azido polymers-energetic binders for solid rocket propellants
TRANSCRIPT

This article was downloaded by: [Indian Institute of Technology Roorkee]On: 10 July 2013, At: 06:33Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Journal of Macromolecular Science, Part C: PolymerReviewsPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/lmsc19
Azido Polymers—Energetic Binders for Solid RocketPropellantsBharti Gaur a , Bimlesh Lochab a , V. Choudhary a & I. K. Varma aa Centre for Polymer Science and Engineering, Indian Institute of Technology Delhi, HauzKhas, New Delhi, IndiaPublished online: 07 Feb 2007.
To cite this article: Bharti Gaur , Bimlesh Lochab , V. Choudhary & I. K. Varma (2003) Azido Polymers—Energetic Bindersfor Solid Rocket Propellants, Journal of Macromolecular Science, Part C: Polymer Reviews, 43:4, 505-545, DOI: 10.1081/MC-120025976
To link to this article: http://dx.doi.org/10.1081/MC-120025976
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) containedin the publications on our platform. However, Taylor & Francis, our agents, and our licensors make norepresentations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of theContent. Any opinions and views expressed in this publication are the opinions and views of the authors, andare not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon andshould be independently verified with primary sources of information. Taylor and Francis shall not be liable forany losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoeveror howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use ofthe Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in anyform to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
JOURNAL OF MACROMOLECULAR SCIENCE�
Part C—Polymer Reviews
Vol. C43, No. 4, pp. 505–545, 2003
Azido Polymers—Energetic Binders for
Solid Rocket Propellants
Bharti Gaur, Bimlesh Lochab, V. Choudhary, and I. K. Varma*
Centre for Polymer Science and Engineering,
Indian Institute of Technology Delhi,
Hauz Khas, New Delhi, India
CONTENTS
1. INTRODUCTION. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 506
2. PREPARATION OF AZIDO POLYMERS. . . . . . . . . . . . . . . . . . . . . . . 508
2.1. Polymerization of Glycidyl Azide (GA) . . . . . . . . . . . . . . . . . . . . . . 508
2.1.1. Derivatization of polyepichlorohydrin (PECH). . . . . . . . . . . 508
2.2. Direct Conversion of Epichlorohydrin to Glycidyl Azide
Polymer (GAP).. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 513
2.3. Simultaneous Degradation and Azidation of PECH and
Its Copolymers. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 514
2.4. Block Copolymers of GAP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 515
2.5. Polymerization of Azidomethyl Oxetanes . . . . . . . . . . . . . . . . . . . . . 515
2.6. Poly(allyl azide) (PAA). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 519
3. PHYSICAL PROPERTIES OF AZIDO POLYMERS . . . . . . . . . . . . . . . 520
4. CURING OF GAP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 522
4.1. Curing Agents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 522
4.2. Cure Monitoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 524
*Correspondence: I. K. Varma, Centre for Polymer Science and Engineering, Indian Institute
of Technology Delhi, New Delhi 110 016, India; Fax: 91-11-26591421; E-mail: ikvarma@
homail.com.
505
DOI: 10.1081/MC-120025976 1532-1797 (Print); 1532-9038 (Online)
Copyright & 2003 by Marcel Dekker, Inc. www.dekker.com
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
4.3. Problems of GAP-Isocyanate Cure . . . . . . . . . . . . . . . . . . . . . . . . . 525
4.4. Characterization of Cured Network. . . . . . . . . . . . . . . . . . . . . . . . . 525
4.5. Curing of Azido Polymers with Dipolarophiles. . . . . . . . . . . . . . . . . 526
5. THERMAL BEHAVIOR OF AZIDO POLYMERS. . . . . . . . . . . . . . . . . 528
5.1. Uncured Azido Polymers—Linear and Branched . . . . . . . . . . . . . . . 528
5.2. Uncured BAMO And AMMO Polymer . . . . . . . . . . . . . . . . . . . . . . 532
5.3. Thermal Behavior of Cured Azido Polymers . . . . . . . . . . . . . . . . . . 533
6. PHOTODECOMPOSITION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 535
7. FORMULATIONS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 536
7.1. Plasticizers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 536
7.2. Oxidizers. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 537
7.3. Pyrolants. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 539
7.4. Ballistic Modifiers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 539
8. CONCLUSION AND FUTURE OUTLOOK . . . . . . . . . . . . . . . . . . . . . 540
ACKNOWLEDGMENT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 540
REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 540
Key Words: Energetic binders; Azide polymers; Poly(allylazide); Oxidizers;
Thermal behavior.
1. INTRODUCTION
High-energy solid rocket propellants are composite materials having abinder [hydroxy terminated polybutadiene (HTPB)], high-energy additives[e.g., ammonium perchlorate (AP)], and pyrolants (metallic powder). HTPB is aninert binder, which has been used in cast-cure propellant systems. Substitution ofHTPB by a more energetic binder may lead to an enhancement in performance ofsuch propellants.
Ammonium perchlorate, the high-energy additive, has its own inherent disad-vantages, as it produces chlorine rich combustion products (30%), which havepotential environmental implications.[1] Further, it produces white smoke trails,which are disadvantageous for specific applications demanding smokeless plume.Ammonium nitrate (AN) has long been considered a highly desirable oxidizer forsolid-fuel rocket propellants because of its extremely low cost, low sensitivity, lowsignature, and absence of halogens. However, it has a crystalline phase stabilityproblem that causes unpredictable ballistic performance in some cases and cata-strophic rocket motor failure in others. This has limited the use of AN withoutthe phase stabilizers.[2]
A new generation of propellants and explosives for missiles and space applica-tion is being developed worldwide these days. The operational trends for futureapplications of these energetic materials are to improve performance, satisfy insen-sitive munitions, comply with environmental aspects, and reduce costs.
The use of energetic additives, mainly binders and plasticizers, is consideredto be one of the practical ways to improve the energy level and other
506 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
technical performances of solid propellants. Recent advances in propellantsresearch, therefore, are aimed at developing high energy binders which give betterperformance than the commonly used HTPB and which are compatible with the eco-friendly oxidizers ammonium nitrate (AN), hydrazinium nitroformate (HNF), orammonium dinitramide (ADN).
Azido polymers have attracted researchers’ attention for the past two decadessince the azido group contributes a positive heat of formation of 313–397 kJ/mol.This is evident from a comparison of heat of formation of ethanol (�278.5 kJ/mol)and 2-azidoethanol, N3CH2CH2OH (þ104 kJ/mol). Compatible propellants withacceptable physical properties could be formulated from azide-containing ingredi-ents.[3] These polymeric azides include poly(glycidyl azide) (GAP) and its copoly-mers, poly(bis-azidomethyl oxetane) [poly(BAMO)] and its copolymers, andpoly(azidomethyl methyloxetane) [poly(AMMO)].[4] These polymers contain azidegroups along the polymer chain as pendant groups, which make them highlyenergetic. The exothermic scission of these –N3 bond releases energy of theorder 685 kJ/mol.[5] The GAP prepolymer is cured by reacting the terminalhydroxy groups with isocyanates, as is done in the state-of-the-art hydroxy termi-nated binder.[6,7] These propellants, in addition to being fuel rich, liberate largeamounts of carbon monoxide, and hydrocarbon on their burning, and are greatlyinsensitive to impact, and provide high burn rates.
The traditional method used for propellant formulation is to mix liquidGAP polymer (Mn<3000) (binder) with curing agents (diisocyanate) and chainextender, trimethylol propane (TMP), high-energy additives, and metallicpowder. However, the energy output of such propellants will be significantlyreduced due to inert properties of diisocyanates and TMP. Another disadvantageof isocyanate cured hydroxy terminated GAP is that the propellant would notbe recyclable, as the crosslinks are irreversibly formed covalent chemical bonds,and thus recovery of energetic ingredients and binders is not possible. Therefore,an elastomeric GAP with Mn>1.4� 105 has been developed to overcome thisproblem. The synthesis and use of recyclable thermoplastic elastomer (TPE)based on azido polymers is gaining importance in the field of energetic binders.
An attractive approach to high energy, low sensitivity propellants involve theuse of energetic oxetane prepolymers. New gun propellants have been developedwhich utilize the thermoplastic elastomer (TPE) [i.e., poly(3,3-bis(azidomethyl),oxetane (BAMO)–3-azidomethyl-methyl oxetane (AMMO)] as the binder and theenergetic solid trimethylenetrinitramine (RDX). These TPE based propellantshave many desirable properties including the ability to be reused, recycled, andreclaimed, an excellent balance of flame temperature and the performance.[8]
Oxetane TPE propellants can readily be mixed and extruded in a twin-screwextruder without aid of any solvents.[9] These binders act as energy partitioningagents by allowing an energetic formulation to maintain a constant energy level ata lower solids percentage.[10]
Although considerable work has been done on high-energy azido plasticizers(molecular weight �500) and binders, a comprehensive review dealing with thestate-of-the-art of these polymers has not been published. This article reviews thesynthesis, characterization, physicochemical, thermal, and mechanical properties ofpolymeric binders containing pendant azido groups.
Azido Polymers 507
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
2. PREPARATION OF AZIDO POLYMERS
2.1. Polymerization of Glycidyl Azide (GA)
The starting monomer, GA is prepared by reacting epichlorohydrin (ECH) withhydrazoic acid to give 1-azido-3-chloro-2-propanol, which is cyclized in the presenceof a base (Sch. 1).
The polymerization of glycidyl azide (GA) was then attempted to be carried outusing the carbocationic method. However, this procedure was unsuccessful as themonomer was found to be unreactive.[3]
2.1.1. Derivatization of Polyepichlorohydrin (PECH)
An alternative route, based on the polymerization of ECH to polyepichloro-hydrin (PECH), followed by the conversion of PECH to GAP by nucleophilicdisplacement of chloride by azide is more successful. Linear or branched hydroxyterminated polyepichlorohydrin has been prepared by ring opening polymerizationof the oxirane group in ECH in the presence of a diol (ethylene glycol) or a triol(1,2,3-propane triol). Azido substitution reaction has been accomplished by reactingwith sodium azide using dimethyl formamide or dimethyl sulfoxide as solvents lead-ing thereby to formation of GAP-diol or GAP-triol (Schs. 2 and 3).
The details of reaction conditions used for the synthesis of these polymers aredescribed below.
H O CH2CH
CH2Cln
O CH2CH2OOCH2CH
CH2Cln
H
H O CH2CH
CH2N3n
O CH2CH2OOCH2CH
CH2N3 n
H
NaN3
HOCH2CH2OHCHCH2
O
CH2Cl +
PECH -DIOL
GAP-DIOL
n
Scheme 2. Preparation of GAP-diol.
CH2
O
CHCH2ClNaN3
HOAcN3CH2CHCH2Cl
OH
OHN3CH2CH CH2
O
-
ECH GA
Scheme 1. Synthesis of GA.
508 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
(a) Synthesis of Polyepichlorohydrin
Polyepichlorohydrin has been synthesized by carrying out the polymerizationof ECH. The conversion depends on the nature of initiator system, monomer toinitiator ratio, solvents, reaction temperature etc. Bronsted acids (e.g., CF3SO3H)as well as Lewis acids (protogens or cationogens) have been used as initiators.[11]
Generally a monomer: initiator ratio in the range of 40� 4:1 has been used. Thepercentage conversion was low when Bronsted acids were used as initiators.Hydrocarbons (toluene), ethers (dioxane, diethyl ether), or chlorinated hydro-carbons (1,2-dichloroethane) have been used as solvents in this polymerization.The effect of catalysts (such as SnCl4, BF3-etherate, or CF3SO3H), solvents,reaction time, and reaction temperature on the percentage yield has been inves-tigated (Table 1).
The polymers thus prepared had low molecular weight and a wide molecularweight distribution. The molecular weight distribution obtained from the GPCchromatograms is given in Table 2.
As is obvious from Table 2, the product distribution is dependent upon theinitiators used. Product obtained when BF3-etherate was used as an initiator hadmolecular masses in the range of 200–2000. When SnCl4 was used as an initiator themolecular mass was considerably high as compared to CF3SO3H or BF3-etherate.When 1,2-dichloroethane or toluene was used as solvent, the reaction was rapid andthe molecular weight distribution was wide. It was noted that as much as 60% of thetetramer was obtained in the system with BF3-etherate in 1,2-dichloroethane. Withethereal solvents, the reaction was slow. High basicity of ethereal solvents is expected
n
n
CH2
O
CH HOCH2CHCH2OH
OH
H O CH2CH
CH2Cl n
OCH2CHCH2O 2CHO
O 2CHO
H
HCH
CH2Cl
CH
CH2Cl
NaN3
n
n
H O CH2CH
CH2N3 n
OCH2CHCH2O 2CHO
O 2CHO
H
HCH
CH2N3
CH
CH2N3
CH2Cl +
Propanetriol
PECH-TRIOL
Scheme 3. Preparation of GAP-triol.
