arachidonic acid induces brain endothelial cell apoptosis via p38-mapk and intracellular calcium...
TRANSCRIPT

1
2
3Q1
4
5678
9
101112
131415
33
34
35
36
37
3839
40
41
42
43
44
45
46
47
48
49
50
51
52
53
Microvascular Research xxx (2014) xxx–xxx
Q2
YMVRE-03424; No. of pages: 15; 4C: 1, 4, 12
Contents lists available at ScienceDirect
Microvascular Research
j ourna l homepage: www.e lsev ie r .com/ locate /ymvre
Arachidonic acid induces brain endothelial cell apoptosis via p38-MAPKand intracellular calcium signaling
OO
FJustin Evans a, YooSeung Ko a, Wilmer Mata a, Muhammad Saquib a, Joel Eldridge a, Aaron Cohen-Gadol c,H. Anne Leaver d, Shukun Wang a, Maria Teresa Rizzo a,b,⁎a Signal Transduction Laboratory, Methodist Research Institute, Indiana University Health, Indiana University School of Medicine, Indianapolis, IN, USAb Department of Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USAc Department of Neurological Surgery, Indiana University School of Medicine, Indianapolis, IN, USAd Division of Clinical Neuroscience, Edinburgh University, Edinburgh, UK
⁎ Corresponding author at: Signal Transduction LaInstitute, Indiana University Health, 1800 North CapitE504E, Indianapolis, IN 46202, USA. Fax: +1 317 962 936
E-mail address: [email protected] (M.T. Rizzo).
http://dx.doi.org/10.1016/j.mvr.2014.04.0110026-2862/© 2014 Published by Elsevier Inc.
Please cite this article as: Evans, J., et al., Arsignaling, Microvasc. Res. (2014), http://dx.d
Ra b s t r a c t
a r t i c l e i n f o161718
19
20
21
22
23
24
25
26
27
Article history:Accepted 27 April 2014Available online xxxx
Keywords:Arachidonic Acidp38-MAPKCalciumBrain endothelial cellsApoptosis
28
29
30
31
32
RECTED PArachidonic acid (AA), a bioactive fatty acid whose levels increase during neuroinflammation, contributes to
cerebral vascular damage and dysfunction. However, the mode of injury and underlying signaling mechanismsremain unknown. Challenge of primary human brain endothelial cells (HBECs) with AA activated a stressresponse resulting in caspase-3 activation, poly(ADP-ribose) polymerase cleavage, and disruption of monolayerintegrity. AA also induced loss of mitochondrial membrane potential and cytochrome c release consistent withactivation of intrinsic apoptosis. HBEC stimulation with AA resulted in sustained p38-MAPK activation andsubsequent phosphorylation of mitogen-activated protein kinase activated protein-2 (MAPKAP-2) kinase andheat shock protein-27 (Hsp27). Conversely, other unsaturated and saturated fatty acids had no effect. Pharmaco-logical and RNA interference-mediated p38α or p38β suppression abrogated AA signaling to caspase-3 andHsp27, suggesting involvement of both p38 isoforms in AA-induced HBEC apoptosis. Hsp27 silencing alsoblocked caspase-3 activation. AA stimulated intracellular calcium release, which was attenuated by inositol1,4,5-trisphosphate (IP3) receptor antagonists. Blockade of intracellular calcium release decreased caspase-3activation, but had no effect on AA-induced p38-MAPK activation. However, inhibition of p38-MAPK or blockadeof intracellular calcium mobilization abrogated AA-induced cytochrome c release. AA-induced caspase-3 activa-tion was abrogated by pharmacological inhibition of lipooxygenases. These findings support a previously unrec-ognized signaling cooperation between p38-MAPK/MAPKAP-2/Hsp27 and intracellular calcium release inAA-induced HBEC apoptosis and suggest its relevance to neurological disorders associated with vascularinflammation.
© 2014 Published by Elsevier Inc.
R
54
55
56
57
58
59
60
61
62
63
64
65
66
UNCOIntroduction
The ability of brain endothelial cells to survive environmentalstressors is a prerequisite for brain development and homeostasis. Afunctional endothelium is essential to ensure the proper metabolic ex-change between blood and the brain parenchyma, preserve the integri-ty of the blood brain barrier (BBB), and support neuroaxonal growth (Jinet al., 2002; Guo et al., 2008; Weiss et al., 2009). Apoptosis constitutes amode of death in the cerebral endothelium and contributes to disrup-tion of endothelial monolayer integrity and subsequent breakdown ofthe BBB, vasogenic edema, and neuronal damage (Rizzo and Leaver,2010).Moreover, apoptotic endothelial cells are impaired in their abilityto sustain neurogenesis or initiate neovascularization, thus delaying
67
68
69
70
71
boratory, Methodist Researchol Avenue, Noyes Bldg, Room9.
achidonic acid induces brainoi.org/10.1016/j.mvr.2014.04
anatomical and functional recovery following insults to the brain. Evi-dence from experimental and clinical studies indicates that injuries tothe cerebral endothelium contribute to the initiation, maintenance,and/or exacerbation of several neurological disorders (Park et al.,2004; Zhou et al., 2004; Rite et al., 2007; Farrall and Wardlaw, 2009;Miyazaki et al., 2011). Thus, targeting the cerebral endothelium mayoffer novel therapeutic strategies to prevent loss of BBB integrity, limitneuronal damage, and enhance brain recovery. Development of suchtherapies, however, awaits a better understanding of the signalingevents that contribute to the destabilization of brain endothelial cellmonolayer integrity and death.
Dysregulated neuroinflammation constitutes a common feature ofseveral neurological disorders (Glass et al., 2010). Arachidonic acid(AA), a biologically active polyunsaturated fatty acid, plays an importantrole in neuroinflammation and vascular dysfunction (Artwohl et al.,2003; Farooqui et al., 2007; Rao et al., 2011). Under resting conditions,AA is esterified at the sn-2 position of membrane phospholipids andexerts structural, signaling, and homeostatic functions (Brash, 2001;
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
TED P
RO
OF
72
73
74Q3
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
A
B
C
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0
Control
AA
A62
0 n
m30 60 90 120 180
Time (min)
Control AA
Control AA
∗∗∗
∗∗∗ ∗∗∗
∗∗
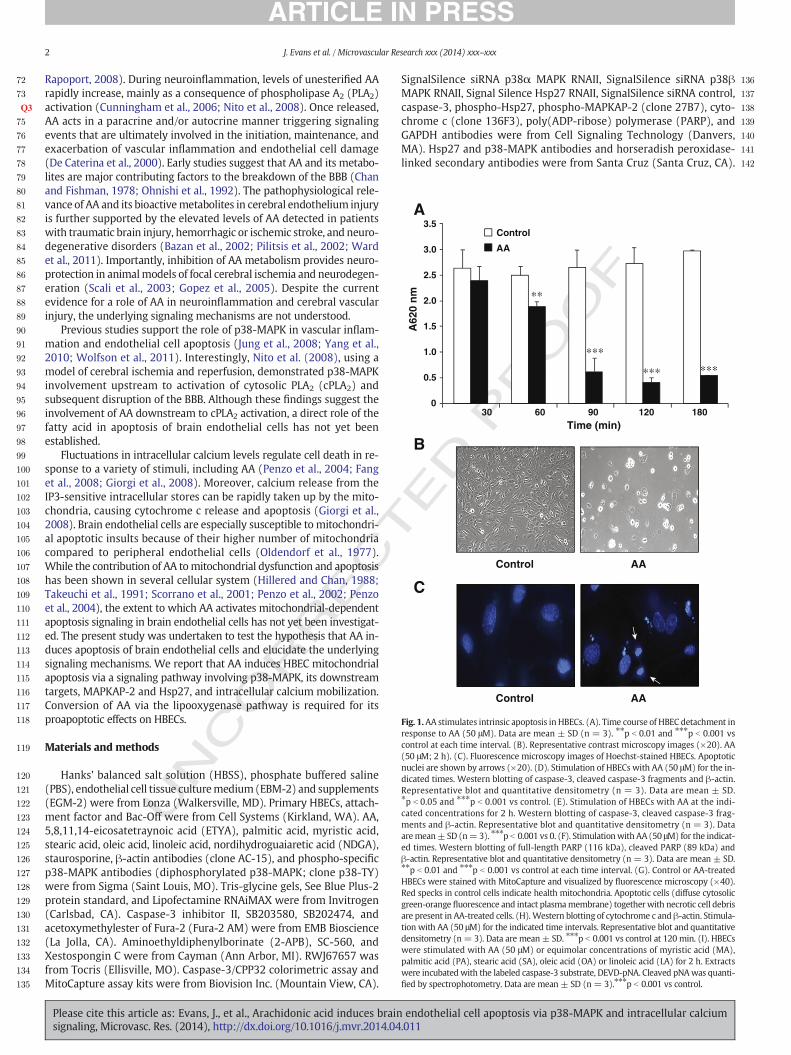
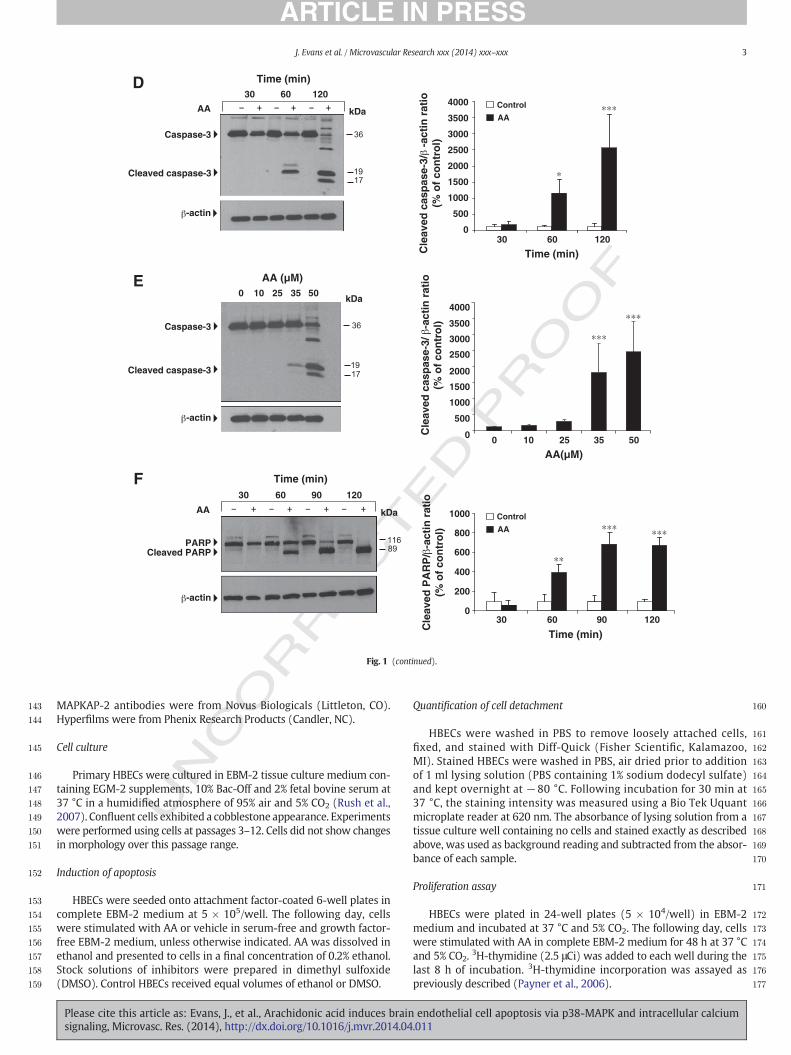
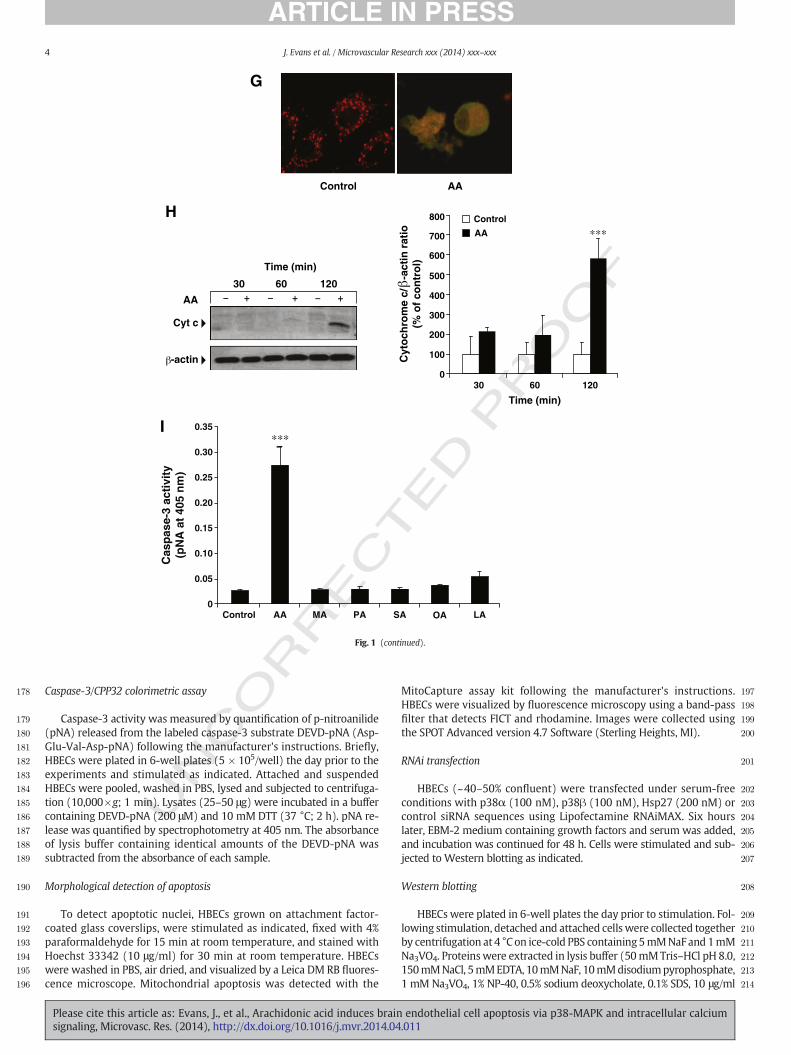
Fig. 1.AA stimulates intrinsic apoptosis in HBECs. (A). Time course of HBEC detachment inresponse to AA (50 μM). Data are mean ± SD (n = 3). ⁎⁎p b 0.01 and ⁎⁎⁎p b 0.001 vscontrol at each time interval. (B). Representative contrast microscopy images (×20). AA(50 μM; 2 h). (C). Fluorescence microscopy images of Hoechst-stained HBECs. Apoptoticnuclei are shown by arrows (×20). (D). Stimulation of HBECs with AA (50 μM) for the in-dicated times. Western blotting of caspase-3, cleaved caspase-3 fragments and β-actin.Representative blot and quantitative densitometry (n = 3). Data are mean ± SD.⁎p b 0.05 and ⁎⁎⁎p b 0.001 vs control. (E). Stimulation of HBECs with AA at the indi-cated concentrations for 2 h. Western blotting of caspase-3, cleaved caspase-3 frag-ments and β-actin. Representative blot and quantitative densitometry (n = 3). Dataaremean± SD (n=3). ⁎⁎⁎p b 0.001 vs 0. (F). Stimulationwith AA (50 μM) for the indicat-ed times. Western blotting of full-length PARP (116 kDa), cleaved PARP (89 kDa) andβ-actin. Representative blot and quantitative densitometry (n = 3). Data are mean ± SD.⁎⁎p b 0.01 and ⁎⁎⁎p b 0.001 vs control at each time interval. (G). Control or AA-treatedHBECs were stained with MitoCapture and visualized by fluorescence microscopy (×40).Red specks in control cells indicate health mitochondria. Apoptotic cells (diffuse cytosolicgreen-orange fluorescence and intact plasmamembrane) togetherwith necrotic cell debrisare present in AA-treated cells. (H).Western blotting of cytochrome c and β-actin. Stimula-tion with AA (50 μM) for the indicated time intervals. Representative blot and quantitativedensitometry (n = 3). Data are mean ± SD. ⁎⁎⁎p b 0.001 vs control at 120 min. (I). HBECswere stimulated with AA (50 μM) or equimolar concentrations of myristic acid (MA),palmitic acid (PA), stearic acid (SA), oleic acid (OA) or linoleic acid (LA) for 2 h. Extractswere incubatedwith the labeled caspase-3 substrate, DEVD-pNA. Cleaved pNAwas quanti-fied by spectrophotometry. Data are mean ± SD (n = 3).⁎⁎⁎p b 0.001 vs control.
