4-monochlorobiphenyl (pcb3) induces mutations in the livers of transgenic fisher 344 rats
TRANSCRIPT

4-Monochlorobiphenyl (PCB3) induces mutations
in the livers of transgenic Fisher 344 rats
Leane Lehmanna, Harald L. Eschb, Patricia A. Kirbyc, Larry W. Robertsonb,
Gabriele Ludewigb*
aUniversity of Karlsruhe (TH), Institute of Applied Biosciences, Section of Food Chemistry and
Toxicology, Kaiserstraße 12, D-76131 Karlsruhe, Germany
bThe University of Iowa, College of Public Health, Dept. of Occupational and Environmental Health
100 Oakdale Campus, Iowa City, IA 52242-5000, USA
cThe University of Iowa, Department of Pathology, Iowa City, IA 52242, USA
*Correspondence:
Dr. Gabriele Ludewig, The University of Iowa,
Dept. of Occupational and Environmental Health
100 Oakdale Campus #234 IREH,
Iowa City, IA 52242-5000
Tel.: +001-319-335 4650; fax +001-319-335 4290
E-mail address: [email protected]
Key Words: 4-monochlorobiphenyl, PCB, BigBlue®, LacI, mutation, Fischer 344 rats
The Author 2006. Published by Oxford University Press. All rights reserved. For permissions, please email: [email protected]
Carcinogenesis Advance Access published August 31, 2006 by guest on A
ugust 2, 2015http://carcin.oxfordjournals.org/
Dow
nloaded from

2
ABBREVIATIONS: A, adenine; ATP, adenosine triphosphate; bp, base pairs; BW, body
weight; EDTA, ethylenediamine tetraacetic acid; C, cytosine; CTP, cytidine triphosphate; G,
guanine; GMP, guanosine monophosphate; HPLC ESI MS/MS, high performance liquid
chromatography electron spray injector/mass spectrometry; lacI, lactose inhibitor; 3-MC, 3-
methylcholanthrene; OD, optical density; PCB, polychlorinated biphenyl, PCB3, 4-
monochlorobiphenyl; pfu, plaque forming unit; PCR, polymerase chain reaction; PND,
postnatal day; T, thymine; TE, Tris EDTA; X-gal, 5-Bromo-4-chloro-3-indolyl β-D-
galactopyranoside.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

3
ABSTRACT
4-Monochlorobiphenyl (PCB3) is found in small amounts in commercial PCB mixtures,
indoor and outdoor air, and in food. In contrast to highly chlorinated congeners that are more
resistant to metabolic attack, PCB3 is more readily converted by xenobiotic-metabolizing
enzymes to monohydroxy-PCBs and further to dihydroxy-metabolites, which can be oxidized
to quinones. Our recent studies demonstrated the initiating action of PCB3 in the livers of
male rats. Therefore we hypothesized that PCB3 and/or its metabolite(s) are mutagenic in rat
livers in vivo. To investigate the mutagenicity and the types of mutations generated by
PCB3, male Fischer 344 BigBlue® rats, transgenic for the lacI gene, were injected
intraperitoneally with PCB3 (600 µmol/kg), 4-hydroxy-PCB3 (4-HO-PCB3, 400 µmol/kg),
3-methylcholanthrene (3-MC, 300 µmol/kg, positive control), or corn oil (negative control)
once per week, for 4 weeks. Animals were killed 10 days after the last injection and the
mutant frequency of the liver lacI gene determined. 3-MC induced a 4-fold increase of the
mutant frequency of the lacI gene in the liver. The mutant frequency in PCB3-treated
animals was also significantly elevated. In contrast, 4-HO-PCB3 induced a non-significant
doubling of the mutant frequency. The mutation spectrum of solvent control mutants was
characterized by transitions, whereas in 3-MC-animals, transversion and frameshift mutations
predominated. The PCB3-induced mutation spectrum was similar to that of the 3-MC-
induced mutants. In contrast, the mutation spectrum of the 4-HO-PCB3 group hardly
differed from that of the control animals. This study demonstrates for the first time the
mutagenicity of a PCB in vivo.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

4
INTRODUCTION
Polychlorinated biphenyls (PCBs) were produced world wide from 1929 to the early
1980s. During this period, an estimated 2 million tons of commercial PCB mixtures were
produced, of which about 0.2 million tons remain in mobile environmental reservoirs [1].
Despite the production ban and heavily restricted use, PCBs can still be found even in the
most remote areas like polar regions and mountain lakes due to atmospheric transport and
precipitation [2-5]. Vaporization of PCBs from landfills [6,7], contaminated surface water [8-
10] and construction material for public buildings [11-15] are main sources for outdoor and
indoor air contamination. Human exposure to PCBs occurs to 90% via food, the inhalation
contributes the remaining 10% [1]. In some populations, however, inhalation exposure was
found to be the major source for daily PCB uptake [16].
Commercial PCBs are mixtures of the 209 possible congeners, which differ in the
number and position of chlorines bound to the biphenyl core. PCB congeners that accumulate
in the food chain are mainly tetra- to hepta-chlorinated, are stored preferably in fatty tissues,
and are only slowly metabolized. In contrast, airborne PCBs are lower chlorinated, and are
susceptible to metabolic attack. They occur in commercial PCB mixtures [17,18], in the
atmosphere [18], especially in cities, in buildings, and near waste sites [19]. They can also be
found in food, however in a lower proportion and more likely in/on food items like
vegetables, possibly through air deposition, in vegetable oils, and in dairy products [20,21].
Congener-specific analysis of indoor air in houses built on soil contaminated with Aroclor
1260, one of the highest chlorinated commercial PCB mixtures, showed that the congeners
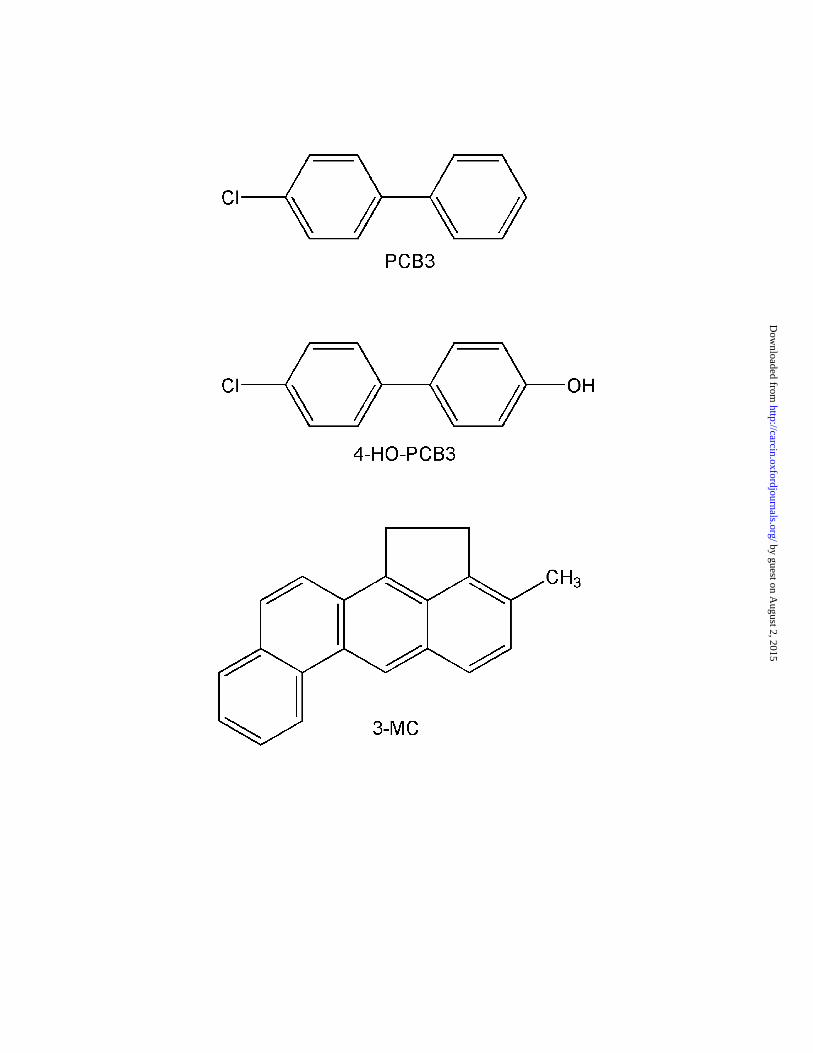
measured in indoor air were mostly 4-monochlorobiphenyl (PCB3, Fig. 1) and 2-
monochlorobiphenyl (PCB1) [22].
PCBs have repeatedly been shown to be complete carcinogens in rodents [23,24]. In
addition, commercial PCB mixtures as well as individual congeners have tumor promoting
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