Azido Polymers 509
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
to suppress the propagation reaction and promote a backbiting reaction to producelow molecular weight cyclic oligomers according to reaction (Sch. 4).
As opposed to typical chain end mechanism mentioned above, the polymeriza-tion of ECH could also proceed in the presence of glycols by activated monomermechanism (AMM), which involves the concept of living polymerization that has notermination or transfer process. In AMM, an activated i.e., protonated or positivelycharged monomer molecule inserts into a growing polymer chain having an -OHgroup at the chain-end (Sch. 5).[12,13] This mechanism leads to a polymer having two-OH groups at the chain ends. Here Mn¼ [ECH]/[glycol]. Side reactions, includingcyclization, are strongly suppressed in the polymerization by AMM and well-definedlinear products are obtained. The activated monomer mechanism operates at low
Table 2. Molecular weight distribution of PECH synthesized under different reaction
conditions (sample designation same as Table 1) (data adapted from Ref.[11]).
Sample
designation n¼ 2 n¼ 3 n¼ 4 n¼ 5 n¼ 6 n¼ 7
A-1 2 0 43 14 6 35
A-2 18 1 4 22 20 35
A-3 1 0 26 12 7 54
A-4 3 2 13 22 19 41
A-5 5 0 60 31 4 0
B-1 5 0 9 11 7 68
B-2 15 0 5 6 6 68
B-3 9 0 12 12 11 56
C-1 45 0 8 21 16 10
C-2 32 0 13 17 14 24
n is the degree of polymerization.
Table 1. Effect of reaction parameters on percent yield of polyepichlorohydrin (data adapted
from Ref.[11]).
Sample
designation Initiator Solvent
Monomer/initiator
ratio
Time
(h)
Yield
(%)
A-1 BF3OEt2 1,2-Dichloroethane 44:1 4.0 98
A-2 Dioxane 42:1 4.0 11
A-3 Toluene 36:1 4.0 88
A-4 Ethyl ether 36:1 4.0 5
A-5 1,2-Dichloroethane 36:1 4.0 19
B-1 SnCl4 1,2-Dichloroethane 54:1 3 75
B-2 Dioxane 43:1 2.5 50
B-3 Toluene 44:1 23.5 73
C-1 CF3SO3H 1,2-Dichloroethane 44:1 7.0 2
C-2 Dioxane 38:1 23.0 12
Temperature �5�C (with dioxane �15�C).
510 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
monomer concentration, which requires slow addition of monomer to the reactionmixture. The PECH-diols obtained under these conditions, had controlled molecularweight up to Mn� 2500 g/mole and with Mw/Mn<1.2. The content of cyclic oligo-mers did not exceed 0.7%. PECH of Mn� 2000 was synthesized by using borontrifluoride–ethylene glycol as catalyst while aluminium triethyl–ethylene glycolyielded PECH of Mn more than 100,000.[12]
(b) Azidation of PECH
Azidation of PECH to GAP has been carried out in an organic or an aqueoussolvent,[3,14] using an ionic azide such as lithium azide, sodium azide, or potassiumazide. A molar excess of ionic azide is needed for this purpose.[14] Complete conver-sion of polyepichlorohydrin to GAP with sodium azide in dimethyl sulphoxide(DMSO) is reported to occur at 90–95�C within 12–18 h, whereas the reaction in
R OH H O R O OH H++
++
""
Initiation:
OH H O O OH H
Propagation:
... + +... + +
""
Scheme 5. Activated monomer mechanism for synthesis of PECH.
The notation ‘‘Hþ’’ means that the proton is not located on any specific oxygenatom, but is rapidly exchanging between various sites.
OCHCH2
CH2Cl
O
CH2Cl
O
CH2Cl
:
OCHCH2-OCHCH2
CH2Cl
O
CH2ClCH2Cl
O..
CH
CH2Cl
CH2
OCH2 CH CH2Cl
CH2
CHO
CH2
CH
O
CH2Cl
ClH2C
O O
CH2Cl
CH2Cl
A
A
A-+ +
polymer
back-biting
+
propagation
+
volatile dimer
-
propagating chain
-ECH + propagating
chain
Scheme 4. Chain growth and backbiting reaction during ECH polymerization.
Azido Polymers 511
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
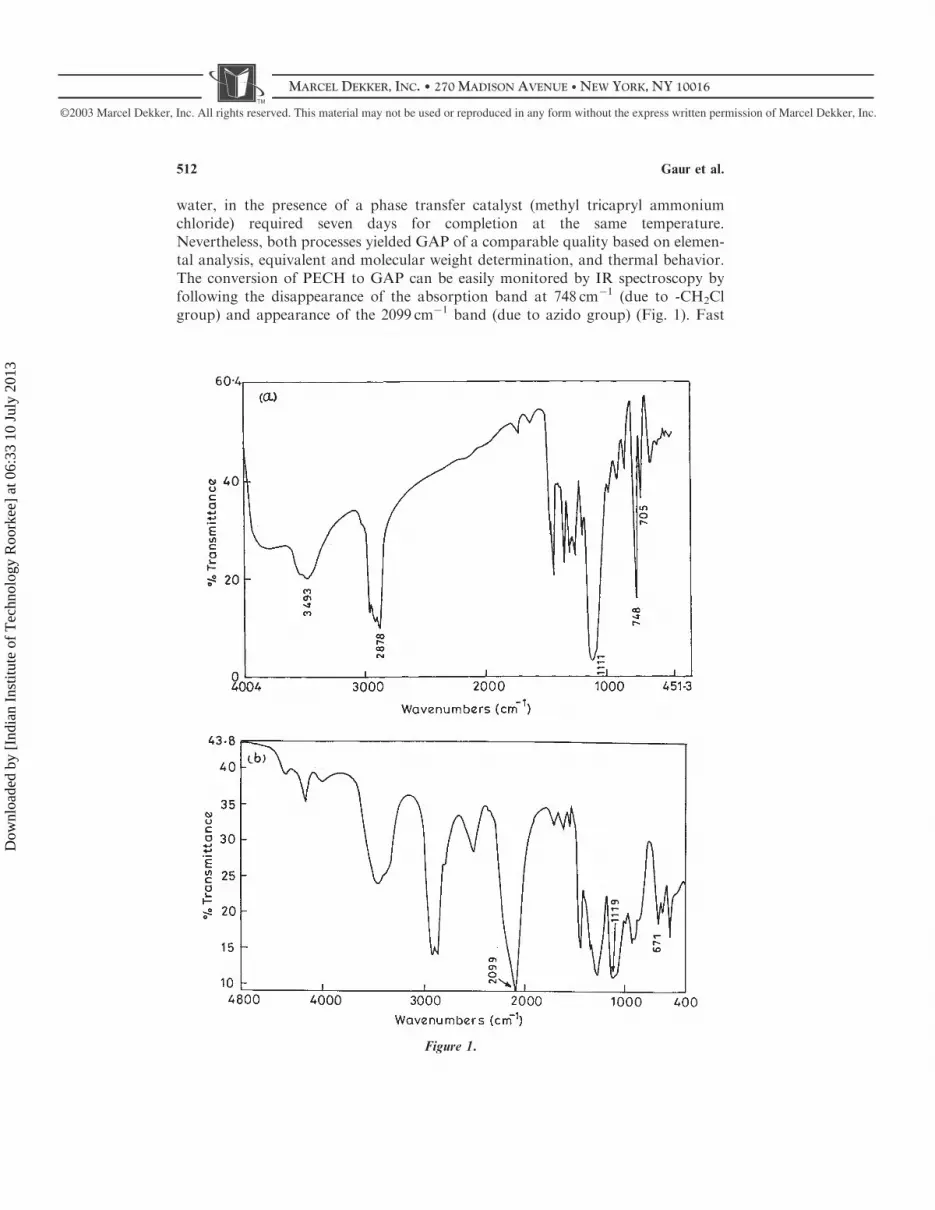
water, in the presence of a phase transfer catalyst (methyl tricapryl ammoniumchloride) required seven days for completion at the same temperature.Nevertheless, both processes yielded GAP of a comparable quality based on elemen-tal analysis, equivalent and molecular weight determination, and thermal behavior.The conversion of PECH to GAP can be easily monitored by IR spectroscopy byfollowing the disappearance of the absorption band at 748 cm�1 (due to -CH2Clgroup) and appearance of the 2099 cm�1 band (due to azido group) (Fig. 1). Fast
Figure 1.
512 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
neutron activation analysis of nitrogen and chlorine in the final product has alsobeen used to monitor the conversion of PECH to GAP.[15]
In the above-mentioned azidation reaction, carried out in solution, one of theproblems is the slowing of the reaction rate above �90% conversion. This decreasein rate, which more than doubles the reaction time from the initial rate, is a con-sequence of the association of the metal cation (e.g., Naþ or Liþ) of the azide saltwith the solvent medium, in which it has limited solubility. In a molten salt method,quaternary ammonium azide such as tetrabutyl ammonium azide and trace amountof water is mixed with PECH and heated at �105�C for about 0.33 h. The resultingliquid product on washing with water and drying showed 85–90% conversion ofPECH to GAP.[16]
Low molecular weight GAP (�500), which could be used mainly as an energeticplasticizer, can be prepared by a one-step method, which involves direct conversionof ECH to GAP.[17] The plasticizer helps in processing of the propellants based onGAP and enhances the stability and mechanical properties. However the hydroxyend-groups of these glycidyl azide polymers react with the isocyanate curing agentleading thereby to a loss of plasticization effect. A diazide terminated azide polymerhas, therefore, been developed to overcome this difficulty. Tosylation of hydroxyterminated polyepichlorohydrin with p-toluene sulfonyl chloride (in presence of pyr-idine) followed by replacement of chlorine and tosyl group by azide (sodium azide inDMF) is a convenient approach for the synthesis of such polymers (Sch. 6).
Azido terminated glycidyl azide polymers (GAP-A) have also been prepared bynitration of hydroxy terminated polyepichlorohydrin and subsequent reaction withsodium azide[18] (Sch. 7).
2.2. Direct Conversion of Epichlorohydrin to
Glycidyl Azide Polymer (GAP)
A single step process for preparation of hydroxy terminated GAP(Mn� 500 g/mol) has been described,[19] which is less time consuming and morecost effective. In this method sodium azide was gradually mixed with ECH (1:1molar ratio), DMF, and EG and the contents were initially stirred at 70�C. Sincethe reaction was exothermic in the initial stages, the temperature was controlled
PECH(OTs)2
NaN3
DMF
OCH2CH2 H
CH2Cl
CH2 CH O
n
H O CH CH2
CH2Cl n
O
PECH(OH)2
OCH2CH2 CH2 CH O
n
O CH CH2
n
O
CH2N3CH2N3
N3N3CH CHCH2CH2
CH2N3 CH2N3
p-toluenesulfonylchloride
pyridine
-1 -1
Scheme 6. Tosylation of PECH followed by azidation.
Azido Polymers 513
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
during the first 30min. The reaction temperature was then raised to 90�C and stirringwas carried out at this temperature for 15 h. The yield was high (�90%).
2.3. Simultaneous Degradation and Azidation of PECH
and Its Copolymers
Branched GAP or copolymer of GAP (70%) with ethylene oxide (30%) (GEC)having variable molecular weight (500–40,000) has been synthesized directlythrough a simultaneous degradation and azidation process by reacting a highmolecular weight rubbery poly(epichlorohydrin) (PECH) or poly(epichlorohydrin-co-ethylene oxide) (PEEC), with the epichlorohydrin (ECH) monomer and sodiumazide (NaN3).
[20,21] Degradation and azidation of a commercial rubbery PECH orPEEC with NaN3 and lithium methoxide (basic cleaving agent) in the presence of apolyol at 120�C in polar organic solvents (DMSO, DMAc, DMF, etc.,) without theuse of ECH monomer has also been reported.[22–24] The polyols used in this synthesiswere 1,2,3-propane triol (PPT), trimethylol propane (TMP), and pentaerythritrol(PE) (Table 3). DMSO is preferred over the other solvents due to the quality ofthe final product.
