university of groningen a potential strategy to treat ... · (kc), and hepatic stellate cells (hsc,...
TRANSCRIPT

University of Groningen
A potential strategy to treat liver fibrosisGonzalo Lázaro, Teresa
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2006
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Gonzalo Lázaro, T. (2006). A potential strategy to treat liver fibrosis: Drug targeting to hepatic stellate cellsapplying a novel linker technology s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 04-12-2018

A potential strategy to treat liver fibrosis:
Drug targeting to hepatic stellate cells applying a
novel linker technology
Teresa Gonzalo Lázaro
PhD Thesis, Groningen University, The Netherlands.

Cover art: Fermín Oliván Ahedo. Adapted from “The Starry night” by
Vincent Van Gogh (1889, located in the Museum of Modern Art, MoMA, New
York), and a fluorescent picture where it is illustrated the purpose of the
present thesis: targeting of drugs to the hepatic stellate cells.
Green/blue color corresponds to the target cell, Hepatic Stellate Cells, within
the fibrotic liver, red color corresponds to losartan-M6PHSA, a drug targeting
conjugate developed during the present thesis project.
Lay out: T. Gonzalo Lázaro
ISBN printed version: 90-367-2720-0 ISBN electronic version: 90-367-2754-5 © 2006 by T. Gonzalo Lázaro. All rights reserved. Neither this book nor its parts may be reproduced or transmitted in any form or by any means without permission of the author. Printed by Gildeprint D rukkerijen, Enschede, The Netherlands.

RIJKSUNIVERSITEIT GRONINGEN
A potential strategy to treat liver fibrosis:
Drug targeting to hepatic stellate cells applying a
novel linker technology
Proefschrift
ter verkrijging van het doctoraat in de Wiskunde en Natuurwetenschappen aan de Rijksuniversiteit Groningen
op gezag van de Rector Magnificus, dr. F. Zwarts, in het openbaar te verdedigen op
vrijdag 29 september 2006 om 13.15 uur
door
Teresa Gonzalo Lázaro
geboren op 29 oktober 1977
te Madrid, Spanje.

Promotores : Prof. dr. K. Poelstra
Prof. dr. D.K.F. Meijer
Copromotor: Dr. R.J. Kok Beoordelingscommissie : Prof. dr. J. Reedijk
Prof. dr. W. Hennink Prof. dr. P. Ginés

A mis padres, Manuel y Cecilia
The old King of Salem said: - “A mysterious force shows you how to realize your destiny.
It prepares your spirit and your will, because there is one great truth on this planet: whoever you are, or whatever it is that you do,
when you really want something, it's because that desire originated in the soul of the universe,
and the universe conspires to help that person to realize his dream. It's your mission on earth”
Paulo Coelho, The Alchemist, 1988.

The research described in this thesis was conducted at the Faculty of
Mathematics and Natural Sciences, Department of Pharmacokinetics and Drug
Delivery, University of Groningen, The Netherlands in collaboration with
Liver Unit (IDIBAPS institute) in the Hospital Clínic in Barcelona, Spain.
This research project was financially supported by SenterNovem (TSGE1083)
and NWO Science Netherlands (R 02-1719, 98-162).
The printing of this thesis was financially supported by grants of:
University of Groningen
Groningen University Institute for Drug Exploration (GUIDE).
Kreatech Biotechnology b.v, Amsterdam.
Paranimfen: Janja Plazar.
Aitana Peire Moráis.

Table of contents
Chapter 1. Aim of the thesis. 1
Chapter 2. General Introduction. 5
A potential strategy to treat liver fi brosis
Chapter 3. Selective targeting of pentoxifylline to hepatic stellate cells 35
using a novel platinum-based linker technology.
Chapter 4. Reduction of advanced liver fi brosis by targeted delivery of the 65
angiotensin II receptor antagonist losartan to hepatic stellate cells.
Chapter 5. Accumulation in hepatic stellate cells and antifi brotic eff ect 97
of losartan-M6PHSA in the CCl4 model of induced liver fi brosis.
Chapter 6. Local inhibition of liver fi brosis by specifi c delivery 125
of a PDGF kinase inhibitor to hepatic stellate cells.
Chapter 7. Summarizing discussion and future perspectives. 155
Chapter 8. Samenvatting. 169
Appendix.
Resumen 176
Abbreviations 182
Acknowledgements 183
Selected color fi gures 188
Curriculum Vitae and Publications 197

Bikes after snow in the garden at my house in Ubbo Emmiussingel,
Groningen, February 2004.

Chapter1Aim of the thesis
“Rest satisfi ed with doing well,
and leave others to talk of you as they please.”Pythagoras (582 BC - 507 BC).

2
Aim of the thesis
Liver fibrosis is the 9th leading cause of death in the world. This chronic disease cannot
be treated successfully with conventional antifibrotic and anti-inflammatory drugs
currently on the market, because they either lack efficacy or cause too many side-effects.
Targeting of antifibrotic agents to hepatic stellate cells is considered a promising strategy
to increase their therapeutic potential. This thesis will present a novel strategy to
synthesize drug targeting conjugates aimed at hepatic stellate cells. The core of our
invention is a novel platinum based a linker system, the so-called Universal Linkage
System (ULS), that allows us to couple a broad range of drugs.
Three different drugs have been employed in the present thesis. Pentoxifylline is an anti-
inflammatory drug that has been suggested as a potential antifibrotic drug once delivered
specifically to hepatic stellate cells (1). Losartan is an angiotensin II receptor antagonist
that is widely used for the treatment of hypertension and renal disease. Its beneficial
effects on the renin-angiotensin system can be applied to improve the liver function
during fibrosis (2). The third drug we have employed is a PDGF-R tyrosine kinase
inhibitor (PTKI), a gleevec-like compound. Due to the role of platelet derived growth
factor (PDGF-B) as the most potent mitogen to the HSC during liver fibrosis (3), PTKI is
regarded as a potential new drug to stop the fibrogenic process.
The aim of the present thesis is therefore to study the therapeutical approach of targeting
antifibrotic drugs specifically to the hepatic stellate cells to improve the liver injury. Three
different drug targeting conjugates are discussed in detailed, as well as the screening of its
efficacy in vitro and in vivo.

Chapter 1
3
Reference List
1. Raetsch,C., Jia,J.D., Boigk,G., Bauer,M., Hahn,E.G., Riecken,E.-O., and
Schuppan,D. 2002. Pentoxifylline downregulates profibrogenic cytokines and
procollagen I expression in rat secondary biliary fibrosis. Gut 50:241-247.
2. Bataller,R., Sancho-Bru,P., Gines,P., Lora,J.M., Al Garawi,A., Sole,M.,
Colmenero,J., Nicolas,J.M., Jimenez,W., Weich,N., Gutierrez-Ramos, J.C., Arroyo,V.,
and Rodes, J. 2003. Activated human hepatic stellate cells express the renin-
angiotensin system and synthesize angiotensin II. Gastroenterology 125:117-125.
3. Pinzani,M. 2002. PDGF and signal transduction in hepatic stellate cells. Front
Biosci. 7:d1720-d1726.
Chapter 1

Modesto, organizado amigo,
t
t
t
i
trabajador profundo, déjame darte el ala de mi canto, el golpe de aire, el salto de mi oda: ella nace de tu invisible máquina, ella vuela desde tu infatigable y encerrado molino, entraña delicada y poderosa, siempre viva y oscura. Mientras el corazón suena y atrae la partitura de la mandolina, allí adentro tú filtras y repartes, separas y divides, multiplicas y engrasas, subes y recoges los hilos y los gramos de la vida, los últimos licores, las ín imas esencias.
Víscera submarina, medidor de la sangre, vives lleno de manos y de ojos, midiendo y trasvasando en tu escondida cámara de alquimista. Amarillo es tu sistema de hidrografía roja, buzo de la más peligrosa profundidad del hombre, allí escondido siempre, sempiterno, en la usina, silencioso.Y todo sentimiento o estímulo creció en tu maquinaria,recibió alguna gota de tu elaboración infatigable, al amor agregaste fuego o melancolía, una pequeña célula equivocada o una fibra gastada en tu trabajo y el aviador se equivoca de cielo, el tenor se derrumba en un silbido, al astrónomo se le pierde un planeta.
Cómo brillan arriba los hechiceros ojos de la rosa, los labios del clavel matutino! Cómo ríe en el río la doncella! Y abajo el filtro y la balanza, la delicada química del hígado, la bodega de los cambios sutiles: nadie lo ve o lo canta, pero, cuando envejece o desgasta su mor ero, los ojos de la rosa se acabaron, el clavel marchitó su den adura y la doncella no cantó en el río. Austera parte o todo de mi mismo, abuelo del corazón, molino de energía: te canto y temo como si fueras juez, metro, fiel implacable, y si no puedo entregarme amarrado a la pureza, si el excesivo manjar o el vino hereditario de mi patria pretendieron perturbar mi salud o el equilibrio de mi poesía, de ti, monarca oscuro, distr buidor de mieles y venenos, regulador de sales, de ti espero justicia: Amo la vida: Cúmpleme! Trabaja! No detengas mi canto. Oda al Hígado. Pablo Neruda, Premio Nobel de Literatura 1971.

Chapter2General Introduction: Potential strategies to treat liver fi brosis
There, inside, you fi lter and apportionYou separate and divide,
You multiply and lubricateYou raise and gather
the threads and the grams of life…from you I hope for justice:
I love life: Do not betray me! Work on!Do not arrest my song.
Ode to the liver, Pablo Neruda, Nobel Prize for literature, 1971.

6
A potential strategy to treat liver fibrosis
Liver
In Greek mythology, Prometheus was punished by the gods for revealing fire to humans
and he was chained to a rock where an eagle, Ethon, pecked out parts of his liver, which
would grow to a complete organ again overnight. Curiously enough, the liver is the only
human internal organ that actually can regenerate itself, a characteristic which apparently
already was known to the Greeks. In fact, the liver is capable of natural regeneration of
lost tissue: as little as 25% of remaining liver can regenerate into a whole liver again (1).
Figure 1. Liver with the right and left lobe. (Obtained from Anatomy of the Human Body.
Fig. 1085 Henry Gray, 1918.)
The adult human liver normally weighs between 1.3 - 3.0 kilograms, and is a soft, pinkish-
brown "boomerang shaped" organ. It is the largest internal organ within the human body.
The liver is essential in keeping the body functioning properly. It plays an important role
in the clearance of compounds from the blood (metabolism, excretion), produces immune
proteins to control infections and directly removes germs and bacteria (innate immune
system) and synthesizes proteins that regulate blood clotting and various other
physiological processes. Furthermore, the liver produces and excretes bile fluid which is
required for food digestion and absorption of fats and fat-soluble vitamins (2). You
cannot live without a proper functioning liver. Due to this complex spectrum of functions
it is not yet possible to produce an artificial organ capable of replacing all functions of the
liver, and the attempts to do so are outside the reach of science in the foreseeable future.

7
Chapter 2
Liver cells network
Classically, the liver was seen as being divided in hexagonal lobules formed by
parenchymal hepatocytes that constitute around 80% of the total liver volume, and also by
three different nonparenchymal cell types: sinusoidal endothelial cells (SEC), Kupffer cells
(KC), and hepatic stellate cells (HSC, formerly known as fat-storing cells, Ito cells,
lipocytes or vitamin A-rich cells). The hepatocytes are important for the high metabolic
activity of the liver and the secretion of compounds into the bile (3;4). The fenestrated
endothelium plays an important role in the filtration of compounds from the blood to the
hepatocyte surface. Like Kupffer cells, SECs have a huge endocytic capacity for many
ligands including glycoproteins and several components of the extracellular matrix (ECM).
SEC are also active in the secretion of cytokines and other mediators of cellular activity
(5;6).
Kupffer cells are local tissue macrophages located in the sinusoids space with a
pronounced endocytic and phagocytic capacity. Kupffer cells control the early phase of
liver inflammation, and thus play an important part in the innate immune defense system
(7). High exposure of Kupffer cells to bacterial products, especially endotoxin
(lipopolysaccharide, LPS), leads to the production of inflammatory mediators and
ultimately to liver injury. Moreover, during liver injury and inflammation, Kupffer cells
secrete enzymes and cytokines that may damage hepatocytes, and they are active in the
remodeling of extracellular matrix (8).
Hepatic stellate cells (HSC) reside in the perisinusoidal space. In the normal liver, HSC are
characterized by storing vitamin A and they control the turnover of extracellular matrix,
and the regulation of the contractility of sinusoids. Acute damage to hepatocytes induces
transformation of the quiescent HSC into activated myofibroblast-like cells and the latter
cells play a key role in the development of inflammatory fibrotic responses (9;10).
Activated HSC transdifferentiate into proliferative, fibrogenic, and contractile
myofibroblasts that initiate further cell proliferation and increased deposition of
extracellular matrix (ECM) components in the process of wound healing (2;11). During
chronic liver injury, the excessive ECM replaces the functional liver, influencing the
Chapter 2

8
A potential strategy to treat liver fibrosis
function of remaining cells and forms a solid mechanical scaffold for cell adhesion and
migration. This matrix consists of collagens, glycoproteins, proteoglycans,
glycosaminoglycans and molecules that are bound specifically to the ECM, such as certain
growth factors/cytokines, matrix metalloproteinases (MMPs) and enzymes such as tissue
transglutaminase and procollagen propeptidases (12). It is a finely tuned ecosystem which
is dysbalanced during chronic liver injury.
Figure 2. Activation of Hepatic Stellate Cells during liver fibrogenesis.
A. Hepatic sinusoid with hepatocytes as parenchymal cells, and the non-parenchymal cells: the hepatic
stellate cells with the vitamin-A droplets (HSC) in the space of Disse, the endothelial cells (EC) and
the Kupffer cells (KC) in the sinusoid.
B. During liver injury, the activation of HSC leads to cell proliferation producing extracellular matrix
components (ECM). Figure adapted from Friedman, J Hepatol2003.
What is liver Fibrosis?
Liver fibrosis is a reaction to chronic liver injury, and it is characterized by an excessive
accumulation of extracellular matrix proteins including collagen. It is a common process
during the majority of chronic liver diseases (13). All liver cell types play their specific role
in liver fibrosis, and there is much evidence now of cross-talk between the different cell
types through the release of a wide variety of key mediators, e.g. nitric oxide, interleukins,
chemokines, growth factors and reactive oxygen species (ROS) (4). The cooperation
between liver cells is better understood due to the knowledge gained in the last decades on
liver fibrosis and other liver diseases.

9
Chapter 2
The fibrogenic process resembles a continuous wound-healing, yet accumulation of huge
amounts of extracellular matrix results in scarring of the tissue and disturbance of blood
supply. The progressive necrotic areas lead to even more inflammation and tissue damage
that needs to be repaired (12). If the liver injury persists, an exacerbation of fibrosis may
lead to the development of cirrhosis (14). Various etiological factors can be responsible
for a perpetuation of the fibrogenic process including alcohol consumption, exposure to
various drugs and toxic chemicals, viral hepatitis, metabolic syndrome, autoimmune
disease and hereditary disorders of metabolism (15). Chronic liver injury finally leads to
cirrhosis and all its complications, portal hypertension, and ultimately liver failure. Liver
transplantation, an extremely costly procedure, is currently the only remedy in this
condition (16).
Central to the liver fibrogenesis is the activation of HSC (17). HSCs are activated by
inflammatory and fibrogenic cytokines such as TGF-β, angiotensin II (18), and PDGF-BB
(19). Cellular changes accompanying HSC activation include morphological changes such
as the appearance of the cytoskeletal protein smooth muscle α -actin (α-SMA), a loss in
the cellular vitamin A stores, and an increase in the appearance of rough endoplasmic
reticulum. An increase in DNA synthesis and cellular proliferation also occurs following
HSC activation. The pattern of gene expression changes and a dramatic increase in types I
and III collagens production occurs (20). TGF-β is the most potent fibrogenic cytokine
described for the HSC activation (21) and the receptor expression for this cytokine is
largely increased following HSC activation. At the same time, HSC proliferate under the
influence of growth factors. Platelet derived growth factor (PDGF-BB) is regarded as the
most potent mitogen for HSCs. The PDGF receptor expression on HSC are also
increased during the liver injury (22;23), leading to a continuous proliferation of these
cells.
The perpetuation of the activated phenotype of HSC is caused by the ongoing cytokine
production and remodeling of extracellular matrix (ECM) (24;25). The collagen
production and the cytokines secreted by activated HSC as well as autocrine and paracrine
stimulation of other liver cell types (injured hepatocytes, Kupffer cells) contribute to the
Chapter 2

10
A potential strategy to treat liver fibrosis
aggravation of the fibrotic process. After prolonged chronic injury, the liver contains high
levels of the matrix proteins collagen and elastin and of other structural glycoproteins,
proteoglycans and pure carbohydrates, i.e. hyaluronan (26).
In conclusion, two major features render the HSC the key fibrogenic cell. Firstly, a
dramatic increase in the synthesis and deposition of extracellular matrix proteins produced
by activated HSC and, secondly, the increased proliferation rate of HSCs which strongly
amplifies the number of fibrogenic cells (27).
Moreover, HSC activation is associated with an increase in cell contractility, which leads to
increased portal pressure via the constriction of individual sinusoids and contraction of
the cirrhotic liver as a whole (28).
Epidemiology of liver fibrosis
Chronic liver disease is responsible for over 1.4 million deaths annually according to data
from the World Health Organization Mortality Database (WHO, World Health Report
2005; http://www.who.int/en/) and in the western world this disease is among the top
ten of disease-related causes of death (CDC, National Center for Health Statistics, 2005).
Overall there has been reported a 13% increase in the death rate from liver-related disease
per year (29). Of the liver-related deaths, 77% were associated with viral hepatitis, 14%
with alcohol abuse, and 9% with hepatocellular carcinoma (30). Many etiological factors
cause fibrosis and eventually lead to cirrhosis. It has been estimated that excessive alcohol
consumption is a major contributor in 41-95 percent of deaths from cirrhosis in some
countries (31). The level and duration of alcohol consumption are important determinants
in the development of liver pathology. As the primary site for detoxification of alcohol
and its metabolites, the liver can go through the following pathological stages: fatty liver,
alcoholic hepatitis, fibrosis and cirrhosis.
Because of the high rates of liver disease, liver transplantation is now considered a
standard therapy for patients with end-stage liver disease, regardless of the cause.
Currently, about 5,000 liver transplants are performed yearly in the United States at more
than 120 medical centers. As a consequence of the limited supply of livers, there are more

11
Chapter 2
than 17,000 persons on the liver transplant waiting list and at least 1,500 will die annually
while waiting (http://liverplan.niddk.nih.gov.).
The concept of Liver fibrosis reversion
Centuries after the greeks suggested it in their mythology, the regeneration of the damaged
liver was finally catalogued by Perez-Tamayo in 1979 (32). In this study, the first evidence
for reversibility of fibrosis and cirrhosis in animal models and human was presented. This
has stimulated researchers to search for potential antifibrotic drugs that could definitely
reverse liver fibrosis. Emerging antifibrotic therapies aim at inhibiting the accumulation of
fibrogenic cells or preventing the deposition of extracellular matrix proteins. Although
various antifibrotic agents are effective in experimental models of liver fibrosis, to date
their efficacy and safety in humans have not been established (33-35). On the other hand,
evidence of fibrosis regression has been documented after treatment of patients with
antivirals for Hepatitis B (36) and Hepatitis C (37) in clinical trials. These studies and
studies in experimental animal models have improved the understanding of mechanisms
of extracellular matrix (ECM) production and degradation. It appeared that liver scar
tissue can be resorbed in fact (38). The accumulated ECM can be degraded through the
action of matrix metalloproteinases, enzymes that digest components of the ECM (39).
The existence of tissue inhibitors of these matrix metalloproteinases (TIMPs) partly
explains the excessive accumulation of ECM in fibrosis and influence the dynamic process
of synthesis and degradation. Interestingly, activated HSC also play a vital role in
orchestrating matrix degradation during liver fibrogenesis. Apart from their participation
in the synthesis of large amounts of extracellular matrix, simultaneously they increase
TIMP-1 levels resulting in decreased matrix degradation. When fibrosis regresses, TIMP-1
levels decline and degradation of ECM increases. This effect is associated with removal of
activated stellate cells through apoptosis (40;41). In contrast, sustained TIMP-1 expression
inhibits protease activity and blocks apoptosis of activated stellate cells (42). Thus, it is
implicit that fibrosis is associated with the massive deposition of extracellular matrix
(ECM), increased levels of inhibitors of matrix metalloproteinases and collagenases
Chapter 2

12
A potential strategy to treat liver fibrosis
(TIMP), and also a significant collagen cross-linking by tisssue transglutaminase activity
(43).
In conclusion, the fibrogenic process can be viewed as a dynamic balance between matrix
degradation and production so that even advanced fibrosis or cirrhosis seem to be
reversible.
Need for antifibrotic therapies
Many studies in the past two decades have shed considerable light on the mechanisms of
liver fibrosis, with particular emphasis on stellate cell biology, and have led to an increased
enthusiasm for treating hepatic fibrosis (44). There is tremendous activity in the field of
drug development for this purpose and ongoing testing of potential antifibrotic agents
(45). Nevertheless, efficient and well-tolerated antifibrotic drugs are still lacking, and
current treatment of hepatic fibrosis is limited to the withdrawal of the harmful agent and
transplantation. In situations in which dealing with the underlying process is not possible,
interference with the liver fibrosis process is essential. A large number of these
approaches have been validated in cultured cells and in animal models, and clinical trials
are underway or anticipated for a growing number of molecules (46).
A successful antifibrotic strategy does not need to eradicate hepatic fibrosis entirely,
because the liver has an enormous functional reserve. Instead, any therapeutical approach
that sufficiently attenuates fibrosis progression to prevent the development of cirrhosis
and/or hepatocellular carcinoma will be viewed as a success (47).
Several antifibrotic therapies have been tried ending with poor or mediocre success (48).
The problems with many potentially antifibrogenic drugs are, among others, the lack of
cell specificity of the drugs in vivo with the occurrence of extrahepatic side effects. For
such drugs, a cell-specific delivery may prove beneficial. The acquired high specificity of a
locally delivered compound would permit the long-term treatment that is required for a
chronic liver disease.
During the past years, several projects concerning target-cell specific antifibrotic therapies
have been started in our department (Table 1). Several promising therapeutic approaches

13
Chapter 2
have been encompassed in our drug delivery strategies, either targeting hepatic
inflammation (dexamethasone, naproxen, losartan)(49) or intracellular signaling and
transcriptional pathways involved in stellate cell activation and ECM turnover
(Pentoxyfilline, gleevec, kinase inhibitors)(50;51), or provoking apoptosis of activated cells
(doxorubicin, gliotoxin)(52;53). Other strategies include the delivery of anti-inflammatory
agents like IL-10. The near future will learn which of the chosen drugs will be most
effective, or which other (combination of) targeted therapies can be envisioned. In the
next section, three drugs will be more extensively discussed: pentoxyfilline, losartan and
the PDGF receptor tyrosine kinase inhibitor (imatinib). These compounds have been
subject of drug targeting strategies in the present thesis.
Chapter 2

14
A potential strategy to treat liver fibrosis
Table 1. Drugs in development for antifibrotic approaches in our group.
Main mechanism Agent Studies (Ref) In our department
Attenuate HSC activation
Pentoxifylline JCR 2006 (54) T. Gonzalo
Interleukin-10 Pharm Res. 2004 (55) H.Rachmawati
Losartan In preparation (56) T. Gonzalo
Reduce inflammation
Prostaglandin In preparation W.Hagens
Promoting apoptosis of HSC
Gliotoxin Liver Int. 2006 (60) W.Hagens
PDGF R inhibitor
Submitted (57) T. Gonzalo
Micophenolic acid
J Hepatol.2005 (58) R.Greupink
Antiproliferative to HSC
Doxorubicin JPET 2006 (59) R.Greupink
p38 MAP kinase inhibitor
JPET 2006 (61) J.Prakash Reduction
inflammation and epithelial-mesenchymal transformation TGF-b kinase
inhibitor In preparation
J.Prakash

15
Chapter 2
Antifibrotic drugs: Pentoxifylline, losartan and PDGF
Tyrosine Kinase Inhibitor (PTKI)
Pentoxifylline
Pentoxifylline (PTX) is a phosphodiesterase inhibitor that is clinically useful for the
treatment of disorders of vascular perfusion and cerebrovascular diseases due to its
favorable effect as a peripheral vasodilator (62). The antifibrogenic effect of PTX on
activated hepatic stellate cells has been extensively reported and demonstrated (63‐65).
Although its mechanism of action remains unclear, it has been suggested that PTX
reduces the transdifferentiation of HSC to myofibroblasts and inhibits HSC proliferation
(11;66;67).
Figure 3. Proposed antifibrotic mechanism of Pentoxifylline action in the cell.
Figure adapted from references (Raetsch,2002; Duncan 1995; Rodriguez-Barbero 2002; Chen 1999)
(11;68-70). Abbreviations: PTX, pentoxifylline; PKA, protein kinase A; cAMP, cyclic adenosine
monophosphate; CTGF, connective tissue growth factor; TGF-β, transforming growth factor β; ERK,
extracellular-regulated kinase; P38 MAP kinase, mitogen-activated kinase; ECM, extracellular matrix.
Chapter 2

16
A potential strategy to treat liver fibrosis
PTX also may reduce the fibrogenic effect of TGF-β on HSC by interference with p38
MAPkinase and ERK1/2 pathways, thereby decreasing hepatic procollagen type 1 mRNA
expression (71). In addition, PTX interferes with cAMP involved in inflammatory
signaling (72). In another study it was shown that PTX blocks NF-kB, one of the
important mediators of HSC, thus preventing the activation and proliferation of HSC
induced by carbon tetrachloride, a rat model of liver fibrosis. In vitro studies with
fibroblasts have shown that PTX potently reduces cell proliferation, stimulates interstitial
collagenase activity, and suppresses the synthesis, secretion, and deposition fibrillar
collagens type I and III, proteoglycans, and fibronectin (73-75). Moreover, Lee et al
demonstrated that PTX downregulates hepatic procollagen type I expression in the bile
duct ligation model of liver fibrosis (76).
Yet, despite beneficial effects in HSC, profibrotic effects of PTX on Kupffer cells have
been reported (11), as well as many effects in other cell types (77;78). Cell-selective
targeting of PTX to HSC seems therefore required to create favorable effects in HSC,
while avoiding the profibrotic effects in Kupffer cells and effects in other organs (79).
Taking into account the antifibrotic properties attributed to PTX, we have developed a
drug targeting conjugate for the delivery of PTX to HSC. In chapter 2 we describe the
development of the novel HSC-directed conjugate PTX-M6PHSA.
Losartan
Losartan is an orally active, nonpeptide angiotensin II (Ang II) receptor antagonist. It was
the first of a new class of drugs introduced for the treatment of hypertension and renal
disease. These angiotensin receptor blockers bind competitively and selectively to the Ang
II type 1 (AT1) receptor, thereby blocking Ang II-induced physiological effects (80).
Systemic hypertension is a complex pathophysiological state that is primarily manifested as
chronic high blood pressure. It is a major risk factor for stroke, ischemic heart disease,
peripheral vascular disease, and progressive renal damage (81). It is well established that a
hyperactive renin angiotensin system (RAS) plays a key role in the development and
maintenance of human primary hypertension. This disorder contributes to at least 10% to
30% of all cases of hypertension by some estimation (82).

17
Chapter 2
RAS blockers are reliable and affordable, and their short duration of action makes them an
excellent drug of choice if reversal of their activity is required. On the other hand, as with
most antihypertensive drugs, their effects are short-lived having to be administered on a
frequent basis with a risk of significant side effects (83).
Recent experimental studies indicate that the RAS also plays an important role in liver
fibrogenesis (84;85). Hepatic stellate cells (HSC) are the main target cell type for the
pathogenic effects of Ang II in liver fibrosis. In the normal human liver, HSC do not
express AT1 receptors nor do they secrete Ang II. Following chronic liver injury however,
HSC transform into myofibroblast-like cells which express both AT1 receptors and
generate mature Ang II, which exerts an array of pro-inflammatory and profibrogenic
actions (86-89). These pathogenic effects can be prevented largely by AT1 receptor
antagonists, as has been demonstrated in different models of experimentally-induced liver
fibrosis (90-92). Based on these data, RAS inhibitors like Angiotensin Converting Enzyme
(ACE) inhibitors or Ang II receptor blockers are currently considered as novel antifibrotic
therapies to treat liver fibrosis. Preliminary clinical data suggest that AT1 receptor
blockers may attenuate the fibrogenic process (93). The use of AT1 receptor blockers
would be particularly useful in conditions characterized by a rapid progression of fibrosis
(i.e. acute alcoholic hepatitis and severe hepatitis C virus reinfection after liver
transplantation). However, in patients with advanced fibrosis, the use of angiotensin
antagonists may be hampered by undesirable effects on the arterial pressure, especially
since patients with cirrhosis are generally associated with low systemic blood pressure.
Indeed, the use of the AT1 receptor blocker losartan in patients with advanced fibrosis
was associated with the risk of hypotensive shock syndrome (94).
We hypothesized that targeting of losartan to HSC could be an effective strategy to
attenuate hepatic fibrosis. Therefore, we have coupled losartan to M6PHSA, a stellate cell-
selective carrier, resulting in losartan-M6PHSA. In addition, this strategy would overcome
side effects such as reduction of blood pressure. Details about this novel approach are
explained in chapters 3 and 4 in the present thesis.
Chapter 2

18
A potential strategy to treat liver fibrosis
PDGF Receptor tyrosine kinase inhibitors
The fundamental role that protein tyrosine kinases appear to play in liver fibrogenesis and
many other diseases, has made them attractive therapeutic targets (95), and has provided
the rationale for the development of specific inhibitors of these enzymes.
PDGFR-β is a receptor tyrosine kinase that consists of an extracellular ligand binding
domain connected to a cytoplasmic domain, responsible for intracellular signal
transduction. Extracellularly, the PDGFRβ receptor binds via the beta subunits only the
isoform PDGF-B (96;97). The binding induces activation of the receptor kinases by
formation of receptor dimers that catalyze the transfer of the γ–phosphate of ATP to
tyrosine residues in protein substrates. This leads to a wide variety of cell responses, e.g,
differentiation, proliferation, migration, angiogenesis and survival (98).
Figure 4. Proposed mechanism of action of PDGFR-β kinase inhibitors interfering with a wide variety of cell
responses. (Adapted from Aaronson 1991).

19
Chapter 2
Activated HSC display high levels of PDGFR-β receptor and release PDGF-BB cytokines
during liver fibrosis (99). It has been demonstrated that PDGFR-β receptors are
upregulated on the cell surface of hepatic stellate cells during fibrosis (100-102). The
fibrogenic process is encouraged by platelet-derived growth factor (PDGF-BB), identified
as the most potent mitogen for HSC (103). Several isoforms of PDGF have been
described in activated HSC have been found. Dimers formed by disulphide bond are
PDGF-AA, AB, BB, CC, or DD polypeptide chains (104). The expression of the PDGF
isoforms is differentially regulated by PDGF-BB itself and by TGF-β1, two mediators that
are produced by HSC themselves during liver fibrogenesis (105;106). New drugs that
inhibit PDGF- β kinase activity have emerged in recent years.
Imatinib mesylate (Gleevec, STI571) was initially designed for use in chronic myeloid
leukemia (CML) as an anti-tumor drug. Imatinib is active against a number of related
tyrosine kinases that are mutated in cancer (107). Since imatinib also inhibits PDGFR-β
kinase activity, it may also be applied as a drug for liver fibrosis.
Other strategies targeting the PDGF pathway involve the use of antibodies against
PDGFR-β kinase or a soluble receptor blocking the natural binding of PDGF cytokine
(108). Blockade of PDGF-BB interaction with its tyrosine kinase receptor may represent a
promising approach for therapeutic intervention in hepatic fibrosis.
We have developed a new construct, PTKI-M6PHSA, in which a PDGF Tyrosine Kinase
Inhibitor (PTKI), related to imatinib mesylate, is coupled to our stellate cell-carrier
M6PHSA. In the present thesis, we tested the impact of PTKI-M6PHSA on liver
fibrogenesis in vitro and in vivo. Chapter 5 describes this approach.
Chapter 2

20
A potential strategy to treat liver fibrosis
Drug targeting technology
The first idea of drug targeting was proposed by Ehrlich in the nineteenth century. He
presented the idea of “the magic bullet” that can bind selectively to specific types of cells
in a manner similar to that of the key and lock approach. Scientists have ever since
worked on the principle of drug targeting based on this idea of specifically delivering
drugs to diseased cells.
Classical drug molecules:
actions in whole body
Side-effects toxicity limits effect
Drug-carrier
no serious side-effects
improved therapeutic effect
Drug targeting approach: locally acting drugs
Drug
limited therapeutic effect
Figure 5. Classical drug administration versus the drug targeting approach.
Drug targeting is defined as selective drug delivery to specific physiological sites, organs,
tissues, or cells where the pharmacological activity of the chosen drug is required (109). In
principle, a drug that distributes throughout the whole body after its administration may
cause side effects at sites other than the pathological tissues. Although that may not create
a problem for the majority of drugs, side-toxicity is one of the limiting events for the
therapeutical use of, for example, cytotoxic agents. Due to these adverse reactions, dose
limitations may prevent effective treatment. Therefore, selective delivery into the target

21
Chapter 2
tissue may allow a higher drug concentration at or in the target cells or even in specific
compartments of the target cells, and thus improve the therapeutic index/safety of such
compounds.
Drug targeting may be classified into two general strategies: passive and active targeting.
Passive targeting is a strategy whereby the physicochemical properties of carrier systems
increase the target/nontarget ratio of the quantity of drug delivered to the target tissues,
organs, or cells. In this way, targeting of drugs would avoid side effects by preventing
major distribution to a particular organ or cell type.
Carriers included in this category are synthetic polymers, some natural polymers such as
albumin, liposomes, micro (or nano) particles, and polymeric micelles. Chemical factors
such as hydrophilicity and positive/negative charge and physical factors such as size and
mass greatly influence the passive targeting efficiency. For example, the cardiotoxicity of
doxorubicin can be decreased by including it in a liposomal formulation (110). However, it
should be stressed that drugs in such preparations should maintain their intrinsic anti-
tumor efficacy and also should not exhibit side effects specifically related to the liposomal
formulations such as liver (macrophage) toxicity.
Active targeting employs specific receptor interactions to increase the delivery of drugs to
a target site where the pharmacological effect of the drug is required. The incorporation of
a homing devices, or site-directed ligands, redirects the construct to specific binding sites
on cell membranes. These interactions include antigen-antibody and ligand-receptor
binding. The homing devices can be carbohydrate ligands, functional groups bearing
antibodies or other peptide ligands. These homing devices may be coupled to drug carriers
like antibodies, or other proteinaceous carriers like albumin or transferrin, polymers,
liposomes or nanoparticles.
Drug Targeting approach to Stellate cells
Targeting of drugs to hepatic stellate cells (HSC) currently represents a challenge for the
scientists involved in the design of a treatment for liver fibrosis. As explained in detail in
Chapter 2

22
A potential strategy to treat liver fibrosis
the previous section, HSC are a crucial target for pharmacological intervention of liver
fibrosis. Various HSC-specific carriers have been developed in our group (79).
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
LOSARTAN
LOSARTAN
HSA M6P
M6P
LOSARTAN
ULS
LOSARTANULS
2
3
4
1
5Stop fibrosis process
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
LOSARTAN
LOSARTAN
HSA M6P
M6P
LOSARTAN
ULS
LOSARTANULS
2
3
4
1
5Stop fibrosis process
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
LOSARTAN
LOSARTAN
HSA M6P
M6P
LOSARTAN
ULS
LOSARTANULS
2
3
4
1
5
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
LOSARTAN
LOSARTAN
HSA M6P
M6P
LOSARTAN
ULS
LOSARTANULS
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
LOSARTAN
LOSARTAN
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
LOSARTAN
LOSARTAN
LOSARTAN
LOSARTAN
M6P R
M6P RHSA
M6P R
M6P RHSA
M6P R
M6P R
M6P R
M6P R
M6P R
M6P R
M6P R
M6P RHSAHSA
LOSARTAN
LOSARTAN
HSA M6P
M6P
LOSARTAN
ULS
LOSARTANULS HSA M6PM6P
M6PM6P
LOSARTAN
ULS
LOSARTAN
ULSULS
LOSARTANULSLOSARTANULSULS
2
3
4
1
5
2
3
4
1 2
3
4
1
5Stop fibrosis process
2
1Induction of M6P/IGFII R
(mannose-6-phosphate insulin growth factor II Receptor).
5
34
Binding of Losartan-M6PHSA
Target cell specific anti-fibrotic actions
Internalization and release of losartan
Liver damage leads to activation of Stellate Cells
2
1Induction of M6P/IGFII R
(mannose-6-phosphate insulin growth factor II Receptor).
5
34
5
34
Binding of Losartan-M6PHSA
Target cell specific anti-fibrotic actions
Internalization and release of losartan
Liver damage leads to activation of Stellate Cells
NKNN
NN
NCl
OH
drug carrierPt
NH2H2NULS=losartanNKNN
NN
NCl
OH
drug carrierPt
NH2H2NULS=NKNN
NN
NCl
OH
drug carrierPt
NH2H2NULS=NKNN
NN
NCl
OH
NKNN
NN
NCl
OH
drug carrierPt
NH2H2NULS=drug carrier
PtNH2H2NULS=
drug carrierPt
NH2H2NULS=losartan
Figure 6. Picture of Losartan-ULS-M6PHSA conjugate targeting to the
Hepatic Stellate cell (star-like cell) via the M6P/IGFII receptor upregulated
during liver injury.
The magic bullet concept can now be applied also for targeting of losartan, among other
drugs, as illustrated in figure 6. In this schematic model, the drug delivery preparation is
composed of three parts: drug, linker and carrier. The applied linkage system is ULS,
which will be explained later in this thesis, and the carrier is M6PHSA, which binds to the
M6P/IGFII-receptor on activated HSC.
Types of carrier
In figure 7 various types of drug carriers are depicted. The type of water-soluble polymeric
carrier also includes apart from the chemically prepared carriers, naturally occurring

23
Chapter 2
polymers. Emulsions comprise small oil droplets stabilized with a monolayer of an
amphiphilic substance on the surface. Nanospheres are solid small particles made from
natural or synthetic polymers. A major difference between droplets in emulsions and
nanospheres is the status of the interior: liquid for emulsions and solid for nanospheres. A
liposome is a vesicle made up with a lipid bilayer that mimics cellular membranes.
Polymeric micelles are an assembly of amphiphilic polymers (typically comprising
multiples, i.e. 10 to 100 of polymeric chains) with a spherical inner core and an outer shell.
Combination strategies are nowadays being employed, for example, antibody-targeted
liposomes, bispecific antibody-mediated viral vectors, ligand-peptide modified plasma
proteins, recombinant proteins, etc. In the present thesis, a modified human serum
albumin protein is being employed as a carrier.
carriers
Polymeric micelleLiposome
Water-soluble polymerProtein carriers
drug
Figure 7. Different types of carriers utilized for drug targeting.
It is generally known that nano-sized carriers are a prerequisite for efficient drug targeting.
Carrier systems of 200 nm diameter or smaller are used for drug targeting, and larger
systems are subjected to nonspecific capture in the reticuloendothelial system (111). On
the other hand, small drug carriers have a short circulation time in the blood stream due to
renal filtration. Therefore, drug carriers with a diameter from 10 nm to 200 nm are used in
drug targeting approaches. These carriers are not largely cleared by renal filtration or by
the reticulo-endothelial system which allows a large amount of delivery to the target sites.
Chapter 2

24
A potential strategy to treat liver fibrosis
M6PHSA as a soluble carrier protein
Albumin is the most abundant plasma protein, and has a biological half-life of 19 days. It
consists of a single chain of 585 aminoacids organized in a tridimensional structure in a
helical conformation. The helices are bound by 17 disulfide bridges, leaving only one free
thiol (Cys34) (112).
Albumin is biodegradable and therefore biocompatible and contains many different
functional groups, i.e. -NH2 of the lysine residues or methionine, which can be used for
conjugation of the homing device, the linker, or the drug. In addition, due to its size and
charge, it is not cleared from the blood by renal filtration.
In our strategy, albumin was modified with sugar mannose-6-phosphate groups on its
surface resulting in M6PHSA (113). M6PHSA has been shown to specifically interact with
mannose 6-phosphate/insulin-like growth factor II (M6P/IGFII) receptors expressed on
the surface of hepatic stellate cells. Due to stellate cell proliferation during liver fibrosis
and a concomitant increase in M6P/IGF II receptor expression on this cell type (100), the
disease process itself may selectively direct the carriers to the diseased tissue. This
targeting strategy may largely contribute to the increased therapeutic concentration of
drug in the target tissue (114).
Linkage between drug and carrier
The concept of the “magic bullet” is very intuitive and appealing and looks as if it can be
easily realized through simply coupling a drug to a carrier. However, the development of
drug targeting constructs is a delicate process and several difficulties have to be overcome
during the synthesis of the conjugate. The linkage between the drug and the carrier system
is a crucial element of the conjugate, as it controls both the stability of the conjugate and
the efficiency of drug release or rate, and eventually, the final therapeutic effect (115). For
bioconjugates, the nature of the linker between the pharmacologic agent and the carrier
often dictates the degree of successful delivery and its outcome. Over decades,
investigators have therefore invested great efforts to find the appropriate linkage system.