5
activity in two stage hepatocarcinogenesis assays [25-27]. Generally, those PCB congeners
that induce cytochrome P-450 dependent monooxygenases (CYPs) in the liver, e.g. PCB77,
PCB126 and PCB153, are efficacious as hepatic tumor promoters [28-30]. PCBs were
reported to have no or only little genotoxic activity in most in vitro genotoxicity tests [24,31]
and their potential as cancer initiators was therefore questioned. Recently, however, evidence
for tumor initiating activity of PCB3 and other lower chlorinated PCBs that can be
metabolically activated (PCB15, PCB52, PCB77) was provided using a modified Solt-Farber
protocol in male Fischer 344 rats [32,33]. Thus PCB mixtures could be complete carcinogens
due to the initiating activity of their lower chlorinated congeners and the promoting activity
of the higher chlorinated congeners.
PCB3 is a substrate for hepatic CYPs and can be activated to electrophiles, namely
arene oxides and quinones [34]. These reactive metabolites can bind to cellular nucleophiles
like glutathione and macromolecules like DNA, RNA, protein and hemoglobin [35-38]. The
generation of reactive oxygen species during oxidative metabolism as well as the resulting
formation of 8-oxo-deoxyguanosine and DNA strand breaks have been demonstrated in vitro
[39-41]. However, the question whether PCB3 or one of its metabolites is mutagenic in vivo
was still unanswered. Therefore, we investigated the mutagenicity of PCB3 in the livers of
transgenic male Fischer 344 (BigBlue®) rats which contain 30-40 copies of the bacterial lacI
gene in every cell as a mutable target sequence. Animals were treated with corn oil (negative
control), the mutagen 3-methylcholanthrene (3-MC, positive control), PCB3, or its main
metabolite 4-hydroxy-PCB3 (4-HO-PCB3) [34]. Mutated lacI sequences were detected after
transfection into E. coli on the basis of a blue plaque phenotype using 5-bromo-4-chloro-3-
indolyl-β-D-galactopyranoside (X-gal), and the type of mutational event was determined by
sequence analysis of the lacI gene. PCB3 as well as the mutagen 3-MC induced a significant
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

6
increase in mutant frequency in the liver of BigBlue® rats, thus demonstrating for the first
time the mutagenicity of PCB3 in vivo.
MATERIALS AND METHODS
Chemical Substances
PCB3 and its hydroxylated metabolite 4-OH-PCB3 [42] were synthesized, purified and
characterized as described [32]. 3-Methylcholanthrene was purchased from Sigma (Cat # M-
6501; St Louis, MO) and 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) from
Research Products International (Prospect, IL). DNA isolation and transpackaging kits were
obtained from Stratagene (La Jolla, CA). All other reagents, media and media supplements
were from Fischer Scientific (Pittsburgh, PA), if not stated otherwise.
Animals and Treatment Protocol
Sixteen male Fisher 344 BigBlue® rats, homozygous for lacI transgene, were purchased from
Stratagene (La Jolla, CA) and obtained from the breeder, Taconic Laboratories, Germantown,
NY) at postnatal day (PND) 30. The animals were kept on a 12-hr light/dark cycle and were
provided with a standard 7013-NIH-13 Modified Open Formula Rat diet and water ad
libitum. After one week of acclimatization animals were weighed and distributed at random
into 4 groups with 4 animals each. Each animal received one weekly i.p. injection on PND
37, 44, 51 and 58 of either 298 µmol (80 mg)/kg bodyweight (BW) 3-MC (positive control),
600 µmol (113 mg)/kg BW PCB3, 400 µmol (82 mg)/kg BW 4-HO-PCB3 in corn oil or 5
ml/kg BW corn oil alone (negative control). The doses of the PCB and metabolites were
selected based on previous studies [32,33]. Throughout the treatment period animals were
monitored daily for well being and weighed twice per week. Seventeen days after the last
injection (PND 75), all animals were euthanized by CO2 asphyxiation followed by cervical
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

7
dislocation. The livers were immediately removed and weighed. Small tissue samples were
quickly excised from the central part of the large lobe of the liver and these samples and the
remaining liver were snap frozen in liquid nitrogen and stored at -80°C until use for analysis
of lacI mutation frequencies and mutant analysis as described below. Parts of the remaining
liver were used to prepare slides for histological analysis. Four micron sections were
prepared, placed on glass slides and stained with conventional hematoxylin and eosin stains.
Slides were then examined using an Olympus BX40 light microscope. All experiments were
conducted with the approval of the University of Iowa Institutional Animal Care and Use
Committee.
DNA Extraction and Packaging
DNA extraction, packaging, and plating for lacI mutant plaque detection were carried out in a
blocked manner (i.e. 1 animal from each group per cycle) to avoid bias from day-to-day
variations following the BigBlue® assay procedure according to the manufacturer�s
instructions (Stratagene). Genomic DNA was extracted from the livers using the BigBlue®
RecoverEase DNA isolation kit according to the manufacturer�s instruction manual. Briefly,
50 mg of liver tissue were homogenized with a Dounce homogenizer at 4°C, and nuclei were
pelleted by centrifugation for 10 min at 1100 g. RNA and proteins in the pellet were digested
with RNase and Proteinase K, and the genomic DNA was purified by dialysis in TE buffer
for 48 h.
For recovery of the lambda transgenic shuttle vector containing the lacI target, i.e.
excision of the lambda vector and packaging into a lambda head, the Lambda Phage
Transpack® packaging kit (Stratagene) was used with 10 µl of genomic DNA per reaction as
described in the manual. The mixture, now containing the lacI transgene packaged into
infectious phages (packaged lacI), was diluted to 1 ml with SM buffer (100 mM NaCl, 8 mM
MgSO4, 50 mM Tris-HCl, 0.004% (w/v) gelatin), and stabilized by addition of 50 µl
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

