celiac disease revisited - karger publishers
TRANSCRIPT

Review Article
GE Port J Gastroenterol 2022;29:111–124
Celiac Disease Revisited
João Calado
a Mariana Verdelho Machado
a, b a
Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal; b Hospital de Vila Franca de Xira, Vila Franca de Xira, Portugal
Received: October 1, 2020Accepted: January 8, 2021Published online: March 17, 2021
Mariana Verdelho MachadoDepartmento de Medicina, Gastrenterologia e Hepatologia, Faculdade de MedicinaUniversidade de Lisboa, Ave. Prof. Egas MonizPT–1649-028 Lisboa (Portugal)mverdelhomachado @ gmail.com
© 2021 Sociedade Portuguesa de Gastrenterologia Published by S. Karger AG, Basel
DOI: 10.1159/000514716
KeywordsCeliac disease · Human leukocyte antigen · Serological tests · Duodenal histology · Diagnostic challenges
AbstractCeliac disease (CD) is a systemic disease triggered by gluten ingestion in genetically predisposed individuals. It manifests primarily as an autoimmune enteropathy associated with specific circulating autoantibodies and a human leukocyte antigen haplotype (HLA-DQ2 or HLA-DQ8). It afflicts roughly 1% of the population, though the majority of patients re-main undiagnosed. Diarrhea and malabsorption are classic manifestations of CD; however, both children and adults can be paucisymptomatic and present extraintestinal manifesta-tions such as anemia, osteoporosis, and abnormal liver tests. CD screening is not recommended for the general popula-tion, and it should be focused on high-risk groups. CD diag-nosis is challenging and relies on serological tests, duodenal histology, and genetic testing. Particularly difficult presenta-tions to manage are seronegative patients, seropositive pa-tients without villus atrophy, and patients who have started a gluten-free diet before the diagnostic workup. The only proven treatment is a lifelong gluten-free diet. We present an in-depth review on the physiopathology and manage-ment of CD, with a particular emphasis on diagnostic chal-lenges. © 2021 Sociedade Portuguesa de Gastrenterologia
Published by S. Karger AG, Basel
Doença celíaca revisitada
Palavras ChaveDoença celíaca · Testes serológicos · Histologia duodenal · Desafios diagnósticos
ResumoA doença celíaca (DC) é uma doença sistémica des-encadeada pela exposição ao glúten em doentes geneti-camente susceptíveis. Manifesta-se maioritariamente por uma enteropatia auto-imune associada a auto-anticorpos e aos haplotipos HLA-DQ2 ou HLA-DQ8. A DC afecta aproximadamente 1% da população mundial admitindo-se, no entanto, que a maioria dos doentes não esteja di-agnosticada. As manifestações clássicas de DC são a diar-reia e sintomas de malabsorção, no entanto tanto crian-ças como adultos podem ser pauci-sintomáticos ou apresentar manifestações extra-intestinais incluindo ane-mia, osteoporose ou alteração das provas hepáticas. O rastreio de base populacional não está recomendado, de-vendo o foco ser nos grupos de risco para DC. O diagnósti-co de DC é um desafio e assenta em três pilares: testes serológicos, histologia duodenal e testes genéticos. Apre-sentações particularmente difíceis de manejar são os doentes sero-negativos, doentes sero-positivos sem atro-fia vilositária e doentes que iniciam dieta sem glúten an-tes da marcha diagnóstica. O único tratamento com eficá-
This is an Open Access article licensed under the Creative Commons Attribution-NonCommercial-4.0 International License (CC BY-NC) (http://www.karger.com/Services/OpenAccessLicense), applicable to the online version of the article only. Usage and distribution for com-mercial purposes requires written permission.

Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124112DOI: 10.1159/000514716
cia comprovada é a dieta sem glúten. Apresentamos uma revisão aprofundada da fisiopatologia e manejo da DC, com particular ênfase nos desafios diagnósticos.
© 2020 Sociedade Portuguesa de GastrenterologiaPublicado por S. Karger AG, Basel
Historical Background
Celiac disease (CD) is a systemic disease triggered by gluten ingestion, in genetically predisposed individuals. It manifests as an autoimmune enteropathy associated with specific circulating autoantibodies and human leu-kocyte antigen haplotype (HLA-DQ2 or HLA-DQ8) [1].
Aretaeus of Cappadocia, in 250 A.D., described a chronic perturbation of “pepsis” (i.e., digestion) and “anadosis” (i.e., absorption) resulting in a general debility which was named “celiac diathesis” [2, 3]. The word “ce-liac” is derived from the Greek “koiliakos,” which means abdominal [4]. However, it was only in 1888 that Samuel Gee [5] published the first modern clinical detailed de-scription of CD. In 1908, in the USA, Christian Herter [6] published a similar description, emphasizing the retarda-tion in growth. For several years, CD was known as Gee-Herter disease [7].
Diet was proposed as a causal contributor since Ara-teus. In the twentieth century, several diets were advo-cated, such as the banana diet [8] and the Fanconi diet based on fruits and vegetables [7]. The causal link to grain consumption (wheat, rye, barley, and, to a lesser extent, oats) was described in the forties by the Dutch pediatri-cian Willem-Karel Dicke. This link came from the obser-vation of the effect of food scarcity on children with CD during the Second World War. Dicke observed that symptoms of children with CD improved when they were not eating bread or grains and worsened after the war ended and these foods reentered their diet [7, 9].
Paulley [10], in 1954, described detailed histological anomalies in the small bowel from surgical specimens (chronic inflammation and atrophy) from patients with CD.
In 1964, Berger et al. [11] reported the presence of se-rum anti-gliadin antibodies (AGA) in CD. It took up to 20 years for serology be considered a diagnostic criterion [12, 13]. More sensitive and specific serological tests have been identified since then [14, 15].
In 1972, Falchuk et al. [16] described the association between a specific HLA genotype and CD and hypothe-sized that CD is a consequence of carrying an abnormal immune response gene to gluten. We know now that
HLA-DQ2/8 is necessary for the development of CD, making HLA determination the third pillar in the diag-nosis of CD [17].
Epidemiology
The prevalence of CD varies according to age, gender, and region. A recent meta-analysis estimated a global prevalence of 1.4% by serological tests and 0.7% by intes-tinal biopsy [18]. This is probably an underestimation of the real prevalence of CD, since it is estimated that only 1 in 5 patients with CD is diagnosed [19]. In some regions, such as Asia and Africa, the number of reported cases of CD is extremely low, even though wheat consumption is increasing [20] and the frequency of CD-associated HLA alleles seems similar to that in Western countries [21]. As such, in Asia, CD is likely to be even less efficiently diag-nosed, with less awareness for the asymptomatic forms of the disease [22]. In this millennium, the prevalence of CD seems to have increased by 33%, for unknown reasons, but it is probably associated with environmental factors [18].
Women are 1.5 times more afflicted than men [18]. The incidence of CD is approximately 2 times more fre-quent in children than in adults, with a second peak in incidence between 50 and 69 years [23].
Genetics are a main factor in the risk for CD, with over 40 genetic loci associations besides the HLA-DQ2/8 hap-lotype [24].
Environmental factors have been studied as risk fac-tors for CD [25–27]. The association between age at glu-ten introduction and CD is controversial [28–32]; how-ever, current recommendations advise gluten introduc-tion between 4 and 12 months of age [33]. The amount and pattern of gluten consumption may have a role and may account for the different prevalence rates of CD across Europe [34].
Breastfeeding does not seem to protect from CD [28, 32, 35–37]. Recurrent respiratory infections (in infants) and gastrointestinal infections (rotavirus and adenovirus in children and Campylobacter in adults) seem to be as-sociated with CD [25, 26, 38], though the evidence is weak [39].
Lastly, high-risk groups for CD include first-degree relatives of CD patients [40] (with a prevalence up to 7.5%) [41], and patients with type 1 diabetes mellitus (T1DM) [42] or other autoimmune diseases [43], IgA de-ficiency [44], and chromosomopathies such as Down syndrome [45] and Turner syndrome [46].

CD Revisited 113GE Port J Gastroenterol 2022;29:111–124DOI: 10.1159/000514716
Pathophysiology
CD results from an intense immune response to gluten leading to small bowel injury with consequent malab-sorption and autoimmune phenomena [47].
Gluten consists of a group of proteins from Gramine-ae of the Triticiae tribe, particularly wheat, rye, and bar-ley. Oats are phylogenetically more distant (Aveneae tribe) but share sufficient similarities to induce symptoms in some patients. Rice, maize, sorghum, and millet are distant enough not to trigger CD [48]. Gluten is the Latin word for “glue,” owing its name to its viscoelastic and ad-hesive properties [26]. The word gluten is widely used to refer to disease-inducing Gramineae proteins; however, strictly speaking, gluten specifically refers to proteins from wheat. Similar proteins in rye are secalins, and in barley they are hordein [49]. Wheat gluten contains 2 ma-jor protein components, i.e., monomeric water-soluble gliadins and multimeric water-insoluble glutenins [50].
Gluten peptides are highly enriched in proline and glu-tamine. Gastrointestinal proteases are deficient in prolyl-endopeptidase activity. As such, the high-proline content makes gluten resistant to gastrointestinal cleavage, allow-ing the subsistence of polypeptides with up to 33 amino acids [51]. This increases gluten’s immunogenicity, since major histocompatibility complex (MHC) II molecules only present peptides at least 9 amino acids long [52].
