2. material und methoden - albert-ludwigs-universität freiburg
TRANSCRIPT

Aus dem Zentrum für Psychische Erkrankungen
Klinik für Psychiatrie und Psychotherapie des Universitätsklinikums Freiburg i.Br.
Modulation von Adenosinrezeptoren
hippocampaler Neurone von Mäusen und Ratten
bei assoziativer Langzeitpotenzierung
INAUGURAL-DISSERTATION
zur Erlangung des Medizinischen Doktorgrades
der Medizinischen Fakultät
der Albert-Ludwigs-Universität
Freiburg i. Br.
Vorgelegt
2017
von Aaron Vogt
geboren in Stuttgart

Dekanin Prof. Dr. Kerstin Krieglstein
Erster Gutachter Prof. Dr. Claus Normann
Zweiter Gutachter Prof. Dr. Josef Bischofberger
Jahr der Promotion 2018

I Inhaltsverzeichnis
Inhaltsverzeichnis
ABBILDUNGSVERZEICHNIS ....................................................................................................... III
TABELLENVERZEICHNIS ............................................................................................................ IV
ABKÜRZUNGSVERZEICHNIS ....................................................................................................... V
1 EINLEITUNG......................................................................................................................... 7
1.1 Synaptische Plastizität .............................................................................................................. 7
1.1.1 Synaptische Langzeitpotenzierung (LTP) ..................................................................... 8
1.1.2 Mechanismen der assoziativen LTP ............................................................................ 10
1.1.3 Neuroplastizitätshypothese der Depression ................................................................ 12
1.2 Schaltregion Hippocampus .................................................................................................... 14
1.3 Adenosin .................................................................................................................................. 15
1.3.1 Adenosinrezeptoren .................................................................................................... 16
1.3.2 Neuromodulation ........................................................................................................ 17
1.3.3 Neuro(patho)physiologische Implikationen ................................................................ 19
1.4 Fragestellung dieser Arbeit .................................................................................................... 20
2 MATERIAL UND METHODEN ...................................................................................... 21
2.1 Lösungen und Substanzen ...................................................................................................... 21
2.1.1 Extrazelluläre Lösungen ............................................................................................. 21
2.1.2 Pipetten-Lösung .......................................................................................................... 21
2.1.3 Substanzen .................................................................................................................. 23
2.2 Versuchstiere ........................................................................................................................... 24
2.3 Hippocampusschnitte ............................................................................................................. 25
2.4 Patch-Clamp-Methode ........................................................................................................... 27
2.5 Aufbau des Patch-Clamp-Messplatzes .................................................................................. 29
2.5.1 Übersicht ..................................................................................................................... 29
2.5.2 Messkammer und optisches System ............................................................................. 29
2.5.3 Messtisch und Mikromanipulator ............................................................................... 30
2.5.4 Pulsstimulator, Verstärker und Software .................................................................... 31
2.6 Pipettenherstellung ................................................................................................................. 32
2.7 Stimulationsprotokoll ............................................................................................................. 32
2.8 Versuchsablauf ........................................................................................................................ 33
2.9 Datenanalyse und -auswertung .............................................................................................. 34

II Inhaltsverzeichnis
3 ERGEBNISSE ..................................................................................................................... 35
3.1 Versuchsreihen mit Mäusen ................................................................................................... 35
3.1.1 Kontrollreihe LTP bei Wildtyp-Mäusen ...................................................................... 35
3.1.2 Messreihe LTP bei transgenen Mäusen ...................................................................... 36
3.2 Versuchsreihen mit Wistar-Ratten ........................................................................................ 37
3.2.1 Kontrollreihe LTP bei Wistar-Ratten .......................................................................... 37
3.2.2 Messreihe mit cCPA .................................................................................................... 38
3.2.3 Messreihe mit DPCPX und cCPA ............................................................................... 39
3.2.4 Messreihe mit NEM und cCPA ................................................................................... 40
3.2.5 Messreihe mit CGS ..................................................................................................... 41
3.2.6 NEM-Zugabe ohne LTP-Induktion .............................................................................. 42
3.3 Übersicht .................................................................................................................................. 42
3.3.1 LTP-Versuchsreihe mit Mäusen .................................................................................. 42
3.3.2 LTP-Versuchsreihe mit Wistar-Ratten ........................................................................ 43
4 DISKUSSION ...................................................................................................................... 45
4.1 Patch-Clamp-Technik an Hirnschnitten ............................................................................... 45
4.2 LTP-Kontrollreihen an Mäusen und Ratten ........................................................................ 46
4.3 LTP-Modulation durch A1R-Überexprimierung ................................................................. 47
4.4 LTP-Modulation durch cCPA ............................................................................................... 48
4.5 LTP-Modulation durch DPCPX + cCPA .............................................................................. 50
4.6 LTP-Modulation durch NEM + cCPA .................................................................................. 51
4.7 LTP-Modulation durch CGS ................................................................................................. 54
4.8 Pathophysiologische Bedeutung ............................................................................................. 56
5 ZUSAMMENFASSUNG ..................................................................................................... 58
LITERATURVERZEICHNIS ......................................................................................................... 59
EIDESSTATTLICHE VERSICHERUNG ..................................................................................... 77
DANKSAGUNG ............................................................................................................................... 78

III Abbildungsverzeichnis
Abbildungsverzeichnis
ABBILDUNG 1: SPIKE TIMING-DEPENDENT PLASTICITY (STDP) ............................................................ 9
ABBILDUNG 2: PHASEN UND MOLEKULARE PROZESSE DER LTP AN EINER
CA3/CA1 SYNAPSE ............................................................................................................. 12
ABBILDUNG 3: SCHEMA SYNAPTISCHER VERBINDUNGEN IM ANGESCHNITTENEN
HIPPOCAMPUS .................................................................................................................... 15
ABBILDUNG 4: SCHEMA DES METABOLISMUS UND SIGNALWEGS VON ADENOSIN ..................... 17
ABBILDUNG 5: SCHEMA DER ÜBEREXPRIMIERUNG DES A1R-GENS ................................................... 24
ABBILDUNG 6: IMMUNOFLUORESZENZMUSTER ANTI-MCHERRY
IM HIPPOCAMPUS-SCHNITT ............................................................................................ 25
ABBILDUNG 7: ANFERTIGEN DER HIRNSCHNITTE .................................................................................. 27
ABBILDUNG 8: PATCHKONFIGURATIONEN ............................................................................................... 28
ABBILDUNG 9: ÜBERSICHT DES PATCH-CLAMP-MESSPLATZES .......................................................... 29
ABBILDUNG 10: MESSTISCH MIT MESSKAMMER WÄHREND MESSUNG ........................................... 30
ABBILDUNG 11: VEREINFACHTES SCHALTBILD EINES PATCH-CLAMP-VERSTÄRKERS ............... 31
ABBILDUNG 12: SCHEMA DES THETA-BURST-PAIRING-PROTOKOLLS .............................................. 33
ABBILDUNG 13: LTP-KONTROLLREIHE WILDTYP-MÄUSE..................................................................... 35
ABBILDUNG 14: LTP-MESSREIHE AN CAMKII-ADOA1R-92-MÄUSEN..................................................... 36
ABBILDUNG 15: LTP-KONTROLLREIHE WISTAR-RATTEN ..................................................................... 37
ABBILDUNG 18: LTP-MESSREIHE MIT NEM UND CCPA AN WISTAR-RATTEN ................................... 40
ABBILDUNG 19: LTP-MESSREIHE MIT CGS AN WISTAR-RATTEN ......................................................... 41
ABBILDUNG 20: EPSP-VERLAUF BEI NEM-ZUGABE OHNE STIMULATIONSPROTOKOLL ............... 42
ABBILDUNG 21: VERGLEICH ALLER LTP-MESSREIHEN MIT DER KONTROLLGRUPPE
IN % DER BASELINE .......................................................................................................... 44
ABBILDUNG 22: EPSP-VERLAUF BEI CCPA-ZUGABE OHNE STIMULATIONSPROTOKOLL ............. 49

IV Tabellenverzeichnis
Tabellenverzeichnis
TABELLE 1: ANGABEN DER LÖSUNGSBESTANDTEILE (IN MMOL/L) .................................................. 22
TABELLE 2: VERÄNDERUNGEN DER EPSP-AMPLITUDEN NACH LTP-INDUKTION IN % DER
BASELINE BEI MÄUSEN ............................................................................................................ 42
TABELLE 3: VERÄNDERUNGEN DER EPSP-AMPLITUDEN NACH LTP-INDUKTION IN % DER
BASELINE BEI RATTEN ............................................................................................................. 43

V Abkürzungsverzeichnis
Abkürzungsverzeichnis
A Ampere
AD Antidepressiva
AMPA α-Amino-3-hydroxy-5-methyl-isoxazol-Propionsäure
ANOVA Analysis of variance
AP Aktionspotenzial
AR Adenosinrezeptor(en)
AxR Ax-Adenosinrezeptor(en); x = 1, 2A, 2B, 3
BDNF Brain derived neurotrophic factor
BPAP Rückgeleitetes Aktionspotenzial (engl.: back propagating action potential)
CA Ammonshorn (lat. Cornu ammonis)
CaMKII Calmodulin-abhängige Proteinkinase II
Ca2+ Kalzium-Ion
CGS 4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-
yl]amino]ethyl]benzenepropanoic acid hydrochloride
CREB cAMP response element-binding protein
cCPA 2-chloro-N(6)-cyclopentyladenosine
DAG Diacylglycerin
DPCPX 8-Cyclopentyl-1,3-dipropylxanthine
EGTA Ethylenglykol-bis(2-aminoethylether)-N,N,N‘,N‘-tetraethansäure
ENT Equilibrativer Nukleosidtransporter
EPSP Exzitatorisches postsynaptisches Potenzial
G Giga (109)
GTP Guanosin-5‘-triphosphat
HEPES 4-(2-Hydroxyethyl)piperazin-1-ehtansulfonsäure
HFS High frequency stimulation
Hz Hertz
IP3 Inositoltriphosphat
K+ Kalium-Ion
LTD Langzeitdepression (engl.: long-term depression)
LTP Langzeitpotenzierung (engl.: long-term potentiation)
µ Mikro (10-9)

VI Abkürzungsverzeichnis
M Mega (106) / mol/l
Mg2+ Magnesium-Ion
ms Millisekunde
mV Millivolt (10-3 Volt)
n nano (10-9) / Anzahl der Experimente
Na+ Natrium-Ion
NEM N-Ethylmaleinimide
NSF N-Ethylmaleinimide-sensitiver Faktor
NMDA N-Methyl-D-Aspartat
Ω Ohm
p Piko (10-12)
PKC Proteinkinase C
PLC Phospholipase C
PSD Postsynaptic density
Rm Membranwiderstand
Rs Serienwiderstand
SNARE Soluble N-Ethylmaleinimide-sensitive-factor attachment receptor
STDP Aktionspotenzial-Intervall-abhängige-Plastizität (engl.: spike-time dependent
plasticity)
TBS Theta-burst stimulation
V Volt
WT Wildtyp
ZNS Zentrales Nervensystem

7 1 Einleitung
1 Einleitung
1.1 Synaptische Plastizität
Das menschliche zentrale Nervensystem (ZNS) verfügt über die erstaunliche Fähigkeit,
neuronale Verschaltungen durch Erfahrungen zu modifizieren. So resultieren aus einer
angepassten neuronalen Aktivität Veränderungen in Verhalten, Gedanken und Gefühlen.
Dieser sogenannten Plastizität liegen Prozesse in struktureller, zellulärer und synaptischer
Veränderbarkeit zugrunde. Während die strukturelle und zelluläre Plastizität funktionelle
Veränderungen sowie Neurogenese und Apoptose bedeuten, moduliert die synaptische
Plastizität die Stärke der Signalübertragung an vorhandenen Synapsen (Davis und Malenka
2002).
Den Begriff der neuronalen Plastizität prägte Ramón y Cajal bereits zu Beginn des 20.
Jahrhunderts, nachdem er die morphologische Neubildung axonaler Kollateralen und Dendriten
beobachtet hatte (Cajal, S. R. Y 1894, 1928; Stahnisch 2003).
Zur selben Zeit vermutete man bereits, dass die Anzahl der Neurone im Erwachsenenalter kaum
schwindet und die Speicherung von Lern- und Gedächtnisinhalten nicht nur auf eine
Neubildung von Neuronen zurückzuführen sei. Schließlich formulierte Donald Hebb 1949 das
Grundprinzip synaptischer Plastizität, welches bis heute gültig ist:
„When an axon of cell A is near enough to excite a cell B and repeatedly or
persistently takes part in firing it, some growth process or metabolic change
takes place in one or both cells such that A's efficiency, as one of the cells firing
B, is increased.“ (Hebb 1949)
Erst einige Jahrzehnte später wiesen Terje Lømo und Timothy Bliss eine solche verstärkte
synaptische Signalübertragung in elektrophysiologischen Experimenten im Hippocampus von
Kaninchen nach (Bliss und Lømo 1973). Dabei registrierten sie nach tetanischer Stimulation
im Tractus perforans eine lang anhaltende Erhöhung der exzitatorischen postsynaptischen
Potenziale (EPSP) im Gyrus dentatus. In ihrer Arbeit nannten sie dieses Phänomen der
Ausbildung einer verstärkten Signaltransduktion Langzeitpotenzierung (engl. long-term
potentiation, LTP).
In darauffolgenden Jahren wurde die Hebb’sche Lernregel erweitert und eine Abschwächung
von EPSP durch asynchrone oder niederfrequente Stimulation beschrieben, folglich

8 1 Einleitung
Langzeitdepression genannt (engl. long-term depression, LTD) (Stent 1973; Levy und Steward
1983).
Obwohl die synaptische Langzeitplastizität das vielversprechendste experimentelle Modell für
das Verständnis von Lernen und Gedächtnis darstellt, braucht es aufgrund der Komplexität
neuronaler Verschaltung nach wie vor den definitiven Nachweis eindeutiger Korrelation (Bliss
und Collingridge 1993; Kandel 2001; Whitlock et al. 2006; Cooke und Bliss 2006). Zwar
konnte anahnd von einigen transgenen Mausmodellen gezeigt werden, dass es Zusammenhänge
zwischen LTP und räumlichem Lernen besonders in Synapsen des Schaffer-
Kollateraltrakts/CA1-Neurone gibt; es bleibt jedoch offen, inwiefern LTP für andere Bereiche
des Lernens verantwortlich gemacht werden kann (Huang et al. 1995; Lynch 2004). Außerdem
konnte gezeigt werden, dass molekulare Prozesse der LTP variieren. Zum einen je nach
Lokalisation, wie beispielsweise in Moosfasern aus dem entorhinalen Kortex im Unterschied
zu Schaffer-Kollateralen, und zum anderen bezogen auf den Entwicklungsstand der Mäuse
(Huang et al. 1995; Yasuda et al. 2003; Luchkina et al. 2014).
Auch wenn die Neurogenese bei Gedächtnisbildung und -erhalt besonders im Hippocampus
eine gewisse Rolle spielt (Deng et al. 2010; Akers et al. 2014), konnten verschiedene Formen
der LTP und LTD in mehreren Gehirnregionen nachgewiesen werden (Fox 2002; Ji et al. 2003;
Purves et al. 2012). Deshalb stellen die Mechanismen der modifizierten synaptischen
Übertragungsstärke nach wie vor ein geeignetes molekulares Erklärungsmodell für Lernen und
Gedächtnis dar (Malenka und Bear 2004; Massey und Bashir 2007).
Von der Minuten bis Stunden andauernden Langzeitplastizität (LTD und LTP) unterscheidet
man die Kurzzeit-Plastizität, welche nur Millisekunden bis Minuten anhält (Abbott und Regehr
2004). Neuere Studien konnten unterschiedliche Mechanismen der Kurzzeit-Potenzierung
(STP) von denen der LTP abgrenzen und damit der STP eine mögliche eigene funktionelle
Bedeutung für das Kurzzeitgedächtnis zuschreiben (Volianskis et al. 2013).
Im Folgenden soll genauer auf die LTP eingegangen werden, da diese der zu untersuchende
Effektparameter aller zugrundeliegenden Experimente ist.
1.1.1 Synaptische Langzeitpotenzierung (LTP)
Wie bereits beschrieben, stellt die synaptische LTP neben der LTD einen wichtigen Teil der
Neuroplastizität dar. Je nach Stimulationsort, -frequenz und -dauer unterscheidet man homo-
von heterosynaptischer Plastizität, bzw. nicht-assoziativer von assoziativer.

9 1 Einleitung
Bei der homosynaptischen Form induziert ausschließlich ein afferenter Stimulus – also ein
präsynaptischer Reiz an einer Synapse – in einem Neuron eine LTP, vorausgesetzt ausreichend
hochfrequente (100-200 Hz) kurzanhaltende Tetani werden appliziert (engl.: high frequency
stimulation, HFS) (Bliss und Gardner-Medwin 1973; Grover und Teyler 1990). Werden
hingegen niederfrequente (1-10 Hz), langanhaltende Stimuli abgegeben, resultiert eine LTD
(engl.: low frequency stimulation, LFS) (Dudek und Bear 1992).
Wird assoziative (heterosynaptische) Langzeitplastizität generiert, kombiniert man in einem
definierten Zeitabstand präsynaptisch stimulierte EPSPs mit einer postsynaptischen
Depolarisation, die am Axonhügel induziert und retrograd über Dendriten fortgeleitet wird (sog.
BPAP, engl.: back propagating action potential) (Stanton und Sejnowski 1989; Brown et al.
1990; Chen et al. 1999; Song et al. 2000).
Neben den Wiederholungen dieser Stimulationspaarungen ist vor allem ihre zeitliche
Reihenfolge entscheidend. Folgt ein BPAP 20 ms oder kürzer auf ein EPSP, entsteht eine LTP,
und folgt umgekehrt ein EPSP auf ein BPAP, eine LTD (Levy und Steward 1983; Song et al.
2000). Außerdem zeigt sich, je kürzer das zeitliche Intervall gewählt wird, desto höhere
Übertragungsstärken resultieren (Markram 1997); (Bi und Poo 1998). Dieser assoziative
Mechanismus wird Aktionspotenzial-Intervall-abhängige Plastizität (engl.: spike timing-
dependent plasticity, STDP) genannt und repräsentiert am wahrscheinlichsten physiologische
Vorgänge hippocampaler Pyramidenzellen (Levy und Steward 1983; Paulsen und Sejnowski
2000; Dan und Poo 2006). Aus diesen Gründen wurde auch in dieser Arbeit eine STDP
induziert.
Abbildung 1: Spike timing-dependent plasticity (STDP)
Änderung der EPSP-Polarität nach Reihenfolge und zeitlichem Abstand von EPSP und BPAP. Je kürzer
(<20 ms) die Abfolge, desto höher bzw. niedriger resultieren EPSP. Modifiziert nach Schmidt et al. (2010).

10 1 Einleitung
1.1.2 Mechanismen der assoziativen LTP
Synapsen der CA3/CA1-Neurone im Hippocampus unterliegen seit Jahrzehnten einem breitem
Forschungsinteresse, weswegen der Mechanismus der LTP an diesem Zellkontakt gut erfasst
ist. Im Folgenden sollen grundlegende Vorgänge der LTP erläutert werden. Zur visuellen
Darstellung molekularer und zellulärer Vorgänge dient Abbildung 2.
Nach Depolarisation der präsynaptischen Membran erfolgt eine Exozytose des Transmitters
Glutamat in den synaptischen Spalt. Glutamat bindet nun an zwei entscheidende Arten
ionotroper Rezeptoren im Bereich erhöhter postsynaptischer Rezeptorendichte, auch genannt
Postsynaptic density (PSD). Zum einen können α-Amino-3-hydroxy-5-methyl-4-
isoxazolpropionat-Rezeptoren (AMPA-Rezeptoren) als ligandengesteuerte Na+/K+-Kanäle die
basale Signalweiterleitung vermitteln, indem sie das negative Ruhemembranpotenzial
anheben und ein EPSP auslösen. Zum anderen kann es zu einer Induktion der LTP kommen,
wenn spannungsabhängige N-Methyl-D-Aspartat-Rezeptoren (NMDA-R) durch folgende
Koinzidenz aktiviert werden: NMDA-R bewirken erst dann einen massiven Ca2+-Einstrom,
wenn ein Mg2+-Block durch Membrandepolarisation gelöst wird und gleichzeitig eine
Glutamatbindung den Kationenkanal aktiviert. Eine Depolarisation kann entweder durch
ausreichend hochfrequente Stimulierung der Präsynapse oder durch ein BPAP ausgelöst
werden. Entscheidend ist dabei die zeitliche Sequenz der STDP, aufgrund welcher am meisten
Ca2+ nach intrazellulär einströmt, wenn der Mg2+-Block kurz nach der relativ langanhaltenden
Glutamatbindung gelöst wird. Es konnte durch eine Blockade der NMDA-R gezeigt werden,
dass auch spannungsabhängige L-Typ Ca2+-Kanäle an dem ausschlaggebenden Ca2+-Einstrom
beteiligt sind, allerdings nur unter bestimmten Voraussetzungen in vitro (Huang und Malenka
1993; Huber et al. 1995; Morgan und Teyler 2001).
Der schnelle Einstrom von Ca2+ in die postsynaptische Zelle mobilisiert zusätzlich
intrazelluläre Ca2+-Speicher und aktiviert im weiteren Verlauf die Kalzium/Calmodulin-
abhängige Proteinkinase II (CaMKII), Proteinkinase C (PKC) und Tyrosinkinasen (Lisman et
al. 2012). CaMKII phosphoryliert und aktiviert zusätzliche AMPA-Rezeptoren und führt zu
einem vermehrten Einbau derselben in die postsynaptische Membran, welche dadurch
wesentlich in ihrer Exzitabilität beeinflusst wird (Citri und Malenka 2008; Lisman et al. 2012).
Silent synapses, in denen bisher keine AMPA-Rezeptoren vorhanden sind, können durch
Einbau von AMPA-Rezeptoren aufgrund benachbarter dendritischer Stimulation zu
exzitatorisch aktiven Synapsen (unsilenced synapses) transformiert werden (Kandel 2013).
Dieser zweite Abschnitt, der zur Aufrechterhaltung der LTP beiträgt, wird als Expression der
LTP verstanden.

11 1 Einleitung
Verschiedene wissenschaftliche Arbeiten gehen davon aus, dass zusätzlich zur
postsynaptischen Sensibilisierung bestimmte Signalmoleküle retrograd zu einer verstärkten
präsynaptischen Transmitterfreisetzung beitragen können. Dazu werden u.a. Lipide (z.B.
Endocannabinoide), Peptide, Wachstumsfaktoren (z.B. brain-derived neurotrophic factor -
BDNF) und insbesondere das Gas NO gezählt. Deren Beitrag zu LTP ist noch nicht ausreichend
geklärt (Regehr et al. 2009; Padamsey und Emptage 2014).
Über die bisher beschriebenen Vorgänge einer early-LTP hinaus, die ein bis drei Stunden
andauert, konnten molekulare Prozesse dargestellt werden, die für eine länger anhaltende Phase
(late-LTP) verantwortlich sind. Hiefür sorgt eine dauerhaft aktivierte Proteinkinase PKMζ
(Sacktor 2010), sowie eine Signalkaskade über cAMP und PKA, welche schlussendlich über
den Transkriptionsfaktor cAMP response element-binding protein (CREB-1) die Synthese
neuer Signalproteine in Gang setzt. Wahrscheinlich werden diese neuen Proteine nicht nur zur
Stabilisierung bestehender Synapsen und Aufrechterhaltung einer erhöhten AMPA-
Rezeptorendichte genutzt, sondern auch zur Ausbildung neuer Synapsen (siehe Abbildung 2;
Yuste und Bonhoeffer 2001; Kandel 2013). Eine strikte Trennung von early- und late-LTP ist
umstritten, zumal gezeigt werden konnte, dass beispielsweise PKA auch während early-LTP
eine essentielle Rolle spielt (Blitzer et al. 1995).
Verschiedene synaptische Eigenschaften unterstreichen die neuronale Anpassungsfähigkeit
eines Lernmodells, bei dem Reize verstärkt, kanalisiert oder nicht weiterverarbeitet werden
müssen. Physiologisch kann ein eigentlich zu schwacher synaptischer Impuls trotzdem zu LTP
führen, wenn er mit einer Depolarisation einer benachbarten Synapse desselben Neurons
gepaart wird. Dieser Vorgang wird als assoziative Konditionierung verstanden. Außerdem
entsteht Gedächtnisbildung zum einen durch Synapsenspezifität, indem nicht-stimulierte
Synapsen keine LTP ausbilden, und zum anderen durch Synapsenkooperativität, bei welcher
sich benachbarte schwache EPSP-Impulse verstärken und LTP triggern (Kandel 2013).
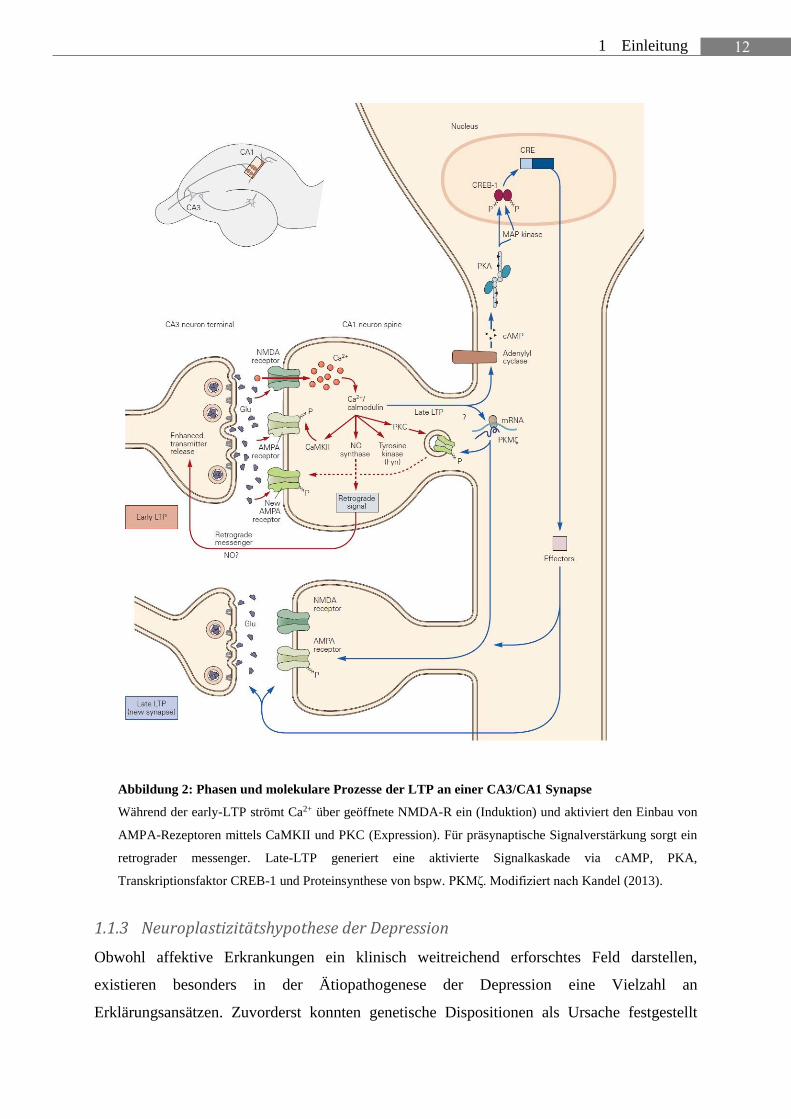
12 1 Einleitung
Abbildung 2: Phasen und molekulare Prozesse der LTP an einer CA3/CA1 Synapse
Während der early-LTP strömt Ca2+ über geöffnete NMDA-R ein (Induktion) und aktiviert den Einbau von
AMPA-Rezeptoren mittels CaMKII und PKC (Expression). Für präsynaptische Signalverstärkung sorgt ein
retrograder messenger. Late-LTP generiert eine aktivierte Signalkaskade via cAMP, PKA,
Transkriptionsfaktor CREB-1 und Proteinsynthese von bspw. PKMζ. Modifiziert nach Kandel (2013).
1.1.3 Neuroplastizitätshypothese der Depression
Obwohl affektive Erkrankungen ein klinisch weitreichend erforschtes Feld darstellen,
existieren besonders in der Ätiopathogenese der Depression eine Vielzahl an
Erklärungsansätzen. Zuvorderst konnten genetische Dispositionen als Ursache festgestellt

