beiträge zum therapeutisch wichtiger barbiturate - eth zürich
TRANSCRIPT

ETH Library
Beiträge zum Nachweistherapeutisch wichtiger Barbiturate
Doctoral Thesis
Author(s):Perlia, Xavier
Publication date:1953
Permanent link:https://doi.org/10.3929/ethz-a-000087723
Rights / license:In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection.For more information, please consult the Terms of use.

Prom. Nr. 2250
Beiträge zum Nachweis
therapeutisch wichtiger Barbiturate
Von der
Eidgenössischen Technischen
Hochschule in Zürich
zur Erlangung
der Würde eines Doktors der
Naturwissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
XAVIER PERLIA
dipl. Apotheker
von Luxemburg
Referent : Herr Prof. Dr. J. Büchi
Korreferent. Herr Prof. Dr. K. Münze!
Juris-Verlag Zürich
1953
Leer - Vide - Empty

MEINEN LIEBEN ELTERN
Leer - Vide - Empty

- 5 -
Meinem hochverehrten Lehrer
Herrn Prof. Dr. J. Büchi
unter dessen Leitung die vorliegende Arbeit am Pharmazeutischen Institut
der Eidgenössischen Technischen Hochschule in Zürich ausgeführt wurde,möchte ich an dieser Stelle für die wertvollen Anregungen und das lebhafte
Interesse, das er meiner Arbeit, sowie für das Wohlwollen, das er mir
persönlich stets entgegenbrachte, herzlich danken.
Herrn P.D.Dr.W.Epprecht verdanke ich bestens seine freundliche
Unterstützung bei den roentgenographischen Untersuchungen.
Herrn R.Schwegler, Verwalter des Pharmazeutischen Institutes
der ETH., bin ich für seine wertvollen Ratschläge sehr zu Dank verpflichtet.

Leer - Vide - Empty

- 7 -
Inhaltsübersicht
Seite
A. Einleitung 9
B. Allgemeiner Teil 11
I. Uebersicht der wichtigsten therapeutisch verwendeten
Barbiturate 11
1. Chemische Uebersicht 11
2. Pharmakologie und Toxikologie 24
3. Therapeutische Uebersicht 35
II. Physikalische und chemische Eigenschaften 40
HI. Isolierung aus Arzneizubereitungen 48
a. Isolierung durch Ausschüttelung 50
b. Isolierung durch Mikrosublimation 52
Allgemeines 52
Begriffsbestimmung 53
Verfahren und Apparaturen 55
Sublimation unter dem Mikroskop 56
Beeinflussung der Bildung und des Aussehens von
Sublimaten 58
Möglichkeiten zur Identifizierung der Mikrosublimate 62
Polymorphic 68
Gewinnung verschiedener Modifikationen 70
Identifikation der verschiedenen Modifikationen 71
IV. Nachweismöglichkeiten der Barbiturate 74
1. Gruppenreaktionen 74
A. Farbreaktionen 74
B. Fällungsreaktionen 79
2. Einzelreaktionen 80
V. Arbeitsplan 88
C. Spezieller Teil 91
I. Verhalten der Barbiturate beim Isolieren aus Arzneizuberei¬
tungen 91
1. Ausschüttelung aus wässerig-weinsaurer Lösung mit
Aether 92
2. Ausschüttelung aus einer Aetherlösung mit 2%igerNatriumkarbonatlösung 93
3. Ausschüttelung aus einer Aetherlösung mit 5%igerNatriumbikarbonatlösung 94
4. Einfluss der Dissoziation der Barbiturate 95
5. Schlussfolgerungen 98

- 8 -
Seite
n. Verhalten der Barbiturate bei der Vakuum-Mikrosublimation 99
À. Abklärung der Sublimationsbedingungen 99
B. Untersuchung der Sublimate 108
m. Ueberprüfung der Gruppenreaktionen 125
A. Farbreaktionen mit Kobalt-II-Salzen 125
B. Farbreaktionen mit Kupfer-H-Salzen 129
IV. Identifikation der einzelnen Derivate 129
V. Vorschläge für den Analysengang zur Isolierung, zum Nach¬
weis der Barbiturat-Gruppe und zur Identifizierung der ein¬
zelnen Barbiturate 143
D. Zusammenfassung 151
E. Literaturzusammenstellung"
153

- 9 -
A. EINLEITUNG
Seit den ältesten Zeiten waren die Menschen sich klar über die grosseund unschätzbare Bedeutung des Schlafes für das körperliche und geistigeWohlbefinden. Im 3. Buch Moses sagt der Herr zu denen, die seinem Gebot
folgen: "Ich will euch Frieden im Lande geben, auf dass ihr in Ruhe schla¬
fet. " Homer preist den Schlaf und nennt einen Schlaftrunk aus Theben, ver¬
mutlich Opium, und in der Ilias wird die Nacht als Mutter der Zwillings¬brüder Thanatos (Tod) und Hypnos (Schlaf) besungen, die als Götter be¬
trachtet wurden. Ebenso wurde der Schlaf in den Aeskulap-Tempeln als
Heilmittel angesehen (Tempelschlaf).
Macbeth sagt im 2. Akt von Shakespeares berühmtem gleichnamigemDrama:
"Mir war, als rief es: 'Schlaft nicht mehr, Macbeth,Mordet den Schlaf." Ihn, den unschuld'gen Schlaf;Schlaf, der des Grams verworr'n Gespinst entwirrt,Den Tod von jedem Lebenstag, das Bad
Der wunden Müh1, den Balsam kranker Seelen,Den zweiten Gang im Gastmahl der Natur,Das nährendste Gericht beim Fest des Lebens".
Bevor man die eigentlichen Schlafmittel kannte, wurde die Schlaf¬
losigkeit mit suggestiven Methoden und mit allgemein hygienischen Mass-
nahmen, sowie mit Opium und Alkohol behandelt.
Das erste richtige Schlafmittel, Chloralhydrat, wurde von Liebig im
Jahre 1832 hergestellt und von Liebreich 1869 eingeführt. Im März 1903berichteten Fischer und von Mehring über eine neue Klasse von Schlafmit¬
teln. Als die wichtigste Verbindung, die sich nicht nur rasch einbürgerte,sondern auch bis heute unvermindert zur Verwendung gelangt, stellte sich
die Diaethylbarbitursäure, das Veronal heraus, das an Intensität der Wir¬
kung alle bisher gebräuchlichen Schlafmittel übertraf.
Von dem im Arzneischatz als Sedativa, Hypnotica und Narcotica viel
gebrauchten Barbituraten und Thiobarbituraten steht in bezug auf Vergif¬tungsfälle Veronal immer noch an erster Stelle. Damit soll aber nicht ge¬
sagt sein, dass andere Barbitursäure-Derivate in dieser Richtung nicht
auch von Bedeutung sind. Viele, durch sie verursachte, Vergiftungen blie¬
ben ihrem Wesen nach nur deshalb unbekannt, weil man kein Isolierungs¬verfahren und keine Identitätsnachweise kannte, oder weil manche dieser
Substanzen im Körper rasch und leicht abgebaut werden. Die aus Blut,Harn, Leberund anderen Geweben isolierten Mengen sind darum oft so ge¬
ring, dass eine Makroanalyse kaum durchführbar ist und der Toxikologeöfters vor schwer zu lösenden Aufgaben steht. Dem Analytiker geht es in
dieser Hinsicht auch nicht besser, wenn er bei Vergiftungsfällen oder
Selbstmordversuchen anhand einer oder zweier nicht gekennzeichneterTabletten, Pulver oder Cachets, Ampullen oder weniger Kubikzentimeter
einer Lösung in einer nicht etikettierten Flasche ein sicheres und defini¬
tives Urteil über die Identität des Giftstoffes geben muss.

- 10 -
Hier kommt dann dem Toxikologen und dem Analytiker die Entwick¬
lung der mikrochemischen Methoden zugute, die in den letzten Jahren in
der allgemeinen Chemie Eingang gefunden haben. In der Toxikologie zeich¬
net sich die Mikrochemie durch einen Minimalverbrauch an Untersuchungs¬material aus und innerhalb der pharmazeutischen Chemie ist sie unter be¬
stimmten Bedingungen besonders geeignet für die Untersuchung von stark
wirkenden und kostbaren Arzneimitteln wie die Alkaloide, Antibiotica,Chemotherapeutica, Fermente, Hormone, Hypnotica, Lokalanaesthetica,Narcotica, Sedativa und Vitamine.
In unseren Untersuchungen bemühten wir uns, einen Beitrag zu leisten
hinsichtlich der Isolierung und Identifizierung kleiner Mengen der thera¬
peutisch am häufigsten gebrauchten Barbiturate. Vor allem beschäftigtenwir uns mit der Ueberprüfung der Isolierung mit Hilfe des Verfahrens von
Stas-Otto, der Reinigung durch Vakuum-Mikrosublimation nach Eder und
der Identifizierung der Mikrosublimate.

hervor.WirkungsdauerderVerkürzung
einegleichzeitigundWirkungderVerstärkungeinerufenSchwefelatom
eindurch2-StellunginSauerstoffatomesdesErsatzderundgruppe
Methyl¬einedurch1-StellunginWasserstoffatomesdesErsatzDer7.
kung.
Wir¬dieverstärktSubstituenteneineminBromvonVorhandenseinDas6.
hervor.WirkungdauerndekürzereinerufenSeitenkettenVerzweigte5.
Wirkung.grössereeinebedingenGruppengesättigteNicht4.
sein.acyclischmusszweitederabersein,alicyclischoderaromatischkann5-StellunginSubstituentenderEiner3.
haben.atomestoff
Kohlen¬2alswenigernichtdarf5-StellunginSubstituentengruppeJede2.
enthalten.
atomeKohlenstoff4-8müssen5-StellunginSubstituentenbeidenDie1.
beachten:Folgendesman
mussallgemeinenImwerden.substituiert5StellunginWasserstoffatome
beidendiemüssengelangen,zuStoffenwirkendenhypnotischzuUm
kommt.BarbitursäurederSalzenentsprechendendenzumanwodurch
ersetzen,MetalleindurchsichlässtDiesessind.bedingt2Stellunginatom
Wasserstoff¬dasdurchwelcheEigenschaften,saurebesitztFormLetztere
II
N-COW
OH.C2,„
C>5V16/\
CO-NHvH^
NH-COH/34\/
0=2CC5
CO-MTH.
(II).Enolformeinerund
(I)Ketoformeinerauftreten:Formenzweiinkannableitet,Derivatenvon
ReiheganzeeineundVeronaldassichdervonBarbitursäure,Die
UebersichtChemische1.
BARBITURATE
VERWENDETENTHERAPEUTISCHWICHTIGSTENDERUEBERSICHTI.
TEILALLGEMEINERB.
-11-

- 12 -
Synthesen
Bei der Synthese von Barbitursäure-Derivaten muss man folgendesbeachten.
1. Die Einführung der substituierten Reste Ri und R2 (gesättigte und unge¬
sättigte Alkyl-, Aryl-, Aralkyl-, alicyclische und heterocyclische Reste)muss Rücksicht nehmen auf die Substitutionsfähigkeit der Wasserstoff¬
atome der freien Barbitursäure und die Reaktionsfähigkeit der Halogen¬verbindungen der einzuführenden Reste.
H ^CO - NH R, CO - NH.
H^ CO-NH Ro^ CO-N^
R, .COOC„H,, R, .CN
^\c/2 5
^c
in iv
2. Die Barbitursäure (I) kann in den meisten Fällen nicht direkt substituiert
-werden; vielmehr müssen vorerst die disubstituierten Derivate des
Malonsäureesters (HI) oder des Cyanessigsäureesters (IV) hergestelltwerden; dies trifft hauptsächlich zu für die Einführung von gesättigtenAlkylresten.
3. Nur sehr reaktionsfähige Halogenalkyle, wie z.B. Allylbromid und Ben-
zylchlorid, lassen sich direkt mit den Na-Verbindungen der Barbitur¬
säure kondensieren (z. B. Dial und Numal).
4. Die Aethylgruppe lässt sich direkt nur in die fertige Barbitursäure ein¬
führen, wenn sie bereits mit einer Phenylgruppe substituiert ist, d. h.
wenn der zu substituierende Wasserstoff aktiviert ist (z.B. Luminal).
5. Die Arylreste lassen sich nicht mit Hilfe ihrer Halogen-Derivate ein¬
führen. So muss der Phenylrest in einer noch früheren Stufe der Syn¬these als für HI und IV eingeführt werden (z.B. Luminal).
6. Die alicyclischen Reste werden vorteilhaft bereits in den Cyanessig-säureaethylester (IV) eingeführt (z.B. Phanodorm).
7. Die heterocyclischen Reste lassen sich in die monosubstituierte Brom-
barbitursäure einführen (z. B. Eldoral).
8. N-alkylierte Barbiturate werden durch Kondensation mit Monoalkylharn-stoff gewonnen (z. B. Evipan).
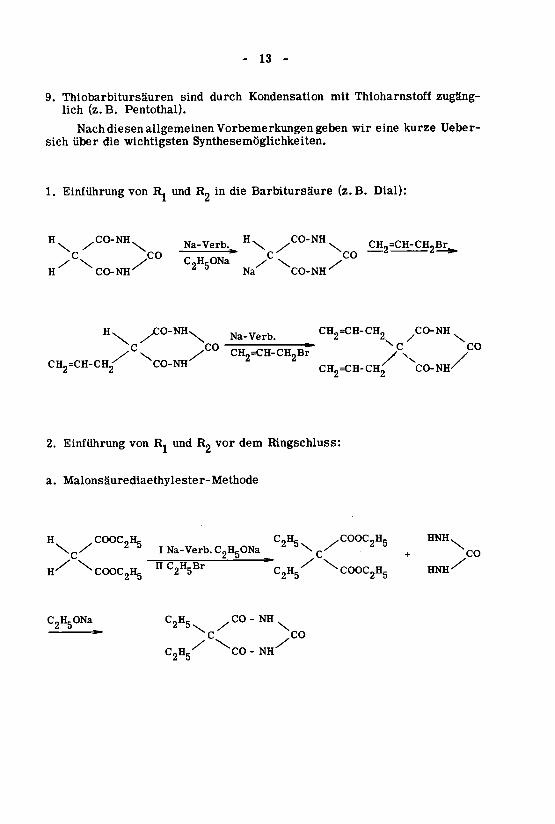
NH-COC2Hg/°°\
""
CO-NHC2HC2H5ONa
HNH^^COOC2H5C2H5nC2H5Br^CCKX^HgH-^\co+Q/5\2
Na-Verb.C2H5ONa3I52
\c/HNH.,COOC,HsC,H,-COOC,H,.H
Malonsäurediaethylester-Methodea.
Ringschluss:demvorR,undRjvonEinführung2.
CO-NH/CH2=CH-CH2CO-NHCH2=CH-CH2
P°P^CH2=CH-CH2Br*/C°//C\^O-NH^C^-CH-CH^
^
Na-Verb./X;0-NHXH\
^CO-NHNa
DZCO-NHXH
'^—2
yCO/<C^ONa"/C0/C\CH,=CH-CH?Br
.CO-NHH\jja-yert,/C0-NHH
Dial):(z.B.BarbitursäuredieinR„undR,vonEinführung1.
Synthesemöglichkeiten.wichtigstendieübersich
Ueber-kurzeeinewirgebenVorbemerkungenallgemeinendiesenNach
Pentothai).B.(z.lich
zugäng¬ThioharnstoffmitKondensationdurchsindThiobarbitursäuren9.
-13-
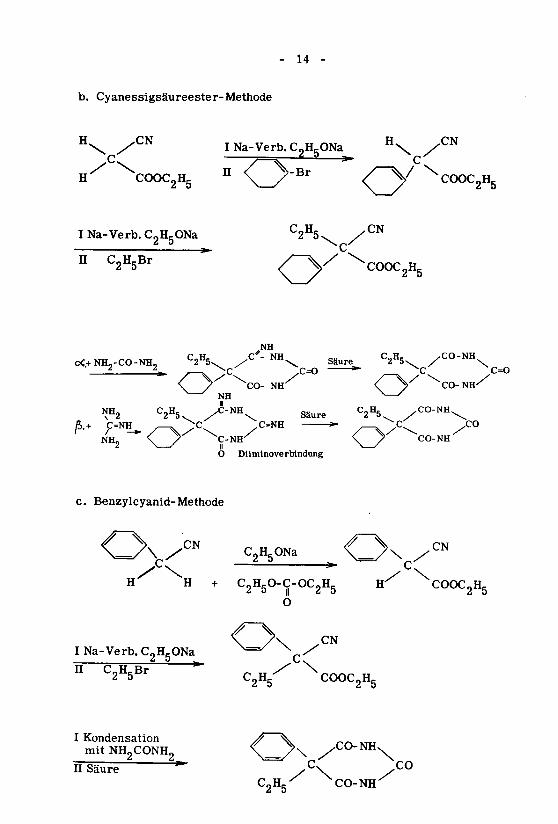
- 14 -
b. Cyanessigsäureester-Methode
INa-Verb.C2H5ONa_ H\
2 ö
C2H5Br
NH2O Diiminoverbindung
c. Benzylcyanid-Methode
C2H5ONa
NH
nh
CO-NH.C2H5^
2 5 |j 2 5 2 5
O
Na-Verb. C " ~ ^=^ x
C2H5Br C2H5 COOC2H5
I Kondensation^ XT„
mitNH9CONH9 \ >\ /CO-NH.
II Säure/ \
C2H5X CO-NH
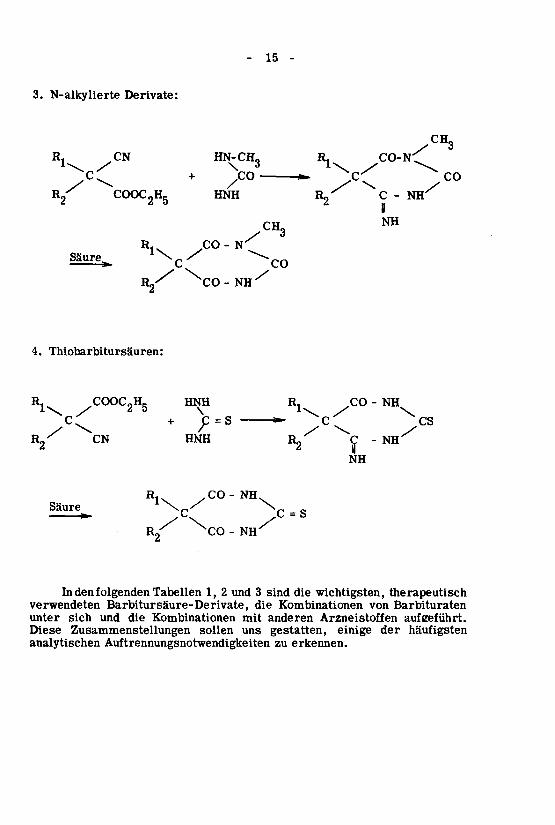
erkennen.zuAuftrennungsnotwendigkeitenanalytischenhäufigstendereinigegestatten,unssollenZusammenstellungenDiese
aufgeführt.ArzneistoffenanderenmitKombinationendieundsichunter
BarbituratenvonKombinationendieBarbitursäure-Derivate,verwendeten
therapeutischwichtigsten,diesind3und21,TabellenfolgendendenIn
NH-^COR£/C~S/CXX^-q^r^Säure
/CO-NHRl\
NH
NH-
ÇR^HNHCNR2XCSC*-C=S+C^
\y*1\\52y1\
NH-COR,HNH^COOC„HRR,
NH
Thiobarbitursäuren:4.
^NH-COHg/
CO^CXSäure>N^-COR/CH3
NH"C^HNHCOOC2H5Kf
CO<^/Co+c^
CO-N'TR,HN-CH,CNR,
3y
CH,
Derivate:N-alkylierte3.
-15-
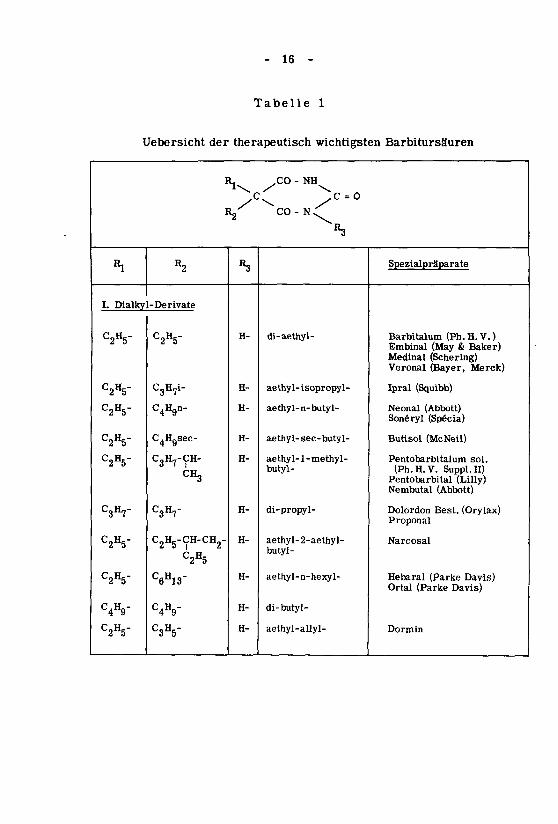
- 16 -
Tabelle 1
Uebersicht der therapeutisch wichtigsten Barbitursäuren
«1
I. Dialky
C2H5-
C2H5"
C2H5-
C2H5-
C3H7-
C2H5"
C2H5"
C4H9"
C2H5-
I
I
R2
-Derivate
C2H5-
C3H7i-
C4Hgn-
C4H9sec-
C3H7-ÇH-CH3
C3H7-
2 5
^2H52
C6H13"
C4H9-
C3H5-
ll\
l/'
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CO - NH
CO- N^«3
di-aethyl-
aethy1- îsopropyl-
aethyl-n-butyl-
aethyl- sec-butyl-
aethyl-1-methyl-butyl-
di-propyl-
aethyl-2-aethyl-butyl-
aethyl-n-hexyl-
di-butyl-
aethyl-allyl-
Spezialpràparate
Barbitalum (Ph. H. V. )Embinal (May & Baker)Medinal (Schering)Veronal (Bayer, Merck)
Ipral (Squibb)
Neonal (Abbott)Sonéryl (Spécia)
Butisol (McNeil)
Pentobarbitalum sol.
(Ph.H.V. Suppl.II)Pentobarbital (Lilly)Nembutal (Abbott)
Dolordon Best. (Orylax)Proponal
Narcosal
Hebaral (Parke Davis)Ortal (Parke Davis)
Dormin
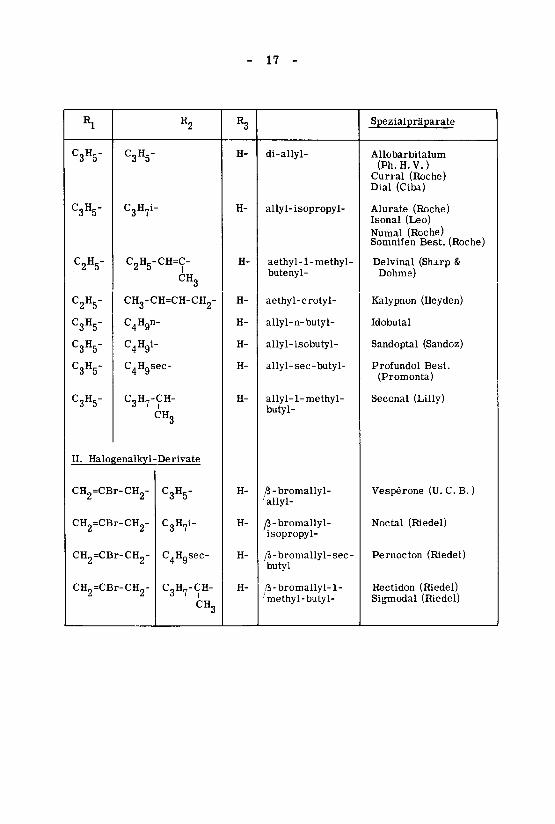
- 17 -
Rl
C3H5"
C.V
C2H5-
C2H5-
C3H5-
C3H5-
C3H5"
C3H5-
R2
C3H5"
Cglty-
C2H5-CH=C-
CH3
CH3-CH=CH-CH2-
C4Hgn-
C4Hgl-
C4Hgsec-
C3H7-CH-
CH3
II. Halogenalkyl-Derivate
CH2=CBr-CH2-
CH2=CBr-CH2-
CH2=CBr-CH2-
CH2=CBr-CH2-
C3H5-
C3H7i-
C4Hgsec-
C3H7-CH-
CH3
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
di-allyl-
allyl-isopropyl-
aethyl-1-methyl-butenyl-
aethyl-crotyl-
allyl-n-butyl-
allyl-isobutyl-
allyl-sec-butyl-
allyl-1-methyl-butyl-
^-bromallyl-'allyl-
y3-bromallyl-îsopropyl-
/î-bromallyl- sec-butyl
/5-bromallyl-l-meth>l-butyl-
Spezialpräparate
Allobarbitalum
(Ph. H. V. )Curral (Roche)Dial (Ciba)
Alurate (Roche)Isonal (Leo)
Numal (Roche)Somnifen Best. (Roche)
Delvinal (Sharp &
Dohme)
Kalypnon (Heyden)
Idobutal
Sandoptal (Sandoz)
Profundol Best.
(Promonta)
Seconal (Lilly)
Vespérone (U. C. B. )
Noctal (Riedel)
Pernocton (Riedel)
Rectidon (Riedel)
Sigmodal (Riedel)
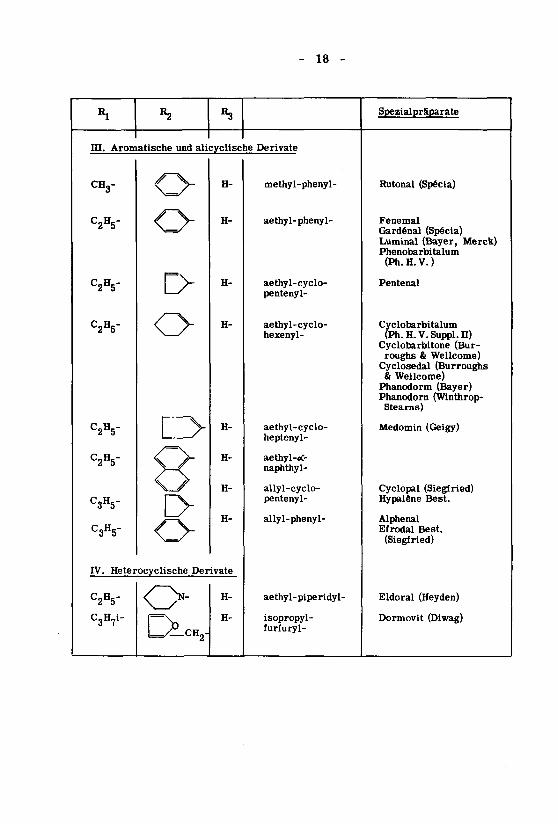
- 18 -
Spezialpräparate
m. Aromatische und aUcyclische Derivate
CHg-
C2H5"
C2H5"
C3H5-
o-
H-
H-
H-
H-
H-
H-
H-
H-
IV. HeterocycUsche Derivate
C3H7i-
O\*rin
H-
H-
methyl-phenyl-
aethyl-phenyl-
aethyl-cyclo-pentenyl-
aethyl-cyclo-hexenyl-
aethyl-cyclo-heptenyl-
aethyl-oC-naphthyl-
allyl-cyclo-pentenyl-
allyl-phenyl-
aethyl-piperidyl-
isopropyl-furiuryl-
Rutonal (Spécia)
Fenemal
Gardénal (Spécia)Luminal (Bayer, Merck)Phenobarbitalum
(Ph. H. V. )
Pentenal
Cyclobarbitalum(Ph. H. V. Suppl. n)
Cyclobarbitone (Bur¬roughs & Wellcome)Cyclosedal (Burroughs& Wellcome)Phanodorm (Bayer)Phanodorn (Winthrop-Steams)
Medomin (Geigy)
Cyclopal (Siegfried)Hypalêne Best.
AlphenalEfrodal Best.
(Siegfried)
Eldoral (Heyden)
Dormovit (Diwag)
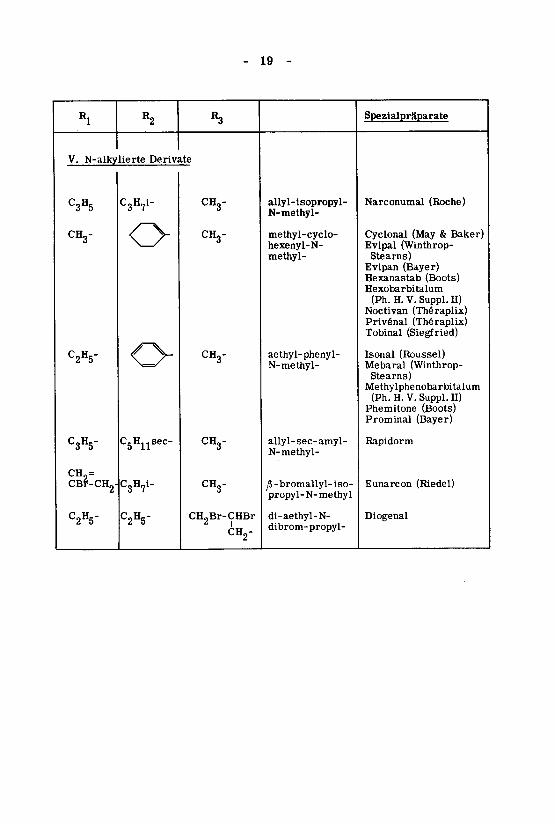
- 19 -
Rl
V. N-alky
C3H5
CH3-
C2H5-
C3H5"
CH,=CBP-CH2-
C2H5-
R2
lierte Deriv<
C3H7i-
o
o
C5Hnsec-
C3H7i-
C2H5"
«3
ite
CH,-0
CH3-
CH3-
CH3-
CH3-
CH2Br-CHBr
CH2-
allyl-isopropyl-N-methyl-
methyl-cyclo-hexenyl-N-methyl-
aethyl-phenyl-N-methyl-
allyl-sec-amyl-
N-methyl-
yS -bromallyl-iso-propyl-N-methyl
di-aethyl-N-dibrom-propyl-
Spezialpräparate
Narconumal (Roche)
Cyclonal (May & Baker)Evipal (Winthrop-Stearns)Evipan (Bayer)Hexanastab (Boots)Hexobarbitalum
(Ph. H. V. Suppl. H)Noctivan (Théraplix)Privénal (Théraplix)Tobinal (Siegfried)
Isonal (Roussel)Mebaral (Winthrop-Stearns)
Methylphenobarbitalum(Ph. H.V. Suppl. H)
Phemitone (Boots)Prominal (Bayer)
Rapidorm
Eunarcon (Riedel)
Diogenal
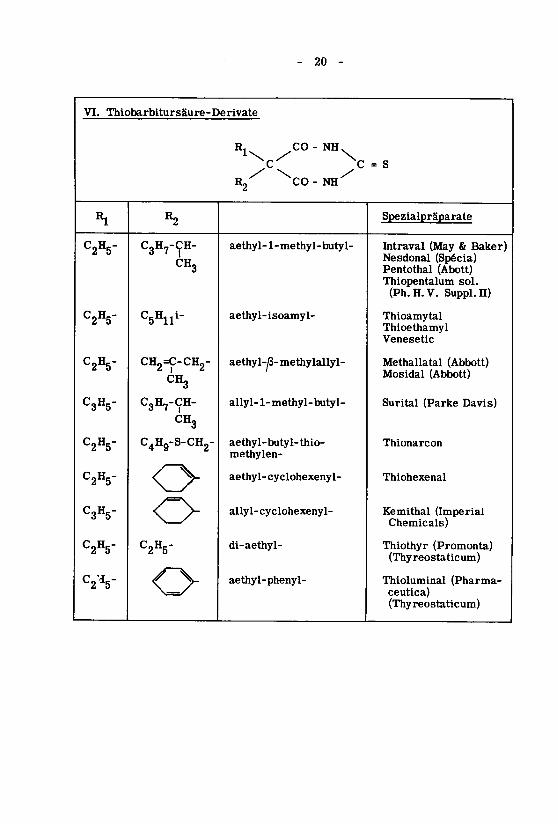
- 20 -
VI. Thiobarbitursäure-Derivate
R CO-NH
C C
R2 CO-NH
C2H5-
C2H5"
C2H5"
C3H5-
C2H5"
C2H5"
C3H5"
C2H5"
C2V
R2
C3H7-ÇH-CHg
CH2=C-CH2-
CH3
C3H7-ÇH-CHg
C^Hg-S-CHg-
Oo
C2H5-
aethyl- 1-methyl-butyl-
aethyl-isoamyl-
aethyl-^3- methylallyl-
allyl-1 - methyl-butyl-
aethyl-butyl-thio-methylen-
aethyl-cyclohexenyl-
allyl- cyclohexenyl-
di-aethyl-
aethyl-phenyl-
= S
Spezialpräparate
Intraval (May & Baker)Nesdonal (Spécia)Pentothal (Abott)Thiopentalum sol.
(Ph.H.V. Suppl.n)
ThioamytalThiœthamylVenesetic
Methallatal (Abbott)Mosidal (Abbott)
Surital (Parke Davis)
Thionarcon
Thiohexenal
Kemithal (ImperialChemicals)
Thiothyr (Promonta)(Thyreostaticum)
Thioluminal (Pharma-ceutica)(Thyreostaticum)

- 21 -
Tabelle 2
Kombinationen von Barbituraten
Di-aethyl-b.
Di-aethyl-b.
Allyl-isopropyl-b.
Allyl- isopropyl- b.
Aüyl-1-methyl-butyl-b.
Aethyl- pheny1- b.
Aethyl-phenyl-b.
Aethyl-cyclohexenyl-b.
+ Allylisopropyl-b.
+ Allylisopropyl-b. +
Br-di-aethyl-b.
+ Di-aethyl-b + Diaethyl-amin
+ Methyl-cyclohexenyl-N-methyl-b.
+ Aethyl-isoamyl-b.
+ Aethyl-1-methyl-butyl-b.
+ Allyl-cyclopentenyl-b.
+ Methyl-cyclohexenyl-N-methyl-b.
Citrophen (Zori)Isamin (Grossmann)Noctifen (Leo)Nyofen (Nyegaard)
Dorman (Paramed)
Escodorm
Somnifen (Roche)
Hypnogen (Leo)
Tuinal (Lilly)
Duotal (Morton)
Dormisan (Reiss)
Kvidorm (Winthrop-Stearns)
Tabelle 3
Kombinationen von Barbituraten mit anderen Arzneistoffen
Di-aethyl-b. + Chinin
+ Codein-diallylbarbiturat
+ Codein + Phenacetin
+ Phenacetin + Antipyrin+ Lactylphenacetin
+ Phenacetin
+ Phenyl-allyl-b. + Allyl-
isobutyl-b. + Scopolamin+ Dihydroergotamin me-thansulfonicum
+ Pyramidon
Chineonal (Merck)
Codeonal
Somnacetin (Weil)
Quadronox (Asta)
Verophen
Plexonal (Sandoz)
Barbamon (Hommels)Veramon (Schering)

- 22 -
Di-propyl-b.
Aethyl-butyl-b.
Aethyl-1-methyl-butyl-b.
Aethyl-isoamyl-b.
Di-allyl-b.
Ally1- isopropyl- b.
Allyl-sec-butyl-b.
Ally1- isobutyl- b.
/î-bromallyl-sec-butyl-b.
+ Pyramidon + Coffein
+ Phenacetin + Codein
+ Phenacetin + Pyramidon
+ Carbromal
+ Aspirin
+ Pyramidon
+ Acetaminophenolallyl-aether
+ Aethylmorphin
+ Bromdiaethylcarbamid +
Di-propyl-b.
+ Codein + Pyramidon
+ Pyramidon
+ Pyramidon + Trasentin
+ Phenacetin
+ Pyramidon
+ Bromdiaethylacetyl-carbamid
+ Pyramidon + Coffein
+ Pyramidon
+ Pyramidon + Dioxycholan-säure
Dolordon (Orylax)
Sonalgin (May & Baker)
Veralgin
Carbrital
Amytal and acetyl-salicylic acid (Lilly)
Amytal and amino-
pyrine (Lilly)
Dialacetin (Ciba)
Didial (Ciba)
Sedoben (Grossmann)
Somnocodal (Sauter)
Barbamin (Streuli)Cibalgin (Ciba)Dimallyl (Hommels)Pyrazonal (Grossmann)
Spasmocibalgin (Ciba)
Nyonal B (Nyegaard)
Allonal (Roche)Barbadon
Profundal (Promonta)
Optalidon (Sandoz)
Doralgin (Riedel)Dormalgin (Riedel)
Dodonal (Riedel)

- 23 -
Phenyl-aethyl-b.
Aethyl-cyclohexenyl-b.
Allyl-cyclopentenyl-b.
Allyl-phenyl-b.
Methyl-phenyl-N-methyl-b.
+ Bellafolin
+ Bellafolin + Gynergen
+ Brom ionogen gebunden
+oC- Bromisovaleriany1-
harnstoff
+ Coffein
+ Chinin + Extr. Crataegi
+ 3-Methyl-5, 5-phenyl-aethyl-hydantoin
+ Phenylaminopropansulfat
+ Pyramidon
+ Pyramidon + Eupaverin+ Pseudotropinbenzil-säureester
+ Theobromin
+ Trasentin
+ Pyramidon
+ Pyramidon + Bromadal
+ Pyramidon
+ Phenyltnethylamino-propanol
+ Diphenylhydantoin
+ Diphenylhydantoin +
aethyl-phenyl-b.
Belladenal (Sandoz)
Bellergal (Sandoz)
Lubrokal (Chem.Werke Albert)
Isonal
Coffeminal (Bayer)
Spasmosédine(Deglande)
Hydantal (Sandoz)
Orténal (Spécia)
Dipheba (Hausmann)Pyraminal
Eupacco (Merck)
Theominal (Bayer)
Neurotrasentin (Ciba)
Hypnon (Brettschneider)
Dormal
Hypalêne
Efrodal (Siegfried)
Comital (Bayer)
Comital L (Bayer)

- 24 -
2. Pharmakologie der Schlafmittel
Allgemein ist zu sagen, dass der für den physiologischen Schlaf gel¬tende Wertmesser: "Je tiefer der Schlaf, um so erquickender, um so gros¬ser die Erholung" (Schoen) (1), nicht für die Schlafmitteltherapie gilt.Denn dem künstlichen Schlaf fehlt wie der Narkose die aufbauende Funktion
des Normalschlafes. Der durch die Schlafmittel herbeigeführte Schlaf wird
nur dann dem natürlichen Schlaf nahekommen, wenn die Schlafmittelwirkungeben ausreicht, um den Schlaf herbeizuführen. Daher ist .eine feine Abstim¬
mung der Schlafmittel nötig und eine individuelle Dosierung, die auch die
individuelle Empfindlichkeit für Schlafmittel berücksichtigt, die je nach der
Art der Erkrankung verändert sein kann.
Da die Physiologie die letzten Ursachen des Schlafes noch nicht zu
ergründen vermocht hat, so ist eine Substitutionstherapie der Schlafstö¬
rungen, die auf physiologischem Wege der Reizausschaltung den Schlaf
herbeiführt, noch nicht möglich. Massgebend für die Eignung eines Stoffes
als Schlafmittel dürften physikalisch - chemische Eigenschaften sein. Die
nicht unangezweifelt gebliebene Lipoidtheorie von Meyer (2) und Over-
ton (3) besagt, dass nur Stoffe, die lipoidlöslich sind, Protoplasma nar¬
kotisieren können. Erfahrungsgemäss sind es bestimmte Atomgruppierungen,die den Charakter als Schlafmittel bestimmen. Ehrhardt (4) bezeichnet
sie als hypnophore Gruppe.
Für die praktische Anwendung erfolgt die Einteilung nicht nach der
chemischen Zusammensetzung, denn zwischen chemischer Konstitution und
biologischer Wirkung lassen sich nicht unbedingt gesetzmässige Beziehungenfestlegen. Ausgehend von den Erkenntnissen von v. Economo (5) über das
SchlafSteuerungszentrum und seine Beziehungen zur Grosshirnrinde haben
Moli tor und Pick (6) eine Einteilung der Schlafmittel nach ihren An¬
griffsarten, entweder an der Hirnrinde oder am Hirnstamm, versucht. Die
Hirnrindenmittel, wie z.B. Chloralhydrat, Avertin, Bromide, bewirken
Schlaf durch Ausschaltung der in der Hirnrinde liegenden schlafhemmenden
Einflüsse. An Stelle eines durch das Schlafzentrum mit seiner hemmenden
Kraft erzwungenen Schlafes narkotisieren und sperren die Rindenschlaf¬
mittel ganz unmittelbar die Hirnzentren und -bahnen, so dass die Empfin¬dungsreize ihre sonst wachhaltende oder auch weckende Kraft verlieren und
somit ein schlafähnlicher Zustand eintritt. Dabei werden wie beim natürli¬
chen Schlaf zuerst bewusst Empfindungen und Wille ausgeschaltet. Es ent¬
steht auf diese Weise künstlich direkter Hirnschlaf. Nach einer grösserenGabe werden auch die tieferen Reflexzentren ausgeschaltet: die Folge ist der
Körperschlaf.
1) Schoen, Erg.Inn. Med. 50, 43(1936)2) Meyer, Arch. Exptl. Path. Pharmakol. 42, 109 (1899)3) Over ton, "Studien über die Narkose, zugleich ein Beitrag zur allge¬
meinen Pharmakologie", Jena (1901)Vjschr. Naturforsch. Ges. Zürich 40, 1 (1895); 44, 88 (1899)
4) Ehrhardt, "Medizin und Chemie", 2, 356(1934)
5) v. Economo, Hdb. Norm. Path. Pysiol. 17, 591(1926)6) Molitor und Pick, Arch.Exptl.Path.PTTarmakol. 115, 318(1926)

- 25 -
Die Hirnstamm-Mittel, zu denen die Barbiturate gehören, verursachen
vornehmlich durch Einwirkung auf das Gebiet des mesodiencephalen Schlaf¬
zentrums den Körperschlaf und, weiter wirkend, dann auch den Hirnschlaf.Die vegetativen Reflexzentren und -bahnen sowie die Schlafzentren werden
angegriffen, indem das sympathische Wachzentrum betäubt und gegen Weck¬
reize abgestumpft wird. Auf diese Weise wird von selbst dem parasympa¬thischen SchlafZentrum das Steuer überlassen. Diese Hypnotica verursachen
also zuerst Körper- und dann unmittelbar durch das Schlafzentrum Hirn¬
schlaf. Bei ausreichender Dosierung kommt zu dieser primären Wirkungauf die vegetative Schlafsteuerung fast stets die Abschwächung und Aufhe¬
bung von Weckreizen und Reflexen, so dass dann die Wirkung der Hirn¬
stamm-Mittel der der Rindenmittel nahekommt. Eine Stütze für die Ein¬
teilung der Hypnotica in Stamm- und Rindenmittel liefert auch die Tatsache,dass in der Nähe des Sehlafsteuerungszentrums liegende Zentren (Wärme¬regulation, Blutdrucksteuerung, Erbrechen, Salz-, Wasser- und Zucker¬
stoffwechsel) nur durch Stamm-, nicht aber durch Rindenmittel angegriffenwerden.
Keeser (7) versuchte durch den chemischen Nachweis der Schlaf¬
mittel eine Lokalisation ihres Angriffes zu ermöglichen. So fand erVeronal
nur im Hirnstamm, Adalin im Gross- und Zwischenhirn. Doch auch diese
Angaben sind nicht unbestritten geblieben, ebenso ist die Richtigkeit der
Angaben von M o 1 i t o r und Pick (6) vielfach angezweifelt worden. Eine
klare Unterscheidung zwischen Stamm- und Rindenmitteln lässt sich heute
nicht mehr aufrecht erhalten. Untersuchungen von Vogt (8) haben ergeben,dass nicht die Verteilung der einzelnen Barbitursäuren auf die verschiede¬
nen Hirnpartien ihre absolute Wirksamkeit bestimmt, sondern die verschie¬
dene spezifische Empfindlichkeit der einzelnen nervösen Gewebe gegenüberein und derselben Konzentration.
Die heute für die Schlafmitteltherapie massgebliche Einteilung der
Schlafmittel ist die nach der Raschheit und Nachhaltigkeit ihrer Wirkung in
Einschlaf-, Durchschlaf- und Dauerschlafmittel. Die Wirkung eines Schlaf¬
mittels wird bestimmt durch die Konzentration im Zentralnervensystem, in
das das Schlafmittel auf dem Blutwege gelangt. Die Konzentration im Blut
ist gegeben durch die Geschwindigkeit der Aufnahme und die Geschwindig¬keit des Gegenspielers, der Entgiftung. Stets wird das Verhältnis der Re¬
sorption zur Elimination einer verabfolgten Menge entscheiden, ob Ein¬
schlafen oder Dauerschlaf erzielt wird.
Je nach der Dauer ihrer Wirkung werden die Barbiturate oft in lang,mittel, kurz und sehr kurz wirkende Stoffe eingeteilt. Im allgemeinen ent¬
spricht der Zeitraum zwischen der Verabfolgung und dem Wirkungseintrittdieser Klassifikation, d. h. bei den kurz wirkenden tritt die Wirkung rasch
ein und bei den lang wirkenden langsam.
Eine Zusammenstellung nach diesem Gesichtspunkt ist in Tabelle 4
aufgeführt.
7) Keeser, Arch. Exptl. Path. Pharmakol. ^35, 251 (1927); 186, 449 (1937)8) Vogt, Arch.Exptl. Path.Pharmakol. 1_78, 603, 608 (1935)
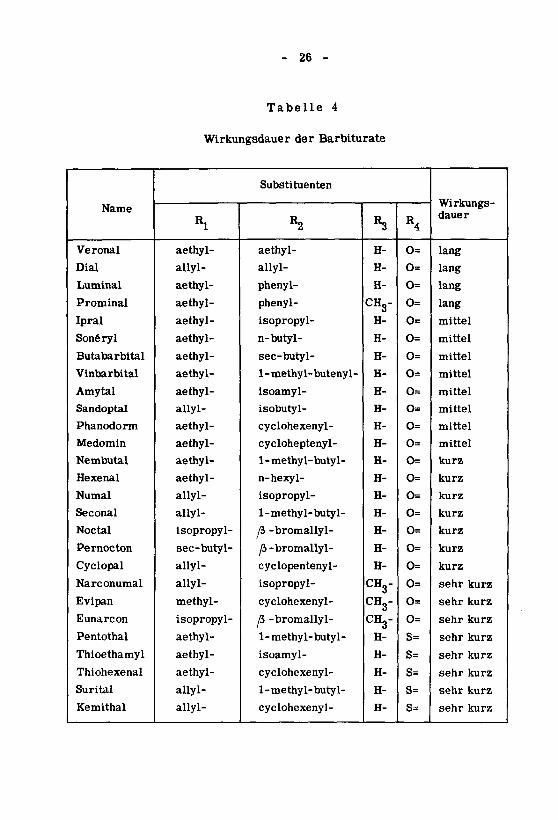
- 26 -
Tabelle 4
Wirkungsdauer der Barbiturate
Name
Veronal
Dial
Luminal
Prominal
Ipral
Sonéryl
Butabarbital
Vinbarbital
Amytal
Sandoptal
Phanodorm
Medomin
Nembutal
Hexenal
Numal
Seconal
Noctal
Pernocton
Cyclopal
Narconumal
Evipan
Eunarcon
Pentothai
Thioethamyl
Thiohexenal
Surital
Kemithal
Rl
aethyl-
allyl-
aethyl-
aethyl-
aethyl-
aethyl-
aethyl-
aethyl-
aethyl-
allyl-
aethyl-
aethyl-
aethyl-
aethyl-
allyl-
allyl-
isopropyl-
sec-butyl-
allyl-
allyl-
methyl-
isopropyl-
aethyl-
aethyl-
aethyl-
aUyl-
allyl-
Substituenten
R2
aethyl-
allyl-
phenyl-
phenyl-
isopropyl-
n-butyl-
sec-butyl-
1 -methy1-buteny1-
isoamyl-
isobutyl-
cyclohexenyl-
cycloheptenyl-
1-methyl-butyl-
n-hexyl-
isopropyl-
1-methyl-butyl-
fi -bromallyl-
jb -bromallyl-
cyclopentenyl-
isopropyl-
cyclohexenyl-
[b -bromallyl-
1-methyl-butyl-
isoamyl-
cyclohexenyl-
1-methyl-butyl-
cyclohexenyl-
H-
H-
H-
CH3-H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CH3-CH3-
CHg-H-
H-
H-
H-
H-
R4
0=
O=
o=
o=
0=
o=
o=
o=
0=
o=
o=
o=
o=
o=
o=
o=
o=
o=
o=
0=
o=
0=
s=
s=
s=
s=
s=
Wirkungs¬dauer
lang
lang
lang
lang
mittel
mittel
mittel
mittel
mittel
mittel
mittel
mittel
kurz
kurz
kurz
kurz
kurz
kurz
kurz
sehr kurz
sehr kurz
sehr kurz
sehr kurz
sehr kurz
sehr kurz
sehr kurz
sehr kurz
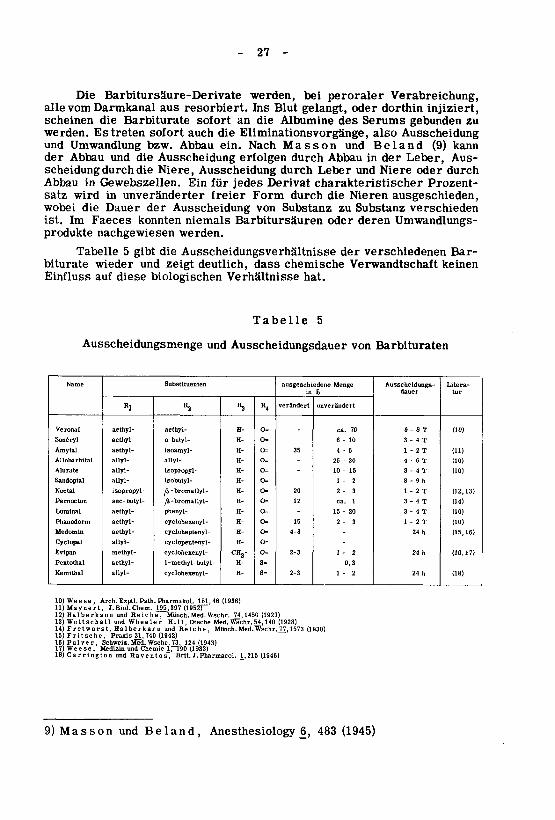
(1945)483£,Anesthesiologyland,BeundonsMas9)
(1946)J.,215J.Pharmacol.Brit.Raventos,undCarnngton18)(1933)17190ChemieundMedizinWeese,17)
124(1943)Schweiz.Med.Wschr.73,Pulver,16)(1942)74031,Praxis15)Fntsche,
(1930)^7,1573Med/Wschr.Münch.Reiche,undHalberkarnFretwurst,14)(1928)54,140Wschr.Med.DtscheHill,1-er-eWheundhallscttWo13)
(1927)74,1450Munch.Med.Wschr.Reiche,undHalberkann12)(1952T"195,397J.Biol.Chem.ll)Maynert,
181,46(1936)Pharmakol.Path.Exptl.Arch.10)Weese,
(18)h241-22-3s=H-cyclohexenyl-allyl-Kemithal
0,3s.H-1-methyl-butyl-aethyl-Pentothai
(10,17)h242-12-3o-CH3-cyclohexenyl-methyl-Evipan
-o=H-cyclopentenyl-allyl-Cyclopal
(15,16)h24-4-8o=H-cycloheptenyl-aethyl-Medomln
(10)T2-13-2150=H-cyclohexenyl-aethyl-Phanodorm
(10)T4-320-15-0-H-phenyl-aethyl-Luminal
(14)T4-31ca.120=H-/X-bromallyl-sec-tautyl-Pernocton
(12,13)T2-13-220o=H--bromallyl-ßisopropyl-Noctal
h9-82-10=H-îsobutyl-allyl-Sandoptal
(10)T4-31510--o=H-îsopropyl-allyl-Alurate
(10)T6-430-25-o=H-allyl-aUyl-Allobarbital
(11)T2-14-6350=H-isoamyl-aethyl-Amytal
T4-36-10o=H-n-butyl-aethyl-Sonéryl
(10)T86-70ca._o=H-aethyl-aethyl-Veronal
unverändertverändertR4«3*2Ri
tur
Litera¬
dauerAusscheidungs¬
*in
MengeausgeschiedeneSubstituentenName
BarbituratenvonAusscheidungsdauerundAusscheidungsmenge
5Tabelle
hat.VerhältnissebiologischendieseaufEinfluss
keinenVerwandtschaftchemischedassdeutlich,zeigtundwiederbiturate
Bar¬verschiedenenderAusscheidungsverhältnissediegibt5Tabelle
werden.nachgewiesenprodukteUmwandlungs¬derenoderBarbitursäurenniemalskonntenFaecesImist.
verschiedenSubstanzzuSubstanzvonAusscheidungderDauerdiewobei
ausgeschieden,NierendiedurchFormfreierunveränderterinwirdsatz
Prozent¬charakteristischerDerivatjedesfürEinGewebszellen.inAbbau
durchoderNiereundLeberdurchAusscheidungNiere,diedurchscheidung
Aus¬Leber,derinAbbaudurcherfolgenAusscheidungdieundAbbauder
kann(9)landBeundonssMaNachein.Abbaubzw.UmwandlungundAusscheidungalsoEliminationsvorgänge,dieauchsoforttretenEswerden.
zugebundenSerumsdesAlbuminedieansofortBarbituratediescheineninjiziert,dorthinodergelangt,BlutInsresorbiert.ausDarmkanalvomalle
Verabreichung,peroralerbeiwerden,Barbitursäure-DerivateDie
-27-
- 28 -
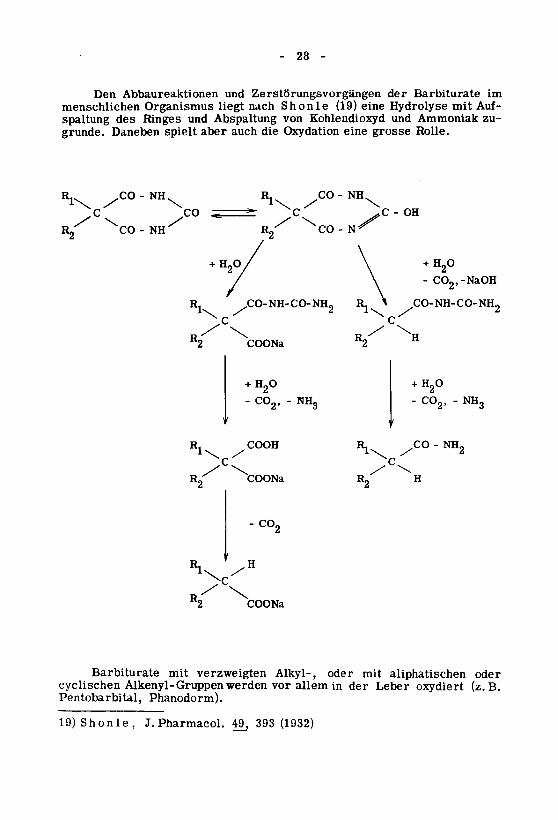
Den Abbaureaktionen und Zerstörungsvorgängen der Barbiturate im
menschlichen Organismus liegt nach Shonle (19) eine Hydrolyse mit Auf¬
spaltung des Ringes und Abspaltung von Kohlendioxyd und Ammoniak zu¬
grunde. Daneben spielt aber auch die Oxydation eine grosse Rolle.
R ^CO-NHXC CO
R2 CO - NH
- OH
- CO2,-NaOH
CO-NH-CO-NH.,
+ H2°- CO2, - NH3
H
COONa
Barbiturate mit verzweigten Alkyl-, oder mit aliphatischen oder
cyclischen Alkenyl-Gruppen werden vor allem in der Leber oxydiert (z.B.Pentobarbital, Phanodorm).
19) Shonle, J. Pharmacol. 49. 393 (1932)
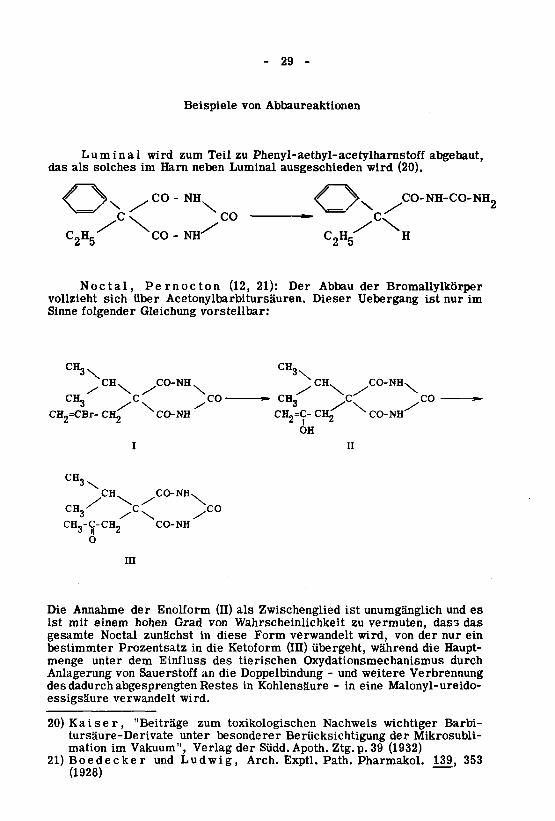
- 29 -
Beispiele von Abbaureaktionen
Luminal wird zum Teil zu Phenyl-aethyl-acetylharnstoff abgebaut,das als solches im Harn neben Luminal ausgeschieden wird (20).
/ \. CO-NH-CO-NH2
C2H5 H
Noctal, Pernocton (12, 21): Der Abbau der Bromallylkörpervollzieht sich über Acetonylbarbitursäuren. Dieser Uebergang ist nur im
Sinne folgender Gleichung vorstellbar:
CH3\ CH3\CHv CO-NH. J-CH\^
C CO CH3 C CO
CH2 CO-NH CH2=C-CHgNCO-NH
OH
CH3\CH
PH ' C3 /U\
CH3-C-CH2 CO-NH
O
III
Die Annahme der Enolform (II) als Zwischenglied ist unumgänglich und es
ist mit einem hohen Grad von Wahrscheinlichkeit zu vermuten, dass das
gesamte Noctal zunächst in diese Form verwandelt wird, von der nur ein
bestimmter Prozentsatz in die Ketoform (III) übergeht, während die Haupt¬menge unter dem Einfluss des tierischen Oxydationsmechanismus durch
Anlagerung von Sauerstoff an die Doppelbindung - und weitere Verbrennungdes dadurch abgesprengten Restes in Kohlensäure - in eine Malonyl-ureido-essigsäure verwandelt wird.
20) Kaiser, "Beiträge zum toxikologischen Nachweis wichtiger Barbi-
tursäure-Derivate unter besonderer Berücksichtigung der Mikrosubli-
mation im Vakuum", Verlag der Südd. Apoth. Ztg. p. 39 (1932)21)Boedecker und Ludwig, Arch. Exptl. Path. Pharmakol. 1J59, 353
(1928)
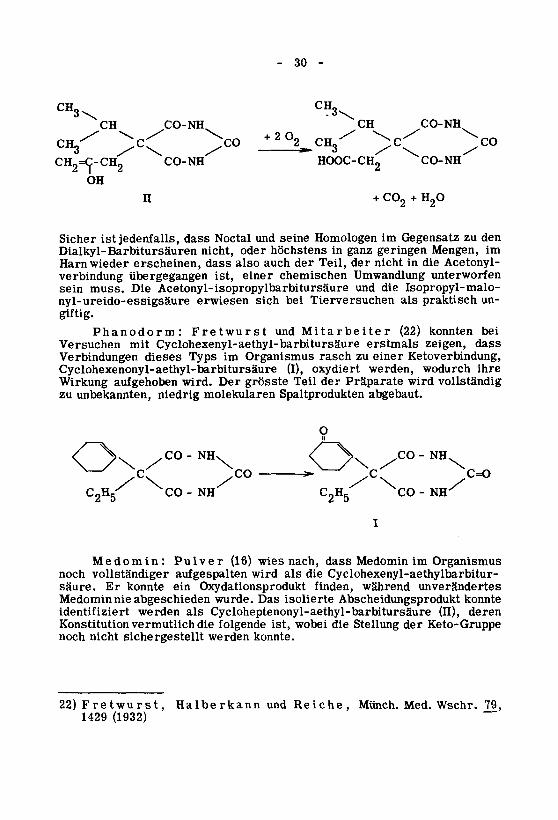
~~
(1932)142979,Wschr.Med.Münch.Reiche,undHalberkannFretwurst,22)
konnte.werdensichergestelltnichtnochKeto-GruppederStellungdiewobeiist,folgendedievermutlichKonstitution
deren(H),Cycloheptenonyl-aethyl-barbitursäurealswerdenidentifiziert
konnteAbscheidungsproduktisolierteDaswurde.abgeschiedennieMedomin
unveränderteswährendfinden,OxydationsprodukteinkonnteErsäure.
Cyclohexenyl-aethylbarbitur-diealswirdaufgespaltenvollständigernoch
OrganismusimMedomindassnach,wies(16)PulverMedomin:
O
CO-NHxC2H5CO-NHCgH^
c=oc^COc o
NH\r"0\/C°NHX
-CO,x
II
O
abgebaut.Spaltproduktenmolekularenniedrigunbekannten,zu
vollständigwirdPräparatederTeilgrössteDerwird.aufgehobenWirkung
ihrewodurchwerden,oxydiert(I),Cyclohexenonyl-aethyl-barbitursäureKetoverbindung,einerzuraschOrganismusimTypsdiesesVerbindungen
dasszeigen,erstmalsCyclohexenyl-aethyl-barbitursäuremitVersuchen
beikonnten(22)MitarbeiterundFretwurstPhanodorm:
giftig.
un¬praktischalsTierversuchenbeisicherwiesennyl-ureido-essigsäure
Isopropyl-malo-dieundAcetonyl-isopropylbarbitursäureDiemuss.sein
unterworfenUmwandlungchemischeneinerist,übergegangenverbindung
Acetonyl-dieinnichtderTeil,derauchalsodasserscheinen,wiederHarn
imMengen,geringenganzinhöchstensodernicht,Dialkyl-Barbitursäuren
denzuGegensatzimHomologenseineundNoctaldassjedenfalls,istSicher
H20+C02+
CO-NHCH2>><
CO-NHCH
HOOC-
CHg^
CH,
022+
n
OH
~"2*2"T
CO-NHCH0=C-CH0
^°°/C\CH3
CO-NHCH
CH3.
-30-
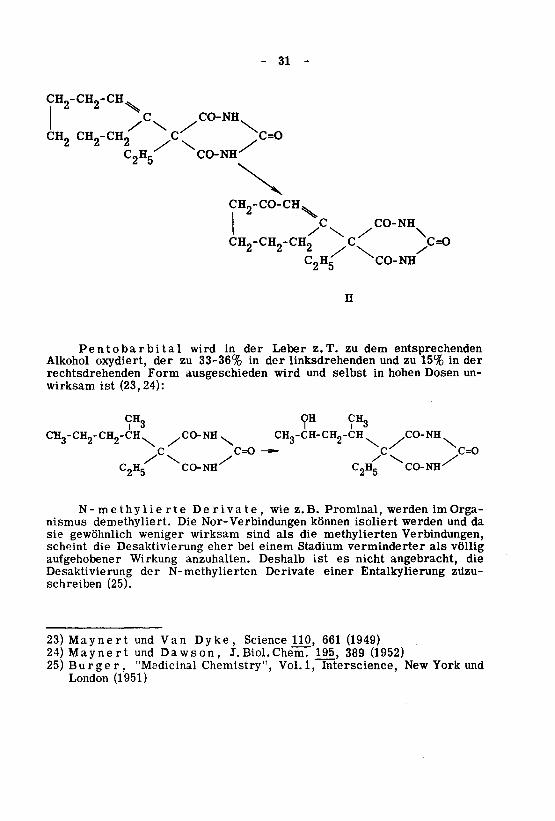
- 31 -
CH„-CH9-CH\
Cx CO-NH
CH, CH„-CH9 <T ^
CH2-CO-CHXJ CO-NH
/ \ / \CH0-CH,-CH, C C=O
* * / \ /
C2Hg XCO-NH
n
Pentobarbital wird in der Leber z.T. zu dem entsprechendenAlkohol oxydiert, der zu 33-36% in der linksdrehenden und zu 15% in der
rechtsdrehenden Form ausgeschieden wird und selbst in hohen Dosen un¬
wirksam ist (23,24):
CHg OH CHg
CH3-CH2-CH2-CH CO-NH^ CHg-CH-CHj-CH CO-NH
c c=o^ /C/ c=o
C2H5 CO-NH C2H5 CO-NH-^
N- me thy lie rte Derivate, wie z.B. Prominal, werden im Orga¬nismus demethyliert. Die Nor-Verbindungen können isoliert werden und da
sie gewöhnlich weniger wirksam sind als die methylierten Verbindungen,scheint die Desaktivierung eher bei einem Stadium verminderter als völligaufgehobener Wirkung anzuhalten. Deshalb ist es nicht angebracht, die
Desaktivierung der N-methylierten Derivate einer Entalkylierung zuzu¬
schreiben (25).
23) Maynert und Van Dyke, Science JUU), 661 (1949)24) Maynert und Dawson, J.Biol.Chem. ^95, 389 (1952)25) Burger, "Medicinal Chemistry", Vol.1, Interscience, New York und
London (1951)
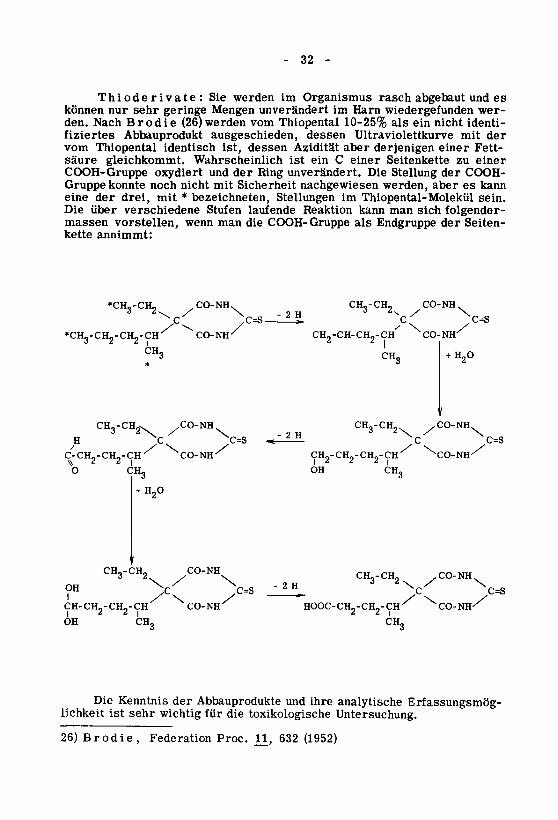
- 32 -
Thioderivate: Sie werden im Organismus rasch abgebaut und es
können nur sehr geringe Mengen unverändert im Harn wiedergefunden wer¬
den. Nach Brodie (26)werden vom Thiopental 10-25% als ein nicht identi¬
fiziertes Abbauprodukt ausgeschieden, dessen Ultraviolettkurve mit der
vom Thiopental identisch ist, dessen Azidität aber derjenigen einer Fett¬
säure gleichkommt. Wahrscheinlich ist ein C einer Seitenkette zu einer
COOH-Gruppe oxydiert und der Ring unverändert. Die Stellung der COOH-
Gruppe konnte noch nicht mit Sicherheit nachgewiesen werden, aber es kann
eine der drei, mit * bezeichneten, Stellungen im Thiopental-Molekül sein.
Die über verschiedene Stufen laufende Reaktion kann man sich folgender-massen vorstellen, wenn man die COOH-Gruppe als Endgruppe der Seiten¬
kette annimmt:
CO-NH. CH,-CH, CO-NH
*CH3-CH2-CH2-ç:iCH,
C-CH2-CH2-CH\> CH3
CO-NH
CO-NIK
- 2 H
CHO=CH-CHO-CH
CHO
CH3-CH2\
>=s
+ H2°
C=S
+ H2O
CH3-CH2^OHICH-CH„-CH„-CH
I 2 2 |OH CH,
CH2-CH2-CH2-CH
OHI
CH,
CO-NH
- 2 H
CO-NH
CH.-CH,3 2
CH„
.CO-NH\
CO-NH'
Die Kenntnis der Abbauprodukte und ihre analytische Erfassungsmög¬lichkeit ist sehr wichtig für die toxikologische Untersuchung.
26) Brodie, Federation Proc. U, 632 (1952)

