1 in 33 newborns is born with a congenital abnormality
TRANSCRIPT


1 in 33 newborns is born with a congenital abnormality: these may be isolated or multiple and range from mild to severe. Between 2%-3% of birth complications and genetic malformations could develop due to maternal conditions during pregnancy, diabetes, viral infections in utero, etc. ◄Multifactorial birth defects are caused by a combination of genes and environmental exposures. An environmental cause can increase the chance for the baby to be born with a birth defect. About 70% of structural malformations are presumed to be multifactorial, meaning the siblings of the affected child could also have a higher risk of being affected, in comparison with healthy population. ◄ 2%-3% of birth defects are caused by the exposure to teratogens or environmental agents (drugs, infections, alcohol, etc.). ◄ 20% of birth defects are associated with single gene mutations. ◄ Chromosome abnormalities cause malformation syndromes with different birth defects. ◄ While 25% of structural malformations are caused by single-gene mutations or chromosomal abnormalities, in many cases a congenital anomaly may have no known cause.◄ Some individuals inherit a gene variant that increases sensitivity to environmental triggers that may cause cleft lip or cleft palate, heart, neural tube and other defects.
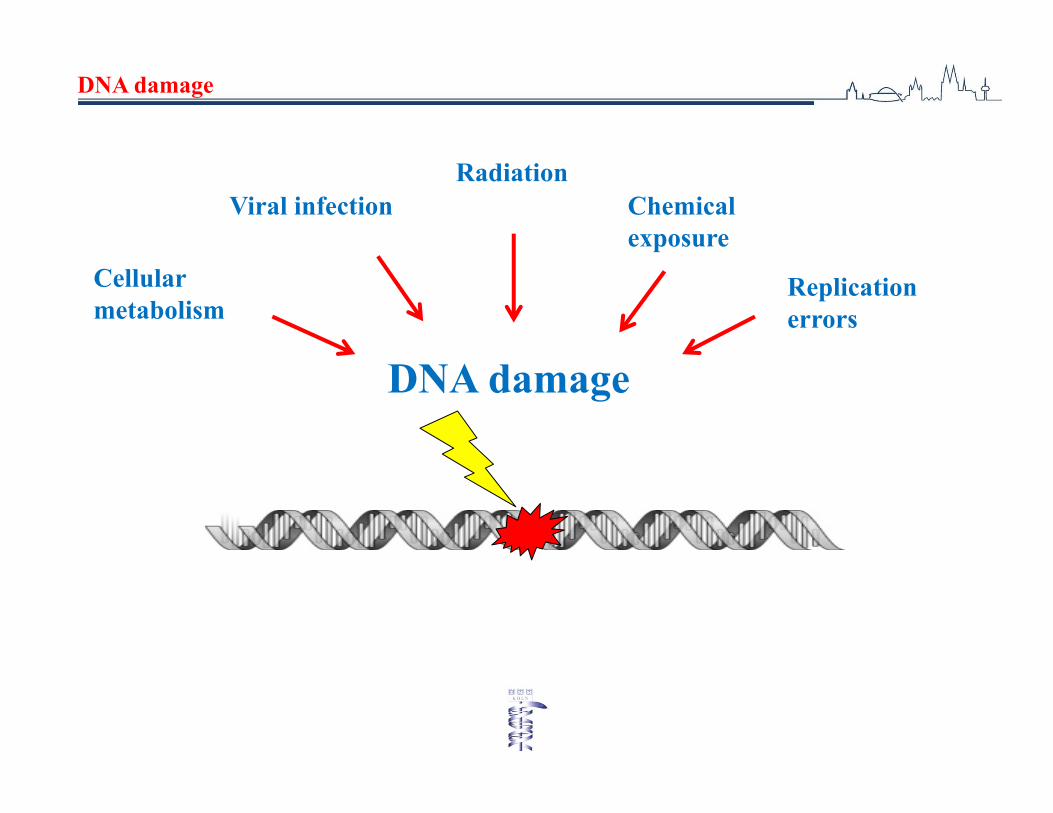
DNA damage
Cellularmetabolism
Viral infectionRadiation
Chemical exposure
Replicationerrors
DNA damage

Mutation(genetic change)
Mutation(genetic change)
ChromosomeChromosome
GeneGene
Multiplegenes +
environmentalfactors
Multiplegenes +
environmentalfactors
• Down syndrome • occuring1/every 150 live births
• Down syndrome • occuring1/every 150 live births
• Cystic fibrosis• Haemophilia• Huntingtondisease
• Cystic fibrosis• Haemophilia• Huntingtondisease
• Cancer• Diabetes• Heart disease
• Cancer• Diabetes• Heart disease
Causes of genetic diseasesCauses of genetic diseases
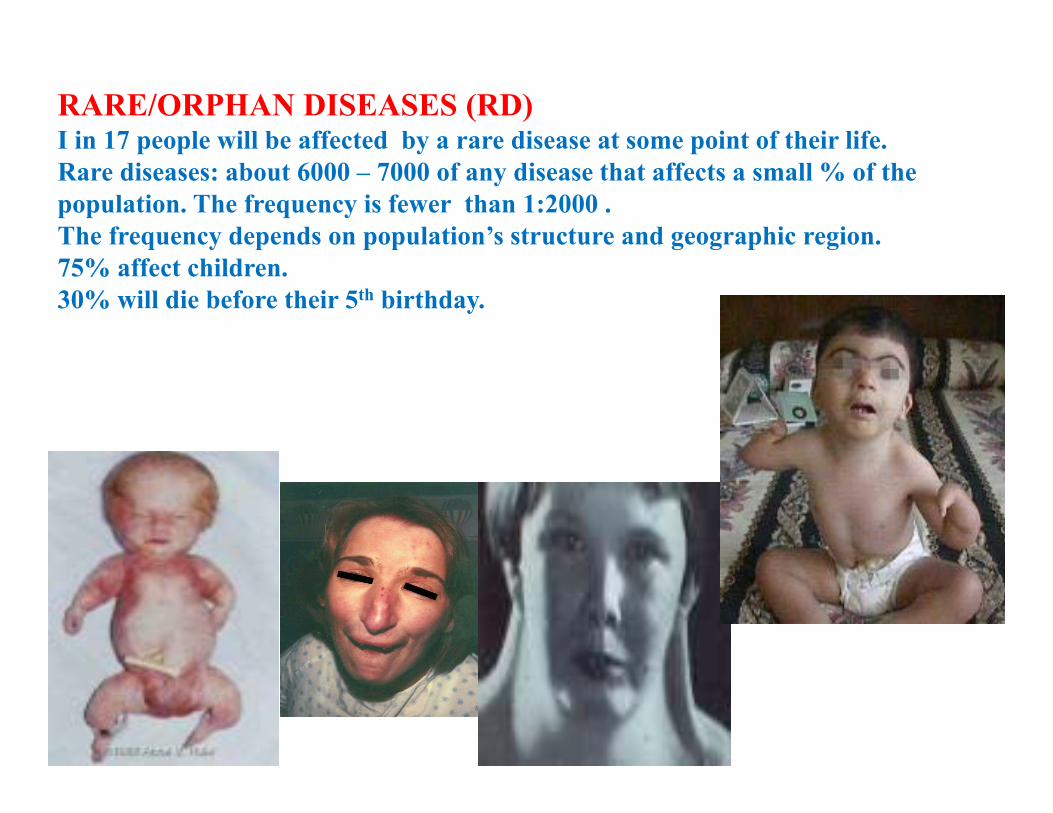
RARE/ORPHAN DISEASES (RD)I in 17 people will be affected by a rare disease at some point of their life. Rare diseases: about 6000 – 7000 of any disease that affects a small % of the population. The frequency is fewer than 1:2000 . The frequency depends on population’s structure and geographic region. 75% affect children.30% will die before their 5th birthday.
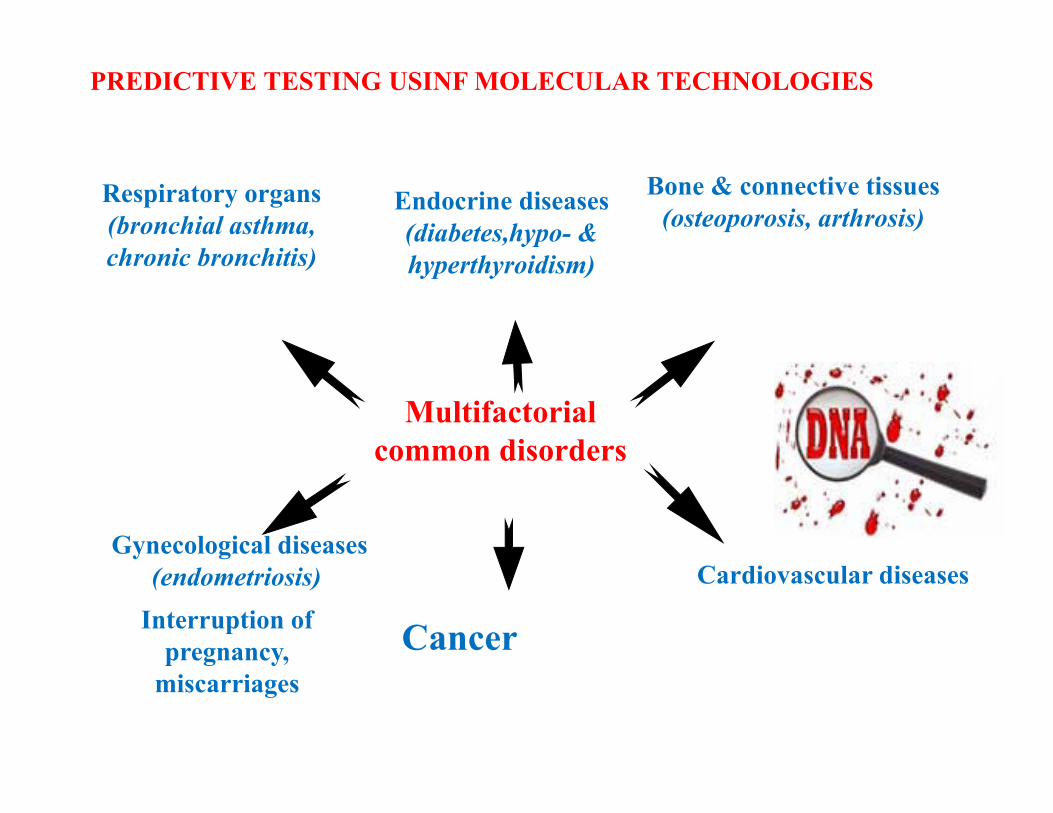
Multifactorialcommon disorders
Respiratory organs(bronchial asthma, chronic bronchitis)
Bone & connective tissues (osteoporosis, arthrosis)
Gynecological diseases(endometriosis) Cardiovascular diseases
Interruption of pregnancy,
miscarriages
Endocrine diseases(diabetes,հypo- & hyperthyroidism)
PREDICTIVE TESTING USINF MOLECULAR TECHNOLOGIES
Cancer
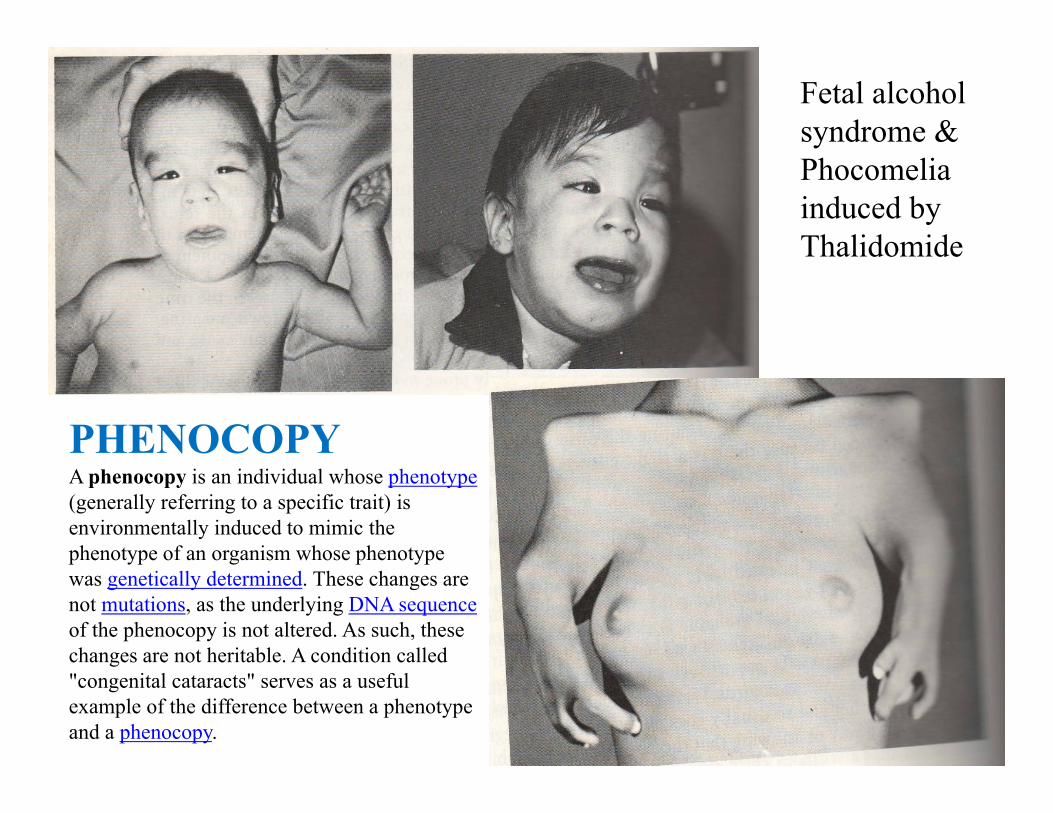
PHENOCOPYA phenocopy is an individual whose phenotype(generally referring to a specific trait) is environmentally induced to mimic the phenotype of an organism whose phenotype was genetically determined. These changes are not mutations, as the underlying DNA sequenceof the phenocopy is not altered. As such, these changes are not heritable. A condition called "congenital cataracts" serves as a useful example of the difference between a phenotype and a phenocopy.
Fetal alcohol syndrome &Phocomelia induced by Thalidomide

GLOSSARYProband – person with genetically determined trait who came to the attention of a
clinician.Sibs – brothers and sisters; Sibship – family of sibs1st degree relatives: 50% common genes: parents/children; brothers/sisters2nd degree relatives: 25% common genes: uncles/aunts; nephews/nieces;
grandparents3rd degree relatives:12.5% common genes: 1st cousins.• A Genetic Locus is a specific position or location on a chromosome.
Frequently, locus is used to refer to a specific gene.• Alleles are alternative forms of a gene, or of a DNA sequence, at a given locus.• Polymorphism means the existence of multiple (two or more) common allelic
forms at a specific locus. • If both alleles at a locus are identical, the individual is Homozygous at that locus
(a Homozygote for that condition).• If the alleles at a locus are different: 1 N and 1 mutant, the individual is
Heterozygous (a Heterozygote carrier for that condition).If the both mutant alleles at a locus are different, the individual is compound
Heterozygous• The Genotype is the genetic composition of an individual, referring to the alleles
at a specific genetic locus.• The Phenotype is the observable expression of the gene; phenotype is influenced
by environmental factors and interactions with other genes.

Three distinct ways in which differences in expression can occur
• Penetrance is the probability that a gene will express the phenotype. When the frequency is less than 100% the gene is said to have reduced penetrance.
• Pleiotropy. When a gene produces diverse phenotypic effects, its expression is said to be pleiotropic.
• Expressivity is the degree (severity) of expression of the phenotype. When the phenotype differs in people with identical genotypes, the phenotype has variable expressivity (allelic heterogenity).

Mendelian (and non-traditional) inheritanceWe all know them already from the school -or do we?
In medical genetics
By "Mendelian inheritance" we mean monogenic disorders. This means diseases that are caused (almost exclusively) by mutations in one gene (pair).
Monogenic inheritance
Autosomal dominantAutosomal recessiveX-linked dominantX-linked recessiveY-linked
The genes are in the chromosomesA normal human being has 46chromosomesThere are 44 (22 pairs) of autosomalchromosomes or autosomesSex-chromosomes are X and Y
In everyday language:Autosomal dominant inheritance =dominant inheritanceAutosomal recessive inheritance =recessive inheritanceX-linked recessive inheritance =X-linked inheritance (and dominant isspecifically mentioned)
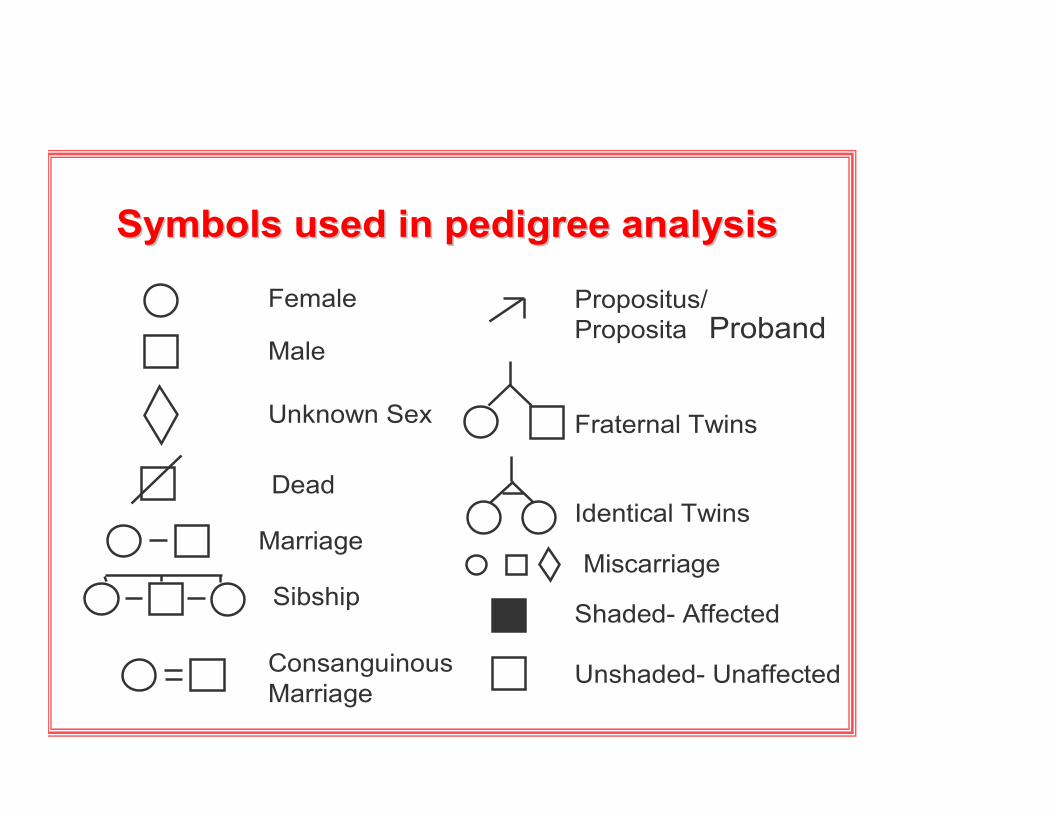
Symbols used in pedigree analysisSymbols used in pedigree analysis
Female
Male
Unknown Sex
Miscarriage
Dead
Marriage
ConsanguinousMarriage
Propositus/Proposita
Fraternal Twins
Identical Twins
Shaded- Affected
Unshaded- Unaffected
Sibship
Proband

Autosomal Dominant inheritance• Usually one of the parents of the affected individual is also affected • Males and females are equally likely to be affected• Males and females are equally likely to transmit the disease to their children• Any child of an affected individual has a 50% risk of inheriting the trait•Aut-dom inheritance is characterised by vertical trasmission of the disease phenotype, a lack of scipped generations.

Variable expressivity: NeurofibromatosisNF type 1: von Recklinghausen disease:mutations of NF1 (neurofibromin) gene on chr. 17q 11.2.Incidence 1:3500 -4000: 90% of all NF casesNeurofibromas (nerve tissue growth tumors): on or under the skin.2 or more neurofibromas; number increase with age. “Cafe au lait” spots Axillary frecklingLisch nodules (affecting the iris)Tumors on the optic nerve (optic gliomas)ScoliosisEpilepsyMacrocephaly on 30-50% of children with NF
NF type 2: “Central NF”: incidence 1:45000: Mutations of NF2 gene Neurofibromin 2: tumor suppressor gene on chromosome 22q12.-Bilateral acoustic neuromas (schvannoma) of cranial nerve 8;-Brain tumors; meningiomas-Hearing loss, balance problems, deafness-Paralysis of face: tumors of cranial nerve 7.
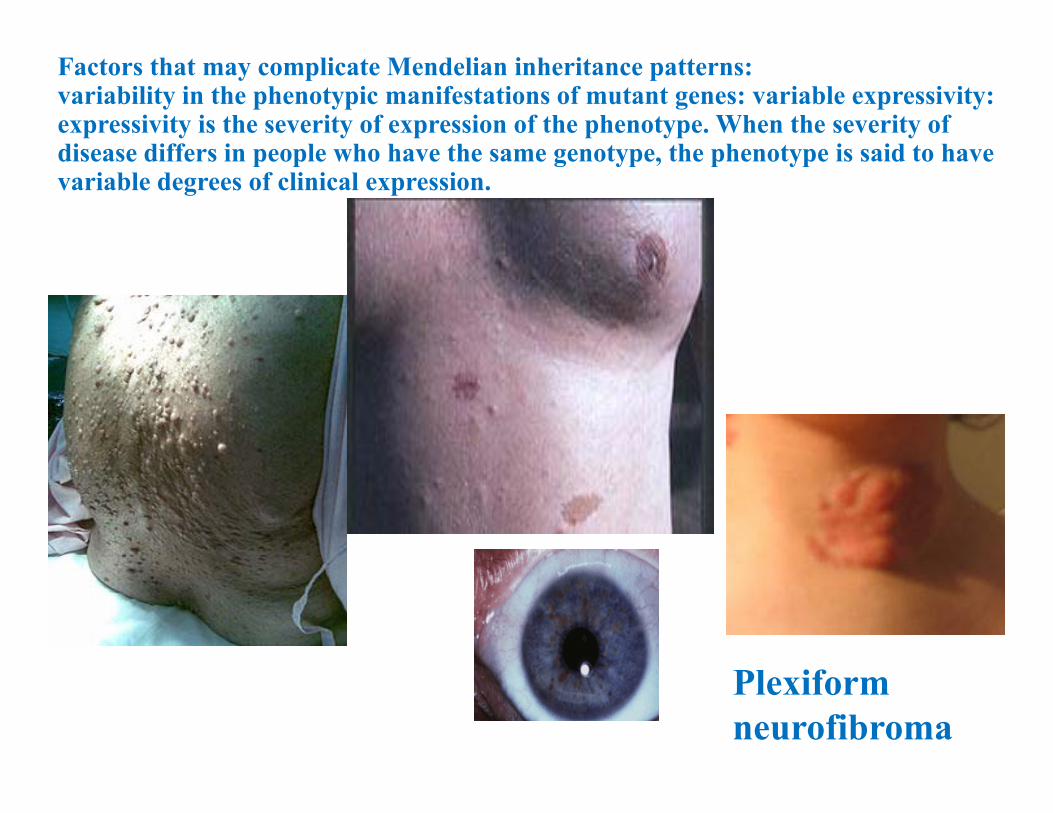
Plexiform neurofibroma
Factors that may complicate Mendelian inheritance patterns:variability in the phenotypic manifestations of mutant genes: variable expressivity: expressivity is the severity of expression of the phenotype. When the severity of disease differs in people who have the same genotype, the phenotype is said to have variable degrees of clinical expression.
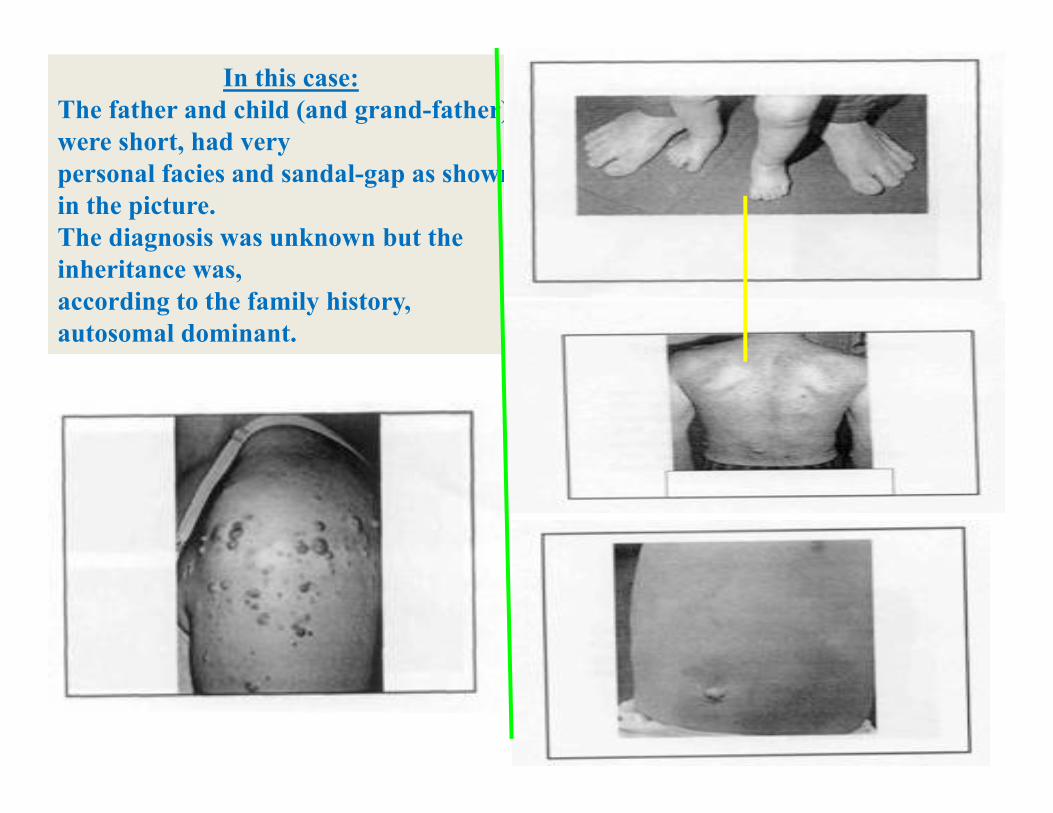
In this case:The father and child (and grand-father) were short, had very personal facies and sandal-gap as shown in the picture.The diagnosis was unknown but the inheritance was, according to the family history, autosomal dominant.

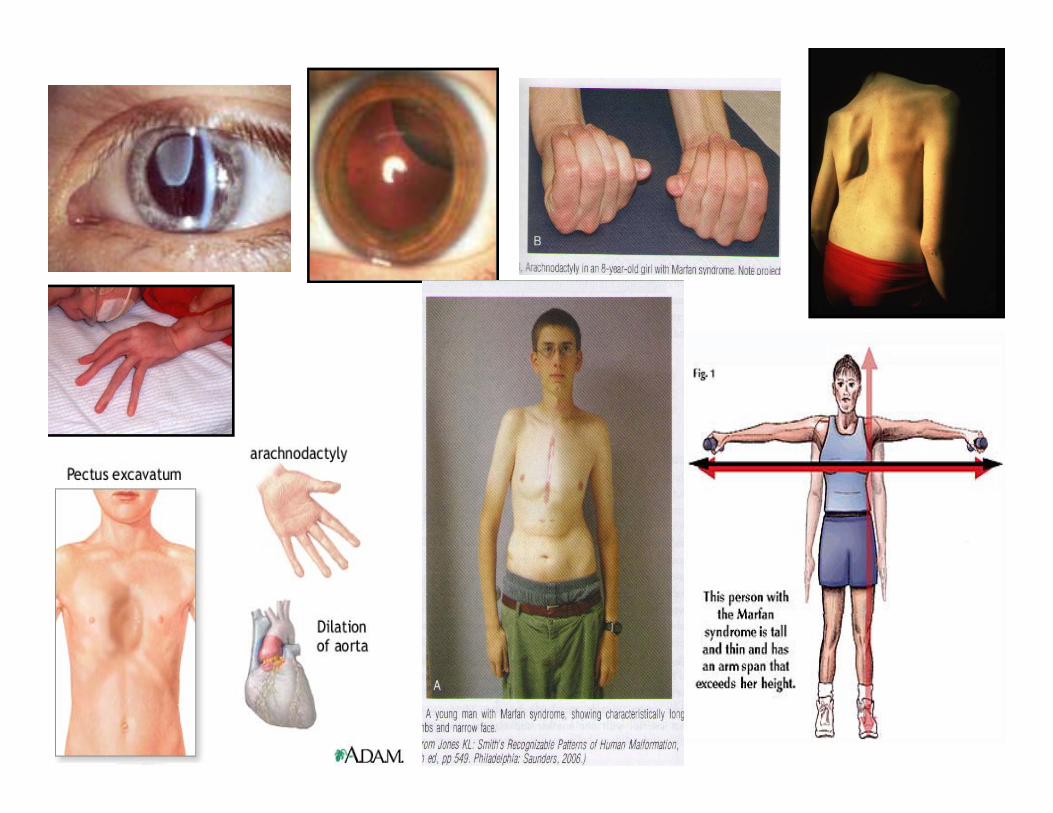
Marfan syndrome: frequency 1:10.000mutation in a gene FIBRILLIN-1 (FBN1): coding fibrillin :connective tissue
protein: mutation alters connective tissue. AD, 30% de novo

Marfan syndrome:example of pleiotropy: defects of 3 major systems:
• CARDIOVASCULAR: prolapse of the mitral valve (1-3% in general population); dilatation of blood vessels, especially aorta (90%)
• SCELETAL: joint hypermobility; pectus excavatum (“hollow chest”); • pectus carinatum (“pigeon chest”)• Tall stature, dolichostenonomelia (long and slender limbs)• Arachnodactyly (spider-like fingers)• OCULAR:• Myopia, ectopia lentis• Hernias, dural ectasia• Pneumothorax• Clinical diagnosis based on the revised Ghent nosology
(Loeys et al, JMG, 2010)
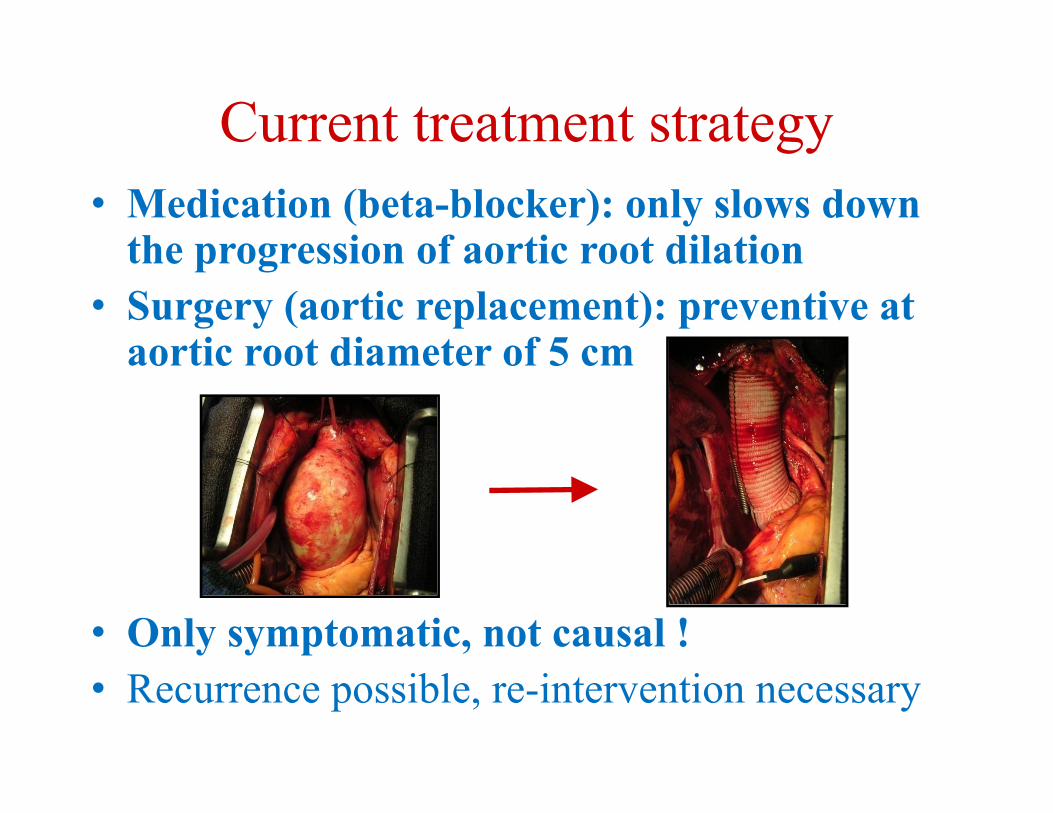
Current treatment strategy• Medication (beta-blocker): only slows down
the progression of aortic root dilation• Surgery (aortic replacement): preventive at
aortic root diameter of 5 cm
• Only symptomatic, not causal !• Recurrence possible, re-intervention necessary

Craniosynostosis
➲Developmental syndrome families with mutations in fibroblast growth factors (responsible for growth and differentiation of mesenchimal and ectodermal cells) receptors genes:FGFR
- Craniosynostosis are the most common craniofacialmalformations with a prevalence of 1 in 2000 births
- Premature fusion of one or more cranial sutures leads toskull deformity and facial asymmetry
- Lack of space for the rapidly growing brain
- A frequent cause of neurological problems
- raised intracranial pressure (ICP)- cranial nerve damage (hearing and visual abnormalities)- learning disabilities / mental retardation
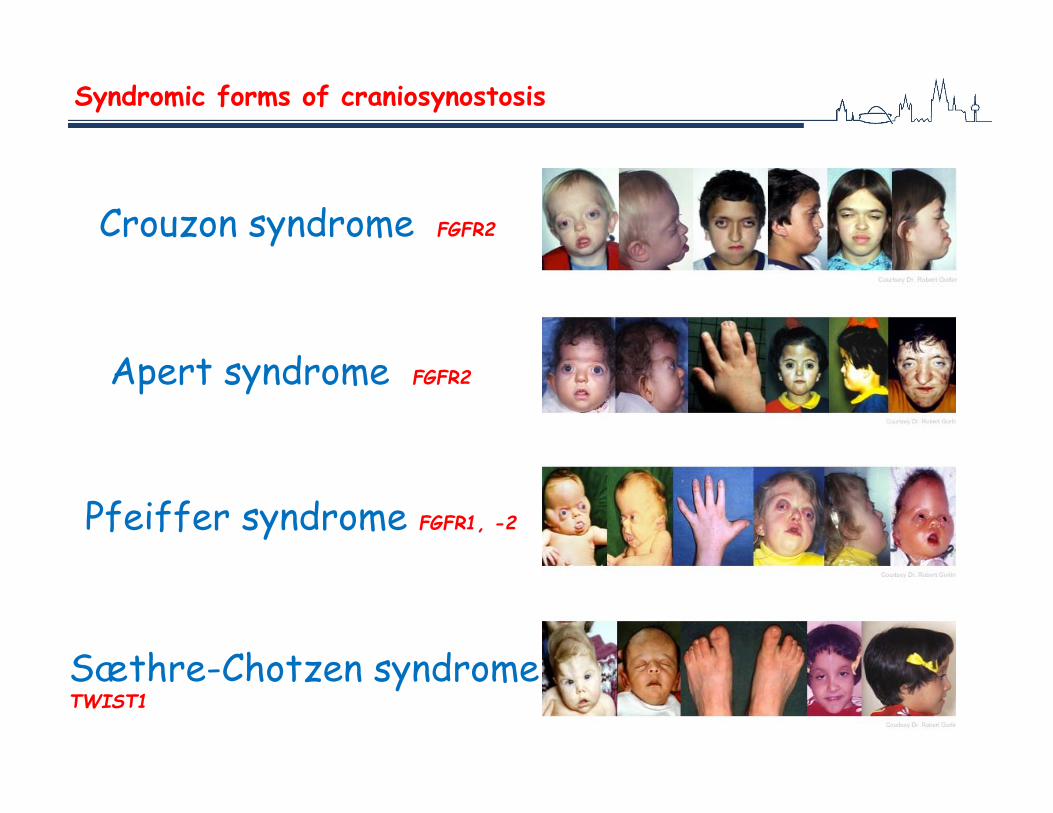
Syndromic forms of craniosynostosis
Crouzon syndrome FGFR2
Apert syndrome FGFR2
Pfeiffer syndrome FGFR1, -2
Sæthre-Chotzen syndrome TWIST1

APERT SYNDROMEgene FGFR2: acrocephalosyndactily
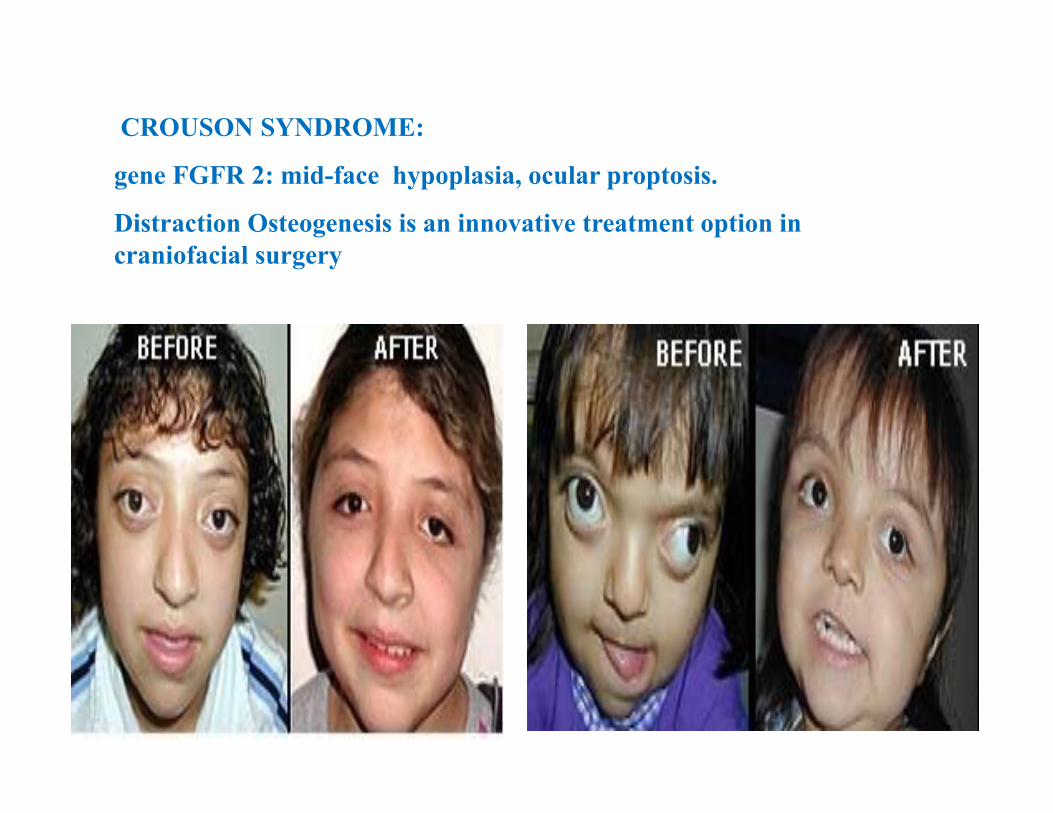
CROUSON SYNDROME:
gene FGFR 2: mid-face hypoplasia, ocular proptosis.
Distraction Osteogenesis is an innovative treatment option in craniofacial surgery
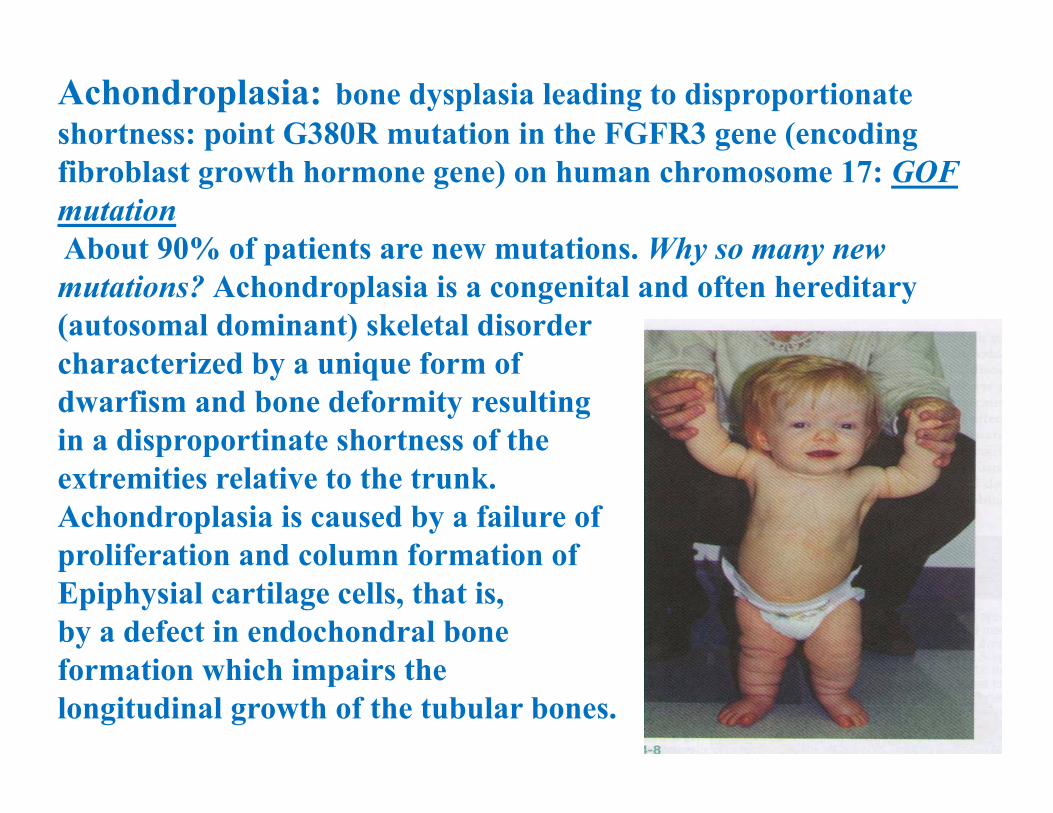
Achondroplasia: bone dysplasia leading to disproportionate shortness: point G380R mutation in the FGFR3 gene (encoding fibroblast growth hormone gene) on human chromosome 17: GOF mutationAbout 90% of patients are new mutations. Why so many new mutations? Achondroplasia is a congenital and often hereditary (autosomal dominant) skeletal disorder characterized by a unique form of dwarfism and bone deformity resulting in a disproportinate shortness of the extremities relative to the trunk.Achondroplasia is caused by a failure of proliferation and column formation of Epiphysial cartilage cells, that is, by a defect in endochondral bone formation which impairs the longitudinal growth of the tubular bones.

To help the child obtain services and support

Autosomal Recessive InheritanceThe diagram below should help explain autosomal recessive inheritance
pattern. There is a 1 in 4 chance of having an affected child if both parents are carriers.

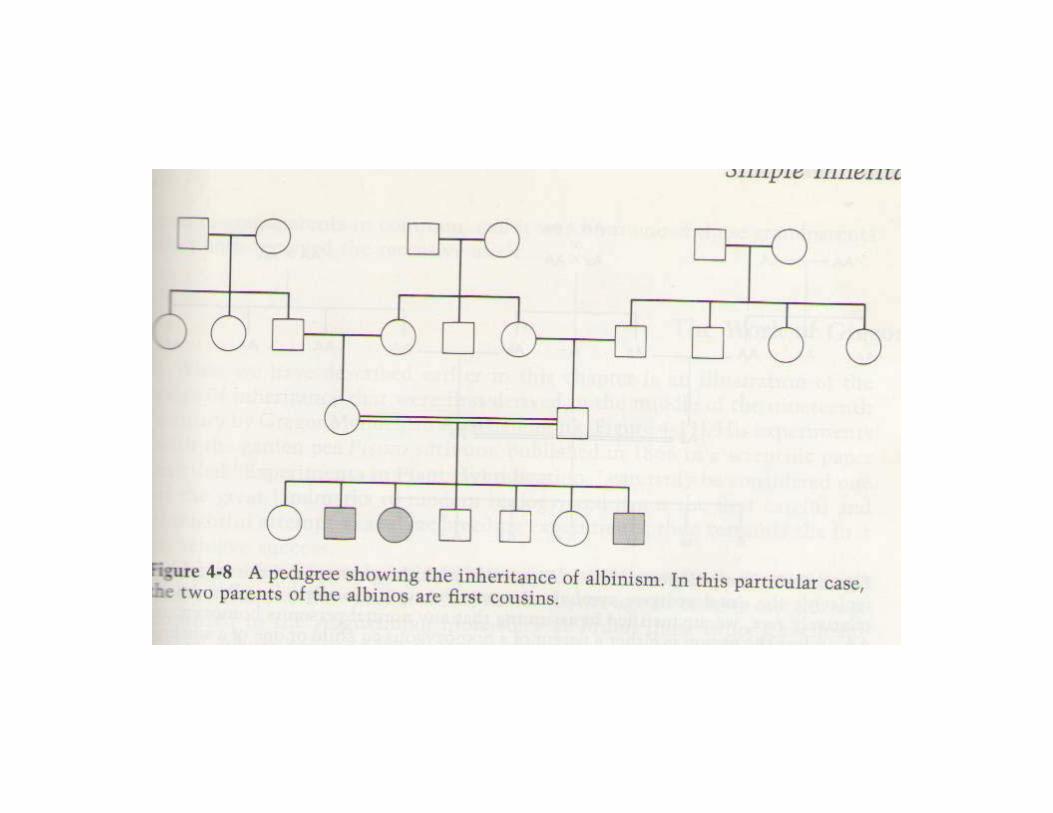
Autosomal -recessive inheritance is typical for cases of gene mutation localized on a non-sex chromosome or autosome. Both copies of the gene need to be defective for the disorder to be expressed. If one copy of the gene is affected but the other is normal then the defect is ‘carried’. Inheritance tends to be horizontal and affects sibs within a family.The disease usually appears only in sibs, not in their parents, offspring or otherrelativesMales and females are equally likely to be affectedThe parents of the affected child may be consanguineousOn the average, one fourth of the sibs of the proband are affected:Recurrence risk: 25%
Typical for recessive inheritance• The phenotype of affected sibling is identical.• However, there are exceptions to this rule•Quasidominant inheritance: recurrence risk 50%: when an affected homozygote mates with a heterozygote.

CONSIDERATIONS:
1. Carrier Frequency: Knowledge of the carrier frequency of a disease andcarrier status of parents are clinically important for genetic counselling as theautosomal recessive disorder must be inherited through both parents. Ex.Cystic fibrosis: frequency of carriers is 1/22 in Caucasians.
2. Genetic isolates: Small groups separated from neighbours by geographic,religious, or linguistic barriers. In isolates the frequency of certain rare recessivegenes is quite different from that in the general population. Although suchpopulations are not strictly consanguineous, the chance of mating with anothercarrier of a particular recessive condition may be as high as that observed incousin marriages. Ex. Tay-Sachs disease in Ashkenazi Jews.

3. Consanguinity is a situation when parents are related and could have inherited the mutations from a single common ancestor.
◄ Consanguinity of the parents of a patient with a genetic disorder is strong evidence for the autosomal recessive inheritance of that condition.
◄ Consanguinity is often the cause of rare autosomal recessive disorders. Ex. Xeroderma pigmentosum (20% of cases result from marriages between cousins) .
◄ Consanguinity increases the chance that a couple will both carry the same disease-causing mutation. It is seen more often in pedigrees involving rare recessive diseases that in those involving common recessive diseases.
◄ At the population level, consanguinity increases the frequency of genetic diseases and mortality. Related individuals have an increased likelihood of sharing alleles that are identical by descent.
• Incest: matings between first-degree relatives (siblings or parent-child): the closer the degree of consanguinity, the greater the increase.



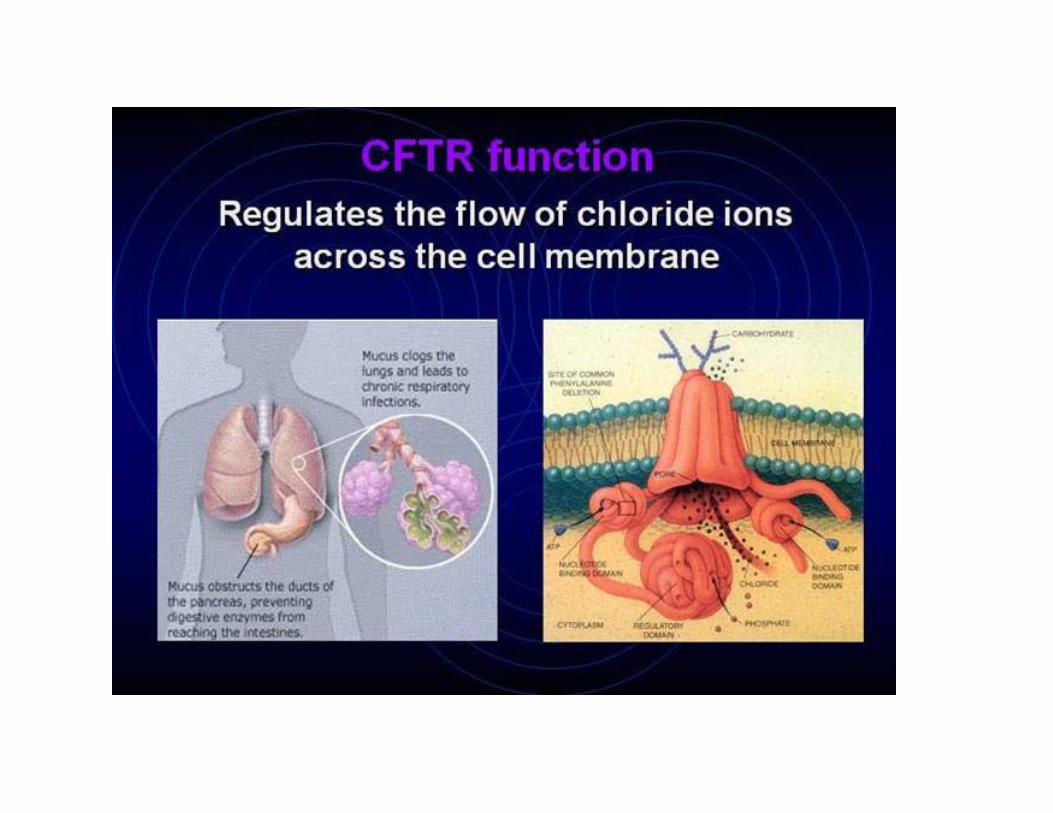
Cystic fibrosis•disturbed function of exocrine glands.• Incidence about 1:2500 in many European countries.
There are more than 1000 different mutations and they lead to different phenotypes (CFTR gene, chromosome 7q).• Different mutations cause interfamilial differences.• However, there are also modifier genes that may cause also intrafamilial differences.MAJOR PHENOTYPIC FEATURESTypical problems: chronical pulmonary infections and pancreatic insufficiency.•Age of onset: Neonatal to adulthood•Progressive pulmonary disease•Exocrine pancreatic insufficiency•Obstructive azoospermia•Growth failure•Meconium ileus•The sweat glands in the skin secrete fluids containing more salt than normal (the basis for the sweat chloride test, used in the diagnosis of CF).

X AND Y LINKED INHERITANCE:
X and Y chromosomesThe X is big (5-6% of genome)with lots of genes(mostly encoding somatic function) markers, and disease-associated mutations.The Y is small (though variable in length)…but it does have ~50 genes.Females have two copies of Xchromosome; males have one X and one Y (hemizygosity).

The Lyon’s Hypothesis: 1 X-chromosome in each cell is randomly inactivated early in the embryonic development of
females.

• The disease affects only males• The parents of the affected male are usually healthy but the mother is a carrier • There may be affected individuals in the maternal side of the family; the affected males are related to one another through females• Any son of a carrier female has a 50% risk of inheriting the trait, any daughter has a 50% risk of being a carrier• The trait is never transmitted from father to son, all daughters of an affected male are carriers
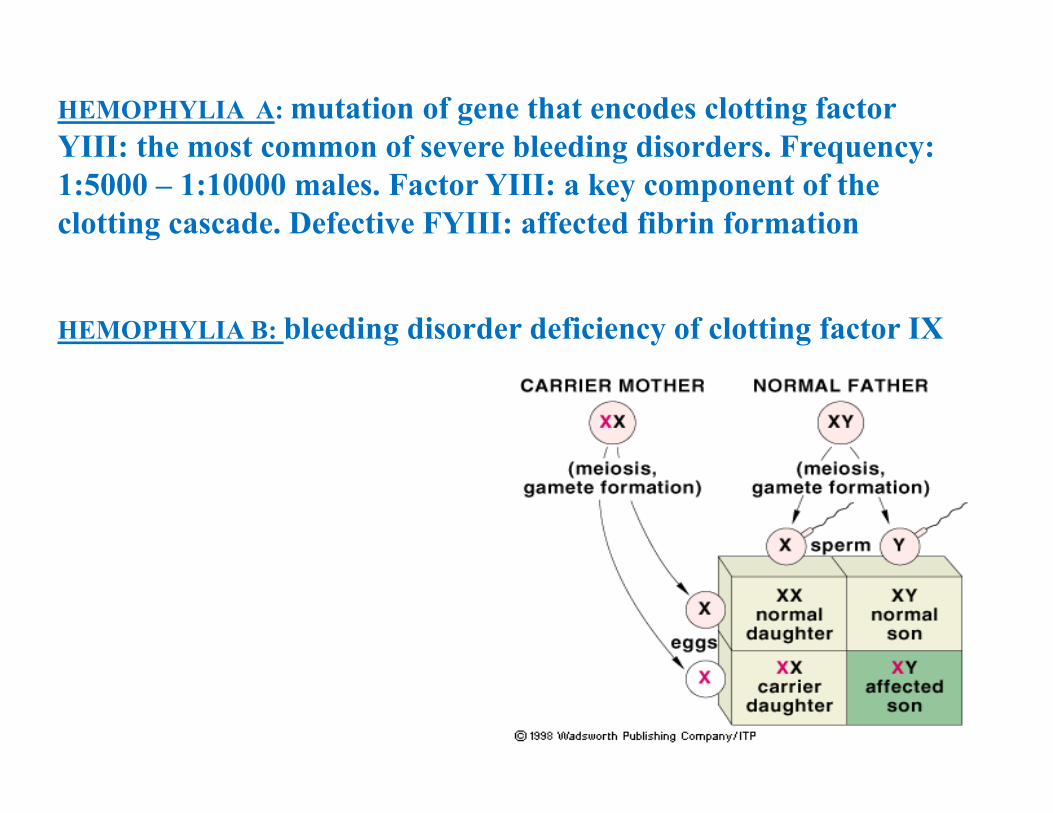
HEMOPHYLIA A: mutation of gene that encodes clotting factor YIII: the most common of severe bleeding disorders. Frequency: 1:5000 – 1:10000 males. Factor YIII: a key component of the clotting cascade. Defective FYIII: affected fibrin formation
HEMOPHYLIA B: bleeding disorder deficiency of clotting factor IX

Queen Victoria’s legacyQueen Victoria was a carrier for hemophilia. Her daughters married into royal families throughout Europe.
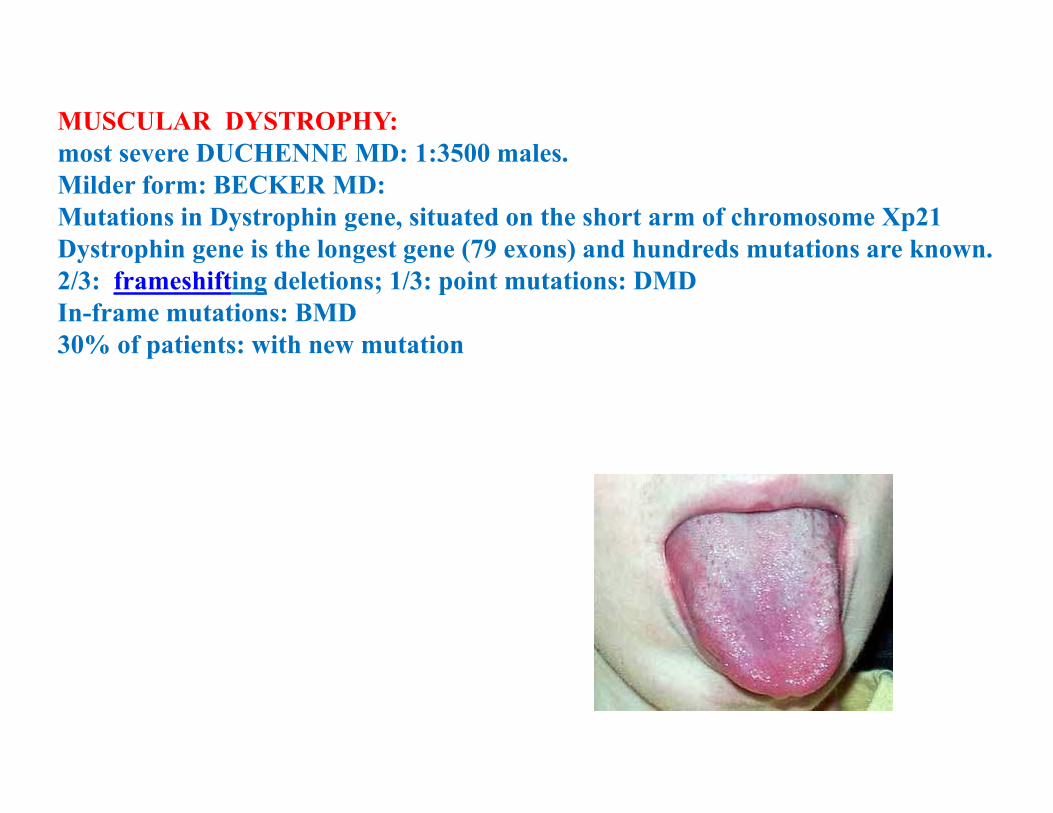
MUSCULAR DYSTROPHY: most severe DUCHENNE MD: 1:3500 males.Milder form: BECKER MD: Mutations in Dystrophin gene, situated on the short arm of chromosome Xp21Dystrophin gene is the longest gene (79 exons) and hundreds mutations are known.2/3: frameshifting deletions; 1/3: point mutations: DMDIn-frame mutations: BMD30% of patients: with new mutation
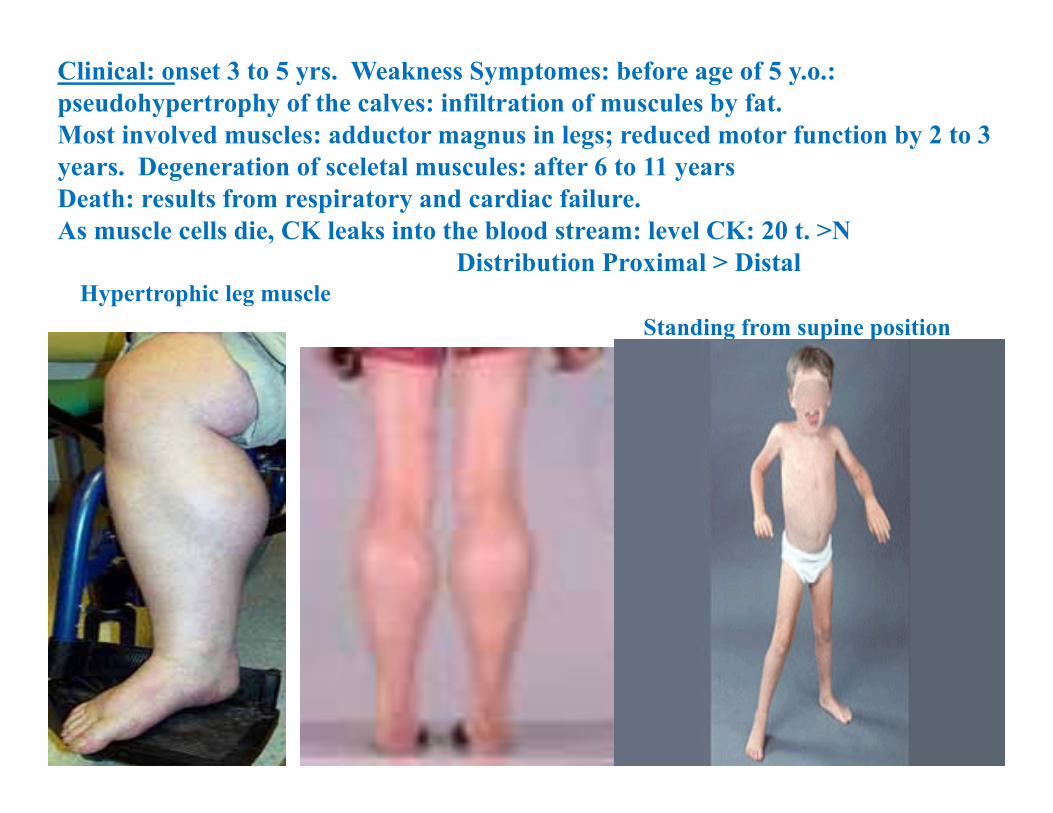
Clinical: onset 3 to 5 yrs. Weakness Symptomes: before age of 5 y.o.: pseudohypertrophy of the calves: infiltration of muscules by fat. Most involved muscles: adductor magnus in legs; reduced motor function by 2 to 3 years. Degeneration of sceletal muscules: after 6 to 11 years Death: results from respiratory and cardiac failure.As muscle cells die, CK leaks into the blood stream: level CK: 20 t. >N
Distribution Proximal > DistalHypertrophic leg muscle
Standing from supine position
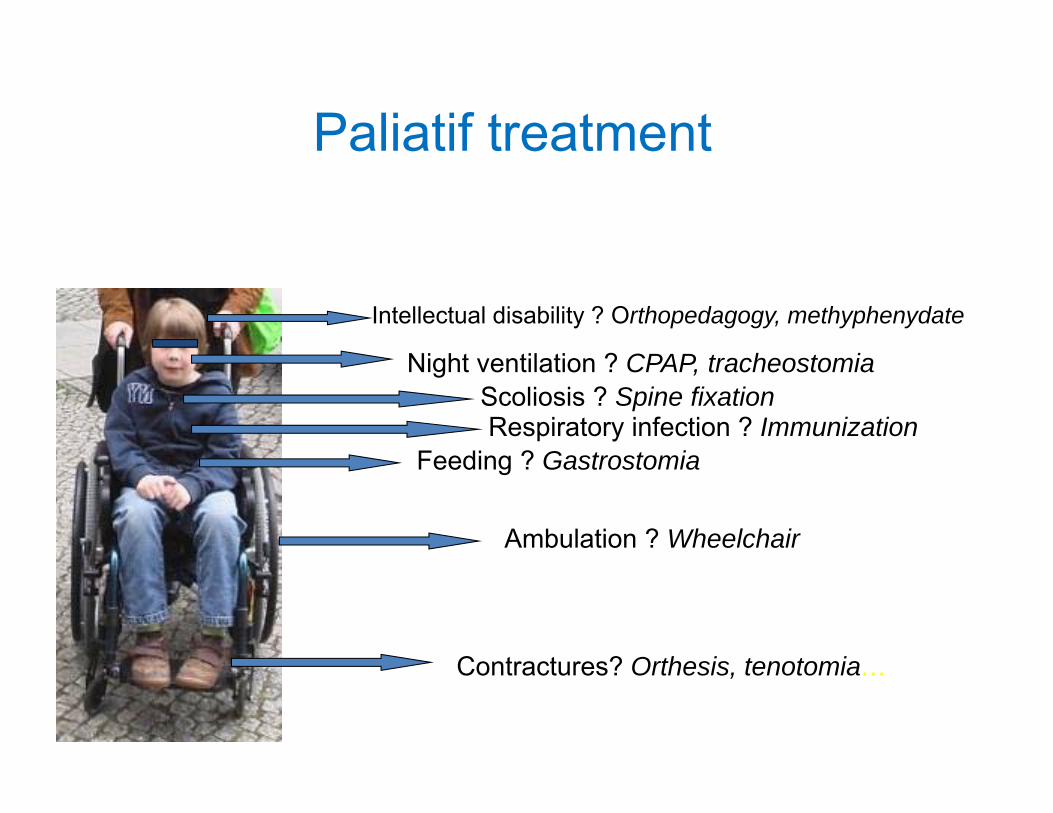
Paliatif treatment
Intellectual disability ? Orthopedagogy, methyphenydate
Night ventilation ? CPAP, tracheostomiaScoliosis ? Spine fixation
Feeding ? Gastrostomia
Ambulation ? Wheelchair
Contractures? Orthesis, tenotomia…
Respiratory infection ? Immunization


X-linked Dominant Inheritance: Characteristics
XhY XHXh
XhXh XHY XhXh XHXh XhY
X-linked dominant diseases display characteristic patterns of inheritance. Ex. Fragile X syndrome.
There are about twice as many affected females as affected males.
Skipped generations are uncommon.
Father-to-son transmission is not seen.
Disease phenotype is seen in generation after generation.
The expression in heterozygous females may be variable.
Often the clinical expression is more severe in hemizygous males than in heterozygous females.
XHXh XhY XHXh XhY XhXh XHY XhY

Hypophosphatemic ricketsVitamin D – resistant rickets The kidneys are impaired in their ability to reabsorb phosphateAbnormal ossificationVitamin D metabolism abnormal.

