unique features of the loblolly pine pinus taeda l...
TRANSCRIPT

INVESTIGATIONHIGHLIGHTED ARTICLE
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn1 John D Liechty Kristian A Stevensdagger Le-Shin WuDagger Carol A Loopstrasect Hans A Vasquez-
Gross William M Doughertydagger Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson Holt
Mark Yandell Aleksey V Zimindaggerdagger James A YorkedaggerdaggerDaggerDagger Marc W Crepeaudagger Daniela Puiusectsect Steven L Salzbergsectsect
Pieter J de Jong Keithanne Mockaitisdaggerdaggerdagger Doreen Mainsectsectsect Charles H Langleydagger and David B NealeDepartment of Plant Sciences and daggerDepartment of Evolution and Ecology University of California Davis California 95616DaggerNational Center for Genome Analysis Support Indiana University Bloomington Indiana 47405 sectDepartment of Ecosystem
Science and Management Texas AampM University College Station Texas 77843 Department of Human Genetics University ofUtah Salt Lake City Utah 84112 daggerdaggerInstitute for Physical Sciences and Technology and DaggerDaggerDepartments of Mathematics and Physics
University of Maryland College Park Maryland 20742 sectsectCenter for Computational Biology McKusick-Nathans Institute ofGenetic Medicine The Johns Hopkins University Baltimore Maryland 21205 Childrenrsquos Hospital Oakland Research InstituteOakland California 94609 daggerdaggerdaggerDepartment of Biology Indiana University Bloomington Indiana 47405 and sectsectsectDepartment of
Horticulture Washington State University Pullman Washington 99163
ABSTRACT The largest genus in the conifer family Pinaceae is Pinus with over 100 species The size and complexity of their genomes(20ndash40 Gb 2n = 24) have delayed the arrival of a well-annotated reference sequence In this study we present the annotation of thefirst whole-genome shotgun assembly of loblolly pine (Pinus taeda L) which comprises 201 Gb of sequence The MAKER-P anno-tation pipeline combined evidence-based alignments and ab initio predictions to generate 50172 gene models of which 15653 areclassified as high confidence Clustering these gene models with 13 other plant species resulted in 20646 gene families of which1554 are predicted to be unique to conifers Among the conifer gene families 159 are composed exclusively of loblolly pine membersThe gene models for loblolly pine have the highest median and mean intron lengths of 24 fully sequenced plant genomes Conifergenomes are full of repetitive DNA with the most significant contributions from long-terminal-repeat retrotransposons In depthanalysis of the tandem and interspersed repetitive content yielded a combined estimate of 82
LOBLOLLY pine is a long-lived diploid member (2n = 24)of the genus Pinus one of 100 species worldwide The
natural range of this primarily outcrossing tree extends overa large portion of the southeastern United States Loblolly
pine is extensively cultivated in this region for timber andpulpwood with plantations growing on 30 million hec-tares and producing 18 of the worldrsquos industrial round-wood (Prestemon and Abt 2002) Loblolly pinersquos preferencefor mild wet climates has made it a model for ecologicalconsiderations relating to carbon sequestration in coastalplain plantations (Noormets et al 2010) The plant biomasson these plantations is thought to contribute to a negativeaccumulation of atmospheric CO2 (Johnsen et al 2001)This species is also under investigation as a potential sourceof sustainable renewable energy both as an independentsource of terpenoids in liquid biofuels and as an intercrop-ping species with other established sources such as switch-grass (Briones et al 2013 Westbrook et al 2013) Theeconomic and ecological importance of this species has ledto several long-term breeding programs and large-scale
Copyright copy 2014 by the Genetics Society of Americadoi 101534genetics113159996Manuscript received November 22 2013 accepted for publication December 13 2013Available freely online through the author-supported open access optionSupporting information is available online at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1Annotation of the genome is presented through the TreeGenes database (Wegrzynet al 2008) It is distributed both via Gbrowse (Stein et al 2002) and WebApollo(httpcodegooglecompapollo-web) In addition flat file access to the FASTAsequences of the genome and transcriptome annotations and gff3 files areavailable through the database FTP site (httploblollyucdavisedubipodftpGenome_DatagenomepinerefseqPita) A masked version of the genome basedon the PIER library 20 is available for download at the same location1Corresponding author Department of Ecology and Evolutionary Biology University ofConnecticut Storrs CT 06269 E-mail jillwegrzynuconnedu
Genetics Vol 196 891ndash909 March 2014 891
studies that have addressed questions about the genetic di-versity and adaptive capabilities (Brown et al 2004 Eckertet al 2010)
In the absence of an affordable and capable technologyto generate and assemble a conifer genome previousinvestigations have relied on other techniques to generatesequence for basic and applied research in pine geneticsFor non-model organisms with large and complex genomestranscriptome resources provide valuable sequence for genediscovery and annotation as well as for comparativegenomics In loblolly pine these were first generated with300000 Sanger-sequenced expressed sequence tags (ESTs)and later as de novo assemblies from next-generation se-quencing technologies (Allona et al 1998 Kirst et al 2003Cairney et al 2006 Lorenz et al 2006 2012) Large-scaleresequencing of the first ESTs and subsequent genotyping ofsingle nucleotide polymorphisms in large populations ex-panded the available molecular marker resources and pro-vided a basis for examining their association with traits ofinterest (Eckert et al 2013) The markers genotyped in thesebreeding populations also improved the density of the geneticlinkage map for loblolly pine (Eckert et al 2009 2010Martiacutenez-Garciacutea et al 2013) The first significant insightinto the genome came from the analysis of 10 bacterialartificial chromosome (BAC) sequences (Kovach et al2010)
Gymnosperms are represented by just four divisionsPinophyta (conifers) Cycadophyta (cycads) Gnetophyta(gnetophytes) and Ginkgophyta (Ginkgo) Their genomesizes range considerably but are generally large between12 and 32 Gbp The pine family (Pinaceae) possesses tre-mendous genome size variation Pines (Pinus) diverged fromspruces (Picea) their closest relatives 85 MYA and possesslarger genomes on average estimated between 22 and 32Gbp (Guillet-Claude et al 2004 Willyard et al 2007) Thesize of these genomes has previously served as a barrier towhole-genome sequencing but recent advances in sequenc-ing technologies and informatics have made assemblingthese megagenomes tractable Unlike many large and com-plex crop genomes there is no evidence to support a whole-genome duplication event The large genome size has primarilybeen attributed to an extensive contribution of interspersedrepetitive content (Morse et al 2009 Kovach et al 2010Wegrzyn et al 2013) Assembly of the Norway spruce ge-nome has shown that LTR retrotransposons in particular arefrequently nested within the long introns of some gene fam-ilies (Nystedt et al 2013) In addition there is evidence forgene duplication pseudogenes and paralogs although theextent of these is not clear (Kovach et al 2010 Pavy et al2012)
To provide a foundation to study the biology of coniferswe annotated the loblolly pine genome the first and largestpine genome assembled to date The whole-genome shotgunsequencing and assembly (Zimin et al 2014) produced tworeference sequences Version 10 the direct output of theMaSuRCA assembler (Zimin et al 2013) was based on
paired-end reads from a haploid megagametophyte andthe matching long insert linking read pairs from diploidneedle tissue Version 101 which applied scaffoldingfrom independent genome and transcriptome assem-blies spans 201 Gbp of sequence and is distributed injust over 144 million scaffolds covering 232 Gbp withan N50 of 669 kb (based on a genome size of 22 Gbp)Our annotation of the loblolly pine genome provides in-sight into the organization of the genome its size con-tent and structure
Materials and Methods
Sequence alignments
Aligning DNA messenger RNA (mRNA) or protein se-quence to the loblolly pine genome presents challenges asthe genome is too large for many common bioinformaticprograms In our approach we sorted the genomic data bydescending scaffold length and partitioned the scaffolds into100 bins such that the genomic sequence for each bincontained roughly the same number of bases This allowedus to parallelize the computations and examine how frag-mentation of the genomic data affected our ability to alignsequence data to the genome We generated initial mappingsto the genome using blat (Kent 2002) with each bin as a tar-get and then used blat utility programs to merge data fromthe 100 bins into a single file and filter that data according toquality metrics In-house scripts were used to parse the blatresults and create input files for exonerate (Slater and Birney2005) which generated the final more refined alignments tothe genome
A set of 83285 de novo-assembled loblolly pine tran-scripts (BioProject PRJNA174450) served as the primarytranscriptome reference These were derived from multipleassemblies of 13 billion RNA-Seq reads selected foruniqueness and putative protein-coding quality and repre-sented samplings from mixed sources of vegetative andreproductive organs seedlings embryos haploid megaga-metophytes and needles under environmental stress(Supporting Information File S1) The transcriptome refer-ence in addition to 45085 sequences generated from300000 reclustered loblolly pine ESTs (Eckert et al2013) were aligned to the genome Sanger-sequencedtranscripts from four other pines available from the Tree-Genes database (Wegrzyn et al 2012) provided additionalalignments Pinus banksiana (13040 transcripts) Pinuscontorta (13570 transcripts) and Pinus pinaster (15648transcripts) Transcriptome assemblies generated via 454pyrosequencing and assembled with Newbler (Roche GSDe novo Assembler) for Pinus palustris (16832 tran-scripts) and Pinus lambertiana (40619 transcripts) (Lor-enz et al 2012) were also aligned Sequence alignmentswere examined at four different cutoffs for the loblollysequence sets and two cutoffs for the other coniferresources Stringent thresholds of 98 identity98
892 J L Wegrzyn et al
coverage served as the starting point for loblolly pinewhile other conifer species were given more permissive(95 identity95 coverage) cutoffs The thresholdswere lowered for all sequence sets to 95 identity50 coverage to further examine the effects of genomefragmentation
To discover orthologous proteins and align them to thegenome we began with 653613 proteins spanning 24species from version 25 of the PLAZA data set (Van Belet al 2012) PLAZA provides a curated and comprehensivecomparative genomics resource for the Viridiplantae includ-ing annotated proteins This set was further curated to ex-clude proteins that were not full length those shorter than21 amino acids and those that had genomic coordinatesthat did not agree with the reported coding sequence (CDS)or did not translate into the reported protein To this set25347 angiosperm proteins reported by the Amborella Ge-nome Project (httpwwwamborellaorg) were addedFrom annotated full-length mRNAs available in GenBanka set of 10793 proteins from Picea sitchensis were includedFrom the Picea abies v10 genome project (Nystedt et al2013) 22070 full-length proteins were included wherethe reported CDS agreed with the translated protein (FileS2 and Table S1) From the loblolly pine transcriptome all83285 transcripts were translated for alignment For thePLAZA protein alignments the target version 101 genomewas hard-masked for repeats for all other alignments thev101 genome was not repeat-masked We accepted analignment when at least 70 of the query sequence wasincluded in the alignment and the exonerate similarity score(ESS) $70
Gene annotation
Annotations for the assembly were generated using theautomated genome annotation pipeline MAKER-P whichaligns and filters EST and protein homology evidenceproduces ab initio gene predictions infers 59 and 39 UTRsand integrates these data to produce final downstream genemodels with quality control statistics (Campbell et al 2014)Inputs for MAKER-P include the Pinus taeda genome assem-bly (v10) Pinus and Picea ESTs conifer transcriptome as-semblies a species-specific repeat library (PIER) (Wegrzynet al 2013) protein databases containing annotated pro-teins for P sitchensis and P abies and version 25 of thePLAZA protein database (Van Bel et al 2012) MAKER-Pproduced ab initio gene predictions via SNAP (Korf 2004)and Augustus (Stanke and Waack 2003) A subset of anno-tations (500 Mb of sequence) were manually reviewed todevelop appropriate filters for the final gene models Giventhe large genome size and potential for spurious annota-tions conservative thresholds for these filters were chosenFilters included the initial removal of single-exon annota-tions because of apparent pseudogene bias annotationsnot containing a recognizable protein domain as interpretedusing InterProScan (Quevillon et al 2005) or annotationsthat do not partially overlap the conifer transcriptome
resources available Among the selected nonoverlappinggene models a subset of high confidence gene models wasfurther analyzed based on the annotation edit distance(AED) score of 020 with canonical start sites and splicesites All gene models identified in v10 were also mapped tothe latest assembly (v101) Functional annotations of thegene models were generated through blastp alignmentsagainst the National Center for Biotechnology Information(NCBI) nr and plant protein databases for significant (E-values 1e-05) and weak (E-value 0001) hits
Alignments of the high-confidence MAKER-P sequencesagainst the genome allowed for identification of largeintrons Introns of lengths 20 50 and 100 kbp andwith 25 gap regions were selected for further analysisClusters from the Markov cluster algorithm (MCL) analysisthat represented gene families with P taeda members andcontained primary introns (first CDS) 20 kbp were alignedand manually reviewed The intronic sequences were inves-tigated for the presence of intron-mediated expression sig-nals (IMEs) IMEs were predicted via sequence motifs usingthe methodology outlined in Parra et al (2007)
Orthologous proteins
The analysis of orthologous genes included a subset of thespecies that were previously selected from PLAZA A total of10 including Arabidopsis thaliana (27403) Glycine max(46324) Oryza sativa (41363) Physcomitrella patens(28090) Populus trichocarpa (40141) Ricinus communis(31009) Selaginella moellendorffii (18384) Theobromacacao (28858) Vitis vinifera (26238) and Zea mays (39172)were used Three external protein sequence sets were alsoincluded Amborella trichopoda (25347) P abies (22070)and P sitchensis (10521) The primary source of the P taedasequence was the complete set of 50172 gene models gen-erated from the MAKER-P pipeline All 14 sequence sets(including the 10 from PLAZA) were clustered to 90 iden-tity within species and combined to generate 399358sequences
The MCL analysis (Enright et al 2002) as implementedin the TRIBE-MCL pipeline (Dongen and Abreu-Goodger2012) was used to cluster the 399358 protein sequencesfrom 14 species into orthologous groups The methodologywas selected due to its robust implementation that avoidsmerging clusters that share only a few edges This leads toaccurate identifications of gene families even in the presenceof lower-quality BLAST hits or promiscuous domains (Frechand Chen 2010) The analysis was performed by runningpairwise NCBI blastp v2227+ (Altschul et al 1990) (E-valuecutoff of 1e-05) against the full set of proteins describedabove In this pipeline the negative log10 of the resultingblastp E-values produces a network graph that serves as in-put to define the orthologous groups The user-supplied in-flation value is used in the second stage to simulate randomwalks in the previously calculated graph A large inflationvalue defines more clusters with lower amount of mem-bers (fine-grained granularity) a small inflation value
Annotation of Loblolly Pine Genome 893
defines not as many clusters but the clusters have moremembers (coarse granularity) A moderate inflation valueof 40 was selected to define the orthologous groups hereFollowing their generation Pfam domains (Punta et al2012) were assigned from the PLAZA annotations of theindividual sequences InterProScan 48 (Hunter et al 2012)was applied to those sequences obtained outside of PLAZA(A trichopoda P sitchensis P abies and P taeda) PFam andGene Ontology (GO) assignments with E-values 1e-05were retained To effectively compare GO annotations theterms were normalized to level four of the classification treeWhen all predicted domains for a given family were classi-fied as retroelements the family was removed After func-tional assessment and filtering custom scripts and Venndiagrams (httpbioinformaticspsbugentbewebtoolsVenn)were applied to visualize gene family membership amongspecies
Gene family gain-and-loss phylogenetics
The DOLLOP v3695 program from the Phylip package(Felsenstein 1989) was used to reconstruct a parsimonioustree explaining the gain and loss of gene families under theDOLLOP parsimony model The DOLLOP algorithm esti-mates phylogenies for discrete character data with twostates (either a 0 or 1) which assumes only one gain butas many losses as necessary to explain the evolutionary pat-tern of states The filtered 8519 gene families (containing112899 sequences representing 13 species the small num-ber of sequences from P sitchensis excluded) were used tocreate a gene family gain-and-loss matrix Gene families thatrepresented all species were dropped from the input to DOL-LOP as their phylogenetic classification would be obfuscatedby a disparate number of starting proteins per species in theanalysis Branch lengths were calculated by counting thenumber of gains or losses for a given node To produce the finalphylogenetic reconstruction a manual curation step was in-troduced to bring the P patens and Selaginella moellendorffiibranches into agreement with known phylogenies DOLLOPwas rerun on this updated tree to reconstruct the final genefamily configurations of the ancestral species Only P patensS moellendorffii and their common ancestor were updatedduring this step The fidelity of the reconstructed trees wasevaluated against a known phylogeny available at Phytozome(Goodstein et al 2012)
Tandem repeat identification
Tandem Repeat Finder (TRF) v407b (Benson 1999) wasrun with the following parameters matching weight of 2mismatch weight of 7 indel penalty of 7 match probabilityof 80 indel probability of 10 minimum score of 50 anda maximum period size (repeating unit) of 2000 Both thegenome and transcriptome were examined and investigatedfor tandem content To accurately assess the overall cover-age and distribution of tandem repeats we filtered overlapsby discriminating against multimeric repeats following pre-viously described approaches (Melters et al 2013) and
excluded those found within interspersed repeats such asthe long terminal ends of LTR transposable elementsMononucleotides (period size of 1) were not scrutinizeddue to the high likelihood of error andor repeat collapsein the assembly process To maintain reproducibility com-mon nomenclature termsmdashsuch as ldquomicrosatellitesrdquo or sim-ple sequence repeats (SSRs) describing 1- to 8-bp periodsldquominisatellitesrdquo describing 9- to 100-bp periods and ldquosat-ellitesrdquo describing 100-bp periodsmdashwere used for classi-fication For comparative analysis these methods wereapplied to the reference genomes of A thaliana v167V vinifera v145 S moellendorffii v100 C sativus v122and P trichocarpa v210 each available through Phyto-zome (Goodstein et al 2012) The draft genome sequencesof P glauca v100 (Birol et al 2013) P abies (Nystedtet al 2013) and A trichopoda v100 were included Boththe filtered TRF output and consensus sequences derivedfrom clustering the filtered TRF output (UCLUST utility at70 identity) were used as queries in similarity searchesUSEARCH (E-value 001) was used to search the Plant-Sat database (Macas et al 2002) Potential centromericsequences were assessed by finding the monomeric tandemarray that covered the largest amount of the genome In-terstitial and true telomeric sequences were isolated fromthe filtered TRF output by searching for (TTTAGGG)n motifs(n 3 and length 1 kbp) The thresholds applied werebased on conservative estimates from telomere restrictionfragment lengths described for P taeda (Flanary andKletetschka 2005)
Homology-based repeat identification
RepeatMasker 330 (RepeatMasker 2013) was used toidentify previously characterized repeats RepeatMaskerwas run using standard settings on the entire genome withPIER 20 as a repeat library (Wegrzyn et al 2013) A maskedversion of the genome was generated at this stage Redun-dancy was eliminated through custom Python scripts bymatching each reference base pair with the highest scoringalignment For full-length estimates we required that thegenomic sequence and the reference PIER elements alignedwith at least 80 identity and 70 coverage Repeat con-tent in introns was independently characterized using thesame methodology High-copy elements were identified bysorting families by full-length copy number High-coverageelements were determined by sorting families by base-paircoverage including both full-length and partial hits MITEHunter (release 112011) (Han and Wessler 2010) wasused to search for miniature inverted-repeat transposableelements (MITEs) within intronic regions
De novo repeat identification
The workflow for characterization and annotation of novelrepeats largely mirrored that described in Wegrzyn et al(2013) REPET 20 (Flutre et al 2011) was used to identifysequences to form the repeat library for CENSOR 4227(Kohany et al 2006) REPETrsquos de novo repeat discovery
894 J L Wegrzyn et al
performs an all-versus-all alignment of the input sequencean unscalable computationally intensive operation As suchthe 63 longest scaffolds in the assembly were used as inputrepresenting 1 of the genome (bin 1) For use in REPETrsquosclassification steps we provided REPET with the PIER data-base in addition to a set of seven conifer repeat librariesdescribed in Nystedt et al (2013) A set of 405109 publiclyavailable full and partial P taeda and Pinus elliottii transcriptswere provided to REPET for host gene identification
Previously characterized elements were removed usingthe PIER and spruce repeat databases and the blastn-blastxstructural filters proposed in Wicker et al (2007) and imple-mented in Wegrzyn et al (2013) Previously classified elementswere identified by blastn alignments passing the 80-80-80threshold (80 identity covering 80 of each sequence forat least 80 bp) Unclassified elements were analyzed usingblastx against a database of ORFs recovered from thePIERspruce nucleotide database with USEARCHrsquos ldquofindorfsrdquoutility at default parameters (Edgar 2010) Elements thatwere classified only to the superfamily level and unclassifiedrepeats were separately clustered with UCLUST (Edgar 2010)at 80 identity Consensus sequences for each cluster werederived from multiple alignments built with MUSCLE(Edgar 2010) and PILER (Edgar and Myers 2005) Finallyseparate clusters were chained into families in which eachclusterrsquos consensus sequence aligned with at least 80identity with at least one other sequence in the familyFurther classification of LTR retroelement families was per-formed using the in-house tool GCclassif (Figure S1 andFile S4)
Results and Discussion
Sequence alignments
Aligning the 83285 de novo-assembled transcripts to thegenome allowed us to infer information about gene regions
including introns and repeats (Table 1) At a stringency of98 query coverage and 98 identity 30993 transcriptsmapped to the genome most of which (29262) mappedwith a unique hit 1731 transcripts mapped to two or moregenomic regions Relaxing the cutoff criteria to 95 identityand 95 query coverage resulted in increases in the numberof transcripts with unique (43972) and non-unique hits(5409) At 95 identity and 50 query coverage the num-ber of transcripts with unique hits to the genome rose to44469 and transcripts aligning to more than one locus to28116 Possible reasons for a transcript aligning to two ormore genomic locations include gene duplications pseudo-genes assembly errors and actively transcribed retroele-ments in the transcriptome assembly
To elucidate how genome fragmentation affected ourability to map sequence to the genome the v101 genomescaffolds were sorted by descending length and divided into100 bins with each bin containing 1 of the genomicsequence Peaks in the gap region content were found inbin 59 (360 ldquoNrdquo bases) and in bin 82 (478 ldquoNrdquo bases)reflecting gaps due to linking libraries (Figure S2A) Whilegap-region content is greatly reduced in bins 86ndash100 thesesame bins show a sharp decrease in scaffold size and hencea sharp increase in the number of scaffolds per bin (FigureS2B) As expected the ability to align transcripts to thegenome decreased as the scaffold length shortened with564 of these transcript mappings occurring in the first25 bins and just 85 in the last 25 bins (Figure S2C)
Using the same techniques we produced transcriptome-to-genome alignments beyond those generated with the denovo transcriptome using existing EST and transcriptomedata for loblolly pine and other closely related species(Table 1) A total of 741 of the loblolly pine reclusteredESTs mapped at 95 query coverage 95 sequence iden-tity Sequencing technology did not affect the percentageof sequence sets aligning to the genome as much as the
Table 1 Mapping ESTtranscriptome resources against P taeda version 101 genome
Project Total sequence Identity Coverage Unique hits Non-unique hits Total mapped
P taeda (reclustered ESTs) 45085 98 98 26700 712 608P taeda (reclustered ESTs) 45085 98 95 29676 1845 6991P taeda (reclustered ESTs) 45085 95 95 31324 2074 7401P taeda (reclustered ESTs) 45085 95 50 29744 5486 7814P taeda (de novo) 83285 98 98 29262 1731 3521P taeda (de novo) 83285 98 95 42822 5130 5758P taeda (de novo) 83285 95 95 43972 5409 5929P taeda (de novo) 83285 95 50 44469 28116 8715P palustris (454) 16832 95 95 11242 719 7106P palustris (454) 16832 95 50 11181 1949 7806P lambertiana (454 + RNASeq) 40619 95 95 13134 317 3311P lambertiana (454 + RNASeq) 40619 95 50 23376 3792 6688P banksiana (TreeGenes clusters) 13040 95 95 9703 513 7834P banksiana (TreeGenes clusters) 13040 95 50 9470 1473 8392P contorta (TreeGenes clusters) 13570 95 95 9575 396 7348P contorta (TreeGenes clusters) 13570 95 50 9534 1083 7824P pinaster (TreeGenes clusters) 15648 95 95 9738 943 6826P pinaster (TreeGenes clusters) 15648 95 50 10221 2491 8124
Annotation of Loblolly Pine Genome 895
phylogenetic relationships among the species P palustrisis most closely related to loblolly pine and obtains map-ping rates similar to the reclustered ESTs at 711 with95 query coverage and 95 identity P lambertiana ismost distant and this is reflected in the minimal mappingsuccess of the transcripts (Table 1)
The common lineage of the angiosperms and the gym-nosperms split 300 MYA (Jiao et al 2011) impedingsearches for nucleic acid sequence homology betweenangiosperms and loblolly pine However we expect a subsetof proteins to remain relatively conserved over multiplegeological periods The results of mapping and aligningthree protein data sets to the loblolly pine v101 genomeare shown in Table 2 where we determined the initialprotein-to-scaffold mapping with blat and created a refinedalignment with exonerate The subset of the PLAZA dataset used includes over half a million proteins from 24 non-gymnosperm plant species 168 of which aligned to theloblolly pine genome Aligning proteins from members ofthe genus Picea (last common ancestor 85 MYA) to theloblolly pine genome proved more fruitful Seventy-fivepercent of 10000 full-length Sitka spruce proteins [themajority predicted from high-quality full-length Sanger-sequenced complementary DNA data (Ralph et al 2008)]aligned to the genome Sixty-nine percent of 22070 P abiesproteins from the Congenie project aligned to the loblollypine genome A total of 84 of the loblolly pine proteinsgleaned from the transcriptome assembly aligned
Fewer than 30 of each PLAZA species protein set alignedto the genome at our initial cutoff (query coverage$70 andESS $70) For 18 species in the PLAZA data set as well asA trichopoda P sitchensis P abies and translated P taedatranscriptome sequences we further break down the protein-to-genome alignments that passed the initial cutoff into threecategories ESS $ 90 90 ESS 80 and 70 ESS 80(Figure 1) In the ESS $ 90 category 452 of Norwayspruce proteins 606 of Sitka spruce proteins and 700of loblolly pine proteins aligned to the genome (File S2)The differences between the two spruce protein sets maybe partially attributed to the sequencing technologiesemployed to generate the original transcriptomes (Sangervs next-generation technologies)
Gene annotation
The MAKER-P annotation pipeline considered the transcriptalignments in addition to ab initio models to predict poten-tial coding regions The initial predictions generated90000 gene models 44 were fragmented or otherwise
indicative of pseudogenes Previous studies of pines haveidentified pseudogene members in large gene families(Wakasugi et al 1994 Skinner and Timko 1998 Gernandtet al 2001 Garcia-Gil 2008) and pseudogene content maybe as much as five times that of functional coding regions(Kovach et al 2010) Conservative filters were thereforenecessary to deduce the actual gene space The multi-exongene model requirement is likely biased against intronlessgenes but it reduces false positives associated with repeatsand pseudogenes Because 20 of Arabidopsis and O sativagenes are intronless (Jain et al 2008) it is likely that ourapproach ignores as much as 20 of the gene space
After applying multi-exon and protein domain filters50172 gene models remained These transcripts rangedfrom 120 bp to12 kbp in length and represented48 Mbpof the genome with an average coding sequence length of965 bp (Table S2) Since a recognizable protein domain wasa requirement the majority of gene models are functionallyannotated (File S3) Functional annotations of the translatedsequences revealed that 49196 (98) of the transcripts havehomology to existing sequences in NCBIrsquos plant protein andnr database A slightly smaller number 48614 (97) alignto a functionally characterized gene product
The full-length gene models that are well supported byaligned evidence and with an AED score 020 total15653 These high-confidence transcripts cover 30 Mbpof the genome and have an average coding sequence lengthof 1295 bp with an average of three introns per gene (TableS2) Once again 98 of these sequences are functionallyannotated 23 of the high-quality alignments are toV vinifera Given the fragmentation of the genome and thelack of intronless genes reported true estimates of genenumber may be as high as 60000 particularly given the83285 unique gene models generated from broad P taedaRNA evidence (File S1) Previous estimates of gene numberin conifers vary widely The gene content of white spruce isestimated to be 32000 (Rigault et al 2011) While com-prehensive evaluation of maritime pine transcriptomicresources from EuroPineDB produced 52000 UniGenes(Fernandez-Pozo et al 2011)
Introns
Plant introns are typically shorter than those found inmammals (Shepard et al 2009) The gene models derivedfrom MAKER-P yielded maximum intron lengths 50 kbpwith several 100 kbp A total of 147425 introns wereidentified in the 50172 transcripts A requirement of25 gap-region content was used to calculate values on
Table 2 Mapping protein sequence against P taeda version 101 genome
Full-length proteins from Total sequence Unique hits Non-unique hits Total mapped Data source
P abies 22070 11580 3638 6895 Nystedt et al (2013)P sitchensis 10793 6516 1574 7495 GenBankP taeda 83285 45656 24427 8415 Current assemblyPLAZA (24 species) 653613 90149 19492 1677 Van Bel et al (2012)
896 J L Wegrzyn et al
144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

