anaplastic large cell lymphoma in leukemic phase: extraordinarily high white blood cell count
TRANSCRIPT

Case Report
Anaplastic large cell lymphoma in leukemic phase:Extraordinarily high white blood cell count
Jacqueline T. Nguyen,1 Michael R. Condron,1 Nghia D. Nguyen,1 Jitakshi De,1 L. Jeffrey Medeiros2 andAnthony Padula2
1Department of Pathology and Laboratory Medicine, University of Texas–Houston Medical School and 2Departmentof Hematopathology, University of Texas M. D. Anderson Cancer Center, Houston, Texas, USA
Anaplastic large cell lymphoma (ALCL) is a distinct type ofT/null-cell non-Hodgkin lymphoma that commonly involvesnodal and extranodal sites. The World Health Organizationof lymphoid neoplasms recognizes two types: anaplasticlymphoma kinase (ALK) positive or ALK negative, theformer as a result of abnormalities involving the ALK geneat chromosome 2p23. Patients with ALCL rarely develop aleukemic phase of disease, either at the time of initial pre-sentation or during the clinical course. Described herein isa patient with ALK+ ALCL, small cell variant, associated withthe t(2;5)(p23;q35), who initially presented with leukemicinvolvement and an extraordinarily high leukocyte count of529 ¥ 109/L, which subsequently peaked at 587 ¥ 109/L.Despite chemotherapy the patient died 21/2 months afterdiagnosis. In the literature review 20 well-documentedcases are identified of ALCL in leukemic phase reportedpreviously, with a WBC ranging from 15 to 151 ¥ 109/L.Leukemic phase of ALCL occurs almost exclusively inpatients with ALK+ ALCL, most often associated with thesmall cell variant and the t(2;5)(p23;q35), similar to thepresent case. Patients with leukemic phase ALK+ ALCLappear to have a poorer prognosis than most patients withALK+ ALCL.
Key words: ALK positive, anaplastic large cell lymphoma, leu-kemic phase, small cell variant, t(2;5)(p23;q35)
Anaplastic large cell lymphoma (ALCL) is recognized in theWorld Health Organization (WHO) classification of lymphoidneoplasms.1,2 These neoplasms are strongly positive for
CD30, usually express T-cell and cytotoxic antigens, andcarry monoclonal T-cell receptor gene rearrangements. In theinitial version of the WHO classification, ALCL was describedas one entity in which anaplastic lymphoma kinase (ALK)+ orALK– subgroups were recognized. The former group is rec-ognized as a relatively homogeneous group with abnormali-ties involving the ALK gene at chromosome 2p23 resulting inALK overexpression. The ALK– group of neoplasms is moreheterogeneous and there has been no clear consensus for itsexistence.3 Savage et al., however, reported that patientswith ALK– ALCL have a prognosis intermediate betweenALK+ ALCL and other types of T-cell lymphomas,4 and thecurrent updated WHO classification now recognizes ALK+and ALK– ALCL as separate entities.1,2
ALK+ ALCL has a number of histological variants.1,3 In themost common or classical variant, the neoplastic cells arelarge and anaplastic with a cohesive growth pattern. Sinusoi-dal involvement is common. Other common variants of ALK+ALCL include the lymphohistiocytic and small cell variants,and there are a number of uncommon variants such as thesarcomatoid variant.1,3 Usually these neoplasms have asubset of neoplastic cells that have distinctive cytologicalfeatures known as hallmark cells. A typical hallmark cell hasa horseshoe-shaped nucleus associated with a perinuclearhof. Hallmark cells can be rare in the small cell variant.
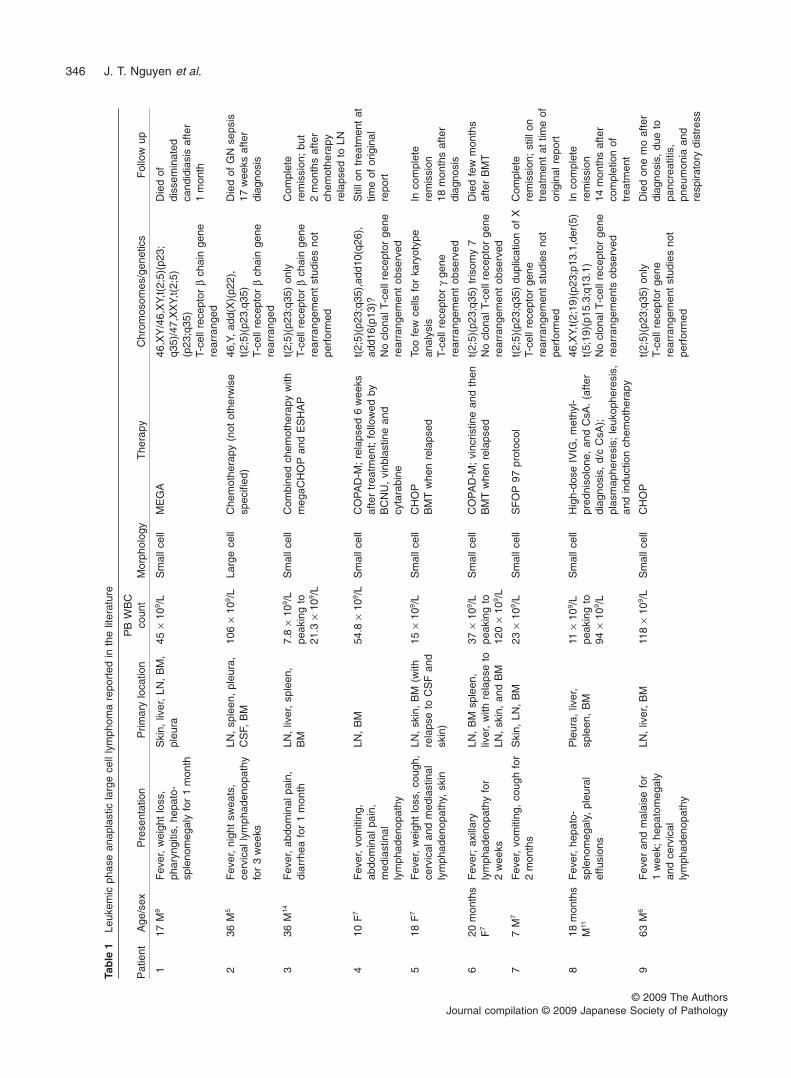
Although a variety of B- and T-cell non-Hodgkin lympho-mas can present with or evolve into a leukemic phase,patients with ALCL in leukemic phase have been reportedonly rarely. In our review of the literature we have identifiedonly 20 cases of ALCL with a leukemic phase (Table 1).5–17
Typically, patients present with an elevated WBC count <100¥ 109/L.
