analyses of the presence of mutations in dystrophin protein to predict their relative influences in...
TRANSCRIPT

Cellular Signalling 26 (2014) 2857–2864
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r .com/ locate /ce l l s ig
Analyses of the presence of mutations in Dystrophin protein to predicttheir relative influences in the onset of Duchenne Muscular Dystrophy
Simanti Bhattacharya, Amit Das, Rakhi Dasgupta, Angshuman Bagchi ⁎Department of Biochemistry and Biophysics, University of Kalyani, Kalyani, Nadia, 741235 West Bengal, India
Abbreviations: Dp, Dystrophin; DMD, DuchenneDystrophin Associated Protein Complex; DG, DystrDystrophy, Dystroglycanopathy, Type C9; OMIM, OnlineABD, Actin Binding Domain; SpR, Spectrin Repeats; CRD,C-terminal region; CH, Calponin Homology; ABS, ActiAccessible Surface Area; PDB, Protein Data Bank.⁎ Corresponding author. Tel.: +91 9051948843; fax: +
E-mail addresses: [email protected], ang(A. Bagchi).
http://dx.doi.org/10.1016/j.cellsig.2014.09.0060898-6568/© 2014 Elsevier Inc. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 22 July 2014Accepted 5 September 2014Available online 16 September 2014
Keywords:Duchenne Muscular DystrophyDystrophin proteinPathogenic mutationsSeverity of mutationsMode of DMD onset
Muscle plays a vital role in the life of vertebrates like humans.Muscle contraction is the only criterion required forlocomotion.Musclefibers also play a vital role as the provider ofmechanical strength and act as a large repositoryof building blocks for protein synthesis in living beings. Muscles function as per the messages received from theextra-cellular signals. One of the central players responsible for capturing and transmission of extra-cellular sig-nals tomaintain the integrity ofmuscle function is the protein calledDystrophin (Dp). However, thewild typeDpprotein accumulates some mutations which lead to a severe disease called Duchenne Muscular Dystrophy(DMD). The disease is so frequent that it is known to affect 1 in 3500 newborns per year. There are a numberof reports that identify themutations leading to DMD. Interestingly, it is also observed that the type ofmutationsaffects the severity of the disease. But the biochemicalmechanismof theDMDonset is still obscure. In the presentscenario, an attempt has beenmade to analyze themutations in the development of the disease.We analyzed thechanges in secondary structure, solvent accessibility and stability of the Dp protein associated with the muta-tions. We tried to correlate the type of mutations with the severity of the disease. So far this is the first reportthat deals with the analyses of the mutations leading to DMD. This study would therefore be essential to comeup with a plausible mechanism of DMD disease onset.
© 2014 Elsevier Inc. All rights reserved.
1. Introduction
1.1. Background
Human growth and development are very much dependent on thestrength of muscle tissues, their masses and maintenance of properintegrity among them. Muscle plays a vital role as the provider ofmechanical strength and acts as a large repository of building blocksfor protein synthesis in living beings [1]. The main characteristic differ-ence of muscle fibers from other tissue types is their ability to contractwhen needed. Muscles in association with other connective tissuescarry out the movement of body parts as well as organs in living beings[2]. Among the three types of major muscle tissues, cardiac musclemaintains the activity of heart; smooth muscle tissue acts as the liningwall to vital organs; and skeletal muscle tissue confers us with theability to move and provides the necessary mechanical strengths to
Muscular Dystrophy; DAPC,oglycan; MDDGC9, MuscularMendelian Inheritance in Man;Cysteine Rich Domain; C-Term,n Binding Site; SASA, Solvent
91 33 2582 [email protected]
perform regular day-to-day activities like walking [3]. It is thereforequite obvious that malfunctioning of muscular tissues will lead to dis-ease conditions with varying degrees of severity. Muscle cell integrityis maintained by a number of factors. Notably among them is Dystro-phin Associated Protein Complex (DAPC) which physically connectsextra-cellular matrix proteins to cellular actin cytoskeleton and therebymaintains muscle cell membrane integrity and actin mediated musclecell contraction [4]. The eminent members of this DAPC complex areDystroglycan (DG), Sarcoglycans, and Dystrophin (Dp) in the orderfrom their places of action from cell-membrane to cytosol. Sarcospan,Syntrophin, Dystrobrevin and other partner proteins also interact withDp to maintain the cascade of flow of extra-cellular signals to the cellinterior [4]. The proteinDG serves as themain gateway for the transmis-sion of the extra-cellular signals through theDAPC complex. DG has twosubunits: alpha Dystroglycan (α-DG), the extra-cellular subunit whichserves as the receptor of extra-cellular signals obtained through itsextra-cellular partner ligand proteins and transmits the signal receivedfrom those partner proteins to the membrane bound subunit betaDystroglycan (β-DG), the second subunit of DG. This β-DG with itsC-terminal poly-proline rich region interacts with the WW domain ofthe cytosolic Dp protein which in turn interacts and regulates actincytoskeleton with its N terminal Actin Binding Domain (ABD). Thetrans-membrane sub-complex of Sarcoglycan with its gamma subunitinteracts with Dp protein whereas its delta subunit makes contactwith DGs. In this way the stem of DAPC forms the bridge linking

2858 S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
extra-cellular matrix to cytosolic actin cytoskeleton [5]. A pointmutation in α-DG, T192M, causes limb girdle muscular dystrophy,MDDGC9 [OMIM: 613818] which affects natural movement of individ-uals along with hampered cognitive inception and other symptoms [6].While looking into themolecular details of disease generation, we havefound that this single substitution of a polar amino acid by a non-polaramino acid (i.e., Threonine192 toMethionine192) has led to an increasein the hydrophobicity of the protein thereby decreasing its extent of in-teractions with its extra-cellular signaling partner, Laminin. This leadsto the onset of disease limb girdle muscular dystrophy. Now this phe-nomenon itself exemplifies the effect of mutations in the system. Onfurther analysis, it has been revealed that mutations in the membersof the DAPC protein complex as well as the mutations in their partnerproteins also cause different types of muscular dystrophies notablyamong them is the Duchenne Muscular Dystrophy (DMD), developedfrom the mutations in DMD gene that encodes Dp protein (427 kDa)[7]. Dp has four functional domains, namely, Actin Binding Domain,Spectrin