Rett syndrome: a childhood onset disorder, affecting females. Period of normal development is between 6-18 months, after: loss of purposeful use of hands, with hand winring and other stereotypic movements; 50% develop seizures, breathing irregularities with apnea, in later age-scoliosis, limitation of mobility.Mutation in gene on the X chromosome, lethal in males.

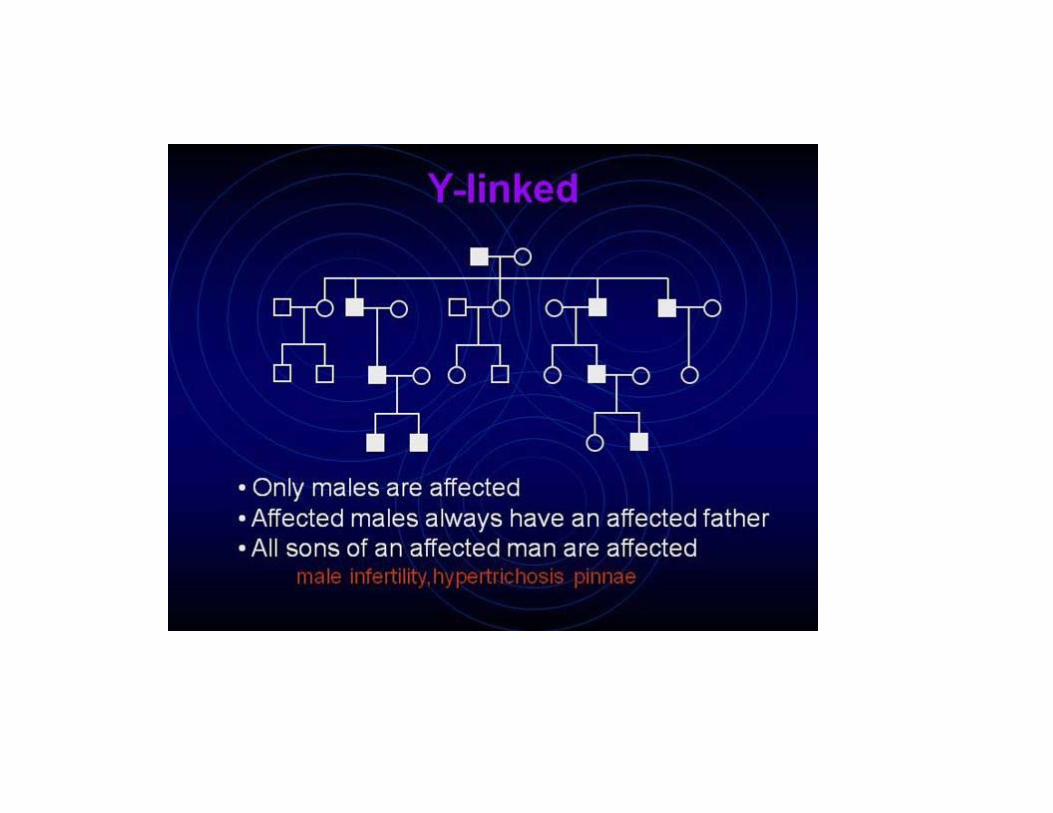
Only males have Y chromosomes • All Y-linked traits are expressed • Passed from father to sons • Approximately three dozen Y-linked raits are known. • Male infertility: SRY gene is responsible for primary hormonal changes, responsible for male development.Azoospermia factor:AZF Hypertrichosis pinnaeRetinitis pigmentosaGenotype XYY: aggression, learning disability, skin problems, obesity
Y CHROMOSOME:

Hypertrichosis pinnae
Retinitis pigmentosa

Mitochondrial Inheritance• Mitochondria are cytoplasmic organelles
important in cellular respiration• Have their own DNA• Carry 37 genes• Transmitted from mother to ALL of her
offspring• No recombination• Males and females equally affected• High mutation rate

МITOCHONDRIAL DISORDERS: MATERNAL TYPE OF INHERITANCE

Reproductive Genetics
◄ 10-15% of pregnancies end in a miscarriage. 3-5 % of one of parent carry a balanced chromosomal abnormality (reciprocal or Robertsonian translocation). ◄ Primary amenorrhea: Turner syndrome (45,X).◄ Androgene intensivity syndrome (AIS) - testicular feminisation syndrome
46, XY, Xq11-mutations of androgene receptor gene in females.◄ Gonadal agenesis/dysgenesis: 20% of XY females have mutated SRY gene (deletion).◄Klinefelter syndrome: 47, XXY: prevalence: 1/500-1/800 male
births.◄ Congenital bilateral aplasia syndrome: mutations of CFTR gene.◄ Yq-microdeletions : in 15% of males with azoospermia. ◄ 46,XX males: due to the translocation of SRY gene into the X-chromosome.

CAH (congenital adrenal hyperplasia): is an autosomal recessive disorder of the adrenal cortex . Incidence 1:10,000–15,000 caused in about 95% of cases by genetic defects in the steroid 21-hydroxylase gene CYP21A2. ◄ CAH summarizes metabolic disorders that lead to inadequate synthesis of adrenal steroid hormones (cortisol, aldosterone). ◄ Clinical manifestations of CAH as disorder of adrenal steroid metabolism symptoms are highly variable and may include virilization of female newborns and life-threatening salt-wasting conditions.◄ Various degrees of genital virilization in females (46,XX) are present at birth, whereas males (46,XY) may appear inconspicuous with hypospadia, etc.◄ Appropriate treatment demands early diagnosis
◄ Neonatal genetic testing is an important confirmatory tool for early and reliable CAH diagnosis.

Dr. Davit Babikyan, PhD
HUMAN GENOME&
GENETIC CHANGES

James Watson & Francis Crick1953

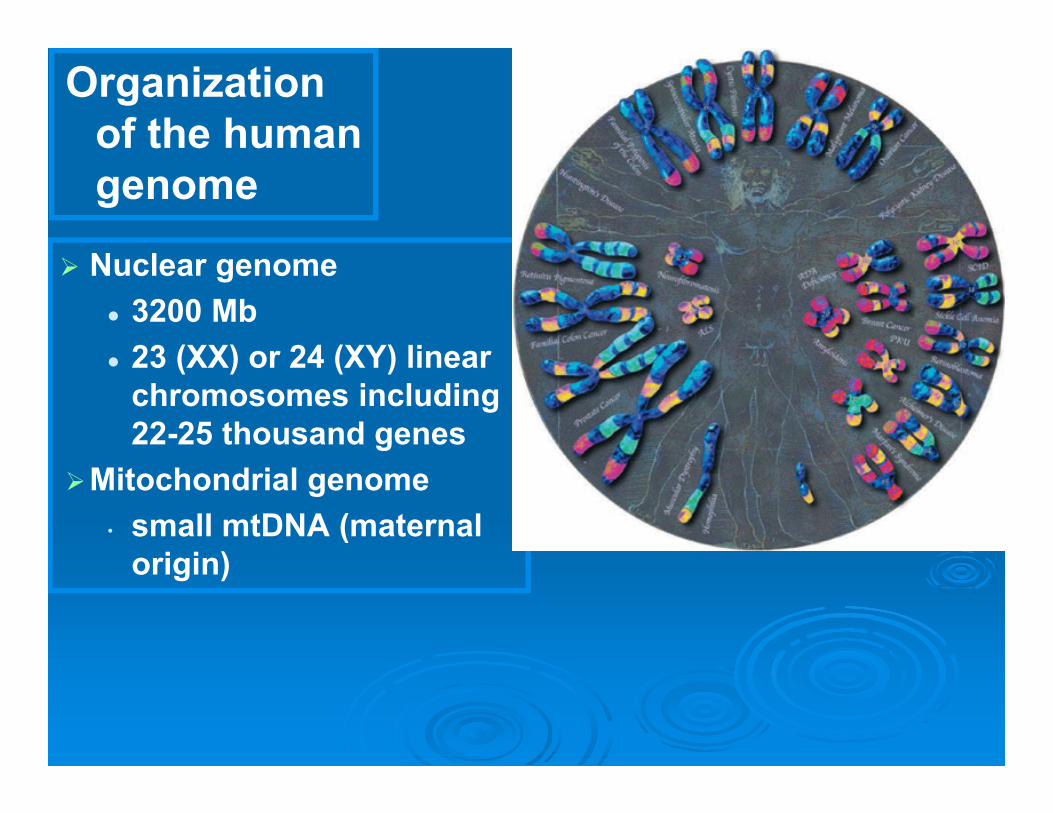
Organization of the human genome
Nuclear genome 3200 Mb 23 (XX) or 24 (XY) linear
chromosomes including 22-25 thousand genes
Mitochondrial genome• small mtDNA (maternal
origin)
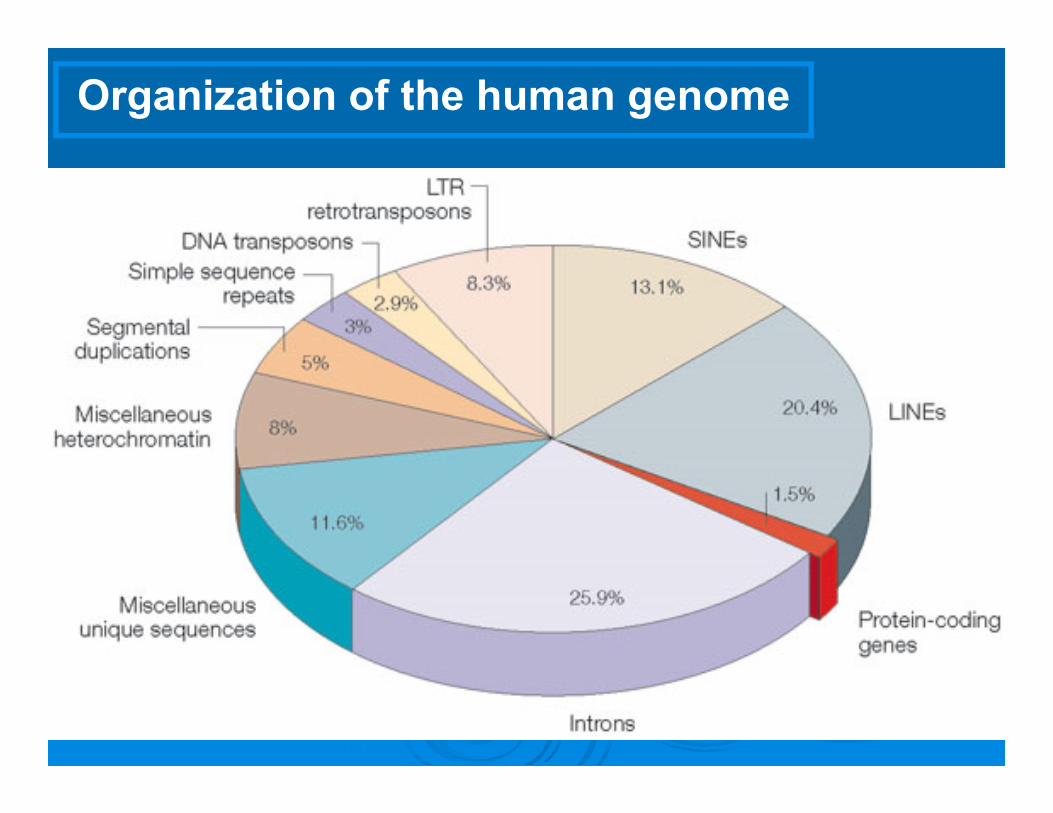
Organization of the human genome

Anatomy of the Gene
DNA
AAAAAAAAA
RNA transcript
mRNA
Promoter Exons Intron
Transcription
Processing
Coding Region
Start
protein
Translation

EXOME: ALL EXONS OF A HUMAN GENOMEabout 1.5% of all coding sequences of human genome.
Total 180.000 exons: 30 Mb in length (transcribed regions): short, functionally important sequences that are translated into proteins.
It is estimated that protein coding regions constitute 85% of the disease-causing mutations.

Genetic ChangesGene-level changes
Chromosomal changes
Genomic changes

Human Genome Plasticity:
Single Nucleotide Polymorphisms (SNPs)
Repetitive DNA sequences
Copy Number Variations (CNVs)

SNPs – Single Nucleotide PolymorphismA polymorphism due to a base substitution or
the insertion or deletion of a single base
TCGAGAGGCTAGGCTAGGA
TCGAGAGGCCAGGCTAGGA
SubstitutionT-allele
C-allele
TCGAGAGGCTAGGCTAGGA
TCGAGAGGCAGGCTAGGA
Insertion/deletion
(+) allele
(-) allele
Occurs on average every 1000 nucleotidesMore than 10 million SNPs in genome
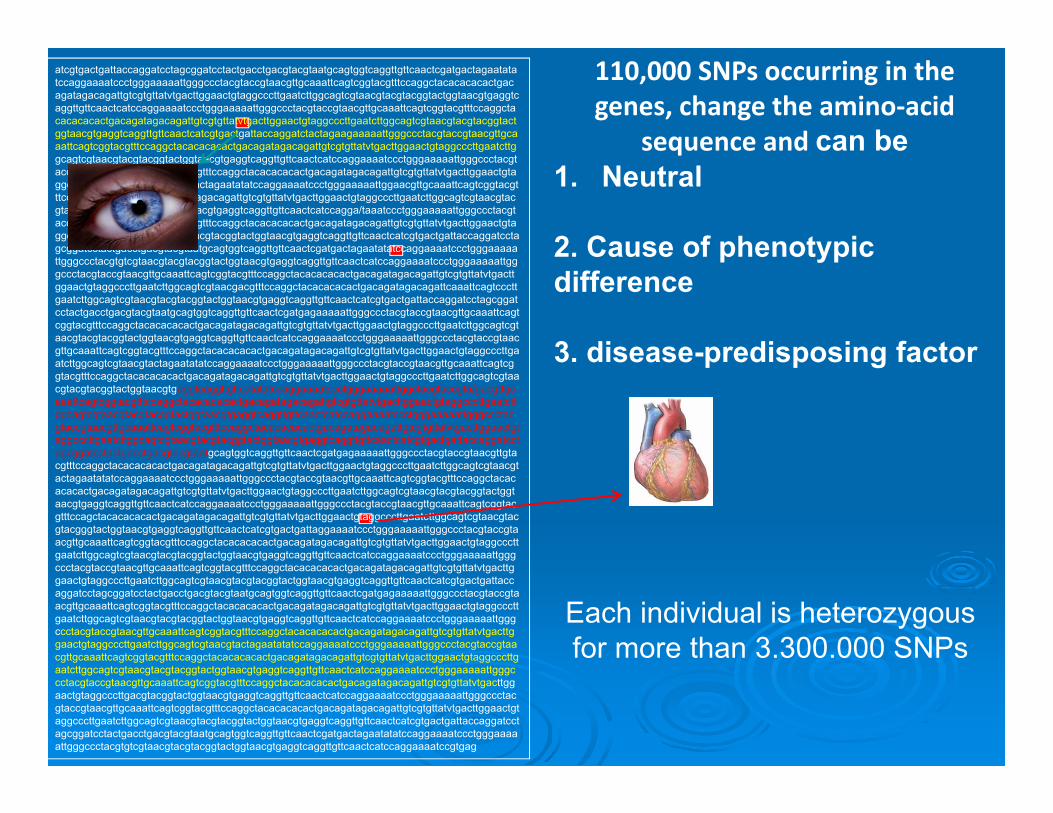
110,000 SNPs occurring in the genes, change the amino‐acid
sequence and can be 1. Neutral
2. Cause of phenotypic difference
3. disease-predisposing factor
atcgtgactgattaccaggatcctagcggatcctactgacctgacgtacgtaatgcagtggtcaggttgttcaactcgatgactagaatatatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaccaggatctactagaagaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtactagaatatatccaggaaaatccctgggaaaaattggaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtagccctacgtaccgtcggtactggtaacgtgaggtcaggttgttcaactcatccagga/taaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaccaggatcctagcggatcctactgacctgacgtacgtaatgcagtggtcaggttgttcaactcgatgactagaatatatccaggaaaatccctgggaaaaattgggccctacgtgtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgacgtttccaggctacacacacactgacagatagacagattcaaattcagtcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaccaggatcctagcggatcctactgacctgacgtacgtaatgcagtggtcaggttgttcaactcgatgagaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtactagaatatatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcatatatccaggaaaatccctgggaaaaattggctacgtaccgta/gacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaccaggatcctagcggatcctactgacctgacgtacgtaatgcagtggtcaggttgttcaactcgatgagaaaaattgggccctacgtaccgtaacgttgtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtactagaatatatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccagctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacgggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaccaggatcctagcggatcctactgacctgacgtacgtaatgcagtggtcaggttgttcaactcgatgagaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtactagaatatatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccctgggaaaaattgggccctacgtaccgtaacgttgcaaattcagtcggtacgtttccaggctacacacacactgacagatagacagattgtcgtgttatvtgacttggaactgtaggcccttgaatcttggcagtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatcgtgactgattaccaggatcctagcggatcctactgacctgacgtacgtaatgcagtggtcaggttgttcaactcgatgactagaatatatccaggaaaatccctgggaaaaattgggccctacgtgtcgtaacgtacgtacggtactggtaacgtgaggtcaggttgttcaactcatccaggaaaatccgtgag
Each individual is heterozygous for more than 3.300.000 SNPs

MicrosatellitesRepeat units of 1-6bp in length (total <few hundred bp), more frequent and polymorphic
CACACACACACA
CACACACACACACACA
6 (CA) allele
8 (CA) allele
Repeat units of 20-70 bp in length (total a few thousand bp)Minisatellites
35 bp 35 bp35 bp
35 bp 35 bp35 bp35 bp
3-repeat allele
4-repeat allele
Repetitive DNA Sequences 45% of Human Genome
Changes in the numbers of repeated DNA sequences arranged in tandem arrays

6 (CA) allele 8 (CA) allele
The individuals genotype for a mircosatellite marker is (6 8)
People have differences in the number of Repeated DNA Sequences of microsatellites and
minisatellites which are transferred to the offspring without changes
Paternal copy Maternal copy
* *
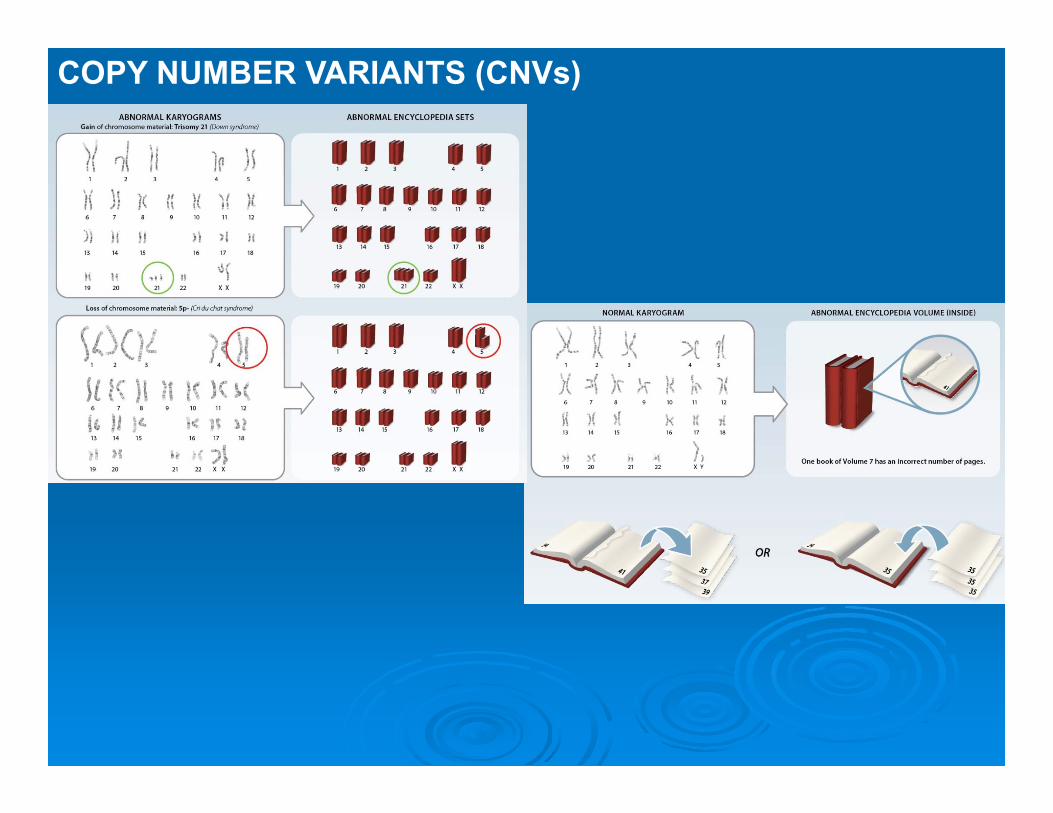
COPY NUMBER VARIANTS (CNVs)

Copy Number Variations (CNVs): Differences in number of repeated DNA sequences longer than 1000 bpThe size of CNVs varies from 100,000 bp to 2,2 million bp
>3,000 CNVs identified consisting >15% of genome
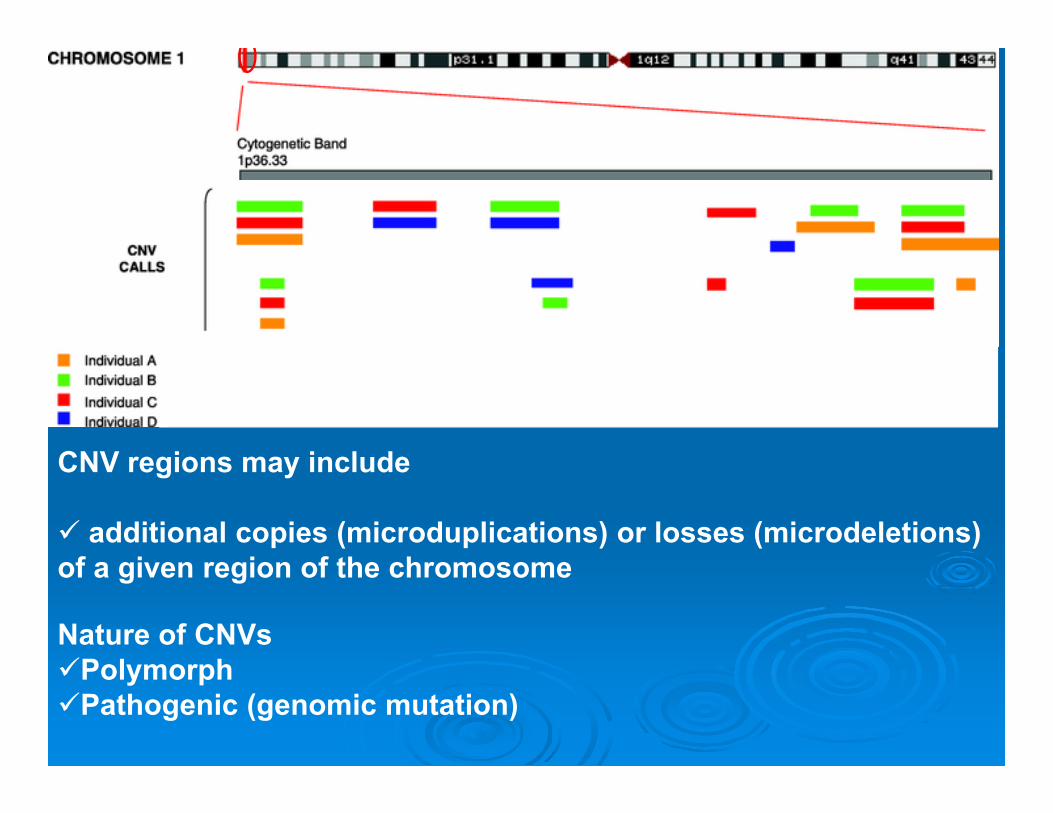
CNV regions may include
additional copies (microduplications) or losses (microdeletions) of a given region of the chromosome
Nature of CNVs PolymorphPathogenic (genomic mutation)

Metabolism of xenobiotics (includ. detoxification of medicines)
Immunity, which are responsible for predisposition of different multifactorial diseases (ex. psoriasis, Crohn’s disease, glomeroulonephritis)
Polymorph CNVscommon in general population(>1%), small in size (<10 kb), andmay include genes which areresponsible for
There is at least 0.2% difference between genomes of every two individuals: 0.08% for SNPs and 0.12% for CNVs

MUTATIONS: Causes of Genetic Disorders
Gene MutationsChromosomal aberrationsGenomic mutations (pathogenic CNVs)

Types of gene mutations Point mutations
Missense (change an amino acid) Nonsense (premature termination) Silent
Deletion Large variation in size
Insertion Duplication Splice site Regulatory Expanded repeat
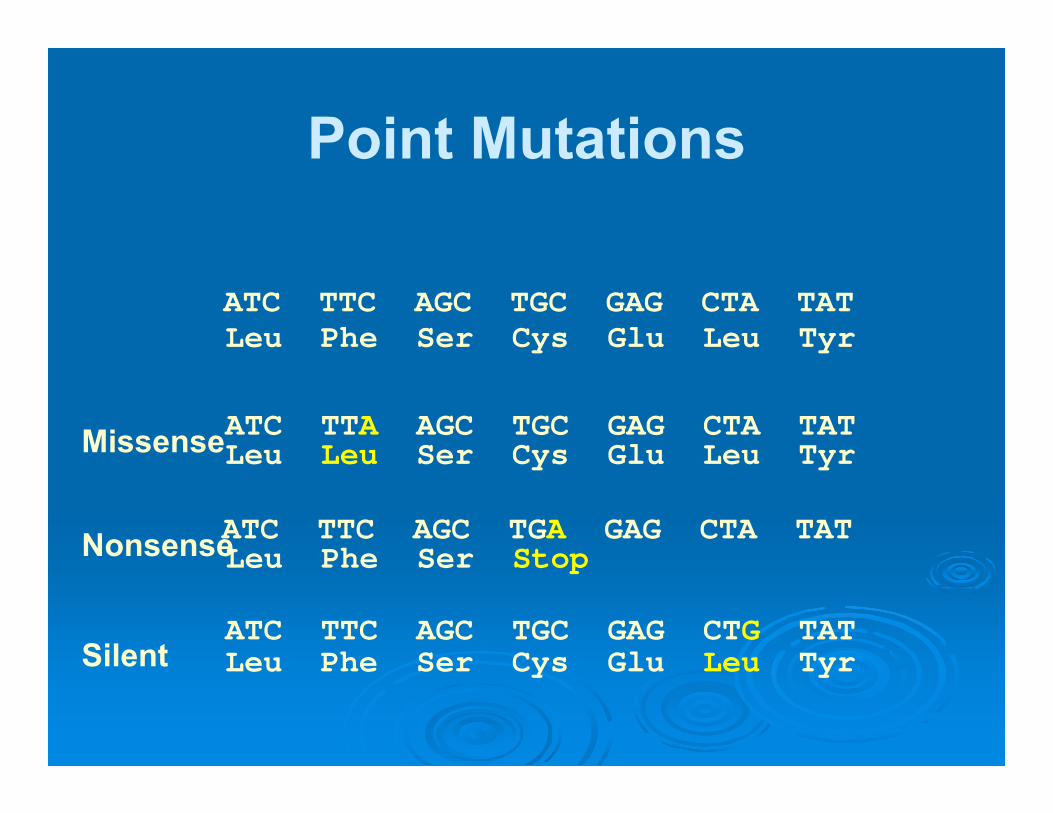
Point Mutations
ATC TTC AGC TGC GAG CTA TAT
ATC TTA AGC TGC GAG CTA TAT
ATC TTC AGC TGA GAG CTA TAT
ATC TTC AGC TGC GAG CTG TAT
Leu Phe Ser Cys Glu Leu Tyr
Leu Phe Ser Stop
Leu Leu Ser Cys Glu Leu Tyr
Leu Phe Ser Cys Glu Leu Tyr
Missense
Nonsense
Silent
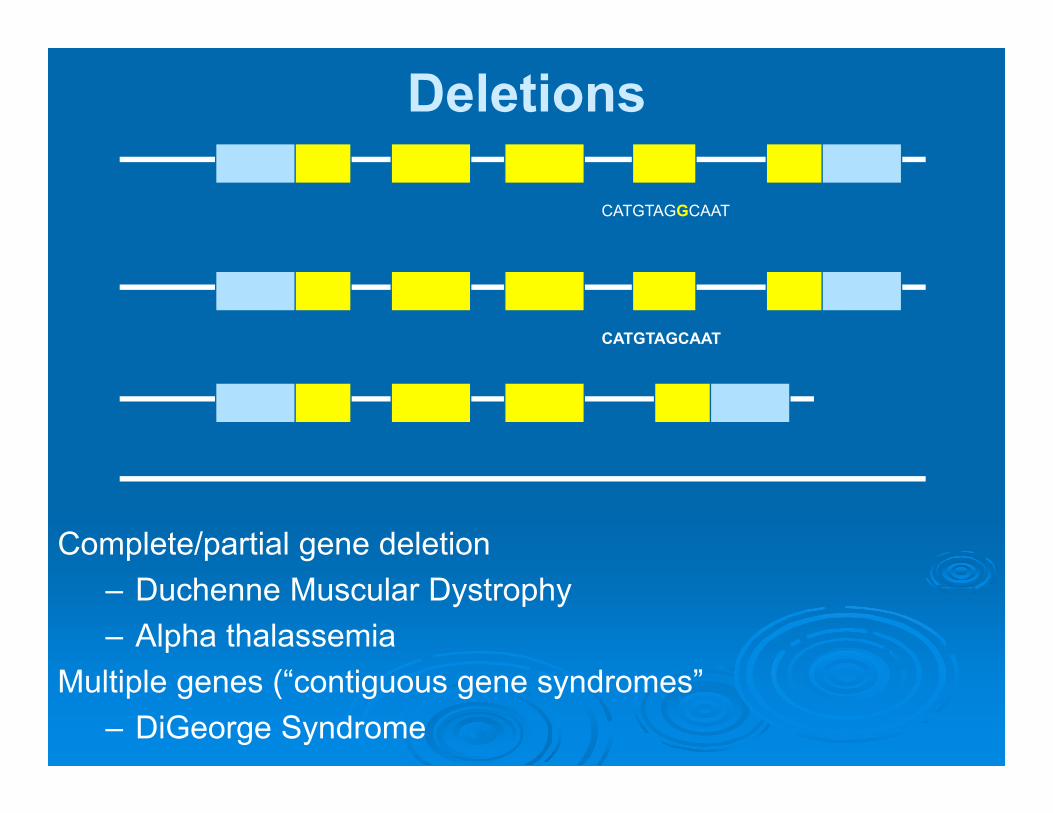
Deletions
CATGTAGGCAAT
CATGTAGCAAT
Complete/partial gene deletion– Duchenne Muscular Dystrophy– Alpha thalassemia
Multiple genes (“contiguous gene syndromes”– DiGeorge Syndrome
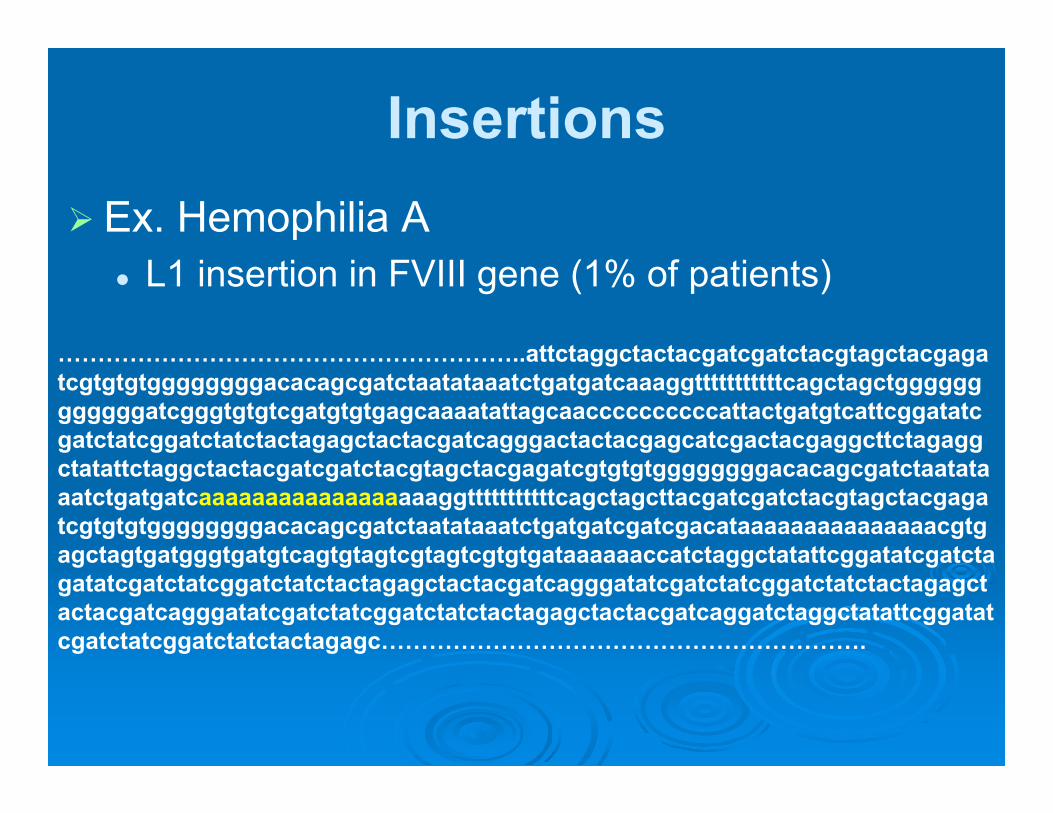
Insertions Ex. Hemophilia A
L1 insertion in FVIII gene (1% of patients)
…………………………………………………..attctaggctactacgatcgatctacgtagctacgagatcgtgtgtggggggggacacagcgatctaatataaatctgatgatcaaaggtttttttttttcagctagctggggggggggggatcgggtgtgtcgatgtgtgagcaaaatattagcaaccccccccccattactgatgtcattcggatatcgatctatcggatctatctactagagctactacgatcagggactactacgagcatcgactacgaggcttctagaggctatattctaggctactacgatcgatctacgtagctacgagatcgtgtgtggggggggacacagcgatctaatataaatctgatgatcaaaaaaaaaaaaaaaaaaggtttttttttttcagctagcttacgatcgatctacgtagctacgagatcgtgtgtggggggggacacagcgatctaatataaatctgatgatcgatcgacataaaaaaaaaaaaaaacgtgagctagtgatgggtgatgtcagtgtagtcgtagtcgtgtgataaaaaaccatctaggctatattcggatatcgatctagatatcgatctatcggatctatctactagagctactacgatcagggatatcgatctatcggatctatctactagagctactacgatcagggatatcgatctatcggatctatctactagagctactacgatcaggatctaggctatattcggatatcgatctatcggatctatctactagagc…………………………………………………….

Unstable trinucleotide repeatsFragile X Syndrome (CGG)n 5’UTHuntington’s syndrome (CAG)n polyglutamineMyotonic dystrophy (CTG)n 3’UTFriedrich’s Ataxia (GAA)n intron 1
Repeat alteration TTCCAG…(CAG)5…CAGCAATTCCAG…(CAG)60…CAGCAA

Pathogenic CNVs (Microduplications and Microdeletions)
Size: 50 kb - 1 Mb
Arise during the development of gametes (de novo)Transferred to a few generations (hereditary)
Pathogenic CNVs are associated with Intellectual DisabilityPhysical developmental delaySchizophreniaAutism
Particularly, neurocognitive disorders are more associated withpathogenic CNVs rather than with other type of mutations.

DNA Diagnostics

PrenatalDiagnostics
Screening
InfectiousDisease
TransplantationMedicine
Genetic Testing • Testing for genetic, including hereditary disorders
• Non-invasive detection of fetal diseases
• Massive early diagnosis of hereditary disorders at childhood for treatment and/or prevention
• Non-invasive, early detection of organ rejection
• Pathogen identification and early detection
DNA Diagnostics For Multiple Areas
Pharmacogenetic Testing
• For prescription and/or dose adjustment of medicines
Presymptomatic Testing
• Diagnosis of hereditary disorders for early detection and/or prevention

Confirmation of a disease’s Diagnosis
Prognosis of a disease’s outcome
Prediction of the development possibility of a disease
Therapeutic prediction of the response to treatment
Practical Use of Genetic Diagnostics

Benefits of Genetic Diagnostics
Better monitoring of diseases
Prescription of an appropriate treatment
Reduction of disease incidence and mortality level
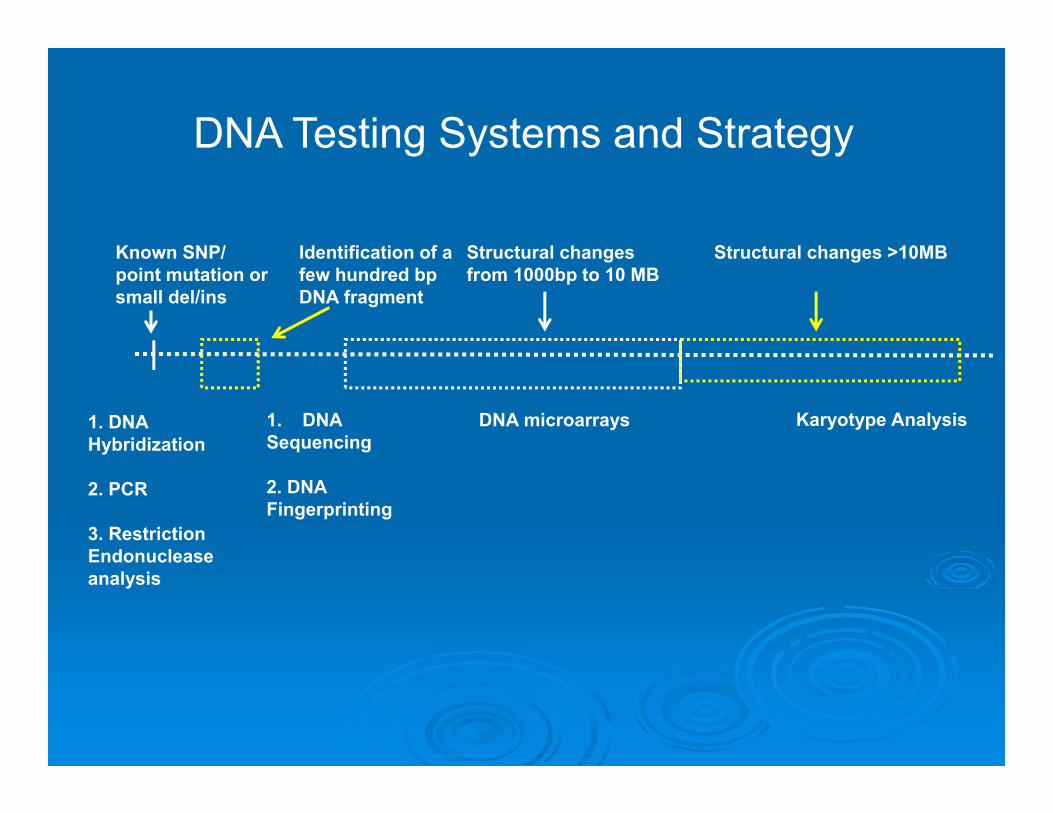
Karyotype Analysis
Structural changes >10MBKnown SNP/ point mutation or small del/ins
1. DNA Hybridization
2. PCR
3. Restriction Endonuclease analysis
Identification of a few hundred bp DNA fragment
1. DNASequencing
2. DNA Fingerprinting
Structural changes from 1000bp to 10 MB
DNA microarrays
DNA Testing Systems and Strategy

DNA ExtractionThe multistep process of isolating DNA from organic
material (cell components, proteins, sugars, etc.).
DNA can be extracted from almost any intact cellular tissue Blood sample (most common for adult testing) Paper cards with blood drops on them Saliva or buccal scrapes Chorionic villus biopsy samples or amniotic fluid (fetal DNA) Muscle tissue, skin, hair, semen, bone marrow One or two cells removed from 8-cell embryo
(in vitro fertilisation) Archived pathological specimens
(tumor samples in paraffin blocks)

Types of Mutations Tested
Disease
Point mutation
Deletions &duplications
Few recurrentmutations?
Many uniquemutations?
Also with pointmutations?
Whole gene?Some exons?
Other mutations

Recurrent MutationsMethods
PCR DNA Hybridization Restriction Endonuclease Analysis

Polymerase chain reaction (PCR):Artificial in vitro replication – amplification of
a short specific DNA sequencePCR process requires 4 main components1. Genomic DNA in small quantity, knowing DNA sequence of the region to
amplify in advance.
2. A large number of free DNA nucleotides (A,G,T,C).
3. DNA polymerase – a thermostable enzyme performing the vital process of DNA replication.
4. Two primers – small DNA sequences (oligonucleotides) which correspond to the DNA sequences immediately adjacent to the sequence of interest of genomic DNA.
5’ aatcgaatgtgcccgtacgattcgatgcgaaactaggagccctatcgat 3’
3’ ttagcttacacgggcatgctaagctacgctttgatcctcgggatagcta 5’
cgggcataactagg
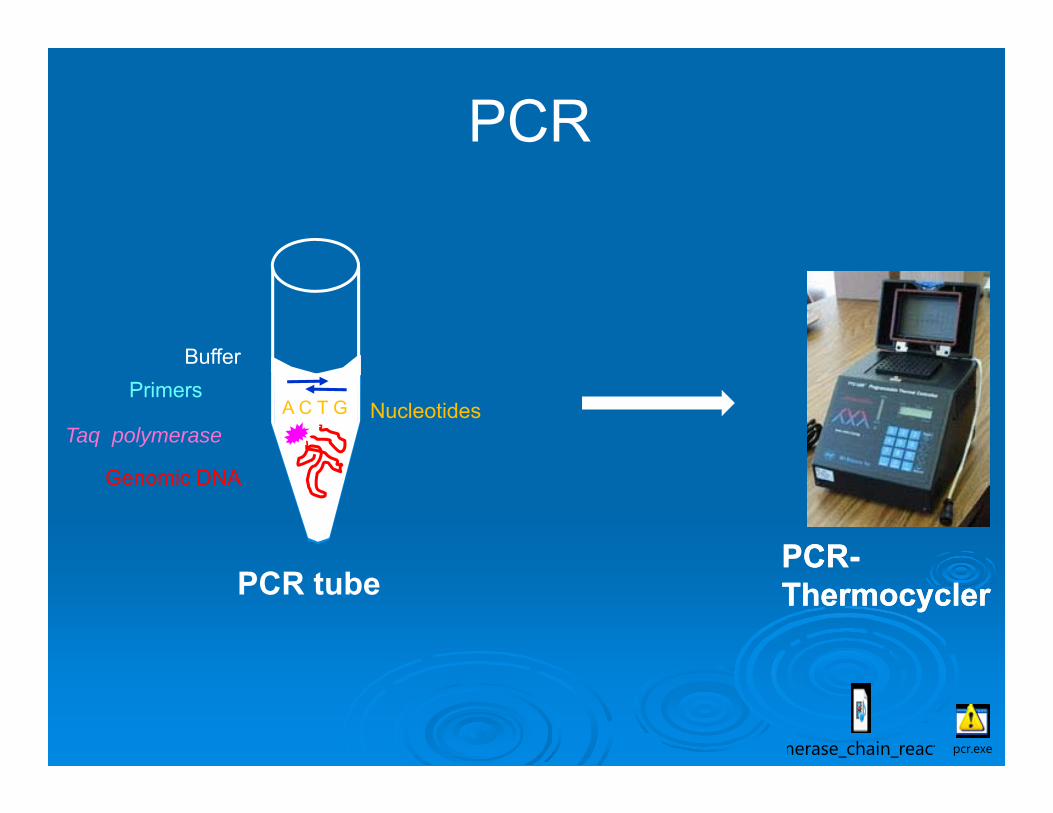
PCR-ThermocyclerPCR-Thermocycler
NucleotidesTaq polymerase
Primers
Genomic DNA
Buffer
A C T G
PCR
PCR tube
merase_chain_react pcr.exe
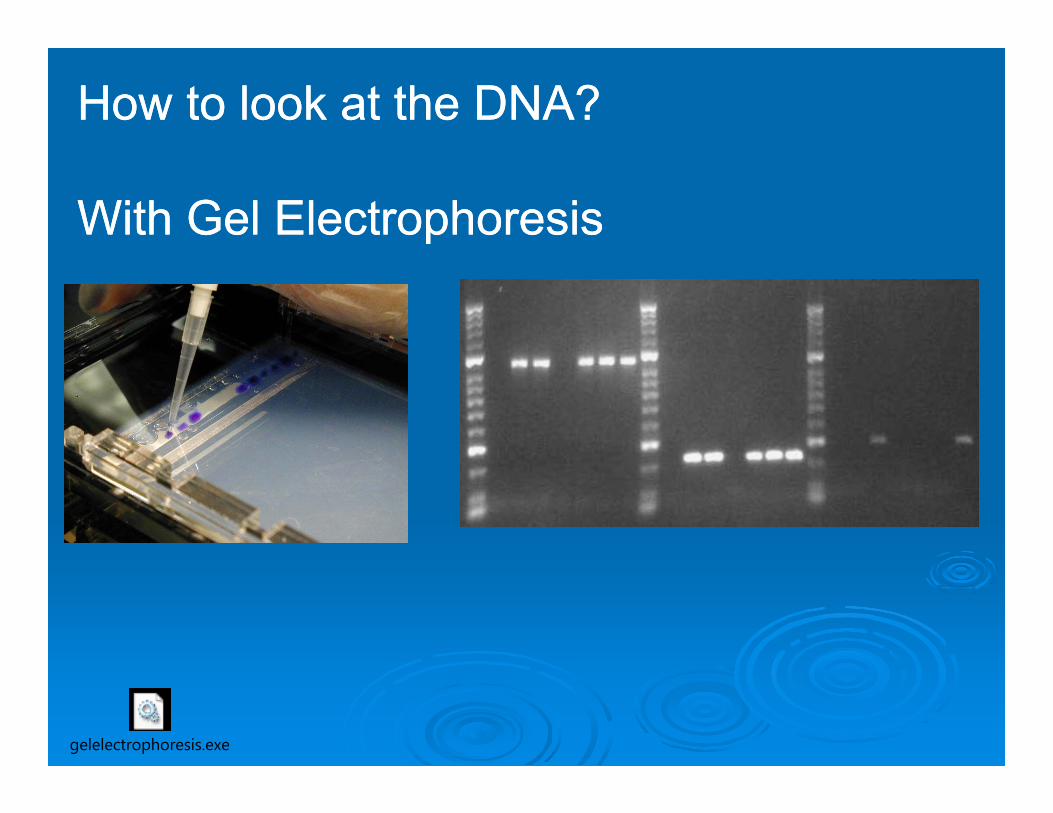
How to look at the DNA?
With Gel Electrophoresis
How to look at the DNA?
With Gel Electrophoresis
gelelectrophoresis.exe

Applications of PCR Detection of mutations Detection of infections Downstream use of DNA Sequencing and DNA
Fingerprinting
644 bp440 bp204 bp
Ex. Prenatal testing of an autosome-recessive disorder

Unique mutations?(when we do not know where is our mutation) Methods of mutation scanning Sequencing - most direct method; Detecting mismatches or heteroduplex DNA
molecules; Detecting of deletions
Structural changes >10MBKnown SNP/ point mutation or small del/ins
Ex. PCR
Scanning of a few hundred bp DNA fragment
DNA Sequencing
Structural changes from 1000bp to 10 MB

cycseq.exe
DNA SequencingDirect DNA Diagnostic Method of Several
Hereditary Disorders in case of absence of major mutations
ExamplesHereditary Breast and Ovarian CancerNeurofibromatosisPhenylketonuria Cystic FibrosisHemophilia B
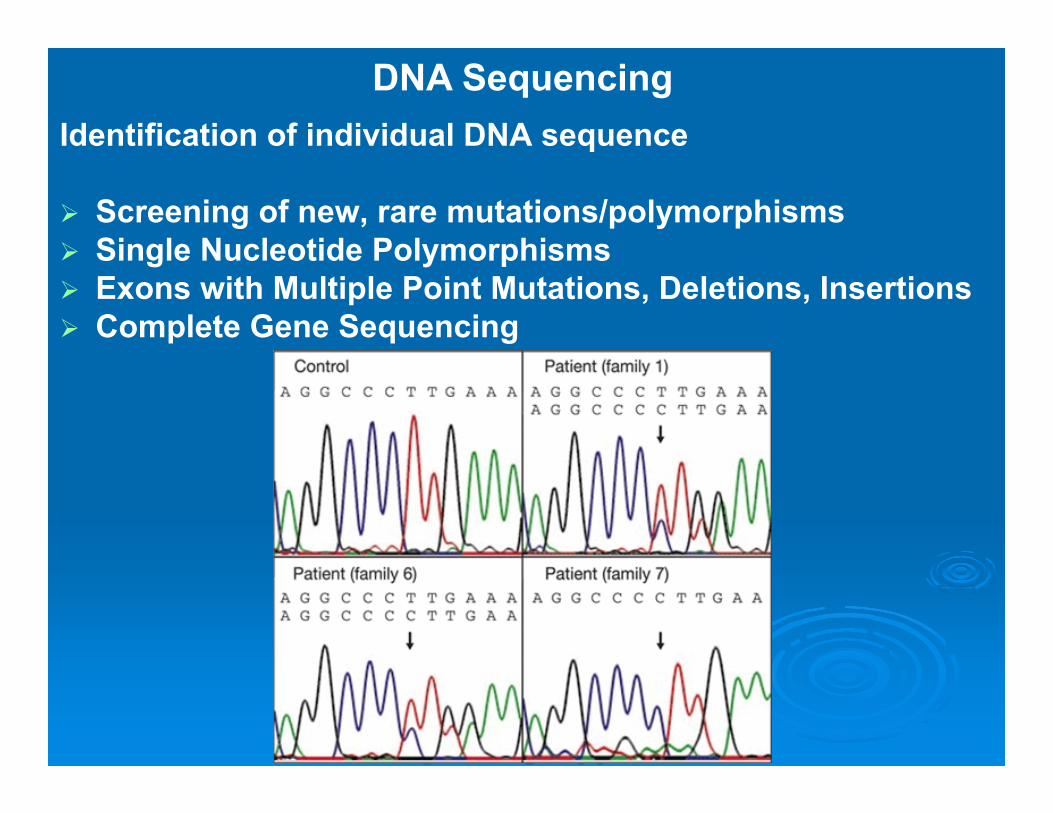
Identification of individual DNA sequence
Screening of new, rare mutations/polymorphisms Single Nucleotide Polymorphisms Exons with Multiple Point Mutations, Deletions, Insertions Complete Gene Sequencing
DNA Sequencing

Problems arising in genetic testingExample: Duchenne muscular dystrophy
Problems:
1. Gene is large, 2,4 Mb, 79 exonsHard to find point mutation
2. High Frequency of new mutations (30% of cases);
3. First mutation carrier is often a mosaic(blood may be not a mutation carrier)
There is a need to use different DNA testing systems and different tissues!!!
60
40
20
Deletion Duplication Point
%
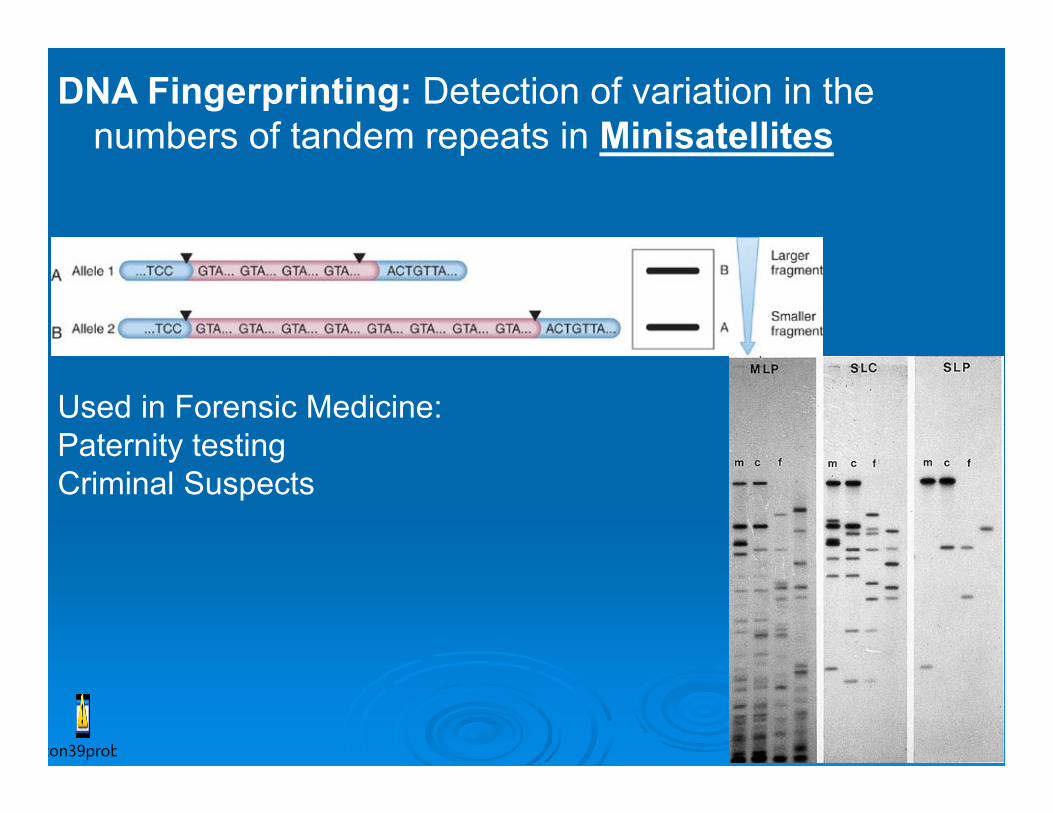
DNA Fingerprinting: Detection of variation in the numbers of tandem repeats in Minisatellites
con39prob
Used in Forensic Medicine: Paternity testing Criminal Suspects
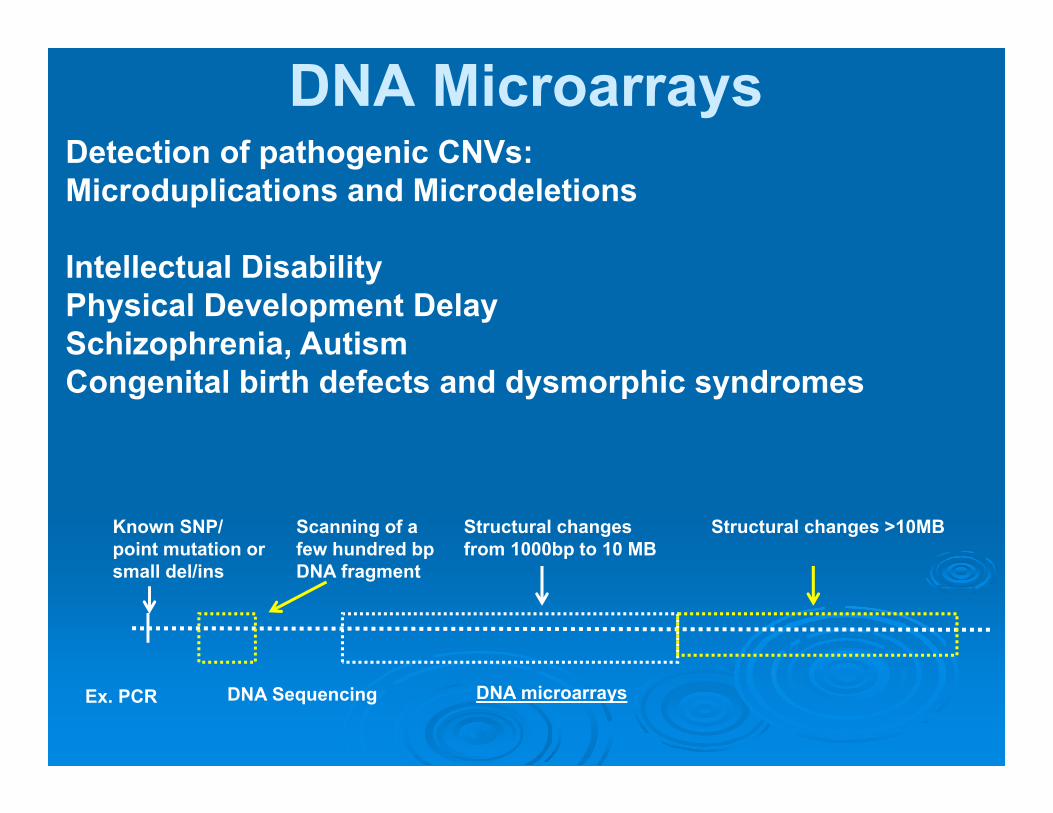
DNA MicroarraysDetection of pathogenic CNVs: Microduplications and Microdeletions
Intellectual DisabilityPhysical Development DelaySchizophrenia, AutismCongenital birth defects and dysmorphic syndromes
Structural changes >10MBKnown SNP/ point mutation or small del/ins
Ex. PCR
Scanning of a few hundred bp DNA fragment
DNA Sequencing
Structural changes from 1000bp to 10 MB
DNA microarrays

Organization of a DNA microarray
• Comparative Genome Hybridization with special matrixes - arrays
• Array-CGH
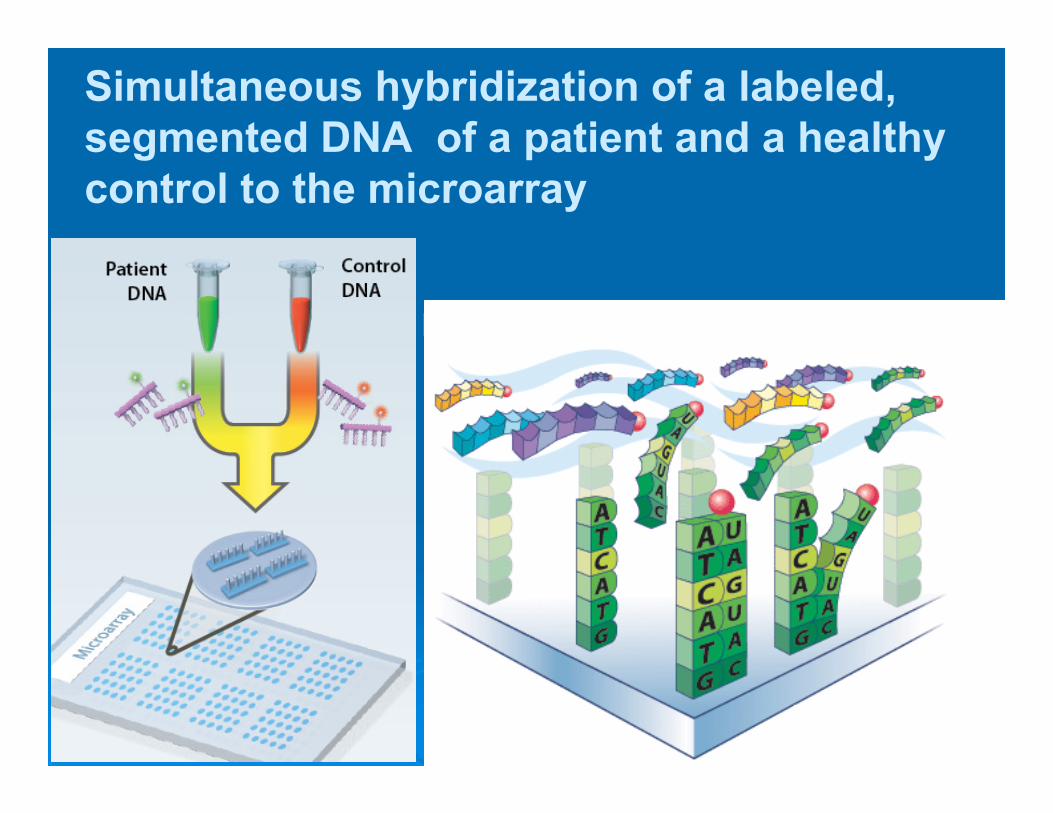
Simultaneous hybridization of a labeled, segmented DNA of a patient and a healthy control to the microarray
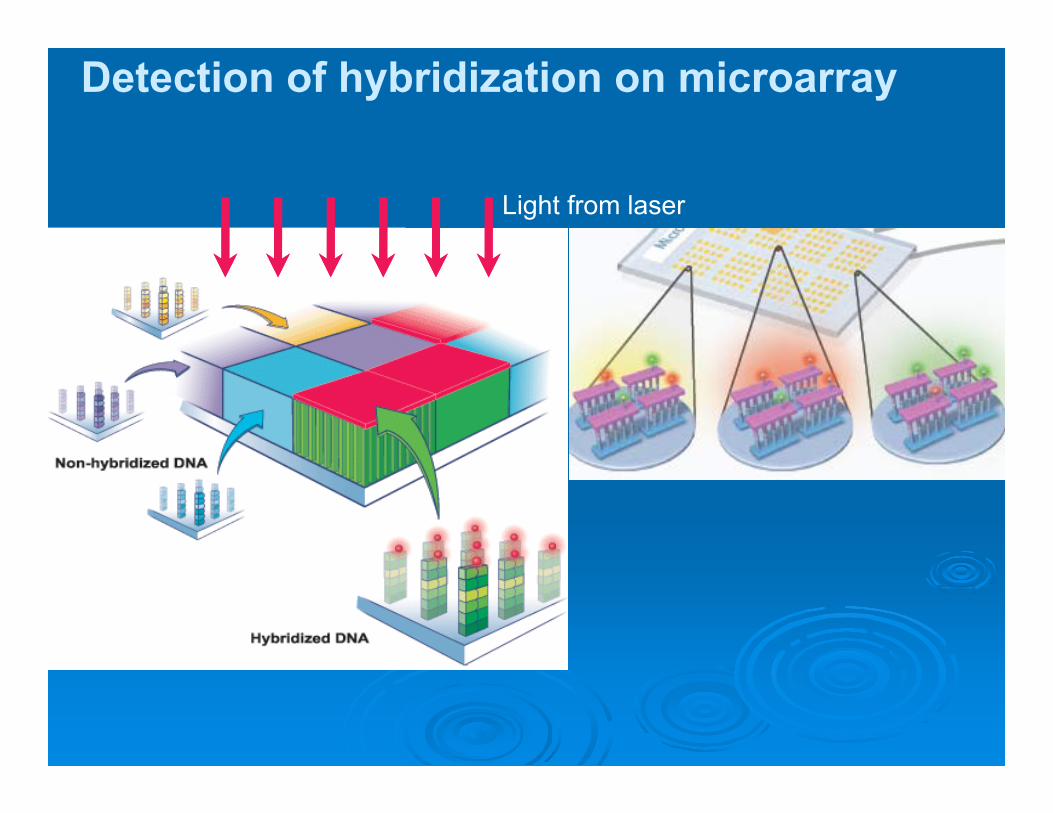
Detection of hybridization on microarray
Light from laser
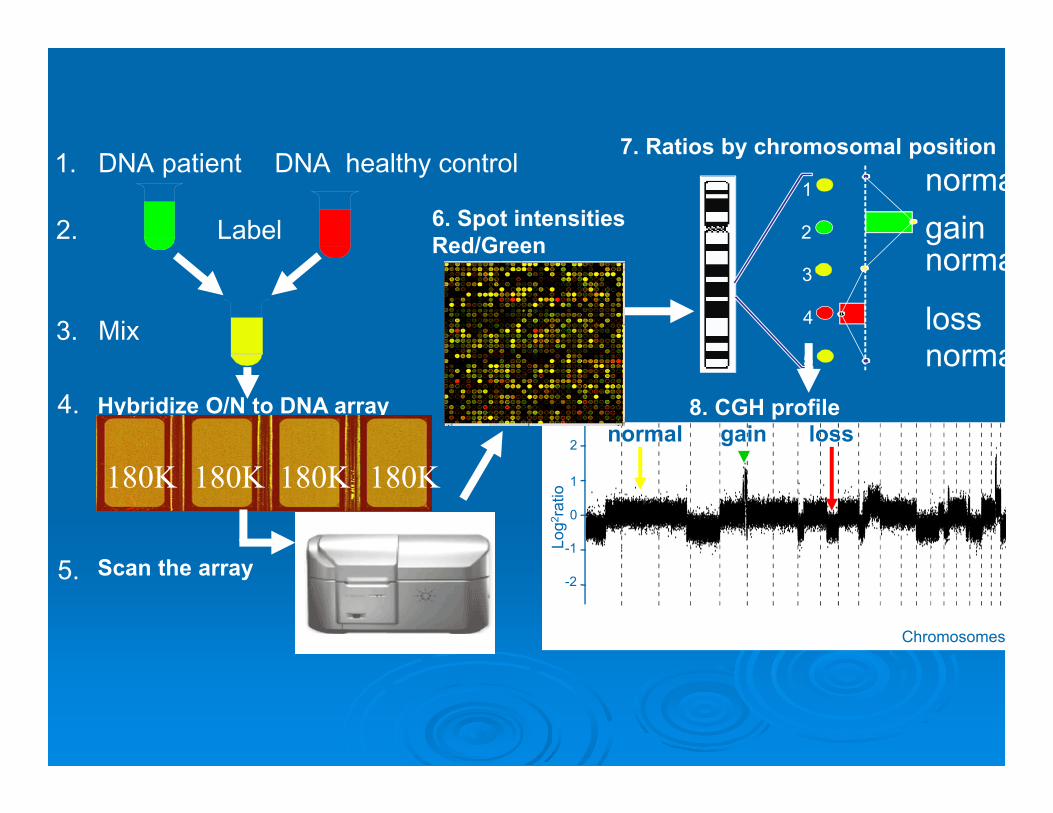
DNA patient DNA healthy control
Mix
1.
2. Label
3.
5. Scan the array
1
2
3
4
5
loss
gain
7. Ratios by chromosomal positionnorma
norma
norma
Hybridize O/N to DNA array4.
180K180K180K180K
6. Spot intensitiesRed/Green
0
1
2
-2
-1Log
2 rat
io
Chromosomes
8. CGH profilegain lossnormal