25
Chapter 2
Linkages used in bioconjugation to proteins
The release of the drug from the carrier is decisive for its pharmacological activity.
Various types of biodegradable linkages have been developed for coupling drugs to
proteins. Amide linkages can be used to conjugate a drug containing carboxylic acid or
amino groups, like mycophenolic acid to M6PHSA (58).
Drugs
NKNN
NN
NCl
OH
Losartan
Pentoxifylline
N
N
N
N
O
O
O
ULS
Schiff
Drug N NH
Carrier
Linkage
Ester
Drug O C carrier
O
drug carrierPt
NH2H2N
M6PHSA carrier
Stellate cell- carrier
N
N
N
N
O
N
N
PTKI
Figure 8. Drugs used in the present thesis, linkers available for coupling these
drugs, and M6PHSA carrier utilized for Stellate cell-directed drug targe ing.
PTKI can be conjugated only via ULS linker. Losartan can be conjugated via Ester or ULS linkage
and Pentoxifylline can be coupled via ULS or Schiff base linkage. ULS reacts with aromatic nitrogens of
the depicted ring (PTKI), tetrazole ring (losartan) or xanthine moiety (pentoxifylline).
t
Chapter 2

26
A potential strategy to treat liver fibrosis
The amide linkage however is not easily split within the target cell. In addition, the ester
linkers may be used to conjugate carboxylic acid groups and hydroxyl groups of drugs, like
in the case of losartan (figure 8). Also mycophenolic acid could be conjugated to
M6PHSA via an ester linkage. However this resulted in less drug molecules coupled to the
carrier as compared to the amide-based conjugate which allowed higher drug loading (58).
Yet, as anticipated, the conjugate synthesized via the ester linkage proved more effective
than with the amide linkage since active drug could be released from the ester linkage.
This example illustrates the crucial role that the linkage plays in drug targeting conjugates
efficiency. The Schiff base hydrazone linkers may form an imino bond between a carbonyl
group of the drug molecule and a hydrazine functionality of the spacer. Such linkages have
been employed with doxorubicin, streptomycin and chlorambucil (116-119). Pentoxifylline
could be also form a Schiff base linkage via its carbonyl group (figure 8). The disulfide
bond is a covalent linkage which arises as a result of the oxidation of two sulfhydryl (SH)
groups of cysteines or other SH-containing material (120). It can be formed with drug
molecules that contain free thiol groups or, alternatively, with drug derivatives in which a
free thiol group has been introduced. For instance, in liver-directed conjugates, gliotoxin
was conjugated to M6PHSA using a disulfide bond (121). A disadvantage of the disulfide
linkage is its relative instability in the bloodstream. Disulfide bonds can be degraded by
reducing enzymes or disrupted chemically by thiol-disulfide exchange with free thiol
compounds such as glutathione (122). Another type of linker is based on polymers.
Multiple drug molecules can be covalently attached to a single functional group of the
carrier when polymeric bridges are used.
The Universal Linkage System (ULS)
As can be appreciated, the finding of the appropriate linkage system is a critical step
during the synthesis of a promising drug carrier construct. Some of the linkers modalities
lack stability in plasma (ester linker), enzymes at non-targeted sites may cleave the linkage
or simply they are not able to react with the drug or the chosen carrier system (115).
A major problem in the synthesis of drug conjugates is that the majority of drugs cannot
be coupled using traditional linking procedures since they lack the appropriate functional

27
Chapter 2
groups (i.e, carbonyl, amino, hydroxyl or thiol groups). In the present thesis, we have
developed conjugates with a new linker technology that allows the coupling of a broader
spectrum of drugs. The ULS (Universal Linker System) is a platinum-based linker that has
been previously applied in Life Sciences for the conjugation of different types of reporter
molecules to DNA and proteins (123;124;128;129).
The ULS linker technology is based on platinum coordination and shows a cis geometry of
the coupled drug and carrier (Figure 8). The importance of this coordination chemistry is
based on the stability and kinetically slow release properties of platinum complexes to
nucleic acids and proteins (125). In general, platinum is found to react with S-donors such
as methionine and the Cys34 residues of albumin, the latter being the most abundant free
thiol group in blood plasma (126). Furthermore, it has been reported extensively that
platinum forms coordination bonds with aromatic Nitrogen groups in DNA, which is
kinetically favored over the reaction with Oxygen groups (127). The most important
feature of the ULS platinum linker in drug delivery derivatives is that the strength of the
drug-linker bond is stable enough to reach the target yet reversible enabling the release of
the drug in the target tissue or cells.
In the presently discussed drug targeting conjugates, different antifibrotic drugs were
linked to ULS and subsequently to the carrier protein M6PHSA. ULS linker bound to
aromatic nitrogens in the drug molecules. Furthermore, ULS may react with the
thiocarbonyl group between the M6P group and the albumin core protein, since it is
known that cisplatinum readily reacts with sulfur containing ligands. However, a reaction
of ULS with other functional groups in the protein like methionine or cysteine residues or
even hydroxyl or amine side chains may also occur. In chapter 2, the possible sites for a
reaction between ULS and M6PHSA are depicted. In the same chapter, characteristics of
drug release from ULS linker are explained in detail. In chapter 3, 4 and 5, we will present
data of drug targeting to HSC, employing the ULS linker during in vivo studies after
single or multiple administrations of the conjugates.
Chapter 2

28
A potential strategy to treat liver fibrosis
Reference List
1. Fausto,N., Campbell,J.S., and Riehle,K.J. 2006. Liver regeneration. Hepatology 43:S45-S53.
2. Friedman,S.L. 2003. liver fibrosis - from bench to bedside. J. Hepatol. 38:S38-S53.
3. Meijer,D.K.F., and Molema,G. 1995. Targeting of drugs to the liver. Semin. Liver Dis. 15:202-256.
4. Kmiec,Z. 2001. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell Biol. 161:III-151.
5. Martinez,I., Sveinbjornsson,B., Vidal-Vanaclocha,F., Asumendi,A., and Smedsrod,B. 1995. Differential cytokine-mediated modulation of endocytosis in rat liver endothelial cells. Biochem. Biophys. Res. Commun. 212:235-241.
6. McGary,C.T., Yannariello-Brown,J., Kim,D.W., Stinson,T.C., and Weigel,P.H. 1993. Degradation and intracellular accumulation of a residualizing hyaluronan derivative by liver endothelial cells. Hepatology 18:1465-1476.
7. West,M.A., and Heaney,M.L. 1992. Regulation of Kupffer cell activation. In Hepatocyte and Kupffer cell interactions. T.R.Billiar, and Curran,R.D., editors. CRC Press. London Tokyo. 209-241.
8. Ramadori,G., and Armbrust,T. 2001. Cytokines in the liver. Eur. J. Gastroenterol. Hepatol. 13:777-784.
9. Rockey,D. 1996. Endothelin in hepatic fibrosis--friend or foe? Hepatol 23:1698-1700.
10. Friedman,S.L. 1999. The virtuosity of hepatic stellate cells. Gastro 117:1244-1246.
11. Raetsch,C., Jia,J.D., Boigk,G., Bauer,M., Hahn,E.G., Riecken,E.-O., and Schuppan,D. 2002. Pentoxifylline downregulates profibrogenic cytokines and procollagen I expression in rat secondary biliary fibrosis. Gut 50:241-247.
12. Schuppan,D., Ruehl,M., Somasundaram,R., and Hahn,E.G. 2001. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 21:351-372.
13. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
14. Giannelli,G., Quaranta,V., and Antonaci,S. 2003. Tissue remodelling in liver diseases. Histol. Histopathol. 18:1267-1274.
15. Dufour,M.C., Stinson,F.S., and Caces,M.F. 1993. Trends in cirrhosis morbidity and
mortality: United States, 1979-1988. Semin. Liver Dis. 13:109-125.
16. Lotersztajn,S., Julien,B., Teixeira-Clerc,F., Grenard,P., and Mallat,A. 2005. Hepatic fibrosis: molecular mechanisms and drug targets. Annu. Rev. Pharmacol. Toxicol. 45:605-628.
17. Friedman,S.L. 1999. The virtuosity of hepatic stellate cells. Gastroenterology 117:1244-1246.
18. Diehl,A.M. 2000. Cytokine regulation of liver injury and repair. Immunol. Rev. 174:160-171.
19. Pinzani,M., Milani,S., Herbst,H., DeFranco,R., Grappone,C., Gentilini,A., Caligiuri,A., Pellegrini,G., Ngo,D.V., Romanelli,R.G., and Gentilini, P. 1996. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 148:785-800.
20. Tsukada,S., Parsons,C.J., and Rippe,R.A. 2006. Mechanisms of liver fibrosis. Clin. Chim. Acta 364:33-60.
21. Gong,W.R., Roth,S., Michel,K., and Gressner,A.M. 1998. Isoforms and splice variant of transforming growth factor β- binding protein in rat hepatic stellate cells. Gastro 114:352-363.
22. Marra,F., Choudhury,G.G., Pinzani,M., and Abboud,H.E. 1994. Regulation of platelet-derived growth factor secretion and gene expression in human liver fat-storing cells. Gastro 107:1110-1117.
23. Pinzani,M., Gentilini,A., Caligiuri,A., De Franco,R., Pellegrini,G., Milani,S., Marra,F., and Gentilini,P. 1995. Transforming growth factor-β1 regulates platelet-derived growth factor receptor β subunit in human liver fat-storing cells. Hepatol 21:232-239.
24. Friedman,S.L. 2000. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 275:2247-2250.
25. Gressner,A.M., Weiskirchen,R., Breitkopf,K., and Dooley,S. 2002. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 7:d793-d807.
26. Gressner,A.M., and Weiskirchen,R. 2006. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J. Cell Mol. Med. 10:76-99.
27. Tsukada,S., Parsons,C.J., and Rippe,R.A. 2006. Mechanisms of liver fibrosis. Clin. Chim. Acta 364:33-60.
28. Rockey,D.C. 2001. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin. Liver Dis. 21:337-349.

29
Chapter 2
29. Lundgren J, Mocroft A, Soriano V, Rochstroh J, Reiss P, Kirk O, de Wit S, Gatell JM, Clotet B, and Phillips A 2005. Is there evidence for an increase in the death rate from liver-related disease in patients with HIV? The EuroSIDA study. European AIDS Clinical Society 10th European AIDS Conference (Abstr.)
30. Weber R, Friis-Moller N, and Sabin C 2005. Liver-related deaths among HIV-infected persons. European AIDS Clinical Society 10th European AIDS Conference (Abstr.)
31. Yoon Y-H, Yi H-Y, and Hilton ME 2005. Liver cirrhosis mortality in the united states, 1970-2002. National Institute on Alcohol abuse and Alcoholism (NIAAA).
32. Perez-Tamayo,R. 1979. Cirrhosis of the liver: a reversible disease? Pathol. Annu. 14 Pt 2:183-213.
33. Garcia,L., Hernandez,I., Sandoval,A., Salazar,A., Garcia,J., Vera,J., Grijalva,G., Muriel,P., Margolin,S., and rmendariz-Borunda,J. 2002. Pirfenidone effectively reverses experimental liver fibrosis. J. Hepatol. 37:797-805.
34. Shimizu,I., Ma,Y.R., Mizobuchi,Y., Liu,F., Miura,T., Nakai,Y., Yasuda,M., Shiba,M., Horie,T., Amagaya,S., and Kawada,N., Hori,H., and Ito,S. 1999. Effects of Sho-saiko-to, a Japanese herbal medicine, on hepatic fibrosis in rats. Hepatology 29:149-160.
35. Siller-Lopez,F., Sandoval,A., Salgado,S., Salazar,A., Bueno,M., Garcia,J., Vera,J., Galvez,J., Hernandez,I., Ramos,M., Aguilar-Cordova,E., and Armendariz-Borunda,J. 2004. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology 126:1122-1133.
36. Kweon,Y.O., Goodman,Z.D., Dienstag,J.L., Schiff,E.R., Brown,N.A., Burchardt,E., Schoonhoven,R., Brenner,D.A., and Fried,M.W. 2001. Decreasing fibrogenesis: an immunohistochemical study of paired liver biopsies following lamivudine therapy for chronic hepatitis B. J. Hepatol. 35:749-755.
37. Poynard,T., McHutchison,J., Manns,M., Trepo,C., Lindsay,K., Goodman,Z., Ling,M.H., and Albrecht,J. 2002. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 122:1303-1313.
38. Friedman,S.L., and Bansal,M.B. 2006. Reversal of hepatic fibrosis -- fact or fantasy? Hepatology 43:S82-S88.
39. Issa,R., Zhou,X., Trim,N., Millward-Sadler,H., Krane,S., Benyon,C., and Iredale,J. 2003.
Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J. 17:47-49.
40. Iredale,J.P., Benyon,R.C., Pickering,J., McCullen,M., Northrop,M., Pawley,S., Hovell,C., and Arthur,M.J.P. 1998. `Mechanisms of spontaneous resolution of rat liver fibrosis - Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 102:538-549.
41. Murphy,F.R., Issa,R., Zhou,XRatnarajah,S., Nagase,H., Arthur,M.J., Benyon,C., and Iredale,J.P. 2002. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J. Biol. Chem. 277:11069-11076.
42. Iredale,J.P. 2001. Hepatic stellate cell behavior during resolution of liver injury. Semin. Liver Dis. 21:427-436.
43. Issa,R., Zhou,X., Constandinou,C.M., Fallowfield,J., Millward-Sadler,H., Gaca,M.D., Sands,E., Suliman,I., Trim,N., Knorr,A., Arthur, M.J., Benyon, R.C., and Iredale,J.P. 2004. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 126:1795-1808.
44. Pinzani,M., Rombouts,K., and Colagrande,S. 2005. Fibrosis in chronic liver diseases: diagnosis and management. J. Hepatol. 42 Suppl:S22-S36.
45. Friedman,S.L. 2004. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat. Clin. Pract. Gastroenterol. Hepatol. 1:98-105.
46. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
47. Bonis,P.A., Friedman,S.L., and Kaplan,M.M. 2001. Is liver fibrosis reversible? N. Engl. J. Med. 344:452-454.
48. Albanis,E., Safadi,R., and Friedman,S.L. 2003. Treatment of hepatic fibrosis: almost there. Curr. Gastroenterol. Rep. 5:48-56.
49. Ramalho,L.N., Ramalho,F.S., Zucoloto,S., Castro-e-Silva Junior, Correa,F.M., Elias,J.J., and Magalhaes,J.F. 2002. Effect of losartan, an angiotensin II antagonist, on secondary biliary cirrhosis. Hepatogastroenterology 49:1499-1502.
Chapter 2

30
A potential strategy to treat liver fibrosis
50. Hernandez,E., Correa,A., Bucio,L., Souza,V., Kershenobich,D., and Gutierrez-Ruiz,M.C. 2002. Pentoxifylline diminished acetaldehyde-induced collagen production in hepatic stellate cells by decreasing interleukin-6 expression. Pharmacol. Res. 46:435-443.
51. Buchdunger,E., Cioffi,C.L., Law,N., Stover,D., Ohno-Jones,S., Druker,B.J., and Lydon,N.B. 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 295:139-145.
52. Kweon,Y.-O., Paik,Y.-H., Schnabl,B., Qian,T., Lemasters,J.J., and Brenner,D.A. 2003. Gliotoxin-mediated apoptosis of activated human hepatic stellate cells. J. Hepatol. 39:38-46.
53. Suzuki,F., Hashimoto,K., Kikuchi,H., Nishikawa,H., Matsumoto,H., Shimada,J., Kawase,M., Sunaga,K., Tsuda,T., Satoh,K., and Sakagami, H. 2005. Induction of tumor-specific cytotoxicity and apoptosis by doxorubicin. Anticancer Res. 25:887-893.
54. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K.F., Molema,G., Poelstra,K., and Kok, R.J. 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
55. Rachmawati,H., Beljaars,L., Reker-Smit,C., van Loenen-Weemaes,A.M., Hagens,W.I., Meijer,D.K.F., and Poelstra,K. 2004. Pharmacokinetic and biodistribution profile of recombinant human interleukin-10 following intravenous administration in rats with extensive liver fibrosis. Pharm. Res. 21:2072-2078.
56. Gonzalo,T., Bataller,R., Sancho-Bru,P., Fontdevilla,C., Swart,J., Beljaars,L., Arroyo,V., Poelstra,K., Gines,P., and Kok,R.J. 2005. Short-term treatment with losartan targeted to activated stellate cells reduces advanced liver fibrogenesis: A new strategy to treat liver fibrosis. Hepatology 42:604A.
57. Gonzalo,T., Poelstra,K., Beljaars,L., Keri,G., Orfi,L., and Kok,R.J. 2005. Specific targeting of a PDGFR tyrosine kinase inhibitor to activated hepatic stellate cells as a promising technology to reduce liver fibrosis. Hepatology 42:733A.
58. Greupink,R., Bakker,H.I., Reker-Smit,C., van Loenen-Weemaes,A.M., Kok,R.J., Meijer,D.K.F., Beljaars,L., and Poelstra,K. 2005. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. 43:884-892.
59. Greupink,R., Bakker,H.I., Bouma,W., Reker-Smit,C., Meijer,D.K., Beljaars,L., and Poelstra,K. 2006. The antiproliferative drug doxorubicin inhibits liver fibrosis in bile duct-ligated rats and can be selectively delivered to hepatic stellate cells in vivo. J. Pharmacol. Exp. Ther.
60. Hagens,W.I., Olinga,P., Meijer,D.K., Groothuis,G.M., Beljaars,L., and Poelstra,K. 2006. Gliotoxin non-selectively induces apoptosis in fibrotic and normal livers. Liver Int. 26:232-239.
61. Prakash,J., Sandovici,M., Saluja,V., Lacombe,M., Schaapveld,R.Q., de Borst,M., vanGoor,H., Henning,R.H., Proost,J.H., Moolenaar,F., Keri,G., Meijer,D.K.F., Poelstra,K., and Kok, R.J. 2006. Intracellular Delivery of the P38 MAPK Inhibitor SB202190 in Renal Tubular Cells: a Novel Strategy to Treat Renal Fibrosis. J. Pharmacol. Exp. Ther.
62. Ward,A., and Clissold,S.P. 1987. Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 34:50-97.
63. Olaso,E., and Friedman,S.L. 1998. Molecular regulation of hepatic fibrogenesis. J. Hepatol. 29:836-847.
64. Windmeier,C., and Gressner,A.M. 1997. Pharmacological aspects of pentoxifylline with emphasis on its inhibitory actions on hepatic fibrogenesis. Gen. Pharmacol. 29:181-196.
65. Pinzani,M., Marra,F., Caligiuri,A., DeFranco,R., Gentilini,A., Failli,P., and Gentilini,P. 1996. Inhibition by pentoxifylline of extracellular signal-regulated kinase activation by platelet-derived growth factor in hepatic stellate cells. Br. J. Pharmacol. 119:1117-1124.
66. Windmeier,C., and Gressner,A.M. 1996. Effect of pentoxifylline on the fibrogenic functions of cultured rat liver fat-storing cells and myofibroblasts. Biochem. Pharmacol. 51:577-584.
67. Desmoulière,A., Xu,G., Costa,A.M.A., Yousef,I.M., Gabbiani,G., and Tuchweber,B. 1999. Effect of pentoxifylline on early proliferation and phenotypic modulation of fibrogenic cells in two rat models of liver fibrosis and on cultured hepatic stellate cells. J. Hepatol. 30:621-631.
68. Rodriguez-Barbero,A., Obreo,J., Yuste,L., Montero,J.C., Rodriguez-Pena,A., Pandiella,A., Bernabeu,C., and Lopez-Novoa,J.M. 2002. Transforming growth factor-beta1 induces collagen synthesis and accumulation via p38 mitogen-activated protein kinase (MAPK) pathway in cultured L(6)E(9) myoblasts. FEBS Lett. 513:282-288.

31
Chapter 2
69. Duncan,M.R., Hasan,A., and Berman,B. 1995. Pentoxifylline, pentifylline, and interferons decrease type I and III procollagen mRNA levels in dermal fibroblasts: evidence for mediation by nuclear factor 1 down-regulation. J. Invest Dermatol. 104:282-286.
70. Chen,Y.M., Wu,K.D., Tsai,T.J., and Hsieh,B.S. 1999. Pentoxifylline inhibits PDGF-induced proliferation of and TGF-beta-stimulated collagen synthesis by vascular smooth muscle cells. J. Mol. Cell Cardiol. 31:773-783.
71. Saati,N., Ravid,A., Liberman,U.A., and Koren,R. 1997. 1,25-dihydroxyvitamin D3 and agents that increase intracellular adenosine 3',5'-monophosphate synergistically inhibit fibroblast proliferation. In Vitro Cell Dev. Biol. Anim 33:310-314.
72. Chen,Y.M., Tu,C.J., Hung,K.Y., Wu,K.D., Tsai,T.J., and Hsieh,B.S. 2003. Inhibition by pentoxifylline of TNF-alpha-stimulated fractalkine production in vascular smooth muscle cells: evidence for mediation by NF-kappa B down-regulation. Br. J. Pharmacol. 138:950-958.
73. Duncan,M.R., Hasan,A., and Berman,B. 1995. Pentoxifylline, pentifylline, and interferons decrease type I and III procollagen mRNA levels in dermal fibroblasts: evidence for mediation by nuclear factor 1 down-regulation. J. Invest Dermatol. 104:282-286.
74. Berman,B., and Duncan,M.R. 1989. Pentoxifylline inhibits normal human dermal fibroblast in vitro proliferation, collagen, glycosaminoglycan, and fibronectin production, and increases collagenase activity. J. Invest Dermatol. 92:605-610.
75. Chang,C.C., Chang,T.C., Kao,S.C., Kuo,Y.F., and Chien,L.F. 1993. Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves' ophthalmopathy and pretibial myxoedema. Acta Endocrinol. (Copenh) 129:322-327.
76. Lee,K.S., Cottam,H.B., Houglum,K., Wasson,D.B., Carson,D., and Chojkier,M. 1997. Pentoxifylline blocks hepatic stellate cell activation independently of phosphodiesterase inhibitory activity. Am. J. Physiol 273:G1094-G1100.
77. Gude,R.P., Binda,M.M., Boquete,A.L., and Bonfil,R.D. 2001. Inhibition of endothelial cell proliferation and tumor-induced angiogenesis by pentoxifylline. J. Cancer Res. Clin. Oncol. 127:625-630.
78. Krakauer,T. 2000. Pentoxifylline inhibits ICAM-1 expression and chemokine production induced by proinflammatory cytokines in human
pulmonary epithelial cells. Immunopharmacology 46:253-261.
79. Beljaars,L., Meijer,D.K.F., and Poelstra,K. 2002. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front. Biosci. 7:E214-E223.
80. Goa,K.L., and Wagstaff,A.J. 1996. Losartan potassium: a review of its pharmacology, clinical efficacy and tolerability in the management of hypertension. Drugs 51:820-845.
81. Hall,W.D. 1999. Risk reduction associated with lowering systolic blood pressure: review of clinical trial data. Am. Heart J. 138:225-230.
82. Stroth,U., and Unger,T. 1999. The renin-angiotensin system and its receptors. J. Cardiovasc. Pharmacol. 33 Suppl 1:S21-S28.
83. Gardon,M.L., Gelband,C.H., Katovich,M.J., and Raizada,M.K. 1999. The potential use of gene therapy in the control of hypertension. Drugs Today (Barc. ) 35:925-930.
84. Wei,H.S., Lu,H.M., Li,D.G., Zhan,Y.T., Wang,Z.R., Huang,X., Cheng,J.L., and Xu,Q.F. 2000. The regulatory role of AT 1 receptor on activated HSCs in hepatic fibrogenesis:effects of RAS inhibitors on hepatic fibrosis induced by CCl(4). World J. Gastroenterol. 6:824-828.
85. Bataller,R., Sancho-Bru,P., Gines,P., Lora,J.M., Al Garawi,A., Sole,M., Colmenero,J., Nicolas,J.M., Jimenez,W., Weich,N., Gutierrez-Ramos, J.C., Arroyo,V., Rodes,J. 2003. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125:117-125.
86. Croquet,V., Moal,F., Veal,N., Wang,J., Oberti,F., Roux,J., Vuillemin,E., Gallois,Y., Douay,O., Chappard,D., and Cales,P. 2002. Hemodynamic and antifibrotic effects of losartan in rats with liver fibrosis and/or portal hypertension. J. Hepatol. 37:773-780.
87. Paizis,G., Gilbert,R.E., Cooper,M.E., Murthi,P., Schembri,J.M., Wu,L.L., Rumble,J.R., Kelly,D.J., Tikellis,C., Cox,A., Smallwood,R.A., and Angus,P.W. 2001. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J. Hepatol. 35:376-385.
88. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Nakatani,T., Tsujinoue,H., and Fukui,H. 2001. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 34:745-750.
89. Ramalho,L.N., Ramalho,F.S., Zucoloto,S., Castro-e-Silva Junior, Correa,F.M.,
Chapter 2

32
A potential strategy to treat liver fibrosis
Elias,J.J., and Magalhaes,J.F. 2002. Effect of losartan, an angiotensin II antagonist, on secondary biliary cirrhosis. Hepatogastroenterology 49:1499-1502.
90. Rimola,A., Londono,M.C., Guevara,G., Bruguera,M., Navasa,M., Forns,X., Garcia-Retortillo,M., Garcia-Valdecasas,J.C., and Rodes,J. 2004. Beneficial effect of angiotensin-blocking agents on graft fibrosis in hepatitis C recurrence after liver transplantation. Transplantation 78:686-691.
91. Sookoian,S., Fernandez,M.A., and Castano,G. 2005. Effects of six months losartan administration on liver fibrosis in chronic hepatitis C patients: a pilot study. World J. Gastroenterol. 11:7560-7563.
92. Yokohama,S., Yoneda,M., Haneda,M., Okamoto,S., Okada,M., Aso,K., Hasegawa,T., Tokusashi,Y., Miyokawa,N., and Nakamura,K. 2004. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 40:1222-1225.
93. Gonzalez-Abraldes,J., Albillos,A., Banares,R., Del Arbol,L.R., Moitinho,E., Rodriguez,C., Gonzalez,M., Escorsell,A., Garcia-Pagan,J.C., and Bosch,J. 2001. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 121:382-388.
94. 2001. Drug Targeting. Organ-Specific Strategies. WILEY-VCH Verlag GmbH. Weinheim.
95. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
96. Hart,C.E., Forstrom,J.W., Kelly,J.D., Seifert,R.A., Smith,R.A., Ross,R., Murray,M.J., and Bowen-Pope,D.F. 1988. Two classes of PDGF receptor recognize different isoforms of PDGF. Science 240:1529-1531.
97. Bergsten,E., Uutela,M., Li,X., Pietras,K., Ostman,A., Heldin,C.H., Alitalo,K., and Eriksson,U. 2001. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 3:512-516.
98. Heldin,C.H., Ostman,A., and Ronnstrand,L. 1998. Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta 1378:F79-113.
99. Wong,L., Yamasaki,G., Johnson,R.J., and Friedman,S.L. 1994. Induction of β-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Invest.1563-1569.
100. De Bleser,P.J., Jannes,P., Van Buul-Offers,S.C., Hoogerbrugge,C.M., Van
Schravendijk,C.F.H., Niki,T., Rogiers,V., Van den Brande,J.L., Wisse,E., and Geerts,A. 1995. Insulinlike growth factor-II/mannose 6-phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transforming growth factor-β in cocultures with sinusoidal endothelial cells. Hepatol 21:1429-1437.
101. Weiner,J.A., Chen,A., and Davis,B.H. 2000. Platelet-derived growth factor is a principal inductive factormodulating mannose 6-phosphate/insulin-like growth factor-II receptorgene expression via a distal E-box in activated hepatic stellate cells. Biochem. J. 345 Pt 2:225-231.
102. Bachem,M.G., Meyer,D., Schäfer,W., Riess,U., Melchior,R., Sell,K.M., and Gressner,A.M. 1993. The response of rat liver perisinusoidal lipocytes to polypeptide growth regulator changes with their transdifferentiation into myofibroblast-like cells in culture. J. Hepatol. 18:40-52.
103. Pinzani,M. 2002. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 7:d1720-d1726.
104. Breitkopf,K., Roeyen,C., Sawitza,I., Wickert,L., Floege,J., and Gressner,A.M. 2005. Expression patterns of PDGF-A, -B, -C and -D and the PDGF-receptors alpha and beta in activated rat hepatic stellate cells (HSC). Cytokine 31:349-357.
105. Pinzani,M. 2002. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 7:d1720-d1726.
106. Breitkopf,K., Lahme,B., Tag,C.G., and Gressner,A.M. 2001. Expression and matrix deposition of latent transforming growth factor beta binding proteins in normal and fibrotic rat liver and transdifferentiating hepatic stellate cells in culture. Hepatology 33:387-396.
107. von Mehren, M. 2005. Targeted therapy with imatinib: hits and misses? J. Clin. Oncol. 23:8-10.
108. Borkham-Kamphorst,E., Herrmann,J., Stoll,D., Treptau,J., Gressner,A.M., and Weiskirchen,R. 2004. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell activation and attenuates liver fibrosis. Lab Invest 84:766-777.
109. Yokoyama,M. 2005. Drug targeting with nano-sized carrier systems. J. Artif. Organs 8:77-84.
110. Batist,G., Harris,L., Azarnia,N., Lee,L.W., and za-Ramirez,P. 2006. Improved anti-tumor response rate with decreased cardiotoxicity of non-pegylated liposomal doxorubicin compared with conventional doxorubicin in first-line treatment of metastatic breast cancer in patients who had received

33
Chapter 2
prior adjuvant doxorubicin: results of a retrospective analysis. Anticancer Drugs 17:587-595.
111. Litzinger,D.C., Buiting,A.M., van,R.N., and Huang,L. 1994. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim. Biophys. Acta 1190:99-107.
112. Bal,W., Christodoulou,J., Sadler,P.J., and Tucker,A. 1998. Multi-metal binding site of serum albumin. J. Inorg. Biochem. 70:33-39.
113. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatol 29:1486-1493.
114. Beljaars,L., Poelstra,K., Weert,B., Bonnema,H., Molema,G., and Meijer,D.K.F. 1998. Mannose 6-phosphate modified albumin accumulates in hepatic stellate cells: potential application as an anti-fibrotic drug carrier. Hepatol 28:233A (Abstr.)
115. Kok,R.J., Ásgeirsdóttir,S.A., and Verweij,W.R. 2001. Development of proteinaceous drug targeting constructs using chemical and recombinant DNA approaches. In Drug Targeting. Organ-Specific Strategies. G.Molema, and Meijer,D.K.F., editors. WILEY-VCH Verlag GmbH. Weinheim. 275-308.
116. Mueller,E.J., Oh,S., Kavalerchik,E., Kappock,T.J., Meyer,E., Li,C., Ealick,S.E., and Stubbe,J. 1999. Investigation of the ATP binding site of Escherichia coli aminoimidazole ribonucleotide synthetase using affinity labeling and site-directed mutagenesis. Biochemistry 38:9831-9839.
117. Rodrigues,P.C., Beyer,U., Schumacher,P., Roth,T., Fiebig,H.H., Unger,C., Messori,L., Orioli,P., Paper,D.H., Mulhaupt,R., and Kratz, F. 1999. Acid-sensitive polyethylene glycol conjugates of doxorubicin: preparation, in vitro efficacy and intracellular distribution. Bioorg. Med. Chem. 7:2517-2524.
118. Di,S.G., Lanza,M., Kratz,F., Merina,L., and Fiume,L. 2004. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur. J. Pharm. Sci. 23:393-397.
119. Kratz,F., Beyer,U., Roth,T., Schutte,M.T., Unold,A., Fiebig,H.H., and Unger,C. 1998. Albumin conjugates of the anticancer drug chlorambucil:
synthesis, characterization, and in vitro efficacy. Arch. Pharm. (Weinheim) 331:47-53.
120. Saito,G., Swanson,J.A., and Lee,K.D. 2003. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 55:199-215.
121. Hagens,W.I., Olinga,P., Meijer,D.K., Groothuis,G.M., Beljaars,L., and Poelstra,K. 2006. Gliotoxin non-selectively induces apoptosis in fibrotic and normal livers. Liver Int. 26:232-239.
122. Arunachalam,B., Phan,U.T., Geuze,H.J., and Cresswell,P. 2000. Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc. Natl. Acad. Sci. U. S. A 97:745-750.
123. Heetebrij,R.J., Talman,E.G., Velzen,M.A., van Gijlswijk,R.P., Snoeijers,S.S., Schalk,M., Wiegant,J., Rijke,F., Kerkhoven,R.M., Raap,A.K., Tanke,H.J., Reedijk,J., and Houthoff, H.J. 2003. Platinum(II)-based coordination compounds as nucleic acid labeling reagents: synthesis, reactivity, and applications in hybridization assays. Chembiochem. 4:573-583.
124. van Gijlswijk,R.P., Talman,E.G., Janssen,P.J., Snoeijers,S.S., Killian,J., Tanke,H.J., and Heetebrij,R.J. 2001. Universal Linkage System: versatile nucleic acid labeling technique. Expert. Rev. Mol. Diagn. 1:81-91.
125. Reedijk,J. 2003. New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc. Natl. Acad. Sci. U. S. A 100:3611-3616.
126. Esposito,B.P., Faljoni-Alario,A., de Menezes,J.F., de Brito,H.F., and Najjar,R. 1999. A circular dichroism and fluorescence quenching study of the interactions between rhodium(II) complexes and human serum albumin. J. Inorg. Biochem. 75:55-61.
127. Reedijk,J. 1999. Why does Cisplatin reach Guanine-n7 with competing s-donor ligands available in the cell? Chem. Rev. 99:2499-2510.
128. US 5,580,990; EP 0,539,466; US 5,714,327; US 5,985,566; US 6,133,038. Kreatech Biotechnology b.v.
129. van Gijlswijk,R.P., Talman,E.G., Peekel,I., Bloem, J., van Velzen,M., Heetebrij,R.J., and Tanke,H.J 2002. Use of horseradish peroxidase- and fluorescein-modified cisplatin derivatives for simultaneous labeling of nucleic acids and proteins. Clin. Chem.. 48:1352-9.
Chapter 2

Martini Tower in Groningen.

Chapter3Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology
Journal of Controlled Release 111: 193-203 (2006).
Teresa Gonzalo1, Eduard G. Talman2, Anke van de Ven1, Kai Temming 1,2,
Rick H.Greupink1, Leonie Beljaars1, Catharina Reker-Smit1 , Dirk K.F. Meijer1,
Grietje Molema3, Klaas Poelstra1 and Robbert J. Kok1
1 Department of Pharmacokinetics and Drug Delivery, Groningen University Institute for Drug Exploration, Groningen, The Netherlands.
2 Kreatech Biotechnology, Amsterdam, The Netherlands.
3 Department of Pathology and Laboratory Medicine, Medical Biology Section, Groningen University Institute for
Drug Exploration, The Netherlands.

36
Targeting Pentoxifylline to hepatic stellate cells
Abstract
Targeting of antifibrotic drugs to hepatic stellate cells (HSC) is a promising strategy to
block fibrotic processes leading to liver cirrhosis. For this purpose, we utilized the neo-
glycoprotein mannose-6-phosphate-albumin (M6PHSA) that accumulates efficiently in
HSC during liver fibrosis. Pentoxifylline (PTX), an antifibrotic compound that inhibits
HSC proliferation and activation in vitro, was conjugated to M6PHSA. We employed a new
type of platinum-based linker, which conjugates PTX via coordination chemistry rather
than via covalent linkage. When incubated in plasma or in the presence of thiol
compounds, free PTX was released from PTX-M6PHSA at a sustained slow rate.
PTX-M6PHSA displayed pharmacological activity in cultured HSC as evidenced by
changes in cell morphology and reduction of collagen I production. PTX-M6PHSA and
platinum coupled PTX did not induce platinum-related toxicity (Alamar Blue viability
assay) or apoptosis (caspase activation and TUNEL staining). In vivo distribution studies in
fibrotic rats demonstrated specific accumulation of the conjugate in nonparenchymal cells
in the fibrotic liver. In conclusion, we have developed PTX-M6PHSA employing a novel
type of platinum linker, which allows sustained delivery of the drug to HSC in the fibrotic
liver.

37
Chapter 3
Introduction
Liver cirrhosis is the end-stage of liver diseases such as viral hepatitis, alcohol abuse, non-
alcoholic steatohepatitis and other diseases in which acute liver damage leads to chronic
inflammation and fibrosis (1). Apart from liver transplantation, no appropriate treatment
is currently available for liver fibrosis. Unfortunately, donor organ shortage and high costs
remain a serious problem for transplantation and, when it occurs, hepatic failure after
transplantation is still burdened by a high mortality rate. Finding a proper
pharmacotherapeutic treatment for liver fibrosis is therefore considered a major challenge.
Selective targeting of antifibrotic drugs to hepatic stellate cells (HSC) has recently been
proposed as a potential new strategy to counteract liver fibrosis at the level of stellate cell
activation (2). HSC have been identified as the key fibrogenic cell type (3;4). During liver
fibrosis, different stimuli induce HSC proliferation, leading to large numbers of activated
HSC that regulate the increased production of extracellular matrix components such as
collagens (5).
We have developed mannose-6-phosphate modified albumin (M6PHSA), which binds
with high affinity to the insulin-like growth factor II/mannose-6-phosphate receptor on
activated HSC (2;6;7). Since the M6PHSA carrier is internalized via receptor-mediated-
endocytosis upon binding to HSC, potent anti-fibrotic compounds can be delivered
intracellularly within the diseased liver, and even within the desired cell-type (8).
Pentoxifylline (PTX) has been suggested for specific targeting to the fibrotic liver (9). Its
antifibrogenic and anti-inflammatory properties on activated HSC have been extensively
reported and demonstrated. PTX reduces the transdifferentiation of HSC to
myofibroblasts and inhibits HSC proliferation (10-12). PTX also reduces the fibrogenic
effect of TGF-β on HSC by interference with p38 MAPkinase and ERK1/2 pathways,
thereby decreasing hepatic procollagen type 1 mRNA expression (13). However, apart
from beneficial effects in HSC, profibrotic effects of PTX on Kupffer cells have been
reported (12), as well as many effects in other cell types (14;15).
Chapter 3

38
Targeting Pentoxifylline to hepatic stellate cells
Cell-selective targeting of PTX to HSC is therefore required to create favorable effects in
HSC, while avoiding the profibrotic effects in Kupffer cells and effects in other organs.
SCH3
M6PHSA carrier (2) Methionine binding
S
NH
NH
M6P M6PHSA carrier
(1)Thiourea binding site:
N
N
N
N
CH3O
OCH3
O
PtNH22HN
S M6PHSA carrier
Schematic structure of PTX-M6PHSA:
Figure 1. General struc ure of PTX-M6PHSA. PTX is coupled via the platinum linker ULS
to thiol groups of the protein. Two possible binding sites in the protein are proposed, i.e. the thiol group of
the M6P homing devices on the modified HSA (1), and the sulfur atom in methionine groups of the
protein (2).
t
In the present paper we describe the development of the novel HSC-directed conjugate
PTX-M6PHSA. To conjugate PTX to M6PHSA, we employed a novel type of platinum
linker chemistry which allows stable coupling of drug molecules to proteins. The new
drug-linking approach is based on the formation of a platinum-ligand coordination bond

39
Chapter 3
with an aromatic nitrogen in PTX, after which the linker is further reacted to electron
donor sites in the carrier protein (Figure 1).
Since the nature of the organoplatinum bond is quite different from the linkages in
classical drug targeting conjugates, we investigated drug-release characteristics and linker-
related toxicity of the conjugates. In addition, we report on cell-specific binding to HSC
and in vitro pharmacological effects of PTX-M6PHSA, as well as its homing to HSC
during liver fibrosis in the rat.
Chapter 3

40
Targeting Pentoxifylline to hepatic stellate cells
Materials and methods
Chemicals and biochemicals
Platinum (ethylenediamine) dichloride (Universal linker System, ULS) and fluorescein-
ULS (FLU-ULS) were prepared as previously described by Kreatech Biotechnology
(Amsterdam, The Netherlands) (16). PTX and cisplatin were obtained from Sigma.
Human Serum Albumin (HSA) was obtained from Sanquin (Amsterdam, The
Netherlands).
Synthesis of conjugates
- Preparation of M6PHSA
M6PHSA was prepared and characterized as described previously (6). Dimeric protein
products which often are present in starting material or formed during reactions were
removed by size-exclusion chromatography using a Hiload 16/60 Superdex 200 column
(Amersham Biosciences). The final monomeric product was extensively dialyzed against
water at 4°C, lyophilized and stored at -20°C until use. Typically, M6PHSA contains 28
M6P groups per albumin.
- Synthesis of PTX-ULS
ULS (82.5 mg, 0.25 mmol) was dissolved in 3 ml of dimethylformamide (DMF) and
converted into the more reactive Pt (en (NO3) Cl (ULS mononitrate) by adding small
aliquots of AgNO3 (38.5 mg, 0.22 mmol, dissolved in 1 ml of DMF) within a time period
of two hours. The precipitated AgCl was removed by centrifugation. PTX (43.2 mg, 0.155
mmol, dissolved in 1 ml of DMF) was added and the resulting mixture was stirred at 60°C
for 2 days. DMF was removed by rotary evaporation, after which the residue was
dissolved in 2 ml of water and again taken to dryness under reduced pressure. The crude
product was dissolved in water (2 ml) and stored overnight at -20°C to precipitate
unreacted ULS as yellow crystals, which were removed by centrifugation (10 minutes at

41
Chapter 3
14,000 rpm). The identity and purity of the final product was checked by HPLC (95%),
NMR and mass spectroscopy. PTX-ULS was stored at 4°C until further use.
Yield: 105 mg (0.166 mmol, 107% yield). Apart from PTX-ULS, the product contained
water and inorganic salts that did not interfere in the next reaction step.
PTX 1H NMR (D2O): δH 1.61 (m, 4H, CH2(CH2)2CH2), 2.24 (s, 3H, CH3CO), 2.64 (t, J =
6.41 Hz, 2H, COCH2), 3.53 (s, 3H, CONCH3), 3.95 (m, 5H, CH2N and CNCH3CH),
7.92 (s, 1H, CH) ppm.
PTX-ULS 1H NMR (D2O): δH 1.62 (m, 4H, CH2(CH2)2CH2), 2.25 (s, 3H, CH3CO), 2.58
(m, 6H, COCH2, H2N(CH2)2NH2), 3.99 (t, J = 6.56 Hz, 2H, CH2N), 4.02 (s, 3H,
CONCH3), 4.55 (s, 3H, CNCH3C), 5.59 (m, 4H, H2N(CH2)2NH2), 8.48 (s, 1H, CH)
ppm.
PTX-ULS 195Pt NMR (D2O): δPt -2471 ppm.
PTX-ULS ionspray MS: Observed product peaks corresponded to PTX-ULS
monochloride (MW = 568.1 Da) and to PTX-ULS in which chloride was replaced by a
hydroxyl anion (MW = 550.1 Da). The latter product is possibly formed during dilution or
ionization of the compound for mass analysis. Both PTX-ULS species will react similarly
in subsequent reaction steps.
- Synthesis of PTX-M6PHSA and other conjugates.
All PTX-ULS-protein and FLU-ULS-protein conjugates listed in Table 1 were prepared
according to the following general protocol, which employs a 10:1 molar ratio of drug-
ULS to protein. Typically, 10 mg of M6PHSA (143 nmol) was dissolved in 1 ml of 20 mM
tricine/NaNO3 buffer pH 8.3. PTX-ULS (1430 nmol, 28.6 µl from a 50 mM stock
solution in water) was added and the pH was checked and corrected if necessary. The
mixture was reacted overnight at 37°C, after which unreacted PTX-ULS was removed by
size-exclusion chromatography using disposable PD10 columns (Amersham Biosciences),
or by dialysis against PBS at 4°C. The final product was sterilized by 0.2 µm filtration and
stored at -20°C.
Chapter 3

42
Targeting Pentoxifylline to hepatic stellate cells
Characterization of drug-protein conjugates
To determine the amount of PTX or FLU conjugated via ULS to the proteins, products
were subjected to various analyses related to either carrier, drug or linker, respectively.
- Protein determination.
The protein content of the conjugates was analyzed by different standard protein assays.
Both Lowry and Bradford assays yielded protein recoveries over 100%, indicating
interference of ULS modification with the protein determination. The bicinchoninic acid
(BCA) assay (Pierce, Rockford, IL, USA) resulted in adequate recovery of freeze-dried
amounts of the products.
- PTX determination.
The amount of PTX coupled to the carrier was analyzed by HPLC after competitive
displacement of the drug by excess of potassium thiocyanate (KSCN), a Pt-coordinating
ligand. In brief, appropriate dilutions of the conjugates corresponding to approximately 10
µM PTX were incubated overnight at 80°C in the presence of 0.5 M KSCN in PBS. After
cooling to room temperature, the samples were stored at -20°C until HPLC analysis. Free,
non-conjugated PTX was determined by HPLC analysis of samples that had not been
incubated with KSCN.
We analyzed PTX and PTX-ULS by reverse-phase HPLC on a Waters system (Waters,
Milford, MA, USA) equipped with an UV detector operated at 274 nm and a thermostated
column oven operated at 40°C. Elutions were performed on a µBondapak Guard-pak C18
precolumn in combination with a 5 µm Hypersil BDS C8 column (250x4.6 mm,
Thermoquest Runcorn, UK). Mobile phase consisted of acetonitrile/water/trifluoracetic
acid (25/75/0.1) at 1 ml/min. PTX and PTX-ULS eluted at 6 min and 5 min after
injection, respectively.
- Characterization of FLU-ULS modified proteins
FLU-ULS coupling ratios were determined spectrophotometrically without destruction of
the conjugates. Appropriate dilutions of the conjugates in PBS were analyzed at 500 nm
and related to a calibration curve of fluorescein sodium.

43
Chapter 3
- Determination of platinum content in conjugates
To assess the number of coupled ULS molecules, appropriate dilutions of the conjugates
corresponding to approximately 1 µM of platinum were analyzed by flameless atomic
absorption spectroscopy (FAAS), as described previously (17).
Table 1. Characterization o conjugates synthesized with PTX-ULS and FLU-ULS. f
Conjugate
Synthesis ratio
drug-ULS : protein
Coupling ratio
drug : protein¶
Coupling ratio
ULS : protein§
PTX-M6PHSA 10:1 6.7 : 1 (±0.7) 6.9 : 1 (±2.7)
PTX-HSA 10:1 1.5 : 1 (±1.0) 3.5 : 1 (±2.1)
FLU-M6PHSA 10:1 6.5 : 1 (±2.0) 6.2: 1 (±2.9)
FLU-HSA 10:1 4.3 : 1 (±2.0) 5.4 : 1 (±0.4)
¶ PTX coupling ratio was determined by HPLC after competitive displacement of the drug
with KSCN at 80°C for 24h. FLU coupling ratio was determined by its absorbance at 500
nm.§ ULS coupling ratio was determined by FAAS.
PTX release studies
The stability of PTX-M6PHSA in biological media was investigated by incubating the
conjugate in pH 7.4 at 37°C up to 7 days in a 1:1 dilution of normal rat serum and PBS.
Released PTX and conjugate bound drug were separated by treating aliquots with
acetonitrile (2:1 v/v) after which the samples were centrifuged at high speed to precipitate
proteins. The clear supernatant was stored at -20°C until HPLC analysis. Calibration
Chapter 3

44
Targeting Pentoxifylline to hepatic stellate cells
curves were made by spiking biological media with free PTX and were treated similar to
PTX-M6PHSA samples.
Thiol-competitive displacement, rather than enzymatic release, most likely facilitates
intracellular release of PTX from the conjugates. We therefore performed drug release
studies by incubating PTX-M6PHSA for 24h at 37°C in PBS pH 7.4 containing 5 mM
glutathione (GSH) or 5 mM dithiothreitol (DTT). PTX release was compared to
incubations in PBS or HSC culture medium, and expressed as the percentage of total
bound PTX.
Isolation and culturing of HSC
HSC were isolated from livers of male Wistar rats (0.3 kg) according to standard
techniques (18). After digestion of the liver with collagenase P (Boehringer, Mannheim,
Germany), pronase (Merck, Darmstadt, Germany), and DNAse (Boehringer), HSC were
separated from other hepatic cells by density-gradient centrifugation and collected at the
top of an 11% Nycodenz-solution (Brunschwig Chemie, Amsterdam). Subsequently, HSC
were cultured in disposable plastic bottles (Nunc, USA), in DMEM (GIBCO) containing
10% fetal calf serum (Hyclone Laboratories Inc., Logan, UT, USA), 100 U/ml penicillin,
and 100 µg/ml streptomycin (Sigma). These culture conditions allow HSC to attach and
proliferate as confirmed by microscopical evaluation. Cells cultured for 2 days after
isolation exhibited the phenotype of quiescent HSC, a compact cell shape with many
cytoplasmic fat droplets, while cells cultured for 10 days after isolation displayed the more
elongated phenotype of activated HSC without the vitamin A containing fat droplets. All
experiments with conjugates were performed with the latter type of activated HSC.
Binding studies of PTX-M6PHSA to target cells
- M6P/IGFII receptor expression
We investigated the specific binding of PTX-M6PHSA to its target receptor in a cell line
that expresses M6P/IGFII receptor. First, immunohistochemical detection of the target
receptor was analyzed. For this purpose, NIH/3T3 cells (2500 cells/well seeded into 96
well-plates) were cultured for 24h at 37°C in DMEM medium (Cambrex, New Jersey)

45
Chapter 3
supplemented with 5% FCS. After washing and acetone-fixation of the cells, expression of
the M6P/IGFII receptor was visualized by immunohistochemical staining (Santa Cruz
Biotechnology,California).
- Radioactive binding studies
In addition, we studied the binding of 125I-radiolabeled PTX-M6PHSA to M6P/IGFII
receptor expressing NIH/3T3 fibroblasts. PTX-M6PHSA was radiolabeled to a specific
activity of 35x106 cpm/µg by a chloramine-T method. Prior to the experiment, the
radiolabeled PTX-M6PHSA was purified by gel filtration on a PD10 column (Amersham
Biosciences). After gel filtration, the radiolabeled PTX-M6PHSA contained less than 5%
free 125I, as determined by precipitation of the protein with 10% trichloroacetic acid.
Binding studies were performed by incubating NIH/3T3 cells (60,000 cells/well seeded in
12-well plates) with 100,000 cpm of radiolabeled PTX-M6PHSA for 4h at 37°C.
Competitive displacement was performed by adding 0.1mg/ml M6PHSA carrier. After
washing of the cells with ice-cold PBS containing 1% bovine serum albumin, cells were
lysed by adding 0.5 ml of 1 M NaOH for 30 min. 125I-radioactivity was counted in an LKB
multichannel counter (LKB, Bromma, Sweden).
- Immunohistochemical binding studies
In addition we performed studies in which binding of PTX-M6PHSA was detected by
immunohistochemical staining of the protein. For this purpose, NIH/3T3 cells were
incubated for 4h at 37°C in the presence of 1 mg/ml M6PHSA or PTX-M6PHSA. After
washing and acetone-fixation of the cells, the binding of the conjugates was detected
immunohistochemically using rabbit-anti-HSA antibody (Cappel ICN Biomedicals,
Zoetermeer, The Netherlands).
Binding/homing studies in BDL rats
Organ distribution studies were performed in BDL rats as described previously (2). All in
vivo experiments were approved by the Local Committee for Care and Use of Laboratory
Animals. Male Wistar rats weighing 250-350 g (Harlan, Zeist, The Netherlands) were
housed under a 12-hour dark/light cycle. Animals had free access to tap water and
Chapter 3

46
Targeting Pentoxifylline to hepatic stellate cells
standard lab chow. Liver fibrosis was induced by ligation of the bile duct under isoflurane
anesthesia (2% isoflurane in 2:1 O2/N2O, 1 L/min) (Abbot Laboratories Ltd.,
Queensborough, UK). Three weeks after BDL, the liver showed severe fibrotic lesions
and excessive matrix deposition, as assessed by immunostaining for collagen III (2). At
this time point, the rats were used for biodistribution studies to determine homing to the
fibrotic liver and subcellular localization within the liver. During biodistribution studies,
rats were kept under isoflurane anesthesia and body temperature was maintained at 37-
38°C.
- Biodistribution of radiolabeled proteins
Rats were i.v. injected via the dorsal penis vein with tracer doses (106 cpm/rat) of either 125I-PTX-M6PHSA or 125I-radiolabeled M6PHSA. At 10 minutes after administration,
blood samples were taken by heart puncture and the organs were excised, washed in
saline, and weighed after which 125I-radioactivity was counted. Urine was recovered from
the bladder and measured. The total radioactivity per organ was calculated and corrected
for blood-derived radioactivity using BDL correction factors.
- Immunohistochemical detection of conjugates
In order to analyze intrahepatic distribution of the conjugates, BDL rats received one i.v.
injection of non-radiolabeled PTX-M6PHSA (2mg/kg). Tissue specimens were processed
for immunohistochemical analysis according to standard procedures. Cryostat sections
(4µm) of the liver were fixed in acetone and stained for the presence of conjugates by anti-
HSA immunodetection. Colocalization of PTX-M6PHSA with HSC was investigated by
double staining of HSA and desmin. To amplify anti-HSA staining, GARPO (peroxidase-
conjugated goat anti- rabbit immunoglobulin) and RAGPO (peroxidase-conjugated rabbit
anti-goat immunoglobulin) secondary antibodies were used in the double staining
protocol. Anti-desmin (Cappel ICN Biomedicals) was used in combination with RAM-AF
(alkaline phosphatase-conjugated rabbit anti-mouse) for detection of activated HSC.

