trabajo de grado - usb
TRANSCRIPT
DESARROLLO DE UN BIOSENSOR A PARTIR DE LA INMOVILIZACIÓN DE LA ENZIMA BIÓXIDO DE
CARBONO DESHIDROGENASA, OBTENIDA DE LA BACTERIA Metanococcus deltae.
.
TRABAJO DE GRADO
CARLOS ANDRES DURAN DUQUE
UNIVERSIDAD DE SAN BUENAVENTURA
2010

TABLA DE CONTENIDO
Pag.
RESUMEN
ABSTRACT
O. INTRODUCCION 1
1. JUSTIFICACION 2
2. OBJETIVOS
2.1. OBJETIVO GENERAL 3
2.2. OBJETIVOS ESPECIFICOS 3
3. MARCO TEORICO 4
3.1 BIOSENSORES 5
3.2 BIOSENSORES EN LA AGROINDUSTRIA 5
3.3 BIOSENSORES CO2 5
3.4 EXTRACTO ENZIMATICO PURIFICADO 6
3.5 BACTERIAS METANOGENICAS 6
3.5.1 METTANOCOCCUS DELTAE 7
3.5.2. TAXONOMIA 8
3.6. CRECIMIENTO BACTERIANO 8
3.7 LISIS 9
3.7.1. METODOS ENZIMATICOS DE RUPTURA CELULAR 9
3.8 ENZIMAS 10
3.8.1. PURIFICACION INICIAL 11
3.8.2. PURIFICACION DE ENZIMAS 12
3.8.3 CROMATOGRAFIAS 12
3.9 ACTIVIDAD ENZIMATICA 13
3.9.1. FACTORES QUE AFECTAN LA ACTIVIDAD ENZIMATICA 14

3.9.2. CALCULO DE LA ACTIVIDAD ENZIMATICA EN ESPECIMENES
BIOLOGICOS Y CUANTIFICACION DE LA CONCENTRACION DE ENZIMAS
CIRCULANTES 14
3.10. ELECTRODOS MODIFICADOS QUIMICAMENTE CON ENZIMAS 14
3.11. APLICACIÓN DE BIOSENSORES 15
3.11.1 SEGURIDAD ALIMENTARIA 15
3.11.2 ADITIVOS 15
3.11.3 FARMACOS 16
3.11.4 RESIDUOS DE PLAGUICIDAS Y FERTILIZANTES 16
3.11.5 COMPONENETES DEL ALIMENTO 16
3.11.6 BIOTOXINAS
16
3.11.7 MICROORGANISMOS PATOGENOS 16
3.11.8 CALIDAD ALIMENTARIA 17
3.11.9 CONTROL DE PROCESOS 17
3.11.10 SENSOR PARA DIOXIDO DE CARBONO, CO2 17
3.12 INMOVILIZACION DE ENZIMAS 19
3.12.1 METODOS DE INMOVILIZACION DE ENZIMAS POR RETENCION
FISICA
3.12.2 ATRAPAMIENTO 19
3.12.3 METODOS DE INMOVILIZACION DE ENZIMAS POR UNION QUIMICA
20
3.12.4 UNION A SOPORTES 20
3.12.5 ADSORCION 21
3.12.6 UNION COVALENTE 22
3.12.7 RETICULADO 22
3.12.8 EFECTOS DE LA INMOVILIZACION 23
3.12.9 EFECTOS DE LA ESTABILIDAD 23
3.12.10 EFECTOS DE LA ACTIVIDAD ENZIMATICA 25

3.12.11 ELECCION DEL METODO DE INMOVILIZACION 27
3.12.12 APLICACIONES DE LAS ENZIMAS INMOVILIZADAS 27
4. METODOLOGIA 29
4.1. LUGAR DE TRABAJO 29
4.2 MATERIAL BIOLÓGICO 29.
4.3 MÉTODOS 29
4.3.1 CULTIVO DE LA BACTERIAS DEL GÉNERO METHANOCOCCUS SP
29
4.3.2 AISLAMIENTO E IDENTIFICACIÓN BACTERIANA 32
4.3.3 PREPARACIÓN DE LOS EXTRACTOS ENZIMÁTICOS OBTENIDOS
POR TRES MÉTODOS DE LISIS CELULAR DIFERENTE 34
4.3.3.1 Lisis celular por Sonicación. 34
4.3.3.2 Lisis celular por Fricción mecánica 35
4.3.3.3 Lisis celular por Agentes Químicos 36
4.3.4.Semipurificación de los extractos enzimáticos obtenidos a partir de la lisis
celular 37
4.3.5 Medición de la actividad enzimática de los extractos celulares obtenidos a
partir de los tres métodos de lisis bacteriana 38
4.3.6. Inmovilización enzimática 39
5. RESULTADOS Y DISCUSIÓN 41
5.1 Cultivo y crecimiento de la cepa bacteriana (M. deltae). 41
5.2 PRUEBAS BIOQUÍMICAS REALIZADAS A LAS BACTERIAS AISLADAS
DEL LAGO. 42
5.3 LISIS BACTERIANA POR TRES DIFERENTES MÉTODOS 44
5.4 MEDICIÓN DE LA ACTIVIDAD ENZIMÁTICA 46
5.5 INMOVILIZACIÓN ENZIMÁTICA Y BIOSENSOR ENZIMÁTICO 48
6. CONCLUSIONES 49
BiBLIOGRAFIA 51