2 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
UNCO
RREC
Rapoport, 2008). During neuroinflammation, levels of unesterified AArapidly increase, mainly as a consequence of phospholipase A2 (PLA2)activation (Cunningham et al., 2006; Nito et al., 2008). Once released,AA acts in a paracrine and/or autocrine manner triggering signalingevents that are ultimately involved in the initiation, maintenance, andexacerbation of vascular inflammation and endothelial cell damage(De Caterina et al., 2000). Early studies suggest that AA and its metabo-lites are major contributing factors to the breakdown of the BBB (Chanand Fishman, 1978; Ohnishi et al., 1992). The pathophysiological rele-vance of AA and its bioactivemetabolites in cerebral endothelium injuryis further supported by the elevated levels of AA detected in patientswith traumatic brain injury, hemorrhagic or ischemic stroke, and neuro-degenerative disorders (Bazan et al., 2002; Pilitsis et al., 2002; Wardet al., 2011). Importantly, inhibition of AA metabolism provides neuro-protection in animalmodels of focal cerebral ischemia and neurodegen-eration (Scali et al., 2003; Gopez et al., 2005). Despite the currentevidence for a role of AA in neuroinflammation and cerebral vascularinjury, the underlying signaling mechanisms are not understood.
Previous studies support the role of p38-MAPK in vascular inflam-mation and endothelial cell apoptosis (Jung et al., 2008; Yang et al.,2010; Wolfson et al., 2011). Interestingly, Nito et al. (2008), using amodel of cerebral ischemia and reperfusion, demonstrated p38-MAPKinvolvement upstream to activation of cytosolic PLA2 (cPLA2) andsubsequent disruption of the BBB. Although these findings suggest theinvolvement of AA downstream to cPLA2 activation, a direct role of thefatty acid in apoptosis of brain endothelial cells has not yet beenestablished.
Fluctuations in intracellular calcium levels regulate cell death in re-sponse to a variety of stimuli, including AA (Penzo et al., 2004; Fanget al., 2008; Giorgi et al., 2008). Moreover, calcium release from theIP3-sensitive intracellular stores can be rapidly taken up by the mito-chondria, causing cytochrome c release and apoptosis (Giorgi et al.,2008). Brain endothelial cells are especially susceptible to mitochondri-al apoptotic insults because of their higher number of mitochondriacompared to peripheral endothelial cells (Oldendorf et al., 1977).While the contribution of AA tomitochondrial dysfunction and apoptosishas been shown in several cellular system (Hillered and Chan, 1988;Takeuchi et al., 1991; Scorrano et al., 2001; Penzo et al., 2002; Penzoet al., 2004), the extent to which AA activates mitochondrial-dependentapoptosis signaling in brain endothelial cells has not yet been investigat-ed. The present study was undertaken to test the hypothesis that AA in-duces apoptosis of brain endothelial cells and elucidate the underlyingsignaling mechanisms. We report that AA induces HBEC mitochondrialapoptosis via a signaling pathway involving p38-MAPK, its downstreamtargets, MAPKAP-2 and Hsp27, and intracellular calcium mobilization.Conversion of AA via the lipooxygenase pathway is required for itsproapoptotic effects on HBECs.
Materials and methods
Hanks' balanced salt solution (HBSS), phosphate buffered saline(PBS), endothelial cell tissue culturemedium(EBM-2) and supplements(EGM-2) were from Lonza (Walkersville, MD). Primary HBECs, attach-ment factor and Bac-Off were from Cell Systems (Kirkland, WA). AA,5,8,11,14-eicosatetraynoic acid (ETYA), palmitic acid, myristic acid,stearic acid, oleic acid, linoleic acid, nordihydroguaiaretic acid (NDGA),staurosporine, β-actin antibodies (clone AC-15), and phospho-specificp38-MAPK antibodies (diphosphorylated p38-MAPK; clone p38-TY)were from Sigma (Saint Louis, MO). Tris-glycine gels, See Blue Plus-2protein standard, and Lipofectamine RNAiMAX were from Invitrogen(Carlsbad, CA). Caspase-3 inhibitor II, SB203580, SB202474, andacetoxymethylester of Fura-2 (Fura-2 AM) were from EMB Bioscience(La Jolla, CA). Aminoethyldiphenylborinate (2-APB), SC-560, andXestospongin C were from Cayman (Ann Arbor, MI). RWJ67657 wasfrom Tocris (Ellisville, MO). Caspase-3/CPP32 colorimetric assay andMitoCapture assay kits were from Biovision Inc. (Mountain View, CA).
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
SignalSilence siRNA p38α MAPK RNAII, SignalSilence siRNA p38βMAPK RNAII, Signal Silence Hsp27 RNAII, SignalSilence siRNA control,caspase-3, phospho-Hsp27, phospho-MAPKAP-2 (clone 27B7), cyto-chrome c (clone 136F3), poly(ADP-ribose) polymerase (PARP), andGAPDH antibodies were from Cell Signaling Technology (Danvers,MA). Hsp27 and p38-MAPK antibodies and horseradish peroxidase-linked secondary antibodies were from Santa Cruz (Santa Cruz, CA).
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
RRECTED P
RO
OF
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
∗∗∗
∗∗∗
∗∗∗
∗
AA
AA
AA
Control
AA
Control
Cle
aved
cas
pas
e-3/
-a
ctin
rat
io(%
of
con
tro
l)C
leav
ed c
asp
ase-
3/
-act
in r
atio
Cle
aved
PA
RP
/ -a
ctin
rat
io(%
of
con
tro
l) ∗∗∗ ∗∗∗
∗∗
906030 120
1000
800
600
400
200
0
(% o
f co
ntr
ol)
Time (min)
0 10 25 35 50
30 60 120
AA(µM)
Time (min)
Time (min)
Time (min)
AA (µM)
D
E
F
30 60 120
0 10 25 35 50
30 60 90 120
Caspase-3
Cleaved caspase-3
Caspase-3
Cleaved caspase-3
Cleaved PARPPARP
-actin
-actin
kDa
kDa
-actin
kDa
4000
3500
3000
2500
2000
1500
1000
500
0
4000
3500
3000
2500
2000
1500
1000
500
0
Fig. 1 (continued).
3J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
UNCOMAPKAP-2 antibodies were from Novus Biologicals (Littleton, CO).
Hyperfilms were from Phenix Research Products (Candler, NC).
Cell culture
Primary HBECs were cultured in EBM-2 tissue culture medium con-taining EGM-2 supplements, 10% Bac-Off and 2% fetal bovine serum at37 °C in a humidified atmosphere of 95% air and 5% CO2 (Rush et al.,2007). Confluent cells exhibited a cobblestone appearance. Experimentswere performed using cells at passages 3–12. Cells did not show changesin morphology over this passage range.
Induction of apoptosis
HBECs were seeded onto attachment factor-coated 6-well plates incomplete EBM-2 medium at 5 × 105/well. The following day, cellswere stimulated with AA or vehicle in serum-free and growth factor-free EBM-2 medium, unless otherwise indicated. AA was dissolved inethanol and presented to cells in a final concentration of 0.2% ethanol.Stock solutions of inhibitors were prepared in dimethyl sulfoxide(DMSO). Control HBECs received equal volumes of ethanol or DMSO.
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
Quantification of cell detachment
HBECs were washed in PBS to remove loosely attached cells,fixed, and stained with Diff-Quick (Fisher Scientific, Kalamazoo,MI). Stained HBECs were washed in PBS, air dried prior to additionof 1 ml lysing solution (PBS containing 1% sodium dodecyl sulfate)and kept overnight at −80 °C. Following incubation for 30 min at37 °C, the staining intensity was measured using a Bio Tek Uquantmicroplate reader at 620 nm. The absorbance of lysing solution from atissue culture well containing no cells and stained exactly as describedabove, was used as background reading and subtracted from the absor-bance of each sample.
Proliferation assay
HBECs were plated in 24-well plates (5 × 104/well) in EBM-2medium and incubated at 37 °C and 5% CO2. The following day, cellswere stimulated with AA in complete EBM-2 medium for 48 h at 37 °Cand 5% CO2. 3H-thymidine (2.5 μCi) was added to each well during thelast 8 h of incubation. 3H-thymidine incorporation was assayed aspreviously described (Payner et al., 2006).
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
RECTED P
RO
OF
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
AAControl
∗∗∗
G
H
100
200
300
400
500
600
700
800
030 60 120
Time (min)
Cyt c
AA
Control
Cyt
och
rom
e c/
-a
ctin
rat
io(%
of
con
tro
l)
-actin
AA
30 60 120
Time (min)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
Control AA MA PA SA OA LA
Cas
pas
e-3
acti
vity
(pN
A a
t 40
5 n
m)
∗∗∗I
Fig. 1 (continued).
4 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
UNCO
R
Caspase-3/CPP32 colorimetric assay
Caspase-3 activity was measured by quantification of p-nitroanilide(pNA) released from the labeled caspase-3 substrate DEVD-pNA (Asp-Glu-Val-Asp-pNA) following the manufacturer's instructions. Briefly,HBECs were plated in 6-well plates (5 × 105/well) the day prior to theexperiments and stimulated as indicated. Attached and suspendedHBECs were pooled, washed in PBS, lysed and subjected to centrifuga-tion (10,000×g; 1 min). Lysates (25–50 μg) were incubated in a buffercontaining DEVD-pNA (200 μM) and 10 mM DTT (37 °C; 2 h). pNA re-lease was quantified by spectrophotometry at 405 nm. The absorbanceof lysis buffer containing identical amounts of the DEVD-pNA wassubtracted from the absorbance of each sample.