13 1 Einleitung
werden (Sullivan et al. 2000). Aus verschiedenen neurobiologischen und psychosozialen
Theorien wie die der erlernten Hilflosigkeit, dysfunktionalen Kognitionen und einem Mangel
an Selbstwert sowie positiven Verstärkern wuchs die Erkenntnis, dass eine Depression am
wahrscheinlichsten aus einem Zusammenspiel vieler prädisponierender Vulnerabilitäten
entsteht (Brakemeier et al. 2008). DeJong-Meyer fasste im Jahr 2005 die genannten
auslösenden Faktoren zu einem sogenannten erweiterten Final-Common-Pathway-Modell der
Depression zusammen.
Es wird angenommen, dass pathophysiologische Mechanismen der Depression mit dem Tier-
Stress-Modell korrelieren. In Experimenten mit Ratten, die chronischem Stress ausgesetzt
waren, konnten hippocampale Veränderungen sowohl in struktureller Plastizität wie
verminderter Neurogenese, als auch in funktioneller Plastizität wie abgeschwächter LTP und
gesteigerter LTD aufgezeigt werden (Castrén 2005; Normann et al. 2007; Holderbach et al.
2007; Serafini 2012). Dies konnte man mittels visuell evozierten Potenzialen (VEP) nicht-
invasiv auch bei Depressiven im Vergleich zu Gesunden feststellen (Normann et al. 2007).
Umgekehrt ist daher naheliegend, dass einige der transkraniellen und invasiven
Hirnstimulationsverfahren (z.B. Elektrokrampftherapie, EKT; repetitive transkranielle
Magnetstimulation, rTMS) zur Behandlung schwerer Depression in ebenjene Neuroplastizität
modulierend eingreifen und einen signifikanten Behandlungserfolg darstellen, auch wenn sie
von kognitiven Nebenwirkungen begleitet sein können (Plewnia und Padberg 2012; Li et al.
2012).
Die Neuroplastizitätshypothese der Depression wird dadurch bestärkt, dass auf zellulärer Ebene
bestimmte Marker im Vergleich zu gesunden Kontrollgruppen ausgemacht werden konnten.
Eine Stressinduktion und die dadurch erhöhte Ausschüttung von Glukokortikoiden kann LTP
insbesondere in CA1-Neuronen von Ratten abschwächen und LTD verstärken (Pavlides et al.
1993; Yamada et al. 2003; Gerges et al. 2004; Krugers et al. 2005; Krugers et al. 2006; Joëls
und Krugers 2007). Der neuroprotektive und für kognitive Funktionen wichtige
Wachstumsfaktor BDNF nimmt zusammen mit dem ihn transkribierenden CREB wohl eine
Schlüsselrolle ein (Nair und Vaidya 2006). Expression von BDNF durch CREB ist bei
Depression bzw. nach Stressinduktion gemindert und verliert damit seine neuronale
Schutzfunktion (Radecki et al. 2005; Sterlemann et al. 2010). Umgekehrt hat eine hohe
Aktivität von CREB einen positiven Einfluss auf Depression (Chen et al. 2001a; Blendy 2006).
Antidepressiva (AD) der Klassen Monoamin-Wiederaufnahmehemmer/-Abbauhemmer
scheinen die Suppression von CREB und BDNF durch Stress im präfrontalen Kortex und

14 1 Einleitung
Hippocampus wieder umzukehren, sodass neben Neurogenese auch zelluläre Anpassungen wie
Dendritenaussprossungen und Synapsenbildung verstärkt werden (Nibuya et al. 1996; Chen et
al. 2001b; Laifenfeld et al. 2005; Warner-Schmidt und Duman 2006). Diese Wirkungsweise
der AD stellt, entgegen der entkräfteten Monoaminmangelhypothese, eine plausiblere
Erklärung dar (Duman 2004; Ruhé et al. 2007; Brunoni et al. 2008; Banasr et al. 2011).
Depressive weisen vermehrt eine linksseitig lokalisierte Atrophie im Hippocampus auf
(Bremner et al. 2000; Campbell et al. 2004; Videbech und Ravnkilde 2004). Eine
Zelldegeneration im Bereich des Hippocampus korreliert wohl mit exekutiven, nicht jedoch mit
kognitiven Dysfunktionen, die im Zusammenhang mit Depression auftreten (Frodl et al. 2006;
Khan et al. 2015).
Zusammenfassend können die beschriebenen Einschränkungen der strukturellen und
funktionellen Neuroplastizität als relevante Faktoren der multifaktoriellen Genese der
Depression betrachtet werden (Normann et al. 2007; Pittenger und Duman 2008; Nissen et al.
2010).
1.2 Schaltregion Hippocampus
Der Hippocampus liegt im medialen Temporallappen und ist Teil des „limbischen Systems“,
dessen Funktion als isolierte Einheit jedoch umstritten ist. Zu der Hippocampusformation im
eigentlichen Sinne zählt man die drei Strukturen Gyrus dentatus, Cornu ammonis
(Ammonshorn, Hippocampus proprius) und Subiculum. Das Ammonshorn (Cornu Ammonis)
kann in die Zellregionen CA1 bis CA4 unterteilt werden. Es zeigt sich histologisch eine
archicorticale Dreischichtung, in der die Pyramidenzellkörper in dem mittleren Stratum
pyramidale liegen und von dort aus Dendriten in das Stratum oriens sowie Stratum radiatum
und lacunosum-moleculare ausbilden (Trepel 2015).
Afferenzen gelangen in den Hippocampus hauptsächlich über den Tractus perforans vom
entorhinalen Kortex zu den Körnerzellen im Gyrus dentatus. Diese glutamatergen Zellen
projizieren über sogenannte Moosfasern in das Stratum radiatum, wo sie glutamaterge CA3-
Pyramidenzellen innervieren. CA3-Zellen wiederum stimulieren CA1-Pyramidenzellen über
Axone, die man Schaffer-Kollateraltrakt nennt. Diese Verschaltungsstelle eignet sich aufgrund
der Unidirektionalität besonders gut für elektrophysiologische Untersuchungen. Zusätzlich
erhalten CA1-Neurone direkte Afferenzen aus dem entorhinalen Kortex. Efferenzen gelangen

15 1 Einleitung
in das Subiculum und bilden dabei eine wichtige Verbindung des Hippocampus über den Fornix
zum „limbischen System“ (Amaral D 2007; Kandel 2013).
Abbildung 3 zeigt schematisch die Binnenverschaltung im Hippocampus.
Abbildung 3: Schema synaptischer Verbindungen im angeschnittenen Hippocampus
a: Informationen gelangen über den Tractus perforans vom entorhinalen Kortex in den Gyrus dentatus.
Körnerzellen projizieren mit ihren Moosfasern in das Cornu ammonis und bilden dort exzitatorische Synapsen
mit CA3-Pyramidenzellen. Letztere sind über den Schaffer-Kollateraltrakt mit CA1-Pyramidenzellen
verbunden, welche einen wichtigen Ausgangspunkt zum Subiculum und zurück zum entorhinalen Kortex
darstellen. b: Die Hippocampusformation liegt im medialen Temporallappen, im limbischen System mit dem
Fornix verbunden. Modifiziert nach Kandel (2013).
Die Funktion des Hippocampus ist noch nicht eindeutig geklärt, da die komplette Darstellung
komplexer neuronaler Netzwerke noch nicht möglich ist. Mit gewisser Wahrscheinlichkeit ist
der Hippocampus jedoch in die Verarbeitung von Emotionen, in Bildung von episodischem
Gedächtnis und räumlicher Orientierung involviert. Möglicherweise dient der Hippocampus
auch dazu, zeitliche und situative Informationen zu kontextualisieren (Neves et al. 2008;
Schiller et al. 2015). Synaptische Plastizität ist eine gut untersuchte Eigenschaft des
Hippocampus und prädestiniert ihn neben der guten anatomischen Abgrenzbarkeit daher zu
einer bevorzugten Grundlage elektrophysiologischer Experimente.
1.3 Adenosin
Adenosin ist ein körpereigenes Nukleosid, bestehend aus der Nukleinbase Adenin und β-D-
Ribose. Es bildet sich aus dem ubiquitären Katabolismus von ATP über die Zwischenstufen
ADP und AMP und kann auch von Neuronen und Gliazellen freigesetzt werden.
Über einen equilibrativen Nukleosidtransporter (ENT) gelangt Adenosin je nach

16 1 Einleitung
Konzentrationsgefälle bidirektional über die Zellmembran. So kann es extrazellulär als
Signalmolekül und Feinmodulator basaler synaptischer Übertragung fungieren, ohne selbst ein
Neurotransmitter zu sein (Sebastião und Ribeiro 2009; Wilson und Mustafa 2009; Dias et al.
2013). Adenosin scheint einen modulierenden Einfluss auf die Neurotransmitter Glutamat,
Dopamin, GABA, Serotonin, Noradrenalin und Acetylcholin zu haben, wenn auch die
Hemmung exzitatorischer Transmitter (z.B. Glutamat) im Vordergrund steht (Shen und Chen
2009). Im Kapitel 1.3.3 wird schließlich auf die Bedeutung von Adenosin bei
neuropathophysiologischen Vorgängen eingegangen.
1.3.1 Adenosinrezeptoren
Wirkung zeigt Adenosin hauptsächlich an den vier bisher bekannten Adenosinrezeptoren (AR),
die an G-Proteine gekoppelt sind (kurz: GPCR) und intrazelluläre Signalkaskaden auslösen.
Von den vier Rezeptor-Subtypen A1, A2A, A2B und A3 kommen im ZNS hauptsächlich A1- und
A2A-Rezeptoren (A1R und A2AR) in hoher Dichte vor, in CA3- und CA1-Neuronen dominieren
A1R (Rebola et al. 2005). Betrachtet man die Acetylcholin-Modulation im Hippocampus,
scheinen A1R in der CA1-Region und A2AR in der CA3-Region jeweils stärker exprimiert zu
sein (Cunha et al. 1994).
Das Guanylnukleotid-bindende Protein (G-Protein) besteht aus den drei Untereinheiten αs bzw.
αi, β und γ. Bindet Adenosin an den Rezeptor, wird die α-Einheit des G-Proteins durch den
Austausch von Guanosindiphosphat (GDP) zu Guanosintriphosphat (GTP) aktiviert. Entweder
wird die nachgeschaltete Adenylatzyklase dadurch stimuliert (αs/Gs) oder inhibiert (αi/Gi) und
setzt bei Stimulation cAMP frei (Kandel 2013). Damit moduliert Adenosin unter anderem die
in Kapitel 1.1.2 beschriebene Kaskade von cAMP, PKA und CREB.
Sowohl der A1R als auch A3-Rezeptor (A3R) sind hauptsächlich Gi-Protein-gekoppelt,
wohingegen die A2A- (A2AR) und A2B-Rezeptoren (A2BR) Gs-Protein-gekoppelt sind. Adenosin
hat eine hohe Affinität zu den Rezeptoren A1, A2A und A3. Die endogene Adenosin-
Konzentration reicht unter physiologischen Bedingungen nicht dazu aus, den ohnehin wenig
exprimierten A2BR zu stimulieren (Fredholm et al. 2001b). Dieser Umstand unterstreicht die
funktionelle Bedeutung der Adenosinkonzentration und Rezeptorendichte, -affinität
sowie -lokalisation.
Außerdem aktiviert der A1R, ebenso wie A2BR und A3R, Phospholipase C (PLC) über die β/γ-
bzw. αq-Einheit, wodurch schließlich über Inositoltriphosphat (IP3) intrazelluläre Ca2+-Speicher

17 1 Einleitung
freigesetzt werden und mittels Diacylglyzerin (DAG) die Proteinkinase C aktiviert wird (Mori
2014). Abbildung 4 skizziert die beschriebenen Signalwege.
Mit der Aktivierung der AR wird zeitgleich deren Desensibilisierung mittels Internalisierung
über β-Arrestin initiiert, wobei der A1R am längsten aktiviert verbleibt (Klaasse et al. 2008).
Abbildung 4: Schema des Metabolismus und Signalwegs von Adenosin
ATP bindet als Signalmolekül an purinerge Rezeptoren (P2X und P2Y) und wird über Hydrolyse zu ADP,
AMP und schließlich Adenosin abgebaut. Adenosin bindet hochaffin an P1-Rezeptoren, vier G-Protein-
gekoppelte Rezeptoren (A1, A2A, A2B, A3) und löst unterschiedliche Signalmechanismen aus. A1R und A3R
(Gi-gekoppelt) hemmen die Adenylatzyklase, sodass kein cAMP-Anstieg erfolgt. Zudem stimulieren sie
zusammen mit A2B die Phospholipase C mit einem Ca2+-Anstieg sowie einer Aktivierung der PKC zur Folge.
A2A und A2BR (Gs-gekoppelt) stimulieren Adenylatzyklase und die nachgeschaltete Kaskade. Über
equilibrative Nukleosidtransporter (ENT) gelangt Adenosin über die Zellmembran. Modifiziert nach Dias et
al. (2013).
1.3.2 Neuromodulation
Verschiedene Wirkungsweisen versuchen den modulierenden Einfluss von AR auf die
synaptische Übertragungsstärke zu erklären, wenngleich diese Mechanismen noch nicht
abschließend erforscht sind und teilweise uneinheitliche Ergebnisse vorliegen.

18 1 Einleitung
Entscheidend für neuromodulatorische Effekte sind die gegensätzlichen Mechanismen vor
allem von A1R und A2AR, die paradoxerweise häufig in einer einzelnen glutamatergen
Pyramidenzelle co-exprimiert werden (Rebola et al. 2005).
Es konnte bisher gezeigt werden, dass LTP über A1R abgeschwächt, jedoch über A2AR verstärkt
wurde (Mendonça und Ribeiro 1996). A1R scheinen NMDA-R – die maßgeblich an LTP-
Induktion beteiligt sind – in hippocampalen Neuronen zu hemmen. Komplexeren und noch
nicht vollständig erfassten Einfluss scheint der A2AR auf den NMDA-R zu haben. Bei
geringerer Aktivierung verstärkt er dessen Wirkung, bei erhöhter Aktivität scheint er diesen
jedoch speziell im Striatum zu hemmen (Klishin et al. 1995; Mendonca et al. 1995; Wirkner et
al. 2000; Rebola et al. 2008; Wilson und Mustafa 2009).
Zusätzlich wird bei Aktivierung von NMDA-R Adenosin freigesetzt, welches als retrograder
messenger über A1R eine präsynaptische Glutamatfreisetzung hemmen kann (Manzoni et al.
1994). A2AR hingegen führt zu einem gegenteiligen Effekt.
Während postsynaptisch die Modulation von NMDA-R im Vordergrund steht, hemmen A1R
präsynaptisch einen Ca2+-Einstrom, der für die exozytotische Freisetzung von Transmittern
benötigt wird (Wu und Saggau 1994).
Sowohl über NMDA-R als auch über A1R werden GIRKs (engl.: G protein-coupled inwardly-
rectifying K+ channels) aktiviert, wodurch das Membranpotenzial hyperpolarisiert und damit
die synaptische Exzitabilität eingeschränkt wird (Chung et al. 2009; Kim und Johnston 2015).
Auch ATP spielt bei HFS präsynaptisch eine Rolle in der Glutamatfreisetzung mittels
purinergen P2X- und P2Y-Rezeptoren. Eine neuere Studie belegt einen direkten, hemmenden
Einfluss auf postsynaptische AMPA-Expression (Pougnet et al. 2014). Außerdem triggern P2-
Rezeptoren eine Adenosinfreisetzung, welche wiederum LTP reguliert (Wieraszko und Ehrlich
1994; Almeida et al. 2003; Rodrigues et al. 2005).
Entscheidend für die konzentrationsabhängige Wirkung von Adenosin ist die vor allem
präsynaptische Heterodimerisierung von A2AR und A1R, die im Striatum von Ciruela et al.
(2006) beschrieben wurde. So führt eine niedrige Adenosinkonzentration über den A1R zu einer
Inhibierung synaptischer Transmission, eine höhere Konzentration zu einer Aktivierung des
A2AR, der wiederum den A1R hemmt und damit zu einer Potenzierung der
Transmitterfreisetzung führt (O'Kane und Stone 1998; Lopes et al. 2000; Brugarolas et al.

19 1 Einleitung
2014). Die beschriebene Feinmodulierung durch den A2AR kommt wahrscheinlich vermehrt in
pathophysiologischen Situationen zum Tragen (Shen und Chen 2009).
1.3.3 Neuro(patho)physiologische Implikationen
Die Rolle von Adenosin im Körper ist weitreichend. Es schwächt Entzündungsgeschehen, ist
an Wundheilung beteiligt, schützt vermutlich Gewebe vor ischämischem Schaden und
vermittelt Vasodilatation, Angiogenese und Neuroprotektion (Fredholm 2007; Haskó et al.
2008; George et al. 2010)
Über den A1R und A2AR werden vermutlich Schlaf-Wachphasen und Schlafinduktion
beeinflusst (McCarley 2007; Huang et al. 2011). Zudem konnte im Mausmodell gezeigt
werden, dass therapeutischer Schlafentzug und Elektrokrampftherapie astrozytäres Adenosin
vor allem im Frontalhirn freisetzten, die A1-Rezeptorendichte erhöhten und somit A1R-
vermittelt antidepressiv wirkten (Elmenhorst et al. 2007; van Calker und Biber 2005). Dies
konnte durch Zugabe von A1-Rezeptoragonisten verstärkt werden (Hines et al. 2013). Die
Arbeit von Kaster et al. (2012) vermutete den antidepressiven Effekt von A1R in der Hemmung
von NMDA-R.
Bekanntermaßen verstärken AR-Antagonisten wie z.B. Coffein neben Wachheit auch
Aufmerksamkeit. Evidenz dafür liefert eine Studie von Mihara et al. (2007). Bei transgenen
Mäusen konnte nachgewiesen werden, dass die Aktivierung von A2AR negativ
Gedächtnisbildung beeinflusst (Wang et al. 2006; Giménez-Llort et al. 2007).
Vergleichbare Knockout-Mäuse schienen in einer anderen Studie weniger depressives
Verhalten zu zeigen. A2A-überexprimierende Ratten zeigten gegenteiliges Verhalten (El
Yacoubi et al. 2001; Coelho et al. 2014). Daraufhin wurden therapeutische, hochselektive
A2AR-Antagonisten (Istradefylline, Preladenant) identifiziert, die im Mausmodell antidepressiv
wirkten (Hodgson et al. 2009). Dieselben Wirkstoffe reduzieren „Off-Phasen“ bei der
Parkinson-Erkrankung und sind bereits in klinischen Phasen der Zulassung (Hickey und Stacy
2012; Vorovenci und Antonini 2015).
Noch nicht hinreichend geklärt ist die Wirkungsweise der Tiefen Hirnstimulation (appliziert
HFS), die bei Parkinson-Erkrankten effektiv Tremor reduziert und auch bei depressiven
Patienten Erfolge zeigt. Dabei scheint der A1R jedenfalls die Tremorreduzierung durch
Hemmung synaptischer Übertragung im Thalamus mitzuverantworten (Bekar et al. 2008).
Durch HFS aktivierte A2AR stimulieren ENTs (Equilibrative Nukleosidtransporter) und führen

20 1 Einleitung
in der Folge zu einer Internalisierung von Adenosin, wodurch die A1R-Aktivität wiederum
gebremst wird (Pinto-Duarte et al. 2005).
Daneben sind der A1R und der A2AR in einigen anderen neurologischen und psychiatrischen
Pathophysiologien involviert, wie beispielsweise in Alzheimer-Demenz, Huntington-
Erkrankung und Epilepsie, auf die in dieser Arbeit jedoch nicht eingegangen wird.
1.4 Fragestellung dieser Arbeit
In dieser Arbeit wird das durch die Arbeitsgruppe etablierte Theta-Burst-Pairing-
Stimulationsprotokoll weiterhin verwendet, um assoziative LTP als Simulation physiologischer
Reize an Mäuse- und Rattenneuronen in vitro zu untersuchen. Viele ältere Arbeiten nutzten
nicht-assoziative Stimulationsprotokolle (HFS).
Grundlage der Experimente ist die Frage danach, inwiefern AR die assoziative LTP modulieren.
Anhand verschiedener Agonisten und Antagonisten der A1R und A2AR soll überprüft werden,
wie sich diese durch extrazelluläre Beimengung auf die LTP an CA3/CA1-Synapsen im
Hippocampus von Wistar-Ratten auswirken. Die Ergebnisse werden jeweils mit einer
Kontrollgruppe verglichen. Es werden zum Teil Experimente von Christopher Landmann
(Arbeitsgruppe, 2015) wiederholt, um sie zu validieren.
Eine zweite Versuchsreihe geht der Frage nach, wie sich eine selektive Überexprimierung des
A1R in der CA1-Region (postsynaptisch) des Hippocampus von C57BL/6-Mäusen in der
assoziativen LTP-Messung zu einer Kontrollgruppe unterscheidet. David Stolz (Arbeitsgruppe,
2015) untersuchte in seinen Experimenten Mäuse mit einer Überexprimierung desselben
Rezeptors in der CA3-Region (präsynaptisch).
Parallel prüfte Julia Knoll (Arbeitsgruppe, noch nicht veröffentlicht) ähnliche Substanzen und
dieselbe transgene Überexprimierung in Mäusen in ihrer Wirkung auf LTD.

21 2 Material und Methoden
2 Material und Methoden
2.1 Lösungen und Substanzen
2.1.1 Extrazelluläre Lösungen
Um die Zellen am Leben zu erhalten, wurden die Hirnschnitte ab Präparation mit künstlicher
extrazellulärer Lösung umspült, der sogenannten aCSF (artificial cerebro-spinal fluid),
wodurch man physiologischen Liquor cerebrospinalis simulierte.
Die genauere Zusammensetzung der Elektrolyte in aCSF ist der Tabelle 1 zu entnehmen.
Mit dem Osmomat 030 (genotec, Deutschland) wurde die Osmolalität von aCSF im Bereich
von 320 ± 3 mosmol/kg gemessen.
Vor den Messungen wurde der aCSF-Lösung zusätzlich Picrotoxin hinzugefügt. Als nicht-
kompetitiver Kanalblocker des GABAA-Rezeptors fördert Picrotoxin indirekt eine
exzitatorische Reizweiterleitung, indem es den inhibitorischen Effekt des GABAA-Rezeptors
aufhebt.
Picrotoxin, als Stammlösung in Ethanol aufbewahrt, wurde der aCSF für eine abschließende
Konzentration von 20 µmol/l hinzugefügt.
Außerdem begann man vor den Messungen bereits damit, die Lösung mit Carbogen (95 % O2,
5 % CO2) zu begasen, um die Hirnschnitte ausreichend zu oxygenieren.
Die Stimulationspipetten wurden mit einer modifizierten aCSF befüllt, welche unter anderem
um die Substanz HEPES (4-(2-Hydroxyethyl)piperazin-1-ehtansulfonsäure) ergänzt wurde.
Auf diese Weise konnte eine optimale Pufferung des pH-Wertes am Ort der Stimulation
gewährleistet werden. Der pH-Wert der Pipettenlösung wurde mit einem pH-Meter (WTW,
InoLab, pH Level 1, Deutschland) kontrolliert und mit NaOH (Natriumhydroxid) auf den
angestrebten pH 7,4 eingestellt.
2.1.2 Pipetten-Lösung
Um einen bestmöglichen Ionenaustausch in der Whole-cell-Zellverbindung zu schaffen,
befüllte man die Patchpipetten mit künstlicher intrazellulärer Lösung; für diese wurde eine
geringgradig niedrigere Osmolalität von 292 mosmol/kg (gemessen mit Osmomat 030, gonotec,
Deutschland) gegenüber der Zytoplasma-Osmolalität verwendet.
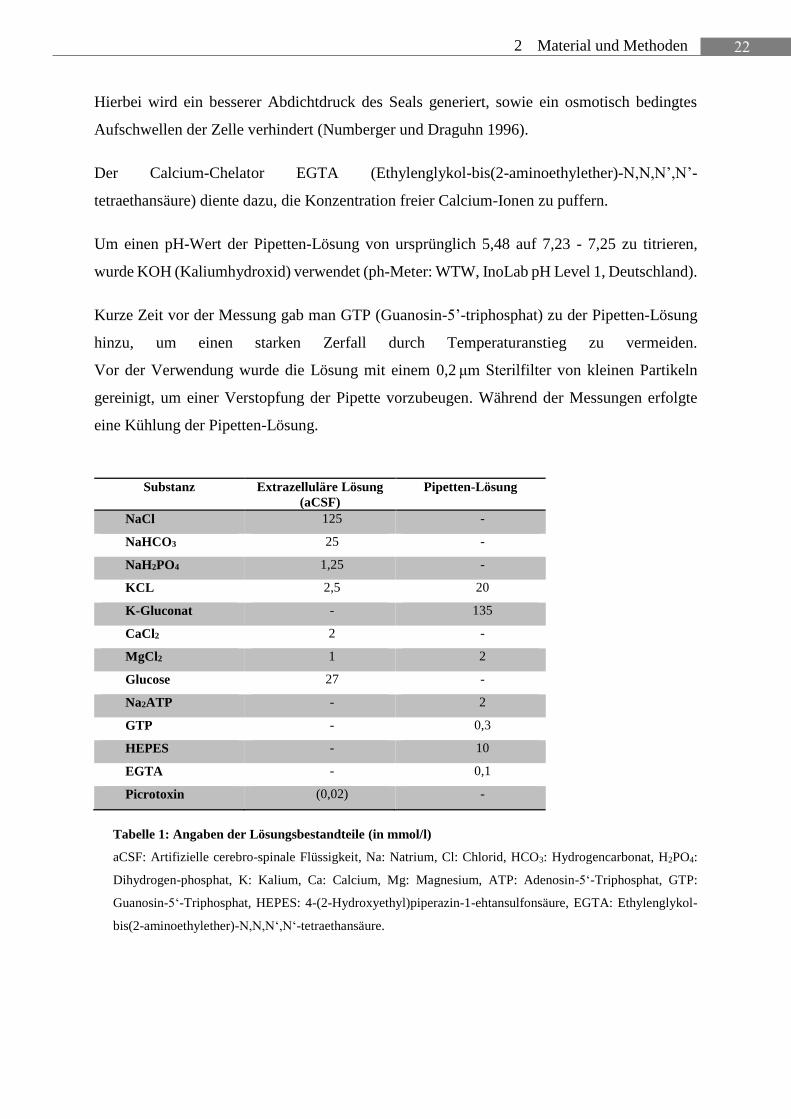
22 2 Material und Methoden
Hierbei wird ein besserer Abdichtdruck des Seals generiert, sowie ein osmotisch bedingtes
Aufschwellen der Zelle verhindert (Numberger und Draguhn 1996).
Der Calcium-Chelator EGTA (Ethylenglykol-bis(2-aminoethylether)-N,N,N’,N’-
tetraethansäure) diente dazu, die Konzentration freier Calcium-Ionen zu puffern.
Um einen pH-Wert der Pipetten-Lösung von ursprünglich 5,48 auf 7,23 - 7,25 zu titrieren,
wurde KOH (Kaliumhydroxid) verwendet (ph-Meter: WTW, InoLab pH Level 1, Deutschland).
Kurze Zeit vor der Messung gab man GTP (Guanosin-5’-triphosphat) zu der Pipetten-Lösung
hinzu, um einen starken Zerfall durch Temperaturanstieg zu vermeiden.
Vor der Verwendung wurde die Lösung mit einem 0,2 μm Sterilfilter von kleinen Partikeln
gereinigt, um einer Verstopfung der Pipette vorzubeugen. Während der Messungen erfolgte
eine Kühlung der Pipetten-Lösung.
Substanz Extrazelluläre Lösung
(aCSF)
Pipetten-Lösung
NaCl 125 -
NaHCO3 25 -
NaH2PO4 1,25 -
KCL 2,5 20
K-Gluconat - 135
CaCl2 2 -
MgCl2 1 2
Glucose 27 -
Na2ATP - 2
GTP - 0,3
HEPES - 10
EGTA - 0,1
Picrotoxin (0,02) -
Tabelle 1: Angaben der Lösungsbestandteile (in mmol/l)
aCSF: Artifizielle cerebro-spinale Flüssigkeit, Na: Natrium, Cl: Chlorid, HCO3: Hydrogencarbonat, H2PO4:
Dihydrogen-phosphat, K: Kalium, Ca: Calcium, Mg: Magnesium, ATP: Adenosin-5‘-Triphosphat, GTP:
Guanosin-5‘-Triphosphat, HEPES: 4-(2-Hydroxyethyl)piperazin-1-ehtansulfonsäure, EGTA: Ethylenglykol-
bis(2-aminoethylether)-N,N,N‘,N‘-tetraethansäure.

