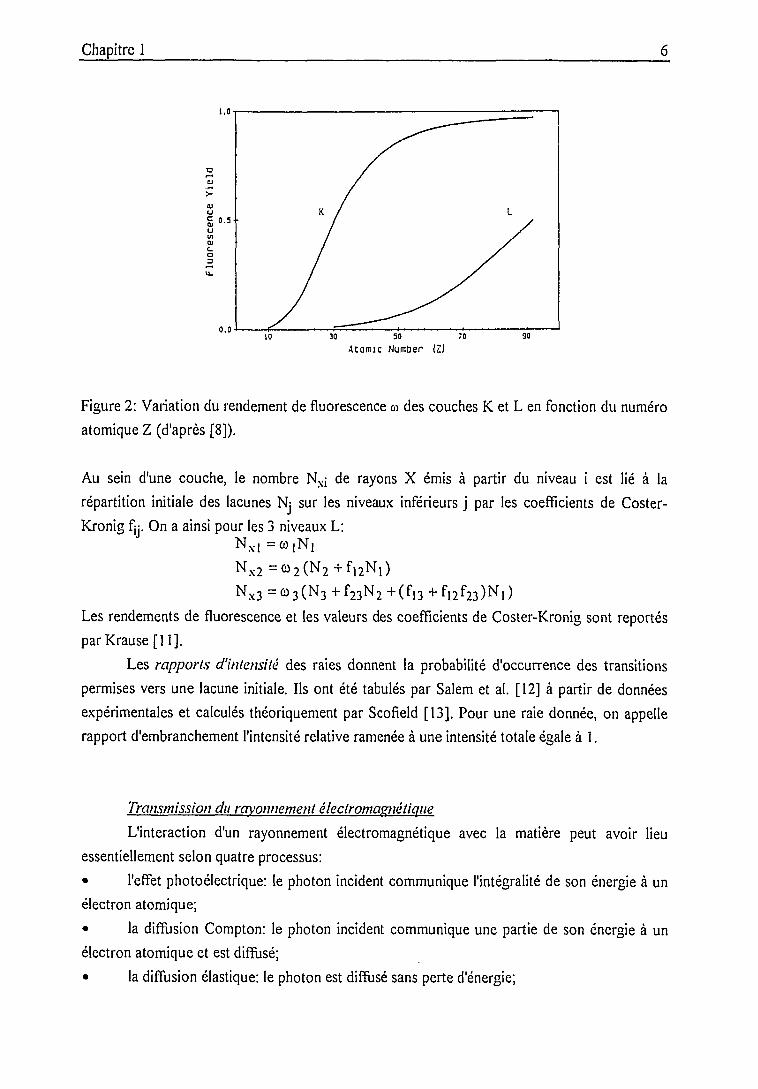
Pour une couche électronique donnée, on appelle rendement de fluorescence © la
probabilité pour qu'une lacune créée dans cette couche donne lieu à l'émission d'un rayon X. La
figure 2 montre la dépendance de © vis-à-vis du numéro atomique Z pour les couches K et L
(valeurs moyennes).
A partir des couches L, cette notion se complique du fait de possibles transitions
internes entre niveaux d'une même couche. Ces transitions, dites de Coster-Kronig, étant plus
rapides que les transitions inter-couches, des lacunes initialement distribuées sur les différentes
sous-couches (e.g. Ll, L2, L3) vont avoir tendance à se concentrer sur celle qui est la plus
externe (i.e. L3) avant que la désexcitation ne se produise.
Chapitre 1
30 50 70
Atomic Number (Z)
Figure 2: Variation du rendement de fluorescence a des couches K et L en fonction du numéro
atomique Z (d'après [8]).
Au sein d'une couche, le nombre Nxj de rayons X émis à partir du niveau i est lié à la
répartition initiale des lacunes N: sur les niveaux inférieurs j par les coefficients de Coster-
Kronig fjj. On a ainsi pour les 3 niveaux L:Nxl=»lNl
Nx2=o2(N2+f12N1)
Les rendements de fluorescence et les valeurs des coefficients de Coster-Kronig sont reportés
parKrause [11].
Les rapports d'intensité des raies donnent la probabilité d'occurrence des transitions
permises vers une lacune initiale. Ils ont été tabulés par Salem et al. [12] à partir de données
expérimentales et calculés théoriquement par Scofield [13]. Pour une raie donnée, on appelle
rapport d'embranchement l'intensité relative ramenée à une intensité totale égale à 1.
Transmission du rayonnement électromagnétique
L'interaction d'un rayonnement électromagnétique avec la matière peut avoir lieu
essentiellement selon quatre processus:
• l'effet photoélectrique: le photon incident communique l'intégralité de son énergie à un
électron atomique;
• la diffusion Compton: le photon incident communique une partie de son énergie à un
électron atomique et est diffusé;
• la diffusion élastique: le photon est diffusé sans perte d'énergie;
Chapitre 1 7.
• pour des rayons y d'énergie supérieure à 1.022 MeV, la génération d'une paire électron
- positron.
Dans le domaine d'énergie des rayons X, [effet photoélectrique est dominant.
La loi de Lambert-Beer donne l'intensité I transmise par une épaisseur x de matériau :
où IQ est l'intensité incidente, p la masse volumique et n le coefficient d'absorption massique
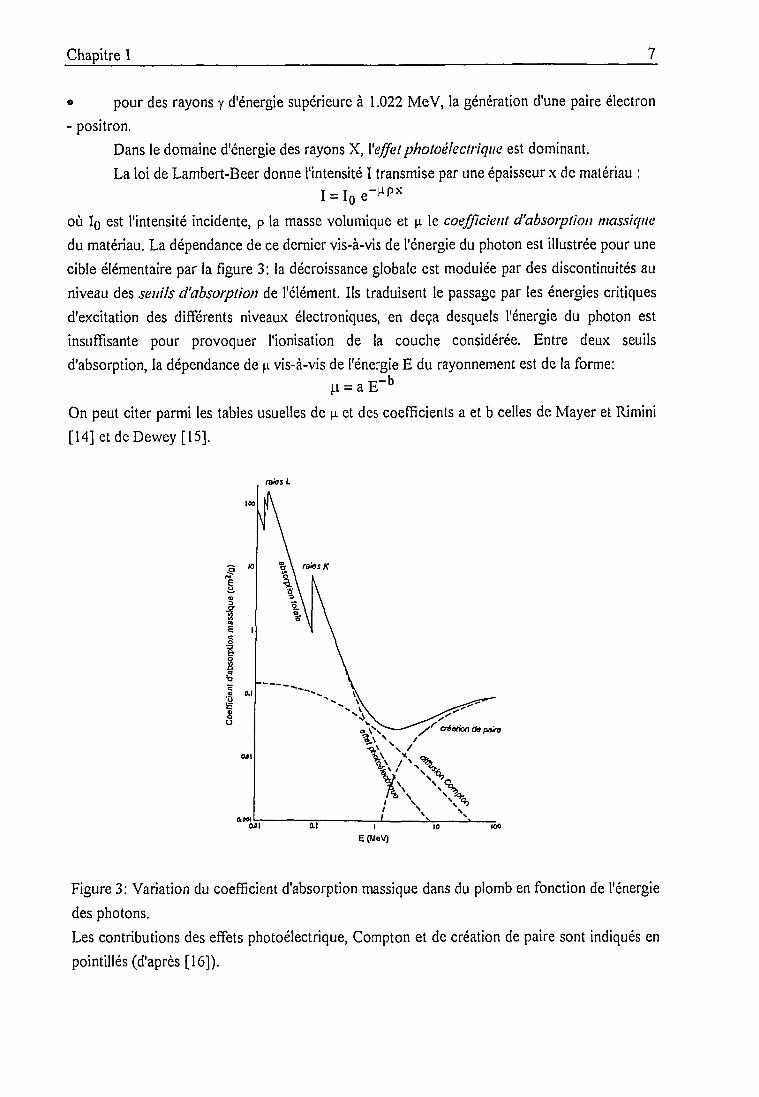
du matériau. La dépendance de ce dernier vis-à-vis de l'énergie du photon est illustrée pour une
cible élémentaire par la figure 3: la décroissance globale est modulée par des discontinuités au
niveau des seuils d'absorption de l'élément. Ils traduisent le passage par les énergies critiques
d'excitation des différents niveaux électroniques, en deçà desquels l'énergie du photon est
insuffisante pour provoquer l'ionisation de la couche considérée. Entre deux seuils
d'absorption, la dépendance de \i vis-à-vis de l'énergie E du rayonnement est de la forme:
u = a E"b
On peut citer parmi les tables usuelles de n et des coefficients a et b celles de Mayer et Rimini
[14] etdeDewey [15].
raies L
E(MeV)
Figure 3: Variation du coefficient d'absorption massique dans du plomb en fonction de l'énergie
des photons.
Les contributions des effets photoélectrique, Compton et de création de paire sont indiqués en
pointillés (d'après [16]).

Chapitre 1 8_
Dans un échantillon multiélémentaire soumis à une source d'excitation, on qualifie
d'attloabsorption la proportion de rayons X issus d'ionisations primaires et non transmis par la
cible. Lorsque ces rayons X provoquent à leur tour des excitations secondaires, on parle de
fluorescence secondaire. Cet effet est rarement pris en compte lors des déterminations
quantitatives par PIXE où il est généralement négligeable, sauf dans des cas bien particuliers
comme celui de certains alliages métalliques.
L'autoabsorption joue un rôle prépondérant dans l'évaluation de la profondeur
d'analyse. La détection des éléments légers dont les rayons X, moins énergétiques, sont plus
fortement absorbés, se limite de ce fait aux premiers microns sous la surface. Pour les éléments
lourds, la profondeur analysée dépend du mode d'excitation: elle sera généralement plus
importante avec des photons (rayons X ou y de faible énergie) qu'avec des particules chargées
dont le parcours est limité par des déflexions (e") et des pertes d'énergie.
Interaction de particules chargées avec la matière
Dans l'interaction d'une particule chargée avec la matière, on peut distinguer:
• les processus de diffusion élastique: l'énergie cinétique est globalement conservée et se
partage entre la particule diffusée et l'énergie de recul de l'atome cible. On définit la section
efficace différentielle da/dQ comme la probabilité d'occurence du phénomène par unité d'angle
solide et le facteur cinématique K comme le rapport de l'énergie cinétique du projectile après
la collision à son énergie incidente. Ces quantités fondamentales sont à la base de la méthode
d'analyse RBS (Rutherford backscattering) [17], Pour un projectile beaucoup plus léger que la
cible, ces quantités sont données dans le système du laboratoire par:
dQ
K =
' / , , xl/2 \ 2
(l-d^sùrej +dcos91 + d
V
(casd « 1 )
où d = Mi/M2,
Mj, Z\, E : masse, numéro atomique, énergie du projectile
M2, Zj : masse, numéro atomique de l'atome cible
e : charge de l'électron
0 : angle de diffusion relatif à la direction incidente
Des électrons incidents de quelques dizaines de keV d'énergie (cas d'une microsonde
électronique ou d'un microscope électronique à balayage (MEB)) subissent de nombreuses
collisions élastiques à grand angle de diffusion. Leur trajectoire dans le matériau n'est de ce fait

Chapitre 1 9
pas rectiligne, contrairement aux protons plus massifs (le rapport des masses est de 1832) et
plus énergiques (quelques MeV) utilisés en PIXE.
• les processus de diffusion inélastique: dans le domaine d'énergie des protons considéré,
le phénomène dominant est la perte d'énergie dE / dx au profit d'une excitation atomique. Elle
s'exprime par la formule de Bethe, soit pour une particule non relativiste:
dE 4rce4 z ? _ T
dx m V2 V
avec:
V : vitesse de la particule incidente
n : densité atomique de l'élément cible
I : potentiel moyen d'ionisation des atomes de la cible
m : masse de l'électron
Le pouvoir d'arrêt S et le parcours moyen R des protons dans le matériau sont définis à
partir de cette quantité:R 0
Cl p 1 — , , • D I p . I — I n Y — I ——
p dx J p •> S
0 Eo
où Eo est l'énergie incidente des protons.
On se réfère en général aux valeurs de pouvoirs d'arrêt et de parcours moyen établies par
Andersen et Ziegler [18].
Typiquement, le parcours moyen de protons de quelques MeV est de l'ordre de
quelques dizaines de jam dans des matériaux silicates, soit un ordre de grandeur de plus que des
électrons dans une microsonde électronique ou un MEB - cette différence étant essentiellement
un effet de la diffusion élastique.
Production de rayons X
La probabilité d'ionisation d'un atome (Z2) par des particules (Zj, E) est donnée pour
un niveau électronique donné par la section efficace d'ionisation a; (E, Z\, Z2). La section
efficace de production ap d'une raie relative à ce niveau s'en déduit par la relation:
où M est le rendement de fluorescence du niveau et b le rapport d'embranchement de la raie.
Sans entrer dans des considérations théoriques, on retiendra qu'à l'heure actuelle c'est la
théorie dite "ECPSSR" (l'abréviation illustre les différents effets pris en compte: Energy loss,
nuclear Coulomb field, Perturbation of the atomic Stationary States, Relativistic effects),
utilisée par Cohen et Harrigan dans leur calcul des sections efficaces pour des protons et des
particules a [19], qui approche au mieux les données expérimentales [20]. A titre indicatif, la
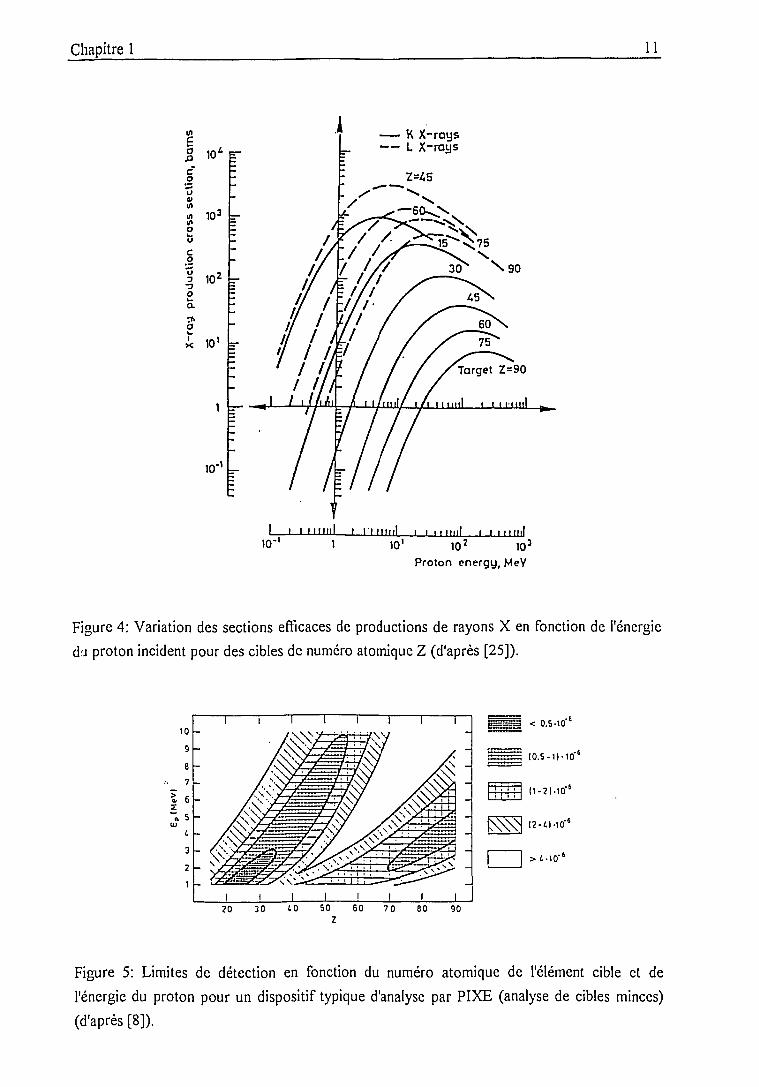
figure 4 montre la variation de ap en fonction de Z2 et de Ej pour des protons (Z]=l). Elle
correspond à des calculs reposant sur le modèle PWBA (Plane Wave Born Approximation)

Chapitre 1 l_0
développé par Merzbacher et Lewis [21] qui est à la base des développements ECPSSR cités.
Dans certains domaines, les valeurs numériques des sections efficaces de la figure 4 sont donc
approximatives puisqu'elles ne tiennent pas compte des perfectionnements ultérieurs mais il est
possible de dégager les tendances générales de leur évolution. Ainsi, il est important de noter
que:
• à une énergie E fixe, a décroît quand Z2 augmente;
• pour Z2 donné, a augmente avec E, passe par un maximum puis diminue. L'énergie
correspondant au maximum est d'autant plus élevée que Z^ est grand;
Pour des projectiles plus lourds que des protons mais tels que Z[/Z2«l , a- est de la
forme:
l V Ai
où E/Aj est l'énergie par nucléon du projectile et P ne dépend que de l'atome cible. La
dépendance de a en (E/Aj)^ explique l'utilisation préférentielle de projectiles légers (protons
plutôt que particules a), les protons nécessitant une énergie beaucoup moins élevée pour une
section efficace équivalente. Pour des projectiles lourds (Zj>6 et Z j ^ - l ) les mécanismes
d'ionisation sont plus complexes du fait de l'interaction des nuages électroniques du projectile
et de l'atome cible (mécanismes "quasi moléculaires", cf. e.g. [22]). De ce fait les applications
analytiques sont plus délicates [23].
Par la suite, nous considérerons uniquement les interactions avec des faisceaux de
protons.
Dans une certaine mesure, la figure 4 peut être mise en parallèle avec une
représentation des limites de détection de la méthode PIXE dans un diagramme (Z,E)
(figure 5): la zone de sensibilité maximale se déplace vers les numéros atomiques élevés
lorsque E croît. Cette zone recouvre plus particulièrement les régions 20 < Z < 35 (excitation
des couches K) et 75 < Z < 85 (excitation des couches L). Par contre, les éléments
relativement lourds tels le Baryum (Z= 56) et les terres rares légères sont nettement
défavorisés. Pour doser ces éléments, il est souvent nécessaire de faire appel à une autre
méthode d'analyse, comme l'activation neutronique ou la fluorescence X induite par
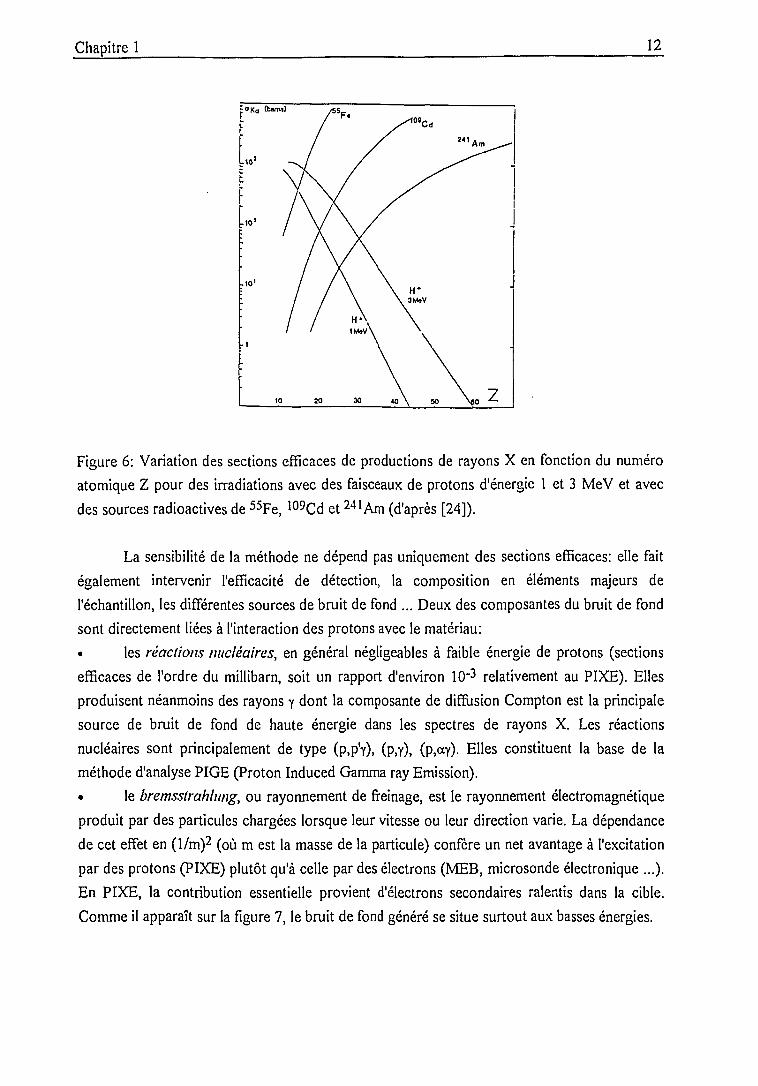
radioisotope (241Am). Pour cette dernière, l'effet de sections efficaces nettement plus élevées,
comme le montre la figure 6, est cependant contrebalancé par le faible flux des sources
disponibles commercialement [24]. A cet inconvénient s'ajoute le fait que les terres rares sont
en général présentes en très faibles concentrations ce qui nécessite des durées d'acquisition des
spectres considérables.
Chapitre 1 11
m
ja 10
o
t>m
s 1°3
oL.<J
O
i
io2
oa
K X-rays— L X-rays
Z=45
'"' " " " i i n nul i l i i l10
Proton energy, MeV
Figure 4: Variation des sections efficaces de productions de rayons X en fonction de l'énergie
du proton incident pour des cibles de numéro atomique Z (d'après [25]).
T I I I I I
20 30 40 50 60 70 80 90
< 0.5-10
10.5-11-10
11-21-10"
> i-10
Figure 5: Limites de détection en fonction du numéro atomique de l'élément cible et de
l'énergie du proton pour un dispositif typique d'analyse par PIXE (analyse de cibles minces)
(d'après [8]).
Chapitre 1 12
Figure 6: Variation des sections efficaces de productions de rayons X en fonction du numéro
atomique Z pour des irradiations avec des faisceaux de protons d'énergie 1 et 3 MeV et avec
des sources radioactives de 55Fe, 109Cd et 24lAm (d'après [24]).
La sensibilité de la méthode ne dépend pas uniquement des sections efficaces: elle fait
également intervenir l'efficacité de détection, la composition en éléments majeurs de
l'échantillon, les différentes sources de bruit de fond ... Deux des composantes du bruit de fond
sont directement liées à l'interaction des protons avec le matériau:
• les réactions nucléaires, en général négligeables à faible énergie de protons (sections
efficaces de l'ordre du millibarn, soit un rapport d'environ 10"3 relativement au PIXE). Elles
produisent néanmoins des rayons y dont la composante de diffusion Compton est la principale
source de bruit de fond de haute énergie dans les spectres de rayons X. Les réactions
nucléaires sont principalement de type (p,p'y), (p.r), (p,ay). Elles constituent la base de la
méthode d'analyse PIGE (Proton Induced Gamma ray Emission).
• le bremsslrahhmg, ou rayonnement de freinage, est le rayonnement électromagnétique
produit par des particules chargées lorsque leur vitesse ou leur direction varie. La dépendance
de cet effet en (1/m)2 (où m est la masse de la particule) confère un net avantage à l'excitation
par des protons (PIXE) plutôt qu'à celle par des électrons (MEB, microsonde électronique ...).
En PIXE, la contribution essentielle provient d'électrons secondaires ralentis dans la cible.
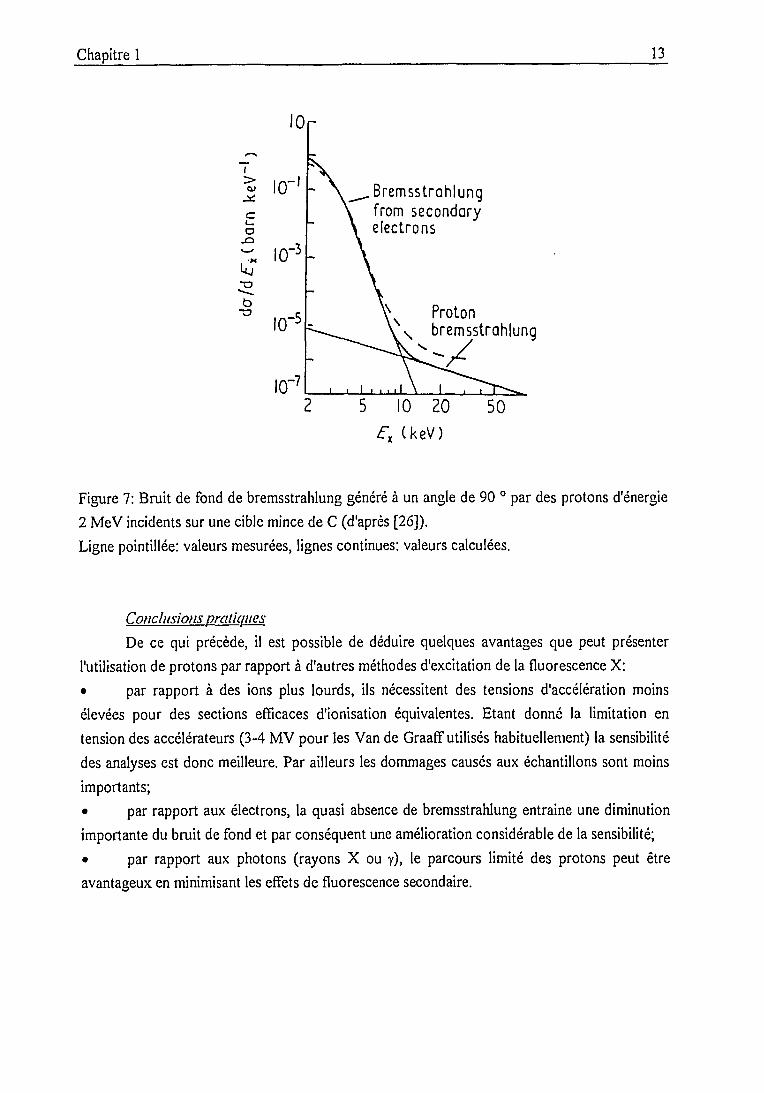
Comme il apparaît sur la figure 7, le bruit de fond généré se situe surtout aux basses énergies.
Chapitre 1 13
aJ3
10"'- .Bremsstrahlungfrom secondaryelectrons
Protonbremsstrahlung
5 10 20
Ex (keV)
Figure 7: Bruit de fond de bremsstrahlung généré à un angle de 90 ° par des protons d'énergie
2 MeV incidents sur une cible mince de C (d'après [26]).
Ligne pointillée: valeurs mesurées, lignes continues: valeurs calculées.
Conclusions pratiques
De ce qui précède, il est possible de déduire quelques avantages que peut présenter
l'utilisation de protons par rapport à d'autres méthodes d'excitation de la fluorescence X:
• par rapport à des ions plus lourds, ils nécessitent des tensions d'accélération moins
élevées pour des sections efficaces d'ionisation équivalentes. Etant donné la limitation en
tension des accélérateurs (3-4 MV pour les Van de Graaff utilisés habituellement) la sensibilité
des analyses est donc meilleure. Par ailleurs les dommages causés aux échantillons sont moins
importants;
• par rapport aux électrons, la quasi absence de bremsstrahlung entraine une diminution
importante du bruit de fond et par conséquent une amélioration considérable de la sensibilité;
• par rapport aux photons (rayons X ou y), le parcours limité des protons peut être
avantageux en minimisant les effets de fluorescence secondaire.

Chapitre 1 14
1.1.1.2. Analyse quantitative
Détermination absolue, cibles minces, cibles épaisses
Considérons un échantillon multiélémentaire soumis à un faisceau de protons. Soit dNjj
le nombre de rayons X de la raie i caractéristique d'un élément j issus d'une épaisseur
élémentaire dx de matériau situé à une distance x de la surface (figure 8):
prcosO
(1)
r
0e
6c
les trois termes entre parenthèses se référant respectivement à la détection, à la transmission
par l'échantillon et à la production des rayons X (par la suite, on appellera T le terme de
transmission). La signification des différents facteurs est la suivante:
parcours des protons dans l'échantillon: x = r cosOe
angle entre la direction du faisceau incident et la normale à l'échantillon
angle de sortie des rayons X relatif à la normale à l'échantillon
efficacité du détecteur pour l'énergie de la raie i
angle solide sous-tendu par le détecteur
concentration pondérale et masse atomique de l'élément j
nombre d'Avogadro
nombre de protons incidents
section efficace de production pour la raie i de l'élément j , à l'énergie E des
protons après un parcours r
: coefficient d'absorption massique de la cible pour la raie i. Les coefficients
élémentaires ayant une structure additive, on aura pour un échantillon composé de
k éléments :
ÇllAn
Cj, Aj
Q/e
Si la cible est une couche mince, il est possible d'utiliser directement l'expression (1) en
assimilant le terme d'absorption T à 1. Dans le cas contraire, une intégration le long du
parcours des protons est nécessaire. Dans le cas de cibles "épaisses", i.e. d'épaisseur supérieure
au parcours des protons:
'U~ e
CN ° T-= 7"i]~8i477 JaPij "S"
R. JL fo Tj dr
00
'' dE
Eo

Chapitre 1 15
(2)
où K est une constante dépendant uniquement de la géométrie de détection.
La valeur de l'intégrale I est calculée numériquement, par approximations successives
des Cfc si la matrice de l'échantillon n'est pas connue. Par la suite, Ny désignera la surface du
pic correspondant à la raie i de l'élément j , ou sa contribution à un pic composite si plusieurs
raies sont superposées.
Dans la pratique, une détermination indirecte par standard externe ou standard interne
est préférable au calcul absolu. Cela permet souvent de limiter les incertitudes expérimentales
(e, Q) et les imprécisions sur certains paramètres fondamentaux (a,...) [27].
faisceau
Figure 8: Représentation schématique de l'interaction des protons dans le matériau et de
l'émission des rayons X.
Standard externe
Le principe consiste à comparer raie à raie les surfaces de pics d'un standard de
composition connue et de l'échantillon à doser. Le rapport correspondant est:
N
N'
ij _ Q C; Ij(E0 ,Ck)(3)
les quantités "primées" se rapportant au standard.

Chapitre 1 ]6_
Les avantages de cette méthode sont:
• l'élimination du terme d'efficacité, dont la variation est souvent mal connue dans la
région des basses énergies (cf. 1.1.2., Paramètres pour la description théorique du détecteur
Si(Li));
• une convergence des C; d'autant plus rapide que le standard a une matrice proche de
celle de l'échantillon (le rapport des intégrales I pouvant être pris égal à 1 en lèrc
approximation). On tend aussi à minimiser ainsi la contribution à l'incertitude des sections
efficaces.
Il faut cependant noter comme inconvénient:
• la fiabilité relative des standards
• la dépendance envers les termes de charge Q, qui peuvent être une source majeure
d'imprécision (cf. 1.1.2. Détermination des concentrations élémentaires, Précision). La solution
qui consiste à effectuer les irradiations dans les même conditions avec la même charge (Q=Q')
n'est valable que si la mesure de la charge est fiable.
Standard interne
On dose indépendamment un des éléments de l'échantillon, les concentrations des
autres éléments s'en déduisant par la relation:
_ C JCJ A J 'S i I j (E 0 ,C k )Nj-j. Cj. Aj s;. I i ' (E 0 ,C k )
les quantités "primées" se référant à la raie de l'étalon interne.
A l'inverse de la méthode du standard externe, on a éliminé ainsi la charge Q, mais les
efficacités interviennent explicitement et les sections efficaces jouent un rôle accru puisqu'elles
ne se rapportent plus au même élément.
Un des moyens de doser l'élément qui sert de standard interne est d'effectuer une
mesure supplémentaire sur un échantillon identique dopé avec une quantité faible et connue de
cet élément.
La méthode du standard interne peut aussi être utilisée alternativement sans étalon
interne dans le cas où tous les éléments majeurs sont analysés. On introduit alors comme
paramètre supplémentaire la relation de fermeture:
]TCj=100%
j

Chapitre 1 ]]_
1.1.2. Mise en oeuvre pour l'analyse sous vide de pâtes céramiques
La méthode d'analyse PIXE peut être mise en oeuvre selon trois types de dispositifsexpérimentaux:• l'analyse sous vide, applicable à des échantillons de petite dimension peu sensibles àl'importante élévation de température induite par le bombardement de protons [28]. Elle estparticulièrement indiquée dans le cas de grandes séries de mesures, car plus aisémentautomatisable;
• l'analyse en faisceau extrait, pour des matériaux fragiles (e.g. parchemins [29]) ou desobjets de grandes dimensions (e.g. peintures de chevalets [30]);• l'analyse en microfaisceau, pour les applications nécessitant une résolution spatiale del'ordre du micron (e.g. analyses de soudures métalliques [31 ]).
Nous nous attacherons à décrire les différentes étapes de la mise en oeuvre de l'analysesous vide de pâtes de céramiques archéologiques. Nous aborderons succintement en annexe lesvariantes apportées pour les autres dispositifs ainsi que les paramètres propres à d'autresmatériaux analysés (glaçures, verres).
De nombreux laboratoires ont utilisé la méthode PIXE pour l'analyse de pâtescéramiques (e.g. [32, 33, 34, 35, 36, 37]). Une procédure d'analyse élémentaire par PIXE sousvide sur cibles épaisses a été établie au Centre de Recherches Nucléaires de Strasbourg (CRN)dès les années 1980 [38, 39]. Elle a été adaptée dans le cadre de ce travail au traitement d'ungrand nombre d'échantillons. A cet effet, le dépouillement des spectres de rayons X et le calculdes concentrations élémentaires ont été réalisés avec le logiciel PIXAN de E. Clayton [40, 41,42], complété d'une interface graphique par T. Calligaro du Laboratoire de Recherches desMusées de France (LRMF). Il existe actuellement de nombreux programmes informatiquesspécifiques à la méthode PIXE: les intercomparaisons attestent de la variété des choix de leursauteurs [43, 44, 45]. L'approche adoptée par PIXAN sera présentée tout au long de cechapitre, ainsi que son adaptation à notre propre dispositif expérimental. Pour le calcul desconcentrations élémentaires, les exigences de précision et de justesse nous ont amené à opterpour une méthode "mixte" basée sur celle du standard externe qui s'affranchit de l'imprécisionsur la charge.
Echantillonnage
Echantillonnage d'une céramique archéologique
Pour des raisons d'encombrement, la majorité des analyses sous vide s'effectue surprélèvement (voir par exemple [38] pour une comparaison entre analyse directe et analyse sur

Chapitre 1 18_
prélèvements). On peut cependant remarquer que les questions soulevées lors de la définition
d'un "bon" échantillonnage peuvent être étendues également au cas des mesures effectuées
directement sur l'objet. Avant de revenir plus en détail sur la nature du matériau céramique en
2.1., citons les facteurs qui interviennent au niveau du prélèvement et auxquels l'interprétation
correcte du résultat de l'analyse est subordonnée:
• \* identification du matériau analysé. Une céramique est souvent constituée de couches
de nature différente: pâte, engobe, glaçure, peinture, ... (cf. annexe 2). Dans le cas d'analyses
directes, il est parfois difficile de distinguer les contributions des différentes couches présentes,
et ce d'autant plus que leurs épaisseurs relativement au parcours des protons dans le matériau
ne sont pas connues [24]. Nous nous intéresserons uniquement ici au "support", à la pâte
céramique,
• la représentativité du prélèvement et du volume analysé. Une pâte céramique n'est pas
un matériau homogène: elle est constituée d'un mélange de particules argileuses fines et de
grains dont les dimensions peuvent être importantes, notamment dans les céramiques dites
"grossières" (cf. e.g. [36]). En pratique, si l'échantillonnage d'une céramique de texture plus
grossière nécessite le prélèvement d'une plus grande quantité de matière pour être
représentatif, la taille des prélèvements de céramiques fines ne peuvent être indéfiniment
réduite. En effet, les céramiques fines peuvent contenir des minéraux en faible proportion dont
la répartition n'est homogène qu'à grande distance et dont la contribution à la composition
globale est prépondérante pour certains éléments traces (e.g. zircons (Zr) [38], chromites
(Cr),...). Le caractère hétérogène est minimisé lorsque l'échantillon est broyé et homogénéisé,
ce qui n'est pas possible lors d'analyses directes.
Il faut bien noter qu'au-delà de la représentativité du prélèvement, c'est celle du volume
analysé qui importe. Ce volume est fonction de la méthode utilisée: l'analyse par activation
neutronique porte sur l'ensemble du volume de l'échantillon, alors qu'en PIXE la profondeur
d'analyse est de quelques \xm à quelques dizaines de ^m (cf. 1.1.1.1.). On peut estimer avec
Swann et Fleming [35] qu'une mesure sera représentative si la dimension du diamètre du
faisceau excède de deux ordres de grandeur la dimension de la plus grande hétérogénéité de
l'échantillon.
Un autre aspect du problème concerne les altérations du matériau céramique lors de
son utilisation [46] et de son enfouissement (cf. e.g. [46, 47, 48, 49, 50]). La plupart de ces
altérations se traduisent par des échanges entre l'objet et le milieu et se manifestent par des
gradients élémentaires entre la surface et le coeur du tesson. On considère que ce dernier est le
plus représentatif de la composition initiale.
• la contamination lors du prélèvement, Parallèlement aux pollutions de surface,
l'opération de prélèvement elle-même est susceptible de contaminer l'échantillon avec les
constituants de l'outil utilisé. On cherchera en général à éviter tout apport ou à défaut on
choisira un outil (mortier d'agate, foret, scie, concasseur, ...) qui produit des contributions
négligeables ou contrôlables: traces d'un élément présent comme élément majeur dans

Chapitre 1 19
l'échantillon, éléments naturellement absents dans les céramiques, éléments dont le dosage n'est
pas accessible par la(les) méthode(s) d'analyse utilisée(s),...
Préparation des cibles
Pour la présente étude, les tessons de céramiques ont été tout d'abord abrasés avec une
meulette en alumine AI2O3 afin de supprimer les éventuels "enduits" (peinture, engobe et
glaçure, cf. 2.3.2) ou d'éliminer une couche superficielle de pâte d'une épaisseur de l'ordre du
mm. Quelques grammes de poudre de céramique sont ensuite prélevés par perçage avec une
mèche au carbure de tungstène; nous reviendrons au paragraphe suivant sur les problèmes
posés par le choix du matériau de la mèche. L'examen des grains obtenus sous microscope
optique permet d'estimer que leurs dimensions sont inférieures à 10-20 ^m, ce qui respecte bien
le rapport de deux ordres de grandeur relativement au diamètre du faisceau (1 ou 2 mm). Le
conditionnement de la poudre en pastilles cylindriques de 1 cm de diamètre sur environ 2 mm
d'épaisseur (cibles épaisses) est effectué sous une pression de 4 tonnes.cm'2. La surface des
pastilles est rendue conductrice par evaporation d'une fine couche de carbone (15-20 (.tg/cm-).
On évite ainsi que le bombardement par le faisceau de protons ne produise des accumulations
de charges à la surface du matériau isolant. Les tensions ainsi créées généreraient un fort
bremsstrahlung des électrons secondaires, et conséquemment un bruit de fond important dans
le spectre. De plus la mesure du courant serait impossible en raison des claquages.
Contamination lors du prélèvement
Nous avons vu qu'une pollution de l'échantillon peut survenir au moment de son
prélèvement. Des échantillons de contrôle ont été constitués par broyage au mortier d'agate
afin de suivre l'évolution de l'apport du foret en carbure de tungstène au fur et à mesure de son
utilisation. Nous avons pu constater qu'une mèche neuve produit une pollution très importante
en Cu et Zn (figure 9a). Cette pollution s'atténue avec l'usage (figure 9b). En limant la mèche
elle peut être réduite drastiquement: en effet, l'apport en Cu et Zn provient de la brasure qui
fixe l'extrémité en carbure de tungstène à son support en acier. Dans ces conditions, seules les
raies du tungstène sont encore visibles (figure 9c). Cet élément n'entrant pas naturellement
dans la composition des gisements argileux en quantité appréciable [51], il ne constitue pas une
gêne directe à l'analyse par PIXE. Par contre, la présence de ses raies caractéristiques dans le
spectre peut être une source d'imprécision dans la déconvolution des pics d'autres éléments. Un
spectre de tungstène pur est présenté sur la figure 10: les raies de plus forte intensité (raies Lpl
à 9.67 keV, Lp2 à 9.96 keV, La à 8.38 keV) sont susceptibles de produire des interférences
dans la région du spectre contenant les raies K de Ni, Cu, Zn; on note aussi des contributions à
proximité des raies K de As (raies Ly de W).
Remarquons que les variations d'intensité des pics de Pb et Zr sur la figure 9 ne sont
pas liées à des pollutions, mais à une distribution hétérogène de ces éléments dans les
échantillons (cf. 2.4.1.1.).
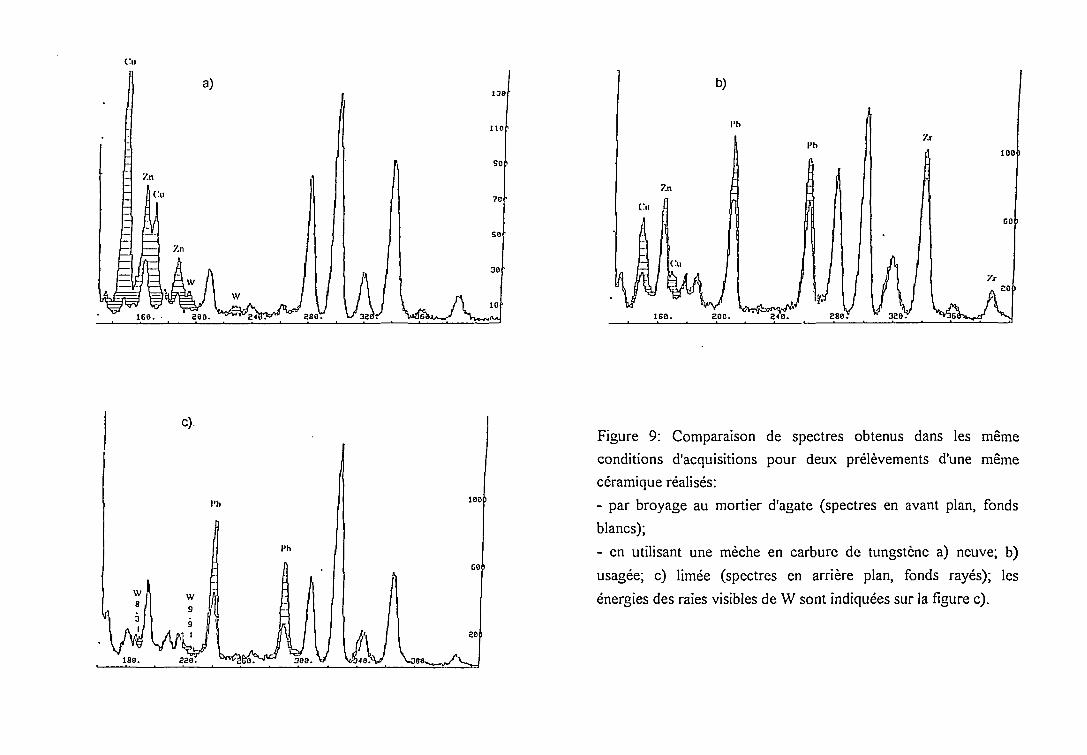
l e e . • sea
Figure 9: Comparaison de spectres obtenus dans les même
conditions d'acquisitions pour deux prélèvements d'une même
céramique réalisés:
- par broyage au mortier d'agate (spectres en avant plan, fonds
blancs);
- en utilisant une mèche en carbure de tungstène a) neuve; b)
usagée; c) limée (spectres en arrière plan, fonds rayés); les
énergies des raies visibles de W sont indiquées sur la figure c).
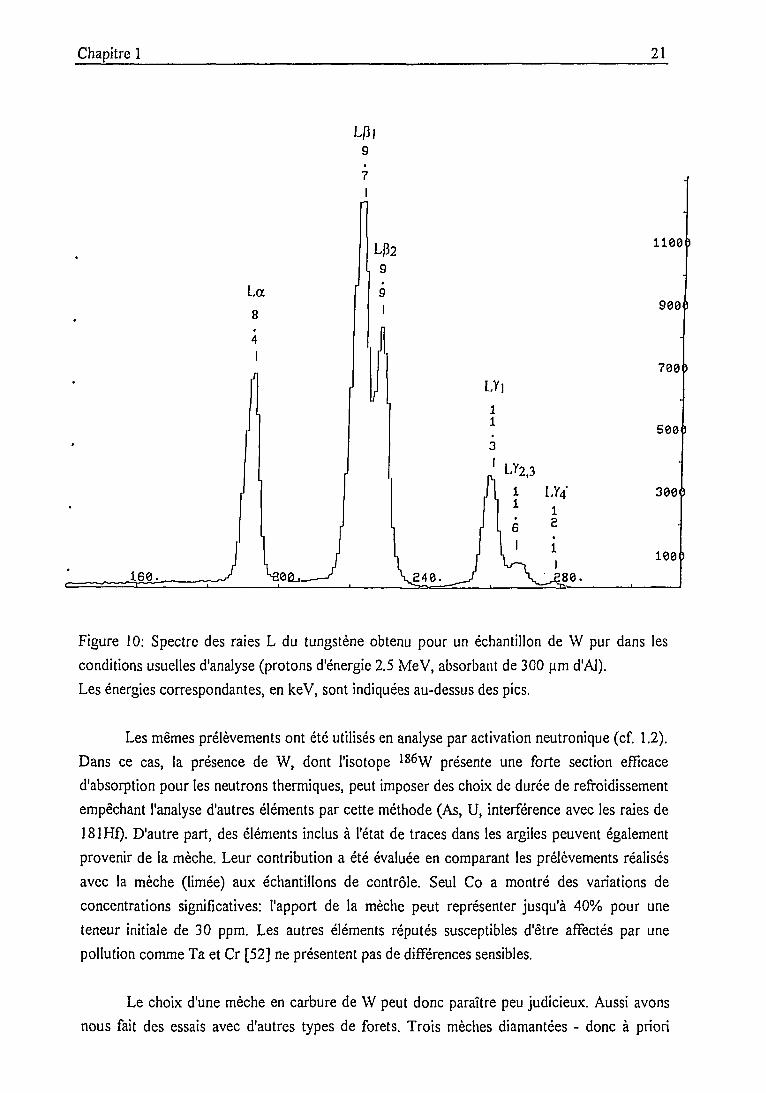
Chapitre 1 21
LJ3|g
La8
4I
L|32g
J.60..
LY|11
LY2.3
LV4'
.S48.
1100
900
700
500
300
100'
Figure 10: Spectre des raies L du tungstène obtenu pour un échantillon de W pur dans les
conditions usuelles d'analyse (protons d'énergie 2.5 MeV, absorbant de 300 u.m d'Al).
Les énergies correspondantes, en keV, sont indiquées au-dessus des pics.
Les mêmes prélèvements ont été utilisés en analyse par activation neutronique (cf. 1.2).
Dans ce cas, la présence de W, dont l'isotope l86W présente une forte section efficace
d'absorption pour les neutrons thermiques, peut imposer des choix de durée de refroidissement
empêchant l'analyse d'autres éléments par cette méthode (As, U, interférence avec les raies de
181Hf). D'autre part, des éléments inclus à l'état de traces dans les argiles peuvent également
provenir de la mèche. Leur contribution a été évaluée en comparant les prélèvements réalisés
avec la mèche (limée) aux échantillons de contrôle. Seul Co a montré des variations de
concentrations significatives: l'apport de la mèche peut représenter jusqu'à 40% pour une
teneur initiale de 30 ppm. Les autres éléments réputés susceptibles d'être affectés par une
pollution comme Ta et Cr [52] ne présentent pas de différences sensibles.
Le choix d'une mèche en carbure de W peut donc paraître peu judicieux. Aussi avons
nous fait des essais avec d'autres types de forets. Trois mèches diamantées - donc à priori

Chapitre 1 22_
exemptes de toute matière polluante - ont ainsi été testées. Nous avons pu constater en
analyse par PIXE que le liant des grains diamantés donnait lieu à des contamination en Zn, Ni
et Cu. Vu le coût relativement élevé et la courte durée de vie de ces mèches, nous n'avons pas
estimé qu'elles étaient nettement plus avantageuses. L'analyse directe, par RIXRF, d'un autre
type de mèche en alumine frittée a également été réalisée: on a noté la présence de traces de
Cu, Zn, Zr.
A défaut de "matériau idéal", nous avons utilisé une mèche en carbure de tungstène
(après élimination de la brasure) en restant vigilants quant aux effets induits.
Dispositif et protocole expérimentaux
Dispositif expérimental au CRN
II est présenté par la figure 11. Les protons sortant d'un accélérateur Van de Graaff de
tension maximale 4 MV sont sélectionnés par un électroaimant à l'énergie nominale. Ils sont
ensuite focalisés par un quadrupole magnétique et collimatés dans la ligne d'analyse maintenue
sous vide (10 -5 Torr). La chambre d'analyse est détaillée sur la figure 12.
Le faisceau est à incidence normale sur une cible placée dans un porte-échantillon
rotatif contenant 8 logements. Le positionnement de la cible et des détecteurs est
reproductible, ce qui permet de travailler à géométrie fixée. Un collimateur en carbone évite la
diffusion de protons sur les matériaux environnant la cible et restreint l'angle solide vu par le
détecteur à cette dernière. Le diamètre du faisceau est défini en amont grâce à un diaphragme
réglable en tantale. Notons que les matériaux des diaphragmes (tantale) et collimateurs
(carbone) ont été choisis de façon à éviter la production de rayons y lors de réactions nucléaires
avec les protons. Le détecteur de rayons X Si(Li) est placé à 135° de la direction du faisceau
incident. Il est séparé de la chambre par une fenêtre de sortie en mylar, à laquelle on peut
adjoindre des absorbants sélectifs. La chaîne d'acquisition des spectres comporte outre le
détecteur un préamplificateur, un amplificateur, un convertisseur d'amplitude et un analyseur
multi-canaux. L'acquisition est pilotée depuis un PC par le logiciel TMCA [53]. Un détecteur
de particules peut également être utilisé pour l'analyse par la méthode RBS. Il est employé en
particulier lors de la détermination de l'efficacité du détecteur Si(Li). Un intégrateur de courant
relié au porte-échantillon métallique isolé du reste de la chambre permet la mesure du courant
donc du nombre de particules incidentes sur la cible.
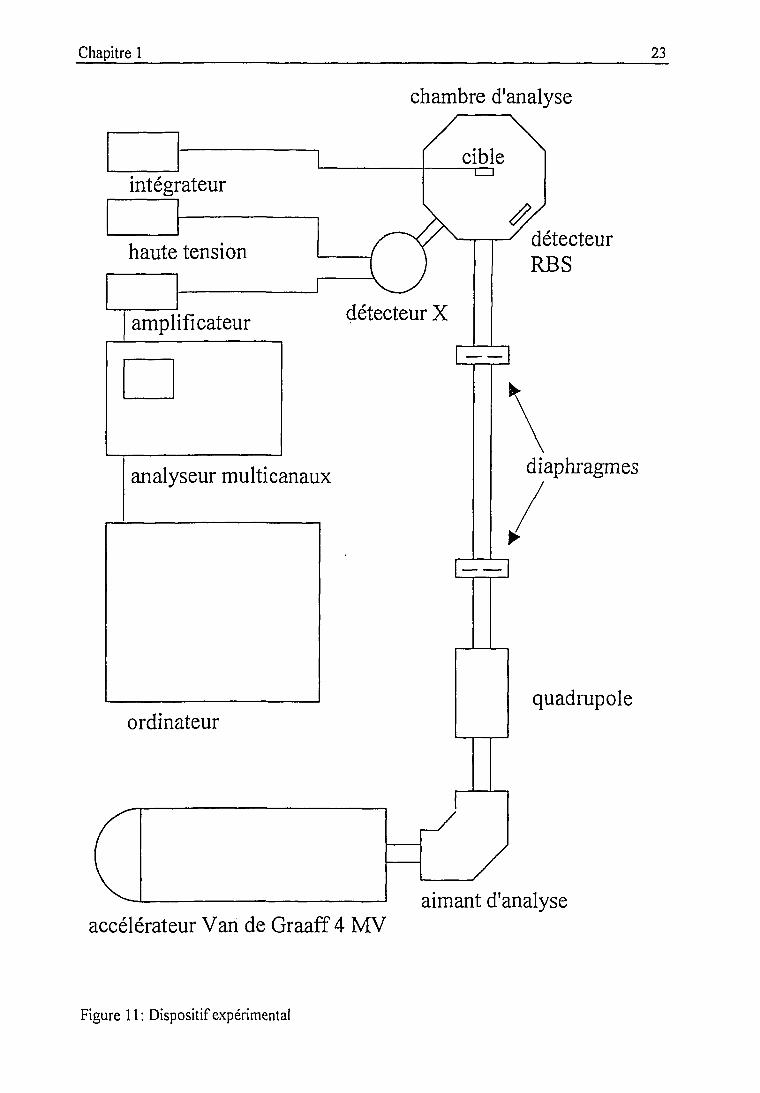
Chapitre 1 23
chambre d'analyse
cibleintégrateur
haute tension
amplificateur
détecteurRBS
analyseur multicanaux diaphragme:
ordinateurquadrupole
accélérateur Van de Graaff 4 MVaimant d'analyse
Figure 11: Dispositif expérimental
Chapitre 1 24
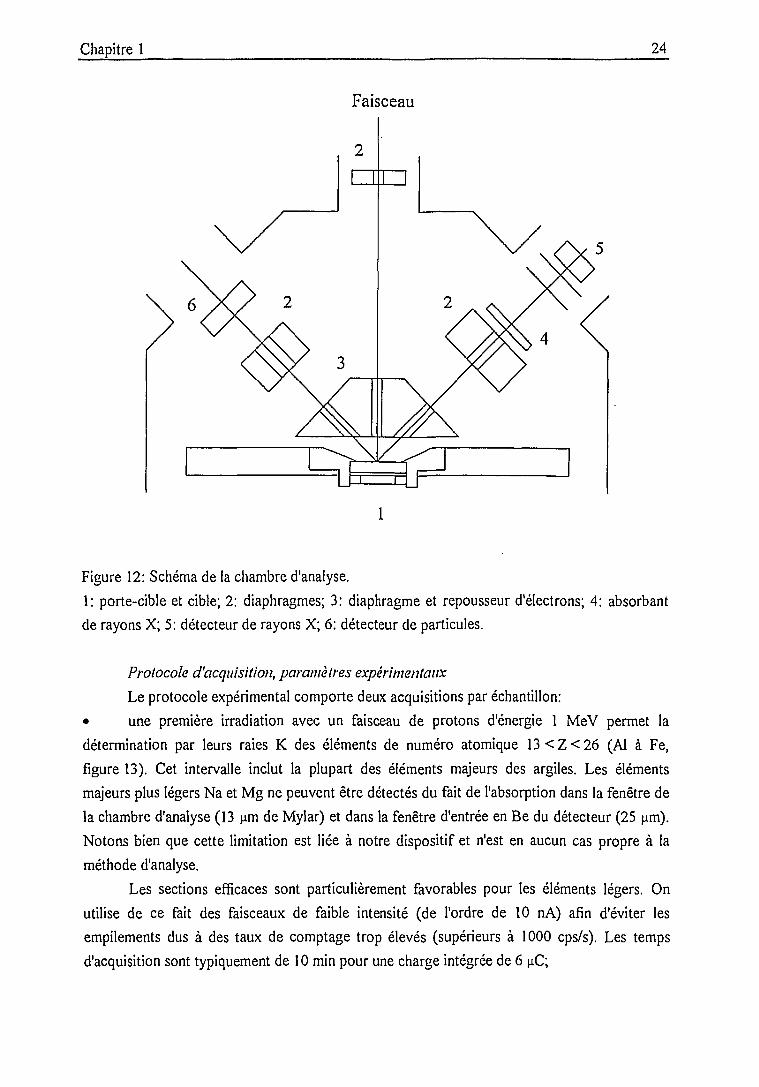
Figure 12: Schéma de la chambre d'analyse.
1: porte-cible et cible; 2: diaphragmes; 3: diaphragme et repousseur d'électrons; 4: absorbant
de rayons X; 5: détecteur de rayons X; 6: détecteur de particules.
Protocole d'acquisition, paramètres expérimentaux
Le protocole expérimental comporte deux acquisitions par échantillon:
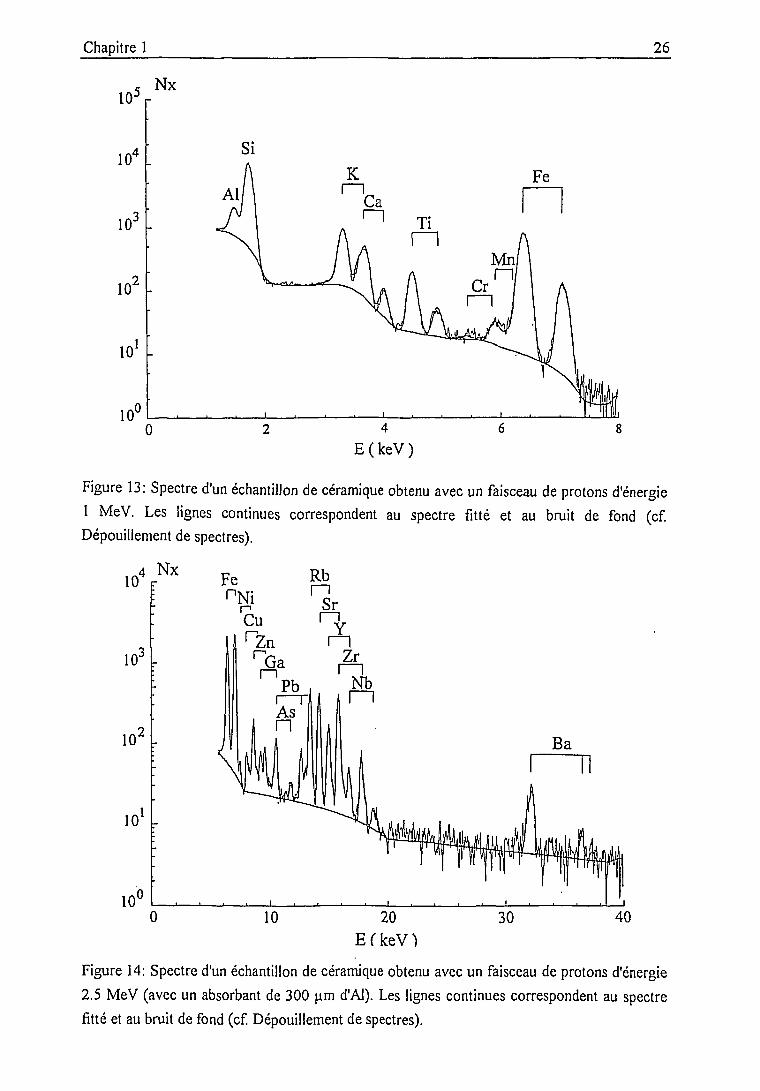
• une première irradiation avec un faisceau de protons d'énergie 1 MeV permet la
détermination par leurs raies K des éléments de numéro atomique 13<Z<26 (Al à Fe,
figure 13). Cet intervalle inclut la plupart des éléments majeurs des argiles. Les éléments
majeurs plus légers Na et Mg ne peuvent être détectés du fait de l'absorption dans la fenêtre de
la chambre d'analyse (13 nm de Mylar) et dans la fenêtre d'entrée en Be du détecteur (25 nm).
Notons bien que cette limitation est liée à notre dispositif et n'est en aucun cas propre à la
méthode d'analyse.
Les sections efficaces sont particulièrement favorables pour les éléments légers. On
utilise de ce fait des faisceaux de faible intensité (de l'ordre de 10 nA) afin d'éviter les
empilements dus à des taux de comptage trop élevés (supérieurs à 1000 cps/s). Les temps
d'acquisition sont typiquement de 10 min pour une charge intégrée de 6 C;

Chapitre 1 25_
• les éléments plus lourds (détection par les raies K de Fe à Ba, figure 14) sont analysés
avec un faisceau de protons d'énergie 2.5 MeV, de forte intensité (~ 200 nA). Des temps
d'acquisition relativement longs (20-30 min, charge intégrée 300-400 pC) sont nécessaires pour
obtenir des statistiques de comptage acceptables sur les raies des éléments les plus lourds qui
sont nettement défavorisés par la chute rapide des sections efficaces avec Z (cf. 1.1.1.1.). Un
filtre de 300|.im d'Al est interposé entre la cible et le détecteur afin de supprimer les rayons X
de faible énergie et à fort taux de comptage produits par les éléments majeurs légers.
Deux échantillons de référence, dont la composition est proche de celle des
échantillons, sont utilisés: une diorite (DR-N) et un basalte (BE-N) fournis par la société
Géostandards de Nancy [54]. On effectue une nouvelle acquisition sur des prélèvement des
standards à chaque série d'analyses ou chaque fois que les conditions expérimentales varient
(instabilités du faisceau,...).
Mesure du courant
Selon l'intensité du faisceau, deux méthodes ont été utilisées:
• à faible courant (10 nA) et faible énergie (1 MeV) de protons, la collection directe des
charges sur la cible pendant l'acquisition - la surface de la cible étant rendue conductrice par
dépôt de carbone (cf. Echantillonnage, Préparation des cibles). Elle nécessite une bonne
isolation du système intégrateur. On préconise en général soit l'intégration sur l'ensemble de la
chambre qui fait office de cage de Faraday, soit l'intégration sur la cible isolée par rapport au
reste de la chambre. Dans ce cas l'emploi d'une contre-tension pour repousser les électrons
secondaires expulsés de la cible qui viendraient à manquer au bilan est nécessaire. Nous
verrons lors des tests de reproductibilité que dans nos conditions expérimentales la mesure
desfaibles courants n'est pas fiable et qu'il est préférable de s'affranchir du paramètre de la
charge (cf. Calcul des concentrations par une méthode "mixte", précision, justesse);
• à fort courant (-200 nA) et forte énergie (2.5 MeV), une détermination indirecte par
des mesures sur cible métallique effectuées avant et après l'acquisition sur l'échantillon. En
effet, la collection directe complète des charges sur des cibles non conductrices n'est plus
assurée. Plusieurs effets contribuent aux pertes de charge et par conséquent à la dispersion des
mesures. Il s'agit en particulier de l'allongement du parcours des protons dans le matériau,
conduisant à la production d'espèces chargées en milieu peu ou pas conducteur loin de la zone
conductrice de carbone, et des dégâts d'irradiation sous fort courant. La méthode indirecte
suppose par contre une relative stabilité du faisceau incident. Notons que les mesures
supplémentaires qu'elle impose la rendent plutôt inadéquate pour les acquisitions rapides à
1 MeV.
Chapitre 1 26
l O 5r
ÎOV
1OJ -
10 -
101 -
Nx
Si
AAl/\
•
•
Ki ii i
Ca
A
ATi 1
Mn/CrM
1—1 1
Fe
AuE ( keV )
Figure 13: Spectre d'un échantillon de céramique obtenu avec un faisceau de protons d'énergie
1 MeV. Les lignes continues correspondent au spectre fitté et au bruit de fond (cf.
Dépouillement de spectres).
10.4 Nx
10'
10'
10 20
E f k e V Ï
30 40
Figure 14: Spectre d'un échantillon de céramique obtenu avec un faisceau de protons d'énergie
2.5 MeV (avec un absorbant de 300 um d'Al). Les lignes continues correspondent au spectre
fitté et au bruit de fond (cf. Dépouillement de spectres).
Chapitre 27
Plusieurs autres méthodes de détermination du courant sont possibles. Citons en
particulier l'approche totalement différente qui consiste à "monitorer" le faisceau par
l'intermédiaire de protons rétrodifrusés sur une feuille mince interposée sur le trajet du faisceau
incident. Le dispositif correspondant sera présenté en annexe dans la description du montage
utilisé en analyse par PIXE en faisceau extrait.
Snectrométrie X. dépouillement de .spectres par le programme PIXAN {subroutine
BATIT)
Dans la gamme des rayons X, les détecteurs les plus couramment utilisés sont des
jonctions en Silicium dopé au Lithium, ou Si(Li). Les détecteurs Ge de haute pureté sont plutôt
réservés à la détection des rayons y. D'autres types de détecteurs, comme les CdTe et les Hgl2
[55], qui fonctionnent à température ambiante, sont encore au stade expérimental.
Une des caractéristiques essentielles d'un détecteur est sa résolution en énergie qui se
traduit par la largeur à mi-hauteur d'une raie de référence. Le Si(Li) employé a une résolution
de 165 à 170 eV pour la raie de 5.9 keV de Mn. Nous définirons dans les paragraphes suivants
son efficacité de détection et sa réponse spectrale, et décrirons leur modélisation dans la
subroutine BATTY de PIXAN [41].
Détermination expérimentale de l'efficacité du délecteur Si(Li)
Une détermination possible consiste à coupler les méthodes PIXE et RBS afin de
s'affranchir de la mesure du courant. On utilise un faisceau de particules a, le facteur
cinématique étant plus favorable que pour les protons. Des cibles minces, si possible
monoélémentaires, sont employées pour éliminer tous les effets liés au ralentissement des
particules et à la transmission des rayons X. Le tableau 1 indique les éléments ou composés
utilisés et les énergies des raies K correspondantes.
cibleénergie (kcV)
Al1.49
Si1.74
Zn2P3
2.01CdS2.31
AgCl2.62
IK3.31
CaF2
3.69
cibleénergie (keV)
Se4.09
Ti4.51
V4.95
Cr5.41
Fe6.40
Ni7.48
Cu8.05
cibleénergie (kcV)
Z112P3
8.64Nb16.62
Ag22.16
In24.21
Sn25.27
Ba32.19
Ce34.72
Tableau 1: Détermination de l'efficacité du détecteur Si(Li): composition des cibles minces
utilisées et énergie de raies K correspondantes; les caractères gras indiquent l'élément considéré
lorsqu'il s'agit d'un composé.

Chapitre 1 28_
Pour une cible mince monoélémentaire, le nombre de rayons X détectés correspondant
à une raie caractéristique est (cf. 1.1.1.1., expression (1)):
où n est le nombre d'atomes par unité de surface de la cible.
De manière analogue, on définit le nombre de particules rétrodiffusées NP par:
NP=7n
da/dQ(0): section efficace différentielle de rétrodiffiision à l'angle de détection 0, en admettant
que cette quantité varie peu sur l'angle solide de détection RBS AQ
sp : efficacité de détection des particules rétrodiffusées; ep = 1 dans le domaine d'énergie
considéré
La position du pic de rétrodiffusion est fixée pour un isotope donné par le rapport
cinématique (cf. 1.1.1.1.).
En faisant le rapport des deux quantités, il vient:
X NP a x Ox
La détection simultanée des rayons X et des particules rétrodiffusées permet ainsi de
s'affranchir de la mesure précise du courant. Les efficacités ont été calculées en utilisant des
sections efficaces ax théoriques de Clayton et Harrigan [19]. Elles servent ensuite à préciser les
paramètres qui décrivent le détecteur dans le programme PIXAN.
Paramètres pour la description théorique du détecteur Si(Li)
On peut décrire l'efficacité de détection de manière élémentaire par:
- la transmission du rayonnement incident par les filtres (fenêtre de sortie en Mylar de la
chambre, filtres additionnels éventuels) et les fenêtres d'entrée du détecteur (fenêtre de Be,
contact électrique en Au, couche morte de Si);
- l'absorption du rayonnement dans le volume sensible du cristal de Si (efficacité intrinsèque):
e = l i - e - ' '»• le filtres
L'efficacité calculée dans la subroutine BATTY de PIXAN selon le modèle proposé par
Hansen [56] et adapté par Cohen [57] considère aussi les paramètres suivants:

Chapitre 1 29_
• un facteur géométrique fg de correction de l'angle solide. Il tient compte du caractère
non ponctuel de la cible et des différences de profondeur d'interaction des photons incidents
dans le détecteur selon leur énergie. Dans notre cas ces effets sont globalement négligeables du
fait de la grande distance entre détecteur et cible (14 cm) imposée par le dispositif
expérimental. La correction apportée ne dépasse pas 1%, sauf pour les rayons X de "haute
énergie" (raies K de 32 keV du Ba) où elle est de l'ordre de 3% [58];
• un facteur de dépendance radiale, qui est parfois modelisé en adjoignant une
composante annulaire à la couche morte de Si. Il n'a pas été pris en compte ici;
• un facteur propre aux filtres percés ("pinhole filters") quelquefois utilisés pour la
détection simultanée des éléments majeurs et traces dans des matériaux dont la matrice est
composée d'éléments légers. La détection des rayons X de faible énergie issus des éléments
majeurs est limitée par l'ouverture du trou, ce qui permet d'atteindre des limites de détection
assez faibles pour des éléments traces plus lourds dont les rayons X ne sont pas absorbés par le
filtre. Cette méthode à priori séduisante est cependant d'application délicate: la fragilité des
filtres courants (Al de quelques 10 a i n c s de |.im d'épaisseur) ne se prête guère à la confection de
"pinhole filters" de rapport surface du trou/surface du détecteur reproductible;
• un facteur tenant compte de filtres hypothétiques supplémentaires correspondant à des
matériaux condensés sur la fenêtre d'entrée du détecteur pendant l'utilisation: hydrocarbures
des pompes à diffusion d'huile, glace. Ces paramètres "incontrôlables" n'ont pas été introduits
dans le calcul.
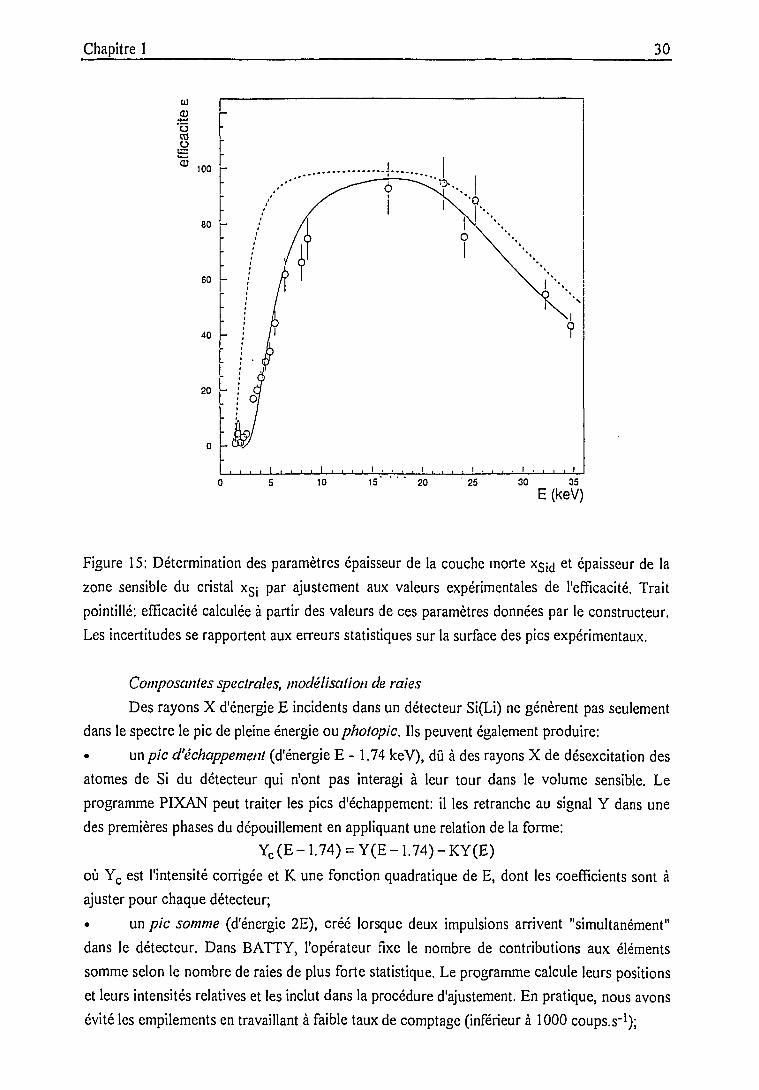
Les paramètres profondeur de la couche morte x^ et épaisseur de la zone sensible du
détecteur xgj, dont les indications données par le constructeur paraissaient les moins fiables ou
les plus sujettes à modifications lors de l'utilisation et des réparations du détecteur, ont été
redéfinis en ajustant la courbe théorique aux efficacités expérimentales (figure 15, annexe 1).
Le fit a été réalisé avec le programme PAW [59].
Il faut noter que la détermination précise de l'efficacité de détection est essentielle pour
le calcul des concentrations par la méthode absolue et par la méthode du standard interne.
Dans le cas de l'utilisation d'un standard externe, elle n'intervient que de façon indirecte au
niveau de la déconvolution des pics dans le spectre de rayons X (cf. Dépouillement de
spectres).
Chapitre 1 30
eu'o.o"S
20 -
o -
20 25 30 35
E (keV)
Figure 15: Détermination des paramètres épaisseur de la couche morte xsid et épaisseur de la
zone sensible du cristal x$j par ajustement aux valeurs expérimentales de l'efficacité. Trait
pointillé: efficacité calculée à partir des valeurs de ces paramètres données par le constructeur.
Les incertitudes se rapportent aux erreurs statistiques sur la surface des pics expérimentaux.
Composantes spectrales, modélisation de raies
Des rayons X d'énergie E incidents dans un détecteur Si(Li) ne génèrent pas seulement
dans le spectre le pic de pleine énergie ou photopic. Ils peuvent également produire:
• un pic d'échappement (d'énergie E - 1.74 keV), dû à des rayons X de désexcitation des
atomes de Si du détecteur qui n'ont pas interagi à leur tour dans le volume sensible. Le
programme PIXAN peut traiter les pics d'échappement: il les retranche au signal Y dans une
des premières phases du dépouillement en appliquant une relation de la forme:
Yc (E - 1.74) = Y(E -1.74) - KY(E)
où Yc est l'intensité corrigée et K une fonction quadratique de E, dont les coefficients sont à
ajuster pour chaque détecteur;
• un pic somme (d'énergie 2E), créé lorsque deux impulsions arrivent "simultanément"
dans le détecteur. Dans BATTY, l'opérateur fixe le nombre de contributions aux éléments
somme selon le nombre de raies de plus forte statistique. Le programme calcule leurs positions
et leurs intensités relatives et les inclut dans la procédure d'ajustement. En pratique, nous avons
évité les empilements en travaillant à faible taux de comptage (inférieur à 1000 coups.s"1);

Chapitre 1 3\_
• des impulsions d'énergie inférieure à E dues à la collection incomplète des porteurs de
charges dans le détecteur: selon le modèle, on considère un photopic gaussien assorti sur son
flan basse énergie d'une exponentielle décroissante ("tailing") et éventuellement d'autres termes
dont un continuum basse énergie ("plateau"). La surface sous ce dernier est souvent assimilée
empiriquement à l'absorption dans la couche morte de Si, ce qui s'accomoderait avec le fait que
le rapport hauteur du plateau / hauteur du photopic varie comme (|Vp)Si avec E. La
modélisation des pics dans PIXAN ne tient pas compte de ce continuum: il est considéré
comme une composante du bruit de fond et l'efficacité se rapporte alors uniquement au
photopic et à son "tailing" basse énergie. Notons que ce modèle est bien adapté pour les
détecteurs de construction récente, mais n'est pas réellement adéquat pour le détecteur utilisé
dans ce travail, qui présente des plateaux marqués.
PIXAN laisse au choix de l'opérateur l'adjonction ou non d'une composante
exponentielle à un pic. Dans l'affirmative, des paramètres supplémentaires sont requis pour
définir la pente et la hauteur maximale de l'exponentielle. Ces paramètres sont soit fixés dans le
programme si leurs valeurs sont connues pour la raie considérée soit "fittés" au cours du
dépouillement. La mauvaise adéquation de la réponse de notre détecteur au modèle ne nous
ayant pas permis une détermination précise, la deuxième possibilité a été adoptée. FA pratique,
seules les raies de plus forte statistique sont concernées: il appartient à l'opérateur de vérifier
que le signal est convenablement réparti entre ses différentes composantes (bruit de fond,
tailing, photopics d'autres raies) et déjuger de la qualité du fit. Cette dernière peut être évaluée
de manière moins subjective par un paramètre dérivé du y} (cf. paragraphe suivant).
Dépouillement de spectres, incertitudes statistiques de comptage, limites de détection
L'approche de BATTY intégre les paramètres physiques fondamentaux dans la
procédure de dépouillement. Le spectre est reconstruit en introduisant pour chaque élément
présent l'ensemble de ses raies; dans chaque série (K, L, M), l'une d'elles sert de référence (en
général la plus intense, ou la mieux isolée) et les autres lui sont rattachées par les rapports
d'intensités théoriques [12, 60] modulés de la fonction d'efficacité et de l'autoabsorption. Pour
les raies L, les valeurs des rapports d'intensité utilisées par PIXAN correspondent à des
protons de 2.5 MeV. Si k composantes contribuent à Intensité de signal Y(E) à l'énergie E:
k
avec:
Aj : intensité de la raie de référence de l'élément j dont la raie k est caractéristique
Rjk : rapport d'intensité de la raie kèmc à la raie de référence
Gjk (E): fonction de forme du pic (cf § précédent)
B(E) : bruit de fond

Chapitre 1 32_
Le calcul des R^ suppose la connaissance de la composition élémentaire de la matrice
du matériau. Dans le cas de céramiques, l'erreur introduite en utilisant les compositions des
standards de référence au lieu de la composition réelle est tout à fait négligeable pour ce qui est
des éléments majeurs. Elle est peu significative compte tenu des fluctuations statistiques de
comptage pour les éléments mineurs et traces.
Pour la détermination du bruit de fond, BATTY propose soit une modélisation
polynomiale, soit un algorithme dont le principe très simple est le suivant:
si pour un canal i, à l'itération n Yi > 0.5 (Yi-i + Yi+i)
alors à l'itération n+1 Yi = 0.5 (Yi-i + Yi+i)
La figure 16 montre l'évolution du fond ainsi calculé avec le nombre d'itérations: les pics sont
progressivement éliminés jusqu'à obtenir un continuum qui suive leur ligne de base. Cette
procédure, qui ne se rattache à aucune notion physique, a l'avantage de pouvoir s'adapter à une
grande variété d'allures de spectres. Elle s'accommode néanmoins moins bien de brusques
discontinuités ("marches"): ceci est typiquement le cas dans les spectres produits par notre
détecteur, où des plateaux précèdent les photopics. Pour les spectres de céramiques, les seules
raies réellement défavorisées par cet effet sont celles des éléments majeurs K et Ca, dont
l'énergie peu supérieure au seuil d'absorption de Si implique un rapport hauteur du plateau /
hauteur du pic élevé. Remarquons que le traitement itératif laisse peu de contrôle à l'opérateur.
Il est parfois utile de subdiviser la région à fitter afin de délimiter des zones où le fond présente
un comportement uniforme.
Une alternative intéressante de traîtement du bruit de fond est utilisée dans le
programme GUPIX [61]: le filtrage digital "top-hat". Il agit sur les composantes de la
transformée de Fourier du signal. Les composantes haute fréquence de fluctuations statistiques
de comptage et basse fréquence de continuum du bruit de fond sont éliminées, isolant les
moyennes fréquences relatives aux photopics.
Classiquement, la qualité du fit est définie par le chi-2 réduit x r2 •
N /-VCXP v \2
2 =±Y--i—__i_L_f. o 2
où N est le nombre de canaux, f le nombre de degrés de liberté, YjCxp et Y. les valeurs
expérimentales et calculées du nombre de coups dans le canal i et a l'incertitude statistique sur
Y. D'après Clayton, qui se base sur les travaux de Sekine et Baba [62] et Isozumi [63], x r2
peut se décomposer en composantes aléatoires et systématiques:
* ? -
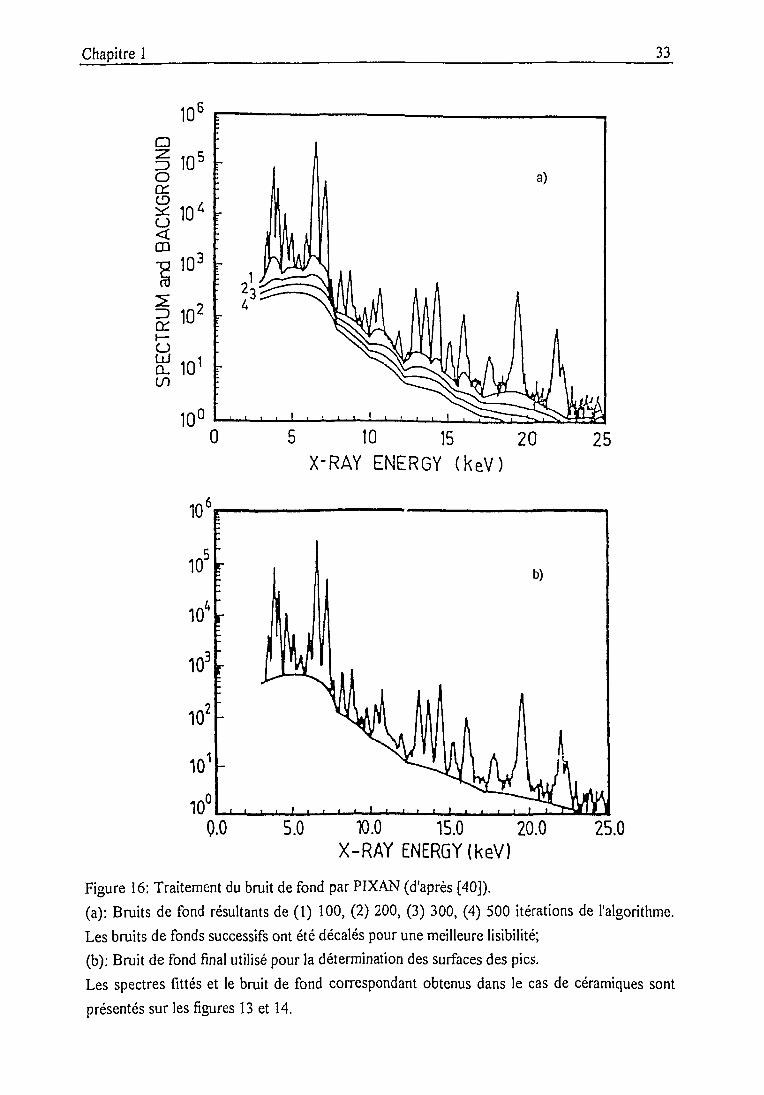
Chapitre 1 33
1Of
Ocm
CKG
<CD
land
^ .ZDC£f—*CJLJLJCLLO
10°
10 4
103
102
101
10(
10 -
1 0 1 -
10'
5 10 15 20X-RAY ENERGY ( keV )
25
5.0 10.0 15.0 20.0X-RAY ENERGY (keV)
25.0
Figure 16: Traitement du bruit de fond par PIXAN (d'après [40]).
(a): Bruits de fond résultants de (1) 100, (2) 200, (3) 300, (4) 500 itérations de l'algorithme.
Les bruits de fonds successifs ont été décalés pour une meilleure lisibilité;
(b): Bruit de fond final utilisé pour la détermination des surfaces des pics.
Les spectres fittés et le bruit de fond correspondant obtenus dans le cas de céramiques sont
présentés sur les figures 13 et 14.
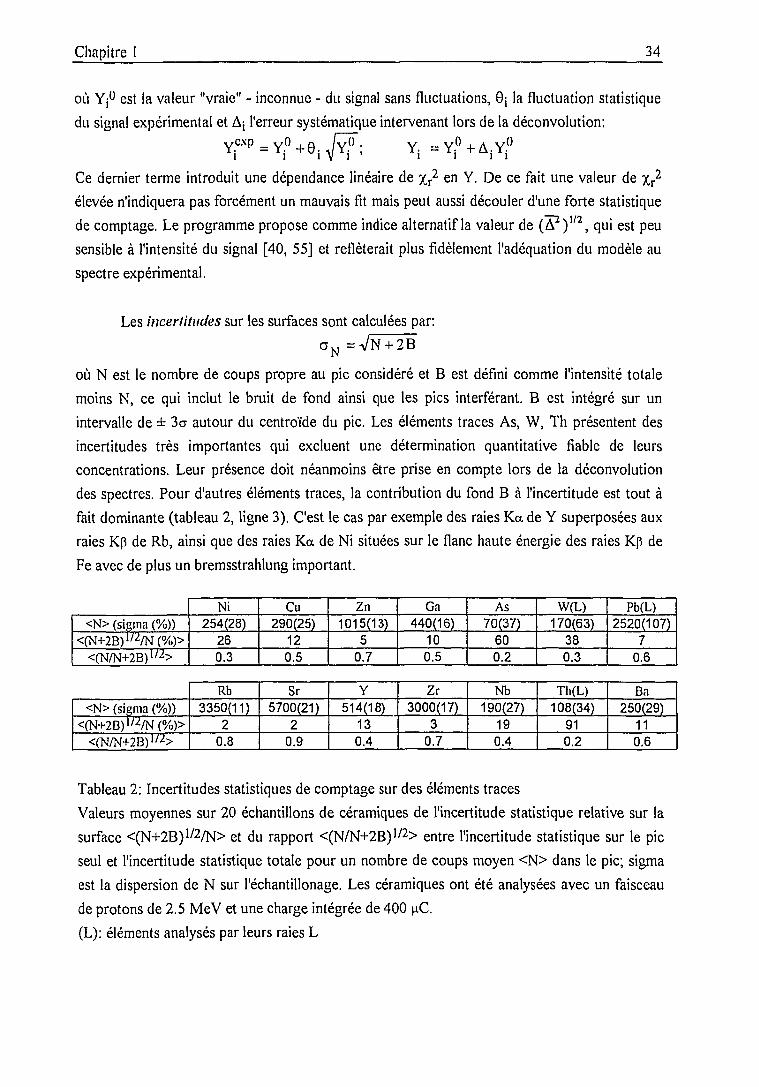
Chapitre 1 34
où Yj° est la valeur "vraie" - inconnue - du signal sans fluctuations, 6; la fluctuation statistique
du signal expérimental et À; l'erreur systématique intervenant lors de la déconvolution:
YCXP — V^ 4-0 / v " • V — V^ 4- A V^I. - ï . +O. yjïi , Ij - I . ï - ^ j ï j
Ce dernier terme introduit une dépendance linéaire de x? en Y. De ce fait une valeur de xr2
élevée n'indiquera pas forcément un mauvais fit mais peut aussi découler d'une forte statistique
de comptage. Le programme propose comme indice alternatif la valeur de (À2)1'2, qui est peu
sensible à l'intensité du signal [40, 55] et refléterait plus fidèlement l'adéquation du modèle au
spectre expérimental.
Les incertitudes sur les surfaces sont calculées par:
a N =VN + 2B
où N est le nombre de coups propre au pic considéré et B est défini comme l'intensité totale
moins N, ce qui inclut le bruit de fond ainsi que les pics interférant. B est intégré sur un
intervalle de ± 3a autour du centroïde du pic. Les éléments traces As, W, Th présentent des
incertitudes très importantes qui excluent une détermination quantitative fiable de leurs
concentrations. Leur présence doit néanmoins être prise en compte lors de la déconvolution
des spectres. Pour d'autres éléments traces, la contribution du fond B à l'incertitude est tout à
fait dominante (tableau 2, ligne 3). C'est le cas par exemple des raies Ka de Y superposées aux
raies Kp de Rb, ainsi que des raies Ka de Ni situées sur le flanc haute énergie des raies Kp de
Fe avec de plus un bremsstrahlung important.
<N> (sigma (%))<(N+2By/2/N (%)>
<(N/N+2B)l/4>
<N> (siema (%))<(N+2Br/'VN (%)>
<(N/N+2B)'^>
Ni254(28)
260.3
Rb3350(11)
20.8
Cu290(25)
120.5
Sr5700(21)
20.9
Zn1015(13)
50.7
Y514(18)
130.4
Ga440(16)
100.5
Zr3000(17)
30.7
As70(37)
600.2
Nb190(27)
190.4
W(L)170(63)
380.3
Th(L)108(34)
910.2
Pb(L)2520(107)
70.6
Ba250(29)
110.6
Tableau 2: Incertitudes statistiques de comptage sur des éléments traces
Valeurs moyennes sur 20 échantillons de céramiques de l'incertitude statistique relative sur la
surface <(N+2B)1/2/N> et du rapport <(N/N+2B)1/2> entre l'incertitude statistique sur le pic
seul et l'incertitude statistique totale pour un nombre de coups moyen <N> dans le pic; sigma
est la dispersion de N sur l'échantillonage. Les céramiques ont été analysées avec un faisceau
de protons de 2.5 MeV et une charge intégrée de 400 nC.
(L): éléments analysés par leurs raies L

Chapitre 35
De ce fait, il est difficile de prévoir "théoriquement" les conditions expérimentales qui
produisent des incertitudes statistiques acceptables bien que l'on puisse parfaitement calculer
des rendements à partir des paramètres fondamentaux (cf. Calcul des rendements par PIXAN
(subroutine THICK) ).
Si certains éléments comme As, W (pollution de la mèche), Th et dans la plupart des
cas Pb sont en concentration inférieure aux limites de détection, l'incertitude statistique conduit
à des limitations importantes de la précision de l'analyse, et ce d'autant plus que l'on utilise une
détermination par standard externe.
Les limites de détection Idd sont définies dans BATTY par référence à Currie [64]
comme:
idd =
A titre d'exemple, le tableau 3 donne les limites de détection moyenne de quelques éléments
traces dans les conditions usuelles d'analyse des céramiques. En général, ces limites dépendent
étroitement de la composition de l'échantillon. Néanmoins pour des céramiques dont les
éléments majeurs ne sont pas plus lourds que Fe et qui ont des teneurs variant dans un
intervalle restreint, les limites de détection pour les éléments traces présentées dans le tableau 3
peuvent être considérées comme représentatives (voir aussi tableau 7). On notera cependant la
valeur moyenne inhabituellement élevée de la concentration en Pb: elle est due à quelques
valeurs très élevées, point sur lequel nous reviendrons en 2.4.1.1. Sur un plan d'ensemble, deux
tendances principales sont en compétition: à haute énergie, la chute des sections efficaces avec
Z (cf. 1.1.1.1., figures 4 et 5) et la baisse d'efficacité sont défavorables mais le bruit de fond est
quasiment absent (cas de Ba), alors qu'à basse énergie le bremsstrahlung et la composante
"plateau" des pics sont importants.
<C> (sigma (%))<ld>
<C> (sigma (%))<ld>
Ni116(28)
64
Rb167(13)
4
Cu61(27)
15
Sr270(20)
3
Zn147(15)
12
Y42(17)
12
Ga27(17)
5
Zr420(18)
20
Asnd7
Nb25(28)
9
W(L)57(75)
32
Th(L)12(35)
18
Pb(L)476(104)
26
Ba802(37)
153
Tableau 3: Limites de détection d'éléments traces (en ppm)
Valeurs moyennes <ld> sur 20 échantillons de céramiques, la concentration moyenne calculée
étant <C> avec une dispersion sigma. Les céramiques ont été analysées avec un faisceau de
protons de 2.5 MeV et une charge intégrée de 400 j C.
(L): éléments analysés par leurs raies L; nd: non déterminé

Chapitre 1 36_
Détermination des concentrai ions élémentaires
Calcul des rendements par PIXAN (subroutine THICK)
Nous avons vu en 1.1.1.2. que l'intensité du signal due à un élément en concentration C
peut s'exprimer sous la forme:N = Q C K I
où Q est la charge de protons incidents d'énergie EQ, K dépend uniquement de l'élément
considéré, de l'énergie de sa raie caractéristique et de la géométrie de détection et I est défini
par:0
JEo
• = H d E
Le rendement Kl calculé par THICK correspond à l'intensité par unité de charge et de
concentration. Les paramètres fondamentaux, sections efficaces o, pouvoir d'arrêt S,
coefficients d'absorption massiques intervenant dans la transmission T sont tirés des références
[19,11, 12], [18], [65, 14].
En analyse par standard externe, le calcul des concentrations se résume à:
r-r ** N
s l N s l I
Les avantages de cette méthode ont été discutés en 1.1.1.2. (Standard externe).
La difficulté consiste à évaluer l'intégrale de correction de matrice I, qui dépend de la
composition - inconnue - de l'échantillon. En choisissant un standard de composition similaire à
celle des argiles étudiées, on peut estimer en première approximation que le rapport des termes
de matrice est 1. Les concentrations obtenues sont utilisées pour calculer I au 1er ordre, et à en
déduire une nouvelle valeur de C. Une itération est en général suffisante dans ces conditions.
Le standard ayant la composition la plus proche, DR-N, sert de référence pour le
dosage des éléments majeurs déterminés avec un faisceau de protons de 1 MeV. Le standard
BE-N joue le rôle d'échantillon de contrôle, ce qui permet d'estimer la qualité de l'analyse (cf.
paragraphe suivant). A 2.5 MeV, il faut utiliser les deux standards "simultanément" pour
couvrir l'ensemble des éléments analysés (concentrations trop faibles ou non certifiées).
Plusieurs spectres obtenus dans les même conditions sont alors sommés afin de minimiser
l'incertitude statistique sur les pics des éléments traces tout en évitant les acquisitions trop
longues (risques d'instabilité du faisceau et de dégâts d'irradiation).
Calcul des concentrations par une méthode "mixte", précision, justesse
Comme il est d'usage lors de test d'une nouvelle procédure expérimentale ou d'un
nouveau protocole, nous avons cherché à évaluer la précision et la justesse des résultats