studies that have addressed questions about the genetic di-versity and adaptive capabilities (Brown et al 2004 Eckertet al 2010)
In the absence of an affordable and capable technologyto generate and assemble a conifer genome previousinvestigations have relied on other techniques to generatesequence for basic and applied research in pine geneticsFor non-model organisms with large and complex genomestranscriptome resources provide valuable sequence for genediscovery and annotation as well as for comparativegenomics In loblolly pine these were first generated with300000 Sanger-sequenced expressed sequence tags (ESTs)and later as de novo assemblies from next-generation se-quencing technologies (Allona et al 1998 Kirst et al 2003Cairney et al 2006 Lorenz et al 2006 2012) Large-scaleresequencing of the first ESTs and subsequent genotyping ofsingle nucleotide polymorphisms in large populations ex-panded the available molecular marker resources and pro-vided a basis for examining their association with traits ofinterest (Eckert et al 2013) The markers genotyped in thesebreeding populations also improved the density of the geneticlinkage map for loblolly pine (Eckert et al 2009 2010Martiacutenez-Garciacutea et al 2013) The first significant insightinto the genome came from the analysis of 10 bacterialartificial chromosome (BAC) sequences (Kovach et al2010)
Gymnosperms are represented by just four divisionsPinophyta (conifers) Cycadophyta (cycads) Gnetophyta(gnetophytes) and Ginkgophyta (Ginkgo) Their genomesizes range considerably but are generally large between12 and 32 Gbp The pine family (Pinaceae) possesses tre-mendous genome size variation Pines (Pinus) diverged fromspruces (Picea) their closest relatives 85 MYA and possesslarger genomes on average estimated between 22 and 32Gbp (Guillet-Claude et al 2004 Willyard et al 2007) Thesize of these genomes has previously served as a barrier towhole-genome sequencing but recent advances in sequenc-ing technologies and informatics have made assemblingthese megagenomes tractable Unlike many large and com-plex crop genomes there is no evidence to support a whole-genome duplication event The large genome size has primarilybeen attributed to an extensive contribution of interspersedrepetitive content (Morse et al 2009 Kovach et al 2010Wegrzyn et al 2013) Assembly of the Norway spruce ge-nome has shown that LTR retrotransposons in particular arefrequently nested within the long introns of some gene fam-ilies (Nystedt et al 2013) In addition there is evidence forgene duplication pseudogenes and paralogs although theextent of these is not clear (Kovach et al 2010 Pavy et al2012)
To provide a foundation to study the biology of coniferswe annotated the loblolly pine genome the first and largestpine genome assembled to date The whole-genome shotgunsequencing and assembly (Zimin et al 2014) produced tworeference sequences Version 10 the direct output of theMaSuRCA assembler (Zimin et al 2013) was based on
paired-end reads from a haploid megagametophyte andthe matching long insert linking read pairs from diploidneedle tissue Version 101 which applied scaffoldingfrom independent genome and transcriptome assem-blies spans 201 Gbp of sequence and is distributed injust over 144 million scaffolds covering 232 Gbp withan N50 of 669 kb (based on a genome size of 22 Gbp)Our annotation of the loblolly pine genome provides in-sight into the organization of the genome its size con-tent and structure
Materials and Methods
Sequence alignments
Aligning DNA messenger RNA (mRNA) or protein se-quence to the loblolly pine genome presents challenges asthe genome is too large for many common bioinformaticprograms In our approach we sorted the genomic data bydescending scaffold length and partitioned the scaffolds into100 bins such that the genomic sequence for each bincontained roughly the same number of bases This allowedus to parallelize the computations and examine how frag-mentation of the genomic data affected our ability to alignsequence data to the genome We generated initial mappingsto the genome using blat (Kent 2002) with each bin as a tar-get and then used blat utility programs to merge data fromthe 100 bins into a single file and filter that data according toquality metrics In-house scripts were used to parse the blatresults and create input files for exonerate (Slater and Birney2005) which generated the final more refined alignments tothe genome
A set of 83285 de novo-assembled loblolly pine tran-scripts (BioProject PRJNA174450) served as the primarytranscriptome reference These were derived from multipleassemblies of 13 billion RNA-Seq reads selected foruniqueness and putative protein-coding quality and repre-sented samplings from mixed sources of vegetative andreproductive organs seedlings embryos haploid megaga-metophytes and needles under environmental stress(Supporting Information File S1) The transcriptome refer-ence in addition to 45085 sequences generated from300000 reclustered loblolly pine ESTs (Eckert et al2013) were aligned to the genome Sanger-sequencedtranscripts from four other pines available from the Tree-Genes database (Wegrzyn et al 2012) provided additionalalignments Pinus banksiana (13040 transcripts) Pinuscontorta (13570 transcripts) and Pinus pinaster (15648transcripts) Transcriptome assemblies generated via 454pyrosequencing and assembled with Newbler (Roche GSDe novo Assembler) for Pinus palustris (16832 tran-scripts) and Pinus lambertiana (40619 transcripts) (Lor-enz et al 2012) were also aligned Sequence alignmentswere examined at four different cutoffs for the loblollysequence sets and two cutoffs for the other coniferresources Stringent thresholds of 98 identity98
892 J L Wegrzyn et al
coverage served as the starting point for loblolly pinewhile other conifer species were given more permissive(95 identity95 coverage) cutoffs The thresholdswere lowered for all sequence sets to 95 identity50 coverage to further examine the effects of genomefragmentation
To discover orthologous proteins and align them to thegenome we began with 653613 proteins spanning 24species from version 25 of the PLAZA data set (Van Belet al 2012) PLAZA provides a curated and comprehensivecomparative genomics resource for the Viridiplantae includ-ing annotated proteins This set was further curated to ex-clude proteins that were not full length those shorter than21 amino acids and those that had genomic coordinatesthat did not agree with the reported coding sequence (CDS)or did not translate into the reported protein To this set25347 angiosperm proteins reported by the Amborella Ge-nome Project (httpwwwamborellaorg) were addedFrom annotated full-length mRNAs available in GenBanka set of 10793 proteins from Picea sitchensis were includedFrom the Picea abies v10 genome project (Nystedt et al2013) 22070 full-length proteins were included wherethe reported CDS agreed with the translated protein (FileS2 and Table S1) From the loblolly pine transcriptome all83285 transcripts were translated for alignment For thePLAZA protein alignments the target version 101 genomewas hard-masked for repeats for all other alignments thev101 genome was not repeat-masked We accepted analignment when at least 70 of the query sequence wasincluded in the alignment and the exonerate similarity score(ESS) $70
Gene annotation
Annotations for the assembly were generated using theautomated genome annotation pipeline MAKER-P whichaligns and filters EST and protein homology evidenceproduces ab initio gene predictions infers 59 and 39 UTRsand integrates these data to produce final downstream genemodels with quality control statistics (Campbell et al 2014)Inputs for MAKER-P include the Pinus taeda genome assem-bly (v10) Pinus and Picea ESTs conifer transcriptome as-semblies a species-specific repeat library (PIER) (Wegrzynet al 2013) protein databases containing annotated pro-teins for P sitchensis and P abies and version 25 of thePLAZA protein database (Van Bel et al 2012) MAKER-Pproduced ab initio gene predictions via SNAP (Korf 2004)and Augustus (Stanke and Waack 2003) A subset of anno-tations (500 Mb of sequence) were manually reviewed todevelop appropriate filters for the final gene models Giventhe large genome size and potential for spurious annota-tions conservative thresholds for these filters were chosenFilters included the initial removal of single-exon annota-tions because of apparent pseudogene bias annotationsnot containing a recognizable protein domain as interpretedusing InterProScan (Quevillon et al 2005) or annotationsthat do not partially overlap the conifer transcriptome
resources available Among the selected nonoverlappinggene models a subset of high confidence gene models wasfurther analyzed based on the annotation edit distance(AED) score of 020 with canonical start sites and splicesites All gene models identified in v10 were also mapped tothe latest assembly (v101) Functional annotations of thegene models were generated through blastp alignmentsagainst the National Center for Biotechnology Information(NCBI) nr and plant protein databases for significant (E-values 1e-05) and weak (E-value 0001) hits
Alignments of the high-confidence MAKER-P sequencesagainst the genome allowed for identification of largeintrons Introns of lengths 20 50 and 100 kbp andwith 25 gap regions were selected for further analysisClusters from the Markov cluster algorithm (MCL) analysisthat represented gene families with P taeda members andcontained primary introns (first CDS) 20 kbp were alignedand manually reviewed The intronic sequences were inves-tigated for the presence of intron-mediated expression sig-nals (IMEs) IMEs were predicted via sequence motifs usingthe methodology outlined in Parra et al (2007)
Orthologous proteins
The analysis of orthologous genes included a subset of thespecies that were previously selected from PLAZA A total of10 including Arabidopsis thaliana (27403) Glycine max(46324) Oryza sativa (41363) Physcomitrella patens(28090) Populus trichocarpa (40141) Ricinus communis(31009) Selaginella moellendorffii (18384) Theobromacacao (28858) Vitis vinifera (26238) and Zea mays (39172)were used Three external protein sequence sets were alsoincluded Amborella trichopoda (25347) P abies (22070)and P sitchensis (10521) The primary source of the P taedasequence was the complete set of 50172 gene models gen-erated from the MAKER-P pipeline All 14 sequence sets(including the 10 from PLAZA) were clustered to 90 iden-tity within species and combined to generate 399358sequences
The MCL analysis (Enright et al 2002) as implementedin the TRIBE-MCL pipeline (Dongen and Abreu-Goodger2012) was used to cluster the 399358 protein sequencesfrom 14 species into orthologous groups The methodologywas selected due to its robust implementation that avoidsmerging clusters that share only a few edges This leads toaccurate identifications of gene families even in the presenceof lower-quality BLAST hits or promiscuous domains (Frechand Chen 2010) The analysis was performed by runningpairwise NCBI blastp v2227+ (Altschul et al 1990) (E-valuecutoff of 1e-05) against the full set of proteins describedabove In this pipeline the negative log10 of the resultingblastp E-values produces a network graph that serves as in-put to define the orthologous groups The user-supplied in-flation value is used in the second stage to simulate randomwalks in the previously calculated graph A large inflationvalue defines more clusters with lower amount of mem-bers (fine-grained granularity) a small inflation value
Annotation of Loblolly Pine Genome 893
defines not as many clusters but the clusters have moremembers (coarse granularity) A moderate inflation valueof 40 was selected to define the orthologous groups hereFollowing their generation Pfam domains (Punta et al2012) were assigned from the PLAZA annotations of theindividual sequences InterProScan 48 (Hunter et al 2012)was applied to those sequences obtained outside of PLAZA(A trichopoda P sitchensis P abies and P taeda) PFam andGene Ontology (GO) assignments with E-values 1e-05were retained To effectively compare GO annotations theterms were normalized to level four of the classification treeWhen all predicted domains for a given family were classi-fied as retroelements the family was removed After func-tional assessment and filtering custom scripts and Venndiagrams (httpbioinformaticspsbugentbewebtoolsVenn)were applied to visualize gene family membership amongspecies
Gene family gain-and-loss phylogenetics
The DOLLOP v3695 program from the Phylip package(Felsenstein 1989) was used to reconstruct a parsimonioustree explaining the gain and loss of gene families under theDOLLOP parsimony model The DOLLOP algorithm esti-mates phylogenies for discrete character data with twostates (either a 0 or 1) which assumes only one gain butas many losses as necessary to explain the evolutionary pat-tern of states The filtered 8519 gene families (containing112899 sequences representing 13 species the small num-ber of sequences from P sitchensis excluded) were used tocreate a gene family gain-and-loss matrix Gene families thatrepresented all species were dropped from the input to DOL-LOP as their phylogenetic classification would be obfuscatedby a disparate number of starting proteins per species in theanalysis Branch lengths were calculated by counting thenumber of gains or losses for a given node To produce the finalphylogenetic reconstruction a manual curation step was in-troduced to bring the P patens and Selaginella moellendorffiibranches into agreement with known phylogenies DOLLOPwas rerun on this updated tree to reconstruct the final genefamily configurations of the ancestral species Only P patensS moellendorffii and their common ancestor were updatedduring this step The fidelity of the reconstructed trees wasevaluated against a known phylogeny available at Phytozome(Goodstein et al 2012)
Tandem repeat identification
Tandem Repeat Finder (TRF) v407b (Benson 1999) wasrun with the following parameters matching weight of 2mismatch weight of 7 indel penalty of 7 match probabilityof 80 indel probability of 10 minimum score of 50 anda maximum period size (repeating unit) of 2000 Both thegenome and transcriptome were examined and investigatedfor tandem content To accurately assess the overall cover-age and distribution of tandem repeats we filtered overlapsby discriminating against multimeric repeats following pre-viously described approaches (Melters et al 2013) and
excluded those found within interspersed repeats such asthe long terminal ends of LTR transposable elementsMononucleotides (period size of 1) were not scrutinizeddue to the high likelihood of error andor repeat collapsein the assembly process To maintain reproducibility com-mon nomenclature termsmdashsuch as ldquomicrosatellitesrdquo or sim-ple sequence repeats (SSRs) describing 1- to 8-bp periodsldquominisatellitesrdquo describing 9- to 100-bp periods and ldquosat-ellitesrdquo describing 100-bp periodsmdashwere used for classi-fication For comparative analysis these methods wereapplied to the reference genomes of A thaliana v167V vinifera v145 S moellendorffii v100 C sativus v122and P trichocarpa v210 each available through Phyto-zome (Goodstein et al 2012) The draft genome sequencesof P glauca v100 (Birol et al 2013) P abies (Nystedtet al 2013) and A trichopoda v100 were included Boththe filtered TRF output and consensus sequences derivedfrom clustering the filtered TRF output (UCLUST utility at70 identity) were used as queries in similarity searchesUSEARCH (E-value 001) was used to search the Plant-Sat database (Macas et al 2002) Potential centromericsequences were assessed by finding the monomeric tandemarray that covered the largest amount of the genome In-terstitial and true telomeric sequences were isolated fromthe filtered TRF output by searching for (TTTAGGG)n motifs(n 3 and length 1 kbp) The thresholds applied werebased on conservative estimates from telomere restrictionfragment lengths described for P taeda (Flanary andKletetschka 2005)
Homology-based repeat identification
RepeatMasker 330 (RepeatMasker 2013) was used toidentify previously characterized repeats RepeatMaskerwas run using standard settings on the entire genome withPIER 20 as a repeat library (Wegrzyn et al 2013) A maskedversion of the genome was generated at this stage Redun-dancy was eliminated through custom Python scripts bymatching each reference base pair with the highest scoringalignment For full-length estimates we required that thegenomic sequence and the reference PIER elements alignedwith at least 80 identity and 70 coverage Repeat con-tent in introns was independently characterized using thesame methodology High-copy elements were identified bysorting families by full-length copy number High-coverageelements were determined by sorting families by base-paircoverage including both full-length and partial hits MITEHunter (release 112011) (Han and Wessler 2010) wasused to search for miniature inverted-repeat transposableelements (MITEs) within intronic regions
De novo repeat identification
The workflow for characterization and annotation of novelrepeats largely mirrored that described in Wegrzyn et al(2013) REPET 20 (Flutre et al 2011) was used to identifysequences to form the repeat library for CENSOR 4227(Kohany et al 2006) REPETrsquos de novo repeat discovery
894 J L Wegrzyn et al
performs an all-versus-all alignment of the input sequencean unscalable computationally intensive operation As suchthe 63 longest scaffolds in the assembly were used as inputrepresenting 1 of the genome (bin 1) For use in REPETrsquosclassification steps we provided REPET with the PIER data-base in addition to a set of seven conifer repeat librariesdescribed in Nystedt et al (2013) A set of 405109 publiclyavailable full and partial P taeda and Pinus elliottii transcriptswere provided to REPET for host gene identification
Previously characterized elements were removed usingthe PIER and spruce repeat databases and the blastn-blastxstructural filters proposed in Wicker et al (2007) and imple-mented in Wegrzyn et al (2013) Previously classified elementswere identified by blastn alignments passing the 80-80-80threshold (80 identity covering 80 of each sequence forat least 80 bp) Unclassified elements were analyzed usingblastx against a database of ORFs recovered from thePIERspruce nucleotide database with USEARCHrsquos ldquofindorfsrdquoutility at default parameters (Edgar 2010) Elements thatwere classified only to the superfamily level and unclassifiedrepeats were separately clustered with UCLUST (Edgar 2010)at 80 identity Consensus sequences for each cluster werederived from multiple alignments built with MUSCLE(Edgar 2010) and PILER (Edgar and Myers 2005) Finallyseparate clusters were chained into families in which eachclusterrsquos consensus sequence aligned with at least 80identity with at least one other sequence in the familyFurther classification of LTR retroelement families was per-formed using the in-house tool GCclassif (Figure S1 andFile S4)
Results and Discussion
Sequence alignments
Aligning the 83285 de novo-assembled transcripts to thegenome allowed us to infer information about gene regions
including introns and repeats (Table 1) At a stringency of98 query coverage and 98 identity 30993 transcriptsmapped to the genome most of which (29262) mappedwith a unique hit 1731 transcripts mapped to two or moregenomic regions Relaxing the cutoff criteria to 95 identityand 95 query coverage resulted in increases in the numberof transcripts with unique (43972) and non-unique hits(5409) At 95 identity and 50 query coverage the num-ber of transcripts with unique hits to the genome rose to44469 and transcripts aligning to more than one locus to28116 Possible reasons for a transcript aligning to two ormore genomic locations include gene duplications pseudo-genes assembly errors and actively transcribed retroele-ments in the transcriptome assembly
To elucidate how genome fragmentation affected ourability to map sequence to the genome the v101 genomescaffolds were sorted by descending length and divided into100 bins with each bin containing 1 of the genomicsequence Peaks in the gap region content were found inbin 59 (360 ldquoNrdquo bases) and in bin 82 (478 ldquoNrdquo bases)reflecting gaps due to linking libraries (Figure S2A) Whilegap-region content is greatly reduced in bins 86ndash100 thesesame bins show a sharp decrease in scaffold size and hencea sharp increase in the number of scaffolds per bin (FigureS2B) As expected the ability to align transcripts to thegenome decreased as the scaffold length shortened with564 of these transcript mappings occurring in the first25 bins and just 85 in the last 25 bins (Figure S2C)
Using the same techniques we produced transcriptome-to-genome alignments beyond those generated with the denovo transcriptome using existing EST and transcriptomedata for loblolly pine and other closely related species(Table 1) A total of 741 of the loblolly pine reclusteredESTs mapped at 95 query coverage 95 sequence iden-tity Sequencing technology did not affect the percentageof sequence sets aligning to the genome as much as the
Table 1 Mapping ESTtranscriptome resources against P taeda version 101 genome
Project Total sequence Identity Coverage Unique hits Non-unique hits Total mapped
P taeda (reclustered ESTs) 45085 98 98 26700 712 608P taeda (reclustered ESTs) 45085 98 95 29676 1845 6991P taeda (reclustered ESTs) 45085 95 95 31324 2074 7401P taeda (reclustered ESTs) 45085 95 50 29744 5486 7814P taeda (de novo) 83285 98 98 29262 1731 3521P taeda (de novo) 83285 98 95 42822 5130 5758P taeda (de novo) 83285 95 95 43972 5409 5929P taeda (de novo) 83285 95 50 44469 28116 8715P palustris (454) 16832 95 95 11242 719 7106P palustris (454) 16832 95 50 11181 1949 7806P lambertiana (454 + RNASeq) 40619 95 95 13134 317 3311P lambertiana (454 + RNASeq) 40619 95 50 23376 3792 6688P banksiana (TreeGenes clusters) 13040 95 95 9703 513 7834P banksiana (TreeGenes clusters) 13040 95 50 9470 1473 8392P contorta (TreeGenes clusters) 13570 95 95 9575 396 7348P contorta (TreeGenes clusters) 13570 95 50 9534 1083 7824P pinaster (TreeGenes clusters) 15648 95 95 9738 943 6826P pinaster (TreeGenes clusters) 15648 95 50 10221 2491 8124
Annotation of Loblolly Pine Genome 895
phylogenetic relationships among the species P palustrisis most closely related to loblolly pine and obtains map-ping rates similar to the reclustered ESTs at 711 with95 query coverage and 95 identity P lambertiana ismost distant and this is reflected in the minimal mappingsuccess of the transcripts (Table 1)
The common lineage of the angiosperms and the gym-nosperms split 300 MYA (Jiao et al 2011) impedingsearches for nucleic acid sequence homology betweenangiosperms and loblolly pine However we expect a subsetof proteins to remain relatively conserved over multiplegeological periods The results of mapping and aligningthree protein data sets to the loblolly pine v101 genomeare shown in Table 2 where we determined the initialprotein-to-scaffold mapping with blat and created a refinedalignment with exonerate The subset of the PLAZA dataset used includes over half a million proteins from 24 non-gymnosperm plant species 168 of which aligned to theloblolly pine genome Aligning proteins from members ofthe genus Picea (last common ancestor 85 MYA) to theloblolly pine genome proved more fruitful Seventy-fivepercent of 10000 full-length Sitka spruce proteins [themajority predicted from high-quality full-length Sanger-sequenced complementary DNA data (Ralph et al 2008)]aligned to the genome Sixty-nine percent of 22070 P abiesproteins from the Congenie project aligned to the loblollypine genome A total of 84 of the loblolly pine proteinsgleaned from the transcriptome assembly aligned
Fewer than 30 of each PLAZA species protein set alignedto the genome at our initial cutoff (query coverage$70 andESS $70) For 18 species in the PLAZA data set as well asA trichopoda P sitchensis P abies and translated P taedatranscriptome sequences we further break down the protein-to-genome alignments that passed the initial cutoff into threecategories ESS $ 90 90 ESS 80 and 70 ESS 80(Figure 1) In the ESS $ 90 category 452 of Norwayspruce proteins 606 of Sitka spruce proteins and 700of loblolly pine proteins aligned to the genome (File S2)The differences between the two spruce protein sets maybe partially attributed to the sequencing technologiesemployed to generate the original transcriptomes (Sangervs next-generation technologies)
Gene annotation
The MAKER-P annotation pipeline considered the transcriptalignments in addition to ab initio models to predict poten-tial coding regions The initial predictions generated90000 gene models 44 were fragmented or otherwise
indicative of pseudogenes Previous studies of pines haveidentified pseudogene members in large gene families(Wakasugi et al 1994 Skinner and Timko 1998 Gernandtet al 2001 Garcia-Gil 2008) and pseudogene content maybe as much as five times that of functional coding regions(Kovach et al 2010) Conservative filters were thereforenecessary to deduce the actual gene space The multi-exongene model requirement is likely biased against intronlessgenes but it reduces false positives associated with repeatsand pseudogenes Because 20 of Arabidopsis and O sativagenes are intronless (Jain et al 2008) it is likely that ourapproach ignores as much as 20 of the gene space
After applying multi-exon and protein domain filters50172 gene models remained These transcripts rangedfrom 120 bp to12 kbp in length and represented48 Mbpof the genome with an average coding sequence length of965 bp (Table S2) Since a recognizable protein domain wasa requirement the majority of gene models are functionallyannotated (File S3) Functional annotations of the translatedsequences revealed that 49196 (98) of the transcripts havehomology to existing sequences in NCBIrsquos plant protein andnr database A slightly smaller number 48614 (97) alignto a functionally characterized gene product
The full-length gene models that are well supported byaligned evidence and with an AED score 020 total15653 These high-confidence transcripts cover 30 Mbpof the genome and have an average coding sequence lengthof 1295 bp with an average of three introns per gene (TableS2) Once again 98 of these sequences are functionallyannotated 23 of the high-quality alignments are toV vinifera Given the fragmentation of the genome and thelack of intronless genes reported true estimates of genenumber may be as high as 60000 particularly given the83285 unique gene models generated from broad P taedaRNA evidence (File S1) Previous estimates of gene numberin conifers vary widely The gene content of white spruce isestimated to be 32000 (Rigault et al 2011) While com-prehensive evaluation of maritime pine transcriptomicresources from EuroPineDB produced 52000 UniGenes(Fernandez-Pozo et al 2011)
Introns
Plant introns are typically shorter than those found inmammals (Shepard et al 2009) The gene models derivedfrom MAKER-P yielded maximum intron lengths 50 kbpwith several 100 kbp A total of 147425 introns wereidentified in the 50172 transcripts A requirement of25 gap-region content was used to calculate values on
Table 2 Mapping protein sequence against P taeda version 101 genome
Full-length proteins from Total sequence Unique hits Non-unique hits Total mapped Data source
P abies 22070 11580 3638 6895 Nystedt et al (2013)P sitchensis 10793 6516 1574 7495 GenBankP taeda 83285 45656 24427 8415 Current assemblyPLAZA (24 species) 653613 90149 19492 1677 Van Bel et al (2012)
896 J L Wegrzyn et al
144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