LISTA DE TABLAS
pág.
Tabla 1. Utilización de biosensores en los distintos campos 5
Tabla 2. Reacción metabólica de Methanococcus deltae 6
Tabla 3. Características generales de las Bacterias Metanogénicas. 7
Tabla 4. Ventajas y desventajas de las enzimas como elementos de
reconocimiento en biosensores. 11
Tabla 5. Comparación de diferentes métodos de inmovilización 27
Tabla 6. Características del gel de Sephadex G-100 38
Tabla 7. Resultados de pruebas bioquímicas realizadas. 42
Tabla 8. Datos del Rompimiiento. 44
Tabla 9. Actividad enzimática especifica de las fracciones
semipurificadas obtenidas. 46

LISTA DE FIGURAS
pág.
Figura 1. Cámara de Neubauer 9
Figura 2. Métodos de Inmovilización. 19
Figura 3. Soportes para la Inmovilización 21
Figura 4. Estabilización de la enzima en la inmovilización. 24
Figura 5. Crecimiento anaeróbico de la bacteria Methanococcus deltae 32
Figura 6. Rompimiento celular por sonicación. 35
Figura 7. . Proceso de ruptura mecánica 37
Figura 8. Columna de DEAE utilizada en la semipurificación enzimática. 37
Figura 9. Electrodo de Platino 40
Figura 10. Coloraciones Diferenciales 41
Figura 11. Curva de crecimiento M. deltae 42
Figura 12. Cinética enzimática. 46

RESUMEN
Hoy en día, las enzimas o extractos enzimáticos son los componentes biológicos más usados en biosensores debido a la diversidad de reacciones bioquímicas que catalizan.
Un extracto celular aislado de la bacteria Methanococcus deltae es el que se pretende utilizar en este proyecto para desarrollar un biosensor, el cual consiste en un electrodo modificado químicamente (EMQ). Esta bacteria utiliza el dióxido de carbono como fuente de carbono y energía para la producción de metano, por medio de una enzima (COX) intracelular que cataliza la transformación del dióxido de carbono en metano. De este modo, este sensor enzimático (Biosensor) se convierte en un ejemplo clásico de un Sistema Químico Integrado (SQI).
Para obtener proteínas intracelulares de los microorganismos, como en este caso de la bacteria Methanococcus deltae existen tres métodos generales: enzimáticos, químicos o físicos. Sin embargo, no todas las metodologías pueden ser utilizadas en procesos a gran escala y más aún estas varían en la efectividad de rompimiento celular, influyendo de una u otra forma en la obtención final de extracto enzimático intracelular. Quizás una de las metodologías más destacadas es la sonicación, el cual se convierte en uno de los métodos más empleados para la obtención de proteínas en el laboratorio. Sin embargo, a gran escala, esta metodología presenta problemas al menos desde el punto de vista teórico debido a la dificultad de transmisión de la intensidad de sonicación a gran volumen así como la eliminación del calor que se genera en el proceso. El presente trabajo es parte de un proyecto de investigación más amplio titulado Elaboración de un biosensor enzimático para bióxido de carbono, realizado por profesores y estudiantes pertenecientes al grupo de investigación de Biotecnología, adscrito al programa de ingeniería agroindustrial de la Universidad de San Buenaventura de Cali; y categorizado en D por Colciencias.
A partir de un proyecto previo se logró determinar que la metodología más eficiente de ruptura del microorganismo era el tratamkento con enzimas tipo liticasa, a partir del cual se obtuvieron altas concentraciones de extracto celular, el cual posteriormente se pasó a varias etapas de purificación.
En una primera etapa se pasó el extracto crudo a través de una columna de Sephadex, y posteriormente por una columna de Ecteolla celulosa, al término del cual se midió la actividad enzimática del extracto por medio de un espectrofotómetro,
En este trabajo de investigación se procedió a la inmovilización de la enzima obtenida posterior a los procesos de purificación enzimática comentado
anteriormente.
Este extracto enzimático se inmovilizará a través del método de unión química, de los que se dispone una mayor información. Como soporte se utilizó un electrodo de platino en el cual se determinó las diferencias en carga cuando se estaba en presencia o ausencia de bióxido de carbono.