Morphological detection of apoptosis
To detect apoptotic nuclei, HBECs grown on attachment factor-coated glass coverslips, were stimulated as indicated, fixed with 4%paraformaldehyde for 15 min at room temperature, and stained withHoechst 33342 (10 μg/ml) for 30 min at room temperature. HBECswere washed in PBS, air dried, and visualized by a Leica DM RB fluores-cence microscope. Mitochondrial apoptosis was detected with the
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
MitoCapture assay kit following the manufacturer's instructions.HBECs were visualized by fluorescence microscopy using a band-passfilter that detects FICT and rhodamine. Images were collected usingthe SPOT Advanced version 4.7 Software (Sterling Heights, MI).
RNAi transfection
HBECs (~40–50% confluent) were transfected under serum-freeconditions with p38α (100 nM), p38β (100 nM), Hsp27 (200 nM) orcontrol siRNA sequences using Lipofectamine RNAiMAX. Six hourslater, EBM-2 medium containing growth factors and serum was added,and incubation was continued for 48 h. Cells were stimulated and sub-jected to Western blotting as indicated.
Western blotting
HBECs were plated in 6-well plates the day prior to stimulation. Fol-lowing stimulation, detached and attached cells were collected togetherby centrifugation at 4 °C on ice-cold PBS containing 5mMNaF and 1mMNa3VO4. Proteinswere extracted in lysis buffer (50mMTris–HCl pH 8.0,150mMNaCl, 5mMEDTA, 10mMNaF, 10mMdisodiumpyrophosphate,1 mM Na3VO4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
TED P
RO
OF
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
0
0.1
0.2
0.3
0.4
0.5
0.6
4
2
0
6
8
10
12
14
Vehicle
Control AA C2-Ceramide
Z-DEVD-FMK
Cas
pas
e-3
acti
vity
(pN
A a
t 40
5 n
m)
3 H-T
hym
idin
e In
corp
ora
tio
n(c
.p.m
.x 1
03 )
3 H-T
hym
idin
e In
corp
ora
tio
n(c
.p.m
.x 1
03 )
A
B
C
∗∗∗
∗∗∗
∗∗∗
∗
∗∗∗
0
5
10
15
20
25
30
∗∗
Control AA Apocynin AA+Apocynin
AA
Control
Z-DEVD-FMK
Vehicle
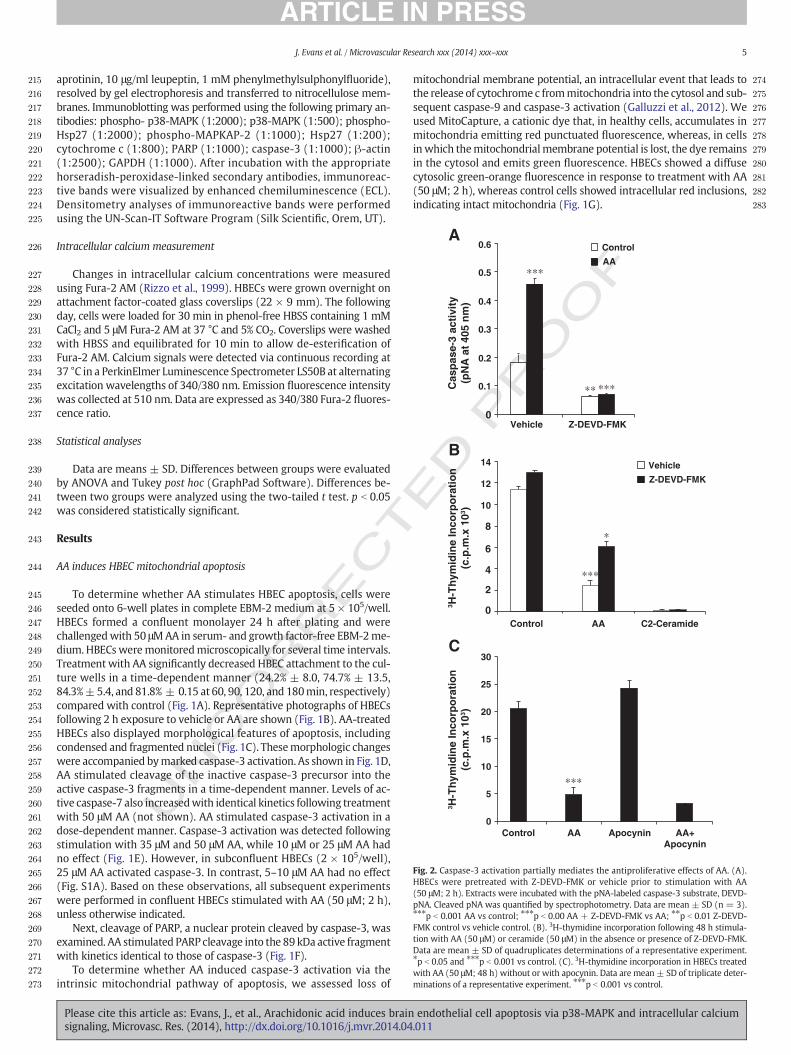
Fig. 2. Caspase-3 activation partially mediates the antiproliferative effects of AA. (A).HBECs were pretreated with Z-DEVD-FMK or vehicle prior to stimulation with AA(50 μM; 2 h). Extracts were incubated with the pNA-labeled caspase-3 substrate, DEVD-pNA. Cleaved pNA was quantified by spectrophotometry. Data are mean ± SD (n = 3).⁎⁎⁎p b 0.001 AA vs control; ⁎⁎⁎p b 0.00 AA + Z-DEVD-FMK vs AA; ⁎⁎p b 0.01 Z-DEVD-FMK control vs vehicle control. (B). 3H-thymidine incorporation following 48 h stimula-tion with AA (50 μM) or ceramide (50 μM) in the absence or presence of Z-DEVD-FMK.Data are mean ± SD of quadruplicates determinations of a representative experiment.⁎p b 0.05 and ⁎⁎⁎p b 0.001 vs control. (C). 3H-thymidine incorporation in HBECs treatedwith AA (50 μM; 48 h) without or with apocynin. Data are mean ± SD of triplicate deter-minations of a representative experiment. ⁎⁎⁎p b 0.001 vs control.
5J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
UNCO
RREC
aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulphonylfluoride),resolved by gel electrophoresis and transferred to nitrocellulose mem-branes. Immunoblotting was performed using the following primary an-tibodies: phospho- p38-MAPK (1:2000); p38-MAPK (1:500); phospho-Hsp27 (1:2000); phospho-MAPKAP-2 (1:1000); Hsp27 (1:200);cytochrome c (1:800); PARP (1:1000); caspase-3 (1:1000); β-actin(1:2500); GAPDH (1:1000). After incubation with the appropriatehorseradish-peroxidase-linked secondary antibodies, immunoreac-tive bands were visualized by enhanced chemiluminescence (ECL).Densitometry analyses of immunoreactive bands were performedusing the UN-Scan-IT Software Program (Silk Scientific, Orem, UT).
Intracellular calcium measurement
Changes in intracellular calcium concentrations were measuredusing Fura-2 AM (Rizzo et al., 1999). HBECs were grown overnight onattachment factor-coated glass coverslips (22 × 9 mm). The followingday, cells were loaded for 30 min in phenol-free HBSS containing 1 mMCaCl2 and 5 μM Fura-2 AM at 37 °C and 5% CO2. Coverslips were washedwith HBSS and equilibrated for 10 min to allow de-esterification ofFura-2 AM. Calcium signals were detected via continuous recording at37 °C in a PerkinElmer Luminescence Spectrometer LS50B at alternatingexcitation wavelengths of 340/380 nm. Emission fluorescence intensitywas collected at 510 nm. Data are expressed as 340/380 Fura-2 fluores-cence ratio.
Statistical analyses
Data are means ± SD. Differences between groups were evaluatedby ANOVA and Tukey post hoc (GraphPad Software). Differences be-tween two groups were analyzed using the two-tailed t test. p b 0.05was considered statistically significant.
Results
AA induces HBEC mitochondrial apoptosis
To determine whether AA stimulates HBEC apoptosis, cells wereseeded onto 6-well plates in complete EBM-2 medium at 5 × 105/well.HBECs formed a confluent monolayer 24 h after plating and werechallengedwith 50 μMAA in serum- and growth factor-free EBM-2me-dium. HBECsweremonitoredmicroscopically for several time intervals.Treatment with AA significantly decreased HBEC attachment to the cul-ture wells in a time-dependent manner (24.2% ± 8.0, 74.7% ± 13.5,84.3%±5.4, and 81.8% ± 0.15 at 60, 90, 120, and 180min, respectively)compared with control (Fig. 1A). Representative photographs of HBECsfollowing 2 h exposure to vehicle or AA are shown (Fig. 1B). AA-treatedHBECs also displayed morphological features of apoptosis, includingcondensed and fragmented nuclei (Fig. 1C). Thesemorphologic changeswere accompanied bymarked caspase-3 activation. As shown in Fig. 1D,AA stimulated cleavage of the inactive caspase-3 precursor into theactive caspase-3 fragments in a time-dependent manner. Levels of ac-tive caspase-7 also increasedwith identical kinetics following treatmentwith 50 μM AA (not shown). AA stimulated caspase-3 activation in adose-dependent manner. Caspase-3 activation was detected followingstimulation with 35 μM and 50 μM AA, while 10 μM or 25 μM AA hadno effect (Fig. 1E). However, in subconfluent HBECs (2 × 105/well),25 μM AA activated caspase-3. In contrast, 5–10 μM AA had no effect(Fig. S1A). Based on these observations, all subsequent experimentswere performed in confluent HBECs stimulated with AA (50 μM; 2 h),unless otherwise indicated.
Next, cleavage of PARP, a nuclear protein cleaved by caspase-3, wasexamined. AA stimulated PARP cleavage into the 89 kDa active fragmentwith kinetics identical to those of caspase-3 (Fig. 1F).
To determine whether AA induced caspase-3 activation via theintrinsic mitochondrial pathway of apoptosis, we assessed loss of
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
mitochondrial membrane potential, an intracellular event that leads tothe release of cytochrome c frommitochondria into the cytosol and sub-sequent caspase-9 and caspase-3 activation (Galluzzi et al., 2012). Weused MitoCapture, a cationic dye that, in healthy cells, accumulates inmitochondria emitting red punctuated fluorescence, whereas, in cellsin which themitochondrialmembrane potential is lost, the dye remainsin the cytosol and emits green fluorescence. HBECs showed a diffusecytosolic green-orange fluorescence in response to treatment with AA(50 μM; 2 h), whereas control cells showed intracellular red inclusions,indicating intact mitochondria (Fig. 1G).
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
6 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
To confirm activation of intrinsic apoptosis, we measured cyto-chrome c release following treatment with 50 μM AA for varioustime intervals (0.5, 1, 2 h). Cytochrome c release significantly in-creased following 2 h stimulation with AA (Fig. 1H). Taken together,these findings implicate the intrinsic pathway of apoptosis in AA-induced HBEC death.
Next, the effect of AA on caspase-3 activity was compared to that ofother fatty acids with various chain length and degree of unsaturation.AA (50 μM; 2 h) stimulated caspase-3 activity, as shown by increasedpNa release from the labeled caspase-3 selective substrate (pNa-DEVD), compared to control (Fig. 1I). In contrast, equimolar concentra-tions of saturated and unsaturated fatty acids, including myristic acid(C14:0), palmitic acid (C16:0), stearic acid (C18:0), oleic acid (C18:1),and linoleic acid (C18:2) were ineffective.
AA-induced inhibition of HBEC proliferation is partially rescued byinhibition of caspase-3
To establish the functional consequences of AA-induced apoptosis,we measured HBEC proliferation in the presence or absence of Z-DEVD-FMK, a cell-permeable irreversible inhibitor of caspase-3. Pre-treatment with Z-DEVD-FMK blocked basal and AA-induced caspase-3activity (Fig. 2A). Moreover, AA-induced inhibition of HBEC prolifera-tion was partially rescued by Z-DEVD-FMK (57.8% ± 8.5 compared tocells treated with AA only). In contrast Z-DEVD-FMK failed to block
UNCO
RRECT
Fig. 3. AA stimulated p38-MAPK, MAPKAP-2 and Hsp27 phosphorylation. (A, B). AA-induced tibody that recognizes p38-MAPK phosphorylated at Tyr-182/Thr-180 (phospho-p38) followed bblot and quantitative densitometry (n=3). Data aremean± SD. ⁎⁎p b 0.01 and ⁎⁎⁎p b 0.001 vsor equimolar concentrations of myristic acid (MA), palmitic acid (PA), stearic acid (SA), oleic acShown is a representative blot and quantitative densitometry (n = 6). Data are mean ± SD. ⁎
stimulation with AA (50 μM; 2 h). Immunoblotting with antibodies against p-MAPKAP-2mean ± SD. ⁎⁎⁎p b 0.001 AA vs control; and ⁎⁎⁎p b 0.001 AA + SB203580 vs AA. (E). HBERepresentative blot and quantitative densitometry (n = 3). Data are mean ± SD. ⁎⁎⁎p b 0
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
RO
OF
the antiproliferative effect of C2-ceramide (Fig. 2B). The antioxidantapocynin also failed to prevent AA-induced inhibition of HBEC prolifer-ation (Fig. 2C).