OCH2CH2 H
CH2Cl
CH2 CH O
n
H O CH CH2
CH2Cl n
O
HNO3 / H2SO4
OCH2CH2
CH2Cl
CH2 CH O
n
O CH CH2
CH2Cl n
O NO2O2N
NaN3
OCH2CH2 CH2 CH O
n
O CH CH2
n
O
CH2N3CH2N3
N3N3CH CHCH2CH2
CH2N3 CH2N3
Hydroxy terminated PECH
Polyepichlorohydrin nitrate
-1 -1
Scheme 7. Nitration of PECH followed by azidation.
514 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
The observed polydispersity index (Mw/Mn) is between 1.70 and 2.10. This typeof molecular weight distribution may be considered relatively low for polymersresulting from a degradation process. GEC had a higher viscosity than GAP witha similar molecular mass. This may be attributed to the presence of ethylene oxidegroups in the polymer chain.
2.4. Block Copolymers of GAP
A triblock copolymer poly(glycidylazide-b-butadiene-b-glycidyl azide) (GAP-PB-GAP) has been reported by Vasudevan and Sundarajan[25] (Sch. 8). In thesynthesis of this block copolymer, HTPB was used as an alcohol and boron trifluo-ride etherate (BF3-etherate) as catalyst. Epichlorohydrin was added to the catalystmixture dropwise. The polymerization was carried out at 0�C for 12 h and then for4 h at room temperature (30�C) under nitrogen atmosphere. Epichlorohydrin adds toboth the hydroxyl groups of HTPB leading to the formation of a secondary alcohol,which is able to react with epichlorohydrin by the same pathway to yield poly-(epichlorohydrin) blocks. Ring opening polymerization was followed by conversionof the -CH2Cl group into -CH2N3 group using sodium azide in DMSO at 105�Cfor 10 h.
2.5. Polymerization of Azidomethyl Oxetanes
Polymerization of substituted four membered oxetanes can produce polymerswith higher molecular weight and higher elongation as compared to GAP.[26] Thefunctionality and molecular weight of the resulting polymer may be controlled easilyas compared to epoxides. Therefore, polymers and copolymers of oxetanes contain-ing azido groups, such as 3-methyl-3-azidomethyl oxetane (AMMO) and 3,3-bis-azidomethyl oxetane (BAMO) have been investigated in the past.[26] The synthesisof poly(BAMO) has been carried out using two different procedures. In one of themethods, 3,3-bis(chloromethyl) oxetane (BCMO) was treated with sodium azide indimethyl formamide (DMF) at 85�C for 2 h. The bisazidomethyl oxetane obtainedwas then homopolymerized or copolymerized with tetrahydrofuran (THF) bycarbocationic ring opening polymerization using BF3-etherate as catalyst in the
Table 3. Solution properties of GAP and copolymers prepared by simultaneous degradation
and azidation in presence of polyols (data adapted from Ref.[20]).
Polymer Polyol [n] (dL/g) Mn Mw
GAP homopolymer PPT 0.122 10,600 18,100
TMP 0.125 11,000 18,500
PE 0.141 11,400 22,700
GEC copolymer PPT 0.185 10,400 20,900
TMP 0.216 14,700 28,500
PE 0.183 11,600 20,400
Azido Polymers 515
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
presence of butanediol as an initiator at �5�C. The other method involved the ringopening polymerization of BCMO or copolymerization with THF usingbutanediol as initiator and BF3-etherate as catalyst at �5�C. The polymer or thecopolymer obtained was then treated with sodium azide in DMF (solvent) at 90�C(Sch. 9). The starting material, BCMO is easily obtained by ring closure of trichloroderivative of pentaerythritol.[27]
Quasi-living cationic polymerization of 3-azidomethyl-3-methyloxetane(AMMO) was achieved with a bis(chlorodimethylsilyl)benzene/Ag hexafluoroanti-monate (AgSbF6) initiating system in methylene chloride at �78�C. The averagemolecular weight (Mn) increased linearly with an increasing monomer/initiator([M]/[I]) ratio and a linear Mn vs. [M ]/[ I] ratio plot passed through the origin.[28]
Both ends of the difunctional initiator are activated.[29]
Block copolymers of BAMO with other cyclic ethers have been synthesized withan aim of developing energetic thermoplastic elastomers (TPE). The TPE thusprepared have high molecular mass, low polydispersity index, low Tg, and goodenergetic characteristics. Block and random copolymers of THF and BAMO havebeen prepared by using triflic anhydride (CF3SO2)2O as a bifunctional initiator.[30]
HO OH
OCH2Cl
BF3-etherate
O OCHCH2CH2CH
CH2ClCH2Cl
OHHO
OCH2Cl
O O CH2CH
CH2ClCH2Cl
O CH2CH
CH2Cl
OHO
CH2Cl
HO
NaN3, DMSO
O OCHCH2CH2CH
CH2N3CH2N3
O CH2CH
CH2N3
OHOCHCH2
CH2N3
HO
CHCH 2CHCH2
n
n
n
n n
n
105ºC, 10h
n
n
n
2
Scheme 8. Preparation of block copolymer poly(GAP-b-PB-b-GAP).
516 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
Homo as well as block copolymers of low polydispersity index (1.23–1.28) could beprepared by using this initiator. In random copolymerization of THF and BAMO, r1and r2 values were found to be 0.33 and 1.12 respectively, showing thereby higherreactivity of BAMO.
Novel energetic oxetane derivatives 3-nitratomethyl-3-methyl oxetane (NMMO)and AMMO could be polymerized by triflic anhydride [(CF3SO2)2O]. The livingcationic characteristics of the polymerization confirmed via a 19FNMR technique.Novel polymers of the A-B-A type with various molecular weight (Mw¼
14,320–40,660) and low polydispersity index (PDI¼ 1.11–1.29) were obtained.[31]
Analysis of the polymer by different techniques (NMR, IR spectroscopy,and DSC) showed that the structure of the polymer was composed of a two-phasedomain structure of amorphous poly(NMMO) phases and crystalline poly(AMMO)phases.
The synthesis of triblock copolymer of BAMO, AMMO, and bis-ethoxymethyloxetane (BEMO) with center block composed of BAMO and AMMO has beenreported by Murphy et al.[32] The copolymerization required 1,4-butanediol andBF3-etherate in the ratio of 1:2. A solution of monomer (AMMO or BAMO) inmethylene chloride was added drop wise to the catalyst system at �10�C. A newmonomer (BAMO or AMMO) was added when more than 95% conversion of theprevious block is reached. The block copolymers are thus sequentially polymerizedin a ‘‘living polymer’’ manner according to activated monomer mechanism.
Synthesis of BAMO and nitratomethyl methyloxetane (NMMO) block copoly-mers has been reported by using 1,4-butanediol/BF3-etherate initiating agent.[33]
A triblock copolymer BAMO–NMMO–BAMO was prepared by using p-bis(a,a-dimethylchloromethyl)benzene ( p-DCC) with silver hexafluoroantimonate(AgSbF6) as initiator.[34] The propagating chain end is a carboxonium ion. Thepolymerization has been carried out in nitrogen atmosphere at low temperature(�70�C) using methylene chloride as solvent. The centre block of NMMO isprepared first, according to Sch. 10.
The cationic copolymerization of AMMO was initiated by a mixtureof BF3�OEt2 and diethylene glycol. The copolymerization of AMMO with3-(azidomethyl)-3-(2,5-dioxaheptyl)oxetane and 3-(azidomethyl)-3-(2,5,8-trioxadecyl)oxetane(II) was investigated in order to obtain (for solid propellants)
OCH2Cl
CH2Cl
NaN3
DMFO
CH2N3
CH2N3CH2N3
CH2N3
BF3.etherateO CH2 C
OCH2Cl
CH2Cl
BF3.etherate CH2Cl
CH2Cl
O CH2 C
CH2
CH2
NaN3
+85ºC , 2 h butanediol n
butanediol n DMF, 90ºC
Scheme 9. Synthetic route for the preparation of poly(BAMO).
Azido Polymers 517
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
materials with less of a crystalization tendency and a lower glass transition tem-perature, (Tg) than poly(AMMO), for example, when AMMO was copolymerizedwith 15wt.% II, it was possible to obtain bifunctional oligomers with Tg �50 to�60�C, without significant loss in the nitrogen content.[35]
Linear ABA triblock and (AB)n segmented block copolymers of energetic mono-mers were synthesized. The rigid and soft blocks are prepared from AMMO andNMMO respectively. Polymerization of AMMO initiated by triethyloxonium tetra-fluoroborate and by spiro (benzoxasilole)/propanediol produced a-mono-hydroxy-poly-AMMO, and o,o0-dihydroxy-poly-AMMO of number averagemolecular weight-16,000 and 2000, respectively and a,o-dihydroxy-poly-NMMO(Mn¼ 13,000).[36] A spiro(benzoxasilole) catalyst, 3,3,30,30-tetrakis(trifluoro-methyl)-1,10-(3H,3H)-spirobis(1,2-benzoxasilole), was used to polymerize 3,3-R,R0-oxetanes: (R,R0
¼ ethoxymethyl; R¼ azidomethyl, R0¼Me; R¼ nitratomethyl,
R0¼Me; and R,R0
¼ azidomethyl) with descending rates in this order. 31PNMRspectra of polymerization mixtures quenched using Bu3P were consistent with anoxonium ion propagating species.[37]
New energetic azide group-containing polyesters have recently beenreported. These are prepared from 2,3-dibromosuccinic acid and 1,2-propanedioland 3-chloropropanediol and succinic/malonic acid. These hydroxyl terminatedpolymers had a functionality of approximately 2.0. The number average molecular
CC
CH3
CH3
CH3
CH3
Cl Cl
:OCH3
CH2NO2
AgSbF6
C
CH3
CC
CH3
CH3
CH3
CH3
CC
CH3
CH3
CH3
CH3
CC
CH3
CH3
CH3
CH3
OCH3
CH2NO2
CC
CH3
CH3
CH3
CH3
O CH2
CH2NO2
CH2O
CH3
CH2NO2
+
++
++
NMMO
++
NMMO
+
Propagating chain
Scheme 10. Preparation of triblock copolymer BAMO–NMMO–BAMO.[34]
518 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
weight of the bromopolyester and chloropolyesters of malonic/succinic acid werefound to be 1280, 1079, and 825, respectively. These halogenated polyesters wereconverted to the corresponding azido polyesters via reaction with sodium azide. Thedecomposition temperatures of these azido polyesters were around 235, 210, and228�C respectively.[38]
Several new energetic polymer systems poly(BAMO-co-GAP) and poly(BAMO-co-glycidyl nitrate (PGN)) and poly(BAMO-co-AMMO) have been evaluated asenergetic thermoplastic elastomeric binders for composite rocket propellants.BAMO-GAP had moderately high density, high heat of formation, and high burningrate, which should make it attractive in formulations requiring high-burning-ratecompounds. BAMO-PGN had a very high density, a favorable oxygen balance,and a reasonably high heat of formation. These properties make BAMO-PGNand BAMO-GAP ideally suited as binders for high-performance energeticcompounds.[39]
2.6. Poly(Allyl Azide) (PAA)
Azidation of poly(allyl chloride) (PAC) is a convenient route for the preparationof poly(allyl azide). However allyl chloride during free radical polymerization under-goes degradative chain transfer yielding polymers of relatively low molecular masses.Poly(allyl chloride) having molecular mass in the range of 2000 g/mol could beobtained by following the cationic route. Allyl chloride (0.5mol) was polymerizedin our laboratories using Lewis acids such as anhydrous TiCl4/FeCl3/AlCl3 andaluminium powder.[40] The concentration of the catalyst was varied from 0.7 to3.2mol % of the monomer. The quantity of aluminium powder was kept constant(2 g). The reaction was carried out at low temperatures (0–20�C) and brown-coloredviscous polymer was formed.
Chlorine in poly(allyl chloride) samples was found to be in the range of 35–40%,which is lower than the calculated value of 46.05%. On the other hand, the carboncontent was higher than the expected value. A discrepancy in chlorine and carboncontent of the polymers may be explained on the basis of branching of poly (allylchloride) (Sch. 11).
