anomeric and exo-anomeric effects in carbohydrate chemistry
TRANSCRIPT

ADVANCES IN CARBOHYDRATE CHEMISTRY AND BIOCHEMISTRY . VOL . 41
ANOMERIC AND EXO-ANOMERIC EFFECTS IN CARBOHYDRATE CHEMISTRY
BY IGOR TVAROSKA* AND TOMAS BLEHAt
* Institute of Chemistry and TPoIymer Institute. Centre of Chemical Research. Slovak Academy of Sciences. *842 38 and f842 36 Bratislava. Czechoslovakia
I . Introduction ........................................................ I1 . Definition of the Anomeric and Exo-anomenc Effects ......................
1 . AnomericEquilibria ............................................... 2 . The Energy of the Anomeric Effect ...................................
111 . Experimental Data on These Effects ..................................... I . Relative Abundance of Isomers ...................................... 2 . Valence Geometry Parameters .......................................
IV . Molecular Orbital Calculations of the Anomeric Effect ...................... I . Conformational Energies ........................................... 2 . The Solvent Effect ................................................. 3 . The Anomeric Effect Energy from MO Calculations ..................... 4 . Coupling of Bond Lengths and Bond Angles to Torsional Angles ........... 5 . Electron Distribution and Lone Pairs .................................
V . The Anomeric and Exo-anomeric Effects in Potential-Function Calculations ... VI . Nature of the Anomeric Effect ..........................................
1 . Electrostatic Interactions ............................................ 2 . Delocalization Interactions .......................................... 3 . Scaling between Electrostatic and Delocalization Interactions . . . . . . . . . . . . . .
VII . Role of the Anomeric Effect in the Reactivity of Carbohydrates . . . . . . . . . . . . . . 1 . Energy of Reaction Paths ........................................... 2 . Lone-Pair Orbital Interactions in Reactivity ............................
45 47 48 53 59 59 71 75 77 88 93 98
101 103 106 107 109 114 116 116 119
I . INTRODUCTION
The concepts of conformational analysis are fundamental to a proper understanding of the relationship between the structure and properties of carbohydrates . The general application of conformational analysis has been stimulated by the relative universality of its fundamental postulates. espe- cially on the qualitative level . The leading notion is the quantification of
Copyright Q 1989 by Academic Ress, Inc . All rights of reproduction in any form reserved . 45

46 IGOR TVAROSKA AND TOMAS BLEHA
steric interactions (bulkiness) of substituents. There is a vast number of examples where the stability of the conformations, their reactivity, or the stereochemistry of products can be explained solely by steric factors.
However, as soon as application of the methods that have been used so successfully in conformational analysis of acyclic and cyclic hydrocarbons to carbohydrates is attempted, it becomes apparent that some additional and quite new factors are at work. They are usually treated as separate conforma- tional effects, and termed by special The most important ofthese factors is the anomeric effect. A basic rule of conformational analysis, de- duced from study of cyclohexane, states that the equatorial position is the energetically favored orientation of a large substituent on a six-membered ring. This principle, however, cannot be applied to polar substituents at an anomeric center in aldopyranose derivatives. Electronegative substituents on the anomeric carbon atom assume a higher abundance of axial positions than could be expected from the analogy with cyclohexane derivatives. This apparently anomalous situation was first discussed by Edward," and it has been termed the anomeric effect by Lemie~x.~ The terms exo-anomeric effect6 and reverse anomeric effect' were later introduced for the orienta- tional preference of the aglycon around the glycosidic C - 0 bond, and for the enhanced trend of the quarternary nitrogen atom to adopt an equatorial orientation, respectively.
The anomeric effect, first identified in carbohydrate chemistry, is now recognized as being of a more-general importance for all molecules having two (or more) heteroatoms linked to the tetrahedral center. The unusual conformational behavior of this class of compounds containing the C - X - C-Y moiety, where X = N, 0, or S, and Y = Br, C1, F, N, 0, or S was denoted as the generalized anomeric effect.* The focal segments of carbohy- drate compounds having oxygen atoms as the heteroatoms X and Y, namely, acetals and hemiacetals, also belong to this class. Instead of the methylene group, some other tetrahedral groups may serve as the anomeric center. This subject has received very extensive experimental and theoretical
( 1 ) E. L. Eliel, N. L. Allinger, S. J. Angyal, and G. A. Momson, Conformational Analysis,
(2) J. F. Stoddart, Stereochemistry of Carbohydrates, Wiley-Interscience, New York, 197 1. (3) N. S. Zefirov, Tetrahedron, 33 (1977) 3193-3202. (4) J. T. Edward, Chem. Ind. (London), (1955) 1102- 1104. (5) R. U. Lemieux, in P. de Mayo (Ed.), Molecular Rearrangements, Vol. 2, Interscience, New
(6) R. U. Lemieux, A. A. Pavia, J. C. Martin, and K. A. Watanabe., Can. J. Chem., 47 (1969)
(7) R. U. Lemieux and A. R. Morgan, Can. J. Chem., 43 (1965) 2205-2213. (8) A. J. de Hoog, H. R. Buys, C. Altona, and E. Havinga, Tetrahedron, 25 (1969) 3365-3375.
Wiley, New York, 1965.
York, 1964, pp. 709-769.
4427 -4439.

ANOMERIC AND EXO-ANOMERIC EFFECTS 41
examination. 1-339-18 From these studies, it has become increasingly apparent that the anomeric effect is a complex phenomenon characterized, apart from the conformational preferences, by unique variations of valence geometry, reactivity, and other properties. This behavior reflects an enhanced stereo- chemical sensitivity of electron distribution of the structural segments in the vicinity of the anomeric center. In this vein, it is proper to speak about the different (that is, energetic, structural, and kinetic) manifestations of the anomeric effect.
The present state of knowledge of the anomeric and exo-anomeric effects in carbohydrates is discussed in this article from the standpoint oftheoretical chemistry. Detailed theoretical argumentation supported by expenmental data is used to rationalize coherently the various manifestations of the ano- meric effect, and to explain its origin. The chapter is divided into seven Sections. Several possibilities of specification of the energy associated with the anomeric effect are demonstrated in Section 11. Section I11 summarizes experimental data concerning the configurational and conformational equi- libria of the anomeric center, and related variations of valence geometry. Section IV presents a survey of molecular orbital calculations of the ano- meric effect based mostly on model compounds. The merits and limitations of semiempirical atom-potential calculations of the anomeric effect are de- scribed in Section V. The nature of the anomenc effect and the prominent role played therein by electron lone-pairs on oxygen atoms are elucidated in Section VI. Finally, in Section VII, some correlations between the stereo- electronic structure and the reactivity of anomers are discussed.
11. DEFINITION OF THE ANOMERIC AND EXO-ANOMERIC EFFECTS
The free-energy aspect of the anomeric effect as displayed by equilibria of isomers is the area wherein the very concept was incepted and developed,
(9) W. A. Szarek and D. Horton (Eds.), TheAnomeric Effect, Origin and Consequences, ACS
( 10) A. J. Kirby, TheAnomeric Effect andRelatedStereoelectronic Efects at Oxygen, Springer-
( I I ) 1. TvaroSka and T. Bleha, Chem. Papers, 39 (1985) 805-847. ( 1 2) I. TvaroSka, in G. Naray-Szabo (Ed.), Theoretical Chemistry of Biological Systems, Else-
(1 3) C. Romers, H. R. Buys, and E. Havinga, in N. L. Allinger and E. L. Eliel (Eds.), Topics in
(14) R. U. Lxmieux, PureAppl. Chem., 25 (1971) 527-548. (15) J. C. Martin, Ann. Chim., 6 (1971) 205-218. (16) R. U. Lemieux, S. Koto, and D. Voisin, in Ref. 9, pp. 17-29. (17) R. U. Lemieux, Ann. N. Y. Acad. Sci., 222 (1973) 915-934. (18) R. U. Lxmieux and S. Koto, Tetrahedron, 30 (1974) 1933- 1944.
Symposium Series, Vol. 87, Washington, 1979.
Verlag, Berlin, 1983.
vier, Amsterdam, 1986, pp. 283-348.
Stereochemistry, Vol. 4, Interscience, New York, 1969, pp. 39-97.

48 IGOR TVAROSKA AND TOMAS BLEHA
and which still dominates its investigation. Obviously, this is because the energy of the anomeric effect is quite amenable to quantification. In the following treatment, the role of the anomeric and related effects on the equilibrium at the anomeric center, and the various choices for definition of the associated Gibbs energy, are briefly described.
1. Anomeric Equilibria
Based on information derived from study of cyclohexane, steric analysis predicts that the most-stable conformation of pyranoses is a chair structure with bulky substituents in equatorial (e) positions. The orientation of a substituent on the anomeric center is, however, an exception, because, at that position, preference for an axial (a) position of polar substituents has been found in several pyranose derivatives. This anomeric effect, the en- hancement in population (relative to cyclohexane) of a position having an electronegative substituent, is illustrated in Fig. 1 by the anomeric equilibria of some a- and P-D-glucopyranose derivatives. Alkoxy, acetoxy, and halogen substituents prefer the a over the e orientation. The only substituent having a preference for the e position is the hydroxyl group: showing 36% of the a isomer. For comparison, there is only 1 1% of the a isomer at equilibrium in the reference cyclohexane derivative, cyclohexanol; that is, D-glucopyranose exhibits about three times more of the axial isomer than could be expected based on steric grounds. This preference for the axial position increases as the electron-withdrawing character of the C- 1 substituent increases, and it also depends on the other ring-substituents and on the solvent (see Table I).
The preference for the axial orientation over the equatorial, displayed by an electronegative substituent at the anomeric carbon atom of pyranoses is equivalent to the preference of the synclinal (sc or gauche) over the antiperi- planar (up or trans) orientation about the ring 0-5 - C- 1 bond in the C-5 -0- 5-C- I-X segment, as shown in Fig. 2. The torsional angle about this and similar bonds in acyclic compounds will be denoted as 8, and the corre- sponding torsional potential as V(8).
a. The Exo-anomeric Effect.-The term exo-anomeric effect was intro- duced to describe an orientational effect on the aglycon part of a glucopyr- anoside,6 arising from the special properties of the acetal moiety. There is no difference in the nature of the anomeric and the exo-anomeric effects; each of them just applies to a different portion of the acetal segment C-5 - 0-5 -C- 1 - 0- 1 - C-i. The anomeric effect is related to the preference of the axial orientation of the aglycon group in glycosides, that is, to the preference of the sc arrangement about the 0 - 5 -C-1 bond, whereas the exo-anomeric effect relates to the preference of the aglycon carbon atom C-i for the sc position at

ANOMERIC AND EXO-ANOMERIC EFFECTS 49
AcO OAc AcO
AcO OAc
88% 12%
(b)
- - HO OH
OH 64%
HoH-
36%
- HO OH
OMe 67% 33%
(4 CH~OAC A c O S c ,
AcO A c : q AcO CI e AcO
94% 6% FIG. 1 .-The Equilibrium Compositions of 0- and /?-D-GluCOpymIOst? Derivatives.
the C- 1 - 0- 1 bond rotational potential specified by the angle a. Obviously, the most important outcome of the exo-anomeric effect concerns the relative stability of mutual orientations of the neighboring saccharide units in oligo- and poly-saccharides. In these cases, the conformational importance of the exo-anomenc effect surpasses that of the anomeric effect.

50 IGOR TVAROSKA AND TOMAS BLEHA
TABLE I Effects of Ring Substituents on the Anomeric Equilibrium of Aldopyranoses and Derivatives
Substituent Axial Compound on C-1 anomer (To) References
D-Glucopyranose HO 32-37 19-25 D-Mannopyranose HO 67-69 19,23,24 D-Allopyranose HO 20 23,24 D-Galactopyranose HO 36 23,24 D-Xylopyranose HO 32-35 19,23,24 D-Lyxopyranose HO 71 23,24 D-Ribopyranose HO 26 23,24 2-Deoxy-~-urubino-hexopyranose HO 41.5 23,24 2-O-Methyl-~-mannopyranose HO 1 5 24 2,3-Di-O-methyl-~-mannopyranose HO 80 24 2,3,4-Tn-O-methyl-~-rnannopyranose HO 86 24 Methyl D-glucopyranoside Me0 67 26,27 Methyl D-mannopyranoside Me0 94 26,27 Methyl D-galactopyranoside MeO 71 26,27 Methyl D-xylopyranoside Me0 69 26,27 1,2,3,4-Tetra-O-acetyI-~-xyIopyranose AcO 8 3" 5 1,2,3,4-Tetra-O-acetyl-~-fucopyranose AcO 83" 5
pyranose. AcO 84' 5
copyranose AcO 86" 5 1,2,3,4,6-Penta-O-acetyl-~-glucopyranose AcO 86" 5
glucopyranose AcO 91" 5
1,2,3,4-Tetra-O-acetyI-6deoxy-6-iodo-~-gluco-
1,2,3,4-Tetra-O-acetyl-6-chloro-6-deoxy-~-glu-
1,2,3,4-Tetra-O-acetyI-6-O-t0syl-~-
Calculated from the Gibbs energy differences given in Ref. 5.
The exo-anomeric effect is illustrated in Fig. 3, which shows three stag- gered orientations for rotation about the glycosidic bond in both the a and p anomer of methyl D-glycopyranoside. These are referred to as (+ sc, + sc), (+ sc, up), and (+ sc, - sc) and (up, + sc), (up, - sc), and (up, ap), respectively, using two torsional angles (6 and a) for specification of orientations of the C-5-0-5-C-1-0-1 -C-i moiety.
(19) H. S. Isbell and W. W. Pigman, J. Res. Nutl. Bur. Stand., 18 (1937) 141 - 194. (20) J. H. Brewster, J. Am. Chem. Soc., 81 (1959) 5475-5483. (2 I ) A. S. Hill and R. S. Shallenberg, Curbohydr. Res., 11 (1969) 541 -545. (22) M. Mathlouthi and D. V. Luu, Curbohydr. Rex, 81 (1980) 203-212. (23) S. J. Angyal, Aust. J. Chem., 21 (1968) 2737-2746. (24) S. J. Angyal, Angew. Chem., Int. Ed. Engl., 8 (1 969) 157 - 167. (25) F. Franks, in J. M. V. Blanshard (Ed.), Polysuccharides in Foods, Butteworth, London,
(26) C. T. Bishop and F. P. Cooper, Cun. J. Chem., 41 (1963) 2743-2758. (27) V. Smirnyagin and C. T. Bishop, Can. J. Chem., 46 (1968) 3085 - 3090.
1979, pp. 33-49.

ANOMERIC AND EXO-ANOMERIC EFFECTS 51
X H
H-1 X
sc aP FIG. 2.-The Orientation Around the 0 - 5 -C-1 Bond in the Axial and Equatorial Forms of
Aldopyranoses.
The qualitative conformational analysis of the two anomers is straightfor- ward. In the axial series, the (+ sc, - sc) conformer is, on steric grounds, very unstable, with the Me group lying below the ring in close proximity to the two axial hydrogen atoms on the C-5 and C-3 atoms. In the equatorial anomer, the (up, + sc) conformer suffers from a repulsion between the Me group and the axial hydrogen atom on C-2. The exo-anomeric effect causes
I 0
\ M e
( +sc, + s c ) + sc. ap) ( + s c , -sc) FIG. 3.-Staggered Orientations of the Aglycon in Methyl a- and PD-Glucopyranoside
Characterized by Torsional Angles 0 and @.

52 IGOR TVAROSKA AND TOMAS BLEHA
preference for the sc conformation; therefore, in both cases, the conformers having the sc position of angle a, that is, (+sc, +sc) and (up, -sc), are expected to be preferred over those having the up position, (+ sc, up) and (up, up) for alkyl glycosides. In fact, analysis of the solid-state structures28-30 revealed that most carbohydrate derivatives adopt the (+sc, -I- sc), or (up, - sc), conformation. A particularly clear illustration of the working of the exo-anomeric effect is exemplified in the structure of a,a-trehalose. In the solid state, this compound exhibits’l an approximate C2 symmetry, with two glycosidic-linkage torsion angles corresponding to the orientation (+ sc, + sc). The preferred conformation of a,a-trehalose in solution is very similar to that in the solid state, and there is no indication of significant proportions of any another conformer^.^^ Also, the simplest acetal, (CH30)2CH2 (di- methoxymethane), which played a key role in the initial development of knowledge of the anomeric effect, has been shown to exist in the (+ sc, + sc) c~nformation.’~ The polymeric analog of dimethoxymethane, namely, poly(oxymethylene), has the analogous synclinal, helical c~nformation.’~
b. The Reverse Anomeric Effect.- Studies of the protonation of N-gly- cosyl-imidazoles and -pyrimidines s h o ~ e d ’ J ~ , ~ ~ that the presence of the posi- tive charge on the nitrogen atom linked to the anomeric center provides a strong driving-force for the aglycon to adopt the equatorial orientation (see Fig. 4). This preference for the e position in excess above the value that ensued from steric analysis of cyclohexane has been termed the reverse anomeric effect.’ The carbamoyl group, having only a partial positive charge on the carbon atom, has a reverse anomeric effect large enough to shift the equilibrium in hexopyranose peracetates toward the e form36 (see Fig. 4).
c. The Generalized Anomeric Effect.- Although the anomeric effect, by its original definition, applies to the properties of pyranoses, it turned out later that this effect is observed in a number of polar, acyclic and cyclic compounds, especially those involving a segment of the general formula - R - X - T- Y - . The group T represents a tetrahedral (anomeric) center of
(28) S. Perez and R. H. Marchessault. Curbohydr. Res.. 65 (1978) 114- 120. (29) I. TvaroSka and T. KoEir, Chem. Zvesri, 35 (1981) 425-440. (30) B. Fuchs, L. Schleifer, and E. Tartakovsky, Nouv. J. Chim., 8 (1984) 275-278. (31) G. A. Jeffrey and R. Nanni, Curbohydr. Res., 137 (1985) 21 -30. (32) K. Bock, J. Defaye, H. Driguez, and E. Bar-Guilloux, Eur. J. Chem., 13 1 (1983) 595-600. (33) E. E. Astrup, Acra Chem. Scund., 25 (1971) 1494-1495. (34) P. de Santis, E. Giglio, A. M. Liquori, and A. Ripamonti, J. Polym. Sci., Purr A . 1 (1963)
(35) H. Paulsen, Z. Gyorgydeak, and M. Friedman, Chem. Ber., 107 (1974) 1590-1613. (36) M. Chmielewski, J. N. &Miller, and D. P. Cerretti, J. Am. Chem. Soc.. 46 (1981) 3903-
1383-1404.
3908.

ANOMERIC AND EXO-ANOMERIC EFFECTS 53
C O N H 2
44%
A c O
AcO b A c
56 ”/.
AcO O A c
FIG. 4.-Examples of Equilibria Involving the Reverse Anomeric Effect.
the types-CHR-, -CH2, -POI--, -Si(CH,)2-, and-SO2-, and X and Y are such heteratoms as N, 0, S, and also halogens in the case of the terminal substituent Y. Acetals, thioacetals, substituted sulfides, phos- phates, siloxanes, and other molecules having heteroatoms in geminal, 1,3 position in the backbone, belong to this group. The general preference for the sc orientation about the T - Y bond in the system R - X - T - Y - has been termed the generalized anomeric effect* and has been reviewed.lOJ *
2. The Energy of the Anomeric Effect
The energy (or, better, enthalpy) and Gibbs energy of the anomeric effect can be deduced from knowledge of the isomer equilibria. It is, however, regrettable that several energy parameters related to sc - up and similar equilibria are tacitly used as an energy measure of the anomeric effect. Consequently, the magnitude of the anomeric effect of a given substituent depends on the procedure applied at its derivation, a fact that makes compar- ison difficult and that can be a source of confusion.
The most frequently used measure of the anomeric effect is based on the comparison of the stability of 2-substituted oxane (tetrahydropyran; THP) and cyclohexane. In general, conformational properties of the oxane ring are similar to those of cyclohexane, with dominance of a chair conformation. It is further presumed that steric interactions in oxane are the same as in cyclohexane, with preference for equatorial positions of bulky substituents. The Gibbs energy of the anomeric effect, AG(AE l), can be expressed as the

54 IGOR TVAROSKA AND TOMAS BLEHA
X
( b ) act" ax X
FIG. 5.- The Standard Equilibrium in (a) Substituted Oxanes and (b) Substituted Cyclohex- anes Used in the Definition of the Anomeric Effect by Eq. I .
difference of the standard, conformational Gibbs energies for substituted oxane (AG:), shown in Fig. 5a, and for the same substituent on cyclohexane, AG:; see Fig. 5b.
AG(AE1) = AGO, - AG! (1)
The term “A parameter” is also used in the literature for the Gibbs energy -AG:. According to the definition in Eq. I, the anomeric effect AG(AE1) depends on the A value of the substituent, and on the temperature and solvent used in measurement of equilibria in Eq. I. This definition can be extended to multisubstituted pyran derivatives, and simple, additivity scheme^^.^^,^^ of steric interactions of substituents are used for the estimation of AG(AE1).
These semiquantitative schemes were based on the assumptions that the pyranoid ring has the same geometry as cyclohexane and that the relative free-energies of each chair form may be obtained by summation of the interaction energies of substituents that are independent of one another, and by taking into account the value of the anomeric effect. The values for interaction energies were obtained experimentally from the equilibria of various cyclitols and p y r a n ~ s e s . ~ ~ , ~ ~ The anomeric equilibrium between a- and P-D-glucopyranose (see Fig. lb) provides a simple illustration of such calculations using a simple additive s ~ h e m e . ~ ~ J ~ The anomers of D - ~ ~ U C O - pyranose differ only in the configuration at the anomeric center; conse- quently, only interactions involving the anomeric hydroxyl group are rele- vant to the conformational equilibrium. From comparison of the steric interactions in each anomer, it is seen that the a anomer has two additional 1,3 diaxial interactions (OH : H). The same two interactions are responsible for the e preference of the hydroxyl group of cyclohexanol. The addition of steric parameters predicts that the a anomer should be 3.8 kJ.mol-’ less