Chapitre 1 37
obtenus. La précision ou reproductibilité est la première exigence à satisfaire: c'est la condition
nécessaire pour que l'ensemble des mesures réalisées soit comparables entre elles. Nous avons
porté une attention particulière à la reproductibilité à long terme des analyses, rarement
présentée dans la "littérature" [66], et qui est pourtant essentielle pour la constitution de bases
de données. La justesse est importante dès lors que l'échange des résultats entre laboratoires
est envisagé: il y a alors lieu en général de faire une série d'inter-comparaisons sur des
standards communs [67, 68, 69] (cf. 1.3.).
Précision
Une approche analytique rigoureuse de la question des incertitudes est malaisée en
PIXE. Une estimation des erreurs commises sur les différents facteurs [27] permet d'obtenir
des ordres de grandeurs [70]. Cependant, cette estimation peut paraître arbitraire pour certains
paramètres, en particulier la charge intégrée. Nous avons préféré apprécier l'incertitude à partir
de mesures de reproductibilité, qui intègrent l'ensemble des facteurs enjeu. La contribution des
fluctuations statistiques de comptage peut être évaluée à partir du tableau 2.
La précision sur une quantité donnée est exprimée comme le rapport (en %) de l'écart
type à la valeur moyenne de plusieurs mesures effectuées sur un même échantillon à plusieurs
mois d'intervalle (reproductibilité à long terme). Les résultats obtenus pour les éléments
majeurs et mineurs légers analysés avec des protons de 1 MeV et pour les éléments traces
dosés à 2.5 MeV sont présentés dans les tableaux 4 et 5 respectivement.
A 1 MeV, nous avons évalué la précision que l'on peut obtenir selon les trois modes
"classiques" de détermination des concentrations élémentaires: calcul absolu, méthodes du
standard externe et du standard interne, ainsi qu'avec une méthode "mixte" qui a été
développée.
o/m (S/Q)al m (S/(S),si)al m (C)aim (Ccorr)
Al13
593
Si90
101
K16102011
Ca12
511
1
Ti10
512
5
Mn86
1113
Fc83
102
Tableau 4: Précision obtenue pour les éléments majeurs et mineurs (rapports (en %) de l'écart-
type a à la valeur moyenne m) sur 13 analyses du standard BE-N avec un faisceau de protons
d'énergie 1 MeV.
S/Q: surface S de la raie caractéristique (en général Ka) d'un élément donné pour une charge
intégrée Q constante; S / (S)s;: rapport de S à la surface du pic de Si; C: concentration
calculée par la méthode du standard externe; Ccorr: concentration obtenue par normalisation
de C à une composition de 100% d'oxides (méthode "mixte").
Dans le cas du calcul absolu, on considère la dispersion des rapports de la surface S des
pics caractéristiques à la charge Q de protons (tableau 4, ligne 1). Cette quantité, directement
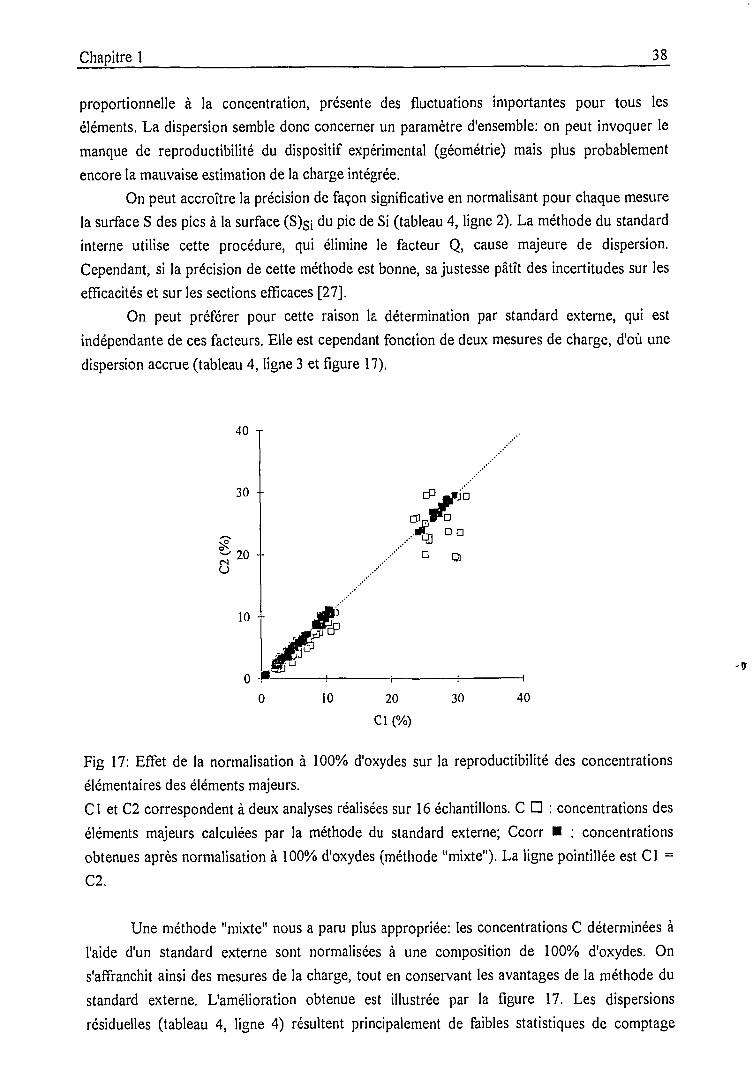
Chapitre 1 38
proportionnelle à la concentration, présente des fluctuations importantes pour tous les
éléments. La dispersion semble donc concerner un paramètre d'ensemble: on peut invoquer le
manque de reproductibilité du dispositif expérimental (géométrie) mais plus probablement
encore la mauvaise estimation de la charge intégrée.
On peut accroître la précision de façon significative en normalisant pour chaque mesure
la surface S des pics à la surface (S)gi du pic de Si (tableau 4, ligne 2). La méthode du standard
interne utilise cette procédure, qui élimine le facteur Q, cause majeure de dispersion.
Cependant, si la précision de cette méthode est bonne, sa justesse pâtît des incertitudes sur les
efficacités et sur les sections efficaces [27],
On peut préférer pour cette raison la détermination par standard externe, qui est
indépendante de ces facteurs. Elle est cependant fonction de deux mesures de charge, d'où une
dispersion accrue (tableau 4, ligne 3 et figure 17).
40 T
esU
30 1
20 +
10 +
cP
Q
10 20
Cl (%)
30 40
Fig 17: Effet de la normalisation à 100% d'oxydes sur la reproductibilité des concentrations
élémentaires des éléments majeurs.
Cl et C2 correspondent à deux analyses réalisées sur 16 échantillons. C • : concentrations des
éléments majeurs calculées par la méthode du standard externe; Ccorr • : concentrations
obtenues après normalisation à 100% d'oxydes (méthode "mixte"). La ligne pointillée est Cl =
C2.
Une méthode "mixte" nous a paru plus appropriée: les concentrations C déterminées à
l'aide d'un standard externe sont normalisées à une composition de 100% d'oxydes. On
s'affranchit ainsi des mesures de la charge, tout en conservant les avantages de la méthode du
standard externe. L'amélioration obtenue est illustrée par la figure 17. Les dispersions
résiduelles (tableau 4, ligne 4) résultent principalement de faibles statistiques de comptage
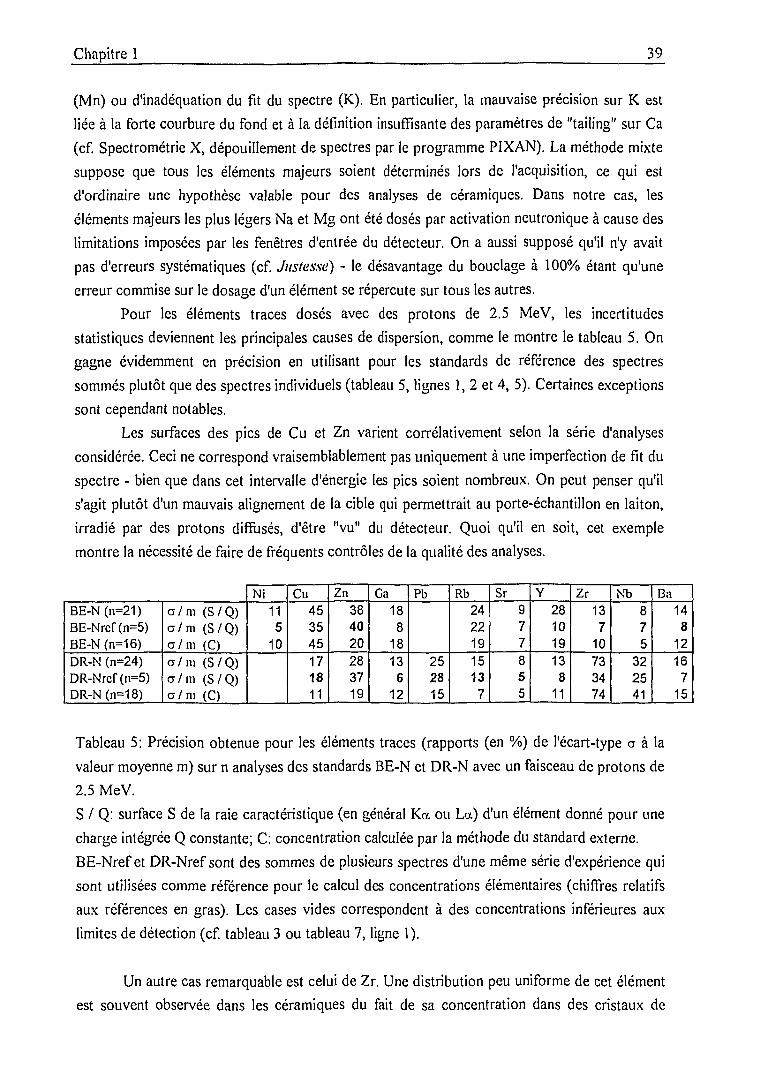
Chapitre 1 39
(Mn) ou d'inadéquation du fit du spectre (K). En particulier, la mauvaise précision sur K est
liée à la forte courbure du fond et à la définition insuffisante des paramètres de "tailing" sur Ca
(cf. Spectrométrie X, dépouillement de spectres par le programme PIXAN). La méthode mixte
suppose que tous les éléments majeurs soient déterminés lors de l'acquisition, ce qui est
d'ordinaire une hypothèse valable pour des analyses de céramiques. Dans notre cas, les
éléments majeurs les plus légers Na et Mg ont été dosés par activation neutronique à cause des
limitations imposées par les fenêtres d'entrée du détecteur. On a aussi supposé qu'il n'y avait
pas d'erreurs systématiques (cf. Justesse) - le désavantage du bouclage à 100% étant qu'une
erreur commise sur le dosage d'un élément se répercute sur tous les autres.
Pour les éléments traces dosés avec des protons de 2.5 MeV, les incertitudes
statistiques deviennent les principales causes de dispersion, comme le montre le tableau 5. On
gagne évidemment en précision en utilisant pour les standards de référence des spectres
sommés plutôt que des spectres individuels (tableau 5, lignes 1, 2 et 4, 5). Certaines exceptions
sont cependant notables.
Les surfaces des pics de Cu et Zn varient corrélativement selon la série d'analyses
considérée. Ceci ne correspond vraisemblablement pas uniquement à une imperfection de fit du
spectre - bien que dans cet intervalle d'énergie les pics soient nombreux. On peut penser qu'il
s'agit plutôt d'un mauvais alignement de la cible qui permettrait au porte-échantillon en laiton,
irradié par des protons diffusés, d'être "vu" du détecteur. Quoi qu'il en soit, cet exemple
montre la nécessité de faire de fréquents contrôles de la qualité des analyses.
BE-N(n=21)BE-Nrcf(n=5)BE-N(n=16)
DR-N (n=24)DR-Nrcf(n=5)DR-N(n=18)
g/m (S/Q)g/m (S/Q)g/m (C)
g/m (S/Q)g/m (S/Q)g/m (C)
Ni11510
Cu453545
171811
Zn384020
283719
Ga18818
13612
Pb
252815
Rb242219
15137
Sr977855
Y281019
13811
Zr13710
733474
Nb875
322541
Ba1481216715
Tableau 5: Précision obtenue pour les éléments traces (rapports (en %) de l'écart-type g à la
valeur moyenne m) sur n analyses des standards BE-N et DR-N avec un faisceau de protons de
2.5 MeV.
S /Q: surface S de la raie caractéristique (en général Ko. ou La) d'un élément donné pour une
charge intégrée Q constante; C: concentration calculée par la méthode du standard externe.
BE-Nref et DR-Nref sont des sommes de plusieurs spectres d'une même série d'expérience qui
sont utilisées comme référence pour le calcul des concentrations élémentaires (chiffres relatifs
aux références en gras). Les cases vides correspondent à des concentrations inférieures aux
limites de détection (cf. tableau 3 ou tableau 7, ligne 1).
Un autre cas remarquable est celui de Zr. Une distribution peu uniforme de cet élément
est souvent observée dans les céramiques du fait de sa concentration dans des cristaux de
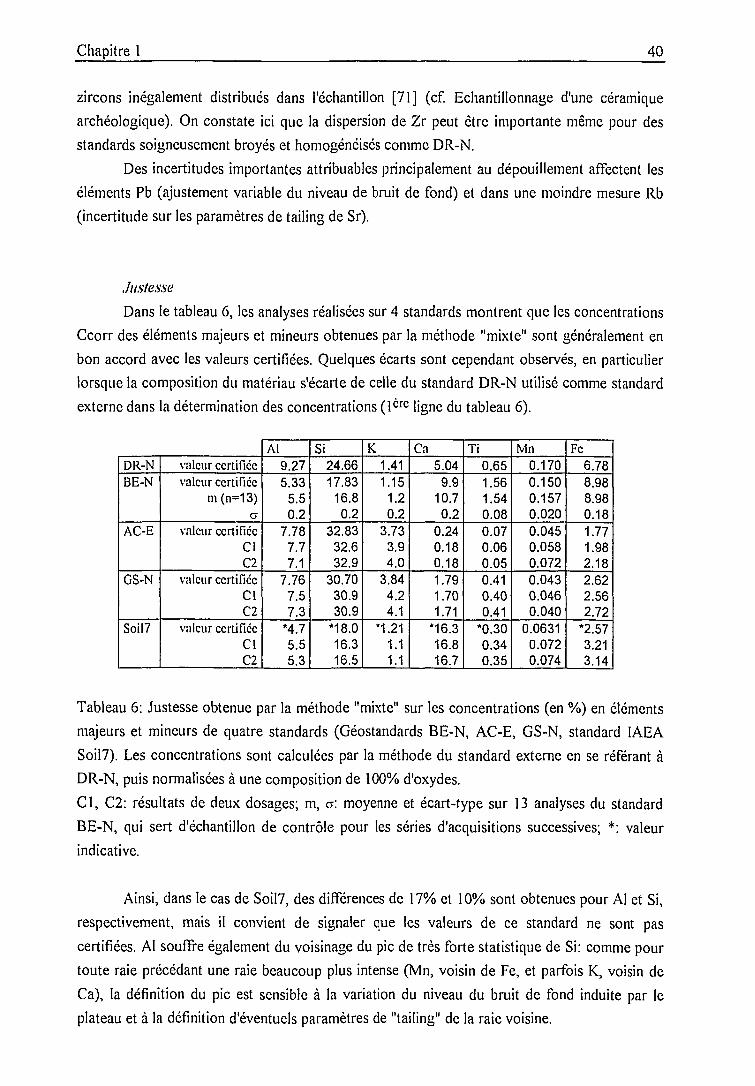
Chapitre 1 40
zircons inégalement distribués dans l'échantillon [71] (cf. Echantillonnage d'une céramique
archéologique). On constate ici que la dispersion de Zr peut être importante même pour des
standards soigneusement broyés et homogénéisés comme DR-N.
Des incertitudes importantes attribuables principalement au dépouillement affectent les
éléments Pb (ajustement variable du niveau de bruit de fond) et dans une moindre mesure Rb
(incertitude sur les paramètres de tailing de Sr).
Justesse
Dans le tableau 6, les analyses réalisées sur 4 standards montrent que les concentrations
Ccorr des éléments majeurs et mineurs obtenues par la méthode "mixte" sont généralement en
bon accord avec les valeurs certifiées. Quelques écarts sont cependant observés, en particulier
lorsque la composition du matériau s'écarte de celle du standard DR-N utilisé comme standard
externe dans la détermination des concentrations (lc r c ligne du tableau 6).
DR-NBE-N
AC-E
GS-N
Soil7
valeur certifiéevaleur certifiée
m(n=13)a
valeur certifiéeClC2
valeur certifiéeClC2
valeur certifiéeClC2
Al9.275.33
5.50.2
7.787.77.1
7.767.57.3
*4.75.55.3
Si24.6617.83
16.80.2
32.8332.632.9
30.7030.930.9
*18.016.316.5
K1.411.15
1.20.2
3.733.94.0
3.844.24.1
*1.211.11.1
Ca5.04
9.910.70.2
0.240.180.181.791.701.71
•16.316.816.7
Ti0.651.561.540.080.070.060.050.410.400.41
*0.300.340.35
Mn0.1700.1500.1570.0200.0450.0580.0720.0430.0460.040
0.06310.0720.074
Fc6.788.988.980.181.771.982.182.622.562.72
•2.573.213.14
Tableau 6: Justesse obtenue par la méthode "mixte" sur les concentrations (en %) en éléments
majeurs et mineurs de quatre standards (Géostandards BE-N, AC-E, GS-N, standard IAEA
Soil7). Les concentrations sont calculées par la méthode du standard externe en se référant à
DR-N, puis normalisées à une composition de 100% d'oxydes.
Cl, C2: résultats de deux dosages; m, a: moyenne et écart-type sur 13 analyses du standard
BE-N, qui sert d'échantillon de contrôle pour les séries d'acquisitions successives; *: valeur
indicative.
Ainsi, dans le cas de SoiI7, des différences de 17% et 10% sont obtenues pour Al et Si,
respectivement, mais il convient de signaler que les valeurs de ce standard ne sont pas
certifiées. Al souffre également du voisinage du pic de très forte statistique de Si: comme pour
toute raie précédant une raie beaucoup plus intense (Mn, voisin de Fe, et parfois K, voisin de
Ca), la définition du pic est sensible à la variation du niveau du bruit de fond induite par le
plateau et à la définition d'éventuels paramètres de "tailing" de la raie voisine.
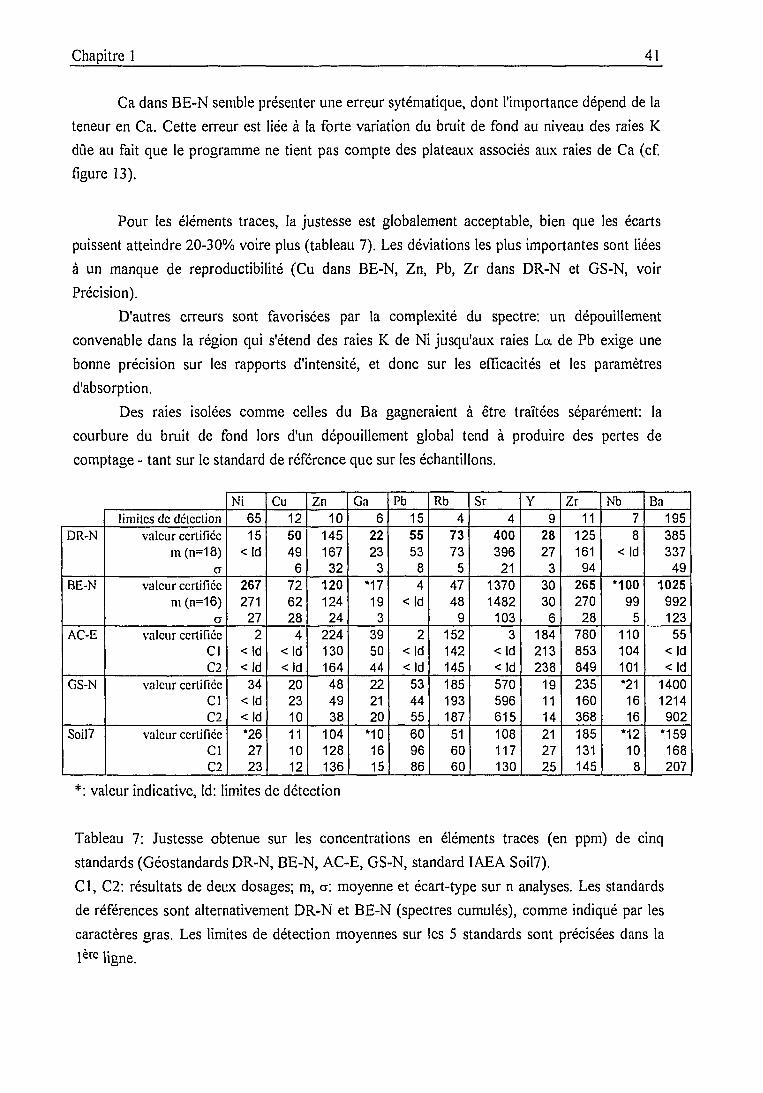
Chapitre 1 41
Ca dans BE-N semble présenter une erreur sytématique, dont l'importance dépend de la
teneur en Ca. Cette erreur est liée à la forte variation du bruit de fond au niveau des raies K
due au fait que le programme ne tient pas compte des plateaux associés aux raies de Ca (cf.
figure 13).
Pour les éléments traces, la justesse est globalement acceptable, bien que les écarts
puissent atteindre 20-30% voire plus (tableau 7). Les déviations les plus importantes sont liées
à un manque de reproductibilité (Cu dans BE-N, Zn, Pb, Zr dans DR-N et GS-N, voir
Précision).
D'autres erreurs sont favorisées par la complexité du spectre: un dépouillement
convenable dans la région qui s'étend des raies K de Ni jusqu'aux raies La de Pb exige une
bonne précision sur les rapports d'intensité, et donc sur les efficacités et les paramètres
d'absorption.
Des raies isolées comme celles du Ba gagneraient à être traitées séparément: la
courbure du bruit de fond lors d'un dépouillement global tend à produire des pertes de
comptage - tant sur le standard de référence que sur les échantillons.
DR-N
BE-N
AC-E
GS-N
Soil7
limites de détectionvaleur certifiée
m(n=18)
<7
valeur certifiéem(n=16)
avaleur certifiée
ClC2
valeur certifiéeClC2
valeur certifiéeClC2
Ni6515
<ld
26727127
2<ld<ld
34<ld<ld*262723
Cu125049
67262284
<ld<ld202310111012
Zn10
14516732
12012424
224130164484938
104128136
Ga6
2223
3*17193
395044222120
*101615
Pb155553
84
<ld
2<ld<ld
534455609686
Rb4
7373
54748
9152142145185193187516060
Sr4
40039621
13701482
1033
<ld<ld570596615108117130
Y9
2827
330306
184213238
191114212725
Zr11
12516194
265270
28780853849235160368185131145
Nb78
<ld
*10099
5110104101*211616
*12108
Ba19538533749
102599212355
<ld<ld
14001214902
*159168207
*: valeur indicative, ld: limites de détection
Tableau 7: Justesse obtenue sur les concentrations en éléments traces (en ppm) de cinq
standards (Géostandards DR-N, BE-N, AC-E, GS-N, standard IAEA Soil7).
Cl, C2: résultats de deux dosages; m, a: moyenne et écart-type sur n analyses. Les standards
de références sont alternativement DR-N et BE-N (spectres cumulés), comme indiqué par les
caractères gras. Les limites de détection moyennes sur les 5 standards sont précisées dans la
lèrc ligne.

Chapitre 1 42
1.2. Analyse élémentaire par activation neutronique
L'activation neutronique est la méthode d'analyse des éléments traces par excellence.
Selon les sections efficaces de capture neutronique des isotopes présents dans un échantillon,
elle peut présenter d'excellentes sensibilités (ppm, voire ppb). Contrairement aux méthodes
atomiques, les sections efficaces n'évoluent pas de manière continue avec Z. Il en résulte que
les sensibilités sont variables d'un élément à l'autre.
Par ailleurs, la méthode repose sur des mesures de radioactivité pour lesquelles les
paramètres temporels jouent un rôle primordial. Un choix adéquat de ces paramètres permet
d'optimiser les conditions d'analyse pour chaque élément.
Enfin, l'échantillon une fois "activé", l'analyse devient indépendante des opérations
ultérieures et en particulier de la pollution. En chimie, ceci permet l'addition d'entraîneurs des
éléments présents en très faibles concentrations. Nous avons utilisé pour la présente étude la
technique dite INAA (Instrumental Neutron Activation Analysis), où les mesures succèdent
aux irradiations sans manipulations chimiques intermédiaires.
L'analyse par activation neutronique connaît de nombreuses applications biomédicales,
environnementales, industrielles, ... [72]. Son utilisation en archéométrie a connu un très fort
développement au cours des années 1970 [73, 74] et la grande quantité de travaux publiés dans
le domaine attestent de son succès [75].
1.2.1. Principe [76]
Production de radioéléments par capture neutronique, schémas de décroissance
L'activation neutronique consiste à générer, par capture neutronique, des radionucléides
dont la désintégration s'accompagne de l'émission de rayons gamma caractéristiques. La
détection de ces rayonnements permet l'identification et la détermination quantitative des
éléments présents.
L'activation neutronique d'un isotope ^X de l'élément X et la désintégration
consécutive du radioisotope formé se fait, dans le cas d'une désintégration P", selon le schéma
suivant:
*+|j +v > z + , Y + y 2 (4)
L'absorption d'un neutron conduit à la formation d'un noyau composé qui se désexcite
en un temps très bref en émettant des rayons gamma "prompts" (Yi). Le noyau résiduel est en
général radioactif et se désintègre le plus souvent par émission P" avec une période T. Cette

Chapitre 1 43_
désintégration conduit à des niveaux excités de l'isotope ' ^ Y de l'élément Y, La désexcitation
se fait par émission de rayons y symbolisés par yj- La détection du rayonnement 72 permet le
dosage de l'élément X. Le noyau '^!,Y peut être stable ou radioactif.
D'autres types de réactions nucléaires et d'autres modes de désintégration sont
également possibles. En particulier, des réactions nucléaires de type (n,p) et (n,a) peuvent
conduire concurremment à la formation d'un même radionucléide que celui généré par une
réaction (n,y). Ainsi, par exemple, le noyau 2 7 Mg peut résulter de trois réactions différentes:2 6 Mg + n 2 7
2 7Al + n >
30Si + n >27Mg
Ces réactions ne sont pas équiprobables et sont caractérisées par des sections efficaces
dépendant de l'énergie des neutrons incidents. Pour des neutrons thermiques, d'énergie 0.025
eV, les réactions conduisant à l'émission de particules chargées (p, a) sont peu probables en
raison de la barrière coulombienne. Les neutrons épithermiques (d'énergie 0.5 eV à 1 MeV) ou
rapides leur sont par contre beaucoup plus favorables. Les sections efficaces présentent dans
ces domaines d'énergie un comportement de type résonnant.
La décroissance du radioisotope formé par émission p+, capture électronique ou
émission a sont plus rares. Le noyau fils Y peut être lui même radioactif comme c'est le cas des
produits de décroissance de U et Th. Les schémas simplifiés correspondants sont les suivants:238u + n >239U 239N >239pu
P~ P"2 3 2Th + n >233Th
Remarquons que 2^Pu et 233TJ soni également instables, mais que leurs longues périodes
(>104 ans) leur donnent une activité spécifique faible par rapport à celles qui sont considérées
en activation neutronique.
Enfin, les noyaux composés de Z élevé sont susceptibles de se désintégrer par fission en
conduisant à la production d'isotopes plus légers. Ces isotopes peuvent être identiques à ceux
résultant de la capture neutronique d'isotopes stables présents au départ dans l'échantillon.
C'est le cas en particulier pour 235U dont la fraction isotopique est de 0.7 % dans l'uranium
naturel.
Equations fondamentales
Considérons le cas le plus courant d'une réaction correspondant au schéma complet
donné par (4):
L'échantillon est soumis à l'irradiation pendant un temps tj, puis laissé au repos durant
un temps tr après lequel les rayons gamma émis sont détectés pendant une durée tc.
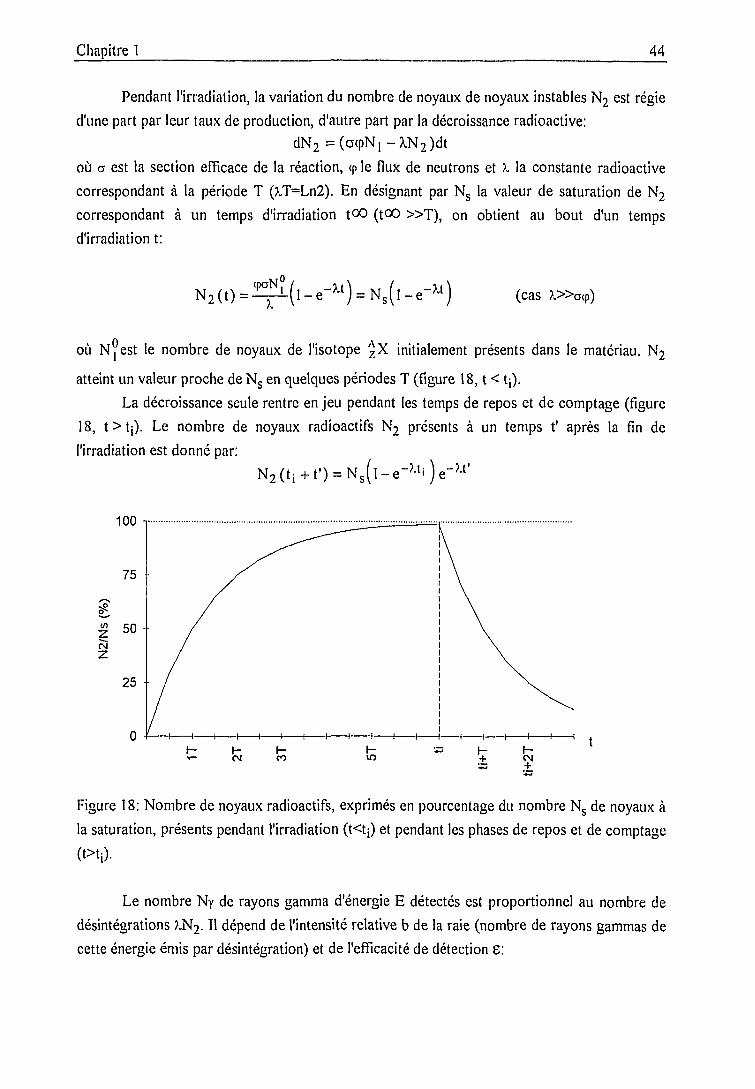
Chapitre 1 44
Pendant l'irradiation, la variation du nombre de noyaux de noyaux instables N2 est régie
d'une part par leur taux de production, d'autre part par la décroissance radioactive:
dN2 =(o<pNi-A.N2)dt
où a est la section efficace de la réaction, <p le flux de neutrons et 7. la constante radioactive
correspondant à la période T (?wT=Ln2). En désignant par Ns la valeur de saturation de N2
correspondant à un temps d'irradiation too (too » T ) , on obtient au bout d'un temps
d'irradiation t:
(cas
où Njest le nombre de noyaux de l'isotope ^X initialement présents dans le matériau. N2
atteint un valeur proche de Ns en quelques périodes T (figure 18, t < tj).
La décroissance seule rentre en jeu pendant les temps de repos et de comptage (figure
18, t>tj). Le nombre de noyaux radioactifs N2 présents à un temps t' après la fin de
l'irradiation est donné par:
100 T
CM
Figure 18: Nombre de noyaux radioactifs, exprimés en pourcentage du nombre Ns de noyaux à
la saturation, présents pendant l'irradiation (t<t,) et pendant les phases de repos et de comptage
Le nombre Ny de rayons gamma d'énergie E détectés est proportionnel au nombre de
désintégrations ?.N2. Il dépend de l'intensité relative b de la raie (nombre de rayons gammas de
cette énergie émis par désintégration) et de l'efficacité de détection 8:

Chapitre 1 45_
le
Ny = e b f\N2dt'o
Ny = e b N s( l - Q~U' ) e~?-1' (1 - e~?ac )
En introduisant les concentrations élémentaires, on obtient l'expression générale:
N =8bCxI^-^(l-e-^)e-^(l-e-^) (5)
avec:
Cx : concentration pondérale de l'élément X de masse atomique Ax
r : rapport isotopique de l'isotope activé '?X
Na : nombre d'Avogadro
m : masse de l'échantillon
Le choix de l'isotope qui permettra le dosage d'un élément donné se fera donc à partir
des paramètres fondamentaux a, r, b et \, en tenant compte des interférences possibles de
rayonnements y émis par d'autres éléments activés simultanément. L'expérimentateur pourra
définir les conditions de l'irradiation et de mesure (tj, tr, tc) afin de favoriser la détermination
d'un élément particulier ou de limiter voire d'éliminer les difficultés résultant de la présence d'un
autre.
En ce qui concerne les argiles, la méthode a une très bonne sensibilité pour la détection
de nombreux éléments trace dans plusieurs régions du tableau périodique: éléments de la
première série de transition, terres rares, ... La détection des éléments majeurs n'est par contre
pas favorisée lors de l'activation par des neutrons thermiques. Pour Si, dosable via la réaction30Si(n,y)31Si, les paramètres sont: r = 3.1 %, a = 0.11 et l'intensité relative b de la seule raie y
1266.2 keV utilisable pour la mesure n'est que de 0.07 % (Si peut cependant être détecté au
moyen d'une seconde réaction avec des neutrons épithermiques [77, 78]). Al, dont l'isotope
unique 27A1 produit par capture neutronique le noyau 28A1 de courte période (2.31 minutes),
n'est pas accessible aux analystes qui ne disposent pas d'une chaîne de spectrométrie auprès des
réacteurs. Le caractère défavorable des paramètres fondamentaux dans le cas des éléments
majeurs peut être conçu comme un avantage, dans la mesure où leurs raies ne dominent pas le
spectre. Il constitue néanmoins une limitation de la méthode pour ce qui est de la connaissance
du matériau.
Analyse quantitative en standard externe, sources d'erreurs
De la même manière qu'en PIXE, plusieurs approches sont possibles pour la
détermination des concentrations élémentaires: le calcul "absolu", qui nécessite la connaissance
de tous les paramètres en jeu, notamment le flux 9 de neutrons, ou le calcul relatif à un
standard, interne ou externe.

Chapitre 1 4£
Nous avons choisi d'utiliser la méthode de détermination par standard externe, qui
élimine plusieurs facteurs multiplicatifs dans (5). La concentration élémentaire Cx d'un élément
X dans l'échantillon s'exprime en fonction de sa concentration C'x dans l'étalon comme:
m (6)
où les quantités "primées" sont relatives au standard.
Les données des périodes et des énergies des raies émises sont de la référence [79].
L'élimination du facteur (p suppose que le flux soit spatialement homogène. Ceci a été
vérifié en irradiant plusieurs prélèvements du même standard en lieu et place des échantillons:
les variations observées ne présentent pas de caractère systématique et ne sont pas
significatives compte tenu des fluctuations statistiques de comptage. D'autre part, certains
éléments comme le cadmium et le bore ont des isotopes dont la section efficace de capture
neutronique est très élevée. Ils sont susceptibles de modifier de façon sensible la distribution du
flux dans le volume contenant les échantillons à analyser en jouant le role d'écran [80].
Cependant, ces éléments ne sont pas naturellement présents dans les matériaux argileux en
quantité suffisante pour produire un effet significatif et nous avons supposé que les
phénomènes de ce type étaient négligeables. Les conditions d'irradiation du standard et de
l'échantillon peuvent donc être considérées comme identiques, ce qui n'est pas le cas en PIXE.
Par ailleurs, l'expression (6) suppose qu'une seule réaction nucléaire (réaction (n,y)) est
à l'origine du radioisotope A+^X. Ceci n'est pas forcément le cas si les neutrons utilisés ne sont
pas bien thermalisés. La contribution d'une réaction concurrente produite par des neutrons
épithermiques (ou rapides) conduirait à la modification suivante:
1 + KO
avec:rw A x
rx A w
où W est l'élément produisant la réaction concurrente. CTt<pt et accpc se rapportent
respectivement à la réaction de X avec les neutrons thermiques et à celle de W avec les
neutrons épithermiques (ou rapides).
La correction n'est nécessaire que si les rapports élémentaires entre échantillons et
standard sont différents. En pratique, le choix d'un standard de même nature que les
échantillons et une proportion de neutrons épithermiques supposée faible nous a amené à
négliger en première approximation la contribution de réactions (n,p) et (n,a). Ceci est
probablement à nuancer pour certains éléments comme Mg, pour lequel la réaction27Al(n,p)27Mg qui fait intervenir l'élément majeur des argiles Al peut représenter une

Chapitre 1 47
contribution importante [81, 75]. L'interférence potentielle due à l'autre composant principal
des argiles, Si, par la réaction 30Si(n,a)27Mg est en général négligeable du fait d'une section
efficace faible.
Une autre possibilité d'interférence réside dans la formation de produits de fission,
essentiellement ceux de 235U. La détermination de Ba, La, Ce et Nd peut être affectée par de
fortes concentrations en U [81].
1.2.2. Mise en oeuvre
Dispositif et protocoles expérimentaux
Dispositif expérimental
Les irradiations et comptages ont été effectuées auprès du Réacteur Universitaire de
Strasbourg. Deux canaux d'irradiation ont été utilisés: le canal thermique central, alimenté par
un système pneumatique, pour les irradiations de longue durée, et un canal latéral muni d'une
chaîne de comptage entièrement automatisée pour les irradiations courtes. Le combustible
(alliage AJ-U enrichi en 235U) a une disposition annulaire autour du canal central et est séparé
de ce dernier par une couche de graphite assurant la thermalisation des neutrons. Le flux de
neutrons est d'environ 1012 neutrons.cm"2.s"1.
La chaîne de spectrométrie gamma comprend un détecteur Ge hyperpur, un
préamplificateur, un amplificateur à rejection d'empilements et un analyseur multicanaux.
L'acquisition est contrôlée depuis un PC par un logiciel d'exploitation.
Protocoles expérimentaux
Un protocole d'analyse multiélémentaire est un compromis entre les facteurs
fondamentaux (a, r, b, X) propres aux éléments en présence et les paramètres expérimentaux (c,
tj, tr, tc). Son but est l'établissement de conditions optimales pour l'analyse d'éléments définis. Il
convient d'ajouter aux divers facteurs cités les paramètres relatifs à la spectrométrie gamma
telles que l'intensité des fonds Compton associés aux rayons gamma ainsi que les taux de
comptage permettant des statistiques adéquates. De nombreux protocoles expérimentaux pour
l'analyse de céramiques archéologiques ont été proposés dans la littérature [75], dont plusieurs
se sont inspirés du travail initial de Perlman et Asaro [73]. Nous avons repris pour l'essentiel
des protocoles déjà testés au réacteur, notamment celui qui a été utilisé pour des mesures
précédentes sur des tessons du Costa-Rica [82].
Le nombre d'irradiations et de comptages a été minimisé en tenant compte du fait que
certains éléments avaient déjà été analysés par PIXE. Le tableau 8 présente les paramètres de
temps d'irradiation tj, repos tr et comptage tc dans une procédure à deux irradiations suivies
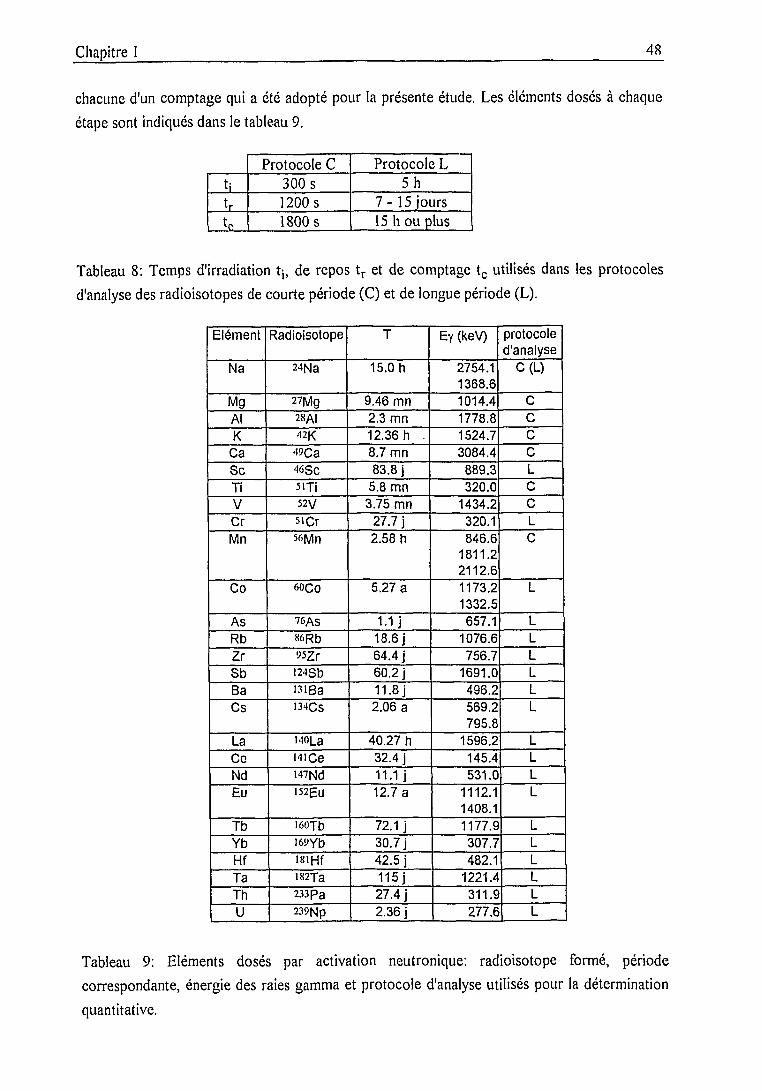
Chapitre 1 48
chacune d'un comptage qui a été adopté pour la présente étude. Les éléments dosés à chaque
étape sont indiqués dans le tableau 9.
tj
tr
Protocole C300 s1200 s1800 s
Protocole L5h
7 - 15 jours15 h ou plus
Tableau 8: Temps d'irradiation tj, de repos tr et de comptage tc utilisés dans les protocoles
d'analyse des radioisotopes de courte période (C) et de longue période (L).
Elément
Na
MgAlK
CaSeTiVCrMn
Co
AsRbZrSbBaCs
LaCeNdEu
TbYbHfTaThU
Radioisotope
24Na
27Mg
28AI
42«
«Ca46SC
siTi52V
siCr56Mn
60CO
™AS86Rb
95Zr
124Sb
i3iBa134CS
MOLa
nice147Nd
152EU
îeoTb169YbI81Hf
i82Ta233Pa
239Np
T
15.0 h
9.46 mn2.3 mn12.36 h .8.7 mn83.8 j
5.8 mn3.75 mn
27.7 j2.58 h
5.27 a
1.1 j18.6 j64.4 j60.2 j11.8 j2.06 a
40.27 h32.4 j
1 1 . 1 J12.7 a
72.1 j30.7 j42.5 j115 j27.4 j2.36 j
Ey (keV)
2754.11368.61014.41778.81524.73084.4
889.3320.0
1434.2320.1846.6
1811.22112.61173.21332.5657.1
1076.6756.7
1691.0496.2569.2795.8
1596.2145.4531.0
1112.11408.11177.9307.7482.1
1221.4311.9277.6
protocoled'analyse
C(L)
CC
CCLCCLC
L
LLLLLL
LLLL
LLLLLL
Tableau 9: Eléments dosés par activation neutronique: radioisotope formé, période
correspondante, énergie des raies gamma et protocole d'analyse utilisés pour la détermination
quantitative.

Chapitre I 49
Le protocole C est destiné à l'analyse via des radioisotopes de courtes périodes, le protocole L
concerne les radioisotopes de longue durée de vie. Les temps de repos sont choisis de manière
à optimiser le rapport signal / bruit. Dans le cas du deuxième protocole, il est suffisamment
long pour que le bruit de fond Compton dû aux isotopes facilement activables de période
intermédiaire (Na, La) soit minime. Cette amélioration globale du rapport signal / bruit se fait
au détriment de la détection de certains isotopes moins bien activables et de période
comparable comme 76As et 239Np.
Les échantillons sont conditionnés sous forme de poudres dans des conteneurs en
polyethylene. La masse moyenne des prélèvements irradiés est de l'ordre de 100 mg, quantité
qui peut être considérée comme représentative pour un matériau céramique [83], Pour le
protocole L, un prélèvement du standard IAEA Soil7 est utilisé comme référence externe pour
chaque lot de quatre échantillons, les cinq containers étant irradiés simultanément lors des
irradiations longues. La mesure du standard est réalisée systématiquement en premier lieu, de
manière à éviter une décroissance trop importante des radioisotopes de périodes moyennes
(24Na, 140La, 76As, 239Np, ...) qui empêcherait leur détermination dans tout le lot
d'échantillons. Dans le cas du protocole C, les irradiations de courte durée sont successives.
Les comptages sont effectués dans les même conditions pour les échantillons et le standard,
dans une géométrie de détection fixe.
Spectrométrie gamma
Le détecteur Ge utilisé a un volume sensible de 77 cm3 et une résolution d'environ 2
keV à 1332 keV. L'efficacité de détection dans la géométrie de comptage usuelle est
représenté sur la figure 19.
Les différentes composantes du spectre gamma sont dues à des phénomènes déjà
évoqués en 1.1.1.1. (Transmission du rayonnement électromagnétique). Pour un rayonnement
gamma incident d'énergie E, on observera dans le spectre:
- le pic de pleine énergie (E);
- la distribution Compton, dont le front correspond à l'énergie maximale de l'électron Compton.
Ce front est situé à E/(l+(mcc2/2E), où mcc
2 est l'énergie de masse de l'électron;
- les pics de simple (E-511 keV) et double échappement (E-1022 keV) lorsque les rayons
gammas incidents ont une énergie suffisante pour générer une paire e"-e+
(E> 2mcc2=1022keV). L'annihilation du positron donne lieu à l'émission de deux rayons
gammas d'énergie 511 keV qui engendrent les pics d'échappement lorsque l'un ou les deux ne
sont pas absorbés dans le volume sensible du détecteur.
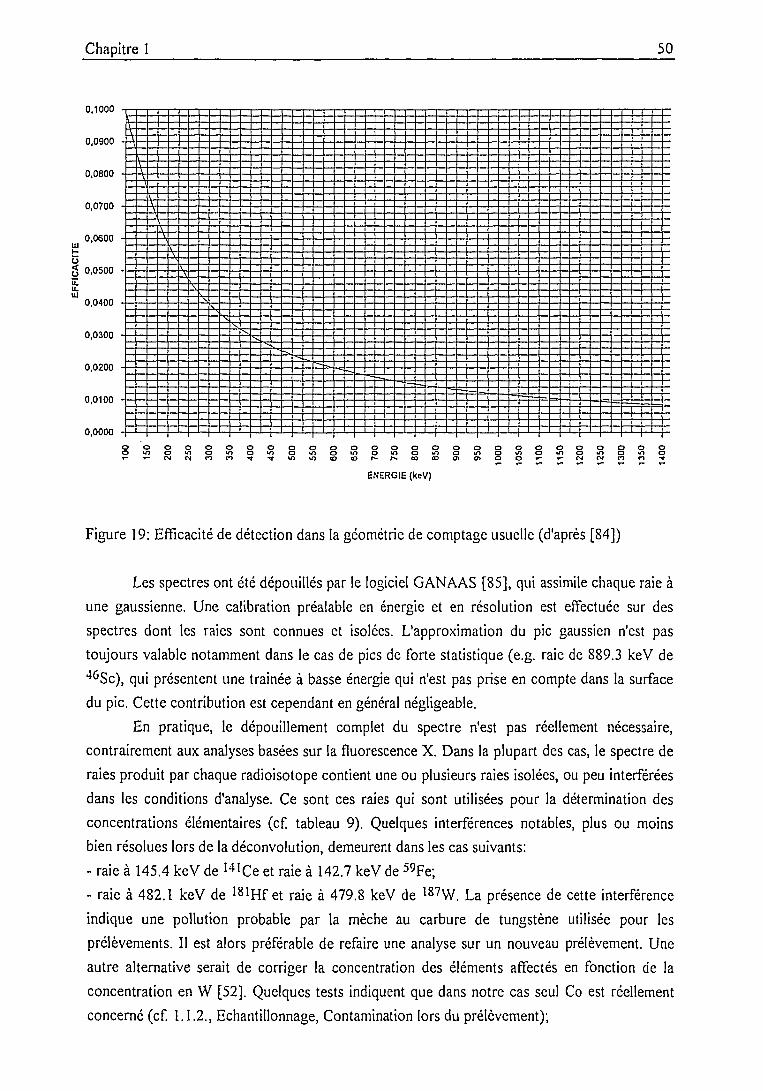
Chapitre
0,1000 -
0,0900 -
0,0800 -
0,0700 -
0,0600 •-
2 0,0500 -u.u.u
0,0400 -
0,0300 -
0,0200 -
0,0100 -
0,0000 -
1 50
1\\
\
V
—
r-
-
-
ENERGIE (keV)
Figure 19; Efficacité de détection dans la géométrie de comptage usuelle (d'après [84])
Les spectres ont été dépouillés par le logiciel GANAAS [85], qui assimile chaque raie à
une gaussienne. Une calibration préalable en énergie et en résolution est effectuée sur des
spectres dont les raies sont connues et isolées. L'approximation du pic gaussien n'est pas
toujours valable notamment dans le cas de pics de forte statistique (e.g. raie de 889.3 keV de46Sc), qui présentent une trainee à basse énergie qui n'est pas prise en compte dans la surface
du pic. Cette contribution est cependant en général négligeable.
En pratique, le dépouillement complet du spectre n'est pas réellement nécessaire,
contrairement aux analyses basées sur la fluorescence X. Dans la plupart des cas, le spectre de
raies produit par chaque radioisotope contient une ou plusieurs raies isolées, ou peu interférées
dans les conditions d'analyse. Ce sont ces raies qui sont utilisées pour la détermination des
concentrations élémentaires (cf. tableau 9). Quelques interférences notables, plus ou moins
bien résolues lors de la déconvolution, demeurent dans les cas suivants:
- raie à 145.4 keV de 14ICe et raie à 142.7 keV de 59Fe;
- raie à 482.1 keV de 18IHf et raie à 479.8 keV de l87W. La présence de cette interférence
indique une pollution probable par la mèche au carbure de tungstène utilisée pour les
prélèvements. Il est alors préférable de refaire une analyse sur un nouveau prélèvement. Une
autre alternative serait de corriger la concentration des éléments affectés en fonction de la
concentration en W [52]. Quelques tests indiquent que dans notre cas seul Co est réellement
concerné (cf. 1.1.2., Echantillonnage, Contamination lors du prélèvement);

Chapitre 1 51
- raie à 846.6 keV de 56Mn et raie à 843.8 de 27Mg, la première étant très nettement
dominante;
- raie à 1112.1 keV de 152Eu et raie à 1115.9 keV de 65Zn, elle-même fortement dominée par
la raie à 1120.5 keV de 46Sc. On obtient cependant en général un bon accord entre les valeurs
des concentrations de Eu déduites de la raie indiquée et de la raie isolée de 152Eu à 1408.1
keV. La concentration finale adoptée est la moyenne des concentrations calculées à partir des
deux raies, sauf en cas d'écart significatif (teneurs inhabituelles en Zn et/ou Se) auquel cas
seule la raie isolée est prise en compte.
Précision, justesse
Bien que des estimations très fines de la précision et de la justesse obtenues en
activation neutronique soient possibles [69], nous nous sommes limités ici à une évaluation de
la précision basée sur les incertitudes statistiques de comptage et à une vérification de la
justesse sur quelques standards. Les incertitudes sur les paramètres fondamentaux, sur les
masses des échantillons et dans la plupart des cas sur l'adéquation du fit peuvent être
considérées comme négligeables devant les fluctuations statistiques sur les surfaces des pics.
Les imprécisions sur la composition des standards ne sont pas prises en compte à ce niveau, ce
qui peut expliquer que certains écarts entre valeurs certifiées et calculées soient supérieurs à
notre estimation de l'incertitude (tableaux 10 et 11). Nous avons évalué l'incertitude sur les
concentrations pour un échantillon "moyen" (teneurs moyennes, tr et tc moyens) et pour un
dosage réalisé dans des conditions défavorables (teneurs faibles, temps de repos trop long par
rapport à la période du radionucléide correspondant ou trop court du point de vue du rapport
signal/bruit) (tableau 10).
A S / S standard (%)A C / C moyen (%)A C / C cas défavorable (%)
A S / S standard (%)A C / C moyen (%)A C / C cas défavorable (%)
Na1.5
33
Mg8
1519
Al589
K122025
Ca26
21
Ti183857
Mn0.7
24
Fc0.5
11
Se0.3
11
V122629
Cr1.1
23
Co1.3
25
Rb47
12
Zr1541
Sb6
1419
Cs58
16
Ba6.6
710
La0.5
24
Ce0.5
22
Nd102427
Eu5
1121
Tb
co
oo o
o
Yb7
1833
Hf135
Ta6
1329
Th0.6
12
U4
1833
Tableau 10: Estimation de l'incertitude sur les concentrations A C / C due aux fluctuations
statistiques de comptage, pour un échantillon "moyen" (conditions moyennes d'analyse) et pour
un dosage réalisé dans des conditions défavorables. L'incertitude statistique moyenne A S / S
sur la surface des pics du standard est indiquée.
Les valeurs données dans le tableau 10 sont indicatives, et le classement des éléments aosés
selon des gammes de précision est probablement plus fondé:
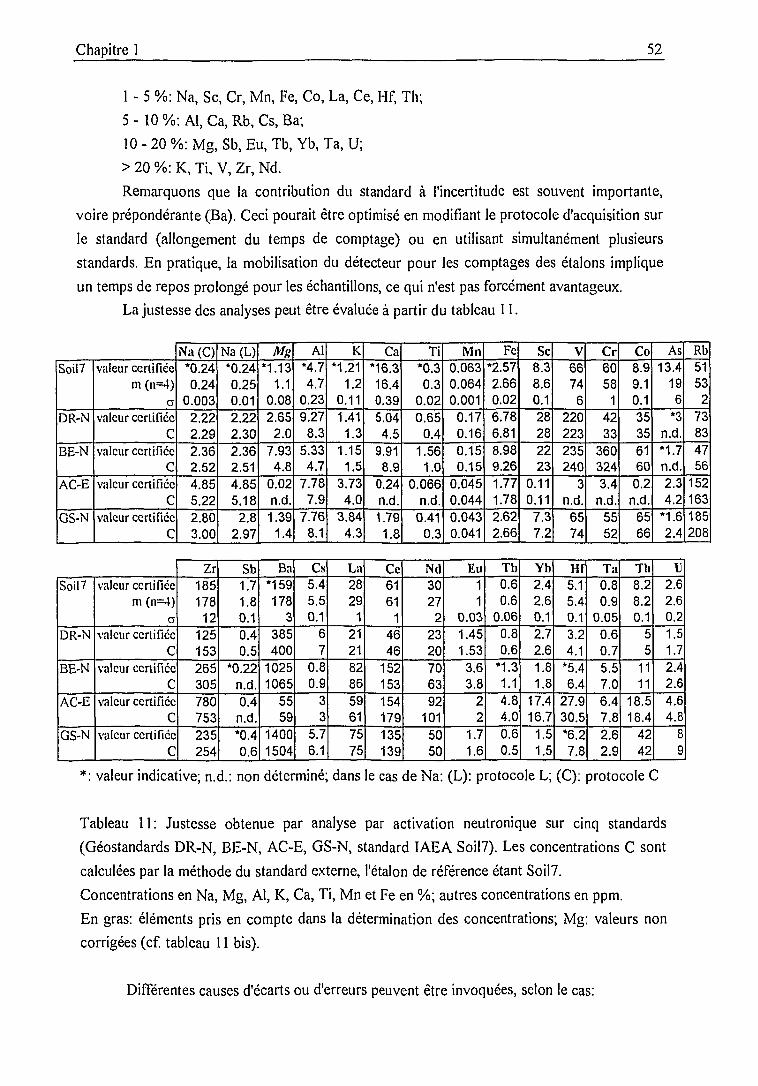
Chapitre 1 52
1 - 5 %: Na, Se, Cr, Mn, Fe, Co, La, Ce, Hf, Th;
5-10 %: Al, Ca, Rb, Cs, Ba;
10-20 %: Mg, Sb, Eu, Tb, Yb, Ta, U;
> 20 %: K, Ti, V, Zr, Nd.
Remarquons que la contribution du standard à l'incertitude est souvent importante,
voire prépondérante (Ba). Ceci pourait être optimisé en modifiant le protocole d'acquisition sur
le standard (allongement du temps de comptage) ou en utilisant simultanément plusieurs
standards. En pratique, la mobilisation du détecteur pour les comptages des étalons implique
un temps de repos prolongé pour les échantillons, ce qui n'est pas forcément avantageux.
La justesse des analyses peut être évaluée à partir du tableau 11.
Soil7
DR-N
BE-N
AC-E
GS-N
valeur certifiéem (n=4)
avaleur certifiée
Cvaleur certifiée
Cvaleur certifiée
Cvaleur certifiée
C
Soil7
DR-N
BE-N
AC-E
GS-N
valeur certifiéem (n=4)
avaleur certifiée
Cvaleur certifiée
Cvaleur certifiée
Cvaleur certifiée
C
Na (C)*0.240.24
0.0032.222.292.362.524.855.222.803.00
Zr185178
12125153265305780753235254
Na (L)*0.240.250.012.222.302.362.514.855.18
2.82.97
Sb1.71.80.10.40.5
*0.22n.d.0.4
n.d.*0.40.6
Mg*1.13
1.10.082.652.0
7.934.8
0.02n.d.1.39
1.4
Ba*159178
3385400
10251065
5559
14001504
Al*4.74.7
0.239.27
8.35.334.7
7.787.9
7.768.1
Cs5.45.50.1
67
0.80.9
33
5.76.1
K*1.21
1.20.111.41
1.31.15
1.53.734.0
3.844.3
La2829
12121828659617575
Ca*16.316.40.395.044.5
9.918.9
0.24n.d.1.791.8
Ce6161
14646
152153154179135139
Ti*0.30.3
0.020.65
0.41.56
1.00.066
n.d.0.41
0.3
Nd3027
22320706392
1015050
Mn0.0630.0640.001
0.170.160.150.15
0.0450.0440.0430.041
Eu11
0.031.451.533.63.8
22
1.71.6
Fc*2.572.660.026.786.818.989.261.771.782.622.66
Tb0.60.6
0.060.80.6
*1.31.14.84.00.60.5
Se8.38.60.128282223
0.110.117.37.2
Yb2.42.60.12.72.61.81.8
17.416.7
1.51.5
V66746
220223235240
3n.d.6574
Hf5.15.40.13.24.1
*5.46.4
27.930.5*6.27.8
Cr6058
14233
3603243.4
n.d.5552
Ta0.80.9
0.050.60.75.57.06.47.82.62.9
Co8.99.10.1353561600.2
n.d.6566
Th8.28.20.1
55
1111
18.518.4
4242
As13.4
196
*3n.d.•1.7n.d.2.34.2
•1.62.4
U2.62.60.21.51.72.42.64.64.8
89
Rb51532
73834756
152163185208
*: valeur indicative; n.d.: non déterminé; dans le cas de Na: (L): protocole L; (C): protocole C
Tableau 11: Justesse obtenue par analyse par activation neutronique sur cinq standards
(Géostandards DR-N, BE-N, AC-E, GS-N, standard IAEA Soil7). Les concentrations C sont
calculées par la méthode du standard externe, l'étalon de référence étant Soil7.
Concentrations en Na, Mg, Al, K, Ca, Ti, Mn et Fe en %; autres concentrations en ppm.
En gras: éléments pris en compte dans la détermination des concentrations; Mg: valeurs non
corrigées (cf. tableau 11 bis).
Différentes causes d'écarts ou d'erreurs peuvent être invoquées, selon le cas:

Chapitre 1 53_
- les concentrations du standard de référence Soil7 sont données avec un certain intervalle de
confiance. On pourrait trouver, par exemple, que la concentration en Ta de BE-N est fort
éloignée de sa valeur certifiée (7 ppm au lieu de 5.5 ppm). Or, la teneur en Ta de Soil7 est
donnée avec un intervalle de confiance de 0.6 - 1.0 ppm, correspondant à une probabilité de
95%, pour une valeur moyenne de 0.8 ppm. Les teneurs correspondantes pour BE-N sont
5.25 ppm - 8.75 ppm, ce qui inclut bien la valeur certifiée. De la même façon, Cr dans DR-N
s'inscrit dans l'intervalle 27 ppm - 41 ppm, ce qui est compatible compte tenu de l'incertitude
statistique sur la mesure avec la valeur certifiée de 42 ppm;
- les interférences dues aux raies de W sont en grande partie responsables des écarts observés
sur les teneurs en Hf pour GS-N et DR-N, qui contiennent respectivement 490 ppm et 130
ppm de W;
- les interférences nucléaires dues à Al sont à l'origine d'erreurs importantes sur les teneurs en
Mg lorsque les rapports de concentrations Al/Mg de Soil7 et des autres standards sont très
différents. L'erreur commise en utilisant la relation (6) (concentration C x nocor calculée par
standard externe, sans correction d'interférences) au lieu de la relation (7) (concentration C x
"réelle" tenant compte des interférences) se déduit de la relation:
x nocor
où Cw est la teneur de l'élément interférant dans l'échantillon, C\v et C'x sont les teneurs dans le
standard. Remarquons que Cx n'est pas forcément inférieure à Cx nocOr: dans une détermination
par standard externe, il est possible d'obtenir des concentrations non corrigées inférieures à la
valeur réelle, ce qui ne serait pas le cas dans une détermination absolue. La valeur de Cx
s'exprime en fonction de Cw, de Cx nocor et du facteur K relatif au rapport entre flux
épithermique et flux thermique par:
Cx = C . \ nocor 1 + K 7 7 L ~ K C wV cx J
K peut être déterminé en réalisant des mesures sur des échantillons d'AJ pur pour lesquels la
concentration apparente en Mg est uniquement due à l'interférence. Nous avons obtenu de
cette façon la valeur:
K = 0.117 ±0.002
L'ordre de grandeur indique que l'approximation d'un flux parfaitement thermalisé dans le canal
latéral (irradiations de courte durée) est plutôt grossière. Le tableau 11 bis compare les
concentrations de Mg obtenues avec et sans correction aux valeurs certifiées de plusieurs
standards.

Chapitre 1 54
Soil7
GS-N
CFA
BCR-1
DR-N
BE-N
Mg ,,ocor
1.4+0.2
1.4 + 0.2
2.2 ±0.4
2.0 + 0.3
4.8 + 0.5
Mg
1.2 + 0.3
0.4 + 0.3
2.5 ±0.6
1.9 ±0.5
6.5 + 0.8
Mg certifié
1.13*
1.39 ±0.12
0.455+0.001
2.10
2.65 ±0.16
7.93 ±0.27
Al/Mg
4.16
5.58
30.8
3.44
3.50
0.67
Tableau 11 bis: Détermination de Mg avec et sans correction de l'interférence due à Al et
incidence de la différence de rapport Al/Mg entre standard de référence Soil7 et échantillons
Mg nocor: calcul en standard externe ne tenant pas compte de l'interférence; Mg: valeur après
correction de l'interférence; Mg certifié: valeur certifiée;
CFA: standard Coal Fly Ash, NBS; BCR-1: standard USGS
La concentration de Mg dans Soil7 est contenue dans l'intervalle de confiance à 95%
(1.10; 1.18) (valeur non certifiée). Les autres incertitudes sont données à 1 a.
Lorsque la valeur de Al/Mg est nettement différente de celle du standard de référence
Soil7, l'application d'une correction est absolument nécessaire pour obtenir des résultats
cohérents, compte tenu de l'incertitude. Lorsque Al/Mg est du même ordre de grandeur, la
correction a peu d'effet et son application a surtout pour conséquence de dégrader la précision
(l'incertitude moyenne passe de 15% à 23%).
Une mesure plus fiable de Mg passerait par l'utilisation du canal pneumatique, non
automatisé mais mieux thermalisé, pour les irradiations. Par ailleurs, l'emploi d'un standard de
teneur en Mg certifiée serait évidemment préférable, mais le choix d'un standard unique de
caractéristiques optimales n'est pas simple. En pratique, les céramiques analysées présentent
pour la plupart des rapports Al/Mg proches de celui de Soil7 et le protocole utilisé peut être
estimé satisfaisant dans ces conditions. On considérera toutefois avec quelques réserves les
valeurs "absolues" de Mg lors de l'interprétation des résultats.

Chapitre 1 . 55
1.3. Bilan analytique et intercomparaisons
1.3.1. Bilan analytique et intercomparaison PIXE - INAA
Nous avons utilisé les méthodes d'analyse PIXE et INAA de manière complémentaire,
et non de manière "concurrentielle". De ce fait, le nombre d'éléments communs dosés dans des
conditions acceptables par les deux méthodes est réduit. En particulier, on peut estimer que la
détermination par PIXE de Mn est peu précise et trop dépendante des paramètres de tailing de
Fe. Nous ne considérerons pas non plus les dosages par INAA de Ti et Zr, dont les statistiques
de comptage sont trop faibles pour fournir des résultats fiables. D'autres éléments présentant
des statistiques de comptage relativement peu favorables en INAA, comme K et Ca, donnent
des résultats plus comparables à ceux obtenus par PIXE.
Dans l'ensemble, l'accord entre les résultats PIXE et INAA présentés sur la figure 20
est satisfaisant. Quelques mesures effectuées par RIXRF [86] sont également données.
On peut remarquer une tendance systématique à la sur-estimation de Ca en PIXE. Cet
effet est très probablement lié à la modélisation de la réponse du détecteur Si(Li) par PIXAN,
qui est mal adaptée aux raies intenses proches du seuil d'absorption de Si.
Les valeurs de Ba sont en général légèrement plus faibles en PIXE que par les autres
méthodes. L'assimilation au baiit de fond de raies de faible statistiques voisines de celles de Ba
(Cs, La, Ce,...) peut être à l'origine d'une sous-estimation de la surface des pics de cet élément.
En ce qui concerne Fe, la valeur finale prise en compte dans les traitements ultérieurs
des données est intermédiaire entre les déterminations par INAA et par PIXE à 1 MeV.
La combinaison des deux méthodes d'analyse PIXE et INAA nous ont permis de
couvrir une large gamme élémentaire. Le tableau 12 propose un récapitulatif des éléments
dosés lors de la mise en oeuvre "de routine" des protocoles d'analyses exposés aux paragraphes
précédents.
La méthode PIXE a une vocation multiélémentaire plus prononcée du fait de la
variation continue des sections efficaces de production des rayons X avec Z. Elle permet de
déterminer les teneurs des éléments majeurs ainsi qu'une dizaine d'éléments traces des argiles.
L'INAA accède à un plus grand nombre d'éléments traces grâce à de très bonnes sensibilités
mais impose des choix expérimentaux plus contraignants. Cet inconvénient est en partie
compensé par une pratique déjà longue de l'INAA en analyse de céramiques archéologiques, ce
qui a permis de proposer des protocoles adéquats. Le dosage de certains éléments majeurs
reste cependant malaisé en analyse de routine (e.g. Si).
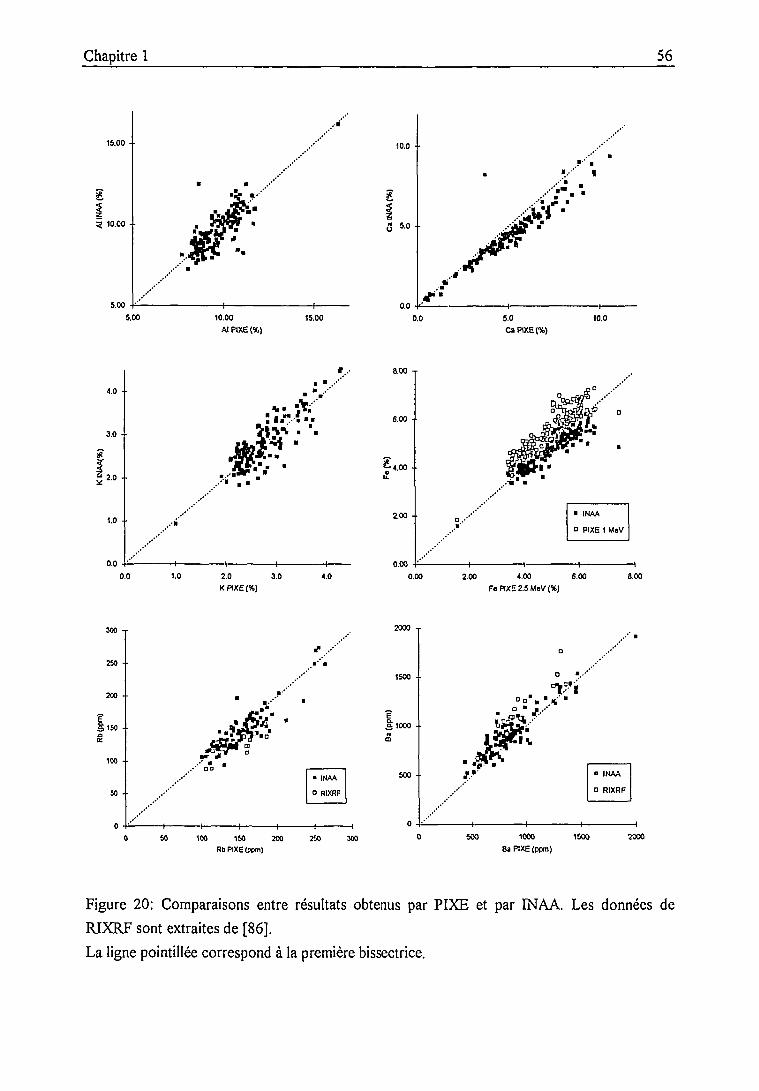
Chapitre 1 56
15.00 •
g
i 10.00 •
5.00 •
• •
•~ j 1
5.00 10.00 15.00
AI pixe ex.)
10.0
g
3 5.0 ••
0.0
" . • • • «
0.0 5.0
CaPIXEC*.)
10.0
4.0 •
3.0
; 2.0 •
1.0 ••
0.0
0.0 1.0
ÏÎAV:V-
2.0 3.0
KPIXE(%)
4.0
8.00
6.00
2.00
0.00 +—0.00
• INAA
o PIXE 1 MoV
2.O0 4.00 6.00
Fe PIXE 2.5 MeVO*)
8 00
250
50 •
• INAA
o RIXRF
H 1 1 1 1-100 150 200 2M MO
RbPIXE(ppm)
2000 T
1500 •
S 1000 •
500 • INAA
D RIXRF
500 1000 1500 2000
BaPIXE(ppm)
Figure 20: Comparaisons entre résultats obtenus par PIXE et par INAA. Les données de
RIXRF sont extraites de [86].
La ligne pointillée correspond à la première bissectrice.

Chapitre 1 57
élément
méthode d'analyse
Na
INAA
(MB)INAA
Al
PIXE
Si
PIXE
K
PIXE
Ca
PIXE
Ti
PIXE
Mn
INAA
Fe
PIXE
Se
INAA
V
INAA
élément
méthode d'analyse
Cr
INAA
Co
INAA
Ni
PIXE
Ga
PIXE
Rb
PIXE
Sr
PIXE
Y
PIXE
Zr
PIXE
Sb
INAA
Cs
INAA
Ba
PIXE
élément
méthode d'analyse
La
INAA
Ce
INAA
(Nd)
INAA
Eu
INAA
Tb
INAA
Yb
INAA
Hf
INAA
Ta
INAA
(Pb)
PIXE
Th
INAA
U
INAA
Tableau 12: Récapitulatif des éléments dosés et de la méthode d'analyse utilisée pour leur
détermination.
Certains paramètres expérimentaux sont difficiles à déterminer avec précision dans les
deux cas: charge intégrée de protons en PIXE, flux et distribution en énergie des neutrons en
activation. Dans ce dernier cas, l'irradiation simultanée du standard et des échantillons
permettrait d'éliminer le facteur flux si les neutrons étaient parfaitement thermalisés. Dans les
réacteurs, la contribution des neutrons épithermiques et rapides aux réactions peut être
appréciable pour quelques éléments (e.g. Mg).
En analyse par activation neutronique, la multiplicité des raies permet en général
d'utiliser des pics isolés lors de l'exploitation du spectre gamma. La spectrométrie des rayons X
exige par contre une déconvolution soigneuse du spectre, la résolution des interférences
nécessitant la connaissance de l'efficacité de détection et de l'autoabsorption. Ces contraintes
font perdre une partie du bénéfice de la détermination par standard externe qui a été mise en
oeuvre pour les deux méthodes.
En PIXE, le calcul des concentrations procède en général par itération à partir d'une
matrice hypothétique. L'absorption dans l'échantillon n'intervenant plus dans le cas des rayons
gamma, l'INAA ne nécessite pas d'hypothèse sur la composition de l'échantillon (à l'exception
des éléments de très forte section efficace de capture neutronique). Par ailleurs,
l'autoabsorption des rayons X et la faible pénétration des protons (quelques dizaines de um
dans les matériaux siliceux) font du PIXE une méthode d'analyse de surface.
Du point de vue pratique, les temps d'analyse sont beaucoup plus longs en INAA.
D'une part, les temps de repos imposent des délais de l'ordre de 15 jours. D'autre part,
l'utilisation d'un seul détecteur pour les comptages de longue durée limite fortement le nombre
d'échantillons que l'on peut traiter (4 échantillons par semaine au lieu d'une cinquantaine en
PIXE).
Malgré les différentes sources d'erreurs, PIXE et INAA sont deux méthodes très
performantes pour l'analyse multiélémentaire des argiles (tableaux 4, 5, 6, 7, 10, 11, figures 17,
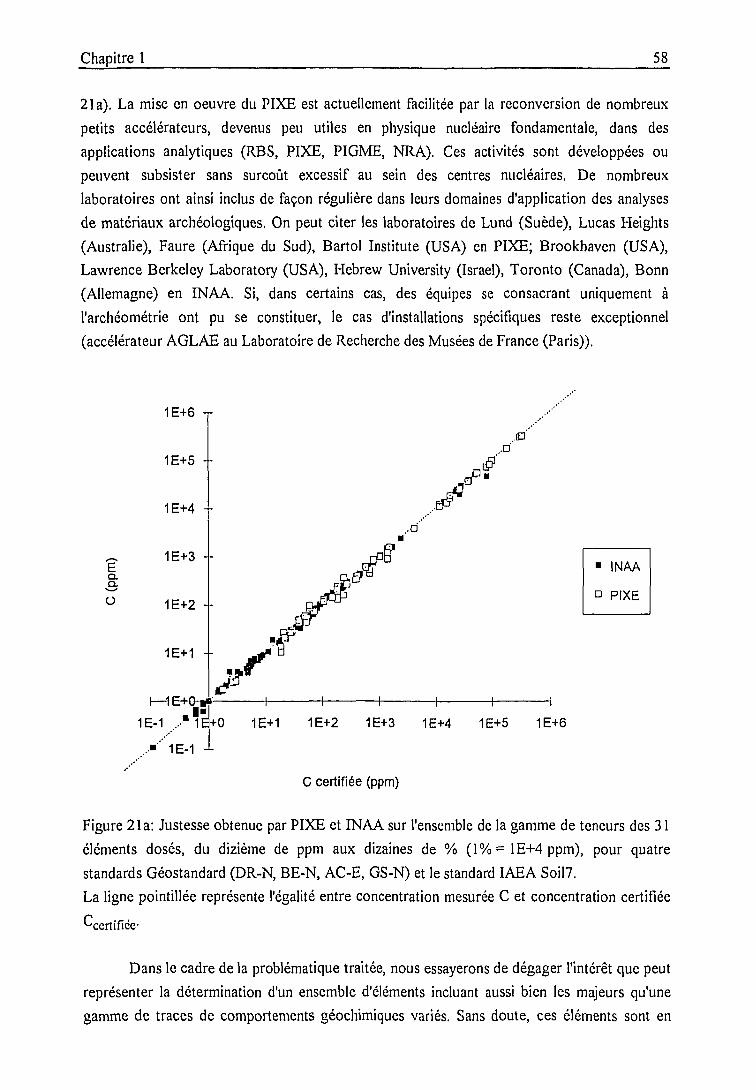
Chapitre 1 58
21a). La mise en oeuvre du PIXE est actuellement facilitée par la reconversion de nombreux
petits accélérateurs, devenus peu utiles en physique nucléaire fondamentale, dans des
applications analytiques (RBS, PIXE, PIGME, NRA). Ces activités sont développées ou
peuvent subsister sans surcoût excessif au sein des centres nucléaires. De nombreux
laboratoires ont ainsi inclus de façon régulière dans leurs domaines d'application des analyses
de matériaux archéologiques. On peut citer les laboratoires de Lund (Suède), Lucas Heights
(Australie), Faure (Afrique du Sud), Bartol Institute (USA) en PIXE; Brookhaven (USA),
Lawrence Berkeley Laboratory (USA), Hebrew University (Israel), Toronto (Canada), Bonn
(Allemagne) en INAA. Si, dans certains cas, des équipes se consacrant uniquement à
l'archéométrie ont pu se constituer, le cas d'installations spécifiques reste exceptionnel
(accélérateur AGLAE au Laboratoire de Recherche des Musées de France (Paris)).
EQ.
a.O
1E+6 j
1E+5
1E+4
1E+3 --
1E+2 --
1E+1 --
f—4E+0-
.D
M-
Jtf1E+5 1E+61E-1 . , " 1E+0 1E+1 1E+2 1E+3 1E+4
...•••" 1E-1
C certifiée (ppm)
Figure 21a: Justesse obtenue par PIXE et INAA sur l'ensemble de la gamme de teneurs des 31
éléments dosés, du dizième de ppm aux dizaines de % (1%= 1E+4 ppm), pour quatre
standards Géostandard (DR-N, BE-N, AC-E, GS-N) et le standard IAEA Soil7.
La ligne pointillée représente l'égalité entre concentration mesurée C et concentration certifiée
^certifiée-
Dans le cadre de la problématique traitée, nous essayerons de dégager l'intérêt que peut
représenter la détermination d'un ensemble d'éléments incluant aussi bien les majeurs qu'une
gamme de traces de comportements géochimiques variés. Sans doute, ces éléments sont en

Chapitre 1 59.
partie accessibles par des méthodes plus "légères" (e.g. fluorescence X) et éventuellement plus
performantes (ICP-MS). Plus qu'à la méthode employée, nous nous attacherons par la suite à
l'interprétation que l'on peut tirer des données élémentaires obtenues, indépendamment des
considérations analytiques ou pratiques. De la même façon, nous n'avons pas mis en
concurrence PIXE et INAA dans le traitement de la problématique. Chacune de ces méthodes
a déjà été utilisée avec succès dans le cadre des études de provenance dans lequel notre travail
s'inscrit (cf. Chapitre 2).
1.3.2. Intercomparaison entre laboratoires
La comparaison de données issues de plusieurs laboratoires n'est pas immédiate. Bien
que l'on s'achemine d'une manière générale vers une meilleure maîtrise des sources d'erreurs
systématiques, des écarts demeurent même entre des déterminations effectuées par les
méthodes réputées les plus sûres comme l'INAA. Des programmes d'intercalibration portant
sur une large gamme d'éléments et de teneurs devraient être un préalable à tout échange de
données entre laboratoires. En pratique, on peut être moins exigeant dans le cas de l'infirmation
d'une hypothèse de provenance. Lorsque la coïncidence des résultats entre deux laboratoires
est bonne pour certains éléments, une absence de correspondance de la teneur de ces éléments
entre un groupe de référence, dosé par l'un des laboratoires, et un groupe de tessons à attribuer
dosé par l'autre sera significative. La proposition d'attribution est par contre plus
contraignante.
Nous avons eu la chance de pouvoir accéder à des données de laboratoires dont les
préoccupations rejoignent la problématique archéologique de la présente étude (cf. 2.4.2,
Groupes de composition de provenance indéterminée).
iMboratoire de céramologie. Lyon
Le laboratoire de céramologie de Lyon a constitué une importante base de données
dans le domaine de la céramique gallo-romaine, en particulier la céramique sigillée (cf. e.g.
[87]), et en céramologie médiévale du pourtour méditerranéen occidental [88]. Dans le cadre
du présent travail, nous nous sommes intéressés aux analyses de céramiques d'ateliers antiques
de la côte Ouest de la Turquie.
La méthode d'analyse utilisée est la fluorescence X en dispersion de longueur d'onde.
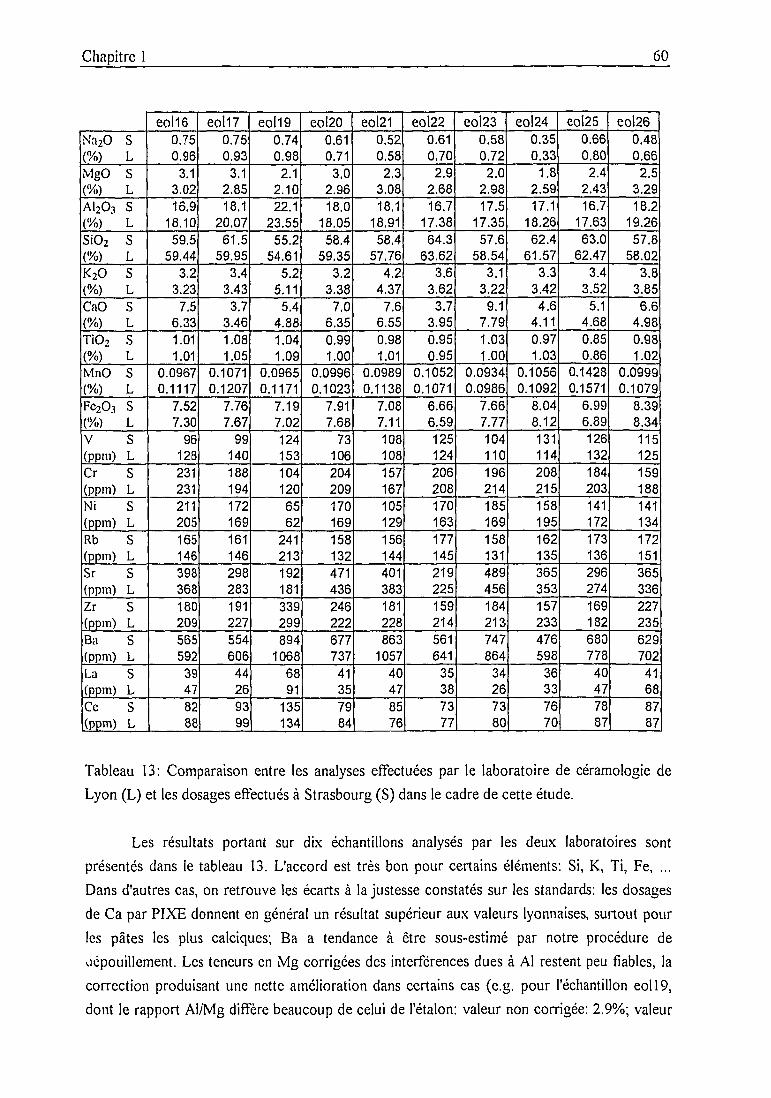
Chapitre 1 60
Na2O S(%) LMgO S(%) LA12O3 S(%) LSiO2 S(%) LK2O S(%) LCaO S(%) LTiO2 S(%) LMnO S(%) LFc2O3 S(%) LV S(ppm) LCr S(ppm) LNi S(ppm) LRb S(ppm) LSr S(ppm) LZr S(ppm) LBa S(ppm) LLa S(ppm) LCe S(ppm) L
eol160.750.963.13.0216.9
18.1059,559.443,23.237.56.331.011.01
0.09670.11177.527.309512823123121120516514639836818020956559239478288
eol170.750.933.1
2.8518.1
20.0761.559.953.43.433.73.461.081.05
0.10710.1207
7.767.679914018819417216916114629828319122755460644269399
eol190.740.982.12.1022.123.5555.254.615.25.115.44.881.041.09
0.09650.11717.197.02124153104120656224121319218133929989410686891135134
eol200.610.713.02.9618.0
18.0558.459.353.23.387.06.350.991.00
0.09960.10237.917.687310620420917016915813247143624622267773741357984
eol210.520.582.33.0818.1
18.9158.457.764.2
4.377.66.550.981.01
0.09890.11387.087.11108108157167105129156144401383181228863105740478576
eol220.610.702.9
2.6816.7
17.3864.363.623.63.623.73.950.950.95
0.10520.10716.666.5912512420620817016317714521922515921456164135387377
eol230.580.722.02.9817.5
17.3557.658.543.13.229.17.791.031.00
0.09340.09867.667.7710411019621418516915813148945618421374786434267380
eol240.350.331.8
2.5917.1
18.2662.461.573.33.424.64.110.971.03
0.10560.10928.048.1213111420821515819516213536535315723347659836337670
eol250.660.802.42.4316.717.6363.062.473.43.525.14.680.850.86
0.14280.15716.996.8912613218420314117217313629627416918268077840477887
eol260.480.662.53.2918.219.2657.858.023.83.856.64.980.981.02
0.09990.10798.398.3411512515918814113417215136533622723562970241688787
Tableau 13: Comparaison entre les analyses effectuées par le laboratoire de ceramologie de
Lyon (L) et les dosages effectués à Strasbourg (S) dans le cadre de cette étude.
Les résultats portant sur dix échantillons analysés par les deux laboratoires sont
présentés dans le tableau 13. L'accord est très bon pour certains éléments: Si, K, Ti, Fe, ...
Dans d'autres cas, on retrouve les écarts à la justesse constatés sur les standards: les dosages
de Ca par PIXE donnent en général un résultat supérieur aux valeurs lyonnaises, surtout pour
les pâtes les plus calciques; Ba a tendance à être sous-estimé par notre procédure de
dépouillement. Les teneurs en Mg corrigées des interférences dues à Al restent peu fiables, la
correction produisant une nette amélioration dans certains cas (e.g. pour l'échantillon eoll9,
dont le rapport Al/Mg diffère beaucoup de celui de l'étalon: valeur non corrigée: 2.9%; valeur

Chapitre 1 6]_
corrigée: 2.1%; valeur donnée par le laboratoire de Lyon: 2.10%), mais également parfois des
dégradations sensibles (e.g. eol24: valeur non corrigée: 2.4%; valeur corrigée: 1.8%; valeur
donnée par le laboratoire de Lyon: 2.59%), la précision étant dans tous les cas médiocre
(incertitudes AMgO de l'ordre de 0.5 - 0.7 % à la). D'autres différences assez marquées sur les
éléments Na, La et V sont considérées comme peu significatives, du moins dans les deux
premiers cas. En effet, la détermination de Na n'est pas considéré à Lyon comme étant très
fiable, et cet élément est rarement utilisé dans l'exploitation des données. Par ailleurs, le dosage
de La sert essentiellement de contrôle d'une éventuelle pollution par un ancien constituant du
fondant, La2Û, qui était utilisé conjointement au borax pour le conditionnement des
échantillons sous forme de perles (cf. annexe 3).
Nous pourrons donc comparer directement nos données avec celles du laboratoire de
céramologie de Lyon pour certains éléments, en particulier Ti. Pour les autres, une
comparaison devrait tenir compte des écaits observés.
Standard Lefkandi brick
Plusieurs standards "maison" sont utilisés de façon interne aux laboratoires. Certains
ont été utilisés à des fins d'intercalibration entre laboratoires. Celui de Perlman et Asaro, du
Lawrence Berkeley Laboratory, a longtemps été le matériau de référence des équipes
impliquées dans l'analyse de céramiques archéologiques [73], Depuis son épuisement, d'autres
standards, en particulier Ohio Red Clay (Brookhaven) [75, 69] et Lefkandi Brick (Fitch
Laboratory) [89, 90], ont été proposés. Grâce à R.E. Jones, nous avons pu obtenir un
échantillonnage de ce dernier, qui a été analysé par plusieurs équipes travaillant dans le
domaine de la céramique byzantine (Fitch Laboratory, Sofia, Venise, Jérusalem) ou sur du
matériel issu de sites de l'Ouest de la Turquie (British Museum). L'ensemble des résultats
obtenus par une quinzaine de laboratoires ayant analysé le standard Lefkandi brick devrait être
publié par R.E. Jones.
Le tableau 14 et la figure 21b comparent les valeurs que nous avons obtenues aux
données publiées ou communiquées par les autres laboratoires. Les écart-types sur nos valeurs
reportés dans le tableau 14 correspondent à la dispersion sur 4 mesures, et sont dans certains
cas assez peu représentatifs des incertitudes moyennes (e.g. K, Ca, Tb). La figure 21b propose
une intercomparaison incluant les équipes aux travaux desquelles nous nous référerons par la
suite. Dans l'ensemble, nos résultats sont tout à fait compatibles avec ceux des autres analystes.
Les décalages relevés précédemment pour Ca et Ba sont également observés ici, et il apparaît
que ces éléments gagnent à être comparés sur les valeurs déterminées par INAA plutôt que par
PIXE. Une bonne correspondance est obtenue avec les données du British Museum Research
Laboratory. Les écarts les plus importants portent sur Cr et Ce, écarts également observés par
rapport au laboratoire de Sofia et de Berlin pour Cr. Plusieurs phénomènes peuvent être une
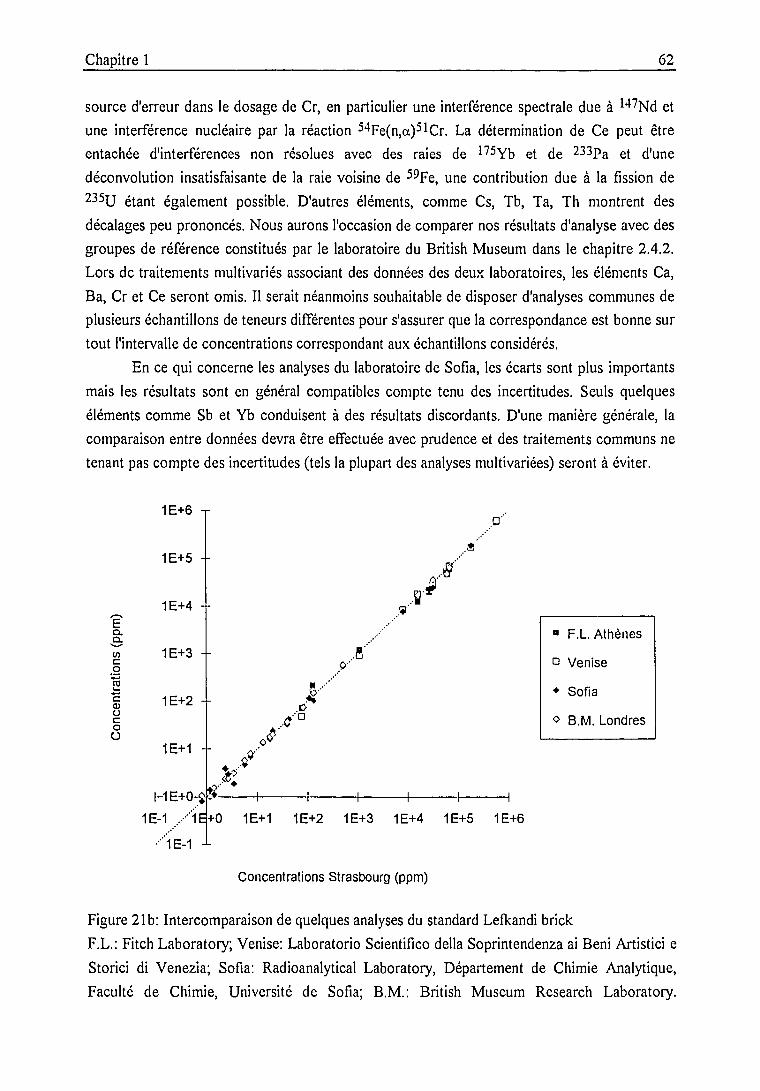
Chapitre 1 62
source d'erreur dans le dosage de Cr, en particulier une interférence spectrale due à 147Nd et
une interférence nucléaire par la réaction 54Fe(n,a)51Cr. La détermination de Ce peut être
entachée d'interférences non résolues avec des raies de 17^Yb et de 233Pa et d'une
déconvolution insatisfaisante de la raie voisine de 59Fe, une contribution due à la fission de235U étant également possible. D'autres éléments, comme Cs, Tb, Ta, Th montrent des
décalages peu prononcés. Nous aurons l'occasion de comparer nos résultats d'analyse avec des
groupes de référence constitués par le laboratoire du British Museum dans le chapitre 2.4.2.
Lors de traitements multivariés associant des données des deux laboratoires, les éléments Ca,
Ba, Cr et Ce seront omis. Il serait néanmoins souhaitable de disposer d'analyses communes de
plusieurs échantillons de teneurs différentes pour s'assurer que la correspondance est bonne sur
tout l'intervalle de concentrations correspondant aux échantillons considérés.
En ce qui concerne les analyses du laboratoire de Sofia, les écarts sont plus importants
mais les résultats sont en général compatibles compte tenu des incertitudes. Seuls quelques
éléments comme Sb et Yb conduisent à des résultats discordants. D'une manière générale, la
comparaison entre données devra être effectuée avec prudence et des traitements communs ne
tenant pas compte des incertitudes (tels la plupart des analyses multivariées) seront à éviter.
Q.0
mo
1O
O
O
1E+6 -r
1E+5 --
1E+4 --
1E+3 --
1E+2 --
1E+1 --
ME+O-$&
,r•
D
•
F.L. Athènes
Venise
Sofia
B.M. Londres
h H
1E-1 / 1E+0 1E+1 1E+2 1E+3 1E+4 1E+5 1E+6
•••"iE-1 - -
Concentrations Strasbourg (ppm)
Figure 21b: Intercomparaison de quelques analyses du standard Lefkandi brick
F.L.: Fitch Laboratory; Venise: Laboratorio Scientifico délia Soprintendenza ai Béni Artistici e
Storici di Venezia; Sofia: Radioanalytical Laboratory, Département de Chimie Analytique,
Faculté de Chimie, Université de Sofia; B.M.: British Museum Research Laboratory.
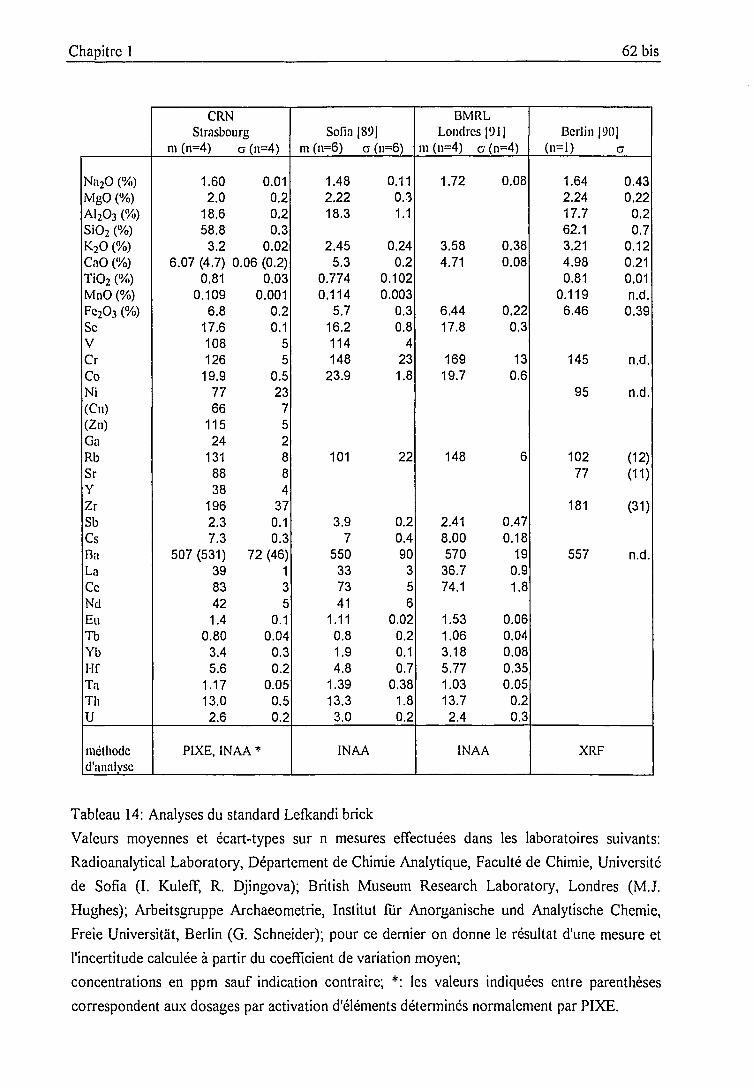
Chapitre 1 62 bis
Na2O (%)MgO (%)A12O3 (%)SiO2 (%)K2O (%)CaO (%)TiO2 (%)MnO (%)FC2O3 (%)SeVCrCoNi(Cu)(Zn)GaRbSrYZrSbCsRaLaCeNdEuTbYbHfTaThU
méthoded'aiinlvsc
CRNStrasbourg
m(n=4) g(n=4)
1.602.0
18.658.8
3.26.07 (4.7) 0.06
0.810.109 C
6.817.6108126
19.97766
11524
1318838
1962.37.3
507 (531) 723983421.4
0.803.45.6
1.1713.02.6
PIXE, INAA *
0.010.20.20.3
0.02(0.2)0.03
J.0010.20.1
55
0.523
752884
370.10.3
.(46)135
0.10.04
0.30.2
0.050.50.2
Sofiam (n=6)
1.482.2218.3
2.455.3
0.7740.114
5.716.2114148
23.9
101
3.97
550337341
1.110.81.94.8
1.3913.3
3.0
[89]g (n=6)
0.110.31.1
0.240.2
0.1020.003
0.30.8
4231.8
22
0.20.490
356
0.020.20.10.7
0.381.80.2
INAA
BMRLLondres [91]
m(n=4) c(n=4)
1.72
3.584.71
6.4417.8
16919.7
148
2.418.00570
36.774.1
1.531.063.185.771.0313.72.4
INAA
0.08
0.380.08
0.220.3
130.6
6
0.470.18
190.91.8
0.060.040.080.350.05
0.20.3
Berlin [90](n=l)
1.642.2417.762.13.214.980.81
0.1196.46
145
95
10277
181
557
XRF
g
0.430.22
0.20.7
0.120.210.01n.d.
0.39
n.d.
n.d.
(12)(11)
(31)
n.d.
Tableau 14: Analyses du standard Lefkandi brick
Valeurs moyennes et écart-types sur n mesures effectuées dans les laboratoires suivants:
Radioanalytical Laboratory, Département de Chimie Analytique, Faculté de Chimie, Université
de Sofia (I. Kuleff, R. Djingova); British Museum Research Laboratory, Londres (M.J.
Hughes); Arbeitsgruppe Archaeometrie, Institut fur Anorganische und Analytische Chemie,
Freie Universitât, Berlin (G. Schneider); pour ce dernier on donne le résultat d'une mesure et
l'incertitude calculée à partir du coefficient de variation moyen;
concentrations en ppm sauf indication contraire; *: les valeurs indiquées entre parenthèses
correspondent aux dosages par activation d'éléments déterminés normalement par PIXE.

chapitre 2.
Caractérisation des pâtes céramiques pour l'étude de laproduction et de la diffusion des céramiques
archéologiques
Application au cas des céramiques byzantines de Pergame

Chapitre 2 ^ 63
2.1. Les études de provenance en céramologie: généralités
La céramique est l'un des vestiges matériels du passé les mieux représentés dans les
fouilles. Elle ne subit en général pas de dégradation importante dans le milieu d'enfouissement,
contrairement à d'autres matériaux tels le métal et le bois. Elle constitue de ce fait un témoin
privilégié des cultures anciennes.
Le but des études de provenance est d'attribuer des céramiques archéologiques
trouvées sur un site donné à leur lieu d'origine. La mise en évidence d'une circulation entre le
site de fabrication et celui de découverte peut avoir de nombreuses implications sur les plans
archéologique et historique: existence de contacts entre deux populations, reconstitution de
routes commerciales, évolution des sources d'approvisionnements, transferts technologiques...
Nous ne donnerons ici que quelques notions de base sur la nature du matériau étudié et
sur les principes des études de provenance. Ces notions sont développées dans les articles et
ouvrages de synthèse (e.g. [92, 66, 75, 74]) ou dans d'autres thèses (e.g. [93, 94, 69]).
Signalons, outre les publications dans des périodiques comme Archaeometry, Journal of
Archaeological Science, Journal of Field Archaeology, Revue d'Archéométrie, ... l'excellente
revue bibliographique Art and Archaeology Technical Abstracts (AATA) qui est la seule à
couvrir le domaine de l'archéométrie à ma connaissance.
Le matériau céramique
Dans son sens commun, le terme "céramique" désigne une terre argileuse cuite. Cette
terre contient deux composantes qui jouent chacune un rôle spécifique: d'une part, les
minéraux argileux dont le caractère plastique permet le façonnage de la céramique et qui sont
responsables de la cohésion de la pièce à la cuisson; d'autre part les éléments non plastiques, ou
dégraissant [95], qui jouent le rôle de "squelette" ou parfois de fondant (matériaux à base
d'alcalins). Cette définition de la céramique est très restrictive et exclut en particulier les
céramiques à pâte siliceuse communes au Moyen-Orient (e.g. [96]). Elle est cependant adaptée
pour la céramique d'époque médiévale qui est considérée dans le cadre de ce travail. Des
définitions plus complètes ont été données dans [97, 98].
Les minéraux argileux sont des silicates d'alumines hydratés composés de particules très
fines (<2um). Ces particules s'assemblent en feuillets entre lesquels des molécules d'eau et des
ions sont susceptibles d'être adsorbés, d'où le caractère plastique. Les minéraux argileux ont
des propriétés complexes [99, 100, 101] et nous n'en évoquerons que quelques unes parmi les
plus importantes pour notre propos. Les minéraux argileux sont classés selon leurs structures
cristallines en plusieurs familles, parmi lesquelles on peut citer les kaolinites, les illites et les
smectites. Du point de vue chimique, leurs compositions se déduisent de celle de la kaolinite:

Chapitre 2 6£
4. L'illite s'en distingue par la présence de cations K+ dans les sites inter-feuillets,
la smectite par celle de Na+ et Ca2+, divers autres ions (Fe, Mn, Mg, Ti,...) pouvant également
être intégrés. Leurs conditions de formation sont différentes. La kaolinite résulte de la
dégradation de roches acides, en milieu acide. L'illite se forme plutôt dans des conditions
alcalines, de fortes concentrations en K et Al constituant des facteurs favorables. La smectite
est issue de l'altération de roches basiques, en particulier de roches volcaniques, dans des
conditions basiques.
La formation des minéraux argileux résulte principalement de l'altération par hydrolyse
des minéraux constitutifs des roches, en particulier des feldspaths. De façon très schématique,
on peut distinguer les argiles des altérations, qui se forment sur le site même de la dégradation
de la roche, les argiles sédimentaires détritiques, qui ont subit un transport aboutissant à un
bassin de sédimentation, et les argiles néoformées, qui précipitent à partir de produits dissouts
dans l'eau [100].
Les dépôts argileux "naturels" sont constitués d'un mélange de minéraux argileux qui
peuvent être de nature minéralogique différente et d'éléments non argileux. Nous appelerons ce
mélange "argile". Le matériau élaboré par le potier à partir de l'argile est nommé "pâte" [95] ou
"pâte céramique". D'après les études ethnographiques, les ressources argileuses utilisées par les
potiers sont en général localisées à proximité des ateliers (moins de 10 km) [92] - ce qui n'est
plus le cas dans les sociétés industrialisées.
Les éléments non argileux présents peuvent être de nature très diverse: fragments de
roches, minéraux (quartz, feldspaths, calcite, oxydes de fer, ...), matériaux d'origine végétale
ou animale (coquilles), matière organique, ... En plus du dégraissant naturel, les pâtes
céramiques peuvent contenir dans certains cas du dégraissant ajouté intentionnellement par le
potier (e.g. dans une argile trop plastique dite "grasse" - d'où le terme de dégraissant [97]).
Certains dégraissants sont typiques d'une intervention humaine, comme la chamotte, terre cuite
broyée [95] dont le rôle est de limiter les phénomènes de retrait à la cuisson.
Divers revêtements de surface peuvent être appliqués sur la pâte céramique: vernis,
engobes, glaçures, peintures, .... Certains sont également de nature argileuse (e.g. engobes),
d'autres s'apparentent à un verre (glaçure) et assurent l'imperméabilité de la pièce. Ces
revêtements peuvent être combinés pour donner lieu à des tffets décoratifs. Les techniques de
décoration, les motifs et les styles font partie des données utilisées dans la description
archéologique des céramiques. L'analyse des revêtements de surface pour l'étude des
technologies anciennes de fabrication constitue un domaine important de l'archéométrie [102,
103, 104].

