serms attenuate estrogen-induced malignant transformation...
TRANSCRIPT

Research Article
SERMs Attenuate Estrogen-Induced MalignantTransformation of Human Mammary Epithelial Cells byUpregulating Detoxification of Oxidative Metabolites
L.P. Madhubhani P. Hemachandra, Hitisha Patel, R. Esala P. Chandrasena, Jaewoo Choi,Sujeewa C. Piyankarage, Shuai Wang, Yijin Wang, Emily N. Thayer, Robert A. Scism,Bradley T. Michalsen, Rui Xiong, Marton I. Siklos, Judy L. Bolton, and Gregory R.J. Thatcher
AbstractThe risk of developing hormone-dependent cancers with long-term exposure to estrogens is attributed
both to proliferative, hormonal actions at the estrogen receptor (ER) and to chemical carcinogenesis elicited
by genotoxic, oxidative estrogen metabolites. Nontumorigenic MCF-10A human breast epithelial cells are
classified as ER� and undergo estrogen-induced malignant transformation. Selective estrogen receptor
modulators (SERM), in use for breast cancer chemoprevention and for postmenopausal osteoporosis, were
observed to inhibit malignant transformation, asmeasured by anchorage-independent colony growth. This
chemopreventive activity was observed to correlate with reduced levels of oxidative estrogen metabolites,
cellular reactive oxygen species (ROS), and DNA oxidation. The ability of raloxifene, desmethylarzoxifene
(DMA), and bazedoxifene to inhibit this chemical carcinogenesis pathway was not shared by 4-hydro-
xytamoxifen. Regulation of phase II rather than phase Imetabolic enzymes was implicatedmechanistically:
raloxifene and DMA were observed to upregulate sulfotransferase (SULT 1E1) and glucuronidase (UGT
1A1). The results support upregulation of phase II metabolism in detoxification of catechol estrogen
metabolites leading to attenuated ROS formation as a mechanism for inhibition of malignant transfor-
mation by a subset of clinically important SERMs. Cancer Prev Res; 7(5); 505–15. �2014 AACR.
IntroductionBreast cancer is the leading cause of cancer death
among women in Western countries. The association ofhormone-dependent cancer with exposure to endogenousestrogens has been known for decades. Of the 2 majormechanisms of estrogen carcinogenesis, the hormonalpathway, mediated via the estrogen receptor (ER), hasbeen extensively studied (1–4). Formation of highly reac-tive estrogen quinone metabolites, which can cause DNAdamage, is believed to be a major contributor to chemicalcarcinogenesis (5–7).In breast epithelial cells, the endogenous estrogens are
metabolized to their 2-OH and 4-OH catechol metabolites,catalyzed by CYP450 1A1 and CYP450 1B1, respectively(Fig. 1). Further oxidation of estrogen catechols to quinones
causes genotoxicity through electrophilic and oxidativeDNA damage, including formation of 8-oxo-7,8-dehydro-20-deoxyguanosine (8-oxo-dG; refs. 8–10). Formation ofreactive oxygen species (ROS) from quinone redox cyclingcan amplify DNAdamage (11, 12). Several lines of evidencestrongly suggest that the estrogen catechols are the proximalcarcinogens in chemical carcinogenesis (13–17). Preven-tion of estrogen-induced chemical carcinogenesis thereforecan theoretically be achieved by "detoxification" of estrogencatechols, via: (i) attenuated formation, (ii) enhanced con-jugative metabolism and clearance, or (iii) trapping ofquinones and ROS (ref. 18; Fig. 1).
Model systems for study of chemical carcinogenesis, aprocess envisioned to develop over many years of exposureto genotoxic insult, represent a challenge. MCF-10 cells arenontumorigenic human breast epithelial cells that undergoestrogen-induced malignant transformation. Owing to lowER levels and lack of proliferative response to estrogens, thecell line is of use in studying chemical carcinogenesis, in theabsence of confounding hormonal proliferative signals(17, 19, 20).
Selective estrogen receptor modulators (SERM) are ERligands that oppose the effects of endogenous estrogens inbreast tissues. In the present study, the potential for pre-vention of estrogen-induced malignant transformation ofMCF-10A cells was studied in response to raloxifene (Ral)and related SERMs. Ral and desmethylarzoxifene (DMA),
Authors' Affiliation: Department of Medicinal Chemistry and Pharmacog-nosy, College of Pharmacy, University of Illinois at Chicago, Chicago,Illinois
Note:Supplementary data for this article are available atCancer PreventionResearch Online (http://cancerprevres.aacrjournals.org/).
Corresponding Author: Gregory R.J. Thatcher, Department of MedicinalChemistry and Pharmacognosy, College of Pharmacy, University of Illinoisat Chicago, 833 S.Wood Street, Chicago, IL 60612. Phone: 312-355-5282;Fax: 312-996-7170, E-mail: [email protected]
doi: 10.1158/1940-6207.CAPR-13-0296
�2014 American Association for Cancer Research.
CancerPreventionResearch
www.aacrjournals.org 505
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296

the activemetabolite of arzoxifene, were observed to inhibitmalignant transformation.
The interconversion of estradiol (E2) with estrone (E1) iscatalyzed by the enzyme 17b–hydroxysteroid dehydroge-nase (17b-HSD; Fig. 1). In MCF-10A cells, as (i) the equi-librium lies strongly toward E1 and (ii) the stability of themethyl ethermetabolites is superior to the catechol estrogenitself, MeOE1 represents a reliable, indirect measurement ofestrogen oxidative metabolism (21). For 3 SERMs, inhibi-tion of malignant transformation of MCF10-A cells wasobserved to correlate with attenuation of estrogen metab-olism as measured by MeOE1. To explain these observa-tions, the response to SERMs ofmediators of estrogen phaseI and II metabolism was studied. "Detoxification" of thecatechol estrogen may be mediated by conjugative metab-olism by sulfotransferase (SULT), UDP-glucuronosyltrans-ferase (UGT), catechol-O-methyl transferase (COMT), andglutathione-S-transferase (GST), or arguably by NAD(P)H:quinoneoxidoreductase (NQO1; refs. 22, 23).Although theexpression of UGT is prominent in hepatic tissues (24), inextra-hepatic tissues such as breast, SULT plays a prominentrole in detoxification (25, 26). The results indicate thatprevention of estrogen-induced transformation by SERMs,resulting fromattenuated estrogenmetabolism, ismediatedby upregulation of SULT1E1 andUGT1A1. Interestingly, ofthe 2 further clinical SERMs, bazedoxifene (Baze) andtamoxifen (Tam), Baze attenuated formation of MeOE1whereas Tam did not. The mechanism of action of Ral andDMA in this model of estrogen-dependentmalignant trans-formation was detoxification of genotoxic estrogen meta-bolites by upregulation of conjugative metabolism andattenuation of oxidative stress. These observations on non-canonical SERM actions, and the outlier nature of Tam, areof therapeutic relevance for an important drug class.
Materials and MethodsChemicals and reagents
All chemicals, reagents, and enzymes were obtained fromSigma or Invitrogen unless stated otherwise. Antibodieswere obtained from Santa Cruz Biotechnology, Cell Signal-
ing Technology, and Sigma. Chemical standards of estrogenmetabolites were obtained from Steraloids Inc. 4-Hydro-xyestrone-1,2,16,16-d4 and 2-methoxyestrone-1,4,16,16-d4 were obtained from CDN Isotopes and used as internalstandards in estrogen metabolism experiments.
Cell lines and cell culture conditionsMCF-10A cells were obtained from American Type Cul-
ture Collection andmaintained in phenol red and estrogen-free Dulbecco’s modified Eagle’s medium and F12medium(DMEM/F12) supplementedwith1%penicillin/streptomy-cin, 5% FBS, cholera toxin (0.1 mg/mL), EGF (20 ng/mL),hydrocortisone (0.5 mg/mL), insulin (10 mg/L), and 5%CO2 at 37�C as described previously (27). MCF-10A cellswere authenticated using single tandem repeat (STR) anal-ysis. MCF-7 cells were obtained from ATCC and used forstandard ERE luciferase assay as described previously (28).TheMDA-MB-231:b41 cell line, ER� cells stably transfectedwith ERb, were a kind gift of Dr D. Tonetti (UIC, Chicago,IL) and used as described for ERE luciferase assay (29).
