bcl2 and hsp27 act at different levels to suppress programmed cell death
TRANSCRIPT

Bcl-2 and Hsp27 act at di�erent levels to suppress programmed cell death
Isabelle Gue nal1,*, Carole Sidoti-de Fraisse1,*, Se bastien Gaumer1 and Bernard Mignotte1,2
1Centre de GeÂneÂtique MoleÂculaire, UPR 9061 du CNRS, F-91198 Gif-sur-Yvette cedex; 2Universite de Versailles-St Quentin,45 avenue des Etats-Unis, F-78035 Versailles cedex, France
Apoptosis and necrosis, two morphologically distinctforms of cell death, can be induced by common stimulidepending on the doses and the cell type. This studycompares the protective e�ect of oncoprotein Bcl-2 andof the small stress protein Hsp27 on these two types ofcell death. We use rat embryo ®broblasts conditionallyimmortalized by the tsA58 mutant of SV40 large Tantigen as parental cells to develop cell lines carryinginducible bcl-2 or hsp27 genes. Two apoptotic stimuliwere used: shift to the restrictive temperature thatinduced p53-mediated apoptosis and treatment with lowdoses of hydrogen peroxide. Necrosis was induced byhigh doses of hydrogen peroxide. Although Bcl-2 andHsp27 protect these cells from necrotic death, only Bcl-2appears capable of preventing apoptotic death. Bcl-2protection is not mediated by a negative e�ect on theinduction of the p53 responsive genes bax or waf1 but itslows down at least two stages of apoptosis: decrease ofmitochondrial membrane potential and subsequent mor-phological changes. In contrast, although Hsp27 hasbeen recently shown to inhibit apoptosis induced byvarious stimuli, its overexpression has no e�ect onapoptosis in this cell system. It should be also noticedthat the apoptotic stimuli (temperature shift or hydrogenperoxide treatment) induce Hsp27, but not Bcl-2accumulation suggesting that, in parental cells, Hsp27might already provide some protection. However, takentogether these results suggest that Hsp27, as well as Bcl-2, acts at several levels to inhibit cell death, but thattheir protective functions only partially overlap.
Keywords: apoptosis; p53; simian virus 40; Bcl-2;Hsp27; necrosis
Introduction
Programmed Cell Death (PCD) is an active process ofcellular self-destruction and serves developmental orhomeostatic functions (Kroemer et al., 1995; Thomp-son, 1995). Apoptosis, which has been de®ned frommorphological considerations (Kerr et al., 1972), is themost often encountered form of PCD, though not theonly one (Clarke, 1990; Schwartz et al., 1993), and canbe induced by a wide range of stimuli. During thesubsequent e�ector phase, the numerous apoptosis-inducing stimuli seem to converge onto a fewstereotypical pathways, and beyond a certain pointthe cells become irreversibly committed to death.During the successive degradation phase, vital struc-
tures and functions are destroyed giving rise to the full-blown phenotype of apoptosis (Kroemer et al., 1995).At the nuclear level, apoptosis is typically accompaniedby condensation and fragmentation of chromatin andnucleus. Cytoplasmic boiling, a marked convolution ofthe cellular surface and the development of peduncu-lated protuberances that separate to produce mem-brane-enclosed apoptotic bodies, is also seen.Apoptosis is morphologically and biochemically
distinct from another type of cell death known asnecrosis. The main features of necrosis are rapidorganelle swelling leading to rupture of the cell andspillage of its content into the extracellular environ-ment. A cell undergoing necrosis can thus provoke anin¯ammatory response. It is generally considered thatthe degenerative process of cell death by necrosis is arelatively uncontrolled and passive death that usually isa result of severe molecular and/or structural damageto cells or of a gross perturbation to the cellularenvironment. Nevertheless, morphological descriptionof necrosis is reminiscent of a type of developmentalcell death referred to by Clarke (1990) as type 3A.Moreover, necrosis can also be induced by physiolo-gical factors as TNF-a and thus may also be one modeof programmed cell death.Genetic studies of PCD in the nematode Caenor-
habditis elegans provided evidence that PCD involves acell-intrinsic death programme (Ellis et al., 1991).Among the 14 identi®ed genes participating in C.elegans developmental PCD, three were found to actduring the e�ector phase: ced-3 and ced-4 which arerequired for PCD and ced-9 which is a suppressor ofPCD. ced-3 is homologous to the interleukin-1-b-converting enzyme (ICE) gene family encodingcysteine proteases involved in mammalian apoptosis(Kumar, 1995), ced-4 has no mammalian homologueidenti®ed so far and ced-9 is homologous to themammalian bcl-2 gene family (Hengartner andHorvitz, 1994).Bcl-2, like Ced-9, is a negative regulator of cell
death, able to prevent cells from undergoing apoptosisinduced by various stimuli in a wide variety of celltypes (Korsmeyer, 1992; Zhong et al., 1993). But themechanism(s) by which Bcl-2 modulates apoptosis isnot yet well known and several con¯icting theorieshave been proposed. bcl-2 is one member of a growingmultigene family, with multiple representatives inmammals, among which is bax. A widely acceptedmodel postulates that homodimers of Bax promoteapoptosis, and that the functional e�ect of Bcl-2 is toform competing heterodimers with Bax that cannotpromote apoptosis (Oltvai et al., 1993; Sedlak et al.,1995). However, in some systems, Bax binding by Bcl-2was not su�cient to prevent apoptosis. Bcl-2 has beenlocalized to the cytoplasmic surfaces of the nuclearenvelope, the endoplasmic reticulum and the outer
Correspondence: B Mignotte*IG and CSF contributed equally to this workReceived 28 October 1996; revised 26 March 1997; accepted1 April 1997
Oncogene (1997) 15, 347 ± 360 1997 Stockton Press All rights reserved 0950 ± 9232/97 $12.00

mitochondrial membrane (Chen et al., 1989; Hock-enbery et al., 1990; Krajewski et al., 1993; Nakai et al.,1993; Akao et al., 1994; de Jong et al., 1994). Thismembrane association is of functional signi®cance asmutant Bcl-2 molecules lacking this membraneanchorage capacity are less e�ective at preventingapoptosis in some systems (Borner et al., 1994; Nguyenet al., 1994). Indeed, recent studies (Zhu et al., 1996)have reported that, in inhibiting apoptosis in MDCKcells, a mutant Bcl-2 molecule whose anchorage istargeted speci®cally to the mitochondria is as e�ectiveas the wild type protein, whereas mutant Bcl-2 targetedto the ER loses this capacity. In contrast, Bcl-2 targetedto the ER in the Rat-1/myc ®broblasts proved to bemore active than when targeted to mitochondria. Thus,Bcl-2 mutants with restricted subcellular location revealdistinct pathways for apoptosis depending on cell type.Alterations of mitochondrial functions have beendescribed as early events of apoptosis in diversemodels (VayssieÁ re et al., 1994; Petit et al., 1995;Zamzami et al., 1995) and it has been shown that Bcl-2 can counteract these events (Shimizu and Eguchi,1996; Zamzami et al., 1996b). Since Bcl-2 blocksapoptosis in cells that do not contain a functionalrespiratory chain (cells lacking mtDNA), it has beenconcluded that the Bcl-2 activity is not directly relatedto mitochondrial respiration (Jacobson et al., 1993). Ithas also been suggested that Bcl-2 can control levels ore�ects of reactive oxygen species (ROS), essentiallygenerated from the respiratory chain at the mitochon-drial level, through an antioxidant pathway (Hock-enbery et al., 1993; Kane et al., 1993). But apoptosiscan proceed normally, and can be prevented by Bcl-2,under anaerobic conditions which minimise theformation of ROS (Jacobson and Ra�, 1995; Shimizuet al., 1995). It is possible that Bcl-2 acts in more thanone way either to prevent the induction of apoptosis bydi�erent stimuli or to control di�erent aspects of theapoptotic e�ector pathway. Nevertheless, taken to-gether and despite the con¯icting theories, all theseresults are in agreement with a central role ofmitochondria in the apoptotic process and bring tothe fore the action of Bcl-2 as possible regulator ofapoptosis at the mitochondrial level (Susin et al., 1996;Wang et al., 1996; Kluck et al., 1997; Yang et al., 1997).Hsp27 belongs to the family of small stress proteins
(hsps) whose synthesis is induced or stimulated by heatshock (Ciocca et al., 1993; Arrigo and Landry, 1994;Arrigo and Mehlen, 1994). It has been observed thatthe small hsps participate in the cellular mechanismsthat protect cells against the deleterious e�ects inducedby thermal injury or other types of stress (Lindquist,1986; Lindquist and Craig, 1988; Huot et al., 1991).For example, in stably transfected murine L929®broblasts, constitutive expression of Hsp27 confersresistance to TNF-a and oxidative stress-inducednecrosis (Mehlen et al., 1995b). In these cells, thekilling induced by TNF-a and oxidative stress isthought to occur through the accumulation ofintracellular ROS, generated from the respiratorychain (Schulze-Ostho� et al., 1992). Cell exposure toTNF-a increases the intracellular concentration of ROSand induces changes in the cellular localisation,structural organisation and phosphorylation of Hsp27(Mehlen et al., 1995a). The protective e�ect of Hsp27against TNF-a appears mediated by an increase in
glutathione (Mehlen et al., 1996a). More recently, ithas been shown that constitutive expression of hsp27increases resistance to drugs-induced apoptosis (Samaliand Cotter, 1996) and blocks Fas/APO1 and staur-osporine-induced apoptosis (Mehlen et al., 1996b).Hence, Hsp27 is able to prevent both apoptotic andnecrotic cell death in di�erent models.The present study was undertaken in order to specify
how Bcl-2 and Hsp27 work to prevent cell death. Wehave used rat embryo ®broblasts conditionallyimmortalised by the tsA58 temperature sensitivemutant of SV40 large T antigen as parental cells todevelop P1-Bcl2 and P1-Hsp27 cell lines carryinginducible bcl-2 or hsp27 genes. These cells wereinduced to apoptosis either by shifting to therestrictive temperature (p53-mediated apoptosis)(Zheng et al., 1994; Gue nal and Mignotte, 1995) orsubjecting them to moderate oxidative stress with lowdoses of hydrogen peroxide. We also used higher dosesof hydrogen peroxide to induce necrotic death of thesecells. The e�ect of Bcl-2 and Hsp27 overexpression oncell death was studied in relation to speci®c geneinduction and mitochondria.