El biosensor enzimático para bióxido de carbono desarrollado se constituye en un implemento de suma importancia para los ingenieros agroindustriales puesto que permite determinar de modo específico concentraciones bajas de dióxido de carbono, además del bajo impacto medioambiental que poseen este tipo de sensores (biosensores) y que tendría la determinación de cantidades bajas de este peligroso gas en la atmósfera o en habitaciones cerradas, por lo cual el enfoque global de estos estudios se encamina al desarrollo de un sistema químico integrado.
ABSTRACT
Nowadays, the enzymes or enzymatic extracts are the biological components most used in biosensors due to the diversity of biochemical reactions that they catalyze.
A cellular extract isolated of the bacterium Methanococcus deltae is that one tries to use in this project for developing a biosensor, which consists of an electrode modified chemically (EMQ). This bacterium uses the carbon dioxide as source of carbon and energy for the production of methane, by means of one enzyme (COX) intracellular that catalyzes the transformation of the carbon dioxide in methane. Thus, this enzymatic sensor (Biosensor) turns into a classic example of a Chemical Integrated System (SQI).
In order to obtain intracellular proteins of the microorganisms, as in this case of the Methanococcus bacterium deltae, exists three general methods: enzymatic, chemical or physical. Nevertheless, all the methodologies cannot on a large scale be used in processes and still more these vary in the effectiveness of cellular breaking, influencing of one or the other form in the final obtaining of intracellular enzymatic extract. Perhaps one of the most outstanding methodologies is the sonicatión, which becomes one of the methods more using for the protein obtaining in the laboratory. Nevertheless, on a large scale, this methodology presents/displays problems at least from the theoretical point of view due to the difficulty of transmission of the intensity of sonicatión to great volume as well as the elimination of the heat that is generated in the process. The present work is part of an investigation project ampler titleholder Elaboration of an enzymatic biosensor for carbon dioxide, realized by professors and students pertaining to the group of investigation of Biotechnology, assigned to the program of agro-industrial engineering of the University of San Buenaventura of Cali; and categorized in D by Colciencias.
In one first stage a crude extract from several líticos methods was obtained and I determine myself what was the most effective method, later went the crude extract through a column of Sephadex, and a column of Cellulous Ecteolla, at the end of which the enzymatic activity of the extract by means of a spectrophotometer was moderate. In this work of investigation it was come to the immobilization of the enzyme obtained in a platinum electrode, subsequent to the processes of commented enzymatic purification previously. This enzymatic extract will be immobilized through method of chemical union, which

greater information is had. As it has supported were used a platinum electrode in which one determined the differences in load when it was in presence or absence of carbon dioxide. The enzymatic biosensor for developed carbon dioxide is constituted in I implement of extreme importance for the agro-industrial engineers since it allows to determine of specific way carbon low dioxide concentrations, besides the low environmental impact that own this type of sensors (biosensors) and that it would have the determination of low amounts of this dangerous gas in the atmosphere or closed rooms, thus the global approach of these studies is directed to the development of an integrated chemical system.
KEY WORDS: enzymes, immobilization, biosensor, carbon dioxide, Methanococcus deltae

2
1. JUSTIFICACIÓN
Colombia, país rico en biodiversidad, es uno de los principales productores de frutas a nivel mundial, debido a su clima netamente tropical y a la fertilidad de sus suelos. De ahí la importancia de desarrollar nuevas tecnologías, implementos y/o equipos que permitan determinar grado de maduración de la fruta, características de la fruta, metabolismo, entre otros.
Uno de los parámetros más importantes en las frutas es la medición de la respiración, el cual permite determinar diversas características metabólicas y fisiológicas de la fruta tal como la maduración. En la respiración los azúcares frutales se convierten en energía y bióxido de carbono, después de pasar por la glucólisis y el ciclo de Krebs. Para las industrias que almacenan, comercian, transportan o tienen algo que ver con las frutas es de gran importancia el desarrollo de implementos que permitan la determinación de cantidades pequeñas en el orden de ppm de bióxido de carbono. Es aquí donde cobra relevancia el desarrollo de biosensores, los cuales son altamente específicos y miden concentraciones muy bajas de Bióxido de Carbono.
El biosensor de bióxido de carbono en este trabajo es importante puesto que la contaminación ambiental, y alimentaria es bastante alta en estos tiempos tanto que se puede observar con los cambios climáticos que se están presentando en el planeta, con este sensor se va a controlar la descomposición de alimentos o detección de co2 en cuartos cerrados de almacenamiento o en casos ambientales en el medio que nos rodea.
Es así como el método de detección de CO2 propuesto en este trabajo está basado en la reacción de oxidorreducción que lleva a cabo la bacteria Methanococcus deltae, bacteria metanogènica de las aguas residuales, por medio de la cual el CO2 en presencia de H2 se reduce a formaldehido y posteriormente a metano (CH4).