8
chloroform. Packaging efficiency was checked by infecting E. coli bacteria, strain SCS-8 (2
ml SCS-8 suspension, OD = 0.5) with 1 µl packaged lacI extract and plating the suspension
with 3 ml top agarose onto 100 mm NZY-agar plates. Infected bacteria result in plaques in
the bacterial lawn.
Determination of lacI Mutant Frequency
For determination of the lacI mutant frequency, a volume of phage extract equivalent
to approximately 12,500 plaque forming units (pfu) was mixed with 2 ml SCS-8 suspension
(OD = 0.5) and incubated at 37°C for 15 min. Fifty µl of each 2 ml SCS-8/phages suspension
were added to 2 ml fresh SCS-8 suspension and mixed with 35 ml top agarose (titer trays).
The rest of the 2 ml SCS-8/phage suspension was mixed with 35 ml top agarose containing
1.5 mg/ml of X-gal. Top agarose containing infected and non-infected SCS-8 was poured
onto 500 mm2 assay trays (Corning, Acton, MA) containing NZY-bottom agar (assay trays).
The trays were vented for 30 min before incubation at 37°C for 20 h and scoring. Packaging
and plating were repeated until 95,000�339,000 pfu were scored for each DNA sample.
Titer trays were scored for pfu and assay trays were inspected for blue plaques. To
confirm the mutant phenotype, and for use in DNA sequence analysis, all putative mutants
from the 500 mm2 assay trays were picked, diluted 1:100 with SM buffer and replated on 100
mm diameter plates with 3.5 ml of top agarose containing 1.5 mg/ml of X-gal. The lacI
mutant frequency was calculated by dividing the number of verified mutant plaques by the
total number of plaques analyzed and presented as number of mutants ± standard error per
106 pfu.
Sequence Analysis of lacI Mutants
The lac I genes of verified mutants were PCR amplified using the following primers
(named according to the manufacturer�s instruction manual) and amplification conditions:
5�-GTATTACCGCCATGCATACTAG-3� (forward PCR primer),
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

9
5�-CGTAATCATGGTCATAGCTG-3� (reverse PCR primer), 30 amplification cycles at
94°C for 30 sec, 53°C for 50 sec, and 72°C for 60 sec. Aliquots of the PCR products were
characterized by agarose gel electrophoresis (1% in TAE buffer, 5 volt/cm). Then, PCR
products were purified using QiaQuick columns (Qiagen, Valencia, CA) and quantified by
their OD at 260 nm. Sixty ng of this purified DNA were used per reaction to sequence the
entire lacI gene (1,083 bp) in both directions using the forward PCR primer, reverse PCR
primer, primer #5 (5�-TCTGGTCGCATTGGGTC-3�) and primer #12 (5�-
AGAACTTAATGGGCCCG-3�) and a dye terminator cycle sequencing kit (Applied
Biosystems, Foster City, CA) according to the manufacturer�s instructions. The sequencing
products were analyzed on an Applied Biosystems automated capillary DNA sequencer
Model 3700 following the procedure outlined by Applied Biosystems. All alterations in DNA
sequence were verified at least once. The same mutation at the same locus in DNA extracted
from the same animal may be due to the same event and was therefore counted only once for
the calculation of the mutation frequency, which is the number of independent mutations
(different locus and/or different type as verified by sequence analysis) per total number of
plaques analyzed and presented as number of mutations ± standard error per 106 pfu.
Statistical Analysis
Gains of body weight were analyzed by Student�s t-test. The lacI mutant frequencies and the
frequencies of recovered mutations were analyzed by one-way ANOVA and Chi-square.
Data are presented as means ± standard error (p< 0.05). Chi-square test and the statistical test
described by [43] using the classes �transition�, �transversion�, and �frameshift� were used to
analyze the lacI mutation profiles of the 4 treatment groups.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

10
RESULTS
The objective of the present study was to clarify whether PCB3 and/or its
monohydroxylated metabolite 4-HO-PCB3 are mutagenic at the lacI gene locus in the liver of
transgenic male Fischer 344 BigBlue® rats. The established mutagen 3-MC (Fig. 1) was used
as a positive control throughout this study.
Development of animal and liver weight
The bodyweights of male BigBlue® rats administered corn oil (negative control
group), 3-MC, PCB3, or 4-HO-PCB3 were monitored from two days before the first injection
(PND 35) until euthanization at PND 75. AT PND 35, bodyweights of all treatment groups
were similar (average 71.5 ± 4.32 g). Bodyweights as well as gains of bodyweight did not
differ significantly during the first 30 experimental days (Figure 2). However, at PND 65 and
75, the gain of bodyweight of animals treated with 3-MC were significantly reduced
compared to the other treatment groups (Figure 2). In contrast, liver weights of all treatment
groups (corn oil, 3-MC, PCB3, and 4-HO-PCB3) did not differ significantly at PND 75 (4.3 ±
0.37 g/100 g bodyweight, 4.7 ± 0.64 g/100 g bodyweight, 4.3 ± 0.26 g/100 g bodyweight, and
4.5 ± 0.33 g/100 g bodyweight, respectively.
Liver Histology
Liver samples were analyzed microscopically. Control animals had normal liver architecture,
one with rare lymphoid aggregates in the parenchyma. PCB3-treated animals had only very
minimal changes with or without small clusters of parenchymal lymphocytes and rare
acidophil bodies. Animals that had received 4-HO-PCB3 showed dilated sinusoid and rare
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

11
acidophil bodies. Three of four animals had signs of minimal portal tract inflammation. 3-
MC-treated animals had mild to severe centrilobular necrosis with various degrees of
lymphocytes and neutrophiles in the necrotic areas. The portal tracts were unaffected.
LacI mutant frequency in the livers of BigBlue® rats
Liver lacI mutant frequencies for BigBlue® rats exposed to corn oil (negative
control), 3-MC (positive control), PCB3, and 4-HO-PCB3 were measured 17 days following
the last injection (PND 75). For control animals, the mutant frequency was 17 ± 4 x 10-6 pfu
(Figure 3 and Supplementary Table 1). Treatment with 3-MC induced a mutant frequency of
88 ± 15 x 10-6 pfu, approximately a fivefold increase over the untreated control (p = 0.004).
The mutant frequency in the liver of rats treated with PCB3 was 48 ± 4 x 10-6 pfu (p = 0.001).
Although elevated more than twofold, the mutant frequency for rats administered 4-HO-
PCB3 (40 ± 12 x 10-6 pfu) was not significantly different from that of the control rats (p =
0.115) using ANOVA for analysis due to the very high variation within this group. When the
Chi-square test was applied, the significance of 3-MC and PCB3 was confirmed and 4-HO-
PCB3 was significant as well (p<0.05).
Sequence analysis of lacI mutants
Sequence analysis of mutants provides information about the kind of mutation
induced spontaneously or by treatment with test compounds. Sixteen of the 17 mutants (95%)
from the vehicle-treated rats (Supplementary Table II), 47 of 56 (85%) from the 3-MC-
treated rats (Supplementary Table III), 24 of 29 (84%) of the mutants from rats treated with
PCB3 (Supplementary Tables IV), and 29 of 34 (83%) of those from rats treated with 4-HO-
PCB3 (Supplementary Table V) provided lacI sequences that were complete, readable, and
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

