biomimetic magnesium–carbonate-apatite nanocrystals endowed with strontium ions as...
TRANSCRIPT

Materials Science and Engineering C 35 (2014) 212–219
Contents lists available at ScienceDirect
Materials Science and Engineering C
j ourna l homepage: www.e lsev ie r .com/ locate /msec
Biomimetic magnesium–carbonate-apatite nanocrystals endowed withstrontium ions as anti-osteoporotic trigger
Michele Iafisco ⁎, Andrea Ruffini, Alessio Adamiano, Simone Sprio, Anna TampieriInstitute of Science and Technology for Ceramics (ISTEC), National Research Council (CNR), Via Granarolo 64, 48018 Faenza, Italy
⁎ Corresponding author at: Institute of Science and TeNational Research Council (CNR), Via Granarolo 64, 48010546699730.
E-mail address: [email protected] (M. Iafisco
0928-4931/$ – see front matter © 2013 Elsevier B.V. All rihttp://dx.doi.org/10.1016/j.msec.2013.11.009
a b s t r a c t
a r t i c l e i n f oArticle history:Received 16 August 2013Received in revised form 30 September 2013Accepted 2 November 2013Available online 14 November 2013
Keywords:ApatiteStrontiumMagnesiumIn vitro bioactivityNanocrystalsOsteoporosis
The present work investigates the preparation of biomimetic nanocrystalline apatites co-substituted with Mg,CO3 and Sr to be used as starting materials for the development of nanostructured bio-devices for regenerationof osteoporotic bone. Biological-like amounts of Mg and CO3 ions were inserted in the apatite structure tomimic the composition of bone apatite, whereas the addition of increasing quantities of Sr ions, from 0 up to12 wt.%, as anti-osteoporotic agent, was evaluated. The chemical–physical features, the morphology, the degra-dation rates, the ion release kinetics as well as the in vitro bioactivity of the as-prepared apatites were fully eval-uated. The results indicated that the incorporation of 12 wt.% of Sr can be viewed as a threshold for the structuralstability of Mg–CO3-apatite. Indeed, incorporation of lower quantity of Sr did not induce considerable variationsin the chemical structure of Mg–CO3-apatite, while when the Sr doping extent reached 12 wt.%, a dramaticallydestabilizing effect was detected on the crystal structure thus yielding alteration of the symmetry and distortionof the PO4. As a consequence, this apatite exhibited the fastest degradation kinetic and the highest amount of Srions released when tested in physiological conditions. In this respect, the surface crystallization of new calciumphosphate phase when immersed in physiological-like solution occurred by different mechanisms and extentsdue to the different structural chemistry of the variously doped apatites. Nevertheless, all the apatites synthe-sized in this work exhibited in vitro bioactivity demonstrating their potential use to develop biomedical deviceswith anti-osteoporotic functionality.
© 2013 Elsevier B.V. All rights reserved.
1. Introduction
Calcium phosphates (CaPs) are widespread in nature because theyconstitute the main inorganic component of the mammalian hard tis-sues [1]. The mineral phase of bone and teeth is commonly assimilatedto synthetic hydroxyapatite (Ca10(PO4)6(OH)2), however, biologicalapatites differ from the stoichiometric ones in several features, includ-ing non-stoichiometry, crystals with dimensions in the nano-scale andlow degree of structural order [2,3]. Moreover, they are Ca and OH defi-cient and they contain significant amounts of foreign ions (such as CO3,Na, K, and Mg), either fully incorporated in the crystal lattice or justinserted in the non-apatitic sites of the surface [4]. The low degree ofcrystallinity, the small crystal sizes and the Ca and OH deficiencies to-getherwith the foreign ion substitution are responsible for the relativelyhigh solubility and specific biological activity of bone apatite withrespect to stoichiometric hydroxyapatite [5].
Materials based on CaP are well known for their excellent biocom-patibility, particularly in establishing tight bone bonding that yieldeffective and rapid osteointegration [6]. Among the CaP compounds
chnology for Ceramics (ISTEC),8 Faenza (RA), Italy. Tel.: +39
).
ghts reserved.
used as bone grafts, themost common is stoichiometric hydroxyapatiteprocessed as dense or porous ceramics, coatings and composites [7–9].It is reasonable to hypothesize that by virtue of their high similaritywith the mineral phase of bone, nano-sized and ionic substituted apa-tites can display better biological performances than coarser crystals[10,11]. Moreover, incorporation of foreign ions yields destabilizationof apatite lattice, hence its degradation is accelerated so that ions rele-vant for biological processes of bone can be released executing specificfunctions [12].
Several previousworks focused on the effect of single ionic substitu-tion in the apatite structure also indicating that these syntheses and therelative structural characterizations are not very complicated tasks[13–16]. On the contrary there is a lack of studies that examinedmulti-ionic substitutions [17,18]. In fact, the co-substitution of two ormore different ions in the hydroxyapatite lattice generates a complexscenario where a competition for the occupation of the crystal sites ofCa and PO4 by the foreign ions is established and where charge imbal-ances may create vacancies in the sites of Ca and OH. Nevertheless itsometimes offers the possibility to introduce and stabilize ions thatare difficult to be incorporated in the apatite structure alone, such asMg ions [19], or to enable substitution of some ions such as silicate ina greater extent [20].