3
2. OBJETIVOS
2.1 GENERAL
Desarrollo de un Biosensor a partir de la inmovilización de la enzima Bióxido de carbono deshidrogenasa.
2.2 ESPECIFICOS
Cultivar y crecer la bacteria Methanococcus Deltae en un medio de cultivo rico en nutrientes.
Realizar una semipurificación del extracto crudo obtenido mediante la utilización de una columna de exclusión por tamaño y una columna de intercambio iónico.
Evaluar la actividad catalítica presentes en el extracto enzimático semipurificado obtenido de la bacteria Methanococcus Deltae.
Inmovilizar enzimas en un electrodo de platino.
Elaboración de un biosensor enzimático para bióxido de carbono.

4
3. MARCO TEORICO
3.1 BIOSENSORES
El término biosensor aparece en la literatura científica a finales de los años 70, aunque el concepto básico e incluso su comercialización comenzó antes. El primer biosensor fue un analizador de glucosa desarrollado por Clark y Lyons en 1962 y comercializado a partir de 1975 por Yellow Springs Instrument Company. Este biosensor se denominó “enzyme electrode” y consistía en una enzima glucosa oxidasa acoplada a un electrodo para oxígeno.
La enzima oxida la glucosa y como consecuencia se produce un descenso proporcional de la concentración de oxígeno en la muestra, que es detectado por el electrodo. En los años siguientes se desarrollaron electrodos enzimáticos para distintas sustancias de interés clínico mediante la unión de enzimas apropiadas a sensores electroquímicos.
El término biosensor comenzó a utilizarse a partir de 1977 cuando se desarrolló el primer dispositivo utilizando microorganismos vivos inmovilizados en la superficie de un electrodo sensible a amonio. Este dispositivo se utilizaba para detectar el aminoácido arginina y sus creadores lo denominaron “sensor bio-selectivo”. Posteriormente para acortar, se denominó “biosensor” y este término ha permanecido desde entonces para designar la unión entre un material biológico y un transductor físico. A partir de ese momento el diseño y las aplicaciones de los biosensores en distintos campos de la química analítica ha continuado creciendo.
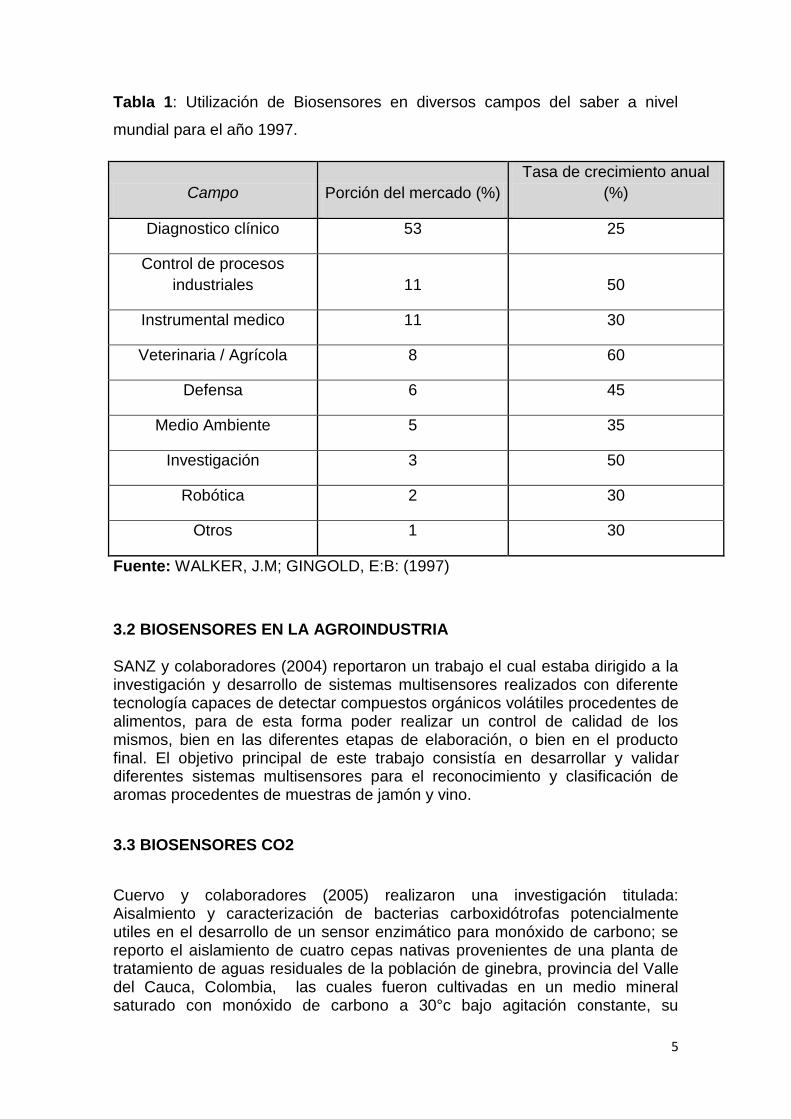
La tecnología de biosensores, han experimentado un notable avance en los últimos años. Estas tecnologías en avanzado estado de madurez han ido transfiriéndose paulatinamente de forma horizontal a otros sectores como el medioambiental, y de forma más incipiente al agroalimentario. En el momento actual los biosensores suponen potentes herramientas de análisis con numerosas aplicaciones a la industria agroalimentaria, apoyándose en los instrumentos de la biotecnología. En la Tabla 1 se muestra como se encontraba para el año de 1997 repartida la utilización de biosensores en diversos campos y sus respectivos crecimientos anuales con lo cual se puede pronosticar el estado actual de la industria de biosensores a nivel mundial.
5
Tabla 1: Utilización de Biosensores en diversos campos del saber a nivel
mundial para el año 1997.
Campo Porción del mercado (%)
Tasa de crecimiento anual
(%)
Diagnostico clínico 53 25
Control de procesos
industriales 11 50
Instrumental medico 11 30
Veterinaria / Agrícola 8 60
Defensa 6 45
Medio Ambiente 5 35
Investigación 3 50
Robótica 2 30
Otros 1 30
Fuente: WALKER, J.M; GINGOLD, E:B: (1997)
3.2 BIOSENSORES EN LA AGROINDUSTRIA
SANZ y colaboradores (2004) reportaron un trabajo el cual estaba dirigido a la investigación y desarrollo de sistemas multisensores realizados con diferente tecnología capaces de detectar compuestos orgánicos volátiles procedentes de alimentos, para de esta forma poder realizar un control de calidad de los mismos, bien en las diferentes etapas de elaboración, o bien en el producto final. El objetivo principal de este trabajo consistía en desarrollar y validar diferentes sistemas multisensores para el reconocimiento y clasificación de aromas procedentes de muestras de jamón y vino.
3.3 BIOSENSORES CO2
Cuervo y colaboradores (2005) realizaron una investigación titulada: Aisalmiento y caracterización de bacterias carboxidótrofas potencialmente utiles en el desarrollo de un sensor enzimático para monóxido de carbono; se reporto el aislamiento de cuatro cepas nativas provenientes de una planta de tratamiento de aguas residuales de la población de ginebra, provincia del Valle del Cauca, Colombia, las cuales fueron cultivadas en un medio mineral saturado con monóxido de carbono a 30°c bajo agitación constante, su

