chapter1 and 2 (part 1) pharchem2
TRANSCRIPT

Pharmaceutical Organic Chemistry
Lecture Presentation 1

Medicinal Chemistry
• is devoted to the discovery and development of new agents for treating diseases.
• Biotechnology-uses the cell’s biochemistry to synthesize new
compounds.Example:• Cell-stimulating factors• Humanized monoclonal antibodies • Fused receptors that intercept immune cell–
generated cytokines.

Cell stimulating factors
• GSF is a glycoprotein that stimulates the bone marrow to produce granulocytes and stem cells and release them into the bloodstream.

Medicinal Chemistry
• is concerned mainly in the organic, analytical, and biochemical aspects of process of discovery and design of clinical agent.
• Opium, belladonna, ephedrine and penicillin.

Lead Compound
• Amantadine protects and treats early influenza A and treat parkinsonian disorders.

Rational Drug design
• X-ray crystallography and nuclear magnetic resonance
• Molecular graphics and computational chemistry

• Why were certain structures selected?
• What modifications were made to produce more focused activity or reduce
adverse reactions or produce better pharmaceutical properties?
• Was the prototypical molecule discovered from random screens, or did the medicinal chemist have a structural
concept of the receptor or an understanding of the disease process?

Drug Design Strategies
Let’s make a change on an existing compound or synthesize a new structure
and see what happens!!!

“Modern Drug”
• Acid–base chemistry is used to aid in formulation and bio-distribution.
• Structural attributes and substituent patterns can be predicted by statistical techniques such as regression analysis.
• Computerized conformational analysis permits the medicinal chemist to predict the drug’s three dimensional (3D) shape that is seen by the receptor

3D figure of a receptor

DRUG DISTRIBUTION


Oral Administration
Factors that affect the drug to be dissolved:
• Chemical structure
• Variation in particle size and particle surface area
• Nature of the crystal form
• Type of tablet coating
• Type of tablet matrix

Example

Example
• (2.5 mg/mL)- bitter taste• (1.05 mg/mL)- suspension

Example

Parenteral Administration
• Water-insoluble methylprednisolone acetate toslightly water-insoluble methylprednisolone to
water-soluble methylprednisolone sodium succinate

Protein Binding
• It may stay in solution, but many drugs will be bound to the serum proteins.
• Drug’solubility, biodistribution, half-life in the body, and interaction with other drugs.

Protein binding
• May limit access to certain body compartments. The placenta is able to block passage of proteins from maternal to fetal circulation.

Protein binding
• Prolong the drug’s duration of action.
• The drug–protein complex is too large to pass through the renal glomerular membranes, preventing rapid excretion
of the drug.
Protein Binding
• Limits the amount of drug available for biotransformation and for interaction with specific receptor sites.

Tissue Deposit
• Neutral fat constitutes some 20% to 50% of body weight and constitutes a depot of considerable importance.
Example: Thiopental

Drug metabolism
• All substances in the circulatory system, including drugs metabolites, and nutrients, will pass through the liver.
“First pass effect”
-Proportion of the drug will be transported into the hepatocyte to inactive chemicals.

Lidocaine
• 60% of this local anesthetic antiarrhythmic agent is metabolized during its initial passage through the liver.
• In contrast to lidocaine’s half-life of less than 2 hours, tocainide’s half-life is approximately 15 hours,
• 40% of the drug excreted unchanged.

Excretion
• The main route of excretion of a drug and its metabolites is through the kidney.
• Nursing mothers must be concerned, because drugs and their metabolites can be excreted in human milk and be ingested by the nursing infant.

THE RECEPTOR
• Synthetic corticosteroids bind to the same receptors as cortisone and hydrocortisone.
• Nonsteroidal anti-inflammatory agents inhibit the prostaglandin-forming enzyme cyclooxygenase.
• The barbiturates act on specific CNS receptors.
• Benzodiazepines combine with the-aminobutyric acid (GABA) receptors.

THE RECEPTOR
• A good ability to fit the receptor favors binding and the desired pharmacological response.
• A poor fit favours the reverse reaction.
• With only a small amount of drug bound to-much smaller pharmacological effect.
• Too small, there may be no discernible response.

“Lock and Key Postulate”
• The initial receptor model was based on a rigid lock-and-key concept, with the drug (key) fitting into a receptor (lock).

New Postulates
• Ligand, use of
• Recombinant DNA
• Genome• mapping

THE RECEPTOR
• Humans (and mammals in general) are very complex organisms that have developed specialized organ systems.
• It is not surprising that receptors are not distributed equally throughout the body.
• Ex. Tamoxifen is used for estrogen-sensitive
breast cancer and for reducing bone loss from osteoporosis

SUMMARY
• Altering the molecule to change the bio-distribution.
• Searching for structures that show increased specificity for the target receptor that will produce the desired pharmacological response while decreasing the affinity for undesired receptors that produce adverse response.
• Attaching the drug to a monoclonal antibody that will bind to a specific tissue antigenic for the antibody.