12
contained a mutation. In some instances the same mutation (location in the sequence and type
of mutation) was identified in several plaques from the same animal. In this case a
conservative approach was used by assuming that these mutations were siblings and
represent a single in vivo mutational event. As a result a total number of 16, 44, 23, and 27
independent mutations from the corn-oil treated control, 3-MC-, PCB3-, and 4-HO-PCB3-
treated groups, respectively, were identified, as listed in Supplementary Tables II�V and
summarized in Table I. A calculation of the frequency for these recovered, sequenced,
independent mutations gave 16 ± 4, 67 ± 13, 38 ± 7, and 30 ± 9 per 106 pfu for animals
treated with corn oil, 3-MC, PCB3, and 4-HO-PCB3, respectively. The significance level
compared to corn oil control for 3-MC, PCB3 and 4-HO-PCB3 was p = 0.01, p = 0.03, and p
= 0.18 using ANOVA and p<0.001, p<0.05, and not significant, respectively, when the Chi-
square test was used. It should be noted that this is a very conservative approach, but even
so, the frequencies of recovered mutations of the 3-MC and PCB3 group were significantly
elevated compared to the control group thus confirming the results obtained with the less
stringent mutant frequencies.
Simple bp substitutions comprised most of the mutations recovered: 13/16 (81%)
from untreated animals, 32/44 (73%) from 3-MC-treated, 17/23 (74%) from PCB3-treated,
and 21/27 (78%) from 4-HO-PCB3-treated rats (Table I). Most of these mutations in
untreated (69%), 3-MC-treated (59%), PCB3-treated (53%) and 4-HO-PCB3-treated (62%)
rats were found within the first 400 bp. Substitutions at G:C accounted for 75% of the
control, 64% of the 3-MC-induced mutations, 61% of he PCB3, and 67% of the 4-HO-PCB3-
induced mutations (Table I and supplementary Tables II-V).
Characterists of lac I mutations in the different treatment groups
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

13
The most frequent type of mutation in the negative control group was G:C→A:T
transition (9/16; 56%), with 22% of these occurring at CpG sites. Also 2 G:C→C:G
transversion (13%), 1 A:T→G:C transition (6%), and 1 G:C→A:T transversion (6%)
mutations were recovered from this group. No A:T→C:G or A:T→T:A transversions were
detected in this group. In addition to bp substitutions, 1 and 2 of the 16 total mutations from
the control group were deletions or insertions, respectively, leading to frameshift mutations
(19% of the total mutations; Table 1 and Supplementary Table II).
Forty-four different mutation sites were discovered in mutants from 3-MC-treated
animals. One mutation was recovered 3 times and another one twice from the same animal,
leaving 44 independent mutations. The predominant mutation types were G:C→T:A
transversions and G:C→A:T transition mutations (27% each). Seven of the 12 transition
mutations (58%) occurred at a CpG site. In addition, there were 7 single-base frameshifts,
three 2-5 bp insertions, and a 7 and a 67 bp deletion, leading to a frameshift mutation rate of
27% of the total mutations in this group (Table 1 and Supplementary Table 3). The total
number of transitions was significantly reduced in the 3-MC-treated animals compared to
solvent controls (Chi-square test, Table I). However, using the test described by Cariello et al.
[43], the 3-MC-induced mutation spectrum in total barely missed being significantly different
from the lacI spectrum of untreated rats (p=0.058).
Of the 17 PCB3-induced single bp substitutions, 3 mutations (18%) occurred at A:T
bp while 14 (82%) were at G:C bp (Table I and Supplementary Table 4). Like the 3-MC
spectrum, the predominant mutations in this group were G:C→A:T transition (30%; 7/23),
with 43% of them occurring at CpG sites, and G:C→T:A transversion (30%; 7/23), followed
by A:T→C:G transversion (9%; 2/23). Also, there was one (4%) A:T→T:A transversion
mutation recovered from this group. Additionally, there were 4 single-base frameshifts, a 4
bp insertion, and a 13 bp deletions, leading to frameshift mutations (26% of the total
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

14
mutations, Table I). The proportion of transitions among PCB3-induced mutations was
significantly different from that of the control group (p < 0.05) and similar to the 3-MC-
induced mutation spectrum. Despite the apparent differences in the proportions of transitions
and transversions recovered from total mutants of the control and the PCB3-treated rats
(Table 1), the PCB3-induced mutation spectrum was not significantly different from the
spectrum of the control animals using the previously described statistical test method [43]
(p=0.117).
Of the 21 4-HO-PCB3-induced single bp substitutions, only 3 mutations (14%)
occurred at A:T bp while 18 (86%) were at G:C bp (Table I). Like the control spectrum, the
predominant mutations in this group were G:C→A:T transition (48%; 13/27), with 23% of
them occurring at CpG sites, followed by G:C→T:A transversions (15%; 4/27), and
A:T→G:C transition (7%; 2/27). There was only one (4%) AT→C:G transversion recovered
from this group and no A:T→T:A transversion. Additionally, there were 2 single-base
frameshifts, three insertions (4-8 bp long), and a 21 bp deletion, leading to frameshift
mutations (22% of the total mutations, Table I). Therefore, the 4-HO-PCB3-induced mutation
spectrum was not significantly different from the spectrum of the control group.
Discussion
The present study demonstrates for the first time that PCB3 is mutagenic in vivo.
Mutant frequencies and mutation spectra of the negative control
In our experiments, the mutant frequency as well as the mutation frequency of the lacI
gene in the liver of untreated BigBlue® rats was 17 ± 4 x 10-6 pfu which is in concordance
with the finding derived from the analysis of 35 independent experiments by Lambert et al.
[44] which yielded an average spontaneous mutation frequency of the lacI gene in the liver of
BigBlue® rats of 35 ± 3 x 10-6 pfu with a range of 10 - 85 x 10-6 pfu.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

15
The dominating kind of mutation in all four groups was bp substitutions (73 � 81%,
compared to 19 - 27% frameshift mutations). Most of these bp substitution mutations in
untreated (69%), 3-MC-treated (59%), PCB3-treated (53%) and 4-HO-PCB3-treated (62%)
rats were found within the first 360 bp of the lac I gene, which is in accordance with
observations of other laboratories [45]. This region contains the DNA binding domain (or
negative complementing region) of the lacI gene [46].
The most frequent type of mutation in the control group was G:C→A:T transition
(56%) which is in agreement with the findings of deBoer and Glickman [45]. Sixtysix
percent (4/6) of the G:C→A:T transitions recovered from our control animals occurred in the
first 400 bp and 50% of these (2/4) occurred at CpG sites (Supplementary Table II).
G:C→A:T transitions are known to be particularly concentrated in the first 400 bp, where
65�85% of them are located at 5�-CpG-3 sites due to spontaneous deamination of methylated
cytosines [47] and the unusual malleability of CpG steps [48,49].
In contrast, deletion events are threefold more common after the first 400 bp [45]. The
3 frameshift mutations recovered from the 16 mutants of the control animals occurred within
this region (Supplementary Table II). Deletion of DNA is frequently mediated by repeated
sequences and both insertions and deletions can be explained by a slipped-mispairing
mechanism [50]. For additional information, we added the location of the frameshift mutation
in Supplementary Tables II-V.
Mutations in 3-MC-treated animals The mutant frequency in the liver of 3-MC-treated rats in our experiments was 88 ±
15 x 10-6 pfu. Sequence analysis revealed that most of them, 93%, were true independent
mutations. If we subtract 7% from those 88 mutants to account for possible siblings we
obtain an estimated mutation frequency for the 3-MC group of about 82 x 10-6 pfu, giving a
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