CO3 is the prevalent anion substitution in the biological apatite(about 4–8 wt.%) [21] and in fact, the bone mineral phase is more
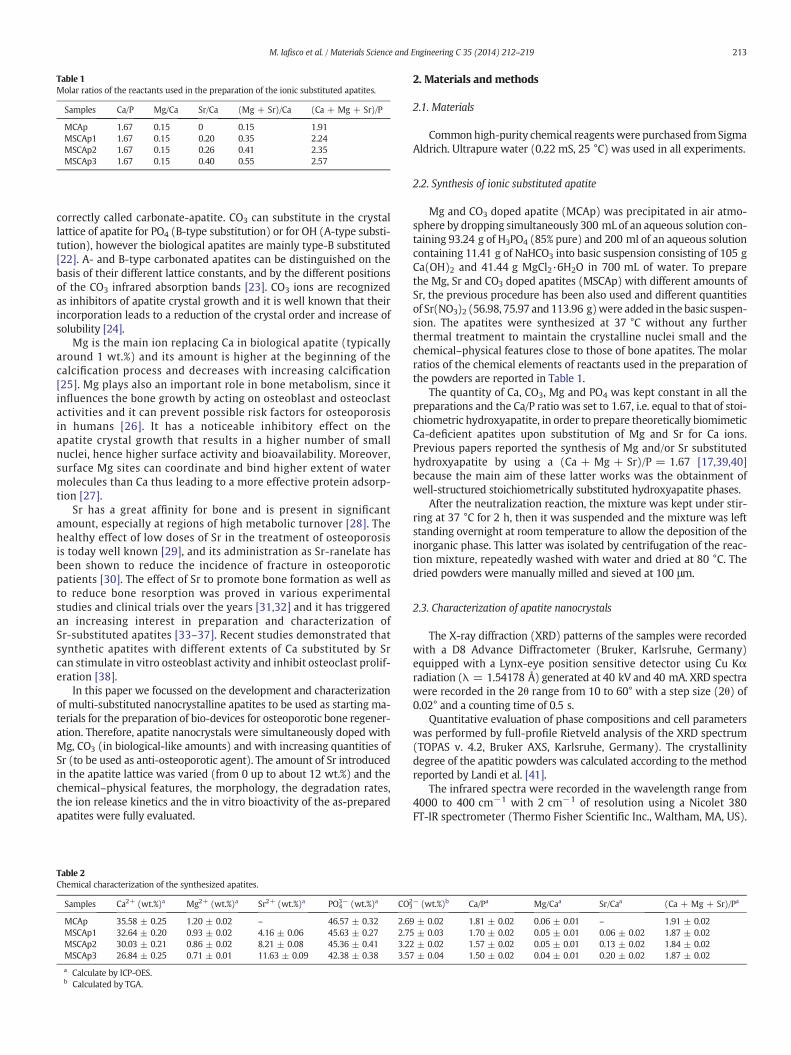
Table 1Molar ratios of the reactants used in the preparation of the ionic substituted apatites.
Samples Ca/P Mg/Ca Sr/Ca (Mg + Sr)/Ca (Ca + Mg + Sr)/P
MCAp 1.67 0.15 0 0.15 1.91MSCAp1 1.67 0.15 0.20 0.35 2.24MSCAp2 1.67 0.15 0.26 0.41 2.35MSCAp3 1.67 0.15 0.40 0.55 2.57
213M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
correctly called carbonate-apatite. CO3 can substitute in the crystallattice of apatite for PO4 (B-type substitution) or for OH (A-type substi-tution), however the biological apatites are mainly type-B substituted[22]. A- and B-type carbonated apatites can be distinguished on thebasis of their different lattice constants, and by the different positionsof the CO3 infrared absorption bands [23]. CO3 ions are recognizedas inhibitors of apatite crystal growth and it is well known that theirincorporation leads to a reduction of the crystal order and increase ofsolubility [24].
Mg is the main ion replacing Ca in biological apatite (typicallyaround 1 wt.%) and its amount is higher at the beginning of thecalcification process and decreases with increasing calcification[25]. Mg plays also an important role in bone metabolism, since itinfluences the bone growth by acting on osteoblast and osteoclastactivities and it can prevent possible risk factors for osteoporosisin humans [26]. It has a noticeable inhibitory effect on theapatite crystal growth that results in a higher number of smallnuclei, hence higher surface activity and bioavailability. Moreover,surface Mg sites can coordinate and bind higher extent of watermolecules than Ca thus leading to a more effective protein adsorp-tion [27].
Sr has a great affinity for bone and is present in significantamount, especially at regions of high metabolic turnover [28]. Thehealthy effect of low doses of Sr in the treatment of osteoporosisis today well known [29], and its administration as Sr-ranelate hasbeen shown to reduce the incidence of fracture in osteoporoticpatients [30]. The effect of Sr to promote bone formation as well asto reduce bone resorption was proved in various experimentalstudies and clinical trials over the years [31,32] and it has triggeredan increasing interest in preparation and characterization ofSr-substituted apatites [33–37]. Recent studies demonstrated thatsynthetic apatites with different extents of Ca substituted by Srcan stimulate in vitro osteoblast activity and inhibit osteoclast prolif-eration [38].
In this paper we focussed on the development and characterizationof multi-substituted nanocrystalline apatites to be used as starting ma-terials for the preparation of bio-devices for osteoporotic bone regener-ation. Therefore, apatite nanocrystals were simultaneously doped withMg, CO3 (in biological-like amounts) and with increasing quantities ofSr (to be used as anti-osteoporotic agent). The amount of Sr introducedin the apatite lattice was varied (from 0 up to about 12 wt.%) and thechemical–physical features, the morphology, the degradation rates,the ion release kinetics and the in vitro bioactivity of the as-preparedapatites were fully evaluated.
Table 2Chemical characterization of the synthesized apatites.
Samples Ca2+ (wt.%)a Mg2+ (wt.%)a Sr2+ (wt.%)a PO43− (wt.%)a CO3
2
MCAp 35.58 ± 0.25 1.20 ± 0.02 – 46.57 ± 0.32 2.69MSCAp1 32.64 ± 0.20 0.93 ± 0.02 4.16 ± 0.06 45.63 ± 0.27 2.75MSCAp2 30.03 ± 0.21 0.86 ± 0.02 8.21 ± 0.08 45.36 ± 0.41 3.22MSCAp3 26.84 ± 0.25 0.71 ± 0.01 11.63 ± 0.09 42.38 ± 0.38 3.57
a Calculate by ICP-OES.b Calculated by TGA.
2. Materials and methods
2.1. Materials
Commonhigh-purity chemical reagentswere purchased fromSigmaAldrich. Ultrapure water (0.22 mS, 25 °C) was used in all experiments.