Acid+ Base
• an acid is a proton donor and a base is a proton acceptor.
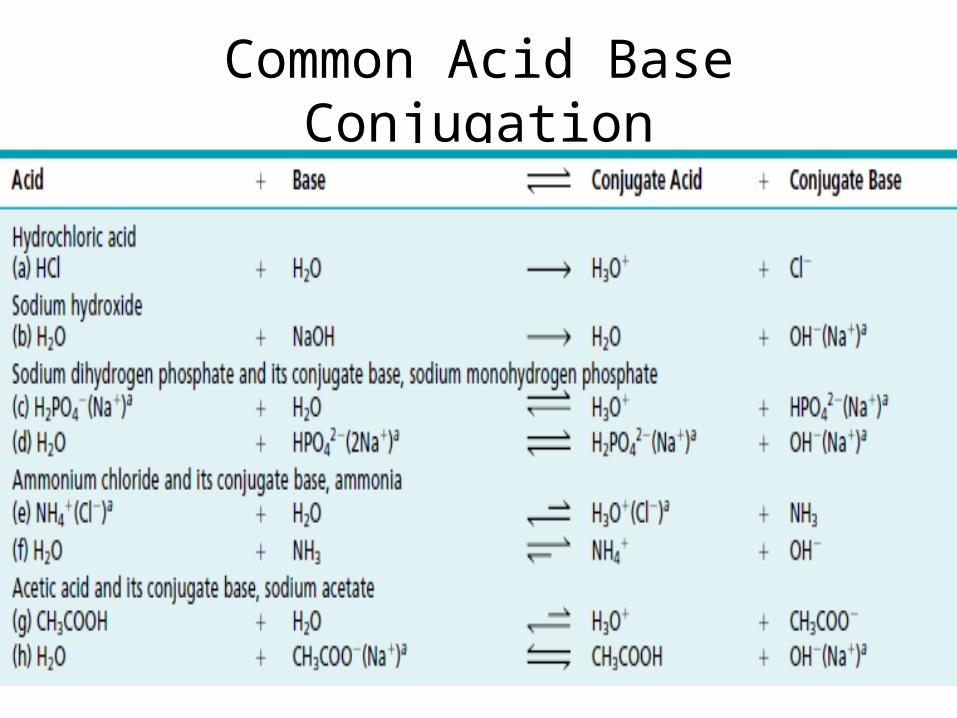
Common Acid Base Conjugation

Acid Base Conjugation
How to predict which direction an acid–base reaction lies?
What extent does the reaction goes to completion?
pKa’s are modified equilibrium constants that indicate theextent to which the acid (proton donor) reacts with water to
form conjugate acid and conjugate base.


Weak acid vs Strong acid

Calculations:

Calculations:

Percent Ionization
Adjust the pH to ensure maximum water solubility (ionic form of the drug) ormaximum solubility in nonpolar media (un-ionic form).

Calculations

Calculations

Calculations

Interpretation
• First, note that when pH = pKa, the compound is 50% ionized (or 50% un-ionized).
When the pKa is equal to the pH, the molar concentration of the acid equals the molar concentration of its conjugate base.


Application
• Phenytoin (HA acid; pKa 8.3) injection must be adjusted to pH 12 with sodium hydroxide to ensure complete ionization and maximize water solubility.

Application
• Tropicamide is an anticholinergic drug administered as eye drops for its mydriatic response during eye examinations.
• With a pKa of 5.2, the drug has to be buffered near pH 4 to obtain more than 90% ionization.

Drug Distribution and pKa
• The pKa can have a pronounced effect on the pharmacokinetics of the drug.
• For HA acids, it is the parent acid that will readily cross these membranes
• For BH acids, the unionized conjugate base (free amine) is the species most readily crossing the non-polar membranes

Drug Distribution and pKa
The drug first encounter the acidic stomach
HA acids with pKa’s of 4 to 5 will tend to be nonionic and be absorbed partially through
the gastric mucosa.

Drug Distribution and pKa
The drug first encounter the acidic stomach
Amines (pKa 9–10) will be protonated (BH acids) in the acidic stomach and usually
will not be absorbed until reaching the mildly alkaline intestinal tract (pH 8).

COMPUTER-AIDED DRUG DESIGN:EARLY METHODS
• Steroidal hormones based on cortisone.
• Adrenergic drugs based on epinephrine.
• Opiate analgesics based on morphine.
• Antibiotics based on penicillin.
• Cephalosporin and tetracycline.

