1h-nmr studies of synthetic polypeptide models ofplasmodium falciparum circumsporozoite protein...
TRANSCRIPT

'H-NMR Studies of Synthetic Polypeptide Models of Plasmodium fakiparum
Circumsporozoite Protein Tandemly Repeated Sequence
G. ESPOSITO, ENIRICERCHE SPA, A. PESSI, and A. S. VERDINI, SCLA VO SPA, 00015 Monterotondo,
Romu, Italy
Synopsis
The major immunodominant region of the coating protein of Plasrmdiwn falnpurwn sporo- mites contains multiple tandem copies of the sequence Asn-Ala-Asn-Pro (NANP). Current efforts for the development of an antisporozoite vaccine are focused on the synthesis of polypeptides reproducing part of the circumsporozoite protein repeat sequence and, in an attempt to relate conformational properties and biological response, 'H-nmr one and two-dimensional studies of the synthetic models (NANP),NA and (NANP), were carried out in water and water/methanol mixtures, at 400 and 500 MHz. In water, (NANP), undergoes fast conformational averaging. At variance, in water/methanol, the molecule appears to adopt an extensive structure, but detailed analysis is impaired by high spectral degeneracy. Based on the results obtained with (NANP),NA and from preliminary experiments in water/trifluoroethanol, an interpretation is suggested for the (NANP), data in water/methanol in terms of a mixed sequence of BI-turns and half-turns (or/and yI-turns) around the positions Ni-l-pi-Ni+l.
INTRODUCTION
Malaria is still the most important parasitic disease worldwide, with an estimated population at risk of more than 1800 million people.
Much of the current effort devoted to the development of an antimalarial vaccine has focused on the sporozoite developmental stage of the parasite, after the pioneering work of R. Nussenzweig and colleagues, who demon- strated that immunization with radiation-attenuated sporozoites could pro- tect mice from subsequent challenge with viable sporozoites'; this finding was then extended to humans2
The immunodominant component of the sporozoite surface coat is the circumsporozoite (CS) protein, and more precisely a tandemly repeated se- quence, characteristic of any single species of plasmodia. In the case of Plasmodium fakiparum, the most diffused and deadly human parasite, the sequence is Asn-Ala-Asn-Pro, or NANP, repeated 41 times, and constitutes more than one third of the whole protein. It has been calculated that this sequence is repeated lo8 times on the sporozoite ~urface.~
Recombinant or synthetic (NANP), peptides as subunit vaccines have already been tried with limited succe8~.~7~
Among the different approaches under current consideration for the devel- opment of antigens capable of inducing high-titer antisporozoite antibodies, which are thought to play a role in protection, it has been proposed that an
Biopolymers, Vol. 28, 225-246 (1989) 8 1989 John Wiley & Sons, Inc. CCC oooS-3525/89/010225-22$04.00

226 ESPOSITO, PESSI, AND VERDINI
effective vaccine could be obtained by Using (NANP), analogues as immuno- gem6
By virtue of the Merent sequential arrangement or deletion of one or two amino acid residues of the tetrapeptide, (NANP), analogues such as (ANNP),, (NNP),, and (NP);-' contain in aqueous media significant amounts of chain segments with locally ordered structures, whose existence has been ascertained for the model polypeptides (NANP),NA and (NAN$'), in water/alcohol mixtures by means of 'H one-dimensional (1D) and two-dimensional (2D) nmr studies, as reported in the present paper.
The existence of common conformational motifs in (NANP), and its ana- logues is confirmed by the observation that antisporozoite antibodies of sera from infected individuals recognize (NANP), as well as all its analogues in an enzyme-linked immunosorbent assay (ELISA) t e S t . ' O
MATERIALS AND METHODS The nmr study here described was mainly carried out on (NANP),NA and
(NANP), was synthesized according to the scheme outlined in Fig. 1 and fractionated on Sephadex G-50 (85 X 2.6 cm column, eluted with 0.1M acetic acid at a flow rate of 36 mL/h): the fractions corresponding to an average MW of 2400 were p l e d and used in the present study; MW determination
(NANP),.
n-rro-osxi b-Am-On _____) & c - A m - r m ~ x l
OCCl/Wt.r.t. b e - Alo-On
Eoc-Alo-Am-Pro-ggxl4 H-Am-PrO-OExl
OCCt/HOEt.r.t. 1 00s-Am-W n-A10-AM-PPO-0sXI- Boc-ABhAlo-ABn-pro-OEx]
OCCl/Wt.r.t.
c i k o n
DCC1.r.t. ~-ABIPAlO-ABn-PrO-OPC- B0C-ABn-Alo-A.n-Pro-On
c1 c1
HC1.W-en-co-m-cn-co-m-cn-co I I I nc C& C C
I I
I I co
nc
co
nc
Fig. 1. Synthesis of (NANP),. Polymerization of HC1. NANP-OPCP in DMSO was initiated by TEA and carried out for 4 days at room temperature; the resulting polymer was precipitated from ethanol, washed, and freeze dried prior to gel chromatography.

'H-NMR OF SYNTHETIC MODELS OF NANP 227
was carried out on a TSK-SW gel column (Toyo-Soda, Tokyo, Japan) in denaturing eluent (6M guanidine chloride) as described in Ref. 11. (NANP),NA was synthesized by solid-phase methods as described in Ref. 12.
Peptide solutions were prepared using H,O/D,O 90 : 10, CD,OH/H,O 90 : 10, 87 : 13, and 85 : 15, and CD,OD/D,O 89 : 11 (the above compositions represent volumetric ratios). D,O and CD,OD came from Aldrich (Milwaukee, WI); CD,OH was from MSD (Montreal, Canada).
H-nmr measurements were carried out at 11,744 T (500.13 MHz) with a Bruker AM 500 spectrometer and at 9 39 T (400.00 MHz) with a Varian VXR 400 spectrometer, in the Fourier mode and quadrature dete~ti0n.l~ Unless otherwise indicated, the experiments were performed at 295 and 300 K.
Peptide concentrations and pH* values (pH*: uncorrected pH meter read- ing) were chosen as follows: 8.7-23 mM, pH* = 3.7 for (NANP),NA in CD,OH/H,O; 6.5-10.4 mM, pH* = 4-5.7 for (NANP), in CD,OH/H,O; 6.5 mM, pH* = 4.0 for (NANP), in CD,OD/D,O; 7.6 mM, pH* = 3.5 for (NANP), in H,O/D,O.
Chemical shift values were referred to sodium 3(trimethylsylil)2,2,3,3-tetra- deuteropropionate for aqueous solutions and to the CD,HOH isotopic impu- rity (3.31 ppm) in water/alcohol mixtures. 1D nmr spectra were obtained using 8,16, or 32 K memory addresses with 10 ppm spectral width (SW). The intense OH resonance was suppressed by 1-1.5 s presaturation, using selective continuous irradiation or DANTE trains.14
Scalar connectivities were obtained from 2D pure phase (pp) double quan- tum filtered (DQF) correlated spectroscopy (COSY)15 and 2D homonuclear total correlation spectroscopy (TOCSY)16 experiments. The latters were run according to the procedure of Bax and David7 and, after them, they will be referred to as 2D HOHAHA (homonuclear Hartmann-Hahn).
2D pp DQF COSY data were acquired collecting 400-500 blocks (2K/block, 32-64 scans/block). Solvent suppression was achieved by selective continuous irradiation during the resuming delay (1.2 s) and, at a reduced power level, also during the evolution time ( t , ) . Quadrature detection in Fl was obtained using the time proportional phase incrementation (TPPI) method.18 Prior to 2D Fourier transform (FT), the experimental matrix was treated with a shifted sine bell function ( ~ / 3 - ~ / 9 radians shift in t , and t,, respectively) and zero filled in t,, in order to obtain a 1K x 1K real matrix, corresponding to 4.7 Hz/point digital resolution in both frequency domains.
2D pp HOHAHA experiments were performed for (NANP),NA in CD,OH/H,O. The OH resonance was presaturated using a DANTE traid4 as recently reported.lg During the mixing time (50 ms) spin-lock was achieved by means of MLEV17l' train, with an effective field of 7.5 KHz. Quadrature detection in F, was accomplished using the TPPI method.18 The experimental matrix dimensions were 700 X 2K (SW, = SW, = 5OOO Hz). Sine bell func- tions, s/6 and s/4 radians shifted in t , and t,, respectively, were applied, followed by zero filling in t,, to obtain, after 2D FT, a final 2K x 2K matrix (digital resolution 4.9 Hz/point). A high-resolution 2D pp COSY experiment was also run for (NANP), in CD,OH/H,O 85 : 15 at 400 MHz, at T = 304 K to measure the amide coupling constants. The experimental matrix dimen- sions were 1400 X 6848 (tl x t,) and 64 scans were collected for each t, increment. The acquisition times were 0.978 s in t, and 0.200 s in tl (i.e., 1.02 and 2.5 Hz resolution). Quadrature detection in was obtained according to
1