ANOMERIC AND EXO-ANOMERIC EFFECTS 55
TABLE I1 Axial Preferences and the Gibbs Energy of the Anomenc Effect AG(AE,)
(in kJ.mol-I) in the 2Substitnted Oxanesa
Group 96" AG: A* AC(AE,) AC(AE,)i References
Br C1 F,CCH,O CI,CCH,O Cl,CHCH,O CICH,CH20 Me0 EtO C,H,O GH9O Me,CHO Me,CO PhO AcO MeS EtS C,H,S C4H9S Me,CHS Me,CS HO
MeHN Me0,C H,NOC
(CH,),N
96d 96d 92' 95e 88' 7 7' 82 80 82 82 75 67 81 73 69 69 69 70 70 70 47 28 18 6 4d
7.5 2.1 7.5 2.1 6.3 7.5 5.0 3.2 3.8 4.21 3.4 4.11 3.8 3.8 2.7 1.7 4.11 3.8 2 . 9 2.5 3.3 2.0 4.28 2.0 2.0 2.1 2.1 2.2
-0.3 4.2 -2.3 6.4* -3.9 5.4 -5.8 5.3 -7.8 5.3'
9.6 9.6
8.0 7.5
5.8 6.7 5.8 6.2
3.9 4. I 1.5
-0.5 - 2.5
10.8 10.8
10.3 9.8
8.1 8.3 7.6 8.5
6.2 7.6 4.4 2.4 0.4
37 38 39 39 39 39 8 8 8 8 8 8
39 41 42 42 42 42 42 42 43 44 44 45 46
In CCI,, unless specified otherwise. The A-values are from Ref. 47, unless otherwise specified. Using A(oxane) values from Q. 2. Neat. In 1,4dioxane.fRef. I . 8 Ref. 48.
For the N(CH,), group, the A value ofthe N(CH,), group was used.-' In pyridine.' For CONH,, the Same value as for the MeO,C group.
(37) G. E. Booth and R. J. Ouellette, J. Org. Chem., 31 (1966) 544-546. (38) C. B. Anderson and D. T. Sepp, J. Org. Chem., 32 (1967) 607-61 1. (39) G . 0. Pierson and 0. A. Runquist, J. Org. Chem., 33 (1968) 2572-2574. (40) H. Booth, T. B. Grindley, and K. A. Khedhair, J. Chem. Soc., Chem. Commun. (1982)
(41) C. B. Anderson and D. T. Sepp, Chem. Ind. (London) (1964) 2054-2056. (42) A. J. de Hoog and E. Havinga, Red. Truv. Chim. Pays-Bus, 89 (1970) 972-979. (43) A. El-Kafrawy and R. Perrand, C. R. Acud. Sci. Ser. C, 280 ( I 975) I2 19 - 122 1. (44) D. Barbry, D. Couturier, and G. Ricard, J. Chem. SOC., Perkin Trans. 2, (1982) 249-254. (45) E. L. Eliel, K. D. Hargrave, K. M. Pietrusiewicz, and M. Manoharan, J. Am. Chem. Soc.,
(46) I. TvaroSka, M. Hricovini, M. Chmielewski, J. Jarosz, and B. Hintze, unpublished results. (47) H.-J. Schneider and V. Hoppen, J. Org. Chem., 43 (1978) 3866-3873. (48) F. R. Jensen, C. H. Bushweller, and B. H. Beck, J. Am. Chem. Soc., 91 (1969) 344-351. (49) H. Booth and M. L. Josefowicz, J. Chem. SOC. Perkin Trans. 2 (1976) 895-901.
1047- 1048.
104 (1982) 3635-3643.

56 IGOR TVAROSKA AND TOMAS BLEHA
TABLE 111 Axial Preferences and the Gibbs Energy of the Anomeric Effect AG(AE,) (in
kJ.moV) in the 4- and 6-Methyl Derivatives of 2-Substituted Oxanesu ~~ ~
Derivative Substituent %a AGZ AqAE,) AG(AE,Y References
4-Methyl HO 58 0.8 5.0 7.3 41 M e 0 82 3.8 8.0 10.3 50 EtO 78 3.1 7.2 9.5 50 Me,CHCH,O 79 3.3' 5 1 AcO 72 2Sd 5.8 7.6 51 Me0,C 1 1 -5.2' 0.1 3.0 52 c1 96 9.0' 11.1 12.3 38 Br 97 11.3 13.4 14.6 38 I 97 11.3 13.0 14.0 38
6-Methyl Me0 77 3.0 7.2 9.5 50 EtO 16 2.9 7.0 9.2 50 Me,CHO 75 2.7 50 Me,CHCH, 78 3.1' 51 Me,O 70 2.1 50 CF,CH,O 80 3.4 50 HC=CCMe,O 71 2.2 50 AcO 75 2.7d 6.0 7.8 51 MeS 64 1.4 5.6 7.9 50 Me,CS 66 1.6 5.8 8.1 50 Me0,C I 1 -5.2e 0.1 3.0 52
In CCl,, unless specified otherwise. Using A(oxane) values from Eq. 2. In 1,4dioxane. In acetic acid. In methanol. / N e a t .
stable than the /? anomer. The experimentally observed Gibbs energy differ- ence in waterz3 is 1.5 kJ.mol-L. The difference of 2.3 kJ.mol-' between the two values represents the magnitude of the anomeric effect AG(AE1).
Examples of variation of the anomeric effect AG(AE 1) with the substitu- ents are shown in Tables I1 and I11 for oxane derivatives. The application of Eq. 1 to the results of the measurements of anomeric and conformational equilibria have establishedZ that the anomeric effect decreases in approxi- mately the following order: halogen > PhCOz > AcO > AcS > RO > RS > HO > NH2 > Me0,CO > imidazolium > pyridinium.
In reality, however, steric interactions in oxane and in a cyclohexane derivative are not the same. Because C-0 bonds are shorter than C-C bonds, repulsive interactions of an axial group on a pyranoid ring are likely to be larger than those of the same group on the cyclohexane ring, and the
(50) E. L. Eliel and C. A. Giza, J. Org. Chem.. 33 (1968) 3754-3758. (5 1 ) C. B. Anderson and D. T. Sepp, Tetrahedron, 24 ( 1968) 1707 - 17 16. (52) C. B. Anderson and D. T. Sepp, J. Org. Chem., 33 (1968) 3272-3276.

ANOMERIC AND EXO-ANOMERIC EFFECTS 57
anomeric effect based on Eq. I is underestimated. F r a n ~ k ~ ~ estimated, in a new way, the steric part of the Gibbs energy difference (AGZ), for an equilib- rium shown in Fig. 5a. This quantity represents the apparent size of the substituent on oxane, or the parameter A(oxane), and correlates with the AGg value.
Eq. 2 shows that the A(oxane) parameters appropriate for oxane should be 50% larger than the values currently used.
Evidently, the use of the (AGZ), term in Eq. I, instead of AGE, brings about a large amplification of the anomeric effect AG(AE 1) as documented in Tables I1 and 111 for substituted oxane. Two values of AG(AE1) in these Tables exemplify an essential drawback of the definition of the anomeric effect by Eq. I: its change in magnitude with the value assigned for the A parameter. The A factors may also vary with the method of their determina- tion. For example, the A value for an OH group in CCl, was reported' to be in the range of 1.2-6.5 k.l.mol-', and this uncertainty is transferred to AG(AE 1). Moreover, because the A parameters are solvent-dependent, so are the AG(AE1) values.
Using Eq. I, even a qualitative decision about the presence of the ano- meric effect can sometimes be ambiguous. For example, from study of 2,3,4-tri-O-acetylpentopyranosylamines, it was concluded35 that the amine group does not exhibit the anomeric effect. However, a correction of the A value for this group,54 5.9 kJ.mo1-I according to Eq. 2, results in -3 kJ.mol-' larger preference of the a form than could be expected on steric grounds, and therefore, the NH2 group should exhibit the anomeric effect.
The other definition implicitly utilized for an estimation of the anomeric effect is based on comparison of the Gibbs energy difference AGZ with the energy AEpF obtained from semiempirical calculations using the atom-po- tential functions. These methods, stemming from classical physics, vary in their complexity from a simple evaluation of steric energy by atom - atom potential^'^.^^ to detailed description of the force field in a molecule by molecular mechanics method^.^' In this concept, the energy of the anomeric effect, AE(AE2), is determined as that part of the potential energy (or Gibbs energy) that is not accounted for by the calculation procedure and is "miss- ing" in AEpF:
(AG:), = A(oxane) = 1.53 X AGE + 0.08 (2)
AGZ AEZ = AEpF + AE(AE2) (3)
(53) R. W. Franck, Tetrahedron, 39 (1983) 3251 -3252. (54) G. W. Buchanan and V. L. Webb, Tetrahedron Lett., (1983) 4519-4520. ( 5 5 ) K. S. Vijayalaksami and V. S. R. Rao, Carbohydr. Res., 22 (1972) 413-424. (56) A. Abe, J. Am. Chem. Soc., 98 (1977) 6477-6480. (57) N. L. Allinger, J. Am. Chem. SOC., 99 (1977) 8127-8134.

58 IGOR TVAROSKA AND TOMAS BLEHA
Furthermore, a questionable approximation of the same entropy and vol- ume of a and e isomers is usually assumed. Obviously, the energy of the anomeric effect, AE(AE2), depends on the quality of the method used for determination of AEpF. The extra function AE(AE2) may differ, depending on whether AGg or AEE has to be matched. Furthermore, if Eq. 3 applies for an equilibrium in a solvent, the extra term AE(AE2) also includes a contri- bution of the solvent effect due to its neglect or an incomplete representation in energy AEpF.
The measures of the anomeric effect, based on Eqs. 1-3 are of relative character, because they are expressed in reference to a standard compound or a computational method. Some absolute measure is needed for theoretical considerations, and it could be simply the positive difference of the energy of the a and e isomers or of the sc and ap conformation in model compounds.58
(4)
This definition does not take into account the usual preference of bulky substituents for the equatorial position in cyclic compounds, and, with the assumption of the same entropy and volume for the a and the e isomer, corresponds to the AG: value in Eq. 1. In this case, a molecule exhibits the anomeric effect if the axial position (sc orientation) is more stable than the equatorial position (up orientation).
All three definitions of the anomeric effect are interrelated, but as the data required for direct recalculation of one definition into another are frequently lacking, a substituent can be characterized by the diverse data about the energy of the anomeric effect. Their comparison for various groups needs caution, and inspection as to how they were originally calculated.
Definitions based on Eqs. I , 3, and 4 should, in principle, also apply for the exo-anomeric and reverse anomeric effects. There are, however, some prob- lems with the practical application of Eq. I in the case of the exo-anomeric effect, because the AGg values are largely not available. For the exo-ano- meric effect, the conformational equilibrium is specified by two dihedral angles, 8 and a, and the value of AGZ is needed for all six individual con- formers shown in Fig. 3. Because rotation around the exocyclic bond by angle 0 is much less restricted in comparison with rotation by angle 8, a mixture of conformers was experimentally observed, with a difficult resolu- tion of AGX into individual components. Ifthe exo-anomenc effect is treated by Eq. 3, the extra term AE(AE2) should be redefined for the whole range of values of the torsional angle a. Due to the lack of experimental data on AGg or AEE, for each conformer in Fig. 3, the energy values calculated “cor- rectly”, for example by some molecular orbital method, are used, instead of
AE(AE3) = E, - E,
(58) S. Wolfe, M. H. Whangbo, and D. J. Mitchel, Curbohydr. Rex, 69 (1979) 1-26.

ANOMERIC AND EXO-ANOMERIC EFFECTS 59
those in Eq. 3. Because the rotational potential V(Q) depends on the orienta- tion around the endocyclic, 0 - C bond, the AE(AE2) term should be consid- ered separately for each anomer. Several functions have been p r ~ p o s e d ~ ~ - ~ ' for AE(AE2), and these are reviewed in Section VI, which is devoted to calculations of potential function.
A negative value of AG(AE1) represents the reverse anomeric effect. It could, perhaps, also be defined in the context of Eq. 4. Here, the reference saturated hydrocarbons exhibit negative energy AE(AE3), and the reverse anomeric effect could be defined as an excess energy (in absolute value) over that for the reference molecules.
111. EXPERIMENTAL DATA ON THESE EFFECTS
A considerable amount of data has been accumulated during the past two decades on the anomeric effect in terms of structure, energy, reactivity, and other properties. In this Section are discussed some pertinent data, and the ensuring generalizations concern the anomeric equilibrium and geometrical structure of isomers, concentrating on pyranoses and their models. In this respect, the multidimensional character of the anomeric effect has to be emphasized. For its full structural description, the torsional angle 8 (and @ for the exo-anomeric effect) has to be supplemented by the data on bond lengths and valence angles in the vicinity of the anomeric center.
1. Relative Abundance of Isomers
Determination of the relative representation of the a and e forms of pyra- noses can be a very difficult and demanding task. In general, the abundance of isomers at equilibrium depends mainly on the type of substituent (agly- con) on the anomeric center, on the other substituents on the ring, and on the solvent. Studies of acyclic and cyclic models are of invaluable assistance in this field (see Fig. 6). Substituted dimethyl ethers bearing an electronegative group X, as in CH30CH2X, are the simplest acyclic compounds wherein the anomeric effect is operative. For C1 and F substituents, the sc orientation, with the 0 -C torsional angle - 69 - 7 1 O , was, from the microwave spectra,62 found to be the most stable. N.m.r. measurement^^^ ofthe C1 derivative gave an energy difference of 6.3 - 8.4 kJ.mol-' between the up and sc forms. For
(59) R. U. Lemieux, K. Bock, L. T. J. Delbaere, S. Koto, and V. S. R. Rao, Can. J. Chem., 58
(60) H. Thragersen, R. U. Lemieux, K. Bock, and B. Meyer, Can. J. Chem.. 60 (1982)44-57. (61) I. TvaroSka, Carbohydr. Res., 125 (1984) 155-160. (62) M. Hyashi and H. Kato, Bull. Chem. Soc. Jpn.. 53 (1980) 2701 -2710. (63) F. A. L. Anet and I. Yavari, J. Am. Chem. SOC., 99 (1977) 6752-6753.
(1980) 631 -653.

60 IGOR TVAROSKA AND TOMAS BLEHA
x
FIG. 6.-Axial-Equatorial Equilibrium in (a) Complex Aldohexopyranoses, (b) a Simple Derivative of Oxane, and (c and d) Corresponding Conformational Equilibria in Acyclic Model Compounds.
such OCH, derivatives as dimethoxymethane, the (+ sc, + sc) conformation with the methyl groups on opposite sides ofthe OCO plane is the most stable. The rotation around each C - 0 bond into the ap position is disfavoredH by -7.1 kJ.mol-'. Qualitatively similar trends are also observed in longer ethers, but, in most cases, the high flexibility of the chains complicates the resolution of individual isomers. Consequently, cyclic models are much more convenient, particularly substituted tetrahydropyrans having an elec- (64) T. Uchida, Y. Kurita, and M. Kubo, J. Polym. Sci., 29 (1956) 365-373.

ANOMERIC AND EXO-ANOMERIC EFFECTS 61
TABLE IV Conformational Equilibria of Tri-Oacetyl and Tri-Obeozoyl PD-Xylopyranose Derivatives as a Function of the Anomeric
Groupa (see Fig. 7)
'C, Conformer at (oh) Equilibrium ~~ ~
Anomeric group R = Ac References R = Bz References
H Me0 AcO BzO AcS F CI Br NH* NHAc NHCOCF, N3 ImidazolyF Pyridiniumc Imidazoliumc
13 19 28 39 28
80 - 90 79
5'5 56 56
20b 65b 95b 956
72 19 72 73 26 73 74 47 75 75 50 74 76 77 90- 100 77 78 98 78
90- 100 79 35 35 35 35 35 35 35
a In acetone unless specified otherwise. In CDCI,. a-D-Xylopyranose de- rivatives.
tronegative group on C-2 (see Fig. 6b). Numerous AGg values for the ano- meric equilibrium in these compounds are available.6,7,37-4a.50-5z,65-7'
F. Sweet and R. K. Brown, Can. J. Chem., 46 (1968) 1543- 1548. N. S. Zefirov, V. S. Blagoveschensky, I. V. Kazimirchik, and N. S . Surova, Zh. Org. Khim., 5(1969) 1150-1151. N. S . Zefirov, V. S. Blagoveschensky, I. V. Kazimirchik, and N. S. Surova, Tetrahedron, 27
C. B. Anderson and M. P. Geis, Tetrahedron, 31 (1975) 1149- 1154. A. J. de Hoog, Org. Magn. Reson.. 6 (1974) 233-235. N. Pothier, D. D. Rowan, P. Deslongchamps, and J. K. Saunders, Can. J. Chem., 59 ( I 98 1)
R. U. Lemieux and J. Hayami, Can. J. Chem., 43 (1965) 2162-2173. P. Luger, G. Kothe, K. Vangehr, H. Paulsen, and F. R. Heiker, Carbohydr. Rex, 68 (1979)
P. L. Durette and D. Horton, Curbohydr. Res., 18 (1971) 403-418. P. L. Durette and D. Horton, J. Org. Chem., 36 (1971) 2658-2669. P. L. Durette and D. Horton, Carbohydr. Rex, 18 (1971) 389-401. P. L. Durette and D. Horton, Carbohydr. Rex. 18 (1971) 419-425. H. Paulsen, P. Luger, and F. R. Heiker, in Ref. 9, pp. 63-79. P. L. Durette and D. Horton, Carbohydr. Rex, 18 (1971) 57-80. P. L. Durette and D. Horton, Carbohydr. Res., 18 (1971) 289-301.
(1971) 31 11 -31 18.
1132- 1139.
207-223.

62 IGOR TVAROSKA AND TOMAS BLEHA
a. The Character of the Ag1ycon.-The equilibrium composition of sev- eral derivatives of oxane and pyranoses are summarized in Tables I-V. Several observations can be made on inspection of these data. The prefer- ence for the axial position increases with the electron-withdrawing character of substituent X, and is most conspicuous for the halogen and alkoxy deriva- tives (see Table 11). An increase in size of the alkoxy group diminishes the preponderance of the a form, such that changing from a methoxyl to tert-bu- toxy group in 2-substituted oxane decreases the abundance of the a form by 15%. This phenomenon was found to be caused by the entropy, rather than the enthalpy, term.* The size of a substituent does not seem to influence the equilibrium of alkylthio derivatives. An enhancement of the electronegativ- ity of X by change from the ethoxy to the trichloroethoxy derivative in- creases the population of the axial form from 80 to 95%. Similarly, in halo- gen derivatives of oxane, the a forms are the sole detectable species (see Table
A tendency toward stabilization of the a form is usually quite pronounced in the acetyl and benzoyl derivatives of fi-D-xylopyranosyl halides (see Fig. 7a) which, in solution, exist mainly or completely in the 'C., conformation, with all substituents in axial position^.^^-*^ This was first pointed outrn for
11).
TABLE V Conformational Equilibria of Tri-O-acetyl- and Tri-0-benzoyl-p
D-ribopyranose Derivatives as a Function of the Anomeric Group"
'C, Conformer at Equilibrium (96)
Anomeric group R = Ac References R = Bz References
H Me0 EtO Me,CHO Me,CO AcO BzO AcS C1 Br NHAc N, S P h ,
24 61 61 62 54 57 56 34 94 95
56
38b 56b
77 79 79 79 79 74 75 76 78 78 35 35 35
46 80 81 78 74 78 77
98
77 79 79 79 79 75 74
78
@ In acetone, unless specified otherwise. In CDCI,.
(80) P. L. Durette and D. Horton, Adv. Carbohydr. Chem. Biochem., 26 (1971) 49- 126. (80a) C. V. Holland, D. Horton, and J. S. Jewell, J. Org. Chem., 32 (1967) 1818- 1820.

ANOMERIC AND EXQANOMERIC EFFECTS 63
X
R = Ac, B z ; X =CI , Br , I
1970 81% FIG. 7.-The Isomer Equilibrium in (a) DXylopyranose Derivatives and (b) a Related
Compound Having the “Anomeric Center” Unsubstituted.
tri-0-acetyl-j3-D-xylopyranosyl chloride. In this case, the anomeric effect prevails over the unfavorable 1,3-diaxial interactions of bulky substituents. At the same time, this result indicates that the 1,3-diaxial interactions of benzoyloxy and acyloxy groups are much weaker than might be expected. This conjecture is supported by the observation72 of a 19% population of the a form in a related compound having an unsubstituted anomeric center, namely, 1,5-anhydro-2,3,4-tri-0-benzoylxylitol (see Fig. 7b), and of 13% in the analogous tri-0-acetyl derivative.
The preference for the axial position diminishes with lowered electronega- tivity of atoms linked to the anomeric center; that is, F > 0 > N > C for the first row of the Periodic Table. For the latter two elements, N and C, the anomeric equilibrium depends on the overall polarity of the substituent. Thus, derivatives of substituted D-arabinopyranose (see Fig. 8) contain - 94% of the isomer having a nitro group in the axial position at equilib- rium.81 N.m.r. measurement^^^ and other s t ~ d i e s ~ J ~ , ~ , ~ ~ of substituted N- pentopyranosyl derivatives showed that the preference for the axial position decreases in the order NO2 > N-PPh, > N, > NHCOCF, > NH2 >
(8 1) B. Aebischer, R. Hollenstein, and A. Vasella, Helv. Chim. Acta, 66 (1 983) 1748 - 1754. (82) P. Finch and A. G. Nagpurkar, Curbohydr. Res., 49 (1976) 275-287.

64 IGOR TVAROSKA AND TOMAS BLEHA
94 Yo 6 % FIG. 8.- The Equilibrium Composition of a Substituted 2-Deoxy-~-arubinc-hexopyranosyl
Nitrate.
95 %
35 70 65 %
A c O i p&& cF3c02-
A c O (62 cF3c02- Ac OAC N
FIG. 9.- Equilibria for D-Xylopyranose Derivatives Having N-Substituents at the Anomeric Center.