Repeats, Cysteine Rich Domain and C-terminal. Mutations inthe functional regions of the Dp protein lead to onset ofmuscular disor-ders with varying degrees of severity. However, among the differenttypes of muscular dystrophies, DMD is the most severe one. DMD, anX-linked disorder, is the most commonmuscular dystrophy worldwideaffecting 1 in 3500 newborns yearly [8]. It has a high rate of morbidityand mortality, with a frequency of 20,000 deaths annually at earlychildhood. The large size of the DMD gene makes it susceptible towardmutations [7]. The mutations in the DMD gene and thereby in the Dpprotein not only causes DMD but also is associated with less severeform Becker Muscular Dystrophy and X-linked Cardiomyopathy [9].However, the severity and the type of muscular dystrophy are depen-dent on the nature of mutation and the region of occurrence of themutations in the Dp protein. The types of mutations leading to DMDhave been categorized as: deletions, duplication and point mutations. Ithas been found that the major contribution to the DMD disease comesfrom deletion types of mutations followed by duplication, both leadingto premature termination of translation of the DMD gene [10]. On theother hand, pointmutations in theDMD gene lead to formation ofmutat-ed protein products causing DMD. Although rigorous works have beendone to identify the mutations leading to DMD, the biochemical mecha-nism behind the onset of the DMD disease due to the point mutationsin the Dp protein is still not clear. In the present scenario, we made anattempt to analyze the point mutations in Dp protein and tried to corre-late the effects of the mutations with disease onset. For collection of themutation data we used information from patients suffering from DMDas mentioned in different databases. We categorized the mutationsas pathogenic or not and analyzed the probable structural changes inthe Dp protein associated with these mutations. Our analysis identifiedthe possible roles of the mutations in the onset of the DMD disease.We also tried to establish the relationship between type of mutationsand severity of the disease. This is so far the first bioinformatic reportthat analyzes the correlations between appearance of mutations andtheir corresponding effects on disease development. This work wouldtherefore be beneficial to explore the molecular details of DMD onset.
2. Materials and methods
2.1. Filtering mutations linked with DMD consequence
The amino acid sequences of the proteins involved in musculardystrophy have been extracted to collect the causative DMDmutations.The mutations have been isolated from three different databases:
1. UMD-DMD (http://www.umd.be/DMD/W_DMD/index.html):This database was built in a joined national effort involvingdifferent diagnostic laboratories in France to provide up-to-dateinformation about mutations in the DMD gene identified inpatients with dystrophinopathies.
2. DMD database for Utah (http://www.genome.utah.edu/DMD/): Thisdatabase provides uswith information about the newmutations. Thedatabase collects information from a number of different subjects.The data from subjects participating in the “Utah DystrophinopathyProject” have detailed historical and phenotypic information. Thedatabase also gathers information from samples received from otherphysicians not directly participating in the “Utah DystrophinopathyProject”. The phenotypes of these patients are also listed in the reportsas received by the physicians [11].
3. e-Dystrophin (http://www.cureduchenne.org/edystrophy.html):This database is basically a database reporting all variants of humanDp produced by in-frame DMD gene mutations. We searched thedatabase thoroughly and selected only those mutations that aredesignated as mis-sense mutations coding for a different aminoacid in Dp protein and have a DMD phenotype.
Somemutations in the record from ‘DMDdatabase for Utah’ showedphenotype B/DMD. Those were also taken into account. A total of 18unique mutations have been collected which are known to cause DMDphenotype in affected individuals. Table 1 presents a detailed list ofthe mutations collected for our analysis.
2.2. Processing of sequence to study the effects of the mutations
To analyze the effects of the mutations, a stretch of amino acids wasextracted from the original sequence of full length Dp protein(UniprotKB P11532) from human. We then incorporated the necessarymutations in the amino acid sequence of the wild type Dp protein oneby one to generate the mutant proteins. To consider the effect of themutation amino acid sequences were considered using a 201 aminoacid window as per [12]. For example if position X is the position ofinterest i.e., amino acid sequence position in the Dp protein wheremutation occurred in patients suffering from DMD, then the sequencestretch considered for our study would be X-100 to X + 100. For eachtype of mutation two sets i.e. wild type set and mutant set wereprepared. The final dataset contained a total of 36 sets of sequences.
2.3. Analysis of the effect of mutations
To assess the effects of the mutations on the structure and functionof the Dp protein we analyzed the following features of the wild typeand mutant Dp proteins:
a) The secondary structures of the protein before and after mutations:For analyzing secondary structure of the wild type and mutant Dpproteins PSI-PRED [13], SPPIDER [http://sppider.cchmc.org/sppider_doc.html] and CFSSP web servers [14] were used. We used a numberof different servers in order to get a consensus result [15].Thismethod is like a cross-validation method. If the mutated residuewas found to reside at turn positions then the type of the turn(whether type I or Type II turn [16]) was further detected withNetTurnP server [17].
b) The stability of the protein before and after the mutation: Thestabilities of the Dpproteins before and aftermutationwere analyzedwith Mupro server [http://www.ics.uci.edu/~baldig/mutation.html].
c) Protein hydropathy profile: This was checked with PROTPARAM[http://web.expasy.org/protparam/] server that employs KyteDolittle algorithm for assigning hydrophobicity score to each aminoacid. The effects of mutations on the variations of solvent accessibil-ities from the amino acid sequences of the wild type and mutantDp proteins were detected by SPPIDER and DSSP software [18].
d) The severity of the mutation: This was checked with Mutation asses-sor [http://mutationassessor.org/], PON-P [19], SIFT [20], Poly Phen2[20], SNAP [21], SNPs&GO servers [http://snps-and-go.biocomp.unibo.it/snps-and-go/] to generate a consensus conclusion. Aminoacid sequences of the wild type andmutant Dp proteins were furthergiven as an input to NCBI-Conserved domain search (http://www.