Analysis of estrogen metabolites in MCF-10A cellsEstrogen metabolites were analyzed in MCF-10A cell as
previously described (27). Briefly, MCF-10A cells wereincubated with E2 (1 mmol/L) in the presence or absenceof SERMs (1 mmol/L) for 6 days. Treatments were renewedevery 3 days. Because DMA and Ral showed a significantinhibition of estrogenmetabolism at 1 mmol/L inMCF-10Acells, a dose response was performed for these 2 SERMs:DMA and Ral (0.1–2.5 mmol/L) were tested in the presenceof E2 (1mmol/L). Samplepreparation andanalysiswasdoneusing the method described by Xu and colleagues (30) withminor modifications as previously described (27). Enzy-matic hydrolysis of cell media was done as previouslydescribed (30) with minor modifications. Briefly, MCF-10A cells were plated in 6-well plates with 3 mL of mediain each well. Cells were treated with E2 (1 mmol/L) in thepresence or absence of SERMs (1 mmol/L) for 6 days andtreatments were renewed every 3 days. Cell media wascollected every 3 days pooled together to get total of 6 mLfor each sample. Standard curves were prepared for 2-
OH
OHOH
OHOH
HO
HO HO HO
O
HO HO
O
E2
E1
HSD17B1-B8
CYP450CYP450 CYP450 1A1
1A11B1
2-OHE1
4-OHE2 4-OHE
12-OHE2
COMT UGT SULTGST
MeOE1 and MeOE
2
metabolites metabolitesGlucuronic acidconjugates
SulfatedGlutathione conjugates
(O)
NQO1
O
O
O
OH O2 DNA
DNA
DNAoxidation
Malignanttransformation
ROS
4-OHE2-Q
Depurinatingadducts
Figure 1. Estrogen metabolismand its relationship to chemicalcarcinogenesis. In breast epithelialcells, several SERMs wereobserved to modulate estrogenmetabolism (as depicted by boxedarrows), in particular viamodulationof detoxification enzymes. GST,glutathione-S-transferase-P1;NQO1, NAPDH:quinoneoxidoreductase; SULT,sulfotransferase; UGT, UDP-glucuronosyltransferase.Catechol-O-methyl transferase(COMT) activity was not perturbedby SERMs. SERMs did notmodulate expression of CYP450sin E2-treated cells.
Hemachandra et al.
Cancer Prev Res; 7(5) May 2014 Cancer Prevention Research506
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296

MeOE1 and 4-MeOE1 using as internal standard 2-MeOE1-d4. The internal standard was also added to each samplebefore further processing. Enzyme hydrolysis buffer wasprepared as previously described (30), which contained L-ascorbic acid, b-glucuronidase, and sulfatase in 0.15 mol/Lsodium acetate buffer (pH¼ 4.6). Equal amounts (6mL) ofhydrolysis buffer was added into each cell media sample (6mL) and incubated overnight (16 hours) at 37�C. Sampleswere extracted into dichloromethane and analyzed usingliquid chromatography/tandem mass spectrometry (LC/MS-MS) as previously described (27). Exemplar amountsof 2-MeOE1 and 4-MeOE1 are provided from standardcurves for experiments in which cells were treated with E2alone: 376� 22 and 319� 95 pmol/L, for 2-MeOE1 and 4-MeOE1, respectively.
ROS formation determined by CM-H2DCFDAMCF-10A cells were grown (4� 103 cells/mL) on each of
8 chambers on a sterile Nunc chambered coverglass andincubated overnight at 37�C with 5% CO2. Cells weretreated with E2 (1 mmol/L) with and without SERMs (1mmol/L) for 6 days. Treatments were renewed after 3 days.Formation of ROS was determined as previously described(31), using CM-H2DCFDA (10 mmol/L) and 0.2 mg/mLHoechst stain for visualization of nuclei.
Detection and measurement of 8-oxo-dG formationMCF-10A cells were plated in 15-cm diameter dishes at a
density of 2� 106 cells per dish in estrogen-freemedia. Cellswere allowed to attach for 1 day and then were treated with4-OHE2 (1 mmol/L) with and without SERMs (1 mmol/L;DMA, Ral, or FDMA) for 72 hours. 8-oxo-dG analysis wasperformed as described previously (17). The native dG wasdetermined by HPLC (UV) scanning at 280 nm. 8-oxo-dGwasdetected bymultiple reactionmonitoring and collision-induced dissociation for the fragmentation pathway of m/z284 ! 168 and m/z 289 ! 173 for (15N5)8-oxo-dG usingpositive ion electrospray. The amount of 8-oxo-dG formedper 106 of dG was plotted. Total 8-oxo-dG per 106 of dGratio for the 4-OHE2–treated sample was taken as 100% forthe purpose of calculation.
Anchorage-independent growth assayAnchorage-independent colony formation cell transfor-
mation assay was performed as previously described (27).Spherical formation of more than 50 cells was taken as acolony. Number of colonies formed in each well werecounted and represented as percentage colony efficiency� SD. Percentage colony efficiency is calculated as thenumber of colonies formed per number of cells plated perwell � 100.
ImmunoblottingMCF-10A cells were treated with E2 (1 mmol/L) in the pre-
sence and absence of SERMs (DMA, FDMA, Ral; 1 mmol/L).Protein expression of CYP450 1B1 and CYP450 1A1 wasanalyzed using Western blot experiments as previouslydescribed (27). Anti-CYP450 1B1 (Sigma; AV51761), anti-
CYP450 1A1 (Santa Cruz; sc-20772), and anti-b-actin (CellSignaling; #4967) antibodies were used as primary antibo-dies. Detoxification enzymes were also analyzed using anti-SULT1 (Santa Cruz; sc-32928), anti-SULT1E1 (Santa Cruz;sc-376009), anti-SULT1A1 (Santa Cruz; sc-130883), anti-GSTpi (Cell Signaling; #3369), anti-NQO1 (Santa Cruz;sc-32793), andantiCOMT(SantaCruz; sc-25844)asprimaryantibodies.Antibodieswerediluted inblocking solution(5%non–fat milk in TBS with 0.1% Tween 20). Blots wereincubated with primary antibody overnight at 4�C and withsecondary antibody for 1 hour at room temperature. Blotswere visualized using chemiluminescence substrate (ThermoScientific). Imagingandanalysiswere doneusingFluroChemsoftware (Cell Biosciences). Each protein band density wasnormalized to the respective b-actin band density and wasrepresented as the relative protein expression. Three inde-pendent experiments were performed and results were repre-sented as average � SD.
RNA isolation and quantification of metabolizingenzyme gene transcripts
MCF-10A cell were plated at a density of 2� 105 cells perwell in a 6-well plate and treated with E2 (1 mmol/L) withandwithout SERMs (1mmol/L) for 24 hours. Total RNAwasisolated from cells using QIAShredder columns and QIA-GEN RNeasy kit (Qiagen Inc.) according to the manufac-turer’s protocol. Total RNA (1 mg) was used to synthesizecDNA using SuperScript III in a 20 mL reaction mixtureaccording to manufacturer’s protocol. Quantitative PCR(qPCR) was done with respective primers. TaqMan FAMprobes and primers (Applied Biosystems) were used for thegene analysis of SULT 1A1, SULT 1E1, and UGT 1A1,whereas human b-actin gene amplification was used as theinternal control. Expression of the gene of interest wasnormalized to the internal control and fold change in geneexpression was calculated. Three independent experimentswere performed in duplicates and the data were representedas an average � SD.
Enzyme activity assaysInhibition of CYP450 1B1 activity was analyzed using
ethoxyresorufin O-dealkylase (EROD) assay as previouslydescribed (27). Inhibition of COMT was assayed by adap-tation of a literaturemethod (32). Recombinant COMT (10mg/mL) was incubated in Tris (10 mmol/L, pH 7.4), MgCl2(1 mmol/L), DTT (1 mmol/L), S-(50-adenosyl)-L-methio-nine (500 nmol/L) with or without Ral, Baze, or DMA (1mmol/L) at 37�C for 5 minutes before initiation of reactionby addition of 6,7-dihydroxycoumarin (5 mmol/L). Reac-tion was monitored by fluorescence (lex ¼ 355nm, lem ¼460 nm).
Statistical analysisThree independent metabolism experiments were per-
formed in triplicates and the data were represented asaverage � SD. The statistical analysis of results consistedof t test or ANOVA using GraphPad Prism version 5 forWindows.
SERMs Upregulate Estrogen Detoxification
www.aacrjournals.org Cancer Prev Res; 7(5) May 2014 507
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296

ResultsDMA,Ral, andBaze, but not 4-OHTam, inhibit estrogenmetabolism in MCF-10A cells
Analysis of E1 methoxy ethers is a useful indirect mea-surement of the formation of catechol metabolites in thepresence of SERMs, as (i) in MCF-10A cells, catechol estro-gens are largely metabolized to methoxyethers that cannotthemselves be directly converted to quinones and (ii)SERMs do not inhibit COMT activity (Supplementary Fig.S1). After 6 days of E2 treatment, higher amounts of E1relative to E2 metabolites and relatively higher amountsof the 2-MeOE1 isomer were observed in all treatments(Supplementary Fig. S2).
