autism exome sequencing: design, data processing and analysis benjamin neale analytic and...
TRANSCRIPT

Autism exome sequencing:design, data processing and analysis
Benjamin NealeAnalytic and Translational Genetics Unit, MGH
&Medical & Population Genetics Program, Broad Institute

Direct Sequencing has Enormous Potential…
• Ng, Shendure: Miller syndrome, 4 cases – exome sequenced reveals causal mutations in DHODH
• Lifton: Undiagnosed congenital chloride diarrhoea (consanguinous)– Exome seq reveals homozygous SLC23A chloride ion transporter mutation– Return diagnosis of CLD (gi) not suspected Bartter syndrome (renal)
• Worthey, Dimmock: 4-year old, severe unusual IBD– exome seq reveals XIAP mutation (at a highly conserved aa) – proimmune disregulation opt for bone marrow transplant over chemo
• Jones, Marra: Secondary lung carcinoma unresponsive to erlotinib– Genome and transcriptome sequencing reveals defects– directs alternative sunitinib therapy
• Mardis, Wilson: acute myelocytic leukaemia but not classical translocation– Genome sequencing (1 week + analysis) reveals PML-RARA translocation– Directs ATRA (all trans retinoic acid) treatment decision

…and tremendous challenges
• Managing and processing vast quantities of data into variation
• Interpreting millions of variants per individual– An individual’s genome harbors
• ~80 point nonsense mutations• ~100-200 frameshift mutations• Tens of splice mutants, CNV induced gene disruptions
For very few of these do we have any conclusive understanding of their medical impact in the population

Successes to date rely on factors that may not apply generally to common endpoints
• Mendelian disorders– Single family rare autosomal recessive (linkage
may target 1% of genome, 2 ‘hits’ in the same gene very unlikely)
– Single (or ‘near single’) gene disorders where nearly all families carry mutations in the same underlying gene
• Somatic or de novo mutations– Extremely rare background rate

Autism exome sequencing
• In progress – ARRA supported by NIMH & NHGRI
• Collaboration between sequencing centers (Baylor & Broad) and Y2 follow-up in autism genetics labs (Buxbaum, Daly, Devlin, Schellenberg, Sutcliffe)
• Targeted production by years end of 1000 cases and 1000 controls (500/500 from each site)

Exome production plan
• Baylor: 1000 samples (Nimblegen capture, SOLiD sequencing)
• Broad: 1000 samples (Agilent capture, Illumina sequencing)
• Predominantly cases and controls pairwise matched with GWAS data (one batch of 50 trios currently being run)
• All samples are available from NIMH repository

Broad Exome Production
• ~700 exomes completely sequenced and recently completed variant calling
• ~300 completed earlier in the Summer and fully analyzed (basis of later analysis slides)
• Main production conducted with matched case-control pairs traveling together through the sequencing lab and computational runs of variant calling
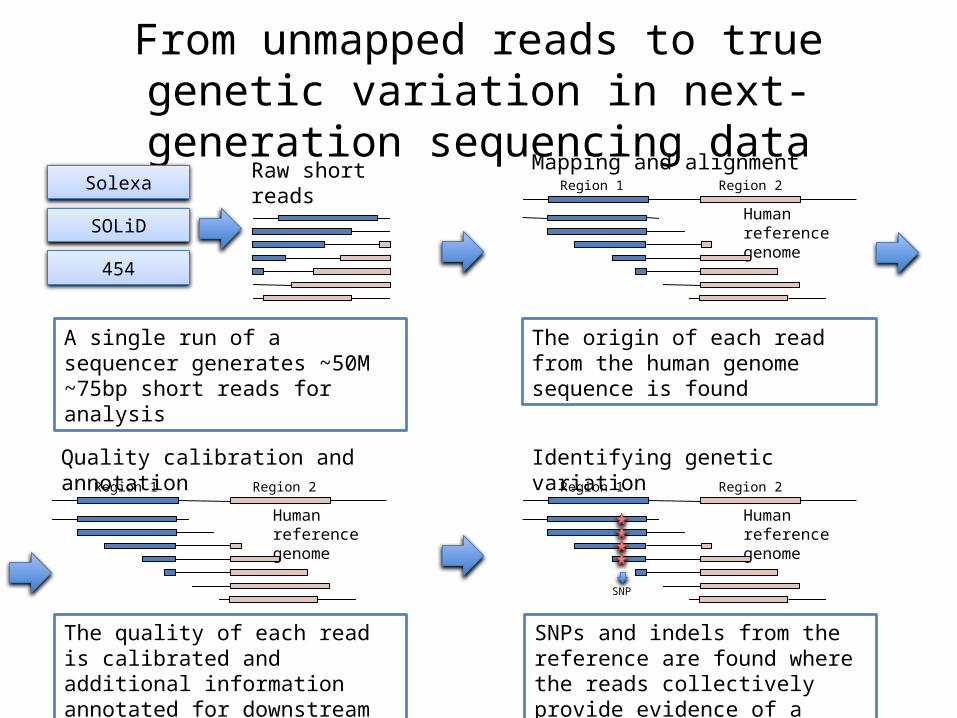
From unmapped reads to true genetic variation in next-generation sequencing data
Raw short reads
Human reference genome
SolexaMapping and alignment
Human reference genome
Quality calibration and annotation
The quality of each read is calibrated and additional information annotated for downstream analyses
The origin of each read from the human genome sequence is found
Human reference genome
Identifying genetic variation
SNPs and indels from the reference are found where the reads collectively provide evidence of a variant
SNP
A single run of a sequencer generates ~50M ~75bp short reads for analysis
SOLiD
454
Region 1 Region 2
Region 1 Region 2 Region 1 Region 2

