activation of the mitochondrial apoptotic pathway produces...
TRANSCRIPT

Research Article
Activation of the Mitochondrial ApoptoticPathway Produces Reactive Oxygen Species andOxidativeDamage inHepatocytesThatContributeto Liver TumorigenesisHayato Hikita, Takahiro Kodama, Satoshi Tanaka, Yoshinobu Saito, Yasutoshi Nozaki,Tasuku Nakabori, Satoshi Shimizu, Yoshito Hayashi, Wei Li, Minoru Shigekawa,Ryotaro Sakamori, Takuya Miyagi, Naoki Hiramatsu, Tomohide Tatsumi, andTetsuo Takehara
Abstract
Chronic hepatitis, including viral hepatitis and steatihepati-tis, is a well-known high-risk condition for hepatocellularcarcinoma. We previously reported that continuous hepatocyteapoptosis drives liver tumors in hepatocyte-specific Bcl-xL orMcl-1 knockout mice. In this study, we further examine theunderlying cellular mechanisms of generating tumors in apo-ptosis-prone liver. In cultured hepatocytes, the administrationof ABT-737, a Bcl-xL/-2/-w inhibitor, led to production ofreactive oxygen species (ROS) as well as activation of caspases.Mitochondria isolated from murine liver, upon administrationof truncated-Bid, a proapoptotic Bcl-2 family protein, releasedcytochrome c and produced ROS, which was dependent onmitochondrial respiration. Hepatic apoptosis, regeneration,accumulation of oxidative damages, and tumorigenesisobserved in hepatocyte-specific Mcl-1 knockout mice weresubstantially attenuated by further deficiency of Bax or Bid,
suggesting that a balance of mitochondrial Bcl-2 family pro-teins governs generation of oxidative stress and other pathol-ogies. Whole-exome sequencing clarified that C>A/G>T trans-version, which is often caused by oxidative DNA damage inproliferating cells, was a frequently observed mutation patternin liver tumors of Mcl-1 knockout mice. The administration ofantioxidant L-N-acetylcysteine did not affect apoptosis, com-pensatory regeneration, or fibrotic responses but significantlyreduced oxidative DNA damage and incidence and multiplicityof live tumors in Mcl-1 knockout mice. In conclusion, activa-tion of the mitochondrial apoptotic pathway in hepatocytesaccumulates intracellular oxidative damages, leading to livertumorigenesis, independently of liver regeneration or fibrosis.This study supports a concept that antioxidant therapy may beuseful for suppressing liver carcinogenesis in patients withchronic liver disease. Cancer Prev Res; 8(8); 693–701. �2015 AACR.
IntroductionHepatocellular carcinoma (HCC) is the third leading cause of
cancer death worldwide. Most HCC develops in patients withchronic hepatitis, including chronic hepatitis C, chronic hep-atitis B, alcoholic liver disease, and nonalcoholic steatohepa-titis (NASH; ref. 1). In the livers of these patients, hepatocyteapoptosis is frequently observed and regarded as one of thecharacteristic features. In clinical trials, the administration oforal caspase inhibitors that specifically block apoptosis signif-icantly reduced serum alanine aminotransferase (ALT) levels inpatients with chronic hepatitis (2, 3), supporting the idea thatelevated serum ALT levels may reflect the severity of hepatocyteapoptosis. Together with many cohort studies showing that a
high level of serum ALT in patients with chronic hepatitis is arisk factor for the development of HCC (4–6), hepatocyteapoptosis may be involved in development of HCC. In con-trast, because apoptosis also functions as a mechanism bywhich damaged cells or malignant-transformed cells are elim-inated, apoptosis may prevent cancer development. Collective-ly, whether the hepatocyte apoptosis that is observed in chronichepatitis is mechanistically linked to HCC development wasunclear.
Apoptosis is regulated by a fine balance of antiapoptotic bcl-2family proteins and proapoptotic bcl-2 family proteins. Underconditions of cellular stress, proapoptotic BH3-only proteins,such as Bid, Bim, and Puma, activate the proapoptotic proteins,Bak andBax. Bak and/or Baxwhen activated homo-oligomerize toforma channel on themitochondrial outermembrane and releasecytochrome c from its intermembrane space to the cytosolicregion, leading to the activation of caspases to execute cell death(7). Antiapoptotic bcl-2 family proteins, including Bcl-2, Bcl-xL,Mcl-1, Bcl-w, and Bfl-1, protect the mitochondrial pathway ofapoptosis by inhibiting Bak and Bax. We previously reported thatamong the antiapoptotic bcl-2 family proteins, Bcl-xL and Mcl-1,are critical molecules that protect hepatocytes from apoptosisbecause hepatocyte-specific Bcl-xL or Mcl-1 knockout (KO) micedisplayed hepatocyte apoptosis over time after birth (8, 9).Recently, we and another group reported that these mice
Department of Gastroenterology and Hepatology, Osaka UniversityGraduate School of Medicine, Suita, Japan.
Note: Supplementary data for this article are available at Cancer PreventionResearch Online (http://cancerprevres.aacrjournals.org/).
Corresponding Author: Tetsuo Takehara, Osaka University Graduate School ofMedicine, 2-2 Yamada-oka, Suita, Osaka 565-0871, Japan. Phone: 81-6-6879-3621; Fax: 81-6-6879-3629; E-mail: [email protected]
doi: 10.1158/1940-6207.CAPR-15-0022-T
�2015 American Association for Cancer Research.
CancerPreventionResearch
www.aacrjournals.org 693
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T

spontaneously develop HCC without any extra stimuli (10, 11).These results strongly support the idea that continuous hepa-tocyte apoptosis is mechanistically linked to liver tumor devel-opment. However, the underlying mechanisms by which theactivation of the mitochondrial pathway of apoptosis leads toliver tumors remains unclear. To clarify this point, we focusedthis study on oxidative stress. We demonstrate that the activa-tion of the mitochondrial pathway of apoptosis enhancesreactive oxygen species (ROS) production without affectingthe mitochondrial inner membrane potential. Among allsomatic mutation patterns, C>A/G>T transversion, which isrelated with oxidative DNA damage, was most frequentlyobserved in liver tumors in Mcl-1 KO mice. The antioxidantL-N-acetylcysteine (NAC) does not affect hepatocyte apoptosisor following compensatory hepatocyte regeneration butreduces oxidative DNA damage and liver tumor development.This report is the first to unveil oxidative stress as an importantmechanistic link that connects hepatocyte apoptosis with laterliver tumor development and suggests the potential use ofantioxidants for the prevention of HCC in patients with chronichepatitis.
Materials and MethodsIn vitro assay
Primary hepatocytes were isolated from C57BL/6J mice(Charles River Japan) by a two-step collagenase–pronaseperfusion of mouse livers as previously described (12). Iso-lated hepatocytes were maintained at 37�C under 5% CO2 inWilliam's Eagle medium containing 10% FCS (Sigma-AldrichJapan), 100 nmol/L dexamethasone, 100 nmol/L insulin(Sigma-Aldrich Japan) and L-glutamine (Invitrogen). BNLCL.2 (CL2) cells were obtained from the ATCC and examinedwithin 6 months after resuscitation. ABT-737, which inhibitsBcl-xL, Bcl-2, and Bcl-w but not Mcl-1, was kindly provided byAbbVie Inc. (North Chicago, IL) and added to primary hepa-tocytes or CL2 cells, as described previously (13). The mea-surement of caspase-3/7 activity and determination of cellviability by WST assay were also described previously (14).Intracellular ROS levels were measured by OxiSelect STA-342(Cell Biolabs, Inc.) according to the manufacturer's instruc-tion. To examine the mitochondrial membrane potential, cellswere stained with JC-10 (AAT Bioquest) and analyzed by afluorescence microscope, FV1000D (Olympus). CL2 cellsincubated with 50 mmol/L FCCP (Abcam) for 30 minuteswere used as a positive control for the loss of the mitochon-drial membrane potential.