Chapitre 2 6S_
Méthodologie en étude de provenance
Deux types de méthodes sont communément mises en oeuvre: les méthodes d'analyse
élémentaire et les méthodes d'analyses minéralogiques et pétrographiques, issues de la
géologie. Dans le premier cas, on considère l'analyse globale de l'argile., i.e. des minéraux
argileux et du dégraissant. Dans le deuxième, on caractérise le dégraissant et la texture de la
pâte, mais les argiles ne peuvent en général être déterminées, leur structure étant détruite à la
cuisson.
Ces méthodes peuvent être utilisée à deux fins: la classification ou l'attribution [105].
Cette dernière opération procède par référence soit à des céramiques de provenance connue,
soit à des matériaux argileux prélevés sur le terrain et de localisation connue, soit encore à des
cartes géologiques.
L'idée de caractériser une céramique par sa composition élémentaire dans le but de
déterminer sa provenance est fort ancienne puisque Harbottle [74] propose de l'attribuer à
Fouqué, archéologue français de la fin du 19èmc siècle. Elle repose sur le postulat que chaque
gisement argileux présente une composition qui lui est propre. En corollaire, deux céramiques
fabriquées en un même lieu de production auraient la même composition; inversement, des
céramiques de compositions différentes ne proviendraient pas du même atelier. La nature
géologique du matériau n'intervient que dans le cadre de la définition d'un "espace de non-
résolution", au sein duquel des formations géologiques uniformes génèrent des argiles
indiscernables [97, 106]. De nombreux laboratoires ont ainsi constitué des bases de données
qui rassemblent les compositions élémentaires de groupes de céramiques de provenance
connue. Les méthodes d'analyse utilisées sont très variées: spectroscopie d'émission optique
(Oxford [107], Fitch Laboratory [217]), fluorescence X (Lyon [87]), activation neutronique
(Lawrence Berkeley Laboratory [73], Brookhaven [74]). L'attribution d'un tesson d'origine
indéterminée à son lieu de production est basée sur la similitude de composition entre le tesson
et le groupe de céramiques représentatif du site de production. La comparaison des
compositions est réalisée soit élément par élément (statistiques univariées [107, 73]), soit au
moyen de techniques statistiques multivariées dont l'utilisation s'impose rapidement dès lors
que le nombre de données en jeu est important [74, 108]. Les principes de certaines d'entre
elles sont exposés en 2.4.1.2.
Les analyses minéralogiques et pétrographiques sont basées sur les propriétés
cristallographiques et optiques des solides cristallisés (cf. annexe 6.A). Les premières sont
réalisées par diffraction des rayons X: la diffraction sur les plans cristallins selon la loi de Bragg
permet l'identification des espèces minérales présentes [109]. Les propriétés cristallographiques
et optiques (indices de réfringence) d'une inclusion minérale déterminent ses caractéristiques en
lumière polarisée et en lumière polarisée et analysée ce qui permet leur identification au
microscope polarisant [110]. Les analyses pétrographiques donnent de nombreuses

Chapitre 2 66
informations relatives à la pâte et aux inclusions qu'elle contient, notamment leur fréquence,
leur distribution et leur nature.
Le principe de l'utilisation des analyses minéralogiques et pétrographiques pour les
études de provenance repose sur la relation entre le dépôt argileux et les roches-mères de
l'altération desquelles il résulte [111, 112, 98]. L'argile utilisée par les potiers résulte en
général de l'altération de roches qui appartiennent à un même bassin de sédimentation: les
inclusions lithiques et minérales présentes dans la pâte reflètent ainsi les ressources géologiques
environnantes. L'application en étude de provenance est illustrée schématiquement sur la
figure 22, empruntée à Maggetti [113].
Figure 22: Représentation schématique du principe de l'application de l'analyse pétrographique
en étude de provenance (d'après [113]).
Les tessons trouvés sur les sites archéologiques A et B et le tesson I trouvé en C ont des
caractéristiques pétrographiques compatibles avec le contexte géologique de leur site de
découverte et peuvent être présumés de fabrication locale. Le tesson II de C contient par
contre des inclusions tout à fait différentes qui ne correspondent à aucune formation
géologique locale. Il en est déduit qu'il s'agit d'une céramique importée sur le site C.
L'approche basée sur l'analyse élémentaire alliée aux techniques statistiques est souvent
opposée à la description des céramiques par leurs caractéristiques minéralogiques et
pétrographiques. En effet, ces dernières sont en général qualitatives et recèlent un caractère
descriptif qui peut être considéré comme subjectif. On remarquera toutefois qu'un traitement
quantitatif des données pétrographiques est possible [114]. D'autre part, certaines techniques
statistiques peuvent s'appliquer à des données qualitatives [115]. Il n'y a donc pas de différence
fondamentale quant à la manière de traiter les données élémentaires ou minéralogiques et
pétrographiques.

Chapitre 2 67_
Certains cas demandent l'usage préférentiel de l'une des approches. Ainsi, lapétrographie est particulièrement bien adaptée à la caractérisation des céramiques grossièresriches en inclusions, dont la composition élémentaire est trop variable pour être distinctive.Inversement, une céramique très fine ne se prêtera pas à l'analyse pétrographique car lesinclusions ne seront pas identifiables, l'analyse élémentaire étant alors plus adéquate.
D'une manière générale, analyses élémentaires, minéralogiques et pétrographiquesgagnent à être utilisées de façon complémentaire (e.g. [116]). Les premières permettent unegestion plus souple d'un grand nombre de données et autorisent des comparaisons rigoureusesentre tessons. Les dernières ont l'avantage de pouvoir être mises en relation avec les donnéesdu terrain et de donner des indications sur les pratiques du potier [117].

Chapitre 2
2.2. La céramique byzantine médiévale:
catégories archéologiques et jalons analytiques
Les fouilles des sites antiques ont longtemps négligé les couches médiévales, moins
prestigieuses que les niveaux gréco-romains. Il n'est donc pas rare que les vestiges byzantins
aient été purement et simplement "décapes" pour dégager les couches inférieures. De ce fait ils
nous parviennent souvent dépourvus de contexte archéologique précis. La céramique de cette
époque a été encore plus profondément affectée du manque d'intérêt des archéologues. On
peut invoquer le peu de prestige de la céramique parmi les arts décoratifs byzantins. Les tables
des cours byzantines la délaissaient pour des ouvrages d'orfèvrerie et des vaisselles de pierres
fines - du moins jusqu'au déclin ultime de l'empire. D'autre part, la supériorité technique des
voisins perses, arabes ou turcs a longtemps relégué la céramique byzantine au rang d'imitation
malhabile de modèles développés par les cultures environnantes. La spécificité de cette
céramique semble être maintenant mieux prise en compte [118, 119, 120], quoique cette
considération concerne surtout les céramiques glaçurées. Les céramiques culinaires ou de
stockage sont encore peu étudiées à l'exception des amphores, qui restent une source
d'information privilégiée sur les échanges et les circuits commerciaux.
Si les études de céramiques byzantines sont actuellement plus nombreuses, elles
s'appuient encore largement sur les travaux de quelques pionniers. Une première synthèse a été
publiée par D. Talbot Rice en 1930 [121], à partir du matériel issu des fouilles anglaises à
Constantinople. On se réfère actuellement plutôt à la classification proposée en 1942 par
C. Morgan [1], dans sa monographie sur les céramiques mises au jour par les Américains à
Corinthe. Ses critères de distinction reposent essentiellement sur les techniques de décoration,
et dans une moindre mesure sur les motifs, le style, la chronologie, la typologie et la couleur de
la pâte céramique (opposition entre pâtes blanches et pâtes rouges). La classification de
Morgan s'articule autour de quatre grandes catégories:
• céramique non glaçurée ("unglazed ware")
• céramique monochrome ("plain-glazed ware")
• céramique peinte ("painted ware")
• céramique décorée par la technique du sgraffito ("sgraffito ware")
les trois dernières désignant implicitement des céramiques glaçurées, et en général également
engobées. Il n'entre évidemment pas dans nos intentions de faire une présentation exhaustive
des types de céramiques isolés par Morgan au sein de chacune de ces catégories. Nous
tenterons uniquement de donner quelques points de repère, et de présenter parmi les types
familiers aux spécialistes ceux qui ont suscité les rares analyses physico-chimiques,
minéralogiques ou pétrographiques de matériel byzantin.

Chapitre 2 69
Si des tentatives emhfyoiinaM£
techniques sont déjà présentas dîin
attendre les duties 1980 p£W voir
céramiques byzantines, dont Utt article
publication de fcW en la maigre [ l ^
prise de conscfance de la p<V1 des
archéométriqu^ dans les rô£Hçrch^
clairement identifié la nature â\t Matériel
fours eux-mêmes - pièces inachevées, \hiih 4% buisson, éléments liés à la production comme les
de deux productions à l'aide d'examens
ie Morgan [1] et de Talbot Rice [122], il faut
line série d'études incluant des analyses de
* Jones qui peut être considéré comme la
semble-t-il dans ces années là à une nette
byzantinistes du potentiel des méthodes
provenance [124], Parallèlement, ils ont alors
propre à attester un atelier en l'absence des
pernettes,..., [12$J. Ces pièces r>e
par les potiers è- proximité des béliers,
Megaw et par K£. Jones, du ffich
d'abord par la. cju&lité indiscutable de
géographique st chronologiq^^ en i
chypriotes, dont l'activité s'é(/h lonne
Elle comporte également des lisons
comme Constantinople et C î
producteurs. $w chaque site £<?tft
représentativité statistique. 1^ Seule
dont le rôle <3e ''groupe t^t'1 pûur
description technique des piè^^ et
bion identifiée permet la comparaison
ainsi que l'on peut espérer
aussi l'influen<?e clss produite
indirectement (\£$ travaux poft0W
intérêt commercial sont en général rejetées
menée conjointement par l'archéologue A.H.S.
âe l'école anglaise d'Athènes, se distingue tout
de référence. Elle atteint une vaste portée
âfà céramiques fabriquées dans sept sites grecs ou
au 19èmc siècle (productions post-byzantines),
dais des centres de "consommation" importants
derniers pouvant d'ailleurs être simultanément
MA vingtaine de tessons, ce qui assure une certaine
est constituée par les céramiques de Paphos,
aWrîbutions explique le caractère composite. La
éventuelle à une catégorie de céramique
s portant sur du matériel analogue. C'est
Voies commerciales la diffusion effective, mais
W 1^ fabrications locales, comme en témoignent
Mr du matériel italien [126, 127, 128] et bulgare
[129, 130, 131, 122]. Regrettas seutëflftflt C\m les analyses présentées par Megaw et Jones
[123] portent sur un nombre restreint *WéW0Mtè (cf. annexe 3), et que la méthode utilisée
(spectroscopiç d'émission ojWdue [§§)) %§i\ relativement peu précise. Elles gagneraient
également à être complétées fW une ( Vdtf W^néralogtque et pétrographique - ce qui a été en
partie réalisé par l/a2zarini et (?£logeftf [\1/4\ Heurs résultats et ceux des autres analystes qui
ont abordé la ç$f#wique byzantine sô^ht dilutes en relation avec les types de céramiques
concernés. Un^ présentation Syïithétify^ q^ itiAtériel analysé est donnée par le tableau 17, la
figure 26 indiquant la locaii^ion d^ X\\£$ (iArrespondant. Des paramètres plus techniques
(éléments dosés, conditions ejtyétfnwtAfos, ,•>,) sont précisés en annexe 3.
Je dirai bien peu de chQ20s sur
céramique non ^teçurée a rMAient
années, peubêite en raison d'une
emières catégories proposées par Morgan. La
Intérêt des archéologues jusqu'à ces dernières
permanence des formes qui rend difficile la

Chapitre 2 70_
corrélation avec un contexte archéologique précis [119]. Cependant, les descriptions de
matériel de fouilles récentes, notamment à Sparte et à Thèbes, accordent une plus large part
aux céramiques non glaçurées, ce qui permet d'en relever les attributs distinctifs du point de
vue de la typologie, des techniques de décoration, de l'aspect macroscopique de la pâte, ...
[133, 134, 135]. En ce qui concerne les recherches de provenance, le postulat est encore bien
répandu selon lequel une céramique "grossière" - le terme étant souvent utilisé, par abus de
langage, par opposition à "glaçuré" - ne peut être que de fabrication locale. Si les céramiques
glaçurées semblent se prêter davantage aux échanges, il ne faut peut-être pas négliger l'attrait
commercial d'un produit caractérisé par des propriétés mécaniques ou thermiques particulières
[133]. Quelques analyses élémentaires de céramiques non glaçurées, issues des sites de
Dhiorios (production), Athènes (production?) et Paphos sont présentées dans la référence
[123]. 11 faudra évidemment attendre de disposer d'un corpus de résultats beaucoup plus
important pour pouvoir en tirer quelque conclusion - et probablement avoir aussi recours à
l'examen pétrographique, qui apporte de nombreuses informations dans le cas de pâtes
"grossières" riches en inclusions. Notons seulement, à propos de la comparaison entre
céramique culinaire et céramique fine d'Athènes, que l'on peut s'attendre à ce que les
compositions de ces deux types de productions ne concordent pas. En effet, les qualités
recherchées sont bien distinctes et motivent couramment sur un même site un traitement
différent de l'argile, voire un choix de matières premières différentes (cf. 2.4.2.). Dans le cas
d'Athènes, la comparaison semble justifiée en raison d'une certaine uniformité des traits
géochimiques caractéristiques des productions de l'Attique (fortes teneurs en Mg, Cr, Ni),
quels que soient leur type et leur datation [66].
Des tessons d'amphores d'Athènes (production?) et de Kounoupi (production) ont
également été examinés [123]. Nous ne traiterons pas ici de la question des amphores, qui
nécessiterait à elle seule une étude spécifique (voir par exemple les nombreux articles dans
[118]). Similaires aux céramiques par leur nature et par les routes commerciales empruntées,
elles en diffèrent par le fait qu'elles sont diffusées non pour leur valeur intrinsèque, mais en tant
que "contenant" de denrées alimentaires.
La céramique byzantine à glaçure monochrome, engobée ou non, présente peu de
paramètres de description supplémentaires à ceux déjà cités pour la céramique non glaçurée.
La gamme de coloris est plutôt restreinte: on trouve surtout du vert foncé, du jaune, du brun et
du brun-mauve. Ces teintes sont très répandues dans le monde médiéval, tant oriental
qu'occidental, et ne présentent de ce fait en elles-même pas un caractère distinctif. Les
colorants correspondants, introduits dans une glaçure de type plombifère, sont respectivement
à base de cuivre, de fer - donnant en concentration croissante des teintes du jaune au brun - et
de manganèse (cf. analyses de glaçures en annexe). Ainsi, bien que les examens techniques de
matériel byzantin fassent jusqu'à présent défaut, on peut acquérir une certaine appréhension des

Chapitre 2 7J_
procédés de fabrication de ces céramiques à glaçures par analogie avec des productions
médiévales comparables [136],
Dans le tableau 17, les tessons de céramique glaçurée monochrome analysés ne sont en
général pas mentionnés lorsqu'ils sont associés à des céramiques peintes ou à sgraffito.
Les céramiques peintes et les céramiques à sgraffito sont celles qui ont suscité le plus
grand nombre d'études, de par la diversité qu'offre la combinaison des motifs et des traitements
de surface. L'un des premiers types de céramiques peintes à se détacher, vers le 7 è m c siècle, des
produits de tradition romaine est nommé "white ware". Il se distingue essentiellement par sa
pâte blanche. Une de ses variétés, caractéristique des 10èmc - l]ème siècles, est renommée
pour ses motifs polychromes peints sous glaçure incolore. Cette variété est connue tant sous
forme de poterie que sous celle de carreaux de pavement ou de panneaux de revêtement
architecturaux. Elle est surtout bien représentée à Constantinople et autour de la mer Noire,
notamment en Crimée et en Bulgarie.
L'abondance de la "white ware" dans les vestiges constantinopolitains a suggéré
l'existence d'un ou de plusieurs centres producteurs à proximité de la capitale. Plusieurs autres
arguments ont été avancés, comme la préexistence locale d'un type de céramiques blanches non
glaçurées de la même famille, la réapparition de céramiques à pâte blanche à Constantinople
après la conquête ottomane - suite à une période dominée par les céramiques à pâte rouge - ,
ou encore la présence non loin d'affleurements d'argile blanche [137].
Pour tester le caractère local de cette production, des analyses élémentaires de tessons
de "white ware" issus des fouilles de Corinthe et de Constantinople ainsi qu'une argile blanche
utilisée actuellement par les potiers d'un faubourg d'Istanbul ont été effectuées [123]. La
similitude de composition (tableau 15) semble à première vue cautionner l'hypothèse d'un lieu
de fabrication unique, qui dans ce cas serait Constantinople. On ne peut cependant conclure
aussi rapidement. En l'absence d'indices archéologiques d'une production locale à
Constantinople - à l'exception (?) d'un raté de cuisson non publié mentionné par Megaw [123]
(fouilles de l'église Kalenderhane Camii) - , il faudrait pouvoir se baser sur des analyses
comparatives confrontant les sites susceptibles d'avoir produit des céramiques "white ware" et
non pas sur une simple ressemblance. Parmi les sites potentiels, Nicée - plus connue sous le
nom d'Iznik pour sa production ottomane, mais où des fours pré-ottomans sont également
attestés [138] - semble se distinguer par une argile nettement plus calcaire [123]. D'autres
pistes sont suggérées par les sources écrites, comme cette mention de "plaques de terres cuites
de Nicomédie" ornant une église de Constantinople [119].
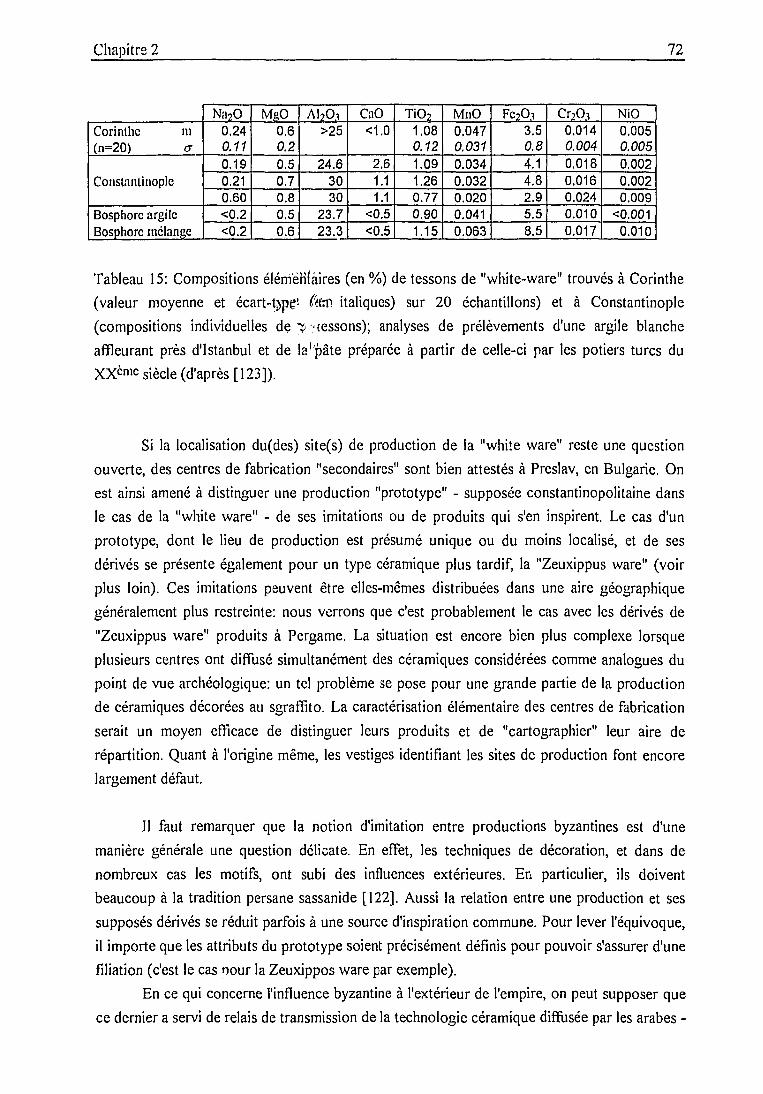
Chapitre 2 72
Corinthc m(n=20) a
Constantinople
Bosphore argile
Bosphore melange
Na2O0.240.110.190.210.60<0.2<0.2
MgO0.60.20.50.70.80.50.6
AI2O.1>25
24.63030
23.723.3
CaO<1.D
2.61.11.1
<0.5<0.5
TiO2
1.080.121.091.260.770.901.15
MnO0.0470.0310.034
0.0320.020
0.0410.063
FC2O33.50.8
4.14.82.95.58.5
Cr2O.T0.0140.0040.0180.0160.0240.0100.017
NiO0.0050.0050.0020.0020.009
<0.0010.010
Tableau 15: Compositions élémentaires (en %) de tessons de "white-ware" trouvés à Corinthe
(valeur moyenne et écart-tjpgî $en italiques) sur 20 échantillons) et à Constantinople
(compositions individuelles de y; cessons); analyses de prélèvements d'une argile blanche
affleurant près d'Istanbul et de la'pâte préparée à partir de celle-ci par les potiers turcs du
è siècle (d'après [123]).
Si la localisation du(des) site(s) de production de la "white ware" reste une question
ouverte, des centres de fabrication "secondaires" sont bien attestés à Preslav, en Bulgarie. On
est ainsi amené à distinguer une production "prototype" - supposée constantinopolitaine dans
le cas de la "white ware" - de ses imitations ou de produits qui s'en inspirent. Le cas d'un
prototype, dont le lieu de production est présumé unique ou du moins localisé, et de ses
dérivés se présente également pour un type céramique plus tardif, la "Zeuxippus ware" (voir
plus loin). Ces imitations peuvent être elles-mêmes distribuées dans une aire géographique
généralement plus restreinte: nous verrons que c'est probablement le cas avec les dérivés de
"Zeuxippus ware" produits à Pergame. La situation est encore bien plus complexe lorsque
plusieurs centres ont diffusé simultanément des céramiques considérées comme analogues du
point de vue archéologique: un tel problème se pose pour une grande partie de la production
de céramiques décorées au sgraffito. La caractérisation élémentaire des centres de fabrication
serait un moyen efficace de distinguer leurs produits et de "cartographier" leur aire de
répartition. Quant à l'origine même, les vestiges identifiant les sites de production font encore
largement défaut.
Il faut remarquer que la notion d'imitation entre productions byzantines est d'une
manière générale une question délicate. En effet, les techniques de décoration, et dans de
nombreux cas les motifs, ont subi des influences extérieures. En. particulier, ils doivent
beaucoup à la tradition persane sassanide [122]. Aussi la relation entre une production et ses
supposés dérivés se réduit parfois à une source d'inspiration commune. Pour lever l'équivoque,
il importe que les attributs du prototype soient précisément définis pour pouvoir s'assurer d'une
filiation (c'est le cas oour la Zeuxippos ware par exemple).
En ce qui concerne l'influence byzantine à l'extérieur de l'empire, on peut supposer que
ce dernier a servi de relais de transmission de la technologie céramique diffusée par les arabes -

Chapitre 2 73_
souvent initialement développée à partir de modèles extrême-orientaux - vers la Méditerranéeoccidentale, en particulier vers l'Italie par l'intermédiaire des marchands vénitiens et génois.Dans ce cadre, la question du rôle de la "white-ware" polychrome comme source de lamajolique italienne a donné lieu à des comparaisons de composition entre des tessons de "whiteware" et de "proto-majolique" exhumés à Corinthe [123]. En distingant une compositionspécifique à chaque groupe, ces analyses prouvent l'absence de relation directe entre les deuxtypes de céramiques. Ceci n'est pas étonnant au vu des différences techniques, comme le fontjustement remarquer Megaw et Jones. En effet, la majolique est une céramique de pâte rouge,couverte d'une glaçure opacifiée à l'étain avec un décor peint sur glaçure, alors que les "white-ware" sont à pâte blanche avec une glaçure transparente, probablement plombifère et nonstannifère, sous ou dans laquelle les pigments ont été appliqués - ces derniers points demandantà être précisés par une étude technique des "white-ware".
Une influence bien moins contestable concerne la transmission de la technique dusgraffito de Byzance vers l'Italie [139]. Le sgraffito - de l'italien graffiare: gratter, égratigner,griffer - consiste à inciser le motif dans un engobe dont on a préalablement recouvert la pâtecéramique. L'effet décoratif est obtenu par contraste entre l'engobe (blanc) et le fond de pâte(rouge) qui apparaît au niveau des incisions. Le tout est en général recouvert d'une glaçureplombifère avec d'éventuelles adjonctions de colorants. Cette technique n'est pourtant paspropre au monde byzantin. Au contraire, elle était déjà utilisée dans le monde islamique auxçcmc. locme siècles. On trouve des coupes décorées au sgraffito rehaussé de pigments verts etbruns depuis Samarcande, en Asie Centrale, où une production de ce type est attestée [140],jusqu'à Fustat, en Egypte, El Zabid, au Yémen, et sur les sites irakiens de Bagdad, Samarra,Bassorah, .... Des analyses de tessons trouvés dans ces derniers contextes montrent une pâtecalcaire couverte d'un engobe quartzeux et d'une glaçure à forte teneur en plomb [35, 96].Dans le monde byzantin, le sgraffito apparaît plus tardivement, vers la fin du 1 l è n i c siècle. Il vadevenir l'une des techniques de décoration les plus répandues de la production byzantine. Lesobjets ornés sont surtout de grandes coupes à pied ou des plats profonds. Le répertoiredécoratif est relativement varié: ornements géométriques, motifs floraux, représentationsd'animaux et, plus rarement, représentations humaines.
On peut distinguer un premier type dit "sgraffito fin", daté de la fin du 1 l è m c siècle etdu début du 12 è n i c siècle. Il est caractérisé par une ornementation à base d'incisions réaliséesexclusivement à la pointe fine, qui forment des motifs particuliers souvent organisés au sein decercles concentriques (figure 23a)). On remarque parmi les motifs des écritures coufiques, quitémoignent d'un certain engouement pour la calligraphie arabe. Le "sgraffito fin" présenteinvariablement une glaçure transparente monochrome qui apparaît en général jaunâtre surl'engobe blanc et rouge-brun sur les parties exposées de la pâte.

Chapitre 2 74
Figure 23: ci-dessus de gauche à droite: céramique décorée par la technique du sgraffito,
correspondant au type "sgraffito fin" (Thèbes, 12ènic siècle) et céramique décorée par la
technique du champlevé (Corinthe, fin 12èmc siècle) [141].
Au cours du 12èmc siècle, la technique du sgraffito va évoluer d'une part avec
l'utilisation d'outils créant des incisions plus larges (gouge), voire un enlèvement de zones
entières d'engobe (technique du champlevé), d'autre part avec l'introduction de pigments dans
la glaçure. En simplifiant la nomenclature de Morgan, j'ai désigné dans le tableau 17 par
"sgraffito incisé" des céramiques au sgraffito dont les incisions ne sont pas toutes fines, le
terme "peint" correspondant à l'adjonction d'au moins une couleur distincte de celle du fond.
Le sgraffito incisé va devenir la forme la plus populaire aux 13èmc - 14ème siècles, notamment
dans sa variante du champlevé où l'engobe est "evidé" autour du motif, un peu comme dans un
bas-relief (figure 23b)). D'une manière générale, on peut noter que le sgraffito est une
adaptation au matériau céramique des techniques de gravure des objets métalliques [142].
Les sites de fabrication de céramiques au sgraffito, au sens large du terme (mais en se
limitant à la production byzantine, de tradition byzantine ou "d'influence byzantine", avec
l'ambiguïté que cela suppose) sont multiples. Les analyses ont à ce jour permis de caractériser
un petit nombre d'entre eux, dont l'activité s'échelonne entre le l l è n i c et le 19èmc siècle:
Constantinople (?) [123, 126] et Sardes (?) [143] en Turquie, Corinthe [123, 126],
Thessalonique [123, 126], Athènes (?) [123, 126], Serres [144] et Didymoteichon [123] en
Grèce, Lemba [123, 126] et Lapithos [123] à Chypre, Venise [126, 127, 128] et Padoue [128]
en Italie, Varna [130, 129, 132] en Bulgarie (cf. tableau 17, les points d'interrogations
s'appliquent à des sites dont le caractère producteur n'est pas pleinement justifié dans les
références citées).
Sur les trois derniers sites, la coexistence de céramiques d'importation d'origine
byzantine et d'imitations locales témoigne de l'influence byzantine au travers des relations

Chapitre 2 75_
commerciales. Si les analyses permettent la distinction entre productions locales et
importations, l'attribution des céramiques importées à leur lieu de production n'est possible que
dans de rares cas [126], faute de groupes de référence. L'hypothèse d'une provenance
métropolitaine repose soit sur des circuits d'échanges connus historiquement, soit sur des
particularismes (monogramme des empereurs Paléologues typiquement constantinopolitain sur
les céramiques des fouilles bulgares). Quoiqu'il en soit, on peut retenir à ce stade l'existence de
courants d'échange. Les découvertes de l'archéologie sous-marinc attestent une
commercialisation intensive de céramiques décorées au sgraffito: par exemple, un navire
naufragé au large de l'île grecque de Pelagonnisos en recelait plusieurs centaines, d'un type un
peu plus tardif que le "sgraffito fin" [118, 119, 123]. La diffusion, moins massive, de certaines
catégories peut être interprétée comme un effet indirect du commerce d'autres denrées ou du
transport de passagers. L'étude de V. François sur les céramiques de l'île de Thasos aux 13 è m e
- I4cmc siècles propose ainsi plusieurs hypothèses expliquant la circulation de petites quantités
de vaisselle par voie maritime selon les usages de la navigation de l'époque [136], Par exemple,
des céramiques proto-majoliques produites à Brindisi ont pu circuler aux 13 c m c - I4cmc siècles
en Méditerranée Orientale dans les effets personnels de passagers des navires marchands
partant de ce port pour la Palestine.
Les comparaisons les plus fructueuses du point de vue des recherches de provenance
sont celles qui portent sur du matériel bien défini du point de vue archéologique. On serait
tenté d'inclure dans cette catégorie le sgraffito fin. Une production en est attestée à Corinthe,
qui se distingue par une pâte fine très calcique, contenant de nombreuses inclusions de calcite
[123, 126]. Des céramiques de même type trouvées à Constantinople [123] présentent des
compositions élémentaires différentes (tableau 16). Pour autant que la notion de sgraffito fin
recouvre bien un type de production spécifique, il est donc assuré que sa fabrication n'était pas
centralisée. On peut d'ailleurs remarquer que ce groupe de tessons de céramiques trouvées à
Constantinople semble relativement hétérogène, si l'on considère l'écart-type des éléments Al et
Fe. Plusieurs facteurs sont susceptibles d'accroître l'écart-type, relevant de la procédure
analytique (incertitudes expérimentales, faible représentativité statistique de
l'échantillonnage,...), du caractère plus ou moins standardisé de la fabrication (quantités
variables de dégraissants) ou encore de paramètres divers, en particulier les phénomènes
d'altérations. On peut se demander lesquels de ces facteurs ont pu intervenir dans ce cas,
concernant un groupe relativement bien défini (20 tessons) et des éléments réputés stables au
cours de l'enfouissement comme Al et Fe. Une autre possibilité réside dans la présence de
plusieurs groupes. Dans ce cas, les sous-structures se distinguent-elles également sur des
critères archéologiques? - ce qui renvoie à l'éventuelle diversité de ce type.
En ce qui concerne l'éventualité que ces céramiques aient été produites localement à
Constantinople, les valeurs données par Lazzarini et Calogero [126] pour des tessons
originaires de la capitale - ou supposés tels (?) - présentent des teneurs en Na et Mg très
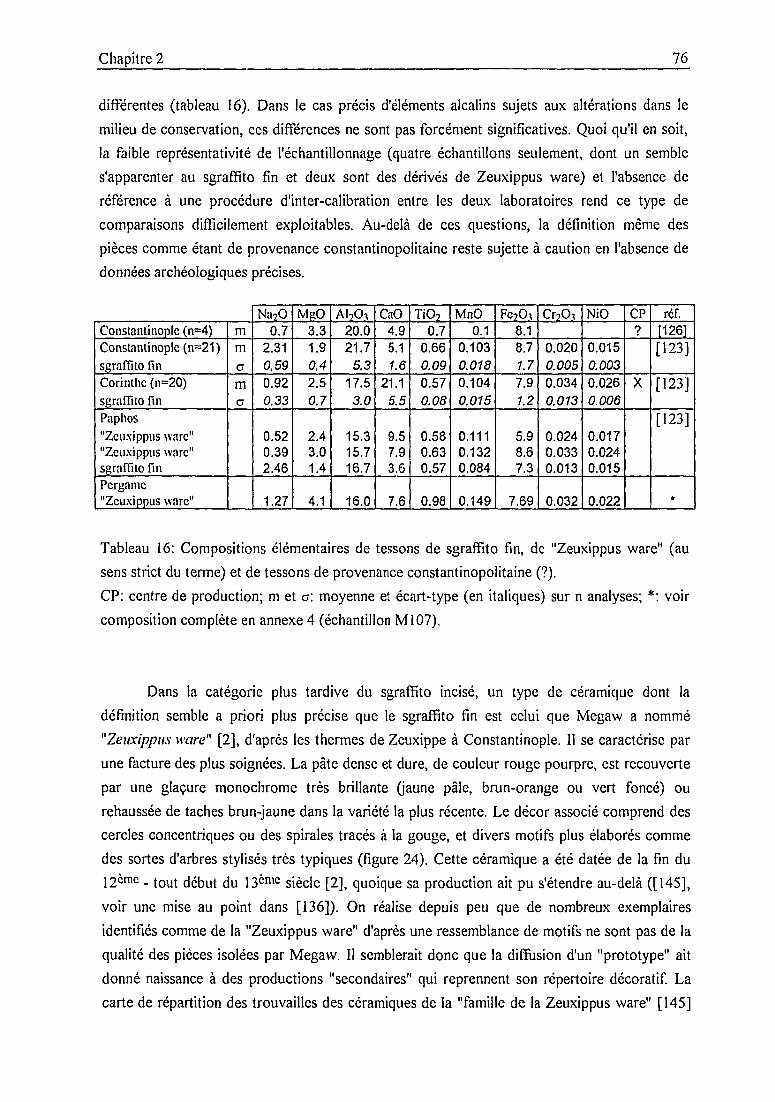
Chapitre 2 76
différentes (tableau 16). Dans le cas précis d'éléments alcalins sujets aux altérations dans le
milieu de conservation, ces différences ne sont pas forcément significatives. Quoi qu'il en soit,
la faible représentativité de l'échantillonnage (quatre échantillons seulement, dont un semble
s'apparenter au sgraffito fin et deux sont des dérivés de Zeuxippus ware) et l'absence de
référence à une procédure d'inter-calibration entre les deux laboratoires rend ce type de
comparaisons difficilement exploitables. Au-delà de ces questions, la définition même des
pièces comme étant de provenance constantinopolitaine reste sujette à caution en l'absence de
données archéologiques précises.
Constantinople (n=4)Constantinople (n=21 )sgraffito finCorinthc (n=20)sgraffito finPaphos"Zeuxippus ware""Zeuxippus ware"sgraffito finPcrganic"Zeuxippus ware"
mmoma
Na2O0.7
2.310.590.920.33
0.520.392.46
1.27
MgO3.31.90.42.50.7
2.43.01.4
4.1
20.021.7
5.317.53.0
15.315.716.7
16.0
CaO4.95.11.6
21.15.5
9.57.93.6
7.6
TiO2
0.70.660.090.570.08
0.580.630.57
0.98
MnO0.1
0.1030.0180.1040.015
0.1110.1320.084
0.149
Fc2Ch8.18.71.77.91.2
5.98.67.3
7.69
Cr7O,
0.0200.0050.0340.013
0.0240.0330.013
0.032
NiO
0.0150.0030.0260.006
0.0170.0240.015
0.022
CP?
X
réf.[126][123]
[123]
[123]
*
Tableau 16: Compositions élémentaires de tessons de sgraffito fin, de "Zeuxippus ware" (au
sens strict du terme) et de tessons de provenance constantinopolitaine (?).
CP: centre de production; m et a: moyenne et écart-type (en italiques) sur n analyses; *: voir
composition complète en annexe 4 (échantillon Ml07).
Dans la catégorie plus tardive du sgraffito incisé, un type de céramique dont la
définition semble a priori plus précise que le sgraffito fin est celui que Megaw a nommé
"Zeuxippus ware" [2], d'après les thermes de Zeuxippe à Constantinople. 11 se caractérise par
une facture des plus soignées. La pâte dense et dure, de couleur rouge pourpre, est recouverte
par une glaçure monochrome très brillante (jaune pâle, brun-orange ou vert foncé) ou
rehaussée de taches brun-jaune dans la variété la plus récente. Le décor associé comprend des
cercles concentriques ou des spirales tracés à la gouge, et divers motifs plus élaborés comme
des sortes d'arbres stylisés très typiques (figure 24). Cette céramique a été datée de la fin du
]2cmc . tout début du 13cmc siècle [2], quoique sa production ait pu s'étendre au-delà ([145],
voir une mise au point dans [136]). On réalise depuis peu que de nombreux exemplaires
identifiés comme de la "Zeuxippus ware" d'après une ressemblance de motifs ne sont pas de la
qualité des pièces isolées par Megaw. Il semblerait donc que la diffusion d'un "prototype" ait
donné naissance à des productions "secondaires" qui reprennent son répertoire décoratif. La
carte de répartition des trouvailles des céramiques de la "famille de la Zeuxippus ware" [145]

Chapitre 2 77
(figure 25) montre un vaste domaine de distribution - qui recouvre pratiquement celui du
sgraffito byzantin dans son ensemble.
Figure 24: Dessin représentant un des motifs typiques de la "Zeuxippus ware" (d'après [121])
Même en distinguant le prototype de ses dérivés, la question des centres de fabrication
de la "Zeuxippus ware" reste plutôt confuse. En ce qui concerne le prototype, l'hypothèse
courante est celle d'un lieu de production unique, dont la localisation à proximité de
Constantinople est accréditée par l'importance numérique des tessons de cette céramique dans
la capitale. Les rares analyses disponibles ne sont guère concluantes puisque seuls deux tessons
trouvés à Paphos identifiés comme de la "véritable Zeuxippus ware" ont été analysés [123] (cf.
tableau 16). A ceux-ci s'ajoute désormais un tesson de Pergame. Les résultats révèlent des
différences de composition assez importantes entre les trois échantillons, mais étant donné le
faible nombre d'échantillons il n'est évidemment pas possible de conclure à ce stade quant au
caractère unique de cette production. Pour ce qui est d'une origine constantinopolitaine, nous
renvoyons au commentaire des données concernant le sgraffito fin. Par ailleurs, la remarque de
Megaw et Jones [123] à propos d'une analogie de composition entre les "Zeuxippus ware"
trouvées à Paphos et un tesson issu du même contexte présentant des motifs typiquement
égéens [4] ne me parait pas devoir impliquer une origine égéenne: cette ressemblance très
relative concernant un échantillon unique n'est probablement pas significative. L'examen
pétrographique, pratiqué malheureusement sur les seuls tessons "de Constantinople", semble
plus informatif en révélant la présence de serpentines. Ces dernières, qui pourraient provenir de
laves basiques de la région de Constantinople [146], constituent un "marqueur" potentiel.
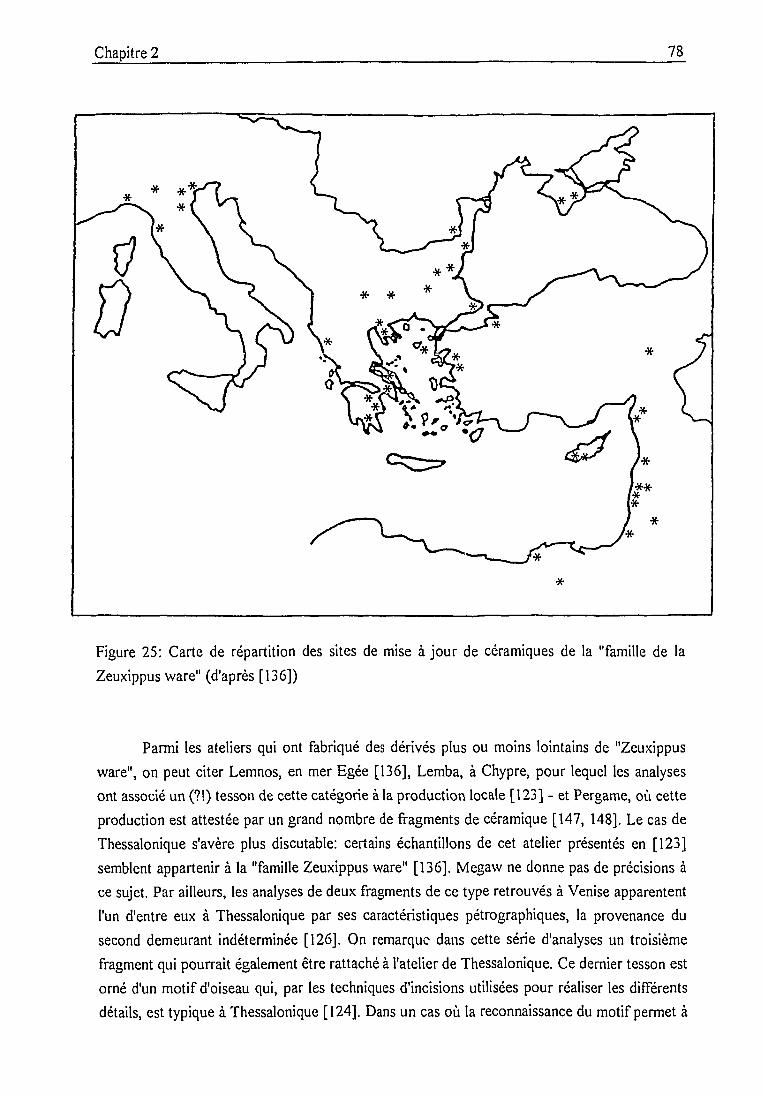
Chapitre 2 78
Figure 25: Carte de répartition des sites de mise à jour de céramiques de la "famille de la
Zeuxippus ware" (d'après [136])
Parmi les ateliers qui ont fabriqué des dérivés plus ou moins lointains de "Zeuxippus
ware", on peut citer Lemnos, en mer Egée [136], Lemba, à Chypre, pour lequel les analyses
ont associé un (?!) tesson de cette catégorie à la production locale [123] - et Pergame, où cette
production est attestée par un grand nombre de fragments de céramique [147, 148]. Le cas de
Thessalonique s'avère plus discutable: certains échantillons de cet atelier présentés en [123]
semblent appartenir à la "famille Zeuxippus ware" [136]. Megaw ne donne pas de précisions à
ce sujet. Par ailleurs, les analyses de deux fragments de ce type retrouvés à Venise apparentent
l'un d'entre eux à Thessalonique par ses caractéristiques pétrographiques, la provenance du
second demeurant indéterminée [126]. On remarque dans cette série d'analyses un troisième
fragment qui pourrait également être rattaché à l'atelier de Thessalonique. Ce dernier tesson est
orné d'un motif d'oiseau qui, par les techniques d'incisions utilisées pour réaliser les différents
détails, est typique à Thessalonique [124]. Dans un cas où la reconnaissance du motif permet à

Chapitre 2 19_
elle seule de conclure, on a la satisfaction de constater que la démarche archéométrique permet
de localiser correctement l'origine du produit ... Pour en revenir aux dérivés de la "Zeuxippus
ware", il est probable que d'autres ateliers restent encore à découvrir, notamment ceux qui ont
fournis les sites francs des côtes du Levant.
Sur ces derniers oi t également été découverts des tessons d'une catégorie de sgraffito
peint dite "Si Symeon ware", datée de la fin du 12èmc - fin du 13èmc siècles. Elle est nommée
ainsi par référence à son centre de production d'Antioche, dont le port (Al Mina) était
surplombé par le monastère de St Syméon. La "St Symeon ware" semble avoir été imitée à
Sardes, où des tessons similaires ont été soumis à des examens pétrographiques [143]. Ceux-ci
présentent un assemblage minéralogique compatible avec la géologie locale, ainsi qu'un
traitement de la pâte qui les apparentent à la facture très uniforme des céramiques trouvées sur
le site, qu'elles soient d'époque archaïque ou byzantine [149]. L'auteur en conclut à une
provenance locale. Une hypothèse est que les potiers d'Al Mina, chassés par la défaite des
Francs devant les Mamelouks, en 1264, ont exportés leur savoir-faire en dehors de la Syrie, et
l'ont transmis notamment aux potiers anatoliens de Sardes [136].
L'identification des centres de production d'un type donné de céramiques et de leurs
aires de diffusion n'est pas la seule "lecture" que l'on puisse faire des analyses. Par exemple, il
serait intéressant de pouvoir suivre l'évolution de la production sur un site, de comparer des
produits différents fabriqués simultanément ou successivement dans un même atelier ou groupe
d'ateliers. Ainsi, en ce qui concerne les céramiques de belle qualité réputées
constantinopolitaines, Lazzarini et Calogero [126] ne donnent qu'une composition moyenne
unique pour un échantillon qui s'apparente au sgraffito fin et deux tessons de la famille de la
"Zeuxippus ware" trouvés à Constantinople - et présentés comme production locale - , ce qui
sous-entend l'homogénéité de l'ensemble (cf. tableau 16). Par contre, au groupe de tessons de
sgraffito fin trouvés à Constantinople analysé par Megaw et Jones ne s'associent pas les deux
échantillons de "Zeuxippus ware" (s.s.) mis au jour à Paphos. Seul l'important écart-type de ce
groupe permet d'envisager le rapprochement d'un échantillon de sgraffito fin issu du même
contexte [123] (cf. tableau 16). Il ressort surtout de ces comparaisons qu'il est difficile
d'interpréter des ressemblances ou dissemblances sur des échantillons en aussi petit nombre, le
problème de l'attribution de pièces individuelles étant d'ailleurs souligné par Megaw et Jones
[123]. La comparaison entre les différents types doit reposer sur un échantillonnage en
importance numérique suffisante pour pouvoir estimer la dispersion des compositions au sein
d'un groupe.
D'autres comparaisons entre types de céramiques au sgraffito peuvent être tentées.
Ainsi, on aurait pu envisager, au vu d'une certaine similarité de composition dans les analyses

Chapitre 2 80^
citées dans [123], que le sgraffito fin du 12°mc trouvé à Constantinople, la cargaison de plats
datés du milieu du 12 c m c siècle du navire naufragé au large de Pelagonnisos et des céramiques
de la fin du 12 è m c trouvées sur l'agora d'Athènes puissent avoir une origine commune. Ces
tessons montrent également une ressemblance de composition avec le groupe de référence de
Thessalonique, centre surtout connu pour sa production aux I 3 è m c - 14 è m c siècles [124]. Il
semblerait qu'un examen pétrographique puisse lever l'indétermination en ce qui concerne
l'éventualité d'une provenance thessalonicienne: Lazzarini et Calogero ont caractérisé cet
atelier par la présence de nombreux pyroxenes, dont des pyroxenes orthorhombiques [126].
Leur propre échantillonnage de sgraffito d'Athènes s'en distingue en ne montrant pas de tels
minéraux.
Pour conclure cette introduction bibliographique, on ne peut que constater la grande
ignorance dans laquelle nous nous trouvons encore quant aux centres de production des
différents types de céramiques byzantines et à leurs aires de diffusion. En dehors des cas bien
attestés de productions influencées par les importations byzantines (Italie, Bulgarie), les sites
de fabrication des prototypes eux-mêmes sont encore inconnus et l'unicité de ces prototypes
est souvent remise en question par des analyses isolées, trop rares pour autoriser à conclure.
On peut espérer que, dans la lignée des travaux de Megaw et Jones [123], la multiplication des
analyses sur des échantillons archéologiquement bien définis permettra de donner des
"définitions statistiques" à chaque type, ce qui rend les comparaisons bien moins hasardeuses
que sur des échantillons individuels. L'approche complémentaire de la minéralogie et de la
pétrographie peut donner des renseignements précieux en révélant des caractéristiques
discriminantes (e.g. la présence de serpentine dans des tessons qui proviendraient de
Constantinople). Il serait intéressant de faire porter les efforts non seulement sur les catégories
prestigieuses de céramiques glaçurées - notamment les exemplaires de "white-ware" et de
"Zeuxippus ware" issus des grandes collections de Constantinople, de Cherson ... - qui sont
susceptibles d'avoir été diffusées à grande distance, mais aussi sur la céramique la plus
communément trouvée sur les sites producteurs - céramique non glaçurée et céramique
glaçurée monochrome. Pour autant que le traitement des argiles soit uniforme sur une certaine
gamme de produits, l'analyse d'un grand nombre de céramiques "courantes" peut permettre de
constituer des groupes de références représentatifs. Toutefois, la localisation même des ateliers
demeure subordonnée à la mise à jour des vestiges correspondants.
Chapitre 2 81
Figure 26: Sites dont le matériel a donné lieu aux analyses présentées dans le tableau 17.
1: Padoue; 2: Venise; 3: Corinthe; 4: Kounoupi - Chinitsa; 5: Athènes; 6: Pélagonnisos
(épave); 7: Thessalonique; 8: Serres; 9: Didymotique; 10: Constantinople; 11: Tscherven; 12:
Schumen; 13: Tzarevetz (Veliko Tirnovo); 14: Baltchik - Kavarna - Kaliakra; 15: Varna; 16:
Pergame; 17: Sardes; 18: Lapithos; 19: Lemba; 20: Paphos; 21: Dhiorios.
Site
Kounoupi,ChinitsaDhioriosConstantinople(Istanbul)Corinthe
Constantinople(Istanbul)Constantinople(Istanbul)
Corinthe
Corinthe
Athènes
Athènes
AthènesLemba
Lemba
Pays
Gr.
Ch.Tu.
Gr.
Tu.
Tu.
Gr.
Gr.
Gr.
Gr.
Gr.Ch.
Ch.
Fig.26n°4
2110
3
10
10
3
3
5
5
519
19
Matériel analysé, type de décor
amphores
céramique culinaire non glaçurée"white ware"
"white ware" (décor estampé,décor incisé, "white-ware"polychrome, "petal ware")*sgraffito fin
sgraffito fin (?), "Constantinopleware" (?), dérivés de "Zeuxippusware"sgraffito fin, ("measles ware"),"slip ware"
sgraffito
céramique engobée glaçuréemonochrome**,sgraffito incisécéramique culinaire non glaçuréeamphoressgraffitosgraffito, sgraffito incisé (peint),dont dérivés de "Zeuxippus ware",sgraffito peint,"slip ware"sgraffito
CP
X
X
X?
X
X
X?
X?X
X
Datation
fin 6ème -début 7è m e
7 (- 8)ème
10ème(?)
lOeme
début 12ème
(?)12 - 13 e m e
fin 11 ôme_l è r e moitié.]2ème
12 - 13eme
fin 12 è m e
fin 12 c m e
12 - 13 e m e
]3eme_ ?
12 - 13cnic
Méthodes d'analyse
OES
OESOES
OES
OES
XRF, XRD, pétrographie
OES
XRF, XRD, pétrographie
OES
OES
XRF, XRD, pétrographieOES
XRF, XRD, pétrographie
Réf
123
123123
123
123
126
123
126
123
123
126123
126
Remarques
quelques échantillons analyséségalement dans [126]
quelques échantillons analyséségalement dans [123]
quelques échantillons analyséségalement dans [126]
quelques échantillons analyséségalement dans [123]
Site
Paphos
Thessalonique
Thessalonique
Serres
Sardes
Sardes
Pergame
Pergame
Pergame
Pays
Ch.
Gr.
Gr.
Gr.
Tu.
Tu.
Tu.
Tu.
Tu.
Fig.26n°20
7
7
8
17
17
16
16
16
Matériel analysé, type de décor
céramique culinaire non glaçurée,"brown and green painted ware",sgraffito (peint), sgraffito incisé(peint), dont "Zeuxippus ware",sgraffito peint "syrien", "slip ware"sgraffito, dont bols à décord'oiseau caractéristique (dit"thessalonicien")sgraffito incisé (peint), dont bols àoiseau "thessalonicien"sgraffito peint
céramique moulée à glaçureturquoise (fabrique de Rayy)
sgraffito, sgraffito peint, (sgraffitoincisé), "St Symeon ware",céramique glaçurée polychromedite "mottled ware", céramique àglaçure turquoisesgraffito incisé, dont dérivés de"Zeuxippus ware","slip ware"céramique (peinte) non glaçurée,sgraffito peintsgraffito fin, sgraffito incisé,sgraffito peint
CP
X
X
X
X?
X
X?
Datation
(?) -début 13 è m e
12- 13 e m c
13 _ Même
2èmc moitié13ème _] 4cmc
2èmc moitiéjoème
fin 1 2 e m c -13ème
12- Hcrnc
12- 14 c m c
12 - I4«nc
Méthodes d'analyse
OES
XRF, XRD, pétrographie
OES
INAA, XRD
pétrographie
pétrographie
PIXE, INAA, pétrographie
PIXE, INAA, pétrographie
PIXE, INAA, pétrographie
Réf
123
126
123
144
143
143
147148
147148148
Remarques
quelques échantillons analyséségalement dans [123]
quelques échantillons analyséségalement dans [1261analyses d'engobes et deglaçures par XRD
analyses d'engobes et deglaçures par pétrographie, XRDet microsonde électroniqueanalyses d'engobes et deglaçures par pétrographie, XRDet microsonde électronique
***
***
***
Site
Pelagonnisos(épave)Venise
lagune deVenise
Varna
Varna
Schumen
Tsherven
Baltchik
Kavarna
Kaliakra
Lapithos
Pays
Tu.
It.
It.
Bu.
Bu.
Bu.
Bu.
Bu.
Bu.
Bu.
Ch.
Fig.26n°6
2
2
15
15
12
11
14
14
14
18
Matériel analysé, type de décor
sgraffito, sgraffito peint
sgraffito, sgraffito peint
sgraffito, (sgraffito fin ?),("measles ware"), sgraffito incisé,dont dérivés de "Zeuxippus ware"et bols à oiseau "thessalonicien"sgraffito, sgraffito peint, sgraffito àmonogrammes
sgraffito, sgraffito peint, sgraffito àmonogrammes
sgraffito, sgraffito peint, sgraffito àmonogrammessgraffito, sgraffito peint, sgraffito àmonogrammessgraffito, sgraffito peint, sgraffito àmonogrammessgraffito, sgraffito peint, sgraffito àmonogrammessgraffito, sgraffito peint, sgraffito àmonogrammessgraffito peint
CP
X?
X
X
Datation
milieu 12 è m e
12 - 1 3 e m e
12 - 13 e m e
13 - 14eme
12- 15 e m e
12 - 14 c m e
12 - 14cn ie
12- 15ème
1 2 - 1 5 e m e
12- 15 è i n c
15cmc
Méthodes d'analyse
OES
XRF (Mg par AAS,microsonde électronique),XRD, pétrographie,spectrométrie MôssbauerXRF (Mg par AAS,microsonde électronique),XRD, pétrographie,spectrométrie MôssbauerINAA (neutrons thermiqueset épithermiques)
INAA (neutrons thermiqueset épithermiques)
INAA (neutrons thermiqueset épithermiques)INAA (neutrons thermiqueset épithermiques)INAA (neutrons thermiqueset épithermiques)INAA (neutrons thermiqueset épithermiques)INAA (neutrons thermiqueset épithermiques)OES
Réf
123
126127
126127
130129132130129132129
129
130132130
130
123
Remarques
Site
Venise
Padoue
Tzarevetz(VelikoTirnovo)Didymotique
Pays
It.
It.
Bu.
Gr.
Fig.26n°2
1
13
9
Matériel analysé, type de décor
sgraffito peint
sgraffito peint
sgraffito, sgraffito peint
céramique engobée glaçuréemonochrome**, (décor estampé)
CP
X
X
X
Datation
15 - 16eme
15 - 16èmc
15- 18èinc
] (jcme
Méthodes d'analyse
analyses élémentaires pardiverses méthodes (dontXRF, spectrométrie deflamme), XRD,pétrographie, spectrométrieMôssbaueranalyses élémentaires pardiverses méthodes (dontXRF, spectrométrie deflamme), XRD,pétrographie, spectrométrieMôssbauerINAA (neutrons thermiqueset épithermiques)
OES
Réf
128
128
131
123
Remarques
analyses d'engobes et deglaçures par XRF etpétrographie
analyses d'engobes et deglaçures par XRF etpétrographie
Tableau 17: Analyses portant sur du matériel byzantin.CP: centre de production; codes pays: Gr.: Grèce, Tu.: Turquie, Ch.: Chypre, It.: Italie, Bu.: Bulgarie; abbreviations: AAS: atomic absorptionspectrophotometry, INAA: instrumental neutron activation analysis, OES: optical emission spectroscopy, PIXE: particle induced X-ray emission, XRD: X-raydiffraction, XRF: X-ray fluorescence (cf annexe 3).
* entre parenthèses: caractère présenté par un nombre restreint d'échantillons de la catégorie (e.g.: sgraffito incisé (peint)) ou précision sur la catégorie (e.g.:céramique moulée à glaçure turquoise (fabrique de Rayy), i.e. identifiée comme production de ce centre persan);** céramiques, engobées ou non, à glaçure monochrome: des tessons de cette catégorie sont également compris dans la plupart des échantillonnages contenantdes céramiques à sgraffito. Elles ne sont en général pas indiquées dans le tableau, sauf si elles présentent une coloration particulière, i.e. autre que vert, jaune,brun ou mauve-brun.*** publications partielles des analyses de Pergame.

Chapitre 2 86^
Tableau 17: Analyses portant sur du matériel byzantin
Conventions adoptées pour la spécification du décor:
- sgraffito: toute pièce décorée par la technique du sgraffito, au sens large du terme;
- sgraffito fin: décor sgraffito exécuté à la pointe fine, désigne plus particulièrement les
productions de la fin du 1 l è m c - début du 12 è m c siècle;
- sgraffito incisé: catégorie qui englobe tout décor sgraffito réalisé en totalité ou en partie avec
un instrument créant des incisions relativement larges (gouge), y compris le champlevé;
- sgraffito peint: décor sgraffito associé à une glaçure polychrome (présence d'au moins une
teinte distincte de celle du fond)
Types de décors non définis dans le texte (d'après [1]):
- "petal ware": céramique engobée glaçurée monochrome à décor en relief disposé en pétales;
10è'»e siècle;
- "green and brown painted ware": céramique engobée glaçurée peinte des deux couleurs vert
et brun; 1 l è m c - 15 è m c siècles;
- "measles ware": céramique engobée glaçurée décorée au sgraffito, où les motifs sont
découpés en écailles avec un point rouge peint au milieu - d'où l'appellation de "measles", i.e.
"rougeole"; fin 1 l è m c - début 12 è m c ;
- " slip ware" ou "slip-painted ware": céramique glaçurée monochrome de pâte rouge peinte à
l'engobe blanche; fin 1 l è m c - 15 è m c siècles;
- "mottled ware" (terme utilisé par J.A. Scott dans [143], cette céramique y est aussi désignée
comme imitation de la céramique T'ang): céramique engobée à glaçure vert clair avec des
taches brun-mauve diffuses; postérieur au milieu du 12 è m c siècle;
- sgraffito à monogrammes: céramique décorée au sgraffito dans le fond de laquelle est incisé
un monogramme, i.e. un ensemble de lettres (grecques, en l'occurence) organisées en motif.
Pour les différentes tentatives d'interprétations, on peut se référer, outre les références citées, à
[121], et pour une approche plus récente à [136];
Ce tableau se limite à des publications portant (quasiment) exclusivement sur du
matériel byzantin, de tradition ou d'influence byzantine, d'époque médiévale et postérieure, par
opposition à l'époque paléochrétienne. On peut supposer que de nombreuses autres analyses
ont été réalisées sur:
- des céramiques paléochrétiennes de tradition romaine (cf. e.g. [150, 151] (analyses
élémentaires));
- des céramiques byzantines retrouvées en Italie - en particulier des "bacini" ornant les édifices
religieux italiens. Plusieurs études pétrographiques ont été réalisées sur du matériel de ce type
[152, 153];

Chapitre 2 87_
- des céramiques franques mises au jour dans les états cotiers du Proche-Orient (e.g. [154]
(analyses élémentaires de glaçures)). A ma connaissance, il existe des études en cours ou non
publiées sur de la céramique non glaçurée de Syrie et de Jordanie, analysée par XRF et
pétrographie (?) par G. Schneider à la Freie Universitât de Berlin [155]. Par ailleurs, des
céramiques glaçurees issues notamment de contextes israéliens ont été analysées par INAA
dans le laboratoire d'archéométrie de la Hebrew University de Jérusalem (J. Yellin) [156]. Il
semblerait que les résultats obtenus aient suscité des interprétations archéologiques
importantes pour des catégories de céramiques comme le sgraffito fin, la Zeuxippus ware
[157], mais les analyses sont malheureusement encore inédites à ce jour dans leur plus grande
partie.
D'autres analyses en cours portent sur le matériel suivant:
- céramique non glaçurée de Corinthe, d'époque franque, étudiée en pétrographie par L.
Joyner à l'école anglaise d'Athènes [158]
- céramiques des fouilles d'Ephèse, dont les analyses sont annoncées par E. Parman [159];
- céramiques des fouilles de Cherson, dont les analyses menées au musée de l'Hermitage sont
signalées (avec quelques réserves?) dans [123 (rapport préliminaire)].
On peut citer le rapport technique, incluant des analyses de pâtes et de glaçures, qui
porte sur du matériel seldjoukide très voisin mis au jour dans l'Est anatolien [160],
Notons également les analyses élémentaires de quelques pièces byzantines, au sein d'un
échantillonnage de céramiques de diverses périodes trouvées à Yalincak (Turquie), qui sont
reportées dans [161a]. Ces auteurs ont aussi analysés des argiles prélevées en Turquie dans les
régions de Canakkale (Ezine, Can), Bilecik (Inhisar), Nevsehir (Aksaray, Avanos), Hatay
(Samandag, Iskenderun, Antalya), et à de nombreux emplacements à proximité d'Ankara [161].
Des verres et des glaçures de céramiques issus d'une épave byzantine ou islamique
échouée au début du llème siècle dans un petit port de la côte Sud de la Turquie ont donné
lieu à des analyses des isotopes du plomb [162].
Enfin, le tableau serait nécessairement incomplet si l'on ne mentionnait les "précurseurs"
en matière d'analyses de céramiques byzantines:
- analyse qualitative par spectrométrie d'emisssion de deux échantillons sgraffito (l'un associé à
la production locale, l'autre de provenance inconnue) issus des fouilles de Corinthe, citée par
Morgan [1];
- diffraction des rayons X sur deux tessons de "white-ware" comparés à un exemplaire de
céramique ottomane d'Iznik de type "Rhodien", dans Talbot Rice [122];

Chapitre 2 88^
- examens de deux tessons de "white-ware" polychrome, par pétrographie, analyse élémentairequalitative et recuit, cités par Coche de la Ferté [163]. Le rapport technique produit par uningénieur de l'Institut de Céramique de Sèvres conclut à des pâtes siliceuses et à des glaçuresplombifères et probablement également alcalines. Les oxydes colorants employés sont le fer, lecuivre, le manganèse, le titane. La température de cuisson est estimée à moins de 800° C.

Chapitre 2 ^ 89
2.3. Les céramiques des fouilles de Pergame
2.3.1. Contexte historique et archéologique [164]
Le site de Pergame est localisé en Asie Mineure, sur un piton rocheux dominant de 300
mètres la vallée du Kaikos (actuel Bakir), à environ 30 kilomètres de la côte égéenne
(figure 27). Il est cerné à l'Ouest par la vallée du Selinous, qui abrite la ville moderne de
Bergama, à l'Est par la vallée du Kestios ou du Kestel en partie recouverte par un lac de
barrage depuis la fin des années 1980.
Bien que les traces les plus anciennes d'occupation du site remontent au 8èmc siècle
avant J.C., le destin historique de Pergame commence après la succession d'Alexandre le
grand, lorsque Philétaire (281 - 263 avant J.C.), général du diadoque Lysimaque, emploie le
trésor de guerre de ce dernier à la fondation de la principauté indépendante de Pergame. Le
site prend une nouvelle envergure avec Attale 1er (241 - 197 avant J.C.), qui vainc les Galates,
peuple celte émigré en Asie Mineure, et conquiert quelque territoire face à ses puissants voisins
Séleucides. C'est sous Eumène II (197 - 159 avant J.C.) que Pergame atteint son apogée. Par
son rayonnement sur le monde hellénistique, elle approche l'image d'une nouvelle Athènes
ambitionnée pour elle par les rois Attalides. Elle s'est alors dotée d'un impressionant ensemble
architectural (temples, théâtre, gymnases, ...) qui défie l'escarpement naturel du site. Sa
bibliothèque concurrence la bibliothèque d'Alexandrie en tant que dépositaire de la culture
grecque. Son école artistique, connue surtout par les frises du monumental autel de Zeus,
marquera l'histoire de l'art antique. Son sanctuaire à Asklepios, dieu guérisseur, attire de
nombreux pèlerins.
Longtemps alliée à l'empire romain, Pergame lui est léguée par testament en 133 avant
J.C. par le dernier roi de la dynastie, Attale III. Au coeur de la province romaine d'Asie, elle va
connaître une période troublée (révolte de Mithridate) avant de retrouver la prospérité sous le
haut Empire. L'agglomération de Pergame s'étend alors vers la plaine bien au-delà de
l'acropole, et abrite une population estimée au 2è m c siècle après J.C. à 160.000 habitants.
L'infrastructure routière développée par les romains relie Pergame aux principales autres villes
de la région: deux voies mènent à Ephèse, via Smyrne et via Sardes, et la route du Nord
permet d'accéder aux sites côtiers ou de rejoindre directement la Propontique au niveau de
Cyzique (figure 27).
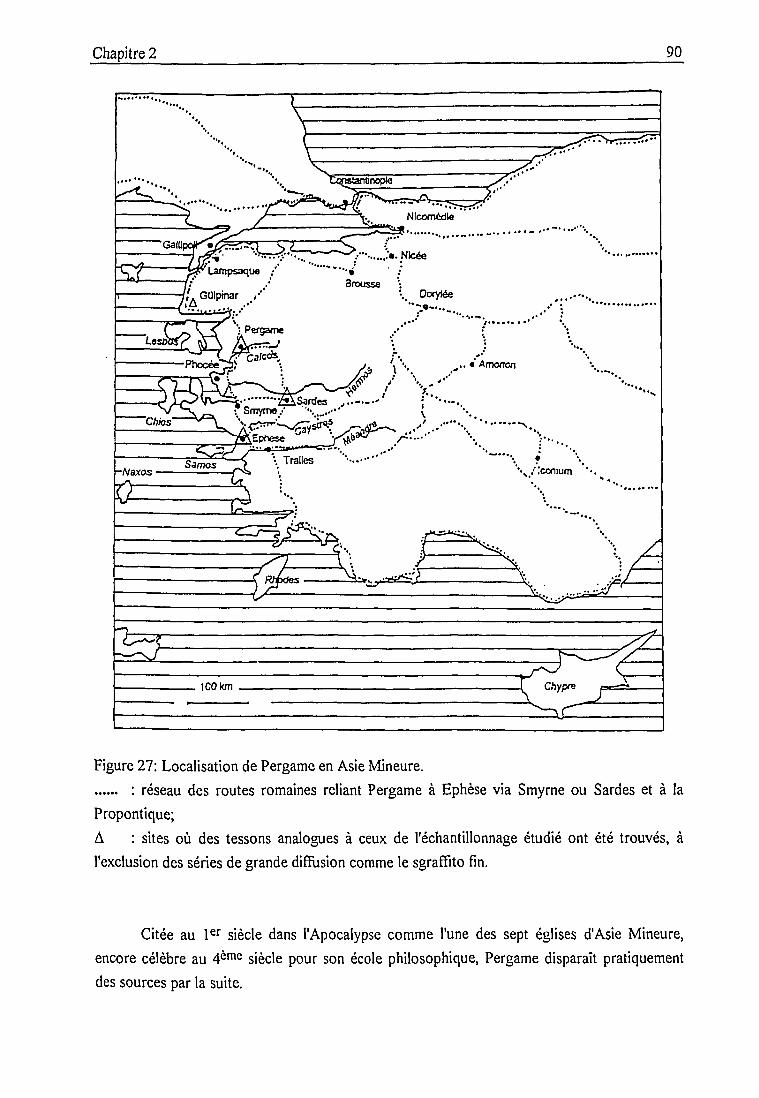
Chapitre 2 90
-A/8X0S •
0= tfa
^ / V— ^ ^ z ^ 7 ^ / ^ - ^ .••"'•-••"•••••• ' "••••.
••N i / . 'conium
~ru.
^7/ Rttotes-
% ..; X3
\ . •
ICO Ion
Figure 27: Localisation de Pergame en Asie Mineure.
: réseau des routes romaines reliant Pergame à Ephèse via Smyrne ou Sardes et à la
Propontique;
À : sites où des tessons analogues à ceux de l'échantillonnage étudié ont été trouvés, à
l'exclusion des séries de grande diffusion comme le sgraffito fin.
Citée au 1er siècle dans l'Apocalypse comme l'une des sept églises d'Asie Mineure,
encore célèbre au 4 è m c siècle pour son école philosophique, Pergame disparaît pratiquement
des sources par la suite.

Chapitre 2 9J_
La Pergame byzantine ne connaîtra plus les fastes des époques hellénistique et romaine.
Les vestiges archéologiques montrent une constaiction publique essentiellement limitée à des
structures défensives (remparts). Pergame ne bénéficie guère de la période de prospérité de
l'empire byzantin sous le règne de Justinien (527-565), mais subit le contrecoup de ses défaites
militaires. A plusieurs reprises, elle doit protéger son acropole par de nouveaux remparts, en
particulier contre les incursions arabes du 8èmc siècle. Bien que les sources mentionnent encore
à cette époque des villes comme Pergame et Sardes comme des "cités" [165], le déclin des
centres antiques de l'Asie Mineure est manifeste. L'ancien réseau routier romain est délaissé au
profit de voies plus septentrionales: les principales routes commerciales rayonnant de
Constantinople, dont la fameuse route de la soie, ne traversent pas la région.
En 1071, la défaite byzantine à Mantzikert, en Anatolie orientale, marque un tournant
avec l'avancée décisive des Turcs seldjoukides en Asie Mineure. Pergame, devenue ville-
frontière, ne retrouve une certaine aisance que sous Manuel 1er Comnène (1143-1180), où elle
aurait peut-être été le centre d'une division administrative [2]. Cette prospérité va se
développer avec le déplacement du pouvoir byzantin des Lascarides en Asie Mineure, suite à la
prise de Constantinople par les croisés (période de l'empire de Nicée, 1204-1261). La
reconquête de la capitale en 1261 est l'amorce du déclin de la Pergame byzantine, qui tombe
aux mains des Turcs ottomans au cours du 1er tiers du 14èmc siècle.
Les fouilles de Pergame débutent dès 1878, sous la direction de l'ingénieur allemand
Cari Humann. Elles vont donner lieu à de vastes campagnes (1878-1886, 1900-1912, 1957-
1968, 1972- ), supervisées par le Musée de Berlin, puis par les Instituts archéologiques
allemands. Depuis 1972, les fouilles sont menées par Wolfgang Radt, deuxième directeur de
l'Institut Archéologique Allemand d'Istanbul.
Elles se sont surtout appliquées à dégager et à restaurer les vestiges des périodes
hellénistique et romaine. Une partie des édifices est reconstituée au Pergamon Museum de
Berlin, dont le célèbre autel de Zeus, trouvé en réemploi dans le rempart byzantin du 8è m c
siècle.
Comme sur la plupart des sites, la culture matérielle est abondamment représentée à
Pergame par de la céramique.
Les céramiques hellénistiques et romaines ont été l'objet de plusieurs études
archéologiques, publiées notamment dans la série des "Pergamenische Forschungen" [166]. De
1978 à 1985, une fouille de sauvetage turque dans la vallée du Kestel mettait au jour des
ateliers de potiers de cette époque, comprenant 26 fours [167]. En première estimation, les
archéologues ont suggéré une exploitation continue entre le 2^mc siècle avant J.C. et le 6^mc
siècle après J.C. Les rebuts et déchets de cuisson montrent une production diversifiée, depuis
des terres cuites "grossières" comme les tuiles et les amphores jusqu'à de fines statuettes

Chapitre 2 92_
moulées [168]. Des tessons de céramiques glaçurées ont également été remarqués [169], ce qui
indique que la technique de la glaçure était pratiquée dans cet atelier. En l'absence de
spécifications supplémentaires, ceci n'implique pas que l'activité de l'atelier se soit poursuivie
au delà de la période paléochrétienne, puisque la céramique plombifère était déjà connue à
Pergame aux époques précédentes [170], En ce qui concerne les ateliers actifs à l'époque
byzantine, la découverte de ratés de cuisson de céramiques byzantines dans le matériel des
fouilles récentes de la ville moyenne de Pergame prouve qu'une production locale a perduré au-
delà de l'époque romaine tardive. La localisation de ces vestiges indique l'existence d'une
officine urbaine. On ignore par contre s'il existait encore à cette période un quartier de potier
analogue à celui de la vallée du Kestel pour les époques hellénistique et romaine. On peut
regretter que la publication de la fouille des ateliers du Kestel se limite actuellement, semble-t-
il, aux deux "communiqués" précités.
La littérature dispose par contre de nombreuses références concernant des objets dits
pergaméniens. Par exemple, on peut citer le cas de certains groupes de statuettes hellénistiques
trouvées dans le site voisin de Myrina. Des considérations stylistiques se référant à la statuaire
monumentale pergaménienne ont conduit les historiens de l'art à les désigner comme issu de
l'atelier de Pergame. Cette dénomination basée sur une analogie stylistique n'implique pas
forcément un lien de provenance. De fait, il semblerait que l'étude pétrographique menée par
J. Gautier [171] associe plutôt les pâtes de ces statuettes aux grands ensembles
métamorphiques situés au Sud-Est de Myrina. Par contre, Pergame serait une origine possible
pour un autre groupe de statuettes, d'un style différent (groupe de Pythodoros). On retrouve
un décalage analogue pour des vases à vernis rouge de "genre pergaménien": des ratés de
cuisson trouvés à Phocée attestent d'une production phocéenne et non pergaménienne [150].
Quoiqu'il en soit, les périodes hellénistique et romaine ont vu se développer sur cette partie de
la côte égéenne un très grand nombre d'ateliers de céramiques. Leurs produits ont parfois
connu une diffusion très importante, en particulier pour les sigillées phocéennes
paléochrétiennes [151], qui reprendraient une tradition céramique antérieurement établie à
Çandarli (ancienne Pitane) [172], le "débouché maritime" de la plaine de Bakir et donc de
Pergame. Plusieurs programmes de prospection ont permis l'échantillonnage et l'analyse de
tessons provenant de ces ateliers en relation avec des céramiques d'époques grecque archaïque
[173], hellénistique tardive et romaine [174] et romaine tardive [150]. Nous aurons l'occasion
de revenir sur les résultats de ces analyses en 2.4.2.
Des céramiques byzantines issues des fouilles de Pergame apparaissent dès le début du
2()ème siècle dans les collections des musées allemands. Quelques photos publiées attireront par
la suite l'attention des spécialistes. Ainsi, un passage de la monographie de Morgan [1] dit:
"That many of the finest wares recovered on various European sites, at Pergamum and
elsewhere, are products of single centres of manufacture is apparent from their uniformity of

Chapitre 2 93_
fabric, but thus far no one of these has been identified." (Que beaucoup des céramiques les plus
fines retrouvées sur divers sites européens, à Pergame et ailleurs, soient les produits de centres
de fabrication uniques est apparent par leur uniformité de fabrique, mais jusqu'à présent aucun
de ces centres n'a été identifié.) On peut déplorer que le constat fait par Morgan soit toujours
d'actualité plus de 50 ans après. Par ailleurs, on peut se demander si le statut apparemment
privilégié de Pergame n'est pas surtout dû à la méconnaissance des vestiges byzantins sur les
autres sites. Quoiqu'il en soit, les fouilles récentes de Pergame ont livré un important matériel
céramique d'époque byzantine, qui est sur le point d'être publié par J.-M. Spieser, professeur
d'histoire de l'art et d'archéologie byzantines à l'université de Strasbourg [5].
Les céramiques étudiées dans sa monographie sont issues des quartiers d'habitations
byzantins de la ville moyenne. Les contextes de fouilles sont peu informatifs: il s'agit surtout de
dépotoirs dont la stratigraphie a été perturbée par des glissements de terrain, occasionnés par
la pente naturelle du site. La numismatique permet néanmoins de proposer une période
d'occupation comprise entre le 12 è m e et le 14 è m c siècle, la seconde moitié du 1 3 è m c siècle étant
la plus représentée. Plusieurs milliers de tessons ont été mis au jour, sans qu'il soit toujours
possible de reconstituer des profils complets, ni même de les rattacher à l'un de ces derniers.
Cette difficulté a motivé l'utilisation de techniques statistiques multivariées pour classifier les
fragments selon leurs données morphométriques (hauteur, diamètre supérieur, diamètre
inférieur, ...), et pour déterminer lesquelles de ces données sont les plus discriminantes [175].
Une telle approche est assez significative de l'ouverture des sciences humaines vers les
mathématiques appliquées, et en particulier vers les méthodes statistiques (cf. e.g. [176]). Elle
n'exclut évidemment pas la description au moyen des critères plus "traditionnels", qui
privilégient les techniques de décoration.
2.3.2. Description des échantillons et classification archéologique (proposée par
Prof. J.-M. Spieser)
Les 160 tessons sélectionnés pour les analyses ont été choisis de manière à représenter
la plupart des catégories de céramiques trouvées à Pergame. Cette représentation ne respecte
toutefois pas la proportionnalité numérique, ce qui permet à des catégories présentes en petites
quantités d'être également étudiées.
Les échantillons ont été répartis en 18 séries, indexées de A à R, à partir de critères
archéologiques. Ces séries recoupent en partie les types "classiques" de la céramique
byzantine: sgraffito fin, "slip-painted ware", "Zeuxippus ware" et dérivés, ... Pour différencier
les autres fragments, de nombreux paramètres ont été pris en compte: forme, couleur ou
association de couleurs de la glaçure, disposition relative des motifs esgrafiés et peints, aspect
et texture de la pâte (grossière / fine, grasse / maigre, couleur, dureté, ...), etc. Les paramètres
relatifs à la pâte ont été l'objet d'une attention particulière, sans toutefois mener à une
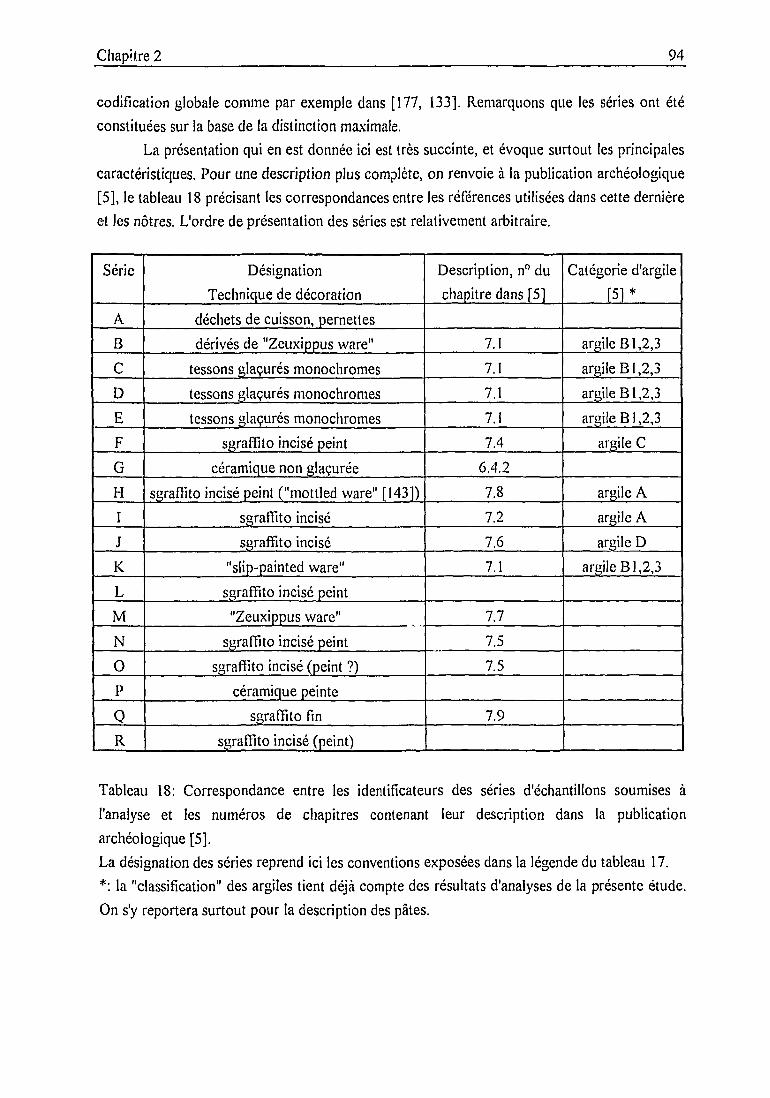
Chapitre 2 94
codification globale comme par exemple dans [177, 133]. Remarquons que les séries ont été
constituées sur la base de la distinction maximale.
La présentation qui en est donnée ici est très succinte, et évoque surtout les principales
caractéristiques. Pour une description plus complète, on renvoie à la publication archéologique
[5], le tableau 18 précisant les correspondances entre les références utilisées dans cette dernière
et les nôtres. L'ordre de présentation des séries est relativement arbitraire.
Série
A
B
C
D
E
F
G
H
I
J
K
L
M
N
0
P
Q
R
Désignation
Technique de décoration
déchets de cuisson, pernettes
dérivés de "Zeuxippus ware"
tessons glaçurés monochromes
tessons glaçurés monochromes
tessons glaçurés monochromes
sgraffito incisé peint
céramique non glaçurée
sgraffito incisé peint ("mottled ware" [143])
sgraffito incisé
sgraffito incisé
"slip-painted ware"
sgraffito incisé peint
"Zeuxippus ware"
sgraffito incisé peint
sgraffito incisé (peint ?)
céramique peinte
sgraffito fin
sgraffito incisé (peint)
Description, n° du
chapitre dans [5]
7.1
7.1
7.1
7.1
7.4
6.4.2
7.8
7.2
7.6
7.1
7.7
7.5
7.5
7.9
Catégorie d'argile
[51*
argile B 1,2,3
argile B 1,2,3
argile B 1,2,3
argile B 1,2,3
argile C
argile A
argile A
argile D
argile B 1,2,3
Tableau 18: Correspondance entre les identificateurs des séries d'échantillons soumises à
l'analyse et les numéros de chapitres contenant leur description dans la publication
archéologique [5].
La désignation des séries reprend ici les conventions exposées dans la légende du tableau 17.
*: la "classification" des argiles tient déjà compte des résultats d'analyses de la présente étude.
On s'y reportera surtout pour la description des pâtes.

Chapitre 2 95_
Série A: échantillons de référence pour l'argile locale (figure 28)
Les trois types d'objets constituant cette série sont incontestablement de provenance
locale, On les trouve typiquement à proximité immédiate des ateliers: trépieds utilisés pour
séparer les pièces dans le four, pièces rejetées car présentant un défaut. Bien qu'aucun vestige
d'atelier n'ait été retrouvé, leur présence est la preuve de l'existence d'une officine urbaine à
Pergame à l'époque byzantine. Il semblerait d'ailleurs que de telles structures existaient déjà
aux époques précédentes [173].
L'échantillonnage se répartit de la manière suivante:
n° 54, 55, 56, 57, 58, 115: pernettes;
n° 111, 123, 158: tessons à pâte très dure et noire, d'aspect surcuit;
n° 73, 81, 109, 154: tessons non glaçurés correspondant à des pièces inachevées - identifiées
comme telles car il existe des céramiques glaçurées de même forme. En particulier, l'échantillon
81 (pied) se rapproche des échantillons de la série F par la typologie et, de manière moins
évidente, par l'aspect de la pâte. La pâte des tessons n° 81 et 109 est de texture plutôt
grossière, celle des n° 154 et 73 est plus fine et micacée.
Série B: famille de la "Zeuxippus ware" (figures 29 et 45)
Ces tessons, très fréquents à Pergame, présentent les attributs de la "Zeuxippus ware".
Ils sont décorés par la technique du sgraffito, avec une combinaison d'incisions fines et plus
épaisses. Leurs motifs sont caractéristiques du type: cercles concentriques (30, 147), chevrons
dans un médaillon central (82), arbres stylisés (30, 147), ... A titre d'exemple, on pourra
comparer l'échantillon 30 au dessin de la figure 24. La présence de rehauts bruns (tirant selon
la concentration en fer soit vers le jaune, soit vers le noir) dans la glaçure jaune clair et de
traces d'arrachement de trépieds les situent dans la classe II de Megaw [2]. Ce dernier cite de
nombreuses céramiques de Pergame issues de fouilles anciennes comme exemples de
"Zeuxippus ware". On peut se demander si les échantillons de la série B leur sont assimilables.
En effet, il faut souligner que les tessons considérés ne présentent pas la pâte fine, rouge
sombre, bien cuite qui est une des marques distinctives du prototype de "Zeuxippus ware". La
glaçure n'est pas toujours non plus de très bonne qualité (e.g. écaillements sur l'échantillon
n° 32, irisations sur le n° 26, . . . ) . Est-ce à dire que l'amalgame entre prototype et dérivés est
présent dès l'article même qui pose la définition de la "Zeuxippus ware"? Il est probable que
Megaw n'a pu observer lui-même les céramiques de Pergame qu'il décrit, et qu'il se base sur les
photos des catalogues allemands qui ne rendent compte que du motif. Quoiqu'il en soit, on
peut supposer qu'une production prototype - unique? - a existé. Il appartient à l'observation
directe de déceler les produits dérivés et à l'analyse de déterminer leur lieu de fabrication.

Chapitre 2 %
Les pâtes sont moyennement fines: de nombreuses inclusions y sont visibles (figure 45),
avec une proportion variable, souvent importante, de micas. Leur couleur va d'un rose orangé
(37) à un rose brun (93), les texture sont également diverses, parfois massives (32) ou, au
contraire, feuilletées (37). La pâte grise des échantillons n° 26 et 82 témoigne d'une
réoxydation (accidentellement?) incomplète suite à la cuisson [97].
La teinte jaune du "fond" de glaçure est plus (37, 39) ou moins soutenue (27), avec une
variante plutôt verte pour le tesson n° 144.
Le cas des échantillons n° 35 et 147 est particulier: incisés, engobés pour l'un (147) et
non glaçurés, ils pourraient aussi bien figurer dans la série de référence en tant que pièces
inachevées. On peut donc d'emblée supposer une fabrication locale pour l'ensemble des pièces
de la série B.
Notons également la présence d'une croûte verdâtre cernant les motifs sur le tesson
n° 147, peut-être destinée à les rehausser par quelque colorant.
Des tessons similaires ont été trouvés dans la région de Troie, à une centaine de km au
Nord-Ouest de Pergame, sur les fouilles de Gùlpinar et de Besik-Tepe [178].
Série C, D, E: tessons engobés glaçurés et sgraffito monochromes (figures 30, 31,
32)
Les séries C, D, E se distinguent entre elles par la couleur de la glaçure, qui est
respectivement jaune, brune et verte. Elles représentent la catégorie de céramiques glaçurées la
plus fréquente et la plus "banale" sur le site. Les tessons sont parfois décorés par la technique
du sgraffito.
Les pâtes présentent des caractéristiques analogues à celles des échantillons de la série
B, sous des aspects encore plus divers: depuis une texture assez grossière et rugueuse (par
exemple, série E: 19, 20, 85, 102) jusqu'à une texture assez fine et lisse (série D: 46, 122; série
E: 118). Cette distinction coïncide souvent avec l'état de conservation de la glaçure: lacunaire
ou altérée (série E: 19), ou au contraire brillante et de bonne qualité (série C: 92; série D: 46,
122; série E: 118). La correspondance n'est toutefois pas univoque, et l'on trouve également
diverses associations intermédiaires (e.g. pâte plutôt grossière et glaçure de bonne qualité: dans
la série C, tesson n° 92).
Le tesson n° 59 de la série E se singularise par une glaçure d'aspect opaque.

Chapitre 2 97
Série F: sgraffito incisé peint à pâte grasse, montrant une relative maîtrise dans
l'application des pigments (figure 33)
Ces céramiques a priori plus tardives que les autres, peut-être même postérieures à la
prise de Pergame par les Ottomans vers 1330, sont très fréquentes à Pergame. Deux critères
ont permis de constituer la série F: d'une part la texture caractéristique de la pâte, grasse et fine
avec quelques grandes inclusions; d'autre part la forme des fragments de pieds (14, 44, 45, 90,
146, ... ), qui est propre à cette série. Les deux critères ne sont pas forcément réalisés
simultanément: ainsi les échantillons 14 et 90 ont une pâte plus dure et sombre qui les
apparente plutôt aux séries B, C, D, E. Notons également que le fragment de pied n° 81 de la
série de référence A présente la même forme que les pieds de la série F. Ce point commun
permet de poser l'hypothèse d'une provenance locale.
Les tessons de la série F correspondent surtout à des pièces de grandes dimensions,
notamment de grands vases.
Les couleurs des échantillons polychromes (plusieurs sortes de vert, brun foncé, brun
clair, mauve brun) sont appliquées en bandes suivant plus ou moins les incisions ou en taches.
La relative maîtrise des contours, le fait que les taches n'aient pas diffusé dans la glaçure
contrastent avec les applications de couleurs moins bien contrôlées des autres séries (B, H,...).
On peut noter quelques particularismes: la glaçure de l'échantillon n° 117 a une texture
très granuleuse; la pâte du n° 14 contient de nombreuses inclusions.
Plusieurs tessons analogues par leur pâte grasse aux échantillons de la série F ont été
observés dans le dépôt des fouilles d'Ephèse [179].
Série G: céramiques non glaçurées (figures 34 et 44)
Cette série ne présente qu'un petit échantillonnage des très nombreuses céramiques non
glaçurées, majoritaires sur le site. Les tessons sélectionnés sont assez variés: fonds de pots,
panses et anses de cruches décorées par diverses techniques: application d'un enduit micacé
(136) ou peint (13), combinaison des deux (77), réseau de lignes parallèles gravées (78), ... Les
pâtes peuvent être totalement ou partiellement grisées (50, 51), e.g. rouges avec un coeur gris
(116). Ceci indique des variations dans la conduite de la cuisson, ou peut-être une maîtrise
imparfaite du procédé. Les pâtes sont plus grossières, avec de nombreuses inclusions comme le
montre une coupe de l'échantillon n° 13 sur la figure 44.