Results
Development of cell lines carrying inducible bcl-2 orhsp27 genes
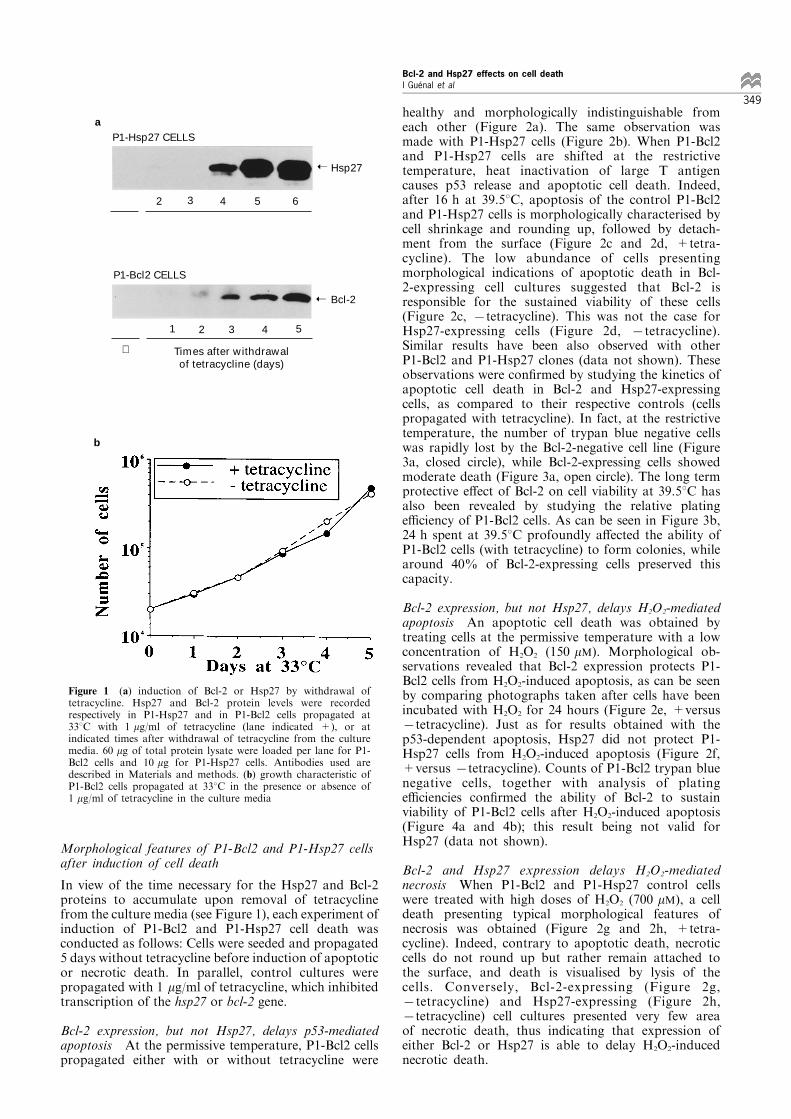
A tetracycline-regulated gene expression system(established by Gossen and Bujard) was used todevelop stable cell lines, using REtsAF rat embryo®broblast cells as the parental cell line. The experi-mental procedure followed is described in Materialsand methods. Brie¯y, the human bcl-2 (or rat hsp27)cDNA was cloned downstream of a minimal promotercarrying bacterial tOp sequences giving rise to plasmidpBMbcl2 (or pBMhsp27). A cytomegalovirus promoterdrives constitutive expression of a tTA fusion protein(plasmid pUHD 15-1). In the absence of tetracycline,the tTA protein binds to tOp and activates transcrip-tion of the bcl-2 (or hsp27) expression construct.Conversely, addition of tetracycline inactivates tTaDNA binding, and thereby transcription of bcl-2 (orhsp27). Distinct clones of P1-Hsp27 and P1-Bcl2 cells,displaying tetracycline-regulated gene expression, wereselected and expanded. Figure 1a represents Westernblots of regulated expression of Hsp27 (and Bcl-2) inone clone of P1-Hsp27 (and one clone of P1-Bcl2) cellsupon removal of tetracycline from the culture media.Basal expression of Hsp27 has been estimated to0.05 ng/mg of total protein when P1-Hsp27 cells arecultivated with tetracycline (the same level is observedin REtsAF parental cells), and accumulation of thisprotein is evident after 5 days of withdrawal oftetracycline from the culture media (evaluated around3 ng/mg of total protein). Basal expression of Bcl-2 isundetectable in the P1-Bcl2 cell line when propagatedwith tetracycline. Upon removal of tetracycline, theaccumulation of Bcl-2 is perceptible from 2 days andincreases until at least 5 days of culture. It should benoted that the growth characteristics at 338C of bothcell lines (population doubling time of approximately24 h) are identical whether cells are cultivated with orwithout tetracycline (Figure 1b).
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
348
Morphological features of P1-Bcl2 and P1-Hsp27 cellsafter induction of cell death
In view of the time necessary for the Hsp27 and Bcl-2proteins to accumulate upon removal of tetracyclinefrom the culture media (see Figure 1), each experiment ofinduction of P1-Bcl2 and P1-Hsp27 cell death wasconducted as follows: Cells were seeded and propagated5 days without tetracycline before induction of apoptoticor necrotic death. In parallel, control cultures werepropagated with 1 mg/ml of tetracycline, which inhibitedtranscription of the hsp27 or bcl-2 gene.
Bcl-2 expression, but not Hsp27, delays p53-mediatedapoptosis At the permissive temperature, P1-Bcl2 cellspropagated either with or without tetracycline were
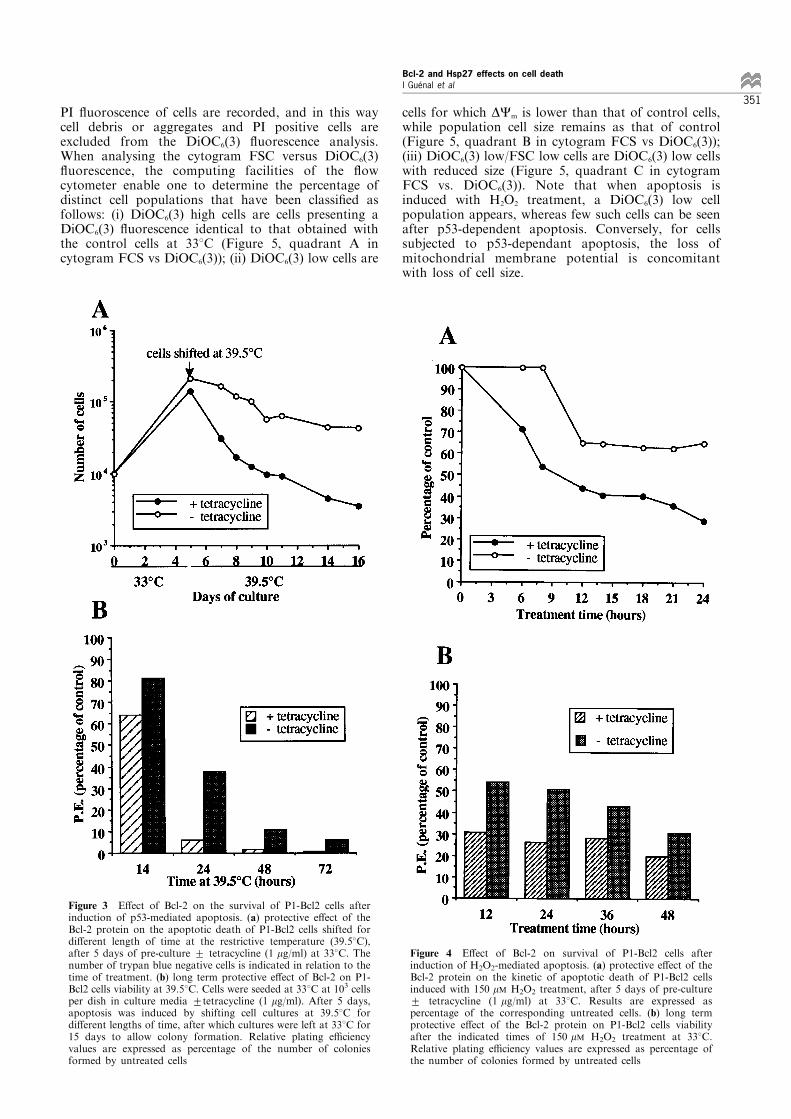
healthy and morphologically indistinguishable fromeach other (Figure 2a). The same observation wasmade with P1-Hsp27 cells (Figure 2b). When P1-Bcl2and P1-Hsp27 cells are shifted at the restrictivetemperature, heat inactivation of large T antigencauses p53 release and apoptotic cell death. Indeed,after 16 h at 39.58C, apoptosis of the control P1-Bcl2and P1-Hsp27 cells is morphologically characterised bycell shrinkage and rounding up, followed by detach-ment from the surface (Figure 2c and 2d, +tetra-cycline). The low abundance of cells presentingmorphological indications of apoptotic death in Bcl-2-expressing cell cultures suggested that Bcl-2 isresponsible for the sustained viability of these cells(Figure 2c, 7tetracycline). This was not the case forHsp27-expressing cells (Figure 2d, 7tetracycline).Similar results have been also observed with otherP1-Bcl2 and P1-Hsp27 clones (data not shown). Theseobservations were con®rmed by studying the kinetics ofapoptotic cell death in Bcl-2 and Hsp27-expressingcells, as compared to their respective controls (cellspropagated with tetracycline). In fact, at the restrictivetemperature, the number of trypan blue negative cellswas rapidly lost by the Bcl-2-negative cell line (Figure3a, closed circle), while Bcl-2-expressing cells showedmoderate death (Figure 3a, open circle). The long termprotective e�ect of Bcl-2 on cell viability at 39.58C hasalso been revealed by studying the relative platinge�ciency of P1-Bcl2 cells. As can be seen in Figure 3b,24 h spent at 39.58C profoundly a�ected the ability ofP1-Bcl2 cells (with tetracycline) to form colonies, whilearound 40% of Bcl-2-expressing cells preserved thiscapacity.