2.2. Synthesis of ionic substituted apatite
Mg and CO3 doped apatite (MCAp) was precipitated in air atmo-sphere by dropping simultaneously 300 mL of an aqueous solution con-taining 93.24 g of H3PO4 (85% pure) and 200 ml of an aqueous solutioncontaining 11.41 g of NaHCO3 into basic suspension consisting of 105 gCa(OH)2 and 41.44 g MgCl2·6H2O in 700 mL of water. To preparethe Mg, Sr and CO3 doped apatites (MSCAp) with different amounts ofSr, the previous procedure has been also used and different quantitiesof Sr(NO3)2 (56.98, 75.97 and 113.96 g)were added in the basic suspen-sion. The apatites were synthesized at 37 °C without any furtherthermal treatment to maintain the crystalline nuclei small and thechemical–physical features close to those of bone apatites. The molarratios of the chemical elements of reactants used in the preparation ofthe powders are reported in Table 1.
The quantity of Ca, CO3, Mg and PO4 was kept constant in all thepreparations and the Ca/P ratio was set to 1.67, i.e. equal to that of stoi-chiometric hydroxyapatite, in order to prepare theoretically biomimeticCa-deficient apatites upon substitution of Mg and Sr for Ca ions.Previous papers reported the synthesis of Mg and/or Sr substitutedhydroxyapatite by using a (Ca + Mg + Sr)/P = 1.67 [17,39,40]because the main aim of these latter works was the obtainment ofwell-structured stoichiometrically substituted hydroxyapatite phases.
After the neutralization reaction, the mixture was kept under stir-ring at 37 °C for 2 h, then it was suspended and the mixture was leftstanding overnight at room temperature to allow the deposition of theinorganic phase. This latter was isolated by centrifugation of the reac-tion mixture, repeatedly washed with water and dried at 80 °C. Thedried powders were manually milled and sieved at 100 μm.
2.3. Characterization of apatite nanocrystals
The X-ray diffraction (XRD) patterns of the samples were recordedwith a D8 Advance Diffractometer (Bruker, Karlsruhe, Germany)equipped with a Lynx-eye position sensitive detector using Cu Kαradiation (λ = 1.54178 Å) generated at 40 kV and 40 mA. XRD spectrawere recorded in the 2θ range from 10 to 60° with a step size (2θ) of0.02° and a counting time of 0.5 s.
Quantitative evaluation of phase compositions and cell parameterswas performed by full-profile Rietveld analysis of the XRD spectrum(TOPAS v. 4.2, Bruker AXS, Karlsruhe, Germany). The crystallinitydegree of the apatitic powders was calculated according to the methodreported by Landi et al. [41].
The infrared spectra were recorded in the wavelength range from4000 to 400 cm−1 with 2 cm−1 of resolution using a Nicolet 380FT-IR spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, US).
− (wt.%)b Ca/Pa Mg/Caa Sr/Caa (Ca + Mg + Sr)/Pa
± 0.02 1.81 ± 0.02 0.06 ± 0.01 – 1.91 ± 0.02± 0.03 1.70 ± 0.02 0.05 ± 0.01 0.06 ± 0.02 1.87 ± 0.02± 0.02 1.57 ± 0.02 0.05 ± 0.01 0.13 ± 0.02 1.84 ± 0.02± 0.04 1.50 ± 0.02 0.04 ± 0.01 0.20 ± 0.02 1.87 ± 0.02

Fig. 1. Section A. XRD diffraction patterns of (a) MCAp, (b) MSCAp1, (c) MSCAp2, and (d) MSCAp3. Section B. Magnification of the spectra in the region of (002) reflections.
Table 3Crystal parameters and specific surface area (SSABET) of the as-synthesized apatites.
Samples Position (2θ°)(I002)
a = b(Å)a
c (Å)a Cell volume(Å3)a
SSABET
(m2g−1)
MCAp 25.88 9.378 (7) 6.889 (4) 524.80 (4) 91 ± 5MSCAp1 25.81 9.417 (2) 6.908 (0) 530.55 (4) 73 ± 4MSCAp2 25.73 9.411 (2) 6.927 (2) 531.34 (7) 52 ± 3MSCAp3 25.61 9.458 (3) 6.952 (7) 538.64 (8) 43 ± 2
a Theoretical values of stoichiometric hydroxyapatite used for the Rietveld refinementare a = b = 9.421(8) Å and c = 6.881(3) Å with a cell volume = 529.02(0) Å3.
214 M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
Powdered sample (approximately 1 mg)wasmixedwith about 200 mgof anhydrous KBr. The mixture was pressed at 10 t pressure into 7 mmdiameter disks. A pure KBr disk was used as blank. Deconvolution bynon-linear fitting of νPO4 bands was performed using GRAMS curve-fitting software.
The specific surface area (SSABET) of the powders was measured bythe Brunauer–Emmett–Teller (BET) gas adsorption method (Sorpty1750, Carlo Erba, Milan, Italy).
The quantification of Ca, Mg, Sr and P was carried out by ICP-OESspectrometers (Liberty 200, Varian, US). Samples were dissolved in1 wt.% ultrapure nitric acid. The analytical emission wavelengthswere: Ca 422.673 nm, Mg 280.270 nm, Sr 421.552 nm and P213.618 nm.
Field emission gun scanning electronmicroscopy (FEG-SEM) (SigmaNTS Gmbh, Carl Zeiss, Oberkochen, Germany) was used to evaluate themorphology and the dimension of the apatites. The powders weresuspended in ethanol, sonicated for 10 min and some drops drippedon aluminum stubs.
The carbonate content was evaluated on dried samples by thermo-gravimetric analysis (TGA) investigations using a Stanton STA 1500(Stanton, London, UK) apparatus. About 10 mg of apatite was weighedin a platinum crucible and heated from room temperature to 1200 °Cunder nitrogen flow. The heating rate was 10 °C/min and alumina wasused as reference standard.
2.4. Ion release
50 mg of apatite powders was added to 10 ml of pH = 7.4 buffersolution (Ca and Mg free Hank's Balanced Salt solution). After 15 s oftreatment in a vortex apparatus, the apatite suspensionwasmaintainedunder shaking at 37 °C. At scheduled times (up to 4 days) the superna-tant (that was well separated from the solid phase by centrifugation)was removed for the Ca, Mg and Sr quantification by ICP-OES.