SNP-Array for whole-genome analysis
Overview of SNP array technology
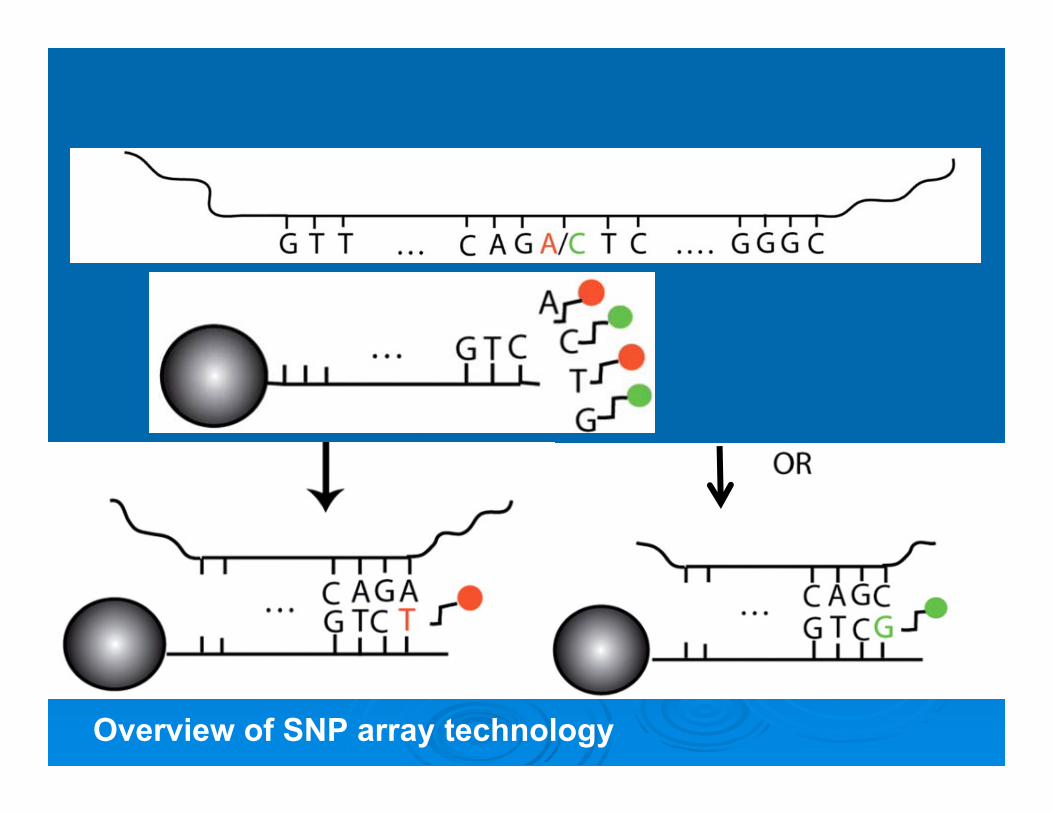
Overview of SNP array technology
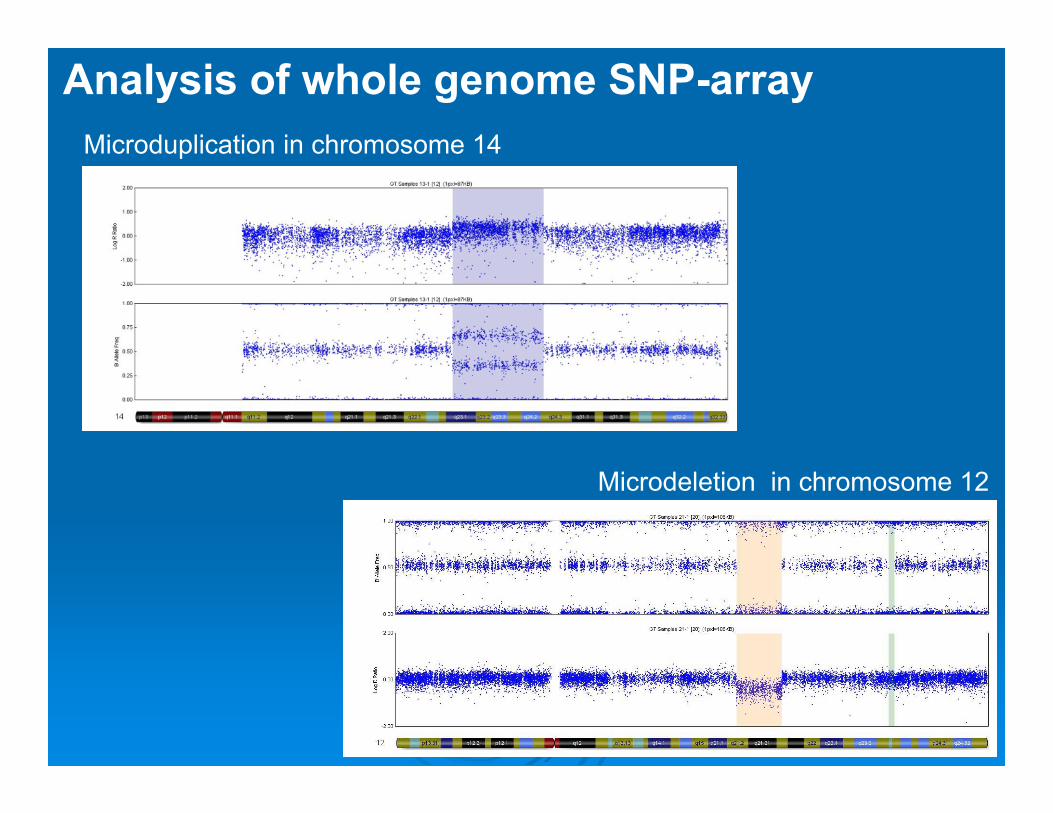
Analysis of whole genome SNP-arrayMicroduplication in chromosome 14
Microdeletion in chromosome 12

Advantages of Array-CGH and SNP-array

Limitations of Array-CGH and SNP-array:
Can not detect

Indications of Array-CGH and SNP-array for Prenatal Diagnostics

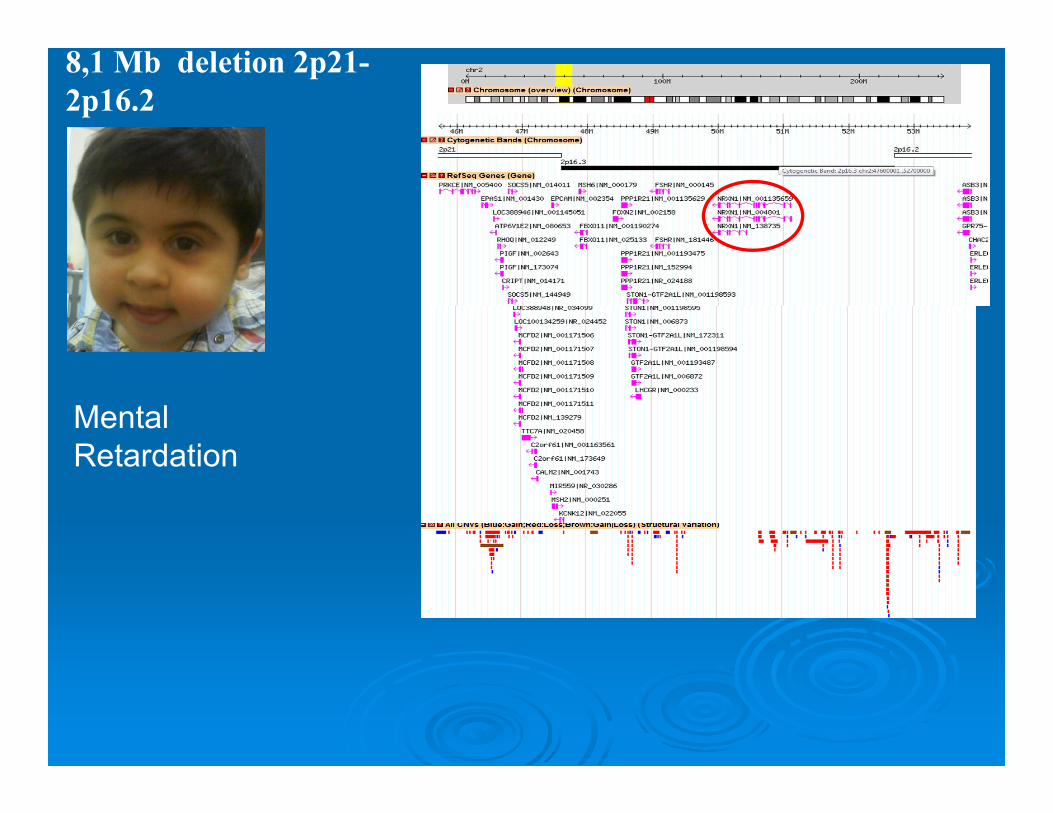
8,1 Mb deletion 2p21-2p16.2
Mental Retardation
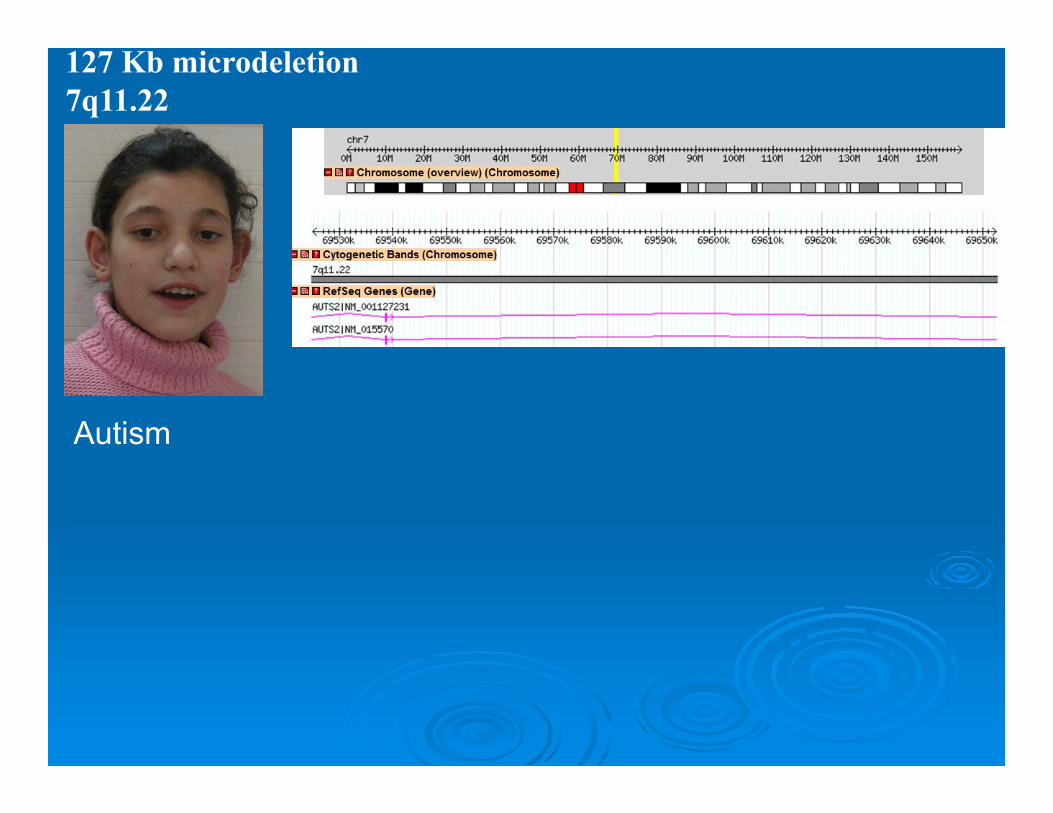
127 Kb microdeletion 7q11.22
Autism

Next‐generation DNA sequencing: High‐throughput and highly parallelized
DNA‐sequencing technologies
High‐throughput: Produces many hundreds of thousands or millions of short reads (25–500 bp) for a low cost and in a short time.
Detection of nucleotide alterations with high sensitivity: Each nucleotide is read many times (generation of huge numbers of data)
Highly parallelized DNA‐sequencing technologies: DNA is sequenced in parallel by synthesis)
Next‐generation DNA sequencing: High‐throughput and highly parallelized
DNA‐sequencing technologies
High‐throughput: Produces many hundreds of thousands or millions of short reads (25–500 bp) for a low cost and in a short time.
Detection of nucleotide alterations with high sensitivity: Each nucleotide is read many times (generation of huge numbers of data)
Highly parallelized DNA‐sequencing technologies: DNA is sequenced in parallel by synthesis)
http://vimeo.com/65886400
http://www.homolog.us/blogs/blog/2012/06/07/animations-for-sequencing-technologies-from-the-web/
http://www.wellcome.ac.uk/Education-resources/Education-and-learning/Resources/Animation/WTX056051.htm

Next Generation Sequencing
Whole Genome Sequencing
Whole Exome Sequencing ‐ The technique of sequencing and analyzing less than 1.5% of the genome
Prenatal DiagnosticsIdentification of new mutations Interpret known monogenic disease genes Identification of structural changes, including CNVsIdentification of new genes associated with rare diseases
Targeted PlatformsCustomized target‐enrichment platforms containing all the genes known to be associated with a given diseaseUseful in the diagnosis of heterogeneous genetic conditions

Kabuki syndromeKabuki syndrome1. Intellectual disability,2. Peculiar face,3. Increased susceptibility to infections and
cancer at childhood
Exome sequencing of unrelated indviduals sharing thesame phenotype
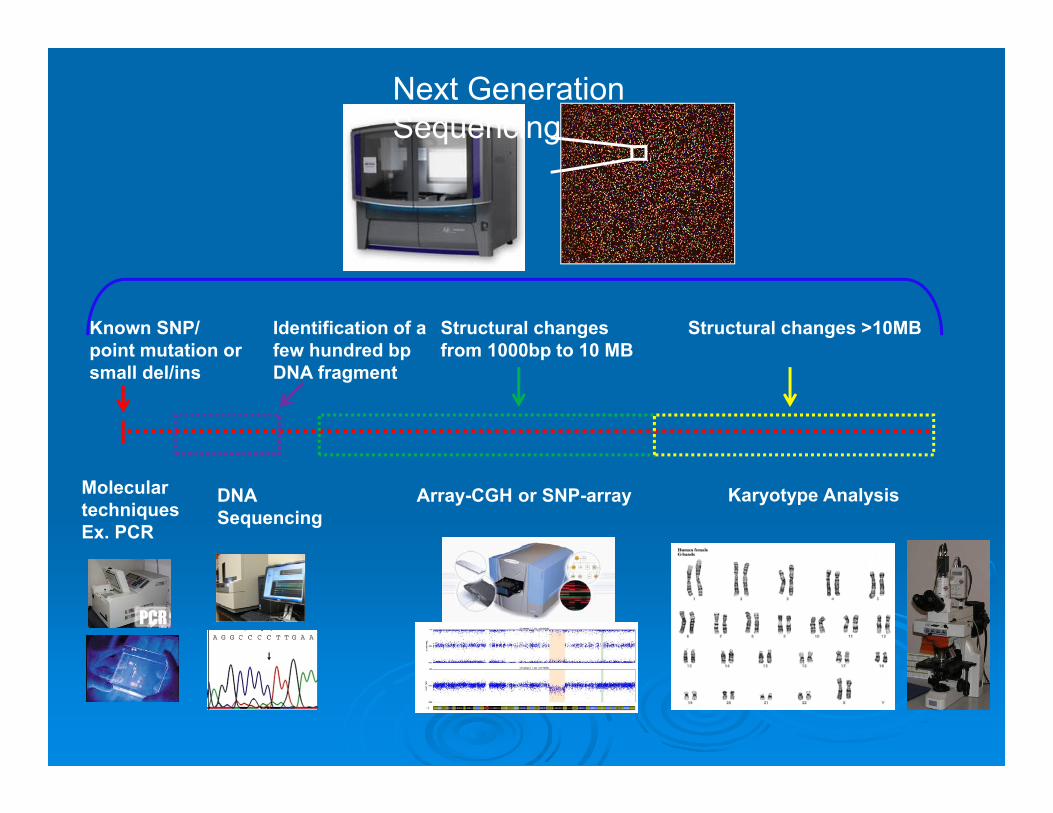
Karyotype Analysis
Structural changes >10MBKnown SNP/ point mutation or small del/ins
Molecular techniquesEx. PCR
Identification of a few hundred bp DNA fragment
DNA Sequencing
Structural changes from 1000bp to 10 MB
Array-CGH or SNP-array
Next Generation Sequencing

CHROMOSOMAL BASIS OF
HUMAN DISEASES
PhD Susanna Midyan

1960: Denver Conference:cytogenetics became a branch of medicine
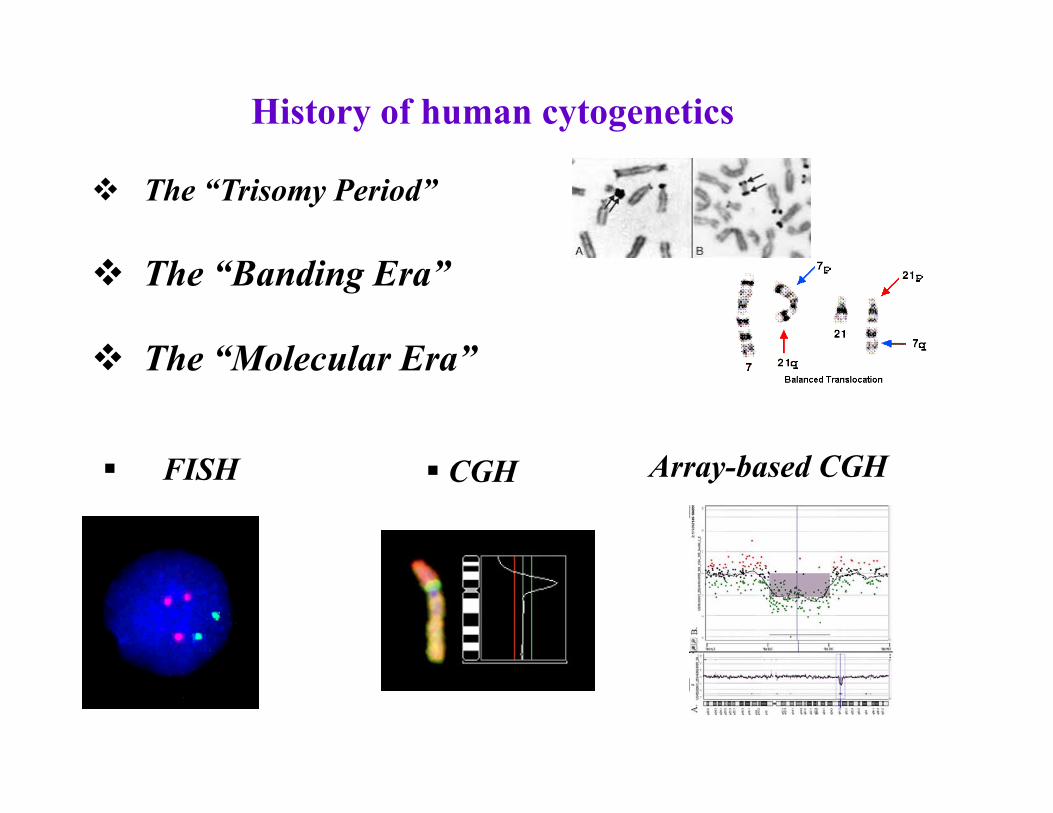
History of human cytogenetics
The “Trisomy Period”
The “Banding Era”
The “Molecular Era”
Аrray-based CGH FISH CGH
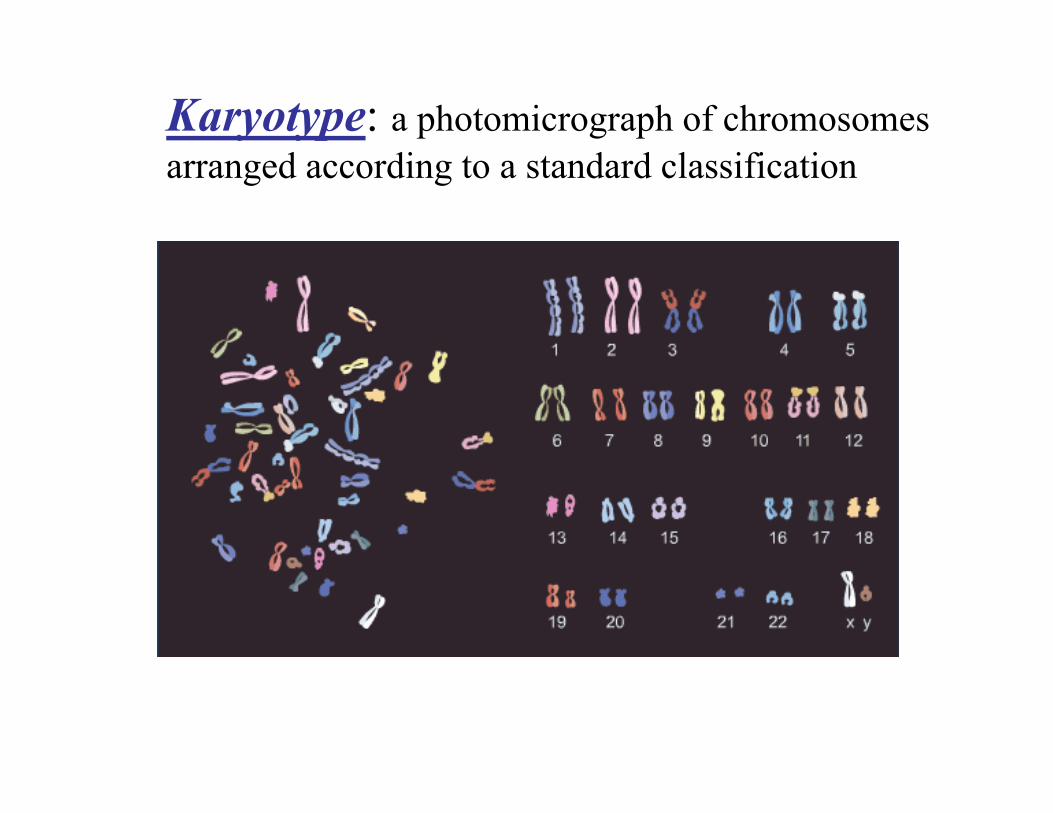
Karyotype: a photomicrograph of chromosomes arranged according to a standard classification

Indications for karyotype study• Diagnose of constitutional disorders
– disorders present at birth (birth defects, ambiguous genitalia)
– intellectual disability and physical development delay – adults with reproductive failure– fetal chromosome diagnosis (preimplantaion/prenatal
diagnostics)• Diagnose of an acquired disorders
– most commonly haematological malignancies and solid tumor
– evaluation of chromosome aberrations in mutagenesis (in vitro)
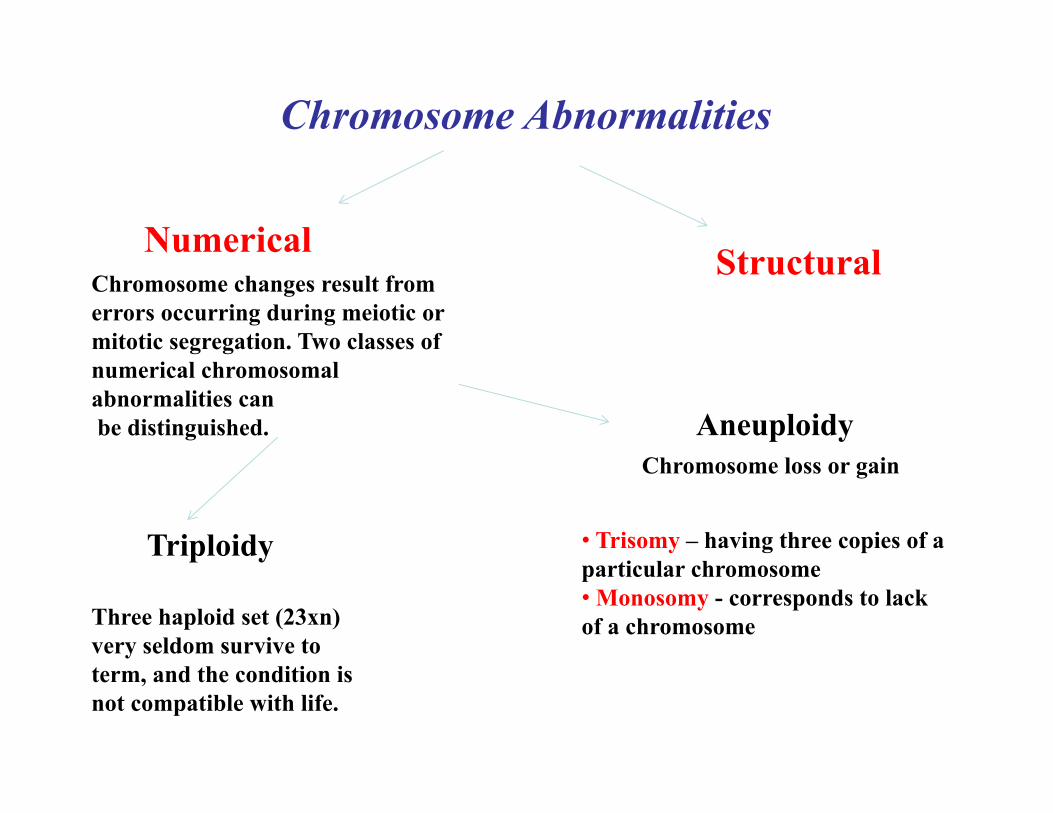
Chromosome Abnormalities
Numerical StructuralChromosome changes result from errors occurring during meiotic or mitotic segregation. Two classes of numerical chromosomal abnormalities can be distinguished. Aneuploidy
Chromosome loss or gain
Triploidy
Three haploid set (23xn) very seldom survive to term, and the condition is not compatible with life.
• Trisomy – having three copies of a particular chromosome • Monosomy - corresponds to lack of a chromosome

• 21 trisomy –Down syndrome ‐ 47,XX,+21
AUTOSOMAL TRISOMIES
• 18‐trisomy – Edwards syndrome ‐ 47,XX,+18
• 13 trisomy – Patau syndrome ‐ 47,XX,+13
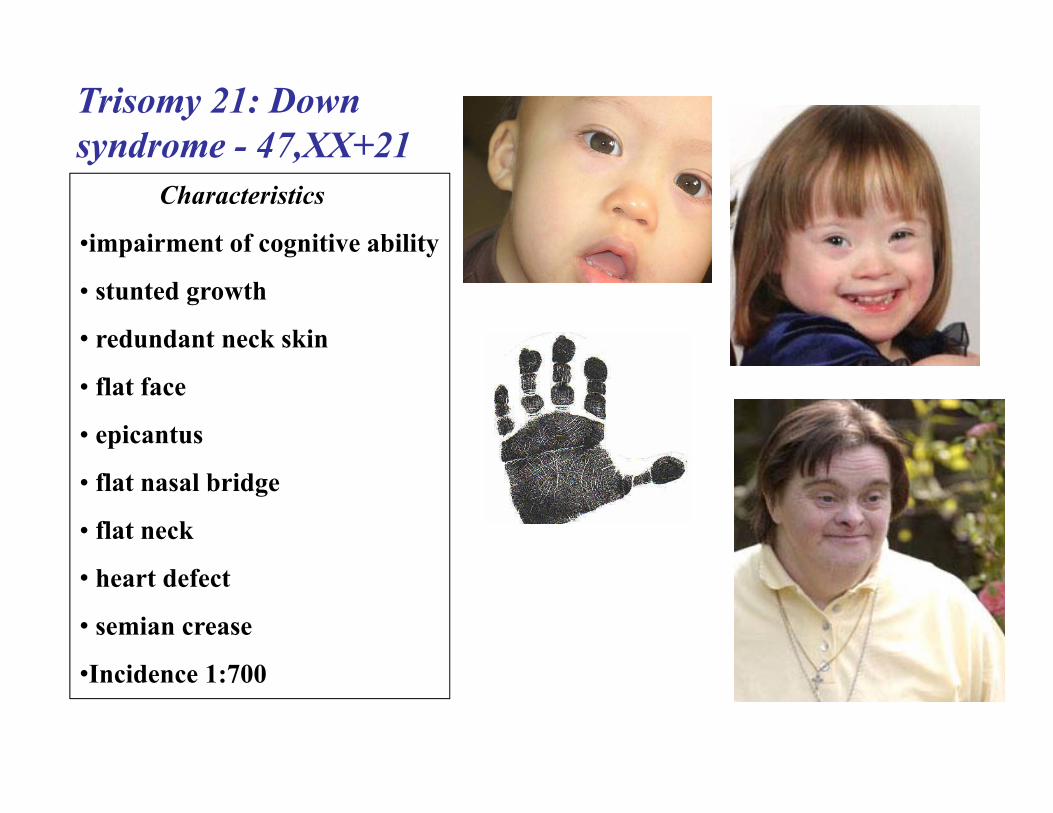
Trisomy 21: Down syndrome - 47,XX+21
Characteristics
•impairment of cognitive ability
• stunted growth
• redundant neck skin
• flat face
• epicantus
• flat nasal bridge
• flat neck
• heart defect
• semian crease
•Incidence 1:700
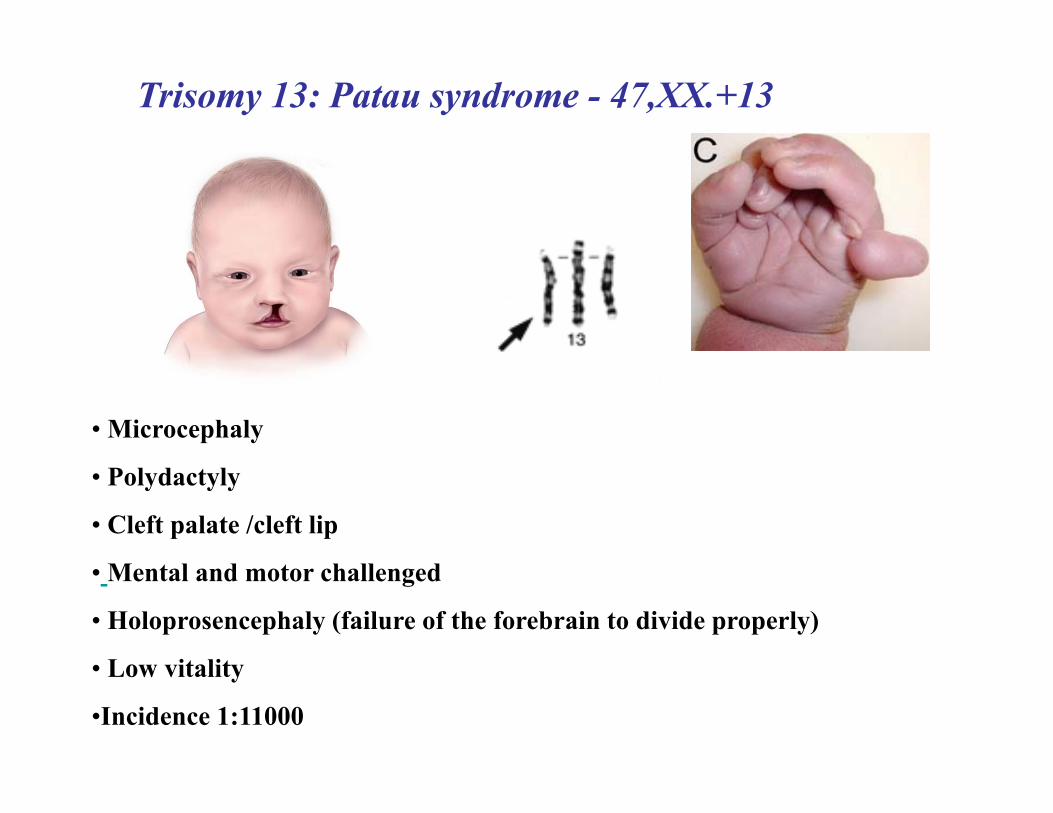
Trisomy 13: Patau syndrome - 47,XX.+13
• Microcephaly
• Polydactyly
• Cleft palate /cleft lip
• Mental and motor challenged
• Holoprosencephaly (failure of the forebrain to divide properly)
• Low vitality
•Incidence 1:11000
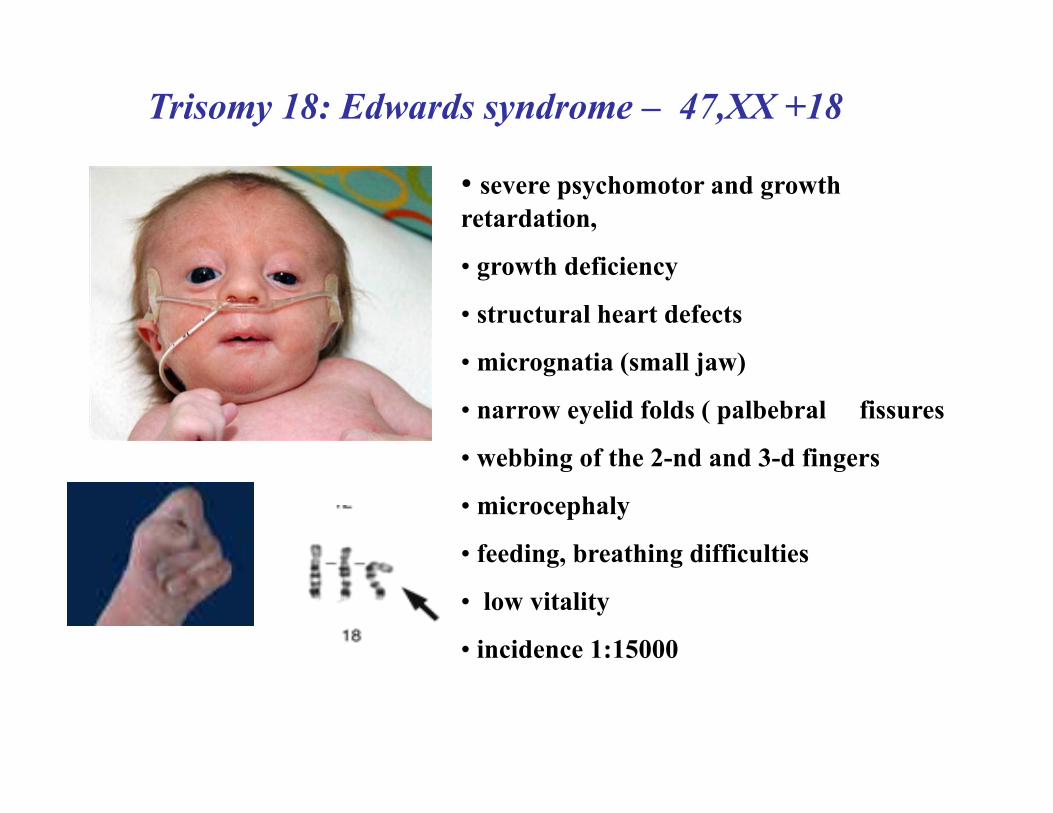
Trisomy 18: Edwards syndrome – 47,XX +18
• severe psychomotor and growth retardation,
• growth deficiency
• structural heart defects
• micrognatia (small jaw)
• narrow eyelid folds ( palbebral fissures
• webbing of the 2-nd and 3-d fingers
• microcephaly
• feeding, breathing difficulties
• low vitality
• incidence 1:15000

X-Y related syndromes• Turner syndrome:
– Occurs when females inherit only one X chromosome; their genotype is X0.
• Triple-X females:– Inherit three X chromosomes; their genotype is XXX or
more rarely XXXX or XXXXX.• Klinefelter syndrome:
– Males inherit one or more extra X chromosomes; their genotype is XXY or more rarely XXXY, XXXXY, or XY/XXY mosaic.
• Jacobs syndrome :– Males inherit an extra Y chromosome; their genotype is XYY.
11
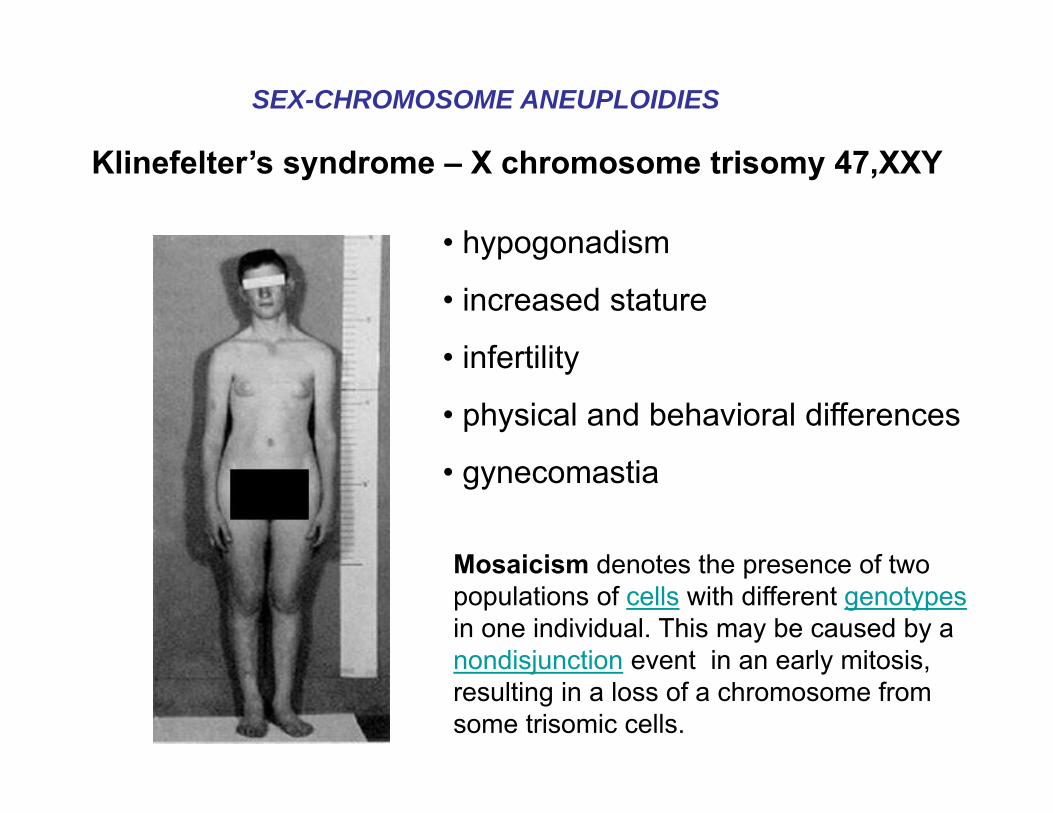
Klinefelter’s syndrome – X chromosome trisomy 47,XXY
• hypogonadism
• increased stature
• infertility
• physical and behavioral differences
• gynecomastia
SEX-CHROMOSOME ANEUPLOIDIES
Mosaicism denotes the presence of two populations of cells with different genotypesin one individual. This may be caused by a nondisjunction event in an early mitosis, resulting in a loss of a chromosome from some trisomic cells.
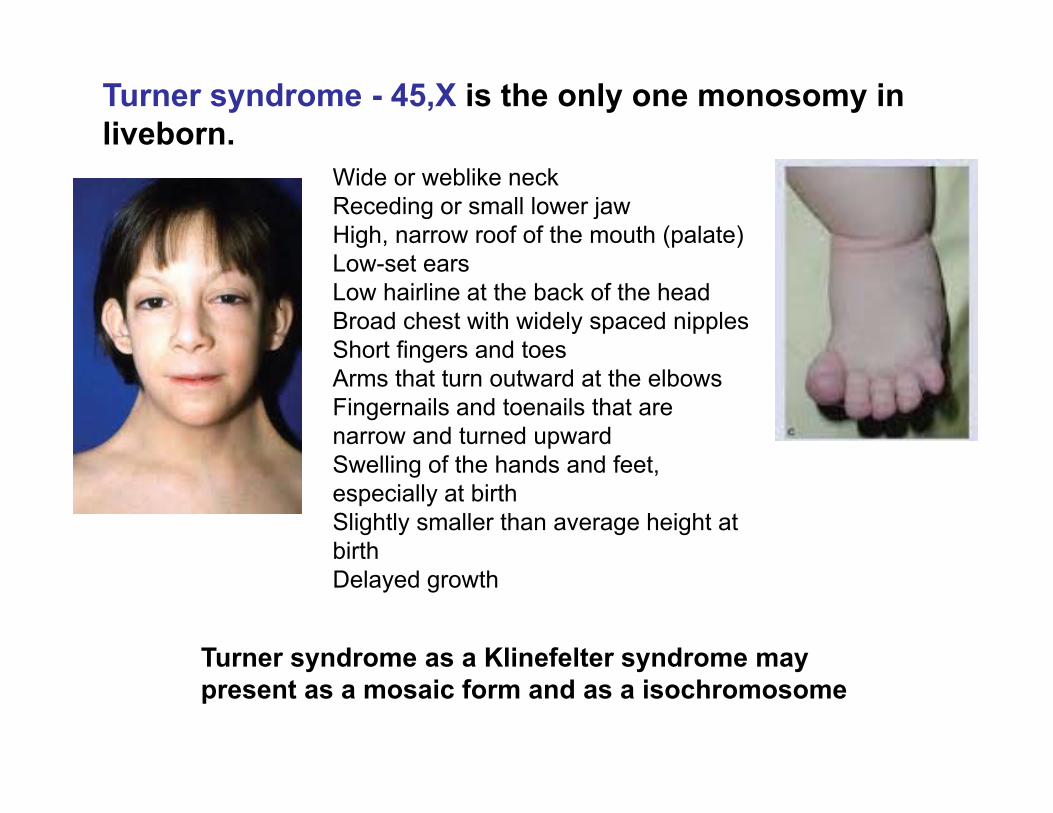
Turner syndrome - 45,X is the only one monosomy in liveborn.
Turner syndrome as a Klinefelter syndrome may present as a mosaic form and as a isochromosome
Wide or weblike neckReceding or small lower jawHigh, narrow roof of the mouth (palate)Low-set earsLow hairline at the back of the headBroad chest with widely spaced nipplesShort fingers and toesArms that turn outward at the elbowsFingernails and toenails that are narrow and turned upwardSwelling of the hands and feet, especially at birthSlightly smaller than average height at birthDelayed growth

Preimplantation stage – 1;5;6;11;19 trisomies
Early miscarrigies – 50% of them are caused by different chromosomes trisomies, particularly:
+16 trisomy
69,ХХХ- triploidy,
After birth - the following 13, 18, 21, Х и Y trisomies and X monosomy occurred in 0,6%.
Numerical chromosome anomalies occurred in different stages of ontogenesis

Structural Chromosome Rearrangements
Balanced Unbalancedis a structural chromosomal abnormalities if there is nogain or loss of chromosomematerial
• inversions • translocations
• reciprocal• robertsonian
there is a gain or loss of chromosome material
• translocations • inversions • insertions • deletions • duplications • ring • dicentric • isochromosomes

ISCN - International System for HumanCytogenetic Nomenclature
• most recent edition pub. 2013• provides standardized “grammatical” rules for the
designation of cytogenetic findings
Example: 46,XY,der(9)(9qter9p23:: ::4q264qter),pat
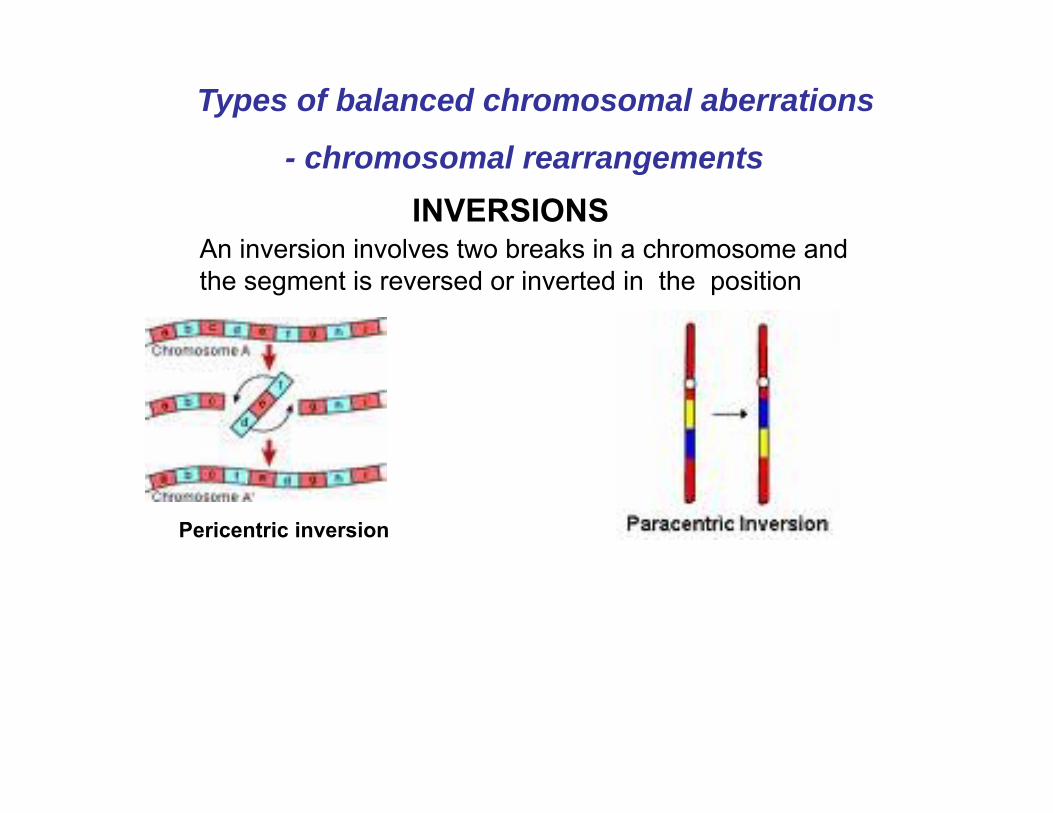
INVERSIONS
Pericentric inversion
An inversion involves two breaks in a chromosome and the segment is reversed or inverted in the position
Types of balanced chromosomal aberrations
- chromosomal rearrangements
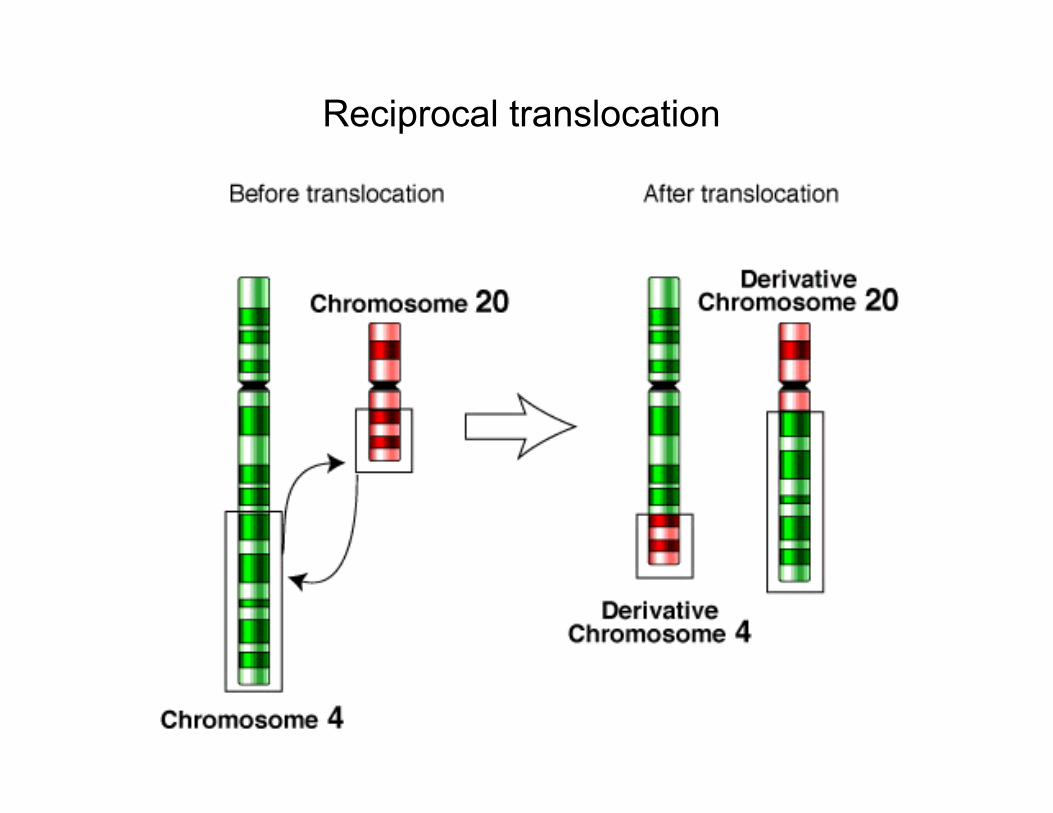
Reciprocal translocation

A carrier of a balanced reciprocal translocation can produce gametes that give rise to an entirely normal child, a phenotypically normal balanced carrier, or various unbalanced karyotypes

CLINICAL CASE
Various ocular abnormalities are presented. Ophthalmologic assessment revealed: •divergent strabismus, •unusually small eyes (microphtalmiа), optic nerve subatrophy,•retina atrophy, •locus oculusum with searching look..receding jaw (microretrognatia), •pointed chin•highly arched palate, •short neck, •nipples asymmetric set with • hypertelorism, •umbilical hernia, brachidactyly, puffy hands, clubfeet, double cryptorchidism, overlapping of 3th and 4th toes
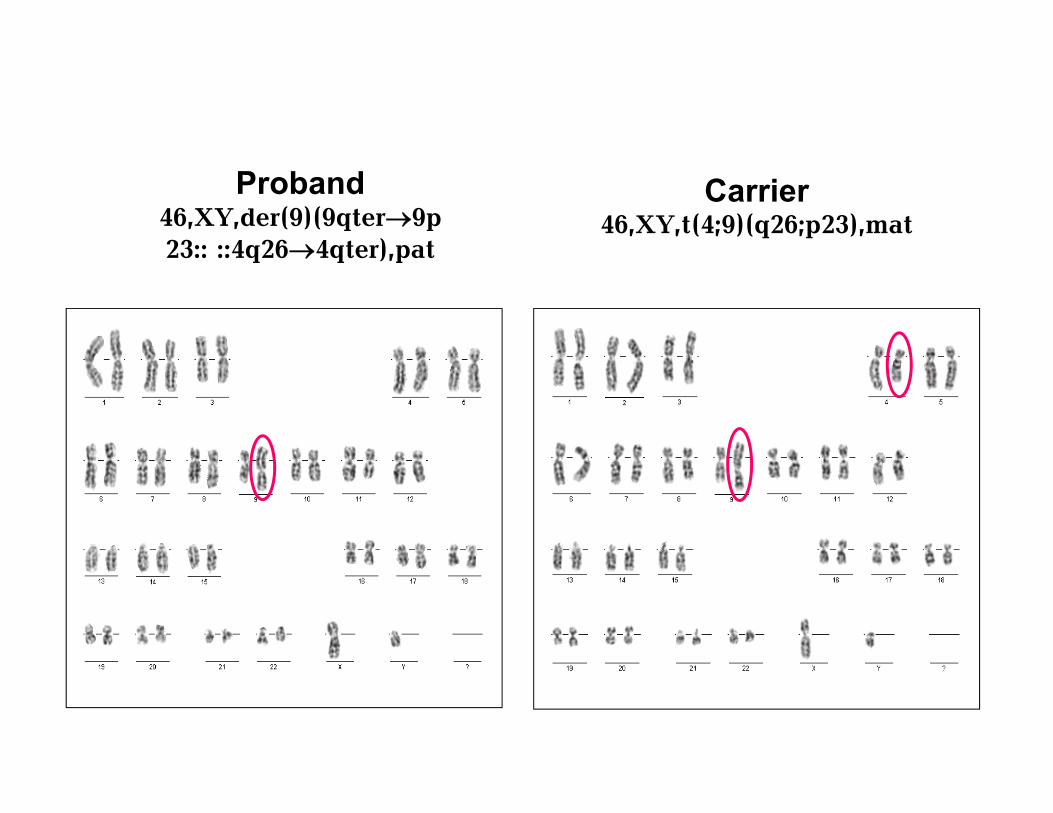
Carrier 46,XY,t(4;9)(q26;p23),mat
Proband 46,XY,der(9)(9qter9p23:: ::4q264qter),pat

Partial trisomy 4q26-qter as a result of a reciprocal translocation in parent
Various ocular abnormalities are presented. Ophthalmologic assessment revealed: •divergent strabismus, •unusually small eyes (microphtalmiа), optic nerve subatrophy,•retina atrophy, •locus oculusum with searching look..receding jaw (microretrognatia), •pointed chin•highly arched palate, •short neck, •nipples asymmetric set with • hypertelorism, •umbilical hernia, brachidactyly, puffy hands, clubfeet, double cryptorchidism, overlapping of 3th and 4th toes
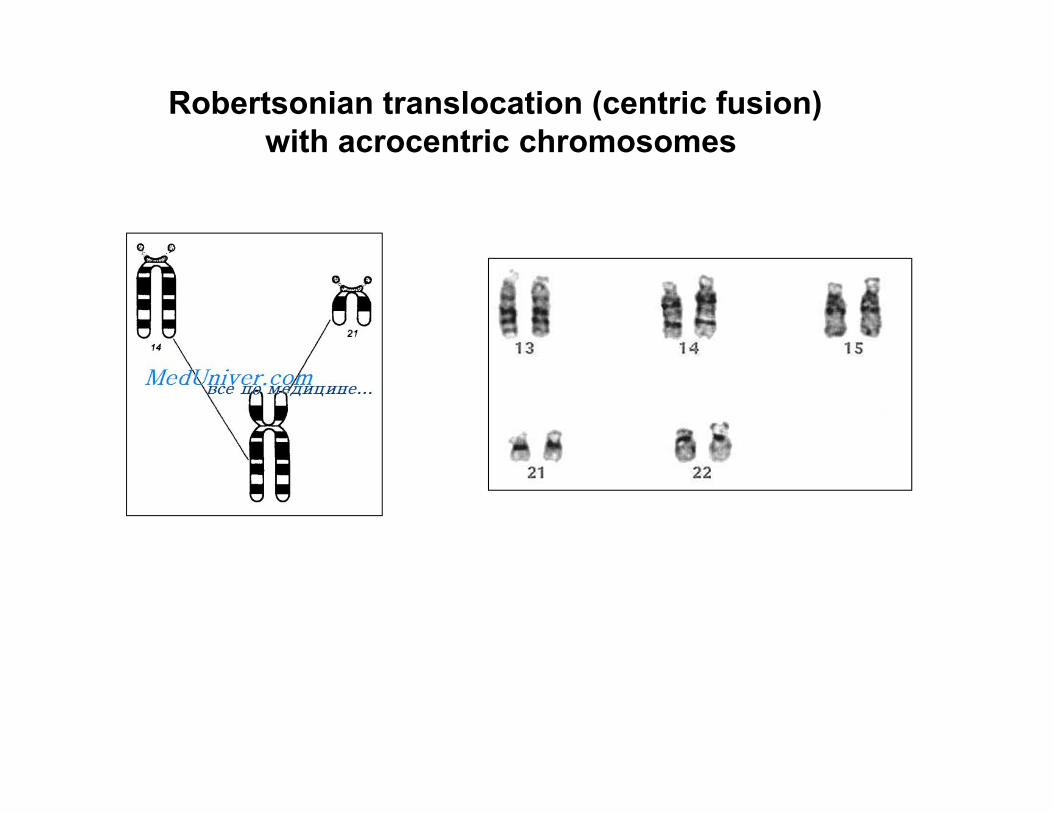
Robertsonian translocation (centric fusion)with acrocentric chromosomes

Carrier 45,XX,t(14;21)(q10;q10)
Proband 46,XX,t(14;21)(q10;q10),+21

DOWN SYNDROME KARYOTYPE VARIANTS
a )47, XX,+21 b) 46,XY,t (13,21),+21c) 46,XX,t (14;21),+21 d) 46,XX,t (15;21),+21e) 46,XY,t(21;21)+21
· A phenotypically normal woman with a 45, XX, -14, -21, +t(14q, 21q) karyotype has a karyotypically normal (46, XY) husband. Among their liveborn offspring, the most likely karyotypes are 46, XY and 46, XX. Which of the following karyotypes is the next most likely among liveborn offspring?
47, XY, +21
46, XY, -14, +t(14q, 21q)
46, XY, -21, +t(14q, 21q)
45, XY, -14, -21, +t(14q, 21q)
45, XY, -14, -21, +16

Unbalanced chromosomal rearrangements encompass several different classes of events: deletions, duplications, inversions..
They result from chromosome breakage with subsequent reunion in a different configuration. Chromosomal rearrangements originating in the germ line, whether inherited from the parents or from a de novo mutation in the gametes, are referred to as constitutional. On the other hand, any changes in the chromosomes which arise during development or during the life of an organism are referred as acquired chromosomal rearrangements
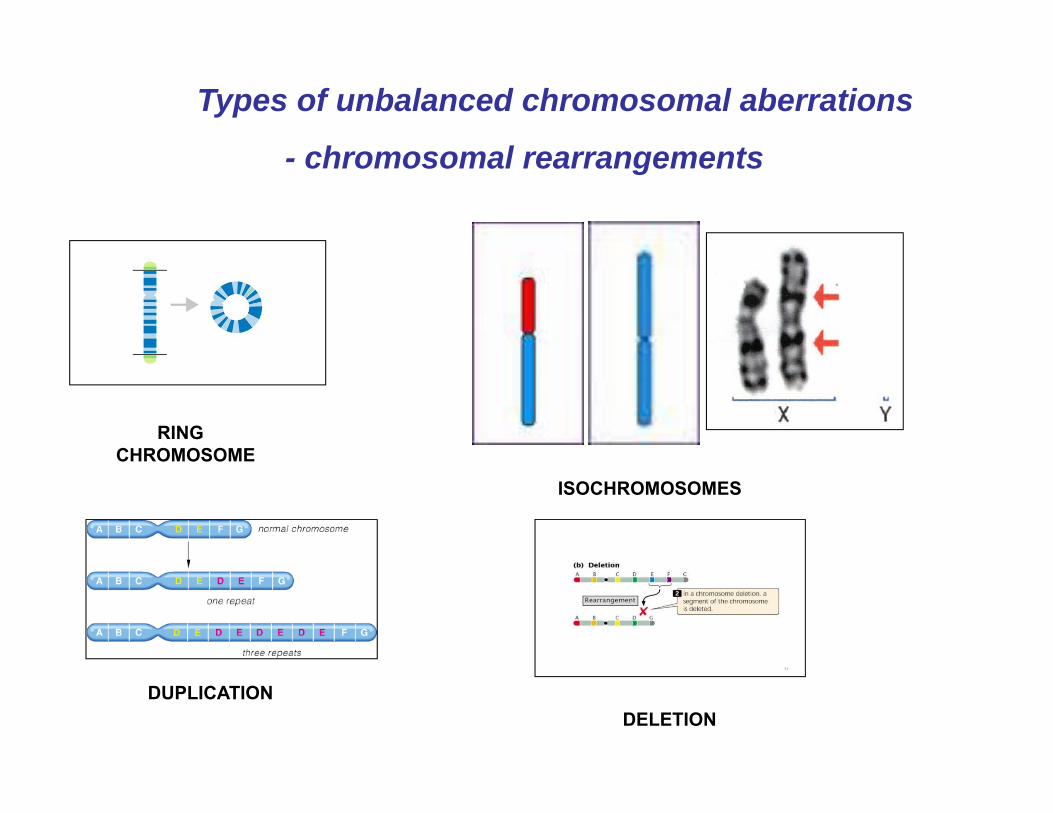
Types of unbalanced chromosomal aberrations
- chromosomal rearrangements
RING CHROMOSOME
ISOCHROMOSOMES
DUPLICATIONDELETION

Tissue samples for chromosome study
• Peripheral blood
• Amniotic fluid
• Chorionic villi
• Fibroblasts from skin biopsy
• Epithelial cells from buccal smear
• Bone marrow (hemoblastosis)
• Solid tumor biopsy28

Advantages1- Enable the entire genome to be viewed at one time.
2- Suitable for mosaicism detection
Disadvantages1- Low resolution. Detect major structural abnormalities
~ 10Mb2- Labor intensive, time consuming and highly dependent
upon operator experience and skills.
Advantages and Disadvantages of conventional cytogenetic technique

Molecular era in cytogenetics
Fluorescent in situ Hybridization(FISH)
Principles andApplications
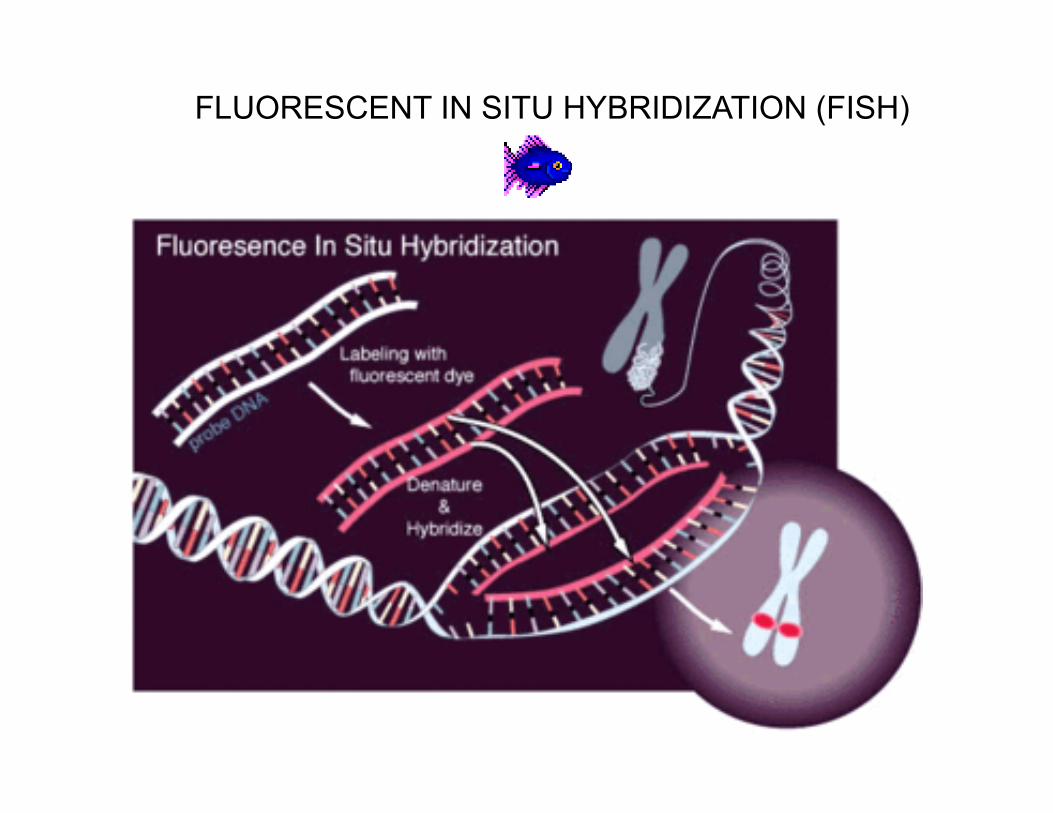
FLUORESCENT IN SITU HYBRIDIZATION (FISH)

FISH (fluorescent in situ hybridization) featuresUses оf a fluorescent probe to hybridize to specific areas of the chromosomes
•The least of chromosomal region disorder that can be detected on karyotype even by the high resolution chromosome analysis comes to 10 Mb from at least 200 genes.•FISH allows to detect changes in chromosome structure, if it consists of 5Mb and less.
•FISH can be used in metaphase cells to detect specific microdeletions beyond the resolution of routine cytogenetics or identify extra material of unknown origin.
•FISH can be used in interphase cells

Locus specific probes to rule out deletions or gains of specific loci
Types of FISH probes
Whole chromosome paint probes to rule out complex rearrangements within chromosomes
Telomere probes to rule out deletions in telomere regions

Interphase-FISH
FISH APPLICATIONS
Myc break point translocation between chromosomes 8 and 14 detected in lymphoblastic leukemia
Preimplantation genetic diagnosis - PGD
Prenatal diagnosis - FISH on uncultured amniocytes

Philadelphian chromosome in CML (Ph)
Translocation lead to the BCR-ABL "fusion" gene creation

Chromosomal microrearrangements
and related syndromes
Non-allelic homologuerecombination (NAHR)
Low copy repeats (LCRs)
Segmental duplication
Mechanism underlying the majority of genomic disorders
Microdeletion and microduplication syndromes

Microdeletion Syndromes Cri-du-chat (5p-).Miller-Dieker syndrome (7q11.23). Smith-Magenis syndrome (17p13.3). Steroid Sulfatase Deficiency (Xp22.3). DiGeorge/Velo-cardio-facial/CATCH-22/ Shprintzen
Syndrome (22q11.2). Kallman Syndrome (Xp22.3).Williams Syndrome (7q11.23).Wolf-Hirsch horn (4p-). Prader-Willi/Angelman Syndrome (15q11.2-13). X-Linked Icthyosis (xp22.3). Retinoblastoma (13q14).