16
5-fold induction of mutations above background levels. Rihn et al. [51] analyzed the
mutagenicity of 3-MC in the liver of male BigBlue® mice. Fourteen days after a single
injection of 80 mg 3-MC/kg BW, a mutation frequency of 203 ± 29 x 10-6 pfu was observed.
This seems to be higher than in our experiment with BigBlue® rats using 4 once a week
injection of 80 mg 3-MC/kg BW, however, the mutation frequency in the liver of the control
animals (76 ± 27 x 10-6 pfu) was also much higher than in our study, resulting in a less than
3-fold induction of mutations by 3-MC. These differences between this mouse and our rat
study may be due to the differing species and treatment protocols.
The predominant mutations among 3-MC-induced total mutations were G:C→T:A
transversion and G:C→A:T transition mutations. Compared to the negative control, the 3-
MC-induced mutation spectrum showed a dramatic increase in G:C→T:A transerversions
(Table 1) and -1 frameshift mutations (Table 1). G:C→T:A transversions were also the
predominant mutations recovered from the 3-MC-induced lambda lacI mutant plaques
sequenced by Rihn et al. [51]. Metabolically activated polycyclic aromatic hydrocarbons are
known to react with the exocyclic amino group of G, A, and - to a lesser extend - C [52],
resulting in G:C→T:A and A:T→T:A transversions and G:C→A:T transition mutations.
Mutant frequencies and mutation spectra of PCB-induced mutants
The purpose of this study was primarily to analyze whether PCB3 and one of its
metabolites, 4-HO-PCB3, induce gene mutations in the livers of male Fischer 344 rats. The
basis for this question was the observation that both compounds and a downstream
metabolite, the 3,4-quinone (see Fig 4) induced γ−glutamyltranspeptidase-positive foci,
which are considered to be pre-neoplastic stages, and adenoma (only PCB3) in the livers of
male Fischer 344 when applied as initiating agent in a modified Solt-Farber protocol [32,33].
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

17
Similarly to the Solt-Farber experiment a slightly lower dose of the 4-HO-PCB was used (4
injections of 400 µmol/kg BW) than of the PCB3 (4 injections of 600 µmol/kg BW) to
account for the fact that 4-HO-PCB3 is expected to be one metabolic step closer to the
proposed ultimate mutagen, the 3,4-quinone of PCB3, and the expected higher toxicity of the
hydroxylated metabolite compared to the parent compound.
In this present study, PCB3 indeed significantly induced mutations in the liver of
transgenic male BigBlue® rats. We did not observe any signs of liver toxicity due to PCB3-
treatment, neither morphologically nor histologically, which argues against indirect effects as
cause for this increase in mutations. Moreover, the mutation spectrum was significantly
different compared to that of control animals, with transitions, which are typical for
spontaneous mutations, contributing significantly less to the PCB3-induced mutation
spectrum than in controls. This characteristic was shared with 3-MC, our positive control and
a known mutagen and carcinogen in rodents. In the 4-HO-PCB3 group on the other hand, the
mutant frequency was elevated twofold, which was not statistically significant. This lack of
significance may in part be due to the large variability in mutation frequency between
animals within this group and the use of only 4 animals per treatment group However, the
mutation spectrum of mutants isolated from animals treated with 4-HO-PCB3, was also not
significantly different from that of the control animals. A slight shift towards more
transversion and frameshift mutations may be present, but it is nowhere near the one seen
with 3-MC or PCB3. Even though the mutant frequency may be underestimated by
inadvertently omitting light colored mutant plaques, the mutational spectra are unlikely to be
biased [45]. Of course, certain deletion or insertion mutations may not be detected in phage
based transgenic rodent systems because of packaging constraints. Thus, agents whose
genotoxicity arises primarily through clastogenic events are less likely to be detected in
transgenic rodent systems [44]. For these compounds existing clastogenicity assays such as
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

18
the in vivo bone marrow micronucleus test may still be more sensitive, although organ
specificity could hamper the detection even in these systems [44]. The results in our
experiments are therefore not sufficient to clearly answer the questions whether 4-HO-PCB3
is mutagenic in the liver of male rats, but they show that the parent compound, PCB3, is a
significantly stronger mutagen with the concentrations and exposure protocol used in this
study.
What is the mechanism of PCB3 activation to a mutagen?
The second goal of this study was to analyze whether the mutation spectra of PCB3
and its metabolite 4-HO-PCB3 could give us any information about the ultimate mutagenic
species. This raises the question of the mechanism of mutation induction involved in the
PCB3-induced mutagenesis. PCB3 is efficiently metabolized by CYPs in the liver to
monohydroxy-PCB3, probably with an epoxide as intermediate, and further to dihydroxylated
metabolites (Figure 4) [34,40,53]. Dihydroxy-PCBs can be further oxidized to the
corresponding semiquinone and quinone. Several reports show metabolic activation of
radiolabeled PCBs to result in protein, RNA, and DNA binding of radioactivity in cell free
systems [54], in cultured cells [55] and in vivo [37]. The formation of such adducts by
monochlorinated biphenyls has been explained by activation through different metabolic
pathways [34,39,56,57].
32P-postlabeling studies with PCB3 and a metabolizing system showed that similar but
also unique adducts were formed depending on whether the incubation environment was
oxidizing or reducing [36]. It was suggested that some DNA adducts of lower chlorinated
biphenyls may be derived from arene oxides (reducing environment), but others may be
formed from the oxidized products of catechol and p-hydroquinone, i,e, semiquinones and
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

19
quinones (oxidizing environment). PCB-quinones form adducts with nitrogen and sulfur
nucleophiles [40,56] as well as with guanosine or isolated DNA in vitro [36,40,53]. Recently,
the structure of the reaction product of PCB3-2,5-quinone with N2 of guanine has been
identified [53]. In addition to the cell-free experiments mentioned above, the induction of
DNA adducts in cultured human hepatocytes by PCB3 has been demonstrated by the 32P-
postlabeling assay [35] and quinonoid protein adducts of 2,5,2�,5�-tetrachlorobiphenyl
(PCB52) were discovered in the liver and brain of treated rats [58].
Besides the reactivity of arene oxide and quinoid PCB metabolites towards DNA, the
induction of oxidative stress might be another possible mechanism for the initiation potential
of PCB3 and some of its metabolites: incubation of isolated DNA with dihalogenated PCB
catechols in the presence of lactoperoxidase and metal ions induced 8-oxo-deoxyguanosine
formation [39]. Several metabolites of PCB3 induce DNA strand breaks in vitro and reactive
oxygen species in vitro as well as in cultured cells [41].
The G:C→T:A transversions recovered from PCB3-induced mutants in the present
study are in concordance with both possible mechanisms: arylalkylating agents such as
activated PCB3 metabolites (e.g. quinones, epoxides) react preferably with the exocyclic
aminogroup of G, A or C [52], which would generate a non-coding DNA lesion. When trying
to interpret such a lesion during transcription, DNA polymerases usually use ATP instead of
CTP resulting in G:C→T:A and A:T→T:A transversion, as well as G:C→A:T transition.
However, G:C→T:A transversion is also the predominant mutation induced by 8-oxo-
deoxyguanosin [59]. Thus both, oxidative stress and/or adduct formation, could have caused
the observed increase in mutations. The fact that -1 frameshift mutations and A:T→C:G and
A:T→T:A transversions also seem to be increased in PCB3-treated animals, similar to 3-MC-
treated animals, may be additional support for arylalkylation as mechanism. Also, the strong
mutagenicity of PCB3 compared to 4-HO-PCB3 seems to argue in favor of an arenoxide as
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