In this report we describe a young man with ALK+ ALCLwho initially presented with an unusually high WBC count of529 ¥ 109/L, which subsequently peaked at 587 ¥ 109/L.Despite therapy the patient died 21/2 months after diagnosis.
Correspondence: Anthony Padula, MD, Department of Hematopa-thology, Box 72, M. D. Anderson Cancer Center, 1515 HolcombeBoulevard, Houston, TX 77030, USA. Email: [email protected]
Received 19 November 2008. Accepted for publication 16 January2009.© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology
Pathology International 2009; 59: 345–353 doi:10.1111/j.1440-1827.2009.02376.x
Tab
le1
Leuk
emic
phas
ean
apla
stic
larg
ece
llly
mph
oma
repo
rted
inth
elit
erat
ure
Pat
ient
Age
/sex
Pre
sent
atio
nP
rimar
ylo
catio
nP
BW
BC
coun
tM
orph
olog
yT
hera
pyC
hrom
osom
es/g
enet
ics
Fol
low
up
117
M9
Fev
er,
wei
ght
loss
,ph
aryn
gitis
,he
pato
-sp
leno
meg
aly
for
1m
onth
Ski
n,liv
er,
LN,
BM
,pl
eura
45¥
109 /
LS
mal
lcel
lM
EG
A46
,XY
/46,
XY,
t(2;
5)(p
23;
q35)
/47,
XX
Y,t(
2;5)
(p23
;q35
)T-
cell
rece
ptor
bch
ain
gene
rear
rang
ed
Die
dof
diss
emin
ated
cand
idia
sis
afte
r1
mon
th
236
M5
Fev
er,
nigh
tsw
eats
,ce
rvic
ally
mph
aden
opat
hyfo
r3
wee
ks
LN,
sple
en,
pleu
ra,
CS
F,B
M10
6¥
109 /
LLa
rge
cell
Che
mot
hera
py(n
otot
herw
ise
spec
ified
)46
,Y,
add(
X)(
p22)
,t(
2;5)
(p23
,q35
)T-
cell
rece
ptor
bch
ain
gene
rear
rang
ed
Die
dof
GN
seps
is17
wee
ksaf
ter
diag
nosi
s
336
M14
Fev
er,
abdo
min
alpa
in,
diar
rhea
for
1m
onth
LN,
liver
,sp
leen
,B
M7.
8¥
109 /
Lpe
akin
gto
21.3
¥10
9 /L
Sm
allc
ell
Com
bine
dch
emot
hera
pyw
ithm
egaC
HO
Pan
dE
SH
AP
t(2;
5)(p
23;q
35)
only
T-ce
llre
cept
orb
chai
nge
nere
arra
ngem
ent
stud
ies
not
perf
orm
ed
Com
plet
ere
mis
sion
;bu
t2
mon
ths
afte
rch
emot
hera
pyre
laps
edto
LN
410
F7
Fev
er,
vom
iting
,ab
dom
inal
pain
,m
edia
stin
ally
mph
aden
opat
hy
LN,
BM
54.8
¥10
9 /L
Sm
allc
ell
CO
PA
D-M
;re
laps
ed6
wee
ksaf
ter
trea
tmen
t;fo
llow
edby
BC
NU
,vi
nbla
stin
ean
dcy
tara
bine
t(2;
5)(p
23;q
35),
add1
0(q2
6),
add1
6(p1
3)?
No
clon
alT-
cell
rece
ptor
gene
rear
rang
emen
tob
serv
ed
Stil
lon
trea
tmen
tat
time
ofor
igin
alre
port
518
F7
Fev
er,
wei
ght
loss
,co
ugh,
cerv
ical
and
med
iast
inal
lym
phad
enop
athy
,sk
in
LN,
skin
,B
M(w
ithre
laps
eto
CS
Fan
dsk
in)
15¥
109 /
LS
mal
lcel
lC
HO
PB
MT
whe
nre
laps
edTo
ofe
wce
llsfo
rka
ryot
ype
anal
ysis
T-ce
llre
cept
org
gene
rear
rang
emen
tob
serv
ed
Inco
mpl
ete
rem
issi
on18
mon
ths
afte
rdi
agno
sis
620
mon
ths
F7
Fev
er;
axill
ary
lym
phad
enop
athy
for
2w
eeks
LN,
BM
sple
en,
liver
,w
ithre
laps
eto
LN,
skin
,an
dB
M
37¥
109 /
Lpe
akin
gto
120
¥10
9 /L
Sm
allc
ell
CO
PA
D-M
;vi
ncris
tine
and
then
BM
Tw
hen
rela
psed
t(2;
5)(p
23;q
35)
tris
omy
7N
ocl
onal
T-ce
llre
cept
orge
nere
arra
ngem
ent
obse
rved
Die
dfe
wm
onth
saf
ter
BM
T
77
M7
Fev
er,
vom
iting
,co
ugh
for
2m
onth
sS
kin,
LN,
BM
23¥
109 /
LS
mal
lcel
lS
FO
P97
prot
ocol
t(2;
5)(p
23;q
35)
dupl
icat
ion
ofX
T-ce
llre
cept
orge
nere
arra
ngem
ent
stud
ies
not
perf
orm
ed
Com
plet
ere
mis
sion
;st
illon
trea
tmen
tat
time
ofor
igin
alre
port
818
mon
ths
M11
Fev
er,
hepa
to-
sple
nom
egal
y,pl
eura
lef
fusi
ons
Ple
ura,
liver
,sp
leen
,B
M11
¥10
9 /L
peak
ing
to94
¥10
9 /L
Sm
allc
ell
Hig
h-do
seIV
IG,
met
hyl-
pred
niso
lone
,an
dC
sA.