Williams-Beuren Syndrome - del 7q11.23
• "elfin" facial appearance with hypercalcemia
• long philtrum
• unusually cheerful demeanor
• wide mouth
• flattened nasal bridge
• cardiovascular problems (supravalvular aortic stenosis)
•It is caused by a deletion of about 26 genes from the long arm of chromosome 7
• prevalence - 10,000-20,000 births
ELN gene deletion
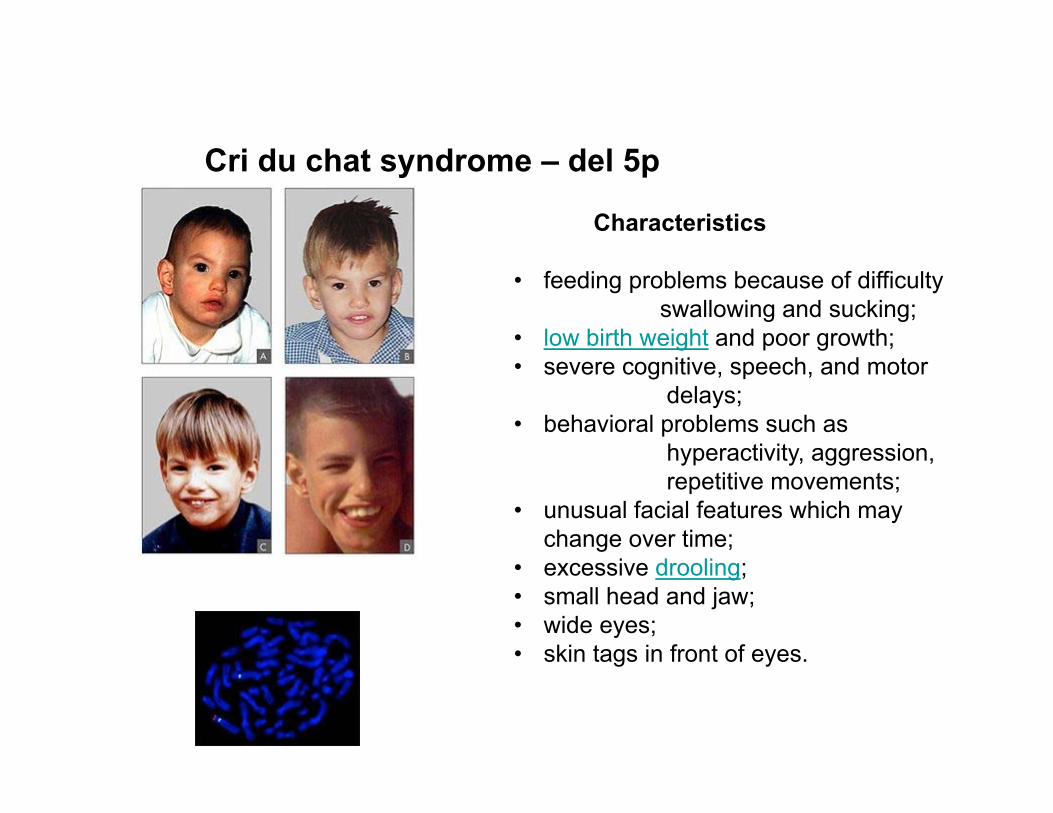
Cri du chat syndrome – del 5p
Characteristics
• feeding problems because of difficulty swallowing and sucking;
• low birth weight and poor growth;• severe cognitive, speech, and motor
delays;• behavioral problems such as
hyperactivity, aggression, repetitive movements;
• unusual facial features which may change over time;
• excessive drooling;• small head and jaw;• wide eyes;• skin tags in front of eyes.

DiGeorge syndrome
Children with DiGeorgesyndrome tend to have the following features:
• a long, narrow face• wide-set, almond-shaped
eyes• a broad nasal bridge and
bulbous nose tip• a small mouth• small, low-set ears that are
folded over at the top• a cleft lip• a cleft palate• an irregular skull shape
Catch -22 gene deletion

Disorders associated with imprinting
• Prader-Willi / Angelman
• Beckwith–Wiedemann syndrome / Silver–Russell dwarfism
• Hydatidiform Moles and Ovarian Teratomas
Parent-of-Origin Effects- For some disorders, expression of disease phenotypes depends on parental origin of mutant allele or abnormal chromosome
- Differences in gene expression b/w allele inherited from mother and allele inherited from father are the result of genomic imprinting.

• It is likely that several dozen or ~ 100 genes show imprinting effects.
• Only one allele (either maternal or paternal) is expressed. Non-imprinted loci (majority of loci) are expressed from both alleles.
Map of imprinted regions in human genome (gray: expressed only from maternal; blue: expressed from paternal)
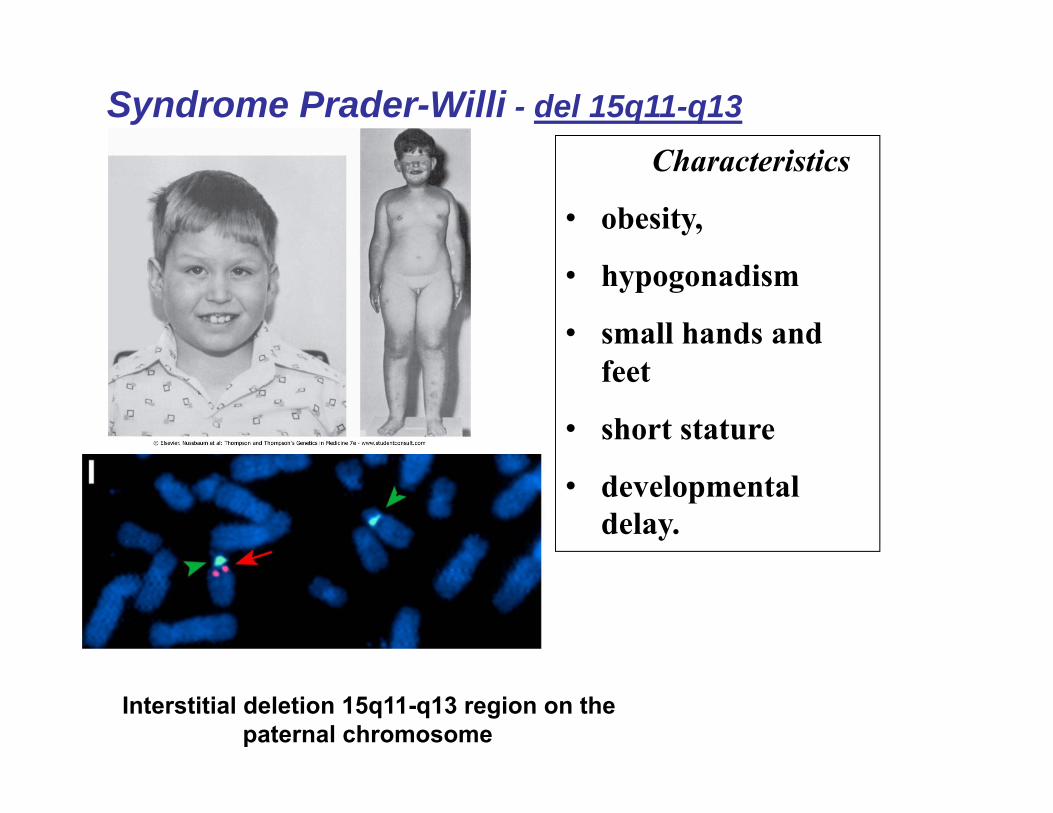
Syndrome Prader-Willi - del 15q11-q13Characteristics
• obesity,
• hypogonadism
• small hands and feet
• short stature
• developmental delay.
Interstitial deletion 15q11-q13 region on the paternal chromosome

Angelman syndrome del 15q11-q13
Characteristics• severe mental retardation
• jerky movements (especially hand flapping)
• seizures
• frequent laughter or smiling, and usually a happy demeanor
Interstitial deletion 15q11-q13 region on the maternal chromosome

Prader-Willy/Angelman Syndromes region
SNRPN D15S10 D15S113D15S63D15S13 UBE3A GABRB3

Another cause of AS is uniparental disomy (UPD)c where the child inherits both copies o chromosome 15 from the father with no copy inhereted from the mother. In this case there is no mutation or deletion, but the child is still missing the active UBE3A gene, because the paternal-derived chromosomes only have brain-inactivated UBE3A genes.
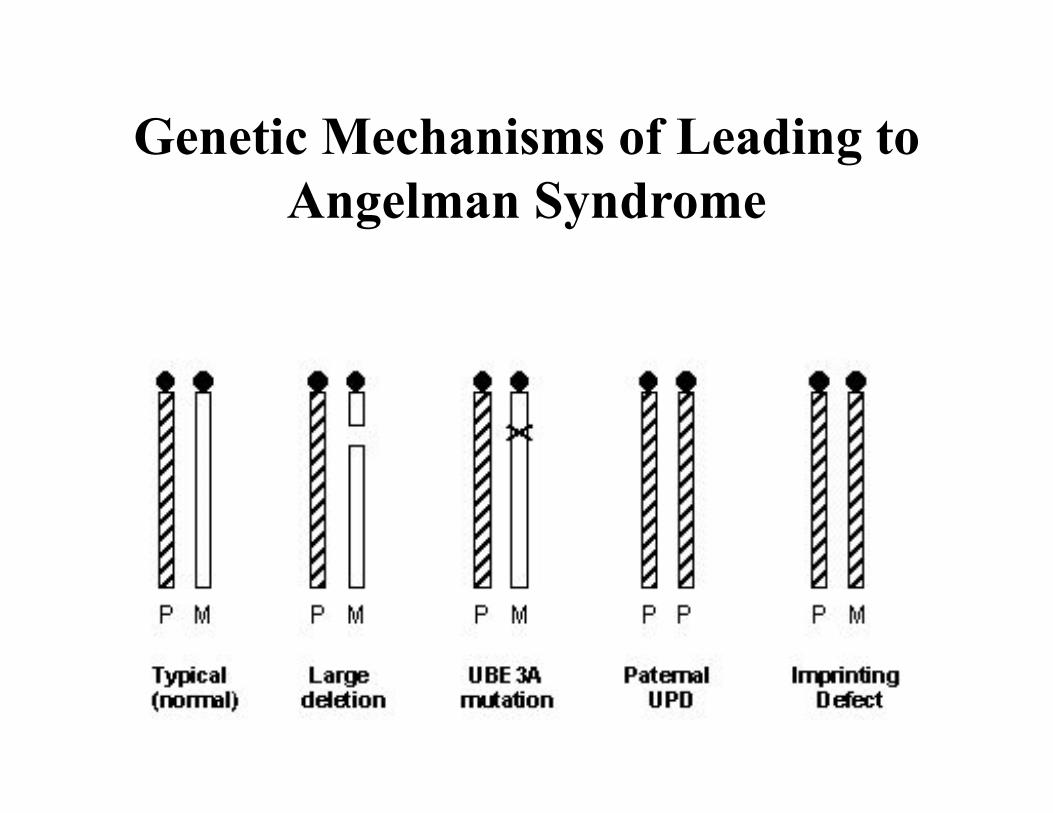
Genetic Mechanisms of Leading to Angelman Syndrome

15q11-q13 region
Prader-Willi syndrome
• 70% - del 15q11-q13 in the paternal chromosome
• 28% – UPD of the maternal chromosome
• 2% - imprinting error
Angelman syndrome
• 70% - del 15q11-q13 in the maternal chromosome
• 28% –UPD of the paternal chromosome
• 2% - imprinting error

FISHAdvantages
• Targeted analysis
• Higher resolution (2Mb-100kb)
Disadvantages• Cannot detect small mutations.• Cannot detect whole genome • Probes are not yet commercially
available for all chromosomal regions.

Further progress in cytogenetics -Comparative Genomic Hybridization (CGH)
• Comparative genomic hybridization (CGH) isdeveloped for the analysis of cryptic geneticimbalances in whole genome or chromosome set.
• Assess the relative copy number of genomic DNAsequences in a comprehensive manner
• Complements karyotyping and provides verysensitive, high resolution genome assessment.
• Balanced translocations and rearrangements can notbe resolved CGH.
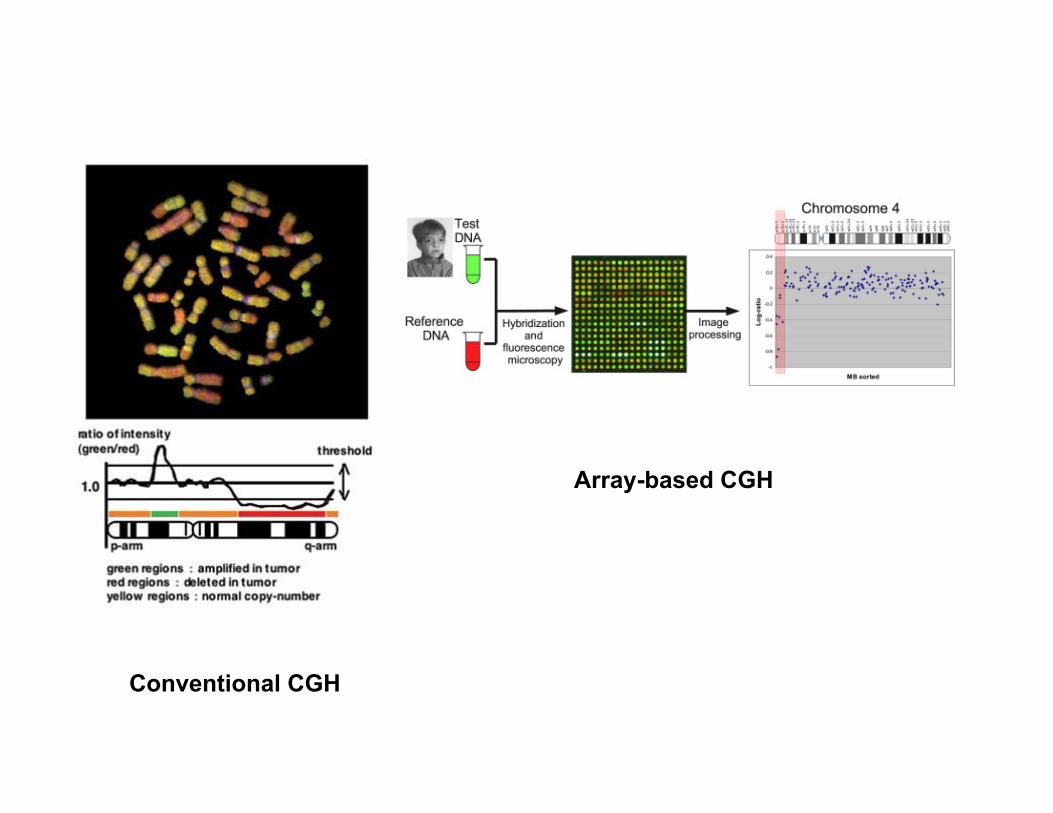
Conventional CGH
Array-based CGH

•Advantages: - Faster - Higher resolution of region’s gain or loss can
be identified than regular chromosome preparation - Useful in tumor analysis, where it is difficult/impossible to obtain a metaphase preparation
•Limitations: - Cannot detect balanced rearrangements - Cannot detect low level of mosaicism

Cancer Genetics
Dr. Davit Babikyan, PhD

Cancer is always geneticCancers result from mutations in the genes that regulate cell growth.
Cancers are associated with mutations that “activate” proteins that stimulate cell growth
DNA damage increases the risk of developing cancer.
Familial cancer syndromes are due to mutations in genes affecting DNA repair or genes that regulate cell growth.
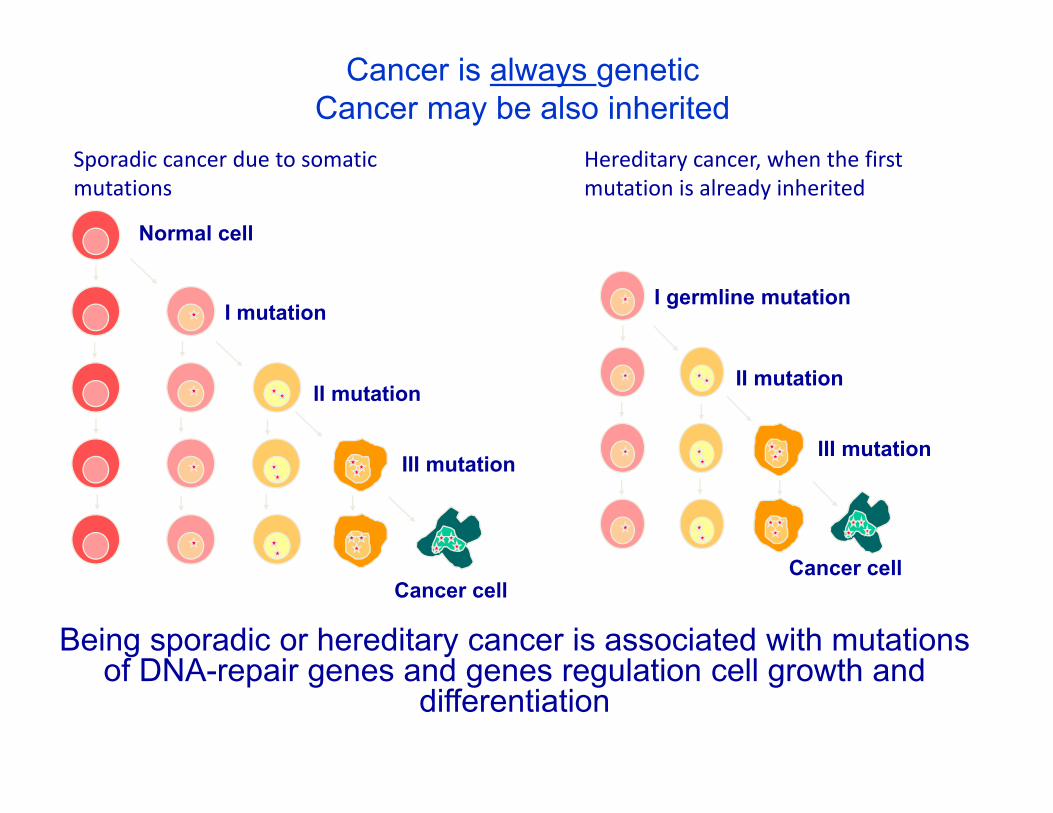
Cancer is always geneticCancer may be also inherited
Normal cell
I mutation
II mutation
III mutation
Cancer cell
I germline mutation
II mutation
III mutation
Cancer cell
Sporadic cancer due to somatic mutations
Hereditary cancer, when the first mutation is already inherited
Being sporadic or hereditary cancer is associated with mutations of DNA-repair genes and genes regulation cell growth and
differentiation

Mutations: Somatic and Germline
Mutation in egg or sperm
Nonheritable
Somatic mutationsOccur in nongermline tissues
Are nonheritable
Somatic mutation(e.g., breast)
Germline mutations
All cells affected in offspring
Present in egg or sperm
Are heritable
Cause cancer family syndrome
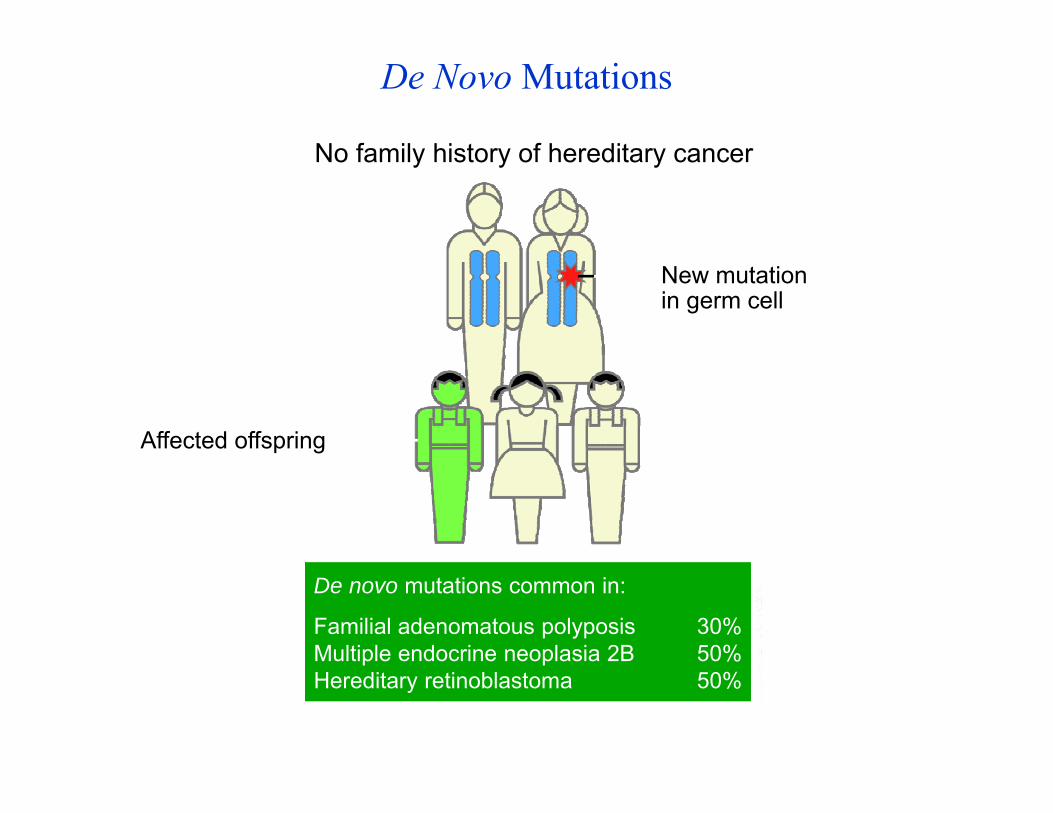
De Novo Mutations
New mutation in germ cell
No family history of hereditary cancer
De novo mutations common in:
Familial adenomatous polyposis 30%Multiple endocrine neoplasia 2B 50%Hereditary retinoblastoma 50%
Affected offspring
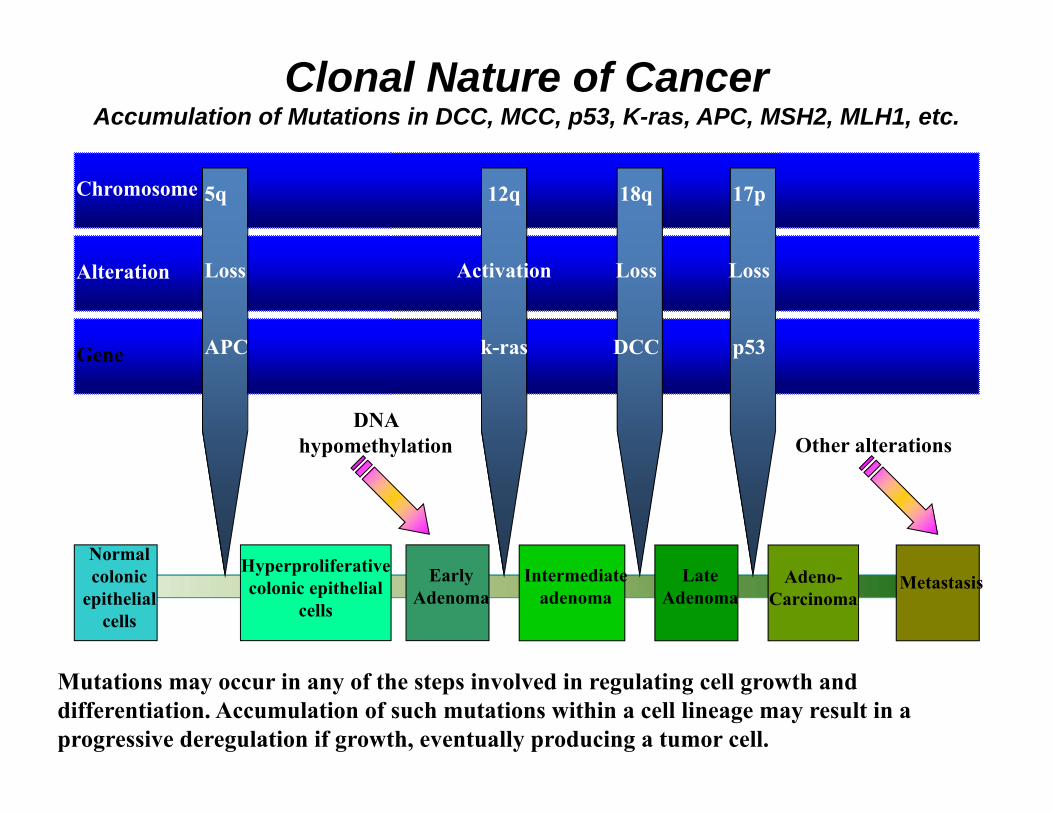
Chromosome
Alteration
Gene
Normal colonic
epithelial cells
5q
Loss
APC
Hyperproliferative colonic epithelial
cells
Early Adenoma
DNA hypomethylation
Intermediate adenoma
Late Adenoma
Adeno-Carcinoma
Metastasis
12q
Activation
k-ras
18q
Loss
DCC
17p
Loss
p53
Other alterations
Mutations may occur in any of the steps involved in regulating cell growth and differentiation. Accumulation of such mutations within a cell lineage may result in a progressive deregulation if growth, eventually producing a tumor cell.
Clonal Nature of CancerAccumulation of Mutations in DCC, MCC, p53, K-ras, APC, MSH2, MLH1, etc.
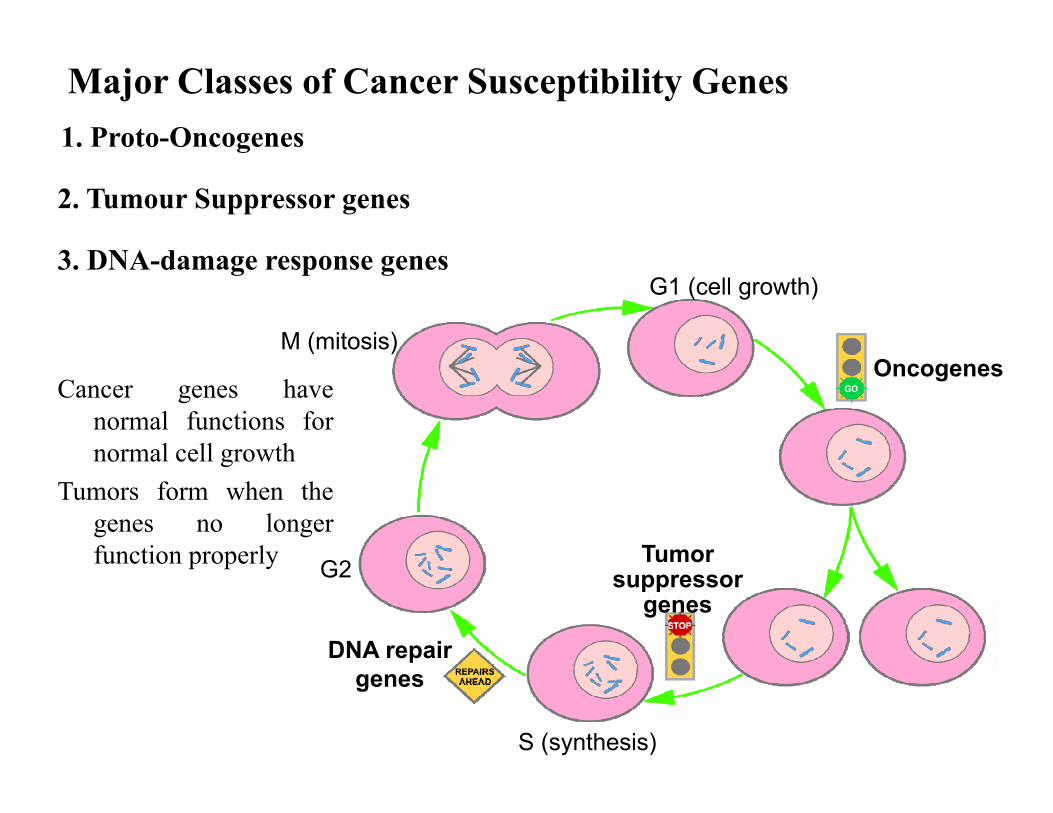
Cancer genes havenormal functions fornormal cell growth
Tumors form when thegenes no longerfunction properly
Major Classes of Cancer Susceptibility Genes1. Proto-Oncogenes
2. Tumour Suppressor genes
3. DNA-damage response genes
M (mitosis)
DNA repair genes
Oncogenes
Tumor suppressor
genesG2
G1 (cell growth)
S (synthesis)

• When mutated in one of the alleles, they may become oncogenes, which can cause cancer
• Do not exhibit germline mutations and often involved in sporadic cancer with somatic mutations
• Rarely involved in inherited cancers
1. Proto-Oncogenes
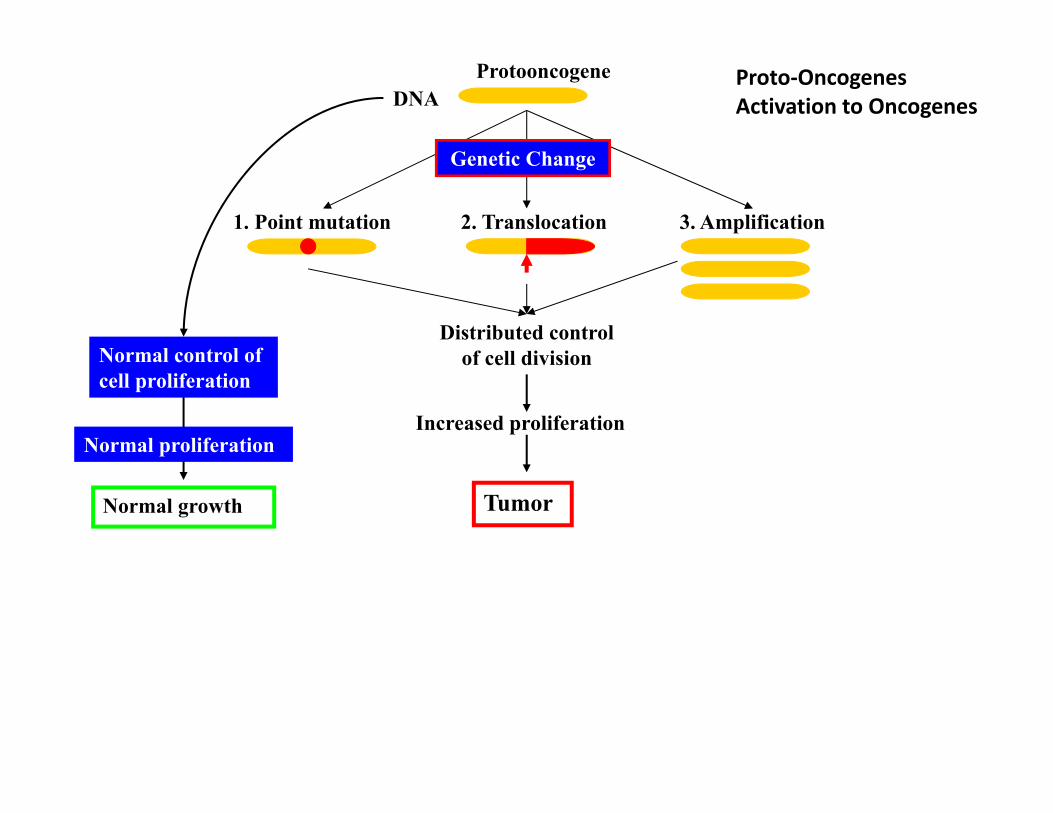
Protooncogene
Normal control of cell proliferation
2. Translocation
Genetic Change
1. Point mutation
Increased proliferation
Distributed control of cell division
Tumor
3. Amplification
DNA
Normal proliferation
Normal growth
Proto‐Oncogenes Activation to Oncogenes

Examples: Genes whose activated products promote cancer are referred to as OncogenesOncogene Cancer
Sis astrocytomaFGF stomach, bladderTGF astocytomas,
hepatomasHGF thyroid cancer
Ras proteins colon, lung, breast, etc.
Examples: Oncogenes involved in hereditary cancerRET Endocrine Neoplasia 2, Familial Meduliar Thyroid Cancer MET Renal Cacrinoma

2. Tumor suppressor genes
• Regulate cell growth even when the cell has only one normal allele of the gene
• Cancer develops only when both alleles are inactivated due to mutations
• Responsible for majority of hereditary cancer syndromes
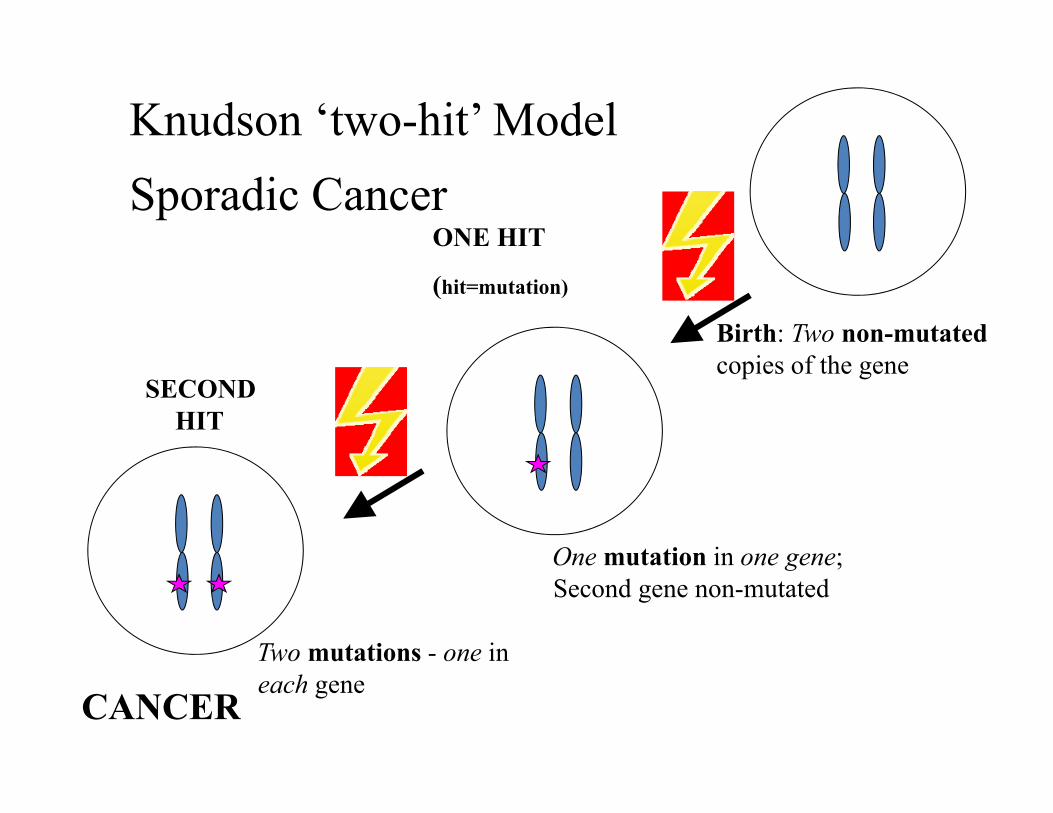
Knudson ‘two-hit’ ModelSporadic Cancer
Birth: Two non-mutatedcopies of the gene
One mutation in one gene; Second gene non-mutated
ONE HIT
(hit=mutation)
SECOND HIT
Two mutations - one in each gene
CANCER

Tumor suppressor genes promote cancer when inactivated
Gene Tumor (somatic mutations)TGFß‐receptor colon, stomachE‐cadherin stomachNF‐1 neuroblastomaNF‐2 schwannoma, meningiomaAPC stomach, colon ,pancreasPTEN endometrial, prostateSMAD2, 4 colon pancreasRB retinoblastoma, breast, lung,
colon, osteosarcomap53 allWT‐1 Wilm’s tumorINK4a kidney, pancreas, breastKFL6 prostate
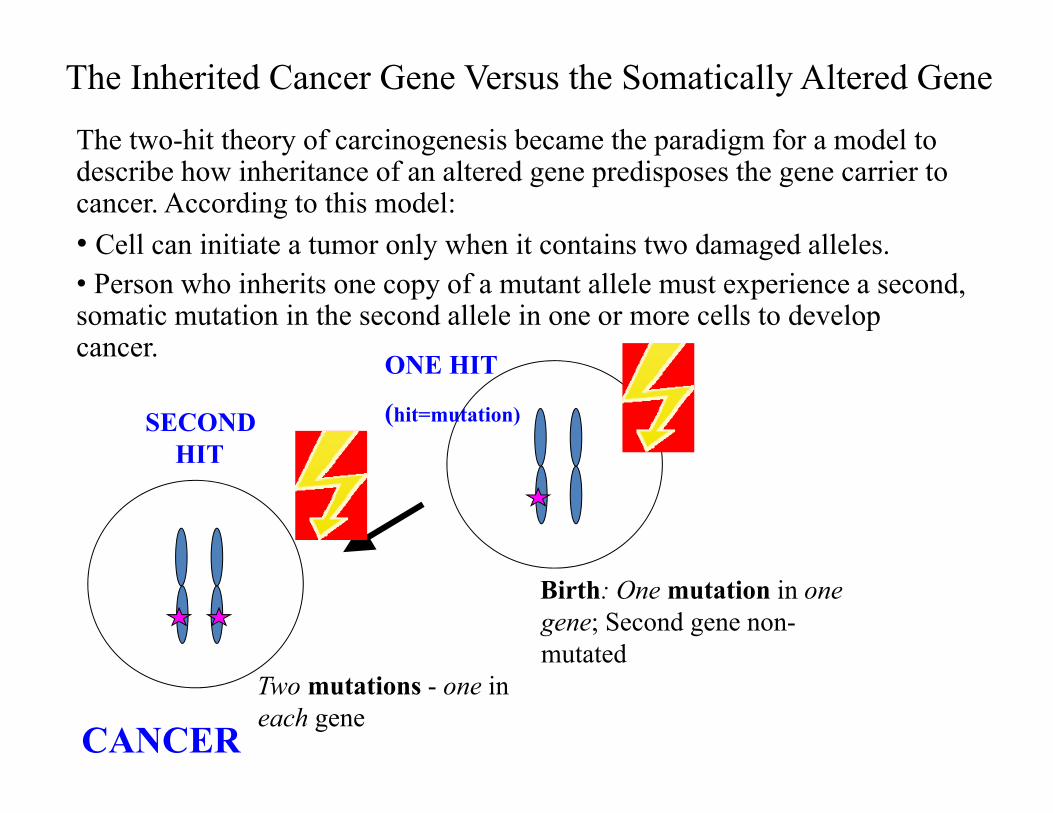
The Inherited Cancer Gene Versus the Somatically Altered GeneThe two-hit theory of carcinogenesis became the paradigm for a model to describe how inheritance of an altered gene predisposes the gene carrier to cancer. According to this model:• Cell can initiate a tumor only when it contains two damaged alleles.• Person who inherits one copy of a mutant allele must experience a second, somatic mutation in the second allele in one or more cells to develop cancer.
Birth: One mutation in one gene; Second gene non-mutated
ONE HIT
(hit=mutation)SECOND HIT
Two mutations - one in each gene
CANCER

Two-Hit Hypothesis of Hereditary Cancer by Tumor Suppressor Genes
If first hit is a germline mutation, second somatic mutation more likely to enable cancer
Somatic mutation
CancerNo cancer
Germline mutation
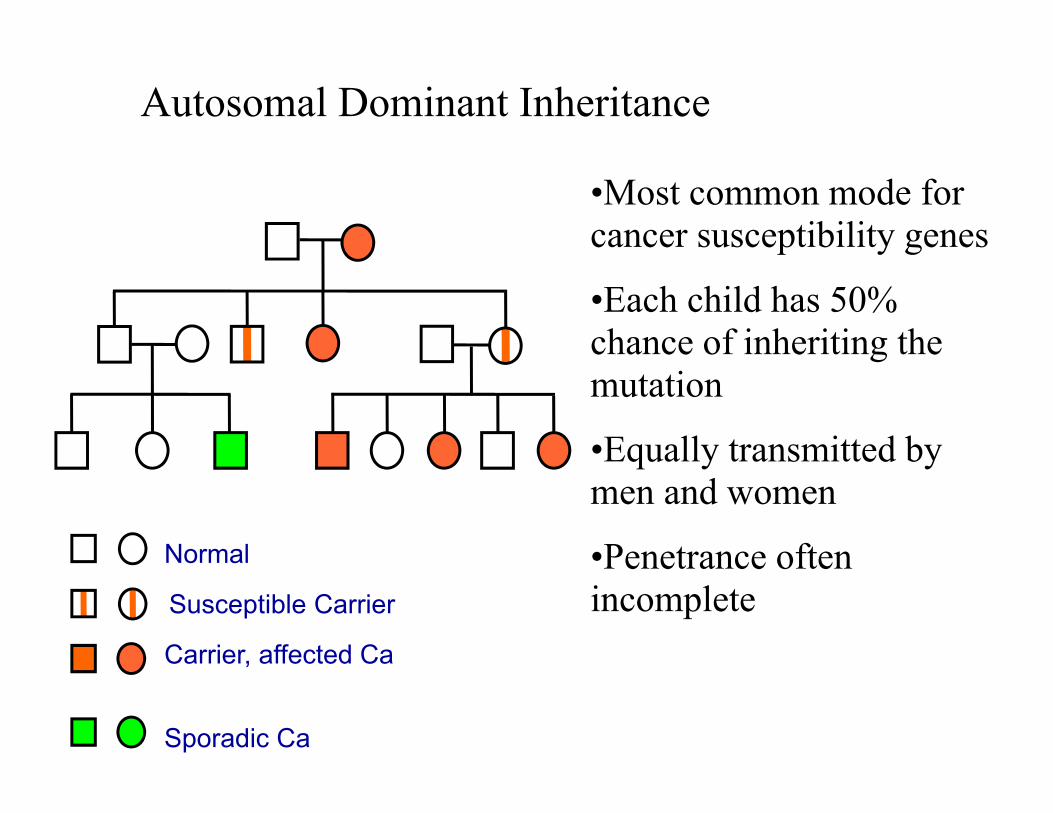
Autosomal Dominant Inheritance
•Most common mode for cancer susceptibility genes
•Each child has 50% chance of inheriting the mutation
•Equally transmitted by men and women
•Penetrance often incomplete
Normal
Carrier, affected Ca
Sporadic Ca
Susceptible Carrier
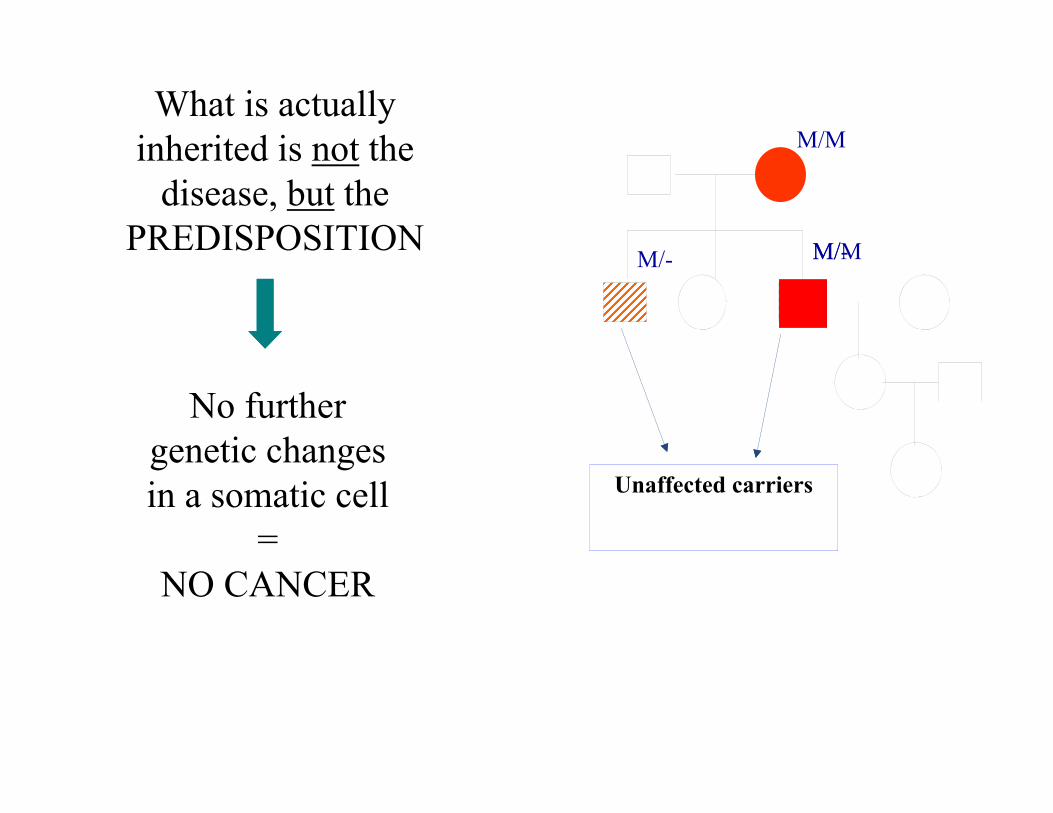
What is actually inherited is not the
disease, but the PREDISPOSITION
Unaffected carriers
No further genetic changes in a somatic cell
=NO CANCER
M/- M/-
M/M
M/M

Tumor Supressor Genes involved in Dominantly Inherited Cancer Syndromes
Syndrome Associated gene
Breast and Ovarian Cancer BRCA1, BRCA2Familial Adenomatous Polyposis (FAP) APCLi Fraumeni P53Familial Retinoblastoma RB1
…and many other

• Repair DNA-damage
• Inherited defects in DNA repair can lead to a high-frequency of somatic mutations
• When these mutations affect pathways that regulate cellular proliferation, a tumor may arise
• Cancer is caused by inactivating mutations in both the alleles
3. DNA-damage Response Genes

DNA damage-response genes involved in Inherited Cancer Syndromes
Type of Inheritance
ATM Ataxia Telangiectasia(lymphoma)
Autosome-Recessive
XP A, B, C, D, E, F, G
Xeroderma Pigmentosum(skin cancer)
Autosome-Recessive
BLM
FANC A,C,D,E
MSH2,6MLH1PMS1,2
Bloom Syndrome (solid tumors)
Fanconi Anemia (leukemia)
Hereditary NonpolyposisColon Cancer
Autosome-Recessive
Autosome-Recessive
Autosome-Dominant
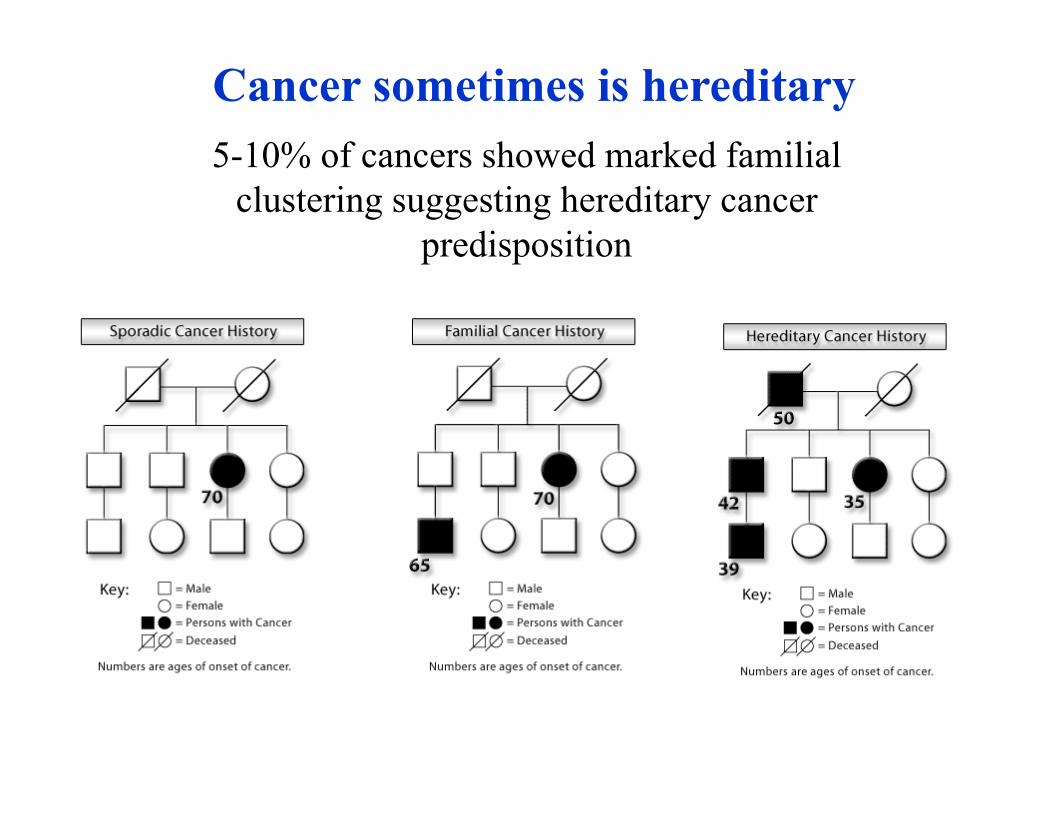
Cancer sometimes is hereditary5-10% of cancers showed marked familial
clustering suggesting hereditary cancer predisposition

Causes of Hereditary Susceptibility to CRC
Sporadic(65%–85%)
Familial (10%–30%)
Hereditary nonpolyposis
colorectal cancer (HNPCC) (2-5%)
Familial adenomatouspolyposis (FAP) (1%)
Rare CRC syndromes
(<0.1%)
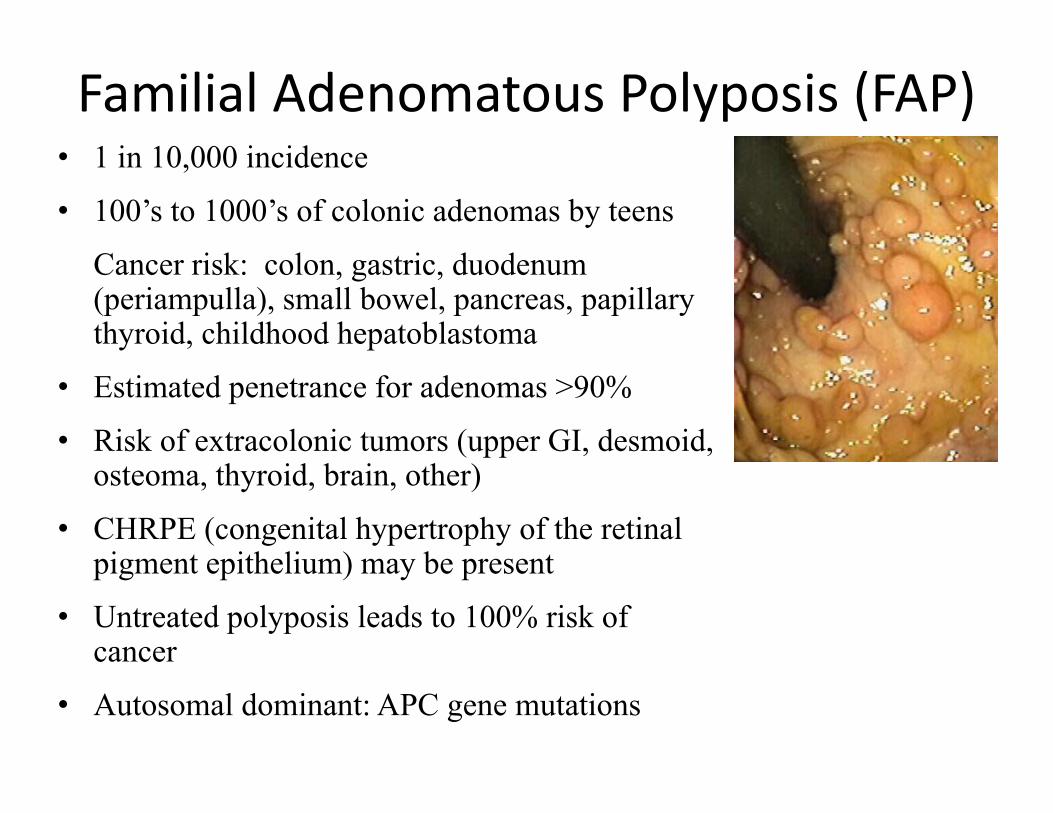
Familial Adenomatous Polyposis (FAP)• 1 in 10,000 incidence • 100’s to 1000’s of colonic adenomas by teens
Cancer risk: colon, gastric, duodenum (periampulla), small bowel, pancreas, papillary thyroid, childhood hepatoblastoma
• Estimated penetrance for adenomas >90%• Risk of extracolonic tumors (upper GI, desmoid,
osteoma, thyroid, brain, other)• CHRPE (congenital hypertrophy of the retinal
pigment epithelium) may be present • Untreated polyposis leads to 100% risk of
cancer • Autosomal dominant: APC gene mutations

Consequences of FAP• Colorectal adenomatous polyps begin to appear at an average
age of 16 years (range 7‐36 years) • Average age at diagnosis: 34‐43 years, when >95% have polyps
Age Individuals with colon cancer
21 7%
45 87%
50 93%
• ~50‐90% develop small bowel polyps– lifetime risk of small bowel malignancy is 4‐12%
• ~50% develop gastric polyps– ~10% gastric cancer
• ~10% develop desmoid tumours
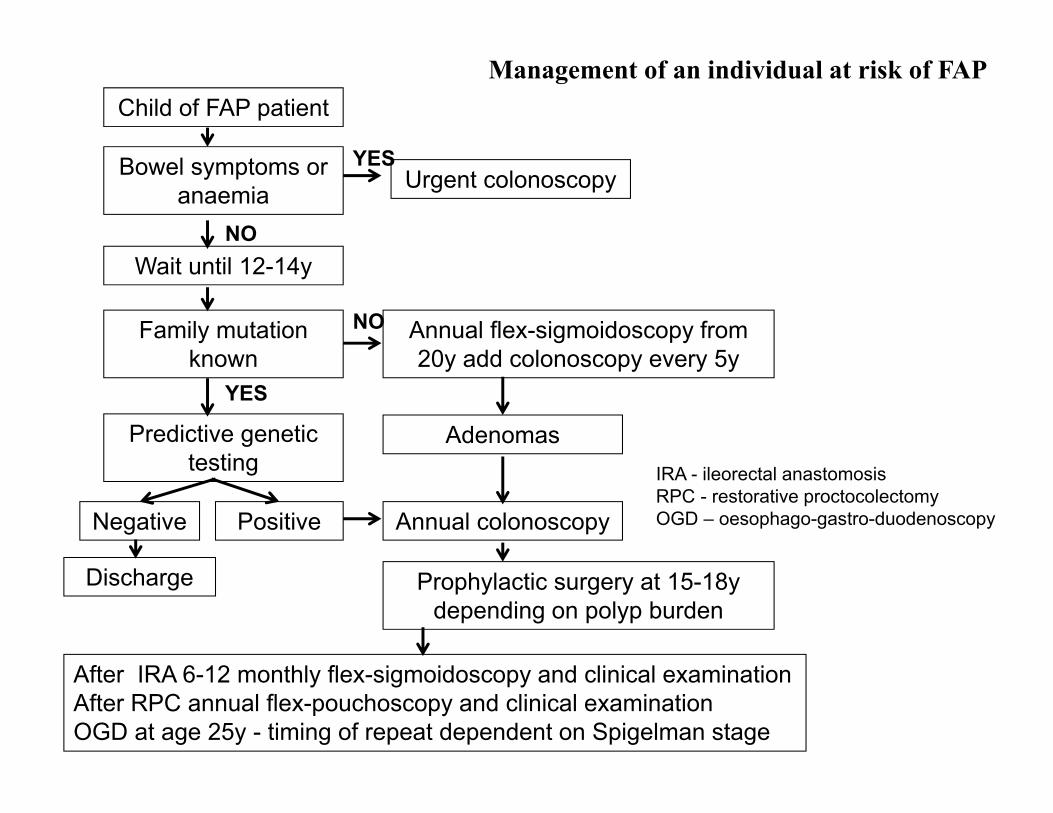
Management of an individual at risk of FAPChild of FAP patient
Bowel symptoms or anaemia
Wait until 12-14y
Family mutation known
Predictive genetic testing
Negative Positive
Urgent colonoscopy
Annual flex-sigmoidoscopy from 20y add colonoscopy every 5y
Adenomas
Annual colonoscopy
Prophylactic surgery at 15-18y depending on polyp burden
Discharge
After IRA 6-12 monthly flex-sigmoidoscopy and clinical examination After RPC annual flex-pouchoscopy and clinical examination OGD at age 25y - timing of repeat dependent on Spigelman stage
NO
YES
YES
NO
IRA - ileorectal anastomosisRPC - restorative proctocolectomyOGD – oesophago-gastro-duodenoscopy

Hereditary Non‐Polyposis Colorectal Cancer (HNPCC) – Lynch Syndrome
• 2-3% of all colorectal cancer cases• Autosomal dominant; penetrance ~80%• Caused by germline mutations in any of one mismatched
repair (MMR) genes: MSH2, MSH6, PMS1 (Chr2), MLH1 (Chr3), PMS2 (Chr7)
• Typical age of CA onset is 40-50 years• Multiple affected generations• 60-70% right-sided/proximal CRC tumors• Polyps may be present, multiple primaries common. Can
overlap with FAP.

HNPCC‐related Cancer Risk to Age 70 Compared to General Population
Cancer General Population Risk
Lynch syn. Risk
Mean Age of Onset in Lynch
Colorectal 7 % 80% 45 years
Endometrium 2.7% 20‐60% 46 years
Stomach <1% 11‐19% 56 years
Ovary 1.5% 9‐12% 42.5 years
Hepatobiliary tract and pancreas
<1% 2‐7% 54 years
Urinary tract <1% 4‐5% ~55 years
Small Bowel <1% 1‐4% 49 years
Brain / CNS <1% 1‐3% 50 years

HNPCC:Revised Clinical Diagnostic Criteria
Patient with colorectal cancer diagnosed <50 y.o. Presence of synchronous or metachronous colon or HNPCC-
related tumors regardless of age Patient with a colorectal cancer with histology of MSI diagnosed
<60 y.o. Patient with colorectal cancer and a I-degree relative with a
HNPCC-associated tumor with one cancer diagnosed <50 y.o. Patient with colorectal cancer with two or more I-degree or II-
degree relatives with a HNPCC-related tumors regardless of age

Laboratory Screening of HNPCCMicrosatellite Instability (MSI) due to Mismatch Repair Failure• 10%–15% of sporadic tumors have MSI• 95% of HNPCC tumors have MSI at multiple loci
Immunohistochemistry (IHC) on tumor tissue can be used to detect the presence or absence of the mismatch repair proteins (MSH2, MLH1, etc.)
“Screen positive” individuals can be offered cancer genetic counseling/assessment and targeted genetic testing

Referral for Healthy Relatives:
• Autosomal dominant pattern of cancers in the family
• Persons with a family history of a known hereditary cancer syndrome, including HNPCC and FAP
• Two or more family members with CRC* at least one <50• Three or more family members with CRC*; any age• Family or personal history of CRC and • One or more 1st degree relative with an HNPCC-related cancer, one diagnosed <50
yrs. *Same side of family
CRC Risk Management
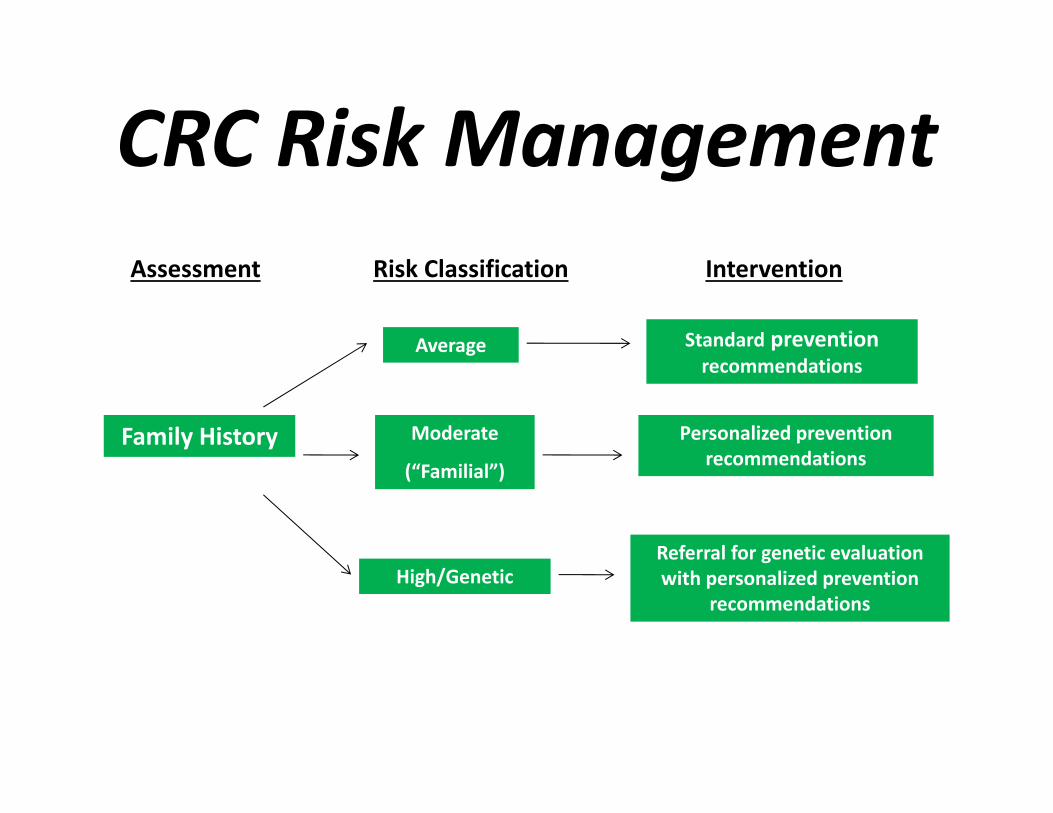
CRC Risk ManagementAssessment Risk Classification Intervention
Family History
Average
Moderate
(“Familial”)
High/Genetic
Personalized prevention recommendations
Standard prevention recommendations
Referral for genetic evaluation with personalized prevention
recommendations

CRC Risk Management
Average Risk1. No family history CRC OR2. One 2nd or 3rd degree relative with CRC Colonoscopy every 10 years
Age to Begin50 years

CRC Risk ManagementModerate/Family history Two 1st degree relatives with CRC any age
or one 1st degree relative with CRC < 60- Colonoscopy every 5 yrs
One 1st degree relative with CRC >60 or two 2nd degree relatives with CRC any age- Average risk screening
* Or 5-10 yrs earlier than earliest case in family
Age to begin
40 years*
40 years

CRC Risk ManagementAge to Begin
HNPCC or suspected HNPCC 20-25 years1. Colonoscopy every 1-2 yrs2. Genetic counseling; consider genetic testing
FAP 10-12 years1. Flex sig or colonoscopy every1-2 yrs2. Genetic counseling; consider genetic testing
20 years1. Full colonoscopy every 5 year

Risk factors influencing penetrance of Breast and Ovarian Cancer
Ionizing radiation Benign neoplasia
Endocrine and Reproductive FactorsFood and alimentation
Familial History
Age
Geographic place
<91.6 / 100.000
Ethnicity
Hereditary Breast and Ovarian Cancer

Family History Is The Main Clue
• BC and / or OC in > 2 close relatives on the same side of the family (maternal or paternal)
• Bilateral breast cancer• Early‐onset breast cancer • Male breast cancer• Associated cancers (ovarian)• Ethnicity (eg Jewish)

BC Pathology Is Another Clue• BC in BRCA1 carriers are usually:
– Basal phenotype CK5/6– Hormone receptor negative (ER‐, PR‐)– HER2 negative– High grade (high mitotic index)
• BRCA2‐associated BCs do NOT have such a specific phenotype.

