a vascular connection to alzheimer's disease
TRANSCRIPT

REVIEW ARTICLE
A Vascular Connection toAlzheimer’s Disease
JOHANNES A. RHODIN* AND TOM THOMAS*†
*Department of Anatomy, College of Medicine, University of South Florida,Tampa, FL, USA; and †Woodlands Medical and Research Center,
Oldsmar, FL, USA
ABSTRACT
Alzheimer’s disease (AD) is characterized by a progressive and debilitating de-mentia in elderly people. The causes of this disease are not known, but major riskfactors include old age and a family history of dementia, Down’s syndrome,female gender, low level of education, and head injury. There is no known curefor Alzheimer’s disease. The disease is characterized by abnormal accumulationof amyloid-b peptide and the protein Tau in the nerve cells and extracellularspace of certain regions of the brain. Cerebral amyloid angiopathy is anothermarker for Alzheimer’s disease. In autopsies, small cerebral arterial blood vesselsand capillaries show signs of inflammation, amyloid accumulations, and a focalbreach of the blood–brain barrier. This review focuses on the results of recentinvestigations of vascular responses to infusion of amyloid-b1-40, the means ofpreventing vascular damage, using a live animal (rat) model, and the combina-tion of intravital video recordings of the mesenteric microvascular bed withelectron microscopic analyses of the same vascular segments. We propose thatthe cerebral vascular changes in patients with Alzheimer’s disease probablyprecede the neuronal damage and dementia. Microcirculation (2001) 8, 207–220.
KEY WORDS: Alzheimer’s disease (AD), antioxidants, amyloid-b, cytokine block-ers, electron microscopic analysis, endothelium, estrogen, inflammation, intra-vital video recording, leukocyte transmigration, microcirculation, selective es-trogen receptor modulator (SERM), superoxide dismutase
INTRODUCTION
Alzheimer’s disease (AD) is characterized by a de-bilitating dementia in elderly people. It is a progres-sive, degenerative disease that affects the brain andresults in impaired memory, thinking, and behavior.It is the most common form of dementia, affectingabout 50% of individuals over the age of 80. Theonset of Alzheimer’s disease is usually slow and in-sidious, and as a result, symptoms emerge slowlyand often go unrecognized. In the final stages of this
disease, patients suffer from severe symptoms in-cluding weight loss, loss of bowel and bladder con-trol, total dependence on caregiver, and secondaryillnesses, ultimately leading to death. Biopsies andpostmortem autopsies show that nerve cells and theextracellular space in certain parts of the brain areinfiltrated by neurofibrillary tangles and plaques ofa toxic protein, amyloid-b, which interferes with themetabolic processes of the nerve cells, and ultimatelydamages the cells (46). The same brain sections alsoshow that brain capillaries, small arteries, and arte-rioles (the end-segment of a small artery, connectingit with the capillary) show signs of vascular inflam-mation and amyloid accumulations (17,25). There isno known cure for Alzheimer’s disease, but certaindrugs that elevate the level of the neurotransmitteracetylcholine in the brain will delay slightly the pro-gression of the dementia.
Supported by National Institute on Aging Grant No. 1PO1AG16223 and Wyeth-Ayerst Pharmaceuticals.For reprints of this article, contact J. Rhodin, M.D., Ph.D., De-partment of Anatomy, College of Medicine, University of SouthFlorida, 12901 Bruce B. Downs Blvd., Tampa, FL 33612, USA;e-mail: [email protected] February 9, 2001; accepted 25 March 2001.
Microcirculation (2001) 8, 207–220© 2001 Nature Publishing Group 1073-9688/01 $17.00www.nature.com/mn

It is not known what causes Alzheimer’s disease, andspecific risk factors for Alzheimer’s disease are dif-ficult to isolate. It is believed that major risk factorsfor this disease include old age and a family historyof Alzheimer’s disease. By age 90, approximately50% of those who have siblings or parents with Alz-heimer’s disease will also have developed this dis-ease. Genetic defects on chromosomes 1, 14, and 21have been associated with a form of Alzheimer’s dis-ease that strikes people early in life. The sporadicform of Alzheimer’s disease that occurs later in lifehas been associated with apolipoprotein (ApoE4allele),a component of a gene on chromosome 19. Other po-tential risk factors include Down’s syndrome (a ge-netic neurological disorder associated with an extracopy of chromosome 21), gender (this disease affectswomen more frequently than men), educational sta-tus (lack of education is an apparent risk factor),and head injury (single or multiple trauma can pro-duce brain abnormalities similar to those seen inAlzheimer’s disease).
All forms of Alzheimer’s disease involve a commonevent: the excessive accumulation in the brain of aprotein, amyloid-b (Ab). It accumulates in the spacebetween the nerve cells, is toxic to nerve cells, andinterferes with the normal function of the brain. It isformed by specific endoproteolytic cleavages of theamyloid-b protein precursor (bAPP), which is en-coded by a gene on chromosome 21 and is con-stitutively secreted by both neuronal and othercells into extracellular fluids throughout life. Theamyloid-b protein precursor is a glycoprotein thatcan exist as both a membrane-bound cell surfacereceptor, and a soluble form (20). The concertedaction of the enzymes b- and g-secretase cleaves thebAPP to form the cytotoxic peptide Ab. Plateletscarry large amounts of amyloid-b and amyloid pro-tein precursor (2,5,6,39). There is uncertaintywhether there is an overproduction of amyloid-b, animpaired clearance, or both (10). The prolonged ex-posure to the nondegradable but potentially toxicamyloid protein probably initiates a continuous im-mune reaction that causes further cell injury andneuronal destruction. All Alzheimer’s disease pa-tients develop neuritic plaques. The neuritic plaquesconsist of extracellular masses of amyloid-b fila-ments intimately associated with dystrophic den-drites and axons, activated microglia, and reactiveastrocytes. Virtually all patients also have manyneurofibrillary tangles, intraneuronal bundles ofpaired helical filaments composed of highly phos-phorylated forms of the microtubule-associated pro-tein Tau.
The initial description of the pathological findings inthe brains of patients suffering from Alzheimer’s dis-ease by Dr. Alois Alzheimer in 1907 clearly indicatedcerebrovascular damage. But the vascular dysfunc-tion in Alzheimer’s disease brains has not receivedmuch attention.
However, cerebrovascular amyloidosis or cerebralamyloid angiopathy has been established as anothermarker for Alzheimer’s disease. The component ofcerebrovascular amyloid is thought to be identicalwith that of senile plaque amyloid. Several investiga-tors (7,9,14,15,17,18,21,28,39,40,42,56,57,61,62)have expressed the opinion that the primary site ofthe onset of Alzheimer’s disease, and the target of thetoxic amyloid, are the small arteries, arterioles, andcapillaries of the central nervous system. They basedtheir hypothesis on the fact that these blood vesselsshow signs of inflammation, amyloid accumulations,and a focal breach of the blood–brain barrier(29,55,60). According to this theory, the blood ves-sels are damaged first, causing the toxic amyloid tobe released and subsequently infiltrate the nervecells. In addition, there is a narrowing of the vascularlumen, leading to a reduction in cerebral blood flow,low oxygen tension, and neuronal damage.
Other investigators (47) used an in vitro technique ofisolating small rings of the rat aorta, exposing therings to amyloid-b for a short time period. The innerlining consists of flat endothelial cells. The exposureto amyloid-b caused damage to the endothelial cells,disrupting the lining and exposing the underlyingsmooth muscle cells and the connective tissue. Theypostulated that the amyloid-b interacts with endo-thelial cells to produce an excess of superoxide radi-cals. The superoxide radical can scavenge endothe-lium-derived relaxing factor and produce potent oxi-dizing agents, which can cause lipid peroxidationand other degenerative changes. Thus, the superox-ide radicals interfere with the normal metabolism ofthe endothelial cells. Thomas and colleagues (47)proved the validity of this theory by exposing theaortic rings to an antioxidant, superoxide dismutase(SOD), which is a scavenger of free radicals, beforeexposing the blood vessels to amyloid-b. The effectwas most remarkable. No damage occurred to theendothelial cells. Similar effects were also observedin the cerebral blood vessels (49).
In an additional experiment, Sutton and colleagues(44) cultured endothelial cells from rat aorta andexposed them to amyloid-b as well as to SOD. Theeffects were similar to the aortic ring experiments.Cells exposed to amyloid-b were disrupted, whereas
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
208

no damage occurred if the cells first were exposed toSOD.