Chapitre 2 98_
Série H: sgraffito incisé peint à pâte fine et à glaçure verte teintée de mauve
(figure 35)
Les échantillons de cette série se distinguent par leur décor: le fond de glaçure vert clair
est rehaussé de grandes zonations mauves, parfois brunes. Ils sont relativement peu fréquents
sur le site. Ils représentent des céramiques de petite dimension (bols sans pieds). Leur pâte est
particulièrement fine et tendre, de couleur claire tirant parfois vers une teinte verdâtre (tesson
n° 4). La plupart des tessons de la série arborent une glaçure très brillante, de très bonne
qualité. Par contraste, celle de l'échantillon n° 131 est terne, et en partie recouverte par une
croûte blanche. La même altération affecte localement le tesson n° 3. On peut se demander si
cette différence est due à des variations dans la composition de la glaçure ou à des phénomènes
d'altération se produisant de manière locale dans le milieu d'enfouissement.
Il est important de remarquer que des tessons similaires ont été trouvés à Sardes (voir
référence [143], figure 5). De la même façon, J. Scott distingue deux variétés dans son
échantillonnage, selon l'état de conservation de la glaçure.
L'échantillon n° 18 est un cas particulier: il se rapproche des autres par le décor, mais
s'en différencie par une pâte et une glaçure plus grossières. On peut soupçonner qu'il s'agit
d'une imitation malhabile de cette catégorie de céramiques.
Série I: sgraffito incisé et champlevé monochromes a pâte fine (figures 36 et 46)
Ces tessons décorés au sgraffito incisé ou au champlevé sont de belle facture. Ils sont
assez peu fréquents sur le site. La pâte très fine, tout comme celle des céramiques de la série
H, présente quelques pores (figure 46) et est apparemment dépourvue d'inclusions. Les motifs
relativement élaborés sont couverts d'une glaçure de bonne qualité. L'impression de bichromie
créée sur certains échantillons (52, 98, 110) provient du contraste de teinte de la glaçure selon
qu'elle apparaît sur fond de pâte ou sur fond d'engobe. On peut remarquer la fraîcheur des
verts et des jaunes-oranges soutenus. Certains fragments de pieds ont une forme peu
représentée par ailleurs.
Le tesson n° 71 présente une glaçure lacunaire, voire absente (?).
Série J : sgraffito incisé monochrome (figure 37)
Cette série relativement mal caractérisée est assez fréquente sur le site. Elle présente
quelque analogie avec la série F, en premier lieu par sa pâte assez pure, avec quelques grandes

, Chapitre 2 99
• inclusions et peu de micas. Cette pâte n'a cependant pas l'aspect gras propre à la série F. Les
j céramiques de la série J sont des vases de grande taille, avec des bords à arête épaisse. La
i glaçure monochrome a une teinte jaune clair.Jt
ii
, Série K: céramiques peintes à l'engobe (slip-pninted ware) (figure 38)
!t
j De nombreux tessons de Pergame sont décorés par cette technique. Leur pâte
s'apparente à celle des séries B à E. Les échantillons n° 6 et 10 présentent une pâte noire qui
les assimile presque à des ratés de cuisson. Deux autres pièces paraissent inachevées: le n° 5 et
le n° 143, non glaçuré.
Les séries qui suivent ne représentent sur le site qu'un petit nombre d'échantillons - à
l'exception de la série composite R.
Série L: sgraffito incisé peint à fine glaçure (figure 39)
Les échantillons de cette série sont très rares sur le site (peut-être une dizaine en tout
sur les environ 4000 tessons collectés lors de la fouille de W. Radt). En dehors des zones
colorées, la glaçure est très fine, voire absente (?). On serait tenté de faire un rapprochement
avec le premier stade de l'évolution de la "green and brown painted ware" tel qu'il est décrit par
P. Armstrong [134]: les couleurs peintes à l'engobe sont appliquées sur le fond d'engobe blanc
et recouvertes par une couche de glaçure transparente très fine, voire presque invisible. Dans le
cas de la série L, l'enduit coloré a l'aspect d'une glaçure.
Série M: "Zeuxippus ware" (figure 39)
Cet unique échantillon représente les quelques très rares tessons de "véritable
Zeuxippus ware" trouvés à Pergame (cf. 2.2. et la discussion suscitée par la série B). La pâte
rouge sombre bien cuite de texture fine, la qualité de l'engobe et de la glaçure en sont les signes
distinctifs, plus encore que le motif "typique" des cercles concentriques tracés à la gouge.

Chapitre 2 100_
Série N: sgraffito incisé peint à motifs verts (figure 40)
Cette série se rapproche un peu par sa pâte grasse de la série F, mais présente beaucoup
plus de micas. Les motifs peints sont d'une nuance de vert assez peu représentée par ailleurs, et
ont tendance à recouper les motifs incisés (80, 129) plutôt que de les suivre.
Série O: sgraffito incisé avec décor au peigne (figure 39)
Cette série est analogue à la précédente par l'aspect de l'argile. L'utilisation du peigne
s'y généralise. La glaçure très lacunaire de l'échantillon n° 23 subsiste dans les incisions.
Série P: céramiques peintes à motifs bruns (figure 39)
La représentation de ces tessons sur le site est très faible. Cette variété de décor peint
de couleur brune leur est tout à fait particulière.
Série Q: sgraffito fin (figure 41)
Cette petite série est bien connue des archéologues (cf. 2.2.): ses incisions fines formant
des motifs caractéristiques, sa glaçure monochrome jaune pâle l'identifient comme du sgraffito
fin. Ce type de céramiques est daté de la fin du 1 l è m c siècle - début du 12 è m c siècle, ce qui en
fait la plus ancienne de l'ensemble de l'échantillonnage. La pâte rouge sombre est moyennement
fine.
Série R: divers (figure 42)
Cette série très composite rassemble des cas particuliers: pièces présentant des
variantes de couleur inhabituelles de la glaçure (dans les verts: 91, 108, 127, 128; couleur
saumon: 29; association des couleurs jaune et orange dans une glaçure de bonne qualité: 42,
105) ou des pâtes particulières (blanche: 134; grasse, associée à une glaçure atypique: 140; 47)
ou encore dans un très mauvais état de conservation (41). Les tessons n° 34 et 134 sont non
glaçurés.

Chapitre 2 10_l_
A l'échantillonnage de tessons d'époque byzantine ont été rajoutées quelques pièces
provenant de l'atelier antique de la vallée du Kestel. Ce site n'est aujourd'hui plus accessible,
car noyé sous les eaux d'un barrage.
Série EOL ou Z (figure 43)
Les échantillons de cette série se présentent principalement sous trois aspects:
céramiques à pâte rouge et vernis rouge (20 à 26), à pâte rouge et vernis noir (16, 17) et
céramiques à parois fines et à pâte noire sans vernis (27, 28: non analysées). Les pâtes des
céramiques vernissées sont assez fines et peu poreuses. Celle du tesson n° 19 se distingue par
une texture plus grasse, associée à un traitement de surface différent.
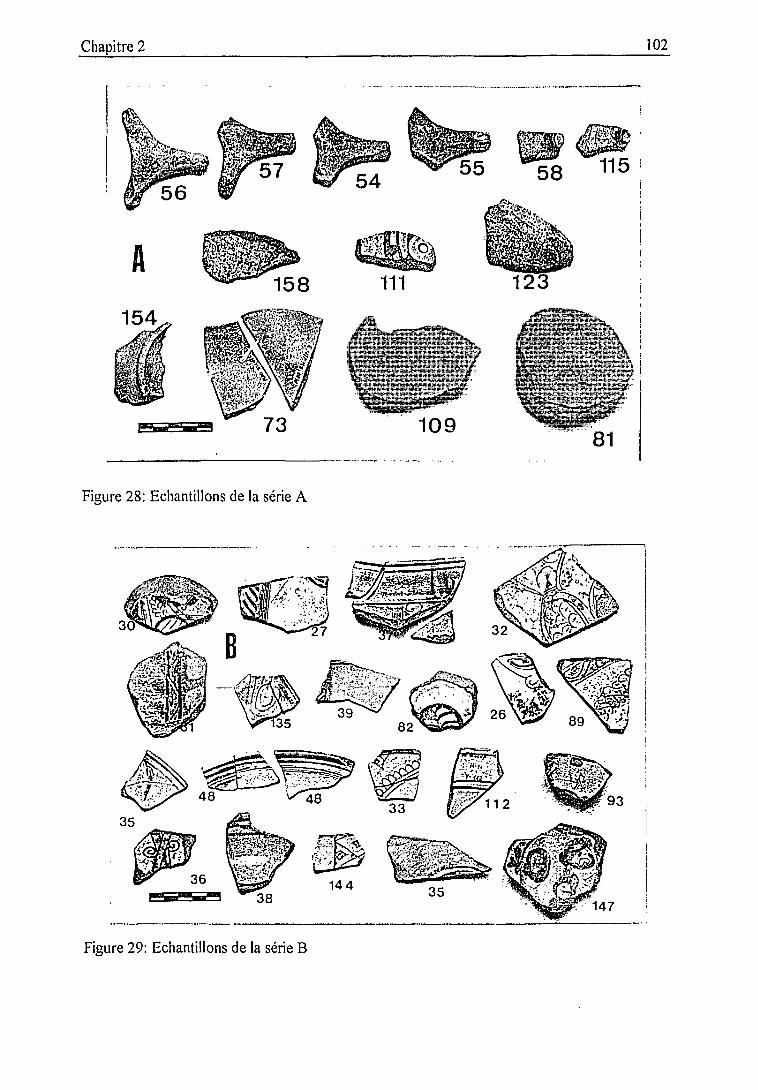
Chapitre 2 102
Figure 28: Echantillons de la série A
Figure 29: Echantillons de la série B

Chapitre 2 103
Figure 30: Echantillons de la série C
Figure 31 : Echantillons de la série D

Chapitre 2 104
104
19
103
102
118
Figure 32: Echantillons de la série E
76 66
Figure 33: Echantillons de la série F
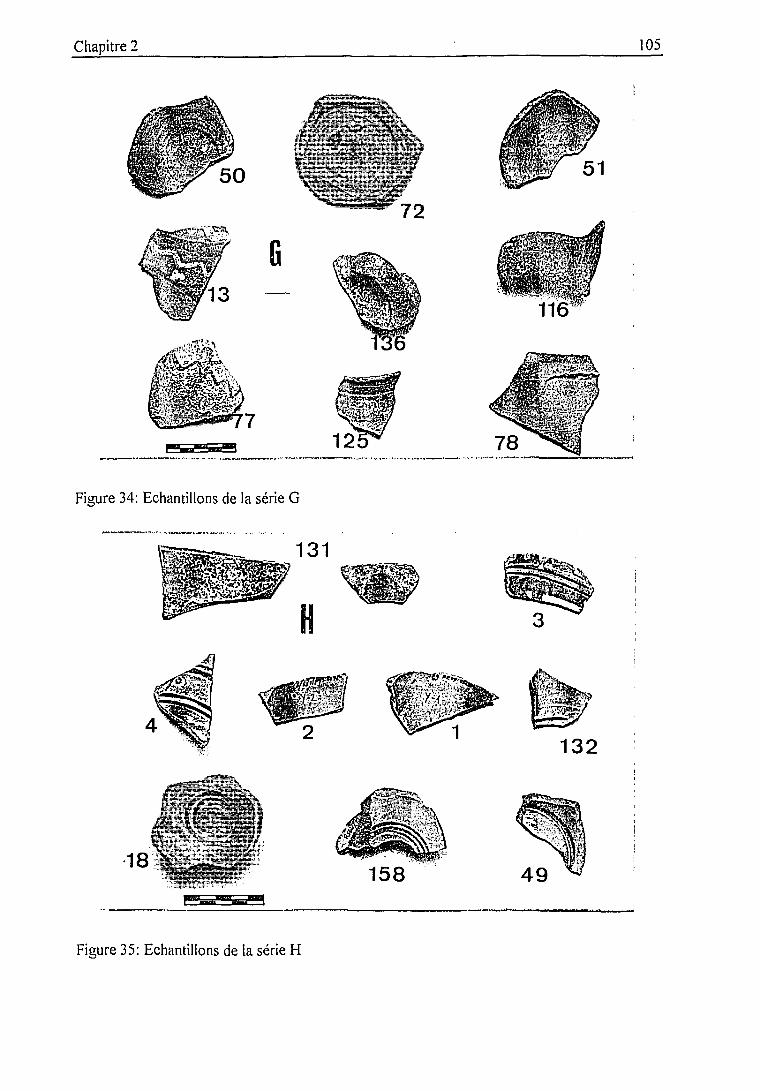
Chapitre 2 105
78
Figure 34: Echantillons de la série G
131
Figure 35: Echantillons de la série H
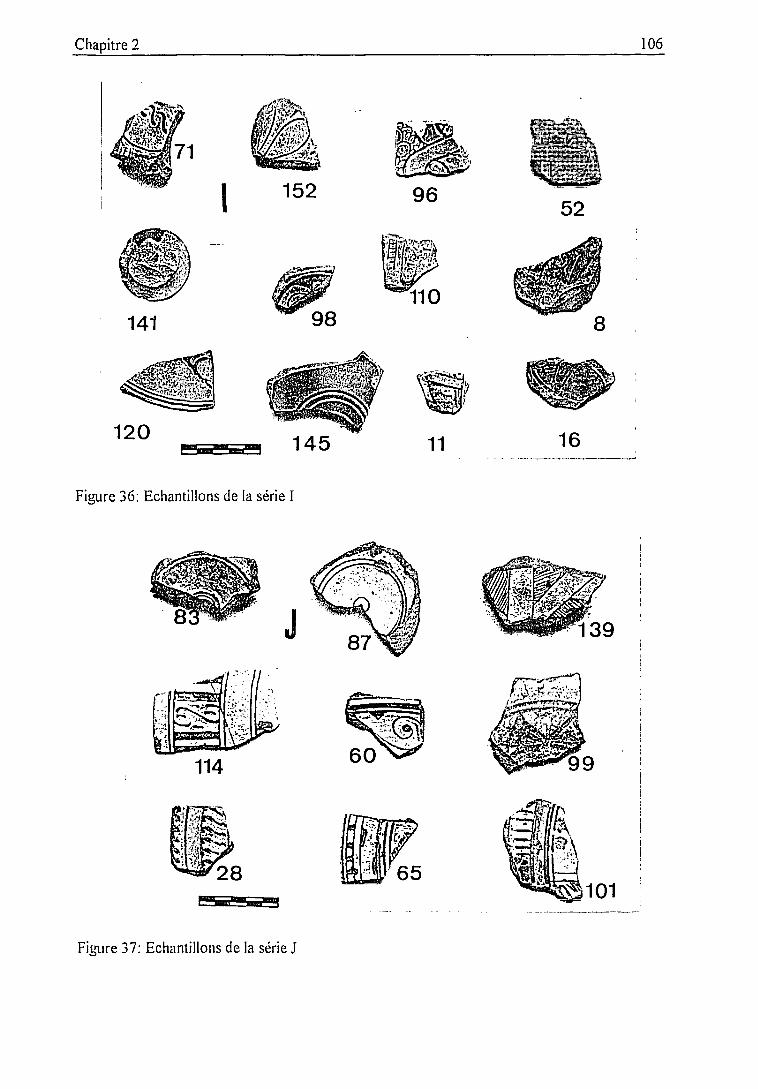
Chapitre 2 106
152
141110
52
120145
Figure 36: Echantillons de la série I
11 16
/ • • • • ' / /
Figure 37: Echantillons de la série J

Chapitre 2 107
10
Figure 38: Echantillons de la série K
148
126
107
149
Figure 39: Echantillons des séries L (haut, gauche), M (haut, droit), 0 (bas, gauche) et P (bas,
droit)
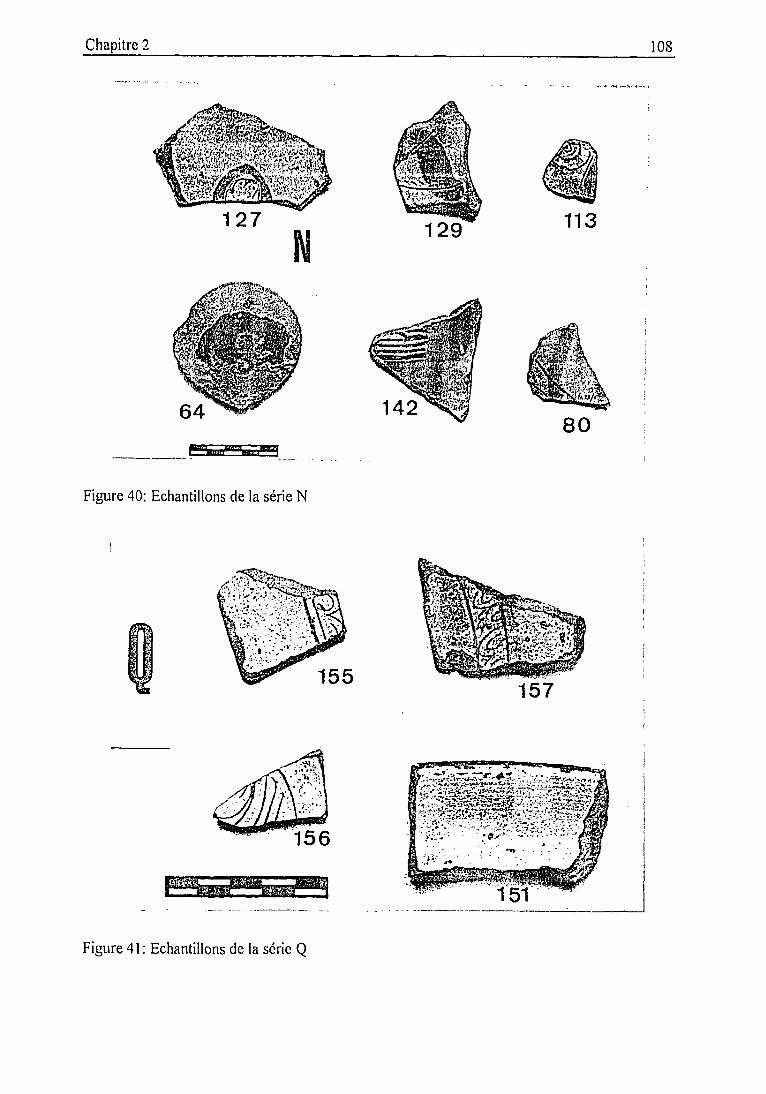
Chapitre 2 108
127 129 113
142
Figure 40: Echantillons de la série N
155 157
W™
156
Figure 41 : Echantillons de la série Q

Chapitre 2 109
Figure 42: Echantillons de la série R
m,18
Figure 43: Echantillons de la série EOL

Chapitre 2 110
Figure 44: Aspect de la pâte sur une coupe fraîche du tesson n°13 de la série G
Figure 45: Aspect de la pâte sur une coupe fraîche du tesson n° 13 5 de la série B
Figure 46: Aspect de la pâte sur une coupe fraîche du tesson n°120 de la série I

Chapitre 2 l_n_
2.3.3. Problématique
Dans une première étape, on cherchera à éprouver la validité de la classication
archéologique. L'homogénéité des séries individuelles est-elle confirmée par l'analyse
élémentaire des pâtes? Certaines séries sont-elles dissociées, et dans ce cas existe-t-il des
paramètres typologiques recoupant cette distinction? Peut-on en regrouper d'autres entre elles,
en particulier celles qui ne se distinguent que sur des facteurs a priori peu significatifs (e.g.
couleur de la glaçure pour C, D, E)?
Ceci nous amène à considérer les relations entre séries, et à nous replacer dans le
contexte d'une production locale à Pergame. Vu dans son ensemble, l'échantillonnage peut se
décomposer de la manière suivante:
- série de référence de la (des) production(s) locale(s): A;
- séries représentant des catégories de céramiques très fréquentes sur le site: B, C, D, E, G, K;
- séries représentant des catégories relativement fréquentes: F, J;
- séries représentant des catégories relativement peu fréquentes: H, I;
- séries représentant des catégories peu fréquentes: L, M, N, 0, P, Q;
- divers: R.
La classification des pièces à partir des résultats d'analyse devrait nous permettre
d'identifier celles qui ont été fabriquées à Pergame comme étant de même composition que les
échantillons de référence. L'importance numérique sur le site, la présence de tessons
d'apparence inachevée ou de surcuits (e.g. au sein des séries B et K) permettent déjà de
présumer quelles séries sont d'origine pergaménienne. Dans d'autre cas, les indices sont plus
ambigus. Ainsi, par exemple, la série tardive F est relativement fréquente et présente des
analogies typologiques avec un des échantillons de référence de la série A. Par ailleurs, la pâte
est nettement distincte de celle des séries de céramiques glaçurées les plus fréquentes sur le site
(C à E). S'agit-il d'une variante dans la production locale? Est-elle obtenue par un traitement
différent de la même argile? Avec une autre argile? D'une manière générale, on essayera de
raisonner suivant le schéma suivant, en se référant aux échantillons de la série A:
pâte identique céramique locale
pâte différente
même argile traitée différemment céramique locale
argile différente
argile locale céramique locale
argile non locale céramique importée
Ce schéma extrêmement simpliste ne nous dit pas comment on distingue une argile locale d'une
argile non locale, ni comment on peut décréter avoir affaire à la même argile traitée
différemment. Ce dernier point est peut-être le moins délicat, car il se ramène souvent au cas

Chapitre 2 m
d'une dilution par des matériaux de composition simple: adjonction d'un dégraissant à base de
quartz, enrichissement en carbonates de calcium .,, Au sens large, il inclut également comme le
suggère le dernier exemple les possibles transformations du tesson au cours de son utilisation
et de son enfouissement, Par contre, exclure la possibilité qu'une argile soit locale demande une
bonne connaissance des ressources disponibles dans la région. A défaut d'une couverture
analytique systématique des gisements argileux, on fera alors fréquemment appel à des
examens pétrographiques, dans l'espoir de déceler des inclusions de nature incompatible avec la
géologie du terrain.
Enfin, on s'interrogera sur le rôle de Pergame en tant que centre de production à
l'époque byzantine, et sur les relations qu'elle entretient avec le reste du monde byzantin:
réseaux d'échanges, influences technologiques, influences stylistiques...
La description archéologique des pièces permet d'identifier des séries de qualité
diffusées à grande distance au sein de l'empire byzantin et des territoires liés commercialement:
sgraffito fin (série Q), "Zeuxippus ware" (série M). Ce dernier type a probablement été une
source d'inspiration pour une production dérivée (série B), comparable par les motifs mais qui
s'en distingue par les caractéristiques macroscopiques de la pâte et de la glaçure.
Des analogies entre le matériel céramique de Pergame et celui trouvé sur d'autres sites,
distants de Pergame de 100-200 km, permet également de supposer des échanges à une échelle
plus régionale:
- à Gùlpinar et à Besik-Tepe, près du site de Troie, ont été retrouvés des tessons similaires à
ceux des séries B à E et K(?) [178];
- à Ephèse, le dépôt des fouilles d'Ayasoluk contient des céramiques semblables à certains
exemplaires de la série F [179];
- à Sardes ont été mis au jour des tessons analogues à ceux de la série H [143].
Ces séries ont-elles été fabriquées à Pergame? Dans l'affirmative, nous ne pourrons pas
en déduire pour autant qu'elles ont été diffusées sur les sites où l'on trouve un matériel
similaire. Pour le prouver, il aurait fallu avoir la possibilité d'analyser également des
échantillons de ce matériel. A l'issue de l'étude, on peut simplement espérer apporter de fortes
présomptions sur la provenance des pièces, qui devront être confirmées par des analyses.
En résumé, nos objectifs dans le cadre de ce travail seront les suivants:
- identifier les séries correspondant à la production de Pergame à l'époque byzantine grâce aux
échantillons de référence;
- caractériser ces séries pour un usage ultérieur dans des études de provenance;
- classifier et caractériser les céramiques importées, sans trop pouvoir espérer proposer une
attribution étant donné le manque de groupes de référence.

Chapitre 2 U3_
L'absence d'analyses comparatives limite évidemment la portée de l'étude, mais l'on peutsouhaiter que celle-ci contribuera justement à édifier un système de références dans le domainetrès peu étudié sur le plan analytique des échanges dans le monde byzantin.

Chapitre 2 114
2.4. Classification et caractérisation des céramiques de Pergame
2.4.1. Traitement statistique des résultats d'analyse élémentaire et classification
des échantillons à partir de leur composition
Les teneurs en 33 éléments ont été déterminées pour les 168 tessons considérés selon
les procédures expérimentales présentées dans le chapitre 1. Les résultats d'analyse sont donnés
dans l'annexe 4. Ces résultats sont ensuite soumis à diverses procédures statistiques afin de
dégager une classification des échantillons.
2.4.1.1. Statistiques univariées
Les statistiques univariées portant sur l'ensemble de l'échantillonnage constituent une
première approche de la structure des données. L'intervalle de variation des teneurs, la forme
des distributions, le repérage des échantillons à valeurs marginales, ... sont autant
d'informations susceptibles d'être visualisées directement sur une représentation en
histogrammes (figure 47). Les statistiques descriptives élémentaires reportées dans le tableau
19 nous donnent les valeurs moyennes, écarts-types et extrémas des concentrations. Elles
indiquent également le coefficient de variation, qui permet d'évaluer la dispersion des données
relativement à la valeur moyenne. Les éléments Pb, Sb, et à un degré moindre la plupart des
alcalins et alcalinos-terreux et certains éléments de transition de la première série comme Cr et
Ni présentent des coefficient de variation élevés. A priori, un coefficient de variation élevé
indique que l'élément a un bon pouvoir discriminant. Mais ceci n'est pas forcément le cas.
Ainsi, l'histogramme de Se, dont le coefficient de variation est plutôt faible révèle une nette
séparation de l'échantillonnage en deux groupes. Inversement, la forte variance de Pb est
essentiellent due à un étalement vers les valeurs élevées qui ne met pas en évidence de
structures de groupes. Il est donc nécessaire de considérer également la forme des
distributions, parmi lesquelles plusieurs types d'allure sont à distinguer:
- les distributions unimodales: V, Ga.
On admet communément que la distribution des teneurs sur un échantillonnage
homogène suit une loi normale (gaussienne) ou log-normale [180]. Inversement, une
distribution d'allure normale n'implique pas forcément la similitude des échantillons. Elle
s'interprète ici plutôt comme la signature d'éléments uniformément répartis sur l'ensemble de
l'échantillonnage, voire dans les matériaux argileux en général. En conséquence, on considère
souvent que ces éléments sont peu aptes à la discrimination entre des céramiques de
provenances différentes, et la tendance est de les éliminer des traitements multivariés.

Chapitre 2 15
Remarquons cependant que certains diagrammes, comme ceux de Zr et Tb, présentent
une allure qui est globalement assez proche d'une distribution normale. On peut se demander
dans quelle mesure elle rend compte de la distribution réelle de l'élément. Certains facteurs tels
la répartition numérique des échantillons au sein des différents groupes de composition, la
précision de l'analyse ou le choix du pas peuvent aussi intervenir. Par conséquent, nous ne
considérerons pas le fait que la distribution soit globalement unimodale comme un critère
suffisant pour justifier l'élimination de la variable correspondante dans les traitements
ultérieurs.
Na2OMgOA12O3
SiO2
K2OCaOTiO2
MnO
Fc2O3
SeVCrCoNiGaRbSrYZrSbCsBaLaCeNdEuTbYbHfTaPbThU
moyenne1.353.118.258.03.47.110.930.118
7.0817.811813025.09725150279331894.217.481944.591.0421.50.833.25.711.462017.44.0
a0.430.61.84.40.63.000.180.016
1.222.824495.850531908483.35.82165.811.770.20.170.60.890.3862.70.8
C.V. (%)
321910818421914
171520382351182132252578332613131713202016212001521
minimum0.141.714.647.72.30.240.600.079
4.908.8654314.1015783315780.65.043530.464.8271.10.552.03.540.882210.72.2
maximum2.364.822.269.05.218.041.310.164
9.4423.019029443.6265372645526338322.734.5147167.6135.3592.01.305.08.532.201020028.08.1
valeurs marginales
R134: 31.0
R134: 1.2
R134: 0.010; G125: 0.017;G50: 0.030; B26: 0.238;B82: 0.236; C80: 0.228R134:2.22R134: 31.4
K7: 594«143:87.1
J28:2000
K143:2.4
B36:25200
R134: 10.6
Tableau 19: Statistiques descriptives sur l'échantillonnage (168 échantillons).
a: écart-type; c.v: coefficient de variation (c.v.= G / m). Les échantillons à valeur extrêmes ont
été éliminés préalablement aux calculs.

Chapitre 2 U6_
- les distributions bimodales ou à dominante bimodale:
On observe une nette dissociation entre deux classes d'effectif comparable pour les
éléments Al, Si, Fe, Se, Cr et de façon moins marquée pour Cs, La, Ce, Eu et U. Nous verrons
par la suite que les variables correspondantes ne sont pas toutes indépendantes. Il existe de
fortes corrélations, par exemple, entre La et Ce, entre Fe et certains éléments de la première
série de transition, entre Si et Al. Pour réaliser la partition en groupes de composition, on
cherchera plutôt à s'appuyer sur un ensemble d'éléments de comportement géochimique
indépendant, et qui ne sont pas liés par des effets de dilution comme Si et Al.
Des distributions de type bimodal, à deux classes d'effectifs beaucoup plus inégaux,
sont observées pour les éléments Ca et Rb. Dans le cas de Rb, un groupe d'échantillons de
teneurs élevées semble isolé du reste de l'échantillonnage. Il est tentant à ce stade de l'assimiler
à un groupe à part entière, surtout si l'on remarque que quatre des six échantillons ainsi
distingués appartiennent à la série archéologique G. Cependant, la singularité et l'homogénéité
de ce groupe sont à tester sur l'ensemble des variables, et ce d'autant plus que les échantillons
concernés sont en petit nombre, donc a priori peu représentatifs de la population parente.
- les distributions polymodales à plus de deux classes: "latentes" dans la plupart des
histogrammes, elles sont parfois assez nettes comme par exemple pour Co et Th.
Un autre type d'allure remarquable est celle des distributions assymétriques, telles
Na2Û, MgO, Ni, Ta, ... Pour un groupe de composition donné, ce comportement est souvent
mis en relation avec un phénomène de pollution. Ceci ne peut évidemment pas être extrapolé à
un diagramme concernant l'ensemble de l'échantillonnage, où une telle allure peut être générée
par la succession de plusieurs groupements. Néanmoins, lorsque les courbes présentent une
forme extrême d'assymétrie avec un étalement vers les valeurs élevées, comme celles de Sb, et
surtout de Pb, nous ferons l'hypothèse qu'un phénomène de ce type affecte l'ensemble ou une
partie de l'échantillonnage. Cette hypothèse est renforcée par le caractère non reproductible des
valeurs de concentrations de Pb.
Remarquons que les céramiques hellénistiques, qui correspondent à une production non
glaçurée, présentent des teneurs en Sb et Pb particulièrement faibles. Les autres tessons ont été
cuits dans des fours servant à la fabrication de céramiques à glaçures plombifères, et l'on peut
supposer que la volatilisation de Pb (et Sb?) à la cuisson est à l'origine d'un apport en ces
éléments dans les pâtes [46], On observe également une légère diffusion de Pb de la glaçure
vers la pâte dans les céramiques glaçurées (cf. analyses à la microsonde, en annexe).
En pratique, nous considérerons qu'il s'agit de "distributions anormales" et éliminerons
les variables Pb et Sb des traitements statistiques multivariés.
Nous éliminerons également de tout traitement ultérieur les échantillons montrant une
composition trop marginale pour un ou plusieurs éléments (cf. tableau 19). Par exemple,

Chapitre 2 1_T7
l'échantillon n° 134 présente plusieurs valeurs extrêmes (AI2O3, K 2 O , Fe 2 O3, MnO, Se, U). Cetesson à pâte blanche est constitué d'une argile de nature très différente de celle des autrestessons, à pâte rouge. Il n'y a pas lieu de pousser la comparaison plus avant, et ce d'autant plusque les valeurs extrêmes sont des sources de perturbation dans les traitements statistiquesmultivariés. Dans d'autres cas, nous pourrons interpréter les écarts et rattacher l'échantillon àun groupe de composit ion sur la base des concentrations des autres éléments.
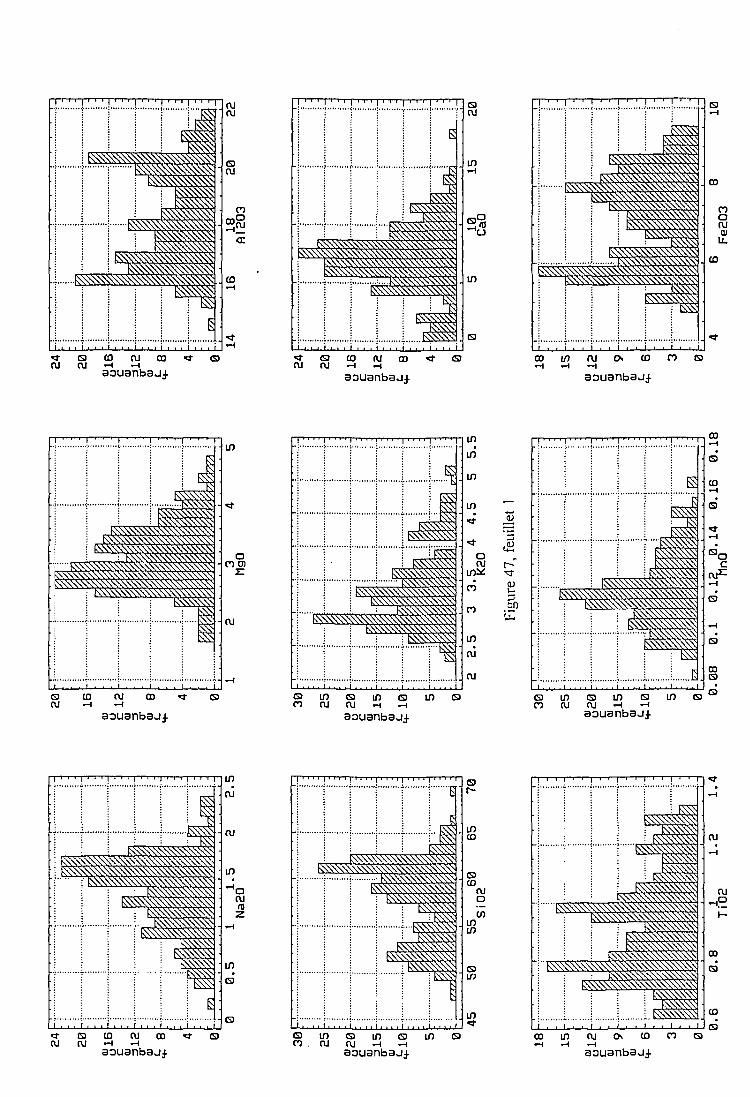
fc
' ' '
• 1 1
r i i
S
n i
^ ^ ^ ^ ^
• • .
1 Ê
<n
CO
J ** f G> CO OJ CO * f 6Jm ni H H
aouanbajj.
r-r-T- i n-[-r-i-r- 1 1 1 -
Ed
... |
wwwwww
\\\\\\\\\\\\\\\\\\\\\\\\N>
; :....kV\SNN\\SW^C .
3°
«t GÎ CO (U 03ni i\i H H
i " i • i i i i i i ' - | i i i i i i
^ ^\\\\\\\\\\\\\\\1
JC3)
00
noru0)
u.CO
oo in ru os co n
aouanbajj-
^\\N\N\\\\\\\\\\\NNN
G) CD OJ CD «•*(XI »H «H
aouanbajj.
ono
OJ
! l • • ' I ' ' • • 1 ' 1 I • • • • I ' ' ' " (
es m ça mO M M H
cs in
aauanbajj-
i . . . . i . . . . i . . i . i . . . . i . . . . i . . . . i
IO
oOJ
rin
inOJ
*~*_o
47. f
eu:
m
Ei
OJ
i • • • ' i ' • • ' i • • ' • r '
ca m es to G)n ru oj -H ~-t
aouanbaj j -
G)
< G> co oj co -*W (U H H
aouanbajj-
I ' ' " i ' ' ' * i " ' ' i * ' ' ' i " " i * ' ' ' i
•0(S
OJo
G) IO C3 IO On , oj oj -< -H
aouanbajj-
I O CS3
-
^
• •
NNSNV
s
1 * \ ' ' !
^^^^^<S^XSS
k\<\x^-s\v<sX\XX
oo m OJ os co^ -H -H
aouanbajj-
OJ
OJo
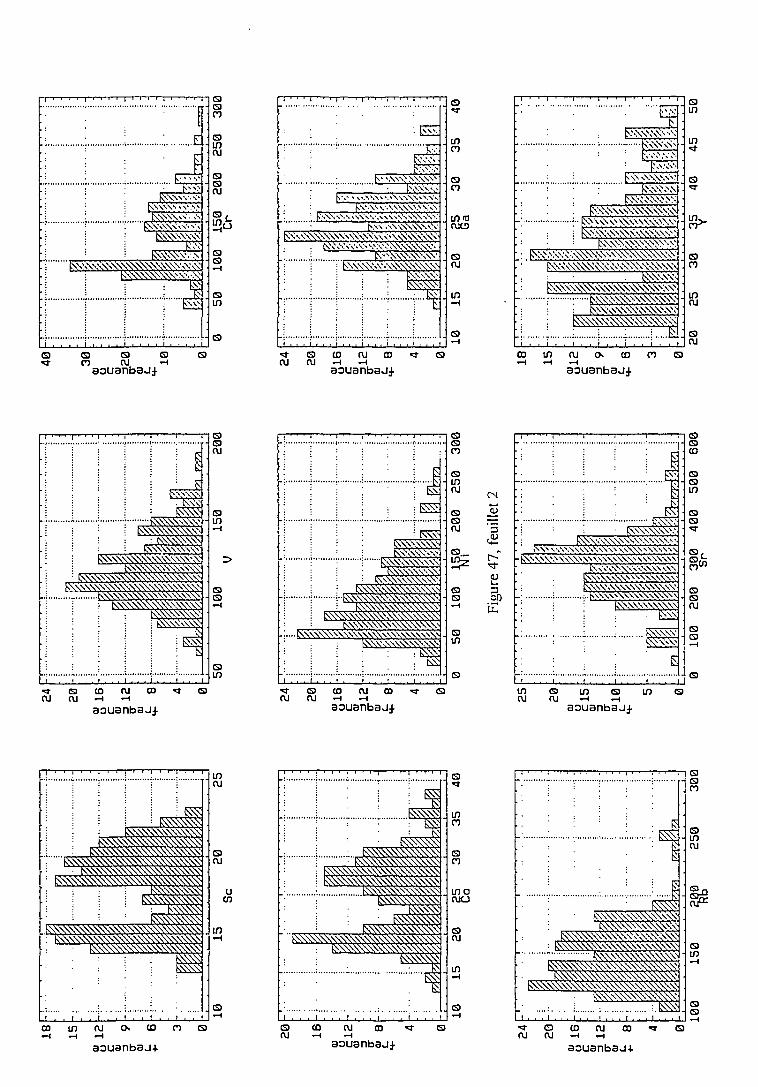
v\\<
1 I , , , , 1
S -
- G>
• ' • ( ' • • ( •
F
—'—'Rv: .vs vy>:y-
•v.wwwww.•
G)n ru
aouanbajj-G) " * S> CO (VI 00 * t
(VI (VI
inn
in oiCVK3
es(VI
Rs\\\\\\\V
C\\\\\\\\\\\\\\\\\\\\\\\VK\\\\\\\\\\\\\\\\\\\\\\\\\V-
ni8 CO (VI CO
1\\\\\\\\\\\V
• K\\\\\\\X\\\\\\\\\V-'K\\\\\\\\\N\\\\\V
t\\\\\\\\\\\\\\s>^\\v^N\\\\\\\\S\\^s\\\,SS'
cs._
ruoai CD ai
es•4—1
jN\\\\\\v.-s s\\\\\Vv\\\\N
>\\\\.\V>V,\\\\\N>\\>\\\\S
N\\\\\\\\\\\\s\\NV
\iNW
\\\\v^\>v
QCO
in
s
C 3 .
roU J
G3O(VI
C3
inru s
(VI
m G> inaouanbajj.
(N\\\\\\NSSS?KNSS$
œ in ru o. ce <s
ino(VO
in
. J Q
a co ru œ(VI «H - I
aouanbajj.co ru oo
33uanbaj+

f requence
acn
frequencet - i - ro ro
•p» co ro co Q •**
frequenceIU
CO
an-*wo
ro
• • • i • • • i
I
s *• œ ro en (s -u
,roHen h' s
) ' ' ' 1 • • • 1 ' • ' 1 ' • •
• ^ w ^ i ;
: g ^ i !
Mili i , . , i , . , • . • . . . i
ro
rnr
oo
ro
frequenceco ro a>
• i ' • • i
ro roi • • • i • • • i
*> SCSVNSN.V\\VJ
\ \ W W N ci
ni—t-
frequencei - t - ro ro to
<3 Ol O O1 (S UI G3CO
rcnQMS
CD
o
r " 'i " "1 ' . . . .
^ I !3 ! I
i . . . . i . . . i . . , . . . .
' ' ' "
f requencet- ro co
o G o Q
cn
CO
cr
cn
\\\\\\\\\\\\\N\\\S\\SS\\\\\\\\\V<v\WJ\\\\\\\\\\\\\\N\\\S\\SS\\\\\\\
frequence
s co CD «o ro cn ooO I I i- i I ' i I • ' I i i I i i i i i i
ro r; ~
<vV- . ..
,SVv "• s\V
f requencet- >- ro ro
en s en s cnfrequence
I H
n:
1
\WWWVwN
AW-1" ^
WWv>
I I , • . I . , . . 1
Q
•-»(3
nroin s>
30
40
e3 *»
vW.s\\N
oo ro CD s t
p3. ] •3 • •
• • 1 ' ' ' 1 t ' ' 1 . . . . 1 1
sS
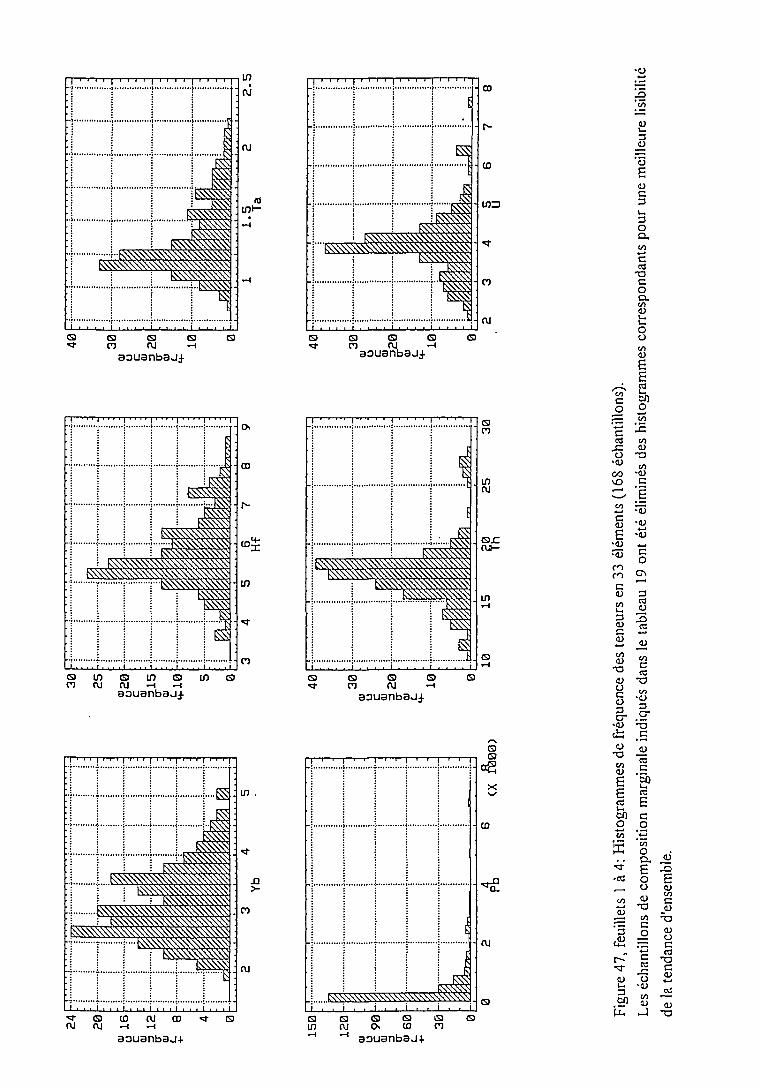
••
I
7Rv
1;
OJ
I ' • ' ' I ' ' ' • I ' ' ' ' I ' ' ' ' TCD
CO
• OJ
ra (S3
re<3 C3 O Q
t re OJ
S\\\\^
1
i.;•;:yr:r:rrr;T:Tr:r::"7i":rr: ~
o<
oa
- r-
• inin
s in s in s 10 essre oj OJ
G> Q G) Q•«r re OJ *H
CS)
1 • . • 1 . 1 . 1 . . . 1 . . . , . . , 1 . . .
ES
i
1 • • • t . . • 1 , , , 1 , 1 , 1 1 , , 1 . , ,
XI
OJ
* t C3 CD OJ 00 «*OJ OJ -I ^
•
j[\\\\\\\\\\\\\\\\\\\\\\\\\\\N
, i 1 . . 1 . • 1 • • I > > I
CSIS)
• CD
- OJ
G3 ^D ^D ^3 Cîin OJ o< co re
3
*CO
_O
CRJ
JZO
*4JCOVO
co+-*C
J• 4 )
enmc4)
3
C4)
• * - *
des
quen
c
'4)
4)•oco4)
SEaenoco
!
CO
4)
3
»4)^-
enft (
4)c3
3Oexco
K)X )CoCl-eo
Ëoo004)
EEueno4-J15
004)
•a0 0
•4)C
'B
éli
•4)-4>
ont
3
3
4)an
s 1
•o
iqué
s
•o.S4)
•—i
ina
en
Eco
' ôô
oQ .
Eoo4)
• a0 0
Ion
c
cha
•4)004)
4)
E4)co
_4>• 7 1
4)OCre!
ten(
4)•a

Chapitre 2 122_
La portée d'une représentation en histogrammes de l'ensemble de l'échantillonnage est
fort limitée pour ce qui est de la classification. Même si sur certains graphiques les échantillons
se distribuent entre plusieurs classes, le nombre apparent de classes a en général peu de rapport
avec le nombre de groupes de composition. Pour accéder à la structure de l'échantillonnage
dans toute sa complexité, il faudrait pouvoir considérer l'ensemble des modalités prises par un
échantillon pour les différentes variables. Difficile à mettre en oeuvre à partir des distributions
univariées, cette approche est aisément réalisable avec des traitements multivariés. Certains
d'entre eux permettent justement de travailler sur une répartition en classes (analyse des
correspondances). En pratique, nous préférerons conserver des variables continues
(concentrations) pour éviter les problèmes liés à la définition arbitraire d'un intervalle propre à
chaque classe.
2.4.1.2. Méthodes multivariées [181, 182]
En analyse des données, on considère des individus (échantillons) définis par les valeurs
qu'ils prennent sur un ensemble de caractères (variables), qualitatifs et quantitatifs. Dans notre
cas, ces variables - les concentrations des 33 éléments dosés - sont quantitatives, mais rien ne
s'oppose formellement à ce que l'on introduise également des paramètres qualitatifs tels la
forme, la couleur, la datation, ... On utilise une représentation sous la forme d'un tableau
numérique (individus x caractères, cf. le tableau de l'annexe 4) qui permet un traitement par le
calcul matriciel. On fait également appel à un support géométrique, en assimilant un échantillon
à un point dans un espace à n dimensions, n étant ici le nombre d'éléments. Le sytème de
coordonnées correspondant associe un élément à chaque axe, la coordonnée du point
représentatif étant la teneur. Dans la plupart des méthodes utilisées, on éprouvera le besoin
d'effectuer un changement de variables lorsque ces dernières sont exprimées dans des unités
différentes ou concernent des quantités d'ordres de grandeur différents - e.g. les teneurs en
éléments majeurs par rapport à celles en éléments traces. Pour se ramener à des quantités sans
dimension et afin que chaque variable intervienne de manière équivalente, nous avons appliqué
une procédure de normalisation couramment utilisée en définissant une nouvelle variable C
par:
C étant la concentration (mesurée) de moyenne C et d'écart-type a.
On se ramène ainsi à un ensemble de variables qui ont toutes une moyenne nulle (variables
dites centrées) et une variance unité (variables dites centrées réduites). Le résultat de cette
procédure dépend de l'échantillonnage choisi. En particulier, on éliminera au préalable les
valeurs trop extrêmes, qui sont susceptibles de décaler la moyenne et d'augmenter

Chapitre 2 \Th_
artificiellement l'écart-type. L'effet de ces valeurs extrêmes est surtout très critique dans des
traitements comme l'analyse en composantes principales (cf. Annexe 5.A). D'autres types de
transformation des données sont également mises en oeuvre dans les études de provenance
[180, 74, 183]: application d'une fonction logarithmique aux teneurs en éléments traces,
division des données centrées par la moyenne plutôt que par l'écart-type,... Remarquons que le
fait de se ramener à une variance unité n'est pas toujours des plus avantageux. Il risque en
particulier de faire surestimer l'influence de teneurs à faible intervalle de variation. Ceci peut
être problématique dans le cas de variables déterminées avec une faible précision. On aura
souvent intérêt à comparer les résultats obtenus avant et après élimination des éléments de ce
type, qui ont tendance à perturber l'information principale.
Plusieurs méthodes sont classiquement mises en oeuvre pour accéder à la structure de
l'échantillonnage. Parmi elles il faut distinguer les analyses factorielles qui ont pour but une
visualisation synthétique des propriétés (ACP, analyse des correspondances,...) des procédures
de classification (CAH, "cluster analysis",...).
Principe des méthodes factorielles
Dans les méthodes factorielles, on effectue un changement de variables linéaire qui
permet de représenter la majeure partie de l'information contenue dans le système en un
minimum de facteurs. Au sein de la gamme des analyses factorielles (analyse des
correspondances, analyse discriminante, ...), nous nous limiterons ici au cadre de l'analyse en
composantes principales (ACP) qui traite uniquement les données quantitatives, et au cas de
variables centrées réduites.
Le but est d'appréhender la répartition de p individus (ou points) dans un espace à n
dimensions. Pour cela, on cherche une base orthonormée telle que leur projection sur le
premier axe, le premier plan, le premier sous-espace à trois dimensions, ... offre une vision la
moins déformée possible, c'est-à-dire telle que les distances entre projections soient les plus
voisines des distances entre les points eux-même. De façon équivalente, le système de
coordonnées recherché exprime à partir de ses premiers axes la plus grande partie de la
variance (ou de l'inertie) du système, i.e. l'allongement global du nuage de points est bien
représenté par une projection sur ces premiers axes nommés axes principaux. Les nouveaux
caractères construits par combinaisons linéaires des variables initiales sont les composantes
principales. Par construction, les composantes principales sont indépendantes et de variance
décroissante. Cette procédure de réduction de la dimension "utile" de l'espace n'est "efficace"
que si les coefficients de corrélation r entre les variables initiales sont non nuls.
On peut montrer que les vecteurs unitaires définissant les axes principaux sont vecteurs
propres de la matrice de corrélation R, dont le terme ry est construit à partir des variances et
covariances des caractères de la matrice X (individus x caractères):

Chapitre 2 124
Sj Sj
k - - - - - - - v " ks? est la variance du caractère i de moyenne Xj et SJ: la covariance des caractères i et j .
Les valeurs propres de R sont les variances des composantes principales.
Une fois les axes et composantes principaux déterminés par diagonalisation de R, deux
types de représentation sont possibles: dans l'espace des individus et dans celui des variables. •
Dans le premier cas, on visualise les projections des points sur les premiers plans factoriels
(axe 1, axe 2), (axe 1, axe 3), ... Les groupements observés sont parfois utilisés comme une
base pour la classification. Il faut toutefois être conscient que l'ACP procède à une
optimisation du plan de projection pour l'ensemble de l'échantillonnage, et que des points
peuvent être individuellement mal représentés. En pratique, on peut évaluer la qualité de la
représentation pour un point donné en calculant le cos2 de l'angle entre vecteur et projection et
en vérifiant que sa valeur est proche de 1. L'interprétation des groupements dans l'espace des
individus fait appel aux propriétés de l'espace des variables. Dans l'espace des variables, chaque
caractère correspond à un vecteur dont les n coordonnées sont les valeurs prises par les n
échantillons. Dans le cas de variables centrées réduites, ces vecteurs sont unitaires et on peut
montrer que le cos de l'angle entre les vecteurs représentatifs de deux caractères est égal à leur
coefficient de corrélation. Lors de la projection sur les plans principaux, ils sont inscrits dans
un cercle de rayon 1, dit cercle des corrélations, qui permet de visualiser directement les
corrélations entre variables, sous réserve qu'elles soient bien représentées, i.e. que les points
correspondants soient proches de la circonférence. Une autre propriété est que la coordonnée
d'une variable par rapport à un axe factoriel est égale à leur coefficient de corrélation. On
obtient ainsi l'interprétation des axes factoriels en identifiant les variables qui leur sont les plus
corrélées (cf. figure 48).
Figure 48: Représentation du résultat d'une ACP dans l'espace des variables, cercle des
corrélations sur les deux premiers plans principaux; variables initiales Vi, composantes
principales Ci.

Chapitre 2 125_
La figure s'interprète de la manière suivante: les variables VI et V2 sont correlées entre elles,
et anticorrelées à V4 (surtout VI, V2 étant moins bien représentée). La première composante
principale Cl , qui est associée à l'axe de plus grande variance du système, correspond à des
fortes valeurs de V4 et de faibles valeurs de VI et V2. V3, pratiquement indépendante des
trois autres variables V, est correlée avec C2.
L'ACP sera principalement utilisée dans ce travail pour son pouvoir interprétatif, en
complément à une procédure de classification automatique (cf. paragraphe suivant). Elle est
également utile en amont des classifications, comme moyen de production d'un système de
variables non correlées dont seules les premières sont significatives. Les informations
redondantes sont ainsi évitées, et les dernières composantes principales, exprimant seulement
quelques fractions de pourcents de la variance du système, sont éliminées en tant que "bruit
statistique".
Principes des méthodes de classification
Ces méthodes ont pour but l'association des p individus en un petit nombre de groupes
homogènes du point de vue des n caractères considérés. La classification est construite au
moyen d'une procédure itérative décrite par un algorithme. Dans un premier temps, il faut
définir un indice qui mesure la ressemblance - ou la dissemblance - entre deux individus ou
entre deux groupes d'individus. Cet indice n'est pas nécessairement une distance au sens
mathématique du terme. Dans le cas de données numériques continues, il est en généralement
défini à partir de la distance euclidienne.
Il existe deux grandes familles de méthodes de classification: les méthodes
hiérarchiques (ascendantes, descendantes) et les méthodes non hiérarchiques.
Classification ascendante hiérarchique (CAH)
Partant de p classes correspondant aux p individus, la CAH constitue p-1 nouvelles
classes par agrégation des deux individus les plus proches (au sens de l'indice de dissimilarité).
A chaque étape, les deux classes les plus proches sont fusionnées en une seule, jusqu'à ne plus
obtenir qu'une classe unique lors de la dernière itération. Les q, q-1, q-2, ..., classes produites
par les partitions successives sont ainsi emboîtées les unes dans les autres, d'où le terme de
"hiérarchique". Inversement, la classification descendante hiérarchique part d'une seule classe
pour parvenir, par dissociations successives, à q individus. Par la suite, nous considérerons
uniquement la CAH.
Une CAH est entièrement définie par son indice de dissimilaritè et par la façon dont on
réalise h fusion entre deux classes. On peut définir ces quantités de manière assez directe et
proche des données elles-même, ce qui permet de garder un meilleur contrôle du processus.

Chapitre 2
C'est le choix qui a été fait par M. Picon, au laboratoire de céramologie de Lyon [108], Lors
d'une fusion, on crée un "pseudo échantillon" qui est le barycentre de tous les individus qui y
contribuent, quel que soit le niveau de l'itération par laquelle ils ont été rattachés au groupe. Ce
type de fusion est dit "en affinités moyennes non pondérées". L'indice de dissimilarité utilisé
est la distance euclidienne.
Nous avons principalement utilisé une méthode alternative, la méthode de Ward, qui
fait appel à la notion d'inertie plutôt qu'à celle de distance. On définit l'inertie I d'un ensemble
d'objets i de poids m; et de distance euclidienne d(i, G) au barycentre G par:
I=5>id2(i,G)i
D'après le théorème de Huyghens, si on répartit l'ensemble des points i en k groupes, de
centres de gravité g , l'inertie totale peut se décomposer en inertie inter-groupes et inertie
intra-groupes:intra-groupes:
k k iek
On conçoit bien que la meilleure partition est celle qui minimise la contribution de l'inertie
intra-classes (groupes les plus homogènes possibles), ce qui est équivalent à rendre maximum
la contribution de l'inertie inter-groupes (groupes nettement séparés).
Dans le cadre d'une CAH, on perd nécessairement de l'inertie inter-groupes en réduisant
le nombre de classes à chaque itération. Le principe de Ward consiste à minimiser cette perte.
Si on considère les classes 1 et 2, de centre de gravité gj et g2 de masse ni] et m2, agrégées en
une classe unique de centre de gravité g et de masse m (m = mi+m2), l'inertie des classes 1 et 2
peut se décomposer comme:
En fusionnant 1 et 2, on réalise une perte de l'inertie inter-groupes ôj2:
m t m 7 7g) = ~-<^ {g\,gi)
mj + m2
Dans la méthode de Ward, on agrégera les deux classes qui présentent non plus la distance
minimale mais la quantité ô12 minimale.
Quel que soit l'algorithme utilisé, on représente les partitions emboîtées d'une CAH
sous une forme plane dite "dendrogramme" (cf. figure 49). En ordonnée est porté le niveau
d'agrégation entre les classes: plus la barre horizontale est basse dans le diagramme, plus les
deux classes qu'elle réunit sont similaires. Les individus sont représentés en "abscisse", au
niveau de fusion le plus bas, dans un ordre qui est uniquement conditionné par la facilité de

Chapitre 2 127
lecture des liens de fusion. Réaliser une partition consiste à choisir à quel niveau on va
sectionner l'arbre: les "branches" qui se trouvent isolées sous le niveau de troncature
constituent les groupes. En pratique, la troncature est effectuée à l'endroit d'une discontinuité
des indices de dissimilarité.
A
5 3
Figure 49: Représentation d'une classification hiérarchique ascendante sous la forme d'un
dendrogramme. La troncature de l'arbre choisie (en pointillés) isole trois classes ((1,4), 6,
(2,5,3)).
Si les deux algorithmes cités sont réputés donner des résultats cohérents, on reproche
parfois à celui de Ward d'amplifier artificiellement les différences entres groupes [184] (ce sont
les carrés des distances qui interviennent), voire de séparer des groupes globalement
homogènes. En contrepartie, il donnera des résultats plus clairs lorsqu'il existe effectivement
des groupements au sein de l'échantillonnage [180].
L'un des inconvénients majeurs de la CAH réside dans le caractère emboîté des
partitions. Une fois une liaison réalisée, elle n'est plus remise en question lors de la construction
des partitions suivantes. C'est pourquoi l'on peut préférer des méthodes non hiérarchiques qui
sont susceptibles de réaliser des réallocations d'individus au sein des classes à chaque itération.
Classification non hiérarchique
Nous en donnerons un exemple en décrivant la procédure de classification autour de
centres mobiles que nous avons utilisée comme alternative à la CAH. Dans ce type de
procédures, on choisit a priori le nombre k de classes dont sera composée la partition, k
individus sont désignés, en général par tirage au sort, comme centres de classes initiaux. Les
autres individus sont attribués chacun au centre de classe le plus proche. Les centres de gravité
de chaque groupe sont déterminés, et servent de nouveaux centres de classes pour l'itération
suivante. On peut décider d'arrêter l'algorithme lorsque l'inertie inter-classes ne varie plus de
manière sensible.

Chapitre 2 128_
La partition obtenue dépend souvent de manière assez critique des centres de classes
initiaux. De plus, le nombre de classes optimal est en général inconnu, Aussi peut-on croiser
plusieurs partitions réalisées en faisant varier k et les centres initiaux afin de déterminer les
formes fortes. Celles ci sont constituées par les groupements stables par les différentes
classifications. Le logiciel SPAD-N propose de compléter la procédure en appliquant une CAH
aux centres de gravité des formes fortes. La partition "définitive" est alors effectuée.
Mise en oeuvre
Nous avons principalement utilisé les logiciels STATGRAPHICS [185] et SPAD-N
[186], avec une préférence pour ce dernier. STATGRAPHICS est un logiciel intégré,
généraliste, qui gère aussi bien les statistiques descriptives, les tests statistiques, ... que les
statistiques multivariées. SPAD-N est plus spécifique aux méthodes factorielles et aux
procédures de classification, et permet de réaliser les deux types de classification - hiérarchique
et non hiérarchique. De plus, la CAH peut être suivie dans SPAD-N d'une étape dite de
consolidation, qui applique la méthode des centres mobiles aux classes obtenues. On peut ainsi
remettre en question des fusions produites à un niveau quelconque de l'algorithme, et procéder
à une réallocation des échantillons "périphériques" aux groupes. SPAD-N est conçu comme un
langage de programmation, bien que les versions développées actuellement pour Mac et PC
soient plus conviviales avec l'introduction d'une interface proposant les options des traitements
statistiques sous forme de menus. De nombreuses procédures d'aide à l'interprétation sont
proposées, mais l'absence de sortie de type graphique sur la version Mac que nous avons
employée reste un inconvénient.
L'étape préliminaire consiste à éliminer les individus à composition anormale (cf.
tableau 19) et à transformer les données d'analyse afin d'obtenir des variables centrées réduites.
On a également écarté des traitements multivariés les éléments Pb et Sb au vu de leur
distribution (cf. Statistiques univariées). Plusieurs essais de classification et d'interprétation des
classes ont ensuite été réalisés.
Classification
La figure 50 présente quatre exemples de dendrogrammes obtenus par CAH avec
l'algorithme de Ward sous différentes conditions. Les trois premiers essais portent sur les
données brutes, i.e. les variables centrées réduites, par opposition au quatrième essai qui est
une classification sur coordonnées factorielles. Entre les trois premiers, seul diffère le choix des
variables.

Chapitre 2 12£
Les paramètres sont les suivants:
* figure 50 a): classification sur l'ensemble des 31 variables;
* figure 50 b): classification sur une sélection de 22 variables.
La question du choix des éléments les plus discriminants dans les classifications de
céramiques est un sujet qui a suscité l'intérêt de nombreux chercheurs. En premier lieu, il parait
nécessaire de déterminer à quel point une mesure de concentration est représentative de la
céramique [187, 188, 189, 190, 47], notamment dans les cas d'hétérogénéité (Ca, éléments de
minéraux lourds inégalement distribués e.g. Zr [38], ...) ou de pollution (Mg (altération en
milieu marin) [48], P (fouilles de dépotoirs) [49], Ba et Sr (enfouissement en terrain filonien)
[50], ...) (voir aussi 1.1.2. Echantillonnage d'une céramique archéologique). En pratique, nous
nous contenterons d'éliminer les échantillons qui présentent des valeurs aberrantes, susceptibles
d'être attribuées à l'un des phénomènes évoqués: K7 (Zr), J28 (Ba), ... et non les variables
elles-même. Le role de ces variables dans la détermination de la partition sera néanmoins
considéré de manière plus critique. Les échantillons écartés pourront éventuellement être
attribués par la suite à l'un des groupes de composition formés.
L'identification des variables les plus significatives s'est souvent appuyée sur leur bonne
séparation de différentes productions, parfois au travers d'une contribution importante à des
fonctions discriminantes. Une compilation des résultats de plusieurs auteurs est présentée dans
[132]. Ces études, menées principalement sur des données obtenues par activation
neutronique, désignent certains alcalins et alcalinos-terreux (Na, Cs, Ca, ...), certains éléments
de transition de la première série (Fe, Co, Cr, Mn, ...), des terres rares (Ce, La, Sm, ...), Ta,
Hf, Th,... comme particulièrement discriminants. Une autre approche comparant, pour des
groupes de composition déterminés au préalable, les dispersions inter- et intra- groupes a
montré qu'il existe une bonne corrélation entre ces deux quantités sur l'ensemble des éléments,
majeurs et traces [191]. Il en ressort que, s'il n'existe pas de "composants miracle" qui
allieraient une faible variation intra-groupes à une forte variation inter-groupes, le caractère
réputé nettement plus discriminant de certains éléments traces comme les terres rares peut être
mis en question. De manière sous-jacente se pose la question du choix de la méthode d'analyse,
et de la nécessité de faire appel à des méthodes "lourdes" et coûteuses comme l'activation
neutronique pour déterminer ces éléments.
Quoiqu'il en soit, nous nous en tiendrons à ce stade à un choix dicté exclusivement par
des considérations de précision, et dont la validité est propre aux procédures analytiques
utilisées et à l'échantillonnage étudié. Le caractère discriminant des éléments pourra être
discuté a posteriori dans le cadre de l'interprétation des classifications par l'ACP (cf.
Caractérisation des groupes de composition).
L'introduction de données déterminées avec une faible précision, qui interviennent dans
l'algorithme de classification au même titre que les autres, est une source de perturbation pour

Chapitre 2 130
l'information recherchée. Comme critère de sélection empirique, on a estimé raisonnable de ne
garder que les variables dont le coefficient de variation c.v. sur l'échantillonnage est largement
supérieur à l'incertitude statistique relative moyenne AC/C (tableau 20). Suivant ce critère, les
variables Mg, V, Ga, Y, Nd, Eu, Tb, U ont été éliminées, ainsi que Co dont la teneur est
sujette à une contamination de la mèche lors du prélèvement (cf. Chapitre 1, 1.1.2,
Echantillonnage, Contamination lors du prélèvement). Bien qu'également déterminé avec peu
de précision, Yb a été conservé comme l'élément dans le cas le moins défavorable parmi les
terres rares lourdes, dont on souhaite conserver un représentant.
C.V. (%)
AC/C (%)
C.V. (%)
AC/C (%)
Na
32
3
Sr
32
3
Mg
19
15
Y
25
19
Al
10
4
Zr
25
4
Si
8
1
Cs
33
8
K
18
3
Ba
26
16
Ca
42
3
La
13
2
Ti
19
5
Ce
13
2
Mn
14
2
Nd
17
24
Fe
17
2
Eu
13
11
Se
15
1
Tb
20
18
V
20
26
Yb
20
18
Cr
38
2
Hf
16
3
Co
23
2
Ta
21
13
Ni
51
38
Th
15
1
Ga
18
14
U
21
18
Rb
21
3
Tableau 20: Comparaison des coefficients de variation sur l'échantillonnage c.v. et des
incertitudes relatives moyennes sur la détermination des concentrations AC/C. Ces dernières
sont calculées uniquement à partir des incertitudes statistiques de comptage (cf. Chapitre 1,
1.1.2., tableau 2, et 1.2.2., tableau 10).
Quelques (rares) auteurs font directement intervenir les incertitudes de mesures dans la
détermination des groupes, de façon "empirique" en faisant varier la position du point dans
l'espace à n dimensions [192] ou de manière plus méthodique en introduisant l'incertitude au
niveau de la définition de la mesure de dissimilarité [193, 194].
* figure 50 c: classification sur les éléments déterminés ou pouvant être déterminés par PIXE -
cette dernière condition s'appliquant à Na, Mg et Mn; les éléments Ga et Y ont été écartés (12
éléments au total).
Il s'agit de tester la capacité de la méthode PIXE à fournir des données suffisantes pour
une classification. Notons qu'un essai analogue pour les données déterminées par INAA ne
serait guère significatif, dans la mesure où l'on a délibérément négligé de doser par INAA les
éléments qui pouvaient l'être par PIXE (en particulier les éléments majeurs).
* figure 50 d: classification sur les 12 premières composantes principales, représentant 90% de
la variance totale de l'échantillonnage.
Une ACP sur les 31 données centrées réduites initiales a été réalisée préalablement à la
classification. Elle a permis de construire de nouvelles variables non correlées, dont les 12

Chapitre 2 131
premières suffisent à exprimer la plus grande partie de la variance du système (cf. figure 52).
Les 19 variables suivantes, qui n'en représentent que 10%, sont assimilées aux fluctuations
statistiques et éliminées. On devrait ainsi pouvoir construire une classification plus stable,
moins sensible à de petites variations des teneurs liées aux erreurs expérimentales. L'annexe
5. A indique les précautions d'emploi de cette procédure.
Les dendrogrammes obtenus (figure 50) ont à première vue la même structure:
opposition franche entre deux groupes principaux, le premier (à gauche) étant homogène dans
sa plus grande partie, le deuxième étant plus composite. C'est surtout sur l'organisation de
cette dernière branche que les quatre diagrammes diffèrent. Le schéma qui semble le plus clair
est celui formé à partir de la CAH sur les coordonnées factorielles: branches mieux espacées
entre elles, discontinuités plus nettes. On constate néanmoins que la suite définie par les
niveaux de fusion ne présente pas de discontinuité qui détermine de manière univoque le niveau
de troncature - en dehors du cas de la partition "triviale" en deux groupes. Dans la plupart des
cas, l'histogramme des indices de niveaux présente des discontinuités générant des partitions en
5-6 ou 8-9 classes (voir un exemple de procédure complète en annexe 5.B). En testant ces
différentes troncatures au regard de la cohérence archéologique, on constate qu'il est difficile
de se satisfaire des groupes obtenus par un niveau de troncature unique sur l'ensemble de
l'échantillonnage. En effet, la scission de groupes dont l'homogénéité du point de vue
archéologique est assurée est considérée comme suspecte. C'est le cas pour la série de tessons
de l'atelier hellénistique de Pergame (à l'exception de Z19, distingué à l'oeil) et pour les tessons
de même caractéristiques techniques et stylistiques des séries F et H. En d'autres termes, on
peut se poser la question de la validité d'une telle procédure, du moins dans le cas où
l'échantillonnage est composé de groupes de céramiques qui n'ont probablement pas la même
homogénéité (pâte grossière/fine, argile calcaire/non calcaire). Ainsi, nous considérerons
surtout par la suite la structure même des dendrogrammes, sans nous référer à un niveau de
troncature donné. En anticipant sur la classification finale (tableau 21), introduisons la
définition des groupes de composition retenus en définitive afin de discuter de la répartition des
échantillons au sein de ces groupes dans les différents dendrogrammes:
- groupe byzl (pour byzantin /ocal, n°l sur la figure 50): on nomme ainsi le groupe de
composition associé à la majeure partie des échantillons de référence locale (série A,
échantillons indiqués par des points noirs sur les figures 50 et 51). Ces tessons de la série A
sont pour la plupart disséminés dans la branche de gauche des dendrogrammes. Le fait qu'ils
soient ainsi dispersés, allié au niveau particulièrement bas des rusions (à l'exception de la partie
droite) permet d'identifier les échantillons associés comme des céramiques de fabrication
locale. En ce qui concerne la partie droite de cette branche, on peut distinguer deux
discontinuités dans les niveaux de fusions sur les diagrammes a) et d), qui correspondent
respectivement au:

a)
mmumiimmntimiMKIIIIUIMuiiiiliiilimilim lllll 1IIUII1 ililmlmlmiiliiillllll iiiuuilMniimliiiiiliilimiill UlluililUUIIlimiUUIUItlUtllllllllllllltltllUllllllllllllllltlllltlllllllllllllllllCllllllllllllllllllllllllllIllllllllllllllIlllltlllllllllllllllilllllllllliltl')
mmiiiiiniummmmiuimimmtmuuliiimliK (in ilimilimiiliuliiimiilnli.mu mlilmlm; mmiuiiiiuii<m(iïlimit<mimmimiuiimumiimmiiimiiiiiimimmmmuimmmiimitmmmiiimimiimiill]:)!!:!!!":::::::!!:::!::::::!:::)!:];::!!::]!)::]:::]::!::)»]::»::::!:]:]::-::!]::)]!;::::;]].]]]!
MiiiiiiniiiiiiiituiMiiiiiiiiiiliiiiiiliiiiiliiMUiliiu lllll iiiiliiiiiiiilnulilltlllllniliilll iliiiulillllll inilliliiiilillliilillUilMIIIMIIIlllllllllllllllllllllllllllllllllllllllllllllllllllilllUltlllllillllllllllllltllllllllllllllllllilllllllltltlllllllllllllllllllilllllllllidllllliltlllfiril
!:!!:]!!!!!::!!!:!!:!!:!!]!::::]::!):!:!::!:::::::)!:!::!::)!:)!:!!!::::]:!!:!::!:]:!:!:!»::!:!::!:::!:!)
intiiiilllllillllUIIIIIUIIIIIIIlliiuiiimiiiiiiiiiiiiiiliiil lllll llillllllillmiiiiiiii milllllim IllllllUllliuiUIIIIIUtimliulilmtniiiilllllllltflllllllllllllllllllllllllllllllllllllllllllillllllllllllllllMllllllUllllllllllllilllllllllllllllllllllllllltllllllinllllllllllliltllillllllllllll
I l l l l l l l l l l l l l l l l l l
Figure 50: Dendrogrammes obtenus par CAH avec l'algorithme de Ward sur: a) données brutes, 31 éléments; b) données brutes, sélection de 22
éléments; c) données brutes, 12 éléments déterminés (ou determinates) par PIXE; d) coordonnées factorielles, 12 premières composantes principales
exprimant 90% de la variance.
Les points noirs indiquent les échantillons de référence locale (série A). Les échantillons constituant les groupes de composition finaux sont reliés par
les barres horizontales numérotées, auquelles viennent se rattacher (en pointillés) des échantillons ou groupes d'échantillons séparés par la procédure
de classification considérée. Identification des groupes de composition (voir définitions dans le texte): 1: byzl; 2: byzp; 3: sgraf; 4: lnop; 5: heil; 6: fhi;
7: g; 8: byzi.

Chapitre 2 133_
- groupe byzp (pour èyrantin "pollué", n°2 sur la figure 50) pour la branche la plus proche du
groupe byzl. Cette dénomination sera justifiée par la suite.
- groupe g (pour céramique "grossière", n°7 sur la figure 50) pour la branche la plus éloignée
de byzl. Notons que cette dernière contient exclusivement des échantillons de la série
archéologique G, et que dans le cas des dendrogramme b) et c) elle est rattachée à l'autre
moitié du dendrogramme. Cette instabilité indique que la filiation entre byzl, byzp et g ne doit
pas être interprétée comme un signe d'origine commune: leur apparente proximité sur les
dendrogrammes a) et d) est surtout due à l'opposition très nette entre les deux branches
principales du dendrogramme.
La structure de la branche principale droite est plus complexe:
- groupes f et hi (parfois cités comme groupe commun fhi, n°6 sur la figure 50) contenant
respectivement tous les échantillons des groupes H et I, à l'exception de H18, et la plupart de
ceux de la série F. Ces échantillons sont toujours associés entre eux dans les dendrogrammes,
groupés ou non avec des tessons d'autres séries. On note en particulier sur le dendrogramme d)
l'association avec trois échantillons de la série A, ce qui pose la question d'une origine locale.
Cette association n'est pas propre au dendrogramme d), elle a été également observée sur
d'autres essais de classification qui ne sont pas présentés ici. Les trois tessons de la série A sont
rattachés de manière stable à deux échantillons des séries B et C, avec lesquels ils constituent
le:
- groupe byzi (pour byzantin local /ndéterminé, n°8 sur la figure 50). Le fait que les
échantillons de byzi soient nettement séparés des autres échantillons de la série de référence A
semble indiquer que la caractérisation de la production byzantine de céramiques glaçurées à
Pergame ne peut pas se résumer à un seul groupe de composition. Reste à examiner si ces
tessons s'écartent du groupe principal byzl pour des raisons "accidentelles" (influence
dominante d'une inclusion rare, pollution,...) ou si ils peuvent correspondre à un autre type de
production pergaménienne, qui utiliserait des techniques de fabrication et/ou des sources de
matières premières différentes.
- groupe sgraf (pour sgra/fito fin, n°3 sur la figure 50): il est composé des échantillons de
sgraffito fin de la série Q, ainsi que du Cl06 qui leur est associé de manière stable.
- groupe hell (pour hellénistique, n°5 sur la figure 50): échantillons hellénistiques locaux (série
EOL, aussi notée Z).
- groupe lnop (n°4 sur la figure 50): échantillons des petites séries L, N, O, P.

Chapitre 2 134
Dans l'ensemble, les échantillons des groupes ci-dessus sont toujours associés sur les
histogrammes. Les échantillons dont la position est instable, et qui ont tendance à s'agréger à
l'un ou l'autre groupe suivant la procédure de classification utilisée, sont considérés comme non
classés. On note cependant des variantes remarquables dans la position relative des groupes
formés: outre les cas déjà évoqués de la branche de rattachement du groupe g, de l'association
occasionnelle de byzi avec fhi, remarquons la tendance à la scission du groupe lnop en deux
sous-groupes. Là encore, le fait que chaque sous-groupe contienne des échantillons de la
série N, homogène par ses caractéristiques archéologiques, incite à considérer cette scission de
façon critique. D'un manière générale, on en revient aux concentrations élémentaires pour
examiner sur quels éléments portent les différences en groupes et sous-groupes. Nous
discuterons dans le paragraphe des associations ou scissions et tenterons de déterminer si elles
peuvent être considérées comme significatives ou non, selon les propriétés des éléments
concernés.
On peut remarquer que le choix des éléments ne semble pas particulièrement critique
dans les limites que l'on s'est imposé dans les exemples présentés (conservation des éléments
majeurs, pas de choix a priori des éléments selon leur concentrations ou leurs propriétés). Du
point de vue de la classification, le dendrogramme résultant des éléments déterminés par la
seule méthode PIXE n'est pas fondamentalement différent des autres (figure 50 c)). Lors d'une
étude préliminaire, une bonne séparation des groupes principaux byzl, fhi et hell avait pu être
obtenue [147]. Par contre, la cohérence des petits groupes comme lnop et byzi est beaucoup
moins évidente. On remarquera également la position très excentrée d'un des échantillons du
groupe fhi (H49), qui se rattache finalement à ce groupe lors de l'étape de consolidation (mais
cette étape suppose dans SPAD-N une troncature préalable, ce qui induit d'autres biais).
Plusieurs autres essais de classification ont été réalisés, en combinant différemment le nombre
de variables initiales et celui des composantes principales retenues. Certains choix d'éléments
ont été faits de manière plus "radicale" en éliminant des groupes d'éléments par famille
géochimique, e.g. alcalins ou alcalinos-terreux plus sensibles aux phénomènes d'altération (e.g.
[195]). Si des distortions importantes sont observées dans certains cas, on retrouve en général
à un niveau de fusion suffisamment élevé (partition en 4 à 6 classes) les groupements stables
que nous avons présentés. Il ne parait cependant pas souhaitable d'éliminer purement et
simplement une famille d'éléments. Au contraire, on aura intérêt à voir représentées autant de
familles géochimiques différentes que possible, afin qu'un groupe d'éléments de même
comportement ne domine pas à lui seul la classification. En ce sens, la procédure qui consiste à
faire précéder la CAH d'une ACP est bénéfique, puisqu'elle réduit à une seule composante des
variables fortement corrélées. On constate néanmoins que le résultat obtenu n'est pas très
différent des autres classifications, et que le traitement préalable ne dispense pas d'une
vérification finale de la cohérence des classes au vu des compositions. De plus, l'ACP nécessite
un choix soigneux des échantillons pour éviter que les axes factoriels ne soient en grande partie

Chapitre 2 135
déterminés par quelques individus de plus forte inertie. Enfin, si la réduction du nombre d'axes
pris en compte peut éviter une trop grande sensibilité à de petits écarts dans les concentrations,
il faut vérifier que les individus soient suffisamment bien représentés dans le sous-espace
considéré (cf. annexe 5.A).
A titre d'exemple des autres types de CAH possibles, la figure 51 montre le
dendrogramme obtenu en utilisant un algorithme différent de celui de Ward. Il a été réalisé au
laboratoire de céramologie de Lyon. Le critère d'aggrégation est ici la distance (euclidienne)
minimale entre classes, l'aggrégation étant effectuée en affinités moyennes non pondérées.
Comme on pouvait le prévoir, les indices de niveau sont nettement moins contrastés qu'avec le
critère basé sur l'inertie (qui dépend du carré de la distance). On observe une grande sensibilité
à de petites variations dans les concentrations: par exemple, la séparation d'un des échantillons
de référence normalement rattaché à byzl (indiqué par le point le plus à droite dans le
dendrogramme) est essentiellement due à une teneur en Zr supérieure aux autres. En
contrepartie, cette procédure risque moins de générer des différences d'indices de niveau
artificiellement élevées, en particulier lors de scissions peu fondées entre groupes. Les résultats
obtenus sont tout à fait comparables à ceux obtenus par la méthode de Ward.
I ...Mi::'i'i !:î=i;i:i"i"i;;il::i'l iii'i1. . .nri^h' i ' l ï
i:::.
, .1.:.:,
:'i!i'il.i.'i.n::i:IT:';';i.';:i:fMlPl1ili1!i; ï lU ' î ' i ï m l
""T".'...,
I—H*MJ 7 ••
Figure 51: Dendrogramme obtenu par CAH par aggregation en affinités moyennes non
pondérées sur les concentrations normalisées de 18 éléments (Na20, MgO, AJ2O3, SiO2, K2O,
CaO, TiO2, MnO, Fe2O3, V, Cr, Ni, Rb, Sr, Zr, Ba, La, Ce).
Les points noirs indiquent les échantillons de référence locale (série A). Les échantillons des
groupes de composition finaux sont reliés par les barres horizontales numérotées, auquelles
viennent se rattacher (en pointillés) des échantillons ou groupes d'échantillons séparés par la
classification considérée. Identification des groupes de composition (voir définitions dans le
texte): 1: byzl; 2: byzp; 3: sgraf; 4: lnop; 5: hell; 6: fhi; 7: g; 8: byzi.
Une méthode de classification non hiérarchique a également été mise en oeuvre
(procédure de classification "mixte" de SPAD-N). Les partitions finales obtenues par
croisement de plusieurs partitions "de base" ne dépendent pas de manière critique des choix a
priori effectués pour ces dernières: nombre de classes (pris nettement supérieur au nombre
supposé de classes finales), individus représentant leurs centres. Les résultats sont analogues à
ceux obtenus précédemment par d'autres méthodes, aux échantillons non classés près.

Chapitre 2 L36.
En conclusion, on constatera que c'est par la confrontation d'un certain nombre d'essaisde classification différant par le choix des éléments, de l'algorithme, par l'utilisation descoordonnées directes ou des coordonnées factorielles, que les groupements stables ont pu êtreidentifiés et la classification finale établie. L'automatisation complète du processus est renduemalaisée par le fait qu'un niveau de troncature unique ne convient pas, du moins lors de lacomparaison de céramiques de finesses différentes et d'homogénéité variable. La validation desclasses obtenues s'appuie en dernier ressort sur les propriétés géochimiques des éléments quisont responsables des associations ou scissions (cf. Caractérisation des classes) et sur lacohérence archéologique. Ce dernier point ne remet pas en question la rigueur de la démarche:on évite simplement de se fier aveuglément à une procédure mathématique qui ne peut prendreen compte le matériau considéré dans toute sa complexité.
Constitution des groupes et confrontation avec la classification archéologique
Suite aux procédures de classification, nous avons finalement distingué six groupes decomposition principaux (byzl+byzp, g, hell, fhi+byzi, sgraf, Inop) constitués de neuf sous-groupes (dissociation de byzp, de byzi, scission de fhi en f et hi) (cf. tableau 21). Quatre d'entreeux: byzl, byzp, byzi, hell contiennent des échantillons de provenance locale attestée (séries Aet EOL). Le groupe le plus important numériquement est byzl. Il inclut la majorité deséchantillons de la série de référence A et des séries archéologiques B, C, D, E, K qui sont lesplus fréquents sur le site, byzl peut donc être considéré comme représentatif de la majeurepartie de la production d'époque byzantine à Pergame. Quelques tessons des séries J et R luisont également associés, mais aucun des séries F, I, L, N, O, P, Q et H à l'exception de H18.Ce dernier, qui se distingue à l'oeil par sa facture moins fine que celle des autres céramiques dela série H, se révèle donc être une imitation locale (si les céramiques de la série H sontimportées) ou une variante locale plus grossière.
byzp et byzi sont des groupes locaux très minoritaires dans l'échantillonnage (5 tessonschacun), peu homogènes, qui rassemblent des tessons issus des mêmes séries que ceux quiconstituent byzl: B, C, D, E et, bien sûr, A. L'existence de ces groupes pose le problème desproductions locales secondaires: les écarts observés sont-ils de nature "accidentelle" (e.g.présence d'une inclusion rare)? sont-ils liés à une pollution? ou correspondent-ils à desproductions qui se distingueraient de byzl par le traitement de la pâte et/ou par le choix de lamatière première? Nous essayerons de répondre à ces questions au paragraphe suivant, par lacaractérisation des groupes. Le nombre très réduit de tessons concernés limite évidemment lavalidité des réponses que l'on pourra apporter.
Les tessons de l'atelier hellénistique pergaménien (série EOL ou Z) constituent legroupe hell. La séparation des productions hellénistiques et byzantines peut être là encore

Chapitre 2 U]_
attribuée a priori à plusieurs facteurs: différence d'ordre technologique, utilisation d'un stock
argileux distinct ou d'une couche différente du même gisement à plus de mille ans d'intervalle.
Cinq tessons de céramique non glaçurée appartenant à la série G constituent le groupe
g. Peu homogène du fait de la texture plus grossière des pâtes, ce groupe se détache pourtant
nettement des autres, notamment par son caractère non calcique. Tous les tessons décorés d'un
enduit micacé ou de traits peints en font partie. Remarquons parmi les échantillons de G le
tesson G72, associé à byzl, qui semble indiquer que l'atelier correspondant ne fabriquait pas
uniquement des céramiques glaçurées. Il faudrait, pour étudier sérieusement les variétés de la
production non glaçurée de Pergame, disposer d'un échantillonnage beaucoup plus fourni et
plus représentatif.
Nous avons déjà mentionné le contenu des groupes sgraf et lnop, qui correspondent
respectivement aux séries archéologiques Q (sgraffito fin) et L, N, 0, P. Le rattachement du
tesson Cl06 à sgraf n'est pas étonnant vu son aspect peu caractéristique (monochrome, sans
décoration). Par contre, lnop est constitué de séries hétérogènes, en particulier P et L n'ont
guère de rapport apparent avec N et 0. Dans certaines classifications, lnop est scindé en deux
groupes (ou plus), les échantillons dissociés étant indiqués entre parenthèses dans le tableau
21. La répartition des échantillons dans les deux sous-groupes ne paraît pas très cohérente a
priori, dans la mesure où elle sépare les échantillons de la série N. Nous verrons qu'il existe
quelques arguments élémentaires qui s'opposent également à une séparation du groupe. La très
faible représentation de chaque série (deux tessons pour chacune des séries L, 0, P) n'autorise
cependant pas à conclure.
Enfin, les groupes f (série F) et hi (séries H et I) sont associés au sein de la plupart des
classifications en un seul groupe, fhi. La distinction de deux sous-ensembles au sein de fhi ne
ressort pas des procédures de classifications, mais est basée sur des différences élémentaires
assez ténues qui seront discutées plus loin. L'unicité du groupe hi est par contre plus assurée,
ce qui permet de conclure à une production unique pour les deux séries de céramiques fines,
pourtant très dissemblables d'aspect. L'association occasionnelle de fhi avec byzi pose le
problème de leur éventuel rattachement à la production locale, qui sera également examiné plus
loia Notons que deux échantillons des séries D (D59) et R (R140) sont associés à hi. D59
présente une glaçure brune opaque (stannifère), qui ne trouve pas d'équivalent au sein de
l'échantillonnage. RI40 est lui aussi un représentant unique, qui se rapprocherait plutôt de la
série F par sa pâte grasse de couleur rose clair mais s'en distingue par le motif et la couleur de
la glaçure. L'association de ces deux tessons atypiques au groupe hi doit être considérée avec
réserve.
Dans l'ensemble, la classification effectuée sur les données de composition élémentaire
est en bon accord avec la classification basée sur des critères archéologiques. Le caractère
local des séries les plus fréquentes sur le site, y compris les dérivés de Zeuxippus ware, est

Chapitre 2 B8_
confirmé par les analyses. Ces dérivés ne peuvent être confondus sur la base des analyses avec
des exemplaires de "véritable Zeuxippus ware", représentés par le tesson M107.
D'autres groupes locaux se distinguent clairement du groupe local principal par leur
composition, comme la série de tessons hellénistiques. On peut peut-être aussi inclure dans ce
cadre le groupe de céramiques byzantines non glaçurées, dont l'origine pergaménienne n'est pas
assurée mais seulement suggérée par son abondance sur le site et par son caractère peu
sophistiqué. Quoi qu'il en soit, il est apparent que l'on ne peut pas définir les céramiques
fabriquées à Pergame par un groupe de composition unique.
Les quelques échantillons de la série de référence A qui ne sont pas rattachés au groupe
byzantin principal permettent ainsi de poser l'hypothèse de l'existence de productions
"secondaires" de céramiques glaçurées à l'époque byzantine. Certains de ces tessons sont
associés de façon occasionnelle avec les séries F, H et I. La question de l'origine locale de ces
céramiques est posée.
Les autres groupes de composition (sgraf, lnop) ne présentent pas d'analogies avec les
tessons des séries locales. Le sgraffito fin (sgraf) constitue un groupe très homogène, alors que
le groupe lnop rassemble des tessons dissemblables par leur technique de décoration dont les
compositions élémentaires sont fort dispersées.
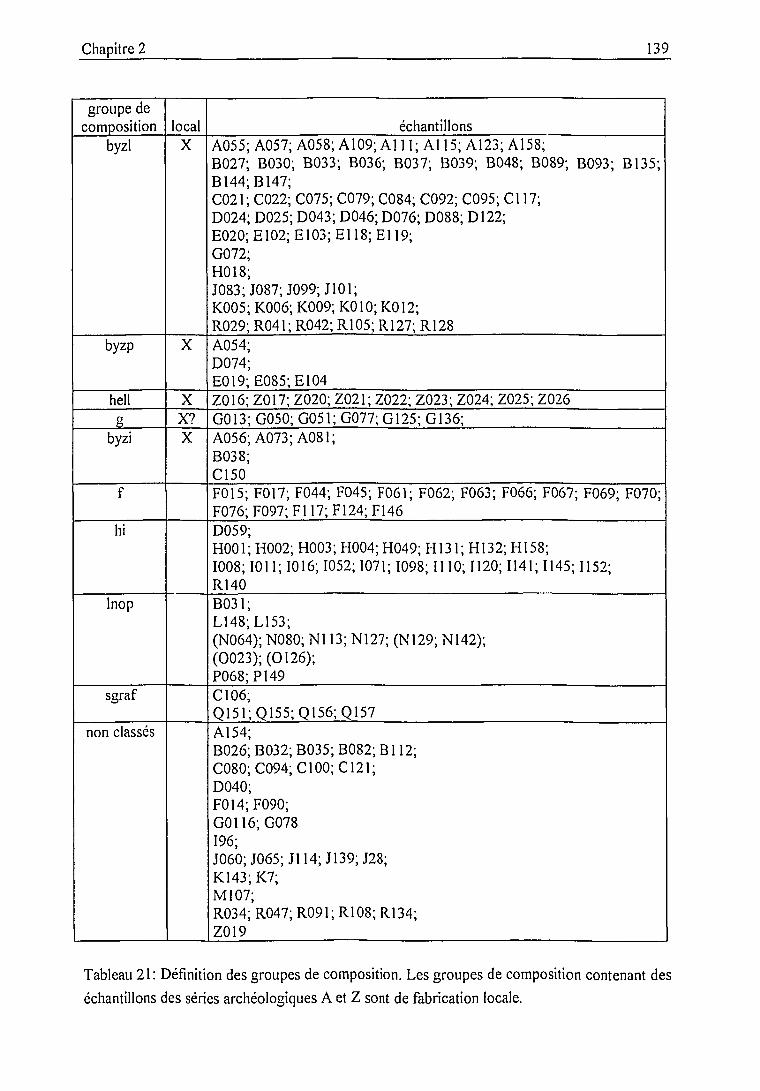
Chapitre 2 139
groupe decomposition
byzl
byzp
hell
byzi
f
hi
lnop
sgraf
non classés
localX
X
XX?X
échantillonsA055; A057; A058; A109; Al 11; Al 15; A123; A158;B027; B030; BO33; B036; B037; B039; B048; B089; B093; B135;B144;B147;C021; C022; C075; C079; C084; C092; C095; Cl 17;D024; D025; D043; D046; D076; D088; D122;E020;E102;E103;E118;E119;G072;HO 18;J083;J087;J099;J101;K005; K006; K009; KO 10; KO 12;R029; R041; R042; R105; R127; R128A054;D074;E019;E085;E104Z016; Z017; Z020; Z021; Z022; Z023; Z024; Z025; Z026G013; G050; G051; G077; G125; G136;A056;A073;A081;B038;C150FOI 5; FOI7; F044; F045; F061; F062; F063; F066; F067; F069; F070;F076;F097;F117;F124;F146D059;HOOl; H002; H003; H004;H049; H131; H132; H158;1008;1011;1016;1052;1071; 1098;II 10;1120;1141; 1145; 1152;R140B031;L148;L153;(N064); N080; NI 13; N127; (N129; N142);(O023);(O126);P068;P149Cl 06;Q151; Q155; Q156; Q157Al 54;B026; B032; B035; B082; Bl 12;C080;C094;C100;C121;D040;F014;F090;G0116;G078196;J060;J065;Jl14;J139; J28;K143; K7;M107;R034; R047; R091; RI08; RI34;Z019
Tableau 21: Définition des groupes de composition. Les groupes de composition contenant des
échantillons des séries archéologiques A et Z sont de fabrication locale.
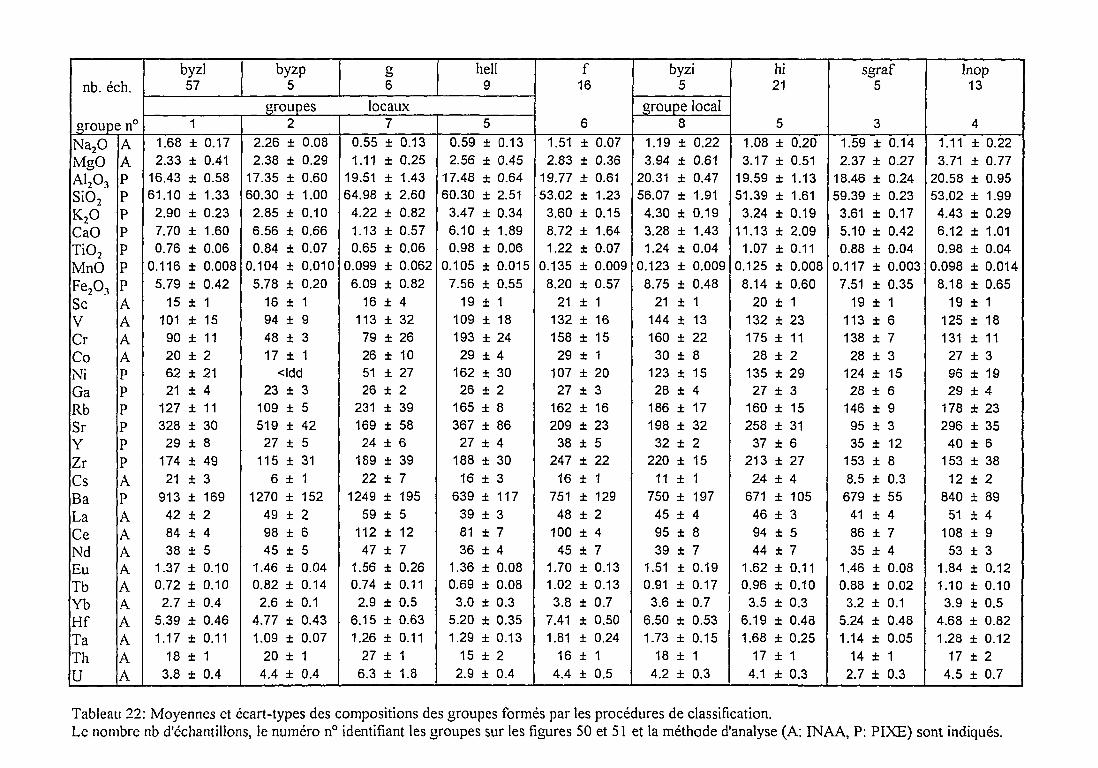
nb. éch.
groupe n°NajOMgOA12O,SiO2
K2OCaOTiO2
MnOFe2O,SeVCrCoNiGaRbSrYZrCsBaLaCeNdEuTbYbHfTaThU
AAPPPPPPPAAAAPPPPPPAPAAAAAAAAAA
byzl
1.682.33
16.4361.10
2.907.700.76
0.1165.79
15
101
90
20
6221
127
32829
17421
913
428438
1.370.72
2.75.391.17
18
3.8
57
1
±±±±±±±±±±±±±±±
±±±
±±±±±±±±±±±±±
0.170.410.581.330.231.600.060.0080.421
1511221
4
11
308
493169
2
4
50.100.100.4
0.460.111
0.4
byzp5
groupes
2.262.38
17.3560.302.856.560.84
0.1045.78
16
94
48
17
2±±±±±±±±±±±±±
0.080.290.601.000.100.660.070.0100.2019
3
1
<ldd23
109
519
27
115
6
127049
98
451.460.82
2.6
4.771.09
20
4.4
±
±±
±±±±±±±±±±±±±±
3
5
425
3111522650.040.140.1
0.430.071
0.4
g6
locaux
0.551.11
19.5164.98
4.221.130.65
0.0996.09
16
113
7926
5126
231
16924
18922
124959
112
47
1.560.74
2.9
6.151.26
27
6.3
7±±±±±±±±±
±
±±±±±±±±±±±±±±
±±
±
±
±
±
±
0.130.251.432.600.820.570.060.0620.824
32
26
10
272
39
58
6
39
7
195
5
12
7
0.260.110.5
0.630.111
1.8
0.592.56
17.4860.30
3.476.100.98
0.1057.56
19
109
193
29
16226
165
36727
188
16
639
39
81
36
1.360.69
3.0
5.201.29
15
2.9
hell9
5
±±±±±±±±±±±±±
±±±±±
±±
±±±±±±
±
±
±
±
±
0.130.450.642.510.341.890.060.0150.55118
24
4302
8
86
4
30
3
117
3
7
40.080.080.3
0.350.1320.4
1.512.83
19.7753.02
3.608.721.22
0.1358.20
21
132
158
29107
27
162
209
38
24716
751
48
100
451.701.02
3.8
7.411.81
16
4.4
f16
6
±
±±±
±±±
±
±±±±±
±±±±±±±±±±±±±±±±±±
0.070.360.611.230.151.640.070.0090.57116
151
203
16
23
522
1
129
2
4
70.130.130.70.500.241
0.5
byzi5
groupe
1.193.94
20.3156.07
4.303.281.24
0.1238.75
21
144
160
30
12328
186
198
32
220
11
750
45
95
391.510.91
3.6
6.501.73
184.2
8
±±±±
±±±±±±±±±±±
±±±±±±±±
±±±±±±±±
local
0.220.610.471.910.191.430.040.0090.48113
22
8
154
1732
215119748
70.190.170.70.530.151
0.3
1.083.17
19.5951.393.24
11.131.07
0.1258.14
20132175
28
13527
160
258
37213
24
671
46
94
441.620.96
3.5
6.191.68
174.1
hi21
5
±±±±
±±±
±±±±±±±±
±±±±±±±±
±±±
±±
±
±
±
0.200.511.131.610.192.090.110.0080.60123
11
2
293
15
31
6
27
4
105
3
5
70.110.100.3
0.480.251
0.3
sgraf
1.592.37
18.4659.39
3.615.100.88
0.1177.51
19113138
28
124
28
146
95
35153
8.5
6794186
351.460.88
3.2
5.241.14
14
2.7
5
3
±±±±
±±±
±±±±±
±
±±±±±
±±±±±
±±±±±±±±
0.140.270.240.230.170.420.040.0030.3516
73
156
9
3
12
8
0.3
55
4
7
40.080.020.1
0.480.051
0.3
lnop
1.113.71
20.5853.024.436.120.98
0.0988.18
19125131279629
178
296
40153
12840
51
108
53
1.841.10
3.9
4.681.28
17
4.5
13
4
±
±±±
±±±
±
±±±±±±±±±±±
±±±±±±
t
±±
±
±
±
0.220.770.951.990.291.010.040.0140.65118113
194
23
356
38
289
49
30.120.100.5
0.820.1220.7
Tableau 22: Moyennes et écart-types des compositions des groupes formés par les procédures de classification.Le nombre nb d'échantillons, le numéro n° identifiant les groupes sur les figures 50 et 51 et la méthode d'analyse (A: INAA, P: PIXE) sont indiqués.