Mitochondrial assayThe protocol for the isolation of mitochondria from murine
liver was described previously (15). Isolated mitochondria werediluted to 1 mg/mL in a reaction buffer [125 mmol/L KCl,0.5 mmol/L MgCl2, 3.0 mmol/L succinic acid, 3.0 mmol/Lglutamic acid, 10 mmol/L HEPES-KOH, pH 7.4, 1 � proteininhibitor cocktail (Nacalai Tesque), 2.5 mmol/L EDTA and BOC-Asp (OMe) CH2F 20 mmol/L] and incubated with 200 nmol/Lrecombinant mouse truncated-Bid (t-Bid; R&D Systems) for 30minutes at 37�C.After incubation, the sampleswere centrifuged at10,000� g for 15 minutes at 4�C. The supernatants were used tomeasure cytochrome c andROSusing anELISAKit (R&DSystems)andOxiSelect STA-347 (Cell Biolabs, Inc.), respectively. To exam-
ine themitochondrial membrane potential, JC-10 (AAT Bioquest,Inc.) was used according to the manufacturer's instruction. Tosuppress mitochondrial respiration, mitochondria were incubat-ed with both 10 mmol/L malonic acid (a complex I inhibitor;Sigma-Aldrich Japan) and 2 mmol/L rotenone (a complex IIinhibitor; Sigma-Aldrich Japan).
MiceHepatocyte-specific Mcl-1 KO mice (mcl-1flox/flox Alb-Cre)
were previously described (9). Bax KO mice (bax�/�) wereobtained from the Jackson Laboratory. Bid KO mice (bid�/�;ref. 16) were kindly provided by Dr. Xiao-Ming Yin (IndianaUniversity, IN). Bax/Mcl-1 double KO mice (bax�/� mcl-1flox/flox
Alb-Cre) and Bid/Mcl-1 double KO mice (bid�/� mcl-1flox/flox
Alb-Cre) were generated by mating the strains. Some mice werecontinuously treated with NAC, an antioxidant, in drinkingwater after weaning. NAC was dissolved in water (10 g/L) andfreshly prepared three times a week. Mice were maintained in aspecific pathogen-free facility and treated with humane carewith approval from the Animal Care and Use Committee ofOsaka University Medical School.
Hepatocyte apoptosis and liver fibrosis assaysSerum ALT levels and caspase-3/-7 activity were measured as
previously described (9). For histological analysis, livers wereformalin-fixed, embedded in paraffin, and thin sliced. The liversections were stained with hematoxylin and eosin (H&E). Todetect cells with oligonucleosomal DNA breaks, terminal deox-ynucleotidyl transferase-mediated deoxyuridine triphosphatenick-end labeling (TUNEL) was performed according to a previ-ously reported procedure (17). For calculating the fibrotic area,liver sections stained with Sirius red were analyzed using imageanalysis software (winROOF visual system; Mitani Co.).
ImmunohistochemistryKi-67, PCNA, and 8-hydroxy-20-deoxyguanosine (8-OHdG)
were labeled in paraffin-embedded liver sections using an anti-ki-67 antibody (Dako), anti-PCNA antibody (Cell SignalingTechnology), and anti-8-OHdG antibody (Nikken Seil), respec-tively. The detection of immunolabeled proteins was performedusing an avidin–biotin complex within the Vectastain ABC Kit(Vector Laboratories).
Real-time reverse-transcription PCR (RT-PCR)Total RNA was prepared using the RNeasy Kit (Qiagen). For
cDNA synthesis, 1 mg of total RNA was reverse-transcribed usingthe High Capacity RNA-to-DNA Master Mix (Applied Biosys-tems). Real-time reverse-transcription PCR (RT-PCR) was per-formed using an Applied Biosystems 7900HT Fast Real-TimePCR System (Applied Biosystems). The following TaqMan GeneExpression Assays were used: mouse-col1a1 (Mm00801666_g1),mouse-col1a2 (Mm00483888_m1), and mouse-b actin(Mm00607939_s1). All expression levels were corrected with thequantified expression level of b-actin mRNA.
Whole-exome sequencing and analysisDNA was extracted from 30 mg of liver tumors and their
surrounding nontumor lesion by Maxwell16 (Promega).Extracted DNA was fragmented by Acoustic Solubilizer (Covaris)and prepared concentrated sequencing library using SireSelectXT
Hikita et al.
Cancer Prev Res; 8(8) August 2015 Cancer Prevention Research694
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T
Regent Kit (Agilent Technologies) and SureSelectXT mouse allexon (Agilent Technologies). Prepared library was sequenced byHiSeq2000 (Illumina). Sequenced data were analyzed by Virmidto detect somatic mutations (18).
Statistical analysisThe data are presented as the mean � standard deviation.
Differences between two groups were determined using anunpaired Student's t test. Multiple comparisons were performedby ANOVA, followed by Fisher post hoc correction. Carcinogenesisrates were analyzed by a x2-test. P < 0.05 was considered statis-tically significant.
ResultsActivation of themitochondrial pathway of apoptosis increasesROS production
To examine the relationship between apoptosis and ROSproduction, we cultured CL2 cells, a nontransformed murineliver cell line, with ABT-737, a Bcl-xL/-2/-w inhibitor. The
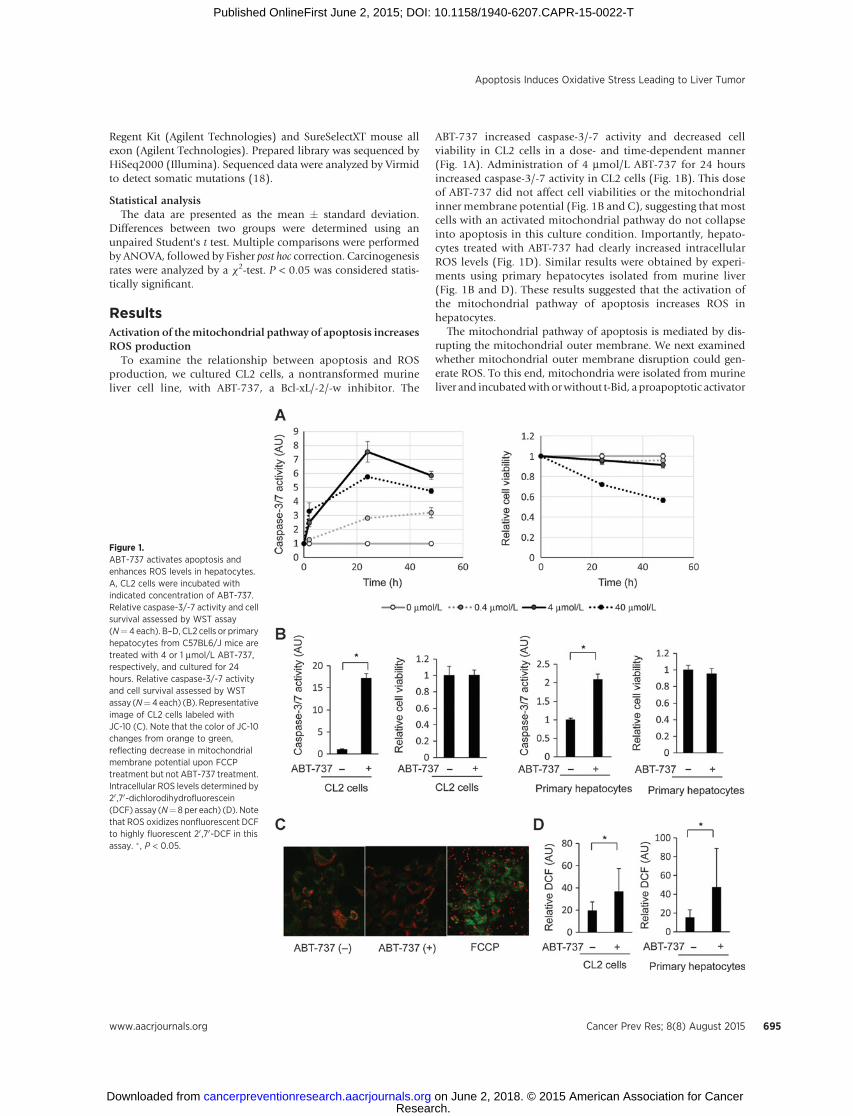
ABT-737 increased caspase-3/-7 activity and decreased cellviability in CL2 cells in a dose- and time-dependent manner(Fig. 1A). Administration of 4 mmol/L ABT-737 for 24 hoursincreased caspase-3/-7 activity in CL2 cells (Fig. 1B). This doseof ABT-737 did not affect cell viabilities or the mitochondrialinner membrane potential (Fig. 1B and C), suggesting that mostcells with an activated mitochondrial pathway do not collapseinto apoptosis in this culture condition. Importantly, hepato-cytes treated with ABT-737 had clearly increased intracellularROS levels (Fig. 1D). Similar results were obtained by experi-ments using primary hepatocytes isolated from murine liver(Fig. 1B and D). These results suggested that the activation ofthe mitochondrial pathway of apoptosis increases ROS inhepatocytes.