Partnership: Genome Sequencing and Analysis (GSA) team @ Broad
• Genome Sequencing and Analysis (GSA) develops core capabilities for genetic analysis – Data processing and analysis methods– Technology development– High-end software engineering– High-throughput data processing for
MPG exome projects with MPG-Firehose
• Staffed by full-time research scientists in MPG– PhDs and BAs in biochemistry,
engineering, computer science, mathematics, and genetics
Group LeaderMark A. DePristo
Analysis TeamKiran Garimella [Lead]Chris HartlCorin Boyko
Development TeamEric Banks [Lead]Guillermo del AngelMenachem FromerRyan Poplin
Software EngineeringMatthew HannaKhalid ShakirAaron McKenna

Developing cutting-edge data processing and analysis methods
Local realignment
Base quality score recalibration
Variation discovery and genotyping
Read-backed phasing
VariantEval
Adaptive error
modeling
Novel SNPs found

Challenges
• Mapping/alignment• Quality score recalibration• Calling variants• Evaluating set of variant sites• Estimating genotypes

Solexa : BWA 454 : SSAHA SOLiD : Corona
• Robust, accurate ‘gold standard’ aligner for NGS
• Developed by Li and Durbin• Recently replaced MAQ, also
by Li and Durbin, used for last 2 years
SAM/BAM files
Region 1
Enormous pile of short reads
from NGS
Detects correct read origin and flags them
with high certainty
Detects ambiguity in the origin of reads and flags
them as uncertain
Reference genome
Mapping and alignment algorithm
Finding the true origin of each read is a computationally demanding and important first step
• Hash-based aligner with high sensitivity and specificity with longer reads
• ABI-designed tool for aligning in color-space
Region 2 Region 3

The SAM file format
• Data sharing was a major issue with the 1000 genomes– Each center, technology and analysis tool used its own
idiosyncratic file formats – no one could exchange data• The Sequence Alignment and Mapping (SAM) file format
was designed to capture all of the critical information about NGS data in a single indexed and compressed file– Becoming a standard and is now used by production
informatics, MPG, and cancer analysis groups at the Broad• Has enabled sharing of data across centers and the
development of tools that work across platforms • More info at http://samtools.sourceforge.net/

Local realignment around indels

SLX GA 454 SOLiD HiSeqComplete Genomics
first of pair readssecond of pair reads first of pair readssecond of pair reads first of pair readssecond of pair reads
Base Quality Score Recalibration

How do indel realignment and base quality recalibration affect SNP
calling?
http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibrationhttp://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels
6.5% of calls on raw reads are likely false positives due to indels
The process doesn’t remove real SNPs

Sensitivity and specificity improved by simultaneous variant calling in 50-100 individuals
• Sensitivity– Greater statistical evidence compiled for true variants
seen in more than one individual • Specificity
– Deviations in metrics that flag false positive sites become much more statistically significant
• Allele balance: Departure from 50:50 (r:nr) in heterozygotes• Strand bias: non-reference allele preferentially seen on one
of the two DNA strands• Proportion of reads with low mapping quality

The Genome Analysis Toolkit (GATK) enables rapid development of efficient and robust analysis tools
Genome Analysis Toolkit (GATK) infrastructure
Analysis tool
Traversal engine
Implemented by userProvided by framework More info: http://www.broadinstitute.org/gsa/wiki/
•All of these tools have been developed in the GATK
•They are memory and CPU efficient, cluster friendly and are easily parallelized
•They are now publically and are being used at many sites around the world
•Supports any BAM-compatible aligner
Initial alignment
MSA realignment
Q-score recalibration
Multi-sample genotyping
SNP filtering
M. DePristo

Additional key advance
• Correcting alignment artifacts and machine-specific biases in read base calling and quality score assignment enables machine-independent variant identification and genotype calling
• 1000 Genomes data even contains individuals with data merged from multiple sequencing platforms!