6
comportamiento se comparo con una cepa de Oligotropha carboxidovorans, adquirida a través de la Type American Culture Collection (ATCC) y estudiada anteriormente en la construcción de un sensor enzimático para CO.
3.4 EXTRACTO ENZIMATICO PURIFICADO.
Cuervo y colaboradores (2005), reportaron un trabajo realizado en la Universidad de San Buenaventura, en la cual se registra la extracción y purificación de la enzima monóxido de carbono deshidrogenasa a partir de la bacteria Oligotropha carboxidovorans, para la obtención de los extractos enzimáticos crudos, los cultivos se sometieron a ruptura por sonicación, solubilizacion con detergente no iónico y centrifugación con el fin de eliminar los restos celulares. El el proceso de purificación, los extractos enzimáticos se pasaron a través de una columna de exclusión molecular y otra de intercambio iónico.
3.5 BACTERIAS METANOGENICAS
Las bacterias metanogénicas abundan en ambientes donde limitan aceptadores de electrones tales como O2, NO3-, Fe3+, y SO42-. Digestores anaerobios, sedimentos anóxicos, suelos de humedales y tractos gastrointestinales son hábitats típicos para encontrar estos microorganismos. Como se muestra en la Tabla 2, la primera reacción es la que se evidencia para el caso de Methanococcus deltae:
TABLA 2: Reacción de Methanococcus deltae útil en la construcción del
biosensor.
FUENTE: http://www.uprm.edu/biology/profs/massol/manual/p4-metanogenesis.pdf
Además la bacteria M. deltae pertenece a la familia Methanococcaceae. En la Tabla 3. se pueden evidenciar algunas de sus características principales.

7
Tabla 3. Características generales de las principales Familias de bacterias metanogénicas.
FUENTE: http://www.uprm.edu/biology/profs/massol/manual/p4-
metanogenesis.pdf.
3.5.1 METHANOCOCCUS DELTAE
La bacteria Methanococcus deltae, es una bacteria gram negativa sin flagelos, perteneciente al grupo de las bacterias metanogénicas, las cuales crecen en ambientes ricos en CO2 y H2, elementos que utilizan para la producción de metano. Por ello, se desarrollan en aguas de desecho y en los deltas de algunos ríos, donde hay gran sedimentación.
En este proyecto, la enzima de interés es una reductasa de Dióxido de carbono. La reacción clave en el metabolismo de esta bacteria es la reducción del CO2 para la producción de CH4, catalizada por una reductasa, la cual posee dos cluster de Fe y S diferentes y un cofactor de molibdeno por cada unidad dimérica.