Young age of onset of breast cancer with BRCA1 phenotype– Hormone receptor negative– HER2 negative– Basal markers CK5/6
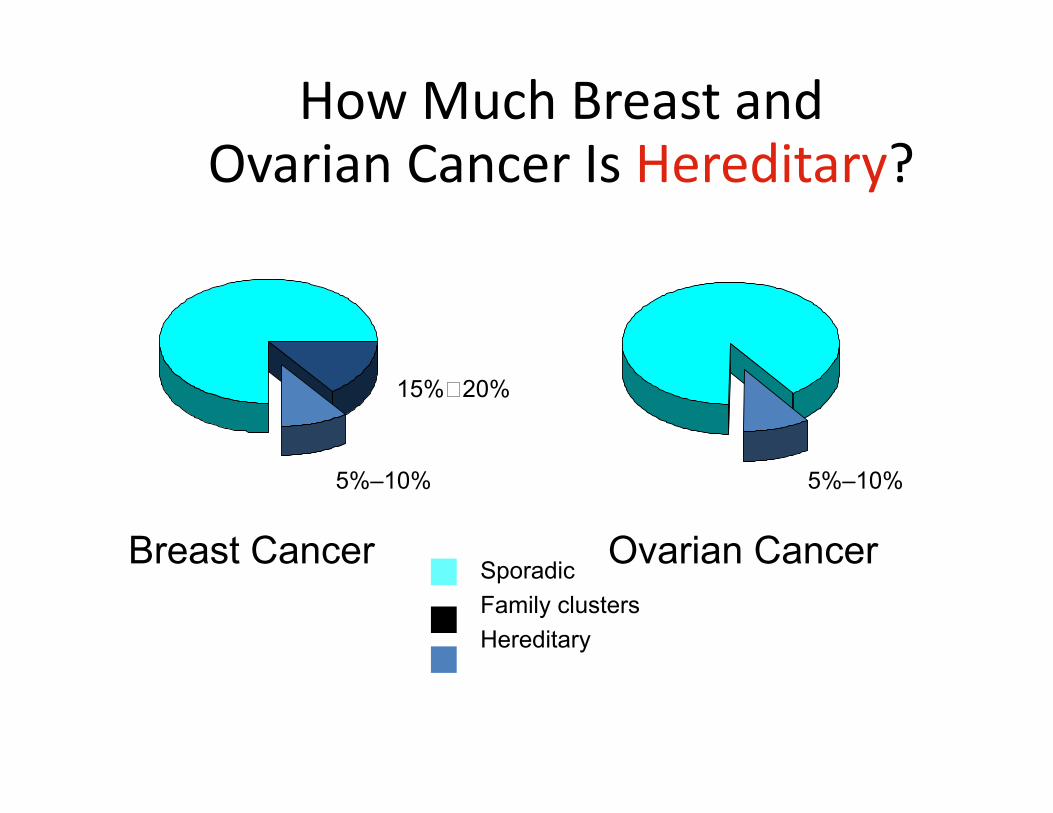
How Much Breast and Ovarian Cancer Is Hereditary?
SporadicFamily clustersHereditary
Ovarian CancerBreast Cancer
5%–10% 5%–10%
15%20%
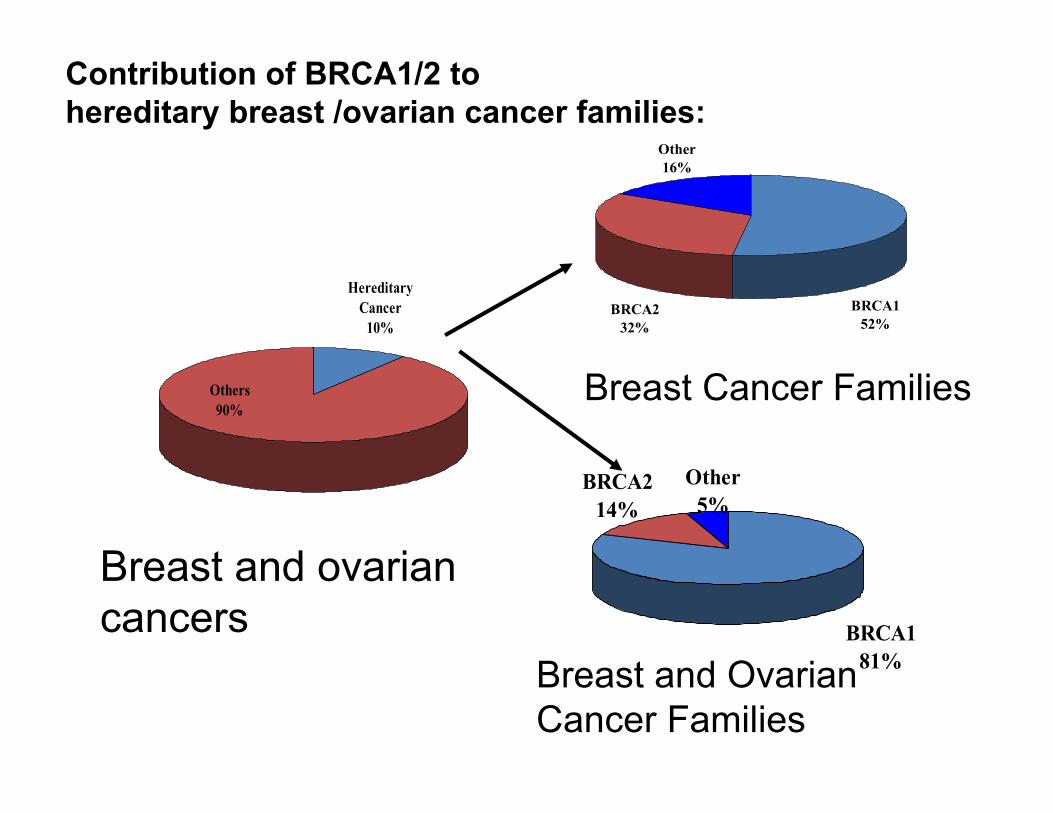
Hereditary Cancer
10%
Others90%
Breast and ovarian cancers
Other16%
BRCA232%
BRCA152%
Breast Cancer Families
Other5%
BRCA214%
BRCA181%Breast and Ovarian
Cancer Families
Contribution of BRCA1/2 to hereditary breast /ovarian cancer families:

BC risk by age BRCA1 BRCA2 General population
40 19% 12%
50 50% 28% 2%
60 64% 48%70 85% 84% 7%
OC risk by age BRCA1 BRCA2 General population
by 70 20-50% 10-30% 1.2%

Characteristics of those families appropriate for consideration for BRCA1 or BRCA2 testing
(including patient’s personal history)
several breast cancers or breast and ovarian cancer two or more ovarian cancers in one family presence of bilateral cancers of the breast or ovary cancers diagnosed at younger than expected agesmultiple affected relatives, demonstrating an autosomal
dominant pattern of inheritance presence of individuals diagnosed with more than one
cancer, e.g. breast and ovarian breast and/or ovarian cancer and Ashkenazi heritagemale breast cancer
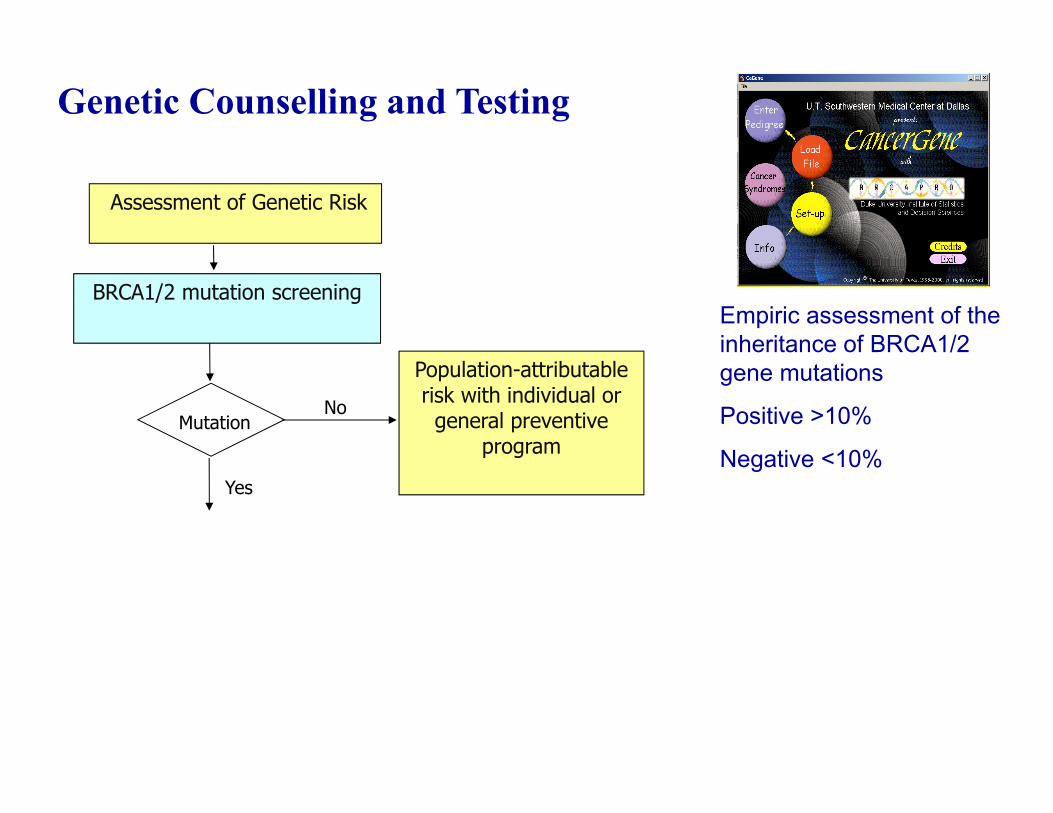
Genetic Counselling and Testing
Empiric assessment of the inheritance of BRCA1/2 gene mutations
Positive >10%
Negative <10%
BRCA1/2 mutation screening
Assessment of Genetic Risk
No
Yes
Mutation
Population-attributable risk with individual or general preventive
program

When a deleterious mutation is found
• Mutation detection is offered to other members of the family to assess risk based on carrier status
• Management strategies discussed based on carrier status
• Mutation analysis can distinguished carriers (highest risk) from non-carriers (lowest risk) and thus improved risk assessment of members of this family

Population-attributable risk with individual or general preventive
programNo
Yes
Genetic Testing of Relatives
BRCA1/2 mutation screening
Mutation
No
Yes
Assessment of Genetic Risk
Monitoring and Prophylactic interventions
Genetic Counselling
Mutation

Relevance to Woman With Recent BC Diagnosis
BRCA1/2 mutation• If pre‐menopausal, HR positive BC, may consider salpingo‐oophorectomy as part of BC treatment, to reduce risk of subsequent BC and OC
• If post‐menopausal, may consider salpingo‐oophorectomy to prevent subsequent OC
BRCA1/2 mutation and/or strong Family History• May consider complete mastectomy and contralateral risk‐reducing mastectomy rather than adjuvant radiation.

Increased Surveillance
Breast Cancer• Clinical examination
every 6 months• Mammogram yearly
beginning age 25-35• MRI (ACS 2007)• Monthly BSE• Prompt evaluation of
abnormal findings
Ovarian Cancer
• Ca-125• Pelvic color-doppler
ultrasound every 6-12 months
• Pelvic examination every 6-12 months

Management of Unaffected Family Members Who Carries a BRCA1/BRCA2 Mutation
• Screening• Self breast examination
- every 1 month• Mammography (MRI?)
– Yearly since 35 y.o.• Pelvic examination, CA125
serum oncomarker test, • Transvaginal ultrasonography
– Yearly
• Cancer prevention• Breast cancer
– Risk reducing mastectomy with cosmesis (>90%)
• Ovarian cancer: – Risk reducing
salpingo-oophorectomy (by 90%)

Strategies for Reducing BC Risk In Mutation CarriersStrategy Risk Reduction LimitationsMastectomy (unilateral or bilateral)
>90% (in case of unilateral mastectomy)
Body image and physiological issues
Premenopausal bilateral salpingo-oophorectomy
by 90%, also decreasing risk of BC by 50% if performed
by age 40-50
Early menopause, infertility
Medical prevention (tamoxifen)
40-50% ? Controversial data, risk of endometrial malignancy and
thromboembolic effectsChemoprevention (oral contraceptives)
by 50% of OC Slight increase of risk of BC

PHARMACOGENOMICS and Personalized Medicine in
Cancer Treatment
• How does a physician know the medicine will work for that patient• Why does someone need twice the standard dose to be effective?• Why does this drug work for only a group of patients?• Why do some patients have side‐effects and others don’t?
The Right Dose of The Right Drug for The Right Indication for The Right Patient at The Right Time.
The Goal of Personalized Medicine

How can Pharmacogenomics help to optimize benefit/risk in Treatment?
• Candidate drug selection based on genetic biomarkers
• Modified dose for patients with variant genotype
• Drugs/Biologics approved for a specific population

DPYD Deficiency and 5FU sensitivity• Variations in gene DPYD can lead to DPD (dihydropyrimidine dehydrogenase) insufficiency.• This results in an inability to inactivate 5-Fluorouracile (5-FU) leading to increased levels of active drug in the system that can
result in 7-fold risk of severe toxicity.•About 7% patients treated with 5-FU have Grade 3-4 toxicity associated with a DPYD gene variation
5-FU Genetic Test
Identifies patient risk for 5-FU/capecitabine toxicity
Enhanced patient monitoring
Dose reduction considerations / Alternate chemotherapies
Modified dose for patients with variant genotype
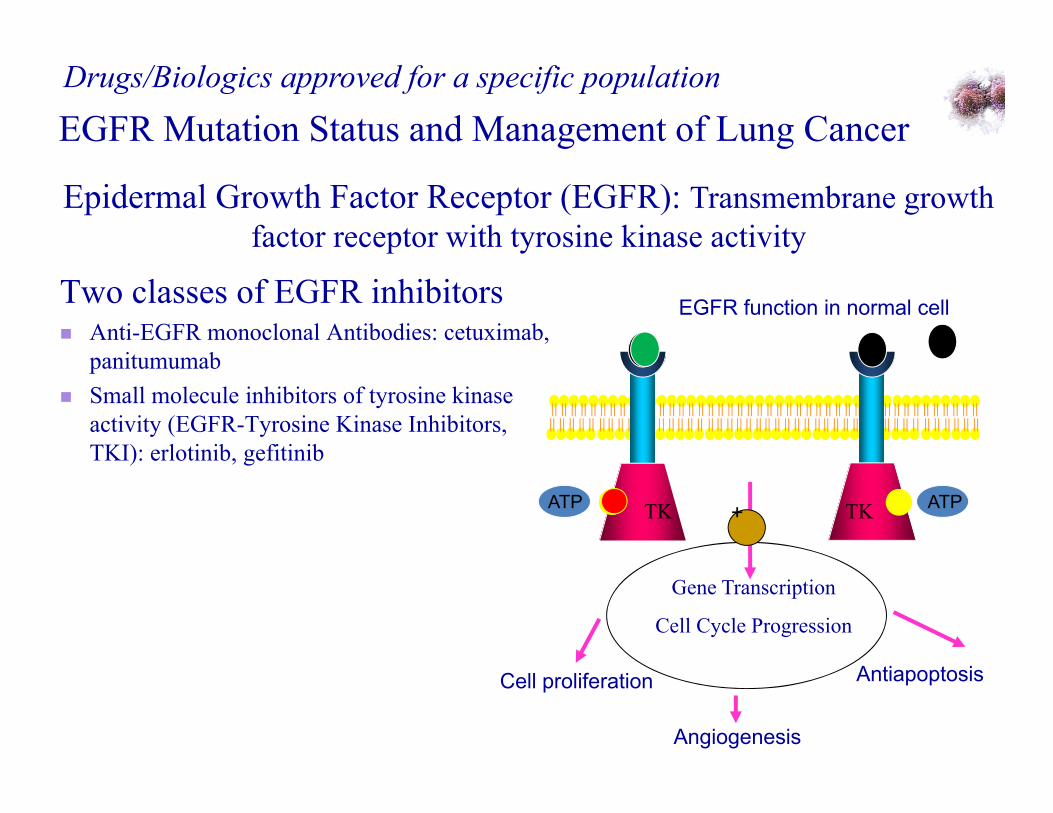
Epidermal Growth Factor Receptor (EGFR): Transmembrane growth factor receptor with tyrosine kinase activity
EGFR Mutation Status and Management of Lung CancerDrugs/Biologics approved for a specific population
TK
EGFR function in normal cell
TKATP ATP
Cell proliferation Antiapoptosis
Angiogenesis
Gene Transcription
Cell Cycle Progression
+
Two classes of EGFR inhibitors Anti-EGFR monoclonal Antibodies: cetuximab,
panitumumab Small molecule inhibitors of tyrosine kinase
activity (EGFR-Tyrosine Kinase Inhibitors, TKI): erlotinib, gefitinib

EGFR Mutations are associated with “Never-Smokers”, gender and Histological subtypes
Mutations are more common in East Asians (30.6%) than in Caucasians (7.6%)
Mutations are more common in non-smokers (34.8%) than in smokers (7.8%)
Mutations are more common in women (26.4%) than in men (9.3%)
Mutations are more common in adenocarcinomas (23.2%) than in other histologies (2.2%)

Responses in patients with or without EGFR mutations
Patients with NSCLC responsive to EGFR-TKI gefitinib or erlotininb are more likely to harbour mutations than not (76.7% vs 23.3%)
EGFR Genetic Test
Identifies patients with EGFR mutation and treatment positive responsiveness to treatment with EGFR-TKI.

EGFR is overexpressed in more than 85% of tumors from patients with metastatic CRC.
Only a subset of patients with CRC achieve a clinical benefit from treatment with EGFR inhibitors.
KRASMutation Status and the Management of Colorectal Cancer (CRC)
Drugs/Biologics approved for a specific population

Another Oncogene KRASMutations are implicated in Colorectal Cancer
Only certain mutations at codons 12&13 of KRAS oncogene lead to constitutive, growth-factor-receptor-independent activation
• KRAS mutations occur early in colorectal carcinogenesis
• Occur in 35% to 40% of CRC
• 95% concordance between paired primary cancers and metastases

KRASMutation as Predictive Marker
Correlation between KRAS mutations and response to anti-EGFR monoclonal antibodies (mAbs) - cetuximab, panitumumab in patients with CRC.
Both drugs are ineffective when the patient's tumor has a KRASmutation, and efficacy limited to patients whose tumors are with wild-type KRAS gene.
KRAS genetic test
Prediction of treatment with anti‐EGFR mAbs (ceuximab/panitumumab) Part of the evaluation of patients withmetastatic CRC.

Herceptin
Herceptin-ը բարդ հակամարմին է, որը1) բլոկադաի է ենթարկում Her-2 ռեցեպտորը և
կանխում է բջիջների աճը և 2) քայքայում է քաղցկեղային բջիջները, որով
դանդաղեցնում է հիվանդության զարգացումը:
Դեղամիջոցներ հաստատված հիվանդների միայն որոշակի խմբի (ենթապոպուլյացիայի) hամար
Her-2 գենի սպիտակուցը կրծքագեղձի էպիթելային բջիջների արտաքին մակերեսի ռեցեպտոր է, որը անհրաժեշտ է բջիջների նորմալ աճի համար
Կրծքագեղձի քաղցկեղի որոշ տեսակներում (30%) Her-2 գենը ենթարկվում է դուպլիկացիայի և ավելի շատ է էքսպրեսավորվում, նպաստելով քաղցկեղային բջիջների աճին և հանդիսանում է հիվանդության զարգացման բացասական պրեդիկտոր

Inborn Errors of Metabolism
Natella Kostandyan, MD

• In 1908 British physician Sir Archibald Garrodpostulated that four inherited conditions of lifelongduration - alkaptonuria, pentosuria, albinism andcystinuria - were caused by defects in specificbiochemical pathways due to the diminished activityor complete lack of a given enzyme. He called thesedisorders “inborn errors of metabolism.” (IEM)
• In 1934 Folling discovered phenylketonuria.
History

• By the mid 1960s, defects that led to the accumulationof metabolic products in the urine, blood, or neuraltissues were identified. These defects were largelyproblems in the catabolism of lipids and amino acidsor in the rapid breakdown of glycogen. Theidentification of the metabolites that accumulated in adisease made possible the identification of the enzymewhose activity was deficient.

• By the mid 1980s, techniques largely had switchedfrom those of the biochemistry of intermediates andenzymes to the identification of mutations in genes.Critical issue in the definition of disease is that havinga genetic defect that may result in a disease is not thesame as having the disease.Having the defect means only having a propensity or arisk of developing the disease. The molecular toolssometimes allow us predict who is likely to develop thedisease and who in the family can neither develop itnor pass it on to offspring.
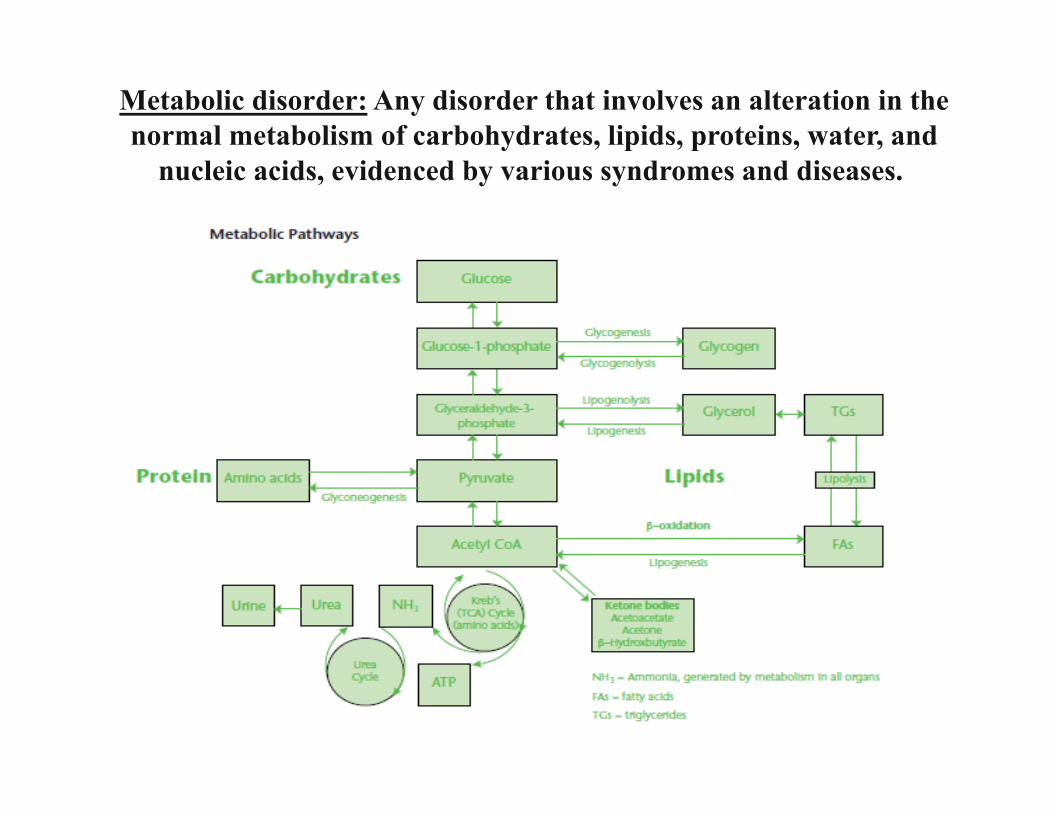
Metabolic disorder: Any disorder that involves an alteration in the normal metabolism of carbohydrates, lipids, proteins, water, and
nucleic acids, evidenced by various syndromes and diseases.

IEMs are inherited
• Autosomal recessive disorders (almost all lysosomalstorage disease)
• X-linked recessive disorders (Fabry and Hunter disease)
• Autosomal dominant disorders (Marfan syndrome, Ehler-Danlos Syndrome)
• Mitochondrial inheritance

• Many individuals previously diagnosed as having
birth injury or atypical forms of psychiatric
disorders or medical diseases, such as multiple
sclerosis, migraines, or stroke, actually have an
undiagnosed IEMs.

Clues to the presence of an IEMs:
• failure to thrive; • dysmorphic features;• abnormalities of hair, skin, skeleton, or all three; • abnormal odor; • organomegaly; • abnormal muscle tone.
Physical examination

Clinical findings in neonates
• Poor feeding• Vomiting, diarrhea, and dehydration• Temperature instability• Tachypnea, apnea• Bradycardia, poor perfusion• Irritability, involuntary movements or posturing,
abnormal tone, • Seizures,• Altered level of consciousness.

Clinical findings in infants and young children
• Recurrent episodes of vomiting;• Recurrent episodes of ataxia, seizures;• Recurrent episodes of lethargy, coma, fulminant
hepatoencephalopathy, or a combination;• Dysmorphic or coarse features, skeletal
abnormalities;• Abnormalities of the hair or skin;• Poor feeding, failure to thrive.

Clinical findings in older children, adolescents, and adults
Common findings include mild-to-profound • mental retardation, • autism, • learning disorders, • behavioral disturbances, • hallucinations.

Neonatal Screening• The purpose of neonatal screening is to detect all
newborns with congenital disorders at a time when these babies are still healthy. The list of screened disorders in Armenia includes congenital hypothyroidism (CH) and phenylketonuria (PKU), and in other countries, many more diseases. Screening allows treating affected, but presymptomatic newborns at an early stage, within the 1st week, and if treated properly, they will stay healthy.
• Many countries now use tandem mass spectrometry, which can screen for over 40 diseases.

The criteria for neonatal screening• There should be an acceptable treatment for patients with
recognized disease.• Facilities for diagnosis and treatment should be available.• There should be a suitable test or examination.• The test should be acceptable to the population.• Case-finding should be a continuing process.• The cost of case-finding
(including diagnosis and treatment of patients diagnosed) should be economically balanced in relation to possible expenditure on medical care as a whole.

Management• Initial treatment of IEMs is aimed at correcting
metabolic abnormalities.• Even the apparently stable patient with mild
symptoms may deteriorate rapidly with progression to death within hours.
• With appropriate therapy patients may completely recover without sequel.
• Start empirical treatment for a potential IEMs as soon as the diagnosis is considered.

Prognosis:
• Prognosis varies based on individual IEMs and may differ for different forms of a particular IEMs.
• A high index of suspicion is critical for early diagnosis and treatment of IEMs.
• Rapid treatment may be life saving and often results in full recovery.

Patient Education:
• Provide counseling (dietary, genetic, psychosocial) as appropriate. Professional and peer support groups exist for many IEMs.
• Provide genetic counseling to discuss prognosis, recurrence risks, screening of other family members, prenatal diagnosis, and support groups.

Medical/Legal Pitfalls:
• Delay in recognition and treatment may result in long-term neurologic impairment or death. Initiate treatment as quickly as possible.
• Consider IEMs in all neonates and young infants with unexplained death.

Categories of IEMs are as follows:
• Amino acid disorders• Carbohydrate disorders• Lysosomal storage disorders• Mitochondrial disorders• Peroxisomal disorders• Purine and pyrimidine disorders• Porphyrias• Metal metabolism disorders

DISORDERS OF AMINO ACID METABOLISM AND TRANSPORT
• Phenylketonuria / Hyperphenylalaninaemia• Tyrosinemia• Albinism• Maple syrup urine disease• Methylmalonic aciduria• Urea cycle disorders• Homocystinemia• Hyperornithinemia etc

Phenylketonuria
• PKU is the most studied and most widespread hereditary disease in group of enzymopathy, connected with inborn metabolic errors of amino acids, particularly phenylalanine (Phe) and tyrosine, as a result of deficiency of enzyme phenylalanine hydroxilase (PAH), characterised mainly by affection of nervous system and cause brain damage.
• The disease is inherited in an autosomal recessive manner.
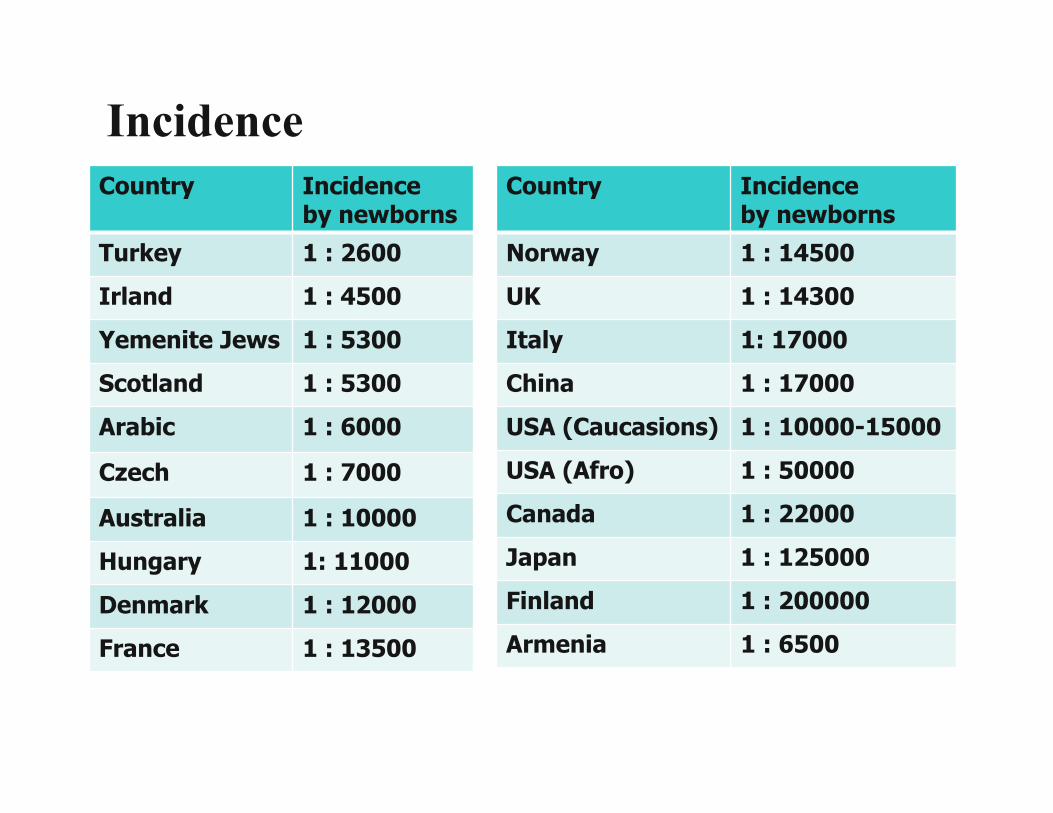
IncidenceCountry Incidence
by newbornsTurkey 1 : 2600Irland 1 : 4500Yemenite Jews 1 : 5300Scotland 1 : 5300Arabic 1 : 6000
Czech 1 : 7000
Australia 1 : 10000Hungary 1: 11000Denmark 1 : 12000France 1 : 13500
Country Incidence by newborns
Norway 1 : 14500UK 1 : 14300Italy 1: 17000China 1 : 17000USA (Caucasions) 1 : 10000-15000USA (Afro) 1 : 50000Canada 1 : 22000Japan 1 : 125000Finland 1 : 200000Armenia 1 : 6500

Pathophysiology• The enzyme phenylalanine hydroxylase normally
converts the amino acid phenylalanine into the aminoacid tyrosine. If this reaction does not take place, phenylalanine accumulates and tyrosine is deficient. Excessive phenylalanine can be metabolized into phenylketones through the minor route, a transaminase pathway with glutamate. Metabolites include phenylacetate, phenylpyruvate and phenyllactate.
• Elevated levels of phenylalanine in the blood and detection of phenylketones in the urine is diagnostic, however most patients are diagnosed via newborn screening.
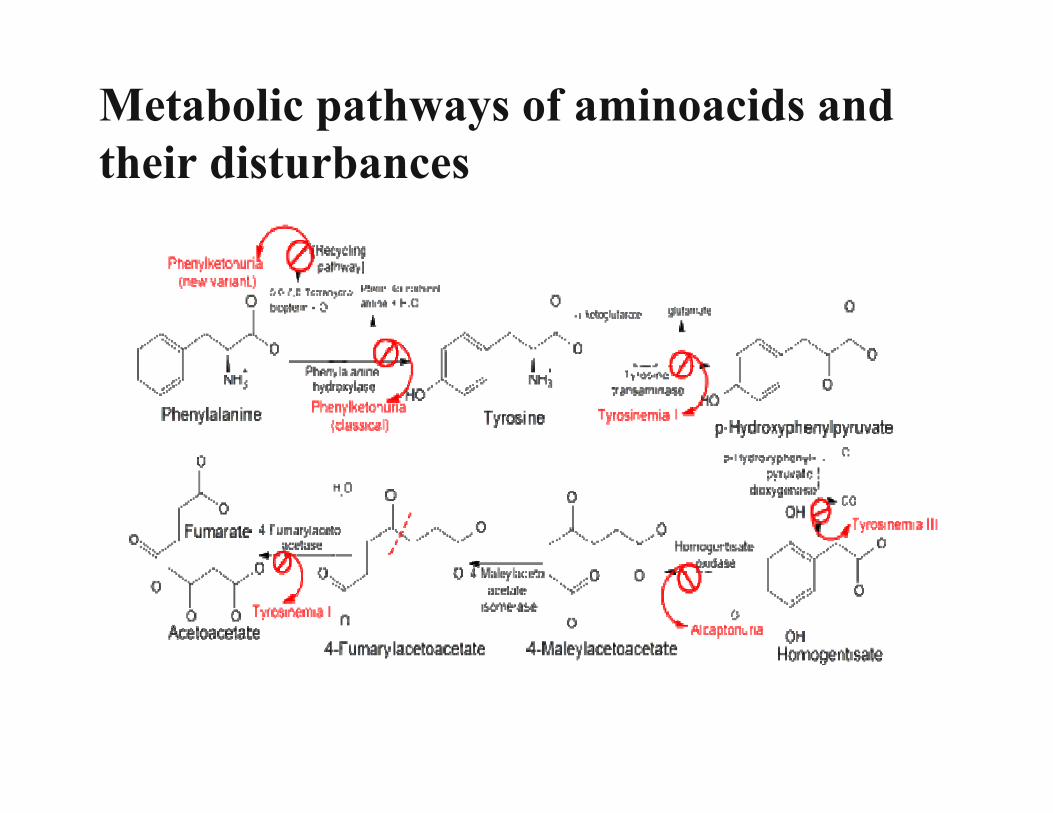
Metabolic pathways of aminoacids and their disturbances
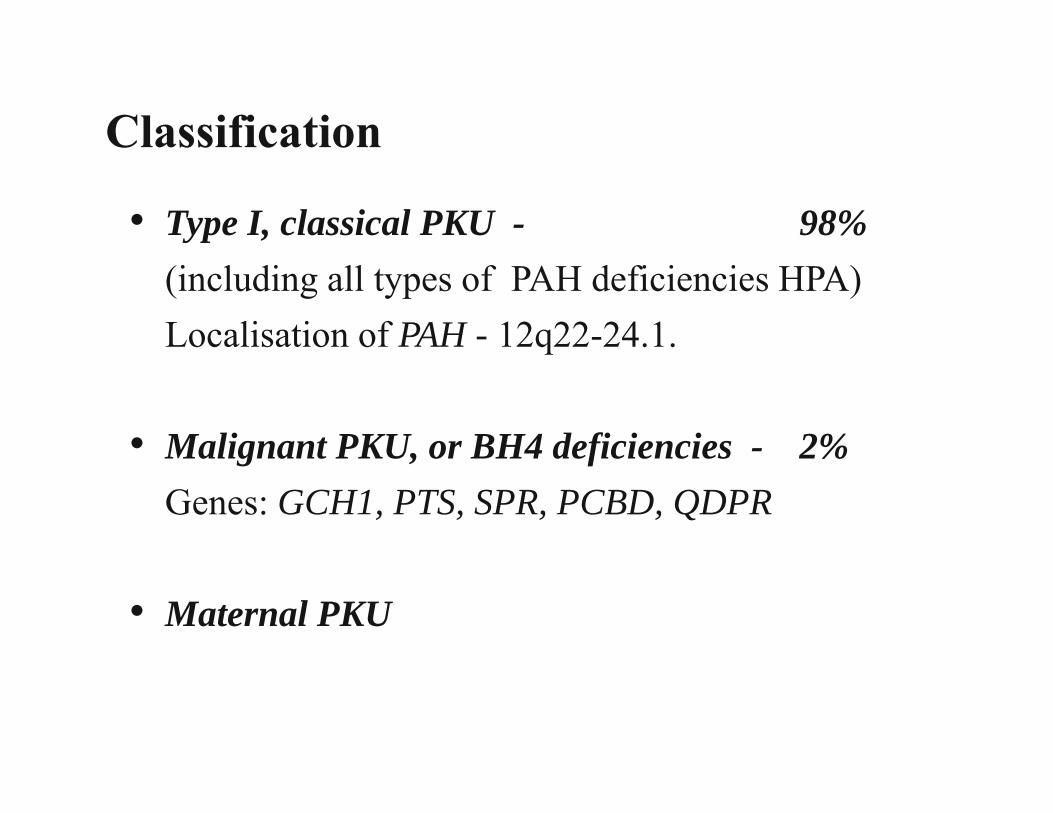
Classification
• Type I, classical PKU - 98% (including all types of PAH deficiencies HPA)Localisation of PAH - 12q22-24.1.
• Malignant PKU, or BH4 deficiencies - 2%Genes: GCH1, PTS, SPR, PCBD, QDPR
• Maternal PKU

Clinical findings (if untreated)• Reduced hair, skin and iris pigmentation • Microcephaly• Neurological impairments
• “floppy baby” syndrome • epilepsy, seizures• tremor, spasticity of the limbs
• Severe mental retardation• Behavioral problems
• hyperactivity, aggressiveness• stereotypy• anxiety and social withdrawal
• Mousy odor from urine and skin• Eczema

Diagnostics• Detection of metabolites in blood and urine
PKU can be easily detected with a simple blood test, PKU screening test for all newborns is a part of the newborn screening. The test is generally done by taking a few drops of blood from the baby before the baby leaves the hospital.
If the initial screening test is positive, further blood tests are required to confirm the diagnosis.
• Definition of enzyme activity
• DNA analysis (also prenatal diagnosis possible)

Treatment• PKU is a treatable disease. Treatment involves a diet
that is extremely low in phenylalanine (in proteins), particularly when the child is growing.
• Phenylalanine is almost in all products, but it is extremely high in diary and meat products, and some cereals. These products must be exclude from everyday diet (red light).
• And any products containing aspartame should be avoided (chewing gum, cola, etc).
• Fruits, vegetables, juices, special low protein products (pasta, sago, flour, etc) can be used without calculation (green light).

Treatment
• To compensate protein needs there are many different
phenylalanine free aminoacid mixture in the world

Maternal PKU• More than 40 years ago it was recognised that the
offspring born to mother with PKU are at risk of
damage from teratogenic effects of high Phe level:
facial dysmorphism, microcephaly, developmental
delay, and congenital heart disease.
• Actually child does not have PKU (he is carrier).
• Mother with PKU needs to keep very strict low
protein diet 1 year before conception, during
pregnancy and breastfeeding.

LYSOSOMAL STORAGE DISORDERS
• The past decade has witnessed major advances in our understanding of the clinical, biochemical, and genetic aspects of lysosomal storage disorders;
• This large and heterogeneous group of almost 50 inherited disorders shares a common pathogenesis;
• Most disorders have both central nervous system andsystemic manifestations, whereas some affect just thecentral nervous system;
• Many patients with lysosomal storage disorders die ininfancy or childhood;
• Patients who survive to adulthood often have adecreased lifespan and significant morbidity.

Presenting signs and symptoms thatsuggest a lysosomal storage disorderObvious physical abnormalities• Nonimmune hydrops fetalis;• Hepatomegaly, splenomegaly, macroglossia;• Coarse facial features, especially progressive;• Skeletal disease: deformations, dysplasias with or
without short stature, skeletal disproportion;• Hirsutism;• Macrocephaly;• Inguinal, umbilical hernias;• Skin lesions – angiokeratomas;• Joint stiffness.

Neurologic• Neurologic degeneration;• Peripheral neuropathy;• Progressive mental retardation/regression;• Progressive dementia;• Behavioral abnormalities, psychosis;• Hyperreactivity, excessive startle;• Seizures;• Hypotonia, weakness;• Spasticity;• Myoclonic jerks;• Ataxia;• Dystonia.
Presenting signs and symptoms thatsuggest a lysosomal storage disorder

Ophthalmologic• Corneal clouding;• Whorled corneal opacity;• Cataract;• Cherry red spot;• Macular degeneration;• Ophthalmoplegia, especially upward gaze palsy;• Optic atrophy;• Strabismus.
Presenting signs and symptoms thatsuggest a lysosomal storage disorder

Cardiac• Unexplained cardiomyopathy• Valvular heart disease• Arrhythmia
Gastrointestinal• Attacks of abdominal pain, diarrhea, constipation
Renal• Proteinuria, isosthenuria• Tubular dysfunction
Presenting signs and symptoms thatsuggest a lysosomal storage disorder

Disease-specific therapy
• Enzyme Replacement Therapy;
• Bone Marrow Transplantation;
• Drug therapy;
• Gene Therapy.

• Mucopolysaccharidoses and oligosaccharidoses
• Mucolipidoses
• Sphingolipidoses
• Gangliosidoses
• Glycogen storage disorders (also can be
classified as the carbohydrate metabolic disorders)
LYSOSOMAL STORAGE DISORDERS

Mucopolysaccharidoses
• MPS I H, S (MPS 1H - Hurler, MPS 1S - Scheie),
• MPS II (Hunter),
• MPS III A, B, C, D (Sanfilippo A, B, C, D),
• MPS IV A, B (Morquio A, B),
• MPS VI (Maroteaux-Lamy),
• MPS VII (Sly).

• MPS I Hurler 1:100 000
• MPS II Hunter 1:150 000
• MPS III Sanfilippo 1: 24 000
• MPS IV Morquio 1:100 000
• MPS VI Maroteaux-Lamy 1:100 000
Incidence

Hurler syndrome (MPS type I)
• It is a rare, inherited disease of metabolism with the deficiency of enzyme lysosomal alpha-L-iduronidasewhich breakdown long chains of sugar molecules called glycosaminoglycans or mucopolysaccharides.
• Without the enzyme, glycosaminoglycans build up and damage organs.
• The disease is inherited in an autosomal recessive manner.

Symptoms:• Early manifestation• Growth retardation• Hepatosplenomegaly• Skeletal abnormalities:
• Spine deformations• Joint disease, including stiffness, claw hand • Thick, coarse facial features
• Cloudy corneas• Deafness• Heart value problems• Mental retardation that gets worse over time• Hernias
Hurler syndrome (MPS type I)
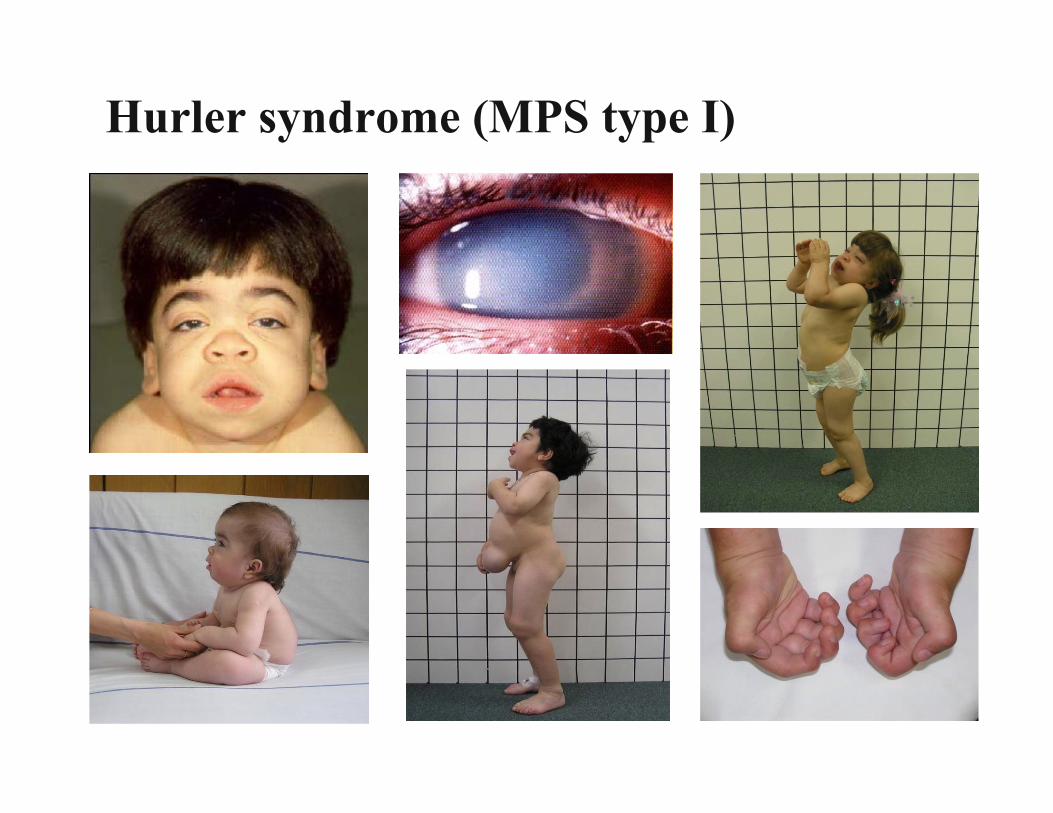
Hurler syndrome (MPS type I)

• The condition is caused by a lack of the enzyme iduronate-2-sulfatase.
• The disease is inherited in a X-linked recessive manner.
Hunter syndrome (MPS type II)
Symptoms• Aggressive behavior, hyperactivity• Mental retardation• Coarse features of the face• Deafness (gets worse over time)• Hypertrichosis• Joint stiffness• Macrocephaly
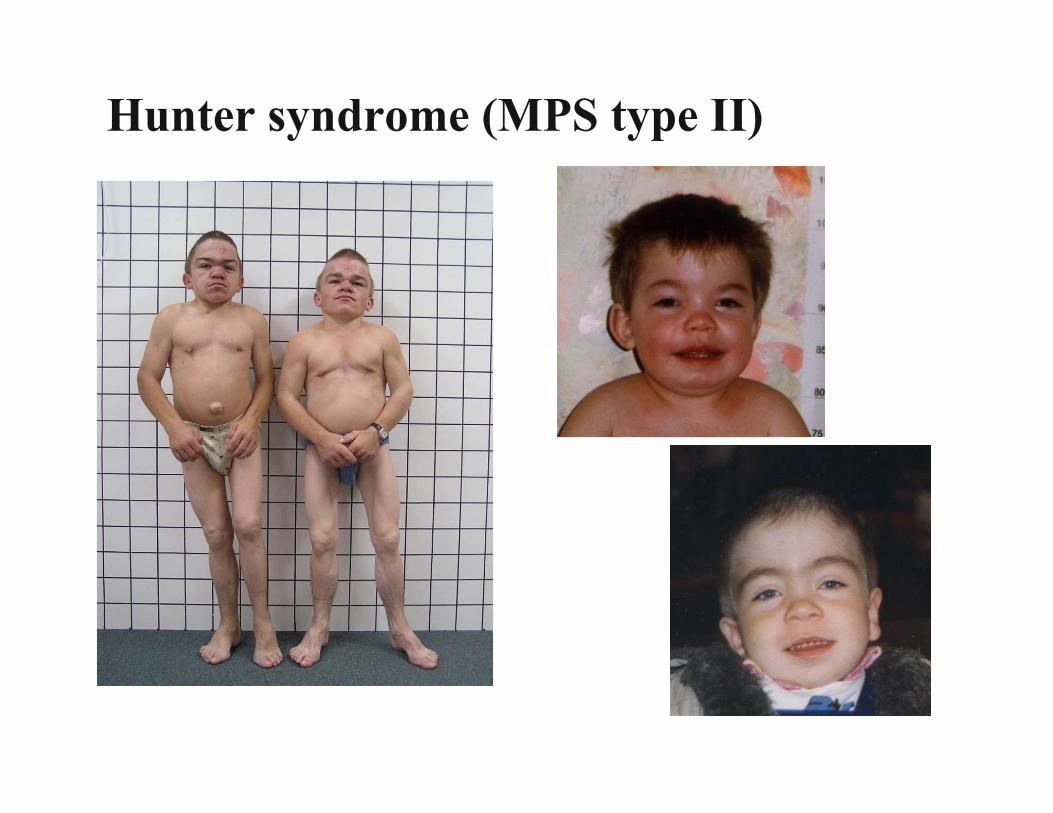
Hunter syndrome (MPS type II)
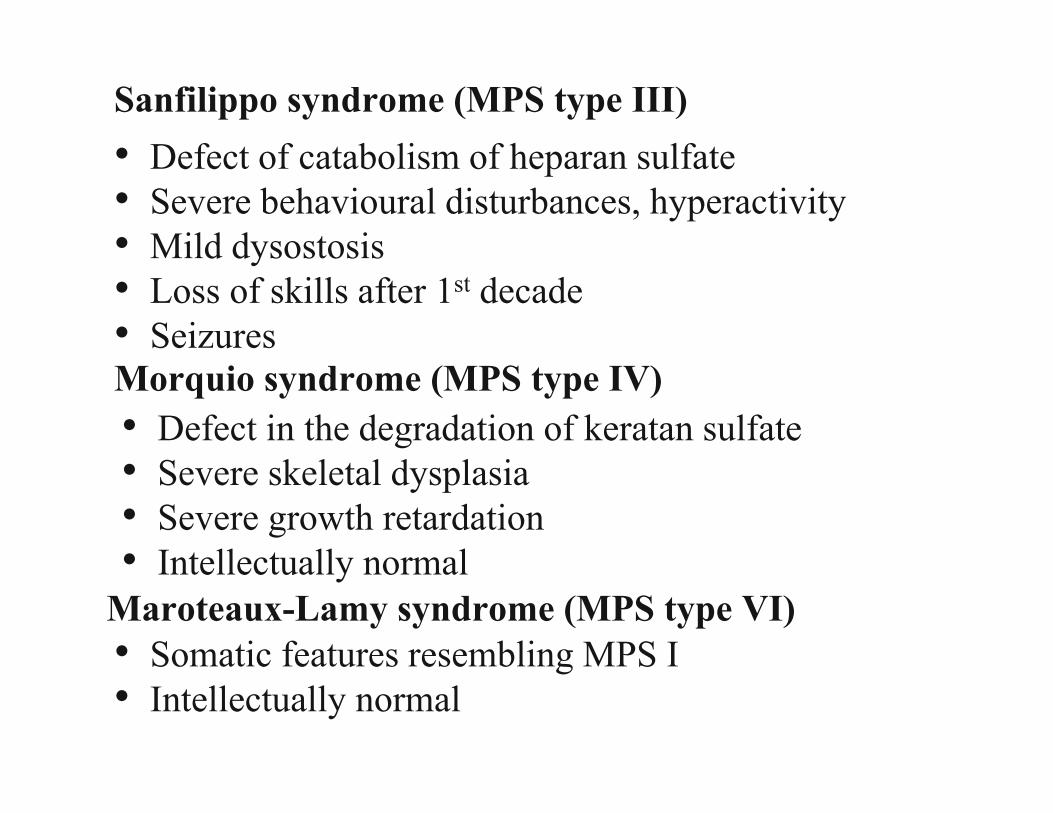
Sanfilippo syndrome (MPS type III)
Morquio syndrome (MPS type IV)
Maroteaux-Lamy syndrome (MPS type VI)
• Defect of catabolism of heparan sulfate • Severe behavioural disturbances, hyperactivity• Mild dysostosis• Loss of skills after 1st decade• Seizures
• Defect in the degradation of keratan sulfate • Severe skeletal dysplasia• Severe growth retardation• Intellectually normal
• Somatic features resembling MPS I• Intellectually normal
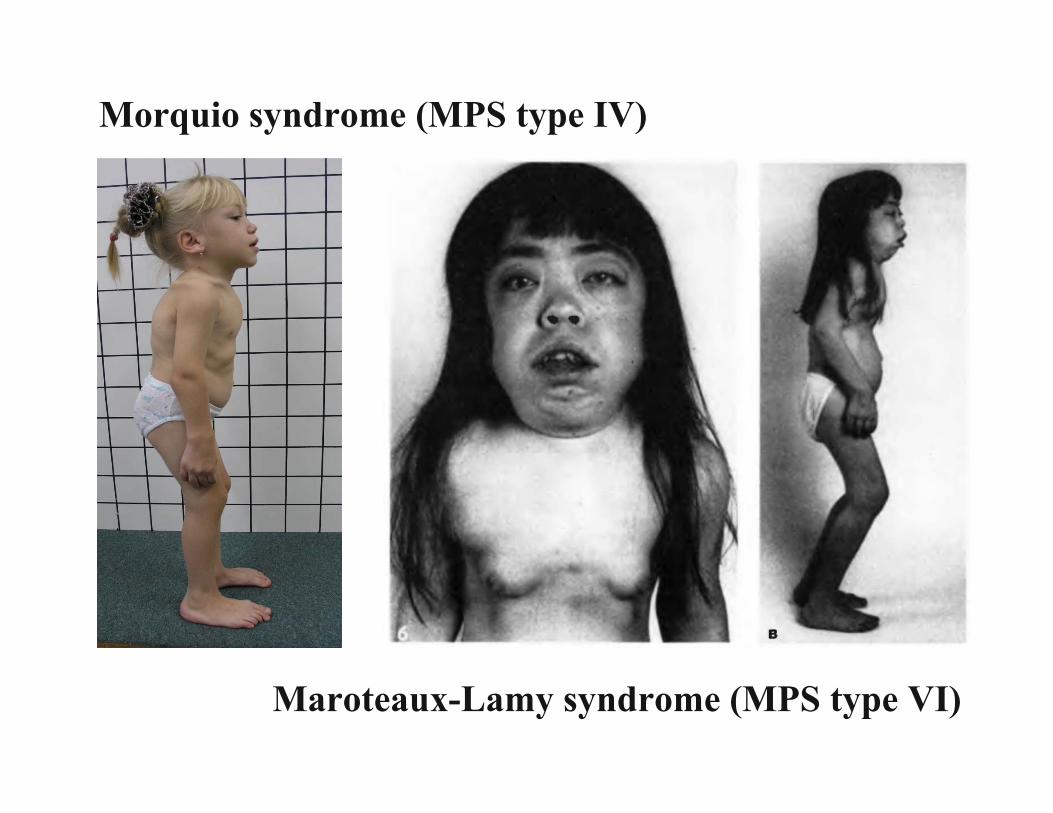
Morquio syndrome (MPS type IV)
Maroteaux-Lamy syndrome (MPS type VI)

• It is a rare (1:40 000 births), inherited and often fatal disorder that disables the heart and skeletal muscles.
• It is caused by mutations in a gene that makes an enzyme called acid alpha-glucosidase (GAA), which normally break down glycogen. The enzyme performs it function in the lysosomes.
• In Pompe disease, mutations in the GAA gene reduce or completely eliminate this essential GAA.
• Excessive amounts of lysosomal glycogen accumulate everywhere in the body, but the cells of the heart and skeletal muscles are the most seriously affected.
• The severity of the disease related to the degree of enzyme deficiency.
Pompe disease (glycogen storage disease type II)
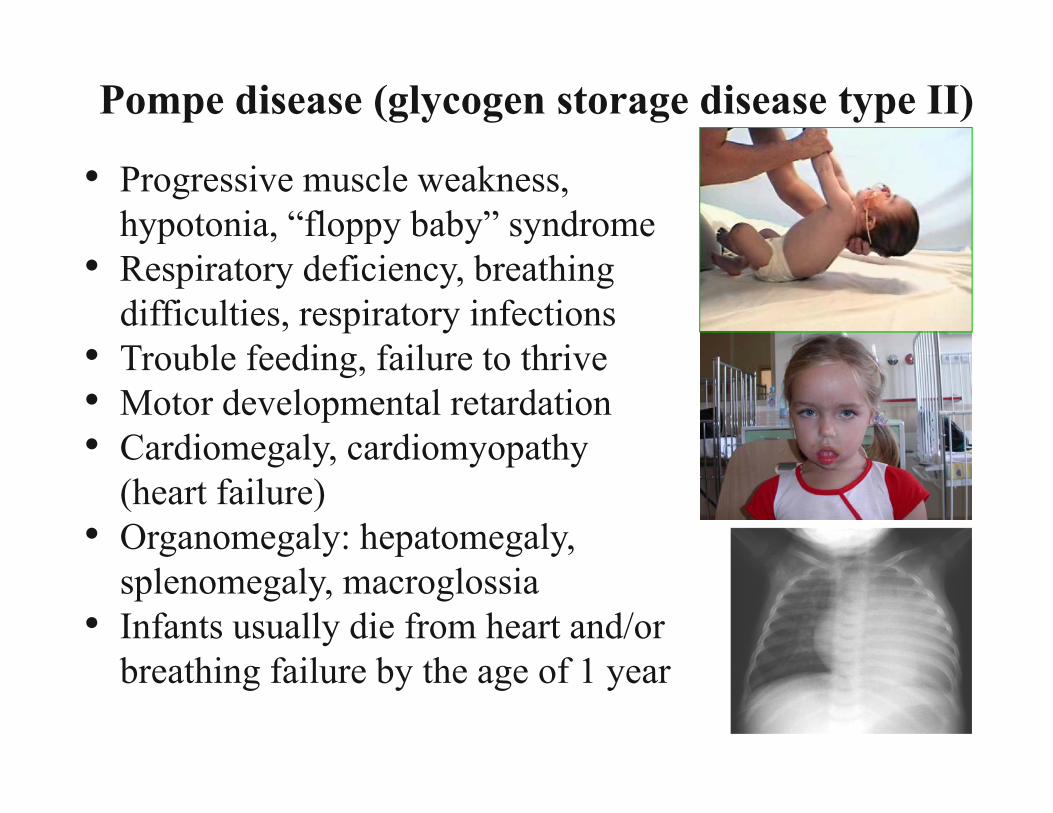
Pompe disease (glycogen storage disease type II)
• Progressive muscle weakness, hypotonia, “floppy baby” syndrome
• Respiratory deficiency, breathing difficulties, respiratory infections
• Trouble feeding, failure to thrive• Motor developmental retardation• Cardiomegaly, cardiomyopathy
(heart failure)• Organomegaly: hepatomegaly,
splenomegaly, macroglossia• Infants usually die from heart and/or
breathing failure by the age of 1 year

• It is a fatal uncurable genetic lipid storage disorder in which harmful quantities of a fatty substance called ganglioside GM2 build up in tissues and nerve cells in the brain.
• The condition is caused by insufficient activity of an enzyme called beta-hexosaminidase A that catalyzes the biodegradation of acidic fatty materials known as gangliosides. Gangliosides are made and biodegraded rapidly in early life as the brain develops.
• Patients and carriers of Tay-Sachs disease can be identified by a simple blood test that measures beta-hexosaminidaseA activity.
• Inheritance: autosomal recessive.• The disease is much more common among people of
Ashkenazi (Eastern European) Jewish descent.
Tay-Sachs disease

Symptoms of Tay-Sachs disease• Infant shows normal development till 5-6 months age• Loosing all gained skills progresively• Deafness• Decreased eye contact, blindness• Decreased musclue tones (loss of muscle strength)• Delayed mental and social skills• Dementia• Increased startle reaction, irritability• Listlessness• Loss of motor skills• Seizures• Slow growth• Death is usually between age 2 and 4 years, often
from pneumonia.

• Fabry disease is caused by the lack of enzyme needed to metabolize lipids. The disease is also called alpha-galactosidase-A deficiency.
• Mutations in the GLA gene cause Fabry disease. The GLA gene provides instructions for making alpha-galactosidase A. This enzyme is active in lysosomes.
• Lipids build up to harmful levels in the eyes, kidneys, autonomic nervous system, and cardiovascular system. Fabry disease is one of several lipids to rage disorders and the only X-linked lipid storage disease.
• Heterozygous females can be symptomatic.
Fabry disease
Symptoms: • Burning sensations in the hands • Acroparesthesia• Small, non-cancerous, raised reddish-purple blemishes
on the skin• Skin angiokeratomas around of a navel, hips, shins • Eye manifestations, especially cloudiness of the cornea• Impaired arterial circulation and increased risk of heart
attack or stroke• Proteinuria, renal failure • Hypohidrosis (decreased sweating), heat intolerance,
fever• Gastrointestinal difficulties: Diarrhea, abdominal pain
Fabry disease

MAIN GOAL
As specific therapies become available, it
will be essential to make the correct
diagnosis early in life, before
irreversible damage has occured.

GENETICS OF NEUROMUSCULAR DISORDERS"Genetics and Neuromuscular Diseases" gives an up-to-date review of genetics information relating to neuromuscular diseases.Neuromuscular disorders affect the peripheral nervous system and muscle. The principle effect of neuromuscular disorders is therefore on the ability to perform voluntary movements. Neuromuscular disorders cause significant incapacity, including, at the most extreme, almost complete paralysis.
What is a neuromuscular disorder? A condition affecting one or more of the following:-Muscles-Nerves-Neuromuscular junction; the area where muscle and nerve make connection-Motor or sensory cell-body
How do we diagnose a neuromuscular disorder? History & Physical Examination Laboratory Tests NCV/EMG MRI Scans Muscle biopsy Nerve biopsy

Neuromuscular diseases include some of the most devastating disorders that afflict mankind, for example motor neuron disease. ND have onset any time from in utero until old age. They are most often genetic. The last 25 years has been the golden age of genetics, with the disease genes responsible for many genetic neuromuscular disorders now identified. ND may be inherited as autosomal dominant, autosomal recessive, or X-linked traits. They may also result from mutations in mitochondrial DNA or from de novo mutations not present in the peripheral blood DNA of either parent. The high incidence of de novo mutation has been one of the surprises of the recent increase in information about the genetics of neuromuscular disorders. The disease burden imposed on families is enormous including decision making in relation to presymptomatic diagnosis for late onset neurodegenerative disorders and reproductive choices. Diagnostic molecular neurogenetics laboratories have been faced with an ever-increasing range of disease genes that could be tested for and usually a finite budget with which to perform the possible testing. Neurogenetics has moved from one known disease gene, the Duchenne muscular dystrophy gene in 1987, to hundreds of disease genes. It can be anticipated that with the advent of next generation sequencing (NGS), most, if not all, causative genes will be identified in the next few years. Any type of mutation possible in human DNA has been shown to cause genetic neuromuscular disorders, including point mutations, small insertions and deletions, large deletions and duplications, repeat expansions or contraction and somatic mosaicism.

Mutations causing neuromuscular disorders affect the largest human proteins for example titin and nebulin. Successful molecular diagnosis can make invasive and expensive diagnostic procedures such as muscle biopsy unnecessary. Molecular diagnosis is currently largely based on Sanger sequencing, which at most can sequence a small number of exons in one gene at a time. NGS techniques will facilitate molecular diagnostics, but not for all types of mutations. For example, NGS is not good at identifying repeat expansions or copy number variations.
Diagnostic molecular neurogenetics is focused on identifying the causative mutation(s) in a patient. In the future, the focus might move to prevention, by identifying carriers of recessive diseases before they have affected children. The pathobiology of many of the diseases remains obscure, as do factors affecting disease severity. The aim of this review is to describe molecular diagnosis of genetic neuromuscular disorders in the past, the present and speculate on the future.

The clinical practice of ND is undergoing enormous change as a result of the wealth of molecular genetic discoveries. The majority of gene discoveries in neurological diseases relate to neuromuscular disorders and a precise DNA based diagnosis is possible. This gives patients accurate prognostic and genetic counselling information, will also facilitate screening programmes for recognised complications such as cardiac or respiratory involvement. Many eligible patients do not benefit from or have access to such diagnostic precision, although this is changing. The discovery of new genes and proteins has opened up avenues of therapies for neuromuscular patients. Therapeutic trials in genetic neuromuscular diseases are in their infancy: a precise DNA-diagnosis is essential and future proven therapies will be contingent upon DNA-diagnosis. It’s no longer acceptable to make “limb-girdle muscular dystrophy” based on histochemistry, a final diagnosis. Immunocytochemistry and protein chemistry with DNA analysis offer the patient the best chance of a precise diagnosis from which accurate prognostication, screening, and genetic counselling will follow.

We often observe differencies in the severity of clinical manifestation within a family due to the causative point mutations, insertions, deletions, duplications, repeat expansions, somatic mosaicism.
Repeat expansion disorders are those in which unstable expansion of tandem nucleotide repeats, ranging from tri-, tetra-, penta- to dodecanucleotide repeats, result in distinct diseases. Such a mechanism is responsible for more than 40 neurological or neuromuscular disorders/phenotypes. Repeat expansion disorders reveal a mendelian inheritance pattern; however, unlike static mutations in mendelian diseases, the repeat mutation process is dynamic. Repeats continue to expand in subsequent generations and even within tissues of individual. Repeat expansions show different genetic properties depending on the location of the repeats. For example, in coding sequencies, such as CAG polyglutamine repeats observe in Huntington disease. Affected individuals in next generations present with a more severe disease phenotype and an earlier age of onset (anticipation).