228 ESPOSITO, PESSI, AND WRDINI
States et d 2 0 ; including 0.65 s selective solvent preirradiation, the total experiment lasted 42 h. Mild resolution enhancement filters were applied and zero filling was performed in both dimensions prior to 2D FT. The size of the final matrix was 4K x 2K reals, with a digital resolution of 0.85 and 1.71 Hz/point in F2 and F,, respectively. Dipolar connectivities were extracted from 2D pp nuclear Overhauser effect spectroscopy (NOESY21 and 2D pp rotating frame NOESY (ROESY)22-24 data, collected at 500 MHz. Quadrature detection in F, was obtained by the TPPI method. In NOESY experiments the solvent was suppressed by selective low-power continuous irradiation during the resuming delay (1.0-1.5 s, usually) and the mixing time (tm), whereas for ROESY a DANTE presaturation train was em~loyed . ’~*~~
For comparison purposes, the temperature and the sizes of all the experi- mental matrices were kept constant with respect to the corresponding scalar correlation experiments. In NOESY experiments, t , was chosen as follows: 95 f 5 ms in water, 189 f 9 ms in CD30H/H20. Limited randomization of t , (10-158) was always allowed to suppress coherent magnetization transfers among scalarly coupled nuclei.25 Special conditions were adopted in the acquisition of NOESY data for (NANP), in CD30H/H20 85 : 15 to be used for quantitative purposes. The experimental resolution was improved in tl (880 increments collected) and the temperature was kept at 293 f 1 K. Two separate data sets were acquired with different t , values (160 + 13% and 220 + 10% ms). Moderate gaussian filters were used in the processing and zero filling was applied only in t,. ROESY spectra were acquired only for (NANP),NA. Two experimental protocols were f ~ l l o w e d , ~ ~ . ~ ~ differing in the spinrlock mixing time procedure: in one case the spin-lock was achieved by a single continuous decoupler pulse (180 ms) with B2 = 5.4 kHz (20b), while in the other case a “chopped” spin-lock pulse sequence was used,24 composed of cycles ( ~ / 6 pulse, 7 , -delay, 10 7 ) of 180 ms overall duration. Reduction of tl and t2 ridges was attained for all the described 2D experiments, according to the suggestions of Ottig et al.26
RESULTS AND DISCUSSION
(NANP),NA Assignments
Table I lists the relevant m parameters for (NANP),NA in CD30H/H20. The assignments were deduced from the analysis of scalar and dipolar
connectivities, following the usual method of Wiuthrich et aL2’ Only the N1 resonances could be unambiguously identified from the 2D HOHAHA spec- trum, due to the lack of backbone amide resonances in this spin system. The remaining assignments were all inferred from ROESY spectra (Fig. 2).
It was necessary to resort to the rotating frame experiments because of the unfavorable tumbling rate of the molecule, T,, which was responsible for the absence of cross peaks in NOESY spectra (at 500 MHz 07, = 1.12, i.e., zero NOE when 7, = 0.32 ns). By following the sequential arNHi+, connectivities, A2 and N3 resonances are readily identified. The assignment of P4 and P8 come from their connectivities with N5 and N9, respectively. In addition, the amall chemical shift difference between P4 and P8 GCH2s is consistent with their correlation to N3 and N7 aCHs, respectively (not shown). A6 can be

TABLE I Relevant 'H-NMR Parameters of (NANP),NA, 11.8 mM in CQOH/H,O 90/10,
Obtained at 500 MHz, T = 295 K, pH* = 3.69"
J (Hz) cp (deg) 8 (ppm) -M/AT (ppb K-')
6.96 4.182 2.94 2.84 7.780 7.103
8.734 4.35 1.374
8.395 4.94 2.95 2.69 7.741 7.147
4.40 2.28 2.03 1.99 3.92 3.86
8.122 4.62 2.77 2.69 7.573 6.94!i
7.784 428 1.376
8.m 4.94 2.94 2.69 7.741 7.101
4.44 2.26 2.04 2.00 3.89 3.84
8.160 4.75 2.81 2.67 7.559 6.929
7.734 4.30 1.428
2.1
4.8 4.6
6.2
7.0
5.9 7.0
3.3
5.1 6.7
2.5
5 2
5.9 7.4
3.6
a-p1 7.4 u-& 5.6
or-NH 6.5 +76; +44; -
u-NH 7.4
U-NH 7.8
a-NH 7.2
u-NH 7.5
a-NH 8.4
- 78; - 162
- 85. - 155
-89; -151
-83; -157
-86; -154
-95; - 145
5.3 7.0
2.4 a-NH 7.3 -85; - - 155
"Some chamid shifts were obtained from 1D spectra (A8 - +O.O006 ppm), but mwt of them came from the 2D HOHAHA spectrum (A8 - fO.O1 ppm). Stereospecific assignments are tentatively given according to the literature (see Refs. 28 and 29).

