a missing link in the transformation from asymmetric to symmetric metallofullerene cages implies a...
TRANSCRIPT

A missing link in the transformation fromasymmetric to symmetric metallofullerene cagesimplies a top-down fullerene formation mechanismJianyuan Zhang1,4, Faye L. Bowles2, Daniel W. Bearden3, W. Keith Ray5, Tim Fuhrer4, Youqing Ye1,4,
Caitlyn Dixon4, Kim Harich5, Richard F. Helm5, Marilyn M. Olmstead2*, Alan L. Balch2*and Harry C. Dorn1,4*
Although fullerenes were discovered nearly three decades ago, the mechanism of their formation remains a mystery. Manyversions of the classic ‘bottom-up’ formation mechanism have been advanced, starting with C2 units that build up to formchains and rings of carbon atoms and ultimately form those well-known isolated fullerenes (for example, Ih-C60). In recentyears, evidence from laboratory and interstellar observations has emerged to suggest a ‘top-down’ mechanism, wherebysmall isolated fullerenes are formed via shrinkage of giant fullerenes generated from graphene sheets. Here, we presentmolecular structural evidence for this top-down mechanism based on metal carbide metallofullerenes M2C2@C1(51383)-C84
(M 5 Y, Gd). We propose that the unique asymmetric C1(51383)-C84 cage with destabilizing fused pentagons is apreserved ‘missing link’ in the top-down mechanism, and in well-established rearrangement steps can form many well-known, high-symmetry fullerene structures that account for the majority of solvent-extractable metallofullerenes.
The discovery of fullerenes1 has opened up new vistas innanoscience, and metallofullerenes have shown greatpromise in photovoltaic2 and biomedical3 applications.
However, their formation mechanism remains unclear. Various ver-sions of the ‘bottom-up’ mechanism have been posited as the mainpathway for fullerene formation4,5, suggesting that fullerenes areformed by consecutively adding C2 units to small carbon nanoclus-ters and cages. In recent years, with the development of graphene6,evidence has emerged to suggest a ‘top-down’ mechanism, wherebyfullerene cages are formed via shrinkage of giant fullerene structuresgenerated from graphene. One piece of direct evidence for the top-down mechanism is the reported laboratory transformation fromgraphene to fullerene7. It has also been demonstrated that fullerenescan be pyrolysed and lose carbon atoms to form smaller fullerenes athigh temperature in an argon stream8. Another strong supportingargument for the top-down mechanism is that fullerenes areformed in the interstellar medium9 by a photochemical process inwhich graphene sheets curve and lose C2 and other fragments10.However, evidence for the top-down mechanism has not beendemonstrated at the molecular level. Here, we present NMR spectro-scopic and X-ray crystallographic structural characterization forM2C2@C1(51383)-C84 (M¼ Y, Gd) metallofullerenes. These mol-ecules have a unique asymmetric cage C1(51383)-C84 that representsthe first characterized preserved intermediate suggesting top-downformation of fullerenes and metallofullerenes.
The proposed top-down mechanism is illustrated in Fig. 1.Under appropriate conditions, graphene sheets can spontaneouslyroll and warp to form other nanostructures11,12, including randomlyformed giant closed carbon networks—the primitive fullerenecages7. In an important computational study, a ‘shrinking hotgiant fullerene’ mechanism that leads to smaller fullerene structures
has been advanced13, which suggests a cascade shrinking process(multiple C2 losses) of giant fullerenes with spontaneous self-assem-bly to appropriate high-symmetry fullerene cages (HSFCs), such asIh-C60 and D5h-C70. Curl attributed the driving force of the shrink-ing process to C2 swapping14. Possible modes for fullerene cagerearrangements (isomerization and/or shrinkage by loss of C2) arealso suggested in Fig. 1. The ‘Stone–Wales’ transformation (SWT)realizes an orthogonal pentagon shift by rotating the central bondof a pyracylene motif. A second mode involves relieving the hightorsional strain in a pentalene motif (fused pentagons) by extrusionof the central C2 unit to form a hexagon15. Another possibility is lossof a C2 unit by extrusion of the central bond of an indene motif, assuggested by Murry and co-workers16. This process has been exper-imentally demonstrated by the capture of heptagon-containingintermediates17,18. Kroto was the first to recognize that final fullerenecages prefer high symmetry without direct pentagon–pentagoncontact (the isolated pentagon rule, IPR)19.
The incorporation of a metal source in the reaction path can leadto the formation of endohedral metallofullerenes (EMFs), in whichparticular HSFCs are stabilized by the presence of encapsulatedatoms or clusters, such as Ih-C80 and C3v-C82. Furthermore, endohe-dral atoms and clusters may cause the rearrangement to stop early,thereby preserving intermediate, unstable fullerene cages. Forexample, some pentalene-containing cages in the formationprocess were found to survive in EMFs20–22. In a cascade toHSFCs, a characteristic feature of an intermediate ‘missing link’ full-erene cage should be a random asymmetric (C1 symmetry), unstable(containing pentalene motifs) structure, but capable of a process ofrearrangement to high-symmetry and IPR-allowed fullerene cages.Although multiple missing links are possible as intermediates indifferent pathways of fullerene formation, no definitive structure
1Virginia Tech Carilion Research Institute, Roanoke, Virginia 24016, USA, 2Department of Chemistry, University of California, Davis, California 95616, USA,3National Institute of Standards and Technology, Chemical Science Division, Hollings Marine Laboratory, Charleston, South Carolina 29412, USA,4Department of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA, 5Department of Biochemistry, VirginiaPolytechnic Institute and State University, Blacksburg, Virginia 24061, USA. *e-mail: [email protected]; [email protected]; [email protected]
ARTICLESPUBLISHED ONLINE: 15 SEPTEMBER 2013 | DOI: 10.1038/NCHEM.1748
NATURE CHEMISTRY | ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemistry 1
© 2013 Macmillan Publishers Limited. All rights reserved.