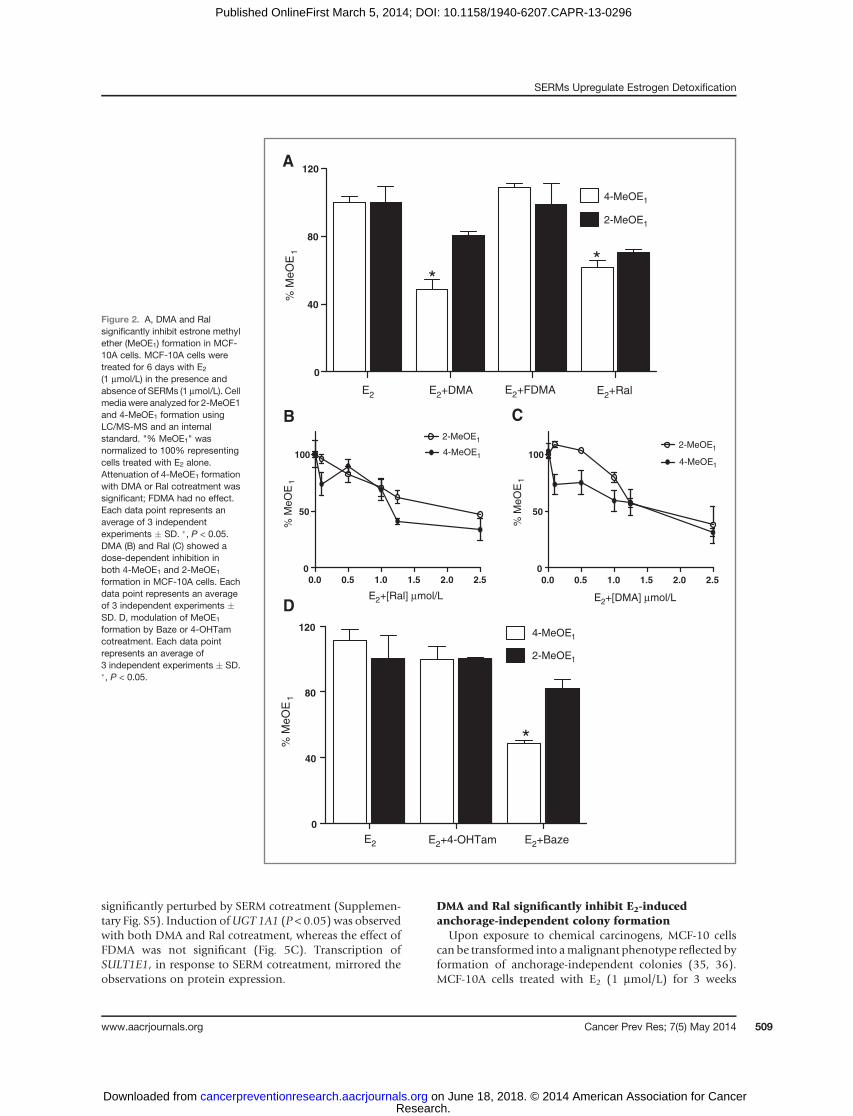
MCF-10A cells incubated with E2 (1 mmol/L) were treatedwith vehicle or SERMs (1 mmol/L) for 6 days and theformation of 4-MeOE1 and 2-MeOE1 was analyzed byLC/MS-MS, which provides a measure of catechol estrogenformation (Fig. 2; refs. 17, 27). Attenuation of MeOE1formation with DMA and Ral reached significance for 4-MeOE1 (P < 0.05) whereas FDMA was without effect (Fig.2A). For Ral and DMA, the reduction in catechol etherformation was found to be concentration-dependent (Fig-ures 2B and C). The effects on estrogen metabolism of theclinical SERMs, Baze and Tam, were also studied. No sig-nificant effect on metabolite formation was observed with4-OHTam, the active metabolite of Tam, whereas Bazeshowed significant inhibition of 4-MeOE1 formation (P <0.05; Fig. 2D).
DMA, Ral, andBaze attenuate estrogen-inducedROS inMCF-10A cells
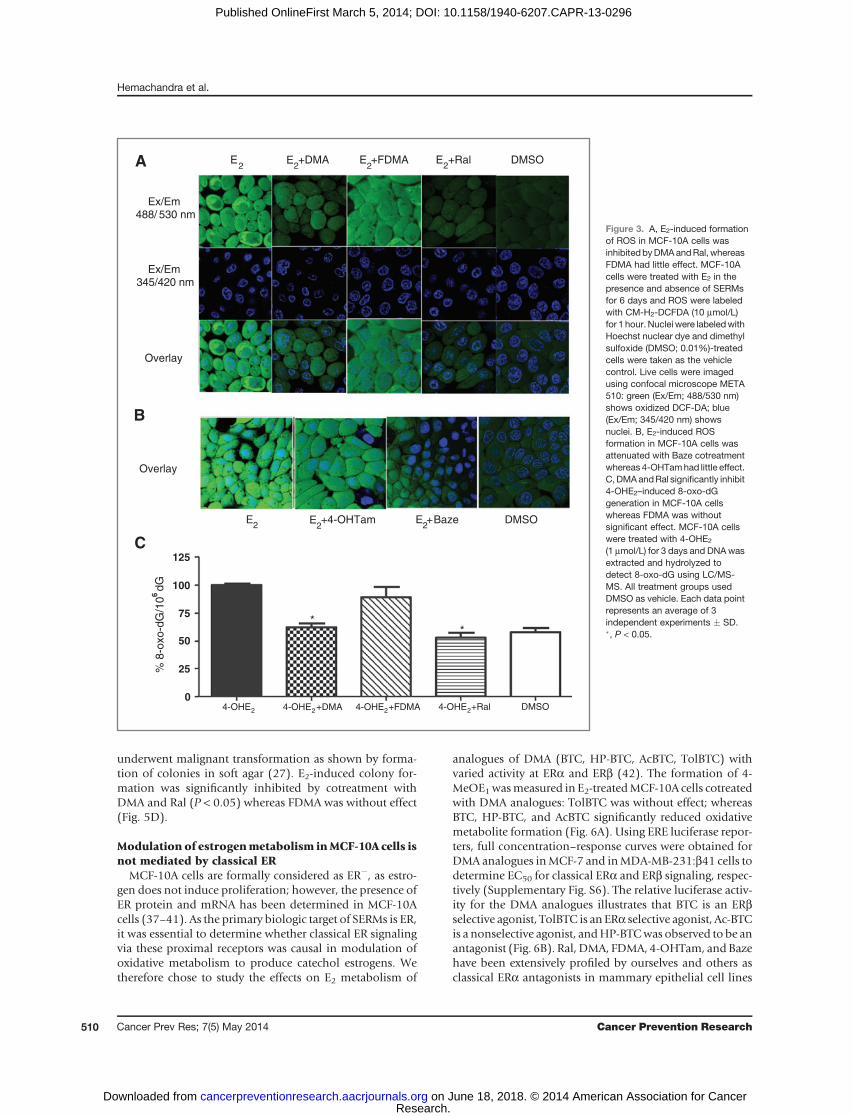
MCF-10A cells incubated with E2 for 6 days and treatedwith the reporter dyeCM-H2DCFDA showed increased ROSlevels compared with the dimethyl sulfoxide (DMSO) vehi-cle control (Fig. 3A). In ERþ cells, localization of ER in thenucleus has been reported to produce nuclear ROS local-ization (31, 33); however, in MCF-10A cells, localizationwas not observed, compatible with the lack of function ofERa as a nuclear transcription factor in this cell line.
E2-induced ROS formation was attenuated in cellscotreated with either DMA or Ral; however, there was nosignificant effect on the formation of ROS with FDMAcotreatment (Fig. 3A). Baze and 4-OHTam were also testedfor their effect on E2-induced ROS formation in MCF-10Acells: no significant effect was observed on 4-OHTam treat-ment; however, Baze attenuated ROS formation (Fig. 3B).
DMA and Ral significantly attenuate 4-OHE2–induced8-oxo-dG formation
Measurement of 8-oxo-dG is routinely used to deter-mine the level of oxidative DNA damage in cells and invivo (34). After 3 days treatment of MCF-10A cells with E2,formation of 8-oxo-dG did not reach significance relativeto DMSO control (data not shown); therefore, MCF-10Acells were treated directly with the catechol estrogenmetabolite, 4-OHE2 (1 mmol/L), for 3 days revealing asignificant increase in 8-oxo-dG relative to DMSO control
(P < 0.001). Cotreatment with either DMA or Ral signif-icantly reduced (P < 0.05) 8-oxo-dG levels induced by4-OHE2. Coadministration of FDMA was again withouteffect (Fig. 3C).
DMA and Ral do not decrease CYP450 expression oractivity
CYP450 enzymes mediate estrogen-induced chemicalcarcinogenesis by catalyzing catechol estrogen formation(Fig. 1). CYP450 expression was analyzed by immunoblot-ting after treatment ofMCF-10A cells with E2 in the presenceor absence of SERMs, showing no effect of SERM cotreat-ment on CYP450 levels (Supplementary Fig. S3). Measure-ment of CYP450 1B1 activity using the ERODassay revealedthe expected inhibition by SERMs at very high concentra-tions, but not at the 1 mmol/L concentration applied to cells(Supplementary Fig. S4).