47
Chapter 3
In vitro effec s of PTX-M6PHSA conjugates t
We tested the pharmacological efficacy of PTX-M6PHSA by incubating activated HSC
for 24h at 37°C with the conjugate or with control compounds relating to the experiments
described below.
- Cytotoxic effects
Cell viability was determined using the Alamar Blue viability assay occoridng to the
protocol of the supplier (Serotec Ltd). Activated HSC (10,000 cells/well seeded in 96 well-
plates, Corning Costar) were incubated for 24h in culture medium spiked with either
cisplatin (100 µM), PTX (100 µM), PTX-ULS (100 µM) or PTX-M6PHSA (1 mg/ml
based in protein concentration, 100uM in platinum concentration).
Induction of apoptosis was determined by caspase 3/7-activity measurement using the
apoptosis Caspase-Glo assay (Promega). Activated HSC (10,000 cells/well seeded in
white-walled 96 well-plates, Corning Costar) were incubated in culture medium spiked
during 24h with either cisplatin (100 µM), PTX (100 µM), PTX-ULS (100 µM) or PTX-
M6PHSA (1 mg/ml based in protein concentration, 100uM in platinum concentration).
Similar studies were performed with HSC (10,000 cells/well in Permanox LAB-TEK
chamber slides, Nalge Nunc Int.) which were subjected to TUNEL staining of fragmented
DNA as read-out parameter of apoptosis induction (19). Quantification per treatment was
performed after a minimum of 100 cells in at least 6 different microscopic fields per
section at magnification of 10x, under the fluorescence microscope. Analysis performed
by two independent observers.
- Antifibrotic effects
HSC (10.000 cells/well in Permanox LAB-TEK chamber slides) were incubated with
culture medium supplemented with either PTX-M6PHSA, PTX-HSA, or FLU-M6PHSA,
all at a concentration of 1 mg/ml. The applied concentration of PTX-M6PHSA
corresponded to 100 µM of conjugated PTX. In addition, HSC were incubated with 1 mM
free PTX. After 24h of incubation at 37°C, HSC were acetone-fixed and stained for the
presence of fibrosis markers according to standard methods using monoclonal antibody
Chapter 3

48
Targeting Pentoxifylline to hepatic stellate cells
against α-smooth muscle actin (Sigma) or polyclonal antibody against collagen type I
(Southern Biotechnology, USA). HSC morphology was analyzed via direct optical
microscopy observation.
Statist cs i
Statistical significance of differences was tested using the two-tailed Student’s t-test,
assuming normal distribution and similar variances of the data. The differences were
considered significant at p-values lower than 0.05. The data are presented as the mean of a
triplicate ± S.D.

49
Chapter 3
Results
Synthesis & characterization
In the present study, we investigated platinum-based coordination chemistry for the
coupling PTX to the M6PHSA carrier protein. This new type of linker binds to aromatic
nitrogens in drug molecules (Figure 1), forming a bond of intermediate binding strength.
To quantify PTX loading onto M6PHSA, the complexed drug was displaced by an excess
of competing platinum-ligand (KSCN). The drug/carrier ratios as shown in Table 1 were
corroborated by determination of platinum/carrier ratios using FAAS. Using a 10:1 initial
synthesis ratio, an average of about 6 to 7 molecules of PTX-ULS was coupled to
M6PHSA. Interestingly, coupling ratios of both PTX-ULS and FLU-ULS with M6PHSA
exceeded those of conjugates prepared with unmodified HSA. Similarly, PTX-ULS
showed also higher drug:carier loading ratio's with other neoglycoproteins like mannose-
HSA (data not shown). Since platinum(II) readily reacts with sulfur containing ligands, we
hypothesized that ULS reacted to the thiocarbonyl group between the M6P ligand and the
albumin core protein, as illustrated in Figure 1. Thiourea reacted rapidly with PTX-ULS at
the applied reaction conditions of the conjugates, as was demonstrated by HPLC analysis
and mass spectrometry (data not shown). However, reaction of ULS to other functional
groups in the protein like methionine or cysteine residues or even hydroxyl or amine side
chains can not be excluded.
Drug release profile of PTX-M6PHSA conjugate
To study the stability of PTX-M6PHSA under physiological conditions, we incubated
PTX-M6PHSA in serum (pH 7.4) at 37°C. About 40% of the conjugated PTX was still
bound to the carrier after 7 days of incubation (Figure 2A). This result indicates that the
linkage between drug and carrier displays adequate stability in plasma, although the drug is
released at a slow rate. PTX-HSA showed similar stability as PTX-M6PHSA (data not
shown).
Chapter 3
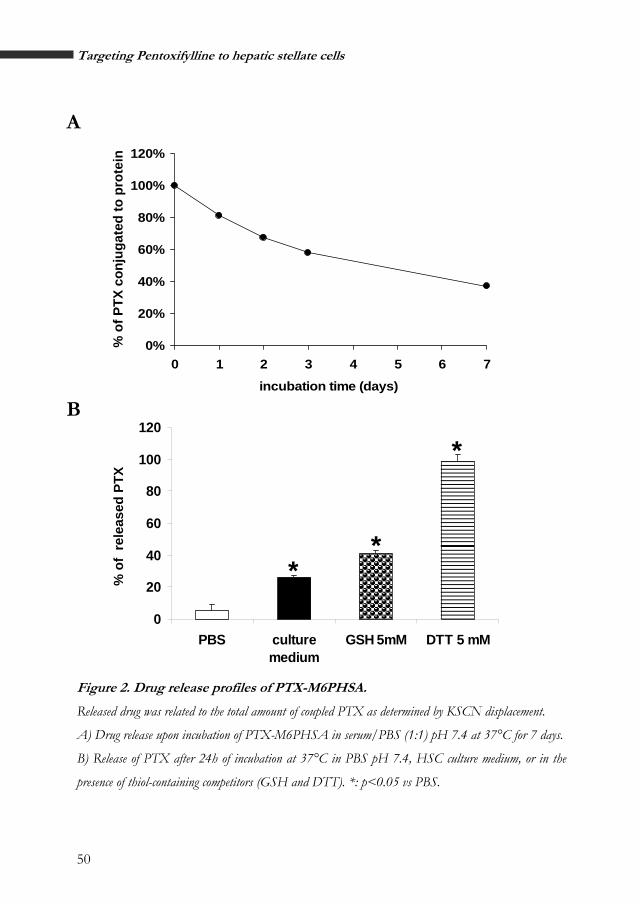
50
Targeting Pentoxifylline to hepatic stellate cells
A
Figure 2. Drug release profiles of PTX-M6PHSA.
Released drug was related to the total amount of coupled PTX as determined by KSCN displacement.
A) Drug release upon incubation of PTX-M6PHSA in serum/PBS (1:1) pH 7.4 at 37°C for 7 days.
B) Release of PTX after 24h of incubation at 37°C in PBS pH 7.4, HSC culture medium, or in the
presence of thiol-containing competitors (GSH and DTT). *: p<0.05 vs PBS.
0%
20%
40%
60%
80%
100%
120%
0 1 2 3 4 5 6 7
incubation time (days)
B
0
20
40
60
80
100
120
PBS culturemedium
GSH 5mM DTT 5 mM
% o
f re
leas
ed P
TX *
**
% o
f PTX
con
juga
ted
to p
rote
in

51
Chapter 3
Since the release of PTX from the platinum is most likely due to competitive displacement
by preferred platinum ligands, such as for instance thiols, we incubated the conjugate with
DTT or GSH. Incubation with the non-physiological molecule DTT resulted in complete
release of the drug from the conjugate (Figure 2B).
The concentration of 5 mM GSH reflects conditions within a cell’s interior. GSH clearly
accelerated the release of PTX from the carrier, as compared to incubation of the
conjugate in PBS or culture medium. This result indicates that free PTX can be generated
from the conjugate inside target cells.
We also incubated PTX-M6PHSA in different buffers at either cytosolic pH (PBS 7.4) or
lysosomal pH (0.1 M sodium acetate buffer, pH 5.0). No differences were observed
between pH5 and pH7 (data not shown). This result indicates that the drug release profile
will not be affected by lysosomal pH.
Binding of PTX-M6PHSA to target cells
Since our characterization reactions revealed that ULS reacted to the thiourea-part of the
M6P groups, binding properties of M6PHSA to M6P/IGFII receptor might be perturbed
after coupling of PTX-ULS.
A
M6P receptor staining Negative control
Figure 3. PTX-M6PHSA binding to target cells.
A) M6P/IGFII Receptor expressing NIH/3T3 fibroblasts were subjected to anti-M6P Receptor
staining (red color) and hematoxillin counterstained (blue color). Note the receptor presence next to cell
nuclei. Magnification 20x.
Chapter 3

52
Targeting Pentoxifylline to hepatic stellate cells
Cel
l-b
oun
d r
adio
acti
vity
(cp
m)
0
50
100
150
200
250
300
350
*
B
125I PTX-M6PHSA + 0,1mg/ml
M6PHSA125I-PTX-M6PHSA + 0.1mg/ml M6PHSA
125I PTX-M6PHSA 125I-PTX-M6PHSA
B) NIH/3T3 cells were incubated with a tracer dose of 125I-PTX-M6PHSA (100.000 cpm/well) in
the absence or presence of excess M6PHSA (0.1 mg/ml) were incubated for 4h at 37°C. *: p<0.05.
C) NIH/3T3 fibroblasts were incubated with PTX-HSA or PTX-M6PHSA at 1 mg/ml for 4h at
37°C, after which the cells were stained for bound products using anti-HSA antibody (red color) and
hematoxillin counterstained (blue color) Magnification 20x.
Negative control PTX-HSA
C
PTX-M6PHSA
After establishing the presence of our target receptor in a NIH/3T3 fibroblast cell line
(figure 3A), we investigated binding of PTX-M6PHSA to these target cells. The observed
binding of 125I-PTX-M6PHSA to the cells could be displaced by competition with excess
of M6PHSA (figure 3B). We therefore concluded that PTX-M6PHSA bound specifically
to the cells via to M6P/IGFII receptor. In addition, non-radiolabeled PTX-M6PHSA was

53
Chapter 3
incubated with 3T3 fibroblasts, after which we visualized binding of PTX-M6PHSA to the
cell surface by anti-HSA immunodetection (Figure 3C). While PTX-M6PHSA showed
intense positive staining, PTX-HSA did not bind to the cells. Thus, binding of PTX-
M6PHSA to target cells was mediated via the M6P homing ligands in the conjugates
Biodistribution of PTX-M6PHSA in rats with liver fibrosis
In addition to experiments with in vitro cultured target cells, we studied the biodistribution
of PTX-M6PHSA in rats with liver fibrosis. Previous studies in our group have
demonstrated that M6PHSA accumulates rapidly and efficiently in HSC within the fibrotic
livers of BDL rats (2). The initial distribution of M6PHSA could be assessed at ten
minutes after injection. In the current experiment, we observed a rapid distribution of 125I-
radiolabeled PTX-M6PHSA and 125I-radiolabeled M6PHSA to the fibrotic liver (Figure 4).
No accumulation in other organs was observed.
0%
10%
20%
%
%
%
%
%
80%
90%
BloodLive
r
SpleenLungs
KidneysHea
rt
Pancreas
thymus
Bladder
Stomach
Small Intestin
e
Large Intestin
e
Coecu
mpenis
Testis Urine
Total reco
very
PTX-M6PHSAM6PHSA
30
40
50
60
70
%of
inje
cted
Figure 4. Organ dis ribution of t 125I-PTX-M6PHSA and 125I-M6PHSA in BDL rats.
Tissue distribution of radiolabeled proteins was determined 10 min after intravenous injection of a tracer
dose (1,000,000 cpm) of the radiolabeled proteins. Total radioactivity in the organs was corrected for
blood-associated radioactivity and expressed as % of the injected dose. white bars: 125I-M6PHSA, and
black bars: 125I-PTX-M6PHSA.
Chapter 3

54
Targeting Pentoxifylline to hepatic stellate cells
Within the fibrotic liver, PTX-M6PHSA displayed an HSC-like distribution pattern, as
demonstrated by colocalization of carrier and desmin (Figure 5). From these results we
concluded that PTX-M6PHSA quickly distributed to the liver of BDL rats, where it
specifically binds to HSC.
A B
20µm 50µm
Figure 5. Colocalization of PTX-M6PHSA with HSC in fibrotic livers of BDL
rats.
Animals were intravenously injected with 2 mg/kg of PTX-M6PHSA and sacrificed 10 min after
administration. Picture A: Double staining for HSA (red) and the HSC marker desmin (blue). Arrows
indicate colocalization of HSA and HSC. Magnification 40x. Picture B: Enlargement of picture A
(100x).
trIn vitro pharmacology of PTX-M6PHSA and con ol compounds
The pharmacological effects of PTX-M6PHSA conjugates were evaluated on activated
HSC. Since cisplatin is a well-known antitumor agent, the use of platinum(II) as a linker
may infer platinum-related toxicity into the drug targeting construct. We therefore
investigated the effects of the conjugates on cell viability and apoptosis. Cell viability
studies did not show any toxicity to the target cells after 24h treatment with PTX, PTX-
ULS or PTX-M6PHSA, in contrast to cisplatin at the same concentration (Figure 6A).
Similarly, treatment of HSC with cisplatin for 24 h induced apoptosis, as reflected in
activation of caspases 3 and 7 (Figure 6B), and the occurrence of DNA strand breaks
assessed by TUNEL staining (Figure 6C). Again, PTX, PTX-ULS and PTX-M6PHSA did

55
Chapter 3
not induce direct activation of caspases or apoptosis (Figure 6B). Quantification of the
TUNEL positive nuclei versus DAPI stained nuclei of HSC showed a significant increase
after incubation with cisplatin, but no increase when cells were incubated with PTX-
M6PHSA (Figure 6D).
One of the hallmarks of liver fibrosis is the induced production of α-smooth muscle actin
(αSMA) and collagen type 1 by activated HSC. We therefore investigated the expression
of these proteins as read-out parameters for antifibrotic effects of PTX. As can be seen in
Figure 7, activated HSC express collagen type I in a granular staining pattern in the
cytoplasm, probably reflecting the presence of procollagen type I, while αSMA stained in
a fiber-like pattern. Treatment with PTX in a concentration of 1 mM reduced the intensity
of both fibrotic markers (Figure 7G,H). Incubation of the cells for 24h with 1 mg/ml
PTX-M6PHSA, which corresponds to 100 µM conjugated PTX, affected collagen type I
expression considerably (Figure 7E). Although the αSMA-staining intensity was not
affected by PTX-M6PHSA, pronounced changes in the morphology of HSC were
observed (figure 7F). HSC reacted to the treatment of PTX-M6PHSA with a loss of
cytoplasmic volume, rounded cell shape and detachment of the cells from the surface of
the plates. These effects were not observed after incubation with equivalent
concentrations of PTX-HSA (Figure 7C,D) or FLU-M6PHSA (data not shown),
indicating that the targeted drug induced these effects.
Chapter 3

56
Targeting Pentoxifylline to hepatic stellate cells
Discussion
In the present paper, we report on a novel drug targeting preparation for the delivery of
the antifibrotic drug PTX to HSC. Considering the important role of HSC in liver fibrosis,
intervening with stellate cell activation seems an attractive approach to counteract liver
fibrosis. We demonstrated accumulation of PTX-M6PHSA in HSC and observed
promising in vitro antifibrotic activity. Furthermore, the novel platinum-linker afforded
straightforward synthesis of conjugates that display slow-release of the coupled drug.
Over the past decades, drug delivery research has invested great efforts to find appropriate
linkage systems for the preparation of drug-carrier conjugates (20;21). Many drug
candidates however lack the appropriate functional groups for commonly applied
conjugation strategies. Although the presently investigated drug, PTX, also offers other
options for conjugation (e.g. hydrazone linkage at its keto functional group), we believe
that the ULS linker has several properties that make it a valuable technology for drug
delivery. First, ULS can be applied for conjugation of drug molecules that contain
aromatic nitrogens. Such groups are present in the chemical structure of many drug
molecules. The ULS linker can therefore facilitate conjugation of drug molecules that can
not be coupled via existing concepts. Second, synthesis of drug-ULS adducts is
straightforward and can be performed at high yields. This may enable synthesis of drug-
carrier conjugates even at low quantities of the drug. An additional advantage of ULS is
that sulfur atoms are reacted readily with the ULS linker (16;17). This provided a
convenient approach for coupling PTX-ULS to M6PHSA, since high drug:carrier ratios
were found in the final product. Lastly, the slow drug release profile is different from
existing drug-carrier conjugates. This may afford continuous drug release during a period
of days, which is not possible with existing linkages.

57
Chapter 3
A
0
20
40
60
80
100
120
control CIS-PT100µM
PTX 100µM PTX-ULS100µM
PTX-M6PHSA100µM
(1mg/ml)
*
Stel
late
Cel
l St
ella
te C
ell
viab
ilit
viab
ility
(
Ala
mar
Blu
e)
B
0
20
40
60
80
100
120
control CIS-PT 100µM PTX 100µM PTX-ULS100µM
PTX-M6PHSA100µM
(1mg/ml)
*
Cas
pas
e 3/
7 ac
tivi
ty
in %
of
RL
U
Figure 6. Effect of PTX-M6PHSA on apoptosis. HSC were incubated for 24h with cisplatin,
PTX, PTX-ULS (100 µM) or PTX-M6PHSA at a concentration of 1 mg/ml, which is equivalent to
100µM cisplatin.
Figure 6. Effect of PTX-M6PHSA on apoptosis. HSC were incubated for 24h with cisplatin,
PTX, PTX-ULS (100 µM) or PTX-M6PHSA at a concentration of 1 mg/ml, which is equivalent to
100µM cisplatin.
A) Alamar Blue viability assay. Fluorescence was corrected for background fluorescence of Alamar Blue
reagent in medium without cells *: p<0.05 vs control HSC.
A) Alamar Blue viability assay. Fluorescence was corrected for background fluorescence of Alamar Blue
reagent in medium without cells *: p<0.05 vs control HSC.
B) Caspase 3/7 activity assay using a luminescent caspase substrate. Emitted light units were normalized
relative to the signal of cisplatin treated HSC. RLU: relative light units, *: p<0.05 vs basal levels.
B) Caspase 3/7 activity assay using a luminescent caspase substrate. Emitted light units were normalized
relative to the signal of cisplatin treated HSC. RLU: relative light units, *: p<0.05 vs basal levels.
Chapter 3

58
Targeting Pentoxifylline to hepatic stellate cells
C) Activated HSC, cisplatin or PTX-M6PHSA induced apoptosis as assayed by TUNEL staining.
Activated Stellate cells (a,b) were incubated with 100 µM cisplatin (c,d) or PTX-M6PHSA at 100
µM (e,f). Nuclei were stained with DAPI (left column) to localize the nuclei of all cells, or for TUNEL
(right column) to visualize nuclei of apoptotic cells. Cisplatin treatment induced apoptosis in almost all
cells, while PTX-M6PHSA treatment is comparable with control HSC. Magnification 10x.
C DAPI TUNEL
Apoptotic nuclei All cell nuclei
a
Control b
d CIS-PT
100 µM c
e f PTX-M6PHSA
100 µM

59
Chapter 3
D
0
20
40
60
80
100
Control CIS-PT 100µM PTX-M6PHSA100µM (1mg/ml)
*%
TU
NE
L p
osit
ive
cells
/D
AP
I
D) Quantification of TUNEL positive cells per treatment *: p<0.05 vs control HSC.
Our in vitro stability/drug-release studies indicated that the release of PTX was driven by
chemical displacement by coordinating ligands for Pt(II). Within a biological matrix, such
ligands are for instance glutathione and ligands containing sulfur atoms (e.g. methionine)
or aromatic nitrogens (nucleotides, histidines). Since platinum reacts fast with thiols but
slower with aromatic nitrogens, we think that competitive displacement by glutathione is a
very likely mechanism for drug release. At present we can only hypothesize on the fate of
the conjugate after internalization. Drug release may occur in the lysosomes, the
compartment to which M6PHSA is routed after internalization (6). Another possibility is
that drug release will occur in multiple steps, in which the albumin is degraded by
lysosomal enzymes first, followed by competitive drug release by thiols in other
intracellular compartments. Although intracellular drug release will be most likely slow,
the rapid uptake of M6PHSA by target cells within the fibrotic liver (Figure 4) will favors
intracellular release of the conjugated PTX from PTX-M6PHSA.An important concern
raised in relation to the use of platinum-derivatives is their potential cisplatin-related
toxicity. Many different platinum compounds have been investigated for their cell-killing
properties in vitro as well as in vivo (22). One generally accepted mechanism of cisplatin
toxicity involves cross-linking of DNA, leading to cell death via apoptotic or necrotic
Chapter 3

60
Targeting Pentoxifylline to hepatic stellate cells
pathways (23). Hence, the toxicity of cisplatin and other platinum(II) compounds is
closely related to the availability of reactive ligand sites at the platina atom. Since in PTX-
M6PHSA the platinum-is fully coordinated, it can only react with endogenous compounds
after release of the drug or carrier. The platinum-ethylendiammine bridge in ULS is
considered very stable. As the release of drug (or carrier) is slow, only low concentrations
of reactive platinum are present in the target cells. Furthermore, cisplatin is readily
detoxified by binding to low-molecular weight thiols and thiol groups in proteins,
especially at lower concentrations. We expect that glutathione adducts will be the major
metabolites resulting from the linker, and we will investigate this more extensively in
future studies. Our present results demonstrate that both PTX-ULS and PTX-M6PHSA
do not produce toxic metabolites and we conclude that novel ULS linker is safe, despite
the cisplatin related structure.
PTX-M6PHSA reduced the production of collagen I notably, an effect that must be
attributed to the delivery of PTX into the HSC, since no effect was observed after
treatment with either PTX-HSA or FLU-M6PHSA. In addition, we observed striking
changes on the morphology of the cells, which were not observed after treatment with
free PTX. Although the observed changes in morphology suggest that the cells may
detach and die (anoikis), we did not observe this in our apoptosis or viability assays.
Moreover, the elimination of fibrogenic HSC by PTX-M6PHSA may be a beneficial effect
in the treatment of liver fibrosis, apart from antifibrotic effects by reducing collagen
deposition. We therefore concluded that PTX-M6PHSA is a promising candidate for
further evaluation as antifibrotic therapy.
Apart from the presently investigated conjugate, PTX-M6PHSA, ULS allows coupling of
many other antifibrotic drug candidates that contain aromatic nitrogen donor groups. In
this respect, we have investigated the delivery of the angiotensin II antagonist losartan,
which shows promising results in animal models of fibrosis and is tested clinically in
cirrhotic patients (24;25). Losartan was coupled to M6PHSA according to similar
protocols as described for PTX (Gonzalo, manuscript in preparation).

61
Chapter 3
α-SMA Collagen type I
D
G H
FE
BA
C
Figure 7. Effects of
PTX-M6PHSA on
fibrosis markers
collagen I and
α-SMA.
HSC were incubated for
24h with PTX-HSA,
PTX-M6PHSA (both at 1
mg/ml, equivalent to 100
µM PTX,) or 1 mM non-
targeted PTX. Cells were
stained for collagen type I or
α-SMA and counterstained
with hematoxillin.
A,B) Stellate cells incubated
with:
C,D) PTX-HSA;
E,F) PTX-M6PHSA;
G,H) non-targeted PTX.
Magnification 10x.
Preliminary results indicate that daily injections with losartan-M6PHSA attenuated BDL
liver fibrosis, while no toxicity was observed. Together with the presently discussed results
on PTX-M6PHSA, this indicates the potential of platinum-coordination chemistry as a
novel modality to prepare drug targeting preparations.
Chapter 3

62
Targeting Pentoxifylline to hepatic stellate cells
Acknowledgements
We kindly acknowledge Anne-Miek van Loenen, Jan Visser, Chantal Pottgens, Hans Pol
and Jan Willem Kok for assistance in different experimental procedures. Marie Lacombe,
Jan Reedijk, Elena Pantoja, and Anne Kalayda are thanked for valuable scientific
discussions relating to the experiments.
Financial support for this project has been granted by SenterNovem (grant TSGE1083)
and by the EU Marie Curie fellowship program (grant HPMI-CT-2002-00218).
Reference List
1. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
2. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatol 29:1486-1493.
3. Bataller,R., and Brenner,D.A. 2001. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin. Liver Dis. 21:437-451.
4. Friedman,S.L. 1999. The virtuosity of hepatic stellate cells. Gastroenterology 117:1244-1246.
5. Friedman,S.L. 2003. Liver fibrosis -- from bench to bedside. J. Hepatol. 38 Suppl 1:S38-S53.
6. Beljaars,L., Olinga,P., Molema,G., De Bleser,P., Geerts,A., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 2001. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver 21:320-328.
7. Beljaars,L., Molema,G., Schuppan,D., Geerts,A., De Bleser,P.J., Weert,B., Meijer,D.K.F.,
and Poelstra,K. 2000. Successful targeting of albumin to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor. J. Biol Chem. 275:12743-12751.
8. Greupink,R., Bakker,H.I., Reker-Smit,C., van Loenen-Weemaes,A., Kok,R.J., Meijer,D.K.F., Beljaars,L., and Poelstra,K. 2005. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. x:x.
9. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
10. Windmeier,C., and Gressner,A.M. 1996. Effect of pentoxifylline on the fibrogenic functions of cultured rat liver fat-storing cells and myofibroblasts. Biochem. Pharmacol. 51:577-584.
11. Desmoulière,A., Xu,G., Costa,A.M.A., Yousef,I.M., Gabbiani,G., and Tuchweber,B. 1999. Effect of pentoxifylline on early proliferation and phenotypic modulation of fibrogenic cells in two rat models of liver fibrosis and on cultured hepatic stellate cells. J. Hepatol. 30:621-631.
12. Raetsch,C., Jia,J.D., Boigk,G., Bauer,M., Hahn,E.G., Riecken,E.-O., and Schuppan,D. 2002. Pentoxifylline downregulates profibrogenic cytokines and procollagen I expression in rat secondary biliary fibrosis. Gut 50:241-247.

63
Chapter 3
13. Chen,Y.M., Wu,K.D., Tsai,T.J., and Hsieh,B.S. 1999. Pentoxifylline inhibits PDGF-induced proliferation of and TGF-beta-stimulated collagen synthesis by vascular smooth muscle cells. J. Mol. Cell Cardiol. 31:773-783.
14. Gude,R.P., Binda,M.M., Boquete,A.L., and Bonfil,R.D. 2001. Inhibition of endothelial cell proliferation and tumor-induced angiogenesis by pentoxifylline. J. Cancer Res. Clin. Oncol. 127:625-630.
15. Krakauer,T. 2000. Pentoxifylline inhibits ICAM-1 expression and chemokine production induced by proinflammatory cytokines in human pulmonary epithelial cells. Immunopharmacology 46:253-261.
16. van Gijlswijk,R.P., Talman,E.G., Peekel,I., Bloem,J., van Velzen,M.A., Heetebrij,R.J., and Tanke,H.J. 2002. Use of horseradish peroxidase- and fluorescein-modified cisplatin derivatives for simultaneous labeling of nucleic acids and proteins. Clin. Chem. 48:1352-1359.
17. Heetebrij,R.J., Talman,E.G., Velzen,M.A., van Gijlswijk,R.P., Snoeijers,S.S., Schalk,M., Wiegant,J., Rijke,F., Kerkhoven,R.M., Raap,A.K. et al 2003. Platinum(II)-based coordination compounds as nucleic acid labeling reagents: synthesis, reactivity, and applications in hybridization assays. Chembiochem. 4:573-583.
18. Geerts,A., Niki,T., Hellemans,K., De Craemer,D., Van den Berg,K., Lazou,J.-M., Stange,G., Van de Winkel,M., and De Bleser,P.J. 1998. Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatol 27:590-598.
19. Gavrieli,Y., Sherman,Y., and Ben Sasson,S.A. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119:493-501.
20. Soyez,H., Schacht,E., and Van Der Kerken,S. 1996. The crucial role of spacer groups in macromolecular prodrug design. Adv. Drug Deliv. Rev. 21:81-106.
21. Kok,R.J., Ásgeirsdóttir,S.A., and Verweij,W.R. 2001. Development of proteinaceous drug targeting constructs using chemical and recombinant DNA approaches. In Drug Targeting. Organ-Specific Strategies. G.Molema, and Meijer,D.K.F., editors. WILEY-VCH Verlag GmbH. Weinheim. 275-308.
22. Ho,Y.P., Au-Yeung,S.C., and To,K.K. 2003. Platinum-based anticancer agents: innovative design strategies and biological perspectives. Med. Res. Rev. 23:633-655.
23. Fuertes,M.A., Castilla,J., Alonso,C., and Perez,J.M. 2002. Novel concepts in the development f platinum antitumor drugs. Curr. Med. Chem. Anti. -Canc. Agents 2:539-551.
24. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
25. Castano,G., Viudez,P., Riccitelli,M., and Sookoian,S. 2003. A randomized study of losartan vs propranolol: Effects on hepatic and systemic hemodynamics in cirrhotic patients. Ann. Hepatol. 2:36-40.
Chapter 3

Sunset at the Golden Gate
Conference by the American Association of Study of the Liver (ASan Francisco, USA, 2005
ASLD),

Chapter4Reduction of advanced liver fi brosis by targeted delivery of the angiotensin II receptor antagonist losartan to hepatic stellate cells
Submitted for publication.
Teresa Gonzalo1, Ramón Bataller2, Pau Sancho-Bru2, Josine Swart1, Jai Prakash1, Constantino Fondevila2, Dirk K.F. Meijer1, Leonie Beljaars1, Marie Lacombe3, Paul vd Hoeven3,4, Vicente Arroyo2, Klaas Poelstra1, Pere Ginès2, Robbert J. Kok1.
1Department of Pharmacokinetics and Drug Delivery, University of Groningen, The Netherlands;
2Liver Unit, Hospital Clínic, IDIBAPS, Barcelona, Catalonia, Spain;
3Kreatech Biotechnology B.V., Amsterdam, The Netherlands; and
4Future Diagnostics BV, Wijchen, The Netherlands

66
Reduction of liver fibrosis by targeted losartan
Abstract
Liver fibrosis often progresses to cirrhosis. To date, no effective therapy
attenuates fibrosis progression in patients with chronic liver diseases. Renin-
angiotensin system inhibitors have been proposed, yet their use in patients with
advanced fibrosis may be hampered by side effects. To improve the balance
between antifibrotic and systemic effects of losartan, we have developed the liver
directed conjugate losartan-M6PHSA (M6PHSA: mannose-6-phosphate modified
human serum albumin), which accumulates in hepatic stellate cells (HSC), the
major fibrogenic cell type in liver fibrosis. Losartan was coupled to M6PHSA via
a platinum-based linker. Cell viability studies revealed that losartan-M6PHSA did
not affect the viability of the hepatic stellate cells. We evaluated the liver homing
and antifibrotic effects of losartan-M6PHSA in rats with liver fibrosis induced by
prolonged bile-duct ligation (BDL) and CCl4 inhalation model. Rats received a
total of four doses of losartan-M6PHSA, M6PHSA alone or oral losartan and
were sacrificed 10 min after the last dose to examine drug distribution. Losartan-
M6PHSA was shown to accumulate in activated HSC in fibrotic rat livers,
amounting 20% of the last administered dose. Losartan-M6PHSA, but not oral
losartan, markedly reduced hepatic collagen and TGFβ1 expression and accumulation of
myofibroblasts and inflammatory cells in the liver, as compared to saline-injected BDL
rats. We conclude that short-term treatment with losartan-M6PHSA targeted to HSC
reduces advanced liver fibrosis and may provide novel means to treat patients with
chronic liver fibrosis.

67
Chapter 4
Introduction
Hepatic fibrosis is a clinical problem for which no adequate therapy is available.
Advanced fibrosis may result in cirrhosis, which is a major cause of death in virtually all
countries(1). Recent experimental studies indicate that the renin-angiotensin system (RAS)
plays an important role in liver fibrogenesis (2). Hepatic stellate cells (HSC) are the main
fibrogenic cell type in the injured liver and are also the main target cell type for the
pathogenic effects of angiotensin II (Ang II) in liver fibrosis. In the normal human liver,
HSC do not express Ang II type 1 (AT1) receptors nor do they secrete Ang II. Following
chronic liver injury, however, HSC transform into myofibroblast-like cells and express
both AT1 receptors and generate mature Ang II, which exerts an array of pro-
inflammatory and profibrogenic actions (3;4). These pathogenic effects can be prevented
largely by AT1 receptor antagonists, as has been demonstrated in different models of
experimentally-induced liver fibrosis (5-8). Based on these data, RAS inhibitors like
Angiotensin Converting Enzyme inhibitors or Ang II receptor blockers are currently
considered as novel antifibrotic therapies to treat liver fibrosis.
AT1 receptor blockers are widely used in patients with arterial hypertension and type II
diabetes to lower blood pressure and prevent the development of cardiac and renal
fibrosis (9;10). Preliminary clinical data suggests that AT1 receptor blockers may also
attenuate liver fibrosis (11-13). The use of AT1 receptor blockers would be particularly
useful in conditions characterized by rapid fibrosis progression (i.e. acute alcoholic
hepatitis and severe hepatitis C virus reinfection after liver transplantation). However, in
these conditions, the use of angiotensin antagonists may be hampered by undesirable
effects on the arterial pressure, especially since patients with cirrhosis are generally
associated with low systemic blood pressure. Indeed, the use of the AT1 receptor blocker
losartan in patients with advanced fibrosis was associated with the development of arterial
hypotension and renal impairment (14). To overcome these limitations, we now propose
an innovative drug delivery approach. This will avoid general effects such as reduction of
blood pressure by lowering systemic drug levels and will increase the efficacy of losartan
Chapter 4

68
Reduction of liver fibrosis by targeted losartan
by achieving high concentrations in the liver. The drug delivery system applied in this
study is based on mannose 6-phosphate modified human serum albumin (M6PHSA), a
carrier that specifically delivers losartan to activated HSC (15),(16). M6PHSA binds to the
mannose-6-phosphate/insulin growth factor type II receptor (M6P/IGII-R), a surface
exposed receptor that is de novo expressed in activated HSC during liver fibrogenesis
(17). Previous studies in our group have demonstrated that drug-M6PHSA conjugates
accumulate rapidly and extensively in the fibrotic liver (18),(19).
To conjugate losartan to M6PHSA, we employed a novel type of platinum linker
chemistry called ULS (Universal Linker System) (20), which can binds losartan via a
coordinative bond at one of the aromatic nitrogen atoms in the tetrazole group of the
drug (figure 1). Application of this novel linker technology was essential for several
reasons. First, coordination chemistry proved straightforward and reliable for linking drug
molecules to the carrier, allowing high synthesis yields in a relative simple approach.
Second, the resulting drug-ULS-carrier conjugates display a unique behavior, in which
drug molecules are released slowly, during a period of days, within the designated target
cells(18). In the present paper, we now describe the pharmacokinetics of losartan-
M6PHSA and its accumulation in fibrotic livers. Furthermore, we report on the
antifibrotic effects of losartan-M6PHSA in rats subjected to bile duct ligation, a well-
characterized model of liver fibrogenesis (21). In addition, we have also evaluated
losartan-M6PHSA efficacy in the CCl4 model of liver fibrosis. Short-term treatment with
losartan-M6PHSA, but not free losartan given orally, reduced inflammation and collagen
deposition in the liver. These promising results suggest that this approach may also be
useful to treat patients with chronic liver diseases.

69
Chapter 4
Experimental protocols
Synthesis of losartan-M6PHSA
Losartan and human serum albumin (HSA) were obtained from Synfine (Ontario,
Canada) and Sanquin (Amsterdam, The Netherlands), respectively. Losartan was first
coupled to Universal Linker System (ULS, Kreatech b.v, The Netherlands), a platinum
complex that forms a co-ordinative bond with the target. Briefly,
platinum(ethylenediamine)dichloride (Pt(en)Cl2; 82.5 mg, 0.253 mmol, dissolved in 3 ml
of dimethylformamide (DMF) was converted into the more reactive Cis-
[Pt(ethylenediamine)nitrate-chloride] Pt(en(NO3)Cl by adding small aliquots of AgNO3
(38.5 mg, 0.227 mmol, dissolved in 1 ml of DMF). The precipitated silver chloride was
removed by centrifugation. The resulting cis-[Pt(ethylenediamine)nitrate-chloride]
solution (32 µmol) or ULS was added to a solution of losartan (32 µmol, 10 mg/ml of the
potassium salt of losartan in DMF). The resulting solution was heated at 60°C for 3 days
during which consumption of the starting material was monitored by analytical HPLC.
Mass spectrometry analysis confirmed the presence of the 1:1 losartan-cisULS species
after completion of the reaction.
Analysis of the product, losartan-ULS, using mass spectrometry, yielded the following
data:
1H NMR of Losartan-cisULS (CD3OD): δH 0.79 (m, 3H, CH3), 1.25 (m, 2H, CH2CH3),
1.48 (m, 2H, CH2CH2CH3), 2.50 (m, 6H, CH2CH2C and CH2NH2), 4.43 (m, 1.8H,
NCH2C), 5.18 (m, 2.2H, CH2OH and remaining NCH2C), 5.42 (m, 4H, NH2); cyclic Hs:
6.82 (m, 0.2 H), 6.87 (m, 1.8 H), 7.04 (t, J = 8.06 Hz, 1H, CHCHCH), 7.18 (m, 0.5H),
7.28 (m, 1H), 7.38 (m, 3H), 7.88 (m, 0.5 H).
195Pt NMR of Losartan-cisULS (CD3OD): Pt -2491 and -2658 ppm.
MS (ESI+) m/z: 695 [M-K+-OH--H]+, 677 [M-K+-Cl--H+]+, 659 [M-K+-Cl--H+-Cl-+OH-
]+.
Chapter 4

7
Reduction of liver fibrosis by targeted losartan
Figure 1. Characterization of losartan-M6PHSA.
(A) Synthesis of losartan-M6PHSA. Presence of Losartan-ULS was confirmed by HPLC analysis
and ion-spray mass spectrometry, which demonstrated the typical isotopic pattern of platinum compounds.
Conjugation of losartan-ULS to M6PHSA was confirmed by HPLC analysis after release of the drug
from the carrier. MonoQ anion exchange chromatography confirmed that the charge of M6PHSA
protein was not affected by losartan coupling. Size exclusion Chromatography showed the monomeric
composition of Losartan-M6PHSA at a retention time of 27 minutes. M6PHSA: mannose-6-
phosphate modified human serum albumin; ULS: Universal Linkage System.
A
N OHCl
N OHCl NH2H2N
Pt
0
NHNN
NN
NH2H2NPt
ClO3N
NHNN
NN Cl
Losartan ULS
Losartan-ULS
NHNN
NN
N OHCl NH2H2N
Pt
M6PHSAcarrier
NHNN
NN
N OHCl NH2H2N
Pt
M6PHSAcarrier
M6PHSAcarrier
Losartan-M6PHSA
M6PHSALosartan-ULS
B Ionspray MS of losartan-ULS
0
100
200
300
400
500
600
700
700 720 740 760
Sign
al (
x103
AU
)
Mass/charge ratio (m/z)
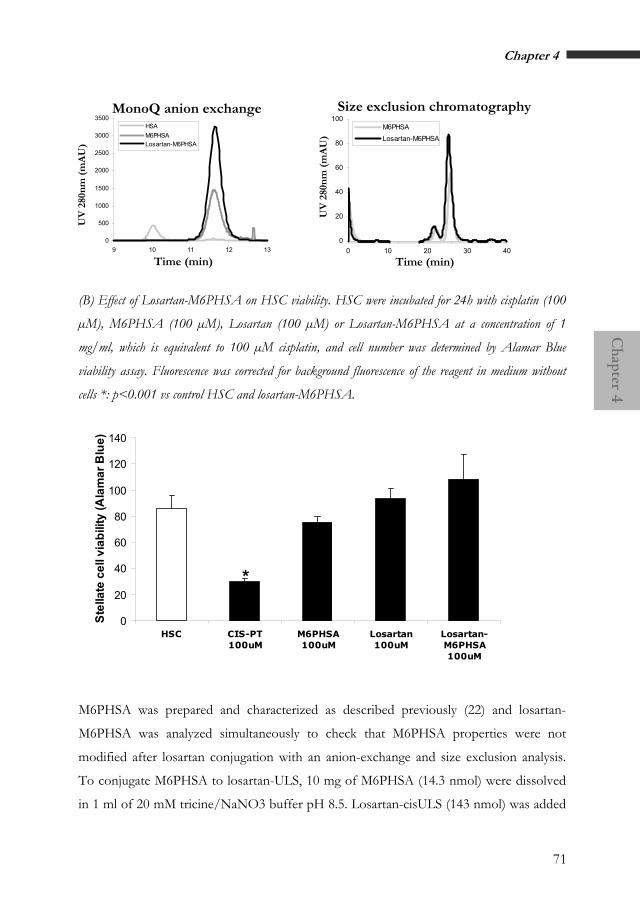
71
Chapter 4
Time (min)
0
20
40
60
80
100
0 10 20 30 40
M6PHSALosartan-M6PHSA
Size exclusion chromatography
0
500
1000
1500
2000
2500
3000
3500
9 10 11 12 13
HSAM6PHSA Losartan-M6PHSA
MonoQ anion exchange
UV
280
nm
(m
AU
)
UV
280
nm
(m
AU
)
Time (min)
(B) Effect of Losartan-M6PHSA on HSC viability. HSC were incubated for 24h with cisplatin (100
µM), M6PHSA (100 µM), Losartan (100 µM) or Losartan-M6PHSA at a concentration of 1
mg/ml, which is equivalent to 100 µM cisplatin, and cell number was determined by Alamar Blue
viability assay. Fluorescence was corrected for background fluorescence of the reagent in medium without
cells *: p<0.001 vs control HSC and losartan-M6PHSA.
0
20
40
60
80
100
120
140
HSC CIS-PT100uM
M6PHSA100uM
Losartan100uM
Losartan-M6PHSA100uM
Stel
late
cel
l via
bilit
y (A
lam
ar B
lue)
*
M6PHSA was prepared and characterized as described previously (22) and losartan-
M6PHSA was analyzed simultaneously to check that M6PHSA properties were not
modified after losartan conjugation with an anion-exchange and size exclusion analysis.
To conjugate M6PHSA to losartan-ULS, 10 mg of M6PHSA (14.3 nmol) were dissolved
in 1 ml of 20 mM tricine/NaNO3 buffer pH 8.5. Losartan-cisULS (143 nmol) was added
Chapter 4