Premutation: in some clinically unaffected individuals, the number of repeats appears to exceed beyong the normal range of general populationin a state called premutation.
√ Premutations show both somatic and germline instability and often can be found in the phenotypically normal antecedent of patients in the family. Premutations often develop into full mutations that contain longer repeat expansion , resulting in disease phenotype.
Neuromuscular disorders include rare uscular dystrophy, athrophy, miotonia, myopathy,neuropathy, amyotrophic sclerosis, SMA, and other motosensor and movemental dysfunctions.
More severe clinical phenotypes, including Duchenne Muscular Dystrophy, with progressive muscular weakness of the hips, thighs, and back causes ddifficulties in walking and in using steps. Lumbar lordosis and enlarged but weak calves (pseudohypertrophy) are visible.

Genetic counsellors can helppeople make decisions aboutgenetic testing and childbearing.
Example of a family tree (pedigree) inspinal muscular atrophy (autosomal recessive)
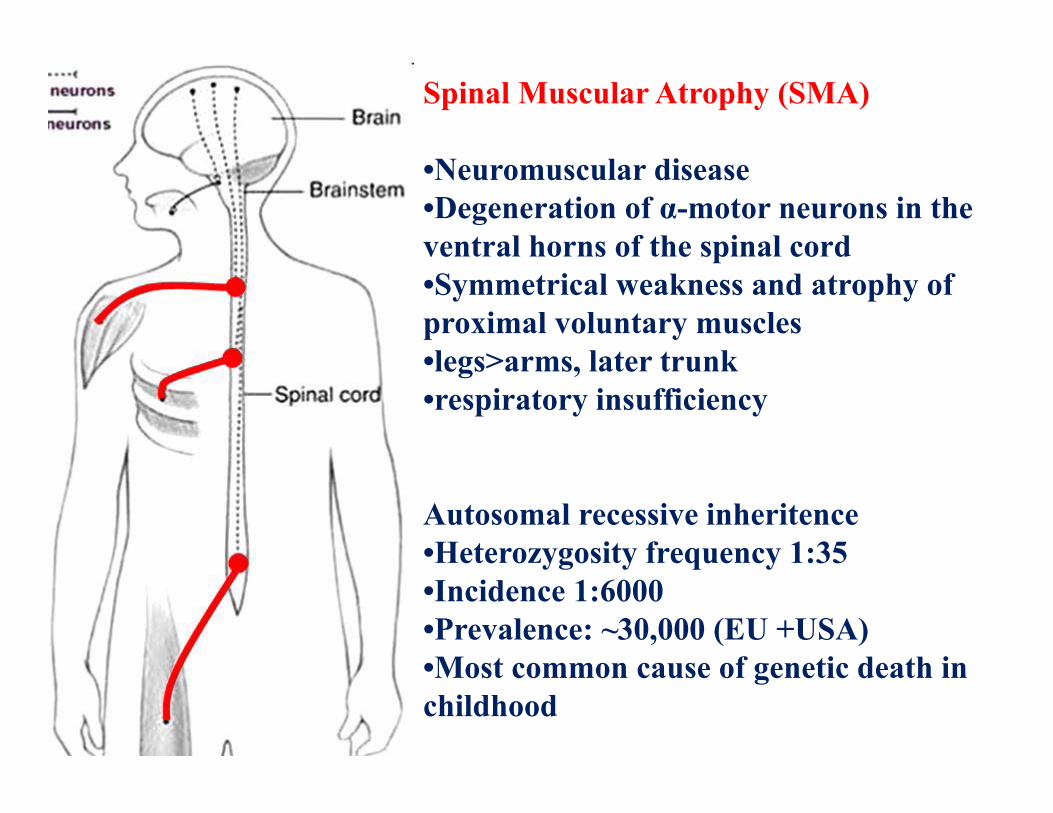
Spinal Muscular Atrophy (SMA)
•Neuromuscular disease •Degeneration of α-motor neurons in the ventral horns of the spinal cord •Symmetrical weakness and atrophy of proximal voluntary muscles •legs>arms, later trunk •respiratory insufficiency
Autosomal recessive inheritence •Heterozygosity frequency 1:35 •Incidence 1:6000 •Prevalence: ~30,000 (EU +USA) •Most common cause of genetic death in childhood

5q12.3-SMA region and SMN genes
SMA which is caused by homozygous mutations in SMN1 is strongly influenced by the SMN2 copy number •SMN2 is alternatively spliced due to a single translationally silent mutation that affects splicing •SMA is the first human genetic disease for which a therapy based on transcription activation and restoration of the correct splicing of a copy gene (SMN2), which is present in all SMA patients has become available. •The first inherited human disorder for which an independent fully protecting modifying gene has been identified •Identification of the SMA modifier, PLS3 helped us to uncover the true molecular pathogenesis of SMA, which is F-actin bundling and dynamics.
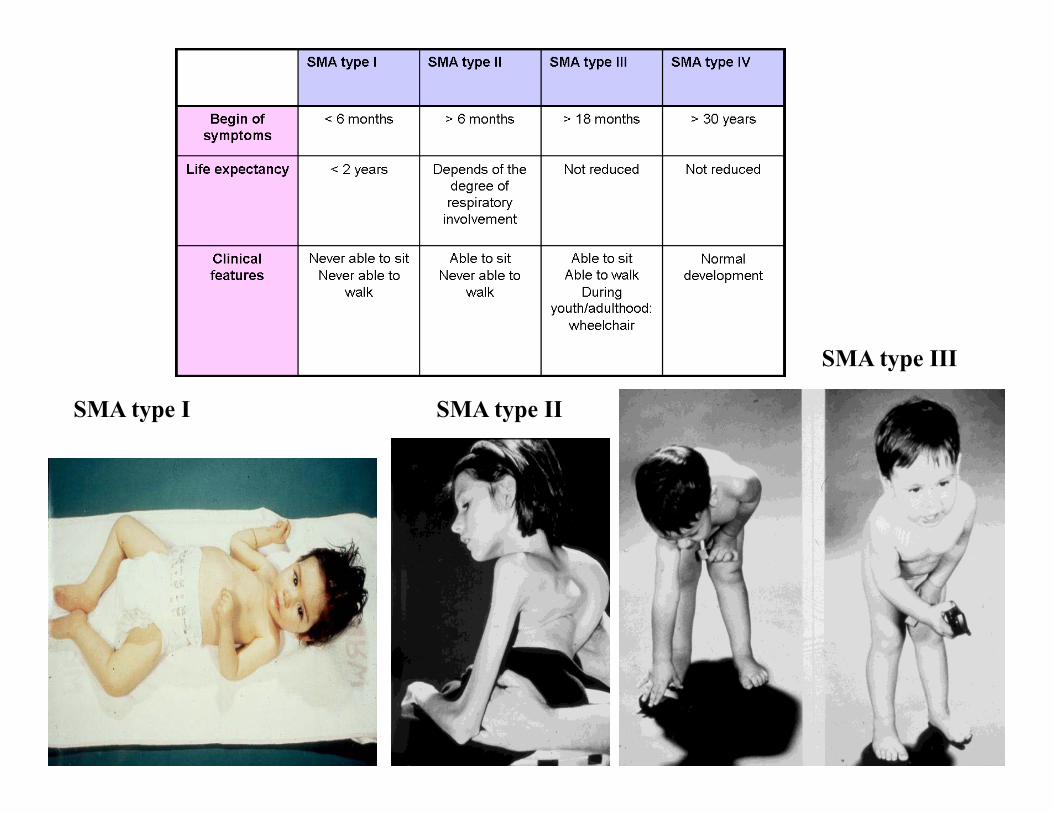
SMA type I SMA type II
SMA type III

The future of SMA therapy •When to start therapy? –Presymptomatic –During maturation of the NMJs Inclusion of SMN1 deletion testing in neonatal screening programs –Intrathecally + systemic application •Which tissues need to be targeted? –Type I SMA •Spinal cord •All other tissues for long term therapy –Type II – IV SMA •Spinal cord while all other tissues might have sufficient SMN levels Intrathecal and systemic application Which drugs? –SMN (ASO or AAV9-SMN gene therapy) –Drugs that stimulates SMN transcription, translation or stabilization –Drugs/Small molecules that counteract the disturbed: •F-actin stabilization •Ca2+ homeostasis •Increased oxidative stress due to hypooxiganation ••Other interventions –Nutrition –Oxigenenation

ANTICIPATION AND REPEAT EXPANSIONan earlier age of onset /more severe expression in generations. More than 20 diseases. Examples:Trinucleotide Repeat Disorders:Fragile - X syndrome (FRAXA)Syndromes (FRAXE, FRAXF, FRAX16A)Huntington DiseaseKennedy DiseaseSpinocerebellar Ataxia types 1 - 10Oculopharyngeal Dystrophy
MYOTONIC DYSTROPHY:DMPK – protein kinase gene (chr19): disease causing mutation: expanded CTG trinucleotide repeats. The number of these repeats strongly correlates with severity:Unaffected persons: 5-37 copies;Mildly affected : 50-100 copies;Severe phenotype: 100 and more copies.
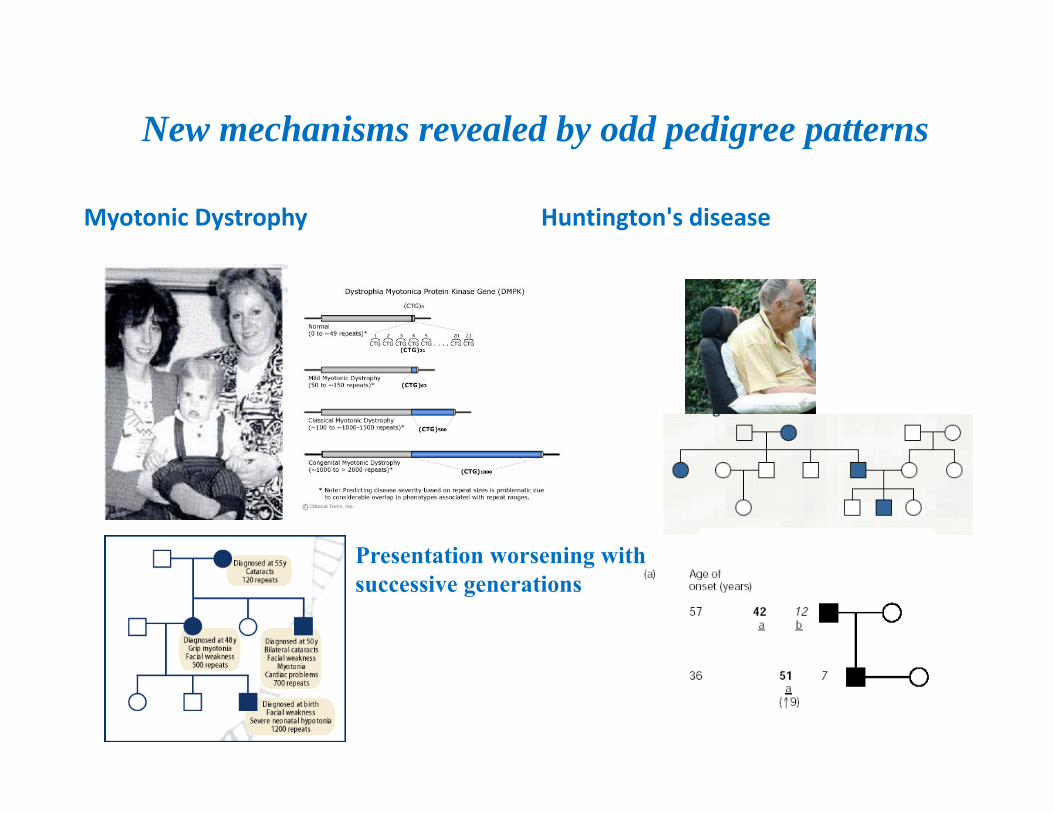
New mechanisms revealed by odd pedigree patterns
Myotonic Dystrophy Huntington's disease
Presentation worsening with successive generations

TRIPLET REPEAT DISORDERS:FRA-X SYNDROME (Xq27.3 REGION CONTAINS A SMALL MICROSATELLITE AREA OF CGG REPEATS)

Fragile X Syndrome
• Age of onset: childhood• Moderate mental retardation• Dysmorphic facies
Caused by mutations (expansions of CGG repeat) in FMR1 gene. Normal alleles (FMR1) - number of CGG 5-50
• Full mutations – number of repeats more than 230• increasing numbers of affected offspring are observed
in later generations of an affected family (genetic anticipation)


Huntington's DiseaseHuntington's is a disease that gives rise to progressive, selective (localized, in the brain), neural cell death associated with a mutation on chromosome 4. Basically it is a inherited Neurodegenerative disorder. Characterized by: Motor disturbance ( both voluntary and involuntary) Cognitive loss Psychiatric manifestations (ie: dementia, and depression) Genetics: The Huntington's Disease gene was discovered in 1993. The gene produces a protein called Huntingtin, which is necessary for normal growth and development and appears active throughout the body. The exact function of this protein is unknown because it is not analogous to any other known proteins. HD gene is the IT-15 and is located on chromosome 4p16.3The IT-15 gene has a highly polymorphic region that consists of (CAG)n - repeats which are located in Exon 1 of the IT-15 gene.

HD is a family disease
HD gene contain a specific section expanded in people with HD. In all people, this stretch of genetic material (DNA), contains a pattern of so-called "trinucleotide repeats". In most people, the repeated pattern "CAG" occurs 30 times or less. In HD, it occurs more than 36 times. There are people with a CAG repeat in the so called "grey zone",between 30 and 36 repeats. At the moment is not sure that somebody "high in the grey zone" may get the disease at a late age (above 60 or later) or not.
Are my children at risk?
Each child of an affected parent has a one in two (or 50%), chance of inheriting the gene that causes HD, and is said to be "at risk". People who carry the gene will eventually develop Huntington's HD does not "skip a generation". Genetic test may be used to help confirm, or ruleout, a diagnosis of HD. By analyzing a person's DNA and counting the number of CAG's it is possible to tell if that person will develop HD.
Treatment for HD takes many forms it is preferable to locate a neurologist with expertise in HD.
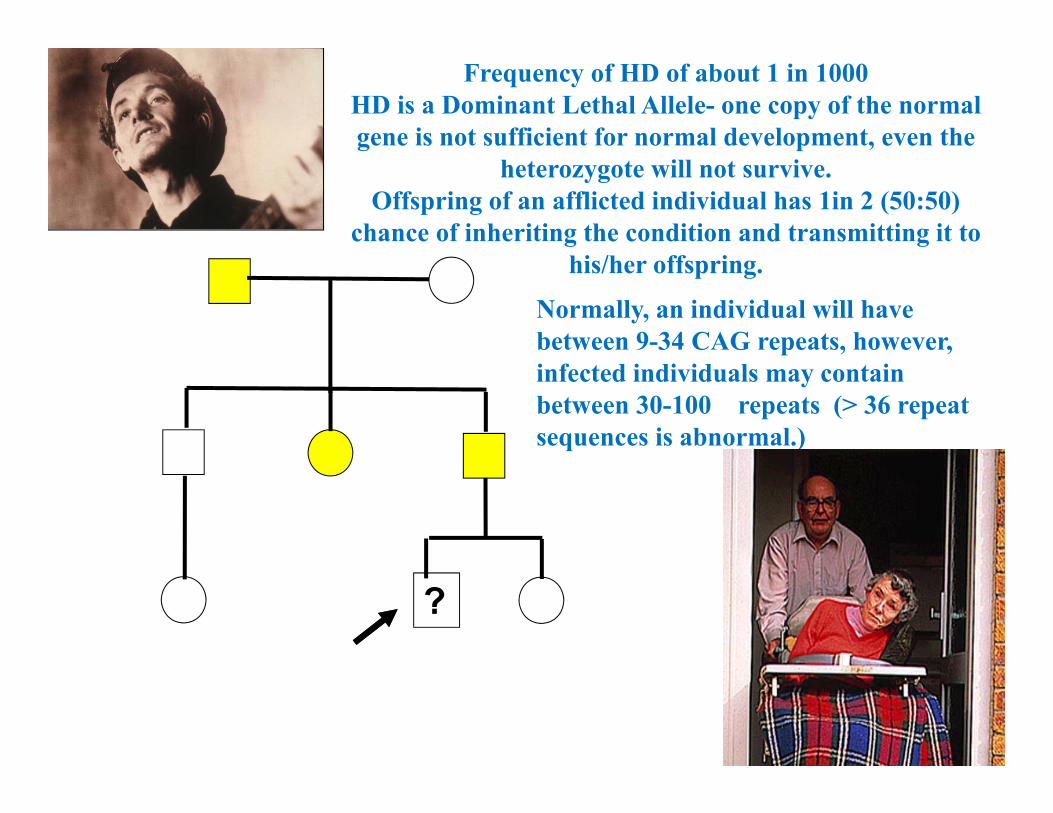
?
Frequency of HD of about 1 in 1000 HD is a Dominant Lethal Allele- one copy of the normal gene is not sufficient for normal development, even the
heterozygote will not survive. Offspring of an afflicted individual has 1in 2 (50:50)
chance of inheriting the condition and transmitting it to his/her offspring.
Normally, an individual will have between 9-34 CAG repeats, however, infected individuals may contain between 30-100 repeats (> 36 repeat sequences is abnormal.)


Charcot‐Marie‐Tooth DiseaseThis is a disease where you have weakness in your feet and lower leg musclesThis disease is one of most common inherited neurological disordersThis disease compromises a group of disorders that affect peripheral nervesIt affects approximately 1 in 2,500 people in the US
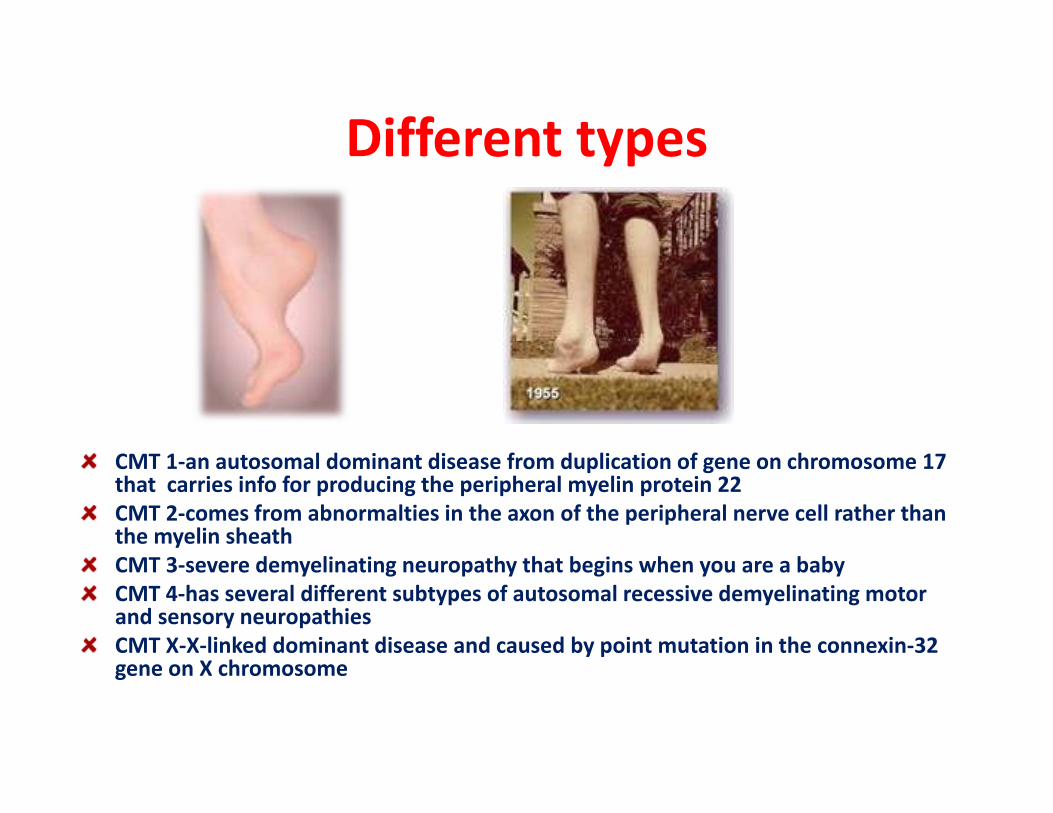
Different types
CMT 1‐an autosomal dominant disease from duplication of gene on chromosome 17 that carries info for producing the peripheral myelin protein 22CMT 2‐comes from abnormalties in the axon of the peripheral nerve cell rather than the myelin sheathCMT 3‐severe demyelinating neuropathy that begins when you are a babyCMT 4‐has several different subtypes of autosomal recessive demyelinating motor and sensory neuropathiesCMT X‐X‐linked dominant disease and caused by point mutation in the connexin‐32 gene on X chromosome

DNA REPAIR DEFICIENCY DISORDERS or a-rec chromosome instability syndromes: are caused by mutations in genes that contribute to DNA replication,repair and genome stability processes. DNA repair defects cause an accelerated aging diseases and an increased risk of cancer: ROTHMUND-THOMSON, XP.Hereditary nonpolyposis colorectal CR is caused by a defective MSH2 gene leading to defective mismatch repair.PROGERIA & Werner, Cockayne syndromes -accelerated aging.
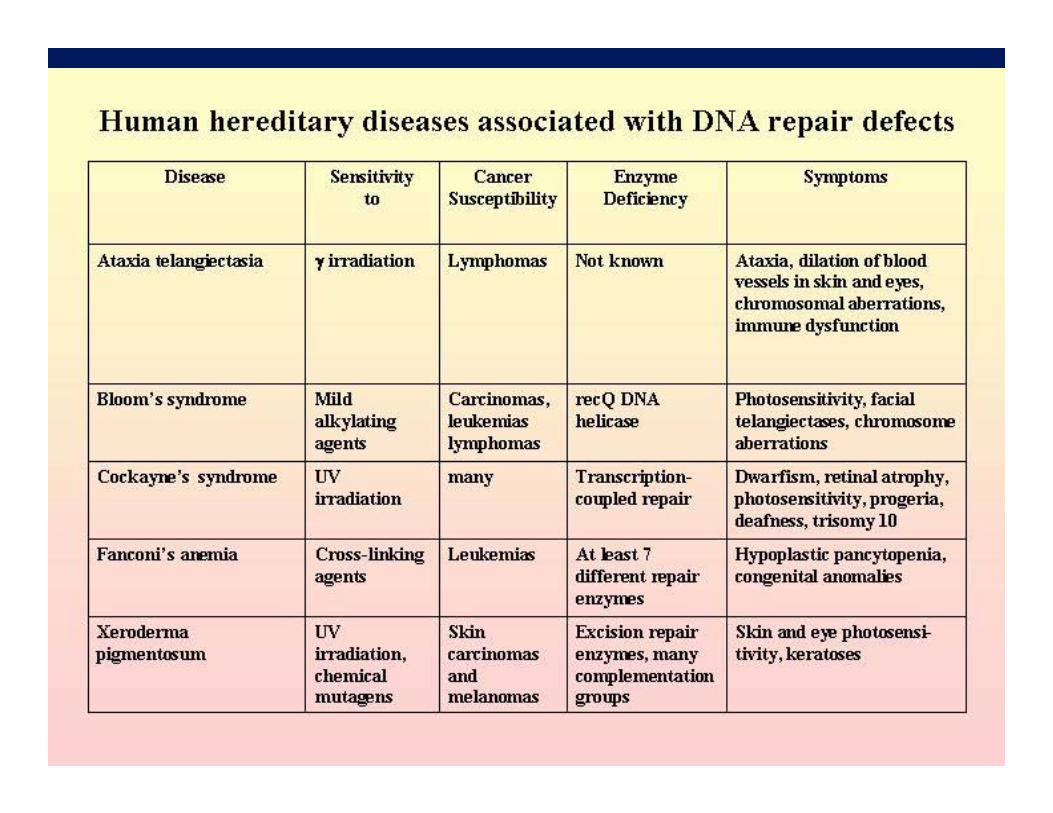


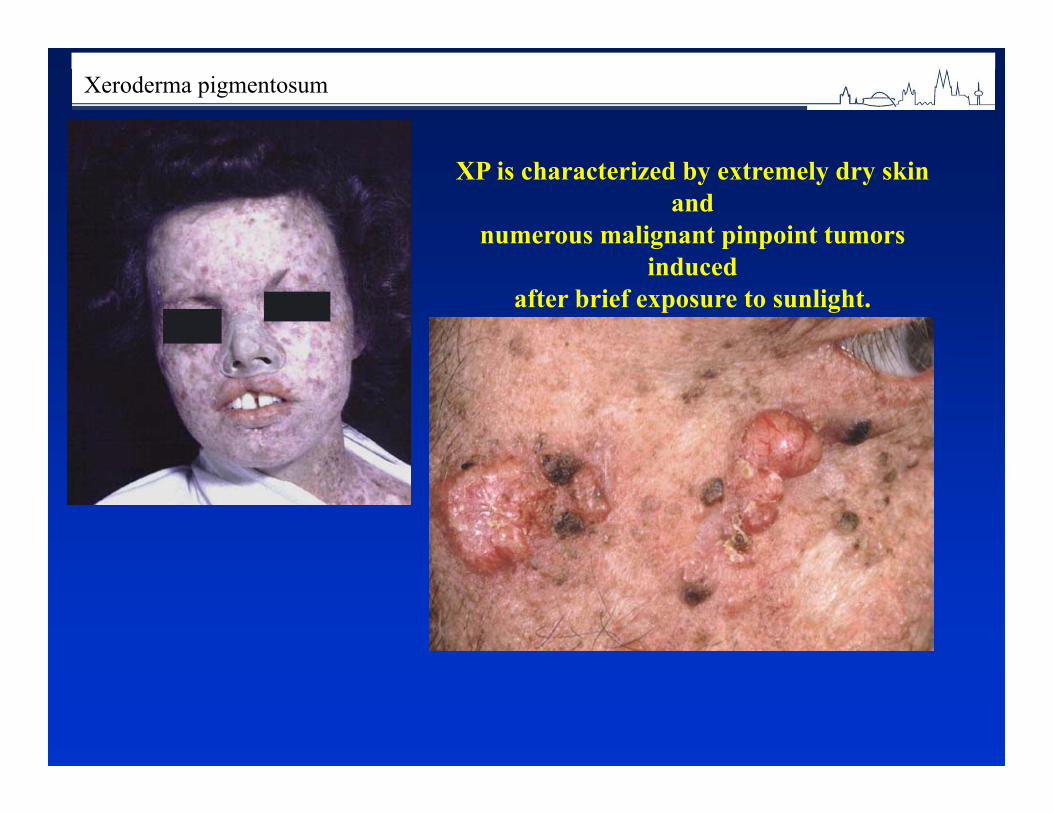
Xeroderma pigmentosum
XP is characterized by extremely dry skin and
numerous malignant pinpoint tumors induced
after brief exposure to sunlight.

• Xeroderma pigmentosum

FANCONI ANEMIA: 16q24.3:11 MUTATIONS in DNA repair gene. Rare genetic condition.
Incidence: 1: 350000 births with a high frequency in Ashkenazi Jews and populations of S.Africa.
-Growth & developmental deficiency
- Bone marrow failure (the mobility to produce blood cells)
- Endocrine problems
- MR
- Congenital defects: short stature, eyes, ears, hand & sceletal: hypoplastic/absent thumbs
-Skin discoloration
-Median age of death: 30 y.

Treatment: Bone marrow or stem cells transplantations;Gene replacement therapy; treatment with hormons and hematopoietic growth factors.
CARRIER TESTING:1 TO 600
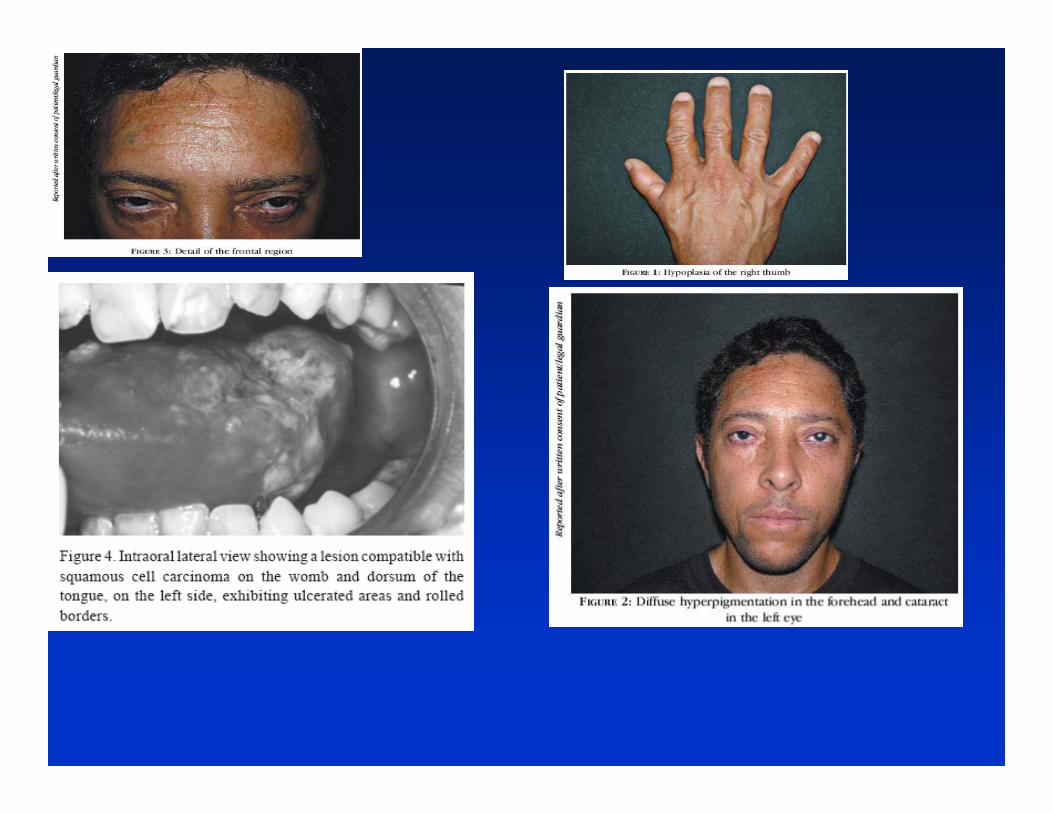

Ataxia-Telangiectasia: Louis-Bar Syndrome
ATM Gene:11q22-23: a rare neurodegenerative syndrome:
Ataxia( difficulty of mouvements and coordination, caused by cerebellum) & Ocular and facial Telangiectasia (small dilated blood vessels)
EXTREME RADIOSENSITIVITY
Immunodeficiency:
predisposition to infections
High risk of cancer:
Haematopoetic malignancy


BLOOM SYNDROME: Congenital Telangiectactic Erythema: Mutations in BLM gene: 15q26.1 (Protein family RecQ helicase: enzyme that bind to DNA and unwind double helix of DNA).Carrier frequency in Eastern European Jewish ancestry:1:100. SYMPTOMS:-Short stature-MR-Microcephaly-Immune deficiency-Cancer predisposition-Face features: a long, narrow face -Micrognathia-Skin hypo- and hyper-pigmented patches: café-au-lait spots, -Telangiectactic erythema of the face-No treatment available
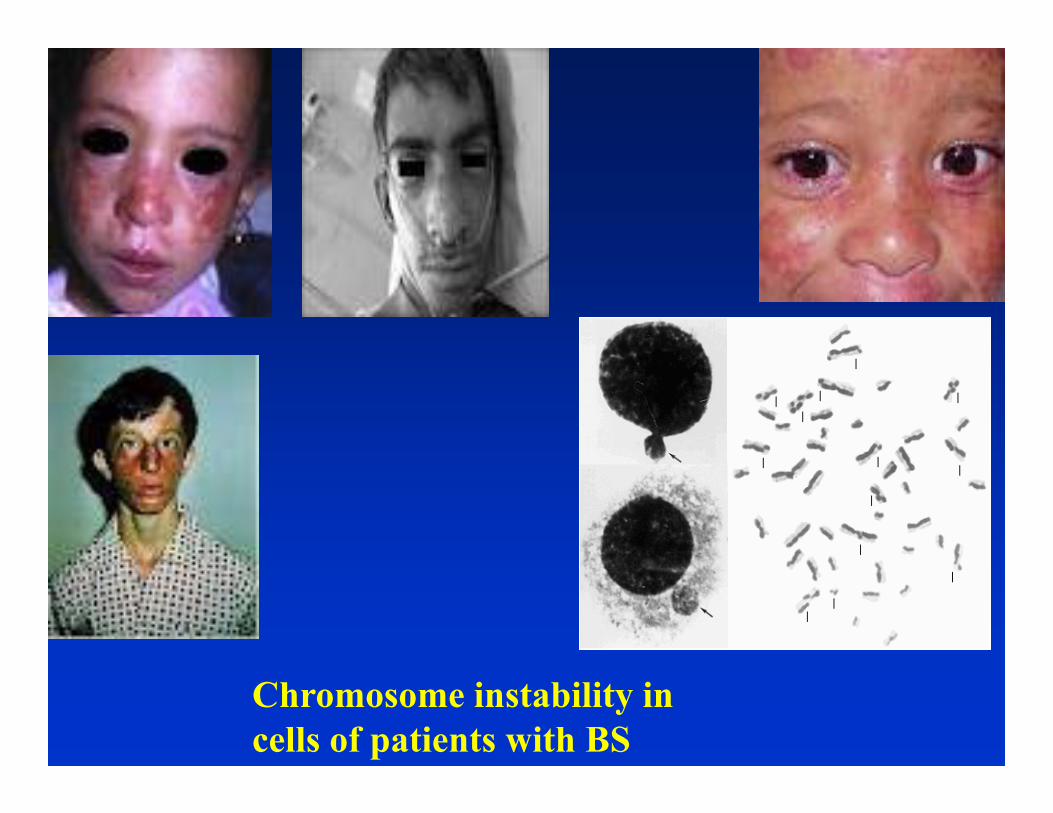
Chromosome instability in cells of patients with BS

Cockayne syndrome
• autosomal recessive inheritance• growth retardation, cachexia• short stature, microcephaly• wizened face• retinal degeneration, optic atrophy• hearing loss• renal failure• kyphosis, vertebral body anomalies• hypoplastic iliac wings• sclerotic phalangeal epiphyses• intellectual disability, seizures• dysmyelinisation, ataxia, tremor• endocrine dysfunction
no skin cancer mutations in:
CSA (ERCC8)CSB (ERCC6)XPBXPDXPG
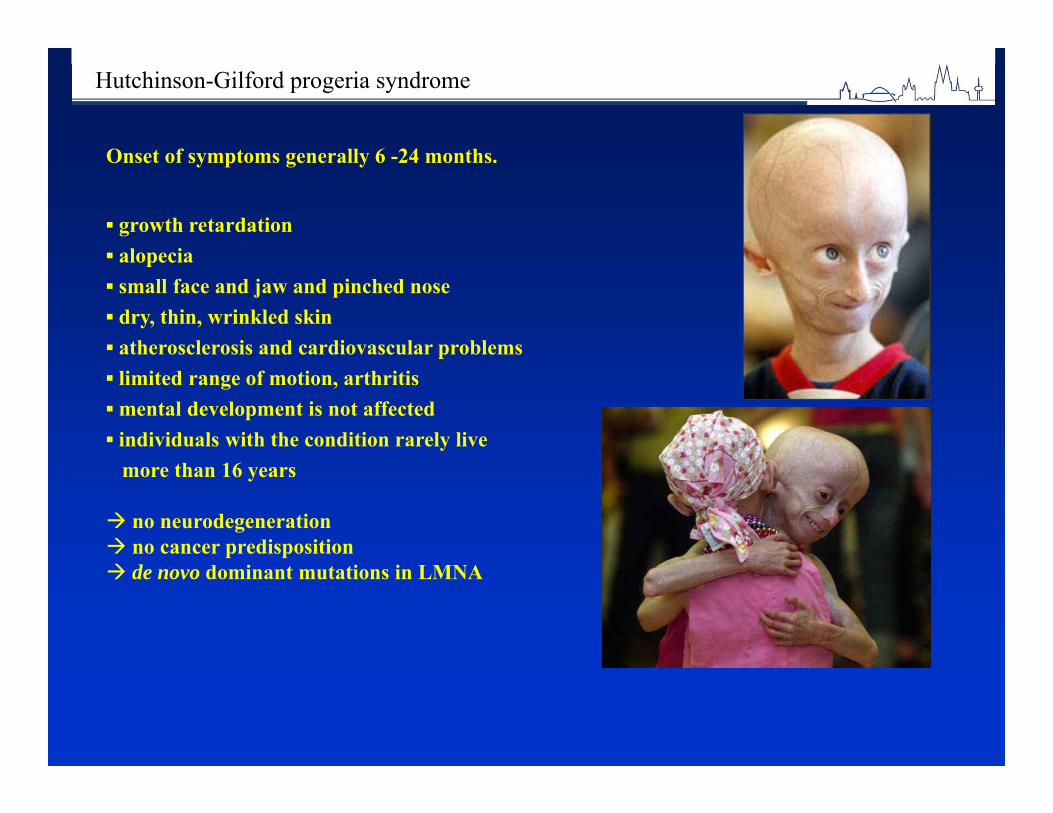
Hutchinson-Gilford progeria syndrome
Onset of symptoms generally 6 -24 months.
▪ growth retardation▪ alopecia ▪ small face and jaw and pinched nose▪ dry, thin, wrinkled skin ▪ atherosclerosis and cardiovascular problems▪ limited range of motion, arthritis▪ mental development is not affected▪ individuals with the condition rarely live
more than 16 years
no neurodegeneration no cancer predisposition de novo dominant mutations in LMNA

Wiedemann -Rautenstr. syn.
Arboleda, AJMG 2011;A155:1712
Hallermann-Streiff syndrome
Cohen, AJMG 1991;41:488
Atypical /non-categorizedcases ofunknown
etiology
Progeria – beyond the Werner and HGP syndromes
Werner syndrome
HGP syndrome
Cockayne syndrome
Xerodermapigmentosum
Kraemer, Neuroscience2007;145:1388
Kraemer, Neuroscience2007;145:1388
Cabanillas, AJMG 2011;A155:2617
Nestor-Guillermo P.
Wrinklyskin syndrome
Rajab, AJMG 2008;A146:965
•Rothmund-Th.•Trichothiodys.•Bloom syndr.•Dyskeratosis c.•Seckel syndr.•Cutis laxa•Gerodermia o.• ……
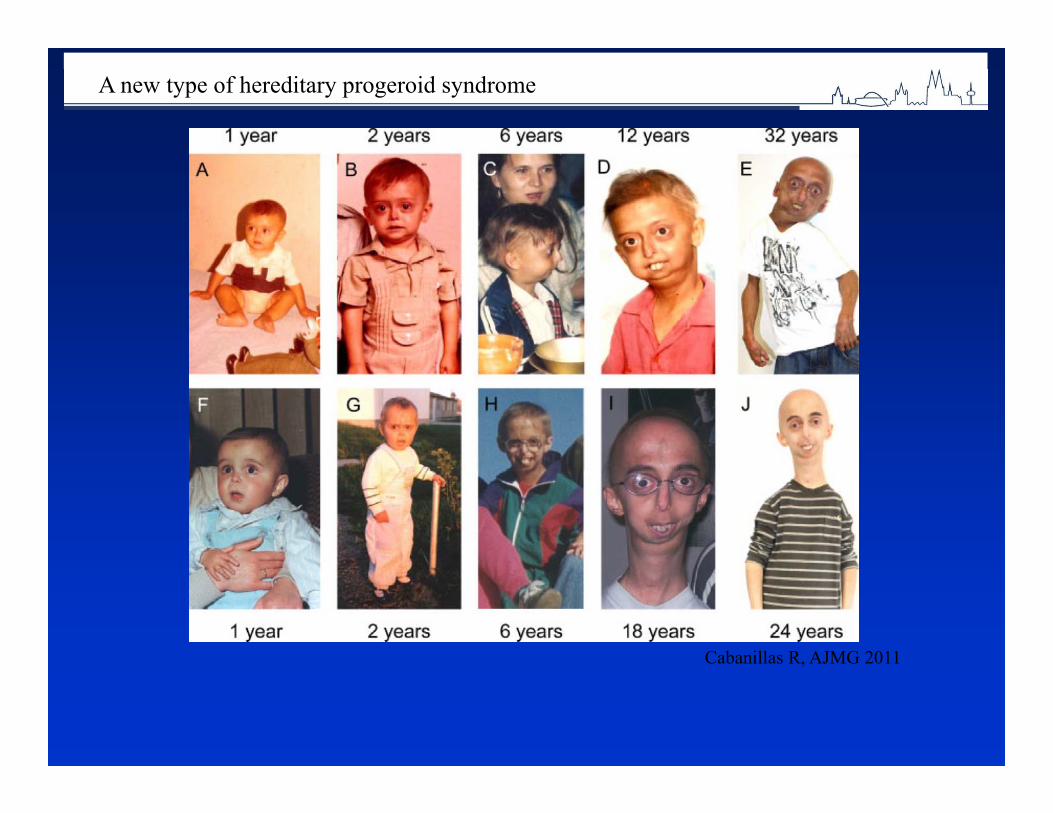
Cabanillas R, AJMG 2011
A new type of hereditary progeroid syndrome
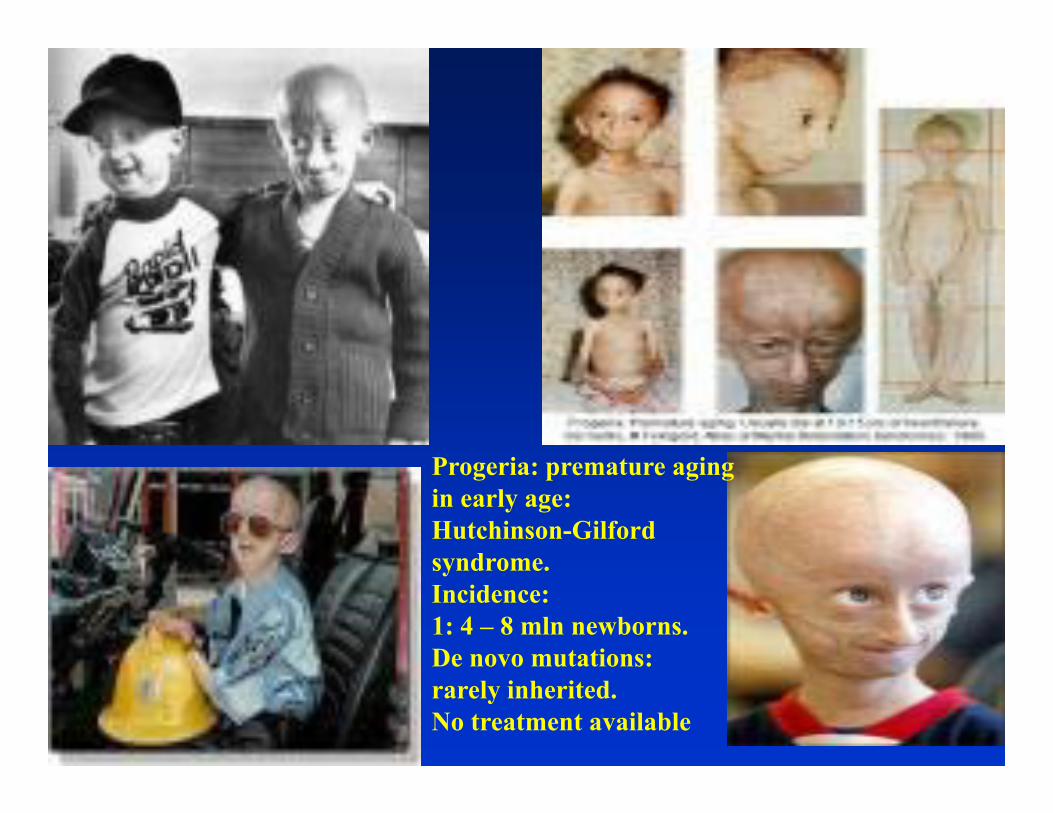
Progeria: premature aging in early age: Hutchinson-Gilford syndrome.Incidence: 1: 4 – 8 mln newborns.De novo mutations: rarely inherited.No treatment available
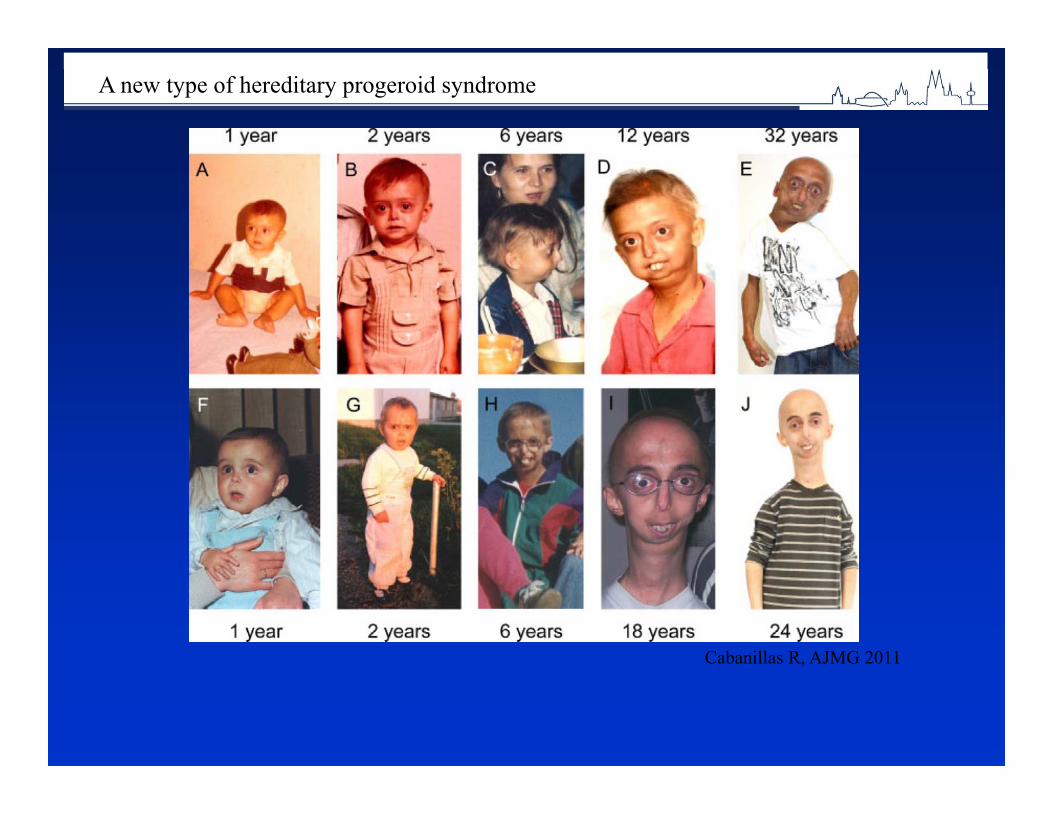
Cabanillas R, AJMG 2011
A new type of hereditary progeroid syndrome

Wiedemann -Rautenstr. syn.
Arboleda, AJMG 2011;A155:1712
Hallermann-Streiff syndrome
Cohen, AJMG 1991;41:488
Atypical /non-categorizedcases ofunknown
etiology
Progeria – beyond the Werner and HGP syndromes
Werner syndrome
HGP syndrome
Cockayne syndrome
Xerodermapigmentosum
Kraemer, Neuroscience2007;145:1388
Kraemer, Neuroscience2007;145:1388
Cabanillas, AJMG 2011;A155:2617
Nestor-Guillermo P.
Wrinklyskin syndrome
Rajab, AJMG 2008;A146:965
•Rothmund-Th.•Trichothiodys.•Bloom syndr.•Dyskeratosis c.•Seckel syndr.•Cutis laxa•Gerodermia o.• ……

Dysmorphology

Etiological factor of hereditary diseases are persistent changes in the hereditary
apparatus of the cell.According to the World Health
Organization’s annual 5-8% of newborneare born with congenital and hereditary
defects.

Birth defects are the leading cause of death in the first year of life.
2-3% of newborn infantsUp to 7% of adult
Clinical investigation of the causes and consequences of birth defects is called
dysmorphology.Dysmorphology literally mean “the study of
abnormal form”.As a medical subspecialty, dismorphology deals
with people who have congenital abnormalities and with their families.

Classification of birth defects
MalformationDisruptionDeformationDisplasia

Malformation A malformation is a primary structural defect of an organ or part of an organ which results from an inherent abnormality in development. This used to be known as a primary or intrinsic malformation. The presence of a malfomation implies that the early development of a particular tissue or organ has been arrested or misdirected.

Common examples of malformations include congenital heart abnormalities such as ventricular or atrial septal defects, cleft lip and/or palate, and neural tube defects such as anencephaly or lumbo-sacral myelomeningocele. Most malformations involving only a single organ show multifactorial inheritance, implying an interaction of many genes with environmental factors.

Disruption The term disruption refers to an abnormal structure
of an organ or tissue as a result of external factors disturbing the normal developmental process. This used to be known as a secondary or extrinsic malformation.
Extrinsic factors which can disrupt normal development include ischaemia, infection and trauma. By definition a disruption is not genetic, although occasionalli genetic factors can predispose to disruptive events.

DeformationA deformation is a defect which results from
an abnormal mechanical force which distorts an otherwise normal structure.
Well recognized examples include dislocation of the hip and mild positional talipes (club feet)both of which can be caused by lack of amniotic fluid (oligohydramnios) or intra-uterine crowding due to twinnin or a structuraly abnormal uterus.

Dysplasia A dysplasia is an abnormal organization
of cells into tissue. Usually the effects are seen in all parts of the body in which that particular tissue is present. For example in a skeletal dysplasia such as thanatophoricdysplasia. Almost all parts of the skeleton are affected. Most dysplasias are caused by single gene defects and associated with high reccurence risks for siblings and/or offspring

Congenital structural abnormalities (major and minor anomalis)
A major anomaly is definet as one which has an adverse outcome on either the function or social acceptability of the individual. Minor abnormalities are of neither
medical nor cosmetic importance.

Minor abnormalities are found in approximately 10% of all newborn babies
If two or more minor abnormalities are present in a newborn infant then there is a risk of between 10% and 20% that the baby will also have a major malformation.

Examples of major congenital structural abnormalities
Ventricular and atrial septal defectsPatent ductus arteriosusFallot s tetralogyAnencephaly, hydrocephaly, microcephalyLumbo-sacral spina bifidaCleft lip/palateOesophageal atresia Imperforate anusBilateral renal agenesis

Examples of minor congenital structural abnormalities
Preauricular pit or tagEpicanthic foldsLacrimal duct stenosisBrushfield spots in the irisLip pitsSingle palmar creaseFifth finger clinodactylySyndactyly between second and third toes

The incidence of structural abnormalities
Incidence (%)Spontaneous miscurriagesFirst trimester 80-85 Second trimester 25All babiesMajor abnormality 4-5Minor abnormality 10

Causis of congenital abnormalitiesGenetic
Chromosomal 6%Single gene 7 %Multifactorial 20-30%
EnvironmentalDrags and chemicals 2%Infections 2%Maternal illness 1%Physical agents 5‐10%

The Skin-angiomas , teleangiectasia, venous network, pigment spots, depigmentation , hypertrichosis hirsutism,lipomas, fibroids, scars, , hyperkeratosis, .

Myotonic dystrophy
Atrophy of the muscles of the face
High foreheadHyperkeratosis

Vaardenburg syndrome
Partial albinism, areas of depigmenta-
tion of the skin.White strand of hair.Heterochromia of the
iris.Hearing loss
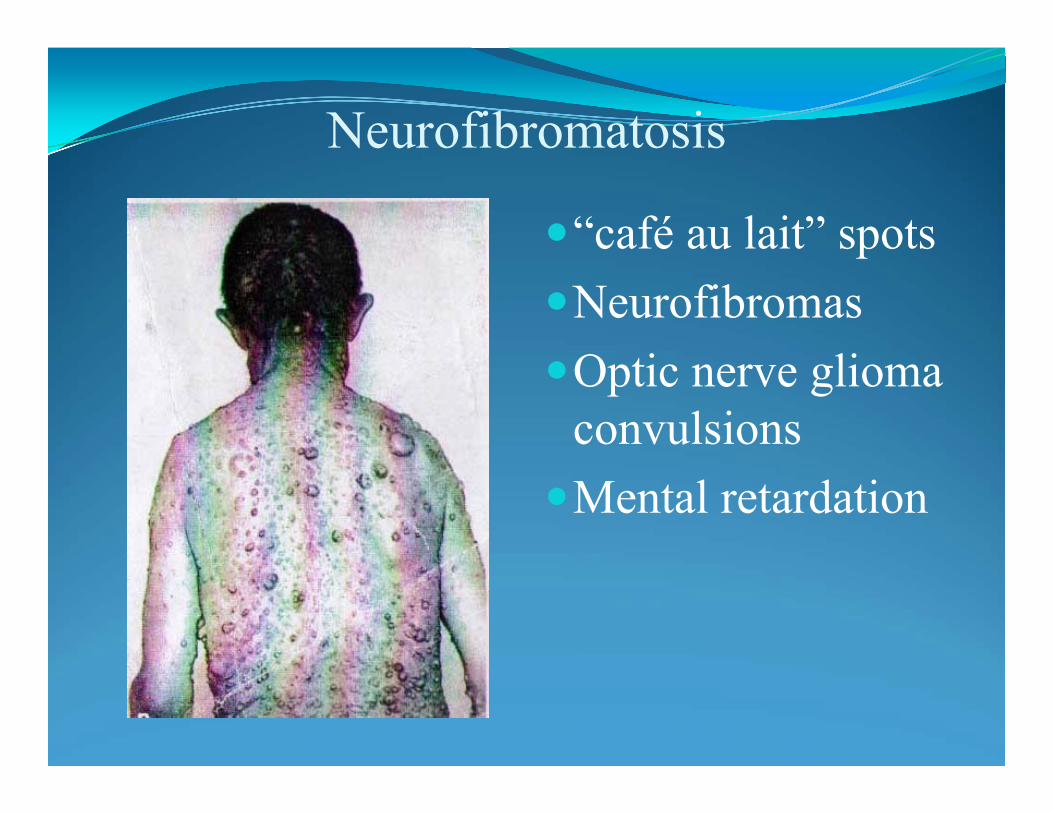
Neurofibromatosis
“café au lait” spotsNeurofibromasOptic nerve glioma
convulsionsMental retardation
The SkullHydrocephalus, microcephaly,
brachycephaly, dolichocephaly, trigonocephaly, flat occiput.
Mental retardationHight nasal bridgeOpen mouthStrabismusJoint hypermobilityMalformed low-set earsChange of teeth
Congenital structural abnormalities
Microtia, micrognathia, mikrostomiyaChange the pattern of the ear
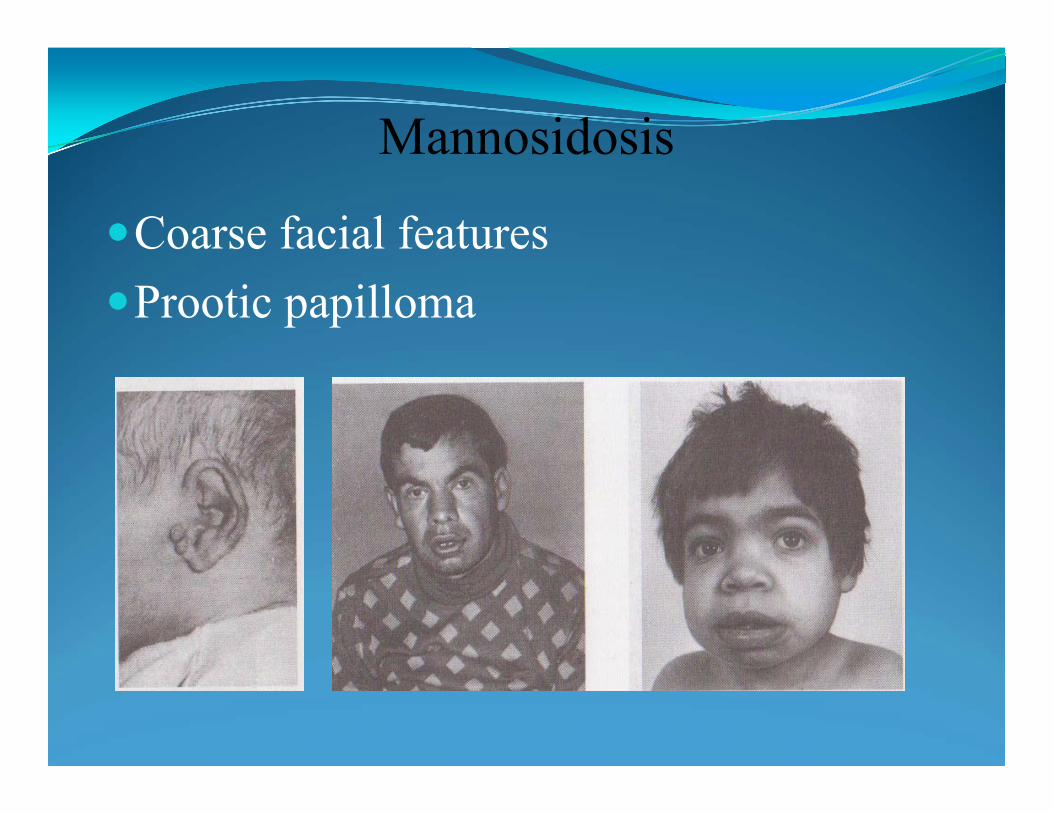
Mannosidosis
Coarse facial featuresProotic papilloma
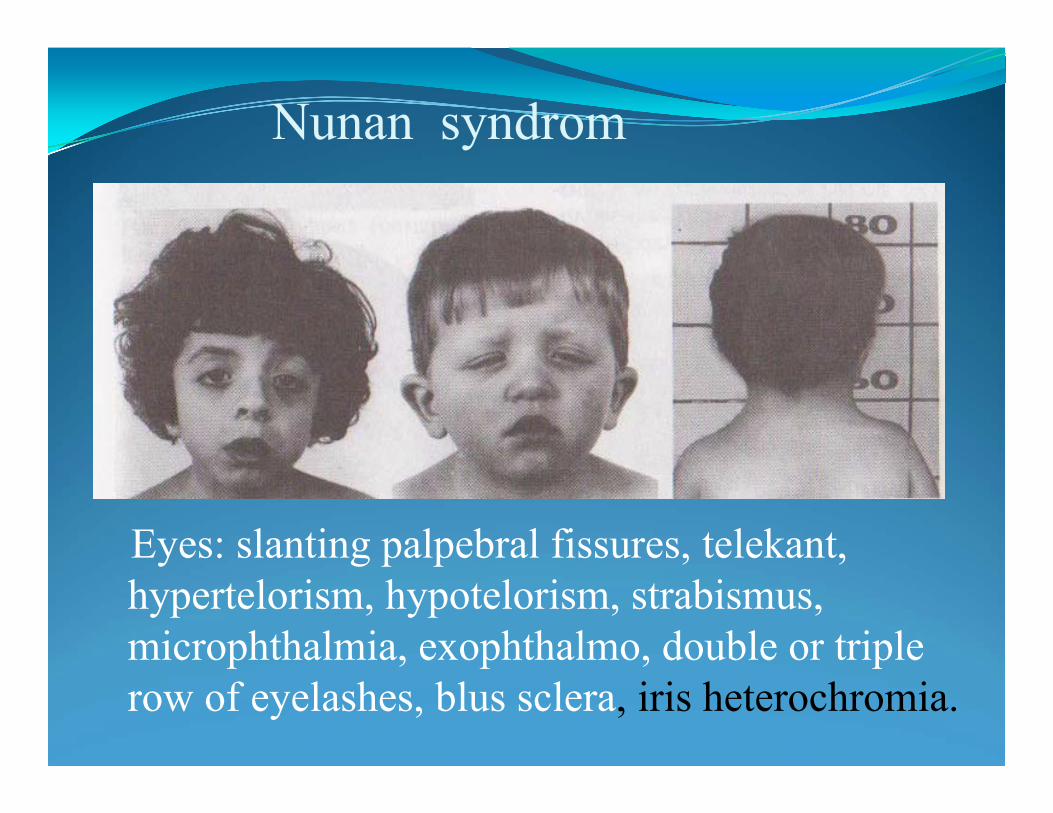
Nunan syndrom
Eyes: slanting palpebral fissures, telekant,hypertelorism, hypotelorism, strabismus, microphthalmia, exophthalmo, double or triple row of eyelashes, blus sclera, iris heterochromia.