Table 1List of mutations. Total 18 mutations have been collected from the threedatabases for DMD and they have been grouped as per their occurrencesin the domains of Dystrophin.
Domains of Dystrophin Mutations
Actin binding domain (ABD) L54RL116P
Spectrin repeats (SpR) F590RD645GN793SE2890K
Cysteine rich domain (CRD) D3187GC3207RR3232HC3313FC3319RK3330TH3331NF3332CD3335YC3340R
C terminus (C-Term) K3383NQ3521K
2859S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and Pfam (http://pfam.xfam.org/) to find the changes in domain organization due tomutation.
2.4. Template search for homology modeling
Weused thewild type andmutant Dp proteins as inputs to search forthe corresponding templates using BLAST [22] against PDB database [23].
2.5. Building of homology model
2.5.1. N-terminal actin binding domain (ABD)The three dimensional structure of this domain is available from the
crystal structure of 1DXX [24]. The structure encompassed majority ofthe active sites residues (9–246 amino acid residues). Thus we usedthis structure as our working model for studying the effects of themutations on the N-terminal Actin Binding Domain (ABD) of the Dpprotein. The stereo-chemical qualities of the structure were checkedusing Procheck [25] and Verify3D [26]. No residues were found to bepresent in the disallowed region of the Ramachandran Plot [27] asobserved from Procheck.We then used the structure to insert themuta-tions which are reported to cause DMD at positions 54 (Leu54Arg) and116 (Leu116Pro) using Accelrys Discovery Studio Package (DS) 2.5. Themutants so generatedwere further energyminimized in explicit solventwith 1000 cycles of Conjugate Gradient algorithm using the CHARMMforce fields [28] until the final root mean squared deviations (RMSD)gradient between the energies of the mutant Dp structures became0.001 Kcal/mole. The stereo-chemical qualities of the built modelswere again checked as mentioned before.
2.5.2. Modeling of amino acid residues 2885–3345There are 10mutations, spanning the amino acid stretch from residue
2885 to residue 3345 of Dp. But unfortunately no single template wasfound for the said amino acid stretch. Sowe employed interactivemodel-ing from Protein model portal tool [http://www.proteinmodelportal.org/?pid=modelling_interactive] to generate the model for this stretchof sequence applying multiple templates. Three independenttemplates have been found to provide the best match for the afore-mentioned stretch of amino acid sequences. The PDB codes for thetemplates are
1EG3 for amino acid residues 3047–3306,1U4Q for amino acid residues 2885–3041 and2E5R for amino acid residue 3312–3334.
Homology model of the entire stretch of amino acids (2885 toresidue 3345) was built using the aforementioned templates and thenvalidated as per the aforementioned protocol.Mutations at their respec-tive positions have been inserted similarly aswasmentioned in the caseof insertions of N terminal mutations. All the structures generated aftermutations were energy minimized and taken for consideration aftertheir structural verification with Procheck and Verify 3D. Only thosemodels were selected which passed Verify3D and Procheck.
2.6. Analysis of the structures
Analyses of hydrogen bond formation, SASA in the protein structureshave been carried out with DS 2.5 software package, VMD 1.9.1 andSDM server [29].
3. Results
In this work we have collected a set of mis-sense point mutationsfrom three different online repositories for DMD patients. We haveselected all themis-sense mutations (Table 1) whichwere clearly iden-tified as the disease causing ones though the extent of the severity ofDMD is not the same for all these mutations. Our basic aim is to classifythe mutations as per the severity of DMD onset and to come up with aplausible biochemical mechanism for the effects of these mutations tocause DMD. These mutations are distributed throughout the entire Dp(Table 1). As we have mentioned earlier that Dp has four distinctfunctional domains: Actin Binding Domain (ABD; spanning amino acidresidues 1–246), Spectrin Repeat Domain (SpR; spanning amino acidresidues 253–3040), Cysteine Rich Domain (CRD; spanning aminoacid residues 3080–3360) and C terminal region (C-term; spanningamino acid residues 3361–3685). We have categorized the mutationsas per their presence in these domains. The following sections willdiscuss about the outcomes of these mutations.