72
Reduction of liver fibrosis by targeted losartan
in 10-fold molar excess at pH 8. The mixture was reacted overnight at 37°C, and purified
by dialysis against PBS at 4°C. The final product was sterilized by filtration and stored at -
20°C. Protein content of the conjugates was assessed by the BCA assay (Pierce, Rockford,
IL). ULS content per losartan-M6PHSA was evaluated by inductive coupled plasma –
atomic emission spectroscopy (ICP-AES) at 214.424nm and at 265.945nm with a VISTA
AX CCD Simultaneous ICP-AES (Varian, Palo Alto, USA). Standards (cisPlatinum) and
unknown samples were spiked with Yttrium as an internal standard (360.074nm). The
amount of losartan coupled to the carrier was analyzed by HPLC after competitive
displacement of the drug from the conjugate by excess of potassium thiocyanate (KSCN).
Free losartan and losartan-ULS were analyzed by reverse-phase HPLC on a Waters
system (Waters, Milford, MA, USA) equipped with an UV detector operated at 269 nm
and a thermostated column oven operated at 40°C. Elutions were performed on a
µBondapak Guard-pak C18 precolumn in combination with a 5 µm Hypersil BDS C8
column (250x4.6 mm, Thermoquest Runcorn, UK) using a mobile phase consisting of
acetonitrile/water/trifluoracetic acid (40/60/0.1, pH 2).
Cytotoxic effects of Losartan-M6PHSA on HSC
HSC were freshly isolated from rat liver and purified with 12% nycodenz gradient
centrifugation. After ten days of culture, HSC are fully activated as detected via αSMA
immunostaining (10,000 cells/well seeded in 96 well-plates, Corning Costar) and were
then incubated for 24h at 37°C in culture medium spiked with either cisplatin (100 µM),
Losartan (100 µM) or Losartan-M6PHSA (1 mg/ml based in protein concentration, 100
µM in platinum concentration). Cell viability was determined using the Alamar Blue
viability assay according to the protocol of the supplier (Serotec Ltd).
Induction of liver fibrosis in rats
BDL model
Common bile duct ligation was used to induce liver inflammation and fibrosis as
described previously (23). Either bile duct ligation or sham-operation was performed in
250 g Male Wistar rats (Harlan, Zeist, The Netherlands). Rats were anesthesized with

73
Chapter 4
isoflurane (2% isoflurane in 2:1 O2/N2O, 1 L/min) (Abbot Laboratories Ltd.,
Queensborough, UK). After midline laparotomy, the common bile duct was doubly
ligated with 4-0 silk and transsected between the two ligations. Sham operation was
performed similarly with exception of ligating and transecting the bile duct.
At days 12-15 after bile duct ligation, rats received intravenous injection of saline,
losartan-M6PHSA (3.3 mg/kg/day, corresponding to 125 µg losartan/kg), M6PHSA
alone (3.3 mg/kg/day), or orally administrated losartan (5 mg/kg/day). Ten rats were
included per group. Animals were sacrificed 10 minutes after the last injection and blood
and organ liver samples were obtained. Animal procedures were approved by the
Committee for Care and Use of Laboratory Animals of the Hospital Clínic, Barcelona.
CCl4 model
Rats (250 g, Male Wistar rats, Harlan) were subjected to CCl4 inhalation during a time
period of 8 weeks as described previously (24). At the beginning of week 9, rats received
four consecutive intravenous injections of saline or losartan-M6PHSA (8 mg/kg,
corresponding to 300 µg losartan/kg). 10 minutes after the fourth injection, animals were
sacrificed and blood and liver samples were obtained. Animal procedures were approved
by the Committee for Care and Use of Laboratory Animals of the Hospital Clínic,
Barcelona.
Serum b ochemical measurements i
i
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin
levels were measured using standard enzymatic procedures by the Hospital Clinic,
Barcelona.
Analysis of losartan-M6PHSA b odistribution
The presence of losartan-M6PHSA or M6PHSA in tissue cryosections was demonstrated
by immunostaining using an anti-HSA antibody (Cappel ICN Biomedicals, Zoetermeer,
The Netherlands), as described elsewhere (37). The co-localization of losartan-M6PHSA
with HSC was assessed by double immunostaining of anti-HSA and anti-desmin (Cappel
Chapter 4

74
Reduction of liver fibrosis by targeted losartan
ICN Biomedicals, Zoetermeer, The Netherlands), a specific marker for rat HSC, as
described previously (16) and the pictures were taking by optical microscope.
For colocalization studies of losartan-M6PHSA and HSC and posterior analysis in the
confocal laser scanning microscope, the mixture of the primary antibodies rabbit anti-
HSA and mouse anti-DESMIN were detected with a mixture of FITC anti-mouse IgG
(1:100) and TRITC anti-rabbit IgG. Images were taken with a confocal laser scanning
microscopy (Leica TCS SP2 AOBS confocal laserscan microscope, Heidelberg, Germany)
equipped with an argon/krypton laser and coupled to a Leitz DM IRB inverted
microscope (Leica). Single-label images were taken sequentially at 488 and 562 nm with
the 40x objetive. Colocalization pictures were obtained by use of Adobe Photoshop.
The amount of losartan in liver tissue homogenates was analyzed by HPLC after a
liquid/liquid extraction with (HPLC)-UV method. Two different procedures were
employed to isolate losartan from tissue homogenates. The first method consisted of
direct extraction from the livers, whereas the second method comprised an additional
incubation of tissues overnight with KSCN 80ºC, in order to release losartan from the
conjugate(18), and subsequent extraction the following day. Liquid–liquid extraction of
losartan from the samples was performed using methyl butyl ether (Riedel-de Haen,
Netherlands). The extraction was performed by adding 3 ml of methyl butyl ether to
200 µl of liver homogenate sample (0.3 g liver/ml of PBS) and vortexing for 5 min. The
mixture was then centrifuged at 900 × g for 5 min and the aqueous layer was frozen in
liquid nitrogen. The upper organic layer was transferred to another borosilicate glass tube
and evaporated completely at 60 °C. The extraction procedure was repeated twice and the
total residue was reconstituted in 200 µl of mobile phase. A 10µl of the reconstituted
sample was injected into the HPLC system.
The liver homogenate samples were estimated at the sensitivity of 0.01 AUFS.
Chromatography was carried out using a C18 (C18, 5µm, 4.6x150mm) reversed-phase
column (Sunfire, Waters Inc., Milford, MA, USA) at 40ºC with an isocratic mobile phase
consisting of acetonitrile–water–trifluoroacetic acid (30:70:0.1, v/v/v; pH 2.0) at a flow
rate of 1ml/min. Drug from liver tissue was measured at 225 nm and evaluated after first

75
Chapter 4
and second method, eluting at 7.4 min. Peak-height ratios of the drug plotted in a
calibration curve were used for the quantification of losartan from the different matrixes.
E
Figure 2. Analysis of biodistribution of losartan-M6PHSA in bile duct ligated rats.
Liver sections were processed for immunohistochemistry and then stained with anti-HSA antibody (see
Experimental procedures). HSA staining was not detected in the lung (A), spleen (B), heart (C) or
kidney (D) (magnification 4x). HSA staining was absent in rats treated with saline (not shown), while it
was detected in the liver within the non-parenchymal cells of rats treated with losartan-M6PHSA (E)
(magnification 4x). Five rats were studied per group.
Quantification of collagen accumulation and infiltration by myofibroblastic
and inflammatory cells
The degree of hepatic fibrosis was estimated as the percentage of area stained with picro
Sirius Red (Sirius Red F3B, Gurr-BDH Lab Supplies, Poole, England). The amount of
Chapter 4

76
Reduction of liver fibrosis by targeted losartan
fibrogenic myofibroblasts was estimated by measuring the percentage of area stained with
anti-smooth muscle α actin (αSMA, DAKO, Carpinteria, CA). Finally, the amount of
inflammatory cells was estimated by measuring the number of cells stained with anti-
CD43 (Serotec, Oxford, United Kingdom) in 10 randomly selected high power fields per
sample. For morphometric assessment of percentage of area with positive staining, an
optic microscope (Nikon Eclipse E600) connected to a high-resolution camera (CC12
Soft-Imaging System, Münster, Germany) was used. Images were analyzed in an
automated image-analysis system (AnalySIS, Soft-Imaging System, Münster, Germany).
Images were captured following automatic white balance and light intensity equilibration
with a 40 × magnification objective and digitized as RGB 24-bit. Each optical image size
at 40x was 88752 µm2 for a 250 x 250 square pixel image, resulting in an optical
resolution of 1,42 µm2/pixel. Image reconstruction was performed using the Multiple
Image Alignment. After shading correction and interactive thresholding, the selected
positive pixels were measured. The positive area was the sum of the area of positive
pixels. The ratio was calculated as the area of positive pixels divided by the total area of
the biopsy. Results are given as percentage of positive area.
Analysis of hepatic gene expression
RNA was isolated from frozen liver samples using Trizol (Life Technologies Inc.,
Rockville, MD). Quantitative PCR was performed with pre-designed Assays-on-Demand
TaqMan probes and primer pairs for rat collagen α1 (II), transforming growth factor β1
(TGFβ1), tissue inhibitor metalloproteinase type 1 (TIMP-1) and ribosome subunit 18s
(Applied Biosystems, Foster City, CA).
Information on these Assay-on-Demand is available at:
http://myscience.appliedbiosystems.com/cdsEntry/Form/gene_expression_keyword.jsp.
TaqMan reactions were carried out in duplicate on an ABI PRISM 7900 machine (Applied
Biosystems).

77
Chapter 4
Statistical analysis
Results are expressed as the mean±s.e.m. Significance was established using t-test, two-
way ANOVA with Bonferroni's post hoc test and Mann-Whitney assay. Differences were
considered significant if P<0.05.
BA
Figure 3. Analysis of colocalization of losartan-M6PHSA within hepatic stellate
cells.
Liver sections were processed for immunohistochemistry and then stained with anti-HSA antibody (see
Experimental procedures). Losartan-M6PHSA co-localized with stellate cells in rat livers (arrows), as
assessed with double immunostaining with anti-HSA and anti-desmin (A). Analysis performed with
confocal laser microscopy verified the colocalization of anti-HSA (red fluorescence color) and anti-desmin
(green fluorescence color) in the regions colored in yellow (arrows) (magnification 40x). Five rats were
studied per group.
Chapter 4

78
Reduction of liver fibrosis by targeted losartan
Results
Synthesis and characterization of losartan-M6PHSA conjugate
i i
Losartan was conjugated to M6PHSA in two synthesis steps. After its reaction to the ULS
linker, the losartan-ULS adduct was conjugated to M6PHSA and the final high-molecular
weight product was extensively purified by dialysis (Fig. 1A). An average of seven
losartan-ULS molecules were coupled per M6PHSA, as assessed by HPLC studies and
confirmed by inductive coupled plasma – atomic emission spectroscopy (ICP-AES) (data
not shown). Conjugation of losartan to M6PHSA did not change the charge or size
features of M6PHSA, as assessed by anion-exchange chromatography and size exclusion
chromatography, respectively (Fig. 1A).
ULS is a derivative of cisplatin and since cisplatin is a well-known antitumor agent, the
use of platinum (II) as a linker may infer unwanted toxicity associated with the drug
targeting construct. We therefore studied the effect of Losartan-M6PHSA on rat HSC
that were cultured and activated in vitro. Cell viability studies did not show toxicity to the
target cells after 24h treatment with Losartan-M6PHSA, in contrast to cisplatin at the
same concentration (Figure 1B).
Pharmacok netics of losartan-M6PHSA in b le-duct ligated rats
We have previously shown that M6PHSA binds to M6P/IGFII-R, which is
predominantly expressed in activated HSC in the fibrotic liver(22). M6PHSA clearly does
not interact with hepatocytes and binds minimally other non-parenchymal cell types, such
as Kupffer cells and sinusoidal endothelial cells. In the current study, we have used this
approach to selectively deliver losartan to HSC in rats with advanced liver fibrosis
induced by bile duct ligation. The animals were administered four consecutive doses of
losartan-M6PHSA (3.3 mg/kg, corresponding to 125 µg losartan/kg) from day 12 to day
15. Control groups were treated with equivalent doses of M6PHSA (3.3 mg/kg), saline, or
free losartan orally at a dosage (5 mg/kg) that has been reported to be effective against
liver fibrosis when given for a prolonged period of time(5-8).

79
Chapter 4
We first assessed whether losartan-M6PHSA preferentially accumulates in the fibrotic rat
liver. Livers as well as other organs (lung, heart, spleen, and kidney) were stained for anti-
HSA to detect the presence of the core protein. As shown in Fig. 2A-2E, losartan-
M6PHSA was only detected in the liver, while it was not present in other organs.
Injection of the carrier alone (M6PHSA) followed a similar distribution pattern (not
shown). Since the animals had been injected with a last dose of the conjugate 10 minutes
prior to their killing, this result clearly illustrates the rapid and selective accumulation of
the product in the liver. Importantly, losartan-M6PHSA co-localized with activated HSC,
as assessed by double immunostaining with anti-HSA and anti-desmin antibodies and
verified after colocalization using fluorescence antibodies anti-HSA and anti-desmin and
detected via confocal laser microscopy (Fig. 3A-3B).
To further demonstrate the selective homing of losartan-M6PHSA to the liver, we
quantified tissue levels of losartan by HPLC. Of note, a 40-fold higher dosage was
administrated in the case of losartan itself compared to losartan-M6PHSA. The dosage of
orally administered losartan was based in previous studies referring to efficacy of
losartan(5), whereas a minor dose of losartan-M6PHSA conjugate was needed after the
efficient delivery of losartan to the HSC. Two different procedures were followed to
isolate losartan from tissue homogenates, either providing free losartan levels or detecting
the presence of conjugated losartan. Following the oral administration of losartan per se,
both analyses yielded an average tissue concentration of 12.1 µg/g liver, which
corresponded to 4% of the cumulative dose (15% of the last dose administered). In
contrast, animals that were given losartan-M6PHSA, exhibited losartan levels of 1.5 µg/g,
corresponding to at least 20% of the cumulative dose (81% of the last injected dose). As
can be appreciated from these results, losartan-M6PHSA demonstrated a preferential liver
distribution. Due to the accumulation of losartan-M6PHSA in the stellate cell, which only
constitutes a small fraction of the whole liver, drug levels in HSC are most likely even
much higher. Oral administered losartan does not show this preferential homing to HSC.
Chapter 4

80
Reduction of liver fibrosis by targeted losartan
t
Saline M6PHSA losartan-M6PHSA Oral losartanSaline M6PHSA losartan-M6PHSA Oral losartan
0
2
4
6
8
10
12
14
16
18
Sham Saline M6PHSA M6PHSA-losartan
Orallosartan
Siriu
s re
d st
aini
ng(%
pos
itive
are
a)
F
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
Sham Saline M6PHSA M6PHSA-losartan
Oral losartan
Proc
olla
gen α1
mR
NA
(2
-∆∆
Ct ) *
#
E
#
*
Figure 4. Effect of differen treatments on the degree of liver fibrosis.
Liver sections were processed for immunohistochemistry and then processed for Sirius Red staining. Severe
bridging was observed in rats receiving saline (A), M6PHSA (B) and oral losartan (D). However, rats
treated with losartan-M6PHSA (C) showed fewer areas with collagen accumulation (magnification 4x).
(E). Quantification of the area with Sirius Red staining in liver specimens (n=10, mean± s.e.m.,
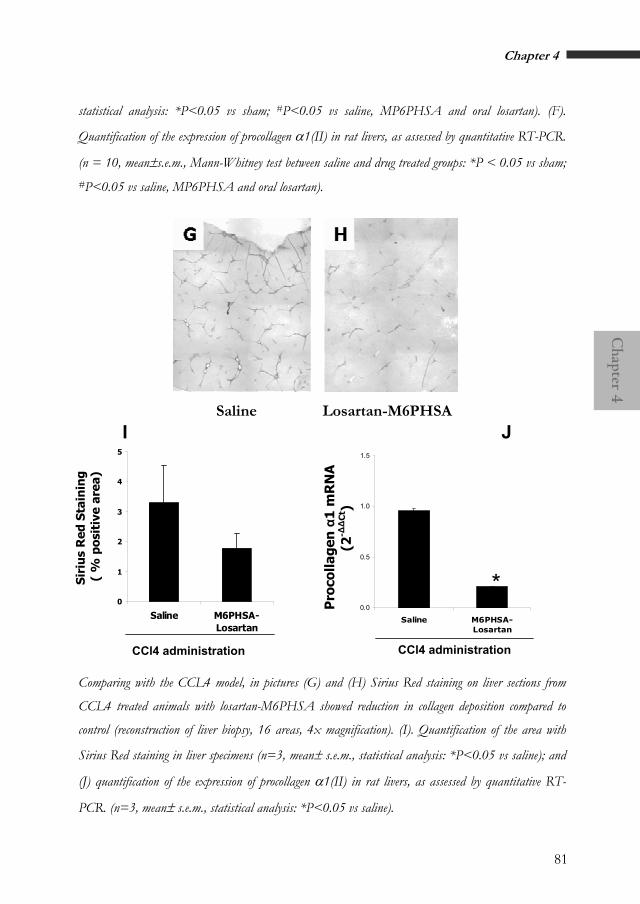
81
Chapter 4
statistical analysis: *P<0.05 vs sham; #P<0.05 vs saline, MP6PHSA and oral losartan). (F).
Quantification of the expression of procollagen α1(II) in rat livers, as assessed by quantitative RT-PCR.
(n = 10, mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P < 0.05 vs sham; #P<0.05 vs saline, MP6PHSA and oral losartan).
I J Saline 6PHSA Losartan-M
0
1
2
3
4
5
Saline M6PHSA-Losartan
Pro
colla
gen
α1
mR
NA
(2-∆∆
Ct )
0.0
0.5
1.0
1.5
Saline M6PHSA-Losartan
*
Siri
us
Red
Sta
inin
g
( %
pos
itiv
e ar
ea)
CCl4 administration CCl4 administration
Comparing with the CCL4 model, in pictures (G) and (H) Sirius Red staining on liver sections from
CCL4 treated animals with losartan-M6PHSA showed reduction in collagen deposition compared to
control (reconstruction of liver biopsy, 16 areas, 4x magnification). (I). Quantification of the area with
Sirius Red staining in liver specimens (n=3, mean± s.e.m., statistical analysis: *P<0.05 vs saline); and
(J) quantification of the expression of procollagen α1(II) in rat livers, as assessed by quantitative RT-
PCR. (n=3, mean± s.e.m., statistical analysis: *P<0.05 vs saline).
Chapter 4

82
Reduction of liver fibrosis by targeted losartan
Short-term t eatment w th losartan M6PHSA but not oral losartan reduces
advanced liver fibrosis
r i -
Although the dosage of losartan-M6PHSA was much lower than of losartan itself, we
hypothesized that the targeting to activated HSC would render the drug more effective in
attenuating advanced liver fibrosis in rats. Previous studies have shown that after 2 weeks,
bile duct ligated rats develop profound changes in the hepatic architecture including
bridging fibrosis (23). As expected, BDL for 15 days resulted in a marked increase in
serum bilirubin and serum levels of enzymes AST and ALT, which was similar in all
treated groups (data not shown). The liver/body weight ratio increased in bile duct ligated
rats and this aspect was not affected by any treatment. These results indicated that short-
term treatment with losartan-M6PHSA or oral losartan did not protect livers from BDL-
induced hepatocellular injury.
We next investigated the degree of liver fibrosis. Following bile duct ligation, rats treated
with saline or M6PHSA alone showed severe septal fibrosis with a marked disruption of
the hepatic architecture (Fig. 4A-3B). Hepatic collagen content, as assessed by
morphometric analysis of Sirius red staining, was markedly increased in these rats as
compared to the sham group (Fig. 4E). In contrast, bile duct ligated rats treated with
losartan-M6PHSA displayed a remarkably decreased collagen deposition with less
frequent formation of bridging fibrosis (Fig. 4C). Importantly, short-term treatment with
oral losartan did not reduce histological fibrosis or the amount of collagen content (Fig.
4D). To confirm these results, hepatic procollagen α1(II) gene expression was quantified.
Procollagen α1(II) was upregulated 10-fold in bile duct ligated rats treated with saline
compared with sham-operated animals (Fig. 4F). Losartan-M6PHSA, but not oral losartan
or M6PHSA alone, reduced procollagen α1(II) by 60% in bile duct ligated rats. These
results strongly indicate that short-term treatment with losartan-M6PHSA, but not oral
losartan, is capable to reduce advanced liver fibrosis in rats with chronic cholestasis.
To further demonstrate that the antifibrotic effect of losartan-M6PHSA was not an
experimental model specific phenomenon, we tested losartan-M6PHSA in the well-
established model of CCl4-induced liver fibrosis (24). After 8 weeks of CCl4 inhalation

83
Chapter 4
initial stages of hepatic fibrosis were reached, at which stage we administered four
consecutive daily doses of losartan-M6PHSA. Both hepatic collagen content, as assessed
by morphometric analysis of Sirius red staining, and hepatic procollagen α1(II) gene
expression were reduced in rats treated with losartan-M6PHSA as compared to saline-
treated rats (Fig. 4G-H).
Mechanisms of reduced liver fibrosis in rats treated with losartan-M6PHSA
To explore the mechanisms involved in the potent therapeutic effect of losartan-
M6PHSA, we first assessed the accumulation of fibrogenic myofibroblasts in bile duct
ligated livers. These cells mostly originate from activated HSC, yet other cell types such as
portal myofibroblasts are also a source of fibrogenic cells in the bile duct ligated livers
(25). We have previously shown that Ang II induces activation and proliferation of HSC
(26). To test whether losartan-M6HSA treatment reduces the accumulation of such cells,
the number of myofibroblasts was assessed in liver specimens by morphometric
quantification of αSMA-positive cells. As shown in Figure 5A, BDL was associated with
the accumulation of abundant αSMA-positive cells in the hepatic parenchyma. These cells
accumulated around proliferating bile ducts as well as in the hepatic sinusoids (Fig. 5E).
Treatment with losartan-M6HSA, but not oral losartan or M6HSA alone, was associated
with a reduction in the accumulation of myofibroblasts (Fig. 5B-5D). Morphometric
analysis of the positive area with αSMA staining confirmed that losartan-M6PHSA
decreased the number of αSMA-positive cells (Fig. 5F). Similar results were obtained after
administration of losartan-M6PHSA to CCl4 fibrotic rats, as shown in figure 5 G-H, and
confirmed after morphometric quantification of αSMA positive cells per liver area (Fig.
5I).
Chapter 4

84
Reduction of liver fibrosis by targeted losartan
Saline M6PHSA losartan-M6PHSA Oral losartanSaline M6PHSA losartan-M6PHSA Oral losartan
t
0
2
4
6
8
10
12
14
16
Sham Saline M6PHSA M6PHSA-losartan
Oral losartan
αSM
A s
tain
ing
(% p
ositi
ve a
rea)
F *
#
Figure 5. Effect of different treatments on the accumula ion of myofibroblasts and
activated hepatic stellate cells, as assessed by smooth muscle α-actin expression
(αSMA).
Liver sections were processed for immunohistochemistry and then stained with anti-αSMA antibody. Rats
receiving saline (A), M6PHSA (B) or oral losartan (D) showed a marked accumulation of αSMA-
positive cells, which co-localized with areas with active fibrogenesis. However, rats treated with losartan-
M6PHSA (C) showed less quantity of αSMA-positive cells (magnification 4x). (E). High
magnification (400x) photomicrograph of a liver from a bile duct ligated rat treated with saline. αSMA
staining was detected in cells located in the sinusoids corresponding to activated hepatic stellate cells (upper
arrow) as well as in myofibroblasts around proliferating bile ducts (lower arrow).

85
Chapter 4
(F). Quantification of the area with αSMA staining in rat liver specimens (n = 10, mean±s.e.m.,
Mann-Whitney test between saline and drug treated groups:*P<0.05 vs sham; #P<0.05 vs saline,
MP6PHSA and oral losartan).
0
1
2
3
4
5
6
7
Saline M6PHSA-Losartan
*
I
CCl4 administration
αSMA staining on liver sections from CCL4 treated animals with losartan-M6PHSA showed less
accumulation of αSMA-positive cells (H) compared to diseased animals treated with saline (G)
(reconstruction of liver biopsy, 16 areas, 4x magnification). (I). Quantification of the area with αSMA
staining in liver specimens (n=3, mean± s.e.m., statistical analysis: *P<0.05 vs saline).
Because TIMP-1 is a survival factor for activated HSC and regulates collagen degradation,
we next explored TIMP-1 hepatic gene expression by quantitative PCR. As shown in Fig.
6A, BDL resulted in approximately 10-fold increase in TIMP-1 hepatic gene expression,
which was not reduced by losartan-M6HSA nor oral losartan. Also, we explored the
hepatic expression of TGFβ1, which is a major fibrogenic cytokine that largely mediates
the fibrogenic actions of Ang II in many organs including the liver (27). Compared to
sham-operated rats, bile duct ligated rats showed a 5-fold increase in TGFβ1 gene
expression, which was reduced by 30% in rats treated with losartan-M6HSA (Fig. 6B).
Chapter 4

86
Reduction of liver fibrosis by targeted losartan
Finally, we explored whether losartan treatment diminished hepatic inflammation, which
is a major event leading to hepatic fibrosis in rats with secondary biliary cirrhosis.
Previous studies have shown that Ang II exerts pro-inflammatory effects in the liver and
that AT1 blockade reduces hepatic inflammation (28) (29). To test this hypothesis, we
quantified the infiltration of inflammatory cells (CD43-positive) in the hepatic
parenchyma by immunohistochemistry. Compared to sham-operated rats, bile duct ligated
rats showed a marked increase in the infiltration of CD43-positive inflammatory cells
(Fig. 7). This effect was blunted by treatment with losartan-M6HSA and, in to lesser
extent, by oral losartan. Overall, these results suggest that the antifibrotic effect of short-
term treatment with losartan-M6HSA may be due to removal of activated HSC, decreased
expression of TGFβ1, and attenuated hepatic inflammation.
t
0.00.20.40.60.81.01.21.41.61.82.0
Sham Saline M6PHSA M6PHSA-losartan
Oral losartan
TIM
P-1
mR
NA
(2
-∆∆
Ct ) *
A
Figure 6. Effect of different treatments on hepatic gene expression from bile duc
ligated rats, as assessed by quantitative RT-PCR.
Rats subjected to bile duct ligation received different treatments, as specified in the figure (n = 10,
mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P<0.05 vs sham).
A. Effect of different treatments on TIMP-1 hepatic gene expression in livers from bile duct ligated rats
(n=10, mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P<0.05 vs sham).

87
Chapter 4
B
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Sham Saline M6PHSA M6PHSA-losartan
Orallosartan
TGF β
1 m
RN
A
(2-∆∆
Ct ) # * #
B. Effect of different treatments on TGFβ1 hepatic gene expression in livers from bile duct ligated rats
(n=10, mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P<0.05 vs sham; #P<0.05 vs saline and M6PHSA).
Chapter 4

88
Reduction of liver fibrosis by targeted losartan
Discussion
The present study demonstrates that advanced liver fibrosis can be successfully treated by
short-term administration of an antifibrotic drug that is selectively targeted to the main
fibrogenic cell type in the liver (i.e. activated HSC). We provide evidence that the delivery
of the AT1 receptor blocker losartan to HSC reduces inflammation and collagen
deposition in two different rat models of liver fibrosis. Importantly, this novel approach
appears to be much more effective than losartan given orally, suggesting that it may be
useful for treating patients with advanced chronic liver diseases. Our data also confirm a
major role for the RAS in the pathogenesis of liver fibrosis.
To date, there is no effective therapy to treat liver fibrosis (1). In view of the high
prevalence of hepatic fibrosis and its potential lethal consequences (i.e. cirrhosis), the
development of safe and effective antifibrotic therapies is an urgent and unmet medical
need in hepatology. Such therapies should be capable of reducing the hepatic fibrogenic
response to injury, even when the causative agent cannot be removed (e.g. chronic hepatic
C not responding to antiviral therapy). Moreover, antifibrotic therapies should be well
tolerated in patients with advanced cirrhosis when used for prolonged periods of time.
Most of the antifibrotic compounds that reduce experimental liver fibrosis do not meet
these requirements (e.g. TGFβ and/or PDGF inhibitors), as they generally display serious
side-effects. The selective delivery of antifibrotic drugs to HSC may theoretically reduce
the undesirable extrahepatic effects while increasing the antifibrotic effect of a given drug.
Macromolecular carriers, such as for instance the presently applied M6PHSA, render such
an approach feasible (16). To test the efficacy of this approach, the antifibrotic compound
chosen to target HSC was an AT1 antagonist. We chose this family of drugs because there
is overwhelming evidence supporting a role for AT1 receptors in the pathogenesis of liver
fibrosis and because AT1 receptors blockers are known to reduce tissue fibrosis in organs
such the kidney and the heart (1) (30) .
The new drug conjugate Losartan-M6PHSA was successfully synthesized by first applying
a novel linker system that binds losartan via a transition-metal coordination bond at the

89
Chapter 4
platinum center to one of the nitrogens in the tetrazole ring of losartan structure. The
resulted Losartan-ULS adduct was subsequently reacted to M6PHSA carrier in a
straightforward reaction, affording an overall reaction efficiency of 70%. Traditionally, the
art of linking drugs to carrier moieties represents a complex issue to deal with (31). A key
property of our platinum linker, ULS, is that it can be applied for conjugation of many
valuable drug molecules containing aromatic nitrogens, forming a bond of intermediate
binding strength (32). The ligand-exchange behaviour of Pt compounds is quite slow,
giving them a high kinetic stability (33). Once in the target cell, ULS does have a strong
thermodynamic preference for binding to S-donor ligands, such as for instance
glutathione or sulphur containing amino acids. As the concentrations platinum in our
conjugates are orders of magnitude lower than applied in cisplatin cancer therapy, one
would predict rapid detoxification of ULS by binding to cytosolic platinophilic
ligands(18). The present HSC viability studies with losartan-M6PHSA (Fig 1B) are in
good agreement with the safety data of other drug-M6PHSA conjugates prepared with the
ULS linker(18).
The main result of the current study is that a short-term treatment with a drug selectively
targeted to HSC is capable of reducing advanced liver fibrosis. In contrast, short-term
treatment with the same drug but given orally only has minor effects. These results
suggest that the selective delivery of drugs to fibrogenic cells may increase the efficacy of
antifibrotic therapy. In previous studies assessing the antifibrotic effect of losartan in rats
with experimentally-induced fibrosis, the drug was given for long periods of time, usually
concomitantly with the initiation of liver injury (5;28;29). Although these studies yielded
positive results, the particular approach used does not really mimic the scenario in
humans. In patients with chronic diseases (e.g. chronic hepatitis C), the onset of liver
fibrosis is a slow process. In most cases, antifibrotic therapies will be initiated when the
liver fibrosis is manifest and the causative stimulus cannot be removed. It is likely that the
efficacy of antifibrotic drugs in this setting is not optimal.
Different mechanisms may explain the strong antifibrotic effect achieved with our drug-
targeting construct. First, targeting losartan to the HSC via the modified albumin,
M6PHSA, increases the fraction of the dose that accumulates within the fibrogenic cells.
Chapter 4

90
Reduction of liver fibrosis by targeted losartan
M6PHSA distributes rapidly and extensively (about 60% of the injected dose) to the liver.
After four administrations of losartan-M6PHSA, losartan levels in the liver accounted for
20% of the cumulative dose, revealing the efficiency of the targeting strategy. We also
showed that M6PHSA mainly distributes to activated HSC located around the hepatic
sinusoid. In contrast, we found 4% of the total dose in the liver after orally administration
of losartan per se, which is in agreement with its distribution over the total body. On the
other hand, the concentrations of losartan in the liver after conventional oral treatment,
which was given in a 40-fold higher dose were higher than after the targeted treatment.
Yet, the antifibrogenic effect of the oral treatment was much lower. Thus, the observed
effects of losartan-M6PHSA cannot be attributed to higher concentrations of losartan per
se, but must be due to the sustained release of the drug in time, or to the subcellular
accumulation within the liver.
Secondly, the activity of losartan-M6PHSA may be enhanced by the specific interaction
that M6PHSA provides. The M6P/IGFII receptor participates in the activation of latent
TGFβ1, which may be affected by M6PHSA (17). The finding that treatment with
M6PHSA alone did not affect fibrosis or inflammation in bile duct ligated rats however,
does not support this hypothesis. Thirdly, we show that targeted losartan rapidly reduces
the accumulation of activated HSC in the fibrotic liver, which is considered a main driven
force in liver fibrogenesis. This is consistent with previous reports showing that Ang II is
a powerful mitogenic agonist for HSC (4). In addition, targeted losartan decreased the
expression of TGFβ1, which is considered the main fibrogenic factor in the liver (34).
This result suggests that locally generated Ang II stimulates TGFβ1 expression in the
liver, similarly to what occurs in other organs such as the kidney (35).
And finally, targeted losartan strongly attenuated infiltration of inflammatory cells in the
liver. This effect was also observed in rats treated with oral losartan, yet to a lesser extent.
This finding may be relevant, since inflammatory cells play a major role in liver
fibrogenesis by releasing fibrogenic cytokines and by directly interacting with HSC (36).
This latter effect is consistent with previous reports showing that Ang II exerts pro-
inflammatory actions both in cultured cells and in vivo (4;23).
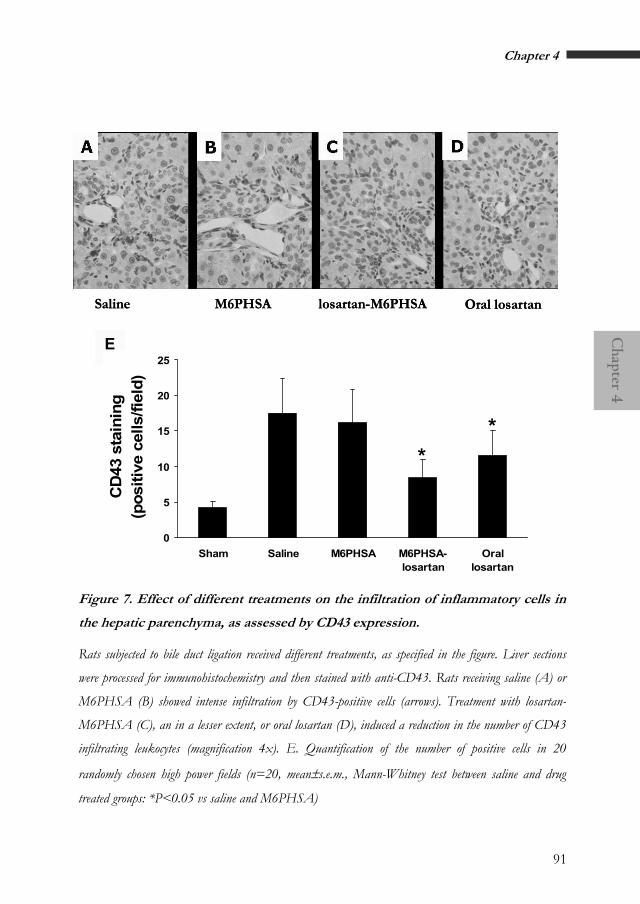
91
Chapter 4
Saline M6PHSA losartan-M6PHSA Oral losartanSaline M6PHSA losartan-M6PHSA Oral losartan
i
E
0
5
10
15
20
25
Sham Saline M6PHSA M6PHSA-losartan
Orallosartan
CD
43 s
tain
ing
(pos
itive
cel
ls/fi
eld)
**
Figure 7. Effect of different treatments on the infiltration of inflammatory cells in
the hepatic parenchyma, as assessed by CD43 express on.
Rats subjected to bile duct ligation received different treatments, as specified in the figure. Liver sections
were processed for immunohistochemistry and then stained with anti-CD43. Rats receiving saline (A) or
M6PHSA (B) showed intense infiltration by CD43-positive cells (arrows). Treatment with losartan-
M6PHSA (C), an in a lesser extent, or oral losartan (D), induced a reduction in the number of CD43
infiltrating leukocytes (magnification 4x). E. Quantification of the number of positive cells in 20
randomly chosen high power fields (n=20, mean±s.e.m., Mann-Whitney test between saline and drug
treated groups: *P<0.05 vs saline and M6PHSA)
Chapter 4

92
Reduction of liver fibrosis by targeted losartan
Our results potentially have implications for the treatment of liver fibrosis. Firstly, we
provide evidence that short-term treatment with a highly active oral compound -losartan-
is capable to attenuate the inflammatory response but it is not strong enough to reduce
liver fibrosis. Therefore, the current assumption that Ang II blockers are highly effective
in attenuating experimental liver fibrosis should be tempered. Second, our results support
the current research to develop innovative systems to deliver fibrogenic drugs to HSC.
This approach would be particularly useful in conditions with rapidly aggressive hepatic
fibrosis (e.g. acute alcoholic hepatitis) in which the use of AT1 receptors blockers may
induce undesirable side effects. Finally, our results indicate the possibility to use drugs
known to attenuate portal pressure in the treatment of patients with acute complications
due to portal hypertension (e.g. acute variceal bleeding). In these patients, the use of oral
losartan is precluded by the development of arterial hypotension, or even hypotensive
shock syndrome (14). Further studies should investigate whether the use of losartan
targeted to HSC lowers portal pressure without reducing systemic arterial pressure.
In conclusion, by using a new platinum-based linker, we successfully coupled losartan to
M6PHSA, a carrier capable to deliver drugs to activated HSC in the fibrotic rat liver. We
provide evidence that the beneficial effects of losartan in liver fibrosis are greatly
enhanced when the drug is selectively targeted to HSC. Importantly, losartan-M6PHSA
was effective in reducing liver fibrosis in two independent experimental models of liver
fibrosis. Further studies should explore whether this novel biopharmaceutical approach is
feasible and safe in patients with liver fibrosis.

93
Chapter 4
Acknowledgements
This study was supported by grants from SenterNovem (TSGE1083), NWO Science
Netherlands (R 02-1719, 98-162), the Ministerio de Ciencia y Tecnología, Dirección
General de Investigación (SAF2005), and from the Instituto de Salud Carlos III
(CO3/02). We thank Montserrat Moreno and Anna Planagumà for their kind help in
animal handling and Elena Juez and Cristina Millan (Hospital Clínic, Barcelona, Spain) for
their excellent technical support. We also thank the department of Pharmaceutical
Analysis (University of Groningen, The Netherlands) for the losartan-ULS mass
spectrometry analysis. Jan Visser (University of Groningen, The Netherlands) is kindly
acknowledged for assistance in HPLC analysis. Klaas Sjollema and Michel Meijer are also
acknowledged for their kind assistance with the confocal pictures at the UMCG
Microscopy and Imaging Center.
Chapter 4

94
Reduction of liver fibrosis by targeted losartan
Reference list
1. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
2. Bataller,R., Sancho-Bru,P., Gines,P., and Brenner,D.A. 2005. Liver fibrogenesis: a new role for the renin-angiotensin system. Antioxid. Redox. Signal. 7:1346-1355.
3. Wei,H.S., Lu,H.M., Li,D.G., Zhan,Y.T., Wang,Z.R., Huang,X., Cheng,J.L., and Xu,Q.F. 2000. The regulatory role of AT 1 receptor on activated HSCs in hepatic fibrogenesis:effects of RAS inhibitors on hepatic fibrosis induced by CCl(4). World J. Gastroenterol. 6:824-828.
4. Bataller,R., Sancho-Bru,P., Gines,P., Lora,J.M., Al Garawi,A., Sole,M., Colmenero,J., Nicolas,J.M., Jimenez,W., Weich,N., Gutierrez-Ramos, J.C., Arroyo, V., and Rodes, J. 2003. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125:117-125.
5. Croquet,V., Moal,F., Veal,N., Wang,J., Oberti,F., Roux,J., Vuillemin,E., Gallois,Y., Douay,O., Chappard,D. et al 2002. Hemodynamic and antifibrotic effects of losartan in rats with liver fibrosis and/or portal hypertension. J. Hepatol. 37:773-780.
6. Paizis,G., Gilbert,R.E., Cooper,M.E., Murthi,P., Schembri,J.M., Wu,L.L., Rumble,J.R., Kelly,D.J., Tikellis,C., Cox,A. et al 2001. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J. Hepatol. 35:376-385.
7. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Nakatani,T., Tsujinoue,H., and Fukui,H. 2001. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 34:745-750.
8. Ramalho,L.N., Ramalho,F.S., Zucoloto,S., Castro-e-Silva Junior, Correa,F.M., Elias,J.J., and Magalhaes,J.F. 2002. Effect of losartan, an angiotensin II antagonist, on secondary biliary cirrhosis. Hepatogastroenterology 49:1499-1502.
9. Burnier,M., and Zanchi,A. 2006. Blockade of the renin-angiotensin-aldosterone system: a key therapeutic strategy to reduce renal and cardiovascular events in patients with diabetes. J. Hypertens. 24:11-25.
10. Sica,D.A., Gehr,T.W., and Ghosh,S. 2005. Clinical pharmacokinetics of losartan. Clin. Pharmacokinet. 44:797-814.
11. Rimola,A., Londono,M.C., Guevara,G., Bruguera,M., Navasa,M., Forns,X., Garcia-
Retortillo,M., Garcia-Valdecasas,J.C., and Rodes,J. 2004. Beneficial effect of angiotensin-blocking agents on graft fibrosis in hepatitis C recurrence after liver transplantation. Transplantation 78:686-691.
12. Sookoian,S., Fernandez,M.A., and Castano,G. 2005. Effects of six months losartan administration on liver fibrosis in chronic hepatitis C patients: a pilot study. World J. Gastroenterol. 11:7560-7563.
13. Yokohama,S., Yoneda,M., Haneda,M., Okamoto,S., Okada,M., Aso,K., Hasegawa,T., Tokusashi,Y., Miyokawa,N., and Nakamura,K. 2004. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 40:1222-1225.
14. Gonzalez-Abraldes,J., Albillos,A., Banares,R., Del Arbol,L.R., Moitinho,E., Rodriguez,C., Gonzalez,M., Escorsell,A., Garcia-Pagan,J.C., and Bosch,J. 2001. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 121:382-388.
15. Beljaars,L., Meijer,D.K., and Poelstra,K. 2002. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front Biosci. 7:e214-e222.
16. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatology 29:1486-1493.
17. De Bleser,P.J., Jannes,P., Van Buul-Offers,S.C., Hoogerbrugge,C.M., Van Schravendijk,C.F.H., Niki,T., Rogiers,V., Van den Brande,J.L., Wisse,E., and Geerts,A. 1995. Insulinlike growth factor-II/mannose 6-phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transforming growth factor-b in cocultures with sinusoidal endothelial cells. Hepatology 21:1429-1437.
18. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K., and Kok,R.J. 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
19. Greupink,R., Bakker,H.I., Reker-Smit,C., van Loenen-Weemaes,A.M., Kok,R.J., Meijer,D.K., Beljaars,L., and Poelstra,K. 2005. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. 43:884-892.