(afte
rdi
agno
sis,
d/c
CsA
);pl
asm
aphe
resi
s;le
ukop
here
sis,
and
indu
ctio
nch
emot
hera
py
46,X
Y,t(
2;19
)(p2
3;p1
3.1,
der(
5)t(
5;19
)(p1
5.3;
q13.
1)N
ocl
onal
T-ce
llre
cept
orge
nere
arra
ngem
ents
obse
rved
Inco
mpl
ete
rem
issi
on14
mon
ths
afte
rco
mpl
etio
nof
trea
tmen
t
963
M6
Fev
eran
dm
alai
sefo
r1
wee
k;he
pato
meg
aly
and
cerv
ical
lym
phad
enop
athy
LN,
liver
,B
M11
8¥
109 /
LS
mal
lcel
lC
HO
Pt(
2;5)
(p23
;q35
)on
lyT-
cell
rece
ptor
gene
rear
rang
emen
tst
udie
sno
tpe
rfor
med
Die
don
em
oaf
ter
diag
nosi
s,du
eto
panc
reat
itis,
pneu
mon
iaan
dre
spira
tory
dist
ress
346 J. T. Nguyen et al.
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

106
F12
Fev
er,
resp
irato
rydi
stre
ssLu
ng,
bila
tera
lki
dney
s,B
M60
¥10
9 /L
peak
ing
to21
6¥
109 /
L
Sm
allc
ell
AP
Oin
itial
ly,
then
MIE
D,
then
alte
rnat
ing
lom
ustin
e,vi
nbla
stin
e,an
dbl
eom
ycin
,an
dlo
mus
tine,
vinb
last
ine,
and
cyta
rabi
ne;
then
BM
T*
46,X
X,t(
2;5)
(p23
;q35
),de
l(10)
(q24
)[17
]/46,
idem
,-de
l(10)
(q26
),+a
dd(1
0)(q
26)[
3]T-
cell
rece
ptor
gene
rear
rang
emen
tst
udie
sno
tpe
rfor
med
Dis
ease
free
17m
onth
saf
ter
diag
nosi
s
119
mon
ths
F12
Fev
er,
coug
h,ly
mph
aden
opat
hyLN
,sp
leen
,liv
er,
lung
,sk
in,
BM
35¥
109 /
Lpe
akin
gto
104
¥10
9 /L
Sm
allc
ell
Initi
altr
eatm
ent
cort
icos
tero
ids,
cycl
opho
spha
mid
e,an
dvi
nbla
stin
e;th
ende
xam
etha
sone
,cy
clop
hosp
ham
ide,
daun
orub
icin
,as
para
gina
sean
din
trat
heca
lmet
hotr
exat
e,fo
llow
edby
cyta
rabi
nean
det
opos
ide;
then
wee
kly
dose
sof
vinb
last
ine
46,X
Y,t(
2;19
)(p2
3;p1
3.1)
,der
(5)
t(5;
19)(
p15.
3;q1
3.1)
T-ce
llre
cept
orge
nere
arra
ngem
ent
stud
ies
not
perf
orm
ed
Die
d9
mon
ths
afte
rdi
agno
sis
1210
M12
Left
alve
olar
ridge
mas
sLN
,C
NS
,B
M,
max
illar
ysi
nus
151
¥10
9 /L
(not
atpr
esen
tatio
nbu
tdu
ring
dise
ase
prog
ress
ion)
Sm
allc
ell
MIE
D;A
PO
;cy
clop
hosp
ham
ide,
met
hotr
exat
e,et
opos
ide;
clad
ribin
e,fo
llow
edby
wee
kly
vinb
last
ine
for
1ye
ar
t(2;
5)(p
23;q
35)
only
T-ce
llre
cept
orge
nere
arra
ngem
ent
stud
ies
not
perf
orm
ed
Die
d2
year
saf
ter
diag
nosi
s
1332
F10
Fev
erfo
r3
wee
ksLN
,sp
leen
,liv
er,
pleu
ra,
CN
S,
BM
76.4
¥10
9 /L
Sm
allc
ell
CH
OP
46,X
X,t(
2;5;
13)(
p23;
q35;
q14)
[6]/4
6,id
em,
add(
19)(
q13.
3)[3
]/46,
XX
[14]
T-ce
llre
cept
orge
nere
arra
ngem
ent
stud
ies
not
perf
orm
ed
Die
dof
resp
irato
rydi
stre
ss2
mon
ths
afte
rdi
agno
sis
1429
M8
Fev
er,
head
ache
,ab
dom
inal
pain
LN,
BM
,liv
er,
sple
en,
pleu
ra,
CN
S
20¥
109 /
Lpe
akin
gto
81.3
¥10
9 /L
Sm
allc
ell
Hyp
er-C
VA
Dw
ithin
trat
heca
lm
etho
trex
ate;
and
late
rifo
sfam
ide
and
carb
opla
tinfo
llow
edby
BM
T
46,X
Y,t(
2;5)
(p23
;q35
)[5]
and
47,id
em,+
X[5
]T-
cell
rece
ptor
gene
rear
rang
emen
tst
udie
sno
tpe
rfor
med
Die
dof
GV
HD
1m
onth
afte
rB
MT
1511
M8
Fev
er,
vom
iting
,di
arrh
ea,
abdo
min
alpa
inLN
,B
M,
liver
,sp
leen
,lu
ng,
kidn
eys,
CN
S
18.3
¥10
9 /L
peak
ing
to23
.8¥
109 /
L
Sm
allc
ell
CC
G-5
941,
then
D-I
CE
46,X
Y,t(
2;5)
(p23
;q35
)[12
]an
d46–
47,
idem
,+7,
+19[
cp2]
Sub
sequ
ent
clon
alev
olut
ion
to46
,X
Y,t(
2;5)
(p23
;q35
),de
r(2)
,t(
2;18
)(p2
3;q1
1.2)
,+7,
add(
17)(
p11.