However, the “acid test” was to use a technique bywhich the effects of amyloid-b on blood vessels canbe studied in the living animal. This was done in aseries of experiments by Thomas and Rhodin andtheir colleagues (32,33,45,48,49,50,51,52).
STUDY IN THE LIVING ANIMAL
The mesenteric membrane holds the intestines inplace and also serves as a conduit for blood vessels,carrying blood to and from the intestines. The mes-enteric membrane is also a place for fat deposition.Before the fat cells develop, a capillary network islaid down, and small arterioles and their venouscounterparts, the venules, give rise to capillarysprouts that fuse with each other and form the cap-illary net. The mesenteric membrane is very thin,and the blood vessels are quite transparent and ar-ranged in a two-dimensional pattern (Fig. 1). This isthe ideal preparation for the study of the reactivity ofsmall blood vessels. The technique was first used byChambers and Zweifach (4), and was subsequentlyrefined by Zweifach (64) and Frasher (11). Rhodin(30,31) further developed this technique and corre-lated the intravital observations with electron micro-scope analyses of identical vascular segments.
In the anesthetized rat, a segment of the small intes-tine is exteriorized, draped over a lucite pedestal,continuously kept moist by a warm saline solution,and placed on the stage of a light microscopeadapted for video recording. At a certain time, theentire mesenteric field can be preserved by a chemi-cal fixative and embedded in liquid plastic that ismade to polymerize. This mesenteric specimen canthen be sectioned and analyzed in the light micro-scope as well as in the transmission electron micro-scope at magnifications ranging from 600× to50,000×.
The effects of amyloid-b on live blood vessels hadnever before been explored until the investiga-tions by Thomas and colleagues (48). Nor had theeffects been studied and recorded, first by direct ob-servations and then by careful analysis of the iden-tical vascular segments in the electron microscope.The accuracy of this technical approach is very pre-cise, and the results remarkable [Figs. 2(A) and2(B)].
This in vivo model provides a rapid method for in-vestigating amyloid-b toxicity, vascular degenera-tion, and inflammatory response.
EFFECTS OF AMYLOID-b INFUSION
In the experiments by Thomas and colleagues(48,49,50,52), male rats and female ovariectomizedrats were given a bolus infusion of amyloid-b1-40(100 ng/g body weight) via a catheter placed in theaorta with its tip near the entrance of one of themajor arteries that supply blood to the small intes-tines. Blood levels of Ab were reported by Sutton andcolleagues (45). Serum amyloid-b levels averaged1.6 ng/mL before infusion. At 30 minutes afterb-amyloid infusion, there was an eightfold increasein b-amyloid levels. Within 1 hour after amyloid-binfusion, 99% of amyloid-b was cleared. Thus, therewas only a brief elevation of Ab achieved. The maxi-mum reached is comparable to Ab levels in amyloiddeposits. Generally, there is no difference in theblood level of Ab between patients and controls, andit is assumed that a certain dysfunction of the blood–brain barrier induces an abnormal transport of Abfrom sera to the parenchyma (24).
Intravital Observations
Within 30–60 minutes, drastic effects were noticedin relation to small arteries and arterioles as well aspostcapillary venules and muscular venules [Figs.3(A) and 3(C)]. White blood cells (granulocytes and
Figure 1. Rat mesenteric microvessels. Intravital record-ing. In this low magnification overview of the typical two-dimensional network of microvessels, the branching of thearterioles (1) and the confluence of venules (2) can easilybe seen. Arterial capillaries (3) and postcapillary venules(4) can also be identified. The flow direction is indicatedby arrows. The direction of the fast flow in the arteriolesis difficult to discern, whereas the flow in the venules isslower and the margination of rolling leukocytes helps toidentify the flow direction. Magnification ×40.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
209
monocytes) slowed down and became attached tothe inner lining of these blood vessels. They alsomoved around locally before they shot out footlikeprocesses (pseudopods) that would penetrate thewall of the vessel. Gradually, the entire leukocytewould go through the wall in a process called “dia-pedesis” and continue to move away from the vesselin the connective tissue of the mesenteric membrane.Platelets became marginated and/or tumbled alongthe arteriolar and venular endothelium, and manywere also attracted to marginated leukocytes [Figs.4(A) and 4(B)]. Aggregations of platelets formed
clots that built up momentarily and then becamedetached again. Some platelets reached the suben-dothelial space. Arteriolar and venular segments of1000 mm were video-recorded for at least 60 min-utes. In the arterioles, there was an average of 20–25leukocytes marginating and adhering to the endo-thelial lining. Approximately one-third would trlans-migrate the wall, whereas two-thirds migrated alongthe endothelium and either became detached or keptmigrating. Subsequent electron microscope analysesof adhering leukocytes showed that about two-thirdswere monocytes and one-third granulocytes. In con-
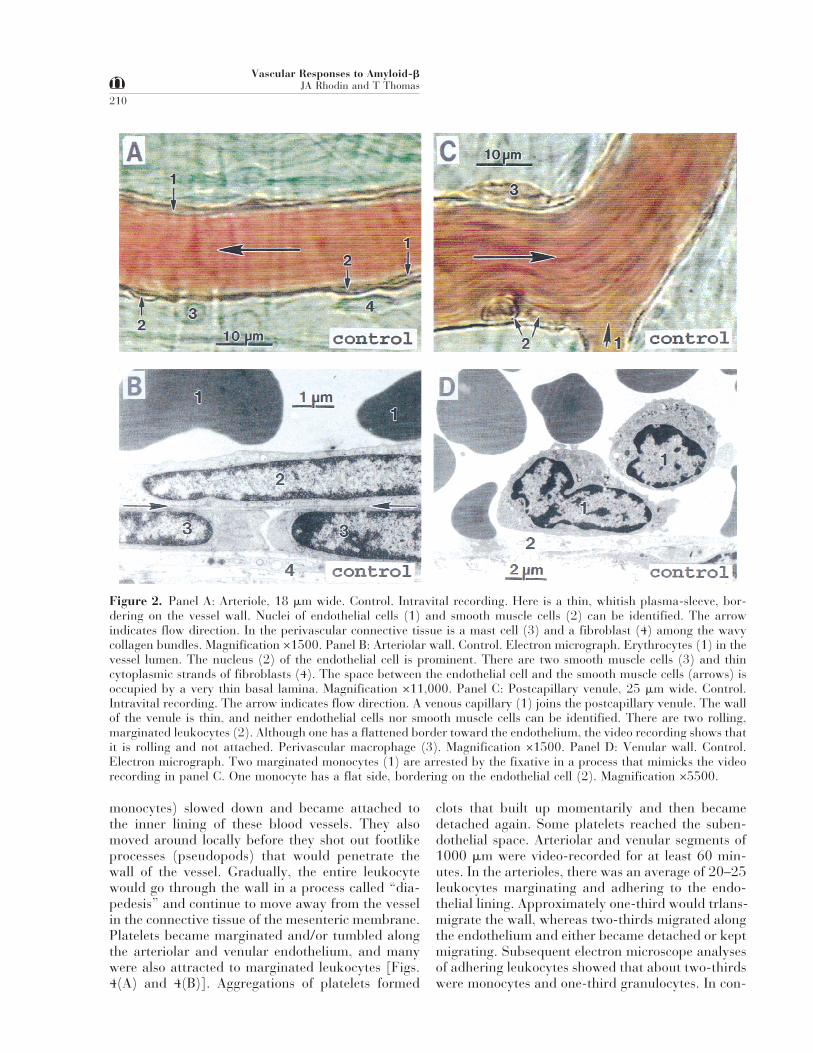
Figure 2. Panel A: Arteriole, 18 mm wide. Control. Intravital recording. Here is a thin, whitish plasma-sleeve, bor-dering on the vessel wall. Nuclei of endothelial cells (1) and smooth muscle cells (2) can be identified. The arrowindicates flow direction. In the perivascular connective tissue is a mast cell (3) and a fibroblast (4) among the wavycollagen bundles. Magnification ×1500. Panel B: Arteriolar wall. Control. Electron micrograph. Erythrocytes (1) in thevessel lumen. The nucleus (2) of the endothelial cell is prominent. There are two smooth muscle cells (3) and thincytoplasmic strands of fibroblasts (4). The space between the endothelial cell and the smooth muscle cells (arrows) isoccupied by a very thin basal lamina. Magnification ×11,000. Panel C: Postcapillary venule, 25 mm wide. Control.Intravital recording. The arrow indicates flow direction. A venous capillary (1) joins the postcapillary venule. The wallof the venule is thin, and neither endothelial cells nor smooth muscle cells can be identified. There are two rolling,marginated leukocytes (2). Although one has a flattened border toward the endothelium, the video recording shows thatit is rolling and not attached. Perivascular macrophage (3). Magnification ×1500. Panel D: Venular wall. Control.Electron micrograph. Two marginated monocytes (1) are arrested by the fixative in a process that mimicks the videorecording in panel C. One monocyte has a flat side, bordering on the endothelial cell (2). Magnification ×5500.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
210
trol animals, only 2–4 leukocytes marginated or ad-hered in arterioles. In the venules, there was an av-erage of 300 marginating and adhering leukocytescompared to about 100 in control animals.