has been experimentally isolated or characterized. In this Article, wereport the first example of a proposed missing link in metallofullereneformation, the chiral C1(51383)-C84 fullerene cage, which is stabilizedby an encapsulated metal carbide cluster. Although there is an earlierreport of a scandium metal carbide metallofullerene Sc2C2@C2v-C68containing a pentalene motif23, the structure of this moleculehas been challenged and suggested to be Sc2@C2v-C70 (ref. 24).The M2C2@C1(51383)-C84 molecules reported herein representthe first unambiguous X-ray structural characterization of apentalene-containing metal carbide metallofullerene, and the firstasymmetric fullerene cage with a pentalene motif. Moreimportantly, it is a starting point in the rearrangement cascade toform smaller HSFCs via simple, well-defined mechanistic steps.
ResultsThe 13C-enriched fullerenes and yttrium metallofullerenes weresynthesized by electric-arc synthesis with graphite rod packedwith 99% 13C amorphous carbon and Y2O3 (average 26 wt% or24 mol% 13C in carbon starting material)25. The gadoliniummetallofullerenes were synthesized by a normal electric-arcprocedure with natural-abundance carbon rods26. The13C-enrichedY2C2@C1(51383)-C84 and natural-abundance Gd2C2@C1(51383)-C84 samples were isolated by multistage high-performance liquidchromatography (HPLC; see Supplementary Figs S1–S6 fordetails) after selective removal of empty cage fullerenes. Both theY2C2@C84 and Gd2C2@C84 exhibit abnormally long, but equal,retention times on a pyrenylethyl (PYE) HPLC column(Supplementary Figs S3, S6). The long retention times for both mol-ecules are consistent with the existence of pentalene motifs27. Thestructure of Gd2C2@C1(51383)-C84 was unambiguously elucidatedby single-crystal analysis, and the structure of Y2C2@C1(51383)-C84
was confirmed by equal retention times, identical UV-visspectra (Supplementary Fig. S7) and 13C NMR chemical shiftcorrelation (Fig. 2b).
In the 13C NMR spectrum for Y2C2@C1(51383)-C84 (Fig. 2a forexpanded regions; Supplementary Fig. S8 for full spectrum), the aro-matic region consists of 84 nearly equal intensity peaks consistentwith low C1 symmetry. The two peaks downfield at 160 ppmconfirm the presence of a pentalene motif. The structure for thispreliminarily studied25 low-symmetry molecule was proposed tobe Y2C2@C1(51383)-C84, based on an independent exhaustivedensity functional theory (DFT) study28. The computationallyderived 13C NMR chemical shifts were in excellent agreementwith the experimental results (Fig. 2b). The carbide resonanceexhibits two different scalar couplings to yttrium (1JY-C¼ 16.6 Hzand 1JY-C¼ 18.8 Hz, Supplementary Fig. S9), indicating thatthe C2 unit is asymmetrically positioned between the two non-equivalent yttrium ions, with one of them associated with thepentalene motif29.
It is well recognized that every fullerene inevitably comprisessome number of hexagons and exactly 12 pentagons. A fullerenecan be imagined to occur as an ‘orange peel’ in which successivehexagons or pentagons are added in a spiral fashion until the cageis closed. If each hexagon and pentagon is numbered in sequence,the ring spiral pattern can be simply represented by the numericalorder of the 12 pentagons alone15. For a fullerene cage made upof 84 carbon atoms, there are 51,568 pentalene-containing and24 IPR isomers. Single-crystal X-ray analysis has been performedfor the Gd2C2@C84 and confirmed that the fullerene site containsa racemic mixture (Fig. 2c) of the chiral pentalene-containingC1 isomer 51383 with the ring spiral 1 2 11 13 16 19 28 31 3335 37 44. The crystallographic results are consistent with the
Graphene
Nanotubes
Primitive fullerene cages
‘Missing links’(low symmetry)
High-symmetry fullerene cages
Ih-C60 D5h-C70
Fullerenes Metallofullerenes
Sc3N@Ih-C80 Y2C2@C3v-C82
Roll/Warp
Possible encapsulationof atoms of clusters
Transformation(loss of nC2)
Transformation(loss of nC2)
a
c b
Figure 1 | Top-down mechanism for fullerene formation. a, Possible spontaneous transformation of graphene into other nanostructures. b, Cascade
shrinkage of fullerenes. In this process, random, giant fullerene structures can shrink and rearrange into high-symmetry small fullerene structures. c, Possible
modes of fullerene isomerization (Stone–Wales transformation) and C2 loss (C2 extrusion from pentalene and indene motif).
ARTICLES NATURE CHEMISTRY DOI: 10.1038/NCHEM.1748
NATURE CHEMISTRY | ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemistry2
© 2013 Macmillan Publishers Limited. All rights reserved.