2),-
18[1
5]
Die
dof
subf
alci
nehe
rnia
tion
and
resp
irato
rydi
stre
ssaf
ter
trea
tmen
tw
ithdr
awal
,3
mon
ths
afte
rdi
agno
sis
1659
F8
Fev
er,
wei
ght
loss
LN,
BM
,liv
er14
9.9
¥10
9 /L
Sm
allc
ell
CH
OP
46,X
X,t(
2;5)
(p23
;q35
)an
d46
,X,a
dd(X
)(p2
2.1)
,t(
2;5)
(p23
;q35
)
Die
d1
mon
thaf
ter
diag
nosi
s
1723
F13
Fev
er,
wea
knes
sLN
,liv
er,
sple
en,
skin
,B
M24
.5¥
109 /
LS
mal
lcel
lC
HO
P;
auto
logo
usB
MT;
CH
OP
agai
naf
ter
lung
rela
pse
t(2;
5;3)
(p23
;q35
;p21
)A
live
attim
eof
repo
rt
ALK+ ALCL in marked leukemic phase 347
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

Tab
le1
Con
tinue
d
Pat
ient
Age
/sex
Pre
sent
atio
nP
rimar
ylo
catio
nP
BW
BC
coun
tM
orph
olog
yT
hera
pyC
hrom
osom
es/g
enet
ics
Fol
low
up
1810
M16
Fev
er,
coug
h,he
pato
sple
nom
egal
y,ly
mph
aden
opat
hy
1.8
¥10
9 /L
peak
ing
to26
.2¥
109 /
L
Sm
allc
ell
Dex
amet
haso
ne,
high
-dos
ecy
tara
bine
,an
dvi
ndes
ine
(NH
L-B
FM
-90)
t(2;
5)(p
23;q
35)
t(4;
11)(
p12;
p15)
der(
10;1
8)(q
10;q
10)
+18,
+22
T-ce
llre
cept
orge
nere
arra
ngem
ent
stud
ies
not
perf
orm
ed
Die
dbe
fore
com
plet
ion
ofch
emot
hera
pydu
eto
Sta
phyl
ococ
cus
aure
usse
ptic
emia
and
diss
emin
ated
intr
avas
cula
rco
agul
atio
n
1925
M17
Wei
ght
loss
,ni
ght
swea
ts,
lym
phad
enop
athy
hepa
to-
sple
nom
egal
y
LN,
sple
en,
liver
,B
M(s
ubse
quen
tly)
Nor
mal
atpr
esen
tatio
n,pe
akin
gto
22¥
109 /
L
Larg
ece
llN
otsp
ecifi
edt(
2;5)
(p23
;q35
)N
umer
ous
othe
rch
rom
osom
eab
norm
aliti
esT-
cell
rece
ptor
bge
nere
arra
ngem
ent
obse
rved
Die
d3
mon
ths
afte
rtr
eatm
ent
2052
M15
Ski
nra
sh,
hepa
to-
sple
nom
egal
y,ly
mph
aden
opat
hy
Ski
n,LN
,sp
leen
,liv
er,
BM
15.5
¥10
9 /L
Larg
ece
llN
otre
port
edC
ompl
exka
ryot
ypic
abno
rmal
ityw
ithch
rom
osom
es1,
2,5,
7,8,
and
15 No
t(2;
5)tr
ansl
ocat
ion;
ALK
-neg
ativ
eD
iagn
osis
base
don
mor
phol
ogy
and
imm
unop
erox
idas
est
udie
sT-
cell
rece
ptor
gene
rear
rang
emen
tst
udie
sno
tpe
rfor
med
Not
repo
rted
2126
M(P
rese
ntca
se)
Fev
er,
abdo
min
alpa
in,
nigh
tsw
eats
LN,
BM
587
¥10
9 /L
Sm
allc
ell
Hyp
er-C
VA
D,
met
hotr
exat
e,A
ra-C
,cy
tara
bine
t(2;
5)(p
23;q
35),
add(
18)(
q21)
T-ce
llre
cept
org
gene
rear
rang
emen
tob
serv
edN
oim
mun
oglo
bulin
heav
ych
ain
(IgH
)ge
nere
arra
ngem
ent
obse
rved
Die
d21 /
2m
onth
saf
ter
diag
nosi
s
AP
O,
doxo
rubi
cin,
vinc
ristin
e,pr
edni
sone
;B
M,
bone
mar
row
;B
MT,
bone
mar
row
tran
spla
nt;
BM
T*,
BM
Tw
ithpr
epre
gim
enco
nsis
ting
ofto
talb
ody
irrad
iatio
n,th
iote
pa,
alem
tuzu
mab
,et
opos
ide,
and
cycl
opho
spha
mid
e;C
CG
-594
1,vi
ncris
tine,
L-as
para
gina
se,
pred
niso
nean
din
trat
heca
lmet
hotr
exat
ein
duct
ion
follo
wed
byco
nsol
idat
ion
with
vinc
ristin
e,et
opos
ide
(VP
-16)
,cy
tosi
near
abin
osid
e(A
ra-C
/VM
-26)
,6-
thio
guan
ine,
high
-dos
em
etho
trex
ate
and
intr
athe
calm
etho
trex
ate,
for
6w
eeks
;C
HO
P,cy
clop
hosp
ham
ide,
adria
myc
in,
vinc
ristin
e,an
dpr
edni
sone
;C
OP
AD
-M,
cycl
opho
spha
mid
e,vi
ncris
tine,
pred
niso
ne,
adria
myc
inan
dm
etho
trex
ate;
CsA
,cy
clos
porin
A;
CS
F,ce
rebr
ospi
nal
fluid
;D
-IC
E,
dexa
met
haso
ne,
ifosf
amid
e,ci
spla
tin,
and
etop
osid
e;E
SH
AP,
etop
osid
e,ci
spla
tin,
pred
niso
ne,a
ndcy
tara
bine
;GN
,Gra
m-n
egat
ive;
GV
HD
,gra
ftve
rsus
host
dise
ase;
hype
r-C
VA
D,c
yclo
phos
pham
ide,
vinc
ristin
e,do
xoru
bici
n,an
dde
xam
etha
sone
;ind
uctio
nch
emot
hera
py,a
dria
myc
in,
cycl
opho
spha
mid
e,cy
tosi
near
abin
osid
e,da
unor
ubic
in,
etop
osid
e,hi
gh-d
ose
met
hotr
exat
e,vi
ncris
tine,
and
intr
athe
cal
cyto
sine
arab
inos
ide
but
excl
udin
gst
eroi
ds;
LN,
lym
phno
de;
ME
GA
,m
ega
chem
othe
rapy
(dos
e-in
tens
ive
prot
ocol