ANOMERIC AND EXO-ANOMERIC EFFECTS 65
NHAc > NHPPh, > imidazole > imidazolium > pyridinium. The groups in the middle of the series, such as NHAc, NHCH, , and aziridine, display a slight preference for the e form. The reverse anomeric effect is typical for imidazole and all groups having positively charged nitrogen. The variation of the anomeric equilibrium with the character of the N-substituent is illus- trated in Fig. 9, and is expressed quantitatively in Tables IV and V. Whereas, in amino-substituted &D-xylopyranose derivatives, the a form is preponder- ant, the e form prevails with the imidazole substituent, and n.m.r. spectros- copy shows only the e form ofthe 'C, conformer in the case ofthe protonated imidazole ring.
Carbon atom substituents on the anomeric center generally favor the equatorial position, but the minimal preference (1.4 kJ.mo1-l) of the e relative to the a position was observed for the ethynyl in oxane. A methoxycarbonyl group45,52 gives a slightly larger population of the e isomer when linked to the oxane ring (relative to cyclohexane). This reversed ano- meric effect is very pronounced for a carbamoyl group. The corresponding derivative of oxane exists mainly (90%) in the equatorial form.& Similarly, in 2,6-anhydroheptonamide~,~~ the carbamoyl group has a strong preference for the equatorial position (see Fig. 10). For example, 3,4,5,7-tetra-O-acetyl- 2,6-anhydro-~-glycero-~-gluco-heptonamide in the ED configuration dis- plays a considerable proportion of the 2C5 conformation (56% in CDcl,), despite extensive 1,3diaxial interactions of four bulky groups. When the more-polar solvent Me2S0 was used, this compound was present almost solely in the *C, conformation.
L - AcO
CONH2
5 C2
CH2OAc I
FIG. 10.- The Anomeric Equilibrium of 3,4,5,7-Tetra-O-acetyI-2,6-anhydro-~-glycero-~- gluco-heptonamide and Its Population in Various Solvents, Illustrating the Reverse Anomeric Effect of the Carbamoyl Group.

66 IGOR TVAROSKA AND TOMAS BLEHA
ACo=oAc - A c O m
AcO AcO AcO
OAc
X = H , CH3, CH21, CH2CI,CH20Ac, or CH20Ts FIG. 1 1 .-Equilibria of D-Xylopyranose Tetraacetate and Its Relatives Listed in Table I.
b. The Other Ring Substituents.- It is well known that the presence and configuration of a hydroxyl group on C-2 of the pyranose ring markedly affects the anomeric Thus, in the case of D-mannopyranose, the axial hydroxyl group on C-2 increases the presence of the a anomer (69%) relative to that for 2-deoxy-~-arabino-hexopyranose (47.5%), which has no hydroxyl group on C-2. Conversely, when the hydroxyl group on C-2 is in the equatorial position, as in D-glucopyranose, the proportion of the a anomer decreases to 36%. These results, summarized in Table I, also show that the same trend, once termed the A2 effect,83 is operative in pentopyran- oses and methyl glycosides. The data for various methylated D-mannoses, given in Table I, indicate that the equilibrium composition changes in favor of the cy anomer as the degree of methylation is increased.24
The electronegativity of the substituent on C-4 also influences the ano- meric equilibrium. Consequently, 2,4-dimethoxyoxane exists in methanol as an equilibrium mixture containing 80% of the isomer having45 an axial methoxyl group on C-2, compared with 67 - 69% for 2-methoxy-4-methyl- oxane . 5 O 3
Finally, some examples of the role of the substituent at C-5 in the ano- meric equilibrium may be mentioned. Anomeric equilibria for a series of substituted pentose and hexose derivatives (see Fig. 1 l), given in Table I, show that an increase in the electronegativity of the equatorial substituent at C-5 increases the axial preference of the acetyl Study of the stereoiso- meric aldopyranose derivative^^^,'^-^ revealed a significant population of both chair conformers, although some limiting cases were observed in which one conformation is very strongly favored. Representative examples are given for the &D-xylopyranose series in Table IV, and for the fi-D-ribopyra- nose series in Table V, respectively. It may be seen that the anomeric effect dominates the conformational preferences, but, in general, the presence of several bulky substituents on the pyranose ring makes the anomeric equilib-
(83) R. E. Reeves, J. Am. Chem. Soc., 72 (1950) 1499- 1506.

ANOMERIC AND EXO-ANOMERIC EFFECTS 67
TABLE VI Influence of the Solvent on the Axial Preferences of the Hydroxyl, Methoxyl,
Methylthio, Aziridinyl, and Carbamoyl Groups in 2-Substituted Oxane Derivativesa
Dielectric constant
O h of the Axial Conformer
Solvent ( E ) H@ MeO' MeSd (CHJ,Ne CONH/
Neat I ,4-Dioxane 2.2 Carbon
tetrachloride 2.2 47 Benzene 2.3 Carbon disulfide 2.6 32 Chloroform 4.8 Pyridine 12.4 55 Acetone 20.7 Methanol 32.7 Acetonitrile 37.5 Dimethyl sulfoxide 46.7 45 Water 78.3 17
72 77
82,83 69 82 66 80
?1,78 59
72 69 54
65,68 50 74 52
31
28 31 29 32 4
4 1 I
23 5 24 8
Data from Ref. 43. From Refs. 6 ,s . 39,50, and 5 I . From Ref. 42. dr From Ref. 44. /From Ref. 46.
ria very intricate, and even the all-axial 'C, form can be ~ b s e r v e d , ~ ~ . ~ ~ as in /3-D-xylopyranose tetraacetate (28%), or in the corresponding tri-0-benzyl- j?-D-xylopyranosyl acetate (47%). As already noted, this form may even preponderate in similar halogen derivatives (see Fig. 7a).
c. The Effect of the Solvent.-The variation ofthe axial preference based on the electronegativity of the ring substituents, and of the aglycon group, as already discussed, suggests that this phenomenon may be sensitive to solva- tion. Table VI shows the abundance of the u form for 2-substituted oxane derivatives (see Fig. 12) in a wide range of solvents. For HO, MeO, and MeS
X
X = OH, OMe, SMe, N(CH2)2, or CONH2 FIG. 12.-Equilibrium for 2-Substituted Oxane Derivatives.
(84) J. B. Lambert and S . M. Wharpy, Curbohydr. Rex, 115 (1983) 33-40,

68 IGOR TVAROSKA AND TOMAS BLEHA
groups, the axial preference is seen to be higher in nonpolar solvents, and lessened in more-polar solvents, although the major difference is between the values in organic solvents and those in water. In dimethyl sulfoxide, the axial preference appears to be higher, as expected on the basis of correlation with the dielectric constant. As may be seen from Table VI, the conforma- tional equilibria of oxane substituted with N(CH2)2 and CONHl groups at C-2 are less sensitive to the solvent than are those having HO, MeO, and MeS aglycon groups. The described trends in solvent effect are also apparent in the data on ~-glucopyranose.~~-~~~*~ The proportion of the a anomer of D-glucopyranose in pyridine is 45%, and in dimethyl sulfoxide it is 44%, as compared with only 32-37% in water. Conversely, in the case of the
TABLE VII Illustrative Examples of the Influence of Solvents and Ring Substituents on the
Gibbs Energy AWAE,) Magnitude (W.mol-') of the Anomeric Effect for Methoxyl, Hydroxyl, and Aziridinyl Groups"
Group Compound Solvent AWAE,) References
Oxane Me0 2-methoxy-
HO
2-methoxy-4-methyl- 2-methoxy-6-methyl- 2-hydroxy-
D-Glucopyranose D-Mannopyranose
2-0-methyl- 2,3-di-O-methyl- 2,3,4,6-tetra-O-methyl-
5-Thio-~-xylose 2-Deoxy-~-arabino-hexopyranose
(CH,),N 2-(Aziridin- 1-yl)oxane
CCl,
CDCI,
HZO CCl, CCl, CCI, CSZ Me,SO H*O HZO H*O HZO H20
HZO H2O HZO CCI, CSZ
CDCI,
C6H6
MeCN
C6H6
Me,SO MeCN
10.3 8 10.3 8 9.7 8 8.0 8 6.7 8
10.0 50 9.6 50 6.2 43 4.5 43 6.0 43 2.6 43 5.0 24 8.4 24 9.2 24
10.1 24 11.0 24 10.8 84 6.3 24 7.5 44 7.6 44 7.8 44 8.0 44 6.9 44 6.9 44
a Based on the constant A(oxane) values of 6.5, 6.5, and 9.9 kJ.mol-I for the MeO, HO, and (CH,XN groups, respectively.

ANOMERIC AND EXO-ANOMERIC EFFECTS 69
N(CH2), group, the polarity of the solvent has little effect on the anomeric ratio, and the proportion of the a anomer is the same in dimethyl sulfoxide and water solution.44
Table VII presents data that illustrate the influence of the solvent and ring substituents on the anomeric effect of methoxyl, hydroxyl, and aziridinyl groups. For the purpose of this Table, the anomeric effect is defined by Eq. I. As noted, this definition takes into account the steric preference of an agly- con for the equatorial position by the A(oxane) value from Eq. 2. The comparison is, however, handicapped by the lack of accurate data on the dependence of the A values on the solvent, even if the increase in the “appar- ent size” of the aglycon due to solvent might be small. For example, the A value ofthe OMegroup increases from 2.5 kJ.mol-I to 4.2 kJ.mol-’ ongoing from CC14 to water.’ It may be clearly seen from Table VII that the anomeric effect of HO and Me0 is higher in less-polar solvents, with dimethyl sulfox- ide being the only exception. These data also document how problematic it could be to characterize the anomeric effect, or the reverse anomeric effect, of a given group by a single universal value which would serve in all cases, regardless of the other ring substituents and the solvent.
In summary, experimental data on the isomeric abundances at anomeric equilibrium reveal that the preference for the axial position depends on several, interconnected factors which were clarified in surveys on carbohy- drate stereochemistry,2*80 and these provided a background for ensuing theo- retical studies. The elucidation of this relationship in complex carbohydrates is greatly facilitated by measurements on the simple derivatives of oxane, and qualitative trends have already been established. Table I1 illustrates several possibilities of the quantification of the energetic aspect of the ano- meric effect. The procedure most frequently used, based on Eq. I, suffers from the ambiguity of the A values for the oxane ring and by their presumed variation with solvent.
d. The Exo-anomeric Equilibria.- Because of the lessened barrier of in- ternal rotation around the exo-anomeric C - 0 bond, characterization of the conformational equilibrium of the type shown in Fig. 3 is very difficult, and complete data have not been reported so far. In contrast to the anomeric torsional angle 8, restricted to a narrow range of values, the exo-anomeric angle (D displays a much broader distribution in all six conformers in Fig. 3. Available information indicates that the (up, + sc) and (+ sc, + sc) orienta- tions are the most favored ones for the e and a isomers, respectively. Mea- sured and calculated dipole moments of alkoxy and alkylthio derivatives of oxane have been ~ o m p a r e d ~ , * , ~ ~ by using the coupling constants of the ano- meric proton for estimation of the abundance of the a and e forms. It was concluded* that, of the six possible conformations shown in Fig. 3, only the

70 IGOR TVAROSKA AND TOMAS BLEHA
(+ sc, + sc) conformation of the u isomer and either the (up, - sc) or (up, + sc) conformation of the equatorial isomer is present. Because the dipole mo- ment calculated for both of the latter conformations had the same value, their relative abundance could not be determined. Lemieux and co- w o r k e r ~ ~ ~ . ' ~ did not detect any appreciable amount of the (up, +sc) con- former in a number ofstructures examined, and concluded that the (up, - sc) and (up, + sc) rotamers are separated by over 8 W.mol- l. On the other hand, the changes in optical rotation for methyl 2,3-dideoxy-c~-~-glycero-pento- pyranoside and its 4,6-ethylidene acetal indicated the presence of the (+ sc, up) conformer for the axial isomer.6
Orientations about the exocyclic, C- 0 bond can be assessed by measuring the vicinal 13C-H coupling constant between the anomeric proton and the a-carbon atom of the aglycon (R) group, provided that the angular depen- dence is known. The vicinal, 13C - H coupling constants for the C - 0 - C - H moiety show a dependence on torsional angle analogous to that for the familiar Karplus equation, and several, fairly complete, Karplus-type curves are available. 16~85-87 Observed values, however, reflect a thermodynamically averaged conformation that does not usually correspond to a physically real one, and separation of the contributions from different conformers cannot be achieved without making questionable assumptions. Therefore, the in- terpretations based on these measurements are very qualitative, and may require revision. Nevertheless, they support the conclusion that (+ sc, + sc) and (up, -sc) are the favored conformations of the axial and equatorial forms, respectively. *6
Additional evidence on the selection of conformations by the exo-ano- meric effect is derived from the solid-state structures of It was earlier observed that the actual orientation of the anomeric alkoxyl group in pyranosides in the solid state corresponds to the (+sc, +sc) or (up, -sc) conformer, and thus proved that these conformers respectively represent the most stable axial and equatorial forms. As already noted, a particularly clear illustration of the operation of the exo-anomeric effect comes from the nonreducing disaccharide a,a-trehalose, in which the most stable orienta- tion about both exocyclic, C - 0 bonds corresponds to the (+sc, +sc) con- former. Analyses of carbohydrate ~ t r u c t u r e s ~ ~ . ~ ~ revealed regularities in the distribution of the torsional angle 0 that are consistent with a restriction of rotation about the exocyclic C-0 bond. The torsional angle for equatorial isomers varies from - 50 O to - 1 10 O, with a mean value of - 79.4 '. For the axial isomers, the range is 30 - 130°, with a mean value of 84.5 O (see Ref. 29).
(85) G. K. Hamer, F. Balm, N. Cyr, and A. S. Perlin, Can. J. Chem., 56 (1978) 3109-31 16. (86) H. Thagersen, Ph.D. Thesis, The Technical University of Denmark, Lyngby, 1977. (87) I. TvaroSka, M. Hricovini, and E. Petrikovi, Carbohydr. Res.. 189 (1989) 359-362.

ANOMERIC AND EXQANOMERIC EFFECTS 71
A subsequent, elaborate survey of 1 1 1 carbohydrate derivativesm confirmed that the axial glycosides occur only in the conformation corresponding to (+ sc, + sc), but the equatorial glycosides show a 3 : 1 distribution in the ratio of (up, -sc) to (up, up) conformers. The (up, up) conformer had eluded scrutiny in previous analyses, and the presence of the (+ sc, up), (+ sc, - sc), and (up, +sc) conformations has not been observed.
Currently, alkyloxy and alkylthio substituents are mainly used as flexible, polar aglycons on the oxane ring. The observation,88 however, that the azido group in the crystal structure of tri-0-acetyl-a-D-arabinopyranosyl a ide is oriented towards the ring-oxygen atom, with a torsion angle of 76 ’, indicates the general character of the exo-anomeric effect. Further experimental effort is needed in order more fully to determine the influence of solvent and pyranose-ring substituents on the exo-anomeric equilibrium.
2. Valence Geometry Parameters
The structural aspects of the anomeric effect manifested in the conforma- tional variation of the valence geometry parameters in hemiacetal and acetal moieties in pyranoses and pyranosides are receiving increased attention. The shortening of the anomeric C - 0 bond relative to its “standard” value was observed, and confirmed to be experimentally significant, some years This shortening is characteristic of any CX, (X = electronegative atom) grouping,w and is apparent, for example, in the structure of fluorometh- a n e ~ , ~ ’ where the carbon- fluorine bond-length decreases from 138.5 pm in CH,F through 135.8 pm in CH,F,, and 132.6 pm in CHF,, to 13 I .7 pm in CF, . In pyranose compounds, the anomeric carbon atom parallels the cen- tral atom, and the two adjacent electronegative atoms are the ring-oxygen atom and the first atom of the aglycon group.
Detailed examination of the available molecular geometry data of carbo- hydrates suggested that there are characteristic patterns of bond lengths and bond angles associated with particular conformation^.^^-^^^^-^^ These (88) P. Luger and H. Paulsen, Chem. Ber.. 107 (1974) 1579- 1589. (89) H. M. Berman, S. S. C. Chu, and G. A. Jeffrey, Science, 157 (1967) 1576- 1577. (90) H. A. Bent, Chem. Rev., 68 (1968) 587-648. (91) D. R. Lide, J. Am. Chem. SOC., 74 (1952) 3548-3552. (92) M. Sundaralingam, Biopolymers, 6 ( 1968) 189 -2 13. (93) G. A. Jeffrey, in L. E. Sutton and M. R. Truter (Us.), Molecular Structure by Diffraction
Methods, Chemical Society, Special Periodical Report, Vol. 6, London, 1978, pp. 183- 221.
(94) G. A. Jeffrey, J. A. Pople, J. S. Binkley, and S. Vishveshwara, J. Am. Chem. Soc., 100 (1978) 373-379.
(95) G. A. Jeffrey and J. H. Yates. J. Am. Chem. SOC., 101 (1979) 820-825. (96) G. A. Jeffrey and J. H. Yates, Carbohydr. Rex, 96 (1981) 205-213. (97) F. Longchambon, Ph.D. Thesis, University of Paris-Nord, Bobigny, France, 1984.

72 IGOR TVAROSKA AND TOMAS BLEHA
TABLE VIII Mean, Hemiacetal and Acetal Geometrieso in a- and /h-Pyraaoses, Methyl a- and
BD-Pyranosides, and Oliosaccharidesw
Methyl Oligo- Pyranos pyranosides saccharides Total
Parameter a B a B a B a B
Number of structures r(C-5 -0-5) r(0-5 -C- 1) r(c-1-0-1) r(0-1 -c-1) (C-5 - 0 - 5 4 - 1 ) (0-5 -C- 1 -0- 1) (C-1-0-1 -c-1) (0-5-C-1-0-1 -C-1)
22 12 10 13 5 22 37 47 144.0 143.4 143.3 143.3 144.0 143.7 143.8 143.5 142.5 142.7 141.4 142.9 142.0 142.0 142.2 142.4 139.8 139.2 139.9 138.1 140.6 139.4 140.0 139.0
142.4 143.0 143.2 143.9 142.6 143.6 114.0 112.1 113.3 112.4 113.9 112.4 113.8 112.3 111.4 107.1 112.4 107.6 11 1.9 107.6 111.8 107.5
113.5 113.7 114.0 115.7 113.6 115.0 69.5 -69.3 65.0 -73.6 73.7 -73.5 69.1 -72.2
Bond lengths in pm; bond angles and torsional angles in degrees.
patterns constitute a convincing manifestation of the anomeric effect, and can be readily discerned from the results of statistical treatment9' of pyra- noses, methyl pyranosides, and oligosaccharide structures summarized in Table VIII.
The most obvious feature of the experimental data on the a- anda-linkage in carbohydrates is a marked difference in the molecular geometry between the two configurations both in bond lengths and bond angles (see Table
143.8 H - 1
0 -5 c -2
113.6O
143 5 I 142.4
0 - 5
' 112 30 10750 115.0° H-1
FIG. 13.-The Mean Valence Geometry Parameters for Aldopyranosides (from Ref. 97).