For our project this is key
• With two centers generating data via distinct experimental and sequencing procedure, harmonizing data is integral to downstream analysis

Stratified analyses
• Because both processes will not afford equivalent coverage of all targets:– Critical that case-control balance and individual
pairings are preserved within center– Final analysis will be stratified by center such that
rare technical differences, lack of coverage on one or the other platform, etc can be managed robustly

Secondary data cleaning is critical
• Identify a quality set of individuals• Identify a quality set of targets• Identifying a quality set of variants

Primary cleaning
• Identify a quality set of individuals• Identify a quality set of targets• Identifying a quality set of variants

Sample composition thus farBatch Case Control Total
1 25 25 50
2 22 21 43
3 41 12 53
4 25 25 50
5 25 25 50
6 25 25 50
Total 164 132 296

Individual Cleaning
• Mean depth of coverage for all targets• Concordance rate with ‘super clean’ SNP Chip
– Contamination checks

Mean Coverage per Sample
Exclude this one

Concordance and contamination checks
• 1/296 samples fails concordance check (genotype call discrepancy) with SNP chip data (sample swap)
• 1/295 samples fails contamination check (proportion of reads calling non-reference alleles at SNP chip homozygous sites indicates >5% DNA from another individual)
• Advance a fully validated set of individuals to downstream analysis

Number of non-referencegenotypes per individual
1500 high frequency sites

Primary cleaning
• Identify a quality set of individuals• Identify a quality set of targets• Identifying a quality set of variants

Distribution of mean target coverage
99.86% targets

Depth vs GC Content
>95% of the targeted exons sequenced between 10 and 500x depth –Defined as successfully evaluated exome
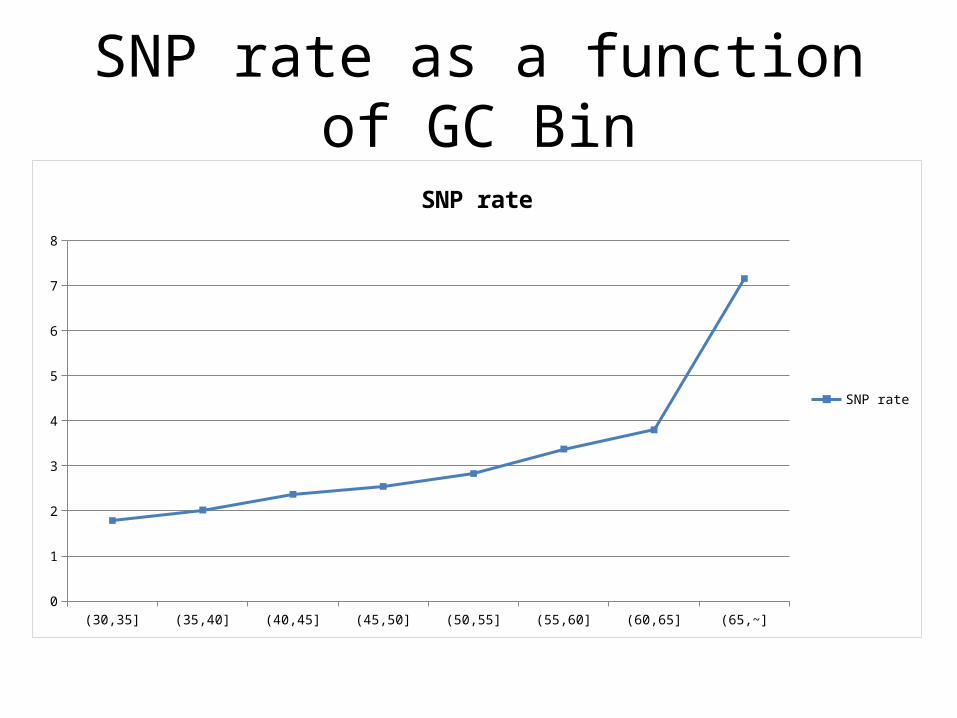
SNP rate as a function of GC Bin
(30,35] (35,40] (40,45] (45,50] (50,55] (55,60] (60,65] (65,~]0
1
2
3
4
5
6
7
8
SNP rate
SNP rate