In the perivascular space, mast cells became acti-vated, as judged by degranulation, and macrophageswere actively moving about, often circling mast cellsin an apparent cell-to-cell signaling.
Electron Microscope Observations
Electron microscope analysis of the arterioles dem-onstrated a variety of endothelial damages. Holesand discontinuities appeared first, followed by en-dothelial cell disruptions and exudation of bloodplasma which often formed cufflike areas of plasma,narrowing of the vascular lumen, as well as en-croachment on and displacement of smooth muscle
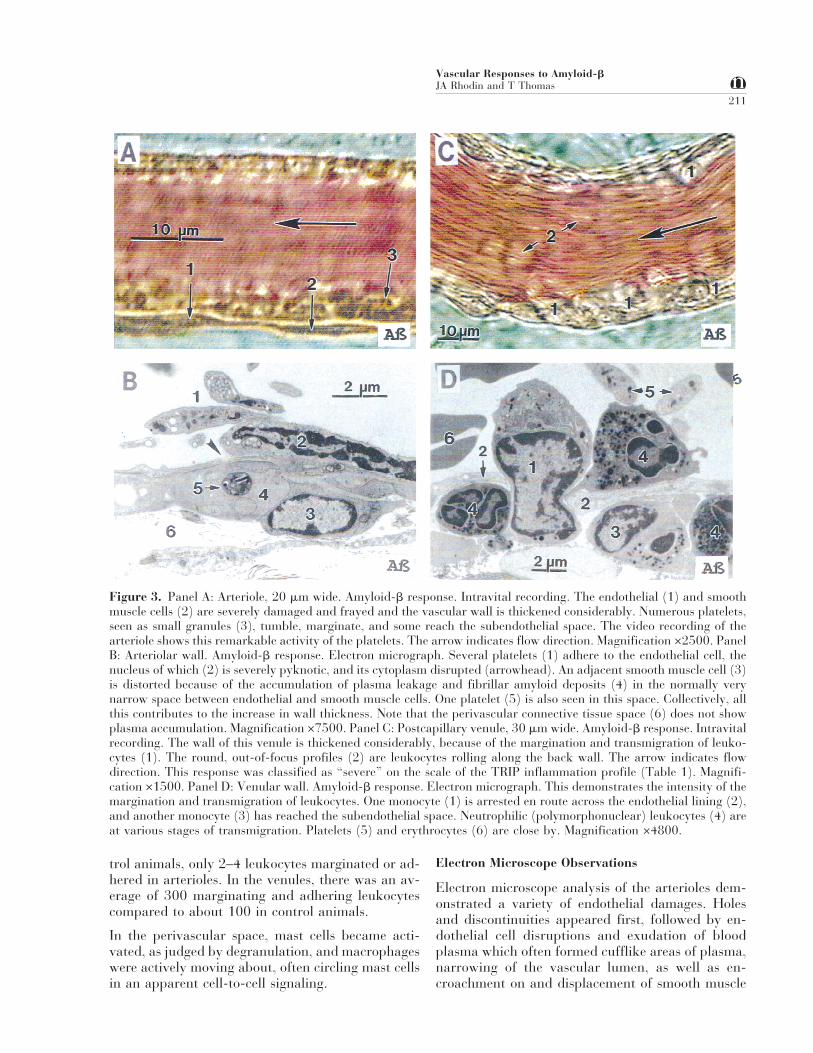
Figure 3. Panel A: Arteriole, 20 mm wide. Amyloid-b response. Intravital recording. The endothelial (1) and smoothmuscle cells (2) are severely damaged and frayed and the vascular wall is thickened considerably. Numerous platelets,seen as small granules (3), tumble, marginate, and some reach the subendothelial space. The video recording of thearteriole shows this remarkable activity of the platelets. The arrow indicates flow direction. Magnification ×2500. PanelB: Arteriolar wall. Amyloid-b response. Electron micrograph. Several platelets (1) adhere to the endothelial cell, thenucleus of which (2) is severely pyknotic, and its cytoplasm disrupted (arrowhead). An adjacent smooth muscle cell (3)is distorted because of the accumulation of plasma leakage and fibrillar amyloid deposits (4) in the normally verynarrow space between endothelial and smooth muscle cells. One platelet (5) is also seen in this space. Collectively, allthis contributes to the increase in wall thickness. Note that the perivascular connective tissue space (6) does not showplasma accumulation. Magnification ×7500. Panel C: Postcapillary venule, 30 mm wide. Amyloid-b response. Intravitalrecording. The wall of this venule is thickened considerably, because of the margination and transmigration of leuko-cytes (1). The round, out-of-focus profiles (2) are leukocytes rolling along the back wall. The arrow indicates flowdirection. This response was classified as “severe” on the scale of the TRIP inflammation profile (Table 1). Magnifi-cation ×1500. Panel D: Venular wall. Amyloid-b response. Electron micrograph. This demonstrates the intensity of themargination and transmigration of leukocytes. One monocyte (1) is arrested en route across the endothelial lining (2),and another monocyte (3) has reached the subendothelial space. Neutrophilic (polymorphonuclear) leukocytes (4) areat various stages of transmigration. Platelets (5) and erythrocytes (6) are close by. Magnification ×4800.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
211
cells, in some cases causing condensation of nuclearcontent (pyknosis) and cell death of endothelial andsmooth muscle cells. Monocytes and granulocyteswere found attached to and between the endothe-lial cell lining and the surrounding smooth musclecells.
It was concluded that amyloid-b had a toxic, inflam-matory effect on the endothelial cytoplasm and or-ganelles, and that the disruption of the endothelial
lining made it possible for the plasma and amyloid-bto escape from the blood. Thus, there is a classicinflammatory response after the infusion of amyloid-b, with the leukocytes decelerating by rolling andadhering to the endothelial cells before migrationacross the vessel wall. The unusual aspect of thisresponse is that it also occurred in small arteries andarterioles [Figs. 4(C) and 4(D)]. Only one publica-tion (23) reported a similar effect on arterioles afterexposure to a laser beam.