2.5. In vitro bioactivity
Apatite pellets were prepared by compacting 300 mg of powder atroom temperature by uniaxial pressing at 30 MPa, followed by coldisostatic pressing at 300 MPa. Each apatite pellet was immersed in2 ml of fetal bovine serum (FBS) (PAA Laboratories GmbH) at 37 °C.At scheduled times (up to 10 days) the supernatant was collected forthe Ca, Mg and Sr quantification by ICP-OES, while the pellets weredried at 40 °C overnight for further characterization. Sodium azide(5 mg/100 ml) was added to FBS as biocide before pellet immersion.Morphology of apatite pellet surface before and after immersion inFBS was analyzed by a scanning electron microscopy (SEM, FEI Quanta200, Eindhoven, The Netherlands) and the element composition of the
surface was evaluated by Energy-Dispersive X-ray Spectroscopy(EDX). The specimens were mounted on aluminum stubs using carbontape and covered with a coating of Au using a coating unit (PolaronSputter Coater E5100, Polaron Equipment, Watford, Hertfordshire,UK). An accelerating voltage of 15 kV and a working distance of10 mm were used for the EDX measurements.
2.6. Statistical analysis
Results are expressed as the mean ± standard deviation (S.D.) of 6replicates.
3. Results
3.1. Apatite characterization
The amount of the chemical elements of the as-synthesized ionicsubstituted apatites, calculated by ICP-OES and TGA is reported inTable 2. The CO3 contentwas evaluated according to theweight loss ob-served between 550 and 950 °C [42]. The Ca/P molar ratio of MCApwashigher than the theoretical one, due to the CO3 substitution in the B-site(see FT-IR characterization). The amount of Sr inside the structure of theapatites increased from 0 to about 12 wt.% as function of the quantity ofSr(NO3)2 present in the basic solution during the syntheses. Previousworks reported that apatite could be doped with Sr in a great extentup to a complete substitution of Ca ions [40,43]. The amount of Mg de-creased from 1.20 to 0.71 wt.% as a function of the quantity of Sr, be-cause of the greater ability of Sr to enter in the structure that reducedthe incorporation of Mg. The CO3 substitution was higher when the12 wt.% of Sr was inserted in the lattice. This fact is a consequence ofthe larger structural strain caused by the partial substitution of Sr forCa, which allows to accommodatemore CO3 ions into the structure [44].It is worth to notice that the amounts ofMg and CO3 incorporated in theapatites (0.7–1.2 wt.% and 2.7–3.6 wt.%, respectively) were in the sameorder of magnitude of those present in biogenic apatites [4]. The com-parison between the Sr/Ca and Mg/Ca molar ratios of the synthesized
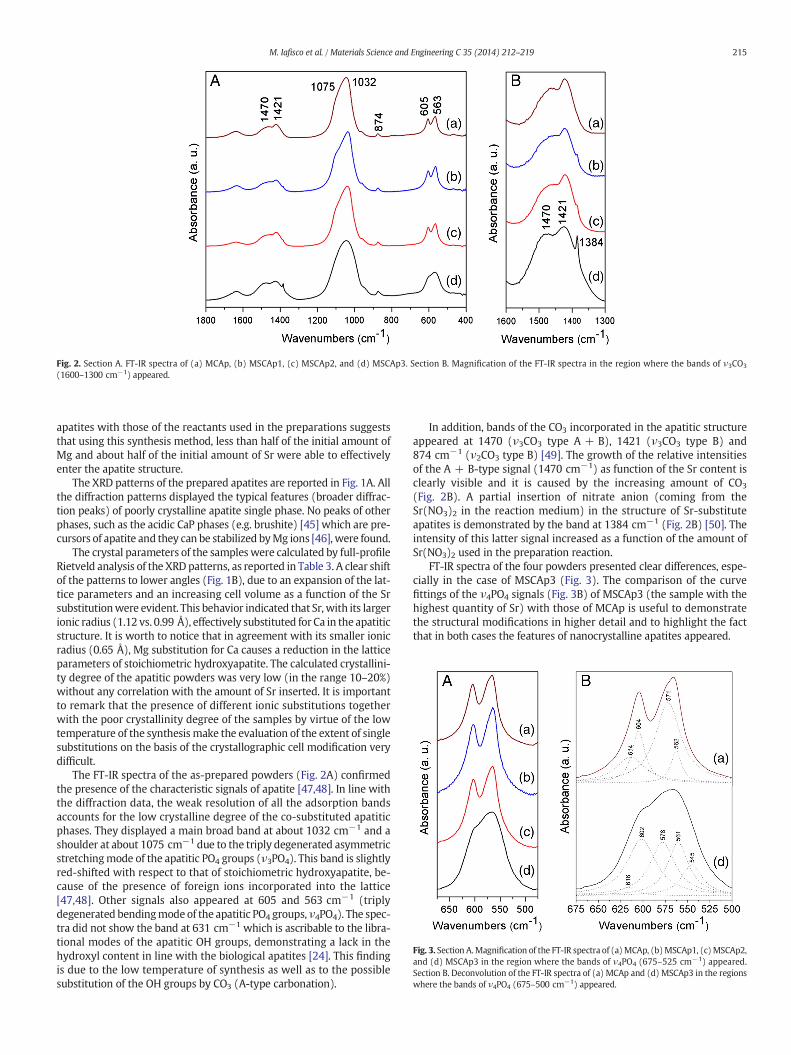
Fig. 2. Section A. FT-IR spectra of (a) MCAp, (b) MSCAp1, (c) MSCAp2, and (d) MSCAp3. Section B. Magnification of the FT-IR spectra in the region where the bands of ν3CO3
(1600–1300 cm−1) appeared.