coverage served as the starting point for loblolly pinewhile other conifer species were given more permissive(95 identity95 coverage) cutoffs The thresholdswere lowered for all sequence sets to 95 identity50 coverage to further examine the effects of genomefragmentation
To discover orthologous proteins and align them to thegenome we began with 653613 proteins spanning 24species from version 25 of the PLAZA data set (Van Belet al 2012) PLAZA provides a curated and comprehensivecomparative genomics resource for the Viridiplantae includ-ing annotated proteins This set was further curated to ex-clude proteins that were not full length those shorter than21 amino acids and those that had genomic coordinatesthat did not agree with the reported coding sequence (CDS)or did not translate into the reported protein To this set25347 angiosperm proteins reported by the Amborella Ge-nome Project (httpwwwamborellaorg) were addedFrom annotated full-length mRNAs available in GenBanka set of 10793 proteins from Picea sitchensis were includedFrom the Picea abies v10 genome project (Nystedt et al2013) 22070 full-length proteins were included wherethe reported CDS agreed with the translated protein (FileS2 and Table S1) From the loblolly pine transcriptome all83285 transcripts were translated for alignment For thePLAZA protein alignments the target version 101 genomewas hard-masked for repeats for all other alignments thev101 genome was not repeat-masked We accepted analignment when at least 70 of the query sequence wasincluded in the alignment and the exonerate similarity score(ESS) $70
Gene annotation
Annotations for the assembly were generated using theautomated genome annotation pipeline MAKER-P whichaligns and filters EST and protein homology evidenceproduces ab initio gene predictions infers 59 and 39 UTRsand integrates these data to produce final downstream genemodels with quality control statistics (Campbell et al 2014)Inputs for MAKER-P include the Pinus taeda genome assem-bly (v10) Pinus and Picea ESTs conifer transcriptome as-semblies a species-specific repeat library (PIER) (Wegrzynet al 2013) protein databases containing annotated pro-teins for P sitchensis and P abies and version 25 of thePLAZA protein database (Van Bel et al 2012) MAKER-Pproduced ab initio gene predictions via SNAP (Korf 2004)and Augustus (Stanke and Waack 2003) A subset of anno-tations (500 Mb of sequence) were manually reviewed todevelop appropriate filters for the final gene models Giventhe large genome size and potential for spurious annota-tions conservative thresholds for these filters were chosenFilters included the initial removal of single-exon annota-tions because of apparent pseudogene bias annotationsnot containing a recognizable protein domain as interpretedusing InterProScan (Quevillon et al 2005) or annotationsthat do not partially overlap the conifer transcriptome
resources available Among the selected nonoverlappinggene models a subset of high confidence gene models wasfurther analyzed based on the annotation edit distance(AED) score of 020 with canonical start sites and splicesites All gene models identified in v10 were also mapped tothe latest assembly (v101) Functional annotations of thegene models were generated through blastp alignmentsagainst the National Center for Biotechnology Information(NCBI) nr and plant protein databases for significant (E-values 1e-05) and weak (E-value 0001) hits
Alignments of the high-confidence MAKER-P sequencesagainst the genome allowed for identification of largeintrons Introns of lengths 20 50 and 100 kbp andwith 25 gap regions were selected for further analysisClusters from the Markov cluster algorithm (MCL) analysisthat represented gene families with P taeda members andcontained primary introns (first CDS) 20 kbp were alignedand manually reviewed The intronic sequences were inves-tigated for the presence of intron-mediated expression sig-nals (IMEs) IMEs were predicted via sequence motifs usingthe methodology outlined in Parra et al (2007)
Orthologous proteins
The analysis of orthologous genes included a subset of thespecies that were previously selected from PLAZA A total of10 including Arabidopsis thaliana (27403) Glycine max(46324) Oryza sativa (41363) Physcomitrella patens(28090) Populus trichocarpa (40141) Ricinus communis(31009) Selaginella moellendorffii (18384) Theobromacacao (28858) Vitis vinifera (26238) and Zea mays (39172)were used Three external protein sequence sets were alsoincluded Amborella trichopoda (25347) P abies (22070)and P sitchensis (10521) The primary source of the P taedasequence was the complete set of 50172 gene models gen-erated from the MAKER-P pipeline All 14 sequence sets(including the 10 from PLAZA) were clustered to 90 iden-tity within species and combined to generate 399358sequences
The MCL analysis (Enright et al 2002) as implementedin the TRIBE-MCL pipeline (Dongen and Abreu-Goodger2012) was used to cluster the 399358 protein sequencesfrom 14 species into orthologous groups The methodologywas selected due to its robust implementation that avoidsmerging clusters that share only a few edges This leads toaccurate identifications of gene families even in the presenceof lower-quality BLAST hits or promiscuous domains (Frechand Chen 2010) The analysis was performed by runningpairwise NCBI blastp v2227+ (Altschul et al 1990) (E-valuecutoff of 1e-05) against the full set of proteins describedabove In this pipeline the negative log10 of the resultingblastp E-values produces a network graph that serves as in-put to define the orthologous groups The user-supplied in-flation value is used in the second stage to simulate randomwalks in the previously calculated graph A large inflationvalue defines more clusters with lower amount of mem-bers (fine-grained granularity) a small inflation value
Annotation of Loblolly Pine Genome 893
defines not as many clusters but the clusters have moremembers (coarse granularity) A moderate inflation valueof 40 was selected to define the orthologous groups hereFollowing their generation Pfam domains (Punta et al2012) were assigned from the PLAZA annotations of theindividual sequences InterProScan 48 (Hunter et al 2012)was applied to those sequences obtained outside of PLAZA(A trichopoda P sitchensis P abies and P taeda) PFam andGene Ontology (GO) assignments with E-values 1e-05were retained To effectively compare GO annotations theterms were normalized to level four of the classification treeWhen all predicted domains for a given family were classi-fied as retroelements the family was removed After func-tional assessment and filtering custom scripts and Venndiagrams (httpbioinformaticspsbugentbewebtoolsVenn)were applied to visualize gene family membership amongspecies
Gene family gain-and-loss phylogenetics
The DOLLOP v3695 program from the Phylip package(Felsenstein 1989) was used to reconstruct a parsimonioustree explaining the gain and loss of gene families under theDOLLOP parsimony model The DOLLOP algorithm esti-mates phylogenies for discrete character data with twostates (either a 0 or 1) which assumes only one gain butas many losses as necessary to explain the evolutionary pat-tern of states The filtered 8519 gene families (containing112899 sequences representing 13 species the small num-ber of sequences from P sitchensis excluded) were used tocreate a gene family gain-and-loss matrix Gene families thatrepresented all species were dropped from the input to DOL-LOP as their phylogenetic classification would be obfuscatedby a disparate number of starting proteins per species in theanalysis Branch lengths were calculated by counting thenumber of gains or losses for a given node To produce the finalphylogenetic reconstruction a manual curation step was in-troduced to bring the P patens and Selaginella moellendorffiibranches into agreement with known phylogenies DOLLOPwas rerun on this updated tree to reconstruct the final genefamily configurations of the ancestral species Only P patensS moellendorffii and their common ancestor were updatedduring this step The fidelity of the reconstructed trees wasevaluated against a known phylogeny available at Phytozome(Goodstein et al 2012)
Tandem repeat identification
Tandem Repeat Finder (TRF) v407b (Benson 1999) wasrun with the following parameters matching weight of 2mismatch weight of 7 indel penalty of 7 match probabilityof 80 indel probability of 10 minimum score of 50 anda maximum period size (repeating unit) of 2000 Both thegenome and transcriptome were examined and investigatedfor tandem content To accurately assess the overall cover-age and distribution of tandem repeats we filtered overlapsby discriminating against multimeric repeats following pre-viously described approaches (Melters et al 2013) and
excluded those found within interspersed repeats such asthe long terminal ends of LTR transposable elementsMononucleotides (period size of 1) were not scrutinizeddue to the high likelihood of error andor repeat collapsein the assembly process To maintain reproducibility com-mon nomenclature termsmdashsuch as ldquomicrosatellitesrdquo or sim-ple sequence repeats (SSRs) describing 1- to 8-bp periodsldquominisatellitesrdquo describing 9- to 100-bp periods and ldquosat-ellitesrdquo describing 100-bp periodsmdashwere used for classi-fication For comparative analysis these methods wereapplied to the reference genomes of A thaliana v167V vinifera v145 S moellendorffii v100 C sativus v122and P trichocarpa v210 each available through Phyto-zome (Goodstein et al 2012) The draft genome sequencesof P glauca v100 (Birol et al 2013) P abies (Nystedtet al 2013) and A trichopoda v100 were included Boththe filtered TRF output and consensus sequences derivedfrom clustering the filtered TRF output (UCLUST utility at70 identity) were used as queries in similarity searchesUSEARCH (E-value 001) was used to search the Plant-Sat database (Macas et al 2002) Potential centromericsequences were assessed by finding the monomeric tandemarray that covered the largest amount of the genome In-terstitial and true telomeric sequences were isolated fromthe filtered TRF output by searching for (TTTAGGG)n motifs(n 3 and length 1 kbp) The thresholds applied werebased on conservative estimates from telomere restrictionfragment lengths described for P taeda (Flanary andKletetschka 2005)
Homology-based repeat identification
RepeatMasker 330 (RepeatMasker 2013) was used toidentify previously characterized repeats RepeatMaskerwas run using standard settings on the entire genome withPIER 20 as a repeat library (Wegrzyn et al 2013) A maskedversion of the genome was generated at this stage Redun-dancy was eliminated through custom Python scripts bymatching each reference base pair with the highest scoringalignment For full-length estimates we required that thegenomic sequence and the reference PIER elements alignedwith at least 80 identity and 70 coverage Repeat con-tent in introns was independently characterized using thesame methodology High-copy elements were identified bysorting families by full-length copy number High-coverageelements were determined by sorting families by base-paircoverage including both full-length and partial hits MITEHunter (release 112011) (Han and Wessler 2010) wasused to search for miniature inverted-repeat transposableelements (MITEs) within intronic regions
De novo repeat identification
The workflow for characterization and annotation of novelrepeats largely mirrored that described in Wegrzyn et al(2013) REPET 20 (Flutre et al 2011) was used to identifysequences to form the repeat library for CENSOR 4227(Kohany et al 2006) REPETrsquos de novo repeat discovery
894 J L Wegrzyn et al
performs an all-versus-all alignment of the input sequencean unscalable computationally intensive operation As suchthe 63 longest scaffolds in the assembly were used as inputrepresenting 1 of the genome (bin 1) For use in REPETrsquosclassification steps we provided REPET with the PIER data-base in addition to a set of seven conifer repeat librariesdescribed in Nystedt et al (2013) A set of 405109 publiclyavailable full and partial P taeda and Pinus elliottii transcriptswere provided to REPET for host gene identification
Previously characterized elements were removed usingthe PIER and spruce repeat databases and the blastn-blastxstructural filters proposed in Wicker et al (2007) and imple-mented in Wegrzyn et al (2013) Previously classified elementswere identified by blastn alignments passing the 80-80-80threshold (80 identity covering 80 of each sequence forat least 80 bp) Unclassified elements were analyzed usingblastx against a database of ORFs recovered from thePIERspruce nucleotide database with USEARCHrsquos ldquofindorfsrdquoutility at default parameters (Edgar 2010) Elements thatwere classified only to the superfamily level and unclassifiedrepeats were separately clustered with UCLUST (Edgar 2010)at 80 identity Consensus sequences for each cluster werederived from multiple alignments built with MUSCLE(Edgar 2010) and PILER (Edgar and Myers 2005) Finallyseparate clusters were chained into families in which eachclusterrsquos consensus sequence aligned with at least 80identity with at least one other sequence in the familyFurther classification of LTR retroelement families was per-formed using the in-house tool GCclassif (Figure S1 andFile S4)
Results and Discussion
Sequence alignments
Aligning the 83285 de novo-assembled transcripts to thegenome allowed us to infer information about gene regions
including introns and repeats (Table 1) At a stringency of98 query coverage and 98 identity 30993 transcriptsmapped to the genome most of which (29262) mappedwith a unique hit 1731 transcripts mapped to two or moregenomic regions Relaxing the cutoff criteria to 95 identityand 95 query coverage resulted in increases in the numberof transcripts with unique (43972) and non-unique hits(5409) At 95 identity and 50 query coverage the num-ber of transcripts with unique hits to the genome rose to44469 and transcripts aligning to more than one locus to28116 Possible reasons for a transcript aligning to two ormore genomic locations include gene duplications pseudo-genes assembly errors and actively transcribed retroele-ments in the transcriptome assembly
To elucidate how genome fragmentation affected ourability to map sequence to the genome the v101 genomescaffolds were sorted by descending length and divided into100 bins with each bin containing 1 of the genomicsequence Peaks in the gap region content were found inbin 59 (360 ldquoNrdquo bases) and in bin 82 (478 ldquoNrdquo bases)reflecting gaps due to linking libraries (Figure S2A) Whilegap-region content is greatly reduced in bins 86ndash100 thesesame bins show a sharp decrease in scaffold size and hencea sharp increase in the number of scaffolds per bin (FigureS2B) As expected the ability to align transcripts to thegenome decreased as the scaffold length shortened with564 of these transcript mappings occurring in the first25 bins and just 85 in the last 25 bins (Figure S2C)
Using the same techniques we produced transcriptome-to-genome alignments beyond those generated with the denovo transcriptome using existing EST and transcriptomedata for loblolly pine and other closely related species(Table 1) A total of 741 of the loblolly pine reclusteredESTs mapped at 95 query coverage 95 sequence iden-tity Sequencing technology did not affect the percentageof sequence sets aligning to the genome as much as the
Table 1 Mapping ESTtranscriptome resources against P taeda version 101 genome
Project Total sequence Identity Coverage Unique hits Non-unique hits Total mapped
P taeda (reclustered ESTs) 45085 98 98 26700 712 608P taeda (reclustered ESTs) 45085 98 95 29676 1845 6991P taeda (reclustered ESTs) 45085 95 95 31324 2074 7401P taeda (reclustered ESTs) 45085 95 50 29744 5486 7814P taeda (de novo) 83285 98 98 29262 1731 3521P taeda (de novo) 83285 98 95 42822 5130 5758P taeda (de novo) 83285 95 95 43972 5409 5929P taeda (de novo) 83285 95 50 44469 28116 8715P palustris (454) 16832 95 95 11242 719 7106P palustris (454) 16832 95 50 11181 1949 7806P lambertiana (454 + RNASeq) 40619 95 95 13134 317 3311P lambertiana (454 + RNASeq) 40619 95 50 23376 3792 6688P banksiana (TreeGenes clusters) 13040 95 95 9703 513 7834P banksiana (TreeGenes clusters) 13040 95 50 9470 1473 8392P contorta (TreeGenes clusters) 13570 95 95 9575 396 7348P contorta (TreeGenes clusters) 13570 95 50 9534 1083 7824P pinaster (TreeGenes clusters) 15648 95 95 9738 943 6826P pinaster (TreeGenes clusters) 15648 95 50 10221 2491 8124
Annotation of Loblolly Pine Genome 895
phylogenetic relationships among the species P palustrisis most closely related to loblolly pine and obtains map-ping rates similar to the reclustered ESTs at 711 with95 query coverage and 95 identity P lambertiana ismost distant and this is reflected in the minimal mappingsuccess of the transcripts (Table 1)
The common lineage of the angiosperms and the gym-nosperms split 300 MYA (Jiao et al 2011) impedingsearches for nucleic acid sequence homology betweenangiosperms and loblolly pine However we expect a subsetof proteins to remain relatively conserved over multiplegeological periods The results of mapping and aligningthree protein data sets to the loblolly pine v101 genomeare shown in Table 2 where we determined the initialprotein-to-scaffold mapping with blat and created a refinedalignment with exonerate The subset of the PLAZA dataset used includes over half a million proteins from 24 non-gymnosperm plant species 168 of which aligned to theloblolly pine genome Aligning proteins from members ofthe genus Picea (last common ancestor 85 MYA) to theloblolly pine genome proved more fruitful Seventy-fivepercent of 10000 full-length Sitka spruce proteins [themajority predicted from high-quality full-length Sanger-sequenced complementary DNA data (Ralph et al 2008)]aligned to the genome Sixty-nine percent of 22070 P abiesproteins from the Congenie project aligned to the loblollypine genome A total of 84 of the loblolly pine proteinsgleaned from the transcriptome assembly aligned
Fewer than 30 of each PLAZA species protein set alignedto the genome at our initial cutoff (query coverage$70 andESS $70) For 18 species in the PLAZA data set as well asA trichopoda P sitchensis P abies and translated P taedatranscriptome sequences we further break down the protein-to-genome alignments that passed the initial cutoff into threecategories ESS $ 90 90 ESS 80 and 70 ESS 80(Figure 1) In the ESS $ 90 category 452 of Norwayspruce proteins 606 of Sitka spruce proteins and 700of loblolly pine proteins aligned to the genome (File S2)The differences between the two spruce protein sets maybe partially attributed to the sequencing technologiesemployed to generate the original transcriptomes (Sangervs next-generation technologies)
Gene annotation
The MAKER-P annotation pipeline considered the transcriptalignments in addition to ab initio models to predict poten-tial coding regions The initial predictions generated90000 gene models 44 were fragmented or otherwise
indicative of pseudogenes Previous studies of pines haveidentified pseudogene members in large gene families(Wakasugi et al 1994 Skinner and Timko 1998 Gernandtet al 2001 Garcia-Gil 2008) and pseudogene content maybe as much as five times that of functional coding regions(Kovach et al 2010) Conservative filters were thereforenecessary to deduce the actual gene space The multi-exongene model requirement is likely biased against intronlessgenes but it reduces false positives associated with repeatsand pseudogenes Because 20 of Arabidopsis and O sativagenes are intronless (Jain et al 2008) it is likely that ourapproach ignores as much as 20 of the gene space
After applying multi-exon and protein domain filters50172 gene models remained These transcripts rangedfrom 120 bp to12 kbp in length and represented48 Mbpof the genome with an average coding sequence length of965 bp (Table S2) Since a recognizable protein domain wasa requirement the majority of gene models are functionallyannotated (File S3) Functional annotations of the translatedsequences revealed that 49196 (98) of the transcripts havehomology to existing sequences in NCBIrsquos plant protein andnr database A slightly smaller number 48614 (97) alignto a functionally characterized gene product
The full-length gene models that are well supported byaligned evidence and with an AED score 020 total15653 These high-confidence transcripts cover 30 Mbpof the genome and have an average coding sequence lengthof 1295 bp with an average of three introns per gene (TableS2) Once again 98 of these sequences are functionallyannotated 23 of the high-quality alignments are toV vinifera Given the fragmentation of the genome and thelack of intronless genes reported true estimates of genenumber may be as high as 60000 particularly given the83285 unique gene models generated from broad P taedaRNA evidence (File S1) Previous estimates of gene numberin conifers vary widely The gene content of white spruce isestimated to be 32000 (Rigault et al 2011) While com-prehensive evaluation of maritime pine transcriptomicresources from EuroPineDB produced 52000 UniGenes(Fernandez-Pozo et al 2011)
Introns
Plant introns are typically shorter than those found inmammals (Shepard et al 2009) The gene models derivedfrom MAKER-P yielded maximum intron lengths 50 kbpwith several 100 kbp A total of 147425 introns wereidentified in the 50172 transcripts A requirement of25 gap-region content was used to calculate values on
Table 2 Mapping protein sequence against P taeda version 101 genome
Full-length proteins from Total sequence Unique hits Non-unique hits Total mapped Data source
P abies 22070 11580 3638 6895 Nystedt et al (2013)P sitchensis 10793 6516 1574 7495 GenBankP taeda 83285 45656 24427 8415 Current assemblyPLAZA (24 species) 653613 90149 19492 1677 Van Bel et al (2012)
896 J L Wegrzyn et al
144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