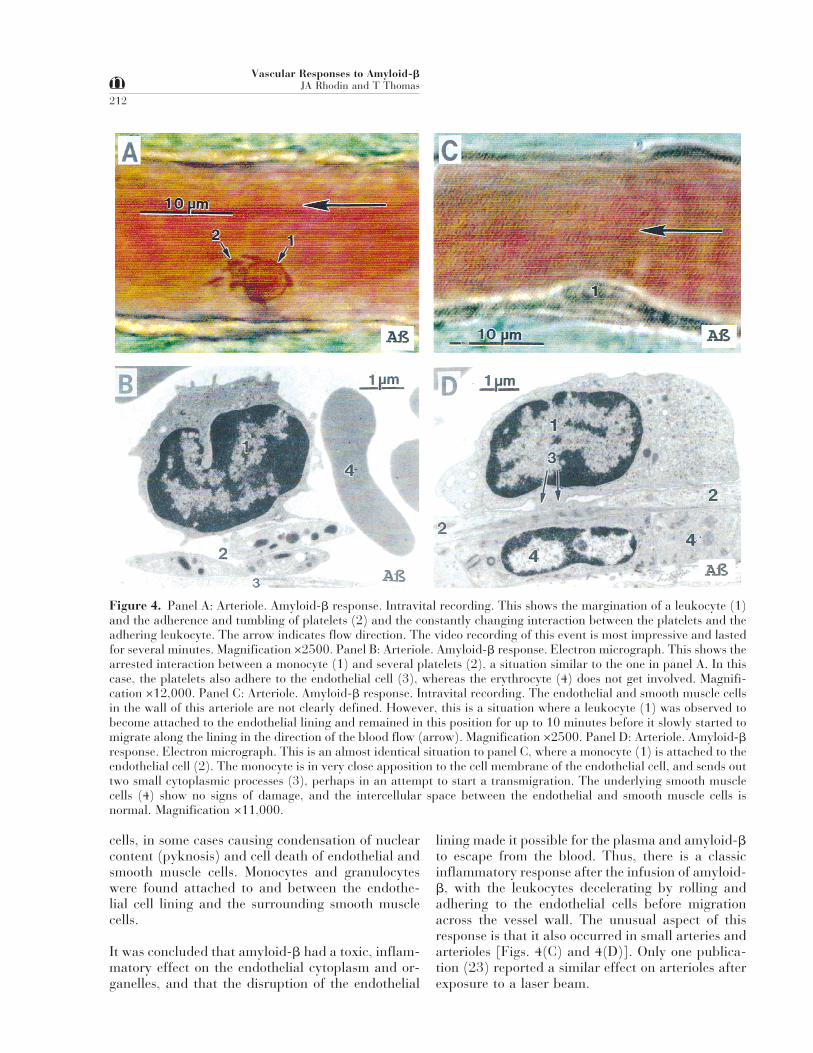
Figure 4. Panel A: Arteriole. Amyloid-b response. Intravital recording. This shows the margination of a leukocyte (1)and the adherence and tumbling of platelets (2) and the constantly changing interaction between the platelets and theadhering leukocyte. The arrow indicates flow direction. The video recording of this event is most impressive and lastedfor several minutes. Magnification ×2500. Panel B: Arteriole. Amyloid-b response. Electron micrograph. This shows thearrested interaction between a monocyte (1) and several platelets (2), a situation similar to the one in panel A. In thiscase, the platelets also adhere to the endothelial cell (3), whereas the erythrocyte (4) does not get involved. Magnifi-cation ×12,000. Panel C: Arteriole. Amyloid-b response. Intravital recording. The endothelial and smooth muscle cellsin the wall of this arteriole are not clearly defined. However, this is a situation where a leukocyte (1) was observed tobecome attached to the endothelial lining and remained in this position for up to 10 minutes before it slowly started tomigrate along the lining in the direction of the blood flow (arrow). Magnification ×2500. Panel D: Arteriole. Amyloid-bresponse. Electron micrograph. This is an almost identical situation to panel C, where a monocyte (1) is attached to theendothelial cell (2). The monocyte is in very close apposition to the cell membrane of the endothelial cell, and sends outtwo small cytoplasmic processes (3), perhaps in an attempt to start a transmigration. The underlying smooth musclecells (4) show no signs of damage, and the intercellular space between the endothelial and smooth muscle cells isnormal. Magnification ×11,000.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
212
It is known that a certain percentage of circulatingleukocytes are normally marginated in venules, andas a response to inflammatory agents will transmi-grate across the walls of the venules. The number ofmarginating and transmigrating leukocytes was in-creased after the introduction of amyloid-b (seeTable 1).
PREVENTION OF AMYLOID-b-INDUCEDVASCULAR DAMAGE
Effects of Antioxidant Infusion
In a series of experiments (49), male rats were in-fused with the antioxidant superoxide dismutase be-fore and subsequent to the infusion of amyloid-b.
The mesenteric vasculature was observed for periodsof at least 60 minutes. There were only few obser-vations of leukocyte adhesion in arterioles (2–4 in a1000-mm segment), and invariably they would be-come detached again and not traverse the arteriolarwall. The electron microscope analysis showed acompletely normal endothelial lining without damageto the endothelial cell cytoplasm and organelles.
Effects of Cytokine Blockers
In a series of experiments, Sutton and colleagues(45) infused in one set of male rats (A) interleukin-1receptor antagonist (IL-1ra) before the infusion ofamyloid-b. In another set of male rats they infused
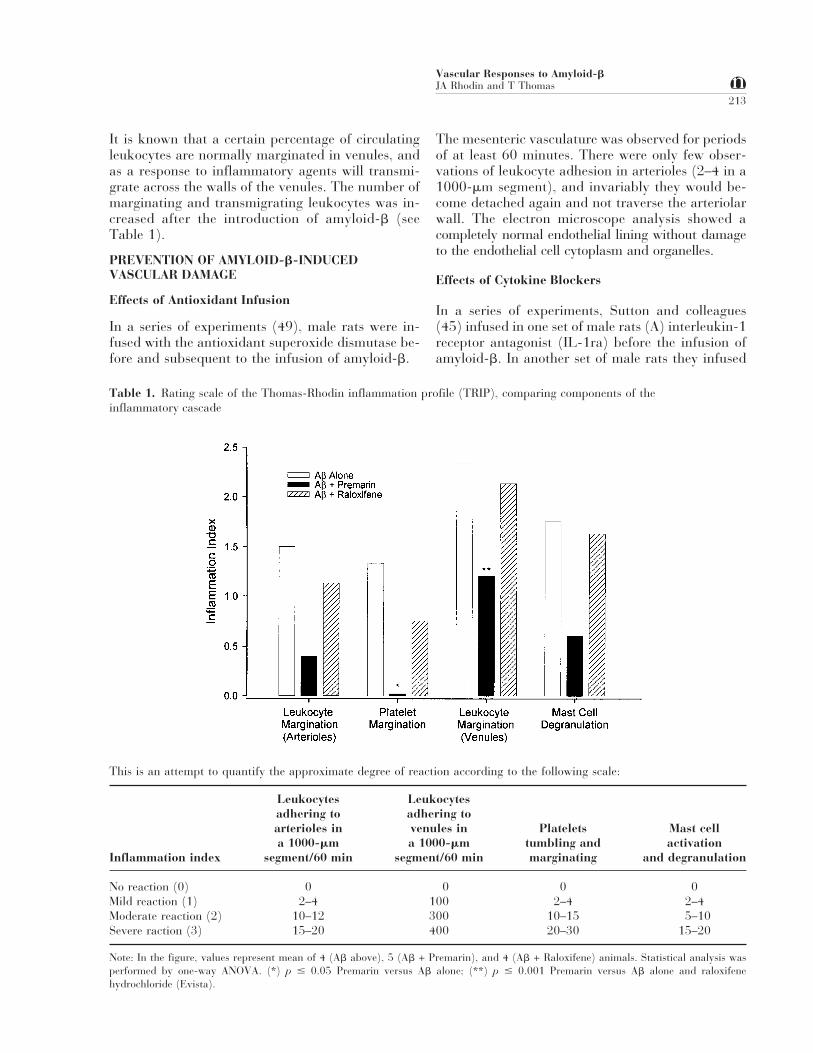
Table 1. Rating scale of the Thomas-Rhodin inflammation profile (TRIP), comparing components of theinflammatory cascade
This is an attempt to quantify the approximate degree of reaction according to the following scale:
Inflammation index
Leukocytesadhering toarterioles ina 1000-mm
segment/60 min
Leukocytesadhering tovenules ina 1000-mm
segment/60 min
Plateletstumbling andmarginating
Mast cellactivation
and degranulation
No reaction (0) 0 0 0 0Mild reaction (1) 2–4 100 2–4 2–4Moderate reaction (2) 10–12 300 10–15 5–10Severe raction (3) 15–20 400 20–30 15–20
Note: In the figure, values represent mean of 4 (Ab above), 5 (Ab + Premarin), and 4 (Ab + Raloxifene) animals. Statistical analysis wasperformed by one-way ANOVA. (*) p # 0.05 Premarin versus Ab alone; (**) p # 0.001 Premarin versus Ab alone and raloxifenehydrochloride (Evista).
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
213

(B) tumor necrosis factor-binding protein (TNF-bp)before the infusion of amyloid-b.