23 2 Material und Methoden
2.1.3 Substanzen
Der aCSF mischte man bei den jeweiligen Experimenten Substanzen bei, welchte unten
angeführte Stoffmengenkonzentrationen der endgültigen Spüllösung enthielten. Die
Substanzen mit höchstem Reinheitsgrad, im Folgenden aufgelistet, stammten von Sigma-
Aldrich (Deutschland) und Tocris (Deutschland).
cCPA 2-chloro-N(6)-cyclopentyladenosine
Sigma Aldrich, Deutschland
Stammlösung in Aqua dest.
Lösung in aCSF: 200 nM
DPCPX 8-Cyclopentyl-1,3-dipropylxanthine
Tocris, Deutschland
Stammlösung in DMSO
Lösung in aCSF: 200 nM
CGS-21680 4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-
yl]amino]ethyl]benzenepropanoic acid hydrochloride
Tocris, Deutschland
Stammlösung in DMSO
Lösung in aCSF: 200 nM
NEM N-Ethylmaleinimide
Sigma Aldrich, Deutschland
Stammlösung in Ethanol
Lösung in aCSF: 250 µM

24 2 Material und Methoden
2.2 Versuchstiere
Alle Tiere wurden mit größter Sorgfalt, umsichtig und respektvoll behandelt. Sowohl während
des Transports, als auch im Labor wurde bestmöglich versucht, die Tiere vor Stressfaktoren
wie Kälte, Lärm und Erschütterung zu schützen.
Für einen Großteil der Versuche wurden weiße, junge Wistar-Ratten P12 (±2 Tage) verwendet.
Bei den anderen Versuchsreihen wurden transgene, junge C57BL/6-Mäuse P11-19 sowie deren
Wildtyp herangezogen. In der Linie CaMKII-AdoA1R-92 wird das A1R-Gen spezifisch in der
CA1-Region des Hippocampus ca. zweifach überexprimiert. Die zugrundeliegende Methode
wurde von Serchov et al. (2015) veröffentlicht. Ein Immunofluoreszenznachweis des
Verteilungsmusters im Hippocampus ist in Abbildung 6 zu sehen.
Um entsprechende transgene Mäuse zu züchten, werden die beiden folgenden Linien gekreuzt,
wobei nur 25 % der F1-Generation die gewünschte Genkombination tragen (siehe
Abbildung 5):
1. CaMKII‐tTA-Linie: Der Promotor CaMKII (Ca2+-Calmodulin-abhängige
Proteinkinase II) exprimiert das Tetracyclin-kontrollierte Transaktivatorgen (tTA) in
Neuronen des Vorderhirns.
2. TRE-A1R-mCherry‐Linie: Das Tetracyclin Response Element (TRE) exprimiert über
einen bidirektionalen Promotor sowohl das Fluoreszenzprotein mCherry-Gen, als auch das
Maus-spezifische A1R-Gen.
Abbildung 5: Schema der Überexprimierung des A1R-Gens
CaMKII exprimiert den Transaktivator (tTA), der Tetracyclin-abhängig (Dox/Doxycylin ist ein stabiles
Tetracyclin-Derivat) an TRE bindet und eine bidirektionale Exprimierung der Gensequenzen mCherry und
mA1R cDNA (A1R-Gen) zur Folge hat. Abbildung nach Serchov et al. (2015).

25 2 Material und Methoden
Abbildung 6: Immunofluoreszenzmuster anti-mCherry im Hippocampus-Schnitt
Grün: Marker für Neuronenkerne; gelb-orange: mCherry-Expression in der CA1-Region.
Abbildung orientiert sich an Serchov et al. (2015).
2.3 Hippocampusschnitte
Die hier beschriebenen Präparationsvorgänge orientieren sich an dem von Edwards und
Bischofberger etablierten Protokoll (Edwards et al. 1989; Bischofberger et al. 2006).
Die Dekapitation der Tiere erfolgte mit einer speziellen Schere. Ab diesem Zeitpunkt wurden
der Schädel, die Großhirnhemisphären und Hirnschnitte fortwährend in halbgefrorener bis
flüssiger, kalter aCSF präpariert und kontinuierlich mit Carbogen (Gasgemisch aus 95 % O2
und 5 % CO2) begast.
Zuerst wurde die Schädelkalotte freigelegt, sodass diese mit einem von dorsal geführten
sagittalen Scherenschnitt median durchtrennt werden konnte. Durch Schnitte, die medio-lateral
auf Höhe der Sutura lambdoidea und rostral der Bulbi olfaktorii gesetzt wurden, konnten die
Kalottenhälften entfernt werden, nachdem die Hirnhäute vorsichtig abgelöst wurden (siehe
Abbildung 7a). Mit einem Skalpell wurde rostral der Bulbi olfaktorii und caudal des
Cerebellums geschnitten, sodass die Großhirnhemisphären mit einem stumpfen Spatel aus der
Schädelhöhle herausgehoben und in eine mit aCSF gefüllte und begaste Petrischale gelegt
werden konnten. Nach drei Minuten wurden die Hemisphären mit einem Schnitt durch den
Corpus callosum getrennt, wobei ein untergelegtes Filterpapier für besseren Halt sorgte. Fortan
wurde für bessere Vergleichbarkeit nur die rechte Hemisphäre verwendet, die linke verworfen.
Auf die mediane Schnittseite gelegt, wurde der kraniale Anteil der rechten Hemisphäre mit
einem zum Lot um 5 ° geneigten, basofrontalen Schnitt entfernt, der ca. 1-2 mm nach apikal
gesetzt wurde (siehe Abbildung 7b). Dies ist der sogenannte „Magic Cut“, dessen Schnittwinkel

26 2 Material und Methoden
so gewählt ist, dass möglichst wenig Schaffer-Kollateralen angeschnitten werden, welche für
die Potenzial-Weiterleitung in den folgenden Experimenten vonnöten sind.
Die daraus entstandene Schnittfläche wurde mit Cyanacrylatkleber (LOCTITE 454, Henkel,
Deutschland) auf die vorgekühlte Schnittkammer des Mikrotoms (Leica VT1200, Deutschland)
geklebt. Ohne Verzug wurde die Kammer mit fast gefrorener aCSF-Lösung aufgefüllt und
weiterhin mit Carbogen begast.
Eingestellt mit einer Vorschubgeschwindigkeit von ca. 1 mm/s stellte das Mikrotom 300 µm
dünne Hippocampusschnitte her, wobei immer eine unbenutzte Rasierklinge (Gilette,
Deutschland) eingespannt wurde. Mit einer gebogenen Injektonsnadel ließ sich jeder Schnitt
von dem haftenden Gewebeblock am Subiculum abtrennen (siehe Abbildung 7c).
Etwa 6 gewonnene transversale Schnitte wurden mit einer Pipette in ein spezielles
Lagerungsgefäß abgelegt. In diesem lagen die Schnitte auf einem Haltering, der mit einer
Mullkompresse bespannt war, sodass ausreichende Oxygenierung durch die umspülende aCSF-
Lösung sichergestellt werden konnte (siehe Abbildung 7d).
Für 20 Minuten wurde das runde Gefäß bei 34,5 °C in einem Wasserbad (Julabo, EC-AP,
Deutschland) inkubiert. Im Anschluss lagerten die Schnitte bei Zimmertemperatur.
Durch die regenerative Phase während der Inkubation wurde die Zellqualität verbessert.
Dadurch konnte eine Durchführung der Messungen für eine Dauer von bis zu 5-6 Stunden
gewährleistet werden.
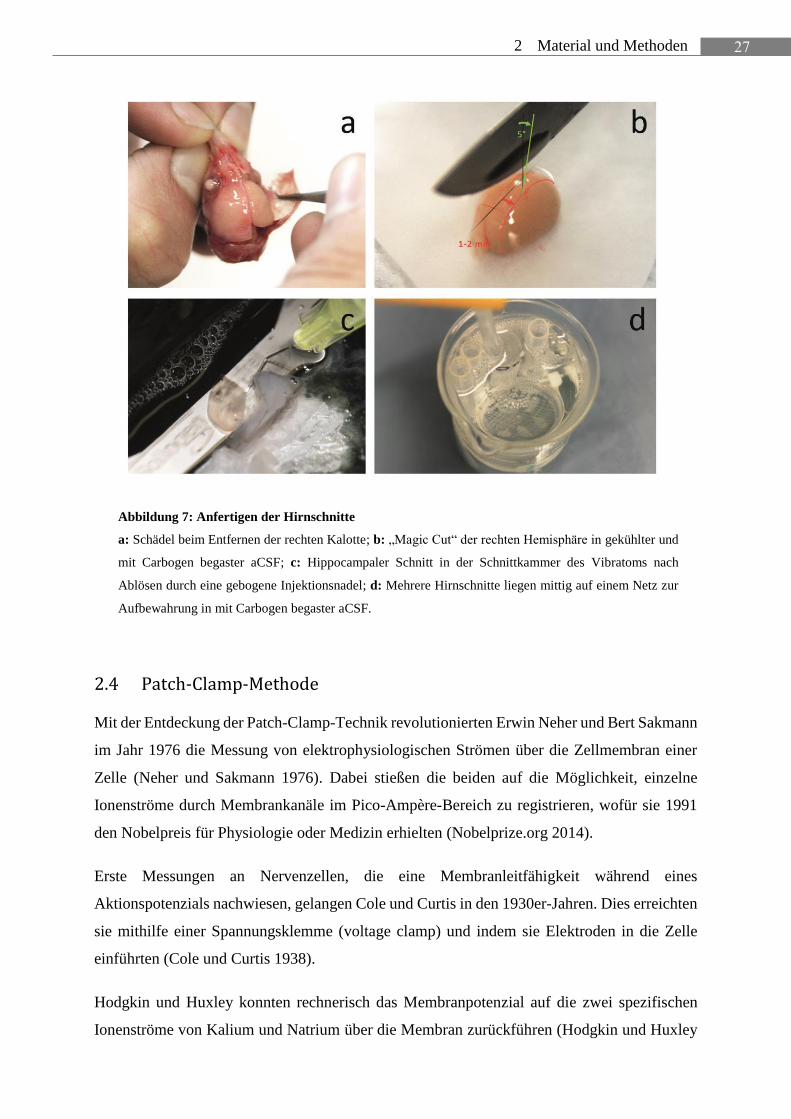
27 2 Material und Methoden
Abbildung 7: Anfertigen der Hirnschnitte
a: Schädel beim Entfernen der rechten Kalotte; b: „Magic Cut“ der rechten Hemisphäre in gekühlter und
mit Carbogen begaster aCSF; c: Hippocampaler Schnitt in der Schnittkammer des Vibratoms nach
Ablösen durch eine gebogene Injektionsnadel; d: Mehrere Hirnschnitte liegen mittig auf einem Netz zur
Aufbewahrung in mit Carbogen begaster aCSF.
2.4 Patch-Clamp-Methode
Mit der Entdeckung der Patch-Clamp-Technik revolutionierten Erwin Neher und Bert Sakmann
im Jahr 1976 die Messung von elektrophysiologischen Strömen über die Zellmembran einer
Zelle (Neher und Sakmann 1976). Dabei stießen die beiden auf die Möglichkeit, einzelne
Ionenströme durch Membrankanäle im Pico-Ampère-Bereich zu registrieren, wofür sie 1991
den Nobelpreis für Physiologie oder Medizin erhielten (Nobelprize.org 2014).
Erste Messungen an Nervenzellen, die eine Membranleitfähigkeit während eines
Aktionspotenzials nachwiesen, gelangen Cole und Curtis in den 1930er-Jahren. Dies erreichten
sie mithilfe einer Spannungsklemme (voltage clamp) und indem sie Elektroden in die Zelle
einführten (Cole und Curtis 1938).
Hodgkin und Huxley konnten rechnerisch das Membranpotenzial auf die zwei spezifischen
Ionenströme von Kalium und Natrium über die Membran zurückführen (Hodgkin und Huxley

28 2 Material und Methoden
1952). Dass für die Membranpermeabilität ein Ionencarrier verantwortlich sei, wie die Autoren
annahmen, konnte allerdings nicht bestätigt werden.
Erst Neher und Sakmann gelang der Nachweis einzelner Ionenkanäle, deren Eigenschaft, sich
zu öffnen oder zu schließen, auf einem spannungsabhängigen Alles-oder-Nichts-Prinzip beruht.
In der verbesserten Patch-Clamp-Technik wird eine Glaspipette verwendet, deren abgerundete
Spitze mit einer minimal großen Öffnung auf der Zellmembran aufsitzt. Durch leichtes
Ansaugen schafft man eine dichte Unterdruckverbindung, wodurch sich Leckströme und
elektrisches Rauschen verringern. Dabei stülpt sich die kreisrunde Zelloberfläche (patch =
„Flicken“) in die Öffnung der Glaspipette und es entstehen bestenfalls Abdichtwiderstände von
einigen Gigaohm (sog. Gigaseal). Ausgehend von dieser Pipetten-Zellverbindung bestehen
mehrere mögliche Patchkonfigurationen (Numberger und Draguhn 1996). Neben der
beschriebenen Cell-attached-Konfiguration (siehe Abbildung 8a) kann durch einen kurzen
Saugimpuls der „patch“ herausgetrennt werden, sodass eine offene Verbindung zum
Zytoplasma hergestellt wird – es ensteht die Whole-cell-Konfiguration (siehe Abbildung 8b).
Möchte man Kanalströme ausschließlich an dem isolierten Membranstück messen, bieten sich
Konfigurationen Inside-out und Outside-out an (siehe Abbildung 8c,d).
Die durchgeführten Experimente wurden ausschließlich mit der Whole-cell-Konfiguration
gemessen. Vorteilhaft dabei ist, dass Ströme über die gesamte Zellmembran registriert und
zusätzlich Stoffe über die Patchpipette in den Intrazellulärraum appliziert werden können.
Abbildung 8: Patchkonfigurationen
a: Cell-attached-Konfiguration; b: Whole-cell-Konfiguation; c: Inside-out-Konfiguration; d: Outside-out-
Konfiguration. Modifiziert nach Numberger und Draguhn (1996).

29 2 Material und Methoden
2.5 Aufbau des Patch-Clamp-Messplatzes
2.5.1 Übersicht
Der Aufbau eines Patch-Clamp-Messplatzes erfordert das Zusammenspiel einzelner Geräte, die
eine Experimentdurchführung ermöglichen, registrieren und dokumentieren. Kernelement stellt
das Mikroskop samt Messkammer, Mikromanipulatoren für die Steuerung der Pipetten und der
Vorverstärker dar. Über eine Kamera und den zugehörigen Videomonitor ist die vergrößerte
Betrachtung des Experiments möglich. Die Einheit aus Pulsstimulator, Verstärker und
Computer ist für eine Durchführung, Analyse und Datenspeicherung des Experiments
unabdingbar.
Abbildung 9: Übersicht des Patch-Clamp-Messplatzes
2.5.2 Messkammer und optisches System
Die präparierten Hirnschnitte wurden nach Inkubation in eine runde Messkammer aus Plexiglas
(Mechanische Werkstätten, Universitätsklinikum Freiburg) überführt, die man auf dem
Mikroskoptisch verankerte. Für ein geringeres Flüssigkeitsvolumen in der Messkammer und
für einen erhöhten Durchfluss sorgte eine angewinkelt vertiefte Plexiglaswanne, deren Boden
ein Deckgläschen bildete. Hierauf wurde der Hirnschnitt gelegt und durch einen mit
Nylonfäden bespannten Platinring an den Rändern fixiert.

30 2 Material und Methoden
Über einen Perfusionszulauf wurde die ständige Versorgung des Hirnschnittes mit frischer
aCSF-Lösung, die beständig mit Carbogen begast wurde, sichergestellt. Eine Vakuumpumpe
(WISA, ASF-Thomas, Deutschland) sorgte für den Ablauf der Flüssigkeit an der
gegenüberliegenden Seite der Messkammer. Der regulierbare Zulauf wurde auf eine
Durchflussrate von ca. 5 ml/min eingestellt.
Abbildung 10: Messtisch mit Messkammer während Messung
a: Messkammer mit Hirnschnitt unter Platinring; b: Patchpipette; c: Stimulationspipette; d: Objektiv; e:
Referenzelektrode; f: Perfusionszulauf; g: Perfusionsablauf
Zur Verbesserung der Optik des verwendeten aufrechten Mikroskops (Axioskop, Zeiss,
Deutschland) wurde die Technik des differentiellen Interferenzkontrasts (DIC) angewendet.
Dies dient der besseren dreidimensionalen Orientierung im Gewebsschnitt während der
Lokalisierung der Zellen. Zusätzlich ermöglichte die Verwendung einer Infrarot-Videotechnik
(IR-Filter und IR-Kamera) eine optimale Lichtausbeute für eine kontrastreiche Darstellung.
Die optische 40-fache Vergrößerung durch ein Wasserimmersionsobjektiv (LUMPLAN FI/IR,
40 x/0.80 W, Olympus, Japan) wurde durch eine zusätzliche elektronische 30-fache
Vergrößerung einer Videokamera (Thomas Völker, Deutschland) erweitert. Sobald ein
geeignetes Neuron in der Mikroskop-Optik aufgefunden wurde, konnte der Pipetten-
Zellkontakt über die Videooptik erschütterungsfrei hergestellt werden.
2.5.3 Messtisch und Mikromanipulator
Um Störeffekte mechanischer Schwingungen und Erschütterungen während der Messungen zu
reduzieren wurde die komplette Messvorrichtung auf einem druckluftgefederten Messtisch
(Newport 3030W-OPT, USA) angebracht.

31 2 Material und Methoden
Für eine präzisere Pipettenplatzierung verwendete man elektrisch betriebene
Mikromanipulatoren (mini 25, Luigs & Neumann, Deutschland). Über ein Bedienpult konnten
so die Stimulations- und Patchpipetten gesteuert werden. An der Patchpipettenhalterung
befestigte man neben dem Vorverstärker einen Schlauch, mit dem Über- oder Unterdruck auf
die Pipette ausgeübt werden konnte.
Leitende Oberflächen von Messtisch, Mikroskop, Mikromanipulatoren und Vorverstärker
wurden elektrisch geerdet und signalleitende Kabel zusätzlich elektrisch abgeschirmt, um
Hintergrundrauschen zu minimieren.
2.5.4 Pulsstimulator, Verstärker und Software
Über eine Stimulationspipette, die einen Ag/AgCl-Elektrodendraht enthielt und mit aCSF-
Lösung befüllt war, wurden Spannungspulse im Schaffer-Kollateraltrakt der CA3-Neurone des
Hippocampus appliziert. Ein Pulsstimulator (A-M SYSTEMS, Model 2100, USA) generierte
diese Spannungspulse, die dann als EPSP-Amplituden über die Patchpipettenelektrode
registriert werden konnten.
Die mit einer Intrazellularlösung befüllte Patchpipette enthielt ebenso einen Ag/AgCl-Draht,
der über einen Vorverstärker am Mikromanipulator mit dem Verstärker (EPC-9, HEKA,
Deutschland) verbunden war.
Der Vorverstärker (Strom-Spannungswandler) maß mittels einer definierten Spannung (sog.
Kommando-/Sollspannung) über der Zellmembran Kompensationsströme, die zur
Aufrechterhaltung dieser Spannung nötig waren. Aufgrund der Verschaltung mit
Rückkopplungswiderstand wurde dieser Strom als Spannung ausgelesen. Dieses Signal, an den
Hauptverstärker weitergeleitet, wurde dort gefiltert und verstärkt.
Abbildung 11: Vereinfachtes Schaltbild eines Patch-Clamp-Verstärkers
OPA: Operationsverstärker; Usoll: Kommando-/Sollspannung; Upip: Pipettenspannung; Uaus:
Ausgangsspannung; Rf: Rückkopplungswiderstand. Modifiziert nach Numberger & Draguhn (1996)

32 2 Material und Methoden
In der Badlösung befand sich ein Silberchlorid-Draht als Referenzelektrode für die Messungen.
Neben dem Modus der Spannungsklemme (voltage clamp) existiert der Modus der
Stromklemme (current clamp). Dabei wird nicht die Spannung, sondern ein definierter Strom
aufrechterhalten und folglich die Änderung der Spannung registriert. Ein solcher
Stromklemmenmodus diente der Messung von EPSPs und Aktionspotenzialen an der Zelle.
Für das Stimulationsprotokoll sowie die Datenmessung und -verarbeitung wurde eine spezielle
Software (Pulse Version 8.77, Heka, Deutschland) verwendet.
2.6 Pipettenherstellung
Für die Herstellung der Stimulations- und Patchpipette dienten Borsilikatkapillaren (Ø außen
2 mm, Ø innen 1,5 mm, Hilgenberg, Deutschland), die in ein Pipetten-Ziehgerät (engl.: puller;
Sutter Instruments Co., P-87, USA) eingespannt und unter vorprogrammierter Zugkraft und
Hitzeeinwirkung auseinandergezogen wurden. So entstanden jeweils zwei identische
Glaspipetten, deren Spitzen die gewünschte Flankenlänge und Öffnungsfläche erhielten. Beide
Faktoren beeinflussten den Pipettenwiderstand. Zielbereich des Öffnungswiderstands der
Patchpipetten war 5-8 MΩ für eine stabile Sealbildung und weniger Lösungsaustausch mit dem
Zytosol. Für Stimulationspipetten genügten lediglich 2-3 MΩ für eine leichtere
Gewebestimulation bei größerer Pipettenöffnung.
Bei der Befüllung der Pipetten half eine Saugvorrichtung (Mechanische Werkstätten,
Uniklinikum Freiburg), mittels der die Pipettenspitze durch Unterdruck mit der gewünschten
Lösung befüllt werden konnte (engl. tip filling).
2.7 Stimulationsprotokoll
Das in dieser Arbeit verwendete assoziative LTP-Stimulationsprotokoll beruht auf einem von
Seifert (2009) etablierten Theta-Burst-Pairing-Modell (siehe Abbildung 12), welches sich
wiederum auf ein Protokoll von Schmidt-Hieber et al. (2004) bezieht.
Hierbei folgt auf ein präsynaptisch stimuliertes EPSP (Stimulationspipette) nach 5 ms ein
postsynaptisch stimuliertes AP (Patchpipette I: 0,7-1,2 nA, Pulsdauer = 3 ms). Durch
fünfmalige Wiederholung dieses Reizpaares mit 100 Hz entsteht eine Salve, welche mit einer
Frequenz von 5 Hz fünfmal wiederholt wird und den sog. Theta-Block formt. Mit einem
Abstand von 10 s wird auch dieser Theta-Block fünfmal wiederholt, sodass insgesamt 125
Reizpaare appliziert werden.

33 2 Material und Methoden
Abbildung 12: Schema des Theta-Burst-Pairing-Protokolls
Bildausschnitt: Sog. Theta-Blöcke mit zeitlich korrelierenden prä- und postsynaptisch, asynchron stimulierten
EPSPs und APs. Insgesamt resultieren 125 Reizpaare. Modifiziert nach Seifert (2009).
2.8 Versuchsablauf
Ein inkubierter Hirnschnitt wurde in der Messkammer mit einem Platinring fixiert, um diese
daraufhin am Messtisch des Mikroskops zu befestigen. Man legte sowohl die Schläuche der
aCSF-Perfusionsanlage, als auch die Erdungselektrode in die Messkammer ein.
In der Mikroskop- und Videomonitorübersicht konnte im CA1-Areal des Hippocampus eine
geeignete Pyramidenzelle ausfindig gemacht werden. Die Stimulationspipette platzierte man in
der Gegend der zuführenden Schaffer-Kollateral-Fasern der CA3-Neurone.
Mit einem Überdruck von ca. 200 mbar in der Patchpipette konnte Verunreinigung der
Pipettenspitze durch Zellmaterial vermieden werden, während man die Patchpipette vorsichtig
auf die Zielzelle absenkte. Sobald der Kontakt durch eine Eindellung (sog. dimpling) sichtbar
wurde, ließ man den Überdruck ab und es bildete sich spontan ein Abdichtwiderstand (sog.
seal) aus. Durch leichten Unterdruck mittels Ansaugen erreichte man ein Giga-Seal im Bereich
einiger GΩ. Zur Überprüfung der Sealqualität diente ein Kommandospannungspuls
(Rechteckpuls +5 mV, 20 ms).
Während dieser Cell-attached-Konfiguration regulierte man die Spannung auf -20 mV,
schließlich auf -70 mV, um daraufhin mit einem kurzen starken Saugimpuls den Membranfleck
(Patch) aus der Membran herauszulösen – daraus resultierte die Whole-cell-Konfiguration.

34 2 Material und Methoden
Um einer Verwechslung zwischen Pyramidenzellen und Interneuronen vorzubeugen, testete
man vor jedem Experiment Aktionspotenzialschwellen und Adaptionseigenschaften von
Pyramidenzellen bei wiederholten Depolarisationen.
Über die Stimulationspipette regulierte man Spannungspulse von 5-95 V und 200 ms Dauer,
sodass EPSPs mit einer Amplitude von 2-7 mV ausgelöst wurden.
Nach jedem Block von 10 EPSP-Messungen erfolgte jeweils ein Testpuls (+5 mV) zur
Bestimmung des aktuellen Membran- und Serienwiderstands.
Jede Baseline-Messung dauerte 5 min, woraufhin das LTP-Induktionsprotokoll durchgeführt
wurde und schließlich EPSP-Messungen (je 30 min) in kurzen Abständen gemessen wurden.
2.9 Datenanalyse und -auswertung
Mit der PulseFit-Software (v8.53 HEKA, Lambrecht, Deutschland) wurden alle Daten erfasst,
welche später mit der Software GraphPad (Prism 6.0, USA) analysiert und graphisch dargestellt
wurden. EPSP-Amplituden der Einzelexperimente wurden als Punkte in einem EPSP/Zeit-
Diagramm skizziert. Für die Darstellung der Messreihe wurden jeweils 6 aufeinanderfolgende
Messwerte gemittelt und mit Fehlerbalken (SEM: standard error of the mean) kombiniert. Der
Baseline entsprechen die Werte vor LTP-Induktion (t = 0).
In der statistischen Auswertung der Experimente mit Mäusen wurden gemittelte Werte der
fünfminütigen Baseline mit denen zwischen der 20. und 30. Minute nach LTP-Induktion mittels
nichtparametrischem Wilcoxon-Rangsummentest auf signifikanten Unterschied (p = 0,05)
untersucht. Bei Experimenten mit Ratten wurde lediglich ein Zeitfenster zwischen der 20. und
25. Minute zum Vergleich gewählt.
Für den Vergleich zweier unabhängier Testreihen wurde ein ungepaarter, nicht-parametrischer
Mann-Whitney-U-Test nach Normalisierung verwendet, bei drei Reihen im Vergleich ein
spezieller ANOVA-Test, der sogenannte Kruskal-Wallis-Test.