8
3.5.2 TAXONOMIA
Dominio: Archaea
Filo: Euryarchaeota
Clase: Methanococci
Orden: Methanococcales
Familia: Methanococcaceae
Género: Methanococcus
3.6 CRECIMIENTO BACTERIANO
Cuando se realizan conteos del número total de células, existen una amplia variedad de métodos para tal efecto, los cuales permiten medir y realizar el seguimiento de un cultivo microbiano. Estos métodos pueden clasificarse en directos e indirectos. Los métodos directos se basan en la medida de la evolución del número de células vivas (técnica de plaqueo) o del número de partículas (técnicas microscópicas y de contadores de partículas). Los métodos indirectos se basan en la medida de algún parámetro del cultivo que nos permite deducir información sobre la evolución del número de microorganismos. Determinar que método emplear en un seguimiento del cultivo en concreto está sujeto a las características del cultivo y del proceso con el cual se esté trabajando.
Uno de los métodos con mayor aceptación para realizar un recuento de microorganismos pueden ser las técnicas de recuento microscópico de células sin fijar usando microscopía de contraste de fase. Para ello se cuenta el número de partículas en un volumen determinado usando una cámara de Petroff-Hauser o de Neubauer (Figura 1). Esta cámara es un portaobjetos modificado que posee una rejilla o cuadricula que nos permite tener un aproximado del número de células presentes en una muestra, a través del conteo en las cuadriculas y un cálculo simple. El procedimiento es rápido y sencillo; pero no permite distinguir células vivas inmóviles de células muertas.

9
Figura 1. Cámara de Neubauer.
Fuente. Castillo Morales G. Instituto Mexicano. 2004
Empleando el microscopio se cuenta el número de células por cada unidad de área de la cuadricula, conociendo el número de células por volumen que determina cada área. La conversión de ese valor a número de células por mililitro de la suspensión original se hizo multiplicándolo por un factor de conversión.
3.7 LISIS
Para lograr un proceso de lisis celular donde el objetivo es obtener una proteína intracelular de algún microorganismo existen tres métodos generales: enzimáticos, químicos o físicos. Teniendo en cuenta que no todas las metodologías pueden ser empleadas en procesos a gran escala.
3.7.1 METODOS ENZIMATICOS DE RUPTURA CELULAR
Comúnmente, se utiliza la lisozima, la cual es una enzima producida comercialmente a partir de la clara de los huevos de gallina; la cual cataliza de forma específica la hidrólisis de enlaces B-1,4-glucosídicos los cuales están presentes en los mucopéptidos de las paredes celulares de las bacterias. Las bacterias gran positivas, que poseen mucopéptidos de pared que les confieren rigidez, son los que presentan mayor sensibilidad a la lisozima. Sin embargo, la ruptura final de la envoltura celular, depende a menudo de la presión osmótica del medio de suspensión una vez que se ha digerido la pared. En las bacterias gran negativas la ruptura de la pared celular raramente se consigue con un tratamiento único con lisozima, peor la adición con EDTA, que actúa

10
como agente quelante de iones metálicos, origina la deseada lisis celular. También es muy común el uso de la enzima liticasa la cual se encarga de digerir la pared celular y hace extremadamente frágiles las células; lo cual desencadena en una eficiencia alta al momento de lograr disrupción celular, su uso se ve limitado en ocasiones por su elevado costo, todo depende de los volúmenes a tratas con esta enzima. (Walker et al, 1997).
3.8 ENZIMAS
Las enzimas son proteínas que catalizan reacciones químicas en los seres vivos. En una reacción catalizada por una enzima se produce una unión del sustrato en una región concreta de la enzima denominada centro activo, que comprende un sitio de unión y un sitio catalítico. Una vez formados los productos la enzima se recupera pudiendo comenzar un nuevo ciclo de reacción. En ocasiones puede ser necesaria la presencia de cofactores para que la enzima pueda regenerarse y estar activa de nuevo. La actividad enzimática está controlada normalmente por el pH, la fuerza iónica, la temperatura y la presencia de cofactores. La estabilidad de las enzimas es un factor limitante para el tiempo de vida de un biosensor de tipo enzimático y se utilizan distintas técnicas para aumentarla, como estabilización química y/o inmovilización. (Trevan et al, 1990).
En algunas ocasiones se utilizan cascadas multienzimáticas, en las que la enzima que actúa como elemento de reconocimiento no actúa directamente sobre el analito, sino sobre algún producto derivado del mismo. Este sistema es muy utilizado en el caso de algunos azúcares en los que se utilizan enzimas que actúan sobre los productos de la hidrólisis de los mismos.
Entre las enzimas disponibles comercialmente las más utilizadas suelen ser las óxidoreductasas. Son enzimas muy estables que catalizan fenómenos de oxidación o reducción utilizando oxígeno o cofactores. Las enzimas pueden acoplarse a transductores de los tipos potenciométrico, amperométrico, optoeléctrico, calorimétrico o piezoeléctrico, y básicamente todas funcionan por inmovilización de la enzima en el propio transductor. Existen enzimas que no se pueden utilizar aisladas debido a que no son suficientemente estables o a que su purificación es difícil o demasiado cara. En estas circunstancias pueden utilizarse orgánulos celulares, células completas o tejidos que contienen las enzimas en una forma natural en un medio más estable.