20
mutagenic intermediate. However, it could also be argued that the 4-HO-PCB3 metabolite
was already too reactive to reach the target organ, the liver, whereas the non-reactive PCB3
was easily transported to the liver and there bioactivated to a mutagenic species with or
without the production of oxidative stress. Thus these in vivo experiments proved for the first
time that PCB3 is indeed mutagenic in the organ where it produced preneoplastic lesions, but
the ultimate mutagenic species and activation mechanism is still elusive.
PCB3 is found in the air of contaminated sites and in urban areas [18]. It can not be
detected in human blood samples [60], indicating its susceptibility to metabolism which
might involve the generation of mutagenic metabolites. In addition, many other lower
chlorinated PCBs which are ubiquitous in our environment may have a similar potential to be
activated to mutagenic and carcinogenic species as our model compound PCB3, as indicated
by the observation that several of them cause preneoplastic foci in rat liver [32,33].
Therefore, the here described mutagenicity of PCB3 in vivo might have strong implication
concerning the potential carcinogenicity of this whole group of ubiquitous environmental
pollutants.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

21
ACKNOWLEDGEMENTS
The authors would like to thank James A. Jacobus for injecting the animals and his help
in the laboratory, Dr. Lehmler for synthesizing and characterizing the study chemicals, and
Dr. Barbara Shane for useful suggestions concerning the assay procedure. This work was
financially supported by NIEHS (ES 07380 and ES13661), DOD (DAMD17-02-1-0241),
EPA (R-82902102-0) and a Hollaender Trainee award to L.L. from the EMS. The content of
this publication is the sole responsibility of the authors and does not necessarily represent the
official views of the funding agencies.
REFERENCES
1. WHO (2003) Polychlorinated biphenyls: Human health aspects. Concise International Chemical Assessment Document. World Health Organisation, Geneva.
2. Fuoco, R., Colombini, M.P., Ceccarini, A. and Abete, C. (1996) Polychlorobiphenyls in Antarctica. Microchem. J., 54, 384-90.
3. Grimalt, J.O., van Drooge, B.L., Ribes, A., Vilanova, R.M., Fernandez, P. and Appleby, P. (2004) Persistent organochlorine compounds in soils and sediments of European high altitude mountain lakes. Chemosphere, 54, 1549-61.
4. Gustafsson, O., Andersson, P., Axelman, J., Bucheli, T.D., Komp, P., McLachlan, M.S., Sobek, A. and Thorngren, J.O. (2005) Observations of the PCB distribution within and in-between ice, snow, ice-rafted debris, ice-interstitial water, and seawater in the Barents Sea marginal ice zone and the North Pole area. Sci. Total. Environ., 342, 261-79.
5. Vilanova, R.M., Fernandez, P. and Grimalt, J.O. (2001) Polychlorinated biphenyl partitioning in the waters of a remote mountain lake. Sci. Total. Environ., 279, 51-62.
6. Hansen, L.G. and O'Keefe, P.W. (1996) Polychlorinated dibenzofurans and dibenzo-p-dioxins in subsurface soil, superficial dust, and air extracts from a contaminated landfill. Arch. Environ. Contam. Toxicol., 31, 271-6.
7. Persson, N.J., Pettersen, H., Ishaq, R., Axelman, J., Bandh, C., Broman, D., Zebuhr, Y. and Hammar, T. (2005) Polychlorinated biphenyls in polysulfide sealants--occurrence and emission from a landfill station. Environ. Pollut., 138, 18-27.
8. Hermanson, M.E. and Hites, R.A. (1989) Long-term measurements of atmospheric polychlorinated biphenyls in the vicinity of Superfund dumps. Environ. Sci. Technol., 23, 1253-1258.
9. Hornbuckle, K.C. and Green, M.L. (2003) The impact of an urban-industrial region on the magnitude and variability of persistent organic pollutant deposition to Lake Michigan. Ambio, 32, 406-11.
10. Schwackhamer, D.L. and Armstrong, D.E. (1986) Estimation of the atmospheric and nonatmospheric contributions and losses of polychlorinated biphenyls for Lake Michigan on the basis of sediment records of remote lakes. Environ. Sci. Technol., 20, 879-833.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

22
11. Digernes, V. and Astrup, E.G. (1982) Are datascreen terminals a source of increased PCB-concentrations in the working atmosphere? Int. Arch. Occup. Environ. Health, 49, 193-7.
12. Gabrio, T., Piechotowski, I., Wallenhorst, T., Klett, M., Cott, L., Friebel, P., Link, B. and Schwenk, M. (2000) PCB-blood levels in teachers, working in PCB-contaminated schools. Chemosphere, 40, 1055-62.
13. Herrick, R.F., McClean, M.D., Meeker, J.D., Baxter, L.K. and Weymouth, G.A. (2004) An unrecognized source of PCB contamination in schools and other buildings. Environ. Health Perspect., 112, 1051-3.
14. Kohler, M., Tremp, J., Zennegg, M., Seiler, C., Minder-Kohler, S., Beck, M., Lienemann, P., Wegmann, L. and Schmid, P. (2005) Joint sealants: an overlooked diffuse source of polychlorinated biphenyls in buildings. Environ. Sci. Technol., 39, 1967-73.
15. Liebl, B., Schettgen, T., Kerscher, G., Broding, H.C., Otto, A., Angerer, J. and Drexler, H. (2004) Evidence for increased internal exposure to lower chlorinated polychlorinated biphenyls (PCB) in pupils attending a contaminated school. Int. J. Hyg. Environ. Health, 207, 315-24.
16. Wilson, N.K., Chuang, J.C. and Lyu, C. (2001) Levels of persistent organic pollutants in several child day care centers. J Expo Anal Environ Epidemiol, 11, 449-58.
17. Hansen, L.G. (1999) The ortho side of PCBs: Occurence and Disposition. Kluwer Academic Publishers, Boston.
18. Uraki, Y., Suzuki, S., Yasuhara, A. and Shibamoto, T. (2004) Determining Sources of Atmospheric Polychlorinated Biphenyls based on Their Fracturing Concentrations and Congener Compositions. Journal of Environmental Science and Health, Part A: Toxic/Hazardous Substances & Environmental Engineering, A39, 2755-2772.
19. Imsilp, K., Schaeffer, D.J. and Hansen, L.G. (2005) PCB disposition and different biological effects in rats following direct soil exposure vs. PCBs off-gassed from the soil. Toxicol. Environ. Chem., 87, 267-285.
20. Duarte-Davidson, R. and Jones, K.C. (1994) Polychlorinated biphenyls (PCBs) in the UK population: estimated intake, exposure and body burden. Sci. Total. Environ., 151, 131-52.
21. Gallani, B., Boix, A., Di Domenico, A. and Fanelli, R. (2004) Occurrence of NDL-PCB in food and feed in Europe. Organohalogen Compounds, 66, 3561-3569.
22. Davis, B., Beach, J., M. Wade, M., Klein, A.K. and Hoch, K. (2002) Risk Assessment of Polychlorinated Biphenyls (PCBs) in Indoor Air. The Toxicologist, Supplement to Toxicological Sciences, 66, 516.
23. Mayes, B.A., McConnell, E.E., Neal, B.H., Brunner, M.J., Hamilton, S.B., Sullivan, T.M., Peters, A.C., Ryan, M.J., Toft, J.D., Singer, A.W., Brown, J.F., Jr., Menton, R.G. and Moore, J.A. (1998) Comparative carcinogenicity in Sprague-Dawley rats of the polychlorinated biphenyl mixtures Aroclors 1016, 1242, 1254, and 1260. Toxicol. Sci., 41, 62-76.
24. Silberhorn, E.M., Glauert, H.P. and Robertson, L.W. (1990) Carcinogenicity of polyhalogenated biphenyls: PCBs and PBBs. Crit. Rev. Toxicol., 20, 440-96.
25. Dean, C.E., Jr., Benjamin, S.A., Chubb, L.S., Tessari, J.D. and Keefe, T.J. (2002) Nonadditive hepatic tumor promoting effects by a mixture of two structurally different polychlorinated biphenyls in female rat livers. Toxicol. Sci., 66, 54-61.
26. Haag-Gronlund, M., Conolly, R., Scheu, G., Warngard, L. and Fransson-Steen, R. (2000) Analysis of rat liver foci growth with a quantitative two-cell model after treatment with 2,4,5,3',4'-pentachlorobiphenyl. Toxicol. Sci., 57, 32-42.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