AA stimulates p38-MAPK, MAPKAP-2, and Hsp27 phosphorylation
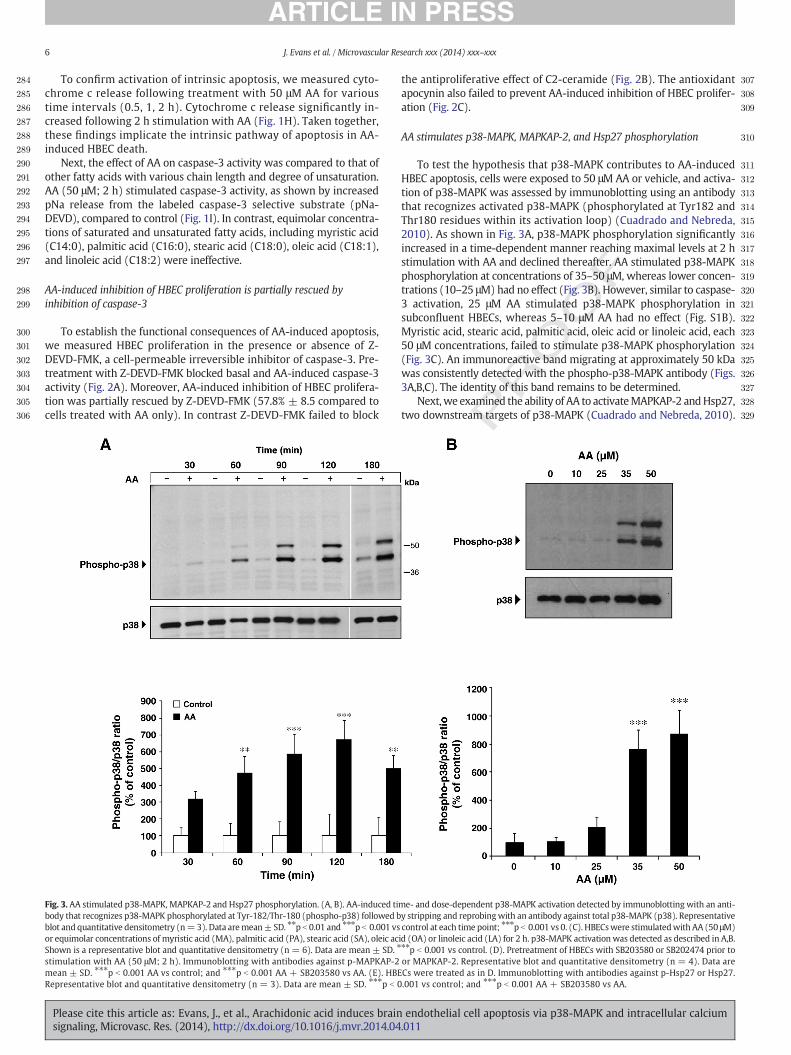
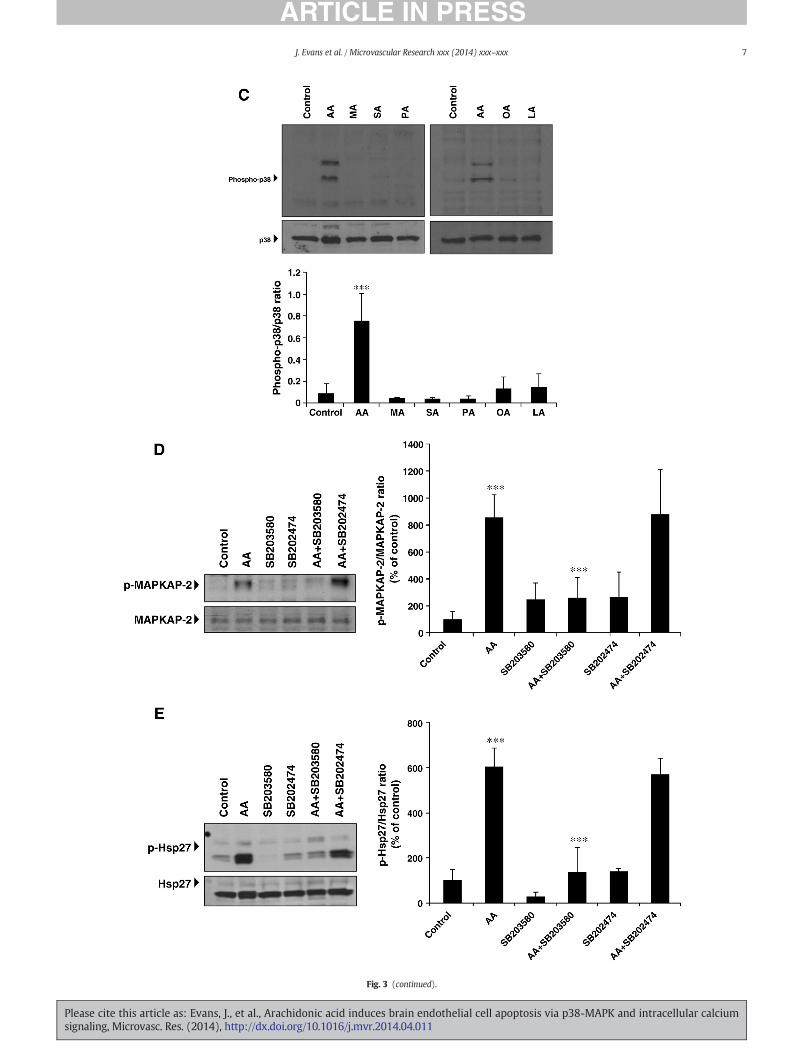
To test the hypothesis that p38-MAPK contributes to AA-inducedHBEC apoptosis, cells were exposed to 50 μM AA or vehicle, and activa-tion of p38-MAPK was assessed by immunoblotting using an antibodythat recognizes activated p38-MAPK (phosphorylated at Tyr182 andThr180 residues within its activation loop) (Cuadrado and Nebreda,2010). As shown in Fig. 3A, p38-MAPK phosphorylation significantlyincreased in a time-dependent manner reaching maximal levels at 2 hstimulation with AA and declined thereafter. AA stimulated p38-MAPKphosphorylation at concentrations of 35–50 μM, whereas lower concen-trations (10–25 μM) had no effect (Fig. 3B). However, similar to caspase-3 activation, 25 μM AA stimulated p38-MAPK phosphorylation insubconfluent HBECs, whereas 5–10 μM AA had no effect (Fig. S1B).Myristic acid, stearic acid, palmitic acid, oleic acid or linoleic acid, each50 μM concentrations, failed to stimulate p38-MAPK phosphorylation(Fig. 3C). An immunoreactive band migrating at approximately 50 kDawas consistently detected with the phospho-p38-MAPK antibody (Figs.3A,B,C). The identity of this band remains to be determined.
Next,we examined the ability of AA to activateMAPKAP-2 andHsp27,two downstream targets of p38-MAPK (Cuadrado and Nebreda, 2010).
ED P
me- and dose-dependent p38-MAPK activation detected by immunoblotting with an anti-y stripping and reprobingwith an antibody against total p38-MAPK (p38). Representativecontrol at each time point; ⁎⁎⁎p b 0.001 vs 0. (C). HBECswere stimulatedwith AA (50 μM)id (OA) or linoleic acid (LA) for 2 h. p38-MAPK activation was detected as described in A,B.⁎⁎p b 0.001 vs control. (D). Pretreatment of HBECs with SB203580 or SB202474 prior toor MAPKAP-2. Representative blot and quantitative densitometry (n = 4). Data areCs were treated as in D. Immunoblotting with antibodies against p-Hsp27 or Hsp27..001 vs control; and ⁎⁎⁎p b 0.001 AA + SB203580 vs AA.
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
UNCO
RRECTED P
RO
OF
Fig. 3 (continued).
7J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
Please cite this article as: Evans, J., et al., Arachidonic acid induces brain endothelial cell apoptosis via p38-MAPK and intracellular calciumsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04.011
330
331
332
333
334
335
336
337
338
339
340
341
342
8 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
AA significantly increasedMAPKAP-2 phosphorylation at the p38-MAPK-dependent site, Thr334 (Fig. 3D). Hsp27phosphorylation at Ser82 also in-creased after AA stimulation (Fig. 3E). Pretreatment with SB203580, ap38-MAPK inhibitor, attenuated AA-induced MAPKAP-2 and Hsp27phosphorylation (70.2% ± 11.3 and 76.3% ± 18.3, respectively). Incontrast, SB202474, the inactive SB203580 analogue, had no effect(Figs. 3D,E).
UNCO
RRECT
Control
AA
SB2035
80
SB2024
74
AA+SB20
3580
Control
AA
RWJ6
7657
AA+RW
J676
57
AA+SB20
2474
0
0.04
0.08
0.12
0.16
0.20
Cas
pas
e-3
acti
vity
(pN
A a
t 40
5 n
m)
AA
+SB
2035
80
AA
+SB
2024
74
AA
Co
ntr
ol
SB
2035
80
SB
2024
74
-actin
∗∗∗
∗∗∗
∗∗∗
∗∗∗
A
B
D
0.5
1.0
0
1.5
2.0
2.5
3.0
Caspase-3
Cleaved caspase-3
A62
0 n
m
Fig. 4. p38-MAPK and Hsp27 are required for AA-induced apoptosis. (A). Pretreatment of HBECblotting of cleaved caspase-3 and β-actin. Representative blot and quantitative densitometry. DAA. (B). Caspase 3-activity monitored by the release of pNA. Data are mean ± SD (n= 3). ⁎⁎⁎pwith SB203580, SB202474, or RWJ67657 (10 μM) prior to stimulationwith AA (50 μM; 2 h). HBcontrol; ⁎⁎⁎p b 0.001 AA + SB203580 vs AA; ⁎⁎⁎p b 0.001 AA + RWJ67657 vs AA. (E). HBECblotting of caspase-3, Hsp27, or GAPDH. Representative blots and quantitative densitometry (n(siRNA control). Lower panel: ⁎⁎⁎p b 0.001 AA (siRNA control) vs control (siRNA control); antransfected with siRNA Hsp27 or siRNA control. Data are mean ± SD (n = 4). ⁎⁎⁎p b 0.001 A(siRNA control).
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
Pharmacological inhibition of p38-MAPK and Hsp27 knockdown abrogateAA-induced apoptosis
To establish the contribution of p38-MAPK to AA-induced apoptosis,HBECs were pretreated with SB203580 or SB202474 prior to measure-ment of AA-induced caspase-3 activation. SB203580 abrogated AA-induced cleavage of caspase-3 and release of the labeled caspase-3
ED P
RO
OF
Control AA
SB2035
80
SB2024
74
AA+SB20
3580
AA+SB20
2474
0.5
1.0
0
1.5
2.0
2.5
3.0
Cle
aved
cas
pas
e-3/
-a
ctin
rat
io(%
of
con
tro
l)
6000
5000
4000
3000
2000
1000
0
∗∗∗
∗∗
Control
AA
SB2035
80
SB2024
74
AA+SB20
3580
AA+SB20
2474
C
∗∗∗
∗∗∗
A62
0 n
m
s with 25 μM SB203580 or SB202474 prior to stimulation with AA (50 μM; 2 h). Westernata are mean± SD (n= 3). ⁎⁎⁎p b 0.001 AA vs control; and ⁎⁎p b 0.01 AA+ SB203580 vsb 0.001 AA vs control; ⁎⁎⁎p b 0.001 AA+ SB203580 vs AA. (C,D). Pretreatment of HBECsEC detachment wasmeasured at 620 nm. Data aremean± SD (n=3). ⁎⁎⁎p b 0.001 AA vss were transfected as indicated and stimulated with AA (50 μM; 2 h) or vehicle. Western= 3). Data are mean ± SD. Upper panel: ⁎⁎⁎p b 0.001 control (siRNA Hsp27) vs controld ⁎⁎⁎p b 0.001 AA (siRNA Hsp27) vs AA (siRNA control). (F). Cell detachment in HBECsA (siRNA control) vs control (siRNA control); and ⁎⁎⁎p b 0.001 AA (siRNA Hsp27) vs AA
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
UNCO
RRECT
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
F
A62
0 n
m
0
0.5
1.0
1.5
2.0
2.5
0
0.5
1.0
1.5
2.0
2.5
Hsp
27/G
AP
DH
rat
io
0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
siRNAControl
siRNAHsp27
siRNAControl
siRNAHsp27
siRNAControl
siRNAHsp27
Cle
aved
cas
pas
e-3/
GA
PD
H r
atio
∗∗∗
∗∗∗∗∗∗
∗∗∗
∗∗∗
∗∗∗
Hsp27
GAPDH
Cleaved caspase-3
siRNA Control Hsp27
E
AA
Control
AA
Control
AA
Control
AA
Fig. 4 (continued).