Bcl-2 expression, but not Hsp27, delays H2O2-mediatedapoptosis An apoptotic cell death was obtained bytreating cells at the permissive temperature with a lowconcentration of H2O2 (150 mM). Morphological ob-servations revealed that Bcl-2 expression protects P1-Bcl2 cells from H2O2-induced apoptosis, as can be seenby comparing photographs taken after cells have beenincubated with H2O2 for 24 hours (Figure 2e, +versus7tetracycline). Just as for results obtained with thep53-dependent apoptosis, Hsp27 did not protect P1-Hsp27 cells from H2O2-induced apoptosis (Figure 2f,+versus 7tetracycline). Counts of P1-Bcl2 trypan bluenegative cells, together with analysis of platinge�ciencies con®rmed the ability of Bcl-2 to sustainviability of P1-Bcl2 cells after H2O2-induced apoptosis(Figure 4a and 4b); this result being not valid forHsp27 (data not shown).
Bcl-2 and Hsp27 expression delays H2O2-mediatednecrosis When P1-Bcl2 and P1-Hsp27 control cellswere treated with high doses of H2O2 (700 mM), a celldeath presenting typical morphological features ofnecrosis was obtained (Figure 2g and 2h, +tetra-cycline). Indeed, contrary to apoptotic death, necroticcells do not round up but rather remain attached tothe surface, and death is visualised by lysis of thecells. Conversely, Bcl-2-expressing (Figure 2g,7tetracycline) and Hsp27-expressing (Figure 2h,7tetracycline) cell cultures presented very few areaof necrotic death, thus indicating that expression ofeither Bcl-2 or Hsp27 is able to delay H2O2-inducednecrotic death.
P1-Hsp27 CELLS
2 3 4 5 6
P1-Bcl2 CELLS
2 3 4 51
⊕ Times after withdrawal of tetracycline (days)
➝
➝
Hsp27
Bcl-2
a
b
Figure 1 (a) induction of Bcl-2 or Hsp27 by withdrawal oftetracycline. Hsp27 and Bcl-2 protein levels were recordedrespectively in P1-Hsp27 and in P1-Bcl2 cells propagated at338C with 1 mg/ml of tetracycline (lane indicated +), or atindicated times after withdrawal of tetracycline from the culturemedia. 60 mg of total protein lysate were loaded per lane for P1-Bcl2 cells and 10 mg for P1-Hsp27 cells. Antibodies used aredescribed in Materials and methods. (b) growth characteristic ofP1-Bcl2 cells propagated at 338C in the presence or absence of1 mg/ml of tetracycline in the culture media
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
349

E�ect of Bcl-2 and Hsp27 on loss of mitochondrialmembrane potential during apoptosis
To study the e�ect of Bcl-2 and Hsp27 on the changesin mitochondrial membrane potential (DCm) inindividual cells, ¯ow cytometric analysis was carriedout using DiOC6(3), a lipophilic cation taken up by
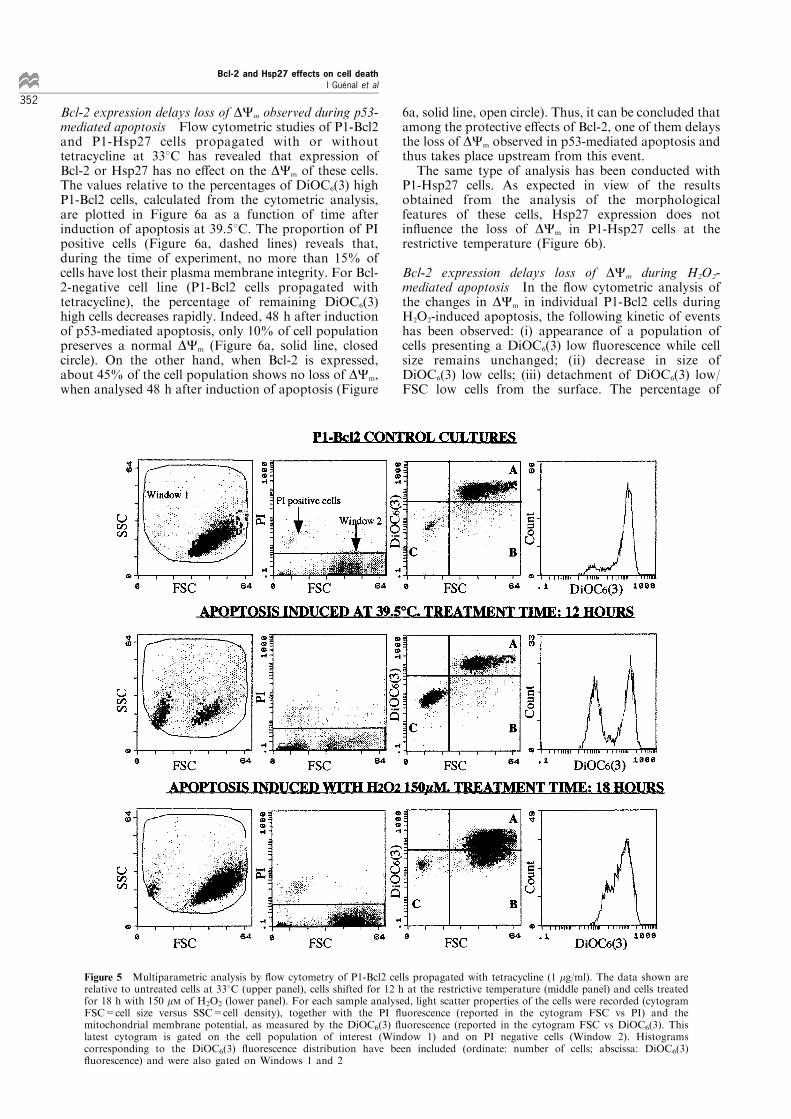
mitochondria in proportion to DCm. In parallel, cellplasma membrane integrity was assessed by stainingcells with PI (see Materials and methods). Figure 5depicts typical cytograms (where each cell analysed isrepresented by a dot) obtained with multiparametric¯ow cytometry analysis of P1-Bcl2 cells. For eachsample analysed, the light scattering properties and the
P1-Bcl2 CELL LINE
CONTROL CULTURES AT 33°C+ tetracycline – tetracycline
APOPTOSIS: CELLS TREATED WITH H2O2 150 µM FOR 24 HOURS
APOPTOSIS: CELLS SHIFTED AT 39.5°C FOR 16 HOURS
NECROSIS: CELLS TREATED WITH H2O2 700 µM FOR 4 HOURS
+ tetracycline
+ tetracycline
+ tetracycline
– tetracycline
– tetracycline
– tetracycline
P1-Hsp 27 CELL LINE
CONTROL CULTURES AT 33°C+ tetracycline – tetracycline
APOPTOSIS: CELLS TREATED WITH H2O2 150 µM FOR 24 HOURS
APOPTOSIS: CELLS SHIFTED AT 39.5°C FOR 16 HOURS
NECROSIS: CELLS TREATED WITH H2O2 700 µM FOR 4 HOURS
+ tetracycline
+ tetracycline
+ tetracycline
– tetracycline
– tetracycline
– tetracycline
e
a b
c d
f
g h
Figure 2 E�ect of Bcl-2 and Hsp27 on the morphological changes of P1-Bcl2 and P1-Hsp27 cells after induction of cell death.Photographs of culture dishes were taken with a phase-contrast microscope and the scale bar equals 200 mm. For both cell lines,following ®ve days of pre-culture + tetracycline (1 mg/ml) at 338C, pictures were taken after 16 h of p53-mediated apoptosis, after24 h of H2O2-mediated apoptosis or after 4 h of induction of necrosis. Photographs of morphological features of the controlcultures at 338C are also included. The pictures shown are representative of three distinct experiments, in each of whichapproximately 20 pictures were examined
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
350
PI ¯uoroscence of cells are recorded, and in this waycell debris or aggregates and PI positive cells areexcluded from the DiOC6(3) ¯uorescence analysis.When analysing the cytogram FSC versus DiOC6(3)¯uorescence, the computing facilities of the ¯owcytometer enable one to determine the percentage ofdistinct cell populations that have been classi®ed asfollows: (i) DiOC6(3) high cells are cells presenting aDiOC6(3) ¯uorescence identical to that obtained withthe control cells at 338C (Figure 5, quadrant A incytogram FCS vs DiOC6(3)); (ii) DiOC6(3) low cells are
cells for which DCm is lower than that of control cells,while population cell size remains as that of control(Figure 5, quadrant B in cytogram FCS vs DiOC6(3));(iii) DiOC6(3) low/FSC low cells are DiOC6(3) low cellswith reduced size (Figure 5, quadrant C in cytogramFCS vs. DiOC6(3)). Note that when apoptosis isinduced with H2O2 treatment, a DiOC6(3) low cellpopulation appears, whereas few such cells can be seenafter p53-dependent apoptosis. Conversely, for cellssubjected to p53-dependant apoptosis, the loss ofmitochondrial membrane potential is concomitantwith loss of cell size.
Figure 3 E�ect of Bcl-2 on the survival of P1-Bcl2 cells afterinduction of p53-mediated apoptosis. (a) protective e�ect of theBcl-2 protein on the apoptotic death of P1-Bcl2 cells shifted fordi�erent length of time at the restrictive temperature (39.58C),after 5 days of pre-culture + tetracycline (1 mg/ml) at 338C. Thenumber of trypan blue negative cells is indicated in relation to thetime of treatment. (b) long term protective e�ect of Bcl-2 on P1-Bcl2 cells viability at 39.58C. Cells were seeded at 338C at 103 cellsper dish in culture media +tetracycline (1 mg/ml). After 5 days,apoptosis was induced by shifting cell cultures at 39.58C fordi�erent lengths of time, after which cultures were left at 338C for15 days to allow colony formation. Relative plating e�ciencyvalues are expressed as percentage of the number of coloniesformed by untreated cells
Figure 4 E�ect of Bcl-2 on survival of P1-Bcl2 cells afterinduction of H2O2-mediated apoptosis. (a) protective e�ect of theBcl-2 protein on the kinetic of apoptotic death of P1-Bcl2 cellsinduced with 150 mM H2O2 treatment, after 5 days of pre-culture+ tetracycline (1 mg/ml) at 338C. Results are expressed aspercentage of the corresponding untreated cells. (b) long termprotective e�ect of the Bcl-2 protein on P1-Bcl2 cells viabilityafter the indicated times of 150 mM H2O2 treatment at 338C.Relative plating e�ciency values are expressed as percentage ofthe number of colonies formed by untreated cells
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
351
Bcl-2 expression delays loss of DCm observed during p53-mediated apoptosis Flow cytometric studies of P1-Bcl2and P1-Hsp27 cells propagated with or withouttetracycline at 338C has revealed that expression ofBcl-2 or Hsp27 has no e�ect on the DCm of these cells.The values relative to the percentages of DiOC6(3) highP1-Bcl2 cells, calculated from the cytometric analysis,are plotted in Figure 6a as a function of time afterinduction of apoptosis at 39.58C. The proportion of PIpositive cells (Figure 6a, dashed lines) reveals that,during the time of experiment, no more than 15% ofcells have lost their plasma membrane integrity. For Bcl-2-negative cell line (P1-Bcl2 cells propagated withtetracycline), the percentage of remaining DiOC6(3)high cells decreases rapidly. Indeed, 48 h after inductionof p53-mediated apoptosis, only 10% of cell populationpreserves a normal DCm (Figure 6a, solid line, closedcircle). On the other hand, when Bcl-2 is expressed,about 45% of the cell population shows no loss of DCm,when analysed 48 h after induction of apoptosis (Figure
6a, solid line, open circle). Thus, it can be concluded thatamong the protective e�ects of Bcl-2, one of them delaysthe loss of DCm observed in p53-mediated apoptosis andthus takes place upstream from this event.The same type of analysis has been conducted with
P1-Hsp27 cells. As expected in view of the resultsobtained from the analysis of the morphologicalfeatures of these cells, Hsp27 expression does notin¯uence the loss of DCm in P1-Hsp27 cells at therestrictive temperature (Figure 6b).