The mitochondrial pathway of apoptosis is mediated by dis-rupting the mitochondrial outer membrane. We next examinedwhether mitochondrial outer membrane disruption could gen-erate ROS. To this end, mitochondria were isolated from murineliver and incubatedwith orwithout t-Bid, a proapoptotic activator
Figure 1.ABT-737 activates apoptosis andenhances ROS levels in hepatocytes.A, CL2 cells were incubated withindicated concentration of ABT-737.Relative caspase-3/-7 activity and cellsurvival assessed by WST assay(N¼ 4 each). B–D, CL2 cells or primaryhepatocytes from C57BL6/J mice aretreated with 4 or 1 mmol/L ABT-737,respectively, and cultured for 24hours. Relative caspase-3/-7 activityand cell survival assessed by WSTassay (N¼4each) (B). Representativeimage of CL2 cells labeled withJC-10 (C). Note that the color of JC-10changes from orange to green,reflecting decrease in mitochondrialmembrane potential upon FCCPtreatment but not ABT-737 treatment.Intracellular ROS levels determined by20 ,70-dichlorodihydrofluorescein(DCF) assay (N¼ 8 per each) (D). Notethat ROS oxidizes nonfluorescent DCFto highly fluorescent 20 ,70-DCF in thisassay. � , P < 0.05.
Apoptosis Induces Oxidative Stress Leading to Liver Tumor
www.aacrjournals.org Cancer Prev Res; 8(8) August 2015 695
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T
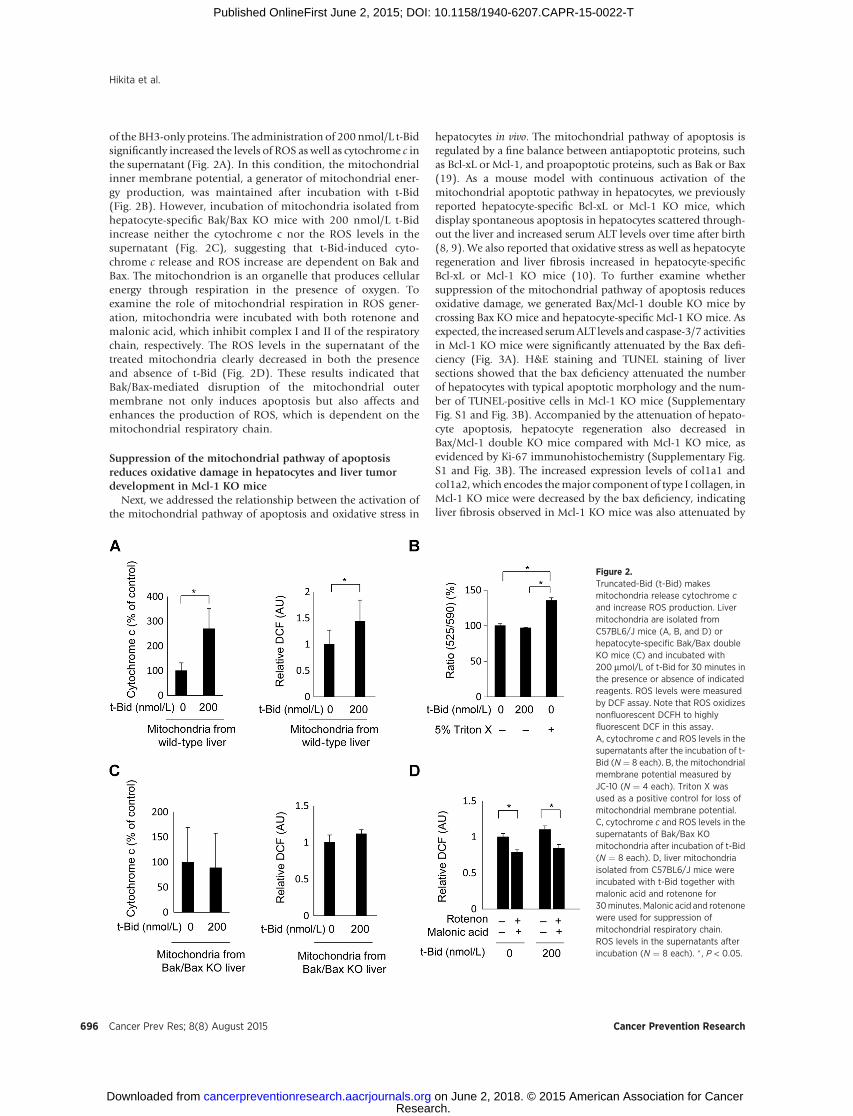
of the BH3-only proteins. The administration of 200 nmol/L t-Bidsignificantly increased the levels of ROS as well as cytochrome c inthe supernatant (Fig. 2A). In this condition, the mitochondrialinner membrane potential, a generator of mitochondrial ener-gy production, was maintained after incubation with t-Bid(Fig. 2B). However, incubation of mitochondria isolated fromhepatocyte-specific Bak/Bax KO mice with 200 nmol/L t-Bidincrease neither the cytochrome c nor the ROS levels in thesupernatant (Fig. 2C), suggesting that t-Bid-induced cyto-chrome c release and ROS increase are dependent on Bak andBax. The mitochondrion is an organelle that produces cellularenergy through respiration in the presence of oxygen. Toexamine the role of mitochondrial respiration in ROS gener-ation, mitochondria were incubated with both rotenone andmalonic acid, which inhibit complex I and II of the respiratorychain, respectively. The ROS levels in the supernatant of thetreated mitochondria clearly decreased in both the presenceand absence of t-Bid (Fig. 2D). These results indicated thatBak/Bax-mediated disruption of the mitochondrial outermembrane not only induces apoptosis but also affects andenhances the production of ROS, which is dependent on themitochondrial respiratory chain.