230 ESPOSITO, PESSI, AND WRDINI
~ _ -
AZP 0 A 2 8 e . ew mmg AW
Nlo 0 AZo
I A2Nu L, --I
AZo 0
N3a .I NQNH
N3
NlbNl
N3.N76NH,
N1.N76NHZ
0
2 .o
PPU
3.0
F1
1.0
5.0
1, 8.0 8.8 8.8 8.4 8.2 6.0 1.8 7.6 7.4 . 7.2 7.0 6.8
ppu $
Fig. 2. ‘H 500 MHz 2D pure phase ROESY contour plot of (NANP),NA, 11.8 mM in CQOH/H,O. Amidealiphatic region connectivities. The corresponding low-field 1D region is also reported. The experiment was performed using a continuous spin-lock mixing period.%
distinguished from A10 (the C-teminal residue) because of the connectivities linking its spin system to the preceding and following N5 and N7. The identification of N9 and A10, just by exclusion, is consistent with the weak connectivity N9a-AlONH. The network of sequential connectivities is con- h e d by examination of the low-field region contour plot [Fig. 3(a)]. The stereospecific assignments of the /?-protons of the asparagine residues, re- ported in Table I, follow from previous evidence.%

'H-NMR OF SYNTHETIC MODELS OF NANP
F1
6.8
7.0
7.2
7.4
7.6
7.8
8.0
8.2
8.4
8.e
6.8
9 .O
F2 0.8 8.0 84 62 Bo 7.8 7.6 7.4 7.2 7.0 6.8 PPM
(a)
I F1
I'
Q
231
F2 88 8S 8 4 8.2 6.0 7.8 7 6 7 4 7.2 7.0 6.8 PPM
(b) Fig. 3. 'H 500 MHz 2D pure phase ROESY contour plot of (NANP),NA, 11.8 mM in
C&OH/H,O. NH-NH ConneCtivitieB. (a) Continuous spin-lock mixing period.= The cross peaks show opposite phase with respect to the diagonal, except for N5 and N96NH cross peaks (+). (b) Chopped spin-lock mixing period. Only sidechain amide exchange cross peaks are detectable, with the same phase as the diagonal.

232 ESPOSITO, PESSI, AND VERDINI
The attribution of the side-chain amide pairs to specific residues was inferred from the observed ROESY connectivities (Fig. 2). In the absence of contrary evidence, it was assumed that the experimental correlation array reflects intraresidue proximity relationships.
The distinction between syn (Z) and anti (E) members of each pair is only tentative and follows from Perrin et al.29
At reasonable contour levels, detectable connectivities were only observed in the continuous spin-lock ROESY spectrum.23 The “chopped” spin-lock proce-
did not give comparable results, as shown in Fig. 3@), despite the same mixing time employed in both experiments. Very weak cross. peaks could only be detected by careful examination of cross sections. Such a finding may be rationalized in terms of the theoretical analysis recently published by B ~ X . ~ The actual effective field in chopped spin-lock experiments is equivalent to the average value over the total duration of the cycle (pulse-delay). This means that in our experiment the actual yB2 was 0.63 KHz. In addition to the lack of sizable cross peaks, the chopped spin-lock ROESY contour plot of Fig. 3(b) also shows interesting features for the exchange cross peaks of the side-chain primary amides. Namely, these all exhibit the same phase (positive) as the diagonal, in contrast with the corresponding data obtained in continuous spin-lock ROESY [Fig. 3(a)]. The latter gave positive side-chain amide cross peaks only for N5 and N9, the remaining being negative (N3 and N7) or missing (Nl).
The result can be ascribed to the relative magnitudes of cross relaxation (a) and syn-anti exchange (k ) rates. Wh$n the latter is slow, the cross relaxation between two amide protons (- 1.73 A apart) may become the predominant mechanism of magnetization transfer in Overhauser spectroscopy. In the rotating frame the sign of cross relaxation is always positive, as opposed to the magnetization transfer by slow chemical exchange, in which the magnetization is simply redistributed. This means that the corresponding cross peaks in 2D rotating frame exchange experiments will manifest opposite signs. The extent to which dipolar interaction will compete with chemical exchange for magneti- zation transfer will be determined by the actual value of u with respect to k. Because of the similar chemical shift range of syn and anti S-amide reso- nances in (NANP),NA (Table I), similar offset effects, i.e., similar a,, for all pairs), it can be concluded that the sign and the intensity changes observed for N3, N7, and N1 amide crow peaks are due to variations of ueff terms in differing spin-lock conditions, the k values being, of course, unaffected be- cause the experimental conditions were kept constant. Quantitative details will be presented elsewhere (Esposito et al., in preparation), but it is clear that the syn-anti S-amide exchange rate is slower for N3, N7, and N1
Table I also reports the values of the J N H u coupling constants, deduced from high-resolution 1D spectra, along with the corresponding values for ‘p
(KN37 KW7 < K N I < KN5, KN9).
angies.31
(NANP),NA: Concentration and Temperature Dependence
The chemical shifts of (NANP),NA (Table I) did not show concentration dependence within the range of 8.7-23 mM. This finding, along with the

'H-NMR OF SYNTHETIC MODELS OF NANP 233
narrow line widths always observed and the absence of NOESY cross peaks, indicates that no aggregation occurs within the investigated range and that the observed species is most likely monomeric.
The thermal coefficients of the amide chemical shifts, measured in the range of 296-316 K, are also listed in Table I. The usual interpretation of these data is based on the behavior in DMSO, but it can be extended to methanolic solutions, as c o n h e d by previous results32 and by isotope exchange rate measurements, carried out with (NANP), (vide infra).
It can be concluded that the N5, N9, A6, and A10 backbone NHs are not solvent exposed. The side-chain NHs do not exhibit particularly small AS/AT values. Although solvent exchange rates for primary amides are larger than for secondary amides, one could expect that their H-bond involvement should still be reflected by the AS/AT value. This does not appear to be necessarily the case with primary amides at the solvent polypeptide interface, because an additional exchange process, the syn-anti interconversion, is superimposed to the solvent exchange. Any temperature increase will also bring about an increase in the syn-anti exchange rate, which may mask (reduce) differences in the intrinsic temperature coefficients of chemical shifts. This is especially true when the rate constants for the two proceases become very close.
In conclusion, the AS/AT value for side-chain primary amides may be scarcely informative about their solvent exposure. As previously discussed, slower syn-anti exchange rates are inferred from ROESY spectra for N1, N3, and N7 side-chain amides, and this line of evidence should represent a more reliable judgment criterion for possible H-bond involvements.
(NANP), Assignments
The nmr spectrum of (NANP), exhibits a very limited chemical shift dispersion in all the investigated solvent systems (Fig. 4). From the connedivi- ties traced in 2D DQF COSY spectra, in addition to the low-field asparagines and to N1, it is only possible to recognize four different spin systems, corresponding to groups of alanines, prolines, and two kinds of asparagines. Such resonance clustering is expected in the absence of secondary shifts due to structural organization, as observed in denatured proteins. The lack of dipolar connectivities in the NOESY spectrum does indicate that this must indeed be the case for (NANP), in water.
At variance, in water/methanol mixtures, the NOESY contour plot re- vealed a number of characteristic cross peaks, quantitatively consistent with an extensive structure.
The limited chemical shift spreading also observed in these conditions (Fig. 4) is likely to reflect a repetitive structure. Table I1 lists the assignments of (NANP), in CD30H/H20. The N1 spin system could be assigned from DFQ COSY. The two downfield-shifted amide resonances (8.27 and 8.21 ppm) belong to asparagine residues. One of them (8.27 ppm) could be tentatively assigned to N3, in analogy with the free tetrapeptide and (NANP),NA. The remaining assignments came from the analysis of NOESY results. Because of resonance overlap, the sequential assignment proceduren did not succeed in the identification of the individual residues of the molecule; it was only possible to follow the sequential connectivities up to P4, and to discriminate

Nxd
NH
f +
A 111
) NH
I
Nv6
NH
f
P(IV
l.9,
Fig. 4
. 'H
400
MH
z 1D
spec
trum
of (
NA
NP)
, in
CD
,OH
/H,O
.