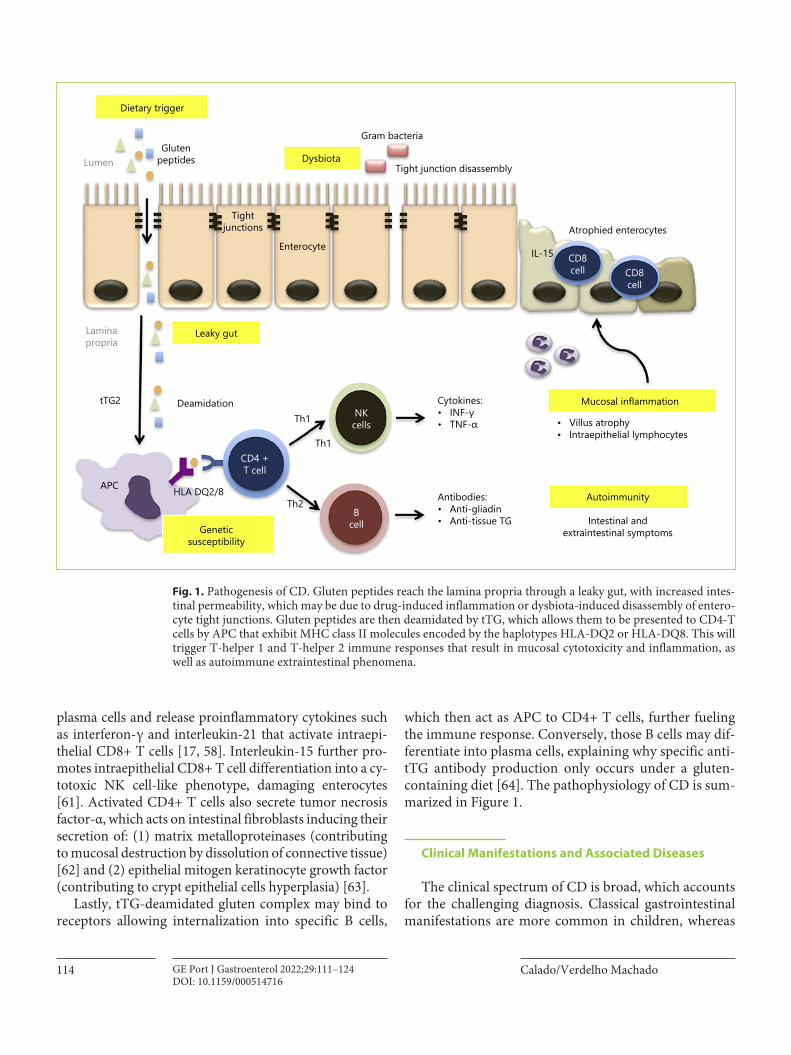
Gluten peptides are not freely absorbed and need a dis-ruption in the epithelial barrier to translocate into the lamina propria, where antigen-presenting cells (APC) re-side. That may occur through a damaged epithelium, in-duced by transient intestinal infection, drug-induced in-flammation (e.g., nonsteroidal anti-inflammatory drugs), or dysbiota-induced disassembly of enterocyte tight junc-tions. Alternatively, gluten can cross the epithelium in a transcellular pathway through binding of gluten-secreto-
ry IgA complexes to the transferrin receptor CD71 or in-side dendritic cells that cross the epithelium [17, 50, 53–55].
MHC-II molecules bind preferentially to peptides with negatively charged amino acids. Even though gluten pep-tides have very few charged amino acids, they are highly susceptible to deamidation on their glutamine residues to negatively charged glutamate by tissue transglutaminase (tTG) [52]. Deamidation significantly increases the sta-bility of the gluten-MHC complex, increasing their im-munogenicity [56].
APC present deamidated gluten peptides bound to a specific MHC class II that map to the HLA-DQ locus. That locus codifies antigen-presenting glycoproteins that are heterodimers constituted by a α-chain (encoded by the DQA1 allele) and a β-chain (encoded by the DQB1 allele). Only HLA-DQ2.5, DQ2.2, and DQ8 (and proba-bly DQ7.5) bind to deamidated gluten peptides, and so their presence is necessary for the development of CD. However, it is not a sufficient condition, since these hap-lotypes are also found in 40% of the general population [53, 57]. Nonetheless, about 90% of CD patients express HLA-DQ2.5 and roughly 10% express HLA-DQ2.2 or HLA-DQ8 [58]. HLA-DQB1*02, present in HLA-DQ2.5 and HLA-DQ2.2, confers a higher risk for CD compared to the presence of HLA-DQB1*03, present in HLA-DQ8. The frequency of HLA-DQB1*02 seems to be even high-er in CD patients with T1DM [59]. Furthermore, there is a dose effect for HLA-DQ2.5, since homozygous individ-uals have a 5-fold increased risk for CD and for severe disease [60]. Haplotypes, HLA, and their risk are shown in Table 1.
APC (dendritic cells and macrophages) present the complex MHC-II-deamidated gliadin to CD4+ T cells, in-ducing a proinflammatory phenotype in T cells [61]. Acti-vated CD4+ T cells promote differentiation of B cells into
Table 1. HLA and the risk of CD
Haplotype HLA alleles Molecules Risk of CD
DQB1* DQA1* DRB1* β chain α chain
DR3-DQ2 02:01 05:01 03 02:01 05:01 cis DQ2.5 highDR5-DQ7 03:01 05:05 11/12 02:02 05:05 trans DQ2.5DR7-DQ2 02:02 02:01 07DR7-DQ2 02:02 02:01 07 02:02 02:01 DQ2.2 lowDR5-DQ7 03:01 05:05 11/12 03:01 05:05 DQ7.5 very lowDR4-DQ8 03:02 03 04 03:02 03 DQ8 low
Adapted from Sollid [57].
Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124114DOI: 10.1159/000514716
plasma cells and release proinflammatory cytokines such as interferon-γ and interleukin-21 that activate intraepi-thelial CD8+ T cells [17, 58]. Interleukin-15 further pro-motes intraepithelial CD8+ T cell differentiation into a cy-totoxic NK cell-like phenotype, damaging enterocytes [61]. Activated CD4+ T cells also secrete tumor necrosis factor-α, which acts on intestinal fibroblasts inducing their secretion of: (1) matrix metalloproteinases (contributing to mucosal destruction by dissolution of connective tissue) [62] and (2) epithelial mitogen keratinocyte growth factor (contributing to crypt epithelial cells hyperplasia) [63].
Lastly, tTG-deamidated gluten complex may bind to receptors allowing internalization into specific B cells,
which then act as APC to CD4+ T cells, further fueling the immune response. Conversely, those B cells may dif-ferentiate into plasma cells, explaining why specific anti-tTG antibody production only occurs under a gluten-containing diet [64]. The pathophysiology of CD is sum-marized in Figure 1.
Clinical Manifestations and Associated Diseases
The clinical spectrum of CD is broad, which accounts for the challenging diagnosis. Classical gastrointestinal manifestations are more common in children, whereas
Dietary trigger
Glutenpeptides
Tightjunctions
Enterocyte IL-15
Atrophied enterocytes
CD8cell CD8
cell
Mucosal inflammation
Autoimmunity
• Villus atrophy• lntraepithelial lymphocytes
Antibodies:• Anti-gliadin• Anti-tissue TG
Cytokines:• INF-γ• TNF-α
NKcells
CD4 +T cell
Geneticsusceptibility
HLA DQ2/8
Deamidation
Leaky gutLaminapropria
tTG2
APC
Bcell
Th1
Th1
Th2Intestinal and
extraintestinal symptoms
Dysbiota
Gram bacteria
Tight junction disassemblyLumen
Fig. 1. Pathogenesis of CD. Gluten peptides reach the lamina propria through a leaky gut, with increased intes-tinal permeability, which may be due to drug-induced inflammation or dysbiota-induced disassembly of entero-cyte tight junctions. Gluten peptides are then deamidated by tTG, which allows them to be presented to CD4-T cells by APC that exhibit MHC class II molecules encoded by the haplotypes HLA-DQ2 or HLA-DQ8. This will trigger T-helper 1 and T-helper 2 immune responses that result in mucosal cytotoxicity and inflammation, as well as autoimmune extraintestinal phenomena.

CD Revisited 115GE Port J Gastroenterol 2022;29:111–124DOI: 10.1159/000514716
adults tend to be paucisymptomatic [25]. Classical mani-festations are chronic diarrhea (in 35%), abdominal pain (28%), and weight loss (22%) [53, 65]. CD can also pres-ent paradoxically with chronic constipation (20%), ab-dominal distension (20%), gastroesophageal reflux (12%), and even obesity [25, 53].
The most common extraintestinal manifestations are decreased bone mineralization (osteopenia in 50–70% and osteoporosis in 5.5% of cases), anemia (32%), arthral-gia (29%), fatigue (26%), and neurological symptoms (20%), particularly gluten ataxia and peripheral neuropa-thy [53, 66]. Gluten ataxia is an autoimmune injury of the cerebellum, induced by gluten ingestion, which manifests with a typical serology and abnormal gait, muscle coordi-nation, and fine control of voluntary movements, as well as cerebellum atrophy on magnetic resonance imaging (up to 60%). The mean age at onset is around 50 years [67] and the effect of a gluten-free diet (GFD) is controversial [68–71]. Gluten neuropathy is a sensitive neuropathy that is associated with serological evidence of CD, which ini-tially affects the hands and feet but usually progresses. The mean age at diagnosis is 55 years and a GFD can im-prove symptoms regardless of the presence or absence of enteropathy [71].
CD can also manifest with hypertransaminasemia (9–14%), recurrent aphthous stomatitis, tooth enamel de-fects, infertility, delayed puberty, and a short stature [53, 66, 72]. Most extraintestinal manifestations improve with a GFD, but an early diagnosis is crucial and some mani-festations, such as enamel defects, may be irreversible [66].
CD is associated with many genetic disorders. The prevalence of CD is higher in patients with chromoso-mopathies (i.e., 5–10% in patients with Down, Turner, and Williams syndromes) [73]. This might be explained by the proinflammatory millieu and impaired function of CD4+ T cells associated with chromosomopathies [46, 74].
Autoimmune glandular diseases, particularly T1DM and thyroid disease, are strongly associated with CD; 10–30% of CD patients have 1 of those 2 autoimmune dis-eases and up to 7% of patients with autoimmune glandu-lar diseases have CD [25, 75]. In fact, those conditions share a genetic background with a tight link to HLA-DQ2/8 and DR3/4 [75]. Interestingly, the prevalence of autoimmune diseases increases with increasing age at di-agnosis, probably as a consequence of a higher duration of exposure to gluten [76].
Herpetiform dermatitis (HD) is a dermatological au-toimmune disease that also shares a genetic background
with CD. Up to 20% of CD patients develop HD and more than 90% of HD patients have CD. HD diagnosis can be confirmed by skin biopsy demonstrating IgA deposits in the papillary dermis adjacent to the lesion. These patients present anti-tTG as well as IgA anti-epidermal transglu-taminase antibodies. HD responds to GFD, although transient treatment with dapsone may be needed [25, 66, 77].
CD patients have an increased risk of hepatic diseases such as steatosis, autoimmune hepatitis, primary biliary cholangitis (at least a 20-fold increase) [78], and primary sclerosing cholangitis (4- to 8-fold increase) [79].
Finally, patients with a selective IgA deficiency present a risk of CD that is 10–20 times higher [80]. The reverse is also true, i.e., IgA deficiency is 10–15 times more fre-quent in patients with CD [81].