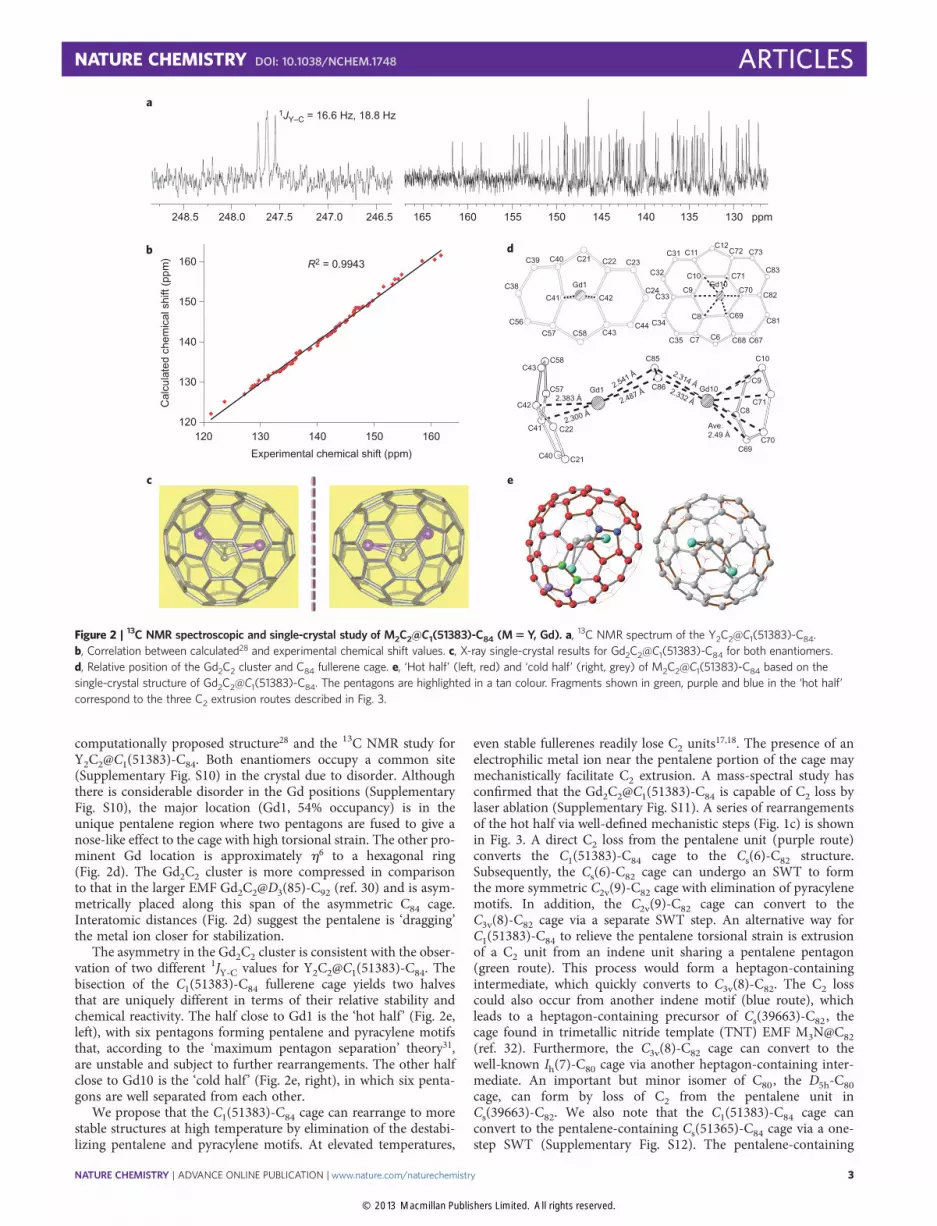
computationally proposed structure28 and the 13C NMR study forY2C2@C1(51383)-C84. Both enantiomers occupy a common site(Supplementary Fig. S10) in the crystal due to disorder. Althoughthere is considerable disorder in the Gd positions (SupplementaryFig. S10), the major location (Gd1, 54% occupancy) is in theunique pentalene region where two pentagons are fused to give anose-like effect to the cage with high torsional strain. The other pro-minent Gd location is approximately h6 to a hexagonal ring(Fig. 2d). The Gd2C2 cluster is more compressed in comparisonto that in the larger EMF Gd2C2@D3(85)-C92 (ref. 30) and is asym-metrically placed along this span of the asymmetric C84 cage.Interatomic distances (Fig. 2d) suggest the pentalene is ‘dragging’the metal ion closer for stabilization.
The asymmetry in the Gd2C2 cluster is consistent with the obser-vation of two different 1JY-C values for Y2C2@C1(51383)-C84. Thebisection of the C1(51383)-C84 fullerene cage yields two halvesthat are uniquely different in terms of their relative stability andchemical reactivity. The half close to Gd1 is the ‘hot half’ (Fig. 2e,left), with six pentagons forming pentalene and pyracylene motifsthat, according to the ‘maximum pentagon separation’ theory31,are unstable and subject to further rearrangements. The other halfclose to Gd10 is the ‘cold half’ (Fig. 2e, right), in which six penta-gons are well separated from each other.