Chapitre 2 141
2.4.2. Caractérisation des groupes de composition par les données élémentaires,
minéralogiqucs et pétrographiques
Une première approche de la structure des groupes et des caractères élémentaires qui
les différencient peut être réalisée au moyen d'une ACP.
La figure 52 représente l'histogramme des valeurs propres de l'ACP sur 30
concentrations élémentaires, centrées et normées, des échantillons constituant les groupes byzl,
byzp, ... à l'exclusion des tessons de céramique "grossière", très nettement différentes des
autres (groupe g, cf. annexe 5.A), des échantillons non classés, et des échantillons à valeurs
"anormales". Elle indique le pourcentage de variance exprimée par chaque composante
principale: les trois premières composantes représentent un pourcentage cumulé d'environ
70%.
! MKEPO !! !
VALECRPBCPFE
P O K E f T . !OMJIE
1234567e9
1011121314151617ia192021222324252627282930
13.96123.77642.92151.73501.08600.89760.79290.69620.53010.50070.38790.36760.31010.29720.24880.22750.20660.19520.16340.13680.11440.10090.07820.06270.05640.04490.04050.03320.02830.0020
46.5412.599.745.783.622.992.642.321.771.671.291.231.030.990.330.760.690.650.540.460.380.340.260.210.190.150.130.110.090.01
46.5459.1368.8674.6578.2781.2683.9086.2287.9989.6690.9592.1893.2194.2095.0395.7996.4897.1397.6798.1398.5198.8599.1199.3299.5099.6599.7999.9099.99
100.00
Figure 52: Histogramme des valeurs propres de l'ACP sur 30 concentrations élémentaires,
montrant le pourcentage de variance exprimé par les composantes principales successives.
Les échantillons correspondent aux groupes de composition déterminés par les CAH (groupe g
excepté).
La comparaison des projections du système sur le premier plan factoriel dans l'espace
des variables et dans celui des individus (figure 53) met en évidence les tendances générales
des différences de composition entre groupes. L'axe 1, qui porte près de 50% de la variance,
oppose nettement les groupes byzl et byzp aux groupes fhi et lnop, sgraf et hell prenant une

Chapitre 2 142_
position intermédiaire. Cette opposition porte sur une variable correlée avec Si et à un degré
moindre avec Na, Sr, Ba, et fortement anticorrélée avec Ai, Fe, Ti, la plupart des éléments de
transition de la série de Fe, et de manière moins marquée avec K, Mg et les terres rares.
Le deuxième axe (13% de la variance) est corrélé avec Th, U et les terres rares légères
La, Ce, (Eu), ainsi qu'avec Ba et Na. Il est anticorrelé avec Cr et Ni. Cet axe sépare byzl de
byzp et fhi de lnop.
Ces informations se vérifient bien dans le tableau 22 par des teneurs nettement plus
élevées en Al, Ti, Fe, Se, Cr, Co, ... et plus faibles en Si pour les groupes fhi et lnop que pour
les groupes byzl et byzp. Les échantillons de lnop se distinguent de ceux de fhi par des
concentrations en K et en terres rares plus importantes, notamment en terres rares légères,
byzp se détache de byzl par ses teneurs en Ba, Sr et Na atteignant les valeurs maxima de
l'échantillonnage. Par ailleurs, byzp présente des teneurs en Cr et Ni remarquablement faibles,
qui l'opposent au groupe hell caractérisé par des teneurs en Cr et Ni plus élevées que la
moyenne et par une faible concentration en Na.
La figure 53 b) éclaire d'un jour nouveau les histogrammes de fréquence des teneurs de
la figure 47, en particulier ceux qui correspondent à des distributions bimodales. Par exemple,
la plupart des variables qui sont très fortement anticorrélées avec l'axe 1 présentent des
distributions de ce type (Fe, Al, Se, Cr,...) qui se résument en fait à une information unique car
toutes ces variables sont corrélées entre elles.
A un deuxième niveau, il serait intéressant de mettre en relation ces corrélations entre
variables avec la nature du matériau, en particulier les corrélations positives selon le concept
des "phases géochimiques" [196, 93]. La variation corrélative d'un groupe d'éléments peut
avoir différentes causes: éléments de même propriétés chimiques, éléments associés au sein
d'une espèce minérale ou de plusieurs espèces participant à un même processus de dépôt (e.g.
phase sableuse à minéraux lourds associant Si, Zr, Hf [93], ...), éléments liés dans un
phénomène de pollution (e.g. Ba, Sr [50]), ... Cette approche n'est toutefois pas aisée à mettre
en oeuvre, car en général un élément donné est susceptible d'entrer dans la composition d'un
grand nombre d'espèces minérales. La variation de l'une d'entre elles peut néanmoins jouer un
rôle prépondérant, mais l'interprétation suppose une certaine connaissance préalable du
matériau. Par exemple, sur la figure 53 b), il est probable que la premier axe représente une
opposition entre une phase argileuse représentée par Al, K et une phase de dégraissant (au sens
large du terme) à base de quartz (Si) et de feldspaths (Na, Sr, Ba). La phase argileuse est
associée à une série d'éléments que l'on peut rapprocher des ferromagnésiens (Fe, Mg, K), des
oxydes de Fe et Ti accompagnés du cortège des éléments de transition de la première série, des
minéraux lourds (Ta, Yb, Tb). Sur le deuxième axe, le groupe formé par les terres rares
légères, U et Th peut être mis en relation avec un minéral accessoire comme l'apatite, qui
concentre ces éléments. Quant à Cr et Ni, ils sont tous deux incorporés préférentiellement à
l'état de traces dans les silicates ferromagnésiens comme l'olivine et le pyroxene.
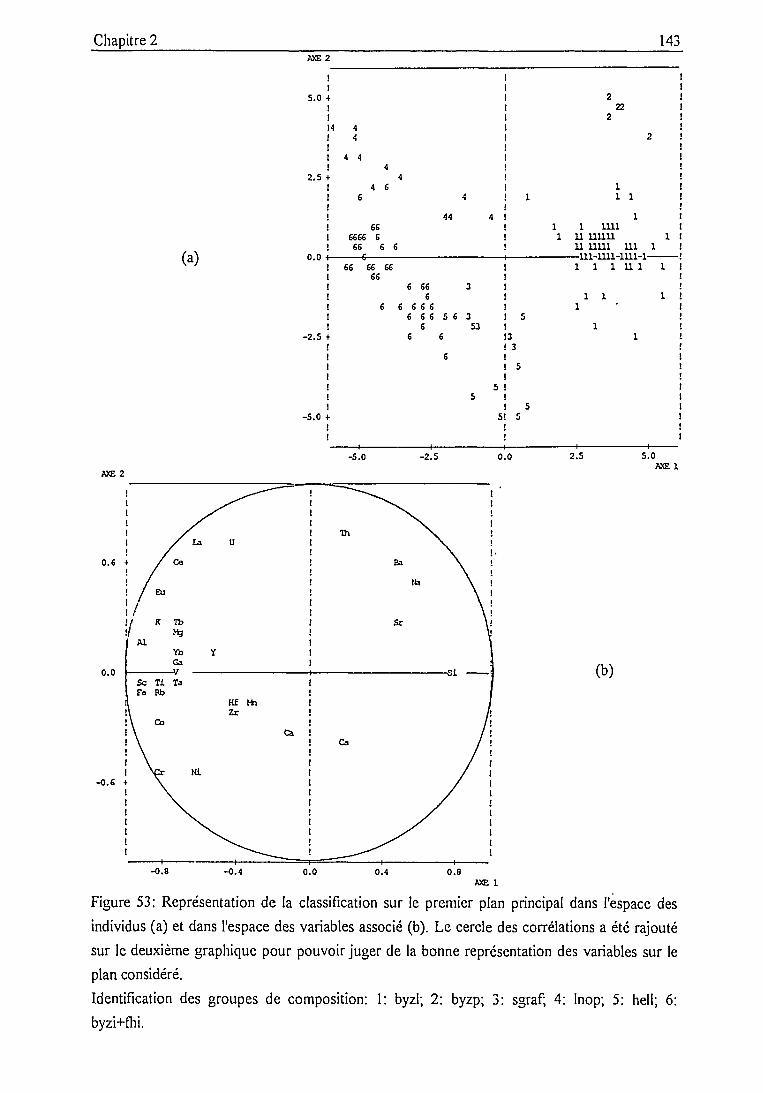
Chapitre 2 143AXE 2
5.0 +
2.5
(a) o.o
î unil îiuii î11 11111 Ul 1îii-iiii-ini-i
1 1 1 U 1 1
-5.0 -2 .5 0.0 5.0
WE 2AXE 1
(b)
-0.8 -0.4 0.0 0.4 o.eAXE 1
Figure 53: Représentation de la classification sur le premier plan principal dans l'espace des
individus (a) et dans l'espace des variables associé (b). Le cercle des corrélations a été rajouté
sur le deuxième graphique pour pouvoir juger de la bonne représentation des variables sur le
plan considéré.
Identification des groupes de composition: 1: byzl; 2: byzp; 3: sgraf; 4: Inop; 5: hell; 6:
byzi+fhi.

Chapitre 2 144_
Une telle interprétation peut paraître très spéculative. Certains points peuvent être
confirmés par les examens minéralogiqucs et pétrographiques. Ainsi, les tessons des groupes hi
et lnop présentent des pâtes fines (phase argileuse dominante) pauvres en dégraissant
quartzeux (SiC^), le groupe lnop étant de plus caractérisé par une abondance de micas blancs
(muscovite riche en K et Al). Les échantillons du groupe byzl sont par contre riches en
minéraux quartzeux, en plagioclases (albitcNaAISi^O^ à anorthite CaAl2Si2Oj$) et contiennent
également des feldspaths alcalins (albite à othoclase KAISi^O^). Lors de leur formation, les
feldspaths sont susceptibles d'incorporer Ba et Sr, ce qui expliquerait la corrélation de ces
éléments aux éléments constitutifs des feldspaths. Le fait que ni K ni Ca ne soient correlés avec
le groupe Na-Sr-Ba est dû à la contribution prépondérante d'une autre espèce minérale
comportant ces éléments (illites, micas, ... pour K, carbonates pour Ca). Cependant, nous
verrons que l'association Ba-Sr peut également être attribuée à une pollution des échantillons
du groupe byzp. L'utilisation de cette méthode pour expliquer la differentiation inter-groupes
est donc d'un usage délicat dès lors que le nombre de groupes est relativement élevé car les
espèces dominantes en concurrence risquent d'être multipliées. Le cadre plus restreint des
causes de variabilité intra-groupes permet de l'utiliser avec moins de réserves, comme nous le
verrons au paragraphe suivant. En général, les oppositions "classiques" entre phases principales
argileuse, siliceuse et carbonatée sont toujours identifiables. Dans le cas de cette ACP, on
remarquera la mauvaise représentation de la phase carbonatée, qui n'est bien corrélée à aucun
des axes principaux.
L'examen visuel des plans factoriels permet ainsi de dégager rapidement un grand
nombre d'informations. La lecture directe des différences entre groupes suppose néanmoins la
bonne représentation sur le plan considéré des variables (points proches de la circonférence du
cercle des corrélations) et des individus (cos2 de l'angle entre vecteur représentatif et
projection proche de 1). Ainsi, le groupe sgraf est mal représenté sur les premiers plans
factoriels, et ne peut être aisément distingué du groupe hell. Cet exemple met en évidence une
des limites de la méthode: les axes déterminés par I'ACP illustrent les tendances principales de
l'ensemble de l'échantillonnage, mais ne prennent guère en compte les petits groupes de
caractéristiques peu saillantes. Une autre limitation réside dans l'interprétation des axes
factoriels. Dans notre cas, les axes 3, 4, ... sont peu correlés avec les variables initiales ce qui
les rend pratiquement inexploitables. Les corrélations entre variables sont parfois explicables en
termes de phases géochimiques. Dans l'absolu, cette méthode devrait permettre de reconnaître
à partir des corrélations entre leurs constituants élémentaires des phases qui ne seraient plus
identifiables par leurs propriétés cristallines et optiques suite à la cuisson de la céramique.
Si l'ACP, en mettant en évidence les contrastes élémentaires les plus marqués, est un
guide très utile dès lors que le volume des données est important, une caractérisation plus
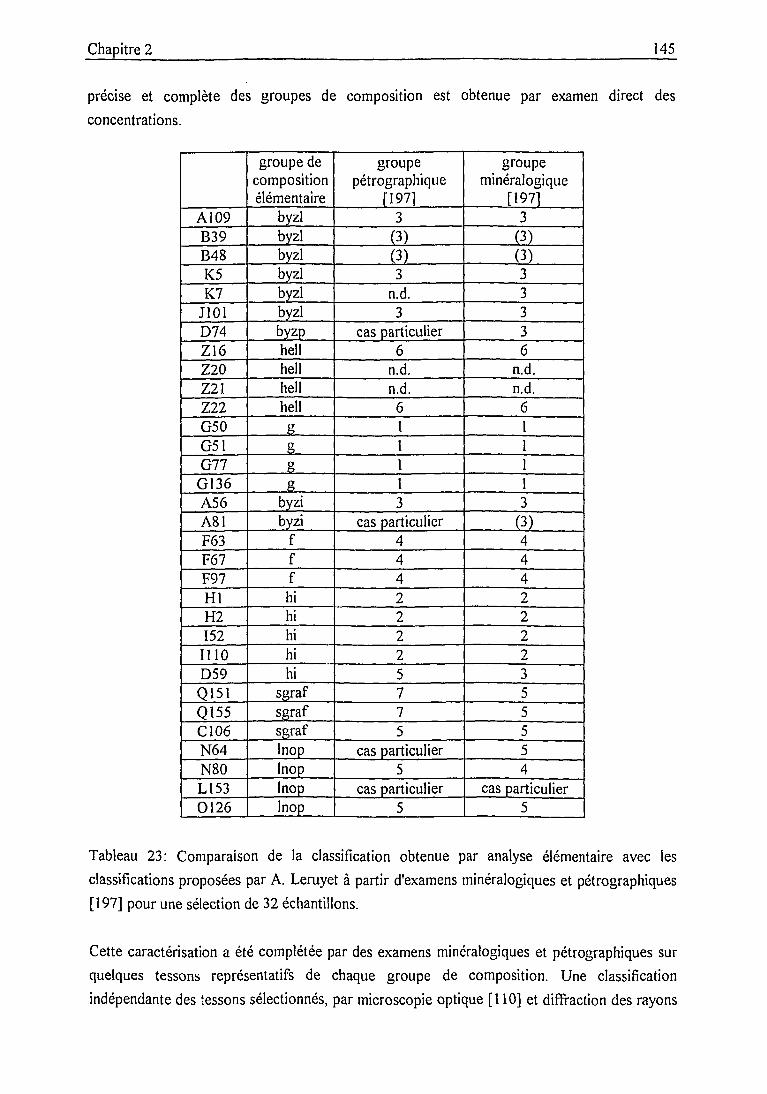
Chapitre 2 145
précise et complète des groupes de composition est obtenue par examen direct des
concentrations.
A109B39B48K5K7
J101D74Z16Z20Z21Z22G50G51G77G136A56A81F63F67F97HlH2152
1110D59Q151Q155C106N64N80L1530126
groupe decompositionélémentaire
byzlbyzlbyzlbyzlbyzlbyzlbyzphellhellhellhellgggg
byzibyzi
fffhihihihihi
sgrafsgrafsgraflnoplnoplnoplnop
groupepétrographique
[197]3
(3)(3)3
n.d.3
cas particulier6
n.d.n.d.
611113
cas particulier44422225775
cas particulier5
cas particulier5
groupeminéralogique
[19713
(3)(3)33336
n.d.n.d.
611113
(3)4442222355554
cas particulier5
Tableau 23: Comparaison de la classification obtenue par analyse élémentaire avec les
classifications proposées par A. Leruyet à partir d'examens minéralogiques et pétrographiques
[197] pour une sélection de 32 échantillons.
Cette caractérisation a été complétée par des examens minéralogiques et pétrographiques sur
quelques tessons représentatifs de chaque groupe de composition. Une classification
indépendante des tessons sélectionnés, par microscopie optique [110] et diffraction des rayons

Chapitre 2 146
X [109] (cf. annexe 6.A) a été l'objet du stage de maîtrise de géologie de A. Leruyet sous la
direction du Professeur J. Honnorez à l'Institut de Géologie de Strasbourg [197], Les résultats
obtenus sont donnés en annexe 6.B et montrent un bon accord entre les différentes méthodes
(tableau 23). Nous utiliserons ici les résultats des analyses par diffraction des rayons X
réalisées dans ce cadre. Les données pétrographiques reprennent pour l'essentiel nos propres
observations au microscope polarisant, complétées par quelques analyses au microscope
électronique à balayage (MEB, cf. annexe 6. A). Remarquons que ces examens caractérisent le
dégraissant minéral des pâtes, les argiles n'étant en général plus identifiables par leurs
propriétés cristallographiques et optiques après la cuisson de la céramique.
Nous avons distingué parmi les groupes de composition deux catégories: ceux qui se
rattachent à la production locale (au sens large du terme, incluant g) et ceux qui ne se
rattachent apparemment pas à la production locale ou pour lesquels le caractère local n'est pas
affirmé (f, hi). On cherchera à déterminer si les céramiques de la première catégorie ont pu être
fabriquées avec la même argile traitée différement, ou si une matière première distincte a été
utilisée. Dans le deuxième cas, on comparera les traits caractéristiques avec ceux de groupes
de références connus par d'autres études qui peuvent être mis en relation du point de vue
archéologique.
Groupes de composition rattachés à la production locak
Groupe byzl
Dans cette étude des céramiques issues des fouilles de Pergame, nous utiliserons le
groupe byzl, représentatif de la majeure partie de la production à l'époque byzantine, comme
référence pour les autres groupes. Le tableau 24 indique les principales différences de
composition.
La variabilité interne du groupe peut être étudiée au moyen d'une ACP. Les deux
premiers axes principaux (exprimant respectivement 20 et 12% de la variance) sont représentés
sur la figure 54. Sur le premier axe, la contribution prédominante est celle des éléments
associés dans des minéraux comme l'apatite (terres rares légères, Th, U), des éléments de
transition de la première série, de Cs, Mg, ..., qui sont opposés à un ensemble d'éléments
caractéristique des feldspaths (Na, Ba, Sr). L'interprétation qui peut en être donnée est que des
proportions variables d'apatite (dont la présence est confirmée par des analyses au MEB) et de
feldspaths sont une cause majeure de dispersion des concentrations élémentaires au sein du
groupe.
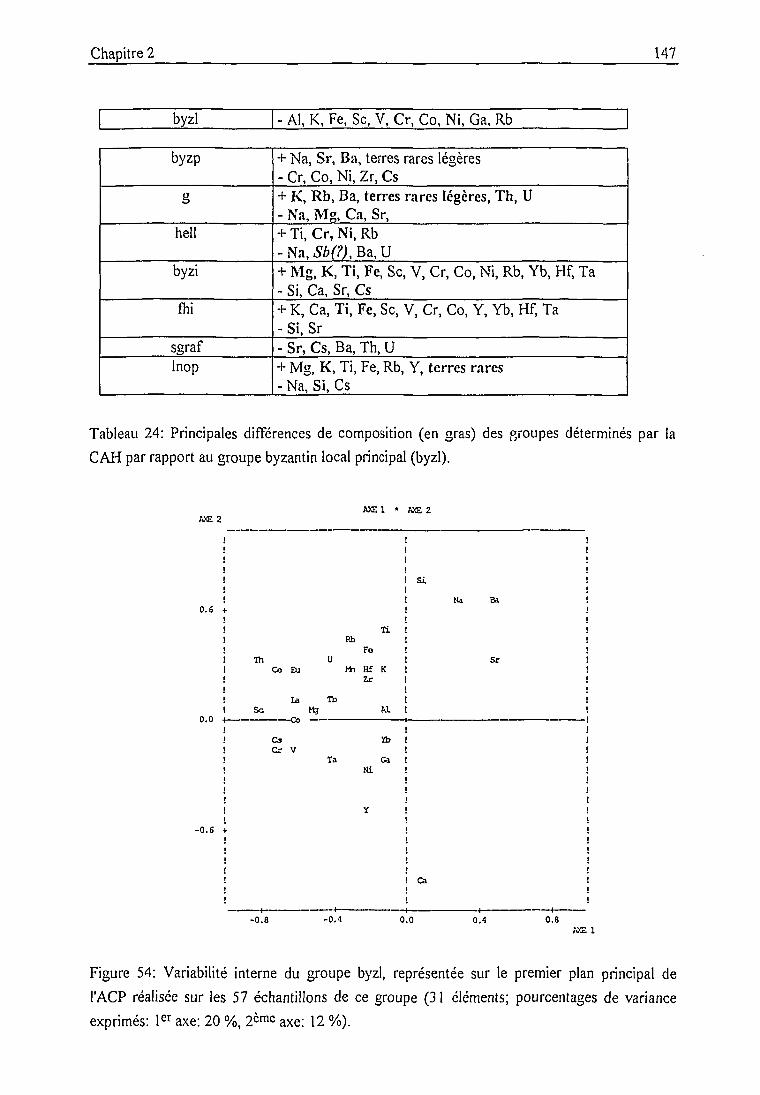
Chapitre 2 147
byzl - Al, K, Fe, Se, V, Cr, Co, Ni, Ga, Rb
byzp
8
hell
byzi
fhi
sgraflnop
+ Na, Sr, Ba, terres rares légères- Cr, Co, Ni, Zr, Cs+ K, Rb, Ba, terres rares légères, Th, U- Na, Mg, Ca, Sr,+ Ti, Cr, Ni, Rb- Na, Sb(?), Ba, U+ Mg, K, Ti, Fe, Se, V, Cr, Co, Ni, Rb, Yb, Hf, Ta- Si, Ca, Sr, Cs+ K, Ca, Ti, Fe, Se, V, Cr, Co, Y, Yb, Hf, Ta- Si, Sr- Sr, Cs, Ba, Th, U+ Mg, K, Ti, Fe, Rb, Y, terres rares- Na, Si, Cs
Tableau 24: Principales différences de composition (en gras) des groupes déterminés par la
CAH par rapport au groupe byzantin local principal (byzl).
AXE 2
0.6
0.0
-0.G
AXE 1 • AXE 2
! Si;!!
t!a
Ti !Rb
FoTh
Ce Eu Mi Hf KZr
TbSe
!!;
!!!
Ai !
CsCr V
Ta GaNi
Ca
-0.8 -0.4 0.0
Ba
Sr
0.4 0.8AXE 1
Figure 54: Variabilité interne du groupe byzl, représentée sur le premier plan principal de
l'ACP réalisée sur les 57 échantillons de ce groupe (31 éléments; pourcentages de variance
exprimés: 1er axe: 20 %, 2 è m c axe: 12 %).

Chapitre 2 148_
Le deuxième axe oppose les deux éléments majeurs Ca et Si qui agissent concuremment
comme "diluants" des autres éléments. Remarquons que les éléments de la phase argileuse Al,
K et le fer lié à l'argile contribuent relativement peu à la variance de byzl. Ils sont représentés
sur le 3 è m e axe principal (9%).
L'étude pétrographique montre une pâte relativement grossière, très hétérométrique
avec quelques inclusions lithiques de plus grande dimension (plus de 500 um). Ces dernières
sont surtout des fragments de roches volcaniques, qui se présentent soit sous une forme
vitreuse complètement amorphe, d'aspect altéré, soit comme une matrice microlithique
contenant des phénocristaux. Les phénocristaux sont principalement des plagioclases, des
biotites et des amphiboles, avec quelques minéraux accessoires comme l'apatite et des oxydes
de Fe-Ti. Le fond microlithique est formé de feldspaths alcalins, identifiés au MEB. Quelques
fragments de roches de nature non volcanique sont également présents, parmi lesquels on
pourrait reconnaître des micaschistes à biotite. Les cristaux isolés dans la pâte sont
principalement des plagioclases (parfois zones), des quartz, des quartz polycristallins. Des
biotites plus ou moins hématisées et des minéraux opaques ont également été observés.
Quelques rares cristaux de feldspaths alcalins sont identifiables par leurs propriétés optiques
(macle double). La diffraction des rayons X révèle une contribution importante de ces
minéraux, qui, d'après A. Leruyet, pourraient être présents sous forme de sanidine [197]. Il est
probable que de nombreux fragments de feldspaths potassiques n'ont pu être identifiés au
microscope en raison de leurs dimensions réduites rendant leurs macles inobservables. Les
rares fragments de hornblende visibles en microscopie sont également mieux mis en évidence
par leurs pics de diffraction. Bien que ces fragments aient une coloration brun-rouge
(pléochroïque) en lumière naturelle, des observations sur des roches prélevées dans la région
de Pergame indiquent qu'il s'agit probablement de hornblende verte qui aurait été rubéfiée à la
cuisson [112, 198]. Remarquons que les échantillons D59, D74, A56 et A81 présentent aussi
en diffraction les pics de la hornblende. Nous reviendrons en détail sur le cas des trois derniers
tessons à propos des groupes byzp et byzi.
Les minéraux et fragments lithiques présentent des formes anguleuses (cf. figure 64 (b))
qui ne sont probablement pas dues à un concassage du dégraissant par le potier étant donné
leur granulométrie très irrégulière. Elles indiquent plutôt des processus de transport limités.
Tous ces matériaux peuvent être rattachés au contexte géologique de la proche région de
Pergame. La carte géologique indique un contexte global volcanique (andésitique), et la
présence locale d'une intrusion de granodiorite, entourée d'une auréole de métamorphisme, qui
domine la vallée du Sélinous (figure 55). Les fragments de roches volcaniques, plagioclases,
biotites et amphiboles observés dans les pâtes céramiques sont probablement issus des roches
andésitiques. Les composantes plus acides: quartz, quartz polycristallin, les feldspaths alcalins
et une contribution de plagioclases et de biotite peuvent provenir des massifs de granodiorite.
Enfin, les roches métamorphiques sont représentées par les rares fragments schisteux.
Granite Y
•*•*•*• Volcanique X • f»oin«. a,c
a . indent»P : timlli
Gneiss, micaschiste
Crétacé
iSijSil Népgàne. continental,' indifférencié
0- Cône volcanique
O Cratère volcanique
Figure 55: Carte géologique schématisée d'une région de la Turquie incluant Pergame, Ephèse et Sardes (d'après [171, 199]).
Les ressources minières à proximité de Pergame sont précisées.

Chapitre 2 150
Nous avons pu constater la grande disparité de compositions élémentaires au sein des
groupes locaux (tableaux 22 et 24, figure 53). Du point de vue pétrographique, des faciès
similaires (groupes byzp et byzi) aussi bien que des faciès tout à fait distincts (groupes hell et
g) sont observés.
Groupe hell
Les tessons de hell montrent une pâte relativement fine, avec de nombreuses inclusions
de très petite dimension (moins de 10 um). Des inclusions de dimension moyenne (200-400
um), en général peu abondantes, sont identifiées comme du quartz mono- ou polycristallin. La
pâte est parfois imprégnée de calcite micritique. On observe de rares fragments de roches
métamorphiques (micaschiste à muscovite). Le trait caractéristique de ce groupe est
l'abondance de minéraux opaques, qui n'est probablement pas sans rapport avec les teneurs
relativement élevées de Cr et Ni (trois fois plus élevées que pour les tessons de byzl). Des
analyses de minéraux lourds ont été effectuées par R. Sauer sur des sédiments argileux prélevés
autour du barrage dont les eaux recouvrent actuellement les ateliers de potiers hellénistiques de
la vallée du Kestel. Elles montrent un assemblage de chromites, de hornblendes et de zircons
[200]. La présence de chromites est en bon accord avec les teneurs en Cr élevées.
Bien qu'il soit fort possible que la pâte de ces céramiques ait été épurée, elle n'a pu
résulter de la même argile que celle utilisée pour les céramiques de byzl. En effet, une
épuration poussée est susceptible de produire des modifications importantes des
concentrations, notamment celle des éléments traces [201], mais il paraît peu vraisemblable
qu'elle ait pu produire un enrichissement sélectif en Cr et Ni. Parmi les éléments majeurs, seules
les teneurs en Na montrent des variations notables par rapport à celles de byzl. Ceci tend à
prouver que les matériaux utilisés pour les deux productions sont différents. Cependant, dans
la mesure où ces dernières sont séparées par un intervalle chronologique de l'ordre de 1500
ans, une hypothèse alternative est qu'elles correspondent à l'exploitation de couches
successives du même gisement de contenus minéralogiques distincts. Le peu de données
archéologiques disponibles sur les ateliers de la vallée du Kestel ne permettent pas de pousser
l'interprétation plus avant
Groupe g
II se distingue nettement de byzl tant par les compositions élémentaires que par les
données pétrographiques. Les échantillons de g ont une pâte grossière, sommairement
travaillée, qui contient de nombreuses inclusions siliceuses caractéristiques dont les dimensions
atteignent plusieurs dizièmes de mm (figures 64 (c) et (d)). D'après les observations au
microscope, elles sont essentiellement constituées de petites "spherules" de quartz (d'environ
5 um). De nombreux opaques de grande dimension sont également présents, ainsi que

Chapitre 2 1_51_
quelques plagioclases et des minéraux accessoires (apatite). La diffraction des rayons X révèle
une contribution importante de feldspaths alcalins. Par rapport à byzl, ceci se traduit sur les
compositions élémentaires par une augmentation des teneurs en K, Rb, Ba (feldspaths
potassiques) et une diminution des teneurs en Na, Ca, Sr (plagioclases).
Les caractéristiques élémentaires les plus saillantes du groupe g sont des teneurs en
CaO et MgO faibles (environ 1% en moyenne) et des teneurs en terres rares légères (La, Ce),
U et Th élevées.
Une faible teneur en CaO n'est pas surprenante dans le cas de céramiques grossières. En
effet, le calcium est présent le plus souvent sous forme de carbonates qui se décomposent au
cours de la cuisson en chaux et gaz carbonique vers 900°C [97] (la température de dissociation
de la calcite pouvant être inférieure ou légèrement supérieure selon la granulométrie et
l'atmosphère de cuisson ([93], [198])). Si la température de cuisson n'a pas été suffisante ou si
elle n'a pas été maintenue élevée suffisamment longtemps pour que le calcium se recombine
avec les autres constituants de la pâte, une réhydratatation de la chaux vive, puis une
recarbonatation ont lieu après la cuisson. Ces transformations s'accompagnent d'un gonflement
qui, lorsque les grains de carbonates sont de grande dimension (cas des cA amiques grossières),
peut menacer la cohésion de la pièce. Pour éviter de tels phénomènes, i! est possible que le
potier ait fait porter son choix sur un matériau spécifiquement non calcaire pour la fabrication
de ces céramiques.
On peut remarquer par ailleurs le comportement distinctif des terres rares, dont le
rapport de concentrations terres rares légères / terres rares lourdes est plus élevé que pour les
autres tessons de l'échantillonnage (figure 56). Cette caractéristique, alliée à de fortes teneurs
en Si, U et Th est typique de la géochimie des roches acides comme les granites et les
rhyolites. Ces roches concentrent la silice et les éléments les plus incompatibles, i.e. ceux qui
ont plus d'affinité pour la phase liquide (magma) que pour la phase solide. Sur la figure 57, le
groupe g a un comportement analogue à celui des granites, alors que byzl est proche de la
moyenne des roches sédimentaires. Bien que nous ne puissions pas affirmer que l'argile
constitutive des céramiques de g dérive de roches magmatiques, nous ferons l'hypothèse que,
dans l'affirmative, ces roches sont plus différenciées que les roches mères qui correspondent
aux tessons de byzl.
Quoi qu'il en soit, il est clair d'après les données élémentaires et pétrographiques que g
et byzl ne peuvent résulter du même matériau traité différemment. Dans l'état actuel de nos
connaissances, il existe plusieurs arguments en faveur d'une production pergaménienne
distincte, dont aucun n'est déterminant. En premier lieu, le "préjugé" selon lequel des
céramiques communes non glaçurées ne circulent guère aux époques historiques - à l'exception
des amphores. De façon plus objective, les formations géologiques variées présentes autour de
Pergame sont susceptibles de produire des matériaux qui se rapprochent du point de vue
pétrographique des fragments lithiques siliceux observés, qu'ils correspondent à des roches
volcaniques (tuffs silicifiés, lahaars [202, 203]) ou à des roches finement grenues.
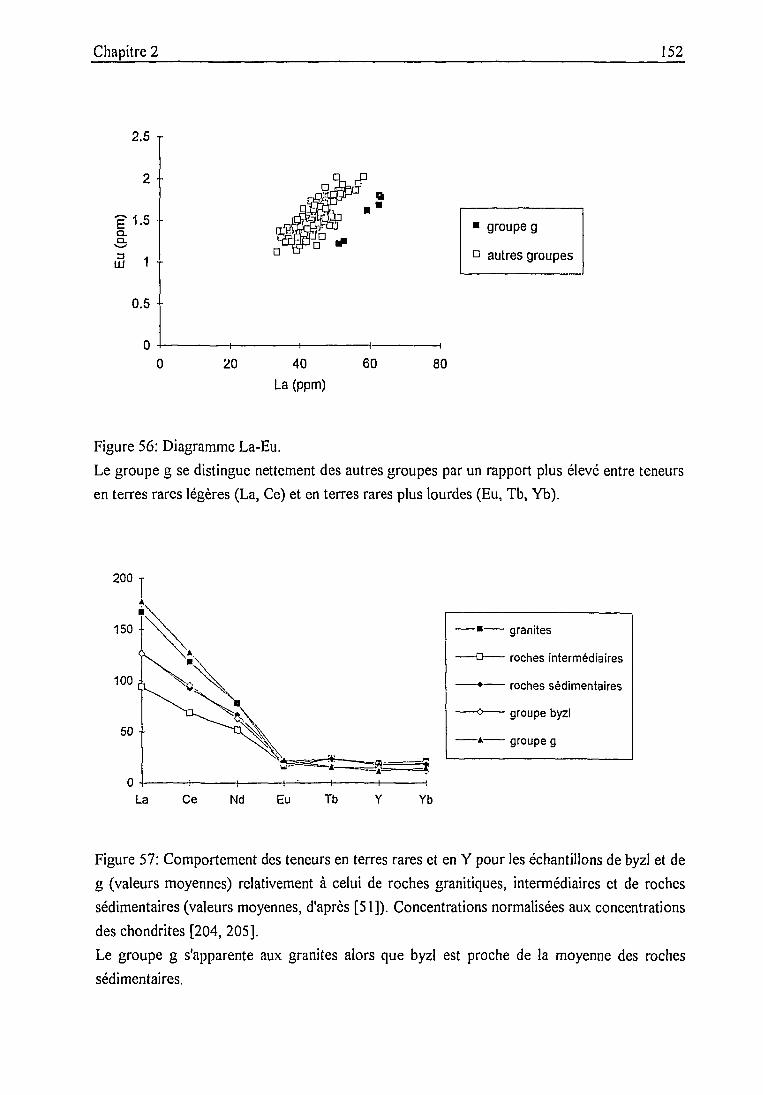
Chapitre 2 152
2.5 y
2
a.a.
ë 1
0.5 -•
20 40
La (ppm)
• groupe g
° autres groupes
60
—i
80
Figure 56: Diagramme La-Eu.
Le groupe g se distingue nettement des autres groupes par un rapport plus élevé entre teneurs
en terres rares légères (La, Ce) et en terres rares plus lourdes (Eu, Tb, Yb).
200
• granites
• roches
• roches
o groupe
* groupe
intermédiaires
sédimentaires
byzl
g
Figure 57: Comportement des teneurs en terres rares et en Y pour les échantillons de byzl et de
g (valeurs moyennes) relativement à celui de roches granitiques, intermédiaires et de roches
sédimentaires (valeurs moyennes, d'après [51]). Concentrations normalisées aux concentrations
des chondrites [204, 205].
Le groupe g s'apparente aux granites alors que byzl est proche de la moyenne des roches
sédimentaires.

Chapitre 2 1_53_
On peut se demander si les caractéristiques des teneurs en terres rares, U, Th et Si et la
présence d'opaques de grande dimension et de roches felsiques seraient compatibles avec un
gisement dérivé des formations siliceuses associées aux zones de minéralisation métallifères
localisées en bordure de l'intrusion de Kozak qui domine la vallée du Selinous [206]. Par
ailleurs, Picon mentionne, suite à ses prospections dans la région, l'utilisation d'une argile non
calcaire à Pergame [150] qui pourrait peut-être correspondre à celle des tessons du groupe g.
Groupe byzp
Un seul échantillon de byzp (D74) a été étudié en lame mince. Il présente les mêmes
types d'inclusions que les tessons de byzl: quartz, plagioclases, fragments de verres
volcaniques, opaques, avec une quantité relativement importante de biotites. Bien qu'il ne soit
pas particulièrement calcique (environ 7% de CaO), ce tesson se distingue par la présence de
carbonates identifiés comme de la calcite en microscopie et par diffraction des rayons X. La
calcite micritique est assez fréquente dans la lame, et on note la présence d'un unique fragment
de calcite sparitique (primaire), de dimensions maximales 600 um environ, entouré d'une sorte
d'auréole foncée qui est probablement liée à la transformation du cristal à la cuisson [198]
(figure 64, (e)).
L'examen du premier plan factoriel de l'ACP sur l'ensemble de l'échantillonnage a révélé
les teneurs particulièrement élevées en Ba et en Sr des tessons de byzp. Ces éléments sont
susceptibles d'être fixés dans des céramiques enfouies en terrain filonien à barytines (BaSO.;) et
célestines (SrSO^ [50, 195, 207]. Or Pergame est située en milieu filonien, et un gisement de
barytine pure est signalé sur la carte géologique à une vingtaine de km à l'Est de Pergame
[199] (figure 55). Des sédiments collectés à proximité du site de Pergame, dans la vallée du
Selinous, contiennent également du baryum sous forme de barytocalcite comme le révèle la
diffraction des rayons X. Il est donc probable que les échantillons de byzp ont subi une
altération consistant en une fixation de Ba et Sr. Elle s'accompagne d'une augmentation de la
concentration en Na2Û (figure 58), les teneurs des autres éléments majeurs de la famille des
alcalins et alcalinos-terreux n'étant pas modifiée de façon notable. Le cas de byzp ne peut être
identifié à celui des céramiques calcaires surcuites étudiées par Picon [50]: les tessons ne sont
pas particulièrement calcaires (teneur moyenne en CaO: 6.6%) et leur température de cuisson
n'excède probablement pas 900°C (cf. la présence de calcite primaire dans D74). Mais un autre
mécanisme de déstabilisation de la phase silicatée, qui reste à identifier, a pu intervenir et
permettre de la même façon la formation de zeolites barytiques secondaires. Remarquons que
les céramiques concernées révèlent à l'examen pétrographique de nombreux fragments de
verres volcaniques, particulièrement instables, et donc susceptibles de fournir le matériel
silicate, qui rentre, avec Al, dans la constitution des zeolites. Nous conclurons à ce stade que
les différences de composition entre les échantillons de byzp et ceux de byzl peuvent être
attribuées à des phénomènes d'altération, et que byzp ne représente donc pas une production
distincte.
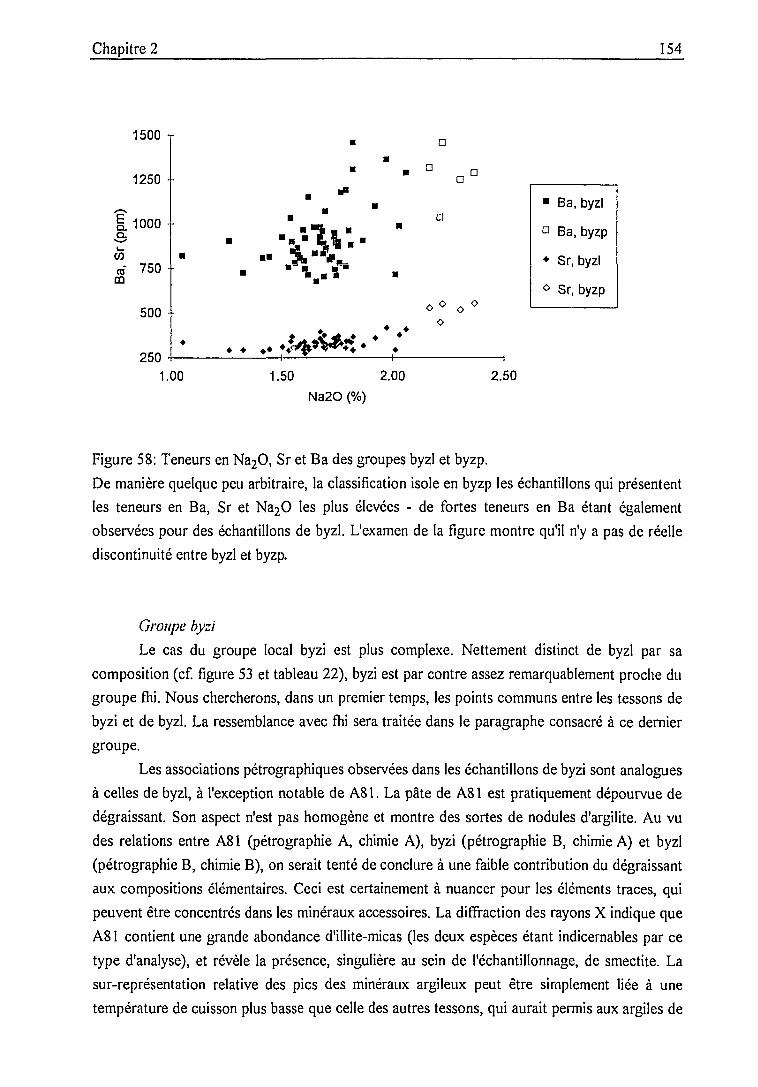
Chapitre 2 154
1500 T
1250 •
1 1000 -S
en 750 |m
500
250• •
• •
1.00 1.50 2.00
Na2O
a
2.50
• Ba, byzl
a Ba, byzp
• Sr, byzl
o Sr, byzp
Figure 58: Teneurs en Na2O, Sr et Ba des groupes byzl et byzp.
De manière quelque peu arbitraire, la classification isole en byzp les échantillons qui présentent
les teneurs en Ba, Sr et Na2O les plus élevées - de fortes teneurs en Ba étant également
observées pour des échantillons de byzl. L'examen de la figure montre qu'il n'y a pas de réelle
discontinuité entre byzl et byzp.
Groupe byzi
Le cas du groupe local byzi est plus complexe. Nettement distinct de byzl par sa
composition (cf. figure 53 et tableau 22), byzi est par contre assez remarquablement proche du
groupe fhi. Nous chercherons, dans un premier temps, les points communs entre les tessons de
byzi et de byzl. La ressemblance avec fhi sera traitée dans le paragraphe consacré à ce dernier
groupe.
Les associations pétrographiques observées dans les échantillons de byzi sont analogues
à celles de byzl, à l'exception notable de A81. La pâte de A81 est pratiquement dépourvue de
dégraissant. Son aspect n'est pas homogène et montre des sortes de nodules d'argilite. Au vu
des relations entre A81 (pétrographie A, chimie A), byzi (pétrographie B, chimie A) et byzl
(pétrographie B, chimie B), on serait tenté de conclure à une faible contribution du dégraissant
aux compositions élémentaires. Ceci est certainement à nuancer pour les éléments traces, qui
peuvent être concentrés dans les minéraux accessoires. La diffraction des rayons X indique que
A81 contient une grande abondance d'illite-micas (les deux espèces étant indicernables par ce
type d'analyse), et révèle la présence, singulière au sein de l'échantillonnage, de smectite. La
sur-représentation relative des pics des minéraux argileux peut être simplement liée à une
température de cuisson plus basse que celle des autres tessons, qui aurait permis aux argiles de

Chapitre 2 155
conserver leur structure cristalline et de donner ainsi lieu à la diffraction. Une cuisson peu
poussée expliquerait également le caractère friable du tesson. Une autre possibilité, qui intègre
les données pétrographiques, serait l'hypothèse d'un mélange d'argiles [197], traduisant peut-
être une tentative d"innovation" technologique. Le caractère délibéré de ce mélange est en fait
discutable: des analyses en diffractions des rayons X de sédiments collectés à proximité de
Pergame révèlent que ces derniers contiennent "naturellement" des illites et des smectites. En
tout cas, les données pétrographiques et minéralogiques du tesson A81 ne trouvent pas
d'équivalent au sein des céramiques de la série F, qui lui sont associées par les caractéristiques
typologiques et par l'aspect macroscopique gras de la pâte.
Par ailleurs, nous avons remarqué parmi les rares inclusions de l'échantillon A81 des
fragments de roches volcaniques. Ces inclusions sont les seuls éléments communs entre A81 et
les autres échantillons locaux de byzi et de byzl. On peut se demander si les inclusions lithiques
des céramiques ne sont pas plus caractéristiques pour les recherches de provenance que la pâte
dans son ensemble. En effet, la composition de la pâte intègre un ensemble de processus,
depuis la formation du gisement argileux jusqu'aux altérations en milieu d'enfouissement, en
passant par les transformations pratiquées par le potier. Les fragments lithiques représentent
plus directement la roche mère (ou du moins l'une des roches mères), bien qu'ils soient
également soumis aux altérations et à des transformations éventuelles lors de la cuisson de la
céramique. La spécificité géologique et géochimique de cette roche mère peut être un
indicateur de provenance, en l'absence de groupes de référence ad hoc [208]. Elle pourrait
également, comme dans le cas de nos groupes locaux de Pergame, constituer un paramètre
d'unité régionale au-delà de la variabilité locale des compositions élémentaires et
minéralogiques de gisements argileux résultant de la dégradation de plusieurs roches mères. En
ce qui concerne les roches volcaniques, on peut mettre en relation, grâce aux propriétés
géochimiques des magmas, des roches émises par des volcans "alimentés" par le même magma
constitutif [204, 209]. Au cours de coulées successives, les roches ont pu subir une
differentiation due à la cristallisation partielle (dite cristallisation fractionnée) du liquide
magmatique lors de son ascension vers le point d'épanchement. Cette differentiation génère des
roches de composition minéralogique et pétrographique différentes, au fur et à mesure que le
liquide s'appauvrit des composants des minéraux ayant cristallisé en profondeur. Cependant, les
éléments qui ont le plus d'affinité pour la phase liquide (éléments dits incompatibles) demeurent
dans les même proportions dans les différentes roches formées - à condition toutefois que le
refroidissement ait été suffisamment rapide pour que des minéraux accessoires concentrant ces
éléments n'aient pu cristalliser. Parmi les éléments les plus incompatibles, citons les terres rares
(en particulier les terres rares légères, de rayon ionique supérieur) et Y, Zr, Nb, Hf, Ta, U et
Th. D'autres éléments moins incompatibles sont incorporés préférentiellement dans certains
minéraux et un appauvrissement important de la phase liquide est observé dès que ces
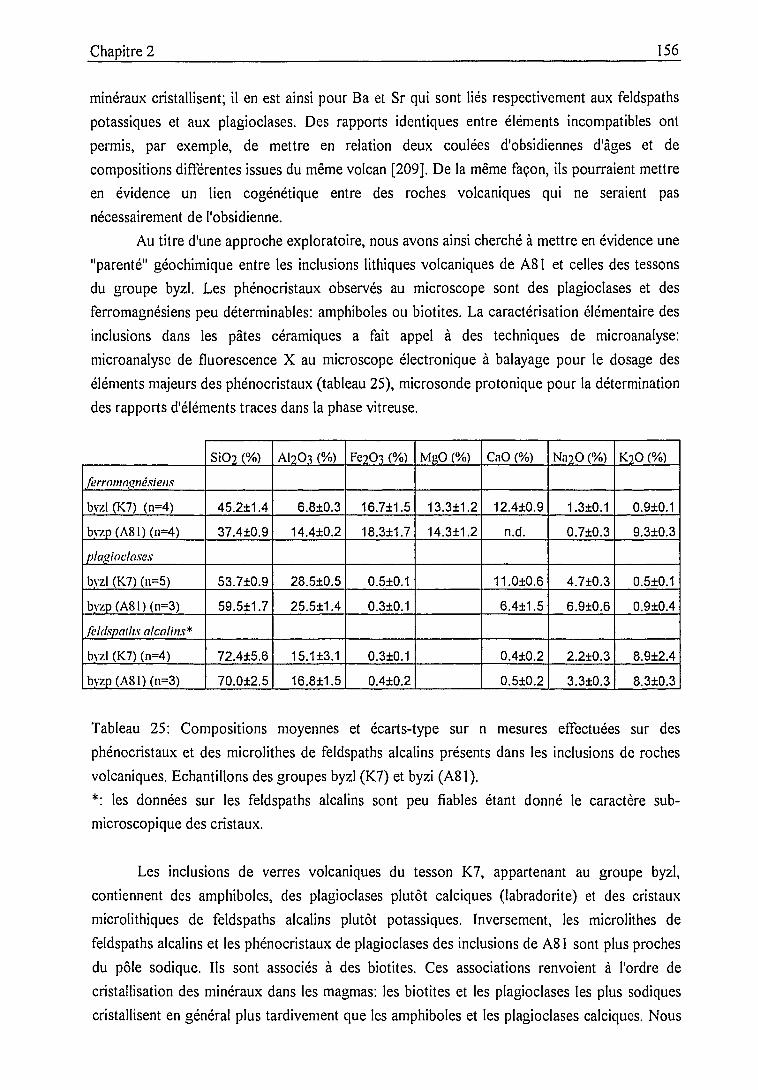
Chapitre 2 156
minéraux cristallisent; il en est ainsi pour Ba et Sr qui sont liés respectivement aux feldspaths
potassiques et aux plagioclases. Des rapports identiques entre éléments incompatibles ont
permis, par exemple, de mettre en relation deux coulées d'obsidiennes d'âges et de
compositions différentes issues du même volcan [209]. De la même façon, ils pourraient mettre
en évidence un lien cogénétique entre des roches volcaniques qui ne seraient pas
nécessairement de l'obsidienne.
Au titre d'une approche exploratoire, nous avons ainsi cherché à mettre en évidence une
"parenté" géochimique entre les inclusions lithiques volcaniques de A81 et celles des tessons
du groupe byzl. Les phénocristaux observés au microscope sont des plagioclases et des
ferromagnésiens peu déterminables: amphiboles ou biotites. La caractérisation élémentaire des
inclusions dans les pâtes céramiques a fait appel à des techniques de microanalyse:
microanalyse de fluorescence X au microscope électronique à balayage pour le dosage des
éléments majeurs des phénocristaux (tableau 25), microsonde protonique pour la détermination
des rapports d'éléments traces dans la phase vitreuse.
ferromngnêsiens
byzl(K7) (n=4)
byzp(A81)(n=4)
plagioclases
byzl (K7) (n=5)
byzp(A81)(n=3)
feldspaths alcalins*
byzl (K7) (n=4)
byzp(A81)(n=3)
SiO2 (%)
45.211.4
37.410.9
53.710.9
59.511.7
72.415.6
70.012.5
AI2O3 (%)
6.810.3
14.410.2
28.510.5
25.511.4
15.113.1
16.811.5
Fc2Oi (%)
16.711.5
18.311.7
0.510.1
0.310.1
0.310.1
0.410.2
MRO (%)
13.311.2
14.311.2
CaO (%)
12.410.9
n.d.
11.010.6
6.411.5
0.410.2
0.510.2
Na2O (%)
1.310.1
0.710.3
4.710.3
6.910.6
2.210.3
3.310.3
K2O (%)
0.910.1
9.310.3
0.510.1
0.910.4
8.912.4
8.310.3
Tableau 25: Compositions moyennes et écarts-type sur n mesures effectuées sur des
phénocristaux et des microlithes de feldspaths alcalins présents dans les inclusions de roches
volcaniques. Echantillons des groupes byzl (K7) et byzi (A81).
*: les données sur les feldspaths alcalins sont peu fiables étant donné le caractère sub-
microscopique des cristaux.
Les inclusions de verres volcaniques du tesson K7, appartenant au groupe byzl,
contiennent des amphiboles, des plagioclases plutôt calciques (labradorite) et des cristaux
microlithiques de feldspaths alcalins plutôt potassiques. Inversement, les microlithes de
feldspaths alcalins et les phénocristaux de plagioclases des inclusions de A81 sont plus proches
du pôle sodique. Ils sont associés à des biotites. Ces associations renvoient à l'ordre de
cristallisation des minéraux dans les magmas: les biotites et les plagioclases les plus sodiques
cristallisent en général plus tardivement que les amphiboles et les plagioclases calciques. Nous

Chapitre 2 1_5J7_
pouvons faire l'hypothèse que les inclusions lithiques de byzi correspondent à des roches plus
différenciées (au sens de la differentiation magmatique) que celles de byzl.
En ce qui concerne les éléments traces, les analyses par PIXE permettent en principe de
de déterminer une gamme d'éléments qui inclut les éléments de transition de la première série
(compatibles pour la plupart), ainsi que Rb, Sr et Ba, qui sont préférentiellement incorporés
dans les feldspaths et les micas, et les incompatibles Zr, Y et Nb. Dans notre cas, ce dernier
élément est en concentration inférieure aux limites de détection (de l'ordre de la dizaine de
ppm). Les rapports d'éléments Rb/Zr, Sr/Zr et Y/Zr ont été mesurés avec une bonne précision
(incertitudes de comptage inférieures au %) dans la phase vitreuse de quatre fragments de
roches volcaniques. Ces quatre fragments sont répartis dans deux tessons, K7 (fragments
K7Z1 et K7Z2) et A81 (fragments A81Z1 et A81Z2), qui appartiennent aux groupes de
composition byzl et byzi, respectivement. Les mesures ont été effectuées sur lames minces, la
dimension des secteurs analysés étant de 100|im2 environ (cf. Annexes 1 et 7B) pour une
profondeur analysée ne dépassant pas l'épaisseur de la préparation.
Les rapports Rb/Zr et Sr/Zr illustrent le comportement relatif des éléments incorporés
dans les feldspaths et les micas par rapport à l'élément Zr, plus incompatible. Nous constatons
que les trois premières séries de mesures (K7Z1, K7Z2, A81Z1) conduisent à des rapports
équivalents, compte tenu des incertitudes (figure 59). La valeur de Rb/Zr plus faible dans
A81Z1 pourrait correspondre à un appauvrissement supplémentaire de la phase vitreuse en Rb
suite à la formation des micas. Pour autant que ces mesures soient significatives, elles indiquent
des propriétés similaires pour les trois fragments de roche analysés. Il n'y aurait pas de
fractionnement différentiel entre les inclusions de byzl et celles de byzi sauf éventuellement en
ce qui concerne les micas.
Il faut certainement nuancer un tel résultat, car de nombreuses facteurs ont été négligés
en première approche. D'une part, en ce qui concerne les propriétés géochimiques des roches
magmatiques, le volcanisme calco-alcalin de la région de Pergame correspond à un cas où la
croûte continentale contribue au matériel magmatique. Il peut y avoir "contamination" du
magma par les éléments de la croûte et modification subséquente des rapports de teneurs
d'éléments incompatibles propres à ce magma. Par ailleurs, la présence d'apatite dans les
tessons de byzl peut indiquer que nous ne sommes plus dans des conditions où les rapports
d'incompatibles sont conservés. Du point de vue expérimental, la définition de la zone à
analyser peut être délicate. Par exemple, on observe des rapports Rb/Zr et Y/Zr plus faibles et
un rapport Sr/Zr plus élevé dans A81Z2. Ces écarts sont très probablement dus à une
délimitation imparfaite de la zone d'analyse: la cartographie élémentaire de Sr révèle une
concentration de cet élément en bord de zone qui correspond selon toute probabilité à un
phénocristal de plagioclase. La présence de ce dernier modifie les rapports de concentrations
d'incompatibles. Cet exemple met en relief un des inconvénients de la microsonde protonique.
Contrairement à un MEB, il n'y a pas de dispositif d'observation direct. Le positionnement de
la zone à analyser demeure très approximatif, et ne peut être ajusté qu'à partir des contrastes
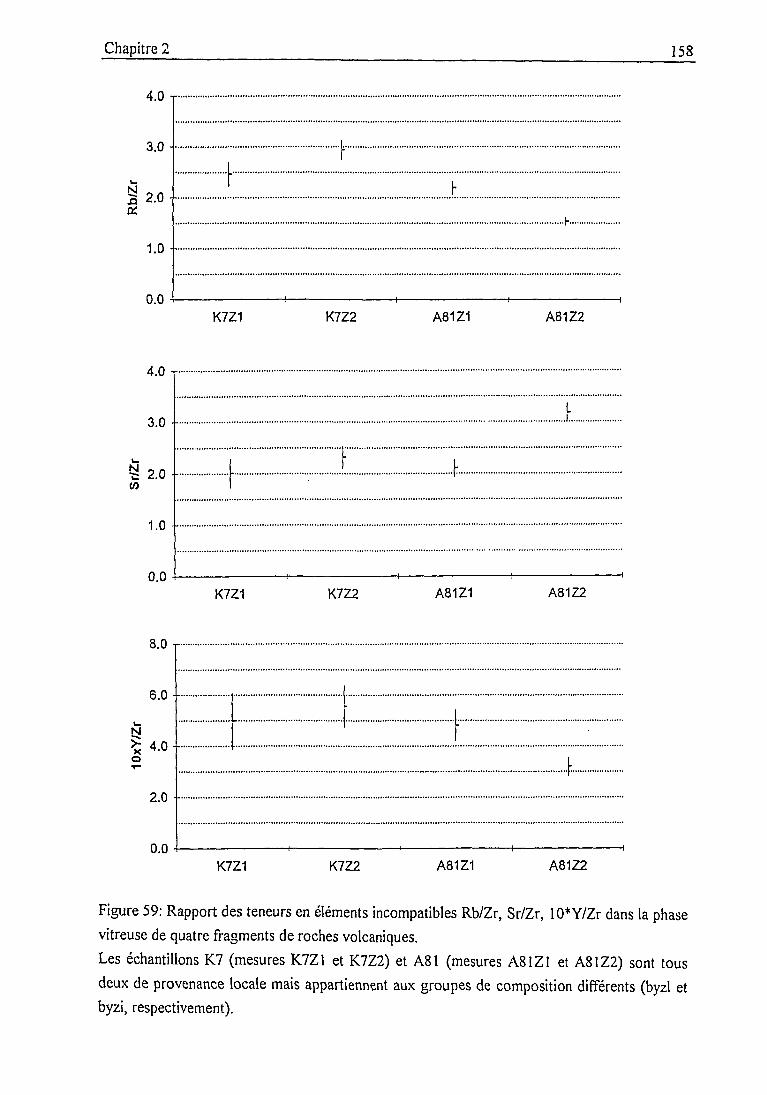
Chapitre 2 158
4.0 r
3.0
S 2.0a:
1 . 0 ••
0.0K7Z1 K7Z2 A81Z1 A81Z2
4.0 T
3.0 -
£ 2 . 0
1.0 -
0.0K7Z1 K7Z2 A81Z1 A81Z2
8.0 r
6.0 •-
N
o4 . 0 •••
2.0 •-
0.0K7Z1 K7Z2 A81Z1 A81Z2
Figure 59: Rapport des teneurs en éléments incompatibles Rb/Zr, Sr/Zr, 10*Y/Zr dans la phase
vitreuse de quatre fragments de roches volcaniques.
Les échantillons K7 (mesures K7Z1 et K7Z2) et A81 (mesures A81Z1 et A81Z2) sont tous
deux de provenance locale mais appartiennent aux groupes de composition différents (byzl et
byzi, respectivement).

Chapitre 2 1_5£
élémentaires observés sur une première acquisition, parfois de longue durée. A un niveau plus
fondamental, la définition de la phase vitreuse dans un matériau très hétérogène comme la
mésostase d'un verre volcanique demeure sujette à caution. De plus, le caractère très
vulnérable des matériaux vitreux vis-à-vis de l'altération rend les dosages d'éléments
mobilisables comme les alcalins peu fiables.
Bien que ces résultats ne soient pas directement exploitables, ils montrent le potentiel
de la microsonde protonique pour la détermination de rapports de teneurs en éléments
incompatibles dans les inclusions volcaniques [210]. La référence directe à des études
géochimiques existantes [211, 212, 213, 203] est cependant malaisée car les analyses publiées
concernent en général la "roche totale" (fraction vitreuse + phénocristaux). Il serait intéressant
d'évaluer l'intérêt de cette technique pour la caractérisation d'inclusions lithiques dans les pâtes
céramiques.
Si nous considérons la production locale dans son ensemble, nous constatons que les
trois groupes locaux (ou supposés tels dans le cas de g): byzl, hell, g, ainsi que le groupe
minoritaire byzi ne représentent pas une unité cohérente, que ce soit du point de vue
élémentaire ou pétrographique. Seul le groupe minoritaire byzp est reconnu comme une
variante de byzl ayant subi un processus d'altération.
Les groupes de composition byzl, hell et g correspondent à des types de céramiques
nettement distincts par leur fonction et leur aspect. Le traitement de la pâte par le potier n'était
probablement pas le même lors de la fabrication de céramiques non glaçurées à pâte grossière
et non calcique destinées à la cuisson ou au stockage (g), de céramiques glaçurées médiévales
à pâte semi-grossière (byzl) ou de céramiques vernissées antiques à pâte épurée (hell).
Cependant, les analyses permettent d'affirmer que les différences observées ne sont pas
uniquement d'ordre technologique. Les transformations d'une même matière première en vue
d'obtenir une pâte plus fine ou, à l'inverse, l'ajout intentionnel de dégraissant, n'ont pu
engendrer de telles modifications des compositions élémentaires ei des faciès pétrographiqucs.
Il est donc vraisemblable que plusieurs gisements argileux distincts ont été exploités à
Pergame. On peut penser qu'ils ont été choisis en fonction des impératifs technologiques
spécifiques à chaque type de production. Un cas analogue de productions spécialisées utilisant
des argiles très différentes est reporté pour le site voisin de Phocée [151].
La grande variabilité géologique et géochimique de la région de Pergame donne la
possibilité de réaliser de tels choix. Elle rend également la définition de caractéristiques
globales propres aux argiles de Pergame très malaisée. Il paraît d'autant plus nécessaire
d'associer à chaque groupe de composition la description archéologique des pièces. Des
comparaisons ultérieures de données analytiques aux groupes de référence de Pergame devront
reposer sur des échantillonnages de même nature ou de même tradition technique, sous peine
de parvenir à des conclusions erronées.

Chapitre 2 160
Groupes de composition de provenance indéterminée
Pour ces groupes qui ne sont pas rattachés à la production pergaménienne (sgraf, lnop),
ou pour lesquels une origine pergaménienne semble douteuse (f, hi), nous avons cherché au-
delà de la caractérisation à mettre en évidence des relations avec des groupes de référence
existant: groupes de référence de céramiques byzantines (cf. 2.2) ou, à défaut, matériel non
contemporain provenant de sites mis en rapport par les hypothèses des archéologues (Ephèse
[214, 215, 174], Sardes [143, 149, 214]). Nous avons également pu considérer des études
relatives à des prospections d'ateliers [150, 173, 216] ou à des analyses de bancs d'argile [161,
174] en Turquie de l'Ouest. Signalons que pour une recherche par localisation géographique, la
compilation réalisée par R.E. Jones [66] qui présente l'ensemble des analyses réalisées (avant
1983) sur des céramiques grecques et chypriotes s'est avérée un outil précieux. Les articles
concernant du matériel supposé pergaménien [171, 217] ont aussi été pris en compte
(cf. 2.3.1).
Groupes/et hi
Les procédures de classification nous ont conduit à considérer un groupe unique, fhi,
occasionnellement associé au groupe local byzi. fhi se compose en réalité de deux sous-
groupes, f et hi
* Différenciation entre f hi et byzi
La ressemblance des compositions élémentaires des groupes fhi et byzi est un argument
en faveur d'une fabrication pergaménienne pour les échantillons des séries archéologiques F, H
et I. Les différences de composition les plus importantes portent sur des alcalins et des
alcalinos-terreux, notamment Ca et Cs (tableau 22). A première vue, ces écarts peuvent donc
paraître peu significatifs puisqu'ils portent sur des éléments susceptibles d'être mobilisés lors
d'une altération. Cependant, plusieurs indices incitent à interpréter les analogies de composition
entre fhi et byzi avec prudence. D'une part, le comportement de la phase carbonatée distingue
les deux ensembles. Sur un diagramme (Ca, Ti), les échantillons des groupes locaux byzi, byzp,
byzi, hell suivent tous approximativement un même alignement alors qu'une corrélation
distincte est observée pour fhi (figure 60a). Ces corrélations négatives ne correspondent pas,
vu leur pente, à un simple phénomène de dilution. Fréquemment observées sur des groupes de
céramiques d'un même atelier, elles peuvent traduire le fait que les dépôts argileux comportent
des séquences calciques et des séquences détritiques, représentées par Ti. La séparation des
deux groupes de corrélation indiquerait des matières premières de nature différente.
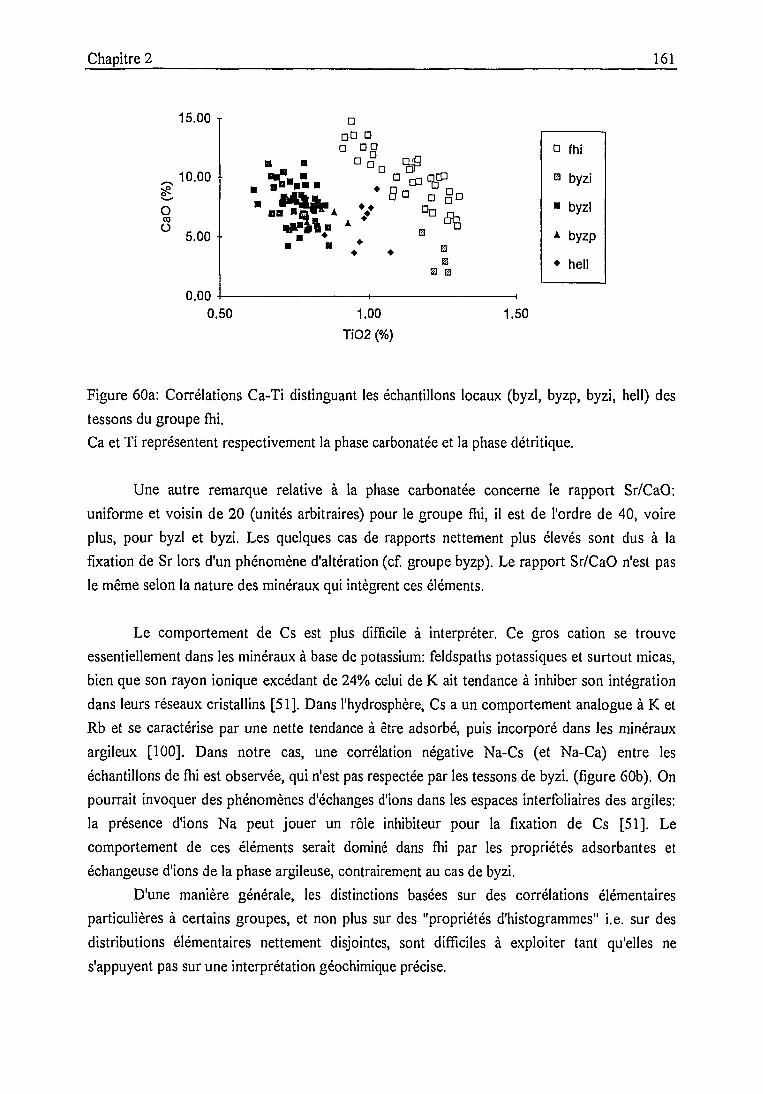
Chapitre 2 161
15.00
10.00
OO
5.00
0.000.50
• Dg
•••
• •
1.00
TiO2 (
1.50
Figure 60a: Corrélations Ca-Ti distinguant les échantillons locaux (byzl, byzp, byzi, hell) des
tessons du groupe fhi.
Ca et Ti représentent respectivement la phase carbonatée et la phase détritique.
Une autre remarque relative à la phase carbonatée concerne le rapport Sr/CaO:
uniforme et voisin de 20 (unités arbitraires) pour le groupe fhi, il est de l'ordre de 40, voire
plus, pour byzl et byzi. Les quelques cas de rapports nettement plus élevés sont dus à la
fixation de Sr lors d'un phénomène d'altération (cf. groupe byzp). Le rapport Sr/CaO n'est pas
le même selon la nature des minéraux qui intègrent ces éléments.
Le comportement de Cs est plus difficile à interpréter. Ce gros cation se trouve
essentiellement dans les minéraux à base de potassium: feldspaths potassiques et surtout micas,
bien que son rayon ionique excédant de 24% celui de K ait tendance à inhiber son intégration
dans leurs réseaux cristallins [51]. Dans l'hydrosphère, Cs a un comportement analogue à K et
Rb et se caractérise par une nette tendance à être adsorbé, puis incorporé dans les minéraux
argileux [100]. Dans notre cas, une corrélation négative Na-Cs (et Na-Ca) entre les
échantillons de fhi est observée, qui n'est pas respectée par les tessons de byzi. (figure 60b). On
pourrait invoquer des phénomènes d'échanges d'ions dans les espaces interfoliaires des argiles:
la présence d'ions Na peut jouer un rôle inhibiteur pour la fixation de Cs [51]. Le
comportement de ces éléments serait dominé dans fhi par les propriétés adsorbantes et
échangeuse d'ions de la phase argileuse, contrairement au cas de byzi.
D'une manière générale, les distinctions basées sur des corrélations élémentaires
particulières à certains groupes, et non plus sur des "propriétés d'histogrammes" i.e. sur des
distributions élémentaires nettement disjointes, sont difficiles à exploiter tant qu'elles ne
s'appuyent pas sur une interprétation géochimique précise.
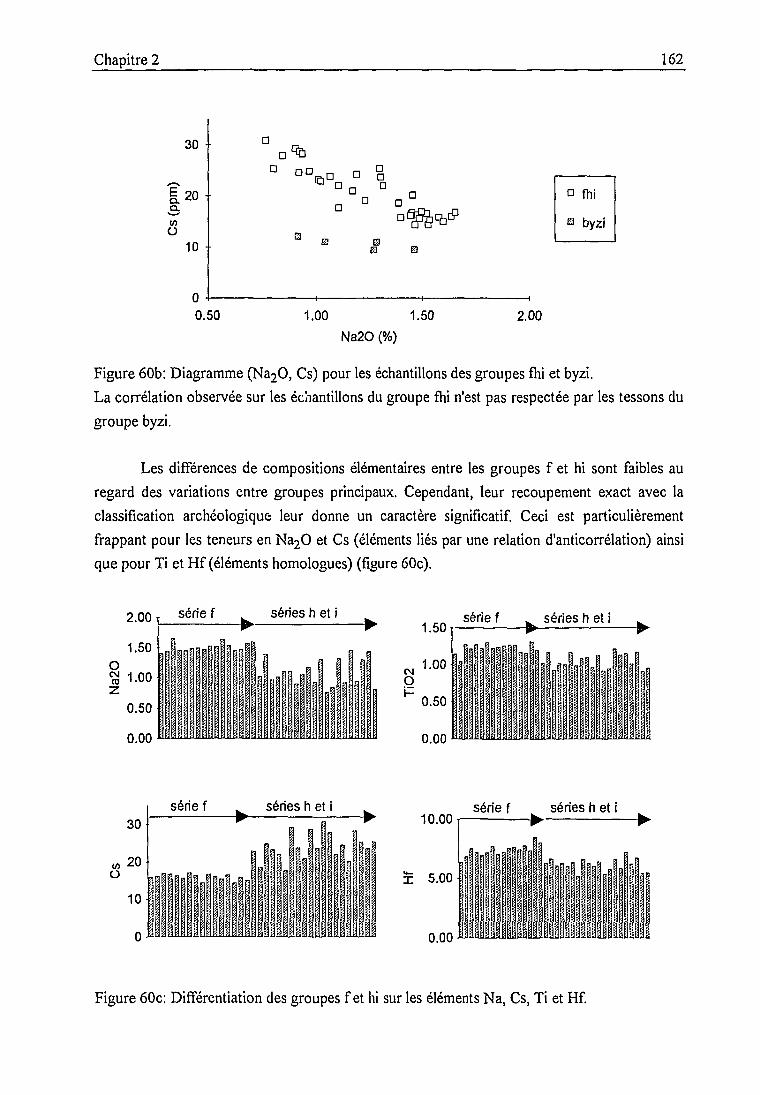
Chapitre 2 162
30 •
I 20 •(f)
o1 0 •
0.50
aa
1.00 1.50
Na20
2.00
Figure 60b: Diagramme (Na2O, Cs) pour les échantillons des groupes fhi et byzi.
La corrélation observée sur les échantillons du groupe fhi n'est pas respectée par les tessons du
groupe byzi.
Les différences de compositions élémentaires entre les groupes f et hi sont faibles au
regard des variations entre groupes principaux. Cependant, leur recoupement exact avec la
classification archéologique leur donne un caractère significatif. Ceci est particulièrement
frappant pour les teneurs en Na2Û et Cs (éléments liés par une relation d'anticorrélation) ainsi
que pour Ti et Hf (éléments homologues) (figure 60c).
2.00
1.50OCMCO
0X1 1.00
0.50
0.00
série f séries h et i
O
1.50
1.00
0.50
0.00
série f
llMSiBBBl J lM J i H Mi ''• f ! '• l & Ê ' 1"
i | : || [i : I | ^ iil \ \ \\ 'Ï'-X \ \v\
séries h et ^
i l i f | i fui!M i l l i iEi f i i
ihl il Jlil ^Ii!il
10.00
I 5.00
0.00
série f séries h et i
\
Figure 60c: Differentiation des groupes f et hi sur les éléments Na, Cs, Ti et Hf.

Chapitre 2 163_
Etant donné la nature des éléments concernés, il paraît difficile de proposer une
interprétation en terme de traitements distinctifs d'une même argile. Par ailleurs, la grande
similitude (intervalles de compositions élémentaires, corrélations inter-éléments) n'incite pas à
considérer des sources argileuses radicalement différentes.
Une differentiation plus marquée est mise en évidence par la diffraction des rayons X:
les échantillons du groupe hi se caractérisent par l'abondance d'un ensemble de raies
attribuables à la famille des pyroxenes (augite). Ces raies ne sont pas présentes en quantité
appréciable dans les échantillons du groupe f ni dans ceux du groupe byzi [197]. Du point de
vue pétrographique, les échantillons des groupes f et hi ont une pâte nettement plus fine que
celle des tessons de byzl et byzi (A81 excepté), et s'en distinguent par la nature des minéraux et
inclusions lithiques. Les inclusions sont de taille moyenne à fine (100-200um) et en abondance
moyenne dans les échantillons de F et dans D59. Dans les tessons de hi, les inclusions sont plus
rares et de plus petite taille (moins de lOOum). Les cristaux identifiables sont des quartz, des
quartz polycristallins et quelques muscovites et minéraux opaques. Des fragments de roches
métamorphiques (quartzites et micaschistes) sont parfois observés. Quelques lames montrent
des pâtes imprégnées de calcite micritique. Les groupes f et hi présentent les pourcentages de
CaO les plus élevés de l'échantillonnage. Du point de vue macroscopique, ceci se traduit pour
certains échantillons de la série H par une teinte claire verdâtre de pâte calcaire bien cuite
[218].
Il faut remarquer une lame mince issue d'un échantillon du groupe F qui recèle une
inclusion lithique singulière (figure 64 (f)) composée d'après des analyses au MEB de
pyroxenes de faciès caractéristique (figure 61), d'albite et de sphène. Cet assemblage
pétrographique pourrait résulter de l'altération hydrothermale et du métamorphisme d'une lave
de type andésitique. Une telle inclusion ne serait donc pas incompatible avec le contexte
géologique de la région de Pergame.
Pour conclure sur la differentiation entre f, hi et byzi, notons que chacun de ces
groupes montre des caractéristiques qui n'apparaissent pas dans les autres. Les differentiations
par les compositions chimiques sont assez ténues, mais coïncident exactement avec les
"familles" archéologiques associant les céramiques à pâte fine (séries H et I) d'une part, les
céramiques à pâte grasse (série F) d'autre part, et les tessons rattachés à la production locale
(groupe byzi). La pétrographie rattache nettement byzi et byzl (à l'exception de A81), alors que
f et hi sont plus proches d'un contexte métamorphique ou sédimentaire (calcaire), à l'exception
notable d'une lame d'un échantillon du groupe F présentant une fragment lithique pouvant
résulter de l'altération d'une roche andésitique. La diffraction de rayons X isole les échantillons
de hi de tous les autres tessons. Les arguments en faveur et en défaveur d'une provenance
locale des échantillons f et hi sont résumés dans le tableau 26.

Chapitre 2 164
Figure 61: Image obtenue au MEB, en mode de contraste de densité électronique (électrons
rétrodifïusés), d'un pyroxene dans l'inclusion caractéristique de l'échantillon F67 (largeur du
"rognon": 100 um)
groupe hi
rroupe f
arguments en faveur d'une provenance
pergaménienne
- compositions élémentaires proches de
celles du groupe local byzi
- compositions élémentaires proches de
celles du groupe local byzi
- inclusion isolée pouvant résulter de
l'altération d'un fragment de roche
andésitique
arguments en défaveur d'une
provenance pergaménienne
- comportement de la phase carbonatée
distincte de celle des échantillons
locaux
- groupe distingué par la diffraction des
rayons X (important spectre de raies de
l'augite)
- pâte fine, sans fragments de roches
volcaniques
- comportement de la phase carbonatée
distincte de celle des échantillons
locaux
- inclusions à caractère métamorphique
(quartzites, micaschistes) fréquentes
Tableau 26: Récapitulatif des arguments en faveur et en défaveur d'une provenance locale des
groupes f et hi.

Chapitre 2 165
* Comparaison des groupes] et hi avec des groupes de références
Dans une première approche, nous avons pu comparer, grâce à M. Picon, directeur du
laboratoire de céramologie de Lyon, les caractéristiques élémentaires des groupes f et hi avec
celles de tessons d'ateliers situés le long du littoral égéen oriental connus pour leur activité au
cours de la période greco-romaine [219], La grande variabilité géochimique de la région de
Pergame est un trait partagé par cette partie de la côte turque: les céramiques d'ateliers situés à
des distances de l'ordre du km peuvent se distinguer par leurs compositions [150, 151]. Les
sites considérés sont, du Nord au Sud: Çandarly, Elée, Grynion, Myrina, Phocée, Clazomène,
Ephèse, Milet, Knydes. Des échantillons des iles de Lesbos, Chio et Samos ont également été
pris en compte.
On constate que les teneurs en TiÛ2 relativement élevées (supérieures à 1%) qui
caractérisent les groupes f et hi sont exceptionnelles dans les ateliers cités. Cet élément étant
dosé de manière équivalente par le laboratoire de Lyon et le nôtre (cf. 1.3), nous pouvons en
conclure qu'il est peu probable que les tessons des groupes f et hi soient des productions
tardives utilisant les gisements exploités par ces ateliers. Si on considère les données dans leur
ensemble, il semblerait que de telles teneurs en Ti ne soient pas propres aux argiles de la région
cotière considérée. Cette observation est en bon accord avec les caractéristiques du volcanisme
à faibles teneurs en Ti qui constitue les massifs couvrant la partie Nord de la région [211,213].
Une provenance régionale n'est toutefois pas exclue, puisque des concentrations en Ti élevées
localisées sont susceptibles de se présenter, comme c'est le cas à Pergame (groupe byzi).
Notons que les concentrations en TiC>2 de f et hi sont également supérieures aux
valeurs publiées pour les céramiques byzantines de Grèce et de Crète [123, 126], à l'exception
de la céramique "white ware" qui n'est pas comparable par ailleurs aux échantillons de
Pergame.
Les groupes f et hi correspondent principalement aux séries archéologiques F, et H, I,
respectivement. Nous avons vu en 2.3.2. que des tessons similaires à ceux de la série F ont été
mis à jour à Ephèse, et que les fouilles de Sardes ont révélé des céramiques analogues à celles
de la série H. Une hypothèse plausible est que les céramiques du groupe f ont été produites à
Ephèse et celles du groupe hi à Sardes.
Des examens pétrographiques et des analyses qualitatives d'engobe et de glaçure ont
été réalisées sur le matériel byzantin de Sardes [143], et une étude est apparemment en cours
sur les tessons byzantins issus du site d'Ayasoluk à Selçuk près d'Ephèse [159]. Il n'existe
toutefois pas à l'heure actuelle de données d'analyse quantitative disponibles sur ces deux
matériels.

Chapitre 2 166
Par contre, plusieurs études ont porté sur les terres cuites antiques de Sardes et
d'Ephèse [214, 215, 174]. Nous avons pu utiliser des données concernant des lampes datées
des époques hellénistique à romaine tardive (6èmc - 7èmc siècles) [214], grâce à M.J. Hughes
du laboratoire du British Museum. Des tessons de datation antérieure fabriqués à Sardes ont
également été analysés dans ce laboratoire. La production de Sardes se caractérise par une
composition homogène quelle que soit l'époque (lydienne à romaine tardive) et le type de
produits considérés (lampes, céramique culinaire, céramique fine), alors que celle d'Ephèse se
répartit en groupes de composition distincts. Ces groupes ne correspondent pas toujours à une
typologie de lampe particulière, mais peuvent rassembler des objets de type et de datation
différents. Si l'on peut définir une composition "sardienne", le cas d'Ephèse est plus complexe
et nécessite la référence à l'un ou l'autre des groupes de composition distingués. Les
échantillons présentés comme provenant soit de Sardes, soit d'Ephèse ont été sélectionnés pour
servir de groupes de référence pour les tessons des groupes f et hi. Les désignations des
groupes sont indiquées dans le tableau 27.
Provenance
Ephèse
Sardes
Groupe
B
E
F
I
J
M
Q
R
S
T
Désignation archéologique, datation, commentaires
lampes de type "Loeschke VIII", 2 A.D. (- 4 A.D.), pâte épurée (?)
lampes de types divers, 5 B.C. -1 A.D.
lampes de types divers, moules de lampes, 3 B.C. - 2 A.D.
lampes de type "Howland 49A", 2 B.C.- 1 A.D.
lampes de type "volute", 1 - 2 A.D.
lampes de types divers, 1 B.C. - 2 A.D.
lampes de types divers, poteries, époque lydienne - 7 A.D.
lampes de types divers, poteries, époque hellénistique - 7 A.D.
lampes de types divers, poteries, époque lydienne - 7 A.D.
poteries, époque lydienne
Tableau 27: Descriptif sommaire des échantillons des groupes de référence de Sardes et
d'Ephèse, d'après [214],
Au chapitre 1.3., nous avons vu que les analyses du standard "Lefkandi brick" réalisées
par le laboratoire du British Museum sont comparables à nos propres analyses, sauf en ce qui
concerne les éléments Ca, Cr, Ba, Ce. En supposant que la bonne coïncidence des résultats
peut être étendue à un certain intervalle élémentaire, les concentrations des échantillons des
groupes de Sardes, d'Ephèse et des groupes f et hi ont été soumises à des CAH (figure 62).
Les éléments Ca, Cr, Ba, Ce, Tb (dosages peu concordants) ainsi que Sb (distributions souvent
non représentatives, cf. 2.4.1.1) et U (données manquantes trop nombreuses) ont été éliminés
au préalable.
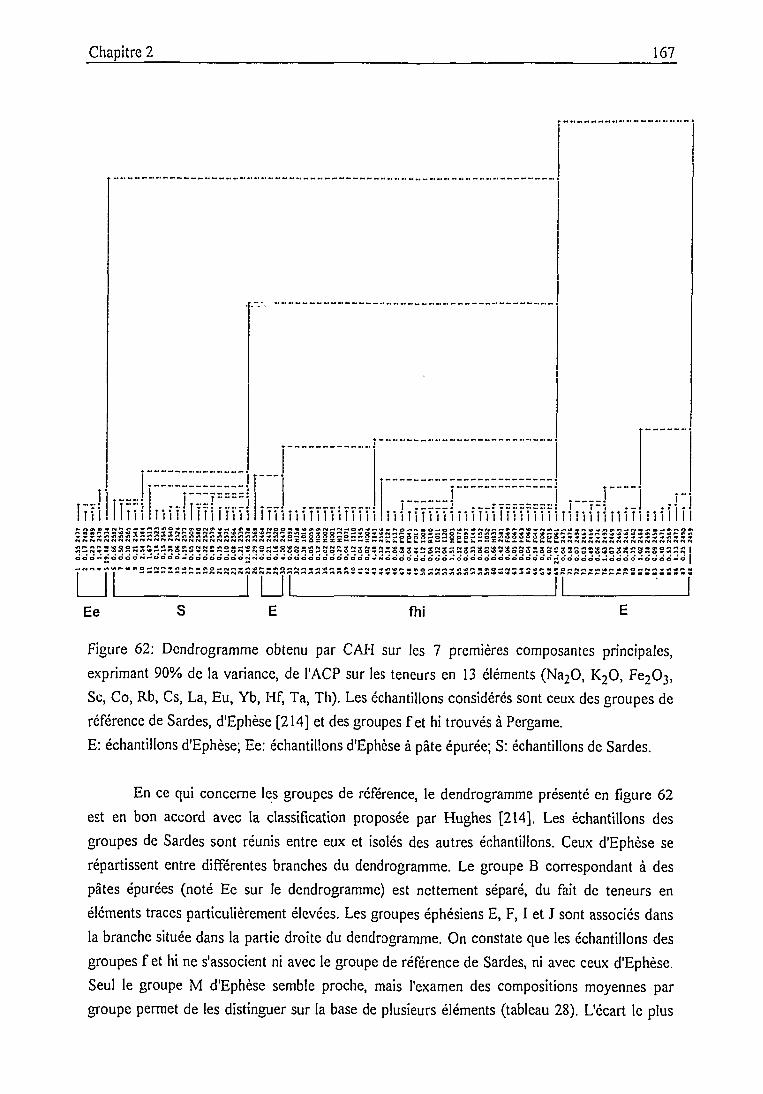
Chapitre 2 167
m ninlmiï î i i 11 n n i T 111111111 m 11 n i i 11111 mïmiîimîmmmiO O » » * ? ? ? * ^ » ^ ' * *
Ee fhi
Figure 62: Dendrogramme obtenu par CAH sur les 7 premières composantes principales,
exprimant 90% de la variance, de l'ACP sur les teneurs en 13 éléments (Na2O, K2O, Fe2O3,
Se, Co, Rb, Cs, La, Eu, Yb, Hf, Ta, Th). Les échantillons considérés sont ceux des groupes de
référence de Sardes, d'Ephèse [214] et des groupes f et hi trouvés à Pergame.
E: échantillons d'Ephèse; Ee: échantillons d'Ephèse à pâte épurée; S: échantillons de Sardes.
En ce qui concerne les groupes de référence, le dendrogramme présenté en figure 62
est en bon accord avec la classification proposée par Hughes [214]. Les échantillons des
groupes de Sardes sont réunis entre eux et isolés des autres échantillons. Ceux d'Ephèse se
répartissent entre différentes branches du dendrogramme. Le groupe B correspondant à des
pâtes épurées (noté Ee sur le dendrogramme) est nettement séparé, du fait de teneurs en
éléments traces particulièrement élevées. Les groupes éphésiens E, F, I et J sont associés dans
la branche située dans la partie droite du dendrogramme. On constate que les échantillons des
groupes f et hi ne s'associent ni avec le groupe de référence de Sardes, ni avec ceux d'Ephèse.
Seul le groupe M d'Ephèse semble proche, mais l'examen des compositions moyennes par
groupe permet de les distinguer sur la base de plusieurs éléments (tableau 28). L'écart le plus

Chapitre 2
important porte sur Cr: le rapport entre les teneurs en Cr du groupe M et des groupes f et hi
est proche de 3. Bien qu'il existe un certain décallage entre les dosages de cet élément réalisés
dans les deux laboratoires (cf. 1.3. et tableau 28), une telle différence peut être considérée
comme significative. D'autres éléments présentent des écarts nettement moins importants,
notamment K et Rb, Cs, La, Ta.
Il semblerait donc à première vue qu'il n'existe pas de relation entre les tessons des
groupes f et hi et les échantillons de référence de Sardes et d'Ephèse. Il convient toutefois de
rester prudent. En particulier, on constate que les compositions des tessons de hi ne sont pas
très différentes des compositions représentatives des céramiques de Sardes. On peut se
demander si ces faibles écarts ne pourraient pas résulter d'un défaut d'inter-calibration, défaut
qu'il est évidemment difficile d'évaluer à partir d'un seul dosage commun.
Une autre approche consiste à faire référence à des prélèvements de terrain. La
comparaison directe de céramiques et d'argiles qui ont pu servir à leur fabrication n'est pas
toujours fructueuse, notamment en raison des transformations effectuées par le potier (cf. e.g.
[66]). Néanmoins, nous avons pu, à titre exploratoire, effectuer quelques prélèvements dans la
vallée du Gediz de part et d'autre de Sardes. De nombreuses briqueteries, situées le long de la
route Izmir-Afyon à hauteur de Salihli, témoignent d'une exploitation intensive de l'argile. Des
argiles micacées naturellement épurées ramassées en surface, des prélèvements dans les amas
argileux de texture plus grossière stockés dans les briqueteries et un prélèvement de sédiment
gréseux issu d'une carrière exploitée, ont constitué un échantillonnage susceptible de
représenter l'argile et le sédiment bruts disponibles dans la vallée du Gediz et le matériau utilisé
pour la confection de briques. Le tableau 29 compare les compositions de ces prélèvements à
celles du groupe fhi et du groupe de référence de Sardes d'après [214]. Les échantillons
d'argile, prélevés à des endroits distants d'une dizaine de km, ont des compositions semblables
ce qui confirmerait l'uniformité relative des argiles de la région de Sardes. Ils sont également
plus homogènes que le matériau des briqueteries. Le sédiment prélevé dans la carrière est
nettement différent des autres échantillons. En comparant l'argile fine avec les tessons de
céramiques, on observe qu'il existe un bon accord avec les teneurs des échantillons du groupe
de référence de Sardes. Les quelques écarts observés peuvent être attribués soit à la procédure
très "approximative" de prélèvement (Na), soit aux écarts analytiques entre les deux
laboratoires (Cr, Tb, Th). Par contre, ces argiles typiques des alentours de Sardes présentent
plusieurs concentrations élémentaires qui se situent en dehors de l'intervalle (m-2a, m+2a)
incluant à 95% de probabilité les teneurs des échantillons du groupe hi. Ces derniers sont plus
riches en Ca, Hf, Ta et moins riches en K, Fe, Mn et à un moindre degré en Si et Al. On ne
peut plus envisager ici que ces différences soient dues à un défaut d'intercalibration.
Il est difficile d'imaginer un traitement qui, partant d'une argile très fine pour aboutir à
une pâte également fine, aurait pu produire ces variations. On retiendra que les argiles
collectées dans la vallée du Gediz et les tessons de référence de Sardes ne semblent pas
s'associer aux échantillons de hi par leur composition élémentaire.