Suppression of the mitochondrial pathway of apoptosisreduces oxidative damage in hepatocytes and liver tumordevelopment in Mcl-1 KO mice
Next, we addressed the relationship between the activation ofthe mitochondrial pathway of apoptosis and oxidative stress in
hepatocytes in vivo. The mitochondrial pathway of apoptosis isregulated by a fine balance between antiapoptotic proteins, suchas Bcl-xL or Mcl-1, and proapoptotic proteins, such as Bak or Bax(19). As a mouse model with continuous activation of themitochondrial apoptotic pathway in hepatocytes, we previouslyreported hepatocyte-specific Bcl-xL or Mcl-1 KO mice, whichdisplay spontaneous apoptosis in hepatocytes scattered through-out the liver and increased serum ALT levels over time after birth(8, 9). We also reported that oxidative stress as well as hepatocyteregeneration and liver fibrosis increased in hepatocyte-specificBcl-xL or Mcl-1 KO mice (10). To further examine whethersuppression of the mitochondrial pathway of apoptosis reducesoxidative damage, we generated Bax/Mcl-1 double KO mice bycrossing Bax KO mice and hepatocyte-specific Mcl-1 KO mice. Asexpected, the increased serumALT levels and caspase-3/7 activitiesin Mcl-1 KO mice were significantly attenuated by the Bax defi-ciency (Fig. 3A). H&E staining and TUNEL staining of liversections showed that the bax deficiency attenuated the numberof hepatocytes with typical apoptotic morphology and the num-ber of TUNEL-positive cells in Mcl-1 KO mice (SupplementaryFig. S1 and Fig. 3B). Accompanied by the attenuation of hepato-cyte apoptosis, hepatocyte regeneration also decreased inBax/Mcl-1 double KO mice compared with Mcl-1 KO mice, asevidenced by Ki-67 immunohistochemistry (Supplementary Fig.S1 and Fig. 3B). The increased expression levels of col1a1 andcol1a2, which encodes themajor component of type I collagen, inMcl-1 KO mice were decreased by the bax deficiency, indicatingliver fibrosis observed in Mcl-1 KO mice was also attenuated by
Figure 2.Truncated-Bid (t-Bid) makesmitochondria release cytochrome cand increase ROS production. Livermitochondria are isolated fromC57BL6/J mice (A, B, and D) orhepatocyte-specific Bak/Bax doubleKO mice (C) and incubated with200 mmol/L of t-Bid for 30 minutes inthe presence or absence of indicatedreagents. ROS levels were measuredby DCF assay. Note that ROS oxidizesnonfluorescent DCFH to highlyfluorescent DCF in this assay.A, cytochrome c and ROS levels in thesupernatants after the incubation of t-Bid (N ¼ 8 each). B, the mitochondrialmembrane potential measured byJC-10 (N ¼ 4 each). Triton X wasused as a positive control for loss ofmitochondrial membrane potential.C, cytochrome c and ROS levels in thesupernatants of Bak/Bax KOmitochondria after incubation of t-Bid(N ¼ 8 each). D, liver mitochondriaisolated from C57BL6/J mice wereincubated with t-Bid together withmalonic acid and rotenone for30minutes. Malonic acid and rotenonewere used for suppression ofmitochondrial respiratory chain.ROS levels in the supernatants afterincubation (N ¼ 8 each). � , P < 0.05.
Hikita et al.
Cancer Prev Res; 8(8) August 2015 Cancer Prevention Research696
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T
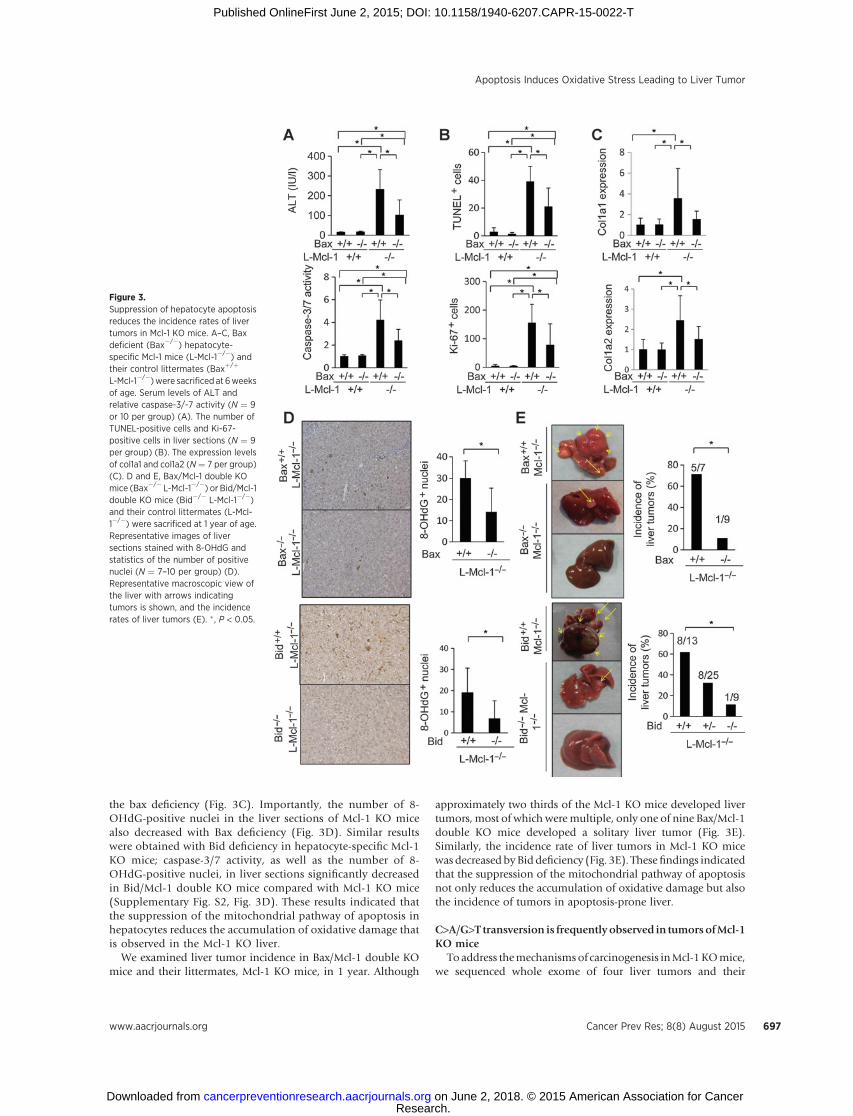
the bax deficiency (Fig. 3C). Importantly, the number of 8-OHdG-positive nuclei in the liver sections of Mcl-1 KO micealso decreased with Bax deficiency (Fig. 3D). Similar resultswere obtained with Bid deficiency in hepatocyte-specific Mcl-1KO mice; caspase-3/7 activity, as well as the number of 8-OHdG-positive nuclei, in liver sections significantly decreasedin Bid/Mcl-1 double KO mice compared with Mcl-1 KO mice(Supplementary Fig. S2, Fig. 3D). These results indicated thatthe suppression of the mitochondrial pathway of apoptosis inhepatocytes reduces the accumulation of oxidative damage thatis observed in the Mcl-1 KO liver.
We examined liver tumor incidence in Bax/Mcl-1 double KOmice and their littermates, Mcl-1 KO mice, in 1 year. Although
approximately two thirds of the Mcl-1 KO mice developed livertumors, most of which weremultiple, only one of nine Bax/Mcl-1double KO mice developed a solitary liver tumor (Fig. 3E).Similarly, the incidence rate of liver tumors in Mcl-1 KO micewas decreased byBid deficiency (Fig. 3E). Thesefindings indicatedthat the suppression of the mitochondrial pathway of apoptosisnot only reduces the accumulation of oxidative damage but alsothe incidence of tumors in apoptosis-prone liver.
C>A/G>T transversion is frequently observed in tumorsofMcl-1KO mice
Toaddress themechanismsof carcinogenesis inMcl-1KOmice,we sequenced whole exome of four liver tumors and their
Figure 3.Suppression of hepatocyte apoptosisreduces the incidence rates of livertumors in Mcl-1 KO mice. A–C, Baxdeficient (Bax�/�) hepatocyte-specific Mcl-1 mice (L-Mcl-1�/�) andtheir control littermates (Baxþ/þ
L-Mcl-1�/�)were sacrificed at 6weeksof age. Serum levels of ALT andrelative caspase-3/-7 activity (N ¼ 9or 10 per group) (A). The number ofTUNEL-positive cells and Ki-67-positive cells in liver sections (N ¼ 9per group) (B). The expression levelsof col1a1 and col1a2 (N¼ 7 per group)(C). D and E, Bax/Mcl-1 double KOmice (Bax�/� L-Mcl-1�/�) or Bid/Mcl-1double KO mice (Bid�/� L-Mcl-1�/�)and their control littermates (L-Mcl-1�/�) were sacrificed at 1 year of age.Representative images of liversections stained with 8-OHdG andstatistics of the number of positivenuclei (N ¼ 7–10 per group) (D).Representative macroscopic view ofthe liver with arrows indicatingtumors is shown, and the incidencerates of liver tumors (E). � , P < 0.05.