We propose that the C1(51383)-C84 cage can rearrange to morestable structures at high temperature by elimination of the destabi-lizing pentalene and pyracylene motifs. At elevated temperatures,
even stable fullerenes readily lose C2 units17,18. The presence of anelectrophilic metal ion near the pentalene portion of the cage maymechanistically facilitate C2 extrusion. A mass-spectral study hasconfirmed that the Gd2C2@C1(51383)-C84 is capable of C2 loss bylaser ablation (Supplementary Fig. S11). A series of rearrangementsof the hot half via well-defined mechanistic steps (Fig. 1c) is shownin Fig. 3. A direct C2 loss from the pentalene unit (purple route)converts the C1(51383)-C84 cage to the Cs(6)-C82 structure.Subsequently, the Cs(6)-C82 cage can undergo an SWT to formthe more symmetric C2v(9)-C82 cage with elimination of pyracylenemotifs. In addition, the C2v(9)-C82 cage can convert to theC3v(8)-C82 cage via a separate SWT step. An alternative way forC1(51383)-C84 to relieve the pentalene torsional strain is extrusionof a C2 unit from an indene unit sharing a pentalene pentagon(green route). This process would form a heptagon-containingintermediate, which quickly converts to C3v(8)-C82. The C2 losscould also occur from another indene motif (blue route), whichleads to a heptagon-containing precursor of Cs(39663)-C82, thecage found in trimetallic nitride template (TNT) EMF M3N@C82(ref. 32). Furthermore, the C3v(8)-C82 cage can convert to thewell-known Ih(7)-C80 cage via another heptagon-containing inter-mediate. An important but minor isomer of C80, the D5h-C80cage, can form by loss of C2 from the pentalene unit inCs(39663)-C82. We also note that the C1(51383)-C84 cage canconvert to the pentalene-containing Cs(51365)-C84 cage via a one-step SWT (Supplementary Fig. S12). The pentalene-containing
1JY–C = 16.6 Hz, 18.8 Hz
248.5 248.0 247.5 247.0 246.5 165 160 155 150 145 140 135 130 ppm
a
C39 C40 C21 C22 C23C32
C38
C56
C57
C58
C57 Gd1 Gd10
C10
C9
C71
C70C69
Ave.2.49 Å
C8
2.383 Å
2.300 Å
2.487 Å2.541 Å 2.314 Å
2.332 ÅC42
C41
C40
C22
C21
C43
C58 C43
C31 C11C12
C72 C73
C83
C82
C81
C67C68
C69
C6C7C35
C85
C86
C34C44
C24C42C41
C8
C33C9 C70
C10 C71Gd10Gd1
c
d
e
R2 = 0.9943160
150
140
Cal
cula
ted
chem
ical
shi
ft (p
pm)
Experimental chemical shift (ppm)
130
120160150140130120
b
Figure 2 | 13C NMR spectroscopic and single-crystal study of M2C2@C1(51383)-C84 (M 5 Y, Gd). a, 13C NMR spectrum of the Y2C2@C1(51383)-C84.
b, Correlation between calculated28 and experimental chemical shift values. c, X-ray single-crystal results for Gd2C2@C1(51383)-C84 for both enantiomers.
d, Relative position of the Gd2C2 cluster and C84 fullerene cage. e, ‘Hot half’ (left, red) and ‘cold half’ (right, grey) of M2C2@C1(51383)-C84 based on the
single-crystal structure of Gd2C2@C1(51383)-C84. The pentagons are highlighted in a tan colour. Fragments shown in green, purple and blue in the ‘hot half’
correspond to the three C2 extrusion routes described in Fig. 3.
NATURE CHEMISTRY DOI: 10.1038/NCHEM.1748 ARTICLES
NATURE CHEMISTRY | ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemistry 3
© 2013 Macmillan Publishers Limited. All rights reserved.

Cs(51365)-C84 is the only C84 cage found for TNT EMF M3N@C84(ref. 33), and loss of C2 from its pentalene will give the C2v(9)-C82isomer. In these routes the missing link C1(51383)-C84 cage is thepoint for the metallofullerene cage to lose chirality and gain symmetry.