Diagnosis
Who to Test?Current guidelines recommend testing for CD pa-
tients with signs, symptoms, or laboratorial evidence of malabsorption, unexplained fatigue, and recurrent mouth ulcers. Furthermore, patients with T1DM or autoim-mune thyroid disease should be regularly tested [82, 83]. Importantly, the presence of symptoms is not required for a diagnosis of CD [84].
CD screening is recommended in patients with irrita-ble bowel syndrome, since these patients are 4 times more likely to have CD than the general population, even pa-
Table 2. Current recommendations on CD screening
Offer serological testing Consider serological testing
– Persistent unexplained abdominal or gastrointestinal symptoms
– Faltering growth– Prolonged fatigue– Unexpected weight loss– Severe or persistent mouth ulcers– Unexplained iron, vitamin B12, or
folate deficiency– T1DM– Autoimmune thyroid disease– Irritable bowel syndrome– First-degree relatives of CD pa-tients
– Unexplained persistent raised liver enzymes– Metabolic bone disorder– Unexplained subfertility or recurrent miscarriage– Dental enamel defects– Unexplained neurologic
symptoms– Down syndrome– Turner syndrome
Adapted from Downey et al. [86].

Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124116DOI: 10.1159/000514716
tients presenting with obstipation [85]. Lastly, first-de-gree relatives of CD patients should be screened, though there are no recommendations regarding the time inter-val for rescreening [73].
The following high-risk groups should also be consid-ered for screening: children and adolescents with chro-mosomopathies and patients with metabolic bone disor-ders, unexplained neurological symptoms, hypertrans-aminasemia or infertility, and dental enamel defects [73]. CD screening recommendations are displayed in Table 2.
Population-based screening is not recommended, since it has not been proven that the diagnosis of asymp-tomatic patients improves their quality of life [87, 88].
Diagnostic ToolsCD diagnosis relies on 3 main pillars, i.e., serological
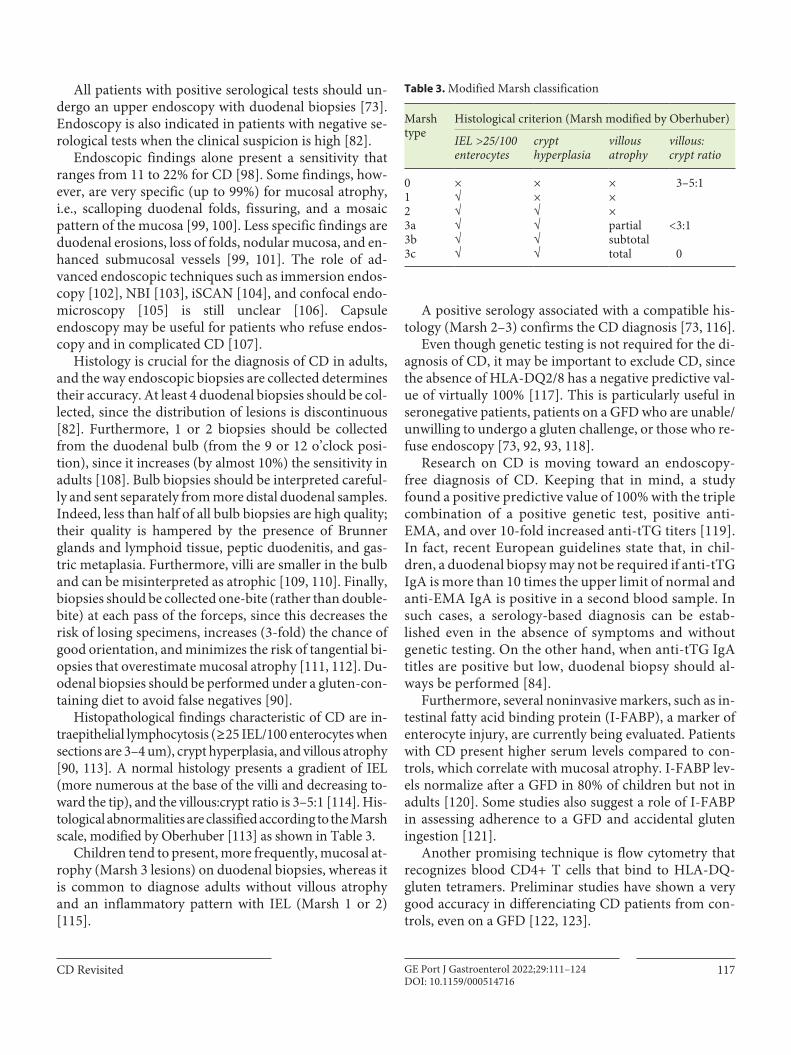
tests, duodenal histology, and genetic testing [89]. The diagnostic algorithm is presented in Figure 2.
Anti-tTG IgA is the recommended first-line serologi-cal test, as it is the most sensitive test (98%) and has a very good specificity (96%) [47]. Anti-tTG is determined through ELISA, allowing quantitation. Anti-endomysial (EMA) IgA reacts to the same antigen of tTG, but bound to tissue, requiring immunofluorescence in tissue from primate oesophagus or human umbilical cord. As such, EMA is more expensive, technically more challenging, and operator dependent, and it only allows qualitative re-sults [90]. The anti-EMA IgA test is the most specific se-
rological test [91] and it should be used as a confirmatory test, especially when the anti-tTG level is lower than 2 times the upper limit of normal [83, 89]. Anti-tTG and anti-EMA IgG have a low sensitivity and should be inter-preted carefully [91].
AGA is not recommended for CD diagnosis due to the low sensitivity and specificity [82, 92]. More recently, an-ti-deamidated gliadin peptides (DGP), which are assessed by an ELISA assay, have been adopted, particularly IgG that is superior to other IgG antibodies (88% sensitivity and 99% specificity) [91]. Anti-DGP IgG is particularly useful in patients with a selective IgA deficiency [83, 93].
Diagnosis should start with measurement of both anti-tTG IgA and IgA serum levels. Positive anti-tTG IgA should prompt duodenal biopsies to confirm the diagno-sis. When the anti-tTG IgA titer is low (4–10 U/mL) with normal IgA levels, anti-EMA IgA should be assessed. Lastly, when IgA < 1 mg/dL, IgG antibodies should be as-sessed (particularly anti-DGP IgG) [90].
All serological tests should be performed under a glu-ten-containing diet to avoid false-negative results [82]. False-positive results may occur with intestinal infections (e.g., Giardia lamblia) [94], chronic liver disease [95], con-gestive heart failure [96], and hypergammaglobulinemia [97]. Of note, serological tests have demonstrated high in-tertest and interlaboratory variability at lower ranges, and this should be confirmed by alternative tests when levels are lower than 10 times the upper limit of normal [84].
tTGIgA+
totalIgA
Upperendoscopy
withduodenalbiopsies
Biopsyconsistentwith CD
(Marsh 2–3)
Biopsy notconsistentwith CD
(Marsh 0–1)
HLA testingif high
suspicion ofCD
Upperendoscopy
with duodenalbiopsies
SeronegativeCD
(Marsh 2–3)
Potential CD
CD confirmed
CDexcluded
CDunlikely
+
+
–
–
Fig. 2. CD diagnosis flowchart.
CD Revisited 117GE Port J Gastroenterol 2022;29:111–124DOI: 10.1159/000514716
All patients with positive serological tests should un-dergo an upper endoscopy with duodenal biopsies [73]. Endoscopy is also indicated in patients with negative se-rological tests when the clinical suspicion is high [82].
Endoscopic findings alone present a sensitivity that ranges from 11 to 22% for CD [98]. Some findings, how-ever, are very specific (up to 99%) for mucosal atrophy, i.e., scalloping duodenal folds, fissuring, and a mosaic pattern of the mucosa [99, 100]. Less specific findings are duodenal erosions, loss of folds, nodular mucosa, and en-hanced submucosal vessels [99, 101]. The role of ad-vanced endoscopic techniques such as immersion endos-copy [102], NBI [103], iSCAN [104], and confocal endo-microscopy [105] is still unclear [106]. Capsule endoscopy may be useful for patients who refuse endos-copy and in complicated CD [107].
Histology is crucial for the diagnosis of CD in adults, and the way endoscopic biopsies are collected determines their accuracy. At least 4 duodenal biopsies should be col-lected, since the distribution of lesions is discontinuous [82]. Furthermore, 1 or 2 biopsies should be collected from the duodenal bulb (from the 9 or 12 o’clock posi-tion), since it increases (by almost 10%) the sensitivity in adults [108]. Bulb biopsies should be interpreted careful-ly and sent separately from more distal duodenal samples. Indeed, less than half of all bulb biopsies are high quality; their quality is hampered by the presence of Brunner glands and lymphoid tissue, peptic duodenitis, and gas-tric metaplasia. Furthermore, villi are smaller in the bulb and can be misinterpreted as atrophic [109, 110]. Finally, biopsies should be collected one-bite (rather than double-bite) at each pass of the forceps, since this decreases the risk of losing specimens, increases (3-fold) the chance of good orientation, and minimizes the risk of tangential bi-opsies that overestimate mucosal atrophy [111, 112]. Du-odenal biopsies should be performed under a gluten-con-taining diet to avoid false negatives [90].
Histopathological findings characteristic of CD are in-traepithelial lymphocytosis (≥25 IEL/100 enterocytes when sections are 3–4 um), crypt hyperplasia, and villous atrophy [90, 113]. A normal histology presents a gradient of IEL (more numerous at the base of the villi and decreasing to-ward the tip), and the villous:crypt ratio is 3–5: 1 [114]. His-tological abnormalities are classified according to the Marsh scale, modified by Oberhuber [113] as shown in Table 3.
Children tend to present, more frequently, mucosal at-rophy (Marsh 3 lesions) on duodenal biopsies, whereas it is common to diagnose adults without villous atrophy and an inflammatory pattern with IEL (Marsh 1 or 2) [115].
A positive serology associated with a compatible his-tology (Marsh 2–3) confirms the CD diagnosis [73, 116].
Even though genetic testing is not required for the di-agnosis of CD, it may be important to exclude CD, since the absence of HLA-DQ2/8 has a negative predictive val-ue of virtually 100% [117]. This is particularly useful in seronegative patients, patients on a GFD who are unable/unwilling to undergo a gluten challenge, or those who re-fuse endoscopy [73, 92, 93, 118].