atV
ande
rbilt
Uni
vers
ity);
meg
aCH
OP,
cycl
opho
spha
mid
e,ad
riam
ycin
,vi
ncris
tine,
and
pred
niso
ne;
MIE
D,
high
-dos
em
etho
trex
ate,
ifosf
amid
e,et
opos
ide,
and
dexa
met
haso
ne;
ND
,no
tdo
ne;
PB
,pe
riphe
ralb
lood
;S
FO
P97
prot
ocol
,vi
nbla
stin
e,ad
riam
ycin
,de
xam
etha
sone
,an
dm
etho
trex
ate.
348 J. T. Nguyen et al.
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

We discuss the clinicopathological, immunophenotypic, andcytogenetic and molecular features of this case, review theliterature, and discuss the differential diagnoses.
CLINICAL SUMMARY
A 26-year-old Hispanic man with no significant past medicalhistory presented with abdominal pain, fever, chills, and nightsweats. Physical examination and radiological imaging indi-cated cervical lymphadenopathy and multiple small mesen-teric and inguinal lymph nodes. The complete blood count atthe time of initial examination was: WBC 529 ¥ 109/L (normalrange, 4–11 ¥ 109/L), platelet count 174 ¥ 109/L (normalrange, 150–440 ¥ 109/L), and hematocrit 40.5% (normalrange, 41.5–50.44%). A manual differential count showed 5%neutrophils, 51% lymphocytes, and 25% atypical lympho-cytes. Bone marrow aspiration and biopsy were performedthat indicated numerous atypical lymphoid cells. Based onthe morphological findings and initial flow cytometry immu-nophenotypic data, a number of different diagnoses wereconsidered before the diagnosis of ALK+ ALCL was estab-lished. The patient was treated with intensive chemotherapycomposed of three cycles of Hyper-CVAD chemotherapy(fractionated cyclophosphamide, vincristine, doxorubicin,and dexamethasone with intrathecal methotrexate and cyt-arabine, alternating cycles every 3 weeks with high-dosemethotrexate and cytarabine).
The patient’s WBC count at the start of chemotherapy was587 ¥ 109/L, which decreased to 72.5 ¥ 109/L 2 weeks aftercycle 2, and reached a nadir of 32 ¥ 109/L after cycle 3. TheWBC increased to 147 ¥ 109/L, however, by the time cycle 4was to be administered. Consideration had been given tointensification with humanized monoclonal antibody to CD52(Alemtuzumab). In addition, the management plan includedevaluating the patient for potential hematopoietic stem celltransplantation, but as an undocumented resident in the USAhe was not eligible. Therefore, the patient decided to return tohis native country and within 1 week was admitted to a localhospital with visual complaints, headache, and obtundation.The patient died 2.5 months after initial presentation.
PATHOLOGICAL FINDINGS
Procedure
Hematology and histology
Peripheral blood and bone marrow aspirate smears were airdried and stained with Wright-Giemsa. A 400-cell differentialcount was performed on bone marrow aspirate smears. Thedecalcified bone marrow core biopsy and the aspirate clot
specimens were fixed in formalin and paraffin-embedded,and histological sections were stained with HE.
Immunohistochemistry
Immunohistochemistry was performed using formalin-fixed,paraffin-embedded tissue sections of the bone marrowaspirate clot specimen. The avidin–biotin peroxidasetechnique was used with either a Ventana Benchmarkautostainer (Tucson, AZ, USA) or a Dako autostainer(Santa Barbara, CA, USA) using antibodies specific forALK-1 (monoclonal), terminal deoxynucleotidyl transferase(TdT; polyclonal), Pax-5 (monoclonal), and CD117 (poly-clonal; Cell Marque, Hot Springs, AR, USA); epithelialmembrane antigen (EMA)/Mc5 (monoclonal, Ventana); andCD30/Ber-H2 (monoclonal) and LMP-1/CS.1-4 (monoclonal;Dako, Glostrup, Denmark). In situ hybridization for EBV-encoded RNA (EBER; PanPath Rembrandt, Amsterdam,The Netherlands) was also performed.