defines not as many clusters but the clusters have moremembers (coarse granularity) A moderate inflation valueof 40 was selected to define the orthologous groups hereFollowing their generation Pfam domains (Punta et al2012) were assigned from the PLAZA annotations of theindividual sequences InterProScan 48 (Hunter et al 2012)was applied to those sequences obtained outside of PLAZA(A trichopoda P sitchensis P abies and P taeda) PFam andGene Ontology (GO) assignments with E-values 1e-05were retained To effectively compare GO annotations theterms were normalized to level four of the classification treeWhen all predicted domains for a given family were classi-fied as retroelements the family was removed After func-tional assessment and filtering custom scripts and Venndiagrams (httpbioinformaticspsbugentbewebtoolsVenn)were applied to visualize gene family membership amongspecies
Gene family gain-and-loss phylogenetics
The DOLLOP v3695 program from the Phylip package(Felsenstein 1989) was used to reconstruct a parsimonioustree explaining the gain and loss of gene families under theDOLLOP parsimony model The DOLLOP algorithm esti-mates phylogenies for discrete character data with twostates (either a 0 or 1) which assumes only one gain butas many losses as necessary to explain the evolutionary pat-tern of states The filtered 8519 gene families (containing112899 sequences representing 13 species the small num-ber of sequences from P sitchensis excluded) were used tocreate a gene family gain-and-loss matrix Gene families thatrepresented all species were dropped from the input to DOL-LOP as their phylogenetic classification would be obfuscatedby a disparate number of starting proteins per species in theanalysis Branch lengths were calculated by counting thenumber of gains or losses for a given node To produce the finalphylogenetic reconstruction a manual curation step was in-troduced to bring the P patens and Selaginella moellendorffiibranches into agreement with known phylogenies DOLLOPwas rerun on this updated tree to reconstruct the final genefamily configurations of the ancestral species Only P patensS moellendorffii and their common ancestor were updatedduring this step The fidelity of the reconstructed trees wasevaluated against a known phylogeny available at Phytozome(Goodstein et al 2012)
Tandem repeat identification
Tandem Repeat Finder (TRF) v407b (Benson 1999) wasrun with the following parameters matching weight of 2mismatch weight of 7 indel penalty of 7 match probabilityof 80 indel probability of 10 minimum score of 50 anda maximum period size (repeating unit) of 2000 Both thegenome and transcriptome were examined and investigatedfor tandem content To accurately assess the overall cover-age and distribution of tandem repeats we filtered overlapsby discriminating against multimeric repeats following pre-viously described approaches (Melters et al 2013) and
excluded those found within interspersed repeats such asthe long terminal ends of LTR transposable elementsMononucleotides (period size of 1) were not scrutinizeddue to the high likelihood of error andor repeat collapsein the assembly process To maintain reproducibility com-mon nomenclature termsmdashsuch as ldquomicrosatellitesrdquo or sim-ple sequence repeats (SSRs) describing 1- to 8-bp periodsldquominisatellitesrdquo describing 9- to 100-bp periods and ldquosat-ellitesrdquo describing 100-bp periodsmdashwere used for classi-fication For comparative analysis these methods wereapplied to the reference genomes of A thaliana v167V vinifera v145 S moellendorffii v100 C sativus v122and P trichocarpa v210 each available through Phyto-zome (Goodstein et al 2012) The draft genome sequencesof P glauca v100 (Birol et al 2013) P abies (Nystedtet al 2013) and A trichopoda v100 were included Boththe filtered TRF output and consensus sequences derivedfrom clustering the filtered TRF output (UCLUST utility at70 identity) were used as queries in similarity searchesUSEARCH (E-value 001) was used to search the Plant-Sat database (Macas et al 2002) Potential centromericsequences were assessed by finding the monomeric tandemarray that covered the largest amount of the genome In-terstitial and true telomeric sequences were isolated fromthe filtered TRF output by searching for (TTTAGGG)n motifs(n 3 and length 1 kbp) The thresholds applied werebased on conservative estimates from telomere restrictionfragment lengths described for P taeda (Flanary andKletetschka 2005)
Homology-based repeat identification
RepeatMasker 330 (RepeatMasker 2013) was used toidentify previously characterized repeats RepeatMaskerwas run using standard settings on the entire genome withPIER 20 as a repeat library (Wegrzyn et al 2013) A maskedversion of the genome was generated at this stage Redun-dancy was eliminated through custom Python scripts bymatching each reference base pair with the highest scoringalignment For full-length estimates we required that thegenomic sequence and the reference PIER elements alignedwith at least 80 identity and 70 coverage Repeat con-tent in introns was independently characterized using thesame methodology High-copy elements were identified bysorting families by full-length copy number High-coverageelements were determined by sorting families by base-paircoverage including both full-length and partial hits MITEHunter (release 112011) (Han and Wessler 2010) wasused to search for miniature inverted-repeat transposableelements (MITEs) within intronic regions
De novo repeat identification
The workflow for characterization and annotation of novelrepeats largely mirrored that described in Wegrzyn et al(2013) REPET 20 (Flutre et al 2011) was used to identifysequences to form the repeat library for CENSOR 4227(Kohany et al 2006) REPETrsquos de novo repeat discovery
894 J L Wegrzyn et al
performs an all-versus-all alignment of the input sequencean unscalable computationally intensive operation As suchthe 63 longest scaffolds in the assembly were used as inputrepresenting 1 of the genome (bin 1) For use in REPETrsquosclassification steps we provided REPET with the PIER data-base in addition to a set of seven conifer repeat librariesdescribed in Nystedt et al (2013) A set of 405109 publiclyavailable full and partial P taeda and Pinus elliottii transcriptswere provided to REPET for host gene identification
Previously characterized elements were removed usingthe PIER and spruce repeat databases and the blastn-blastxstructural filters proposed in Wicker et al (2007) and imple-mented in Wegrzyn et al (2013) Previously classified elementswere identified by blastn alignments passing the 80-80-80threshold (80 identity covering 80 of each sequence forat least 80 bp) Unclassified elements were analyzed usingblastx against a database of ORFs recovered from thePIERspruce nucleotide database with USEARCHrsquos ldquofindorfsrdquoutility at default parameters (Edgar 2010) Elements thatwere classified only to the superfamily level and unclassifiedrepeats were separately clustered with UCLUST (Edgar 2010)at 80 identity Consensus sequences for each cluster werederived from multiple alignments built with MUSCLE(Edgar 2010) and PILER (Edgar and Myers 2005) Finallyseparate clusters were chained into families in which eachclusterrsquos consensus sequence aligned with at least 80identity with at least one other sequence in the familyFurther classification of LTR retroelement families was per-formed using the in-house tool GCclassif (Figure S1 andFile S4)
Results and Discussion
Sequence alignments
Aligning the 83285 de novo-assembled transcripts to thegenome allowed us to infer information about gene regions
including introns and repeats (Table 1) At a stringency of98 query coverage and 98 identity 30993 transcriptsmapped to the genome most of which (29262) mappedwith a unique hit 1731 transcripts mapped to two or moregenomic regions Relaxing the cutoff criteria to 95 identityand 95 query coverage resulted in increases in the numberof transcripts with unique (43972) and non-unique hits(5409) At 95 identity and 50 query coverage the num-ber of transcripts with unique hits to the genome rose to44469 and transcripts aligning to more than one locus to28116 Possible reasons for a transcript aligning to two ormore genomic locations include gene duplications pseudo-genes assembly errors and actively transcribed retroele-ments in the transcriptome assembly
To elucidate how genome fragmentation affected ourability to map sequence to the genome the v101 genomescaffolds were sorted by descending length and divided into100 bins with each bin containing 1 of the genomicsequence Peaks in the gap region content were found inbin 59 (360 ldquoNrdquo bases) and in bin 82 (478 ldquoNrdquo bases)reflecting gaps due to linking libraries (Figure S2A) Whilegap-region content is greatly reduced in bins 86ndash100 thesesame bins show a sharp decrease in scaffold size and hencea sharp increase in the number of scaffolds per bin (FigureS2B) As expected the ability to align transcripts to thegenome decreased as the scaffold length shortened with564 of these transcript mappings occurring in the first25 bins and just 85 in the last 25 bins (Figure S2C)
Using the same techniques we produced transcriptome-to-genome alignments beyond those generated with the denovo transcriptome using existing EST and transcriptomedata for loblolly pine and other closely related species(Table 1) A total of 741 of the loblolly pine reclusteredESTs mapped at 95 query coverage 95 sequence iden-tity Sequencing technology did not affect the percentageof sequence sets aligning to the genome as much as the
Table 1 Mapping ESTtranscriptome resources against P taeda version 101 genome
Project Total sequence Identity Coverage Unique hits Non-unique hits Total mapped
P taeda (reclustered ESTs) 45085 98 98 26700 712 608P taeda (reclustered ESTs) 45085 98 95 29676 1845 6991P taeda (reclustered ESTs) 45085 95 95 31324 2074 7401P taeda (reclustered ESTs) 45085 95 50 29744 5486 7814P taeda (de novo) 83285 98 98 29262 1731 3521P taeda (de novo) 83285 98 95 42822 5130 5758P taeda (de novo) 83285 95 95 43972 5409 5929P taeda (de novo) 83285 95 50 44469 28116 8715P palustris (454) 16832 95 95 11242 719 7106P palustris (454) 16832 95 50 11181 1949 7806P lambertiana (454 + RNASeq) 40619 95 95 13134 317 3311P lambertiana (454 + RNASeq) 40619 95 50 23376 3792 6688P banksiana (TreeGenes clusters) 13040 95 95 9703 513 7834P banksiana (TreeGenes clusters) 13040 95 50 9470 1473 8392P contorta (TreeGenes clusters) 13570 95 95 9575 396 7348P contorta (TreeGenes clusters) 13570 95 50 9534 1083 7824P pinaster (TreeGenes clusters) 15648 95 95 9738 943 6826P pinaster (TreeGenes clusters) 15648 95 50 10221 2491 8124
Annotation of Loblolly Pine Genome 895
phylogenetic relationships among the species P palustrisis most closely related to loblolly pine and obtains map-ping rates similar to the reclustered ESTs at 711 with95 query coverage and 95 identity P lambertiana ismost distant and this is reflected in the minimal mappingsuccess of the transcripts (Table 1)
The common lineage of the angiosperms and the gym-nosperms split 300 MYA (Jiao et al 2011) impedingsearches for nucleic acid sequence homology betweenangiosperms and loblolly pine However we expect a subsetof proteins to remain relatively conserved over multiplegeological periods The results of mapping and aligningthree protein data sets to the loblolly pine v101 genomeare shown in Table 2 where we determined the initialprotein-to-scaffold mapping with blat and created a refinedalignment with exonerate The subset of the PLAZA dataset used includes over half a million proteins from 24 non-gymnosperm plant species 168 of which aligned to theloblolly pine genome Aligning proteins from members ofthe genus Picea (last common ancestor 85 MYA) to theloblolly pine genome proved more fruitful Seventy-fivepercent of 10000 full-length Sitka spruce proteins [themajority predicted from high-quality full-length Sanger-sequenced complementary DNA data (Ralph et al 2008)]aligned to the genome Sixty-nine percent of 22070 P abiesproteins from the Congenie project aligned to the loblollypine genome A total of 84 of the loblolly pine proteinsgleaned from the transcriptome assembly aligned
Fewer than 30 of each PLAZA species protein set alignedto the genome at our initial cutoff (query coverage$70 andESS $70) For 18 species in the PLAZA data set as well asA trichopoda P sitchensis P abies and translated P taedatranscriptome sequences we further break down the protein-to-genome alignments that passed the initial cutoff into threecategories ESS $ 90 90 ESS 80 and 70 ESS 80(Figure 1) In the ESS $ 90 category 452 of Norwayspruce proteins 606 of Sitka spruce proteins and 700of loblolly pine proteins aligned to the genome (File S2)The differences between the two spruce protein sets maybe partially attributed to the sequencing technologiesemployed to generate the original transcriptomes (Sangervs next-generation technologies)
Gene annotation
The MAKER-P annotation pipeline considered the transcriptalignments in addition to ab initio models to predict poten-tial coding regions The initial predictions generated90000 gene models 44 were fragmented or otherwise
indicative of pseudogenes Previous studies of pines haveidentified pseudogene members in large gene families(Wakasugi et al 1994 Skinner and Timko 1998 Gernandtet al 2001 Garcia-Gil 2008) and pseudogene content maybe as much as five times that of functional coding regions(Kovach et al 2010) Conservative filters were thereforenecessary to deduce the actual gene space The multi-exongene model requirement is likely biased against intronlessgenes but it reduces false positives associated with repeatsand pseudogenes Because 20 of Arabidopsis and O sativagenes are intronless (Jain et al 2008) it is likely that ourapproach ignores as much as 20 of the gene space
After applying multi-exon and protein domain filters50172 gene models remained These transcripts rangedfrom 120 bp to12 kbp in length and represented48 Mbpof the genome with an average coding sequence length of965 bp (Table S2) Since a recognizable protein domain wasa requirement the majority of gene models are functionallyannotated (File S3) Functional annotations of the translatedsequences revealed that 49196 (98) of the transcripts havehomology to existing sequences in NCBIrsquos plant protein andnr database A slightly smaller number 48614 (97) alignto a functionally characterized gene product
The full-length gene models that are well supported byaligned evidence and with an AED score 020 total15653 These high-confidence transcripts cover 30 Mbpof the genome and have an average coding sequence lengthof 1295 bp with an average of three introns per gene (TableS2) Once again 98 of these sequences are functionallyannotated 23 of the high-quality alignments are toV vinifera Given the fragmentation of the genome and thelack of intronless genes reported true estimates of genenumber may be as high as 60000 particularly given the83285 unique gene models generated from broad P taedaRNA evidence (File S1) Previous estimates of gene numberin conifers vary widely The gene content of white spruce isestimated to be 32000 (Rigault et al 2011) While com-prehensive evaluation of maritime pine transcriptomicresources from EuroPineDB produced 52000 UniGenes(Fernandez-Pozo et al 2011)
Introns
Plant introns are typically shorter than those found inmammals (Shepard et al 2009) The gene models derivedfrom MAKER-P yielded maximum intron lengths 50 kbpwith several 100 kbp A total of 147425 introns wereidentified in the 50172 transcripts A requirement of25 gap-region content was used to calculate values on
Table 2 Mapping protein sequence against P taeda version 101 genome
Full-length proteins from Total sequence Unique hits Non-unique hits Total mapped Data source
P abies 22070 11580 3638 6895 Nystedt et al (2013)P sitchensis 10793 6516 1574 7495 GenBankP taeda 83285 45656 24427 8415 Current assemblyPLAZA (24 species) 653613 90149 19492 1677 Van Bel et al (2012)
896 J L Wegrzyn et al
144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

performs an all-versus-all alignment of the input sequencean unscalable computationally intensive operation As suchthe 63 longest scaffolds in the assembly were used as inputrepresenting 1 of the genome (bin 1) For use in REPETrsquosclassification steps we provided REPET with the PIER data-base in addition to a set of seven conifer repeat librariesdescribed in Nystedt et al (2013) A set of 405109 publiclyavailable full and partial P taeda and Pinus elliottii transcriptswere provided to REPET for host gene identification
Previously characterized elements were removed usingthe PIER and spruce repeat databases and the blastn-blastxstructural filters proposed in Wicker et al (2007) and imple-mented in Wegrzyn et al (2013) Previously classified elementswere identified by blastn alignments passing the 80-80-80threshold (80 identity covering 80 of each sequence forat least 80 bp) Unclassified elements were analyzed usingblastx against a database of ORFs recovered from thePIERspruce nucleotide database with USEARCHrsquos ldquofindorfsrdquoutility at default parameters (Edgar 2010) Elements thatwere classified only to the superfamily level and unclassifiedrepeats were separately clustered with UCLUST (Edgar 2010)at 80 identity Consensus sequences for each cluster werederived from multiple alignments built with MUSCLE(Edgar 2010) and PILER (Edgar and Myers 2005) Finallyseparate clusters were chained into families in which eachclusterrsquos consensus sequence aligned with at least 80identity with at least one other sequence in the familyFurther classification of LTR retroelement families was per-formed using the in-house tool GCclassif (Figure S1 andFile S4)
Results and Discussion
Sequence alignments
Aligning the 83285 de novo-assembled transcripts to thegenome allowed us to infer information about gene regions
including introns and repeats (Table 1) At a stringency of98 query coverage and 98 identity 30993 transcriptsmapped to the genome most of which (29262) mappedwith a unique hit 1731 transcripts mapped to two or moregenomic regions Relaxing the cutoff criteria to 95 identityand 95 query coverage resulted in increases in the numberof transcripts with unique (43972) and non-unique hits(5409) At 95 identity and 50 query coverage the num-ber of transcripts with unique hits to the genome rose to44469 and transcripts aligning to more than one locus to28116 Possible reasons for a transcript aligning to two ormore genomic locations include gene duplications pseudo-genes assembly errors and actively transcribed retroele-ments in the transcriptome assembly
To elucidate how genome fragmentation affected ourability to map sequence to the genome the v101 genomescaffolds were sorted by descending length and divided into100 bins with each bin containing 1 of the genomicsequence Peaks in the gap region content were found inbin 59 (360 ldquoNrdquo bases) and in bin 82 (478 ldquoNrdquo bases)reflecting gaps due to linking libraries (Figure S2A) Whilegap-region content is greatly reduced in bins 86ndash100 thesesame bins show a sharp decrease in scaffold size and hencea sharp increase in the number of scaffolds per bin (FigureS2B) As expected the ability to align transcripts to thegenome decreased as the scaffold length shortened with564 of these transcript mappings occurring in the first25 bins and just 85 in the last 25 bins (Figure S2C)
Using the same techniques we produced transcriptome-to-genome alignments beyond those generated with the denovo transcriptome using existing EST and transcriptomedata for loblolly pine and other closely related species(Table 1) A total of 741 of the loblolly pine reclusteredESTs mapped at 95 query coverage 95 sequence iden-tity Sequencing technology did not affect the percentageof sequence sets aligning to the genome as much as the
Table 1 Mapping ESTtranscriptome resources against P taeda version 101 genome
Project Total sequence Identity Coverage Unique hits Non-unique hits Total mapped
P taeda (reclustered ESTs) 45085 98 98 26700 712 608P taeda (reclustered ESTs) 45085 98 95 29676 1845 6991P taeda (reclustered ESTs) 45085 95 95 31324 2074 7401P taeda (reclustered ESTs) 45085 95 50 29744 5486 7814P taeda (de novo) 83285 98 98 29262 1731 3521P taeda (de novo) 83285 98 95 42822 5130 5758P taeda (de novo) 83285 95 95 43972 5409 5929P taeda (de novo) 83285 95 50 44469 28116 8715P palustris (454) 16832 95 95 11242 719 7106P palustris (454) 16832 95 50 11181 1949 7806P lambertiana (454 + RNASeq) 40619 95 95 13134 317 3311P lambertiana (454 + RNASeq) 40619 95 50 23376 3792 6688P banksiana (TreeGenes clusters) 13040 95 95 9703 513 7834P banksiana (TreeGenes clusters) 13040 95 50 9470 1473 8392P contorta (TreeGenes clusters) 13570 95 95 9575 396 7348P contorta (TreeGenes clusters) 13570 95 50 9534 1083 7824P pinaster (TreeGenes clusters) 15648 95 95 9738 943 6826P pinaster (TreeGenes clusters) 15648 95 50 10221 2491 8124
Annotation of Loblolly Pine Genome 895
phylogenetic relationships among the species P palustrisis most closely related to loblolly pine and obtains map-ping rates similar to the reclustered ESTs at 711 with95 query coverage and 95 identity P lambertiana ismost distant and this is reflected in the minimal mappingsuccess of the transcripts (Table 1)
The common lineage of the angiosperms and the gym-nosperms split 300 MYA (Jiao et al 2011) impedingsearches for nucleic acid sequence homology betweenangiosperms and loblolly pine However we expect a subsetof proteins to remain relatively conserved over multiplegeological periods The results of mapping and aligningthree protein data sets to the loblolly pine v101 genomeare shown in Table 2 where we determined the initialprotein-to-scaffold mapping with blat and created a refinedalignment with exonerate The subset of the PLAZA dataset used includes over half a million proteins from 24 non-gymnosperm plant species 168 of which aligned to theloblolly pine genome Aligning proteins from members ofthe genus Picea (last common ancestor 85 MYA) to theloblolly pine genome proved more fruitful Seventy-fivepercent of 10000 full-length Sitka spruce proteins [themajority predicted from high-quality full-length Sanger-sequenced complementary DNA data (Ralph et al 2008)]aligned to the genome Sixty-nine percent of 22070 P abiesproteins from the Congenie project aligned to the loblollypine genome A total of 84 of the loblolly pine proteinsgleaned from the transcriptome assembly aligned
Fewer than 30 of each PLAZA species protein set alignedto the genome at our initial cutoff (query coverage$70 andESS $70) For 18 species in the PLAZA data set as well asA trichopoda P sitchensis P abies and translated P taedatranscriptome sequences we further break down the protein-to-genome alignments that passed the initial cutoff into threecategories ESS $ 90 90 ESS 80 and 70 ESS 80(Figure 1) In the ESS $ 90 category 452 of Norwayspruce proteins 606 of Sitka spruce proteins and 700of loblolly pine proteins aligned to the genome (File S2)The differences between the two spruce protein sets maybe partially attributed to the sequencing technologiesemployed to generate the original transcriptomes (Sangervs next-generation technologies)
Gene annotation
The MAKER-P annotation pipeline considered the transcriptalignments in addition to ab initio models to predict poten-tial coding regions The initial predictions generated90000 gene models 44 were fragmented or otherwise
indicative of pseudogenes Previous studies of pines haveidentified pseudogene members in large gene families(Wakasugi et al 1994 Skinner and Timko 1998 Gernandtet al 2001 Garcia-Gil 2008) and pseudogene content maybe as much as five times that of functional coding regions(Kovach et al 2010) Conservative filters were thereforenecessary to deduce the actual gene space The multi-exongene model requirement is likely biased against intronlessgenes but it reduces false positives associated with repeatsand pseudogenes Because 20 of Arabidopsis and O sativagenes are intronless (Jain et al 2008) it is likely that ourapproach ignores as much as 20 of the gene space
After applying multi-exon and protein domain filters50172 gene models remained These transcripts rangedfrom 120 bp to12 kbp in length and represented48 Mbpof the genome with an average coding sequence length of965 bp (Table S2) Since a recognizable protein domain wasa requirement the majority of gene models are functionallyannotated (File S3) Functional annotations of the translatedsequences revealed that 49196 (98) of the transcripts havehomology to existing sequences in NCBIrsquos plant protein andnr database A slightly smaller number 48614 (97) alignto a functionally characterized gene product
The full-length gene models that are well supported byaligned evidence and with an AED score 020 total15653 These high-confidence transcripts cover 30 Mbpof the genome and have an average coding sequence lengthof 1295 bp with an average of three introns per gene (TableS2) Once again 98 of these sequences are functionallyannotated 23 of the high-quality alignments are toV vinifera Given the fragmentation of the genome and thelack of intronless genes reported true estimates of genenumber may be as high as 60000 particularly given the83285 unique gene models generated from broad P taedaRNA evidence (File S1) Previous estimates of gene numberin conifers vary widely The gene content of white spruce isestimated to be 32000 (Rigault et al 2011) While com-prehensive evaluation of maritime pine transcriptomicresources from EuroPineDB produced 52000 UniGenes(Fernandez-Pozo et al 2011)
Introns
Plant introns are typically shorter than those found inmammals (Shepard et al 2009) The gene models derivedfrom MAKER-P yielded maximum intron lengths 50 kbpwith several 100 kbp A total of 147425 introns wereidentified in the 50172 transcripts A requirement of25 gap-region content was used to calculate values on
Table 2 Mapping protein sequence against P taeda version 101 genome
Full-length proteins from Total sequence Unique hits Non-unique hits Total mapped Data source
P abies 22070 11580 3638 6895 Nystedt et al (2013)P sitchensis 10793 6516 1574 7495 GenBankP taeda 83285 45656 24427 8415 Current assemblyPLAZA (24 species) 653613 90149 19492 1677 Van Bel et al (2012)
896 J L Wegrzyn et al
144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