Research on CD is moving toward an endoscopy-free diagnosis of CD. Keeping that in mind, a study found a positive predictive value of 100% with the triple combination of a positive genetic test, positive anti-EMA, and over 10-fold increased anti-tTG titers [119]. In fact, recent European guidelines state that, in chil-dren, a duodenal biopsy may not be required if anti-tTG IgA is more than 10 times the upper limit of normal and anti-EMA IgA is positive in a second blood sample. In such cases, a serology-based diagnosis can be estab-lished even in the absence of symptoms and without genetic testing. On the other hand, when anti-tTG IgA titles are positive but low, duodenal biopsy should al-ways be performed [84].
Furthermore, several noninvasive markers, such as in-testinal fatty acid binding protein (I-FABP), a marker of enterocyte injury, are currently being evaluated. Patients with CD present higher serum levels compared to con-trols, which correlate with mucosal atrophy. I-FABP lev-els normalize after a GFD in 80% of children but not in adults [120]. Some studies also suggest a role of I-FABP in assessing adherence to a GFD and accidental gluten ingestion [121].
Another promising technique is flow cytometry that recognizes blood CD4+ T cells that bind to HLA-DQ-gluten tetramers. Preliminar studies have shown a very good accuracy in differenciating CD patients from con-trols, even on a GFD [122, 123].
Table 3. Modified Marsh classification
Marsh type
Histological criterion (Marsh modified by Oberhuber)
IEL >25/100 enterocytes
crypt hyperplasia
villous atrophy
villous: crypt ratio
0 × × × 3–5:11 √ × ×2 √ √ ×3a √ √ partial <3:13b √ √ subtotal3c √ √ total 0

Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124118DOI: 10.1159/000514716
Diagnostic ChallengesSeronegative-CD refers to a compatible histology and
HLA, with a negative serology, and corresponds to 2% of CD patients [124, 125]. To confirm seronegative CD, the histology must improve after GFD. However, GFD is ad-vised only after excluding other diagnoses, since seroneg-ative CD accounts for < 30% of seronegative villous atro-phy or epithelial lymphocytosis [90, 126, 127]. As such, these should be excluded: autoimmune enteropathy (an-ti-enterocyte antibody positive), common variable im-munodeficiency, Crohn disease, eosinophilic gastroen-teritis, infectious diseases (Whipple disease, G. lamblia, tuberculosis, HIV-associated enteropathy, and tropical sprue), bacterial overgrowth, lymphoproliferative diseas-es, and drug-associated enteropathy. The drugs most fre-quently implicated are nonsteroidal anti-inflammatory drugs, immunosuppressors (azathioprine, mycopheno-late mofetil, and methotrexate), and angiotensin receptor antagonists, in particular olmesartan, which is responsi-ble for one fifth of seronegative duodenal-atrophy cases in the USA [116, 128–130]. In CD, IEL is composed sole-ly of CD8+ T cells, whereas in non-CD villous atrophy IEL has mixed CD8+ and CD4+ T cells. CD-associated lymphocytosis is also suggested when over 5% of T-cell receptors in IEL are γ/δ and when the base-tip decrescen-do gradient is lost [90, 114, 131]. Importantly, non-CD mucosal atrophy reverts spontaneously, without GFD, in over two thirds of patients [126].
Compared to seropositive patients, seronegative CD patients tend to be older (age 49 vs. 36 year) but they pres-ent more frequently the classic phenotype [125]. The physiopathology of seronegative CD is not yet clear, but some studies have suggested a high antibody-antigen binding affinity entrapping antibodies in the lamina pro-pria away from the bloodstream. Accordingly, detection of tTG-anti-tTG immunocomplexes in the mucosa can help to identify these patients [132]. Other explanations for seronegative CD are immune system immaturity [133], selective IgA deficiency, a diet poor in gluten, treat-ment with immunossupressors, and refractory long-term CD [126].
Potential CD (PCD) refers to seropositive patients with a normal duodenal mucosa (Marsh 0) or intraepi-thelial lymphocytosis (Marsh 1) without crypt hyperpla-sia or villous atrophy. PCD accounts for 10% of CD pa-tients [93]. Whereas over 80% of children are asymptom-atic [134, 135], the majority of adults (79%) with PCD are symptomatic, mostly with a nonclassic phenotype [136]. Symptomatic PCD patients should be kept on a GFD, since it results in clinical improvement. The management
of asymptomatic PCD patients is less straightforward, since the progression rate to overt CD is low, i.e., 13% in 10 years [137]. Asymptomatic PCD patients may main-tain a gluten-containing diet, with evaluation every 6 months for symptoms and serology and a duodenal bi-opsy every 2 years if there is a persistently positive serol-ogy [136].
Patients on a GFD prior to CD diagnosis may be tested for HLA since the absence of HLA-DQ2/8 excludes CD. In the presence of HLA-DQ2/8, patients should repeat the diagnostic workup after a gluten challenge [138]. Tradi-tionally, a gluten challenge consists of consumption of 7.5 g/day of gluten for 6–8 weeks; however, 3 g/day of gluten (equivalent to 2 slices of bread) is as effective. For patients who cannot tolerate a long gluten challenge, recent studies suggest that 2 weeks may be enough. One caveat is that, whereas histology can be performed immediately after the challenge, serology must be postponed 2 more weeks after the 2-week challenge. This approach allows a correct di-agnosis in more than 75% of CD patients [138].
Non-celiac gluten sensitivity (NCGS) is a functional disorder that must be differentiated from CD. NCGS is 6 times more prevalent than CD [139], and it is more fre-quent in females in their second or third decade [140]. Clinical manifestations are elicited by gluten ingestion and they are similar to CD [141]; however, the kinetics between gluten ingestion and symptoms is much faster; intestinal or extra-intestinal symptoms develop and re-solve hours to days after gluten ingestion or eviction [142, 143]. Furthermore, NCGS is not associated with autoim-mune disorders. NCGS diagnosis is clinical with demon-stration of symptoms resolution with a GFD, and recur-rence after rechallenge, and requiring exclusion of CD and wheat allergy [141, 144]. The pathogenesis is un-known, though it is probably multifactorial, resulting from an interplay between the environment (other com-ponents of wheat), intestinal barrier dysfunction, gut dys-biota, and diregulated innate imune responses [140].
Management
A lifelong GFD is the only proven treatment and is recommended for classical, nonclassical, seronegative CD, symptomatic PCD, and HD or gluten ataxia [93]. GFD is not recommended for asymptomatic adults with PCD, since only a minority of these patients will develop villus atrophy [136].
A GFD consists of a strict elimination of wheat (and its gluten-containing derivatives bulgur, couscous, and seit-

CD Revisited 119GE Port J Gastroenterol 2022;29:111–124DOI: 10.1159/000514716
an) [89], rye, and barley [145]. Elimination of oats is not so straightforward. In fact, oats contain avenin, a peptid related to gluten that may elicit similar immune reactions. Oats can also induce symptoms by an increase in fiber content. Hence, the oat intake should not exceed 50–60 g/day and patients should be clinically and serologically monitored. Oats should be avoided in severe disease [146].
The tolerable amount of gluten is variable, but as little as 1/100th of a slice of bread (around 50 mg of gluten) is sufficient to induce mucosal atrophy. Gluten-free is de-fined as < 20 ppm of gluten (around 6 mg/day) [147, 148]. Patients should be aware of nondietary potential sources of gluten contamination such as tooth paste and lipstick.
Gastrointestinal symptoms improve after 1 month and usually disappear after 6 months on a GFD [149]. Most patients become seronegative after 6 months on a GFD and only 17% remain seropositive after 1 year [150, 151], suggesting gluten contamination [73]. Anti-tTG IgA is the preferred serological test to monitor GFD adherence. Histological normalization takes longer, particularly in adults, in whom it takes 2–5 years [152]. Only 66% of pa-tients on a GFD achieve a total histological recovery after 1 year, in contrast with the expected recovery in 95% of children [153].
A GFD can also improve extraintestinal manifesta-tions and CD-related conditions. However, some mani-festations such as enamel defects and osteopenia may be irreversible or just partially corrected [66, 154].
The GFD should be lifelong, even if the patient ac-quires a clinical tolerance to gluten. Although 20% of pa-tients maintain histological remission after gluten rein-troduction, IEL and a positive serology tend to remain, and those patients are at an increased risk for extraintes-tinal manifestations and late relapse [155]. Importantly, strict adherence to a GFD is low, i.e., 17–48% [156, 157], and mortality seems to increase 5-fold in patients who do not comply with a GFD [158].
Alternative therapies are under investigation but none has shown sufficient efficacy yet to enter clinical practice. Investigational drugs include genetically modified less immunogenic wheat strains, prolyl endopeptidases, non-absorbable polymers with a high affinity for gliadin, drugs that act on intestinal permeability gluten deamination, and HLA inhibitors, among others [159].
Patients should be monitored at 6 months and then yearly for GFD adherence, symptoms, serology, a micro-nutrient deficiency, and associated conditions. Laborato-rial tests should include anti-tTG IgA, a full blood count, iron, folic acid, vitamin B12, calcium, vitamin D, thyroid function, and anti-thyroid antibodies [26, 152]. Follow-
up endoscopy is advised for persistent or relapsing symp-toms despite a GFD [82]. Osteodensitometry should be assessed every 1–2 years [89]. Lastly, vaccination against pneumococci, Haemophilus influenza, and meningococci are strongly recommended [73].
About 1.5% of CD patients progress to refractory CD (RCD), defined as persistence of clinical malabsorption and villous atrophy, after 1 year on GFD, after exclusion of other causes for villous atrophy or malignancy [116]. The main cause of persistent villous atrophy is inadver-tent gluten ingestion. Other conditions, i.e., lactose intol-erance, irritable bowel syndrome, small bowel bacterial overgrowth, pancreatic insufficiency, and microscopic colitis, should be excluded [83].
RCD is subclassified into types I and II, according to phenotype and clonality of IEL. In type I RCD, IEL are phenotypically normal with polyclonality of the T-cell re-ceptor, whereas in typeII T cells are aberrant, lacking sur-face CD8 and CD3 expression while expressing intracy-toplasmatic CD3, and presenting a monoclonal receptor rearrangement. The distinction of these 2 entities is cru-cial because the treatment and prognosis are different [89, 160, 161]. Type I RCD usually responds to steroids and budesonide or immunomodulators such as azathioprine. Type II RCD is more aggressive and it is associated with ulcerative jejunoileitis, severe malabsorption, a high risk of progression to enteropathy-associated T-cell lympho-ma (EATL) (50%, in 5–10 years) [154, 162], and a 5-year survival rate of 44–58% [154, 160]. Type II RCD does not respond to steroids, should not be treated with azathio-prine because of concerns of an increase in the risk for EATL, and may require treatment with cladribine or an autologous/allogenic bone marrow transplant. Targeting of IL-15 is a promising therapeutic strategy [163].