Fig. 3. Section A.Magnification of the FT-IR spectra of (a)MCAp, (b)MSCAp1, (c)MSCAp2,and (d) MSCAp3 in the region where the bands of ν4PO4 (675–525 cm−1) appeared.Section B. Deconvolution of the FT-IR spectra of (a) MCAp and (d) MSCAp3 in the regionswhere the bands of ν4PO4 (675–500 cm−1) appeared.
215M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
apatites with those of the reactants used in the preparations suggeststhat using this synthesis method, less than half of the initial amount ofMg and about half of the initial amount of Sr were able to effectivelyenter the apatite structure.
The XRD patterns of the prepared apatites are reported in Fig. 1A. Allthe diffraction patterns displayed the typical features (broader diffrac-tion peaks) of poorly crystalline apatite single phase. No peaks of otherphases, such as the acidic CaP phases (e.g. brushite) [45] which are pre-cursors of apatite and they can be stabilized byMg ions [46], were found.
The crystal parameters of the sampleswere calculated by full-profileRietveld analysis of the XRD patterns, as reported in Table 3. A clear shiftof the patterns to lower angles (Fig. 1B), due to an expansion of the lat-tice parameters and an increasing cell volume as a function of the Srsubstitutionwere evident. This behavior indicated that Sr,with its largerionic radius (1.12 vs. 0.99 Å), effectively substituted for Ca in the apatiticstructure. It is worth to notice that in agreement with its smaller ionicradius (0.65 Å), Mg substitution for Ca causes a reduction in the latticeparameters of stoichiometric hydroxyapatite. The calculated crystallini-ty degree of the apatitic powders was very low (in the range 10–20%)without any correlation with the amount of Sr inserted. It is importantto remark that the presence of different ionic substitutions togetherwith the poor crystallinity degree of the samples by virtue of the lowtemperature of the synthesis make the evaluation of the extent of singlesubstitutions on the basis of the crystallographic cell modification verydifficult.
The FT-IR spectra of the as-prepared powders (Fig. 2A) confirmedthe presence of the characteristic signals of apatite [47,48]. In line withthe diffraction data, the weak resolution of all the adsorption bandsaccounts for the low crystalline degree of the co-substituted apatiticphases. They displayed a main broad band at about 1032 cm−1 and ashoulder at about 1075 cm−1 due to the triply degenerated asymmetricstretchingmode of the apatitic PO4 groups (ν3PO4). This band is slightlyred-shifted with respect to that of stoichiometric hydroxyapatite, be-cause of the presence of foreign ions incorporated into the lattice[47,48]. Other signals also appeared at 605 and 563 cm−1 (triplydegenerated bendingmode of the apatitic PO4 groups, ν4PO4). The spec-tra did not show the band at 631 cm−1 which is ascribable to the libra-tional modes of the apatitic OH groups, demonstrating a lack in thehydroxyl content in line with the biological apatites [24]. This findingis due to the low temperature of synthesis as well as to the possiblesubstitution of the OH groups by CO3 (A-type carbonation).
In addition, bands of the CO3 incorporated in the apatitic structureappeared at 1470 (ν3CO3 type A + B), 1421 (ν3CO3 type B) and874 cm−1 (ν2CO3 type B) [49]. The growth of the relative intensitiesof the A + B-type signal (1470 cm−1) as function of the Sr content isclearly visible and it is caused by the increasing amount of CO3
(Fig. 2B). A partial insertion of nitrate anion (coming from theSr(NO3)2 in the reaction medium) in the structure of Sr-substituteapatites is demonstrated by the band at 1384 cm−1 (Fig. 2B) [50]. Theintensity of this latter signal increased as a function of the amount ofSr(NO3)2 used in the preparation reaction.
FT-IR spectra of the four powders presented clear differences, espe-cially in the case of MSCAp3 (Fig. 3). The comparison of the curvefittings of the ν4PO4 signals (Fig. 3B) of MSCAp3 (the sample with thehighest quantity of Sr) with those of MCAp is useful to demonstratethe structural modifications in higher detail and to highlight the factthat in both cases the features of nanocrystalline apatites appeared.

216 M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
In most nanocrystalline apatites and especially in the biological onesadditional bands are observed which do not appear in well-crystallizedhydroxyapatite and which have been designated as “non-apatitic envi-ronments” of the mineral ions [4]. These “non-apatitic” phosphateenvironments have been shown to emerge more clearly in the ν4PO4
domain [51]. Bands at about 603, 575 and 562 cm−1 (ν4PO4) and at616 cm−1 (non-apatitic PO4) are visible in both spectra, whereas onlyin the MSCAp3 the band at 545 cm−1 (apatitic HPO4) appeared. Thislatter signal is present only in the sample with the highest substitutionof Sr (data not shown). This finding is in agreementwith theworks of Liet al. [36] and Kikuchi et al. [52] confirming that a high quantity of Srsubstituted for Ca induces the accommodation of HPO4 groups in thecrystalline structure. Taking into account these characterizations wecan consider that the incorporation of 12 wt.% of Sr had a dramaticallydestabilizing effect on the apatite structure thus altering the symmetryand yielding a distortion of the tetrahedral PO4.
In Fig. 4 the FEG-SEM images of the apatitic powders are depicted.They appeared as aggregates of plate-shaped nanocrystals withsizes of about 40–50 nm. The morphology and the dimension of thenanocrystals were not influenced by the amount of Sr, whereas the ten-dency to form aggregates upon freeze-drying seemed to increase as afunction of the quantity of Sr. This trend can explain the decrease oftheir specific surface area as a function of the Sr content (Table 3).