95
Chapter 4
20. van Gijlswijk,R.P., Talman,E.G., Janssen,P.J., Snoeijers,S.S., Killian,J., Tanke,H.J., and Heetebrij,R.J. 2001. Universal Linkage System: versatile nucleic acid labeling technique. Expert. Rev. Mol. Diagn. 1:81-91.
21. Paizis,G., Cooper,M.E., Schembri,J.M., Tikellis,C., Burrell,L.M., and Angus,P.W. 2002. Up-regulation of components of the renin-angiotensin system in the bile duct-ligated rat liver. Gastroenterology 123:1667-1676.
22. Beljaars,L., Olinga,P., Molema,G., De Bleser,P., Geerts,A., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 2001. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver 21:320-328.
23. Bataller,R., Gabele,E., Parsons,C.J., Morris,T., Yang,L., Schoonhoven,R., Brenner,D.A., and Rippe,R.A. 2005. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology 41:1046-1055.
24. Claria J, and Jimenez,W. 1999. Renal dysfunction and ascites in carbon tetrachloride-induced liver cirrhosis in rats. Pathogenesis, diagnosis and treatment. V.Arroyo, Gines,P., Rodes,J., and Schrier,R., editors. Blackwell Science Inc. Malden.M; USA. USA. 379-396.
25. Magness,S.T., Bataller,R., Yang,L., and Brenner,D.A. 2004. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology 40:1151-1159.
26. Bataller,R., Gabele,E., Schoonhoven,R., Morris,T., Lehnert,M., Yang,L., Brenner,D.A., and Rippe,R.A. 2003. Prolonged infusion of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am. J. Physiol Gastrointest. Liver Physiol 285:G642-G651.
27. Ishizaka,N., Saito,K., Noiri,E., Sata,M., Ikeda,H., Ohno,A., Ando,J., Mori,I., Ohno,M., and Nagai,R. 2005. Administration of ANG II induces iron deposition and upregulation of TGF-beta1 mRNA in the rat liver. Am. J. Physiol Regul. Integr. Comp Physiol 288:R1063-R1070.
28. Wei,Y.H., Jun,L., and Qiang,C.J. 2004. Effect of losartan, an angiotensin II antagonist, on hepatic fibrosis induced by CCl4 in rats. Dig. Dis. Sci. 49:1589-1594.
29. Yang,Y.Y., Lin,H.C., Huang,Y.T., Lee,T.Y., Hou,M.C., Lee,F.Y., Liu,R.S., Chang,F.Y., and Lee,S.D. 2002. Effect of 1-week losartan
administration on bile duct-ligated cirrhotic rats with portal hypertension. J. Hepatol. 36:600-606.
30. Gross,O., Schulze-Lohoff,E., Koepke,M.L., Beirowski,B., Addicks,K., Bloch,W., Smyth,N., and Weber,M. 2004. Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol. Dial. Transplant. 19:1716-1723.
31. Kok,R.J., Ásgeirsdóttir,S.A., and Verweij,W.R. 2001. Development of proteinaceous drug targeting constructs using chemical and recombinant DNA approaches. In Drug Targeting. Organ-Specific Strategies. G.Molema, and Meijer,D.K.F., editors. WILEY-VCH Verlag GmbH. Weinheim. 275-308.
32. Heetebrij,R.J., Talman,E.G., Velzen,M.A., van Gijlswijk,R.P., Snoeijers,S.S., Schalk,M., Wiegant,J., Rijke,F., Kerkhoven,R.M., Raap,A.K., Tanke, H.J., Reedijk, J., and Houthoff, H.J. 2003. Platinum(II)-based coordination compounds as nucleic acid labeling reagents: synthesis, reactivity, and applications in hybridization assays. Chembiochem. 4:573-583.
33. Reedijk,J. 2003. New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc. Natl. Acad. Sci. U. S. A 100:3611-3616.
34. Qi,Z., Atsuchi,N., Ooshima,A., Takeshita,A., and Ueno,H. 1999. Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc. Natl. Acad. Sci. U. S. A 96:2345-2349.
35. Huang,Y., Wongamorntham,S., Kasting,J., McQuillan,D., Owens,R.T., Yu,L., Noble,N.A., and Border,W. 2006. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 69:105-113.
36. Marra,F. 2002. Chemokines in liver inflammation and fibrosis. Front Biosci. 7:d1899-d1914.
37. Beljaars,L., Poelstra,K., Molema,G., and Meijer,D.K.F. 1998. Targeting of sugar- and charge-modified albumins to fibrotic rat livers: the accessibility of hepatic cells after chronic bile duct ligation. J. Hepatol. 29:579-588
Chapter 4

Koh Pha Ngan
Thailand, September 2004

Chapter5Accumulation in hepatic stellate cells and antifi brotic eff ect of losartan-M6PHSA in the CCL4 model of induced liver fi brosis
Manuscript in preparation.
Teresa Gonzalo1, Ramón Bataller2, Pau Sancho-Bru2, Dirk K.F. Meijer1, Leonie Beljaars1, Marie Lacombe3, Frank Opdam3, Vicente Arroyo2, Klaas Poelstra1, Pere Ginès2, Robbert J. Kok1,4.
1Department of Pharmacokinetics and Drug Delivery, University of Groningen, The Netherlands.
2Liver Unit, Hospital Clínic, IDIBAPS, Barcelona, Catalonia, Spain.
3Kreatech Biotechnology B.V., Amsterdam, The Netherlands.
and 4Department of Pharmaceutics, Utrecht University, The Netherlands.

98
Losartan-M6PHSA in CCL4 model of liver fibrosis
Abstract
The renin-angiotensin system (RAS) plays a fundamental role in liver fibrogenesis and
hence the use of losartan is proposed as an attractive antifibrotic strategy. However, the
use of RAS blockers can worsen the development of systemic hypotension associated with
liver fibrosis. To avoid the systemic interactions of losartan and improve its liver
accumulation, we have developed losartan-M6PHSA, a drug targeting preparation that
selectively accumulates in hepatic stellate cells (HSC), the main fibrogenic cell type in liver
fibrosis. We investigated the liver accumulation of losartan-M6PHSA and its antifibrotic
effects in the carbon tetrachloride (CCL4) model of rat liver fibrosis. After 9 weeks of
CCL4 inhalation, rats were treated with four daily doses of losartan-M6PHSA (8mg/kg,
containing 0.3 mg losartan/kg), free losartan (0.3mg/kg) or M6PHSA carrier alone
(8mg/kg). Expression of M6P/IGF II target receptors and accumulation of losartan-
M6PHSA in the fibrotic liver were confirmed immunohistochemically. We determined
losartan levels in the liver by HPLC, demonstrating a liver accumulation of 13.2 ±6.4% vs.
5.4±0.2% of the cumulative dose for losartan-MPHSA and free losartan, respectively, at
10 minutes after the last dose. Hepatic collagen deposition, as assessed by morphometric
analysis of Sirius red staining, was markedly lower in rats treated with losartan-M6PHSA
whereas treatment with losartan alone had no effect (p<0.05) Accumulation of α-smooth
muscle actin-positive cells was reduced in rats treated with M6PHSA-losartan or losartan
(p<0.05), but not after treatment with M6PHSA carrier. Finally, losartan-M6PHSA and
losartan reduced procollagen α1(II) gene expression by 80% and 70%, respectively. We
conclude that losartan acts as a potent antifibrogenic drug in the CCL4 model of liver
fibrosis, and that targeting of losartan to HSC strongly potentiates its effects.

99
Chapter 5
Introduction
Hepatic fibrosis is a dynamic process caused by chronic liver injury due to various inciting
stimuli including viral hepatitis (especially hepatitis B and C), alcohol abuse, obesity,
toxins, autoimmune attack of hepatocytes or bile duct epithelium, metabolic disease, or
congenital abnormalities (1;2). Advanced liver fibrosis eventually results in cirrhosis, which
represent a huge and global healthcare burden and a major cause of death (3).
Consequently, there is an urgent need for antifibrotic therapies that can retard or reverse
the ongoing fibrogenic processes in the liver.
Hepatic stellate cells (HSC) are considered as the main collagen-producing cells in the
injured liver, and key fibrogenic factors have been identified in HSC (4). In recent years
the renin-angiotensin system (RAS) was found to play a major role. Angiotensin II (Ang
II) mediated myofibroblast proliferation, infiltration of inflammatory cells, and
extracellular matrix (ECM) deposition (5-7). Neither in the normal human liver, HSC do
not express Angiotensin II type 1 (AT1) receptors nor secrete angiotensin II (Ang II).
During chronic liver injury, however, activated HSC express functional angiotensin
receptors and generate mature Ang II, which can activate the profibrogenic processes in
an autocrine or paracrine manner (8;9). The biological effects of Ang II in cultured human
and rodent HSC can be prevented by preincubation of HSC with angiotensin receptor
antagonists (8). Several studies in different models of liver fibrosis have established that
treatment with compounds that block RAS activity ameliorate experimentally induced
liver fibrosis (10-12). Therefore, inhibition of RAS system may be a relevant strategy to
prevent fibrosis progression during chronic liver diseases (13). The angiotensin receptor
antagonist Losartan has been proposed as a potential candidate to be tested in clinical
trials in patients suffering rapid progression of liver fibrosis (i.e. acute alcoholic hepatitis
and severe hepatitis C virus reinfection after liver transplantation) (14). However, patients
with advanced cirrhosis and activation of the systemic RAS show hypotension or reduced
systemic blood pressure. In this situation, it is dangerous to apply antihypertensive agents
because it can cause a further decrease in arterial pressure and renal impairment. Specific
Chapter 5

100
Losartan-M6PHSA in CCL4 model of liver fibrosis
targeting of losartan to activated HSC in the fibrotic liver might circumvent this
compromised situation. Such a liver-selective therapeutic drug would increase losartan
concentrations locally in the fibrotic liver, especially in activated HSC, whereas the low
drug concentrations in other organs may not elicit adverse systemic effects such as
reduction of blood pressure.
Table 1. Characterization and structure of losartan-M6PHSA.
Conjugate
synthesis ratio
losartan-ULS : protein
coupling ratio
losartan : protein¶
coupling ratio
ULS : protein§
losartan-M6PHSA
10:1
7 : 1 (±1.9)
7 : 1 (±2.7)
¶ Losartan coupling ratio was determined by HPLC after competitive displacement of the drug by excess of
potassium thiocyanate with KSCN at 80°C for 24h. § Universal Linkage System (ULS) coupling ratio
was determined by atomic emission spectroscopy (ICT-AES). These values were obtained from three
independent losartan-M6PHSA synthesized conjugates. M6P: mannose-6-phosphate; HSA: human
serum albumin; ULS: Universal linkage system.
Schematic structure of losartan-M6PHSA.
NKNN
NN
N OHCl
PtNH2H2N
M6PHSA carrier

101
Chapter 5
In recent years, the HSC-selective drug carrier mannose 6-phosphate modified human
serum albumin (M6PHSA) was developed (15). After systemic administration, M6PHSA
binds to surface-exposed M6P/IGFII receptors, which are de novo expressed in activated
HSC during liver fibrosis. In the present study, we investigated the expression of the
M6P/IGFII target receptors in the CCL4 inhalation model of rat liver fibrosis. Expression
of the M6P/IGFII receptor has been described in the first stages (hours) during acute
liver injury (16), but little is known about its expression during the slow and ongoing
disease progression. Furthermore, we assessed the accumulation of losartan-M6PHSA in
the CCL4-fibrotic livers and studied whether losartan-M6PHSA is more effective than
non-targeted losartan in attenuating the fibrogenic process.
Chapter 5

102
Losartan-M6PHSA in CCL4 model of liver fibrosis
Materials and methods
Synthesis of osartan-M6PHSA l
Losartan and human serum albumin (HSA) were obtained from Synfine (Ontario, Canada)
and Sanquin (Amsterdam, The Netherlands), respectively. Losartan was first coupled to
Universal Linker System (ULS™) (Kreatech Biotechnology, Amsterdam, The
Netherlands), a platinum complex that forms a co-coordinative bond with the protein
backbone (17). Briefly, cis-platinum(ethylenediamine)nitrate-chloride (cis-Pt(en)NO3Cl,
from now on referred to as ULS) was prepared by treating cis-Pt(en)dichloride with 0.9
equivalent of AgNO3 (18). The resulting solution of ULS (32 µmol in DMF) was added to
a solution of losartan (32 µmol, 10 mg/ml of the potassium salt of losartan in DMF), and
the mixture was heated at 60°C for 3 days during which consumption of the starting drug
was monitored by analytical HPLC. Mass spectrometry analysis confirmed the presence of
the 1:1 losartan-ULS adducts after completion of the reaction.
Analysis of the product, losartan-ULS, using mass spectrometry, yielded the following
data:
1H NMR of Losartan-ULS (CD3OD): δH 0.79 (m, 3H, CH3), 1.25 (m, 2H, CH2CH3), 1.48
(m, 2H, CH2CH2CH3), 2.50 (m, 6H, CH2CH2C and CH2NH2), 4.43 (m, 1.8H, NCH2C),
5.18 (m, 2.2H, CH2OH and remaining NCH2C), 5.42 (m, 4H, NH2); cyclic Hs: 6.82 (m,
0.2 H), 6.87 (m, 1.8 H), 7.04 (t, J = 8.06 Hz, 1H, CHCHCH), 7.18 (m, 0.5H), 7.28 (m,
1H), 7.38 (m, 3H), 7.88 (m, 0.5 H).
195Pt NMR of Losartan-ULS (CD3OD): -2491 and -2658 ppm.
MS (ESI+) m/z: 695 [M-K+-OH--H]+, 677 [M-K+-Cl--H+]+, 659 [M-K+-Cl--H+-Cl-+OH-
]+.
M6PHSA was prepared and characterized as described previously (19). To conjugate
M6PHSA to losartan-ULS, 10 mg M6PHSA (14.3 nmol) was dissolved in 1 ml of 20 mM
tricine/NaNO3 buffer pH 8.5.

103
Chapter 5
SHAM
group
CCL4
+ control
saline
CCL4
+losartan-
M6PHSA
CCL4
+ IV
losartan
CCL4
+M6PHSA
Number of rats 5 3 3 3 3
Body weight (g) 472.17± 17.8 375.3 ± 29.8 379.7 ± 4.5 367.3 ± 28.4 396.7 ± 48.6
Administration route
----- IV IV IV IV
Equivalent
losartan
----- ----- 0.3 mg/kg 0.3 mg/kg ----- Dosage
Equivalent protein
----- ------ 8 mg/kg ----- 8 mg/kg
Liver/body ratio
(g/g)
0.037 ±
0.003
0.034 ±
0.001
0.036 ±
0.004
0.037 ±
0.002
0.038 ±
0.004
Bilirubin (umol/l)
0.1 ± 0.06 0.2 ± 0.01 0.3 ± 0.2 0.2 ± 0.01 0.2 ± 0.01
AST (U/l) 67.8 ± 12.28 122.3 ± 8.4 117.3 ± 24.5 121.0 ± 11.4 97.3 ± 10.1
ALT (U/l) 54.5 ± 10.0 94.3 ± 8.5 75.0 ± 14.0* 101.0 ± 12.7 67.3 ± 8.0
Table 2. Dosage regimens, animal data and fibrotic parameters
Data are shown as mean ± SD.CCL4, carbon tetrachloride ; AST, aspartate aminotransferase. ALT, Alanin aminotransferase. * p< 0.05 vs Losartan.
Chapter 5

104
Losartan-M6PHSA in CCL4 model of liver fibrosis
Losartan-ULS (143 nmol) was added in 10-fold molar excess at pH 8. The mixture was
reacted overnight at 37°C, and purified by dialysis against PBS at 4°C. The final product
was sterilized by filtration and stored at -20°C. Protein content of the conjugates was
assessed by the BCA assay (Pierce, Rockford, IL). The amount of losartan coupled to the
carrier was analyzed by HPLC after competitive displacement of the drug from the
conjugate by excess of potassium thiocyanate (KSCN) as described before (20). ULS
content per losartan-M6PHSA conjugate was evaluated by inductive coupled plasma –
atomic emission spectroscopy (ICP-AES) at 214.424nm and at 265.945 nm with a VISTA
AX CCD Simultaneous ICP-AES (Varian, Palo Alto, USA). Standards (cisPlatinum) and
unknown samples were spiked with Yttrium as an internal standard (360.074 nm).
Free losartan and losartan-ULS were analyzed by reverse-phase HPLC on a Waters system
(Waters, Milford, MA, USA) equipped with an UV detector operated at 269 nm and a
thermostated column oven operated at 40°C. Elutions were performed on a µBondapak
Guard-pak C18 precolumn in combination with a 5 µm Hypersil BDS C8 column
(250x4.6 mm, Thermoquest Runcorn, UK) using a mobile phase consisting of
acetonitrile/water/trifluoracetic acid (40/60/0.1, pH 2).
Evaluation of losartan-M6PHSA in the CCL4 inhalation model of liver fibrosis
The liver fibrosis model of CCL4 inhalation was used to induce liver inflammation and
fibrosis as described previously (21) in 250 g Male Wistar rats (Harlan, Zeist, The
Netherlands). Rats were subjected to CCl4 inhalation during 8 weeks and and
phenobarbital administration in drinking water (0.3g/l).
In the 9th week, rats were treated with four consecutive daily intravenous injections of
saline, losartan-M6PHSA (8 mg/kg, corresponding to 0.3 mg losartan/kg), M6PHSA
alone (8 mg/kg), or free of losartan (0.3 mg losartan/kg). Compounds were administered
under anesthesia with isoflurane (2% isoflurane in 2:1 O2/N2O, 1 L/min) (Abbot
Laboratories Ltd., Queensborough, UK). Ten minutes after the last injection, animals
were sacrificed and blood and liver samples were obtained. Animal procedures were
approved by the Committee for Care and Use of Laboratory Animals of the Hospital
Clínic, Barcelona.

105
Chapter 5
Serum b ochemical measurements i
i
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin
levels were measured using standard enzymatic procedures by the Hospital Clinic,
Barcelona.
Presence of M6P/IGFII receptor in CCL4 induced fibrotic livers
The presence of M6P/IGFII receptor was demonstrated by immunostaining on frozen
liver sections (4µM) using an anti-M6P/IGFII antibody (K-21, sc-14413, IGF-II R, Goat
polyclonal IgG, Santa Cruz Biotechnology) (19).
Analysis of losartan-M6PHSA b odistribution
The presence of losartan-M6PHSA or M6PHSA was demonstrated by immunostaining
using an anti-HSA antibody (Cappel ICN Biomedicals, Zoetermeer, The Netherlands), as
described elsewhere (22). The co-localization of losartan-M6PHSA with HSC was
assessed by double immunostaining of anti-HSA and anti-desmin (Sigma), a specific
marker for rat HSC, as described previously (23).
Figure 1. Detection of M6P/IGF II receptor in rat liver with hepatic fibrosis
induced by CCl4 inhalation. A. General view, magnification of 20x, B. M6P/IFGII receptor
staining in a sinusoidal pattern, magnification of 40x.
Chapter 5

106
Losartan-M6PHSA in CCL4 model of liver fibrosis
The amount of losartan in liver tissue homogenates (0.3 g liver/ml of PBS, prepared by
Turrax homogenization) was analyzed by HPLC-UV methods. Two different procedures
were employed to extract losartan from tissue homogenates. The first method consisted in
direct extraction of losartan from liver homogenates, whereas with the second method the
extraction of losartan was preceded by the incubation of tissues overnight with KSCN
80ºC, in order to release losartan from the conjugate (24), and subsequent liquid–liquid
extraction of losartan using methyl butyl ether (Riedel-de Haen, Netherlands). The
extraction was performed by adding 3 ml of methyl butyl ether to 200 µl of liver
homogenate and vortexing for 5 min. Layers were separated by centrifugation at 900 × g
for 5 min and the aqueous layer was frozen in liquid nitrogen. The upper organic layer was
transferred to another borosilicate glass tube and evaporated completely at 60 °C. The
extraction procedure was repeated twice and the total residue was reconstituted in 200 µl
of mobile phase. Ten µl of the reconstituted sample was injected into the HPLC system.
The liver homogenate samples were analyzed at the sensitivity of 0.01 AUFS.
Chromatography was carried out using a C18 (C18, 5µm, 4.6x150mm) reversed-phase
column (Sunfire, Waters Inc., Milford, MA, USA) at 40ºC with an isocratic mobile phase
consisting of acetonitrile–water–trifluoroacetic acid (30:70:0.1, v/v/v; pH 2.0) at a flow
rate of 1ml/min. Losartan concentration in liver tissue was measured at 225 nm at a
retention time of 7.4 min. Peak-height ratios of the drug plotted in a standard calibration
curve were used for the quantification of losartan from the different matrixes.
Quantification of collagen accumulation and detec ion of activated
myofibroblast cells
t
Hepatic collagen deposition was estimated by assessment of the percentage of area stained
with picro Sirius Red on paraffin-embedded liver sections (Sirius Red F3B, Gurr-BDH
Lab Supplies, Poole, England). The amount of fibrogenic myofibroblasts was estimated by
measuring the percentage of area stained with anti-smooth muscle-α actin on paraffin-
embedded liver sections (αSMA, DAKO, Carpinteria, CA). For morphometric assessment
of percentage of area with positive staining, an optic microscope (Nikon Eclipse E600)
connected to a high-resolution camera (CC12 Soft-Imaging System, Münster, Germany)

107
Chapter 5
was used. Images were analyzed with an automated image-analysis system (AnalySIS, Soft-
Imaging System, Münster, Germany). Images were captured following automatic white
balance and light intensity equilibration with a 40 × magnification objective and digitized
as RGB 24-bit. Each optical image size at 40x was 88752 µm2 for a 250 x 250 square pixel
image, resulting in an optical resolution of 1,42 µm2/pixel. Image reconstruction was
performed using the Multiple Image Alignment. After shading correction and interactive
thresholding, the selected positive pixels were measured. The positive area was the sum of
the area of positive pixels. The ratio was calculated as the area of positive pixels divided by
the total area of the biopsy. Results are given as percentage of positive area.
Analysis of hepatic gene expression
RNA was isolated from frozen liver samples using Trizol (Life Technologies Inc.,
Rockville, MD). Quantitative PCR was performed with pre-designed Assays-on-Demand
TaqMan probes and primer pairs for rat collagen α1 (II), tissue inhibitor of
metalloproteinase 1 (TIMP-1) and ribosome subunit 18s (Applied Biosystems, Foster City,
CA). Information on these Assays-on-Demands is available at:
http://myscience.appliedbiosystems.com/cdsEntry/Form/gene_expression_keyword.jsp.
TaqMan reactions were carried out in duplicate on an ABI PRISM 7900 machine (Applied
Biosystems).
Statistical analysis
Results are expressed as the mean±sd. Significance was established using t-test.
Differences were considered significant if P<0.05.
Chapter 5

108
Losartan-M6PHSA in CCL4 model of liver fibrosis
Results
Synthesis of Losartan-M6PHSA conjugate
Losartan-M6PHSA was successfully synthesized in a straightforward protocol that did not
require extensive purification steps of intermediate products. An average of seven
molecules of losartan were coupled to M6PHSA as assessed by HPLC studies and
confirmed by FAAS analysis of the ULS linker (Table 1). The net negative charge of the
modified protein, reflected by the increased retention time after anion exchange
chromatography, was not changed in Losartan-M6PHSA in comparison with M6PHSA.
Moreover, the size exclusion chromatography showed the existence of a monomeric
preparation of losartan-M6PHSA with absence of protein aggregations (Table 1).
tFigure 2. Organ distribution of losar an-M6PHSA.
Localization of Losartan-M6PHSA in different organs was demonstrated by anti-HSA
immunostaining. Colocalization of losartan-M6PHSA and HSC was detected by anti-HSA/ anti-
DESMIN double immunostaining.
A. Anti-HSA immunostaining in rat liver. B. Colocalization of losartan-M6PHSA with HSC within
a fibrotic liver. Arrows indicate the colocalization of red color, presence of carrier, with blue color, desmin,
a marker of activated HSC. 40x magnification.

109
Chapter 5
Lung (C), heart (D), spleen (E) and kidney (F) sections stained with anti-HSA after Losartan-
M6PHSA injection in CCl4 treated animals. Images were obtained at 20x magnification per organ
tissue.
Clinical and biochemical parameters
The induction of hepatic fibrosis by administration of CCl4 together with oral
phenobarbital was associated with increases in body weight and serum liver enzymes (AST
and ALT), as compared with the sham group. ALT was significantly decreased by the
treatment with losartan-M6PHSA in comparison with losartan alone (Table 2). Plasma
bilirrubin levels indicated normal levels in all groups and there were no changes in the
liver/body ratio.
M6P/IGF II receptor expression in fibrotic liver induced by CCl4
First, we investigated the presence of M6P/IGFII receptor in liver sections at 9 weeks of
inhalation of CCL4. We observed the staining for the receptor throughout the fibrotic
liver, mainly associated extracellularly with hepatic stellate cells in a non-parenchymal
distribution pattern (Figure 1). Receptor expression was also observed in hepatocytes, but
this receptor was present intracellularly. Previous studies showed that this cell type did not
bind the carrier M6PHSA (19).
Chapter 5

110
Losartan-M6PHSA in CCL4 model of liver fibrosis
Accumulation and distribution of losartan-M6PHSA in the fibrotic liver
Previous studies in our group have demonstrated that M6PHSA accumulated rapidly and
efficiently in various stages of liver fibrosis induced by bile duct ligation (22;25). The
fibrotic process in the CCl4 inhalation model has completely different characteristics from
the BDL model, both in underlying pathophysiology and speed of progression. Since this
may affect the extent of M6PHSA accumulation in the liver, we investigated the organ
distribution of losartan-M6PHSA by immunohistochemical techniques and by HPLC
analysis of losartan. To be able to detect the cellular distribution of losartan-M6PHSA in
tissue sections of the organs, rats were sacrificed 10 minutes after the last administration
of the conjugate. We observed a preferential distribution of losartan-M6PHSA to the
fibrotic liver (Figure 2A), whereas the staining in the other organs was negative (Figure
2C-F). Within the fibrotic liver, losartan-M6PHSA displayed an HSC-like distribution
pattern, as demonstrated by colocalization of carrier (anti-HSA staining) and the stellate
cell marker desmin (Figure 2B). From these results we concluded that losartan-M6PHSA
rapidly distributed to the liver 9 weeks after the induction of fibrosis in the CCl4 model,
where it selectively binds and is taken up into HSC.
Quantification of losartan in liver tissue by HPLC
After demonstrating that Losartan-M6PHSA distributed in the CCL4 fibrotic liver, we
evaluated the accumulation of the drug in the liver tissue. For this purpose, tissue levels of
losartan were analyzed by HPLC (Figure 3). The measured losartan levels in livers of rats
treated with losartan-M6PHSA corresponded to 13.2%±6.4% of the cumulative dose.
In contrast, animals that were given free losartan did not show such a preferential liver
accumulation, as only 5.4%±0.2% of the total administrated dose was found in the liver
(P<0.05). These data indicate the efficiency of the drug targeting approach. Since a 25-
fold higher dose of free losartan was administered, higher levels of losartan in the liver
tissue were found compared to losartan-M6PHSA. However, losartan targeted results in a
higher percentage of accumulation after administration, avoiding the body distribution of
the free losartan non-targeted.

111
Chapter 5
0%
5%
10%
15%
20%
25%
Losartan-M6PHSA0,3mg/kg
losartan 8 mg/kg
% o
f los
arta
n of
the
cum
ulat
ive
dose
Figure 3. Quantitation of losartan levels in the livers of CCL4 rats.
Losartan levels in the liver were determined by HPLC as described in the materials and methods section.
The accumulation was related to the cumulative doses administered to the animal.
Losartan-M6PHSA effect on liver fibrosis
Morphometric analysis of hepatic collagen deposition is considered to be the gold
standard for quantification of liver fibrosis (26). We employed Sirius Red staining on liver
tissues for the assessment of fibrous collagen deposition. CCl4 rats treated with either
saline, M6PHSA or losartan showed similar levels of collagen deposition, as shown in
Figure 4-A, C, D. In contrast, rats treated with losartan-M6PHSA displayed a reduction in
the amount of fibrotic area (Figure 4B). Morphometric analysis of the Sirius red staining
showed a significant reduction of the fibrotic area in rats treated with losartan-M6PHSA
compared those treated with saline, losartan or M6PHSA (1.8±0.5%, 3.3±1.3%, 3.2±0.9%
and 3.5±0.1% of positive area, respectively; p<0.05) (Figure 4E).
M6PHSA (2.5±1.1%, positive area per liver area) or losartan (2.6±0.6%), compared to
saline treated (4.9±1.0%) or M6PHSA-treated (3.5±0.1%) fibrotic rats. (p<0.05, Figure 5).
Chapter 5

112
Losartan-M6PHSA in CCL4 model of liver fibrosis
losartan Saline M6PHSAlosartan-M6PHSA
E
*
0
20
40
60
80
100
120
140
CCL4 (9w eeks)control
CCl4 L
Siriu
s R
ed s
tain
ing
(%
pos
itive
are
a)
Figure 4. Liver fibrosis histology af
treated with: A. Saline, B. M6PHSA, C.
stained with Sirius Red. Images represent reco
performed with AnalySIS Image Processing
deposition was analysed per liver area of rat l
M6PHSA and losartan. Quantification
magnification per liver biopsy performed with
losartan-M6PHSA versus CCL4 control, los
OS-M6PHSAIV
CCl4 M6PHSA IV CCl4 losartan IV
ter losartan-M6PHSA treatment. CCL4 rats were
losartan-M6PHSA and D. losartan. Liver sections were
nstruction of 16 images of 4x magnification per liver biopsy
. E. Quantification of Sirius Red positive area. Collagen
iver tissue treated with: control saline, losartan-M6PHSA,
is performed over reconstruction of 16 images of 4x
AnalySIS Image Processing. *p<0.05 significant values of
artan and M6PHSA groups.

113
Chapter 5
Since the fibrogenic process is correlated with transformation of hepatic stellate cells into
myofibroblasts, we investigated the expression of the myofibroblast marker alfa-smooth
muscle actin (SMA) in livers. SMA staining was reduced in rats treated with losartan-
Hepatic gene expression analysis via quantitative RT-PCR
In addition to the protein expression of fibrotic markers, we studied gene expression
levels in treated and untreated fibrotic rats. Analysis of procollagen type 1α2, and TIMP-1
gene expression was conducted to elucidate the antifibrotic mechanism utilized by
losartan-M6PHSA. Procollagen type 1α2 gene expression was reduced by 80% in CCl4
rats treated with losartan-M6PHSA (p<0.001), (Figure 6A) and 70% after losartan
treatment. No effect was observed after free losartan treatment on TIMP-1 gene
expression while an upregulation of TIMP-1 expression after targeted losartan treatment
was observed (Figure 6B).
Chapter 5

114
Losartan-M6PHSA in CCL4 model of liver fibrosis
Discussion
Many studies have demonstrated the antifibrotic effects of RAS blockers during
experimental liver fibrosis. Interference with the renin-angiotensin system may therefore
become a new tool to stop the fibrotic process (27-30). By antagonizing the angiotensin
AT-1 receptor in the fibrotic liver, losartan can prevent the production of fibrogenic
mediators whose expression is enhanced by the activated RAS system (31). During these
studies, losartan was administered during 9 to 12 weeks starting at the time of the
induction of liver injury in the CCL4 animal model (32;33). Such prolonged administration
of losartan was needed in order to achieve a reduction of experimentally induced liver
fibrosis.
We now report on a new preparation, losartan-M6PHSA, which delivers the angiotensin
antagonist losartan selectively to HSC in the fibrotic liver. The potential benefit of such a
conjugate is the increased liver selectivity, thereby lowering systemic side actions of the
drug, which enhances its effect at a relatively low dose. The delivery of losartan to HSC
thus leads to an improved safety and improved efficacy towards the resolution of liver
fibrosis. In the present study we demonstrate that a pronounced effect can be achieved by
losartan-M6PHSA after only 4 days of treatment, in a model in which fibrosis has
progressed for already 9 weeks.
Drug targeting of antifibrotic agents to the liver has been often investigated in the BDL
model. The extrahepatic obstruction of the bile duct leads to an accumulation of excessive
bile salts and other molecules that provoke an inflammatory and fibrogenic process in the
liver and bile duct proliferation (34). The CCL4 model of liver fibrosis is induced by
administration of CCl4 and phenobarbital and is pathogenically completely different from
the BDL model. CCl4 requires bioactivation by cytochrome p450, yielding the reactive
metabolite, the trichloromethyl radical, which initiates lipid peroxidation, resulting in
toxin-induced liver damage provoking extensive liver fibrosis (35). BDL is a rapid and
progressive model (36), while CCL4 model is relatively slow.

115
Chapter 5
Saline losartan-M6PHSA M6PHSA osartan l
E
**
0
1
2
3
4
5
6
7
CCL4 (9w eeks)control
CCl4 LOS-M6PHSA IV
CCl4 M6PHSA IV CCl4 losartan IV
S
MA
Sta
inin
g
( %
pos
itive
are
a)
Figure 5. SMA immunostaining of rat livers.
A. SMA positive cells on rat liver. CCL4 rats were treated with: A. Saline, B. M6PHSA, C. losartan-
M6PHSA and D. losartan and immunostained for α-SMA. Images represent one area of 4x
magnification per liver biopsy performed with AnalySIS Image Processing.
E. Quantification of SMA positive cells per liver area. CCL4 rats were treated with: control (saline),
losartan-M6PHSA, M6PHSA and losartan. Quantification is performed by reconstruction of 16
images of 4x magnification per liver biopsy performed with AnalySIS Image Processing. *p<0.05
significant values of losartan-M6PHSA versus losartan and M6PHSA groups.
Chapter 5

116
Losartan-M6PHSA in CCL4 model of liver fibrosis
Consequently, CCL4 may reflect the human situation better, in which the slow fibrogenic
process can be ongoing for several years before treatment is initiated. The demonstration
of the efficacy of losartan-M6PHSA in the CCL4 model of liver fibrosis thus strengthens
its potential as a new antifibrotic modality.
M6PHSA carriers bind to the M6P/IGII receptor on activated HSC (19). Bleser et al (37)
have demonstrated the expression of the M6P/IGII receptor during the early phases of
the CCL4 model. We have now identified the presence of our target receptor at later
stages, i.e. at week 9 after the onset of liver fibrosis. Subsequent targeting of losartan-
M6PHSA confirmed the cell-selective delivery, showing a rapid accumulation in the CCl4-
fibrotic liver. Furthermore, the association of losartan-M6PHSA with activated HSC
verified the specific delivery of our construct to the key fibrogenic cell during disease.
Specific targeting to the liver is desired during the fibrotic process caused by a disturbed
balance between the portal pressure and the systemic blood pressure in patients, which
eventually may be lethal. There is an enhanced portal pressure while at the same time the
splanchnic blood pressure is reduced. This excludes the use of systemic vasoactive agents
because the reduction of portal pressure should be achieved strictly locally. Losartan acts
locally on the angiotensin AT-1 receptors and has also direct anti-fibrotic effects on HSC,
the most important cell during liver fibrosis (1). These actions render losartan as an ideal
antifibrotic agent, but its systemic vasoactive effects and renal effects probably will
compromise its chronic administration in patients with liver fibrosis.
Drug targeting to specific target cells will increase the fraction of the drug that distributes
to the target cells (38), optimizing drug concentrations in the liver. Our results reveal the
efficacy of the targeting strategy of losartan-M6PHSA to activated HSC during the
experimental fibrosis condition. It has been shown that, apart from the HSC, other
sinusoidal liver cell types, such as Kupffer and endothelial cells, also take up M6PHSA
conjugates (39). As has been previously described, those cells also express the AT-1
receptor and are actively involved during liver fibrosis (40;41). Partial delivery to these
sinusoidal cells might be additionally beneficial.

117
Chapter 5
The most striking result of the present study was the efficiency of losartan-M6PHSA in
preventing extracellular matrix deposition after only 3 days of losartan-M6PHSA
administration in rats with developed fibrosis. Since collagen deposition constitutes the
major component of the extracellular matrix (ECM) (42), the reduction of scar liver tissue
after treatment with losartan-M6PHSA was remarkable. Similar results with losartan-
M6PHSA in BDL rats have been observed (Gonzalo, manuscript in preparation).
A
0.0
0.5
1.0
1.5
CCL4 (9 weeks)control
M6PHSA-Losartan
M6PHSA Losartan
* *
*
Proc
olla
gen α1
mR
NA
(2-∆∆
Ct )
B
TIM
P-1
mR
NA
(2
-∆∆
Ct )
0.0
5
1.0
1.5
2.0
2.5
CCL4 (9w eeks) control
M6PHSA-Losartan
M6PHSA Losartan
*
0.
Figure 6. Hepatic gene expression.
A. Quantification of procollagen α1(II) gene expression. Procollagen α1(II) expression was analyzed in
RT-PCR from rat liver tissues treated with control saline, losartan-M6PHSA, M6PHSA and losartan.
*p< 0.05, vs CCL4 treated animals.
Chapter 5

118
Losartan-M6PHSA in CCL4 model of liver fibrosis
Furthermore, Losartan-M6PHSA treated fibrotic animals showed significant reduction in
mRNA expression of procollagen. This may represent a major aspect of the antifibrotic
effect of losartan in the fibrotic liver. Once present in the liver tissue, losartan acts as an
effective antifibrogenic drug, blocking the proliferation of activated HSC (43). In addition,
the antagonism of AT-1 receptor may prevent the upregulation of the procollagen gene
(44;45) a gene crucial in the fibrogenic process.
Although procollagen gene expression was also downregulated by non-targeted losartan,
collagen protein accumulation was not reduced after this treatment. This contradictory
result may underline the difficulty to generally correlate expression of certain genes with
post-regulation and protein assembly events. However, this may also be explained by
losartan concentration patterns that may largely differ between the losartan-M6PHSA and
non-targeted losartan treatment. As previously hypothesized for pentoxifylline-M6PHSA
conjugates (46), losartan-M6PHSA could act as a depot, intracellularly releasing losartan in
a slow-release manner, thereby providing sustained drug levels during a prolonged period
of time. In contrast, after administration of losartan non-targeted, drug concentrations are
only shortly elevated.
Following oral administration, systemic bioavailability of losartan is limited to 33% (47).
In our previous studies, after oral administration of losartan we did not detect any effect
on procollagen gene expression (Gonzalo, manuscript in preparation). In contrast, we
now observe a reduction of procollagen gene expression after intravenous administration
of losartan, probably due to higher bioavailability of the drug.
Administration of M6PHSA itself also produced a reduction of procollagen gene
expression. By interacting with M6P/IGII receptor, M6PHSA may prevent the processing
of latent TGF-β, locally present in the liver tissue. Thus, the action of TGF-β on its HSC
receptors may also be impeded and procollagen gene activation via this pathway may be
reduced within the stellate cell (48). On the other hand, we observed an opposite effect of
losartan-M6PHSA on TIMP-1 gene expression. Thus, the antifibrotic effect observed by
losartan-M6PHSA in reducing collagen production does not employ TIMP-1 inhibition.

119
Chapter 5
Liver fibrosis is in principle a reversible process in which the stellate cells have been
identified as the key fibrogenic cells (1). Exposed to toxicants, HSC undergo
morphological transition and activation and leading to the production of large amounts of
ECM components (49). Consequently, resolution of stellate cell activation represents an
essential step towards reversion of liver fibrosis. In the present study, losartan-M6PHSA
and non-targeted losartan treatment both led to a significant reduction of activated HSC.
The effects observed in this study by losartan-M6PHSA and non-targeted losartan
indicate a similar effect of both compounds in vivo. However, the efficiency of losartan-
M6PHSA is superior considering the large amount of free losartan administered versus
the low dose of losartan-M6PHSA.
In conclusion, we have demonstrated the homing of losartan-M6PHSA to the CCl4
induced fibrotic liver and clear therapeutic effects during only 3 days of treatment, where
losartan-M6PHSA was superior to non-targeted losartan. The observed effects might
reflect an actual reversion of the fibrosis process. This novel drug delivery concept is
consistent with previous results in another model of liver fibrosis, and in our opinion
warrants further dose-regimen efficacy studies in animal models and humans.
Acknowledgements
This study was supported by grants from SenterNovem (TSGE1083), NWO Science
Netherlands (R 02-1719, 98-162), the Ministerio de Ciencia y Tecnología, Dirección
General de Investigación (SAF2005), and from the Instituto de Salud Carlos III
(CO3/02). We thank Montserrat Moreno and Elena Juez for their kind help in animal
handling and Annemiek van Loenen, Catharina Reker-Smit, Jan Visser and Cristina for
their excellent technical support. Colleagues at Kreatech are acknowledged for critical
reading of the manuscript.
Chapter 5

120
Losartan-M6PHSA in CCL4 model of liver fibrosis
Reference List
1. Friedman,S.L. 2003. Liver fibrosis -- from bench to bedside. J. Hepatol. 38 Suppl 1:S38-S53.
2. Pinzani,M. 1999. Liver fibrosis. Springer Semin. Immunopathol. 21:475-490.
3. Fallowfield,J.A., and Iredale,J.P. 2004. Targeted treatments for cirrhosis. Expert. Opin. Ther. Targets. 8:423-435.
4. Reeves,H.L., and Friedman,S.L. 2002. Activation of hepatic stellate cells--A key issue in liver fibrosis. Front. Biosci. 7:D808-D826.
5. Bataller,R., Gines,P., Nicolas,J.M., Gorbig,M.N., Garcia-Ramallo,E., Gasull,X., Bosch,J., Arroyo,V., and Rodes,J. 2000. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 118:1149-1156.
6. Bataller,R., Gabele,E., Parsons,C.J., Morris,T., Yang,L., Schoonhoven,R., Brenner,D.A., and Rippe,R.A. 2005. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology 41:1046-1055.
7. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Yanase,K., Namisaki,T., Yamazaki,M., Tsujinoue,H., Imazu,H. et al 2003. Angiotensin-II induces the tissue inhibitor of metalloproteinases-1 through the protein kinase-C signaling pathway in rat liver fibrosis development. Hepatol. Res. 27:51-56.
8. Wei,H.S., Li,D.G., Lu,H.M., Zhan,Y.T., Wang,Z.R., Huang,X., Zhang,J., Cheng,J.L., and Xu,Q.F. 2000. Effects of AT1 receptor antagonist, losartan, on rat hepatic fibrosis induced by CCl4. World J. Gastroenterol. 6:540-545.
9. Bataller,R., Sancho-Bru,P., Gines,P., Lora,J.M., Al Garawi,A., Sole,M., Colmenero,J., Nicolas,J.M., Jimenez,W., Weich,N. et al 2003. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125:117-125.
10. Croquet,V., Moal,F., Veal,N., Wang,J., Oberti,F., Roux,J., Vuillemin,E., Gallois,Y., Douay,O., Chappard,D. et al 2002. Hemodynamic and antifibrotic effects of losartan in rats with liver fibrosis and/or portal hypertension. J. Hepatol. 37:773-780.
11. Paizis,G., Gilbert,R.E., Cooper,M.E., Murthi,P., Schembri,J.M., Wu,L.L., Rumble,J.R., Kelly,D.J., Tikellis,C., Cox,A. et al 2001. Effect of angiotensin II type 1 receptor blockade on
experimental hepatic fibrogenesis. J. Hepatol. 35:376-385.
12. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Nakatani,T., Tsujinoue,H., and Fukui,H. 2001. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 34:745-750.
13. Bataller,R., Sancho-Bru,P., Gines,P., and Brenner,D.A. 2005. Liver fibrogenesis: a new role for the renin-angiotensin system. Antioxid. Redox. Signal. 7:1346-1355.
14. Gonzalez-Abraldes,J., Albillos,A., Banares,R., Del Arbol,L.R., Moitinho,E., Rodriguez,C., Gonzalez,M., Escorsell,A., Garcia-Pagan,J.C., and Bosch,J. 2001. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 121:382-388.
15. Beljaars,L., Geerts,A., De Bleser,P.J., Molema,G., Meijer,D.K.F., and Poelstra,K. 1999. The binding and uptake of hepatic stellate cell-selective drug carriers. Hepatology 30:556A (Abstr.)
16. De Bleser,P.J., Scott,C.D., Niki,T., Xu,G., Wisse,E., and Geerts,A. 1996. Insulin-like growth factor II/mannose 6-phosphate-receptor expression in liver and serum during acute CCl4 intoxication in the rat. Hepatology 23:1530-1537.
17. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K. et al 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
18. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K. et al 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
19. Beljaars,L., Olinga,P., Molema,G., De Bleser,P., Geerts,A., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 2001. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver 21:320-328.
20. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K. et al 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.

121
Chapter 5
21. Claria J, and Jimenez,W. 1999. Renal dysfunction and ascites in carbon tetrachloride-induced liver cirrhosis in rats. Pathogenesis, diagnosis and treatment. V.Arroyo, Gines,P., Rodes,J., and Schrier,R., editors. Blackwell Science Inc. Malden.M; USA. USA. 379-396.
22. Beljaars,L., Poelstra,K., Molema,G., and Meijer,D.K.F. 1998. Targeting of sugar- and charge-modified albumins to fibrotic rat livers: the accessibility of hepatic cells after chronic bile duct ligation. J. Hepatol. 29:579-588.
23. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatology 29:1486-1493.
24. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K. et al 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
25. Greupink,R., Bakker,H.I., Reker-Smit,C., van Loenen-Weemaes,A., Kok,R.J., Meijer,D.K.F., Beljaars,L., and Poelstra,K. 2005. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. x:x.
26. Lin,X.Z., Horng,M.H., Sun,Y.N., Shiesh,S.C., Chow,N.H., and Guo,X.Z. 1998. Computer morphometry for quantitative measurement of liver fibrosis: comparison with Knodell's score, colorimetry and conventional description reports. J. Gastroenterol. Hepatol. 13:75-80.
27. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
28. Wei,H.S., Lu,H.M., Li,D.G., Zhan,Y.T., Wang,Z.R., Huang,X., Cheng,J.L., and Xu,Q.F. 2000. The regulatory role of AT 1 receptor on activated HSCs in hepatic fibrogenesis:effects of RAS inhibitors on hepatic fibrosis induced by CCl(4). World J. Gastroenterol. 6:824-828.
29. Croquet,V., Moal,F., Veal,N., Wang,J., Oberti,F., Roux,J., Vuillemin,E., Gallois,Y., Douay,O., Chappard,D. et al 2002. Hemodynamic and antifibrotic effects of losartan in rats with liver fibrosis and/or portal hypertension. J. Hepatol. 37:773-780.
30. Paizis,G., Gilbert,R.E., Cooper,M.E., Murthi,P., Schembri,J.M., Wu,L.L., Rumble,J.R.,
Kelly,D.J., Tikellis,C., Cox,A. et al 2001. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J. Hepatol. 35:376-385.
31. Ramalho,L.N., Ramalho,F.S., Zucoloto,S., Castro-e-Silva Junior, Correa,F.M., Elias,J.J., and Magalhaes,J.F. 2002. Effect of losartan, an angiotensin II antagonist, on secondary biliary cirrhosis. Hepatogastroenterology 49:1499-1502.
32. Croquet,V., Moal,F., Veal,N., Wang,J., Oberti,F., Roux,J., Vuillemin,E., Gallois,Y., Douay,O., Chappard,D. et al 2002. Hemodynamic and antifibrotic effects of losartan in rats with liver fibrosis and/or portal hypertension. J. Hepatol. 37:773-780.
33. Ishizaka,N., Saito,K., Noiri,E., Sata,M., Ikeda,H., Ohno,A., Ando,J., Mori,I., Ohno,M., and Nagai,R. 2005. Administration of ANG II induces iron deposition and upregulation of TGF-beta1 mRNA in the rat liver. Am. J. Physiol Regul. Integr. Comp Physiol 288:R1063-R1070.
34. Nan,J.X., Park,E.J., Lee,S.H., Park,P.H., Kim,J.Y., Ko,G.N., and Sohn,D.H. 2000. Antifibrotic effect of Stephania tetrandra on experimental liver fibrosis induced by bile duct ligation and scission in rats. Arch. Pharm. Research 23:501-506.
35. Chapman,K., Prabhudesai,M., and Erdman,J.W., Jr. 1992. Effects of ethanol and carbon tetrachloride upon vitamin A status of rats. Alcohol Clin. Exp. Res. 16:764-768.
36. Kountouras,J., Billing,B.H., and Scheuer,P.J. 1984. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br. J. Exp. Pathol. 65:305-311.
37. De Bleser,P.J., Jannes,P., Van Buul-Offers,S.C., Hoogerbrugge,C.M., Van Schravendijk,C.F.H., Niki,T., Rogiers,V., Van den Brande,J.L., Wisse,E., and Geerts,A. 1995. Insulinlike growth factor-II/mannose 6-phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transforming growth factor-b in cocultures with sinusoidal endothelial cells. Hepatology 21:1429-1437.
38. Kok,R.J., Ásgeirsdóttir,S.A., and Verweij,W.R. 2001. Development of proteinaceous drug targeting constructs using chemical and recombinant DNA approaches. In Drug Targeting. Organ-Specific Strategies. G.Molema, and Meijer,D.K.F., editors. WILEY-VCH Verlag GmbH. Weinheim. 275-308.
39. Greupink,R., Bakker,H.I., Reker-Smit,C., van Loenen-Weemaes,A.M., Kok,R.J., Meijer,D.K.,
Chapter 5

122
Losartan-M6PHSA in CCL4 model of liver fibrosis
Beljaars,L., and Poelstra,K. 2005. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. 43:884-892.
40. Wei,Y.H., Jun,L., and Qiang,C.J. 2004. Effect of losartan, an angiotensin II antagonist, on hepatic fibrosis induced by CCl4 in rats. Dig. Dis. Sci. 49:1589-1594.
41. Leung,P.S., Suen,P.M., Ip,S.P., Yip,C.K., Chen,G., and Lai,P.B. 2003. Expression and localization of AT1 receptors in hepatic Kupffer cells: its potential role in regulating a fibrogenic response. Regul. Pept. 116:61-69.
42. Pinzani,M., Rombouts,K., and Colagrande,S. 2005. Fibrosis in chronic liver diseases: diagnosis and management. J. Hepatol. 42 Suppl:S22-S36.
43. Gross,O., Schulze-Lohoff,E., Koepke,M.L., Beirowski,B., Addicks,K., Bloch,W., Smyth,N., and Weber,M. 2004. Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol. Dial. Transplant. 19:1716-1723.
44. Bataller,R., Gabele,E., Schoonhoven,R., Morris,T., Lehnert,M., Yang,L., Brenner,D.A., and Rippe,R.A. 2003. Prolonged infusion of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am. J. Physiol Gastrointest. Liver Physiol 285:G642-G651.
45. Qi,Z., Atsuchi,N., Ooshima,A., Takeshita,A., and Ueno,H. 1999. Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc. Natl. Acad. Sci. U. S. A 96:2345-2349.
46. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K. et al 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
47. Oparil,S. 2000. Newly emerging pharmacologic differences in angiotensin II receptor blockers. Am. J. Hypertens. 13:18S-24S.
48. Rodriguez-Barbero,A., Obreo,J., Yuste,L., Montero,J.C., Rodriguez-Pena,A., Pandiella,A., Bernabeu,C., and Lopez-Novoa,J.M. 2002. Transforming growth factor-beta1 induces collagen synthesis and accumulation via p38 mitogen-activated protein kinase (MAPK) pathway in cultured L(6)E(9) myoblasts. FEBS Lett. 513:282-288.
49. Gressner,A.M., Weiskirchen,R., Breitkopf,K., and Dooley,S. 2002. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 7:d793-d807.