The eyesHypertelorism
Slanting palpebralfissures
Flat nose
TelekantEpikant
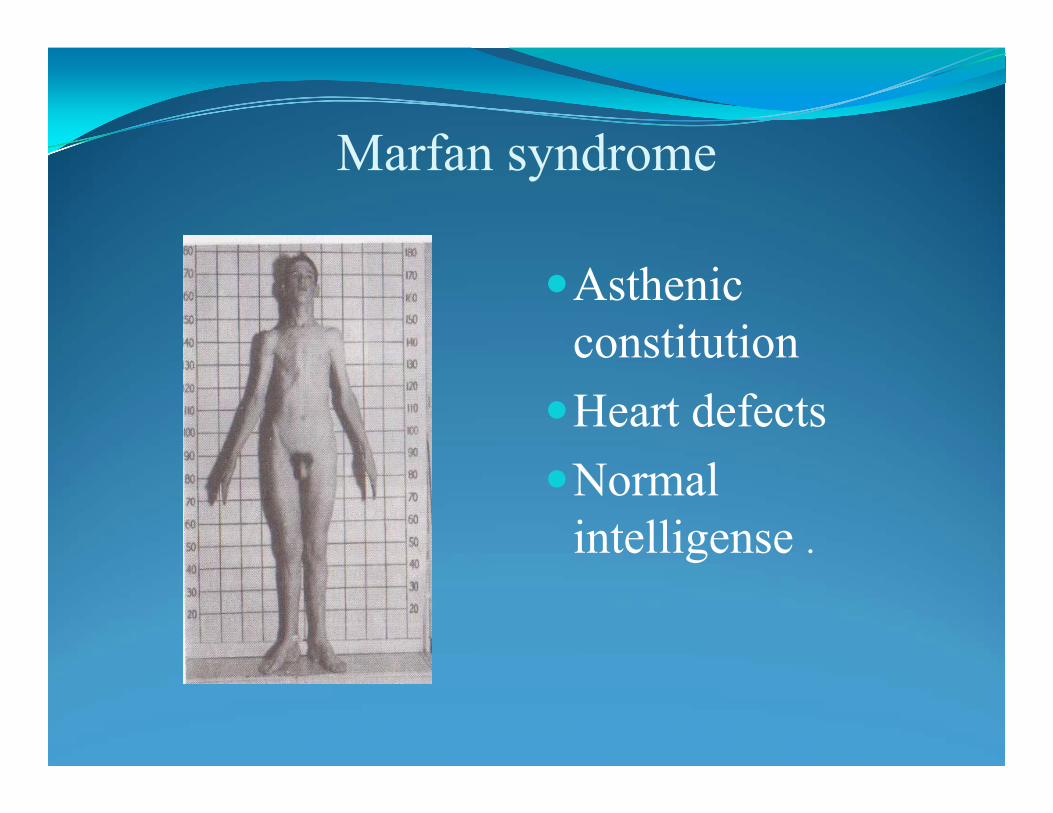
Marfan syndrome
AsthenicconstitutionHeart defectsNormal
intelligense .
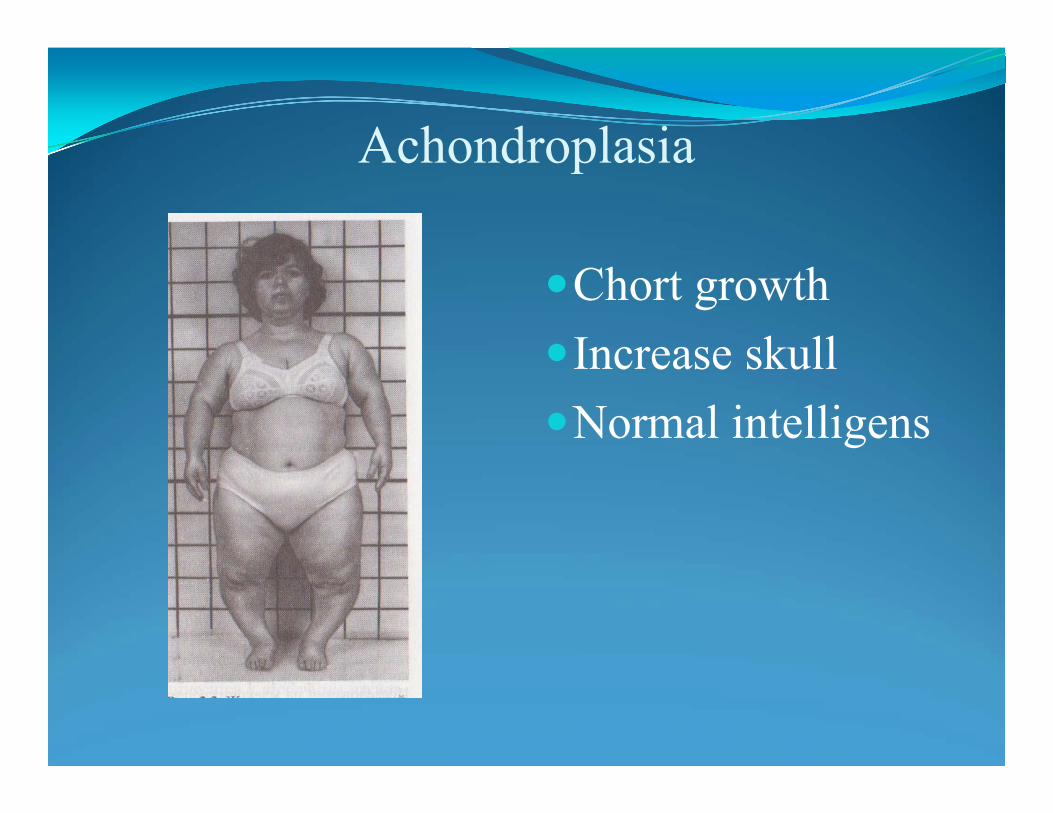
Achondroplasia
Chort growthIncrease skullNormal intelligens
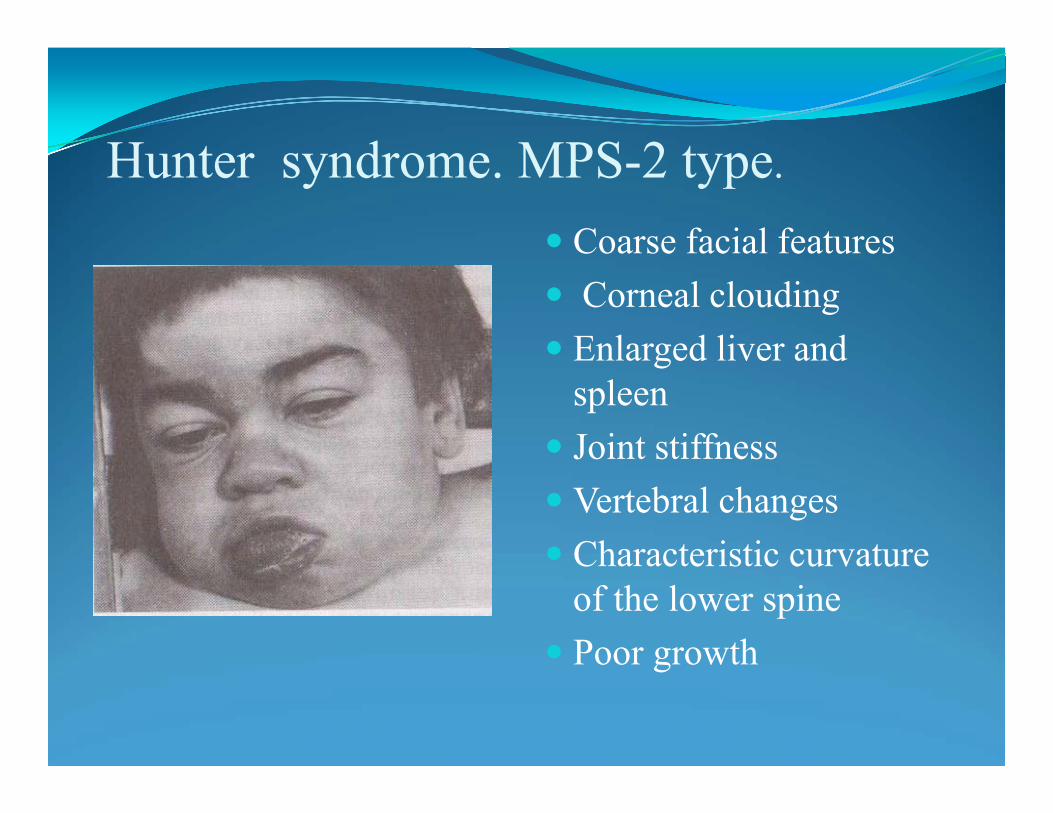
Hunter syndrome. MPS-2 type.
Coarse facial features Corneal clouding Enlarged liver and
spleen Joint stiffness Vertebral changes Characteristic curvature
of the lower spine Poor growth

Morquio syndrome. MPS 4 type. Sceletal abnormalities Chort body, neck and
limbs Hypermobility and
dislocations of joints Lumbar lordosis Heart defects Hearing loss, deafness Normal intelligence

Maroteaux-Lamy sindrome. MPS-6Coarse facial
featuresBone abnormalities Hearing lossCorneal cloudingNormal
intelligence
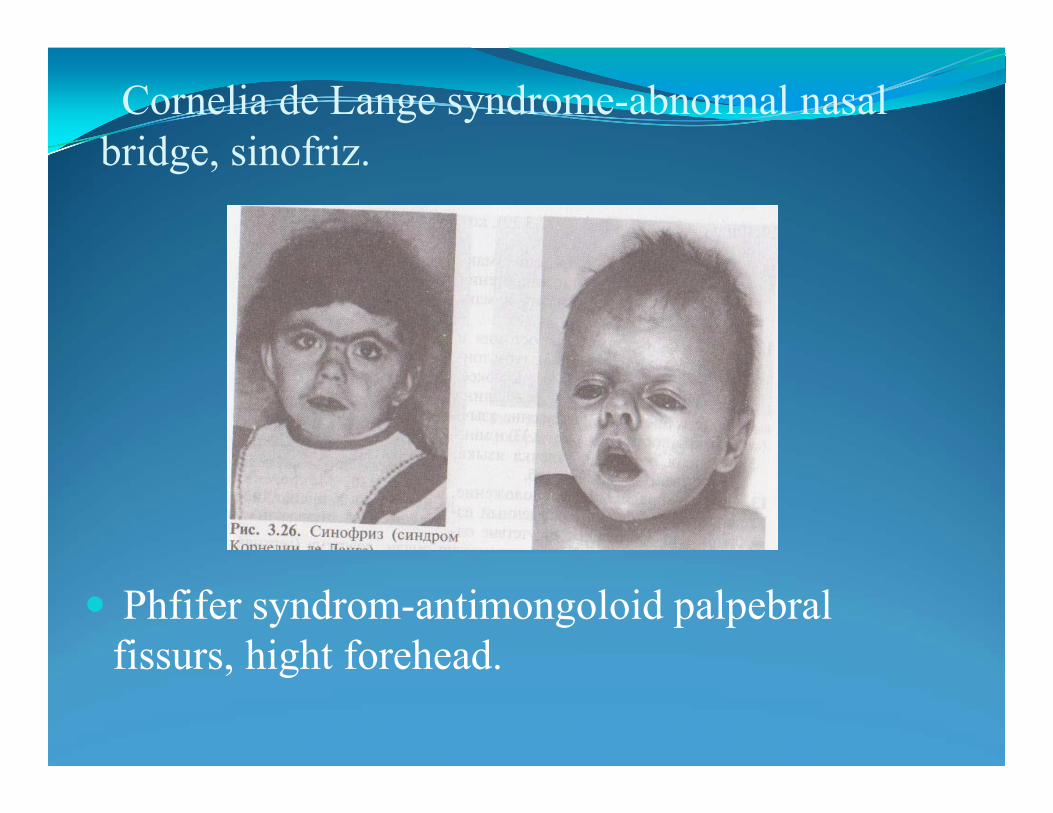
Cornelia de Lange syndrome-abnormal nasal bridge, sinofriz.
Phfifer syndrom-antimongoloid palpebralfissurs, hight forehead.

•Diabetic embryopathy
• Congenital heart disease
• Neural tube defects• Femoral hypoplasia• Sirenomelia
(“mermaidism

Aarskog syndrom
DwarfismHypertelorism, ptosisBrachydactylyBone changesCryptorchidismLow-set, malformed
ears
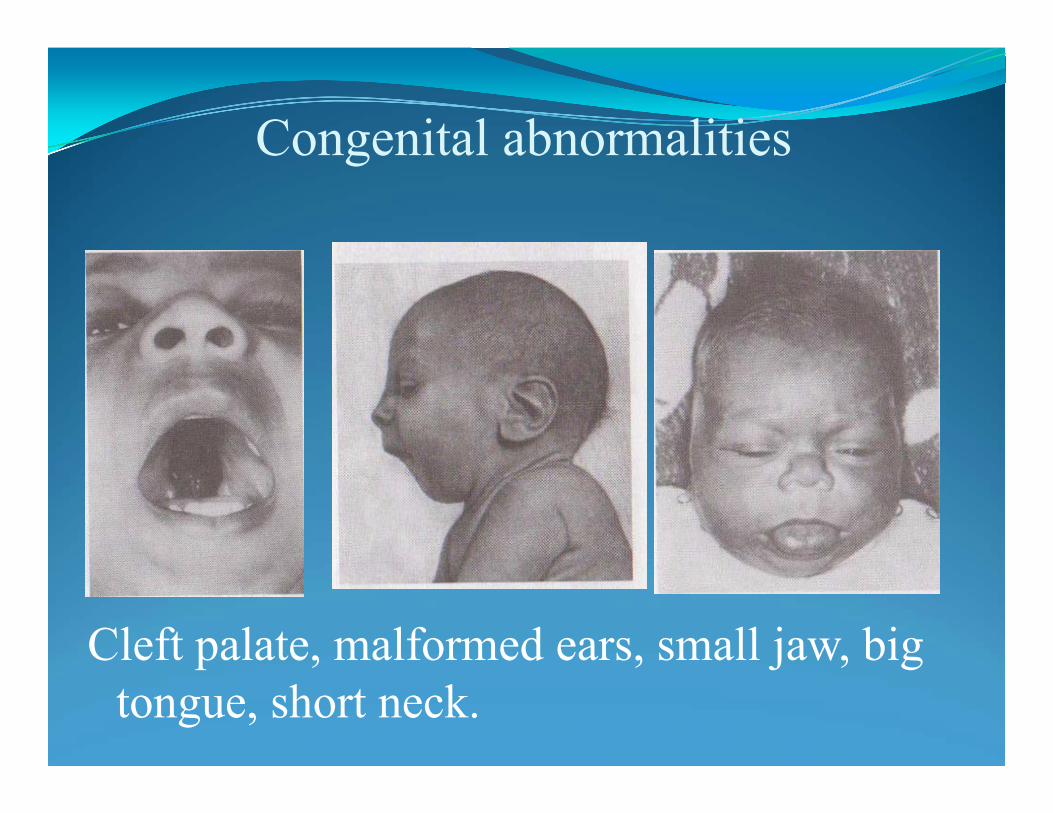
Congenital abnormalities
Cleft palate, malformed ears, small jaw, big tongue, short neck.
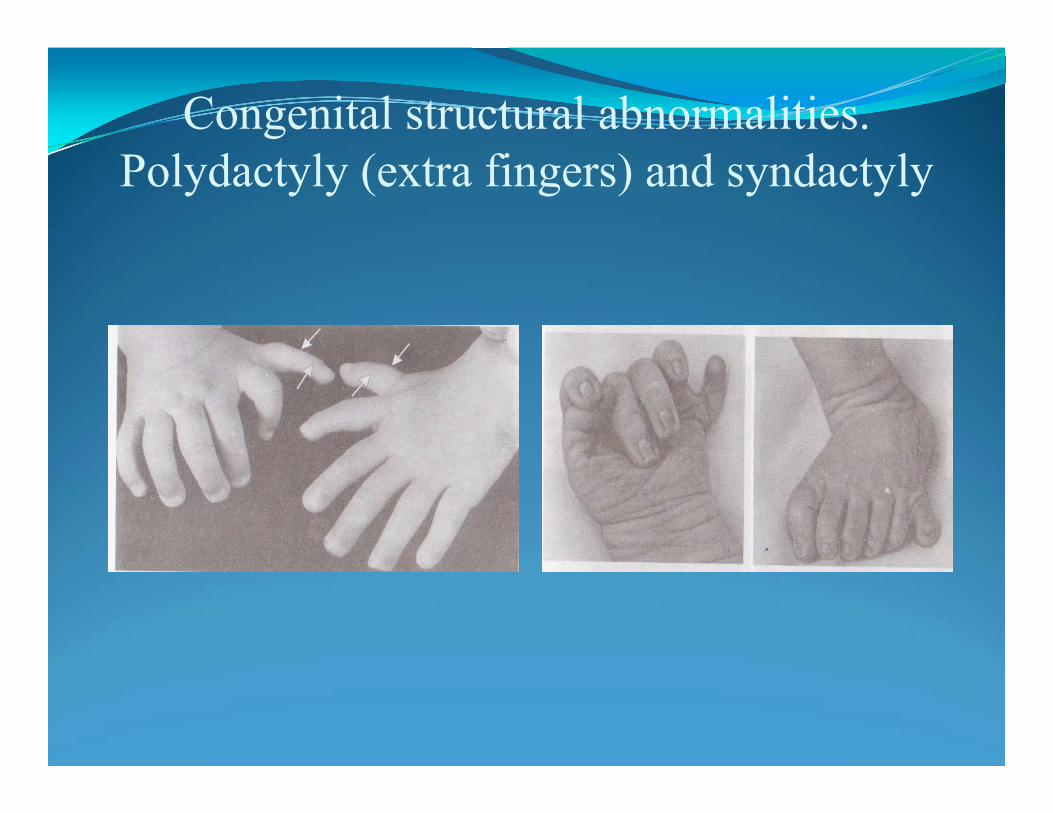
Congenital structural abnormalities. Polydactyly (extra fingers) and syndactyly
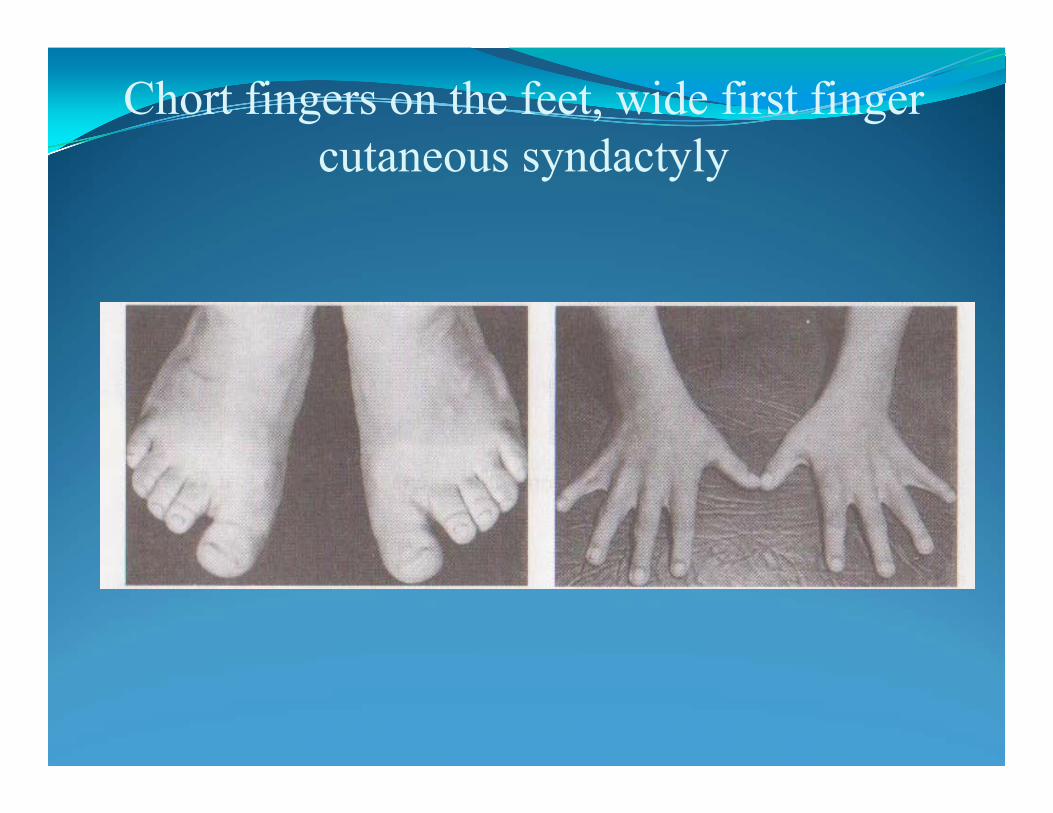
Chort fingers on the feet, wide first finger cutaneous syndactyly

Syndactyly
The hand of a person with Down syndrome

A child with thalidomide embryopathy. There is absence of the upper limbs (amelia) The lower limbs chow phocomelia and polydactyly.
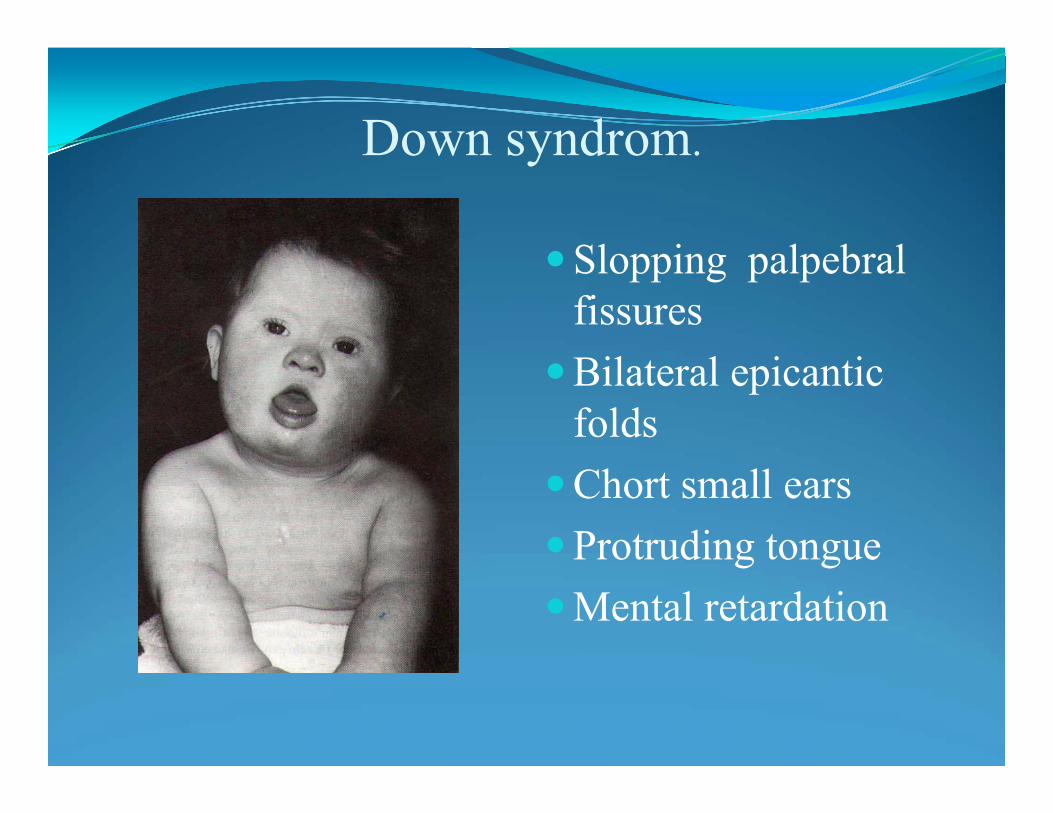
Down syndrom.
Slopping palpebralfissures
Bilateral epicanticfolds
Chort small ears Protruding tongueMental retardation
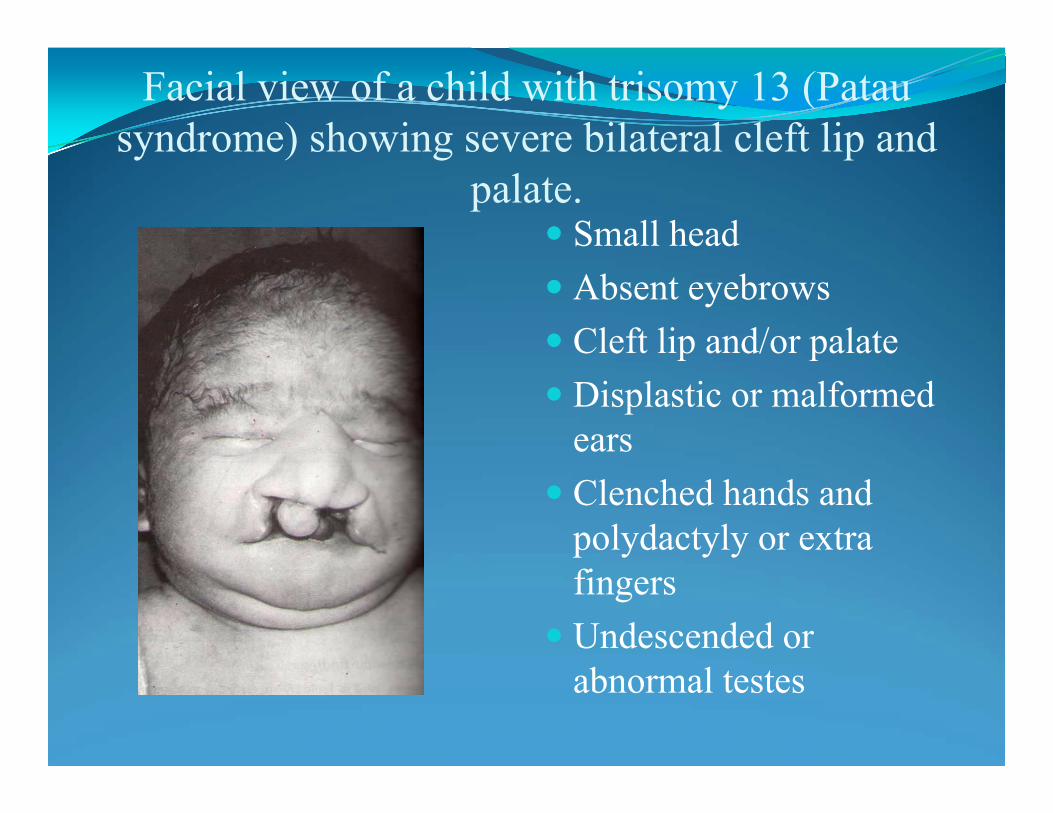
Facial view of a child with trisomy 13 (Patausyndrome) showing severe bilateral cleft lip and
palate. Small head Absent eyebrows Cleft lip and/or palate Displastic or malformed
ears Clenched hands and
polydactyly or extra fingers
Undescended or abnormal testes
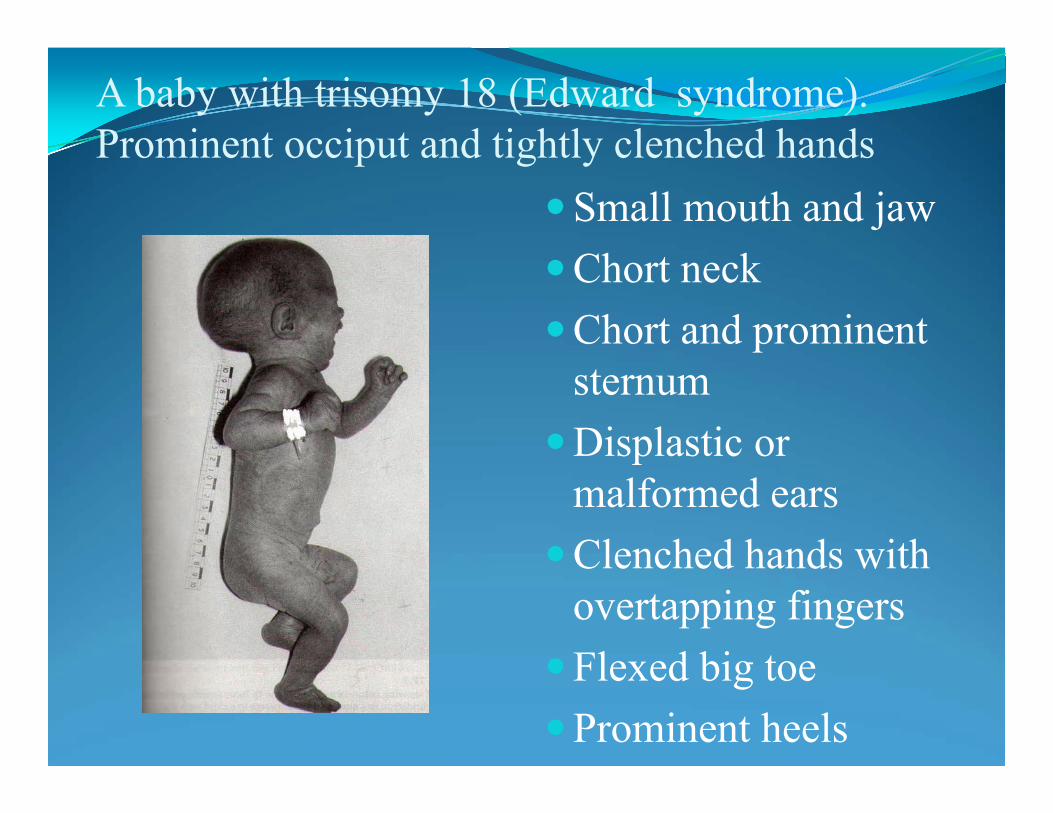
A baby with trisomy 18 (Edward syndrome). Prominent occiput and tightly clenched hands
Small mouth and jawChort neckChort and prominent
sternumDisplastic or
malformed earsClenched hands with
overtapping fingersFlexed big toeProminent heels

Facial view of a 2 year-old boy with the cri-du-chat syndrome (5p-).
Severe mental retardation.Characteristic cat-
like cry.

Facial view of a 3-year old child with the Wolf-Hirchhorn syndrome (4p-)
Mental retardationBone abnormalitiesRenal patologyHeart defectsDelay physical
developmentConvulchons
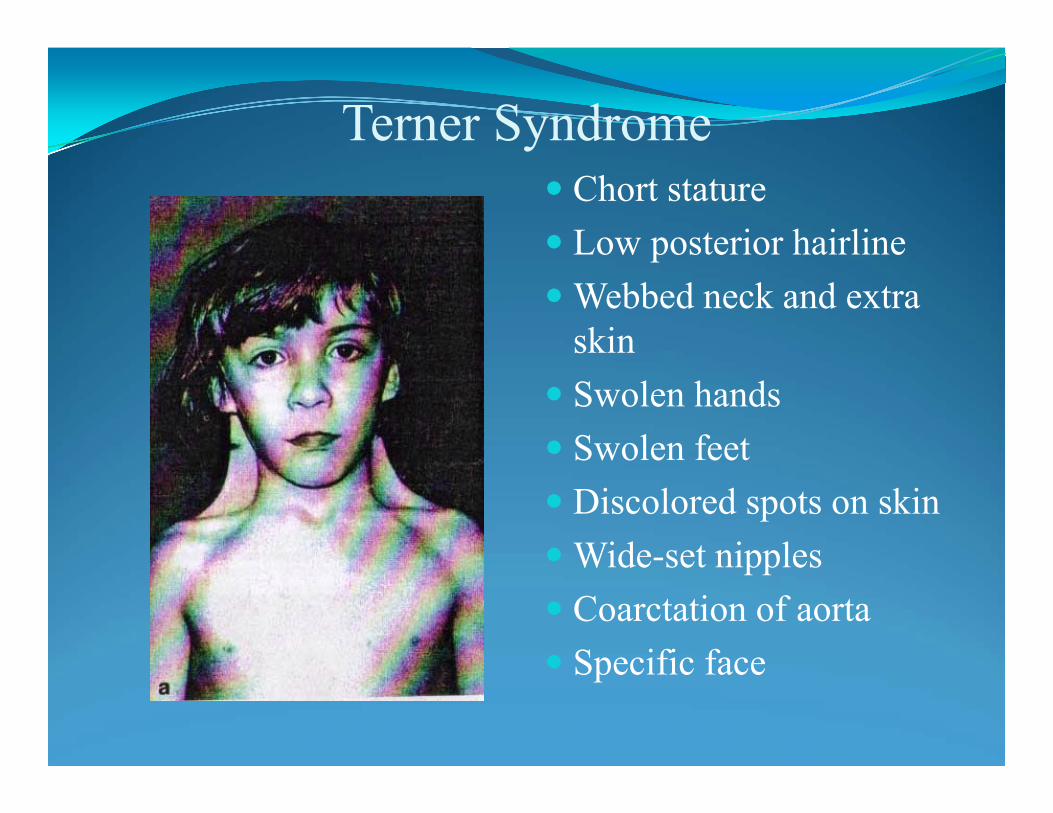
Terner Syndrome Chort stature Low posterior hairline Webbed neck and extra
skin Swolen hands Swolen feet Discolored spots on skin Wide-set nipples Coarctation of aorta Specific face
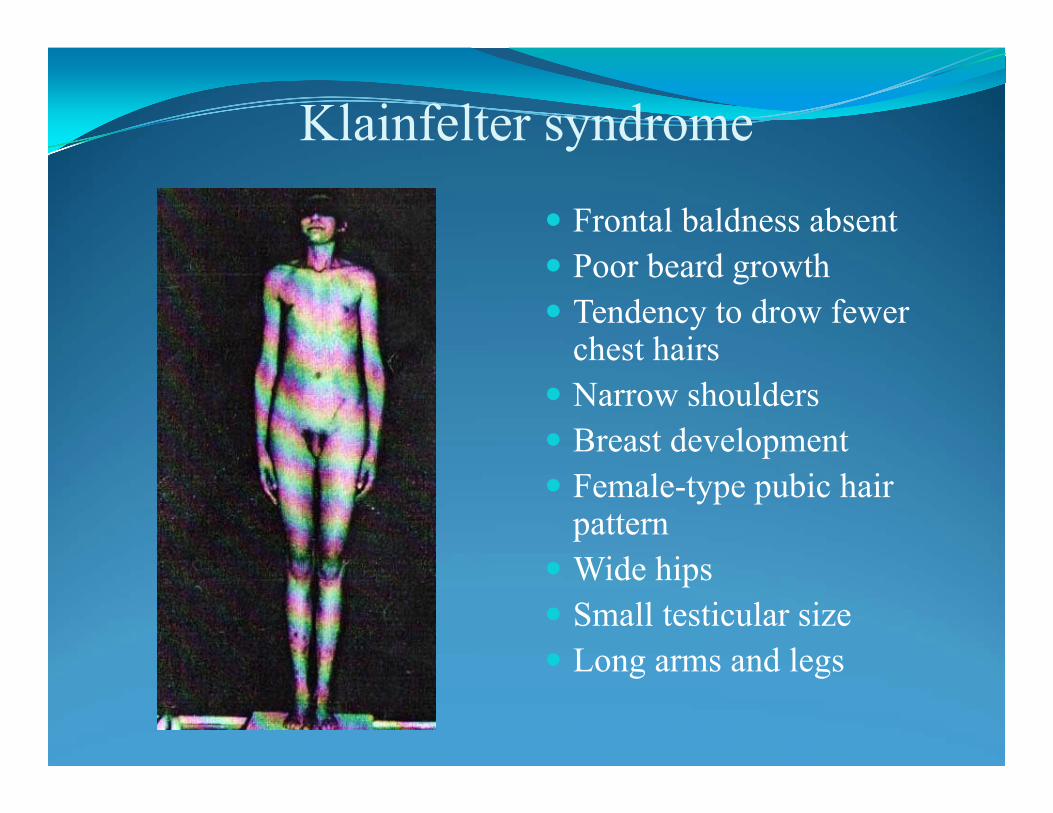
Klainfelter syndrome
Frontal baldness absent Poor beard growth Tendency to drow fewer
chest hairs Narrow shoulders Breast development Female-type pubic hair
pattern Wide hips Small testicular size Long arms and legs
The Skull
The Skull

Oral cavity

Pathology of eye
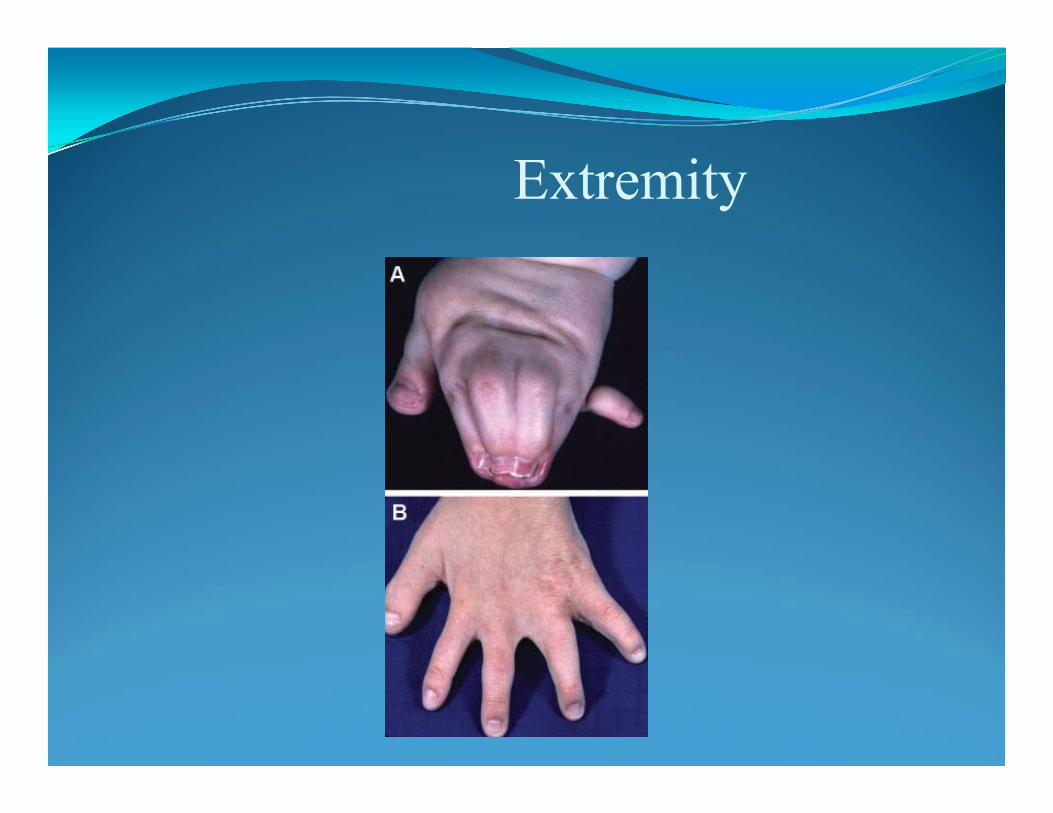
Extremity
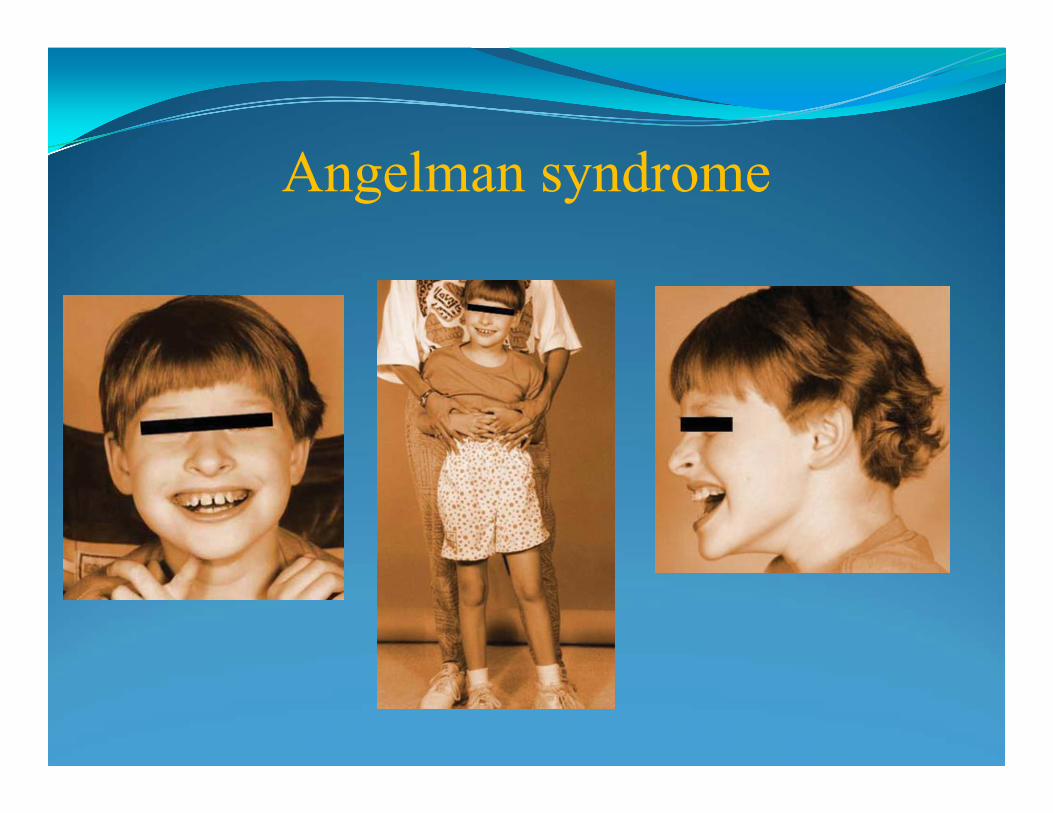
Angelman syndrome

Congenital aplasia of the skin
Thelangectazia

Beckwith-Wiedemann syndrome
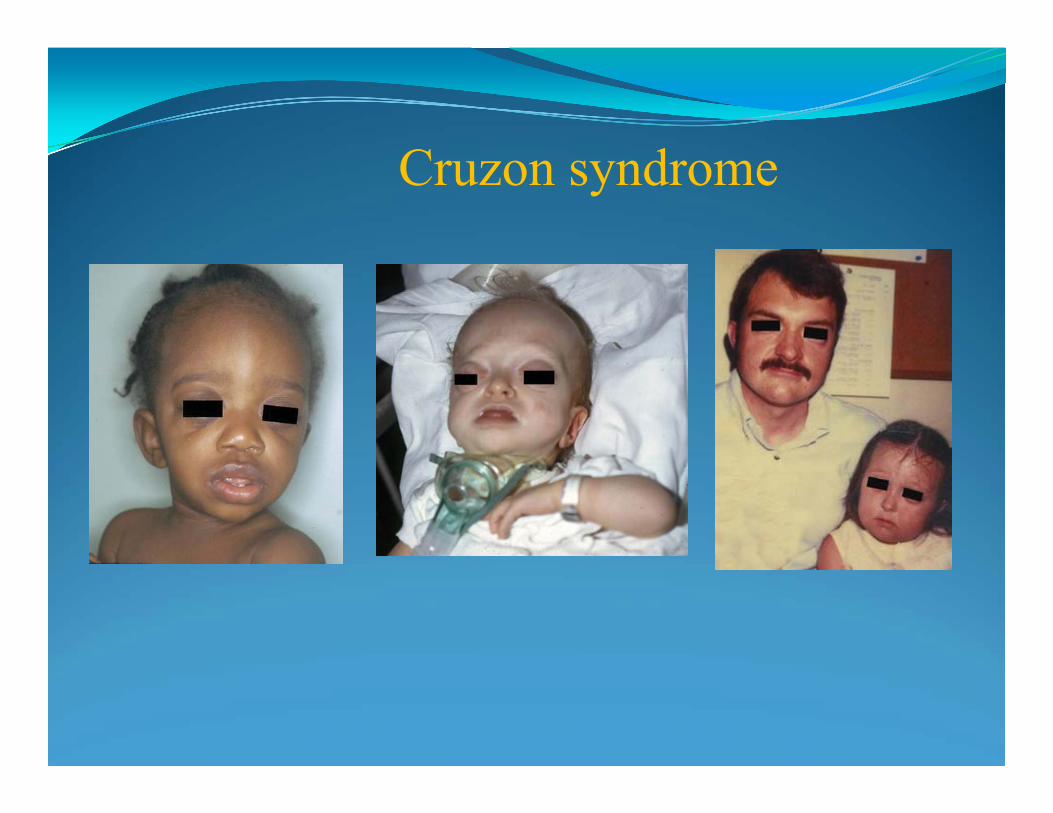
Cruzon syndrome
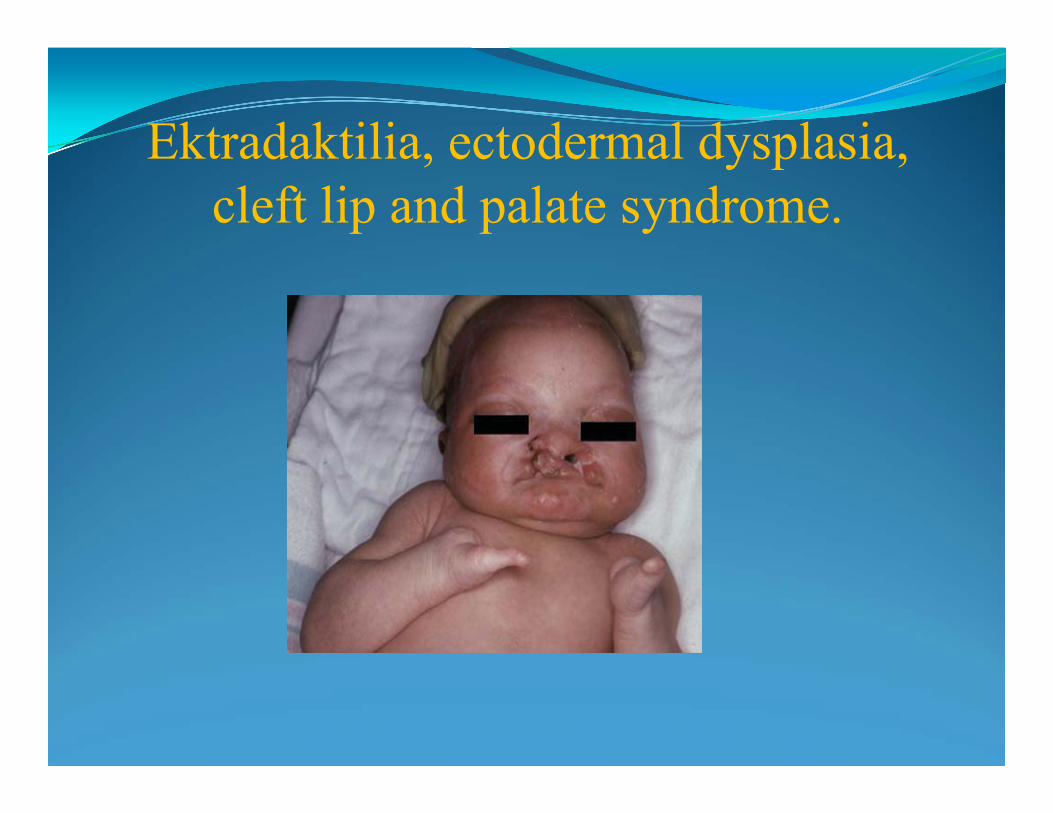
Ektradaktilia, ectodermal dysplasia,cleft lip and palate syndrome.
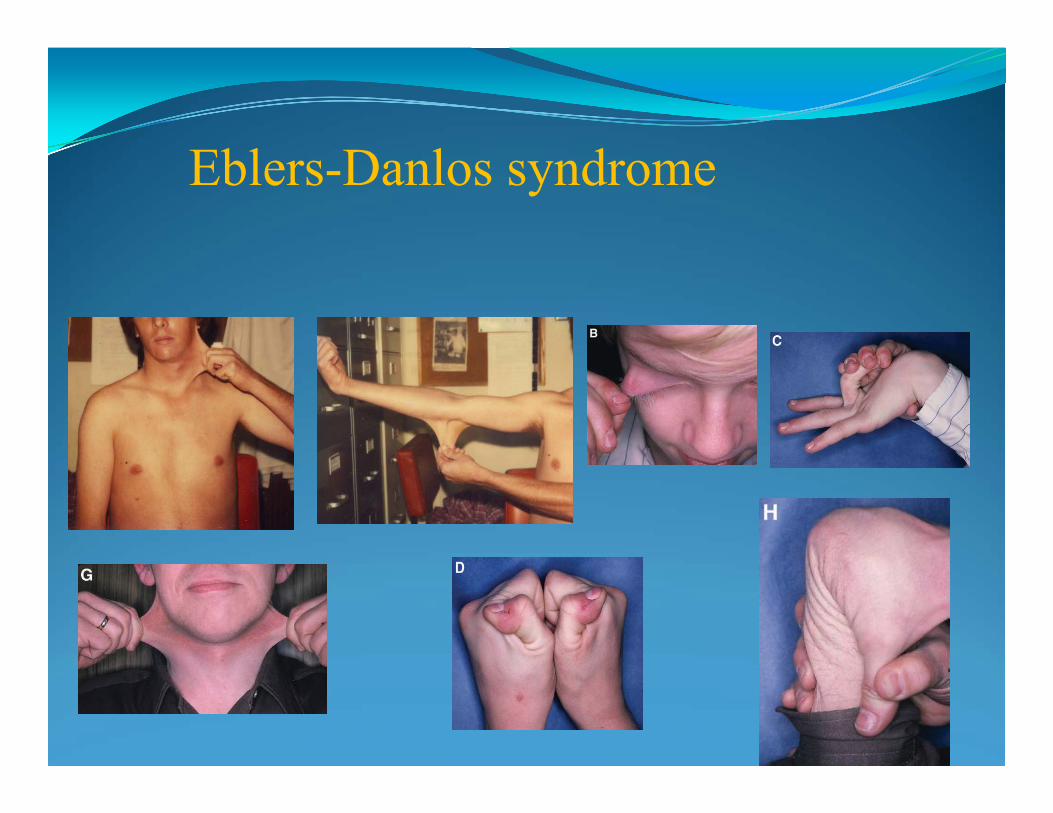
Eblers-Danlos syndrome

X-chromosome fragile syndrome
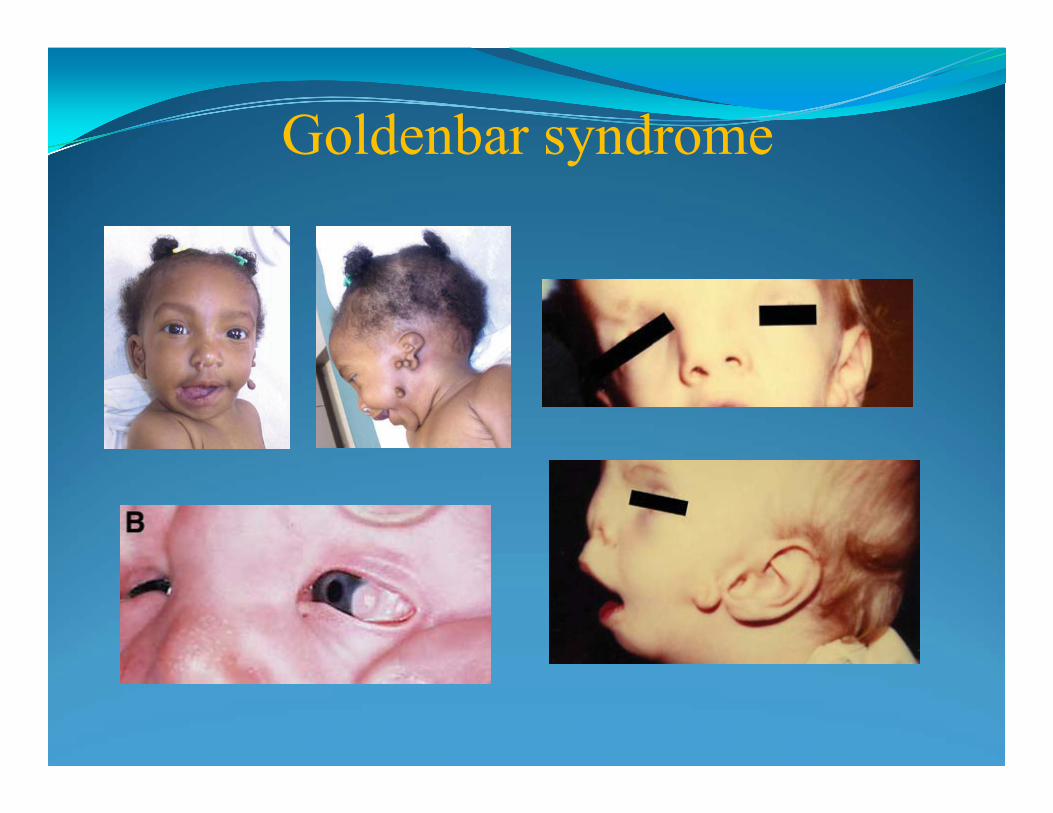
Goldenbar syndrome

Holt-Oram syndrome
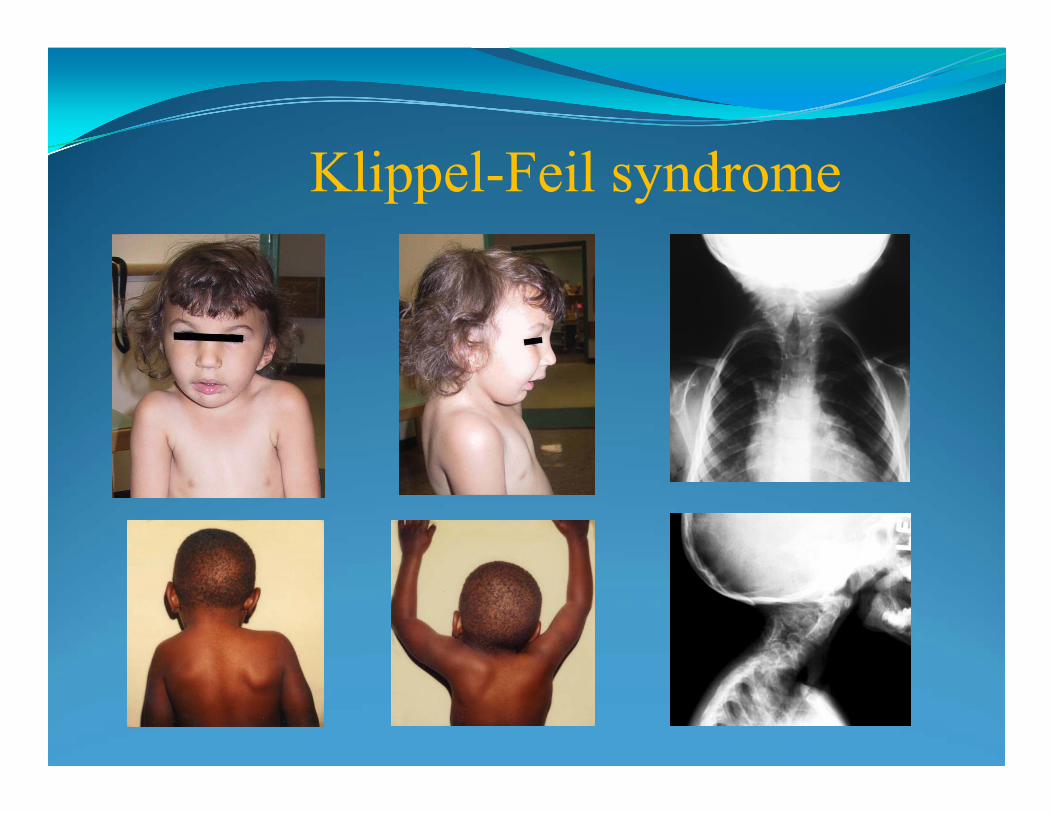
Klippel-Feil syndrome
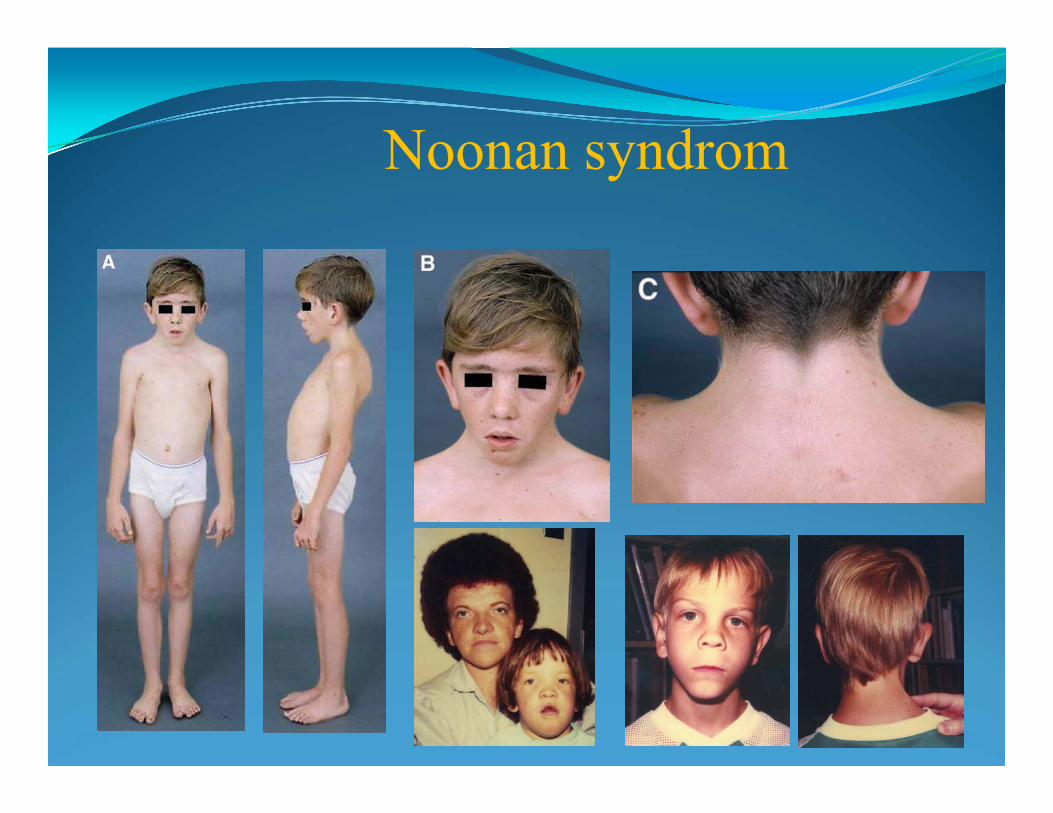
Noonan syndrom
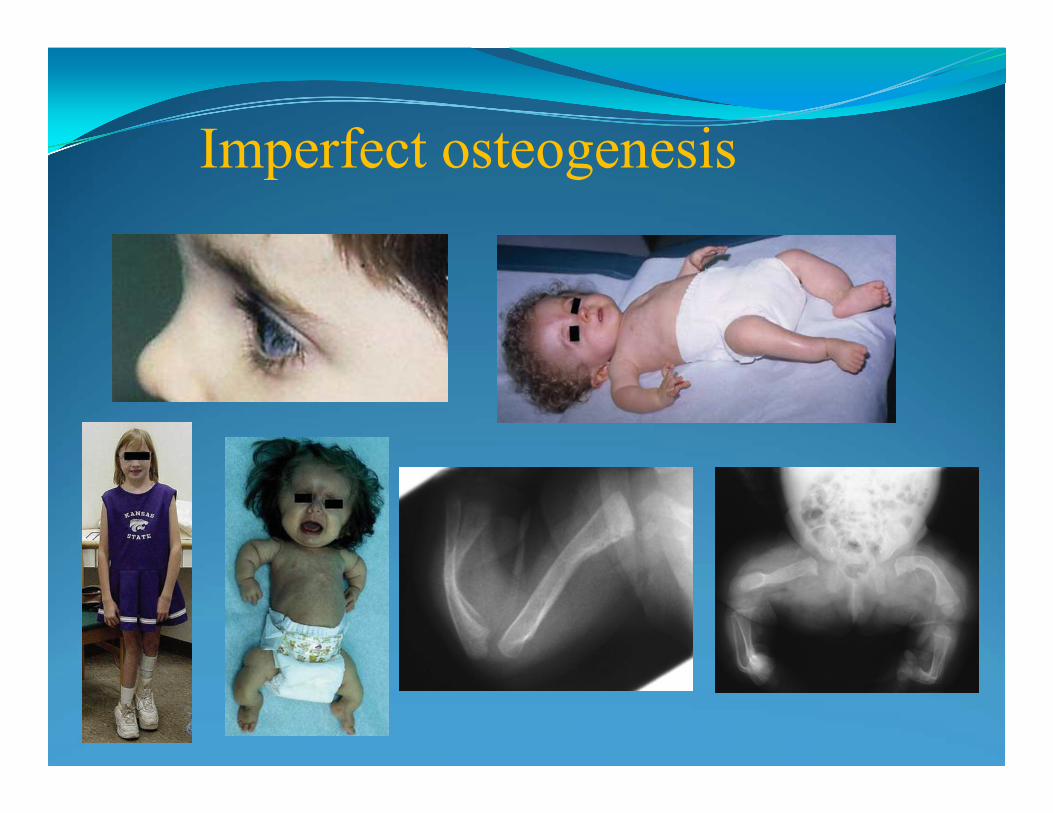
Imperfect osteogenesis
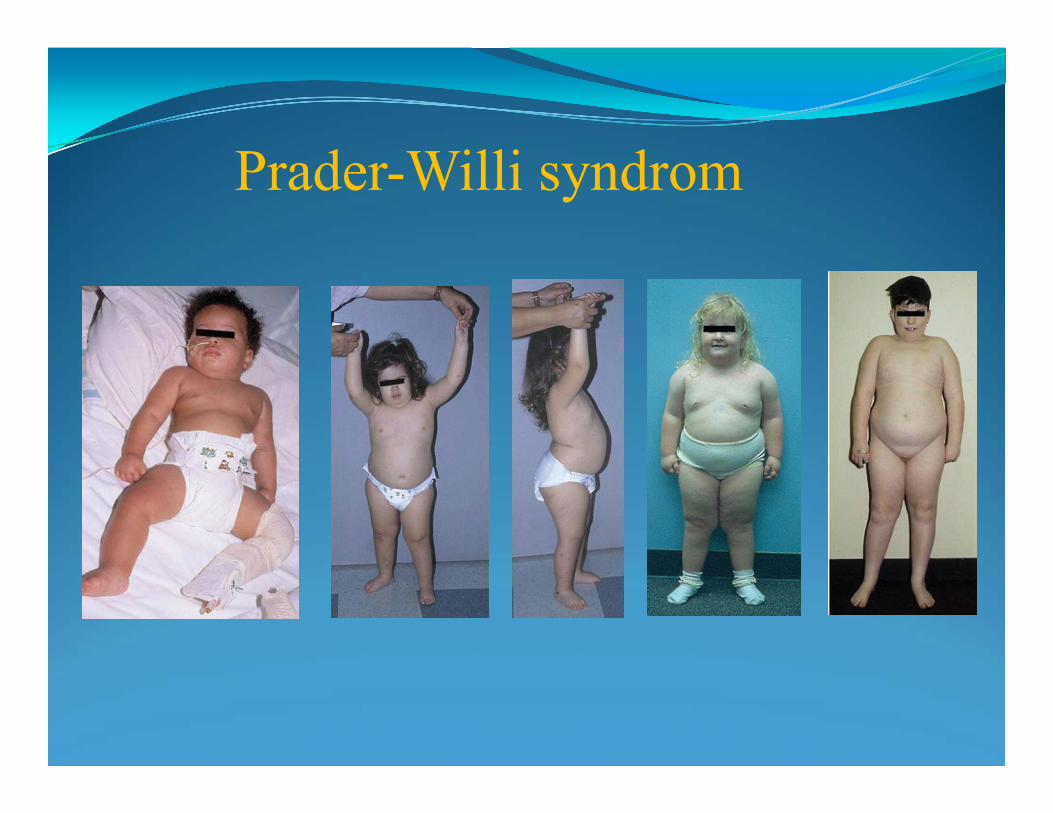
Prader-Willi syndrom
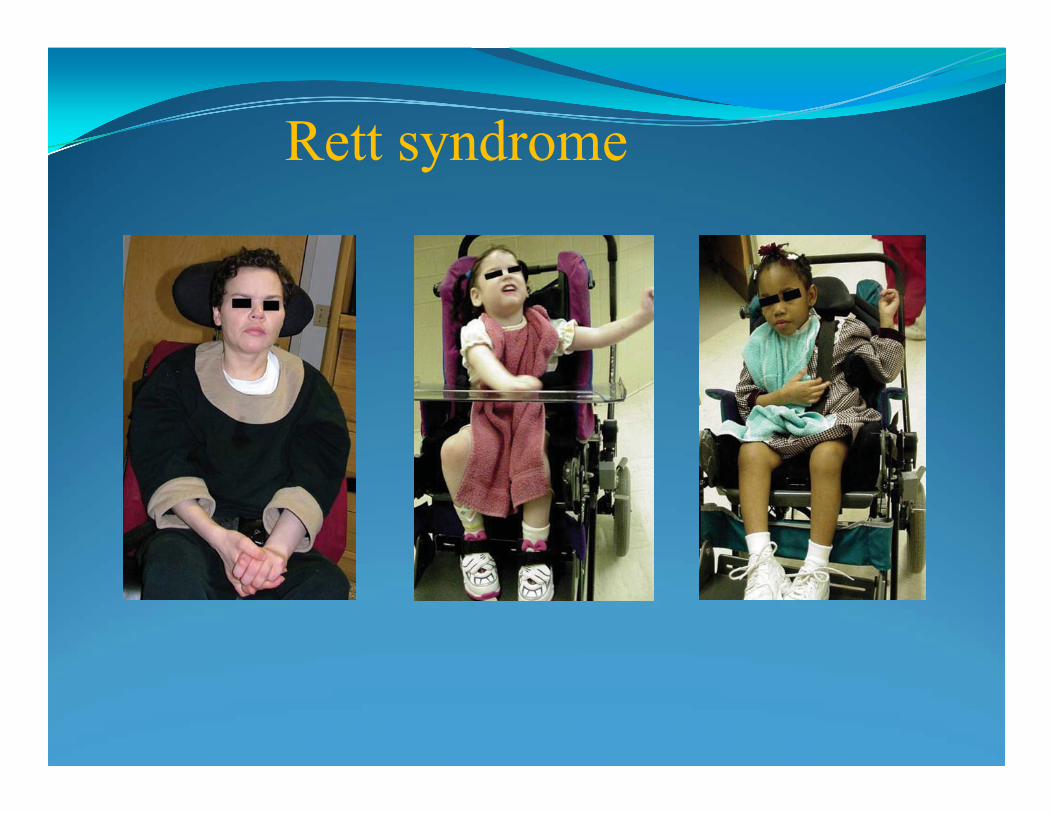
Rett syndrome
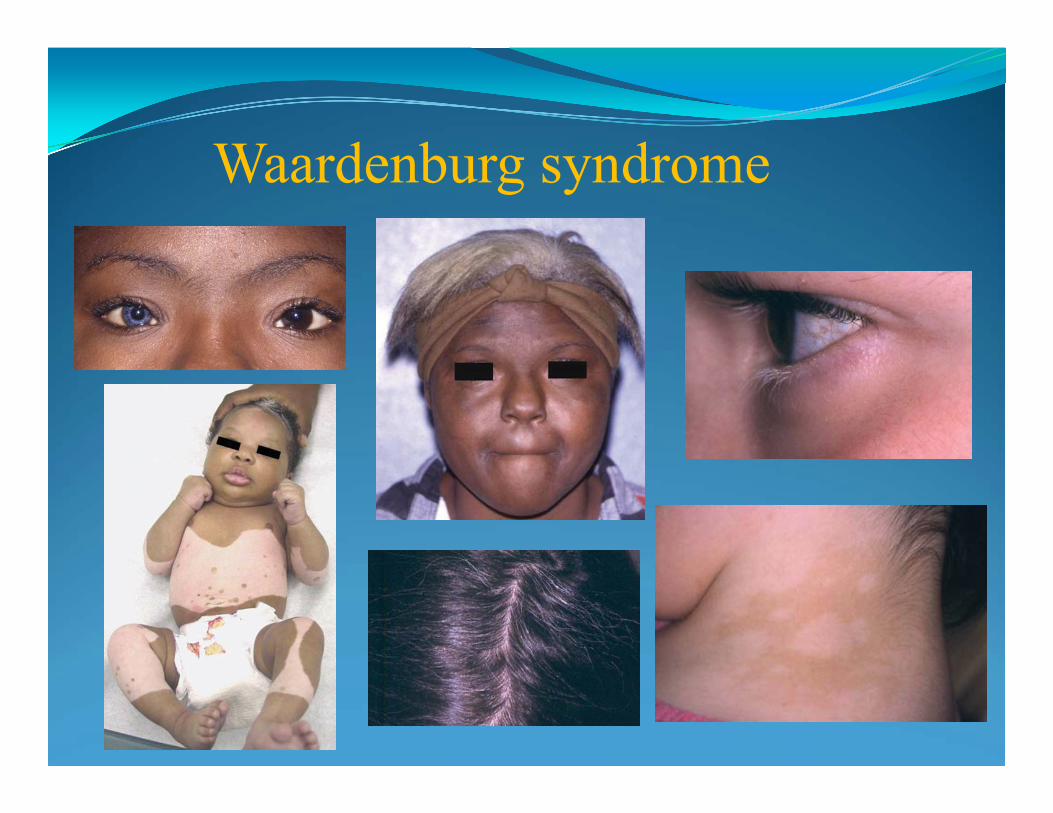
Waardenburg syndrome
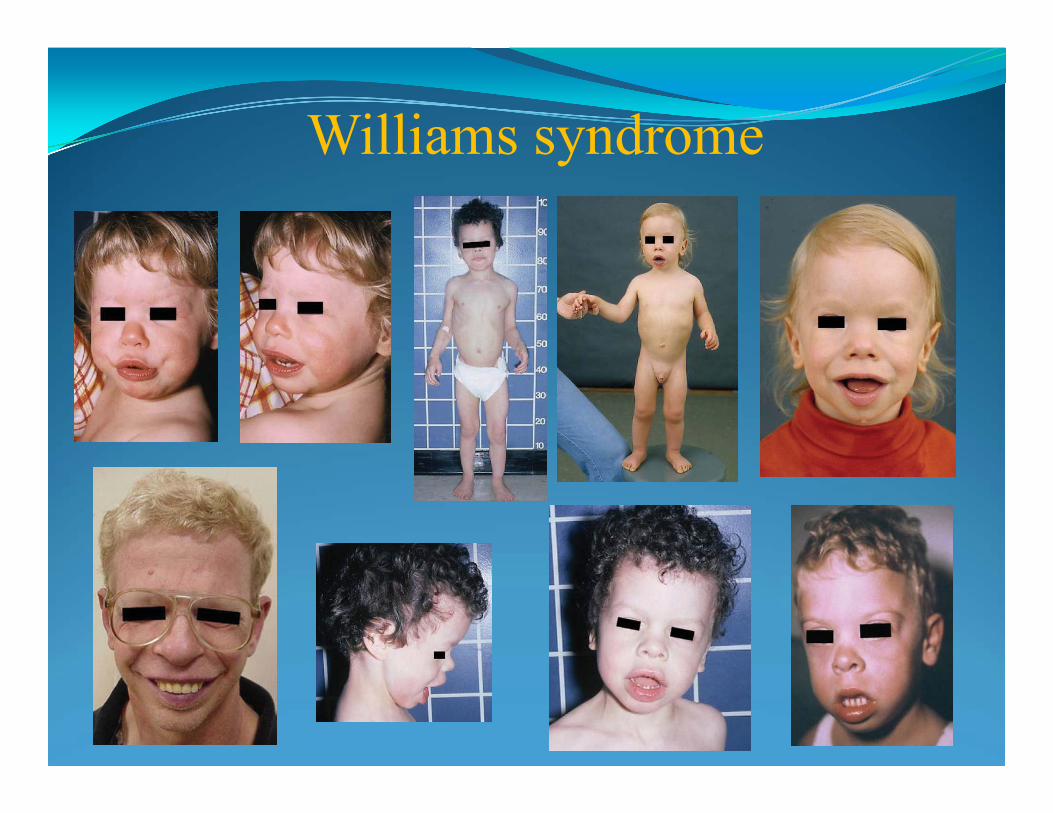
Williams syndrome

PREVENTION OF GENETIC DISORDERS, PRENANATAL DIAGNOSTICS AND
GENETIC SCREENING PROGRAMES
HowGeneticTestingCanSaveChild'sLife

Clinical genetic service see their role as providing support to families suffering from genetic disease in the
way of prevention of symptoms developing or prenatal diagnostics
and birth prevention.