Apoptosis Induces Oxidative Stress Leading to Liver Tumor
www.aacrjournals.org Cancer Prev Res; 8(8) August 2015 697
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T
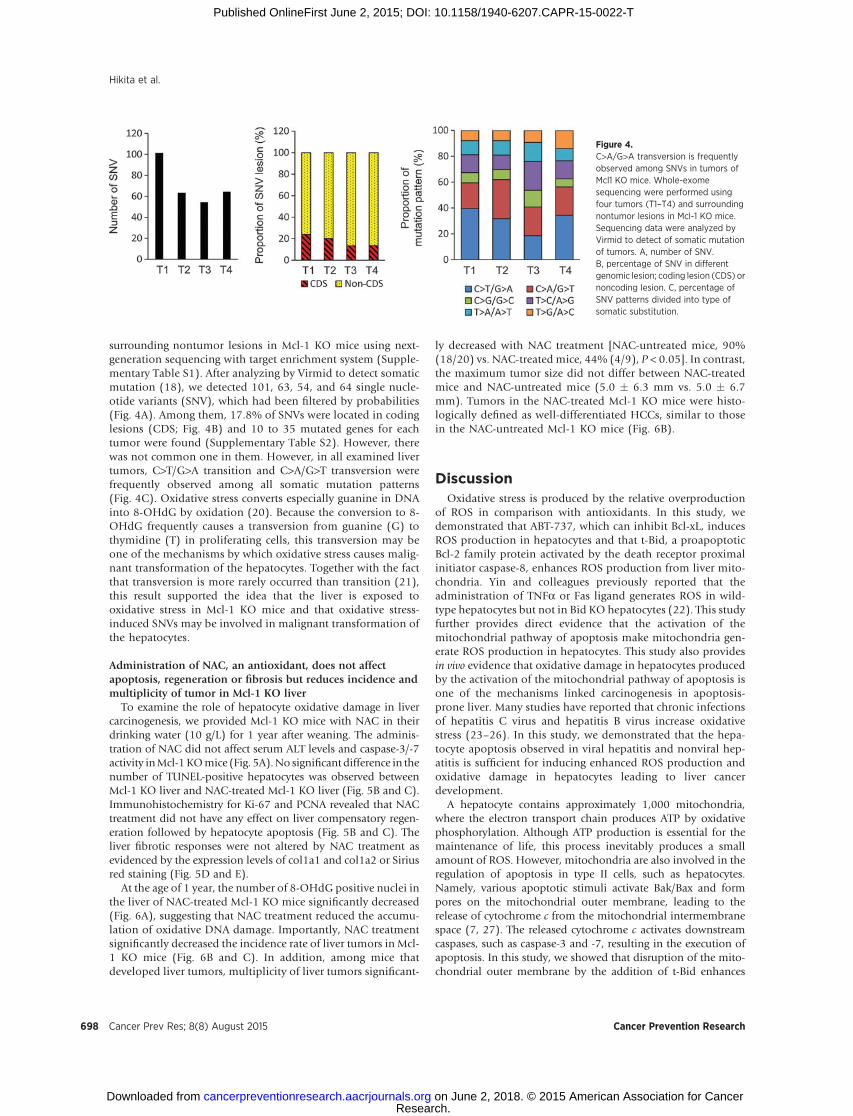
surrounding nontumor lesions in Mcl-1 KO mice using next-generation sequencing with target enrichment system (Supple-mentary Table S1). After analyzing by Virmid to detect somaticmutation (18), we detected 101, 63, 54, and 64 single nucle-otide variants (SNV), which had been filtered by probabilities(Fig. 4A). Among them, 17.8% of SNVs were located in codinglesions (CDS; Fig. 4B) and 10 to 35 mutated genes for eachtumor were found (Supplementary Table S2). However, therewas not common one in them. However, in all examined livertumors, C>T/G>A transition and C>A/G>T transversion werefrequently observed among all somatic mutation patterns(Fig. 4C). Oxidative stress converts especially guanine in DNAinto 8-OHdG by oxidation (20). Because the conversion to 8-OHdG frequently causes a transversion from guanine (G) tothymidine (T) in proliferating cells, this transversion may beone of the mechanisms by which oxidative stress causes malig-nant transformation of the hepatocytes. Together with the factthat transversion is more rarely occurred than transition (21),this result supported the idea that the liver is exposed tooxidative stress in Mcl-1 KO mice and that oxidative stress-induced SNVs may be involved in malignant transformation ofthe hepatocytes.
Administration of NAC, an antioxidant, does not affectapoptosis, regeneration or fibrosis but reduces incidence andmultiplicity of tumor in Mcl-1 KO liver
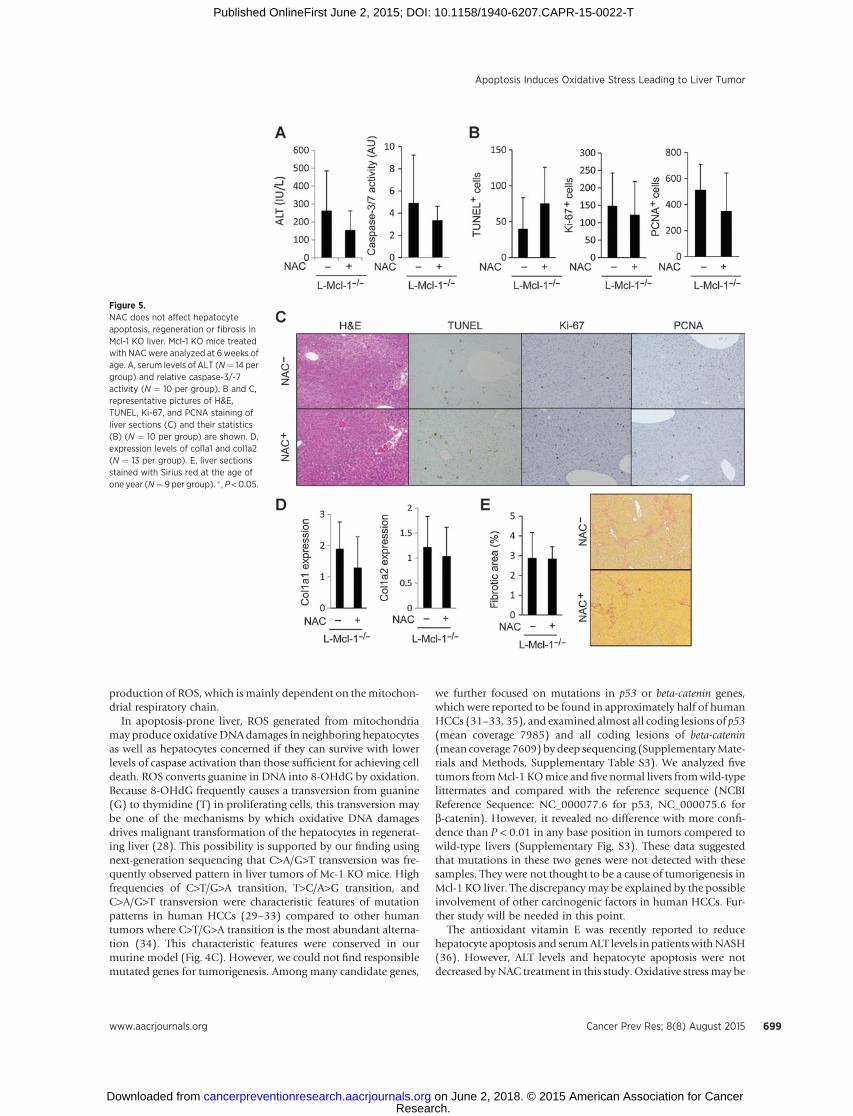
To examine the role of hepatocyte oxidative damage in livercarcinogenesis, we provided Mcl-1 KO mice with NAC in theirdrinking water (10 g/L) for 1 year after weaning. The adminis-tration of NAC did not affect serum ALT levels and caspase-3/-7activity inMcl-1KOmice (Fig. 5A).No significant difference in thenumber of TUNEL-positive hepatocytes was observed betweenMcl-1 KO liver and NAC-treated Mcl-1 KO liver (Fig. 5B and C).Immunohistochemistry for Ki-67 and PCNA revealed that NACtreatment did not have any effect on liver compensatory regen-eration followed by hepatocyte apoptosis (Fig. 5B and C). Theliver fibrotic responses were not altered by NAC treatment asevidenced by the expression levels of col1a1 and col1a2 or Siriusred staining (Fig. 5D and E).