- 33 -
Das zeitliche Missverhältnis zwischen Resorption und Elimination
führt zu Kumulation, also zu einer sukzessiven Anreicherung des Wirk¬
stoffes im Organismus. Die Kumulation ist im allgemeinen für Schlafmittel
eine unerwünschte und sogar gefährliche Erscheinung. Von einem brauch¬
baren Schlafmittel wird deshalb verlangt, dass seine Wirkung am Morgenbeim Erwachen des Patienten völlig abgeklungen ist. In den Fällen, in denen
man in dem Schlafmittel eine Dauerwirkung erzielen will, kann die Kumu¬
lation vorteilhaft sein, so bei der Behandlung von ErregungszuständenGeisteskranker.
Eine Frage, die den Arzt immer wieder interessiert, ist die der Ge¬
wöhnung an ein Schlafmittel. Klinische Erfahrungen und experimentelleUntersuchungen haben ergeben, dass man nicht von einer Gewöhnung schlecht¬
hin sprechen kann, sondern nur von einer Gewöhnung bezüglich eines be¬
stimmten Wirkungsmechanismus. So reagieren an Phanodorm gewöhnteKatzen völlig normal auf Evipan. Es lässt sich ausserdem zeigen, dass bei
verschiedenen Teilwirkungen eines Mittels die Gewöhnung sich nicht auf
alle gleichmässig erstreckt.
Gewöhnung
Es ist wenig bekannt, dass Schlafmittel auch als "Stimulantien" im
populären Sinn des Wortes verwendet werden können. Wenn die Schlafmittel
auch auf die meisten Menschen eine schlaferzeugende Wirkung ausüben, so
gibt es doch Leute, bei denen bestimmte Schlafmittel geistige und körper¬liche Müdigkeit zum Verschwinden bringen und die Sorgen des täglichenLebens in den Hintergrund treten lassen. Dies bewirkt ein gewisses psy¬chisches Wohlbehagen, ja manchmal eine Munterkeit, die den Schlafeintritt
verhindert. Es kann auch vorkommen, dass dieser behagliche Zustand von
Schlaf abgelöst wird; denn die peinigenden Gedanken werden verjagt und
die Sinne finden Ruhe. Diese Menschen neigen dazu, bei WiderwärtigkeitenSchlafmittel zu nehmen. Aber die Versuchung ist auch dort gross, wo das
Medikament nur Schlaf und keine gehobene Stimmung oder ein lustbetontes
Wohlbehagen (Euphorie) bewirkt; es genügt diesen Menschen, den bedrük-
kenden Gedanken zu entfliehen.
Im allgemeinen verursachen Barbitursäure-Präparate keine nennens¬
werte Gewöhnung. Es gibt jedoch Leute, welche die Dosis rasch steigern,aber die meisten Gewohnheitsverbraucher halten sich jahrelang an die
gleiche Dosis. Wenn einem Patienten das Schlafmittel plötzlich weggenom¬men wird, das er Abend für Abend einzunehmen pflegte, so kann es zu un¬
angenehmen und manchmal gefährlichen Zuständen kommen; aber diese
Abstinenzerscheinungen sind niemals so stark, wie beim chronischen Ko¬
kainismus oder Morphinismus. Diese Symptome sind auch bei Patienten
festzustellen, die Schlafmittel als "Stimulantien" verwenden. In diesen
Fällen besteht oft ein besonders starker Medizinhunger, der sich in einem
quälenden Bedürfnis nach dem gewohnten Stoff äussert, genau wie bei einem
passionierten Tabakraucher, der einen ähnlichen Hunger nach Tabak empfin¬det. Diese Abstinenzsymptome äussern sich durch Rastlosigkeit, Angst und
innere Unruhe, Schlaflosigkeit, Appetitlosigkeit und Herzklopfen, quälendeUnruhe oder Schmerzempfindung in den Armen und den Beinen, sowie ab
und zu in richtigen Bewusstseinstrübungen und Verwirrtheit, eventuell be-

- 34 -
gleitet von Halluzinationen und von Hunger nach dem Medikament, dem
Hauptsymptom dafür, dass der Patient dem Stoff wirklich verfallen ist.
Bei der Entwöhnung soll man die Dosis langsam verringern, um die
Abstinenzsymptome zu vermeiden. Das Resultat der Entwöhnung ist hier,wie bei allen Fällen von Medizinmissbrauch, abhängig von Charakter, Ge¬
sundheitszustand und äusseren Verhältnissen des Patienten, aber auch von
der Dauer der Kur (27).
Vergiftung
Hier kann man auch unterscheiden zwischen einer akuten und einer
chronischen Vergiftung.
Akute Vergiftung: Sie wird mit Veronal und anderen oft gebrauch¬ten Barbitursäuren häufig beobachtet, besonders bei Selbstmordversuchen.
Das Vergiftungsbild entspricht vollständig dem der Schlafmittelvergiftung.Die Vergiftung gleicht einer mehr oder weniger tiefen Narkose, aber sie
dauert lange wegen des langsamen Abbaues und Ausscheidung der Schlaf¬
mittel. Nach Einnahme grosser Dosen tritt rasch Bewusstlosigkeit ein,nachher kommt es zu einer reflexlosen Narkose. Nach sehr grossen Dosen
tritt der Tod infolge Atemlähmung im Laufe von wenigen Stunden ein. In
der Regel zieht sich die Vergiftung aber in die Länge. Die Bewusstlosigkeitdauert 1-2, eventuell sogar 3 Tage, während welcher sich infolge der auf¬
gehobenen Hustenreflexe und der verminderten Ventilation der Lungen(Zyanose) eine rasch verlaufende Bronchopneumonie entwickelt, die bis¬
weilen sogar die einzige Ursache des letalen Ausganges sein kann. Durch
ihre lähmende Wirkung auf die Medulla oblongata bewirken die Schlafmitteleine mehr oder weniger ausgesprochene zentral bedingte Gefässinsuffizienz,die nicht selten in das typische Bild des Schocks übergeht und in vielen Fäl¬
len zum Tode führt.
Chronische Vergiftungen können auftreten:
1. Durch ständigen Gebrauch therapeutischer Dosen.
Diese sieht man am häufigsten bei Veronal, Luminal und Dial, die bei wie¬
derholter Einnahme kumulieren. In leichteren Fällen entwickeln sich
schleichend neurasthenische Erscheinungen, u.a. auch Schlaflosigkeit, die
häufig als Verschlimmerung des Krankheitsbildes gedeutet und durch Er¬
höhung der Schlafmitteldosis behandelt werden. Bei fortgeschrittener Ver¬
giftung sieht man eine bunte Reihe verschiedener neurologischer Symptome,mitunter auch Hauterscheinungen wie Urticaria, scarlatiniformes Erythemoder Purpura, die mit Fieber einhergehen können. Sehr selten tritt eine
Dermatitis exfoliativa auf, die mitunter letal verlaufen kann.
2. Bei ständiger Einnahme grosser Dosen zur Erzielung eines Rauschzu¬
standes und einer Euphorie.Hier ist die psycnische Bereitschaft zu einer Narcoticasucht das Wesentli¬che (siehe oben).
27) Miller, "Rauschgifte und Genussmittel", Schwabe, Basel (1951)

- 35 -
Behandlung der Vergiftung
Der Magen wird durch Aspiration geleert. Eine Magenspülung darf
nur vorgenommen werden, wenn der Oberkörper des Patienten nach unten
gelagert werden kann, da sonst das Spülwasser in die Lungen gelangen kann.
Durch Verabreichung einer grossen Dosis eines zentral wirkenden Ana-
lepticums wirkt man einer Lähmung des Zentralnervensystemes entgegen.Strychnin bietet wegen seiner langdauernden Wirkung gegenüber Cardiazol
und Coramin beträchtliche Vorteile. Neben Strychnin wird auch besonders
Picrotoxin empfohlen. Neben zentralwirkenden Analeptica gibt man Sympa-tol und andere Mittel der Adrenalin-Gruppe, um eine kontrahierende Wir¬
kung auf die Gefässe auszuüben. Mit Vorteil wendet man Benzedrin an, das
sowohl eine zentralerregende als auch eine periphere gefässkontrahierendeWirkung ausübt.
Bei allen ernsteren Fällen mit Bewusstlosigkeit gibt man prophylaktischein leicht lösliches Sulfonamid oder Penicillin, um einer eventuellen Pneu¬
monie entgegenzuwirken.
Sowohl bei leichten Vergiftungen wie auch bei beginnendem Erwachen
aus der Bewusstlosigkeit sieht man häufig eine starke motorische Unruhe
(Exzitationsstadium). Selbst im tiefen Coma kann man gelegentlich tonische
Krämpfe oder eine starke Muskelstarre der Extremitäten beobachten. Dies
kann mitunter zu Irrtümern Anlass geben, indem der behandelnde Arzt an
eine Vergiftung mit einem zentralerregenden Gift denkt, daher eine Barbi-
tursäure verabreicht und so zum tödlichen Ausgang beiträgt (28).
3. Therapeutische Uebersicht der Barbiturate
Die Barbiturate zeigen zahlreiche und wichtige therapeutische Indi¬
kationen. Sie werden aber hauptsächlich wegen ihrer schlafmachenden und
sedativen Wirkung angewendet.
Hypnotica
Bei Schlafstörungen unterscheidet man zwei verschiedene Erschei¬
nungen. Bei der ersten ist das Einschlafen schwierig, aber wenn man ein¬
mal eingeschlafen ist, ist der Schlaf ruhig. Bei der zweiten ist das Ein¬
schlafen leicht, aber der Schlaf ist durch häufiges Erwachen gestört. Bei
Störungen der ersten Art ist ein kurz wirkendes Barbiturat das Mittel der
Wahl, das den Schlaf binnen einer halben Stunde herbeiführt und dessen
Wirkung auf vier bis sechs Stunden beschränkt ist. Für die zweite Art
wendet man ein mittellang wirkendes Barbiturat an, dessen Wirkung weni¬
ger schnell eintritt, dafür aber länger, sechs bis etwa acht Stunden anhält.
28) Miller, "Pharmakologie als theoretische Grundlage einer rationellen
Pharmakotherapie", Schwabe, Basel (1947)

- 36 -
Nach diesen Gesichtspunkten sind die Barbiturate auch eingeteilt wor¬den in
Einschlafmittel,Durchschlafmittel und
Oauerschlafmittel.
Diese Einteilung steht aber auch in engem Zusammenhang mit der Resor-
bierbarkeit, dem Abbau und der Ausscheidung der Barbiturate im menschli¬
chen Organismus.
Die Einschlafmittel sind die schnell resorbierbaren und eliminierba¬
ren Stoffe, die ihre Wirkung auf die kurze Zeit des Schlafeintrittes be¬
schränken und am nächsten Morgen keine Nachwirkungen zeigen. Muss die
Ruhepause aus äusseren Gründen abgebrochen werden, so soll der Patient
nach dem Erwachen frei von jeder Nachwirkung des angewandten Mittels
sein. Ein typisches Einschlafmittel ist z.B. Evipan. Es wird zum Teil be¬
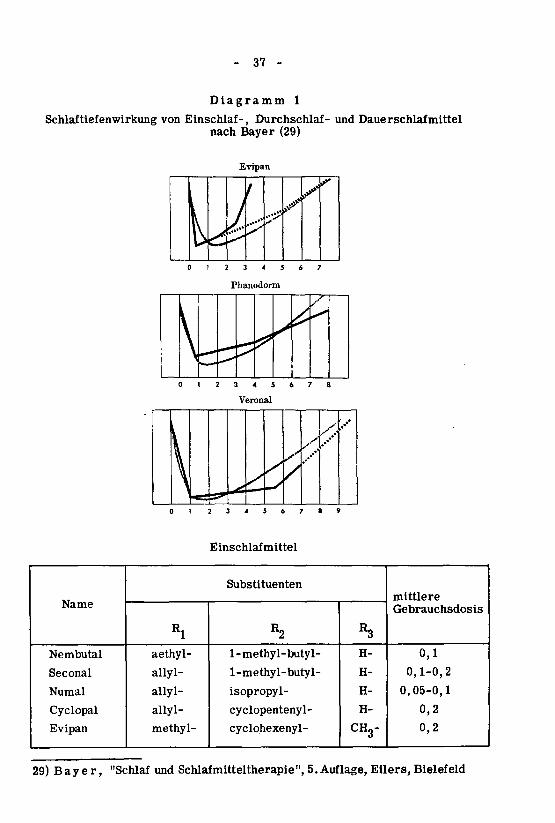
reits vom Magen resorbiert und nach den Untersuchungen von Wee se (10)über die Leber abgebaut. Das Bild der Schlaftiefenkurve (Diagramm 1)veranschaulicht deutlich seine Eignung. Die Kurve erreicht rasch ihren
tiefsten Punkt, d. h. sehr schnell tritt tiefer Schlaf ein. Im allgemeinen be¬
ginnt die Wirkung schon nach 5-10 Minuten. Sehr rasch setzt aber auch
wieder eine Abnahme der Wirkung ein, die Kurve steigt erst langsamer,nach 2-3 Stunden aber steil an. Die Wirkung klingt ab, der durch das Mittel
herbeigeführte Schlaf geht in den physiologischen Schlaf über.
Zur Behandlung von Durchschlafstörungen sind Mittel notwendig, die
im Körper lange wirken, bei denen also der Abbau langsamer vor sich gehtals bei den Einschlafmittein. Allerdings muss der Abbau doch so rasch er¬
folgen, dass der Patient nach einer ruhig und tief durchschlafenen Nacht
mit dem Gefühl der Frische und Erholung erwacht. Am Morgen dürfen sich
keine hypnotischen Nachwirkungen mehr geltend machen. Wie die Kurve
(Diagramm 1) über die Schlaftiefenwirkung zeigt, lehnt sich die Schlaf¬
tiefenkurve sehr eng an die normale Schlaftiefenkurve an. Das besagt schon,dass Phanodorm die Voraussetzungen für ein gutes Durchschlafmittel er¬
füllt.
Bei Schlafstörungen schwerer Art wird es im einzelnen nicht leicht
zu unterscheiden sein, ob es sich um eine Einschlaf- oder um eine Durch¬
schlafstörung handelt. Vielfach erstrecken sich diese schweren Schlafstö¬
rungen auf die ganze Schlafpériode. Man kann, wenn die EinschlafStörungüberwiegt, ein Einschlafmittel in erhöhter Dosis mit einem Durchschlaf¬
mittel kombinieren. Reicht eine solche Kombination nicht aus, so müssen
stärkere, sogenannte Dauerschlafmittel angewandt werden. Sie zeichnen
sich durch eine langsamere Resorption und eine sich auf einige Tage hin¬
ziehende Ausscheidung aus. Die Wirkung tritt nach ca. einer Stunde ein und
dauert sechs bis etwa zwölf Stunden. Ein Blick auf die Schlaftiefenkurve
(Diagramm 1) zeigt, dass Veronal einen besonders tiefen Schlaf von langerDauer herbeiführt. Es sind deshalb ganz bestimmte Indikationen für eine
Dauerwirkung, bei denen Veronal zu verabreichen ist.
Für die tägliche Praxis gibt somit die Differenzierung in der Wirkungder Schlafmittel dem Arzt die Möglichkeit, je nach Art und Schwere der
Schlafstörung das für den einzelnen Kranken geeignete Mittel zu verordnen.
In den Tabellen 6, 7 und 8 sind die Barbiturate entsprechend ihren Ei¬
genschaften als Einschlaf-, Durchschlaf- und Dauerschlafmittel aufgeführt.
- 37 -
Diagramm 1
Schlaftiefenwirkung von Einschlaf-, Durchschlaf- und Dauerschlafmittel
nach Bayer (29)
Evipan
A
0 2 3 4 S
Plianodorm
t 7
0 1 2 A 5 7 »
Veronal
\IJ y
yy
/
01 234567a»
Einschlafmittel
Name
Nembutal
Seconal
Numal
Cyclopal
Evipan
Substituenten
Rl
aethyl-
allyl-
allyl-
allyl-
methyl-
R2
1-methyl-butyl-
1-methyl-butyl-
isopropyl-
cyclopentenyl-
cyclohexenyl-
R3
H-
H-
H-
H-
CH3-
mittlere
Gebrauchsdosis
0,1
0,1-0,2
0,05-0,1
0,2
0,2
29) Bayer, "Schlaf und Schlafmitteltherapie", 5. Auflage, Ellers, Bielefeld
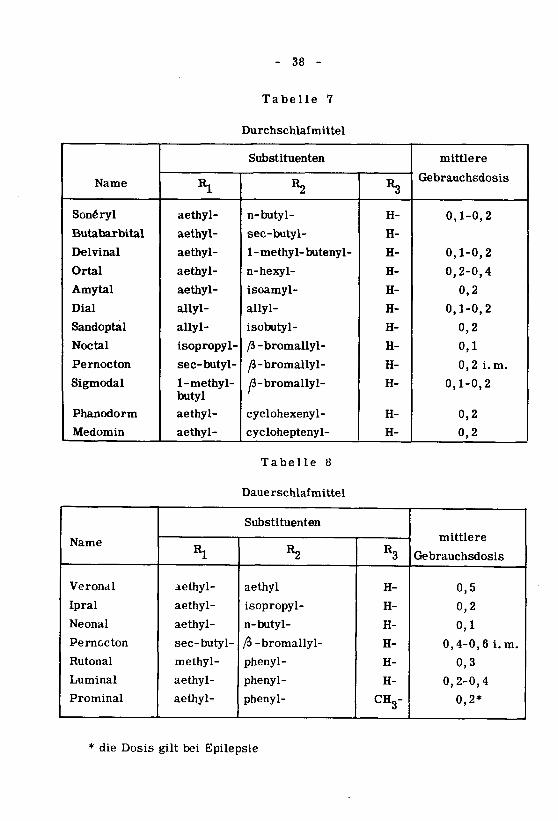
- 38 -
Tabelle 7
Durchschlafmittel
Name
Sonéryl
Butabarbital
Delvinal
Ortal
Amytal
Dial
Sandoptal
Noctal
Pernocton
Sigmodal
Phanodorm
Medomin
Substituenten
«1
aethyl-
aethyl-
aethyl-
aethyl-
aethyl-
allyl-
allyl-
isopropyl-
sec-butyl-
1-methyl-butyl
aethyl-
aethyl-
R2
n-butyl-
sec-butyl-
1 - methyl-buteny1-
n-hexyl-
isoamyl-
allyl-
isobutyl-
/3-bromallyl-
^-bromallyl-
/J-bromallyl-
cyclohexenyl-
cycloheptenyl-
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
mittlere
Gebrauchsdosis
0,1-0,2
0,1-0,2
0,2-0,4
0,2
0,1-0,2
0,2
0,1
0,2 i.m.
0,1-0,2
0,2
0,2
Tabelle 8
Daue rschlafmitte1
Name
Verondl
Ipral
Neonal
Pernocton
Rutonal
Luminal
Prominal
Rl
aethyl-
aethyl-
aethyl-
sec-butyl-
methyl-
aethyl-
aethyl-
Substituenten
R2
aethyl
isopropyl-
n-butyl-
ß -bromallyl-
phenyl-
phenyl-
phenyl-
R3
H-
H-
H-
H-
H-
H-
CH3-
mittlere
Gebrauchsdosis
0,5
0,2
0,1
0,4-0,6 i.m.
0,3
0,2-0,4
0,2*
* die Dosis gilt bei Epilepsie

- 39 -
Sedativa
Als Beruhigungsmittel sind die Barbiturate den Bromiden überlegen;denn ihr Wirkungseintritt ist viel rascher als derjenige der Bromide. Die
sedative Dosis beträgt zwischen 1/3 und 1/4 der hypnotischen Dosis. Als
Sedativa werden die Barbiturate unter anderem gebraucht bei Aufregungszu-ständen, Hyperthyroidie, essentieller Hypertension, Uebelkeit und See¬
krankheit, Chorea, Keuchhusten. Bei hyperthyroiden Patienten vermindert
die sedative Wirkung der Barbiturate die nervöse Uebererregbarkeit. Bei
akuten manischen Stadien, besonders bei Delirium tremens, sind sie von
grossem Wert und man kann sie dann auch in hypnotischen oder sogaranaesthetiscjien Dosen parenteral anwenden. Von der sedativen Wirkungmacht man auch Gebrauch bei der Behandlung von Süchtigen.
Analgetica
Die analgetische Wirkung von Salicylsäure-, Paraminophenol- und
Pyrazolon-Derivaten wird durch Barbiturate erhöht. Hierzu eignen sich
besonders die kurz wirkenden Derivate, wobei die Dosierung etwa 1/3 der
hypnotischen Dosis ausmacht. Barbiturate allein, in Dosen welche keine
Bewusstlosigkeit hervorrufen, wirken nicht analgetisch und sie können in
Gegenwart von Schmerzen keine Beruhigung und keinen Schlaf herbeiführen,sondern in solchen Fällen kann Delirium auftreten. Bei Abwesenheit von
Schmerzen hingegen potenzieren Analgetica die hypnotische Wirkung der
Barbiturate nicht.
Krampfstillende Wirkung
Die Barbitursäure-Derivate sind von grossem Wert bei der sympto¬matischen Behandlung von Krämpfen, wie sie bei Tetanus, Eklampsie,Epilepsie und Gehirnblutung auftreten, und bei Krämpfen, die durch Kokain,Picrotoxin- und Strychnin-Vergiftungen hervorgerufen werden. Hier wendet
man vor allem mittellang oder kurz wirkende Barbiturate an. Bei der Be¬
handlung von Epilepsie sind vor allem Luminal und besonders Prominal von
grosser Bedeutung.
Anaesthetica
Zu Anaesthesiezwecken werden die Barbiturate angewendet als Nar¬
cotica, rektale Anaesthetica, Praenarcotica und in der Geburtshilfe. Die
Anwendung als Narcotica beschränkt sich nur auf Operationen geringerDauer, da die Barbiturat-Narkose im Gegensatz zur Inhalationsnarkose
nicht steuerbar ist. Als Narcotica und Praenarcotica mit nachfolgenderInhalationsnarkose werden N-methylierte und Thiobarbiturate angewendet.In Tabelle 9 findet sich eine Zusammenstellung der als Narcotica und Prae¬
narcotica gebräuchlichen Barbiturate.
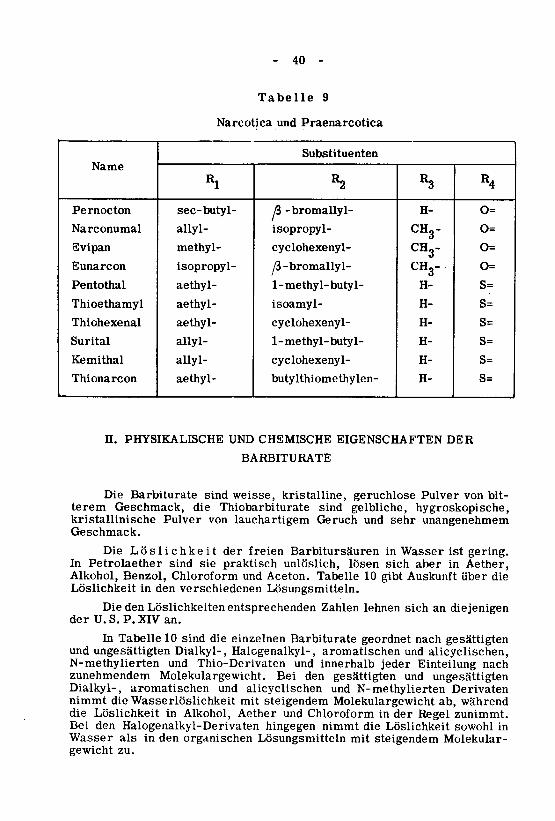
- 40 -
Tabelle 9
Narcotica und Praenarcotica
Name
Pernocton
Narconumal
Svipan
Eunarcon
Pentothai
Thioethamyl
Thiohexenal
Surital
Kemithal
Thionarcon
Substituenten
«1
sec-butyl-
allyl-
methyl-
isopropyl-
aethyl-
aethyl-
aethyl-
allyl-
allyl-
aethyl-
R2
ß -bromallyl-
isopropyl-
cyclohexenyl-
/3-bromallyl-1- methyl-butyl-
isoamyl-
cyclohexenyl-
1- methyl-butyl-
cyclohexenyl-
butylthiomethylen-
*3
H-
CH3"
CH3-CH3-H-
H-
H-
H-
H-
H-
R4
O=
O=
o=
o=
s=
s=
s=
s=
s=
s=
H. PHYSIKALISCHE UND CHEMISCHE EIGENSCHAFTEN DER
BARBITURATE
Die Barbiturate sind weisse, kristalline, geruchlose Pulver von bit¬
terem Geschmack, die Thiobarbiturate sind gelbliche, hygroskopische,kristallinische Pulver von lauchartigem Geruch und sehr unangenehmemGeschmack.
Die Löslichkeit der freien Barbitursäuren in Wasser ist gering.In Petrolaether sind sie praktisch unlöslich, lösen sich aber in Aether,Alkohol, Benzol, Chloroform und Aceton. Tabelle 10 gibt Auskunft über die
Löslichkeit in den verschiedenen Lösungsmitteln.
Die den Löslichkeiten entsprechenden Zahlen lehnen sich an diejenigender U.S. P.XIV an.
In Tabelle 10 sind die einzelnen Barbiturate geordnet nach gesättigtenund ungesättigten Dialkyl-, Halogenalkyl-, aromatischen und alicyclischen,N-methylierten und Thio-Derivaten und innerhalb jeder Einteilung nach
zunehmendem Molekulargewicht. Bei den gesättigten und ungesättigtenDialkyl-, aromatischen und alicyclischen und N-methylierten Derivaten
nimmt die Wasserlöslichkeit mit steigendem Molekulargewicht ab, während
die Löslichkeit in Alkohol, Aether und Chloroform in der Regel zunimmt.Bei den Halogenalkyl-Derivaten hingegen nimmt die Löslichkeit sowohl in
Wasser als in den organischen Lösungsmitteln mit steigendem Molekular¬
gewicht zu.
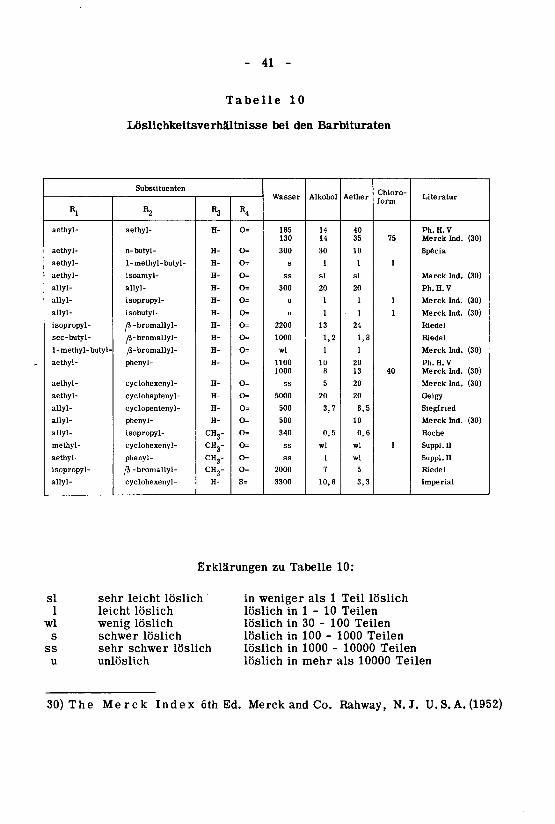
- 41 -
Tabelle 10
Loslichkeitsverhältnisse bei den Barbituraten
Substitue nten
aethyl-
aethyl-
aethyl-
aethyl-
allyl-
allyl-
allyl-
isopropyl-
sec-butyl-
1-methyl-butyl-
aethyl-
aethyl-
aethyl-
allyl-
allyl-
allyl-
methyl-
aethyl-
lsopropyl-
allyl-
«2
aethyl-
n-butyl-
1-methyl-butyl-
îsoamyl-
allyl-
îsopropyl-
isobutyl-
jh -bromallyl-
/3-bromallyl-
ß-bromallyl-
phenyl-
cyclohexenyl-
cycloheptenyl-
cyclopentenyl-
phenyl-
isopropyl-
cyclohexenyl-
phenyl-
ß -bromallyl-
cyclohexenyl-
«3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CH3-
CH3-CH3-
CH3-H-
R4
0=
O=
o=
0=
0=
0=
0=
0=
o=
o=
0=
o=
o=
0=
0=
0=
0=
o=
0=
s=
Wasser
185
130
300
s
SS
300
u
u
2200
1000
wl
1100
1000
SS
5000
500
580
340
SS
SS
2000
3300
Alkohol
14
14
30
1
sl
20
1
1
13
1,2
1
10
8
5
20
3,7
0,5
wl
1
7
10,6
Aether
40
35
10
1
si
20
1
1
24
1,8
1
20
13
20
20
6,5
10
0,6
wl
wl
5
3,3
Chloro¬
form
75
1
1
1
40
1
Literatur
Ph. H. V
Merck Ind. (30)
Spécia
Merck Ind. (30)
Ph. H. V
Merck Ind. (30)
Merck Ind. (30)
Riedel
Riedel
Merck Ind. (30)
Ph. H. V
Merck Ind. (30)
Merck Ind. (30)
Geigy
Siegfried
Merck Ind. (30)
Roche
Suppl.n
Suppl. II
Riedel
Imperial
Erklärungen zu Tabelle 10:
sl sehr leicht löslich
1 leicht löslich
wl wenig löslich
s schwer löslich
ss sehr schwer löslich
u unlöslich
in weniger als 1 Teil löslich
löslich in 1 - 10 Teilen
löslich in 30 - 100 Teilen
löslich in 100 - 1000 Teilen
löslich in 1000 - 10000 Teilen
löslich in mehr als 10000 Teilen
30) The Merck Index 6th Ed. Merck and Co. Rahway, N.J. U.S.A. (1952)

- 42 -
Durch ihren sauren Charakter, der den an den Stickstoffatomen ge¬bundenen Wasserstoffatomen zugeschrieben werden muss, lösen sie sich
CO-NH
C C - OH
^ cb - nS
unter Salzbildung in alkalischen Flüssigkeiten, wie Natron- und Kali¬
lauge, Ammoniak, Natriumkarbonat usw., daneben auch in organischenBasen, wie z.B. Diaethylamin, Pyridin und Piperidin. Therapeutisch wich¬
tig sind besonders die Natrium-, Calcium- und Diaethylamin-Salze. Die
Salze reagieren in wässeriger Lösung alkalisch. In Tabelle 11 sind die pH-Werte einiger Natriumsalze von Barbituraten aufgeführt. Thiopental und
Surital enthalten noch eine bestimmte Menge Natriumkarbonat.
Tabelle 11
pH-Werte einiger Barbituratsalze
BarbiUl Na
Butabarbital Na
Vinbarbital Na
Phénobarbital Na
Hexobarbital Na
Thiopental Na
Surital Na
Kemithal Na
Substituenten
«1
aethyl-
aethyl-
aethyl-
aethyl-
methyl-
aethyl-
allyl-
allyl-
B2
aethyl-
n-butyl-
1-methyl-butenyl-
phenyl-
cyclohexenyl-
1-methyl-butyl-
1-methyl-butyl-
cyclohexenyl-
%
H-
H-
H-
H-
CH3-
H-
H-
H-
B4
0-
0-
o=
o=
0=
s-
s=
Konzentration
0,1m
1%
1%
10%
2,5%10 %
2,5%
10%
pH
9,4
9,0-10,2
8,5- 9,5
9,3
11,5
10,510,6
10,5
10,7-10,9
10,6
Literatur
Herck Index (30)
Merck Index (30)
Herck Index (30)
Herck Index (30)
Herck Index (30)Carnngton &
Raventos (17)
Herck Index (30)Carnngton &
Raventos (17)Spéc la
Parke Davis
Carrington &
Raventos (17)
- 43 -
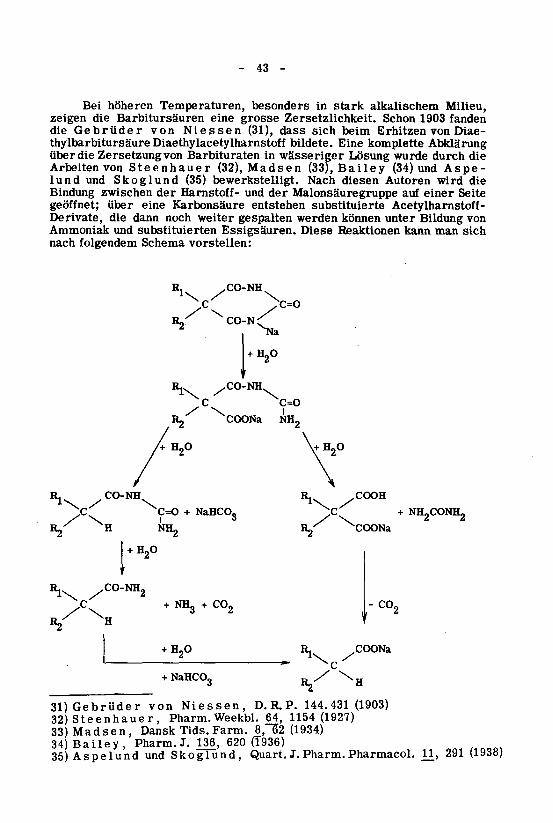
Bei höheren Temperaturen, besonders in stark alkalischem Milieu,zeigen die Barbitursäuren eine grosse Zersetzlichkeit. Schon 1903 fanden
die Gebrüder von Niessen (31), dass sich beim Erhitzen von Diae-
thylbarbitursäure Diaethylacetylharnstoff bildete. Eine komplette Abklärungüber die Zersetzung von Barbituraten in wässeriger Lösung wurde durch die
Arbeiten von Steenhauer (32), Madsen (33), Bailey (34) und Aspe-lund und Skoglund (35) bewerkstelligt. Nach diesen Autoren wird die
Bindung zwischen der Harnstoff- und der Malonsäuregruppe auf einer Seite
geöffnet; über eine Karbonsäure entstehen substituierte Acetylharnstoff-Derivate, die dann noch weiter gespalten werden können unter Bildung von
Ammoniak und substituierten Essigsäuren. Diese Reaktionen kann man sich
nach folgendem Schema vorstellen:
. CO-NH
IL
^c'
CO-NH
/CO-NHC=O
COONa NH2
C=O + NaHCO,3 NH2CONH2
H2°+ H2
NH3
+ CO2 - C0o
COONa
+ NaHCO,
31) Gebrüder von Niessen, D. R. P. 144.431 (1903)
32)Steenhauer, Pharm.Weekbl. 64, 1154(1927)
33) Madsen, Dansk Tids. Farm. 8, 62 (1934)
34) Bailey, Pharm. J. 136, 620 (T936)
35) Aspelund und Skoglund, Quart. J. Pharm. Pharmacol. n_, 291 (1938)

44 -
Nielsen (36)fand, dass sich freie Säure in wässerigen Lösungen der
Natriumsalze bilde, wenn die Löslichkeit der freien Säure gering ist, wie
z. B. bei Phénobarbital.
Die Zersetzungsgeschwindigkeit und der -grad von aethyl - phenyl-barbitursauremNatrium nimmt nach Nielsen (36), Tomski und Waller
(37) und Berasain und Vitali (38) mit zunehmender Temperatur und
pH zu. Dasselbe gilt nach Madsen (33) und Bailey (34) für diaethyl-barbitursaures Natrium.
Als Stabilisatoren der wässerigen Barbitursäure-Lösungen kommen
verschiedene Alkohole und Glykole, Paraldehyd, Glycerin, stickstoffhaltigeVerbindungen wie Urethan, Harnstoff, und Amine in Frage.
Husa und Jatul (39) untersuchten den Einfluss von Zeit, Tempe¬ratur, Konzentration, pH und das Vorhandensein von Stabilisatoren auf Lö¬
sungen von aethyl-phenylbarbitursaurem Natrium. Sie fanden, dass Hitze
zu Zersetzungen führt, deren Ausmass abhängig ist von der Temperaturund der Zeit des Erhitzens. Das Herabsetzen des pH vermindert den Grad
der Zersetzung, aber nicht genügend, um von praktischem Wert zu sein.
Bei pH-Werten von 8,8 und weniger fällt Aethyl-phenylbarbitursäure aus.
Weiter fanden sie, dass wässerig-alkoholische Lösungen stabiler gegen¬über Hitze sind als wässerige Lösungen und dass Lösungen mit einem hohen
Gehalt an Propylen-glykol haltbarer sind als wässerige.
Die Schmelzpunkte der von uns näher studierten Barbiturate lie¬
gen zwischen 60° und 190° und sind in Tabelle 12 zusammengestellt.
Die Schmelzpunkte in Tabelle 12 sind geordnet nach zunehmendem
Molekulargewicht der einzelnen Verbindungen.
Die Differenzen der einzelnen Werte bei derselben Verbindung be¬
ruhen auf folgenden Gründen: Definition des Schmelzpunktes, Methoden und
Apparaturen zur Bestimmung und Korrektur des gefundenen Schmelzpunk¬tes.
Die von Brandstätter, Kofier, Fischer und Reimers aufgeführtenWerte wurden auf dem Kofier-Mikroschmelzpunktapparat bestimmt.
36) Nielsen, Dansk Tids. Farm. 1_, 137 (1933)37) Tom ski und Waller, Pharm. J. 139, 421 (1937)38) Berasain und Vitali, Rev. FarmTBl, 463 (1939)39) Husa und Jatul, J.Am.Pharm.Assoc. JJ3, 217 (1944)
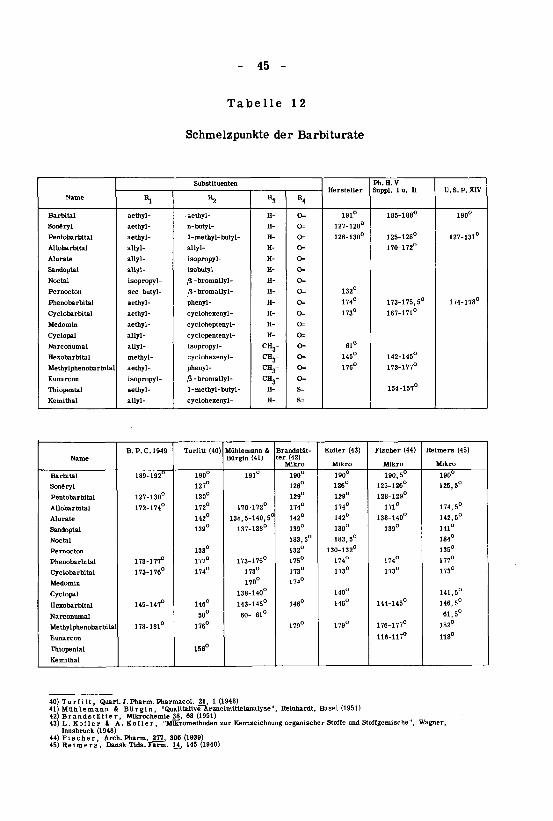
- 45 -
Tabelle 12
Schmelzpunkte der Barbiturate
Name
Barbital
Sonéryl
Pentobarbital
Allobarbital
Alurate
Sandoptal
Noctal
Pernocton
Phénobarbital
Cyclobarbltal
Medomin
Cyclopal
Narconumal
Hexobarbital
Methylphenobarbital
Eunarcon
Thiopental
Kemithal
*1
aethyl-
aethyl-
aethyl-
allyl-
allyl-
allyl-
isopropyl-
sec-butyl-
aethyl-
aethyl-
aethyl-
allyl-
allyl-
methyl-
aethyl-
îsopropyl-
aethyl-
allyl-
Substituenten
aethyl-
n-butyl-
1-methyl-butyl-
allyl-
îsopropyl-
îsobutyl-
ß -bromallyl-
ß-bromallyl-
phenyl-
cyclohexenyl-
cycloheptenyl-
cyclopentenyl-
îsopropyl-
cyclohexenyl-
phenyl-
/3-bromallyl-1-methyl-butyl-
cyclohexenyl-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CH3-CHj-
CH3-
CHj-H-
H-
B4
0=
0=
Cte
0=
O=
O=
O=
0=
0=
O=
0=
0-
0-
O=
o=
0=
s=
s=
nersteiie r
191°
127-128°
126-130°
132°
174°
173°
61°
145°
176°
Ph. H.V
Suppi. i u. n
185-188°
125-128°
170-172°
173-175,5°167-171°
142-145°
173-177°
154-157°
U S P XIV
190°
127-131°
174-178°
Name
Barbital
Sonéryl
Pentobarbllal
Allobarbital
Alurate
Sandoptal
Noctal
Pernocton
Phénobarbital
Cyclobarbital
Medomin
Cyclopal
Hexobarbital
Narconumal
Methylphenobarbital
Eunarcon
Thiopental
Kemithal
B. P.C.1949
189-192°
127-130°
172-174°
173-177°
173-176°
145-147°
178-181°
Turfitt (40)
190°
127°
130°
172°
142°
139°
133°
177°
174°
146°
50°
176°
156°
Muhlemann &
BUrgln (41)
191°
170-172°
138,5-140,5°137-138°
173-175°
173°
170°
138-140°
143-145°
60- 61°
Brandstät-
ter (42)Mikro
190°
126°
129°
174°
142°
139°
183,5°132°
175°
173°
174°
146°
179°
Kofier (43)
Mikro
190°
126°
129°
174°
142°
139°
183,5°130-132°
174°
173°
140°
146°
179°
Fischer (44)
Mikro
190,5°125-126°
128-129°
171°
138-140°
139°
174°
173°
144-145°
176-177°
116-117°
Reimers (45)
Mikro
190°
125,5°
174,5°142,5°141°
184°
135°
177°
173°
141,5°146,5°
61,5°182°
118°
40) Turfitt, Quart. J.Pharm.Pharmacol. 21, 1 (1948)41) Mühlemann & Bürgin, "Qualttatlvê~Arzneimittelanalyse", Reinhardt, Basel (1951)
42) Brandstätter, Mikrochemie 38, 68(1951)43)L.Kofier & A.Kofier, "Mlk"romethoden zur Kennzeichnung organischer Stoffe und Stoffgemische ', Wagner,
Innsbruck (1948)44) Fischer, Arch. Pharm. 277, 306 (1939)45) Reimers, Dansk Tids.Firm. 14, 145(1940)

- 46 -
Als Schmelzpunkt gilt:
nach Ph. H. V das Temperaturintervall vom Beginn der Tröpf¬chenbildung ("Schwitzen der Substanz") bis zum Zusammenflies-
sen der Substanz.
nach U. S. P. XIV: the melting range or temperature of a so¬
lid is defined as those points of temperature within which or at
which the solid coalesces and is completely melted.
nach Brit. Ph: 1948: the temperature at which liquefactionoccurs is regarded as the melting point of the substance.
Diese drei Arzneibücher benützen die Kapillarmethode zur Bestim¬
mung des Schmelzpunktes, jedoch bestehen Unterschiede bei der Ausfüh¬
rung.
Nach Ph. H. V wird das Röhrchen mit der Substanz derart am Ther¬
mometer befestigt, dass die Substanz sich in mittlerer Höhe des Queck-
silbergefässes des Thermometers befindet. Die Badflüssigkeit wird vor¬
sichtig erwärmt und die Temperatur von 10° unterhalb des zu erwartenden
Schmelzpunktes ab so langsam gesteigert, dass zur Erhöhung um 2° min¬
destens 1 Minute erforderlich ist.
Nach U. S. P. XIV wird das Röhrchen mit der Substanz neben das
Quecksilbergefäss des Thermometers befestigt. Das Bad wird dann bis
30° unterhalb des zu erwartenden Schmelzpunktes erhitzt, das Röhrchen
eingeführt, das Heizen derart fortgesetzt, dass die Temperatur 3° pro
Minute steigt bis 3° unterhalb des zu erwartenden Schmelzpunktes. Hier¬
auf wird so geregelt, dass die Temperatur 1° pro Minute steigt. Die Tem¬
peratur bei welcher die Kolonne der Substanz gegen die Wände zusammen-
fliesst gilt als Beginn und die Temperatur, wo die Substanz flüssig wird,als Ende des Schmelzpunktes.
Nach Brit. Ph. 1948 wird das Bad so geheizt, dass die Tempe¬ratur 3° pro Minute steigt. Wenn die Temperatur 10° unterhalb des zu er¬
wartenden Schmelzpunktes ist, wird das Röhrchen eingeführt, so dass das
untere Ende in der Mitte des Quecksilbergefässes des Thermometers ist.
Nach Kofier liest man als Schmelzpunkt jene Temperatur ab, bei
der die kleineren Kristalle und Partikelchen vollständig zerflossen und von
den grösseren in den Schmelztropfen nur noch Reste zu sehen sind. Bei
der Mikroschmelzpunktbestimmung lässt man die Temperatur im Anfangrasch ansteigen und richtet es so ein, dass innerhalb der letzten 10° vor
dem zu erwartenden Schmelzpunkt der Anstieg in der Minute etwa 2° be¬
trägt.
Die Angaben über Schmelzpunkte von Mühlemann und Bürginwurden im Eder 'sehen Schmelzpunktapparat erhalten (46).
Die freien Barbitursäure-Derivate lassen sich im Gegensatz zu ihren
Salzen leicht und ohne Zersetzung sublimieren. Das Sublimat ein und
derselben Ausgangssubstanz zeigt unter dem Mikroskop verschiedene Kri¬
stallformen, die sich nicht nur durch den äusseren Habitus, sondern auch
durch den Schmelzpunkt unterscheiden (siehe S. 68).
46) Eder, Bull. Féd. Int. Pharm. 8, 86 (1927)