ANOMERIC AND EXO-ANOMERIC EFFECTS 73
142.8 145.0
109.90 109.80 112.60
FIG. 14.-The Valence Geometry Parameters in Two Forms of D-Xylopyranosyl Fluoride Derivatives (from Ref. 98).
VIII). The ring 0 - 5 -C- 1 and anomeric C-1-0-1 bond lengths inp-D glyco- sides differ appreciably. The length of the 0-5 -C- l bond approaches the standard value of 142.5 pm, and the C-1-0-1 bond is much shorter ( 139.0 pm). The two external bond-lengths are longer than normal, and are almost equal. The bond angle at the anomeric carbon atom is 107.5 O , that is, less than tetrahedral, and of the two angles on the oxygen atom, the glyco- sidic C- l - 0- l -C angle is much the larger, l l 5 .O O . In the a-D-glycosides, the anomeric bond is still the shortest C-0 bond, but the difference is - 1 pm less. There is no difference between the two C-0-C bond angles, and the bond angle at the anomeric carbon atom is - 4" greater than in p-~-glyco- sides (see Fig. 13). The data on methyl pyranosides show similar features. In pyranoses, in contrast, the only significant differences observed between the a- and p-D anomers are in the bond angles at the ring-oxygen atom and the anomeric carbon atom, amounting to 1 14.0 and 1 1 1.4", respectively, in the a anomers, whereas they are smaller in the panomers, namely, 1 12.1 and 107.1 '. The data observed for oligosaccharides are less precise, because of the greater complexity of their structures, but they show a pattern similar to that of the methyl glycosides.
Several pyranosyl halides have been studied as acetylated or benzoylated derivatives. In both the fluorides and chlorides, the equatorial carbon- halogen bonds are shorter than the axial bonds, with the data for fluorides9* illustrated in Fig. 14. The C- F bond-lengths in both derivatives ofj?-D-xylo- pyranosyl fluoride are significantly shorter than the 143.2 pm found for the non-anomeric C - F bond in 1,3,4-tri-~-acetyl-2-deoxy-2-fluoro-~-xylo- p y r a n ~ s e . ~ ~ There is also a variation of 4.4 pm in the C- 1 - 0-5 bond-lengths.
Some 1 11 carbohydrate derivatives have been statistically treated,30 and coupling of the C - 0 bond-lengths and C - 0 - C bond angles to the orienta- tion about the exocyclic C-1-0-1 bond (exo-anomeric effect) was demon- strated. These results are given in Table IX. The differences in bond angles and bond lengths show a small but significant variation with the torsion
(98) G. Kothe, P. Luger, and H. Paulsen, Actu Crystallogr., Sect. B, 35 (1979) 2079-2087. (99) G. Kothe, P. Luger, and H. Paulsen, Actu Crystallogr., Sect. B, 32 (1976) 2710-2714.
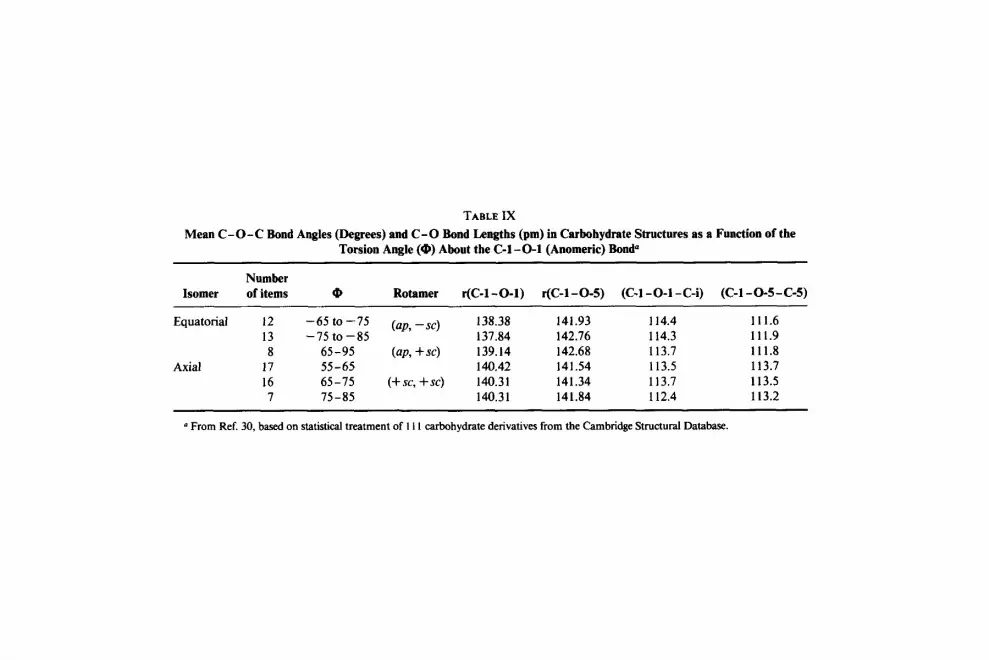
TABLE IX Mean C - 0 - C Bond Angles (Degrees) and C - 0 Bond Lengths (pm) in Carbohydrate Structures as a Function of the
Torsion Angle (@) About the C - 1 - 0 4 (Anomeric) Bond"
Number Isomer of items @ Rotamer r(C-1-0-1) r(C-1-0-5) (C-1-0-1 -C-i) (C-1-0-5-C-5)
Equatorial 12 -65 to -75 -Sc) 138.38 141.93 1 14.4 111.6 13 -75t0-85 137.84 142.76 114.3 1 1 1.9 8 65-95 (UP, +SC) 139.14 142.68 113.7 111.8
Axial 17 55-65 140.42 141.54 113.5 113.7 16 65-75 (+sc, +SC) 140.31 141.34 113.7 113.5 7 75-85 140.31 141.84 112.4 113.2
From Ref. 30, based on statistical treatment of I 1 I carbohydrate derivatives from the Cambridge Structural Database.

ANOMERIC AND EXO-ANOMERIC EFFECTS 75
angle @. In both equatorial groups, the - sc and + sc torsional minima of Q, are located at angles larger than - 60 and + 60 O , respectively.
IV. MOLECULAR ORBITAL CALCULATIONS OF THE ANOMERIC EFFECT
The description and understanding of the nature of stereoelectronic ef- fects is an appropriate field for the application of organic quantum chemis- try. Molecular orbital (MO) methods* can describe the electron distribution in molecules, and the changes in internal rotation. In principle, they give the total potential energy of individual conformers completely, without the necessity to correct for various “effects.” Quantum chemical calculations offer a deeper insight into the orbital interactions in the molecule, and reveal the factors responsible for the stabilization of any conformation.
The best description of the stereochemical behavior of an isolated mole- cule is achieved by nonempirical ab initio calculations with the sufficiently extended basis of the atomic orbitals, for example, 6-31G. However, an investigation of the conformational properties of a molecule having only two torsional angles, such as 0 and Q,, resulting in the energy map E(0, CP), represents the multiple (even one hundredfold) repetition of a routine calcu- lation of the energy. Therefore, in current practice, study is mostly confined to the less time-consuming methods either of ab initio methods with a minimal basis of orbitals (STO-3G), or to the semiempirical MO methods (PCILO, CND0/2, and MNDO). In both cases, a cautious approach is necessary, entailing careful comparison of computed properties for a given group of molecules with experimental data. It should be pointed out that there exists an inclination to consider ab initio results, even with the re- stricted or minimal basis set, as automatically superior to those of any semiempirical MO method. Calculations of a conformational energy for molecules exhibiting the anomeric effect (see later) give several examples of the deceptiveness of this claim.
An optimal choice of the quantum chemical method does not solve all of the problem, however. The isolated molecule calculations can be performed by the complete or partial optimization of the molecular geometry, or by assuming fixed bond-lengths and valence angles, with torsional angles as the only degrees of freedom. Although the optimization considerably extends the computing time, it is often unavoidable, owing to the relatively large
* MO = molecular orbital; STO-3G = the nonempirical (ub initio) method with the mini- mal basis using three Gaussian functions for one Slater atomic orbital: 4-3 IG, 6-3 IG, and so forth are the nonempirical methods with the extended basis, using Gaussian functions split into two groups; the semiempirical MO methods, CNDO/2 and MNDO (Complete Neglect, and Modified Neglect, of Differential Overlap, respectively); and PCILO, Pertur- bative Configurational Interaction using Localized Orbitals.

76 IGOR TVAROSKA AND TOMAS BLEHA
changes of structural parameters in glycosides that are attributable to inter- nal Finally, it is important to recognize that energy calcu- lations refer to isolated molecules, whereas conformational equilibria are generally measured in the liquid phase, where intermolecular interactions and solvent effects can be substantial. Therefore, the calculations should be supplemented by a procedure accounting for the influence of environment, before comparing with experimental data, especially those for aqueous solu- tions.
180
60
60 180 -60 0 (degrees)
FIG. 15.-The Conformational Energy Map of Dimethoxymethane,loo With Energy Con- tour in kJ.mol.-l. Two Conformations of Dimethoxymethane Corresponding to a and e forms of 2-Methoxyoxane Are Also Shown.

ANOMERIC AND EXO-ANOMERIC EFFECTS 77
1. Conformational Energies
Owing to the complexity of the internal motion of carbohydrate mole- cules, the elucidation of their conformational properties by MO calculations requires a lessening of dimensionality to manageable proportions. Several small acyclic molecules have therefore been used as models for ab initio or semiempirical MO studies on the structural segments of carbohydrates. On the whole, calculations reproduce all of the main structural trends and con- formational preferences observed experimentally in the crystal structures of carbohydrates and in solution.
a. Acyclic Model Compounds.- The anomeric effect has been studied in several simple acyclic molecules having the general formula YCH2X, where Y is OH, OCH, , SH, or SCH, , and X is an electronegative group, such as C1, F, 3 H , OCH, , SCH, , NH2, and NHf , by both ab initio and semiempirical methods. Special attention has been devoted to the - 0 - C - 0 - segment, and methanediol, methoxymethanol, and dimethoxymethane were used for the modelling of the acetal and hemiacetal moieties of carbohydrate
A complete description of the energy of these molecules as a function of one, or two, torsion angles 0 and Q, characterizing rotations about the C - 0 or C - S bonds is given by a one- or two-dimensional torsional potential. As an example, the CND0/2 calculated potential energy surface E(0, @) for dimethoxymethanelW is shown in Fig. 15. The relevant conformations of dimethoxymethane with torsional angles fixed at 60 and 180”, correspond- ing to the axial and equatorial isomers of an oxane acetal, are also illustrated. The possibility of internal rotation about two C - 0 bonds is responsible for the “double” presence of the anomeric effect, resulting in the stabilization of two conformations with the methyl groups on opposite sides of the -0-C- 0 - plane, (+ sc, + sc) and (- sc, - sc). The forms (- sc, + sc) and (+ sc, - sc), havingadjacent methyl groups on the same side ofthe -0-C-0- plane are energetically unfavorable, owing to the 1,3-diaxial type of steric interactions.
molecules.11,58,94,100-108
(100) I. TvaroSka and T. Bleha, J. Mol. Struct., 24 (1975) 249-259. (101) G. A. Jeffrey, J. A. Pople, and L. Radom, Carbohydr. Rex, 25 (1972) 117- 131. (102) G. A. Jeffrey, J. A. Pople, and L. Radom, Carbohydr. Rex, 38 (1974) 81-95. (103) S. Vishveshwara, Chem. Phys. Lett., 59 (1978) 30-32. (104) L. Radom, W. J. Hehre, and J. A. Pople, J. Am. Chem. Soc., 93 (1971) 289-300. (105) D. G. Gorenstein and D. Kar, J. Am. Chem. Soc., 99 (1977) 627-677. (106) C. van Alsenoy, L. Schafer, J. N. Scarsdale, and J. 0. Williams, J. Mol. Struct. Thee
(107) P. Bonnet, D. Rinaldi. and J. P. Marchal, J. Chem. Phys., 70 (1974) 298-302. (108) I. TvaroSka and T. Bleha, Collect. Czech. Chem. Commun., 45 (1980) 1883- 1895.
chem., 86(1981) 111-117.

78 IGOR TVAROSKA AND TOMAS BLEHA
TABLE X Calculated Relative Energies (kJ.moI-') of Stable
Conformers of R'OCHzORz with Respect to the (sc, sc) Rotamer Compared with Experimental Values for
Dimethox ymetbane ~ _ ___~ ~~
R1 Rz Method (sc, up) (up, up) References
H H STO-3G
4-31G
6-31G H CH, 4-31G CH, CH, STO-3G
4-21G
CNDO/2
PCILO MNDO Exp.
11.7 9.4
10.6" 19.7 18.4" 19.8 18.8 15.7 12.6 - 1.0
6.6 19.P 10.8 10.00 13.3" 4.6 4.9 3.8 5.0 5.2 7.1 5.0 8.1' 6.2d 6.3c
28.3 58 27.6 103 26.9 103 46.9 104 46.1 101 49.2 94 44.8 103 43.4 94 43.4 102 9.7 105
13.9 105 22.2 106 32.2 106 31.1 106
I06 15.8 LOO 14.9 105 10.9 107 10.9 108 17.2 I 1 14.2 64
109 18.8 107 12.4 107 13.0 107
a Optimization of bond lengthsor bond angles. Complete optimiza- Liquid phase. CSolution in 1 : 1 tion of geometry. 'Gas phase.
dimethoxymethane- heptane.
Furthermore, there are four minima on the conformational surface repre- senting the conformations of the type (up, sc), and one minimum corre- sponding to the (up, up) conformation. The map calculated for methanediol is similar, but, in contrast to that for dimethoxymethane, the (up, up) con- former of methanediol is found to be a local maximum.1oL A potential surface has also been reportedlo2 for methoxymethanol, a structural inter- mediate between dimethoxymethane and methanediol, with lessened sym- metry due to the presence of two different rotors. For example, conforma- tions (up, sc) and (sc, up) can be distinguished in this case, and both are only doubly degenerate.
A comparison of relative energies of dimethoxymethane, methanediol,

ANOMERIC AND EXO-ANOMERIC EFFECTS 79
TABLE XI Calculated Relative Energies (W.mol-l) of Stable
Conformers of ROCH,X with Respect to (sc) or (sc, sc) Orientations
R X Method AEl AEz References ~
H F STO-3G 12.7 58 13.1 95
4-3 1 G 26.0 95 23.4 111 26.8" 111
4-21G 14.9b 112 6-31G 25.5 95
18.8' 95 2 I .4" 95 21.8" 95
CNDO/2 10.0 11 H C1 STO-3G 18.6 58
20. I 95 4-31G 22.6 95 6-31G 21.3 95
CH, C1 4-31G 5.9 95 15.5" 95 12.1" 95
CNDO/2 7.7 I13 exp. 6.3-8.4 63
CH, CH, CNDO/2 -2.1 I 1 H NH, STO-3G - 1 . 1 13.8 114
4-21G -5.7b -1.0 112 4-31G -2.6 -8.8 104
-5.5" -9.1 114 CH, NH, CNDO/2 -3.1 I 1 CH, NH; CNDO/2 - 3.8 11
CH, F 4-31G 18.0 95
- * Optimization of bond lengths or bond angles. Complete optimi-
zation of geometry.
and methoxymethanol conformations calculated by different methods with experimental values is shown in Table X. It is seen that conformations of the (sc, sc) type of dimethoxymethane are predicted as preferred by both ab initio and semiempirical MO calculations, and the calculated energies are generally consistent with experimental evidence about the stability of con- formers. On the basis of electron diffraction studies,33 dipole moment, and Kerr constant measurements,64J09J lo it was established that (sc, sc) with both
(109) M. Sakakibara, Y. Yonemura, H. Matsuura, and H. Murata, J. MoI. Sfrucf., 66 (1980)
(1 10) R. J. W. Le Fevre, A. Sundaram, and R. K. Pierens, J. Chem. Soc., (1963) 479-488. 333-337.

80 IGOR TVAROSKA AND TOMAS BLEHA
angles identical, 66.3", is the most stable conformer. The data in Table X suggest that the anomeric stabilization by the two consecutive rotations about the C - 0 bonds is coupled, and not additive. The first rotation, from (up, up) to (up, sc) in dimethoxymethane, apparently leads to greater stabili- zation than the second rotation, from (up, sc) to (sc, sc).
The results of the energy calculations for the remaining ROCH,-X m0~ecU~eSll,58,95,103,104,111-114 are presented in Table XI. The relative energies are given by the difference between the (sc) and (up) rotamers for X = F, C1, and by the differences between the (sc, sc) and (sc, up) conformers (AEl), and between the (sc, sc) and (up, up) conformers (AE,) for X = CH,, NH,, and NHf . With the exception of the NH2 group, the preference for the sc orienta- tion is confirmed by the calculations, and it decreases in the order F > OH > C1> OCH, for the 4-3 1G basis set. The energy differences calculated by the semiempirical methods for substituted-dimethyl ethers CH,OCH, - X are lower than those estimated from the ub initio calculations on HOCH2- X molecules. In methyl and amino derivatives, the preference for the up orientation increases in the order NH; > CH, > NH2. All of these results are consistent with the experimentally observed anomeric prefer- ences in substituted oxane and in pyranoses (see Tables I - VI).
A few MO calculations have been reported for the acyclic molecules RSCH2X (see Table XII), where R is H or CH,, and X is F, C1, OH, SH, OCH,, and SCH,, used as the models of the thioacetal moiety in thio sugar^.^^^' 15-' l9 The calculated potential surfaces for HSCH2SH, CH,SCH,SCH, , HSCH,OH, and CH3SCH20CH,' 149' 16r1 l9 are, in their gross features, similar to those previously obtained for methanediol and dimeth- oxymethane. Table XI1 shows differences in energies of RSCH,X con- formers. In the case of the thioacetal segment 0 - C- s, the lowest energy is found for the (sc, sc) conformer. Energies of(sc, up) and (up, up) conformers, relative to (sc, sc), are lower than corresponding values in oxygen analogs. The modelling of the dithioacetal moiety is more complicated, and the results obtained are contradictory. It can be deduced from the data, however, that the preference for the sc orientation is lessened in the rotation about the
( 1 1 1 ) L. Radom, W. J. Hehre and J. A. Pople, J. Am. Chem. Soc., 94 (1972) 2371-2381. ( I 12) L. Schafer, C. van Alsenoy, J. 0. Williams, and J. N. Scarsdale, J. Mol. Struct. Theo-
(1 13) I. TvaroSka and T. Bleha, Tetrahedron Lett., (1975) 249-252. (1 14) P. Kaliannan, S. Vishveshwara, and V. S. R. Rao, J. Mol. Struct. Theochem., 105 (1983)
(1 15) S. Vishveshwara and V. S. R. Rao, Curbohydr. Rex, 104 (1982) 2 1 - 32. ( I 16) M. Ohsaku and H. Murata, J. Mol. Struct. Theochem., 85 (1981) 125- 131. ( I 17) I. TvaroSka, Chem. Zvesti, 38 (1984) 189- 197. ( 1 18) L. Nerskov-Lauritsen, F. S. Jerrgensen, and J. W. Jaroszewski, Curbohydr. Rex, 123
( I 19) I. TvaroSka, Collect. Czech. Chem. Commun., 49 (1984) 345-354.
chem., 76 (1981) 349-361.
359-374.
(1983) 1-11.

ANOMERIC AND EXO-ANOMERIC EFFECTS 81
TABLE XI1 Calculated Relative Energies (kJ.mol-') with Respect to (sc) or (sc, sc)
Orientations for the Stable Conformers of RSCH,X Molecules
H X Method (sc,up) (up,sc) (up,up) References
H F STO3G H C1 STO-3G H SH STO-3G
4-31G CNDO/2
CH, SH CNDO/2 CH, SCH, STO-3G
PClLO MNDO
H OH STO-3G
CH, OCH, PClLO MNDO
11.0 11.1 0.8 4.3' 0.5" 9.0
- 1.3 - 3.7
4.2 - 3.4
5.0" 2.8 4.9" 2.06 1.7 9.6
~~
58 58
4.1 115 9.2 115 3.5 115
19.0 115 5.0 116
5.5 11.3 I16 7.4 115
- 3.0 117 11.1 117,118
14.1 115 16.6 I15 13.9 115 5.5 119
17.5 119
a Complete optimization of geometry.. Optimization of bond lengths or bond angles.
C - S bond in comparison with that for the C- 0 bond. Experimental studies for these molecules120-123 gave the (sc, sc) conformation as the most stable in the solid state. The latter conformation also dominates in the liquid state, where additional conformations, (sc, up), (up, sc), and (up, up), are also present. A lessened stabilization of the sc with respect to the up orientation on rotation about the C-S bond in comparison with the C - 0 bond is supported by the dipole moment and measurements of the Kerr constant of dithioacetals. 124~125 These results suggest, however, that the preferred confor- mation of some dithioacetal derivatives in CC14 is the (sc, up).
In summary, it appears that MO methods successfully predict the energy of the conformers in substituted ethers and acetals. At the same time, it is interesting that the agreement of calculated results with the available experi- mental data (see Table X) seems to be better for the selected semiempirical methods than for some ub initio calculations, apparently owing to fortituous compensations.
(120) M. Ohsaku, Y. Shiro, and H. Murata, Bull. Chem. SOC. Jpn., 45 (1972) 113- 121. (121) M. Ohsaku, Bull. Chem. Soc. Jpn., 47 (1974) 965-975. (122) H. Matsuura, K. Kimura, and H. Murata, J. Mol. Struct., 64 (1980) 281 -284. (123) H. Matsuura, H. Murata, and M. Sakakibara, J. Mol. Sfruct.. 96 (1983) 267-275. (124) 0. Exner, V. JehliEka, and J. Firil, Collect. Czech. Chem. Commun., 37 (1972) 466-477. ( 1 25) A. N. Vereshchagin and 0. Exner, Collect. Czech. Chem. Commun.. 38 (1973) 690-696.