phylogenetic relationships among the species P palustrisis most closely related to loblolly pine and obtains map-ping rates similar to the reclustered ESTs at 711 with95 query coverage and 95 identity P lambertiana ismost distant and this is reflected in the minimal mappingsuccess of the transcripts (Table 1)
The common lineage of the angiosperms and the gym-nosperms split 300 MYA (Jiao et al 2011) impedingsearches for nucleic acid sequence homology betweenangiosperms and loblolly pine However we expect a subsetof proteins to remain relatively conserved over multiplegeological periods The results of mapping and aligningthree protein data sets to the loblolly pine v101 genomeare shown in Table 2 where we determined the initialprotein-to-scaffold mapping with blat and created a refinedalignment with exonerate The subset of the PLAZA dataset used includes over half a million proteins from 24 non-gymnosperm plant species 168 of which aligned to theloblolly pine genome Aligning proteins from members ofthe genus Picea (last common ancestor 85 MYA) to theloblolly pine genome proved more fruitful Seventy-fivepercent of 10000 full-length Sitka spruce proteins [themajority predicted from high-quality full-length Sanger-sequenced complementary DNA data (Ralph et al 2008)]aligned to the genome Sixty-nine percent of 22070 P abiesproteins from the Congenie project aligned to the loblollypine genome A total of 84 of the loblolly pine proteinsgleaned from the transcriptome assembly aligned
Fewer than 30 of each PLAZA species protein set alignedto the genome at our initial cutoff (query coverage$70 andESS $70) For 18 species in the PLAZA data set as well asA trichopoda P sitchensis P abies and translated P taedatranscriptome sequences we further break down the protein-to-genome alignments that passed the initial cutoff into threecategories ESS $ 90 90 ESS 80 and 70 ESS 80(Figure 1) In the ESS $ 90 category 452 of Norwayspruce proteins 606 of Sitka spruce proteins and 700of loblolly pine proteins aligned to the genome (File S2)The differences between the two spruce protein sets maybe partially attributed to the sequencing technologiesemployed to generate the original transcriptomes (Sangervs next-generation technologies)
Gene annotation
The MAKER-P annotation pipeline considered the transcriptalignments in addition to ab initio models to predict poten-tial coding regions The initial predictions generated90000 gene models 44 were fragmented or otherwise
indicative of pseudogenes Previous studies of pines haveidentified pseudogene members in large gene families(Wakasugi et al 1994 Skinner and Timko 1998 Gernandtet al 2001 Garcia-Gil 2008) and pseudogene content maybe as much as five times that of functional coding regions(Kovach et al 2010) Conservative filters were thereforenecessary to deduce the actual gene space The multi-exongene model requirement is likely biased against intronlessgenes but it reduces false positives associated with repeatsand pseudogenes Because 20 of Arabidopsis and O sativagenes are intronless (Jain et al 2008) it is likely that ourapproach ignores as much as 20 of the gene space
After applying multi-exon and protein domain filters50172 gene models remained These transcripts rangedfrom 120 bp to12 kbp in length and represented48 Mbpof the genome with an average coding sequence length of965 bp (Table S2) Since a recognizable protein domain wasa requirement the majority of gene models are functionallyannotated (File S3) Functional annotations of the translatedsequences revealed that 49196 (98) of the transcripts havehomology to existing sequences in NCBIrsquos plant protein andnr database A slightly smaller number 48614 (97) alignto a functionally characterized gene product
The full-length gene models that are well supported byaligned evidence and with an AED score 020 total15653 These high-confidence transcripts cover 30 Mbpof the genome and have an average coding sequence lengthof 1295 bp with an average of three introns per gene (TableS2) Once again 98 of these sequences are functionallyannotated 23 of the high-quality alignments are toV vinifera Given the fragmentation of the genome and thelack of intronless genes reported true estimates of genenumber may be as high as 60000 particularly given the83285 unique gene models generated from broad P taedaRNA evidence (File S1) Previous estimates of gene numberin conifers vary widely The gene content of white spruce isestimated to be 32000 (Rigault et al 2011) While com-prehensive evaluation of maritime pine transcriptomicresources from EuroPineDB produced 52000 UniGenes(Fernandez-Pozo et al 2011)
Introns
Plant introns are typically shorter than those found inmammals (Shepard et al 2009) The gene models derivedfrom MAKER-P yielded maximum intron lengths 50 kbpwith several 100 kbp A total of 147425 introns wereidentified in the 50172 transcripts A requirement of25 gap-region content was used to calculate values on
Table 2 Mapping protein sequence against P taeda version 101 genome
Full-length proteins from Total sequence Unique hits Non-unique hits Total mapped Data source
P abies 22070 11580 3638 6895 Nystedt et al (2013)P sitchensis 10793 6516 1574 7495 GenBankP taeda 83285 45656 24427 8415 Current assemblyPLAZA (24 species) 653613 90149 19492 1677 Van Bel et al (2012)
896 J L Wegrzyn et al
144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

144425 introns (Table 3) The average length of these in-trons was 27 kbp with a maximum length of 318 kbpCompared with mapping the loblolly pine transcriptomeagainst the genome we identified only 3350 sequences(13) with one or more introns Among these sequences10991 introns were present an average of 328 per se-quence Their maximum reported lengths are 150 kbp Thenumber of genes in the transcriptome containing introns ismuch lower than in other eukaryotes although this is likelyskewed by the requirement of full-length genes againsta fragmented genome For this reason we focused the anal-ysis on our high-confidence set of 15653 MAKER-derivedtranscripts which had 48720 usable introns with an averagelength of 24 kbp The longest reported intron in this set is158 kbp In total 1610 introns were between 20 and 49kbp in length 143 between 50 and 99 kbp and 18 100kbp in length (File S5) When compared with the proteinsequences from 22 other plant species aligned to theirgenomes only A trichopoda and Z mays reported similarmaximum values (Figure 2A) The median and mean val-ues for P taeda introns are comparable to the other plantspecies (Figure 2B) although it is likely that our intronlengths are an underestimate due to the fragmentation ofthe assembly Estimates from the recent genome assemblyof P abies reported intron lengths20 kbp and a maximumlength of 68 kbp (Nystedt et al 2013) Long intron lengthshave been estimated for pines for some time and are notedto contribute to their large genome sizes (Ahuja and Neale2005)
Long intron sizes delay the production of protein prod-ucts and increase the error rate in intron splicing in animals(Sun and Chasin 2000) In plants increasing intron length ispositively correlated with gene expression (Ren et al 2006)In most eukaryotes the first (primary) intron is usually thelongest (Bradnam and Korf 2008) and is generally in the 59UTR The first intron in the CDS region is also generallylonger than distal introns (Bradnam and Korf 2008) Thisis largely true for loblolly pine as well
IME refers to specific well-conserved sequences in intronsthat enhance expression IMEs in Arabidopsis and O sativaintrons near the transcription start site have greater signalthan those more distal (Rose et al 2008 Parra et al 2011)We applied the word-based discriminator IMEter designedto identify these signals in intronic sequences to the firstintrons in the CDS (as a comprehensive set of introns in the59 UTR was not available) We identified 400 primary CDSintrons between 20 and 49 kbp 38 between 50 and 99 kbpand 8 100 kbp in length all with a IMEter score 10and several with a score 20 which suggests strong en-hancement of expression Transcripts in this category includethose annotated as transcription factors (WRKY) cysteinepeptidases (cathepsins) small-molecule transporters (non-aspanins) transferases and several that are less character-ized (File S5)
Orthologous proteins
A comparison of the 47207 clustered loblolly pine genemodels to the 352151 proteins curated from 13 plant
Figure 1 Orthologous proteins derivedfrom PLAZA and mapped to the loblollypine genome (101) at various similarityscores Also included are proteins basedon Picea sitenchis sequence from GenBankP abies proteins from the Congenie Ge-nome project and proteins from theAmborella Genome project These datawere generated by examining the pro-teins for which at least 70 of the pro-tein was included in the local alignmentWe then generated for each speciesfour categories based on the ESS s90(100 ESS 90) s80(90 ESS 80)s70 (80 ESS 70) and none (70 ESS)
Annotation of Loblolly Pine Genome 897
species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

species resulted in 20646 unique gene families (afterfiltering the domains that exclusively annotate as transpos-able elements) representing 361433 (905) sequences withan average of 17 genes per family (File S6) As with most denovo clustering methods these predictions are likely to be anoverrepresentation of family size and number since clustersare formed for all non-orphan predictions by MCL (Bennetzenet al 2004) The families range in size from 5229 membersfrom 14 species to 2 members from one species
P taeda genes belong to 7053 gene families (File S7)Distribution of gene families among conifers (P abiesP sitchensis and P taeda) a basal angiosperm speciesA trichopoda mosses (S moellendorffii and P patens)monocots (O sativa and Z mays) and dicots (A thalianaG max P trichocarpa T cacao R communis and V vinifera)are shown in Figure 3 We identified 1554 conifer-specificgene families that contain at least one sequence in the threeconifer species (P taeda P abies and P sitchensis) (File S8)slightly higher than the 1021 reported by the P abies genomeproject (Nystedt et al 2013) Some of the largest familiesannotated include transcription factors (Myb WRKY andHLH) oxidoreductases (ie cytochrome p450) disease resis-tance proteins (NB-ARC) and protein kinases Among theconifer-specific families 159 were unique to P taeda 32 ofwhich had 5 or more members (Neale et al 2014)
Sixty-six unique molecular function terms apply to the8795 (4245) gene families annotated in 14 species Atotal of 24 molecular function terms describe 241 of the1554 conifer-specific gene families (Figure 4) When com-paring the GO distribution of all 14 species against theconifer-specific families four of the top five GO assign-ments are the same (protein binding nucleic acid bind-ing ion binding and hydrolase activity) with conifershaving an additional large contribution from small-moleculebinding (31 families) The 11686 gene families with nocontributions from conifers are described by 43 molecularfunction terms (Figure 4) The largest categories in thisgroup include hydrolase activity nucleic acid binding per-oxidase activity and lyase activity Of the 7053 gene familieswith a P taeda member 6094 (864) have at least oneprotein domain assignment and 5249 (7442) have molec-ular function GO term assignments Since these families havemembers in most of the plant species analyzed they are likelyconserved across eukaryotes thus reflecting their higher an-notation rate The largest categories containing P taeda in-clude protein binding transferase activity and nucleic acidbinding similar to recent findings for P glauca and Piceamariana (Pavy et al 2012) Examination of these large genefamilies across species also provides a preview of intron ex-pansion examples of which are seen in several gene familiesincluding those involved in lipid metabolism ATP bindingand hydrolase activity (Figure S3 and File S9)
Gene family gain-and-loss phylogenetics
Using DOLLOP we constructed a single maximum parsi-mony tree from all TRIBE-MCL clusters containing five orTa
ble
3Su
mmaryofintronstatistics
Total
sequen
ces
Totalno
ofintrons
Intronswith
minim
algap
regions
Longest
intron(bp)
Ave
rageintron
length
(bp)
Noofintrons
20ndash49
kbp
Nooffirst
introns20
ndash49
kbp
Noofintrons
50ndash99
kbp
Nooffirst
introns50
ndash99
kbp
Noof
introns10
0kb
pNooffirst
introns10
0kb
p
50172
14742
514
457
931
852
427
4154
6018
4069
934
310
863
15653
49720
48720
15887
823
9618
1059
214
384
1813
898 J L Wegrzyn et al
more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

more genes This represented 10877 gene families witha mean size of 31 genes and a total of 336571 splice formsSmaller gene families contributed noise to phylogeneticreconstruction that may be due to overclassification of thesmaller family sizes The final phylogenetic matrix contained13 of the 14 species in the original MCL analysis with 8519gene families of size $5 P sitchensis was removed from theanalysis because of the partial sequence representation
The resulting tree presents our best estimate of genefamily evolution for the 13 focal species (Figure 5 and TableS3) Note that the gymnospermndashangiosperm split (Pavyet al 2012 Nystedt et al 2013) is reproduced and representsthe longest internal edge in the reconstruction The gym-nosperm common ancestor is separated from the rest of thetree by a large number of gains and losses Similarly thereare also many lineage-specific gains and losses from theancestral gymnosperm to the two extant gymnospermsNotably there are more gene families not present in
gymnosperms than present in angiosperms We observe thisas many more losses than gains on the gymnosperm side ofthe gymnospermndashangiosperm split
Tandem repeat identification
Tandem repeats are the chief component of telomeres andcentromeres in higher order plants and other organismsThey are also ubiquitous across heterochromatic pericen-tromeric and subtelomeric regions (Richard et al 2008Navajas-Perez and Paterson 2009 Cavagnaro et al 2010Leitch and Leitch 2012) Tandem repeats affect variationthrough remodeling of the structures that they constitutethey modify epigenetic responses on heterochromatin andalter expression of genes through formation of secondarystructures such as those found in ribosomal DNA (Jeffreyset al 1998 Richard et al 2008 Gemayel et al 2010) Pres-ent at 56 million loci (Table S4) tandem repeats seemunusually abundant in loblolly pine However these loci
Figure 2 Intron lengths were compared for 23 species which include 21 species curated by the PLAZA project A trichopoda and P taeda (A)Comparison of maximum intron lengths for the first four intron positions in the CDS (B) Comparisons of median intron lengths for the same species forthe first four intron positions in the CDS Species codes are the following Al (Arabidopsis lyrata) Am (A trichopoda) At (A thaliana) Bd (B distachyon)Cp (Carica papaya) Fv (Fragaria vesca) Gm (G max) Md (Malus domestica) Me (Manihot esculenta) Mt (Medicago truncatula) Oi (O sativa sspindica) Oj (O sativa ssp japonica) Pi (P taeda) Pp (P patens) Pt (P trichocarpa) Rc (R communis) Sb (S bicolor) Sm (S moellendorffii) Tc (T cacao)Vv (V vinifera) and Zm (Z mays)
Annotation of Loblolly Pine Genome 899
represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

represent only 286 of the genome consistent with pre-vious studies of loblolly pine (Hao et al 2011 Magbanuaet al 2011 Wegrzyn et al 2013) and spruce (271 inP glauca and 240 in P abies) Conifers are in the middle ofthe spectrum of tandem content among nine plant speciesevaluated with estimates ranging from 153 in C sativusto 429 in A trichopoda (File S10)
Clustering at 70 identity cuts the number of uniquetandem repeat loci to less than a quarter its original size(from 5696347 to 1411119 sequences) with the largestcluster having 4999 members This suggests an abundanceof highly diverged yet somewhat related repeats as hasbeen described by others (Kovach et al 2010 Wegrzyn et al2013) When accounting for overlap with interspersed re-peat content loblolly pinersquos total tandem repeat coveragefalls to 078 of the genome (Table S4 and Table S5) Only338 of tandem content is found outside of interspersedrepeats including the LTR retrotransposons that make upthe majority of conifer genomes (Nystedt et al 2013 Wegrzynet al 2013) Further blurring the line between these twoclasses of repeats retrotransposons have been shown topreferentially insert into pericentromeric heterochromatinof rice and SSR-rich regions of barley (Ramsay et al 1999Kumekawa et al 2001)
Considering microsatellites the P taeda genome has thedensest and highest coverage of microsatellites of any co-nifer to date (012 467040 loci) although not by a widemargin (Figure 6A and Table S4) Heptanucleotides are sta-tistically more represented in loblolly pine than in the othertwo conifers (chi square test x2 = 7367 df = 2 P =00251) (Figure 6A) AT-rich di- and trinucleotides arecommon in dicot plants (Navajas-Perez and Paterson2009 Cavagnaro et al 2010) Among conifers (AGTC)n
is the most common dinucleotide in loblolly pine while(ATTA)n and (TGAC)n are favored in P abies andP glauca respectively The most common trinucleotidesare (AAG)n in loblolly pine (ATT)n in P abies and (ATG)nin P glauca (File S11) These findings support the notionsthat microsatellites are unstable and that microsatellite sec-ondary structures are likely more conserved than their spe-cific sequences (Jeffreys et al 1998 Richard et al 2008Navajas-Perez and Paterson 2009 Gemayel et al 2010Melters et al 2013) The CDS of loblolly pine is depletedin microsatellites compared to the full genome but isdenser in hexanucleotides and almost equal in trinucleoti-des (Figure 6B) Multiples of three (eg trinucleotideshexanucleotides) are normally conserved in coding regionsdue to selection against frameshift mutations (Cavagnaroet al 2010 Gemayel et al 2010)
Minisatellites (period length of 9ndash100) are also slightlymore represented in loblolly pine at 47 million loci re-presenting 176 of the genome compared to 172 inP glauca and 153 in P abies (Table S4 and Table S5)Satellites (period length of 100+) follow a similar pattern(P taeda 098 P abies 077 and P glauca 096)(Table S4) Minisatellites seem to make up the overwhelm-ing majority of the tandem repeat content Different assem-blies andor sequencing technologies alter the quantity oftandem repeat content by as much as twofold as can beseen by comparing the microsatellite density between theloblolly BAC assemblies fosmid assemblies and the full ge-nome (Figure 6B) (Wegrzyn et al 2013) Tandem repeatsare troublesome for the assembly of large genomes whichare often partial toward dinucleotide and 9- to 30-bp periods(Figure S4) (Navajas-Perez and Paterson 2009) In fact thetop three period sizes in coverage of the genome are 27 21
Figure 3 Results of the TRIBE-MCL analysis that distin-guishes orthologous protein groups The Venn diagramdepicts a comparison of protein family counts of fiveplant classifications gymnosperms (P abies P sitchensisand P taeda) monocots (O sativa and Z mays) mosses(P patens and S moellendorffii) dicots (A thalianaG max P trichocarpa R communis T cacao andV vinifera) and a basal angiosperm (A trichopoda)
900 J L Wegrzyn et al
and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

and 20 bp in that order Minisatellites especially those 20bp in length contribute as much or more to the genomethan microsatellites This trend extends to other conifers(Table S5) as well as to angiosperms including C sativusA thaliana and P trichocarpa In total two consensussequences and five monomer sequences representing ap-proximately five different species from PlantSat share ho-mology with loblolly pine satellites and minisatellites Theaverage coverage however is low (45) with the mostabundant hits to centromeric sequences of Pinus densifloraand Z mays (Table S6)
The telomeric sequence (TTTAGGG)n first identified inA thaliana (Richards and Ausubel 1988) has the most loci(23926) of any tandem repeat motif and the most overallsequence coverage (at 21 Mbp or 001 of the ge-nome) Its instances range from 104 to 95 kbp in lengthand from 2 to 317 in numbers of copies of the monomer perlocus Variants like those from tomato [TT(TA)AGGG]n(Ganal et al 1991) and multimers with those variants werenot assessed so our estimate of the amount of telomericsequence is likely low These estimates include the true telo-meres which are on the longer side of the spectrum inlength and copies along with ITRs The longest locus is15 kbp Telomeres appear especially long in pine (eg 57kbp in Pinus longaeva) (Flanary and Kletetschka 2005) and
in our assembly could be positively affected from the mega-gametophyte source as has been shown in Pinus sylvestris(Aronen and Ryynanen 2012) These ITRs are remnants ofchromosomal rearrangements that occupy large sections ofgymnosperm chromosomes (Leitch and Leitch 2012)A potential centromere monomer TGGAAACCCCAAATTTTGGGCGCCGGG (27 bp) is moderately high in fre-quency across the scaffolds and represents the secondhighest fraction of the genome covered with 5183 locicovering 18 Mbp (0009 of the genome) Its periodsize is substantially shorter than the 180-bp averagefrom other plants (Melters et al 2013) About 03 ofthe scaffolds contain this potential centromeric sequenceand these scaffolds average 779 bp in length A close var-iant TGGAAACCCCAAATTTTGGGCGCCGCA (21 bp) alsohigh in coverage and frequency shares homology (E-value =3e-9 100 identity) with the repetitive sequence of an881-bp probe (GenBank accession AB051860) developedto hybridize to centromeric and pericentromeric regions ofP densiflora (Hizume et al 2001) Perhaps this secondvariant forms a type of ldquolibraryrdquo that facilitates centromereevolution by forming a higher-order repeat structure withthe first variant Whether the centromere is evolving inpines cannot be deduced by assembling short reads as theyfail to accurately capture such structures (Melters et al
Figure 4 Gene Ontology distribution normalized for molecular function The orthologous groups defined are exclusive to the angiosperms and conifersrespectively The angiosperm set includes A trichopoda A thaliana G max P trichocarpa P patens S moellendorffii R communis O sativa Tcacao V vinifera and Z mays The conifer set includes P abies P sitchensis and P taeda
Annotation of Loblolly Pine Genome 901
2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

2013) It is also important to note that the actual coverage ofcentromeric sequence is likely much higher than reported asthese two variants likely do not represent the entirety of thecentromere Homologs were not found in P abies or P glaucaconsistent with it being specific to the subgenus Pinus(Hizume et al 2001) The presence of the Z mays centro-meric sequence along with the Pinus centromeric sequenceconceivably supports the hypothesis that although certainsatellites are conserved across species their localization isnot (Leitch and Leitch 2012)
Homology-based repeat identification
RepeatMasker with PIER as a repeat library identified 14Gbp (5858) of the genome as retroelements 240 Mbp(104) as DNA transposons and 12 Gbp (506) asderived from uncategorized repeats (Table 4) Consideringonly full-length repeats there are 179367 (561) retroele-ments 11026 (013) DNA transposons and 56024(048) uncategorized repeats Most studies in angiospermshave found a surplus of class I compared to class II content(Kumar and Bennetzen 1999 Civaacuteň et al 2011) and lob-lolly pine continues this trend with or without filtering forfull-length elements The ratio of uncategorized repeats toall repeats is 112 for the genome as seen in a previousBAC and fosmid study (Wegrzyn et al 2013) Introns con-tained 3452 LTRs 5428 retrotransposons and 352
DNA transposons (Figure S5 and File S12) First introns20kbp have the lowest amount of repetitive content at 5026while distal introns 100 kbp have the highest amount at7375 We expected higher repetitive content in longerintrons since intron expansion can be partially attributedto proliferation of repeats
LTR retroelements make up 97 Gbp (4168) of thegenome sequence of which 25 Gbp (1098) are Gypsyelements and 21 Gbp (914) are Copia elements (TableS7) Gypsy and Copia LTR superfamilies are found across theplant phylogeny and are often a significant portion of therepetitive content (Kejnovsky et al 2012) Partial and full-length alignments have a 121 ratio of Gypsy to Copia ele-ments full-length elements show a slightly more skewedratio of 131 lower than the previous estimate of 191 inP taeda (Wegrzyn et al 2013) Estimates in other conifersare on average 21 with the exception of Abies sibiricawhich was reported 321 (Nystedt et al 2013) Estimatesin angiosperms represent a wide range 181 in V vinifera13 in P trichocarpa 281 in A thaliana and 112 inC sativus (Wegrzyn et al 2013) The large number of LTRsthat cannot be sorted into superfamilies may be due to thepresence of long autonomous retrotransposon derivatives(LARDs) and terminal-repeat retrotransposons in miniature(TRIMs) among LTR content GCclassif was able to detectprotein domains on many LTRs but fewer than half were
Figure 5 Parsimonious tree predicted by DOLLOP with protein families derived from the MCL analysis of size $5 The gains and losses of 13 species(A thaliana A trichopoda G max O sativa P patens P trichocarpa P abies P taeda R communis S moellendorffii T cacao V vinifera andZ mays) are indicated on tree nodes and branches
902 J L Wegrzyn et al
classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

classifiable based on the ordering of their domains TheseLTRs may be nonfunctional requiring actively transposing ele-ments to supply the genes needed for proliferation similar toLARDs and TRIMs (Witte et al 2001 Kalendar et al 2004)
Non-LTR retroelement content is usually found at lowerfrequencies in conifers (Friesen et al 2001) Long inter-spersed nuclear elements (LINEs) cover 546 Mbp (235)of the genome (Table S7) higher than the 071 previouslyrecorded (Wegrzyn et al 2013) and higher than inP sylvestris (052) and P abies (096) (Nystedt et al2013) Some angiosperms show comparable coverage ofLINEs 296 in Brachypodium distachyon (Jia et al 2013)282 in Brassica rapa (Wang et al 2011) and 34 in Csativus (Huang et al 2009) LINEs are thought to have playedroles in telomerase and gene evolution (Schmidt 1999)Short interspersed nuclear elements (SINEs) cover 268kbp (0001) a miniscule portion of the genome as pre-viously reported (Wegrzyn et al 2013) SINE content issimilarly negligible in P sylvestris and P abies (Nystedtet al 2013) Low ratios of non-LTR to LTR retroelementsare also seen in most angiosperms (Jia et al 2013)
DNA transposons make a negligible contribution to in-terspersed element content Terminal inverted repeats (TIRs)
cover 186 Mbp (080) of the genome and helitrons cover22 Mbp (010) of the genome (Table S7) Repeats withinintrons are involved in exon shuffling (Bennetzen 2005)and epigenetic silencing (Liu et al 2004) As expectedintrons are richer in DNA transposons at 352 comparedto 104 across the genome and 93 of intronic DNAtransposon content is composed of TIRs compared to 77across the genome (File S12) MITEs have been identified inloblolly pine BACs (Magbanua et al 2011) and are prefer-entially located near genes in rice (Zhang and Hong 2000)However the methodologies that we applied extracted onlytwo MITE sequences across 25 Mb of intron sequence ClassII content is lower in most conifers including P sylvestrisP abies (Nystedt et al 2013) and Taxus maireri (Hao et al2011) This has generally been the case in angiosperms as well(Civaacuteň et al 2011) although there are exceptions (Feschotteand Pritham 2007)
De novo repeat identification
Considering a smaller subset of the entire genome sequenceallowed us to compare de novo and similarity-based repeatannotation methods In the 63 longest scaffolds REPET dis-covered 15837 putative repeats (Table S8) forming just
Figure 6 (A) Microsatellite density for three conifer genomes (green) one clubmoss genome (purple) and five angiosperm genomes (orange) (loci permegabase) (B) Microsatellite density (loci per megabase) of the coding sequence of the loblolly pine genome compared to the v10 genome and twoother loblolly genomic data sets (BACs and fosmids)
Annotation of Loblolly Pine Genome 903
under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