- 47 -
Tabern (47) hat die Verteilungskoeffizienten (Tabelle 13)einiger Barbiturate zwischen Wasser und einem speziell gereinigten, fett¬
säurefreiem Olivenöl bei 20° bestimmt, indem er gleiche Mengen Oel und
wässerige Lösung einige Stunden kontinuierlich schüttelte und anschliessendden Stickstoffgehalt der wässerigen Lösung nach Kjeldahl bestimmte. Die
Tabelle zeigt, dass bestimmte Parallelen zwischen Verteilungskoeffizientund hypnotischer Wirkung bestehen. Beide nehmen vom Dimethyl-Derivatan zu und erreichen ein Maximum bei den Aethyl-(1-methy1-butyl)-, sec-
Butyl-allyl- und sec-Butyl-ß-bromallyl-barbitursäuren. Die einzige Aus¬
nahme bildet die Diallyl-Verbindung, die wie die meisten ungesättigtenGlieder etwas wirksamer ist als der Verteilungskoeffizient erwarten lässt.
Erhöht man die C-Zahl der Substituenten noch mehr und damit auch die Li-
poidlöslichKeit, so kommt man zu Produkten geringerer Wirksamkeit. Dies
ist ein Beweis, dass neben der Lipoidlöslichkeit noch andere Faktoren eine
wesentliche Rolle spielen. Wenn übrigens die Lipotropie keine rein physi¬kalische Eigenschaft ist, da sie ja vom Chemismus der Verbindung abhängt,so muss doch für die Ursachen der Wirkung auch an Faktoren gedacht wer¬
den, die mit dem Chemismus in keinen einfachen Zusammenhang gebrachtwerden können.
Das osmotische Verhalten gelöster Stoffe hängt nach Traube (48)in erster Linie von der Oberflächenspannung ab. Je mehr ein Stoffdie Oberflächenspannung vermindert, um so leichter gelangt er durch
Membranen. Von dieser Schnelligkeit hängt die Wirkung von hypnotischenStoffen ab, da bei zu langsamer Resorption oder zu langsamem Eindringenin die Zellen die für die Reaktion nötige Schwelle eventuell nicht erreicht
werden kann, weil ja gleichzeitig Abbau und Ausscheidung erfolgen. Ta¬
bern (47) und Graham (49) haben die Oberflächenspannungen einigerwässeriger Lösungen von Barbitursäure-Derivaten bestimmt. Diese, in
Tabelle 13 eingetragenen Werte lassen erkennen, dass eine Herabsetzungder Oberflächenspannung in gewissen Grenzen eine Wirkungssteigerungverspricht.
47) Tabern und Shelberg, J.Am.Chem.Soc. 55, 328 (1933)
48) Traube, B. 17, 2294 (1884)49) Graham, Am. J. Pharm. _106, 295 (1934)
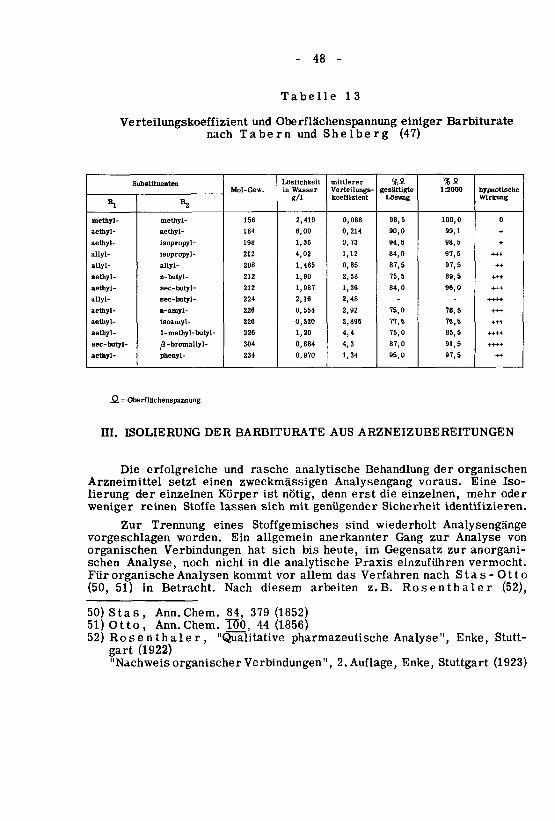
- 48 -
Tabelle 13
Verteilungskoeffizient und Oberflächenspannung einiger Barbiturate
nach Tabern und Shelberg (47)
Substituent»
«1
methyl-
aethyl-
aethyl-
allyl-
allyl-
aethyl-
aethyl-
allyl-
aethyl-
aethyl-
aethyl-
sec-butyl-
aelhyl-
«2
methyl-
aethyl-
isopropyl-
isopropyl-
allyl-
n-butyl-
sec-butyl-
sec-butyl-
n-amyl-
isoamyl-
1-methyl-butyl-
fi -bromallyl-
phenyl-
Mol-Gew.
156
184
198
212
208
212
212
224
226
226
226
304
234
Löslichkeit
in Wasser
s/i
2,419
6,00
1,36
4,02
1,465
1,90
1,987
2,16
0,554
0,530
1,20
0,684
0,970
mittlerer
Verteilungs¬koeffizient
0,066
0,214
0,73
1,12
0,85
2,58
1,36
2,48
2,92
2,895
4,4
4,3
1,34
%2gesättigte
Lösung
98,5
90,0
94,5
84,0
87,5
75,5
84,0
-
75,0
77,5
75,0
87,0
95,0
1:2000
100,0
99,1
98,5
97,5
97,5
89,5
96,0
-
76,5
76,5
85,5
91,5
97,5
hypnotischeWirkung
0
+
+
+++
++
+++
+++
++++
+++
+++
++++
++++
Q_= Oberflächenspannung
HI. ISOLIERUNG DER BARBITURATE AUS ARZNEIZUBEREITUNGEN
Die erfolgreiche und rasche analytische Behandlung der organischenArzneimittel setzt einen zweckmässigen Analysengang voraus. Eine Iso¬
lierung der einzelnen Körper ist nötig, denn erst die einzelnen, mehr oder
weniger reinen Stoffe lassen sich mit genügender Sicherheit identifizieren.
Zur Trennung eines Stoffgemisches sind wiederholt Analysengängevorgeschlagen worden. Ein allgemein anerkannter Gang zur Analyse von
organischen Verbindungen hat sich bis heute, im Gegensatz zur anorgani¬schen Analyse, noch nicht in die analytische Praxis einzuführen vermocht.
Für organische Analysen kommt vor allem das Verfahren nach Stas- Otto
(50, 51) in Betracht. Nach diesem arbeiten z.B. Rosenthaler (52),
50)Stas, Ann.Chem. 84, 379(1852)51) Otto, Ann.Chem. TÜO, 44 (1856)52) Rosenthaler, "Qualitative pharmazeutische Analyse", Enke, Stutt¬
gart (1922)"Nachweis organischer Verbindungen", 2. Auflage, Enke, Stuttgart (1923)

- 49 -
Sabalitschka (53), Gadamer (54), Winterfeld (55), Dietzel und
Mitarbeiter (56). Autenrieth - Bauer (57), Mühlemann und
Bürgin (41), Bamann-Ullmann (58) und Staudinger (59).
Zur Identifizierung sind in den oben genannten Anleitungen physikali¬sche und chemische Eigenschaften der isolierten Körper angeführt. Zu¬
sätzliche Angaben, vor allem chemische Reaktionen, sind unter anderem
zu finden bei: Vieböck (60), Schoorl (61), während bei Feigl (62)und Mayrhofer (63) besonders die Mikroreaktionen beschrieben sind.
53) Sabalitschka, "Anleitung zum chemischen Nachweis der Gifte",Berlin und Wien (1923)
54) Gadamer, "Lehrbuch der chemischen Toxikologie", Göttingen (1924)55) Winte rf eld, "Praktikum der organisch-präparativen pharmazeuti¬
schen Chemie und Leitfaden der chemischen Arzneimittelanalyse", 3.
Auflage, Steinkopf, Dresden und Leipzig (1950)56) Dietzel, Paul und Tunmann, Süddtsche Apoth. Ztg. 80, 335, 347,
353 (1940)Dietzel und T u n m a n n
, "Anleitung zur Analyse organischer Arz¬
neimittel", 2. Auflage, Schmiedel, Stuttgart (1949)57) Autenrieth und Bauer, "Die Auffindung der Gifte und starkwir¬
kender Arzneistoffe zum Gebrauch in chemischen Laboratorien", 6.
Auflage, Steinkopf, Dresden und Leipzig (1943)58) Bamann und Ullmann, "Chemische Untersuchungen von Arznei¬
gemischen, Arzneispezialitäten und Giftstoffen", Jantsch, Günzburg (1951)59) Staudinger, "Anleitung zur organischen qualitativen Analyse", 5.
Auflage, Springer, Berlin (1948)60) Vieböck, "Analysengang zur Erkennung von Arzneimitteln", 2. Auf¬
lage, Deuticke, Wien (1949)61) Schoorl, "Organische Analyse", N. V.D. B.Centen's Uitgevers Mij.
Amsterdam (1941)62) Feigl, "Qualitative Analyse mit Hilfe von Tüpfelreaktionen", 3. Auf¬
lage, Akademische Verlagsgesellschaft, Leipzig (1938)63) May rhof e r
,"Mikrochemie der Arzneimittel und Gifte", Urban &
Schwarzenberg, Berlin und Wien (1928)
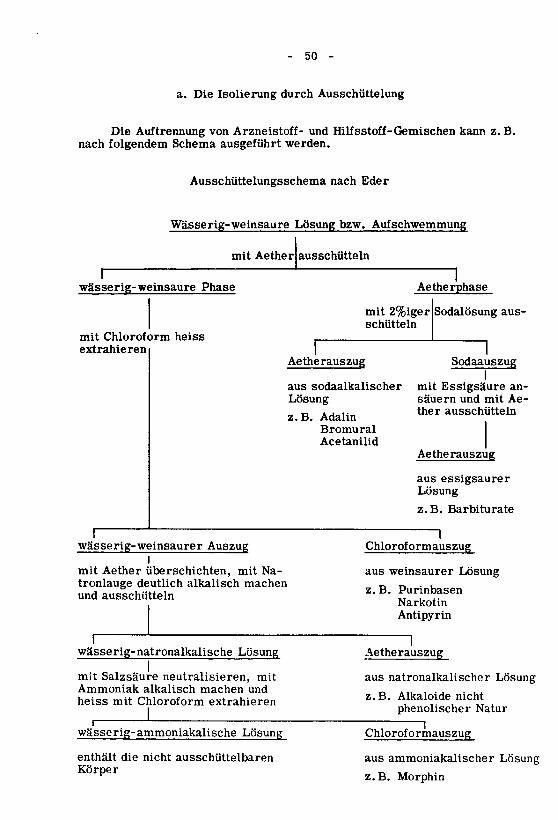
- 50 -
a. Die Isolierung durch Ausschuttelung
Die Auftrennung von Arzneistoff- und Hilfsstoff-Gemischen kann z.B.
nach folgendem Schema ausgeführt werden.
Ausschüttelungsschema nach Eder
Wässerig-weinsaure Lösung bzw. Aufschwemmung
mit Aether ausschütteln
I
wässerig-weinsaure PhaseI
Aetherphase
mit 2%igerschütteln
mit Chloroform heiss
extrahieren IAetherauszug
aus sodaalkalischer
Lösung
z.B. Adalin
Bromural
Acetanilid
Sodalösung aus-
ISodaauszug
Imit Essigsäure an¬
säuern und mit Ae¬
ther ausschütteln
Aetherauszug
aus essigsaurerLösung
z. B. Barbiturate
wässerig-weinsaurer AuszugI
mit Aether überschichten, mit Na¬
tronlauge deutlich alkalisch machen
und ausschütteln
I
Chloroformauszug
aus weinsaurer Lösung
z. B. Purinbasen
Narkotin
Antipyrin
wässerig-natronalkalische Lösung
mit Salzsäure neutralisieren, mit
Ammoniak alkalisch machen und
heiss mit Chloroform extrahieren
Aetherauszug
aus natronalkalischer Lösung
z.B. Alkaloide nicht
phenolischer Natur
wässerig-ammoniakalische Lösung
enthält die nicht ausschüttelbaren
Körper
Chloroformauszug
aus ammoniakalischer Lösung
z.B. Morphin

- 51 -
Die Trennung der Stoffgemische beruht auf folgender Ueberlegung:Organische Substanzen, Säuren und Basen, liegen in wässeriger Lösungz.T. in dissoziierter, z.T. in nicht dissoziierter Form vor, was durch
folgenden Gleichgewichtszustand veranschaulicht wird:
AcO'* + H+ <
^AcOH
Die dissoziierte Form ist wasserlöslich, aber in den meisten organischenLösungsmitteln unlöslich, die nicht dissoziierte dagegen löst sich in orga¬
nischen Lösungsmitteln. Schüttelt man die wässerige Lösung einer orga¬
nischen Säure oder Base mit einem organischen Lösungsmittel aus, so gehtder nicht dissoziierte Anteil in das organische Lösungsmittel. Da durch
Erhöhung der Wasserstoffionenkonzentration die Dissoziation zurückge¬
drängt wird, können organische Säurebildner (Säuren, Phenole, Enole etc.)durch Vermehrung der Wasserstoffionen-Konzentration in die ausschüttel¬
bare Nichtelektrolyt-Form gedrängt werden.
AcO" + H+(aetherlöslich)
Das gleiche gilt sinngemäss für die organischen Basenbildner, die durch
Steigerung der Hydroxylionen-Konzentration in die ausschüttelbare Nicht¬
elektrolyt-Form gedrängt werden. Stärkere Säuren werden nur dann gutausschüttelbar sein, wenn man die Lösung mit einer starken Mineralsäure
sehr sauer macht, umgekehrt ist bei stärkeren Basen starke Alkalisierung
nötig. Bei einigen organischen Basen ist indessen die basenbildende Eigen¬schaft so schwach, dass ihre Salze in schwach saurer Lösung praktischvöllig hydrolysiert sind. Es ist also die Art und Menge der zu verwenden¬
den Säure, ob Salzsäure, Schwefelsäure oder Weinsäure, und des Alkalis
bei der Ausschüttelung nach Stas-Otto dem jeweilig vorliegenden Falle an¬
zupassen. Aus einer weinsauren, wässerigen Lösung gehen somit u. a. die
Barbiturate in das organische Lösungsmittel, in unserem Falle in den
Aether.
Zur Auftrennung wird das Untersuchungsmaterial mit verdünnter
Schwefelsäure oder mit Weinsäure angesäuert und dann nacheinander mit
Aether und Chloroform ausgeschüttelt. Nach dem Abtrennen der Aether-
bzw. Chloroformlösungen werden dieselben mit einem Alkali ausgeschüttelt.Dietzel (56) und Staudinger (59) schüttelten mit einer 2%igen Na¬
triumkarbonatlösung aus, Winterfeld (55) und Bamann - Ullmann
(58) mit einer gesättigten Natriumbikarbonatlösung; Mühlemann und
Bürgin (41) (64) schütteln die Aetherlösung nacheinander mit 5%iger Na¬
triumbikarbonat-, 5%iger Natriumkarbonat- und 0,5-l%iger Natriumhy¬droxydlösung aus, um eine Trennung in starke Säuren, stark saure Phenole
und schwache Säuren und in schwach saure Phenole zu erzielen. Daneben
bringen sie die wässerige Phase immer wieder auf ein bestimmtes pH.Nach der Abtrennung der organischen Lösungsmittel werden die wässerigenalkalischen Lösungen angesäuert und wiederholt mit Aether ausgeschüttelt.Nach dem Abtrennen wird der Aether auf dem Wasserbad abdestilliert und
der Rückstand wird dann auf Barbiturate geprüft.
* AcO" = anionisches Säureradikal, z.B. CHqCOO~64) Bürgin, J.Pharm. Belg. 7, 3(1952)

- 52 -
Warren (65) arbeitet bei der Ausschüttelung entweder mit Chloro¬
form (bei Aethyl-isopropyl-, Aethyl-n-hexyl-, Allyl-1-methyl-butyl-, Iso-
propyl-ß-bromallyl-, sec-Butyl-ß -bromallyl-, Allyl-phenyl-barbitursäureund Aethyl - 1 - methyl - butyl- und Aethyl-isoamylthiobarbitursäure) oder
einem Chloroform-Aether-Gemisch4:1 (Aethyl-isopropyl-, Aethy1-n-butyl-,Aethyl-1-methyl-butyl-, Aethyl-isoamyl-, Aethyl-n-hexyl-, Allyl-allyl-,Allyl-isopropyl-, Allyl-1-methyl-butyl-, Isopropyl-^-bromallyl-, Aethyl-cyclohexenyl-, Allyl-phenyl- und Methyl-cyclohexenyl-N-methyl-barbitur-säure, Aethyl-1-methyl-butyl- und Aethyl-isoamyl-thiobarbitursäure). In¬
folge der schlechten Löslichkeit von Isopropy1-/3 -bromallyl- und sec-Butyl-
ß-bromallyl- barbitursäure in Chloroform ist die Verwendungeines Lö¬
sungsmittelgemisches vorteilhaft, jedoch nicht unbedingt notwendig. Des
weiteren fand er keinen besonderen Vorteil bei einer der beiden Methoden.
Bei der quantitativen Auftrennung und Bestimmung eines Arzneimit¬
telgemisches muss man sich bewusst sein, dass es zu einer gegenseitigenBeeinflussung der Löslichkeit kommen kann.
Der an unserem Institut verwendete Analysengang zur Durchführungpharmazeutischer Analysen lehnt sich im wesentlichen an die Vorschlägevon Autenrieth-Bauer (57) und Dietzel (56) an. Das zu unter¬
suchende Gemisch wird in Wasser gelöst oder mit Wasser aufgeschwemmt,mit Weinsäure auf ein pH von ca. 2 gebracht und wiederholt mit Aether aus¬
geschüttelt. Die vereinigten Aetherauszüge werden dann mit 2%iger Soda¬
lösung extrahiert. Der Sodaauszug wird abgetrennt, mit Essigsäure ange¬säuert und wiederholt mit Aether ausgezogen. Die vereinigten Aetherlö-
siingen werden mit wasserfreiem Natriumsulfat getrocknet und der Aether
auf dem Wasserbad abdestilliert. Der Rückstand enthält dann die Barbitur-
säure-Derivate. Um nun zur weiteren Untersuchung und besonders zur
Identifikation möglichst reine Stoffe zu haben, hat es sich als vorteilhaft
erwiesen, eine Sublimation oder besser eine Mikrosublimation im Vakuum
durchzuführen.
Im folgenden soll deshalb eine kurze Uebersicht über diese Verfahren,die Sublimationsbedingungen und über die Möglichkeiten der Identifizierungvon Sublimaten gegeben werden.
b. Isolierung durch Mikrosublimation
Allgemeines
Die Sublimation erfreut sich in der technischen Chemie als einfaches
Reinigungs- und Isolierungsverfahren grosser Beliebtheit und kann auch in
der Mikrochemie wertvolle Dienste leisten. Zuerst in der Pharmakognosiezur Isolierung von Inhaltsstoffen aus den Drogen verwendet, hat sie auchin der pharmazeutischen und toxikologischen Chemie Eingang gefunden.Der Grund dazu darf wohl darin zu suchen sein, dass die Sublimation ge¬stattet, mit kleinen und kleinsten Mengen Substanz zu arbeiten und auf ein¬
fache Art möglichst reine Stoffe zu isolieren. Die Sublimate wiederum
eignen sich für die Untersuchungen unter dem Mikroskop vorzüglich, seien
65) Warren, J. Assoc. Official Agr. Chem. 25, 799(1942): 27. 352(1944);26, 101 (1943)
— —

- 53 -
es die kristalloptischen Untersuchungen an den oft sehr gut ausgebildetenSinzelkristallen, die Mikroschmelzpunktbestimmung oder die mikrochemi¬
schen Reaktionen. Unter den untersuchten Arzneistoffen war es die Gruppeder Alkaloide, die durch Eder (66) eine eingehende Bearbeitung fand.
Haas (67), Fischer (68), St rebel (69) und andere wandten sich den
immer wichtiger werdenden synthetischen Arzneimitteln, wie z.B. den
Antipyretica, Chemotherapeutica, Lokalanaesthetica, Narcotica, Hypnotica,Sedativa zu. Es musste in den verschiedenen Gruppen für die pharmazeu¬tisch-chemische und die toxikologische Analyse nach Isolierungsmöglich¬keiten sowie Identifizierungsmethoden gesucht werden. Schwierigkeit bietet
in den einzelnen Arzneistoffgruppen der sichere Nachweis eines Einzelstof¬
fes, da sich jene vielfach durch ähnlichen chemischen Bau und damit zu¬
sammenhängend ähnliche Reaktionsfähigkeit auszeichnen. Die Mikrosubli-
mation stellt eine sehr gute Isolierungsmethode dar, und die Sublimate kön¬
nen weiter zur Identifizierung der Substanz verwendet werden.
Begriffsbestimmung
Unter Sublimation versteht man die Erscheinung, dass viele feste
Substanzen bei erhöhter Temperatur sich direkt verflüchtigen und auf käl¬
teren Vorlagen in festem Zustande wieder niederschlagen. Unter Mikro-
sublimation fasst man nach Kempf (70) alle Sublimationsverfahren zusam¬
men, bei denen die Sublimate zum Zwecke mikroskopischer Betrachtung auf
einer ebenen Glasplatte, gewöhnlich Objektträger oder Deckglas, aufgefan¬gen werden.
Der Zweck des Sublimierens im chemischen Laboratorium ist vor
allem die Reinigung eines Rohproduktes von anhaftenden Verunreinigungenoder die Trennung der Einzelbestandteile eines Gemisches. Das gleichebezweckt auch die Mikrosublimation. Darüber hinaus wird aber in der Mi¬
krochemie die Sublimation häufig unmittelbar als Reaktion auf einen be¬
stimmten Stoff herangezogen, indem man trachtet, möglichst gut ausge¬bildete Kristalle zu erhalten, die durch ihre Form, ihre optischen Eigen¬schaften, den Mikroschmelzpunkt, Löslichkeit und mikrochemische Reak¬
tionen identifiziert werden. Auch die Temperatur, bei der das Sublimat ent¬
standen ist, wird mitunter für die Diagnose herangezogen.
66) Eder, "Ueber die Mikrosublimation im luftverdünnten Raum", Diss.
ETH. Zürich (1912)67) Haas, "Ueber Vakuummikrosublimation synthetischer Arzneistoffe
und Identifizierung der Sublimate auf kristalloptischem Wege", Diss.
ETH. Zürich (1930)68) Fischer, Mikrochemie 10, 409 (1931) Arch. Pharm. 271., 466(1933);
277, 306 (1939)69) S t r e be 1, "Untersuchungen über die Vakuum-Mikrosublimation neuerer
synthetischer Arzneistoffe und die Identifizierung der Sublimate", Diss.
ETH. Zürich (1950)70) Kempf, in Houben, "Methoden der organischen Chemie", 3.Auflage,
I.Bd.p. 693 (1925)

- 54 -
Sublimieren im eigentlichen Sinne ist das Ueberführen einer Substanz
aus dem festen unmittelbar in den dampfförmigen Aggregatzustand durch
Wärmezufuhr und die Rückverwandlung der Dämpfe durch Wärmeentzug
wieder unmittelbar in den festen Zustand. Die Sublimation verläuft also
nach dem Schema fest —»dampfförmig -»fest, während bei der Destilla¬
tion dem dampfförmigen Aggregatzustand eine flüssige Phase vorausgehtund nachfolgt. Bei der Mikrosublimation schiebt sich nicht selten zwischen
den dampfförmigen und festen Aggregatzustand eine flüssige Phase ein, so
dass also der Vorgang in dieser Weise verläuft: fest—»dampfförmig—»
flüssig—»fest. Das wichtigste Kennzeichen der Sublimation, der Ueber-
gang vom dampfförmigen unmittelbar in den festen Zustand, ist demnach
hier entfallen.
In der Praxis hält man trotzdem auch in diesem Falle an dem Aus¬
druck Mikrosublimation fest, weil der Zweck der Operation in der Ueber-
führung des dampfförmigen Aggregatzustandes in die feste Phase besteht
und weil die sich dazwischen schiebende flüssige Phase häufig nicht be¬
obachtet wird.
Theoretisch sind alle festen Stoffe schon bei Zimmertemperatur su¬
bi mierbar. Bei den meisten Körpern ist jedoch bei dieser Temperatur die
Su ilimationsgeschwindigkeit so gering, dass die Sublimation nicht wahr-
nenmbar ist. In der Praxis spricht man daher nur dann von Sublimierbar-
keit eines Stoffes, wenn unterhalb seines Schmelzpunktes in absehbarer
Zeit die Verflüchtigung wahrnehmbarer Mengen erfolgt. Die "wahrnehm¬
bare Menge" ist dabei ein sehr dehnbarer Begriff. Bei der Makrosublima¬
tion werden darunter meist wägbare, bei der Mikrosublimation mikrosko¬
pisch sichtbare Mengen verstanden. Um praktisch eine Sublimation durch¬
zuführen, ist bei den meisten Körpern eine Erhöhung der Temperatur und
bei manchen auch eine Herabsetzung des Luftdruckes notwendig. Tempera¬tur und Luftdruck sind aber nicht die einzigen Faktoren, die die Sublima¬
tionsgeschwindigkeit beeinflussen. Es spielen unter anderem die Grosse
der Oberfläche des Sublimationsgutes, der von den Dämpfen zu überwindende
Höhenunterschied, d.h. der Sublimationsabstand, und die Temperaturdif¬ferenz zwischen dem Sublimationsgut und der Vorlage eine Rolle.
Aus all dem geht hervor, dass man nicht von einem Sublimations¬
punkt eines Stoffes sprechen kann als Ausdruck für die Temperatur des
Auftretens des ersten wahrnehmbaren Sublimates. Obwohl die Bezeichnung
"Sublimationspunkt und Sublimationstemperatur" schon seit langem wieder¬
holt verpönt wurde (z.B. von Kempf (71) und Eder (66)), erscheinen
auch in der neueren Literatur immer wieder solche Angaben. Man kann
nicht einmal von einer Sublimationstemperatur schlechtweg sprechen. Un¬
ter Sublimationstemperatur verstehen wir immer ein Temperaturintervall,innert welchem eine bestimmte Substanz gut ausgebildete Kristalle bei der
Sublimation unter den bestimmten Versuchsbedingungen liefert. Trotzdem
spielt praktisch die Sublimationstemperatur eines; Körpers eine Rolle, sie
bezieht sich dann aber immer auf eine bestimmte Versuchsanordnung. Da¬
mit hängt es zusammen, dass man im Schrifttum recht abweichende Subli¬
mationstemperaturen für ein und denselben Stoff finden kann. Man muss
sich bewusst sein, dass jede dieser Angaben nur für eine ganz bestimmte
Arbeitsweise Berechtigung besitzt.
71) Kempf, J. Prakt. Chem. 78, 201 (1908)

- 55 -
Verfahren und Apparaturen
Eine Betrachtung über die Methoden und die Sublimationsapparate im
allgemeinen würde hier zu weit führen. Eder (66) hat in seiner Disserta¬
tion einen ausgezeichneten historischen Ueberblick zu geben vermocht, und
die neueren Verfahren zur Mikrosublimation sind von Fischer (72) und
Kofier (73) in übersichtlicher Art und Weise zusammengestellt. Eine
kurze Zusammenfassung der wichtigsten Methoden für analytische Zwecke
findet sich bei St rebel (69). In neuerer Zeit wurde von Gettler, Um-
berger und Goldbaum (74) eine Mikrosublimationsapparatur beschrie¬
ben, die die Trennung von Stoffgemischen durch Sublimation erlaubt. Die
Sublimate werden auf einen austauschbaren durchsichtigen Film abgelagertund können darauf direkt mikroskopisch und chemisch untersucht werden.
Die von uns verwendete Methode ist das von St rebel (69) verbesserte
Verfahren nach Eder (66). Die Apparatur (siehe Abb. 1) besteht aus einem
Abbildung 1
72) Fischer, Mikrochemie ^5, 247(1934)73) L. Kof 1 e r und A
.Ko f 1 e r
, "Mikroskopische Methoden in der Mikro¬
chemie", Haim & Co., Wien und Leipzig (1936)74) Gettler, Umberger und Goldbaum, Anal.Chem. 22, 600 (1950)

- 56 -
etwa 2-2,5 cm weiten Rohr aus Jenaerglas, dessen unteres Ende sich stu¬
fenweise zu einem Näpfchen von 1 cm Tiefe und 0,5 cm lichter Weite ver¬
engt. In das Näpfchen, das mehr oder weniger tief in ein Oelbad taucht,kann das fein gepulverte Sublimationsgut gegeben werden. Unmittelbar über
das Näpfchen wird ein Deckglas von 18 mm Durchmesser gelegt, auf wel¬
chem sich das Sublimat kondensieren kann. Den Abschluss nach oben bildet
ein durchbohrter, mit Glasrohr versehener Gummistopfen, und das Ganze
kann durch eine Wasserstrahlpumpe bequem evakuiert werden. Die Tempe¬ratur des mit einem Mikrobrenner erwärmten Heizbades kann mit einem
Thermometer kontrolliert werden, und der Druck lässt sich an einem da¬
zwischen geschalteten Manometer ablesen. Der mit der Wasserstrahlpumpeerhältliche verminderte Druck beträgt mit einigen Schwankungen, die durch
die Wassertemperatur bedingt sind, ca. 10-12 mm Hg.
Das Näpfchen taucht mit seinem untersten Teil in ein Oelbad, welches
aus einem Becherchen von ca. 7 cm Höhe und 4 cm lichter Weite besteht.
Zur Einhaltung einer konstanten Badtemperatur wurde nach St rebel (69)der Apparatur ein kleines Rührwerk beigefügt. Der untere, in das Heizbad
ragende Teil eines Glasstabes wurde rechtwinklig abgebogen und zu einem
in das Becherchen passenden Kreis geschlossen. Der obere Teil des Glas¬
stabes wird durch eine Führung geleitet und oben zu einem kleinen Kreis
gebogen, an welchem eine Schnur befestigt ist. Diese wird durch ein recht¬
winklig abgebogenes Glasrohr, das als Führung dient, gezogen und auf einer
mit einem Motor in Verbindung stehenden Spule mittels eines kleinen Draht¬
hakens exzentrisch befestigt, so dass eine periodische Zugwirkung ausge¬übt wird. Die Gegenwirkung kommt durch das Gewicht des Glasstabes zu¬
stande. Mit dieser Vorrichtung, bei der mehrere Rührer, in unserem Falle
vier oder fünf, gleichzeitig in Betrieb genommen werden können, ist es
möglich, unter genau gleichen Druckverhältnissen mehrere Sublimationen
bei verschiedenen Temperaturen nebeneinander durchzuführen. Die Tempe¬ratur des Heizbades weist dabei nur Schwankungen innerhalb 1-2° auf.
Sublimation unter dem Mikroskop
Bei dem Eder'sehen Mikrosublimationsapparat ist die zur mikrosko¬
pischen Kontrolle immer wieder notwendige Unterbrechung des Versuches
besonders umständlich. Man muss dann z.B. von 10 zu 10° unterbrechen,das Deckglas aus dem Apparat nehmen und unter dem Mikroskop betrach¬
ten. Ohne Zweifel ist jede Unterbrechung mit einer Störung der Kristall¬
bildung verbunden.
Gegenüber dieser Arbeitsweise ist die Vakuum-Mikrosublimation un¬
mittelbarunter dem Mikroskop wesentlich einfacher und zugleich leistungs¬fähiger. Kofier und Dernbach (75) benutzen einen Apparat, bestehend
aus einer quadratischen Glasplatte mit 3,5 cm Seitenlänge, auf die eine
Mikrovakuumglocke aufgeschliffen ist. Die Mikrovakuumglocke besitzt ein
seitlich eingesetztes Röhrchen zum Anschluss an eine Wasserstrahlpumpeoder an einen Hochvakuumapparat. Die Höhe der Glocke beträgt 6 mm, ihre
75) Kofier und Dernbach, Mikrochemie _9. 345 (1931)76)Pyrkosch, Pharm. Ztg. Nachr. 88, 178(1952)

- 57 -
obere Wand muss parallel der unteren Glasplatte und planparallel sein, da¬
mit die mikroskopische Beobachtung nicht gestört ist. Die Mikrosublima-
tion mit dieser Vorrichtung wird unmittelbar auf der Heizplatte des Mikro-
schmelzpunktapparates vorgenommen. In dieser Richtung hat auch Fi¬
scher (72) Sublimationsvorrichtungen auf einem Mikroschmelzpunktapparatmit Vakuum- oder Kühlvorrichtung entwickelt. Eine andere Anordnung hat
Pyrkosch (76) gewählt. In einer runden Messingplatte findet sich in der
Mitte eine Bohrung zur Aufnahme des Sublimationsgutes. Zur Veränderungdes Sublimationsabstandes können in der Bohrung Messingscheiben ver¬
schiedener Dicke eingelegt werden. Zum Auffangen des Sublimates dienen
runde Deckgläschen, die auf die Bohrung in eine Vertiefung eingelegt werden
können. Soll im Vakuum gearbeitet werden, so wird ein Aufsatz aus Messingbenützt, derauf die Sublimationsplatte aufgeschliffen und mit Vakuumfett ab¬
gedichtet ist. Diesem Aufsatz, der aus zwei verschraubten Platten besteht und
hohl ist, sind in der Mitte, auf der Ober-und Unterseite, zwei runde Glasplat¬ten eingekittet. An der unteren Glasplatte des Aufsatzes, welche sich über
dem Sublimationsraum befindet, wird mit Glycerin ein rundes Deckgläschen,zum Auffangen des Sublimates, befestigt.
Für unsere Untersuchungen der Sublimationsvorgänge unter dem Mi¬
kroskop verwendeten wir eine Modifikation des Eder'schen Sublimations¬
apparates. Der wesentliche Unterschied zu diesem besteht darin, dass er
ein seitlich eingesetztes Röhrchen zum Anschluss an die Wasserstrahl¬
pumpe trägt und dass der obere Teil einen Planschliff hat und mit einer
4 mm dicken planparallelen runden Glasplatte bedeckt ist (Abb. 2). Die Ab¬
dichtung erfolgt mit Vakuumfett. So ist es nun möglich, die Kristallbildungund eventuelle Umwandlungen der einzelnen Kristalle während der Subli¬
mation unter dem Mikroskop zu verfolgen.
Abbildung 2

- 58 -
Die Beeinflussung der Bildung und des Aussehens von Sublimaten
Als wesentliche Faktoren, welche die Bildung und das Aussehen der
Sublimate beeinflussen, sind von Eder (66), Haas (67), L. Kofier und
A. Kofier (73,77), Fischer (72) und Strebel (69) auf Grund theore¬
tischer Betrachtungen und anhand praktischer Erfahrungen u.a.
die Druckverhältnisse,die Temperaturverhältnisse,der Sublimationsabstand,die Sublimationsdauer und
die Beeinflussung des Sublimates
erkannt worden.
1. Druckverhältnisse
Der äussere Druck spielt auch bei der Sublimation eine grosse Rolle,indem die Sublimationsgeschwindigkeit annähernd umgekehrt proportionalist mit dem äusseren Druck. Es lässt sich daher im luftverdünnten Raum
in der Regel mit grösserer Geschwindigkeit sublimieren als unter gewöhn¬lichem Druck. Da beim Arbeiten im Vakuum ausserdem die Sublimations¬
temperatur herabgesetzt wird, lässt sich die Zersetzung vieler Substanzen
vermeiden oder doch wesentlich vermindern. Bei niedrigeren Temperaturenwerden auch schönere und für kristalloptische Untersuchungen geeignetereKristalle erhalten.
Der Einfluss des Druckes auf die Sublimation ist noch wenig unter¬
sucht worden. Aus Untersuchungen von Janot und Chaigneau (78) an
zahlreichen Alkaloiden geht hervor, dass trotz Verminderung des Druckes
auf 0,01-0,03 mm Tröpfchen-Sublimate erhalten wurden, wie sie auch von
Eder (66) bei Anwendung von 8-12 mm beobachtet wurden. Um festzustel¬
len, ob der Druck die Sublimation beeinflusst, sublimierten wir verschie¬
dene Substanzen bei 720 mm, 50 mm, 10 mm, 1 mm und 0,05 mm, aber
bei sonst überall gleichbleibenden Bedingungen.
77) L. Kofier und A. Kofier, "Mikromethoden zur Kennzeichnung or¬
ganischer Stoffe und Stoffgemische", Wagner, Innsbruck (1948)78) Janot und Chaigneau, C. R. Acad.Sci. j!25, 1371 (1947)

- 59 -
2. Temperaturverhältnisse
Von jeher hat es die Bearbeiter der Mikrosublimation interessiert,ob den einzelnen sublimierbaren Stoffen bestimmte Sublimationstempera¬turen als Konstanten zugewiesen werden könnten. Um dies zu überprüfen,gingen sie zur Verwendung"von Apparaten über, welche exakte Temperatur¬messungen erlauben. Als einfachste Messung wurde jene der Bad- oder
Blocktemperatur eingeführt. In der allerdings unzutreffenden Annahme,diese Temperatur übertrage sich gleichmässig auf das Sublimationsgut,wurde sie von den meisten Autoren als Sublimationstemperatur bezeichnet.
Nach den bisher gemachten Beobachtungen können die sublimierbaren
Arzneistoffe bei den verschiedensten Temperaturen und innerhalb von gros-sen Temperaturbereichen Sublimate bilden. Stoffe mit relativ grossem
Dampfdruck sind schon weit unterhalb ihres Schmelzpunktes und dann
meistens in einem weiten Bereichsublimierbar (z.B. Benzoesäure, Koffein,Naphthalin, Theobromin, etc.). Daneben gibt es Substanzen, welche erst
kurz unterhalb ihres Schmelzpunktes und zwar in einem enger begrenztenBereich sublimieren, Eder (66) konnte in Bezug auf die Bildung schöner
Kristallisate bei den von ihm bearbeiteten Alkaloiden beobachten, dass um
so bessere Resultate erhalten werden, je grosser der Dampfdruck ist und
je tiefer die Sublimationstemperatur unter dem Schmelzpunkt liegt. In der
Literatur variieren nun die Angaben über die beobachteten Sublimations¬
temperaturen ausserordentlich stark. Kofier (77) hat z.B. die von ver¬
schiedenen Autoren bestimmten Werte für Theobromin zusammengetragenund dabei die Feststellung gemacht, dass sie zwischen 91° und 300° ange¬
geben wurden. Mit Recht macht Kofier (77) darauf aufmerksam, dass
die Temperatur, bei welcher ein Sublimat auftritt, abhängig ist vom Zu¬
stand des Sublimationsgutes, vom äusseren Druck, vom Sublimationsab¬
stand und anderen Faktoren. Die Angaben über Sublimationstemperaturensind besser weit zu fassen. Von einigem Wert können sie nur dann sein,wenn die exakten Versuchsbedingungen angegeben sind. Von einem Subli¬
mationspunkt sollte überhaupt nicht gesprochen werden, obwohl derartigeAngaben etwa in der Literatur zu finden sind.
Die Angaben über die Sublimationstemperatur können niemals den
Wert von Konstanten besitzen. Es darf ihnen lediglich eine Bedeutung als
Arbeitsanleitung zugesprochen werden. Ihre Bestimmung ist ungenau und
gibt ausserdem einen sehr schlechten Einblick in die vorherrschenden
Temperaturverhältnisse bei der Sublimation. Für den Sublimationsvorgang,der sich aus verschiedenen Phasen zusammensetzt (Verdampfung des Su¬
blimationsgutes, Wanderung des Dampfes zum Kristallisationsort und
Kristallisation) interessieren die nachfolgend aufgeführten Temperaturen:
Bad-Temperatur (BT),Temperatur des Sublimationsgutes (SGT),Temperatur des Dampfraumes (DRT) und
Temperatur des Kristallisationsortes (ST).

- 60 -
Bereits Eder (66) hatte den Versuch unternommen, in dem von ihm
entwickelten Vakuum - Mikrosublimationsapparat die Temperaturverhält¬nisse zu erforschen, durch Messung der Bad-, Plättchen- und Innentempe¬ratur. St rebel (69) befasste sich dann eingehend mit den Beziehungenzwischen der Bad-, Sublimationsgut-, Dampfräum- und Sublimat-Tempe¬ratur, unter anderem dem sogenannten Temperaturgefalle, worunter man
die Differenz SGT - ST versteht.
Von grösstem Interesse hinsichtlich Einfluss der Temperaturbedin¬gungen ist die Frage, ob unter sonst gleichen Bedingungen bei verschiede¬
nen Sublimationstemperaturen Kristallisate von unterschiedlichem Habitus
resultieren. Dies ist nun tatsächlich der Fall, und zwar bei polymorphenSubstanzen. Die vermehrte Bildung instabiler Formen bei polymorphenSubstanzen infolge grösserer Temperaturgefälle ist vor allem durch Kof¬
ier (79) bei der Mikrosublimation von p-Oxybenzoesäuremethylester bei
gewöhnlichem Druck festgestellt worden und hat uns bei den Barbituraten
ebenfalls beschäftigt.
3. Sublimationsabstand
Das Temperaturgefälle ist auch weitgehend abhängig vom Sublima¬
tionsabstand, d.h. von der Distanz zwischen dem Sublimationsgut und dem
als Rezipienten für die Kristallisate dienenden Deckgläschen. Eine Ver-
grösserung des Sublimationsabstandes vergrössert ausser dem Tempera¬turgefälle auch die Steighöhe der Dämpfe und den Dampfräum. Auswirkungendavon sind notwendigerweise höhere Sublimationstemperaturen, eine Ver¬
längerung der Sublimationszeit und die Inkaufnahme vermehrter Zersetzun¬
gen. Einzig für die fraktionierte Sublimation dürfte sich ein vergrösserterSublimationsabstand günstig auswirken, indem er erlaubt, eine bessere
Trennung von Stoffgemischen zu erreichen.
Systematische Untersuchungen über den Einfluss des Sublimations¬
abstandes auf das Temperaturgefälle und die Bildung der Sublimate sind,soweit uns bekannt wurde, bis heute noch nicht durchgeführt worden. Wir
überprüften deshalb die Vakuum-Mikrosublimation nach Eder in der Weise
auf den Einfluss des Sublimationsabstandes, dass wir Eder'sche Sublima-
tionsgefässe mit einem Abstand von 5,5; 9,0; 12,5; 17,0 und 24,0 mmzwischen Näpfchenboden und Deckgläschen herstellen Hessen (siehe Abb. 3)und Sublimate von Cyclobarbital und Methylphenobarbital gewannen.
79) L. Kofier, Mikrochemie 9, 45 (1931)

- 61 -
Abbildung 3
4. Sublimationsdauer
Die Sublimationsdauer ist nach den praktischen Erfahrungen abhängigvom äusseren Druck, der Sublimationstemperatur und dem Sublimations¬
abstand. Geringer Druck, hohe Temperatur und kleiner Abstand kürzen
sie ganz erheblich ab. Bei lang dauernder Sublimation ist die Beobachtunggemacht worden, dass Tröpfchen, Tröpfchenkristalle und kleine Einzel¬
kristalle allmählich wieder verschwinden. Kofier (73) zeigte das Auf¬
fressen von Tröpfchen durch Kristallaggregate bei Dial und das Wachstum
von Salicylsäurekristallen auf Kosten sehr feiner Einzelkristalle. Ent¬
sprechende Beobachtungen machten wir bei Allobarbital, wo sich vorerst
instabile Formen bilden, welche während der über zwei Stunden ausgedehn¬ten Sublimation bei derselben Badtemperatur verschwanden unter Vermeh¬
rung der stabilen Form. Wir können uns diese Erscheinung dadurch erklä¬
ren, dass die verschwindend kleineren Formen dank grösserer Oberfläche
einen höheren Dampfdruck besitzen.

- 62 -
5. Beeinflussung der Sublimate
Da sich die Tröpfchensublimate und die kleinen Kristalle nicht zur
Identifikation eignen, ist immer wieder versucht worden, die Sublimate so
zu beeinflussen, dass die Tröpfchen kristallisieren und gut ausgebildeteEinzelkristalle erhalten werden. Möglichkeiten dafür sind das Kratzen der
Vorlage, das Kratzen der Tröpfchensublimate, das Impfen der Vorlage,die Kristallisation des Sublimates aus einem organischen Lösungsmittelund die Umsublimierung. Bei der Impfung tritt im Sublimat aber immer
die Modifikation auf, mit der geimpft wurde.
Möglichkeiten zur Identifizierung der Mikrosublimate
1. Kristallform
Von manchen Forschern und Praktikern wird das Aussehen und der
Habitus der Sublimate, wenn nicht sogar als völlig hinreichendes, so doch
als wichtige s Merkmal für die Identifizierung eines Stoffes anerkannt. Schon
zahlreiche Forscher, vor allem auch Kofier (73) und Fischer (68,72)haben immer darauf hingewiesen, dass dieses Vorgehen nicht richtig ist
und zu schweren Irrtümern bei der mikroskopischen Untersuchung von
organischen Verbindungen geführt hat. Eine grosse Mannigfaltigkeit in der
Ausbildung von Sublimaten führt immer wieder dazu, dass bei zahlreichen
Arzneistoffen ähnliche Einzelkristalle und Kristallaggregate auftreten. Bei
manchen Sublimaten bilden sich ferner Stellen mit stark differierendem
Kristallhabitus aus. Diese überraschende Variabilität der Ausbildungs¬formen von Einzelkristallen und Kristallaggregaten lässt sich nach Kofier
(73) erklären durch die gleichzeitige Auswirkung "innerer" und "äusserer"
Faktoren während der Kristallbildung, welche die Keimbildung, das Wachs¬
tum der Einzelkristalle (hinsichtlich der Bevorzugung bestimmter Wachs¬
tumsrichtungen) und die Art der Vereinigung von Einzelkristallen zu Aggre¬gaten bedingen. Als "innere" Faktoren werden die beeinflussenden Eigen¬schaften bezeichnet, welche einer bestimmten Substanz innewohnen. Die
"äusseren" Faktoren dagegen werden weitgehend bestimmt durch die Ver¬
suchsbedingungen, welche Verhältnisse schaffen, die vom Analytiker kaum
eindeutig beherrscht werden können. Das Kristallsublimat ist immer das
Resultat der Auswirkungen beider Faktoren; bei den bisher näher unter¬
suchten Arzneistoffen haben sich die "äusseren" Faktoren als wesentlich
formbestimmend erwiesen.
Für uns hat das Mikrosublimat andere Bedeutung. Es ist ein weit¬
gehend gereinigtes Isolierungsprodukt aus Arzneistoffgemischen, Arznei¬
formen und toxikologischem Material, das sich für die sichere Identifizie¬
rung mit Hilfe physikalischer und chemischer Methoden eignet und dieser
Auswertung in vermehrtem Mass zugeführt werden muss.

- 63 -
2. Kristalloptische Methoden
Die Möglichkeit der Identifizierung der Mikrosublimate mit Hilfe von
kristalloptischen Methodenwurden in unseren Untersuchungen nicht benützt.
Angaben über kristalloptische Eigenschaften der Barbiturate finden sich
u.a. bei: Haas (67), Fischer (44), Castle (80), Poe und Mitarbei¬
ter (81), Winters (82). Die kristalloptischen Eigenschaften der ver¬
schiedenen Modifikationen einzelner Barbiturate sind bei Fischer (83),Fischer und Kofier (84), Lindpaintner (85), Kofier (86), Kofier
und Fischer (87) beschrieben.
3. Mikroschmelzpunktbestimmung
Der grosse Vorteil der Mikroschmelzpunktbestimmung besteht darin,dass der Schmelzvorgang sehr geringer Substanzmengen, selbst einzelner
Kristalle, unter dem Mikroskop beobachtet werden kann. Ausser dem
Schmelzpunkt lassen sich dabei noch andere charakteristische Eigenschaftenfeststellen, welche der Identifizierung einer Substanz dienlich sein können.
Für unsere bei der Mikrosublimation im Vakuum erhaltenen Sublimate hiel¬
ten wir diese Methode, aus oben erwähnten Gründen, als die einzig richtigefür die weitere Untersuchung.
Wir benutzten den, dem Koflerblock (88,89) nachgebildeten Balzer¬
block mit Thermoelement. Das Instrument wurde geeicht mit Testsubstan¬
zen. Als Schmelzpunkt ermittelten wir das Temperaturintervall vom Tau¬
punkt bis zum Klarschmelzpunkt der Substanz. Bei der Bestimmung des
einfachen Mikroschmelzpunktes machte sich bei allen Sublimaten ihre Ei¬
genschaft der Sublimation störend bemerkbar, indem das zudeckende Deck¬
gläschen bis in die Nähe des Schmelzpunktes mit feinen Kristallen oder
Sublimattröpfchen bedeckt wurde und oft die Beobachtung des Schmelzpunk¬tes erschwerte. Bei den polymorphen Substanzen konnten die einzelnen
Schmelzpunkte, sowie Umwandlung von instabiler zu stabiler Modifikation
beobachtet werden. Die Identität der Stoffe wird vorteilhaft erhärtet durch
die Bestimmung eines Mikro-Mischschmelzpunktes. Der Schmelzpunkt soll
bei Identität der Stoffe scharf und praktisch im gleichen Temperaturbereichliegen.
80) Castle, J.Am. Pharm. Assoc. 38, 47 (1949)81) Poe, Witt und Snodgrass, Mikrochemie 34, 235 (1949)82) Winters, "Bijdrage tot de opsporing der barbitalen bij het toxicolo-
gisch onderzoek", Diss. Leyden (1936)83) Fischer, Arch. Pharm. ^70, 149 (1932)84) Fischer und A. Kolfer, Arch.Pharm. 270, 207 (1932)85) Lindpaintner, Mikrochemie 27, 21 (193T»T86) A. Kofier, Mikrochemie 33, 4~{l947)87) A. Kofier und Fischer, Arch. Pharm. 273, 483 (1935)88) L. Kofier und Hilbck, Mikrochemie 9, "3iT(1931)89) L. Kofier, Mikrochemie ^5, 242 (1934)"
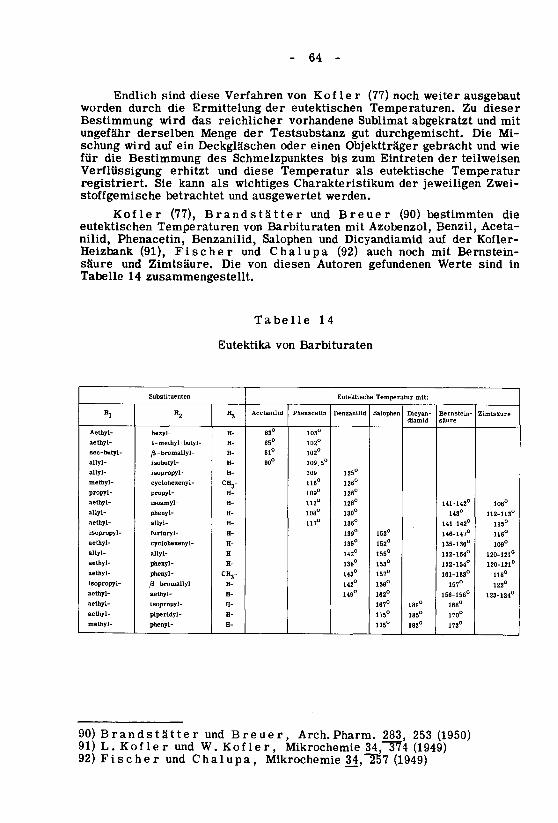
- 64 -
Endlich sind diese Verfahren von Kofier (77) noch weiter ausgebautworden durch die Ermittelung der eutektischen Temperaturen. Zu dieser
Bestimmung wird das reichlicher vorhandene Sublimat abgekratzt und mit
ungefähr derselben Menge der Testsubstanz gut durchgemischt. Die Mi¬
schung wird auf ein Deckgläschen oder einen Objektträger gebracht und wie
für die Bestimmung des Schmelzpunktes bis zum Eintreten der teilweisen
Verflüssigung erhitzt und diese Temperatur als eutektische Temperaturregistriert. Sie kann als wichtiges Charakteristikum der jeweiligen Zwei¬
stoffgemische betrachtet und ausgewertet werden.
Kofier (77), Brandstätter und Breuer (90) bestimmten die
eutektischen Temperaturen von Barbituraten mit Azobenzol, Benzil, Aceta-
nilid, Phenacetin, Benzanilid, Salophen und Dicyandiamid auf der Kofler-
Heizbank (91), Fischer und Chalupa (92) auch noch mit Bernstein¬
säure und Zimtsäure. Die von diesen Autoren gefundenen Werte sind in
Tabelle 14 zusammengestellt.
Tabelle 14
Eutektika von Barbituraten
Substituenten
"l
Aethyl-
aethyl-
sec-butyl-
allyl-
allyl-
methyl-
propyl-
aethyl-
allyl-
aethyl-
isopropyl-
aethyl-
allyl-
aethyl-
aethyl-
isopropyl-
aethyl-
aethyl-
aethyl-
methyl-
R2
hexyl-
1-methyl-butyl-
/3-bromallyl-isobutyl-
lSopropyl-
cyclohexenyl-
propyl-
lsoamyl-
phenyl-
allyl-
furfuryl-
cyclohexenyl-
allyl-
phenyl-
phenyl-
ß -bromallyl-
aethyl-
isopropyl-
pipendyl-
phenyl-
"3
H-
H-
H-
H-
H-
CH3-H-
H-
H-
H-
H-
H-
H-
H-
CB3-H-
H-
H-
H-
H-
Eutektische Temperatur mit:
Acetamhd
83°
85°
81°
90°
Phenacetin
103°
102°
102°
109,5°109
116°
109°
112°
108°
117°
Benzanilid
125°
126°
126°
128°
130°
136°
139°
135°
142°
138°
143°
142°
149°
Salophen
153°
152°
155°
153°
157°
158°
162°
167°
175°
175°
Dicyan¬diamid
181°
185°
183°
Bernstein¬
säure
141-142°
143°
141-142°
146-147°
135-136°
152-154°
152-154°
161-163°
157°
156-158°
168°
170°
173°
Zimtsäure
106°
112-113°
115°
116°
109°
120-121°
120-121°
118°
122°
123-124°
90) Brandstätter und Breuer, Arch. Pharm. 283, 253 (1950)91) L. Kofier und W. Kofier, Mikrochemie 34,~3T4 (1949)92) Fischer und Chalupa, Mikrochemie 3^,"2^7 (1949)

- 65 -
Leider sind die eutektischen Temperaturen zur Differenzierung weniggeeignet. Wie aus Tabelle 14 ersichtlich ist, liegen die Eutektika der einzel¬
nen Barbiturate mit derselben Testsubstanz ziemlich nahe beieinander, so
dass es zur Charakterisierung eines bestimmten Derivates mehr als ein
Eutektikum braucht. Auch dann ist eine Unterscheidung noch schwierig und
nicht immer zuverlässig, denn diese Eutektika liegen dann auch in ziemlich
engen Grenzen, so z.B. die Eutektika von Isopropyl-furfuryl- bis Isopro-pyl-/3-bromallyl-barbitursäure mit Benzanilid und Salophen liegen einer¬
seits zwischen 135° und 142° und anderseits zwischen 152° und 158°. Man
kann dies auf die grosse Verwandtschaft der Barbiturate zurückführen.
4. Brechungsexponenten von Schmelzen
Nach Kofier (93) lässt sich auf dem Mikroschmelzpunktapparatdurch Beimengung von Glaspulvern mit bekannten Brechungsexponenten in
einfacher Weise eine Bestimmung des Brechungsexponenten der Schmelze
einer Substanz an die Mikroschmelzpunktbestimmung anschliessen und da¬
durch eine weitere, für die physikalische Charakterisierung wertvolle
Konstante gewinnen. Nach Brandstätter (42) bietet diese Methode vor¬
zügliche Dienste zur Identifizierung von Barbituraten. Die teilweise starke
Unterkühlbarkeit der Schmelzen von Barbitursäure - Derivaten bietet den
Vorteil, mit einer geringen Anzahl von Glaspulvern zur Bestimmung der
Lichtbrechung auszukommen, so dass in der Regel nach der Bestimmungdes Schmelzpunktes nur mit einem oder zwei Gläsern Versuche angestelltwerden müssen, um die Lichtbrechung zu ermitteln und damit die Substanz
zu identifizieren. In Tabelle 15 sind die von Brandstätter (42) ermit¬
telten Temperaturen für Luftbrechungsgleichheit von Barbituraten mit Glas¬
pulvern verschiedener Brechungsindices aufgeführt.
In der Tabelle 15 sind die Barbiturate nach zunehmendem Schmelz¬
punktgeordnet, wobei Verbindungen mit ähnlichen Schmelzpunkten zu enge¬ren Gruppen zusammengefasst sind und wobei angestrebt wurde, in einer
Gruppe mit einem oder zwei Gläsern auszukommen. Z.B. sind fünf Ver¬
bindungen mit Schmelzpunkten von 172° bis 175° angeführt, von denen sich
vier mit dem Glas 1,4937 bestimmen lassen. Die Temperatur der Licht¬
brechungsgleichheit differiert jeweils um mindestens 10°. Brandstätter
(42) kommt zum Schluss, dass die exakte Mikroschmelzpunktbestimmungin Kombination mit der Bestimmung der Lichtbrechung der Schmelzen ein
ausserordentlich einfaches und sicheres Mittel zur Unterscheidung von
Barbitursäure-Derivaten darstellt.
93) L. Kofier, Mikrochemie 22, 241 (1937)
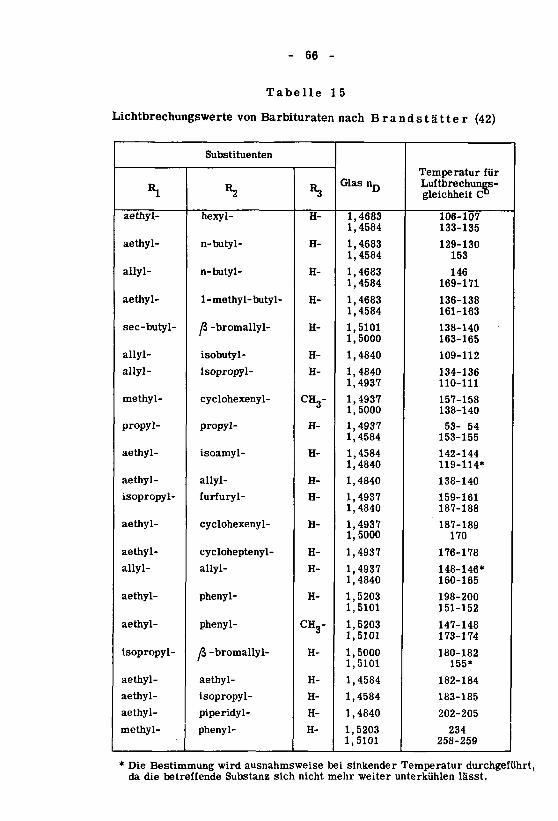
- 66 -
Tabelle 15
Lichtbrechungswerte von Barbituraten nach Brandstätter (42)
Substituenten
«1
aethyl-
aethyl-
allyl-
aethyl-
sec-butyl-
allyl-
allyl-
methyl-
propyl-
aethyl-
aethyl-
isopropyl-
aethyl-
aethyl-
allyl-
aethyl-
aethyl-
isopropyl-
aethyl-
aethyl-
aethyl-
methyl-
hexyl-
n-butyl-
n-butyl-
1-methyl-butyl-
/i -bromallyl-
isobutyl-
isopropyl-
cyclohexenyl-
propyl-
isoamyl-
allyl-
furfuryl-
cyclohexenyl-
cycloheptenyl-
allyl-
phenyl-
phenyl-
& -bromallyl-
aethyl-
isopropyl-
piperidyl-
phenyl-
«3
H-
H-
H-
H-
H-
H-
H-
CH3-
H-
H-
H-
H-
H-
H-
H-
H-
CH3-
H-
H-
H-
H-
H-
Glas nD
1,46831,4584
1,46831,4584
1,46831,4584
1,46831,4584
1,51011,5000
1,4840
1,48401,4937
1,49371,5000
1,49371,4584
1,45841,4840
1,4840
1,49371,4840
1,49371,5000
1,4937
1,49371,4840
1,52031,5101
1,52031,5101
1,50001,5101
1,4584
1,4584
1,4840
1,52031,5101
Temperatur für
Luftbrechungs¬gleichheit C°
106-107
133-135
129-130
153
146
169-171
136-138
161-163
138-140
163-165
109-112
134-136
110-111
157-158
138-140
53- 54
153-155
142-144
119-114*
138-140
159-161
187-188
187-189170
176-178
148-146*
160-165
198-200
151-152
147-148
173-174
180-182
155*
182-184
183-185
202-205
234
258-259
* Die Bestimmung wird ausnahmsweise bei sinkender Temperatur durchgeführt,da die betreffende Substanz sich nicht mehr weiter unterkühlen lässt.