Genetic counselling is an integral part
of the management of patients and families
with genetic disorders

Genetic Counseling Contexts Reproductive Issues
Preconception counseling
Prenatal
Infertility
Pediatrics Newborn Screening
Specialty Clinics
Adult-Onset conditions Specialty Clinics
Pre-symptomatic testing: Breast and Colon Cancer, Huntington’s disease

Principles of CounselingPre-screening, counseling should include:
Range of symptoms/severity of each disease Review of family and medical history Diagnosis of the disease Pattern of inheritance and genetic risk assessment Risk of carrier status & affect on offspring Meaning of positive and negative results Factors to consider in decision-making Further testing necessary for prenatal diagnosis Screening programs

Types of Genetic Screening
Carrier testing can reduce the frequency of homozygous and double heterozygous states
Prenatal diagnosis can reduce the frequency of birth defects, chromosomal abnormality and gene homozygousity in fetal
Newborn screening can reduce symptoms manifestation in a child

Preventiveservicesingeneticscanbedividedintoprimary,secondaryandtertiary levels.
STAGES OF GENETIC DISORDERS PREVENTION

Primary prevention of genetic diseases:
• Carrier detection
• Premarital
• Preconceptional primary prevention
All women of childbearing age can take steps even before conception to improve their chances of having a healthy baby and minimize the incidence of neural tube defects in newborn. These include: taking the B vitamin and folic acid daily; adopting a healthy lifestyle;
Preimplantaion diagnostics
The advent of human in vitro fertilization and developments in molecular science have made it possible to diagnose genetic disease in fetuses at a very early stage of development (before implantation) and getting a pre-pregnancy health checkup

Secondary - addressed to prevention of a sick child birth with chromosomal abnormalities via prenatal diagnosis and abortion

“Screening & invasive diagnostic testing for aneuploidy should be available to all women who present for prenatal care before 20 wks of gestation regardless of age”
Women should be counseled regarding the differences between screening and invasive diagnostic testing.”

Primary and secondary levels of prevention are the intrauterine selection оf the defective embryos
Prenatal diagnostics is effective way for the genetic disorders prevention:
• non-direct (non- invasive) -
maternal serum screening
(first and second trimester of pregnancy)
• direct (invasive) – chorion biopsy and amniocentesis
(first and second trimester of pregnancy)
• US examination

Preimplantationgenetic diagnosis
Chorion villisampling
cordocentesis
amniocentesis
Prenatal diagnosis

METHODS FOR PRENATAL DIAGNOSISI trimester
Non-invasive methods: Ultrasound detection - nuchal translucency thickness, depressed nasal bridge|Maternal serum screening - PAPP-A, beta hCG,
to identify pregnancy with higher risks
of Downs syndrome. Invasive method: transcervical chorion villous byopsy for fetal karyotype study or molecular genetic testing
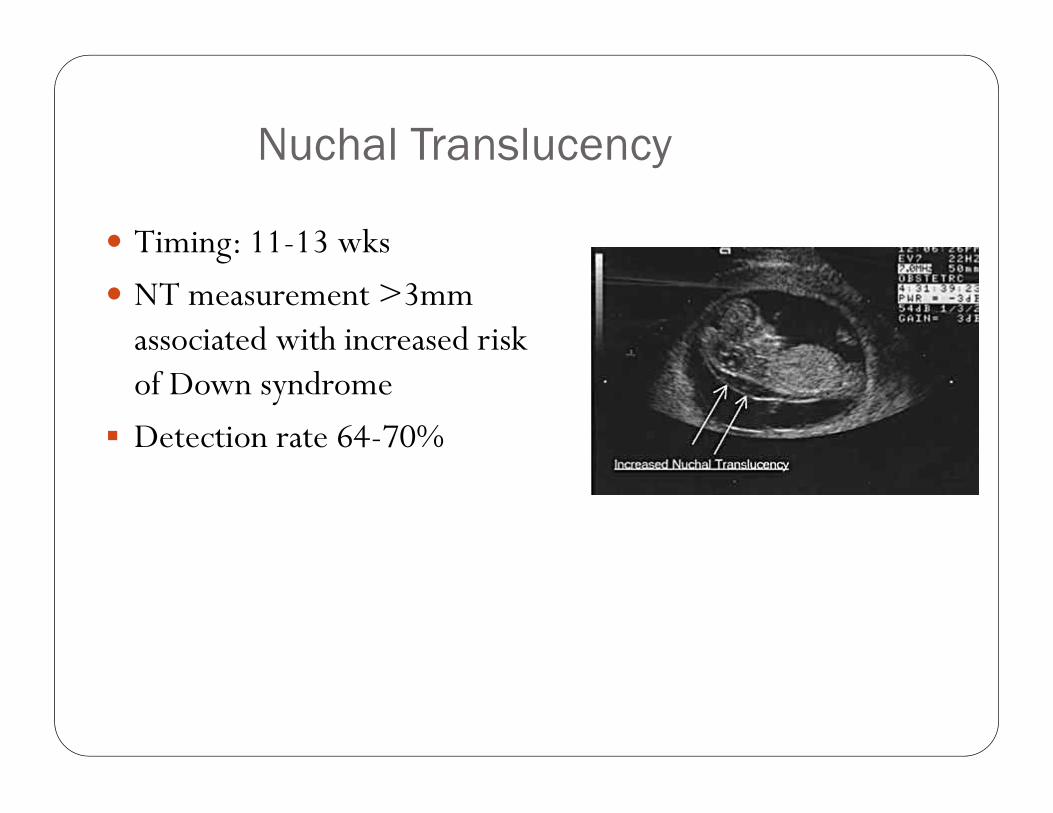
Nuchal Translucency
Timing: 11-13 wks
NT measurement >3mm associated with increased risk of Down syndrome
Detection rate 64-70%

METHODS FOR PRENATAL DIAGNOSIS2st Trimester
Screening - non-invasive methods: Alpha-fetoprotein (AFP) Unconjugated estriol (uE3) Beta-human chorionic gonadotropin (b-HCG)
Detection rates Trisomy 21: 75-80% Trisomy 18: 60 % NTD: 75-80 %
Diagnostics - invasive method: Amniocentesis for karyotype study, biochemical and DNA testing.
Results – in 3 days by FISH for 21,13,18 and X/Y chromosomes in uncultured amniocytes and in 3-4 weeks for karyotyping

WHAT IS AFP?
AFP is a glycoprotein of 590 amino acids
is normally produced by the fetal yolk sac, the fetal gastrointestinal tract
Elevated levels are seen in multiple gestation as well as in a neural tube defects including spina bifida, anencephaly, and abdominal wall defects.
Low levels of maternal serum AFP are associated with Down syndrome and trisomy 18., fetal death
Genetic Ultrasound Fetal anatomy screen -Timing: 18-
20 wks
• Evaluate for major structural anomalies and minor markers hydrops,oligohydramnios,intrauterine growth retardation, central nerbvous system disorders, craniofacial disorders,chestdisorders, abdomen/pelvis disorders, skeletal system disorders

Ultrasound finding
May/may not be associated with chromosome condition or known genetic condition
Offered: Amniocentesis (possibly CVS)

Recommendations
Combined 1st trimester screening is an effective screening test, better than NT alone
Women with positive first trimester screens should be offered counseling and an option of CVS (chorion villus sampling) or 2nd trimester amniocentesis
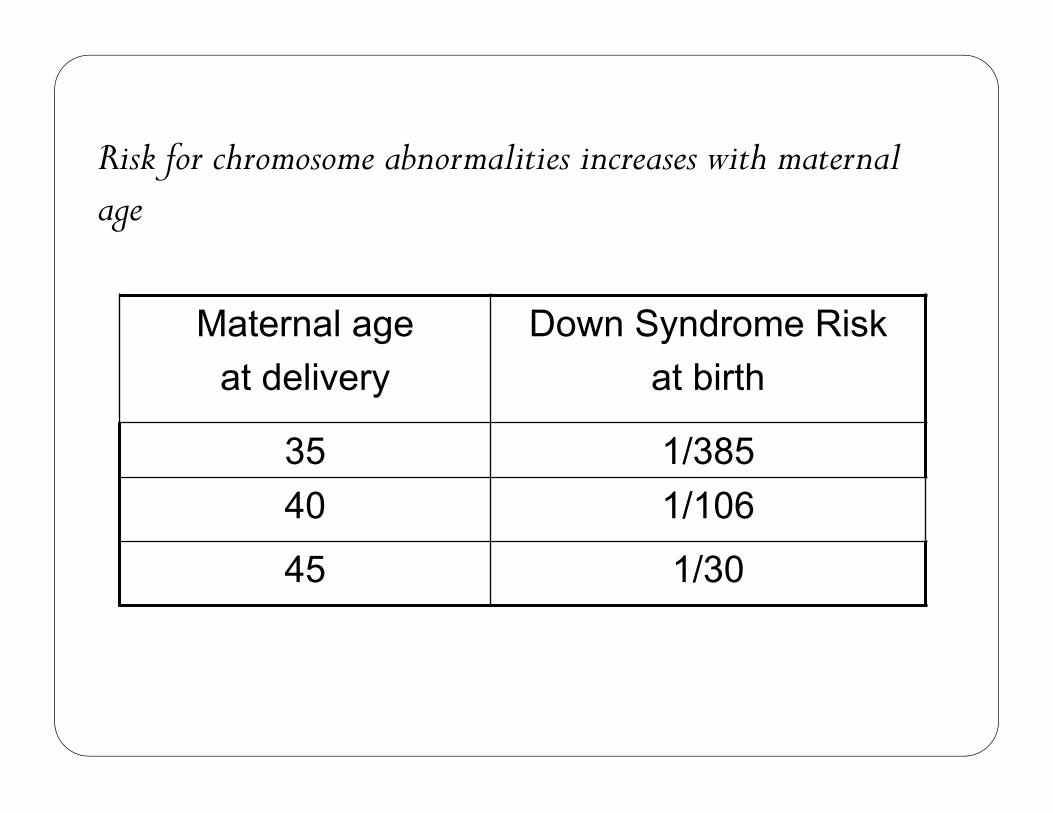
Risk for chromosome abnormalities increases with maternal age
Maternal age at delivery
Down Syndrome Risk at birth
35 1/38540 1/106
45 1/30

Tertiary level of prevention is aimed at the phenotypic correction of mutant genes clinical manifestation via:

REQUIREMENTS FORNewborn genetic screening:
•Gene is identified and pathogenesis isunderstood•Disease is accompanied with severephenotype•Disease incidence mist not be low than1:10000•Treatment strategy for the disease isdeveloped

Newborn genetic screening
Prevention of Congenital Hypothyroidism with hormone replacement therapy Phenylketonuria screening with dietary restriction of phenylalanine Congenital adrenal hyperplasia Galactosemia

Genetics of Familial Mediterranean Fever (FMF)• FMF is the prototypic and probably most common hereditary periodic fever
(HPF) syndrome, also known as recurrent hereditary polyserositis.• HPFs are a group of autoinflammatory disorders characterized by seemingly
unprovoked fever and localized inflammation in lack of high-titer autoantibodies antigen-specific T-cells• FMF is a recessively-inherited disorder that affects people of Mediterranean
ancestry, especially Armenians, North African Jews, Arabs, and Turks.• FMF classically presents in childhood or adolescence with 1-3 day episodes of
fever often accompanied by severe abdominal pain, pleurisy, monoarticular arthritis, or an erythematous rash on the ankle or foot known as erysipeloid erythema. The symptoms vary among affected individuals, sometimes even among members of the same family. Amyloidosis, which can lead to renal failure, is the most severe complication of FMF.
• During attacks there is usually neutrophilia and brisk acute-phase response -between attacks, patients feel well (biochemical evidence for inflammation may persist)
• Treatment with daily oral colchicine reduces the frequency and severity of FMF attacks and prevents the development of amyloidosis.
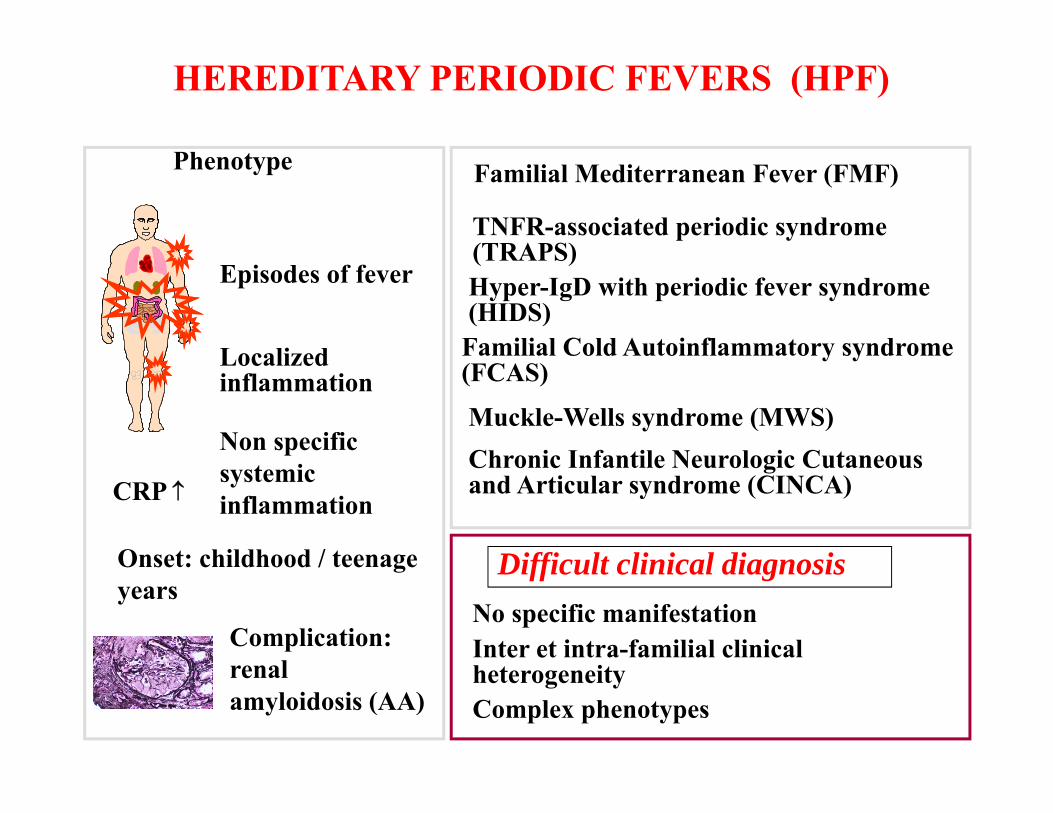
HEREDITARY PERIODIC FEVERS (HPF)
Familial Mediterranean Fever (FMF)
TNFR-associated periodic syndrome (TRAPS)
Muckle-Wells syndrome (MWS)
Familial Cold Autoinflammatory syndrome (FCAS)
Chronic Infantile Neurologic Cutaneous and Articular syndrome (CINCA)
Hyper-IgD with periodic fever syndrome (HIDS)
Non specific systemic inflammation
Episodes of fever
Localized inflammation
Complication: renal amyloidosis (AA)
CRP
Phenotype
Difficult clinical diagnosisNo specific manifestationInter et intra-familial clinical heterogeneityComplex phenotypes
Onset: childhood / teenage years

Clinical entities
FMF
FCASMWSCINCA
HIDS
TRAPS
AR
AR
MEFV:Chr 16p13.3
NLRP3/CIAS1: Chr 1q44
TNFRSF1A:Chr 12p13.2
AD
MVK: Chr 12q24.11
AD
Mode of inheritanceGenes
Gene families responsable for Hereditary Periodic Fever syndromes
CAPS (Cryopyrin -associated periodic syndromes)
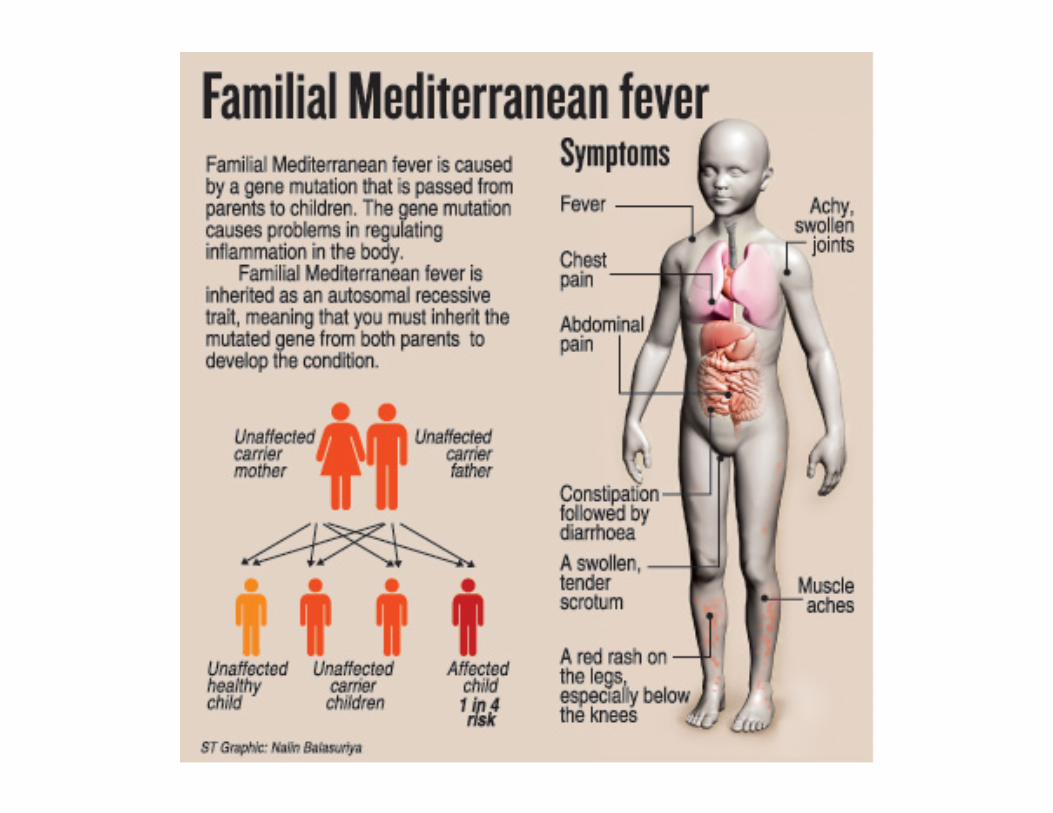

First mutations related to FMF seems to be appeared about 25000 years ago and to have spread firstly from eastern Mediterranean to Spain and then to North Africa, Turkey, Iraq, Armenia as a result of migrations of populations. Nowdays FMF cases have been documented worldwide.
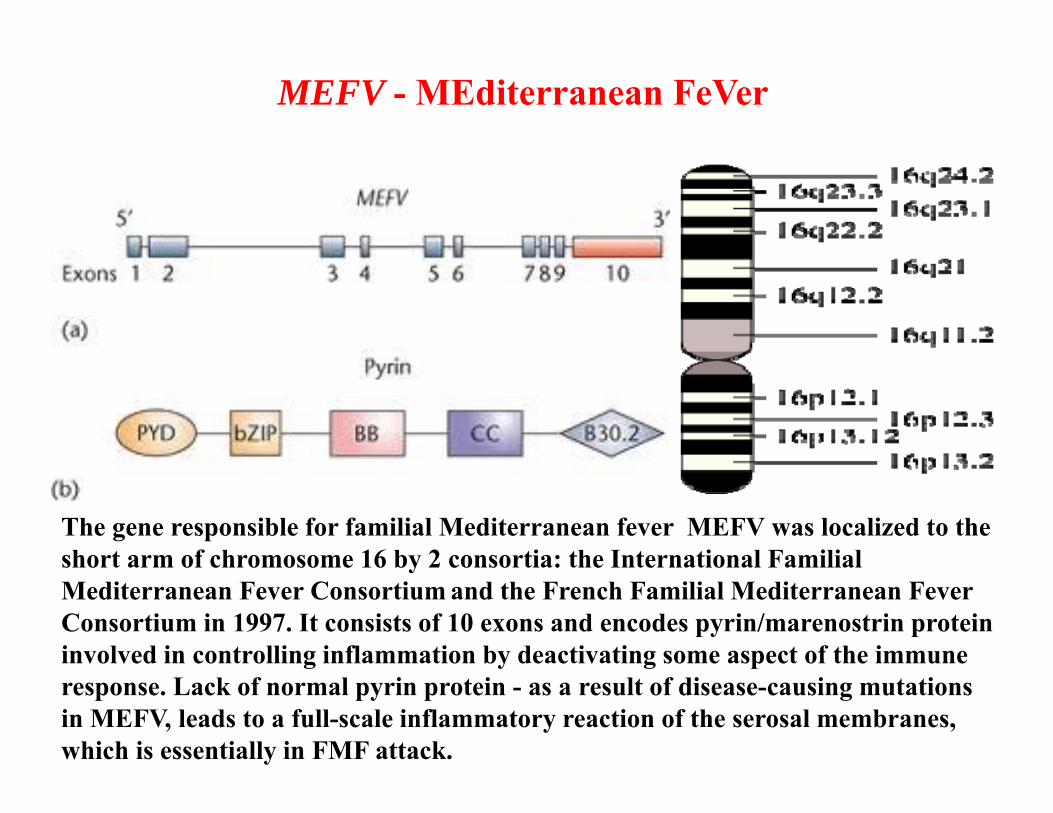
MEFV - MEditerranean FeVer
The gene responsible for familial Mediterranean fever MEFV was localized to the short arm of chromosome 16 by 2 consortia: the International Familial Mediterranean Fever Consortium and the French Familial Mediterranean Fever Consortium in 1997. It consists of 10 exons and encodes pyrin/marenostrin protein involved in controlling inflammation by deactivating some aspect of the immune response. Lack of normal pyrin protein - as a result of disease-causing mutations in MEFV, leads to a full-scale inflammatory reaction of the serosal membranes, which is essentially in FMF attack.

Mutation Nucleotide change Aminoacid changes
M694V ATG GTG Меthionine Valine
V726A GTT GCT ValineAlanine
M680I ATG ATC Methionine Isoleucine
MEFV MUTATIONS SYMBOLS

GENETIC TESTING OF FMF The diagnosis of FMF is based on a combination of clinical and molecular genetic findings
• Clinical diagnosis : according to the established clinical criteria by Livneh et al. (1997-2000). In order to determine the clinical differences between patients the criteria of severity scores are used (Tel Hashomer, 1997; 2000; Mor A., 2005).
• DNA: from blood samples • Molecular investigation of MEFV mutations: genetic testing for confirmation
of FMF diagnosis with two MEFV mutations homozygote genotype – both mutant allels are the same compaund-heterozygote genotype – 2 different mutant alleles one from
each parent • Laboratory testings: USI, blood and urine tests, ESR, fibrinogen, CRP• Interpretation of molecular investigation results, taking decision for
patients/family further follow up. With early diagnosis and colchicine-treatment the prognosis of FMF is good.
• For patients with clinical manifistation but with one or no MEFV mutation, clinical diagnosis remains important for furhter management.

DISTRIBUTION OF MOST COMMON MEFV MUTATIONS IN 450 HEALTHY INDIVIDUALS
AND 15424 FMF PATIENTS
EXON MUTATION HEALTHY FMF
EXON 10: M694V 4.7% 50.6%V726A 4.6% 22.3%M680I 1.8% 18.7%R761H 0.2% 3.2%M694I ━ 0.4%
EXON 5: F479L 0.4% 1.3%EXON 3: P369S 4.9% ━
EXON 2: E148Q 3.4% 2.2%
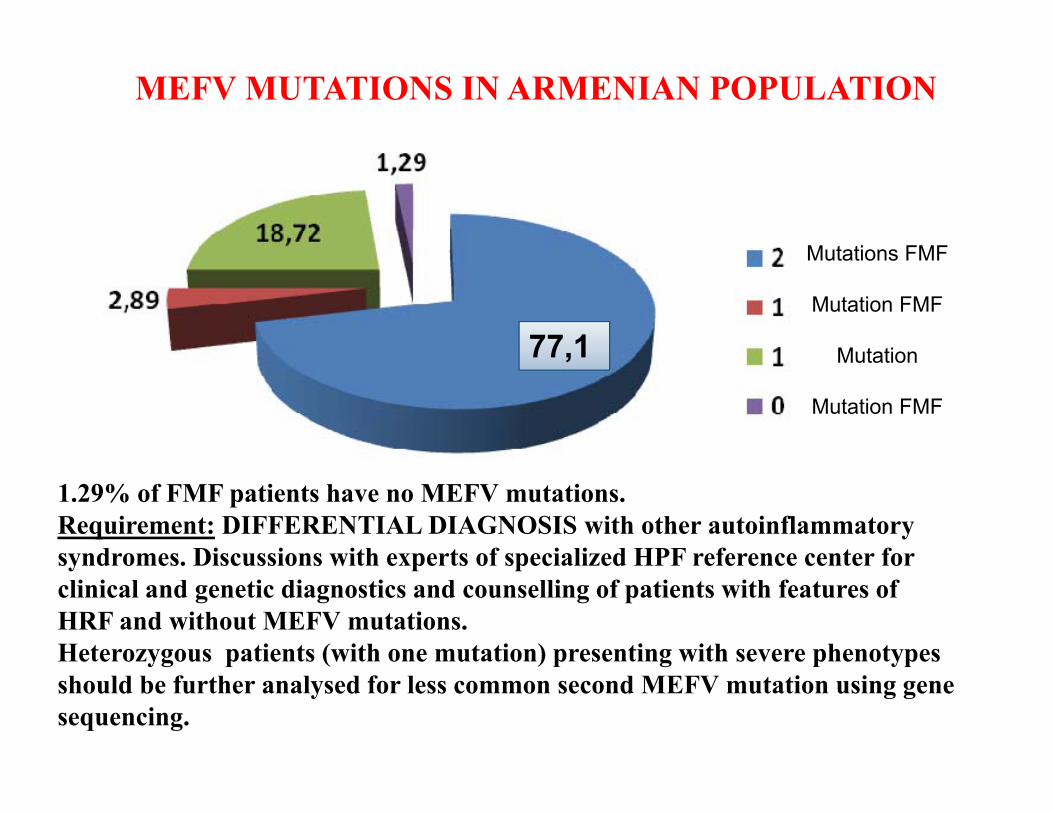
MEFV MUTATIONS IN ARMENIAN POPULATION
Mutations FMF
Mutation FMF
Mutation
Mutation FMF
77,1
1.29% of FMF patients have no MEFV mutations.Requirement: DIFFERENTIAL DIAGNOSIS with other autoinflammatorysyndromes. Discussions with experts of specialized HPF reference center forclinical and genetic diagnostics and counselling of patients with features ofHRF and without MEFV mutations.Heterozygous patients (with one mutation) presenting with severe phenotypesshould be further analysed for less common second MEFV mutation using genesequencing.

FREQUENCY OF MEFV MUTATIONS IN FMF PATIENTS:
98.71% WITH 12 COMMON (92.39% IN EXON 10) MUTATIONS
M694V 40.9%
V726A 27,9%
M680I 18.8%
Others 12.28%
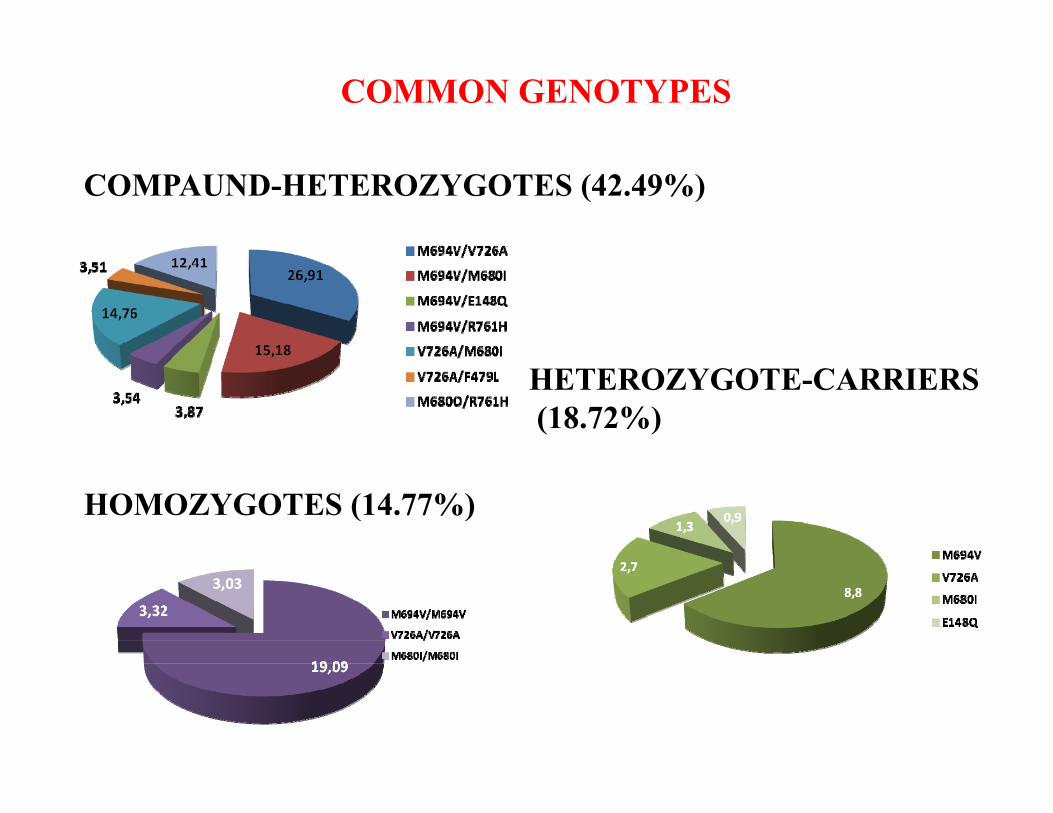
COMPAUND-HETEROZYGOTES (42.49%)
HETEROZYGOTE-CARRIERS(18.72%)
COMMON GENOTYPES
HOMOZYGOTES (14.77%)
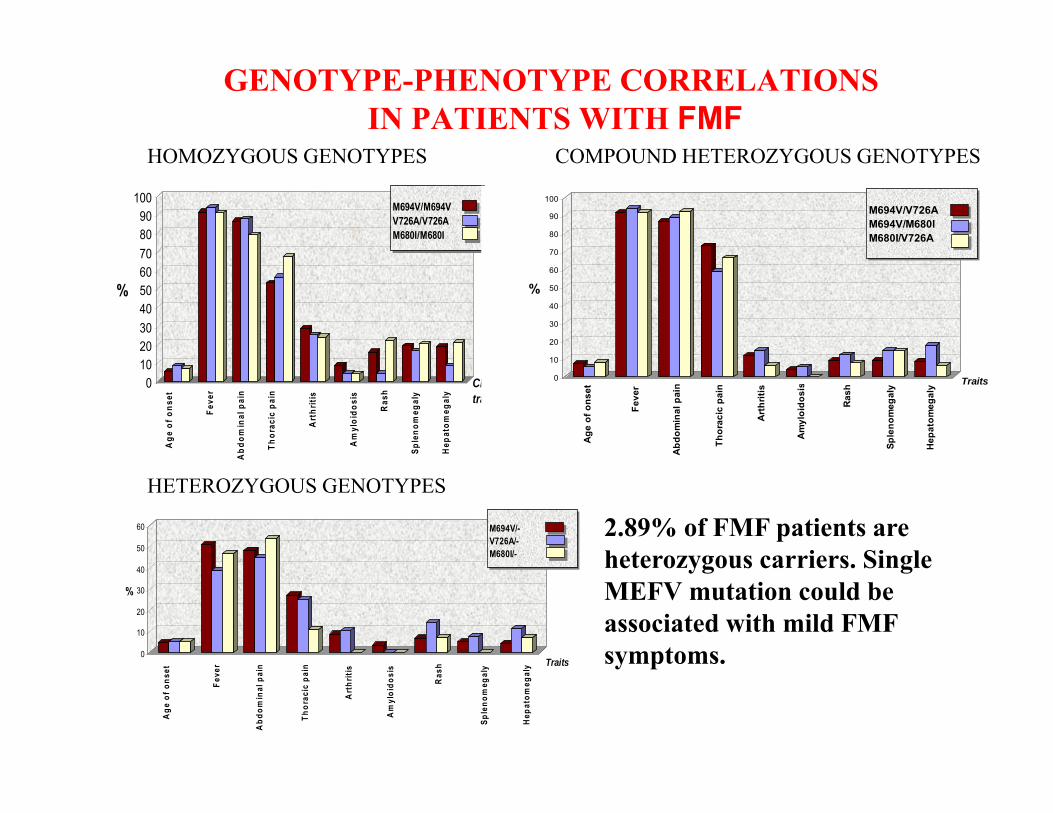
0
10
20
30
40
50
60
%
M694V/-V726A/-M680I/-
Age
of o
nset
Feve
r
Abd
omin
al p
ain
Thor
acic
pai
n
Art
hriti
s
Am
yloi
dosi
s
Ras
h
Sple
nom
egal
y
Hep
atom
egal
y Traits
0102030405060708090
100
%
M694V/M694VV726A/V726AM680I/M680I
Age
of o
nset
Feve
r
Abd
omin
al p
ain
Thor
acic
pai
n
Art
hriti
s
Am
yloi
dosi
s
Ras
h
Sple
nom
egal
y
Hep
atom
egal
y
Clinical traits
0
10
20
30
40
50
60
70
80
90
100
%
M694V/V726AM694V/M680IM680I/V726A
Age
of o
nset
Feve
r
Abdo
min
al p
ain
Thor
acic
pai
n
Arth
ritis
Amyl
oido
sis
Rash
Sple
nom
egal
y
Hep
atom
egal
y Traits
COMPOUND HETEROZYGOUS GENOTYPES HOMOZYGOUS GENOTYPES
2.89% of FMF patients are heterozygous carriers. Single MEFV mutation could be associated with mild FMF symptoms.
GENOTYPE-PHENOTYPE CORRELATIONSIN PATIENTS WITH FMF
HETEROZYGOUS GENOTYPES
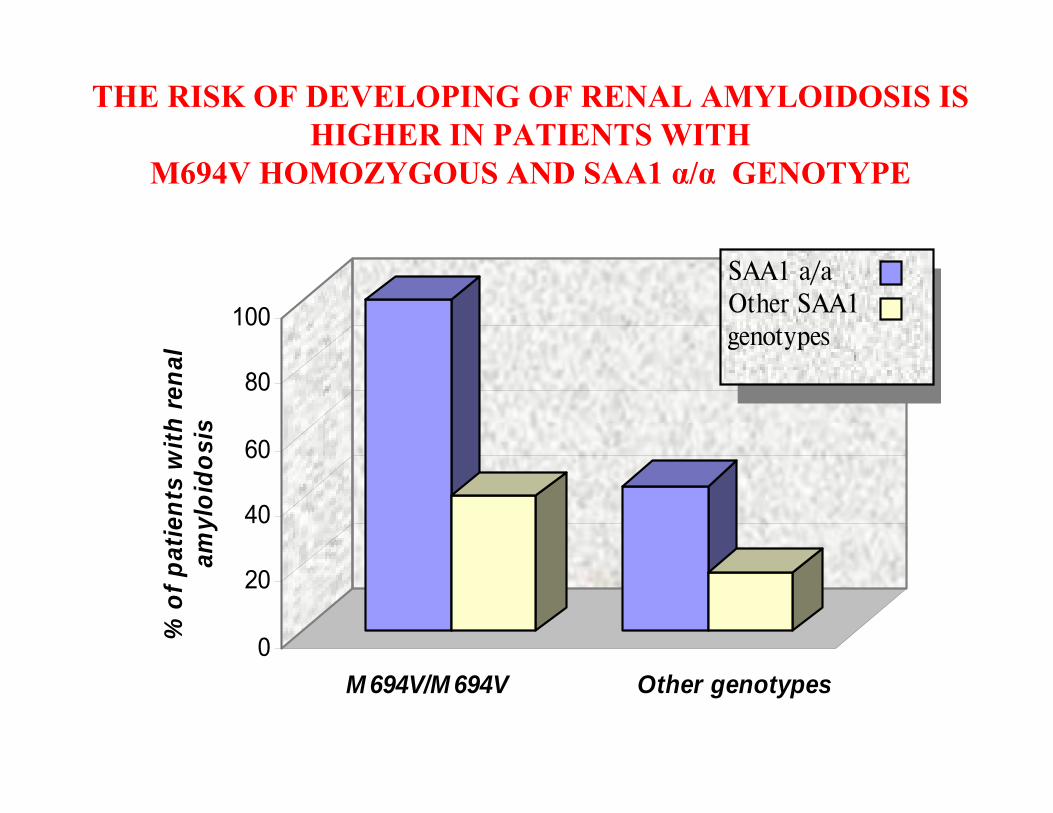
THE RISK OF DEVELOPING OF RENAL AMYLOIDOSIS IS HIGHER IN PATIENTS WITH
M694V HOMOZYGOUS AND SAA1 α/α GENOTYPE
0
20
40
60
80
100
% o
f pat
ient
s w
ith re
nal
amyl
oido
sis
M 694V/M 694V Other genotypes
SAA1 a/aOther SAA1 genotypes

• MOLECULAR GENETIC ANALYSIS improves early and correct diagnosis of FMF, and allows to commence lifelong treatment of affected individuals with colchicine. Diagnosis is based on clinical features, response to treatment with colchicine, and genetic analysis.
• TESTING OF ASYMPTOMATIC INDIVIDUALS – for estimation of carriers. As frequency of MEFV gene mutations is extremely high in Armenians (1:4) genotyping is recommended for asymptomatic family members (especially siblings of affected children).
• Molecular study of NON-TYPICAL FMF to reveal other autoinflammatorysyndromes.
• TESTING OF FAMILY MEMBERS with any clinical symptoms of FMF.
• GENETIC COUNSELLING of FMF patients and families based on the clinical and genetic investigations.

Genetics Counseling
Natella Kostandyan

Main Genetic Problems
• Part of population suffering or at high risk of hereditary disorders
• Genetic Disorders and Congenital Defects• Newborns 1‐3%
– Hospitalised children 30%
• Chronic Disorders with Genetic Background– 10% of Adult Population

The Structure of Genetic Services
Clinical Genetics• Therapeutist• Physician‐geneticist
Cytogenetics Molecular Genetics
General Physician

Common Indications For Genetics Referral1. Person with mental retardation or developmental delay2. Family history of a hereditary condition (FraX, diabetes, cystic
fibrosis)3. Presence of multiple congenital anomalies, or isolated birth defect
(neural tube defect)4. Person at risk for a genetic condition, including late-onset
disorders (cancer)5. Person or family with questions about the genetic aspects of any
medical condition6. Couples with a history of recurrent miscarriages7. Consanguinity in a couple8. Teratogen counseling9. Preconceptional counseling and risk factor counseling, including
advanced maternal age10.Positive newborn test (PKU)

http://www.ncbi.nlm.nih.gov/sites/GeneTests

www.orphanet.org
Diagnosis and Counseling of more than 3000 rare (Orphan) genetic disorders

What is Genetic Counseling• Genetic counseling is a process of
communication that deals with the human problems associated with the occurrence, or the risk of recurrence, of a genetic disorder in a family.
• It is intended to provide such individuals and families with information about their condition, to explore the personal consequences of this information, and to aid families at risk to make informed reproductive decisions.

Goals of genetic counseling are to help individuals and families to
• Comprehend the medical facts including diagnosis, prognosis and management
• Appreciate the way heredity contributes to the disorder and risk of recurrence
• Understand the alternatives for dealing with these risks
• Choose the course of action that seems appropriate for the individual or family being counseled
• Make the best possible adjustment to the condition or the risk of its recurrence

Process of Genetic Counseling
• Information Gathering– Family and Medical History– Genealogical Analysis– Physical Examination
• Risk Assessment– Actual risk vs. perceived risk
• Genetic Tests• Information Giving• Psychosocial Counseling

Ordering Genetic Testing
Genetic tests may be ordered by any physician.
• Choosing a laboratory• Pretest counseling and informed consent• Sample logistics and supporting documentation• Test result interpretation and follow-up

EUROGENTEST.ORG

Testing Strategy: Lesson from Science
Most genes have 100s of disease-causing mutations
Genes can have benign sequence variants (polymorphisms) which have no effect on health
Sometimes it is unclear whether a sequence variant is disease-causing or benign
The disease-causing mutation must be known before relatives at-risk can be tested
Genetic tests usually cannot detect all disease-causing mutations in a gene
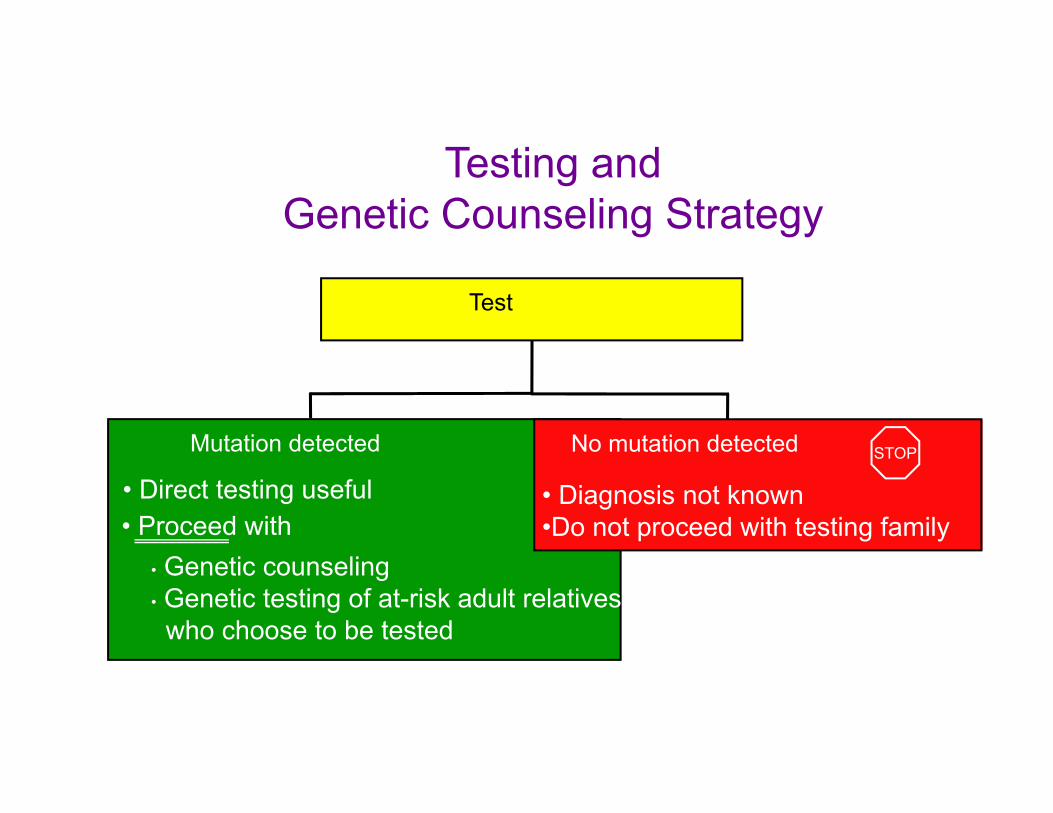
Testing andGenetic Counseling Strategy
Test
Mutation detected
• Direct testing useful• Proceed with
• Genetic counseling• Genetic testing of at-risk adult relatives who choose to be tested
• Diagnosis not known•Do not proceed with testing family
No mutation detected STOP

Testing Strategies. • Single gene disease w/ only recurrent mutations
(e.g. Achondroplasia)– Test for recurrent mutation– Positive result
• penetrance known– Negative result
• False negative rate known. – Phenotypic testing, if indicated.

Testing Strategies.
• Single gene ds w/recurrent and private mutations (e.g., CFTR, thalassemias).
• test for “ethnic” recurrent mutation(s)• If positive, significance known• If negative, and index case or relative, perform
“mutation scanning” test.

The magnitude of the risk of occurrence or recurrence
• when genotypes are known• when alternative Genotypes are possible• of disorders with incomplete penetrance• of disorders with late age of onset• when counseling for consanguinity• when the gene has not been identified
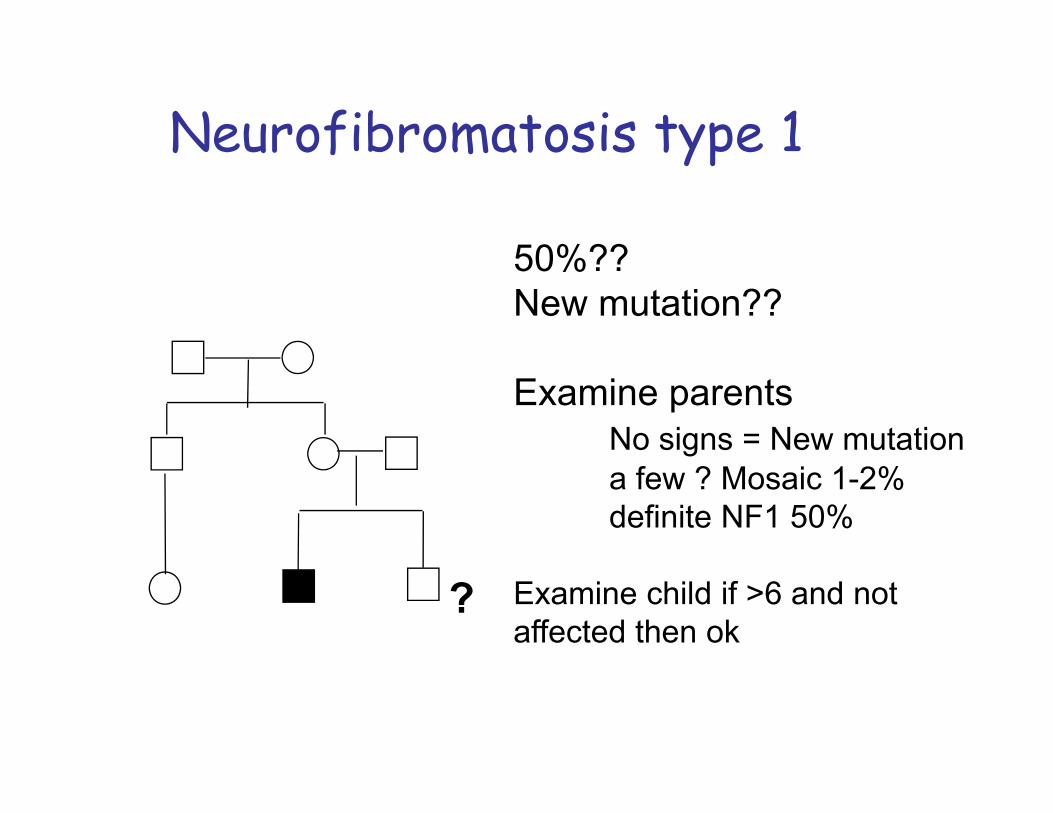
?
50%??New mutation??
Examine parentsNo signs = New mutationa few ? Mosaic 1-2%definite NF1 50%
Examine child if >6 and not affected then ok
Neurofibromatosis type 1

Carrier frequency 1/25Many different mutations (>700)Common mutation F508 (3bp deletion))
1/25 X 1/2 X 1/4 = 1/200
1/21/251/4
A-R Cystic Fibrosis
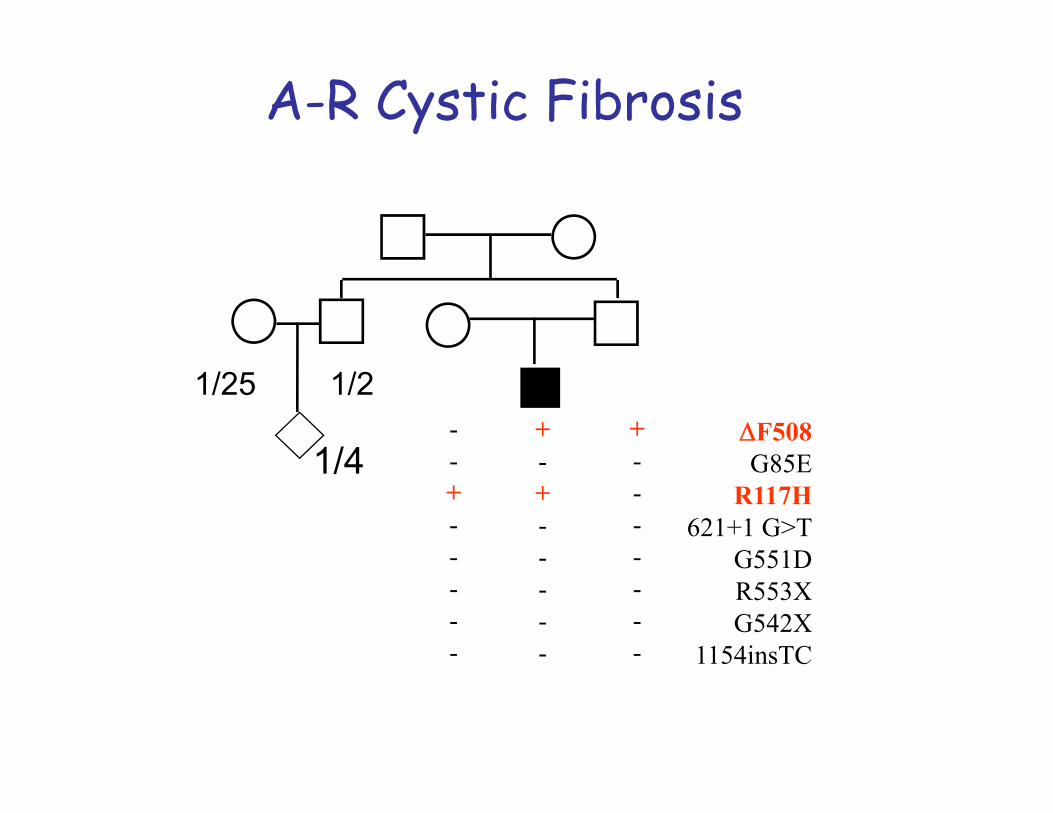
1/21/25F508G85E
R117H621+1 G>T
G551DR553XG542X
1154insTC
+-+-----
+-------
--+-----
A-R Cystic Fibrosis
1/4
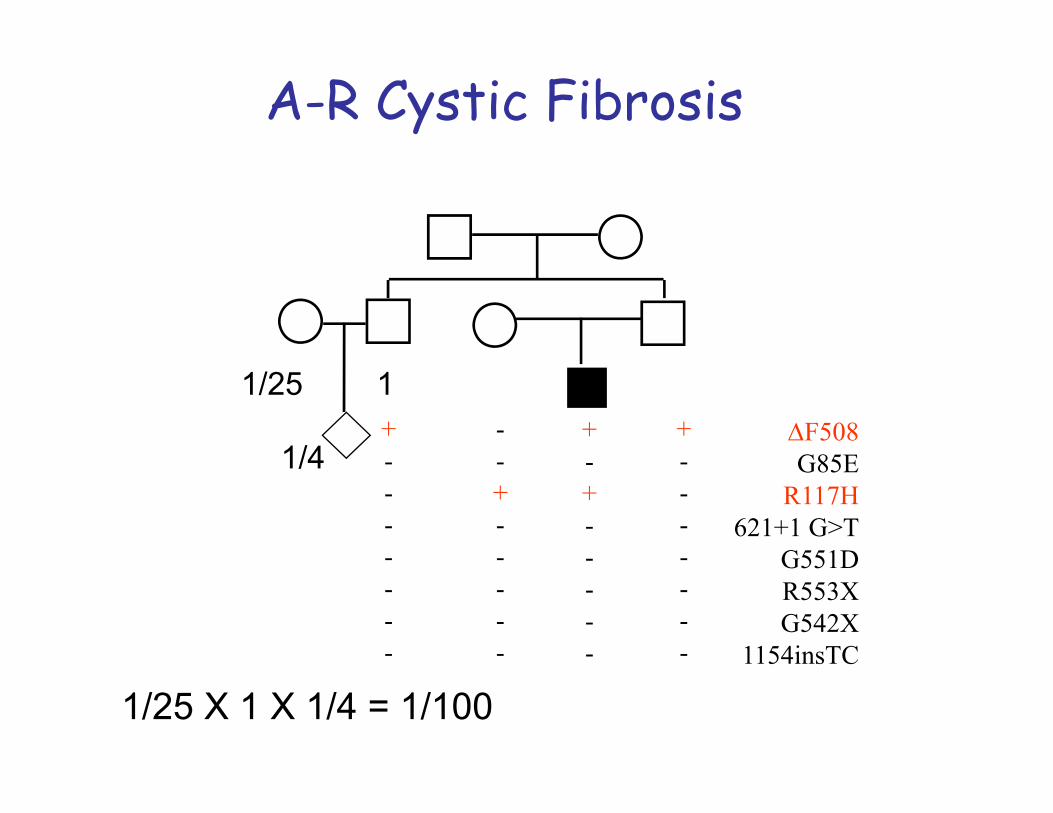
F508G85E
R117H621+1 G>T
G551DR553XG542X
1154insTC
+-+-----
+-------
--+-----
+-------
1/25 1
1/25 X 1 X 1/4 = 1/100
1/4
A-R Cystic Fibrosis
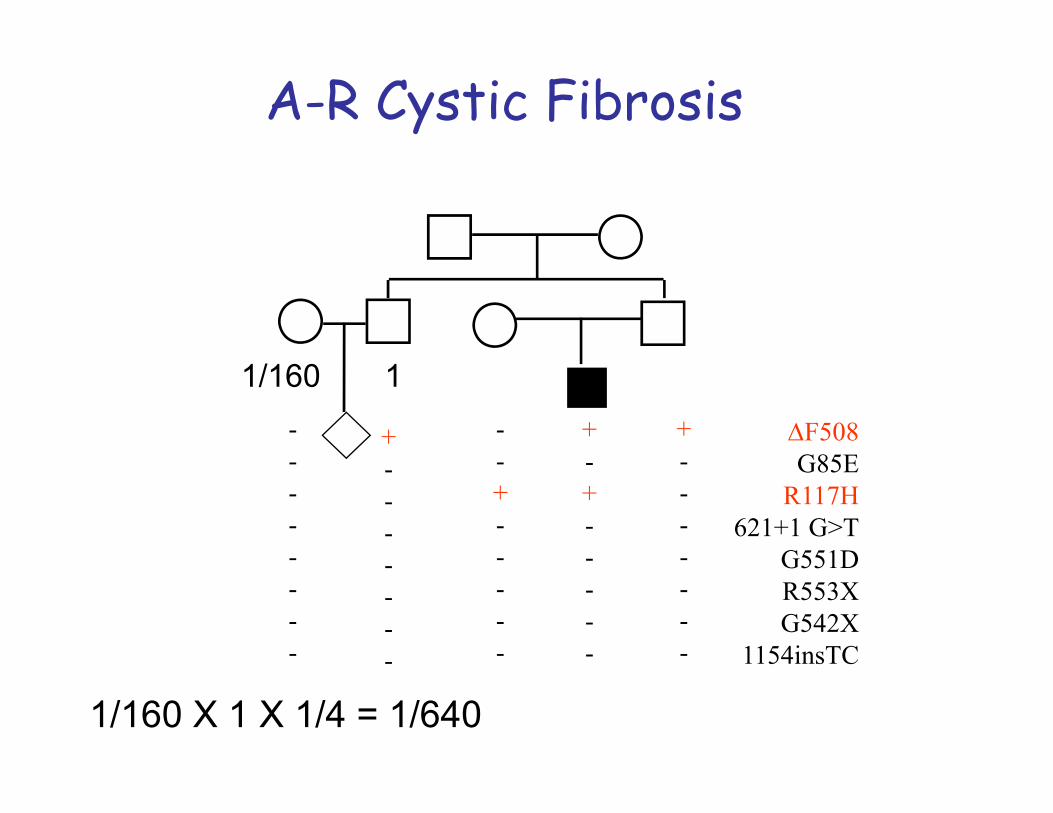
F508G85E
R117H621+1 G>T
G551DR553XG542X
1154insTC
+-+-----
+-------
--+-----
--------
+-------
11/160
1/160 X 1 X 1/4 = 1/640
A-R Cystic Fibrosis

Reduced penetrance
• Penetrance = proportion of heterozygotes who show evidence of the effects of mutation.
• Expressed as %?
retinoblastoma
Risk = ½ x P = 0.4

Presymptomatic testing
• Individual has no signs of disease• Has FH of disease• Wants to know if will develop it• Mainly done for Huntington’s disease and
some familial cancer/cardiac syndromes

Case study Diagnostic testing for Huntington’s disease
Diagnosis has implications for children as now at 50% risk
Need to consider wider family

Carrier testing
• Autosomal recessive , X linked recessive or chromosomal rearrangements
• Only implications are for reproduction• General ethical principle is against testing in
childhood (autonomy, labelling and stigma, ensuring appropriate counselling)
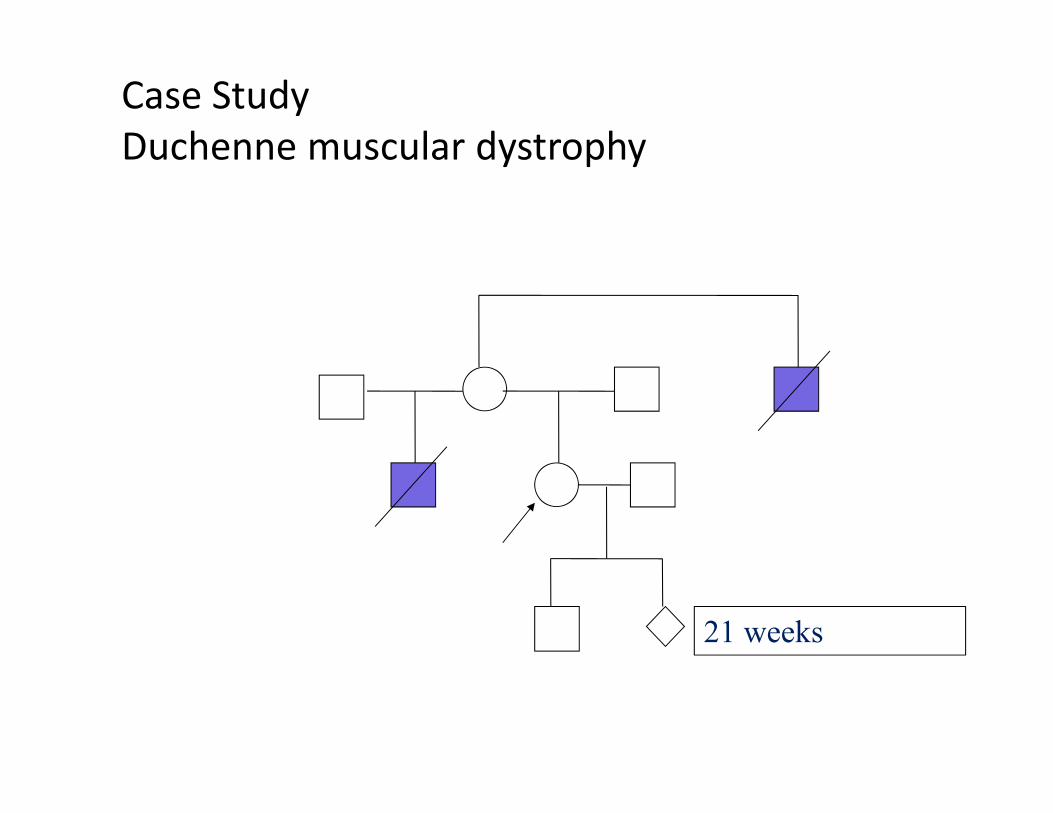
Case StudyDuchenne muscular dystrophy
21 weeks

Genetic CouselingPrinciple of Nondirectiveness: Clinical geneticists subscribe to the principle of nondirectivenes: information about risks, natural history, treatment, and outcome are presented in a balanced and neutral manner, but dicisions about actions (ex. reproduction) are left to the family.

GENETIC SCREENING

Genetic Screening Definition
The goal of screening is early recognition of a disorder so that intervention will prevent or reverse disorder process or so that informed reproductive decisions can be made.
Screening does not equal testing. The methods are the same, the difference is “why” the tests are done.

Objectives of Screening• Therapy
• Prevention
• Identify at risk individuals / couples

Some examples of genetic screening
• Prenatal screening• Newborn screening• Carrier screening

Prenatal Screening• This can detect a disorder before a baby is born.• An ultrasound test is used to determine if the fetus is at a
high or low risk from a genetic disorder.• Disorders are diagnosed by examining a small amount of
fetal cells. This carries a small risk to the fetus.• If diagnosed early in the pregnancy, there is still the
possibility of abortion.

Newborn Screening An effective public health strategy for treatable disorders such as • congenital hypothyroidism, • cystic fibrosis, • phenylketonuria, and • galactosemia

Carrier Screening• This involves testing prospective parents for diseases
that they show no symptoms of, but may carry a recessive gene for.
• A blood sample or cheek cell sample is analysed to determine whether either parent carries a faulty gene.
• If both parents carry a specific faulty gene, the chance of the fetus receiving the gene from both parents is 25%, and the chance of being a carrier is 50%.
• If both parents carry a faulty gene, they may decide to have prenatal testing on the fetus.

Principles of Screening
The basic elements of a test’s validity includes its
• Sensitivity : proportion of true positives detected
• Specificity : proportion of true negatives detected.
• When sensitivity is increased, specificity decreases, and vice versa.

Levels of Screening• Population Screening: The presumptive identification of an
unrecognised disease/defect by the application of tests/examinations (Maternal serum alpha-fetoprotein testing)
• Genetic Screening: The search in a population for persons possessing certain genotypes that
• are already associated with disease or predisposition to disease
• may lead to disease in their descendents

Population Screening• Condition sufficiently frequent in screened population• Condition serious or fatal without intervention• Condition must be treatable or preventable• Effective follow-up program possible• Screening and management must be cost-effective• Specimens must be easy to collect• Analysis must be simple, reliable, reproducible, and lend
itself to mass screening• Analysis should have high sensitivity

Selected Population Screening • Heterozygote (carrier)
• parents of child with CF• mothers of boys with Fragile-X syndrome
• Ethnic specific: Certain ethnic groups are at such a high risk for a particular condition that selected screening is warranted (low frequency in general population)
• Mediterranean • Hemoglobinopathies• Familial Mediterranean Fever