% discovered variants that are singletons
50-250x - half the data, median coverage, singleton plateau at 34%
Lowest bin badly deficient in singletons – but higher rate ofcalled variants overall…
------- 95% of targets covered between 10 and 500x ----------

Primary cleaning
• Identify a quality set of individuals• Identify a quality set of targets• Identifying a quality set of variants

Defining the set of variant sites
• Define the technical parameters of true polymorphisms using a core set of ‘gold-standard’ true positive variant sites:– Are sites contained in a reference sample (e.g., dbSNP, another
exome or genome study)– High quality target depth range (50-250) – no true sites should
be missed and no excess coverage suggesting mapping concerns• Define distribution of technical properties (balance of
reference/non-reference alleles; balance of non-reference alleles on +/- strand; read mapping quality)– Filter non-dbSNP, non-ideal coverage calls based on these
distributions

Allele Balance Example
1% 99%95%5%
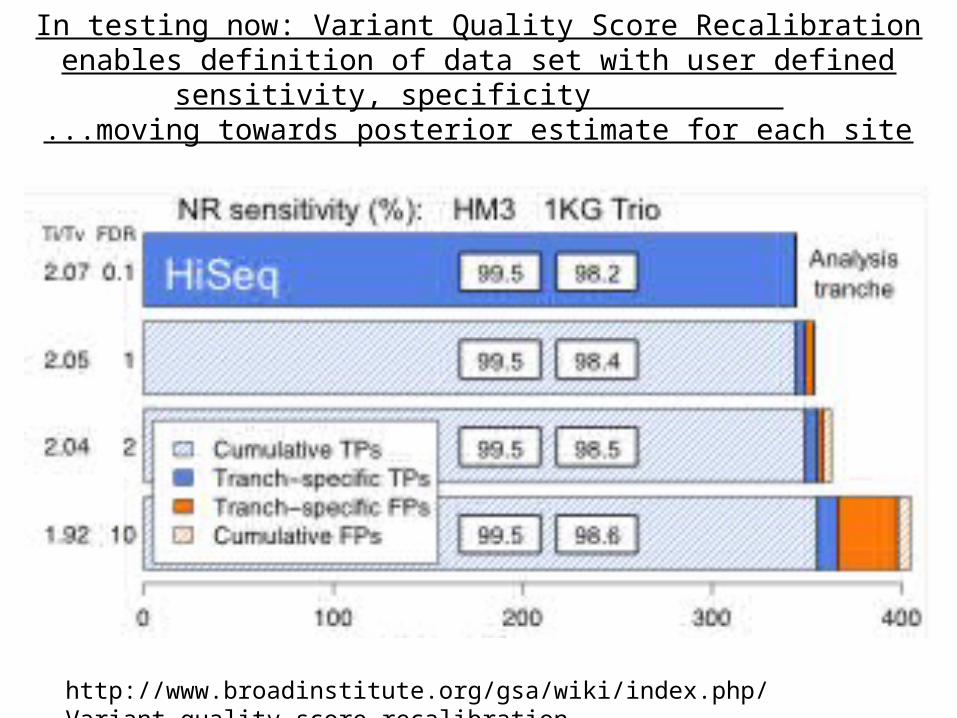
In testing now: Variant Quality Score Recalibration enables definition of data set with user defined sensitivity,
specificity ...moving towards posterior estimate for each site
http://www.broadinstitute.org/gsa/wiki/index.php/Variant_quality_score_recalibration

On to analysis…
• 204,123 variants pass all filters across 294 samples…number of all variants & singletons continue to increase as data is added
• How do we assess how this QC process performed downstream? Is the experiment working?