DMA and Ral detoxify estrogen metabolites via actionon phase II enzymes
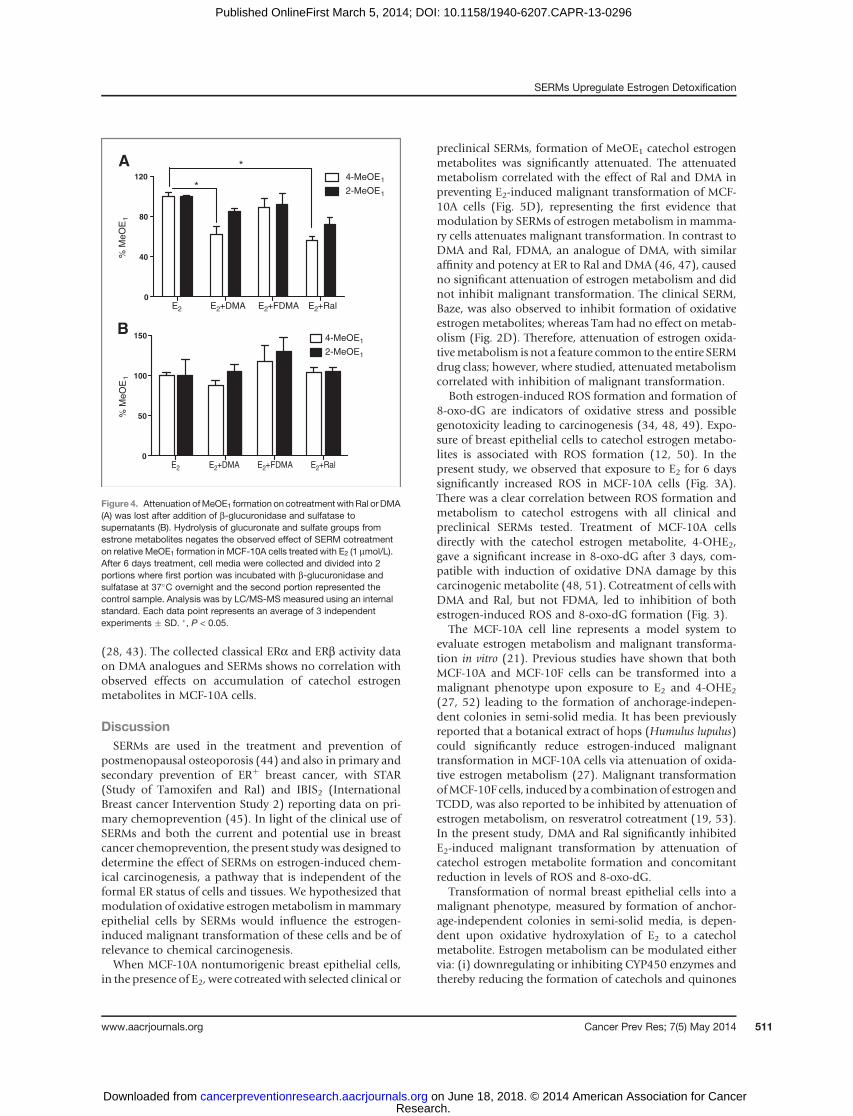
Sulfation and glucuronidation play key roles in conju-gative detoxification; therefore, levels of estrogen metabo-lites in cell media were measured in the presence of asulfatase/b-glucuronidase cocktail that causes enzymatichydrolysis of conjugates. The attenuation of 4-MeOE1 and2-MeOE1 formation by SERMs was completely lost underthese conditions (Fig. 4), leading to the conclusion thatconjugative metabolism is responsible for the attenuationof catechol estrogen metabolite formation caused by SERMcotreatment.
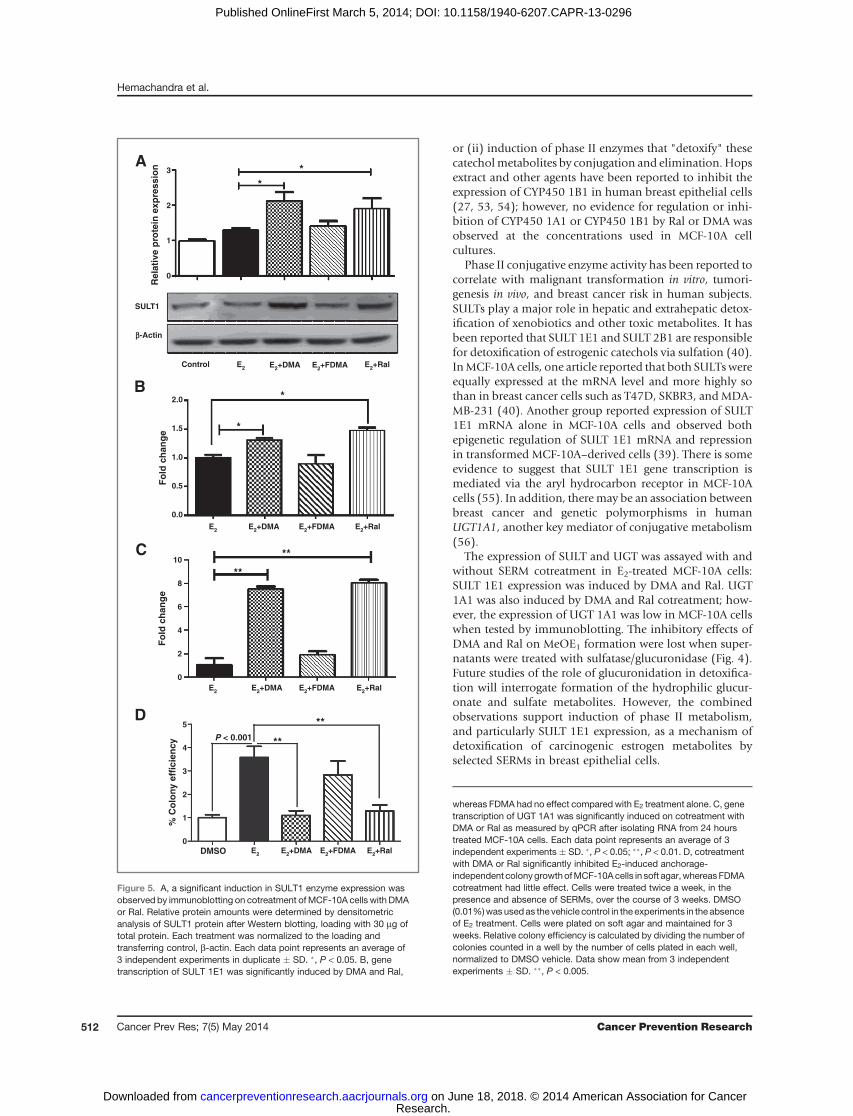
Extending this observation, cotreatment of MCF-10Acells with E2 and either DMA or Ral significantly elevatedimmunoreactivity of SULT1 family enzymes (Fig. 5A).Further analysis indicated that expression of SULT 1E1was induced by cotreatment with DMA or Ral (Supple-mentary Fig. S5) but that SULT 1A1 expression was notsignificantly changed (Supplementary Fig. S5). E2 itselfdid not modulate SULT1 expression and once againFDMA was unable to mimic the effects of Ral and DMA(Fig. 5A). Reduction of quinones by NQO1 is able tomaintain a reducing cellular environment, unless thisactivity contributes to redox cycling (11, 22) (Fig. 1).The reduction in NQO1 expression after treatment ofMCF-10A cells with E2 was negated or reversed by cotreat-ment with SERMs (Supplementary Fig. S4). Similar anal-yses of COMT and GST-P1 expression showed no effectfrom cotreatment with SERMs (Supplementary Fig. S5).The lack of sensitivity of COMT to E2 and drug treatmentsfurther supports measurements of MeOE1 as reflective ofcatechol estrogen formation.
Because immunoblotting showed induction of SULT1family proteins after cotreatment with DMA or Ral, qPCRexperiments were conducted to examine the effect of SERMson SULT1E1 and SULT1A1. A significant increase in genetranscription of SULT1E1was observedwith cotreatment ofDMA andRal (P < 0.05)whereas the effect of FDMAwas notsignificant (Fig. 5B). There was an induction of SULT1A1gene transcription in E2 incubations, which was not
Hemachandra et al.
Cancer Prev Res; 7(5) May 2014 Cancer Prevention Research508
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296
significantly perturbed by SERM cotreatment (Supplemen-tary Fig. S5). Induction ofUGT 1A1 (P < 0.05) was observedwith both DMA and Ral cotreatment, whereas the effect ofFDMA was not significant (Fig. 5C). Transcription ofSULT1E1, in response to SERM cotreatment, mirrored theobservations on protein expression.
DMA and Ral significantly inhibit E2-inducedanchorage-independent colony formation
Upon exposure to chemical carcinogens, MCF-10 cellscan be transformed into amalignant phenotype reflected byformation of anchorage-independent colonies (35, 36).MCF-10A cells treated with E2 (1 mmol/L) for 3 weeks
0.0 0.5 1.0 1.5 2.0 2.5
0
50
100
2-MeOE1
4-MeOE1
E2+[Ral] µmol/L E2+[DMA] µmol/L
% M
eO
E1
% M
eO
E1
0.0 0.5 1.0 1.5 2.0 2.5
0
50
1002-MeOE1
4-MeOE1
B C
E2 E2+4-OHTam E2+Baze
0
40
80
1204-MeOE1
2-MeOE1
*
D
E2 E2+DMA E2+FDMA E2+Ral
0
40
80
120
4-MeOE1
2-MeOE1
**
% M
eO
E1
% M
eO
E1
A
Figure 2. A, DMA and Ralsignificantly inhibit estrone methylether (MeOE1) formation in MCF-10A cells. MCF-10A cells weretreated for 6 days with E2
(1 mmol/L) in the presence andabsence of SERMs (1 mmol/L). Cellmedia were analyzed for 2-MeOE1and 4-MeOE1 formation usingLC/MS-MS and an internalstandard. "% MeOE1" wasnormalized to 100% representingcells treated with E2 alone.Attenuation of 4-MeOE1 formationwith DMA or Ral cotreatment wassignificant; FDMA had no effect.Each data point represents anaverage of 3 independentexperiments � SD. �, P < 0.05.DMA (B) and Ral (C) showed adose-dependent inhibition inboth 4-MeOE1 and 2-MeOE1
formation in MCF-10A cells. Eachdata point represents an averageof 3 independent experiments �SD. D, modulation of MeOE1
formation by Baze or 4-OHTamcotreatment. Each data pointrepresents an average of3 independent experiments � SD.�, P < 0.05.