under 7000 novel repeat families Full-length repeats cover64 Mbp (282) of the sequence set and full-length andpartial de novo sequences combined account for 180 Mbp(793) This ratio is markedly different from the fraction offull-length repeats in the full genome determined by simi-larity alone Full-length sequences identified as previouslyclassified or which could be classified to the superfamilylevel cover 113 Mbp (498) of the bin 1 sequence accord-ing to de novo analysis Of these 66 Mbp overlap withRepeatMaskerrsquos similarity-based annotation of the bin Full-length novel repeats cover 529 Mbp (2326) of the bin 1sequence in the de novo analysis Similarity analysis over-lapped 74 Mbp of these repeats As expected REPETrsquos de novocontribution was left largely undiscovered by RepeatMaskerrsquossimilarity-based annotation method In total we see anoverlap of 14 Mbp between our similarity and de novo an-notation methods and a total combined full-length repeatcoverage of 697 Mbp
Highly represented repeat families
High-coverage elements varied between similarity and denovo approaches PtAppalachian PtRLC_3 PtRLG_13 andPtRLX_3423 are shared among the top high-coverage ele-ments (Table S9) Many of the high-coverage elementsfound are not highly represented in Wegrzyn et al (2013)which used a similar approach This corroborates a high rateof divergence and a high incidence of incomplete retrotrans-poson content PtRLG_Conagree the highest-coverage re-peat annotated 075 of the genome covering 174 Mbp(Table S9) TPE1 a previously characterized Copia elementhas the second-highest coverage with 114 Mbp (049)Other highly represented elements include PtRLG_OuachitaPtRLG_Talladega PtRLX_Piedmont PtRLG_Appalachianand PtRLG_Angelina which were previously characterizedas part of a set of high coverage de novo repeat families
(Wegrzyn et al 2013) PtIFG7 a Gypsy element covers 90Mbp (039) (Table S9) As previously noted in Kovachet al (2010) no single family dominates the repetitive con-tent with the highest-coverage element PtRLG_Conagreeaccounting for 1 of the genome (Table S9) Gymnythought to occupy 135 Mb in the genome with full-lengthelements (Morse et al 2009) comprises only 33 Mbp IFG7thought to occupy up to 58 of the genome (Magbanuaet al 2011) occupies only 039 of the genome The top100 highest-coverage elements account for 20 of thegenome (Figure 7A) Among high-coverage intronic repeatsPtRLX_106 and PtRLG_Appalachian were identified in bothbin 1 and in the genome Side-by-side comparisons of high-copy and high-coverage families show a much lower contri-bution per family in the introns compared to the genome(Table S8 and Table S9)
The highly divergent nature of most of the transposableelements seems to conflict with the uniformity seen in thosethat are highly represented A total of 4397 full-lengthPtAppalachian sequences and 2835 PtRLG_13 sequenceswere 80 similar to their reference sequences and 1559and 237 sequences align at 90 identity and 90 cover-age respectively These alignments suggest that many of thehighly represented elements may be actively transposingdespite the diverged nature of many transposable elementfamilies
Repetitive content summary
In total 153 Gbp (6534) of genomic sequence waslabeled as interspersed content via homology (Table 4)while de novo estimates place total interspersed content at793 (similarity and de novo combined in bin 1 yield8181) This is consistent with the de novo figures reportedby Wegrzyn et al (2013) at 86 and by Kovach et al (2010)at 80 but is higher than in P sylvestris at 52 (Nystedt
Table 4 Summary of interspersed repeats from homology-based identification
Full-length All repeats
Class Order SuperfamilyNo of
elements Length (bp) genomeNo ofelements Length (bp) genome
I LTR Gypsy 49183 264644712 114 5127514 2544140822 1098I LTR Copia 36952 207086169 089 4385545 2119375506 914
Total LTR LTR 179367 962249326 415 17432917 9660836674 4168I DIRS (Dictyostelium
transposableelement)
4935 26820856 012 596008 335540558 145
I Penelope 4966 13456930 006 422276 188350501 081I LINE 15353 53411263 023 906403 545648705 235I SINE 137 68023 000 670 182264 000
Total RT(Retrotransposon)
262028 1299761701 561 25166637 13577984814 5858
II TIR 7932 20499522 009 420769 185871618 080II Helitron 1105 3500491 002 47003 22396711 010
Total DNA 11026 31035610 013 519708 240545369 104Uncategorized 56024 110604852 048 3164894 1172394606 506
Total interspersed 336037 1458952566 629 29249206 15145555948 6534Tandem repeats 210810342 091 210810342 091
Total 1669762908 720 15356366290 6625
904 J L Wegrzyn et al
et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

et al 2013) P glauca at 40ndash60 (Hamberger et al 2009Liu et al 2011 Nystedt et al 2013) P abies at 70 (Nystedtet al 2013) Sorghum bicolor at 63 (Paterson et al 2009)and Secale cereale at 693 (Bartos et al 2008) Masking allrepeat content results in the complete masking of 9 millionscaffolds covering 32 Gbp of sequence Full-length ele-ments cover only 720 of the genome much lower than
the 2598 previously estimated in BAC and fosmid sequen-ces (Wegrzyn et al 2013)
Strict filters as described in Wegrzyn et al (2013) en-sured the quality of the discovered de novo sequence andthe repeat library used in similarity analysis PIER was de-rived from de novo content in loblolly pine BACs and fosmidsdiscovered with the same methodology It is surprising then
Figure 7 (A) Repeat family cover-age Repeat families on the x-axisare ordered by coverage in des-cending order Solid lines illustratecumulative coverage as more fam-ilies are considered Dashed linesrepresent the total repetitive con-tent for that data set (B) Compar-ison of bin 1 repetitive content forboth partial and full-length anno-tations ldquoFull-length + Partialrdquo refersto all full-length and partial hits andldquoPercentage of datasetrdquo is a func-tion of the total length annotatedby each classification
Annotation of Loblolly Pine Genome 905
that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

that similarity analysis characterized 66 of the genomesequence as repetitive while de novo analysis characterized793 of bin 1 as repetitive suggesting a plethora of furtherhigh-quality novel repeat content One possibility is thatthe large number of new repeat sequences discovered denovo may be due to TRIMs and LARDs the latter for whichtransposition events yield high sequence variability evenamong mRNA transcripts (Kalendar et al 2004) LARDsare a subset of LTRs and class I retrotransposons and couldeasily inflate the figures for these two categories while re-ducing the number and coverage of known superfamiliesHigh variability in retrotransposition is common amongangiosperms and gymnospermsmdashnot surprising as reversetranscription is known to be highly error-prone (Gabrielet al 1996) A single burst of retrotransposition can poten-tially result in hundreds of repeats all independently di-verging (El Baidouri and Panaud 2013) One such eventmay have occurred long ago in loblolly pine resulting inthe many ancient diverged single-copy repeat families beingidentified Finally we note that repetitive elements are a com-mon obstacle in genome assembly scaffold resolution mayhave decreased the amount of detectable repetitive content asrepeats located on a terminal end of a sequence may becollapsed upon assembly With this level of repetitive contentthe likelihood of terminal repeat collapse is markedly higherthan in smaller and less repetitive genomes
Conclusion
We have presented a comprehensive annotation of thelargest genome and first pine The size of the genome andthe absence of well-characterized sequence for close rela-tives presented significant challenges for annotation Theinclusion of a comprehensive transcriptome resource gener-ated from deep sequencing of loblolly pine tissue types notpreviously examined was key to identifying the gene spaceThis resource along with other conifer resources provideda platform to train gene-prediction algorithms The largergene space enabled us to better quantify and examine thethose that are unique to conifers and potentially to gym-nosperms The long scaffolds available in our sequenceassembly facilitated the identification of long introns pro-viding a resource to study their role in gene regulation andtheir relationship to the high levels of repetitive content inthe genome To characterize sequence repeats we applieda combined similarity and de novo approach to improveupon our existing repeat library and to better define thecomponents of the largest portion of the genome This an-notation will not only be the foundation for future studieswithin the conifer community but also a resource for a muchlarger audience interested in comparative genomics and theunique evolutionary role of gymnosperms
Acknowledgments
The support and resources from the Center for High Perfor-mance Computing at the University of Utah are gratefully
acknowledged Funding for this project was made avail-able through the US Department of AgricultureNationalInstitute of Food and Agriculture (2011-67009-30030) awardto DBN at the University of California Davis This work usedthe Extreme Science and Engineering Discovery Environmentsupported by National Science Foundation grant OCI-1053575and in particular High Performance Computing resources ofthe partner centers at The University of Texas at Austin andIndiana University (grant ABI-1062432)
Note added in proof See Zimin et al 2014 (pp 875ndash890)in this issue for a related work
Literature Cited
Ahuja M R and D B Neale 2005 Evolution of genome size inconifers Silvae Genet 54 126ndash137
Allona I M Quinn E Shoop K Swope S St Cyr et al1998 Analysis of xylem formation in pine by cDNA sequenc-ing Proc Natl Acad Sci USA 95 9693ndash9698
Altschul S F W Gish W Miller E W Myers and D J Lipman1990 Basic local alignment search tool J Mol Biol 215 403ndash410
Aronen T and L Ryynanen 2012 Variation in telomeric repeats ofScots pine (Pinus sylvestris L) Tree Genet Genomes 8 267ndash275
Bartos J E Paux R Kofler M Havrankova D Kopecky et al2008 A first survey of the rye (Secale cereale) genome compo-sition through BAC end sequencing of the short arm of chromo-some 1R BMC Plant Biol 8 95
Bennetzen J L 2005 Transposable elements gene creation andgenome rearrangement in flowering plants Curr Opin GenetDev 15 621ndash627
Bennetzen J L C Coleman R Y Liu J X Ma andW Ramakrishna2004 Consistent over-estimation of gene number in complexplant genomes Curr Opin Plant Biol 7 732ndash736
Benson G 1999 Tandem repeats finder a program to analyzeDNA sequences Nucleic Acids Res 27 573ndash580
Birol I A Raymond S D Jackman S Pleasance R Coope et al2013 Assembling the 20 Gb white spruce (Picea glauca) ge-nome from whole-genome shotgun sequencing data Bioinfor-matics 29 1492ndash1497
Bradnam K R and I Korf 2008 Longer first introns are a generalproperty of eukaryotic gene structure PLoS ONE 3 e3093
Briones K M J A Homyack D A Miller and M C Kalcounis-Rueppell 2013 Intercropping switchgrass with loblolly pinedoes not influence the functional role of the white-footed mouse(Peromyscus leucopus) Biomass Bioenergy 54 191ndash200
Brown G R G P Gill R J Kuntz C H Langley and D B Neale2004 Nucleotide diversity and linkage disequilibrium in lob-lolly pine Proc Natl Acad Sci USA 101 15255ndash15260
Cairney J L Zheng A Cowels J Hsiao V Zismann et al2006 Expressed sequence tags from loblolly pine embryos re-veal similarities with angiosperm embryogenesis Plant MolBiol 62 485ndash501
Campbell M S M Law C Holt J C Stein G D Mogue et al2014 MAKER-P an annotation pipeline and genome-databasemanagement tool for second-generation genome projects PlantPhysiol 164 513ndash524
Cavagnaro P F D A Senalik L Yang P W Simon T T Harkinset al 2010 Genome-wide characterization of simple sequencerepeats in cucumber (Cucumis sativus L) BMC Genomics 11 569
Civaacuteň P M Svec and P Hauptvogel 2011 On the coevolution oftransposable elements and plant genomes J Bot doi1011552011893546
906 J L Wegrzyn et al
Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

Dongen S and C Abreu-Goodger 2012 Using MCL to extractclusters from networks pp 281ndash295 in Bacterial Molecular Net-works edited by J Helden A Toussaint and D ThieffrySpringer Berlin Heidelberg Germany New York
Eckert A J B Pande E S Ersoz M H Wright V K Rashbrooket al 2009 High-throughput genotyping and mapping of sin-gle nucleotide polymorphisms in loblolly pine (Pinus taeda L)Tree Genet Genomes 5 225ndash234
Eckert A J A D Bower S C Gonzalez-Martinez J L WegrzynG Coop et al 2010 Back to nature ecological genomics ofloblolly pine (Pinus taeda Pinaceae) Mol Ecol 19 3789ndash3805
Eckert A J Wegrzyn J Liechty J Lee W Cumbie et al2013 The evolutionary genetics of the genes underlying phe-notypic associations for loblolly pine (Pinus taeda Pinaceae)Genetics 195 1353ndash1372Edgar R C 2010 Search and clus-tering orders of magnitude faster than BLAST Bioinformatics26 2460ndash2461
Edgar R C and E W Myers 2005 PILER identification andclassification of genomic repeats Bioinformatics 21 i152ndashi158
El Baidouri M and O Panaud 2013 Comparative genomic pa-leontology across plant kingdom reveals the dynamics of TE-driven genome evolution Genome Biol Evol 5 954ndash965
Enright A J S Van Dongen and C A Ouzounis 2002 An effi-cient algorithm for large-scale detection of protein familiesNucleic Acids Res 30 1575ndash1584
Felsenstein J 1989 PHYLIP Phylogeny Inference Package (Ver-sion 32) Cladistics 5 164ndash166
Fernandez-Pozo N J Canales D Guerrero-Fernandez D P VillalobosS M Diaz-Moreno et al 2011 EuroPineDB a high-coverageweb database for maritime pine transcriptome BMC Genomics12 366
Feschotte C and E J Pritham 2007 DNA transposons andthe evolution of eukaryotic genomes Annu Rev Genet 41331ndash368
Flanary B E and G Kletetschka 2005 Analysis of telomerelength and telomerase activity in tree species of various life-spans and with age in the bristlecone pine Pinus longaeva Bio-gerontology 6 101ndash111
Flutre T E Duprat C Feuillet and H Quesneville2011 Considering transposable element diversification in denovo annotation approaches PLoS ONE 6 e16526
Frech C and N Chen 2010 Genome-wide comparative genefamily classification PLoS ONE 5 e13409
Friesen N A Brandes and J Heslop-Harrison 2001 Diversityorigin and distribution of retrotransposons (gypsy and copia) inconifers Mol Biol Evol 18 1176ndash1188
Gabriel A M Willems E H Mules and J D Boeke1996 Replication infidelity during a single cycle of Ty1 retro-transposition Proc Natl Acad Sci USA 93 7767ndash7771
Ganal M W N L V Lapitan and S D Tanksley 1991 Macrostruc-ture of the tomato telomeres Plant Cell 3 87ndash94
Garcia-Gil M R 2008 Evolutionary aspects of functional andpseudogene members of the phytochrome gene family in Scotspine J Mol Evol 67 222ndash232
Gemayel R M D Vinces M Legendre and K J Verstrepen2010 Variable tandem repeats accelerate evolution of codingand regulatory sequences Annu Rev Genet 44 445ndash477
Gernandt D S A Liston and D Pinero 2001 Variation in thenrDNA ITS of Pinus subsection Cembroides implications formolecular systematic studies of pine species complexes MolPhylogenet Evol 21 449ndash467
Goodstein D M S Shu R Howson R Neupane R D Hayeset al 2012 Phytozome a comparative platform for greenplant genomics Nucleic Acids Res 40 D1178ndashD1186
Guillet-Claude C N Isabel B Pelgas and J Bousquet 2004 Theevolutionary implications of knox-I gene duplications in coni-fers correlated evidence from phylogeny gene mapping and
analysis of functional divergence Mol Biol Evol 21 2232ndash2245
Hamberger B D Hall M Yuen C Oddy B Hamberger et al2009 Targeted isolation sequence assembly and characteriza-tion of two white spruce (Picea glauca) BAC clones for terpenoidsynthase and cytochrome P450 genes involved in conifer defencereveal insights into a conifer genome BMC Plant Biol 9 106
Han Y J and S R Wessler 2010 MITE-Hunter a program fordiscovering miniature inverted-repeat transposable elementsfrom genomic sequences Nucleic Acids Res 38 e199
Hao D L Yang and P Xiao 2011 The first insight into theTaxus genome via fosmid library construction and end sequenc-ing Mol Genet Genomics 285 197ndash205
Hizume M F Shibata Y Maruyama and T Kondo 2001Cloning of DNA sequences localized on proximal fluorescentchromosome bands by microdissection in Pinus densifloraSieb amp Zucc Chromosoma 110 345ndash351
Huang S W R Q Li Z H Zhang L Li X F Gu et al 2009 Thegenome of the cucumber Cucumis sativus L Nat Genet 411275ndash1281
Hunter S P Jones A Mitchell R Apweiler T K Attwood et al2012 InterPro in 2011 new developments in the family anddomain prediction database Nucleic Acids Res 40(Databaseissue) D306ndashD312
Jain M P Khurana A K Tyagi and J P Khurana 2008 Genome-wide analysis of intronless genes in rice and Arabidopsis FunctIntegr Genomics 8 69ndash78
Jeffreys A J D L Neil and R Neumann 1998 Repeat instabil-ity at human minisatellites arising from meiotic recombinationEMBO J 17 4147ndash4157
Jia J Z S C Zhao X Y Kong Y R Li G Y Zhao et al2013 Aegilops tauschii draft genome sequence reveals a generepertoire for wheat adaptation Nature 496 91ndash95
Jiao Y N N J Wickett S Ayyampalayam A S Chanderbali LLandherr et al 2011 Ancestral polyploidy in seed plants andangiosperms Nature 473 97ndash113
Johnsen K H D Wear R Oren R O Teskey F Sanchez et al2001 Carbon sequestration and southern pine forests J For99 14ndash21
Kalendar R C M Vicient O Peleg K Anamthawat-Jonsson ABolshoy et al 2004 Large retrotransposon derivatives abun-dant conserved but nonautonomous retroelements of barleyand related genomes Genetics 166 1437ndash1450
Kejnovsky E J Hawkins and C Feschotte 2012 Plant transpos-able elements biology and evolution pp 17ndash34 in Plant Ge-nome Diversity edited by J F Wendel J Greilhuber JDolezel and I J Leitch Springer Berlin Heidelberg GermanyNew York
Kent W J 2002 BLAT the BLAST-like alignment tool GenomeRes 12 656ndash664
Kirst M A F Johnson C Baucom E Ulrich K Hubbard et al2003 Apparent homology of expressed genes from wood-form-ing tissues of loblolly pine (Pinus taeda L) with Arabidopsisthaliana Proc Natl Acad Sci USA 100 7383ndash7388
Kohany O A J Gentles L Hankus and J Jurka 2006 Annota-tion submission and screening of repetitive elements in Re-pbase RepbaseSubmitter and Censor BMC Bioinformatics7 474
Korf I 2004 Gene finding in novel genomes BMC Bioinformatics5 59
Kovach A J L Wegrzyn G Parra C Holt G E Bruening et al2010 The Pinus taeda genome is characterized by diverse andhighly diverged repetitive sequences BMC Genomics 11 420
Kumar A and J L Bennetzen 1999 Plant retrotransposonsAnnu Rev Genet 33 479ndash532
Kumekawa N N Ohmido K Fukui E Ohtsubo and H Ohtsubo2001 A new gypsy-type retrotransposon RIRE7 preferential
Annotation of Loblolly Pine Genome 907
insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

insertion into the tandem repeat sequence TrsD in pericentro-meric heterochromatin regions of rice chromosomes MolGenet Genomics 265 480ndash488
Leitch A R and I J Leitch 2012 Ecological and genetic factorslinked to contrasting genome dynamics in seed plants NewPhytol 194 629ndash646
Liu J Y H He R Amasino and X M Chen 2004 siRNAs tar-geting an intronic transposon in the regulation of natural flow-ering behavior in Arabidopsis Genes Dev 18 2873ndash2878
Liu W S Thummasuwan S K Sehgal P Chouvarine and D GPeterson 2011 Characterization of the genome of bald cy-press BMC Genomics 12 553
Lorenz W W F Sun C Liang D Kolychev H Wang et al2006 Water stress-responsive genes in loblolly pine (Pinustaeda) roots identified by analyses of expressed sequence taglibraries Tree Physiol 26 1ndash16
Lorenz W W S Ayyampalayam J M Bordeaux G T HoweK D Jermstad et al 2012 Conifer DBMagic a database hous-ing multiple de novo transcriptome assemblies for 12 diverseconifer species Tree Genet Genomes 8 1477ndash1485
Macas J T Meacuteszaacuteros and M Nouzovaacute 2002 PlantSat a special-ized database for plant satellite repeats Bioinformatics 18 28ndash35
Magbanua Z V S Ozkan B D Bartlett P Chouvarine C A Saskiet al 2011 Adventures in the enormous a 18 million clone BAClibrary for the 217 Gb genome of loblolly pine PLoS ONE 6 e16214
Martiacutenez-Garciacutea P J K Stevens J Wegrzyn J Liechty M Crepeauet al 2013 Combination of multipoint maximum likelihood(MML) and regression mapping algorithms to construct a high-density genetic linkage map for loblolly pine (Pinus taeda L)Tree Genet Genomes 9 1529ndash1535
Melters D K Bradnam H Young N Telis M May et al2013 Comparative analysis of tandem repeats from hundredsof species reveals unique insights into centromere evolutionGenome Biol 14 R10
Morse A D Peterson M Islam-Faridi K Smith Z Magbanuaet al 2009 Evolution of genome size and complexity in PinusPLoS ONE 4 e4332
Navajas-Perez R and A H Paterson 2009 Patterns of tandemrepetition in plant whole genome assemblies Mol Genet Ge-nomics 281 579ndash590
Neale D B J L Wegrzyn K A Stevens A V Zimin D Puiuet al 2014 Decoding the massive genome of loblolly pineusing haploid DNA and novel assembly strategies GenomeBiol 15 R59
Noormets A M J Gavazzi S G Mcnulty J C Domec G Sunet al 2010 Response of carbon fluxes to drought in a coastalplain loblolly pine forest Glob Change Biol 16 272ndash287
Nystedt B N R Street A Wetterbom A Zuccolo Y C Lin et al2013 The Norway spruce genome sequence and conifer ge-nome evolution Nature 497 579ndash584
Parra G K Bradnam and I Korf 2007 CEGMA a pipeline toaccurately annotate core genes in eukaryotic genornes Bioinfor-matics 23 1061ndash1067
Parra G K Bradnam A B Rose and I Korf 2011 Comparativeand functional analysis of intron-mediated enhancement signalsreveals conserved features among plants Nucleic Acids Res 395328ndash5337
Paterson A H J E Bowers R Bruggmann I Dubchak J Grimwoodet al 2009 The Sorghum bicolor genome and the diversificationof grasses Nature 457 551ndash556
Pavy N B Pelgas J Laroche P Rigault N Isabel et al 2012 Aspruce gene map infers ancient plant genome reshuffling andsubsequent slow evolution in the gymnosperm lineage leadingto extant conifers BMC Biol 10 84
Prestemon J P and R C Abt 2002 Southern Forest ResourceAssessment highlights The Southern Timber Market to 2040 JFor 100 16ndash22
Punta M P C Coggill R Y Eberhardt J Mistry J Tate et al2012 The Pfam protein families database Nucleic Acids Res40 D290ndashD301
Quevillon E V Silventoinen S Pillai N Harte N Mulder et al2005 InterProScan protein domains identifier Nucleic AcidsRes 33 W116ndashW120
Ralph S G H J Chun D Cooper R Kirkpatrick N Kolosovaet al 2008 Analysis of 4664 high-quality sequence-finishedpoplar full-length cDNA clones and their utility for the discoveryof genes responding to insect feeding BMC Genomics 9 57
Ramsay L M Macaulay L Cardle and M Morgante S degliIvanissevich et al 1999 Intimate association of microsatelliterepeats with retrotransposons and other dispersed repetitiveelements in barley Plant J 17 415ndash425
Ren X Y O Vorst M W Fiers W J Stiekema and J P Nap2006 In plants highly expressed genes are the least compactTrends Genet 22 528ndash532
RepeatMasker 2013 Available at httpwwwrepeatmaskerorgAccessed July 22 2013
Richard G F A Kerrest and B Dujon 2008 Comparative ge-nomics and molecular dynamics of DNA repeats in eukaryotesMicrobiol Mol Biol Rev 72 686ndash727
Richards E J and F M Ausubel 1988 Isolation of a highereukaryotic telomere from Arabidopsis thaliana Cell 53 127ndash136
Rigault P B Boyle P Lepage J E Cooke J Bousquet et al2011 A white spruce gene catalog for conifer genome analy-ses Plant Physiol 157 14ndash28
Rose A B T Elfersi G Parra and I Korf 2008 Promoter-proximalintrons in Arabidopsis thaliana are enriched in dispersed signalsthat elevate gene expression Plant Cell 20 543ndash551
Schmidt T 1999 LINEs SINEs and repetitive DNA non-LTR ret-rotransposons in plant genomes Plant Mol Biol 40 903ndash910
Shepard S M McCreary and A Fedorov 2009 The peculiaritiesof large intron splicing in animals PLoS ONE 4 e7853
Skinner J S and M P Timko 1998 Loblolly pine (Pinus taeda L)contains multiple expressed genes encoding light-dependentNADPH protochlorophyllide oxidoreductase (POR) Plant CellPhysiol 39 795ndash806
Slater G S and E Birney 2005 Automated generation of heu-ristics for biological sequence comparison BMC Bioinformatics6 31
Stanke M and S Waack 2003 Gene prediction with a hiddenMarkov model and a new intron submodel Bioinformatics 19Ii215ndashIi225
Stein L D C Mungall S Q Shu M Caudy M Mangone et al2002 The Generic Genome Browser a building block fora model organism system database Genome Res 12 1599ndash1610
Sun H Z and L A Chasin 2000 Multiple splicing defects in anintronic false exon Mol Cell Biol 20 6414ndash6425
Van Bel M S Proost E Wischnitzki S Movahedi C Scheerlincket al 2012 Dissecting plant genomes with the PLAZA compar-ative genomics platform Plant Physiol 158 590ndash600
Wakasugi T J Tsudzuki S Ito K Nakashima T Tsudzuki et al1994 Loss of all ndh genes as determined by sequencing theentire chloroplast genome of the black pine Pinus thunbergiiProc Natl Acad Sci USA 91 9794ndash9798
Wang X W H Z Wang J Wang R F Sun J Wu et al2011 The genome of the mesopolyploid crop species Brassicarapa Nat Genet 43 1035ndash1039
Wegrzyn J L J M Lee B R Tearse and D B Neale2008 TreeGenes a forest tree genome database Int J PlantGenomics 2008 412875
Wegrzyn J L D Main B Figueroa M Choi J Yu et al2012 Uniform standards for genome databases in forest andfruit trees Tree Genet Genomes 8 549ndash557
908 J L Wegrzyn et al
Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