Bcl-2 expression delays loss of DCm during H2O2-mediated apoptosis In the ¯ow cytometric analysis ofthe changes in DCm in individual P1-Bcl2 cells duringH2O2-induced apoptosis, the following kinetic of eventshas been observed: (i) appearance of a population ofcells presenting a DiOC6(3) low ¯uorescence while cellsize remains unchanged; (ii) decrease in size ofDiOC6(3) low cells; (iii) detachment of DiOC6(3) low/FSC low cells from the surface. The percentage of
i
i
i
ii
i
Figure 5 Multiparametric analysis by ¯ow cytometry of P1-Bcl2 cells propagated with tetracycline (1 mg/ml). The data shown arerelative to untreated cells at 338C (upper panel), cells shifted for 12 h at the restrictive temperature (middle panel) and cells treatedfor 18 h with 150 mM of H2O2 (lower panel). For each sample analysed, light scatter properties of the cells were recorded (cytogramFSC=cell size versus SSC=cell density), together with the PI ¯uorescence (reported in the cytogram FSC vs PI) and themitochondrial membrane potential, as measured by the DiOC6(3) ¯uorescence (reported in the cytogram FSC vs DiOC6(3). Thislatest cytogram is gated on the cell population of interest (Window 1) and on PI negative cells (Window 2). Histogramscorresponding to the DiOC6(3) ¯uorescence distribution have been included (ordinate: number of cells; abscissa: DiOC6(3)¯uorescence) and were also gated on Windows 1 and 2
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
352
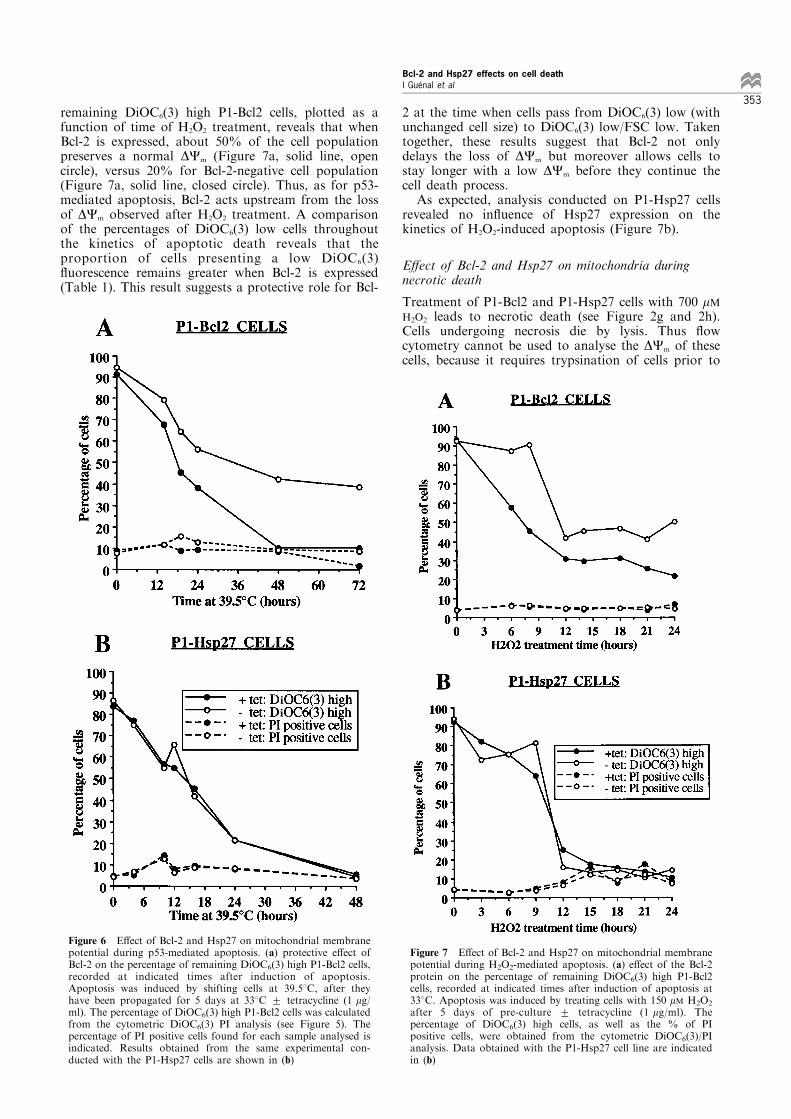
remaining DiOC6(3) high P1-Bcl2 cells, plotted as afunction of time of H2O2 treatment, reveals that whenBcl-2 is expressed, about 50% of the cell populationpreserves a normal DCm (Figure 7a, solid line, opencircle), versus 20% for Bcl-2-negative cell population(Figure 7a, solid line, closed circle). Thus, as for p53-mediated apoptosis, Bcl-2 acts upstream from the lossof DCm observed after H2O2 treatment. A comparisonof the percentages of DiOC6(3) low cells throughoutthe kinetics of apoptotic death reveals that theproportion of cells presenting a low DiOC6(3)¯uorescence remains greater when Bcl-2 is expressed(Table 1). This result suggests a protective role for Bcl-
2 at the time when cells pass from DiOC6(3) low (withunchanged cell size) to DiOC6(3) low/FSC low. Takentogether, these results suggest that Bcl-2 not onlydelays the loss of DCm but moreover allows cells tostay longer with a low DCm before they continue thecell death process.As expected, analysis conducted on P1-Hsp27 cells
revealed no in¯uence of Hsp27 expression on thekinetics of H2O2-induced apoptosis (Figure 7b).
E�ect of Bcl-2 and Hsp27 on mitochondria duringnecrotic death
Treatment of P1-Bcl2 and P1-Hsp27 cells with 700 mMH2O2 leads to necrotic death (see Figure 2g and 2h).Cells undergoing necrosis die by lysis. Thus ¯owcytometry cannot be used to analyse the DCm of thesecells, because it requires trypsination of cells prior to
ii
Figure 6 E�ect of Bcl-2 and Hsp27 on mitochondrial membranepotential during p53-mediated apoptosis. (a) protective e�ect ofBcl-2 on the percentage of remaining DiOC6(3) high P1-Bcl2 cells,recorded at indicated times after induction of apoptosis.Apoptosis was induced by shifting cells at 39.58C, after theyhave been propagated for 5 days at 338C + tetracycline (1 mg/ml). The percentage of DiOC6(3) high P1-Bcl2 cells was calculatedfrom the cytometric DiOC6(3) PI analysis (see Figure 5). Thepercentage of PI positive cells found for each sample analysed isindicated. Results obtained from the same experimental con-ducted with the P1-Hsp27 cells are shown in (b)
ii
Figure 7 E�ect of Bcl-2 and Hsp27 on mitochondrial membranepotential during H2O2-mediated apoptosis. (a) e�ect of the Bcl-2protein on the percentage of remaining DiOC6(3) high P1-Bcl2cells, recorded at indicated times after induction of apoptosis at338C. Apoptosis was induced by treating cells with 150 mM H2O2
after 5 days of pre-culture + tetracycline (1 mg/ml). Thepercentage of DiOC6(3) high cells, as well as the % of PIpositive cells, were obtained from the cytometric DiOC6(3)/PIanalysis. Data obtained with the P1-Hsp27 cell line are indicatedin (b)
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
353
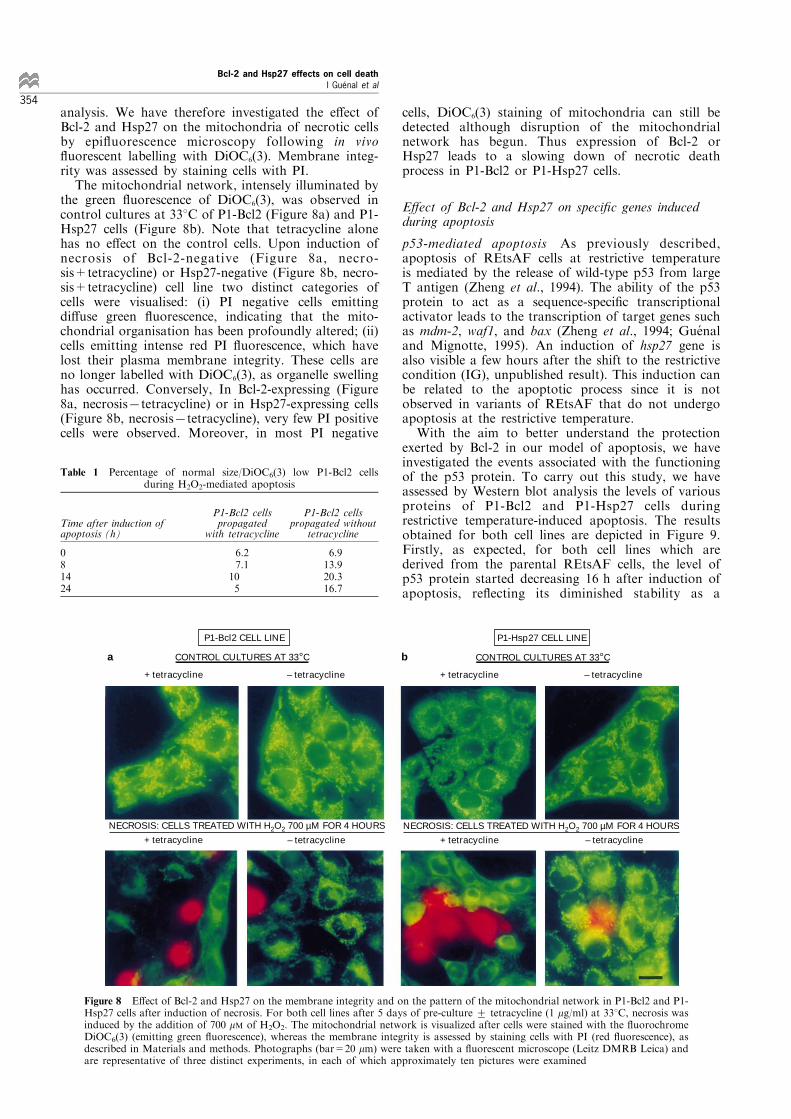
analysis. We have therefore investigated the e�ect ofBcl-2 and Hsp27 on the mitochondria of necrotic cellsby epi¯uorescence microscopy following in vivo¯uorescent labelling with DiOC6(3). Membrane integ-rity was assessed by staining cells with PI.The mitochondrial network, intensely illuminated by
the green ¯uorescence of DiOC6(3), was observed incontrol cultures at 338C of P1-Bcl2 (Figure 8a) and P1-Hsp27 cells (Figure 8b). Note that tetracycline alonehas no e�ect on the control cells. Upon induction ofnecrosis of Bcl-2-negative (Figure 8a, necro-sis+tetracycline) or Hsp27-negative (Figure 8b, necro-sis+tetracycline) cell line two distinct categories ofcells were visualised: (i) PI negative cells emittingdi�use green ¯uorescence, indicating that the mito-chondrial organisation has been profoundly altered; (ii)cells emitting intense red PI ¯uorescence, which havelost their plasma membrane integrity. These cells areno longer labelled with DiOC6(3), as organelle swellinghas occurred. Conversely, In Bcl-2-expressing (Figure8a, necrosis7tetracycline) or in Hsp27-expressing cells(Figure 8b, necrosis7tetracycline), very few PI positivecells were observed. Moreover, in most PI negative
cells, DiOC6(3) staining of mitochondria can still bedetected although disruption of the mitochondrialnetwork has begun. Thus expression of Bcl-2 orHsp27 leads to a slowing down of necrotic deathprocess in P1-Bcl2 or P1-Hsp27 cells.