11
TABLA 4. Ventajas y desventajas de las enzimas como elementos de reconocimiento en biosensores.
ENZIMAS
Ventajas Desventajas
Elevada de sensibilidad
Sensibilidad frente a
condiciones
Respuesta rápida
ambientales ( pH, temperatura
o
Autoregenerables fuerza iónica)
Permiten monitorización continua
En ocasiones requieren
presencia de
No requieren pasos de lavado Cofactores
Gran variedad de enzimas
disponibles
Pueden ser inhibidos por
sustancias
Permiten detectar tóxicos
desconocidos de la muestra
que inhiben enzimas Tiempo de vida limitado
Permiten amplificar señales
Diseño sencillo
Fácil construcción
Bajo costo
Manejo sencillo
Fuente: Hall, R. H. (2002)
3.8.1 PURIFICACION INICIAL
Una vez provocada la ruptura celular, la primera etapa en la purificación de una enzima intracelular es la separación de los restos celulares. La separación de sólidos de líquidos es una operación clave en el aislamiento de una enzima y normalmente se lleva a cabo por centrifugación o filtración. Existen diversas centrifugas para la purificación enzimática, con rangos de capacidad desde las de menos de 1 ml hasta aquellas que superan varios litros, capaces de aplicar una fuerza centrifuga superior a 100.000 g. Sin embargo, para la eliminación de células bacterianas, de restos celulares o de los precipitados proteicos es suficiente disponer de 200.000 g. Existen en el mercado muchas centrifugas

12
de este tipo adecuadas para preparaciones en una escala intermedia. (Walker et al, 1997).
3.8.2 PURIFICACION DE ENZIMAS Aislar una proteína determinada de un estimado de 10 000 proteínas diferentes presentes en una célula típica es una tarea ardua y complicada (Lodish et al, 2002). Debido a que las enzimas son proteínas que forman parte de la maquinaria metabólica de los seres vivos, para su extracción y utilización en biosensores se hace necesario un procedimiento de purificación que permita obtener la enzima libre de otros compuestos biológicos presentes normalmente en los seres vivos, tales como otras enzimas, restos de pared celular, membranas, etc. Cualquier molécula, sea proteína, hidrato de carbono o ácido nucleico, puede ser separada de otras moléculas sobre la base en las diferencias de algunas características físicas. Si bien la secuencia de aminoácidos de una proteína determina su función, la característica física más útil para la separación de proteínas es el tamaño (Lodish et al, 2002). Normalmente el proceso de purificación comprende varias etapas: eliminación de ácidos nucleicos, eliminación de restos celulares, eliminación de contaminantes no deseados. Concentración, etc. Cuando se realiza la tarea de purificación de una enzima no debe perderse de vista el principio general de que cuanto menos pasos haya, menor será la pérdida en cuanto a cantidad de proteína, sin embargo, a medida que aumenta el número de pasos en la purificación es de esperar que aumente también el nivel de pureza de la enzima obtenida. 3.8.3 CROMATOGRAFIAS Una técnica de uso común para la separación de proteínas es la cromatografia, la cual está basada en el principio de que las moléculas disueltas en una solución interactúan (se unen y disocian) con una superficie sólida. Si se permite que la solución fluya a través de la superficie, las moléculas que interactúan con frecuencia con la superficie permanecerán más tiempo ligadas a ella, por lo que se desplazarán a menor velocidad que las que interactúen poco con la superficie. La cromatografia líquida se lleva a cabo en una columna en las que hay perlas esféricas en un empaquetamiento denso. La naturaleza de estas perlas determina si la separación de las proteínas depende de diferencias de masa, carga o afinidad de unión (Lodish et al, 2002). Todos los tipos de cromatografía normalmente usados en el laboratorio pueden emplearse también en la industria. Las cromatografías más usadas son filtración en geles (exclusión molecular), intercambio iónico y la cromatografía de afinidad. Las técnicas cromatografías más antiguas, tales como las basadas en Sephadex .y DEAE- Sephadex, vienen siendo reemplazados gradualmente por los medios de la nueva generación, como las basadas en celulosa y