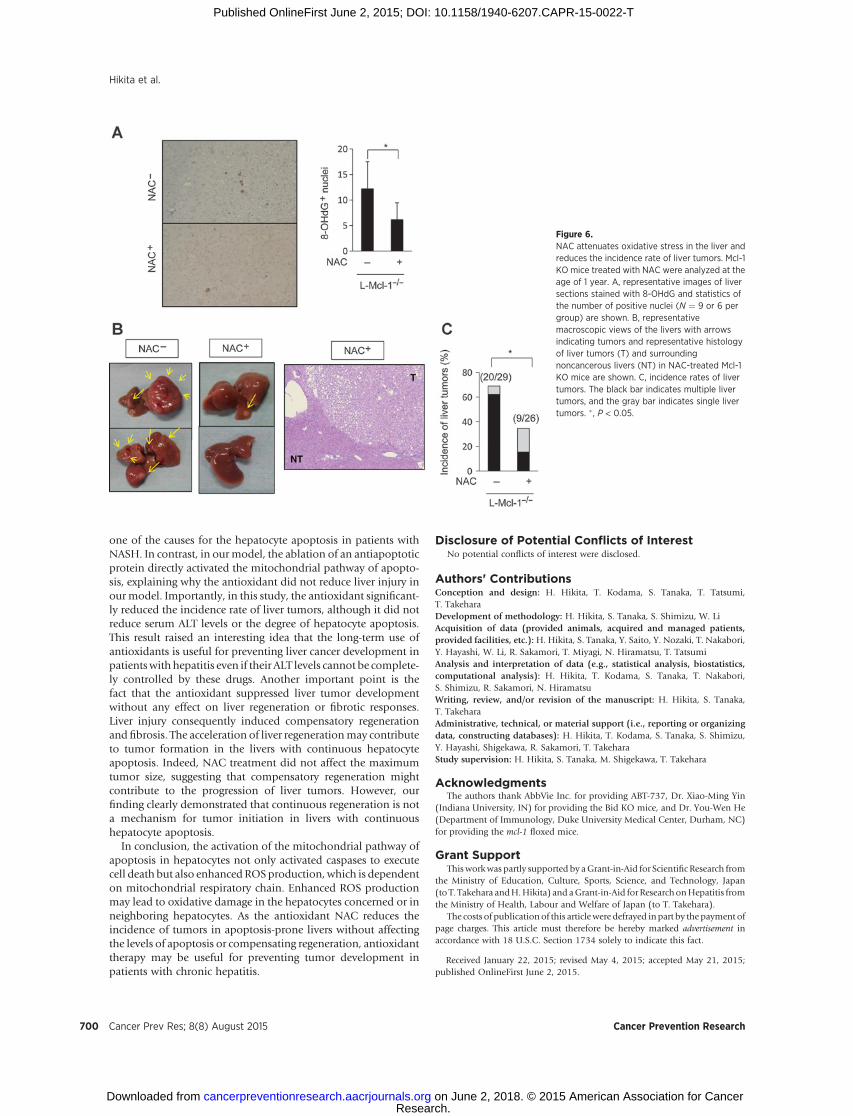
At the age of 1 year, the number of 8-OHdG positive nuclei inthe liver of NAC-treated Mcl-1 KO mice significantly decreased(Fig. 6A), suggesting that NAC treatment reduced the accumu-lation of oxidative DNA damage. Importantly, NAC treatmentsignificantly decreased the incidence rate of liver tumors in Mcl-1 KO mice (Fig. 6B and C). In addition, among mice thatdeveloped liver tumors, multiplicity of liver tumors significant-
ly decreased with NAC treatment [NAC-untreated mice, 90%(18/20) vs. NAC-treated mice, 44% (4/9), P < 0.05]. In contrast,the maximum tumor size did not differ between NAC-treatedmice and NAC-untreated mice (5.0 � 6.3 mm vs. 5.0 � 6.7mm). Tumors in the NAC-treated Mcl-1 KO mice were histo-logically defined as well-differentiated HCCs, similar to thosein the NAC-untreated Mcl-1 KO mice (Fig. 6B).
DiscussionOxidative stress is produced by the relative overproduction
of ROS in comparison with antioxidants. In this study, wedemonstrated that ABT-737, which can inhibit Bcl-xL, inducesROS production in hepatocytes and that t-Bid, a proapoptoticBcl-2 family protein activated by the death receptor proximalinitiator caspase-8, enhances ROS production from liver mito-chondria. Yin and colleagues previously reported that theadministration of TNFa or Fas ligand generates ROS in wild-type hepatocytes but not in Bid KO hepatocytes (22). This studyfurther provides direct evidence that the activation of themitochondrial pathway of apoptosis make mitochondria gen-erate ROS production in hepatocytes. This study also providesin vivo evidence that oxidative damage in hepatocytes producedby the activation of the mitochondrial pathway of apoptosis isone of the mechanisms linked carcinogenesis in apoptosis-prone liver. Many studies have reported that chronic infectionsof hepatitis C virus and hepatitis B virus increase oxidativestress (23–26). In this study, we demonstrated that the hepa-tocyte apoptosis observed in viral hepatitis and nonviral hep-atitis is sufficient for inducing enhanced ROS production andoxidative damage in hepatocytes leading to liver cancerdevelopment.
A hepatocyte contains approximately 1,000 mitochondria,where the electron transport chain produces ATP by oxidativephosphorylation. Although ATP production is essential for themaintenance of life, this process inevitably produces a smallamount of ROS. However, mitochondria are also involved in theregulation of apoptosis in type II cells, such as hepatocytes.Namely, various apoptotic stimuli activate Bak/Bax and formpores on the mitochondrial outer membrane, leading to therelease of cytochrome c from the mitochondrial intermembranespace (7, 27). The released cytochrome c activates downstreamcaspases, such as caspase-3 and -7, resulting in the execution ofapoptosis. In this study, we showed that disruption of the mito-chondrial outer membrane by the addition of t-Bid enhances
Figure 4.C>A/G>A transversion is frequentlyobserved among SNVs in tumors ofMcl1 KO mice. Whole-exomesequencing were performed usingfour tumors (T1–T4) and surroundingnontumor lesions in Mcl-1 KO mice.Sequencing data were analyzed byVirmid to detect of somatic mutationof tumors. A, number of SNV.B, percentage of SNV in differentgenomic lesion; coding lesion (CDS) ornoncoding lesion. C, percentage ofSNV patterns divided into type ofsomatic substitution.
Hikita et al.