35 3 Ergebnisse
3 Ergebnisse
3.1 Versuchsreihen mit Mäusen
3.1.1 Kontrollreihe LTP bei Wildtyp-Mäusen
Als Referenz für Experimente mit transgenen CaMKII-AdoA1R-92-Mäusen wurden Wildtyp-
Mäuse desselben Stammes C57BL/6 verwendet. Die mittlere EPSP-Amplitude der Baseline
betrug 4,01 ± 0,31 mV. Im Vergleichszeitfenster (20-30 min nach LTP-Induktion) erhöhten
sich die Werte sehr signifikant auf einen mittleren EPSP-Wert von 6,74 ± 0,75 mV bzw. auf
166,9 ± 10,05 % der Baseline (p = 0,0039, n = 9).
-5 0
3
6
9
1 2
1 5
0 1 0 2 0 3 0
Z e i t [ m in ]
EP
SP
- A
mp
litu
de
[m
V]
I n d u k t io na b
c
-5 0
0
2
4
6
8
1 0
0
4 0
8 0
1 2 0
0 1 0 2 0 3 0
R s
In d u k tio n R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs [M
] / R
m [-p
A]
B a s e lin e n a c h I n d u k t io n
0
2
4
6
8
EP
SP
- A
mp
litu
de
[m
V]
* *
Abbildung 13: LTP-Kontrollreihe Wildtyp-Mäuse
a: Übersicht der gesamten Messreihe; b: Vergleich der mittleren EPSP-Amplituden vor LTP-Induktion und
20-30 min danach. Der Unterschied ist sehr signifikant (**); c: Beispiel eines Einzelexperiments
(20140116_s3c1), neben EPSP-Werten sind auch Membran- und Serienwiderstand (Rm, Rs) dargestellt.
36 3 Ergebnisse
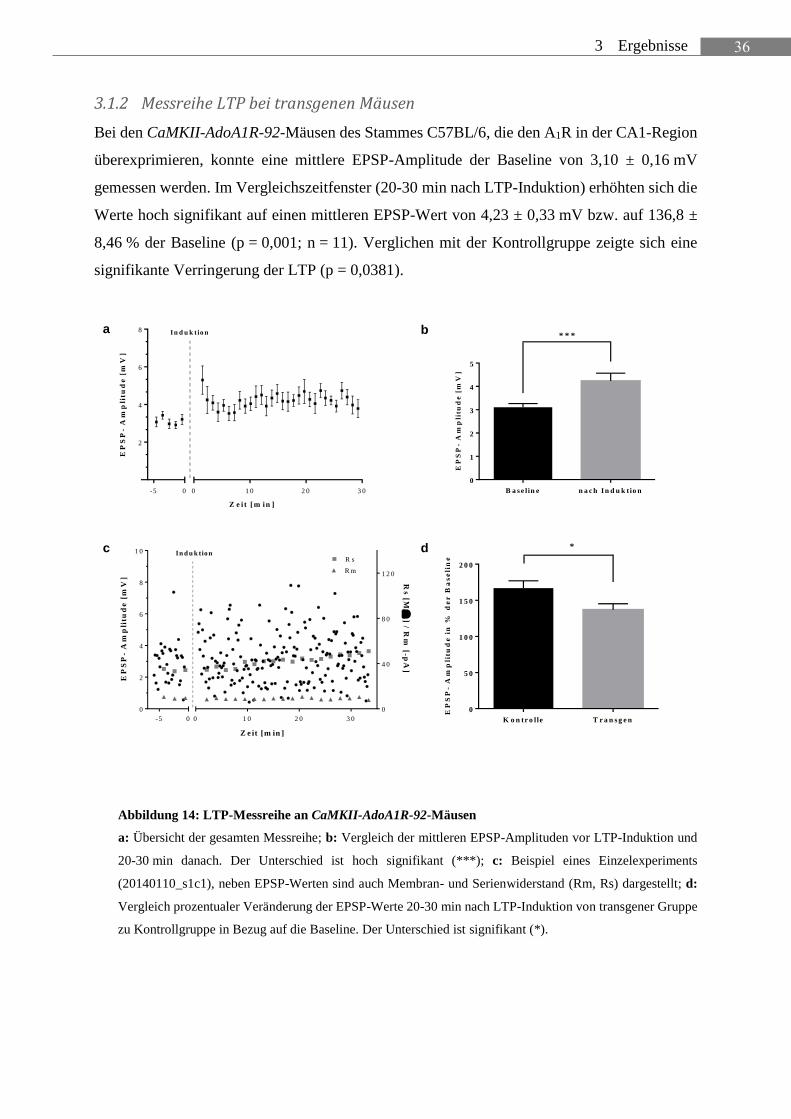
3.1.2 Messreihe LTP bei transgenen Mäusen
Bei den CaMKII-AdoA1R-92-Mäusen des Stammes C57BL/6, die den A1R in der CA1-Region
überexprimieren, konnte eine mittlere EPSP-Amplitude der Baseline von 3,10 ± 0,16 mV
gemessen werden. Im Vergleichszeitfenster (20-30 min nach LTP-Induktion) erhöhten sich die
Werte hoch signifikant auf einen mittleren EPSP-Wert von 4,23 ± 0,33 mV bzw. auf 136,8 ±
8,46 % der Baseline (p = 0,001; n = 11). Verglichen mit der Kontrollgruppe zeigte sich eine
signifikante Verringerung der LTP (p = 0,0381).
-5 0
2
4
6
8
0 1 0 2 0 3 0
Z e i t [ m in ]
EP
SP
- A
mp
litu
de
[m
V]
I n d u k t io na b
c
-5 0
0
2
4
6
8
1 0
0
4 0
8 0
1 2 0
0 1 0 2 0 3 0
R s
R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs
[M
] / Rm
[-pA
]
In d u k tiond
B a s e lin e n a c h I n d u k t io n
0
1
2
3
4
5
EP
SP
- A
mp
litu
de
[m
V]
* * *
K o n tr o lle T r a n s g e n
0
5 0
1 0 0
1 5 0
2 0 0
EP
SP
- A
mp
litu
de
in
% d
er
Ba
se
lin
e
*
Abbildung 14: LTP-Messreihe an CaMKII-AdoA1R-92-Mäusen
a: Übersicht der gesamten Messreihe; b: Vergleich der mittleren EPSP-Amplituden vor LTP-Induktion und
20-30 min danach. Der Unterschied ist hoch signifikant (***); c: Beispiel eines Einzelexperiments
(20140110_s1c1), neben EPSP-Werten sind auch Membran- und Serienwiderstand (Rm, Rs) dargestellt; d:
Vergleich prozentualer Veränderung der EPSP-Werte 20-30 min nach LTP-Induktion von transgener Gruppe
zu Kontrollgruppe in Bezug auf die Baseline. Der Unterschied ist signifikant (*).
37 3 Ergebnisse
3.2 Versuchsreihen mit Wistar-Ratten
3.2.1 Kontrollreihe LTP bei Wistar-Ratten
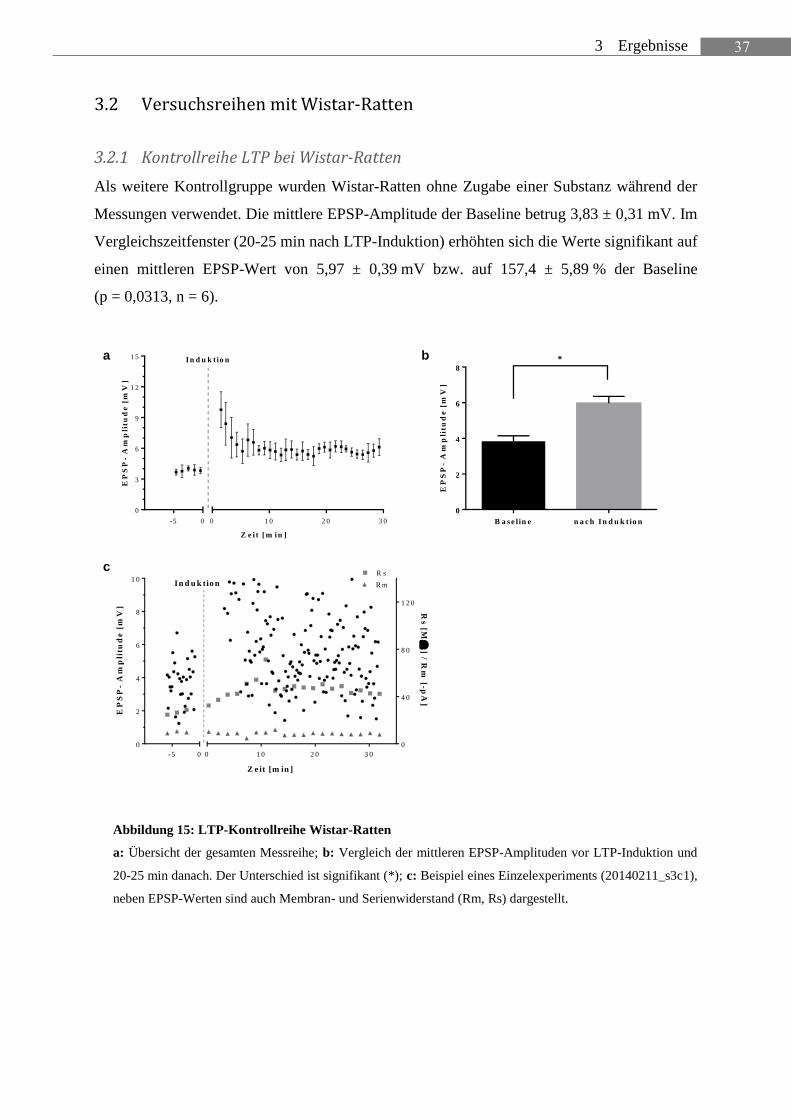
Als weitere Kontrollgruppe wurden Wistar-Ratten ohne Zugabe einer Substanz während der
Messungen verwendet. Die mittlere EPSP-Amplitude der Baseline betrug 3,83 ± 0,31 mV. Im
Vergleichszeitfenster (20-25 min nach LTP-Induktion) erhöhten sich die Werte signifikant auf
einen mittleren EPSP-Wert von 5,97 ± 0,39 mV bzw. auf 157,4 ± 5,89 % der Baseline
(p = 0,0313, n = 6).
-5 0
0
3
6
9
1 2
1 5
0 1 0 2 0 3 0
Z e i t [ m in ]
EP
SP
- A
mp
litu
de
[m
V]
I n d u k t io n
B a s e lin e n a c h I n d u k t io n
0
2
4
6
8
EP
SP
- A
mp
litu
de
[m
V]
*a b
c
-5 0
0
2
4
6
8
1 0
0
4 0
8 0
1 2 0
0 1 0 2 0 3 0
R s
R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs
[M
] / Rm
[-pA
]
I n d u k tio n
Abbildung 15: LTP-Kontrollreihe Wistar-Ratten
a: Übersicht der gesamten Messreihe; b: Vergleich der mittleren EPSP-Amplituden vor LTP-Induktion und
20-25 min danach. Der Unterschied ist signifikant (*); c: Beispiel eines Einzelexperiments (20140211_s3c1),
neben EPSP-Werten sind auch Membran- und Serienwiderstand (Rm, Rs) dargestellt.
38 3 Ergebnisse
3.2.2 Messreihe mit cCPA
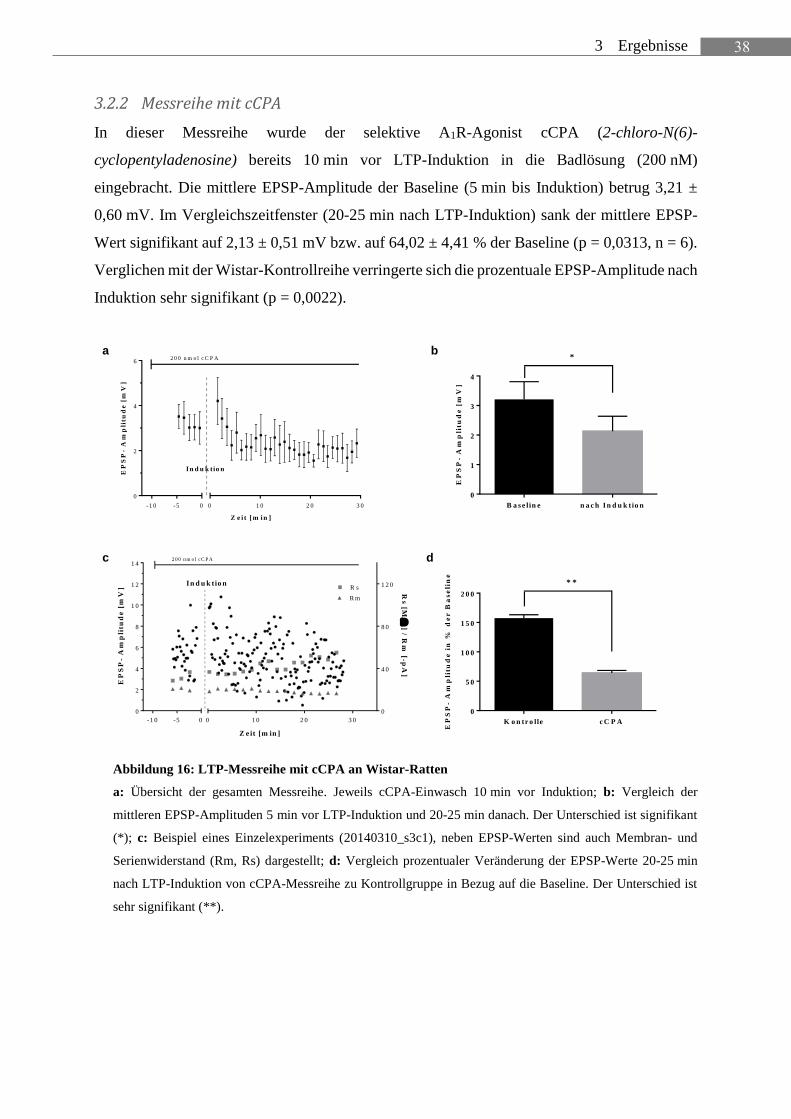
In dieser Messreihe wurde der selektive A1R-Agonist cCPA (2-chloro-N(6)-
cyclopentyladenosine) bereits 10 min vor LTP-Induktion in die Badlösung (200 nM)
eingebracht. Die mittlere EPSP-Amplitude der Baseline (5 min bis Induktion) betrug 3,21 ±
0,60 mV. Im Vergleichszeitfenster (20-25 min nach LTP-Induktion) sank der mittlere EPSP-
Wert signifikant auf 2,13 ± 0,51 mV bzw. auf 64,02 ± 4,41 % der Baseline (p = 0,0313, n = 6).
Verglichen mit der Wistar-Kontrollreihe verringerte sich die prozentuale EPSP-Amplitude nach
Induktion sehr signifikant (p = 0,0022).
-1 0 -5 0
0
2
4
6
0 1 0 2 0 3 0
Z e i t [ m in ]
EP
SP
- A
mp
litu
de
[m
V]
I n d u k t io n
2 0 0 n m o l c C P Aa b
c d
B a s e lin e n a c h I n d u k t io n
0
1
2
3
4
EP
SP
- A
mp
litu
de
[m
V]
*
K o n tr o lle c C P A
0
5 0
1 0 0
1 5 0
2 0 0
EP
SP
- A
mp
litu
de
in
% d
er
Ba
se
lin
e
* *
-1 0 -5 0
0
2
4
6
8
1 0
1 2
1 4
0
4 0
8 0
1 2 0
0 1 0 2 0 3 0
R s
R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs
[M
] / Rm
[-pA
]
I n d u k tio n
2 0 0 n m o l cC P A
Abbildung 16: LTP-Messreihe mit cCPA an Wistar-Ratten
a: Übersicht der gesamten Messreihe. Jeweils cCPA-Einwasch 10 min vor Induktion; b: Vergleich der
mittleren EPSP-Amplituden 5 min vor LTP-Induktion und 20-25 min danach. Der Unterschied ist signifikant
(*); c: Beispiel eines Einzelexperiments (20140310_s3c1), neben EPSP-Werten sind auch Membran- und
Serienwiderstand (Rm, Rs) dargestellt; d: Vergleich prozentualer Veränderung der EPSP-Werte 20-25 min
nach LTP-Induktion von cCPA-Messreihe zu Kontrollgruppe in Bezug auf die Baseline. Der Unterschied ist
sehr signifikant (**).
39 3 Ergebnisse
3.2.3 Messreihe mit DPCPX und cCPA
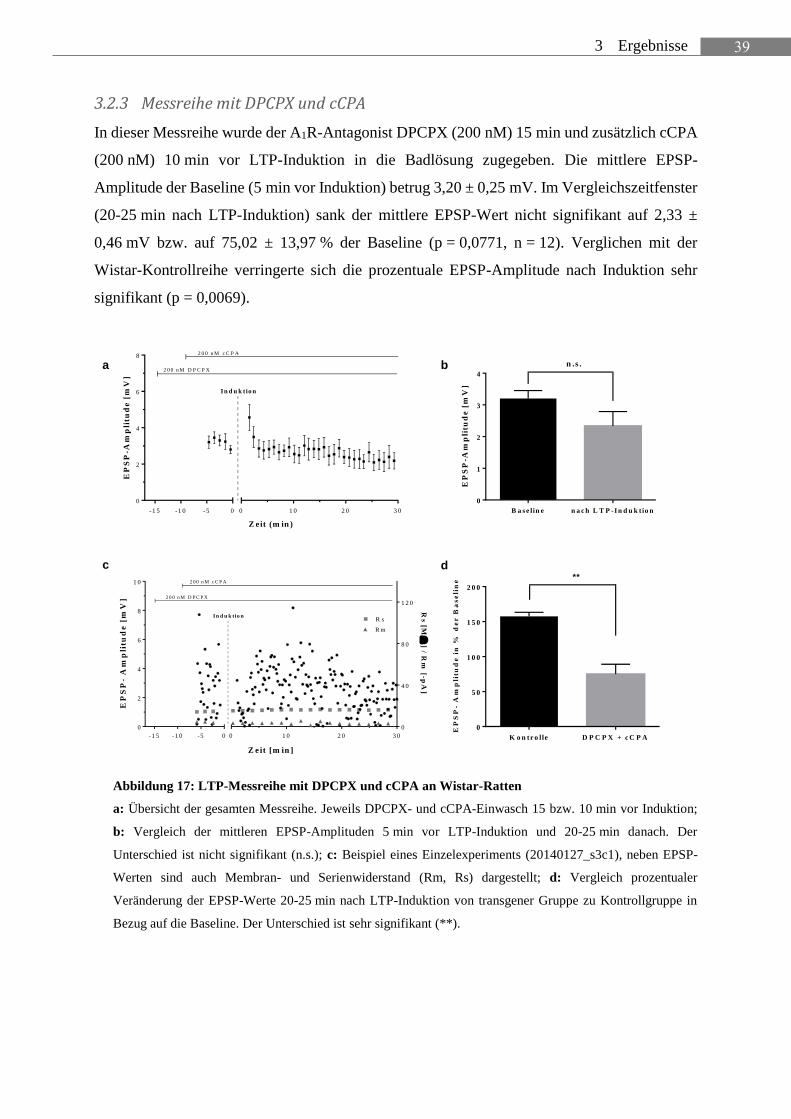
In dieser Messreihe wurde der A1R-Antagonist DPCPX (200 nM) 15 min und zusätzlich cCPA
(200 nM) 10 min vor LTP-Induktion in die Badlösung zugegeben. Die mittlere EPSP-
Amplitude der Baseline (5 min vor Induktion) betrug 3,20 ± 0,25 mV. Im Vergleichszeitfenster
(20-25 min nach LTP-Induktion) sank der mittlere EPSP-Wert nicht signifikant auf 2,33 ±
0,46 mV bzw. auf 75,02 ± 13,97 % der Baseline (p = 0,0771, n = 12). Verglichen mit der
Wistar-Kontrollreihe verringerte sich die prozentuale EPSP-Amplitude nach Induktion sehr
signifikant (p = 0,0069).
-1 5 -1 0 -5 0
0
2
4
6
8
0 1 0 2 0 3 0
Z eit (m in )
EP
SP
-Am
pli
tud
e [
mV
]
I n d u k t io n
2 0 0 n M c C P A
2 0 0 n M D P C P X
B a s e lin e n a c h L T P -I n d u k t io n
0
1
2
3
4
EP
SP
-Am
pli
tud
e [
mV
]
n .s .
K o n tr o lle D P C P X + c C P A
0
5 0
1 0 0
1 5 0
2 0 0
EP
SP
- A
mp
litu
de
in
% d
er
Ba
se
lin
e
**
a b
dc
-1 5 -1 0 -5 0
0
2
4
6
8
1 0
0 1 0 2 0 3 0
0
4 0
8 0
1 2 0
R s
R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs [M
] / R
m [-p
A]
I n d u k tio n
2 0 0 n M c C P A
2 0 0 n M D P C P X
Abbildung 17: LTP-Messreihe mit DPCPX und cCPA an Wistar-Ratten
a: Übersicht der gesamten Messreihe. Jeweils DPCPX- und cCPA-Einwasch 15 bzw. 10 min vor Induktion;
b: Vergleich der mittleren EPSP-Amplituden 5 min vor LTP-Induktion und 20-25 min danach. Der
Unterschied ist nicht signifikant (n.s.); c: Beispiel eines Einzelexperiments (20140127_s3c1), neben EPSP-
Werten sind auch Membran- und Serienwiderstand (Rm, Rs) dargestellt; d: Vergleich prozentualer
Veränderung der EPSP-Werte 20-25 min nach LTP-Induktion von transgener Gruppe zu Kontrollgruppe in
Bezug auf die Baseline. Der Unterschied ist sehr signifikant (**).
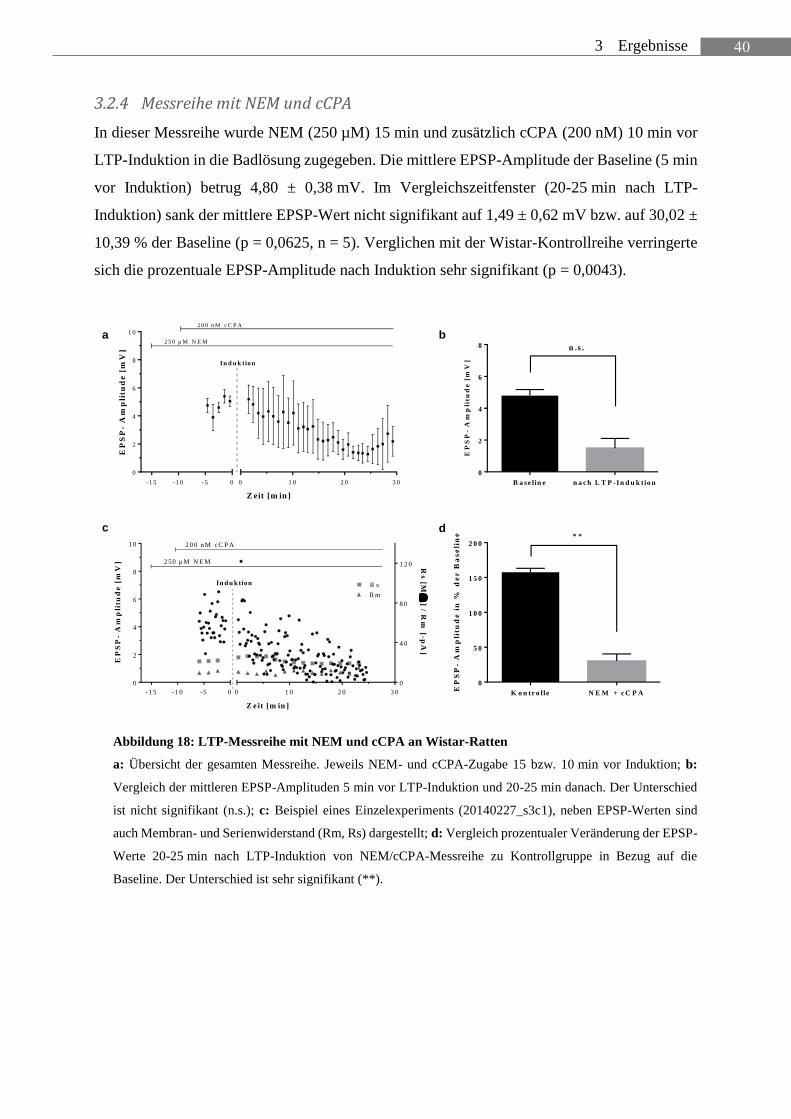
40 3 Ergebnisse
3.2.4 Messreihe mit NEM und cCPA
In dieser Messreihe wurde NEM (250 µM) 15 min und zusätzlich cCPA (200 nM) 10 min vor
LTP-Induktion in die Badlösung zugegeben. Die mittlere EPSP-Amplitude der Baseline (5 min
vor Induktion) betrug 4,80 ± 0,38 mV. Im Vergleichszeitfenster (20-25 min nach LTP-
Induktion) sank der mittlere EPSP-Wert nicht signifikant auf 1,49 ± 0,62 mV bzw. auf 30,02 ±
10,39 % der Baseline (p = 0,0625, n = 5). Verglichen mit der Wistar-Kontrollreihe verringerte
sich die prozentuale EPSP-Amplitude nach Induktion sehr signifikant (p = 0,0043).
-1 5 -1 0 -5 0
0
2
4
6
8
1 0
0 1 0 2 0 3 0
Z eit [m in ]
EP
SP
- A
mp
litu
de
[m
V]
I n d u k t io n
2 0 0 n M c C P A
2 5 0 µ M N E M
K o n tr o lle N E M + c C P A
0
5 0
1 0 0
1 5 0
2 0 0
EP
SP
- A
mp
litu
de
in
% d
er
Ba
se
lin
e * *
B a s e lin e n a c h L T P -I n d u k t io n
0
2
4
6
8
EP
SP
- A
mp
litu
de
[m
V]
n .s .
a b
dc
-1 5 -1 0 -5 0
0
2
4
6
8
1 0
0
4 0
8 0
1 2 0
0 1 0 2 0 3 0
R s
R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs [M
] / R
m [-p
A]
In d u k tion
2 0 0 n M cC P A
2 50 µ M N E M
Abbildung 18: LTP-Messreihe mit NEM und cCPA an Wistar-Ratten
a: Übersicht der gesamten Messreihe. Jeweils NEM- und cCPA-Zugabe 15 bzw. 10 min vor Induktion; b:
Vergleich der mittleren EPSP-Amplituden 5 min vor LTP-Induktion und 20-25 min danach. Der Unterschied
ist nicht signifikant (n.s.); c: Beispiel eines Einzelexperiments (20140227_s3c1), neben EPSP-Werten sind
auch Membran- und Serienwiderstand (Rm, Rs) dargestellt; d: Vergleich prozentualer Veränderung der EPSP-
Werte 20-25 min nach LTP-Induktion von NEM/cCPA-Messreihe zu Kontrollgruppe in Bezug auf die
Baseline. Der Unterschied ist sehr signifikant (**).