The mesenteric vasculature was video-recorded for60 minutes. In either case (A or B), the effect onleukocyte margination and adherence was similar.An average of 2–4 leukocytes marginated and ad-hered in an observed 1000-mm arteriolar segmentper 60 minutes. None was recorded to transmigrate,and all became detached. There was no marginationor tumbling of platelets. There was some mast celldegranulation, but to a much lesser degree than inb-amyloid alone. The margination of leukocytes inthe postcapillary venules was within normal range.Electron microscope analyses showed in both casesof cytokine blockers that the configuration of thecells of the arteriolar wall and the ultrastructure oftheir organelles were completely normal. The vascu-lar actions of the cytokines IL-1b and TNF-a wereinvestigated by Rhodin and colleagues (32) using thesame mesenteric animal (rat) model. It was shownthat these two proinflammatory cytokines trigger anendothelial and smooth muscle dysfunction, verysimilar to the effect of amyloid-b. TNF-a produceda more severe effect than IL-1b, but in both cases,there were endothelial damage, leukocyte margin-ation (average 12–17) and transmigration, plateletmargination, smooth muscle necrosis, and mast cellactivation and degranulation. It was concluded thatthe cytokines IL-1b and TNF-a mediate disruptionand inflammatory response initiated by amyloid-b.
Effects of Estrogen and Selective EstrogenReceptor Modulators
Using both male rats and ovariectomized femalerats, Thomas and colleagues (50,52) investigatedthe possibility that orally administered conjugatedequine estrogen (Premarin) and a selective estrogenreceptor modulator, raloxifene hydrochloride(Evista), would have a protective effect against thevascular damage caused by amyloid-b.
Premarin 2 mg/kg per day was given orally for 2weeks to a set of rats, and Evista 3 mg/kg per daywas given orally for 2 weeks to a second set of rats.In both cases, at the end of the second week, the ratswere infused with freshly prepared amyloid-b(1-40),100 ng/g body weight, diluted with 0.25 mL isotonicsaline.
Premarin showed remarkable anti-inflammatory ac-tion, whereas Evista had no significant beneficial ef-fect. In the case of Premarin, the reactivity and ul-trastructure of both arterioles and venules were com-pletely normal, whereas in the case of Evista there
were more than the normal number of leukocytesmarginating in arterioles, and a considerable num-ber of leukocytes marginating in the postcapillaryvenules.
In rats that had received a daily oral administrationof Premarin for 2 weeks, the Premarin treatmentwas discontinued for 1 week, followed by an infusionof amyloid-b. The protective influence of the conju-gated estrogen was lost, as both arterioles and post-capillarty venules reacted similarly to the situationwith amyloid-b alone.
A rating scale was developed (Table 1) to quantifythe myriad of cellular responses involved in the in-flammatory process. This scale allows comparison ofthe inflammatory response under various experi-mental and treatment conditions.
IMPORTANCE AND INTERPRETATIONS OFOBSERVATIONS IN LIVE RATS
Effects of Amyloid-b
The observations demonstrate that b-amyloid has atoxic effect on endothelial cells in the living animal,confirming that the effects are similar to those pre-viously observed in the in vitro experiments.
There are several factors that cooperate in bringingon the toxic effects of amyloid-b (Fig. 5). It is as-sumed that the circulating, soluble amyloid-b inter-acts with receptor sites on endothelial cells, so-calledRAGE (receptor for advanced glycation end prod-uct) (22,63), generating reactive oxygen species(ROS). The superoxide radicals interact with nitricoxide (NO) to generate toxic peroxynitrite, whichleads to lipid peroxidation, membrane degeneration,cytoplasmic disruption, plasma leakage, smoothmuscle cell damage, and fibrillar amyloid deposits.
There is ample evidence to show that Ab-inducedfree-radical production and oxidative stress have amajor role in the pathology of Alzheimer’s disease(AD). Activation of NADPH oxidase forming super-oxide radicals is observed in AD brains (37). Nitricoxidase activity is also elevated in AD (12). Peroxy-nitrite, the product of the reaction between NO andsuperoxide, is a strong oxidizing and nitrating agentthat can interact with biomolecules. Ab and proin-flammatory cytokines enhance peroxynitrite produc-tion (53). A role for peroxynitrite in the pathology ofAD is indicated by a number of reports (1,3,27,54).Therefore, Ab-mediated superoxide and peroxyni-trite production plays an important role in the pa-thology of AD.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
214
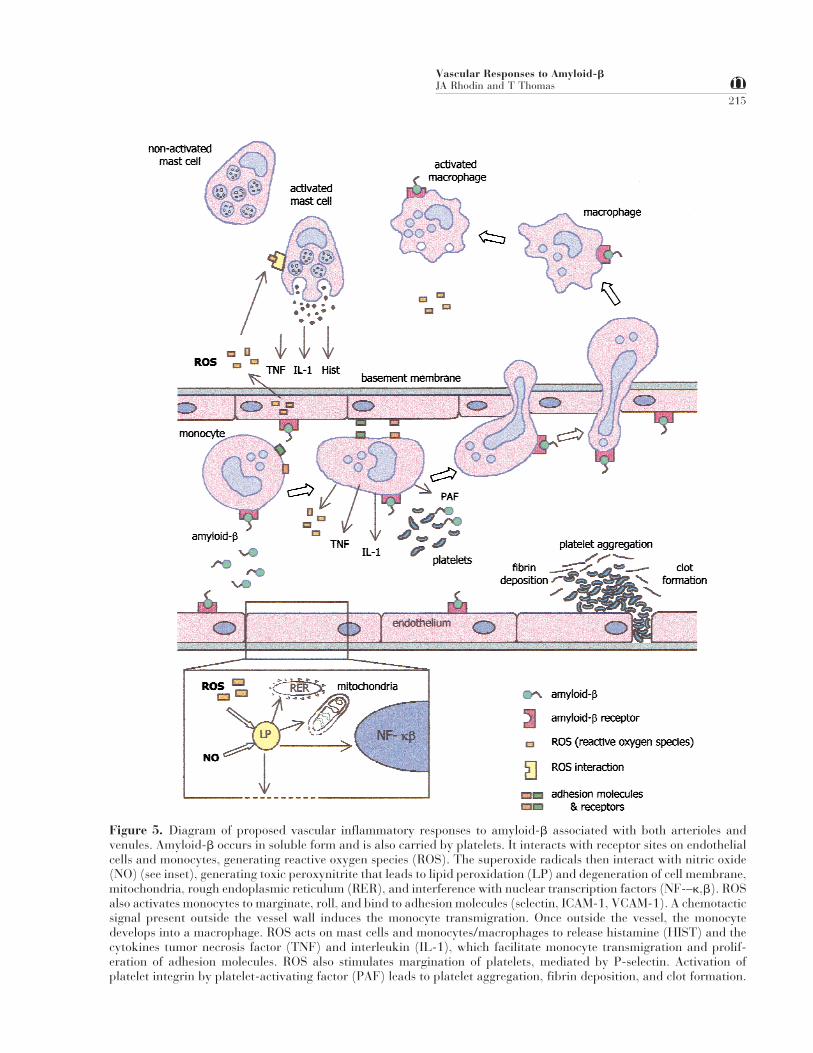
Figure 5. Diagram of proposed vascular inflammatory responses to amyloid-b associated with both arterioles andvenules. Amyloid-b occurs in soluble form and is also carried by platelets. It interacts with receptor sites on endothelialcells and monocytes, generating reactive oxygen species (ROS). The superoxide radicals then interact with nitric oxide(NO) (see inset), generating toxic peroxynitrite that leads to lipid peroxidation (LP) and degeneration of cell membrane,mitochondria, rough endoplasmic reticulum (RER), and interference with nuclear transcription factors (NF-–k,b). ROSalso activates monocytes to marginate, roll, and bind to adhesion molecules (selectin, ICAM-1, VCAM-1). A chemotacticsignal present outside the vessel wall induces the monocyte transmigration. Once outside the vessel, the monocytedevelops into a macrophage. ROS acts on mast cells and monocytes/macrophages to release histamine (HIST) and thecytokines tumor necrosis factor (TNF) and interleukin (IL-1), which facilitate monocyte transmigration and prolif-eration of adhesion molecules. ROS also stimulates margination of platelets, mediated by P-selectin. Activation ofplatelet integrin by platelet-activating factor (PAF) leads to platelet aggregation, fibrin deposition, and clot formation.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
215

ROS activate leukocytes to decelerate, roll, and mar-ginate, binding to endothelial adhesion molecules.Activated leukocytes now have an increased affinityof their b2-integrin receptors for endothelial inter-cellular adhesion ligands (ICAM-1, VCAM-1). As aresult, the leukocytes transmigrate the vessel wall.ROS also activate mast cells and macrophages torelease histamine and cytokines TNF-a and IL-1b.The cytokines facilitate leukocyte migration as wellas proliferation of endothelial cell adhesion mol-ecules. ROS also stimulate platelet tumbling andmargination, mediated by P-selectin, and very likelycause degranulation of platelets and release of amy-loid-b, sequestered in the platelets.