- 67 -
5. Mikrochemische Reaktionen
Nach Durchführung der oben besprochenen Methoden zur Identifikation
von Sublimaten bilden die mikrochemischen Färb- und vor allem die Kri¬
stallfällungsreaktionen ein weiteres Glied in der Kette der Möglichkeiten,einen Stoff zu identifizieren.
Van Itallie und Steenhauer (94) arbeiteten mit Ammonium¬
phosphat, ammoniakalischer Silbernitratlösung, Bariumhydroxyd, Essig¬säure, Thalliumacetat und ammoniakalischer Kupferlösung als Fällungs¬mittel für Barbiturate. Rosenthaler (95,96) fällt mit Ammoniumchlorid,Ammoniumsulfat, Kaliumsulfat und Natriumsulfat, Fischer (44) mit
Ammoniumphosphat, ammoniakalischer Silbernitratlösung, Jodjodkalium,Barytwasser und Magnesiamixtur, Van Zijp (97) verwendet Jodjodkalium,Jodjodnatrium und Berberinsulfat, Strzyzowski und Dé ver in (98)ammoniakalischeSilbernitratlösung, Beck (99)beschreibt Kristallfällungenmit Yttriumnitrat, Aquopentammin-, Chloronitrotetrammin- und Diaquo-tetramminkobaltiaken, Wagenaar (100) Kristallfällungen mit einer Mi¬
schung von Aethylendiamin und Kupfersulfat.
In neuerer Zeit findet besonders die von Lüdy-Tenger (101)einge¬führte und von Lang und Stephan (102) und von Kaiser und Lang(103) weiter verbreiteten Methode mit Schwermetallkomplexen grössereAnwendung.
Den Kristallfällungsreaktionen soll sich aber, wo immer möglich,eine Bestimmung des Schmelzpunktes oder einer anderen physikalischenKonstante anschliessen. Denn der Habitus der gefällten Kristalle allein ist
nicht immer ein sicherer Beweis für das Vorhandensein eines bestimmten
Stoffes, weil die einzelnen Fällungsreaktionen in hohem Masse von den
Versuchsbedingungen abhängig sind.
94) Van Itallie und Steenhauer, Pharm.Weekbl. 617, 977 (1930)95) Rosenthaler, Mikrochemie 18, 50 (1935)96) Rosenthaler, Apoth. Ztg. 48~793 (1933)97) Van Zijp, Pharm.Weekbl. 7T, 1075 (1934); 73, 764 (1936)98) Strzyzowski und Déverin, Helv.Chim.Acta 16, 1288 (1933)
99) Beck, Mikrochemie ^9, 206(1941)100) Wagenaar, Pharm.Weekbl. 78, 345 (1941)101) Lüdy-Tenger, Pharm.Acta Helv. _19, 3&5 (1944)102) Lang und Stephan, Süddtsche Apoth. Ztg. 90, 739 (1950)103) Kaiser und Lang, Süddtsche Apoth. Ztg. 9|7428 (1952)
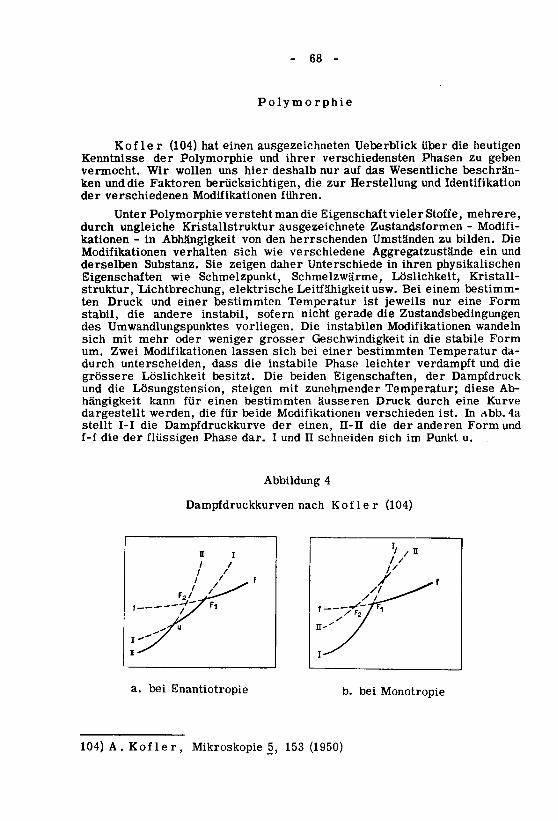
- 68 -
Polymorphie
Kofier (104) hat einen ausgezeichneten Ueberblick über die heutigenKenntnisse der Polymorphie und ihrer verschiedensten Phasen zu gebenvermocht. Wir wollen uns hier deshalb nur auf das Wesentliche beschrän¬
ken und die Faktoren berücksichtigen, die zur Herstellung und Identifikation
der verschiedenen Modifikationen führen.
Unter Polymorphie versteht man die Eigenschaft vieler Stoffe, mehrere,durch ungleiche Kristallstruktur ausgezeichnete Zustandsformen - Modifi¬
kationen - in Abhängigkeit von den herrschenden Umständen zu bilden. Die
Modifikationen verhalten sich wie verschiedene Aggregatzustände ein und
derselben Substanz. Sie zeigen daher Unterschiede in ihren physikalischenEigenschaften wie Schmelzpunkt, Schmelzwärme, Löslichkeit, Kristall¬
struktur, "Lichtbrechung, elektrische Leitfähigkeit usw. Bei einem bestimm¬
ten Druck und einer bestimmten Temperatur ist jeweils nur eine Form
stabil, die andere instabil, sofern nicht gerade die Zustandsbedingungendes Umwandlungspunktes vorliegen. Die instabilen Modifikationen wandeln
sich mit mehr oder weniger grosser Geschwindigkeit in die stabile Form
um. Zwei Modifikationen lassen sich bei einer bestimmten Temperatur da¬
durch unterscheiden, dass die instabile Phase leichter verdampft und die
grössere Löslichkeit besitzt. Die beiden Eigenschaften, der Dampfdruckund die Lösungstension, steigen mit zunehmender Temperatur; diese Ab¬
hängigkeit kann für einen bestimmten äusseren Druck durch eine Kurve
dargestellt werden, die für beide Modifikationen verschieden ist. In Abb. 4a
stellt I-I die Dampfdruckkurve der einen, II-II die der anderen Form und
f-f die der flüssigen Phase dar. I und II schneiden sich im Punkt u.
Abbildung 4
Dampfdruckkurven nach Kofier (104)
a. bei Enantiotropie b. bei Monotropie
104) A. Kofier, Mikroskopie^, 153(1950)

- 69 -
Erwärmt man die bei Raumtemperatur beständige Form II über u hinaus,so wird sie, da sie jetzt den grösseren Dampfdruck besitzt, unbeständigund wandelt sich in Modifikation I um, deren Schmelzpunkt bei Fj liegt.Gelingt es jedoch, II ohne Umwandlung zu erwärmen, so tritt Verflüssigungbei F2, das ist bei einer niedrigeren Temperatur als I, ein. Geht man um¬
gekehrt von der geschmolzenen Substanz aus, so wandelt sich die aus Rest-
kristallen entstandene Form I bei Abkühlung über u in II zurück. Man nennt
derartiges Verhalten polymorpher Substanzen enantiotrop (105).
Die Dampfdruckkurven können auch so charakterisiert sein, dass der
Umwandlungspunkt oberhalb des Schmelzpunktes zu liegen kommt und da¬
durch virtuell wird (Abb. 4b). Bei solchen Stoffen, es ist die grössere Zahl,ist Modifikation I mit dem stets niedriger bleibenden Dampfdruck stabil;daher kann sich nur II in I umwandeln und nicht umgekehrt. Beim Erwär¬
men schmilzt I bei F\, ehe der virtuelle Umwandlungspunkt u erreicht
werden kann. Man nennt solche Modifikationen monotrop (105).
Zur Bezeichnung der Modifikationen erwiesen sich die römischen
Ziffern als zweckmässig, wobei die stabile, das ist jeweils die höchst¬
schmelzende Form mit I, die instabilen in der Reihenfolge ihrer abneh¬
menden Schmelzpunkte mit II, IQ, IV usw. gekennzeichnet werden.
Instabile Modifikationen, die sich bei den gegebenen Bedingungenverhältnismässig schwer in die stabile Form umwandeln, heissen haltbar
(metastabil). Bemerkenswert ist, dass die Haltbarkeit mancher instabiler
Formen durch geringfügige Verunreinigungen stark gesteigert werden
kann, so dass bei Reinsubstanzen manchmal bestimmte instabile Formen
nur sehr schwer entstehen und sich rasch in die stabilen Formen umwandeln,während sie bei unreinen Präparaten ohne Umwandlung zum Schmelzen ge¬bracht werden können.
Der Uebergang aus instabilen in stabilere Zustände kann in verschie¬
dener Weise erfolgen: 1. schlagartig, 2. allmählich. Im ersten Fall findet
mit grosser Geschwindigkeit ein "Umklappen" ganzer Netzebenen eines
Gitters in die Orientierung des anderen Gitters statt. Diese Umwandlungerfolgt nicht durch thermischen Platzwechsel der Kristallbausteine, sondern
ist eine durch innere Spannungen ausgelöste Umlagerung. Dabei entsteht
meist aus einem Einzelkristall nicht wieder ein einziger Kristall, sondern
ersterer zerfällt in mehr oder weniger zahlreiche Einzelkristalle. Beider
zweiten Art der Umlagerung, die auch als Umbau-Umwandlung bezeichnet
wird, erfolgt die Bewegung nicht für alle Bausteine eines zusammenhän¬
genden Gitterbereiches gleichzeitig, sondern durch Platzwechselvorgänge(Diffusion). Diese verlaufen daher mit messbarer, temperaturabhängigerGeschwindigkeit und kommen wie die Diffusion bei tiefen Temperaturenzum Stillstand. Die Umwandlung ist keimbedingt. Das neu gebildete Aggre¬gat kann bezüglich der Korngrösse feiner, gleich oder gröber sein als das
ursprüngliche Kristallisat.
105) Lehmann, "Molekularphysik", Leipzig (1888)

- 70 -
Gewinnung verschiedener Modifikationen
Nach Kofier (43,77,104) können zur Gewinnung instabiler Formen
die Mikrosublimation, die Kristallisation aus der Schmelze und die Kri¬
stallisation aus verschiedenen organischen Lösungsmitteln angewendet wer¬
den.
Die Keimbildung in einer Schmelze kann freiwillig oder unfreiwilligerfolgen. Die spontane Keimbildung insbesondere instabiler Formen wird
durch stärkere Unterkühlung gefördert. Zur mikroskopischen Untersuchungschmilzt man ein Objektträger - Deckglas - Präparat durch, legt es auf den
kalten Mikroskoptisch und beobachtet den Erstarrungsvorgang. Häufig ent¬
stehen bei polymorphen Substanzen spontan Herde verschiedener Modifika¬
tionen nebeneinander, meist in Form von Sphärolithen, seltener als Einzel¬
kristalle oder als Kristalldrusen. In vielen Fällen kann schon auf Grund
von Unterschieden im Habitus auf das Vorliegen mehrerer Modifikationen
geschlossen werden. Die Kristallisation einer Schmelze kann auch durch
unfreiwillige Keimbildung (Keiminduktion) erzwungen werden. Dies kann
durch unspezifische oder spezifische Kristallisationserreger erfolgen. Als
unspezifische Kristallisationserreger gelten Druck, Erschütterung und
elektromagnetische Felder. Es handelt sich dabei entweder um ein direktes
isomorphes Fortwachsen oder um eine durch geringere gitterstrukturelleVerwandtschaft bedingte Keimbildungserleichterung.
Eine zweite aussichtsreiche Methode zur Herstellung instabiler For¬
men ist die Sublimation, wobei instabile Formen wegen des höheren Dampf¬druckes eher bei normalem als bei vermindertem Druck auftreten. Die da¬
bei erhaltenen Kristalle sind je nach Temperatur, Dauer der Sublimation,Sublimationsabstand und somit Temperaturdifferenz zwischen dem ver¬
dampfenden Material und der auffangenden Fläche von verschiedener Grosse
und Ebenmässigkeit. Infolge der isolierten Lage ist in vielen Fällen die
Schmelzpunktbestimmung eher durchführbar, da die Gefahr der Infektionmit stabilen Keimen geringer ist.
Die Mikrosublimation erwies sich uns für die Gewinnung verschiede¬
ner Modifikationen von Barbituraten als vorteilhafter. Sie erlaubte uns zu
zeigen, wieviel Modifikationen die eine in Frage kommende Substanz auf¬
weist. Zwei oder mehr verschiedene Kristallformen können gewöhnlich auf
ein und demselben Deckglas beobachtet werden. EinNachteil dieser Methode
besteht jedoch darin, dassman mit ihr nicht immer nur eine bestimmte Mo¬
difikation, vor allem instabile, in grösserer Menge herstellen kann.
Aus Lösungsmitteln fällt im allgemeinen eher die bei der entsprechen¬den Temperatur beständige Form aus. Jedoch kommt die gleichzeigite Aus¬
bildung von instabilen Formen häufig vor. In seltenen Fällen kristallisiert
aus einem bestimmten Lösungsmittel stets eine bestimmte instabile Form
aus.

- 71 -
Identifikation der verschiedenen Modifikationen
Chemische Methoden zur Identifizierung der einzelnen Modifikationen
können nicht ausgeführt werden, da sie dasselbe chemische Verhalten zei¬
gen. Aus diesem Grunde spielen die physikalischen Methoden eine wichtigeRolle. Die Bestimmung des Schmelzpunktes instabiler Modifikationen gelingtin vielen Fällen ohne weiteres, in anderen Fällen begegnet man jedoch gros-sen Schwierigkeiten. Nach Kof le r (104) bestehen für die Schmelzpunktbe¬stimmung an instabilen Formen folgende Möglichkeiten:
1. die direkte Bestimmunga. an Kristallisäten aus der Schmelze,b. an Sublimaten.
2. die indirekte Bestimmunga. auf Grund der Schmelzkurven im Zweistoffsystem,b. mittels der eutektischen Temperatur mit geeigneten Testsub¬
stanzen.
Bei der Sublimation besteht, infolge der isolierten Lage der Einzel¬
kristalle, eher die Möglichkeit, ohne Umwandlung den Schmelzpunkt zu er¬
reichen.
Eine Zusammenstellung der in der Literatur angegebenen Schmelzpunkt-te der stabilen und instabilen Formen der Barbitursäure-Derivate findet
sich in Tabelle 16.
Das Polarisationsmikroskop erlaubt uns, ein vollständiges Bild über
die optischen Eigenschaften, wie z. B. die Brechungsindices, die Art der
Auslöschung, die Symmetrie, die optischen Achsenwinkel zu erhalten. Ver¬
schiedene Modifikationen zeigen verschiedene optische Charakteristika, so
dass die optischen Eigenschaften für die Identifikation der einzelnen Modifi¬
kationen genügen könnten, wenn nicht die erhaltenen Werte hie unddaunsi--
cher wären.
Die sicherste Methode zur Identifizierung von Modifikationen ist die
Untersuchung mit Hilfe von Roentgenstrahlen, wozu man jedoch grössereMengen der zu untersuchenden Modifikation zur Verfügung haben muss.
Kristalluntersuchungen mit Hilfe von Roentgenstrahlen
Man kennt drei Methoden, um Kristalle mit Hilfe von Roentgenstrah¬len zu untersuchen:
1. Methode nach Laue mit polychromatischen Strahlen
2. Methodenach Debye und Scherrer mit monochromatischen Strahlen
(Pulvermethode)
3. Drehkristallmethode mit monochromatischen Strahlen
Die Methoden basieren auf dem Bragg'schen Gesetz, welches die Wellen¬
länge Xder Roentgenstrahlen mit dem Einfallwinkel resp. Reflexionswin¬
kel 0 der Roentgenstrahlen und der Aequidistanz d der reflektierenden
Atomebenen verbindet.
- 72 -
Tabelle 16
Schmelzpunkte polymorpher Barbiturate
Substituent«!
»1
Aethyl-
aethyl-
aethyl-
n-propyl-
aethyl-
n-propyl-
aethyl-
allyl-
ieopropyl-
methyl-
aethyl-
allyl-
aethyl-
isopropyl-
«2
aethyl-
propyl-
lsoamyl-
isopropyl-
1-methyl-butyl-
allyl-
isopropyl-
1$ -bromallyl-
pheiiyl-
phenyl-
cydopentenyl-
plperidyl-
aUyl-
«3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H
CH,-
I
190,5°191°190°190°190°
146°
156-157°
175°
128-129°
148°
158-160°
142°142°O
183,5
285-227°
177°
140°
222-223°
64°
n
184°184°183°183°183°
132°
150-151°
155°
114°
146°
128-130°
138-140°
139°139°180°
199-200°
174°174°174°
214-216°217°
61,5°
m
180°
181°181°
112°
133-134°
s?179°
166-167°167°
166-167°
210°
IV
176°176°176°
130°130° ?
156-157°157°
156-157°
Literatur
Fischer (44)Plicnêr & Koüer (84)Lindpalntner (85)Koller {WlTsö^Yueh Huang (106)
Brandatätter (107)
Fischer (44)
Brandstätter (107)
Fischer (44)
Reimers (45)
Fischer (44)
Fischer (44)
Reimers (45)Tso-Vueh Huang (106)Holler (108)
Fischer (44)
Fischer (44)KoflerA Fischer (87)Tso-ïueh Huang (1U6) *
Koner (43) ••
Fischer (44)Kofler (43)
Reimers (45)
.Tso-Yueh Huang (106) besteht noch eine fünfte Modifikation vom Schmelzpunkt 162-163°.Kofi er (43) ist die Allyl-cyclopentenyl-barbiturBäure polymorph.
Abbildung 5
Bragg'sche Gleichung
G E L M
B
106) Tso-Yueh Huang, Acta Pharm.Int.2, 43, 95, 173(1951)107) Brandstätter, Z.Physik. Chem. Abf. A 191, 227(1942)

- 73 -
In der Figur der Abb. 5 stellen AB und CD zwei gleichwertige, paralleleAtomebenen des bestrahlten Kristalles dar, EF und GH zwei paralleleStrahlen derselben Wellenlänge X, die mit einem Winkel 0 auf die Atom¬
ebenen einfallen. Die unter dem Winkel © reflektierten Strahlen sind FL
resp. HM. Die Strecke GHM ist um die Distanz JH + HK = 2 JH = 2 d sin0
grosser als die Strecke EFL. Die reflektierten Strahlen verstärken sich
gegenseitig nur dann, wenn sie sich in Phase befinden, d.h. wenn die zu¬
sätzliche Strecke des zweiten Strahles eine ganze Zahl von Wellenlängendarstellt. Hieraus ergibt sich die Bragg'sche Gleichung:
n X = 2 d sin ©
Die Reflexion von Roentgenstrahlen bekannter Wellenlänge an einem
Kristall erlaubt somit die Berechnung der Netzebenenabstände d zwischen
gleichwertigen parallelen Atomebenen des untersuchten Kristalles. In
einem Kristallgitter kommen zahlreiche, verschieden liegende Scharen
von unter sich parallelen Netzebenen vor, wobei jeder ein bestimmter,für die betreffende Schar konstanter Netzebenenabstand d entspricht. Eine
genaue Analyse der Netzebenenanordnung und der Intensität der durch die
Gitterebenen verursachten Roentgenreflexe lässtdie Bestimmung der Atom¬
anordnung im Kristallgitter zu.
Debye undScherrer (108) zeigten dass man nicht nur mit Ein¬
kristallen, sondern auch mit Pulvern die Netzebenenabstände bestimmen
kann. Diese Methode beruht auf dem Prinzip, dass in dem aus zahlreichen
Kristallenen bestehenden Präparat immer eine Anzahl Kristallenen vor¬
handen sind, deren Orientierung gerade so ist, dass die Reflexionsbedin¬
gung für die einfallenden Strahlen an einer Netzebenenschar erfüllt ist.
Greift man z. B. eine bestimmte Netzebenenschar Ex vom Abstand dx aus
der grossen Zahl der Netzebenenscharen des Kristallgitters heraus, so
reflektiert diese Schar lange nicht in jedem Körnchen des Pulvers. Dies
ist nur dann der Fall, wenn n\= 2 dx sin ©x gilt, mit anderen Worten,wenn die Schar Ex mit dem Einfallsstrahl den Winkel © einschliesst. Die¬
se Bedingung erfüllen sämtliche Netzebenen, welche einen Kegel tangieren,dessen halber Oeffnungswinkel ©_ ist und dessen Achse demEinfallstrahl
entspricht. Die Reflexstrahlen all dieser Gitterebenen liegen auf einem
zweiten um den Einfallstrahl als Achse zu denkenden Kegel, dessen halber
Oeffnungswinkel 2©x
beträgt. Diese Reflexkegel werden auf einem photo¬
graphischen Film aufgenommen. Aus ihrem Oeffnungswinkel (= 4©)lässtsich der Netzebenenabstand der betreffenden Netzebenenscharen berechnen.
Jede Substanz gibt bei solchen Aufnahmen ein für sie charakteristi¬
sches Diagramm, d.h. eine Abfolge von mehr oder weniger intensiven Re¬
flexlinien. Substanzen, welche dasselbe Kristallgiter, aber verschiedene
äussere Formen (Habitus) haben, ergeben dasselbe Diagramm; Substanzen
mit gleichem Chemismus, aber mit verschiedener Kristallstruktur (poly¬
morphe Substanzen) geben hingegen unterschiedliche Diagramme.
Wir benützten die Pulvermethode bei der Untersuchung von Allobar-
bital-Sublimaten (siehe S.112).
108) Debye undScherrer, Physik. Z.H, 277 (1916); 18, 291(1917)

- 74 -
IV. NACHWEISMOEGLICHKEITEN DER BARBITURATE
Nach der Isolierung durch Ausschüttelung und eventueller Reinigungdurch Sublimation soll der Rückstand auf Anwesenheit von Barbitursäure-
Derivaten geprüft werden. Man greift dabei zuerst zu einer Gruppenreaktionund bei positivem Ausfall dieser Reaktion handelt es sich darum, das ein¬
zelne Barbiturat mit Hilfe von Einzelreaktionen zu identifizieren.
1. Gruppenreaktionen
Beim Nachweis der Barbitursäure-Derivate als Gruppe kommen
Färb- und Fällungsreaktionen in Frage. Im folgenden soll eine Uebersicht
über die zahlreichen Gruppenreaktionen und deren Verbesserungsvorschlä¬
ge gegeben werden. Des weiteren soll an Hand dieser Uebersicht nachgese¬hen werden, ob eine dieser Gruppenreaktionen noch eine Unterscheidungs¬möglichkeit zwischen Dialkyl-, Halogenalkyl-, N-methylierten und Thio-
Derivaten zulässt.
A. Farbreaktionen
Grosse Bedeutung erlangte die von Parri (109) 1924 erstmalig be¬
schriebene und 1931 von Zwikker (110) in die auch heute noch vielfach
übliche Anwendungsformen gebrachte Kobaltfarbreaktion. Der Nachweis be¬
ruht im Prinzip auf der Bildung einer blauvioletten Komplexverbindung des
Barbiturates mit einem Co-H-Salz und einer basischen Komponente in ei¬
nem wasserfreien Lösungsmittel. Zwikker (111) stellte verschiedene Ko¬
baltkomplexe der Diaethyl-barbitursäure rein dar, und zwar als rote, brau¬
ne und blaue Kobaltverbindung.
Die rote Kobaltverbindung stellt ein feurig rotviolettes Pulver dar, das
aus luftbeständigen, langgestreckten prismatischen Kristallen besteht und
sehr empfindlich ist gegen Wasser. Es wird als ein Dibarbital-Kobaltodiam-
min (I) folgender Formel charakterisiert:
<*>** NH00 C2H5
109) Parri, Boll. Chim. Farm. 63, 401 (1924)110) Zwikker, Pharm.Weekbl.~B8, 975 (1931)111) Zwikker, Pharm.Weekbl. ÏÏ9", 1178 (1932)

- 75 -
Die braune Verbindung, Hexamminkobaltibarbitalhydroxyd, ist ein
lockeres, braunes, glitzerndes Kristallpulver, von Ammoniakgeruch, und
hat folgende Zusammensetzung (n):
iCom(NH3)
1^(Barbital)2n
Diese Verbindung gibt freiwillig 1 Mol Ammoniak ab, wobei der Ammoniak¬
geruch verschwindet und sich Pentamminhydroxycobaltibarbital (HI) bildet:
-l OH
Com(NH3)5 T
ni
Die blaue Kobaltverbindung hat folgende Zusammensetzung:
25% Dichlorodibarbital-Cobalto-Kalium [ci2(Barbital)2 Co]k275% Chlorohydroxydobarbital-Cobalto-Kalium [Cl(OH)(Barbital)2Co] Kj
Bei der Ausführung der Kobaltfarbreaktion muss man darauf achten,dass das Testmaterial und die verwendeten Lösungen vollkommen wasser¬
frei sind und Barbiturat, Kobaltsalz und basische Komponente in einem op¬timalen Verhältnis zueinander stehen. Die Beständigkeit und Intensität der
Färbung hängen wesentlich von der Art der verwendeten Base ab. Nach
Kozelka und Tatum (112) besteht eine stöchiometrische Beziehung zwi¬
schen Kobalt und Barbiturat in der Weise, dass sich vermöge der acht ko-
ordinativen Valenzen des Kobalts acht Di-imid-barbiturat-Moleküle bzw.
vierMono-imid-barbiturat-Moleküle mit einem Kobaltatom zu einem farbi¬
gen Komplex verbinden. Die Funktion der Base besteht nach Ansicht dieser
Autoren lediglich darin, durch Einstellen eines bestimmten pH-Wertes eine
verschiedene Stabilität der Komplexe zu bedingen.
Riley und Mitarbeiter (113) wiesen nach, dass von möglichenAbbauprodukten des Amytals (Aethyl-isoamyl-barbitursäure) nur Isoamyl-aethylessigsäure mit Kobaltacetat und Isopropylamin eine Färbung gab. Da
dieser Körper keine Imidgruppierung enthält, ist es nach Ansicht dieser
Autoren klar, dass die Färbung in diesem Falle nicht auf die Imidgruppezurückzuführen ist.
Aus Untersuchungen von Go m ah r und Kresbach (114) über die
Zusammensetzung des gefärbten Reaktionsproduktes geht hervor, dass die
Reaktion nicht nach den von Zwikker (111) und Kozelka und Tatum
(112) gegebenen Erklärungen verläuft. Sie erhielten bei der direkten Um¬
setzung von Allyl - isopropyl-N-methyl-barbitursaurem und diaethylbarbi-tursaurem Natrium mit wässeriger Kobalt-H-Lösung einen Niederschlag des
112) Kozelka und Tatum, J. Pharmacol. Exptl.Therap. 59, 54(1937)113) Riley, Krause, Steadman, Hunter und Hodge, Proc. Soc.
Exptl. Biol.Med. 45, 424(1940)114)Gomahr und Kresbach, Sc.Pharm. 19, 148(1951)

- 76 -
entsprechenden schwerlöslichen Co-ü-Cg-dialkyl-barbiturates, Co (Allyl-isopropyl - N - methyl - barbitursäure)2 bzw. Co (Diaethylbarbitursäurejo.
Beim Behandeln mit einer organischen Base, wie z. B. Pyridin, des Co-rf-
C5-Dialkylbarbiturates bildet sich offenbar eine Verbindung zweiter Ord¬
nung, ein tiefgefärbter Komplex, der mit wasserfreien organischen Lö¬
sungsmitteln entsprechend gefärbte Lösungen gibt und somit für eine Farb¬
reaktion sehr gut brauchbar ist. Dieser Komplex wird besonders leicht
durch Hydrolyse zerstört, wodurch die besondere Empfindlichkeit der Nach¬
weisreaktion gegenüber wasserhaltigen Lösungsmitteln erklärt erscheint.
Es ist nach den Autoren die Annahme berechtigt, dass es sich bei der Ko¬
balt-Reaktion der Barbiturate um die Bildung eines Komplexes handelt, in
dem die Barbiturat-Moleküle durch Hauptvalenzen an das Zentralatom Ko¬
balt gebunden sind, die basische Komponente bei der Nachweisreaktion
aber die Liganden der ersten Sphäre liefert. Der Austausch der Ligandenbewirkt eine Aenderung des Farbtones, was die mannigfaltigen Ergebnissebei der Verwendung verschiedener Basen erklärt. Die Notwendigkeit, die
Reaktionspartner in einem gewissen optimalen Verhältnis zur Reaktion zu
verwenden, erklären sich die Autoren so, dass sich bei einem Ueberschuss
an anorganischer Base basisches Kobaltsalz bzw. -hydroxyd, bei einem
Ueberschuss an organischer Base Verbindungen von der Art des Dipyridin-cobaltochlorids o.a. bilden, d.h. es ist nötig, die Reaktionspartner in
einem Verhältnis zu verwenden, welches der stöchiometrischen Zusam¬
mensetzung des Komplexes Kobalt(Base)n. (Barbitrat^ entspricht. Der Un¬
terschied im Verhalten der N-substituierten Barbiturate zu dem der nur
C5 - disubstituierten wird darin zu suchen sein, dass bei letzteren zwei
koordinative Valenzen des Kobalts bereits in Art eines innerkomplexenSalzes an den Iminogruppen der zwei Barbiturat-Moleküle abgesättigt wer¬
den.
Parri (109) benützte zum Nachweis von Veronal ein Kobalt-Et-Salz
und Ammoniak in alkoholischer Lösung, wobei eine rote, etwas nach violett
tendierende Farbe auftrat. Zwikker (110) verwendete als Lösungsmittelwasserfreies Methanol und als Base eine gesättigte Lösung von Barium¬
oxyd. Bodendorf (115) ersetzte das Methanol durch absoluten Aethylal-kohol und die Base durch Barium-, Kalium- oder Natriumhydroxyd. Für
die quantitative Bestimmung benutzte er als Base jedoch Piperidin, während
Rosenthaler (116) hierzu Piperazin vorzog. Oettel (117) anderseits
arbeitete mit methanolischen Kobaltacetat- und Lithiumhydroxyd-Lösungen-Flo tow (118) kocht 1-2 mg des Barbitursäure-Derivates mit etwa 1 cm
einer 0, 2%igen Kobalt - Methanol - Lösung auf, setzt etwas Borax zu und
kocht nochmals auf. Es entsteht eine veilchenblaue, schwach fluoreszie¬
rende Färbung, die recht beständig ist und auf Zusatz von einem TropfenPyridin in ein kräftiges Rotviolett umschlägt.
115) Bodendorf, Arch.Pharm. 270, 290(1932)116) Rosenthaler, "ToxikologiscFe Mikroanalyse" (1935)117) Oettel. Arch.Pharm. 274, 1(1936)118) Flotow, Pharm. ZenträlKT 88, 198 (1949)

- 77 -
Mit Isopropylamin als Alkalisierungsmittel arbeiten Kopp any i und
Mitarbeiter (119), Raventos (120), Riley und Mitarbeiter (113),Cohen (121), Matt son (122). Einige von diesen Autoren verwenden diese
Reaktion ebenfalls zur quantitativen Bestimmung. Rasmussen und
Jerslev (123) verwenden als Base Isobutylamin, als Lösungsmittel Chlo¬
roform und absolutes Methanol und wasserfreies Kobaltacetat. Go m ah r
und Kresbach (114) untersuchten das Verhalten der Barbiturate unter
Verwendung von Kobaltnitrat und verschiedenen anorganischen und organi¬schen Basen, wobei sich, besonders zu photometrischen Messungen, eine
Mischung von 5 T. 10%igem Piperidin und 1 T. 5%igem Ammoniak in ab¬
solutem Methanol als sehr günstig erwies.
Um die Beständigkeit der Farbreaktion zu erhöhen, verfuhr Robles
(124) folgendermassen: Das Barbiturat wird in Chloroform gelöst, 3 Tropfeneiner 5%igen wässerigen Lösung von Kobaltnitrat, 1 Tropfen 0,1 n Kali¬
umcyanid-Lösung und 3 Tropfen Pyridin zugesetzt. Beim Stehen bildet sich
eine kirschrote Färbung, die 30 Tage stabil blieb und durch Wasserzusatz
nur unwesentlich beeinflusst wurde. Einen Zusatz von Kaliumcyanid bei der
Kobaltreaktion verwenden auch Paget und Desodt (125,126) und Per-
rotti (127).
Um die Erfassungsgrenze von Barbitursäure-Derivaten mit Hilfe der
Kobalt-Reaktion zu erhöhen, verwendet Seiles (128) Filterpapierstreifen,die mit 1 %iger alkoholischer Kobaltnitrat-Lösung getränkt und dann ge¬
trocknet werden. Der Streifen wird dann in eine weisse Porzellanschale
gebracht. Mit einer Kapillarpipette tropft man dann einige Tropfen der in
Alkoholoder Aether gelösten Substanz darauf und gibt daneben einige Tropfen5- oder 10%iges Ammoniak. Bei Gegenwart von einem Barbiturat tritt die
charakteristische Färbung auf.
Kresbach und Gomahr (129) tropfen in vorgezeichnete Kreise
auf Filterpapier einen Tropfen einer l%igen Lösung von Kobaltnitrat in ab¬
solutem Methanol. Die, die zu untersuchende Substanz enthaltende, Chloro-
formlösung wird mit Pyridin bis zu einem Gehalt von 0, 5 % versetzt und
im Vakuum auf 0,5 cm^ eingeengt. Diese Menge wird nun vorsichtig auf
die Reaktionsflächen aufgetropft, wobei man zum Zweck einer weitgehendenAnreicherung Tropfen für Tropfen eintrocknen lässt. Nach kurzem Trock¬
nen setzt man diese Stellen der Einwirkung von Ammoniakdämpfen aus. In
Gegenwart von Barbituraten tritt eine Blauviolettfärbung ein.
119) Koppanyi, Dille, Murphy und Krop, J.Am.Pharm.Assoc. 23,1074 (1934)
~
120) Raventos, Brit.J. Pharmacol. 1, 210(1946)121) Cohen, Am.J.Pharm. 118, 40 (T946)122) Mattson und Holt, J."£m.Pharm.Assoc. 38, 55 (1949)123) Rasmussen und Jerslev, Dansk Tids. Farm. 25, 29(1951)124)Gurmendi Robles, Bol. Soc.Quim.Peru 15, 7ÖT1949)125) Paget und Desodt, J.Pharm.Chim. 18, 2TT7 (1933)126) Desodt, "Les barbituriques, leur toxicologie", thèse Nancy (1933)127) Perrotti, Boll. Chim. Farm. 78, 497(1939)128) Selles, Anales Soc. Espan. FésTQuim. 36, 115(1940)129) Kresbach und Gomahr, Wr. Med. Ws"chr. 63, 476(1951)

- 78 -
Neben den intensiv gefärbten Kobalt-Barbiturat-Komplexen findet die
Reaktion der Barbiturate mit Kupfer-II-Salzen in Gegenwart von organi¬schen Basen Anwendung bei toxikologischen Analysen. Zwikker (110)verwendete eine Mischung von Pyridin, 10%iger Kupfersulfatlösung und
Wasser. Die hierbei entstehenden rotvioletten Kupfer-Barbiturat-Pyridin-
Komplexe, für die er die Zusammensetzung Cu(Pyridin)2. (Barbiturat^feststellte, benützte er ursprünglich nur zur Reinigung von Rohbarbitura-
ten, wie sie bei toxikologischen Arbeiten anfallen. Rosenthaler (116)macht von dieser Reaktion in mikrochemischer Hinsicht Gebrauch. Von
Wagenaar (100) wird in einer Modifikation der Zwikker'sehen Reaktion
5%ige Kupfersulfatlösung und Aethylendiamin zur Identifikation der Barbi¬
turate verwendet. Jon s s on (130) arbeitet mit Kupferacetessigester und
Pyridin.
Flotow (118) stellte fest, dass die Kupfer-Pyridin-Komplexverbin-dungen von Zwikker (110) mit kräftig violetter Farbe von guter Bestän¬
digkeit in Chloroform löslich sind. Da sich die Nachweisbarkeitsgrenzedurch die Chloroformausschüttelung bis zu 0,1 mg Verona! steigern lässt,hält Flotow (118) diese Reaktion zur Vornahme von kolorimetrischen
Barbiturat-Bestimmungen für besser geeignet als die Kobaltreaktion. Go-
mahr und Kresbach (131) haben die Zwikker'sehe Reaktion in Form
einer Ausschüttelungsreaktion zum qualitativen Nachweis der Barbiturate
und anderer ähnlich gebauter pharmazeutischer Körper erprobt. Die Reak¬
tion wird so ausgeführt, dass zu einigen mg der zu untersuchenden Sub¬
stanz, die in einer Pyridin-Chloroform-Mischung gelöst werden, Kupfer¬sulfatlösung zugesetzt wird. Nach dem Umschütteln trat bei allen den Auto¬
ren zugänglichen Barbituraten - mit Ausnahme der Thiobarbiturate, die eine
Grünfärbung.geben - eine Violettfärbung der Chloroformschicht ein.
In Analogie zur Komplexbildung mit Kobalt-II-Salzen bilden Verbin¬
dungen, welche die Gruppe -CO-NH-CO- oder -CO-NH-CS- enthalten, auch
mit Kupfer-II-Salzen in alkalischem Milieu typisch gefärbte Lösungen von
Komplexverbindungen. Barbiturate mit der Gruppe - CO - NH - CO - gebenViolettfärbung, Thiobarbiturate und Thiourazile mit der Gruppe -CO-NH-
CS- geben grüngefärbte Chloroformlösungen; Hydantoine, die als cyclischeUreide der Glykolsäure die Gruppe -CO-NH-CO- nur unsymmetrisch auf¬
weisen, geben eine intensive Blaufärbung.
Neben der Kobalt-Reaktion verwenden Turfitt (40), Mot ha (132)und Parkes (133) als Gruppenreaktion auf Barbiturate selenige Säure.
Beim Erhitzen eines Gemisches von Barbiturat und seleniger Säure mit
konzentrierter Schwefelsäure tritt eine Grünfärbung auf. Man unterbricht
dann das Erhitzen und gibt in einer Porzellanschale zu dieser Lösung einigeTropfen Alkohol; es bildet sich dabei eine schön rote Trübung.
Dialkyl-, Halogenalkyl-, N-methylierte und Thio-Derivate lassen sich
mit der Kobaltfarbreaktion nicht unterscheiden, während es mit der Kupfer-sulfat-Pyridin-Chloroform-Reaktion möglich ist, zwischen Barbituraten und
Thio-Derivaten zu differenzieren.
130) Jonsson, Svensk Farm.Tids. 35, 659(1931)131) Gomahr und Kresbach, Sc.Ffiarm. 19, 154(1951)132) Motha, Boll.Chim. Farm. 73, 259 (1934T133) Parkes, Analyst 75, 448 (IÏÏ50)

- 79 -
B. Fällungsreaktionen
Als Fällungsreaktion kann ebenfalls die von Zwikker (110) einge¬führte und von Rosenthaler (116), Jon s son (130) und Wagenaar(100) modifizierte Kupferkomplex - Reaktion gebraucht werden. Daneben
finden sich besonders noch Fällungsreaktionen mit Quecksilbersalzen, von
denen Millon- und Denigès-Reagens die wichtigsten sind. Ueber die Nach¬
weismöglichkeiten von Barbituraten mit Hilfe von Millons Reagens berich¬
teten u.a. Paget und Desodt (134), Tilly (135), Paget und Tilly(136), Chatfield (137) und Stainier (138), wobei die Zusammensetzungdes Reagens bei den einzelnen Autoren nicht immer die gleiche ist. DenigèsReagens verwenden u.a. Stainier (138), Hargreaves und Nixon
(139) und Mühlemann und Bürgin (41).
Motley (140) erhält auf Zusatz von Mercuronitrat zu einer Lösungeines Barbiturates einen weissen bis grauen Niederschlag, der auf Zusatz
von Kaliumjodidlösung eine grünlich gefärbte kolloide Lösung gibt. Cohen
(121) gebraucht zum Nachweis von Barbitursäure-Derivaten bei toxikolo¬
gischen Analysen folgende Tests: Mercurinitrat-, Silbernitrat-, Mercuro¬
nitrat-, Mercurinitrat-Kaliumjodid- und Mercurinitrat-Ferrichlorid-Test.
Die mit Millons und Denigês' Reagens erhaltenen Niederschläge sind
weisslich bis grau und flockig oder kristallin. Einzelne Autoren verwenden
diese Reaktionen ebenfalls als Einzelreaktion auf bestimmte Barbitursäure-
Derivate, und zwar je nach Löslichkeit des Niederschlages in einem Ueber-
schuss des Reagens oder in Säuren je nach Farbe und Aussehen des Nie¬
derschlages.
Weitere Fällungsreaktionen, besonders Mikroreaktionen, wurden bei
den Möglichkeiten zur Identifikation von Mikrosublimaten (siehe S. 67) be¬
sprochen.
Von den aufgeführten Fällungsreaktionen erlaubt einzig die Reaktion
nach Wagenaar (100) mit Kupfersulfat und Aethylendiamin eine Unter¬
scheidungsmöglichkeit, indem mit diesem Reagens alle Barbiturate, mit
Ausnahme der N-methylierten, eine Fällung geben.
134) Paget und Desodt, Bull.Sc.Pharmacol. 39, 532(1932)135) Tilly, Bull.Sc.Pharmacol. 43^ 587(1936)136) Paget und Tilly, J.PharmTChim. 25, 222(1937)137) Chatfield, Pharm.J. 143, 346 (19337138) Stainier, J. Pharm. BeTgT 5, 26(1950)139) Hargreaves und Nixon, ~J.Am.Pharm.Assoc. 22, 1250(1933)140) Motley, J.Pharmacol. 72, 30(1941)

- 80 -
2. Einzelreaktionen
Hat man an Hand der Kobalt- oder Kupferfarbreaktion oder mit einer
Fällungsreaktion den bei der Ausschüttelung erhaltenen Rückstand als Bar-
bitursäure-Abkömmling gekennzeichnet, so muss sich hieran die Identifi¬
zierung des Barbiturates anschliessen.
Neben der Bestimmung von physikalischen Konstanten wie z.B.
Schmelzpunkt, kristalloptische Eigenschaften, Eutektika, die bei den Mög¬lichkeiten zur Identifizierung von Sublimaten eingehend behandelt wurden,können noch chemische Reaktionen zum Nachweis eines bestimmten Barbi¬
turates beitragen.
Fischer (44) führt das Verhalten der Barbiturate gegenüber Kalium-
permanganat in saurer Lösung, das Vorhandensein von Brom oder Schwefel
im Barbiturat-Molekül auf und kann so in Gruppen auftrennen.
De Wolff (141,142) schildert eine systematische Untersuchungs¬methode zur Identifizierung der gebräuchlichen Barbiturate nach folgendenGesichtspunkten:
1. je nach dem Vorhandensein von Brom, Schwefel oder keinem
dieser Elemente im Molekül kann man bereits drei Hauptgrup¬pen unterscheiden;
2. beim Erhitzen der Natriumsalze der Barbiturate in wässerigerLösung bilden sich unlösliche Acetylharnstoff - Derivate. Die
Zersetzungsgeschwindigkeit bei den einzelnen Derivaten ist
verschieden gross, was man als Unterscheidungsmerkmal be¬
nutzen kann;
3. der Kristallhabitus der Acetylharnstoff-Derivate kann zur Iden¬
tifikation ebenfalls herangezogen werden;
4. die Geschwindigkeit der Entfärbung von Permanganatlösung in
neutraler und saurer Lösung;
5. die Oxydation von Barbituraten, die eine Phenylgruppe enthal¬
ten, liefert Benzoesäure;
6. beim Erhitzen mit Lauge liefern die Barbiturate substituierte
Malonsäuren und Harnstoff; letzterer unterliegt weiterer Zer¬
setzung zu Ammoniak. Ist aber eine am Stickstoff gebundeneMethylgruppe zugegen, dann entsteht neben Ammoniak Mono-
methylamin, das mit Tetrachlorchinon nachgewiesen werden
kann.
Zur Kennzeichnung der einzelnen Barbiturate können die Substituentenin C-5-Stellung mit Hilfe chemischer Reaktionen identifiziert werden:
141) de Wolff, Pharm.Weekbl. 84, 537(1949)142) de Wolff, ActaPharm.Int.T, 107(1951)

- 81 -
Ungesättigte Radikale
Neben der Reduktion von Kaliumpermanganat in saurer Lösung, wie
sie Fischer (44) und de Wolff (141) verwenden, wird auch das Ver¬
halten von Kaliumpermanganat in alkalischer Lösung zum Nachweis von un¬
gesättigten Gruppen herangezogen. Mühlemann und Bürgin (41) re¬
duzieren in natronalkalischer Lösung.
Das Allylradikal CH2 = CH - CH2 - kann auf Grund der Doppel¬bindung zwei Halogenatome fixieren (143), z.B. Brom, um ein dibromier-
tes Derivat zu geben. Bei der Hydrolyse dieses Stoffes erhält man eine
Kette mit zwei alkoholischen Hydroxylgruppen, und zwar eine primäre und
eine sekundäre.
CH2Br - CHBr - CH2- CHgOH - CHOH - CH2 -
Die alkoholischen Gruppen lassen sich dann oxydieren, besonders unter
Einwirkung von Brom in der Wärme, und man erhält ein Derivat, das sich
vom Glyoxal ableiten lässt:
CHO - CO - CH2 -
Glyoxal und seine Derivate lassen sich leicht mit Farbreaktionen charakte¬
risieren. So erhält man mit phenolischen Körpern in Gegenwart von kon¬
zentrierter Schwefelsäure Kondensationsprodukte, die sich durch ihre
schöne Färbung kennzeichnen. Pesez (143) fand, dass Guaiacol, Salicyl-säure und Kreosotsäure-Derivate die charakteristischen Färbungen ergeben.
Ferner lässt sich das Allylradikal noch mit Aldehyden in Gegenwartvon konzentrierter Schwefelsäure kondensieren. Lagarce (144) und Tur-
fitt (40) verwenden Vanillin, Pesez (145) Formol, Salicylaldehyd, Pe¬
sez (145) und Turfitt (40) p-Dimethylaminobenzaldehyd.
Cyclisches Polymethylenradikal
Nach Pesez (146) geben Cyclopentenyl- und Cyclohexenyl-Derivatecharakteristische Farbreaktionen mit hydratisierter Schwefelsäure, Vanil¬
lin, Piperonal, p-Dimethylaminobenzaldehyd, Salicylaldehyd, Benzaldehyd,Furfurol, Phenol, Resorcin in salzsaurem und schwefelsaurem Milieu.
Deshusses (147), (148) nitriert zuerst den Ring und identifiziert dann das
nitrierte Produkt mit Hydroxylamin und mit m-Nitrobenzaldehyd. Die letzte
Reaktion erlaubt eine Differenzierung zwischen Phenyl- und cyclischemPolymethylenradikal.
143) Pesez, J.Pharm.Chim 25, 508(1937)144) Lagarce, J.Pharm.Chim. 12, 364(1930)145) Pesez, J.Pharm.Chim. 27,T47 (1938)146) Pesez, J.Pharm.Chim. '28", 379 (1938)147) Deshusses, Pharm.ActäTHelv. 19, 358(1944)148) Deshusses, Pharm.Acta Helv. ZlT 199(1945)
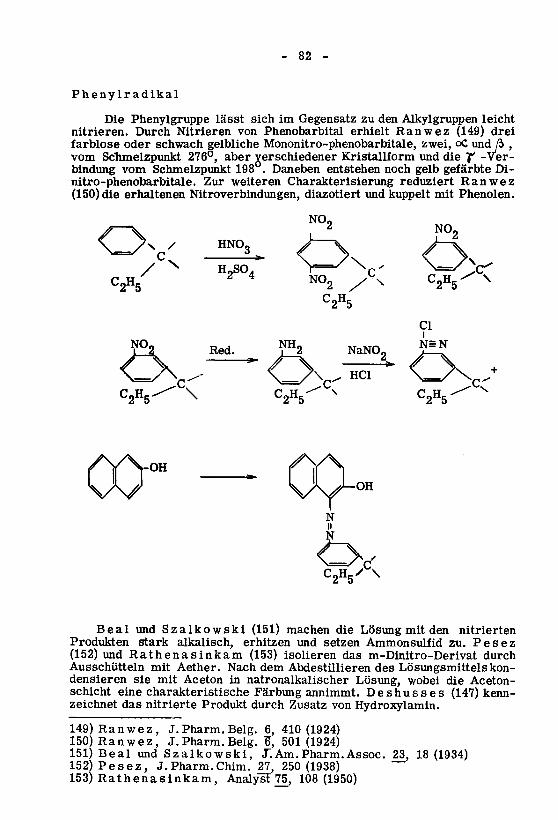
- 82 -
Phenylradikal
Die Phenylgruppe lässt sich im Gegensatz zu den Alkylgruppen leicht
nitrieren. Durch Nitrieren von Phénobarbital erhielt Ranwez (149) drei
farblose oder schwach gelbliche Mononitro-phenobarbitale, zwei, oC undJb ,
vom Scnmelzpunkt 276°, aber verschiedener Kristallform und die f -Ver¬
bindung vom Schmelzpunkt 198 . Daneben entstehen noch gelb gefärbte Di-
nitro-phenobarbitale. Zur weiteren Charakterisierung reduziert Ranwez
(150) die erhaltenen Nitroverbindungen, diazotiert und kuppelt mit Phenolen.
SS
C2H5
Beal und Szalkowski (151) machen die Lösung mit den nitrierten
Produkten stark alkalisch, erhitzen und setzen Ammonsulfid zu. Pesez
(152) und Rathenasinkam (153) isolieren das m-Dinitro-Derivat durch
Ausschütteln mit Aether. Nach dem Abdestülieren des Lösungsmittels kon¬densieren sie mit Aceton in natronalkalischer Lösung, wobei die Aceton-
schicht eine charakteristische Färbung annimmt. Deshusses (147) kenn¬
zeichnet das nitrierte Produkt durch Zusatz von Hydroxylamin.
149) Ranwez, J.Pharm. Belg. 6, 410(1924)150) Ranwez, J.Pharm. Belg. ïï, 501 (1924)151) Beal und Szalkowski, J".Am.Pharm.Assoc. 23, 18 (1934)152) Pesez, J.Pharm.Chim. 27, 250(1938)153) Rathenasinkam, Analyst 75, 108(1950)