82 IGOR TVAROSKA AND TOMAS BLEHA
b. Fourier Component Analysis of Torsional Potentials.- In order to facilitate the interpretation of conformational equilibria for rotation about the C - 0 bond in compounds having the general formula CH30CH2X, it is useful to expand the potential function as a truncated Fourier expansion.111
v(e) = 0.5 vy(i - cos 8) + 0.5 v:(i - cos 28) (5)
(4)
+0.5 vg(i - cos 381,
v(e) = v,(e) + v,(e) + v3(e). or
For such an asymmetric structure as dimethoxymethane with one torsion angle fixed at 60 O , additional sine terms are necessary, in order to account for the lack of symmetry about 8 = 180" (see Ref. 94). The individual compo- nents V,(8), V,(8), and V3(8) of the total potential function V(8) can be identified with specific physical effects of similar periodicity. For example, the onefold term, V,(8) = 0.5 Vy( 1 - cos O), moves from a maximum value to a minimum value as 8 changes by 180". The same variation with torsion shows dipolar or steric interactions. The twofold term, V2(8), changes from a maximum to a minimum as 8 changes by 90". This periodicity frequently corresponds to the change of delocalization interactions. Finally, the three- fold term, V3(8), moves from a maximum value to a minimum value as it changes by 60". This is generally attributed to the intrinsic torsion potential.
The onefold, twofold, and threefold components all contribute to the location of the resultant maxima and minima at the torsional potential, as illustrated in Fig. 16 for dimethoxymethane and chloromethoxymethane.113 Values of Vg for both molecules are negative, suggesting a preference for the staggered XCOC up and sc conformations. The positive V(: indicates a pref- erence for the synperiplanar (sp, 0 = 0") conformation over the up confor- mation. This is consistent with a simple dipole-dipole argument which favors the sp conformer, with the opposed dipoles of segments C - 0 - C and C- X, over the up conformer having parallel dipoles (see Fig. 17). In chloro- methoxymethane, the Vy term is lowered, owing to steric interactions be- tween the methyl group and the chlorine atom in the sp position. The V: term, associated with delocalization interactions, is negative for chloro- methoxymethane, favoring orthogonal conformation (8 = 90') over up and sp conformations, and is small and positive for dimethoxymethane. The Fourier decomposition of the potential energy reveals that the dominance of the V, term in dimethoxymethane and that of the V, term in chloromethox- ymethane is responsible for the preference for the sc conformation. The torsional potential constants of Eq. 5 for substituted dimethyl ethers CH,0CH2X, determined from the potential energy calculated by the se- miempirica111J'7 and ub initio MO method^,^^.^'^ are summarized in Table

83 ANOMERIC AND EXO-ANOMERIC EFFECTS
Vp= 14.7
v;= 1.5
v;=-,.s
I I I 60" 120"
4
0
- 4
-8
- 12
3"
V t = - 6 . 6
V,"- - 6.4
I I I 60" 120" 180" 3
(b) Torsion angle (0) (4
FIG. 16.-Torsional Potential (V) in (a) Dimethoxymethane and (b) Chloromethoxymeth- ane, and Their Decomposition by Fourier Expansion, Eq. 5 (from Ref. 113).
XIII. A qualitative rationalization of the conformational preference in CH,OCH,X, where X = F, SCH,, CH, , NH,, and NHf , can be advanced along the same lines as for dimethoxymethane and chloromethoxymethane, with the contributions of the dipolar, delocalization, and intrinsic torsional terms.
c. Calculations on Cyclic Model Compounds.-In most cases, the MO calculations on simple acyclic molecules correctly describe the preference for the sc over the up orientation. Nevertheless, the acyclic models have, besides a lack of experimental data, several evident shortcomings in order fully to represent the conformational behavior of the cyclic carbohydrate structures. To clarify this behavior further, calculations have been carried

84 IGOR TVAROSKA AND TOMAS BLEHA
H3C---
.-.%
CH3 H'I H
f
H
FIG. 17.- Dipole- Dipole Interactions in Dimethoxymethane and in Chloromethoxy- methane.
out on 2-substituted oxane12,46J26-131 derivatives. In Figs. 18 and 19, the PCILO potential of rotation about the exocyclic C- 0 bond for two forms of 2 - m e t h o ~ y o x a n e ~ ~ ~ - ~ * ~ (MTHP) is compared with corresponding energies for dimethoxymethane calculated by the ub initkP4 and CND0/2 methods. loo
Figs. 18 and 19 display the existence of five minima for chair conformers of 2-methoxyoxane. For the u form, the lowest minimum appeared at 63", and the next one, having -6.2 kJ.mo1-I higher energy, at 152.2". These minima correspond to the (+ sc, + sc) and (+ sc, ap) conformers shown in Fig. 3. In the region ofthe third staggered position (+ sc, - sc), there is a broad maximum in Fig. 18. For the e form, Fig. 19 shows three minima; the lowest energy is found for the (up, -sc) conformer which lies 3.1 kJ.mol-' higher than the (+ sc, + sc) conformer of the u form of 2-methoxyoxane. Confor- mations (up, +sc) and (up, ap) are disfavored by 5.7 and 6.8 kJ.mol-', respectively, relative to (ub, -sc). Based on the energies of the individual conformers, the equilibrium ratios 70.8 : 6.0 : 19.9 : 2.0 : 1.3 have been calcu- latedL2' for the distribution of the (+ sc, + sc), (+ sc, up), (up, - sc), (up, + sc), and (up, up) conformers, respectively. The calculated u to e ratio of
(126) T. Koiarand I. TvaroSka, Theor. Chim. Actu, 53 (1979) 9-19. (127) I. TvaroSka and T. Koiir, J. Am. Chem. Soc., 102 (1980) 6929-6936. (128) I. TvaroSka and T. Koiar, Curbohydr. Rex, 90 (1981) 173- 185. (129) I. TvaroSka and T. Koiar, Znt. J. Quantum Chem., 23 (1983) 765-778. (130) I. TvaroSka and T. KoiBr, J. Mol. Srruct. Theochem., 123 (1985) 141 - 154. (131) L. Guibe, J. Augk, S. David, and 0. Eisenstein, J. Chem. Phys., 58 (1973) 5579-5583.

ANOMERIC AND EXO-ANOMERIC EFFECTS 85
TABLE XI11 Calculated Rotational Potential Constants (kJ.mol-') Describing Internal Rotation About the C - 0 Bond' in
Substituted Dimethyl Ethers, XCH,OCH,
X Method q q V! References
OCH, CNDO/2
F CNDO/2
C1 CNDO/2
SCH, X I L O NH, CND0/2 CH, CNDO/2 NH: CNDO/2
4-31G
4-31G
4-3 IG
14.7 1.5 -3.8 113 20.0 -20.0 - 10.0 103 13.3 0.0 -4.4 1 1 6.7 -17.4 -16.3 103 3.6 -6.6 -6.4 1 1
38.8 -46.7 -28.7 103 4.0 -9.3 -7.1 117
-9.1 6.3 -2.7 11 -6.2 0.1 -3.8 11 10.0 5.0 -6.2 I 1
a In OCH, and SCH, derivatives, the methyl group is in the ap posi- tion relative to the C - 0 bond.
76.8 : 23.2% is in agreement with 77-83% ofthe a form of 2-methoxyoxane derivatives measured in nonpolar solvents (see Tables I1 and 111). The pre- ponderance of conformations having the methyl group in the sc position relative to the ring-oxygen atom is a clear demonstration of the working of the exo-anomeric effect. This preference is not limited to the chair forms, but has also been found from computation in all pyranose ring forms of 2-meth- o x y o ~ a n e . ~ ~ ~ , ~ ~ ~
The MO calculations of the conformational equilibria of 2-chloro-oxane and 2-fluoro-oxane have been camed The ab initio STO-3G calcu- lations for 2-chloro-o~ane~~~ confirmed the stabilization of the a position of the C1 atom relative to the e one. An energy difference of 5.0 kJ.mol-' is found if the C - C1 bond-length 177 pm is assumed for both conformers, and 15.5 kJ.mol-' ifthe axial C-Cl bond is lengthened to 182 pm. These results are in qualitative agreement with the energy difference of 9 kJ.mol-l ob- served in the pure The axial preference of both the 2-fluoro- and 2-chloro-oxane is also corroborated by the results ofthe PCILO and MNDO methods.I2 For the fluoro derivative, both methods predict an energy differ- ence of 6.7 kJ.mol-'. For the chloro derivative, the PCILO and MNDO methods give energies of 10.1 kJ.mo1-' and 13.2 kJ.mol-', respectively. A PCILO calculationM for 2-carbamoyloxane showed that the conformation having the CONH, group in the e position is preferred over the a position. The calculated energy difference of - 7 kJ.mol-' indicates a strong, reverse anomeric effect.

86
I - E 7 Y
W 9
u
30.C
20.1
1O.L
IGOR TVAROSKA AND TOMAS BLEHA
I I I I I I I I I I I I I I I I I I I I I \ I
6 'CH3
I i a
I I I I
@ (degrees)
FIG. 18.-Torsional Potential of the Exocyclic C - 0 Bond in the Axial Form of 2-Methox- yoxane Calculated by the KILO Method (Full Line) and Corresponding Potential in Dimeth- oxymethane (Dashed Line) Calculated by the CNDO/2 Method (Curve a)lw and by the ab initio Method (Curve b).%

ANOMERIC AND EXOANOMERIC EFFECTS 81
I I I I I
60 120 180 240 300 @ (degrees)
FIG. I9.-The Same as in Fig. 18, but for the Equatorial Form of 2-Methoxyoxane.

88 IGOR TVAROSKA AND TOMAS BLEHA
As already noted, a,&-trehalose assumes, in the solid state2’ and in solu- t i ~ n , ~ ~ the sc conformations of the two glycosidic C - 0 bonds. The confor- mational properties of the three trehaloses were studied132 by using 2-(oxan- 2-y1oxy)oxane as a model in three isomeric forms (a, a), (e, e), and (a, e). In these compounds, the anomeric and exo-anomeric effects influence the properties of four C - 0 bonds in the C-5 - 0 - 5 -C- 1 - 0 - 1 -C-1’-0-5’-C-5’ moiety. The energy of the rotation about the C-1-0-1 or 0-1 -C-1’ bond (torsional angles and a’) depends on the orientation of the adjacent C-0 bonds (8 and 0’ angles), and resembles the potential profiles calculated for the a and e forms of 2-methoxyoxane. The preferred form is the (a, a), the next is the (a, e) form, and the (e, e) form has the highest energy. The most stable conformer ofall three forms is the conformer ofthe a, a form, where all four C - 0 bonds are in the + sc position.
d. Saccharides.- In a few cases, the anomeric equilibrium of monosac- charides has been treated by MO computation. The majority of calculation^^^^-^^^ on isolated D-glucopyranose incorrectly predict that the p anomer is the preferred form, and the energy difference between the anomers lies in the interval of 2 - 38 kJ.mol-’, according to the applied MO method. The PCILO ~alculated’~’ a : panomeric ratio for isolated D-glucopyranose of 76 : 24 differs considerably from these values, and is close to the ratio of the a : e forms of 77 : 23 for isolated 2-methoxyoxane (see earlier).
Similar PCILO calculations on methyl a- and p-D-glucopyranoside estab- l i ~ h e d l ~ ~ that the a anomer is the more stable, and the calculated energy difference is -4.2 kl.mol-’. A preference for the sc orientation about the C- 1 - 0 - 1 bond was suggested by calculations for both anomers. The ap arrangements about the anomeric C- 1 -0- 1 bond in the a and Panomers are 4.0 and 4.6 kJ.mo1- ’, respectively, higher in energy than the corresponding sc position.
2. The Solvent Effect
It is important to recognize that the energy calculations discussed refer to isolated molecules in the gas phase, whereas experimental values are mea-
(132) I. TvaroSka and i. Vaclavik, Carbohydr. Rex, 170 (1986) 137- 149. (1 33) Yu. A. Zhdanov, V. I. Minkin, Yu. A. Ostroumov, and G. N. Dorofeenko, Curbohydr.
(134) W. B. Neely,J. Med. Chem., 12(1969) 16-17. (135) N. Cyr, A. S. Perlin, and M. A. Whitehead, Can. J. Chem., 50 (1972) 814-820. ( 1 36) S. Melberg, K. Rasmussen, R. Scordamaglia, and C. Tosi, Curbohydr. Rex, 76 ( 1 979)
(137) I. TvaroSka and T. KoiBr, Theor. Chim. Actu, 70 (1986) 99- 114. (138) I. TvaroSka and T. KoiBr, Chem. Papers, 41 (1987) 501 -510.
Rex, 7 (1968) 156- 160.
23-37.

ANOMERIC AND EXO-ANOMERIC EFFECTS 89
sured in solution where the effect of the medium can be substantial. In fact, significant differences in conformer population have been observed for oxane derivatives by n.m.r. in various solvents (see Section 111). As already mentioned, >77% of 2-methoxyoxane exists in the a form in nonpolar solvents, but only 52% in water (see Table VI).
There exist several approaches for theoretical prediction of the effect of the solvent on conf~rma t ion . '~~J~ One possible procedure is based on inclusion of the solute and several solvent molecules in "supermolecule" quantum chemical calculations. Such an approach might be useful in providing infor- mation about optimal solvation sites, but the evaluation of the overall energy of solvation can hardly be obtained in this way, because of the necessity to include many solvent molecules and to perform a complete energy minimi- zation. An alternative approach is to treat the solvent as a dielectric contin- uum. Computer simulations of a solution by the Monte Carlo or molecular dynamic methods, explicitly including the carbohydrate molecule and many solvent molecules, could present the ultimate treatment of solvent effect. However, at present, such an approach to accurate evaluation of solvation energies is still far too expensive, owing to the complexity and high flexibility of carbohydrates.
A quite reliable estimate of the solvent effect results from theories that combine micro- and macro-scopic parameters of solute and solvent. For example, in the solvophobic theory,14' the energy of a solute molecule in solvent, EWln , is given as the sum of isolated molecule energy, E,, and the solvation term, E,, . The latter term encompasses the energy of cavity for- mation in the solvent to accommodate the solute, E,,, and the energy of subsequent solvent - solute interactions, Ei,. The interaction part is com- posed of the energy of dispersion, Edisp, and electrostatic, EeM , interactions. The final expression for E,, can be written as
E-1, = Ei, + E, + EeM + E w . (7)
Calculations of the effect of the solvent upon the conformational proper- ties of dimethoxymethaneLo8 based on Eq. 7 indicate that, in highly polar solvents, the ap orientation about the C - 0 bond might even be preferred. For example, the (sc, sc) conformation still prevails when parameters of CCl, are assumed in Eq. 7, with only the energy difference between (sc, sc) and (ap, sc) lowered by 2 kJ.mol-' in comparison to the isolated molecule. In water,
( 1 39) B. Pullman (Ed.), Environmental Efects on Molecular Structure and Properties. Jerusa-
(140) M. Berndt and J. S. Kwiatkowski, in G. Naray-Szabo (Ed.), Theoretical Chemistry of
(141) 0. Sinanoglu in B. Pullman (Ed.), Molecular Association in Biology, Academic Press,
lem Symp. Quantum Chem. Biochem., Vol. 8, Reidel, Dordrecht, 1976.
Biological Systems. Elsevier, Amsterdam, 1986, pp. 349 -422.
New York, 1976, pp. 427-445.

90 IGOR TVAROSKA AND TOMAS BLEHA
however, the (up, up) conformation has been found as the minimum-energy structure from calculations.
The analysis of individual terms of solvation energy reveal that electro- static interactions constitute the dominant term in the solvation energy of dimethoxymethane. The lessening of the preference for the sc position with rising polarity of the medium is supported by the results of dielectric mea- surements of dimethoxymethane in the gaseous and liquid phases, com- bined, by CNDO/2 calculations.107 It was found that the Gibbs energy dif- ference of the (up, up) and (up, sc) conformations with respect to (sc, sc) gradually decreases in the succession: gaseous phase, 1: 1 (v/v) dimethoxymethane- heptane, and neat liquid (see Table X). Similarly, as in dimethoxymethane, the effect of polar solvents brings about stabilization of the up position in thio analogs of dimeth0xymethane.l l 7* l I9
The extent to which this kind of calculation is able to predict the effect of the solvent on conformational properties of carbohydrates has been thor- oughly tested on 2-substituted oxane derivatives, 12~127 ~-glucopyranose,~~~ and methyl a- and P-D-glucopyranoside. 138 In the model applied,'27 the cavity term in Eq. 7 is based on an expression taken from the Scaled Particle Theory,'42 and the electrostatic term is calculated according to the reaction field theory.143 The dispersion term takes into account both attractive and repulsive nonbonding interactions by using a combination of London dis- persion energy and Born-type rep~1sion.l~~
The effect of the solvent on the abundance of the conformers of 2-meth- oxyoxane is demonstrated in Table XIV, where molar fractions of the axial form are compared with available experimental data already shown in Table V1. For a number of solvents, the agreement is remarkably good. Although the results indicate a decreased abundance of the a form of 2-methoxyoxane with increase in the dielectric constant of the solvent, the dependence is not a simple one. The calculations also reproduce such subtle factors as the pro- nounced effect of chloroform when compared with other solvents of similar polarity and, conversely, a relatively weak effect of dimethyl sulfoxide in comparison to less-polar solvents. The analysis of the role of individual solvation energy terms in the total energy suggests that the conformationally most important term is the contribution of electrostatic interactions that stabilize the up conformations. Conversely, the dispersion term shows only a slight conformational dependence.
The calculated abundance of the five conformers of 2-methoxyoxane quantitatively describes, for the first time, the exo-anomeric equilibrium about the C - 0 bond for the a and e forms in various solvents. At present,
(142) R. A. Pierrotti, Chem. Rev., 76 (1976) 717-726. (143) R. J. Abraham and E. Bretschneider, in W. J. Onville-Thomas (Ed.), ZnternalRotation in
Molecules, Academic Press, London, 1974, pp. 48 1 - 584.

TABLE XIV Calculated Molar Compositions (in %) of Conformers of 2-Methoxyoxane (at 298.2 K), and Comparison of the Calculated and Experimental Molar Compositions, x,, of the Axial Form in the Isolated State and in Solution
~~
Solvent E" (+ SC, + sc) (+ sc, UP) (+ sc, - sc) (UP, - sc) (UP, UP) x,(calc.) x.(exp.Y
Neat 2-methoxyoxane 1 ,4-Dioxane Carbon tetrachloride Benzene Carbon disulfide Chloroform Fluorobenzene Oxolane Octanol Pyridine Acetone Ethan o I Methanol Acetonitrile Dimethyl sulfoxide Water
70.8 2.21 67.4 2.24 69.7 2.28 69.6 2.64 68.8 4.43 63.4 5.42 66.5 7.58 64.4
10.34 67.7 12.40 61.5 20.70 62.6 24.55 60.7 32.70 55.9 37.50 58.7 46.68 61.2 78.30 35.8
6.0 6.6 6.2 6.3 6.5 7.3 6.8 1.3 6.7 7.7 7.6 8.0 8.9 8.4 8.0
12.0
19.9 21.8 20.3 20.2 20.7 24.2 22.3 23.3 21.4 25.1 24.2 25.1 27.4 26.0 24.7 34.0
2.0 2.4 2.2 2.3 2.3 2.7 2.4 2.7 1.8 2.9 2.9 3.1 3.6 3.3 3.1 5.7
1.3 76.8 1.8 74.0 1.6 75.9 1.6 75.9 1.7 75.3 2.4 70.7 2.0 73.3 2.3 71.7 2.4 74.4 2.8 69.2 2.7 70.2 3.1 68.7 4.2 64.8 3.6 67. I 3.0 69.2
12.5 47.8
82,83 82 80
71,78
72
69 65,68
74 52
~~
a 6 = dielectric constant. * From Refs. 6 and 8.

92 IGOR TVAROSKA AND TOMAS BLEHA
such detailed data on the conformer population are not available from experiments. The results given in Table XIV show that, for the u form, the abundance of the (+sc, ap) conformer in nonpolar solvents is <8%. It increases with increase in the solvent polarity, and reaches a maximum of 12% in water, which corresponds to - 26% of the population of the u form. Similarly, the abundance of the (up, up) conformer in the e form does not exceed 3% in nonpolar solvents, and amounts to 12.5% in water. The latter figure means that, in water, the (up, up) conformation comprises - 24% of the e form.
The preferences for the a form in 2-chloro-o~ane'~ 2-fluoro-o~ane,'~ and D-glucopyranose, '37 calculated by the method mentioned127 in various sol- vents, are presented in Table XV. In all of these compounds, the axial preference decreases with increase in the polarity of the solvent as well. In water, the e form is favored for 2-fluoro-oxane and D-glucopyranose. The values calculated for 2-chloro-oxane satisfactorily reproduce the observed axial preference of 93-96% in acetonitrile and acetone.38 The anomeric ratio calculated for D-glucopyranose (see Table XV) illustrates how compar- ison of the theoretical values referring to the isolated molecules with experi- mental data in aqueous solution might be misleading. Although, for the isolated molecule, the PCILO calculations predict 76% of the axial form, the correction for solvent brings the data in Table XV to very satisfactory agree- ment with the composition in pyridine (45% of the a form), in dimethyl sulfoxide (44%), and in water (32 - 37%). Similar results of solvent polarity have been reported for methyl a- and &D-gluc~pyranoside.'~~ Whereas, for an isolated molecule, the energy difference between the an- omers is - 4.2 kJ.mol-', for a methanol solution, the calculation predicts a
TABLE XV The Calculated Preference of the (I Form of D-Glucopyranose,
2-Chlorooxane, and 2-Fluoraaxane in Various solvent^^^^^^
Solvent D-
Glucopyranose 2-Chlorooxane 2-Fluorooxane
Isolated molecule 1,4-Dioxane Carbon
Chloroform Pyridine Acetone Methanol Acetonitrile Dimethyl sulfoxide Water
tetrachloride
76.0 68.5
71.4 62.5 48.9 49.9 37.3 41.9 45.7 32.2
98.3 97.8
98.0 96.9 96.3 96.5 94.1 95.1 95.9 78.4
94.0 90.6
91.7 87.5 85.6 86.1 77.6 81.0 84.1 37.0

ANOMERIC AND EXO-ANOMERIC EFFECTS 93
1.3 kJ,mol-’ energy differen~e.’~~ This corresponds to a 6390 preference for the a anomer, in agreement with the experimental value26*27 of 67%.
Calculation of the effect of the solvent on the abundance of the 2-(oxan-2- y1oxy)oxane conf~rmers’~~ revealed that the solution behavior of the (a, a) form is markedly different from that of other saccharides where large differ- ences in the solvent effect of aqueous and nonaqueous solution on the equilibrium around the glycosidic linkage have been f o ~ n d . ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ” ~ ~ ~ ~ The calculated population of the most stable conformer is 96.5%, in both the isolated molecule and in solution. The equilibrium composition of con- formers in the other two forms, (a, e) and (e, e), depends on the solvent, and the solvent - solute electrostatic interactions (Eq. 7) are mainly responsible for the shift of equilibrium in s01ution.l~~
The influence of the ring and anomeric oxygen hydration on the axial preference of an aglycon has been studied for the five most stable conformers of 2-methoxyo~ane.~~~ Both the intramolecular structure and the energy of a supermolecule formed with 2-methoxyoxane and water molecules were cal- culated by the PCILO method. It was concluded that the oxygen atoms in the acetal segment act as a weak monobase in water, and that hydrogen bonding does not influence the axial preference of the methoxyl group.
3. The Anomeric Effect Energy from MO Calculations
The energy difference provided by MO calculations, with or without the inclusion of solvent, serves as a direct measure of the anomeric energy, AE(AE3), when its “absolute” definition by Eq. 4 is used. However, the more-frequent use of a “relative” definition by Gibbs energy difference in Eq. I warrant an attempt to recalculate the AE(AE3) data to the values AG(AE 1). Such a procedure is, of necessity, an approximation, because the assumption that AG: = AE(AE3) neglects the entropy and volume changes of conformers owing to absence of suitable information, and the cyclohex- ane-based and solvent-independent A values must be used.
The Gibbs energies, AG(AE 1 ), estimated by the foregoing procedure from PCILO calculations taking s01vent’~J~~J~’ into account for three oxane de- rivatives and D-glucopyranose, are listed in Table XVI. The constant values of A(oxane), namely, 3.2, 1.6,6.5, and 6.5 kJ.mo1-l corrected for the oxane ring from the A-values by Eq. 2 were used for the C1, F, CH3, and OH groups, respectively. The dependence of the anomeric effect upon the solvent follows the trends in equilibrium composition discussed in the previous Sections. The anomeric effect is maximized in the isolated molecules; the Gibbs en- ergy, AG(AE l), gradually decreases in more-polar solvents. The major dif- ference appears between the effect in organic solvents and in water, and, in
(144) I . TvaroSka, Biopolymers, 21 (1982) 1887- 1897. (145) I. TvaroSka, Biopolymers, 23 (1984) 1951 - 1960.