Flow cytometry immunophenotyping
Immunophenotypic analysis of peripheral blood and bonemarrow aspirate samples was performed using a whole bloodlysis method followed by three-color flow cytometry on aFACSort analyzer (Becton Dickinson, San Jose, CA, USA)using the CD45 versus side-scatter gating technique. Mono-clonal antibodies, conjugated to fluorescein isothiocyanate orphycoerythrin, were used that were specific for CD3 (Leu-4,clone SK7), CD4 (Leu-3a, clone SK3), CD5 (clone 17F12),CD7 (clone 4H9), CD8 (Leu-2a, clone SK1), CD10 (cloneHI10a), CD11c (clone S-HCL-3), CD13 (clone L138), CD14(clone MFP9), CD16 (Leu-11b, clone GO22), CD19 (Leu-12,clone 4G7), CD20 (clone L27), CD22 (clone S-HCL-1), CD23(clone EBVCS-5), CD25 (clone 2A3), CD33 (clone P67.6),CD34 (clone My10), CD45 (anti–Hle-1, clone 2D1), CD52(clone CF1D12), CD56 (clone MY31), human leukocyteantigen (HLA)-DR (clone L243), and surface immunoglobulinkappa (clone TB28-2) and lambda (clone 1-155-2) lightchains. All antibodies were supplied by Becton Dickinsonexcept CD52, which was supplied by Caltag Laboratories(Invitrogen, Carlsbad, CA, USA).
Cytogenetics and molecular analysis
Conventional cytogenetic analysis was performed on thebone marrow aspirate material. Three metaphases wereanalyzed from one 24 h unstimulated culture, and anadditional 17 metaphases were examined from a 72 hlipopolysaccharide-stimulated culture.
Fluorescence in situ hybridization (FISH) was performedon bone marrow aspirate smears using the LSI ALK dualcolor breakapart DNA probe (Vysis, Downers Grove, IL,USA) which hybridizes to the 2p23 locus according to themanufacturer’s instructions.
ALK+ ALCL in marked leukemic phase 349
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

Molecular analysis for T-cell receptor gamma chain generearrangements was performed using a peripheral bloodspecimen and a polymerase chain reaction (PCR)-basedmethod utilizing primers specific for the variable (V) andjoining (J) regions of the T-cell receptor gamma chain gene.The amplified products were analyzed by capillary electro-phoresis.
Molecular analysis for immunoglobulin heavy chain immu-noglobulin gene rearrangements was performed using aperipheral blood specimen and a PCR-based method usingprimers specific for the frameworks 1, 2, and 3 of the immu-noglobulin heavy chain gene V regions and primers specificfor the J region. The amplified products were analyzed bycapillary electrophoresis.
RESULTS
Morphological findings
The peripheral blood smear (Fig. 1) showed normocytic nor-mochromic anemia with marked leukocytosis and lymphocy-tosis. The white blood cell count was 529 ¥ 109/L with 51%lymphocytes. Most of the lymphocytes were atypical, small tomedium in size, with irregular nuclear contours. A smallernumber, <5%, were larger with vacuolated and basophiliccytoplasm. Rare cells with an irregularly shaped nucleusresembling cloverleaf and horseshoe shapes were observed.
The bone marrow aspirate smears showed numerousatypical lymphocytes admixed with maturating bone marrowelements. The bone marrow aspirate clot specimen (Fig. 2)was hypercellular (90%) and also showed a predominantpopulation of atypical lymphoid cells, representing approxi-mately 60% of all cells. The bone marrow biopsy specimenwas of very poor quality, being extensively fragmented withaspiration artifact and almost no hematopoietic bone marrow.
Immunophenotype
Flow cytometry immunophenotyping of peripheral blood andbone marrow aspirate specimens showed an abnormal popu-lation of cells positive for CD4, CD5, CD13, CD25, CD45,CD52, and HLA-DR, and negative for CD3, CD7, CD8,CD10, CD11C, CD14, CD16, CD19, CD20, CD22, CD23,CD33, CD34, CD56, and immunoglobulin kappa and lambdalight chains.
Immunohistochemistry performed on the bone marrowaspirate clot specimen showed that the cells were positive forALK-1, CD30, and EMA. The CD30 pattern of expressionwas membranous and paranuclear (Fig. 3). The pattern ofALK-1 was nuclear and cytoplasmic (Fig. 4). The neoplasticcells were negative for CD117, TdT, PAX5, and EBV latent
membrane protein type 1. In situ hybridization for EBER wasnegative.
Chromosome and molecular studies
Conventional cytogenetics of the bone marrow aspirate speci-men (Fig. 5) showed eight normal and 12 abnormal meta-phases with the following karyotype: 46,XY,t(2;5)(p23;q35),add(18)(q21). FISH showed disruption of the ALK gene on2p23.
Molecular studies of a peripheral blood specimen showeda monoclonal T-cell receptor gamma chain gene rearrange-ment and no evidence of monoclonal immunoglobulin heavychain gene rearrangement.
DISCUSSION
Leukemic involvement is highly unusual in patients withALCL. Including the present case, we have identified a totalof 21 cases of ALCL in leukemic phase reported in the litera-ture (Table 1). Almost all of these cases, 20 of 21 (95.2%),were ALK+ ALCL. Eighteen of the 20 (95%) ALK+ ALCL weredescribed as or designated as the small cell variant andtherefore we agree with Onciu et al. that this variant seems tobe particularly prone to entering the blood in large numbers.12
Although the explanation for why small cell variant of ALCLcommonly presents with a leukemic phase is unknown, cyto-genetic abnormalities in addition to the t(2;5) translocationmay provide a clue. Twenty cases reported have availablecytogenetic data. Most cases of leukemic phase ALK+ ALCLhave been associated with the t(2;5)(p23;q35): 17 of 20(85%). Two neoplasms reported had three-way translo-cations: t(2;5;13)(p23;q35;q14) and t(2;5;3)(p23;q35;p21).Of the three cases without the t(2;5), one case involvedt(2;19)(p23;p13.1),11 one case involved poorly describedabnormalities of chromosome 2, 5, and 20,12 and one case ofALK– ALCL involved a complex karyotype with abnormalitiesof chromosomes 1, 2, 5, 7, 8, and 15.15 Because the t(2;5)occurs in approximately 80% of all ALK+ ALCL, it does notappear that the t(2;5) is preferentially associated with theleukemic phase of disease.