SERMs Upregulate Estrogen Detoxification
www.aacrjournals.org Cancer Prev Res; 7(5) May 2014 509
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296
underwent malignant transformation as shown by forma-tion of colonies in soft agar (27). E2-induced colony for-mation was significantly inhibited by cotreatment withDMA and Ral (P < 0.05) whereas FDMA was without effect(Fig. 5D).
Modulation of estrogenmetabolism inMCF-10A cells isnot mediated by classical ER
MCF-10A cells are formally considered as ER�, as estro-gen does not induce proliferation; however, the presence ofER protein and mRNA has been determined in MCF-10Acells (37–41). As the primary biologic target of SERMs is ER,it was essential to determine whether classical ER signalingvia these proximal receptors was causal in modulation ofoxidative metabolism to produce catechol estrogens. Wetherefore chose to study the effects on E2 metabolism of
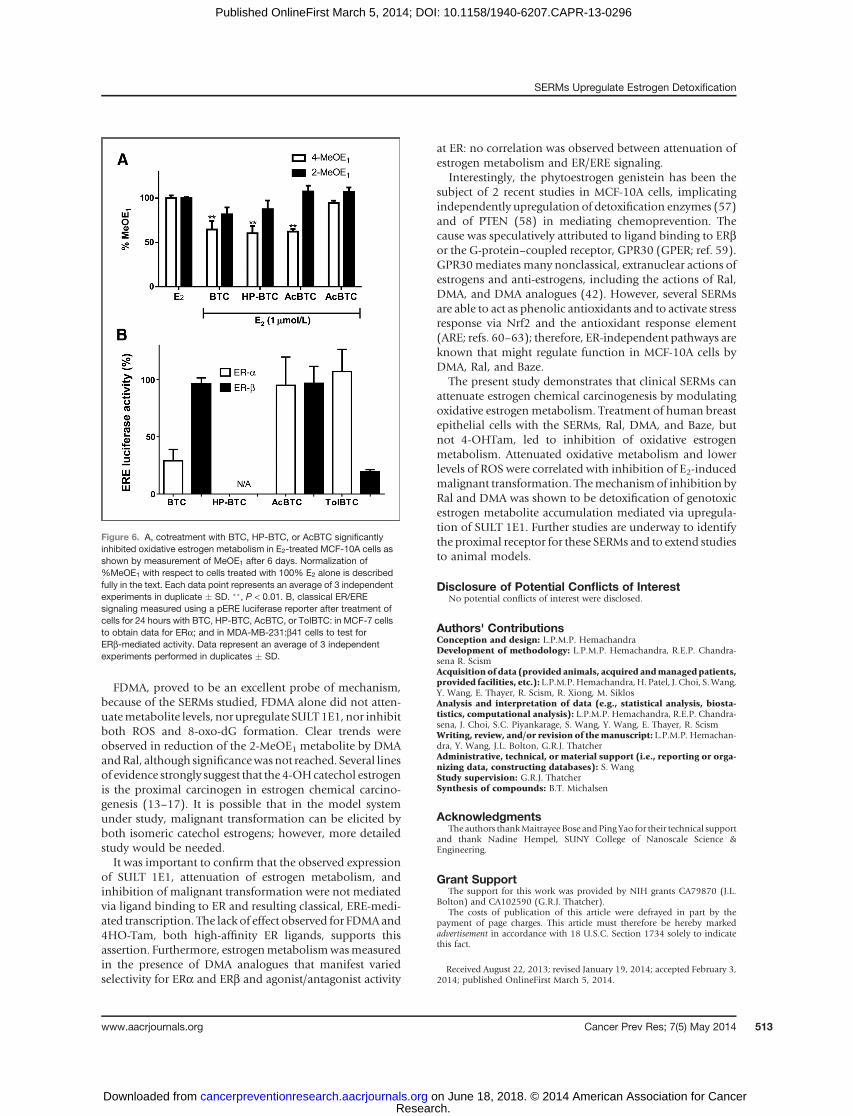
analogues of DMA (BTC, HP-BTC, AcBTC, TolBTC) withvaried activity at ERa and ERb (42). The formation of 4-MeOE1wasmeasured in E2-treatedMCF-10A cells cotreatedwith DMA analogues: TolBTC was without effect; whereasBTC, HP-BTC, and AcBTC significantly reduced oxidativemetabolite formation (Fig. 6A). Using ERE luciferase repor-ters, full concentration–response curves were obtained forDMA analogues inMCF-7 and inMDA-MB-231:b41 cells todetermine EC50 for classical ERa and ERb signaling, respec-tively (Supplementary Fig. S6). The relative luciferase activ-ity for the DMA analogues illustrates that BTC is an ERbselective agonist, TolBTC is an ERa selective agonist, Ac-BTCis a nonselective agonist, andHP-BTCwas observed to be anantagonist (Fig. 6B). Ral, DMA, FDMA, 4-OHTam, andBazehave been extensively profiled by ourselves and others asclassical ERa antagonists in mammary epithelial cell lines
E2
E2+DMA E
2+FDMA E
2+Ral DMSO
Ex/Em488/ 530 nm
Overlay
Ex/Em345/420 nm
A
E2
E2+4-OHTam E
2+ Baze DMSO
Overlay
C
4-OHE2 4-OHE2+DMA 4-OHE2+FDMA 4-OHE2+Ral DMSO0
25
50
75
100
125
**
% 8
-oxo-d
G/1
06dG
B
Figure 3. A, E2-induced formationof ROS in MCF-10A cells wasinhibitedbyDMAandRal, whereasFDMA had little effect. MCF-10Acells were treated with E2 in thepresence and absence of SERMsfor 6 days and ROS were labeledwith CM-H2-DCFDA (10 mmol/L)for 1 hour. Nuclei were labeledwithHoechst nuclear dye and dimethylsulfoxide (DMSO; 0.01%)-treatedcells were taken as the vehiclecontrol. Live cells were imagedusing confocal microscope META510: green (Ex/Em; 488/530 nm)shows oxidized DCF-DA; blue(Ex/Em; 345/420 nm) showsnuclei. B, E2-induced ROSformation in MCF-10A cells wasattenuated with Baze cotreatmentwhereas 4-OHTamhad little effect.C, DMAandRal significantly inhibit4-OHE2–induced 8-oxo-dGgeneration in MCF-10A cellswhereas FDMA was withoutsignificant effect. MCF-10A cellswere treated with 4-OHE2
(1 mmol/L) for 3 days and DNAwasextracted and hydrolyzed todetect 8-oxo-dG using LC/MS-MS. All treatment groups usedDMSO as vehicle. Each data pointrepresents an average of 3independent experiments � SD.�, P < 0.05.
Hemachandra et al.
Cancer Prev Res; 7(5) May 2014 Cancer Prevention Research510
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296
(28, 43). The collected classical ERa and ERb activity dataon DMA analogues and SERMs shows no correlation withobserved effects on accumulation of catechol estrogenmetabolites in MCF-10A cells.
DiscussionSERMs are used in the treatment and prevention of
postmenopausal osteoporosis (44) and also in primary andsecondary prevention of ERþ breast cancer, with STAR(Study of Tamoxifen and Ral) and IBIS2 (InternationalBreast cancer Intervention Study 2) reporting data on pri-mary chemoprevention (45). In light of the clinical use ofSERMs and both the current and potential use in breastcancer chemoprevention, the present study was designed todetermine the effect of SERMs on estrogen-induced chem-ical carcinogenesis, a pathway that is independent of theformal ER status of cells and tissues. We hypothesized thatmodulation of oxidative estrogenmetabolism inmammaryepithelial cells by SERMs would influence the estrogen-induced malignant transformation of these cells and be ofrelevance to chemical carcinogenesis.When MCF-10A nontumorigenic breast epithelial cells,
in the presence of E2, were cotreatedwith selected clinical or
preclinical SERMs, formation of MeOE1 catechol estrogenmetabolites was significantly attenuated. The attenuatedmetabolism correlated with the effect of Ral and DMA inpreventing E2-induced malignant transformation of MCF-10A cells (Fig. 5D), representing the first evidence thatmodulation by SERMs of estrogen metabolism in mamma-ry cells attenuates malignant transformation. In contrast toDMA and Ral, FDMA, an analogue of DMA, with similaraffinity and potency at ER to Ral and DMA (46, 47), causedno significant attenuation of estrogen metabolism and didnot inhibit malignant transformation. The clinical SERM,Baze, was also observed to inhibit formation of oxidativeestrogenmetabolites; whereas Tam had no effect onmetab-olism (Fig. 2D). Therefore, attenuation of estrogen oxida-tivemetabolism is not a feature common to the entire SERMdrug class; however, where studied, attenuated metabolismcorrelated with inhibition of malignant transformation.