9J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
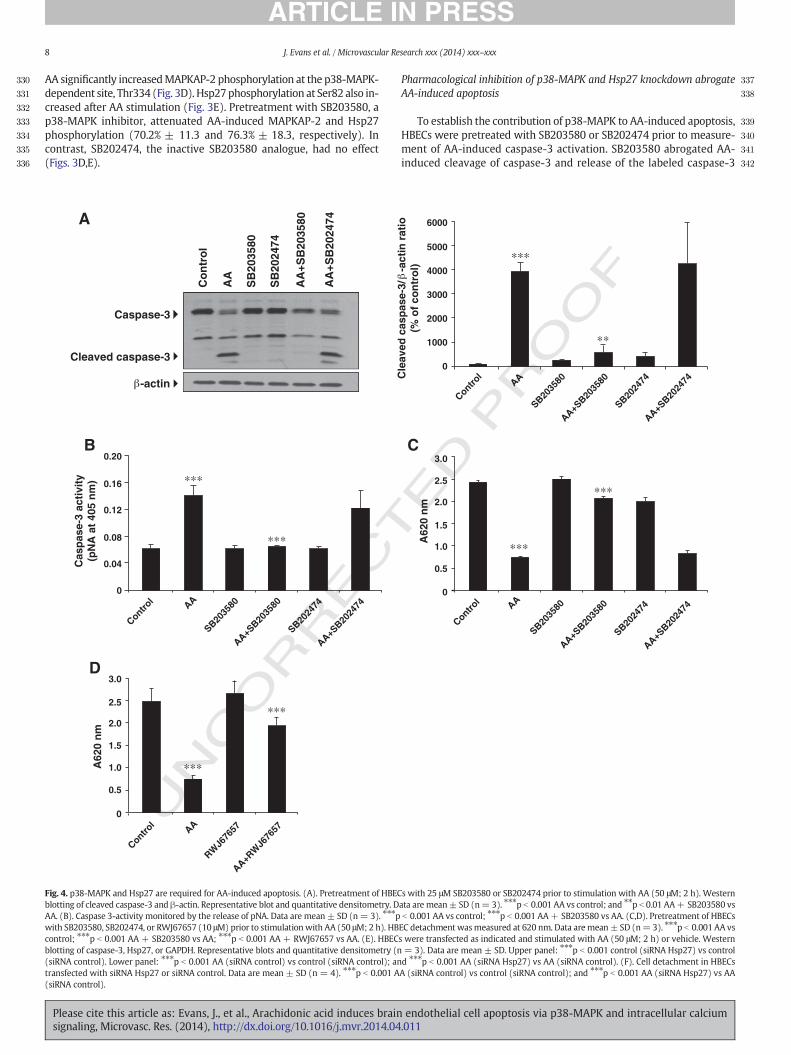
substrate, whereas SB202474 had no effect (Figs. 4A,B). SB203580or RWJ67657, another selective p38-MAPK inhibitor, significantly de-creased AA-induced HBEC detachment (78.9% ± 1.9 and 67.2% ± 22respectively), whereas SB202474 did not (Figs. 4C,D).
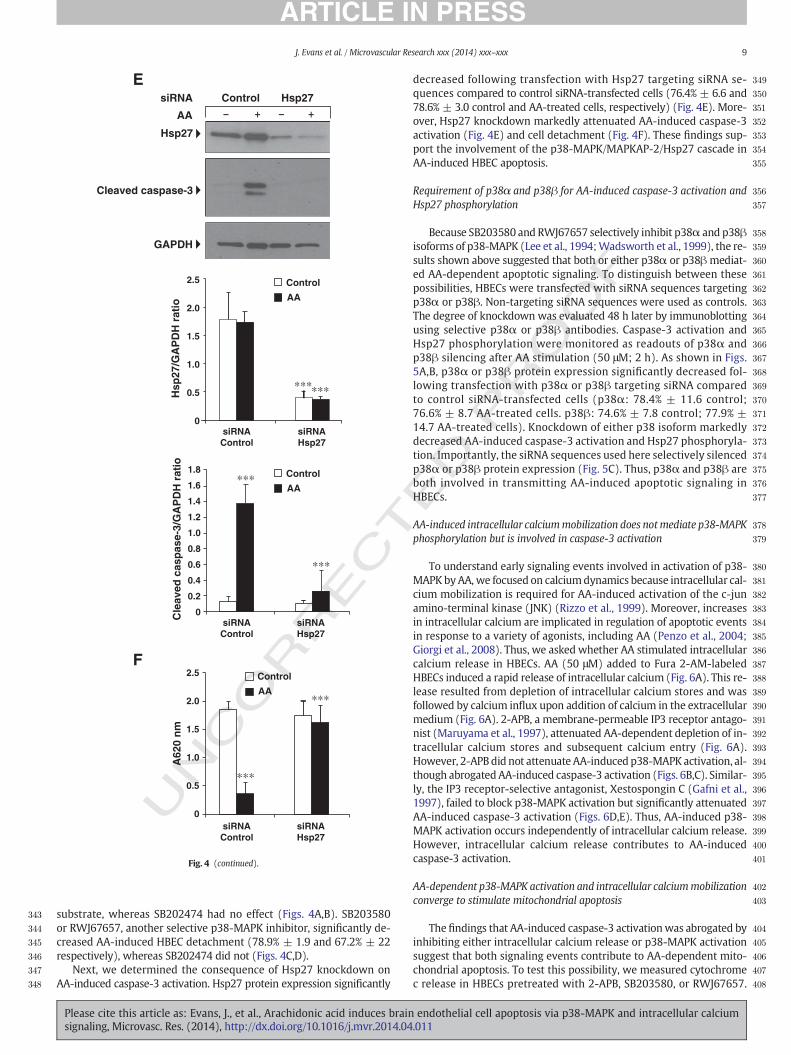
Next, we determined the consequence of Hsp27 knockdown onAA-induced caspase-3 activation. Hsp27 protein expression significantly
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
ED P
RO
OF
decreased following transfection with Hsp27 targeting siRNA se-quences compared to control siRNA-transfected cells (76.4% ± 6.6 and78.6% ± 3.0 control and AA-treated cells, respectively) (Fig. 4E). More-over, Hsp27 knockdown markedly attenuated AA-induced caspase-3activation (Fig. 4E) and cell detachment (Fig. 4F). These findings sup-port the involvement of the p38-MAPK/MAPKAP-2/Hsp27 cascade inAA-induced HBEC apoptosis.
Requirement of p38α and p38β for AA-induced caspase-3 activation andHsp27 phosphorylation
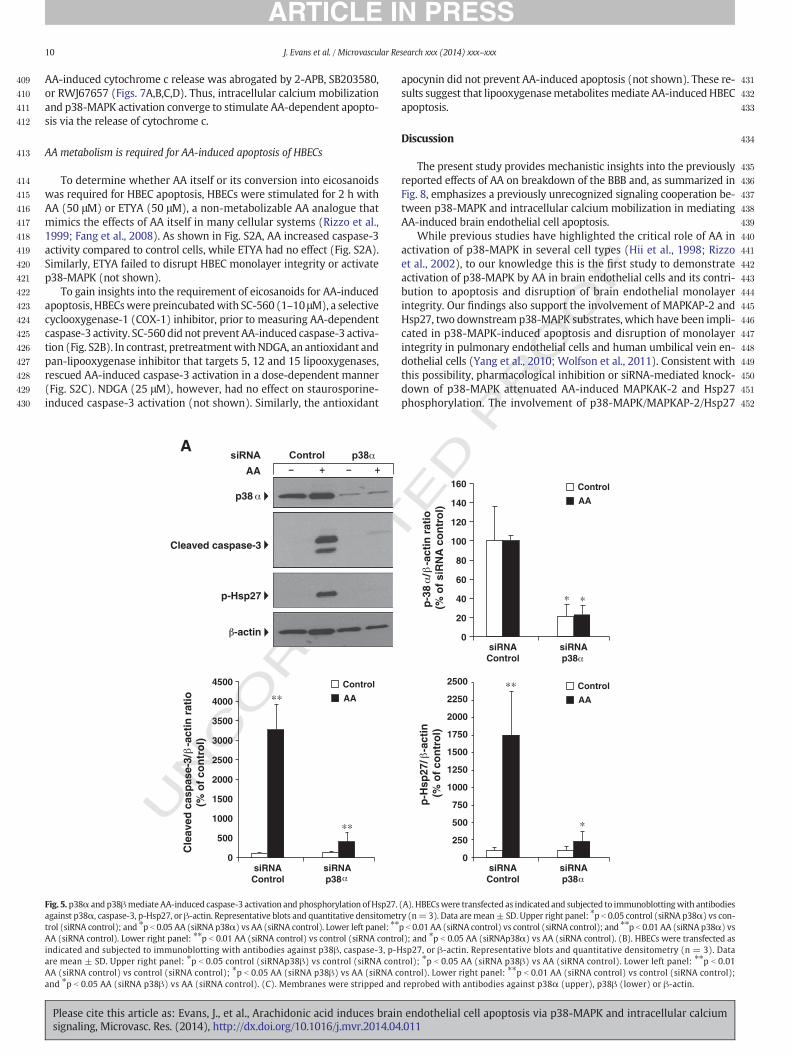
Because SB203580 and RWJ67657 selectively inhibit p38α and p38βisoforms of p38-MAPK (Lee et al., 1994;Wadsworth et al., 1999), the re-sults shown above suggested that both or either p38α or p38βmediat-ed AA-dependent apoptotic signaling. To distinguish between thesepossibilities, HBECs were transfected with siRNA sequences targetingp38α or p38β. Non-targeting siRNA sequences were used as controls.The degree of knockdown was evaluated 48 h later by immunoblottingusing selective p38α or p38β antibodies. Caspase-3 activation andHsp27 phosphorylation were monitored as readouts of p38α andp38β silencing after AA stimulation (50 μM; 2 h). As shown in Figs.5A,B, p38α or p38β protein expression significantly decreased fol-lowing transfection with p38α or p38β targeting siRNA comparedto control siRNA-transfected cells (p38α: 78.4% ± 11.6 control;76.6% ± 8.7 AA-treated cells. p38β: 74.6% ± 7.8 control; 77.9% ±14.7 AA-treated cells). Knockdown of either p38 isoform markedlydecreased AA-induced caspase-3 activation and Hsp27 phosphoryla-tion. Importantly, the siRNA sequences used here selectively silencedp38α or p38β protein expression (Fig. 5C). Thus, p38α and p38β areboth involved in transmitting AA-induced apoptotic signaling inHBECs.
AA-induced intracellular calciummobilization does notmediate p38-MAPKphosphorylation but is involved in caspase-3 activation
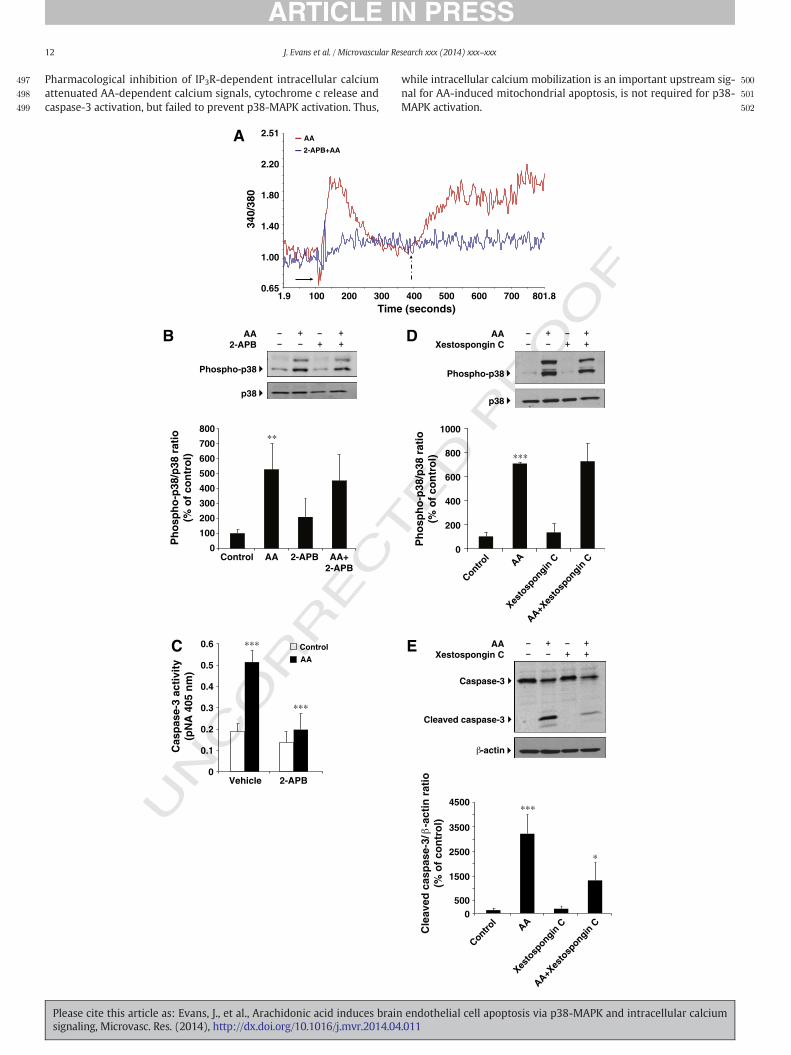
To understand early signaling events involved in activation of p38-MAPK byAA, we focused on calciumdynamics because intracellular cal-cium mobilization is required for AA-induced activation of the c-junamino-terminal kinase (JNK) (Rizzo et al., 1999). Moreover, increasesin intracellular calcium are implicated in regulation of apoptotic eventsin response to a variety of agonists, including AA (Penzo et al., 2004;Giorgi et al., 2008). Thus, we asked whether AA stimulated intracellularcalcium release in HBECs. AA (50 μM) added to Fura 2-AM-labeledHBECs induced a rapid release of intracellular calcium (Fig. 6A). This re-lease resulted from depletion of intracellular calcium stores and wasfollowed by calcium influx upon addition of calcium in the extracellularmedium (Fig. 6A). 2-APB, a membrane-permeable IP3 receptor antago-nist (Maruyama et al., 1997), attenuated AA-dependent depletion of in-tracellular calcium stores and subsequent calcium entry (Fig. 6A).However, 2-APB did not attenuate AA-induced p38-MAPK activation, al-though abrogated AA-induced caspase-3 activation (Figs. 6B,C). Similar-ly, the IP3 receptor-selective antagonist, Xestospongin C (Gafni et al.,1997), failed to block p38-MAPK activation but significantly attenuatedAA-induced caspase-3 activation (Figs. 6D,E). Thus, AA-induced p38-MAPK activation occurs independently of intracellular calcium release.However, intracellular calcium release contributes to AA-inducedcaspase-3 activation.