E�ect of Bcl-2 and Hsp27 on speci®c genes inducedduring apoptosis
p53-mediated apoptosis As previously described,apoptosis of REtsAF cells at restrictive temperatureis mediated by the release of wild-type p53 from largeT antigen (Zheng et al., 1994). The ability of the p53protein to act as a sequence-speci®c transcriptionalactivator leads to the transcription of target genes suchas mdm-2, waf1, and bax (Zheng et al., 1994; Gue naland Mignotte, 1995). An induction of hsp27 gene isalso visible a few hours after the shift to the restrictivecondition (IG), unpublished result). This induction canbe related to the apoptotic process since it is notobserved in variants of REtsAF that do not undergoapoptosis at the restrictive temperature.With the aim to better understand the protection
exerted by Bcl-2 in our model of apoptosis, we haveinvestigated the events associated with the functioningof the p53 protein. To carry out this study, we haveassessed by Western blot analysis the levels of variousproteins of P1-Bcl2 and P1-Hsp27 cells duringrestrictive temperature-induced apoptosis. The resultsobtained for both cell lines are depicted in Figure 9.Firstly, as expected, for both cell lines which arederived from the parental REtsAF cells, the level ofp53 protein started decreasing 16 h after induction ofapoptosis, re¯ecting its diminished stability as a
Table 1 Percentage of normal size/DiOC6(3) low P1-Bcl2 cellsduring H2O2-mediated apoptosis
P1-Bcl2 cells P1-Bcl2 cellsTime after induction of propagated propagated withoutapoptosis (h) with tetracycline tetracycline
081424
6.27.1
105
6.913.920.316.7
a CONTROL CULTURES AT 33°C
NECROSIS: CELLS TREATED WITH H2O2 700 µM FOR 4 HOURS
+ tetracycline – tetracycline + tetracycline – tetracycline
+ tetracycline – tetracycline + tetracycline – tetracycline
P1-Bcl2 CELL LINE P1-Hsp27 CELL LINE
b
NECROSIS: CELLS TREATED WITH H2O2 700 µM FOR 4 HOURS
CONTROL CULTURES AT 33°C
Figure 8 E�ect of Bcl-2 and Hsp27 on the membrane integrity and on the pattern of the mitochondrial network in P1-Bcl2 and P1-Hsp27 cells after induction of necrosis. For both cell lines after 5 days of pre-culture + tetracycline (1 mg/ml) at 338C, necrosis wasinduced by the addition of 700 mM of H2O2. The mitochondrial network is visualized after cells were stained with the ¯uorochromeDiOC6(3) (emitting green ¯uorescence), whereas the membrane integrity is assessed by staining cells with PI (red ¯uorescence), asdescribed in Materials and methods. Photographs (bar=20 mm) were taken with a ¯uorescent microscope (Leitz DMRB Leica) andare representative of three distinct experiments, in each of which approximately ten pictures were examined
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
354
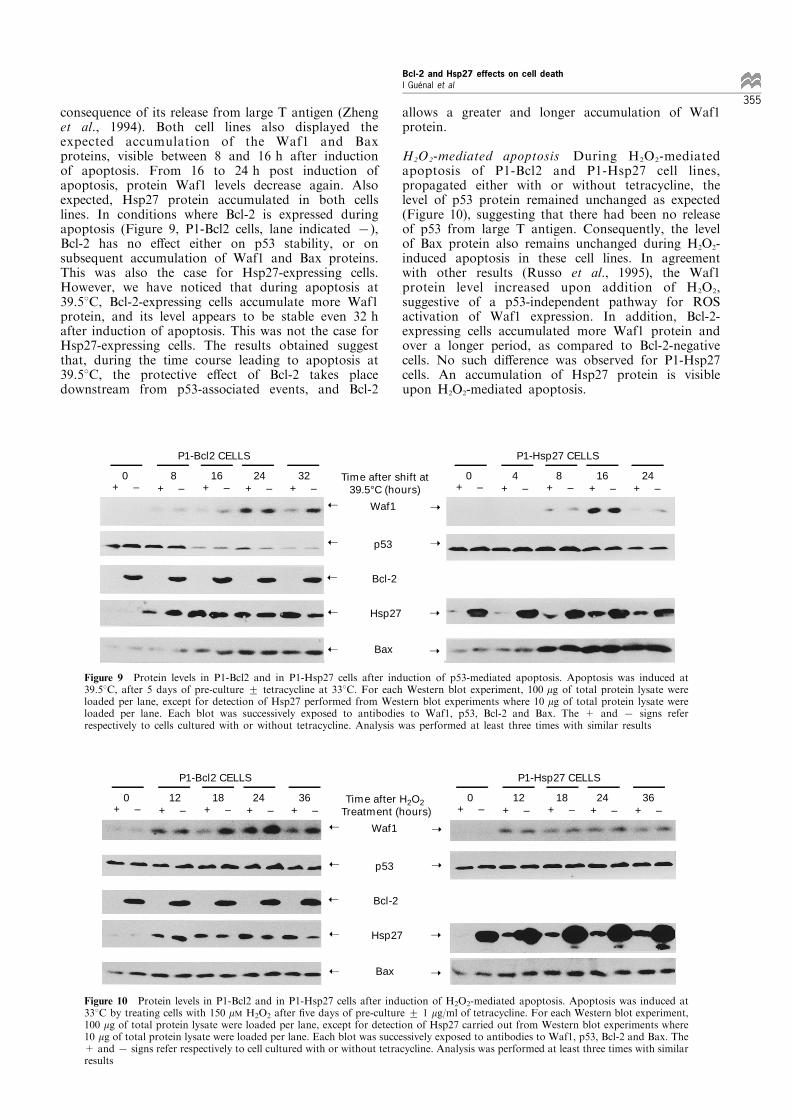
consequence of its release from large T antigen (Zhenget al., 1994). Both cell lines also displayed theexpected accumulation of the Waf1 and Baxproteins, visible between 8 and 16 h after inductionof apoptosis. From 16 to 24 h post induction ofapoptosis, protein Waf1 levels decrease again. Alsoexpected, Hsp27 protein accumulated in both cellslines. In conditions where Bcl-2 is expressed duringapoptosis (Figure 9, P1-Bcl2 cells, lane indicated 7),Bcl-2 has no e�ect either on p53 stability, or onsubsequent accumulation of Waf1 and Bax proteins.This was also the case for Hsp27-expressing cells.However, we have noticed that during apoptosis at39.58C, Bcl-2-expressing cells accumulate more Waf1protein, and its level appears to be stable even 32 hafter induction of apoptosis. This was not the case forHsp27-expressing cells. The results obtained suggestthat, during the time course leading to apoptosis at39.58C, the protective e�ect of Bcl-2 takes placedownstream from p53-associated events, and Bcl-2
allows a greater and longer accumulation of Waf1protein.
H2O2-mediated apoptosis During H2O2-mediatedapoptosis of P1-Bcl2 and P1-Hsp27 cell lines,propagated either with or without tetracycline, thelevel of p53 protein remained unchanged as expected(Figure 10), suggesting that there had been no releaseof p53 from large T antigen. Consequently, the levelof Bax protein also remains unchanged during H2O2-induced apoptosis in these cell lines. In agreementwith other results (Russo et al., 1995), the Waf1protein level increased upon addition of H2O2,suggestive of a p53-independent pathway for ROSactivation of Waf1 expression. In addition, Bcl-2-expressing cells accumulated more Waf1 protein andover a longer period, as compared to Bcl-2-negativecells. No such di�erence was observed for P1-Hsp27cells. An accumulation of Hsp27 protein is visibleupon H2O2-mediated apoptosis.