2
agra fort with extra anterior page
Diwan-i-Am (Hall of Public Audience), Red Fort of Agra
India, December 2005.

Chapter6Local inhibition of liver fi brosis by specifi c delivery of a PDGF kinase inhibitor to hepatic stellate cells
Submitted for publication.
1,Teresa Gonzalo, 1Leonie Beljaars, 1Marja van de Bovenkamp, 1Anne-Miek van Loenen, 1Catharina Reker-Smit, 1Dirk K.F. Meijer, 2Marie Lacombe, 2Frank Opdam, 3György Kéri, 3László Orfi , 1Klaas Poelstra, 1,4Robbert J Kok.
1Department of Pharmacokinetics and Drug Delivery, Groningen University Institute for Drug Exploration, University of Groningen, The Netherlands;
2Kreatech Biotechnology B.V., Amsterdam;
3Department of Medical and Pharmaceutical Chemistry, Semmelweis University, Budapest, Hungary;
4Department of Pharmaceutics, Utrecht University, The Netherlands.

126
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
Abstract
Liver fibrosis is characterized by an excessive proliferation and activation of Hepatic
Stellate Cells (HSC). Platelet-derived growth factor (PDGF-BB) is the most potent
mitogen for HSC and inhibition of PDGF signalling via specific delivery of a PDGF
kinase inhibitor to HSC might therefore be an attractive strategy to counteract the
progression of liver fibrosis. The HSC-selective carrier M6PHSA was equipped with a
PDGF receptor tyrosine kinase inhibitor (PTKI, an imatinib derivative) by means of a
novel platinum-based linker (ULSTM).
Culture-activated rat HSC and precision-cut liver slices from fibrotic rats were incubated
with PTKI-M6PHSA and fibrosis markers were evaluated by quantitative RT-PCR. The
gene expression of α-smooth muscle actin and α1-(I)-procollagen were reduced by 50%
in both in vitro systems after treatment with PTKI-M6PHSA (0.1 mg/ml, corresponding to
10µM of PTKI) and free PTKI showed similar effects.
Next, we examined the homing and antifibrotic effects of PTKI-M6PHSA in bile duct
ligated (BDL) rats. Male Wistar rats at day 10 after BDL were injected intravenously with a
single dose of 3.3 mg/kg of PTKI-M6PHSA and compared with non-treated BDL rats.
PTKI-M6PHSA was detected in the liver 2h after administration in a non-parenchymal
distribution pattern. The antifibrotic effects of PTKI-M6PHSA were analysed by Sirius
Red and α-smooth muscle actin stainings. Both parameters were reduced 24h and 48h
after the single dose of PTKI-M6PHSA (p<0.01, p<0.05. resp), in comparison with non-
treated rats. We subsequently conducted a multiple dose study administering PTKI-
M6PHSA to BDL rats and untargeted PTKI, where we found no effect of either
treatment.
In summary, PTKI-M6PHSA showed an antifibrogenic effect in cultured HSC and
fibrotic liver slices. This effect was also demonstrated in vivo after direct targeting of the
PDGFR kinase inhibitor to activated HSC during liver fibrosis after single administration.
Upon longer treatment however, PDGF kinase inhibition can not block the fibrotic

Chapter 6
process. We therefore conclude that delivery of a PDGF-kinase inhibitor to HSC is a
promising technology to attenuate liver fibrogenesis, although other activating pathways
may need to be inhibited in parallel.
Theoretical
synthesis ratio Coupling ratio Yield of synthesis#
A
NHl
O
C127
PTKI-ULS: protein
PTKI : protein¶
PTKI-M6PHSA
10:1
8 : 1 (±1.9)
84%
0
100
200
300
400
500
600
700
800
900
10 15 20 25 30
PTKI-M 6PHSA
M 6PHSA
NH
N NCH3
NPt
2+M6PHSA carrier
UV
280
nm
(m
AU
)
MonoQ anion exchange
UV
280
nm
(m
AU
)
0
200
400
600
800
00
1200
1400
00
9 10 11 12 13
HSAM6PHSAPTKI-M6PHSA
16B
10
Size exclusion chromatography
Time (min) Time (min)
Figure 1. Characteristics of PTKI-M6PHSA drug targeting conjugate.
A. Synthesis of PTKI-M6PHSA. PTKI was conjugated via the platinum based linker ULS to the
stellate cell selective carrier M6PHSA. Coordination bonds between drug-linker and linker-carrier are
depicted as dotted lines. ¶ PTKI/M6PHSA coupling ratio was determined by HPLC after competitive
displacement of the drug with potassium thiocyanate (KSCN) at 80°C for 24h. # Based in the BCA
assay or protein concentration. Values obtained from three independent PTKI-M6PHSA synthesized
conjugates. B. MonoQ anion exchange chromatography confirmed that the charge of the protein was not
affected. Size exclusion chromatography showed the monomeric composition of PTKI-M6PHSA.
M6PHSA: mannose-6-phosphate modified human serum albumin; ULS: Universal Linkage System.
Chapter 6

128
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
Introduction
Liver fibrosis is a proliferative disease that may be initiated by a variety of factors
including chronic hepatitis, virus infections, alcohol drinking, and drug abuse. It has been
extensively documented that activated hepatic stellate cells (HSC) play a fundamental role
in the development of liver fibrosis (1;2). During liver fibrosis, activated HSC proliferate
and deposit extracellular matrix proteins, a process that is driven by an array of cytokines
and growth factors. Among these, platelet-derived growth factor (PDGF-BB) has been
identified as the most potent mitogen for HSC (3). Activated HSC produce PDGF (4) and
PDGFR-β receptors are highly upregulated on the cell surface of hepatic stellate cells
during fibrosis (5-7).
Imatinib (STI 571, Gleevec) is employed in the treatment of chronic myelogenous
leukaemia (CML) and gastrointestinal stromal tumors (GISTs) (8). It inhibits several
tyrosine kinases that are mutated during cancer development. In addition, imitanib is a
potent inhibitor of PDGF-B kinase. Consequently, imatinib has been tested for its
antifibrotic effect in cultured HSC (9) and has recently been evaluated in different animal
models of liver fibrosis (10-13). The fundamental role that PDGF signalling appears to
play in liver fibrogenesis has made it an attractive therapeutic target for the treatment of
liver fibrosis (14).
In the present study, we have investigated whether the antifibrotic effects of a PDGF
tyrosine kinase inhibitor (PTKI), a drug structurally associated to imatinib (15), can be
enhanced by local delivery to HSC in the fibrotic liver. PDGF tyrosine kinase activity
plays a role in many more processes than HSC proliferation, so it is reasonable to expect
side effects of its inhibition. Drug targeting strategy can improve the effect of the drug by
increasing local concentrations at the target site and by providing slow local drug release.
It will also prevent side effects in other tissue or organs (16).
To effectuate local delivery and effects within HSC in the liver, we have developed a new
drug targeting construct, PTKI-M6PHSA, in which PTKI is coupled to the HSC-directed
carrier protein mannose-6-phosphate-human serum albumin (M6PHSA). M6PHSA is a

129
Chapter 6
well-established carrier that binds to the M6P/IGFII receptor on HSC and accumulates
rapidly and extensively in the liver of fibrotic rats (17). To conjugate PTKI to M6PHSA,
we have employed a novel type of platinum linker chemistry called ULSTM (Universal
Linker System)(18). ULS allows stable coupling of drug molecules to proteins based on
the formation of a platinum-ligand coordination bond (19). Application of this novel
linker technology was essential since it appears to be straightforward and reliable for
linking PTKI molecules to the carrier, allowing high synthesis yields in a relatively simple
approach. Second, the resulting PTKI-ULS-M6PHSA conjugates display a unique
behavior of slow release of drug molecules during a period of days within the designated
target cells.
In the present study, we describe the development of PTKI-M6PHSA and its impact on
liver fibrogenesis in vitro and in vivo. Culture of HSC and fibrotic liver slices were employed
as in vitro systems to prove the antifibrotic effect of PTKI-M6PHSA. In addition, PTKI-
M6PHSA was tested in a model of liver fibrosis to study the distribution and effects on
the development of liver fibrosis.
Chapter 6

130
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
BDL
day 10
control saline
BDL
day 11
control saline
BDL
day 12
control saline
BDL
day 10
+PTKI-M6PHSA
BDL
day 11
+PTKI-M6PHSA
BDL
day 12
+PTKI-M6PHSA
Number of rats 5 5 5 3 5 5
Body weight (g) 250.2 ± 46.7 311.2 ± 14.7 273.4 ± 16.4 280.7 ± 10.1 301.4 ± 14.1 306.4 ± 13.5
Liver/body ratio (g/g) 0.055 ± 0.009 0.062 ± 0.007 0.065 ± 0.004 0.061 ± 0.002 0.060 ± 0.006 0.065 ± 0.009
Dose Saline Saline Saline 3.3mg/kg 3.3mg/kg 3.3mg/kg
Bilirubin (umol/l) 183.7 ± 35.8 178.0 ± 48.2 228.5 ± 18.7 252.3 ± 55.9 136.0 ± 10.0
183.5 ± 27.9
ALT (U/l) 86.3 ±30.9 79.8 ±46.1 79.3 ±11.2 96.5 ± 4.9 72.7 ±10.6 81.0 ±17.0
Alkaline Phosphatase
(U/l) 391.2 ±46.4 476.0 ±162.6 358.3 ±234.1 458.0 ±11.2 426.0±101.3 511.3±121.4
Table 1. Dose regimens, animal data and biochemical parameters from PTKI-M6PHSA single injection study. Data are shown as mean ± SD. BDL, bile duct ligated animals; ALT, alanine aminotransferase.

131
Chapter 6
Materials and methods
Materials
The Protein Tyrosine Kinase Inhibitor (PTKI, 4-Chloro-N-[4-methyl-3-(4-pyridin-3-yl-
pyrimidin-2-ylamino)-phenyl]-benzamide) was kindly provided by György Kéri (Vichem
Chemie Research Ltd., Budapest, Hungary). M6PHSA was prepared as described
previously (19). Cis-[Pt(ethylenediamine)nitrate-chloride] (cisULS) was prepared as
previously described (19).
Synthesis of PTKI-ULS-M6PHSA
PTKI-ULS was synthesized and purified by Kreatech Biotechnology (Amsterdam, The
Netherlands). In brief, PTKI (7.2 µmol, 3 mg; 10 mg/ml in DMF) was mixed with an
equimolar amount of cisULS (7.2 µmol, 2.4 mg; 20 mM in DMF). The reaction mixture
was heated at 37°C for 24h after which consumption of the starting material was
monitored by analytical HPLC. An additional amount of cisULS was added (0.5
equivalent, 3.6 µmol) and the reaction was continued for 48h at 37°C. The crude mixture
was concentrated under reduced pressure and dissolved in methanol (600 µl). The crude
product was purified by preparative HPLC and the collected peaks of the main product
were taken to dryness under reduced pressure. The resulting white solid was treated with
water to remove anorganic salts and dried. Yield: 0.9 mg (20%). Mass spectrometry
analysis confirmed the presence of the 1:1 PTKI-ULS species.
1H NMR of PTKI (CD3OD): δH 2.33 (s, 3H, CH3), 7.26 (d, J = 8.28 Hz, 1H, CCH3CH),
7.37 (m, 2H, CHCl), 7.52 (m, 3H, N(CH)2CCCH), 7.93 (d, J = 8.60 Hz, 2H, CHCHCl),
8.22 (s, 1H, NHCCHC), 8.47 (d, J = 5.23 Hz, 1H, CHNCNH), 8.64 (m, 2H, CH(CH)2C
and CHCHCNH), 9.29 (s, 1H, NCHC) ppm.
1H NMR of PTKI-ULS (CD3OD): δH 2.27 (m, 3H, CH3), 2.66 (m, 2H, CH2), 2.74 (m,
2H, CH2), 7.15 (m, 3H, CHCCH3 and CHCl), 7.47 (m, 3H, N(CH)2CCCH), 7.84 (d, J =
5.23 Hz, 1H, CHCHCNH), 7.89 (m, 2 H, CHCHCl), 8.39 (d, J = 3.95 Hz, 1H,
CHNCNH) ppm.
Chapter 6

132
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
Mass spectrometry of PTKI-ULS (ESI+): calculated mass: 705 (m/z); detected masses:
706 [M]+, 688 [M-Cl-+OH-]+, 670 [M-Cl--H+]+, 669 [M-Cl--H+]+, 651 [M-Cl--Cl-+OH-
+H]+.
HPLC analysis: Separations were performed on a Luna2 C18 column that was maintained
at 40°C. The mobile phase consisted of a binary solvent system of triethylammonium
acetate (100mM pH 5.0):acetonitrile 90:10 (solvent A) and triethylammonium acetate
(100mM pH 5.0):acetonitrile 70:30 (solvent B). The column was eluted at a flow rate of
1.1 mL/min. Compounds were eluted at a stepwise gradient (0%B from 0-4 min; 0-46%B
from 4-17 min; 46-100%B from 17-19 min; 100%B from 19-25 min; 100-0%B from 25-
27 min; 0%B from 27-34 min). PTKI eluted at 21.2 min (60.8%B) and PTKI-ULS eluted
at 11.5 min (26.5%B).
PTKI-ULS was conjugated to M6PHSA according to a general protocol that has been
described elsewhere for the synthesis of Pentoxifylline-ULS-M6PHSA (19). Briefly,
PTKI-ULS (143 nmol, 1.6 mg that was dissolved in DMF/H2O at 6.7 mg/ml) was added
in 10-fold molar excess to M6PHSA (14.3 nmol, 10 mg, dissolved in 1 ml of 20 mM
tricine/NaNO3 buffer pH 8.3). The pH was checked and adjusted to pH 8 if necessary.
The mixture was incubated overnight at 37°C, and dialysed against PBS at 4°C. The final
product was sterilized by filtration via a 0.2 µm filter and stored at -20°C. Protein content
was assessed by the BCA assay (Pierce, Rockford, IL, USA). PTKI-M6PHSA and
M6PHSA were analyzed by size-exclusion chromatography and anion exchange
chromatography as described before (17) to verify that coupling of PTKI-ULS did not
alter the properties of the M6PHSA protein. The amount of PTKI coupled to M6PHSA
was analyzed by isocratic HPLC after competitive displacement of the drug by overnight
incubation at 80°C with excess of potassium thiocyanate (KSCN, 0.5M in PBS). Elutions
were performed on a Waters system (Waters, Milford, MA, USA) equipped with a 5 µm
Hypersil BDS C8 column (250x4.6 mm, Thermoquest Runcorn, UK), a thermostated
column oven operated at 40°C and an UV detector operated at 269 nm. The mobile phase
consisted of acetonitrile/water/trifluoracetic acid (40/60/0.1, pH 2) at a flow rate of 1.0
ml/min with a sensitivity of 0.01. Retention times: PTKI: 7 min; PTKI-ULS: 5 min.

133
Chapter 6
Figure 2. Activated HSC incubated with PTKI-M6PHSA.
A. Effect of PTKI-M6PHSA and PTKI on HSC cell viability, as determined by Alamar Blue viability
assay. Indicated concentrations reflect the platinum content of the conjugate, or equivalent amounts
cisplatin, PTKI or M6PHSA. Cultured HSC were incubated for 24h with the compounds (*P<0.01).
A. Effect of PTKI-M6PHSA and PTKI on HSC cell viability, as determined by Alamar Blue viability
assay. Indicated concentrations reflect the platinum content of the conjugate, or equivalent amounts
cisplatin, PTKI or M6PHSA. Cultured HSC were incubated for 24h with the compounds (*P<0.01).
B. Activated HSC gene expression after incubation with PTKI-M6PHSA, PTKI and M6PHSA.
Concentrations denote the amount of PTKI (10 µM) or the corresponding amount of M6PHSA carrier
(0.1mg/ml). Gene expressions levels were normalized to the expression of GAPDH and subsequently
normalized to the relative expression of control cells (*P<0.01 and #P<0.05).
B. Activated HSC gene expression after incubation with PTKI-M6PHSA, PTKI and M6PHSA.
Concentrations denote the amount of PTKI (10 µM) or the corresponding amount of M6PHSA carrier
(0.1mg/ml). Gene expressions levels were normalized to the expression of GAPDH and subsequently
normalized to the relative expression of control cells (*P<0.01 and
Activated HSC
Activated HSC
cisPlatinum 100µM
cisPlatinum 100µM
PTKI PTKI -M6PHSA -M6PHSA
100µM 100µM PTKI PTKI 100µM 100µM
M6PHSA 100µM
M6PHSA 100µM
Stel
late
cel
l via
bilit
y (A
lam
ar b
lue)
St
ella
te c
ell v
iabi
lity
(Ala
mar
blu
e)
**
0
20
40
60
80
100
120A
B
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
HSCcontrol PTKI-M6PHSA 10µM(0.1mg/ml)
PTKI 10uM M6PHSA 0.1mg/ml
GAPDH TIMP-1 PDGFR-B Collagen 1a2 SMA
*
#
**
Rel
ativ
e ge
ne e
xpre
ssio
n
#P<0.05).
Chapter 6

134
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
Cells
Hepatic stellate cells were isolated from male Wistar rat livers by the pronase-collagenase
method followed by a density centrifugation on a 12% Nycodenz gradient according to
Geerts et al (20;21)(Hepatol 1998). The isolated stellate cell fraction was cultured in 6 well-
plates (Corning) in Dulbecco’s Modified Eagle’s Medium (Gibco, Life technologies Ltd.)
supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 mg/ml
streptomycin in a 5% CO2 humidified atmosphere at 37°C. Cells were split after 3 days
and cultured until day 10 to obtain the activated HSC phenotype, and these culture
activated HSC were used for the experiments described below.
Cell viability studies
Activated HSC (10,000 cells/well seeded in 96 well-plates, Corning) were washed with
serum-free medium and incubated for 24h in medium supplemented with 100 µM PTKI,
PTKI-M6PHSA (1mg/ml, corresponding to 100 µM of PTKI) or M6PHSA (1mg/ml).
During the last 2h of the incubations, Alamar blue reagent (Serotec, Oxford, UK) was
added in a proportion of 10% of the volume per well. Viability of the cells was determined
fluorimetrically according to the supplier’s instructions.
Effects on gene expression
The potential antifibrotic activity of PTKI-M6PHSA and PTKI was evaluated in cultured
HSC and in precision-cut liver slices of fibrotic rat livers.
Activated HSC were incubated as described above with PTKI (10 µM), PTKI-M6PHSA
(0.1mg/ml, corresponding to 10 µM of PTKI), or M6PHSA (0.1 mg/ml) for 24h, after
which they were processed for RNA analysis as described below. A total of four
independent experiments were performed in HSC cultures from four different Wistar
Male rats.
Precision-cut liver slices were prepared as described elsewhere (22;23). Briefly, precision-
cut liver slices (8mm diameter, 250µm) from fibrotic livers of BDL3 rats (week 3 after
BDL) were prepared using a Krumdieck tissue slicer and stored in University of
Winconsin preservation solution (UW) on ice until further use.

135
Chapter 6
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
BDL liver slice + PBS PTKI-M6PHSA 10µM(0.1mg/ml)
PTKI 10uM M6PHSA 0.1mg/ml
GAPDH TIMP-1 PDGFR-B Collagen 1a2 SMA
*#
* *
#
**
Rel
ativ
ege
neex
pres
sion
Figure 3. Effect on gene expression of PTKI-M6PHSA on BDL3 liver slices.
Concentrations denote the amount of PTKI (10 µM) or the corresponding amount of M6PHSA carrier
(0.1mg/ml). Gene expression levels of control slices were normalized versus GAPDH and subsequently
normalized versus the expression levels in control fibrotic liver slices (*P<0.01 and #P<0.05).
Slices were preincubated for 2 h in William’s medium E (Gibco, Life technologies Ltd.,
Paisley, Scotland, UK) supplemented with D-glucose (25mM) and gentamycin (50 mg/ml)
and saturated with 95% O2, 5% CO2 at 37°C. Slices were transferred into fresh medium
and incubated individually in six-well plates with PTKI-M6PHSA (0.1 mg/ml,
corresponding to 10 µM of PTKI), PTKI (10 µM) or M6PHSA (0.1 mg/ml). After 24 h
of incubation, slices were snap-frozen in liquid nitogen (real-time PCR analysis). Each
measurement was performed on three liver slices from the same liver and the experiment
was repeated on livers from three different BDL3 rats.
Chapter 6

136
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
Animal experiments
All animal studies were approved by the local committee for care and use of laboratory
animals at Groningen University, and were performed according to strict governmental
and international guidelines on animal experimentation. Animals had free access to tap
water and standard lab chow and were housed in a 12h/12h light/dark cycle. All the
animals included in these studies were monitored by analysis of body weight (BW) and
biochemical parameters reflecting liver functions like serum bilirubin levels, AST, ALT,
AP and gamma-GT levels. These analyses were performed at the University Medical
Center Groningen by standard biochemical procedures.
BDL model
Liver fibrosis was induced in male Wistar rats (250 g, Harlan, Zeist, The Netherlands) by
bile duct ligation as described previously (24). Briefly, rats were anesthesized with
isoflurane (2% isoflurane in 2:1 O2/N2O, 1 L/min) (Abbot Laboratories Ltd.,
Queensborough, UK). After midline laparotomy, the common bile duct was ligated by
double ligature with 4-0 silk and transected between the two ligations. Animals were
allowed to recover and carefully observed until final sacrifice at the end of the
experiments.
Distr bution study i
At day 10 after BDL, rats received a single intravenous injection of PTKI-M6PHSA (3.3
mg/kg, corresponding to 150 µg PTKI/kg). Control animals were injected with an
equivalent volume of the vehicle (saline, 250 µl). Animals were sacrificed 2h post injection
of the compounds. Organs were harvested and processed for immunohistochemical
detection of the conjugate as described below.
Single dose effect study
At day 10 after BDL, rats received a single intravenous injection of 3.3 mg/kg PTKI-
M6PHSA or saline. Animals were sacrificed day 11 or 12 post injection of the
compounds. Blood and organs were harvested and processed for analysis of serum
markers, RNA isolation and immunohistochemical analysis as described below.

Chapter 6
A PTKI-M6PHSA after 2h Control saline
100µM 100µM
Figu
Immu
2hour
Immu
points
Mult
Starti
M6PH
saline
subse
cyclo
1M H
every
(24h
137
100µM 100µM
100µM 100µM
re 4. Localization of PTKI-M6PHSA in different organs and liver.
nostaining with Anti-HSA of different organs from rats treated with PTKI-M6PHSA after
s of PTKI-M6PHSA single administration. A. liver; B. heart; C. kidney; D. lung; E. spleen.
nostaining with Anti-HSA of liver tissue from rats treated with PTKI-M6PHSA at different time
after single administration: control saline; 2h; 24h; 48h. Magnification, 20x.
iple dose study
ng at day 10 after BDL, rats received four daily intravenous injections of PTKI-
SA (3.3 mg/kg, corresponding to 150 µg PTKI/kg), free PTKI (150 µg/kg) or
. PTKI was dissolved first in DMSO at a concentration of 15 mg/ml and
quently diluted to a concentration of 0.25 mg/ml with Hydroxy propyl-B-
dextrin (ENCAPSIN, Janssen Biotech 30-222-55) 20% w/v (pH adjusted to 3.0 with
Cl). The final dosage of DMSO was less than 0.05%. Animals were treated again
24h, and received a total of 4 doses until they were sacrificed at day14 after BDL
post injection of the last dose). A control group of animals was sacrificed at day 10,
Chapter 6

138
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
i.e. just before start of the treatments. Blood and organs were harvested and processed
similar as for single dose effect studies.
i
BDL day 10
control saline
BDL day 14
control saline
BDL day 14
+ PTKI-M6PHSA
BDL day 14
+ PTKI
Number of rats 5 6 5 6
Body weight (g) 250.2 ± 46.7 290.4 ± 9.6 273.4 ± 16.4 280.7 ± 10.1
Liver/body ratio (g/g)
0.055 ± 0.009 0.060 ± 0.009 0.063 ± 0.011 0.060 ± 0.004
Dose saline saline
150 ug/kg (PTKI conc) or
3.3mg/kg (prot conc)
150 ug/kg
Bilirubin (umol/l)
183.7 ± 35.8 152.0 ± 14.2 187.4 ± 35.7 156.5 ± 32.8
ALT (U/l) 86.3 ±30.9 118.5 ±46.9 117.6 ±32.3 121.3 ±34.2
Alkaline Phosp
(U/l) 391.2 ±46.4 405.2 ±46.0 478.6 ±132.9 410.0 ±91.2
Table 2. Dose regimens, animal data and biochem cal serum parameters from
PTKI-M6PHSA multiple administration study. Data are shown as mean ± SD. BDL, bile
duct animals.
RNA isolation and gene expression analysis
Total RNA was isolated from cells or liver slices using an RNA isolation kit (Qiagen
GmbH, Hilden,Germany) according to the manufacturer’s procedures. Isolation of RNA
from liver tissue was performed using Trizol (manufacturer). The amount of RNA was
estimated with a Nanodrop system (ND-1000 Spectrophotometer, NanoDrop
Technologies, Wilmington, DE). cDNA was synthesized from similar amounts of RNA

139
Chapter 6
using Superscript III first strand synthesis kit (Invitrogen life technology, Carlsbad, CA).
The reverse transcriptase reaction was performed for 5min at 65°C, 10 min at 25°C, 50
min at 50°C and 5 min at 85°C with random primers. Quantitative Real-time RT-PCR was
performed in duplicate on a ABI 7900HT system (Applied Biosystems, Foster City, CA)
using SYBR green primers. 1µl of cDNA solution corresponding to approximately 0.5µg
was added in each PCR reaction as well as 10µl of SYBR Green PCR Master Mix (Applied
Biosystems) and the forward and reverse primers (50 µM). For each sample, 1 µl of
cDNA was mixed with 0.4 µl of each gene-specific primer (50 µM), 0.8 µl DMSO, 8.4 µl
water and 10 µl SYBR Green PCR Master Mix. The housekeeping gene Glyceraldehyde-3-
phosphate dehydrogenase (GAPDH) was used as a reference gene and for normalization
of the other genes, while ultrapure water was used as a negative control.
The following primers were used to investigate antifibrotic responses: rat smooth muscle
α actin forward, 5'-GACACCAGGGAGTGATGGTT -3’, reverse, 5'-
GTTAGCAAGGTCGGATGCTC-3’ (product size 202 bp); rat collagen type 1 a1
(collagen 1 a1) forward, 5’-AGCCTGAGCCAGCAGATTGA-3’, reverse, 5’-
CCAGGTTGCAGCCTTGGTTA-3’, (product size 145 bp); rat PDGFR-β forward, 5'-
TGTTCGTGCTATTGCTCCTG -3’, reverse, 5'-TCAGCACACTGGAGAAGGTG -3’,
(product size 201 bp); rat timp-1 forward, 5'-GAGAGCCTCTGTGGATATGT -3’,
reverse, 5'-CAGCCAGCACTATAGGTCTT -3’, (product size 334 bp); and rat GAPDH
forward 5’-CGCTGGTGCTGAGTATGTCG-3’, reverse, 50-CTGTG
GTCATGAGCCCTTCC-30, (product size 179 bp). PCR reactions consisted of 40 cycles
(denaturation 15 s 95°C, annealing 15 s 56°C, extension 40 s 72°C). The formation of
single products was confirmed by analyzing the dissociation step at the end of each PCR
reaction.
Analysis of hepatic gene expression
RNA was isolated from frozen liver samples using an RNA isolation kit (Qiagen GmbH,
Hilden,Germany). Quantitative PCR was performed as described above with primers for
rat collagen α1 (II), smooth muscle α-actin, PDGFR-β, tissue inhibitor metalloproteinase
type 1 (TIMP-1) and GAPDH (Applied Biosystems, Foster City, CA). Real-time PCR was
Chapter 6

140
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
carried out on an ABI PRISM 7900HT. Data were analyzed with the SDS 2.1 software
program (Applied Biosystems). The relative amount of the designated PCR product was
calculated by the comparative threshold cycle (CT) method and referred to control
treatment.
Immunohistochemical analyses
Cryostat sections (4 µm) of liver, heart, kidney, lung and spleen were acetone-fixed and
stained for the presence of the PTKI-M6PHSA conjugate with an antibody directed
against HSA (Cappel ICN Biomedicals, Zoetermeer, The Netherlands) as described
elsewhere (24).
The degree of hepatic fibrosis was estimated as the percentage of area of each section
(4µm) stained positive with picro Sirius Red in saturated picric acid (both from Sigma).
The amount of fibrogenic myofibroblasts was estimated by measuring the percentage of
area stained with anti-smooth muscle α actin (αSMA, Sigma, Gillingham, UK) in 10
randomly selected high power fields per sample. An optic microscope (Olympus BX40)
connected to a high-resolution camera (Olympus Camedia C-5050 zoom) was used for
morphometric assessment of percentage of area with positive staining. Microphotographs
were taken at an original magnification of 4x10. The fibrotic area per liver section was
quantified by morphometric analysis of the sections using the Image J software package
(NIH, Bethesda, ML, USA) and images were captured following automatic white balance
and light intensity equilibration with a 40 × magnification objective and digitized as RGB
24-bit. After shading correction and interactive thresholding, the selected positive pixels
were measured. The positive area was the sum of the area of positive pixels per liver
biopsy. Results were calculated as the average area of positive pixels per liver biopsy and
divided into each group of animal treatment.
Statistical analysis
Results are expressed as the mean of at least three independent experiments ± SD, unless
otherwise indicated. Statistical analysis was performed with an unpaired Student’s t-test
and differences were considered significant at p<0.05.

141
Chapter 6
Results
Synthesis and characterization of PTKI-M6PHSA conjugate
When observing the structure of PTKI (Figure 1A) or the related kinase inhibitor
imitanib, it is clear that the structure is mainly composed of aromatic rings lacking
functional groups that can be used for drug linking purposes. In general, drug conjugation
protocols often start with the preparation of a drug-linker adduct at carboxyl, hydroxyl,
thiol or primary amino groups. Since this is not possible for PTKI, we applied a novel
type of platinum-based drug linker that conjugates the drug via a coordinative linkage at
one of the pyridyl nitrogens (Figure 1A). HPLC analysis and mass spectrometry indicated
complete derivatization of the drug into the drug-ULS 1:1 product. The PTKI-ULS
adduct was subsequently conjugated to M6PHSA and the final high-molecular weight
product was extensively purified by dialysis, which also removed free PTKI-ULS
molecules not linked to the carrier (HPLC analysis, data not shown). Summarizing the two
reaction steps, an overall yield of 84% was reached for the synthesis of PTKI-M6PHSA
(Figure 1A). An average of eight PTKI-ULS molecules was coupled per M6PHSA, as
determined by HPLC after release of the drug from the carrier. Conjugation of PTKI to
M6PHSA did not change the charge or size features of M6PHSA, as assessed by anion-
exchange chromatography and size exclusion chromatography, respectively (Figure 1B).
Effect of PTKI-M6PHSA on cultured stellate cells
The use of ciplatinum as a linker may introduce platinum-associated effects in the drug
targeting preparation. We therefore evaluated PTKI-ULS-M6PHSA for its effect on HSC
viability. In contrast to free cisplatin, which clearly displayed toxic effects, PTKI-
M6PHSA, PTKI and M6PHSA did not reduce the viability of HSC after 24h treatment
(Figure 2A).
To examine the potential antifibrotic effects of PTKI-M6PHSA conjugate and non-
conjugated PTKI on activated HSC, we investigated the expression of fibrosis related
genes in cultured HSC by quantitative RT-PCR. PTKI-M6PHSA downregulated the
Chapter 6

142
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
expression of α-SMA and collagen α1 (II) by 80% and 60% respectively, and PTKI
reduced the expression of the same genes with a similar potency (figure 2B). In contrast,
M6PHSA did not affect the expression of these genes in HSC. The other genes that were
examined, PDGFR-β or TIMP-1, were not significantly reduced by either treatment.
Effect of PTKI-M6PHSA on BDL3 liver slices
In addition to cultured cells, we also used another in vitro system, which encompasses all
liver cell types (hepatocytes, HSC, Kupffer cells and liver endothelial cells) in the context
of their natural environment and extracellular matrix (22). We incubated precision-cut
slices from fibrotic BDL rat livers (BDL3 slices) with PTKI-M6PHSA, PTKI or
M6PHSA and measured the impact of the compounds on gene expression by real-time
PCR.
Interestingly, PTKI-M6PHSA and PTKI had similar effects in liver-slices treatment and
cultured HSC (Figure 3). A significant suppression of collagen α1 (II), α-smooth muscle
actin, PDGFR-β and TIMP-1 mRNA levels by PTKI (80%; 70%; 60%, p<0.01 and 50%,
p<0.05, respectively) and PTKI-M6PHSA (70%; 70%, p<0.01; 40%, p<0.05, respectively)
was found after evaluation of mRNA expression levels.
Animal studies
Table 1 shows the animal characteristics after a single injection study of PTKI-M6PHSA
or vehicle. Administration of PTKI-M6PHSA did not affect the body weight of the
treated BDL rats versus non-treated. None of the biochemical parameters analyzed were
significantly changed.
Distr bution of PTKI-M6PHSA in f brotic ratsi i
Previous studies in our laboratory have demonstrated that M6PHSA binds to and is taken
up by activated HSC in the fibrotic liver. Typically, about 60% of the injected dose
accumulated in the fibrotic liver already within 10 min after administration, in a non-
parenchymal distribution pattern (17). The present study confirms this accumulation of
PTKI-M6PHSA in the liver 2h after administration (Figure 4A), while the construct was
undetectable in other organs like heart, kidney, lung and spleen. (Figure 4B-E). As shown

143
Chapter 6
in Figure 4A (right panel), the staining in the liver reflects non-parenchymal cell
distribution, with hardly any accumulation of the product in the hepatocytes. These results
suggest that the coupling of PTKI-ULS did not alter the distribution of the HSC-selective
carrier in vivo.
0
5000
10000
15000
20000
25000
30000
BDL + saline BDL + PTKI-M6PHSA
Sir
ius
Red
stai
ning
: ar
ea s
tain
ed p
er li
ver
bios
y
bdl day 10 bdl day 11bdl day 12
#
*
A
t
B
BD
L da
y 11
B
DL
day
12
Figure 5. Effect of PTKI-M6PHSA single adminis ration to BDL rats on the
deposition of collagen in the liver, as assessed by Sirius Red staining.
A. Quantification of the area with Sirius Red staining in rat liver specimens (n = 25, mean±sd ttest
between saline and drug treated groups:*P<0.01 and #P<0.05 vs. BDL saline control). B. Liver
sections were processed for immunohistochemistry and then stained with Sirius Red. Rats receiving saline
(left column) showed a marked deposition of collagen, as assessed with Sirius Red, which co-localized with
areas with active fibrogenesis. However, rats treated with PTKI-M6PHSA (right column) showed
Chapter 6

144
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
attenuated liver fibrosis development. Magnification (40x) photomicrograph of liver sections from bile duct
ligated rats on day 10 and 11 post BDL (i.e. 24h and 48h after administration, respectively).
fiEffects of PTKI-M6PHSA in brotic rats
Antifibrotic effects of PTKI-M6PHSA were studied in two different experimental set-ups.
First, we investigated whether a single dose of the product would affect fibrotic gene
expression, in a similar approach as described above for cultured HSC and precision-cut
slices. We did not observe changes in the expression of the chosen reporter genes for
fibrosis (data not shown). Immunohistochemical staining of fibrotic markers, however,
clearly indicated pharmacological activity of the delivered drug. Sirius Red staining for
collagen showed a continuous increment in deposited extracellular matrix components
between day 10 to 12 after bile duct ligation (Figure 5A). Treatment with a single dose of
PTKI-M6PHSA significantly attenuated collagen deposition both 24h and 48h after
administration (p<0.01; 0<0.05, respectively) (Figure 5B). Apart from reduced staining
intensity, the antifibrotic effect of PTKI-M6PHSA was also illustrated by a reduced
portal-portal bridging in fibrotic areas.
Smooth muscle α-actin staining (αSMA) showed an increased number of αSMA-positive
cells between days 10 and 12, which correlates with the exacerbation of liver fibrosis upon
prolonged BDL (Figure 6A). Treatment of BDL rats with PTKI-M6PHSA significantly
attenuated the number of activated HSC stained areas at 24h and 48h respectively (Figure
6B, p<0.01).
After completion of the single dose study, we continued our pharmacological evaluation
with a multiple dose study in the same model. Apart from testing of the conjugate PTKI-
M6PHSA, we now also included the free drug at a dose equivalent to the amount of PTKI
in the conjugate (150 µg/kg). Such a low dosage will not necessarily be therapeutically
effective, in view of the necessary dose of 5 to 20 mg/kg of imitanib in other studies
(12;25). Animal characteristics of the multiple dose study are included in Table 2.
We analyzed mRNA expression levels for collagen-α1 (II), smooth muscle α-actin,
PDGFR-β and TIMP-1 and found no significant differences between treated and

145
Chapter 6
untreated animals of study. Also collagen deposition and α- SMA expression was not
different between those groups (Table 3).
t
l
0
5000
10000
15000
20000
25000
30000
BDL + saline BDL + PTKI-M6PHSA
SM
A st
aini
ng:
area
sta
ined
per
live
r bio
sy bdl day 10 bdl day 11bdl day 12
**
A
B
BD
L da
y 11
B
DL
day
12
Figure 6. Effect of PTKI-M6PHSA single adminis ration to BDL rats on the
accumulation of myofibrob asts and activated HSC, as assessed by smooth muscle
α-actin expression (αSMA).
A. Quantification of the area with αSMA staining in rat liver specimens (n = 25, mean±sd ttest
between saline and drug treated groups:*P<0.01 vs. BDL saline control).
B. Liver sections were processed for immunohistochemistry and then stained with anti-αSMA antibody.
Rats receiving saline (left panels) showed a marked accumulation of αSMA-positive cells, which co-
localized with areas with active fibrogenesis. However, rats treated with PTKI-M6PHSA (right panels)
showed less quantity of αSMA-positive cells. Magnification (40x) photomicrograph of liver sections from
bile duct ligated rats on day 10 and 11 post BDL (i.e. 24h and 48h after administration, respectively)
Chapter 6

146
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
Discussion
Liver fibrosis is in principle a reversible process in which the stellate cells have been
identified as the key fibrogenic cells (26). PDGF is the most potent mitogen for HSC in
vitro, and it plays an important role in the transformation of HSC into myofibroblast-like
cells in vivo (27). Targeting the PDGF signalling cascade therefore represents a promising
antifibrotic approach. In the present study, we employed a kinase inhibitor with a closely
related structure to imatinib (Gleevec, Novartis). The antifibrogenic properties of the
latter well-known kinase inhibitor have already been described (10;12). On the other hand,
concerns have been raised in relation to the effectiveness of imatinib as an antifibrogenic
drug (13), especially in later stages of fibrosis. The new product PTKI-M6PHSA was
designed to effectively and specifically deliver the inhibitor to HSC, in order to improve
its antifibrotic properties. In the present study, we demonstrate the antifibrogenic effects
of PTKI-M6PHSA in two different and validated in vitro systems and in the BDL model
of liver fibrosis.
We confirmed that PTKI showed a similar antifibrotic potential as imatinib by incubating
the drug with culture-activated HSC and precision-cut liver slices. In both test systems,
PTKI potently inhibited the gene expression of fibrotic markers. The observed effect is
presumably attributed to the blockade of PDGF’s profibrogenic and mitogenic actions
since the growth factor is synthesized in situ by the activated HSC in culture or in slices
(28). Furthermore, PTKI is a very potent inhibitor of PDGFR-kinase with an IC50 of 10
nM (15). We also found that PTKI reduced PDGFR-β and TIMP-1 expression. TIMP-1
expression is increased during liver fibrosis and plays an important role in liver
fibrogenesis by modulating extracellular matrix remodelling (29). More importantly,
PTKI-M6PHSA had similar pharmacological effects as PTKI in activated HSC and
fibrotic liver slices. These results suggest that the drug is effectively released from the
carrier during the incubation period, and is capable of inhibiting the PDGF-R kinase
pathway.