1HNMR spectrum of poly(allyl chloride) is shown in Fig. 2. The chloromethylprotons in poly(allyl chloride) were observed at 4.03 ppm. The vinyl proton reso-nance signals of allyl chloride at 5.22, 5.32, and 5.94 ppm were absent in poly(allylchloride). Additional resonance signals were observed at 0.88–2.27 ppm. The inte-gration of the resonance signals at 0.88–2.27 ppm did not correspond to the numberof methyl and methylene protons in the polymer backbone for a linear-chain poly-mer. The loss of chlorine, as indicated by elemental analysis results, also suggests theformation of a branched polymer. Therefore, on the basis of elemental analysis and1HNMR it was concluded that poly(allyl chloride) was a branched polymer. Theazidation of poly(allyl chloride) was carried out using sodium azide and dimethylsulphoxide as solvent at 100�C for 12 h. The conversion of PAC to PAA was mon-itored by IR spectroscopy by following the decrease in the intensity of 736 cm�1
absorption band due to -CH2Cl and appearance of a strong band due to -CH2N3 at2095 cm�1 (Fig. 3).
Azido Polymers 519
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
3. PHYSICAL PROPERTIES OF AZIDO POLYMERS
Depending on the backbone structure and molecular mass, the Tg of linear azidopolymers range from �20 to �70�C. The glass transition temperature of GAP wasfound to be �45�C. Tg of branched GAP and branched glycidyl azide-ethylene oxidecopolymer is in the range of �60 to �70�C.[20] The physical properties of variousglycidyl azide polymers are given in Table 4.[41,42] Since GAP is in a rubbery state at
Figure 2.
CH2 CH
CH2Cl
CH2 CH
CH2Cl
AlCl3 FeCl3 CH2 CH CH2 CH
CH2ClCH2
AlCl4FeCl4
CH2 CH
CH2
CH2 CH
CH2Cl
CH2
CH
CH2Cl
CH2 CH CH2Cl
or
+
+- -or
Scheme 11. Probable side reaction during the synthesis of poly(allyl chloride).
520 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
temperatures above Tg, which is lower than the lowest operational temperature(�40�C) for rocket monitors, it can be considered a mechanically safe binder forcomposite propellant.[43] Tg values of GAP increased with increase in the quantityof decomposed -N3 groups. Branched GAP cured with IPDI has a Tg �23� 3�C.
Figure 3.
Azido Polymers 521
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
The presense of ethylene oxide units in branched GAP reduced the Tg to�40� 2�C.[21]
The Tg of poly(AMMO) (molecular weight 2800–6700 g/mol) was determined byDSC and was in the range �51.5 to �45.5�C.[44] Poly(AMMO-b-NMMO-b-AMMO) had a Tm¼ 82�C and Tg¼�3�C was phase separated in the melt. Thiscopolymer decomposed at 224�C evolving 0.69 kJ/g of heat. It has excellentmechanical properties with elongation of 683% at break, 5.25MPa stress, and82% recovery. The poly(AMMO-b-NMMO)n copolymer has similar thermal prop-erties and spectroscopic characteristic, but is inferior to tri block copolymer in allrheological and mechanical properties.[36]
BAMO homopolymer has a Tg of �28�C and Tm of 76–80�C.[26] The Tg isreduced to �60�C in the copolymers of BAMO and THF.[26] The viscosity is inthe range of 500–5000 cps, depending on the molecular mass (500–5000 g/mol).The functionality of the linear polymer is 1.5–2.0 and for the branched version itvaried from 5.0–7.0. The burning rate of GAP is very sensitive to the amount of thecuring agent. The material (88.5% GAP) self-extinguishes at ambient pressure andburns at 1.96 cm/s at 6.89MPa.[3]
A maximum burn rate of 1.68 cm/s has been reported for GAP cured with TDIor isophorone diisocyanate.[6] Burning rate of BAMO polymer is less than thatof GAP.[4]
4. CURING OF GAP
4.1. Curing Agents
Hydroxy terminated azido polymers are cross-linked by polyisocyanates via aurethane forming reaction. Functionality greater than two must be used to ensurereticulation. In such systems, different mechanical properties of the binder can beobtained by adjusting the parameters of the curing reaction and the componentconcentrations, which result in varying cross-link density of the matrix. A schematic
Table 4. Characteristics of azide polymers (data adapted from Refs.[41,42]
Sample designation
Properties
GAP-DIOL
hydroxy
terminated
GAP-TRIOL
hydroxy
terminated
GAP-A
azide
terminated
�Hf (kJ/g) 1.170 1.170 2.299
�Hr (kJ/g) 1.839 1.630 —
Density (g/cc) 1.29 1.29 1.27
Appearance Light yellow
liquid
Light yellow
liquid
Light yellow
liquid
Tg (�C) �45 �45 �56 to �69
�Hf¼ heat of formation, �Hr¼ heat of reaction.
522 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
representation of the preparation of polyurethanes by such a polyaddition procedureis shown in Sch. 12.
The choice of an appropriate curing agent is of prime importance to meet theminimum strain, stress, and hardness levels of mechanical properties, whichcrucially affect the performance of the propellant.[45] Commonly used isocyanatesare isophorone diisocyanate (IPDI), toluene diisocyanate (TDI), 4,40-diisocyanatodiphenylmethane (MDI), 4,40-dicyclohexyl methane diisocyanate (H12MDI),Desmodur N-100. The molecular formulae of some of these curing agents[7,46] aregiven in Sch. 13.
The polyol reactants, coupling agents or chain extenders, which are generallyexplored in the synthesis of polyurethanes, are triethanolamine, hexanetriol,trimethylol propane,[47] glycerol, pentaerythritol , pyrogallol.[21] Their main function
CH 2NCO
NCOOCN
H 12 MD I
NCOOCNH 3 C
IPDI
NCO
N
O
O
NH
NH
NCONCO
Desmodur N-100
CH 3
NCO
NCO
CH 3
NCOOCN
80% + 20% TDI
Scheme 13. Structure of polyisocyanates used for curing azido polymers.
R OH
N C O
N C O
OH O O CN H R'R O O CN H NH C O
HO O C N
R'
NH CO O R
O R N C O
R'
N C O
n + n
n - 1
Scheme 12. Urethane network formation.
Azido Polymers 523
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
is to make the PU well cured. They have a noticeable effect on the curing reactionof azido polymer with the isocyanate and thus affect the mechanical properties.[23]
The network structure obtained by reacting glycidyl azide polymer (GAP) withDesmodur N-100 and pentaerythritol can be schematically represented as follows(Sch. 14).
Catalysts are often required to obtain complete curing and also to speed up thereaction, although the amount and type of catalyst depends on the nature of reac-tants. The most common types of catalysts used are organometallics (e.g., organotincompounds) and tertiary amines (triethyl amine, TEA). Dibutyltin dilaurate (DBTL)is known to be the most suitable catalyst for the urethane formation between longchain diols including GAP[41] and isocyanates. The curing time of GAP is reducedfrom three weeks to 5–6 days at 60�C in the presence of DBTL (30–75 ppm).[48]
When composite catalyst, triphenyl bismuth (TPB) and DBTL are used for curingGAP-diisocyanate in the presence of trimethylol propane (TMP), curing time of 3–4days is required.[47–49] This composite catalyst suppresses CO2 formation andpromotes cross-link density.
4.2. Cure Monitoring
The curing of GAP is a slow process. The mixtures having a NCO/OH ratio of 0.7or 0.8 require a longer curing time (about three weeks) for a complete curing than dothe samples with higher NCO/OH ratio (0.9–1.2) (about two weeks). The curingreaction has been monitored by gel-time determination, viscosity and hardness mea-surements, FTIR spectroscopy, and DSC studies. Gel-time, which determines the potlife of a propellant and is related to the extent of cross-linking, affects the processing ofan uncured propellant. Propellant slurry should have a reasonably low viscosity andlong enough pot life to make it castable. Gel-time can be determined by viscositymeasurements and is strongly dependent on the amount of curing catalyst,[48] natureof isocyanate, and ratio of NCO/OH. 4,40-Diisocyanato diphenylmethane (MDI),exhibited highest rate of curing while IPDI reflected lowest rate of reaction.[21]
The hardness increases as the curing reaction proceeds and reaches a plateau ofconstant value, thereby indicating the completion of curing reaction of GAP. Theultimate hardness of the cured GAP increased with an increase in NCO/OH ratio.The propellants become harder upon solid loading. The binder with a NCO/OHratio of 0.8 had the lowest value of hardness and therefore may be considered to bemore suitable for composite propellant applications.[49] Monitoring the hardnessgives an apparent curing completion but not a true chemical completion. There
GAP
OCOHN
OCOHNO N
O
HN
HN ONHOCO
O
O
O
Scheme 14. Network structure of cured GAP using Desmodur N-100.
524 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
might be a significant number of unreacted GAP molecules, which could notparticipate in curing due to the diffusion-controlled nature of the reaction.
The Tg of pure GAP, which is around �45� 1�C[48] increases on curing by 6–9�C and depends on the NCO/OH ratio. An increase in the extent of curing (byincrease in NCO/OH ratio) leads to an increase in Tg values. Addition of plasticizers(i.e., bis-2,2-dinitropropyl acetal and bis-2,2-dinitropropyl formal (BDNPA/F) didnot affect the curing reaction but reduced Tg to �47�C which is low enough toproduce a rubbery propellant.[48]
Kinetics of polyurethane formation between GAP and N-100 of catalyzed aswell as uncatalyzed reaction has been studied in the bulk state by quantitative FTIRspectroscopy by following a decrease in the intensity of NCO stretching band at2270 cm�1 and appearance of new band at 1726 cm�1. The enthalpy and entropy ofactivation for uncatalyzed reaction were found to be 44.1� 0.5 kJ/mol and�196�2 J/Kmol, respectively. The rate constant for the catalyzed and uncatalyzedreaction at 60�C was found to be 4.37, and 3.88� 10�6 Lmol�1s�1 respectively. Asignificant rate enhancement is observed in the presence of a catalyst at operatingconditions.[45]
4.3. Problems of GAP-Isocyanate Cure
There are certain problems, which are encountered while using isocyanatesas curing agents for hydroxy-terminated binders. The first and the most seriousproblem is the gas evolution, when GAP is cured with diisocyanates, because the-N¼C¼O group, reacts rapidly with water to release CO2 and forms numerous voidsin the cured explosive, resulting in a decrease of loading density, mechanicalstrength, performance, and safety. Hydroxyl terminated GAP, itself has inherenttrapped moisture due to hydroxyl groups, which is needed to be expelled beforecuring with isocyanates in order to prevent bubble formation. Void-free propellantsrequire de-gassing before curing. Addition of deflagerating additives eliminates thisde-gassing step.[50] A second problem is a significant decrease in the energy output ofplastic bonded explosives (PBXs) or propellants, because of the inert properties ofdiisocyanates and chain extenders (TMP). A third problem is the loss of plasticizingeffect during curing of the hydroxyl groups of GAP with isocyanates. Curing causesan increase in Tg of GAP and it increases with increasing in NCO/OH ratio.[50]
In order to avoid this problem, it is desirable either to add a plasticizer or to providea plasticizer without hydroxyl groups i.e., azide terminated GAP (GAPA). Inorder to keep both high-energy density and improved mechanical properties, useof elastomeric GAP (average molecular weight>1.4� 105) along with multi-hydroxy-functional branched GAP (B-GAP) has been reported.[47]
4.4. Characterization of Cured Network
Network structure of elastomers can be characterized by swelling, solubility, andmechanical measurements. The average molecular mass of the chain sectionsbetween cross-links, Mc, was calculated using Flory-Rehner equation from the swel-
Azido Polymers 525
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
ling studies of GAP elastomers (Desmodur N-100, IPDI/TMP, and H12MDI/TMP)in tetrahydrofuran solvent at room temperature. The volume of the swollen networkprepared with the same ratio of NCO/OH depended on the structure of isocyanatesand was highest with IPDI and lowest with Desmodur N-100. Desmodur N-100cured elastomers for the same ratio of NCO/OH have smaller values of Mc thanthose of IPDI/TMP and H12MDI/TMP cured elastomers. A linear relationshipbetween sol fraction and Mc values was observed.
[7]
In order to assess the effect of residual DMSO in B-GAP on the properties ofcured energetic binders, small amounts of DMSO (2, 4, and 6% w/w) to that ofB-GAP were intentionally added to the curing mixtures NCO/OH ratio as 1.2using TDI as the curing agent. The cured samples containing 6% (w/w) ofDMSO, appeared less transparent and much softer than the other samples. Thishas been attributed to some degree of phase separation caused by the presence ofDMSO.[21]
NCO-terminated energetic azide can itself be used as a curing agent for OH-terminated aliphatic polymers to obtain an elastomer for solid rocket propellants.[51]
4.5. Curing of Azido Polymers with Dipolarophiles
Curing of azido polymers has also been done with multifunctional dipolaro-philes e.g., multifunctional acrylic or acetylenic esters (dimethylene glycol diacrylate,tetraethylene glycol diacrylate, ethylene glycol diacrylate, hexanediol diacrylate,pentaerythritol tri/tetraacrylate, hexane diol diacrylate) or acrylic amide molecules(hexane diamine bis-acrylamide, methylene bis-acrylamide).