+ – + – + – + – + – 0 8 16 24 32
P1-Bcl2 CELLS
+ – + – + – + – + – 0 4 8 16 24
P1-Hsp27 CELLS
Time after shift at 39.5°C (hours)
➝
➝
➝
➝
➝
➝
➝
➝
➝
Waf1
p53
Bcl-2
Hsp27
Bax
Figure 9 Protein levels in P1-Bcl2 and in P1-Hsp27 cells after induction of p53-mediated apoptosis. Apoptosis was induced at39.58C, after 5 days of pre-culture + tetracycline at 338C. For each Western blot experiment, 100 mg of total protein lysate wereloaded per lane, except for detection of Hsp27 performed from Western blot experiments where 10 mg of total protein lysate wereloaded per lane. Each blot was successively exposed to antibodies to Waf1, p53, Bcl-2 and Bax. The + and 7 signs referrespectively to cells cultured with or without tetracycline. Analysis was performed at least three times with similar results
+ – + – + – + – + – 0 12 18 24 36
P1-Bcl2 CELLS
+ – + – + – + – + – 0 12 18 24 36
P1-Hsp27 CELLS
Time after H2O2 Treatment (hours)
➝
➝
➝
➝
➝
➝
➝
➝
➝
Waf1
p53
Bcl-2
Hsp27
Bax
Figure 10 Protein levels in P1-Bcl2 and in P1-Hsp27 cells after induction of H2O2-mediated apoptosis. Apoptosis was induced at338C by treating cells with 150 mM H2O2 after ®ve days of pre-culture + 1 mg/ml of tetracycline. For each Western blot experiment,100 mg of total protein lysate were loaded per lane, except for detection of Hsp27 carried out from Western blot experiments where10 mg of total protein lysate were loaded per lane. Each blot was successively exposed to antibodies to Waf1, p53, Bcl-2 and Bax. The+ and7 signs refer respectively to cell cultured with or without tetracycline. Analysis was performed at least three times with similarresults
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
355

Discussion
The experiments reported here have been performed inorder to locate Bcl-2 and Hsp27 action in relation togene induction (Gue nal and Mignotte, 1995) and to thedecrease of mitochondrial transmembrane potential(DCm) occurring during the apoptotic process. Apop-tosis of P1-Bcl2 and P1-Hsp27 cells, carrying induciblebcl-2 or hsp27 genes and derived from the parentalREtsAF cells, was induced by two di�erent means:shift to the restrictive temperature and moderateoxidative stress. Upon a shifting to the restrictivetemperature these cells undergo p53-dependent apop-tosis (Zheng et al., 1994). Early in this process, cellsexhibit a decrease of DCm occurring simultaneouslywith cell shrinkage (VayssieÁ re et al., 1994). In contrast,during hydrogen peroxide-induced apoptosis, thedecrease of DCm preceeds the reduction in cell size,suggesting that mitochondrial membranes are one ofthe primary targets of hydrogen peroxide (this paper).In agreement with this suggestion, when yeast cells aregiven similar doses of H2O2, they display a decrease inDCm that is not followed by an apoptotic death butonly by a transient cell cycle arrest (SG, unpublishedresults).Some results suggested that Bcl-2, in addition to its
possible functions in the endoplasmic reticulum (Lamet al., 1994), nuclear envelope (Ryan et al., 1994) andmitochondria (Zamzami et al., 1996a), may alsooperate at the chromosomes of mitotic nuclei (Lu etal., 1994) and may counteract apoptosis by inhibitingexpression of speci®c nuclear genes (Ivanov et al.,1995; Grimm et al., 1996). A study performed withbreast epithelial cells reported that Bcl-2 suppressesexpression of the p53-target genes and suggested thatBcl-2 may inhibit p53 functional activity. Thus, it hasbeen proposed that Bcl-2 is involved in the regulationof an early commitment step determining whether cellsproliferate or die (Upadhyay et al., 1995). Our resultsobtained with P1-Bcl2 cells during p53-mediatedapoptosis do not support this hypothesis: overexpres-sion of bcl-2 does not abolish the induction of thep53-target genes waf1 and bax. A similar observationhas also been made during apoptosis induced by atemperature sensitive p53 (Guillouf et al., 1995; Wanget al., 1995). Furthermore, our results show that Bcl-2might even allow an apparently longer waf1 induction,which suggests that Bcl-2 may permit cells expressinghigh levels of Waf1 to survive longer in culture.Alternatively, Bcl-2 could indirectly upregulate Waf1and permit growth arrest rather than apoptosis(Guillouf et al., 1995; Polyak et al., 1996). Whateverthe case, Bcl-2 can protect P1-Bcl2 cells from p53-mediated apoptosis without inhibiting induction ofp53 target genes and thus acts exclusively downstreamfrom p53. Bcl-2 also protects P1-Bcl2 cells fromapoptosis induced by hydrogene peroxide. Duringthis process, although p53 remains inactivated bylarge T antigen, as evidenced by the lack of p53 leveldecrease and Bax induction, Waf1 level increase. Asimilar p53-independent pathway for activation ofwaf1 expression has been observed following anoxidative stress (Russo et al., 1995). Similar to theresults obtained during p53-mediated apoptosis, Bcl-2does not inhibit accumulation of Waf1 and may allowa sustained survival of Waf1-expressing cells during
H2O2-mediated apoptosis. Thus, whatever the stimu-lus, Bcl-2 protects cells without signi®cantly inhibitingp53-target genes induction, suggesting that it does notplay a role during the induction phases of apoptosis.Stable overexpression of bcl-2 has been shown to
increase DCm of L929 cells and this e�ect has beensuggested to be linked to its anti-apoptotic activitysince the ionophore nigericin, known to increase DCm
has a similar e�ect (Hennet et al., 1993). However, weshow here that bcl-2 overexpression in P1-Bcl2 cells atthe permissive temperature is not associated with anincrease in DCm. This discrepancy may result fromdi�erences between the two cell lines. Alternatively, theincrease in DCm observed in L929 cells may be a longterm consequence of bcl-2 overexpression. The kineticanalysis of events occurring during P1-Bcl2 apoptosisinduced either at the restrictive temperature or byhydrogen peroxide shows, in agreement with otherresults (Shimizu and Eguchi, 1996; Susin et al., 1996;Zamzami et al., 1996b), that Bcl-2 delays the decreaseof DCm occurring during early apoptosis. Furthermore,during hydrogen peroxide-induced apoptosis thekinetics of passage from cells with low DCm andnormal size to cells with low DCm and small size is alsoslowed down by bcl-2 overexpression. Thus, Bcl-2 alsoinhibits changes occurring after the DCm decrease,suggesting that it could either inhibit cytoskeletonrearrangement occurring during apoptosis or couldallow cells to endure a low DCm for a longer timebefore the cell death process continues.We have found that the cell death induced by
150 mM hydrogen peroxide is apoptotic, while a 700 mMtreatment leads to necrotic death. This result is inagreement with previous works showing that the samephysiological signals (TNF-a) (Laster et al., 1988) orthe same chemical or physical stresses (Lennon et al.,1991) may induce apoptosis or necrosis depending onthe dose and/or the cell type. Bcl-2 protects againstH2O2-induced apoptotic death but also necrotic celldeath, which is in agreement with previous resultsshowing that Bcl-2 may protect cells from necrosis(Kane et al., 1995; Subramanian et al., 1995; Shimizuet al., 1996). It suggests that Bcl-2 can counteract theoxidative stress following exposure to H2O2 and/or thatone step of apoptosis controlled by Bcl-2 is alsoinvolved in necrosis.In contrast, in this cell system, Hsp27 does not delay
apoptosis induced by p53 activation or moderateoxidative stress. No e�ect of hsp27 overexpression isfound either at the level of the pattern of geneinduction or at the level of the decrease in DCm.Thus, although hsp27 expression is known to mediatean increase in glutathione capable of protecting cellsagainst oxidative stress or TNFa-induced necrotic celldeath (Mehlen et al., 1995b, 1996a), its overexpressiondoes not delay the decrease of DCm observed duringapoptosis of REtsAF-derived cells induced by oxidativestress. However, we show that Hsp27 can delaynecrosis induced by H2O2 showing that it is indeedcapable of inhibiting oxidative stress-induced celldeath. It could seem contradictory that, in our cellsystem, Hsp27 inhibits cell death induced by 700 mM ofH2O2 but is ine�cient in preventing cell death inducedby lower doses. This suggests that Hsp27 is indeedcapable of preventing cellular damages produced byH2O2 but that it is not able to prevent early irreversible
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
356

events of apoptosis occurring in P1-Hsp27 cells. Inagreement with this hypothesis some P1-Hsp27 cellsprotected from necrosis by Hsp27 seem to ®nally dieby apoptosis (data not shown). It has been shown thatHsp27 blocks Fas/APO-1-based and staurosporine-induced apoptosis of L929 cells (Mehlen et al.,1996b) while Bcl-2 inhibits staurosporine-inducedapoptosis (Jacobson et al., 1994; Jacobson and Ra�,1995) but only weakly Fas/APO-1-mediated apoptosis(Itoh et al., 1993; Reed, 1994). We show here that Bcl-2, but not Hsp27, delays p53-mediated apoptosis ofREtsAF-derived cells which suggest that apoptosis ofthese cells involves an Hsp27-independent pathway.However, it remains possible that the basal level ofHsp27, although very low in REtsAF-derived cells,confers maximum protection against apoptotic eventsinhibitable by Hsp27. Furthermore, the fact that theapoptotic stimuli used in this study (temperature shiftor hydrogen peroxide treatment) induce Hsp27accumulation suggest that this increase in Hsp27 levelmay be enough to provide a protection to REtsAFcells. Whatever the case, these results show that Hsp27and Bcl-2 act at di�erent levels to prevent apoptosis.Oxidative stress can damage cells by lipid peroxida-
tion and alteration of protein and nucleic acidstructure. To prevent oxidative damage, mammaliancells have developed an antioxidant defence system thatincludes nonenzymatic antioxidants (eg. glutathione) aswell as enzymatic activities (eg catalase, superoxidedismutase) (Pinkus et al., 1996). However, ROS alsoplay a role in physiological systems: they were shownto be responsible for the inducible expression of genesassociated with in¯ammatory and immune responses.Current evidence indicates that di�erent stimuli useROS as signalling messengers to activate transcriptionfactors, such as AP-1 and NF-kB, and induce geneexpression (Pinkus et al., 1996). Moreover, severalobservations suggest that ROS might mediate apopto-sis (Buttke and Sandstrom, 1994). Although the role ofROS in the execution of the cell death programme islargely con¯icting, there is now compelling evidencethat ROS can activate apoptosis (Jacobson, 1996). Onepossible explanation of the e�ect of Hsp27 onapoptosis and necrosis is that necrosis is mainly theconsequence of cellular damage that can be minimizedby overexpression of Hsp27, while low doses aresu�cient to activate an Hsp27-independent apoptoticprogramme.In conclusion, depending on the cell system and/or
of the stimuli, Bcl-2 and/or Hsp27 can protect cellsfrom apoptotic death. In addition, both are capable ofprotecting cells against oxidative damages leading tonecrosis. Taken together, these results suggest that Bcl-2 and Hsp27 act at several levels to protect cells againstapoptosis but that their function does not totally (ifany) overlap.