Patients with CD, especially long-standing and un-treated patients, present a higher risk for EATL and small intestine adenocarcinoma compared to the general popu-lation. The 5-year survival rate for EATL is 11%. The risk of developing other malignancies is still an unanswered topic [89, 164–166].
CD patients seem to have a 20% increase in mortality, particularly in those diagnosed as young adults and in the first 2 years after the diagnosis. Of note, increased mortal-ity in CD patients seems to occur even 10 years after the diagnosis [167]. The mortality rate is probably influenced not only by the age at diagnosis but also by the severity of the presentation, the adherence to a GFD, the ammount of gluten intake, and associated conditions [168].
Specific mortality increases in lymphoproliferative disorders, with a 2-fold increase particularly in women

Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124120DOI: 10.1159/000514716
over 50 years of age and in the 2 years following the diag-nosis. In fact, in a Finnish study on 12,803 CD patients followed for 7 years, 55% of CD patients died from T-cell lymphoma, compared to 3% of the reference population [169]. Globally, however, no increased risk for cancer-related mortality has been proven [166]. Importantly, CD patients seem to have a 5-fold increased risk of dying from infections, particularly sepsis [169].
Conclusions
CD is still an underdiagnosed entity that poses a diag-nostic challenge despite having been described, for the first time, almost 2 thousand years ago. It presents a wide range of unspecific signs and symptoms, both gastroin-testinal and extraintestinal. Adults tend to be paucisymp-tomatic, presenting nonclassical symptoms which can also occur in children. As such, even though population-based screening is not recommended, physicians should use an active case-finding strategy with a low threshold for screening.
Diagnosis requires highly accurate serological tests and a compatible duodenal histopathology, although in children duodenal biopsy may be avoided if the anti-tTG is higher than 10 times the upper limit of normal and anti-EMA is positive in a second blood sample. The pres-ence of HLA-DQ2/8 is mandatory for the development of
CD, being particularly helpful in excluding CD. Typical histology findings such as villous atrophy and crypt hy-perplasia are unspecific and other diseases, such as Crohn disease, infections, and drug-induced duodenitis, must be excluded.
A lifelong GFD is the only treatment with proven ef-ficacy; however, adherence to this diet is very low. As such, a GFD must be emphasized and monitored regu-larly. CD patients seem to have an increased mortality, most likely if left untreated.
Current and future research on CD should address en-doscopy-free diagnostic algorithms, easier monitoring of dietary gluten contamination, and alternative nondietary therapeutic strategies.
Conflict of Interest Statement
The authors have no conflict of interests to declare.
Funding Sources
The authors received no financial support for the research, au-thorship, or publication of this article.
Author Contributions
J.C. wrote this review. M.V.M. wrote and corrected this review.
References
1 Ludvigsson JF, Leffler DA, Bai JC, Biagi F, Fa-sano A, Green PH, et al. The Oslo definitions for coeliac disease and related terms. Gut. 2013 Jan; 62(1): 43–52.
2 Dowd B, Walker-Smith J. Letter: samuel Gee, Aretaeus, and the coeliac affection. Br Med J. 1974 May; 2(5916): 442.
3 Gasbarrini GB, Mangiola F, Gerardi V, Ianiro G, Corazza GR, Gasbarrini A. Coeliac disease: an old or a new disease? History of a patholo-gy. Intern Emerg Med. 2014 Apr; 9(3): 249–56.
4 Freeman HJ. Celiac disease: a disorder emerg-ing from antiquity, its evolving classification and risk, and potential new treatment para-digms. Gut Liver. 2015 Jan; 9(1): 28–37.
5 Gee S. On the coeilac affection. St Barthole-mews Hosp Rep. 1888; 24: 17.
6 Herter CA. On infantilism from chronic in-testinal infection, characterized by the over-growth and persistence of flora of the nursling period. New York: MacMillan; 1909. p. 416.
7 Yan D, Holt PR. Willem Dicke. Brilliant clin-ical observer and translational investigator. Discoverer of the toxic cause of celiac disease. Clin Transl Sci. 2009 Dec; 2(6): 446–8.
8 Haas SV. The value of the banana in the treat-ment of coeliac disease. Am J Dis Child. 1924;
24: 421–37. 9 van Berge-Henegouwen GP, Mulder CJ. Pio-
neer in the gluten free diet: Willem-Karel Dicke 1905-1962, over 50 years of gluten free diet. Gut. 1993 Nov; 34(11): 1473–5.
10 Paulley JW. Observation on the aetiology of idiopathic steatorrhoea; jejunal and lymph-node biopsies. BMJ. 1954 Dec; 2(4900): 1318–21.
11 Berger E, Burgin-Wolff A, Freudenberg E. Di-agnostisehe Bewertung des Nachweises yon Gliadin-AntikSrpern bei Cöliakie. Klinische Wochenschrift. 1964; 42: 788–90.
12 Kilander AF, Dotevall G, Fällström SP, Gill-berg RE, Nilsson LÅ, Tarkowski A. Evalua-tion of gliadin antibodies for detection of co-eliac disease. Scand J Gastroenterol. 1983 May; 18(3): 377–83.
13 Signer E, Bürgin-Wolff A, Berger R, Birbau-mer A, Just M. Antibodies to gliadin as a screening test for coeliac disease. A prospec-tive study. Helv Paediatr Acta. 1979 Feb;
34(1): 41–52.
14 Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997 Jul; 3(7): 797–801.
15 Chorzelski TP, Beutner EH, Sulej J, Tchorze-wska H, Jablonska S, Kumar V, et al. IgA anti-endomysium antibody. A new immunologi-cal marker of dermatitis herpetiformis and coeliac disease. Br J Dermatol. 1984 Oct;
111(4): 395–402.16 Falchuk ZM, Rogentine GN, Strober W. Pre-
dominance of histocompatibility antigen HL-A8 in patients with gluten-sensitive enteropa-thy. J Clin Invest. 1972 Jun; 51(6): 1602–5.
17 Parzanese I, Qehajaj D, Patrinicola F, Aralica M, Chiriva-Internati M, Stifter S, et al. Celiac disease: from pathophysiology to treatment. World J Gastrointest Pathophysiol. 2017 May; 8(2): 27–38.
18 Singh P, Arora A, Strand TA, Leffler DA, Ca-tassi C, Green PH, et al. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. 2018 Jun; 16(6): 823–836.e2.

CD Revisited 121GE Port J Gastroenterol 2022;29:111–124DOI: 10.1159/000514716
19 Lionetti E, Gatti S, Pulvirenti A, Catassi C. Ce-liac disease from a global perspective. Best Pract Res Clin Gastroenterol. 2015; 29: 365–79.
20 Yuan J, Gao J, Li X, Liu F, Wijmenga C, Chen H, et al. The tip of the “celiac iceberg” in Chi-na: a systematic review and meta-analysis. PLoS One. 2013 Dec; 8(12):e81151.
21 Poddighe D, Turganbekova A, Baymukashe-va D, Saduakas Z, Zhanzakova Z, Abdra-khmanova S. Genetic predisposition to celiac disease in Kazakhstan: potential impact on the clinical practice in Central Asia. PLoS One. 2020 Jan; 15(1):e0226546.
22 Poddighe D, Rakhimzhanova M, Marchenko Y, Catassi C. Pediatric celiac disease in central and east asia: current knowledge and preva-lence. Medicina (Kaunas). 2019 Jan; 55(1): 1–8.
23 West J, Fleming KM, Tata LJ, Card TR, Crooks CJ. Incidence and prevalence of celiac disease and dermatitis herpetiformis in the UK over two decades: population-based study. Am J Gastroenterol. 2014 May; 109(5):
757–68.24 Withoff S, Li Y, Jonkers I, Wijmenga C. Un-
derstanding Celiac Disease by Genomics. Trends Genet. 2016 May; 32(5): 295–308.
25 McAllister BP, Williams E, Clarke K. A Com-prehensive Review of Celiac Disease/Gluten-Sensitive Enteropathies. Clin Rev Allergy Im-munol. 2019 Oct; 57(2): 226–43.
26 Lebwohl B, Sanders DS, Green PH. Coeliac disease. Lancet. 2018 Jan; 391(10115): 70–81.
27 Amil Dias J. Celiac Disease: What Do We Know in 2017? GE Port J Gastroenterol. 2017 Nov; 24(6): 275–8.
28 Vriezinga SL, Auricchio R, Bravi E, Castillejo G, Chmielewska A, Crespo Escobar P, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med. 2014 Oct; 371(14): 1304–15.
29 Aronsson CA, Lee HS, Liu E, Uusitalo U, Hummel S, Yang J, et al.; TEDDY STUDY GROUP. Age at gluten introduction and risk of celiac disease. Pediatrics. 2015 Feb; 135(2):
239–45.30 Silano M, Agostoni C, Sanz Y, Guandalini S.
Infant feeding and risk of developing celiac disease: a systematic review. BMJ Open. 2016 Jan; 6(1):e009163.
31 Pinto-Sánchez MI, Verdu EF, Liu E, Bercik P, Green PH, Murray JA, et al. Gluten Introduc-tion to Infant Feeding and Risk of Celiac Dis-ease: Systematic Review and Meta-Analysis. J Pediatr. 2016 Jan; 168: 132–143.e3.
32 Lionetti E, Castellaneta S, Francavilla R, Pul-virenti A, Tonutti E, Amarri S, et al.; SIGENP (Italian Society of Pediatric Gastroenterology, Hepatology, and Nutrition) Working Group on Weaning and CD Risk. Introduction of gluten, HLA status, and the risk of celiac dis-ease in children. N Engl J Med. 2014 Oct;
371(14): 1295–303.33 Fewtrell M, Bronsky J, Campoy C, Domellöf
M, Embleton N, Fidler Mis N, et al. Comple-mentary feeding: A position paper by the Eu-ropean Society for Paediatric Gastroenterol-ogy, Hepatology, and Nutrition (ESPGHAN)
committee on nutrition. J Pediatr Gastroen-terol Nutr. 2017 Jan; 64(1): 119–32.
34 Weile B, Cavell B, Nivenius K, Krasilnikoff PA. Striking differences in the incidence of childhood celiac disease between Denmark and Sweden: a plausible explanation. J Pediatr Gastroenterol Nutr. 1995 Jul; 21(1): 64–8.
35 Szajewska H, Shamir R, Mearin L, Ribes-Koninckx C, Catassi C, Domellöf M, et al. Gluten introduction and the risk of coeliac disease: A position paper by the european so-ciety for pediatric gastroenterology, hepatol-ogy, and nutrition. J Pediatr Gastroenterol Nutr. 2016 Mar; 62(3): 507–13.