Azides are 1,3-dipolar compounds and readily undergo a [3þ 2] cycloadditionwith olefinic or acetylinic esters and amides to yield a five-membered heterocyclicring (e.g., 1,2,3-triazoline or 1,2,3-triazole.[52] The kinetics and mechanism of suchcycloaddition has been reported in the literature.[53] A concerted nonsynchronusmechanism with partial charges at the transition state (charge imbalance) has beensuggested for such cycloaddition which are always cis and usually regiospecific.[54,55]
The triazoline thus synthesized is thermally labile and looses N2 to yield aziridines.Therefore, the reactions are carried out at low temperatures and may take weeks forcompletion. Rigid alkenes such as norbornene, react with azide on heating to formexo-adduct. For example, norbornene: aryl azide adducts have been prepared byrefluxing in petroleum ether (60–90�C) for 3–4 h.[55]
The use of highly functional dipolarophiles can reduce the relative amount ofcross-linking agent required to achieve a desired degree of cross-linking, therebymaintaining a higher percentage of high energy chemical moieties in the cross-linked polymer network. Further reaction between an azido polymer and a multi-functional dipolarophile is not stoichiometrically limited by the amount of hydroxy/azido groups contained in the azido polymer. An azido polymer has a relatively largeamount of pendant azido groups, and a multifunctional dipolarophile can react withsome or all of these azido groups. This allows for extensive control of the degree ofcross-linking of the azido polymer thus providing reaction products having a varietyof morphologies and/or hardness properties.[50] The typical reaction between anazido group and a multifunctional dipolarophile[56] is depicted in Sch. 15.
526 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
The above-mentioned reaction is insensitive to moisture and hence there is noneed to avoid moisture in reactants (e.g., drying of fillers etc.). Dipolarophiles haveadvantages in producing cross-linked high-energy materials. Cross-linking at azidogroups with these dipolarophiles changes the azido group to a triazoline or a triazolegroup, which can ensure stability[56] and enhances the burn rate of the cross-linkedpolymer.[51] Azido polymers with improved burn rate have been prepared by usingazido polymers, an acrylic and an acetylenic ester or amide groups.[56]
The curing of poly(allyl azide) (PAA) using ethylene glycol dimethacrylate(EGDMA) has also been reported.[40] The reaction can be depicted according tothe reaction (Sch. 16).
N3 HC C C
O
O R CC
O
O CH
C
O
O R
NN
N
C
O
OH
NN
N
H
Polymer +
Polymer Polymer
2
Scheme 15. Reaction scheme for cross-linking of azido polymers by difunctional
dipolarophiles.
CH2 CH2O OC CC
CH3CH3
CH2CH2
O O
C
CH2 CH2O OC C C
CH3CH3
CH2CH2
O O
C
N
NN N N
N
N3+Polym er
Polym erPolym er
Scheme 16. Curing of PAA using EGDMA.
Azido Polymers 527
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
Preferred amounts of the multifunctional dipolarophile are in the range of20–100 pph (parts by weight of an ingredient/100 parts by weight of the azido poly-mer). Such an additional reaction can be easily carried out at ambient conditions inabout 1–24 h. The reaction can be carried out in the temperature range of 0�C toabout 90�C. However, it is preferable to use temperatures between 25–40�C. Thereaction rate is typically hindered by the presence of solvent.[50]
Azides are known to react readily with both electron-deficient as well aselectron-withdrawing dipolarophiles. Therefore, studies on highly electron-deficientdipolarophiles such as bismaleimides, bisitaconimides, or endo-5-norbornene-2,3-dicarboximide (bisnadimides) with allyl azide and glycidyl azide were carried outby Varma et al.[57] The curing of azide could be carried out at 40–60�C to yieldflexible products with improved thermal properties.
5. THERMAL BEHAVIOR OF AZIDO POLYMERS
Thermal stability and degradation of uncured and cured azido polymers hasbeen investigated by DSC and thermogravimetric analysis in nitrogen atmosphere.Analysis of gases generated by the decomposition has been done by gas chromatog-raphy and IR spectroscopy.
5.1. Uncured Azido Polymers—Linear and Branched
In the DSC scans of GAP a single exothermic peak is observed in the tempera-ture range of 186–273�C with exothermic peak temperature 247� 5�C due to elim-ination of nitrogen from the azide group.[57] The energy liberated at this stage isaround 1.9 kJ/g.[58,59] Sahu et al.[59] however have reported a bimodal exotherm inGAP polymers of molecular weight 1150 and 2150 g/mol, with exothermic peaktemperatures at 218, 242, and 222�C, 262�C, respectively. The DSC scan (Fig. 4)of poly(allyl azide) showed an exothermic transition in the temperature range of155–274�C, with the exothermic peak temperature at 231�C. The energy liberatedwas 1.099 kJ/g.[40]
The thermal decomposition of polyglycidyl azide (GAP) and bis(azidomethyl)-oxetane-tetrahydrofuran copolymer (BAMO/THF) showed overall first-orderkinetics. Additonal azide groups at the terminal positions in GAP decomposedindependently and increased the rate of decomposition. However, the decompositonkinetics was less affected by the additional azide groups in the main chain. Thedecompositon properties of aged polymer samples were the same as those ofunaged ones.[60]
The DSC traces of azido polymers have also been recorded using different ramprates (0.2–10�C/min). A ramp rate higher than 5�C/min causes violent detonation inthe DSC cell (Fig. 5). Such explosions are also observed when more than 5mg ofsample is used.[40]
In the TG trace of GAP recorded in nitrogen atmosphere, two mass loss stepswere observed. The first decomposition corresponds to nearly 36% mass loss inthe temperature range of 210–270�C. The second stage is completed around
528 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
410�C. The temperature of the maximum rate of mass loss in the first step ofdecomposition was 250�C, which coincides with the exothermic peak temperaturesin DSC scans. Therefore, the first step of mass loss in TG can be attributed toelimination of N2, which theoretically should have been around 30% in GAP.
Figure 5.
Figure 4.
Azido Polymers 529
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
However, a mass loss of 36% was observed and may be due to breakdown ofpolymer backbone at higher temperature.
The thermal decomposition of poly(allyl azide) also proceeded in two steps.[40]
The first step of the decomposition occurred in the temperature range of 160–322�C.This decomposition corresponded to 28.3% mass loss (Fig. 6). The exothermictransition (observed in DSC) and accompanied by a mass loss (�28%) in the TGtrace can be attributed to the breakdown of azido group. Theoretically the decom-position of the azido group in a linear PAA should have resulted in a mass loss of33.7%. The poly(allyl azide) thus had lower azido content, which may be attributedto the branched structure of PAA (Sch. 11).
Major mass loss in GAP and PAA was observed above 400�C (�59%) andwas due to breakdown of the polymer backbone leading to the formation ofhydrogen, carbon monoxide, carbon dioxide, methane, ammonia, hydrogencyanide gas, and other higher hydrocarbons.[61] Small molecules are formed byBAMO and GAP, but some large fragments are evolved by AMMO.[61] A charresidue of 15% at 600�C and 7% at 800�C was obtained and was attributed tothe formation of cross-linked structure by imine intermediate (inter- as well asintra-molecular) (Sch. 17).
The N3 group elimination in GAP has been followed by IR spectroscopy. Adecrease in the intensity of N3 groups at 2100 and 1280 cm�1 was observed onheating to 170�C and new peaks appeared at 1529 (NH bending), 1677(C¼N-C-H stretch), 1725 (C¼N-C-C stretch), and 3350 cm�1 (NH stretching).The intensity of the C-O-C (ether) at 1410 cm�1 remained unchanged duringthe initial stages of decomposition supporting the view that in the initial stagesof decomposition only the side chain breakdown was the main process. Intra- andintermolecular bridge formation (Sch. 17) has been proposed to account for the
Figure 6.
530 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
insolubility of partially degraded samples and for the appearance of -C¼N-C-type of structures in IR.[58]
After the elimination of N2 from azido group of GAP further reaction of nitreneintermediate proceed according to Sch. 18.[61]
The combustion characteristics (burning rates, temperature profiles) and kineticparameters (order of reaction, activation energy, pre-exponential factor of rate con-stants) for the thermal decomposition of GAP together with the composition of theproducts of both the combustion and decomposition of uncrosslinked GAP (with amolecular weight of 350 or 2000) and cured GAP were studied. The final tempera-ture of a flame of GAP was measured at 1000 to 1100K. However, approximately47% of the mass of the combustion products was volatile gases N2, H2, CO, CO2,CH4, C2H4, C2H6, NH3, H2O, acetonitrile, acrylonitrile, and furane, as obtained bymass-spectrometry by using freezing/thawing in a liquid nitrogen trap.[62]
HO (CH2 CH
CH2
N3
O CH2 CH
CH2
N3
O) H HO (CH2 CH O CH2 CH O) H
C-H
N-H
C-H
N-H
N2
CH2
CH
O CH2 CHCH
NN
O
CH
CH2CH
N
O OCH
CH
CH2
CH2
CH2CH O OCHCH2
N
CH
CH2
n n + 2n
IntramolecularcrosslinkingIntermolecular
crosslinking
Scheme 17. Intra- and inter-molecular reactions of the imino groups.
OH CH CH2
CH2N
CH CH2
CH2N
O CH CH2
CH2N
O CH CH2
CH2N
O
HCN C2H4
HCN CO CH4
NH3 CO CH2CO
NH3 CH2O HCCOO + +
+
+ +
+ +
(I)
(II)
(III)
(IV)
:
:
:
:
Scheme 18. Decomposition patterns of the nitrene backbone after N2 has been liberated
from GAP.
Azido Polymers 531
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
5.2. Uncured BAMO and AMMO Polymer
A mass spectroscopic study on the thermal decomposition of BAMO andother azide polymers was made by Faber and Srivastava.[63] During isothermalexperiments, the melting temperature of BAMO was found to be 75�C. Slightdecomposition began at 130�C, and at that temperature the release of N2 fromazide groups was observed to be the primary decomposition path, where the rateof N2 release increased as the temperature is increased. The three-carbon backboneof the polymer remains intact until a higher temperature, approximately 160�C,when the mass spectrometer (MS) showed fragments of HCN, H2, CH2, CH3, O,OH, H2O, H2CO, CH2OH, and NO2 in addition to N2. These species showed theinitiation of secondary decomposition. BAMO monomer and polymer were investi-gated by Brill and his coworkers[64,65] with the rapid scanning Fourier infrared(RSFTIR) technique to characterize the slow (5K/min) and rapid (50–255K/min)thermolysis of these compounds and with the simultaneous mass and temperaturechange (SMATCH)/FTIR spectroscopy to determine the kinetics of mass-lossprocess of the compounds.
The TGA data revealed that the first stage mass loss occurred rapidly at about187�C and was completed at about 247�C with the approximately 35% mass loss.This stage was predominantly related to the elimination of N3 bond in the infrared(IR) spectra of the sample. The second stage mass loss process occurred slowlywithout heat liberation and was caused by the decomposition of remaining frag-ments as detected by the strong IR absorption of C¼N, C-H, C-O bonds. The rate ofgradual weight loss was compared under isothermal and nonisothermal conditionsfor AMMO, BAMO, and GAP by TGA during the initial 20–40% of mass losswhere decomposition of the azide group dominates.[65] Isothermal rate constants, k,obtained for different azide polymers are given in Table 5. The following order ofrates of isothermal decomposition was observed: GAP>BAMO>AMMO. BAMOpolymer and copolymer with tetrahydrofuran (THF) were studied by Miyazaki andKubota.[66]
Thermal stability of aged THF copolymers with AMMO or NMMO decreasedin the order poly(THF)>poly(AMMO-THF-AMMO,) poly(NMMO-THF-NMMO.[67] DSC studies revealed that the copolymers of THF with BAMO,AMMO, NMMO have low Tg and large decomposition enthalpies which werebrought about by attached azido groups.[68] NMMO based polymers exhibitedlower thermal stability and relatively higher decomposition enthalpies. Thermal
Table 5. Isothermal TGA rate constants (s�1) for azide polymers (data adapted from
Ref.[65]).