- 83 -
Zur Erkennung des Phenylradikales oxydiert de Wolff (141) die am
C-5 mit einer Phenylgruppe substituierten Barbiturate in alkalischer Lösungmit Kaliumpermanganat; die dabei gebildete Benzoesäure trennt er durch
Sublimation ab und weist sie mikroskopisch nach.
Ekkert (154) unterscheidet zwischen Barbital und Phénobarbital
durch Behandeln mit Formol und Schwefelsäure. In Gegenwart von Phéno¬
barbital tritt in der Wärme eine weinrote Färbung auf.
S-haltige Derivate
Neben der Aufschlussmethodenach Lassaigne und Nachweis des Schwe¬
fels mit Bleiacetat oder mit Nitroprussidnatrium erhält man beim Ansäuern
einer alkalischen Lösung eines Thio - Derivates und Nitroprussidnatriumnach Laubie (155) einen roten, in Amylalkohol löslichen Farbstoff.
Eine andere Methode zur Identifizierung der einzelnen Barbiturate ist
die Darstellung von Derivaten. Derivate von o-, m- und p-Brombenzyl- und
Chlorbenzyl-Halogeniden wurden von Poe und Mitarbeitern (156) und
Castle und Poe (157) hergestellt. p-Nitrobenzyl-Derivate gewannen u.a.
Lyons und Dox (158), Hargreaves und Nixon (138), Jespersenund Larsen (159), Hultquist und Poe (160), Hultquist, Poe und
Witt (161,162), Castle und Poe (157), Poe und Mitarbeiter (156),Lillmann (163) und Reimers (164).
p-Nitrobenzyl-Derivate
Die Kondensation zwischen Barbiturat und p-Nitrobenzylchlorid ist nicht
nur bei den Imidowasserstoffatomen in Stellung 1 und 3 möglich, sondern auch
bei den MethylenwasserStoffatomen in Stellung 5.
H CO-NH.
H CO - NIK
Hieraus ergibt sich, dass bei der reinen Barbitursäure die Bildung eines
Tetraderivates möglich ist, nämlich 1,3, 5, 5-tetra-p-Nitrobenzyl-barbitur-säure folgender Formel
154) Ekkert, Pharm. Zentralh. 67, 481 (1926)155) Laubie, Bull.Trav.Soc.Pharm.Bordeaux fasc. 1(1949)156) Poe, Witt und Snodgrass, Mikrochemie 34, 235(1949)157) Castle und Poe, J.Am.Chem.Soc. 66, 1440~(1944)158) Lyons und Dox, J. Am. Chem.Soc. 5T7 288 (1929)159) Jespersen und Larsen, Dansk Tf3s.Farm. 8, 212(1934)160) Hultquist und Poe, Ind. Eng.Chem. Anal. Ed. 1_, 398(1935)161) Hultquist, Poe und Witt, Ind. Eng.Chem. Anal.Ed. 14. 219(1942)162) Hultquist, Poe und Witt, J. Am.Chem.Soc. 67, 688(1945)163) Lillmann, Analyst 75, 626 (1950)164) Reimers, Dansk Ti3s. Farm. 15, 281(1941)
- 84 -
R CO - N-R
C C = -CH2-C6H4-NO2IT CO - N - R
Bei den Monoalkylbarbitursäuren besteht die Möglichkeit zur Bildung eines
Tri- und bei den Dialkylbarbitursäuren eines Disubstitutionsproduktes. Bei
den N-methylierten Derivaten besteht nur die Möglichkeit einer Monosubsti¬
tution.
Um bei den Dialkylbarbituraten die Möglichkeit einer Monosubstitution
auszuschliessen, bringt man einerseits die zur vollständigen Substitution
aequivalente Menge inAnwendung und anderseits behandelt man das Reaktions¬
produkt mit Natronlauge, worin die Monoderivate mit freiem Imidowasserstoff
löslich sind, während die Disubstitutionsprodukte hierin unlöslich sind.
Von dentherapeutisch verwendeten Barbitursäure-Derivaten geben dem¬nach alle, sowohl die Dialkyl- als auch die Dialkyl-N-methylierten Derivate
mit p-Nitrobenzylchlorid Di-resp. Monosubstitutionsprodukte.
Die p-Nitrobenzyl-Derivate der Barbiturate sind weisse, kristalline
Pulver, löslich in Chloroform und schwer löslich in Wasser und Weingeist.Die von den verschiedenen Autoren bestimmten Schmelzpunkte dieser Ver¬
bindungen sind in Tabelle 17 zusammengestellt.
Tabelle 17
Schmelzpunkte der p-Nitrobenzyl-Derivate
Substltuenten
Rl
Aethyl-
aethyl-
aethyl-
aethyl-
aethyl-
propyl-
aethyl-
aethyl-
allyl-
allyl-
allyl-
allyl-
allyl-
isopropyl-
sec-butyl -
1 -methyl -
butyl -
methyl -
aethyl-
aethyl-
allyl-
allyl-
methyl-
aethyl-
R2
aethyl-
isopropyl-
n-butyl-
1-methyl-butyl-
isoamyl-
propyl-
allyl-
1 -methyl-bu-
tenyl-
allyl-
isopropyl-
n-butyl-
Isobutyl-
1-methyl-butyl-
ß -bromallyl-
ß -bromallyl-
^-bromallyl-
phenyl-
phenyl-
cyclohexenyl-
cyclopentenyl-
phenyl-
cyclohexenyl-
phenyl-
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CH3-
CH3-
Jespersen u.
Larsen(159)
korr.
193,5°
148,5°
145,5°
182,3°196,3°
192, 5°
192,0°
127, 5°
200,5°191,5°
197,0°183,5°
196,0°
152,0°114,5°114,5°
Hargreavea u.
Nixon (139)
unkorr.
192°
160°
146°
142°
138°
190°
191°
182°
195°
Lyons u.
Dox (158)
korr.
193,5°
148, 5°
145, 5°
196,3°
192, 5°
192,0°
127,5°
200,5°
191, 5°
183, 5°
197,0°
114, 5°
114, 5°
Reimers
(164)
mikro
195°
157, 5°
150,5°
150°
184, 5°
199°
195°
193°
134°
179,5°
206,5°196°
188°
184°
198, 5°
185°
154, 5°
Hultquis u.
Mitarb. (160)
unkc
a
193°
157°
128°
151°
172°
192°
178°
204°
184°
rr.
b
192°
157°
129°
153°
169°
191°
180°
206°
182,5°
Castle u.
Poe (157)
korr.
a
131,5°
158°
172°
181,5°
b
132, 5°
163°
178°
185°
a : Block
b : Röhrchen
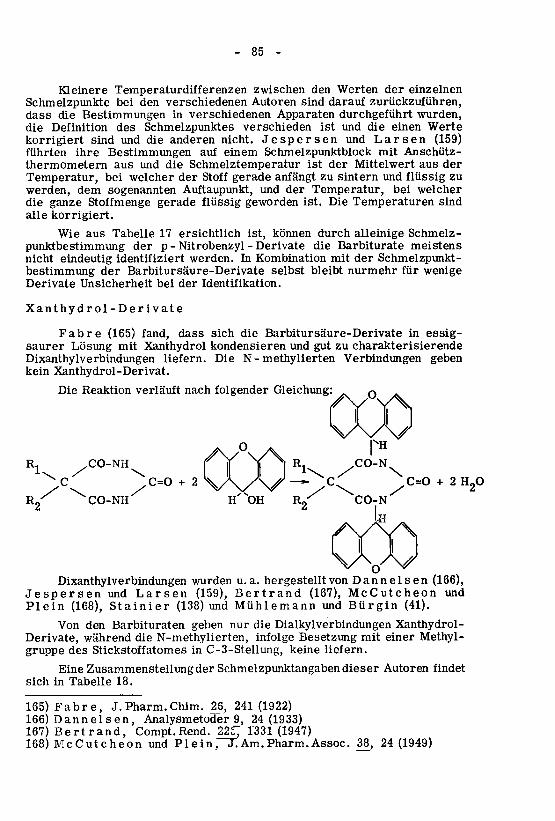
- 85 -
Kleinere Temperaturdifferenzen zwischen den Werten der einzelnen
Schmelzpunkte bei den verschiedenen Autoren sind darauf zurückzuführen,dass die Bestimmungen in verschiedenen Apparaten durchgeführt wurden,die Definition des Schmelzpunktes verschieden ist und die einen Werte
korrigiert sind und die anderen nicht. Jespersen und Larsen (159)führten ihre Bestimmungen auf einem Schmelzpunktblock mit Anschütz-
thermometern aus und die Schmelztemperatur ist der Mittelwert aus der
Temperatur, bei welcher der Stoff gerade anfängt zu sintern und flüssig zu
werden, dem sogenannten Auftaupunkt, und der Temperatur, bei welcher
die ganze Stoffmenge gerade flüssig geworden ist. Die Temperaturen sind
alle korrigiert.
Wie aus Tabelle 17 ersichtlich ist, können durch alleinige Schmelz¬
punktbestimmung der p - Nitrobenzyl - Derivate die Barbiturate meistens
nicht eindeutig identifiziert werden. In Kombination mit der Schmelzpunkt¬bestimmung der Barbitursäure-Derivate selbst bleibt nurmehr für wenigeDerivate Unsicherheit bei der Identifikation.
Xanthydrol-Derivate
Fabre (165) fand, dass sich die Barbitursäure-Derivate in essig¬saurer Lösung mit Xanthydrol kondensieren und gut zu charakterisierende
Dixanthylverbindungen liefern. Die N - methylierten Verbindungen gebenkein Xanthydrol-Derivat.
Die Reaktion verläuft nach folgender Gleichung:
o
Dixanthylverbindungen wurden u.a. hergestellt von Dannelsen (166),Jespersen und Larsen (159), Bertrand (187), McCutcheon und
Plein (168), Stainier (138) und Mühlemann und Bürgin (41).
Von den Barbituraten geben nur die Dialkylverbindungen Xanthydrol-
Derivate, während die N-methylierten, infolge Besetzung mit einer Methyl¬
gruppe des Stickstoffatomes in C-3-Stellung, keine liefern.
Eine Zusammenstellung der Schmelzpunktangaben dieser Autoren findet
sich in Tabelle 18.
165) Fabre, J.Pharm.Chim. 26, 241 (1922)166) Dannelsen, Analysmetoder £, 24(1933)167) Bertrand, Compt.Rend. 22b, 1331(1947)168) McCutcheon und Plein,T.Am.Pharm.Assoc. 38, 24(1949)
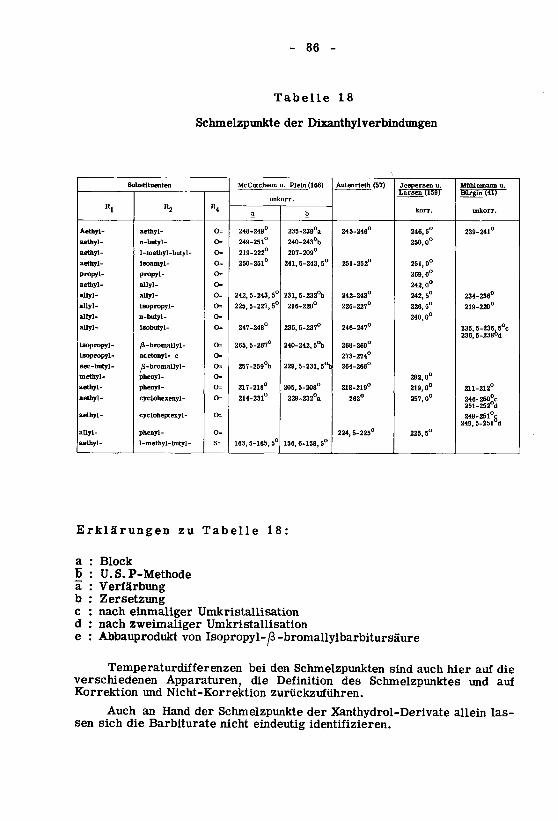
- 86 -
Tabelle 18
Schmelzpunkte der Dixanthylverbindungen
Substltuenten
"l
Aethyl-
aethyl-
aethyl-
aethyl-
propyl-
aethyl-
allyl-
allyl-
allyl-
allyl-
Isopropyl-
lsopropyl-
sec-butyl-
methyl-
aethyl-
aethyl-
aethyl-
allyl-
aethyl-
"2
aethyl-
n-butyl-
1-methyl-butyl-
isoamyl-
propyl-
allyl-
aUyl-
isopropyl-
n-butyl-
isobutyl-
ß -bromallyl-
acetonyl- e
y3-bromallyl-phenyl-
phenyl-
cyclohexenyl-
cycloheptenyl-
phenyl-
1-methyl-butyl-
R4
0=
0-
O=
0«
0=
0-
O«
0=
0=
Cfe
O=
O=
0.
O=
O=
0=
(k
O=
s=
McCutcheon j. Plein (168)
unkorr.
a
248-249°
249-251°
219-222°
250-251°
242,5-243,5°225, 5-227, 5°
247-248°
265, 5-267°
257-259°b
217-218°
214-231°
163, 5-165, 5°
b
235-239°a
240-243°b
207-209°
241, 5-243, 5°
231, 5-232°b
216-220°
235, 5-237°
240-242, 5°b
229, 5-231,5°t
205,5-208°229-232°a
156, 6-158, 5°
Autenrleth (57)
245-246°
251-252°
242-243°
226-227°
246-247°
268-269°
273-274°
264-266°
218-219°
263°
224, 5-225°
Jespersen u.
Larsen u->uj
korr.
246,5°
250,0°
251,0°
269,0°242,0°242,5°
226,5°
240,0°
282,0°219,0°
257,0°
225,5°
MüMemann u.
unkorr.
239-241°
234-236°
219-220°
235, 5-236, 5°c236,5-238°d
211-212°
246-250°c251-252°d
248-251°c249,5-251°d
Erklärungen zu Tabelle 18:
Block
U. S. P-Methode
VerfärbungZersetzungnach einmaliger Umkristallisation
nach zweimaliger Umkristallisation
Abbauprodukt von Isopropyl-/3 -bromallylbarbitursäure
Temperaturdifferenzen bei den Schmelzpunkten sind auch hier auf dieverschiedenen Apparaturen, die Definition des Schmelzpunktes und aufKorrektion und Nicht-Korrektion zurückzuführen.
Auch an Hand der Schmelzpunkte der Xanthydrol-Derivate allein las¬
sen sich die Barbiturate nicht eindeutig identifizieren.

- 87 -
Osazone
Zur Charakterisierung der Diaethylbarbitursäure stellte Parri (109)das Phenylhydrazon dar und gab einige Farbreaktionen an, mit denen man
dieses Derivat identifizieren kann.
Identifikation mit Hilfe von Roentgenstrahlen
Die einzelnen Methoden und das Prinzip der Pulvermethode nach
De bye und Scherrer (108) sind auf Seite 71 beschrieben. Obschon Hull
(169) schon 1919 zeigte, dass man Roentgendiagramme zur Identifikation
von Substanzen mit gutem Erfolg anwenden kann, dauerte es ziemlich lange,bis diese Methoden allgemein angewandt wurden.
Tso-Yueh Huang (106) untersuchte mit Hilfe von RoentgenstrahlenPhénobarbital, Allobarbital, Cyclobarbital, Hexobarbital, Allyl-isopropyl-barbitursäure, Barbital, die Modifikationen der polymorphen Substanzen
und das Verhalten eines Allobarbital - Allyl - isopropylbarbitursäure-Ge¬misches. Als Charakteristika führt er die sin^ ©-Werte und die relative
Intensität der Reflexlinien auf.
Papierchromatographische Trennung und Identifizierung
Bei der Extraktion von Barbitursäure - Derivaten aus biologischemMaterial müssen gewöhnlich Mikromethoden zu ihrer Identifikation ange¬
wendet werden. Wickström und Salvesen (170) führten den Mikro-
nachweis mit Hilfe der Papierverteilungschromatographie durch. Sie trenn¬
ten acht Barbiturate und zwei Thiobarbiturate mit drei verschiedenen Lö¬
sungssystemen und geben die beobachteten Rp-Werte an. Mit Hilfe ver¬
schiedener Spray - Reagenzien, wie ammoniakalische Silbernitraüösung,Diaet ylamin in Methanol und gesättigte Kupfersulfat - Methanol - Lösung,differenzieren sie die Barbiturate, welche ungefähr die gleichen Rp-Werteunter denselben Bedingungen zeigen. Die Autoren halten diese Resultate für
vorteilhaft, wenn die Barbiturate aus biologischem Material isoliert wur¬
den und der Mikronachweis nötig wird.
Algeri und Walker (171) untersuchten zehn der gebräuchlichstenBarbiturate mittels einer Modifikation der aufsteigenden Methode von Wil¬
liams und Kirby (172). Als Lösungsmittel diente frisch bereiteter, mit
5 n Ammoniak gesättigter n - Butylalkohol. Die Identifizierung erfolgtemittels UV-Licht (Pentothai), l%iger Silberacetatlösung und 0, l%igemThiodiphenylcarbazon in 95%igem Alkohol, mit Lemaire - Reagens und
Thiodiphenylcarbazon (Amytal, Allobarbital, Pentobarbital, Ortal, Seconal,Phénobarbital) oder mit 10%iger Kaliumpermanganat-Lösung (Hexobarbital,Allobarbital, Seconal, Pentothai). Daneben wurden noch Barbital und Neonal
untersucht und die R„-Werte der einzelnen Barbiturate werden angegeben.r
169) Hull, J.Am.Chem.Soc. 41, 1168 (1919)170) Wickström und Salvesen, J.Pharm.Pharmacol. 4, 98 (1952)171) Algeri und Walker, Am.J.Clin.Path. 22, 37 (19527172) Williams und Kirby, Science New York~l07, 481 (1948)

- 88 -
V. ARBEITSPLAN
1. Auswahl der zu bearbeitenden Barbiturate
Gegenstand der nachfolgenden Untersuchungen ist die analytische Be¬
arbeitung jener Barbiturate, die bis heute eine bedeutende Stellung in der
Schlafmittel-Therapie einzunehmen vermochten. Die Wahl einer beschränk¬
ten Gruppe aus der grossen Zahl der zur Verfügung stehenden Derivate
erfolgte nach folgenden Gesichtspunkten:
1. Es wurden nur die Verbindungen berücksichtigt, die sich in der
Therapie gut eingeführt haben und die heute auf Grund ihrer Ei¬
genschaften als bewährte Narcotica, Hypnotica und Sedativa gelten.
2. Berücksichtigt wurde ferner eine Auswahl C-5-substituierter Bar¬
biturate in dem Sinne, dass Derivate mit Dialkyl-, Halogenalkyl-,aromatischen und alicyclischen und N-methylierten Substituenten
sowie Thioderivate zur Untersuchung gelangten.
In der Wahl einer Verbindung wurde auch auf ihre Bedeutung in der
modernen Literatur, wie auch auf ihre Berücksichtigung in den offiziellen
Arzneibüchern, Ergänzungswerken, wie Pharmakopöen, British Pharma¬
ceutical Codex, New and Nonofficial Remedies etc., abgestellt.
Hieraus ergibt sich folgende Auswahl:
Bezeichnung
Barbital
SonérylPentobarbital
Allobarbital
Alurate
SandoptalNoctal
Pernocton
Phénobarbital
CyclobarbitalMedomin
CyclopalNarconumal
Hexobarbital
Methylpheno-barbital
Eunarcon
ThiopentalKemithal
Rl
aethyl-aethyl-aethyl-allyl-allyl-allyl-isopropyl-sec-butyl-aethyl-aethyl-aethyl-allyl-allyl-methyl-
aethyl-
isopropyl-aethyl-allyl-
Substituent en
R2
aethyl-n-butyl -
1-methyl-butyl-allyl-isopropyl-isobutyl-ß -bromallyl-
ß -bromallyl-phenyl-cyclohexenyl-cycloheptenyl-cyclopentenyl-isopropyl-cyclohexenyl-
phenyl-
ß -bromallyl-1-methyl-butyl-cyclohexenyl-
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CH,
CHg
CHgCH,H^H-
R4
O=
O=
o=
0=
0=
0=
o=
o=
o=
o=
0=
o=
0=
o=
o=
o=
s=
s=

- 89 -
2. Arbeitsprogramm
Die Untersuchungen wurden nach folgendem Arbeitsprogramm durch¬
geführt:
I. Isolierung der Barbiturate aus Arzneizubereitungendurch
1. Ausschüttelung einer wässerig - weinsauren Lösung oder Auf¬
schwemmung mit Aether:
Hier handelt es sich darum, zu untersuchen, ob man die Barbitur-
säure - Derivate aus einer wässerig - weinsauren Lösung quantitativ mit
Aether extrahieren kann und ob die Konzentration der Weinsäure (pH) und
die Menge der wässerigen Phase bei der Aetherextraktion eine wesentliche
Rolle spielen.
2. Ausschüttelung der Aetherlösung mit verschiedenen Alkalien:
Zur weiteren Abtrennung der Barbiturate von anderen Arzneistoffen
wird die Aetherlösung mit 2%iger Natriumkarbonatlösung ausgeschüttelt.Hierbei stellte sich die Frage, ob:
a. die Konzentration von 2% und die verwendeten Mengen Na¬
triumkarbonatlösung (20, 20 und 15 cm«*) zu einer quantitativenExtraktion führen,
b. man eine stärkere Konzentration oder sogar ein stärkeres
Alkali verwenden muss oder
c. sogar ein schwächeres Alkali die quantitative Extraktion er¬
möglicht.
Aus dem Verhalten der Barbiturate beim Ausschütteln mit verschie¬
den starken Alkalien war zu beurteilen, ob eine gewisse Auftrennung auf
diesem Wege möglich ist.
II. Verhalten der Barbiturate bei der Mikrosublimation
im Vakuum
1. Abklärung der Sublimationsbedingungen2. Untersuchung und Identifizierung der Sublimate
III. Ueberprüfung der Gruppenreaktionen
Um einen sicheren Beweis für das Vorhandensein eines Barbiturates
zu haben, sollen die in der Literatur angegebenen Gruppenreaktionen auf
ihre Brauchbarkeit geprüft werden. Von grossem Wert wäre eine Reaktion,die nur auf Barbiturate anspricht und nicht auch auf andere, in derselben
Ausschüttelungsgruppe auftretende Arzneistoffe.

90 -
IV. Ueberprüfung von Einzelreaktionen
Da in manchen Fällen die Identifikation eines bestimmten Barbitur-
säure-Derivates mit einfachen physikalischen Methoden, wie z. B. Schmelz¬
punktbestimmung, infolge eines nur sehr geringen Unterschiedes zu anderen
Derivaten nicht möglich ist und mikrochemische Reaktionen und Manipula¬tionen nicht immer durchgeführt werden können, soll man auf chemische
Reaktionen zurückgreifen können. Farbreaktionen können befriedigendeResultate liefern, doch ist gewöhnlich eine Unsicherheit in der Beurteilungdes Farbtones vorhanden. Die Herstellung von Derivaten, vorausgesetzt,dass genügend Substanz zur Verfügung steht, eignet sich deshalb besser zur
Identifizierung eines bestimmten Derivates.
Anhand der Untersuchungsresultate sollte es möglich werden, einen
brauchbaren Analysengang zur Isolierung und zum qualitativen Nachweis
der bearbeiteten Barbiturate vorzuschlagen.

- 91 -
C. SPEZIELLER TEIL
I. VERHALTEN DER BARBITURATE BEIM ISOLIEREN AUS
ARZNEIZUBEREITUNGEN
Um die Barbiturate aus Arzneigemischen und -Zubereitungen zu iso¬
lieren, verwendeten wir das Ausschüttelungsverfahren. Wir lösten das be¬
treffende Barbiturat in Wasser oder schwemmten es mit Wasser auf und
fügten Weinsäurelösung zu. In wässeriger Lösung dissoziieren die Barbi¬
turate in
Ketoform
Rl
^co-
co-
Enolform
N
OH
C C -O + H+
Ro CO - W
wobei die dissoziierte Form wasserlöslich, aber in organischen Lösungs¬mitteln unlöslich ist, die nicht dissoziierte Form hingegen in Wasser un¬
löslich, in organischen Lösungsmitteln löslich ist und somit mit diesen
ausschüttelbar ist. Um nun die Extraktion quantitativ ausführen zu können,müssen wir die Dissoziation zurückdrängen, was durch Erhöhung der Was¬
serstoffionenkonzentration möglich ist. Durch Säurezusatz drängen wir die
Barbiturate in die ausschüttelbare Nichtelektrolytform und können dann
quantitativ extrahieren. Hierbei ist die Stärke der zuzusetzenden Säure von
derjenigen der auszuschüttelnden abhängig.
Die wässerig-weinsaure Lösung schüttelten wir dann mit Aether aus
und extrahierten hierauf die Aetherlösung mit alkalischer Lösung, um die
Barbiturate von anderen Stoffen abzutrennen. Diese Extraktion erfolgt nach
dem Eder'sehen Analysengang mit einer 2%igen Sodalösung. Da andere
Autoren teils mit einem schwächeren, teils mit einem stärkeren Alkali
arbeiten, überprüften wir nun die Ausschüttelbarkeit einer Aetherlösungder Barbiturate mit verschiedenen Alkalien, um festzustellen, welches eine
quantitative Extraktion ermöglicht.

- 92 -
1. Ausschüttelung aus wässerig-weinsaurer Lösung
mit Aether
Eine genau gewogene Menge des Barbiturates wurde in 15 cm*' Wasser
gegeben und hierauf 15 cm^ 5%iger Weinsäurelösung zugesetzt und einigeMale umgeschüttelt. Dann wurde diese Lösung dreimal mit je 20 cm3 Aether
ausgeschüttelt. Die vereinigten Aetherlösungen wurden mit wasserfreiem
Natriumsulfat getrocknet und der Aether abdestilliert. Die Rückstände er¬
gaben die in Aether übergegangene Menge des Barbiturates, die in Tabelle
19 in Prozent der Einwaage aufgeführt ist.
Tabelle 19
Aus weinsaurer Lösung in Aether übergsgangene Menge Barbiturat
in % der Finwaage
Barbital
SonérylPentobarbital
Allobarbital
Alurate
SandoptalNoctal
Pernocton
Phénobarbital
CyclobarbitalMedomin
CyclopalNarconumal
Hexobarbital
MethylphenobarbitalEunarcon
ThiopentalKemithal
95,78%94, 20%95,87%94,83%96, 99%
97,07%98,43%97,40%96,81%96, 48%97,46%97,44%97, 36%95, 25%98,11%98, 23%93,18%94, 73%
Bei Verwendung von Natriumsalzen der Barbiturate verfuhren wir
gleich wie oben angegeben ist. Auf Zusatz derselben Menge Weinsäurelö¬
sung zu der wässerigen Lösung des Natriumsalzes fielen innerhalb kurzer
Zeit die freien Säuren aus, mit Ausnahme von Barbital, das in Lösungblieb. Die Ausschüttelungmit Aether lieferte uns Werte derselben Grössen-
ordnung wie mit den freien Säuren.
Für diese Versuche fanden wir, wie aus Tabelle 19 ersichtlich ist.
die verwendete Menge Weinsäure (Gesamtkonzentration von 2, 5%, pH 2,05)als ausreichend für die quantitative Extraktion der Barbiturate mit Aether.
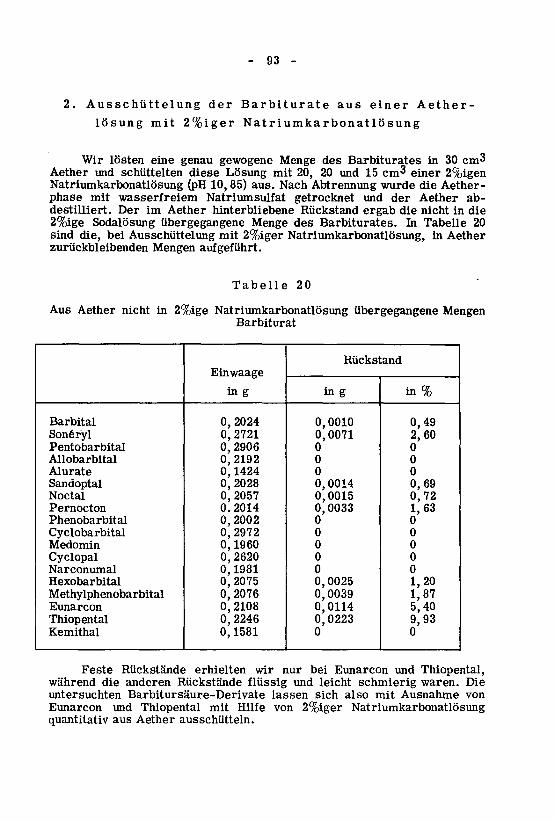
- 93 -
2. Ausschüttelung der Barbiturate aus einer Aether-
lösung mit 2%iger Natriumkarbonatlösung
Wir lösten eine genau gewogene Menge des Barbiturates in 30 cm3Aether und schüttelten diese Lösung mit 20, 20 und 15 cm^ einer 2%igenNatriumkarbonatlösung (pH 10,85) aus. Nach Abtrennung wurde die Aether-
phase mit wasserfreiem Natriumsulfat getrocknet und der Aether ab¬
destilliert. Der im Aether hinterbliebene Rückstand ergab die nicht in die
2%ige Sodalösung übergegangene Menge des Barbiturates. In Tabelle 20
sind die, bei Ausschüttelung mit 2%iger Natriumkarbonatlösung, in Aether
zurückbleibenden Mengen aufgeführt.
Tabelle 20
Aus Aether nicht in 2%ige Natriumkarbonatlösung übergegangene MengenBarbiturat
Barbital
SonérylPentobarbital
Allobarbital
Alurate
SandoptalNoctal
Pernocton
Phénobarbital
CyclobarbitalMedomin
CyclopalNarconumal
Hexobarbital
MethylphenobarbitalEunarcon
ThiopentalKemithal
Einwaage
in g
0, 2024
0,27210, 2906
0,21920,14240, 2028
0, 2057
0.2014
0, 2002
0, 2972
0,19600, 2620
0,19810,20750, 2076
0,21080,22460,1581
Rückstand
in g
0,00100,00710
0
0
0,00140,00150.0033
0
0
0
0
0
0,00250,00390,01140,02230
in%
0,492,600
0
0
0,690,721,630
0
0
0
0
1,201,875,409,930
Feste Rückstände erhielten wir nur bei Eunarcon und Thiopental,während die anderen Rückstände flüssig und leicht schmierig waren. Die
untersuchten Barbitursäure-Derivate lassen sich also mit Ausnahme von
Eunarcon und Thiopental mit Hilfe von 2%iger Natriumkarbonatlösungquantitativ aus Aether ausschütteln.

- 94 -
3. Ausschüttelung der Barbiturate aus einer Aether-
lösung mit 5 %iger Natriumbikarbonatlösung
Eine genau gewogene Menge des entsprechenden Barbiturates wurde
in 30 cm3 Aether gelöst und diese Lösung hierauf mit 20, 20 und 15 cm>>
einer 5%igen Natriumbikarbonatlösung (pH 8,08) ausgeschüttelt. Die Aether-
phase wurde nach Abtrennung mit wasserfreiem Natriumsulfat getrocknetund der Aether abdestilliert. Aus dem Aetherrückstand berechneten wir die
in 5%ige Natriumbikarbonatlösung übergegangenen Mengen Barbiturat, die
in Tabelle 21 zusammengestellt sind.
Tabelle 21
Aus Aether in 5%ige Natriumbikarbonatlösung übergegangeneMengen Barbiturat
Barbital
SonérylPentobarbital
Allobarbital
Alurate
SandoptalNoctal
Pernocton
Phénobarbital
CyclobarbitalMedomin
CyclopalNarconumal
Hexobarbital
MethylphenobarbitalEunarcon
ThiopentalKemithal
55,92%24, 40%24, 60%49, 67%33,44%32, 60%27, 55%29, 38%43, 74%42, 64%27,88%41,04%30,76%34,34%33,99%44,40%14,15%32,18%
Wie aus Tabelle 21 ersichtlich ist, ist eine quantitative Extraktion
aus Aether mit Natriumbikarbonatlösung nicht möglich. Im allgemeinenkönnen wir sagen, dass die Ausschüttelbarkeit mit der Löslichkeit des be¬
treffenden Barbiturates in Wasser zunimmt. Die Verwendung einer ge¬
sättigten Bikarbonatlösung (pH 8,15) lieferte uns ebenfalls keine günstigerenResultate.

- 95 -
4. Einfluss der Dissoziation der Barbiturate
Da wir sowohl bei der Ausschiittelung der Aetherlösung mit 2%igerNatriumkarbonat- als auch mit 5%iger Natriumbikarbonatlösung gewisseUnterschiede im Verhalten der Barbiturate feststellten, interessierte es
uns, zu erfahren, ob dieses unterschiedliche Verhalten durch Unterschiede
in den Dissoziationseigenschaften der einzelnen Barbitursäure - Derivate
erklärt werden könnte. Dies veranlasste uns, weil keine Angaben über die
Dissoziationskonstanten der von uns untersuchten Barbiturate zu finden
waren, diese Konstanten selbst zu bestimmen.
a. Theoretisches
Die Säure HA zeigt in Wasser folgendes Gleichgewicht:
HA + HÖH *.* HgO+ + A"
und die Gleichgewichtskonstante wird durch die Gleichung
a H3O
gegeben. Da nun aHqO+ &leicn ist der Wasserstoffionenkonzentration, so
kann der Einfachheit3 halber geschrieben werden:
Eine Methode zur Bestimmung der Dissoziationskonstanten beruht auf
der Messung der Wasserstoffionenkonzentration in einer Lösung, welche
eine bekannte Menge der Säure und ihres Salzes mit einer starken Base
enthält. Für eine Säure HA wird die Dissoziationskonstante K gegebendurch
+xaA- VxCA- fH^xfA-aH+xaAK x
^a aHA CHA ÏHA
wobei H das Wasserstoffion, wie es in Lösung ist, darstellt.
In einer Mischung von a Mol einer schwachen Säure und b Mol ihres
stark ionisierten Salzes kann letzteres als vollständig in A"-Ionen dissoziiert
angesehen werden, so dass C^- = b ist. Desweiteren wird der Ueber-
schuss an Anionen, gemäss dem Gleichgewichtsgesetz, die Ionisierung der
Säure HA zurückdrängen, so dass letztere als vollständig nichtionisiert an¬
gesehen werden kann und Cha = a gesetzt werden kann. Diese Voraus¬
setzungen gelten aber nur bei einer schwachen Säure. Mit Hilfe einer ge¬
eigneten Wasserstoffelektrode bestimmt man eine Menge, welche als a^+angesehen wird, so dass alle Glieder der obigen Gleichung mit Ausnahme

- 96 -
von f^- und fijA zugänglich sind. Letzteres kann als Einheit genommen
werden und ersteres kann bestimmt oder von der Debye-Hückel'schenGleichung abgeleitet werden, und so kann K^ bestimmt werden. Für den
speziellen Fall nun, wo die Lösung gleich viel Säure wie Salz enthält, oder
wenn die schwache Säure zur Hälfte mit einer starken Base neutralisiert
ist, gilt:
CHA = CA~
Die Wasserstoffionenkonzentration ist dann ungefähr gleich der Dissozia¬
tionskonstante
CH+ = Ka
Da man ferner den negativen Logarithmus der Wasserstoffionenkonzen¬
tration allgemein mit pH bezeichnet, kann man, unter Berücksichtigungder obigen Konzentrationsbedingungen, schreiben:
-pH = -pKa
Die Formel beruht, wie aus ihrer Herleitung hervorgeht, auf Näherungen,die nur dann genügend genau sind, wenn es sich um schwache Säuren oder
schwache Basen handelt. Somit kann man bei schwachen Elektrolyten bei
geeigneter Konzentrationswahl aus der pH-Messung die Dissoziations¬
konstanten berechnen (173).
Kolthoff (174) hat kolorimetrisch die Titrationskurven zahlreicher
Alkaloide bestimmt und daraus die Wasserstoffionenkonzentration berech¬
net. Die Dissoziationskonstanten können daneben noch mit Hilfe der Leit¬
fähigkeitsmessung ermittelt werden.
b. Bestimmung der Dissoziationskonstanten
Die Bestimmungen wurden nach den Angaben von Glasstone (173)und Stoffel (175) ausgeführt. Infolge der schlechten Wasserlöslichkeit
der Barbiturate erfolgte ein Zusatz von 10% Alkohol. Es wurden 0,02n-Lösungen der einzelnen Barbitursäure-Derivate hergestellt und davon je20 cm' mit der gleichen Menge 0,0In-Natronlauge gemischt, so dass also
das Barbiturat zu 50% als Säure und zu 50% als Salz in einer Gesamt¬
konzentration von 0,01 in der zu untersuchenden Flüssigkeit vorhanden war.
Dann wurde mit dem Metrohm-Messgerät das pH gemessen (Temperatur20°).
173) Glasstone, "Textbook of physical chemistry", 2nd edition, 5
printing, B. Van Nostrand Co. Inc. New York (1948)174) Kolthoff, "Der Gebrauch von Farbindikatoren. Dire Anwendung in
der Neutralisationsanalyse und bei der kolorimetrischen Bestimmungder Wasserstoffionenkonzentration", Springer, Berlin (1923)
175) Stoffel, "Synthese und physikalisch-chemische Ueberprüfung einigerlokalanästhetisch wirksamer aliphatischer Carbonsäureester", Diss.
ETH. Zürich (1949)
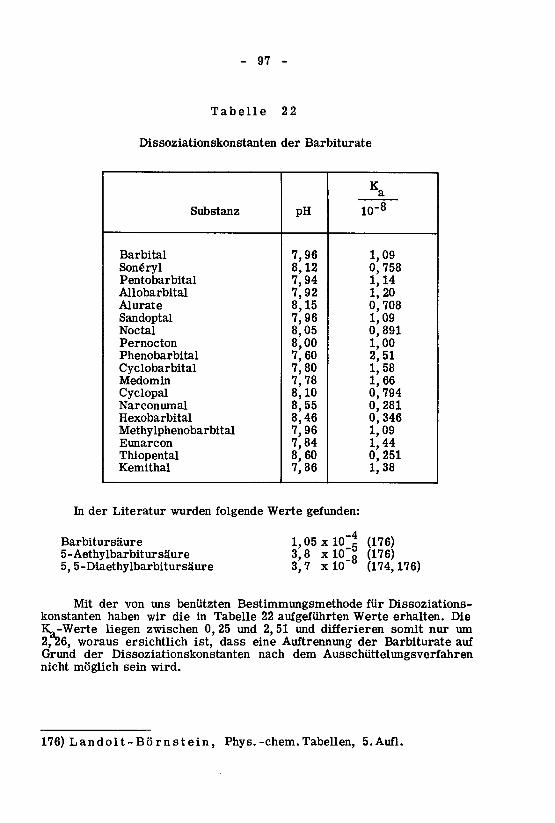
- 97 -
Tabelle 22
Dissoziationskonstanten der Barbiturate
Substanz
Barbital
SonérylPentobarbital
Allobarbltal
Alurate
SandoptalNoctal
Pernocton
Phénobarbital
CyclobarbitalMedomin
CyclopalNarconumal
Hexobarbital
MethylphenobarbitalEunarcon
ThiopentalKemithal
pH
7,968,127,947,928,157,968,058,007,607,807,788,108,558,467,967,848,607,86
Ka
10"8
1,090,7581,141,200,7081,090,8911,002,511,581,660,7940,2810,3461,091,440,2511,38
In der Literatur wurden folgende Werte gefunden:
Barbitursäure
5-Aethylbarbitursäure5, 5-Diaethylbarbitursäure
1,05x10"! (176)3,8 xlO'X (176)3,7 xlO"0 (174,176)
Mit der von uns benützten Bestimmungsmethode für Dissoziations¬
konstanten haben wir die in Tabelle 22 aufgeführten Werte erhalten. Die
IC-Werte liegen zwischen 0, 25 und 2,51 und differieren somit nur um
2, z6, woraus ersichtlich ist, dass eine Auftrennung der Barbiturate auf
Grund der Dissoziationskonstanten nach dem Ausschüttelungsverfahrennicht möglich sein wird.
176) Landolt-Börnstein, Phys.-ehem.Tabellen, 5. Aufl.

- 98 -
5. Schlussfolgerungen
a. Ausschüttelung aus weinsaurer Lösung mit Aether
Hier können wir eine praktisch quantitative Extraktion der Barbi¬
turate aus einer 2,5%igen wässerig-weinsauren Lösung (pH 2,05) mit
Aether annehmen. Die Dissoziation der einzelnen Barbitursäure-Derivate
wird hier so weit zurückgedrängt, dass sie kaum die Ausschüttelung be-
einflusst. Wenn wir trotzdem keine 100%ige Extraktion haben, so ist dies
wahrscheinlich eher der Adsorption an Natriumsulfat oder sonstigen Ver¬
lusten bei der Ausschüttelung zuzuschreiben als der Wasserlöslichkeit.
Auf Grund der Resultate könnte man trotzdem sagen, dass die Extraktion
bei den schlecht wasserlöslichen Barbituraten im allgemeinen etwas bes¬
ser ist als die bei den relativ gut wasserlöslichen.
b. Ausschüttelung einer Aetherlösung mit Alkalien
Da wir mit 2%iger Sodalösung (pH 10,85) eine quantitative Extraktion
aus Aether erreichten, erübrigte sich eine Ausschüttelung mit stärkeren
Alkalien. Extrahierten wir hingegen mit einem schwächeren Alkali (Na¬triumbikarbonat), so verlief die Ausschüttelung nicht mehr quantitativ.Die Erklärung ist darin zu suchen, dass erstens die Salzbildung mit einer
Bikarbonatlösung (pH 8,08 bis 8,15) nicht quantitativ ist, dass also der
undissoziierte Anteil in der Lösung den dissoziierten überwiegt, und dass
zweitens die Wasserlöslichkeit in diesem Falle eine grössere Rolle spieltund daher nicht zu vernachlässigen ist.
Unsere Ausschüttelungsergebnisse stehen mehr oder weniger im
Gegensatz zu den Angaben von Mühlemann und Bürgin (41) über die
Auftrennung der einzelnen Stoffe und Stoffgemische. Nach diesen Autoren
ist die Salzbildung der Barbitursäure-Derivate sowohl mit Bikarbonat als
auch mit Karbonat nicht quantitativ. Bei der Ausschüttelung erhalten sie
die Barbiturate in folgenden Gruppen: Cyclopal, Medomin und z. T. Bar-
bital bei der bikarbonatalkalischen Extraktion, Phénobarbital hauptsäch¬lich bei der karbonatalkalischen und Allobarbital, Alurate, Sandoptal,Cyclobarbital, Hexobarbital und Narconumal bei der natronalkalischen,
Dagegen erhielten wir mit allen untersuchten Barbituraten mit Sodalösungeine quantitative Extraktion, während wir mit Bikarbonat im besten Falle
(Barbital) 55,92%, Cyclopal zu 41,04% und Medotiin zu 27,88% extrahie¬
ren konnten. Von einer Auftrennung der Barbiturate durch Ausschütteln
mit verschieden starken Alkalien kann unseres Erachtens keine Rede sein.

- 99 -
n. VERHALTEN DER BARBITURATE BEI DER VAKUUM¬
MIKROSUBLIMATION
A. Abklärung der Sublimationsbedingungen
Ehe wir uns dem Verhalten der Barbiturate im einzelnen bei der
Vakuum-Mikrosublimation zuwenden, wollen wir noch die Faktoren unter¬
suchen, welche die Bildung und das Aussehen der Sublimate beeinflussen.
Es sind dies vor allem
die Druckverhältnisse,die Temperaturverhältnisse,der Sublimationsabstand,die Sublimationsdauer und
die Beeinflussung des Sublimates.
1. Druckverhältnisse
Um den Einfluss des Druckes auf die Sublimatbildung zu überprüfen,sublimierten wir Cyclobarbital und Methylphenobarbital bei 720 mm,50 mm, 10 mm, 1 mm und 0,05 mm. Diese Versuche führten zu den fol¬
genden Beobachtungen (siehe Seite 100 und 101).
Als Resultat dieser Untersuchungen darf man zusammenfassen:
1. Je niedriger der Sublimationsdruck ist, um so tiefer ist die Subli¬
mationstemperatur und desto rascher treten die Sublimate auf.
2. Die Ausbildung brauchbarer Sublimate ist bei gewöhnlichem Druck
schwieriger als bei vermindertem. Die Sublimate fallen bei Cyclo¬barbital und Methylphenobarbital in der Regel vorerst als Tröpfchen-Sublimate an, welche Anlass geben zur Ausbildung erzwungener Kri¬
stallaggregate, und das Auftreten instabiler Formen bei polymorphenSubstanzen ist häufiger.
3. Mit Abnahme des Druckes und dank der tieferen Temperaturen und
kürzeren Sublimationsdauer ist weniger mit Zersetzung zu rechnen.
Es kommt dabei zur Ausbildung schöner, derber, für die kristall¬
optischen Untersuchungen geeigneter Einzelkristalle. Das Optimumder besten Kristallbildung fanden wir zwischen 11 und 1 mm Hg.
4. Die bei den verschiedenen äusseren Drucken bei derselben mono-
morphen Substanz gewonnenen Sublimate unterscheiden sich hinsicht¬
lich Kristallhabitus (Tröpfchen, Einzelkristalle, Anordnung der Ag¬gregate) nicht wesentlich voneinander.
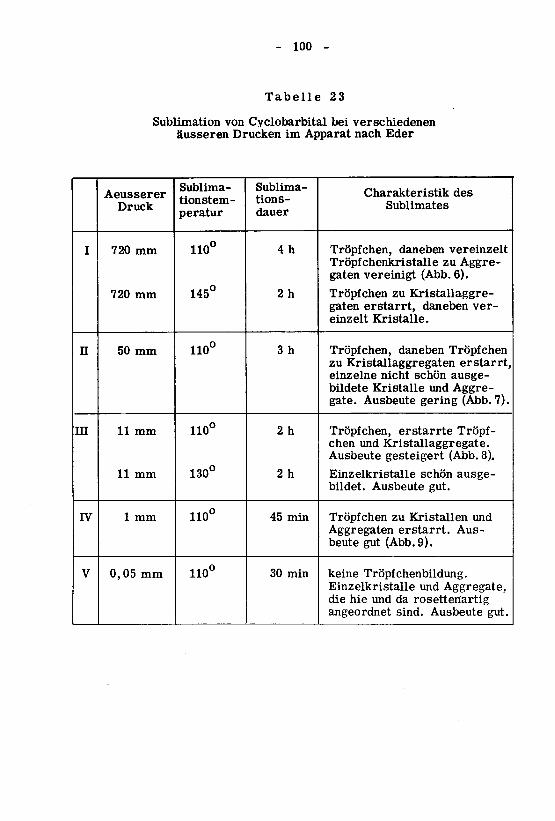
- 100 -
Tabelle 23
Sublimation von Cyclobarbital bei verschiedenen
äusseren Drucken im Apparat nach Eder
I
n
in
rv
V
Aeusserer
Druck
720 mm
720 mm
50 mm
11 mm
11 mm
1 mm
0,05 mm
Sublima¬
tionstem-
peratur
110°
145°
110°
110°
130°
110°
110°
Sublima¬
tions-
dauer
4h
2h
3h
2h
2 h
45 min
30 min
Charakteristik des
Sublimates
Tröpfchen, daneben vereinzelt
Tröpfchenkristalle zu Aggre¬gaten vereinigt (Abb. 6).
Tröpfchen zu Kristallaggre¬gaten erstarrt, daneben ver¬
einzelt Kristalle.
Tröpfchen, daneben Tröpfchenzu Kristallaggregaten erstarrt,einzelne nicht schön ausge¬
bildete Kristalle und Aggre¬gate. Ausbeute gering (Abb. 7).
Tröpfchen, erstarrte Tröpf¬chen und Kristallaggregate.Ausbeute gesteigert (Abb. 8).
Einzelkristalle schön ausge¬
bildet. Ausbeute gut.
Tröpfchen zu Kristallen und
Aggregaten erstarrt. Aus¬
beute gut (Abb. 9).
keine Tröpfchenbildung.Einzelkristalle und Aggregate,die hie und da rosetteriartigangeordnet sind. Ausbeute gut.
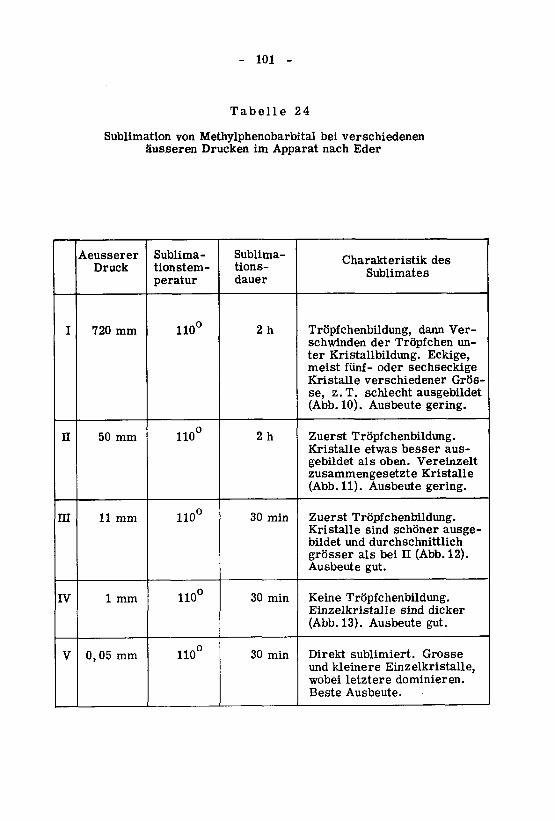
- 101 -
Tabelle 24
Sublimation von Methylphenobarbital bei verschiedenen
äusseren Drucken im Apparat nach Eder
I
n
in
IV
V
Aeusserer
Druck
720 mm
50 mm
11 mm
1 mm
0,05 mm
Sublima¬
tionstem-
peratur
110°
110°
110°
110°
110°
Sublima¬
tions-
dauer
2h
2h
30 min
30 min
30 min
Charakteristik des
Sublimates
Tröpfchenbildung, dann Ver¬
schwinden der Tröpfchen un¬
ter Kristallbildung. Eckige,meist fünf- oder sechseckigeKristalle verschiedener Grös-
se, z.T. schlecht ausgebildet(Abb. 10). Ausbeute gering.
Zuerst Tröpfchenbildung.Kristalle etwas besser aus¬
gebildet als oben. Vereinzelt
zusammengesetzte Kristalle
(Abb. 11). Ausbeute gering.
Zuerst Tröpfchenbildung.Kristalle sind schöner ausge¬bildet und durchschnittlich
grosser als bei II (Abb. 12).Ausbeute gut.
Keine Tröpfchenbildung.Einzelkristalle sind dicker
(Abb. 13). Ausbeute gut.
Direkt sublimiert. Grosse
und kleinere Einzelkristalle,wobei letztere dominieren.
Beste Ausbeute.
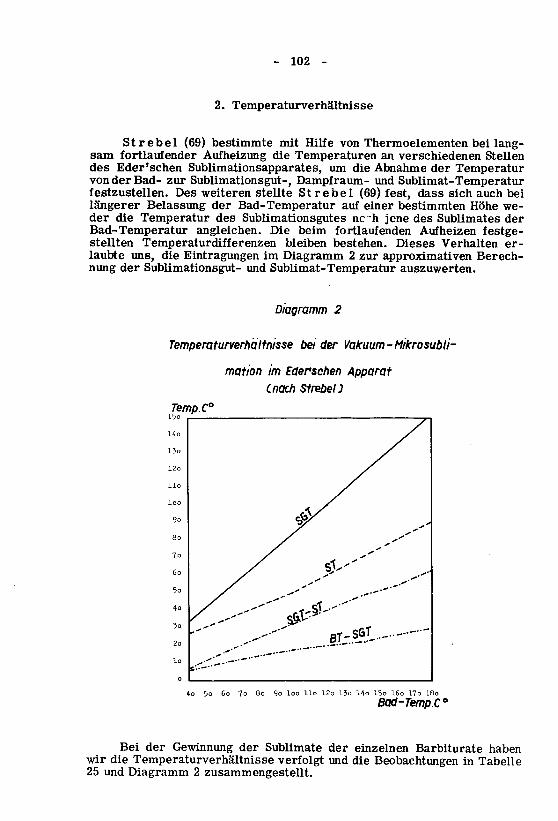
- 102 -
2. Temperaturverhältnisse
St re bel (69) bestimmte mit Hilfe von Thermoelementen bei lang¬sam fortlaufender Aufheizung die Temperaturen an verschiedenen Stellen
des Eder'schen Sublimationsapparates, um die Abnahme der Temperaturvon der Bad- zur Sublimationsgut-, Dampfräum- und Sublimat-Temperaturfestzustellen. Des weiteren stellte St rebel (69) fest, dass sich auch bei
längerer Belassung der Bad-Temperatur auf einer bestimmten Höhe we¬
der die Temperatur des Sublimationsgutes nc^h jene des Sublimates der
Bad-Temperatur angleichen. Die beim fortlaufenden Aufheizen festge¬stellten Temperaturdifferenzen bleiben bestehen. Dieses Verhalten er¬
laubte uns, die Eintragungen im Diagramm 2 zur approximativen Berech¬
nung der Sublimationsgut- und Sublimat-Temperatur auszuwerten.
Diagramm 2
Temperaturverhaltnisse bei der Vakuum-Mikrosubli-
motion im Ederschen Apparat
Cnach Streben
Temp.C
13o
12o
llo
loo
9o
80
7o
60
5o
4o
3o
2o
lo
0
//
r
^'-''' ^§&-"
^
_.-'"'" §Tr.
4o 5o 6o 7o 8o 9o loo llo 12o 13o 14o 15o 16o 17o
Bad-Temp.C
Bei der Gewinnung der Sublimate der einzelnen Barbiturate habenwir die Temperaturverhältnisse verfolgt und die Beobachtungen in Tabelle25 und Diagramm 2 zusammengestellt.
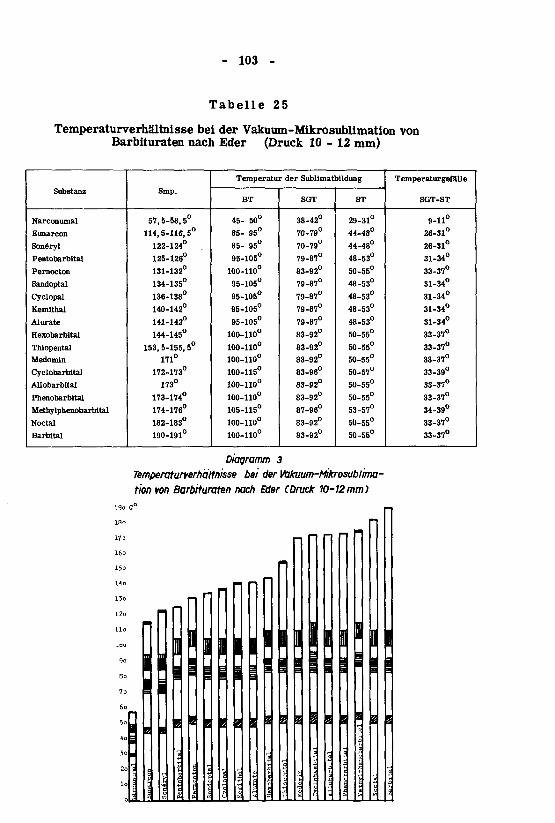
- 103 -
Tabelle 25
Temperaturverhältnisse bei der Vakuum-Mikrosublimation von
Barbituraten nach Eder (Druck 10 - 12 mm)
Substanz
Narconumal
Eunarcon
Sonéryl
Pentobarbital
Fernoctoii
Sandoptal
Cyclopal
Kemlthal
Alurate
Hexobarbital
Thlopental
Medomln
Cyclobarbital
Aüobarbital
Phénobarbital
Methylphenobarbital
Noctal
Barbltal
Smp.
57, 5-58, 5°
114,5-116,5°122-124°
125-126°
131-132°
134-135°
136-138°
140-142°
141-142°
144-145°
153, 5-155, 5°
171°
172-173°
173°
173-174°
174-176°
182-183°
190-191°
Temperatur der Sublimatbildung
BT
45- 50°
85- 95°
85- 95°
95-105°
100-110°
95-105°
95-105°
95-105°
85-105°
100-110°
100-110°
100-110°
100-115°
100-110°
100-110°
105-115°
100-110°
100-110°
SGT
38-42°
70-79°
70-79°
79-87°
83-92°
79-87°
79-87°
79-87°
79-87°
83-92°
83-92°
83-92°
83-96°
83-92°
83-92°
87-96°
83-92°
83-92°
ST
29-31°
44-48°
44-48°
48-53°
50-55°
48-53°
48-53°
48-53°
48-53°
50-55°
50-55°
50-55°
50-57°
50-55°
50-55°
53-57°
50-55°
50-55°
Temperaturgefalle
SGT-ST
9-11°
26-31°
26-31°
31-34°
33-37°
31-34°
31-34°
31-34°
31-34°
33-37°
33-37°
33-37°
33-39°
33-37°
33-37°
34-39°
33-37°
33-37°
Diagramm 3
Temperaturvertiältnisse bei der Vakuum-Mikrosublima¬
tion von Barbituraten nach Eder Cûruck 10-12 mm}

- 104 -
Aus Tabelle 25 und Diagramm 3 lässt sich entnehmen, dass die
Bad-Temperatur während des Sublimationsvorganges bei den niedrigschmelzenden Barbituraten ziemlich nahe unterhalb des Schmelzpunktesder sublimierten Substanz liegt, bei den höher schmelzenden Substanzen
jedoch eine ständig sich vergrössernde Temperaturdifferenz von 20-80°
vorhanden ist.
Die Temperatur des Sublimationsgutes liegt bei allen
Barbituraten, mit Ausnahme von Narconumal, 5-10° tiefer als die Bad-
Temperatur und die Sublimat-Temperatur liegt zwischen 44-57°,also innerhalb eines recht engen Temperaturbereiches. Bei der wiederhol¬
ten Sublimation derselben Substanz konnten mit geringen Abweichungenimmer wieder dieselben Temperaturverhältnisse gefunden werden, doch
darf man auch hier nicht diese Temperaturen als dem betreffenden Stoff
zugeordnete, exakte physikalische Konstanten betrachten. Sie haben nur
den Charakter von orientierenden Angaben für die Durchführung der Mikro¬
sublimation im Eder'schen Sublimationsapparat. Sie können insofern ein
schätzbares Hilfsmittel der Mikrosublimation sein, als sie voraussehen
lassen, ob zwei Stoffe durch fraktionierte Mikrosublimation getrennt wer¬
den können und bei welchen Temperaturen die Substanzen zweckmässigsublimiert werden.
Bei den polymorphen Barbituraten treten bei gleicher Bad-Tempera¬tur und bei verschiedener Bad-Temperatur Kristallisate von verschiede¬
nem Habitus auf, d.h. einzelne Modifikationen treten nebeneinander auf.
Hierzu ist zu bemerken, dass die instabilen Formen in der Regel bei
niedrigeren, die stabilen bei höheren Temperaturen auftreten.
Genaue Angaben zur Darstellung einer bestimmten Modifikation in
Abhängigkeit der Temperatur können nicht gegeben werden, da die gefunde¬nen Werte nur für die Mikrosublimation im Eder'schen Apparat gelten und
neben der Temperatur auch noch andere Faktoren eine wesentliche Rolle
spielen.
3. Sublimationsabstand
Um den Einfluss des Sublimationsabstandes auf das Temperaturge¬fälle und auf die Bildung der Sublimate zu überprüfen, sublimierten wir
Cyclobarbital und Methylphenobarbital in modifizierten Eder'schen Subli-
mationsgefässen, die einen Abstand, von 5,5; 9,0; 12,5; 17,0 und 24,0 mmzwischen Näpfchenboden und Deckgläschen aufwiesen (Abb. 3).