AA-dependent p38-MAPK activation and intracellular calciummobilizationconverge to stimulate mitochondrial apoptosis
The findings that AA-induced caspase-3 activationwas abrogated byinhibiting either intracellular calcium release or p38-MAPK activationsuggest that both signaling events contribute to AA-dependent mito-chondrial apoptosis. To test this possibility, we measured cytochromec release in HBECs pretreated with 2-APB, SB203580, or RWJ67657.
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
10 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
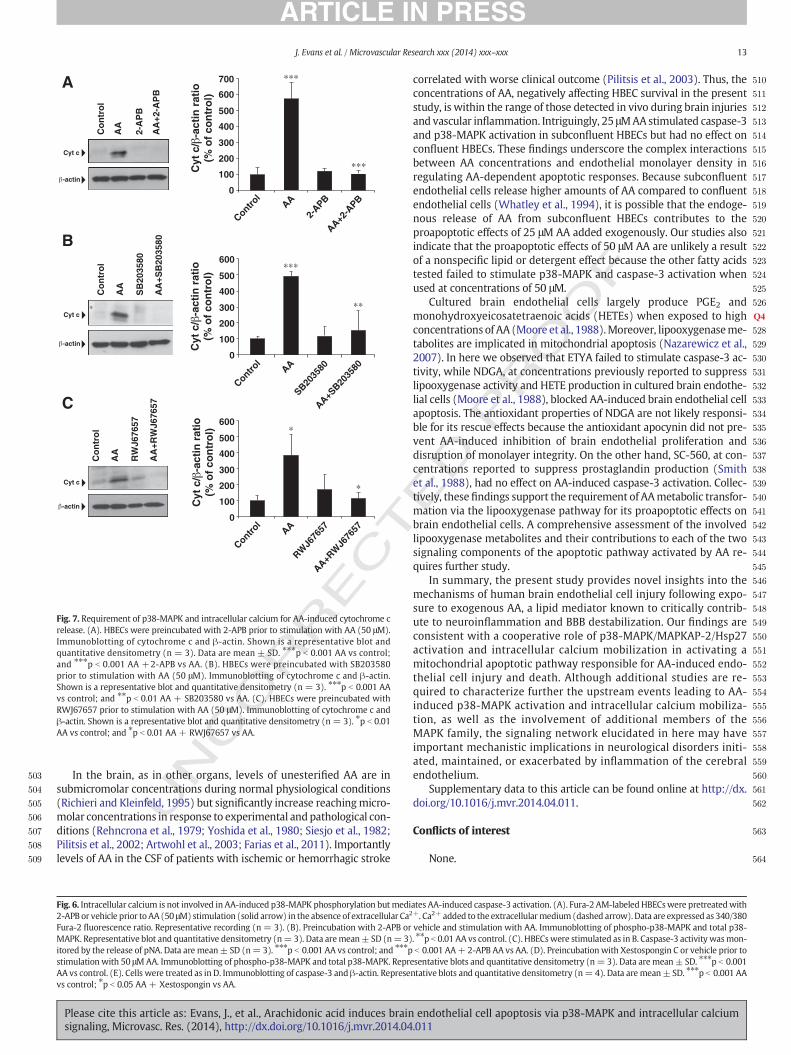
AA-induced cytochrome c release was abrogated by 2-APB, SB203580,or RWJ67657 (Figs. 7A,B,C,D). Thus, intracellular calcium mobilizationand p38-MAPK activation converge to stimulate AA-dependent apopto-sis via the release of cytochrome c.
AA metabolism is required for AA-induced apoptosis of HBECs
To determine whether AA itself or its conversion into eicosanoidswas required for HBEC apoptosis, HBECs were stimulated for 2 h withAA (50 μM) or ETYA (50 μM), a non-metabolizable AA analogue thatmimics the effects of AA itself in many cellular systems (Rizzo et al.,1999; Fang et al., 2008). As shown in Fig. S2A, AA increased caspase-3activity compared to control cells, while ETYA had no effect (Fig. S2A).Similarly, ETYA failed to disrupt HBEC monolayer integrity or activatep38-MAPK (not shown).
To gain insights into the requirement of eicosanoids for AA-inducedapoptosis, HBECswere preincubatedwith SC-560 (1–10 μM), a selectivecyclooxygenase-1 (COX-1) inhibitor, prior to measuring AA-dependentcaspase-3 activity. SC-560 did not prevent AA-induced caspase-3 activa-tion (Fig. S2B). In contrast, pretreatmentwithNDGA, an antioxidant andpan-lipooxygenase inhibitor that targets 5, 12 and 15 lipooxygenases,rescued AA-induced caspase-3 activation in a dose-dependent manner(Fig. S2C). NDGA (25 μM), however, had no effect on staurosporine-induced caspase-3 activation (not shown). Similarly, the antioxidant
UNCO
RRECT
∗∗
∗∗
Cle
aved
cas
pas
e-3/
-a
ctin
rat
io(%
of
con
tro
l)
p38
Cleaved caspase-3
p-Hsp27
-actin
4500
4000
3500
3000
2500
2000
1500
1000
500
0
AA
Control
siRNAp38
siRNAControl
AA
A
Fig. 5. p38α and p38βmediate AA-induced caspase-3 activation and phosphorylation ofHsp27.against p38α, caspase-3, p-Hsp27, or β-actin. Representative blots and quantitative densitomettrol (siRNA control); and ⁎p b 0.05 AA (siRNA p38α) vs AA (siRNA control). Lower left panel: ⁎⁎
AA (siRNA control). Lower right panel: ⁎⁎p b 0.01 AA (siRNA control) vs control (siRNA controindicated and subjected to immunoblotting with antibodies against p38β, caspase-3, p-Hare mean ± SD. Upper right panel: ⁎p b 0.05 control (siRNAp38β) vs control (siRNA conAA (siRNA control) vs control (siRNA control); ⁎p b 0.05 AA (siRNA p38β) vs AA (siRNA cand ⁎p b 0.05 AA (siRNA p38β) vs AA (siRNA control). (C). Membranes were stripped and
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
RO
OF
apocynin did not prevent AA-induced apoptosis (not shown). These re-sults suggest that lipooxygenasemetabolitesmediate AA-inducedHBECapoptosis.
Discussion
The present study provides mechanistic insights into the previouslyreported effects of AA on breakdown of the BBB and, as summarized inFig. 8, emphasizes a previously unrecognized signaling cooperation be-tween p38-MAPK and intracellular calcium mobilization in mediatingAA-induced brain endothelial cell apoptosis.
While previous studies have highlighted the critical role of AA inactivation of p38-MAPK in several cell types (Hii et al., 1998; Rizzoet al., 2002), to our knowledge this is the first study to demonstrateactivation of p38-MAPK by AA in brain endothelial cells and its contri-bution to apoptosis and disruption of brain endothelial monolayerintegrity. Our findings also support the involvement of MAPKAP-2 andHsp27, two downstream p38-MAPK substrates, which have been impli-cated in p38-MAPK-induced apoptosis and disruption of monolayerintegrity in pulmonary endothelial cells and human umbilical vein en-dothelial cells (Yang et al., 2010; Wolfson et al., 2011). Consistent withthis possibility, pharmacological inhibition or siRNA-mediated knock-down of p38-MAPK attenuated AA-induced MAPKAK-2 and Hsp27phosphorylation. The involvement of p38-MAPK/MAPKAP-2/Hsp27
ED P
∗∗
∗
∗ ∗
20
0
40
60
80
100
120
140
160
2500
2250
2000
1750
1500
1250
1000
750
500
250
0
siRNAControl
siRNAControl
siRNAp38
siRNAp38
AA
Control
AA
Control
p-H
sp27
/ -a
ctin
(%
of
con
tro
l)p
-38
/
-act
in r
atio
(%
of
siR
NA
co
ntr
ol)
(A). HBECswere transfected as indicated and subjected to immunoblottingwith antibodiesry (n=3). Data aremean± SD. Upper right panel: ⁎p b 0.05 control (siRNA p38α) vs con-p b 0.01 AA (siRNA control) vs control (siRNA control); and ⁎⁎p b 0.01 AA (siRNA p38α) vsl); and ⁎p b 0.05 AA (siRNAp38α) vs AA (siRNA control). (B). HBECs were transfected assp27, or β-actin. Representative blots and quantitative densitometry (n = 3). Data
trol); ⁎p b 0.05 AA (siRNA p38β) vs AA (siRNA control). Lower left panel: ⁎⁎p b 0.01ontrol). Lower right panel: ⁎⁎p b 0.01 AA (siRNA control) vs control (siRNA control);reprobed with antibodies against p38α (upper), p38β (lower) or β-actin.
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
RECTED P
RO
OF
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
470
471
472
473
474
475
476
477
478
479
480
481
482
483
484
485
486
487
488
489
490
491
492
493
494
495
496
siRNA
∗
∗ ∗
siRNAControl
siRNAp38
B
∗
∗∗ ∗∗
AA
Control
AA
Control
AA
Control
0
20
40
60
80
100
120
140
0
200
400
600
800
1000
0
1000
1600
1400
1200
1000
800
600
400
200p
-38
/
-act
in r
atio
(% o
f si
RN
A c
on
tro
l)p
-Hsp
27/
-act
in
(% o
f co
ntr
ol)
siRNAControl
siRNAp38
siRNAControl
siRNAp38
Cle
aved
cas
pas
e-3/
-a
ctin
rat
io(%
of
con
tro
l)
Cleaved caspase-3
p-Hsp27
p38
-actin
Control p38
AA
siRNA Control p38p38
p38
p38
C
-actin
-actin
Fig. 5 (continued).
11J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
UNCO
R
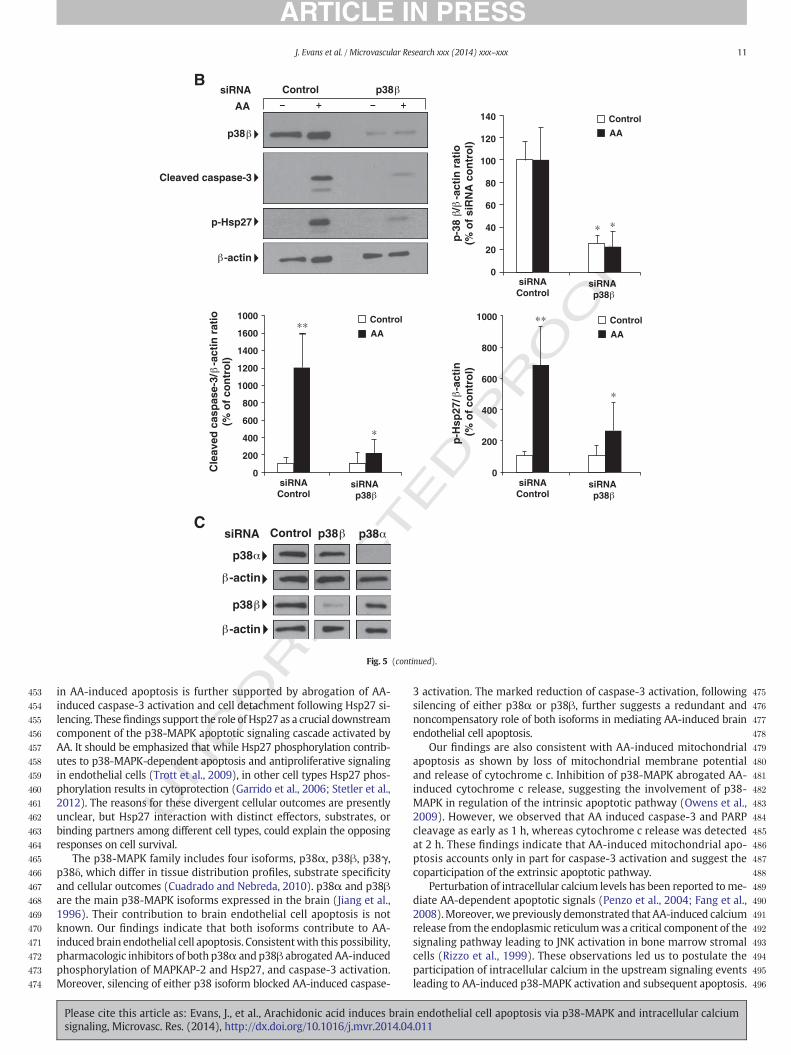
in AA-induced apoptosis is further supported by abrogation of AA-induced caspase-3 activation and cell detachment following Hsp27 si-lencing. Thesefindings support the role of Hsp27 as a crucial downstreamcomponent of the p38-MAPK apoptotic signaling cascade activated byAA. It should be emphasized that while Hsp27 phosphorylation contrib-utes to p38-MAPK-dependent apoptosis and antiproliferative signalingin endothelial cells (Trott et al., 2009), in other cell types Hsp27 phos-phorylation results in cytoprotection (Garrido et al., 2006; Stetler et al.,2012). The reasons for these divergent cellular outcomes are presentlyunclear, but Hsp27 interaction with distinct effectors, substrates, orbinding partners among different cell types, could explain the opposingresponses on cell survival.