Chapitre 2 169
Un autre argument allant dans le même sens est donné par les résultats de la diffraction
des rayons X (cf. annexe). Les sédiments de la vallée du Gediz, qui dérivent des massifs
métamorphiques du Menderes, sont dominés d'une manière générale par les illites et micas.
L'observation macroscopique révèle que les micas blancs sont particulièrement abondants.
Dans les argiles, les analyses en diffraction indiquent également un forte proportion de calcite,
du quartz, de la chlorite et des pyroxenes en proportion moyenne, et des feldspaths en faible
proportion. Les matériaux de briqueterie diffèrent des argiles par une très forte contribution de
quartz. Parmi les prélèvements de terrain, seul le sédiment de carrière contient des plagioclases
en proportion importante. De la dolomite est présente dans certains échantillons.
Les diffractogrammes des échantillons du groupe hi sont caractérisés par une forte
proportion d'augite et de plagioclases, une proportion moyenne de quartz et par la faible
abondance, voire l'absence d'illites-micas et de feldspaths potassiques. Malgré les modifications
apportées par la cuisson (en particulier la destruction des structures des argiles), il paraît
difficile de constituer une pâte présentant ces caractéristiques à partir des matériaux bruts
considérés. En effet, d'après les analyses en diffraction X effectuées sur nos prélèvements, une
forte proportion de pyroxenes et de plagioclases n'est pas représentative des terrains sardiens.
L'absence des micas blancs serait aussi étonnante, bien qu'une température de cuisson de
l'ordre de 900-1000 °C soit susceptible de détruire la muscovite [93] et que le caractère
phylliteux des micas rende une détermination fiable en diffraction des rayons X moins aisée
[171].
Plusieurs indices semblent donc infirmer l'hypothèse d'une provenance sardienne pour
les céramiques de hi. Cependant, les défauts d'intercalibration gênant la référence à des groupes
de composition d'origine connue et le caractère très préliminaire des procédures de
prélèvement sur le terrain nous permettent difficilement de conclure. Un programme complet
d'intercomparaisons entre les laboratoires et une prospection de terrain effectuée en
collaboration avec des géologues connaissant la géologie locale seraient l'amorce d'un réel
traitement de la question. De plus, la confrontation avec des données d'analyse acquises sur les
céramiques byzantines trouvées à Sardes, de type similaire aux tessons de la série H [143],
seraient probablement nécessaires pour proposer une conclusion définitive en ce qui concerne
cette catégorie de céramiques.
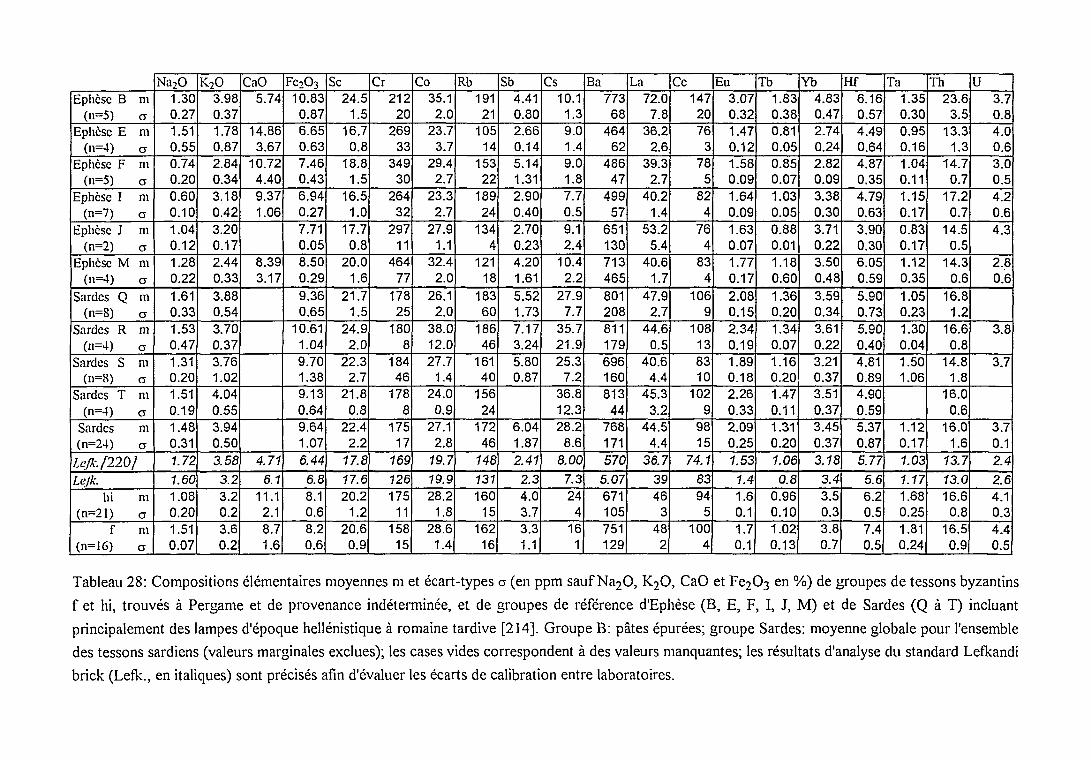
Ephcse B m(n=5) CT
Ephcsc E m(n=4) a
Ephcsc F ni(11=5) CT
Ephèse I m(n=7) CT
Ephcsc J ni(n=2) CT
Ephcsc M m(n=4) a
Sardes Q m(n=8) CT
Sardes R m(11=4) CT
Sardes S m(11=8) CT
Sardes T m(n=4) CT
Sardes m(n=24) CT
Lejk. [220]Lejk.
hi m(n=21) CT
f ni(n=16) CT
Na2O1.300.271.510.550.740.200.600.101.040.121.280.221.610.331.530.471.310.201.510.191.480.311.72
1.601.080.201.510.07
K2O3.980.371.780.872.840.343.180.423.200.172.440.333.880.543.700.373.761.024.040.553.940.503.58
3.23.20.23.60.2
CaO5.74
14.863.67
10.724.409.371.06
8.393.17
4.71
6.111.12.18.71.6
Fc2O3
10.830.876.650.637.460.436.940.277.710.058.500.299.360.65
10.611.049.701.389.130.649.641.076.44
6.88.10.68.20.6
Se24.5
1.516.7
0.818.81.5
16.51.0
17.70.8
20.01.6
21.71.5
24.92.0
22.32.7
21.80.8
22.42.2
17.8
17.620.2
1.220.6
0.9
Cr212
20269
33349
30264
32297
11464
7717825
1808
18446
1788
17517
169
126175
11158
15
Co35.1
2.023.7
3.729.4
2.723.3
2.727.9
1.132.4
2.026.1
2.038.012.027.7
1.424.0
0.927.1
2.819.7
19.928.2
1.828.6
1.4
Rb191
21105
14153
22189
24134
4121
18183
6018646
16140
15624
17246
148
131160
15162
16
Sb4.410.802.660.145.141.312.900.402.700.234.201.615.521.737.173.245.800.87
6.041.872.41
2.34.03.73.31.1
Cs10.11.39.01.49.01.87.70.59.12.4
10.42.2
27.97.7
35.721.925.3
7.236.812.328.2
8.68.00
7.324
4161
Ba77368
46462
48647
49957
65113071346580120881117969616081344
768171570
5.07671105751129
La72.07.8
36.22.6
39.32.7
40.21.4
53.25.4
40.61.7
47.92.7
44.60.5
40.64.4
45.33.2
44.54.4
36.73946
3482
Ce1472076
378
5824
764
834
1069
108138310
1029
9815
74.18394
5100
4
Eu3.070.321.470.121.580.091.640.091.630.071.770.172.080.152.340.191.890.182.260.332.090.257.531.41.60.11.70.1
Tb1.830.380.810.050.850.071.030.050.880.011.180.601.360.201.340.071.160.201.470.111.310.201.060.8
0.960.101.020.13
Yb4.830.472.740.242.820.093.380.303.710.223.500.483.590.343.610.223.210.373.510.373.450.373.18
3.43.50.33.80.7
Hf6.160.574.490.644.870.354.790.633.900.306.050.595.900.735.900.404.810.894.900.595.370.875.77
5.66.20.57.40.5
Ta1.350.300.950.161.040.111.150.170.830.171.120.351.050.231.300.041.501.06
1.120.171.031.171.680.251.810.24
Th23.6
3.513.31.3
14.70.7
17.20.7
14.50.5
14.30.6
16.81.2
16.60.8
14.81.8
16.00.6
16.01.6
13.713.016.60.8
16.50.9
U3.70.84.00.63.00.54.20.64.3
2.80.6
3.8
3.7
3.70.12.4
2.64.10.34.40.5
Tableau 28: Compositions élémentaires moyennes m et écart-types CT (en ppm sauf Na2Û, K^O, CaO et Fe2Û3 en %) de groupes de tessons byzantins
f et hi, trouvés à Pergame et de provenance indéterminée, et de groupes de référence d'Ephèse (B, E, F, I, J, M) et de Sardes (Q à T) incluant
principalement des lampes d'époque hellénistique à romaine tardive [214]. Groupe B: pâtes épurées; groupe Sardes: moyenne globale pour l'ensemble
des tessons sardiens (valeurs marginales exclues); les cases vides correspondent à des valeurs manquantes; les résultats d'analyse du standard Lefkandi
brick (Lefk., en italiques) sont précisés afin d'évaluer les écarts de calibration entre laboratoires.
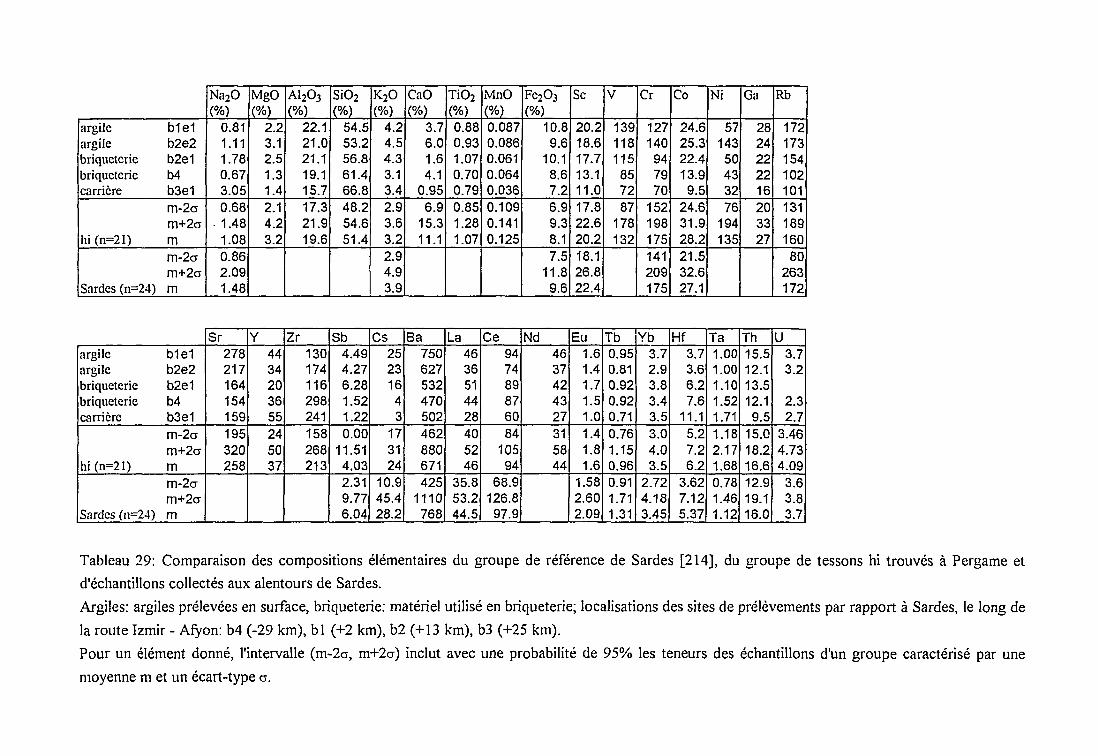
argile b1e1argile b2e2briqueterie b2e1briqueterie b4carrière b3e1
m-2am+2<j
hi (n=21) m
m-2am+2a
Sardes (n=24) m
Na2O
0.811.111.780.673.050.681.481.080.862.091.48
MgO
2.23.12.51.31.42.14.23.2
A12O3
22.121.021.119.115.717.321.919.6
SiO2
54.553.256.861.466.848.254.651.4
K20
4.24.54.33.13.42.93.63.22.94.93.9
CaO
3.76.01.64.1
0.956.9
15.311.1
TiO2
0.880.931.070.700.790.851.281.07
MnO
0.0870.0860.0610.0640.0360.1090.1410.125
Fe2O3
10.89.6
10.18.67.26.99.38.17.5
11.89.6
Se
20.218.617.713.111.017.822.620.218.126.822.4
V
139118115857287
178132
Cr
127140947970
152198175141209175
Co
24.625.322.413.99.5
24.631.928.221.532.627.1
Ni
5714350433276
194135
Ga
2824222216203327
Rb
17217315410210113118916080
263172
argile b1e1argile b2e2briqueterie b2e1briqueterie b4carrière b3e1
m-2<7
m+2ahi(n=21) m
m-2crm+2a
Sardes (n=24) m
Sr278217164154159195320258
Y4434203655245037
Zr130174116298241158268213
Sb4.494.276.281.521.220.00
11.514.032.319.776.04
Cs25231643
173124
10.945.428.2
Ba750627532470502462880671425
1110768
La4636514428405246
35.853.244.5
Ce947489876084
10594
68.9126.897.9
Nd4637424327315844
Eu1.61.41.71.51.01.41.81.6
1.582.602.09
Tb0.950.810.920.920.710.761.150.960.911.711.31
Yb3.72.93.83.43.53.04.03.5
2.724.183.45
Hf3.73.66.27.6
11.15.27.26.2
3.627.125.37
Ta1.001.001.101.521.711.182.171.680.781.461.12
Th15.512.113.512.19.5
15.018.216.612.919.116.0
U3.73.2
2.32.7
3.464.734.09
3.63.83.7
Tableau 29: Comparaison des compositions élémentaires du groupe de référence de Sardes [214], du groupe de tessons hi trouvés à Pergame et
d'échantillons collectés aux alentours de Sardes.
Argiles: argiles prélevées en surface, briqueterie: matériel utilisé en briqueterie; localisations des sites de prélèvements par rapport à Sardes, le long de
la route Izmir - Afyon: b4 (-29 km), bl (+2 km), b2 (+13 km), b3 (+25 km).
Pour un élément donné, l'intervalle (m-2a, m+2a) inclut avec une probabilité de 95% les teneurs des échantillons d'un groupe caractérisé par une
moyenne m et un écart-type a.
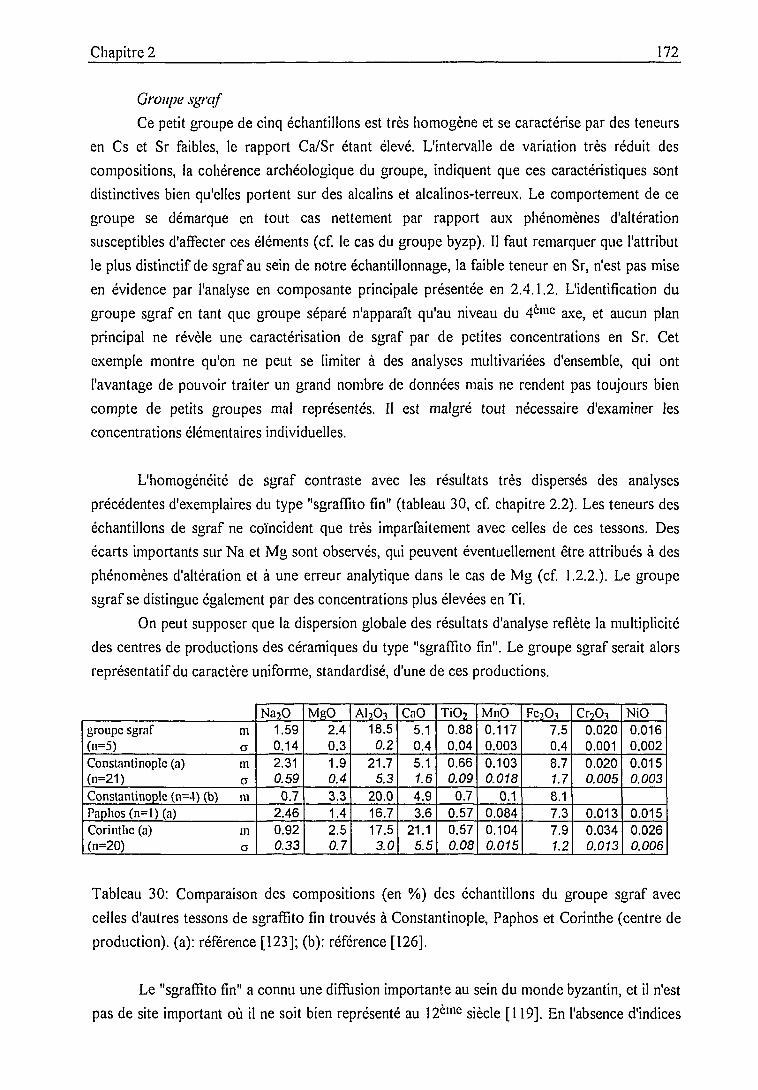
Chapitre 2 172
Groupe sgraf
Ce petit groupe de cinq échantillons est très homogène et se caractérise par des teneurs
en Cs et Sr faibles, le rapport Ca/Sr étant élevé. L'intervalle de variation très réduit des
compositions, la cohérence archéologique du groupe, indiquent que ces caractéristiques sont
distinctives bien qu'elles portent sur des alcalins et alcalinos-terreux. Le comportement de ce
groupe se démarque en tout cas nettement par rapport aux phénomènes d'altération
susceptibles d'affecter ces éléments (cf. le cas du groupe byzp). Il faut remarquer que l'attribut
le plus distinctif de sgraf au sein de notre échantillonnage, la faible teneur en Sr, n'est pas mise
en évidence par l'analyse en composante principale présentée en 2.4.1.2. L'identification du
groupe sgraf en tant que groupe séparé n'apparaît qu'au niveau du 4 è m c axe, et aucun plan
principal ne révèle une caractérisation de sgraf par de petites concentrations en Sr. Cet
exemple montre qu'on ne peut se limiter à des analyses multivariées d'ensemble, qui ont
l'avantage de pouvoir traiter un grand nombre de données mais ne rendent pas toujours bien
compte de petits groupes mal représentés. Il est malgré tout nécessaire d'examiner les
concentrations élémentaires individuelles.
L'homogénéité de sgraf contraste avec les résultats très dispersés des analyses
précédentes d'exemplaires du type "sgraffito fin" (tableau 30, cf. chapitre 2.2). Les teneurs des
échantillons de sgraf ne coïncident que très imparfaitement avec celles de ces tessons. Des
écarts importants sur Na et Mg sont observés, qui peuvent éventuellement être attribués à des
phénomènes d'altération et à une erreur analytique dans le cas de Mg (cf. 1.2.2.). Le groupe
sgraf se distingue également par des concentrations plus élevées en Ti.
On peut supposer que la dispersion globale des résultats d'analyse reflète la multiplicité
des centres de productions des céramiques du type "sgraffito fin". Le groupe sgraf serait alors
représentatif du caractère uniforme, standardisé, d'une de ces productions.
groupe sgraf m(n=5) aConstantinople (a) m(n=21) CT
Constantinople (n=4) (b) mPaphos(n=l)(a)Corinthc (a) m(n=20) G
Na2O1.590.142.310.59
0.72.460.920.33
MgO2.40.31.90.43.31.42.50.7
Al2O3
18.50.2
21.75.3
20.016.717.5
3.0
CaO5.10.45.11.64.93.6
21.15.5
TiO2
0.880.040.660.09
0.70.570.570.08
MnO0.1170.0030.1030.018
0.10.0840.1040.015
Fc2O}7.50.48.71.78.17.37.91.2
Cr2O,0.0200.0010.0200.005
0.0130.0340.013
NiO0.0160.0020.0150.003
0.0150.0260.006
Tableau 30: Comparaison des compositions (en %) des échantillons du groupe sgraf avec
celles d'autres tessons de sgraffito fin trouvés à Constantinople, Paphos et Corinthe (centre de
production), (a): référence [123]; (b): référence [126].
Le "sgraffito fin" a connu une diffusion importante au sein du monde byzantin, et il n'est
pas de site important où il ne soit bien représenté au 12emc siècle [119]. En l'absence d'indices

Chapitre 2 173_
archéologiques, il paraît difficile de faire une hypothèse a priori d'un lieu ou d'une région de
production pour les tessons de sgraf. Ces derniers se distinguent immédiatement des
céramiques issues du seul centre de production attesté, Corinthe, par une argile beaucoup
moins calcique (tableau 30). Nous avons comparé la composition des céramiques de sgraf avec
celles données par Megaw et Jones [123] pour les autres centres ayant fabriqué de la
céramique byzantine - toutes époques et tous styles confondus. En considérant également les
dosages présentés par Lazzarini et Calogero [126], on constate que le groupe qui montre la
meilleure concordance avec sgraf est celui de Thessalonique. Dans le tableau 31, les
compositions de sgraf sont mises en parallèle avec les dosages d'échantillons provenant de
Thessalonique réalisés par différents auteurs. La concordance est particulièrement remarquable
par rapport aux concentrations en éléments majeurs données par Lazzarini et Calogero [126],
les teneurs en Cr et Ni étant en bon accord avec les valeurs tirées de [123]. Par contre, les
résultats des dosages par INAA du laboratoire de Sofia ne coïncident pas sur plusieurs
éléments. Certains écarts peuvent être imputés à un défaut d'intercalibration (K, Sb) car ils
correspondent aux décalages observés sur les dosages du standard commun Lefkandi brick.
D'autres différences de teneurs ne rentrent pas dans ce cadre, et semblent infirmer l'hypothèse
d'une provenance thessalonicienne. C'est le cas en particulier pour Fe, Se, Cs, Ba, Nd, Tb. On
regrette vivement qu'aucun auteur n'ait analysé l'élément Sr, qui paraît être le plus discriminant
dans le cas du groupe sgraf.
Il faut également remarquer que les compositions des échantillons provenant de
Thessalonique sont peu homogènes, tant pour les éléments majeurs que pour les traces. Nous
avons pu disposer, grâce à I. Kuleff, de données individuelles pour les tessons analysés au
laboratoire de Sofia. Les valeurs indiquées dans le tableau 31 ont été calculées après
élimination de plusieurs valeurs extrêmes différant parfois d'un ordre de grandeur, afin
d'obtenir une moyenne et un écart-type représentatif de la plus grande partie des échantillons.
On peut se demander si plusieurs groupes de composition homogène, correspondant à
différentes productions thessaloniciennes, pourraient être constitués. L'échantillonnage
considéré est trop réduit pour en juger.
L'approche pétrographique peut apporter des arguments plus décisifs. Lazzarini et
Calogero [126] caractérisent les tessons de Thessalonique par la présence de pyroxenes
orthorombiques typiques. Ceux-ci sont associés à des cristaux anguleux ou sub-anguleux de
quartz, à des feldspaths alcalins et plagioclases, des minéraux opaques, des fragments de
schistes ardoisiers, dans une pâte pourvue d'un dégraissant abondant de taille moyenne à fine.
Illite et mullite sont aussi présentes.
Les pâtes des échantillons du groupe sgraf montrent des faciès pétrographiques
beaucoup plus variables que ne le laisseraient supposer l'uniformité des compositions
élémentaires. Elles contiennent des quantités diverses de dégraissant, de granulométrie
hétérométrique, avec une fraction fine (moins de 100 um) dominante. Les grains peuvent être
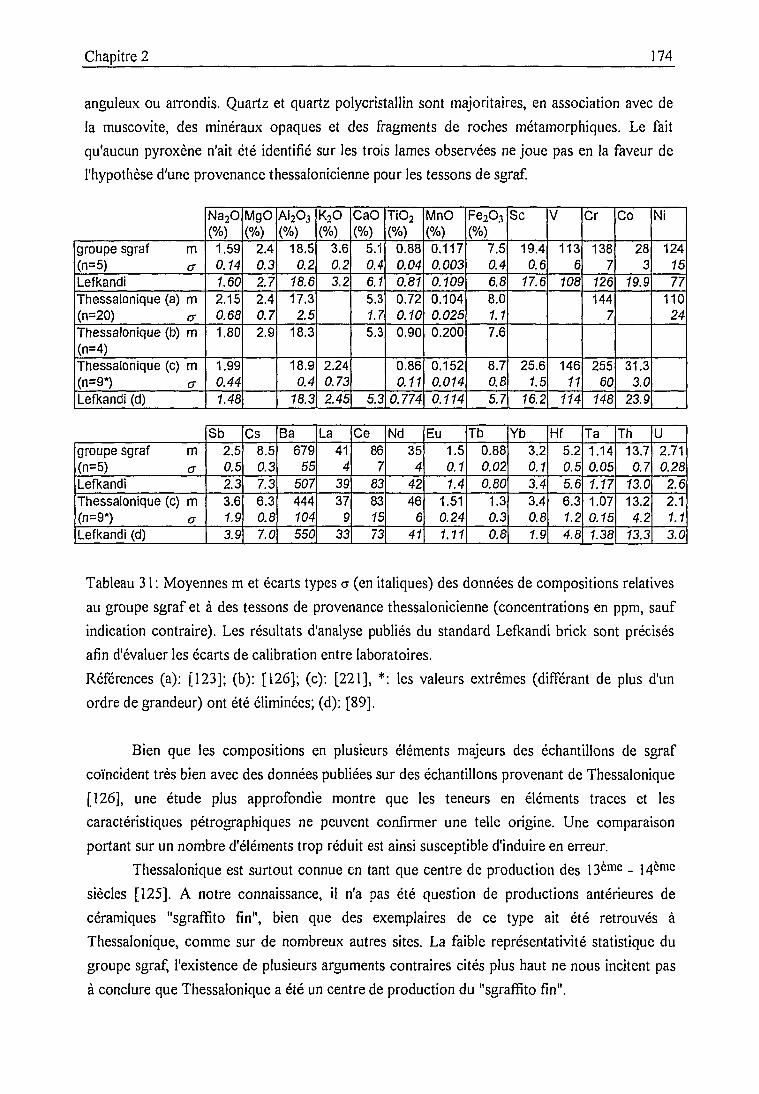
Chapitre 2 174
anguleux ou arrondis. Quartz et quartz polycristallin sont majoritaires, en association avec de
la muscovite, des minéraux opaques et des fragments de roches métamorphiques. Le fait
qu'aucun pyroxene n'ait été identifié sur les trois lames observées ne joue pas en la faveur de
l'hypothèse d'une provenance thessalonicienne pour les tessons de sgraf.
groupe sgraf m(n=5) aLefkandiThessalonique (a) m(n=20) aThessalonique (b) m(n=4)Thessalonique (c) m(n=9*) aLefkandi (d)
Na2O(%)
1.590.141.602.150.681.80
1.990.441.48
MgO(%)
2.40.32.72.40.72.9
AI2O3
(%)18.50.2
18.617.32.5
18.3
18.90.4
18.3
K2O(%)
3.60.23.2
2.240.732.45
CaO(%)
5.10.46.15.31.75.3
5.3
TiO2
(%)0.880.040.810.720.100.90
0.860.11
0.774
MnO(%)0.1170.0030.1090.1040.0250.200
0.1520.0140.114
Fe2O3
(%)7.50.46.88.01.17.6
8.70.85.7
Se
19.40.6
17.6
25.61.5
16.2
V
1136
108
14611
114
Cr
1387
126144
7
25560
148
CO
283
19.9
31.33.0
23.9
Ni
1241577
11024
groupe sgraf m(n=5) a
LefkandiThessalonique (c) m(n=9*) a
Lefkandi (d)
Sb2.50.52.33.61.93.9
Cs8.50.37.36.30.87.0
Ba679
55507444104550
La41
43937
933
Ce86
783831573
Nd354
4246
641
Eu1.50.11.4
1.510.241.11
Tb0.880.020.80
1.30.30.8
Yb3.20.13.43.40.81.9
Hf5.20.55.66.31.24.8
Ta1.140.051.171.070.151.38
Th13.70.7
13.013.24.2
13.3
U2.710.282.62.11.13.0
Tableau 31 : Moyennes m et écarts types a (en italiques) des données de compositions relatives
au groupe sgraf et à des tessons de provenance thessalonicienne (concentrations en ppm, sauf
indication contraire). Les résultats d'analyse publiés du standard Lefkandi brick sont précisés
afin d'évaluer les écarts de calibration entre laboratoires.
Références (a): [123]; (b): [126]; (c): [221], *: les valeurs extrêmes (différant de plus d'un
ordre de grandeur) ont été éliminées; (d): [89],
Bien que les compositions en plusieurs éléments majeurs des échantillons de sgraf
coïncident très bien avec des données publiées sur des échantillons provenant de Thessalonique
[126], une étude plus approfondie montre que les teneurs en éléments traces et les
caractéristiques pétrographiques ne peuvent confirmer une telle origine. Une comparaison
portant sur un nombre d'éléments trop réduit est ainsi susceptible d'induire en erreur.
Thessalonique est surtout connue en tant que centre de production des 13èmc - 14èmc
siècles [125]. A notre connaissance, il n'a pas été question de productions antérieures de
céramiques "sgraffito fin", bien que des exemplaires de ce type ait été retrouvés à
Thessalonique, comme sur de nombreux autres sites. La faible représentativité statistique du
groupe sgraf, l'existence de plusieurs arguments contraires cités plus haut ne nous incitent pas
à conclure que Thessalonique a été un centre de production du "sgraffito fin".

Chapitre 2
On remarquera qu'aucune des argiles turques dont les analyses sont reportées par
Birgùl et al. [161] ne présente des teneurs en Sr comparables à celles des échantillons de sgraf.
Les prélèvements réalisés dans la région de Çanakkale présentent un intérêt particulier,
puisqu'une hypothèse basée sur des considérations archéologiques a proposé cette région
comme origine potentielle d'une production de sgraffito fin [137]. Nous pouvons affirmer qu'il
n'y a pas de correspondance entre le sgraffito fin correspondant à sgraf et le gisement considéré
par Birgiil et al. [161], localisé à Ezine. Ceci ne signifie pas que la région de Çanakkale ne soit
pas une source possible pour une production de sgraffito fin. D'une part, nous ne pouvons pas
évaluer, en l'absence d'autres données, à quel point les prélèvements considérés sont
représentatifs des ressources argileuses de l'ensemble de la région. D'autre part, il est possible
que sgraf ne représente qu'une des catégories de sgraffito fin et que plusieurs ateliers ont
fabriqués ce type de céramiques.
Groupe Itwp
Ce groupe est peu homogène comme en témoigne une tendance à la scission dans les
classifications. Il correspond aux séries archéologiques L, 0 , P et N, qui diffèrent par leurs
traitements de surface (glaçure "de fond" absente ou lacunaire pour L, décor au peigne pour O,
teinte brune distinctive des glaçures pour P). Chacune des trois premières séries n'est
représentée que par deux échantillons, mais ces deux échantillons sont de composition très
similaire. Les tessons de la série N ont par contre des compositions nettement plus variables, et
on peut douter de l'unité de la série. La cohérence du groupe lnop lui-même parait assez
spéculative, impression renforcée par son caractère très composite et le faible nombre
d'échantillons de chaque série. Il est fort probable que des sous-structures apparaîtraient sur un
échantillonnage plus représentatif.
Les tessons du groupe lnop présentent cependant des caractéristiques communes qui
sont propres à ce groupe: abondance des micas (surtout micas blancs) dont l'alignement
résultant du montage des pièces au tour donne un aspect orienté aux pâtes à l'examen
pétrographique, teneurs relativement élevées en Al, K et en éléments ferro-magnésiens (qui en
découlent), richesse en terres rares par rapport aux autres céramiques glaçurées. Ce dernier
trait est plus marqué pour les terres rares légères (La) et pour Eu que pour les terres rares
lourdes comme Yb (figure 63). Les pâtes sont plutôt fines et contiennent outre les micas des
quartz mono- et polycristallins et des minéraux opaques. L'un des tessons (N64) présente une
grande plage singulière (d'environ 2 mm) dans laquelle des plagioclases et des micas sont
cimentés par de la calcite micritique. Les deux tessons de la série P présentent également
quelques particularités. Ils se distinguent par des teneurs en Hf - et dans une moindre mesure
en Zr - plus faibles que celles des autres échantillons de lnop. Par ailleurs, une analyse
qualitative par fluorescence X induite par radioisotope (241Am) des motifs bruns foncés qui
ornent ces tessons montre que Fe et Cu y sont présents en quantités comparables,
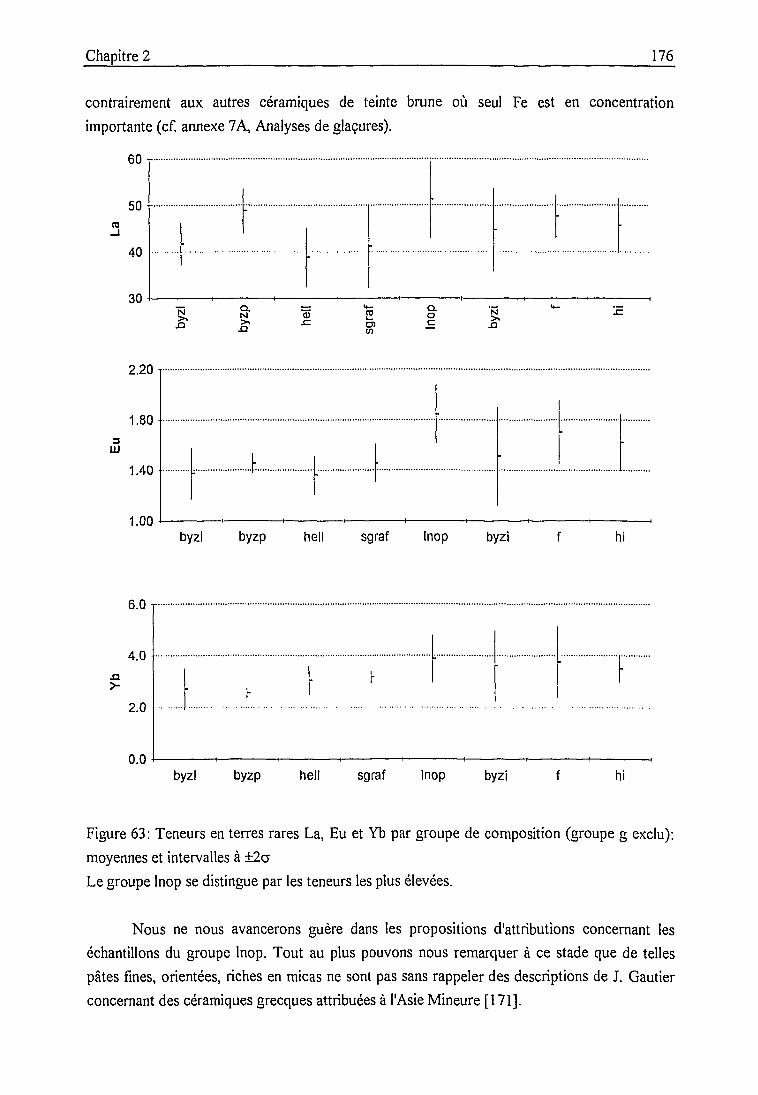
Chapitre 2 176
contrairement aux autres céramiques de teinte brune où seul Fe est en concentration
importante (cf. annexe 7 A, Analyses de glaçures).
60 T
50 -
ra
40
30N>>£1
Q.
g, 2Q.o
2.20 T
1.80
1.40
1.00byzl byzp hell sgraf Inop byzi hi
byzl byzp hell sgraf lnop byzi hi
Figure 63: Teneurs en terres rares La, Eu et Yb par groupe de composition (groupe g exclu):
moyennes et intervalles à ±2o
Le groupe lnop se distingue par les teneurs les plus élevées.
Nous ne nous avancerons guère dans les propositions d'attributions concernant les
échantillons du groupe lnop. Tout au plus pouvons nous remarquer à ce stade que de telles
pâtes fines, orientées, riches en micas ne sont pas sans rappeler des descriptions de J. Gautier
concernant des céramiques grecques attribuées à l'Asie Mineure [171].
(a)(c)(e)
(b)(d)(0
cf. légende page suivante

Chapitre 2 177
Figure 64: Vues de lames minces de pâtes céramiques au microscope polarisant:
(a) Echantillon D74, lumière polarisée et analysée, dimensions du cadre: 3x2 mm.
Assemblage courant dans les pâtes céramiques correspondant à la production byzantine
de Pergame: verres volcaniques, quartz polycristallins, grands cristaux de biotite, feldspath
altéré. Des plagioclases (non représentés) sonr en général également présents, ainsi que de
rares fragments de schistes.
(b) Echantillon A56, lumière polarisée, dimensions du cadre: 3x2 mm.
Fragment de roche volcanique de forme anguleuse, contenant des phénocristaux de
ferromagnésiens (amphiboles) et des minéraux accessoires.
(c) Echantillon G77, lumière polarisée, dimensions du cadre: 3x2 mm.
Texture de la pâte d'une céramique non glaçurée.
(d) Echantillon G77, lumière polarisée et analysée, dimensions du cadre: 3x2 mm.
Roches felsiques et opaques constituent la majorité des inclusions de ces céramiques.
(e) Echantillon D74, lumière polarisée, dimensions latérales de l'inclusion: 0.6 mm.
Cristal de calcite primaire entouré d'un cerne gris.
(f) Echantillon F67, lumière polarisée, dimensions latérales de l'inclusion: 0.7 mm (cliché A.
Leruyct)
Inclusion contenant des baguettes de plagioclases, des cristaux de pyroxenes (cf.
figure 61 ) et de sphène.

Conclusion 178
Conclusion
Par l'analyse élémentaire et l'utilisation de techniques statistiques multivariées, notre
travail a permis d'identifier les catégories de céramiques fabriquées à Pergame. Des groupes de
référence pergaméniens, utilisables pour des études de provenance ultérieures, ont été
constitués et caractérisés par leur composition élémentaire, ainsi que par des données
minéralogiques et pétrographiques.
Les méthodes PIXE et INAA d'analyse élémentaire ont été mises en oeuvre de façon
complémentaire afin de doser une trentaine d'éléments, incluant les éléments majeurs des pâtes
céramiques ainsi qu'un grand nombre d'éléments traces de comportement géochimique
différent. Précision et justesse ont été évalués. Le dosage du standard "Lefkandi brick" analysé
par plusieurs laboratoires a permis d'estimer la capacité de nos résultats a être échangés.
Les concentrations élémentaires obtenues ont été soumises à différentes procédures de
classification afin de distinguer des groupes de composition similaire au sein de
l'échantillonnage. En particulier, l'association d'une classification ascendante hiérarchique à une
analyse par composantes principales préalable peut être avantageuse. L'application de la
classification uniquement aux premières composantes principales permet ainsi de s'affranchir
des corrélations inter-élémentaires.
Quelle que soit la procédure utilisée, six groupes principaux peuvent être définis
compte tenu des propriétés géochimiques des éléments et de la cohérence archéologique.
Trois de ces groupes sont rattachés à la production locale.
Le groupe principal contient la plupart des pièces inachevées, ratés de cuisson et
trépieds qui constituent la référence locale. Il comprend en particulier la série de tessons qui
s'apparentent par leurs motifs au type archéologique "Zeuxippus ware". Ces pièces sont des
imitations locales d'un prototype de qualité supérieure dont les exemplaires les plus
spectaculaires ont été mis au jour à Istanbul et à Chersonese. En effet, la présence à Pergame
de quelques tessons de "véritable Zeuxippus ware", qui se distinguent nettement par leur
composition, supporte l'hypothèse que les ateliers pergaméniens ont subi l'influence stylistique
de ce centre, mais que Pergame ne peut être considérée comme l'origine d'une production de
Zeuxippus ware au sens strict du terme. Le centre de production du ou des prototype(s)
demeure à ce jour encore inconnu. On peut supposer que les imitations fabriquées à Pergame

Conclusion
ont été diffusées à l'échelle régionale, puisque l'on a trouvé des tessons similaires de la même
facture relativement grossière à une centaine de km au Nord-Ouest de Pergame (Gùlpinar).
Un autre groupe local correspond aux tessons de l'atelier hellénistique. Il présente des
compositions élémentaire et minéralogique distinctes de celle du groupe principal. Les
différences observées ne peuvent être attribuées ni à des variations techniques dans le
traitement de l'argile, ni à des altérations, mais impliquent l'utilisation de matières premières
différentes.
Un troisième groupe qui comprend uniquement des céramiques non glaçurées, de pâte
plus grossière, peut être supposé de provenance locale. Des compositions qui correspondent à
celles de roches mères plus acides et des caractéristiques pétrographiques particulières
témoignent de l'exploitation d'un gisement argileux distinct des précédents.
Nous suggérons donc qu'à Pergame plusieurs sources de matières premières ont été
exploitées, selon la catégorie de céramiques et l'époque de fabrication. On peut penser qu'elles
ont été choisies en fonction des impératifs technologiques spécifiques à chaque type de
production. La grande diversité géologique de la région de Pergame donne la possibilité de
réaliser de tels choix. Elle rend également la définition de caractéristiques globales propres aux
argiles de Pergame très malaisée. Il paraît d'autant plus nécessaire d'associer à chaque groupe
de composition la description archéologique des pièces. Des comparaisons ultérieures de
données analytiques aux groupes de référence de Pergame devront reposer sur des
échantillonnages de même nature ou de même tradition technique, sous peine de parvenir à des
conclusions erronées.
L'attribution des céramiques qui ne se rattachent pas à la production locale est plus
délicate du fait de la rareté des données analytiques concernant les céramiques byzantines.
Les quelques tessons de "sgraffito fin" trouvés à Pergame montrent une composition
remarquablement uniforme. Pour ce type de céramiques, produites en quantités importantes au
12cme siècle et diffusées à grande distance par plusieurs ateliers, il est difficile de proposer une
hypothèse de provenance a priori. La comparaison des nos données avec les groupes de
référence de matériel byzantin connus par des études précédentes, toute époque et tout style
confondus, montre une bonne correspondance sur les teneurs en éléments majeurs avec l'atelier
de Thessalonique. Cette coïncidence n'est cependant pas confirmée par les teneurs en éléments
traces, ni par les données pétrographiques. Nous ne pouvons donc pas en conclure que l'atelier
de Thessalonique, connu pour son activité aux 13èmc - 14èmc siècles, ait produit
antérieurement du "sgraffito fin".

Conclusion 180
Deux autres séries de céramiques, de composition proche, peuvent être mises en
relation avec les sites d'Ephèse et de Sardes sur la base de critères archéologiques. En l'absence
d'analyses de matériel contemporain, nous nous sommes référés à des données concernant des
céramiques et des lampes gréco-romaines. Nous n'avons pas pu observer de correspondance
satisfaisante. Cependant, la grande diversité des argiles de la région d'Ephèse ne permet pas
d'exclure ce site comme provenance possible. Par contre, les argiles de Sardes qui montrent
des compositions peu variables à différentes périodes d'exploitation, ne semblent pas
correspondre aux céramiques trouvées à Pergame. Ces résultats demandent à être validés par
des programmes d'intercalibration entre laboratoires et par des études réalisées sur le terrain.
L'étude par des moyens analytiques de la céramique byzantine n'en est qu'à ses débuts.
L'obtention de résultats exploitables du point de vue archéologique est rendue très malaisée du
fait de la rareté des groupes de référence d'ateliers connus. En caractérisant les productions de
Pergame, nous espérons avoir pu contribuer à une meilleure connaissance des relations entre
centres producteurs et sites de consommation au sein du monde byzantin.

Références 181
Références
[I] Morgan, C, Corinth, Results of Excavations, vol.XI: The Byzantine Pottery, Harvard
University Press (1942)
[2] Megaw, A.H.S., Zeuxippus ware, Annual of the British School at Athens, 63, 67-88,
pi. 14-21 (1968)
[3] Lane, A., Medieval finds at Al Mina in North Syria, Archaeologia, 87, 45-53 (1937)
[4] Megaw, A.H.S., An early thirteenth century aegean glazed ware, dans: Studies in Memory
of David Talbot Rice, Edinburgh, 34-45 (1975)
[5] Spieser, J.-M., Die byzantinische Keramik in der Wohnstadtgrabung, Pergamenische
Forschungen, à paraître
[6] Ion beam for material analysis, Bird, J.R., Williams, J.S., éds., Academic Press, New York
(1989)
[7] Johansson, T.B., Akselsson, R., Johansson, S.A.E., X-ray analysis: elemental trace analysis
at the 10"12 g level, Nucl. Instr. andMetk, 84, 141-143 (1970)
[8] Johansson, S.A.E., Campbell, J.L., PIXE: A novel technique for elemental analysis, John
Wiley & Sons, New York (1988)
[9] Bearden, J.A., X-ray wavelengths, Rev. Mod. Phys., 39, 1, 78-124 (1967)
[10] Tertian, R., Claisse, F., Principles of quantitative X-ray fluorescence analysis, Heyden,
Londres (1982)
II1] Krause, M.O., Atomic radiative and radiationless yields for K and L shells, J. Phys. Chem.
Ref. Data, 8,307-327 (1979)
[12] Salem, S.I., Panossian, S.L., Krause, R.A., Experimental K and L relative X-ray emission
rates, At. Data and Nucl. Data Tables, 14, 2, 91-109 (1974)
[13] Scofield, J.H., Relativistic Hartree-Slater values for K and L X-ray emission rates, At.
Data andNucl. Data Tables, 14, 2, 121-137 (1974)
[14] Ion beam handbook for material analysis, Mayer, J.W., Rimini, E., éds, Academic Press,
New York (1977)
[15] Dewey, J.D., Handbook of X-ray and microprobe data, Elion, H.A., Stewart, D.C., éds.,
Pergamon Press, New York (1966)
[16] Evans, R.D., The Atomic nucleus, Me Graw-Hill, New York (1955)
[17] Chu, W.-K., Mayer, J.W., Nicolet, M.A., Backscattering spectrometry, Academic Press,
New York (1978)
[18] Andersen, H.H., Ziegler, J.F., The stopping and ranges of ions in matter, vol. 3,
Pergamon Press, New York (1977)

Références 182
[19] Cohen, D.D., Harrigan, M., K- and L-shell ionization cross sections for protons and
helium ions calculated in the ECPSSR theory, At. Data and Nttcl. Data Tables, 33, 2,
255-343 (1985)
[20] Heitz, C, Costa, G.J., Cailleret, J., Lagarde, G., Sections efficaces de productions de
rayons X-K, L par 'H et 4He, rapport interne CRN-PN, 82-24, Physique Nucléaire,
Centre de Recherches Nucléaires, Strasbourg (1982)
[21] Merzbacher, E., Lewis, H.W., X-ray production by heavy charged particles, Encyclopedia
of Physics, vol. 34, Springer Verlag, Berlin, 166-192 (1958)
[22] Heitz, Ch., Analysis by heavy ion X-ray emission, Progress in Crystal Growth and
Characterization, 8, 131-150 (1984)
[23] Heitz, Ch., Kwadow, M., Tenorio, D., Surface analysis by argon ion induced X-ray
fluorescence, Nucl. Instr. andMeth., 149, 483-488 (1978)
[24] Heitz, C, Lagarde, G., Pape, A., Tenorio, D., Zarate, C, Menu, M., Scotee, L., Jaidar,
A., Alviso, R., Gonzalez, D., Gonzalez, V., Radioisotope induced X-ray emission - a
complementary method to P1XE analysis, Nucl. Instr. andMeth. B, 14, 93-98 (1986)
[25] Gordon, B.M., Kramer, H.W., On the development of a system for trace element analysis
in the environment by charged particle X-ray fluorescence,./. Radioanal. Chem., 12, 181
(1972)
[26] Folkmann, F., Gaarde, C, Huus, T., Kemp, K., Proton Induced X-ray Emission as a tool
for trace element analysis, Nucl. Instr. andMeth., 116, 487-499(1974)
[27] Clayton, E., Uncertainties in theoretical thick target PIXE yields, Nucl. Instr. andMeth.,
191,567-572(1981)
[28] Cahill, T.A., McColm, D.W., Kusko, B.H., Control of temperature in thin samples during
ion beam analysis, Nucl. Instr. andMeth. B, 14, 38-44 (1986)
[29] Cahill, T.A., Kusko, B.H., Schwab, R.N., Analyses of inks and papers in historical
documents through external beam PIXE techniques, Nucl. Instr. andMeth., 181, 205-208
(1981)
[30] Tuurnala, T., Hautojarvi, A., Harva, K., Examination of oil paintings by using a
PIXE/PIGME combination, Nucl. Instr. andMeth. B, 14, 70-75 (1986)
[31] Demortier, G., Review of recent applications of high energy microprobes in arts and
archeology, Nucl. Instr. andMeth. B, 54, 334-345 (1991)
[32] Peisach, M., Pineda, C.A., Jacobson, L., Thick target PIXE analysis of coastal and inland
Namibiam pottery, Nucl. Instr. andMeth. B, 49, 309-312 (1991)
[33] Boulle, G.J., Peisach, M., Trace element analysis of archaeological materials and the use
of pattern recognition methods to establish identity, J. Radioanal. Chem., 50, 1-2, 205-
215(1979)
[34] Duerden, P., Clayton, E., Bird, J.R., Cohen, D.D., Recent ion beam analysis studies on
archaeology and art, Nucl. Instr. andMeth. B, 14, 50-57 (1986)

Références 183
[35] Fleming, S.J., Swann, C.P., Recent applications of PIXE spectrometry in archaeology
II.Characterization of Chinese pottery exported to the Islamic world, Nucl. Instr. and
Math. B, 64, 528-537 (1992)
[36] Houdayer, A., Lessard, L., Brissaud, I., Quantitative PIXE analysis of powdered pottery
samples bound in an organic matrix, Nucl. Instr. andMeth. B, 3, 412-415 (1984)
[37] Baijot-Stroobants, J., Bodart, F., Ancient pottery analysis by proton induced
bombardment and Mossbauer spectroscopy, Nucl. Instr. andMeth., 142, 293-300 (1977)
[38] Lagarde, G., Brissaud, I., Cailleret, J., Fontes, P., Heitz, Ch., Thick target PIXE analysis
of clays for archaeological studies, Nucl. Instr. andMeth. B, 1, 45-50 (1984)
[39] Lagarde, G., Cailleret, J., Heitz, C , Quantitative analysis of thick targets containing light
elements (Z < 12) by PIXE, Nucl. Instr. andMeth., 205, 545-553 (1983)
[40] Clayton, E., PIXAN - The Lucas Heights PIXE analysis package, Australian Atomic
Energy Report, AAEC-M113 (1986)
[41] Clayton, E., BATTY83: a computer program for thick target PIXE analysis, Nucl. Instr.
andMeth., 218, 221-224 (1983)
[42] Clayton, E., Cohen, D.D., Duerden, P., Thick target PIXE analysis and yield curve
calculations, Nucl. Instr. andMeth., 180, 541-548 (1981)
[43] Bird, J.R., X-ray spectrum processing, interpretation and computerisation in Amsel, G.,
Bird, J.R., Heitz, Ch., Lahanier, Ch., Menu, M., Overview of the discussions at the
Premontres workshop, Nucl. Instr. andMeth. B, 14, 162-167 (1986)
[44] Watjen, U., Currently used computer programmes for PIXE analysis, Nucl. Instr. and
Meth.B, 22, 29-33(1987)
[45] Campbell, J.L., Maenhaut, W., Bombelka, E., Clayton, E., Malmqvist, K., Maxwell, J.A.,
Pallon, J., Vandenhaute, J., An intercomparison of spectral data processing techniques in
PIXE, Nucl. Instr. andMeth. B, 14, 204-220 (1986)
[46] Bearat, H., Etude de quelques altérations physico-chimiques des céramiques
archéologiques, Thèse de doctorat, Université de Caen (1990)
[47] Vitali, V., Franklin, U.M., Hancock, R.G.V., La stabilité des céramiques par rapport à
l'environnement, Revue d'Archéométrie, 8, 41-44 (1984)
[48] Lemoine, C, Meille, E., Poupet, P., Barraudon, J.N., Borderie, B., Etude de quelques
altérations de composition chimique de céramiques en milieu marin et terrestre, Actes du
XXème symposium international d'archéométrie, Revue d'Archéométrie, supplément
1981, vol III, 349-360(1981)
[49] Lemoine, C , Picon, M., La fixation du phospore par les céramiques lors de leur
enfouissement et ses incidences analytiques, Revue d'Archéomélrie, 6, 101-112 (1982)
[50] Picon, M., La fixation du baryum et du strontium par les céramiques, Revue
d'Archéométrie, 11,41-47(1987)
[51] Handbook of Geochemistry, Wedepohl, K.H., éd., Springer Verlag, Berlin (1978)

Références 184
[52] Attas, M., Fossey, J.M., Yaiïe, L., Corrections for drill-bit contamination in sampling
ancient pottery for neutron activation analysis, Archaeometry, 26, 1, 104-107 (1984)
[53] Manuel d'utilisation et de programmation des cartes TMCA2-TISA-LJSA, Aries target
system electronic (1991)
[54] Geostandard Newsletters, 13, Special Issue (1989)
[55] Liu-Xu, X., Conception et réalisation d'un instrument de mesure "EDXRF" utilisant des
détecteurs Hgl2, Thèse de doctorat, Université Strasbourg I (1994)
[56] Hansen, J.S., McGeorge, J.C., Nix, D., Schmidt-Ott, W.D., Unus, I., Fink, R.W.,
Accurate efficiency calibration and properties of semiconductor detectors for low-energy
photons, Nucl. Instr. and Me th., 106, 365-379 (1973)
[57] Cohen, D.D., A radially dependent photopeak efficiency model for Si(Li) detectors, Nucl.
Instr. andMeth., 178, 481-490 (1980)
[58] Aspiazu, J., Mise au point d'un programme informatique pour l'analyse quantitative pat-
la méthode FIXE, Thèse de doctorat, Université Strasbourg l (1991)
[59] PAW - Physics Analysis Workstation, CERN, Geneva, Switzerland, version 2.02/06
(1993)
[60] Cohen, D.D., Harrigan, M., Calculated L-shell X-ray line intensities for proton and helium
ion impact, At. Data andNucl. Data Tables, 34, 3, 393-414 (1986)
[61] Maxwell, J.A., Campbell, J.L., Teesdale, W.J., The Guelph PIXE software package, Nucl.
Instr. andMeth. B, 43, 218-230 (1989)
[62] Sekine, T., Baba, H., A measure of the degree of fit between the calculated and observed
spectra, Nucl. Instr. andMeth., 133, 171-173 (1976)
[63] Isozumi, Y., Uncertainties in the y} fit for nuclear radiation spectra, Nucl. Instr. and
Meth. A, 235, 164-173(1985)
[64] Currie, L.A., Limits for qualitative detection and quantitative determination, Anal. Chem,
40,586-593(1968)
[65] Theisen, R., Vollath, D., Tables of X-ray mass attenuation coefficients, Verlag
Stahleisen, M.B.H. Dusseldorf (1967)
[66] Jones, R.E., Greek and Cypriot pottery: A review of scientific studies, Fitch Laboratory
Occasional Paper, 1, British School at Athens, Athènes (1986)
[67] Harbottle, G., Provenience studies using neutron activation analysis: the roie of
standardization, dans: Archaeological ceramics, Olin, J.S., Franklin, A.D., éds.,
Smithsonian Institution Press, Washington, D.C., 67-77 (1982)
[68] Yellin, J., Perlman, I., Asaro, F., Michel, H.V., Mosier, D.F., Comparison of neutron
activation analysis from the Lawrence Berkeley Laboratory and the Hebrew University,
Archaeometry, 20, 1, 95-100 (1978)
[69] Sabir, A., Etude de justesse et de précision dans une méthode automatique d'analyse par
activation neutronique non destructive. Cas des matrices siliceuses. Application à la

Références 185
recherche de l'origine de céramiques archéologiques, Thèse de Doctorat, Université
Paris-Sud Orsay (1986)
[70] Kadom-Al-Neami, A., Mesure des sections efficaces de production des rayons XL des
éléments 56Ba, 57La et 5sCe par des protons de 1 MeV -3.5 MeV, Thèse de doctorat,
Université Strasbourg I (1988)
[71] Fontes, P., Brissaud, I., Lagarde, G., Leblanc, J., Person, A., Saliège, J.-F., Heitz, Ch.,
PIXE analysis and provenance study of archaeological potsherds from West Africa, Nucl.
Instr. andMeih. B, 3, 404-407 (1984)
[72] Parry, S., Activation spectrometry in chemical analysis, Chemical Analysis, 119, Wiley-
Interscience, New York (1991)
[73] Perlman, I., Asaro, F., Pottery analysis by neutron activation, Archaeometry, 11, 21-52
(1969)
[74] Harbottle, G., Activation analysis in archaeology, Radiochemistry, 3, 33-72 (1976)
[75] Kuleff, I., Djingova, R., Activation analysis in archaeology, dans: Activation analysis,
Alfassi, Z.B., éd., CKC-Press, Florida, vol.2, 427-489 (1990)
[76] De Soete, D., Gijbels, R., Hoste, J., Neutron activation analysis, Chemical Analysis, 34,
Wiley-Interscience, New York (1972)
[77] Alfassi, Z.V., Lavi, N., Simultaneous determination of sodium, magnesium, aluminium,
silicon and phosphorus by instrumental neutron activation analysis using reactor and
epithermal neutrons, Analyst, 109, 959-962 (1985)
[78] Penev, I., KulefY, I., Djingova, R., Simultaneous determination of aluminium, magnesium,
and silicon in rocks, glasses, and pottery, J. Radioanal. Nucl. Chem. Letters, 93, 3, 219-
232(1985)
[79] Erdtmann, G., Soyka, W., Gamdat'78, Zentralabteilung fur Chemische Analysen der
Kernforschungsanlage Julich GmbH (1978)
[80] Williams, C.T., Wall, F., An INAA scheme for the routine determination of 27 elements in
geological and archaeological samples, dans: Neutron activation and plasma emission
spectrometry analysis in archaeology. Techniques and applications, Hughes, M.J., Cowell,
M.R., Hook, D.R., éds., British Museum occasional paper, 82, 105-119 (1991)
[81] Gilmore, G.R., Sources of uncertainty in the neutron activation analysis of pottery, dans:
Neutron activation and plasma emission spectrometry analysis in archaeology. Techniques
and applications, Hughes, M.J., Cowell, M.R., Hook, D.R., éds., British Museum
occasional paper, 82, 105-119 (1991)
[82] Rossini, I., Application de l'analyse multiélémentaire par fluorescence X et activation
neutronique à la caractérisation d'un site archéologique, Thèse de doctorat, Strasbourg I
(1991)

Références 186
[83] Carrot, F., Dardenne, C, Deschamps, N., Lahanier, C, Revel, G., Determination of
optimum conditions for INAA of archaeological clay figurines, ./. Radioanal. Nitcl.
Chem., 168, 2, 287-296 (1993)
[84] Hiver, P., Rapport de stage de CES, Réacteur Universitaire, Université Strasbourg I
(1994)
[85] GANAAS/SPEDAC, Nuclear analysis software, IAEA, Vienne (1991)
[86] Oweijane, F., Analyse de céramiques byzantines par fluorescence X induite par le
rayonnement y de 59.5 keV de ^^m et comparaison des résultats avec ceux obtenus par
PfXE et par AAN, Rapport de stage de DEA de physique nucléaire, Université
Strasbourg 1(1994)
[87] Picon, M., Vichy, M., Meille, E., Composition of the Lezoux, Lyon and Arezzo Samian
ware, Archaeometry, 13, 2, 191-208 (1971)
[88] Articles dans: La ceramica médiévale net Medilerraneo Occidentale, Florence (1986); La
céramique médiévale de la Méditerranée Occidentale, Paris (1980)
[89] Djingova, R., Kuleff, I., Penev, I., Instrumental neutron activation analysis of reference
materials for archaeometric investigations of pottery,./. Radioanal. Nucl. Chem. Letters,
144,6,397-406(1990)
[90] Schneider, G., Wirz, E., Chemical answers to archaeological questions - Roman terracotta
lamps as documents of economic history, Documents et travaux IGAL, 16, 13-48 (1992)
[91] Hughes, M.J., communication personnelle
[92] Rice, P.M., Pottery analysis. A sourcebook, The University of Chicago Press, Chicago
(1987)
[93] Walter, V., Etude pétrographique, minèralogique et géochimique d'amphores gauloises
découvertes dans le Nord-Est de la France. Contribution à l'étude des matériaux, des
technologies et des provenances, Thèse de doctorat, Université Strasbourg II (1988)
[94] Attas, M., Analyse par activation neutron/que de la céramique de Lerne (Grèce) à l'âge
du bronze ancien: productions locales et échanges commerciaux, Thèse de 3 è m c cycle,
Université Paris-Sud Orsay (1980)
[95] Colloque sur le vocabulaire technique céramique en archéologie, Compte rendu, Annales
duLRMF, 74-79(1971)
[96] Mason, R.B., Tite, M.S., The beginnings of stonepaste technology, Archaeometry, 36, 1,
77-91 (1994)
[97] Picon, M., Introduction à l'étude technique des céramiques sigillées de Lezoux, Centre de
Recherches sur les Techniques Gréco-Romaines, 2, Université de Dijon (1973)
[98] Echallier, J.C., Eléments de technologie céramique et d'analyse des terres cuites
archéologiques, Documents d'archéologie méridionale, Série méthodes et techniques, 3,
5-38(1984)
[99] Caillère, S., Hénin, S., Minéralogie des argiles, Masson, Paris (1968)

Références 187
[100] Millot, G., Géologie des argiles, Masson, Paris (1964)
[101] Deer, W.A., Howie, R.A., Zussman, J., Rock forming minerals, vol.3: Sheet silicates,
Longmans, Londres(1962)
[102] Tite, M.S., Freestone, I.C., Meeks, N.D., Bimson, M. The use of the scanning electron
microscopy in the technological examination of ancient ceramics, dans: Archaeological
ceramics, Olin, J.S., Franklin, A.D., éds., Smithsonian Institution Press, Washington,
D.C., 109-120(1982)
[103] Vandiver, P., Les glaçures des céramiques anciennes, Pour la science, 152, 78-86
(1990)
[104] Rafaillac-Desfosses, C, Céramiques glaçurées médiévales. Recherche de données
physiques sur les techniques de fabrication et sur l'altération, Thèse de doctorat,
Université Bordeaux fil (1994)
[105] Picon, M., La céramologie de laboratoire, Nouvelles de l'archéologie, 1, 37-45 (1979)
[106] Widemann, F., L'analyse par activation neutronique des céramiques anciennes:
groupements et différenciation, Latomus, 160, 47-71 (1979)
[107] Catling, H.W., Richards, E.E., Blin-Stoyle, A.E., Correlation between composition and
provenance of Mycenaean and Minoan pottery, Annual of the British School at Athens,
58,94-115(1963)
[108] Picon, M., Le traitement des données d'analyse, PACT, 10, 379-399 (1984)
[109] Eberhart, J.-P., Analyse structurale et chimique des matériaux, Dunod, Paris (1989)
[110] Roubault, M., Détermination des minéraux des roches au microscope polarisant,
Lamarre-Poinat, Paris (1982)
[111] Shepard, A.O., Ceramics for the archaeologist, Carnegie Institution of Washington,
Washington, D.C. (1965)
[112] Courtois, L., Examen au microscope pétrographique des céramiques archéologiques,
Notes et monographies techniques, C.R.A., 8 (1976)
[113] Maggetti, M., Phase analysis and its significance for technology and origin, dans:
Archaeological ceramics, Olin, J.S., Franklin, A.D., éds., Smithsonian Institution Press,
Washington, D.C., 121-133 (1982)
[114] Schmitt, A., Apports et limites de la pétrographie quantitative: application au cas des
amphores de Lyon, Revue d'Archéométrie, 17, 51-63 (1993)
[115] Echallier, J.C., Jallot, L., Le matériel céramique de Moulin Villard (Caissargues, Gard).
Remarques sur la formalisation et l'analyse des données provenant d'observations
microscopiques et résultats, Revue d'Archéométrie, 16, 71-87 (1992)
[116] Méry, S., Emergence et développement de la production céramique de la péninsule
d'Oman à l'âge du bronze en relation avec l'Asie moyenne, Thèse de doctorat, Université
Paris I, 2 volumes (1991)

Références 188
[117] Day, P., Technology and ethnography in pétrographie studies of ceramics, dans:
Archaeometry, Proc. 25 th Int. Archaeometry Symposium (Athens 1986), Maniatis, Y.,
éd., 139-147(1989)
[118] Recherches sur la céramique byzantine, Actes du colloque organisé par l'école française
d'Athènes et l'université de Strasbourg II, Athènes 8-10 avril 1987, Déroche, V., Spieser,
J.-M., éds., Bulletin de correspondance hellénique, supplément XVIII (1989)
[119] Spieser, J.-M., La céramique byzantine médiévale, dans: Hommes et richesses de
l'Empire byzantin, II, VIIIe - XVe siècle, Kravari, V., Lefort, J., Morrisson, C, éds.,
Réalités byzantines, 3, Paris (1989)
[120] Spieser, J.-M., Le monde byzantin. La céramique: nouvelles approches, dans: Le grand
allas de l'archéologie, Encyclopaedia Universalis, 144-145 (1985)
[121] Talbot Rice, D., Byzantine glazed pottery, Clarendon Press, Oxford (1930)
[122] Talbot Rice, D., The Pottery of Byzantium and the Islamic World, dans: Studies in
Islamic Art and Architecture in honour of K.A.C. Creswell (Mélanges Creswell), Le
Caire, 194-236(1965)
[123] Megaw, A.H.S., Jones, R.E., Byzantine and allied pottery: a contribution by chemical
analysis to the problem of origin and distribution, Annual of the British School at Athens,
78, 235-263, pi. 24-30(1983)
présentation préliminaire dans:
Megaw, A.H.S., Jones, R.E., Spectrographic analyses of Byzantine and allied pottery,
Jahrbuch der Osterreichischen Byzantinistik, 32, 3, 577-585 (1982)
aussi cité dans:
Jones, R.E., Greek and Cypriot Pottery: a review <.-/ scientific studies, Fitch Laboratory
Occasional Paper, 1, British School at Athens, Athènes (1986)
[124] Herrin, J., La céramique, dans: Splendeur de Byzance, catalogue d'exposition Europalia
1982, Bruxelles, Musées royaux d'art et d'histoire, 225-239 (1982)
[125] Bakirtzis, C, Papanikola-Bakirtzis, D., De la céramique byzantine en glaçure à
Thessalonique, Byzanlino-Bulgarica, VII, 421-434 (1981)
[126] Lazzarini, L., Calogero, S., Early local and imported sgraffito ware in Venice: a
characterization and provenance study, dans: Archaeometry, Proc. 25th Int. Archaeometry
Symposium (Athens 1986), Maniatis, Y., éd., 571-584 (1989)
[127] Calogero, S., Lazzarini, L., Caratterizzazione Chimico-Fisica di Ceramiche Graffite
Bizantine e Veneziane Arcaiche trovate nella Laguna Veneta, Faenza, 69, 60-70, pi. 11
(en italien) (1983)
description des échantillons dans:
Lazzarini, L., Canal, E., Ritrovamenti di Ceramica Graffita Bizantina in Laguna e la
Nascita del Graffito Veneziano, Faenza, 69, 19-59, pi. 2-10 (en italien) (1983)

Références 189
[128] Lazzarini, L., Calogero, S., Burriesci, N., Petrera, M., Chemical, mineralogical and
Môssbauer studies of Venetian and Paduan Renaissance sgraffito ceramics, Archaeometry,
22, 1,57-68(1980)
[129] KulefF, I., Djingova, R., Djingov, G., Provenance study of sgraffito ceramics (XIII-
XIV c.) from Shumen, Varna and Tcherven (North-Eastern Bulgaria), dans:
Archaeometry, Proc. 25th Int. Archaeometry Symposium (Athens 1986), Y. Maniatis, éd.,
533-542(1989)
description des échantillons et résultats détaillés dans:
Kuleff, I., Djingova, R., Penev, I., Chemische zusammensetzung des mittelalterichen
sgraffitokeramiks III. Eine untersuchung von archaeologischen funde aus Schumen und
Tscherven (12 - 14 jh.), ,/ahrbuch der Museen in Nordbulgarien, 17, 289-311 (en bulgare,
résumé allemand) (1991)
[130] Kuleff, I., Djingova, R., Djingov, G., Provenance study of pottery by INAA, dans: Proc.
7th Int. Conf. Modern Trends in Activation Analysis, Copenhague, 787-791 (1986)
description des échantillons et résultats détaillés dans:
Djingova, R., Kuleff, I., Penev, I., Chemical content of medieval sgraffito ceramic findings
II. Investigation of archaeological findings from Kalikra, Kavarna and Baltchik (XII -
XIV a), Izv. Archaeol. Museum, Varna (en bulgare, résumé anglais) (sous presse)
et:
Kuleff, I., Djingova, R., Djingov, G., Ann. Univ. Sofia, Fac. Chim., 81 (en bulgare) (sous
presse)
[131] Kuleff, I., Djingova, R., Penev, I., Instrumental Neutron Activation Analysis of pottery
for provenience study,./. Radioanal. Mud. Chem. Articles, 99, 2, 345-358 (1986)
[132] Djingova, R., KulefF, I., Provenance study of pottery and glass by INAA, dans: Neutron
Activation and Plasma Emission Spectrometric Analysis in Archaeology, Techniques and
Applications, Hugues, M.J., Cowell, M.R., Hook, D.R., éds., British Museum occasional
paper, 82, 121-141 (1991)
[133] Sanders, G.D.R., Excavations at Sparta: the Roman stoa, 1988-91. Preliminary report,
part 1, Medieval pottery, Annual of the British School at Athens, 88, 251-286, pi. 23-26
(1993)
[134] Armstrong, P., Byzantine Thebes: excavations on the Kadmeia, 1980, Annual of the
British School at Athens, 88, 295-335, pi. 29-36 (1993)
[135] Vassi, 0., An unglazed ware pottery workshop in twelfth-century Lakonia, Annual of
the British School at Athens, 88, 287-293, pi. 27-28 (1993)
[136] François, V., La céramique byzantine de Thasos et un échantillonnage des productions
médiévales orientales et occidentales du XIHe au XVe siècle, Thèse de doctorat,
Université Strasbourg II, 3 volumes (1990)

Références 190
[137] Hayes, J.W., Excavations al Saraçhane in Istanbul, vol. 2: The Pollery, Princeton
University Press (1992)
[138] Aslanapa, O., Yetkin, S., Altun, A., The Iznik tile kilns excavations (The second round:
1981-1988), Istanbul (1989)
[139] La ceramica ne/ monda bizantino Ira XI e XV se cola e suai rapport i con I'Italia,
Gelichi, S., éd., Florence (1993)
[140] Terres secrètes de Samarcande, céramiques du VIIIe au XIIIe siècle, catalogue
d'exposition, Paris, Institut du Monde Arabe (1992)
[141] Nikolakopoulos, G., Considérations sur l'esthétique de la céramique byzantine, dans:
Recherches sur la céramique byzantine, Déroche, V., Spieser, J.-M., éds., Bulletin de
correspondance hellénique, supplément XVIII, 317-326 (1989)
[142] Vogt, C, Les arts de la table à Byzance, dans: Art byzantin, Les dossiers d'archéologie,
176,40-43(1992)
[143] Scott, J.A., Kamilli, D.C., Late Byzantine glazed pottery from Sardis, Actes du XVe
Congrès International des Etudes Byzantines (Athènes 1976), II, Athènes, 679-696
(1981)
[144] Wisseman, S., The materials analysis of Byzantine pottery, dans: Papanikola-Bakirtzis,
D., Dauterman Maguire, E., Maguire, H., Ceramic art from Byzantine Serres, Illinois
Byzantine Studies, 3, 66-69 (1992) (rapport préliminaire)
[145] Megavv, A.H.S., Zeuxippus ware again, dans: Recherches sur la céramique byzantine,
Déroche, V., Spieser, J.-M., éds., Bulletin de correspondance hellénique, supplément
XVIII, 277-289(1989)
[146] Yilmaz, Y., Professseur à la Istanbul Teknik Universitesi, communication personnelle
[147] Waksman, S. Y., Pape, A., Heitz, C, FIXE analysis of Byzantine ceramics, Nucl. Inslr.
andMelh. B, 85, 824-829 (1994)
[148] Waksman, S.Y., Rossini, I., Heitz, C, Byzantine Pergamon: characterization of the
ceramics production centre, hoc. 29th Int. Archaeometry Symposium (Ankara 1994) (à
paraître)
[149] Kamilli, D.C., Mineral Analysis of the Clay Bodies, dans: Ramage, A., Lydian houses
and architectural terracottas, Archaeological Exploration of Sardis, Monograph 5,
Harvard University Press, Cambridge Mass., 12-14 (1978)
[150] Empereur, J.-Y., Picon, M., A propos d'un nouvel atelier de "Late Roman C", Figlina,
7, 143-146(1986)
[151] Mayet, F., Picon, M., Une sigillée phocéenne tardive ("Late Roman C ware") et sa
diffusion en Occident, Figlina, 7, 129-142 (1986)
[152] D'Ambrosio, B., Mannoni, T., Sfrescola, S., Stato délie richerche mineralogiche sulle
ceramiche mediterranee, dans: La ceramica médiévale ne/ mediterraneo occidentale,
Florence, 601-609(1986)

Références 191
[153] Marinoni, T., Provenienze ed analisi petrografiche interpretate. L'esempio delle
ceramiche bizantine, dans: La ceramica nel mondo bizantino Ira XI e XV secolo e suoi
rapporti con l'Italia, Gelichi, S., éd., Florence, 341-345 (1993)
[154] Frierman, J.D., The physical and chemical properties of some crusaders ceramics, Israel
Exploration Journal, 17, 153-157(1967)
[155] Schneider, G., communication personnelle
[156] Yellin, J., communication personnelle
[157] Boas, A., The import of Western ceramics to the Latin kingdom of Jerusalem, Israel
Exploration Journal, 44, 1-2 (1994)
[158] Day, P., communication personnelle
[159] Parman, E., The pottery from Saint John's Basilica at Ephesos, dans: Recherches sur la
céramique byzantine, Déroche, V., Spieser, J.-M., éds., Bulletin de correspondance
hellénique, supplément XVIII, 277-289 (1989)
[160] Bakirer Ö., The Medieval pottery and baked clay objects, dans: Korucutepe, Final
Report on the Excavation of the Universities of Chicago, California (Los Angeles) and
Amsterdam in the Keban Resen'oir, Eastern Anatolia 1968-1970, Studies in ancient
civilization, vol. 3, 189-249 (1980)
[161a] Birgül, 0., Diksic, M., Yaffe, L., Activation analysis of Turkish and Canadian clays and
Turkish pottery,./. Radioanal. Chem., 39, 45-62 (1977)
[161] Birgül, 0., Diksic, M., Yaffe, L., X-ray fluorescence analysis of Turkish clays and
pottery, Archaeometry, 21, 2, 203-218 (1979)
[162] Barnes, I.L., Brill, R.H., Deal, E.C., Piercy, G.V., Lead isotope studies of some of the
finds from the Serçe Liman shipwreck, Proc. 24th Intern. Archaeometry Symposium, Olin,
J.S., Blackman, M.J., éds., Smithsonian Inst. press, Washington D.C., 1-12 (1986)
[163] Coche de la Ferté, E., Décors en céramique byzantine au musée du Louvre, Cahiers
archéologiques, 9, 187-217 (1957)
[164] Radt, W., Pergamon, DuMont Buchverlag, Cologne (1988)
[165] Foss, C, Byzantine and Turkish Sardis, Archaeological Exploration of Sardis,
monograph 4, Harvard University Press, Cambridge Mass. (1976)
[166] Pergamenische Forschungen, Berlin
voir par exemple plusieurs articles dans le volume 1 (Band I, 1972), ainsi que:
Band II: Hellenistische Keramik aus Pergamon, Schäfer, J. (1968)
Band III: Terrakotten aus Pergamon, Töpperwein, E. (1976)
voir aussi, dans la collection Altertümer von Pergamon, Band XIV, Keramik (1984)
[167] Erdemgil, S., Le quartier des potiers à Pergame, Le monde de la bible, 11, 33-34 (1992)
[168] Erdemgil, S., Ozenir, S., Preliminary report on the kilns excavated in Ketios valley,
Rivista di Archaeologia, 6, Tecnologia nell'Antichità, 2, 109 (1982)
[169] Picon, M., communication personnelle

Références 192
[170] Pinkwart, D., Hellenistisch-rômische Bleiglasurkeramik aus Pergamon, Pergamenische
Forschungen, I, 140-163 (1972),
[171] Gautier, J., Myrina: centre de production ou de consommation?, dans: Etudes
céramiques en archéologie, Documents et Travaux IGAL, Paris, 9, 27-93 (1985)
[172] Kazanski, M., Morrisson, C, Sodini, J.-P., Le monde byzantin. La culture matérielle:
productions et échanges, dans: Le grand allas de l'archéologie, Encyclopaedia
Universalis, 138-139(1985)
[173] Dupont, P., Classification et provenance des céramiques orientales d'Istros, Dacia, 27,
1/2, 19-43(1983)
[174] Sauer, R., Outschar, U., Schneider, G., Combined petrography and geochemical studies
in the determination of pottery production centers - 'the case of Ephesos'
et:
Sauer, R., Outschar, U., Schneider, G., Late Hellenistic and Roman ceramics along the
western coast of Asia Minor: local production centers - historical and archaeological
requirements
tous deux à paraître dans: Proc. 29 th Int. Archaeometry Symposium (Ankara 1994)
[175] Spieser, J.-ML, Informatique et céramique: l'exemple de Pergame, dans: Recherches sur
la céramique byzantine, Déroche, V., Spieser, J.-M., éds., Bulletin de correspondance
hellénique, supplément XVIII, 291-302 (1989)
voir aussi:
Spieser, J.-M., Céramique byzantine de Pergame, Jahrbuch der Osterreichischen
Byzantinistik, 32/3, 561-576 (1982)
[176] Archéologie et calcul, 10/18, Paris (1978)
[177] Armstrong, P., Some Byzantine and later settlements in Eastern Phokis, Annual of (he
British School at Athens, 84, 1-47, pi. 1-11 (1989)
[178] Yenisehirlioglu, F., La céramique glaçurée de Gùlpinar, dans: Recherches sur la
céramique byzantine, Déroche, V., Spieser, J.-M., éds., Bulletin de correspondance
hellénique, supplément XVIII, 303-315 (1989)
[179] Spieser, J.-M., Etude de la céramique byzantine d'Asie Mineure, Rapport des missions
effectuées en 1983, Activité byzantine, Fondation Européenne de la Science, vol. II, Paris,
307-329(1987)
[180] Pollard, A.M., Data analysis, dans: Jones, R.E., Greek and Cypriot Pottery: a review of
scientific studies, Fitch Laboratory Occasional Paper, 1 (1986)
[181] Bouroche, J.-M., Saporta, G., L'analyse des données, P.U.F., Collection "Que sais-je?"
(1989)
[182] Lebart, L., Morineau, A., Fénelon, J.-P., Traitement des données statistiques. Méthodes
et programmes, Dunod, Paris (1982)

Références 193
[183] Peisach, M., Ion beam analytical methods for attempts to establish the provenance of
archaeological objects in the absence of reference artefacts, Nucl Instr. and Math. B, 14,
99-115(1986)
[184] Roux, M., Algorithmes de classification, Masson, Paris (1985)
[185] STATGRAPHICS, STSC, Inc., Rockville MD 20852 (1988)
[186] Lebart, L., Morineau, A., Lambert, T., Pleuvret, P., SPAD.N, CISIA, Saint-Mandé
(1991)
[187] Ricq, J.C., Essai de sélection des éléments chimiques particulièrement propres à
caractériser un groupe de céramiques, Notes et Monographies Techniques du C.R.A., 5
(1974)
[188] Diebolt, J., Ricq, J.C., An attempt to select suitable elements to characterize an ancient
ceramic by neutron activation analysis, J. Radioanal. Nucl. Chem., 39, 9-20 (1977)
[189] Dufournier, D., Etude comparative de plusieurs tessons d'une même poterie,
Archéologie Médiévale, 2, 305-323 (1972)
[190] Franklin, U.M., Hancock, R.G.V., The influence of post-burial conditions on trace
element composition of ancient sherds, Actes du XXème symposium international
d'archéométrie, Revue d'Archéomélrie, supplément 1981, vol III, 111-119 (1981)
[191] Picon, M., L'analyse par activation neutronique est-elle la meilleure méthode que l'on
puisse employer pour déterminer l'origine des céramiques?, Revue d'Archéométrie, 15, 95-
101 (1991)
[192] Peisach, M., Jacobson, L., Boulle, G.J., Gihwala, D., Underhill, L.G., PIXE analysis of
archaeological material and the use of multivariate analysis for characterisation, Nucl.
Instr. andMeth., 193, 337-341 (1982)
[193] Beier, T., Mommsen, H., Modified Mahalanobis filters for grouping pottery by chemical
composition, Archaeometry, 36, 2, 287-306 (1994)
[194] Mommsen, H., Kreuser, A., Weber, J., A method for grouping pottery by chemical
composition, Archaeometry, 30, 1, 47-57 (1988)
[195] Picon, M., Quelques observations complémentaires sur les altérations de composition
des céramiques au cours du temps: cas de quelques alcalins et alcalinos-terreux, Revue
d'Archéométrie, 15, 117-122 (1991)
[196] Besnus, Y., Etude géochimique sur des faciès marnes noires du Trias du Sud-Est du
Massif Central (France), Sciences de la Terre, 25, 2, 219-239 (1982)
[197] Leruyet, A., Etude pétrographique, minéralogique et géochimique de céramique
byzantine provenant d'Asie Mineure. Contribution à l'étude des matériaux et de leurs
provenances, Rapport de stage de maîtrise de géologie, Institut de Géologie, EOPGS,
Strasbourg (1994)

Références 194
[198] Echallier, J.-C, Méry, S., L'évolution minéralogique et physico-chimique des pâtes
calcaires au cours de la cuisson: expérimentation en laboratoire et application
archéologique, Documents et Travaux IGAL, 16, 87-120 (1992)
[199] Geological map of Turkey, Izmir, 500.000èinc, Mineral Research and Exploration
Institute (M.T.A.), Ankara (1973)
[200] Sauer, R., communication personnelle
[201] Attas, M., Yaffe, L., Fossey, J.M., Neutron activation analysis of early bronze age
pottery from lake Vouliagmeni, Perakhora, central Greece, Archaeometry, 19, 1, 33-43
(1977)
[202] Carte géologique de la Turquie, feuillet Balikesir (G4), 100.000ènic, Minerai Research
and Exploration Institute (M.T.A.), Ankara (1989)
[203] Dikili-Bergama, Jeotermal Araçtirma, Sahasina Uiçkin, MTA Rapor, 5444 (1972)
[204] Me Birney, Igneous Petrology, Oxford University Press (1984)
[205] Haskin, L.A., Haskin, M.A., Frey, F.A., Wildeman, T.R., Relative and absolute
terrestrial abundances of the rare earths. Origin and distribution of the elements, Ahrens,
L.H., éd., Intern. Ser. Monographs Earth ScL, 30, 889-912 (1968)
[206] Brinkmann, R., Izdar, E., Excursion to Bergama (Pergamon) and Kinik, dans: Geology
and history of Turkey, Campbell, A.S., éd., Petroleum Exploration Society, Tripoli, 509-
511(1971)
[207] Picon, M., Un exemple de pollution aux dimensions kilométriques: la fixation du baryum
par les céramiques, Revue d'Archèomèlrie, 9, 27-29 ( 1985)
voir aussi:
Picon, M., Ricq, J.-C, Un exemple d'altération de la composition des amphores
massaliètes: le cas d'Olbia, Lettre d'information du CRA, 30, Archéologie du Midi
Méditerranéen, 12, 65-67 (1986)
[208] Thierrin-Michael, G., Amphores italiques et analyses à la microsonde nucléaire (MSN),
rapport destiné au groupe de travail "caractérisation" du GDR "Méthodes nucléaires en
archéologie"(réunion du 03/12/92) (1992)
[209] Cauvin, M.-C, Besnus, Y., Tripier, J., Montigny, R., Nouvelles analyses d'obsidiennes
du proche-Orient: modèle de géochimie des magmas utilisé pour la recherche
archéologique, Palèorient, 17, 2, 5-20 (1991)
[210] Mosbah, M., Piccot, D., Clocchiatti, R., Metrich, M., Gosset, J., Joron, J.-L., Micro-
PIXE within magmatic inclusions using GUPIX software, Nucl. Inslr. and Me th. B, 82,
139-145(1993)
[211] Innocenti, F., Manetti, P., Mazzuoli, R., Pasquaré, G., Villari, L., Anatolia and north-
western Iran, dans: Andésites, Thorpe, R.S., éd., Wiley & Sons, 327-349 (1982)
[212] Fytikas, M., Innocenti, F., Manetti, P., Mazuoli, R., Peccerillo, A., Villari, L., Tertiary to
quaternary evolution of volcanism in the Aegean region, dans: The geological evolution of

Références 195
the Eastern Mediterranean, Special Publication of the Geological Society, 17, 687-699
(1985)
[213] Borsi, S., Ferrara, G., Innocenti, F., Mazzuoli, R., Geochronology and petrology of
recent volcanics in the eastern Aegean sea (West Anatolia and Lesvos island), Bulletin de
volcanologie, 36, 473-496 (1972)
[214] Hughes, ML, Leese, M.N., Smith, R.J., The analysis of pottery lamps mainly from
Western Anatolia, including Ephesus, by neutron activation analysis, dans: Bailey, D.M., A
catalogue of lamps in the British Museum, III, Roman provincial lamps, British Museum
publications, Londres, 461-485 (1988)
[215] Hughes, M.J., Leese, M.N., Neutron activation analysis of pottery lamps from Ephesus
dating from the Archaic to the late Roman period, Proc. 22nd Symposium on
Archaeometry (Bradford 1982), Bradford, 363-376 (1983)
[216] Dupont, P., Recherches de laboratoire sur les céramiques grecques orientales archaïques,
Revue d'Archéomélrie, 1, 105-114 (1977)
[217] Calamiotou, M., Filipakkis, S.E., Jones, R.E., Kassab, D., X-ray and spectrographic
analyses of terracotta figurines from Myrina: an attempt to characterize workshops, J.
Archaeological Science, 11, 103-117 (1984)
[218] Picon, M., Vertet, H., La composition des premières sigillées de Lezoux et le problème
des céramiques calcaires, RAECE, 21, 1-2, 207-218 (1970)
[219] Picon, M., communication personnelle
[220] Hughes, M.J., communication personnelle
[221] Kuleff, L, communication personnelle
[222] Trouslard, P., Tirira, J., PYROLE, Laboratoire de l'accélérateur Van de Graaff,
I.N.S.T.N., CE. Saclay, 91191 Gif sur Yvette, France
[223] Hughes, M.J., Cowell, M.R., Hook, D.R., Neutron activation procedures at the British
Museum research laboratory, dans: Neutron activation and plasma emission spectrometry
analysis in archaeology. Techniques and applications, Hughes, M.J., Cowell, M.R., Hook,
D.R., éds., British Museum occasional paper, 82, 29-46 (1991)
[224] Mando, P. A., Advantages and limitations of external beams in applications to arts &
archaeology, geology, and environmental problems, Nucl. Instr. andMeth. B, 85, 815-823
(1994)
[225] Keutgen, T., Applications de la méthode P1XE pour l'analyse non destructive de
glaçures et de céramiques anciennes. Interprétation de l'émission XL non shiftée lors de
la collision Kr sur rfh, Au et Ta, Rapport de stage de DE A, Université Catholique de
Louvain(1993)
[226] Gratuze, B., Soulier, I., Barrandon, J.N., Foy, D., De l'origine du cobalt dans les verres,
Revue d'Archéométrie, 16, 97-108 (1992)

Références 196
[227] Fleming, S.J., Swann, C.P., Color additives and trace elements in ancient glasses:
specialized studies using PIXE spectrometry, Nucl. Inslr. and Meth. B, 22, 411-418
(1987)
[228] Brun, N., Etude de verres opaques celtiques et gallo-romains, Thèse de doctorat,
Université Paris-Sud Orsay (1991)
[229] Bordas, M., Possibilités offertes par l'utilisation simultanée des méthodes PIXE et
PIGE en analyse élémentaire, Thèse de doctorat, Université Strasbourg I (1988)
[230] Menu, M., Calligaro, T., Salomon, J., Amsel, G., Moulin, J., The dedicated accelerator-
based IB A facility AGLAE at the Louvre, Nucl. Instr. and Meth. B, 45, 610-614 (1990)
[231] Vittel, C, Pâtes et glaçures céramiques, Denges (1982), cité dans: François, V., La
céramique byzantine de Thasos et un échantillonnage des productions médiévales
orientales et occidentales du XlIIe au XVe siècle, Thèse de doctorat, Université
Strasbourg II, 3 volumes (1990)
[232] Lôvestam, G., Swietlicki, E., An external beam setup for the Lund proton microprobe,
Nucl. Instr. and Meth. B, 43, 104-111 (1989)
[233] Lôvestam, G., Swietlicki, E., Off-line data evaluation of elemental maps obtained from
scanning nuclear microprobe analyses, Scanning microscopy, 6, 3, 607-624 (1992)
[234] Lôvestam, G., Calligaro, T., Duval, A., Salomon, J., The AGLAE scanning nuclear
microprobe setup, Nucl. Inslr. and Meth. B, 77, 66-70 (1993)
[235] Lôvestam, G., Development of a scanning proton microprobe - computer-control,
elemental mapping and applications, Thèse de doctorat publiée, Lund Institute of Science
and Technology, Lund (1989)
[236] Jund, J., Etude minèralogique et technique des céramiques de l'Alsace du Nord: terre
cuite vernissée de Soufflenheim et grès de Betschdorf Thèse de 3ème cycle, Université
Strasbourg 1(1979)
[237] Balfet, H., Fauvet Berthelot, M.F., Monzon, S., Lexique et typologie des poteries. Pour
la normalisation de la description des poteries, Presses du C.N.R.S. (1989)
[238] Papanikola-Bakirtzis, D., Dauterman Maguire, E., Maguire, H., Ceramic art from
Byzantine Serres, Illinois Byzantine Studies, 3 (1992)

Annexe 1 1
ANNEXE 1
PARAMETRES EXPERIMENTAUX POUR L'ANALYSE ELEMENTAIRE PAR LA
METHODE PIXE
1. Analyses sous vide de pâtes céramiques
Efficacité de détection
• épaisseurs des filtres fixes, des fenêtres d'entrée du détecteur Si(Li) et du cristal
(données du constructeur):xMylar= 1 3 ^ m
xgc = 25 urn
x A u = 200 Â
xSid # 0. l nm
x$j = 5 mm
• valeurs de paramètres obtenues par un ajustement aux efficacités expérimentales:
x S i d = 13.9 + 0.3 jim
xsi = 3.72 ±0.09 mm
• paramètres géométriques pour le calcul de l'angle solide de détection:
dist = 140 mm distance détecteur - cible
d = 6 mm diamètre du cristal
Paramètres expérimentaux
• acquisition 1
énergie des protons: 1 MeV
diamètre du faisceau: 1 mm
courant: 6-10 nA
taux de comptage: < 1000 cps.s"'
temps d'acquisition: « 10 min
charge intégrée: 6 uC
filtres additionnels: néant
intervalle d'énergie des rayons X: 1 - 8 keV
éléments dosés: Al, Si, K, Ca, Ti, (Mn), Fe

Annexe 1
• acquisition 2
énergie des protons: 2.5 MeV
diamètre du faisceau: 2 mm
courant: « 200 nA
taux de comptage: * 50 cps.s"1
temps d'acquisition: 20-30 min
charge intégrée: 200-400 uC
filtres additionnels: 300 um Al
intervalle d'énergie des rayons X: 6 - 40 keV
éléments dosés: Fe, Ni, (Cu, Zn), Ga, (As, Pb), Rb, Sr, Y, Zr, (Nb), Ba
2. Analyses en faisceau extrait de glaçures plombifères
Efficacité de détection
• épaisseurs des filtres fixes, des fenêtres d'entrée du détecteur Si(Li) et du cristal
(données du constructeur):
xBc = 8 nm
xAu = 200 Â
x s i d#0.1 um
xgj = 5.07 mm
• paramètres géométriques pour le calcul de l'angle solide de détection:
dist = 2 cm distance détecteur - cible
d = 6 mm diamètre du cristal
Paramètres expérimentaux
énergie nominale des protons: 2.94 MeV
énergie des protons sur la cible: 2.5 MeV, après traversée de 4 um de Zr (fenêtre de sortie de
la ligne) et de 1.95 cm d'air. Les pertes d'énergie des protons dans les différents absorbants
sont calculées avec le logiciel PYROLE [222];
diamètre du faisceau: = 1 mm
courant: « 0.1 nA
taux de comptage: 1000-2000 cps.s"1
temps d'acquisition: 10-15 min
charge intégrée: « 0.1 uC

Annexe 1
filtres additionnels: 2 cm d'air, 50 um Be
intervalle d'énergie des rayons X: 1.5 - 17 keV
éléments dosés: Si, K, Ca, Ba (raies L), éléments de transition de la première série, Pb (raies L
(et M))
3. Analyses d'inclusions de verres volcaniques à la microsonde protonique
Efficacité de détection
cf. 2.
Paramètres expérimentaux
énergie des protons: 3 MeV
diamètre du faisceau: 3 (am
courant: 0.5-1 nA (mesuré dans le pipe)
taux de comptage: « 100 cps.s^1
temps d'acquisition: 3-4 heures
temps d'irradiation par pixel: 10 us (par passage)
filtres additionnels: 50 um Mylar, 40 um Al
intervalle d'énergie des rayons X: 3 - 40 keV
définition: 64x64 pixels
pas de balayage: 0.24 um
dimension des zones analysées: 100-150 u2 (dimensions maximales de l'ordre du mm2)
éléments considérés: éléments de la première série de transition, de Rb, Sr, Ba et Y, Zr et Ti

Annexe 2
ANNEXE 2
PETIT GLOSSAIRE DE TERMES TECHNIQUES EN CERAMOLOGIE
céramique, poterie, terre cuite: "objet en argile qui a subi une déshydratation par cuisson"[237]
argile: "matériau résultant de la décomposition de différentes roches; dans ses élémentsprincipaux, c'est un silicate d'alumine hydraté (SiO2, AI2O3, H2O)" [237]; "matériau ayantdes qualités plastiques et donc apte à être façonné" [95]; matériau naturel plastique résultantde la décomposition de différentes roches et principalement composé de minéraux argileux
minéraux argileux: phyllosilicates d'alumine hydratés composés de particules de dimensionsinférieures à 2 uni
dégraissant: "matériau non plastique, inclus naturellement dans l'argile ou sciemment ajouté parle potier et qui a pour effet de compenser une trop grande plasticité de la pâte" [95]
pâte: "matériau constituant une poterie: la pâte préparée par pétrissage avec de l'eau estdestinée à être façonnée à l'état plastique et fixée par la cuisson" [237]
plasticité: propriété d'un matériau qui lui permet d'être façonné quand il est humide et degarder sa forme après le façonnage (d'après [92])
tesson: fragment de poterie
engobe(G.B.: slip): "pâte fine en suspension dans l'eau. Il s'applique avant la cuisson sur lematériau d'une poterie dont on veut masquer l'aspect grossier ou la couleur, et peut être à labase d'effets décoratifs. Il est parfois lui-même recouvert d'une glaçure" [95];
glaçure: "terme générique des revêtements vitrifiés, quelle que soit leur origine, et leur degréde transparence ou d'opacité" [95]
pernette ou trépied: petit support à trois pieds, à base d'argile, placé entre deux céramiquesglaçurées lors de l'enfournement. Son utilisation permet d'accroître le nombre de piècescuites simultanément tout en évitant au maximum le contact entre les céramiques
sgraffito: technique d'incision ou de gravure à travers une couche d'engobe qui fait apparaîtrela pâte sous-jacente. L'effet décoratif est créé par contraste entre l'engobe (blanc ou crème)et la pâte (rouge) qui apparaît au niveau des incisions (d'après [238])
incision: "action d'entailler l'argile crue. Le décor qui en résulte" [237]
gravure: "action d'entailler l'argile cuite ou complètement sèche" [237]
champlevé: technique qui consiste à exciser des surfaces importantes d'engobe en faisantapparaître la pâte sous-jacente. L'effet décoratif est créé par contraste entre le motif ajouréen engobe (blanc ou crème) se détachant sur le fond de pâte (rouge) (d'après [238, 237])
excision: "sur une poterie raffermie, action d'enlever de l'argile par arrachement ou découpage"[237]
majolique: terre cuite recouverte d'une glaçure opaque stannifère [92]

Annexe 3 I
ANNEXE 3
PROCEDURES EXPERIMENTALES
de laboratoires impliqués dans des programmes d'analyse de céramiques byzantines (Chapitre
2, 2.2., tableau 17) ou dont les travaux sont cités au Chapitre 2 (2.4.2.)
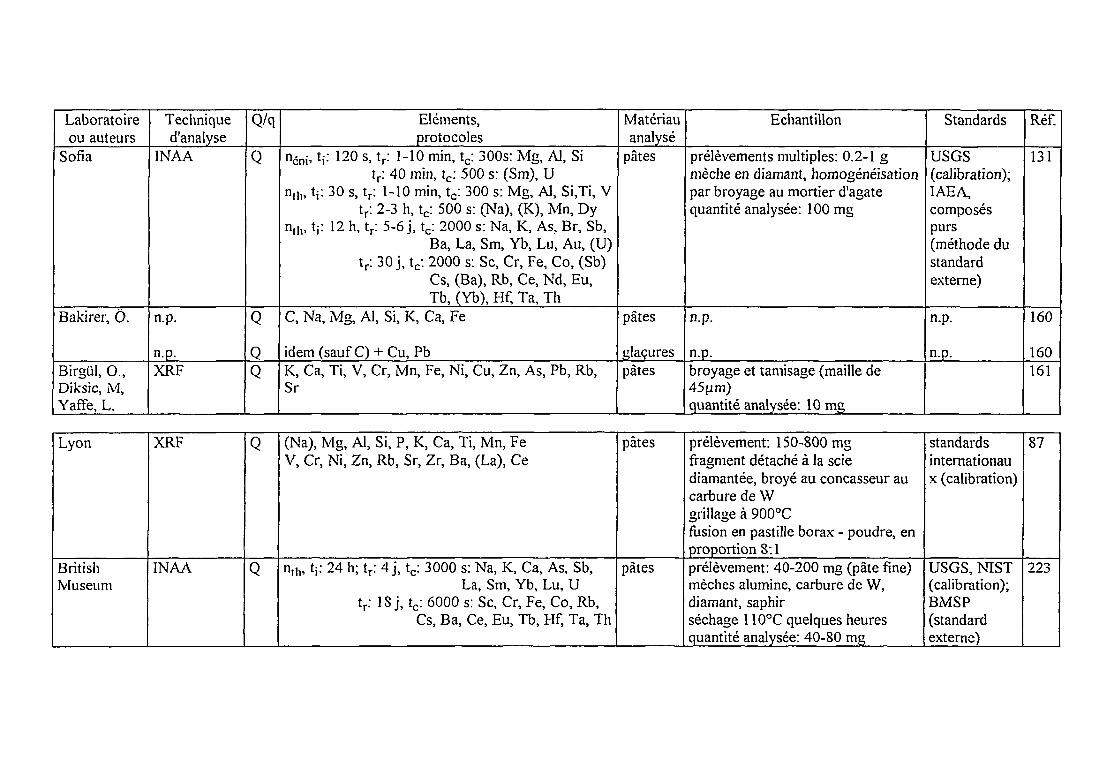
Laboratoireou auteurs
Sofia
Bakirer, 0.
Birgûl, 0.,Diksic, M,Yaffe, L.
Techniqued'analyse
INAA
n.p.
n.p.XRF
Q/q
Q
Q
Eléments,protocoles
néni, t,-: 120 s, tr: 1-10 min, tc: 300s: Mg, Al, Sitr: 40 min, tc: 500 s: (Sm), U
n)h, t;: 30 s, tr: 1-10 min, tc: 300 s: Mg, Al, Si,Ti, Vtr: 2-3 h, tc: 500 s: (Na), (K), Mn, Dy
nth, t;: 12 h, tr: 5-6 j , tc: 2000 s: Na, K, As, Br, Sb,Ba, La, Sm, Yb, Lu, Au, (U)
tr: 30 j , tc: 2000 s: Sc, Cr, Fe, Co, (Sb)Cs, (Ba), Rb, Ce, Nd, Eu,Tb, (Yb), Hf, Ta, Th
C, Na, Mg, Al, Si, K, Ca, Fe
idem (sauf C) + Cu, PbK, Ca, Ti, V, Cr, Mn, Fe, Ni, Cu, Zn, As, Pb, Rb,Sr
Matériauanalysé
pâtes
pâtes
glaçurespâtes
Echantillon
prélèvements multiples: 0.2-1 gmèche en diamant, homogénéisationpar broyage au mortier d'agatequantité analysée: 100 mg
n.p.
n.p.broyage et tamisage (maille de45 u m)quantité analysée: 10 mg
Standards
USGS(calibration);IAEA,composéspurs(méthode dustandardexterne)
rn.p.
n.p.
Réf.
131
160
160161
Lyon
BritishMuseum
XRF
INAA
Q
Q
(Na), Mg, Al, Si, P, K, Ca, Ti, Mn, FeV, Cr, Ni, Zn, Rb, Sr, Zr, Ba, (La), Ce
n,h, tj: 24 h; tr: 4 j , tc: 3000 s: Na, K, Ca, As, Sb,La, Sm, Yb, Lu, U
tr: 18 j , tc: 6000 s: Sc, Cr, Fe, Co, Rb,Cs, Ba, Ce, Eu, Tb, Hf, Ta, Th
pâtes
pâtes
prélèvement: 150-800 mgfragment détaché à la sciediamantée, broyé au concasseur aucarbure de Wgrillage à 900°Cfusion en pastille borax - poudre, enproportion 8:1prélèvement: 40-200 mg (pâte fine)mèches alumine, carbure de W,diamant, saphirséchage 110°C quelques heuresquantité analysée: 40-80 mg
standardsinternationaux (calibration)
USGS, NIST(calibration);BMSP(standardexterne)
87
223

Annexe 3 IV
Légende:
Q/q: quantitatif/qualitatif; n.p.: non précisé
Abréviations:
méthodes d'analyse: AAS: atomic absorption spectrophotometry, INAA: instrumental
neutron activation analysis, OES: optical emission spectroscopy, XRF: X-ray fluorescence;
protocoles d'analyse par INAA: n^: neutrons de réacteurs ("thermiques"), népi:
neutrons épithermiques, tj: temps d'irradiation, tr: temps de repos, tc: temps de comptage;
standards: USGS: United States Geological Survey, NBS/NIST: National Bureau of
Standards (ancienne dénomination)/ National Institute of Standards and Technology (nouvelle
dénomination), IAEA: International Atomic Energy Agency, BMSP: British Museum Standard
Pottery.