3.2. Ion release
In Fig. 5 the releasedmass percentages of Ca,Mg and Sr from thepre-pared samples at pH 7.4 as a function of time are reported. The extent ofCa release can be considered as an index of the apatite dissolution.Because of the destabilizing effect of the foreign ions in the crystallattice, the apatite doped with the highest amount of Sr had a fastest
Fig. 4. FEG-SEM images of (A) MCAp, (B) M
degradation kinetic in physiological conditions (Fig. 5A). In this way, itwas confirmed that the Sr doping accelerates the apatite degradationrate and that this ion can be released during the dissolution matrixthus being able to execute its therapeutic function. In the same way ofCa, the Sr released from the apatites increased as a function of Sr content(Fig. 5C), while this phenomenon did not occur in the case ofMg release(Fig. 5B). Indeed, the amount ofMg released fromMCAp (Sr 0 wt.%) andMCAp (Sr 12 wt.%) gave the same result; hence, intermediate amountsof Sr doping ionswere found to have a stabilizing effect onMg inside theapatite. It can be supposed that the bigger size of Sr compensates for thesmaller size of Mg as compared to Ca, therefore stabilizing Mg in theapatite lattice. This effect occurred only when the amount of Sr is lessthan 12 wt.%, confirming that the latter value can be viewed as a thresh-old for the structural stability of apatite.
3.3. In vitro bioactivity
In vitro bioactivity is usually evaluated by examining the ability ofbiomaterials to induce the crystallization of new CaP phase on itssurface when immersed in simulated body fluid (SBF) [53]. In thiswork fetal bovine serum (FBS) (pH 7.4) was used because it is morecomparable to the in vivo situation [54]. The surface Ca/P ratios of theapatite pellets before and after immersion at different times in FBS,quantified by EDX are reported in Table 4. Before the immersion inFBS, the surface Ca/P ratio of the pellets decreased as a function of theSr content in line with the bulk Ca/P trend evaluated on the powdersby ICP-OES analyses. The Ca/P ratio of MCAp, MSCAp1 and MSCAp2,after 4 and 7 days of immersion in FBS, increased to values close tothat of stoichiometric hydroxyapatite, indicating the possible depositionof CaP phase on the surface of the pellets. Whereas, after 10 days theCa/P ratio slightly decreased probably because of the initial dissolution
SCAp1, (C) MSCAp2 and (D) MSCAp3.

Fig. 5. Released mass percentages of (A) Ca, (B) Mg and (C) Sr ions from the synthesizedapatite at pH 7.4 as a function of time.
Table 4Surface Ca/P ratio (quantified by EDX) of apatite pellets before and after immersion in FBSat different times.
Before immersion in FBS After immersion in FBS
4 days 7 days 10 days
MCAp 1.61 ± 0.08 1.69 ± 0.08 1.64 ± 0.08 1.52 ± 0.06MSCAp1 1.51 ± 0.09 1.68 ± 0.07 1.67 ± 0.08 1.55 ± 0.06MSCAp2 1.47 ± 0.07 1.68 ± 0.09 1.65 ± 0.07 1.57 ± 0.06MSCAp3 1.24 ± 0.07 1.08 ± 0.05 1.07 ± 0.06 0.72 ± 0.03
Fig. 6. Ca amount of FBS solutions after the contact with apatite pellets at different times.The initial Ca concentration of FBS was 21 ppm.
217M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
of the pellets as well as of the new formed CaP phases. On the contrary,the Ca/P ratio of MSCAp3 pellets after contact with FBS decreased withtime because of the highest and fastest degradation of this kind ofco-substituted apatite at physiological pH in comparison with theother apatites synthesized in this work.
In Fig. 6 the Ca concentrations of FBS solution after contact with theapatitic pellets at different times are reported. The initial Ca amount ofFBS was calculated to be about 21 ppm. After the contact with theMCAp,MSCAp1 andMSCAp2 pellets for 4, 7 and 10 days, the Ca concen-tration of the FBS solution decreased with respect to the initial one, inagreement with the crystallization of new CaP phase on the pellets
surface. The Ca concentration of FBS slightly decreased as a function ofcontact time and in particular appreciable differences can be viewedcomparing the amount of Ca after 4 days and 10 days. It is also impor-tant to underline that no considerable variationswere found comparingthe FBS solutions after the contact with MCAp, MSCAp1 and MSCAp2pellets. In contrast, the Ca amount of FBS after contact with MSCAp3pellets dramatically increased in consequence of the apatite dissolution.
The SEM images of the pellets surface before and after immersion inFBSwere acquired to evaluate themorphology of the CaP phase formedon their surface. No differences have been found comparing the surfaceof the MCAp, MSCAp1 and MSCAp2 pellets in terms of roughness, mor-phology and amount of the newCaPphase precipitated. Some represen-tative images of theMCAp1 pellets surface before and after contact withFBS are reported in Fig. 7. Before the immersion, the pellet surfaces ap-peared smooth (Fig. 7A), whereas after 4 days of immersion (Fig. 7B)they appeared rougher and globular-like structures started to form.After 10 days (Fig. 7C) the roughness of the surfaces increased and thesize of globular deposits increased. The globular-like morphology ofthe CaP phase formed on the pellet surface (Fig. 7D) closely resembledthat shown in similar works [55,56].
The SEM images of the surface of the MSCAp3 pellets before andafter immersion in FBS are reported in Fig. 8. Before the immersion inFBS the pellet surface appeared smooth (Fig. 8A) similar to that ofother apatites synthesized in this work. After 10 days of immersionsome globular-like deposits appeared (Fig. 8B), indicating that also inthis case the formation of new apatite took place.
Two possible mechanisms were proposed to explain the formationof new CaP on the surface of apatite-based materials in SBF. The firstone is based on the surface charge [57]. According to this mechanism,the negatively charged surface sites of apatite (PO4 and OH groups) in-teract with the positively charged Ca ions of SBF forming Ca-rich amor-phous calcium phosphates (ACPs) on the surface. These Ca-rich ACPsthen interact with the negatively charged PO4 ions of SBF to formCa-deficient ACP, which can crystallize into bone-like apatite deposits

Fig. 7. SEM images of MSCAp1 pellets surface (A) before and after immersion in FBS for (B) 4 and (C) 10 days. (D) Magnification of the globular-like deposits.
218 M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
on apatite surface. The second mechanism is related to the dissolutionproperties of apatite [58]. The dissolution of apatite at physiologicalconditions releases Ca and PO4 ions into the surrounding fluid raisingits supersaturation and resulting in the precipitation of CaP on itssurface. Consequently, the ionic substitution has an important role onthe in vitro bioactivity of apatite [15,59,60] because they influence thesurface charge, the dissolution kinetic and the chemical species releasedduring the degradation.