36 Szajewska H, Shamir R, Chmielewska A, Pieścik-Lech M, Auricchio R, Ivarsson A, et al.; PREVENTCD Study Group. Systematic review with meta-analysis: early infant feed-ing and coeliac disease—update 2015. Ali-ment Pharmacol Ther. 2015 Jun; 41(11):
1038–54.37 Mearin ML. The prevention of coeliac disease.
Best Pract Res Clin Gastroenterol. 2015; 29:
493–501.38 Stene LC, Honeyman MC, Hoffenberg EJ,
Haas JE, Sokol RJ, Emery L, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitu-dinal study. Am J Gastroenterol. 2006 Oct;
101(10): 2333–40.39 Ludvigsson JF, Murray JA. Epidemiology of
Celiac Disease. Gastroenterol Clin North Am. 2019; 48: 1–18.
40 Nellikkal SS, Hafed Y, Larson JJ, Murray JA, Absah I. High Prevalence of Celiac Disease Among Screened First-Degree Relatives. Mayo Clin Proc. 2019 Sep; 94(9): 1807–13.
41 Singh P, Arora S, Lal S, Strand TA, Makharia GK. Risk of celiac disease in the first- and sec-ond-degree relatives of patients with celiac disease: A systematic review and meta-analy-sis. Am J Gastroenterol. Am J Gastroenterol. 2015 Nov; 110(11): 1539–48.
42 Pham-Short A, Donaghue KC, Ambler G, Phelan H, Twigg S, Craig ME. Screening for celiac disease in type 1 diabetes: A systematic review. Pediatrics. 2015 Jul; 136(1):e170–6.
43 Elli L, Bonura A, Garavaglia D, Rulli E, Flori-ani I, Tagliabue G, et al. Immunological com-orbity in coeliac disease: associations, risk fac-tors and clinical implications. J Clin Immu-nol. 2012 Oct; 32(5): 984–90.
44 Meini A, Pillan NM, Villanacci V, Monafo V, Ugazio AG, Plebani A. Prevalence and diag-nosis of celiac disease in IgA-deficient chil-dren. Ann Allergy Asthma Immunol. 1996 Oct; 77(4): 333–6.
45 Carnicer J, Farré C, Varea V, Vilar P, Moreno J, Artigas J. Prevalence of coeliac disease in Down’s syndrome. Eur J Gastroenterol Hepa-tol. 2001 Mar; 13(3): 263–7.
46 Mårild K, Størdal K, Hagman A, Ludvigsson JF. Turner syndrome and celiac disease: A case-control study. Pediatrics. 2016 Feb;
137(2):e20152232.47 Guandalini S, Assiri A. Celiac disease: a re-
view. JAMA Pediatr. 2014 Mar; 168(3): 272–8.
48 Kagnoff MF. Celiac disease: pathogenesis of a model immunogenetic disease. J Clin Invest. 2007 Jan; 117(1): 41–9.
49 Kagnoff MF. Overview and pathogenesis of celiac disease. Gastroenterology. 2005 Apr;
128(4 Suppl 1):S10–8.50 Balakireva AV, Zamyatnin AA. Properties of
gluten intolerance: gluten structure, evolu-tion, pathogenicity and detoxification capa-bilities. Nutrients. 2016 Oct; 8(10): 8.
51 Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in Celiac Sprue. Science. 2002 Sep;
297(5590): 2275–9.52 Stamnaes J, Sollid LM. Celiac disease: autoim-
munity in response to food antigen. Semin Immunol. 2015 Sep; 27(5): 343–52.
53 Leonard MM, Sapone A, Catassi C, Fasano A. Celiac disease and nonceliac gluten sensitivi-ty: A review. JAMA -. JAMA. 2017 Aug;
318(7): 647–56.54 Lammers KM, Lu R, Brownley J, Lu B, Gerard
C, Thomas K, et al. Gliadin induces an in-crease in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008 Jul; 135(1):
194–204.e3.55 Rescigno M, Di Sabatino A. Dendritic cells in
intestinal homeostasis and disease. J Clin In-vest. 2009 Sep; 119(9): 2441–50.
56 Xia J, Sollid LM, Khosla C. Equilibrium and ki-netic analysis of the unusual binding behavior of a highly immunogenic gluten peptide to HLA-DQ2. Biochemistry. 2005 Mar; 44(11): 4442–9.
57 Sollid LM. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics. 2017 Aug; 69(8-9):
605–16.58 du Pré MF, Sollid LM. T-cell and B-cell im-
munity in celiac disease. Best Pract Res Clin Gastroenterol. 2015 Jun; 29(3): 413–23.
59 Poddighe D, Rebuffi C, De Silvestri A, Capit-tini C. Carrier frequency of HLA-DQB1*02 allele in patients affected with celiac disease: A systematic review assessing the potential ra-tionale of a targeted allelic genotyping as a first-line screening. World J Gastroenterol. 2020 Mar; 26(12): 1365–81.
60 Karinen H, Kärkkäinen P, Pihlajamäki J, Ja-natuinen E, Heikkinen M, Julkunen R, et al. Gene dose effect of the DQB1*0201 allele con-tributes to severity of coeliac disease. Scand J Gastroenterol. 2006 Feb; 41(2): 191–9.
61 Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009 Dec; 9(12): 858–70.
62 Pender SL, Tickle SP, Docherty AJ, Howie D, Wathen NC, MacDonald TT. A major role for matrix metalloproteinases in T cell injury in the gut. J Immunol. 1997 Feb; 158(4): 1582–90.
63 Bajaj-Elliott M, Poulsom R, Pender SL, Wa-then NC, MacDonald TT. Interactions be-tween stromal cell—derived keratinocyte growth factor and epithelial transforming growth factor in immune-mediated crypt cell hyperplasia. J Clin Invest. 1998 Oct; 102(8):
1473–80.

Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124122DOI: 10.1159/000514716
64 Sollid LM, Molberg O, McAdam S, Lundin KE. Autoantibodies in coeliac disease: tissue transglutaminase—guilt by association? Gut. 1997 Dec; 41(6): 851–2.
65 Fernández A, González L, de-la-Fuente J. Co-eliac disease: clinical features in adult popula-tions. Rev Esp Enferm Dig. 2010 Jul; 102(8):
466–71.66 Laurikka P, Nurminen S, Kivelä L, Kurppa K.
Extraintestinal manifestations of celiac dis-ease: early detection for better long-term out-comes. Nutrients. 2018 Aug; 10(8): 1–14.
67 Hadjivassiliou M, Sanders DD, Aeschlimann DP. Gluten-related disorders: gluten ataxia. Dig Dis. 2015; 33(2): 264–8.
68 Bushara KO. Neurologic presentation of ce-liac disease. Gastroenterology. 2005 Apr;
128(4 Suppl 1):S92–7.69 Freeman HJ. Neurological disorders in adult
celiac disease. Can J Gastroenterol. 2008 Nov;
22(11): 909–11.70 Hadjivassiliou M, Rao DG, Grìnewald RA,
Aeschlimann DP, Sarrigiannis PG, Hoggard N, et al. Neurological dysfunction in coeliac disease and non-coeliac gluten sensitivity. Am J Gastroenterol. 2016 Apr; 111(4): 561–7.
71 Mearns ES, Taylor A, Thomas Craig KJ, Pug-lielli S, Leffler DA, Sanders DS, et al. Neuro-logical manifestations of neuropathy and ataxia in celiac disease: A systematic review. Nutrients. 2019 Feb; 11(2): 380.
72 Reunala T, Salmi TT, Hervonen K, Kaukinen K, Collin P. Dermatitis herpetiformis: A com-mon extraintestinal manifestation of coeliac disease. Nutrients. 2018 May; 10(5): 1–9.
73 Husby S, Koletzko S, Korponay-Szabó IR, Mearin ML, Phillips A, Shamir R, et al.; ESP-GHAN Working Group on Coeliac Disease Diagnosis; ESPGHAN Gastroenterology Committee; European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. European Society for Pediatric Gastroenterol-ogy, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gas-troenterol Nutr. 2012 Jan; 54(1): 136–60.
74 Pellegrini FP, Marinoni M, Frangione V, Te-deschi A, Gandini V, Ciglia F, et al. Down syn-drome, autoimmunity and T regulatory cells. Clin Exp Immunol. 2012 Sep; 169(3): 238–43.
75 Kahaly GJ, Frommer L, Schuppan D. Celiac disease and endocrine autoimmunity - the ge-netic link. Autoimmun Rev. 2018 Dec; 17(12):
1169–75.76 Ventura A, Magazzù G, Greco L; SIGEP Study
Group for Autoimmune Disorders in Celiac Disease. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. Gastroenterology. 1999 Aug; 117(2): 297–303.
77 Rodrigues F, Bachmeyer C. Coeliac disease and dermatitis herpetiformis. Lancet. 2018 Sep; 392(10151): 916.
78 Sørensen HT, Thulstrup AM, Blomqvist P, Nørgaard B, Fonager K, Ekbom A. Risk of pri-mary biliary liver cirrhosis in patients with co-eliac disease: danish and Swedish cohort data. Gut. 1999 May; 44(5): 736–8.
79 Ludvigsson JF, Elfström P, Broomé U, Ekbom A, Montgomery SM. Celiac disease and risk of liver disease: a general population-based study. Clin Gastroenterol Hepatol. 2007 Jan;
5(1): 63–69.e1.80 Korponay-Szabó IR, Dahlbom I, Laurila K,
Koskinen S, Woolley N, Partanen J, et al. El-evation of IgG antibodies against tissue trans-glutaminase as a diagnostic tool for coeliac disease in selective IgA deficiency. Gut. 2003 Nov; 52(11): 1567–71.
81 Kumar V, Jarzabek-Chorzelska M, Sulej J, Karnewska K, Farrell T, Jablonska S. Celiac disease and immunoglobulin a deficiency: how effective are the serological methods of diagnosis? Clin Diagn Lab Immunol. 2002 Nov; 9(6): 1295–300.
82 Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA; American College of Gastro-enterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gas-troenterol. 2013 May; 108(5): 656–76.
83 Al-Toma A, Volta U, Auricchio R, Castillejo G, Sanders DS, Cellier C, et al. European So-ciety for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United European Gastro-enterol J. 2019 Jun; 7(5): 583–613.