Both estrogen-induced ROS formation and formation of8-oxo-dG are indicators of oxidative stress and possiblegenotoxicity leading to carcinogenesis (34, 48, 49). Expo-sure of breast epithelial cells to catechol estrogen metabo-lites is associated with ROS formation (12, 50). In thepresent study, we observed that exposure to E2 for 6 dayssignificantly increased ROS in MCF-10A cells (Fig. 3A).There was a clear correlation between ROS formation andmetabolism to catechol estrogens with all clinical andpreclinical SERMs tested. Treatment of MCF-10A cellsdirectly with the catechol estrogen metabolite, 4-OHE2,gave a significant increase in 8-oxo-dG after 3 days, com-patible with induction of oxidative DNA damage by thiscarcinogenic metabolite (48, 51). Cotreatment of cells withDMA and Ral, but not FDMA, led to inhibition of bothestrogen-induced ROS and 8-oxo-dG formation (Fig. 3).
The MCF-10A cell line represents a model system toevaluate estrogen metabolism and malignant transforma-tion in vitro (21). Previous studies have shown that bothMCF-10A and MCF-10F cells can be transformed into amalignant phenotype upon exposure to E2 and 4-OHE2(27, 52) leading to the formation of anchorage-indepen-dent colonies in semi-solid media. It has been previouslyreported that a botanical extract of hops (Humulus lupulus)could significantly reduce estrogen-induced malignanttransformation in MCF-10A cells via attenuation of oxida-tive estrogen metabolism (27). Malignant transformationofMCF-10F cells, inducedby a combinationof estrogen andTCDD, was also reported to be inhibited by attenuation ofestrogen metabolism, on resveratrol cotreatment (19, 53).In the present study, DMA and Ral significantly inhibitedE2-induced malignant transformation by attenuation ofcatechol estrogen metabolite formation and concomitantreduction in levels of ROS and 8-oxo-dG.
Transformation of normal breast epithelial cells into amalignant phenotype, measured by formation of anchor-age-independent colonies in semi-solid media, is depen-dent upon oxidative hydroxylation of E2 to a catecholmetabolite. Estrogen metabolism can be modulated eithervia: (i) downregulating or inhibiting CYP450 enzymes andthereby reducing the formation of catechols and quinones
E2 E2+DMA E2+FDMA E2+Ral0
40
80
120 4-MeOE1
2-MeOE1*
*
% M
eO
E1
E2 E2+DMA E2+FDMA E2+Ral0
50
100
150 4-MeOE1
2-MeOE1
% M
eO
E1
A
B
Figure 4. Attenuation ofMeOE1 formation on cotreatment withRal orDMA(A) was lost after addition of b-glucuronidase and sulfatase tosupernatants (B). Hydrolysis of glucuronate and sulfate groups fromestrone metabolites negates the observed effect of SERM cotreatmenton relative MeOE1 formation in MCF-10A cells treated with E2 (1 mmol/L).After 6 days treatment, cell media were collected and divided into 2portions where first portion was incubated with b-glucuronidase andsulfatase at 37�C overnight and the second portion represented thecontrol sample. Analysis was by LC/MS-MS measured using an internalstandard. Each data point represents an average of 3 independentexperiments � SD. �, P < 0.05.
SERMs Upregulate Estrogen Detoxification
www.aacrjournals.org Cancer Prev Res; 7(5) May 2014 511
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296
or (ii) induction of phase II enzymes that "detoxify" thesecatecholmetabolites by conjugation and elimination. Hopsextract and other agents have been reported to inhibit theexpression of CYP450 1B1 in human breast epithelial cells(27, 53, 54); however, no evidence for regulation or inhi-bition of CYP450 1A1 or CYP450 1B1 by Ral or DMA wasobserved at the concentrations used in MCF-10A cellcultures.
Phase II conjugative enzyme activity has been reported tocorrelate with malignant transformation in vitro, tumori-genesis in vivo, and breast cancer risk in human subjects.SULTs play a major role in hepatic and extrahepatic detox-ification of xenobiotics and other toxic metabolites. It hasbeen reported that SULT 1E1 and SULT 2B1 are responsiblefor detoxification of estrogenic catechols via sulfation (40).InMCF-10A cells, one article reported that both SULTs wereequally expressed at the mRNA level and more highly sothan in breast cancer cells such as T47D, SKBR3, and MDA-MB-231 (40). Another group reported expression of SULT1E1 mRNA alone in MCF-10A cells and observed bothepigenetic regulation of SULT 1E1 mRNA and repressionin transformed MCF-10A–derived cells (39). There is someevidence to suggest that SULT 1E1 gene transcription ismediated via the aryl hydrocarbon receptor in MCF-10Acells (55). In addition, theremay be an association betweenbreast cancer and genetic polymorphisms in humanUGT1A1, another key mediator of conjugative metabolism(56).
The expression of SULT and UGT was assayed with andwithout SERM cotreatment in E2-treated MCF-10A cells:SULT 1E1 expression was induced by DMA and Ral. UGT1A1 was also induced by DMA and Ral cotreatment; how-ever, the expression of UGT 1A1 was low in MCF-10A cellswhen tested by immunoblotting. The inhibitory effects ofDMA and Ral on MeOE1 formation were lost when super-natants were treated with sulfatase/glucuronidase (Fig. 4).Future studies of the role of glucuronidation in detoxifica-tion will interrogate formation of the hydrophilic glucur-onate and sulfate metabolites. However, the combinedobservations support induction of phase II metabolism,and particularly SULT 1E1 expression, as a mechanism ofdetoxification of carcinogenic estrogen metabolites byselected SERMs in breast epithelial cells.
3
2
1
0
Re
lati
ve
pro
tein
ex
pre
ss
ion *
*
SULT1
b-Actin
Control E2
E2
E2
E2
E2+DMA
E2+DMA
E2+DMA
E2+DMA
E2+FDMA
E2+FDMA
E2+FDMA
E2+FDMA
E2+Ral
E2+Ral
E2+Ral
E2+Ral
Fo
ld c
ha
ng
eF
old
ch
an
ge
*
*
2.0
1.5
1.0
0.5
0.0
10
8
6
4
2
0
5
4
3
2
1
0
% C
olo
ny
eff
icie
ncy P < 0.001
DMSO
**
**
**
**
A
B
C
D
Figure 5. A, a significant induction in SULT1 enzyme expression wasobserved by immunoblotting on cotreatment ofMCF-10A cells with DMAor Ral. Relative protein amounts were determined by densitometricanalysis of SULT1 protein after Western blotting, loading with 30 mg oftotal protein. Each treatment was normalized to the loading andtransferring control, b-actin. Each data point represents an average of3 independent experiments in duplicate � SD. �, P < 0.05. B, genetranscription of SULT 1E1 was significantly induced by DMA and Ral,
whereas FDMA had no effect compared with E2 treatment alone. C, genetranscription of UGT 1A1 was significantly induced on cotreatment withDMA or Ral as measured by qPCR after isolating RNA from 24 hourstreated MCF-10A cells. Each data point represents an average of 3independent experiments � SD. �, P < 0.05; ��, P < 0.01. D, cotreatmentwith DMA or Ral significantly inhibited E2-induced anchorage-independent colonygrowthofMCF-10Acells in soft agar,whereasFDMAcotreatment had little effect. Cells were treated twice a week, in thepresence and absence of SERMs, over the course of 3 weeks. DMSO(0.01%)wasused as the vehicle control in the experiments in the absenceof E2 treatment. Cells were plated on soft agar and maintained for 3weeks. Relative colony efficiency is calculated by dividing the number ofcolonies counted in a well by the number of cells plated in each well,normalized to DMSO vehicle. Data show mean from 3 independentexperiments � SD. ��, P < 0.005.
Hemachandra et al.
Cancer Prev Res; 7(5) May 2014 Cancer Prevention Research512
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296
FDMA, proved to be an excellent probe of mechanism,because of the SERMs studied, FDMA alone did not atten-uatemetabolite levels, nor upregulate SULT1E1, nor inhibitboth ROS and 8-oxo-dG formation. Clear trends wereobserved in reduction of the 2-MeOE1 metabolite by DMAandRal, although significancewasnot reached. Several linesof evidence strongly suggest that the 4-OHcatechol estrogenis the proximal carcinogen in estrogen chemical carcino-genesis (13–17). It is possible that in the model systemunder study, malignant transformation can be elicited byboth isomeric catechol estrogens; however, more detailedstudy would be needed.It was important to confirm that the observed expression
of SULT 1E1, attenuation of estrogen metabolism, andinhibition of malignant transformation were not mediatedvia ligand binding to ER and resulting classical, ERE-medi-ated transcription. The lack of effect observed for FDMAand4HO-Tam, both high-affinity ER ligands, supports thisassertion. Furthermore, estrogenmetabolismwasmeasuredin the presence of DMA analogues that manifest variedselectivity for ERa and ERb and agonist/antagonist activity
at ER: no correlation was observed between attenuation ofestrogen metabolism and ER/ERE signaling.
Interestingly, the phytoestrogen genistein has been thesubject of 2 recent studies in MCF-10A cells, implicatingindependently upregulation of detoxification enzymes (57)and of PTEN (58) in mediating chemoprevention. Thecause was speculatively attributed to ligand binding to ERbor the G-protein–coupled receptor, GPR30 (GPER; ref. 59).GPR30mediates many nonclassical, extranuclear actions ofestrogens and anti-estrogens, including the actions of Ral,DMA, and DMA analogues (42). However, several SERMsare able to act as phenolic antioxidants and to activate stressresponse via Nrf2 and the antioxidant response element(ARE; refs. 60–63); therefore, ER-independent pathways areknown that might regulate function in MCF-10A cells byDMA, Ral, and Baze.