Thus, the amyloid-b-induced vascular endothelialdysfunction produces an inflammatory reaction,mimicking the signs of vascular inflammation seenin biopsies and postmortem autopsies of individualswith Alzheimer’s disease (57).
Effects of Antioxidant Superoxide Dismutase
Superoxide dismutase is an antioxidant enzymewhose normal function is to detoxify excessive levelsof oxygen-radical superoxide. The endothelial dys-function and inflammatory responses to amyloid-bwere prevented by the oxygen-radical-scavengingenzyme SOD. The antioxidant SOD may, therefore,be used to preserve normal vascular integrity andfunction by preventing, retarding, or reversing thevascular damage induced by amyloid-b.
Effects of Cytokine Blockers
Cytokine blockade with either IL-1ra or TNF-bp be-fore infusion of amyloid-b resulted in greatly dimin-ished margination and adherence of leukocytes withno transmigration of either leukocytes or platelets.Electron microscopy showed no damage to the vesselwall, and the endothelial and smooth muscle cellsappeared normal.
Therefore, the cytokines TNF-a and IL-1b seem tomediate the vascular disruption and inflammatoryresponse initated by amyloid-b (35). Antagonism ofthese proinflammatory cytokines may offer new av-enues for therapy in Alzheimer’s disesase.
Effects of Estrogen and Selective EstrogenReceptor Modulator
Conjugated equine estrogen, orally administered for2 weeks, prevented vascular deposition of amyloid-b, endothelial and vessel wall disruption withplasma leakage, platelet and mast cell activation, aswell as adhesion and transmigration of leukocytes.
It is assumed that estrogen blocks the endothelial cellamyloid-b receptors and regulates agents such asnitric oxide. Estrogen, therefore, has a potent anti-oxidant activity. This beneficial effect was lost whenestrogen treatment was discontinued.
The observations illustrate novel actions of conju-gated estrogens in protecting against amyloid-b-induced vascular damage and inflammatory re-sponse. Thus, these observations support the conceptthat estrogen therapy may retard the developmentand severity of dementia in postmenopausal womenif initiated early before cellular damage is irrevers-ible (26).
Currently available selective estrogen receptormodulators such as raloxifene hydrochloride(Evista) may have only a limited range of targetedactivity and may not provide the broad range of ben-efits accorded by the conjugated equine estrogenPremarin. The long-term clinical benefits of selectiveestrogen receptor modulators relative to estrogen re-placement therapy, especially with regard to cardio-vascular disease and dementia, remains to be estab-lished.
A schematic flow diagram (Fig. 6) summarizes ourview of how different factors are related to eachother in Alzheimer’s disease, and how they interactto mediate Ab-induced microvascular dysfunction.
EFFECTS OF AMYLOID-b ON CENTRAL NERVOUSSYSTEM BLOOD VESSELS
Only the local effects of amyloid-b on systemic vas-culature were investigated in the studies reviewedhere, because the vasculature of the central nervoussystem (CNS) is not as readily available for intravitalobservation. The architecture of the central nervoussystem arterial walls is similar to peripheral vesselsexcept for the presence of tight junctions and a re-duced number of pinocytic vesicles in the endothelialcells. A similar action of amyloid-b in rat cerebralvasculature may be expected. This assumption issupported by an investigation of amyloid-b-mediated vasoactivity and vascular endothelial dam-age in isolated bovine brain artery rings, studied invitro by Thomas and colleagues (49). An investiga-tion by Jancso and colleagues (13) in rats given anintracarotid infusion of amyloid-b(1-42) peptide. Itdemonstrated an impaired blood–brain function, us-ing a quantitative Evans blue method.
DISCUSSION
For the first time, it has been demonstrated thatadministration of amyloid-b rapidly initiates an in-
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
216
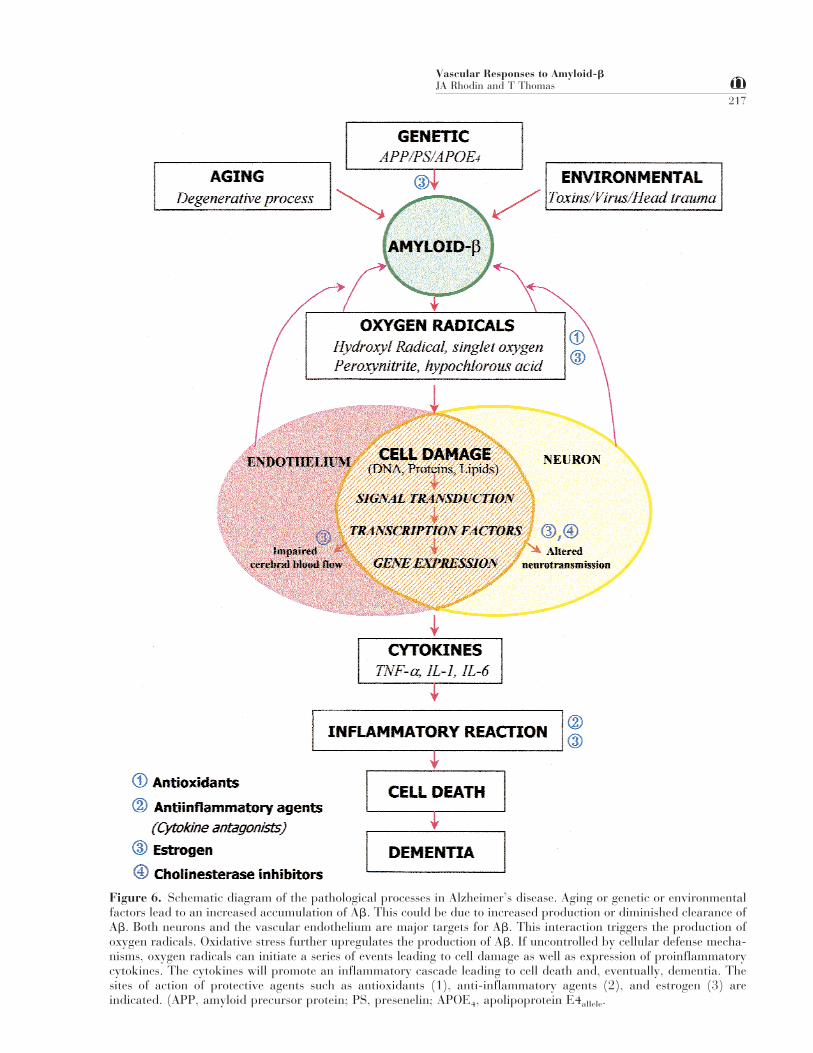
Figure 6. Schematic diagram of the pathological processes in Alzheimer’s disease. Aging or genetic or environmentalfactors lead to an increased accumulation of Ab. This could be due to increased production or diminished clearance ofAb. Both neurons and the vascular endothelium are major targets for Ab. This interaction triggers the production ofoxygen radicals. Oxidative stress further upregulates the production of Ab. If uncontrolled by cellular defense mecha-nisms, oxygen radicals can initiate a series of events leading to cell damage as well as expression of proinflammatorycytokines. The cytokines will promote an inflammatory cascade leading to cell death and, eventually, dementia. Thesites of action of protective agents such as antioxidants (1), anti-inflammatory agents (2), and estrogen (3) areindicated. (APP, amyloid precursor protein; PS, presenelin; APOE4, apolipoprotein E4allele.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
217

flammatory reaction in both arterioles and venulesof the rat mesentery. The reaction has been video-recorded and the cellular responses analyzed by elec-tron microscopy. It is proposed that the amyloid in-teracts with receptors on the endothelial cells, gen-erating reactive oxygen species. This triggers acascade of inflammatory events including leukocyteand platelet margination and transmigration,smooth muscle and endothelial cell degeneration,leakage of plasma, and deposition of fibrillar amy-loid.The vascular effect of amyloid-b is rapid and pro-duced by low concentrations of soluble amyloid. Theneurotoxic properties of amyloid-b require higherconcentrations of the aggregated peptide and takelonger periods (days) of exposure.It has also been demonstrated that the toxic effect ofamyloid-b is mediated by the cytokines TNF-a andIL-1b, and an inhibition of these cytokines preventsthe vascular effects of amyloid-b.Also, it has been demonstrated that the vasculardamage by amyloid-b can be prevented by an anti-oxidant such as superoxide dismutase and by conju-gated equine estrogens.Areas of Uncertainty and Controversy
The mesenteric animal model offers a rapid means oftesting the intravital effects of amyloid-b. In vitroexperiments with rat aortic rings and bovine cerebralarteries have demonstrated damage to these vesselssimilar to those in live mesenteric vessels. It remainsto be shown by intravital techniques (cranial win-dow) that cerebral and meningeal blood vessels reactthe same way to amyloid-b.