'H-NMR OF SYNTHETIC MODELS OF NANP 235
TABLE I1 'H-NMR knsignments for (NANP),, 6.5 mM in C%OH/H,O 87/13, T = 300 K,
pH* = 5.4, Obtained at 500 MHz" ~~~
NX(I) x = 1 5 9,13,17,21 NH - 8.21 8.12 8.10 8.09 a 4.17 4.63 4.58 4.56 4.56 81 ProR 2.95 2.83 2.76 2.76 2.76 &Pros 2.83 2.78 2.69 2.69 2.69
NH 7.74 7.78 7.76 7.73 7.66 a 4.24 4.21 4.22 4.21 4.26 8 1.34 1.39 1.38 1.36 1.38
AX(W x = 2 6,10,14,18,22
x = 3 7,11,15,19,23 NX(II1) NH 8.27 7.98 7.96 7.92 7.90 a 4.85 4.89 4.89 4.94 4.97 81 ProR 2.88 2.88 2.88 2.95 3.01 82 Pros 2.74 2.74 2.74 2.71 2.90
81 2.30 82 1.98
4, s, 3.84
PX(Iv) X = 4,8,12, 16,20,24 a 4.44
Yl, Y2 2.03
Side-chain amides SNH, 7.78 7.75 7.68 7.58 7.56 6NHz 7.12 7.12 7.14 6.93 6.89
a c h e m i d shifts in ppm; A6 = kO.01 ppm. The stereospecific assignments are tentatively given according to Ref. 28. The identification of syn (6NH,) and anti (6NHE) amide protons follows from Ref. 29.
between asparagine residues preceding and following proline residues, hence- forth referred to as N(II1) and N(I), respectively. Analogously, the unresolved proline and alanine resonances will be referred to as P(1V) and A(I1). Figure 5 illustrates the relevant NOESY connectivities. The identification of N5 relies on the observation of weak NOESY cross peaks to ANH and Pi3 (A6 and P4) at 293 K. The same pattern might arise for N21, but resolution of the remaining C-terminal residues would be expected in this case. Nor can it arise from N23, because the characteristic NOES to the preceding alinine P-methyls were not observed [as opposed to N3 and N(II1) patterns]. An increased flexibility of the N-terminal moiety, compared to the rest of the molecule, is suggested by the weak intensity of the relative connectivities (undetectable at the contour level of Fig. 5), in agreement with the variation of the NOESY pattern observed at 300 K.
(NANP),: Aggregation in Water / Methanol
(NANP), chemical shift values were substantially constant within the investigated ranges of pH , concentration, and solvent composition. The resonance line widths in water/methanol were very much similar to those observed in pure water.
The conformational flexibility of the molecule in the latter solvent, inferred from the lack of NOE effects, is hardly conceivable for a stable aggregate, suggesting that this is also the case in water/methanol.
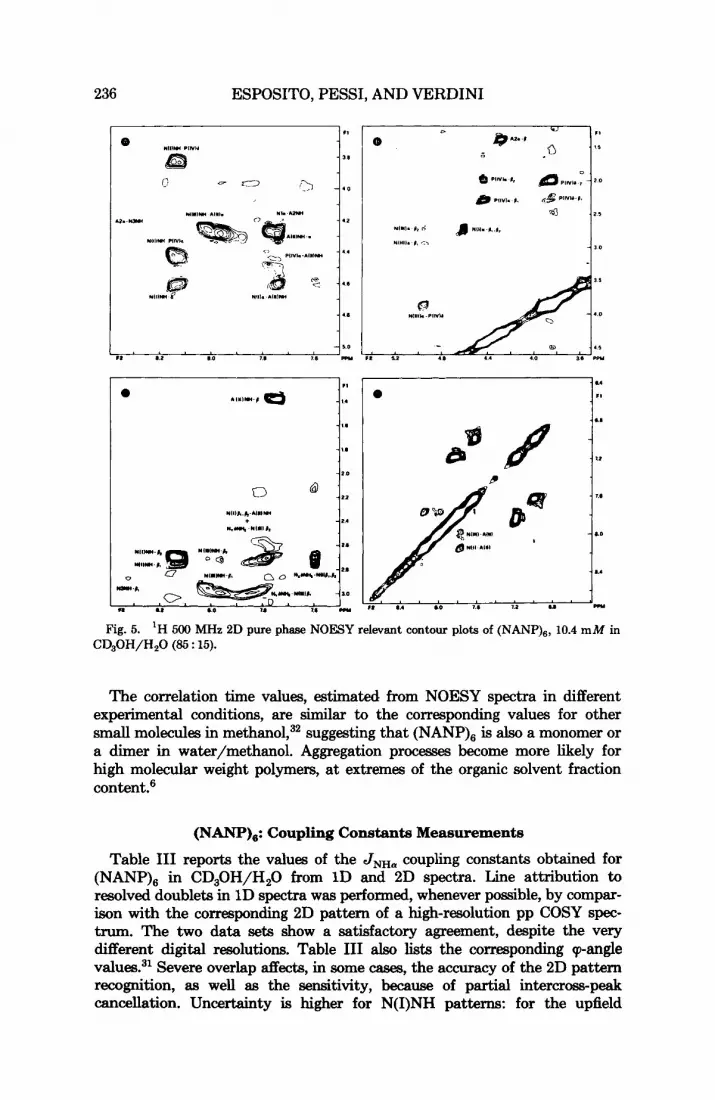
236 ESPOSITO, PESSI, AND VERDINI
I I *l
1
b
Fig. 5. 'H 500 MHz 2D pure phase NOESY relevant contour plots of (NANP),, 10.4 mM in CD,OH/H,O (85 : 15).
The correlation time values, estimated from NOESY spectra in different experimental conditions, are similar to the corresponding values for other s m d molecules in methanol,32 suggesting that (NmP), is also a monomer or a dimer in water/methanol. Aggregation processes become more likely for high molecular weight polymem, at extremes of the organic solvent fraction content?
(NANP),: Coupling Constants Measurements
Table I11 reports the values of the JNHn coupling constants obtained for (NANP), in CD30H/H20 from 1D and 2D spectra. Line attribution to resolved doublets in 1D spectra was performed, whenever possible, by compar- ison with the corresponding 2D pattern of a high-resolution pp COSY spec- trum. The two data sets show a satisfactory agreement, despite the very different digital resolutions. Table I11 also lists the corresponding cp-angle ~alues .~ ' Severe overlap affects, in some cases, the accuracy of the 2D pattern recognition, as well as the sensitivity, because of partial intercross-peak cancellation. Uncertainty is higher for N(1)NH patterns: for the upfield