- 105 -
Tabelle 26
Sublimationsversuche mit Cyclobarbital
bei 11 mm Druck, einer Sublimationstemperatur von 110°, einer Sublimations¬
dauer von 45 Minuten und bei verschiedenem Sublimationsabstand
I
n
in
rv
V
Sublimations -
abstand
5, 5 mm
9,0 mm
12, 5 mm
17,0 mm
24,0 mm
Sublimationsverlauf
Kristallhabitus
Es bilden sich direkt Tröpfchen, in denen
nach einigen Minuten Höfe auftraten, die
kleine leistenförmig zugespitzte Kristalle
aufwiesen. Daneben fanden sich noch längereKristalle, die an den beiden Schmalseiten
eingekerbt waren, und einzelne Aggregate(Abb. 14). Sublimatbildung nur am Deckgläs¬chen.
Zuerst trat Tröpfchenbildung ein. In den Her¬
den bildeten sich kurzprismatische und leisten¬
förmig zugespitzte Kristalle, die zu Aggre¬gaten zusammengelagert waren. Grössere
Einzelkristalle waren selten (Abb. 15). Subli¬
matbildung nur am Deckgläschen.
Auch hier erhielten wir zuerst Tröpfchen.Die Kristallformen waren gleich wie bei H,doch ziemlich grosser (Abb. 16). Sublimatbil¬
dung nur am Deckgläschen.
Auf dem Deckgläschen bildeten sich nach eini¬
ger Zeit Tröpfchen, während Kristalle direkt
ins Ansatzröhrchen sublimierten. Nach 45 Mi¬
nuten fanden sich auf dem Deckgläschen ver¬
einzelt viereckige Kristalle neben Tröpfchen(Abb. 17).
Innerhalb 45 Minuten erhielten wir auf dem
Deckgläschen nur Tröpfchen, während Kri¬
stalle direkt ins Ansatzröhrchen sublimierten.
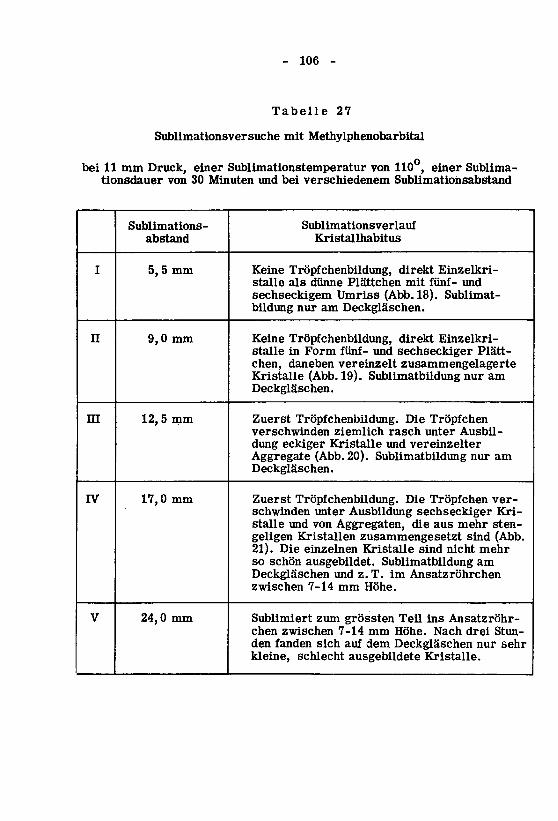
- 106 -
Tabelle 27
Sublimationsversuche mit Methylphenobarbital
bei 11 mm Druck, einer Sublimationstemperatur von 110°, einer Sublima¬
tionsdauer von 30 Minuten und bei verschiedenem Sublimationsabstand
I
n
m
IV
V
Sublimations¬
abstand
5, 5 mm
9,0 mm
12,5 mm
17,0 mm
24,0 mm
Sublimationsverlauf
Kristallhabitus
Keine Tröpfchenbildung, direkt Einzelkri¬
stalle als dünne Plättchen mit fünf- und
sechseckigem Umriss (Abb. 18). Sublimat¬
bildung nur am Deckgläschen.
Keine Tröpfchenbildung, direkt Einzelkri¬
stalle in Form fünf- und sechseckiger Plätt¬
chen, daneben vereinzelt zusammengelagerteKristalle (Abb. 19). Sublimatbildung nur amDeckgläschen.
Zuerst Tröpfchenbildung. Die Tröpfchenverschwinden ziemlich rasch unter Ausbil¬
dung eckiger Kristalle und vereinzelter
Aggregate (Abb. 20). Sublimatbildung nur am
Deckgläschen.
Zuerst Tröpfchenbildung. Die Tröpfchen ver¬
schwinden unter Ausbildung sechseckiger Kri¬
stalle und von Aggregaten, die aus mehr sten¬
geligen Kristallen zusammengesetzt sind (Abb.21). Die einzelnen Kristalle sind nicht mehr
so schön ausgebildet. Sublimatbildung amDeckgläschen und z. T. im Ansatzröhrchen
zwischen 7-14 mm Höhe.
Sublimiert zum grössten Teil ins Ansatzröhr¬
chen zwischen 7-14 mm Höhe. Nach drei Stun¬
den fanden sich auf dem Deckgläschen nur sehr
kleine, schlecht ausgebildete Kristalle.

- 107 -
Bei Cyclobarbital bildeten sich bei allen Versuchen zuerst Tröpfchen,während bei Methylphenobarbital bei einem Sublimationsabstand von 5,5und 9,0 mm sich die Kristalle direkt bildeten. Der Habitus änderte sich
insofern mit zunehmendem Abstand, als die "Aggregate" häufiger auftra¬
ten und die Ausbildung schöner Kristalle eher abnahm. Die besten Er¬
fahrungen hinsichtlich der Ausbildung schöner Kristalle machten wir bei
einem Abstand von 9 bis 12, 5 mm. Bei Versuch V konnten wir durch Ein¬
tauchen des Sublimationsgefässes (Abstand Badniveau : Deckgläschen =
ca. 10 mm) ebenfalls gut ausgebildete Kristalle erhalten.
4. Sublimationsdauer
Wie schon bei den Temperaturverhältnissen angegeben wurde, ist
die Darstellung einer bestimmten Modifikation nicht nur allein von der
Sublimations-Temperatur, dem Druck und dem Sublimationsabstand ab¬
hängig, sondern auch noch von der Sublimationsdauer. Bei Verlängerungder Sublimationsdauer, bei sonst aber gleichbleibenden Bedingungen, bilden
sich bei der Sublimation von Cyclobarbital und Methylphenobarbital, ge¬nerell bei monomorphen Substanzen, keine wesentlich veränderten Kri¬
stalle. Bei der Sublimation von polymorphen Barbituraten hingegen können
Veränderungen des Kristallbildes im Verlauf der Sublimation festgestelltwerden. So bilden sich bei der Sublimation von Allobarbital zuerst Tröpf¬chen und instabile Formen, welche dann bei der über zwei Stunden dauern¬
den Sublimation unter Vermehrung der stabilen Modifikation verschwinden
(Abb. 32, 33, 34 und 35).
5. Beeinflussung der Sublimate
Erhielten wir bei der Sublimation eines Stoffes meist nur tröpfchen-förmige oder schlecht ausgebildete kristalline Sublimate, so verwendeten
wir die Methode des Impfens des Deckgläschens mit der zu sublimieren-
den Reinsubstanz vor der Sublimation. Zu diesem Zweck brachten wir auf
ein gut gereinigtes Deckgläschen etwas Substanz, verrieben sie dort und
wischten dann das Deckgläschen mit einem faserfreien Tuch wieder blank.
Wir hatten demnach schon Kristallkeime und die Kristallisation konnte
leichter einsetzen. Die Sublimate bildeten sich bei einer etwas tieferen
Temperatur und die Kristalle sind kleinere, aber gleichmässig entwickelte
und in ihrer Kristallform oft sehr schön ausgebildete Einzelkristalle.
Bei der Impfung tritt aber immer diejenige Kristallform (Modifi¬kation) auf, mit der wir geimpft haben.

- 108 -
B. Untersuchung der Sublimate
Als Möglichkeiten zur Identifizierung der Sublimate der einzelnen
Barbiturate und ihrer Modifikationen zogen wir den Kristallhabitus, die
Mikroschmelzpunktbestimmung und die Mikroreaktionen zur Untersuchung
heran, verzichteten aber auf kristalloptische Bestimmungen. Hierbei wa¬
ren wir uns aber bewusst, dass der Kristallhabitus der Sublimate an und
für sich kein hinreichendes Merkmal für die Identifizierung eines Stoffes
ist und daher nur orientierende Bedeutung haben kann, z.B. in dem Sinne,dass man an Hand der Kristallbilder bei der Identifizierung verschiedene
Barbiturate ausschalten und so die Anzahl der in Frage kommenden ver¬
kleinern kann.
Die Bestimmung des Mikroschmelzpunktes war in unse¬
rem Falle besonders günstig, da sie uns erlaubte, bei polymorphen Sub¬
stanzen in den meisten Fällen die Schmelzpunkte der einzelnen Modifika¬
tionen direkt zu bestimmen. Der von uns benützte Balzerblock, in seiner
Ausführung dem Koflerblock nachgebildet, besteht aus einer elektrisch
heizbaren Metallplatte, in deren Mitte eine kleine Oeffnung von 1,5 mmden Durchtritt des Lichtes ermöglicht. Auf der Platte ruht eine weitere
kleinere, rechteckige, aber dünnere Metallplatte in der Grosse eines
Objektträgers, die durch einen wärmeisolierten Metallstab während der
Schmelzpunktbestimmung verschoben werden kann. Gegen die umgebendeLuft ist der Apparat durch einen an der Peripherie der Metallplatte ange¬
brachten 6 mm hohen Metallring, auf dem eine Glasplatte aufgelegt ist,geschützt. Die ganze Apparatur ruht auf dem Mikroskoptisch. Die Heiz¬
platte steht mit einem gut regulierbaren Widerstand in Verbindung. Zur
Temperaturmessung dient ein Kupfer-Konstaatan-Thermoelement und ein
Millivoltmeter. Die Eichung des Instrumentes wurde mit Reinsubstanzen
von bekanntem Schmelzpunkt in der gleichen Weise zwischen zwei Deck¬
gläschen durchgeführt, wie dies später mit den Sublimaten geschah. Das
MilliVoltmeter ergab uns eine Messgenauigkeit von ± 1°. Die Temperatur¬angaben beziehen sich nicht auf den Klarschmelzpunkt, sondern auf das
Temperaturintervall vom Taupunkt bis zum Klarschmelzpunkt, wie es
auch die Ph.H.V angibt. Bei den Sublimaten machte sich die bei ihrer Her¬
stellung erwünschte Eigenschaft der Sublimation störend bemerkbar, in¬
dem das aufliegende Deckgläschen bis in die Nähe des Schmelzpunktes mit
feinen Kristallen oder Sublimationströpfchen bedeckt wurde und oft die
Beobachtung des Schmelzpunktes erschwerte oder fast verunmöglichte.
Der Schmelzpunkt der einzelnen Barbiturate wurde daneben noch auf
dem Maquenne-Block und auf der Kofierheizbank bestimmt. Diese besteht
aus einem einseitig geheizten bandförmigen Metallkörper, der ein lineares
Temperaturgefälle besitzt. Zur Bestimmung des Schmelzpunktes wird die
Substanz mit Hilfe einer Lanzettnadel auf die Oberfläche der Bank aufge¬streut und mit Hilfe eines Zeigers, der an einer Skala verschiebbar ist,die Schmelztemperatur ermittelt. Die Eichung des Apparates erfolgt mit
Testsubstanzen.
Als mikrochemische Reaktionen überprüften wir besonders die Fäl¬
lungen mit Schwermetallkomplexen. Folgende Fällungsmittel wurden
verwendet:

- 109 -
Eisen-Reagens
FerrichloridlosungSalzsäure konz.
KaliumjodidWasser
Kupfer-Reagens
Kupfer sulfat
Salzsäure konz.
KaliumjodidWasser
Wismut-Reagens
Basisches Wismutkarbonat
Salzsäure konz.
KaliumjodidWasser
ad
ad
ad
3,01,03,0
10,0
0,31,03,010,0
0,51,03,0
10,0
Für die Untersuchungen wurden vorerst die Möglichkeiten und Re¬aktionsverhältnisse an der reinen Substanz studiert. Zu diesem Zweckewurde sehr wenig Stoff auf einen Objektträger gebracht, ein Tropfen des
entsprechenden Reagens zugesetzt, mit einem grossen Deckglas zugedecktund dann vorsichtig auf kleiner Flamme (Mikrobrenner) erwärmt, bis sich
der Niederschlag gelöst hatte oder die Flüssigkeit aufkochte. Hierbei ist
zu beachten, dass der nicht gelöste Anteil durch die auftretenden Blasen
nicht unter dem Deckglas herausgeschleudert wird. Beim Erkalten kann
man dann unter dem Mikroskop die Kristallbildung beobachten. Fielen die
Fällungen einigermassen charakteristisch aus, so wurden die Mikroreak-
tionen auf die durch Sublimation erhaltenen Sublimate übertragen. Bei der
Auswertung der Kristallformen der so gefällten Komplexverbindungen mussman Vorsicht walten lassen, denn sie hängen weitgehend von den Versuchs¬
bedingungen ab.
Barbital (5-Aethyl-5-aethyl-barbitursäure)
Smp. der Reinsubstanz (Block): 186-188° *
Smp. des Sublimates (mikro): I 190-191°, H 183°, HI 181°, IV 176°
Sublimatbildung und -eigenschaften: Die Sublimation im
Eder'sehen Apparat geht recht gut. Den ersten Anflug eines Sublimates
beobachteten wir bei ca. 80° Bad-Temperatur. Bei langsam ansteigenderTemperatur bildete sich das Sublimat schön aus, ohne dass Tröpfchenbil¬dung beobachtet wurde. Die günstigste Sublimationstemperatur lag bei
100-110°. Ein so gewonnenes Sublimat enthält aber fast immer Kristalle
verschiedener Modifikationen (Abb. 22). Die einzelnen Modifikationen durch
Sublimation mehr oder weniger rein darzustellen, gelingt nicht immer
ganz gut, doch kann man durch Aenderung der Bad-Temperatur, aber sonst
gleichbleibende Bedingungen, die Bildung der einzelnen Modifikationen
beeinflussen.
* Alle Schmelzpunkte sind korrigiert

- 110 -
Die stabile Modifikation bildet sich bei einer Bad-Temperatur von
100-110° in Form von Nadeln oder stengeligen Kristallen (Abb. 23).
Modifikation II erhielten wir bei einer Bad-Temperatur von ca. 110°in Form von prismatischen Kristallen mit zugespitzten Enden (Abb. 24).Nach A. Kof 1er (86) bestehen die im Handel unter dem Namen Veronal
oder Diaethylbarbitursäure erhältlichen Präparate aus verschiedenen Lö¬
sungsmitteln kristallisiert fast ausschliesslich aus dieser Modifikation.
Modifikation in erhielten wir in geringen Mengen bei einer Bad-
Temperatur von ca. 90°. Beim spontanen Erstarren der Schmelze bildete
sie sich in Form von Nadeln (Abb. 25).
Die quadratischen und rechteckigen aus Zwillingslamellen zusammen¬
gesetzten Blättchen der Modifikation IV erhielten wir sehr schön bei einer
Bad-Temperatur von ca. 85° (Abb. 26). Nach A. Kofi er (86) sind Modi¬
fikation m und IV enantiotrop mit einem Umwandlungspunkt bei 135°.
Schmelzpunkt: Die einzelnen Modifikationen liessen sich mit
Ausnahme von Modifikation HI direkt im Sublimat bestimmen. Da diese
Form unter 130° die grösste Flüchtigkeit besitzt, kann sie nicht direkt
zum Schmelzen gebracht werden. Wir bestimmten deshalb den Smp. dieser
Modifikation in der erstarrten Schmelze, wo sie bei spontaner Kristalli¬
sation fast immer auftritt.
Bei der Bestimmung des Smp. auf der Kofler-Heizbank sublimierten
Nadeln, die bei 190° schmolzen.
Eisenkomplex: Einige Kriställchen Barbital, mit dem Eisen-
Reagens zusammen erhitzt, gaben beim Erkalten dunkelbraune, lange,gerade Nadeln, die meistens einen gemeinsamen Mittelpunkt zeigten. Die
Bildung und das Wachstum dieser Nadeln lässt sich unter dem Mikroskopsehr schön verfolgen (Abb. 61).
Kupferkomplex: Mit dem Kupfer-Reagens gab Barbital grosse
violettbraun gefärbte derbe rhombische Kristalle (Abb. 62).
S o n é r y 1 (5-Aethyl-5-n-butyl-barbitursäure)
Smp. der Reinsubstanz (Block): 122-124°
Smp. des Sublimates (mikro): 122-124°
Sublimatbildung und -eigenschaften: Bei der Sublimation
bildeten sich zuerst Tröpfchen; bei 90-95° erhielten wir Nadeln, die oft
zu gekrümmten Garben zusammengesetzt waren (Abb. 27).
Eisenkomplex: Beim Erkalten bildeten sich kleine, zum Teil
seesternförmige, zum Teil gerade, wenig charakteristische Formen.
Kupferkomplex: Hier erhielten wir Kristallgebilde mit lanzett¬
förmigem Umriss, deren Ränder stark gezackt sind und hie und da eine
Mittellinie erkennen lassen.
Pentobarbital (5-Aethyl-5-(l-methylbutyl)-barbitursäure)
Smp. der Reinsubstanz (Block): 125-126°
Smp. des Sublimates (mikro): 1125-126°, H 110, 5°

- Ill -
Sublimatbildung und -eigenschaf ten: Bei der Sublimation
im Eder'schen Apparat bildeten sich zuerst Tröpfchen. Bei einer Bad-
Temperatur von ca. 85° erschienen im Sublimat Kristalle; sie stellten
Aggregate von Rauten und flachen Plättchen mit verschiedenem Umriss
dar (Abb. 28). Steigerten wir die Temperatur, so bildeten sich bei ca. 100°
dünne, verzweigte Nadeln und Büschel (Abb. 29).
Schmelzpunkt: Die verschiedenen Kristallformen gehören den
beiden Modifikationen des Pentobarbitals an. Bei der Mikroschmelzpunkt-bestimmung schmelzen die flachen Plättchen und Rauten zum Teil bei
110, 5°, zum Teil wandeln sie sich in kleine Nadeln um, die bei 125-126°
schmelzen. Die grösseren Nadeln (Abb. 29) sublimieren bei der Schmelz¬
punktbestimmung öfters noch einmal zu kleineren Nadeln um, die bei 125-
126° schmelzen.
Eisenkomplex: Beim Erkalten bilden sich kreuzweise gelagertegrössere und kleinere Nadeln, in deren Schnittpunkt sich kleine mehr oder
weniger charakteristische Gebilde befinden.
Kupferkomplex: Hier erhielten wir Nadeln, die öfters zu grös¬seren blätterförmigen Gebilden auswuchsen.
Allobarbital (5-Allyl-5-allyl-barbitursäure)
Smp. der Reinsubstanz (Block): 169-171°
Smp. des Sublimates (mikro): 1173°, n 124,5-126,5°
Sublimatbildung und -eigenschaften: Zuerst erhielten
wir Tröpfchen, dann bildeten sich bei einer Bad-Temperatur von ca. 80
dünne, längere nadeiförmige oder auch stengelige Kristalle (Abb. 30).Steigerten wir die Bad-Temperatur langsam, so bildeten sich dicke, pla¬stische Kristalle mit rhomboidischem Umriss (Abb. 31).
Die ersteren Kristalle bildeten sich jedoch nur bei Temperaturenunter 95° und bei kurz dauernder Sublimation. Bei Temperatursteigerungoder bei lang dauernder Sublimation verschwanden diese Kristalle unter
Vermehrung der dicken plastischen Kristalle. Bei der Sublimation unter
dem Mikroskop konnten wir dieses Verhalten ziemlich gut beobachten. Zu¬
erst bildeten sich, bei langsam ansteigender Bad-Temperatur, die dünnen,nadeiförmigen Kristalle. Wir beliessen nun die Bad-Temperatur, auf ca.
90-100° und konnten feststellen, dass diese Kristalle im Anfang noch wei¬
ter wuchsen, dann aber, bei gleichzeitiger Vermehrung der dicken pla¬stischen Kristalle, nach zwei Stunden gänzlich verschwunden waren
(Abb. 32, 33, 34 und 35).
Schmelzpunkt: Die dicken plastischen Kristalle schmolzen bei
173,die nadeiförmigen bei 124, 5-126, 5°. Der Schmelzpunkt der beiden
Formen ist ziemlich verschieden und so konnten wir annehmen, dass es
sich um zwei Modifikationen handelt. Um nun genau feststellen zu können,ob die bei der Sublimation erhaltenen verschiedenartigen Kristalle zwei
Modifikationen darstellen, wurden mit verschiedenen Roentgenstrahlen(Cr und Cu) und mit verschiedenen Kameras Aufnahmen gemacht.

- 112 -
Roentgenographische Untersuchungen (*): Es zeigte sich
nun hierbei, dass die Aufnahmen mit Cr-Strahlung (1KK = 2, 2896 A) und
einer Kamera mit einem Durchmesser von 57, 2 mm sich als die geeignete¬sten für diese Substanzen erwiesen.
Von den folgenden Substanzen wurden Aufnahmen gemacht:
Substanz A: dicke, plastische Kristalle (Sublimat) (Abb. 31)Substanz B: Ausgangssubstanz, Smp. 169-171°
QSubstanz C: 5, 5-Diallyl-barbitursäure (Kopenhagen), Smp. 172
Substanz D: dünne, nadeiförmige Kristalle (Sublimat) (Abb. 30)
Die Belichtungsdaten für die mit Cr-K-Strahlung hergestellten Auf¬
nahmen sind in Tabelle 28 wiedergegeben.
Tabelle 28
Belichtungsdaten der Aufnahmen von 5,5-Diallylbarbitursäure
Cr-Strahlung XK = 2, 2896 Â
Substanz
A
B
C
D
Belichtungszeit
6 h
6 h
2,5 h
3 h
Röhrenspannung
35 kV
35 kV
37 kV
37 kV
Stromstärke
15 mA
15 mA •
9 mA
9 mA
Diese an den Präparaten A, B, C und D hergestellten Aufnahmen
ergaben die in Abb. 36 wiedergegebenen Diagramme.
*) Herrn Privatdozent Dr.W.Epprecht, unter dessen Leitung die Aufnahmenam roentgenographischen Institut derETH. gemacht und ausgewertet wur¬
den, sei auch an dieser Stelle herzlichst gedankt.

- 113 -
Abbildung 36
Roentgendiagramme von 5, 5-Diallylbarbitursäure
Cr-Strahlung XK, = 2, 2896 Â ; Kameradurchmesser 57, 2 mm
A
B
C
D
dicke, plastische Kristalle, Smp. 173 (Sublimat) ' * ^
Ausgangssubstanz, Smp. 169-171°
5, 5-Diallylbarbitursäure Kopenhagen, Smp. 172°dünne, nadeiförmige Kristalle, Smp. 124, 5-126, 5° (Sublimat)
Die Auswertung der Aufnahmen ergab die in Tabelle 29 festgehalte¬nen Netzebenenabstände.
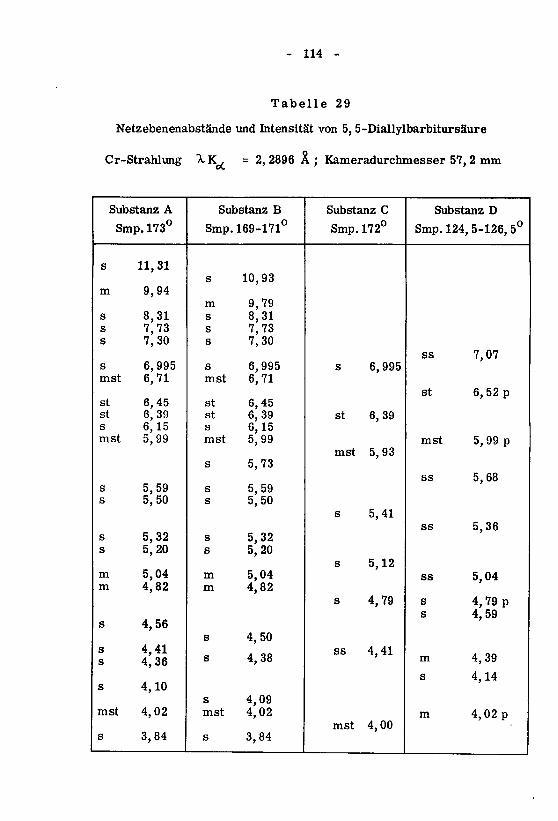
- 114 -
Tabelle 29
Netzebenenabstände und Intensität von 5, 5-Diallylbarbitursäure
Cr-Strahlung XK, = 2,2896 Â; Kameradurchmesser 57,2 mm
Substanz A
Smp. 173°
s
m
s
s
s
s
mst
st
st
s
mst
s
s
s
s
m
m
s
s
s
s
mst
s
11,31
9,94
8,317,737,30
6,9956,71
6,456,396,155,99
5,595,50
5,325,20
5,044,82
4,56
4,414,36
4,10
4,02
3,84
Substanz B
Smp.
s
m
s
s
s
s
mst
st
st
s
mst
s
s
s
s
s
m
m
s
s
s
mst
s
169-171°
10,93
9,798,317,737,30
6,9956,71
6,456,396,155,99
5,73
5,595,50
5,325,20
5,044,82
4,50
4,38
4,094,02
3,84
Substanz C
Smp.
s
st
mst
s
s
s
ss
mst
172°
6,995
6,39
5,93
5,41
5,12
4,79
4,41
4,00
Substanz D
Smp. 124,
ss
st
mst
ss
ss
ss
s
s
m
s
m
5-126, 5°
7,07
6,52p
5,99 p
5,68
5,36
5,04
4,79p4,59
4,39
4,14
4,02p
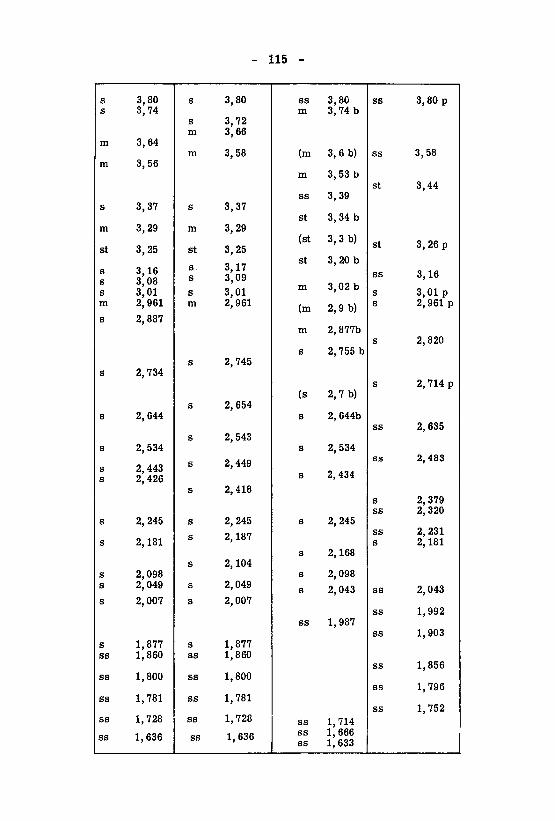
- 115 -
s
s
m
m
s
m
st
s
s
s
m
s
s
s
s
s
s
s
s
s
s
s
s
ss
ss
ss
ss
ss
3,803,74
3,64
3,56
3,37
3,29
3,25
3,163,083,012,961
2,887
2,734
2,644
2,534
2,4432,426
2,245
2,181
2,0982,049
2,007
1,8771,860
1,800
1,781
1,728
1,636
s
s
m
m
s
m
st
s
s
s
m
s
s
s
s
s
s
s
s
s
s
s
ss
ss
ss
ss
ss
3,80
3,723,66
3,58
3,37
3,29
3,25
3,173,09
3,012,961
2,745
2,654
2,543
2,449
2,418
2,245
2,187
2,104
2,049
2,007
1,8771,860
1,800
1,781
1,728
1,636
ss
m
(m
m
ss
st
(st
st
m
(m
m
s
(s
s
s
s
s
s
s
s
ss
ss
ss
ss
3,803,74 b
3,6 b)
3,53 b
3,39
3,34 b
3,3 b)
3,20 b
3,02 b
2,9 b)
2,877b
2,755 b
2,7 b)
2,644b
2,534
2,434
2,245
2,168
2,098
2,043
1,987
1,7141,6661,633
ss
ss
st
st
ss
s
s
s
s
ss
ss
s
ss
ss
s
ss
ss
ss
ss
ss
ss
3,80 p
3,58
3,44
3,26p
3,16
3,01 p
2,961 p
2,820
2,714p
2,635
2,483
2,3792,320
2,2312,181
2,043
1,992
1,903
1,856
1,796
1,752

- 116 -
Die relative Intensität der Beugungslinien ist durch folgende willkür¬
liche Bezeichnung wiedergegeben:
SS
s
m
mst
st
sehr schwach
schwach
mittel
mittelstark
stark
Die Beschaffenheit einzelner Linien ist noch durch folgende Bezeich¬
nung charakterisiert:
p punktiertb breite Linie
Substanz B ist darnach mit den dicken plastischen Kristallen der
Substanz A in bezug auf ihre KristallStruktur identisch; Substanz C ist in
ihrem Kristallzustand leicht gestört, kann aber ebenfalls als identisch mit
A und B angesehen werden. Die Substanzen A, B und C gehören somit ein
und derselben Modifikation I an, welche äusserlich dicke, plastische Kri¬
stalle mit rhomboidischem Umriss zeigt.
Substanz D ist hingegen als eine eigene Modifikation anzusehen. Sie
ergab ein Diagramm, welches ausser den für sie charakteristischen Inter¬
ferenzlinien noch als punktierte Linien die intensivsten Reflexe der Modi¬
fikation I zeigt. Dies rührt davon her, dass die Sublimate mit Modifikation
II immer ein wenig mit Modifikation I verunreinigt sind.
Unsere Resultate der roentgenographischen Untersuchungen lassen
somit sicher erkennen, dass Allobarbital im Gegensatz zu den Befunden
von Tso-Yueh Huang (106) polymorph ist.
Eisenkomplex: Beim Erkalten scheiden sich ziemlich grosse,
lange, gefiederte Nadeln aus, die gewöhnlich nach einem gemeinsamenMittelpunkt orientiert sind (Abb. 63).
Kupferkomplex: Auch hier bilden sich grosse, lange Nadeln aus
(Abb. 64).
Alurate (5-Allyl-5-isopropyl-barbitursäure)
Smp. der Reinsubstanz (Block): 138-139°Smp . des Sublimates (mikro): 1141-1420, n 138
,m 133, 5-134, 5
,IV 130°
Sublimatbildung und -eigenschaften: Bei der Sublimation
entstand erstmals ein schwacher Anflug bei etwa 70°, der fast nur aus
Tröpfchen bestand. Bei höherer Temperatur bildeten sich zunächst auch
wieder Tröpfchen, die sich dann zu schön entwickelten Kristallen umwan¬
delten. Die günstigste Sublimationstemperatur lag bei 100-105°. Sublimate,die bei dieser Temperatur gewonnen wurden, zeigten Kristalle verschie¬
dener Modifikationen. Fischer (83) unterscheidet zwei Modifikationen:

- 117 -
1. dünne, flächenartig ausgebildete Kristalle, manchmal leisten-
förmig, zugespitzt, und parallel verwachsene Aggregate, Smp. 133,5-134, 5Ö.
2. die stabile bei 138,5-140, 5° schmelzende Modifikation zeigt zu¬
meist körperliche Kristalle, entweder in Form von kurzen Säulchen mit
der Umrissform eines Rhomboids oder lange stengelige Kristalle mit ab¬
geschrägten Enden, ferner selten regelmässige rhombische Kristalle.
Brand stätter (107) und Reimers (45) identifizierten an Hand
der Schmelzpunkte vier verschiedene Modifikationen mit den respektivenSchmelzpunkten: 1142°, II139°, 111133° und IV ca. 130°. Tso-Yueh
Huang (106) führt in seiner Arbeit über Roentgenstrahlenuntersuchungenan Barbitursäure-Derivaten folgende Modifikationen auf:
Modifikation I, Smp. 142°, Nadeln, sehr stabil
Modifikation n, Smp. 139Q, prismatische Nadeln, metastabil
Modifikation m, Smp. 133,dünne hexagonale Plättchen, instabil
Modifikation IV, Smp. 130°, konnte nicht erhalten werden. Er er¬
hielt zwar eine andere Kristallform, aber es scheint, dass es sich nur
um ein Aggregat von III handelt. Trotzdem der Autor einen Unterschied
im Schmelzpunkt fand, waren die Roentgendiagramme beider Formen
gleich.
Schmelzpunkt: Bei unseren Versuchen erhielten wir bei einer
Bad-Temperatur von ca. 100° einzelne oder zusammengelagerte Nadeln,die bei 141-142° schmolzen (Abb. 37) und die die stabile Modifikation dar¬
stellen.
Sublimierten wir Alurate bei ca. 85°, so konnten wir im Sublimat
zweierlei Kristallformen beobachten. Die eine bildete plastische Kristalle
in Form von kurzen Säulchen (Abb. 38), während die andere mehr oder
weniger lange, an beiden Enden schwalbenschwanzförmig eingekerbte,Kristalle darstellte (Abb. 39). Bei der Mikroschmelzpunktbestimmungsublimierte gewöhnlich ein Teil zu langen Nadeln um, die bei 141-142°
schmolzen, während die andern Formen bei 138° resp. 130° schmolzen.
Die aus einer wässerigen Alkohollösung von uns kristallisierten fla¬
chen, dünnen, an beiden Enden zugespitzten Plättchen (Abb. 40) zeigteneinen Schmelzpunkt von 133-134°.
Eisenkomplex: Beim Erkalten bildeten sich zuerst Tröpfchen,aus denen nach einiger Zeit flache, etwas breite Nadeln herauswuchsen.
Kupferkomplex: Hier erhielten wir beim Abkühlen elliptische
Gebilde, die jedoch nicht immer sehr schön ausgebildet waren.
Sandoptal (5-AUyl-5-isobutyl-barbitursäure)
Smp. der Reinsubstanz (Block): 134-135°Smp. des Sublimates (mikro): 134-135
Sublimatbildung und -eigenschaften: Auch hier bildeten
sich zuerst Tröpfchen und dann Kristalle. Bei einer Sublimationstempera¬tur von ca. 100° erschienen im Sublimat leistenförmig zugespitzte Kri¬
stalle, die oft zu Aggregaten zusammengewachsen waren (Abb. 41).

- 118 -
Eisenkomplex: Beim Erkalten entstehen im Anfang zarte Nadel¬
bäumchen und kleine feine verzweigte Nadeln, die dann noch weiter wuch¬
sen.
Kupferkomplex: Die anfangs ausgefallenen Nadeln mit einem
gemeinsamen Mittelpunkt wuchsen stark aus und bildeten dann breitere
Nadeln, die sich voneinander loslösten.
Noctal (5-Isopropyl-5-(ß-bromallyl)-barbitursäure)Smp. der Reinsubstanz (Block): 178, 5-179,5°Smp. des Sublimates (mikro): 1182-183°, n 180-181
,HI 177-178°
Sublimatbildung und -eigenschaften: Bei der Sublimation
im Eder'sehen Apparat bildeten sich direkt Kristalle ohne die Tröpfchen¬vorstufe. In dem bei einer Bad-Temperatur von 100-110° gewonnenen
Sublimat Hessen sich verschiedene Kristallformen feststellen.
Schmelzpunkt: Die prismatischen, zugespitzten Nadeln (Abb.42)wuchsen bei der Schmelzpunktbestimmung weiter aus und schmolzen bei
182-183°. Die dünnen, flachen, sechseckigen Kristalle (Abb. 43) schmol¬
zen bei 177-178°, die rechteckigen Kristalle (Abb. 44) aber bei 180-181°.
Eisenkomplex: Der Komplex fiel als kleine, schwarze Nadeln
aus, die grösstenteils einzeln vorliegen, zum Teil auch sternartig über¬
einander gelagert sind.
Kupferkomplex: Beim Erkalten bildeten sich direkt farblose
rhombische Kristalle.
Nach Lang und Stephan (102) ist hier der Silberkomplexam reaktionsfähigsten und liefert die grössten und ausgeprägtesten For¬
men und ist zum Nachweis von Noctal am geeignetesten.
Pernocton (5-Sec-butyl-5-(/3 -bromallyl)-barbitursäure)
Smp. der Reinsubstanz (Block): 130-131°Smp. des Sublimates (mikro): 131-132
Sublimatbildung und -eigenschaften: Die Sublimation von
Pernocton gestaltete sich gegenüber den anderen Barbituraten schwieri¬
ger. Es bildeten sich zuerst nur Tröpfchen, die erst nach löstündigerSublimationsdauer zu Kristallen erstarrten. Das bei einer Bad-Tempe¬ratur von ca. 110° gewonnene Sublimat bestand aus etwas spitzen Nadeln,die in Form von Büscheln und Garben angeordnet waren (Abb. 45). Impftenwir das Deckgläschen, um die Sublimationsdauer zu verkürzen, so er¬
hielten wir kleinere, meistens übereinander gelagerte Nadeln und sechs¬
eckige Kristalle (Abb. 46).
Schmelzpunkt: Beide Formen zeigten einen Schmelzpunkt von
131-132°.
Eisenkomplex: Der in der Wärme gut lösliche Komplex gehtbeim Erkalten direkt in die Tröpfchenstufe über. Aus den Tröpfchen bil¬
deten sich nach einiger Zeit Nadelkreuze.
Kupfer- und Wismutkomplex: bildeten sich nur in Form
von Tröpfchen.

- 119 -
Phénobarbital (5-Aethyl-5-phenyl-barbitursäure)
Smp. der Reinsubstanz (Block): 172-173°Smp .
des Sublimates (mikro): I 173-174°, H 165, 5-167, 5°, m 155, 5-
156,5°
Sublimatbildung und -eigenschaf ten: Bei der Sublimation
erhielten wir zuerst Tröpfchen, dann bildeten sich bei ca. 100° Bad-
Temperatur Kristalle. Die dabei erhaltenen Sublimate zeigten verschiedene
Kristallformen, die den einzelnen Modifikationen angehörten.
Nach Fischer (68) und A. Kofi er und Fischer (87) kann man
dreierlei Kristallformen unterscheiden:
Stabile Form: Smp. 174°, rhomboidische Tafeln und Kristallplätt-
chen, oft fächerförmig verwachsen und auf die Kante gestellt oder zu
sphärolithisehen Aggregaten vereinigt.
Metastabile Form: Smp. 156-157, dünne, lange, nadeiförmige,
auch stengelige Kristalle, an den Enden schief abgeschnitten und garben¬artig verwachsen.
Metastabile Form: 166-167°, Komplexe von kurzprismatischen Kri¬
stallen, am Ende gerade abgeschnitten und seitlich abgeschrägt. Beim
Erhitzen kann Schmelzen und Umwandlung der metastabilen Formen be¬
obachtet werden. Die metastabile Form des Smp. 156-157° wandelt sich
sehr leicht, oft spontan ohne ersichtlichen Grund in die stabile Form um,während die metastabile Form des Smp. 166-167° relativ beständiger ist
und häufiger zum Schmelzen gebracht werden kann.
Bei Untersuchungen mit Roentgenstrahlen fand Tso-Yueh Huang(106) noch zwei weitere Modifikationen von Phénobarbital. Die eine Mo¬
difikation vom Smp. 177° erhielt er durch Erhitzen des käuflichen Pro¬
duktes bis etwas unter den Schmelzpunkt von der Modifikation mit Smp.174°. Hierbei stellte er fest, dass die Roentgendiagramme der beiden
Formen verschieden waren. Die andere Modifikation, vom Smp. 162-163°,erhielt er durch Abkühlung einer Schmelze zwischen zwei Deckgläschen,während bei der Abkühlung auf einem Deckgläschen sich immer die Mo¬
difikation mit einem Smp. 166-167° bildete. Nach Tso-Yueh Huang(106) geht die Umwandlung einer Form in die andere nach folgendemSchema vor sich:
IV (Smp. 156-157°) ca. 90°^ m (Smp. 166-167°) ca-126°»r.0
n (Smp. 174°) ca-160
» I (Smp. 177°)
Hingegen bildet sich Modifikation IVa nur unter bestimmten Bedingungen.

- 120 -
Schmelzpunkt: Bei einer Bad-Temperatur von 100-105° erhiel¬
ten wir die in Abb. 47 dargestellten Kristalle. Bei der Mikroschmelzpunkt-bestimmungwandelten sich bei 135-140° die Büschel zu kleinen Tafeln und
Plättchen um und schmolzen bei 173-1740. Rhomboidische Tafeln erhiel¬
ten wir bei einer Temperatur von ca. 120°, wo sie zu Aggregaten zusam¬
mengelagert sind. Bei der Kristallisation aus der Schmelze bildeten sie
sich schöner und regelmässiger aus (Abb. 48). Sublimierten wir Phéno¬
barbital bei einer Temperatur von ca. 130°, so bildeten sich die in Abb.49 dargestellten Kristalle, die bei 165, 5-167, 5° schmolzen. Bei Atmos¬
phärendruck und einer Temperatur von ca. 150° erhielten wir das in Abb.50 gezeigte Büd. Diese Kristalle schmolzen bei 155,5-156,5°. Abb. 51
zeigt ein Sublimat, das wir bei einer Bad-Temperatur von ca. 110° erhiel¬
ten. Bei der Mikroschmelzpunktbestimmung erfolgte, bei einer Tempera¬tur von 130-140°, teilweise Umwandlung zu Tafeln und Plättchen, die dannbei 173-174° schmolzen, während die nicht umgewandelten Kristalle den
Smp. 155, 5-156, 5° zeigten.
Eisenkomplex: Nach dem Erhitzen scheiden sich beim Erkalten
braune, ziemlich derbe Nadelbüschel, daneben kleinere sternförmige Ge¬bilde ab (Abb. 65).
Kupferkomplex: Beim Erkalten scheiden sich feinere Nadel¬
büschel und daneben einzelne Nadeln ab (Abb. 66).
Wismutkomplex: Beim Abkühlen erhält man langgestreckte,schiefwinkelige Prismen und Nadeln.
Cyclobarbital (5-Aethyl-5-cyclohexenyl-barbitursäure)
Smp. der Reinsubstanz (Block): 171-172°Smp. des Sublimates (mikro): 172-173
Sublimatbildung und - eigenschaf ten: Hier lag die Subli¬mations - Anfangstemperatur bei ca. 80°. Der entstandene Anflug war
durchwegs aus Tröpfchen gebildet. Bei Steigerung der Bad-Temperaturbildeten sich kurzprismatische Kristalle, die dann oft parallel zu flachen
Aggregaten verwachsen waren, und leistenförmig zugespitzte Kristalle(Abb. 16).
Eisenkomplex: Der Komplex bildet sich sehr leicht. Beim Er¬kalten zeigen sich zuerst kleine Gebilde, die dann rasch zu grossen brau¬
nen, zum Teil segeiförmigen Platten auswachsen (Abb. 67).
Kupferkomplex: Der sich erst allmählich beim Erkalten zei¬
gende Komplex besteht aus fast schwarzen, laubmoosförmigen Kristallen(Abb. 68).
Medomin (S-Aethyl-S-cycloheptenyl-barbitursäure)
Smp. der Reinsubstanz (Block): 171-173°Smp. des Sublimates (mikro): 171°

- 121 -
Sublimatbildung und - eigenschaf ten : Bei der Sublimation
bildeten sich zuerst Tröpfchen, die dann zu Kristallaggregaten erstarrten.
Bei der günstigsten Sublimationstemperatur von 100-110° bildeten sich
flache, viereckige oder mehreckige Platten, die zu grösseren Gebilden
zusammengelagert waren (Abb. 52). Daneben fanden sich eckige Platten,die halbmondförmig zusammengelagert waren (Abb. 53).
Schmelzpunkt: Beide Formen schmolzen bei 171°.
Eisenkomplex: Der Komplex bildete sich gut aus und bestand aus
unregelmässig begrenzten Rhomben und Aggregaten.
Kupferkomplex: zeigt dasselbe Bild wie der Eisenkomplex.
Cyclopal (5-Allyl-5-cyclopentenyl-barbitursäure)
Smp. der Reinsubstanz (Block): 136-138°Smp. des Sublimates (mikro): 1136-138
,H 124,5-126,5
Sublimatbildung und -eigenschaften: Das erste Auftreten
eines Sublimates beobachteten wir bei einer Bad-Temperatur von ca. 90°.Beim Steigern der Temperatur verstärkte sich die Sublimatbildung. Das
Sublimat, das sich zunächst in Tröpfchenform zeigte, kristallisierte bei
der günstigsten Sublimationstemperatur von 100-105° zu langen, ver¬
zweigten Nadeln und Spiessen aus (Abb. 54). Neben dieser Kristallform
fanden sich in den Sublimaten noch ziemlich grosse prismatische, an bei¬
den Enden zugespitzte Kristalle (Abb. 55).
Schmelzpunkt: Die Nadeln, die öfters zu Garben zusammenge¬
lagert waren, zeigten den Smp. 136-138°, während die andern Kristalle
bei 124, 5-126,5° schmolzen.
Eisenkomplex: Beim Erkalten bildeten sich zuerst dunkelbraune
Tröpfchen. Neben unverändert ausgeschiedenem Cyclopal fanden sich nach
einiger Zeit rötliche, lange Nadeln.
Kupferkomplex: Dieser Komplex kristallisiert besser als der
Eisenkomplex; wir erhielten hier dünne Nadeln, die an einem Ende fächer¬
förmig ausgebreitet waren.
Narconumal (5-Aethyl-5-isopropyl-N-methyl-barbitursäure)
Smp. der Reinsubstanz (Block): 63,5-64°Smp. des Sublimates (mikro): 57, 5-58, 5
Sublimatbildung und -eigenschaf ten: Infolge des niedrigenSmp. von Narconumal war die Sublimation hier schwieriger. Bei einer
Bad-Temperatur von 45° bildeten sich nur Tröpfchen, die erst nach einigerSublimationsdauer erstarrten und Nadeln zeigten, die mehr oder wenigergarbenförmig angeordnet waren (Abb. 56). Stiegen wir mit der Bad-Tempe¬ratur auf 50°, so erhielten wir auch Tröpfchen, erkannten aber an einigenStellen im Sublimat die Entstehung eines Hofes, in dessen Zentrum sich
ein Kristallherd gebildet hatte. Dieser bestand aus übereinander gelager¬ten Nadeln, die öfters büschelförmig angeordnet waren.
Schmelzpunkt: Die erstarrten Kristalle und die Nadeln schmolzen
bei 57, 5-58,5°.

- 122 -
Eisenkomplex: Beim Erkalten schieden sich Tröpfchen aus. Aus
ihnen bildeten sich nach einiger Zeit schmale, einzeln liegende, lange Na¬
deln, die hie und da verzweigt waren.
Kupferkomplex: Auch hier bildeten sich aus der Tröpfchenvor¬
stufe Nadeln.
Hexobarbital (S-Methvl-S-cyclohexenyl-N-methyl-barbitursaare)
Smp. der Reinsubstanz (Block): 142-143°Smp. des Sublimates (mikro): 144-145
Sublimatbildung und -eigenschaften: Bei der günstigsten
Sublimationstemperatur von 100-110° erhielten wir flache Nadeln, deren
Enden eingekerbt sind. Sie sind oft verwachsen und am Rande stufenför¬
mig abgesetzt (Abb. 57).
Eisenkomplex: Der Komplex fiel beim Erkalten zuerst tropf-
chenförmig aus und es bildeten sich erst nach einiger Zeit lange, aus den
Tröpfchen herausragende, Nadeln aus.
Kupferkomplex: Hier erhielten wir beim Abkühlen feine, farb¬
lose Nadeln, die einen gemeinsamen Mittelpunkt besitzen und an den En¬
den verzweigt sind. Nach Lang und Stephan (102) sind diese "Evipan-
Sonnen" besonders gut zur Unterscheidung des Hexobarbitals geeignet.
Methylphenobarbital (5-Aethyl-5-phenyl-N-methyl-barbitur-säure)
Smp. der Reinsubstanz (Block): 175-176°
Smp. des Sublimates (mikro): 174-176
Sublimatbildung und - eigenschaften: Es bildeten sich
hier zuerst Tröpfchen, dann Kristalle. Die günstigste Sublimationstempe¬
ratur lag bei ca. 110°. Die bei dieser Temperatur erhaltenen Sublimate
bestanden aus dünnen, fünf- und sechseckigen Plättchen, daneben fanden
sich auch solche mit rhomboidischem und trapezförmigem Umriss (Abb.12).
Methylphenobarbital gab mit den einzelnen Schwermetallreagenzienkeine Fällungen.
Eunarcon (5-Isopropyl-5-(fi-bromallyl)-N-methyl-barbitursäure)
Smp. der Reinsubstanz (Block): 99-102° Z.
Smp. des Sublimates (mikro): 114,5-116,5°
Sublimatbildung und -eigenschaften: Bei einer Bad-
Temperatur von ca. 90° erhielten wir zuerst Tröpfchen, die dann bald zu
Kristallaggregaten erstarrten. Die Aussenränder dieser Aggregate waren
ziemlich scharf abgezeichnet und gerade. Sie scheinen aus stufenförmigübereinander gelagerten Nadeln oder Tafeln zusammengesetzt zu sein
(Abb. 58). Die günstigste Sublimationstemperatur lag zwischen 85-95°.