3.1. Mutations in actin binding domain (ABD)
The N-terminal Actin Binding Domain (ABD) of Dp protein com-prises amino acid residues 1–246 and harbors two Calponin Homology(CH) domains, viz., CH1 spanning amino acid residues 15–119 and CH2spanning amino acid residues 134–237. TheDpABDacts as dimer and inthe dimeric form two such ABDs orient themselves in an antiparallelmanner thereby protecting the hydrophobic linker region that linksCH1 and CH2. This CH1 has Actin Binding Site1 (ABS1) comprisingamino acid residues 21–27 and ABS2 comprising of amino acid residues88–116. Similarly, CH2 has ABS3 comprising amino acid residues131–157. At the time of actin binding, the ABD from the dimeric Dpprotein opens up to make ABSs accessible to actin [24]. From our setof collected mutations, L54R and L116P fall in the CH1 of ABD. TheLeu116 is one of the main actin binding residues residing at ABS2.Interestingly, both of the aforementioned mutations have been identi-fied as highly pathogenic (supplementary file1). These two mutations(L54R and L116P) have not been found to cause any significant changesin the secondary structures of the Dp protein (supplementary file2).However, the mutation L54R generates a turn at amino acid residue55. This substitution of a non-polar Leucine (L) with polar Arginine(R) has also caused the site to become more hydrophilic than its wildtype form (supplementary file3).We then searched the NCBI conserveddomain database (CDD) for identification of possible domain architec-ture (in addition to the existing ABD) in Dp with a window of 100amino acids keeping the 54 L amino acid at the center as mentionedin the Materials and methods section. The search result revealed thepresence of Sac6 domain which has been reported to bind and regulateactin. Interestingly, the appearance of this Sac6 domain is abolishedwhen 54 L is mutated to R. The mutations also decreased the stabilityof the Dp dimer by lowering the total numbers of intermolecular hydro-gen bonds (Fig. 1A) formed between the dimmers (343in wild type
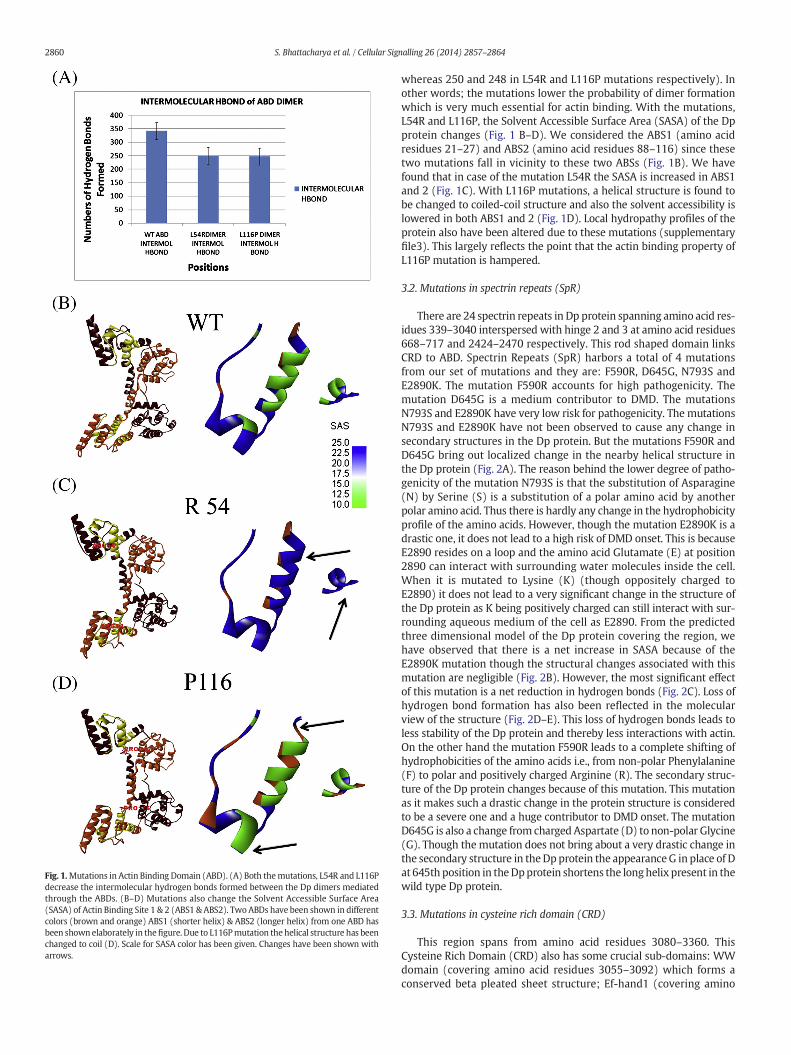
Fig. 1.Mutations in Actin Binding Domain (ABD). (A) Both themutations, L54R and L116Pdecrease the intermolecular hydrogen bonds formed between the Dp dimers mediatedthrough the ABDs. (B–D) Mutations also change the Solvent Accessible Surface Area(SASA) of Actin Binding Site 1 & 2 (ABS1 & ABS2). Two ABDs have been shown in differentcolors (brown and orange) ABS1 (shorter helix) & ABS2 (longer helix) from one ABD hasbeen shown elaborately in thefigure. Due to L116Pmutation thehelical structure has beenchanged to coil (D). Scale for SASA color has been given. Changes have been shown witharrows.
2860 S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
whereas 250 and 248 in L54R and L116P mutations respectively). Inother words; the mutations lower the probability of dimer formationwhich is very much essential for actin binding. With the mutations,L54R and L116P, the Solvent Accessible Surface Area (SASA) of the Dpprotein changes (Fig. 1 B–D). We considered the ABS1 (amino acidresidues 21–27) and ABS2 (amino acid residues 88–116) since thesetwo mutations fall in vicinity to these two ABSs (Fig. 1B). We havefound that in case of the mutation L54R the SASA is increased in ABS1and 2 (Fig. 1C). With L116P mutations, a helical structure is found tobe changed to coiled-coil structure and also the solvent accessibility islowered in both ABS1 and 2 (Fig. 1D). Local hydropathy profiles of theprotein also have been altered due to these mutations (supplementaryfile3). This largely reflects the point that the actin binding property ofL116P mutation is hampered.