41 3 Ergebnisse
3.2.5 Messreihe mit CGS
In dieser Messreihe wurde der A2A-Rezeptoragonist CGS-21680 (200 nM) 10 min vor LTP-
Induktion in die Badlösung zugegeben. Die mittlere EPSP-Amplitude der Baseline (5 min vor
Induktion) betrug 3,62 ± 0,32 mV. Im Vergleichszeitfenster (20-25 min nach LTP-Induktion)
sank der mittlere EPSP-Wert nicht signifikant auf 2,38 ± 0,71 mV bzw. auf 72,36 ± 23,01 %
der Baseline (p = 0,3215, n = 6). Verglichen mit der Wistar-Kontrollreihe verringerte sich die
prozentuale EPSP-Amplitude nach Induktion sehr signifikant (p = 0,0087).
-1 0 -5 0
0
2
4
6
8
0 1 0 2 0 3 0
Z e i t [ m in ]
EP
SP
- A
mp
litu
de
[m
V]
I n d u k t io n
2 0 0 n m o l C G S
B a s e lin e n a c h I n d u k t io n
0
1
2
3
4
5
EP
SP
- A
mp
litu
de
[m
V]
n .s .
K o n tr o lle C G S
0
5 0
1 0 0
1 5 0
2 0 0
EP
SP
- A
mp
litu
de
in
% d
er
Ba
se
lin
e **
a b
c d
-1 0 -5 0
0
2
4
6
8
1 0
0
4 0
8 0
1 2 0
0 1 0 2 0 3 0
R s
R m
Z e it [m in ]
EP
SP
- A
mp
litu
de
[m
V]
Rs [M
] / R
m [-p
A]
2 0 0 n m o l C G S
In d u k tion
Abbildung 19: LTP-Messreihe mit CGS an Wistar-Ratten
a: Übersicht der gesamten Messreihe. Jeweils CGS-Einwasch 10 min vor Induktion; b: Vergleich der mittleren
EPSP-Amplituden 5 min vor LTP-Induktion und 20-25 min danach. Der Unterschied ist nicht signifikant (n.s.);
c: Beispiel eines Einzelexperiments (20140304_s2c1), neben EPSP-Werten sind auch Membran- und
Serienwiderstand (Rm, Rs) dargestellt; d: Vergleich prozentualer Veränderung der EPSP-Werte 20-25 min
nach LTP-Induktion von CGS-Messreihe zu Kontrollgruppe in Bezug auf die Baseline. Der Unterschied ist
sehr signifikant (**).
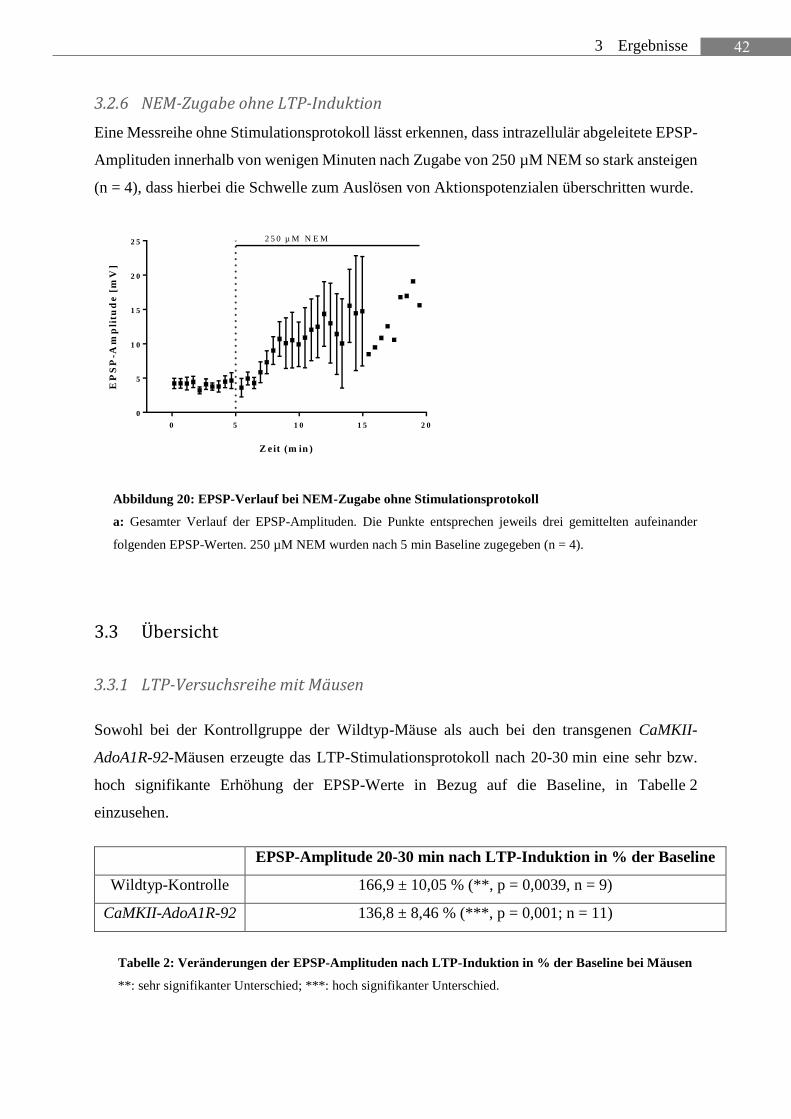
42 3 Ergebnisse
3.2.6 NEM-Zugabe ohne LTP-Induktion
Eine Messreihe ohne Stimulationsprotokoll lässt erkennen, dass intrazellulär abgeleitete EPSP-
Amplituden innerhalb von wenigen Minuten nach Zugabe von 250 µM NEM so stark ansteigen
(n = 4), dass hierbei die Schwelle zum Auslösen von Aktionspotenzialen überschritten wurde.
0 5 1 0 1 5 2 0
0
5
1 0
1 5
2 0
2 5
Z eit (m in )
EP
SP
-Am
pli
tud
e [
mV
]
2 5 0 µ M N E M
Abbildung 20: EPSP-Verlauf bei NEM-Zugabe ohne Stimulationsprotokoll
a: Gesamter Verlauf der EPSP-Amplituden. Die Punkte entsprechen jeweils drei gemittelten aufeinander
folgenden EPSP-Werten. 250 µM NEM wurden nach 5 min Baseline zugegeben (n = 4).
3.3 Übersicht
3.3.1 LTP-Versuchsreihe mit Mäusen
Sowohl bei der Kontrollgruppe der Wildtyp-Mäuse als auch bei den transgenen CaMKII-
AdoA1R-92-Mäusen erzeugte das LTP-Stimulationsprotokoll nach 20-30 min eine sehr bzw.
hoch signifikante Erhöhung der EPSP-Werte in Bezug auf die Baseline, in Tabelle 2
einzusehen.
EPSP-Amplitude 20-30 min nach LTP-Induktion in % der Baseline
Wildtyp-Kontrolle 166,9 ± 10,05 % (**, p = 0,0039, n = 9)
CaMKII-AdoA1R-92 136,8 ± 8,46 % (***, p = 0,001; n = 11)
Tabelle 2: Veränderungen der EPSP-Amplituden nach LTP-Induktion in % der Baseline bei Mäusen
**: sehr signifikanter Unterschied; ***: hoch signifikanter Unterschied.

43 3 Ergebnisse
Verglichen mit der Kontrollgruppe zeigte sich eine signifikante Verringerung der LTP bei den
transgenen Tieren (p = 0,0381). Abbildung 14d in Kapitel 3.1.2 verdeutlicht dies grafisch.
3.3.2 LTP-Versuchsreihe mit Wistar-Ratten
In der Wistar-Kontrollgruppe konnte eine LTP mit signifikantem Unterschied der EPSP-Werte
20-25 min nach LTP-Induktion in Bezug auf die Baseline erreicht werden.
Trotz LTP-Stimulationsprotokoll verringerte sich die EPSP-Amplitude signifikant bei
Messungen mit cCPA. In den Messreihen mit DPCPX + cCPA, NEM + cCPA sowie CGS
konnten keine signifikanten Unterschiede festgestellt werden, obwohl sich tendenziell
Verringerungen der EPSP-Amplituden abzeichneten. Tabelle 3 gibt dazu eine Übersicht.
EPSP-Amplitude 20-25 min nach LTP-Induktion in % der Baseline
Wistar-Kontrolle 157,4 ± 5,89 % (*, p = 0,0313, n = 6)
cCPA 64,02 ± 4,41 % (*, p = 0,0313, n = 6)
DPCPX + cCPA 75,02 ± 13,97 % (n.s., p = 0,0771, n = 12)
NEM + cCPA 30,02 ± 10,39 % (n.s., p = 0,0625, n = 5)
CGS 72,36 ± 23,01 % (n.s., p = 0,3215, n = 6)
Tabelle 3: Veränderungen der EPSP-Amplituden nach LTP-Induktion in % der Baseline bei Ratten
Unterschiede: n.s.: nicht signifikant; *: signifikant
Im paarweisen Vergleich der normalisierten EPSP-Amplituden nach Induktion mittels Mann-
Whitney-U-Test unterscheiden sich die Messreihen mit cCPA (p = 0,0022), DPCPX + cCPA
(p = 0,0069), NEM + cCPA (p = 0,0043) und CGS (p = 0,0087) jeweils sehr signifikant von
der Kontrollgruppe.
Im Vergleich der drei Messreihen mit cCPA, DPCPX + cCPA und NEM + cCPA im
Zeitintervall 20-25 min nach Induktion untereinander ergibt sich mit dem Kruskal-Wallis-Test
und Dunns multiplem Vergleichstest kein signifikanter Unterschied (p = 0,0819).
Die folgende Abbildung 22 veranschaulicht diese Ergebnisse.
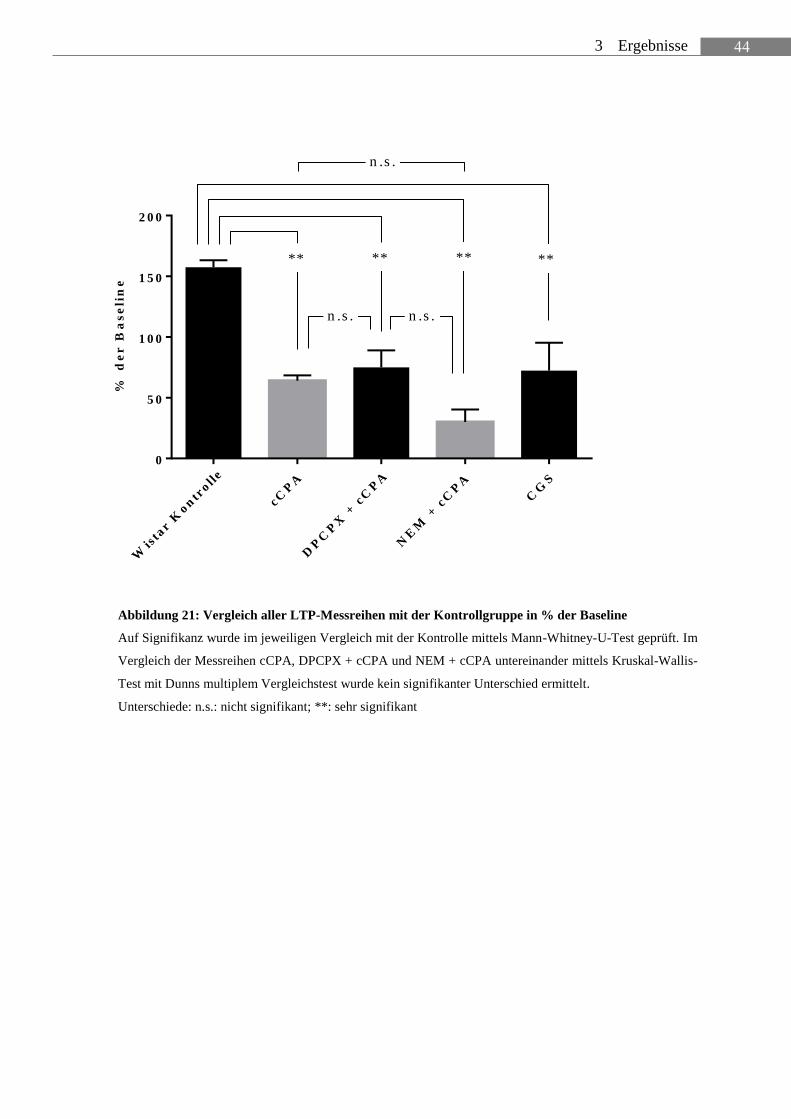
44 3 Ergebnisse
Wis
tar K
on
troll
e
cCP
A
DP
CP
X +
cC
PA
NE
M +
cC
PA
CG
S
0
5 0
1 0 0
1 5 0
2 0 0
% d
er
Ba
se
lin
e
** **** **
n .s .
n .s . n .s .
Abbildung 21: Vergleich aller LTP-Messreihen mit der Kontrollgruppe in % der Baseline
Auf Signifikanz wurde im jeweiligen Vergleich mit der Kontrolle mittels Mann-Whitney-U-Test geprüft. Im
Vergleich der Messreihen cCPA, DPCPX + cCPA und NEM + cCPA untereinander mittels Kruskal-Wallis-
Test mit Dunns multiplem Vergleichstest wurde kein signifikanter Unterschied ermittelt.
Unterschiede: n.s.: nicht signifikant; **: sehr signifikant

45 4 Diskussion
4 Diskussion
4.1 Patch-Clamp-Technik an Hirnschnitten
Entgegen vieler Vorteile der Patch-Clamp-Technik, wie z.B. intrazelluläre physiologische
Messungen an einzelnen Zellen in vitro durchführen zu können, resultieren einige Störfaktoren
aus dem komplexen und sensiblen methodischen Vorgehen.
Trotz vorsichtiger Präparation des Gehirns und der Anfertigung der Hirnschnitte werden
Neurone zwangsläufig einem unnatürlichen, mechanischen und hypoxischen Stress ausgesetzt,
der die weiteren Experimente verfälschen kann (Hershkowitz et al. 1993; Kirov et al. 1999;
Taubenfeld et al. 2002). Dadurch kann sich u.a. der Adenosinspiegel unkalkulierbar im
Schnittgewebe erhöhen, worauf Neurone in Inkubation jedoch mit rascher Regeneration und
beschleunigter Desensibilisierung der A1R antworten (Winn et al. 1981; Fowler; Fowler 1993;
Frenguelli et al. 2003; Zur Nedden et al. 2011). Auch wenn durch die Isolation des
Hippocampusschnitts afferente (und efferente) synaptische Verbindungen zu außerhalb gelegen
Hirnregionen gekappt werden, verbleiben die zu untersuchenden Zellen in ihrem engeren
physiologischem Zellverband.
Obgleich in der Whole-cell-Konfiguration bestmögliche physiologische Lösungs-
zusammensetzungen in Pipetten verwendet wurden, konnten Elektrolytverschiebungen
schlussendlich nicht vermieden werden. Dasselbe gilt für aCSF als osmotisch wirksame
Nährlösung. Zusätzlich zu einer optischen Vorauswahl unter dem Mikroskop unterstützt die
Messung von Serienwiderstand und Ruhemembranpotenzial die Kontrolle der Zellqualität.
Weitere mögliche Störfaktoren sind Erschütterungen des Messplatzes, fehlerhafte Erdung
leitender Oberflächen, elektrische Ströme durch benachbarte Geräte, Verschleißerscheinungen
der Silberdrahtelektrode in Mess- und Stimulationspipette, unterschiedliche Wanddicke und
Öffnungswiderstand der Pipetten, die allesamt konstante Messergebnisse manipulieren können.
Nagetiere wie Mäuse und Ratten sind nur eingeschränkt als Modell der vorliegenden
Experimente auf den Menschen übertragbar, da die genetische Ähnlichkeit der
Adenosinrezeptor-Subtypen zwar stark, dennoch nicht identisch ist (Fredholm et al. 2000).

46 4 Diskussion
4.2 LTP-Kontrollreihen an Mäusen und Ratten
Als Referenz für die Versuche mit transgenen Mäusen und Stoffzugaben bei Ratten in den
beiden Kontrollgruppen, konnte jeweils eine – im Vergleich zu anderen Experimentalserien der
Arbeitsgruppe – schwach ausgeprägte, dennoch stabile und langanhaltende LTP gemessen
werden. So unterschied sich die LTP der Wildtyp-Mäuse des Stammes C57BL/6 mit 166,9 ±
10,05 % sehr signifikant von der Baseline. Bei der Kontrollgruppe der Wistar-Ratten erhöhte
sich die Baseline signifikant auf eine LTP mit 157,4 ± 5,89 %.
Bisher wurde mit einem identischen Theta-Burst-Stimulationsprotokoll (siehe Kapitel 2.7) in
derselben Arbeitsgruppe assoziative LTP an Wistar-Ratten in Höhe von 279,5 ± 22,2 % (Wolff
2007), 168.9 ± 3.22 % (Holderbach 2006), 214,7 ± 34,18 % (Seifert 2009) induziert. In einer
anderen, am selben Messstand stattfindenden Arbeit lieferte die Wistar-Kontrollreihe eine
vergleichbar geringer ausfallende Erhöhung der LTP auf 179 % (Ehrenberger, noch nicht
veröffentlicht). Ein reduziertes Stimulationsprotokoll mit nur 25 Reizpaaren lieferte dennoch
eine LTP von 182,10 ± 22,79 % (Dratz 2013).
An Mäusen des Stammes C57BL/6 wurden höhere LTP-Niveaus von 234,1 ± 51,44 %
(Landmann 2015) und 225,6 ± 31,22 % (Stolz 2015) gemessen.
Eine Ursache der relativ niedrig gemessenen LTP in den Kontrollreihen von Mäusen und Ratten
könnte an der gemeinsamen Nutzung des Schlauchsystems liegen, über das die Testsubstanzen
zugeführt wurden. Diesem Problem wurde durch regelmäßige Spülungen mit destilliertem
Wasser nach jedem Arbeitstag entgegengewirkt. Eventuell hätte die ca. 5-minütige Spülung
zwischen den einzelnen Messungen verlängert werden müssen, insbesondere wenn vorher mit
cCPA gemessen wurde.
Die in Kapitel 4.1 beschriebenen, sehr fehleranfälligen Versuchsbedingungen müssen ebenso
als mögliche Erklärung für die schwach ausgeprägten LTP-Messungen in Betracht gezogen
werden.
Des Weiteren sollte beachtet werden, dass in den vorliegenden Experimenten bei Mäusen ein
Zeitintervall von 20-30 min nach Induktion mit der Baseline verglichen wurde; bei Wistar-
Ratten das Intervall von 20-25 min nach Induktion – der besseren Vergleichbarkeit geschuldet,
da ein Teil der Messreihen mit Substanzen nicht über 25 min hinaus gemessen wurde.

47 4 Diskussion
Gegenüber einem Deckeneffekt bei einer sehr hohen LTP erweist sich eine niedrigere LTP
vorteilig, sofern LTP-verstärkende Substanzen verwendet werden.
Zusammenfassend stellen die signifikanten Erhöhungen im Vergleich zur Baseline dennoch
langanhaltende und stabile LTP-Messungen dar, sodass diese als Referenz für die weiteren
Experimente herangezogen werden können.
4.3 LTP-Modulation durch A1R-Überexprimierung
Aus dem signifikanten Unterschied der LTP von 166,9 ± 10,05 % in der WT-Kontrolle zu 136,8
± 8,46 % in der transgenen A1R -Maus kann man schließen, dass endogenes Adenosin über den
postsynaptischen A1R an der CA3/CA1-Synapse des Hippocampus der Maus assoziative LTP
abschwächt.
Dieses Ergebnis bestärkt grundsätzlich die Ergebnisse anderer Arbeiten, welche eine
verminderte LTP mit der Aktivierung des A1R in Zusammenhang bringen, auch wenn andere
Versuchsbedingungen, beispielsweise Rezeptoragonisten oder -antagonisten, nicht-assoziative
Stimulationsprotokolle, extrazelluläre Ableitungen, andere Spezies unterschiedlichen Alters
oder andere Lokalisationen angewandt wurden (Larson et al. 1993; Mendonça und Ribeiro
1994; Forghani und Krnjevic 1995; Abraham und Huggett 1997; Rex et al. 2005).
Die in Kapitel 4.4 diskutierten Messergebnisse mit cCPA bestätigen zusätzlich die Wirkung des
A1R auf die LTP.
Mit einer A1R-Überexprimierung in der CA3-Region, vereinzelt auch in der CA1-Region,
konnte Stolz (2015) mit einem vergleichbaren Versuchsaufbau eine Reduktion der LTP auf
46,61 ± 11,70 % (p = 0,125; n = 5) in Bezug auf die Baseline messen – eine hoch signifikante
Absenkung zu dessen LTP-Kontrollreihe (p = 0,0043).
Betrachtet man die Wirkung der erhöhten Expression von prä- (CA3) und postsynaptischen
(CA1) A1R auf die LTP bei Mäusen, scheint Adenosin stärker an der Präsynapse zu wirken,
wenngleich der quantitative Vergleich von zwei unterschiedlichen Arbeiten als kritisch zu
bewerten ist.
Ob der hemmende Einfluss von Adenosin über A1R auf die präsynaptische
Transmitterfreisetzung mittels Blockierung von Ca2+-Kanälen größer ist als auf die
postsynaptische Hemmung von NMDA- und AMPA-Rezeptoren, bleibt weiterhin offen. Einige
Arbeiten haben die präsynaptische Hemmung via A1R bereits hervorgehoben (Dunwiddie und

48 4 Diskussion
Haas 1985; Wu und Saggau 1994). Der Mechanismus, über den A1R Ca2+-Kanäle hemmen,
bleibt weiterhin unklar, wobei im Nucleus accumbens G-Proteine daran beteiligt zu sein
scheinen (Chieng und Bekkers 2001). Gundlfinger et al. (2007) führten diesen präsynaptischen
molekularen Mechanismus an der Moosfaser/CA3-Synapse auf eine Hemmung
spannungsabhängiger Ca2+-Kanäle zurück.
Womöglich besteht ein Unterschied prä- oder postsynaptischer Hemmung aber auch aufgrund
einer alternierenden Expression des A1R oder aufgrund divergierender endogener
Adenosinkonzentrationen innnerhalb der Hippocampusregionen (Zur Nedden et al. 2011; Trieu
et al. 2015).
Grundsätzlich muss dennoch davon ausgegangen werden, dass die Aktivierung des A1R sowohl
prä- als auch postsynaptisch (CA3/CA1) zu einer effektiven Hemmung der assoziativen LTP
führt.
4.4 LTP-Modulation durch cCPA
In der Messreihe mit dem selektiven A1R-Agonist cCPA (200 nM) wurde eine signifikante
LTP-Reduktion auf 64,02 ± 4,41 % der Baseline 20-25 min nach LTP-Induktion gemessen. Es
besteht ein sehr signifikanter Unterschied zur LTP der Wistar-Kontrollgruppe.
In der vorliegenden Messreihe wurde cCPA (200 nM) bereits 10 min vor LTP-Induktion in die
Badlösung eingebracht, wobei nach den ersten 5 min die Baseline korrigiert wurde.
Aus einer von Julia Knoll durchgeführten Messreihe (noch nicht veröffentlicht) ohne
Stimulationsprotokoll geht hervor, dass intrazellulär abgeleitete EPSP-Amplituden ca. 5 min
nach Zugabe von 200 nM cCPA stetig abfallen. Abbildung 22 veraunschaulicht dies.
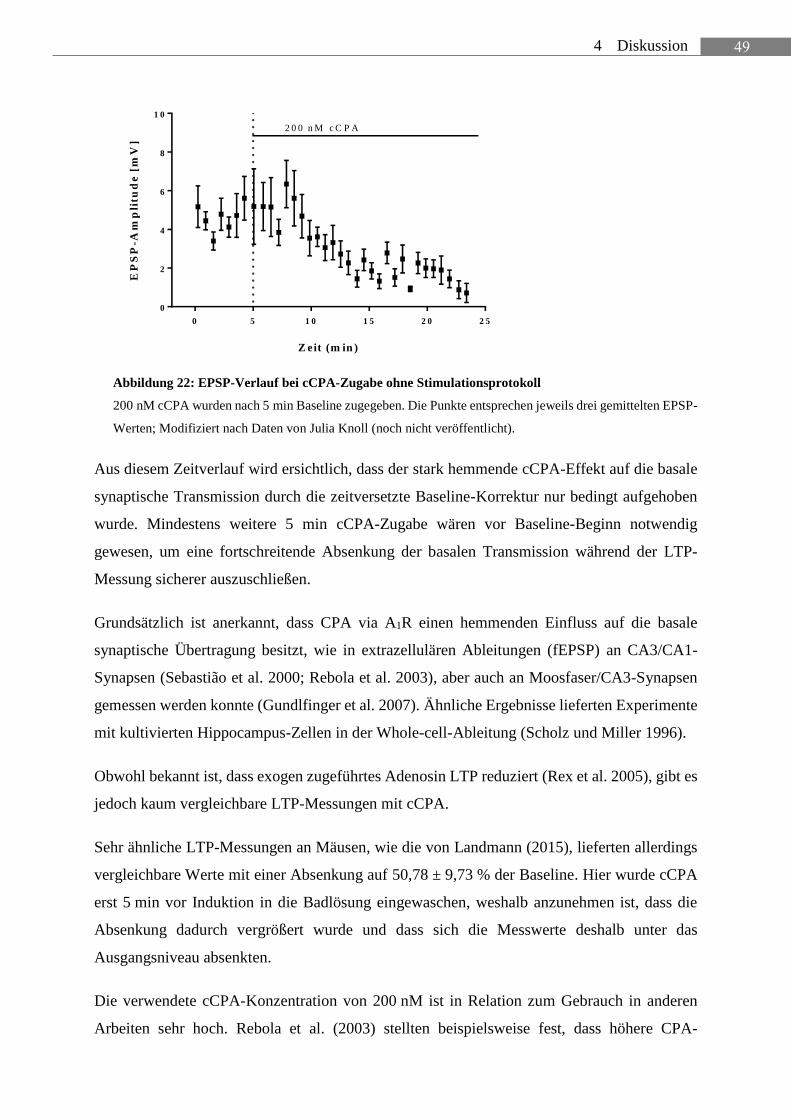
49 4 Diskussion
0 5 1 0 1 5 2 0 2 5
0
2
4
6
8
1 0
Z eit (m in )
EP
SP
-Am
pli
tud
e [
mV
]2 0 0 n M c C P A
Abbildung 22: EPSP-Verlauf bei cCPA-Zugabe ohne Stimulationsprotokoll
200 nM cCPA wurden nach 5 min Baseline zugegeben. Die Punkte entsprechen jeweils drei gemittelten EPSP-
Werten; Modifiziert nach Daten von Julia Knoll (noch nicht veröffentlicht).
Aus diesem Zeitverlauf wird ersichtlich, dass der stark hemmende cCPA-Effekt auf die basale
synaptische Transmission durch die zeitversetzte Baseline-Korrektur nur bedingt aufgehoben
wurde. Mindestens weitere 5 min cCPA-Zugabe wären vor Baseline-Beginn notwendig
gewesen, um eine fortschreitende Absenkung der basalen Transmission während der LTP-
Messung sicherer auszuschließen.
Grundsätzlich ist anerkannt, dass CPA via A1R einen hemmenden Einfluss auf die basale
synaptische Übertragung besitzt, wie in extrazellulären Ableitungen (fEPSP) an CA3/CA1-
Synapsen (Sebastião et al. 2000; Rebola et al. 2003), aber auch an Moosfaser/CA3-Synapsen
gemessen werden konnte (Gundlfinger et al. 2007). Ähnliche Ergebnisse lieferten Experimente
mit kultivierten Hippocampus-Zellen in der Whole-cell-Ableitung (Scholz und Miller 1996).
Obwohl bekannt ist, dass exogen zugeführtes Adenosin LTP reduziert (Rex et al. 2005), gibt es
jedoch kaum vergleichbare LTP-Messungen mit cCPA.
Sehr ähnliche LTP-Messungen an Mäusen, wie die von Landmann (2015), lieferten allerdings
vergleichbare Werte mit einer Absenkung auf 50,78 ± 9,73 % der Baseline. Hier wurde cCPA
erst 5 min vor Induktion in die Badlösung eingewaschen, weshalb anzunehmen ist, dass die
Absenkung dadurch vergrößert wurde und dass sich die Messwerte deshalb unter das
Ausgangsniveau absenkten.
Die verwendete cCPA-Konzentration von 200 nM ist in Relation zum Gebrauch in anderen
Arbeiten sehr hoch. Rebola et al. (2003) stellten beispielsweise fest, dass höhere CPA-

50 4 Diskussion
Konzentrationen als 100 nM keine größere Suppression der fEPSP hervorrufen. Dies könnte
erklären, weshalb EPSP-Werte nach LTP-Induktion sehr stark unter das Baseline-Level
abfallen. Zusammenfassend kann davon ausgegangen werden, dass die Aktivierung von A1R
eine assoziative LTP verhindert und sogar eine ausgeprägte Umkehrung hervorrufen kann.
4.5 LTP-Modulation durch DPCPX + cCPA
In dieser Messreihe mit Einwasch des A1R-Antagonist DPCPX (200 nM) 15 min und zusätzlich
cCPA (200 nM) 10 min vor LTP-Induktion sank der mittlere EPSP-Wert nicht signifikant auf
75,02 ± 13,97 % der Baseline. Der Unterschied zu der Wistar-Kontrollreihe ist sehr signifikant
(p = 0,0069).
Damit konnte gezeigt werden, dass eine LTP ausbleibt, wenn die Wirkung des A1R-
Antagonisten DPCPX durch den zeitversetzt applizierten A1R-Agonisten cCPA aufgehoben
wird. Im Vergleich zu der Messreihe mit ausschließlich cCPA zeigt sich im Kruskal-Wallis-
Test kein signifikanter Unterschied.
Man kann davon ausgehen, dass DPCPX die tonische A1R-vermittelte Adenosin-Wirkung auf
die LTP demaskiert, bzw. eine LTP dadurch erhalten bleibt (Forghani und Krnjevic 1995;
Rebola et al. 2003; Rex et al. 2005; Stolz 2015). Messungen ohne LTP-Stimulation lieferten
dosisabhängige Resultate für die Veränderung der basalen synaptischen Transmission. In einer
Studie von Caruana et al. (2012) führte die Applikation von 10 nM DPCPX bei CA3- und CA1-
Pyramidenzellen zu keiner, bei CA2-Neuronen hingegen zu einer starken Erhöhung der fEPSP.
Erst höhere Konzentrationen von 50 nM (Forghani und Krnjevic 1995; Rebola et al. 2003) und
500 nM DPCPX (Rex et al. 2005; Trieu et al. 2015) schienen fEPSP-Werte an der CA3/CA1-
Synapse zu erhöhen. Derselbe Effekt konnte an der Moosfaser/CA3-Synapse beobachtet
werden (Gundlfinger et al. 2007).
In der vorliegenden Messreihe erfolgt der Einwasch von DPCPX 15 min vor LTP-Induktion
daher womöglich zu spät, um eine Verstärkung der basalen Übertragung während der Messung
komplett auszuschließen.
Wie im vorigen Kapitel bereits erläutert, wirkt cCPA entgegensetzt zu DPCPX hemmend auf
die basale Übertragungsstärke.
Der Einwasch von cCPA in Kombination mit DPCPX invertierte die LTP beinahe ebenso stark
wie der alleinige cCPA-Einwasch. Aufgrund ähnlich hoher Bindungsaffinitäten beider

51 4 Diskussion
Liganden, aber wesentlich höheren Selektivität von cCPA für den A1R ist denkbar, dass cCPA
mit einem Ki-Wert von 0,4 nM (0,2-0,7 nM) in der Lage ist, DPCPX (Ki-Wert: 0,3 nM;
0,49 ± 0,06 nM) innerhalb von Minuten an den Rezeptorbindungsstellen zumindest teilweise
zu verdrängen (Lohse et al. 1988; Suzuki et al. 1992). Bindungsaffinität und Selektivität
genannter A1R-Liganden bei Ratten sind beim Menschen geringer ausgeprägt und daher nicht
übertragbar (Wilson und Mustafa 2009).
Eine Arbeit konnte zeigen, dass DPCPX mit 1 µM imstande ist, den hemmenden Effekt von
CPA mit 100 nM auf eine EPSP-Messung in der Substantia nigra von Ratten komplett
aufzuheben (Shen und Johnson 1997). In anderen Experimenten zeigte sich, dass DPCPX die
Wirkung von CPA bei Konzentrationen von 50 nM vs. 30 nM bzw. 200 nM vs. 1 µM bei
gealterten Ratten blockieren konnte (Sebastião et al. 2000; Rebola et al. 2003). Berücksichtigt
man die höhere Affinität von cCPA für den A1R im Verlgeich zu CPA, ist die ohnehin schwach
ausgeprägte Fähigkeit von DPCPX, CPA zu verdrängen, in den vorliegenden Experimenten mit
cCPA noch verminderter zu erwarten (Fredholm et al. 2001a).
Es bleibt festzuhalten, dass cCPA in der vorliegenden Konzentration einen stark hemmenden
Einfluss auf die LTP besitzt, der durch vorherige Applikation von DPCPX in wahrscheinlich
zu geringer Konzentration bzw. mit schwächerer Bindungsaffinität/Selektivität nicht
aufzuheben ist. Dies verdeutlicht dennoch die Abhängigkeit der assoziativen LTP von der A1R-
Aktivität.
4.6 LTP-Modulation durch NEM + cCPA
Während des Einwaschs von N-Ethylmaleimide (NEM) und dem Einstellen der Baseline wurde
ein Anstieg der EPSP-Amplituden verzeichnet, der in gesonderten Experimenten ohne LTP-
Stimulation veranschaulicht wurde (vgl. Kapitel 3.2.7). Innerhalb weniger Minuten konnten
massiv verstärkte EPSP-Amplituden an der CA3/CA1-Synapse beobachtet werden. Eine
vergleichbare Erhöhung mit Peak nach ca. 10 min der fEPSP bei extrazellulär beigemengtem
NEM (200 µM) konnte an der Moosfaser/CA3-Synapse nachgewiesen werden (Moore et al.
2003).
In einer LTP-Messreihe mit Zugabe von NEM (15 min vor LTP-Induktion) und cCPA (10 min
vor LTP-Induktion) wurde die Baseline-Messung bei stabilen EPSP-Werten erst 5 min vor
LTP-Induktion begonnen.