Has the matching worked?
• Matched samples based on MDS distance from combined GWAS data
• Consider the set of doubletons (two copies in the dataset)
• Overall, we should see that there are comparable numbers of variants seen in 2 cases or 2 controls versus 1 case and 1 control; and we should see an excess of 1 case:1 control variants in matched pairs
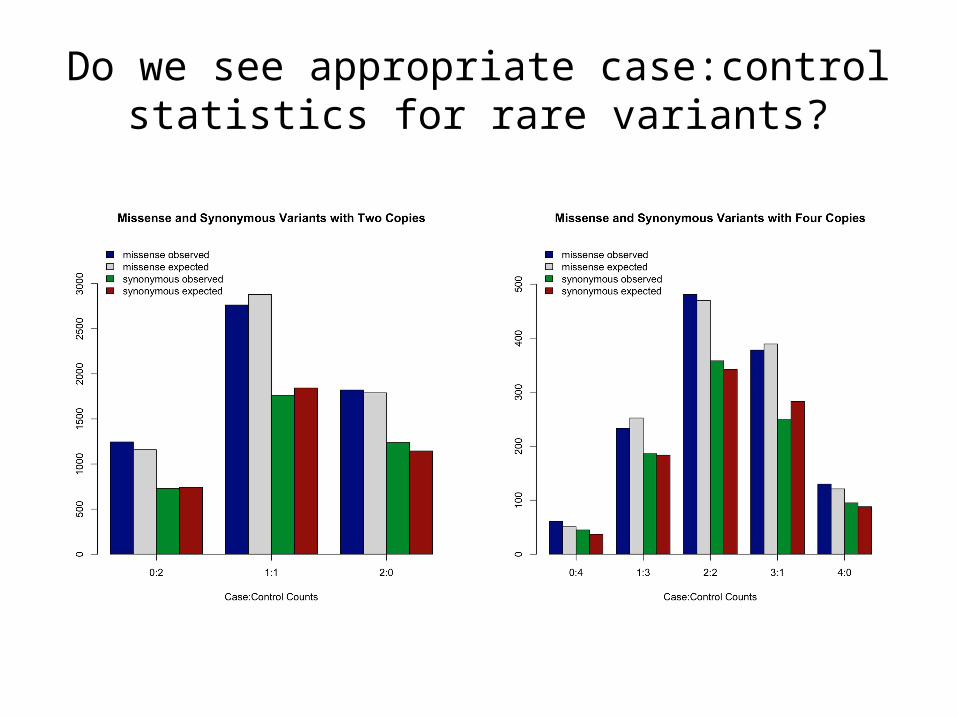
Do we see appropriate case:control statistics for rare variants?

Visual Representation
Case Control1234… …118
Case Control
Case Control
Case Control
Case Control
VS.
Case Control
Case Control
Case Control
Case Control
Case Control
Case Control
Case Control
Case Control
Case Control
Case Control
or
Expectation: 1/235 for within pair doubles 15,829 doubletons -> ~67.4Observed: 163 instances observed,
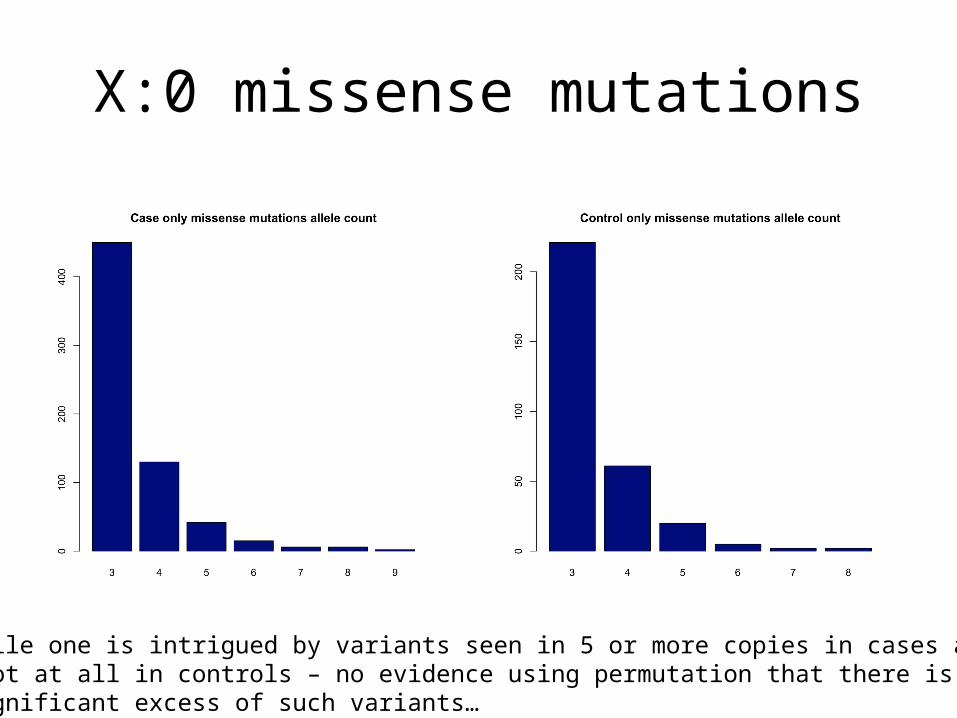
X:0 missense mutations
While one is intrigued by variants seen in 5 or more copies in cases and not at all in controls – no evidence using permutation that there is asignificant excess of such variants…

Specific nonsense mutations in cases or controls only
Impossible to pick out which might be relevant

Aggregations of rare nonsense mutations in single genes, all in cases only
• Encouraging – many genes where 1-3% of cases and no controls harbor nonsense mutations – best case scenario?