The present study demonstrates that clinical SERMs canattenuate estrogen chemical carcinogenesis by modulatingoxidative estrogen metabolism. Treatment of human breastepithelial cells with the SERMs, Ral, DMA, and Baze, butnot 4-OHTam, led to inhibition of oxidative estrogenmetabolism. Attenuated oxidative metabolism and lowerlevels of ROS were correlated with inhibition of E2-inducedmalignant transformation. Themechanismof inhibition byRal and DMA was shown to be detoxification of genotoxicestrogen metabolite accumulation mediated via upregula-tion of SULT 1E1. Further studies are underway to identifythe proximal receptor for these SERMs and to extend studiesto animal models.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L.P.M.P. HemachandraDevelopment of methodology: L.P.M.P. Hemachandra, R.E.P. Chandra-sena R. ScismAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): L.P.M.P.Hemachandra,H. Patel, J. Choi, S.Wang,Y. Wang, E. Thayer, R. Scism, R. Xiong, M. SiklosAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis): L.P.M.P. Hemachandra, R.E.P. Chandra-sena, J. Choi, S.C. Piyankarage, S. Wang, Y. Wang, E. Thayer, R. ScismWriting, review, and/or revision of themanuscript: L.P.M.P. Hemachan-dra, Y. Wang, J.L. Bolton, G.R.J. ThatcherAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): S. WangStudy supervision: G.R.J. ThatcherSynthesis of compounds: B.T. Michalsen
AcknowledgmentsThe authors thankMaitrayee Bose andPing Yao for their technical support
and thank Nadine Hempel, SUNY College of Nanoscale Science &Engineering.
Grant SupportThe support for this work was provided by NIH grants CA79870 (J.L.
Bolton) and CA102590 (G.R.J. Thatcher).The costs of publication of this article were defrayed in part by the
payment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received August 22, 2013; revised January 19, 2014; accepted February 3,2014; published OnlineFirst March 5, 2014.
Figure 6. A, cotreatment with BTC, HP-BTC, or AcBTC significantlyinhibited oxidative estrogen metabolism in E2-treated MCF-10A cells asshown by measurement of MeOE1 after 6 days. Normalization of%MeOE1 with respect to cells treated with 100% E2 alone is describedfully in the text. Each data point represents an average of 3 independentexperiments in duplicate � SD. ��, P < 0.01. B, classical ER/EREsignaling measured using a pERE luciferase reporter after treatment ofcells for 24 hours with BTC, HP-BTC, AcBTC, or TolBTC: in MCF-7 cellsto obtain data for ERa; and in MDA-MB-231:b41 cells to test forERb-mediated activity. Data represent an average of 3 independentexperiments performed in duplicates � SD.
SERMs Upregulate Estrogen Detoxification
www.aacrjournals.org Cancer Prev Res; 7(5) May 2014 513
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296

References1. Russo J, Russo IH. Biological and molecular bases of mammary
carcinogenesis. Lab Invest 1987;57:112–37.2. Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcino-
genesis 1996;17:2279–84.3. Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogen-
esis 2000;21:427–33.4. Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer.
N Engl J Med 2006;354:270–82.5. Bolton JL, Pisha E, Zhang F, Qiu S. Role of quinoids in estrogen
carcinogenesis. Chem Res Toxicol 1998;11:1113–27.6. Bolton JL, Thatcher GRJ. Potential mechanisms of estrogen quinone
carcinogenesis. Chem Res Toxicol 2008;21:93–101.7. Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I,
Higginbotham S, et al. Molecular origin of cancer: Catechol estro-gen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad SciU S A 1997;94:10937–42.
8. Iida T, Furuta A, Kawashima M, Nishida J, Nakabeppu Y, Iwaki T.Accumulation of 8-oxo-20-deoxyguanosine and increased expressionof hMTH1 protein in brain tumors. Neurol Oncol 2001;3:73–81.
9. Roszkowski K, JozwickiW,BlaszczykP,Mucha-MaleckaA, SiomekA.Oxidative damageDNA: 8-oxoGua and8-oxodGasmolecularmarkersof cancer. Med Sci Monit 2011;17:CR329–33.
10. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidativestress and DNA damage in human carcinogenesis. Mutat Res2011;711:193–201.
11. Wang Z, Chandrasena ER, Yuan Y, Peng KW, van Breemen RB,Thatcher GR, et al. Redox cycling of catechol estrogens generatingapurinic/apyrimidinic sites and 8-oxo-deoxyguanosine via reactiveoxygen species differentiates equine and human estrogens. ChemRes Toxicol 2010;23:1365–73.
12. Fussell KC, Udasin RG, Smith PJS, Gallo MA, Laskin JD. Catecholmetabolites of endogenous estrogens induce redox cycling and gen-erate reactive oxygen species in breast epithelial cells. Carcinogenesis2011;32:1285–93.
13. Li JJ, Li SA. Estrogen carcinogenesis in Syrian hamster tissues: role ofmetabolism. Fed Proc 1987;46:1858–63.
14. Zhu BT, Bui QD, Weisz J, Liehr JG. Conversion of estrone to 2- and 4-hydroxyestrone by hamster kidney and liver microsomes: implicationsfor the mechanism of estrogen-induced carcinogenesis. Endocrinol-ogy 1994;135:1772–9.
15. Han X, Liehr JG. Microsome-mediated 8-hydroxylation of guaninebases of DNA by steroid estrogens: correlation of DNA damage byfree radicals with metabolic activation to quinones. Carcinogenesis1995;16:2571–4.
16. Zhao Z, Kosinska W, Khmelnitsky M, Cavalieri EL, Rogan EG, Chak-ravarti D, et al. Mutagenic activity of 4-hydroxyestradiol, but not 2-hydroxyestradiol, in BB rat2 embryonic cells, and the mutationalspectrum of 4-hydroxyestradiol. Chem Res Toxicol 2006;19:475–9.
17. Kastrati I, Edirisinghe PD, Hemachandra LP, Chandrasena ER, Choi J,Wang YT, et al. Raloxifene and desmethylarzoxifene block estrogen-induced malignant transformation of human breast epithelial cells.PLoS One 2011;6:e27876.
18. Rogan EG, Badawi AF, Devanesan PD, Meza JL, Edney JA,West WW,et al. Relative imbalances in estrogen metabolism and conjugation inbreast tissue of women with carcinoma: potential biomarkers ofsusceptibility to cancer. Carcinogenesis 2003;24:697–702.
19. Lu F, Zahid M,Wang C, SaeedM, Cavalieri EL, Rogan EG. Resveratrolprevents estrogen-DNA adduct formation and neoplastic transforma-tion in MCF-10F cells. Cancer Prev Res 2008;1:135–45.
20. Soule HD, Maloney TM, Wolman SR, Peterson WD, Brenz R, McGrathCM, et al. Isolation and characterization of a spontaneously immor-talized human breast epithelial cell line, MCF-10. Cancer Res 1990;50:6075–86.
21. Russo J, Hasan LareefM, Balogh G, GuoS, Russo IH. Estrogen and itsmetabolites are carcinogenic agents in human breast epithelial cells.J Steroid Biochem Mol Biol 2003;87:1–25.
22. Chandrasena RE, Edirisinghe PD, Bolton JL, Thatcher GRJ. Problem-atic detoxification of estrogen quinones by NAD(P)H-dependent
quinone oxidoreductase and glutathione-S-transferase. Chem ResToxicol 2008;21:1324–9.
23. Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS forNQO1-catalyzed reduction of estrogen ortho-quinones. Free RadicBiol Med 2007;43:1289–98.
24. Buckley DB, KlaassenCD. Tissue- and gender-specificmRNAexpres-sion of UDP-glucuronosyltransferases (UGTs) in mice. Drug MetabDispos 2007;35:121–7.
25. SpinkBC,KatzBH,HussainMM,PangS,Connor SP, AldousKM, et al.SULT1A1 catalyzes 2-methoxyestradiol sulfonation in MCF-7 breastcancer cells. Carcinogenesis 2000;21:1947–57.
26. Tamura HO, Taniguchi K, Hayashi E, Hiyoshi Y, Nagai F. Expressionprofiling of sulfotransferases in human cell lines derived from extra-hepatic tissues. Biol Pharm Bull 2001;24:1258–62.
27. Hemachandra LP, Madhubhani P, Chandrasena R, Esala P, ChenSN, Main M, et al. Hops (Humulus lupulus) inhibits oxidative estro-gen metabolism and estrogen-induced malignant transformation inhuman mammary epithelial cells (MCF-10A). Cancer Prev Res2012;5:73–81.
28. Overk CR, Peng KW, Asghodom RT, Kastrati I, Lantvit DD, Qin Z, et al.Structure-activity relationships for a family of benzothiophene selec-tive estrogen receptor modulators including raloxifene and arzoxifene.Chem Med Chem 2007;2:1520–6.
29. Tonetti DA, Rubenstein R, DeLeon M, Zhao H, Pappas SG, BentremDJ, et al. Stable transfection of an estrogen receptor beta cDNAisoform into MDA-MB-231 breast cancer cells. J Steroid BiochemMol Biol 2003;87:47–55.