Polymer
180�C
(x� 106)
185�C
(x� 106)
190�C
(x� 106)
195�C
(x� 106)
200�C
(x� 106)
205�C
(x� 106)
210�C
(x� 106)
AMMO — 4.85 8.21 13.00 19.27 33.36 50.20
BAMO — 12.76 24.13 38.65 64.88 85.62 138.10
GAP 12.98 22.88 35.18 59.50 82.45 133.10 —
532 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
stability of these polymers was further improved by curing with isocyanateand pentaerythritol. Poly(BAMO) and poly(BAMO-THF) degrade exothermicallywith Tmax of 237�C and 241�C respectively; and the activation energy of 39 and40 kcal/mol.[29]
5.3. Thermal Behavior of Cured Azido Polymers
The TGA showed a two-stage decomposition pattern in the case of poly (allylazide).[40] A mass loss of 20–25% was observed in the temperature range of161–324�C and may be due to the elimination of nitrogen from the azido group.The second stage decomposition was in the temperature range of 313–525�Caccounting for the mass loss of 52–61%. This is due to the thermal degradation ofthe main chain along with the 1,2,3-triazoline group formed by the cross-linkingreaction of poly(allyl azide) with ethylene glycol dimethacrylate (EGDMA)(5–45 phr). The TG trace of poly(allyl azide) cured with EGDMA is given inFig. 7. Violent detonation was observed when a high heating rate and samplemass of more than �5mg was used (Fig. 8).
The combustion wave structure and thermal decomposition process of GAPpropellent, cured with hexamethylene diisocyanate and crosslinked with trimethylol-propane to formulate the propellant were studied to determine the parameters thatcontrol the burning rate. The burning rate of the propellant is high even though theadiabatic flame temperature is less than that of conventional solid propellants. Theenergy released at the burning surface of this propellant is caused by the scission ofN–N2 bond which produces gaseous N2. The heat flux transferred back from the gasphase to the burning surface is very small compared with the heat generated at theburning surface. The activation energy of the decomposition of the burning surfaceof this propellant was 87 kJ/mol.[69]
Figure 7.
Azido Polymers 533
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
The Arrhenius activation energies (Ea) of decomposition of uncrosslinked andcured GAP are mostly clustered in the 150.5–183.9 kJ/mol range, which is similar tothe RN–N2 bond strength of 146.3–171.4 kJ/mol (Table 6).
An attempt has been made to address several of the discrepancies in thechemistry of GAP. There is broad consistency among the global values of Ea fordecomposition of GAP and general agreement that the rate is dominated by thedecomposition of the azide group, discrepancies exist in the occurrence of later stageproducts. For example, the decomposition of the alkane imine or nitrene after therelease of N2 is found to produce NH3 and HCN in significant amounts in severalstudies. It is found that NH3 content increases with the -OH content of GAP, whichsuggests that end-chain azide groups mostly form NH3. The HCN/NH3 ratioincreases with increasing temperature. However, NH3 is also formed in isocyanatecured GAP and GAP plasticizer, which possess no -OH groups. Some differences areexpected from the use of different types of samples, e.g., different MW, terminalgroups, or cure. The activation energy obtained from DSC and TGA for branchedGAP (B-GAP) is in the range of 138–145 kJ/mol, which is lower than that for GAP-TRIOL (160–178 kJ/mol), indicating that B-GAP decomposes more easily ascompared to GAP-TRIOL.[74]
The burning rate and thermochemical measurements have been conducted toobtain information on the effects of pressure, initial temperature, and catalyst onammonium perchlorate/3-azidomethyl-3-methyloxetane polymer (AP/AMMO) andammonium perchlorate/hydroxy-terminated polybutadiene propellants. Pressureinsensitive burning (i.e., plateau burning) was observed when the HTPB binderwas replaced with the AMMO binder for the AP composite propellants. The
Figure 8.
534 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
temperature sensitivity of both AP/AMMO and AP/HTPB propellants increases aspressure increases. The addition of ferric oxide accelerates the burning rate of bothAP/AMMO and AP/HTPB propellants. The temperature sensitivity of the gas-phasereaction is increased significantly when the HTPB binder is replaced with theAMMO binder.[70]
Measurements of gaseous species and temperature profiles for RDX/BAMOpseudo-propellants were performed by Lee et al.[76] The propellants were madefrom a physical mixture of RDX and BAMO in weight ratio of 80:20.Experiments were conducted at atmospheric pressure in argon with heat fluxes of100 and 400 W/cm2 delivered by a CO2 laser. The products of each ingredient, RDXand BAMO, were found to exist simultaneously throughout the gas phase; however,primary reaction chemistry in the gas phase was dominated by RDX.
The accelerated aging of BAMO-HMX propellent was conducted at 347K forseveral weeks. BAMO-HMX propellants for a very low cross-linking ratio formed acavity between HMX and BAMO binder by evolution of N2, CO2, and H2O duringaccelerated aging. An exotherm, generated by the decomposition of azide binder,initiated and accelerated the thermal decomposition of HMX. The burning rate ofBAMO-HMX propellant was larger than those of BAMO binder and HMX, respec-tively. However, the propellant could not maintain the combustion at low pressure,at which its burning rate was equal to that of BAMO binder.[77]
6. PHOTODECOMPOSITION
Photosensitivity of polymers with pendant azide groups is well known. Suchpolymers are used as negative photoresists mixed with an unsaturated hydrocar-bon polymer under intense UV radiation.[59] The azide group undergoes decom-position, producing a nitrene radical, which quenches by abstracting H fromanother polymer, reacting with itself forming an azo group or by reacting witha double bond of another polymer forming an aziridine linkage. In photoresistapplications, the advantage of aziridine formation by nitrene is generally realizedleading to cross-linking and photo insolubility. Scheme 19 describes the photo-decomposition of the azide group and the reactions of the nitrene radical thusformed.
Table 6. Literature Arrhenius kinetics for the decomposition of GAP (data adapted from
Ref.[61]).
Method Ea (kJ/mole) A (s�1) Temp. range (K) Comments Ref. no.
DSC 166–177 7� 1015–
8.9� 1015520–540 uncured 43, 70&71
DSC 152.1 2� 1013 520–535 cured 70
DSC 178 5.0� 1017 — cured 43
TGA 154–164 — 400–473 — 65, 72&73
A is the frequency factor.
Azido Polymers 535
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
Acceleration of the photoinsolubility of GAPs can be achieved by the additionof an unsaturated hydrocarbon polymer [e.g., poly(ethylene-propylene-ethylene-norbornene)], where the nitrene radical of GAPs reacted with >C¼C< resultingin cross linking through the formation of an aziridine ring.
7. FORMULATIONS
As mentioned earlier, a typical propellent formulation comprises of severalingredients. For example, a solid rocket motor propellant consists of 70–85wt%solids chosen from energetic solids and oxidizers and 15–30wt% of a binder suchas polyoxetane (poly(BAMO/AMMO)) or GAP and an isocyanate curing agent. Thedetails of these ingredients are described in the subsequent text. A plasticizer, such astriethylene glycol dinitrate (TEGDN), N-butyl-2-nitratoethyl nitramine (BuNENA),trimethylolethane trinitrate (TMETN), and butanetriol trinitrate (BTTN), is usedwith the polyoxetane binder, preferably at a 0–3.0:1 plasticizer–polymer weight ratio.The energetic solids and oxidizers can include such material as particulate NH4ClO4,Al, HMX, etc. A propellant formulation with acceptable mechanical propertiescontained BAMO-AMMO-Desmodur N 100 copolymer 12.5, TEGDN 12.5,ammonium perchlorate 48.0, HMX 5.0, and Al 22.0wt%.[78]
7.1. Plasticizers
The curing and solid loading increases the Tg of the polymers. Therefore, forbeing used as binder in composite propellants, the Tg of GAP should be lowered tocompensate the reverse effect of curing and solid loading on the Tg. The Tg valuesof gum stocks upon curing can be compensated by using a plasticizer. Variousplasticizers which are used in order to improve the poor mechanical properties ofGAP at low temperature are iso-decyl pelargonate (IDP), dioctyl adipate (DOA),
R N3 R N N2
R N R N N R
R N H C R NH C
CH CR N R NH
R N R N
+
+
:
:2
:
++:
: +
.
Scheme 19. Photodecomposition of azido polymer under UV irradiation.
536 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
dimethylene glycol dibenzoate (DMGDB),[21] dioctyl azelate (DOZ), dioctyl seba-cate (DOS), diethyl phthalate (DEP), 1,2,4-butanetriol trinitrate (BTTN), trimethyl-olethane trinitrate (TMETN), polyethylene glycol (PEG), bis-2,2-dinitropropylacetal and bis-2,2-dinitropropyl formal (BDNA/F).[42] The plasticizer had no effecton the decomposition characteristics of GAP. The addition of a plasticizer affects themechanical properties (e.g., ultimate hardness) of the gum stocks significantly.
The Tg of GAP is decreased from �45 to �65�C with bis-2,2-dinitropropylacetal/formal (BDNPA/F) and to �48.4�C with dioctyl adipate (DOA) at a 25/75plasticizer/polymer weight ratio. The decomposition properties remain practicallyunchanged, while the Tg increased to �36.8�C upon curing.[43]
Nitroplasticizer (BDNA/F) has been found to be more compatible with ener-getic binder GAP than PEG.[79] Nitrate plasticizer, TMETN, played an importantrole in improvement of not only combustion properties but also insensitivities ofGAP/AN propellants.[80]
Low molecular mass GAPs can be exploited as an energetic plasticizer.[18] GAPwith a molecular mass of 500 would be useful as a plasticizer in composites andpropellant compositions.[17] An azide-terminated azido polymer (GAP-A) acts asplasticizer for propellants.[18] The replacement of nitroglycerin plasticizer by GAPin composite-modified double-base propellants results in greater safety in handling,reduced detonation sensitivity, higher burning rate and specific impulse, andimproved mechanical properties.[81]
Cycloaddition of azido group containing polyoxyalkylenes by reaction withdiethenyl benzene to form triazole crosslinked, energetic plasticizers for compositespropellants has been reported recently. Thus, repeating units of GAP, (AMMO),(BAMO-NMMO), and (BAMO-AMMO) are effective as energetic plasiticizerswhen cross-linked with 1,4-bis(ethynyl)benzene, 1,4-bis(ethynylcarbonyl)benzene,or 1,3-bis(cyanoethynyl) benzene.[82]
7.2. Oxidizers
The primary function of an oxidizer in a propellant is to provide oxygen forthe combustion of hydrocarbon fuel species. Higher loading of oxidizer increases thespecific impulse (Isp) of a propellant but simultaneously it adversely affects themechanical properties.[1] Positive oxygen balance, high heat formation, high density,and high thermal stability are the other important criteria for an energeticoxidizer. Most commonly used oxidizers are crystalline fine particles of ammoniumperchlorate (AP), ammonium dinitramide (ADN), glycerol trinitrate (NG), cyclote-tramethylene tetranitramine (HMX), cyclotrimethylene trinitramine (RDX),(TAGN), hydrazinium nitroformate (HNF), hexanitro hexaaza iso-wurzitane, orHNIW (CL-20). Sorguyl, bicyclo HMX, and tetranitro-bicyclononanone (K56) aresome examples of nitraza polycycles, which are gaining importance. Sorguyl andK56 are hydrolytically unstable whereas bicyclo HMX is extremely difficult tosynthesize.
AP and HMX are two solid ingredients often used in modern solid propellants,either composite propellent or composite modified double base propellants. Duringthe past decade, the search for more energetic propellants has led to the development
Azido Polymers 537
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
of CL-20, ADN, HNF, etc. Formulation of BAMO/AMMO with CL20, RDX andthe combination of the two leads to potentially attractive high energy TPE gunpropellants.
Structure and characteristics of some of these oxidizers are given in Sch. 20 andTable 7, respectively.
ONO2
ONO2
ONO2
NG
N N
N
NO2
NO2O2N
RDX
N
NN
N
NO2
NO2O2N
O2N
HMX
N
N N
N
N
N
O2N NO2
NO2O2N
CL-20
NO2
NO2
ADN
NH4+ N-
NH2NH3+ C
NO2
NO2
NO2
HNF
N
N N
N
NO2NO2
NO2NO2
Bicyclo HMX
N
N N
NO O
NO2NO2
NO2NO2
Sorguyl
N
N N
NO
NO2NO2
NO2NO2
K56
Scheme 20. Representative structure of oxidizers.[83,84]
Table 7. Characteristics of oxidizers (data adapted from Refs.[83,85])
Oxidizer Formula
Mol. wt.