'H-NMR OF SYNTHETIC MODELS OF NANP 237
TABLE I11 JNHa Coupling Constants and Corresponding 'p Torsion Angles for
(NANP), in CD30H/H,0
IDa 2Db
N3 N5 N(I)
N(II1)
7 .O 8.2 7.9
7.7 5.4' 7.2
7.3 7.7 7.6 7.7 7.9 7.3
7.2 8.3
8.2
5.7'
7.2
7.6 7.6 7.7
7.2
-82; -158 -92; -148 -90; -150
-88; - 152 +29; +91; - -84; - 156
-85; - 155 -88, - 152 -88, -152 -88, -152 -90, -150 -85; -155
-70; - 170
~~ ~ ~
a V a l ~ e s obtained from a 6.5 mM solution in CD30H/H,0 87/13, T = 299 K, pH' = 4.0.
bValues obtained from a 10.4 mM solution in CD30H/H,0 85/15, T = 304 K, pH* = 5.7.
'Only for some upfield components. Because of resonance overlap, the identification of the
A J = k0.2 Hz.
A J = kO.85 Hz.
corresponding pattern is not unambiguous.
resonances the experimental data do not allow us to distinguish unambigu- ously between two different values of JNHa (7.2 and 5.4 Hz).
(NANP),: Quantitative Analysis of Dipolar Effects
The cross-relaxation information encoded in NOESY cross peaks may be exploited to calculate internuclear distances. The underlying theory has been described in detail elsewhere.= Internuclear distances evaluated from NOESY spectra represent the experimental set for restrained molecular dynamics simulations.34 We shall be concerned with this topic in a forthcoming paper.
The extensive overlap of homologous residue resonances in (NANP), spec- tra poses some problems for the quantitative analysis of NOESY data. Linear polypeptide chains are often characterized by conformational flexibility. Therefore the dipolar effeds observed with (NANP), might mainly reflect the local order of partially structured molecules. Even small differences in local conformation may, in fact, signiscantly alter the extent of cross relaxation while leaving the average chemical shift substantially unaffected. Thus the evaluation of the cross-peak build-up rate% may be misleading for structural conclusions in this case. For these reasons we have used the diagonal/cross- peak intensity ratios.% When the diagonal peaks are sufficiently resolved, this method avoids the propagation of systematic errors arising from possible differences in the number of homologous site contributing to individual cross Peaks.

238 ESPOSITO, PESSI, AND VERDINI
Of course, the problem of different extents of cross relaxation within individual groups of unresolved resonances cannot be overcome. The calcu- lated distances are acceptable average estimates only over narrow conforma- tional distributions.
Two NOESY data sets, collected at different t , (160-220 ms), were exam- ined, and the integrals of 185 diagonal and cross peaks from F, cross sections were measured. The proline P-proton diagonal and cross peaks were employed for the evaluation of T~ and the obtained average value, 0.76 & 0.05 ns, was found in agreement with previous determinations for molelcules of similar size.32 The results of the analysis confirm that (NANP), is extensively structured in water/methanol. The intensity ratios could not assume the observed values for a partially structured molecule because the unstructured moiety resonances would still contribute to the diagonal.
(NANP),: Isotope Exchange Experiments
The observation of proton-deuterium exchange in 1D spectra of 6.5 mM (NANP), in CD,OD/D,O at 300 K proved scarcely informative. A detailed map of the slowly exchanging amides is barely defined when most of the labile proton resonances disappear within a few seconds from dissolution. Only peaks from the N(1)NHs and the GNHs at 7.14-7.12 ppm could be observed; also, these resonances did not last very long (3-4 min). Successive experiments were therefore performed at low temperatures.
We did not follow the time evolution of the residual integrals of the NHs resonances at constant temperatures. Rather, we measured the temperature dependence of the NH resonance integrals at equidistant time intervals (30 min), in the range of 266.8-289.4 K. Regardless of the actual kinetic mecha- nism (EX1 or EX2),r’ the polypeptide amide exchange follows an apparent first-order kinetic law:
if sufficient time has elapsed between dissolution and measurement.% In Eq. (1) [NH], the concentration of the protonated species, is measured from the residual amide integrals, and [NH],, the initial concentration, may be evalu- ated from the integrals of suitable nonlabile proton resonances [P(IV) PI in our case]. No preirradiation of the OH isotopic impurity was performed, to avoid systematic errors arising from saturation transfer. In our experimental conditions we may write d(ln a)/d(l/T) = - EJR, where T is the tempera- ture, E,, is the exchange activation energy, R the gas constant and In a = (l/t’) In ([NH],/[NH]) (t’ is the constant time interval). The E, values for free amide exchange are known (17.5 kcal/mol at pH > 3 and 15.0 kcal/mol at pH < 3),% and their increase corresponds to the additional energy met necessary to break amide hydrogen bonds or, more generally, to enable the amide protons to exchange.
For (NANP), in water/methanol, the free amide exchange activation en- ergy iS likely to be similar to the corresponding value in water, as the insolubility of the m o l d e in pure methanol suggests that (NmP), is preferentially solvated by water in mixed aqueous solutions. This is in fact the