Materials and methods
Plasmids construction
Plasmid pUHD15-1 is a tet repressor-herpes simplex virustransactivator protein VP16 (tTA) fusion-protein expres-sion construct; plasmid pUHD10-3, tTA-responsive con-struct, contains tet operator (tOp) sequences. These twoplasmids were derived from those described by Gossen and
Bujard (1992) and were provided to us by Dr B Henglein(Institut Curie, Paris, France), after he has modi®ed theseconstructs by cloning (i) the puromycin resistant gene(1.2 kb) into the XhoI site of pUHD15-1 and (ii) a SV40polyadenylation sequence into the XhoI site of pUHD10-3(Schulze et al., 1995). The cDNA encoding human bcl-2(nucleic acid sequence from 1 to 1855) was subcloned fromplasmid SFFV-Bcl-2 nl, provided to us by Dr SJKorsmeyer (Howard Hughes Medical Institute, St Louis,USA), into the EcoRI site of pUHD10-3, giving rise topBMbcl2. Plasmid pBMhsp27 was constructed by subclon-ing the cDNA encoding rat heat shock hsp27 from plasmidpBSlzap-hsp27 (I Gue nal, unpublished results) between theEcoRI and XhoI sites of pUHD10-3.
Development of Bcl-2 and Hsp27-expressing cell lines
The temperature-sensitive REtsAF cell line was selectedand isolated at 338C at low density from a rat embryo®broblast culture infected with a tsA58 mutant of SV40.When cultures are shifted at the non-permissive tempera-ture (39.58C), cells undergo apoptotic death. REtsAF cellswere stably transfected (Lipofectin technique-Life Tech-nologies) with the tTA expression plasmid pUHD15-1,providing resistance to puromycin. Cells were selected inmedium containing puromycin (1 mg/ml, Gibco) andtetracycline (1 mg/ml, Sigma). Resistant colonies wereclones and expanded yielding tTa producing REtsAFcells. This cell line was named P1.
One of these clones was stably cotransfected with PSVtk-neob (providing resistance to geneticin) and either pBMbcl2or pBMhsp27 at a 1 : 10 ratio. Transfected cells were selectedin medium containing puromycin (1 mg/ml), G418 (Geneticin250 units/ml, Gibco) and tetracycline (1 mg/ml). Severalresistant P1-Bcl2 and P1-Hsp27 clones were isolated andtested respectively for the expression of Bcl-2 and Hsp27proteins, following removal of tetracycline from the culturemedia.
All constructs were active in tTA-producing cells in atetracycline-dependent manner.
The P1-Bcl2 clone used for this study gives an increase inBcl-2 level of at least 10 ± 20-fold after 5 days of induction.Quantitative estimation has not been done since puri®ed Bcl-2 is not commercially available.
For the P1-Hsp27 clone selected for this work,quantitative analysis performed with murine puri®edHsp27 protein (StressGen) indicates that the basal level ofHsp27 in this cell line propagated with tetracycline isaround 0.05 ng/mg of total protein. After 5 days ofinduction without tetracycline, this level reaches about3 ng/mg of total protein.
Cell culture conditions
P1-Bcl2 and P1-Hsp27 cell lines were propagated inDMEM/HAMF12 (vol/vol, Gibco) medium supplementedwith 2% serum substitute (Ultroser-G, Gibco) pluspenicillin (100 mg/ml) and streptomycin (100 U/ml) under5% CO2).
Cells were routinely maintained at 338C in the presence ofpuromycin and tetracycline (both at 1 mg/ml ®nal concentra-tion) and were regularly screened for the absence ofmycoplasma.
Induction of cell death: apoptosis or necrosis
Before each experiment of induction of cell death, P1-Bcl2cells were seeded in 60 mm dishes at a concentration of 2to 66104 cells and propagated for ®ve days withouttetracycline in the culture media, in order to allowaccumulation of the Bcl-2 protein. In parallel, controlP1-Bcl2 cultures were propagated in the presence of
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
357

1 mg/ml of tetracycline, which prevents the association ofthe transactivator tTa with the tetO sequence and henceinhibits transcription of bcl-2.
This same procedure was followed when studying the P1-Hsp27 cell line.
For both cell lines after ®ve days of `pre-culture' with orwithout tetracycline, apoptotic cell death was induced eitherby shifting cell cultures at the restrictive temperature (39.58C)or by treating cells at the permissive temperature with 150 mMhydrogen peroxide (H2O2 3%, Sigma).
Necrosis was obtained by treating cells at the permissivetemperature (after 5 days of pre-culture with or withouttetracycline) with hydrogen peroxide at a ®nal concentrationof 700 mM.
Kinetics of cell death
Inhibition of cell proliferation was evaluated in relation tothe time after induction of cell death. At the end of eachtreatment time, the e�ect was quanti®ed by determining theproportion of live cells (estimated by trypan blue dyeexclusion), expressed as percentage of the untreated cellpopulation.
At di�erent times after induction of cell death, the long-term cytotoxic e�ect was evaluated by studying the relativeplating e�ciency of treated cells. To carry out this study,16103 treated and untreated cells were seeded into 100 mmdishes and incubated at 338C for 15 days. At the end of thisincubation time, the colonies were stained with Coomassieblue (Coomassie blue 0.1%, acetic acid 10%, methanol50%). Relative plating e�ciency values were expressed aspercentage of the number of colonies formed by untreatedcells.
Conditions of staining with DiOC6(3) and PI
Propidium iodide (PI, Sigma) is a non-speci®c DNAintercaling agent which is excluded by the plasma membraneof living cells. Stock solution of PI was prepared in distilledwater at 1 mg/ml and kept in the dark at 7208C. PI (l Exmax 540 nm; l Em max 625 nm) was used at 10 mg/ml ®nalconcentration and the ¯uorescence was immediately eval-uated by ¯ow cytometry.
DiOC6(3) (3.3'-Diethyloxacarbocyanine, Molecular Probes)is a lipophilic dicarbocyanine dye. It is a positively chargedmolecule that passes through the plasma membrane andpartition into the lipid phase of all intracellular membranes.At low doses, the positive charge of the dye causes it toaccumulate into mitochondria under the in¯uence of negativemitochondrial membrane potential (Korchak et al., 1982;Koning et al., 1993; Suzuki et al., 1994). The stock solutionof DiOC6(3) (l Ex max 484 nm; l Em max 501 nm) wasprepared at 0.5 mM in ethanol and kept in the dark at7208C.For mitochondrial membrane potential measurements, cells(106 per ml) were incubated in culture media with 0.1 mM ofDiOC6(3) for 30 min at 338C, before the addition of PI andanalysis of the sample by ¯ow cytometry.
With PI, cells were considered positive when their¯uorescence intensity was higher than that of control cells.With DiOC6(3), cells were considered `low' (DiOC6(3) lowcells) when their ¯uorescence intensity was lower than those
of control cells (DiOC6(3) high cells), while keeping normallight scatter properties.
Flow cytometry
Flow cytometric analysis were performed on a ELITE ESP¯ow cytometer (COULTER). Fluorescence excitation wasobtained through the blue line (488 nm) of an argon ionlaser operating at 15 mW. Green ¯uorescence of DIOC6(3)was collected with a 525 nm band pass ®lter and red¯uorescence of PI with a 610 nm band pass ®lter. Analysiswere performed on 10 000 cells and data were stored inlistmode. Light scatter values were measured on linearscale of 1024 channels and ¯uorescence intensities on alogarithmic scale of ¯uorescence of four decades of log.
Western blot analysis
Cells were rinsed twice in cold PBS, collected with ascraper and lysed by the addition of 0.5% of Nonidet-P40.Crude extracts (60 to 100 mg of protein; 10 mg for detectionof Hsp27 protein) were analysed by SDS ± PAGE in 15%acrylamide, 0.2% bisacrylamide. For Western blotting,proteins were transferred to nitrocellulose membrane(Schleicher & Schuell) according to Towbin et al. (1979).Blots were exposed to the ®rst antibody (mouse or rabbitIg) overnight at 48C and then to horseradish peroxydase-conjugated anti-mouse (or rabbit) immunoglobulin serum(Biosystem) for 1 h at room temperature. The immuno-reactivity was revealed using the Amersham ECL kit.
To carry out this study, blots were successively exposed tothe following antibodies: anti-Waf1 (C-19); anti-p53 (clonepAb122); anti-Bcl-2 (clone 100) and anti-Bax (N-19). DuplicateWestern blots loaded with 10 mg of protein from crude extractswere performed and exposed to anti-Hsp27 (SPA-801). Allantibodies were purchased from Santa Cruz, with the exceptionof anti-p53 (generous gift from Dr E May, Villejuif, France)and anti-Hsp27 (StressGen Biotechnologies Corp.).
In vivo ¯uorescent labelling of mitochondria
P1-Bcl2 and P1-Hsp27 cells were seeded on glass coverslipsand propagated with or without tetracycline (1 mg/ml) at338C for 5 days before the induction of cell death, afterwhich they were stained with DIOC6(3) and PI as describedabove. Coverslips were placed on a slide chamber contain-ing fresh medium and cells were then immediatelyexamined by epi¯uorescence and photographed under aLeitz DMRB Leica microscope.
AcknowledgementsWe thank Drs JL VayssieÁ re and P Arrigo and V Rinchevalfor helpful discussions and Drs R Karess, M Laurent andC Thermes for their critical reading of the manuscript. Thiswork was supported in part by grants from the Associationpour la Recherche contre le Cancer (no. 6960), from theLigue Nationale Contre le Cancer and from the MinisteÁ rede l`Education Nationale de l`Enseignement Supe rieur et dela Recherche (ACC-SV4). IG and CSF were supported by afellowship from the Ligue Nationale Contre le Cancer.
References
Akao Y, Otsuki Y, Kataoka S, Ito Y and Tsujimoto Y.(1994). Cancer Res., 54, 2468 ± 2471.
Arrigo AP and Landry J. (1994). In The biology of heat shockproteins and molecular chaperones, Morimoto R, TissieÁ resA and Georgopoulos C (eds.). Cold Spring Harbor press:New York, pp. 335 ± 373.