Cancer Prev Res; 8(8) August 2015 Cancer Prevention Research698
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T
production of ROS, which is mainly dependent on themitochon-drial respiratory chain.
In apoptosis-prone liver, ROS generated from mitochondriamay produce oxidativeDNAdamages in neighboring hepatocytesas well as hepatocytes concerned if they can survive with lowerlevels of caspase activation than those sufficient for achieving celldeath. ROS converts guanine in DNA into 8-OHdG by oxidation.Because 8-OHdG frequently causes a transversion from guanine(G) to thymidine (T) in proliferating cells, this transversion maybe one of the mechanisms by which oxidative DNA damagesdrives malignant transformation of the hepatocytes in regenerat-ing liver (28). This possibility is supported by our finding usingnext-generation sequencing that C>A/G>T transversion was fre-quently observed pattern in liver tumors of Mc-1 KO mice. Highfrequencies of C>T/G>A transition, T>C/A>G transition, andC>A/G>T transversion were characteristic features of mutationpatterns in human HCCs (29–33) compared to other humantumors where C>T/G>A transition is the most abundant alterna-tion (34). This characteristic features were conserved in ourmurine model (Fig. 4C). However, we could not find responsiblemutated genes for tumorigenesis. Among many candidate genes,
we further focused on mutations in p53 or beta-catenin genes,which were reported to be found in approximately half of humanHCCs (31–33, 35), and examined almost all coding lesions of p53(mean coverage 7985) and all coding lesions of beta-catenin(mean coverage 7609) bydeep sequencing (SupplementaryMate-rials and Methods, Supplementary Table S3). We analyzed fivetumors fromMcl-1 KOmice andfive normal livers fromwild-typelittermates and compared with the reference sequence (NCBIReference Sequence: NC_000077.6 for p53, NC_000075.6 forb-catenin). However, it revealed no difference with more confi-dence than P < 0.01 in any base position in tumors compered towild-type livers (Supplementary Fig. S3). These data suggestedthat mutations in these two genes were not detected with thesesamples. They were not thought to be a cause of tumorigenesis inMcl-1 KO liver. The discrepancymay be explained by the possibleinvolvement of other carcinogenic factors in human HCCs. Fur-ther study will be needed in this point.
The antioxidant vitamin E was recently reported to reducehepatocyte apoptosis and serumALT levels in patients withNASH(36). However, ALT levels and hepatocyte apoptosis were notdecreased byNAC treatment in this study. Oxidative stressmay be
Figure 5.NAC does not affect hepatocyteapoptosis, regeneration or fibrosis inMcl-1 KO liver. Mcl-1 KO mice treatedwith NAC were analyzed at 6 weeks ofage. A, serum levels of ALT (N¼ 14 pergroup) and relative caspase-3/-7activity (N ¼ 10 per group). B and C,representative pictures of H&E,TUNEL, Ki-67, and PCNA staining ofliver sections (C) and their statistics(B) (N ¼ 10 per group) are shown. D,expression levels of col1a1 and col1a2(N ¼ 13 per group). E, liver sectionsstained with Sirius red at the age ofone year (N¼ 9 per group). � , P <0.05.
Apoptosis Induces Oxidative Stress Leading to Liver Tumor
www.aacrjournals.org Cancer Prev Res; 8(8) August 2015 699
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T
one of the causes for the hepatocyte apoptosis in patients withNASH. In contrast, in our model, the ablation of an antiapoptoticprotein directly activated the mitochondrial pathway of apopto-sis, explaining why the antioxidant did not reduce liver injury inour model. Importantly, in this study, the antioxidant significant-ly reduced the incidence rate of liver tumors, although it did notreduce serum ALT levels or the degree of hepatocyte apoptosis.This result raised an interesting idea that the long-term use ofantioxidants is useful for preventing liver cancer development inpatientswith hepatitis even if their ALT levels cannot be complete-ly controlled by these drugs. Another important point is thefact that the antioxidant suppressed liver tumor developmentwithout any effect on liver regeneration or fibrotic responses.Liver injury consequently induced compensatory regenerationand fibrosis. The acceleration of liver regenerationmay contributeto tumor formation in the livers with continuous hepatocyteapoptosis. Indeed, NAC treatment did not affect the maximumtumor size, suggesting that compensatory regeneration mightcontribute to the progression of liver tumors. However, ourfinding clearly demonstrated that continuous regeneration is nota mechanism for tumor initiation in livers with continuoushepatocyte apoptosis.
In conclusion, the activation of the mitochondrial pathway ofapoptosis in hepatocytes not only activated caspases to executecell death but also enhancedROSproduction, which is dependenton mitochondrial respiratory chain. Enhanced ROS productionmay lead to oxidative damage in the hepatocytes concerned or inneighboring hepatocytes. As the antioxidant NAC reduces theincidence of tumors in apoptosis-prone livers without affectingthe levels of apoptosis or compensating regeneration, antioxidanttherapy may be useful for preventing tumor development inpatients with chronic hepatitis.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: H. Hikita, T. Kodama, S. Tanaka, T. Tatsumi,T. TakeharaDevelopment of methodology: H. Hikita, S. Tanaka, S. Shimizu, W. LiAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): H. Hikita, S. Tanaka, Y. Saito, Y. Nozaki, T. Nakabori,Y. Hayashi, W. Li, R. Sakamori, T. Miyagi, N. Hiramatsu, T. TatsumiAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): H. Hikita, T. Kodama, S. Tanaka, T. Nakabori,S. Shimizu, R. Sakamori, N. HiramatsuWriting, review, and/or revision of the manuscript: H. Hikita, S. Tanaka,T. TakeharaAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): H. Hikita, T. Kodama, S. Tanaka, S. Shimizu,Y. Hayashi, Shigekawa, R. Sakamori, T. TakeharaStudy supervision: H. Hikita, S. Tanaka, M. Shigekawa, T. Takehara
AcknowledgmentsThe authors thank AbbVie Inc. for providing ABT-737, Dr. Xiao-Ming Yin
(Indiana University, IN) for providing the Bid KO mice, and Dr. You-Wen He(Department of Immunology, Duke University Medical Center, Durham, NC)for providing the mcl-1 floxed mice.
Grant SupportThisworkwas partly supported by aGrant-in-Aid for Scientific Research from
the Ministry of Education, Culture, Sports, Science, and Technology, Japan(to T. Takehara andH.Hikita) and aGrant-in-Aid for ResearchonHepatitis fromthe Ministry of Health, Labour and Welfare of Japan (to T. Takehara).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received January 22, 2015; revised May 4, 2015; accepted May 21, 2015;published OnlineFirst June 2, 2015.
Figure 6.NAC attenuates oxidative stress in the liver andreduces the incidence rate of liver tumors. Mcl-1KO mice treated with NAC were analyzed at theage of 1 year. A, representative images of liversections stained with 8-OHdG and statistics ofthe number of positive nuclei (N ¼ 9 or 6 pergroup) are shown. B, representativemacroscopic views of the livers with arrowsindicating tumors and representative histologyof liver tumors (T) and surroundingnoncancerous livers (NT) in NAC-treated Mcl-1KO mice are shown. C, incidence rates of livertumors. The black bar indicates multiple livertumors, and the gray bar indicates single livertumors. �, P < 0.05.
Cancer Prev Res; 8(8) August 2015 Cancer Prevention Research700
Hikita et al.
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T

References1. Dhanasekaran R, Limaye A, Cabrera R. Hepatocellular carcinoma: current
trends in worldwide epidemiology, risk factors, diagnosis, and therapeu-tics. Hepat Med 2012;4:19–37.
2. Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G.Clinical trial: the efficacy and safety of oral PF-03491390, a pancaspaseinhibitor—a randomized placebo-controlled study in patients with chron-ic hepatitis C. Aliment Pharmacol Ther 2010;31:969–78.