Annexe 4 I
ANNEXE 4
COMPOSITIONS ELEMENTAIRES DES PATES DES 168 CERAMIQUES ANALYSEES
On se référera:
• au Chapitre 1, 1.3. (Bilan analytique), pour la méthode d'analyse utilisée pour la
détermination de chaque élément;
• au Chapitre 2, 2.3.2., pour la description des échantillons;
• au Chapitre 2, 2.4.1.2.(Composition des groupes et confrontation avec ia classification
archéologique), pour la répartition des échantillons au sein des groupes de composition.

w
XJ
a:
CO
o
z
oO
o
>
o(J)
MnO |Fe2O3
CM
g
CaO
K2O
SiO2
AI2O3
MgO
OCMCO
Z
447|
o
CD
enCM
coCO
CN
m
cn
cn
mm
103
o
in
o
CO
m
CDCM
ooco
•(-Is-
coCN
.22
CN
A054
335
mCO
COCO
COCO
n
m
108
o
o
CN00
Is-CN
CO
CD
CO
co
COCN
".Si
A055
184
inCD
CDCM
120
CO
N
169
CO
CM
T—
CO
o
CMCNT"
OJ
CM•51-
Tj"
00in
00CD
coCO
.46
990V
313
CD
IN
CD
m
T-
OJCN
30
00
m
124
o
cnIs-
o
coIs-
coCM
CO
CO
co
CNCM
.65
A057
335
COCO
CNOJ
a
T-
coCN
enoo
00
o
co
T-
co
o
o
CO
! • -
OJ
m5
T-
CD
Is-CM
.76
A058
216
»
00CM
135
CO
43.
180
CDCO
cn
CM
00
n
o
CD
^~
TJ-
m
Tf
CN
54
o
20
00•si-
.91
A073
173
CN00
COCN
00CD
COCOCN
138
?
oCM
inCO
114
o
\—CMT~
OCN
CN
Is-
mCO
oCM
oCO
.27
A081
337
CD
CD
in
N
105
CD
in
CMCO
TCCN
o
inCO
o
CD
tnCM
oT-CD
Is-eo
•«*•
CN
eno
<
339
CNCN
N
CO
m
,-00•r-
oooo
TJ-cn
om
o
o
in00
eoCN
menin
Is-h-
CNCM
.60
<
300
CO
CN
CNCD
O^—CN
cn
3)
CMCO
114
o
CO
o
Is-Is-
00CN
co
co
T-
co
LOCN
.70
in
<
352
o
COCN
00
CN
CD
CD
om
113
o
CDCD
O
T—
T—
Tl-OJ
menin
oCD
CNCN
.67
COCN
<
330]
m
CM
S
T -
CDCN
293
inCM
in
CN
117
o
CDCO
o
eoOO
COCO
CNr—in
CO
Is-
• < -
CO
.30
in
<
356
CN
O)
Olin
CO
*-
CO
co
oen
eoroT-
CD•<t
109
o
eoo
en00
Is-OJ
eooeo
ooeo
CD
"73
CO
m<
192
N
•«tCM
168
t-
r>
206
coco
oCOCM
CO00
238
o
ooCD
o
CM
enCM
oomin
•<fr
CO
COCO
.96
B026
319
oTf
in
COM-
CO00
o
m
T~
m
en
o
O
CN00
CNCO
Is-
61 61
in
mCM
.72
B027
295
CD
M-
m
v-CD
O
cn
CDCM
CO
inin
o
»
o
CN
CDCN
CO
m
oco
coCM
.45
B030
332)
mCOCM
cô
T—
comCN
127
CDCO
r-»CO
095
o
CDO
eo
in
oCN
m
CN
o
Is-
.99
B031
243
CM
CD
164
CD
mCM
192
CD
CO
134
o
CMCD
O
CO
r-
CDCM
cnCO
co
COCO
.34
B032
312
00
CDCO
OO
cn
CD
mr
CO
mm
122
o
co
o
CNCD
OCO
ooco
in
m
oCO
.62
B033
262
CD
CMCM
CO
m
oCDCM
o
o
CO
CN
T-
co
152
o
j _
T"~
CMCN
mco
Is-oeo
ocn
cnCM
enCO
B035
, 291
oCO
inCM
n
20
CD
enen
CM
inin
123
a
CD
o
oCO
CDCM
Is-
CD
aCD
COCM
.53
B036
320
om
inCO
CD
CDO
CN
T -
m
110
o
00
coo
CNCD
CO
co
CO
oCD
oCD
oCN
.73
B037
CN
inCM
135
CO00CM
178
CM
co
CO
CN
•<!•
en
123
o
coCM
CDCM
m•3-
CD
m
CMo
CN
.28
CO
cc
CC
319
COTi-
en
I-.en
incn
CMCO
in
00in
121
o
CO00
o
CDIs-
coCO
CN
co
COCD
v—
CN
.74
B039
302
N
"51-
m
CNCD
CO00
CN
00
inin
111
o
no
o
COCO
Is-
5
oCD
in
CN
.79
B048
189
o• < *
N
157
r-mCO
207
T—
M-
~>|CM
TJ-CO
236
o
COCD
o
oCO
T—
CO
T -
inm
oCO
o• < *
h-CD
B082
307
coTj-
CO
in
enen
cn
CDti-
en'D~
CNin
121
o
CN00
O
COCD
CMCO
T-
CD
m
coCD
oCO
.56
B089
324
o
CD
cnco
CDCD
ooCD
CD
of)
r-.in
119
o
p;o
CD
CMCO
in
CO
ooin
•«}•
CM
.74
B093
239
D
CM
111
CMOJCN
128
CD
o
CD
eo
117
o
CD
eno
COIs-
t—
• ^ •
T-
eom
~CO
CO
Is-co
B112
S3£
COCM
COin
ooCD
COCO
o
T—
co
112
o
CO00
o
coin
• < *
CO
COCMCD
mco
inCN
.74
B135
287
eo•>«•
CO
CN
co
CNCD
131
COCD
CM
COin
113
o
o
o
CDCM
CO00
m
T-
CO
COCO
.42
B144
294
inm
o>
co
CM
130
00
oCD
125
o
ooo
o
in
coCO
oCOCD
oCO
COCM
.60
B147
291
CMCO
r)
CDin
a
COCO
CD
v-
COCD
103
o
CDCO
o
CO• < *
oCO
CNCMCD
COIs-
00v-
.82
C021
363
inco
CM
co
Ti-
co
CDto
154
o
eo00
o
COin
coCM
CDCM
co
co
oCN
.82
330O
342
o
N
O
COCD^~
o
103
COf
CD
m
110
o
en
o
CD
m
ooCM
ooCO
co
CNCD
T -
CN
.54
C075
327
Si
N
CNin
17.0
101
CD
m
102
o
O
Is-CD
OCO
CNCD
Is-CO
CDr—
Is-Is-;
C079

CO
A01
CO
CD
2
oO
O
>
oCO
Fe2
O3
MnO
CM
oi -
CaO
K2O
SiO
2A
I2O
3M
gO
OCMCO
Z
155
COCM
00CN
co
CDTfCO
183
132
TfOCM
OO)
833
o
CO
o
oCD
enCM
in
in
COh-
Tf
00
CD
eno
IC08
029
3
coCM
CM
COCD
T-
oCM
O)
150
T~
CD
COCD
118
o
rao
COf»
(3)CM
CMOCD
T-
CO
COco
.01
CM
|C08
437
3 i
CMco
enT—
CO
oen
oCO
107
Tf
CMCO
114
o
75
o
m00
r-CM
CD
oCD
enLO
CO
co
.69
36001
246
00co
CO
105
COCNCM
116
135
CDt~~
O00
079
o
.89
o
CDLO
T -
Tf
enTfin
ot—
COCN
.15
IC09
431
6en
CO
m
enen
CO
r-
T—
oTf
LOin
106
o
.73
o
CDOl
CM
CO
CD
CMin
COCM
.61
IC09
529
2•>-.
CDCMCM
107
142
CDCD
COCD
098
o
.97
o
Tf
r-
Tf
CO
00
r-m
CMCO
Tf
CM
.23
ooO
enen
mm
mCM
enen
enco
144
105
CMO
Tf
r»
118
o
95
o
Tf
enco
Tf
enm
mco
en
.44
cooO
309j
enCM
en
coen•T—
Oen
h-oT-
CMin
COCD
112
o
LOt»~o
CMf-
CM
CMCMCD
OCD
h-CM
.57
301
00
m
LOCM
00CD
CDCDCM
enen
144
CMoo
Tf
r~-
Tf<3)
oo
89
o
Tf
Tf
LO
CO
00
sm
LOCM
.29
CM
O
245
oo
LOco
CDCNT-
O
mCM
cocoT"
130
r--00
oO)
123
o
CDCM
OTf
Tf
Tf
OTf
m
o
T-
.04
omLO
3431
coCM
VZ
coTf
00
CM
•c—O
in00
m
CMco
114
o
CM
O
00LO
r-CM
00
CM
co
T-
h-
coCM
.80
ID02
431
1 j
N
OLO
in
M
ooen
103
Tf
CO
co
r~
O
or-o
or--
r-CM
CO
CD
00t--
T-
CM
.74
ID02
523
51
CD
m
00CM
co
oo
2514
915
2
O)en
00
r-
T"*00
o
Tf
00
co
cor»m
00
O)
CMCO
.44
D04
031
3|
coT—
00CM
OO
co
oen
en
102
LOTf
r--in
120
o
r -CDo
T -
o
mCM
OO
com
coCD
coCN
.58
D04
3
S00
CD
COCN
O
ooenr-
Tfen
113
CDm
com
123
o
77
o
r-t~-
CM
ooCD
Tf
1-
T -
CM
.55
ID04
621
7
enco
coCM
enen
ooKl
194
enoT—
CDO)
in
124
o
00
oT—
Tf
co
CNCO
00
Tf
m
00
O)
mCM
.19
D05
954
9
T—
00CM
OCM
COTf
COTf
m00
r-Tf
CO
m
097
o
.88
o
•«-r-
CM
CMenm
o00
t—
CM
.23
CM
ID07
441
633
6
en r-»CM.CM
CMCM
03CO
COCO
Tf
co
en03
coTf
CMCD
en
o
.82
o
enLO
enCM
LO
CD
enCD
oCM
COTf
enen
CMen
Tf
en
TfTf
Oco
121
o
f-i^
o
f-eo
o00
CO
CMCD
oCD
CO|CM
CM
.97
ID07
6
CM
.74
00[00
18
350|
00CM
enm
T -
cn
T—CO
115
enco
CDin
109
o
£1o
Tf
en
CDCM
COenm
eo
coCM
.75
ID12
251
8]
S
ooCM
O
CDt'-
enTf
110
CDCD
I--
m
121
o
Tf00
o
Tf
cnCM
menin
r-eo
coCM
s—coCM
IE01
937
7|
CM
en
n
CDr-
3)
CDCO
-,-CO
in
105
o
N.O
en
co
enoco
coCD
en
.03
CM
0303]
528
in
O
CMh-
cnTf
men
co
oCD
103
o
eni-»
o
coCD
enCM
moco
ooo
oCM
r-CM
E08
540
81
cô
coCM
inco
Tf
en
oco
Tfen
enTf
CO
m
116
o
CMCO
O
coCD
enCM
m
CD
CDCD
Tf
CM
.06
CM
CMOL3
357
inCM
m
CDTf
Tf
en
»
108
CDTf
r-LO
131
o
CM00
o
CNCO
f-CM
eo
coCD
••CM
.67
T—
coo
LU
|399
o
coCM
O
TfCD
00Tf
OO)
oCD
enin
00en
o
coen
o
CMCD
ooCM
moCD
o
mCM
.37
CM
t'O
LU
3271
TfOO
cof-»
CDen
oen
116
Tf
LO
00
m
CD
O
75
o
ooCO
enCM
COenm
enco
r-CM
758
00
LU
394
:M
CM
•n
mr~-
o00
co00
enco
r--m
102
o
enCD
o
oo
O)CM
enenm
Tf
in
mCM
.67
en
LU
210
CMTf
00CM
240
co
234
enLO
coCMCM
en
coTf
O
.06
coin
coCM
00
00
m
CMOO
co
.04
Tf
OLL
198
CD
COCN
115
Tf
170
co
oCN
T-
00
135
o
.22
00
00
Tf
CO
oCO
m
Tf
OCM
enCM
.45
m
oLL
195
»
N
143
enenCM
205
coTf
CD
CM
Oen
135
o27
•*—co
00
00
COCM
m
-,-oCM
enCM
.45
i~-
oLL
215j
enm
mCM
en
r>~enCM
m
moCM
enr-
O)00
o
CMCM
or-»
coco
Tf
Tf
m
OO
en
coco
.49
t't'Od
|
195|
N
coen
coenCM
Tf
m
Tf00
mCM
Tf00
139
o
oco
Tf
CD
r~00
Tf
TfLO
cooCN
h-CM
.50
F04
518
2|
S3
CO
Tf
:N
CD
••o
CM
oCM
co
122
o
00CN
coen
mco
T-
00LO
Tf
enT—
OCO
.47
|F06
123
0
ro
co
106
Tf
00CM
00Tf
T—
m
enen
r-co
138
o
TfCN
OO
coco
CN
in
Tf
oCM
coCN
.53
F06
2I7
4|
234|
co
132
menCM
TfLO
m
CM
CM
m00
135
o
CDCN
o
co
coCM
m
oo
8|
18
CM
.52
|F06
3

w
.a
a:
CO
o
z
o
o
o
Fe2O3
MnO
egO
CaO
K2O
20!S
AI2O3
MgO
OegCO
Z
194
mco
coeg
cooo
oooco
o
CO•<*•
N
CMOO
152
o
.28
0000
toco
ooeg
m
ooCM
h-CM
to
F066
205
3
O)eg
146
co
m
VI
o\Z
coCO
126
o
.27
co
co
CM
m
CMoeg
CM
CO
m
F067
992
n
oCO
00CD
r».COeg
CM
r-
o
CM00
123
o
.17
to
CDCO
COom
ooeg
oeg
in
F069
5)
CO
m
toCM
coCDCM
O)
COCM
0000
00
123
o
.15
toO)
COCO
COTJ-m
18
CMCM
OZO
d]
211
mco
L9
CM3
L9
CM
mL2
CMO)
147
o
CO
00
toCO
inCM
m
CO
20
00CM
00in
F076
198
\Z
CM
SIZ
ininCO
rl-in
a
••*\Z
CO00
138
o
.06
CMin
00CM
r-oCD
00CD
inCO
IF090
602
CO
M
CD
O00CM
CO
CMCO
moCM
00
129
o
.20
CDCO
CO
in
COCD
•tfCO
.60
F097
249
mCM
119
xi-COCM
com
CO
1-
Ooo
o
.14
o
CO
T-
în
CO6I-
COCO
.40
ZH
-dl
214
CM
\Z
COCD
T-
CM
CO
G)CO
CO81o
132
o
.05
r--o
Tt-CO
com
81-
CDCM
.45
F124
170
egeg
o>00
mO)CM
a
COCO
m\Z
CM
142
o
.28
coto
COCO
•<t
COin
CO
oCM
CMCO
into
IF146
161
m
N
G)
m
otoco
CD
CO
h~
G>
00co
143
o
toto
o
O)
o
toCMco
TTOCM
T—
53
o
G013
CD
TJ-
CM
eg
H
"*
3)
00CM
in
r>
o
.60
o
O)
o
m
>«ttoto
T -
G>
O)O
00coo
0909|
co
mCM
o00
x—Oco
r-
eg
O)CO
mco
156
o
.77
o
o
CMco
T-
into
O)
o>
in
COCO
o
L909|
294
CDco
COCM
OO)
oG)
COG)
CD
CMCD
118
o
o
O)
m
O)CM
inCMCD
CDCD
LOCM
CMCD
G072
LO
OOeg
o00
egooCO
102
CO
m
m81-
CDCD
150
o
.65
o
CDo
00
O)
to
oCJ)
O)o
.59
o
G077
106
toCO
toCM
COCO
COo>CM
139
Tt-
CMCM
o00
153
o
.88
o
ooo
O)CM
1-
oto
CM
COCM
.27
IG078
co
r-
00CO
O)CM
•o
Tf
co
145
toCO
VZ
O)
163
o
.91
o
CDo
oCO
COT—to
CMoCM
meg
.24
G116
992
mCD
COCM
co
o81CO
m
mco
CO00
T-
m
017
o
.63
o
00
CMCO
oG)co
eg
O)
o
.66
o
92I-9|
o
COeg
CO
CD
CM
S
too
egCO
egco
098
o
.61
o
CDT—
CO
CO
CD
mCM
•>*
.51
o
G136
| 264'
1-9
LOCM
144
o:M
toto
LOCM
co81-
co
117
o
.02
CDCM
OCO
T-
CM
m
O)
CO
.02
l-OO
Hl
270
45
meg
134
Tf
f2
o
to61-
egCM
o
.00
CDCO
O)CM
CMCMLO
r~-
egCO
.10
H002
260
O)
N
112
TJ-COCM
LOto
LO
mÏZ
N.CO
COCO
o
.17
CO
coCO
T-
m
rj-G)
1 -
CO
o
H003
303
D
CDCM
215
m00CM
egO)
COeg
o
rg
CD00
114
o
.98
o
coeg
egCO
1-9
-a-CO
COCO
.90
o
H004
L 358
COCM
ineg
in• < *
egCO
CD
00
?
00
m
115
o
.73
o
COLO
oCO
coco
CDLO
COCM
.92
H018
247,
o
eg
T-
inLOCM
COCO
CM
ooO)
oCO
118
o
.09
oCO
COCO
eginin
to00
oCO
.07
H049
239
O)CO
COeg
CDCO
m3
t'Z I
ooCD
T-
egeg
o00
co
o
.16
CO•<-
egCO
coI-9
oen
coCO
.39
H131
257
CDin
mCM
158
r--COCM
3)
CM
CDCO
O00
119
o
.92
o
CO
oCO
mCMin
r»-
TT
CO
.97
o
H132
273
3)
00eg
137
co
CM
O
?
moo
CO00
109
o
.02
COv-
coCO
T-
egin
o>00
inCO
.01
H158
272
o
toCO
157
o3
D
3)
00T*"
eg
TfO)
136
o
.17
O)
CO
co
Oeg
TJ-
eg
oCO
*"
I008
COCOCM
19
O)CM
141
COCOCM
o
en
co
N
CO
130
o
.10
oo
TTCO
LOoin
moCM
COCO
.17
1011
253
CM00
coeg
156
T -
meg
CMCD
CO
oo
o>co
117
o.13
to00
oCO
T-
CMLO
oCMCM
inCM
.92
o
1016
234
3
YZ
118
moco
COCO
toto
coT-eg
CO
138
o.15
00
o
coCO
00o>•tt-
CDoeg
oCO
.46
I052
244
57
CDCM
107
CO00eg
o
toCM
h»
N
122
o
.01
T-
coCO
ooom
CMoCM
TT
co
.94
o
1071
161
COeg
129
•«a-
Si
3)
o
CD
T-
901-
o
O)
o
ooo
coCM
h-
00CO
.44
II096
257
CD
Si
140
CDCDeg
o
155
LO
130
o
.95
o
toCO
CMCO
O)O)
CDO)
tô"CM
.80
o
I098
272
5
ÔÔ"eg
eg00CM
124
o
N
CM00
125
o
.96
o
T—
CMCO
CM
LO
OOCM
CDCM
.92
o
1111
0
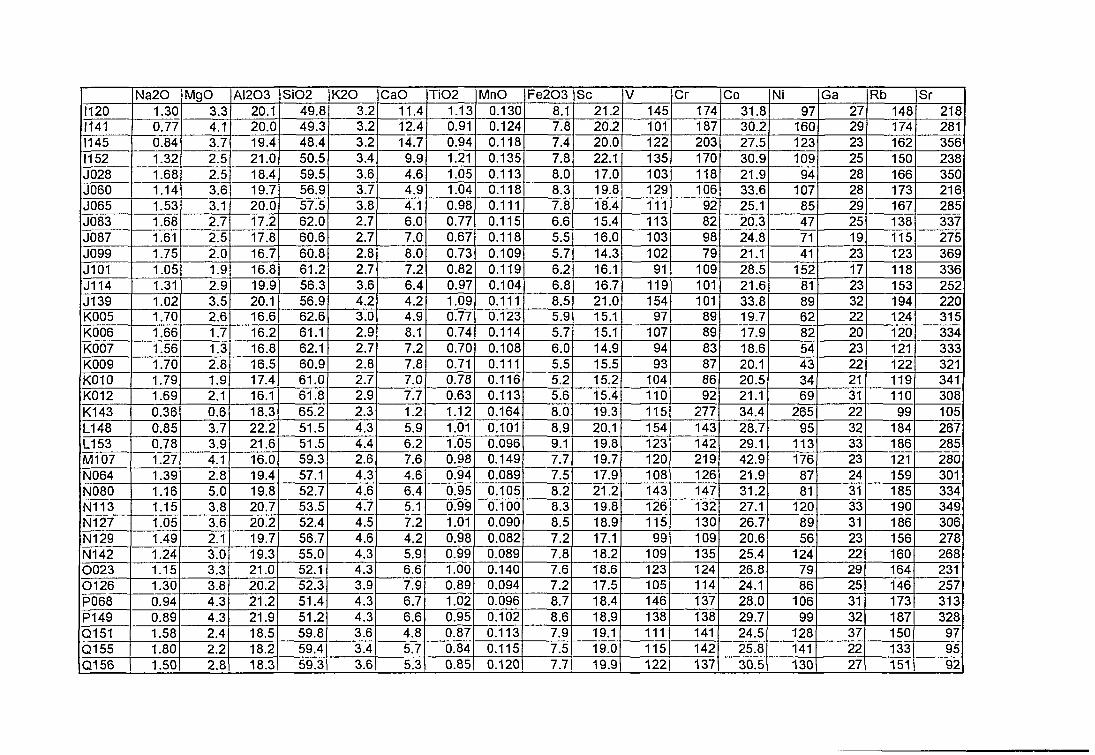
1120114111451152J028J060J065J083J087J099J101J114J139K005K006K007K009K010K012K143L148L153M107N064N080N113N127N129N142O023O126P068P149Q151Q155Q156
Na2O1.300.770.841.321.681.141.531.681.611.751.051.311.021.701.661.561.701.791.690.360.850.781.271.391.161.151.051.491.241.151.300.940.891.581.801.50
MgO3.34.13.72.52.53.63.12.72.52.01.92.93.52.61.71.32.81.92.10.63.73.94.12.85.03.83.62.13.03.33.84.34.32.42.22.8
AI2O320.120.019.421.018.419.720.017.217.816.716.819.920.116.616.216.816.517.416.118.322.221.616.019.419.820.720.219.719.321.020.221.221.918.518.218.3
SiO249.849.348.450.559.556.957.562.060.660.861.256.356.962.661.162.160.961.061.865.251.551.559.357.152.753.552.456.755.052.152.351.451.259.859.459.3
K2O3.23.23.23.43.63.73.82.7
in2.82.73.64.23.02.92.72.82.72.92.34.34.42.64.34.64.74.54.64.34.33.94.34.33.63.43.6
CaO11.412.414.7
9.94.64.94.16.07.08.07.26.44.24.98.17.27.87.07.71.25.96.27.64.66.45.17.24.25.96.67.96.76.64.85.75.3
TiO21.130.910.941.211.051.040.980.770.670.730.820.971.090.770.740.700.710.780.631.121.011.050.980.940.950.991.010.980.991.000.891.020.950.870.840.85
MnO0.1300.1240.1180.1350.1130.1180.1110.1150.1180.1090.1190.1040.1110.1230.1140.1080.1110.1160.1130.1640.1010.0960.1490.0890.1050.1000.0900.0820.0890.1400.0940.0960.1020.1130.1150.120
Fe2O38.17.87.47.88.08.37.86.65.55.76.26.88.55.95.76.05.55.25.68.08.99.17.77.58.28.38.57.27.87.67.28.78.67.97.5
Sc21.220.220.022.117.019.818.415.416.014.316.116.721.015.115.114.915.515.215.419.320.119.819.717.921.219.818.917.118.218.617.518.418.919.119.0
7.7| 19.9
V145101122135103129111113103102
91119154
97107
9493
104110115154123120108143126115
99109123105146138111115122
Cr174187203170118106
92829879
109101101
898983878692
277143142219126147132130109135124114137138141142137
Co31.830.227.530.921.933.625.120.324.821.128.521.633.819.717.918.620.120.521.134.428.729.142.921.931.227.126.720.625.426.824.128.029.724.525.830.5
Ni97
160123109
9410785477141
1528189628254433469
26595
113176
8781
1208956
1247986
10699
128141130
Ga272923252828292519231723322220232221312232332324313331232229253132372227
Rb148174162150166173167138115123118153194124120121122119110
99184186121159185190186156160164146173187150133151
Sr218281356238350216285337275369336252220315334333321341308105267285280301334349306278268231257313328
979592
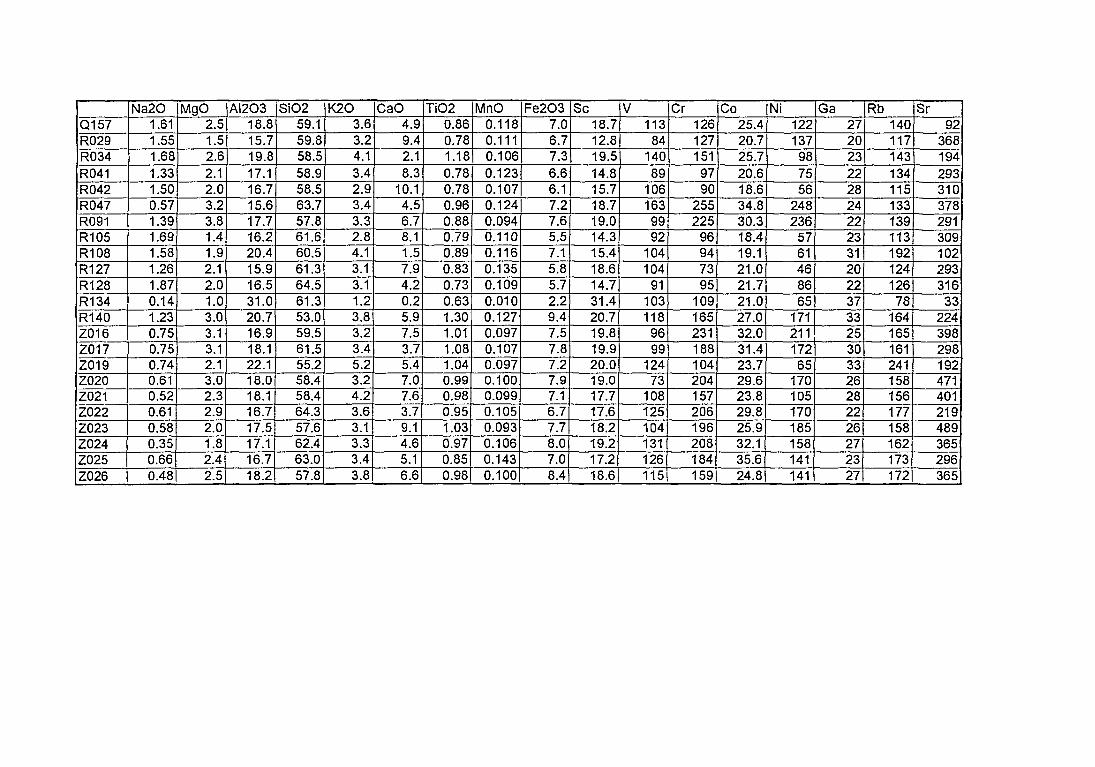
Q157R029R034R041R042R047R091R105R108R127R128R134R140Z016Z017Z019Z020Z021Z022Z023Z024Z025Z026
Na201.611.551.681.331.500.571.391.691.581.261.870.141.230.750.750.740.610.520.610.580.350.660.48
MgO2.51.52.62.12.03.23.81.41.92.12.01.03.03.13.12.13.02.32.92.01.82.42.5
AI2O318.815.719.817.116.715.617.716.220.415.916.531.020.716.918.122.118.018.116.717.517.116.718.2
SiO259.159.858.558.958.563.757.861.660.561.364.561.353.059.561.555.258.458.464.357.662.463.057.8
K2O3.63.24.13.42.93.43.32.84.13.13.11.23.83.23.45.23.24.23.63.13.33.43.8
CaO4.99.42.18.3
10.14.56.78.11.57.94.20.25.97.53.75.47.07.63.79.14.65.16.6
TiO20.860.781.180.780.780.960.880.790.890.830.730.631.301.011.081.040.990.980.951.030.970.850.98
MnO0.1180.1110.1060.1230.1070.1240.0940.1100.1160.1350.1090.0100.1270.0970.1070.0970.1000.0990.1050.0930.1060.1430.100
Fe2O37.06.77.36.66.17.27.65.57.15.85.72.29.47.57.87.27.97.16.77.78.07.08.4
Sc18.712.819.514.815.718.719.014.315.418.614.731.420.719.819.920.019.017.717.618.219.217.218.6
V11384
14089
1061639992
104104
911031189699
12473
108125104131126115
Cr1261271519790
255225
96947395
109165231188104204157206196208184159
Co25.420.725.720.6
Ni1221379875
18.6| 5634.830.318.419.121.021.721.027.032.031.423.729.623.829.825.932.135.624.8
248236
5761468665
17121117265
170105170185158141141
Ga2720232228242223312022373325303326282226272327
Rb14011714313411513313911319212412678
164165161241158156177158162173172
Sr92
36819429331037829130910229331633
224398298192471401219489365296365
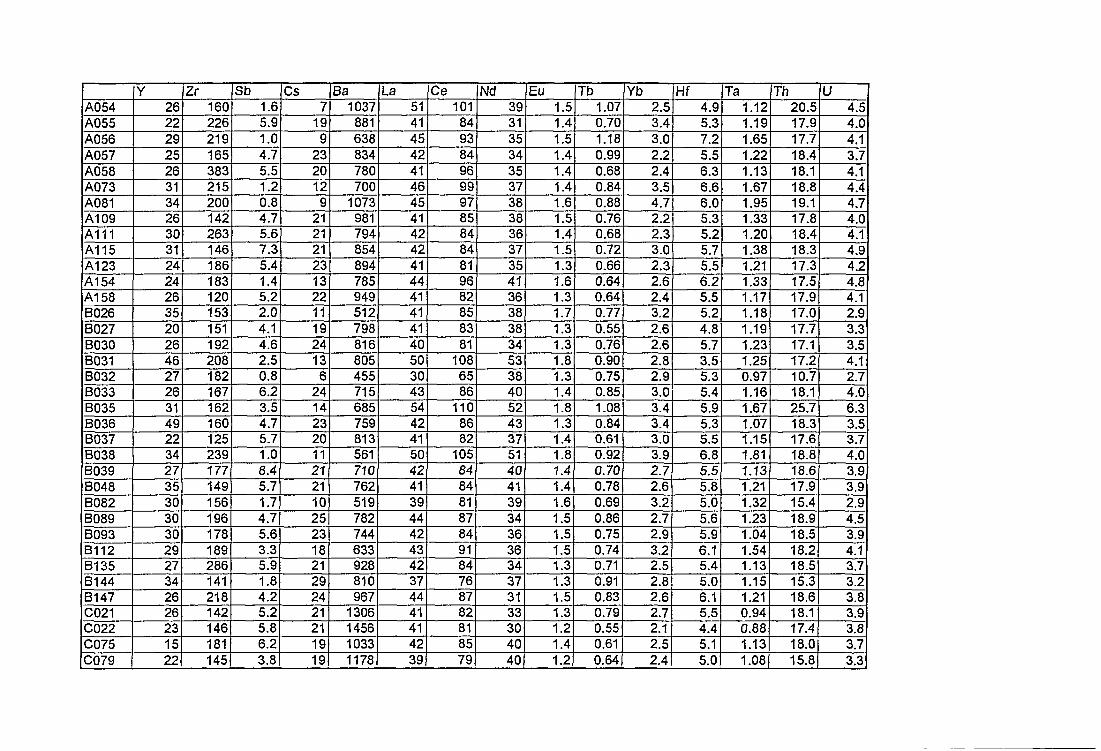
A054A055A056A057A058A073A081A109A111A115A123A154A158B026B027B030B031B032B033B035B036B037B038B039B048B082B089B093B112B135B144B147C021C022C075C079
Y262229252631342630312424263520264627263149223427353030302927342626231522
Zr160226219165383215200142263146186183120153151192208182167162160125239177149156196178189286141218142146181145
Sb1.65.91.04.75.51.20.84.75.67.35.41.45.22.04.14.62.50.86.23.54.75.71.08.45.71.74.75.63.35.91.84.25.25.86.23.8
Cs71992320129212121231322111924136241423201121211025231821292421211919
Ba103788163883478070010739817948548947859495127988168054557156857598135617107625197827446339288109671306145610331178
La514145424146454142424144414141405030435442415042413944424342374441414239
Ce1018493849699978584848196828583811086586110868210584
L_ 848187849184768782818579
Nd393135343537383836373541363838345338405243375140413934363634373133304040
Eu1.51.41.51.41.41.41.61.51.41.51.31.61.31.71.31.31.81.31.41.81.31.41.81.41.41.61.51.51.51.31.31.51.31.21.41.2
Tb1.070.701.180.990.680.840.880.760.680.720.660.640.640.770.550.760.900.750.851.080.840.610.920.700.780.690.860.750.740.710.910.830.790.550.610.64
Yb2.53.43.02.22.43.54.72.22.33.02.32.62.43.22.62.62.82.93.03.43.43.03.92.72.63.22.72.93.22.52.82.62.72.12.52.4
Hf4.95.37.25.56.36.66.05.35.25.75.56.25.55.24.85.73.55.35.45.95.35.56.85.55.85.05.65.96.15.45.06.15.54.45.15.0
Ta1.121.191.651.221.131.671.951.331.201.381.211.331.171.181.191.231.250.971.161.671.071.151.811.131.211.321.231.041.541.131.151.210.940.881.131.08
Th20.517.917.718.418.118.819.117.818.418.317.317.517.917.017.717.117.210.718.125.718.317.618.818.617.915.418.918.518.218.515.318.618.117.418.015.8
U4.54.04.13.74.14.44.74.04.14.94.24.84.12.93.33.54.12.74.06.33.53.74.03.93.92.94.53.94.13.73.23.83.93.83.73.3
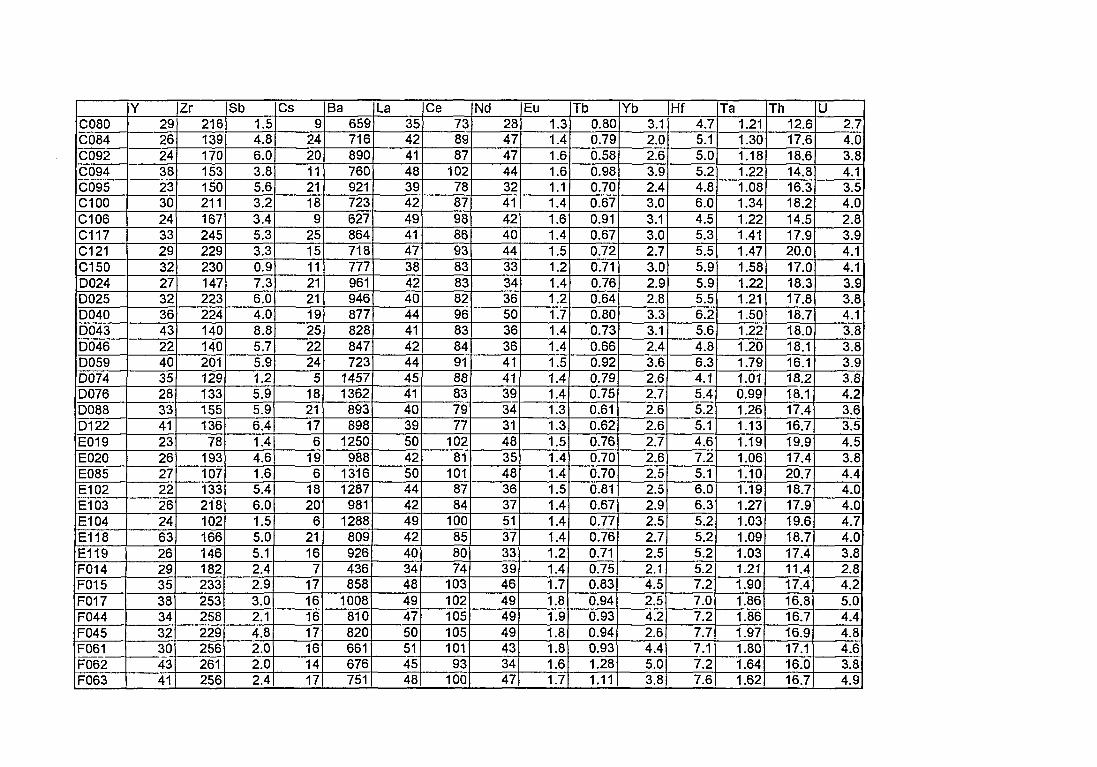
C080C084C092C094C095C100C106C117C121C150D024D025D040D043D046D059D074D076D088D122E019E020E085E102E103E104E118E119F014F015F017F044F045F061F062F063
Y |Zr292624382330243329322732364322403528334123262722262463262935383432304341
21613917015315021116724522923014722322414014020112913315513678193107133218102166146182233253258229256261256
Sb1.54.86.03.85.63.23.45.33.30.97.36.04.08.85.75.91.25.95.96.41.44.61.65.46.01.55.05.12.42.93.02.14.82.02.02.4
Cs92420112118925151121211925222451821176196182062116717161617161417
Ba659716890j
7609217236278647187779619468778288477231457136289389812509881316128798112888099264368581008810820661676751
La354241483942494147384240444142444541403950425044424942403448494750514548
Ce7389871027887988693838382968384918883797710281101878410085807410310210510510193100
Nd284747443241424044333436503636414139343148354836375137333946494949433447
Eu1.31.41.61.61.11.41.61.41.51.21.41.21.71.41.41.51.41.41.31.31.51.41.41.51.41.41.41.21.41.71.81.91.81.81.61.7
Tb0.800.790.580.980.700.670.910.670.720.710.760.640.800.730.660.920.790.750.610.620.760.700.700.810.670.770.760.710.750.830.940.930.940.931.281.11
Yb3.12.02.63.92.43.03.13.02.73.02.92.83.33.12.43.62.62.72.62.62.72.62.52.52.92.52.72.52.14.52.54.22.64.45.03.8
Hf4.75.15.05.24.86.04.55.35.55.95.95.56.25.64.86.34.15.45.25.14.67.25.16.06.35.25.25.25.27.27.07.27.77.17.27.6
Ta1.211.301.181.221.081.341.221.411.471.581.221.211.501.221.201.791.010.991.261.131.191.061.101.191.271.031.091.031.211.901.861.861.971.801.641.62
Th12.617.618.614.816.318.214.517.920.017.018.317.818.718.018.116.118.218.117.416.719.917.420.718.717.919.618.717.411.417.416.816.716.917.116.016.7
U2.74.03.84.13.54.02.83.94.14.13.93.84.13.83.83.93.84.23.63.54.53.84.44.04.04.74.03.82.84.25.04.44.84.63.84.9
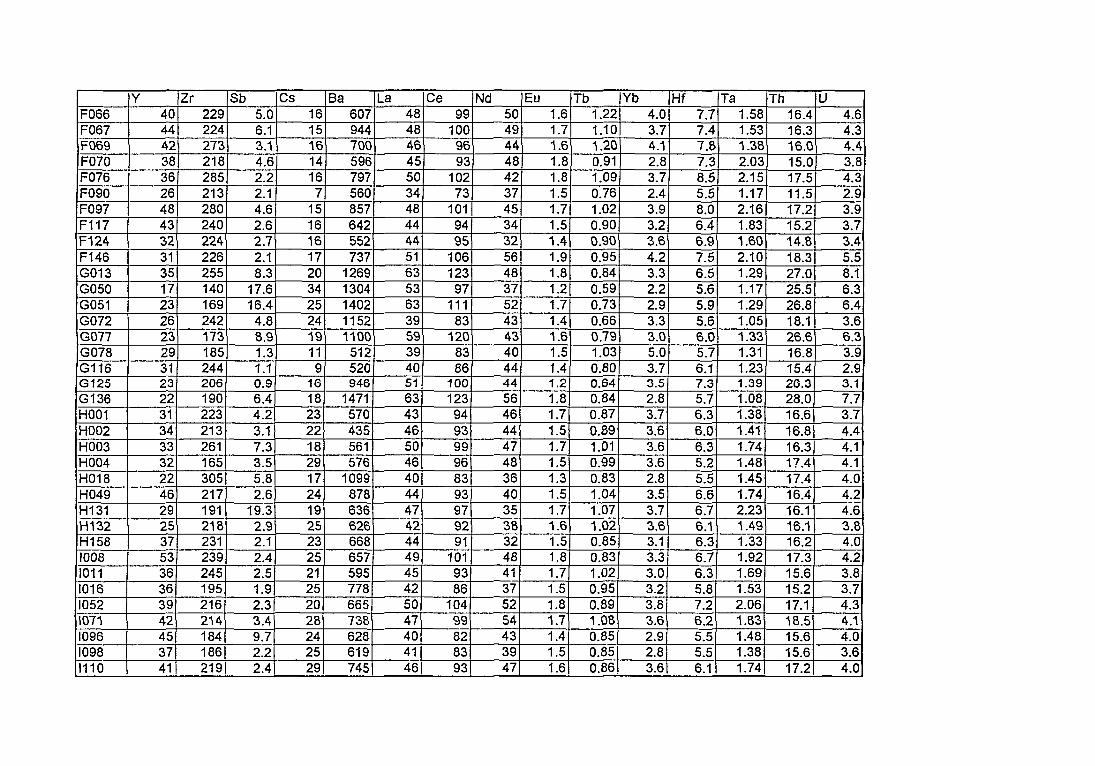
F066F067F069F070F076F090F097F117F124F146G013G050G051G072G077G078G116G125G136H001H002H003H004H018H049H131H132H158I00810111016I0521071I096I0981110
Y404442383626484332313517232623293123223134333222462925375336363942453741
Zr229224273218285213280240224226255140169242173185244206190223213261165305217191218231239245195216214184186219
Sb5.06.13.14.62.22.14.62.62.72.18.317.616.44.88.91.31.10.96.44.23.17.33.55.82.619.32.92.12.42.51.92.33.49.72.22.4
Cs1615161416715161617203425241911916182322182917241925232521252028242529
Ba6079447005967975608576425527371269130414021152110051252094614715704355615761099878636626668657595778665738628619745
La484846455034484444516353633959394051634346504640444742444945425047404146
Ce99100969310273101949510612397111831208386100123949399968393979291101938610499828393
Nd504944484237453432564837524343404444564644474836403538324841375254433947
Eu1.61.71.61.81.81.51.71.51.41.91.81.21.71.41.61.51.41.21.81.71.51.71.51.31.51.71.61.51.81.71.51.81.71.41.51.6
Tb1.221.101.200.911.090.761.020.900.900.950.840.590.730.660.791.030.800.64
0.840.870.891.010.99
0.831.041.071.020.850.83
1.020.950.891.080.850.850.86
Yb4.03.74.12.83.72.43.93.23.64.23.32.22.93.33.05.03.73.52.83.73.63.63.62.83.53.73.63.13.33.03.23.83.62.92.83.6
Hf7.77.47.87.38.55.58.06.46.97.56.55.65.95.66.05.76.17.35.76.36.06.35.25.56.66.76.16.36.76.35.87.26.25.55.56.1
Ta1.581.531.382.032.151.172.161.831.602.101.291.171.291.051.331.311.231.391.081.381.411.741.481.451.742.231.491.331.921.691.532.061.831.481.381.74
Th16.416.316.015.017.511.517.215.214.818.327.025.526.818.126.616.815.426.328.016.616.816.317.417.416.416.116.116.217.315.615.217.118.515.615.617.2
U4.64.34.43.84.32.93.93.73.45.58.16.36.43.66.33.92.93.17.73.74.44.14.14.04.24.63.84.04.23.83.74.34.14.03.64.0
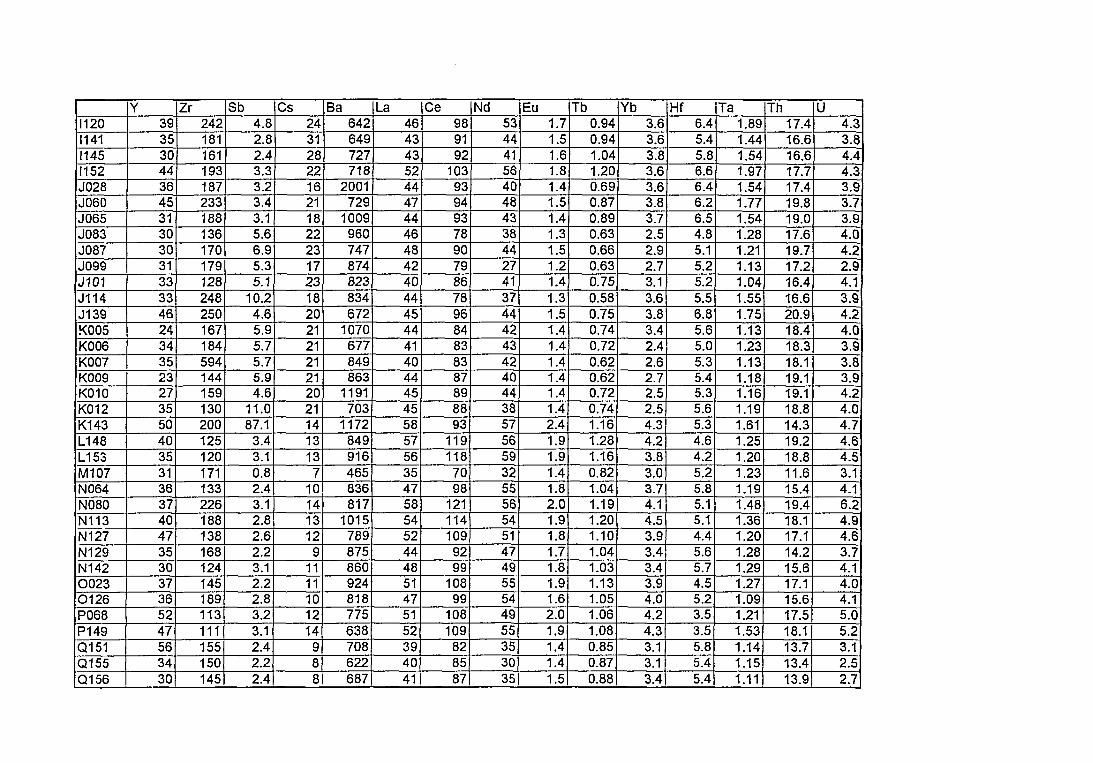
1120114111451152J028J060J065J083J087J099J101J114J139K005K006K007K009K010K012K143L148L153M107N064N080N113N127N129N142O023O126P068P149Q151Q155Q156
Y393530443645313030313333462434352327355040353136374047353037365247563430
Zr242181161193187233188136170179128248250167184594144159130200125120171133226188138168124145189113111155150145
Sb4.82.82.43.33.23.43.15.66.95.35.110.24.65.95.75.75.94.611.087.13.43.10.82.43.12.82.62.23.12.22.83.23.12.42.22.4
Cs2431282216211822231723182021212121202114131371014131291111101214988
Ba642649727718
200172910099607478748238346721070677849863119170311728499164658368171015789875860924818775638708622
La
6871
464343524447444648424044454441404445455857563547585452444851475152394041
Ce989192103939493789079867896848383878988931191187098121114109929910899108109828587
Nd534441564048433844274137444243424044385756593255565451474955544955353035
Eu1.71.51.61.81.41.51.41.31.51.21.41.31.51.41.41.41.41.41.42.41.91.91.41.82.01.91.81.71.81.91.62.01.91.41.41.5
Tb0.940.941.041.200.690.870.890.630.660.630.750.580.750.740.720.620.620.720.741.161.281.160.821.041.191.201.101.041.031.131.051.061.080.850.870.88
Yb3.63.63.83.63.63.83.72.52.92.73.13.63.83.42.42.62.72.52.54.34.23.83.03.74.14.53.93.43.43.94.04.24.33.13.13.4
Hf6.45.45.86.66.46.26.54.85.15.25.25.56.85.65.05.35.45.35.65.34.64.25.25.85.15.14.45.65.74.55.23.53.55.85.45.4
Ta1.891.441.541.971.541.771.541.281.211.131.041.551.751.131.231.131.181.161.191.611.251.201.231.191.481.361.201.281.291.271.091.211.531.141.151.11
Th17.416.616.617.717.419.819.017.619.717.216.416.620.918.418.318.119.119.118.814.319.218.811.615.419.418.117.114.215.617.115.617.518.113.713.413.9
U4.33.84.44.33.93.73.94.04.22.94.13.94.24.03.93.83.94.24.04.74.64.53.14.16.24.94.63.74.14.04.15.05.23.12.52.7
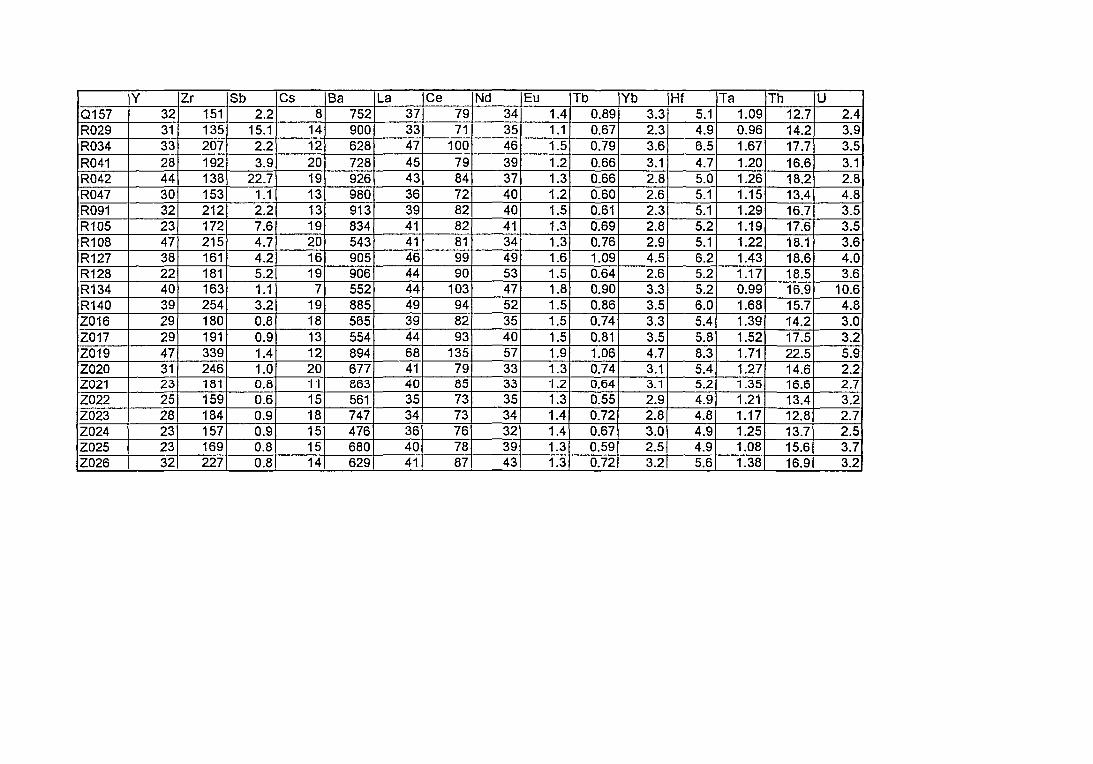
Q157R029R034R041R042R047R091R105R108R127R128R134R140Z016Z017Z019Z020Z021Z022Z023Z024Z025Z026
Y3231332844303223473822403929294731232528232332
Zr151135207192138153212172215161181163254180191339246181159184157169227
Sb2.215.12.23.9
22.71.12.27.64.74.25.21.13.20.80.91.41.00.80.60.90.90.80.8
Cs81412201913131920161971918131220111518151514
Ba752900628728926980913834543905906552885565554894677863561747476680629
La3733474543363941414644444939446841403534364041
Ce7971100798472828281999010394829313579857373767887
Nd3435463937404041344953475235405733333534323943
Eu1.41.11.51.21.31.21.51.31.31.61.51.81.51.51.51.91.31.21.31.41.41.31.3
Tb0.89
0.67
0.79
0.66
0.66
0.60
0.610.69
0.761.090.640.90
0.86
0.740.811.060.740.640.550.720.670.590.72
Yb3.32.33.63.12.82.62.32.82.94.52.63.33.53.33.54.73.13.12.92.83.02.53.2
Hf5.14.96.54.75.05.15.15.25.16.25.25.26.05.45.88.35.45.24.94.84.94.95.6
Ta1.090.961.671.201.261.151.291.19
1.221.431.170.991.681.391.521.71
1.271.351.211.171.251.081.38
Th12.714.217.716.618.213.416.717.618.118.618.516.915.714.217.522.5
14.616.6
13.412.8
13.715.616.9
U2.43.93.53.12.84.83.53.53.64.03.610.6
4.83.03.25.92.22.73.22.72.53.73.2

Annexe 5 I_
ANNEXE 5
A. PROCEDURE DE SELECTION DES ECHANTILLONS POUR UNE ACP.
ENCHAINEMENT ACP - CAH
Lors d'un traitement des données par une ACP, on souhaite en général dégager des
tendances d'ensemble sur l'échantillonnage. Il n'est donc pas souhaitable que des individus de
comportement particulier jouent un rôle prépondérant dans la détermination des premiers plans
principaux. L'élimination des individus isolés dont la contribution à l'inertie est importante
permet d'éviter que les premiers plans factoriels traduisent essentiellement le caractère distinctif
de ces échantillons au détriment d'informations concernant la structure d'ensemble.
L'inspection des histogrammes élémentaires permet déjà d'éliminer un certain nombre
de ces échantillons. Ils ne se distinguent cependant pas toujours par des valeurs extrêmes. Un
moyen de les mettre en évidence est de réaliser d'une première ACP, préalable à l'ACP
principale.
La procédure DEFAC du logiciel SPAD-N trie les individus par ordre de coordonnées
décroissantes sur un axe factoriel, et permet ainsi d'identifier les individus de plus forte inertie
sur cet axe. DEFAC réalise également des tris analogues sur les variables continues (variables
les plus correlées avec le facteur examiné) ou sur les variables qualitatives (modalités les plus
significatives).
Cette procédure est appliquée ci-dessous pour les deux premiers axes principaux d'une
ACP portant sur 31 concentrations élémentaires des 168 échantillons considérés dans cette
étude. Ces axes représentent respectivement 36 % et 16 % de la variance. Les 32 individus
présentant les coordonnées les plus extrêmes sont mentionnés (la valeur du seuil étant
ajustable).
Sur le premier axe, seul l'échantillon Z19 se distingue par une coordonnée minimale
relativement isolée, les valeurs des autres coordonnées se suivant sans discontinuité marquée.
Sur le deuxième axe, cinq échantillons de la série G montrent des valeurs très positives. Ces
échantillons de la série G, ainsi que Z19 et R134, sont séparés des autres par une discontinuité.
Ces observations illustrent deux cas typiques:
D'une part, celui de l'individu Z19, qui ne correspond pas à une catégorie d'échantillons
mais au contraire à un exemplaire unique ayant un comportement singulier. On aura intérêt à
éliminer Z19 de l'ACP principale, car il contribue de manière relativement importante aux deux
premiers axes et risque ainsi de provoquer une "distortion" en favorisant des directions de
l'espace sur lesquelles les autres individus ne seront pas forcément bien représentés.

Annexe 5 II
sen LE TKXSJR l CES
i I! HO-EPO 
Annexe 5 IT[
D'autre part, les échantillons de la série G qui apparaissent groupés et nettement distincts des
autres échantillons par leur compositions élémentaires. Il est en général intéressant de
conserver de tels échantillons dans l'ACP principale: interprétables selon leur modalités (dans
notre cas, la série archéologique), leurs propriétés en tant que groupe de composition seront
bien mises en évidence sur l'axe 2. Cependant dans le cas de l'ACP présentée au Chapitre 2,
2.4.2., figure 53, nous avons éliminé ces échantillons de forte inertie pour privilégier les
différences moins marquées existant entre les autres groupes de composition.
L'enchaînement ACP - CAH peut être avantageux car il permet d'appliquer la
classification à des variables non correlées. La restriction aux premières composantes
principales nécessite cependant certaines précautions (appliquer la CAH à la totalité des
composantes principales revient à travailler sur les données brutes). En premier lieu, il est
nécessaire d'éliminer les individus singuliers, par exemple au moyen de la procédure ci-dessus.
Le choix du nombre d'axes à prendre en compte est effectué au vu de l'histogramme des
valeurs propres de l'ACP (cf. Chapitre 2, 2.4.2., figure 52). On peut décider qu'un pourcentage
de variance cumulée de 90% est une représentation très satisfaisante, les axes correspondant
aux 10% restant étant assimilés à du bruit et éliminés. Il convient à notre avis de considérer
également les valeurs individuelles de la somme des cos2 des angles entre vecteur représentatif
et projection. Par exemple, pour une valeur inférieure à 0.75 (correspondant à un angle de
30°), le point considéré sera relativement mal représenté et son affectation par la classification
ne pourra pas être considérée comme significative. Le logiciel STATGRAPHICS propose un
"output" explicite de la somme des cos2, contrairement à SPAD-N qui donne le cos2 pour
chaque axe.
B. EXEMPLE DE PARTITIONS OBTENUES PAR TRONCATURE D'UN
DENDROGRAMME RESULTANT D'UNE CAH
Le dendrogramme considéré est présenté Chapitre 2, 2.4.2., fig 50 d). Il a été réalisé
par classification ascendante hiérarchique, l'algorithme d'agrégation étant celui de Ward. On a
fait précéder la CAH d'une analyse par composantes principales sur les 31 variables de
composition, centrées et réduites, dont les 12 premières composantes ont été conservées pour
la classification. Celles-ci expriment 90% de la variance du système.
La partition recherchée est celle qui divise l'échantillonnage en groupes de composition
homogènes. On cherche donc à identifier une (ou plusieurs) discontinuité(s) de l'indice de
fusion entre classes de la CAH, qui est interprétée comme la transition d'une variabilité inter-
groupes à une variabilité intra-groupes.

Annexe 5
OASSIFICAXICH ASOMÏOTE EESAK1U0UE : CES3IPTICN CES €0 tCETCS D'DJDIGS y^5 PUCS
IV
ELEVES
NCM. ADJE EEHJ EcT. Pons INDICE CES DOKES DE KIVEAD
2552572S8259260261262263264265266267263269270271272273274275276277278279280281282283284285286287283289290291292293294295296297298299300301302303304305306307
308309310311
312313
314315
198249
1993
D23
22429
243258236250253231254212262
6424624526727226625122823912022026027627027826528228356
248287268289290277261133295292247302296303285293
284257304297
311303
312314
84584691
23024020620925252
2027722
207241244197156101269
7223818722727S139203256137
6894
274280259242279273255288286271264291136140232293263281294237300
299305301307
306309
310313
3522366563776
1354825
198
119
1125
436496
1428
58
126
193411U17182
128
222716611431
6662448
6272
86158
3.005.002.002.003.006.006.005.006.003.007.007.006.00
13.005.004.008.002.005.00
19.008.00
U.009.00
11.0025.00
4.003.006.004.009.006.00
14.0028.00S.008.00
12.00G.00
19.0034.001J..0012.0017.0018.002.00
12.008.00
22.0027.0016.0061.0014.0031.00
6.0066.0024.0048.00
62.0072.00
86.00158.00
0.038800.039760.040110.041780.042720.042790.043620.043910.045180.045950.047280.048350.051160.052960.052970.054730.05622 '0.057330.06065 <0.06889 '0.06981 '0.07074 '0.07133 <0.07570 '0.07612 *0.07866 •0.07909 *0.08174 •0.08495 •0.08632 •0.09734 •0.10522 •0.10729 *0.11086 •0.11204 •0.13002 *0.14288 *0.14471 •0.15531 •0.16165 •0.17383 •0.18164 •0.18389 •0.19001 *0.21167 «0.21181 *0.25576 •0.30667 •0.39226 •0.40802 •0.41909 •0.46576 •
0.74735 *0.78193 *0.78777 •0.87519 *
1.761B5 •2.83076 •
3.16656 *9.11298 •
******•• * •
r*«*
*************************
trcnaitiiga s
La visualisation de l'histogramme des indices (ci-dessus) permet souvent de repérer les
discontinuités de façon plus commode que sur le dendrogramme lui-même (Chapitre 2, 2.4.2.,
fig 50 d)).
Nous avons choisi deux niveaux de troncature qui correspondent à un changement
relativement net des indices de dissimilarité. Les troncatures trop "triviales" (e.g. en 2, 3...
classes) n'ont pas été retenues.

Annexe 5 V
On obtient deux partitions, en 5 et 9 classes respectivement, dont le contenu est donné
ci-dessous:
ODCPOE ' a ' CE L'ARBBE EN 5 CLASSES
EESCHIPTICN KMSUEE
!!11I!
+•
CLASSE
aalaaa2aaa3aaa4aaa5a
! EFEïEriF !
! 66 !i 6 !! 24 !! 14 !! 48 !
H 1-
poms
66.006.00
24.0014.0048.00
CCNTENCJ
1 A 66 !67 A 72 !73 A 96 !97 A 110 !
I l l A 158 !
CCMECSmCN DE : CDOPCFE 'a ' DE L'ARERE EN 5 CLASSES
• OASSS 1 / 5
A055 A057 A058 A109 A U 1 A115 A123 A158 B027 B030 B033 B036 B037 B039 B048 B089 B093 B135 B144 B147. CO21 C022 O075 C079 C084C092 C095 Q 1 7 D024 D025 D043 D046 D076 D088 D122 E020 E102 Q 0 3 E118 E119 G072 B318 J083 J087 JO99 J101 K005 KD06 K009 K010K012 KJ29 R041 R042 R105 R127 R128 AO54 D074 E019 E085 E104 B112 a 0 0 0 2 1 J114
• CLASSE 2 / 5
G013 G050 G051 G077 G125 0 3 6
CLASSE 3 / S —
O 0 6 0151 0155 Q156 Q157 Z016 Z017 Z020 Z021 Z022 Z023 ZO24 Z025 2026 A154 B032 P014 F090 M107 RO47 R091 R108 G078 G116
CLASSE 4 / 5
B031 U 4 8 L153 H064 N080 N113 N127 N129 KL42 0023 0126 PO68 P149 C094
CLASSE 5 / 5
A056 AO73 A081 B038 C150 D059 F015 F017 FO44 F045 F061 F062 F063 F066 F067 F069 F070 F076 F097 F117 E124 E146 BX)1 H002 H003H004 H049 H131 KL32 H158 1008 1011 1016 1052 1071 1098 1110 n 2 0 1141 1145 1152 R140 B035 D040 JD€O 006S J139 RO34
A ce niveau de troncature, on trouve encore des associations "contre nature" comme
celles de la série hellénistique pergamenienne (échantillons Z) et de la série de sgraffito fin
(échantillons Q). Cette dernière série a été fabriquée en quantité importante puisqu'elle a connu
une large diffusion à l'époque byzantine, et a du laisser de nombreux débris dans le(s) site(s) de
production correspondant. Or les communiqués de fouille des ateliers hellénistiques de
Pergame ne mentionnent pas de céramiques de ce type. L'absence de parenté réelle entre les
deux séries peut être aisément vérifiée en examinant les concentrations élémentaires (cf.
tableau en annexe 4, concentrations en Cr, Ni, Sr ...).

Annexe 5 VI
COTtHE 'b 1 CE L'MEFE EU 9 CLASSES
CESCRLPnCH
! CLASSE !
1 bblb !! yx>^ !! bb3b !1 fcb4b !! bb5b !! bbëb !1 tb7b !! bb8b !! bb9b !
EFFECTIF
a5248
16143117
POIDS
a.ooS.002.004.008.00
16.0014.0031.0017.00
! CCKIEJO !
1 B62 B67 B69 B73 B81 B97 B
111 B142 A
61 !66 t68 !72 !80 !96 1
110 !141 !158 !
DE : OXVWË "b1 DE VPPEPZ, EH 9 CLASSES
• CLASSE 1 / 9
AÛ55 M57 A0S8 A109 A l l l A11S A123 A1S8 B027 B030 B033 B036 B037 E039 E04B B089 E093 B135 B144 B147 C021 C022 C07S CO79 C084C092 C095 a i 7 DO24 D02S D043 EC46 C076 D088 D122 E020 Q 0 2 E0.03 E118 E119 G072 H018 J083 J0B7 JO99 JlOl KDOS KD06 K009 KOlOK012 KD29 R041 P042 BIOS R127 R128 B112 C100 0 2 1 J114
CLASSE 2 / 9
AÛ54 DO74 E019 E08S E104
CLASSE 3 / 9
G050 GL2S
CLASSE 4 / 9
C013 O3S1 O077 d 3 6
CLASSE S / 9
CI.06 Q151 0155 Q156 0157 R108 G078 G116
CLASSE 6 / 9
Z016 Z017 Z020 Z021 Z022 Z023 Z024 Z02S Z026 A154 B032 F014 F090 M107 PJM7 PD91
• CLASSE 7 / 9 —
B031 L148 U 5 3 N064 N080 H113 K127 KL29 NI42 0023 0126 P068 P149 C094
CLASSE 8 / 9
AQ56 A073 N381 B038 C150 FOIS F017 F044 F04S F06X F062 F063 F066 F067 F069 F076 F097 F146 H003 E D I 1008 1052 1120 1152 R140B03S D040 J060 J065 JX39 PO34
• CLASSE 9 / 9
D059 F070 F117 F124 H001 HD02 H004 H049 H132 E058 IOU 1016 1071 1099 IUO 1141
On constate que les séries Z et Q sont maintenant bien séparées. Quelques échantillons,
de position instable lors de petites variations dans les conditions choisies pour réaliser la CAH,
leur sont associées de manière non significative ("non classés"). Si cette scission est cohérente
du point de vue du contexte archéologique, il n'en est pas de même pour celle des échantillons
des séries F, H et I (groupe fhi). Cette dernière ne correspond à aucune distinction
typologique, et surtout sépare des céramiques de caractéristiques très semblables: par exemple
le tesson F70 est isolé de l'ensemble de tessons de la série F auquel il est rattaché.

Annexe 6 I
ANNEXE 6
A. PRINCIPES DES ANALYSES MINERALOGIQUES, PETROGRAPHIQUES ET DE LA
MICROSCOPE ELECTRONIQUE A BALAYAGE
Extrait des annexes techniques de:Etudes céramiques en archéologie, Documents et Travaux IGAL, Paris, 9, 123-125 (1985)N.B. : Les travaux présentés dans ce numéro des
Documents et Travaux de 1'ICAL n'étant pas unique-ment destinés à des lecteurs spécialistes desjelences de la terre, mais également à des archéo-logues, 11 nous a paru nécessaire d'expliciter som-mairement les techniques analytiques mentionnéespar les différents auteurs.
Ces notes techniques ne peuvent donner qu'unaperçu schématique et Incomplet des méthodes et desrésultats que l'on peut obtenir et nous prions lesspécialistes de bien vouloir nous en excuser.
LA MICROSCOPIE PETROCRAPIIIQUE
Cette technique, d'un usage quotidien dans lessciences de la terre, permet d'étudier les rochespar transparence à l'aide d'un microscope spécialdit "microscope polarisant". Ce microscope diffèred'un microscope ordinaire (du type de ceux utilisésen biologie) par l'adjonction, sur le trajet desrayons lumineux, d'un dispositif de polarisationcomposé de deux éléments : le polarlseur et l'ana-lyseur. Le polnrlseur, placé entre la source lumi-neuse et la préparation étudiée, transforme la lu-mière ordinaire en lumière polarisée qui ne vibreplus que selon un seul plan. En apparence, cettelumière polarisée a les mêmes propriétés que lalumière naturelle. Elle permet l'étude directe dela structure de l'échantillon et la reconnaissancede certaines caractéristiques des minéraux (forme,couleur, clivages, indice de réfraction, etc.) etla structure générale de la roche.
En traversant un minéral transparent la lumièrepolarisée est modifiée : le plan de vibration estplus ou moins décalé et les longueurs d'ondes quicomposent la lumière subissent des retards diffé-rents entraînant un déphasage. Ces modifications nesont pas naturellement perceptibles à l'oeil hu-main, mais l'interposition de l'analyseur (qui esten fait un second polarlseur orienté à 90* du pre-mier) sur le trajet des rayons lumineux les met enévidence. Elles se manifestent en particulier pardes phénomènes d'extinction et d1éclairement ainsique par des colorations spécifiques (teintes depolarisation), qui sont Indépendantes de la couleurpropre du minéral. L'échantillon analysé étant mon-té sur une platine tournante graduée, II est possi-ble de mesurer la variation de ces déformations dela lumière pour diverses orientations des minéraux,par rapport au plan de vibration de la lumière po-larisée émise.
Les modifications observées étant directementinduites par la structure cristalline du corps étu-dié, leur détermination permet l'identification decette structure et par vote de conséquence la dé-termination du minéral.
Pour pouvoir analyser des fragments de roche aumicroscope polarisant II faut auparavant avoir con-fectionné une plaque mince à partir de l'échantil-lon. Cette plaque mince est une fine lame de. rochecollée sur un support de verre et amincie par po-lissage jusqu'à l'épaisseur standard, de 3O/4. Eneffet, à cette épaisseur la majorité des minérauxsont transparents et peuvent donc être étudiés enmicroscopic. Par ailleurs, à cette épaisseur lequartz, minéral très fréquent dans les roches, aune teinte de polarisation parfaitement blanche.Comme les modifications de la lumière observée sont
directement dépendantes de l'épaisseur traverséel'adoption d'une épaisseur standard facilite la.détermination. La teinte de polarisation particu-lière du quartz à 30 /* permet un ajustage rigoureuxde l'épaisseur de la lame mince par simple contrôleoptique.
References
BORDET P. - 1968 - Précis d'optique cristalline.-Masson, Paris.
COURTOIS L. - 1976 - Examen au microscope pétrogra-phlque des céramiques archéologiques.- Notes etmonographies techniques, n* 8, C.R.A.-C.N.R.S.
JUNG J. - 1977 - Précis de pétrographie.- Masson,Parts.
ROUBAULT H. - 1982 - Détermination des minéraux desroches au microscope polarisant.- Lamarre-Polnat, Paris.
LA MICROSCOPIE ELECTRONIQUEA BALAYAGE (M.E.B.)
Le principe de la microscopic électronique àbalayage avait été énoncé par Knoll dès 1935. Ce-pendant, II a fallu attendre les travaux deCasslett en 1965 et les progrès des techniques detélévision pour que ce procédé d'observation puissedevenir opérationnel et se développer. En effet,contrairement au microscope optique ou au microsco-pe électronique à transmission, le grandlsscmentn'est pas obtenu par l'action de lentilles optiquesou magnétiques mais par le traitement des signauxservant à former l'image.
Un M.E.B. se compose de trois parties essentiel-les :- un système de production d'électrons ;- un système de balayage ;- un système de traitement de l'Information et deformation de l'image.
Dans une enceinte sous vide poussé un canon àélectrons produit un faisceau dirigé vers l'objet àanalyser. Un ensemble de lentilles électro-magnéti-ques focalise ce faisceau d'électrons sur la surfa-ce de l'échantillon, sur une largeur variant de 5 à100 A. Un système de deflection magnétique permetde balayer à l'aide de ce fin faisceau électroniquetoute la surface étudiée.
Le bombardement électronique de l'échantillonprovoque, par Interaction avec les atomes de lamatière bombardée, l'émission d'un certain nombrede rayonnements (électrons secondaires ou rétro-dlffusés, rayons X, etc.). Ces rayonnements sontporteurs d'Informations sur la matière d'où ilssont Issus et selon le type d'information désiréeon sélectionnera l'un ou l'autre des rayonnements.Pour la production d'Images on utilise habituelle-ment les électrons secondaires.
Le rayonnement choisi est détecté, amplifié puistransmis, sous forme de signal électrique, à unécran de télévision dont le balayage est synchronede celui du faisceau d'électrons émis en directionde l'échantillon. Le rapport entre la largeur dubalayage objet et celle du balayage écran déterminele grandtssement. Un point de l'échantillon ayant

Annexe 6 II
un pouvoir émlsslf élevé apparaîtra très brillantsur l'écran. Au contraire un point faiblement émls-slf apparaîtra sombre. Comme cette émlsslvlté esten partie fonction de l'orientation par rapport àl'organe de détection, on obtiendra sur l'écran uneImpression de relief, avec une grande profondeur dechamp et un contraste Important. Le grandlssementpeut varier de quelques fols a 50 000 ou 100 000fols avec une limite actuelle de résolution de100 X (soit 0,00001 mm).
Le M.E.B. permet d'étudier la surface d'échan-tillons massifs. Il fournit les détails de structu-re en relief mats pas la couleur. Les Images obte-nues sur l'écran peuvent être photographiées direc-tement.
En analysant d'autres rayonnements on peut obte-nlr -en même temps que l'Image et soit sur la tota-lité de celle-ci, soit sur un secteur plus llmi.cc-un spectre de rayons X ou une analyse chimique del'objet étudié. Le M.E.B. fonctionne alors en micro-sonde et II en existe divers types fournissant desrenseignements différents.
Pour être étudié l'échantillon doit être Intro-duit dans l'appareil sur une platine orientablespéciale (ce qui limite sa taille). Mais 11 doitsurtout être préalablement métallisé pour augmentersa conductibilité électrique. Cette metallisationse fait généralement par vaporisation d'or, en cou-che tris mince (quelques Â), sur la surface.
Références
EBERHART J.P. - 1976 - Méthodes physiques d'étudedes minéraux et des matériaux solides.- Doln,Paris.
MAURICE F., MENY L. et TIXIER R. - 1981 -Micro-analyse et microscopic électronique à balayage.-Les éditions de physique, Les Ulls.
LA DIFFRACTION DE RAYONS X
Les rayons X, découverts par Roentgen en 1895,sont, comme la lumière visible, des radiationsélectro-magnétiques. Ils en diffèrent par leurslongueurs d'ondes beaucoup plus courtes (0t2 à 2 A)et donc par leur énergie plus Importante. Ils sonthabituellement classés dans les rayonnements demoyenne énergie.
Le baron Von Laue et W.L. Bragg entre autres ontmontre en 1912 que ces rayonnements pouvaient êtredlffractês par un réseau cristallin selon la loi deBragg : nA =• 2d sln 6, dans laquelle n » ordre dela diffraction, A • longueur d'onde des rayons Xutilisés, d » distance entre les plans rétlculalresdu cristal, et 8 = angle sous lequel se fait ladiffraction. SI un rayonnement X de longueur d'ondeconnue est dirigé sur un monocristal tournant surlui-même, ce rayonnement va se dlffracter sur tousles plans du réseau cristallin. En mesurant, pourchaque position de diffraction, l'angle 9 (angle deBragg) on peut calculer les distances des diffé-rents plans rétlculalres. Ces angles et distancesétant caractéristiques dans chaque cas d'un réseaucristallin, lui-même caractéristique d'un type deminéral, on peut en déduLre la nature du minéralétudié.
Tous les corps cristallisés connus et en parti-culier les minéraux, ont ainsi été caractérisés.Ceci a permis de constituer des fichiers donnantles paramètres crlstallographlques de toutes lessubstances cristallines, ce qui évite d'avoir à lesre-calculer à chaque analyse.
Dans la pratique II est rare de pouvoir travail-ler sur des mono-cristaux Isolés. Aussi utlllse-t-
on la méthode dite "des poudres". Dans cette métho-de l'échantillon est broyé très finement, puiscompacté sur' un porte-objet de façon à ce que Upoudre présente une surface bien plane, qui seraexposée au faisceau de rayons X. Ainsi préparél'échantillon présente un grand nombre de crlstal-lltes dont l'orientation est aléatoire et qui, sca-tlstlqucment, vont présenter aux rayonnements tous-les plans réticulalrcs existant dans le cristal. Ladétermination se fait ensuite en mesurant les va-leurs angulaires pour toutes les diffractions, com-me dans le cas des mono-cristaux.
Cette méthode permet en outre d'analyser desmélanges de corps cristallisés (ce qui est le casdes roches par exemple). Le spectre définitif obte-nu est alors la somme de tous les spectres descorps présents. Il est évident que plus le mélangesera complexe, plus 'le dépouillement du spectredeviendra ardu.
La diffraction de rayons X fournit de très bonsrésultats sur le plan qualitatif mais est très mé-diocre sur le plan quantitatif. En effet, dans lesmélanges, des diffractions pourront se cumuler etcertains corps peuvent en masquer d'autres. C'estpar exemple le cas des carbonates pour les argiles.
Dans la pratique l'échantillon et son porte-échantillon sont placés, au centre d'un goniomètre,dans l'axe du faisceau de rayons X dont l'émissionest continue. Un appareil de mesure du rayonnementdlffracté se déplace, à vitesse déterminée, sur lecercle du goniomètre, enregistrant ainsi la valeurde l'Intensité dlffractée pour tous les angles. Unappareil d'enregistrement retranscrit sur bande lesvaleurs mesurées. Sur ces diagrammes s'enregistrentà la fols les valeurs angulaires de diffraction 9(en réalité 9/2) et les diffractions sous forme depics dont la hauteur est proportionnelle à l'In-tensité du rayonnement dlffracté. On considère dansla pratique que cinq pics principaux (qui sontl'équivalent des raies sur les films) suffisentpour déterminer une espèce cristallisée.
Pour affiner la sensibilité de détection on sé-lectionne une gamme de longueurs d'ondes très pré-cise en choisissant la nature de l'anti-cathode dutube émetteur de rayons X. Les anti-cathodes cuivre(Cu) sont de plus en plus souvent remplacées pardes anti-cathodes cobalt (Co) dans les analysesmlnéraloglques. La connaissance de la nature decette anti-cathode et donc de la longueur d'ondes,est absolument nécessaire pour procéder au dépouil-lement du spectre. Les enregistrements photographi-ques sur film sont par ailleurs de plus en plusdélaissés au profit des enregistrements en bandesdonnant des diagrammes plus aisés à dépouiller etplus précis.
En fonction du type de corps à analyser on faitgénéralement subir un certain nombre de prépara-tions en laboratoire à l'échantillon afin d'amélio-rer la finesse de l'analyse. C'est le cas par exem-ple pour les argiles.
Les corps non cristallisés, dits amorphes (commele verre) ne peuvent pas être étudiés par diffrac-tion de rayons X.
References
BROUN C. - 1961 - The X-ray Identification andcrystal structures of clay minerals.- Mlneralo-glcal society, Londres.
EBERHART J.P. - 1976 - Méthodes physiques d'étudedes minéraux et des matériaux solides.- Doln,Paris.
ZUSSMAN J. - 1967 - Physical methods In determina-tive mineralogy.- Academic Press.
1974 - Selected powder diffraction data for mine-rals.- Joint Committee on powder diffractionstandards, Philadelphie.

Annexe 6 III
B. CLASSIFICATION PROPOSEE PAR A. LERUYET A PARTIR DES ANALYSES
MINERALOGIQUES ET PETROGRAPHIQUES DE 32 ECHANTILLONS
Extrait de:
Leruyet, A., Etude pêtrographique, mineralogique et géochimique de céramique byzantine
provenant d'Asie Mineure. Contribution à l'étude des matériaux et de leurs provenances,
Rapport de stage de maîtrise de géologie, Institut de Géologie, EOPGS, Strasbourg (1994)
IV - CLASSIFICATIONS .
Les résultats vont être présentés sous forme de deux tableaux : le premier ayant rapport à l'examenpctrographique, le second à l'examen minéralogique.
1 - GROUPES FORMES A PARTIRDE L'EXAMENPETROGRAPHIOUE .
1 : HY1 - HY2 - IY52 - IY110 caractérisé par :- une pâte assez claire (brune), présentant une structure très fine.- une porosité assez homogène.- des opaques quasi absents.- des dégraissants en faible quantité et de petite taille (50 ums) et plutôt anguleux.- pas de roches felsiques, peu de micas (quelques individus de biotite).- calcite secondaire présente au bord des pores.- décoration fine avec les deux couches d'engobe et de glaçure d'épaisseur bien
régulière. Les échantillons HY sont même décorés des deux côtés.
2 : GY50 - GY51 - GY77 - GY136 caractérisé par :- une pâte foncée, amorphe qui contient pas mal de minéraux opaques.- beaucoup de dégraissants (plus de 50%), de roches felsiques et d'éléments
accessoires en inclusion dans ces roches (apatite).- plagioclases et sanidines assez remarquables.- pas de calcite secondaire, ni de décoration en surface (quoique l'échantillon GY77
présente un rebord micacé et une autre face encroûtée, c'est à dire bordée de calcite dont l'épaisseur estvariable).
- les micas sont isolés et noirs.
3 : OY126 - NY80 - DY59 -CY106 caractérisé par :

Annexe 6 IV
- une pâte assez claire, de structure assez fine et semblant être orientée.- une porosité faible surtout pour DY59 et NY80.- une quantité de dégraissants relativement moyenne, par contre ces derniers sont
de petites tailles et les fragments sont souvent non identifiables (sauf pour les quartzites).- les micas sont très souvent insérés dans la pâte. Ils sont représentés par la
muscovite qui est de petite taille (baguettes de 3 nms sur 80 ^ms dans DY59).- pas de calcite secondaire sauf pour OY126 et CYI06.
4 : KY5 - AY56 - AY109 - JY101 surtout caractérisé par la présence de hornblende (voir tabl pour autrescaractéristiques). Ces échantillons ne sont pas décorés et une quantité assez forte de dégraissants (surtout pourKY5). La présence de fragments de roches volcaniques rend ce groupe assez homogène (sauf en JY101). Il estaussi caractériser par l'abondance assez forte des biotites et des dégraissants de tailles très variées.
4b : BY48 - et dans une moindre mesure BY3 9 caractérisé par :- des micas non insérés dans la pâte et représentés surtout par la biotite.- une quantité de dégraissants assez forte, mais ces derniers sont moins grossier que
dans le groupe 2.- une structure de la pâte plutôt grossière.- pas mal d'éléments accessoires comme de l'apatite ou des zircons et c'est pour cela
que ce groupe se rapproche assez du groupe des GY (mais dégraissants moins grossiers et roches felsiques bienmoins abondantes). Cependant il a été repéré dans ces échantillons des roches volcaniques d'où la volonté derapprocher ce groupe avec le groupe 4.
- Dans BY39 a été trouvé en bel exemple de sanidine U-.
5 : FY63 - FY67 - FY97 est un groupe qui est à rapprocher du précédent par la présence moins élevée ici deroches volcaniques (sauf en FY63? - superbes exemples en FY67). Les autres traits de ce groupe sont:
- présence d'une engobe et d'une glaçure.- aspect fin de la pâte.- présence constante de fragments de quartzite et de micashiste.- pas de calcite secondaire.
Enfin quelques échantillons ne peuvent être rattachés à l'un de ces groupes sans hétérogérùser ces derniers.C'est le cas pour les QY, marqués par une assez forte quantité de dégraissants de forme anguleuse àsubanguleuse et de petite taille (environ 100 ums) et l'absence de roches felsiques et pyroclastiques.
Le groupe des EOL a aussi des caractéristiques différentes (voir tableau). Les autres échantillonsparaissent singuliers ou tout au moins difficilement classable :
- AY81 n'a rien en commun avec les autres lames minces : sa pâte est claire etsemble être un mélange de deux argiles, l'une très rousse et l'autre moins. Il y a très peu. de dégraissants sauf encertains endroits bien précis où la matice se trouve enrichie en fragments anguleux, cassés et de petite taille : onpeut penser alors que le potier a rajouté des morceaux d'argilites (c'est à dire des fragments de céramiques déjàcuits ) pour renforcer l'ossature de sa poterie. Cet échantillon reste assez riche en quartz et contient desmorceaux de calcite primaire (indices pour déterminer la température de cuisson, basse dans ce cas).
- DY74 aurait pu être rattaché au groupe 4 ou 4b s'il n'y avait pas une assez forteteneur en calcite.
- LY153 est particulièrement riche en muscovites insérées dans la pâte et l'absencede fragments de roches volcaniques fait qu'on ne peut la rattacher au groupe des FY.
- NY64.2 présente une singularité au niveau d'un pore de 2mms de long environ : un fragment deroche riche en grands cristaux de micas, de plagioclases et de quartz et à matrice apparemment carbonatée sedifférencie assez nettement de tout ce qu'on a vu auparavant. Cependant il est difficile de penser que cefragment ait pu être amené pendant l'utilisation de la poterie sans que celui-ci ne la déchire autour, c'estpourquoi il est délicat de parler de pollution.