There is great similarity between the cerebral vascu-lar damage seen in autopsies of patients that sufferedfrom Alzheimer’s disease and the damage producedexperimentally in rat mesenteric vessels. However,the all-important issue here is: Does the vasculardamage occur before or after the appearance of neu-rofibrillary tangles, neuritic plaques, and neuronaldeath?
Are blood vessels exposed to Ab from the luminal orabluminal side? Amyloid-b is produced by all cellsincluding neurons, endothelial cells, and smoothmuscle cells. Ab normally circulates in the blood.Both APP and Ab are primarily (more than 90%)sequestered in platelets (39). Platelet activationcauses release of amyloid.
The general consensus is that vascular amyloid con-tributes to the amyloid deposits in the brain(7,9,14,15,17,18,28). There are other investigators
(34,38,58,59) who maintain that Ab is derived fromthe brain alone; that it accumulates in the interstitialfluid; and that there is a failure of, or a reduction in,the elimination of the Ab peptides along perivascularinterstitial fluid (lymphatic) drainage pathways ofthe brain. This would lead to plaque formation in theextracellular space, near capillaries, and in the wallsof arterioles and arteries. However, this hypothesisdoes not account for the process leading to formationof neurofibrillary tangles within axons and den-drites. Thomas and colleagues (47,49) administeredAb on the surface of the blood vessels in both theanimal model and in vitro blood vessel preparations.The response was similar whether Ab was appliedoutside the blood vessel or injected into the circula-tion.
Amyloid-ß rapidly diffuses in and out of the brainand can alter the permeability of brain capillaries(43). Blood–brain barrier dysfunction is evident inthe brains of AD patients (16). This will enable fac-tors from the peripheral circulation, including Aß, togain access to the brain. Amyloid-b is also rapidlytransported from brain across the blood–brain bar-rier by LDL receptor-mediated protein-1 (36).
It is the opinion of the authors of this review, as wellas a steadily increasing number of investigators(8,19), that the vascular dysfunction may precedethe neuronal damage and dementia. Thus, preven-tion of amyloid-b-induced cerebrovascular damagemay provide new avenues for Alzeimer’s diseasetherapy.
ACKNOWLEDGMENTS
We would like to gratefully acknowledge the invalu-able and expert technical support of Ms. Margie Bry-ant, Ms. Linda Clark, and Ms. Amanda Garces.
REFERENCES
1. Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y,Shimizu K, Umegae N, Hayase N, Shiono H, Kabaya-shi S. (2000). Nitration of manganese superoxide dis-mutase in cerebrospinal fluids is a marker for perox-initrite-mediated oxidative stress in neurodegenera-tive diseases. Ann Neurol 47:524–527.
2. Bush AL, Martins RN, Rumble B, Moir R, Fuller S,Milward E, Currie J, Ames D, Weidemann A, FischerP, Multhaup G, Beyreuther K, Masters LL. (1990).The amyloid precursor protein of Alzheimer’s diseaseis released by human platelets. J Biol Chem 265:15977–15983.
3. Chabrier P-E, Demerle-Pallardy C, Auguet M.(1999). Nitric oxide synthases: targets for therapeuticstrategies in neurological diseases. Cell Mol Life Sci55:1029–1035.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
218

4. Chambers R, Zweifach BW. (1944). The topographyand function of mesenteric capillary circulation. Am JAnat 75:173–205.
5. Chen M, Inestrosa NC, Ross GS, Fernandez HL.(1995). Platelets are the primary resource of amyloidbeta-peptide in human blood. Biochem Biophys ResCommun 213:96–103.
6. Davies TA, Billingslea A, Johnson R, Greenberg S,Ortiz M, Long H, Sgro K, Tibbles H, Seetoo K, Rath-bun W, Schonhorn J, Rimons ER. (1997). Stimulusresponses and amyloid precursor protein processingin DAMI megakaryocytes. J Lab Clin Med 130:21–32.
7. de la Torre JC. (1994). Impaired brain microcircula-tion may trigger Alzheimer’s disease. Neurosci Biobe-hav Rev 18:397–401.
8. de la Torre JL, Hachinski V. (Eds.) (1997). Cerebro-vascular pathology in Alzheimer’s disease. Ann NYAcad Sci 826:1–523.
9. de la Torre JC, Mussivand T. (1993). Can disturbedbrain circulation cause Alzheimer’s disease? NeurolRes 15:146–153.
10. Frangione B. (1989). Systemic and cerebral amyloid-osis. Ann Med 21:69–72.
11. Frasher WG Jr. (1973). Attached floating cat mesen-tery preparation for vital microscopy. Microvasc Res5:376–383.
12. Hensley K, Maidt ML, Yu Z, Sang H, MarkesberyWR, Floyd RA. (1988). Electrochemical analysis ofprotein nitrotyrosine and dityrosine in the Alzheimerbrain indicates region-specific accumulation. J Neu-rosci 18:8126–8132.
13. Jancso G, Domoki F, Santha P, Varga J, Fischer J,Orosz K, Penke B, Becskei A, Dux M, Toth L. (1998).b-Amyloid (1-42) peptide impairs blood-brain bar-rier function after intracarotid infusion in rats. Neu-rosci Lett 253:139–141.
14. Kalaria RN. (1993). Cerebral microvasculature andimmunological factors in Alzheimer’s disease. ClinNeurosci 1:204–211.
15. Kalaria RN. (1996). Cerebral vessels in ageing andAlzheimer’s disease. Pharmacol Ther 72:193–214.
16. Kalaria RN. (1999). The blood-brain barrier and ce-rebrovascular pathology in Alzheimer’s disease. AnnNY Acad Sci 893:113–125.
17. Kalaria RN, Hedera P. (1995). Differential degenera-tion of the cerebral microvasculature in Alzheimer’sdisease. NeuroReport 6:477–480.
18. Kalaria RN, Hedera P. (1996). b-amyloid vasoactiv-ity in Alzheimer’s disease. Lancet 347:1492–1493.
19. Kalaria RN, Ince P. (Eds.) (2000). Vascular factors inAlzheimer’s disease. Ann NY Acad Sci 903:1–556.
20. Kang J, Lemaire H, Unterbeck A, Salbaum JM, Mas-ters CL, Grzeschik K, Multhaup K, Beyreuther K,Muller-Hill B. (1987). The precursor of Alzheimer’sdisease amyloid A4 protein resembles a cell-surfacereceptor. Nature 325:733–736.
21. Knopman D, Boland LL, Mosley T, Howard G, LiaoD, Szoklo M, McGovern P, Folsom AR. (2001). Car-
diovascular risk factors and cognitive decline inmiddle-aged adults. Neurology 56:42–48.
22. Mattson MP, Rydel RR. (1996). Amyloid ox-toxtransducers. Nature 382:674–675.
23. Mayrovitz HN, Tuma RF, Wiedemann MP. (1980).Leukocyte adherence in arterioles following extravas-cular tissue trauma. Microvasc Res 20:264–272.
24. Miyakawa T, Kimura T, Hirata S, Fujise N, Ono T,Ishizuka K, Nakabayashi J. (2000). Role of blood ves-sels in producing pathological changes in the brainwith Alzheimer’s disease. Ann NY Acad Sci 903:46–54.
25. Naslund J, Haroutunian V, Mohs R, Davis KL, DaviesP, Greengard P, Buxbaum JD. (2000). Correlationbetween elevated levels of amyloid-b peptide in thebrain and cognitive decline. JAMA 283:1571–1577.
26. Ohkura T, Isse K, Akazawa K, Hamamoto M, Yaoi Y,Hagino N. (1994). Evaluation of estrogen treatmentin female patients with dementia of the Alzheimertype. Endocr J 41:361–367.
27. Paris D, Parker TA, Town T, Suo Z, Fang C, Hum-phrey J, Crawford F, Mullan M. (1998). Role of per-oxynitrite in the vasoactive and cytotoxic effects ofAlzheimer’s b-amyloid 1-40 peptide. Exp Neurol152:116–122.