52 4 Diskussion
Statt einer LTP-Erniedrigung mit cCPA alleine auf 64,02 ± 4,41 % der Baseline, wurde ein
noch geringerer LTP-Verlauf von 30,02 ± 10,39 % mit NEM und cCPA gemessen. Allerdings
ist der Unterschied beider Messreihen im Kruskal-Wallis-Test mit Dunn’s multiplem
Vergleichstest nicht signifikant.
Hieraus wird ersichtlich, dass NEM nicht in der Lage ist, den hemmenden Effekt des durch
cCPA agonisierten A1R nach LTP-Stimulation aufzuheben. Womöglich hemmt NEM die LTP
ebenso und führt dadurch zu einem tendenziellen Summeneffekt mit cCPA.
NEM ist imstande, den A1R von dem zugehörigen Gi/o-Protein zu entkoppeln, sodass dessen
vorranging präsynaptische tonische Hemmung der Transmitterfreisetzung durch endogenes
Adenosin aufgehoben wird (Nakajima 1990; Shapiro 1994; Jeong 2002). Unsicher bleibt, ob
NEM über den SNARE-Komplex (siehe unten) auch die astrozytäre Exozytose von
ATP/Adenosin hemmen kann, um damit eine zusätzliche Enthemmung durch Abschwächung
des A1R-Effekts herbeizuführen, sofern sich dieser überhaupt entfalten kann (Pascual 2005;
Florian et al. 2011; Fujita et al. 2014).
Damit ließe sich die erwähnte EPSP-Erhöhung der Messreihe mit NEM ohne LTP-Stimulation
bereits erklären – ungeachtet der Eigenschaften von NEM, die im Folgenden noch erläutert
werden.
Eine kleine LTP-Messreihe mit NEM alleine, die hier nicht zur Darstellung kommt, zeigte
allerdings einen Trend hin zu einer starken LTD anstelle einer LTP – ähnlich den Messungen
mit NEM + cCPA.
Die Arbeit von Lledo (1998) zeigte, dass eine postsynaptische Applikation von lediglich 5 nM
NEM in CA1-Neuronen bereits zu einer signifikanten Reduktion der LTP führte. In zeitlich
folgenden Untersuchungen wurde zudem eine Reduktion der basalen Transmission durch NEM
und NEM-ähnlichen Peptiden aufgezeigt, die ebenso postsynaptisch appliziert wurden
(Nishimune et al. 1998; Lüscher et al. 1999).
Neben der G-Protein-Entkopplung bewirkt NEM auch die potente Hemmung des NEM-
sensitiven Faktors (NSF), einer zytosolischen ATPase, die mit einigen
Membranfusionsproteinen (SNARE-Proteine) interagiert (Glick und Rothman 1987; Hanson et
al. 1997). Damit vermittelt NSF vor allem Recycling und Endozytose der GluR2-enthaltenden
AMPA-R in der postsynaptische Membran (Noel et al. 1999; Jurado et al. 2013). Unterstützt
wird die NSF/GluR2-Interaktion durch die Exprimierung von PKMζ, welche für die

53 4 Diskussion
Aufrechterhaltung der late-LTP verantwortlich ist (Yao et al. 2008). Kontrovers diskutierte
Forschungsergebnisse kommen zu dem Schluss, dass GluR1-enthaltende AMPA-R aus
endosomalen Reservepools einem perisynaptischen Austausch unterworfen sind und zu einem
Großteil der LTP-Aufrechterhaltung dienen, wohingegen die GluR2-Domäne direkt
Endozytose aus der PSD vermittelt (Passafaro et al. 2001; Beretta et al. 2005; Yang et al. 2008).
Über passive laterale Membranbewegung wandern AMPA-R bei LTP-Induktion in den Bereich
der PSD und führen zu einer stärkeren Exzitabilität der Postsynapse (Borgdorff und Choquet
2002; Yudowski et al. 2007; Makino und Malinow 2009; Kneussel und Hausrat 2016).
Da early-LTP auf Reservepools von AMPA-R angewiesen ist, könnte NEM über Blockade
endosomaler Pools von GluR2-AMPA-R zu einer zeitversetzten LTP-Abschwächung führen,
solange noch extrasynaptische Pools von GluR1-AMPA-R verfügbar sind (Granger et al. 2012).
Dies könnte darauf hindeuten, dass die starke Erhöhung der basalen Transmission auf eine frühe
Entkopplung des A1R von dem zugehörigen Gi-Protein durch NEM zurückgeführt werden
könnte. Womöglich spielt die Wirkung von NEM auf NSF hierbei eine geringere Rolle, da NSF
vorwiegend in der PSD exprimiert wird und somit die Aufhebung der basalen A1R-bedingten
Hemmung der Transmitterfreisetzung mehr Gewicht erfahren könnte (Walsh und Kuruc 1992).
Es bleibt offen, inwieweit die Agonisierung des A1R durch die hohe sättigende Konzentration
von cCPA an der Verminderung der LTP mittels möglicher Restaktivität beteiligt ist, oder ob
diese Agonisierung infolge einer kompletten G-Protein-Entkopplung des A1R konkomitant ist.
Aufgrund eines offentsichtlich raschen Wirkungseintritts innerhalb weniger Minuten scheint
das lipophile NEM problemlos über Zellmembranen an dessen spezifische zytoplasmatische
Wirkorte zu gelangen. Ob eine sich über den Zeitverlauf verändernde Wirkung von NEM
existiert, müsste in weiteren Experimenten nachgewiesen werden. Um die Funktionsweise der
cCPA-Hemmung noch besser verstehen zu können, müssten Vergleichsmessungen mit NEM
alleine über einen Zeitverlauf über 20 min hinaus, sowie eine aussagekräftige LTP-Messreihe
mit NEM alleine durchgeführt werden. Die breite Wirkung und extrazelluläre Gabe von NEM
erschweren allerdings spezifische Interpretationen.

54 4 Diskussion
4.7 LTP-Modulation durch CGS
Mit dem A2A-Rezeptoragonist CGS-21680 (200 nM), 10 min vor LTP-Induktion in die
Badlösung gegeben, reduzierte sich die mittlere EPSP-Amplitude nicht signifikant auf 72,36 ±
23,01 % der Baseline (p = 0,3215, n = 6). Der Unterschied zur Wistar-Kontrollreihe im
Paarvergleich ist sehr signifikant (p = 0,0087).
In verschiedenen Arbeiten verstärkten sich allerdings LTP-Messungen mit CGS (10-30 nM),
welches durch einmalige oder kontinuierliche Applikaton eingewaschen wurde (Mendonça und
Ribeiro 1994; Cunha und Ribeiro 2000; Almeida et al. 2003; Dias et al. 2012).
Da A2AR in der Lage sind, ENTs zu steuern und so zu einem extrazellulären
Konzentrationsabfall von Adenosin zu führen, wird die hemmende A1R-Wirkung zusätzlich
reduziert (Pinto-Duarte et al. 2005).
Es stellt sich jedoch die Frage, ob der Effekt von CGS auf die LTP konzentrationsabhängig ist.
Verschiedenen Untersuchungen zu zufolge erhöht sich die basale EPSP/AMPA-Amplitude
ohne LTP-Stimulation bei niedrigen Konzentrationen bis 30 nM; 100 nM CGS führten jedoch
zu keiner Erhöhung (Lopes et al. 2002; Dias et al. 2012). Dies geht einher mit Experimenten,
in denen Konzentrationen im Bereich von µM die fEPSP-Amplituden dosisabhängig
verkleinern (Lupica et al. 1990). Aufgrund der Affinitätseigenschaften ist CGS-21680 bei
Ratten hochselektiv für A2AR (Ki-Wert: 22 nM) und bindet erst im Bereich von µM an A1R,
dann auch als A1R-Agonist wirkend (Hutchison et al. 1989). CGS scheint erst bei sehr hohen
Konzentrationen den cAMP-Spiegel zu erhöhen, was zu der These führte, dass die Aktivierung
des A2AR bereits bei niedrigen Konzentrationen cAMP-unabhängig PKA und AMPA-R
(unabhängig von NMDA-R) aktiviert und so eine postsynaptische Verstärkung der
Transmission erreicht (Kessey und Mogul 1997; Dias et al. 2012). Entgegen dieser Annahme
zeigten Cunha und Ribeiro (2000), dass die Wirkung des A2AR wahrscheinlicher mit der PKC
als mit der PKA assoziiert ist. Gleichzeitig deaktiviert PKC den A1R und damit dessen tonische
Hemmung. In diesem Zusammenhang erfährt eine mögliche A1R/A2AR-Heterodimerisierung
eine besondere Bedeutung bei präsynaptischer Transmitterfreisetzung, wie sie im Stiatum
bereits beschrieben wurde (Ciruela et al. 2006). Demnach ist die Wirkung von CGS bei
niedrigen Konzentrationen auf die early-LTP über PKC plausibler, da die PKA vor allem in
late-LTP involviert ist.

55 4 Diskussion
Wie schon von Lopes et al. (2002) vermutet, gibt es vermehrt Hinweise dafür, dass die
Aktivierung des A2AR erst dann Wirkung zeigt, wenn zuvor ein Ligand an den A1R gebunden
hat (Hernández-González et al. 2014). Dies könnte unter anderem ein Grund dafür sein,
weswegen CGS beispielsweise bei geringer extrazellulärer Adenosinkonzentration oder als
alleinige Applikation in dieser Arbeit keine LTP begünstigte.
Vermutlich wird die postsynaptische Exzitabilität dadurch erhöht, dass A2AR über mGluR5 auf
die NMDA-R-Aktivität wirkt (Tebano et al. 2005; Mouro 2012; Kouvaros und
Papatheodoropoulos 2016). Dagegen schienen höhere Konzentrationen von CGS im Striatum
die Leitfähigkeit von NMDA-R herabzusetzen. Dies geschieht vermutlich ohne Beteiligung der
PKC oder PKA, sondern mithilfe des PLC/IP3/CaMKII-Signalwegs (Nörenberg et al. 1997;
Wirkner et al. 2000; Gerevich et al. 2002).
Ein ähnlicher Effekt könnte im Hippocampus auftreten, da eine wahrscheinliche Interaktion des
A2AR mit Gi/Go-Proteinen festgestellt wurde, wodurch eine Signalkaskade via PLC auch hier
erklärbar wäre (Cunha et al. 1999). Dies könnte für die vorliegende LTP-Messreihe mit CGS
ursächlich sein, die eher einer LTD gleichkommt.
Unklare Bindungseigenschaften bestehen außer den soeben genannten auch aufgrund der
Vermutung, dass CGS-21680 eine zweite hochaffine Bindungsstelle (G-Protein-gekoppelt)
neben A2AR im Hippocampus besitzen könnte (Cunha et al. 1999).
Da A1R erst bei wesentlich höheren Konzentrationen affin und A2BR/A3R nicht affin für CGS-
21680 sind, könnten noch unbekannte, zuvor diskutierte Signalkaskaden für die LTP-
hemmende Wirkung von 200 nM CGS vermutlich mitverantwortlich sein.
Eine konzentrationsabhängige Wirkung von CGS-21680 auf AC, PLC und den Effekt auf LTP
im Hippocampus bleibt weiter zu evaluieren.
Physiologisch betrachtet leuchtet ein, dass der A2AR die Wirkung des A1R abhängig von
endogener Adenosinkonzentration herabsetzen oder unterstützen kann, um damit LTP
feinmodulieren zu können.

56 4 Diskussion
4.8 Pathophysiologische Bedeutung
Evaluiert man adenosinerge Effekte auf die LTP, so bleibt festzuhalten, dass im Besonderen
die Exzitabilität unter Wirkung des A1R herabgesetzt wird. Diese Ergebnisse entsprechen
gemäß dem aktuellen Forschungsstand einer antidepressiven Wirkungsweise und stehen
bislang im Gegensatz zu der Neuroplastizitätstheorie der Depression.
Die LTP-hemmende Wirkung von aktivierten AR geht einher mit der slow-wave-activity
während Tiefschlafphasen (non-REM) bzw. langanhaltender Wachheit wie beispielsweise einer
Schlafentzugstherapie. Daraus lassen sich wichtige Funktionen wie Schlafinduktion, global
downscaling für anschließendes Konsolidieren neuer Gedächtnisinhalte, aber auch kognitive
Defizite ableiten (Diekelmann und Born 2010).
Mit Züchtung der auch in dieser Arbeit verwendeten A1R-überexprimierenden Maus (CaMKII-
AdoA1R-92-Linie) konnte Serchov et al. (2015) einen anxiolytischen und antidepressiven
Effekt, jedoch keine Auswirkungen auf Spontanaktivität, exploratives Verhalten, räumliches
Lernen und Gedächtnis nachweisen. Dies deckt sich mit Ergebnissen einer A1R-Knockout-
Maus, die umgekehrt verstärktes ängstliches Verhalten aufwies (Lang et al. 2003; Giménez‐
Llort et al. 2002; Giménez‐Llort et al. 2005). Zudem scheint ein Zusammenhang zwischen A1R-
und A2AR-Genpolymorphismen und der zerebralen Verfügbarkeit von A1R und damit einer
Anfälligkeit für Angststörungen bei menschlichen Individuen in vivo zu bestehen (Hohoff et al.
2014).
Es konnte außerdem gezeigt werden, dass sowohl erhöhte A1R-Aktivität, ausgelöst durch
besispielsweise Schlafentzug, als auch pharmakologische Behandlungen mit Imipramin oder
Ketamin in eine gemeinsame Endstrecke münden: die Hochregulierung des Proteins Homer1A
(Serchov et al. 2015). Vermutlich verhält es sich bei anderen antidepressiven Therapieformen
wie ECT, DBS und eventuell rTMS ähnlich, da diese wahrscheinlich ebenso mit einer erhöhten
A1R-Aktivität bzw. Adenosinkonzentration einhergehen (Gleiter et al. 1989; Bekar et al. 2008).
Ähnlich der Wirkung von CGS-21680 in dieser Arbeit, konnte auch Fontinha et al. (2009) mit
dem hochselektiven A2AR-Agonist SCH-58261 eine effektive Hemmung der LTP an
CA3/CA1-Synapsen aufzeigen. Gleichzeitig wurde anhand verschiedener
Verhaltensexperimente mit Ratten die entscheidende Bedeutung der A2AR-Modulation für
assoziatives Lernen bewiesen.

57 4 Diskussion
Klärungsbedarf besteht insbesondere bei Rezeptor-Rezeptor-Interaktionen, die dem A2AR
vermutlich dessen feinmodulierende Rolle verleihen und welche interessante Angriffspunkte
für pharmakologische Eingriffe bei verschiedenen Pathologien bieten (Coelho et al. 2014). Ob
dabei die in dieser Arbeit vermutete dosisabhängige Wirkung des A2AR-Agonisten CGS-21680
eine Rolle spielt, bleibt weiter zu erfoschen. Wie aber bereits an dem Parkinson-Medikament
Istradefylline deutlich wurde, werden in Zukunft spezifische A2AR-Antagonisten aufgrund
neuroprotektiver Eigenschaften sicherlich eine gewichtigere Rolle spielen – ungeachtet des
schon gebräuchlichen, alltäglichen Koffeinkonsums.

58 5 Zusammenfassung
5 Zusammenfassung
Adenosin hält nicht nur die Homöostase der Schlaf-Wachphasen aufrecht, sondern spielt unter anderem
eine gewichtige Rolle bei der Hebb’schen Plastizität, welche für die Speicherung neuer
Gedächtnisinhalte verantwortlich ist. Verschiedene neuropsychiatrische Krankheiten wie Depression,
Angststörungen, M. Alzheimer oder M. Parkinson weisen eine Beeinflussung oder Störung des
adenosinergen Systems bzw. entsprechender Adenosinrezeptoren auf.
Die vorliegende Arbeit untersuchte, wie die Modulation der A1- und A2AR die assoziative LTP – als Teil
der synaptischen Langzeitplastizität – beeinflusst. An der CA3/CA1-Synapse des Hippocampus wurden
etablierte LTP-Stimulationsprotokolle mit TBS appliziert und EPSP-Veränderungen mittels Patch-
Clamp-Technik in der Wholecell-Konfiguration gemessen.
Durch Gabe des A1R-Agonisten cCPA in Hirnschnitten von Wistar-Ratten konnte keine LTP ausgelöst
werden.
Zudem bestätigte und präzisierte sich diese Blockade in anderen Experimenten. Demgemäß zeigte sich
bei Mäusen mit einer Überexprimierung des A1R-Gens im CA1-Bereich des Hippocampus, dass
postsynaptische A1R an der Suppression der LTP signifikant beteiligt sind. Aus Messreihen von Stolz
(2015) mit transgenen Mäusen derselben Ausprägung im CA3-Bereich ging bereits eine präsynaptische
Hemmung der LTP hervor.
Mithilfe weiterer Stoffbeimengungen konnte weder DPCPX (ein A1R-Antagonist), noch NEM (ein
GPCR-Entkoppler) die cCPA-Hemmung der LTP aufheben. Unterschiede der
Konzentrationsverhältnisse und Bindungseigenschaften sind hierfür in Erwägung zu ziehen, ebenso eine
direkte Wirkung von NEM auf AMPA-R.
Abgeschwächte LTP-Messungen mit dem A2AR-Agonisten CGS-21680 geben Hinweise auf noch
unbekannte bzw. potenzielle konzentrationsabhängige Wirkungsweisen. Hierbei könnten die ohnehin
unterschätzte Heterodimerisierung der A2AR/A1R und der dadurch stattfindende feinmodulierende
Crosstalk ausschlaggebend sein.
Auch wenn NEM und CGS-21680 über teils unbekannte Mechanismen verfügen, sollten deren
interessante Auswirkungen auf die LTP Gegenstand weiterer Forschung sein.
Weitergehende Untersuchungen sind außerdem nötig, um zelluläre Prozesse in Prä- und Postsynapse
unter Modulierung des Adenosinlevels und der AR in Bezug auf early-LTP zu präzisieren und um die
konzentrationsabhängige Funktion des Dimers A1R/A2AR besser verstehen zu können.
Fügte sich diese Arbeit auch nicht in die Neuroplastizitätshypothese der Depression ein, liefert sie
dennoch Gründe für eine inverse Interpretation ebendieser. Überdies stellen die Ergebnisse eine
schlüssige Verbindung zu der Hypothese des synaptic downscaling während des non-REM-Schlafs her.

59 Literaturverzeichnis
Literaturverzeichnis
Abbott, L.F.; Regehr, W.G. (2004): Synaptic computation. In: Nature 431 (7010), S. 796–
803. DOI: 10.1038/nature03010.
Abraham, W.C.; Huggett, A. (1997): Induction and reversal of long‐term potentiation by
repeated high‐frequency stimulation in rat hippocampal slices. In: Hippocampus 7 (2),
S. 137–145. DOI: 10.1002/(SICI)1098-1063(1997)7:2<137::AID-HIPO3>3.0.CO;2-K.
Akers, K.G.; Martinez-Canabal, A.; Restivo, L.; Yiu, A.P.; Cristofaro, A. de; Hsiang, H.-L.L.
et al. (2014): Hippocampal neurogenesis regulates forgetting during adulthood and
infancy. In: Science 344 (6184), S. 598–602. DOI: 10.1126/science.1248903.
Almeida, T.; Rodrigues, R.J.; Mendonca, A. de; Ribeiro, J.A.; Cunha, R.A. (2003): Purinergic
P2 receptors trigger adenosine release leading to adenosine A2A receptor activation
and facilitation of long-term potentiation in rat hippocampal slices. In: Neuroscience
122 (1), S. 111–121.
Amaral D, L.P. (2007): Hippocampal neuroanatomy. In: The hippocampus book. Hg. v. Per
Andersen. Oxford: Oxford University Press. 37-114.
Banasr, M.; Dwyer, J.M.; Duman, R.S. (2011): Cell atrophy and loss in depression: reversal
by antidepressant treatment. Cell differentiation, Cell division, growth and death. In:
Current Opinion in Cell Biology 23 (6), S. 730–737. DOI: 10.1016/j.ceb.2011.09.002.
Bekar, L.; Libionka, W.; Tian, G.-F.; Xu, Q.; Torres, A.; Wang, X. et al. (2008): Adenosine is
crucial for deep brain stimulation-mediated attenuation of tremor. In: Nature medicine
14 (1), S. 75–80. DOI: 10.1038/nm1693.
Beretta, F.; Sala, C.; Saglietti, L.; Hirling, H.; Sheng, M.; Passafaro, M. (2005): NSF
interaction is important for direct insertion of GluR2 at synaptic sites. In: Molecular
and Cellular Neuroscience 28 (4), S. 650–660. DOI: 10.1016/j.mcn.2004.11.008.
Bi, G.Q.; Poo, M.M. (1998): Synaptic modifications in cultured hippocampal neurons:
dependence on spike timing, synaptic strength, and postsynaptic cell type. In: J
Neurosci 18 (24), S. 10464–10472.
Bischofberger, J.; Engel, D.; Li, L.; Geiger, Jörg R P; Jonas, P. (2006): Patch-clamp recording
from mossy fiber terminals in hippocampal slices. In: Nature protocols 1 (4), S. 2075–
2081. DOI: 10.1038/nprot.2006.312.
Blendy, J.A. (2006): The role of CREB in depression and antidepressant treatment. In:
Biological psychiatry 59 (12), S. 1144–1150. DOI: 10.1016/j.biopsych.2005.11.003.
Bliss, T.V.; Collingridge, G.L. (1993): A synaptic model of memory: long-term potentiation
in the hippocampus. In: Nature 361 (6407), S. 31–39. DOI: 10.1038/361031a0.

60 Literaturverzeichnis
Bliss, T.V.; Gardner-Medwin, A.R. (1973): Long-lasting potentiation of synaptic transmission
in the dentate area of the unanaesthetized rabbit following stimulation of the perforant
path. In: The Journal of Physiology 232 (2), S. 357–374. DOI:
10.1113/jphysiol.1973.sp010274.
Bliss, T.V.; Lømo, T. (1973): Long-lasting potentiation of synaptic transmission in the
dentate area of the anaesthetized rabbit following stimulation of the perforant path. In:
J Physiol 232 (2), S. 331–356.
Blitzer, R.D.; Wong, T.; Nouranifar, R.; Iyengar, R.; Landau, E.M. (1995): Postsynaptic
CAMP pathway gates early LTP in hippocampal CA1 region. In: Neuron 15 (6), S.
1403–1414. DOI: 10.1016/0896-6273(95)90018-7.
Borgdorff, A.J.; Choquet, D. (2002): Regulation of AMPA receptor lateral movements. In:
Nature 417 (6889), S. 649–653. DOI: 10.1038/nature00780.
Brakemeier, E.-L.; Normann, C.; Berger, M. (2008): Ätiopathogenese der unipolaren
Depression. Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz. In:
Bundesgesundheitsbl. 51 (4), S. 379–391. DOI: 10.1007/s00103-008-0505-x.
Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. (2000):
Hippocampal volume reduction in major depression. In: The American journal of
psychiatry 157 (1), S. 115–118. DOI: 10.1176/ajp.157.1.115.
Brown, T.H.; Kairiss, E.W.; Keenan, C.L. (1990): Hebbian synapses: biophysical mechanisms
and algorithms. In: Annu Rev Neurosci 13, S. 475–511.
Brugarolas, M.; Navarro, G.; Martínez‐Pinilla, E.; Angelats, E.; Casadó, V.; Lanciego, J.L.;
Franco, R. (2014): G‐Protein‐Coupled Receptor Heteromers as Key Players in the
Molecular Architecture of the Central Nervous System. In: CNS neuroscience &
therapeutics 20 (8), S. 703–709. DOI: 10.1111/cns.12277.
Brunoni, A.R.; Lopes, M.; Fregni, F. (2008): A systematic review and meta-analysis of
clinical studies on major depression and BDNF levels: implications for the role of
neuroplasticity in depression. In: The international journal of
neuropsychopharmacology / official scientific journal of the Collegium Internationale
Neuropsychopharmacologicum (CINP) 11 (8), S. 1169–1180. DOI:
10.1017/S1461145708009309.
Cajal, S. R. Y (1894): The Croonian Lecture: La Fine Structure des Centres Nerveux. In:
Proceedings of the Royal Society of London 55 (331-335), S. 444–468. DOI:
10.1098/rspl.1894.0063.
Cajal, S. R. Y (1928): Degeneration and Regeneration of the Nervous System. English
translation by May R.M. et al., eds. (1991). New York: Oxford University Press.
Campbell, S.; Marriott, M.; Nahmias, C.; MacQueen, G.M. (2004): Lower hippocampal
volume in patients suffering from depression: a meta-analysis. In: The American
journal of psychiatry 161 (4), S. 598–607. DOI: 10.1176/appi.ajp.161.4.598.
Caruana, D.A.; Alexander, G.M.; Dudek, S.M. (2012): New insights into the regulation of
synaptic plasticity from an unexpected place: Hippocampal area CA2. In: Learn. Mem.
19 (9), S. 391–400. DOI: 10.1101/lm.025304.111.