3.2. Mutations in spectrin repeats (SpR)
There are 24 spectrin repeats in Dp protein spanning amino acid res-idues 339–3040 interspersed with hinge 2 and 3 at amino acid residues668–717 and 2424–2470 respectively. This rod shaped domain linksCRD to ABD. Spectrin Repeats (SpR) harbors a total of 4 mutationsfrom our set of mutations and they are: F590R, D645G, N793S andE2890K. The mutation F590R accounts for high pathogenicity. Themutation D645G is a medium contributor to DMD. The mutationsN793S and E2890K have very low risk for pathogenicity. The mutationsN793S and E2890K have not been observed to cause any change insecondary structures in the Dp protein. But the mutations F590R andD645G bring out localized change in the nearby helical structure inthe Dp protein (Fig. 2A). The reason behind the lower degree of patho-genicity of the mutation N793S is that the substitution of Asparagine(N) by Serine (S) is a substitution of a polar amino acid by anotherpolar amino acid. Thus there is hardly any change in the hydrophobicityprofile of the amino acids. However, though the mutation E2890K is adrastic one, it does not lead to a high risk of DMD onset. This is becauseE2890 resides on a loop and the amino acid Glutamate (E) at position2890 can interact with surrounding water molecules inside the cell.When it is mutated to Lysine (K) (though oppositely charged toE2890) it does not lead to a very significant change in the structure ofthe Dp protein as K being positively charged can still interact with sur-rounding aqueous medium of the cell as E2890. From the predictedthree dimensional model of the Dp protein covering the region, wehave observed that there is a net increase in SASA because of theE2890K mutation though the structural changes associated with thismutation are negligible (Fig. 2B). However, the most significant effectof this mutation is a net reduction in hydrogen bonds (Fig. 2C). Loss ofhydrogen bond formation has also been reflected in the molecularview of the structure (Fig. 2D–E). This loss of hydrogen bonds leads toless stability of the Dp protein and thereby less interactions with actin.On the other hand the mutation F590R leads to a complete shifting ofhydrophobicities of the amino acids i.e., from non-polar Phenylalanine(F) to polar and positively charged Arginine (R). The secondary struc-ture of the Dp protein changes because of this mutation. This mutationas it makes such a drastic change in the protein structure is consideredto be a severe one and a huge contributor to DMD onset. The mutationD645G is also a change from charged Aspartate (D) to non-polar Glycine(G). Though themutation does not bring about a very drastic change inthe secondary structure in theDpprotein the appearanceG in place of Dat 645th position in theDpprotein shortens the longhelix present in thewild type Dp protein.
3.3. Mutations in cysteine rich domain (CRD)
This region spans from amino acid residues 3080–3360. ThisCysteine Rich Domain (CRD) also has some crucial sub-domains: WWdomain (covering amino acid residues 3055–3092) which forms aconserved beta pleated sheet structure; Ef-hand1 (covering amino
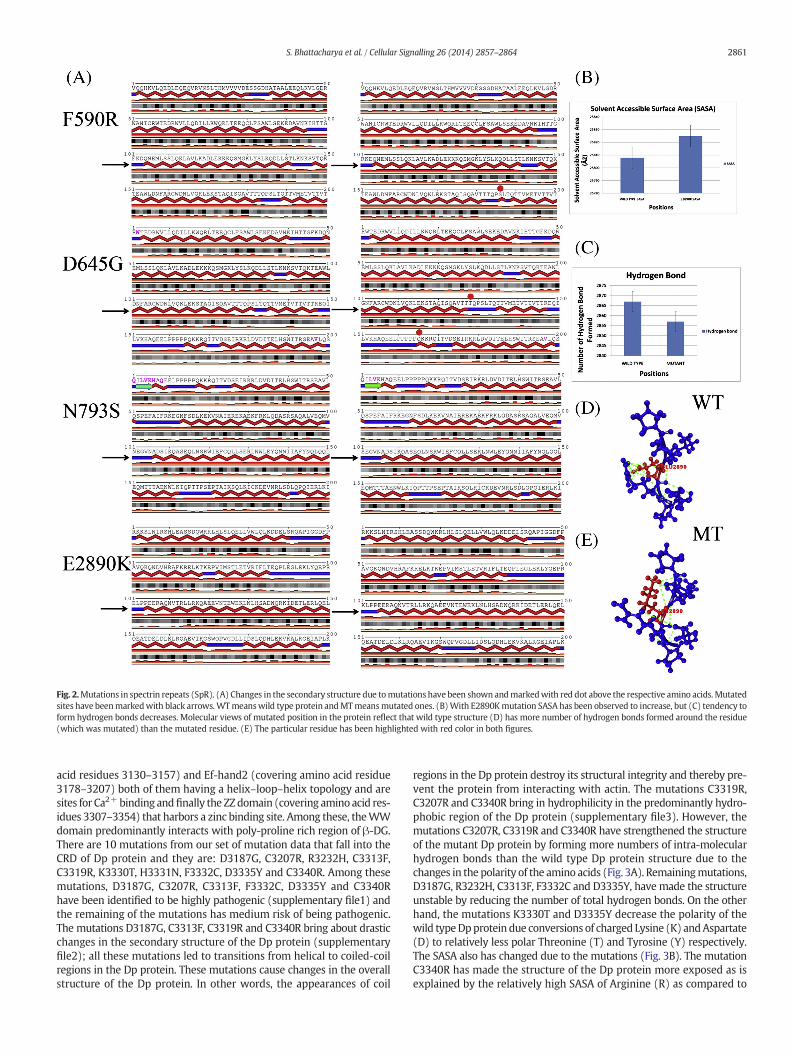
Fig. 2.Mutations in spectrin repeats (SpR). (A) Changes in the secondary structure due tomutations have been shown andmarkedwith red dot above the respective amino acids. Mutatedsites have beenmarkedwith black arrows.WTmeanswild type protein andMTmeansmutated ones. (B)With E2890Kmutation SASA has been observed to increase, but (C) tendency toform hydrogen bonds decreases. Molecular views of mutated position in the protein reflect that wild type structure (D) has more number of hydrogen bonds formed around the residue(which was mutated) than the mutated residue. (E) The particular residue has been highlighted with red color in both figures.