Arrigo AP and Mehlen P. (1994). In Heat shock or cell stressproteins in the nervous system. Meyer J and Brown I (eds.).Academic press: pp. 145 ± 167.
Borner C, Martinou I, Mattmann C, Irmler M, Schaerer E,Martinou JC and Tschopp J. (1994). J. Cell Biol., 126,1059 ± 1068.
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
358

Buttke TM and Sandstrom PA. (1994). Immunol. Today, 15,7 ± 10.
Chen LZ, Nourse J and Cleary ML. (1989). Mol. Cell. Biol.,9, 701 ± 710.
Ciocca DR, Oesterreich S, Chamness GC, McGuire WL andFuqua SA. (1993). J. Natl. Cancer Inst., 85, 1558 ± 1570.
Clarke PGH. (1990). Anat. Embryol., 181, 195 ± 213.de Jong D, Prins FA, Mason DY, Reed JC, van Ommen GBand Kluin PM. (1994). Cancer Res., 54, 256 ± 260.
Ellis RE, Yuan JY and Horvitz HR. (1991). Annu. Rev. CellBiol., 7, 663 ± 698.
Gossen M and Bujard H. (1992). Proc. Natl. Acad. Sci. USA,89, 5547 ± 5551.
Grimm S, Bauer MK, Baeuerle PA and Schulze-Ostho� K.(1996). J. Cell Biol., 134, 13 ± 23.
Gue nal I and Mignotte B. (1995). FEBS Lett., 374, 384 ± 386.Guillouf C, Grana X, Selvakumaran M, De Luca, GiordanoA, Ho�man B and Liebermann DA. (1995). Blood, 85,2691 ± 2698.
Hengartner MO and Horvitz HR. (1994). Cell, 76, 665 ± 676.Hennet T, Bertoni G, Richter C and Peterhans E. (1993).
Cancer Res., 53, 1456 ± 1460.Hockenbery D, Nunez G, Milliman C, Schreiber RD andKorsmeyer SJ. (1990). Nature, 348, 334 ± 336.
Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL andKorsmeyer SJ. (1993). Cell, 75, 241 ± 251.
Huot J, Roy G, Lambert H, Chretien P and Landry J. (1991).Cancer Res., 51, 5245 ± 5252.
Itoh N, Tsujimoto Y and Nagata S. (1993). J. Immunol., 151,621 ± 627.
Ivanov VN, Deng G, Podack ER and Malek TR. (1995). Int.Immunol., 7, 1709 ± 1720.
Jacobson MD. (1996). Trends Biochem. Sci., 21, 83 ± 86.Jacobson MD, Burne JF, King MP, Miyashita T, Reed JCand Ra� MC. (1993). Nature, 361, 365 ± 369.
Jacobson MD, Burne JF and Ra� MC. (1994). EMBO J., 13,1899 ± 1910.
Jacobson MD and Ra� MC. (1995). Nature, 374, 814 ± 816.Kane DJ, Ord T, Anton R and Bredesen DE. (1995). J.
Neurosci Res., 40, 269 ± 275.Kane DJ, Sara®an TA, Anton R, Hahn H, Gralla EB,Valentine JS, Ord T and Bredesen DE. (1993). Science,262, 1274 ± 1277.
Kerr JFR, Wyllie AH and Currie AR. (1972). Br. J. Cancer,26, 239 ± 257.
Kluck RM, Bossy-Wetzel E, Green DR and Newmeyer DD.(1997). Science, 275, 1132 ± 1136.
Koning AJ, Lum PY, Williams JM and Wright R. (1993).Cell. Motil. Cytoskeleton, 25, 111 ± 128.
Korchak HM, Rich AM and Wilkenfeld C. (1982). Biochem.Biophys. Res. Commun., 108, 1495 ± 1501.
Korsmeyer SJ. (1992). Blood, 80, 879 ± 886.Krajewski S, Tanaka S, Takayama S, Schibler MJ, FentonWand Reed JC. (1993). Cancer Res., 53, 4701 ± 4714.
Kroemer G, Petit PX, Zamzami N, VayssieÁ re JL andMignotte B. (1995). FASEB J., 9, 1277 ± 1287.
Kumar S. (1995). Trends Biochem. Sci., 20, 198 ± 202.Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL andDistelhorst CW. (1994). Proc. Natl. Acad. Sci. USA, 91,6569 ± 6573.
Laster SM, Wood JG and Gooding L. (1988). J. Immunol.,141, 2629 ± 2634.
Lennon SV, Martin SJ and Cotter TG. (1991). Cell Prolif.,24, 203 ± 214.
Lindquist S. (1986). Annu. Rev. Biochem., 55, 1151 ± 1191.Lindquist S and Craig EA. (1988). Annu. Rev. Genet., 22,631 ± 677.
Lu QL, Hanby AM, Nasser HM, Gschmeissner SE, Lu PJ,Taylor PJ, Krajewski S, Reed JC and Wright NA. (1994).J. Cell Sci., 107, 363 ± 371.
Mehlen P, Kretz-Remy C, Briolay J, Fostan P, Mirault MEand Arrigo AP. (1995a). Biochem. J., 312, 367 ± 375.
Mehlen P, Kretz-Remy C, Preville X and Arrigo AP. (1996a).EMBO J., 15, 2695 ± 2706.
Mehlen P, Preville X, Chareyron P, Briolay J, Klemenz Rand Arrigo AP. (1995b). J. Immunol., 154, 363 ± 374.
Mehlen P, Schulze-Ostho� K and Arrigo AP. (1996b). J.Biol. Chem., 271, 16510 ± 16514.
Nakai M, Takeda A, Cleary ML and Endo T. (1993).Biochem. Biophys. Res. Commun. 196, 233 ± 239.
Nguyen M, Branton PE, Walton PA, Oltvai ZN, KorsmeyerSJ and Shore GC. (1994). J. Biol. Chem., 269, 16521 ±16524.
Oltvai ZN, Milliman CL and Korsmeyer SJ. (1993). Cell, 74,609 ± 619.
Petit PX, Lecoeur H, Zorn E, Dauguet C, Mignotte B andGougeon ML. (1995). J. Cell Biol., 130, 157 ± 167.
Pinkus R, Weiner LM and Daniel V. (1996). J. Biol. Chem.,271, 13422 ± 13429.
Polyak K, Waldman T, He TC, Kinzler KW and VogelsteinB. (1996). Genes Dev., 10, 1945 ± 1952.
Reed JC. (1994). J. Cell. Biol., 124, 1 ± 6.Russo T, Zambrano N, Esposito F, Ammendola R, CiminoF, Fiscella M, Jackman J, O'Connor PM, Anderson CWand Appella E. (1995). J. Biol. Chem., 270, 29386 ± 29391.
Ryan JJ, Prochownik E, Gottlieb CA, Apel IJ, Merino R,Nunez G and Clarke MF. (1994). Proc. Natl. Acad. Sci.USA, 91, 5878 ± 5882.
Samali A and Cotter TG. (1996). Exp. Cell Res., 223, 163 ±170.
Schulze A, Zerfass K, Spitkovsky D, Middendorp S, BergesJ, Helin K, Jansen DP and Henglein B. (1995). Proc. Natl.Acad. Sci. USA, 92, 11264 ± 11268.
Schulze-Ostho� K, Bakker AC, Vanhaesebroeck B, BeyaertR, Jacob WA and Fiers W. (1992). J. Biol. Chem., 267,5317 ± 5323.
Schwartz LM, Smith SW, Jones MEE and Osborne BA.(1993). Proc. Natl. Acad. Sci. USA, 90, 980 ± 984.
Sedlak TW, Oltvai ZN, Yang E, Wang K, Bouse LH,Thompson CB and Korsmeyer SJ. (1995). Proc. Natl.Acad. Sci. USA, 92, 7834 ± 7838.
Shimizu S and Eguchi Y. (1996). Oncogene, 13, 21 ± 29.Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y,Matsuda H and Tsujimoto Y. (1996). Oncogene, 12,2045 ± 2050.
Shimizu S, Eguchi Y, Kosaka H, Kamiike W, Matsuda Hand Tsujimoto Y. (1995). Nature, 374, 811 ± 813.
Subramanian T, Tarodi B and Chinnadurai G. (1995). Cell.Growth Di�er., 652, 131 ± 137.
Susin SA, Zamzani N, Castedo M, Hirsh T, Marchetti P,Macho A, Daugas E, Geuskens M and Kroemer G. (1996).J. Exp. Med., 184, 1 ± 11.
Suzuki K, Ehara T, Osafune T, Kuroiwa H, Kawano S andKuroiwa T. (1994). Eur. J. Cell Biol., 63, 280 ± 288.
Thompson CB. (1995). Science, 267, 1456 ± 1462.Towbin H, Staehelin T and Gordon J. (1979). Proc. Natl.
Acad. Sci. USA, 76, 4350 ± 4354.Upadhyay S, Li G, Liu H, Chen YQ, Sarkar FH and KimHR. (1995). Cancer Res., 55, 4520 ± 4524.
VayssieÁ re JL, Petit PX, Risler Y and Mignotte B. (1994).Proc. Natl. Acad. Sci. USA, 91, 11752 ± 11756.
Wang Y, Okan I, Szekely L, Klein G andWiman KG. (1995).Cell Growth Di�er., 6, 1071 ± 1075.
Wang H, Rapp UR and Reed JC. (1996). Cell, 87, 629 ± 638.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, PengTI, Jones DP and Wang X. (1997). Science, 275, 1129 ±1132.
Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA,Masse B and Kroemer G. (1996a). FEBS Lett., 384, 53 ±57.
Zamzami N, Marchetti P, CastedoM, Zanin C, VayssieÁ re JL,Petit PX and Kroemer G. (1995). J. Exp. Med., 181,1661 ± 1672.
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
359

Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M and Kroemer G. (1996b). J. Exp.Med., 183, 1533 ± 1544.
Zheng DQ, VayssieÁ re JL, Lecoeur H, Petit PX, Spatz A,Mignotte B and Feunteun J. (1994). Oncogene, 9, 3345 ±3351.
Zheng LT, Sara®an T, Kane DJ, Charles AC, Mah SP,Edwards RH and Bredesen DE. (1993). Proc. Natl. Acad.Sci. USA, 90, 4533 ± 4537.
Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B andAndrews DW. (1996). EMBO J., 15, 4130 ± 4141.
Bcl-2 and Hsp27 effects on cell deathI GueÂnal et al
360