30. Xu X, Keefer LK, Ziegler RG, Veenstra TD. A liquid chromatography-mass spectrometry method for the quantitative analysis of urinaryendogenous estrogen metabolites. Nat Protoc 2007;2:1350–5.
31. Wang Z, Wijewickrama GT, Peng KW, Dietz BM, Yuan L, van BreemenRB, et al. Estrogen receptor {alpha} enhances the rate of oxidativeDNAdamage by targeting an equine estrogen catechol metabolite to thenucleus. J Biol Chem 2009;284:8633–42.
32. Kurkela M, Siiskonen A, Finel M, Tammela P, Taskinen J, Vuorela P.Microplate screening assay to identify inhibitors of human catechol-O-methyltransferase. Anal Biochem 2004;331:198–200.
33. Peng KW, Wang H, Qin Z, Wijewickrama GT, Lu M, Wang Z, et al.Selective estrogen receptor modulator delivery of quinone warheadsto DNA triggering apoptosis in breast cancer cells. ACS Chem Biol2009;4:1039–49.
34. Loft S, Deng XS, Tuo J, Wellejus A, Sorensen M, Poulsen HE. Exper-imental study of oxidative DNA damage. Free Radic Res 1998;29:525–39.
35. Kim DW, Sovak MA, Zanieski G, Nonet G, Romieu-Mourez R, Lau AW,et al. Activation of NF-kappaB/Rel occurs early during neoplastictransformation of mammary cells. Carcinogenesis 2000;21:871–9.
36. Calaf G, Russo J. Transformation of human breast epithelial cells bychemical carcinogens. Carcinogenesis 1993;14:483–92.
37. Kastrati I, Edirisinghe PD, Wijewickrama GT, Thatcher GR. Estrogen-induced apoptosis of breast epithelial cells is blocked by NO/cGMPand mediated by extranuclear estrogen receptors. Endocrinology2010;151:5602–16.
38. Spink DC, Spink BC, Cao JQ, DePasquale JA, Pentecost BT, FascoMJ, et al. Differential expression of CYP1A1 and CYP1B1 in humanbreast epithelial cells and breast tumor cells. Carcinogenesis 1998;19:291–8.
39. Fu J, Weise AM, Falany JL, Falany CN, Thibodeau BJ, Miller FR, et al.Expression of estrogenicity genes in a lineage cell culture model ofhuman breast cancer progression. Breast Cancer Res Treat 2010;120:35–45.
40. Hevir N, Trost N, Debeljak N, Rizner TL. Expression of estrogen andprogesterone receptors and estrogen metabolizing enzymes indifferent breast cancer cell lines. Chem Biol Interact 2011;191:206–16.
41. Wang J, Gildea JJ, Yue W. Aromatase overexpression induces malig-nant changes in estrogen receptor alpha negative MCF-10A cells.Oncogene 2013;32:5233–40.
Cancer Prev Res; 7(5) May 2014 Cancer Prevention Research514
Hemachandra et al.
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296

42. Abdelhamid R, Luo J, Vandevrede L, Kundu I, Michalsen B, Litosh VA,et al. Benzothiophene selective estrogen receptor modulators provideneuroprotection by a novel GPR30-dependent mechanism. ACSChem Neurosci 2011;2:256–68.
43. Miller CP,ColliniMD, TranBD,HarrisHA,KharodeYP,Marzolf JT, et al.Design, synthesis, and preclinical characterization of novel, highlyselective indole estrogens. J Med Chem 2001;44:1654–7.
44. Komm BS, Chines AA. An update on selective estrogen receptormodulators for the prevention and treatment of osteoporosis. Matur-itas 2012;71:221–6.
45. Dutertre M, Smith CL. Molecular mechanisms of selective estrogenreceptor modulator (SERM) action. J Pharmacol Exp Ther 2000;295:431–7.
46. Liu H, Bolton JL, Thatcher GRJ. Chemical modification modulatesestrogenic activity, oxidative reactivity, and metabolic stability in 40F-DMA, a new benzothiophene selective estrogen receptor modulator.Chem Res Toxicol 2006;19:779–87.
47. Yu B, Dietz BM, Dunlap T, Kastrati I, Lantvit DD, Overk CR, et al.Structural modulation of reactivity/activity in design of improved ben-zothiophene selective estrogen receptor modulators: induction ofchemopreventive mechanisms. Mol Cancer Ther 2007;6:2418–28.
48. Okoh V, Deoraj A, Roy D. Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim BiophysActa 2011;1815:115–33.
49. Loft S, Poulsen HE. Cancer risk and oxidative DNA damage in man.J Mol Med (Berl) 1996;74:297–312.
50. Chen Z-H, Na H-K, Hurh Y-J, Surh Y-J. 4-Hydroxyestradiol inducesoxidative stress and apoptosis in human mammary epithelial cells:possible protection by NF-[kappa]B and ERK/MAPK. Toxicol ApplPharmacol 2005;208:46–56.
51. Markides CS, Roy D, Liehr JG. Concentration dependence of proox-idant and antioxidant properties of catecholestrogens. Arch BiochemBiophys 1998;360:105–12.
52. Park S-A, Na H-K, Kim E-H, Cha Y-N, Surh Y-J. 4-Hydroxyestradiolinduces anchorage-independent growth of humanmammaryepithelialcells via activation of kB kinase: potential role of reactive oxygenspecies. Cancer Res 2009;69:2416–24.
53. ZahidM, SaeedM, Beseler C, Rogan EG, Cavalieri EL. Resveratrol andN-acetylcysteine block the cancer-initiating step in MCF-10F cells.Free Radic Biol Med 2011;50:78–85.
54. Chen ZH, Hurh YJ, NaHK, Kim JH, Chun YJ, KimDH, et al. Resveratrolinhibits TCDD-induced expression of CYP1A1 and CYP1B1 andcatechol estrogen-mediatedoxidativeDNAdamage in cultured humanmammary epithelial cells. Carcinogenesis 2004;25:2005–13.
55. Fu J, Fang H, Paulsen M, Ljungman M, Kocarek TA, Runge-Morris M.Regulation of estrogen sulfotransferase expression by confluence ofMCF10A breast epithelial cells: role of the aryl hydrocarbon receptor.J Pharmacol Exp Ther 2011;339:597–606.
56. Shatalova EG, Walther SE, Favorova OO, Rebbeck TR, Blanchard RL.Genetic polymorphisms in human SULT1A1 and UGT1A1 genesassociate with breast tumor characteristics: a case-series study.Breast Cancer Res 2005;7:R909–21.
57. Steiner C, Peters WH, Gallagher EP, Magee P, Rowland I, Pool-ZobelBL. Genistein protects human mammary epithelial cells from benzo(a)pyrene-7,8-dihydrodiol-9,10-epoxide and 4-hydroxy-2-nonenal gen-otoxicity bymodulating the glutathione/glutathione S-transferase sys-tem. Carcinogenesis 2007;28:738–48.
58. Rahal OM, Simmen RC. PTEN and p53 cross-regulation induced bysoy isoflavone genistein promotesmammary epithelial cell cycle arrestand lobuloalveolar differentiation. Carcinogenesis 2010;31:1491–500.
59. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptorGPER in health and disease. Nat Rev Endocrinol 2011;7:715–26.
60. Bolton JL, Yu L, Thatcher GRJ. Quinoids formed from estrogens andantiestrogens. Methods Enzymol 2004;378:110–23.
61. Dowers TS, Qin ZH, Thatcher GRJ, Bolton JL. Bioactivation of selec-tive estrogen receptor modulators (SERMs). Chem Res Toxicol2006;19:1125–37.
62. Yu L, Liu H, Li W, Zhang F, Luckie C, van Breemen RB, et al. Oxidationof raloxifene to quinoids: potential toxic pathways via a diquinonemethide and o-quinones. Chem Res Toxicol 2004;17:879–88.
63. Liu H, Liu J, van Breemen RB, Thatcher GRJ, Bolton JL. Bioactivationof the selective estrogen receptor modulator desmethylated arzox-ifene to quinoids: 40-fluoro substitution prevents quinoid formation.Chem Res Toxicol 2005;18:162–73.
www.aacrjournals.org Cancer Prev Res; 7(5) May 2014 515
SERMs Upregulate Estrogen Detoxification
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296

2014;7:505-515. Published OnlineFirst March 5, 2014.Cancer Prev Res L.P. Madhubhani P. Hemachandra, Hitisha Patel, R. Esala P. Chandrasena, et al. Oxidative MetabolitesHuman Mammary Epithelial Cells by Upregulating Detoxification of SERMs Attenuate Estrogen-Induced Malignant Transformation of
Updated version
10.1158/1940-6207.CAPR-13-0296doi:
Access the most recent version of this article at:
Material
Supplementary
1
http://cancerpreventionresearch.aacrjournals.org/content/suppl/2014/03/05/1940-6207.CAPR-13-0296.DCAccess the most recent supplemental material at:
Cited articles
http://cancerpreventionresearch.aacrjournals.org/content/7/5/505.full#ref-list-1
This article cites 63 articles, 10 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerpreventionresearch.aacrjournals.org/content/7/5/505To request permission to re-use all or part of this article, use this link
Research. on June 18, 2018. © 2014 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst March 5, 2014; DOI: 10.1158/1940-6207.CAPR-13-0296