Annexe 6
2 - GROUPES FORMES A PARTIR DE L'EXAMEN MINERALOGIOUE .
1 : HY1 - HY2 - IY52 - IY110 caractérisé par :- une teneur en quartz relativement basse (ce qui est à lier avec la faible quantité de
dégraissants).- l'absence remarquable de feldspaths alcalins.- un taux relativement très faible de micas, ce qui est un peu contradictoire avec les
observations pétrographiques.- la présence élevée d'augite, non observée au microscope. Il faut mettre en parallèle
que ces échantillons présentent les plus belles glaçures et que l'influence du sel de plomb n'est peut être paspour rien dans le fait que les pics à 2.99 et 2.96À soient d'intensité élevée.
2 : Le groupe des GY est aussi bien homogène et caractérisé par une teneur en quartz et en sanidine assezforte, la sous-abondance des plagioclases. Seul GY136 présente le pic à 3.70Â dont on ne connaît pas la valeursélective. La présence plus que douteuse d'augite est encore une fois à mettre en parallèle avec l'absence dedécoration.
3 : La présence de hornblende, qui est bien mieux détectable par diffractions des R.X, même s'il est en faiblequantité, permet de construire un groupe assez hétérogène, constitué des échantilons KY5, KY7.2, BY39,AY56, DY59, DY74, AY81, JY101 et AY109. Tous ces échantillons sont marqués par une abondancerelativement basse des quartz (sauf pour JY101), mais plus élevée des felspaths plagioclases et potassiques (saufpour AY56). En ce qui concerne le pic à 3.70Â, il n'est pas signicatif du groupe, tout comme la teneur encalcite et micas. C'est pourquoi, en fait, on isolera les poudres :
- AY81 très riche en micas et illite. Sur le diffractogramme apparaît très nettementaussi la présence d'une argile gonflante ou smectite, ce qui appuie bien l'hypothèse d'un mélange.
- DY74 est très riche en calcite.- BY39 qui est la seule poudre à âtre plus riches en plagioclases qu'en quartz.- JY101 est trop riche en quartz et peu en felspaths.
Si ce n'est la cause de la présence faible de la hornblende, DY59 pourrait être rattaché au groupeFY63, FY67, FY97, NY80 qui n'a pas de fortes particularités et qui de ce fait marque sa singularité.
Les échantillons QY et OY, ainsi que NY64.2 et CY106 peuvent être associés par la forte teneur enquartz et la faibe abondance relative de feldspaths. Si le pic à 3.70Â est représentatif, il faut remarquer qu'il estbien présent dans ces échantillons (sauf pour CY106). La teneur en hématite est aussi assez forte dans cegroupe, bien qu'au microscope on n'ait pas vu beaucoup de biotites.
Enfin quelques échantillons sont délicats à intégrer dans l'un de ces groupes:- BY48 pourrait être compris dans le groupe 3, mais la hornblende justement y est
absente.- le groupe des EOL se distingue de tous les autres ce qui est en accord avec les
données archéologiques, puisque il correspond en fait aux poteries "hellénistiques". Il est quand même assezhétérogène : l'échantillon EOL22 se trouve être énormément riche en quartz, richesse d'ailleurs non observéeen pétrographie.
- LY153 est trop fortement marqué par les micas.
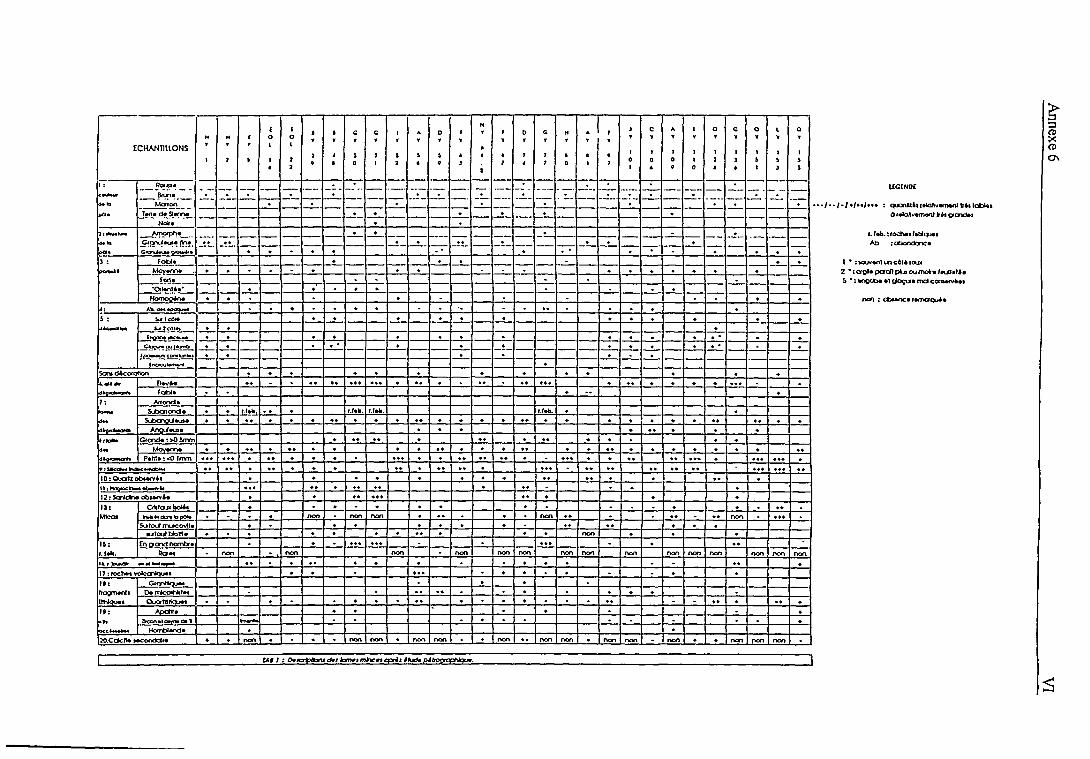
ECHANTILLONS
y.
, .
S :
SanacMcota
7:
fîouu«
ManonTwr* d« 3«on«
Nok*
A/TKvph«
Fatal*
fort*'Otont***
Homooért*
Ab. « M *paE>>*M
V* lcûtt
SurJoî**!
f r X» « u *Ctoçv*auf*xra
«on
Ajtond»
Subcna*J*u»«
&cnd«:>oeaynMov*rrt*
P«R1* : <0 Invn
10: Quartz ob**rvii
13:
15:
14: f ^Mtâ»
I I :fragment i
19:
n c c t i i n t i i
Ofafaaxboli*
Vrioirf nxacovtt*
En grand nombrePar*
ApaM*
HV
*
HT
—
*
*
«
non
*
r
•
•
• •
-
non
1ot
—
•
—
•
*
i
i
_
•
•
non
-
1V
—
*
•
•
• •
•
" 7 "
*• *
*
*
•
*
•
c
— • -
•
»
•
•
•
-
•
•
non
G
*
*
•
f.feh.
* •
*
• » • •
•
•
non
*
*
*•
non
•
*
-
*
• •
non
—
*
•
•
• •
non
•
- .
*
*
•
•
non
-
-
•
N
»
— •
*
•
Y
»
•
•
•
•
non
•
-
•
non
—!"
•
•
—~^~
non
•
• •
•
TÛT
. .
• • »
• •
-
• • •
•
non
—
*
*
• •
non*
•
non
*
*
*
non
non
*
•
*
**
*
• •
•
-
•
*
•
non
•
•
non
•
non
t
*
*
•
*
•
*
-
—
*
•
*
non
•
*
non
- - -
*
*
*
• • *
non
*
*
# «
*
*
non
*
*
—
*
-7-1
*+ •
• •
•
-
non
— -
*
*
• •
*
non
non
*
•
• • •
non
non
*
* »
non
*
X
- / . - / - / • / • • / • * • : quanBtitt»lanv«Twn!ti4ilcfc>l«iâ t*iaSv*moril té* grand*!
r. f «b. : roch*i f *UC*J«IAb rabondanc*
1 * :touv«nîmc6!tfoux2 " :S * :
non : ct»*nc» i
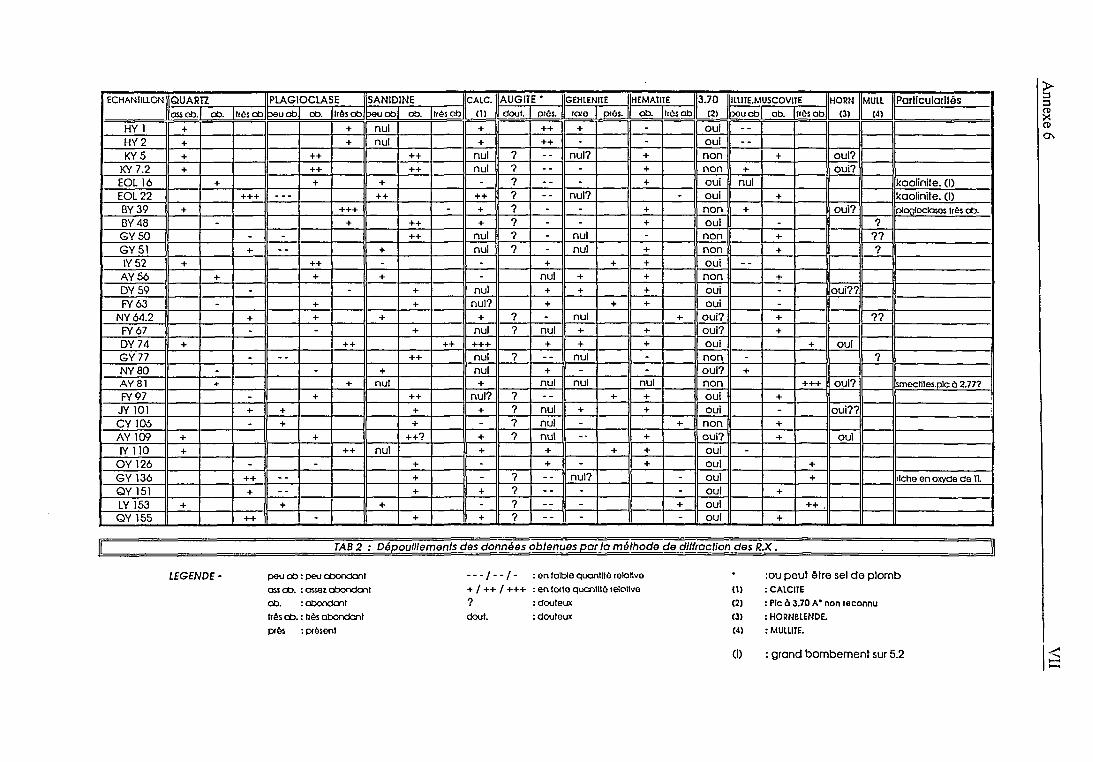
ECHANTILLON
HYlHY2KY5
KY7.2EOL16EOL22BY 39BY 48GY50GY51IY52
AY 56DY59FY63
NY 64.2FY67DY74GY77NY 80AY 81FY97
JY101CY 106AY 109IY110
OY126GY136QY151LY153QY155
QUARTZossob.
++++
+
+
+
++
+
Ob. |trèsob
+
-
+
-+
+++
-+
-
+-
-
+-
-+++
++
PLAGIOCLASEjeuob
-
- -
- -
++
- -- -+
ot>. |tfôsob
+++++
+++
++-
-
+
+
-
-
++
++++
-
++
+
++
SANIDINE5euob| ot>. jtrêsob
nulnul
+++
+-+
+
+nul
nul
+
++++
++++
++
+
++
++++
++?
+++
+
-
++
CAIC.(1)++
nulnul-
++++
nulnul--
nulnul?
+nul+++nulnul+
nul?+-++--+-+
AUGITE 'dout. | ptèi.
????????
??
?
????
????
++++- -- -- -- -----+
nul++-
nul+- -+
nul- -nulnulnul++- -- -- -- -
GEHLENITE
raie
+-
nul?--
nul?--
nulnul
++
nul++
nul-
nul
+--•
-nul?
---
près.
+
+
+
+
HEMATITE
ob. |lrès ob
-
-
+++
++-+++++
++--
nul++
+++
-
+
+
--+-
3.70(2)
ouiouinonnonOUI
OUI
nonouinonnonou!nonOUI
ouioui?oui?ouinonoui?nonouiouinonoui?OUI
ouiouiouiouioui
IILITE.MUSCOVITE
Deuob
- -
- -
+nul
+
- -
-+
-
at>.
+
+
++
+--++
+-++
+
+
liés ob
+
+++
++
++
HORN(3)
oui?oui?
oui?
oui??
oui
oui?
oui??
oui
MULL
M
????
??
?
Particularités
kaolinite. (1)kaolinite. CI)plogloclosGS liés at>.
smeclltes.plc à 2.77?
riche en oxyde cie Tl.
| TAB 2 : Dépouillements des données obtenues par la méthode de diffraction des R.X. . |
LEGENDE - peu ob : peu abondantossab. : assez abondantab. : abondanttiês ab. : très abondantprès : présent
/ - - / - :en faible quantité relative+ / ++ / +++ : en forto quantité lelollve? : douteuxdout. : douteux
0)(2)
(3)(4)
:ou peut être sel de plomb: CALC1TE
: Pic à 3.70 A* non reconnu
: HORNBLENDE.
: MULUTE.
(I) : grand bombement sur 5.2

Annexe 7 I
ANNEXE 7
A. MISE EN OEUVRE DE L'ANALYSE PAR PIXE EN FAISCEAU EXTRAIT
Vers un protocole d'analyse non destructive de glaçures plombifères par PIXE et PIGE
Nous avons mentionné au Chapitre 1 (1.1.2.) l'intérêt de l'analyse par faisceau extrait
dans le cas d'objets volumineux ou fragiles. Elle est toute indiquée pour des objets de musées
(e.g. [224]) ne pouvant être ni échantillonnés, ni modifiés par quelque traitement préalable (e.g.
application d'une couche de matériau conducteur). Elle permet également d'éviter ou de limiter
les dégâts d'irradiation. A titre exploratoire, nous avons mis en oeuvre cette technique pour
l'analyse des glaçures de quelques tessons byzantins, avec l'objectif de jetter les bases d'un
protocole d'analyse applicable pour tout matériau vitreux plombifère. Ce travail a été en partie
réalisé à l'occasion du stage de DEA de Thomas Keutgen, étudiant de l'Université catholique
de Louvain, au Laboratoire de Recherches des Musées de France (LRMF) [225]. Nous
insistons sur son caractère très préliminaire.
La glaçure est un matériau élaboré qui s'apparente à un verre. Elle résulte de la fusion
(parfois partielle seulement) d'un matériau formateur siliceux, d'un ou de plusieurs fondants
(alcalins, plomb, bore) qui déterminent le type de glaçure (plombifère, alcaline,...), d'éléments
stabilisants (e.g. Al) et éventuellement d'autres "ingrédients" comme des colorants (oxydes
métalliques) et des opacifiants (Sn).
Les techniques de fabrication des glaçures anciennes sont l'objet de nombreuses études
(e.g. [103, 104]). En raison de leur caractère composite, les matériaux vitreux sont rarement
abordés dans des problématiques de provenance, sauf pour la caractérisation spécifique de l'un
des constituants: produit colorant contenant des éléments traces signant son origine [226],
rapports isotopiques du plomb propres à une région [162]. Une indication indirecte sur la
provenance peut cependant être donnée par l'analyse élémentaire en déterminant la nature et la
proportion des différents éléments entrant dans la "recette" utilisée sur un site de production
donné. On peut cependant se demander quelle est la spécificité de cette "recette" dans un cadre
géochronologique défini.
Nous avons cherché à compléter la caractérisation des céramiques byzantines de
Pergame par l'analyse de leurs glaçures. Suite à la classification des tessons par la composition
élémentaire de leurs pâtes, quelques échantillons représentatifs des groupes de composition les
plus fournis (groupe byzantin local (byzl), groupe de céramiques à pâte grasse (f), groupe de
céramiques à pâte fine (hi)) ont été sélectionnés.
La méthode PIXE est plutôt bien adaptée à l'analyse de matériaux vitreux plombifères
[227, 228]. Elle permet d'une part de déterminer tous les éléments majeurs des matrices
vitreuses (éventuellement par couplage avec la méthode PIGE [229]), contrairement par
exemple à l'INAA par laquelle la détermination de Si est difficile (cf. Chapitre 1, 1.2.) et celle
de Pb impossible. Elle est d'autre part plus sensible que les autres techniques basées sur

Annexe 7 II
l'excitation de la fluorescence X pour le dosage des éléments de transition de la première série
qui constituent les principaux colorants des verres et glaçures.
Expérimentation
Les analyses ont été réalisées auprès de l'accélérateur tandem de 2 MV "AGLAE" du
LRMF [230]. Une ligne de faisceau à 30° est réservée à l'analyse en faisceau extrait.
L'extrémité de la ligne a été dotée au cours de cette série d'expériences d'un dispositif de
mesure de courant sur la fenêtre de sortie. Le schéma du dispositif est indiqué ci-dessous: les
particules rétrodiffùsées sur la fenêtre de sortie sont détectées par un détecteur à barrière de
surface, les particules transmises constituant le faisceau incident sur l'échantillon à analyser. Le
signal RBS intégré sur un intervalle d'énergie défini permet de "monitorer" le faisceau, ou de
déterminer la charge intégrée sur l'échantillon cible si une calibration par une mesure de
courant sur un conducteur placé sur la fenêtre de sortie a été effectuée au préalable. Une
fenêtre en Kapton (épaisseur 8 u.m) couverte d'une fine couche d'Au (100 nm) et une fenêtre
de Zr (épaisseur 4 um) ont été successivement utilisées. La première offre l'avantage d'une
bonne résistance aux radiations pour un matériau organique, alliée à un straggling faible [224].
La deuxième est de résistance supérieure mais produit des rayons X qui peuvent être vus du
détecteur Si(Li).
45'
Kapton
Dispositif d'extraction du faisceau à l'air.
La cible est placée à 2 cm de la fenêtre de sortie de la ligne et à 2 cm également du
Si(Li) positionné à 135° de la direction du faisceau incident. Un filtre de 50 um de Be est
interposé entre la cible et le détecteur afin d'éviter la pénétration de protons diffusés dans ce
dernier. L'ionisation de l'air sur le trajet du faisceau permet l'écoulement des charges et évite
leur accumulation sur la cible.

Annexe 7 III_
L'air joue aussi le rôle d'absorbant des rayons X émis, ce qui empêche la détermination
des éléments plus légers que Si dans la configuration adoptée. Ces éléments peuvent être dosés
soit par leurs raies X, en faisant baigner le dispositif dans une atmosphère d'hélium plutôt que
dans l'air [224], soit par les raies y résultant des réactions nucléaires induites par les protons.
Cette dernière méthode est nommée PIGE (Particle Induced Gamma-ray Emission) [6], Elle
est particulièrement intéressante dans le cas de glaçures sodiques car l'isotope 23Na présente
des sections efficaces élevées à basse énergie (résonance vers 1.4 MeV). Les résultats
préliminaires obtenus par cette méthode pour le dosage de Na, Al, Mg et Si par des réactions
de type (p,p'y) principalement sont rapportés par Keutgen [225]. Notons seulement que la mise
en oeuvre simultanée du PIXE et du PIGE est délicate, le PIGE nécessitant en général des
courants plus intenses et des énergies plus élevée en raison de la différence importante des
sections efficaces respectives (le rapport des sections efficaces d'émission entre rayons X et y
est de l'ordre de 103). D'autre part les fonctions d'excitation des réactions nucléaires (variation
des sections efficaces en fonction de l'énergie des particules incidentes) présentent des
résonances. A moins que l'utilisation de cet effet ne soit mise à profit pour des études de
profils, le choix de l'énergie incidente peut être critique, surtout si des cibles de composition
très différentes sont étudiées. Dans le cas d'acquisitions multiples sur un même échantillon,
l'analyse directe nécessite un dispositif spécial de positionnement afin de définir le point
d'impact. Un tel dispositif a été réalisé depuis au LRMF.
Pour l'analyse des glaçures plombifères par PIXE, des protons d'énergie 2.5 MeV
permettent de déterminer une gamme d'éléments dont l'énergie des raies caractéristiques varie
entre 1.7 et 16 keV environ, incluant les principaux constituants Si et Pb (dosé par ses raies L),
les fondants "secondaires" K, Ca et les colorants usuels des glaçures médiévales (Fe, Cu, Mn,
Co). De faibles courants (environ 0.1 nA) sont utilisés afin d'éviter les empilements. Des
incertitudes statistiques sur les surfaces des pics de moins de 10% sont obtenues avec des
temps d'acquisition de 10-15 min. Le détail des conditions expérimentales est donné en
annexe 1. Une deuxième aquisition avec un courant plus intense et un absorbant de 150 um
d'Al permet de déterminer les concentrations d'éléments comme Sn, Sb, Ba par leurs raies K.
Le travail à une énergie plus élevée (3.5 MeV) permet l'utilisation de la méthode PIGE dans
des conditions plus favorables (cf. [225]).
Les standards de verres au plomb Brill C (Corning), BCR 126 (Corning) et Oxford 4
(Oxford), mis à notre disposition par M. Eveno du LRMF et B. Velde de l'ENS, ont été utilisés
pour déterminer les concentrations par la méthode du standard externe. Les concentrations ont
été obtenues après deux itérations du terme d'effet de matrice calculé par la subroutine THICK
du programme BATTY, les éléments non détectés étant assimilés à de l'oxygène (cf. Chapitre
1, 1.1.2., Calcul des rendements par PIXAN (subroutine THICK)).
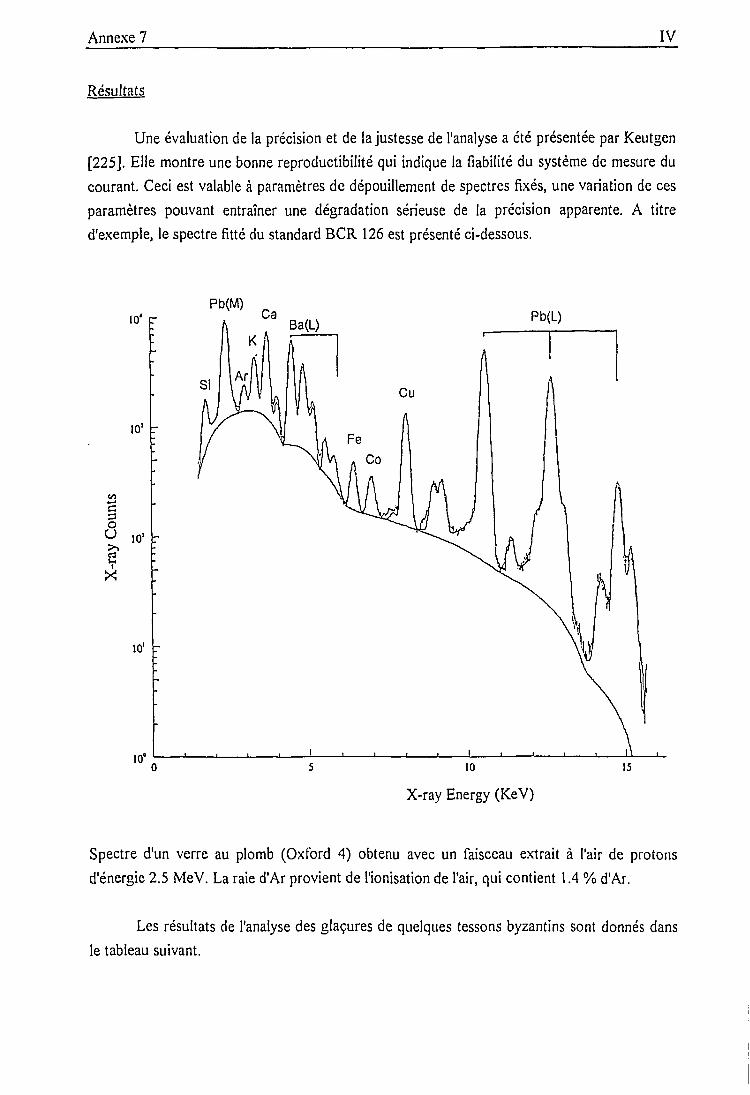
Annexe 7 IV
Résultats
Une évaluation de la précision et de la justesse de l'analyse a été présentée par Keutgen
[225]. Elle montre une bonne reproductibilité qui indique la fiabilité du système de mesure du
courant. Ceci est valable à paramètres de dépouillement de spectres fixés, une variation de ces
paramètres pouvant entraîner une dégradation sérieuse de la précision apparente. A titre
d'exemple, le spectre fitté du standard BCR 126 est présenté ci-dessous.
Pb(L)
10°15
X-ray Energy (KeV)
Spectre d'un verre au plomb (Oxford 4) obtenu avec un faisceau extrait à l'air de protons
d'énergie 2.5 MeV. La raie d'Ar provient de l'ionisation de l'air, qui contient 1.4 % d'Ar.
Les résultats de l'analyse des glaçures de quelques tessons byzantins sont donnés dans
le tableau suivant.
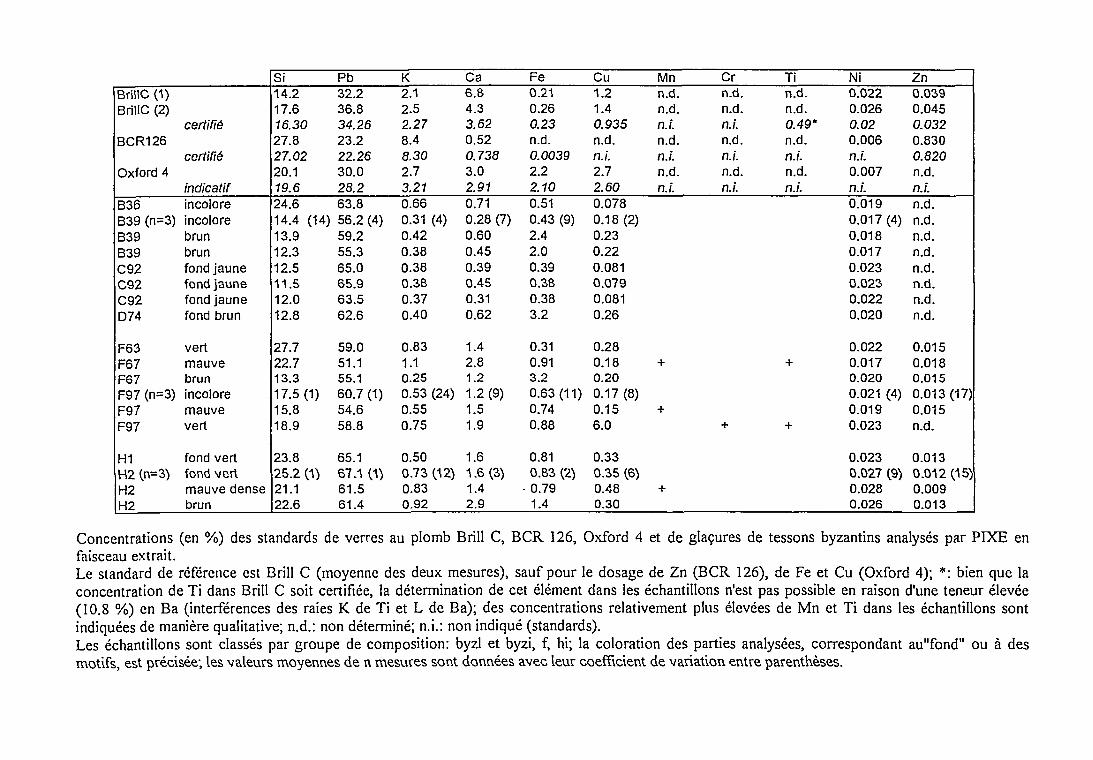
BrillC (1)BrillC (2)
BCR126
Oxford 4
B36B39 (n=3)B39B39C92C92C92D74
F63F67F67F97 (n=3)F97F97
H1H2 (n=3)H2H2
certifié
certifié
indicatifincoloreincolorebrunbrunfond jaunefond jaunefond jaunefond brun
vertmauvebrunincoloremauvevert
fond vertfond vertmauve densebrun
Si14.217.616.3027.827.0220.119.624.614.4 (14)13.912.312.511.512.012.8
27.722.713.317.5(1)15.818.9
23.825.2 (1)21.122.6
Pb32.236.834.2623.222.2630.028.263.856.2 (4)59.255.365.065.963.562.6
59.051.155.160.7 (1)54.658.8
65.167.1 (1)61.561.4
K2.12.52.278.48.302.73.210.660.31 (4)0.420.380.380.380.370.40
0.831.10.250.53 (24)0.550.75
0.500.73 (12)0.830.92
Ca6.84.33.620.520.7383.02.910.710.28 (7)0.600.450.390.450.310.62
1.42.81.21.2(9)1.51.9
1.61.6(3)1.42.9
Fe0.210.260.23n.d.0.00392.22.100.510.43 (9)2.42.00.390.380.383.2
0.310.913.20.63(11)0.740.88
0.810.83 (2)
• 0.791.4
Cu1.21.40.935n.d.n.i.2.72.600.0780.18(2)0.230.220.0810.0790.0810.26
0.280.180.200.17 (8)0.156.0
0.330.35 (6)0.480.30
Mnn.d.n.d.n.i.n.d.n.i.n.d.n.i.
+
+
+
Crn.d.n.d.n.i.n.d.n.i.n.d.n.i.
+
Tin.d.n.d.0.49*n.d.n.i.n.d.n.i.
+
+
Ni0.0220.0260.020.006n.i.0.007n.i.0.0190.017(4)0.0180.0170.0230.0230.0220.020
0.0220.0170.0200.021 (4)0.0190.023
0.0230.027 (9)0.0280.026
Zn0.0390.0450.0320.8300.820n.d.n.i.n.d.n.d.n.d.n.d.n.d.n.d.n.d.n.d.
0.0150.0180.0150.013(17)0.015n.d.
0.0130.012(15)0.0090.013
Concentrations (en %) des standards de verres au plomb Brill C, BCR 126, Oxford 4 et de glaçures de tessons byzantins analysés par PIXE enfaisceau extrait.Le standard de référence est Brill C (moyenne des deux mesures), sauf pour le dosage de Zn (BCR 126), de Fe et Cu (Oxford 4); *: bien que laconcentration de Ti dans Brill C soit certifiée, la détermination de cet élément dans les échantillons n'est pas possible en raison d'une teneur élevée(10.8 %) en Ba (interférences des raies K de Ti et L de Ba); des concentrations relativement plus élevées de Mn et Ti dans les échantillons sontindiquées de manière qualitative; n.d.: non déterminé; n.i.: non indiqué (standards).Les échantillons sont classés par groupe de composition: byzl et byzi, f, hi; la coloration des parties analysées, correspondant au'Tond" ou à desmotifs, est précisée; les valeurs moyennes de n mesures sont données avec leur coefficient de variation entre parenthèses.

Annexe 7 VI_
Pour les standards, matériaux homogènes dont les concentrations en Pb sont relativement
faibles et proches de celles du standard de référence, les valeurs obtenues sont en général
voisines des valeurs certifiées. Le dosage des échantillons de glaçures donne probablement des
valeurs beaucoup plus approximatives, deux raisons principales pouvant être invoquées. D'une
part, ces glaçures ont des teneurs en Pb beaucoup plus élevées que celle du standard de
référence. Malgré la double itération sur l'effet de matrice, il peut subsister une erreur sur la
détermination de Pb qui a des incidences importantes sur les autres éléments en raison de la
forte absorption des rayons X, en particulier pour les éléments légers comme Si. Les variations
de la concentration de Si au sein d'un même groupe d'échantillons (e.g. B36 dans le premier
groupe, F67 dans le deuxième), voire d'un même échantillon (F67), peuvent probablement être
attribuées à une mauvaise estimation de la teneur en Pb. D'autre part, cette estimation est
d'autant plus délicate que la glaçure n'est pas forcément homogène sur l'épaisseur analysée, en
raison notamment des altérations au cours de l'enfouissement [104, 46]. Ceci constitue un
obstacle majeur à la définition quantitative de sa composition. Nous reviendrons sur ce point,
dans la partie B de cette annexe, à propos de la distribution de Pb dans l'épaisseur de la
glaçure. Notons qu'une certaine "homogénéité latérale" est observée, puisque des mesures
effectuées sur un même échantillon en différents points d'impact dans les glaçures "de fond"
donnent des résultats similaires. Il est probable que l'épaisseur de la glaçure est en chaque
point supérieure à la profondeur d'analyse, et que l'engobe et la pâte sous-jacentes ne
contribuent pas au signal. Pour une glaçure contenant plus de 30% de Pb, la profondeur
analysée varie d'environ 5 um pour Si à 15 um pour Pb (raies L), le parcours moyen de
protons de 2.5 MeV étant de 35 um.
De ces résultats essentiellement qualitatifs peuvent être tirées quelques informations.
D'une part, Pb joue le rôle de fondant principal, voire exclusif. Les alcalins Ca et K sont
présents en faible proportion. Les concentrations en Na ne dépassent pa;; 1% d'après les
résultats du PIGE sur les échantillons du premier groupe (B36 à D74), et des analyses au MEB
sur deux échantillons des deuxième et troisième groupes (Hl et F67) indiquent une somme des
concentrations en Na2O et MgO inférieure à 1%. Par ailleurs, ces glaçures ne sont pas
stannifères. La détermination précise de Sn serait difficile dans les conditions présentes
d'analyse, car elle devrait se faire par l'intermédiaire des raies L interférant avec les raies K de
Ca et K. Une deuxième acquisition permettant le dosage de Sn par ses raies K serait alors
préférable.
La comparaison des résultats obtenus pour les différents groupes de tessons n'est pas
aisée du fait de teneurs en Si et Pb peu fiables. On peut remarquer que Ca et l'élément trace Zn
semblent être présents en proportion relativement plus importantes dans les matrices des
échantillons des deux derniers groupes (f et hi) - Zn étant en quantités inférieures aux limites
de détection dans le premier groupe. Il serait intéressant de savoir si ces variations peuvent
correspondre à l'utilisation de matières premières différentes. Une bonne connaissance des

Annexe 7 VH_
matériaux utilisés pour la fabrication des glaçures permettrait probablement de répondre sur ce
point.
Par ailleurs, les glaçures des céramiques des séries archéologiques F (groupe f) et H
(groupe hi) ont des matrices de compositions assez similaires pour les éléments dosés. Elles
sont pourtant d'aspect très différent. Dans le cas des tessons de la série F, l'application de la
couleur est relativement bien maîtrisée et sa diffusion est limitée (voir en particulier les traits de
couleur brun foncé dans F69 (figure 33)). Au contraire, dans les échantillons de la série H, la
teinte mauve est répartie de façon très "floue" (figure 35). Ceci ne traduit probablement pas un
manque de maîtrise dans la répartition des pigments mais au contraire la recherche d'un rendu
particulier. Quoi qu'il en soit, les résultats obtenus par analyse directe par PIXE ne mettent pas
en évidence l'utilisation de "recettes" différentes au niveau des matrices vitreuses. Une
détermination précise des éléments responsables de la viscosité du mélange lors de la cuisson
de la glaçure (e.g. Si, Al) et de l'ensemble des alcalins serait nécessaire pour être fixé sur ce
point. Il est également possible que les matrices vitreuses soient semblables et que les
dissemblances portent sur la partie colorée elle-même. Ceci ne semble pas être le cas pour les
échantillons examinés. Une autre explication réside dans une différence d'ordre technologique
qui ne se traduise pas par des variations élémentaires. En particulier, une première cuisson des
couleurs qui précéderait celle de la glaçure [136] a pu être pratiquée dans le cas des
céramiques de la série F (voir aussi à ce sujet les céramiques de la série L).
En ce qui concerne les éléments qui sont à l'origine de la coloration des glaçures, des
concentrations relativement élevées de Fe, Cu (de l'ordre du pourcent) et de Mn sont en
général associées aux couleurs brune, verte et mauve, respectivement. Certaines teintes de vert
ne correspondent cependant pas à des teneurs en Cu élevées. C'est en particulier le cas des
glaçures de "fond" vert clair des tessons de la série H. Les principaux autres éléments pouvant
donner une teinte verte aux glaçures plombifères (Cr et Ni suite à une cuisson en atmosphère
oxydante, Fe suite à une cuisson en atmosphère réductrice [231]) ne présentent pas non plus de
concentrations particulièrement élevées, mais une quantité minime peut être suffisante pour
générer la coloration.
Les analyses directes par PIXE en faisceau extrait doivent être considérés comme
qualitatives pour des matériaux peu homogènes comme les glaçures. Le type de glaçure et,
éventuellement, la nature des colorants peuvent être identifiés. Une analyse sur coupe sera plus
à même de donner un résultat fiable, pour autant qu'il soit possible de définir une composition
caractéristique (i.e. de séparer la glaçure "saine" des couches d'altération et des couches
d'interfaces). Les résultats très préliminaires obtenus semblent indiquer que les trois groupes de
céramiques analysés se distinguent non seulement par la composition élémentaire de leurs
pâtes, mais aussi par la nature des matières premières utilisées et par la technique de coloration
des glaçures. Une investigation plus poussée permettrait peut-être d'identifier des techniques
spécifiques à certaines de ces productions.

Annexe 7 VIII
B. MISE EN OEUVRE DE LA MICROSONDE PROTONIQUE POUR L'ANALYSE PAR
LA METHODE PIXE
Application à l'étude de l'interface argile - glaçure et à l'analyse d'inclusions de verres
volcaniques dans les pâtes céramiques
Ces dernières années ont connu un développement rapide des techniques de
microanalyse dans les laboratoires pratiquant les méthodes d'analyse par faisceaux d'ions - en
particulier le PIXE. La microsonde protonique a été notamment utilisée pour analyser des
matériaux archéologiques aussi divers que la céramique, le métal, le papier, les os... (cf. e.g.
[31]). A titre d'exemple, un dispositif de microfaisceau extrait réalisé à Lund a permis, grâce
aux techniques de traitements statistiques multivariés, de reconstituer le texte effacé d'un
manuscrit grec [232].
Les microfaisceaux sont souvent couplés avec un système de balayage selon le schéma
présenté ci-dessous. En chaque point d'analyse, ou pixel, le signal détecté est stocké avec les
coordonnées du point. Ceci permet non seulement de définir un spectre par pixel, mais
également de déterminer la répartition spatiale des éléments dans la zone analysée. Ce dernier
type d'utilisation correspond à la cartographie élémentaire. D'autres traitement des données
permettent également de réaliser des profils de concentrations ou encore des profils en énergie
des rayons X [233].
focusing scanningmagnets device
LJ target
elementalmap
scanningsystem
computer
Schéma de principe de la cartographie élémentaire en analyse par microsonde protonique
(d'après [233]). Le faisceau focalisé sur la cible est couplé à un système de balayage. Le
spectre de rayons X acquis en chaque point est associé aux coordonnées spatiales
correspondantes (x,y). La sélection d'une raie caractéristique d'un élément permet de
déterminer sa répartition sur l'ensemble de la zone analysée.

Annexe 7 LX
Une microsonde protonique a été récemment implantée auprès de l'accélérateur
AGLAE du LRMF, dans un dispositif d'analyse sous vide [234], Elle atteint une résolution
spatiale de l'ordre du micron, pour une définition limitée actuellement à 64x64 pixels. Le
logiciel d'exploitation MICROSYS [232] permet de traiter les données "on-line" (i.e. pendant
l'acquisition). Il est couplé au logiciel MICROSTAT [233] destiné aux traitements statistiques
"off-line".
Le positionnement de l'échantillon est contrôlé à l'aide d'une loupe binoculaire visant le
point d'impact du faisceau à 45°. Lorsque la structure de l'échantillon est connue, la zone
d'analyse peut être redéfinie de manière plus précise à partir des contrastes élémentaires
obtenus lors d'une première acquisition de courte durée. L'opération s'effectue par modification
des coordonnées du point origine et de la distance (nombre de pas de balayage) entre chaque
pixel. Le pas, qui est fonction de l'énergie des protons et du champ électromagnétique régnant
entre les bobines déflectrices, est déterminé en analysant une grille de périodicité connue. Un
channeltron détectant les électrons secondaires donne une image topographique de
l'échantillon, avec une résolution qui n'est en aucun cas comparable à celle d'un MEB (de
l'ordre du um, au mieux, contre quelques nm pour ce dernier). Cette image permet cependant
de repérer les principales irrégularités de surface qui sont à l'origine de contrastes
"élémentaires" artificiels. Une analyse plus fiable se fera sur échantillon poli.
Les photographies de la page suivante illustrent, à titre d'exemple, certaines possibilités
du système d'analyse. L'échantillon examiné est une coupe de céramique (tesson ¥67), la
glaçure étant située dans la partie droite et la pâte céramique dans la partie gauche. La
photographie du haut présente un enchaînement de procédures courantes. Le spectre global de
l'ensemble de la zone analysée (en haut, à gauche) est utilisé pour la calibration et la définition
des intervalles d'énergies correspondant aux raies caractéristiques des éléments à cartographier.
Par exemple, le signal intégré sur un intervalle encadrant les raies La de Pb, pointées sur le
spectre par le curseur, permet de générer la cartographie de cet élément (en haut à droite). Le
contraste, qui se traduit par une différence de relief et de couleur, permet de distinguer de
façon nette la glaçure, riche en plomb, de la pâte. Il faut remarquer que les cartes obtenues
correspondent à des contrastes d'intensité, et non à des contrastes élémentaires. Outre les
concentrations, des facteurs comme le niveau de bruit de fond et l'épaisseur de l'échantillon
interviennent également. Ces facteurs peuvent être pris en compte par le programme
d'exploitation [233], mais une cartographie portant réellement sur les concentrations demande
des traitements supplémentaires [235]. Nous avons ensuite défini une zone dans la pâte
céramique (en bas, à droite), dont le spectre est présenté dans le dernier cadre (en bas, à
gauche). La sommation sur l'ensemble des pixels de cette zone permet d'obtenir une bonne
statistique, ce qui n'est pas le cas pour un pixel unique. Nous remarquons que les raies
caractéristiques de Pb sont présentes dans le spectre de la pâte. Cet élément n'est pourtant pas

Annexe 7 X
ttnn» (T JIAW worn wn n H^HVI-WIA.
Spectre de l'ensemble de la zone analysée (en haut à gauche) et spectre d'une région de la pâte
céramique (en bas, à gauche) définie (en bas, à droite) à partir de la cartographie élémentaire
de Pb (en haut, à droite).
:7
Cartographie élémentaire de Pb à l'interface pàte-glaçure montrant l'hétérogénéité de la glaçure
et la diftusion de Pb de la glaçure vers la pâte (correspondance entre intensités I et couleurs:
blanc: 250<I; jaune: 200<I<250; orange: 150<I<200; bnin: 100<I<150; vert: 10<I<100; bleu:
3<I<10); dimensions: 500x500 \xm environ; définition "gonflée" à 256x256 pixels, définition
réelle 64x64 pixels.
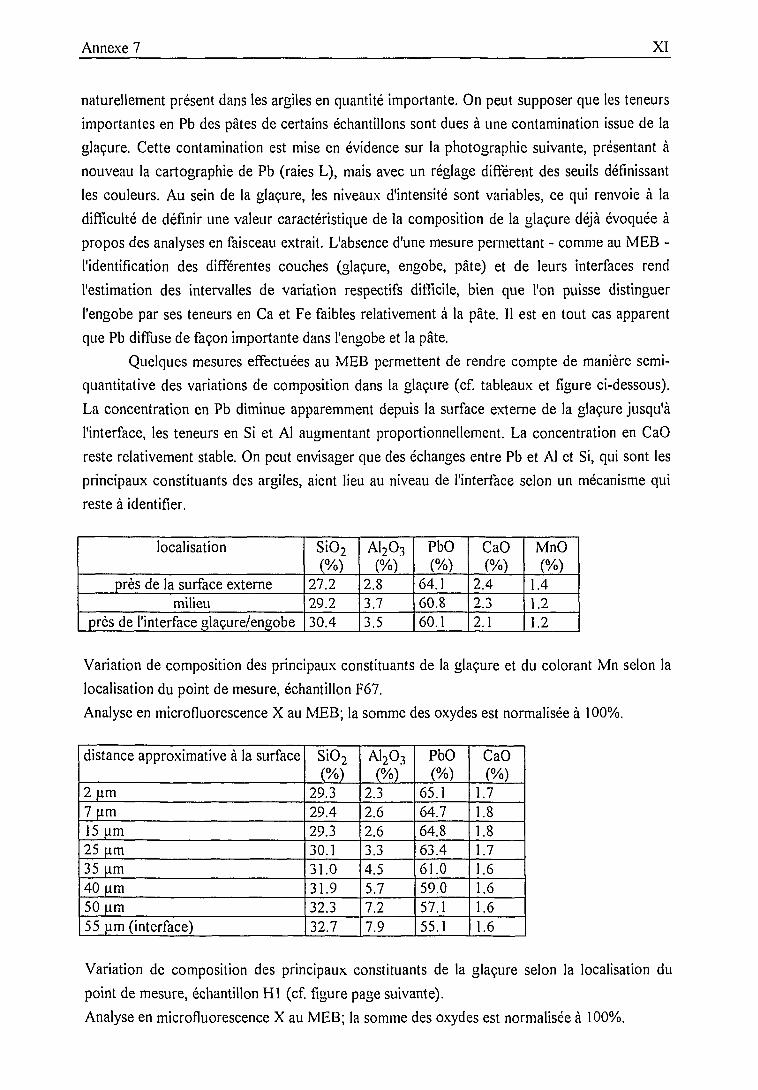
Annexe 7 XI
naturellement présent dans les argiles en quantité importante. On peut supposer que les teneurs
importantes en Pb des pâtes de certains échantillons sont dues à une contamination issue de la
glaçure. Cette contamination est mise en évidence sur la photographie suivante, présentant à
nouveau la cartographie de Pb (raies L), mais avec un réglage different des seuils définissant
les couleurs. Au sein de la glaçure, les niveaux d'intensité sont variables, ce qui renvoie à la
difficulté de définir une valeur caractéristique de la composition de la glaçure déjà évoquée à
propos des analyses en faisceau extrait. L'absence d'une mesure permettant - comme au MEB -
l'identification des différentes couches (glaçure, engobe, pâte) et de leurs interfaces rend
l'estimation des intervalles de variation respectifs difficile, bien que l'on puisse distinguer
l'engobe par ses teneurs en Ca et Fe faibles relativement à la pâte. Il est en tout cas apparent
que Pb diffuse de façon importante dans l'engobe et la pâte.
Quelques mesures effectuées au MEB permettent de rendre compte de manière semi-
quantitative des variations de composition dans la glaçure (cf. tableaux et figure ci-dessous).
La concentration en Pb diminue apparemment depuis la surface externe de la glaçure jusqu'à
l'interface, les teneurs en Si et Al augmentant proportionnellement. La concentration en CaO
reste relativement stable. On peut envisager que des échanges entre Pb et Al et Si, qui sont les
principaux constituants des argiles, aient lieu au niveau de l'interface selon un mécanisme qui
reste à identifier.
localisation
près de la surface externemilieu
près de l'interface glaçure/engobe
SiO2
(%)27.229.230.4
A12O3
(%)2.83.73.5
PbO(%)
64.160.860.1
CaO(%)
2.42.32.1
MnO(%)
1.41.21.2
Variation de composition des principaux constituants de la glaçure et du colorant Mn selon la
localisation du point de mesure, échantillon F67.
Analyse en microfluorescence X au MEB; la somme des oxydes est normalisée à 100%.
distance approximative à la surface
2 um7 um15 um25 um35 um40 um50 um55 um (interface)
SiO2
(%)29.329.429.330.131.031.932.332.7
A12O3(%)
2.32.62.63.34.55.77.27.9
PbO(%)
65.164.764.863.461.059.057.155.1
CaO(%)
1.71.81.81.71.61.61.61.6
Variation de composition des principaux constituants de la glaçure selon la localisation du
point de mesure, échantillon Hl (cf. figure page suivante).
Analyse en microfluorescence X au MEB; la somme des oxydes est normalisée à 100%.
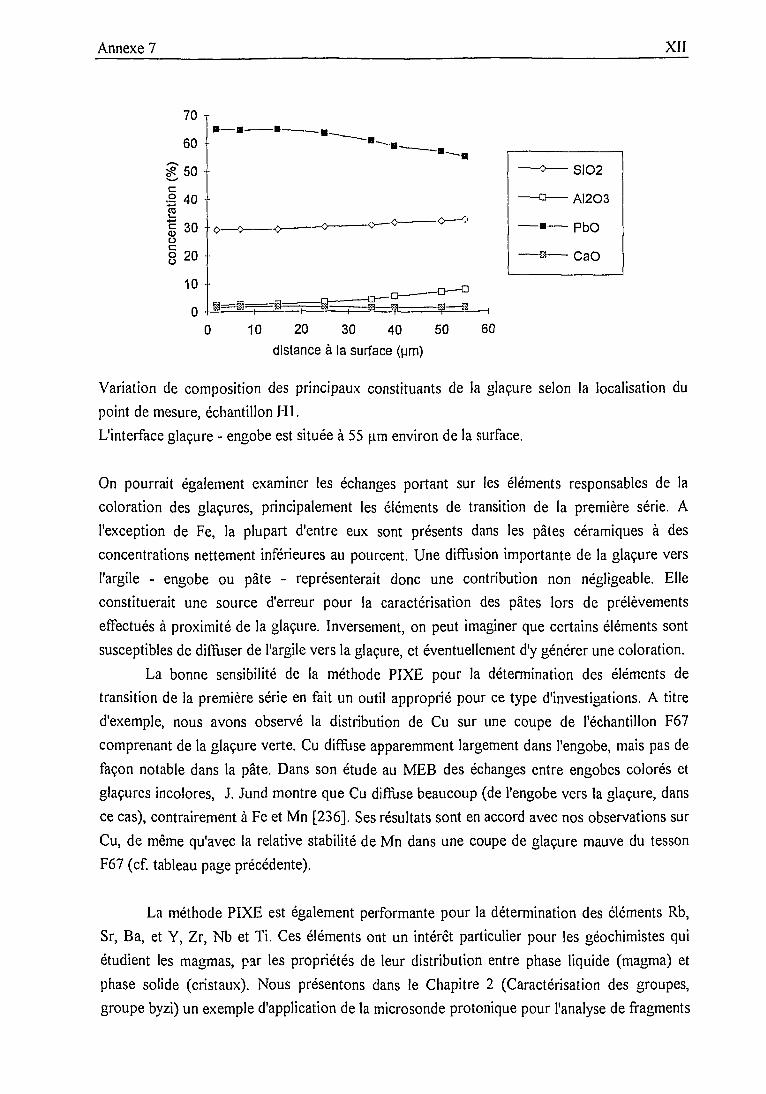
Annexe 7 XII
70 !
60
g 50 +.2 40Sg 30+O-o§ 20
10
0
SI
SiO2
AI2O3
PbO
CaO
10 20 30 40 50
distance à la surface (um)
60
Variation de composition des principaux constituants de la glaçure selon la localisation du
point de mesure, échantillon Hl.
L'interface glaçure - engobe est située à 55 um environ de la surface.
On pourrait également examiner les échanges portant sur les éléments responsables de la
coloration des glaçures, principalement les éléments de transition de la première série. A
l'exception de Fe, la plupart d'entre eux sont présents dans les pâtes céramiques à des
concentrations nettement inférieures au pourcent. Une diffusion importante de la glaçure vers
l'argile - engobe ou pâte - représenterait donc une contribution non négligeable. Elle
constituerait une source d'erreur pour la caractérisation des pâtes lors de prélèvements
effectués à proximité de la glaçure. Inversement, on peut imaginer que certains éléments sont
susceptibles de diffuser de l'argile vers la glaçure, et éventuellement d'y générer une coloration.
La bonne sensibilité de la méthode PIXE pour la détermination des éléments de
transition de la première série en fait un outil approprié pour ce type d'investigations. A titre
d'exemple, nous avons observé la distribution de Cu sur une coupe de l'échantillon F67
comprenant de la glaçure verte. Cu diffuse apparemment largement dans l'engobe, mais pas de
façon notable dans la pâte. Dans son étude au MEB des échanges entre engobes colorés et
glaçures incolores, J. Jund montre que Cu diffuse beaucoup (de l'engobe vers la glaçure, dans
ce cas), contrairement à Fe et Mn [236]. Ses résultats sont en accord avec nos observations sur
Cu, de même qu'avec la relative stabilité de Mn dans une coupe de glaçure mauve du tesson
F67 (cf. tableau page précédente).
La méthode PIXE est également performante pour la détermination des éléments Rb,
Sr, Ba, et Y, Zr, Nb et Ti. Ces éléments ont un intérêt particulier pour les géochimistes qui
étudient les magmas, par les propriétés de leur distribution entre phase liquide (magma) et
phase solide (cristaux). Nous présentons dans le Chapitre 2 (Caractérisation des groupes,
groupe byzi) un exemple d'application de la microsonde protonique pour l'analyse de fragments

Annexe 7 M
de verres volcaniques inclus dans les pâtes céramiques. Dans cet exemple, le rapport des
teneurs en Rb, Sr, Y et Zr de zones définies dans la matrice vitreuse est considéré. L'efficacité
de détection variant peu dans cette région du spectre, nous avons considéré qu'en première
approximation les rapports des surfaces des pics caractéristiques (corrigées des interférences)
sont équivalents aux rapports des concentrations. Les conditions expérimentales permettant la
détermination simultanée des éléments Ca à Ba par leurs raies K sont données en annexe 1. Le
dosage de ces éléments nécessiterait un dispositif de mesure du courant et la connaissance de la
courbe d'efficacité du détecteur.
La microsonde sert ici essentiellement à définir les zones d'analyse. L'utilisation des
cartographies élémentaires permet de ne pas inclure de phénocristaux dans ces zones. Les
phénocristaux sont surtout dans notre cas des plagioclases et des ferromagnésiens. Les
premiers sont repérables par leurs teneurs importantes en Ca, les deuxièmes par leurs teneurs
importantes en Fe.
L'utilisation de la microsonde protonique pour l'analyse par la méthode PIXE est de
mise en oeuvre délicate. D'une part, l'absence de dispositif d'observation directe à la
microsonde d'AGLAE au moment des expériences rend le positionnement de l'échantillon et
l'interprétation des structures fines difficile. Une bonne connaissance de la répartition des
concentrations élémentaires ou un repérage préalable au MEB permettraient probablement une
exploitation plus fiable des données. D'autre part, on peut mettre en évidence des contrastes
d'intensité, mais la réalisation de cartographies élémentaires au sens strict du terme
nécessiterait une détermination précise du courant ainsi qu'un étalonnage de la gamme de
contrastes à l'aide de dépouillements "off-line" [235].

Annexe 8
ANNEXE 8
BibliographieIndex alphabétique
Alfassi, Z.V., Lavi, N., Simultaneousdetermination of sodium, magnesium,aluminium, silicon and phosphorus byinstrumental neutron activation analysisusing reactor and epithermal neutrons,Analyst, 109, 959-962 (1985)
Alter tiimer von Pergamon, DeutschesArchàologisches Institut, Walter de Gruyter&Co, BerlinKeramik, Band XIV, 123-143, planches 23-29 (1984)
Andersen, H.H., Ziegler, J.F., The stoppingarid ranges of ions in matter, vol. 3,Pergamon Press, New York (1977)
Archéologie et calcul, 10/18, Paris (1978)
Armstrong, P., Byzantine Thebes:excavations on the Kadmeia, 1980, Annualof the British School at Athens, 88, 295-335, pi. 29-36(1993)
Armstrong, P., Some Byzantine and latersettlements in Eastern Phokis, Annual of theBritish School at Athens, 84, 1-47, pi. 1-11(1989)
Aslanapa, 0., Yetkin, S., Altun, A., TheIznik tile kilns excavations (The secondround: 1981-1988), Istanbul (1989)
Aspiazu, J., Mise au point d'un programmeinformatique pour l'analyse quantitativepar la méthode FIXE, Thèse de doctorat,Université Strasbourg I (1991)
Attas, M., Yaffe, L., Fossey, J.M., Neutronactivation analysis of early bronze agepottery from lake Vouliagmeni, Perakhora,central Greece, Archaeometry, 19, 1, 33-43(1977)
Attas, M., Analyse par activationneutronique de la céramique de Lerne(Grèce) à l'âge du bronze ancien:productions locales et échangescommerciaux, Thèse de 3 è m e cycle,Université Paris-Sud Orsay (1980)
Attas, M., Fossey, J.M., Yaffe, L.,Corrections for drill-bit contamination insampling ancient pottery for neutronactivation analysis, Archaeometry, 26, 1,104-107 (1984)
Baijot-Stroobants, J., Bodart, F., Ancientpottery analysis by proton inducedbombardment and Môssbauer spectroscopy,Nucl. Instr. andMeth., 142, 293-300 (1977)
Bakirer Ô., The Medieval pottery andbaked clay objects, dans: Korucutepe, FinalReport on the Excavation of theUniversities of Chicago, California (LosAngeles) and Amsterdam in the KebanReservoir, Eastern Anatolia 1968-1970,Studies in ancient civilization, vol. 3, 189-249(1980)
Bakirtzis, C, Papanikola-Bakirtzis, D., Dela céramique byzantine en glaçure àThessalonique, Byzantino-Bulgarica, VII,421-434(1981)
Balfet, H., Fauvet Berthelot, M.F.,Monzon, S., Lexique et typologie despoteries. Pour la normalisation de ladescription des poteries, Presses duC.N.R.S. (1989)
Barnes, I.L., Brill, R.H., Deal, E.C., Piercy,G.V., Lead isotope studies of some of thefinds from the Serçe Liman shipwreck,Proc. 24th Intern. ArchaeometrySymposium, Olin, J.S., Blackman, M.J.,éds., Smithsonian Inst. press, WashingtonD.C., 1-12 (1986)

Annexe 8 II
Bearat, H., Etude de quelques altérationsphysico-chimiques des céramiquesarchéologiques, Thèse de doctorat,Université de Caen (1990)
Bearden, J.A., X-ray wavelengths, Rev.Mod. Phys., 39, 1. 78-124 (1967)
Beier, T., Mommsen, H., ModifiedMahalanobis filters for grouping pottery bychemical composition, Archaeometry, 36, 2,287-306(1994)
Besnus, Y., Etude géochimique sur desfaciès marnes noires du Trias du Sud-Est duMassif Central (France), Sciences de laTerre, 25, 2, 219-239 (1982)
Bird, J.R., X-ray spectrum processing,interpretation and computerisation dans:Amsel, G., Bird, J.R., Heitz, Ch., Lahanier,Ch., Menu, M., Overview of the discussionsat the Premontres workshop, Nucl. Instr.andMeth. B, 14, 162-167 (1986)
Birgùl, 0., Diksic, M., Yaffe, L., X-rayfluorescence analysis of Turkish clays andpottery, Archaeometry, 21, 2, 203-218(1979)
Birgul, 0., Diksic, M., Yaffe, L., Activationanalysis of Turkish and Canadian clays andTurkish pottery, J. Radioanal. Chem., 39,45-62(1977)
Boas, A., The import of Western ceramicsto the Latin kingdom of Jerusalem, IsraelExploration Journal, 44, 1-2 (1994)
Bordas, M., Possibilités offertes parl'utilisation simultanée des méthodes PIXEet PIGE en analyse élémentaire, Thèse dedoctorat, Université Strasbourg I (1988)
Borsi, S., Ferrara, G., Innocenti, F.,Mazzuoli, R., Geochronology and petrologyof recent volcanics in the eastern Aegeansea (West Anatolia and Lesvos island),Bulletin de volcanologie, 36, 473-496(1972)
Boulle, G.J., Peisach, M., Trace elementanalysis of archaeological materials and theuse of pattern recognition methods toestablish identity, J. Radioanal. Chem., 50,1-2, 205-215 (1979)
Bouroche, J.-M., Saporta, G., L'analysedes données, P.U.F., Collection "Que sais-je?" (1989)Brinkmann, R., Izdar, E., Excursion to
Bergama (Pergamon) and Kinik, dans:Geology and history of Turkey, Campbell,A.S., éd., Petroleum Exploration Society,Tripoli, 509-511(1971)
Brun, N., Etude de verres opaquesceltiques et gallo-romains, Thèse dedoctorat, Université Paris-Sud Orsay (1991)
Cahill, T.A., Kusko, B.H., Schwab, R.N.,Analyses of inks and papers in historicaldocuments through external beam PIXEtechniques, Nucl. Instr. and Meth, 181,205-208(1981)
Cahill, T.A., McColm, D.W., Kusko, B.H.,Control of temperature in thin samplesduring ion beam analysis, Nucl. Instr. andMeth. B, 14, 38-44(1986)
Caillère, S., Hénin, S., Minéralogie desargiles, Masson, Paris (1968)
Calamiotou, M., Filipakkis, S.E., Jones,R.E., Kassab, D., X-ray and spectrographicanalyses of terracotta figurines from Myrina:an attempt to characterize workshops, J.Archaeological Science, 11, 103-117(1984)
Calogero, S., Lazzarini, L., Caratteriz-zazione Chimico-Fisica di CeramicheGraffite Bizantine e Veneziane Arcaichetrovate nella Laguna Veneta, Faenza, 69,60-70, pi. 11 (en italien) (1983)
Campbell, J.L., Maenhaut, W., Bombelka,E., Clayton, E., Malmqvist, K., Maxwell,J.A., Pallon, J., Vandenhaute, J., Anintercomparison of spectral data processingtechniques in PIXE, Nucl. Instr. and Meth.B, 14,204-220(1986)

Annexe 8 III
Carrot, F., Dardenne, C, Deschamps, N.,Lahanier, C, Revel, G., Determination ofoptimum conditions for INAA ofarchaeological clay figurines, J. Radioanal.Nucl. Chenu, 168, 2, 287-296 (1993)
Carte géologique de la Turquie, feuilletBalikesir (G4), 100.000èmc, MineraiResearch and Exploration Institute(M.T.A.), Ankara (1989)
Catling, H.W., Richards, E.E., Blin-Stoyle,A.E., Correlation between composition andprovenance of Mycenaean and Minoanpottery, Annual of the British School atAthens, 58, 94-115(1963)
Cauvin, M.-C, Besnus, Y., Tripier, J.,Montigny, R., Nouvelles analysesd'obsidiennes du proche-Orient: modèle degéochimie des magmas utilisé pour larecherche archéologique, Paléorient, 17, 2,5-20(1991)
Chu, W.-K., Mayer, J.W., Nicolet, M.A.,Backscatteting spectromelry, AcademicPress, New York (1978)
Clayton, E., PIXAN - The Lucas HeightsPIXE analysis package, Australian AtomicEnergy Report, AAEC-M113 (1986)
Clayton, E., BATTY 83: a computerprogram for thick target PIXE analysis,Nucl. Instr. andMeth., 218, 221-224 (1983)
Clayton, E., Uncertainties in theoreticalthick target PIXE yields, Nucl. Instr. andMeth., 191, 567-572 (1981)
Clayton, E., Cohen, D.D., Duerden, P.,Thick target PIXE analysis and yield curvecalculations, Nucl. Instr. and Meth., 180,541-548(1981)
Coche de la Ferté, E., Décors en céramiquebyzantine au musée du Louvre, Cahiersarchéologiques, 9, 187-217 (1957)
Cohen, D.D., Harrigan, M., Calculated L-shell X-ray line intensities for proton andhelium ion impact, At. Data and Nucl. DataTables, 34, 3, 393-414(1986)
Cohen, D.D., Harrigan, M., K- and L-shellionization cross sections for protons andhelium ions calculated in the ECPSSRtheory, At. Data and Nucl. Data Tables, 33,2,255-343 (1985)
Cohen, D.D., A radially dependentphotopeak efficiency model for Si(Li)detectors, Nucl. Instr. andMeth., 178, 481-490(1980)
Colloque sur le vocabulaire techniquecéramique en archéologie, Compte rendu,Annales du LRMF, 74-79 (1971)
Courtois, L., Examen au microscopepétrographique des céramiquesarchéologiques, Notes et monographiestechniques, C.R.A., 8 (1976)
Currie, L.A., Limits for qualitativedetection and quantitative determination,Anal. Chem, 40, 586-593 (1968)
D'Ambrosio, B., Mannoni, T., Sfrescola, S.,Stato délie richerche mineralogiche sulleceramiche mediterranee, dans: La ceramicamédiévale nel mediterraneo occidentale,Florence, 601-609(1986)
Day, P., Technology and ethnography inpétrographie studies of ceramics, dans:Archaeometry, Proc. 25 th Int.Archaeometry Symposium (Athens 1986),Maniatis, Y., éd., 139-147 (1989)
De Soete, D., Gijbels, R., Hoste, J.,Neutron activation analysis, ChemicalAnalysis, 34, Wiley-Interscience, New York(1972)
Deer, W.A., Howie, R.A., Zussman, I ,Rock forming minerals, vol.3: Sheetsilicates, Longmans, Londres (1962)

Annexe 8 IV
Demortier, G., Review of recentapplications of high energy microprobes inarts and archeology, NucL Inslr. and Melh./Î, 54,334-345 (1991)
Dewey, J.D., Handbook of X-ray andmicroprobe data, Elion, H.A., Stewart,D.C., éds., Pergamon Press, New York(1966)
Diebolt, J., Ricq, J.C., An attempt to selectsuitable elements to characterize an ancientceramic by neutron activation analysis, J.Radioanal. Nucl. Chem., 39, 9-20 (1977)
Dikili-Bergama, Jeotermal Araçtirma,Sahasina Iliçkin, M'l'A Rapor, 5444 (1972)
Djingova, R., Kuleff, I., Penev, I., Chemicalcontent of medieval sgraffito ceramicfindings II. Investigation of archaeologicalfindings from Kaliakra, Kavarna andBaltchik (XII - XIV c), Izv. Archaeol.Museum, Varna (en bulgare, résumé anglais)(sous presse)
Djingova, R., Kuleff. I., Provenance studyof pottery and glass by INAA, dans:Neutron Activation and Plasma EmissionSpectrometric Analysis in Archaeology,Techniques and Applications, Hugues, M.J.,Cowell, M.R., Hook, D.R., éds., BritishMuseum occasional paper, 82, 121-141(1991)
Djingova, R., Kuleff, I., Penev, I.,Instrumental neutron activation analysis ofreference materials for archaeometricinvestigations of pottery, J. Radioanal.Nucl. Chem. Letters, 144, 6, 397-406(1990)
Duerden, P., Clayton, E., Bird, J.R.,Cohen, D.D., Recent ion beam analysisstudies on archaeology and art, Nucl. Inslr.andMeth. B, 14, 50-57 (1986)
Dufournier, D., Etude comparative deplusieurs tessons d'une même poterie,Archéologie Médiévale, 2, 305-323 (1972)
Dupont, P., Classification et provenancedes céramiques orientales d'Istros, Dacia,27, 1/2, 19-43 (1983)
Dupont, P., Recherches de laboratoire surles céramiques grecques orientalesarchaïques, Revue d'Archéométrie, 1, 105-114(1977)
Eberhart, J.-P., Analyse structurale etchimique des matériaux, Dunod, Paris(1989)
Echallier, J.-C, Méry, S., L'évolutionminéralogique et physico-chimique des pâtescalcaires au cours de la cuisson:expérimentation en laboratoire et applicationarchéologique, Documents et TravauxIGAL, 16,87-120(1992)
Echallier, J.C., Jallot, L., Le matérielcéramique de Moulin Villard (Caissargues,Gard). Remarques sur la formalisation etl'analyse des données provenantd'observations microscopiques et résultats,Revue d'Archéométrie, 16, 71-87 (1992)
Echallier, J.C., Eléments de technologiecéramique et d'analyse des terres cuitesarchéologiques, Documents d'archéologieméridionale, Série méthodes et techniques,3, 5-38 (1984)
Empereur, J.-Y., Picon, M., A propos d'unnouvel atelier de "Late Roman C", Figlina,7, 143-146(1986)
Erdemgil, S., Le quartier des potiers àPergame, Le monde de la bible, 11, 33-34(1992)
Erdemgil, S., Ozenir, S., Preliminary reporton the kilns excavated in Ketios valley,Rivista di Archaeologia, 6, Tecnologianeli'Antichita, 2, 109(1982)
Erdtmann, G., Soyka, W., Gamdat'78,Zentralabteilung fur Chemische Analysender Kernforschungsanlage Julich GmbH(1978)

Annexe 8
Evans, R.D., The Atomic nucleus,Graw-Hill, New York (1955)
Me
Fleming, S.J., Swann, C.P., Recentapplications of PIXE spectrometry inarchaeology II.Characterization of Chinesepottery exported to the Islamic world, Nucl.Inslr. and Meth. B, 64, 528-537 (1992)
Fleming, S.J., Swann, C.P., Color additivesand trace elements in ancient glasses:specialized studies using PIXEspectrometry, Nucl. Inslr. and Meth. B, 22,411-418(1987)
Folkmann, F., Gaarde, C, Huus, T., Kemp,K., Proton Induced X-ray Emission as a toolfor trace element analysis, Nucl. Instr. andMeth.,U6, 487-499(1974)
Fontes, P., Brissaud, I., Lagarde, G.,Leblanc, I , Person, A., Saliège, J.-F., Heitz,Ch., PIXE analysis and provenance study ofarchaeological potsherds from West Africa,Nucl. Instr. and Meth. B, 3, 404-407 (1984)
Foss, C, Byzantine and Turkish Sardis,Archaeological Exploration of Sardis,monograph 4, Harvard University Press,Cambridge Mass. (1976)
François, V., La céramique byzantine deThasos et un échantillonnage desproductions médiévales orientales etoccidentales du XHIe au XVe siècle, Thèsede doctorat, Université Strasbourg II, 3volumes (1990)
Franklin, U.M., Hancock, R.G.V., Theinfluence of post-burial conditions on traceelement composition of ancient sherds,Actes du XXème symposium internationald'archéomélrie, Revue d'Archéométrie,supplément 1981, vol III, 111-119 (1981)
Frierman, J.D., The physical and chemicalproperties of some crusaders ceramics,Israel Exploration Journal, 17, 153-157(1967)
Fytikas, M., Innocenti, F., Manetti, P.,Mazuoli, R., Peccerillo, A., Villari, L.,Tertiary to quaternary evolution ofvolcanism in the Aegean region, dans: Thegeological evolution of the EasternMediterranean, Special Publication of theGeological Society, 17, 687-699(1985)
GANAAS/SPEDAC, Nuclear analysissoftware, IAEA, Vienne (1991)
Gautier, J., Myrina: centre de productionou de consommation?, dans: Etudescéramiques en archéologie, Documents etTravaux IGAL, Paris, 9, 27-93 (1985)
Geological map of Turkey, Izmir,500.000èinc, Mineral Research andExploration Institute (M.T.A.), Ankara(1973)
Geostandard Newsletters, 13, Special Issue(1989)
Gilmore, G.R., Sources of uncertainty inthe neutron activation analysis of pottery,dans: Neutron activation and plasmaemission spectrometry analysis inarchaeology. Techniques and applications,Hughes, M.J., Cowell, M.R., Hook, D.R.,éds., British Museum occasional paper, 82,105-119(1991)
Gordon, B.M., Kramer, H.W., On thedevelopment of a system for trace elementanalysis in the environment by chargedparticle X-ray fluorescence, J. Radioanal.Chenu, 12, 181 (1972)
Gratuze, B., Soulier, I., Barrandon, J.N.,Foy, D., De l'origine du cobalt dans lesverres, Revue d'Archéométrie, 16, 97-108(1992)
Handbook of Geochemistry, Wedepohl,K.H., éd., Springer Verlag, Berlin (1978)

Annexe 8 VI
Hansen, J.S,, McGeorge, J.C, Nix, D.,Schmidt-Ott, W.D., Unus, I., Fink, R.W.,Accurate efficiency calibration andproperties of semiconductor detectors forlow-energy photons, Nucl. Instr. and Meth.,106,365-379(1973)
Harbottle, G., Provenience studies usingneutron activation analysis: the role ofstandardization, dans: Archaeologicalceramics, Olin, J.S., Franklin, A.D., éds.,Smithsonian Institution Press, Washington,D.C., 67-77(1982)
Harbottle, G., Activation analysis inarchaeology, Radiochemistry, 3, 33-72(1976)
Haskin, L.A., Haskin, M.A., Frey, F.A.,Wildeman, T.R., Relative and absoluteterrestrial abundances of the rare earths.Origin and distribution of the elements,Ahrens, L.H., éd., Intern. Ser. MonographsEarth Sci., 30, 889-912 (1968)
Hayes, J.W., Excavations at Saraçhane inIstanbul, vol. 2: The Pottery, PrincetonUniversity Press (1992)
Heitz, C, Lagarde, G., Pape, A., Tenorio,D., Zarate, C, Menu, M., Scotee, L., Jaidar,A., Alviso, R., Gonzalez, D., Gonzalez, V.,Radioisotope induced X-ray emission - acomplementary method to PIXE analysis,Nucl. Instr. and Meth. B, 14, 93-98 (1986)
Heitz, Ch., Analysis by heavy ion X-rayemission, Progress in Crystal Growth andCharacterization, 8, 131- 150 (1984)
Heitz, C, Costa, G.J., Cailleret, 1,Lagarde, G., Sections efficaces deproductions de rayons X-K, L par 'H et4He, rapport interne CllN-PN, 82-24,Physique Nucléaire, Centre de RecherchesNucléaires, Strasbourg (1982)
Heitz, Ch., Kwadow, M., Tenorio, D.,Surface analysis by argon ion induced X-rayfluorescence, Nucl. Instr. and Meth., 149,483-488(1978)
Herrin, J., La céramique, dans: Splendeurde Byzance, catalogue d'expositionEuropalia 1982, Bruxelles, Musées royauxd'art et d'histoire, 225-239 (1982)
Hiver, P., Rapport de stage de CES,Réacteur Universitaire, UniversitéStrasbourg 1(1994)
Houdayer, A., Lessard, L., Brissaud, I.,Quantitative PIXE analysis of powderedpottery samples bound in an organic matrix,Nucl. Instr. andMeth. B, 3, 412-415 (1984)
Hughes, M.J., Cowell, M.R., Hook, D.R.,Neutron activation procedures at the BritishMuseum research laboratory, dans: Neutronactivation and plasma emission spectrometryanalysis in archaeology. Techniques andapplications, Hughes, M.J., Cowell, M.R.,Hook, D.R., éds., British Museumoccasional paper, 82, 29-46 (1991)
Hughes, M.J., Leese, M.N., Smith, R.J.,The analysis of pottery lamps mainly fromWestern Anatolia, including Ephesus, byneutron activation analysis, dans: Bailey,D.M., A catalogue of lamps in the BritishMuseum, III, Roman provincial lamps,British Museum publications, Londres, 461-485 (1988)
Hughes, M.J., Leese, M.N., Neutronactivation analysis of pottery lamps fromEphesus dating from the Archaic to the lateRoman period, Proc. 22nd Symposium onArchaeometry (Bradford 1982), Bradford,363-376(1983)
Innocenti, F., Manetti, P., Mazzuoli, R.,Pasquaré, G., Villari, L., Anatolia andnorth-western Iran, dans: Andésites,Thorpe, R.S., éd., Wiley & Sons, 327-349(1982)
Ion beam for material analysis, Bird, J.R.,Williams, J.S., éds., Academic Press, NewYork (1989)
Ion beam handbook for material analysis,Mayer, J.W., Rimini, E., éds, AcademicPress, New York (1977)

Annexe 8 VII
Isozumi, Y., Uncertainties in the y} fit fornuclear radiation spectra, Nucl. Instr. andMeth. A, 235, 164-173(1985)
Johansson, S.A.E., Campbell, J.L., PIXE: Anovel technique for elemental analysis,John Wiley & Sons, New York (1988)
Johansson, T.B., Akselsson, R., Johansson,S.A.E., X-ray analysis: elemental traceanalysis at the 10'12 g level, Nucl. Instr. andMeth., 84, 141-143(1970)
Jones, R.E., Greek and Cypriot pottery: Areview of scientific studies, FitchLaboratory Occasional Paper, 1, BritishSchool at Athens, Athènes (1986)
Jund, J., Etude minéralogique et techniquedes céramiques de l'Alsace du Nord: terrecuite vernissée de Soufflenheim et grès deBetschdorf Thèse de 3 è m c cycle, UniversitéStrasbourg 1(1979)
Kadom-Al-Neami, A., Mesure des sectionsefficaces de production des rayons X^ deséléments 5(fia, $-jLa et ^8Ce par desprotons de J MeV -3.5 MeV, Thèse dedoctorat, Université Strasbourg I (1988)
Kamilli, D.C., Mineral Analysis of the ClayBodies, dans: Ramage, A., Lydian housesand architectural terracottas,Archaeological Exploration of Sardis,Monograph 5, Harvard University Press,Cambridge Mass., 12-14 (1978)
Kazanski, M., Morrisson, C, Sodini, J.-P.,Le monde byzantin. La culture matérielle:productions et échanges, dans: Le grandatlas de l'archéologie, EncyclopaediaUniversalis, 138-139(1985)
Keutgen, T., Applications de la méthodePIXE pour l'analyse non destructive deglaçures et de céramiques anciennes.Interprétation de l'émission XL non shiftéelors de la collision Kr sur Th, Au et Ta,Rapport de stage de DEA, UniversitéCatholique de Louvain (1993)
Krause, M.O., Atomic radiative andradiationless yields for K and L shells, ./.Phys. Chetn. Ref. Data, 8, 307-327 (1979)
Kuleff, I., Djingova, R., Djingov, G., Ann.Univ. Sofia, Fac. Chim., 81 (en bulgare)(sous presse)
Kuleff, I., Djingova, R., Penev, I.,Chemische zusammensetzung desmittelalterichen sgraffitokeramiks III. Eineuntersuchung von archaeologischen fundeaus Schumen und Tscherven (12 - 14 jh.),Jahrbuch derMuseen in Nordbulgarien, 17,289-311 (en bulgare, résumé allemand)(1991)
Kuleff, I., Djingova, R., Activation analysisin archaeology, dans: Activation analysis,Alfassi, Z.B., éd., CRC-Press, Florida,vol.2, 427-489 (1990)
Kuleff, I., Djingova, R., Djingov, G.,Provenance study of sgraffito ceramics(XIII-XIVc.) from Shumen, Varna andTcherven (North-Eastern Bulgaria), dans:Archaeometry, Proc. 25 th Int.Archaeometry Symposium (Athens 1986),Maniatis, Y., éd., 533-542 (1989)
Kuleff, I., Djingova, R., Djingov, G.,Provenance study of pottery by INAA,dans: Proc. 7th Int. Conf. Modern Trends inActivation Analysis, Copenhague, 787-791(1986)
Kuleff, I., Djingova, R., Penev, I.,Instrumental Neutron Activation Analysis ofpottery for provenience study, J. Radioanal.Nucl. Chem. Articles, 99, 2, 345-358 (1986)
La ceramica médiévale nel MéditerranéeOccidentale, Congresso Internazionale déliaUniversita degli studi di Siena, (Sienne,Faenza, 1984), Florence (1986)
La céramique médiévale de laMéditerranée Occidentale Xe - XVe siècles,Congrès Internationaux du CNRS n°584(Valbonne 1978), Editions du CNRS, Paris(1980)

Annexe 8 VIII
La ceramica nel mondo bizantino ira XI eXV secolo e suoi rapport! con /'Italia,Gelichi, S., éd., Florence (1993)
Lagarde, G., Brissaud, I., Cailleret, J.,Fontes, P., Heitz, Ch., Thick target PIXEanalysis of clays for archaeological studies,Nucl. Instr. andMeth. B, 1, 45-50 (1984)
Lagarde, G., Cailleret, J., Heitz, C,Quantitative analysis of thick targetscontaining light elements (Z < 12) by PIXE,Nucl. Instr. andMeth., 205, 545-553 (1983)
Lane, A., Medieval finds at Al Mina inNorth Syria, Archaeologia, 87, 45-53(1937)
Lazzarini, L., Calogero, S., Early local andimported sgraffito ware in Venice: acharacterization and provenance study,dans: Archaeometry, Proc. 25th Int.Archaeometry Symposium (Athens 1986),Maniatis, Y., éd., 571-584 (1989)
Lazzarini, L., Canal, E., Ritrovamenti diCeramica Graffita Bizantina in Laguna e laNascita del Graffito Veneziano, Faenza, 69,19-59, pi. 2-10 (en italien) (1983)
Lazzarini, L., Calogero, S., Burriesci, N.,Petrera, M., Chemical, mineralogical andMôssbauer studies of Venetian and PaduanRenaissance sgraffito ceramics,Archaeometry, 22, 1, 57-68 (1980)
Lebart, L., Morineau, A., Lambert, T.,Pleuvret, P., SPAD.N, CISIA, Saint-Mandé(1991)
Lebart, L., Morineau, A., Fénelon, J.-P.,Traitement des données statistiques.Méthodes et programmes, Dunod, Paris(1982)
Lemoine, C, Picon, M., La fixation duphospore par les céramiques lors de leurenfouissement et ses incidences analytiques,Revue d'Archéomélrie, 6, 101-112 (1982)
Lemoine, C, Meille, E., Poupet, P.,Barrandon, J.N., Borderie, B., Etude dequelques altérations de compositionchimique de céramiques en milieu marin etterrestre, Actes du XXème symposiuminternational d'archéométrie, Revued'Archéométrie, supplément 1981, vol III,349-360(1981)
Leruyet, A., Etude pétrographique,minéralogique et géochimique decéramique byzantine provenant d'AsieMineure. Contribution à l'étude desmatériaux et de leurs provenances, Rapportde stage de maîtrise de géologie, Institut deGéologie, EOPGS, Strasbourg (1994)
Liu-Xu, X., Conception et réalisation d'uninstrument de mesure "EDXRF" utilisantdes détecteurs Hgh, Thèse de doctorat,Université Strasbourg I (1994)
Lôvestam, G., Calligaro, T., Duval, A.,Salomon, J., The AGLAE scanning nuclearmicroprobe setup, Nucl. Instr. andMeth. B,77,66-70(1993)
Lôvestam, G., Swietlicki, E., Off-line dataevaluation of elemental maps obtained fromscanning nuclear microprobe analyses,Scanning microscopy, 6, 3, 607-624 (1992)
Lôvestam, G., Development of a scanningproton microprobe - computer-control,elemental mapping and applications, Thèsede doctorat publiée, Lund Institute ofScience and Technology, Lund (1989)
Lôvestam, G., Swietlicki, E., An externalbeam setup for the Lund proton microprobe,Nucl. Instr. and Meth. B, 43, 104-111(1989)
Maggetti, M., Phase analysis and itssignificance for technology and origin, dans:Archaeological ceramics, Olin, J.S.,Franklin, A.D., éds., Smithsonian InstitutionPress, Washington, D.C., 121-133 (1982)

Annexe 8 IX
Mando, P.A., Advantages and limitations ofexternal beams in applications to arts &archaeology, geology, and environmentalproblems, Nucl. Instr. and Meth. B, 85,815-823(1994)
Mannoni, T., Provenienze ed analisipetrografiche interpretate. L'esempio delleceramiche bizantine, dans: La ceramica nelmondo bizantino tra XI e XV secolo e suoirapporti con l'Italia, Gelichi, S., éd.,Florence, 341-345(1993)
Mason, R.B., Tite, M.S., The beginnings ofstonepaste technology, Archaeometry, 36,1, 77-91 (1994)
Maxwell, J.A., Campbell, J.L., Teesdale,W.J., The Guelph PIXE software package,Nucl. Instr. and Meth. B, 43, 218-230(1989)
Mayet, F., Picon, M., Une sigilléephocéenne tardive ("Late Roman C ware")et sa diffusion en Occident, Figlina, 7, 129-142(1986)
Me Birney, Igneous Petrology, OxfordUniversity Press (1984)
Megaw, A.H.S., Zeuxippus ware again,dans: Recherches sur la céramiquebyzantine, Déroche, V., Spieser, J.-M., éds.,Bulletin de correspondance hellénique,aupplément XVIII, 277-289 (1989)
Megaw, A.H.S., Jones, R.E., Byzantine andallied pottery: a contribution by chemicalanalysis to the problem of origin anddistribution, Annual of the British School atAthens, 78, 235-263, pi. 24-30 (1983)
Megaw, A.H.S., Jones, R.E.,Spectrographic analyses of Byzantine andallied pottery, Jahrbuch derÖsterreichischen Byzantinistik, 32, 3, 577-585(1982)
Megaw, A.H.S., An early thirteenth centuryaegean glazed ware, dans: Studies inMemory of David Talbot Rice, Edinburgh,34-45 (1975)
Megaw, A.H.S., Zeuxippus ware, Annualof the British School at Athens, 63, 67-88,pi. 14-21 (1968)
Menu, M., Calligaro, T., Salomon, J.,Amsel, G., Moulin, J., The dedicatedaccelerator-based IBA facility AGLAE atthe Louvre, Nucl. Instr. and Meth. B, 45,610-614(1990)
Mery, S., Emergence et développement dela production céramique de la péninsuled'Oman à l'âge du bronze en relation avecl'Asie moyenne, Thèse de doctorat,Université Paris I, 2 volumes (1991)
Merzbacher, E., Lewis, H.W., X-rayproduction by heavy charged particles,Encyclopedia of Physics, vol. 34, SpringerVerlag, Berlin, 166-192 (1958)
Millot, G., Geologie des argiles, Masson,Paris (1964)
Mommsen, H., Kreuser, A., Weber, J., Amethod for grouping pottery by chemicalcomposition, Archaeometry, 30, 1, 47-57(1988)
Morgan, C, Corinth, Results ofExcavations, vol.XI: The Byzantine Pottery,Harvard University Press (1942)
Mosbah, M., Piccot, D., Clocchiatti, R.,Metrich, M., Gösset, J., Joron, J.-L., Micro-PIXE within magmatic inclusions usingGUPIX software, Nucl. Instr. and Meth. B,82, 139-145 (1993)
Nikolakopoulos, G., Considérations surl'esthétique de la céramique byzantine, dans:Recherches sur la céramique byzantine,Déroche, V., Spieser, J.-M., éds., Bulletinde correspondance hellénique, supplémentXVm, 317-326(1989)

Annexe 8 X
Oweijane, F., Analyse de céramiquesbyzantines par fluorescence X induite par lerayonnement y de 59.5 keV de ^^Am etcomparaison des résultats avec ceuxobtenus par FIXE et par AAN, Rapport destage de DEA de physique nucléaire,Université Strasbourg I (1994)
Papanikola-Bakirtzis, D., DautermanMaguire, E., Maguire, H., Ceramic art fromByzantine Serres, Illinois Byzantine Studies,3(1992)
Parman, E., The pottery from Saint John'sBasilica at Ephesos, dans: Recherches sur lacéramique byzantine, Déroche, V., Spieser,J.-M, éds., Bulletin de correspondancehellénique, supplément XVIII, 277-289(1989)
Parry, S., Activation spectrometry inchemical analysis, Chemical Analysis, 119,Wiley-Interscience, New York (1991)
PAW - Physics Analysis Workstation,CERN, Geneva, Switzerland, version2.02/06(1993)
Peisach, M, Pineda, C.A., Jacobson, L.,Thick target PIXE analysis of coastal andinland Namibiam pottery, Nucl. Instr. andMeth.B, 49, 309-312(1991)
Peisach, M., Ion beam analytical methodsfor attempts to establish the provenance ofarchaeological objects in the absence ofreference artefacts, Nucl. Instr. and Meth.5,14,99-115(1986)
Peisach, M., Jacobson, L., Boulle, G.J.,Gihwala, D., Underhill, L.G., PIXE analysisof archaeological material and the use ofmultivariate analysis for characterisation,Nucl. Instr. and Meth., 193, 337-341 (1982)
Penev, I., Kuleff, I., Djingova, R.,Simultaneous determination of aluminium,magnesium, and silicon in rocks, glasses,and pottery, ,/. Radioanal. Nucl. Chem.Letters, 93, 3, 219-232 (1985)
Pergamenische Forschungen, Pergamon.Gesammelte Aufsàtze, DeutschesArchaologisches Institut, Walter de Gruyter&Co, Berlin
Schâfer, J., Hellenistische Keramik ausPergamon, Band II (1968)Tôpperwein, E., Terrakotten aus Pergamon,Band III (1976)
Perlman, I., Asaro, F., Pottery analysis byneutron activation, Archaeometry, 11, 21-52(1969)
Picon, M., L'analyse par activationneutronique est-elle la meilleure méthodeque l'on puisse employer pour déterminerl'origine des céramiques?, Revued'Archèomètrie, 15, 95-101 (1991)
Picon, M., Quelques observationscomplémentaires sur les altérations decomposition des céramiques au cours dutemps: cas de quelques alcalins et alcalinos-terreux, Revue d'Archèomètrie, 15, 117-122(1991)
Picon, M., La fixation du baryum et dustrontium par les céramiques, Revued'Archèomètrie, 11, 41-47(1987)
Picon, M., Ricq, J.-C, Un exempled'altération de la composition des amphoresmassaliètes: le cas d'Olbia, Lettred'information du CRA, 30, Archéologie duMidi Méditerranéen, 12, 65-67 (1986)
Picon, M., Un exemple de pollution auxdimensions kilométriques: la fixation dubaryum par les céramiques, Revued'Archèomètrie, 9, 27-29 (1985)
Picon, M., Le traitement des donnéesd'analyse, PACT, 10, 379-399 (1984)
Picon, M., La céramologie de laboratoire,Nouvelles de l'archéologie, 1, 37-45 (1979)
Picon, M., Introduction à l'étude techniquedes céramiques sigillées de Lezoux, Centrede Recherches sur les Techniques Gréco-Romaines, 2, Université de Dijon (1973)

Annexe 8 XI
Picon, M., Vichy, M., Meille, E.,Composition of the Lezoux, Lyon andArezzo Samian ware, Archaeometry, 13, 2,191-208(1971)
Picon, M., Vertet, H., La composition despremières sigillées de Lezoux et le problèmedes céramiques calcaires, RAECE, 21, 1-2,207-218(1970)
Pinkwart, D., Hellenistisch-rômischeBleiglasurkeramik aus Pergamon,Pergamenische Forschungen, I, 140-163(1972),
Pollard, A.M., Data analysis, dans: Jones,R.E., Greek and Cypriot Pottery: a reviewof scientific studies, Fitch LaboratoryOccasional Paper, 1 (1986)
Radt, W., Pergamon, DuMont Buchverlag,Cologne (1988)
Rafaillac-Desfosses, C, Céramiquesglaçurées médiévales. Recherche dedonnées physiques sur les techniques defabrication et sur l'altération, Thèse dedoctorat, Université Bordeaux III (1994)
Recherches sur la céramique byzantine,Actes du colloque organisé par l'écolefrançaise d'Athènes et l'université deStrasbourg II, Athènes 8-10 avril 1987,Déroche, V., Spieser, J.-M, éds., Bulletinde correspondance hellénique, supplémentXVHI(1989)
Rice, P.M., Pottery analysis. A sourcebook,The University of Chicago Press, Chicago(1987)
Ricq, J.C., Essai de sélection des élémentschimiques particulièrement propres àcaractériser un groupe de céramiques, Noteset Monographies Techniques du C.R.A., 5(1974)
Rossini, I., Application de l'analysemultièlëmeniaire par fluorescence X etactivation neutronique à la caractérisationd'un site archéologique, Thèse de doctorat,Strasbourg 1(1991)
Roubault, M., Détermination des minérauxdes roches au microscope polarisant,Lamarre-Poinat, Paris (1982)
Roux, M., Algorithmes de classification,Masson, Paris (1985)
Sabir, A., Etude de justesse et de précisiondans une méthode automatique d'analysepar activation neutronique non destructive.Cas des matrices siliceuses. Application àla recherche de l'origine de céramiquesarchéologiques, Thèse de Doctorat,Université Paris-Sud Orsay (1986)
Salem, S.I., Panossian, S.L., Krause, R.A.,Experimental K and L relative X-rayemission rates, At. Data and Nucl. DataTables, 14,2,91-109(1974)
Sanders, G.D.R., Excavations at Sparta:the Roman stoa, 1988-91. Preliminaryreport, part 1, Medieval pottery, Annual ofthe British School at Athens, 88, 251-286,pi. 23-26 (1993)
Sauer, R., Outschar, U., Schneider, G.,Combined petrography and geochemicalstudies in the determination of potteryproduction centers - 'the case of Ephesos', àparaître dans: Proc. 29th Int. ArchaeometrySymposium (Ankara 1994)
Sauer, R., Outschar, U., Schneider, G.,Late Hellenistic and Roman ceramics alongthe western coast of Asia Minor: localproduction centers - historical andarchaeological requirements, à paraître dans:Proc. 29th Int. Archaeometry Symposium(Ankara 1994)
Schmitt, A., Apports et limites de lapétrographie quantitative: application au casdes amphores de Lyon, Revued'Archéomélrie, 17, 51-63 (1993)
Schneider, G., Wirz, E., Chemical answersto archaeological questions - Romanterracotta lamps as documents of economichistory, Documents et travaux IGAL, 16,13-48 (1992)

Annexe 8 XII
Scofield, J.H., Relativistic Hartree-Slatervalues for K and L X-ray emission rates, At.Data and Nucl. Data Tables, 14, 2, 121-137 (1974)
Scott, J.A., Kamilli, D.C., Late Byzantineglazed pottery from Sardis, Actes du XVeCongrès International des EtudesByzantines (Athènes 1976), II, Athènes,679-696 (1981)
Sekine, T., Baba, H., A measure of thedegree of fit between the calculated andobserved spectra, Nucl. Instr. and Me th.,133, 171-173 (1976)
Shepard, A.O., Ceramics for thearchaeologist, Carnegie Institution ofWashington, Washington, D.C. (1965)
Spieser, J.-M., Die byzantinische Keramikin der Wohnstadtgrabung, PergamenischeForschungen, à paraître
Spieser, J.-M., Informatique et céramique:l'exemple de Pergame, dans: Recherches surla céramique byzantine, Déroche, V.,Spieser, J.-M., éds., Bulletin decorrespondance hellénique, supplémentXVIII, 291-302(1989)
Spieser, J.-M., La céramique byzantinemédiévale, dans: Hommes et richesses del'Empire byzantin, II, VIIIe - XVe siècle,Kravari, V., Lefort, J., Morrisson, C, éds.,Réalités byzantines, 3, 249-260, pi. XI-XII(1989)
Spieser, J.-M., Etude de la céramiquebyzantine d'Asie Mineure, Rapport desmissions effectuées en 1983, Activitébyzantine, Fondation Européenne de laScience, vol. II, Paris, 307-329 (1987)
Spieser, J.-M., Le monde byzantin. Lacéramique: nouvelles approches, dans: Legrand atlas de l'archéologie, EncyclopaediaUniversalis, 144-145(1985)
Spieser, J.-M., Céramique byzantine dePergame, Jahrbuch der OslerreichischenByzantinistik, 32/3, 561-576 (1982)
STATGRAPHICS, STSC, Inc., RockvilleMD 20852 (1988)
Talbot Rice, D., The Pottery of Byzantiumand the Islamic World, dans: Studies inIslamic Art and Architecture in honour ofK.A.C. Creswell (Mélanges Creswell), LeCaire, 194-236(1965)
Talbot Rice, D., Byzantine glazed pottery,Clarendon Press, Oxford (1930)
Terres secrètes de Samarcande,céramiques du VIIIe au XIIIe siècle,catalogue d'exposition, Paris, Institut duMonde Arabe (1992)
Tertian, R., Claisse, F., Principles ofquantitative X-ray fluorescence analysis,Heyden, Londres (1982)
Theisen, R., Vollath, D., Tables of X-raymass attenuation coefficients, VerlagStahleisen, M.B.H. Dusseldorf (1967)
Thierrin-Michael, G., Amphores italiques etanalyses à la microsonde nucléaire (ivISN),rapport destiné au groupe de travail"caractérisation" du GDR "Méthodesnucléaires en archéologie"(réunion du03/12/92) (1992)
Tite, M.S., Freestone, I.C., Meeks, N.D.,Bimson, M. The use of the scanningelectron microscopy in the technologicalexamination of ancient ceramics, dans:Archaeological ceramics, Olin, J.S.,Franklin, A.D., éds., Smithsonian InstitutionPress, Washington, D.C., 109-120 (1982)
Trouslard, P., Tirira, J., PYROLE,I.N.S.T.N., CE. Saclay, Gif sur Yvette
Tuurnala, T., Hautojàrvi, A., Harva, K.,Examination of oil paintings by using aPIXE/PIGME combination, Nucl. Instr. andMelh. B, 14, 70-75 (1986)
Vandiver, P., Les glaçures des céramiquesanciennes, Pour la science, 152, 78-86(1990)

Annexe 8 XIII
Vassi, 0., An unglazed ware potteryworkshop in twelfth-century Lakonia,Annual of the British School at Athens, 88,287-293,'pi. 27-28(1993)
Vitali, V., Franklin, U.M., Hancock,R.G.V., La stabilité des céramiques parrapport à l'environnement, Revued'Archéométrie, 8, 41-44 (1984)
Vogt, C, Les arts de la table à Byzance,dans: Art byzantin, Les dossiersd'archéologie, 176, 40-43 (1992)
Waksman, S.Y., Rossini, I., Heitz, C,Byzantine Pergatnon: characterization of theceramics production centre, Proc. 29th Int.Archaeometry Symposium (Ankara 1994) (àparaître)
Waksman, S.Y., Pape, A., Heitz, C, PIXEanalysis of Byzantine ceramics, Mitel. Instr.andMeth. B, 85, 824-829 (1994)
Walter, V., Etude pétrographique,minéralogique et géochimique d'amphoresgauloises découvertes dans le Nord-Est dela France. Contribution à l'étude desmatériaux, des technologies et desprovenances, Thèse de doctorat, UniversitéStrasbourg II (1988)
Wàtjen, U., Currently used computerprogrammes for PIXE analysis, Nucl. Instr.andMeth. B, 22, 29-33 (1987)
Widemann, F., L'analyse par activationneutronique des céramiques anciennes:groupements et différenciation, Latomus,160,47-71(1979)
Williams, C.T., Wall, F., An INAA schemefor the routine determination of 27 elementsin geological and archaeological samples,dans: Neutron activation and plasmaemission spectrometry analysis inarchaeology. Techniques and applications,Hughes, M.J., Cowell, M.R., Hook, D.R.,éds., British Museum occasional paper, 82,105-119(1991)
Wisseman, S., The materials analysis ofByzantine pottery, dans: Papanikola-Bakirtzis, D., Dauterman Maguire, E.,Maguire, H., Ceramic art from ByzantineSerres, Illinois Byzantine Studies, 3 ,66-69(1992)
Yellin, J., Perlman, I., Asaro, F., Michel,H.V., Mosier, D.F., Comparison of neutronactivation analysis from the LawrenceBerkeley Laboratory and the HebrewUniversity, Archaeometry, 20, 1, 95-100(1978)
Yenisehirlioglu, F., La céramique glaçuréede Gulpinar, dans: Recherches sur lacéramique byzantine, Déroche, V., Spieser,J.-M., éds., Bulletin de correspondancehellénique, supplément XVTII, 303-315(1989)

RÉSUMÉ
Les fouilles de Pcrgamc, en Asie Mineure, ont mis au jour de nombreuses céramiquesdatées du 12èmo - I4èmc siècles. Parmi celles-ci, pièces inachevées, ratés de cuisson etpernettes attestent d'une production locale sur le site à l'époque byzantine.
Un échantillonnage représentatif constitué de 160 tessons a été soumis à l'analyseélémentaire par les méthodes PIXE (Particle Induced X-ray Emission) et INAÀ (InstrumentalNeutron Activation Analysis). L'application de techniques statistiques multivariées auxrésultats d'analyse a permis de classer les tessons par groupes de composition similaire et dedistinguer les céramiques fabriquées à Pergame' des céramiques importées. Plusieurs groupesde productions locales sont attestés, correspondant à des céramiques de catégorie et d'époquede fabrication différentes. La caractérisation géochimique des pâtes, complétée par desexamens pétrographiques et minéralogiqucs, indique que des matières premières spécifiquesont été utilisées pour fabriquer chacun de ces produits. Les données analytiques relatives auxcéramiques produites à Pergame pourront servir de référence pour des études de provenanceultérieures.
Du fait de la rareté de tels groupes de référence, les céramiques importées à Pergamene peuvent être attribuées à leur lieu d'origine, Parmi les types de céramiques diffusées àgrande distance dans le monde byzantin, des importations de quelques exemplaires de"sgraffito fin" et de "Zeuxippus ware" sont identifiés. Ce dernier type a été une sourced'influence stylistique pour les ateliers de Pergame, car les analyses montrent que des imitationsde "Zeuxippus ware" y ont été fabriquées. Ces imitations ont probablement clles-même connuune diffusion régionale.
SUMMARY
An important ceramics material dated back to the 12th-14th centuries has beenexcavated in Pergamon (Turkey). Among these findings, wasters, tripod stilts and unfinishedware attest to local production in the Byzantine period.
Elemental analysis by the methods PIXE (Particle Induced X-ray Emission) and INAA(Instrumental Neutron Activation Analysis) has been performed on a representative samplingof 160 sherds, including attested local material. Multivariate statistical techniques were used toclassify the sherds into groups of similar composition and thus to distinguish ceramics made inPergamon from imported wares. Several groups of local production have been constituted,which correspond to wares differing in date and fabric. The geochemical characterization ofthe pastes, complemented with petrographical and mineralogical data, shows that specific rawmaterials'have been used to manufacture each ware. The analytical data related to ceramicsmade in Pergamon will serve as reference data for future provenance studies.
Such reference groups of Byzantine ceramics are very rare, and therefore the ceramicsimported into Pergamon cannot be attributed as to their origin. Among the ceramics widelydiffused in the Byzantine world, some importations belonging to the "fine sgraffito" and"Zeuxippus ware" types have been identified. The latter type has been a source of stylisticalinfluence for the workshops of Pergamon, since the analyses show that imitated "Zeuxippusware" has been produced there. These imitations were probably themselves diffused on aregional scale.
MOTS CLÉSEmpire Byzantin 12èmc-14èmc siècles; Pergame; céramique; archéométrie; étude deprovenance; PIXE; activation neutronique; analyse multivariée.