2861S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
acid residues 3130–3157) and Ef-hand2 (covering amino acid residue3178–3207) both of them having a helix–loop–helix topology and aresites for Ca2+binding andfinally the ZZdomain (covering amino acid res-idues 3307–3354) that harbors a zinc binding site. Among these, theWWdomain predominantly interacts with poly-proline rich region of β-DG.There are 10 mutations from our set of mutation data that fall into theCRD of Dp protein and they are: D3187G, C3207R, R3232H, C3313F,C3319R, K3330T, H3331N, F3332C, D3335Y and C3340R. Among thesemutations, D3187G, C3207R, C3313F, F3332C, D3335Y and C3340Rhave been identified to be highly pathogenic (supplementary file1) andthe remaining of the mutations has medium risk of being pathogenic.The mutations D3187G, C3313F, C3319R and C3340R bring about drasticchanges in the secondary structure of the Dp protein (supplementaryfile2); all these mutations led to transitions from helical to coiled-coilregions in the Dp protein. These mutations cause changes in the overallstructure of the Dp protein. In other words, the appearances of coil
regions in the Dp protein destroy its structural integrity and thereby pre-vent the protein from interacting with actin. The mutations C3319R,C3207R and C3340R bring in hydrophilicity in the predominantly hydro-phobic region of the Dp protein (supplementary file3). However, themutations C3207R, C3319R and C3340R have strengthened the structureof the mutant Dp protein by forming more numbers of intra-molecularhydrogen bonds than the wild type Dp protein structure due to thechanges in the polarity of the amino acids (Fig. 3A). Remainingmutations,D3187G, R3232H, C3313F, F3332C and D3335Y, have made the structureunstable by reducing the number of total hydrogen bonds. On the otherhand, the mutations K3330T and D3335Y decrease the polarity of thewild typeDpprotein due conversions of charged Lysine (K) andAspartate(D) to relatively less polar Threonine (T) and Tyrosine (Y) respectively.The SASA also has changed due to the mutations (Fig. 3B). The mutationC3340R has made the structure of the Dp protein more exposed as isexplained by the relatively high SASA of Arginine (R) as compared to
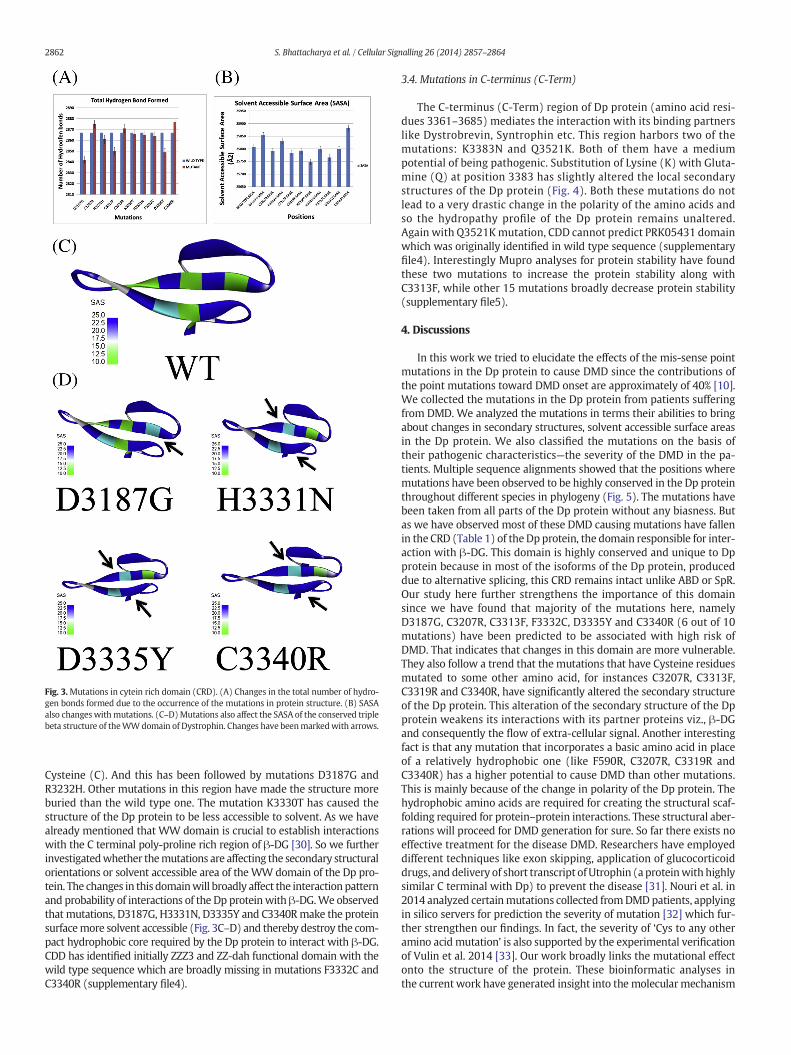
Fig. 3.Mutations in cytein rich domain (CRD). (A) Changes in the total number of hydro-gen bonds formed due to the occurrence of the mutations in protein structure. (B) SASAalso changeswithmutations. (C–D)Mutations also affect the SASA of the conserved triplebeta structure of theWWdomain of Dystrophin. Changes have beenmarkedwith arrows.