Genes with multiple nonsense mutations seen only in cases or controls
Many genes with 2 or more nonsense mutations seen only in cases –not appreciably different from rate in controls suggesting vast majority is simply the background rate at which such variants occur…

Challenges of connecting rare variation to complex phenotype
• Variation must be considered in aggregate per gene (or pathway…) rather than individually
• In phenotypically relevant genes, many rare variants will be neutral (i.e., background rate is high)
• Many documented cases exist where gain and loss of function mutations in same gene have opposite effects on phenotype
• Polygenicity will not be our friend here…realistically, no reason to think much smaller samples than in GWAS will be required
• at this point, the best case scenarios of highly penetrant rare mutations (that would have escaped prior large linkage and GWAS studies), aggregations of very rare alleles that explain 1% of the overall variance in risk, etc – cannot be distinguished from the background distribution of test statistics
• Some opportunistic models (.1%-.5% variants with OR~5-10, high penetrance recessive subtypes) may be able to be detected and confirmed through follow-up soon…no reason to have assurance these exist however

Parallel analysis tracks will be taken
• High MZ/DZ ratio suggests potential recessive component: search for excess autozygosity, IBD=2 sharing with affected sibs then homozygosity for rare allele; compound heterozygosity for rare alleles
• Highly deleterious alleles: Identify all non-synonymous/nonsense/splice variants unique to the study, not seen in 1000 Genomes or external control exome data (mostly singletons, very rare and de novo variants – perhaps ranked by predicted impact/PolyPhen) and compare burden in cases versus controls (testing a severe Mendelian mutation model)
• Heritable low frequency: Take all standing variation, observed two or more times in the study and perform sensitive test of gene-wide variation using C-alpha test of overdispersion (testing for effect of rare and low frequency variants of modest/intermediate penetrance across the gene)

Flexible, extensible data representation (variants, genotypes and meta-data)
A number of ways to use the librarycommand line, R, web, C/C++
Efficient random-access for very large datasets
Key references datasetsdbSNP, 1000G, PolyPhen2, gene transcript and sequence information
Large suite of up-to-date methods availableMadsen-Browning (+/- variable threshold), Li-Leal, C-alpha, etc.
Tools for analysis of variation data from next-generation sequencing platforms: the PLINK/Seq library
http://pngu.mgh.harvard.edu/purcell/pseq/
Shaun Purcell

PSEQ Features
• Individual statistics• Variant statistics • Single-locus association• Regional association• Incorporation of annotation information

Data Sharing
• Autism exome data made available
– Providing the gold-standard calibration for variant calling in other contemporaneous Broad exome studies
– Control variable sites and counts made available for comparison with other Broad exome studies
– First batch of raw data deposited to dbGAP exchange area – NIMH controls broadly consented for general medical use

Data Sharing
• Summary database of sites discovered, non-reference allele counts and target by target coverage provide invaluable cross-study information:– Technical validation of low frequency sites– Ability to evaluate whether specific sites or categories of
variants in certain genes are commonly, rarely or never seen
– Greatly enhance selection for follow-up– No individual level data or phenotype information need
be exchanged
Already in use across autism, schizophrenia, released 1000G studies - would love to collaborate with NHLBI & cancer studies at this level

Credits
• ARRA Autism sequencing– Mark Daly– Christine Stevens– Stacey Gabriel– Broad Sequencing Platform
• Jen Baldwin, Jane Wilkinson
Joe Buxbaum, Bernie Devlin, Richard Gibbs, Jerry Schellenberg, Jim Sutcliffe
NIMH, NHGRI
• Broad GSA team– Mark DePristo– Eric Banks– Kiran Garimella– Ryan Poplin
• Exome analysis– Mark Daly– Manny Rivas– Jared Maguire– Ben Voight– Shaun Purcell
– Kathryn Roeder & Bernie Devlin