'H-NMR OF SYNTHETIC MODELS OF NANP 239
TABLE IV Apparent Activation Energy of H-D Amide Exchange for (NANP),, 6.3 mM in
CQOD/&O 89/11, pH* = 4.0, Obtained in the Temperature Range of 266.8-289.4 K"
E, apparent (kcal mol-') r
N3NH 14.0 - 0.982 N5NH 16.2 - 0.995 N(1)NH 22.3 - 0.971 N(I)NHcarr 17.5 - 0.983 N(II1)NH 15.9 - 0.972 N(III)NH, 12.4 - 0.986 A(I1)NH + NXGNHE 13.4 - 0.994
N,SNH, (7.78-7.68 ppm) 10.1 - 0.967
N,GNH, (7.14-7.12 ppm) 15.2 - 0.994 N,GNH, (6.93-6.89 ppm) 11.1 - 0.982
A(I1)NH 17.4 - 0.988
Ny GNHE (7.58-7.56 ppm) 10.4 - 0.969
"At low temperatures, N(1)NH and N(II1)NH resonances exhibit partial overlap. Kinetic parameters were therefore calculated accounting for this overlap [N(I)NH, and N(III)NHcorr] and neglecting it [N(I)NH and N(III)NH], i.e., considering only the resonances resolved through- out the entire temperature range inveatigated. Asparaghe side-chain amidea are indicated using their chemical shift range at 300 K. r is the correlation coefficient of the least-square fitting.
case with N3NH and some GNHs. From our data the apparent activation energy for N3NH exchange is 14.0 kcal/mol whereas a value of 10-11 kcal/mol is obtained for resolved NySNHE and NySNH, resonances, respec- tively, consistent with the expected difference between primary and secondary amides. In other case^ also, the spectral overlap affects the accuracy of the apparent
activation energy determination, as outlined in Table IV. Some conclusions may, however, be drawn, just considering the apparent En values with respect to the limiting values obtained for free primary and secondary amides. First, N(1)NH and N,GNH, (7.14-7.12 ppm) exhibit an average 45% increase of the apparent En, suggesting their involvement in H bonds; second, in the reso- nance envelope including A(I1)NH and NXGNHE, the low-field component [A(II)NH] shows some 30% increase in the apparent En value, while the upfield resonances give the same apparent En as the free-exchanging side-chain amides. This suggests that at least some of the A(1I)NHs are hydrogen bonded; last, a slight increase (15%) of the apparent En is found for N5NH. This, presumably, reflects the weakness of the H bond involving a residue homologous to N(I)NH, located in a flexible region of the molecule.
(NANP),NA. Structural Interpretation
Figure 6 summarize the relevant nmr structural data obtained for (NANP),NA. The intensities depicted in this diagram were directly deduced from inspection of the cross-peak contour levels. The dipolar connectivity pattern of (NANP),NA exhibits repeating features in homologous sequence fragments. In particular, the folding around P4N5 and P8N9 seems rather

240 ESPOSITO, PESSI, AND VERDINI
4 8
N A N P N A N P N A
1 d g c a -
65 7.4 7.8 7.2 7.5 8.4 7.3 JN,
slow m. m 8 exchange
Fig. 6. Relevant nmr structural information for (NANP),NA in water/methanol.
similar, with a pattern reminiscent of tight 8-turns. A BII-turn can be ruled out because of the J N H u value of N5 and N9.
The experimental data indicate values around - 90" or - 150" for cp5 and cp9, incompatible with a PI1 folding (where = +go", i.e., J N H u = 5 Hz). The same evidence ( J N H u # 5 Hz) has been recently used by Mcharfi and co-workersm to reject a BII-type folding for the N- and C-protected dipep- tides Pro-Asp(OMe), Pro-Asp and Pro-Asn, in CDC1,. Based also on addi- tional data, they proposed a PI-turn. For (NANP),NA in water/methanol, it is possible to rule out a PI-type turn, too, because of the observation of P4a-N5NH and P8a-NSNH ROES (Fig. 2), instead of the expected Pi& Ni+,NH connectivity. We conclude therefore that in the chosen experimental conditions, (NANP),NA adopts a rather extended conformation with two half-turns located at P4-N5 and P8-N9. Both proline residues are found in trans conformation.
The torsion angles expected for a half-turn at Pi-Ni+, are 'pi = -6O", I l / i = +120", 'pi+, = -90", and Il/i+l = 0". The observed PiP2-Ni+?NH con- nectivities (Fig. 2) could be due to the combined effects of pirrohdine ring puckering and deviation of the I l / i value (130°/1400). The proximity of Pi& and Ni+,NH, however, rules out the possibility of a yI-turn, where theu expected separation would be larger than 0.4 nm. Mirror @-turns (PI' and PII") can also be excluded, because of the rigid proline cp angle and the observed J N H a couplings.41
The presence of half-tums in (NANP),NA deserves some further comment. This unusual structural motif has been previously identified in rabbit liver metallothionein-2,41 using the same nmr arguments discussed above. In that case, however, the half-turn was somehow maintained by the folding of a 61-residue protein, heavily constrained by seven disulfide bridges.
In (NANP),NA the half-turns are maintained by hydrogen bonds. ROESY data indicate that the N3 and N7 pros P-protons (the upfield ones) are closer than the proR P 's to the a-proton (not shown), while the NH-P separation is approximately the same for both P ' s (Fig. 2). This is consistent with the values of the a-p coupling constants in N3 and N7, obtained by simulation of the corresponding multiplets ( JpsR = 9.4 Hz; J,,, = 4.6 Hz). Thus xf = x\ = 180". In view of the extended backbone conformation of N3 and N7 (from ROESY connectivities (p, = (p7 = -150" and J/, = t,b7 = +120°), this side- chain torsion angle is particularly well suited for H-bond formation. The P4 and P8 carbonyls are, in fact, close to the SNHs of the preceding asparagine residues. Therefore, the slow syn-anti exchange inferred from ROESY results

'H-NMR OF SYNTHETIC MODELS OF NANP 241
\- Y I
I
Fig. 7. Conformation of (NANP),NA as inferred from nmr data.
for N3 and N7 6NH may be rationalized in terms of H bond to the P4 and P8 carbonyls.
Along the same lines, the conformation of the Nl side chain can be deduced. In this case xi = +60"/+ 80", and favourable interactions, such as N16NHE-A!2C0, N16NHz-NlCO, and NlNHi-Nly CO, may conceivably arise from local conformational fluctuations. On the other hand, the low-tem- perature coefficients of N5, A6, N9, and A10 NHs can be accounted for in terms of reduced solvent accessibility, due to either H-bond formation to N5yCO and N9yCO or steric hindrance. For N9 the available evidence suggests an average side-chain conformation (x', = - So"), while for N5 limit- ing xi values of -60" and +So" are deduced. It is worth mentioning that, analogously to the latter features, all the /?I-turns found by Mcharfl et al. in PN- and PD-protected dipeptides were further stabilized by intraresidue hydrogen bonding in N- or D-residue (CXNH-~CO).~ Figure 7 shows the conformation of (NANP),NA inferred from the present analysis. The confi- dence limits of the proposed structure are mainly due to the stereospecific assignments of the asparagine /?-protons and to possible scalar transfers and offset effects in ROESY spectra.3o In view of the dimensions of the examined molecule, a more realistic picture should also account for some conformational flexibility. The extent of the fluctuation appears, however, limited, as judged from the consistency of independent nmr parameters.
(NANP),: Structural Interpretation
Figure 8 shows the relevant nmr structural parameters obtained for (NANP), in water/methanol. By comparison with Fig. 6, reporting the corresponding data for (NANP),NA, a number of analogies can be readily seen. There is,