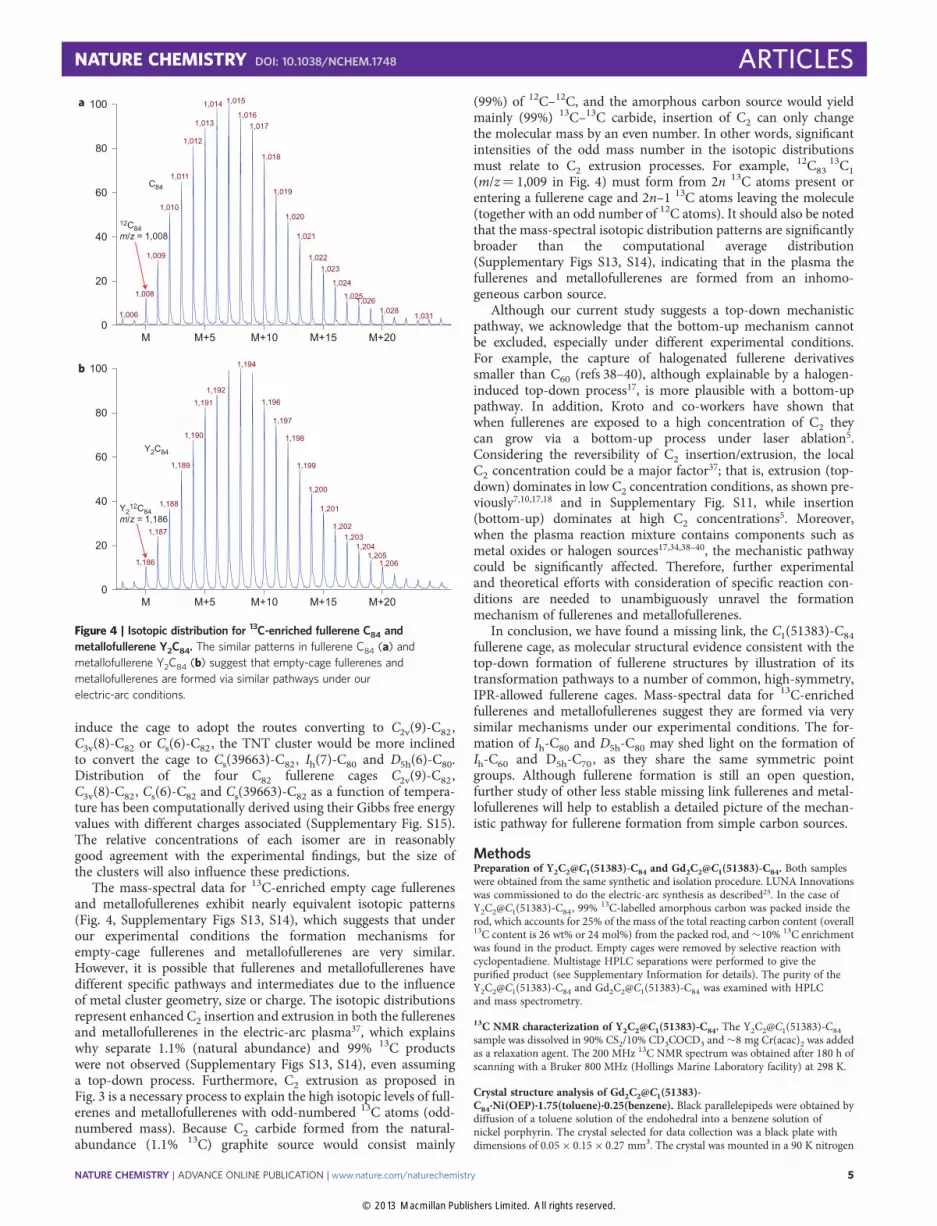
High-resolution mass-spectrometry is a powerful tool for inves-tigating the formation process of fullerenes and metallofullerenes5,34.The isotopic distribution of 13C-enriched fullerenes and metalloful-lerenes was studied in respective chromatographic fractions(Supplementary Figs S13, S14). By careful examination of the isoto-pic distribution (see examples in Supplementary Tables S1, S2), wedetermined that the 13C content in empty cage products is�9.5 mol% (Supplementary Fig. S13). The 13C content is10.5 mol% in metallofullerenes Y2C84 and 10.2 mol% in Y2C86,respectively (Supplementary Fig. S14). The subtle differencebetween the overall 13C content in empty-cage fullerenes andmetallofullerenes could be due to the geometry of the carbon rod,in which the spatial location of the metal source is more intimatelyassociated with the 13C-labelled amorphous carbon (inside) thannatural-abundance graphite (outside). More importantly, emptycage fullerenes and metallofullerenes exhibit very similar overall13C isotopic patterns (Supplementary Figs S13, S14). As a directcomparison (Fig. 4), empty cage fullerene C84 and metallofullereneY2C84 (same number of carbon atoms) share a nearly identical iso-topic pattern except for a slight difference in 13C mol% content.
DiscussionThe fullerene cages described in Fig. 3 and Supplementary Fig. S12involve a large fraction of the known metallofullerenes and cover allclasses of encapsulated atoms and clusters, including traditionalmetallic fullerenes, metal nitride (the TNT EMF), metal carbide,metal sulfide, metal oxide, metal cyanide and methanide clusterful-lerenes, as shown in Supplementary Table S3 (ref. 35). These cagesaccount for the majority of the solvent-extractable yield of metallo-fullerenes. Metallofullerenes containing large endohedral groupsneed a large cage initially in order to fit the cluster inside withouthuge energy expenditure, and the more plausible explanation forthe distorted endohedral clusters (for example, metal carbidecluster25) is that the corresponding metallofullerenes are formedfrom top-down cage shrinkage. In particular, the Cs(6)-C82,C2v(9)-C82 and C3v(8)-C82 cages represent the three cage isomersfound in the metal carbide C82 EMFs36. Furthermore, as suggestedby Shinohara, the presence of endohedral carbide clusters (M2C2)can be viewed as a result of a favourable ‘envelopment’ process byencapsulated metal atoms, as opposed to extrusion of C2 from thefullerene cage36, making metal carbide clusters an especially impor-tant family in the cascade process. Depending on the size and chargeof the endohedral units, the fullerene cage will choose differentroutes in the transformation scheme. For example, while themetal carbide cluster can stabilize the C1(51383)-C84 cage or
[5,6] C2 loss
[5,6] C2 loss
[5,5] C2 loss
C84
C82
C80
[5,5] C2 loss
C1(51383)-C84
[5,6] C2 loss
SWT
SWT SWT
SWTSWT
C3v(8)-C82 C2v(9)-C82 Cs(6)-C82
Ih(7)-C80D5h(6)-C80
Cs(39663)-C82
Figure 3 | The fullerene structural rearrangement map starting from the missing link C1(51383)-C84 cage. Many well-known metallofullerene cages are
involved in this process. Depending on the size and charge of the encapsulated atom(s) or clusters, the cage may prefer a certain sequence of the
transformation map. Colours are used to visualize the motifs involved in steps indicated by matching coloured arrows.