(g/mol)
Density
(g/cm3)
�Hf
(kJ/mol)
AP ClH4NO4 117.5 1.95 �296
AN H4N2O3 80 1.72 �365
ADN H4N4O4 124 1.81 �150
HNF CH5N7O6 183 1.86–1.93 �71 to �72
RDX C3H6N6O6 222 1.805 þ70
HMX C4H8N8O8 296 1.91 þ84
Bicyclo HMX C4H6N8O8 294 1.87 þ125
Sorguyl C4H2N8O8 322 2.035 �292
K56 C5H6N8O9 322 1.975 �144
CL-20 C6H6N12O12 438 1.92–2.04 þ339 (g,a,b)þ372 (")
538 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
Ammonium perchlorate is the most widely used oxidizer in composite and com-posite modified double base (CMDB) rocket propellants. AP has two inherent dis-advantages, (1) it produces chlorine rich combustion products (30%), posingenvironmental hazards such as ozone depletion and acid rain and (2) it produceswhite smoke trail during inclemental weather, which is detrimental for specific appli-cations demanding smokeless plume. Among other oxidizers, AN and ADN offerschlorine free formulations. However, AN has drawbacks of hygroscopic nature andmultiple transition phases in the temperature range of practical importance andcomparatively low energy. ADN and HNF have attracted global attention. CL-20is also being evaluated as a rocket propellant component.[41,83] HNF has certainadvantages over ADN such as its simple method of synthesis, nonhygroscopicnature, higher density, and melting point. The addition of crystalline particlessuch as AP and nitramine particles decreases the burning rate of GAP significantlyeven though the combustion temperature increases.[86] The particle size of the oxidi-zer also plays an important factor in determining the performance of the propel-lants.[83,85,87] The energetic composites of GAP/nitroplasticizer (BDNA/F) andpolyethylene glycol with HMX in various particle sizes have been studied andcompared with HTPB composites.[79]
The flame structure of GAP propellants is altered by the nature of the oxidizer.The flame is highly heterogeneous in GAP/AP propellants but homogeneous inGAP/HMX and GAP/TAGN propellants.[85]
7.3. Pyrolants
The addition of metal particles i.e., Al, Mg, Ti, B, Zr, etc., to GAPforms energetic materials (GAP/metal pyrolants). These are also called fuel materialparticulates. The metal particles mixed within GAP react with the nitrogen, C, COproduced by the thermal decomposition of GAP. The most probable chemical reac-tions, which are likely to take place are formation of aluminium or magnesiumnitride (AlN, Mg3N2) and titanium carbide (TiC). GAP-coated amorphous-boron-based fuel-rich propellants exhibit more vigorous phenomena, high burning rates,and a lesser extent of residue agglomeration than the uncoated baseline propellant.Moreover, reaction mechanisms were proposed to elucidate the combustion pro-ducts.[33] However, the reaction process is highly dependent on the type of metalparticles mixed. Thus the burning rates and thermal decomposition vary with thetype of metals mixed. Though GAP contains no oxidizer fragments in its products,addition of metal particles increases the energy of GAP.[89]
The flame temperatures of GAP/metal pyrolants increases with increase in con-centration of pyrolants. The addition of B or Al generates higher flame temperaturecompared with Mg, Ti, or Zr.
7.4. Ballistic Modifiers
In general, ballistic modifiers bring down the maximum decomposition tempera-ture. They are also known as burning rate modifiers. Their main function is to
Azido Polymers 539
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
catalyze the burn rates. Commonly exploited ballistic modifiers are copper chromite(Cu(CrO2)2), ferric oxide, butyl ferrocene, catocene (C27H32Fe2). The mixture ofcatocene and copper chromite in BAMO/NMMO copolymers augmented the burn-ing rate 1.6 times with decreasing temperature sensitivity.[33] The combustion rate ofGAP-HMX propellant was increased by addition of 2% lead citrate and 0.6% C.New molybdenum oxide/vanadium oxide burning modifiers, with pure AN(atomized from melt) has resulted in improved combustion behavior and stabilityof GAP/AN-propellants.[84]
8. CONCLUSION AND FUTURE OUTLOOK
Energetic fuel binders based on cyclic ethers (oxirane, oxetane derivatives) andhaving pendant azido group have been investigated in the past two decades for gunand rocket propellants. These polymers (e.g., GAP, BAMO, AMMO) are promisingcandidates for energetic binders in future composite propellants having minimumsmoke, reduced pollution, and sensitivity. Such propellants in addition to being fuelrich, liberate large amounts of H2, N2, CO, and gaseous hydrocarbons on burning inthe primary chamber, are insensitive to impact and provide high burn rates.
Inspite of all the advantages mentioned above, there are certain limitations ofthese azide binders. The decomposition products of cured GAP contain HCN, whichis highly toxic. Attempts have to be made to suppress the HCN evolution. The Tg ofGAP increases on curing and by presence of oxidizers, pyrolants, etc. There is a needto develop energetic binders with lower Tg.
Energetic binders based on hydrocarbon polymers containing pendant azidegroup may provide an alternative approach for achieving this goal.
This review focused on the synthesis, characterization, physical properties, andthermal behavior of these energetic azido binders. Preliminary studies on poly(allylazide) have also been included. It is obvious that one can tailor the properties ofthese polymers by changing their backbone structure and by copolymerization.More studies are needed for developing energetic binders using structuralmodifications.
ACKNOWLEDGMENT
The Armament Research Board, Government of India, Ministry of Defenceis gratefully acknowledged for providing the financial assistance for carrying outthis work.
REFERENCES
1. Aggarwal, J.P.; Walley, S.M.; Field, J.E. High speed photographic study ofthe impact response of ammonium dinitramide and glycidyl azido polymer.J. Propulsion and Power 1997, 13, 463.
2. Borman, S. Advanced energetic binders for military and space applications.Military Technology 1994, 72, 18.
540 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
3. Frankel, M.B.; Grant, L.R.; Flanagan, J.E. Historical development ofGAP. J. Propulsion and Power 1992, 8, 560.
4. Kubota, N.J. Combustion of energetic azide polymers, Propulsion and Power1995, 11, 677.
5. Madhavan, A.; Ramasubramanian, T.S.; Hariharasubramanian, A.; Kannan,K.G.; Ninan, K.G. A Redox Method for the Estimation of Nitrogen inGlycidyl Azide Polymer. In Second International High Energy MaterialsConference and Exhibit IIT-Chennai, India, Dec. 8–10, 1998.
6. Sahu, S.K.; Chavan, M.K.; Thakur, J.V.; Kulkarni, S.G.; Panda, S.P.Performance Evaluation of Glycidyl Azide Polymers as Integrated RamRocket Propellants. In Second International High Energy MaterialsConference and Exhibit IIT-Chennai, India, Dec. 8–10, 1998.
7. Eroglu, M.S.; Guven, O. Characterisation of network structure of poly(glycidylaxide) elastomers by swelling, solubility and mechanical measurements.Polymer 1998, 39, 1173.
8. Braithwaite, P.C.; Sanderson, A.C.; Wardle, R.B.; Peters, S. Optimization ofBAMO-AMMO for gun propellants, (JANNAF 29th Propellant Development& Characterization Subcommittee Meeting, 2000), CPIA Publication 2000, cfCA 2000, 135, 109317.
9. Braithwaite, P.C.; Haaland, A.C.; Wardle, R.B.; Harris, L.E.; Manning, T.;Prickett, S. Development of improved artillery gun propellants. InternationalAnnual Conference of ICT, cf CA 1999, 131, 118115.
10. Wardle, R.B.; Braithwaite, P.C.; Haaland, A.C.; Hartwell, J.A.; Hendrickson,R.R.; Lott, V.; Wallace, I.A.; Zisette, C.B. High energy oxetane/Hnw propel-lants, International Annual Conference of ICT 1996, 27th(Energetic Materials),52.1–52.7.
11. Ito, K.; Usami, N.; Yamashita, Y. Cationic oligomerisation of epichlorohydrin.Polym. J. 1979, 11, 171.
12. Panda, S.P.; Sahu, S.K.; Thakur, J.V.; Kulkarni, S.G.; Kumbhar, C.G.;Sadafule, D.S. Synthesis and characterisation of hydroxyl terminated poly-epichlorohydrin and polyglycidyl azide. Def. Sci. J. 1996, 46, 399.
13. Biedron, T.; Kubisa, P.; Penczek, S. Polyepichlorohydrin diols free of cyclics:synthesis and characterization. J. Polym. Sci.: Part A: Polym. Chem. 1991, 29,619.
14. Frankel, M.B. Aqueous process for the quantitative conversion of polyepi-chlorohydrin to glycidyl azide polymer US Patent 4,379,894, 1990.
15. Panda, S.P.; Kulkarni, S.G.; Sahu, S.K.; Bhoraskar, V.N.; Dokhale, P.A.J. Fastneturon activation analysis of high energy materials and polymers. EnergeticMaterials 1998, 16, 309.
16. Wagner, R.I. Glycidyl azide polymer synthesis by molten salt method. USPatent 5,055,600, 1991.
17. Ampleman, G. Synthesis of a diazido terminated energetic plasticizer. USPatent 5,124,463, 1992.
18. Wilson, E.R.; Frankel, M.B. Azide-terminated azido compound as energeticplasticizer for propellants. US Patent 4,781,861, 1988.
19. Ahad, E. Process for the preparation of allyl azide substituted hydroxyl termi-nated polyethers. US Patent 4, 891, 438, 1990.
Azido Polymers 541
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
20. Bui, V.T.; Ahad, E.; Rheaume, D.; Raymond, M.P. Energetic polyurethanesfrom branched glycidyl azide polymer. J. Appl. Polym. Sci. 1996, 62, 27.
21. Bui, V.T.; Ahad, E.; Rheaume, D.; Whitehead, R. Evaluation of banchedglycidyl azide polymer purified by solvent extraction. Ind. Eng. Chem. Res.1997, 36, 2219.
22. Ahad, E. Branched azido copolymers. US Patent 5, 130, 381, 1992.23. Ahad, E. Branched energetic polyether elastomers. US Patent 5, 214,110, 1993.24. Nazare, A.N.; Asthana, S.N.; Patwardhan, S.S.; Singh, H. Comparative studies
on GAP formed from degradation of high molecular weight PECH and onestage conversion from ECH. In 4th International Symposium on ExplosiveTechnology and Ballistics (NIXT-92), South Africa, 1992.
25. Vasudevan, V.; Sundarajan, G. Synthesis of GAP-PB-GAP triblock copolymerand application as modifier in AP/HTPB composite propellant. Propellants,Explosives and Pyrotechnics 1999, 24, 295.
26. Ou, Y.; Chen, B.; Yan, H.; Jia, H.; Li, J.; Dong, S. Development of energeticadditives for propellants. J. Propulsion and Power 1995, 11, 838.
27. Nair, J.K.; Satpute, R.S.; Polke, B.G.; Mukundan, T.; Asthana, S.N.; Singh, H.Synthesis and characterisation of bis-azido methyl oxetane and its polymer andcopolymer with tetrahydrofuran. Def. Sci. J. 2002, 52, 147.
28. Talukder, M.A.H. Ring-opening polymerization of oxetanes by cationicinitiators: polymerization of 3-azidomethyl-3-methyloxetane with bis(chlorodi-methylsilyl)benzene/silver hexafluoroantimonate initiating system. Makromol.Chem., Macromolecular Symposia 1991, 501.
29. Talukder, M.A.H. Ring-opening polymerization of oxetanes by cationicinitiators: polymerization of azidomethyloxetane with bis(chlorodimethylsilyl)-benzene/silver hexafluoroantimonate initiating system. ACS Polymer Preprints(Division of Polymer Chemistry), 1990, 31, 93.
30. Hsiue, G.; Liu Y.-L.; Chiu, Y.-S. Triblock copolymer based on cyclic ethers:preparation and properties of tetrahydrofuran and 3,3-bis(azidomethy) oxetanetriblock copolymers. J. Polym. Sci.: Part A: Polym. Chem. 1994, 32, 2155.
31. Liu, Ying-Ling; Hsiue, Ging-Ho; Chiu, Yie-Shun. Studies on the polymeriza-tion mechanism of 3-nitratomethyl-3-methyloxetane and 3-azidomethyl-3-methyloxetane and the synthesis of their respective triblock copolymers withtetrahydrofuran. J. Polym. Sci.: Part A: Polym. Chem. 1995, 33, 1607.