84 Husby S, Koletzko S, Korponay-Szabó I, Kurppa K, Mearin ML, Ribes-Koninckx C, et al. European Society Paediatric Gastroenter-ology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J Pedi-atr Gastroenterol Nutr. 2020 Jan; 70(1): 141–56.
85 Irvine AJ, Chey WD, Ford AC. Screening for Celiac Disease in Irritable Bowel Syndrome: An Updated Systematic Review and Meta-analysis. Am J Gastroenterol. 2017 Jan; 112(1):
65–76.86 Downey L, Houten R, Murch S, Longson D.;
Guideline Development Group. Recognition, assessment, and management of coeliac dis-ease: summary of updated NICE guidance. BMJ. 2015 Sep; 351:h4513.
87 Ludvigsson JF, Card TR, Kaukinen K, Bai J, Zingone F, Sanders DS, et al. Screening for ce-liac disease in the general population and in high-risk groups. United European Gastroen-terol J. 2015 Apr; 3(2): 106–20.
88 Leffler DA, Kelly CP. The cost of a loaf of bread in symptomless celiac disease. Gastro-enterology. 2014 Sep; 147(3): 557–9.
89 Elli L, Ferretti F, Orlando S, Vecchi M, Mon-guzzi E, Roncoroni L, et al. Management of celiac disease in daily clinical practice. Eur J Intern Med. 2018; 61: 15–24.
90 Kowalski K, Mulak A, Jasińska M, Paradows-ki L. Diagnostic challenges in celiac disease. Adv Clin Exp Med. 2017 Jul; 26(4): 729–37.
91 Schyum AC, Rumessen JJ. Serological testing for celiac disease in adults. United European Gastroenterol J. 2013 Oct; 1(5): 319–25.
92 Korponay-Szabó IR, Troncone R, Discepolo V. Adaptive diagnosis of coeliac disease. Best Pract Res Clin Gastroenterol. 2015 Jun; 29(3):
381–98.
93 Lau MS, Sanders DS. Optimizing the diag-nosis of celiac disease. Curr Opin Gastroen-terol. 2017 May; 33(3): 173–80.
94 Hanevik K, Wik E, Langeland N, Hausken T. Transient elevation of anti-transglutamin-ase and anti-endomysium antibodies in Giardia infection. Scand J Gastroenterol. 2018; 53(7): 809–12.
95 Vecchi M, Folli C, Donato MF, Formenti S, Arosio E, de Franchis R. High rate of positive anti-tissue transglutaminase antibodies in chronic liver disease. Role of liver decom-pensation and of the antigen source. Scand J Gastroenterol. 2003 Jan; 38(1): 50–4.
96 Peracchi M, Trovato C, Longhi M, Gasparin M, Conte D, Tarantino C, et al. Tissue trans-glutaminase antibodies in patients with end-stage heart failure. Am J Gastroenterol. 2002 Nov; 97(11): 2850–4.
97 Castillo NE, Theethira TG, Leffler DA. The present and the future in the diagnosis and management of celiac disease. Gastroenterol Rep (Oxf). 2015 Feb; 3(1): 3–11.
98 Emami MH, Karimi S, Nemati A. Do endo-scopic markers still play a role in the diagno-sis of celiac disease? Indian J Gastroenterol. 2008 Sep-Oct; 27(5): 183–5.
99 Dickey W. Endoscopic markers for celiac disease. Nat Clin Pract Gastroenterol Hepa-tol. 2006 Oct; 3(10): 546–51.
100 Ianiro G, Gasbarrini A, Cammarota G. En-doscopic tools for the diagnosis and evalua-tion of celiac disease. World J Gastroenterol. 2013 Dec; 19(46): 8562–70.
101 Balaban DV, Popp A, Vasilescu F, Haidautu D, Purcarea RM, Jinga M. Diagnostic yield of endoscopic markers for celiac disease. J Med Life. 2015 Oct-Dec; 8(4): 452–7.
102 Cammarota G, Pirozzi GA, Martino A, Zuc-calà G, Cianci R, Cuoco L, et al. Reliability of the “immersion technique” during routine up-per endoscopy for detection of abnormalities of duodenal villi in patients with dyspepsia. Gastrointest Endosc. 2004 Aug; 60(2): 223–8.
103 Dutta AK, Sajith KG, Shah G, Pulimood AB, Simon EG, Joseph AJ, et al. Duodenal villous morphology assessed using magnification narrow band imaging correlates well with histology in patients with suspected malab-sorption syndrome. Dig Endosc. 2014 Nov;
26(6): 720–5.104 Cammarota G, Ianiro G, Sparano L, La Mura
R, Ricci R, Larocca LM, et al. Image-en-hanced endoscopy with I-scan technology for the evaluation of duodenal villous pat-terns. Dig Dis Sci. 2013 May; 58(5): 1287–92.
105 Leong RW, Nguyen NQ, Meredith CG, Al-Sohaily S, Kukic D, Delaney PM, et al. In vivo confocal endomicroscopy in the diag-nosis and evaluation of celiac disease. Gas-troenterology. 2008 Dec; 135(6): 1870–6.
106 Penny HA, Mooney PD, Burden M, Patel N, Johnston AJ, Wong SH, et al. High defini-tion endoscopy with or without I-Scan in-creases the detection of celiac disease during routine endoscopy. Dig Liver Dis. 2016 Jun;
48(6): 644–9.

CD Revisited 123GE Port J Gastroenterol 2022;29:111–124DOI: 10.1159/000514716
107 Lewis SK, Semrad CE. Capsule Endoscopy and Enteroscopy in Celiac Disease. Gastro-enterol Clin North Am. 2019; 48: 73–84.
108 McCarty TR, O’Brien CR, Gremida A, Ling C, Rustagi T. Efficacy of duodenal bulb bi-opsy for diagnosis of celiac disease: a system-atic review and meta-analysis. Endosc Int Open. 2018 Nov; 6(11):E1369–78.
109 Taavela J, Popp A, Korponay-Szabo IR, Ene A, Vornanen M, Saavalainen P, et al. A Prospective Study on the Usefulness of Duodenal Bulb Biopsies in Celiac Disease Diagnosis in Children: urging Caution. Am J Gastroenterol. 2016 Jan; 111(1): 124–33.
110 Voutilainen M, Juhola M, Färkkilä M, Sip-ponen P. Gastric metaplasia and chronic in-flammation at the duodenal bulb mucosa. Dig Liver Dis. 2003 Feb; 35(2): 94–8.
111 Padda S, Shah I, Ramirez FC. Adequacy of mucosal sampling with the “two-bite” for-ceps technique: a prospective, randomized, blinded study. Gastrointest Endosc. 2003 Feb; 57(2): 170–3.
112 Latorre M, Lagana SM, Freedberg DE, Lewis SK, Lebwohl B, Bhagat G, et al. Endoscopic biopsy technique in the diagnosis of celiac disease: one bite or two? Gastrointest En-dosc. 2015 May; 81(5): 1228–33.
113 Oberhuber G. Histopathology of celiac dis-ease. Biomed Pharmacother. 2000 Aug;
54(7): 368–72.114 Brown I, Mino-Kenudson M, Deshpande V,
Lauwers GY. Intraepithelial lymphocytosis in architecturally preserved proximal small intestinal mucosa: an increasing diagnostic problem with a wide differential diagnosis. Arch Pathol Lab Med. 2006 Jul; 130(7): 1020–5.
115 Ciccocioppo R, Kruzliak P, Cangemi GC, Pohanka M, Betti E, Lauret E, et al. The spec-trum of differences between childhood and adulthood celiac disease. Nutrients. 2015 Oct; 7(10): 8733–51.
116 Ludvigsson JF, Bai JC, Biagi F, Card TR, Ciacci C, Ciclitira PJ, et al.; BSG Coeliac Dis-ease Guidelines Development Group; Brit-ish Society of Gastroenterology. Diagnosis and management of adult coeliac disease: guidelines from the British Society of Gas-troenterology. Gut. 2014 Aug; 63(8): 1210–28.
117 Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol. 2005 Sep; 3(9):
843–51.118 Sciurti M, Fornaroli F, Gaiani F, Bonaguri C,
Leandro G, Di Mario F, et al. Genetic suscep-tibilty and celiac disease: what role do HLA haplotypes play? Acta Biomed. 2018 Dec; 89 9-S: 17–21.
119 Fuchs V, Kurppa K, Huhtala H, Laurila K, Mäki M, Collin P, et al. Serology-based cri-teria for adult coeliac disease have excellent accuracy across the range of pre-test proba-bilities. Aliment Pharmacol Ther. 2019 Feb;
49(3): 277–84.
120 Adriaanse MP, Tack GJ, Passos VL, Damoi-seaux JG, Schreurs MW, van Wijck K, et al. Serum I-FABP as marker for enterocyte damage in coeliac disease and its relation to villous atrophy and circulating autoantibod-ies. Aliment Pharmacol Ther. 2013 Feb;
37(4): 482–90.121 Adriaanse MP, Leffler DA, Kelly CP, Schup-
pan D, Najarian RM, Goldsmith JD, et al. Se-rum I-FABP Detects Gluten Responsiveness in Adult Celiac Disease Patients on a Short-Term Gluten Challenge. Am J Gastroenter-ol. 2016 Jul; 111(7): 1014–22.
122 Sarna VK, Lundin KE, Mørkrid L, Qiao SW, Sollid LM, Christophersen A. HLA-DQ-Gluten Tetramer Blood Test Accurately Identifies Patients With and Without Celiac Disease in Absence of Gluten Consumption. Gastroenterology. 2018 Mar; 154(4): 886–896.e6.
123 Sarna VK, Skodje GI, Reims HM, Risnes LF, Dahal-Koirala S, Sollid LM, et al. HLA-DQ:gluten tetramer test in blood gives better detection of coeliac patients than biopsy af-ter 14-day gluten challenge. Gut. 2018 Sep;
67(9): 1606–13.124 Schiepatti A, Biagi F, Fraternale G, Vattiato
C, Balduzzi D, Agazzi S, et al. Short article: mortality and differential diagnoses of vil-lous atrophy without coeliac antibodies. Eur J Gastroenterol Hepatol. 2017 May; 29(5):
572–6.125 Volta U, Caio G, Boschetti E, Giancola F,
Rhoden KJ, Ruggeri E, et al. Seronegative ce-liac disease: shedding light on an obscure clinical entity. Dig Liver Dis. 2016 Sep; 48(9):
1018–22.126 Aziz I, Peerally MF, Barnes JH, Kandasamy
V, Whiteley JC, Partridge D, et al. The clini-cal and phenotypical assessment of seroneg-ative villous atrophy; a prospective UK cen-tre experience evaluating 200 adult cases over a 15-year period (2000-2015). Gut. 2017 Sep; 66(9): 1563–72.