The amounts of Ca ions in the FBS solutions after contact with theMCAp, MSCAp1 and MSCAp2 pellets were lower than the initial oneindicating that the new CaP phase on these materials was formed
Fig. 8. SEM images of MSCAp3 pellets surface (A) be
according to the first mechanism. Nevertheless, the slight reduction ofCa/P ratio of the pellets after 10 days of immersion can suggest thatthe phenomena of dissolution and re-precipitation can occur at longertimes. On the contrary, the higher amounts of Ca in the FBS solutionsafter contact with the MCAp3 pellets implied that in this case the newCaP was formed through the second mechanism. The lower amount ofglobular deposits on the MCAp3 pellets surfaces with respect to theother synthesized apatites can be due not only to the different forma-tion mechanism but also by virtue of the higher release of Sr and Mgions (data not shown) which reduced the extent of CaP precipitationin physiological conditions [61,62].
fore and (B) after immersion in FSB for 10 days.

219M. Iafisco et al. / Materials Science and Engineering C 35 (2014) 212–219
4. Conclusions
Ionic substitution could be a powerful tool to increase the biomedi-cal performances of apatite as well as to improve its chemical similaritywith the biological one. Basing on this approach, in this work thepreparation of nanocrystalline apatites co-substituted with Mg, CO3
and Sr has been evaluated. To the best of our knowledge this workrepresents the first attempt to prepare biomimetic nano-sized apatitesco-substituted with these ions as building-block materials to developbio-devices with anti-osteoporotic effect.
In this respect, it was found that the incorporation of 12 wt.% of Srcan be viewed as a threshold for the structural stability of Mg–CO3-apatite, thus resulting in a pronounced alteration of the crystal symme-try and in an increased degradation kinetic, with the highest amount ofSr ions released in physiological conditions. In consequence, the pelletsmade of apatite doped with the Sr which amounts lower than 12 wt.%have a good tendency to form a newCaPphase on their surface probablydue to the interactions of their surface negative groups with the posi-tively charged Ca ions of FBS. On the other hand, due to high dissolutionrates, the surface formation of new CaP on the 12 wt.% Sr-doped apatitepellets was reduced. Finally, although occurring by different mecha-nisms and extents, all the apatites synthesized in this work exhibitedin vitro bioactivity demonstrating their potential use to develop bio-medical devices with anti-osteoporotic functionality.
Acknowledgments
The authorswould like to acknowledge the PNR-CNRAging Program2012–2014 and the Europeanprojects SMILEY (FP7-NMP-2012-SMALL-6-310637) and OPHIS (FP7-NMP-2009-SMALL-3-246373).
References
[1] L. Wang, G.H. Nancollas, Chem. Rev. 108 (2008) 4628–4669.[2] M.J. Olszta, X.G. Cheng, S.S. Jee, R. Kumar, Y.Y. Kim, M.J. Kaufman, E.P. Douglas, L.B.
Gower LB, Mater. Sci. Eng. R. Rep. 58 (2007) 77–116.[3] N. Roveri, B. Palazzo, M. Iafisco, Expert Opin. Drug Deliv. 5 (2008) 861–877.[4] J. Gómez-Morales, M. Iafisco, J.M. Delgado-López, S. Sarda, C. Drouet, Prog. Cryst.
Growth Charact. Mater. 59 (2013) 1–46.[5] S.V. Dorozhkin, Acta Biomater. 6 (2010) 715–734.[6] M. Navarro, A. Michiardi, O. Castaño, J.A. Planell, J. R. Soc. Interface 5 (2008)
1137–1158.[7] E. Champion, Acta Biomater. 9 (2013) 5855–5875.[8] J. Juhasz, S. Best, J. Mater. Sci. 47 (2012) 610–624.[9] K.E. Tanner, J. R. Soc. Interface 7 (2010) S541–S557.
[10] Y. Cai, R. Tang, J. Mater. Chem. 18 (2008) 3775–3787.[11] Z. Evis, T.J. Webster, Adv. Appl. Ceram. 110 (2011) 311–320.[12] S. Sprio, A. Tampieri, E. Landi, M. Sandri, S. Martorana, G. Celotti, G. Logroscino,
Mater. Sci. Eng. C 28 (2008) 179–187.[13] C. Drouet, M.T. Carayon, C. Combes, C. Rey, Mater. Sci. Eng. C 28 (2008) 1544–1550.[14] E. Landi, G. Logroscino, L. Proietti, A. Tampieri, M. Sandri, S. Sprio, J. Mater. Sci. Mater.
Med. 19 (2008) 239–247.[15] E. Boanini, M. Gazzano, A. Bigi, Acta Biomater. 6 (2010) 1882–1894.[16] J. Ma, Y. Wang, L. Zhou, S. Zhang, Mater. Sci. Eng. C 33 (2013) 440–445.[17] V. Aina, G. Lusvardi, B. Annaz, I. Gibson, F. Imrie, G. Malavasi, L. Menabue, G. Cerrato,
G. Martra, J. Mater. Sci. Mater. Med. 23 (2012) 2867–2879.[18] A. Büyükaksoy, N.C. Köseoglu, M.H. Aslan, A.Y. Oral, Adv. Eng. Mater. 11 (2009)
B77–B81.
[19] I.R. Gibson, W. Bonfield, J. Mater. Sci. Mater. Med. 13 (2002) 685–693.[20] N.Y. Mostafa, H.M. Hassan, O.H. Abd Elkader, J. Am. Ceram. Soc. 94 (2011)
1584–1590.[21] C. Rey, V. Renugopalakrishman, B. Collins, M. Glimcher, Calcif. Tissue Int. 49 (1991)
251–258.[22] R. Zapanta-Legeros, Nature 206 (1965) 403–404.[23] M. Iafisco, J.G. Morales, M.A. Hernandez-Hernandez, J.M. Garcia-Ruiz, N. Roveri, Adv.