Quantitativestructure–activity relationships
• To quantify the effect of a structural change on a definedpharmacological response are used to:(a) Predict biological activity in untested compounds(b) Define the structural requirements required for a good fit
between the drug molecule and the receptor(c) Design a test set of compounds to maximize the amount
of information concerning structural requirements for activity from a minimum number of compounds tested.

QSAR
• 1865 to 1870 by Crum-Brown and Fraser
-Gradual chemical modification in the molecular structure of a series of poisons produced some important differences
in their action.

QSAR
Biological response can be predicted
from physical chemical properties such as:
Vapor pressure
Water solubility
Electronic parameters
Steric descriptors
Partition coefficients

QSAR
The most lethal compound in this assay was chlorpromazine, with a BR (LD100) of only 0.00000631 mmol; and the least active was ethanol, with a BR of 0.087096 mmol.
It takes about 13,800 times as many millimoles of ethanol than of chlorpromazine to kill 100% of the test subjects

Partition Coefficient
Drug will go into series of partitioning steps.
(a) leaving the aqueous extracellular fluids,
(b) passing through lipid membranes, and
(c) Entering other aqueous environments before reaching the receptor

Partition Coefficient
• The partition coefficient (P) is the ratio of the molar concentration of chemical in the
non-aqueous phase versus that in the
aqueous phase.
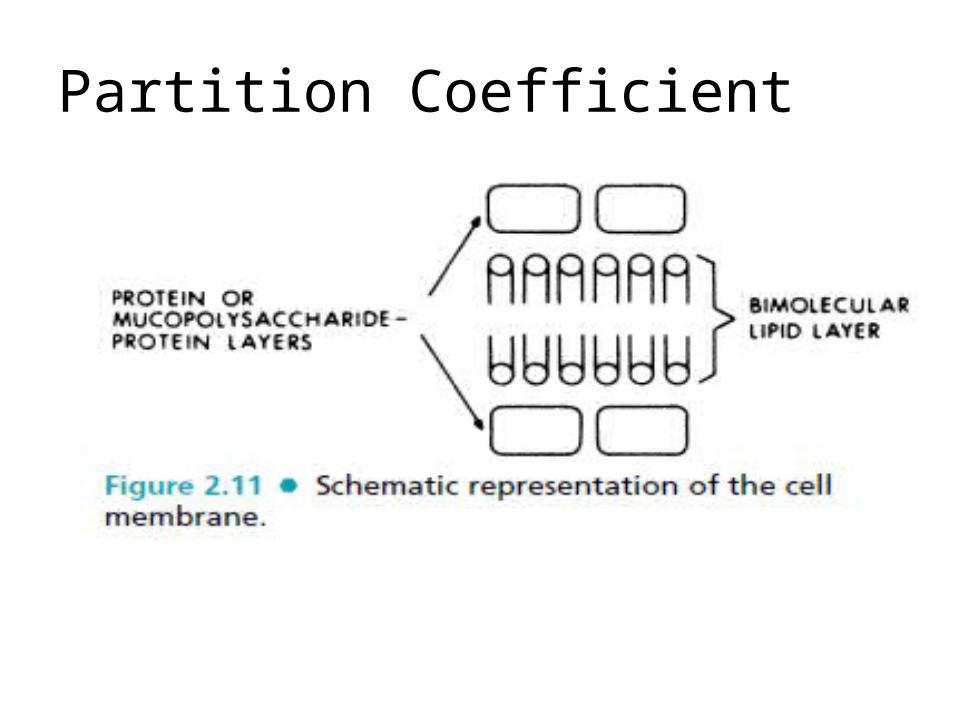
Partition Coefficient

The two outer layers, one facing the interior and the other facing theexterior of the cell, consist of the polar ends of the bifunctional lipids. These surfaces are exposed to an aqueous polar environment. The
polar ends of the charged phospholipids and other bifunctional lipids are solvated by the water molecules.
There are also considerable amounts of charged proteins and mucopolysaccharides present on the surface


Computer Aided DesignNEWER METHOD
• Through the use of computer graphics, structures of organic molecules can be entered into a computer and manipulated in many ways.
• Calculate molecular properties and generate pharmacophore hypotheses.
• Once a pharmacophore hypothesis has been developed, structural databases (commercial, corporate, and/or public) of 3D structures can be searched rapidly for hits (i.e., existing compounds that are available with the required functional groups and permissible spatial orientations as defined by the search query).
• It has become popular to carry out in silico (computer as opposed to biological) screening of drug–receptor candidateinteractions, known as virtual high-throughput screening(vHTS), for future development.