28. Perlmutter LS, Chang Chui H. (1990). Microangiop-athy, the vascular basement membrane and Alzhei-mer’s disease: a review. Brain Res Bull 24:677–686.
29. Petito CK, Levy DE. (1980). The importance of ce-rebral arterioles in alterations of the blood-brain bar-rier. Lab Invest 43:262–268.
30. Rhodin JAG. (1986). Perfusion and superfusion fixa-tion effects on rat mesentery microvascular beds. In-travital and electron microscope analyses. J Submi-crosc Cytol Pathol 18:453–470.
31. Rhodin JAG, Lim SS. (1979). Combined intravitalmicroscopy and electron microscopy of the blind be-ginnings of the mesenteric lymphatic capillaries of therat mesentery. Acta Physiol Scand Suppl 463:51–58.
32. Rhodin J, Thomas T, Bryant MW, Clark L, SuttonET. (1999). Animal model of vascular inflammation.J Submicrosc Cytol Pathol 31:305–311.
33. Rhodin J, Thomas T, Bryant MW, Sutton ET. (2000).Animal model of Alzheimer-like vascular pathologyand inflammatory reaction. Vascular factors in al-zheimer’s disease. Ann NY Acad Sci 903:345–352.
34. Roher AE. (1993). b-amyloid (1-42) is a major com-ponent of cerebrovascular amyloid deposits: implica-tions for the pathology of Alzheimer’s disease. ProcNatl Acad Sci USA 90:10836–10840.
35. Sheng JG, Ito K, Skinner RD, Mark RE, Rovnaghi CR,Van Eldik LJ, Griffin ST. (1996). In vivo and in vitroevidence supporting a role for the inflammatory cy-tokine interleukin-1 as a driving force in Alzheimerpathogenesis. Neurobiol Aging 17:761–766.
36. Shibata M, Yamada S, Kumar SR, Calero M, BadingJ, Frangione B, Holtzman DM, Miller CA, StricklandDK, Ghiso J, Zlokovic BV. (2000). Clearance of Alz-
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
219

heimer’s amyloid-b 1-40 peptide from brain by LDLreceptor-related protein-1 at the blood-brain barrier.J Clin Invest 106:1489–1499.
37. Shimohama S, Tanino H, Kawakami N, Okamura N,Kodama H, Yamaguchi T, Hayakawa T, NunomuraN, Chiba S, Perry G, Smith MA, Fujimoto S. (2000).Activation of NADPH oxidase in Alzheimer’s diseasebrains. Biochem Biophys Res Commun 273:5–9.
38. Shinkai Y, Yoshimura M, Ito Y, Odaka A, Suzuki N,Yanagisawa K, Ihara Y. (1995). Amyloid beta-proteins 1-40 and 1-42 (43) in the soluble fraction ofextra- and intracranial blood vessels. Ann Neurol 38:421–448.
39. Smith CCT. (1997). Stimulated release of the b-amy-loid protein of Alzheimer’s disease by normal humanplatelets. Neurosci Lett 235:157–159.
40. Sparks DL. (1997). Coronary artery disease, hyper-tension, ApoE, and cholesterol: a link to Alzheimer’sdisease? Ann NY Acad Sci 826:128–146.
41. Sparks DL. (1999). Vascular related and mediatedalterations in Alzheimer’s disease. Cereb Cortex14:733–772.
42. Stewart PA, Hayakawa K, Akers MA, Vinters HV.(1992). A morphometric study of the blood-brainbarrier in Alzheimer’s disease. Lab Invest 67:734–742.
43. Strazielle N, Ghersi-Egea J-F, Ghiso J, Dehouck M-P,Frangione B, Patlak C, Fenstermacher J, Gorevic P.(2000). In vitro evidence that b-amyloid peptide 1-40diffuses across the blood-brain barrier and affects itspermeability. J Neuropath Exp Neurol 59:29–38.
44. Sutton ET, Hellerman GR, Thomas T. (1997).b-amyloid induced endothelial necrosis and inhibi-tion of nitric oxide production. Exp Cell Res 230:368–376.
45. Sutton ET, Thomas T, Bryant MW, Landon CS, New-ton CA, Rhodin JAG. (1999). Amyloid-b peptide in-duced inflammatory reaction is mediated by the cy-tokines tumor necrosis factor and interleukin-1. JSubmicrosc Cytol Pathol 31:313–323.
46. Tanzi, RE. (1989). Molecular genetics of Alzheimer’sdisease and the amyloid-b peptide precursor gene.Ann Med 21:91–94.
47. Thomas T, Thomas G, McLendon C, Sutton T, Mul-lan M. (1996). b-amyloid-mediated vasoactivity andvascular endothelial damage. Nature 380:168–171.
48. Thomas T, Sutton ET, Bryant MW, Rhodin JAG.(1997). In vivo vascular damage, leukocyte activationand inflammatory response induced by b-amyloid. JSubmicrosc Cytol Pathol 29:293–304.
49. Thomas T, McLendon C, Sutton ET, Thomas G.(1997). Cerebrovascular endothelial dysfunction me-diated by b-amyloid. NeuroReport 8:1387–1391.
50. Thomas T, Rhodin JAG, Sutton ET, Bryant MW,Price JM. (1999). Estrogen protects peripheral andcerebral blood vessels from toxicity of Alzheimer pep-
tide amyloid-b and inflammatory reaction. J Submi-crosc Cytol Pathol 31:571–579.
51. Thomas T, Rhodin J. (2000). Vascular actions of es-trogens and Alzheimer’s disease. Vascular factors inAlzheimer’s disease. Ann NY Acad Sci 903:501–509.
52. Thomas T, Bryant MW, Clark L, Garces A, Rhodin J.(2001). Estrogen and raloxifene activities on amy-loid-b induced inflammatory reaction. Microvasc Res61:28–39.
53. Torreilles F, Salman-Tabcheh S, Gur–eacute;rinM-C, Torreilles J. (1999). Neurodegenerative disor-ders: the role of peroxynitrite. Brain Res Rev 30:153–163.
54. van Dyke K. (1997). The possible role of peroxyni-trite in Alzheimer’s disease: a simple hypothesis thatcould be tested more thoroughly. Med Hypotheses 48:375–380.
55. Vinters HV, Pardridge WM. (1986). The blood-brainbarrier in Alzheimer’s disease. Can J Neurol Sci 13:446–448.
56. Vinters HV. (1987). Cerebral amyloid angiopathy. Acritical review. Stroke 18:311–324.
57. Vinters HV, Secor DL, Read SL, Frazee JG, Tomi-yasu U, Stanley TM, Ferreiro JA, Akers MA. (1994).Microvasculature in brain biopsy specimens from pa-tients with Alzheimer’s disease: an immunohisto-chemical and ultrastructural study. UltrastructPathol 18:333–348.
58. Weller RO, Massey A, Newman TA, Hutchings M,Kuo Y-M, Roher AE. (1998). Cerebral amyloid angi-opathy: b-amyloid accumulates in putative intersti-tial fluid drainage pathways in Alzheimer’s disease.Am J Pathol 153:725–734.
59. Weller RO, Massey A, Kuo Y-M, Roher AE. (2000).Cerebral amyloid angiopathy: accumulation of Ab ininterstitial fluid drainage pathways in Alzheimer’sdisease. Ann NY Acad Sci 903:110–117.
60. Wisniewski HM, Kozlowski PB. (1982). Evidence forblood-brain barrier changes in senile dementia of theAlzheimer type (SDAT). Ann NY Acad Sci 396:119–129.
61. Wisniewski HM, Wegiel J, Wang KC, Lach B. (1992).Ultrastructural studies of the cells forming amyloid inthe cortical vessel wall in Alzheimer’s disease. ActaNeuropathol 84:117–127.
62. Yamashita K, Miyakawa T, Katsuragi S. (1991). Vas-cular changes in brains with Alzheimer’s disease. JpnJ Psychiatry Neurol 45:79–84.
63. Yan DS, Chen X, Zhu H, Roher A, Slattery T, Zhao L,Nagashima M, Morser J, Migheli A, Nawroth P, SternD, Schmidt AM. (1996). RAGE and amyloid-b pep-tide neurotoxicity in Alzheimer’s disease. Nature 382:685–691.
64. Zweifach BW. (1954). Direct observation of the mes-enteric circulation in experimental animals. Anat Rec120:277–288.
Vascular Responses to Amyloid-bJA Rhodin and T Thomas
220