127 Schiepatti A, Sanders DS, Biagi F. Seronega-tive coeliac disease: clearing the diagnostic dilemma. Curr Opin Gastroenterol. 2018 May; 34(3): 154–8.
128 Elli L, Branchi F, Sidhu R, Guandalini S, As-siri A, Rinawi F, et al. Small bowel villous atrophy: celiac disease and beyond. Expert Rev Gastroenterol Hepatol. 2017 Feb; 11(2):
125–38.129 Jansson-Knodell CL, Hujoel IA, Rubio-
Tapia A, Murray JA. Not All That Flattens Villi Is Celiac Disease: A Review of Enter-opathies. Mayo Clin Proc. 2018 Apr; 93(4):
509–17.130 Kamboj AK, Oxentenko AS. Clinical and
Histologic Mimickers of Celiac Disease. Clin Transl Gastroenterol. 2017 Aug; 8(8):e114.
131 Walker MM, Murray JA, Ronkainen J, Aro P, Storskrubb T, D’Amato M, et al. Detec-tion of celiac disease and lymphocytic enter-opathy by parallel serology and histopathol-ogy in a population-based study. Gastroen-terology. 2010 Jul; 139(1): 112–9.
132 Salmi TT, Collin P, Korponay-Szabó IR, Lau-rila K, Partanen J, Huhtala H, et al. Endomysi-al antibody-negative coeliac disease: clinical characteristics and intestinal autoantibody deposits. Gut. 2006 Dec; 55(12): 1746–53.
133 Ierardi E, Losurdo G, Piscitelli D, Giorgio F, Sorrentino C, Principi M, et al. Seronegative celiac disease: where is the specific setting? Gastroenterol Hepatol Bed Bench. 2015;
8(2): 110–6.134 Tosco A, Salvati VM, Auricchio R, Maglio
M, Borrelli M, Coruzzo A, et al. Natural His-tory of Potential Celiac Disease in Children. Clin Gastroenterol Hepatol. 2011; 9: 320–5.
135 Auricchio R, Tosco A, Piccolo E, Galatola M, Izzo V, Maglio M, et al. Potential celiac chil-dren: 9-year follow-up on a gluten-contain-ing diet. Am J Gastroenterol. 2014 Jun;
109(6): 913–21.136 Volta U, Caio G, Giancola F, Rhoden KJ,
Ruggeri E, Boschetti E, et al. Features and Progression of Potential Celiac Disease in Adults. Clin Gastroenterol Hepatol. 2016;
14: 686-693.e1.137 Lionetti E, Castellaneta S, Francavilla R, Pul-
virenti A, Naspi Catassi G, Catassi C; SI-GENP Working Group of Weaning and CD Risk. Long-Term Outcome of Potential Ce-liac Disease in Genetically at-Risk Children: The Prospective CELIPREV Cohort Study. J Clin Med. 2019 Feb; 8(2): 186.
138 Leffler D, Schuppan D, Pallav K, Najarian R, Goldsmith JD, Hansen J, et al. Kinetics of the histological, serological and symptomatic re-sponses to gluten challenge in adults with co-eliac disease. Gut. 2013 Jul; 62(7): 996–1004.
139 Sapone A, Bai JC, Ciacci C, Dolinsek J, Green PH, Hadjivassiliou M, et al. Spectrum of gluten-related disorders: consensus on new nomenclature and classification. BMC Med. 2012 Feb; 10(1): 13.
140 Volta U, De Giorgio R, Caio G, Uhde M, Manfredini R, Alaedini A. Nonceliac Wheat Sensitivity: An Immune-Mediated Condi-tion with Systemic Manifestations. Gastro-enterol Clin North Am. 2019 Mar; 48(1):
165–82.141 Catassi C, Elli L, Bonaz B, Bouma G, Carroc-
cio A, Castillejo G, et al. Diagnosis of non-celiac gluten sensitivity (NCGS): the salerno experts’ criteria. Nutrients. 2015 Jun; 7(6):
4966–77.142 Schuppan D, Pickert G, Ashfaq-Khan M,
Zevallos V. Non-celiac wheat sensitivity: dif-ferential diagnosis, triggers and implica-tions. Best Pract Res Clin Gastroenterol. 2015 Jun; 29(3): 469–76.
143 Volta U, Caio G, De Giorgio R, Henriksen C, Skodje G, Lundin KE. Non-celiac gluten sensitivity: A work-in-progress entity in the spectrum of wheat-related disorders. Best Pract Res Clin Gastroenterol. 2015; 29: 477–91.
144 Mansueto P, Seidita A, D’Alcamo A, Carroc-cio A. Non-celiac gluten sensitivity: litera-ture review. J Am Coll Nutr. 2014; 33(1): 39–54.

Calado/Verdelho MachadoGE Port J Gastroenterol 2022;29:111–124124DOI: 10.1159/000514716
145 Bascuñán KA, Vespa MC, Araya M. Celiac disease: understanding the gluten-free diet. Eur J Nutr. 2017 Mar; 56(2): 449–59.
146 Dennis M, Lee AR, McCarthy T. Nutritional Considerations of the Gluten-Free Diet. Gas-troenterol Clin North Am. 2019; 48: 53–72.
147 Akobeng AK, Thomas AG. Systematic re-view: tolerable amount of gluten for people with coeliac disease. Aliment Pharmacol Ther. 2008 Jun; 27(11): 1044–52.
148 Hischenhuber C, Crevel R, Jarry B, Mäki M, Moneret-Vautrin DA, Romano A, et al. Re-view article: safe amounts of gluten for patients with wheat allergy or coeliac disease. Aliment Pharmacol Ther. 2006 Mar; 23(5): 559–75.
149 Oxentenko AS, Murray JA. Celiac Disease: Ten Things That Every Gastroenterologist Should Know. Clin Gastroenterol Hepatol. Elsevier, Inc; 2015. p. 13.
150 Rashid M, Lee J. Serologic testing in celiac disease: practical guide for clinicians. Can Fam Physician. 2016 Jan; 62(1): 38–43.
151 Zanini B, Lanzarotto F, Mora A, Bertolazzi S, Turini D, Cesana B, et al. Five year time course of celiac disease serology during glu-ten free diet: results of a community based “CD-Watch” program. Dig Liver Dis. 2010 Dec; 42(12): 865–70.
152 Mulder CJ, Wierdsma NJ, Berkenpas M, Ja-cobs MAJM, Bouma G. Preventing compli-cations in celiac disease: Our experience with managing adult celiac disease. Best Pract Res Clin Gastroenterol. 2015; 29: 459–68.
153 Galli G, Esposito G, Lahner E, Pilozzi E, Cor-leto VD, Di Giulio E, et al. Histological re-covery and gluten-free diet adherence: a pro-spective 1-year follow-up study of adult pa-tients with coeliac disease. Aliment Pharmacol Ther. 2014 Sep; 40(6): 639–47.
154 Malamut G, Cellier C. Complications of co-eliac disease. Best Pract Res Clin Gastroen-terol. 2015; 29: 451–8.
155 Matysiak-Budnik T, Malamut G, de Serre NP, Grosdidier E, Seguier S, Brousse N, et al. Long-term follow-up of 61 coeliac patients diagnosed in childhood: evolution toward latency is possible on a normal diet. Gut. 2007 Oct; 56(10): 1379–86.
156 Hall NJ, Rubin G, Charnock A. Systematic review: adherence to a gluten-free diet in adult patients with coeliac disease. Aliment Pharmacol Ther. 2009 Aug; 30(4): 315–30.
157 Leffler DA, Edwards-George J, Dennis M, Schuppan D, Cook F, Franko DL, et al. Fac-tors that influence adherence to a gluten-free diet in adults with celiac disease. Dig Dis Sci. 2008 Jun; 53(6): 1573–81.
158 Corrao G, Corazza GR, Bagnardi V, Brusco G, Ciacci C, Cottone M, et al. Mortality in patients with coeliac disease and their rela-tives: a cohort study. Lancet. 2001; 358: 356–61.
159 Costes LM, Meresse B, Cerf-Bensussan N, Samsom JN. The role of animal models in unravelling therapeutic targets in coeliac disease. Best Pract Res Clin Gastroenterol. 2015 Jun; 29(3): 437–50.
160 Malamut G, Cellier C. Refractory celiac dis-ease: epidemiology and clinical manifesta-tions. Dig Dis. 2015; 33(2): 221–6.
161 Roshan B, Leffler DA, Jamma S, Dennis M, Sheth S, Falchuk K, et al. The incidence and clinical spectrum of refractory celiac disease in a north american referral center. Am J Gastroenterol. 2011 May; 106(5): 923–8.
162 Nijeboer P, van Wanrooij R, van Gils T, Wi-erdsma NJ, Tack GJ, Witte BI, et al. Lym-phoma development and survival in refrac-tory coeliac disease type II: histological re-sponse as prognostic factor. United European Gastroenterol J. 2017 Mar; 5(2):
208–17.163 Malamut G, Cellier C. Refractory Celiac Dis-
ease. Gastroenterol Clin North Am. 2019; 48:
137–44.164 Catassi C, Bearzi I, Holmes GK. Association
of celiac disease and intestinal lymphomas and other cancers. Gastroenterology. 2005 Apr; 128(4 Suppl 1):S79–86.
165 Ilus T, Kaukinen K, Virta LJ, Pukkala E, Col-lin P. Incidence of malignancies in diag-nosed celiac patients: a population-based es-timate. Am J Gastroenterol. 2014 Sep; 109(9):
1471–7.166 Tio M, Cox MR, Eslick GD. Meta-analysis:
coeliac disease and the risk of all-cause mor-tality, any malignancy and lymphoid malig-nancy. Aliment Pharmacol Ther. 2012 Mar;
35(5): 540–51.167 Lebwohl B, Green PH, Söderling J, Roels-
traete B, Ludvigsson JF. Association Be-tween Celiac Disease and Mortality Risk in a Swedish Population. JAMA. 2020 Apr;
323(13): 1277–85.168 Biagi F, Corazza GR. Do different patients
with coeliac disease have different mortality rates? Gut. 2015 Aug; 64(8): 1187–8.
169 Koskinen I, Virta LJ, Huhtala H, Ilus T, Kaukinen K, Collin P. Overall and Cause-Specific Mortality in Adult Celiac Disease and Dermatitis Herpetiformis Diagnosed in the 21st Century. Am J Gastroenterol. 2020 Jul; 115(7): 1117–24.