242 ESPOSITO, PESSI, AND VERDINI
N1 A 2 N3 P4 N5 ... . C d n - d NM - dP Jan 7.5 7.0 0.2
slow 0 exchange
- N I An NllC PIP
+ *
Fig. 8. Reievant nmr structural information for (NANP), in water/methanol. ( E) only at 300 K (0) partially slow exchange; (+) N,GNH, overlap; (*) N(I)NH-P(IV)& or N(1)NH-P(1V)y.
however, one remarkable difference, concerning the apparent incongruency in (NANP), of the N(1) and P(1V) connectivities. The observed P(1V)a-N(1)NH and P(1V)Q-N(1)NH NOES cannot arise from the same pair of consecutive residues. The distances evaluated from the cross peak to P(1V)a and P(1V)Q (and vice versa) are, respectively, 0.25 and 0.32 nm. No conformation is compatible with such values, even accounting for the overlap of P(1V)G proton cross peaks. One might assign the P(1V)a-N(1)NH NOE to consecutive pairs and the P(1V)G-N(1)NH to nonconsecutive pairs, or vice versa. Another possibility could be to assume that the observed correlations arise from every other P(1V)-N(1) pair (not necessarily alternated, however). In this case the calculated distances should be corrected. The resonances assigned to N(1)NH should account for four protons. If only two N(1)NHs are close to the preceding proline a-protons, their distance from the Q-protons of the same prolines is large (> 0.5 nm), leading to negligible contribution to the cross peak. Conversely, if the Gi-NHi,, separations of the remaining consecutive P(1V)-N(1) pairs are smaller than 0.32 nm, the ar and the NH,,, protons cannot be closer than 0.34 nm, and the corresponding contribution to the cross peaks should be small. The lower distance limits become then 0.22/0.55 nm and 0.34/0.28 nm for P(1V)a-N(I)NH/P(IV)Q-N(1)NH from different consec- utive pairs. An iterative procedure might be applied to reproduce the observed cross-peak intensities. The final results, however, will be only slightly different from the above values.
Within this framework, a number of structural proposals become plausible, because one has to decide about the distribution of secondary structure elements throughout the molecule, without any hint about their precise location along the primary sequence (a crucial point for high-resolution nmr studies of bipolymers).
The most reliable confonnational conclusion we can draw is based on the results obtained with (NANP),NA. Figure 9 illustrates the interpretation of the nmr structural data of Fig. 8 in terms of a mixed conformation, with alternated PI- and half-turns (and/or y,-turns). The alternation is arbitrary. No evidence in favor of B-sheet structures or cis proline has been detected. The JNHP values (Table 111) and the NOESY evidence are consistent with the mixed /3I/half-turn structure. The observation of weak P(1V)a-A(I1)NH cross peaks, expected in both PI (0.36 nm) and half-turn (0.33 nm), might lead

'H-NMR OF SYNTHETIC MODELS OF NANP 243
N I A I I N E t P I K N I AILNII IPIP
+ slow 6 .. exchange
ha If- t u rn ( X'
BI
Fig. 9. Interpretation of the nmr structural data of (NANP), (Fig. 8) in terms of a mixed sequence of BI-turns and half-turns (and/or yI). (+) Not for yI; (0) uncertain.
us to exclude the yI turn possibility, but an unambiguous conclusion cannot be reached because of the overlap of some A(I1)NH and N,G-NH, reso- nances.
The NH resonance of N5, a residue homologous to N(I), exhibits the connectivity to P(1V)S at 293 K and the connectivity to P(1V)a at 300 K. This result, while reflecting the flexibility of the N-terminal moiety, agrees with the interpretation of the P(1V)-N(1) NOES supporting the mixed BI/half-turn conformation (Fig. 9).
The amide slow exchanges, observed in isotope exchange experiments, can be rationalized in terms of H bonds.
In BI-turns the alanine NHs are hydrogen bound to the preceding N(II1) mbonyls, while in half-turns, again, we find one of the N(II1)S NHs H bonded to the next-door proline carbonyl. As previously seen with (NANP),NA, the asparagine side-chain conformation should be analyzed, to account for most of the hydrogen bonds. In (NANP),, with an in- creased number of overlapping NB multiplets, a detailed analysis is impossi- ble. We may notice that the asparagine side-chain NOESY pattern changes with respect to (NANP),NA. For N(I), the observed connectivities might arise from two different conformations, x1 = +60° and -6o", reflecting the N(1)NH-N(1)yCO hydrogen bond in BI-turns, and the simultaneous N(1)y-CO H bond to N(1)NH and A(I1)NH in half-turns [or the N(1II)CO-N(1)NH and the N(1)yCO-A(I1)NH H bonds in y,-turns]. On the other hand, for N(III), the pattern supporting a x1 = 180" is clearly distin- guished only at 300 K.
The mixed BI/half-turn conformation is reported in Fig. 10. Alternative interpretations are possible. If the P(V)a-N(1)NH connectivi-
ties are assigned to Pi-Ni+l pairs and the P(1V)G-N(1)NH connectivities are attributed to Ni+, - Pi+8 (Pz+4 is not possible because of the remaining experimental restraints), one ends up with a structure with tight turns at each Pi-Ni+, location. The experimental evidence could support turns^^ (yKturns are inverse type y-turns, similar to yI) and/or BII-tums.
preliminary results obtained in water/trifiuoroethanol suggest that (NANP), adopts mainly a BI folding at P,-Ni+, locations. This finding, along

244 ESPOSITO, PESSI, AND VERDINI
Fig. 10. Propod conformation for (NANP), in water/methanol.
with the experimental results obtained for (NANP),NA, leads us to favor the mixed BI/half-tum conformation for (NANP), in water/methanol.
CONCLUSIONS
The nmr spectrum of (NANP), exhibited a very narrow chemical shift dispersion for the resonances of homologous residues in all the investigated experimental conditions, suggesting either the lack of definite structure or a highly repetitive conformational arrangement. In aqueous solution, the absence of any intraresidue Overhauser effect in
the 2D NOESY spectrum indicates that the molecule undergoes fast confor- mational averaging.
At variance, characteristic and regular NOE patterns, quantitatively consis- tent with an extensive structure, were observed in water/methanol mixtures, but detailed analysis was impaired by spectral degeneracy.
The conformational motifs featured by the NOESY results of (NANP), were checked with the model peptide (NANP),NA in water/methanol solu- tion. Dipolar connectivities were observed in the 2D ROESY spectrum, and