94 IGOR TVAROSKA AND TOMAS BLEHA
TABLE XVI Solvent Dependence of the Anomeric Effect, AG(AE,), Recalculated from AE(AE,), of the
Chlorine, Fluorine, Methoxyl, and Hydroxyl Groupsa
Solvent Clb Fb OCH,b OH'
Isolatedmolecule 13.3 8.3 9.5 9.4 1 ,4-Dioxane 12.6 7.2 9.1 8.4 Carbon
tetrachloride 12.9 7.6 9.4 8.8 Carbondisulfide 12.6 7.2 9.3 8.5 Chloroform 11.7 6.4 8.7 7.8 F'yridine 11.3 6.0 8.5 6.4 Acetone 11.4 6.1 8.6 6.5 Methanol 10.1 4.7 8.0 5.2 Acetonitrile 10.6 5.2 8.3 5.7 Dimethyl sulfoxide 11.0 5.7 8.4 6.1 Water 6.4 0.3 6.3 4.6
*Based on the constant A(oxane) values of 3.2, 1.6, 6.5, and 6.5 kJ.mol- for the c1, F, OCH, , and OH groups, respec- tively. In 2-substituted oxane. In ~-glucopyranose.
the isolated molecule, the Gibbs energy of the anomeric effect decreases in the order of C1> OCH, = OH > F.
In the exo-anomeric effect, identification of its value from MO calcula- tions is slightly more complicated when using the definition of Eq. 4. Be- cause there exist two energetically nonequivalent sc positions for the tor- sional angle @, in principle both could be used in Eq. 4. In practice, however, one of the sc positions is disfavored by steric interactions with the rest of molecule; for example, + sc in the e form or - sc in the u form of 2-methox- yoxane (see Fig. 3). The energy differences between conformations calcu- lated for dimethoxymethane and related acyclic compounds," for 2-meth- oxyoxane conformers of different pyranoid-ring shapes, 127~1u) and for the model compounds of the three t r e h a l o s e ~ ~ ~ ~ make it possible to estimate quantitatively the magnitude of the exo-anomeric effect in various solvents. These estimates seem to be the only data on the energy of the exo-anomeric effect defined by Eq. 4 thus far available in the literature. Its magnitude, AE(EAE3), calculated as the difference in energies of the conformers having the aglycon group in the sc and the up position (for the angle @) varies in the isolated molecules between 5 and 10 kJ.mol-'. The exo-anomeric energy, AE(EAE3), may exceed the anomeric-effect energy, AE(AE3), when rota- tion by the angle @ brings about a larger stabilization of the sc position than rotation by the angle 8.
For the transition from AE(EAE3) values to Gibbs energy AG(EAEl), a reference molecule has to be selected that should serve as a benchmark to

ANOMERIC AND EXO-ANOMERIC EFFECTS 95
assess steric interactions. 2-Ethyloxane seems to be the natural choice, but the detailed conformational energetics of this molecule is not available. A simple acyclic model, ethyl methyl ether, therefore has to be used in order to estimate the "A" values appropriate for the exo-anomeric effect.
The ap orientation is favored in ethyl methyl ether'&; from its 80% popu- lation in the gas phase, as determined by electron diffraction, this implies that the Gibbs energy difference between the sc and ap rotamers is 5.1 kJ.mol-'. This value represents the steric preference ofthe methyl group for the ap orientation about the C - 0 bond, and serves as a rough estimate of an A-value for the rotation about the exocyclic, anomeric C -0 bond. Such an assumption neglects the differences in the orientational equilibrium about the C - 0 bond in cyclic and acyclic ethers, but may be used as a first approximation. The Gibbs energy ofthe exo-anomeric effect, AG(EAE 1 ), is, therefore, defined as the sum of the energy difference for the sc and ap rotamers about the exocyclic C - 0 bond, AE(EAE3), and the A-value for the corresponding equilibrium in ethyl methyl ether. For example, the energy data for 2-methoxyoxane and the trehalose models analyzed earlier in this Section, combined with the A-value of 5.1 kJ.mol-', give an estimate of the exo-anomeric effect in isolated molecules of between 10 and 15 kJ.mol-'.
For solutions, the data for the various ring-forms of the axial 2-methox- yoxane given in Table XVII show that AG(EAE1) for the exo-anomeric effect is minimal in water, and higher in less-polar media. The results for the model of a,a-trehalose, namely, the (a, a) form of 2-(oxan-2-yloxy)oxane, indicate a different behavior. 132 The magnitude of the exo-anomeric effect calculated for this compound is - 15 kJ.mol-', and is not sensitive to solva-
TABLE XVII The Effect of Solvent upon the Exo-anomeric Effect" (W.mol-L),
AWEAE,), for Selected Ring-Shapes for the Axial Form of 2-Metho~yoxane'~
Solvent 'CI 'C, 'So "S, U B B,
Isolated molecule Carbon tetrachloride Chloroform Acetone Methanol Acetonitrile Dimethyl sulfoxide Water
11.2 12.1 14.0 11.1 14.2 11.8 11.1 11.7 13.5 10.9 13.9 11.4 10.4 11.1 12.8 9.7 13.1 10.7 10.3 10.8 12.6 9.8 12.8 10.3 9.7 10.1 11.8 9.3 11.9 9.4 9.9 10.3 12.1 9.6 12.2 9.7
10.2 10.6 12.4 9.9 12.5 10.0 7.8 8.0 9.7 7.5 9.4 7.1
Based on an estimate of the A value of 5. I kJ.mol-' from ethyl methyl ether.
(146) K. Oyanagi and K. Kuchitsu, Bull. Chem. SOC. Jpn., 51 (1978) 2237-2242.

96 IGOR TVAROSKA AND TOMAS BLEHA
TABLE XVIII Observed and Calculated Bond Lengths (pm) and Bond Angles (degrees) for Substituted Dimethyl
Ethers (CH,OCH,-X)
X 0 0 Method r(C-0) r(0-C) r(C-X) (C-0-C) (0-C-X) References
F sc
c1 sc
a p
NH, up sc
a p ap
NH; up sc
U P U P
CH, up sc
a p a p
SCH, a p sc
a p UP sc sc
Exp. 4-31G 4-21G CNDO/2 MNDO 4-31G CNDO/2 MNDO Exp. 4-31G 4-21G" MNDO
MNDO CND0/2 MNDO CNDO/2 MNDO CNDO/2 MNDO CNDO/2 MNDO CNDO/2 MNDO CNDO/2 MNDO MNDO MNDO MNDO
4-31G
142.4 144.7 145.2 137.0 140.6 143.7 137.0 140.3 142.1 144.9 145.0 140.9 143.9 140.6 137.9 140.1 138.0 140.1 136.9 142.1 136.4 141.9 138.2 140.3 138. I 140.3 i 39.2 138.9 138.5
136.2 139.6 139.8 137.3 139.8 140.5 137.4 140.4 136.2 140.5 139.9 137.2 141.0 138.5 137.1 140.6 137.0 140.7 137.6 136.4 138.0 137.6 137.0 140.2 137.1 140.1 140.5 140.6 140.7
138.5 139.5 140.7 134.9 135.3 137.6 135.2 134.7 182.2 188.6 184.4 183.2 183.7 181.1 141.2 146.5 141.0 146.3 144.1 156.6 144.6 154.7 146.6 154.8 146.7 154.4 175.0 176.1 176.2
113.5
115.2 104.6 122.2
100.5 120.0 1 14.0
116.3 122.1
120.3 101.7 121.4 107.7 120.4 110.9 123.4 100. 1 122.1 106.0 120.7 107.7 119.6 120.1 1 19.6 121.4
111.3
110.8 I 10.6 108.6
106.0 105.0 112.9
112.5 112.0
107.3 113.4 110.4 117.1 109.3 112.1 106.0 105.7 103.5 116.7 1 1 1.9 110.4 109.7 110.4 105.9 114.5
62 95
147 1 1 11 95 1 1 1 1 62 95
147 1 1 95 1 1 11 1 1 1 1 11 11 11 11 11 11 11 1 1 1 1
119 119 119
a Chlonne atom basis set contafns d functions.
tion. The considerable values of the exo-anomeric effect mentioned document its important role in conformational equilibria of glycosidically linked compounds containing pyranoid rings. These results also explain why the oligo- and poly-saccharides in the solid state28 show a distinct preference for the sc position.
(147) J. E. B o w , M. Altman, F. R. Cordell, and Y. Dai, J. Mol. Struct. THEOCHEM., 94
(148) I. TvaroSka and T. Bleha, Can. J. Chem., 57 (1979) 424-435. (1983) 373-390.

TABLE XIX Comparison of the Calculated Bond Lengths (pm) and Bond Angles (degrees) with Experimental Values for Dimethoxymethane
Conformer Method r(C-5-0-5) r ( 0 - 5 4 - 1 ) r(C-1-0-1) r(0-1 -C-i) a(6) a(]) /3 References
(sc, sc) Exp. Exp." 4-3 lG 4-21G CNDO/2 PCILO MNDO
(an sc) Expa 4-31C 4-21G CNDO/2 K I L O MNDO
(UP, UP) 4-3 1 C 4-21G CNDO/2 K I L O MNDO
140.3 143.5 144.4 144.9 137.9 138.5 140.5 143.3 143.4 144.2
138.4 140.2 143.5 144.4
138.4 140.3
140.3 141.6 142.3 142.2 137.6 138.4 139.9 142.8 142.5 143.2 137.9 138.5 140.5 140.6 142.0 137.8 138.4 140.0
140.3 140.4 142.3 142.2 137.6 138.4 139.9 138.3 140.0 140.9 137.7 138.4 139.3 140.6 142.0 137.8 138.4 140.0
140.3 143.1 144.4 144.9 137.9 138.5 140.5 142.7 144.3 144.8
138.5 140.6 140.6 144.4
138.4 140.3
1 14.2 113.4 I 15.9 114.5
106.3 123.5 111.4 115.8 114.3
105.5 120.0 143.5 114.4
105.8 119.3
112.6 114.2 33 112.3 113.1 94 113.9 115.9 94 112.4 114.5 I12 114.9 I48 113.2 106.3 I 1 113.6 123.5 I I 107.9 113.4 94 110.9 116.1 94 109.5 114.9 112 110.0 148 110.9 107.5 I 1 108.2 123.4 I I
94 105.9 114.0 112 104.6 148 105.8 105.8 I ! 103.1 119.3 1 1
a Mean values from X-ray structures of methyl aldopyranosides.

98 IGOR TVAROSKA AND TOMAS BLEHA
4. Coupling of Bond Lengths and Bond Angles to Torsional Angles
The analysis of a large body of structural information concerning the detailed geometry of the acetal and related groups in carbohydrates (see Section 111) established that there are small but significant differences in the molecular geometry, depending on the orientation of these groups. The characteristic patterns of the variations in bond lengths and valence angles are correctly reproduced by both ab initio and semiempirical MO calcula- tions. Most of the available results for substituted ethers CH,OCH,X are summarized in Tables XVIII and XIX, and those for 2-substituted oxane derivatives are given in Table XX.
The calculated bond lengths and angles shown in Tables XVIII and XIX agree rather well with experiment, although some differences are apparent. Upon closer inspection, it is suggested that neither method can be preferred in the prediction of absolute values of bond lengths and bond angles. For example, ab initio calculations at the 4-31G or 4-21G level predict C - 0 bond lengths that are -2-4 pm longer, and the semiempirical PCILO method, - 2 pm shorter, than those found experimentally. At the same time, the PCILO calculated variations of bond lengths are less striking than the variation of experimental values. However, the trends, if not the absolute values, are reproduced correctly, and agreement is good when considering that the calculations refer to an isolated, model molecule, and experimental data are usually collected for more-complex molecules as crystalline solids.
The most noticeable feature of the geometry relaxation of CH,OCH,X compounds that occurs during rotation around the C - 0 bond is the reversal of the C - 0 and C - X bond lengths. The C - 0 bond in the sc orientation is shortened, compared with its length in the ap orientation, whereas the C- X bond in the ap orientation becomes longer than in the sc orientation. In bond lengths, the most pronounced changes occur in the 0-C-X angle. The 0-C-0 bond angle in dimethoxymethane calculated by an ab initio method with 4-21G basis set falls from 112.4" for (sc, sc) to 109.5' for (sc, ap), and to 105.9" for the (ap, ap) conformation.122 The PCILO calculations result in similar values of 113.2, 110.9, and 105.8", respectively. The bond angle 0 - C - X increases in other compounds by 1 to 5 O on going from the ap to the sc orientation.
Because dimethoxymethane has a 2-fold axis of symmetry in the (sc, sc) and (ap, ap) conformations, the two outer, and similarly, the two inner, C- 0 bonds are indistinguishable. In 2-methoxyoxane, the calculations suggest that all four C - 0 bond-lengths at the acetal center are also different in the (149) P. Luger, G. Kothe, and H. Paulsen, Chem. Ber., 109 (1976) 1850- 1855. (150) G. Kothe, P. Luger, and H. Paulsen, Carbohydr. Rex, 37 (1974) 283-292. ( 1 5 1) I. TvaroSka, unpublished results. (1 52) S. Perez and C. Vergelati, Acta Crystallogr., Sect. B, 40 (1984) 294-299.

TABLE XX PCILO Calculated Bond Lengths (pm) and Bond Angles (degrees) for 2-Substituted Oxane Derivatives, Compared with
Observed Data on Carbohydrates
Group B 9 r(C-5-0-5) 40-5-C-1) r(C-1 -X) a(C-5-0-5-C-1) a(0-5-C-1 -X) References
F sc
ap
CI sc
up
OCH, sc sc
sc up up -sc
up sc
ap ap SCH, sc sc
-sc -sc sc up up -sc
up sc UP ap
139.7 145.0 139.6 142.8 139.8 145.1 139.8 142.7 139.6 143.3 139.6 139.5 143.3 139.5
139.5 139.7 144.6 139.6 139.7 139.7 144.1 139.7 139.7
138.9 136.2 139.2 140.6 137.5 138.3 138.4 141.5 139.9 141.4 139.9 140.0 142.9 140.0 142.7 139.8 138.5 144.6 138.4 138.4 138.9 143.5 139.0 138.9
136.6 138.9 136.3 136.7 183.5 185.9 182.3 175.4 139.4 139.9 139.7 139.2 138.1 139.4 139.1 139.4 191.5 179.9 191.9 191.9 190.6 180.4 190.8 190.9
110.9 112.6 110.1 109.9 1 12.9 113.0 108.2 11 1.1 11 1.9 113.3 111.4 1 12.4 112.4 1 11.9 113.7 112.7 113.3
114.9 112.9 110.8
110.7 110.8
106.8 110.7 103.2 105.7 107.8 107.9 103.4 107.1 110.5 1 12.4 106.4 106.6 107.6 107.2
102.3 110.5 11 1.9 110.0 108.7 105.2 108.8 105.0 104.1
12 98" 12 98" 12
149" 12
150" 151 97"
151 151
151
151 151 152" 151 151 151 152" 151 151
97"
30"
* Experimental value.

IGOR TVAROSKA AND TOMAS BLEHA
\ \ \ \
I / /
I I I
- 60 @ (degrees)
60 180
FIG. 20.-The Variation ofBond AnglesC-5-0-5-C-1 (CurvesDenoted bya)and0-5-C- 1 -0-1 (Denoted by b) with Torsional Angle in the Axial Form (Dashed Line) and Equatorial Form (Full Line) of 2-Methoxyoxane.
(sc, sc) and (up, up) conformations. In agreement with experiment, the endocyclic C-5 -0-5 -C-1 bond is longer than the corresponding exocyclic C- I - 0- 1 - C bond (see Table XX).
A striking interdependence of the bond angles and the orientation of the anomeric bond is made clear in Fig. 20 by presenting a plot of the C-5 - 0-5 - C- 1 and 0 - 5 - C- 1 - 0- 1 bond angles versus the angle @ for the a and e forms of 2-methoxyoxane. The former bond-angle displays the maximum varia- tion,changingintherangesof 105.5 to 115.5" and 101.5 to 108.5" forthea and e forms, respectively. As may be seen, this angle is -4" larger in the a than in the e form. In both cases, the minimum value ofthe angle pertains to

ANOMERIC AND EXOANOMERIC EFFECTS 101
the ap orientation. All of the calculated trends are in accord with the experi- mental data reviewed in Section I11 on the influence of conformation on the acetal ge~met ry ,~~* '~* and confirm a significant function of the anomeric and exo-anomeric effects in specification of the geometry parameters of saccha- rides.
As a corollary to the foregoing discussion, the computations also provide an answer to the controversial question of the influence of assumed geome- try on the calculated, conformational-energy differences. The results present evidence of the necessity of at least a partial optimization, including the main bond lengths and bond angles, in the theoretical calculations of molecules exhibiting the anomeric and exo-anomeric e f f e ~ t ~ . ~ ~ J ~ ~ J ~ ~ J ~ ~
5. Electron Distribution and Lone Pairs
Besides the energy and equilibrium geometry of conformers, the MO methods provide a great deal of useful information on the distribution of electronic charge in molecules, and on the degree of localization of individ- ual MO's on the atomic centers. A relative excess or deficit ofelectrons on the atom is represented by net charges Q1. Whereas most MO's are widely delocalized by their nature, some prominent MOs, such as those involving lone-pair electrons, are centered on one atom. Symmetry of two lone-pairs on oxygen atoms in the acetal and related moieties is a crucial point in the rationalization of the nature of the anomeric effect (see Section VI).
The traditional, most familiar picture of the lone-pair orbitals on the oxygen in water, alcohols, and ethers assumes two localized orbitals of tetra- hedral symmetry and of equal energy. There is, however, experimental evi- dence concerning the energetic non-equivalence of the two lone-pair orbitals in saturated, oxygen-containing molecules155J56 that supports an alternative description of lone pairs. In this representation, one lone-pair orbital having x symmetry is perpendicular to the R -0 - R plane, and the second lone-pair orbital, having c7 symmetry in the R-0-R plane, is - 1-2 eV lower in energy. Both representations are shown in Fig. 2 1. Similarly, two different pictures can be used for three lone-pair orbitals of halogen^.'^' The energetic and directional non-equivalence of lone-pair orbitals is an important factor for any situation where the symmetry of the interaction, or the energy of the lone-pair electrons, is critical, as in examination of the conformational pref- erences.
An experimental approach to the elucidation of the character of lone-pair
(153) R. U. Lemieux and K. Bock, Arch. Biochem. Biophys., 221 (1983) 125- 134. (154) I. TvaroSka and S. Perez, Curbohydr. Rex, 149 (1986) 389-410. (155) D. W. Sweigart and D. W. Turner, J. Am. Chem. Soc.. 94 (1972) 5599-5603. (156) S. Cradock and R. A. Whiteford, J. Chem. SOC. Furuduy Trans. 2,68 (1972) 28 1-288. ( 1 57) 0. Eisenstein, N. T. Anh, Y . Jean, A. Devaquet, J. Cantacuzene, and L. Salem, Tetrahe-
dron, 30(1974) 1717-1723.

102 IGOR TVAROSKA AND TOMAS BLEHA
(a) (b) FIG. 2 1 .- Lone-Pair Electrons ofoxygen Shown in (a) u-z Representation, and (b) sp3-Hy-
bridization Representation.
electrons consisted in a measurement of electron densities by X-ray and neutron diffraction techniques.I5* These difficult, but highly accurate, mea- surements have been carried out to date for two sugar molecules.'s9J60 The electron density maps computed for the ring and anomeric oxygen atoms of j?-DL-arabinopyranose'60 suggest that the spatial distribution of lone-pair orbitals on each oxygen atom corresponds to the sp3 type of orbital, rather than to a and a lone-pair orbitals. The four lone-pair orbitals are not, how- ever, identical with the lone-pair orbital of the anomeric oxygen atom, which is ap to the C- 1 - 0-5 bond, thus differing from the other three orbitals.
In carbohydrates, the anomeric carbon atom is bonded to two hetero- atoms having higher electronegativity than carbon. The electronegativity differences and the presence of lone-pair electrons are reflected in the elec- tron distribution in the molecule. The ring-oxygen atom on one side, and the aglycon (X) on the other, lessen the electron density at the anomeric carbon atom, and the C - 0 and C - X a-bonds are polarized in the direction of the more electronegative atoms. On the other hand, a delocalization of electrons from lone-pair orbitals of the oxygen aglycon towards the anomeric carbon atom could take place (see Section VI). Because the spatial relationships of the lone-pair orbitals and the bonds at the anomeric center change with the conformation, there are also differences in the electron distribution between the conformers.
It was established from calculations of substituted dimethyl etherI2 that the largest difference in net charge occurs at the central carbon atom and on its substituent; that is, on the atoms where a delocalization interaction occurs. A pronounced enhancement of negative charge on the oxygen atom ( 1 58) P. Coppens, in J. M. Robertson (Ed.), MTP Int. Rev. Sci. Php . Chem., Ser. 2, Vol. 1 1,
(159) J . C. Hanson, L. C. Sieker, and L. H. Jensen, Acfa Crystallogr., Sect. B, 29 (1973)
(160) F. Longchambon, H. Gillier-Pandraud, R. Weist, B. Rees, A. Mitschler, R. Feld, M.
Butterworth, London, 1975, pp. 21-54.
797 - 808.
Lehmann, and P. Becker, Acta Crystallogr., Sect. B, 41 (1985) 47-56.