3. Ratziu V, SheikhMY, Sanyal AJ, Lim JK, ConjeevaramH, Chalasani N, et al.A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012;55:419–28.
4. Kasahara A, Hayashi N, Mochizuki K, Takayanagi M, Yoshioka K, KakumuS, et al. Risk factors for hepatocellular carcinoma and its incidence afterinterferon treatment in patients with chronic hepatitis C. Osaka LiverDisease Study Group. Hepatology 1998;27:1394–402.
5. Oze T, Hiramatsu N, Yakushijin T, Miyazaki M, Yamada A, Oshita M, et al.Post-treatment levels of a-fetoprotein predict incidence of hepatocellularcarcinoma after interferon therapy. Clin Gastroenterol Hepatol 2014;12:1186–95.
6. Lee MH, Yang HI, Liu J, Batrla-Utermann R, Jen CL, Iloeje UH, et al.Predictionmodels of long-term cirrhosis andhepatocellular carcinoma riskin chronic hepatitis B patients: risk scores integrating host and virusprofiles. Hepatology 2013;58:546–54.
7. Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in themitochondria. J Cell Physiol 2003;195:158–67.
8. Takehara T, Tatsumi T, Suzuki T, Rucker Er, Hennighausen L, Jinushi M,et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepa-tocyte apoptosis and liver fibrotic responses. Gastroenterology 2004;127:1189–97.
9. Hikita H, Takehara T, Shimizu S, Kodama T, LiW,Miyagi T, et al. Mcl-1 andBcl-xL cooperatively maintain integrity of hepatocytes in developing andadult murine liver. Hepatology 2009;50:1217–26.
10. Hikita H, Kodama T, Shimizu S, Li W, Shigekawa M, Tanaka S, et al. Bakdeficiency inhibits liver carcinogenesis: a causal link between apoptosisand carcinogenesis. J Hepatol 2012;57:92–100.
11. Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S, et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1triggers proliferation and hepatocarcinogenesis in mice. Hepatology2010;51:1226–36.
12. Kodama T, Takehara T, Hikita H, Shimizu S, Shigekawa M, Tsunematsu H,et al. Increases in p53 expression induce CTGF synthesis by mouse andhuman hepatocytes and result in liver fibrosis in mice. J Clin Invest2011;121:3343–56.
13. Hikita H, Takehara T, Shimizu S, Kodama T, Shigekawa M, Iwase K, et al.The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppressesgrowth of hepatoma cells in combination with sorafenib. Hepatology2010;52:1310–21.
14. Shimizu S, Takehara T,HikitaH,KodamaT,Miyagi T,HosuiA, et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafe-nib-induced apoptosis in human hepatocellular carcinoma. J Hepatol2010;52:698–704.
15. Hikita H, Takehara T, Kodama T, Shimizu S, Hosui A, Miyagi T, et al. BH3-only protein bid participates in the Bcl-2 network in healthy liver cells.Hepatology 2009;50:1972–80.
16. Yin X,Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, et al. Bid-deficientmiceare resistant to Fas-induced hepatocellular apoptosis. Nature 1999;400:886–91.
17. Takehara T, Hayashi N, Tatsumi T, Kanto T, Mita E, Sasaki Y, et al.Interleukin 1beta protects mice from Fas-mediated hepatocyte apoptosisand death. Gastroenterology 1999;117:661–8.
18. Kim S, Jeong K, Bhutani K, Lee J, Patel A, Scott E, et al. Virmid: accuratedetection of somatic mutations with sample impurity inference. GenomeBiol 2013;14:R90.
19. Wei M, Zong W, Cheng E, Lindsten T, Panoutsakopoulou V, Ross A, et al.Proapoptotic BAX and BAK: a requisite gateway tomitochondrial dysfunc-tion and death. Science 2001;292:727–30.
20. David SS, O'Shea VL, Kundu S. Base-excision repair of oxidative DNAdamage. Nature 2007;447:941–50.
21. Lynch M. Rate, molecular spectrum, and consequences of human muta-tion. Proc Natl Acad Sci U S A 2010;107:961–8.
22. Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependentgeneration of oxygen radicals promotes death receptor activation-inducedapoptosis in murine hepatocytes. Hepatology 2004;40:403–13.
23. Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, et al.Mitochondrial injury, oxidative stress, and antioxidant gene expressionare induced by hepatitis C virus core protein. Gastroenterology2002;122:366–75.
24. Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, Majano PL, Ben-edicto I, Rosado JA, et al. Hepatitis C virus NS5A and core proteins induceoxidative stress-mediated calcium signalling alterations in hepatocytes.J Hepatol 2009;50:872–82.
25. Koike K. Hepatitis B virus X gene is implicated in liver carcinogenesis.Cancer Lett 2009;286:60–8.
26. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV-and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 2013;13:123–35.
27. Hikita H, Takehara T, Kodama T, Shimizu S, Shigekawa M, Hosui A, et al.Delayed-onset caspase-dependent massive hepatocyte apoptosis upon Fasactivation in Bak/Bax-deficient mice. Hepatology 2011;54:240–51.
28. van LoonB,Markkanen E,H€ubscherU.Oxygen as a friend and enemy: howto combat the mutational potential of 8-oxo-guanine. DNA Repair (Amst)2010;9:604–16.
29. Totoki Y, TatsunoK, Yamamoto S, Arai Y,Hosoda F, Ishikawa S, et al. High-resolution characterization of a hepatocellular carcinoma genome.Nat Genet 2011;43:464–9.
30. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al.Whole-genome sequencing of liver cancers identifies etiological influencesonmutation patterns and recurrentmutations in chromatin regulators. NatGenet 2012;44:760–4.
31. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al.Integrated analysis of somatic mutations and focal copy-number changesidentifies key genes and pathways in hepatocellular carcinoma. Nat Genet2012;44:694–8.
32. Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al.Trans-ancestry mutational landscape of hepatocellular carcinoma gen-omes. Nat Genet 2014;46:1267–73.
33. SchulzeK, ImbeaudS, Letouz�e E, Alexandrov LB,Calderaro J, Rebouissou S,et al. Exome sequencing of hepatocellular carcinomas identifies newmutational signatures and potential therapeutic targets. Nat Genet2015;47:505–11.
34. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al.Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153–8.
35. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genomesequencing identifies recurrent mutations in hepatocellular carcinoma.Genome Res 2013;23:1422–33.
36. Hoofnagle JH, Van Natta ML, Kleiner DE, Clark JM, Kowdley KV, LoombaR, et al. Vitamin E and changes in serum alanine aminotransferase levels inpatients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther2013;38:134–43.
www.aacrjournals.org Cancer Prev Res; 8(8) August 2015 701
Apoptosis Induces Oxidative Stress Leading to Liver Tumor
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T

2015;8:693-701. Published OnlineFirst June 2, 2015.Cancer Prev Res Hayato Hikita, Takahiro Kodama, Satoshi Tanaka, et al. That Contribute to Liver TumorigenesisReactive Oxygen Species and Oxidative Damage in Hepatocytes Activation of the Mitochondrial Apoptotic Pathway Produces
Updated version
10.1158/1940-6207.CAPR-15-0022-Tdoi:
Access the most recent version of this article at:
Material
Supplementary
C1
http://cancerpreventionresearch.aacrjournals.org/content/suppl/2015/06/04/1940-6207.CAPR-15-0022-T.DDC2http://cancerpreventionresearch.aacrjournals.org/content/suppl/2015/06/11/1940-6207.CAPR-15-0022-T.Access the most recent supplemental material at:
Cited articles
http://cancerpreventionresearch.aacrjournals.org/content/8/8/693.full#ref-list-1
This article cites 36 articles, 3 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerpreventionresearch.aacrjournals.org/content/8/8/693To request permission to re-use all or part of this article, use this link
Research. on June 2, 2018. © 2015 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst June 2, 2015; DOI: 10.1158/1940-6207.CAPR-15-0022-T