ARTICLES NATURE CHEMISTRY DOI: 10.1038/NCHEM.1748
NATURE CHEMISTRY | ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemistry4
© 2013 Macmillan Publishers Limited. All rights reserved.
induce the cage to adopt the routes converting to C2v(9)-C82,C3v(8)-C82 or Cs(6)-C82, the TNT cluster would be more inclinedto convert the cage to Cs(39663)-C82, Ih(7)-C80 and D5h(6)-C80.Distribution of the four C82 fullerene cages C2v(9)-C82,C3v(8)-C82, Cs(6)-C82 and Cs(39663)-C82 as a function of tempera-ture has been computationally derived using their Gibbs free energyvalues with different charges associated (Supplementary Fig. S15).The relative concentrations of each isomer are in reasonablygood agreement with the experimental findings, but the size ofthe clusters will also influence these predictions.
The mass-spectral data for 13C-enriched empty cage fullerenesand metallofullerenes exhibit nearly equivalent isotopic patterns(Fig. 4, Supplementary Figs S13, S14), which suggests that underour experimental conditions the formation mechanisms forempty-cage fullerenes and metallofullerenes are very similar.However, it is possible that fullerenes and metallofullerenes havedifferent specific pathways and intermediates due to the influenceof metal cluster geometry, size or charge. The isotopic distributionsrepresent enhanced C2 insertion and extrusion in both the fullerenesand metallofullerenes in the electric-arc plasma37, which explainswhy separate 1.1% (natural abundance) and 99% 13C productswere not observed (Supplementary Figs S13, S14), even assuminga top-down process. Furthermore, C2 extrusion as proposed inFig. 3 is a necessary process to explain the high isotopic levels of full-erenes and metallofullerenes with odd-numbered 13C atoms (odd-numbered mass). Because C2 carbide formed from the natural-abundance (1.1% 13C) graphite source would consist mainly
(99%) of 12C–12C, and the amorphous carbon source would yieldmainly (99%) 13C–13C carbide, insertion of C2 can only changethe molecular mass by an even number. In other words, significantintensities of the odd mass number in the isotopic distributionsmust relate to C2 extrusion processes. For example, 12C83
13C1(m/z¼ 1,009 in Fig. 4) must form from 2n 13C atoms present orentering a fullerene cage and 2n–1 13C atoms leaving the molecule(together with an odd number of 12C atoms). It should also be notedthat the mass-spectral isotopic distribution patterns are significantlybroader than the computational average distribution(Supplementary Figs S13, S14), indicating that in the plasma thefullerenes and metallofullerenes are formed from an inhomo-geneous carbon source.
Although our current study suggests a top-down mechanisticpathway, we acknowledge that the bottom-up mechanism cannotbe excluded, especially under different experimental conditions.For example, the capture of halogenated fullerene derivativessmaller than C60 (refs 38–40), although explainable by a halogen-induced top-down process17, is more plausible with a bottom-uppathway. In addition, Kroto and co-workers have shown thatwhen fullerenes are exposed to a high concentration of C2 theycan grow via a bottom-up process under laser ablation5.Considering the reversibility of C2 insertion/extrusion, the localC2 concentration could be a major factor37; that is, extrusion (top-down) dominates in low C2 concentration conditions, as shown pre-viously7,10,17,18 and in Supplementary Fig. S11, while insertion(bottom-up) dominates at high C2 concentrations5. Moreover,when the plasma reaction mixture contains components such asmetal oxides or halogen sources17,34,38–40, the mechanistic pathwaycould be significantly affected. Therefore, further experimentaland theoretical efforts with consideration of specific reaction con-ditions are needed to unambiguously unravel the formationmechanism of fullerenes and metallofullerenes.
In conclusion, we have found a missing link, the C1(51383)-C84fullerene cage, as molecular structural evidence consistent with thetop-down formation of fullerene structures by illustration of itstransformation pathways to a number of common, high-symmetry,IPR-allowed fullerene cages. Mass-spectral data for 13C-enrichedfullerenes and metallofullerenes suggest they are formed via verysimilar mechanisms under our experimental conditions. The for-mation of Ih-C80 and D5h-C80 may shed light on the formation ofIh-C60 and D5h-C70, as they share the same symmetric pointgroups. Although fullerene formation is still an open question,further study of other less stable missing link fullerenes and metal-lofullerenes will help to establish a detailed picture of the mechan-istic pathway for fullerene formation from simple carbon sources.
MethodsPreparation of Y2C2@C1(51383)-C84 and Gd2C2@C1(51383)-C84. Both sampleswere obtained from the same synthetic and isolation procedure. LUNA Innovationswas commissioned to do the electric-arc synthesis as described25. In the case ofY2C2@C1(51383)-C84, 99% 13C-labelled amorphous carbon was packed inside therod, which accounts for 25% of the mass of the total reacting carbon content (overall13C content is 26 wt% or 24 mol%) from the packed rod, and �10% 13C enrichmentwas found in the product. Empty cages were removed by selective reaction withcyclopentadiene. Multistage HPLC separations were performed to give thepurified product (see Supplementary Information for details). The purity of theY2C2@C1(51383)-C84 and Gd2C2@C1(51383)-C84 was examined with HPLCand mass spectrometry.
13C NMR characterization of Y2C2@C1(51383)-C84. The Y2C2@C1(51383)-C84sample was dissolved in 90% CS2/10% CD3COCD3 and �8 mg Cr(acac)2 was addedas a relaxation agent. The 200 MHz 13C NMR spectrum was obtained after 180 h ofscanning with a Bruker 800 MHz (Hollings Marine Laboratory facility) at 298 K.