23
27. Haag-Gronlund, M., Kato, Y., Fransson-Steen, R., Scheu, G. and Warngard, L. (1997) Promotion of enzyme altered foci in female rat 2,3,3',4,4',5-hexachlorobiphenyl. Toxicol. Appl. Pharmacol., 147, 46-55.
28. Buchmann, A., Kunz, W., Wolf, C.R., Oesch, F. and Robertson, L.W. (1986) Polychlorinated biphenyls, classified as either phenobarbital- or 3-methylcholanthrene-type inducers of cytochrome P-450, are both hepatic tumor promoters in diethylnitrosamine-initiated rats. Cancer Lett., 32, 243-53.
29. Buchmann, A., Ziegler, S., Wolf, A., Robertson, L.W., Durham, S.K. and Schwarz, M. (1991) Effects of polychlorinated biphenyls in rat liver: correlation between primary subcellular effects and promoting activity. Toxicol. Appl. Pharmacol., 111, 454-68.
30. Glauert, H.P., Robertson, L.W. and Silberhorn, E.M. (2001) PCBs and tumor promotion. PCBs, Recent Advances in Environmental Toxicology and Health Effects, 355-371.
31. Ludewig, G. (2001) Cancer initiation by PCBs. The University Press of Kentucky, Lexington, KY.
32. Espandiari, P., Glauert, H.P., Lehmler, H.J., Lee, E.Y., Srinivasan, C. and Robertson, L.W. (2003) Polychlorinated biphenyls as initiators in liver carcinogenesis: resistant hepatocyte model. Toxicol. Appl. Pharmacol., 186, 55-62.
33. Espandiari, P., Glauert, H.P., Lehmler, H.J., Lee, E.Y., Srinivasan, C. and Robertson, L.W. (2004) Initiating activity of 4-chlorobiphenyl metabolites in the resistant hepatocyte model. Toxicol. Sci., 79, 41-6.
34. McLean, M.R., Bauer, U., Amaro, A.R. and Robertson, L.W. (1996) Identification of catechol and hydroquinone metabolites of 4-monochlorobiphenyl. Chem. Res. Toxicol., 9, 158-64.
35. Borlak, J., Hock, A., Hansen, T. and Richter, E. (2003) DNA adducts in cultures of polychlorinated biphenyl-treated human hepatocytes. Toxicol. Appl. Pharmacol., 188, 81-91.
36. McLean, M.R., Robertson, L.W. and Gupta, R.C. (1996) Detection of PCB adducts by the 32P-postlabeling technique. Chem. Res. Toxicol., 9, 165-71.
37. Pereg, D., Tampal, N., Espandiari, P. and Robertson, L.W. (2001) Distribution and macromolecular binding of benzo[a]pyrene and two polychlorinated biphenyl congeners in female mice. Chem Biol Interact, 137, 243-58.
38. Tampal, N., Myers, S. and Robertson, L.W. (2003) Binding of polychlorinated biphenyls/metabolites to hemoglobin. Toxicol Lett, 142, 53-60.
39. Oakley, G.G., Devanaboyina, U., Robertson, L.W. and Gupta, R.C. (1996) Oxidative DNA damage induced by activation of polychlorinated biphenyls (PCBs): implications for PCB-induced oxidative stress in breast cancer. Chem. Res. Toxicol., 9, 1285-92.
40. Oakley, G.G., Robertson, L.W. and Gupta, R.C. (1996) Analysis of polychlorinated biphenyl-DNA adducts by 32P-postlabeling. Carcinogenesis, 17, 109-14.
41. Srinivasan, A., Lehmler, H.J., Robertson, L.W. and Ludewig, G. (2001) Production of DNA strand breaks in vitro and reactive oxygen species in vitro and in HL-60 cells by PCB metabolites. Toxicol. Sci., 60, 92-102.
42. Tampal, N., Lehmler, H.J., Espandiari, P., Malmberg, T. and Robertson, L.W. (2002) Glucuronidation of hydroxylated polychlorinated biphenyls (PCBs). Chem. Res. Toxicol., 15, 1259-66.
43. Cariello, N.F., Piegorsch, W.W., Adams, W.T. and Skopek, T.R. (1994) Computer program for the analysis of mutational spectra: application to p53 mutations. Carcinogenesis, 15, 2281-5.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