2862 S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
Cysteine (C). And this has been followed by mutations D3187G andR3232H. Other mutations in this region have made the structure moreburied than the wild type one. The mutation K3330T has caused thestructure of the Dp protein to be less accessible to solvent. As we havealready mentioned that WW domain is crucial to establish interactionswith the C terminal poly-proline rich region of β-DG [30]. So we furtherinvestigatedwhether themutations are affecting the secondary structuralorientations or solvent accessible area of the WW domain of the Dp pro-tein. The changes in this domainwill broadly affect the interaction patternand probability of interactions of the Dp proteinwith β-DG.We observedthat mutations, D3187G, H3331N, D3335Y and C3340Rmake the proteinsurfacemore solvent accessible (Fig. 3C–D) and thereby destroy the com-pact hydrophobic core required by the Dp protein to interact with β-DG.CDD has identified initially ZZZ3 and ZZ-dah functional domain with thewild type sequence which are broadly missing in mutations F3332C andC3340R (supplementary file4).
3.4. Mutations in C-terminus (C-Term)
The C-terminus (C-Term) region of Dp protein (amino acid resi-dues 3361–3685) mediates the interaction with its binding partnerslike Dystrobrevin, Syntrophin etc. This region harbors two of themutations: K3383N and Q3521K. Both of them have a mediumpotential of being pathogenic. Substitution of Lysine (K) with Gluta-mine (Q) at position 3383 has slightly altered the local secondarystructures of the Dp protein (Fig. 4). Both these mutations do notlead to a very drastic change in the polarity of the amino acids andso the hydropathy profile of the Dp protein remains unaltered.Again with Q3521Kmutation, CDD cannot predict PRK05431 domainwhich was originally identified in wild type sequence (supplementaryfile4). Interestingly Mupro analyses for protein stability have foundthese two mutations to increase the protein stability along withC3313F, while other 15 mutations broadly decrease protein stability(supplementary file5).
4. Discussions
In this work we tried to elucidate the effects of the mis-sense pointmutations in the Dp protein to cause DMD since the contributions ofthe point mutations toward DMD onset are approximately of 40% [10].We collected the mutations in the Dp protein from patients sufferingfrom DMD. We analyzed the mutations in terms their abilities to bringabout changes in secondary structures, solvent accessible surface areasin the Dp protein. We also classified the mutations on the basis oftheir pathogenic characteristics—the severity of the DMD in the pa-tients. Multiple sequence alignments showed that the positions wheremutations have been observed to be highly conserved in the Dp proteinthroughout different species in phylogeny (Fig. 5). The mutations havebeen taken from all parts of the Dp protein without any biasness. Butas we have observed most of these DMD causing mutations have fallenin the CRD (Table 1) of the Dp protein, the domain responsible for inter-action with β-DG. This domain is highly conserved and unique to Dpprotein because in most of the isoforms of the Dp protein, produceddue to alternative splicing, this CRD remains intact unlike ABD or SpR.Our study here further strengthens the importance of this domainsince we have found that majority of the mutations here, namelyD3187G, C3207R, C3313F, F3332C, D3335Y and C3340R (6 out of 10mutations) have been predicted to be associated with high risk ofDMD. That indicates that changes in this domain are more vulnerable.They also follow a trend that the mutations that have Cysteine residuesmutated to some other amino acid, for instances C3207R, C3313F,C3319R and C3340R, have significantly altered the secondary structureof the Dp protein. This alteration of the secondary structure of the Dpprotein weakens its interactions with its partner proteins viz., β-DGand consequently the flow of extra-cellular signal. Another interestingfact is that any mutation that incorporates a basic amino acid in placeof a relatively hydrophobic one (like F590R, C3207R, C3319R andC3340R) has a higher potential to cause DMD than other mutations.This is mainly because of the change in polarity of the Dp protein. Thehydrophobic amino acids are required for creating the structural scaf-folding required for protein–protein interactions. These structural aber-rations will proceed for DMD generation for sure. So far there exists noeffective treatment for the disease DMD. Researchers have employeddifferent techniques like exon skipping, application of glucocorticoiddrugs, and delivery of short transcript of Utrophin (a proteinwith highlysimilar C terminal with Dp) to prevent the disease [31]. Nouri et al. in2014 analyzed certainmutations collected fromDMDpatients, applyingin silico servers for prediction the severity of mutation [32] which fur-ther strengthen our findings. In fact, the severity of ‘Cys to any otheramino acid mutation’ is also supported by the experimental verificationof Vulin et al. 2014 [33]. Our work broadly links the mutational effectonto the structure of the protein. These bioinformatic analyses inthe current work have generated insight into the molecularmechanism

Fig. 4.Mutations in the C-terminal region (C-Term). Changes in the secondary structure due to mutations have been shown and marked with red dot above the respective amino acids.Mutated sites have been marked with black arrow head. WT means wild type protein and MT means mutated protein.
2863S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
of disease onset by these different types of mis-sense mutations.These structural elaborations will definitely be helpful in developingnew therapeutic approaches with targeted delivery to combat this dis-ease that causes high rate of morbidity in early childhood of affectedindividuals.
5. Conclusions
In our current study, we have tried to explore the effect of mis-sensepoint mutations on the structures of the Dp protein. The point muta-tions we have gathered here also have been observed to affect theprotein structure in various ways which may lead to disturb the mode
Fig. 5. Conservation of the mutated region throughout phylogeny. The positions where mis
of interactions of the Dp protein. One set of mutations have beenobserved to affect the secondary structure whereas other sets ofmutations have been observed to affect the hydropathy profile of theDp protein by either making it more accessible to water or by buryingthe local area deep inside the protein core and thereby altering thesolvent accessible surface area. But in all the cases, these effectswill per-turb the structure of the protein and thereby hamper the interaction ofthe mutated protein with its partner proteins. And this may ultimatelylead to an interruption in the signal transduction process crucial forcell development. This molecular insight generated from our extensivestudywill definitely shed some light on developing effective therapeuticapproach against this disease.
-sense point mutations have occurred are highly conserved throughout the phylogeny.