Crystal structure analysis of Gd2C2@C1(51383)-C84
.Ni(OEP).1.75(toluene).0.25(benzene). Black parallelepipeds were obtained bydiffusion of a toluene solution of the endohedral into a benzene solution ofnickel porphyrin. The crystal selected for data collection was a black plate withdimensions of 0.05 × 0.15 × 0.27 mm3. The crystal was mounted in a 90 K nitrogen
100a
b
80
C84
12C84m/z = 1,008
Y212C84
m/z = 1,186
Y2C84
60
40
20
1,006
1,186
1,187
1,188
1,189
1,190
1,191
1,192
1,194
1,196
1,197
1,198
1,199
1,200
1,201
1,2021,203
1,2041,205
1,206
1,008
1,009
1,011
1,010
1,012
1,013
1,014 1,015
1,016
1,017
1,018
1,019
1,020
1,021
1,022
1,023
1,024
1,0251,026
1,0281,031
0M M+5 M+10 M+15 M+20
100
80
60
40
20
0M M+5 M+10 M+15 M+20
Figure 4 | Isotopic distribution for 13C-enriched fullerene C84 and
metallofullerene Y2C84. The similar patterns in fullerene C84 (a) and
metallofullerene Y2C84 (b) suggest that empty-cage fullerenes and
metallofullerenes are formed via similar pathways under our
electric-arc conditions.
NATURE CHEMISTRY DOI: 10.1038/NCHEM.1748 ARTICLES
NATURE CHEMISTRY | ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemistry 5
© 2013 Macmillan Publishers Limited. All rights reserved.

cold stream provided by an Oxford Cryostream low-temperature apparatus on thegoniometer head of a Bruker D8 diffractometer equipped with an ApexII charge-coupled device detector. Data were collected with the use of synchrotron radiation(l¼ 0.77490 Å) at Beamline 11.3.1 at the Advanced Light Source, LawrenceBerkeley Laboratory. The structure was solved by direct methods (SHELXS97) andrefined by full-matrix least-squares on F2 (SHELXL97). Crystallographic data havebeen deposited in the Cambridge Crystallographic Data Center (CCDC 928943).
Received 26 March 2013; accepted 7 August 2013;published online 15 September 2013
References1. Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F. & Smalley, R. E. C60-
buckminsterfullerene. Nature 318, 162–163 (1985).2. Ross, R. B. et al. Endohedral fullerenes for organic photovoltaic devices. Nature
Mater. 8, 208–212 (2009).3. Shultz, M. D. et al. Metallofullerene-based nanoplatform for brain tumor
brachytherapy and longitudinal imaging in a murine orthotopic xenograftmodel. Radiology 261, 136–143 (2011).
4. Curl, R. F. On the formation of the fullerenes. Phil. Trans. R. Soc. Lond. A 343,19–32 (1993).
5. Dunk, P. W. et al. Closed network growth of fullerenes. Nature Commun. 3,855 (2012).
6. Geim, A. K. & Novoselov, K. S. The rise of graphene. Nature Mater. 6,183–191 (2007).
7. Chuvilin, A., Kaiser, U., Bichoutskaia, E., Besley, N. A. & Khlobystov, A. N.Direct transformation of graphene to fullerene. Nature Chem. 2, 450–453 (2010).
8. Cross, R. J. & Saunders, M. Transmutation of fullerenes. J. Am. Chem. Soc.127, 3044–3047 (2005).
9. Cami, J., Bernard-Salas, J., Peeters, E. & Malek, S. E. Detection of C60 and C70in a young planetary nebula. Science 329, 1180–1182 (2010).
10. Berne, O. & Tielens, A. G. G. M. F. Formation of buckminsterfullerene (C60)in interstellar space. Proc. Natl Acad. Sci. USA 109, 401–406 (2012).
11. Li, Z., Cheng, Z., Wang, R., Li, Q. & Fang, Y. Spontaneous formation ofnanostructures in graphene. Nano Lett. 9, 3599–3602 (2009).
12. Shenoy, V. B., Reddy, C. D. & Zhang, Y.-W. Spontaneous curling of graphenesheets with reconstructed edges. ACS Nano 4, 4840–4844 (2010).
13. Irle, S., Zheng, G., Wang, Z. & Morokuma, K. The C60 formation puzzle ‘solved’:QM/MD simulations reveal the shrinking hot giant road of the dynamicfullerene self-assembly mechanism. J. Phys. Chem. B 110, 14531–14545 (2006).
14. Curl, R. F., Lee, M. K. & Scuseria, G. E. C60 buckminsterfullerene high yieldsunraveled. J. Phys. Chem. A 112, 11951–11955 (2008).
15. Fowler, P. W. & Manolopolous, D. E. in An Atlas of Fullerenes 2007 edn.(Dover Publications, 2007).
16. Murry, R. L., Strout, D. L., Odom, G. K. & Scuseria, G. E. Role of sp3 carbonand 7-membered rings in fullerene annealing and fragmentation. Nature366, 665–667 (1993).
17. Troshin, P. A. et al. Isolation of two seven-membered ring C58 fullerenederivatives: C58F17CF3 and C58F18. Science 309, 278–281 (2005).
18. Ioffe, I. N. et al. Chlorination of C86 to C84Cl32 with nonclassical heptagon-containing fullerene cage formed by cage shrinkage. Angew. Chem. Int. Ed.49, 4784–4787 (2010).
19. Kroto, H. W. The stability of the fullerenes C24, C28, C32, C36, C50, C60 and C70.Nature 329, 529–531 (1987).
20. Wang, C. R. et al. C66 fullerene encaging a scandium dimer. Nature 408,426–427 (2000).
21. Stevenson, S. et al. A stable non-classical metallofullerene family. Nature408, 427–428 (2000).
22. Tan, Y.-Z., Xie, S.-Y., Huang, R.-B. & Zheng, L.-S. The stabilization of fused-pentagon fullerene molecules. Nature Chem. 1, 450–460 (2009).