ANOMERIC AND EXO-ANOMERIC EFFECTS 103
TABLE XXI PCILO Calculated Net Atomic Charges0 and Dipole Momentsb of
the Most Stable Conformers of 2-Metho~yoxane'~~
(+ sc, + sc)
291.8 -21.0 -45.0
0.4 160.0 100.6
- 170.5 - 194.2 - 78.0
0.28
294.0 - 30.8
44.7 0.6
160.0 97.8
- 171.3 -181.4 - 75.4
2.17
299.9 -25.3
40.9 0.1
156.5 102.6
- 160.1 - 187.5 -96.5
2.13
(UP, +sc)
293.8 - 34.5
42.6 - 0.6 156.5 104.6
- 169.1 - 186.9 - 76.9
2.36
(UP, UP)
299.0 -35.2
43.4 -0.6 154.7 95.3
- 154.2 - 168.3 -94.9
3.18
"Q(i), in lo-C. b,u, in D, ID= 13.33563 X mAs.
and the heteroatoms (X) was found in ethers upon conversion of the up into the sc orientation. Similarly, dependence on conformation was inferred from net charges of selected atoms and dipole moments of 2-methoxyox- aneIz8 (see Table XXI). With the exception of the atoms of the aced seg- ment, the changes in electron distribution by rotation about the C- 1 - 0- 1 bond are small. The minimum dipole moment (0.28D) is exhibited by the (+ sc, + sc) conformer, where dipole moments of lone-pair orbitals on the ring-oxygen atom are oriented antiparallel to those at the anomeric oxygen atom. The largest dipole moment (3.18D) is associated with the (ap, up) conformers, with lone-pair orbitals on both oxygen atoms being oriented parallel.
V. THE ANOMERIC AND EXO-ANOMERIC EFFECTS IN POTENTIAL- FUNCTION CALCULATIONS
Although several MO methods successfully describe the stereochemical behavior of pyranose models, their application to the more-complex carbo- hydrates and oligosaccharides is limited at present, due to reasons of econ- omy. An alternative, more empirical approach has to be pursued, based on the summation of suitable potential functions (PF) accounting for intramo- lecular interactions. The intramolecular energy terms usually represent con- tributions from nonbonded interactions, electrostatic interactions, hydro- gen bonding, torsional terms, bond stretching, and valence-angle bending terms. The first attempts to calculate the stability of monosaccharide isomers by the additive have shown the necessity of introduction of an extra anomeric-effect parameter, in addition to the parameters of interaction

1 04 IGOR TVAROSKA AND TOMAS BLEHA
between substituents. The extra parameter amounted to 1.8 -6.3 kJ.mol-’, depending on the chemical structure of the monosaccharides and on the selected set of parameters. An identical problem arises when PF calculations are carried out on saccharides and similar molecules. In conformity with Eq. 3, an extra energy contribution, AE(AE2), is assumed in order to “convert” the results of PF calculations into agreement with experiment. Obviously, the anomeric-effect correction, AE(AEZ), in the PF method depends on the set of potential functions used, and thus it is not universal. For example, extra energy, 1.7 W.mol-l, was needed in order to account correctly for equilibrium in hexo- and pento-pyran~ses,~~ and PF calculationss6 of rota- tion around the C - 0 bond in 2-methoxyoxane required the addition of 4.6 kJ.mol- of stabilization energy in order to reproduce the experimental data.
These and numerous other studies make clear that direct application of the PF method, developed primarily for nonpolar molecules, to saccharides and their models requires its modification, especially in a way to account properly for the anomeric effect. In the first stage, the modifications con- cerned mainly the energy of the anomeric effect, and, in later development, the valence geometry aspect. MO studies of the model compounds were instrumental in achieving the required improvement of the PF method. It was evident from MO calculations of torsional energetics of acetals. 113~161
that the PF procedure must be modified by the proper incorporation of the lone-pair electrons into calculation of intramolecular energy. The lone-pairs bring about the anisotropy of electron distribution in a molecule, such that the majority of the charge density is localized in their direction. The lone pairs may be regarded as pseudoatoms having a size and dipole moment, and they may be incorporated into the PF scheme. A simple PF method was successfully modified161 in this way, by consideration of the lone-pair dipoles in calculation of the electrostatic term. The magnitude and the localization of the lone-pair dipoles can be transferred from MO calculations of simple compounds or, alternatively, by using the universal procedure16* where the modulus and direction ofthe lone-pair dipoles is expressed in dependence on hybridization (bond angle) on the heteroatom. By using the procedure, the PF method correctly reproduced conformational energies of dimethoxy- methane; that is, the extra energy term, AE(AE2), was zero in this case.’61
The development ofthe PF method culminated in molecular mechanics, a reliable method of prediction of conformational energy and equilibrium structure of nonpolar molecules. For molecules having several heteroatoms, a modified parameterization was developed, with the lone-pair parameters as a part of the force field, and with their inclusion into all types of intramo-
(161) I. TvaroSka and T. Bleha, Collect. Czech. Chem. Commun., 43 (1978) 922-931. (162) I. TvaroSka and T. Bleha, Biopolymers, 18 (1979) 2537-2547.

ANOMERIC AND EXO-ANOMERIC EFFECTS 105
lecular interaction^.'^^ Using the MM2 method parameterized in this way, reasonable conformational energies were computed for pyranose models, provided that interactions of the bond dipoles were assumed in the electro- static term.'64 Computations with net charges on atoms only, that is, lacking lone-pair dipoles, gave conformational energies in disagreement with the experimental data.
An alternative way of adaptation of the PF computational scheme to the molecules exhibiting the anomeric effect consists in the addition of a proper, preferably simple, potential term. In other words, an attempt is made in this approach to establish the extra energy term, AE(AE2), beforehand, and then to incorporate it into the PF method. In one of the first trials, an additional, twofold torsional term for C - 0 rotation in the C-0-C-0 segment was assumed,'65 with a bamer of - 7.6 kJ.mol-l at -90". In the next attempt to calculate the torsional potential around the glycosidic bond by the simple PF method, nonbonded interactions were combined,59 with the ab initio 4-3 1G torsional potential of dimethoxymethane serving as an extra energy term. Because, in such a procedure, some interactions were evidently counted twice, the corrected approach known as HSEA (Hard Sphere Exo-Ano- meric) was suggested,60 where the extra energy, AE( EAE2), is expressed as a simple function of the torsional angle Q, for each anomer separately. A similar approximation of the latter extra energy was developed6' that was in analogy with Eq. I, from MO calculations for cyclic models, as the difference between the torsional potential of 2-methoxyoxane and that of 2-ethylox- ane. The resulting different expressions for a and p anomers, as a function of the angle a, were subsequently used as additional energy contributions in the PF studies of oligosaccharide conformations. 153~154
All of the amendments of the PF method so far discussed were centered on the conformational energy. The development of empirical computational methods able to account for the valence geometry changes on the anomeric center is much less straightforward. Sophisticated force-fields are required, with the numerous parameters exactly reproducing the interdependence of the bond lengths, bond angles, and torsional angles. The first force-field developed for saccharides166 correctly described the overall saccharide struc- ture, but failed to imitate the specific variations of geometry in the vicinity of the anomeric carbon atom with different configurations of substituents. In contrast to the conformational energy, the incorporation of lone airs^','^^ into the molecular mechanics schemes MM 1 and MM2 did not bring about improvement in the description of the anomeric center geometry. Modifica- (163) N. L. Allinger and D. Y. Chung, J. Am. Chem. Soc., 98 (1976) 6798-6803. (164) T. Koiir, A. Sarko, and I. TvaroSka, unpublished results. ( 165) U. Burkert, Tetrahedron, 35 ( 1979) I945 - 195 1. (166) S. Melberg and K. Rasmussen, J. Mol. Srrucr., 57 (1979) 215-239.

106 IGOR TVAROSKA AND TOMAS BLEHA
tion of the MM 1 by separate parameters for the ring-oxygen atom, the glycosidic oxygen atom, and the anomeric carbon atom, which, more- over, differ for the a and p anomers in the calculation of the energy terms, constituted the first successful attempt. The MM2 method has been amended in a similar way, and used to calculate the structure of various conformers of trehalose models,132 c e l l o b i ~ s e , ~ ~ ~ and maltose.154 The com- putations demonstrate the ability of the method to render the variations of disaccharide geometry with the orientation around the glycosidic bonds. In spite of the success, the modifications described were handicapped by the necessity to introduce additional parameters to an already large set of param- eters. To avoid this problem, the standard C - 0 bond length in the MM2 method has been expressed168 as a function of the orientation of both of the central C - 0 bonds in the acetal segment. Further testing in the future should assess the merits and shortcomings of this and other modifications of force fields. A full reproduction of the anomeric and exo-anomenc effect in conformational energies and valence geometry of saccharides is an ultimate goal of this effort.
VI. NATURE OF THE ANOMERIC EFFECT
In previous Sections, we have presented the experimental evidence as to the various manifestations of the anomeric effect, and have discussed the results of quantum-mechanical calculations made on model compounds and carbohydrates. In this Section, the nature of this effect will be discussed. It is obvious that any explanation of the anomeric and the exo-anomeric effect should clarify both the conformational preference, and the variations of the bond lengths and bond angles. Various rationalizations of the ano- meric effect have been offered over the years; they may be roughly divided into two main groups, namely, the electrostatic and the delocalization ra- tionalization of the anomeric effect. Besides these two dominant concepts, the anomeric effect was regarded as a result of a fine equilibrium between the electron - electron repulsion and the nucleus - electron a t t ra~t ion , '~~ of a barrier of internal rotation about the C-X bond170 or of the Jahn-Teller effect.171 However, neither of these explanations has gained wide recogni- tion, and, therefore, they will not be discussed here. The two prevailing rationalizations, electrostatic and delocalization, involve interactions of the
(167) G. A. Jeffrey and R. Taylor, J. Comput. Chem., 1 (1980) 99- 109. (168) L. Nerskov-Launtsen and N. L. Allinger, J. Comput. Chem., 5 (1984) 326-335. ( 169) S. Woife, A. Rauk, L. M. Tel, and 1. G. Csizmadia, J. Chem. Soc., B, ( I 97 1) 136 - 145. (170) Yu. A. Zhdanov, R. M. Minayaev, and V. I. Minkin, J. Mol. Struct., 16 (1973) 357-364. (171) R. Ponec and V. Chvalovsky, Collect. Czech. Chem. Commun., 39 (1974) 2613-2615.

ANOMERIC AND EXO-ANOMERIC EFFECTS 107
H t X
(a) (6 1 FIG. 22.- Dipolar Interactions in 2-Substituted Oxane Derivatives.
lone-pair electrons of oxygen (or other hetero atom). The character of those lone pairs (hybridization) is thus a very important element in the discussion.
1. Electrostatic Interactions
In the first interpretation, the anomeric effect was discussed4 in terms of electrostatic repulsion between the carbon-substituent dipole and the result- ant dipole of the lone-pair orbitals on the ring-oxygen atom (see Fig. 22). The repulsive interactions are maximized in the equatorial conformer when the dipoles are parallel, and account for the preference for the axial conformer. The difference in dipole-dipole interaction energies between the axial and equatorial conformer of 2-chloro- and 2-bromo-4-methyloxane has been estimated38 at 11.3 and 10.0 kJ.mol-I by use of classical electrostatics. In dimethoxymethane, the differences in electrostatic interactions calculated between the (up, up) and (up, sc) and between the (up, sc) and (sc, sc) conformers are - 7 and 2 kJ.mol-', respectively.161 If electrostatic interac- tions between the dipoles along the axes of the sp3-type lone-pair orbitals on the oxygen atoms are a s ~ u m e d , ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ * ~ ~ ~ ~ ~ ~ this approach was picture- squely termed the rabbit-ear effect.173 Thus, the most stable conformer of acyclic and cyclic acetals is that having on oxygen atoms the minimum number of syn-axial, lone-pair interactions that can cause dipolar repulsion, with an energy of - 4 kJ.mo1-I assigned to each of such interactions. The syn-axial, lone-pair orbital interactions in the acetal segment of 2-methox- yoxane are illustrated in Fig. 23 (compare, also, Fig. 3). 2-Methoxyoxane having an equatorial methoxyl group has two conformers, (up, - sc) and (up, + sc), that have a single, syn-axial, lone-pair orbital interaction, and a third, (up, up), which has two such interactions. On the other hand, in the axial form, two conformers, (+ sc, - ap) and (+ sc, - sc), have one interaction, and
(172) R. 0. Hutchins, L. D. Kopp, and E. L. Eliel, J. Am. Chem. Sm., 90 (1968) 7174-7175. (173) E. L. Eliel, Angew. Chem., 17 (1972) 779-791.

108 IGOR TVAROSKA AND TOMAS BLEHA
(+sc , -sc ) (ap, ap) FIG. 23.-The Rabbit-ear Effect Interactions in the Staggered Conformations of an A c e d
Segment.
the third, (+ sc, + sc), has no lone-pair orbital interactions. It may be pre- dicted, therefore, that the axial will be more stable than the equatorial con- former and that the (+ sc, + sc) conformer should be the most stable of the axial conformers, and the (up, - sc) conformer the most stable of the equato- rial conformers, owing to unfavorable interactions between the methyl group and the axial hydrogen atom on C-2 in the (up, +sc) conformer.
The electrostatic concept predicts the stability of conformers in qualitative agreement with experimental observation, but quantitative accord cannot

ANOMERIC AND EXO-ANOMERIC EFFECTS 109
be expected from such a crude approach. The largest support for this ration- alization comes from an observed lessening of the anomeric effect in more- polar solvents (see Tables VI, VII, and XIV). Also, the analysis of the individ- ual terms in the intramolecular energy of dimethoxymethane s h o ~ ~ ~ ~ ~ , ~ ~ ~ that the preference for the sc orientation about the C - 0 bond originates in the electrostatic repulsion of the electron densities located on the two oxygen and associated bonds. However, the electrostatic concept is unable to ration- alize variations of the bond lengths and bond angles that are characteristic for the anomeric effect.
2. Delocalization Interactions
A second, perhaps more popular, rationalization arose from investiga- tions of a-halo ether s t r u c t ~ r e s . ~ ~ J ~ ~ * ~ ~ ~ In these compounds, preference for the sc orientation is associated with a characteristic lengthening of the carbon - halogen bond, whereas the adjacent C -0 bond is shorter.I3 A ster- eoelectronic explanation is illustrated for chloromethoxymethane in Fig. 24. A stabilization of the sc orientation is attributed to delocalization or back- donation of the a-type, lone-pair orbital on oxygen to the antibonding or- bital of the adjacent carbon - halogen bond. The n-type lone-pair orbital is oriented perpendicular to the C- 0- C plane, and therefore the best orienta- tion of the carbon-chlorine bond for this delocalization is in the orthogonal (og) orientation where both orbitals are in the same plane. This interaction produces the following consequences; the lengthening of the C- C1 bond by populating of its a* antibonding orbital, the contraction ofthe C- 0 bond by increasing its double-bond character, and an opening of the 0- C-C1 bond- angle compared to its tetrahedral value, because of the increased double- bond character at C.
Some insight into these interactions may be obtained from the distribu- tion of oxygen lone-pair electrons calculated by the ab initio method with 4-3 1G basis for methanol, fluoromethanol, and methanedi01.I’~ In metha- nol itself, the a-type lone-pair electrons are subjected to back-donation into antibonding-type orbitals of suitable symmetry on the methyl group. This leads to a decrease of the n-orbital population from the value of 2.00, appro- priate to water, 1.97. A comparable electron displacement occurs in fluoro- methanol when the C-F bond is in the nodal plane of the oxygen lone-pair orbital. If, however, the C-F bond and lone-pair orbital are in the same plane (in the og orientation), the back-donation lowers the population to
( 1 74) N. Gresh, P. Clavene, and A. Pullman, Theor. Chim. Acta, 66 (1984) 1-20. (175) E. A. C. Lucken, J. Chem. Soc., (1959) 2954-2960. (176) G. Baddeley, Tetrahedron Lett., (1973) 1645- 1648. (177) J. A. Pople, Tetrahedron, 30 (1974) 1605-1615.

110 IGOR TVAROSKA AND TOMAS BLEHA
R" CI
I I I I I
;I &-
CH3 / O \ i' CH3 +?j\ & H- C #QC'
H c C
I 01 H H
FIG. 24.-Back-donation of Lone-pair Orbital on Oxygen into the Adjacent C - 0 Bond in Chloromethoxymethane.
1.94, and leads to additional stabilization. Similarly, the back-donation from the oxygen lone-pair orbital to the C-0 antibonding orbital in the (sc, sc) conformation of methanediol is 0.05, which is significantly larger than the 0.03 in methanol. In resonance terms, this corresponds to double-bond- no- bond structures 0 - - C = O+. This simple, delocalization approach has been used to rationalize differences in n.q.r. frequency in a-chloro e t h e r ~ . ' ~ ' J ~ ~
The delocalization concept has been further advanced by stressing the necessity for analysis of the orbital interactions of both lone-pair orbitals on an oxygen atom, one with n-symmetry and one with o - ~ y m m e t r y . ~ * ~ ~ ~ ~ J ~ ~ Even though the lone-pair orbital of the o-symmetry is more strongly bonded, its interactions with appropriate antibonding orbitals cannot be neglected. It has been shown157 that, if the energetic and directional non-
( 1 78) S. David, 0. Eisenstein, W. J. Hehre, L. Salem, and R. Hoffmann, J. Am. Chem. SOC., 95 (1973) 3806-3807.

ANOMERIC AND EXO-ANOMERIC EFFECTS 1 1 1
X H
(4 ( b ) FIG. 25.-Orientation of Two Non-equivalent Lone-pairs of the u and A Type in the a and e
Forms of 2-Substituted Oxane Derivatives.
equivalence of the two oxygen lone-pair orbitals is accounted for, their interactions with the antibonding C- X orbital leads to the axial preference of the electronegative substituent X. The orientations of the pertinent or- bitals in the two competing conformations of 2-substituted derivatives of oxane are shown in Fig. 25. A qualitative estimate for X = C1 that the axial has a larger stabilization than the equatorial conformer. The incor- poration of two nonequivalent, lone-pair orbitals into the analysis of the nature of the anomeric effect was a step of considerable importance, but qualitative reasoning still prevailed at that time.
Detailed perturbation calculation of all delocalization interactions of lone-pairs has been performed for dirneth~xymethane'~~ ; to date, this is the only one conducted. The two lone-pair orbitals on oxygen atoms are engaged in mutual interactions by a through-space mechanism and in interactions with appropriately oriented antibonding orbitals of the CH2 group having n-symmetry and with o*-antibonding orbitals of two central C - 0 bonds by a through-bond mechanism. 179 The relevant molecular orbitals of dimeth- oxymethane are illustrated in Fig. 26, but, for the sake of simplicity, only one (179) R. Hoffman, Acc. Chern. Res., 4(1971) 1-9.

112 IGOR TVAROSKA AND TOMAS BLEHA
CH) CH3
Cii,
(aPJ ap) (“p D ‘ 9 ) FIG. 26.--Interactions of Lone-pair Orbitals on Oxygen Atoms with (a) Antibonding Or-
bitals of CH, Group and (b) Antibonding Orbital of Adjacent C - 0 Bond, for (up, up) and (up, og) Conformations of Dimethoxymethane. [For the sake of simplicity, only one oftwo perpen- dicular antibonding orbitals of the CH, group and the antibonding orbital of one C - 0 bond are shown.]

ANOMERIC AND EXO-ANOMERIC EFFECTS 113
of two perpendicular, n-antibonding orbitals of the CH, group and one a*-antibonding orbital of two central C - 0 bonds are included. These or- bitals are separately shown in Figs., 26a and 26b. The magnitude of the individual contributions to delocalization interactions in dimethoxymeth- ane depends on the mutual orientation of pertinent orbitals, and, therefore, on the orientation about the two inner C - 0 bonds.
In the (up, ap) conformation, the lone-pair orbitals on oxygen atoms having the same symmetry are colinear and, therefore, interactions between the two lone-pairs on oxygen atoms by the through-space mechanism are optimal, as well as interactions with the Ir*-antibonding orbitals of the CH, group by a through-bond mechanism. The back-donation into a a*-anti- bonding orbital of central C - 0 bonds is possible only from the a-type of lone-pair, because the n-type of lone-pair is perpendicular to the 0-5 -C- 1 - 0 - 1 plane.
In the (up, og) conformation, owing to rotation about one central C - 0 bond, the spatial orientations of two lone-pair orbitals on the corresponding oxygen atom are interchanged. Consequently, the a-type of lone-pair orbital interacts by a through-space mechanism with the n-type of lone-pair orbital of the second oxygen atom, and vice versu. Similarly, interactions of these lone-pair orbitals with antibonding orbitals of the CH, group are reversed. The back-donation of lone-pairs on the oxygen atom into the a*-antibond- ing orbital of the C - 0 bond in the (up, og) conformation is reversed in comparison to the (up, ap) conformation. The n-type of lone-pair is in the same plane as the relevant C - 0 bond, and the back-donation into the antibonding orbital is maximal, whereas the back-donation of the a-type of lone-pair disappears.
The energy of the aforementioned interactions as a function of rotation about the C - 0 bond has been calculated by perturbation MO theory.148 The results show that the overall stabilization energy is rather large, -37 kJ.mol-I in the (up, up) conformation, but its conformational dependence is not pronounced. The stabilization-energy curve resembles the V, term of Fourier expansion (see Eqs. 5 and 6), with the ogposition being - 1 kJ.mo1-1 less stable than the up position.148 This situation arises as a consequence of reverse conformational dependence on the n- and a-type lone-pair interac- tions. The foregoing analysis does not confirm the delocalization concept as being the origin of the energetic manifestation of the anomeric or exo-ano- menc effects in acetals. On the other hand, the electron transfer associated with lone-pair orbital interactions is in complete accord with the trends observed, and it explains all of the changes in the bond lengths and valence angles, including the shortening of C - 0 bonds in (up, up) conformations, which is not obvious from the double bond-no bond interpretation of the

114 IGOR TVAROSKA AND TOMAS BLEHA
anomeric effect. A study of trans- 1,8-dioxadecalin by photoelectron spec- troscopylsO confirmed the results of theoretical analysis. An acetal segment fixed in the (ap, up) conformation shows two well resolved, low ionizations, at 9.08 and 9.93 eV. The difference of ionization potentials of 0.85 eV is the largest observed for acetals, 155 and indicates a strong, through-space interac- tion of lone-pairs in the (up, up) conformation.
3. Scaling between Electrostatic and Delocalization Interactions
The foregoing discussion shows clearly that neither the electrostatic nor the delocalization concept is able to describe all of the peculiarities of the anomeric effect in saccharides. The electrostatic concept, sometimes naively represented by the rabbit-ears effect, properly describes the preference for the sc orientation and the decrease of this preference with increasing solvent polarity, but fails to reproduce changes in valence geometry. On the other hand, the delocalization concept, assuming non-equivalent, oxygen lone- pair orbitals, completely interprets the changes observed in bond lengths and bond angles, but does not account for the energy preference of the sc orienta- tion. It is apparent, therefore, that only the combination of both concepts can give a coherent explanation of all of the phenomena mentioned. A decomposition of the calculated torsional potential into components of the Fourier expansions (Eqs. 5 and 6) provides a simple technique to illustrate the balance of both concepts. As has been noted, although the decomposi- tion is rather formal, the simple physical meaning can be ascribed to the expansion coefficients. The terms V,, V,, and V, are identified with the inherent, torsional barrier for single-bond rotation, with the delocalization interactions of lone-pair orbitals and with the interaction of dipoles in the rotating segments, respectively. 111 Obviously, the shape of the torsional po- tential and the positions of the energy minima depend on the magnitude of the expansion coefficients. In Section IV, 1, the torsional potential of the C - 0 bond in CH,OCH,X molecules calculated by MO methods has already been analyzed (see Table XIII).
The energy difference between the up and sc orientations can be ex- pressedgL3 by using Eq. 5.
AE(AE3) = 0.75(V? - Vs) (8)
Hence, the anomeric effect [positive value of AE(AE3)l can appear at a suitable combination of the V? and Vq coefficients. Because the V, term is associated with the dipole-dipole interactions of polar groups, and the V, term with delocalization interactions of lone-pair orbitals on heteroatoms, it may be concluded that the latter equation also effectively describes a balance
( 180) F. S. Jerrgensen and L. Nerrskov-huntsen, Tetrahedron Left., (1982) 522 1 - 5224.