Eng. Mater. 12 (2010) B218–B223.[24] C. Rey, C. Combes, C. Drouet, H. Sfihi, A. Barroug, Mater. Sci. Eng. C 27 (2007)
198–205.[25] J. Burnell, E. Teubner, A. Miller, Calcif. Tissue Int. 31 (1980) 13–19.[26] J. Chou, S.M. Valenzuela, J. Santos, D. Bishop, B. Milthorpe, D.W. Green, M. Otsuka, B.
Ben-Nissan, J. Tissue Eng. Regen. Med. (2012), http://dx.doi.org/10.1002/term.1576.[27] L. Bertinetti, C. Drouet, C. Combes, C. Rey, A. Tampieri, S. Coluccia, G. Martra,
Langmuir 25 (2009) 5647–5654.[28] S.G. Dahl, P. Allain, P.J. Marie, Y. Mauras, G. Boivin, P. Ammann, Y. Tsouderos, P.D.
Delmas, C. Christiansen, Bone 28 (2001) 446–453.[29] P.J. Marie, Curr. Opin. Pharmacol. 5 (2005) 633–636.[30] G. Boivin, A. Doublier, D. Farlay, J. Trace Elem. Med. Biol. 26 (2012) 153–156.[31] E. Bonnelye, A. Chabadel, F. Saltel, P. Jurdic, Bone 42 (2008) 129–138.[32] S. Peng, X.S. Liu, G. Zhou, Z. Li, K.D.K. Luk, X.E. Guo, W.W. Lu, J. Bone Miner. Res. 26
(2011) 1272–1282.[33] N.D. Ravi, R. Balu, T.S. Sampath Kumar, J. Am. Ceram. Soc. 95 (2012) 2700–2708.[34] E. Landi, A. Tampieri, G. Celotti, S. Sprio, M. Sandri, G. Logroscino, Acta Biomater. 3
(2007) 961–969.[35] E. Landi, S. Sprio, M. Sandri, G. Celotti, A. Tampieri, Acta Biomater. 4 (2008) 656–663.[36] Z.Y. Li, W.M. Lam, C. Yang, B. Xu, G.X. Ni, S.A. Abbah, K.M.C. Cheung, K.D.K. Luk, W.W.
Lu, Biomaterials 28 (2007) 1452–1460.[37] J. Terra, E.R. Dourado, J.-G. Eon, D.E. Ellis, G. Gonzalez, A.M. Rossi, Phys. Chem. Chem.
Phys. 11 (2009) 568–577.[38] E. Boanini, P. Torricelli, M. Fini, A. Bigi, J. Mater. Sci. Mater. Med. 22 (2011)
2079–2088.[39] Y. Cai, S. Zhang, X. Zeng, Y. Wang, M. Qian, W. Weng, Thin Solid Films 517 (2009)
5347–5351.[40] A. Bigi, E. Boanini, C. Capuccini, M. Gazzano, Inorg. Chim. Acta 360 (2007)
1009–1016.[41] E. Landi, A. Tampieri, G. Celotti, S. Sprio, J. Eur. Ceram. Soc. 20 (2000) 2377–2387.[42] M. Iafisco, E. Varoni, E. Battistella, S. Pietronave, M. Prat, N. Roveri, L. Rimondini, Int.
J. Artif. Organs 33 (2010) 765–774.[43] Y. Lin, Z. Yang, J. Cheng, L. Wang, J. Wuhan Univ. Technol. Mater. Sci. Ed. 23 (2008)
475–479.[44] T. Kanno, J.-I. Horiuchi, M. Kobayashi, Y. Motogami, T. Akazawa, J. Mater. Sci. Lett. 18
(1999) 1343–1345.[45] M. Iafisco, M. Marchetti, J. Gómez Morales, M.A. Hernández-Hernández, J.M. García
Ruiz, N. Roveri, Cryst. Growth Des. 9 (2009) 4912–4921.[46] P.N. Kumta, C. Sfeir, D.-H. Lee, D. Olton, D. Choi, Acta Biomater. 1 (2005) 65–83.[47] S.-C. Liou, S.-Y. Chen, H.-Y. Lee, J.-S. Bow, Biomaterials 25 (2004) 189–196.[48] S. Koutsopoulos, J. Biomed. Mater. Res. 62 (2002) 600–612.[49] F. Ren, Y. Ding, Y. Leng, J. Biomed. Mater. Res. A (2013), http://dx.doi.org/
10.1002/jbm.a.34720.[50] C.M. Bender, J.M. Burlitch, D. Barber, C. Pollock, Chem. Mater. 12 (2000) 1969–1976.[51] C. Rey, Biomaterials 11 (1990) 13–15.[52] M. Kikuchi, A. Yamazaki, R. Otsuka, M. Akao, H. Aoki, J. Solid State Chem. 113 (1994)
373–378.[53] T. Kokubo, H. Takadama, Biomaterials 27 (2006) 2907–2915.[54] F. Yao, J.P. LeGeros, R.Z. LeGeros, Acta Biomater. 5 (2009) 2169–2177.[55] W. Chrzanowski, W.J. Yeow, R. Rohanizadeh, F. Dehghani, RSC Adv. 2 (2012)
9214–9223.[56] Y. Wang, Z. Sam, X. Zeng, C. Kui, Q. Min, W. Weng, Mater. Sci. Eng. C 27 (2007)
244–250.[57] H.M. Kim, T. Himeno, T. Kokubo, T. Nakamura, Biomaterials 26 (2005) 4366–4373.[58] R.Z. Legeros, Clin. Mater. 14 (1993) 65–88.[59] F. Balas, J. Pérez-Pariente, M. Vallet-Regí, J. Biomed. Mater. Res. A 66 (2003)
364–375.[60] M.H. Fathi, E. Mohammadi Zahrani, J. Cryst. Growth 311 (2009) 1392–1403.[61] B. Bracci, P. Torricelli, S. Panzavolta, E. Boanini, R. Giardino, A. Bigi, J. Inorg. Biochem.
103 (2009) 1666–1674.[62] A.L. Oliveira, R.L. Reis, P. Li, J. Biomed. Mater. Res. B 83B (2007) 258–265.