61 Literaturverzeichnis
Castrén, E. (2005): Is mood chemistry? In: Nature reviews. Neuroscience 6 (3), S. 241–246.
DOI: 10.1038/nrn1629.
Chen, A.C.-H.; Shirayama, Y.; Shin, K.-H.; Neve, R.L.; Duman, R.S. (2001a): Expression of
the cAMP response element binding protein (CREB) in hippocampus produces an
antidepressant effect. In: Biological psychiatry 49 (9), S. 753–762. DOI:
10.1016/S0006-3223(00)01114-8.
Chen, B.; Dowlatshahi, D.; MacQueen, G.M.; Wang, J.-F.; Young, L. (2001b): Increased
hippocampal bdnf immunoreactivity in subjects treated with antidepressant
medication. In: Biological psychiatry 50 (4), S. 260–265. DOI: 10.1016/S0006-
3223(01)01083-6.
Chen, H.-X.; Otmakhov, N.; Lisman, J. (1999): Requirements for LTP Induction by Pairing in
Hippocampal CA1 Pyramidal Cells. In: Journal of Neurophysiology 82 (2), S. 526–
532.
Chieng, B.; Bekkers, J.M. (2001): Inhibition of calcium channels by opioid‐ and adenosine‐receptor agonists in neurons of the nucleus accumbens. In: British Journal of
Pharmacology 133 (3), S. 337–344. DOI: 10.1038/sj.bjp.0704072.
Chung, H.J.; Qian, X.; Ehlers, M.; Jan, Y.N.; Jan, L.Y. (2009): Neuronal activity regulates
phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying
potassium channels. In: PNAS 106 (2), S. 629–634. DOI: 10.1073/pnas.0811615106.
Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M. et al. (2006):
Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1–
A2A Receptor Heteromers. In: J. Neurosci. 26 (7), S. 2080–2087. DOI:
10.1523/JNEUROSCI.3574-05.2006.
Citri, A.; Malenka, R.C. (2008): Synaptic plasticity: multiple forms, functions, and
mechanisms. In: Neuropsychopharmacology : official publication of the American
College of Neuropsychopharmacology 33 (1), S. 18–41. DOI:
10.1038/sj.npp.1301559.
Coelho, J.E.; Alves, P.; Canas, P.M.; Valadas, J.S.; Shmidt, T.; Batalha, V.L. et al. (2014):
Overexpression of Adenosine A2A Receptors in Rats: Effects on Depression,
Locomotion, and Anxiety. In: Frontiers in psychiatry 5, S. 67. DOI:
10.3389/fpsyt.2014.00067.
Cole; Curtis (1938): Electrical Impedance of Nerve During Activity. In: Nature <London>
142 (3587), S. 209–210. Online verfügbar unter http://dx.doi.org/10.1038/142209b0.
Cooke, S.F.; Bliss (2006): Plasticity in the human central nervous system. In: Brain : a
journal of neurology 129 (Pt 7), S. 1659–1673. DOI: 10.1093/brain/awl082.
Cunha, R.A.; Constantino, M.D.; Ribeiro, J.A. (1999): G Protein coupling of CGS 21680
binding sites in the rat hippocampus and cortex is different from that of adenosine A1
and striatal A2A receptors. In: Naunyn-Schmiedeberg's Arch Pharmacol 359 (4), S.
295–302. DOI: 10.1007/PL00005355.

62 Literaturverzeichnis
Cunha, R.A.; Milusheva, E.; Vizi, E.S.; Ribeiro, J.A.; Sebastião, A.M. (1994): Excitatory and
Inhibitory Effects of A1 and A2A Adenosine Receptor Activation on the Electrically
Evoked [3H]Acetylcholine Release from Different Areas of the Rat Hippocampus. In:
Journal of Neurochemistry 63 (1), S. 207–214. DOI: 10.1046/j.1471-
4159.1994.63010207.x.
Cunha, R.A.; Ribeiro, J.A. (2000): Adenosine A2A receptor facilitation of synaptic
transmission in the CA1 area of the rat hippocampus requires protein kinase C but not
protein kinase A activation. In: Neuroscience letters 289 (2), S. 127–130.
Dan, Y.; Poo, M.-M. (2006): Spike timing-dependent plasticity: from synapse to perception.
In: Physiological reviews 86 (3), S. 1033–1048. DOI: 10.1152/physrev.00030.2005.
Davis, Kenneth L.; Malenka, Robert C. (2002): Neuropsychopharmacology. The fifth
generation of progress : an official publication of the American College of
Neuropsychopharmacology. Chapter 11. Philadelphia: Lippincott/Williams & Wilkins.
DeJong-Meyer, R. (2005): Depressive Störungen: Ätiologie. In Baumann U. & Perrez M.
(Hrsg.). Lehrbuch klinische Psychologie - Psychotherapie. 3., vollständig überarb.
Aufl. Bern: Hans Huber. 862-877.
Deng, W.; Aimone, J.B.; Gage, F.H. (2010): New neurons and new memories: how does adult
hippocampal neurogenesis affect learning and memory? In: Nature reviews.
Neuroscience 11 (5), S. 339–350. DOI: 10.1038/nrn2822.
Dias, R.B.; Ribeiro, J.A.; Sebastiao, A.M. (2012): Enhancement of AMPA currents and
GluR1 membrane expression through PKA-coupled adenosine A(2A) receptors. In:
Hippocampus 22 (2), S. 276–291. DOI: 10.1002/hipo.20894.
Dias, R.B.; Rombo, D.M.; Ribeiro, J.A.; Henley, J.M.; Sebastião, A.M. (2013): Adenosine:
setting the stage for plasticity. In: Trends in Neurosciences 36 (4), S. 248–257. DOI:
10.1016/j.tins.2012.12.003.
Diekelmann, S.; Born, J. (2010): The memory function of sleep. In: Nature reviews.
Neuroscience 11 (2), S. 114–126. DOI: 10.1038/nrn2762.
Dratz, E. (2013): Modulation assoziativer LTP im Hippocampus. Med. Dissertation,
Universität Freiburg i. Br.
Dudek, S.M.; Bear, M.F. (1992): Homosynaptic long-term depression in area CA1 of
hippocampus and effects of N-methyl-D-aspartate receptor blockade. In: Proceedings
of the National Academy of Sciences 89 (10), S. 4363–4367. DOI:
10.1073/pnas.89.10.4363.
Duman, R.S. (2004): Depression: a case of neuronal life and death? In: Biological psychiatry
56 (3), S. 140–145. DOI: 10.1016/j.biopsych.2004.02.033.
Dunwiddie, T.V.; Haas, H.L. (1985): Adenosine increases synaptic facilitation in the in vitro
rat hippocampus: evidence for a presynaptic site of action. In: The Journal of
Physiology 369, S. 365–377.

63 Literaturverzeichnis
Edwards, F.A.; Konnerth, A.; Sakmann, B.; Takahashi, T. (1989): A thin slice preparation for
patch clamp recordings from neurones of the mammalian central nervous system. In:
Pflugers Arch 414 (5), S. 600–612.
El Yacoubi, M.; Ledent, C.; Parmentier, M.; Bertorelli, R.; Ongini, E.; Costentin, J.;
Vaugeois, J.M. (2001): Adenosine A2A receptor antagonists are potential
antidepressants: evidence based on pharmacology and A2A receptor knockout mice.
In: British Journal of Pharmacology 134 (1), S. 68–77. DOI: 10.1038/sj.bjp.0704240.
Elmenhorst, D.; Meyer, P.T.; Winz, O.H.; Matusch, A.; Ermert, J.; Coenen, H.H. et al.
(2007): Sleep deprivation increases A1 adenosine receptor binding in the human brain:
a positron emission tomography study. In: The Journal of neuroscience : the official
journal of the Society for Neuroscience 27 (9), S. 2410–2415. DOI:
10.1523/JNEUROSCI.5066-06.2007.
Florian, C.; Vecsey, C.G.; Halassa, M.M.; Haydon, P.G.; Abel, T. (2011): Astrocyte-derived
adenosine and A1 receptor activity contribute to sleep loss-induced deficits in
hippocampal synaptic plasticity and memory in mice. In: The Journal of neuroscience
: the official journal of the Society for Neuroscience 31 (19), S. 6956–6962. DOI:
10.1523/JNEUROSCI.5761-10.2011.
Fontinha, B.M.; Delgado-García, J.M.; Madroñal, N.; Ribeiro, J.A.; Sebastião, A.M.; Gruart,
A. (2009): Adenosine A(2A) receptor modulation of hippocampal CA3-CA1 synapse
plasticity during associative learning in behaving mice. In: Neuropsychopharmacology
: official publication of the American College of Neuropsychopharmacology 34 (7), S.
1865–1874. DOI: 10.1038/npp.2009.8.
Forghani, R.; Krnjevic, K. (1995): Adenosine antagonists have differential effects on
induction of long-term potentiation in hippocampal slices. In: Hippocampus 5 (1), S.
71–77. DOI: 10.1002/hipo.450050109.
Fowler, J.C.: Changes in extracellular adenosine levels and population spike amplitude during
graded hypoxia in the rat hippocampal slice. In: Naunyn-Schmiedeberg's Arch
Pharmacol 347 (1), S. 73–78. DOI: 10.1007/BF00168775.
Fowler, J.C. (1993): Changes in extracellular adenosine levels and population spike amplitude
during graded hypoxia in the rat hippocampal slice. In: Naunyn-Schmiedeberg's
archives of pharmacology 347 (1), S. 73–78.
Fox, K. (2002): Anatomical pathways and molecular mechanisms for plasticity in the barrel
cortex. In: Neuroscience 111 (4), S. 799–814. DOI: 10.1016/S0306-4522(02)00027-1.
Fredholm, B.B. (2007): Adenosine, an endogenous distress signal, modulates tissue damage
and repair. In: Cell Death & Differentiation 14 (7), S. 1315–1323. DOI:
10.1038/sj.cdd.4402132.
Fredholm, B.B.; Arslan, G.; Halldner, L.; Kull, B.; Schulte, G.; Wasserman, W. (2000):
Structure and function of adenosine receptors and their genes. In: Naunyn-
Schmiedeberg's archives of pharmacology 362 (4-5), S. 364–374.
Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. (2001a):
International Union of Pharmacology. XXV. Nomenclature and classification of
adenosine receptors. In: Pharmacological reviews 53 (4), S. 527–552.

64 Literaturverzeichnis
Fredholm, B.B.; Irenius, E.; Kull, B.; Schulte, G. (2001b): Comparison of the potency of
adenosine as an agonist at human adenosine receptors expressed in Chinese hamster
ovary cells. In: Biochemical pharmacology 61 (4), S. 443–448.
Frenguelli, B.G.; Llaudet, E.; Dale, N. (2003): High‐resolution real‐time recording with
microelectrode biosensors reveals novel aspects of adenosine release during hypoxia
in rat hippocampal slices. In: Journal of Neurochemistry 86 (6), S. 1506–1515. DOI:
10.1046/j.1471-4159.2003.01957.x.
Frodl, T.; Schaub, A.; Banac, S.; Charypar, M.; Jäger, M.; Kümmler, P. et al. (2006): Reduced
hippocampal volume correlates with executive dysfunctioning in major depression. In:
Journal of Psychiatry and Neuroscience 31 (5), S. 316–325.
Fujita, T.; Chen, M.J.; Li, B.; Smith, N.A.; Peng, W.; Sun, W. et al. (2014): Neuronal
transgene expression in dominant-negative SNARE mice. In: The Journal of
neuroscience : the official journal of the Society for Neuroscience 34 (50), S. 16594–
16604. DOI: 10.1523/JNEUROSCI.2585-14.2014.
George, E.M.; Cockrell, K.; Adair, T.H.; Granger, J.P. (2010): Regulation of sFlt-1 and
VEGF secretion by adenosine under hypoxic conditions in rat placental villous
explants. In: American journal of physiology. Regulatory, integrative and comparative
physiology 299 (6), S. R1629-33. DOI: 10.1152/ajpregu.00330.2010.
Gerevich, Z.; Wirkner, K.; Illes, P. (2002): Adenosine A2A receptors inhibit the N-methyl-D-
aspartate component of excitatory synaptic currents in rat striatal neurons. In:
European journal of pharmacology 451 (2), S. 161–164.
Gerges, N.Z.; Aleisa, A.M.; Schwarz, L.A.; Alkadhi, K.A. (2004): Reduced basal CaMKII
levels in hippocampal CA1 region: possible cause of stress-induced impairment of
LTP in chronically stressed rats. In: Hippocampus 14 (3), S. 402–410. DOI:
10.1002/hipo.10193.
Giménez-Llort, L.; Schiffmann, S.N.; Shmidt, T.; Canela, L.; Camón, L.; Wassholm, M. et al.
(2007): Working memory deficits in transgenic rats overexpressing human adenosine
A2A receptors in the brain. In: Neurobiology of Learning and Memory 87 (1), S. 42–
56. DOI: 10.1016/j.nlm.2006.05.004.
Giménez‐Llort, L.; Fernández‐Teruel, A.; Escorihuela, R.M.; Fredholm, B.B.; Tobeña, A.;
Pekny, M.; Johansson, B. (2002): Mice lacking the adenosine A1 receptor are anxious
and aggressive, but are normal learners with reduced muscle strength and survival rate.
In: European Journal of Neuroscience 16 (3), S. 547–550. DOI: 10.1046/j.1460-
9568.2002.02122.x.
Giménez‐Llort, L.; Masino, S.A.; Diao, L.; Fernández‐Teruel, A.; Tobeña, A.; Halldner, L.;
Fredholm, B.B. (2005): Mice lacking the adenosine A1 receptor have normal spatial
learning and plasticity in the CA1 region of the hippocampus, but they habituate more
slowly. In: Synapse 57 (1), S. 8–16. DOI: 10.1002/syn.20146.
Gleiter, C.H.; Deckert, J.; Nutt, D.J.; Marangos, P.J. (1989): Electroconvulsive Shock (ECS)
and the Adenosine Neuromodulatory System. Effect of Single and Repeated ECS on
the Adenosine A1 and A2 Receptors, Adenylate Cyclase, and the Adenosine Uptake
Site. In: J Neurochem 52 (2), S. 641–646. DOI: 10.1111/j.1471-4159.1989.tb09168.x.

65 Literaturverzeichnis
Glick, B.S.; Rothman, J.E. (1987): Possible role for fatty acyl-coenzyme A in intracellular
protein transport. In: Nature 326 (6110), S. 309–312. DOI: 10.1038/326309a0.
Granger, A.J.; Shi, Y.; Lu, W.; Cerpas, M.; Nicoll, R.A. (2012): LTP requires a reserve pool
of glutamate receptors independent of subunit type. In: Nature 493 (7433), S. 495–
500. DOI: 10.1038/nature11775.
Grover, L.M.; Teyler, T.J. (1990): Two components of long-term potentiation induced by
different patterns of afferent activation. In: Nature 347 (6292), S. 477–479. DOI:
10.1038/347477a0.
Gundlfinger, A.; Bischofberger, J.; Johenning, F.W.; Torvinen, M.; Schmitz, D.; Breustedt, J.
(2007): Adenosine modulates transmission at the hippocampal mossy fibre synapse via
direct inhibition of presynaptic calcium channels. In: The Journal of Physiology 582
(Pt 1), S. 263–277. DOI: 10.1113/jphysiol.2007.132613.
Hanson, P.I.; Roth, R.; Morisaki, H.; Jahn, R.; Heuser, J.E. (1997): Structure and
Conformational Changes in NSF and Its Membrane Receptor Complexes Visualized
by Quick-Freeze/Deep-Etch Electron Microscopy. In: Cell 90 (3), S. 523–535. DOI:
10.1016/S0092-8674(00)80512-7.
Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. (2008): Adenosine receptors: therapeutic
aspects for inflammatory and immune diseases. In: Nature reviews. Drug discovery 7
(9), S. 759–770. DOI: 10.1038/nrd2638.
Hebb, D. O. (1949): The organization of behavior;. A neuropsychological theory. New York:
Wiley (Wiley's books in clinical psychology).
Hernández-González, O.; Hernández-Flores, T.; Prieto, G.A.; Pérez-Burgos, A.; Arias-García,
M.A.; Galarraga, E.; Bargas, J. (2014): Modulation of Ca2+-currents by sequential and
simultaneous activation of adenosine A1 and A2A receptors in striatal projection
neurons. In: Purinergic Signalling 10 (2), S. 269–281. DOI: 10.1007/s11302-013-
9386-z.
Hershkowitz, N.; Katchman, A.N.; Veregge, S. (1993): Site of synaptic depression during
hypoxia: a patch-clamp analysis. In: Journal of Neurophysiology 69 (2), S. 432–441.
Hickey, P.; Stacy, M. (2012): Adenosine A2A Antagonists in Parkinson’s Disease: What’s
Next? Current Neurology and Neuroscience Reports. In: Curr Neurol Neurosci Rep 12
(4), S. 376–385. DOI: 10.1007/s11910-012-0279-2.
Hines, D.J.; Schmitt, L.I.; Hines, R.M.; Moss, S.J.; Haydon, P.G. (2013): Antidepressant
effects of sleep deprivation require astrocyte-dependent adenosine mediated signaling.
In: Translational Psychiatry 3 (1), S. e212. DOI: 10.1038/tp.2012.136.
Hodgkin, A.; Huxley, A. (1952): A quantitative description of membrane current and its
application to conduction and excitation in nerve. In: Bulletin of Mathematical Biology
52 (1-2), S. 25–71.

66 Literaturverzeichnis
Hodgson, R.A.; Bertorelli, R.; Varty, G.B.; Lachowicz, J.E.; Forlani, A.; Fredduzzi, S. et al.
(2009): Characterization of the Potent and Highly Selective A2A Receptor
Antagonists Preladenant and SCH 412348 [7-[2-[4-2,4-Difluorophenyl]-1-
piperazinyl]ethyl]-2-(2-furanyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-
amine] in Rodent Models of Movement Disorders and Depression. In: J Pharmacol
Exp Ther 330 (1), S. 294–303. DOI: 10.1124/jpet.108.149617.
Hohoff, C.; Garibotto, V.; Elmenhorst, D.; Baffa, A.; Kroll, T.; Hoffmann, A. et al. (2014):
Association of adenosine receptor gene polymorphisms and in vivo adenosine A1
receptor binding in the human brain. In: Neuropsychopharmacology : official
publication of the American College of Neuropsychopharmacology 39 (13), S. 2989–
2999. DOI: 10.1038/npp.2014.150.
Holderbach, R.; Clark, K.; Moreau, J.-L.; Bischofberger, J.; Normann, C. (2007): Enhanced
long-term synaptic depression in an animal model of depression. In: Biological
psychiatry 62 (1), S. 92–100. DOI: 10.1016/j.biopsych.2006.07.007.
Holderbach, R.S. (2006): Synaptische Plastizität und Neurogenese in einem Depressions-
Tiermodell. Med. Dissertation, Universität Freiburg i.Br.
Huang, Y.Y.; Kandel, E.R.; Varshavsky, L.; Brandon, E.P.; Qi, M.; Idzerda, R.L. et al.
(1995): A genetic test of the effects of mutations in PKA on mossy fiber LTP and its
relation to spatial and contextual learning. In: Cell 83 (7), S. 1211–1222.
Huang, Y.Y.; Malenka, R.C. (1993): Examination of TEA-induced synaptic enhancement in
area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the
induction of LTP. In: The Journal of neuroscience : the official journal of the Society
for Neuroscience 13 (2), S. 568–576.
Huang, Z.-L.; Urade, Y.; Hayaishi, O. (2011): The Role of Adenosine in the Regulation of
Sleep. In: CTMC 11 (8), S. 1047–1057. DOI: 10.2174/156802611795347654.
Huber, K.M.; Mauk, M.D.; Kelly, P.T. (1995): Distinct LTP induction mechanisms:
contribution of NMDA receptors and voltage-dependent calcium channels. In: Journal
of Neurophysiology 73 (1), S. 270–279.
Hutchison, A.J.; Webb, R.L.; Oei, H.H.; Ghai, G.R.; Zimmerman, M.B.; Williams, M. (1989):
CGS 21680C, an A2 selective adenosine receptor agonist with preferential
hypotensive activity. In: The Journal of pharmacology and experimental therapeutics
251 (1), S. 47–55.
Jeong, H.-J. (2002): Adenosine A1 Receptor-Mediated Presynaptic Inhibition of GABAergic
Transmission in Immature Rat Hippocampal CA1 Neurons. In: Journal of
Neurophysiology 89 (3), S. 1214–1222. DOI: 10.1152/jn.00516.2002.
Ji, R.-R.; Kohno, T.; Moore, K.A.; Woolf, C.J. (2003): Central sensitization and LTP: do pain
and memory share similar mechanisms? In: Trends in Neurosciences 26 (12), S. 696–
705. DOI: 10.1016/j.tins.2003.09.017.
Joëls, M.; Krugers, H.J. (2007): LTP after stress: up or down? In: Neural plasticity 2007, S.
93202. DOI: 10.1155/2007/93202.

67 Literaturverzeichnis
Jurado, S.; Goswami, D.; Zhang, Y.; Molina, A.J.M.; Südhof, T.C.; Malenka, R.C. (2013):
LTP Requires a Unique Postsynaptic SNARE Fusion Machinery. In: Neuron 77 (3), S.
542–558. DOI: 10.1016/j.neuron.2012.11.029.
Kandel, E.R. (2001): The molecular biology of memory storage: a dialogue between genes
and synapses. In: Science 294 (5544), S. 1030–1038.
Kandel, Eric R. (2013): Principles of neural science. 5th ed. New York: McGraw-Hill.
Kaster, M.P.; Machado, D.G.; Santos, A.R.S.; Rodrigues, A.L.S. (2012): Involvement of
NMDA receptors in the antidepressant-like action of adenosine. In: Pharmacological
reports : PR 64 (3), S. 706–713.
Kessey, K.; Mogul, D.J. (1997): NMDA-Independent LTP by adenosine A2 receptor-
mediated postsynaptic AMPA potentiation in hippocampus. In: Journal of
Neurophysiology 78 (4), S. 1965–1972.
Khan, S.A.; Ryali, V.; Bhat, P.S.; Prakash, J.; Srivastava, K.; Khanam, S. (2015): The
hippocampus and executive functions in depression. In: Industrial psychiatry journal
24 (1), S. 18–22. DOI: 10.4103/0972-6748.160920.
Kim, C.S.; Johnston, D. (2015): A1 adenosine receptor-mediated GIRK channels contribute to
the resting conductance of CA1 neurons in the dorsal hippocampus. In: Journal of
Neurophysiology 113 (7), S. 2511–2523. DOI: 10.1152/jn.00951.2014.
Kirov, S.A.; Sorra, K.E.; Harris, K.M. (1999): Slices Have More Synapses than Perfusion-
Fixed Hippocampus from both Young and Mature Rats. In: J. Neurosci. 19 (8), S.
2876–2886. Online verfügbar unter http://www.jneurosci.org/content/19/8/2876.full.
Klaasse, E.C.; IJzerman, A.P.; Grip, W.J. de; Beukers, M.W. (2008): Internalization and
desensitization of adenosine receptors. In: Purinergic Signalling 4 (1), S. 21–37. DOI:
10.1007/s11302-007-9086-7.
Klishin, A.; Lozovaya, N.; Krishtal, O. (1995): A1 adenosine receptors differentially regulate
the N-methyl-d-aspartate and non-N-methyl-d-aspartate receptor-mediated
components of hippocampal excitatory postsynaptic current in a Ca2+/Mg2+-
dependent manner. In: Neuroscience 65 (4), S. 947–953.
Kneussel, M.; Hausrat, T.J. (2016): Postsynaptic Neurotransmitter Receptor Reserve Pools for
Synaptic Potentiation. In: Trends in Neurosciences 39 (3), S. 170–182.
Kouvaros, S.; Papatheodoropoulos, C. (2016): Major dorsoventral differences in the
modulation of the local CA1 hippocampal network by NMDA, mGlu5, adenosine
A2A and cannabinoid CB1 receptors. In: Neuroscience 317, S. 47–64. DOI:
10.1016/j.neuroscience.2015.12.059.
Krugers, H.J.; Alfarez, D.N.; Karst, H.; Parashkouhi, K.; van Gemert, N.; Joëls, M. (2005):
Corticosterone shifts different forms of synaptic potentiation in opposite directions. In:
Hippocampus 15 (6), S. 697–703. DOI: 10.1002/hipo.20092.

68 Literaturverzeichnis
Krugers, H.J.; Goltstein, P.M.; van der Linden, S.; Joëls, M. (2006): Blockade of
glucocorticoid receptors rapidly restores hippocampal CA1 synaptic plasticity after
exposure to chronic stress. In: The European journal of neuroscience 23 (11), S.
3051–3055. DOI: 10.1111/j.1460-9568.2006.04842.x.
Laifenfeld, D.; Karry, R.; Grauer, E.; Klein, E.; Ben-Shachar, D. (2005): Antidepressants and
prolonged stress in rats modulate CAM-L1, laminin, and pCREB, implicated in
neuronal plasticity. In: Neurobiology of disease 20 (2), S. 432–441. DOI:
10.1016/j.nbd.2005.03.023.
Landmann, Björn Christopher (2015): Pharmakologische Modulation der assoziativen
synaptischen Langzeitplastizität durch Adenosinrezeptoren. Med. Dissertation.
Universität Freiburg i. Br.
Lang, U.E.; Lang, F.; Richter, K.; Vallon, V.; Lipp, H.-P.; Schnermann, J.; Wolfer, D.P.
(2003): Emotional instability but intact spatial cognition in adenosine receptor 1 knock
out mice. In: Behavioural Brain Research 145 (1–2), S. 179–188. DOI:
10.1016/S0166-4328(03)00108-6.
Larson, J.; Xiao, P.; Lynch, G. (1993): Reversal of LTP by theta frequency stimulation. In:
Brain research 600 (1), S. 97–102.
Levy, W.B.; Steward, O. (1983): Temporal contiguity requirements for long-term associative
potentiation/depression in the hippocampus. In: Neuroscience 8 (4), S. 791–797.
Li, W.; Liu, L.; Liu, Y.-y.; Luo, J.; Lin, J.-y.; Li, X. et al. (2012): Effects of electroconvulsive
stimulation on long-term potentiation and synaptophysin in the hippocampus of rats
with depressive behavior. In: The journal of ECT 28 (2), S. 111–117. DOI:
10.1097/YCT.0b013e31824a47ca.
Lisman, J.; Yasuda, R.; Raghavachari, S. (2012): Mechanisms of CaMKII action in long-term
potentiation. In: Nature reviews. Neuroscience 13 (3), S. 169–182. DOI:
10.1038/nrn3192.
Lledo, P. (1998): Postsynaptic Membrane Fusion and Long-Term Potentiation. In: Science
279 (5349), S. 399–403. DOI: 10.1126/science.279.5349.399.
Lohse, M.J.; Klotz, K.N.; Schwabe, U.; Cristalli, G.; Vittori, S.; Grifantini, M. (1988): 2-
Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine
receptors. In: Naunyn-Schmiedeberg's archives of pharmacology 337 (6), S. 687–689.
Lopes, L.V.; Cunha, R.A.; Kull, B.; Fredholm, B.B.; Ribeiro, J.A. (2002): Adenosine A2A
receptor facilitation of hippocampal synaptic transmission is dependent on tonic A1
receptor inhibition. In: Neuroscience 112 (2), S. 319–329. DOI: 10.1016/S0306-
4522(02)00080-5.
Lopes, L.V.; Cunha, R.A.; Ribeiro, J.A. (2000): Cross talk between A1 and A2A adenosine
receptors in the hippocampus and cortex of young adult and old rats. J Neurophysiol.
In: Journal of Neurophysiology 82 (6), S. 3196–3203. Online verfügbar unter
https://www.researchgate.net/profile/Luisa_Lopes3/publication/12700869_Cross_talk
_between_A1_and_A2A_adenosine_receptors_in_the_hippocampus_and_cortex_of_y
oung_adult_and_old_rats_J_Neurophysiol/links/0f3175367fee699f99000000.pdf.