ANOMERIC AND EXO-ANOMERIC EFFECTS I15
of the two origins of the anomeric effect, that is, of electrostatic and delocali- zation interactions. Depending on the character of the heteroatoms, one or the other factor may prevail. A definite appraisal can be obtained only by calculation of the V, and V, terms for a given molecule. For example, comparison of the Vy and Vq values for dimethoxymethane and chloro- methyl ether (see Table XIII) showed that this balance in dimethoxymeth- ane is considerably shifted toward electrostatic interactions, whereas, in chloromethyl ether, the preference for the sc rotamer is determined mainly by delocalization interactions. A dominance of delocalization interactions reflects a strong tendency of the chlorine substituent for back-donation,181 and explains the more-pronounced differences of bond lengths and bond angles observed between a- and P-D-pyranosyl chlorides than in other pyra- nose derivatives (see Table XX).
The balance of electrostatic and delocalization interactions in an isolated molecule may be perturbed by the influence of the solvent. In calculations based on Eq. 7, the analysis of solvation-energy terms suggested'*' that the electrostatic contribution stabilizing the up orientation of the acetal segment is the conformationally dominant term. For example, in 2-methoxyoxane, the difference in energy of the (ap, ap) and (up, sc) conformers in water, compared to that in the isolated molecule, caused by solute -solvent electro- static interactions alone, amounts to - 4 kJ.mol-*. Accordingly, the inter- and intra-molecular, electrostatic interactions operate in reverse directions in acetals. Whereas the intramolecular, electrostatic interactions are respon- sible, together with delocalization interactions, for the appearance of the anomeric, reverse anomeric, and exo-anomeric effects, the solute - solvent electrostatic interactions lessen their magnitude, and may even cancel them. Of course, the solvent may also influence the electron distribution and energy of MO's in a molecule. In this way, the orbital interactions of lone- pairs and delocalization contributions to the anomeric effect may be scaled by the solvent, but this mechanism of the environmental effect is, in most cases, of only minor importance.
Finally, a less conventional explanation of the origin of the anomeric effect may be mentioned. In a communication,4o it was stated that, contrary to the results from m e a ~ u r e m e n t s ~ , ~ ~ - ~ * ~ ~ - ~ ~ and theoretical calcula-
the equilibrium constant for axial - equatorial equilibrium in 2-methoxyoxane derivatives is independent of variation in the temperature. Both anomers are therefore isoenthalpic, and the Gibbs energy difference is largely determined by the entropic term, the axial isomer having 11.3 J. K- mol- greater entropy.
(181) R. C. Bingham, J. Am. Chem. Soc., 97 (1975) 9743-9746.

116 IGOR TVAROSKA AND TOMAS BLEHA
VII. ROLE OF THE ANOMERIC EFFECT IN THE REACTIVITY OF
CARBOHYDRATES
In the previous Sections, we have discussed the consequences of the ano- meric effect for the ground state of aldopyranoses as reflected in the stability of conformers, valence structure, and electron distribution. It may be ex- pected that intramolecular interactions involved in the anomeric effect, apart from the ground state, also influence the other points on the general- ized reaction-hypersurface. As a matter of fact, it is common belief in carbo- hydrate chemistry that the anomeric and related stereoelectronic effects modify the reactivity of saccharides. Their actual influence on the course of reaction is, however, far from being understood. Comprehension is com- plicated by numerous factors, such as the flexibility of the pyranose ring, the variability of the type and localization of substituents and reacting groups on the ring, the concentration of reactants, and the character of the solvent. 182-186 Intensive exploration of this problem has resulted in formula- tion of qualitative rules'87 allowing interpretation of the course of the whole range of reactions by using the concept of the anomeric effect. A complete description of progress in understanding the reactivity on the anomeric center lies outside the scope ofthe present article, and the reader is referred to excellent reviews. Instead, we shall focus our attention on some consequences ensuing, for reactivity, from quantum-chemical studies of saccharides and their models.
1. Energy of Reaction Paths
The observation of the significant stereoelectronic effect on reactivity (that is, a higher reaction rate of one conformer than another for stereoelec- tronic reasons) presumes the condition that the barrier of transition, AEL, from the less reactive (A) to the more reactive (B) conformer exceeds the activation energy of reaction, AE& , of the B conformer. When this condition is not satisfied, the two conformers rapidly interchange, because of the low
(182) J. N. BeMiller, Adv. Carbohydr. Chem.. 22 (1967) 25- 108. (183) B. Capon, Chem. Rev., 69 (1969) 407-498. (184) E. H. Cordes and H. G. Bull, Chem. Rev., 74 (1974) 581 -603. ( 185) J. Szejtli, Saurehydrolyse der glykosidischer Bindungen, Akademiai Kiado, Budapest,
(186) C. Schuerch, in Ref. 9, pp. 80-94. (187) P. Deslongschamps, Tetrahedron, 31 (1976) 2463-2490. ( 188) P. Deslongschamps, Stereoelectronic Eflectts in Organic Chemistry, Pergamon, Oxford,
(189) V. G. S. Box, J. Heterocycl. Chem., 19 (1982) 1939- 1966. (190) V. G. S. Box, J. Heterocycl. Chem., 20 (1983) 1641 - 1653.
1976.
1983.

ANOMERIC AND EXO-ANOMERIC EFFECTS 117
barrier, AEL, and they can react through the same transition state, that ofthe reactive conformer B; and also, their relative reactivity is determined solely by the difference of ground-state energies. However, if the barrier, AEL, is larger than the activation energy of the reactive conformer, AEg, but is smaller than the analogous activation energy of the less reactive conformer A, both conformers can again react through the transition state of conformer A, but the conformational interconversion through barrier AEL is the rate- determining step10J91-193 for conformer A. The overall reaction of A will thus be slower than that of B, as a result of the higher activation energy, AEL, for direct reaction, although the observed rate has no direct relationship to the size ofthis barrier. Traditionally, stereoelectronic effects are looked for in the ground-state energies only, but they also operate in the transition states of reactions and conformational interconversions.
What are the energy relations in the six-membered ring of pyranoid sac- charides? The energy difference of the axial and equatorial forms on the anomeric center is low, in the interval of - 0- 1 1 lcJ.mol-l, dependingon the medium (see Tables 11,111, and VII). The interconformational (pseudorota- tional) barrier, AEL, in six-membered rings is -42-46 kJ.mo1-I. Some authors lS7 assumed an additional increase of the pseudorotational bamer due to the anomeric effect, but, in contrast, data for the ring inversion in 2,2-dimethoxyoxane 194 indicate a significant decrease of the barrier by - 6 - 9 kJ.mol-l relative to the rings, with the absence of the anomeric effect. The activation bamers in reactions are usually higher than the foregoing figures for AEL, and that explains why it is very difficult to observe the stereoelec- tronic effect in the reactivity of various conformers of saccharides. In order to assess the magnitude of this effect, systems conformationally much more rigid than those of the majority of saccharides have to be used. Various complex, model compounds have been d e ~ i g n e d ' ~ ~ - l ~ ~ wherein the rigidity of a proper structural segment is secured by chemical fixation.
It is generally accepted that the transition-state theory provides the most convenient framework for calculation of the rate constants. Its rigorous application, however, needs a knowledge of the energy hypersurface of a
(191) N. S. Zefirov, Tetrahedron, 33 (1977) 2719-2722. (192) J. I. Seeman, Chem. Rev., 83 (1983) 83-134. (193) D. F. DeTar, J. Org. Chem., 5 1 (1986) 3749-375 1. (194) C. L. Perrin and 0. Nunez, J. Chem. SOC., Chem., Commun., (1984) 333-334. (195) S. Chandrasekhar, A. J. Kirby, and R. J. Martin, J. Chem. Soc., Perkin Trans. 2, (1983)
(196) A. J. Kirby and R. J. Martin, J. Chem. Soc., Perkin Trans. 2, (1983) 1627-1632. (197) A. J. Kirbyand R. J. Martin, J. Chem. Soc., Perkin Trans. 2, (1983) 1633-1636. (198) A. J. Briw, C. M. Evans, R. Glenn, and A. J. Kirby, J. Chem. SOC., Perkin Trans. 2,
1619- 1626.
(1983) 1637-1640.

118 IGOR TVAROSKA AND TOMAS BLEHA
given reaction, mostly not available (with the exception of some simple, few-atom systems). From the point of view of the stereoelectronic effect, it is sufficient to compare its involvement in the ground states versus transition states of various reaction-channels. For instance, in spite of a large absolute value of the anomeric effect, no difference in reactivity might be observed when its contributions to the ground and transition states compensate each other.
Estimates of the anomeric effect in the ground state and transition state, or in intermediates, have been used to explain the different reaction-rates of two anomers in some reactions by employing the established reaction-mech- anism. Especially suitable in this respect are reactions wherein the identical transition state is assumed for the reaction of both anomers. In this case, the axial and equatorial conformers of a given compound react at different rates, owing to a different stabilization of the ground states by the anomeric effect. Acid hydrolysis of 2-oxanyl acetals and of methyl glucosides are probably examples. The stabilization of the axial isomer in the ground state retards its reaction, by enhancement of the reaction bamer relative to the equatorial isomer, which is then hydrolyzed the faster.lS3- lS5
Actually, this reasoning lay behind the first observation of the anomeric effect? Similar stabilization of the reaction intermediates by the anomeric effect explains, qualitatively, the kinetics of conversion of derivatives of 8-D-galacturonic acid into the corresponding a-L-altruronic acid deriva- t i v e ~ . ' ~ ~ Hydrolysis of the a is a few times faster than that of the p anomer of aryl glycoside~,'~~ and this can probably be explained similarly. In this case, the hydrolysis is affected by the initial, proton-transfer equilibrium. Because of the reverse anomeric effect, the relative stability of the resulting conjugate acids is reversed with respect to the neutral molecule, and thepanomer is the more This means that the reactivity of the axial anomer of the conjugate acid increases (and vice versa for the equatorial anomer).
The paucity of information on the mechanism of reactions, and on the structure of the transition state, and the role of the anomeric effect in its stabilization, constitutes the main reason why qualitative interpretation of reactivity as shown in the aforementioned examples is still very rare. An alternative, more-popular estimation of the relative reaction-rates of con- formers is based on the lone-pair orbital interactions, and their symmetry and energy in the ground state, and could be loosely associated with the perturbation theory of chemical reactivity.200
( 1 99) P. KovaE, J. Hirsch, I. TvaroSka, R. PalovEik, V. KovaEik, and T. Sticzay, Collect. Czech.
(200) G. Klopman (Ed.), Chemical Reactivity and Reaction Paths, Wiley-Interscience, New Chem. Commun., 41 (1976) 3804-3811.
York, 1974.

ANOMERIC AND EXO-ANOMERIC EFFECTS 119
2. Lone-Pair Orbital Interactions in Reactivity
The lone pairs of oxygen atoms are the least strongly bound atomic or- bitals, and thus are localized prevailingly in the highest-occupied MOs. Hence, it is natural that lone pairs play a central role in various interpreta- tions of stereochemical dependence of reactivity in carbohydrates. In this respect, the most successful approach is the so-called antiperiplanar, lone- pair hypothesis, derived from study of the hydrolysis of esters and a m i d e ~ . ' ~ ~ J ~ ~ This theory is formally similar to the simple delocalization concept of the anomeric effect involving the back-donation of the oxygen lone-pair into the up antibonding C - X orbital. The fundamental presump- tion of the theory is that any reactive conformer must have on each oxygen atom a lone pair oriented up to the leaving group. The concept, based on sp3 hybridization on oxygen, was originally developed in order to account for the experimentally observed ratio of products in the oxidation of acetals. At present, this scheme is widely accepted, and it has been applied to the whole range of reactions on the anomeric enter.'^.^^^- 198~201
However, experimental evidence of a marked stereoelectronic effect on the reactivity of saccharides is very difficult to obtain, because of the flexibil- ity ofthe pyranoid ring. For example, the equatorial isomer can probably not only react through its chair conformations, but also by way of conformations close to the boat forms in which the sp3 lone-pair on the ring-oxygen atom is antiperiplanar to the exocyclic C - 0 bond.I0 The largest stereoelectronic control of reactivity observed to date is found in the hydrolysis of the axial and equatorial pair of 1 -(2,4-dinitrophenoxy)-9-oxabicyclo(3.3.l)non- ane,198 where the conformation of the equatorial form is fixed by the ring junction. As a result, the lone pair on oxygen cannot facilitate cleavage ofthe C-OR bond. This compound is hydrolyzed - l O I 3 times slower than the corresponding, axial 2,4-dinitrophenoxy acetal.
The antiperiplanar, lone-pair hypothesis is based on the presumption that the lifetime of a tetrahedral intermediate in hydrolysis is shorter than the average time of rotation about the C - 0 bond188; that is, the barrier of conformational transition exceeds the reaction barrier. However, a simple intermediate (hydrogen orthoester) has been detected,202 and on this basis, the antiperiplanar, lone-pair theory was questioned.203 Similarly, studies on the hydrolysis of saccharide derivative^^@'*^^^ indicated that the departure of
(201) V. G. S. Box, J. Heterocycl. Chem., 22 (1984) 891 -905. (202) B. Capon, J. H. Gall, and D. M. A. Grieve, J. Chem. SOC., Chem. Commun., (1976)
(203) C. L. Pemn and G. M. L. Arrhenius, J. Am. Chem. Soc., 104 (1982) 2839-2842. (204) L. Hosie, P. J. Marshall, and M. L. Sinnott, J. Chem. Soc., Perkin Trans. 2, (1984)
(205) A. J. Bennet and M. L. Sinnott, J. Am. Chem. SOC., 108 (1986) 7287-7294.
1034- 1035.
1121-1131.

120 IGOR TVAROSKA AND TOMAS BLEHA
the aglycon from the anomeric oxygen atom does not require the conforma- tions wherein a leaving group is in the up position relative to the lone pair on the ring-oxygen atom. Apparently, the interpretation of the reactivity differ- ences based on the selection of only one type of lone-pair interaction is not satisfactory for all cases.
Although interactions with the antibonding orbital are implicitly invoked in the antiperiplanar, lone-pair hypothesis, additional, coexisting, lone-pair interactions are the basis of another qualitative approach to rationalizing many reactions of the monosaccharides.189Jw~201 This approach utilizes the fact that the energy of the most reactive MOs increases with their interac- tions.
The magnitude of orbital interactions can only indirectly be evaluated from experiment, through geometrical changes, or photoelectronic spectra; however, they can be calculated by quantum chemistry. Perturbation MO calculations for dimetho~ymethane,'~~ elaborated in Section VI, showed that the dominant components in the frontier orbitals of dimethoxymethane are the oxygen lone-pairs combined with the antibonding orbitals of the CH,X group. This is illustrated in Fig. 27 for three highest-occupied (HO) molecular orbitals.
The theoretical analysis of orbital interactions in dimethoxymethane 148 is supported by photoelectron spectra of lY8-dioxadecalin, ls0 which predict the decreasing order of lone-pair interactions in the conformational sequence as being (up, up) > (up, sc) > (sc, sc). In all of the conformers, the energy of HOMO exceeds the energy of parent orbitals, and decreases in the same order as the energy of interaction. The enhancement of nucleophilicity of oxygen atoms relative to the noninteracting ones, and its variation with conformation, are the consequences of orbital interactions on the reactivity of molecules having an acetal segment. For example, the increase of electro- negativity of the glycosidic oxygen atom by some aglycons decreases the energy of oxygen lone-pairs and their interactions. As a result, changes in the valence geometry can be observed, as, for example, in axial and equatorial aryl-2-oxanyl acetals, where a linear correlation was found between the C - 0 bond length and the pK, value of the conjugated acid of the aglycon.206
Calculations 148 predicted that lone-pair interactions in the (up, up) con- formation corresponding to /3-glycosides are larger than in the (sc, sc) con- formation representing a-glycosides, and thus, the increase of nucleophilic- ity in the former isomers should be higher. The enhancement of oxygen nucleophilicity can be demonstrated in several, simple reactions. A suitable measure of nucleophilicity of the 0 - 1 atom is its rate of protonation in
(206) A. J. Briggs, R. Glenn, P. G. Jones, A. J. Kirby, and P. Ramaswamy, J. Am. Chem. SOC., 106 (1984) 6200-6206.

('p 8 ' 9 ) FIG. 27.- Symmetry of the Three Highest-Occupied MO Orbitals in Dimethoxymethane.'"

122 IGOR TVAROSKA AND TOMAS BLEHA
dimethyl sulfoxide, which is systematically higher in &aldopyranoses than in the corresponding a a n ~ m e r s . ~ ~ ~ Similarly, a higher rate of scission of the C- 1 - H- 1 bond was observed in the e form of 2-methoxyoxane than in the a form.z0s This rate is even lower in oxane, where lone-pair interactions are absent.
Lone-pair interactions by the through-space mechanism, and the increase of the nucleophilicity of the participating atoms, have been implicated in mechanistic proposals for the oxidation of acetals, and for some reductions of glyc~loses. '~~ On the other hand, the relative reactivities of the anomers in S N ~ processes has been discussed in terms of through-bond interactions of the lone pairs.lW Summing up the available evidence, it is clear that both types of lone-pair interaction must be recognized, in order to rationalize the stereoreactivity of saccharides.
According to the perturbation theory of chemical reactivity,200 the sym- metry and energy of the frontier orbitals determine the stereospecifity and the rate of reaction. We have shown that the mutual interactions of lone pairs of oxygen atoms, and their interactions with the properly oriented antibond- ing orbitals, actually affect several molecular properties of acetals, including the energy of the frontier MO and of orbitals in their vicinity. A more-rigor- ous account of the stereoreactivity differences should start from the com- plete picture of electron distribution and energies of molecular orbitals that are connected to relative rate-constants by the relations derived in the per- turbation theory of chemical reactivity.z00 Quantum chemical calculations made by following this procedure should help to clarify the role of the anomeric effect in the reactivity of saccharides.
In closing this short and incomplete, theoretical excursion into the field of reaction stereoselectivity in carbohydrates, we have also to mention that some other aspects in this article have been treated only partially; for in- stance, the anomeric effect in rings other than six-membered, or in carbohy- drate radicals. It is hoped that the present article has shown that many questions on the nature and various manifestations of the anomeric and related effects have already been answered. The problems still remaining to be solved will attract growing attention.zw-z'2 We believe that this survey,
(207) B. Gillet, D. J. Nicole, and J.-J. Delpuech, J. Chem. Soc., Perkin Trans. 2, (1981)
(208) V. Malatesta and K. U. Ingold, J. Am. Chem. Soc., 103 (1981) 609-614. (209) J. P. Praly and R. U. Lemieux, Can. J. Chem., 65 (1987) 213-223. (210) A. Cosse-Barbi and J. E. Dubois, J. Am. Chem. Soc., 109 (1987) 1503-151 1. (2 1 1 ) P. Aped, Y. Apeloig, A. Ellencweig, B. Fuchs, I. Goldberg, M. Karni, and E. Tartakovsky,
(212) D. G. Gorenstein, Chem. Rev., 87 (1987) 1047- 1077.
1329-1335.
J. Am. Chem. Soc., 109 (1987) 1486- 1495.

ANOMERIC AND EXO-ANOMERIC EFFECTS I23
based on theoretical conformational analysis, will contribute to the final solution of the puzzle named the anomeric effect.
ACKNOWLEDGMENT
In retrospect, the authors gratefully acknowledge the friendly and inspiring atmosphere in the laboratory of Prof. Robert H. Marchessault at the UniversitC de MontrCal, where, during their stay in the late seventies, the first thoughts of writing this article crystallized.