Wegrzyn J L B Lin J Zieve W Dougherty P Martiacutenez-Garciacutea et al 2013 Insights into the loblolly pine genomecharacterization of BAC and fosmid sequences PLoS ONE8 e72439
Westbrook J W M F R Resende P Munoz A R Walker J LWegrzyn et al 2013 Association genetics of oleoresin flow inloblolly pine discovering genes and predicting phenotype forimproved resistance to bark beetles and bioenergy potentialNew Phytol 199 89ndash100
Wicker T F Sabot A Hua-Van J L Bennetzen P Capy et al2007 A unified classification system for eukaryotic transpos-able elements Nat Rev Genet 8 973ndash982
Willyard A J Syring D S Gernandt A Liston and R Cronn2007 Fossil calibration of molecular divergence infers a mod-erate mutation rate and recent radiations for pinus Mol BiolEvol 24 90ndash101
Witte C P Q H Le T Bureau and A Kumar 2001 Terminal-repeat retrotransposons in miniature (TRIM) are involved inrestructuring plant genomes Proc Natl Acad Sci USA 9813778ndash13783
Zhang X L and G F Hong 2000 Preferential location of MITEsin rice genome Acta Biochim Biophys Sin (Shanghai) 32223ndash228
Zimin A G Marais D Puiu M Roberts S Salzberg et al2013 The MaSuRCA genome assembler Bioinformatics 292669ndash2677
Zimin A K Stevens M Crepeau A Holtz-Morris M Korablineet al 2014 Sequencing and assembly the 22-Gb loblolly pinegenome Genetics 196 875ndash890
Communicating editor M Johnston
Annotation of Loblolly Pine Genome 909
GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

GENETICSSupporting Information
httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
Unique Features of the Loblolly Pine(Pinus taeda L) Megagenome Revealed
Through Sequence AnnotationJill L Wegrzyn John D Liechty Kristian A Stevens Le-Shin Wu Carol A Loopstra Hans A Vasquez-
Gross William M Dougherty Brian Y Lin Jacob J Zieve Pedro J Martiacutenez-Garciacutea Carson HoltMark Yandell Aleksey V Zimin James A Yorke Marc W Crepeau Daniela Puiu Steven L Salzberg
Pieter J de Jong Keithanne Mockaitis Doreen Main Charles H Langley and David B Neale
Copyright copy 2014 by the Genetics Society of AmericaDOI 101534genetics113159996
2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

2 SI J L Wegrzyn et al
Figure S1 Flowchart for GCclassif Circles represent states in GCclassif in which the arrows containing the proper input represent transitions to the next state AP is aspartic protease RT is reverse transcriptase RH is RNAse H and INT is integrase Epsilons allow states to be skipped
J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 3 SI
Figure S2 The genome sorted by descending scaffold size and placed in 100 bins (A) The total number of bases in each bin is in pink while the proportion of gap regions is shown in purple (B) Number of scaffolds per bin (C) The de-novo transcriptome data aligned to the genome where each bar indicates the number of transcripts that were aligned to sequence in each bin and where the transcript was required to align to a unique location in the genome with 98 query coverage and 98 identity
4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

4 SI J L Wegrzyn et al
Figure S3 Multiple Sequence Alignment between genes predicted to be within a gene family where the units are measured in base pairs Introns (green) coding regions (white) and alignment gaps (orange) are depicted for both families (A) Cermaidase Physcomitrella patens Arabidopsis thaliana Ricinus communis Populus trichocarpa Glycine max Vitis vinifera and Pinus taeda (B) Hydrolase Selaginella moellendorffii and Pinus taeda
J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 5 SI
Figure S4 Tandem repeat content of the genomic sequence in bin 1 of the genome Each locus (tandem array) is plotted by its period (x-axis) and the frequency of that particular period (y-axis) The size of each locus corresponds to the number of copies of the tandem repeat The color corresponds to the total length (in bp) of that locus with red being the largest and green being the smallest
6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

6 SI J L Wegrzyn et al
Figure S5 Intronic repeats Introns were separated into primary and distal categories in addition to length categories (20-49 Kbp 50-99 Kbp and greater than 100 Kbp) The intronic sequence was analyzed via homology-based methods against the PIER database
J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 7 SI
Table S1 Orthologous proteins from the PLAZA project aligned to the Pinus taeda versions 101 genome
Species Protein count Query protein
count Aligned
uniquely Percent aligned
uniquely Aligned non-
uniquely
Arabidopsis lyrata 32657 30892 4720 153 1029
Arabidopsis thaliana 27407 27160 4482 165 1018
Brachypodium distachyon 26632 26200 4099 156 880
Carica papaya 26954 23199 4098 177 793
Chlamydomonas reinhardtii 16784 13001 384 30 107
Fragaria vesca 34748 34702 3412 98 752
Glycine max 46324 37388 8336 223 1788
Malus domestica 63515 46856 6257 134 1356
Manihot esculenta 30748 25474 5937 233 1231
Medicago truncatula 45197 44699 3803 85 888
Micromonas sp RCC299 9985 8934 389 44 101
Oryza sativa ssp indica 48788 48629 3787 78 846
Oryza sativa ssp japonica 41363 41186 4300 104 915
Ostreococcus lucimarinus 7769 6322 382 60 105
Ostreococcus tauri 7994 6762 231 34 64
Physcomitrella patens 28090 22807 3302 145 812
Populus trichocarpa 40141 36393 7185 197 1530
Ricinus communis 31009 28113 4475 159 910
Selaginella moellendorffii 18384 13661 1436 105 329
Sorghum bicolor 33117 26513 3880 146 842
Theobroma cacao 28858 28136 4457 158 877
Vitis vinifera 26238 25663 5096 199 1069
Volvox carteri 15520 13118 478 36 147
Zea mays 39172 37805 5223 138 1103
PLAZA proteins (24 species) 727394 653613 90149 127 19492
For 24 species in the PLAZA data set a subset of proteins that passed additional quality checks (denoted lsquoQuery proteinsrsquo comprised the set of proteins we attempted to align to the loblolly pine genome by first mapping with blat and then a performing a final alignment exonerate The minimum query coverage is 70 and the minimum similarity is an exonerate-calculated similarity score of 70 Proteins that aligned once or more than once at this criteria were counted as lsquoAligned uniquelyrsquo or lsquoAligned non-uniquelyrsquo respectively
8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

8 SI J L Wegrzyn et al
Table S2 Summary of MAKER Gene Annotations
Total
sequence
Average
length (bp)
(CDS)
Median
length (bp)
(CDS)
Shortest
Sequence
(bp)
Longest
sequence
(bp)
Total
sequence
(bp)
GC
()
Total
introns
Avg
intronsgene
50172 965 727 120 12657 48440991 4256 147425 2
15653 1290 1067 150 12657 20190331 4356 49720 3
J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 9 SI
Table S3 GainLoss Protein Matrix Table
Arabidopsis thaliana
Amborella trichopoda
Glycine max
Picea abies
Picea sitchensis
Pinus taeda
Physcomitrella patens
Populus trichocarpa
Ricinus communnis
Selaginella moellendorffii
Theobroma cacao
Vitis vinifera
Arabidopsis thaliana 0 314 -53 3003 3991 1988 1857 -339 -334 3084 -369 347
Amborella trichopoda -314 0 -367 2689 3677 1674 1543 -653 -648 2770 -683 33
Glycine max 53 367 0 3056 4044 2041 1910 -286 -281 3137 -316 400
Picea abies -3003 -2689 -3056 0 988 -1015 -1146 -3342 -3337 81 -3372 -2656
Picea sitchensis -3991 -3677 -4044 -988 0 -2003 -2134 -4330 -4325 -907 -4360 -3644
Pinus taeda -1988 -1674 -2041 1015 2003 0 -131 -2327 -2322 1096 -2357 -1641
Physcomitrella patens -1857 -1543 -1910 1146 2134 131 0 -2196 -2191 1227 -2226 -1510
Populus trichocarpa 339 653 286 3342 4330 2327 2196 0 5 3423 -30 686
Ricinus communnis 334 648 281 3337 4325 2322 2191 -5 0 3418 -35 681
Selaginella moellendorffii
-3084 -2770 -3137 -81 907 -1096 -1227 -3423 -3418 0 -3453 -2737
Theobroma cacao 369 683 316 3372 4360 2357 2226 30 35 3453 0 716
Vitis vinifera -347 -33 -400 2656 3644 1641 1510 -686 -681 2737 -716 0
10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

10 SI J L Wegrzyn et al
Table S4 Summary of tandem repeat content in Pinus taeda
Microsatellites (2-8 bp) Total number of loci Total number of copies Variants Total length (bp) of sequence sets
Dinucleotide 136469 31423955 10 6107780 003
Trinucleotide 59777 10853759 61 3210065 001
Tetranucleotide 22748 2963773 205 1160245 001
Pentanucleotide 32605 789203 609 3901223 002
Hexanucleotide 47314 4951429 2159 2956985 001
Heptanucleotide 145992 12949958 2448 8931410 004
Octanucleotide 22135 1359453 3903 1066823 000
Total 467040 72394357 9395 27334531 012
Minisatellites (9-100 bp)
9-30 3205612 106328482 1446648 205169621 091
31-50 853241 22561807 635066 84379493 037
51-70 377512 10237267 338958 59129899 026
71-100 247280 5706828 235021 47737215 021
Total 4683645 144834384 2655693 396416228 176
Satellites (gt100 bp)
101-200 351264 7807707 345179 106605726 047
201-300 79942 1719339 79699 42421186 019
301-400 28494 616477 28406 20617800 009
gt400 35428 787146 35329 51362778 023
Total 495128 10930669 488613 221007490 098
Grand Total 5645813 22815941 3153701 644758249 286
J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 11 SI
Table S5 A comparison of the most common tandem period in Picea abies Picea glauca and Pinus taeda
Pinus taeda v101 Picea glauca v10 Picea abies v10
Microa Minib Satc Micro Mini Sat Micro Mini Sat
Most frequent
period size 7 21 123 2 21 102 2 50 101
Cumulative length
(Mbp) 893 2009 866 226 870 489 1038 2900 525
Num of loci 145992 361356 27422 62592 179716 23256 255380 285648 24895
Most frequent
period () 004 009 004 001 004 002 005 015 003
Total cumulative
length (Mbp) 2733 39642 22101 748 35790 19883 1863 29925 15181
Total 012 176 098 004 172 096 010 153 077
Total overall
content 286 271 240
aMicrosatellites bMinisatellites cSatellites
12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

12 SI J L Wegrzyn et al
Table S6 Summary of hits to the PlantSat database
Family
Genbank Accessions Number of hits Localization Best hit Identity () Best hit length (bp) Best hit coverage () Best hit e-value
Consensus Sequences
Citrus_limon_90 M38369 3 B+---- 982 56 622 31e-20
Zea_mays_MBsC216 AF139910 2 NA 895 38 176 75e-05
Monomer Sequences
Pinus_PCSR AB051860 67 B+---- 1000 27 1000 26e-06
Zea_mays_MBsC216 AF139910 2 B+---- 895 38 176 750E-05
Cucurbita_160 X82944 5 NA 100 23 136 00027
Citrus_limon_90 M38369 3 B+---- 982 56 622 310E-20
Lens_Lc30 AJ401232 1 B-+++- 96 25 4464 00032
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence
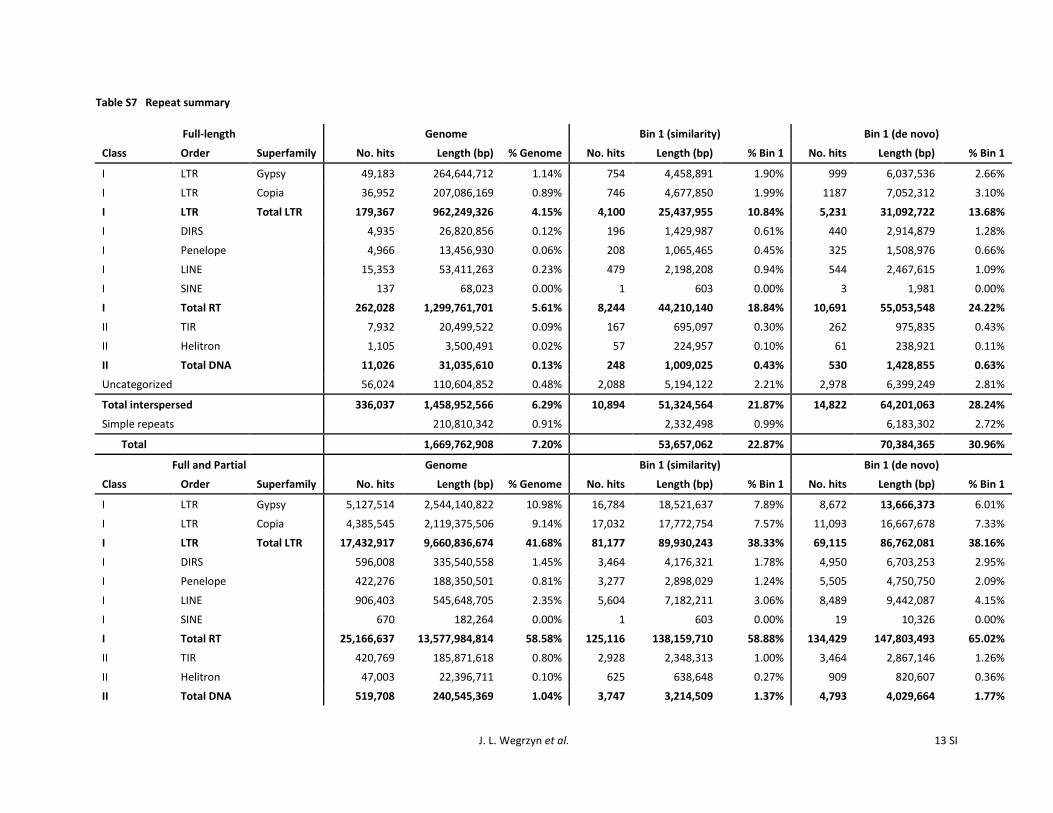
J L Wegrzyn et al 13 SI
Table S7 Repeat summary
Full-length Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 49183 264644712 114 754 4458891 190 999 6037536 266
I LTR Copia 36952 207086169 089 746 4677850 199 1187 7052312 310
I LTR Total LTR 179367 962249326 415 4100 25437955 1084 5231 31092722 1368
I DIRS
4935 26820856 012 196 1429987 061 440 2914879 128
I Penelope
4966 13456930 006 208 1065465 045 325 1508976 066
I LINE
15353 53411263 023 479 2198208 094 544 2467615 109
I SINE
137 68023 000 1 603 000 3 1981 000
I Total RT
262028 1299761701 561 8244 44210140 1884 10691 55053548 2422
II TIR
7932 20499522 009 167 695097 030 262 975835 043
II Helitron
1105 3500491 002 57 224957 010 61 238921 011
II Total DNA 11026 31035610 013 248 1009025 043 530 1428855 063
Uncategorized 56024 110604852 048 2088 5194122 221 2978 6399249 281
Total interspersed 336037 1458952566 629 10894 51324564 2187 14822 64201063 2824
Simple repeats 210810342 091 2332498 099 6183302 272
Total 1669762908 720 53657062 2287 70384365 3096
Full and Partial Genome Bin 1 (similarity) Bin 1 (de novo)
Class Order Superfamily No hits Length (bp) Genome No hits Length (bp) Bin 1 No hits Length (bp) Bin 1
I LTR Gypsy 5127514 2544140822 1098 16784 18521637 789 8672 13666373 601
I LTR Copia 4385545 2119375506 914 17032 17772754 757 11093 16667678 733
I LTR Total LTR 17432917 9660836674 4168 81177 89930243 3833 69115 86762081 3816
I DIRS
596008 335540558 145 3464 4176321 178 4950 6703253 295
I Penelope
422276 188350501 081 3277 2898029 124 5505 4750750 209
I LINE
906403 545648705 235 5604 7182211 306 8489 9442087 415
I SINE
670 182264 000 1 603 000 19 10326 000
I Total RT
25166637 13577984814 5858 125116 138159710 5888 134429 147803493 6502
II TIR
420769 185871618 080 2928 2348313 100 3464 2867146 126
II Helitron
47003 22396711 010 625 638648 027 909 820607 036
II Total DNA 519708 240545369 104 3747 3214509 137 4793 4029664 177
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence
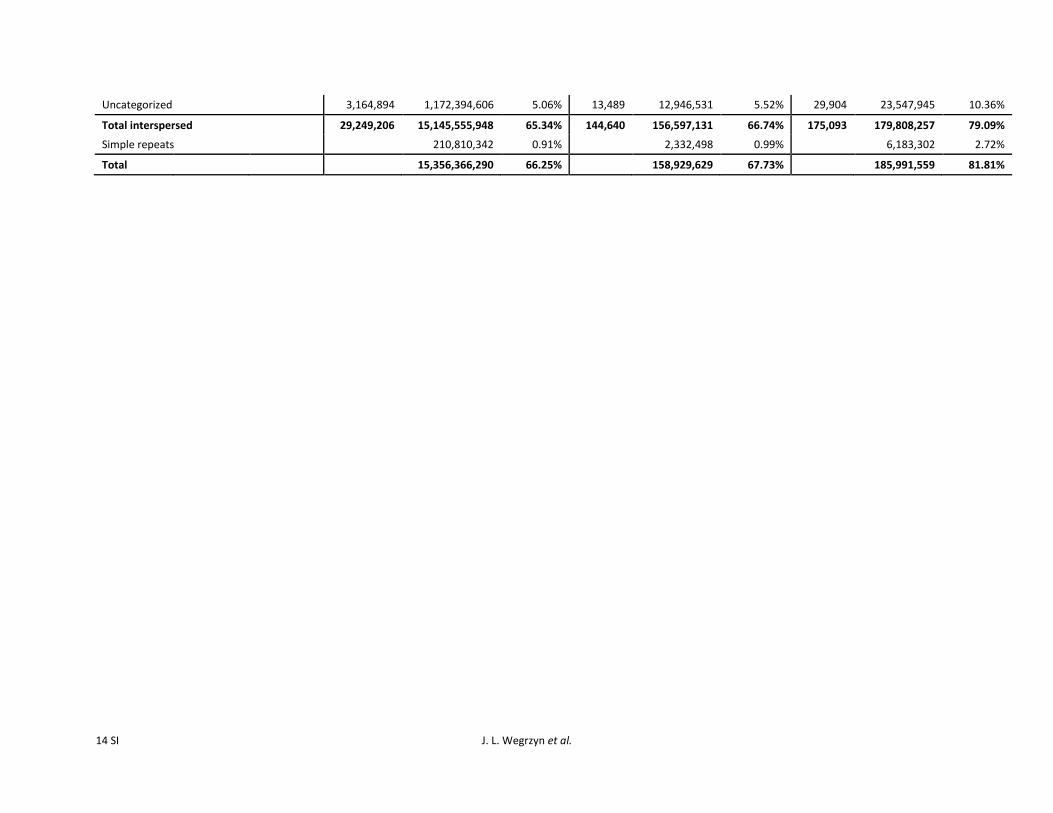
14 SI J L Wegrzyn et al
Uncategorized 3164894 1172394606 506 13489 12946531 552 29904 23547945 1036
Total interspersed 29249206 15145555948 6534 144640 156597131 6674 175093 179808257 7909
Simple repeats 210810342 091 2332498 099 6183302 272
Total 15356366290 6625 158929629 6773 185991559 8181
J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 15 SI
Table S8 High Copy Full-Length Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family No full-length Family No full-length Family No full-length Family No full-length
PtRLG_13 49 PtRXX_107 174 PtAppalachian 4347 PtRLX_2461 1925
PtAppalachian 41 PtRLC_3 70 PtRLG_13 2835 PtRXX_4292 1506
PtRLX_3765 29 PtRLX_3765 65 PtRLX_1912 2638 PtRLX_3423 1452
PtRLX_1912 29 PtAppalachian 62 PtRLC_283 1882 PtRLX_3902 1249
PtRLC_782 26 IFG7_I 57 PtRLX_291 1810 PtRLC_601 1247
PtRLX_3423 24 PtRXX_2933 55 PtRLX_11 1630 PtRLX_2504 928
PtRXX_2933 24 PtRLX_3423 43 PtRLC_334 1535 PtRLX_3298 910
PtRLX_291 23 PtPineywoods 39 PtRLC_782 1407 PtRLX_106 891
PtPineywoods 19 PtRLG_13 38 PtPineywoods 1386 PtRLX_2798 847
PtRLX_3902 19 PtRLX_2461 33 PtRLG_432 1361 PtRLX_140 844
PtRLX_2461 18 PtNoCat_2490 31 PtRLX_106 1285 PtRLG_476 826
PtRLC_283 17 PtRLX_291 30 PtRLC_488 1187 PtDTX_135 816
PtRLG_854 17 PtRLX_3871 30 PtRLG_673 1103 PtDTX_145 786
PtRXX_3669 17 PtRLC_494 29 PtRLG_854 1097 PtRLX_2789 785
PtRLX_14 16 PtRLX_3435 29 PtRLC_514 1090 PtRLC_860 723
PtRLX_11 16 PtRYX_183 27 PtRLG_504 1071 PtRLG_933 717
PtRIX_403 16 PtNoCat_2560 24 PtRLG_10 1040 PtRLC_822 714
PtNoCat_2560 15 PtRLX_3902 24 PtRXX_121 1036 PtRYX_173 713
PtRLG_504 15 PtRXX_3669 24 PtRXX_109 1030 PtRXX_2933 684
PtRYX_183 15 PtRXX_4898 24 PtRLG_537 1004 PtRLX_2876 676
PtRLX_106 15 PtRLX_3892 23 PtRLC_577 964 PtRLC_616 672
PtRXX_109 15 PtRLX_2487 22 PtRIX_403 957 PtDTX_114 670
PtRLC_334 14 PtRLX_14 21 PtRLG_18 950 PtRLG_688 653
PtRLC_482 14 PtDXX_123 20 PtRLX_1335 929 PtRLX_3104 634
PtRLG_673 13 PtRLX_11 20 PtRLX_3008 887 PtRLX_3184 627
PtRLG_432 13 PtRLC_617 19 TPE1 875 PtRLG_445 627
PtRIX_31 13 PtRLC_665 19 PtRLC_390 864 PtRLC_617 625
PtRXX_121 13 PtRLX_119 19 PtRLX_119 863 PtRLC_824 621
PtRPX_11 12 PtRLX_90 19 PtRIX_13 859 PtDTX_24 595
PtRLC_577 12 PtRIX_403 18 PtRLX_3765 858 PtRLX_3719 593
PtRLC_617 12 PtRLC_482 18 PtRLG_551 848 PtRLC_621 589
PtRLG_551 12 PtRLC_857 18 PtRLG_708 840 PtDTX_72 585
PtRLX_3435 12 PtRXX_4980 18 PtRLX_2005 824 PtRXX_1672 582
PtRIX_13 12 PtRYX_88 18 PtRLG_440 823 PtRLX_90 575
TPE1 12 Contig5_LTR 17 PtRLC_346 804 PtPotentialGene_173 564
PtRLX_3871 11 PtRLC_528 17 PtRPX_11 786 PtRLC_692 558
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence
16 SI J L Wegrzyn et al
PtRXX_4898 11 PtRLG_18 17 PtRLX_28 772 PtDTX_121 556
PtRLG_885 11 PtRLG_854 17 PtRLX_14 762 PtRLG_673 553
PtRPX_174 11 PtRLG_885 17 PtRLX_194 703 PtRLC_346 542
PtRLC_709 11 PtRXX_3144 17 PtRLG_1 701 PtRXX_27 536
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence
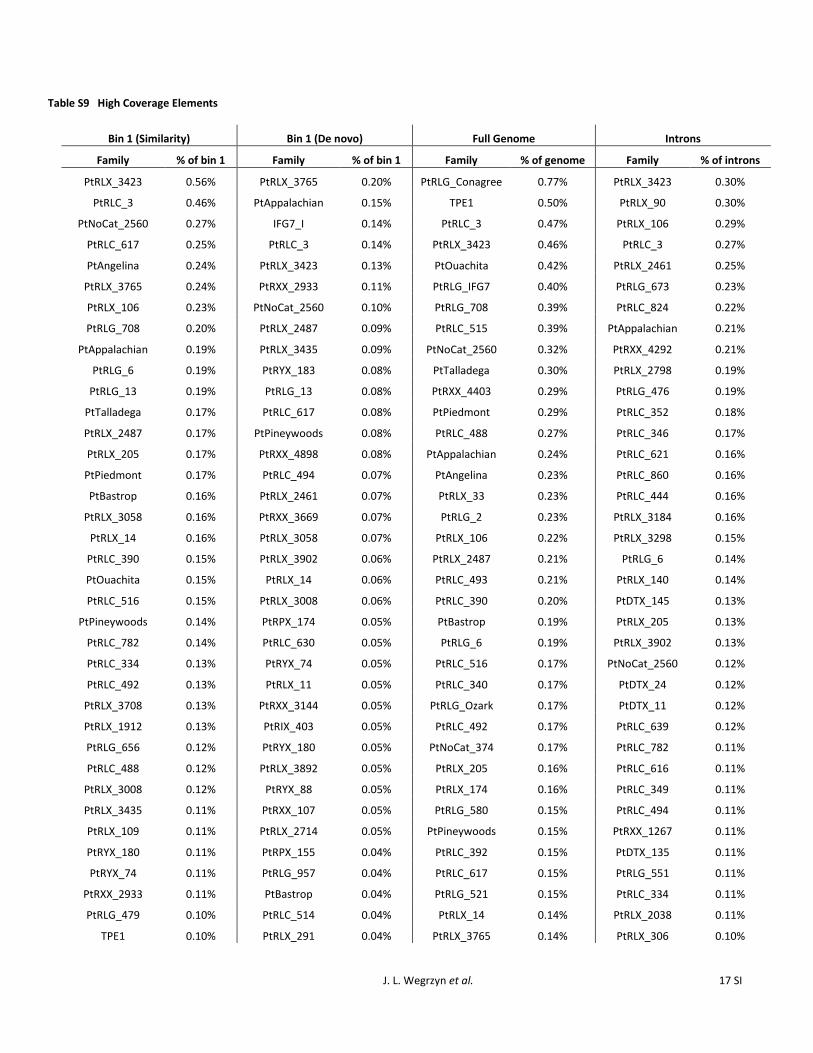
J L Wegrzyn et al 17 SI
Table S9 High Coverage Elements
Bin 1 (Similarity) Bin 1 (De novo) Full Genome Introns
Family of bin 1 Family of bin 1 Family of genome Family of introns
PtRLX_3423 056 PtRLX_3765 020 PtRLG_Conagree 077 PtRLX_3423 030
PtRLC_3 046 PtAppalachian 015 TPE1 050 PtRLX_90 030
PtNoCat_2560 027 IFG7_I 014 PtRLC_3 047 PtRLX_106 029
PtRLC_617 025 PtRLC_3 014 PtRLX_3423 046 PtRLC_3 027
PtAngelina 024 PtRLX_3423 013 PtOuachita 042 PtRLX_2461 025
PtRLX_3765 024 PtRXX_2933 011 PtRLG_IFG7 040 PtRLG_673 023
PtRLX_106 023 PtNoCat_2560 010 PtRLG_708 039 PtRLC_824 022
PtRLG_708 020 PtRLX_2487 009 PtRLC_515 039 PtAppalachian 021
PtAppalachian 019 PtRLX_3435 009 PtNoCat_2560 032 PtRXX_4292 021
PtRLG_6 019 PtRYX_183 008 PtTalladega 030 PtRLX_2798 019
PtRLG_13 019 PtRLG_13 008 PtRXX_4403 029 PtRLG_476 019
PtTalladega 017 PtRLC_617 008 PtPiedmont 029 PtRLC_352 018
PtRLX_2487 017 PtPineywoods 008 PtRLC_488 027 PtRLC_346 017
PtRLX_205 017 PtRXX_4898 008 PtAppalachian 024 PtRLC_621 016
PtPiedmont 017 PtRLC_494 007 PtAngelina 023 PtRLC_860 016
PtBastrop 016 PtRLX_2461 007 PtRLX_33 023 PtRLC_444 016
PtRLX_3058 016 PtRXX_3669 007 PtRLG_2 023 PtRLX_3184 016
PtRLX_14 016 PtRLX_3058 007 PtRLX_106 022 PtRLX_3298 015
PtRLC_390 015 PtRLX_3902 006 PtRLX_2487 021 PtRLG_6 014
PtOuachita 015 PtRLX_14 006 PtRLC_493 021 PtRLX_140 014
PtRLC_516 015 PtRLX_3008 006 PtRLC_390 020 PtDTX_145 013
PtPineywoods 014 PtRPX_174 005 PtBastrop 019 PtRLX_205 013
PtRLC_782 014 PtRLC_630 005 PtRLG_6 019 PtRLX_3902 013
PtRLC_334 013 PtRYX_74 005 PtRLC_516 017 PtNoCat_2560 012
PtRLC_492 013 PtRLX_11 005 PtRLC_340 017 PtDTX_24 012
PtRLX_3708 013 PtRXX_3144 005 PtRLG_Ozark 017 PtDTX_11 012
PtRLX_1912 013 PtRIX_403 005 PtRLC_492 017 PtRLC_639 012
PtRLG_656 012 PtRYX_180 005 PtNoCat_374 017 PtRLC_782 011
PtRLC_488 012 PtRLX_3892 005 PtRLX_205 016 PtRLC_616 011
PtRLX_3008 012 PtRYX_88 005 PtRLX_174 016 PtRLC_349 011
PtRLX_3435 011 PtRXX_107 005 PtRLG_580 015 PtRLC_494 011
PtRLX_109 011 PtRLX_2714 005 PtPineywoods 015 PtRXX_1267 011
PtRYX_180 011 PtRPX_155 004 PtRLC_392 015 PtDTX_135 011
PtRYX_74 011 PtRLG_957 004 PtRLC_617 015 PtRLG_551 011
PtRXX_2933 011 PtBastrop 004 PtRLG_521 015 PtRLC_334 011
PtRLG_479 010 PtRLC_514 004 PtRLX_14 014 PtRLX_2038 011
TPE1 010 PtRLX_291 004 PtRLX_3765 014 PtRLX_306 010
18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