Clinically, the age range of affected patients is wide, from7 months to 63 years, but 15 of 21 patients (71.4%) wereyounger than 30 years of age. There is a male predominance(61.9%). These findings are in keeping with the tendency ofALK+ ALCL to affect younger male patients. Most patientshave B-type symptoms, with fever most common, reported in18 of 21 patients (85.7%). Weight loss and night sweats areless common, reported in fewer than one-third of patients.Patients with leukemic phase ALK+ ALCL typically havewidespread involvement of many organ systems at the time
350 J. T. Nguyen et al.
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

of diagnosis, as shown by physical examination and radio-logical studies. The degree of leukocytosis is variable, but isusually under 100 ¥ 109/L in most patients. Four patientshave had WBC counts >100 ¥ 109/L: 106, 118, 149.9, and151 ¥ 109/L. The present case was highly unusual, involvinga peak WBC of 587 ¥ 109/L, almost fourfold higher than in anyother case reported.
Patients with ALK+ ALCL are usually younger and have amore favorable clinical course than patients with other typesof T-cell lymphoma when treated with appropriate multi-agentchemotherapy. This is true despite the fact that patients withALK+ ALCL commonly present with advanced disease and Bsymptoms.1,3 Patients with leukemic phase ALK+ ALCL,however, appear to be a poor prognostic subset. At least 13patients with leukemic phase ALK+ ALCL have died ofdisease, usually within a few months, and additional patientshad relapsed by the time their case was reported. Thissuggestion has been made previously by others,12 and thepresent case supports this interpretation.
Figure 2 Bone marrow clot specimen (original magnification ¥500)showing sheets of lymphoma cells, mostly small–intermediate, withscattered large cells having irregular nuclei and occasional promi-nent nucleoli, admixed with normal hematopoietic elements. A mitoticfigure is observed.
Figure 3 Bone marrow clot specimen (original magnification ¥500).Immunohistochemical staining for CD30 is strongly positive in thesubset of large neoplastic cells (as is usually the case in this variant)in a membranous and paranuclear dot-like pattern.
Figure 4 Bone marrow clot specimen (original magnification ¥500).Immunohistochemical staining for anaplastic lymphoma kinase high-lights the neoplastic cells in a nuclear and cytoplasmic pattern. Thelarger cells show a stronger staining pattern, as is usually the case inthis variant.
Figure 1 (a) Peripheral blood smear(original magnification ¥1000) showingsmall–intermediate sized lymphomacells with irregular nuclei and a giant cellwith hallmark-like features. (b) Periph-eral blood smear (original magnification¥1000) showing smaller lymphoma cellsand occasional large cells, one with aprominent mitosis.
a b
ALK+ ALCL in marked leukemic phase 351
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

Microscopically, most of the neoplastic cells of ALK+ ALCLin a peripheral blood smear often have ‘flower-like’ or irregu-lar nuclear shapes and do not resemble the common appear-ance of ALK+ ALCL cells in tissue biopsy specimens. Rarelarge cells with horseshoe shaped nuclei, however, cansometimes be identified. The presence of these large cells isa clue to the correct diagnosis, although the predominantsmall cell features and the rarity of leukemic presentationcreate a diagnostic challenge.
Other entities that are part of the morphological differentialdiagnosis include adult T-cell leukemia/lymphoma, T lympho-blastic leukemia/lymphoma, Sezary syndrome, and T-cellprolymphocytic leukemia. The circulating neoplastic cells ofadult T-cell leukemia/lymphoma are well known for theirflower-like nuclear appearance, and patients often presentsimilarly to the present patient, with a high WBC count andwidespread disease. Adult T-cell leukemia/lymphoma alsocan express CD30 in a subset of cases. This possibilitywas excluded in the current case by serological testing forhuman T-cell lymphotropic virus (HTLV)-1 and HTLV-2, whichwas negative. In addition, cases of adult T-cell leukemia/lymphoma do not express cytotoxic markers, the pattern andintensity of CD30 expression is usually variable and lessintense, and these neoplasms lack ALK expression andt(2;5)(p23;q35). The neoplastic cells of T-lymphoblasticleukemia/lymphoma typically have more blastic chromatinand they have an immature T-cell immunophenotype, includ-ing expression of TdT. T-lymphoblastic leukemia/lymphomais negative for CD30 and ALK. Sezary syndrome is charac-terized by the presence of larger Sezary cells and smallerLutzner type cells that can appear multilobate or flower-like inthe peripheral blood smear. The present patient did not haveerythroderma or skin lesions to support the diagnosis of
Sezary syndrome. The neoplastic cells of Sezary syndromerarely express CD30 or cytotoxic antigens and are negativefor ALK. Lastly, T-cell prolymphocytic leukemia was initiallyconsidered as a potential diagnosis. Patients with T-cell pro-lymphocytic leukemia (T-PLL) often present with clinicallyaggressive disease, a high WBC count, and extensive bonemarrow disease, as seen in the present patient. AlthoughT-cell prolymphocytes usually have a distinct central nucleo-lus, a subset of cases designated as small cell variant ofT-PLL can have Sezary cell-like features. Unlike the presentpatient, T-cell prolymphocytic leukemia usually affects olderpatients and the neoplastic cells do not express CD30 orALK.
The flow cytometry immunophenotypic findings in thepresent case also caused some initial confusion because theneoplastic cells expressed CD13, a myeloid-associatedmarker, which led to the diagnosis of acute biphenotypicleukemia of mixed T-cell and myeloid lineage being consid-ered. Recent flow cytometry immunophenotyping of ALCL,however, has shown that ALK+ ALCL commonly expressesCD13.18,19 This is now a recognized pitfall in establishing thediagnosis of ALCL by flow cytometry immunophenotyping.