The p38-MAPK family includes four isoforms, p38α, p38β, p38γ,p38δ, which differ in tissue distribution profiles, substrate specificityand cellular outcomes (Cuadrado and Nebreda, 2010). p38α and p38βare the main p38-MAPK isoforms expressed in the brain (Jiang et al.,1996). Their contribution to brain endothelial cell apoptosis is notknown. Our findings indicate that both isoforms contribute to AA-induced brain endothelial cell apoptosis. Consistentwith this possibility,pharmacologic inhibitors of both p38α and p38β abrogated AA-inducedphosphorylation of MAPKAP-2 and Hsp27, and caspase-3 activation.Moreover, silencing of either p38 isoform blocked AA-induced caspase-
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
3 activation. The marked reduction of caspase-3 activation, followingsilencing of either p38α or p38β, further suggests a redundant andnoncompensatory role of both isoforms in mediating AA-induced brainendothelial cell apoptosis.
Our findings are also consistent with AA-induced mitochondrialapoptosis as shown by loss of mitochondrial membrane potentialand release of cytochrome c. Inhibition of p38-MAPK abrogated AA-induced cytochrome c release, suggesting the involvement of p38-MAPK in regulation of the intrinsic apoptotic pathway (Owens et al.,2009). However, we observed that AA induced caspase-3 and PARPcleavage as early as 1 h, whereas cytochrome c release was detectedat 2 h. These findings indicate that AA-induced mitochondrial apo-ptosis accounts only in part for caspase-3 activation and suggest thecoparticipation of the extrinsic apoptotic pathway.
Perturbation of intracellular calcium levels has been reported tome-diate AA-dependent apoptotic signals (Penzo et al., 2004; Fang et al.,2008).Moreover, we previously demonstrated that AA-induced calciumrelease from the endoplasmic reticulumwas a critical component of thesignaling pathway leading to JNK activation in bone marrow stromalcells (Rizzo et al., 1999). These observations led us to postulate theparticipation of intracellular calcium in the upstream signaling eventsleading to AA-induced p38-MAPK activation and subsequent apoptosis.
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
497
498
499
500
501
502
12 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
Pharmacological inhibition of IP3R-dependent intracellular calciumattenuated AA-dependent calcium signals, cytochrome c release andcaspase-3 activation, but failed to prevent p38-MAPK activation. Thus,
UNCO
RRECT
AA 2-APB AA+2-APB
∗∗∗
∗∗∗
∗∗
Ph
osp
ho
-p38
/p38
rat
io(%
of
con
tro
l)
Phospho-p38
p38
Control
2-APBVehicle
B
C
AA2-APB
800
700
600
500
400
300
200
100
0
Cas
pas
e-3
acti
vity
(pN
A 4
05 n
m)
0.6
0.5
0.4
0.3
0.2
0.1
0
AA
Control
AA2.51
2.20
1.80
1.40
1.00
0.651.9 100 200 300
Time
2-APB+AA
A
340/
380
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
while intracellular calcium mobilization is an important upstream sig-nal for AA-induced mitochondrial apoptosis, is not required for p38-MAPK activation.
ED P
RO
OF
AAXestospongin C
AAXestospongin C
∗
∗∗∗
∗∗∗
Cle
aved
cas
pas
e-3/
-a
ctin
rat
io(%
of
con
tro
l)
Ph
osp
ho
-p38
/p38
rat
io(%
of
con
tro
l)1000
800
600
400
200
0
AAXes
tosp
ongin C
Control
AAXes
tosp
ongin C
AA+Xes
tosp
ongin C
AA+Xes
tosp
ongin C
Control
Phospho-p38
p38
D
E
Caspase-3
Cleaved caspase-3
-actin
4500
3500
2500
1500
5000
400 500 600 700 801.8 (seconds)
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011
CO
RRECT
503
504
505
506
507
508
509
510
511
512
513
514
515
516
517
518
519
520
521
522
523
524
525
526
527Q4
528
529
530
531
532
533
534
535
536
537
538
539
540
541
542
543
544
545
546
547
548
549
550
551
552
553
554
555
556
557
558
559
560
561
562
563
564
AA
∗∗∗
∗∗
∗
∗
Cyt c
-actin
Cyt c
-actin
Cyt c
-actin
Co
ntr
ol
2-A
PB
AA
+2-A
PB
AA
Co
ntr
ol
SB
2035
80
AA
+SB
2035
80
AA
Co
ntr
ol
RW
J676
57
AA
+RW
J676
57
A
B
C
∗∗∗
∗∗∗
0
100
200
300
400
500
600
0
100
200
300
400
500
600
0
100
200
300
400
500
600
700
Cyt
c/
-act
in r
atio
(% o
f co
ntr
ol)
Cyt
c/
-act
in r
atio
(% o
f co
ntr
ol)
Cyt
c/
-act
in r
atio
(% o
f co
ntr
ol)
AA
2-APB
Control
AA+2-A
PB
AASB20
3580
Control
AA+SB20
3580
AARW
J676
57
Control
AA+RW
J676
57
Fig. 7. Requirement of p38-MAPK and intracellular calcium for AA-induced cytochrome crelease. (A). HBECs were preincubated with 2-APB prior to stimulation with AA (50 μM).Immunoblotting of cytochrome c and β-actin. Shown is a representative blot andquantitative densitometry (n = 3). Data are mean ± SD. ⁎⁎⁎p b 0.001 AA vs control;and ⁎⁎⁎p b 0.001 AA +2-APB vs AA. (B). HBECs were preincubated with SB203580prior to stimulation with AA (50 μM). Immunoblotting of cytochrome c and β-actin.Shown is a representative blot and quantitative densitometry (n = 3). ⁎⁎⁎p b 0.001 AAvs control; and ⁎⁎p b 0.01 AA + SB203580 vs AA. (C). HBECs were preincubated withRWJ67657 prior to stimulation with AA (50 μM). Immunoblotting of cytochrome c andβ-actin. Shown is a representative blot and quantitative densitometry (n = 3). ⁎p b 0.01AA vs control; and ⁎p b 0.01 AA + RWJ67657 vs AA.
13J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
UNIn the brain, as in other organs, levels of unesterified AA are in
submicromolar concentrations during normal physiological conditions(Richieri and Kleinfeld, 1995) but significantly increase reachingmicro-molar concentrations in response to experimental andpathological con-ditions (Rehncrona et al., 1979; Yoshida et al., 1980; Siesjo et al., 1982;Pilitsis et al., 2002; Artwohl et al., 2003; Farias et al., 2011). Importantlylevels of AA in the CSF of patients with ischemic or hemorrhagic stroke
Fig. 6. Intracellular calcium is not involved in AA-induced p38-MAPK phosphorylation butmedi2-APB or vehicle prior to AA (50 μM)stimulation (solid arrow) in the absence of extracellular Ca2
Fura-2 fluorescence ratio. Representative recording (n = 3). (B). Preincubation with 2-APB orMAPK. Representative blot and quantitative densitometry (n=3). Data aremean± SD (n=3)itored by the release of pNA. Data are mean± SD (n= 3). ⁎⁎⁎p b 0.001 AA vs control; and ⁎⁎⁎pstimulation with 50 μMAA. Immunoblotting of phospho-p38-MAPK and total p38-MAPK. ReprAA vs control. (E). Cells were treated as in D. Immunoblotting of caspase-3 and β-actin. Represevs control; ⁎p b 0.05 AA + Xestospongin vs AA.
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
ED P
RO
OF
correlated with worse clinical outcome (Pilitsis et al., 2003). Thus, theconcentrations of AA, negatively affecting HBEC survival in the presentstudy, is within the range of those detected in vivo during brain injuriesand vascular inflammation. Intriguingly, 25 μMAA stimulated caspase-3and p38-MAPK activation in subconfluent HBECs but had no effect onconfluent HBECs. These findings underscore the complex interactionsbetween AA concentrations and endothelial monolayer density inregulating AA-dependent apoptotic responses. Because subconfluentendothelial cells release higher amounts of AA compared to confluentendothelial cells (Whatley et al., 1994), it is possible that the endoge-nous release of AA from subconfluent HBECs contributes to theproapoptotic effects of 25 μM AA added exogenously. Our studies alsoindicate that the proapoptotic effects of 50 μM AA are unlikely a resultof a nonspecific lipid or detergent effect because the other fatty acidstested failed to stimulate p38-MAPK and caspase-3 activation whenused at concentrations of 50 μM.
Cultured brain endothelial cells largely produce PGE2 andmonohydroxyeicosatetraenoic acids (HETEs) when exposed to highconcentrations of AA (Moore et al., 1988).Moreover, lipooxygenaseme-tabolites are implicated in mitochondrial apoptosis (Nazarewicz et al.,2007). In here we observed that ETYA failed to stimulate caspase-3 ac-tivity, while NDGA, at concentrations previously reported to suppresslipooxygenase activity and HETE production in cultured brain endothe-lial cells (Moore et al., 1988), blocked AA-induced brain endothelial cellapoptosis. The antioxidant properties of NDGA are not likely responsi-ble for its rescue effects because the antioxidant apocynin did not pre-vent AA-induced inhibition of brain endothelial proliferation anddisruption of monolayer integrity. On the other hand, SC-560, at con-centrations reported to suppress prostaglandin production (Smithet al., 1988), had no effect on AA-induced caspase-3 activation. Collec-tively, thesefindings support the requirement of AAmetabolic transfor-mation via the lipooxygenase pathway for its proapoptotic effects onbrain endothelial cells. A comprehensive assessment of the involvedlipooxygenase metabolites and their contributions to each of the twosignaling components of the apoptotic pathway activated by AA re-quires further study.
In summary, the present study provides novel insights into themechanisms of human brain endothelial cell injury following expo-sure to exogenous AA, a lipid mediator known to critically contrib-ute to neuroinflammation and BBB destabilization. Our findings areconsistent with a cooperative role of p38-MAPK/MAPKAP-2/Hsp27activation and intracellular calcium mobilization in activating amitochondrial apoptotic pathway responsible for AA-induced endo-thelial cell injury and death. Although additional studies are re-quired to characterize further the upstream events leading to AA-induced p38-MAPK activation and intracellular calcium mobiliza-tion, as well as the involvement of additional members of theMAPK family, the signaling network elucidated in here may haveimportant mechanistic implications in neurological disorders initi-ated, maintained, or exacerbated by inflammation of the cerebralendothelium.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.mvr.2014.04.011.
Conflicts of interest
None.
ates AA-induced caspase-3 activation. (A). Fura-2 AM-labeled HBECswere pretreatedwith+. Ca2+ added to the extracellularmedium (dashed arrow).Data are expressed as 340/380vehicle and stimulation with AA. Immunoblotting of phospho-p38-MAPK and total p38-. ⁎⁎p b 0.01 AA vs control. (C). HBECswere stimulated as in B. Caspase-3 activity wasmon-b 0.001 AA+ 2-APB AA vs AA. (D). Preincubation with Xestospongin C or vehicle prior to
esentative blots and quantitative densitometry (n= 3). Data aremean± SD. ⁎⁎⁎p b 0.001ntative blots and quantitative densitometry (n= 4). Data aremean± SD. ⁎⁎⁎p b 0.001 AA
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011

TD P
RO
OF
565
566
567
568
569
570Q5
571
572573574575576577578579580581582583584585586587588589590591592593594595596597598599600601602
603604605606607608609610611
Apoptosis
Disruption of monolayer integrity
IP3R
Ca2+
Arachidonic Acid
Cytochrome cHsp-27
MAPKAP-2
p38MAPKα/β
P
P
PLOXs
Caspase-3
P
Fig. 8. Proposed signaling pathwaymediating AA-inducedHBEC apoptosis. Stimulation of HBECswith AA triggers signaling events characterized by intracellular calciummobilization andphosphorylation of p38-MAPK, MAPKAP-2 and Hsp27. Intracellular calcium and activated p38-MAPK/MAPKAP-2/Hsp27-dependent signaling converge into the mitochondria to inducecaspase-3-dependent apoptosis and cell detachment. Conversion of AA by lipooxygenases (LOXs) is required for AA-dependent apoptotic signaling. The solid and dashed arrows indicatedirect and indirect experimental evidence in support of the proposed targets/pathways, respectively. Pharmacological or siRNA-mediatedmanipulation of the proposed targets/pathwaysis indicated. Arrowheads: ►activation: │ inhibition.