- 123 -
Schmelzpunkt: Bei der Bestimmung des Smp. auf dem Block trat
unter Braunfärbung Zersetzung ein, während das Sublimat ziemlich schön
bei 114, 5-116, 5° schmolz.
Eisen- und Kupferkomplexe: Sie konnten nur in der Tröpf¬chenstufe erhalten werden.
Thiopental (5-Aethyl-5-(l-methylbutyl)-2-thio-barbitursäure)
Smp . der Reinsubstanz (Block): 155, 5-156, 5°Smp. des Sublimates (mikro): 153,5-155,5
Sublimatbildung und - eigenschaften : Das erste Auftreten
eines Sublimates beobachteten wir bei ca. 85°, wo es jedoch nur aus Tröpf¬chen bestand. Beim Steigern der Bad-Temperatur auf 100-110° bildeten
sich Kristalle. Diese waren zu Aggregaten zusammengelagert und nicht
immer schön ausgebildet (Abb. 59).
Schmelzpunkt: Der Smp. betrug 153, 5-155,5 .
Eisen- und Kupferkomplexe: Sie bildeten sich nur in Form
von Tröpfchen aus.
Kemithal (5-Allyl-5-cyclohexenyl-2-thio-barbitursäure)
Smp. der Reinsubstanz (Block): 120-122° / 140-141°Smp. des Sublimates (mikro): 140-142
Sublimatbildung und -eigenschaften: Die Sublimation von
Kemithal geht etwas schwieriger als die der anderen Derivate. Bei einer
Bad-Temperatur von 90-95° fanden sich nach 15-stündiger Dauer im Su¬
blimat fast nur Tröpfchen, daneben vereinzelte Kristalle. Steigerten wir
die Temperatur, so bildete sich das Sublimat leichter, aber es waren ne¬
ben Kristallen auch noch Tröpfchen vorhanden. Die günstigste Sublima¬
tionstemperatur von 100-105° lieferte jedoch nach längerer Zeit ein kri¬
stallines Sublimat, das aus prismatischen Kristallen mit abgerundetenEcken und Kristallaggregaten bestand (Abb. 60).
Schmelzpunkt: Die aus dem Natriumsalz mit Säure in Freiheit
gesetzte Allyl-cyclohexenyl-thiobarbitursäure zeigte bei der Schmelzpunkt¬bestimmung ein verschiedenartiges Verhalten. Wurde die Säure im Schwe-
felsäureexsikkator oder bei ca. 80° im Trockenschrank getrocknet, so
floss die Substanz bei der Schmelzpunktbestimmung bei 120-122° zu einer
undurchsichtigen Masse zusammen, die sich bei Steigern der Temperaturaufhellte und klar wurde. Wurde die Säure jedoch im Hochvakuum oder
längere Zeit bei gewöhnlichem Vakuum getrocknet, so erhielten wir einen
scharfen und klaren Schmelzpunkt von 140-141°. Dieser Smp. stimmt mit
dem von Raventos (120) und der Herstellerfirma angegebenen Schmelz¬
punkt überein. Dieses verschiedenartige Verhalten lässt sich dadurch er¬
klären, dass die niedrigschmelzende Form wahrscheinlich noch Kristall¬
wasser enthält.
Eisen- und Kupferkomplex: Es konnten nur tropfchenförmigeFällungen erhalten werden.
In Tabelle 30 sind die gefundenen Schmelzpunkte zusammengestellt.
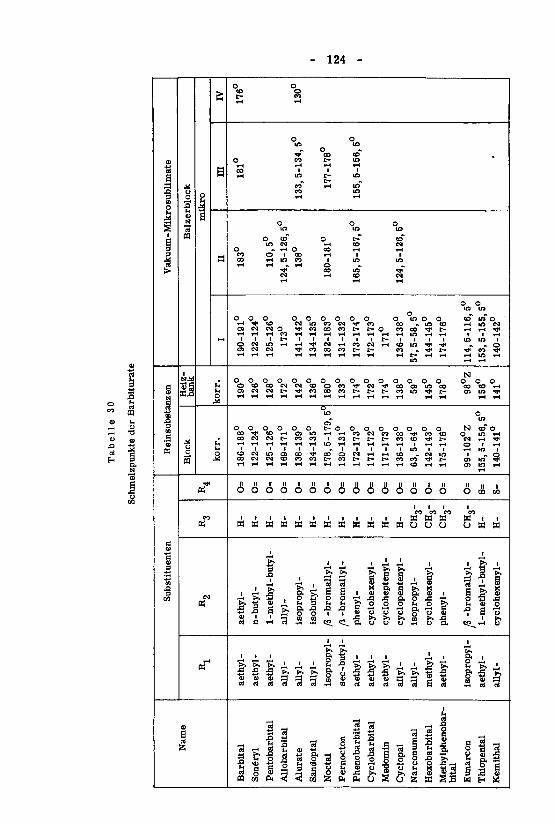
WHWSSXZOOQifUf\<<PienCQ
emithalhlopentalunarcon3"8
S*Q.J=
gSI«sui
j
exobarbltalarconumalyclopalMedomin|
yclobarbitalal
bit
baro
henernoctonoctalSandoptal|
luratellobarbitalentobarbitalDnérylarbitalName
allyl--
aethylisopropyaethyl--
methylallyl-allyl-aethyl--
aethylaethyl-sec-butyisopropyallyl-allyl-allyl--
aethylaethyl--
aethyltii-t
111
fi>>uU
>>Ua
43
^"S.CQ.saiH
i
Ci1
cd0)
sa•?0)G
Uisop"wO"o1
SI1
Xioja§•SS-C
o»ubOuooOhust*_L0)
but%ti
43oaj:43
enylooo
,MS.CMw hexthy]om;1S"ÈGPi
ffiK
iaa2*£t
1§
enylitenyteny:enylallylallylistitu enyl-L-butyallyl-11111i
l-butyV
tlnten
xmoOUOXXXXXXKxxSBXXtitXXXX1iiiiiii111eo
eo
i
coeo
t
co
i
OOOoOOOOOOOOOOoti CO(QOnuMiiIIII11IIiiIfIIII11IIIlIIIIII-<*
1-H
1-Hmos
^most-^eocoE-r-t-cor-coeocoCMCM00a8o~lmCMCDfifiCMOCO•<*coeninCMCOqo
tmfi1fin111t111u«f*>OuQ>
^1-HCMt-^coeoc-t-r-eoieoCOC-CMCM00M
H»hinOCDCO*00coCMCOftmC»i-HCD^00Oco"N
in
o
oooOooOO79,5
ooOOOosubsti
oanz
^most-*meot-c-r-eo00CO^*t-CMCNO)
kor1-CCO0000mOScoTr»CMTfeooCOCMCM00COo^
xS0)s
OOOoOOoOoOOoOOOOOo2%N
fi1-H
c-*
in
eoC-c-eoCOeo*PCMCMOSo.,-**incoi-HCMeo1-1CM*<**fifimNo
imm111t-i1ii1t-111fi
tH11i-Hi-Hin1-1wftr-ii-HftinfiCOi-Hi-Hfin*ftftc-^rcocoot-c-eo00CO-«t*oCMCMOScmini-icoin00co<•*«CMeomCMCO-*fioincoooinOoooooOOoo
ommoo
fi1-Hfi
CMco1-1CMt*m00"#>
Ofi»i-H1-1mm1eoinfi00a
1ii-lCO1cTCO
a3
126,55
167,81°otH
cm
co"
m
in
o
o
BaloooM
mik;erbkro:t->m
i-ii-ioO^
meoCJ43
in*i-Heo">s•m
m77-m
i
1-1
COSa
ft1-Hi-ii-H
int-COoV
co00Vo
inm
oo
130176fcoO
-124-

- 125 -
m. UEBERPRUEFUNG DER GRUPPENREAKTIONEN
Nach der Isolierung durch Ausschüttetung und nach eventueller Reini¬
gung des Rückstandes durch Sublimation ist es zweckmässig, die Barbi-
turatgruppe als solche zu charakterisieren, um damit die Anwesenheit
oder Abwesenheit von Barbitursäure - Derivaten in der Ausschüttelungs-gruppe festzustellen. Von den in der Literatur als Gruppenreaktion auf
Barbiturate aufgeführten Reaktionen überprüften wir vor allem die Farb¬
reaktionen mit Kobalt-H-Salzen und Kupfer-II-Salzen auf ihre Spezifität.Daneben sollte festgestellt werden, ob eine bestimmte Reaktion auf alle
Barbitursäure-Derivate (Dialkyl-, Halogenalkyl-, aromatische und cycli-
sche, N-methylierte und Thio-Verbindungen) oder nur auf einige dieser
Verbindungen anspricht und so eine Differenzierung innerhalb der Barbi-
turatgruppe ermöglicht.
A. Farbreaktionen mit Kobalt-II-Salzen
Die Reaktion beruht im Prinzip auf der Bildung eines violetten Kom¬
plexes des Barbiturates mit einem Co-II-Salz und einer basischen Kompo¬nente in einem wasserfreien Lösungsmittel. Die einzelnen Reaktionspartnermüssen in einem optimalen Verhältnis verwendet werden, das der stöchio-
metrischen Zusammensetzung des Komplexes entsprechen muss. Bei einem
Ueberschuss an anorganischer Base fällt basisches Kobaltsalz bzw. -hy-droxyd aus.
Als basische Komponenten prüften wir anorganische und organischeBasen auf ihre Verwendbarkeit bei der Kobalt-Farbreaktion.
1. Kalilauge
Die einzelnen Barbiturate wurden in 1-2 cm"* absolutem Alkohol ge¬
löst, 0, 5 cm3 einer l%igen Lösung von Kobaltnitrat in absolutem Alkohol
und 0, 5 cm^ einer l%igen Lösung von Kaliumhydroxyd in absolutem Alko¬
hol zugesetzt.
Die Dialkyl-, Halogenalkyl- und die aromatischen und cyclischen De¬
rivate ergaben eine blauviolette Färbung, die Thio-Derivate waren mehr
violett gefärbt, während die N-methylierten Derivate, mit Ausnahme von
Narconumal, nicht reagierten. Bei Narconumal trat eine blauviolette
Färbung auf, die jedoch rasch verblasste. Die Färbung ist nicht stabil und
verschwand innerhalbkurzer Zeit unter Abscheidung eines Niederschlages.

- 126 -
2. Ammoniak
Wir lösten die Barbiturate in 2 cm3 absolutem Methanol auf, fügten2 Tropfen einer 3%igen Lösung von Kobaltnitrat in absolutem Methanol und
2 Tropfen einer 10%igen Lösung von Ammoniak in absolutem Methanol zu.
Alle Barbiturate gaben eine violette Färbung, die ca. 10 Minuten be¬
ständig war.
3. Bariumoxyd
Zu einer Lösung von etwas Barbiturat in 1 cm3 absolutem Methanol
fügten wir 3 Tropfen einer 2%igen Lösung von Kobaltchlorid in absolutem
Methanol und 2 Tropfen einer gesättigten Lösung von Bariumoxyd in abso¬
lutem Methanol zu.
Alle Derivate, mit Ausnahme von Hexobarbital und Methylphenobar-bital, gaben eine blauviolette Färbung, die nach einigen Minuten, unter
Abscheidung eines blauvioletten Niederschlages, verblasste.
4. Natriummethanolat
Verwendeten wir als basische Komponente eine 5%ige Lösung von
Natriummethanolat in absolutem Methanol, so gaben alle Barbitursäure-
Derivate eine Violettfärbung, die bei Hexobarbital und Methylphenobarbitaljedoch schnell verblasste.
5. Piperidin-Ammoniak
Wir lösten wenig Substanz in 1 cm^ absolutem Methanol, setzten
0, 5 cm3 einer l%igen Lösung von Kobaltnitrat in absolutem Methanol und
dann 0, 2 cm3 einer Mischung von 5 T. 10%igem Piperidin und 1T. 5%igemAmmoniak in absolutem Methanol zu.
Die Dialkyl-, Halogenalkyl-, die aromatischen und cyclischen, die
Thio - Derivate und Narconumal gaben eine violettrötliche Färbung, die
nach drei Tagen noch vorhanden war, aber im Vergleich zu der frisch ge¬färbten Lösung abgeschwächt war; Hexobarbital, Methylphenobarbital und
Eunarcon waren schwächer gefärbt, und die Färbung war nur ca. eine
Stunde haltbar.
Gebrauchten wir anstelle von Kobaltnitrat getrocknetes Kobaltacetat
und anstelle von absolutem Methanol als Lösungsmittel der Barbiturate
absolutes Chloroform oder Isopropanol, so war die Färbung tiefer und
über drei Tage haltbar.

- 127 -
6. Isobutylamin
gZu einer Lösung des Barbiturates in 1,5 cm Chloroform fügten wir
0, 25 cm;> einer 0, Olm-Lösung von Kobaltacetat in absolutem Methanol und
0, 25 cmJ einer molaren Lösung von Isobutylamin in Chloroform zu.
Bei den Barbituraten trat eine rötlichviolette Färbung auf, währenddie Thio-Derivate mehr violett gefärbt waren.
Verwendeten wir anstelle von getrocknetem Kobaltacetat Kobaltnitrat,Co(NC>3)o.6 H2O, so ist die Violettfärbung intensiver, und das Reagensallein, das im ersten Fall sehr leicht violett gefärbt ist, ist gelbgrünlich.Die mit den Barbituraten erhaltene Färbung ist, im offenen Reagensglasaufbewahrt, über 5 Tage haltbar. Die mehr violettblaue Färbung der Thio-
Derivate geht allmählich in Bordeauxrot über.
7. Isopropylamin
3 3Wir lösten wenig Barbiturat in 1, 2 cm Chloroform, setzten 0,2 cm
l%ige Kobaltacetatlösung in absolutem Methanol und 0,6 cm^ 10%ige Iso-
propylaminlösung in Chloroform zu.
Alle Barbiturate zeigten eine violettrötliche Färbung, die sich von
der leichten Violettfärbung des Reagenzgemisches unterschied.
Mit 0,01m- und mit 0, 6%iger Kobaltacetatlösung, sowie mit l%igerKobaltnitratlösung in absolutem Methanol gaben die Barbiturate diesselbeviolettrötliche Färbung, während die Thiobarbiturate mit Co(NC<3)2.6 H2Oetwas mehr violettblau gefärbt waren.
q
Daneben lösten wir noch etwas Barbiturat in 1 cm einer Mischung
von 1 T. Isopropylamin und 9 T. Chloroform, setzten darauf 1 cm' einer
l%igen wässerigen Kobaltnitratlösung zu und schüttelten gut durch. Beim
Stehenlassen nimmt die Chloroformschicht eine violette Färbung in Gegen¬wart aller geprüften Barbitursäure-Derivate an, während die wässerigeSchicht einen blaugrünen Niederschlag aufwies. Beim Reagenzgemisch,ohne Barbiturat, ist die Chloroformschicht gelblichgrün gefärbt und die
wässerige Schicht zeigt einen blauen Niederschlag.
Tüpfelreaktion
Um die Empfindlichkeit der Kobaltreaktion zu steigern, führten wir
die Reaktion auch auf Filterpapier durch. Zu diesem Zweck lösten wir et¬
was Barbiturat in Chloroform auf und brachten diese Lösung tropfenweiseauf einen Streifen Filterpapier und zwar in der Weise, dass wir jedenTropfen eintrocknen Hessen, ehe wir den nächsten zugaben. Hierauf fügtenwir 1-2 Tropfen einer l%igen Kobaltnitratlösung in absolutem Methanol
zu, trockneten und setzten die so behandelte Stelle Ammoniakdämpfen aus,wobei sich ein violettblauer Fleck bildete. Auf diese Weise konnten wir,je nach Barbiturat, 5-20 "$ nachweisen, während die Empfindlichkeitsgrenzeim Reagenzglas bei ca. 0,1 mg lag.

- 128 -
Besprechung
Das Kobaltsalz an und für sich spielt beim qualitativen Nachweis von
Barbituraten keine Rolle, denn ob wir ein kristallwasserhaltiges oder ein
wasserfreies Salz verwenden, fällt, in Gegenwart von Barbitursäure-
Derivaten, die Reaktion positiv aus. Bei quantitativen Bestimmungen hin¬
gegen dürfte das wasserfreie Kobaltacetat vorteilhafter sein.
Als Lösungsmittel eignet sich ein Alkohol-Chloroform-Gemisch bes¬
ser als nur ein Alkohol allein. Die besten Resultate erhielten wir mit
einem 10-12,5%igen (v/v) Methanol-Chloroform-Gemisch, dessen Kon¬
zentration auch derjenigen von Rasmussen und Jerslev (123) und
Koppanyi und Mitarb. (119) entspricht. Methanol kann auch durch
absolutes Aethanol oder Isopropanol ersetzt werden.
Ist in dem Lösungsmittelgemisch ein mit Wasser mischbares Lö¬
sungsmittel vorhanden, so wird, in Gegenwart von Wasser, der Komplexdurch Hydrolyse gespalten, was die besondere Empfindlichkeit der Nach¬
weisreaktion gegenüber wasserhaltigen Lösungsmitteln zu erklären scheint.
Verwendeten wir hingegen nur ein, und mit Wasser nicht mischbares Lö¬
sungsmittel, z.B. Chloroform, und eine wässerige Kobaltsalzlösung, so
bildete sich der Komplex dennoch, d. h. die Chloroformschicht zeigte die
charakteristische Violettfärbung, während sich in der wässerigen Schicht
ein blauer Niederschlag von basischem Kobaltsalz bzw. -hydroxyd bildete.
Die basische Komponente kann anorganisch oder organisch sein, doch
sind die aliphatischenAmine den starken anorganischen Basen vorzuziehen.
Mit steigender Amin-Konzentration nimmt auch die Farbintensität zu und
erreicht nach Rasmussen und Jerslev (123) ihr Maximum bei 100
Mol Amin auf 1 Mol Kobaltsalz, doch ist, bei der qualitativen Analyse,auch schon eine geringere Konzentration genügend.
Von den übrigen, in derselben Gruppe des Analysenganges nach Eder
anfallenden Stoffen reagieren Theophyllin und Theobromin Natrium sa-
licylicum mit der Kobaltreaktion schwach positiv. Daneben geben auch
einige Sulfonamide eine schwach positive Reaktion. Man kann diese Re¬
aktion demnach als mehr oder weniger spezifisch für die Barbitursäure-
Derivate ansehen.
Eine Differenzierung innerhalb der Barbituratgruppe mit Hilfe der
Kobaltreaktion ist nur zwischen den N-methylierten Derivaten Hexobar¬
bital und Methylphenobarbital einerseits und den übrigen Derivaten ander¬
seits unter der Bedingung möglich, dass man als basische Komponenteeine starke anorganische Base (KOH, NaOH, BaO) verwendet. Unter die¬
sen Voraussetzungen reagieren Hexobarbital und Methylphenobarbital nicht,Narconumal und Eunarcon schwach, aber doch deutlich positiv. In Gegen¬wart von Ammoniak und von organischen Basen (Piperidin, Pyridin, ali¬
phatischen Aminen) hingegen geben alle Barbitursäure-Derivate eine ge¬färbte Komplexverbindung, die jedoch bei Hexobarbital und Methylpheno¬barbital weniger haltbar ist.

- 129 -
B. Farbreaktionen mit Kupfersalzen
Ebenso wie die Kobalt-II-Salze geben die Kupfer-II-Salze in Gegen¬wart einer basischen Komponente mit Barbitursäure-Derivaten in einem
organischen Lösungsmittel gefärbte Komplexverbindungen.
Als Base verwendeten wir Pyridin und Isopropylamin und als Lösungs¬mittel Chloroform und führten die Reaktion folgendermassen aus:
o
Wir lösten wenig Barbiturat in 1 cm des Pyridin- bzw. Isopropyla-min-Chloroform-Gemisches (1 T. Pyridin bzw. Isopropylamin und 9 T.
Chloroform), setzten dann 1 cm3 einer l%igen wässerigen Kupfersulfat¬lösung zu und schüttelten gut durch. Nach dem Absetzenlassen zeigte die
Chloroformschicht eine violette Färbung in Gegenwart von Barbituraten
und mit Thiobarbituraten eine Grünfärbung. Die wässerige Schicht war bei
allen blau gefärbt.
Von den übrigen, in derselben Gruppe des Analysenganges nach Eder
anfallenden Stoffen war die Chloroformschicht mit Salicylsäure grünblau,Aspirin und Hydantoinen blau, Theophyllin, Theobromin Natrium salicyli-cum und Coffein Natrium salicylicum und Thiourazilen grün, mit Persedon
und Sedulon nicht gefärbt.
Innerhalb der Barbitursäure-Gruppe können wir mit dieser Reaktion
zwischen Barbituraten und Thiobarbituraten unterscheiden. Ferner erlaubt
sie uns eine Differenzierung zwischen Barbituraten, Thiobarbituraten,Hydantoinen, Thiourazilen, Purin-Derivaten und Persedon und Sedulon,und eignet sich demnach vorteilhaft als Gruppenreaktion auf diese Körper¬klassen.
IV. IDENTIFIKATION DER EINZELNEN DERIVATE
Hat man an Hand der Kobalt- oder Kupferfarbreaktion den zu unter¬
suchenden Stoff als ein Barbitursäure-Derivat gekennzeichnet, so müssen
wir nun das Barbiturat selbst identifizieren.
Neben den physikalischen Konstanten, wie z. B. Schmelzpunkt- und
Mischschmelzpunktbestimmung, müssen öfters auch noch chemische Re¬
aktionen zum Nachweis beitragen, denn die Schmelzpunkte der einzelnen
Derivate liegen zum Teil ziemlich nahe aneinander, so dass eine Unter¬
scheidung an Hand des Schmelzpunktes allein nicht ausreicht. Innerhalb
der Barbituratgruppe kann man, je nachdem Brom, Schwefel oder keines
dieser Elemente im Molekül vorhanden ist, bereits drei Hauptgruppenunterscheiden. Diese Gruppen können wir dann noch weiter unterteilen in
solche mit gesättigten, mit aromatischen und cyclischen Radikalen. Als
weitere Identifizierungsmöglichkeit der einzelnen Barbiturate stellten wir
Derivate von p-Nitrobenzylchlorid und von Xanthydrol her, um so mit
Hilfe ihrer Schmelzpunkte die Barbiturate zu unterscheiden.

- 130 -
Brom-Nachweis (Beilstein'sche Probe)
Ein Kupferdraht wird ausgeglüht, bis er die Flamme nicht mehr grünfärbt. Nach dem Erkalten bringt man ihn in die zu untersuchende Substanz
und erhitzt ihn dann am Rande der nicht leuchtenden Flamme. Halogen-haltige Substanzen bewirken eine Grünfärbung.
Diese Probe ist positiv bei Noctal, Pernocton und Eunarcon.
Schwefel-Nachweis
a. Lassaigne'sche Probe:
In einem Glühröhrchen wird etwas zu untersuchende Substanz mit
einem sauberen Stückchen Natriummetall erhitzt und zum Schmelzen ge¬bracht. Man erhitzt dann noch kurz zur Rotglut und bringt das Glühröhr¬
chen noch heiss in 2-3 cm-* destillierten Wassers. Den Schwefel kann man
nun mit Bleiacetat nach Ansäuern mit Essigsäure oder durch Tüpfeln der
alkalischen Lösung mit Nitroprussidnatrium - Violettfärbung in Gegenwartvon Schwefel - nachweisen.
b. Neben dieser Aufschlussmethode kann man die Thio-Derivate
noch folgendermassen nachweisen. Man versetzt etwas Substanz mit 1-2cm3 Wasser, fügt 2 Tropfen konzentrierte Natronlauge und 2 Tropfen5%ige Nitroprussidnatrium-Lösung zu. Es bildet sich eine orangerötlicheFärbung. Man lässt eine Stunde stehen und fügt dann 10-12 Tropfen Salz¬
säure zu. Die Farbe der Lösung geht über grün in rot; dieser rote Farb¬
stoff ist in Amylalkohol löslich.
Diese beiden Reaktionen sind positiv bei Thiopental und Kemithal.
Nachweis von ungesättigten Radikalen (Permanganat-Reaktion)
In alkalischer Lösung reduzieren ungesättigte Reste das Permanga-nat-Ion zum grünen Manganat-Ion. Je nachdem welches Radikal vorliegtgeht die Reduktion schneller oder langsamer, d.h. die Farbe wechselt
direkt von violett zu grün oder geht von violett über blau nach grün. Zur
Ausführung dieser Reaktion lösten wir wenig Substanz in 1 cm3 2n-Natron-
lauge und fügten dann 1 Tropfen 0, ln-Kaliumpermanganat-Lösung zu. Es
wirken reduzierend: Allobarbital, Alurate, Sandoptal, Noctal, Pernocton,Cyclobarbital, Medomin, Cyclopal, Hexobarbital, Narconumal, Eunarcon,Kemithal und Thiopental. Letzteres reduziert ebenfalls Kaliumpermanga-nat, obschon es keine ungesättigte Gruppe enthält.
Verwendeten wir anstelle von Natronlauge eine 5%ige Bikarbonat¬
lösung, so wurde Kaliumpermanganat von Allobarbital, Alurate, Sandoptal,Cyclopal, Narconumal und Thiopental reduziert. Bei Noctal, Pernocton,Cyclobarbital, Medomin, Hexobarbital, Eunarcon und Kemithal trat die
Grünfärbung erst nach einiger Zeit auf.

- 131 -
Formaldehyd-Schwefelsäure-Reaktion
In einem Reagenzglas werden zu etwas Substanz 2 cm konzentrierte
Schwefelsäure und dann 4 Tropfen Formaldehyd Ph. H. V zugesetzt und
während 2 bis 3 Minuten in einem siedenden Wasserbad erhitzt. Barbital
und Sonéryl zeigten keine Reaktion, Phénobarbital und Methylphenobarbitalwiesen eine weinrote Färbung auf, Cyclopal schon auf Zusatz von Schwe¬
felsäure eine Braunfärbung, und alle übrigen Derivate zeigten eine grüneFluoreszenz.
Mit dieser Reaktion kann man Phénobarbital und Methylphenobarbital,sowie Barbital und Sonéryl von den übrigen Barbituraten unterscheiden.
Salicylaldehyd-Schwefelsäure-Reaktion
o
In einem Reagenzglas werden zu etwas Substanz 2 cm konzentrierte
Schwefelsäure zugesetzt. Hierauf fügt man einen Tropfen einer 5%igenalkoholischen Salicylaldehydlösung zu und erhitzt einige Minuten in einem
siedenden Wasserbad. Beim Erhitzen entsteht in Gegenwart von Allobar-
bital eine Rotfärbung, während alle übrigen Derivate nicht reagierten,sondern nur die Gelbfärbung des Reagens zeigten.
m-Nitrobenzaldehyd-Schwefelsäure-Reaktion
10 Tropfen einer l%igen Lösung von m-Nitrobenzaldehyd in kon¬
zentrierter Schwefelsäure wurden mit wenig Substanz eine halbe Minute
auf dem Wasserbad erwärmt. Die Derivate mit einem Cyclohexenylrestim Molekül zeigten eine dunkelrote Färbung beim Erhitzen; Medomin
wurde violettrot und Thiopental rot gefärbt.
Vanillin-Schwefelsäure-Reaktion
Als Reagenzien verwendeten wir hier eine 5%ige alkoholische Va¬
nillinlösung und hydratisierte Schwefelsäure, die wir durch Mischen von
2 T. konzentrierter Schwefelsäure und 1 T. destillierten Wassers her¬
stellten. In einem Reagenzglas lösten wir etwas Barbiturat in 5 Tropfen
Vanillinlösung, setzten dann 2 cm^ hydratisierte Schwefelsäure zu und er¬
hitzten während 3 Minuten in einem siedenden Wasserbad. Cyclopal zeigteschon in der Kälte eine schöne smaragdgrüne Färbung, Kemithal war grün¬lichgelb gefärbt, und alle anderen Barbiturate zeigten die gelbe Farbe des
Reagens. Beim Erhitzen wurde Cyclopal dunkelblau, Cyclobarbital, Me¬
domin, Hexobarbital und Kemithal rot gefärbt, während die übrigen gelbblieben. Fügten wir nun 2 cm"> Wasser zu, so blieb bei Cyclopal die dun¬
kelblaue Farbe bestehen, bei Cyclobarbital, Medomin, Hexobarbital und
Kemithal schlug die rote Farbe in violett um.

- 132 -
Vanillin-Salzsäure-Reaktion
Wir lösten etwas Barbiturat in 5 Tropfen einer 5%igen alkoholischen
Vanillinlösung, setzten 2 cm' Salzsäure zu und erhitzten auf dem Wasser¬
bad. Alle Barbiturate zeigten die gelbliche Färbung des Reagenzgemischesmit Ausnahme von Cyclopal, das in der Kälte blaugrün, beim Erhitzen
dunkelblau und nach Verdünnen mit 2 cm3 Wasser blau gefärbt war.
Mandelin's Reagens
Wir benützten ein Reagens folgender Zusammensetzung: 0,1 g fein
zerriebenes vanadinsaures Ammonium wurden kalt in 20 g konzentrierter
Schwefelsäure gelöst.
Dieses Reagens gab mit den einzelnen Barbituraten keine charakte¬
ristischen Färbungen.
Mecke's Reagens
Beim vorsichtigen Erhitzen von wenig Substanz mit 0, 5-1 cnr* Rea¬
gens (0,1 g selenige Säure in 20 cm3 konzentrierter Schwefelsäure) über
freier Flamme bildete sich bei fast allen Barbituraten eine Grünfärbung.
Obschon diese Reaktion bei unseren Versuchen nicht immer sehr
eindeutig war, benützen Turfitt (40), Motha (132) und Parkes (133)diese Reaktion als Gruppenreaktion auf Barbiturate.
p-Dimethylaminobenzaldehyd-Schwefelsäure-Reaktion
Zu etwas Substanz in einem Reagenzglas fügten wir 0,5-1 cm^ einer
5%igen Lösung von p-Dimethylaminobenzaldehyd in konzentrierter Schwe¬felsäure zu.
Pentobarbital und Thiopental zeigten eine leicht bräunliche Färbungund eine grüne Fluoreszenz.
Cyclobarbital, Hexobarbital und Kemithal waren rot gefärbt, während
Cyclopal eine mehr braunrote Färbung aufwies.
Medomin zeigte eine auffallende Erscheinung. Bei durchfallendemLicht war die Lösung rot und bei auffallendem Licht betrachtet zuerst grünund nach einiger Zeit blau.
Die übrigen Derivate waren farblos oder zeigten nur eine leicht
gelbliche, nicht charakteristische Färbung.
Erhitzten wir die einzelnen Gemische während einer Minute in einem
siedenden Wasserbad, so konnten wir folgendes feststellen:

- 133 -
Pentobarbital und Thlopental nahmen eine dunkelrote Färbung an und
zeigten eine grüne Fluoreszenz.
Bei Cyclobarbital, Hexobarbital und Kemithal wurde die rote Färbungdunkler und ausgeprägter, während Cyclopal die braunrote Farbe behielt.
Medomin nahm eine dunkelviolette Färbung an.
Alurate und Narconumal zeigten beim Erhitzen eine rotviolette Fär¬
bung und Phemiton eine rötliche.
Wir führten diese Reaktion dann noch als Tüpfelreaktion aus und er¬
hielten dabei folgendes:
Cyclobarbital und Hexobarbital: ViolettfärbungKemithal: zuerst orange, dann rot
Cyclopal: braunrot
Medomin: grün, dann blau
Allobarbital: gelb, oliv, grün, violett
Verwendeten wir anstelle der 5%igen eine 2, 5%ige Lösung von p-
Dimethylaminobenzaldehyd in konzentrierter Schwefelsäure, so zeigtendie einzelnen Barbiturate dieselben Färbungen wie bei der 5%igen Lösung.
/5-Naphthol-Schwefelsäure-Reaktion
Etwas Barbiturat lösten wir in 5 Tropfen einer 5%igen alkoholischen
Lösung von ß -Naphthol und fügten dann 2 cm3 hydratisierte Schwefel¬
säure zu. Mit Cyclopal erhielten wir in der Kälte eine himbeerrote Farbe
und nach dem Erhitzen auf dem Wasserbad eine blaue. Alle übrigen Bar¬
biturate zeigten keine Färbungen, ausser Cyclobarbital, Hexobarbital und
Kemithal, die nach dem Erhitzen eine gelbgriine Opaleszenz aufwiesen.
In einem Reagenzglas lösten wir wenig Barbiturat in 5 Tropfen 5%igeralkoholischer Lösung von ß -Naphthol, fügten 2 cm3 konzentrierte Schwe¬
felsäure zu und erhitzten auf dem Wasserbad. Die Derivate mit einem
Allylrest im Molekül zeigten eine schwache grüne Fluoreszenz, die bei
Allobarbital aber stark ausgeprägt war. Die anderen Derivate zeigtenkeine charakteristischen Färbungen.
Benzaldehyd-Schwefelsäure-Reaktion
Zu einer Lösung von wenig Barbiturat in 6 Tropfen 5%iger alkoholi¬
scher Benzaldehydlösung fügten wir 2 cm3 hydratisierter Schwefelsäure
zu. Kemithal gab in der Kälte eine rosaviolette Färbung. Beim Erhitzen
auf dem Wasserhad zeigte Cyclopal eine dunkelviolette Färbung, die auf
Zusatz von 5 cm3 Wasser in grün umschlug. Die anderen Derivate waren,wie das Reagens, leicht gelbstichig.

- 134 -
Furfurol-Schwefelsäure-Reaktion
Wir lösten wenig Barbiturat in 6 Tropfen 5%iger alkoholischer Fur-
furollösung, fügten 2 cm^ hydratisierte Schwefelsäure zu und erhitzten
während 2 Minuten auf dem Wasserbad.
Cyclopal gab in der Kälte eine dunkelviolette Färbung, die beim Er¬
hitzen auf dem Wasserbad beständig war und auch beim Verdünnen mit
Wasser erhalten blieb.
Hexobarbital, Cyclobarbital und Medomin zeigten nach dem Erhitzen
eine Violettfärbung.
Kemithal war dunkelrot gefärbt; diese Farbe schlug beim Verdünnen
mit Wasser nach violettrot um.
Die anderen Barbiturate verhielten sich wie das Reagens.
Piperonal-Schwefelsäure-Reaktion
Auch hier ist es wieder nur Cyclopal, das auf Zusatz von 6 Tropfeneiner 5%igen alkoholischen Lösung von Piperonal und 2 cm«* konzentrier¬
ter Schwefelsäure eine eindeutige Reaktion ergab. Es bildete sich eine
smaragdgrüne Farbe, die auch beim Erhitzen bestehen blieb.
Phenol-Schwefelsäure-Reaktion
In einem Reagenzglas fügten wir zu etwas Substanz einige Tropfeneiner 5%igen wässerigen Phenollösung und 2 cm3 hydratisierte Schwefel¬
säure zu und erhitzten auf dem Wasserbad.
Cyclopal war in der Kälte goldgelb gefärbt, in der Hitze wurde die
Farbe mehr orangerötlich. Bei Zusatz von Wasser nahm die Lösung eine
rosa Färbung an, die auf Zusatz von 1 cm^ konzentriertem Ammoniaküber violett nach blau ging.
Cyclobarbital, Hexobarbital und Kemithal waren nach dem Erhitzen
rosa gefärbt; beim Verdünnen mit Wasser wurde die Lösung rosa-violett
und auf Zusatz von Ammoniak farblos.
Die anderen Barbiturate wiesen nur farblose Lösungen auf.

- 135 -
Schwefelsäure-Reaktion
q
Zu ca. 0,1 g Substanz setzten wir 2 cm konzentrierte Schwefelsäurezu und beobachteten bei den einzelnen Barbituraten die folgenden Farbver¬
änderungen:
Barbital, Sonéryl, Alurate, Sandoptal, Phénobarbital, Narconumal
und Methylphenobarbital lösten sich farblos auf.
Allobarbital löste sich zuerst farblos, dann gelblich und nach einigerZeit mit rötlicher Farbe vollständig auf.
Noctal, Pernocton und Eunarcon zeigten eine gelborange Färbung,die nach und nach mehr nach orange neigte.
Pentobarbital und Thiopental lösten sich mit gelber Farbe auf.
Cyclobarbital und Hexobarbital waren zuerst gelb bis gelbbraun ge¬
färbt, dann ging die Farbe langsam nach rotbraun über.
Kemithal löste sich mit orange Farbe auf, die nach einiger Zeit in
rötlichbraun überging.
Medomin wurde zuerst orange gefärbt, dann rotorange.
Cyclopal färbte sich auf Zusatz von Schwefelsäure braun.
In Tabelle 31 sind die wichtigsten Färbungen der Barbiturate mit den
einzelnen Reagenzien aufgeführt.
Die gesättigten Dialkyl-Derivate geben mit keinen Reagenzien Fär¬
bungen, mit Ausnahme von Pentobarbital, das auch nur mit p-Dimethyl-aminobenzaldehyd reagiert und so von den übrigen Derivaten unterschieden
werden kann.
Bei den ungesättigten Dialkyl-Derivaten können wir Allobarbital von
Alurate und Sandoptal besonders mit Hilfe von Salicylaldehyd unterscheiden.
Die halogenierten Barbiturate lassen sich mit diesen Reaktionen nicht
untereinander differenzieren, können aber durch die Anwesenheit von
Brom im Molekül von den anderen unterschieden werden.
Phénobarbital und Methylphenobarbital, beide mit einem Phenylrest,können wohl von den anderen Verbindungen mit einem aromatischen Rest,nicht aber voneinander unterschieden werden.
Barbiturate mit einem cyclischen Rest im Molekül geben die schön¬
sten Färbungen und lassen sich mit diesen Reaktionen, mit Ausnahme von
Cyclobarbital und Hexobarbital, voneinander differenzieren.
Die beiden Thio-Derivate lassen sich ebenfalls unterscheiden, wobei
Thiopental, trotzdem es kein ungesättigtes Radikal enthält, Kaliumper-manganatin natronalkalischer Lösung zu grünem Manganatsalz reduziert.
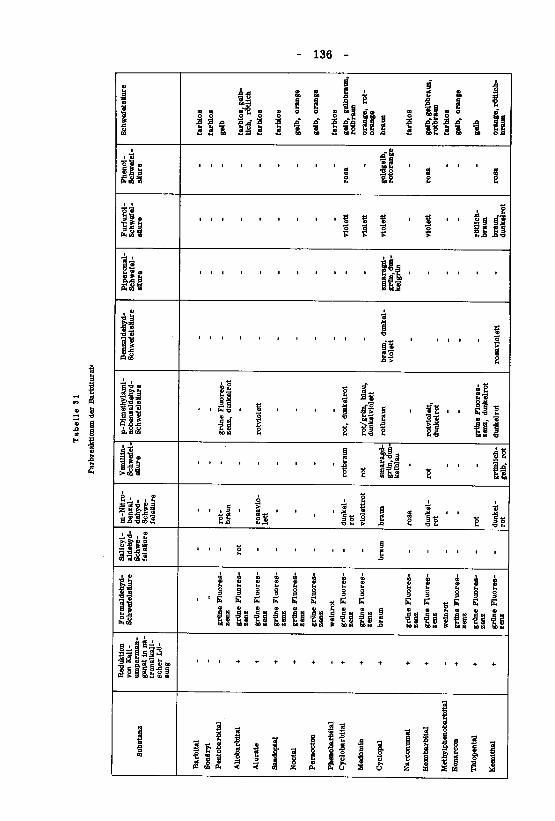
braun
rötlich-
orange,
gelb
roaa
dunkelrot
braun,
braun
rotlich-
-
rosavlolett
dunkelrot
dunkelrot
zenz,
Fluores¬
grüne
rot
gelb,
grünlich¬
rot
dunkel-
rot
-
zenz
Fluores¬
grüne
zenz
Fluores¬
grüne
++
Kemlthal
Thiopental
orange
gelb
,-
--
--
--
-
zenz
Fluores¬
grüne
+Eunarcon
farblos
rotbraun
gelbbraun,
gelb
,rosa
violett
-dunkelrot
rotviolett,
rot
rot
dunkel¬
~
rot
wein
zenz
Fluores¬
grüne
+
Metfaylphenobarbltal
Hexobarbital
farblos
--
--
--
rosa
-
zenz
Fluores¬
grüne
+Narconumal
braun
rotorange
gold
gelb
,violett
kelgründun¬
grün,
smaragd¬
violett
dunkel-
braun,
rotbraun
kelblaudun¬
grün
,smaragd¬
braun
braun
braun
+Cyclopal
orange
rot¬
orange,
-violett
--
violett
dunkel
blau,
rot/
grün
,rot
violettrot
-
zenz
Fluores¬
grüne
+Medomin
rotbraun
gelbbraun,
gelb
,rosa
violett
--
dunkelrot
rot,
rotbraun
rot
-dunkel
-
zenz
Fluores¬
grüne
+Cyclobarbital
farblos
--
--
--
--
rot
wein
-Phénobarbital
orange
gelb
,-
--
--
--
-
zenz
Fluores¬
grüne
+Pernocton
orange
gelb
,-
--
--
--
-
zenz
Fluores¬
grüne
+Noctal
farblos
--
--
--
--
zenz
Fluores¬
grüne
+Sandoptal
farblos
--
--
rotviolett
-
lett
rosavio¬
-
zenz
Fluores¬
grüne
+Alurate
rötlich
lich
,gelb¬
farblos,
--
--
--
-rot
zenz
Fluores¬
grüne
+AUobarbltal
gelb
--
--
dunkelrot
zenz,
Fluores¬
grüne
-
braun
rot¬
-
zenz
Fluores¬
grüne
-Pentobarbital
farblos
--
--
--
--
--
Sonéryl
farblos
Barbital
Schwefelsäure
säure
Schwefel¬
Phenol-
säure
Öl¬
Schwef
Furfurol-
säure
el-
Schwef
Piperonal-
Schwefelsäure
Benzaldehyd-
Schwefelsäure
nobenzaldehyd-
p-Dimethylaml-
säure
Öl¬
Schwef
Vanlllln-
säure
fel
Schwe-
dehyd-
benzal-
m-Nitro-
felsäure
Schwe-
aldehyd-
Sali
cyl-
Schwefelsäure
Formaldehyd-
sung
Lö¬
scher
tronalkali¬
na¬
in
ganat
umperman¬
Kali¬
von
Reduktion
Substanz
Barbiturat«
der
Farbreaktionen
31
Tabelle

- 137 -
Phenyl- und cyclische Radikale
Um das Phenylradikal und cyclische Polymethylenradikale zu cha¬
rakterisieren, stellten wir die entsprechenden Nitroverbindungen her. Zu
diesem Zwecke fügten wir zu ca. 0, 5 g des entsprechenden Barbiturates
5 cm3 konzentrierte Schwefelsäure und 10 Tropfen konzentrierte Salpeter¬säure zu und erhitzten 5-6 Minuten in einem siedenden Wasserbad. Nach
dem Erhitzen wurde das Gemisch in Eiswasser gekühlt und dann vorsichtigmit 20 cm3 Wasser versetzt. Der sich bildende Niederschlag wurde abge¬trennt.
Phénobarbital und Methylphenobarbital gaben beim Versetzen mit
Wasser einen gelben Niederschlag.
Die Lösungen von Cyclobarbital und Hexobarbital nahmen auf Zusatz
von Wasser eine grüne Farbe an, die bei weiterem Zusatz von Wasser
nach gelb umschlug und wobei sich dann auch ein gelber Niederschlagbildete.
Medomin nahm auf Zusatz der Nitrierflüssigkeit beim Erhitzen eine
olivbraune Farbe an, die sich beim Abkühlen aufhellte, auf Zusatz von
wenig Wasser in grün überging und auf weiteren Zusatz von Wasser gelbwurde. Hier bildete sich kein Niederschlag, sondern nur eine leichte Trü¬
bung, weshalb wir die wässerige Flüssigkeit 2 mal mit je 10 cm3 Aether
extrahierten, die Aetherlösungen abtrennten, mit wasserfreiem Natrium¬
sulfat trockneten und den Aether abdestillierten. Es hinterblieb ein
schmieriger Rückstand.
Kemithal wurde durch das Nitriergemisch rotbraun gefärbt. Auf Zu¬
satz von Wasser nahm die Lösung eine orangegelbe Färbung an und zeigtenur eine leichte Trübung. Auch hier extrahierten wir mit Aether und er¬
hielten einen etwas schmierigen Rückstand.
Cyclopal nahm beim Versetzen mit dem Schwefelsäure-Salpeter¬säure-Gemisch eine braune Farbe an. Bei Zusatz von Wasser hellte sich
die Flüssigkeit etwas auf und es entstand eine leichte Trübung, die wir
mit Aether extrahierten.
Die Niederschläge bzw. Rückstände der einzelnen nitrierten Barbi¬
turate wurden im Schwefelsäure-Exsikkator getrocknet.
Mit den erhaltenen Nitroverbindungen führten wir verschiedene Re¬
aktionen aus.

- 138 -
Diazo-Reaktion
In einem Reagenzglas fügten wir zu etwas Substanz ein Körnchen rei¬
nen Zink und ca. 2 cm3 verdünnte Schwefelsäure zu und erwärmten auf dem
Wasserbad, bis die Reaktion in Gang war. Wir Hessen so lange einwirken,bis sich die Nitroverbindungen vollständig aufgelöst hatten, filtrierten ab
und kühlten durch Einstellen in Eis ab. Zu dieser eisgekühlten Lösungfügten wir lern3 einer 10%igen Kaliumnitritlösung und dann eine alkalische
Jl -Naphthollösung zu.
Phénobarbital, Methylphenobarbital und Cyclobarbital gaben einen
schönen roten Niederschlag.
Bei Hexobarbital und Kemithal war der Niederschlag mehr orangerotgefärbt.
Medomin und Cyclopal zeigten einen orangegelblichen Niederschlag.
Mit cC -Naphthol erhielten wir dieselben Ergebnisse.
Reaktion mit Hydroxylamin und Ammoniak
Etwas Substanz erhitzten wir in einem Reagenzglas mit einigen Kri-
stellen Hydroxylaminhydrochlorid und ca. 2 cm3 konzentriertem Ammoniak
auf dem Wasserbad.
Mit Phénobarbital und Methylphenobarbital bildete sich direkt eine
dunkel rotviolette Färbung.
Hexobarbital, Cyclobarbital und Kemithal gaben nach einiger Zeit
eine mehr rotbraune Färbung.
Medomin und Cyclopal färbten sich orangebraun in der Wärme.
Kondensation mit Aceton in alkalischer Lösung
o
In einem Reagenzglas lösten wir wenig Substanz in 2 cm Aceton,fügten 2 cm3 verd. Natronlauge zu und schüttelten gut durch. Nach demAbsetzen konnten wir bei den einzelnen Verbindungen mehr oder wenigercharakteristische Färbungen feststellen, die in Tabelle 32 wiedergegebensind.
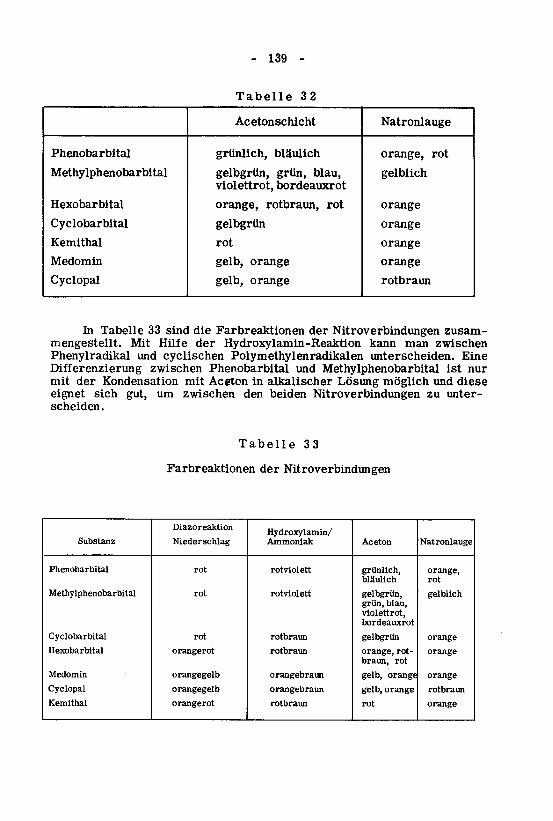
- 139 -
Tabelle 32
Phénobarbital
Methylphenobarbital
Hexobarbital
Cyclobarbital
Kemithal
Medomin
Cyclopal
Acetonschicht
grünlich, bläulich
gelbgrün, grün, blau,violettrot, bordeauxrot
orange, rotbraun, rot
gelbgrün
rot
gelb, orange
gelb, orange
Natronlauge
orange, rot
gelblich
orange
orange
orange
orange
rotbraun
In Tabelle 33 sind die Farbreaktionen der Nitroverbindungen zusam¬
mengestellt. Mit Hilfe der Hydroxylamin-Reaktion kann man zwischen
Phenylradikal und cyclischen Polymethylenradikalen unterscheiden. Eine
Differenzierung zwischen Phénobarbital und Methylphenobarbital ist nur
mit der Kondensation mit Aceton in alkalischer Lösung möglich und diese
eignet sich gut, um zwischen den beiden Nitroverbindungen zu unter¬
scheiden .
Tabelle 33
Farbreaktionen der Nitroverbindungen
Substanz
Phénobarbital
Methylphenobarbital
Cyclobarbital
Hexobarbital
Medomin
Cyclopal
Kemithal
Diazoreaktion
Niederschlag
rot
rot
rot
orangerot
orangegelb
orangegelb
orangerot
Hydroxylamin/Ammoniak
rotviolett
rotviolett
rotbraun
rotbraun
orangebraun
orangebraun
rotbraun
Aceton
grünlich,bläulich
gelbgrün,grün, blau,violettrot,bordeauxrot
gelbgrün
orange,rot¬braun, rot
gelb, orange
gelb, orange
rot
Natronlauge
orange,rot
gelblich
orange
orange
orange
rotbraun
orange

- 140 -
p-Nitrobenzyl-Derivate
Bei der Kondensation von p-Nitrobenzylchlorid mit Dialkyl-, Halo-
genalkyl-, aromatischen, alicyclischen, N-methylierten und Thio-Deri-
vaten bilden sich unter Austritt von Chlorwasserstoff die entsprechendenDi- resp. Mono-Derivate.
Darstellung: 1/400 Mol des Barbitursäure - Derivates wurde in
10 cm3 Wasser mittels Natriumkarbonat gelöst. Die erforderliche MengeNatriumkarbonat ist abhängig von der Anzahl substituierbarer Wasser¬
stoffatome; auf jedes ersetzbare Wasserstoffatom bringt man 1/800 Mol
Natriumkarbonat in Anwendung. Nach Auflösung fügte man eine Lösungvon p-Nitrobenzylchlorid in 10 cm3 Alkohol zu, indem für jedes ersetzbare
Wasserstoffatom 1/400 Mol p-Nitrobenzylchlorid zur Reaktion gebrachtwurde. Die Lösung, in der sich beim Zusatz der alkoholischen p-Nitro-benzylchlorid-Lösung ein weisser Niederschlag gebildet hatte, wurde eine
halbe Stunde unter Rückflusskühlung auf dem Wasserbad erwärmt. Beim
Erhitzen ging der Niederschlag zuerst in Lösung, fiel aber nach ca. 10
Minuten wieder aus, wobei sich die überstehende Flüssigkeit bei allen
Derivaten im Anfang gelblich bis grünlich verfärbte.
Nach dem Abkühlen wurde der Niederschlag abgenutscht, mit Alkohol
und mit Wasser gewaschen, dann mit 10 cm3 n-Natronlauge geschüttelt,um eventuell gebildetes Monosubstitut und nicht umgesetztes Barbiturat
zu entfernen, wieder abgenutscht und mit Wasser gewaschen. Hierauf
wurde der Niederschlag in Chloroform gelöst, die Chloroformlösung mit
Natriumsulfat getrocknet und auf dem Wasserbad auf ein kleines Volumen
eingeengt. Durch Zusatz von Alkohol wurden die p-Nitrobenzyl-Derivatewieder ausgefällt, abgenutscht, mit Alkohol gewaschen und im evakuierten
Schwefelsäure-Exsikkator getrocknet.
Die p-Nitrobenzyl-Derivate der Dialkylbarbitursäuren fielen leicht
schön kristallin aus, während diejenigen der N-methylierten und Thio-
Barbiturate zuerst als eine Art Oel ausfielen. Die ölige Schicht wurde ab¬
getrennt, in Chloroform gelöst und wie oben angegeben weiter behandelt.
Die kristallinen Niederschläge bildeten sich nach 24-48stündigem Stehen.
Die p-Nitrobenzyl-Derivate sind weisse bis gelbliche, die der Thio-
barbiturate orange-rötliche kristalline Pulver, löslich in Chloroform und
schwer löslich in Alkohol.
Sie sublimieren weit schwieriger als die Barbiturate. Bei der Va-
kuum-Mikrosublimation im Eder'schen Apparat lag, bei einem Druck von
10-12 mm, die Sublimationstemperatur bei allen Derivaten ca. 10-20° un¬
terhalb dem Schmelzpunkt. Bei den meisten Derivaten erhielten wir beider Sublimation Tröpfchen und Kristalle, wobei die Tröpfchen auch nach
12-18-stündiger Sublimationsdauer nicht verschwanden. Impften wir das
Deckgläschen, so bildeten sich die Kristalle leichter, waren ausgeprägterund auch besser ausgebildet.
In Tabelle 34 sind die Schmelzpunkte der p-Nitrobenzyl-Derivateund der entsprechenden Sublimate zusammengestellt. Zu dieser Tabelleist folgendes zu bemerken:

- 141 -
Der Schmelzpunkt des Pentobarbital-Derivates, auf dem Block be¬
stimmt, betrug 147-1490, das Sublimat schmolz bei 120, 5-122, 5°, während
das geimpfte Produkt bei 146-148° schmolz. Das Sublimat des Allobarbi-
tal-Derivates zeigte zwei verschiedene Kristallformen, von denen eine
bei 190-191° und die andere bei 172,5-173,5° schmolz. Das geimpfte und
das nicht geimpfte Sublimat des Thiopental-Derivates schmolz bei 155,5-156, 5°, dem Schmelzpunkt der freien Aethyl-1-methylbutyl-thiobarbitur-säure, während der Schmelzpunkt des p-Nitrobenzyl-Derivates, auf dem
Block bestimmt, 148-150° betrug.
Eine sichere Identifizierung der einzelnen Barbiturate mit den ent¬
sprechenden p-Nitrobenzyl-Derivaten ist nicht immer möglich, da die
Schmelzpunkte oft ziemlich nahe aneinander liegen und so eine Unterschei¬
dung ausgeschlossen ist. Infolge der mehr oder weniger schlechten Subli-
mierbarkeit und der schlechten Ausbildung der Sublimate hinsichtlich Kri¬
stallhabitus sind die Sublimate dementsprechend von geringem Wert.
Tabelle 34
Schmelzpunkte der p-Nitrobenzyl-Derivate
Name
Barbltal
Sonéryl
Pentobarbita]
AUobarbltal
Alurate
Sandoptal
Noctal
Pernocton
Phénobarbital
Cyclobarbital
Medomin
Cyclopal
Narconumal
Hexobarbital
Methylpheno-barbital
Eunarcon
Thiopental
Kemithal
Substituenten
Rl
aethyl-
aethyl-
aethyl-
allyl-
allyl-
allyl-
isopropyl-
sec-butyl-
aethyl-
aethyl-
aetbyl-
allyl-
allyl-
methyl-
aethyl-
isopropyl-
aethyl-
allyl-
R2
aethyl-
n-butyl-
1-methyl-butyl-
allyl-
isopropyl-
isobutyl-
ß -bromallyl-
ß -bromallyl-
phenyl-
cyclohexenyl-
cycloheptenyl-
cyclopentenyl-
isopropyl-
cyclohexenyl-
phenyl-
ß -bromallyl-
1-methyl-butyl-
cyclohexenyl-
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
CH3-CH3-
CH3-
CH3-H-
H-
R4
O=
0=
0=
0=
o=
o=
0-
o=
o=
o=
o=
o=
o=
o=
o=
o=
s=
s=
Block
korr.
193-194°
144-145°
147-149°
193-194°
191,5-193°
175, 5-176,5°202,5-203,5°191-192°
182,5-184°
195-196, 5°
161,5-164°180-182°
87- 89°
110-112°
112-113°
112-114,5°148-150°
nicht kristallin
Balzerblock
mikro
189-191°
142-143°
146-148° 120,5-122,5°190-191° 172,5-173, 5°
183-185°
172,5-174,5°198-199°
177-179°
180-182°
191-193°
160,5-161,5°177-178°
91°
110,5°111,5°
114,5°
155, 5-156, 5°
-