18 SI J L Wegrzyn et al
PtRLX_33 010 PtRLG_916 004 PtRLG_585 014 PtRLX_334 010
PtRIX_403 010 PtRLG_643 004 PtRLG_13 014 PtRLG_13 010
PtRPX_174 010 PtRLX_3871 004 PtRLC_334 014 PtRLX_2504 010
J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 19 SI
File S1
Methodology of the loblolly transcriptome
Total RNA was prepared from 27 loblolly pine collections including vegetative and reproductive organs seedlings needles seeds and isolated megagametophytes from a variety of individuals Polyadenylated RNA was enriched from total RNA and strand-specific libraries prepared by dUTP exchange in a manner similar to Zhong et al (2011) using Illumina TruSeq mRNA indexed adaptors (Mockaitis lab Indiana University) Indexed libraries were pooled and sequenced as 102 nt x 2 paired reads on a HiSeq2000 instrument using TruSeq SBS v3 reagents (Scheffler lab Genomics and Bioinformatics Research Unit USDA ARS Stoneville MS) A total of 134 billion reads (671 million pairs) were used for sample-specific and grouped assemblies RNA assemblies their quality analyses and selection for use will be described in detail (Fuentes-Soriano Loopstra Wu Gilbert and Mockaitis in preparation) Briefly reads were trimmed to 89 nts based on equivalent base representation and quality scores gtQ20 using a custom Perl script Trinity (Grabherr et al 2011) versions 20121005 and 20130225 as well as Velvet-Oases (0208) were used to generate parallel assemblies of read sets that were then classified for protein coding completeness length and uniqueness of sequence using software of the EvidentialGene suite (Gilbert 2012) 87241 of 14 M transcripts were determined to be the longest of aligned unique sequences that contained complete CDS and as such were classified as primary Any potential contaminant transcripts derived from sampling were retained Of the set 83285 mapped to the loblolly pine genome assembly with no coverage or mismatch filters and these were used as evidence for annotating putative gene loci described here The selected transcripts and trimmed reads files are available as TSA and SRA entries within NCBI BioProject PRJNA174450 References Gilbert D 2012 EvidentialGene httparthropodseugenesorgEvidentialGene Grabherr M G B J Haas M Yassour J Z Levin D A Thompson et al 2011 Full-length transcriptome assembly from RNA-
Seq data without a reference genome Nature Biotechnol 29 644-U130 Schulz M H D R Zerbino M Vingron and E Birney 2012 Oases robust de novo RNA-seq assembly across the dynamic range
of expression levels Bioinformatics 28 1086-1092 Zhong S J-G Joung Y Zheng Y-r Chen B Liu et al 2011 High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation Cold Spring Harb Protoc 2011 pdbprot5652
File S2
Accession numbers and additional details for alignment data sets
Available for download as an Excel file at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

20 SI J L Wegrzyn et al
File S3
An explanation of PIER and GCclassif
The Pine Interspersed Element Resource (PIER) v20 includes 5280 elements belonging to six families (plnrep grasrep mcotrep
dcotrep oryrep and athrep) from Repbase Update 1707 (Jurka et al 2005) five additional elements previously characterized
in the literature and 9415 elements characterized in loblolly pine BAC and fosmid sequences (Wegrzyn et al 2013) We
developed an open-source classification tool GCclassif which analyzes the pol region characteristic of LTR retroelements to
classify LTR retrotransposons into either Gypsy or Copia superfamilies based on the ordering of the protein domains (Figure S1)
First ORFs of an unclassified LTR are identified using the findorfs utility in USEARCH with default parameters HMMER 30
(Eddy 2011) follows with hmmsearch using selected Pfam profiles as a library against the generated ORFs Finally HMMER hits
are sorted and chained based so that all combinations of the alignments are considered After all possible protein family
orderings have been constructed the highest-scoring (log-odds) chain is selected as the correct classification If there is
insufficient evidence the element remains unclassified Source code can be found at httpgithubcombylinnealelab-
scriptsblobmasterGCclassifpy
With the subsequently developed GCclassif tool 498 LTR retroelements were reclassified into the Gypsy superfamily and 416
LTR retroelements were reclassified into the Copia superfamily 2430 additional sequences were classified as LTR
retrotransposons at the order level but further classification was made difficult by their lack of detectable internal domains or
structures and lack of alignments to known orders or superfamilies Although portions of protein domains can be detected in
1782 of these sequences insufficient evidence kept them from being reclassified into Gypsy or Copia superfamilies The new
classifications were incorporated into PIER prior to analysis of genomic repetitive content
Files S4-S8
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1
File S4 Annotations for MAKER-derived gene models
File S5 Transcript notations including introns gt 20Kbp
File S6 Gene Families Greater than or equal to 2
File S7 Genes with Annotations
File S8 Conifer-Specific families
J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 21 SI
File S9
Multiple Sequence Alignments related to Figure S2 GeneFamily 3795 gtPP00070G01110 MAGEVFTPSVTQGFWGPITASTEWCEMNYQVTSLVAEFYNTISNIPGIILAFLGVYYSIS QKFERRFSVLHLSTIALGIGSILFHATLKYAQQQSDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQIHYVFLALLCLPRMYKYYMYTKDPLARKLAHLY VLCLALGAICWLADRHLCSWICKLKVNPQGHALWHILEGFNSYFGNTFLQYCRAQQLNWN PRIDYLLGVVPYVKVQKGDTERKEQ gtPP00188G00180 MAGEALTSSTAQGFWGPITASTEWCEKNYEVTPLVAEFYNTISNIPGIVLAFIGVYYSIS QKFERRFSALHLSTIALGMGSILFHATLRYAQQQCDETPMVWAMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAILHSQFRFVAGFQVHYVFLALLCLPRMYKYYMYTKDPLVRKLAHRY VLCLALGAICWLADRHLCSWICKLKVNPQGHGLWHVLVGFNSYFGNTFLQYCRAQQLNWN PRIEYSLGVLPYVKVERSDNDRKEE gtPP00055G00680 -MAGEPSSGWLQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILAIIGLYYAIS QKFERRFSVLHLSTIALCIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTIFAVLHSQFRFVVGFQLHLVLLAVLCLPRMYKYYIHTKDPAVRKLAHKY ILFLVLGGMCWLADRHLCNQISKLRVNPQGHALWHVLMGFNSYIGTTFLLYCRAEQLNWN PKVEYVLGLLPYVKVQKSESERKEQ gtPP00452G00100 -MAEKISPGSHQGYWGPITASTEWCEKNYEVTPMVAEFYNTISNVPGIILALIGLYYAIS QKFERRFSVLHLSTIALSIGSSLFHATLKYAQQQSDETPMVWVMLLYIYVLYSPDWHYRS TMPTVLFLYGTVFAILHSQFRFVVGFQLHFVLLALLCLPRMYKYYIHTTDPAVRKLARKY VLFLVMGAICWLVDRHLCNQVSKLRINPQGHALWHVFMGYISYLGNTFLQYCRAEQLNWN PKVEHVLGVLPYVKVQKSDRDRKEQ gtPita_1A_I9_VO_Locus_71152_Transcript_1_4_Confidence_0500_Length_1856 -----MASQRVDSFWGPVTSTTDWCEKNYAVSAYIVEFFNTISNIPCIILAFIGLINSLR QRFEKRFSVLHLSNMALAIGSMIFHATLQHAQQQSDETPMVWEMLLYIYVLYSPDWHYTY TMPTFLFLYGAAFATFHSLFRFDLGFKIHYIISAGLCLPRMYKYYIHTTEPAAKRLAHLY ILTLILGGMCWLLDRTFCDTVSTWYINPQGHALWHIFMGFNAYFANAFLQFCRAQQREWR PEIRHVLGL-PYVKIFKVKSE---- gtVV05G06510 ---------MISNI----------------------KFLNTVSNVPGIVLGLFGLINALR QGFEKRFSVLHISNIILAIGSILHHSSLQRLQQQSDETPMVWEMLLYIYILHSPDWHYQS TMPTFLFLYGAAFAIVHSQVHFGIGFKIHYVILCLLCIPRMYKYYIHTQDMSAKRLAKLH LGTLFIGSLCWLSHRLSHKDSSHWYFSLQGHALWHVLMGFNSYFANAFLMFCRAQQREWN PKVVHFLGL-PYVKIQKPKIQ---- gtPT01G31720 -----MAE-AISSFWGPVTSA-EWCEKNYVYSSYIAEFFNTVSIIPGILLALIGLINALR QRFEKRFSVLHISNMILAIGSMLYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS VMPTFLFLYGAAFAIFHALVRFEIGFKVHYVILCLLCVPRMYKYYIYTKDASAKRLAKLY LATITTGSLCWLFDRLFCNNISQWYFNPQGHALWHVLMGFNSYFANTFLMFWRAQQLGWN PKVAHFMGFFPYVKIQKPKTQ---- gtRC29904G00190 -----MAEGGISSFWGPVTSP-EWCEKNYAYSSYIAEFFNTISNVPGILLAFIGLINALR QRFEKRFSVLHISNMILGIGSISYHATLQRMQQQGDETPMVWEMLLYFYILYSPDWHYRS TMPTFLFFYGAAFAVFHSLVRFGIGFKVHYAILCLLCVPRMYKYYIYTNDVSAKRLAKLY VG---------------------WSFNPQGHALWHVLMGFNSYFANTFLMFCRAQQLGWN PKVVDLLGFFPYVKVRKPKTQ---- gtGM07G39500 -----MAE-SISSFWGPVTSTKECCEINYAYSSYIAEFFNTISNIPTILLALIGLINALR QRFEKRFSVLHVSNMTLAIGSMLYHATLQHVQQQSDETPMVWEVLLYMYILYSPDWHYRS TMPIFLFVYGALFAVAHSVFHFGIGFKVHYIILILLCVPRMYKYYIYTQDVSAKRLAKLF LGTFVLGSLFGFCDRVFCKEISRWPINPQGHALWHVFMGFNSYFANTFLMFCRAQQRGWS PKVVHLMGV-PYVKIEKPKSQ---- gtAT4G22330 -----MAD-GISSFWGPVTSTIECCEMNYAYSSYIAEFYNTISNVPGILLALIGLVNALR
22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

22 SI J L Wegrzyn et al
QRFEKRFSILHISNMILAIGSMLYHATLQHVQQQSDETPMVWEILLYMYILYSPDWHYRS TMPTFLFLYGAAFAIVHAYLRFGIGFKVHYVILCLLCIPRMYKYYIHTEDTAAKRIAKWY VATILVGSICWFCDRVFCKTISQWPVNPQGHALWHVFMSFNSYCANTFLMFCRAQQRGWN PKVKYFLGVLPYVKIEKPKTQ---- gtPT11G00510 -----MAEGGISSFWGPVTSTTECCEKNYAYSSYIAEFHNTISNIPCIVLALVGLINALR QRFEKRFSVLHISNMILAIGSMIFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPIFLFLYGAVFAAVHSVVRLGIGFKVHYAILCLLCIPRMYKYYIYTQDASAKRLAKMY VATLLIGTLCWLFDRIFCKEISSWPINPQGHALWHVFMGFNSYLANTFLMFCRARQRGWS PKVVHFMGVLPYVKIEKPKAQ---- gtRC29842G00070 -----MAD-GISSFWGPVTSTTECCEKNYAHSSYIAEFYNTISNIPCILLALIGLINALR QRFEKRFSVLHISNMILAIGSMFFHATLQRVQQQSDETPMVWEMLLYMYILYSPDWHYRS TMPTFLFLYGAVFAIVHSVVHFGIGFKVHYVILCLLCIPRMYKYYIYTQDAAAKWLAKLY VATLFIGSLCWLCDRIFCKKISNWPINPQGHALWHVFMGFNSYFANTFLMFCRAEQRGWS PKVVCFAGILPYVKIEKPKVQ---- GeneFamily 10970 gtPita_2A_all_VO_Locus_12631_Transcript_1_3_Confidence_0167_Length_3462 MPVEKGVCGSIRPAAAMAEVKLTDSQLNTICAPIDRPLMILAAAGSGKTLVICHRILHLI SKGVSPKDILAVTFTRRAGQDLLHRLQCIAAAQQCFGGEGSLDVVGIRVGTFHSFCLSVL RAFPNHAGLAPDFVVFTPKMQLDLLENLVEEWHTQRRHEINGQSMQFQDKLARKSKQLQG RHQSTAEKKAFFKAGAYRLFQHLRTSQFVKSQEGAADVELNDS-HGDGFSKWVFHQYHEH LLQANGIDFNSFVRYTLDMLKKWPNALESAGLKAQYIFVDEFQDTDVSQFELLKILCKDH AHITVVGDDDQQIYSWRGATGLYNIKNFENVFKGGITTKLEQNFRSTGAIVASARSLISK NQSRMPKTVRTASPTGTLMTICECRNDQCEVTAIVDFIISLKKQGVPLQEIAILYRLQRI GLEVQHNLQAQGIECHIKGSGSGSTGNQCLVANGGQVGTGLGDVFHDILAVLRLAICESD DLASKHLLDLFCPSMSLVIKDCLSYLKLQKGLSLLQAIKSTRSHLVGLLVCDDLSSYAPA KSIAILSESDHLTLEGMHSILKLLAATKNDSKNMGLRDVIFNILQQISYVRGSRDVPQAL KADLDGRKGYNQQLVSSSGKVDTNSLNSLQYAGIKALLKEAALFDAEIRTGSQNQNSCSI KESNSATPQTSYMDLKQHRSRKRGL-NLSPDKPGFAGNIDFSLDAKSLDKLRIFLDRVTL KMHDSELGENNASIPIRPVQSVPSSSACDTGVTLSTIHQAKGLEWEGVILVRANDGIIPV RDSDFVVGPTTPNTPNIIDYGTTRNEDVITREAGNKNNLENNDISLLGPPILETRGKFSS GPNREGIEEERRLMYVALTRAKRFVLVTHVMMEGGQQMTPSCFLADIPPGLVRRTTCYES KIPDPGLLLDSVTPSPISTKSVSSANDEAVKGKTKDPSGIFYSQNKDTNVKDDMQYSSKI LKSCTPNMRRKTNLQVNASELSLEMASTGSMSDCHLRKYSSKKTGNVVEISKSDECGSQS DDSEFEKPVRKGRKRKQKQVICSEGEE gtSM00010G01000 ------------------MVELTESQVAAVTAPISCPLMILAAAGSGKTLVLCYRILHLI RNGVPAKEILAVT----------------------------------------------- ---------------------LELLQELVYEWHLMRSN---------------------G QKCEGSLKKQLFRTEAYRMMKQI--GQMQRKHDTAA---LNSAKKGGGMLDWVLERYHSH LQNINAIDFNSFTRRTNDVLESFP--------------VDEFQDTDTGQFKLLQALSTRH HHVTVVGDDDQQIYSWRGAAGIQNLESFSSTFGGATVVKLERNFRSTGAIVAAARSVISR NESRMRKSIGTTAPTGKLVAMCECRNVLCEVAAISNFISSSRAEGVKLSEIAVLYRVHRV GVELQHHLQAIGVPCKMKVTGG---SDDSTVSAKGFSST--GEALADILGVLRLILNESD DYACKRLMLLFCPDGRSEVLSCVMQLQQAGSSSLLQSLKTLRSHVLG-MSKDSLSAPWIQ PVVEKLGVLVKEVIKGAQVLMKIMADTKEELQRLGMKDVVMNLLQQIGPYR-----EEAR TASLQ---------ISSTANLSGGS----DFAGIKALLKEAAAFDGDNAFVMAKTSA--- -QTASKSKNASYKDMRRLQKRDPGQGTVRSSQSDFKDHT---------QRLRVFLDQVAL KLHEYELGEDN--------KTLSSPSDC---VTLSTIHQAKGLEWPAIVLARANEGVLPV FDTDMV----------------SDAQDILEEE---------------------------- --------------------------ITYLMKDSGQQALPSRFLAEIPRGLVKRIVSYDH QV---------------------------------------------------------- -----------TNITATTNTI--------------------------------------- -----------GRKREERG--------
J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence

J L Wegrzyn et al 23 SI
Files S10-S12
Available for download as Excel files at httpwwwgeneticsorglookupsuppldoi101534genetics113159996-DC1 File S10 Overview of tandem content in Arabidopsis thaliana Vitis vinifera Selaginella moellendorffii Cucumis sativus Populus trichocarpa Picea glauca Picea abies and Amborella trichopoda File S11 Tritetra nucleotide motifs and frequency File S12 Full Repeat Summary for Similarity Search Applied to Intronic Sequence