14 J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
EC
Acknowledgments
We thank Dr. Dino Rotondo for the critical suggestions, Dr. RafatSiddiqui for the use of the spectrometer, Elaine Bammerline for theillustrations, Marilyn Michael Yurk for the editorial assistance, JamieHeitzenrater and Andrea Doell for the technical assistance. This workwas supported by Institutional Funds to M.T.R.
612613614615616617618619620621622623624625626627628629630631632633634635636637638639640641642643644645646647
UNCO
RRReferences
Artwohl, M., et al., 2003. Free fatty acids trigger apoptosis and inhibit cell cycle progres-sion in human vascular endothelial cells. FASEB J. 18, 146–148.
Bazan, N.G., et al., 2002. Prostaglandins and other lipid mediators in Alzheimer's disease.Prostaglandins Other Lipid Mediat. 68–69, 197–210.
Brash, A.R., 2001. Arachidonic acid as a bioactive molecule. J. Clin. Invest. 107, 1339–1345.Chan, P.H., Fishman, R.A., 1978. Brain edema: induction in cortical slices by polyunsaturat-
ed fatty acids. Science 201, 358–360.Cuadrado, A., Nebreda, A.R., 2010. Mechanisms and functions of p38 MAPK signaling.
Biochem. J. 429, 403–417.Cunningham, T.J., et al., 2006. Inhibition of secreted phospholipase A2 by neuron survival
and anti-inflammatory peptide CHEC-9. J. Neuroinflammation 11, 3–25.De Caterina, R., et al., 2000. Fatty acid modulation of endothelial activation. Am. J. Clin.
Nutr. 71, 213S–223S.Fang, K.-M., et al., 2008. Arachidonic acid induces both Na+ and Ca2+ entry resulting in
apoptosis. J. Neurochem. 104, 1177–1189.Farias, S.E., et al., 2011. Lipid mediators in cerebral spinal fluid of traumatic brain injured
patients. J. Trauma 71, 1211–1218.Farooqui, A.A., et al., 2007. Modulation of inflammation in brain: a matter of fat. J.
Neurochem. 101, 577–599.Farrall, A.J., Wardlaw, J.M., 2009. Blood–brain barrier: ageing and microvascular disease—
systematic review and meta-analysis. Neurobiol. Aging 30, 337–352.Gafni, J., et al., 1997. Xestospongins: potent membrane permeable blockers of the inositol
1,4,5-trisphosphate receptor. Neuron 19, 723–733.Galluzzi, L., et al., 2012. Mitochondrial control of cellular life, stress, and death. Circ. Res.
111, 1198–1207.Garrido, C., et al., 2006. Heat shock proteins 27 and 70: anti-apoptotic proteins with
tumorigenic properties. Cell Cycle 5, 2592–2601.Giorgi, C., et al., 2008. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 8,
119–130.Glass, C.K., et al., 2010. Mechanisms underlying inflammation in neurodegeneration. Cell
140, 918–934.
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
EGopez, J.J., et al., 2005. Cyclooxygenase-2-specific inhibitor improves functional outcomes,provides neuroprotection and reduces inflammation in a rat model of traumatic braininjury. Neurosurgery 56, 590–604.
Guo, S., et al., 2008. Neuroprotection via matrix-trophic coupling between cerebral endo-thelial cells and neurons. Proc. Natl. Acad. Sci. U. S. A. 105, 7582–7587.
Hii, C.S., et al., 1998. Stimulation of p38 phosphorylation and activity by arachidonic acidin HeLa cells, HL60 promyelocytic leukemic cells, and human neutrophils. J. Biol.Chem. 273, 19277–19282.
Hillered, L., Chan, P.H., 1988. Effects of arachidonic acid on respiratory activities in isolatedbrain mitochondria. J. Neurosci. Res. 19, 94–100.
Jiang, Y., et al., 1996. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J. Biol. Chem. 271, 17920–17926.
Jin, K., et al., 2002. Vascular endothelial growth factor (VEGF) stimulates neurogenesisin vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 99, 11946–11950.
Jung, Y.-S., et al., 2008. Cadmium induces apoptotic cell death through p38 MAPK in brainmicrovessel endothelial cells. Eur. J. Pharmacol. 578, 11–18.
Lee, J.C., et al., 1994. A protein kinase involved in the regulation of inflammatory cytokinebiosynthesis. Nature 372, 739–746.
Maruyama, T., et al., 1997. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrablemodulator of Ins (1,4,5)P3-induced Ca2+ release. J. Biochem. 122, 498–505.
Miyazaki, K., et al., 2011. Disruption of neurovascular unit prior to motor neuron degen-eration in amyotrophic lateral sclerosis. J. Neurosci. Res. 5, 718–728.
Moore, S.A., et al., 1988. Eicosanoid metabolism in cerebromicrovascular endothelium.Am. J. Physiol. 254, C37–C44 (Cell Physiol. 23).
Nazarewicz, R.R., et al., 2007. 12(S)-hydroperoxyeicosatetraenoic acid (12-HETE) in-creases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch.Biochem. Biophys. 468, 114–120.
Nito, C., et al., 2008. Role of the p38 mitogen-activated protein kinase/cytosolic phospho-lipase A2 signaling pathway in blood–brain barrier disruption after focal cerebralischemia and reperfusion. J. Cereb. Blood Flow Metab. 28, 1686–1696.
Ohnishi, T., et al., 1992. Vasogenic brain edema induced by arachidonic acid: role of extra-cellular arachidonic acid in blood–brain barrier dysfunction. Neurosurgery 30,545–551.
Oldendorf, W.H., et al., 1977. The large apparent work capability of the blood–brain bar-rier: a study of the mitochondrial content of capillary endothelial cells in brain andother tissues of the rat. Ann. Neurol. 1, 409–417.
Owens, T.W., et al., 2009. Apoptosis commitment and activation of mitochondrial Baxduring anoikis is regulated by p38MAPK. Cell Death Differ. 16, 1551–1562.
Park, S., et al., 2004. Neurovascular protection reduces early brain injury after subarach-noid hemorrhage. Stroke 35, 2412–2417.
Payner, T., et al., 2006. Microsomal prostaglandin E synthase-1 regulates human gliomacell growth via prostaglandin E(2)-dependent activation of type II protein kinase A.Mol. Cancer Ther. 5, 1817–1826.
Penzo, D., et al., 2002. Effects of fatty acids on mitochondria: implications for cell death.Biochem. Biophys. Acta 1555, 160–165.
endothelial cell apoptosis via p38-MAPK and intracellular calcium.011

648649650651652653654655656657658659660661662663664665666667668669670671672673674675676677678679680
681682683684685686687688689690691692693694695696697698699700701702703704705706707708709
15J. Evans et al. / Microvascular Research xxx (2014) xxx–xxx
Penzo, D., et al., 2004. Arachidonic acid released by phospholipase A(2) activation triggersCa(2+)-dependent apoptosis through themitochondrial pathway. J. Biol. Chem. 279,25219–25225.
Pilitsis, J.G., et al., 2002. Free fatty acids in human cerebrospinal fluid following subarach-noid hemorrhage and their potential role in vasospasm: a preliminary observation. J.Neurosurg. 97, 272–279.
Pilitsis, J.G., et al., 2003. Measurement of free fatty acids in cerebrospinal fluid from pa-tients with hemorrhagic and ischemic stroke. Brain Res. 985, 198–201.
Rao, J.S., et al., 2011. Increasedneuroinflammatory and arachidonic acid cascademarkers, andreduced synaptic proteins, in brain of HIV-1 transgenic rats. J. Neuroinflammation 9, 19.
Rapoport, S.I., 2008. Arachidonic acid and the brain. J. Nutr. 138, 2515–2525.Rehncrona, S., et al., 1979. Recovery of brain mitochondrial function in the rat after com-
plete and incomplete cerebral ischemia. Stroke 10, 437–446.Richieri, G.V., Kleinfeld, A.M., 1995. Unbound free fatty acid levels in human serum. J. Lipid
Res. 36, 229–240.Rite, I., et al., 2007. Blood–brain barrier disruption induces in vivo degeneration of nigral
dopaminergic neurons. J. Neurochem. 101, 1567–1582.Rizzo, M.T., Leaver, H.A., 2010. Brain endothelial cell death: modes, signaling pathways and
relevance to neutral development, homeostasis and disease. Mol. Neurobiol. 42, 52–63.Rizzo, M.T., et al., 1999a. Induction of apoptosis by arachidonic acid in chronic myeloid
leukemia cells. Cancer Res. 59, 5047–5053.Rizzo, M.T., et al., 1999b. Arachidonic acid induces mobilization of calcium stores and c-
jun gene expression: evidence that intracellular calcium release is associated withc-jun activation. Prostaglandins Leukot. Essent. Fat. Acids 60, 187–198.
Rizzo, M.T., et al., 2002. Specificity of arachidonic acid-induced inhibition of growth andactivation of c-jun kinases and p38mitogen-activated protein kinase in hematopoiet-ic cells. Prostaglandins Leukot. Essent. Fat. Acids 66, 31–40.
Rush, S., et al., 2007. c-jun amino-terminal kinase and mitogen activated protein kinase 1/2mediate hepatocyte growth factor-inducedmigration of brain endothelial cells. Exp. CellRes. 313, 121–132.
Scali, C., et al., 2003. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses braininflammation and protects cholinergic neurons from excitotoxic degenerationin vivo. Neuroscience 117, 909–919.
UNCO
RRECT
Please cite this article as: Evans, J., et al., Arachidonic acid induces brainsignaling, Microvasc. Res. (2014), http://dx.doi.org/10.1016/j.mvr.2014.04
PRO
OF
Scorrano, L., et al., 2001. Arachidonic acid causes cell death through the mitochondrialpermeability transition. Implications for tumor necrosis factor-alpha apoptotic signal-ing. J. Biol. Chem. 276, 12035–12040.
Siesjo, B.K., et al., 1982. The influence of bicuculline-induced seizures of free fatty acidconcentrations in cerebral cortex, hippocampus, and cerebellum. J. Neurochem. 39,796–802.
Smith, C.J., et al., 1988. Pharmacological analysis of cyclooxygenase-1 in inflammation.Proc. Natl. Acad. Sci. U. S. A. 95, 13313–13318.
Stetler, R.A., et al., 2012. Phosphorylation of HSP27 by protein kinase D is essential for me-diating neuroprotection against ischemic neuronal injury. J. Neurosci. 32, 2667–2682.
Takeuchi, Y., et al., 1991. A possible mechanism of mitochondrial dysfunction during ce-rebral ischemia: inhibition of mitochondrial respiration activity by arachidonic acid.Arch. Biochem. Biophys. 289, 33–38.
Trott, D., et al., 2009. Effect of phosphorylated hsp27 on proliferation of human endothe-lial and smooth muscle cells. Proteomics 9, 3383–3394.
Wadsworth, S.A., et al., 1999. RWJ 67657, a potent, orally active inhibitor of p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 291, 680–687.
Ward, N.C., et al., 2011. Cytochrome P450 metabolites of arachidonic acid are elevated instroke patients compared with healthy controls. Clin. Sci. 121, 501–507.
Weiss, N., et al., 2009. The blood–brain barrier in brain homeostasis and neurologicaldiseases. Biochim. Biophys. Acta 1788, 842–857.
Whatley, R.E., et al., 1994. Proliferation-dependent changes in release of arachidonic acidfrom endothelial cells. J. Clin. Invest. 94, 1889–1900.
Wolfson, R.K., et al., 2011. HMGB1 induces human lung endothelial cell cytoskeletal rear-rangement and barrier disruption. Microvasc. Res. 81, 189–197.
Yang, D., et al., 2010. Induction of MAPK phosphatase-1 by hypothermia inhibits TNF-alpha-induced endothelial barrier dysfunction and apoptosis. Cardiovasc. Res. 85,520–529.
Yoshida, S., et al., 1980. Effects of transient ischemia on free fatty acids and phospholipidsin the gerbil. J. Neurosurg. 53, 323–331.
Zhou, C., et al., 2004. Caspase inhibitors prevent endothelial apoptosis and cerebral vaso-spasm in dog model of experimental subarachnoid hemorrhage. J. Cereb. Blood FlowMetab. 24, 419–431.
EDendothelial cell apoptosis via p38-MAPK and intracellular calcium.011