147
Chapter 6
Having established the antifibrotic potential of the conjugate, we confirmed in vivo homing
of PTKI-M6PHSA from the circulation to the fibrotic liver. Similar to findings with other
drug-M6PHSA conjugates (19;30) we detected its presence in non-parenchymal cells by
anti-HSA immunostaining. These findings suggest that PTKI-M6PHSA is mainly
delivered within the fibrotic organ in BDL rats and therefore is associated with HSC.
Assuming that PTKI-M6PHSA behaves similar to other drug targeting preparations that
encompass the ULS linker, we can expect a slow release of PTKI during days rather than
hours, providing continuous local drug levels (19) (Prakash, SB-LZM paper2006,
Temming RGD-PEG-SB-HSA paper2006).
Several approaches for a blockade of the PDGF signaling have been investigated as a
strategy to block the ongoing liver fibrotic process. Some papers showed that monoclonal
antibodies directed against the extracellular domain of the PDGF receptor can prevent
binding of the PDGF ligand, thereby inhibiting mitogenic signaling (31;32). Consistent
with this, Kinnman and colleagues (9) described that imatinib inhibited PDGF-BB
induced proliferation of HSC in vitro, but a short-term treatment in vivo did not completely
blunt HSC proliferation 48 hours after BDL. In a different strategy, Borkham-Kamphorst
and colleagues produced a soluble antifibrogenic protein drug, sPDGFRB, and observed a
reduction in ECM deposition after daily administration fourteen days after BDL (33).
According to Neef and colleagues (13) this effect of imatinib is limited to the early phase
of fibrogenesis, due to the lack of efficiency in advanced liver injury, i.e., 3 to 4 weeks
after BDL. In contrast to this latter observtions, there is another study where a
pronounced inhibition of SMA-positive cells is found after 8 weeks of imatinib
administration (12). These authors found a marked attenuation of the liver fibrosis in the
pig-serum induced model of liver fibrosis, characterized by a slow progression of
fibrogenesis, resembling the human situation. Thus, it might be a relevant therapy in
patients with early stages of liver fibrosis.
Our strategy differs completely from the above listed studies in which the free drug
imatinib is used (10;12). We employed a carrier, M6PHSA that is internalized by the target
HSC and releases the kinase inhibitor inside the cells.
Chapter 6

148
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
After single administration, PTKI-M6PHSA markedly reduced collagen deposition
together with a suppression of αSMA-positive cells number which reflects activated HSC
(34). In contrast to the effects on protein levels, PTKI-M6PHSA administered to BDL
animals did not affect the gene expression of the fibrotic markers. This discrepancy either
reflects different sensitivity of the assays, or corresponds to a greater reduction of collagen
and αSMA at the protein level than at gene expression level. This divergence has
previously been described in experiments investigating the inhibition of hepatic fibrosis
(35), and might be due to post-transcriptional regulation of collagen expression in cultured
HSC (36).
The observation that antifibrotic effects are discernable even 48h after a single dose
suggests that effective PTKI levels are present during a prolonged period of time. In
previous studies with other ULS-based conjugates, we have observed a slow drug release
pattern from the ULS linker that may afford continuous drug release during a period of
days (19). The proposed mechanism is competitive displacement of the drug by S-donor
ligands like glutathione and ligands containing sulfur atoms (e.g. methionine) or aromatic
nitrogens (nucleotides, histidine). After binding of PTKI-M6PHSA to HSC and its
internalization, the release of PTKI from the conjugate is a critical point for exerting the
inhibitory effect on PDGF-R kinase. The release of PTKI may occur in the lysosomes, the
compartment to which M6PHSA is routed after internalization (37). Hence, a continued
release of PTKI from the conjugate would be available and a sustained blockade of
PDGF kinase activity may be achieved. In the present study, PTKI-M6PHSA produced a
significant effect on the fibrotic process 24 to 48 hours after its administration to BDL
rats. These results are in good agreement with the previously hypothesized release kinetics
of our drug-conjugates. Furthermore, it suggests the possibility of employing PTKI-
M6PHSA as a depot from which the drug is released for a prolonged period of time.
Our data are in good agreement with the studies of Neef and colleagues. Yet, after
multiple dose administration of PTKI-M6PHSA, the collagen and αSMA protein
expression was not decreased while the results mentioned above show that the process of
liver fibrosis development is clearly decreased by a single dose of PTKI-M6PHSA. How

149
Chapter 6
can this unexpected lack of effect after repeated administration of our drug-targeting
conjugate be explained? We propose that this may be due to the activation of alternative
cytokine pathways that compensates for the inhibition of PDGF-R cascade (38).
Parameter analyzed
BDL day 10 + control
saline
BDL day 14 + control
saline
BDL day 14 + PTKI-M6PHSA
BDL day 14 + PTKI
GAPDH 1.1 ± 0.4 1.3 ± 0.6 0.8 ± 0.2 1.3 ± 0.4
α-SMA 0.8 ± 0.4 1.1 ± 0.7 1.2 ± 0.3 1.1 ± 0.3
Collagen
0.8 ± 0.3 1.3 ± 0.3 1.2 ± 0.4 1.2 ± 0.5
Gene expression
(fold induction)
PDGF Receptor 1.0 ± 0.3 3.2 ± 2.4 3.2 ± 1.9 2.6± 0.6
Sirius Red staining ++ +++ +++ +++
Protein expression
(staining intensity)
αSMA staining
++ +++ +++ +++
Table 3. Effect of PTKI-M6PHSA multiple administration to BDL rats. Data are
shown as mean ± SD.
Redundancy of regulatory pathways is a common feature in the fibrotic process.
Especially if the causative agent of liver fibrosis is not removed, the specific inhibition of
PDGF-R on the hepatic stellate cells may temporarily affect the proliferation of HSC and
collagen deposition, as we observed after single injection of PTKI-M6PHSA, but not after
prolonged administration of PDGF-R inhibitor. Long-term administration of PTKI-
M6PHSA may allow enough time for the activation of alternative pathways, such as TGF-
Chapter 6

150
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
β, that may compensate for the inhibition of PDGF cascade (13). Albeit other
compensatory mechanisms are also possible, this would also suggest that the combined
elimination of more than one pathway could lead to an effective inhibition of hepatic
fibrosis (33). For example, simultaneous inhibition of PDGF and TGF-β cascade might
be more appropriate strategy to attenuate the process of liver fibrosis. Recent experiments
explaining this approach showed that it was more effective than inhibition of either
pathway alone (39). A TGF-β receptor kinase inhibitor has been described and cell-
selective delivery of this construct to the kidney is now being examined in our laboratory
(Prakash, manuscript in preparation). Consequently, targeting both cascades would be an
eligible approach in future investigations.
In conclusion, the data presented in the present study provide evidence that PTKI-
M6PHSA is effective in vitro and specifically taken up by non-parenchymal cells in the
liver, during fibrosis. Single dose administration of PTKI-M6PHSA proved to be an
effective antifibrotic strategy in vivo, although prolonged administration did not reveal
significant effects. Cell-selective elimination of multiple signalling pathways may provide a
tool to design novel strategies.
Acknowledgements
This study was supported by grants from SenterNovem (TSGE1083), NWO Science
Netherlands (R 02-1719, 98-162). Jan Visser and Jai Prakash (University of Groningen,
The Netherlands) are kindly acknowledged for assistance in HPLC analysis and colleagues
at Kreatech for critical reading of the manuscript.

151
Chapter 6
Reference List
1. Tsukada,S., Parsons,C.J., and Rippe,R.A. 2006. Mechanisms of liver fibrosis. Clin. Chim. Acta 364:33-60.
2. Friedman,S.L. 1999. The virtuosity of hepatic stellate cells. Gastroenterology 117:1244-1246.
3. Pinzani,M. 2002. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 7:d1720-d1726.
4. Wong,L., Yamasaki,G., Johnson,R.J., and Friedman,S.L. 1994. Induction of b-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Invest.1563-1569.
5. De Bleser,P.J., Jannes,P., Van Buul-Offers,S.C., Hoogerbrugge,C.M., Van Schravendijk,C.F.H., Niki,T., Rogiers,V., Van den Brande,J.L., Wisse,E., and Geerts,A. 1995. Insulinlike growth factor-II/mannose 6-phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transforming growth factor-b in cocultures with sinusoidal endothelial cells. Hepatol 21:1429-1437.
6. Weiner,J.A., Chen,A., and Davis,B.H. 2000. Platelet-derived growth factor is a principal inductive factormodulating mannose 6-phosphate/insulin-like growth factor-II receptorgene expression via a distal E-box in activated hepatic stellate cells. Biochem. J. 345 Pt 2:225-231.
7. Bachem,M.G., Meyer,D., Schäfer,W., Riess,U., Melchior,R., Sell,K.M., and Gressner,A.M. 1993. The response of rat liver perisinusoidal lipocytes to polypeptide growth regulator changes with their transdifferentiation into myofibroblast-like cells in culture. J. Hepatol. 18:40-52.
8. von,M.M. 2005. Targeted therapy with imatinib: hits and misses? J. Clin. Oncol. 23:8-10.
9. Kinnman,N., Goria,O., Wendum,D., Gendron,M.C., Rey,C., Poupon,R., and Housset,C. 2001. Hepatic stellate cell proliferation is an early platelet-derived growth factor-mediated cellular event in rat cholestatic liver injury. Lab Invest 81:1709-1716.
10. Buchdunger,E., Cioffi,C.L., Law,N., Stover,D., Ohno-Jones,S., Druker,B.J., and Lydon,N.B. 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 295:139-145.
11. Buchdunger,E., Zimmermann,J., Mett,H., Meyer,T., Muller,M., Regenass,U., and Lydon,N.B.
1995. Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc. Natl. Acad. Sci. U. S. A 92:2558-2562.
12. Yoshiji,H., Noguchi,R., Kuriyama,S., Ikenaka,Y., Yoshii,J., Yanase,K., Namisaki,T., Kitade,M., Masaki,T., and Fukui,H. 2005. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am. J. Physiol Gastrointest. Liver Physiol 288:G907-G913.
13. Neef,M., Ledermann,M., Saegesser,H., Schneider,V., Widmer,N., Decosterd,L.A., Rochat,B., and Reichen,J. 2006. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J. Hepatol. 44:167-175.
14. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
15. Zimmermann,J., Buchdunger,E., Mett,H., Meyer,T., and Lydon,N.B. 1997. Potent and selective inhibitors of the abl-kinase:phenylamino-pyrimidine (PAP) derivatives. Bioorganic & Medicinal Chemistry Letters 7:187-192.
16. Beljaars,L., Meijer,D.K., and Poelstra,K. 2002. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front Biosci. 7:e214-e222.
17. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatol 29:1486-1493.
18. van Gijlswijk,R.P., Talman,E.G., Janssen,P.J., Snoeijers,S.S., Killian,J., Tanke,H.J., and Heetebrij,R.J. 2001. Universal Linkage System: versatile nucleic acid labeling technique. Expert. Rev. Mol. Diagn. 1:81-91.
19. Gonzalo,T., Talman,E.G., van,d., V, Temming,K., Greupink,R., Beljaars,L., Reker-Smit,C., Meijer,D.K., Molema,G., Poelstra,K. et al 2006. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control Release 111:193-203.
20. Beljaars,L., Weert,B., Geerts,A., Meijer,D.K.F., and Poelstra,K. 2003. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue. Biochem. Pharmacol. 66:1307-1317.
21. Geerts,A., Niki,T., Hellemans,K., De Craemer,D., Van den Berg,K., Lazou,J.-M., Stange,G., Van de Winkel,M., and De Bleser,P.J.
Chapter 6

152
Local inhibition of liver fibrosis by targeting a PDGF kinase inhibitor to stellate cells
1998. Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatol 27:590-598.
22. Olinga,P., Merema,M.T., de Jager,M.H., Derks,F., Melgert,B.N., Moshage,H., Slooff,M.J., Meijer,D.K., Poelstra,K., and Groothuis,G.M. 2001. Rat liver slices as a tool to study LPS-induced inflammatory response in the liver. J. Hepatol. 35:187-194.
23. van de,B.M., Groothuis,G.M., Draaisma,A.L., Merema,M.T., Bezuijen,J.I., van Gils,M.J., Meijer,D.K., Friedman,S.L., and Olinga,P. 2005. Precision-cut liver slices as a new model to study toxicity-induced hepatic stellate cell activation in a physiologic milieu. Toxicol. Sci. 85:632-638.
24. Beljaars,L., Poelstra,K., Molema,G., and Meijer,D.K.F. 1998. Targeting of sugar- and charge-modified albumins to fibrotic rat livers: the accessibility of hepatic cells after chronic bile duct ligation. J. Hepatol. 29:579-588.
25. Neef,M., Ledermann,M., Saegesser,H., Schneider,V., Widmer,N., Decosterd,L.A., Rochat,B., and Reichen,J. 2006. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J. Hepatol. 44:167-175.
26. Friedman,S.L. 2003. Liver fibrosis -- from bench to bedside. J. Hepatol. 38 Suppl 1:S38-S53.
27. Pinzani,M., Milani,S., Grappone,C., Weber,F.L., Gentilini,P., and Abboud,H.E. 1994. Expression of Platelet-Derived Growth Factor in a Model of Acute Liver Injury. Hepatol 19:701-707.
28. Pinzani,M. 2002. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 7:d1720-d1726.
29. Iredale,J.P., Benyon,R.C., Pickering,J., McCullen,M., Northrop,M., Pawley,S., Hovell,C., and Arthur,M.J.P. 1998. `Mechanisms of spontaneous resolution of rat liver fibrosis - Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 102:538-549.
30. Greupink,R., Bakker,H.I., Reker-Smit,C., van Loenen-Weemaes,A.M., Kok,R.J., Meijer,D.K.F., Beljaars,L., and Poelstra,K. 2005. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. 43:884-892.
31. Heldin,C.-H. 1997. Simultaneous induction of stimulatory and inhibitory signals by PDGF. FEBS Lett. 410:17-21.
32. Shulman,T., Sauer,F.G., Jackman,R.M., Chang,C.N., and Landolfi,N.F. 1997. An antibody reactive with domain 4 of the platelet-derived growth factor beta receptor allows BB binding while inhibiting proliferation by impairing receptor dimerization. J. Biol. Chem. 272:17400-17404.
33. Borkham-Kamphorst,E., Herrmann,J., Stoll,D., Treptau,J., Gressner,A.M., and Weiskirchen,R. 2004. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell activation and attenuates liver fibrosis. Lab Invest 84:766-777.
34. Olaso,E., and Friedman,S.L. 1998. Molecular regulation of hepatic fibrogenesis. J. Hepatol. 29:836-847.
35. Yata,Y., Gotwals,P., Koteliansky,V., and Rockey,D.C. 2002. Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: implications for antifibrotic therapy. Hepatology 35:1022-1030.
36. Stefanovic,B., Hellerbrand,C., Holcik,M., Briendl,M., Aliebhaber,S., and Brenner,D.A. 1997. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol. Cell Biol. 17:5201-5209.
37. Beljaars,L., Olinga,P., Molema,G., De Bleser,P., Geerts,A., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 2001. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver 21:320-328.
38. Kinnman,N., Hultcrantz,R., Barbu,V., Rey,C., Wendum,D., Poupon,R., and Housset,C. 2000. PDGF-mediated chemoattraction of hepatic stellate cells by bile duct segments in cholestatic liver injury. Lab Invest 80:697-707.
39. Yoshiji,H., Kuriyama,S., Noguchi,R., Ikenaka,Y., Yoshii,J., Yanase,K., Namisaki,T., Kitade,M., Yamazaki,M., Asada,K. et al 2006. Amelioration of liver fibrogenesis by dual inhibition of PDGF and TGF-beta with a combination of imatinib mesylate and ACE inhibitor in rats. Int. J. Mol. Med. 17:899-904.


2
costa da mote
boektitel
In ancient times, Romans called the Galician coast in Spain along the n
tip of the Iberia
orthwestern
n Peninsula " Finisterrae" (End of the Land), and in modern
osta da Morte, Finisterre, La Coruña
times this coastal area is known as "Costa da Morte" (The Dead Coast).
Severe storms have destroyed many ships over the centuries.
C
Galicia, España.

Chapter7Summarizing discussion, conclusions and future perspectives

156
Summarizing discussion, conclusions and perspectives
Liver fibrosis is a progressive disease that ultimately leads to cirrhosis and this occurs
when the ongoing injury and the subsequent excessive production of extracellular matrix
are not stopped (1). The cause of liver fibrosis can be very diverse: alcoholism, chronic
infection by virus Hepatitis B or C, diabetes, obesity, as well as autoimmune diseases. A
considerable number of people are affected by liver fibrosis, presently being the 9th
leading cause of death in the western world. Since most of the proclaimed antifibrotic
therapies have failed in clinical testing, liver transplantation remains the only relevant and
successful treatment. This invasive procedure however constitutes severe risks for the
patient, and is associated with high costs and lack of donor organs. Consequently, liver
fibrosis is an unmet medical need and novel antifibrotic therapies will have a huge impact
on the future treatment of liver diseases (2).
Recent insights in the molecular mechanisms of liver fibrosis have provided novel
approaches in mechanistically-based antifibrotic strategies. One of the most important
findings is the recognition of activated hepatic stellate cells (HSC) as major collagen-
producing cells in the injured liver, and their identification as a major target cell for
antifibrotic therapy (3;4). In the present PhD research project we exploited this knowledge
and studied the possibility of treating liver fibrosis by targeting antifibrotic drugs to these
HSC. By using a cell-selective delivery of drugs in the body, we can improve drugs that
exhibit unacceptable side-effects that limit their clinical applicability. To deliver these
potential antifibrotic drugs to HSC, mannose-6-phosphate-modified human serum
albumin (M6PHSA) was employed as a drug carrier (5). Although this technology has
been applied for various drugs in recent years as a novel therapeutic approach in the liver
fibrosis field, not all promising antifibrotic drugs possess appropriate chemical groups for
their conjugation to M6PHSA. In the present study, we therefore explored a new linker
technology for the preparation of HSC-directed conjugates of antifibrotic drugs, which
allowed the targeting of a broader range of drugs. We show that the application of the
platinum-based linkage system ULS™ as drug linker, permitted a straightforward and
rapid method for the development of drug-M6PHSA conjugates, and afforded the
investigation of three new products in the chosen liver fibrosis models. We hypothesized

157
Chapter 7
that cell-specific distribution of these products and the fact that they accumulate inside the
target stellate cells will improve the efficacy of the conjugated drugs greatly, and will also
prevent undesired side effects.
Chapter 1 presents the scope of the present thesis. Chapter 2 briefly reviews the function
of the liver and the problems associated with liver fibrosis. In the same chapter, we also
describe the pharmacological actions of the drugs employed in the present thesis. The
selection of the antifibrotic drug candidates was guided by two main criteria:
1. The candidate drug should be a promising antifibrotic agent to treat liver fibrosis or
acts on a crucial HSC process in this respect, but may exhibit severe side effects in
other tissues than the liver. The candidate drug may not only exert antifibrotic actions
in HSC but also profibrotic actions in other cell types, so that cell-selective drug
targeting will confine its activity to HSC. Pentoxifylline (PTX, chapter 3) is a
representative of this latter class of compounds, while losartan (chapter 4&5) can be
regarded a compound with a safety risk in fibrotic patients, despite its well-
documented safety in hypertensive patients. The third compound (PTKI, chapter 6), a
PDGF kinase inhibitor with great similarity to the anticancer agent Gleevec
(Novartis), is an experimental compound of which the safety profile has not been
investigated. Yet, the widespread involvement of PDGF receptor systems in various
physiological processes implies that its inhibition will evoke intra or extrahepatic side
effects, and that this drug may strongly benefit from targeting.
2. The candidate drug lacks appropriate chemical groups that allow its conjugation to
the M6PHSA carrier via the common covalent coupling reactions. Yet, it should have
a chemical structure that enables a coordinative bond with the ULS linker. These
specifications are discussed in the second part of chapter 2.
In chapter 3, we describe the development of the first generation of ULS-based
conjugates, PTX-M6PHSA, and we demonstrate that the application of the ULS
linker is straightforward and safe, despite its cisplatin-related structure. Several
Chapter 7

158
Summarizing discussion, conclusions and perspectives
chemical and biological characteristics of the product were examined in various in vitro
assays, to demonstrate the feasibility of platinum-based linkage chemistry for drug delivery
purposes. We investigated conjugate stability and biodegradability, safety, and
pharmacological efficacy, and concluded that the conjugate possessed unique drug-
releasing properties. We also observed promising antifibrotic effects of PTX-M6PHSA in
vitro. The production of collagen was reduced notably and we observed striking changes in
the morphology of HSC, which were not observed after treatment with free PTX.
Although the observed changes in morphology suggest that the cells may detach and die
“anoikis”, we did not observe this in our apoptosis or viability assays. An elimination of
fibrogenic HSC by PTX-M6PHSA may be a beneficial effect during liver fibrosis, apart
from antifibrotic effects by reducing collagen deposition. We therefore concluded that
PTX-M6PHSA is a promising candidate for further evaluation as an antifibrotic drug.
We also showed that advanced liver fibrosis can be successfully treated with
losartan-M6PHSA, the second novel conjugate presented in this thesis. Chapters 4
& 5 describe the development, characterization and in vivo properties of losartan-
M6PHSA. The effect of losartan-M6PHSA was extensively studied in two different rat
models of liver fibrosis. Importantly, this novel drug targeting preparation appeared to be
far more effective than free losartan, despite the lower dosage of losartan-M6PHSA
administered, compared to free losartan. This observation indicated that drug targeting
may enhance the intrinsic efficacy of antifibrotic agents. This improvement was attributed
to an altered biodistribution of the drug in the organism and we demonstrated an
enhanced delivery of our construct to HSC. We examined several steps in the cellular
handling of the conjugate: First, after binding to M6P/IGFII receptor, losartan-M6PHSA
was internalized via the endocytotic route (5). Degradation of the conjugate will therefore
occur in the lysosomes, the compartment to which M6PHSA is routed after
internalization (6). We postulate that drug release occurs in multiple steps, in which the
albumin is first degraded by lysosomal enzymes, followed by actual release of the drug
from ULS in other intracellular compartments, e.g. in the cytosol. This chemical bond
between the drug and the ULS can not be degraded by proteinases or low pH present

159
Chapter 7
within the lysosomes. More likely, release of the drug will occur by competitive agents that
bind to the ULS and displace the drug. Glutathione may be such a compound (7). The
cytosol contains high levels of glutathione, so we hypothesize that this compound
competitively displaces losartan from the platinum linker, a process that we have already
demonstrated in vitro for PTX-M6PHSA. Since the intracellular drug release rate was
shown to be relatively slow, a local depot of losartan is formed in the target cells that will
generate continuous drug levels at the target site.
The mechanism of action of losartan-M6PHSA during liver fibrosis is unknown, and
pharmacodynamic differences with the parent drug can not be excluded. The effect of
losartan is mediated by binding to the angiotensin II type 1 receptor (AT1). Interaction of
losartan with this AT1 receptor after binding of losartan-M6PHSA to the M6P/IGFII
receptor may occur via receptor cross-talk in the dynamic plasma membrane. In this
context, the so- called “rafts”, enriched domains where various receptors reside (8), or the
dynamic membrane invaginations in the early endosomes may facilitate such a process. In
this case, we propose a model where cell-membrane associated losartan-M6PHSA might
bind to other putative receptors on the plasma membrane. Another possibility is that
Losartan-M6PHSA may act as an antagonist of the AT1 receptor of a neighbouring cell.
Subsequently, the involvement of angiotensin II (Ang II) in the modulation of TGF-β
ligand expression may be important for the antifibrotic action of losartan (9). Ang II
increased TGF-β gene expression in a dose-dependent manner in activated HSCs in vitro
(10). TGF-β plays a crucial role in HSC activation and thereby regulates the production of
matrix molecules during liver fibrosis. The increase of intrahepatic levels of TGF-β,
induced by bile duct ligation, was attenuated in AT1 receptor knockout mice (11).
Inhibition of the release of Ang II by losartan therefore can markedly suppress the
development of liver fibrosis (12;13). So, Ang II seems to be an important pro-fibrotic
factor. Thus, the selective delivery of losartan to the HSC by means of losartan-M6PHSA
may also interfere in the TGF-β cascade and therefore account for the observed
antifibrotic effect.
Chapter 7

160
Summarizing discussion, conclusions and perspectives
Drug targeting of antifibrotic agents to the liver has been frequently investigated in the
bile duct ligation (BDL) model. The extrahepatic obstruction of the bile duct leads to an
accumulation of bile salts and other molecules that provoke an inflammatory and
fibrogenic process in the liver and bile duct proliferation (14). In the CCl4 model of liver
fibrosis, the injury is induced by administration of CCl4 and phenobarbital and the
pathogenesis of fibrosis in this model is completely different from the BDL model. BDL
is a very rapid and progressive model (15), while the CCl4 model of liver fibrosis is
relatively slow. Consequently, the CCl4 model may better reflect human fibrotic disease, in
which the slow fibrogenic process can be ongoing for several years before the disease
becomes manifest and treatment is initiated.
The demonstration of the efficacy of losartan-M6PHSA in both the BDL model and the
CCl4 model of liver fibrosis thus strengthens its potential as a new antifibrotic modality.
In Chapter 6, we present studies showing that the delivery of a PDGF-kinase-
inhibiting drug to HSC may be a novel strategy to attenuate liver fibrogenesis.
Targeting the PDGF signaling cascade represents a promising antifibrotic approach since
PDGF is regarded as the most potent mitogen for HSC in vitro, and it plays an important
role in the transformation of HSC into myofibroblast-like cells in vivo (16). Recent studies
have reported antifibrogenic effects of the BCR-ABL kinase inhibitor imatinib (Gleevec),
which potently inhibits PDGF-BB kinase (17;18). Kinase inhibitors represent an
important and challenging class of novel therapeutics. They seem especially suitable for
specific interference with the activation or transformation of cells induced by growth
factors or inflammatory mediators. The HSC-selective carrier M6PHSA was therefore
equipped with the PDGF receptor tyrosine kinase inhibitor (PTKI, a compound closely
related to imatinib) by means of the ULS-based linkage strategy. PTKI-M6PHSA induced
antifibrogenic effects in cultured HSC and in fibrotic liver slices, and also displayed
significant antifibrotic effects after a single dose in the BDL model of liver fibrosis.
However, the administration of multiple dosages did not lead to a significant
enhancement of the effect. Thus, while PTKI-M6PHSA proved most effective in vitro,
losartan-M6PHSA was the most effective compound in animal models. Of note, there is

161
Chapter 7
an evident increase of PDGF receptor expression in the BDL model of liver fibrosis from
day 10 to day 14. This receptor system apparently could not be efficiently blocked by the
dose of the targeted PTKI that we administered. We suggest that a higher dose could
improve the effect of PTKI via an effective blockade of the PDGF receptor kinase, which
is so abundantly expressed in activated HSC.
The observed differences in effectiveness may be also explained by the complex nature of
liver fibrosis and HSC activation. Especially if the causative agent of liver fibrosis is not
removed, specific inhibition of PDGF-R in HSC may only temporarily affect HSC
proliferation (19). Chronic treatment regimens might allow enough time for the activation
of alternative pathways and cytokines that may compensate for the inhibition of the
PDGF cascade (20). Thus, only inhibiting PDGF would not be sufficient to stop HSC
proliferation, activation and subsequent production of extracellular matrix constituents.
Parallel pro-fibrogenic mechanisms, such as TGF-β-mediated activation, could be
responsible for triggering the fibrogenic process within the liver despite a complete
blockade of the PDGF receptor pathway.
Future perspectives.
Our strategy offers multiple opportunities for the development of novel HSC- directed
therapeutics. For instance, the coupling of different classes of kinase inhibitors that
presented problems with conjugation to M6PHSA-like carriers in the past. This strategy
profoundly changes the body distribution of these drugs and thus leading to enhanced
efficacy and reduced side-effects which are important especially for the long-term
treatment with this kind of drugs.
An interesting new concept that may originate from the studies with PTKI-M6PHSA is
the use of so-called “cocktail treatments”. Such combination therapies are already
common in anticancer research, but could also be applicable to liver fibrosis.
Chapter 7

162
Summarizing discussion, conclusions and perspectives
What cocktail would be the most effective one, can only be speculated but a combined
inhibition of TGF-β and PDGF cascades might significantly contribute to an effective
inhibition of hepatic fibrosis (21). Kinase inhibiting drugs may represent a valuable asset
in such an approach. In a similar linkage strategy as described for the other drugs in this
thesis, a TGF-β receptor kinase inhibitor (ALK5 inhibitor, 3-(Pyridin-2-yl)-4-(4-quinonyl)-
1H-pyrazole) was conjugated to a renal targeting system (Prakash, manuscript in
preparation), and this approach is also applicable to the M6PHSA carrier. Combined
administration of the ALK5-inh-M6PHSA conjugate with PTKI-M6PHSA can be
explored to counteract PDGF and TGF kinase cascades in HSC, but other rational
combinations of inhibitors are feasible as well. Interestingly, the combination of two
antifibrotic agents intervening in PDGF signaling (imitanib) and the renin-angiotensin
pathway (the angiotensin converting enzyme inhibitor, perindopril) has recently been
investigated and proved more effective than inhibition of either pathway alone (22). The
participation of angiotensin II (Ang II) in modulating TGF-β expression has been already
discussed. Ang II is a potent inducer of TGF-β synthesis in cultured HSCs in vitro (10)
and intervention with the renin-angiotensin system (RAS) appears to be very effective if
this is combined with an inhibition of the mitogenic action of PDGF. In this respect, a
combination therapy with losartan-M6PHSA and PTKI-M6PHSA seems an attractive
approach, and both conjugates are currently available in our department. Thus, targeting
of both intracellular cascades in the HSC could be the eligible approach in future
investigations for an effective anti-fibrotic therapy.
Another possibility is to target anti-angiogenic drugs to the fibrotic liver, in order to
prevent the development of hepatocarcinoma in patients with liver fibrosis. It has been
already reported that neovascularization is increased during the development of liver
fibrosis, and suppression of angiogenic signaling attenuates liver fibrogenesis (23). The
ongoing destruction of healthy liver tissue and tissue remodeling during liver fibrosis
constitutes a serious risk factor for the development of liver cancer (24). Among others,
liver fibrosis is associated with hypoxia, which is one of the strongest inducers of vascular
endothelial growth factor (VEGF)(25). The elevated expression of VEGF and other

163
Chapter 7
growth factors favors the progression of tumor growth and angiogenesis. Delivery of a
VEGF receptor kinase inhibitor to the fibrotic liver thus seems an interesting approach,
and the delivery of anti-angiogenic agents to endothelial cells was shown to be a feasibile
option in our lab. We have recently developed drug-carrier conjugates of the VEGF-
receptor kinase inhibitor PTK787 (vatalanib) and RGD-equipped albumins (Temming,
manuscript in preparation). The presently discussed ULS-based conjugation strategy was
also applied for preparation of these PTK787-HSA conjugates. Another interesting aspect
of PTK787 is that it also inhibits the PDGR receptor kinase, apart from its VEGF
receptor kinase inhibition. Therefore, the delivery of PTK787 to the fibrotic liver by
means of a PTK787-M6PHSA conjugate would interfere with the formation of new blood
vessels and simultaneously affect the proliferative actions of PDGF on HSC, thus
combining antiangiogenic activity with antifibrotic actions.
Another quite relevant question is which specific pathways need to be blocked at specific
phases of the fibrotic process. Various profibrotic pathways become activated at different
time points during the progression of liver fibrosis (26), so the timing of an appropriate
therapy is crucial for its efficacy. Antifibrotic agents can either serve as a preventive
therapy for the development of fibrosis in newly affected areas, or they may act in areas
where the fibrosis is already established. A specific treatment might evolve in a
personalized therapy, in which the disease progression and therapeutic response should
both be closely monitored in time. Future experiments may therefore focus on the
activation of kinase pathways and expression profiles of profibrotic factors by inspecting
biopsies from patients suffering from liver fibrosis. The availability of appropriate
biomarkers will enable disease monitoring and the proper selection of antifibrotic drugs
that subsequently can be delivered specifically to the fibrotic liver by means of drug
conjugates.
Apart from developing a novel linker technology, we also have developed a very
promising compound, losartan-M6PHSA, which showed antifibrotic effects in two
different models of liver fibrosis. The optimization of losartan-M6PHSA conjugates
should be part of future research, including the investigation of long-term administration
Chapter 7

164
Summarizing discussion, conclusions and perspectives
and safety before the clinical trials. In addition, it would be interesting to explore the
application of losartan-M6PHSA to treat the portal hypertension occurring in liver
fibrosis. Cirrhotic patients suffer from portal hypertension which, as a consequence, leads
to systemic hypotension. The specific delivery of losartan to the liver would avoid its
hypotensive actions in peripheral vascular beds, and by this reduce the risk of a
hypotensive shock. Testing of this hypothesis in an animal model of advanced liver
fibrosis, including measurement of both systemic and portal blood pressure seems one of
the most challenging ideas. In addition, follow-up studies of losartan-M6PHSA in
different animal models of liver fibrosis are required to validate its therapeutic applicability
in this disease.
Furthermore, we have demonstrated that ULS-based products are not-toxic per se, as
demonstrated by Alamar blue studies, caspase 3/7 activation and TUNEL assays in vitro. In
sight of a prolonged administration of ULS conjugates to treat liver fibrosis the question
that remains unanswered is the toxicity of cisplatinum after drug release. There are various
reasons that validate its potential application in therapy. Compared to the high
administration of cisplatinum in antitumour therapy, the dose that it would be
administered to stop liver fibrosis is rather low. First, because the fibrogenesis in the liver
is developed during ten to twenty years and the therapy would aim to a monthly
administration of a potent antifibrotic drug targeted to HSC, which would prevent the
formation of extracellular matrix and scar tissue. Secondly, ULS’s reactive groups are
covered with the targeted drug and the carrier. After degradation of the carrier and release
of the drug, ULS linker would be readily detoxified with many intracellular molecules that
are available in the cell containing amino or thiol groups. Taking into consideration that
this is one of the reasons explaining the cell-resistance phenomenon to cisplatinum in
anticancer therapy, the ULS strategy remains applicable. In any case, a favorable treatment
for the patients with liver fibrosis would prevail always considering the suitable balance of
the beneficial treatment versus possible side effects. In this regard, more extensive safety
testing is required during further development of this technology.

165
Chapter 7
Conclusions
The research presented in this thesis demonstrates that platinum coordination chemistry
can be employed for the conjugation of potential drug candidates to target the fibrotic
liver. Drug-ULS conjugates display adequate stability and bioreversibility, and a unique
slow release profile of the coupled drug upon prolonged incubation. This feature can be
attributed to the platinum-coordination bond and has been observed for all tested
compounds. We demonstrated that our approach of HSC-directed drug targeting is
effective in animal models of liver fibrosis, both when examining hepatic drug levels and
the pharmacological outcome. Eventually, the present work may generate a versatile
approach for linking various antifibrotic agents and other classes of drugs to targeting
devices, leading to effective conjugates that can stop or reverse the ongoing process of
liver fibrosis.
7
Chapter 7

166
Summarizing discussion, conclusions and perspectives
Reference List
1. Friedman,S.L. 2003. Liver fibrosis -- from bench to bedside. J. Hepatol. 38 Suppl 1:S38-S53.
2. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
3. Friedman,S.L. 1999. The virtuosity of hepatic stellate cells. Gastroenterology 117:1244-1246.
4. Beljaars,L., Meijer,D.K., and Poelstra,K. 2002. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front Biosci. 7:e214-e222.
5. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatol 29:1486-1493.
6. Beljaars,L., Olinga,P., Molema,G., De Bleser,P., Geerts,A., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 2001. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver 21:320-328.
7. Reedijk,J. 2003. New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc. Natl. Acad. Sci. U. S. A 100:3611-3616.
8. Pike,L.J. 2006. Rafts Defined. J. Lipid Res.
9. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Nakatani,T., Tsujinoue,H., and Fukui,H. 2001. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 34:745-750.
10. Bataller,R., Sancho-Bru,P., Gines,P., Lora,J.M., Al Garawi,A., Sole,M., Colmenero,J., Nicolas,J.M., Jimenez,W., Weich,N. et al 2003. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125:117-125.
11. Yang,L., Bataller,R., Dulyx,J., Coffman,T.M., Gines,P., Rippe,R.A., and Brenner,D.A. 2005. Attenuated hepatic inflammation and fibrosis in angiotensin type 1a receptor deficient mice. J. Hepatol. 43:317-323.
12. George,J., Roulot,D., Koteliansky,V.E., and Bissell,D.M. 1999. In vivo inhibition of rat stellate cell activation by soluble transforming growth
factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc. Natl. Acad. Sci. U. S. A 96:12719-12724.
13. Nakamura,T., Sakata,R., Ueno,T., Sata,M., and Ueno,H. 2000. Inhibition of transforming growth factor beta prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 32:247-255.
14. Nan,J.X., Park,E.J., Lee,S.H., Park,P.H., Kim,J.Y., Ko,G.N., and Sohn,D.H. 2000. Antifibrotic effect of Stephania tetrandra on experimental liver fibrosis induced by bile duct ligation and scission in rats. Arch. Pharm. Research 23:501-506.
15. Kountouras,J., Billing,B.H., and Scheuer,P.J. 1984. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br. J. Exp. Pathol. 65:305-311.
16. Pinzani,M., Milani,S., Grappone,C., Weber,F.L., Gentilini,P., and Abboud,H.E. 1994. Expression of Platelet-Derived Growth Factor in a Model of Acute Liver Injury. Hepatol 19:701-707.
17. Yoshiji,H., Noguchi,R., Kuriyama,S., Ikenaka,Y., Yoshii,J., Yanase,K., Namisaki,T., Kitade,M., Masaki,T., and Fukui,H. 2005. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am. J. Physiol Gastrointest. Liver Physiol 288:G907-G913.
18. Buchdunger,E., Cioffi,C.L., Law,N., Stover,D., Ohno-Jones,S., Druker,B.J., and Lydon,N.B. 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 295:139-145.
19. Kinnman,N., Hultcrantz,R., Barbu,V., Rey,C., Wendum,D., Poupon,R., and Housset,C. 2000. PDGF-mediated chemoattraction of hepatic stellate cells by bile duct segments in cholestatic liver injury. Lab Invest 80:697-707.
20. Neef,M., Ledermann,M., Saegesser,H., Schneider,V., Widmer,N., Decosterd,L.A., Rochat,B., and Reichen,J. 2006. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J. Hepatol. 44:167-175.
21. Borkham-Kamphorst,E., Herrmann,J., Stoll,D., Treptau,J., Gressner,A.M., and Weiskirchen,R. 2004. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell

167
Chapter 7
activation and attenuates liver fibrosis. Lab Invest 84:766-777.
22. Yoshiji,H., Kuriyama,S., Noguchi,R., Ikenaka,Y., Yoshii,J., Yanase,K., Namisaki,T., Kitade,M., Yamazaki,M., Asada,K. et al 2006. Amelioration of liver fibrogenesis by dual inhibition of PDGF and TGF-beta with a combination of imatinib mesylate and ACE inhibitor in rats. Int. J. Mol. Med. 17:899-904.
23. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Hicklin,D.J., Wu,Y., Yanase,K., Namisaki,T., Yamazaki,M. et al 2003. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut 52:1347-1354.
24. Park,Y.N., Yang,C.P., Cubukcu,O., Thung,S.N., and Theise,N.D. 1997. Hepatic stellate cell activation in dysplastic nodules: evidence for an alternate hypothesis concerning human hepatocarcinogenesis. Liver 17:271-274.
25. Kim,K.R., Moon,H.E., and Kim,K.W. 2002. Hypoxia-induced angiogenesis in human hepatocellular carcinoma. J. Mol. Med. 80:703-714.
26. Friedman,S.L., and Bansal,M.B. 2006. Reversal of hepatic fibrosis -- fact or fantasy? Hepatology 43:S82-S88.
Chapter 7

Cafe Central with its gothic vaults and quality coffee is probably the most famous coffeehouse in Vienna. At the turn of the century, it was a meeting place for intellectuals such as Victor Adler and Otto Bauer, writer Peter Altenberg and Leon Trotsky.
Annual meeting & Exposition of the Controlled Release Society
Vienna, Austria, 2006.

Chapter8Samenvatting
Don’t take life too seriously; you will never get out of it alive.
(Elbert Hubbard, 1856-1915).

170
Samenvatting
Lever fibrose is een chronische ziekte waarbij de leverfunctie steeds verder achteruitgaat
door beschadiging van gezonde delen van de lever en de daaropvolgende productie van
littekenweefsel (extracellulaire matrix). Veel voorkomende oorzaken van lever fibrose zijn:
alcoholisme, infecties met het Hepatitis B of C virus, diabetes, obesitas en autoimmuun
ziektes. Lever fibrose is de 9e doodsoorzaak door ziekte in de wereld, maar komt in
toenemende mate voor door stijging van het aantal mensen met overgewicht en met
hepatitis C infecties.
Op dit moment zijn er nog geen goede therapieën voor lever fibrose en veel van de
nieuwe experimentele middelen falen in klinische studies. Transplantatie van de lever is op
dit moment de enige mogelijke behandeling voor cirrhotische patiënten, maar deze
invasieve procedure brengt grote risico’s met zich mee voor de patiënt, en is ook zeer
kostbaar. Daarnaast is er een groot tekort aan geschikte transplantatie levers. De
ontwikkeling van nieuwe therapeutische middelen die lever fibrose kunnen remmen is dan
ook een belangrijke medische vraagstelling.
Recente inzichten in de onderliggende mechanismen van lever fibrose hebben geleid tot
nieuwe aangrijpingspunten voor antifibrotische geneesmiddelen. Eén van de belangrijke
ontdekkingen is de sleutelrol die geactiveerde Hepatische Stellaat Cellen (HSC) spelen bij
fibrotische processen. In dit proefschrift worden studies beschreven waarin antifibrotische
geneesmiddelen naar de stellaat cellen gestuurd worden middels cel-specifieke drug
targeting. Deze gerichte sturing kan de werking van een geneesmiddel verbeteren: een
hogere concentratie geneesmiddel in de stellaat cel leidt mogelijk tot een sterker
antifibrotisch effect. Daarnaast kan drug targeting de bijwerkingen van een geneesmiddel
verminderen, omdat er minder van de werkzame stof in andere weefsels terecht komt. Dit
is met name relevant voor antifibrotische geneesmiddelen, die wel werkzaam zijn in
klinische studies, maar tegelijkertijd onacceptabele bijwerkingen hebben, omdat
extracellulaire matrix vorming een normaal proces is in alle weefsels.
Om de geneesmiddelen af te leveren in stellaat cellen en te zorgen dat ze pas daar
beginnen te werken, worden ze gekoppeld aan het eiwit mannose-6-fosfaat-HSA
(M6PHSA; HSA is humaan serum albumine). Deze geneesmiddel drager (‘carrier’) is in

171
Chapter 8
onze groep ontwikkeld en wordt al een aantal jaren succesvol toegepast. In dit proefschrift
is daarbij een nieuwe koppelingstechniek toegepast, het zogeheten Universal Linkage
System (ULS™). De kern van het ULS is een platina atoom dat coördinatie bindingen kan
vormen met het geneesmiddel en de carrier. De reden dat we voor deze nieuwe
koppelingsstrategie gekozen hebben is dat veel therapeutisch interessante geneesmiddelen
niet geschikt zijn om met de bestaande koppelingschemie aan een geneesmiddeldrager te
binden. De synthese van de preparaten is dan technisch moeilijk of zelfs geheel
onmogelijk. Dit proefschrift toont aan dat de ULS-techniek een effectieve methode is
voor de synthese van geneesmiddel-M6PHSA conjugaten. In een kort tijdsbestek werden
drie nieuwe conjugaten met antifibrotische geneesmiddelen ontwikkeld: de
geneesmiddelen pentoxifylline, losartan en een tyrosine kinase remmer werden gekoppeld
aan de stellaat cel-specifieke geneesmiddeldrager. Naast de chemische en
physicochemische karakterisatie werden de conjugaten getest op gekweekte stellaatcellen.
Een belangrijk onderdeel van het proefschrift is de evaluatie van de nieuwe producten in
diermodellen van leverfibrose.
In hoofdstuk 1 wordt de doelstelling van het proefschrift toegelicht. De basishypothese is
dat geneesmiddelen via ULS aan M6PHSA gekoppeld kunnen worden, en dat de werking
van antifibrotische geneesmiddelen verbeterd kan worden middels cel-specifieke
aflevering in stellaat cellen.
Hoofdstuk 2 behandelt de achtergronden van lever fibrose en de geneesmiddelen die in
dit proefschrift onderzocht zijn. Verder worden de achterliggende principes van ULS
coördinatie chemie en drug targeting middels M6PHSA behandeld.
Hoofdstuk 3 beschrijft de ontwikkeling van het conjugaat PTX-M6PHSA. Pentoxifylline
(PTX) is een geneesmiddel dat in de lever zowel gunstige (antifibrotische) als ongunstige
(profibrotische) activiteit heeft. Drug targeting naar stellaat cellen zou de profibrotische
effecten in Kupffer cellen kunnen voorkomen. Omdat PTX-M6PHSA het eerste
ontwikkelde preparaat met ULS is, hebben we dit product uitvoerig getest op stabiliteit en
geneesmiddel afgifte. In deze proeven bleek al snel dat het geneesmiddel met een
langzame snelheid uit het conjugaat vrijkomt. Dit zogeheten vertraagde afgifteprofiel kan
Chapter 8

172
Samenvatting
gunstig zijn bij de behandeling van chronische ziekteprocessen. Daarnaast hebben we de
mogelijke toxiciteit van het ULS onderzocht, vanwege de overeenkomsten met
cytostatische platinaverbindingen. Een groot verschil met zulke cytostatica is echter dat de
reactieve groepen van ULS al gebruikt zijn voor de koppeling van het geneesmiddel aan
de geneesmiddeldrager, en dus niet meer kunnen reageren met andere producten. Platina
toxiciteit van de conjugaten was dan ook niet aantoonbaar. Wel werd er een duidelijk
farmacologisch effect van het gekoppelde PTX aangetoond in gekweekte stellaat cellen,
waarmee het basisprincipe van dit proefschrift werd aangetoond.
Hoofdstuk 4 & 5 behandelen het conjugaat losartan-M6PHSA. Losartan is een remmer
van angiotensine II, en wordt veel toegepast bij de behandeling van hoge bloeddruk.
Recent is ontdekt dat angiotensine II ook een belangrijke rol speelt bij de activatie van
stellaatcellen, en wordt losartan getest als antifibrotisch geneesmiddel. Het gebruik van
losartan door leverpatiënten is echter niet zonder gevaar omdat leverfibrose vaak gepaard
gaat met een systemisch verlaagde bloeddruk. Wij hebben daarom onderzocht of losartan
specifiek in de lever afgeleverd kan worden, om zo de stellaat cel-specifieke effecten te
versterken. Losartan-M6PHSA is onderzocht in twee verschillende diermodellen van
leverfibrose, het galgangligatie model (bile duct ligation, BDL) en het CCl4 inhalatie
model. In beide modellen vertoonde losartan-M6PHSA een superieur effect, ondanks de
veel lagere dosis van het conjugaat in vergelijking met vrij losartan (niet getarget losartan).
Deze observatie toont aan dat de intrinsieke werkzaamheid van een geneesmiddel
verbeterd kan worden middels drug targeting.
Hoofdstuk 6 behandelt het derde product, PTKI-M6PHSA. De afkorting PTKI staat
voor PDGF Tyrosine Kinase Inhibitor, en verwijst naar een verbinding die sterk verwant
is met het geneesmiddel imitanib (Gleevec). PDGF (Platelet Derived Growth Factor) is
één van de belangrijkste factoren die aanzetten tot leverfibrose en wordt beschouwd als de
meest krachtige factor in stellaat cel proliferatie. Het remmen van PDGF-geïnduceerde
effecten in stellaatcellen is dan ook een interessante aanpak van leverfibrose, en kinase
remmers zoals imitanib en PTKI zijn daarvoor zeer geschikt. PTKI biedt geen
aangrijpingspunten voor de koppeling aan M6PHSA middels klassieke

173
Chapter 8
koppelingsstrategieën. De synthese via het ULS was echter wel succesvol, en het
ontwikkelde product werd vervolgens getest in gekweekte stellaatcellen en in
weefselplakjes (slices) van levers van fibrotische ratten. In beide systemen was PTKI-
M6PHSA effectief. Ook in de daaropvolgende studies in ratten met lever fibrose was het
product werkzaam, hoewel de effectiviteit paradoxaal genoeg afnam bij meerdaagse
toediening. Een mogelijke verklaring hiervoor is dat de gekozen dosis te laag geweest is.
Daarnaast spelen ook andere groeifactoren een rol in lever fibrose, en het kan zijn dat
remming van PDGF alleen niet voldoende is om het complexe fibrotische proces te
stoppen. Remming van meerdere groeifactoren middels een combinatie van
geneesmiddel-M6PHSA conjugaten zou hiervoor een interessante aanpak zijn.
Samenvattend kan gesteld worden dat platina coördinatie chemie een veelbelovende
techniek is voor de koppeling van geneesmiddelen aan drug targeting preparaten.
Geneesmiddel-ULS conjugaten zijn stabiel maar tegelijkertijd ook bioreversibel, en
vertonen een uniek vertraagd afgifteprofiel. De werkzaamheid van de ontwikkelde
preparaten is aangetoond in stellaatcellen en in diermodellen, en werd onderbouwd met
gegevens over de orgaandistributie van de conjugaten. Verder is aangetoond dat ULS-
conjugaten niet per definitie toxisch zijn, hoewel verdere en langduriger studies
betreffende de veiligheid van de linker nog wel nodig zijn. De toevoeging van een nieuw
linker systeem om geneesmiddelen aan eiwitten te koppelen vormt een belangrijke
bijdrage in het huidige onderzoek naar de ontwikkeling van effectieve geneesmiddelen die
leverfibrose kunnen tegengaan. Dankzij deze techniek kunnen vele verschillende soorten
geneesmiddelen worden gekoppeld and cel-specifieke geneesmiddeldragers.
Chapter 8

The author.