69 Literaturverzeichnis
Luchkina, N.V.; Huupponen, J.; Clarke, V.R.J.; Coleman, S.K.; Keinänen, K.; Taira, T.;
Lauri, S.E. (2014): Developmental switch in the kinase dependency of long-term
potentiation depends on expression of GluA4 subunit-containing AMPA receptors. In:
Proceedings of the National Academy of Sciences of the United States of America 111
(11), S. 4321–4326. DOI: 10.1073/pnas.1315769111.
Lupica, C.R.; Cass, W.A.; Zahniser, N.R.; Dunwiddie, T.V. (1990): Effects of the selective
adenosine A2 receptor agonist CGS 21680 on in vitro electrophysiology, cAMP
formation and dopamine release in rat hippocampus and striatum. In: The Journal of
pharmacology and experimental therapeutics 252 (3), S. 1134–1141.
Lüscher, C.; Xia, H.; Beattie, E.C.; Carroll, R.C.; Zastrow, M. von; Malenka, R.C.; Nicoll,
R.A. (1999): Role of AMPA Receptor Cycling in Synaptic Transmission and
Plasticity. In: Neuron 24 (3), S. 649–658. DOI: 10.1016/S0896-6273(00)81119-8.
Lynch, M.A. (2004): Long-term potentiation and memory. In: Physiol Rev 84 (1), S. 87–136.
DOI: 10.1152/physrev.00014.2003.
Makino, H.; Malinow, R. (2009): AMPA Receptor Incorporation into Synapses during LTP.
The Role of Lateral Movement and Exocytosis. In: Neuron 64 (3), S. 381–390. DOI:
10.1016/j.neuron.2009.08.035.
Malenka, R.C.; Bear, M.F. (2004): LTP and LTD: an embarrassment of riches. In: Neuron 44
(1), S. 5–21. DOI: 10.1016/j.neuron.2004.09.012.
Manzoni, O.; Manabe, T.; Nicoll, R. (1994): Release of adenosine by activation of NMDA
receptors in the hippocampus. In: Science 265 (5181), S. 2098–2101. DOI:
10.1126/science.7916485.
Markram, H. (1997): Regulation of Synaptic Efficacy by Coincidence of Postsynaptic APs
and EPSPs. In: Science 275 (5297), S. 213–215. DOI: 10.1126/science.275.5297.213.
Massey, P.V.; Bashir, Z.I. (2007): Long-term depression: multiple forms and implications for
brain function. In: Trends Neurosci 30 (4), S. 176–184.
McCarley, R.W. (2007): Neurobiology of REM and NREM sleep. In: Sleep medicine 8 (4), S.
302–330. DOI: 10.1016/j.sleep.2007.03.005.
Mendonca, A. de; Sebastiao A.M.; Ribeiro J.A. (1995): Inhibition of NMDA receptor-
mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor
activation. In: Neuroreport 6 (8), S. 1097–1100.
Mendonça, A.d.; Ribeiro, J.A. (1996): Adenosine and neuronal plasticity. In: Life Sciences 60
(4–5), S. 245–251. DOI: 10.1016/S0024-3205(96)00544-9.
Mendonça, A. de; Ribeiro, J.A. (1994): Endogenous adenosine modulates long-term
potentiation in the hippocampus. In: Neuroscience 62 (2), S. 385–390. DOI:
10.1016/0306-4522(94)90373-5.

70 Literaturverzeichnis
Mihara, T.; Mihara, K.; Yarimizu, J.; Mitani, Y.; Matsuda, R.; Yamamoto, H. et al. (2007):
Pharmacological Characterization of a Novel, Potent Adenosine A1 and A2A
Receptor Dual Antagonist, 5-[5-Amino-3-(4-fluorophenyl)pyrazin-2-yl]-1-
isopropylpyridine-2(1H)-one (ASP5854), in Models of Parkinson's Disease and
Cognition. In: J Pharmacol Exp Ther 323 (2), S. 708–719. DOI:
10.1124/jpet.107.121962.
Moore, K.A.; Nicoll, R.A.; Schmitz, D. (2003): Adenosine gates synaptic plasticity at
hippocampal mossy fiber synapses. In: PNAS 100 (24), S. 14397–14402. DOI:
10.1073/pnas.1835831100.
Morgan, S.L.; Teyler, T.J. (2001): Electrical Stimuli Patterned After the Theta-Rhythm
Induce Multiple Forms of LTP. In: Journal of Neurophysiology 86 (3), S. 1289–1296.
Online verfügbar unter http://jn.physiology.org/content/jn/86/3/1289.full.pdf.
Mori, A. (2014): Adenosine Receptors in Neurology and Psychiatry: Elsevier Science. Online
verfügbar unter https://books.google.de/books?id=pgZ0AwAAQBAJ.
Mouro, F. (2012): Modulation of NMDA receptor activity through adenosine A2A receptors
in the hippocampus. Med. Diss. Universität Lissabon.
Nair, A.; Vaidya, V.A. (2006): Cyclic AMP response element binding protein and brain-
derived neurotrophic factor: Molecules that modulate our mood? Journal of
Biosciences. In: J Biosci 31 (3), S. 423–434. DOI: 10.1007/BF02704114.
Nakajima, T. (1990): N-ethylmaleimide uncouples muscarinic receptors from acetylcholine-
sensitive potassium channels in bullfrog atrium. In: The Journal of General
Physiology 96 (4), S. 887–903. DOI: 10.1085/jgp.96.4.887.
Neher, E.; Sakmann, B. (1976): Single-channel currents recorded from membrane of
denervated frog muscle fibres. In: Nature 260 (5554), S. 799–802.
Neves, G.; Cooke, S.F.; Bliss, T.V.P. (2008): Synaptic plasticity, memory and the
hippocampus: a neural network approach to causality. In: Nature Reviews
Neuroscience 9 (1), S. 65–75. DOI: 10.1038/nrn2303.
Nibuya, M.; Nestler, E.J.; Duman, R.S. (1996): Chronic antidepressant administration
increases the expression of cAMP response element binding protein (CREB) in rat
hippocampus. In: J. Neurosci. 16 (7), S. 2365–2372. Online verfügbar unter
http://www.jneurosci.org/content/16/7/2365.full.pdf.
Nishimune, A.; Isaac, J.T.; Molnar, E.; Noel, J.; Nash, S.; Tagaya, M. et al. (1998): NSF
Binding to GluR2 Regulates Synaptic Transmission. In: Neuron 21 (1), S. 87–97.
DOI: 10.1016/S0896-6273(00)80517-6.
Nissen, C.; Holz, J.; Blechert, J.; Feige, B.; Riemann, D.; Voderholzer, U.; Normann, C.
(2010): Learning as a model for neural plasticity in major depression. In: Biological
psychiatry 68 (6), S. 544–552. DOI: 10.1016/j.biopsych.2010.05.026.
Nobelprize.org (2014): The Nobel Prize in Physiology or Medicine 1991. Nobel Media AB.
Online verfügbar unter
http://www.nobelprize.org/nobel_prizes/medicine/laureates/1991/, zuletzt geprüft am
05.05.2016.

71 Literaturverzeichnis
Noel, J.; Ralph, G.; Pickard, L.; Williams, J.; Molnar, E.; Uney, J.B. et al. (1999): Surface
Expression of AMPA Receptors in Hippocampal Neurons Is Regulated by an NSF-
Dependent Mechanism. In: Neuron 23 (2), S. 365–376. DOI: 10.1016/S0896-
6273(00)80786-2.
Nörenberg, W.; Wirkner, K.; Illes, P. (1997): Effect of adenosine and some of its structural
analogues on the conductance of NMDA receptor channels in a subset of rat
neostriatal neurones. In: British Journal of Pharmacology 122 (1), S. 71–80. DOI:
10.1038/sj.bjp.0701347.
Normann, C.; Schmitz, D.; Fürmaier, A.; Döing, C.; Bach, M. (2007): Long-term plasticity of
visually evoked potentials in humans is altered in major depression. In: Biological
psychiatry 62 (5), S. 373–380. DOI: 10.1016/j.biopsych.2006.10.006.
Numberger, Markus; Draguhn, Andreas (1996): Patch-Clamp-Technik. Heidelberg [u.a.]:
Spektrum, Akad. Verl. (Labor im Fokus).
O'Kane, E.; Stone, T.W. (1998): Interaction between adenosine A1 and A2 receptor-mediated
responses in the rat hippocampus in vitro. In: European journal of pharmacology 362
(1), S. 17–25. DOI: 10.1016/S0014-2999(98)00730-4.
Padamsey, Z.; Emptage, N. (2014): Two sides to long-term potentiation: a view towards
reconciliation. In: Philosophical transactions of the Royal Society of London. Series B,
Biological sciences 369 (1633), S. 20130154. DOI: 10.1098/rstb.2013.0154.
Pascual, O. (2005): Astrocytic Purinergic Signaling Coordinates Synaptic Networks. In:
Science 310 (5745), S. 113–116. DOI: 10.1126/science.1116916.
Passafaro, M.; Piëch, V.; Sheng, M. (2001): Subunit-specific temporal and spatial patterns of
AMPA receptor exocytosis in hippocampal neurons. In: Nat Neurosci 4 (9), S. 917–
926. DOI: 10.1038/nn0901-917.
Paulsen, O.; Sejnowski, T.J. (2000): Natural patterns of activity and long-term synaptic
plasticity. In: Current opinion in neurobiology 10 (2), S. 172–179.
Pavlides, C.; Watanabe, Y.; McEwen, B.S. (1993): Effects of glucocorticoids on hippocampal
long‐term potentiation. In: Hippocampus 3 (2), S. 183–192. DOI:
10.1002/hipo.450030210.
Pinto-Duarte, A.; Coelho, J.E.; Cunha, R.A.; Ribeiro, J.A.; Sebastiao, A.M. (2005):
Adenosine A2A receptors control the extracellular levels of adenosine through
modulation of nucleoside transporters activity in the rat hippocampus. In: J
Neurochem 93 (3), S. 595–604. DOI: 10.1111/j.1471-4159.2005.03071.x.
Pittenger, C.; Duman, R.S. (2008): Stress, depression, and neuroplasticity: a convergence of
mechanisms. In: Neuropsychopharmacology : official publication of the American
College of Neuropsychopharmacology 33 (1), S. 88–109. DOI:
10.1038/sj.npp.1301574.
Plewnia, C.; Padberg, F. (2012): Transkranielle und invasive Hirnstimulationsverfahren bei
Depression. Der Nervenarzt. In: Nervenarzt 83 (8), S. 1006–1012. DOI:
10.1007/s00115-012-3573-y.

72 Literaturverzeichnis
Pougnet, J.-T.; Toulme, E.; Martinez, A.; Choquet, D.; Hosy, E.; Boué-Grabot, E. (2014):
ATP P2X receptors downregulate AMPA receptor trafficking and postsynaptic
efficacy in hippocampal neurons. In: Neuron 83 (2), S. 417–430. DOI:
10.1016/j.neuron.2014.06.005.
Purves, Dale; Mooney, Richard D.; Platt, Michael L. (2012): Neuroscience. 5th ed.
Sunderland (MA): Sinauer Associates.
Radecki, D.T.; Brown, L.M.; Martinez, J.; Teyler, T.J. (2005): BDNF protects against stress-
induced impairments in spatial learning and memory and LTP. In: Hippocampus 15
(2), S. 246–253. DOI: 10.1002/hipo.20048.
Rebola, N.; Coelho, J.E.; Costenla, A.R.; Lopes, L.V.; Parada, A.; Oliveira, C.R. et al. (2003):
Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the
hippocampus of kindled rats. In: European Journal of Neuroscience 18 (4), S. 820–
828. DOI: 10.1046/j.1460-9568.2003.02815.x.
Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. (2008): Adenosine A2A receptors are essential
for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. In:
Neuron 57 (1), S. 121–134. DOI: 10.1016/j.neuron.2007.11.023.
Rebola, N.; Rodrigues, R.J.; Lopes, L.V.; Richardson, P.J.; Oliveira, C.R.; Cunha, R.A.
(2005): Adenosine A1 and A2A receptors are co-expressed in pyramidal neurons and
co-localized in glutamatergic nerve terminals of the rat hippocampus. In:
Neuroscience 133 (1), S. 79–83. DOI: 10.1016/j.neuroscience.2005.01.054.
Regehr, W.G.; Carey, M.R.; Best, A.R. (2009): Activity-dependent regulation of synapses by
retrograde messengers. In: Neuron 63 (2), S. 154–170. DOI:
10.1016/j.neuron.2009.06.021.
Rex, C.; Kramár, E.; Ll, C.; B, L.; Cm, G.; G, L. (2005): Long-term potentiation is impaired
in middle-aged rats: regional specificity and reversal by adenosine receptor
antagonists. In: J Neurosci 25 (25), S. 5956–5966. DOI: 10.1523/JNEUROSCI.0880-
05.2005.
Rodrigues, R.J.; Almeida, T.; Richardson, P.J.; Oliveira, C.R.; Cunha, R.A. (2005): Dual
presynaptic control by ATP of glutamate release via facilitatory P2X1, P2X2/3, and
P2X3 and inhibitory P2Y1, P2Y2, and/or P2Y4 receptors in the rat hippocampus. In:
The Journal of neuroscience : the official journal of the Society for Neuroscience 25
(27), S. 6286–6295. DOI: 10.1523/JNEUROSCI.0628-05.2005.
Ruhé, H.G.; Mason, N.S.; Schene, A.H. (2007): Mood is indirectly related to serotonin,
norepinephrine and dopamine levels in humans: a meta-analysis of monoamine
depletion studies. In: Molecular psychiatry 12 (4), S. 331–359. DOI:
10.1038/sj.mp.4001949.
Sacktor, T.C. (2010): How does PKMζ maintain long-term memory? In: Nat Rev Neurosci 12
(1), S. 9–15. DOI: 10.1038/nrn2949.
Schiller, D.; Eichenbaum, H.; Buffalo, E.A.; Davachi, L.; Foster, D.J.; Leutgeb, S.;
Ranganath, C. (2015): Memory and Space: Towards an Understanding of the
Cognitive Map. In: The Journal of neuroscience : the official journal of the Society for
Neuroscience 35 (41), S. 13904–13911. DOI: 10.1523/JNEUROSCI.2618-15.2015.

73 Literaturverzeichnis
Schmidt, Robert F.; Lang, Florian; Heckmann, Manfred (Hg.) (2010): Physiologie des
Menschen. S. 95. 31., überarb. und aktualisierte Aufl. Heidelberg: Springer-Medizin-
Verl. (Springer-Lehrbuch).
Schmidt-Hieber, C.; Jonas, P.; Bischofberger, J. (2004): Enhanced synaptic plasticity in newly
generated granule cells of the adult hippocampus. In: Nature 429 (6988), S. 184–187.
DOI: 10.1038/nature02553.
Scholz, K.P.; Miller, R.J. (1996): Presynaptic inhibition at excitatory hippocampal synapses:
development and role of presynaptic Ca2+ channels. In: Journal of Neurophysiology
76 (1), S. 39–46. Online verfügbar unter
http://jn.physiology.org/content/jn/76/1/39.full.pdf.
Sebastião, A.; Ribeiro, J. (2009): Tuning and Fine-Tuning of Synapses with Adenosine. In:
Current Neuropharmacology 7 (3), S. 180–194. DOI: 10.2174/157015909789152128.
Sebastião, A.M.; Cunha, R.A.; Mendonça, A. de; Ribeiro, J.A. (2000): Modification of
adenosine modulation of synaptic transmission in the hippocampus of aged rats. In:
British Journal of Pharmacology 131 (8), S. 1629–1634.
Seifert, Gabriel (2009): Langzeitpotenzierung in der CA1-Region. Verschiedene
Modulatoren. Med. Dissertation. Universität Freiburg i.Br.
Serafini, G. (2012): Neuroplasticity and major depression, the role of modern antidepressant
drugs. In: World journal of psychiatry 2 (3), S. 49–57. DOI: 10.5498/wjp.v2.i3.49.
Serchov, T.; Clement, H.-W.; Schwarz, M.K.; Iasevoli, F.; Tosh, D.K.; Idzko, M. et al.
(2015): Increased Signaling via Adenosine A1 Receptors, Sleep Deprivation,
Imipramine, and Ketamine Inhibit Depressive-like Behavior via Induction of
Homer1a. In: Neuron 87 (3), S. 549–562. DOI: 10.1016/j.neuron.2015.07.010.
Shapiro, M. (1994): Angiotensin II inhibits calcium and M current channels in rat sympathetic
neurons via G proteins. In: Neuron 12 (6), S. 1319–1329. DOI: 10.1016/0896-
6273(94)90447-2.
Shen, H.-Y.; Chen, J.-F. (2009): Adenosine A2A Receptors in Psychopharmacology:
Modulators of Behavior, Mood and Cognition. In: Current Neuropharmacology 7 (3),
S. 195–206. DOI: 10.2174/157015909789152191.
Shen, K.; Johnson, S.W. (1997): Presynaptic GABAB and adenosine A1 receptors regulate
synaptic transmission to rat substantia nigra reticulata neurones. In: The Journal of
Physiology 505 (1), S. 153–163. DOI: 10.1111/j.1469-7793.1997.153bc.x.
Song, S.; Miller, K.D.; Abbott, L.F. (2000): Competitive Hebbian learning through spike-
timing-dependent synaptic plasticity. In: Nature neuroscience 3 (9), S. 919–926. DOI:
10.1038/78829.
Stahnisch, F.W. (2003): Making the brain plastic: early neuroanatomical staining techniques
and the pursuit of structural plasticity, 1910-1970. In: Journal of the history of the
neurosciences 12 (4), S. 413–435. DOI: 10.1076/jhin.12.4.413.27917.

74 Literaturverzeichnis
Stanton, P.K.; Sejnowski, T.J. (1989): Associative long-term depression in the hippocampus
induced by hebbian covariance. In: Nature 339 (6221), S. 215–218. DOI:
10.1038/339215a0.
Stent, G.S. (1973): A physiological mechanism for Hebb's postulate of learning. In: Proc Natl
Acad Sci U S A 70 (4), S. 997–1001.
Sterlemann, V.; Rammes, G.; Wolf, M.; Liebl, C.; Ganea, K.; Müller, M.B.; Schmidt, M.V.
(2010): Chronic social stress during adolescence induces cognitive impairment in aged
mice. In: Hippocampus 20 (4), S. 540–549. DOI: 10.1002/hipo.20655.
Stolz, S. D. (2015): Synaptische Langzeitplastizität bei A1-Rezeptor‐Knockout und -
überexprimierenden Mäusen. Med. Dissertation. Universität Freiburg i. Br.
Sullivan, P.F.; Neale, M.C.; Kendler, K.S. (2000): Genetic epidemiology of major depression:
review and meta-analysis. In: The American journal of psychiatry 157 (10), S. 1552–
1562. DOI: 10.1176/appi.ajp.157.10.1552.
Suzuki, F.; Shimada, J.; Mizumoto, H.; Karasawa, A.; Kubo, K.; Nonaka, H. et al. (1992):
Adenosine A1 antagonists. 2. Structure-activity relationships on diuretic activities and
protective effects against acute renal failure. In: J. Med. Chem. 35 (16), S. 3066–3075.
DOI: 10.1021/jm00094a022.
Taubenfeld, S.M.; Stevens, K.A.; Pollonini, G.; Ruggiero, J.; Alberini, C.M. (2002): Profound
molecular changes following hippocampal slice preparation: loss of AMPA receptor
subunits and uncoupled mRNA/protein expression. In: Journal of Neurochemistry 81
(6), S. 1348–1360. DOI: 10.1046/j.1471-4159.2002.00936.x.
Tebano, M.T.; Martire, A.; Rebola, N.; Pepponi, R.; Domenici, M.R.; Grò, M.C. et al. (2005):
Adenosine A2A receptors and metabotropic glutamate 5 receptors are co‐localized and
functionally interact in the hippocampus: a possible key mechanism in the modulation
of N‐methyl‐d‐aspartate effects. In: Journal of Neurochemistry 95 (4), S. 1188–1200.
DOI: 10.1111/j.1471-4159.2005.03455.x.
Trepel, Martin (2015): Neuroanatomie. Struktur und Funktion - mit StudentConsult-Zugang.
6. Aufl. München: Urban & Fischer in Elsevier.
Trieu, B.H.; Kramár, E.A.; Cox, C.D.; Jia, Y.; Wang, W.; Gall, C.M.; Lynch, G. (2015):
Pronounced differences in signal processing and synaptic plasticity between piriform‐hippocampal network stages: a prominent role for adenosine. In: The Journal of
Physiology 593 (13), S. 2889–2907.
van Calker, D.; Biber, K. (2005): The Role of Glial Adenosine Receptors in Neural Resilience
and the Neurobiology of Mood Disorders. Neurochemical Research. In: Neurochem
Res 30 (10), S. 1205–1217. DOI: 10.1007/s11064-005-8792-1.
Videbech, P.; Ravnkilde, B. (2004): Hippocampal volume and depression: a meta-analysis of
MRI studies. In: The American journal of psychiatry 161 (11), S. 1957–1966. DOI:
10.1176/appi.ajp.161.11.1957.

75 Literaturverzeichnis
Volianskis, A.; Bannister, N.; Collett, V.J.; Irvine, M.W.; Monaghan, D.T.; Fitzjohn, S.M. et
al. (2013): Different NMDA receptor subtypes mediate induction of long-term
potentiation and two forms of short-term potentiation at CA1 synapses in rat
hippocampus in vitro. In: The Journal of Physiology 591 (Pt 4), S. 955–972. DOI:
10.1113/jphysiol.2012.247296.
Vorovenci, R.J.; Antonini, A. (2015): The efficacy of oral adenosine A2A antagonist
istradefylline for the treatment of moderate to severe Parkinson's disease. In: Expert
review of neurotherapeutics 15 (12), S. 1383–1390. DOI:
10.1586/14737175.2015.1113131.
Walsh, M.J.; Kuruc, N. (1992): The Postsynaptic Density. Constituent and Associated
Proteins Characterized by Electrophoresis, Immunoblotting, and Peptide Sequencing.
In: J Neurochem 59 (2), S. 667–678. DOI: 10.1111/j.1471-4159.1992.tb09421.x.
Wang, J.H.; Ma, Y.Y.; van den Buuse, M. (2006): Improved spatial recognition memory in
mice lacking adenosine A2A receptors. In: Experimental Neurology 199 (2), S. 438–
445. DOI: 10.1016/j.expneurol.2006.01.005.
Warner-Schmidt, J.L.; Duman, R.S. (2006): Hippocampal neurogenesis: opposing effects of
stress and antidepressant treatment. In: Hippocampus 16 (3), S. 239–249. DOI:
10.1002/hipo.20156.
Whitlock, J.R.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. (2006): Learning induces long-term
potentiation in the hippocampus. In: Science 313 (5790), S. 1093–1097.
Wieraszko, A.; Ehrlich, Y.H. (1994): On the role of extracellular ATP in the induction of
long-term potentiation in the hippocampus. In: Journal of Neurochemistry 63 (5), S.
1731–1738.
Wilson, Constance N.; Mustafa, S. Jamal (2009): Adenosine receptors in health and disease.
Dordrecht, New York: Springer (Handbook of experimental pharmacology, 0171-
2004, v. 193).
Winn, H.R.; Rubio, R.; Berne, R.M. (1981): Brain adenosine concentration during hypoxia in
rats. In: The American journal of physiology 241 (2), S. H235-42.
Wirkner, K.; Assmann, H.; Köles, L.; Gerevich, Z.; Franke, H.; Nörenberg, W. et al. (2000):
Inhibition by adenosine A2A receptors of NMDA but not AMPA currents in rat
neostriatal neurons. In: British Journal of Pharmacology 130 (2), S. 259–269. DOI:
10.1038/sj.bjp.0703234.
Wolff, C. von (2007): Die Wirkung selektiver Serotonin-Wiederaufnahme-Hemmer auf die
assoziative synaptische Langzeitplastizität im Hippocampus der Ratte. Med.
Dissertation, Universität Freiburg i. Br.
Wu, L.G.; Saggau, P. (1994): Adenosine inhibits evoked synaptic transmission primarily by
reducing presynaptic calcium influx in area CA1 of hippocampus. In: Neuron 12 (5),
S. 1139–1148.
Yamada, K.; McEwen, B.; Pavlides, C. (2003): Site and time dependent effects of acute stress
on hippocampal long-term potentiation in freely behaving rats. Experimental Brain
Research. In: Exp Brain Res 152 (1), S. 52–59. DOI: 10.1007/s00221-003-1519-0.

76 Literaturverzeichnis
Yang, Y.; Wang, X.-b.; Frerking, M.; Zhou, Q. (2008): Delivery of AMPA receptors to
perisynaptic sites precedes the full expression of long-term potentiation. In:
Proceedings of the National Academy of Sciences 105 (32), S. 11388–11393. DOI:
10.1073/pnas.0802978105.
Yao, Y.; Kelly, M.T.; Sajikumar, S.; Serrano, P.; Tian, D.; Bergold, P.J. et al. (2008): PKM
Maintains Late Long-Term Potentiation by N-Ethylmaleimide-Sensitive
Factor/GluR2-Dependent Trafficking of Postsynaptic AMPA Receptors. In: Journal of
Neuroscience 28 (31), S. 7820–7827. DOI: 10.1523/JNEUROSCI.0223-08.2008.
Yasuda, H.; Barth, A.L.; Stellwagen, D.; Malenka, R.C. (2003): A developmental switch in
the signaling cascades for LTP induction. In: Nature neuroscience 6 (1), S. 15–16.
DOI: 10.1038/nn985.
Yudowski, G.A.; Puthenveedu, M.A.; Leonoudakis, D.; Panicker, S.; Thorn, K.S.; Beattie,
E.C.; Zastrow, M. von (2007): Real-Time Imaging of Discrete Exocytic Events
Mediating Surface Delivery of AMPA Receptors. In: Journal of Neuroscience 27 (41),
S. 11112–11121. DOI: 10.1523/JNEUROSCI.2465-07.2007.
Yuste, R.; Bonhoeffer, T. (2001): Morphological changes in dendritic spines associated with
long-term synaptic plasticity. In: Annual review of neuroscience 24, S. 1071–1089.
DOI: 10.1146/annurev.neuro.24.1.1071.
Zur Nedden, S.; Hawley, S.; Pentland, N.; Hardie, D.G.; Doney, A.S.; Frenguelli, B.G.
(2011): Intracellular ATP Influences Synaptic Plasticity in Area CA1 of Rat
Hippocampus via Metabolism to Adenosine and Activity-Dependent Activation of
Adenosine A1 Receptors. In: J. Neurosci. 31 (16), S. 6221–6234. DOI:
10.1523/JNEUROSCI.4039-10.2011.

77 Eidesstattliche Versicherung
Eidesstattliche Versicherung

78 Danksagung
Danksagung
An erster Steller gilt mein herzlicher Dank für das Ermöglichen dieser Arbeit Herrn Prof. Dr.
Claus Normann. Während der Arbeit im Labor und während des Verfassens der
Dissertationsschrift konnte ich auf seine kompetente und geduldige Begleitung bauen und hatte
jederzeit einen guten Ansprechpartner.
Ebenso möchte ich mich bei Herrn Prof. Dr. Josef Bischofberger für die Übernahme des
Zweitgutachtens sehr bedanken.
Den Laborkollegen Konstantin Ehrenberger, Julia Knoll und Max Horn sei Dank, dass das
Lösen von technischen Problemen ein Leichtes wurde und dass mir die gemeinsame Zeit sehr
viel Freude bereitet hat.
Die Züchtung und Bereitstellung der transgenen Tiere übernahm dankenswerterweise Tsvetan
Serchov. Auch dem Team des Tierstalls im BioMed-Zentrum, im Besonderen Tanja Richter,
möchte ich für die Bereitstellung der gezüchteten Ratten danken.
Schließlich möchte ich besonders meinen Eltern für die weitreichende Unterstützung danken,
wodurch mir eine solche Arbeit überhaupt erst ermöglicht wurde.
Alisa danke ich neben Korrektur dieser Arbeit vor allem für ihre geduldige Unterstützung und
Begleitung.