32. Murphy, E.A.; Ntozakhe, T.; Murphy, C.J.; Fay, J.J.; Sperling, L.H.Characterization of poly[3,3-bis(ethoxymethyl)oxetane] and poly[3,3-bis(azido-methyl)oxetane] and their block copolymers. J. Appl. Polym. Sci. 1989, 37, 267.
33. Kimura, E.; Oyumi, Y. Effects of copolymerization ratio of BAMO/NMMOand catalyst on sensitivity and burning rate of HMX propellant. Propellants,Explosives and Pyrotechnics 1995, 20, 215.
34. Talukder, M.A.H.; Lindsay, G.A. Synthesis and the preliminary analysis ofblock copolymers of 3,30-bis(azidomethyl)oxetane and 3-nitratomethyl-30-methyloxetane. J. Polym. Sci.: Part A: Polym. Chem. 1990, 28, 2393.
35. Cheradame, H.; Gojon, E. Synthesis of polymers containing pseudohalidegroups by cationic polymerization. 2. Copolymerization of 3,3-bis(azidomethyl)-oxetane with substituted oxetanes containing azide groups. Makromol. Chem.1991, 192, 919.
542 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
36. Xu, B.; Lin, Y.G.; Chien, J.C.W. Energetic ABA and (AB)n thermoplasticelastomers. J. Appl. Polym. Sci. 1992, 46, 1603.
37. Xu, B.; Lillya, C.P.; Chien, J.C.W. Spiro(benzoxasilole)catalyzed polymerizationof oxetane derivatives. J. Polym. Sci.: Part A: Polym. Chem. 1992, 30, 1899.
38. Athar, J.; Borah, P.; Mukundan, T.; Sarwade, D.B.; Asthana, S.N.Spiro(benzoxasilole) catalyzed polymerization of oxetane derivatives. J.Polym. Mater. 2002, 19, 183.
39. Braithwaite, P.; Edwards, W.; Sanderson, A.J.; Wardle, R.B. The synthesis andcombustion of high energy thermoplastic elastomer binders. InternationalAnnual Conference of ICT, 2001, cf CA 2001, 135:373693.
40. Gaur, B.; Lochab, B.; Choudhary, V.; Varma, I.K. Thermal behaviour ofpoly(allylazide). J. Therm. Anal. Cal. 2003, 71, 467.
41. Finck, B.; Graindorge, H. NewMolecules for High Energetic Materials. In 27thInt. Annu. ICT Conf. (Energetic Materials) 1996, 23.
42. Nazare, A.N.; Asthana, S.N.; Singh, H.J. Glycidyl azide polymers (GAP)—Anenergetic component of advanced solid rocket propellants—A review. EnergeticMaterials 1992, 10, 43.
43. Selim, K.; Ozkar, S.; Yilmaz, L. Thermal characterization of glycidyl azidepolymer (GAP) and GAP-based binders for composite propellants. J. Appl.Polym. Sci. 2000, 77, 538.
44. Mitarai, Y.; Anan, T.; Irie, A.; Ohuchi, S.; Tamura, N. Physicochemical prop-erties of AMMO polymer, Kogyo Kayaka 1990, 51, 240; cf CA 1991, 114,188532.
45. Keskin, S.; Ozkar, S. Kinetics of polyurethane formation between glycidyl azidepolymer and a triisocyanate. J. Appl. Polym. Sci. 2001, 81, 918.
46. Panda, S.P.; Sahu, S.K.; Thakur, J.V.; Kulkarni, S.G.; Kumbhar, C.G.;Sadafule, D.S. On decomposition mechanism and explosive properties ofGAP based RAM rocket propellants. American Institute of Aeronautics andAstronautics 1996, IS-267, 1,
47. Wang, P.; Xia, Z.; Zhou, Y.; Li, C. Investigation of High Molecular WeightGAP. In 27th Int. Annu. ICT Conf. (Energetic Materials) 1996, 25.
48. Kasicki, H.; Pekel, F.; Ozkar, S. Curing characteristics of glycidyl azide poly-mer-based binders. J. Appl. Polym. Sci. 2001, 80, 65.
49. Russel, R., Jr.; Chan, L.M. Propellant binders cure catalyst. US Patent4,379,903, 1983.
50. Russel, R., Jr.; Chan, L.M. Glycidyl azide propellant with antigassing addi-tives. US Patent 5,092,945, 1992.
51. Frankel, M.B.; Wilson, E.R.; Woolery, D.O. Energetic azido curing agents. USPatent 4,96,2213, 1990.
52. Katritzky, A.R.; Singh, S.K. Synthesis of C-carbamoyl-1,2,3-triazoles bymicrowave-induced 1,3-dipolar cycloaddition of organic azides to acetylenicamides. J. Org. Chem. 2002, 67, 9077.
53. Al-Sader, B.H.; Kadri, M. Kinetics and mechanism of the 1,3-dipolar cyclo-addition of phenyl azides to methyl 3-pyrrolidinoacrylate. Tetrahedron Letter1985, 26, 466.
54. Huisgen, R. Mechanism of 1,3-dipolar cycloadditions. Reply, J. Org. Chem.1968, 33, 2261.
Azido Polymers 543
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
55. Scheiner, P.; Schomaker, J.H.; Deming, S.; Libbey, W.I.; Nowack, G.P. Theaddition of aryl azides to norbornene. A kinetic investigation. J. Am. Chem.Soc. 1965, 87, 306.
56. Manzara, A.P. Azido polymers having improved burn rates. US Patent5,681,904, 1997.
57. Varma, I.K.; Choudhary, V.; Gaur, B.; Lochab, B.; Oberoi, S. A process for theproduction of energetic binder (azide polymer), Indian Patent Appl. 1316/Del/2002, December 31, 2002.
58. Eroglu, M.S.; Guven, O. Thermal decomposition of poly(glycidyl azide) asstudied by high-temperature FTIR and thermogravimetry. J. Appl. Polym.Sci. 1996, 61 (5), 201.
59. Sahu, S.K.; Panda, S.P.; Sadafule, D.S.; Kumbhar, C.G.; Kulkarni, S.G.;Thakur, J.V. Thermal and photodegradation of glycidyl azide polymers.Polym. Degrad. and Stab. 1998, 62, 495.
60. Oyumi, Y. Thermal decomposition of azide polymers. Propellents, Explosives,Pyrotechnics 1992, 17, 226.
61. Arisawa, H.; Brill, T.B. Thermal decomposition of energetic materials 71:Structure-decomposition kinetics relationships in flash pyrolysis of glycidylazide polymer (GAP). Combustion and Flame 1998, 112, 533.
62. Korobeinichev, O.P.; Kuibida, L.V.; Volkov, E.N.; Shmakov, A.G. Mass spec-trometric study of combustion and thermal decomposition of GAP.Combustion and Flame 2002, 129, 136.
63. Faber, M.; Srivastva, R.D. Mass spectrometric kinetic studies on several azidopolymers. Combustion and Flame 1984, 55, 203.
64. Oyumi, Y.; Brill, T.B. Thermal decomposition of energetic materials 12;Infrared spectral and rapid thermolysis studies of azide containing monomersand polymers. Combustion and Flame 1986, 65, 533.
65. Chen, J.K.; Brill, T.B. Thermal decomposition of energetic materials. 54.Kinetics and near-surface products of azide polymers AMMO, BAMO, andGAP in simulated combusion. Combustion and Flame 1991, 87, 157.
66. Miyazaki, T.; Kubota, N. Energetics of BAMO polymer. Propellants,Explosives and Pyrotechnics 1992, 17, 5.
67. Chang, T.C.; Wu, K.H.; Chen, H.B.; Ho, S.Y.; Chiu, Y.S. Thermal degradationof aged polytetrahydrofuran and its copolymers with 3-azidomethyl-30-methyl-oxetane and 3-nitratomethyl-30-methyloxetane by thermogravimetry. J. Polym.Sci.: Part A: Polym. Chem. 1996, 34, 3337.
68. Liu, Ying-Ling; Hsiue, Ging-Ho; Chiu, Yie-Shun. Thermal characteristics ofenergetic polymers based on tetrahydrofuran and oxetane derivatives. J. Appl.Polym. Sci. 1995, 58, 579.
69. Kubota, N.; Sonobe, T. Combustion mechanism of azide polymers.Propellants, Explosives, Pyrotechnics, 1988, 13, 172.
70. Goshgarian, J.E.; Beckstead, M.W. Air Force Rocket Propulsion Laboratory:Edwards AFB, CA, June, 1982.
71. Lengelle, G.; Fourest, B.; Godon, J.C.; Gain, C. In 29th Propulsion ConferenceMonterey, CA, 1993, AIAA-92-2413.
72. Mishra, I.B.; Juncau, G.P.; Groom, T. 11th JANNAF Propulsion Meeting,1984, 7.
544 Gaur et al.
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
73. Dhar, S.S.; Asthana, S.N.; Singh, H.; Natu, G.N. Fizika Goreniya iVzryva,Thermal decomposition of GAP and GAP based double base propellents.Combustion, Explosives and Shock waves, 29 (3), 276, 1993, CfCA 119:229468.
74. Feng, H.T.; Mintz, K.J.; Augsten, R.A.; Jones, D.E.G. Thermal analysis ofbranched GAP. Thermochim. Acta 1998, 311, 105.
75. Bazaki, H. Combusion mechanism of 3-azidomethyl-3-methyloxetane(AMMO) composite propellants. Progress in Astronautics and Aeronautics2000, 185, 439.
76. Lee, Y.; Tang, Ching-Jen; Litzinger, T.A. Thermal decomposition of RDX/BAMO pseudo-propellants. Combustion and Flame 1999, 117, 795.
77. Oyumi, Y.; Inokami, K.; Kiyotaka, Y.; Kazuhiro, M.K. Thermal decomposi-tion of BAMO/HMX propellants. Propellants, Explosives, Pyrotechnics 1993,18, 62.
78. Hatch, R.L.; Wardle, R.B.; Lund, G.K.; Hamilton, S.R. Solid rocketpropellant formulations contg. isocyanate-cured (azidomethyl)methyloxetane-bis(azidomethyl)oxetane [BAMO/AMMO] copolymer energetic binders. PCTInt. Appl. 1995, 21. WO 94-US 7945 19940714; cf CA 1995, 123, 87608.
79. Shen, S.M.; Leu, A.L.; Chen, S.I.; Yeh, H.C. Thermal characteristics of GAP,GAP/BDNPA/BDNPF and PEG/BDNPA/BDNPF and the energetic compo-sites. Thermochim. Acta 1991, 180, 251.
80. Oyumi, Y.; Kimura, E. IM characteristics of GAP/AN composite propellants.Kayaku Gakkaishi 1996, 57.
81. Sayles, D.C. Non nitroglycerin-containing composite-modified double-basepropellant. US Patent 4,707,199, 1987.
82. Reed, R., Jr. Cycloaddition of azide group-containing polyoxyalkylenes byreaction with aryl diacetylenes to form triazole-crosslinked energetic plasticizersfor composite propellants. US 97-880 710; cf CA 2000, 133, 152741.
83. Golifier, M.; Graindorge, H.; Longevialle, Y.; Mace, H. New EnergeticMolecules and their Applications in Energetic Materials. In 29th Int. Annu.ICT Conf. (Energetic Materials), 1998, 3.
84. Nielsen, A.T. In ‘‘Chemistry of Energetic Materials’’, ‘‘Polycyclic AmineChemistry’’; ed. G. Olah and D. R. Squire, Academic Press Inc. 1991,Chap. 5, pp 95.
85. Dendage, P.S.; Sarwade, D.B.; Asthana, S.N.; Singh, H. Hydrazinium nitro-formate (HNF) and HNF based propellants: A review. J. Energetic Materials2001, 9, 41.
86. Kuwahara, T.; Takizuka, M.; Onda, T.; Kubota, N. Combustion of GAP basedenergetic pyrolents. Propellants, Explosives, Pyrotechnics 2000, 25, 112.
87. Martin, W.B.; William, D.V. Azide polymers derivatized with dipolarophilesfor aqueous coatings producing transparent films. Eur. Patent 6,33,280, 2000.
88. Kubota, N.; Sonobe, T.; Yamamoto, A.; Shimizu, J. Burning rate character-istics of GAP propellants. Propulsion and Power 1990, 6, 686.
89. Klaus, M.; Jutta, B.-M.; Hiltmar, S. Propellants, Explosives, Pyrotechnics1996, 21, 139.
Azido Polymers 545
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3

©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
Dow
nloa
ded
by [
Indi
an I
nstit
ute
of T
echn
olog
y R
oork
ee]
at 0
6:33
10
July
201
3