23. Shi, Z. Q., Wu, X., Wang, C. R., Lu, X. & Shinohara, H. Isolation andcharacterization of Sc2C2@C68: a metal-carbide endofullerene with a non-IPRcarbon cage. Angew. Chem. Int. Ed. 45, 2107–2111 (2006).
24. Zheng, H., Zhao, X., Wang, W.-W., Yang, T. & Nagase, S. Sc2@C70 rather thanSc2C2@C68: density functional theory characterization of metallofullereneSc2C70. J. Chem. Phys. 137, 014308 (2012).
25. Zhang, J. et al. Nanoscale fullerene compression of an yttrium carbide cluster.J. Am. Chem. Soc. 134, 8487–8493 (2012).
26. Fu, W. et al. Gd2@C79N: isolation, characterization, and monoadductformation of a very stable heterofullerene with a magnetic spin state of S¼ 15/2.J. Am. Chem. Soc. 33, 9741–9750 (2011).
27. Zhang, J. et al. Enhanced dipole moments in trimetallic nitride templateendohedral metallofullerenes with the pentalene motif. J. Am. Chem. Soc.135, 3351–3354 (2013).
28. Yang, T., Zhao, X., Li, S.-T. & Nagase, S. Is the isolated pentagon rule alwayssatisfied for metallic carbide endohedral fullerenes? Inorg. Chem. 51,11223–11225 (2012).
29. Fu, W. et al. 89Y and 13C NMR cluster and carbon cage studies of an yttriummetallofullerene family, Y3N@C2n (n¼ 40–43). J. Am. Chem. Soc. 131,11762–11769 (2009).
30. Yang, H. et al. Detection of a family of gadolinium-containing endohedralfullerenes and the isolation and crystallographic characterization of one memberas a metal carbide encapsulated inside a large fullerene cage. J. Am. Chem. Soc.130, 17296–17300 (2008).
31. Rodriguez-Fortea, A., Alegret, N., Balch, A. L. & Poblet, J. M. The maximumpentagon separation rule provides a guideline for the structures of endohedralmetallofullerenes. Nature Chem. 2, 955–961 (2010).
32. Mercado, B. Q. et al. Is the isolated pentagon rule merely a suggestion forendohedral fullerenes? The structure of a second egg-shaped endohedralfullerene—Gd3N@Cs(39663)-C82. 130, 7854–7855 (2008).
33. Beavers, C. M. et al. Tb3N@C84: an improbable, egg-shaped endohedralfullerene that violates the isolated pentagon rule. J. Am. Chem. Soc. 128,11352–11353 (2006).
34. Tan, Y.-Z. et al. Carbon arc production of heptagon-containing fullerene C68.Nature Commun. 2, 420 (2011).
35. Popov, A. A., Yang, S. & Dunsch, L. Endohedral fullerenes. Chem. Rev. 113,5989–6113 (2013).
36. Inoue, T. et al. Trapping a C2 radical in endohedral metallofullerenes:synthesis and structures of (Y2C2)@C82 (isomers I, II, and III). J. Phys. Chem. B108, 7573–7579 (2004).
37. Saha, B., Irle, S. & Morokuma, K. Hot giant fullerenes eject and capture C2molecules: QM/MD simulations with constant density. J. Phys. Chem. C115, 22707–22716 (2011).
38. Xie, S. Y. et al. Capturing the labile fullerene 50 as C50Cl10. Science 304,699 (2004).
39. Tan, Y.-Z. et al. An entrant of smaller fullerene: C56 captured by chlorinesand aligned in linear chains. J. Am. Chem. Soc. 130, 15240–15241 (2008).
40. Ziegler, K., Mueller, A., Amsharov, K. Y. & Jansen, M. Capturing the most-stableC56 fullerene cage by in situ chlorination. Chem. Asian J. 6, 2412–2418 (2011).
AcknowledgementsThe authors acknowledge support from the National Science Foundation (grants CHE-0938043 to H.C.D. and CHE-1011760 to A.L.B. and M.M.O.) and the Hollings MarineLaboratory NMR Facility. The authors also thank the Advanced Light Source, supported bythe Director, Office of Science, Office of Basic Energy Sciences, of the US Department ofEnergy (contract no. DE-AC02-05CH11231) for beam time, and S.J. Teat and C.M. Beaversfor assistance.
Author contributionsJ.Z., Y.Y. and C.D. prepared the samples. F.L.B. and M.M.O. performed the crystal study.D.W.B. performed NMR characterization. T.F. performed DFT computations. W.K.R.,R.F.H. and K.H. provided mass-spectral analysis. J.Z., M.M.O., A.L.B. and H.C.D analysedthe results. J.Z. and H.C.D composed the manuscript and modified it based on criticalcomments from M.M.O. and A.L.B. The entire project was supervised by A.L.B. and H.C.D.
Additional informationSupplementary information is available in the online version of the paper. Reprints andpermissions information is available online at www.nature.com/reprints. Correspondence andrequests for materials should be addressed to M.M.O., A.L.B. and H.C.D.
Competing financial interestsThe authors declare no competing financial interests.
ARTICLES NATURE CHEMISTRY DOI: 10.1038/NCHEM.1748
NATURE CHEMISTRY | ADVANCE ONLINE PUBLICATION | www.nature.com/naturechemistry6
© 2013 Macmillan Publishers Limited. All rights reserved.