2864 S. Bhattacharya et al. / Cellular Signalling 26 (2014) 2857–2864
Conflict of interest
The authors declare no conflict of interests.
Acknowledgment
The authors are really grateful to the BIF Center, Dept of Biochemis-try and Biophysics, University of Kalyani for providing the necessaryequipments and workstation to carry out the experiments. SB and ADalso are thankful to UGC, India and CSIR, India for their respectivefellowships. The authors would like to acknowledge the DST-PURSEprogram 2012–2015 going on in the Department of Biochemistry andBiophysics, University of Kalyani and the DBT (project no. BT/PR6869/BID/7/417/2013) for the support.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.cellsig.2014.09.006.
References
[1] T.A. Churchward-Venne, C.H. Murphy, T.M. Longland, S.M. Phillips, Amino Acids 45(2013) 231–240.
[2] A. Uezumi, M. Ikemoto-Uezumi, K. Tsuchida, Front. Physiol. 5 (2014) 68.[3] C. Batters, C. Veigel, E. Homsher, J.R. Sellers, Front. Physiol. 5 (2014) 90.[4] J. Ehmsen, E. Poon, K. Davies, J. Cell Sci. 115 (2002) 2801–2803.[5] H.L. Sweeney, E.R. Barton, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 13464–13466.[6] S. Bhattacharya, A. Das, S. Ghosh, R. Dasgupta, A. Bagchi, Gene 537 (2014) 108–114.[7] E.P. Hoffman, R.H. Brown Jr., L.M. Kunkel, Cell 51 (1987) 919–928.[8] P. Morris, Paediatr. Anaesth. 7 (1997) 1–4.[9] A. Holland, S. Carberry, K. Ohlendieck, Curr. Protein Pept. Sci. 14 (2013) 680–697.
[10] E.P. Hoffman, A. Bronson, A.A. Levin, S. Takeda, T. Yokota, A.R. Baudy, E.M. Connor,Am. J. Pathol. 179 (2011) 12–22.
[11] K.M. Flanigan, D.M. Dunn, A. von Niederhausern, P. Soltanzadeh, E. Gappmaier, M.T.Howard, J.B. Sampson, J.R. Mendell, C. Wall, W.M. King, A. Pestronk, J.M. Florence,A.M. Connolly, K.D. Mathews, C.M. Stephan, K.S. Laubenthal, B.L. Wong, P.J.Morehart, A. Meyer, R.S. Finkel, C.G. Bonnemann, L. Medne, J.W. Day, J.C. Dalton, M.K. Margolis, V.J. Hinton, Hum. Mutat. 30 (2009) 1657–1666, http://dx.doi.org/10.1002/humu.21114.
[12] J.Y. Chen, E. Youn, S.D. Mooney, Methods Mol. Biol. 541 (2009) 449–461.[13] D.W.A. Buchan, F. Minneci, T.C.O. Nugent, K. Bryson, D.T. Jones, Nucleic Acids Res. 41
(W1) (2013) W340–W348.[14] A.T. Kumar, Wide Spectr. 1 (2013) 15–19.[15] A. Bagchi, Gene 25 (2013) 207–210.[16] C.M. Wilmot, J.M. Thornton, J. Mol. Biol. 203 (1988) 221–232.[17] B. Petersen, C. Lundegaard, T.N. Petersen, PLoS One 5 (2010) e15079.[18] W. Kabsch, C. Sander, Biopolymers 22 (1983) 2577–2637.[19] A. Olatubosun, J. Väliaho, J. Härkönen, J. Thusberg, M. Vihinen, Hum. Mutat. 33
(2012) 1166–1174.[20] S.E. Flanagan, A.M. Patch, S. Ellard, Genet. Test. Mol. Biomark. 14 (2010) 533–537.[21] Y. Bromberg, G. Yachdav, B. Rost, Bioinformatics 24 (2008) 2397–2398.[22] S.F. Altschul, W. Gish, W. Miller, E.W. Myers, D.J. Lipman, J. Mol. Biol. 215 (1990)
403–410.[23] H.M. Berman, Acta Crystallogr. A 64 (2008) 88–95.[24] F.L. Norwood, A.J. Sutherland-Smith, N.H. Keep, J. Kendrick-Jones, Structure 8 (2000)
481–491.[25] R.A. Laskowski, M.W. MacArthur, D.S. Moss, J.M. Thornton, J. Appl. Crystallogr. 26
(1993) 283–291.[26] D. Eisenberg, R. Lüthy, J.U. Bowie, Methods Enzymol. 277 (1997) 396–404.[27] G.N. Ramachandran, C. Ramakrishnan, V. Sasisekharan, J. Mol. Biol. 7 (1963) 95–99.[28] B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, M. Karplus,
J. Comput. Chem. 4 (1983) 187–217.[29] C.L. Worth, R. Preissner, T.L. Blundell, Nucleic Acids Res. 39 (Web Server issue)
(2011) W215–W222.[30] Z. Salah, A. Alian, R.I. Aqeilan, Front. Biosci. (Landmark Ed) 17 (2012) 331–348.[31] E. Mercuri, F. Muntoni, Curr. Opin. Pediatr. 25 (2013) 701–707.[32] N. Nouri, E. Fazel-Najafabadi, M. Behnam, N. Nouri, O. Aryani, M. Ghasemi, J. Nasiri,
M. Sedghi, Gene 535 (2014) 250–254.[33] A. Vulin, N. Wein, D.M. Strandjord, E.K. Johnson, A.R. Findlay, B. Maiti, M.T. Howard,
Y.J. Kaminoh, L.E. Taylor, T.R. Simmons, W.C. Ray, F. Montanaro, J.M. Ervasti, K.M.Flanigan, Hum. Mutat. 35 (2014) 257–264.