AppendixResumenAbbreviationsAcknowledgementsSelected color picturesCurriculum Vitae and publications
Nobody said it was easy,No one ever said it would be this hard…
I was just guessing, at numbers and fi gures, pulling the puzzles apart…Question of science, science and progress…
Do not speak as loud as my heart.
“The Scientist”,
A Rush of Blood to the Head, Coldplay, 2002.

176
Una nueva estrategia para tratar la fibrosis hepática
La fibrosis hepática es el resultado de un proceso inflamatorio y de reparación de tejido en
respuesta a un daño hepático prolongado. Se define como la acumulación de tejido
conectivo en el hígado debido al desequilibrio entre producción y degradación de matriz
extracelular. Mientras que la fibrosis hepática es un proceso reversible, su fase final, la
cirrosis, está considerada como una fase irreversible de la enfermedad (1).
Las causas que provocan la fibrosis hepática son diversas: alcoholismo, infección crónica
por virus de la Hepatitis B o C, diabetes, obesidad y enfermedades autoinmunes. La
fibrosis hepática representa la novena causa de muerte en el mundo occidental. La mayoría
de las terapias antifibróticas han fallado a nivel clínico y el transplante de hígado continúa
siendo el único tratamiento relevante de éxito. Sin embargo, este procedimiento invasivo
implica riesgos graves para el paciente, está sujeto a la existencia de donantes de órganos y
es muy costoso. Consecuentemente, la fibrosis hepática es una enfermedad que
actualmente no tiene tratamiento, por lo que una nueva terapia antifibrótica tendría un
gran impacto en el tratamiento futuro de las enfermedades del hígado.
Recientemente se han descubierto mecanismos moleculares que intervienen en el proceso
de fibrosis hepatica, proporcionando nuevas alternativas en la búsqueda de estrategias
antifibróticas. Uno de los descubrimientos más importantes es el reconocimiento de las
células estrelladas hepáticas (Hepatic Stellate Cells, HSC) como las mayores productoras
de colágeno en el hígado dañado (2), y su identificación como célula diana en la terapia
antifibrótica (3).
En este proyecto de tesis doctoral hemos investigado la posibilidad de tratar la fibrosis
hepática mediante el transporte específico de fármacos con actividad antifibrótica a las
células estrelladas del hígado. Mediante dicho transporte, podemos mejorar la eficacia de
fármacos en el organismo que provocan graves efectos secundarios limitando su
aplicabilidad en la clínica. Para este transporte específico a las células estrelladas,
utilizamos como transportador de fármacos la albúmina humana modificada con un
azúcar manosa-6-fosfato (M6PHSA) (4). Este transportador se utiliza por su unión
específica al receptor M6P/IGFII que se sobreexpresa en las células estrelladas durante el
desarrollo la etapa de fibrosis hepática, tal como se ha demostrado en nuestro laboratorio.

177
Resumen
De este modo, la droga unida a M6PHSA será transportada específicamente a las células
estrelladas en el hígado durante la fase fibrótica. A pesar de que esta tecnología ha sido
utilizada con varios fármacos como una nueva estrategia terapéutica en el campo de la
fibrosis hepática, la elección de la droga estaba restringida por su estructura química que
permite, o no, su conjugación con M6PHSA.
En esta tesis doctoral, hemos desarrollado una nueva tecnología de enlace de fármacos
con el transportador específico a las células estrelladas, M6PHSA. De este modo,
podemos preparar complejos fármaco-transportador que tienen HSC como célula diana
con la posibilidad de conjugar diferentes clases de medicamentos potencialmente
antifibróticos.
En el presente proyecto, hemos demostrado que el uso del sistema de enlace de
medicamentos basado en platina, ULS™ linker (Universal Linkage System TM), permite el
desarrollo rápido de complejo droga-M6PHSA que hemos aplicado en tres fármacos
antifibróticos. Además, la acumulación de estos complejos en la célula estrellada diana va a
mejorar considerablemente la eficacia de los fármacos transportados y va a prevenir
efectos secundarios no deseados.
En el capítulo 1 se expone el objetivo de esta tesis doctoral. En el capítulo 2 se revisa
brevemente la función del hígado y las complicaciones derivadas de la fibrosis hepática.
En el mismo capítulo se describe el mecanismo de acción de los fármacos utilizados en
este proyecto de investigación. La selección de los fármacos antifibróticos está
basado en dos criterios fundamentales:
1. El fármaco candidato debe tener potencial antifibrótico en el hígado, actuando en
una fase crucial en el proceso fibrótico llevado a cabo por las células estrelladas. Sin
embargo, puede tener efectos opuestos a lo esperado, tales como provocar acciones
profibróticas en otros tejidos o tipos celulares. Al unirse a un transportador
específico, se posibilitará el efecto farmacológico únicamente en la célula estrellada.
2. El fármaco candidato carece de grupos químicos apropiados para conjugarse con el
transportador M6PHSA utilizando los enlaces covalentes tradicionales. No obstante,
la droga debe poseer una estructura química apropiada que permita un enlace de
Appendix

178
Una nueva estrategia para tratar la fibrosis hepática
coordinación con el ULS linker. Estas especificaciones se tratan en detalle en la
segunda parte del capitulo 2.
En el capitulo 3, detallamos el desarrollo de la primera generación de compuestos
basados en ULS linker, PTX-M6PHSA, y demostramos que el uso de ULS linker
es sencillo y seguro, a pesar de su estructura parecida al cisplatino.
Se han examinado varias características químicas y biológicas de PTX-M6PHSA en
diferentes ensayos in vitro, para demostrar la viabilidad del uso del enlace químico de
platino como objetivo para construir complejos droga-M6PHSA de liberación específica
en la célula diana.
En nuestro laboratorio, investigamos la estabilidad, biodegradabilidad, seguridad y eficacia
farmacológica de PTX-M6PHSA y concluimos que posee propiedades únicas relacionadas
con la liberación controlada de la droga PTX. Además observamos un interesante efecto
antifibrótico después de analizar el efecto de PTX-M6PHSA in vitro. La producción de
colágeno se redujo notablemente y observamos cambios sorprendentes en la morfología
de las células estrelladas, que no se observaron después del tratamiento con PTX sin
conjugar. A pesar de que los cambios observados en la morfología sugieren que las células
pueden separarse y morir “anoikis”, no observamos este efecto en los ensayos de
apoptosis o viabilidad celular. La eliminación de células estrelladas fibrógenicas mediada
por PTX-M6PHSA puede constituir un efecto beneficioso durante la fase de la fibrosis
hepática, máxime si se suma a la reducción de la deposición de colágeno. En conclusión,
proponemos PTX-M6PHSA como candidato potencial para su evaluación posterior como
droga antifibrógenica.
En los capítulos 4 & 5 de la presente tesis, demostramos que la liberación
específica de losartán a las células estrelladas mediante losartán -M6PHSA, es más
efectivo que el losartán oral en el tratamiento de la fibrosis hepática avanzada.
Losartán-M6PHSA es el segundo compuesto que sintetizamos con ULS linker, y
en estos capítulos describimos su desarrollo, caracterización y efecto antifibrótico
in vivo.

179
Resumen
El efecto de losartán-M6PHSA ha sido ampliamente investigado en dos diferentes
modelos de fibrosis hepática. El efecto de losartán consiste en el bloqueo antagonista del
receptor de angiotensina-II tipo 1 (AT1) (5). Es importante destacar que este nuevo
compuesto de liberación específica resultó ser mucho más efectivo que losartán oral, a
pesar de la dosis tan baja administrada en el compuesto losartán-M6PHSA en
comparación con losartán oral. Estas investigaciones indicaron que esta nueva estrategia
puede mejorar la eficacia intrínseca de losartán mediante la alteración de su
biodistribución en el organismo y así provocar la liberación especifica de losartán a las
células estrelladas.
La demostración de la eficacia de losartán-M6PHSA en dos modelos de fibrosis hepática,
ligadura de conducto biliar (BDL) y de tetracloruro de carbono (CCl4), refuerza su
potencial como nueva estrategia para el tratamiento de la fibrosis del hígado.
En el Capitulo 6, se presentan los estudios que demuestran que la liberación a las
células estrelladas de una droga inhibidora del receptor de PDGF puede ser una
estrategia innovadora para atenuar la fibrogénesis en el hígado dañado.
La cascada de señales de PDGF es una diana terapéutica crítica y una interesante estrategia
antifibrótica, desde que PDGF está considerado como el más potente mitógeno para las
células estrelladas in vitro, y desempeñan una función crucial en la transformación de las
células estrelladas a miofibroblastos in vivo (6). Estudios recientes han descrito efectos
antifibrógenicos del inhibidor de kinasa Imatinib (Gleevec), que inhibe potencialmente la
kinasa PDGF-BB (7). El transportador selectivo para células estrelladas, M6PHSA, se
unió al inhibidor de la tirosina kinasa del receptor de PDGF (PTKI, compuesto similar a
imatinib) mediante el ULS linker.
PTKI-M6PHSA promovió efectos antifibrogénicos en células estrelladas cultivadas y en
un sistema in vitro que consiste en capas de tejido fibrótico hepático (fibrotic liver slices).
Además, PTKI-M6PHSA demostró efectos antifibróticos significativos después de una
sola administración en el modelo animal BDL de fibrosis hepática. Sin embargo, la
administración de más dosis no produjo una mejora del efecto observado.
Appendix

180
Una nueva estrategia para tratar la fibrosis hepática
Las diferencias observadas en la efectividad de los tratamientos podrían ser explicadas por
la naturaleza compleja de la fibrosis hepática y la activación de las células estrelladas (9).
Especialmente, si el agente que causa la fibrosis hepática no se elimina, la inhibición
específica de un determinado receptor en las células estrelladas podria afectar sólo
temporalmente a la fase de fibrogénesis hepática. Por lo tanto, es necesaria la inhibición
simultánea de mecanismos cruciales durante la activación de células estrelladas en la
fibrosis hepática (8).
Perspectivas de futuro.
La estrategia descrita proporciona muchas oportunidades para el desarrollo de nuevos
compuestos que liberen drogas a las células estrelladas. Por ejemplo, la unión de diferentes
clases de inhibidores de kinasas que presentaban problemas de conjugación con M6PHSA
transportadores en el pasado. Esta estrategia modifica la distribución en el organismo de
estos medicamentos mejorando su eficacia y reduciendo los efectos secundarios que son
vitales especialmente para el tratamiento a largo plazo con este tipo de medicamentos.
Un concepto interesante es el uso de “tratamientos cocktail”. Estas terapias de
combinación de drogas son comunes en investigación contra el cáncer, pero podría ser
también aplicable a la fibrosis hepática.
Sólamente podemos especular qué tratamiento cocktail sería el más efectivo, pero una
inhibición combinada de la cascada de TGF-β y PDGF podría contribuir
significativamente a una inhibición eficaz de la fibrosis hepática (8). En este caso, una
terapia combinando losartán-M6PHSA y PTKI-M6PHSA promete ser una estrategia
atractiva y ambos compuestos han sido sintetizados y están disponibles en nuestro
laboratorio. Por lo tanto, la liberación de fármacos inhibiendo ambas cascadas
intracelulares en las células estrelladas podría ser la estrategia apropiada en investigaciones
futuras para una efectiva terapia antifibrótica.

181
Resumen
Conclusiones
La investigación presentada en esta tesis doctoral demuestra que la química de
coordinación del cisplatino puede ser utilizada para la conjugación de drogas
potencialmente efectivas para tratar la fibrosis hepática. Los compuestos droga-ULS han
demostrado una estabilidad adecuada y bioreversibilidad, y un perfil único de liberación
controlada del medicamento. Esta característica puede ser atribuida a la unión platino y ha
sido observada en todos los compuestos investigados. Se ha demostrado que nuestra
estrategia de liberación específica de medicamentos a las células estrelladas es efectiva en
modelos animales de fibrosis hepática.
Finalmente, el trabajo presentado puede generar una estrategia versátil para unir varios
agentes antifibróticos y otras clases de drogas a transportadores específicos, formando
compuestos efectivos que pueden detener o revertir el proceso de fibrosis hepática.
Lista de referencias
1. Bataller,R., and Brenner,D.A. 2005. Liver fibrosis. J. Clin. Invest 115:209-218.
2. Friedman,S.L. 1999. The virtuosity of hepatic stellate cells. Gastroenterology 117:1244-1246.
3. Beljaars,L., Meijer,D.K., and Poelstra,K. 2002. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front Biosci. 7:e214-e222.
4. Beljaars,L., Molema,G., Weert,B., Bonnema,H., Olinga,P., Groothuis,G.M.M., Meijer,D.K.F., and Poelstra,K. 1999. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatol 29:1486-1493.
5. Yoshiji,H., Kuriyama,S., Yoshii,J., Ikenaka,Y., Noguchi,R., Nakatani,T., Tsujinoue,H., and Fukui,H. 2001. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 34:745-750.
6. Pinzani,M., Milani,S., Grappone,C., Weber,F.L., Gentilini,P., and Abboud,H.E. 1994. Expression of Platelet-Derived Growth Factor in a Model of Acute Liver Injury. Hepatol 19:701-707.
7. Yoshiji,H., Noguchi,R., Kuriyama,S., Ikenaka,Y., Yoshii,J., Yanase,K., Namisaki,T., Kitade,M., Masaki,T., and Fukui,H. 2005. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am. J. Physiol Gastrointest. Liver Physiol 288:G907-G913.
8. Yoshiji,H., Kuriyama,S., Noguchi,R., Ikenaka,Y., Yoshii,J., Yanase,K., Namisaki,T., Kitade,M., Yamazaki,M., Asada,K. et al 2006. Amelioration of liver fibrogenesis by dual inhibition of PDGF and TGF-beta with a combination of imatinib mesylate and ACE inhibitor in rats. Int. J. Mol. Med. 17:899-904.
9. Friedman,S.L., and Bansal,M.B. 2006. Reversal of hepatic fibrosis -- fact or fantasy? Hepatology 43:S82-S88.
Appendix

182
Abbreviations
List of abbreviations
α-SMA α-smooth muscle actin
ALT Alanine transaminase
Ang II Angiotensin II
AT1 R Angiotensin II receptor type 1
AST Aspartate transaminase
BCA Bicinchoninic acid
BDL Bile duct ligated
CCL4 Carbon tetrachloride
CTGF connective tissue growth factor
cAMP cyclic-adenosine monophosphate
cDNA complementary deoxirrubonucleotide
DMF Dimethylformamide
DTT Dithiothreitol
EC Endothelial cells
ECM Extracellular matrixs
ERK Extracellular-regulated kinase
FAAS Flameless atomic absorption spectroscopy
FLU-ULS Fluorescein and ULS platinum linker
GAPDH Glyceraldehyde-3-phosphate dehydrogenase
GSH Glutathione
HPLC High Pressure Liquid Chromatrography
HSA Human serum albumin
HSC Hepatic stellate cell
ICP-AES Inductive coupled plasma – atomic emission spectroscopy
KC Kupffer cells
KSCN potassium thiocyanate
LPS Lipopolysaccharide
LOS Losartan
M6PHSA Mannose-6-phosphate modified human serum albumin
M6P/IGFIIR Mannose-6-phosphate insulin growth factor II Receptor
MS Mass Spectrometry
MMPs Matrix metalloproteinases
NF-kB Nuclear Factor-kB
NMR Nuclear magnetic resonance
PC Parenchymal cells
PCR Polimerase chain reaction
PKA Protein kinase A
P38 MAP kinase p38 mitogen-activated kinase
PDGF-BB Platelet-derived growth factor
PDGFR Platelet-derived growth factor Receptor
PTKI PDGF receptor tyrosine kinase inhibitor
PTX Pentoxifylline
PTX-ULS PTX and ULS platinum linker
TIMP-1 Tissue Inhibitor of Metalloproteinases-1
TGF-β Transforming Growth Factor-β
TUNEL TdT-mediated dUTP Nick End Labeling
RAS Renin Angiotensin System;
ROS Reactive oxygen species
ULSTM Universal Linker System (cisPlatinum (ethylenediamine) linker

183
Acknowledgements
When does it all begin? The story goes like that…
My first memory about Holland, ever, is related to Gerben Koning at the train station
with a map of Utrecht, willing to welcome me in a completely new country for me, The
Netherlands. I want to thank specially you, Gerben, for trusting in me. You gave me the wings
I needed to possibly think in dedicating the next 5 years doing scientific research.
I also want to express my honest gratitude to Gert Storm for asking me to stay longer
in Utrecht in order to work in a new research project. Thanks also to Enrico, Raymond,
Marieke and Carmen for the good companion in those days that was enough to convince me
that the spirit is important while doing the experiments in the lab!
Starting my PhD in Groningen was easy having already some knowledge of The
Netherlands: habits and meals….tough! Robbert, as my supervisor, I would like to thank you
for supporting me till the end in order to successfully finalize my thesis, and for your always-
open-for-discussion dynamism and scientific views that stimulated me to find the correct
answers.
Specially thanks to Klaas, my promotor. You have this enthusiasm and positive view
of the situation that is contagious, and your spirit was very valuable for the moments that I was
down. From the stressful moments I have learnt from you to handle it better producing a calm
environment, which allowed me to say goodbye to sleepless nights!!
Dick Meijer, thanks for trusting in me, and encourage me to fight for my own ideas.
You also made it possible the connection with Barcelona group via Joan Rodes and always
encouraged me to pursue it.
Ingrid, the great Ingrid. I am really happy to have met you because you inspire and
encourage me. Whenever we got to a conversation, it was rare the day I did not learn
something new. Either science or life, it was always interesting. I hope that you and Nicolien
make it all the way to Santiago de Compostela (Saint James) to do the pilgrimage and enjoy
some Spanish wine!
Geni, I would like to thank you for your bright comments during the friday meetings,
and for your notorious implication in science: you always try to find the appropriate way based
in strict scientific argumentation. I have learnt a lot from you.
I also want to express my sincere thanks to the members of the reading committee,
Prof. Jan Reedijk, Prof. Wim Hennink and Prof. Pére Ginés, for their careful reading and
quick judgement of the thesis.
Appendix

184
Agradecimientos
Naturally, I would like to thank my students, Anke and Josine, who did an excellent
work in the development of these new conjugates! I enjoyed a lot working together with you
and I wish you a bright future in your careers. I also should thank Gerrit Scherphof for your
friendly welcome to Groningen, and to Jan Kamps and Sigga for your participation in the early
stages on my thesis project.
I wish to thank all my colleages of the department of Pharmacokinetics and Drug
Delivery. Leonie, thanks for your smart comments and fruitful brainstorming during the
meetings. Anne-Miek, thanks for our lively conversations and your help during in vivo
experiments. Catharina requires special thanks because she has been my roommate during all
this four years and we have shared sad moments together with very happy moments
(Thomas!!), thanks for your faithful support. Hester, thank you for sharing also the room with
me and for your nice help and support all this time together. Alie, I want to thank you for your
generous help in the lab and above all, your beautiful smile making life more beautiful!. “Ome”
Jan, now more “ome” than ever, million thanks for your warm help in many things: paperwork
and in the HPLC lab, and thanks for smiling in the difficult moments. Gillian and Ingrid,
thanks for your kind help at the secretary office in solving the thousand little problems. Bert,
thanks for your help to learn the mysteries of Reference Manager. Hans, thanks for sharing
your knowledge about pharmacokinetics with the rest of the group. Frits, thanks for your
contagious young spirit!
Special thanks to the “human lab”, Inge, Marieke, Peter, Annelies, Ansar and
Marjolijn for creating a very nice atmosphere in the department. And to the neighbouring
department, Therapeutic Gene Modulation, thanks to Marianne, Hidde Anna-Rita, Antoine,
Gera and Willemijn for their smiles, the how-are-you and good mornings on the corridors.
For your support in you-can-make-the-PhD & smile, I would like to thank Esther,
Asia, Jai, Joanna, Kai, Marja, Anne-Marie, Rick, Werner and Janja for all our conversations and
healthy laughs at thee time or bier time at the Gouden Zweep. Esther, thanks for your honesty
and healthy spirit, please never change that, I wish you the very best in your future! Asia,
thanks for your warm support from the beginning of my PhD thesis, you have been always
there for me. To Jai, thanks for the interminable talks in the lab at around 7 p.m, trying to
solve the world problems or just solve the last experiments crisis! I will never forget your
wedding in New Delhi, and the 4 a.m ring of fire ceremony of compromise with Ruchi. I wish
you the best for the future because you deserve this happiness. To the one who brought a

185
Acknowledgements
good sense of humour (finally) to the lab, Kai, and shared the Indian adventure at 4 a.m (yes, it
was tough). Marja, thanks for sharing good moments in the conferences, New York-Boston-
San Francisco! I wish you good luck in your new adventure, Canada. To Hilde thanks for our
funny talks at the beginning of my PhD, I miss specially the moments we used to laugh so
much that we decided not to share a room in order to be able to finish our projects.
My short research visit to Barcelona was great. Ramón Bataller offered his lab
without hesitance and he adopted me as one of his students. Ramón, I would like to thank you
for your openness, good heart and young spirit, which I hope you never loose. Your brought
scientific expertise made it pleasant from you to learn. It was the best working with friendly
and helpful colleagues at Bataller’s lab: Pau, Montse, Elena, Jordi and Esther made my research
project and stay in a delightful familiar environment. I got to learn there the “you can make
it” attitude from Pau, the innocent and brighten mind from Montse, and the generous help
from Elena. They shared emotions, laughs, tasty catalan meals, and their computers with me,
and above all, the support at the stabulari…Moltes Gracies!
Really special welcome to Barcelona from my favourite Madrilenian and Catalans
waiting for me just arriving from Groningen with a typical home made meal. Laurita i., Eva y
Laura e., pedazo de cenas que nos hemos pegao,ehh, todo eran risas y buen rollo…hay que repetirlo! Those
made my spare time in Barcelona full of laughing and good times, especially when singing and
dancing Brasilian songs see pictures). Moltes gracies, muchas gracias.
To my friends made in Utrecht. A Rocío, gracias guapetona, por ofrecerme siempre tu casa para
dormir y por las tertuliejas con mojitos, y por aquel inolvidable Queensday 2006 con Laura, ya sabes mas
aventuras vendrán y nos seguiremos riendo, eso que no falte. A Dani, por tus consejillos en ciertos momentos y
por tu incombustible energía, como lo haces, tío? To Dan, your amazing enthusiasm and spirit makes
immediately a good atmosphere around you, thanks for all your support these years. To
Miriam, thanks for our times in Utrecht that I will never forget and that finally I will see you
again soon! A Pilar por nuestras aventuras y esas empanadas tan buenas! To Paola, for our amazing
adventures together! To David, Pachi, Ranier, Iciar Jerome, Lorenzo and Jorg thanks for
making Utrecht home every time I come back.
To my friends in Madrid and Bilbao. A Montse, Leticia y Raquel, gracias por mantener
nuestra amistad, por animarme en todo momento, y sobre todo por querer tenerme de vuelta en los madriles
pronto! Elenilla, gracias por todos los momentos que hemos pasado juntas, nuestras charlas y por estar
literalmente siempre ahí si te necesitaba, en Leiden o ahora en Madrid, te mereces lo mejor porque eres una
Appendix

186
Agradecimientos
luchadora y puedes con todo. Ana! que para nosotras no hay distancia porque desde Ameland, pasando por
Australia, y ahora en Bilbao, estamos siempre en contacto y me encanta ir a Bilbao a visitarte, cuenta con ello.
A Puy, Ruth, Inés y Laura gracias por compartir una amistad desde siempre, pasando por cualquier época y
cambios en nuestras vidas. A Puy, Ruth e Inés, gracias por venir a visitarme a Groningen, anda que no nos lo
pasamos bien! Gracias a Maria Victoria, Carolina y Olivia por acogerme en Burgos con alegría.
To my family in Groningen, especially in the moments when I needed support and
hug… Irenilla!! Encanto, jo, muchas gracias por las cenas durante los dias frios de invierno, las tertuliejas y
las risas...te dejo en tu casa-palacio de Groningen, pero te espero en La Coruña! To Harry, thanks for the
incontable dinners together enjoying your company, and next time it will be in different
locations/countries, exciting! Isn’t it? Delphine, thanks for your vitality and your smile, good
luck in Nice!! Antonella, thanks for all the moments together, I wish you the best in your new
adventure in Italy…To Tim& Doretta, heyyy we will see each other in La Coruña, I will miss
you guys and the Zuiping on thursdays. A Marian, chiquilla, que no te queda nada y que vas a
conseguirlo tu también, el doctorado, claro, mucha suerte y muchos ánimos. To Niko, lots of success in
Cambridge!. A Irene y Jon y Pablo, que me encanta veros y que mil gracias por sex in the city, me esta
salvando la vida durante la escritura de esta tesis...no lo sabéis bien! To Magdalena& Dan, I wish you the
best. To Virginia, Kacper, Kieran, Damiano, Alexio, Eleonora, Kengo and Ivan, for the funny
times together. To Astrid & Erika, genial haberos conocido, y os deseo mucha suerte con el futuro: la
galería de arte y la abogada. To Primoz, thanks for your incredible energy and for the delicious
dinners made by you with Slovenian products. To Giorgo, thanks for our lunches and
adventures together and the past in the Ubbo Emmiussingel house!! To Ilir, thanks for
dinners/parties and all the fun together with jokes listening to the music/chun-chun, etc…To
Margot, thanks for taking care of me, sometimes like my dutch mother! I will never forget my
four years spent in the attic apartment of the Ubbo Emmiussingel.
To all my family in Spain. A toda mi familia en España por apoyarme y darme ánimos, en las
Nocheviejas en Santa Cruz, en Santa Pola, San Juan, Alicante, Sevilla, Ciudad Real, Valencia,
Guadalupe...
Janja, thanks a lot for many things. First, for coming to Gronigen, you were a great
surprise because you enlighten all what surrounds you! You brought a positive energy and a
generous heart from Slovenia to our department… and you appeared in the precise moment,
(let’s be soulmates)…and secondly I want to tell you that is great having you as my friend and my
paranimph.

187
Acknowledgements
To Aitana, my second paranimph, I should thank you for the millions of dinner
together, for talking-solving/or not- discussing the problems and for your invariable support.
You have been a pillar for me in Groningen, without you…no sé no sé…bueno, guapa, anda
que…no tengo que decirte yo cosas ni nada….pero sencillamente, gracias por estar ahí incontables veces para ese
abrazillo, para preparar la cena (sabiendo que mis cenas...pfff), para hablar de loquesea....que nos vemos en
Londres-La Coruña de fijo.
To my parents. They have supported me from the beginning: because of them I
obtain the courage and strength to be able to make it. A mis padres: que me habéis apoyado desde el
principio. De vosotros he aprendido a tener la fuerza y el coraje para terminar esta tesis. Gracias, porque no
habéis dejado ni un día de llamarme, de quererme y de apoyarme, y por todos esos momentos buenos y malos,
que nos han unido mucho más. To my brother Pablo. Gracias por tu apoyo y por hacerte 8horas en tren
Karlsruhe-Groningen, dejarlo todo para estar con tu hermana Teresa. Nunca olvidaré aquello. To my sister
Maria. Gracias por tu alegría, buen rollo y optimismo, que no falte! Y por tener esos tres soletes, Javier, Carlos
y Alicia y solazo, Manel. To my sister Clara. Por estar siempre conmigo.
For the one who has been invariably supporting me, Fermín, and for all those
moments that you have been taking care of me, a pesar de la distancia…the distance, instead of
dividing us, has brought us nearer than ever. Now we know that we can cope with everything.
Gracias por todo, mi amor.
Y la vida siguió, como siguen las cosas que no tienen mucho sentido…
Y la sangre al galope por mis venas, y una nube de arena,
dentro de mi corazón...
Donde habita el Olvido, Joaquín Sabina, 1999, 19 Días y500 noches.
Appendix

Selected color pictures
Selected color pictures
Chapter 3
Figure 3. PTX-M6PHSA binding to target cells.
Negative control PTX-HSA PTX-M6PHSA
C) NIH/3T3 fibroblasts were incubated with PTX-HSA or PTX-M6PHSA at 1 mg/ml for 4h at 37°C, after which the cells were stained for bound products using anti-HSA antibody (red color) and hematoxillin counterstained (blue color) Magnification 20x.
α-SMA Collagen type I
D
G H
FE
BA
C
Figure 7. Effects of PTX-M6PHSA on fibrosis markers
collagen I and α-SMA. HSC were incubated for 24h withPTX-HSA, PTX-M6PHSA (both at1 mg/ml, equivalent to 100 µM PTX,)or 1 mM non-targeted PTX. Cells werestained for collagen type I or α-SMAand counterstained with hematoxillin. A,B) Stellate cells incubated with: C,D) PTX-HSA; E,F) PTX-M6PHSA; G,H) non-targeted PTX. Magnification 10x.
188

189
Selected color pictures
Chapter 4
t
Figure 3. Analysis of colocalization of losartan-M6PHSA within hepatic stellatecells. Liver sections were processed for immunohistochemistry and then stained with anti-HSA antibody (see Experimental procedures). Losartan-M6PHSA co-localized with stellate cells in rat livers (arrows), as assessed with double immunostaining with anti-HSA and anti-desmin (A). Analysis performed with confocal laser microscopy verified the colocalization of anti-HSA (red fluorescence color) and anti-desmin (green fluorescence color) in the regions colored in yellow (arrows) (magnification 40x). Five rats were studied per group.
Saline M6PHSA losartan-M6PHSA Oral losartanSaline M6PHSA losartan-M6PHSA Oral losartan
Figure 4. Effect of differen treatments on the degree of liver fibrosis.
Liver sections were processed for immunohistochemistry and then processed for Sirius Red staining. Severe bridging was observed in rats receiving saline (A), M6PHSA (B) and oral losartan (D). However, rats treated with losartan-M6PHSA (C) showed fewer areas with collagen accumulation (magnification 4x). (n = 10, mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P < 0.05 vs sham; #P<0.05 vs saline, MP6PHSA and oral losartan).
BA
Appendix

190
Selected color pictures
Chapter 4
Saline M6PHSA losartan-M6PHSA Oral losartanSaline M6PHSA losartan-M6PHSA Oral losartan
Figure 5. Effect of different treatments on the accumula ion of myofibroblasts and activated hepatic stellate cells, as assessed by smooth muscle α-actinexpression (αSMA).
t
0
2
4
6
8
10
12
14
16
Sham Saline M6PHSA M6PHSA-losartan
Oral losartan
αSM
A s
tain
ing
(% p
ositi
ve a
rea)
F *
#
Liver sections were processed for immunohistochemistry and then stained with anti-αSMA antibody. Rats receiving saline (A), M6PHSA (B) or oral losartan (D) showed a marked accumulation of αSMA-positive cells, which co-localized with areas with active fibrogenesis. However, rats treated with losartan-M6PHSA (C) showed less quantity of αSMA-positive cells (magnification 4x). (E). High magnification (400x) photomicrograph of a liver from a bile duct ligated rat treated with saline. αSMA staining was detected in cells located in the sinusoids corresponding to activated hepatic stellate cells (upper arrow) as well as in myofibroblasts around proliferating bile ducts (lower arrow). (F). Quantification of the area with αSMA staining in rat liver specimens (n = 10, mean±s.e.m., Mann-Whitney test between saline and drug treated groups:*P<0.05 vs sham; #P<0.05 vs saline, MP6PHSA and oral losartan).

191
Selected color pictures
Chapter 4
Saline M6PHSA losartan-M6PHSA Oral losartanSaline M6PHSA losartan-M6PHSA Oral losartan
E
0
5
10
15
20
25
Sham Saline M6PHSA M6PHSA-losartan
Orallosartan
CD
43 s
tain
ing
(pos
itive
cel
ls/fi
eld)
**
Figure 7. Effect of different treatments on the infiltration of inf ammatory cells in the hepatic parenchyma, as assessed by CD43 expression.
l
Rats subjected to bile duct ligation received different treatments, as specified in the figure. Liver sections were processed for immunohistochemistry and then stained with anti-CD43. Rats receiving saline (A) or M6PHSA (B) showed intense infiltration by CD43-positive cells (arrows). Treatment with losartan-M6PHSA (C), an in a lesser extent, or oral losartan (D), induced a reduction in the number of CD43 infiltrating leukocytes (magnification 4x). (n=20, mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P<0.05 vs saline and M6PHSA) E. Quantification of the number of positive cells in 20 randomly chosen high power fields (n=20, mean±s.e.m., Mann-Whitney test between saline and drug treated groups: *P<0.05 vs saline and M6PHSA)
Appendix

192
Selected color pictures
Chapter 5
tFigure 2. Organ distribution of losar an-M6PHSA.
Localization of Losartan-M6PHSA in different organs was demonstrated by anti-HSA immunostaining. Colocalization of losartan-M6PHSA and HSC was detected by anti-HSA/ anti-DESMIN double immunostaining.
A. Anti-HSA immunostaining in rat liver. B. Colocalization of losartan-M6PHSA with HSC within a fibrotic liver. Arrows indicate the colocalization of red color, presence of carrier, with blue color, desmin, a marker of activated HSC. 40x magnification.
Lung (C), heart (D), spleen (E) and kidney (F) sections stained with anti-HSA after Losartan-M6PHSA injection in CCl4 treated animals. Images were obtained at 20x magnification per organ tissue.

193
Selected color pictures
Chapter 5
losartan Saline M6PHSAlosartan-M6PHSA
Figure 4. Liver fibrosis histology after losartan-M6PHSA treatment. CCL4 rats were treated with: A. Saline, B. M6PHSA, C. losartan-M6PHSA and D. losartan. Liver sections were stained with Sirius Red. Images represent reconstruction of 16 images of 4x magnification per liver biopsy performed with AnalySIS Image Processing. E. Quantification of Sirius Red positive area. Collagen deposition was analysed per liver area of rat liver tissue treated with: control saline, losartan-M6PHSA, M6PHSA and losartan. Quantification is performed over reconstruction of 16 images of 4x magnification per liver biopsy performed with AnalySIS Image Processing. *p<0.05 significant values of losartan-M6PHSA versus CCL4 control, losartan and M6PHSA groups.
losartan-M6PHSA M6PHSASaline osartan l
Figure 5. SMA immunostaining of rat livers.
A. SMA positive cells on rat liver. CCL4 rats were treated with: A. Saline, B. M6PHSA, C. losartan-M6PHSA and D. losartan and immunostained for α-SMA. Images represent one area of 4x magnification per liver biopsy performed with AnalySIS Image Processing. E. Quantification of SMA positive cells per liver area. CCL4 rats were treated with: control (saline), losartan-M6PHSA, M6PHSA and losartan. Quantification is performed by reconstruction of 16 images of 4x magnification per liver biopsy performed with AnalySIS Image Processing. *p<0.05 significant values of losartan-M6PHSA versus losartan and M6PHSA groups.
Appendix

194
Selected color pictures
Chapter 6
0
5000
10000
15000
20000
25000
30000
BDL + saline BDL + PTKI-M6PHSA
Sir
ius
Red
stai
ning
: ar
ea s
tain
ed p
er li
ver
bios
y
bdl day 10 bdl day 11bdl day 12
#
*
BD
L da
y 12
B
DL
day
11
A
B
i
Figure 5. Effect of PTKI-M6PHSA single admin stration to BDL rats on the deposition of collagen in the liver, as assessed by Sirius Red staining.
A. Quantification of the area with Sirius Red staining in rat liver specimens (n = 25, mean±sd ttest between saline and drug treated groups:*P<0.01 and #P<0.05 vs. BDL saline control).
B. Liver sections were processed for immunohistochemistry and then stained with Sirius Red. Rats receiving saline (left column) showed a marked deposition of collagen, as assessed with Sirius Red, which co-localized with areas with active fibrogenesis. However, rats treated with PTKI-M6PHSA (right column) showed attenuated liver fibrosis development. Magnification (40x) photomicrograph of liver sections from bile duct ligated rats on day 10 and 11 post BDL (i.e. 24h and 48h after administration, respectively).

195
Selected color pictures
Chapter 6
ii
0
5000
10000
15000
20000
25000
30000
BDL + saline BDL + PTKI-M6PHSA
SM
A st
aini
ng:
area
sta
ined
per
live
r bio
sy bdl day 10 bdl day 11bdl day 12
**
A
B
BD
L da
y 11
B
DL
day
12
Figure 6. Effect of PTKI-M6PHSA single admin stration to BDL rats on the accumulation of myof broblasts and activated HSC, as assessed by smooth muscle α-actin expression (αSMA).
A. Quantification of the area with αSMA staining in rat liver specimens (n = 25, mean±sd ttest between saline and drug treated groups:*P<0.01 vs. BDL saline control).
B. Liver sections were processed for immunohistochemistry and then stained with anti-αSMA antibody. Rats receiving saline (left panels) showed a marked accumulation of αSMA-positive cells, which co-localized with areas with active fibrogenesis. However, rats treated with PTKI-M6PHSA (right panels) showed less quantity of αSMA-positive cells. Magnification (40x) photomicrograph of liver sections from bile duct ligated rats on day 10 and 11 post BDL (i.e. 24h and 48h after administration, respectively).
Appendix

196

197
Curriculum Vitae
Teresa Gonzalo Lázaro was born on 29th October 1977 in Madrid, Spain. She
grew up in Madrid and attended secondary and high school there.
In September 1995 she started as a student of Pharmacy in Universidad de Alcalá,
Madrid. In the summer of 1998, she went to Iowa, EEUU, to do a practicum in a
pharmacy office organized by the International Pharmaceutical Students Federation
(IPSF). In the period 1999-2000, she got a research grant to study the relationship
between aggressive behaviour and the serotonin in humans at the Department of Pharmacology,
under the supervision of. Dr. Betés. In the last year of her studies, she went to the
Netherlands to work on a research project dealing with targeting genes to ovarian carcinoma
cells using lipoplexes under the supervision of Dr. Gerben Koning and Prof. Dr. Gert
Storm, Department of Pharmaceutics at the University of Utrecht. She graduated
from Universidad de Alcalá in September 2001 with a Master’s degree in Pharmacy.
In October 2001, she started a short research project dealing with TAT-
mediated uptake of liposomes by ovarian carcinoma cells in the Department of Pharmaceutics
at the University of Utrecht under the supervision of Dr. Gerben Koning, Dr. Enrico
Mastrobattista and Prof. Dr. Gert Storm.
In September 2002, she started as a PhD student in the Department of
Pharmacokinetics and Drug Delivery at the University of Groningen, under the
supervision of Dr. Robbert Jan Kok, Prof. Dr. Ingrid Molema, Prof. Dr. Dick Meijer
and Prof. Dr. Klaas Poelstra. During the spring of 2005 she was a visiting student at
the IDIBAPS institute in Hospital Clínic in Barcelona, Spain, under the supervision of
Dr. Ramón Bataller. The results of her research project are presented in this thesis
book.
Appendix

198
List of Publications
Articles
T. Gonzalo, E.G. Talman, A. A. van de Ven, K. Temming, R. Greupink, L. Beljaars, C.
Reker-Smit, D.K.F.Meijer, G. Molema, K. Poelstra, and R. J. Kok.
Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker
technology. Journal of Controlled Release, 2006 ; 111: 193-203.
T. Gonzalo, R. J. Kok, P. Sancho-Bru, J. Swart, J. Prakash, C.Fondevila, D.K.F. Meijer, L.
Beljaars, M. Lacombe, P. vd Hoeven, V. Arroyo, K. Poelstra, P. Ginès and R. Bataller.
Reduction of advanced liver fibrosis by targeted delivery of the angiotensin II receptor
antagonist losartan to hepatic stellate cells. Submitted for publication.
T. Gonzalo, R. Bataller, P. Sancho-Bru, D.K.F. Meijer, L. Beljaars, M. Lacombe, F. Opdam,
V. Arroyo, K. Poelstra, P. Ginès and R. J. Kok.
Accumulation in hepatic stellate cells and antifibrotic effect of losartan-M6PHSA in the CCL4
model of induced liver fibrosis. Manuscript in preparation.
T. Gonzalo, L. Beljaars, M. Van de Bovenkamp, A-M van Loenen, C. Reker-Smit,
D.K.F.Meijer, M. Lacombe, F. Opdam, G. Keri, L. Orfi, K. Poelstra, and R. J. Kok.
Local inhibition of liver fibrosis by specific delivery of a PDGF kinase inhibitor to hepatic
stellate cells. Submitted for publication.
K. Temming, M. Lacombe, P.van der Hoeven, J.Prakash, T. Gonzalo, E.C.F.Dijkers, L.Orfi,
G.Keri, K. Poelstra, G. Molema, and R. J. Kok.
Delivery of the p38 MAPkinase inhibitor SB202190 to angiogenic endothelial cells:
development of novel RGD-equipped and pegylated drug-albumin conjugates using platinum
(II) based drug linker technology. Bioconjugate Chemistry. In press, 2006.

199
Curriculum Vitae
Selected abstracts
T. Gonzalo, A. A. van de Ven, L. Beljaars, C. Reker-Smit, D.K.F.Meijer, G. Molema, K.
Poelstra, and R. J. Kok.
Delivery of pentoxifylline to hepatic stellate cells as a new tool to attenuate liver fibrosis.
Hepatology, 2004; 40(4):610A-611A.
American Association for Study of the Liver.( AASLD, 2004), Boston, USA.
T. Gonzalo, R. Bataller, P. Sancho-Bru, C. Fontdevilla, J. Swart, L. Beljaars, V. Arroyo, K.
Poelstra, P. Gines, and R. J. Kok.
Stellate cell-targeted losartan, but not oral losartan, reduces liver fibrosis.
FIGON Dutch Medicine Days (2005). Lunteren, The Netherlands.
T. Gonzalo, R. Bataller, P. Sancho-Bru, C. Fontdevilla, J. Swart, L. Beljaars, V. Arroyo, K.
Poelstra, P. Gines, and R. J. Kok.
Short-term treatment with losartan targeted to activated stellate cells reduces advanced liver
fibrogenesis: A new strategy to treat liver fibrosis, Hepatology, 2005; 42:604A.
American Association for Study of the Liver. (AASLD, 2005), San Francisco, USA.
T. Gonzalo, K. Poelstra, L. Beljaars, G. Keri, L.Orfi and R. J. Kok.
Specific targeting of a PDGFR tyrosine kinase inhibitor to activated hepatic stellate cells as a
promising technology to reduce liver fibrosis, Hepatology, 2005; 42: 733A-733A.
American Association for Study of the Liver. (AASLD, 2005), San Francisco, USA.
T. Gonzalo, R. Bataller, P. Sancho-Bru, C. Fontdevilla, J. Swart, L. Beljaars, J. Prakash,
M.Lacombe, Paul vd Hoeven, V. Arroyo, K. Poelstra, P. Gines, and R. J. Kok.
Selective delivery of losartan targeted to hepatic stellate cells using losartan-M6PHSA
successfully reduces advanced liver fibrogenesis.
Annual meeting & Exposition of the Controlled Release Society.(CRS, July 2006), Vienna,
Austria.
Appendix

200
List of Publications
Selected oral presentations
Liposome meeting. (March, 2002), Oberjoch, Germany.
Groningen University Institute for Drug Exploration (GUIDE) early summer meeting.
(June, 2003), Groningen, The Netherlands.
Spanish Asociation for the Study of the Liver (AEEH, Feb 2006), Madrid, Spain.
The Center for Liver Diseases in Groningen, Retraite CLDS (May, 2006), The Netherlands.
Groningen University Institute for Drug Exploration (GUIDE) early summer meeting.
(June, 2006), Groningen, The Netherlands.
Awards
First prize award to the poster giving the best means and angles of improving
communication between various pharmaceutical disciplines. FIGON Dutch Medicine Days
(October, 2005).
Controlled Release Society PhD Student Travel Grant award, for participating in the
Annual meeting & Exposition of the Controlled Release Society. (July, 2006), Vienna,
Austria.
Patent
Participation in the patent: Method of conjugating therapeutic compounds to cell targeting devices via metal
complexes.(nr.of patent P72797EP00) (owned by Kreatech Biotechnology and Groningen
University).