- 142 -
Xanthydrol-Derivate
Bei der Kondensation von Dialkyl-, Halogenalkyl-, aromatischen, ali-
cyclischen und Thio-Derivaten mit Xanthydrol in Eisessig bilden sich unter
Wasseraustritt die entsprechenden Dixanthyl-Verbindungen (S.85).
Darstellung: Zu einer Lösung von 2 g Xanthydrol in 15 cm Eis¬
essig wurden 1,2 g des entsprechenden Barbitursäure-Derivates gegebenund die Mischung während 30 Minuten auf 80-85° erhitzt. Beim Erhitzen
nahm die Lösung eine gelbe Farbe an und es bildete sich nach ca. 10 Minu¬
ten, mit Ausnahme von Allylisopropyl-, Aethylcyclohexenyl-barbitursäureund Aethyl-1-methylbutyl-thiobarbitursäure, ein weisser kristalliner Nie¬
derschlag. Bei den drei genannten Derivaten fiel der Niederschlag erst
nach dem Erkalten der Lösung aus. Die gebildeten Dixanthylverbindungenwurden abgenutscht, mitheissem Alkohol gewaschen und in 10-20 cm3 heis-
sem Benzol gelöst. Diese Lösung wurde mit der fünffachen Menge sieden¬
dem Alkohol versetzt, rasch durch Watte filtriert und auskristallisieren
gelassen. Die ausgeschiedenen Xanthydrol-Derivate wurden abgenutscht,zuerst 2-3 Stunden bei 40° und dann 48 Stunden im evakuierten Schwefelsäu-
re-Exsikkator getrocknet. Die Ausbeuten betrugen ungefähr 50%.
Die dargestellten Xanthydrol-Derivate sind weisse kristalline Pulver,löslich in Benzol und Dioxan und schwer löslich in Alkohol. In Tabelle 35
sind die Schmelzpunkte der erhaltenen Derivate zusammengestellt. Die
Schmelzpunkte liegen, mit Ausnahme der Dixanthyl-Verbindung von Aethyl-1-methylbutyl-thiobarbitursäure, alle ziemlich hoch und ein Teil der Ver¬
bindungen schmilzt unter Zersetzung. Weiter ist zu beachten, dass einzel¬
ne Schmelzpunkte ziemlich nahe aneinander liegen und die Identifizierungder Barbiturate mit Hilfe der Xanthydrol-Derivate nicht in allen Fällen ein
eindeutiges Resultat liefert. Die Xanthydrol-Derivate sublimieren schwie¬
riger als die Barbiturate. Bei der Vakuum-Mikrosublimation im Eder'sehen
Apparat lag, bei einem Druck von 10-12mm, die Sublimationstemperatur30-40° unterhalb dem Schmelzpunkt. Bei allen Derivaten bildeten sich zu¬
erst Tröpfchen und dann Kristalle. Diese waren aber immer sehr klein und
sehr wenig differenziert, so dass die Sublimate nicht von grossem Wertfür weitere Untersuchungen sind.
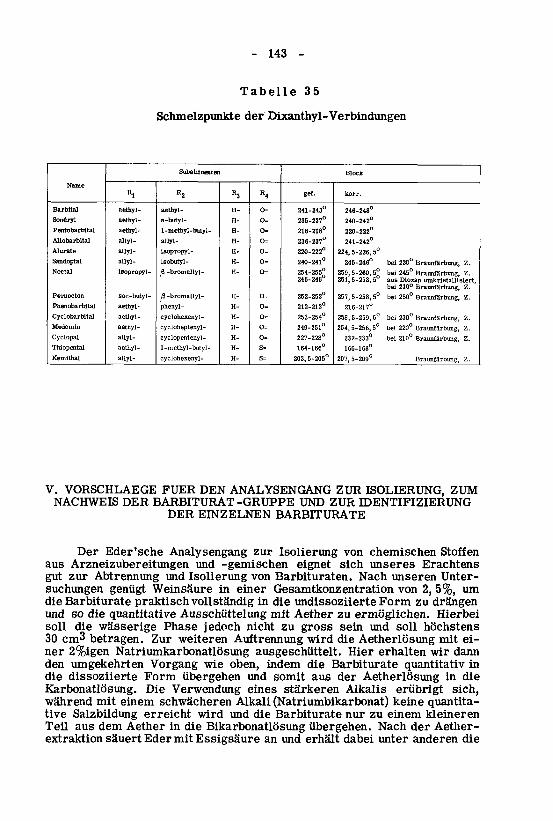
- 143 -
Tabelle 35
Schmelzpunkte der Dixanthyl-Verbindungen
Name
Barbltal
Sonéryl
PentobarWtal
AUobarbital
Alurate
Sandoptal
Noctal
Pernocton
Phénobarbital
Cyclobarbital
Medomin
Cyclopal
Thiopental
Kemithal
Substituenten
«i
aethyl-
aethyl-
aetliyl-
allyl-
allyl-
allyl-
isopropyl-
sec-butyl-
aethyl-
aethyl-
aethyl-
allyl-
aethyl-
allyl-
R2
aethyl-
a-butyl-
1 -methyl-butyl-
allyl-
isopropyl-
isobutyl-
jh -bromallyl-
ß -bromallyl-
phenyl-
cyclohexenyl-
cycloheptenyl-
cyclopentenyl-
1-methyl-butyl-
cyclohexenyl-
R3
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
H-
R4
0-
O=
o=
o=
o=
0=
o=
0«
0=
0=
0=
0«
s>
s=
Block
gef
241-243°
235-237°
216-218°
236-237°
220-222°
240-241°
254-255°246-248°
252-253°
212-213°
253-254°
249-251°
227-228°
164-166°
203, 5-205°
korr.
246-248°
240-242°
220-222°
241-242°
224,5-226,5°245-246° bei 230° Bpaimfàrbung, Z.
259, 5-260, 5° bei 245° Braunfärbimg, Z
251, 5-253, 5 aus Dioxan umkristallisiert,bei 210° Braunfärbung, Z
257, 5-258, 5° bei 250° Braunfàrbung, Z
216-217°
258, 5-259, 5° bei 230° Braunfärbung, Z
254,5-256, 5° bei 220° Braunfàrbung, Z
232-233° bei 210° Braunfàrbung, Z.
166-168°
207, 5-209° Braunfàrbung, Z
V. VORSCHLAEGE FUER DEN ANALYSENGANG ZUR ISOLIERUNG, ZUM
NACHWEIS DER BARBITURAT-GRUPPE UND ZUR IDENTIFIZIERUNG
DER EINZELNEN BARBITURATE
Der Eder'sche Analysengang zur Isolierung von chemischen Stoffen
aus Arzneizubereitungen und -gemischen eignet sich unseres Erachtens
gut zur Abtrennung und Isolierung von Barbituraten. Nach unseren Unter¬
suchungen genügt Weinsäure in einer Gesamtkonzentration von 2, 5%, um
die Barbiturate praktisch vollständig in die undissoziierte Form zu drängenund so die quantitative Ausschüttelung mit Aether zu ermöglichen. Hierbei
soll die wässerige Phase jedoch nicht zu gross sein und soll höchstens
30 cm3 betragen. Zur weiteren Auftrennung wird die Aetherlösung mit ei¬
ner 2%igen Natriumkarbonatlösung ausgeschüttelt. Hier erhalten wir dann
den umgekehrten Vorgang wie oben, indem die Barbiturate quantitativ in
die dissoziierte Form übergehen und somit aus der Aetherlösung in die
Karbonatlösung. Die Verwendung eines stärkeren Alkalis erübrigt sich,während mit einem schwächeren Alkali (Natriumbikarbonat) keine quantita¬tive Salzbildung erreicht wird und die Barbiturate nur zu einem kleineren
Teil aus dem Aether in die Bikarbonatlösung übergehen. Nach der Aether-
extraktion säuert Eder mit Essigsäure an und erhält dabei unter anderen die

- 144 -
Barbitursäure-Derivate. Unserer Meinung nach würde man hier anstelle
der Essigsäure besser Weinsäure oder Schwefelsäure verwenden, um auch
hier die Dissoziation soweit wie möglich zurückzudrängen, um die Extrak¬
tion quantitativ zu gestalten und Verluste zu vermeiden.
Den isolierten Rückstand prüfen wir nun direkt auf die Barbiturat-
Gruppe. Von den Gruppenreaktionen schlagen wir diejenigen vor, die mit
allen Barbituraten positiv reagieren. Von den überprüften Kobaltreaktionen
eignen sich besonders jene, die als basische Komponente ein aliphatischesAminhaben und als Lösungsmittel ein Alkohol-Chloroform-Gemisch haben.
Daneben kommt als Gruppenreaktion noch vor allem die Ausschüttelungs-methode mit einer wässerigen Kupfersulfat- oder Kobaltsalzlösung und ei¬
nem Alkali-Chloroform-Gemisch in Frage. Als alkalische Komponentekann man hier ein aliphatisches Amin, Pyridin oder Piperidin benützen.
Welcher von diesen Reaktionen den Vorzug zu geben ist schwer zu sagen,doch hat die zweite einige Vorteile, die darin bestehen, dass man keine ab¬
soluten Lösungsmittel (Alkohol) braucht und dass die Gegenwart von Was¬
ser nicht stört. Um ganz sicher zu sein, kann man immer noch beide Reak¬
tionen ausführen.
Hat man nun die Barbiturat-Gruppe als solche nachgewiesen, so han¬
delt es sich jetzt noch darum, das einzelne Barbiturat selbst zu identifizie¬
ren.
An Hand der bei der Sublimation erhaltenen Kristalle, des Schmelz¬
punktes und der eventuellen Umwandlungen bei der Schmelzpunktbestimmungerhält man schon gewisse Anhaltspunkte über die Identität des betreffenden
Barbiturates. Der Kristallhabitus der bei der Sublimation erhaltenen Kri¬
stalle ist von vielen Faktoren abhängig und genügt in den meisten Fällen
nicht, während der Schmelzpunkt, besonders bei polymorphen Substanzen,zu Irrtümern Anlass geben kann. Die Schwermetall-Fällungen mit dem Su¬
blimat sind nur dann von Bedeutung, wenn man unter den gleichen Bedingun¬gen Vergleichsreaktionen ausführt.
Zur besseren Uebersicht teilen wir die Barbiturate in Schmelzpunkt-Gruppen ein, um so die Identifizierung zu erleichtern.
Nach Bestimmung des Schmelzpunktes der zu untersuchenden Sub¬
stanz prüft man mit Kaliumpermanganat in natronalkalischer Lösung auf
die Anwesenheit oder Abwesenheit von ungesättigten Gruppen im Molekül.
Steht genügend Substanz zur Verfügung, so kann man Derivate herstellen,deren Schmelzpunkte, mit anderen Reaktionen zusammen, in den meisten
Fällen eine genaue Identifizierung zulassen. Ist dies nicht der Fall, so kann
man an Hand von Farbreaktionen die Identifizierung vornehmen.
Wir wollen nun an Hand des in Tabelle 36 aufgestellten Schemas die
Identifizierungsmöglichkeiten der Barbiturate kurz besprechen.
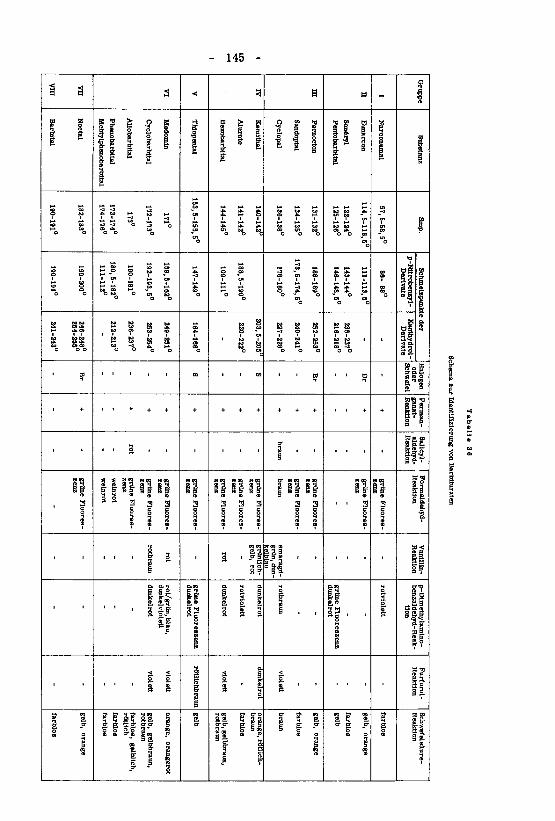
farblos orange
gelb,
_.
_
zenz
Fluores¬
grüne
_
+Br
241-243°
254-255°
246-248°
190-191°
199-200°
190-191°
182-183°
Barbital
Noctal
vm
vn
farblos
farblos
rötlich
gelblich,
farblos,
rotbraun
gelbbraun,
gelb,
orangerot
orange,
violett
violett
dunkelrot
violett
dunkel
blau,
rot/grün,
rotbraun
rot
weinrot
rot
wein
zenz
Fluores¬
grüne
zenz
Fluores¬
grüne
zenz
Fluores¬
grüne
rot
+ + +
-
212-213°
236-237°
253-254°
249-251°
111-112°
180,5-182°
190-191°
192-193,5°
159,5-162°
174-176°
173-174°
173°
172-173°
171°
Mehtylphenobarbital
Phénobarbital
Allobarbital
Cyclobarbltal
Medomin
VI
gelb
rötlichbraun
dunkelrot
Fluoreszenz
grüne
-
zenz
Fluores¬
grüne
-+
S164-166°
147-149°5°
5-155,
153,
Thiopental
V
rotbraun
gelbbraun,
gelb,
farblos
braun
rötlich-
orange,
violett
dunkelrot
dunkelrot
rotviolett
dunkelrot
rot
rot
gelb,grünlich-
zenz
Fluores¬
grüne
zenz
Fluores¬
grüne
zenz
Fluores¬
grüne
-
+ + +S
220-222°
5-205"
203,
109-111°
188,5-190°
144-145°
141-142°
140-142°
HezDbarbital
Alurate
Kemithal
rv
braun
farblos orange
gelb,
violett
rotbraun
dun-
grün,
smaragd¬
braun
zenz
Fluores¬
grüne
zenz
Fluores¬
grüne
braun
+ + +Br
227-228°
240-241°
252-253°
178-180°
5-174,5°173, 188-189°
136-138°
134-135°
131-132°
Cyclopal
Sandoptal
Pernocton
m
gelb
farblos orange
gelb,
-
dunkelrot
Fluoreszenz
grüne
-
zenz
Fluores¬
grüne
_
+Br
216-218°
235-237°
5°
146-148,
143-144°
111-113,5°
125-126°
122-124°
114,5-116,5°
Pentobarbital
Sonéiyl
Eunarcon
n
farblos
-rotviolett
-
zenz
Fluores¬
grüne
-+
--
88°86-
57,5-58,5°Narconumal
I
Reaktion
Derivate
Xanthydrol-
Derivate
p-Nltrobenzyl-
Reaktion
Schwefelsäure-
Reaktlon
Furfurol-
tion
benzaldehyd-Reak-
p-Dimethylamlno-
Reaktion
Vanillin-
Formaldehyd¬
Sallcyl-
Reaktlon
ganat-
Perman-
Schwefel
oder
Halogen
der
Schmelzpunkte
Smp.
Substanz
Gruppe
in
Barbituraten
von
Identifizierung
zur
Schema
36
Tabelle

- 146 -
Gruppe I
Infolge seines tiefen Schmelzpunktes lässt sich Narconumal leicht
identifizieren.
Gruppe II
Von den drei hier aufgeführten Barbituraten ist Eunarcon durch die
Anwesenheit von Brom gekennzeichnet und unterscheidet sich dadurch von
Sonéryl und Pentobarbital. Sonéryl reagiert mit keiner der angeführtenFarbreaktionen, während Pentobarbital mit p-Dimethylaminobenzaldehydin der Wärme eine dunkelrote Färbung gibt. Die p-Nitrobenzyl- und die
Xanthydrol-Derivate lassen ebenfalls eine Unterscheidung zu.
Gruppe in
Hier kann man Pernocton auch durch die Anwesenheit von Brom von
den anderen Verbindungen dieser Gruppe unterscheiden. Cyclopal gibt im
Gegensatz zu Sandoptal mit allen Reagenzien schöne Färbungen. Die
Schmelzpunkte der p-Nitrobenzyl-Derivate von Cyclopal und Sandoptal lie¬
gen nahe aneinander, während diejenigen der Xanthydrol-Derivate eher ei¬
ne Differenzierung zulassen.
Gruppe IV
Kemithal unterscheidet sich von Alurate und Hexobarbital durch das
Vorhandensein von Schwefel im Molekül. Alurate zeigt nur mit zwei Rea¬
genzien Färbungen, während Kemithal und Hexobarbital mit den meisten
gefärbt werden. An Hand der Schmelzpunkte der p-Nitrobenzyl-Derivatekann gut zwischen Alurate und Hexobarbital unterschieden werden.
Gruppe V
In dieser Gruppe findet sich nur das schwefelhaltige Thiopental.

- 147 -
Gruppe VI
Die Schmelzpunkte der Barbitursäure-Derivate liegen hier nahe anein¬
ander und erlauben kaum eine genaue Identifizierung. Mit den p-Nitrobenzyl-Derivaten kann man zwischen Medomin, Phénobarbital und Methylphenobar-bital einerseits und Cyclobarbital und AUobarbital anderseits unterscheiden.Die Xanthydrol-Derivate sind hier besser zur Unterscheidung der einzel¬
nen Barbiturate geeignet. Phénobarbital und Methylphenobarbital geben die
Kaliumpermanganatreaktion nicht und werden als einzige mit Formaldehyd-Schwefelsäure weinrot gefärbt. Medomin lässt sich schön mit p-Dimethyl-aminobenzaldehyd und AUobarbital mit Salicylaldehyd-Schwefelsäure nach¬
weisen. p-Dimethylaminobenzaldehyd lässt ebenfalls eine Differenzierungzwischen Cyclobarbital einerseits und Medomin und AUobarbital anderseits
7u. Die grösste Schwierigkeit bietet wohl die Unterscheidung von Phénobar¬
bital und Methylphenobarbital. Neben der Identifizierung mit Hilfe der p-Ni-trobenzyl- und Xanthydrol-Derivate bietet unseres Erachtens der Kristallha¬
bitus des Sublimates ein sicheres Unterscheidungsmerkmal.
Gruppe VII und VHI
Noctal ist durch die Anwesenheit von Brom im Molekül gekennzeich¬net und Barbital gibt keine von den aufgeführten Farbreaktionen.
Im folgenden soll eine kurze Anleitung zum Nachweis und zur Identi¬
fizierung der einzelnen Barbiturate gegeben werden.
ANLEITUNG ZUM NACHWEIS UND ZUR IDENTIFIZIERUNG
DER BARBITURATE
I. GRUPPENREAKTION
1. Die zu untersuchende Substanz wird in 1,5 cm Chloroform gelöst,
0,25 cm3 einer 0,01m-Lösungvon Co(NO3)2-6 H2O in absolutem Me¬
thanol und 0, 25 cm3 einer m-Lösung von Isobutylamin in Chloroform
zugesetzt. Alle Barbitursäure-Derivate bewirken Violettfärbung.

- 148 -
g2. a. Die zu untersuchende Substanz wird in 1 cm einer Mischung
von 1 T. Isobutylamin und 9 T. Chloroform gelöst und 1 cm*einer l%igen wässerigen Lösung von Co(NO3)2-6 H2O zugesetzt.Bei Gegenwart von Barbituraten tritt Violettfärbung der Chloro¬
formschicht auf.
b. Wird anstelle von Kobalt nitrat eine l%ige wässerige Kupfersul¬fatlösung verwendet, so tritt bei Barbituraten eine Violett-, bei
Thiobarbituraten eine Grünfärbung der Chloroformschicht auf.
U. VAKUUM-MIKROSUBLIMATION
Der Kristallhabitus des Sublimates kann zur Identifizierung beitragen,ist aber allein kaum ein sicheres Merkmal. Bei der Mikroschmelzpunktbe-stimmung ist auf die hierbei eventuell auftretenden Umwandlungen der Kri¬
stalle zu achten. Bei mehr oder weniger reinen Substanzen ist der Schmelz¬
punkt oft ein sicheres Merkmal.
Vor der weiteren Untersuchung der betreffenden Substanz prüft manauf die Anwesenheit von Brom und Schwefel.
A. Anwesenheit von Brom
In Frage kommen:
1. Eunarcon Smp. 114,5-116,5°p-Nitrobenzyl-Derivat: Smp. 111-113,5°
2. Pernocton Smp. 131-132°p-Nitrobenzyl-Derivat: Smp. 188-189
3. Noctal Smp. 182-183°p-Nitrobenzyl-Derivat: Smp. 199-200
Die drei bromierten Derivate geben keine unterschiedlichen Farbreak¬
tionen, doch sollte eine Identifizierung an Hand der Schmelzpunkte der rei¬
nen Substanz und der p-Nitrobenzyl-Derivate möglich sein.
B. Anwesenheit von Schwefel
In Frage kommen:
1. Kemithal Smp. 140-142°a. p-Dimethylaminobenzaldehyd: dunkelrot
b. Furfurol: dunkelrot

- 149 -
2. Thiopental Smp. 153,5-155,5°a. p-Nitrobenzyl-Derivat: Smp. 147-149°b. p-Dimethylaminobenzaldehyd: grüne Fluoreszenz,
dunkelrot
Hat man weder Brom noch Schwefel nachweisen können, so prüft manmit Kaliumpermanganat in natronalkalischer Lösung, wobei ungesättigteRadikale das Permanganat zu grünem Manganatsalz reduzieren.
C. Reduzierend wirken:
1. Narconumal Smp. 57,5-58,5°a. p-Nitrobenzyl-Derivat: Smp. 86-88°b. p-Dimethylaminobenzaldehyd: rotviolett
2. Sandoptal Smp. 134-135°a. p-Nitrobenzyl-Derivat: Smp. 173,5-174,5b. Vanillin: negativ
3. Cyclopal Smp. 136-138°a. p-Nitrobenzyl-Derivat: Smp. 178-180
b. Vanillin: smaragdgrün, dunkelblau
c. Furfurol: violett
d. Piperonal: smaragdgrün
4. Alurate Smp. 141-142°a. p-Nitrobenzyl-Derivat: Smp. 188,5-190b. p-Dimethylaminobenzaldehyd: rotviolett
c. Vanillin: negativ
5. Hexobarbital Smp. 144-145°a. p-Nitrobenzyl-Derivat: Smp. 109-111
b. p-Dimethylaminobenzaldehyd: dunkelrot
c. Vanillin: rot
6. Medomin Smp. 171°a. p-Nitrobenzyl-Derivat: Smp. 159,5-162b. p-Dimethylaminobenzaldehyd: rot/grün, blau,
dunkelviolett
c. Vanillin: rot
7. Cyclobarbital Smp. 172-173°a. p-Nitrobenzyl-Derivat: Smp. 192-193,5b. p-Dimethylaminobenzaldehyd: dunkelrot
c. Vanillin: rotbraun
8. Allobarbital Smp. 173°0
a. p-Nitrobenzyl-Derivat: Smp. 190-191
b. Salicylaldehyd: rot

- 150 -
D. Nicht reduzierend wirken:
1. Sonéryl Smp. 122-124°a. p-Nitrobenzyl-Derivat: Smp. 143-144°b. p-Dimethylaminobenzaldehyd: negativ
2. Pentobarbital Smp. 125-126°a. p-Nitrobenzyl-Derivat: Smp. 146-146,5°b. p-Dimethylaminobenzaldehyd: grüne Fluoreszenz,
dunkel rot
3. Phénobarbital Smp. 173-174°a. p-Nitrobenzyl-Derivat: Smp. 180,5-182°b. Formaldehyd: weinrot
c. Nitro-Verbindung: Kondensation mit Aceton
4. Methylphenobarbital Smp. 174-176°a. p-Nitrobenzyl-Derivat: Smp. 111-112
b. Formaldehyd: weinrot
c. Kristallhabitus
d. Nitro-Verbindung: Kondensation mit Aceton
,o5. Barbital Smp. 190-191^
a. p-Nitrobenzyl-Derivat: 190-191
b. Formaldehyd: negativ
o

- 151 -
D. ZUSAMMENFASSUNG
1. Von den wichtigsten therapeutisch verwendeten Barbitursäure-De¬rivaten wurde eine chemische, pharmakologische und therapeutische Ueber-sicht gegeben und ihre physikalischen und chemischen Eigenschaften zusam¬
mengestellt. Von diesen Barbituraten wurden folgende achtzehn Vertreteranalytisch bearbeitet:
Barbital
SonérylPentobarbital
Allobarbital
Alurate
SandoptalNoctal
Pernocton
Phénobarbital
CyclobarbitalMedomin
CyclopalNarconumal
Hexobarbital
MethylphenobarbitalEunarcon
ThiopentalKemithal
2. Die Isolierungsmöglichkeiten der Barbiturate aus Arzneigemischenund -Zubereitungen wurden gestreift und dabei besonders die Ausschütte-
lungsmethode beachtet. Es zeigte sich, dass die Barbitursäure-Derivatesich aus wässerig-weinsaurer Lösung mit Aether und aus Aether mit Na¬
triumkarbonatlösung quantitativ extrahieren lassen. Zwecks weiterer Auf¬
trennung ist im Eder'sehen Analysengang bei der Ausschüttelung die Essig¬säure durch eine stärkere Säure zu ersetzen.
3. Das Verhalten der Barbiturate bei der Mikrosublimation im Vakuumund die einzelnen Sublimationsbedingungen, wie Druck, Temperatur, Ab¬stand und Temperaturgefälle, wurden untersucht. Die Identifizierung der
Sublimate wurde an Hand des Kristallhabitus, des Schmelzpunktes und mit
Hilfe von Mikroreaktionen durchgeführt. Die Vakuum-Mikrosublimation mit
anschliessender Schmelzpunktbestimmung der Sublimate hat sich als sehr
günstig beim qualitativen Nachweis der Barbiturate erwiesen, während der
Kristallhabitus der Sublimate allein kaum zur Identifizierung genügt.
4. Von den in der Literatur erwähnten Gruppenreaktionen wurden die
Farbreaktionen mit Kobalt-II- und Kupfer-II-Salzen näher untersucht. Zum
qualitativen Nachweis sind besonders die Kobaltreaktionen geeignet, die als
basische Komponente ein aliphatisches Amin und als Lösungsmittel ein Me¬
thanol-Chloroform-Gemisch aufweisen. Hierbei ist Isobutylamin dem Iso-
propylamin wegen seiner geringeren Flüchtigkeit vorzuziehen.
5. Die Identifizierung der einzelnen Barbitursäure-Derivate erfolgteweiter noch mittels Farbreaktionen, durch Nachweis einer bestimmten
Gruppe im Molekül und durch Darstellung von p-Nitrobenzyl- und von Xan-
thydrol-Derivaten.

- 152 -
6. An Hand der Schmelzpunkte der Barbiturate, der p-Nitrobenzyl-und Xanthydrol-Derivate, sowie von mehr oder weniger charakteristischen
Farbreaktionen können die Barbitursäure-Derivate identifiziert werden.
7. Es wurde eine Anleitung zum Nachweis und zur Identifizierungder in dieser Arbeit behandelten Barbiturate gegeben.

- 153 -
E. LITERATURZUSAMMENSTELLUNG
1) Schoen, Erg.Inn.Med. 50, 43 (1936)2) Meyer, Arch. Exptl. PatKTPharmakol. 42, 109 (1899)3)Overton, "Studien über die Narkose, zugleich ein Beitrag zur allge¬
meinen Pharmakologie", Jena (1901). Vjschr. Naturforsch. Ges. Zürich
40, 1 (1895); 44, 88 (1899)4) "ETirhardt, "Medizin und Chemie 2, 356 (1934)5)v.Economo, Hdb.Norm.Path.Pnysiol. 17, 591(1926)6) Molitor und Pick, Arch. Exptl. Path. PEärmakol. 115, 318(1926)7) Keeser, Arch. Exptl. Path. Pharmakol. 135, 251 (1927)7186, 449 (1937)8) Vogt, Arch. Exptl. Path. Pharmakol. 17871*03, 608(1935)9)Masson und Beland, Anesthesiology~6, 483(1945)
10) Wee se, Arch. Exptl. Path. Pharmakol. fSl, 46(1936)11) Maynert, J.Biol.Chem. 195, 397 (1952T12) Halberkann und Reiche7~Münch.Med.Wschr. 74, 1450(1927)13) Wottschall und Wheeler-Hill, Dtsche Med. Wschr. 54, 140(1928)14) Fretwurst, Halberkann und Reiche, Münch. Med.~Wschr. 77,
1573 (1930)—
15)Fritsche, Praxis 31, 740(1942)16) Pulver, Schweiz.Mid.Wschr. 73, 124(1943)17) W e e s e, Medizin und Chemie 1,190 (1933)18) Carrington und Raventos, Brit.J.Pharmacol. 1, 215(1946)19)Shonle, J.Pharmacol. 49, 393(1932)20) Kaiser, "Beiträge zumTöxikologisehen Nachweis wichtiger Barbitur-
säure-Derivate unter besonderer Berücksichtigung der Mikrosubli-
mation im Vakuum", Verlag der Südd. Apoth. Ztg., p.39 (1932)21) Boedecker und Ludwig, Arch. Exptl. Path. Pharmakol. 139, 353
(1928)22) Fretwurst, Halberkann und Reiche, Münch. Med. Wschr. 79,
1429 (1932)—
23) Maynert und Van Dyke, Science 110, 661 (1949)24) Maynert und Dawson, J.Biol.Chem. 195, 389 (1952)25) Burger, "Medicinal Chemistry", Vol. l,TSlerscience, New York und
London (1951)26)Brodie, Federation Proc. 11, 632(1952)27) Miller, "Rauschgifte und üenussmittel", Schwabe, Basel (1951)28) M/zfller, "Pharmakologie als theoretische Grundlage einer rationel¬
len Pharmakotherapie", Schwabe, Basel (1947)29) Bayer, "Schlaf und Schlafmitteltherapie", 5.Auflage, Eilers, Biele¬
feldt.
30) The Merck Index, 6 Ed., Merck & Co., Rahway, N.J./U.S.A.(1952)
31) Gebrüder von Niessen, D.R.P. 144.431 (1903)32) Steenhauer, Pharm.Weekbl. 64, 1154(1927)33) M ad s en, Dansk Tids. Farm. 8,~B2 (1934)34) Bailey, Pharm.J. 136, 620(1936)35) Aspelund und SkogTünd, Quart. J.Pharm.Pharmacol. 11, 291(1938)36) Nielsen, Dansk Tids. Farm. 7, 137(1933)
~~
37) To m ski und Waller, Pharm.J. 139, 421 (1937)38) Berasain und Vitali, Rev.FarmTBl, 463 (1939)

- 154 -
39)Husa und Jatul, J.Am.Pharm.Assoc. 33, 217(1944)40) Turfitt, Quart.J.Pharm.Pharmacol. 2Ç~1 (1948)41) Mühle mann und Bürgin, "Qualitative Arzneimittelanalyse", Rein¬
hardt, Basel (1951)42) Brandstätter, Mikrochemie 38, 68(1951)43) L. Kofi er und A. Kofier, ""^Mikromethoden zur Kennzeichnung
organischer Stoffe und Stoffgemische", Wagner, Innsbruck (1948)44) Fischer, Arch.Pharm. 2TT 306(1939)45) Reimers, Dansk Tids. Farm. 14, 145(1940)46)Eder, Bull.Féd.Int.Pharm. 8,~EB (1927)47) Tabern und Shelberg, J.Äm.Chem.Soc. 55, 328 (1933)48) Traube, B.17, 2294(1884)
~~
49) Graham, AmTJ.Pharm. 106, 295(1934)50)Stas, Ann.Chem. 84, 379~(IB52)51) Otto, Ann.Chem. TÜO, 44 (1856)52) Rosenthaler, "Qualitative pharmazeutische Analyse", Enke
,Stutt¬
gart (1922). "Nachweis organischer Verbindungen", 2. Auflage, Enke,Stuttgart (1923)
53) Sabalitschka. "Anleitung zum chemischen Nachweis der Gifte",Ber¬lin und Wien (1923)
54)Gadamer, "Lehrbuch der chemischen Toxikologie", Göttingen (1924)55) Winterfeld, "Praktikum der organisch-präparativen pharmazeuti¬
schen Chemie und Leitfaden der chemischen Arzneimittelanalyse",3. Auflage, Steinkopf, Dresden und Leipzig (1950)
56) Dietzel, Paul und Tunmann, Süddtsche Apoth. Ztg. 80, 335, 347,353 (1940)Dietzel und Tunmann, "Anleitung zur Analyse organischer Arz¬
neimittel", 2. Auflage, Schmiedel, Stuttgart (1949)57) Autenrieth und Bauer, "Die Auffindung der Gifte und stark wir¬
kender Arzneistoffe zum Gebrauch in chemischen Laboratorien", 6.
Auflage, Steinkopf, Dresden und Leipzig (1943)58) Bamann und Ulimann, "Chemische Untersuchungen von Arznei¬
gemischen, Arzneispezialitäten und Giftstoffen", Jantsch, Günzburg(1951)
59) Staudinger, "Anleitung zur organischen qualitativen Analyse", 5.
Auflage, Springer, Berlin (1948)60)Vieböck, "Analysengang zur Erkennung von Arzneimitteln", 2. Auf¬
lage, Deuticke, Wien (1949)61)Schoorl, "Organische Analyse", N.V.D.B.Centen's Uitgevers Mij.
Amsterdam (1941)62) Feigl, "Qualitative Analyse mit Hilfe von Tüpfelreaktionen", S.Auf¬
lage, Akademische Verlagsgesellschaft, Leipzig (1938)63) Mayrhof er, "Mikrochemie der Arzneimittel und Gifte", Urban &
Schwarzenberg, Berlin und Wien (1928)64) Bürgin, J.Pharm. Belg. 7, 3(1952)65) Warren, J.Assoc.Official Agr.Chem. 25, 799 (1942); 26, 101 (1943);
27, 352 (1944)~ ~
66)TTder, "Ueber die Mikrosublimation im luftverdünnten Raum", Diss.
ETH. Zürich (1912)67) Haas, "Ueber Vakuummikrosublimation synthetischer Arzneistoffe
und Identifizierung der Sublimate auf kristalloptischem Wege", Diss.
ETH. Zürich (1930)68) Fischer, Mikrochemie 10^ 409 (1931); Arch.Pharm. 271, 466 (1933);
277, 306 (1939)

- 155 -
69) St r e b e 1, "Untersuchungen über die Vakuum-Mikrosublimation neuerer
synthetischer Arzneistoffe und die Identifizierung der Sublimate", Diss.
ETH. Zürich (1950)70)Kempf, in Houben, "Methoden der organischen Chemie", 3. Auflage,
Bd. I, p. 693 (1925)71)Kempf, J.Prakt.Chem. 78, 201(1908)72) Fischer, Mikrochemie IF, 247(1934)73) L .Kofier und A.Kolf er, "Mikroskopische Methoden in der Mikro¬
chemie", Haim & Co., Wien und Leipzig (1936)74) Gettler, Umberger und Goldbaum, Anal.Chem. 22, 600(1950)75) L. Kofier und Dernbach, Mikrochemie 9, 345 (193ÏF76)Pyrkosch, Pharm.Ztg.Nachr. 88, 178 (1052)77) L. Kofier und A. Kofier, "Mlkromethoden zur Kennzeichnung
organischer Stoffe und Stoffgemische", Wagner, Innbruck (1948)78)Janot und Chaigneau, C.R.Acad.Sci. 225, 1371(1947)79) L. Kofi er, Mikrochemie 9, 45 (1931)80) Castle, J.Am. Pharm. Assoc. 38, 47(1949)81) Poe, Witt und Snodgrass, "Mikrochemie 34, 235 (1949)82) Winters, "Bijdrage tot de opsporing der bafbltalen bij het toxicolo-
gisch onderzoek", Diss. Leyden (1936)83) Fischer, Arch.Pharm. 270, 149(1932)84) Fischer und A. Kofler7~Ârch.Pharm. 270, 207 (1932)85)Lindpaintner, Mikrochemie 27, 21 (193ÏÏT"86) A. Kofier, Mikrochemie 33^ 471947)87) A. Kof 1er und Fis eher,"Arch. Pharm. 273, 483 (1935)88) L. Kofier und Hilbck, Mikrochemie 9, ~38"(1931)89) L. Kofier, Mikrochemie 15, 242 (1934)"90) Brandstätter und Breuer, Arch.Pharm. 283, 253(1950)91) L. Kofier und W. Kofier, Mikrochemie 34,"~374 (1949)92) Fischer und Chalupa, Mikrochemie 34,"257 (1949)93) L. Kofier, Mikrochemie 22, 241 (1937)""94) Van Itallie und Steenhauer, Pharm.Weekbl. 67, 977(1930)95) Rosenthaler, Mikrochemie 18, 50(1935)96)Rosenthaler, Apoth. Ztg. 487~793 (1933)97) Van Zijp, Pharm.Weekbl. 71, 1075(1934); 73, 764(1936)98) Strzyzowski und Déverin, Helv. Chim. Acïâ 16, 1288(1933)99) Beck, Mikrochemie 29, 206 (1941)
100) Wagenaar, Pharm .'Weekbl. 78, 345(1941)101) Lüdy-Tenger, Pharm.Acta"Helv. 19, 385(1944)102) Lang und Stephan, Süddtsche ApothTZtg. 90, 739 (1950)103) Kaiser und Lang, Süddtsche Apoth. Ztg. 9"27 428 (1952)104) A. Kofier, Mikroskopie 5, 153 (1950)105) Lehmann, "Molekularphysik", Leipzig (1888)106)Tso-Yueh Huang, Acta Pharm.Int. 2, 43, 95, 173(1951)107) Brandstätter, Z. Physik.Chem.Abt.Ä 191, 227(1942)108)Debye und Scherrer, Physik. Z. 17, 27T(1916); 18,291 (1917)109)Parri, Boll.Chim.Farm. 63, 401(1924)110)Zwikker, Pharm.Weekbl.~B8, 975(1931)111) Zwikker, Pharm.Weekbl. TO, 1178 (1932)112) Kozelka und Tatum, J. PHärmacol. Exptl. Therap.59, 54 (1937)113)Riley, Krause, Steadman, Hunter und Hodge, Proc. Soc.
Exptl. Biol.Med. 45, 424(1940)114)Gomahr und KFësbach, Se.Pharm. 19, 148(1951)

- 156 -
115) Bodendorf, Arch.Pharm. 270, 290(1932)116) Rosenthaler, "Toxikologiscne Mikroanalyse" (1935)117)Oettel, Arch.Pharm. 274, 1(1936)118) Flotow, Pharm. ZentràThT 88, 198(1949)119) Koppanyi, Dille, Murphy und Krop, J. Am. Pharm. Assoc.j!3,
1074 (1934)120) Raventos, Brit.J.Pharmacol. 1, 210(1946)121) Cohen, Am.J.Pharm. 118, 40(1946)122)Mattson und Holt, J.~5m.Pharm.Assoc. 38, 55 (1949)123) Rasmussen und Jerslev, Dansk Tids.Farm. 25, 29(1951)124) Gurrrendi Robles, Bol.Soc.Quim. Peru 15, 7011949)125)Paget und Desodt, J. Pharm.Chim. 18, 2Ü7 (1933)126)Desodt, "Les barbituriques, leur toxicologie", thèse Nancy (1933)127) Perrotti, Boll. Chim. Farm. 78, 497 (1939)128) Selles, Anales Soc. Espan. Fés.Quim. 36, 115(1940)129) Kresbach und Gomahr, Wr.Med.Wschr. 63, 476 (1951)130) Jonsson, Svensk Farm.Tids. 35, 659(1931)131) Gomahr und Kresbach, Sc.PHarm. 19, 154(1951)132)Motha, Boll.Chim. Farm. 73, 259 (1934T133) Parkes, Analyst 75, 448 (IÏÏ50)134) Paget und DesodT, Bull.Se. Pharmacol. 39, 532 (1932)135) Tilly, Bull.Se.Pharmacol. 43, 587(1936)136) Paget und Tilly, J.PharmTChim. 25, 222(1937)137) Chatfield, Pharm.J. 14JL 346(19397138) Stainier, J.Pharm.BelgTJ, 26(1950)139) Hargr eaves und Nixon, J. Am. Pharm. Assoc. 22, 1250(1933)140) Motley, J.Pharmacol. 72, 30(1941)141) de Wolff, Pharm.WeekT5I. 84, 537(1949)142) de Wolff, Acta Pharm. Int."?, 107(1951)143) Pesez, J. Pharm.Chim. 25, "508 (1937)144) Lagarce, J.Pharm.ChimT 12, 364(1930)145) Pesez, J.Pharm.Chim. 27,~247 (1938)146) Pesez, J.Fharm.Chim. 28", 379 (1938)147)Deshusses, Pharm.Acti~Helv. 19, 358(1944)148) Deshusses, Pharm. Acta Helv. 20", 199(1945)149)Ranwez, J. Pharm.Belg. 6, 410 TD24)150) Ranwez, J.Pharm.Belg. 5, 501 (1924)151) Beal und Szalkowski, J. Am. Pharm. Assoc. 23, 18(1934)152) Pesez, J.Pharm.Chim. 27, 250(1938)
~
153) Rathenasinkam, Analyst 75, 108(1950)154) Ekkert, Pharm. Zentralh. GT, 481 (1926)155) Laubie, Bull. Trav.Soc. Pharm. Bordeaux fasc.l (1949)156) Poe, Witt und Snodgrass, Mikrochemie 34, 235(1949)157) Castle und Poe, J.Am.Chem.Soc. 66, 144Ü~(1944)158) Lyons und Dox, J.Am.Chem.Soc. 5T/ 288 (1929)159) Jespersen und Lars en, Dansk TicTs. Farm. 8, 212(1934)160) Hultquist und Poe, Ind.Eng.Chem. Anal. Ed."7, 398(1935)161) Hultquist, Poe und Witt, Ind. Eng. Chem. Anal. Ed. 14, 219(1942)162) Hultquist, Poe und Witt, J. Am.Chem.Soc. 67, 688(1945)163) L ill mann, Analyst 75, 626(1950)164) Reimers, Dansk Ti3s. Farm. 15, 281(1941)165)Fabre, J.Pharm.Chim. 26, 24T~(1922)166) Dannelsen, AnalysmetoHer 9, 24(1933)

- 157 -
167) Bertrand, Compt.Rend. 225, 1331(1947)168) McCutcheon und Plein,T.Am.Pharm.Assoc. 38, 24(1949)169) Hull, J.Am.Chem.Soc. 41, 1168 (1919)
~
170) Wickström und Salvesen, J.Pharm.Pharmacol. 4, 98(1952)171) Algeri und Walker, Am. J. Clin. Path. 22, 37 (1952T172) Williams und Kirby, Science New YorFl07, 481(1948) ..
173) Glas s tone, "Textbook of physical chemîsîry", 2nd edition, 5n
printing, B. Van Nostrand Co. Inc., New York (1948)174) Ko It hoff, "Der Gebrauch von Farbindikatoren. Dire Anwendung in
der Neutralisationsanalyse und bei der kolorimetrischen Bestimmungder Wasserstoffionenkonzentration", Springer, Berlin (1923)
175) Stoffel, "Syntheseund physikalisch-chemische Ueberprüfung einigerlokalanästhetisch wirksamer aliphatischer Carbonsäureester", Diss.
ETH. Zürich (1949)176) Landolt-Börnstein, Phys.-chem.Tabellen, 5.Auflage

- 158 -
Die nachfolgenden Firmen stellten uns die für diese Arbeit benötigtenSubstanzen sowie Publikationen zur Verfügung. Für ihr Entgegenkommendanken wir ihnen an dieser Stelle noch bestens.
Abbott Laboratories, North ChicagoCibaA.G., Basel
Farbenfabriken Bayer, Leverkusen
J.R. Gelgy & Co. A. G., Basel
F. Hoffmann - La Roche & Co. A. G., Basel
Imperial Chemical (Pharmaceuticals) Ltd., Manchester
May & Baker Ltd., DagenhamJ.D.Riedel - E.de Haen A. G., Berlin
A.G. vorm. B.Siegfried, ZofingenSociété Parisienne d'Expansion Chimique, Paris

- 159 -
Curriculum vitae
Ich wurde am 16. Dezember 1923 in Luxemburg als Sohn des Louis
Perlia und der Marguerite Gönner geboren. Dort besuchte ich die Primar¬
schulen und das Gymnasium. Nach einem kriegsbedingten Unterbruch in
den Jahren 1941-1944 erhielt ich im Winter 1944 das Reifezeugnis. Nach
Absolvierung der naturwissenschaftlichen Oberkurse und des zweijährigenPraktikums in Luxemburg studierte ich zwei Semester an der Universität
Lausanne. Die vier Semester des Fachstudiums absolvierte ich am Phar¬
mazeutischen Institut der Eidg. Technischen Hochschule und bestand im
Frühjahr 1951 die Diplomprüfung. Hierauf begann ich unter Leitung von
Herrn Prof. Dr. J. Büchi die vorliegende Promotionsarbeit, die ich im
Februar 1953 zu Ende führte.

\Abb 6
Cyclobarbital 110° 720 mm
Abb 7
Cyclobarbital 110° 50 mm
Abb 8 Abb 9
Cyclobarbital 110° 11mm Cyclobarbital 110/1 mm
Sublimation von Cyclobarbital bei verschiedenen Drucken
(90 x)

LAbb 10 Abb 11
Methylphenobarbital 110"/720mm Methylphenobarbital 110° ' 50 mm
*&
o
o
0 CN9»
Abb. 12 Abb 13
Methylphenobarbital 110°'11 mm Methylphenobarbital 110°/1 mm
Sublimation von Methylphenobarbital bei verschiedenen Drucken
(90 x)
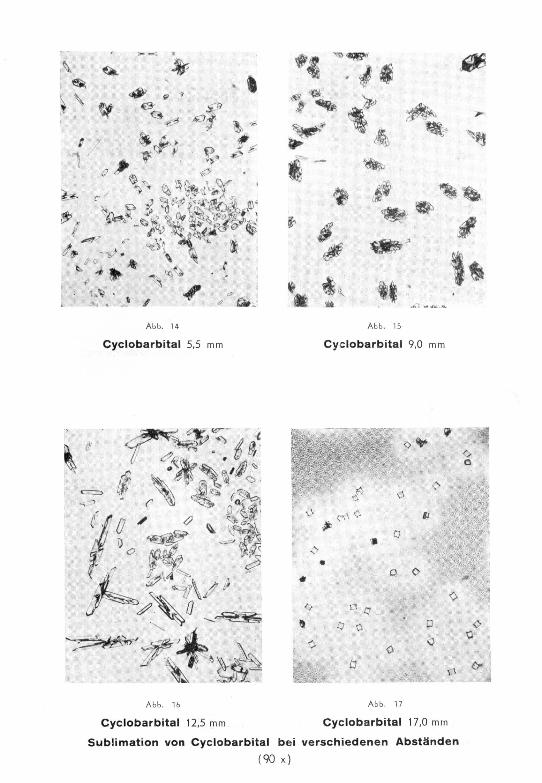
k*- r
feu
Abb 14
Cyclobarbital 5,5 mm
*.
Abb 15
Cyclobarbital 9,0 mm
Abb 16
Cyclobarbital 12,5 mm
M«
0
O'
Q O
'1
0
Abb 17
Cyclobarbital 17,0 mm
Sublimation von Cyclobarbital bei verschiedenen Abständen
(90 x)
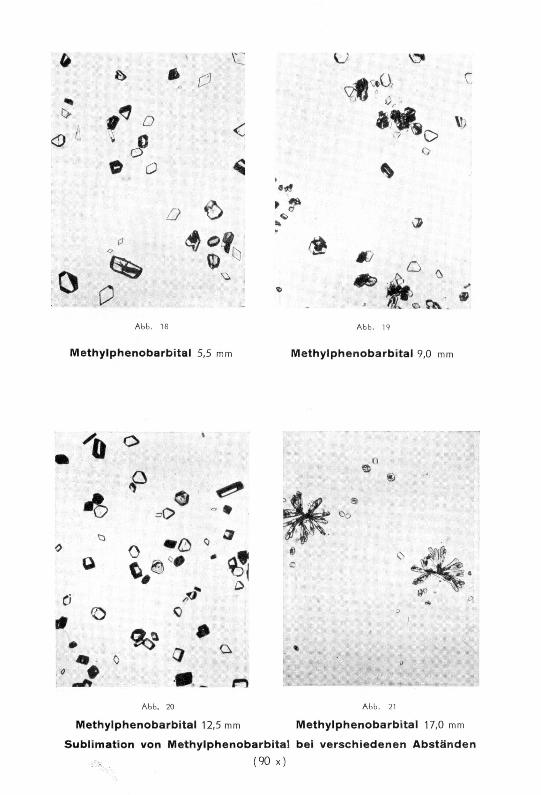
0
v
V
r
o
o >
Abb 18
Methylphenobarbital 5,5 mm
Os
© 6K-
Abb 19
Methylphenobarbital 9,0 mm
0 0
û
Abb. 20 Abb 21
Methylphenobarbital 12,5 mm Methylphenobarbital 17,0 mm
Sublimation von Methylphenobarbital bei verschiedenen Abständen
(90 x)

I
I
o.
0
/
V
Abb 22
Barbital, Ubersichtsbild (150 x)
Abb 23
Barbital I (90x)
Abb 24
Barbital II (360 x)
Abb 25
Barbital III (150 x)

O
o
O o
o
Abb 26
Barbital IV (150 x)
Abb 27
Sonéryl (90x)
A
Abb. 28
Pentobarbital II (150 x)
Abb 29
Pentobarbital I (150 x)
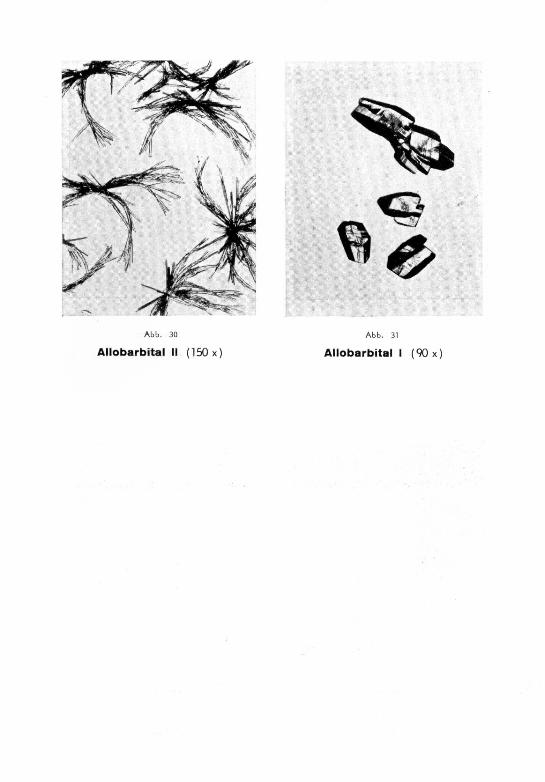
Abb 30
Allobarbital II (150x)
Abb 31
Allobarbital I (90x)
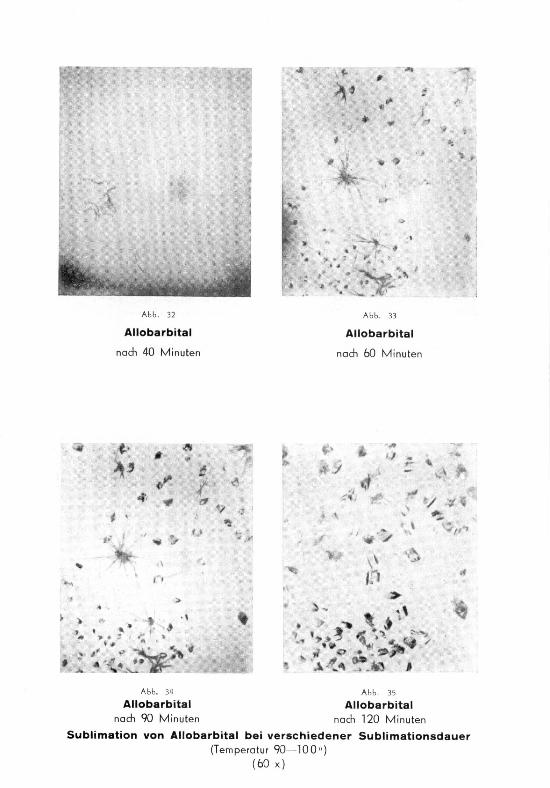
Wf
*ï
Abb 32
Allobarbital
nach 40 Minuten
Abb 33
Allobarbital
nach 60 Minuten
*0
I /
-\
HUUk A
Abb
#
A'
34
Allobarbital
nach 90 Minuten
• •>;.• *. * "\
Abb 35
Allobarbital
nach 120 Minuten
Sublimation von Allobarbital bei verschiedener Sublimationsdauer
(Temperatur 90- 100»)
(60 x)
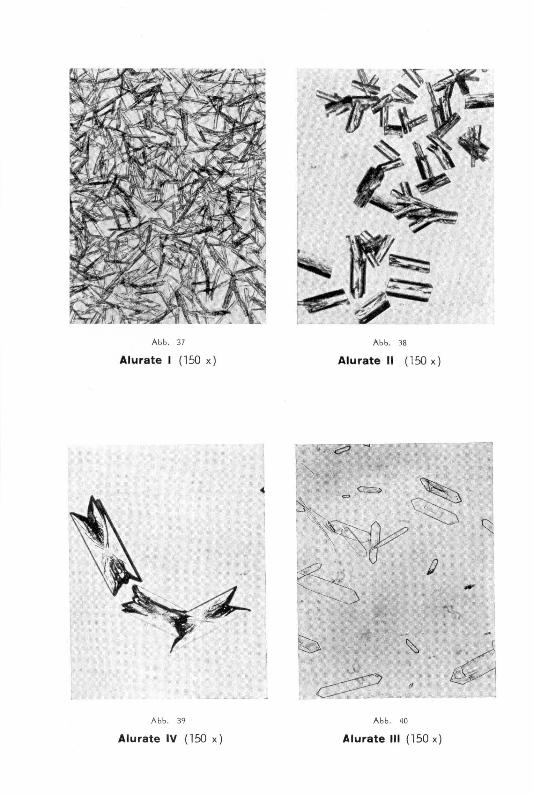
Abb 37
Alurate I (150 x)
Abb 38
Alurate II (150 x)
Abb 39
Alurate IV (150 x)
Abb 40
Alurate III (150 x)
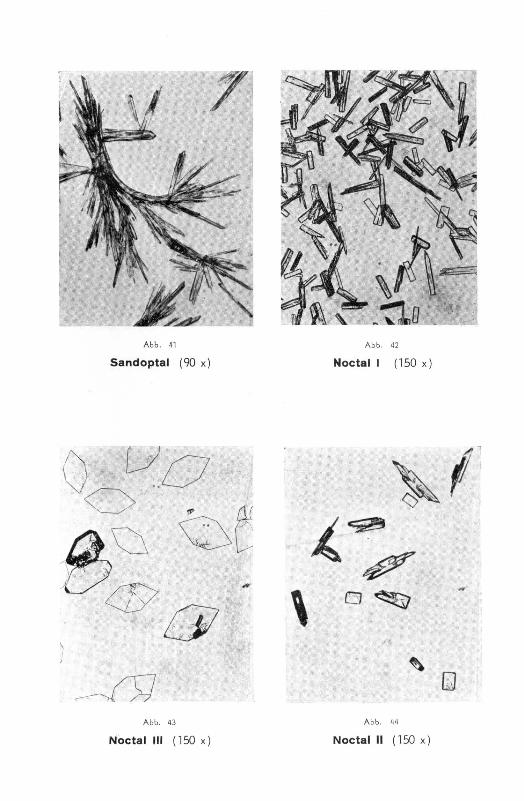
\Abb. 41
Sandoptal (90 x)
Abb 42
Noctal I (150 x)
\ a
Abb. 43
Noctal III (150 x)
Abb 44
Noctal II (150 x)

Abb 41
Pernocton (90x)
Abb 46
Pernocton (90x)
Abb 47
Phénobarbital I (90 x)
Abb 48
Phénobarbital I (150 x)

Abb 49
Phénobarbital 11 (150 x)
Abb 50
Phénobarbital III (150 x)
^\ (>
Abb 51
Phénobarbital III (150x)
Abb 52
Medomin (150 x)

Abb. 53
Medomin (150 x)
Abb. 54
Cyclopal I (90 x)
Abb. 55
Cyclopal II (150 x)
Abb. 56
Narconumal (90 x)
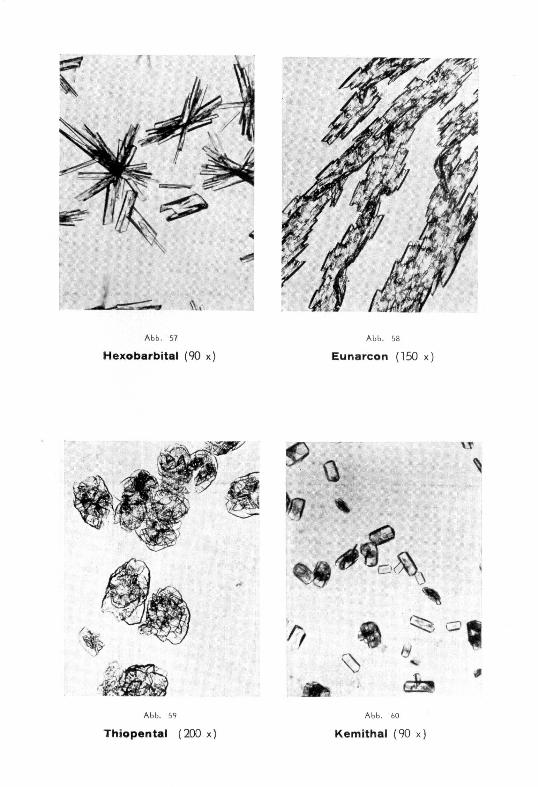
2S?
%
Abb 57
Hexobarbital (90 x)
Abb 58
Eunarcon (150 x)
Abb 59
Thiopental (200 x)
Ci
Abb 60
Kemithal (90 x)

Abb 61
Barbital-Fe-Komplex (90 x)
Abb. 62
Barbital -Cu- Komplex (90 x)
Abb. 63 Abb 64
Allobarbital - Fe. Komplex (90 x) Allobarbital-Cu-Komplex (90 x)
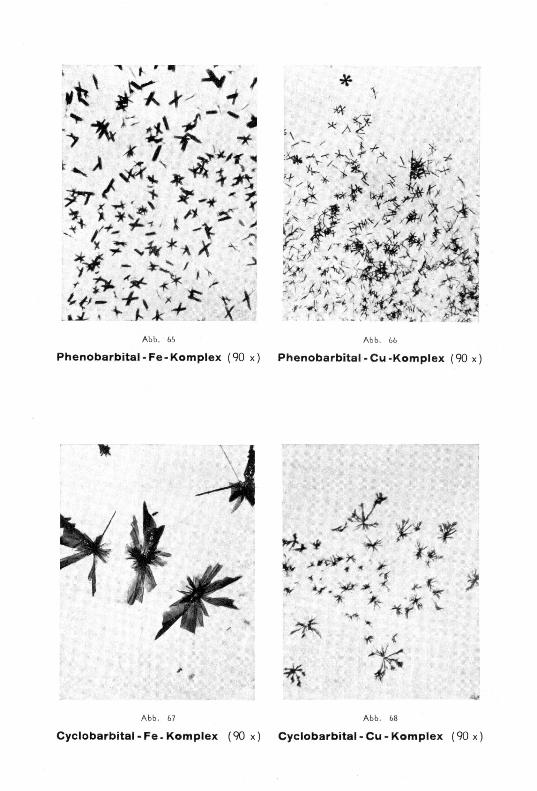
•si
s
Abb 65 Abb 66
Phénobarbital-Fe-Komplex (90 x) Phénobarbital-Cu-Komplex (90x)
1,% *
"*1
Abb 67 Abb 68
Cyclobarbital-Fe. Komplex (90 x) Cyclobarbital-Cu-Komplex (90 x)