'H-NMR OF SYNTHETIC MODELS OF NANP 245
the sequence-specific assignments as well as the conformation of this decapep- tide could be determined. (NANP),NA adopts, as a whole, an extended structure, with two half-tums located around N3P4N5 and N7P8N9, with the side-chain amides of N3 and N7 hydrogen bound to the P4 and P8 carbonyls, respectively. In addition, the N5 and N9 side chains are involved in H bonds to N5, A6, and N9, A10 backbone NHs. In view of these results, a possible interpretation for the nmr structural
data of (NANP), in water/methanol could suggest the presence of a mixed sequence of BI- and half-turns (or/and y,-tums) around the locations Ni-l-PiNi+i.
No evidence in favor of 8-sheet structures has been detected. Alternatively, the experimental data could also be interpreted in terms of
consecutive BII- or/and y,-turns, with contacts between two consecutive octapeptide units.
The latter interpretation is weakened by preliminary results obtained for (NANP), in water/trifluoroethanol, suggesting an increase of the BI pattern at the expense of the half-turn in the backbone, along with conformational changes of the asparagine side chains. Thus the overall structure seems rather to evolve toward a left-handed spiral, with proline residues at the edges of tight turns, in agreement with previous molecular mechanics predictions.43
The changes in dipolar connectivity patterns, observed with the increase of the molecular weight of the peptide and/or the organic character of the solvent, are accompanied by conformational variations of the asparagine side chains. The details of such variations are currently being investigated, but it can be anticipated that, in a molecule devoid of particularly relevant hy- drophobic clusters, the hydrophilic interactions of the asparagine side chains are likely to play a pivotal role in determining the conformation of the whole molecule.
We thank Prof. W. A. Gibbons for the uae of the Bruker AM 500 spectrometer, and Dr. A. Pastore for help with the computer graphics. We also thank M. Ehrbini, R. Richichi, A. Ludovici, S. Mercuri, and F. Bonelli for their assistance.
References 1. Nussenzweig, R., Vanderberg, J. & Most, H. (1969) Milit. Med. 134, 1176-1182. 2. Clyde, D. F., McCarthy, V. C., Miller, R. M. & Woodward, W. E. (1975) Am. J. Trop. Med.
3. Miller, L. H., Howard, R. J., Carter, R.,%ood, M. F., Nussenzweig, V. & Nussenzweig, R.
4. Ballou, W. R., H o b , S. L., Sherwood, J. A., Hollingdale, M. R., Neva, F. A., Hockmeyer, W. T., Gordon, T. M., Schneider, I., Wirtz, R. A., Young, J. F., Wanserman, F., Reeve, P., Digga,
5. Herrington, D. A., Clyde, D. F., Losonsky, G., Cortesia, M., Murphy, J. R. Davis, J., Baqar, S., Felix, A. M., Heimer, E. P., Gillessen, D., Nardin, E., Nussenzweig, R., Nussenzweig, V., Hollingdale, M. & Levine, M. M. (1987) Nahve 328,257-259.
6. Verdini, A. S. (1987) paper presented at the 1st Conference Jacques Monod: Molecular Approaches to Vaccination against Parasitic Diseases, Roscoff, France, September 1-4.
7. Verdini, A. S., Bonelli, F. & Pessi, A. (1987) Italian Patent Application 22948. 8. Verdini, A. S., P d , A. & Bonelli, F. (1987) Italian Patent Application 21892. 9. Verdini, A. S., Pessi, A. & Bonelli, F. (1988) Italian Patent Application 20888.
Hyg. 24, 397-401.
(1986) science 234,1349-1356.
C. L. & Choulay, J. D. (1987) Lancet 1, 1277-1281.
10. Verdini, A. S., Bonelli, F. & Pessi, A. To be published.

246 ESPOSITO, PESSI, AND VERDINI
11. Kato, Y., Komiya, K., Sasaki, H. & Hashimoto, T. (1980) J. Chromatog. 193, 458-463. 12. Togna, A. R., Del Giudice, G., Verdini, A. S., Bonelli, F., P&, A., Engers, H. & Corradin,
13. Hoult, D. I. & Richards, R. E. (1975) Proc. Roy. SOC. London, Ser. A 344, 311-340. 14. Morris, G. A. & Freeman, R. (1978) J. Magn. Reson. 29, 433-462. 15. Piantini, U., hensen , 0. W. & Emst, R. R. (1982) J. Am. Chem. SOC. 104, 6800-6801. 16. MUer, L. & Emst, R. R. (1979) MoZ. Phys. 38, 963-992. 17. Bax, A. & Davis, D. (1985) J. Magn. Reson. 65, 355-360. 18. Marion, D. & Wiithrich, K. (1983) B k h e m . Biophys. Res. Commun. 113,967-974. 19. Esposito, G., Gibbons, W. A. & Bazzo, R. (1988) J. Magn. Reson., in press. 20. States, D. J., Haberkom, R. A. & Ruben, B. J. (1982) J. Magn. Reson. 48, 286-292. 21. Jeener, J., Meier, B. H., Bachmann, P. & Emst, R. R. (1979) J. Chem. Phys. 7 l , 4546-4553. 22. Bother-By, A. A., Stephens, R. L., Lee, J., Warren, C. D. & Jeanloz, R. W. (1984) J. Am.
23. Bax, A. &Davis, J. (1985) J. Magn. Reson. 63, 207-213. 24. Kessler, H., Griesinger, C., Kesserbaum, R., Wagner, K. & Emst, R. R. (1987) J. Am.
25. Macura, S., Wiithrich, K. & Emst, R. R. (1982) J . Magn. Res. 46, 269-282. 26. Ottig, G., Widmer, H., Wagner, G. & Wiithrich, K. (1986) J. Magn. Reson. 66, 187-193. 27. Wiithrich, K., Wider, G., Wagner, G. & Braun, W. (1982) J. Mol. Biol. 166, 311-319. 28. Kainmho, M. & Ajisaka, K. (1975) J. Am. Chem. SOC. 97,5630-5631. 29. Perrin, C. L., Jonhston, E. R., Lollo, C. P. & Kobrin, P. A. (1987) J. Am. Chem. SOC. 103,
30. Bax, A. (1988) J. Mogn. Reson. 77, 134-147. 31. Pardi, A., Billeter, M. & Wiithrich, K. (1984) J. MoZ. Biol. 180,741-751. 32. Esposito, G., Carver, J. A., Boyd, J. & Campbell, I. D. (1987) Biochemistry, 26, 1043-1050. 33. Macura, S. & Ernst, R. R. (1980) MoZ. Phys. 41, 95-117. 34. Kaptein, R., Zuiderwerg, E. R. P., Scheek, R. M., W e n s , R. & van Gunsteren, W. F. (1985)
35. Anil Kumar, Wagner, G., Emst, R. R. & Wuthrich, K. (1981) J. Am. Chem. SOC. 103,
36. Espoaito, G. & Pastore, A. (1988) J. Magn. Reson. 76, 331-336. 37. Hvidt, A. & Nielsen, S. 0. (1966) Ado. Protein Chem. 21, 287-386. 38. Wedin, R. E., Delepierre, M., Dobson,>C. M. & Poulsen, F. M. (1982) Biochemistry 2l,
39. Eglander, S. W. & Kallenbach, N. R. (1984) Quart. Rev. Biophys. 16,521-655. 40. M c M , M., Aubry, A., Boussard, G. & Marraud, M. (1986) Eur. Biophys. J. 14,43-51. 41. Wagner, G., Neuhaus, D., Worgotter, E., Vasak, M., Kagi, J. H. R. & Wuthrich. K. (1986)
42. Bandekar, J. & Krimm, S. (1985) Znt. J. Peptide Protein Res. 26,407-415. 43. Gibson, K. D. & Scheraga, H. A. (1986) Proc. Natl. A d . Sci. USA 83, 5649-5653.
G. (1986) J. Z m m ~ n ~ l . 137, 2956-2960.
Chem. SOC. 106,811-813.
Chem. SOC. 109,607-609.
4691 -4696.
J . Mol. Bbl . 182,179-182.
3654-3658.
1098-1103.
J. Mol. Biol. 187, 131-135.
Received July 28, 1988 Accepted August 1,1988