24
44. Lambert, I.B., Singer, T.M., Boucher, S.E. and Douglas, G.R. (2005) Detailed review of transgenic rodent mutation assays. Mutat. Res., 590, 1-280.
45. de Boer, J.G. and Glickman, B.W. (1998) The lacI gene as a target for mutation in transgenic rodents and Escherichia coli. Genetics, 148, 1441-51.
46. Adler, K., Beyreuther, K., Fanning, E., Geisler, N., Gronenborn, B., Klemm, A., Muller-Hill, B., Pfahl, M. and Schmitz, A. (1972) How lac repressor binds to DNA. Nature, 237, 322-7.
47. Ehrlich, M., Zhang, X.Y. and Inamdar, N.M. (1990) Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat Res, 238, 277-86.
48. el Antri, S., Mauffret, O., Monnot, M., Lescot, E., Convert, O. and Fermandjian, S. (1993) Structural deviations at CpG provide a plausible explanation for the high frequency of mutation at this site. Phosphorus nuclear magnetic resonance and circular dichroism studies. J Mol Biol, 230, 373-8.
49. Lefebvre, A., Mauffret, O., Hartmann, B., Lescot, E. and Fermandjian, S. (1995) Structural behavior of the CpG step in two related oligonucleotides reflects its malleability in solution. Biochemistry, 34, 12019-28.
50. Jego, N., Thomas, G. and Hamelin, R. (1993) Short direct repeats flanking deletions, and duplicating insertions in p53 gene in human cancers. Oncogene, 8, 209-13.
51. Rihn, B.H., Bottin, M.C., Coulais, C., Rouget, R., Monhoven, N., Baranowski, W., Edorh, A. and Keith, G. (2000) Genotoxicity of 3-methylcholanthrene in liver of transgenic big Blue mice. Environ. Mol. Mutagen., 36, 266-73.
52. Dipple, A. (1995) DNA adducts of chemical carcinogens. Carcinogenesis, 16, 437-41. 53. Zhao, S., Narang, A., Ding, X. and Eadon, G. (2004) Characterization and
quantitative analysis of DNA adducts formed from lower chlorinated PCB-derived quinones. Chem Res Toxicol, 17, 502-11.
54. Wyndham, C. and Safe, S. (1978) In vitro metabolism of 4-chlorobiphenyl by control and induced rat liver microsomes. Biochemistry, 17, 208-15.
55. Wong, A., Basrur, P. and Safe, S. (1979) The metabolically mediated DNA damage and subsequent DNA repair by 4-chlorobiphenyl in Chinese hamster ovary cells. Res Commun Chem Pathol Pharmacol, 24, 543-50.
56. Amaro, A.R., Oakley, G.G., Bauer, U., Spielmann, H.P. and Robertson, L.W. (1996) Metabolic activation of PCBs to quinones: reactivity toward nitrogen and sulfur nucleophiles and influence of superoxide dismutase. Chem Res Toxicol, 9, 623-9.
57. Borlakoglu, J.T., Haegele, K.D., Reich, H.J., Dils, R.R. and Wilkins, J.P. (1991) In vitro metabolism of [14C]4-chlorobiphenyl and [14C]2,2',5,5'-tetrachlorobiphenyl by hepatic microsomes from rats and pigeons. Evidence against an obligatory arene oxide in aromatic hydroxylation reactions. Int J Biochem, 23, 1427-37.
58. Lin, P.H., Sangaiah, R., Ranasinghe, A., Upton, P.B., La, D.K., Gold, A. and Swenberg, J.A. (2000) Formation of quinonoid-derived protein adducts in the liver and brain of Sprague-Dawley rats treated with 2,2',5, 5'-tetrachlorobiphenyl. Chem Res Toxicol, 13, 710-8.
59. Le Page, F., Margot, A., Grollman, A.P., Sarasin, A. and Gentil, A. (1995) Mutagenicity of a unique 8-oxoguanine in a human Ha-ras sequence in mammalian cells. Carcinogenesis, 16, 2779-84.
60. DeCaprio, A.P., Johnson, G.W., Tarbell, A.M., Carpenter, D.O., Chiarenzelli, J.R., Morse, G.S., Santiago-Rivera, A.L. and Schymura, M.J. (2005) Polychlorinated biphenyl (PCB) exposure assessment by multivariate statistical analysis of serum congener profiles in an adult Native American population. Environmental Research, 98, 284-302.
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from
25
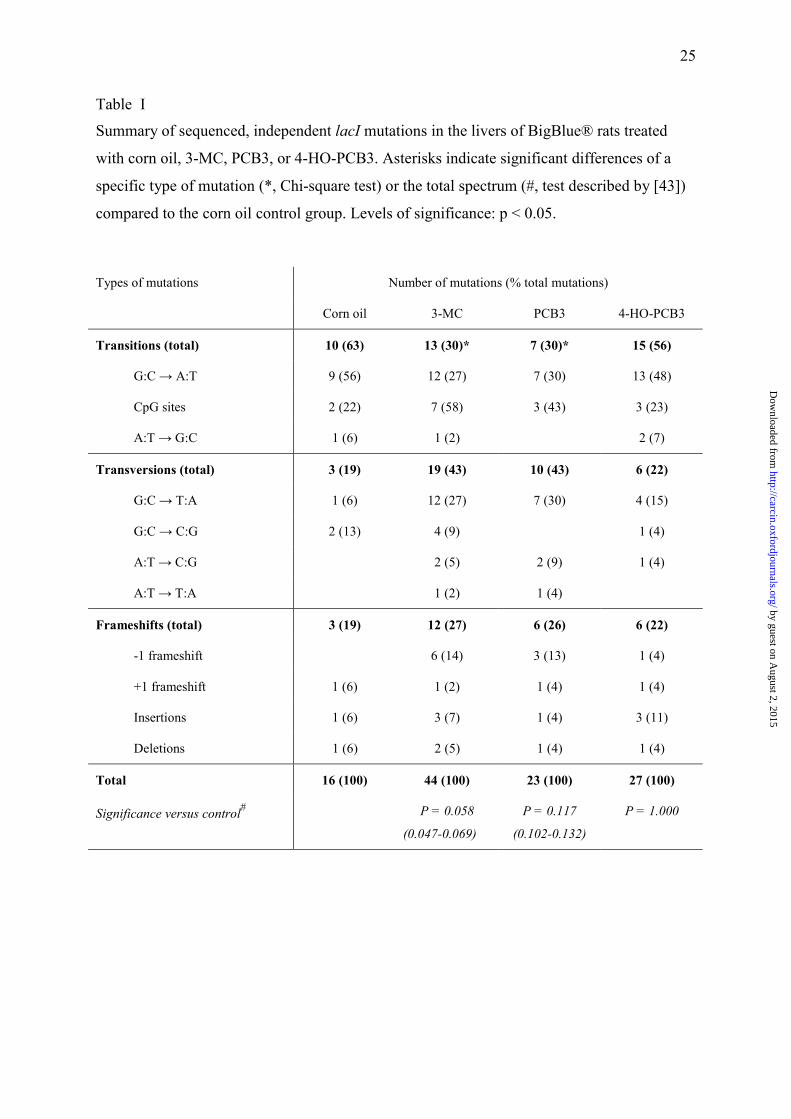
Table I
Summary of sequenced, independent lacI mutations in the livers of BigBlue® rats treated
with corn oil, 3-MC, PCB3, or 4-HO-PCB3. Asterisks indicate significant differences of a
specific type of mutation (*, Chi-square test) or the total spectrum (#, test described by [43])
compared to the corn oil control group. Levels of significance: p < 0.05.
Types of mutations Number of mutations (% total mutations)
Corn oil 3-MC PCB3 4-HO-PCB3
Transitions (total) 10 (63) 13 (30)* 7 (30)* 15 (56)
G:C → A:T 9 (56) 12 (27) 7 (30) 13 (48)
CpG sites 2 (22) 7 (58) 3 (43) 3 (23)
A:T → G:C 1 (6) 1 (2) 2 (7)
Transversions (total) 3 (19) 19 (43) 10 (43) 6 (22)
G:C → T:A 1 (6) 12 (27) 7 (30) 4 (15)
G:C → C:G 2 (13) 4 (9) 1 (4)
A:T → C:G 2 (5) 2 (9) 1 (4)
A:T → T:A 1 (2) 1 (4)
Frameshifts (total) 3 (19) 12 (27) 6 (26) 6 (22)
-1 frameshift 6 (14) 3 (13) 1 (4)
+1 frameshift 1 (6) 1 (2) 1 (4) 1 (4)
Insertions 1 (6) 3 (7) 1 (4) 3 (11)
Deletions 1 (6) 2 (5) 1 (4) 1 (4)
Total 16 (100) 44 (100) 23 (100) 27 (100)
Significance versus control# P = 0.058
(0.047-0.069)
P = 0.117
(0.102-0.132)
P = 1.000
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

26
FIGURE LEGENTS
Figure 1. Chemical structures of PCB3, 4-HO-PCB3, and 3-MC.
Figure 2. Weight gain of male BigBlue® rats treated with corn oil (negative control), 3-MC
(positive control), PCB3, and 4-HO-PCB3. Asterisks indicate significant differences to the
untreated control group. Levels of significance: *, p < 0.05; **, p < 0.01; (Student�s unpaired
t-test). Arrows, injection, PND, post natal day.
Figure 3. Mutant frequencies in the liver after treatment of male transgenic BigBlue® rats
with 5 ml/kg BW corn oil (negative control), 298 µmol/kg BW 3-MC (positive control), 600
µmol/kg BW PCB3, or 400 µmol/kg BW 4-HO-PCB3. Data represent mean of 4 animals ±
standard error (p<0.5). Asterisks indicate significant differences to the control group. Levels
of significance: **, p < 0.01 (One-way ANOVA).
Figure 4. Metabolic activation of PCB3 to quinones (Q), modified from [32].
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from
For Peer Review
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from

For Peer Review
by guest on August 2, 2015
http://carcin.oxfordjournals.org/D
ownloaded from