In summary, we report a patient with ALK+ ALCL whopresented with an extraordinarily high WBC of 529 ¥ 109/Lthat subsequently peaked at 587 ¥ 109/L before therapy.Leukemic phase ALCL is uncommon, with only 19 cases ofALK+ ALCL reported in the literature. The present case wasunusual in that the degree of leukocytosis far exceeded pre-viously reported cases. Patients who present with leukemicphase ALCL are a diagnostic challenge, particularly initially,because morphological and flow cytometry immunopheno-typing can suggest other possible diagnoses. Consideringthe possibility of leukemic phase of ALCL, and performing anappropriate workup including immunohistochemical stains(e.g. CD30, ALK) and cytogenetic studies readily leads to thecorrect diagnosis.
REFERENCES
1 Delsol G, Jaffe ES, Falini B et al. Anaplastic large cell lym-phoma (ALCL), ALK-positive. In: Swerdlow SH, Campo E,Harris NL et al., eds. WHO Classification of Tumours of Hae-matopoietic and Lymphoid Tissues, 4th edn. Lyon: InternationalAgency for Research on Cancer, 2008; 312–16.
2 Mason DY, Campo E, Harris NL et al. Anaplastic large celllymphoma, ALK-negative. In: Swerdlow SH, Campo E, HarrisNL et al., eds. WHO Classification of Tumours of Haematopoi-etic and Lymphoid Tissues, 4th edn. Lyon: International Agencyfor Research on Cancer, 2008; 317–19.
3 Medeiros LJ, Elenitoba-Johnson KS. Anaplastic large cell lym-phoma. Am J Clin Pathol 2007; 127: 707–22.
4 Savage K, Harris NL, Vose JM et al. International PeripheralT-cell Lymphoma Project. Blood 2008; 111: 5496–504.
5 Anderson MM, Ross CW, Singleton TP, Sheldon S, Schnitzer B.Ki-1 anaplastic large cell lymphoma with a prominent leukemicphase. Hum Pathol 1996; 27: 1093–5.
Figure 5 Karyotype showing the t(2;5) translocation with additionalchromatin material on the long arm of chromosome 18.
352 J. T. Nguyen et al.
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology

6 Awaya N, Mori S, Takeuchi H et al. CD30 and the NPM-ALKfusion protein (p80) are differentially expressed between periph-eral blood and bone marrow in primary small cell variant ofanaplastic large cell lymphoma. Am J Hematol 2002; 69: 200–4.
7 Bayle C, Charpentier A, Duchayne E et al. Leukaemic presen-tation of small cell variant anaplastic large cell lymphoma:Report of four cases. Br J Haematol 1999; 104: 680–88.
8 Grewal JS, Smith LB, Winegarden JD et al. K-positive anaplas-tic large cell lymphoma with a leukemic phase and multi-organinvolvement: A report of three cases and a review of the litera-ture. Ann Hematol 2007; 86: 499–508.
9 Kinney MC, Collins RD, Greer JP, Whitlock JA, Sioutos N, KadinME. A small-cell-predominant variant of primary Ki-1 (CD30)+T-cell lymphoma. Am J Surg Pathol 1993; 17: 859–68.
10 Kong SY, Cho HJ, Suk JH et al. A novel complext(2;5;13)(p23;q35;q14) in small cell variant type anaplastic largecell lymphoma with peripheral involvement. Cancer Genet Cyto-genet 2004; 154: 183–5.
11 Meech SJ, McGavran L, Odom LF et al. Unusual childhoodextramedullary hematologic malignancy with natural killer cellproperties that contains tropomyosin 4-anaplastic lymphomakinase gene fusion. Blood 2001; 98: 1209–16.
12 Onciu M, Behm FG, Raimondi SC et al. ALK-positive anaplasticlarge cell lymphoma with leukemic peripheral blood involvementis a clinicopathologic entity with an unfavorable prognosis.Report of three cases and review of the literature. Am J ClinPathol 2003; 120: 617–25.
13 Sano F, Tasaka T, Nishimura H et al. Small cell variant ofanaplastic large cell lymphoma diagnosed by a novel chromo-somal abnormality t(2;5;3)(p23;q35;p21) of bone marrow cells.Pathol Int 2008; 58: 494–7.
14 Villamor N, Rozman M, Esteve J et al. Anaplastic large-celllymphoma with rapid evolution to leukemic phase. Ann Hematol1999; 78: 478–82.
15 Dalal BI, Chhanabhai M, Horsman DE, LeHuquet J, CouplandR. Anaplastic large-cell lymphoma presenting as acute leuke-mia. Am J Hematol 2005; 79: 164–5.
16 Takahashi D, Nagatoshi Y, Nagayama J et al. Anaplastic largecell lymphoma in leukemic presentation: A case report and areview of the literature. J Pediatr Hematol Oncol 2008; 30:696–700.
17 Fischer P, Nacheva E, Mason DY et al. Ki-1 (CD30)-positivehuman cell line (Karpas 299) established from a high-gradenon-Hodgkin’s lymphoma, showing a 2;5 translocation and rear-rangement of the T-cell receptor beta-chain gene. Blood 1988;72: 234–40.
18 Kesler MV, Paranjape GS, Asplund SL, McKenna RW, Jamal S,Kroft SH. Anaplastic large cell lymphoma: A flow cytometricanalysis of 29 cases. Am J Clin Pathol 2007; 128: 314–22.
19 Muzzafar T, Wei EX, Lin P, Medeiros LJ, Jorgensen JL. Flowcytometric immunophenotyping of anaplastic large cell lym-phoma. Arch Pathol Lab Med 2009; 133: 49–56.
ALK+ ALCL in marked leukemic phase 353
© 2009 The AuthorsJournal compilation © 2009 Japanese Society of Pathology