13
agarosa entrecruzadas, copolímeros de poliacrilamida y agarosa, etc. Todos estos materiales han sido diseñados para lograr partículas más pequeñas y rígidas, que proporcionan mayor velocidad de flujo y además mayor poder de resolución (Copeland R, 1993). En la cromatografía por filtración en geles, es posible separar proteínas que difieren en su masa. En esta técnica la columna está compuestas por perlas porosas de poliacrilamida, dextran (como el Sephadex), o agarosa. Las proteínas fluyen alrededor de las perlas esféricas de la cromatografía por filtración en gel. Sin embargo, la superficie de las perlas está perforada por grandes orificios, por lo que las proteínas pasan cierto tiempo dentro de esos orificios. Dado que las proteínas más pequeñas pueden penetrar en los orificios con mayor facilidad que las más grandes, se desplazan por la columna de filtración en gel con mayor lentitud que las más voluminosas. El volumen total de líquido requerido para eluir la proteína de la columna depende de su masa, cuanto menor sea la masa mayor será el volumen de elución. En consecuencia, una mezcla de proteínas se separa en función del tamaño, eluyéndose primero las proteínas más grandes y a continuación las más pequeñas. Las cuales son retardadas por su accesibilidad a un volumen mayor de disolvente (Lodish et al. 2002).
Otra cromatografía ampliamente utilizada es la cromatografía de intercambio iónico. Cuando una disolución que contiene iones negativos, se pone en contacto con una resma aniónica (Ecteolla Cellulose), se produce un intercambio de aniones entre la disolución y la columna, según su constante de intercambio (como cualquier reacción ácido-base), fijándose fuertemente los aniones a la columna. El grado de retención o retardo de una proteína por una resma de intercambio jónico depende de la magnitud de la carga de la proteína al pH o a la concentración del eluyente utilizado (Lodish et al.2002). Cabe anotar que algunas técnicas cromatográficas, aunque son muy antiguas, tienen todavía validez, teniendo en cuenta que son métodos sencillos y eficientes en donde la inversión es menor y se logran conseguir los resultados esperados.
3.9 ACTIVIDAD ENZIMATICA
Las enzimas, debido a que se encuentran en pequeñas cantidades en líquidos corporales, no pueden ser cuantificadas, por lo que con frecuencia se mide su actividad. La actividad enzimática se mantiene a concentraciones relativamente constantes por un equilibrio entre la síntesis de las enzimas y la degradación de estas. Una pequeña reacción de algunas de las enzimas pasa sin interrupción a través de las membranas celulares y puede comprobarse su actividad.

14
3.9.1 FACTORES QUE AFECTAN LA ACTIVIDAD ENZIMATICA.
Debido a que la velocidad de las reacciones catalizadas por enzimas depende de la temperatura (T), de la concentración de sustrato (S) y de la enzima, del pH y de otros factores, como los inhibidores, y la condición de las muestras, es indispensable seguir exactamente todos los detalles del procedimiento para cuantificar la actividad enzimática.
Casi todas las enzimas con significados clínico tienen actividad optima cerca del pH neutro (6 a 8), aunque algunas la inhiben a un pH inferior o superior; por ejemplo, la fosfatasa acida o fosfatasa alcalina.
3.9.2 CALCULO DE LA ACTIVIDAD ENZIMATICA EN ESPECIMENES BIOLOGICOS Y CUANTIFICACION DE LA CONCENTRACION DE ENZIMAS CIRCULANTES.
En 1961, la Enzyme Comission (EC) propuso la unidad internacional de actividad enzimática (u), que se define como la cantidad de enzima que transforma 1 umol (1 umol = 10-6 mol) de sustrato por minuto en condiciones estándares de pH, temperatura y concentración de sustrato; en los casos en que una molécula de productos formados. En el laboratorio clínico, la unidad internacional de actividad enzimática se expresa como U/L (unidad/litro), y también, como milunidades /mililitro (mU/ml). El Sistema Internacional (SI) de Unidades ha propuesto una nueva unidad de actividad enzimática, denominada catal, que la Organización Mundial de la Salud (OMS) ha adoptado. Un catal es la cantidad catalítica de enzima que cataliza una reacción con velocidad de un mol por segundo (mol/s) y se expresa como catal/litro (cat/L).
3.10 ELECTRODOS MODIFICADOS QUIMICAMENTE CON ENZIMAS.
Hoy por hoy, el desarrollo de biosensores (sensores basados en principios del comercio se encuentran en la actualidad sensores enzimáticos para la determinación de glucosa, sacarosa, lactato, etanol, metanol y demanda bioquímica reconocimiento molecular inherente a muchas reacciones bioquímicas en las que participan enzimas y anticuerpos) ha tomado un auge cada vez mayor.
El sensor electroquímico enzimático es un electrodo metálico con una superficie la cual contiene una enzima de oxido-reducción inmovilizada. Estas enzimas pueden ser aisladas a partir de microorganismos, tales como bacterias, protozoarios, hongos, entre otros o a partir de tejidos animales o vegetales. Además, el avance en las tecnologías actuales y las técnicas de separación permiten el aislamiento y purificación de estas enzimas por métodos físico-químicos que proveen una relativa pureza. En este tipo de sensor el electrodo actúa como una fuente o sumidero de electrones para la reacción enzima-sustrato específico, naturalmente todo esto sui realmente ocurre una comunicación sea directa o indirecta entre el electrodo y los centros activos de la enzima.

15
Dependiendo de la concentración del sustrato, así será la cantidad de corriente eléctrica que pasa a través del electrodo modificado. Generalmente, para aumentar la eficiencia entre el electrodo y el componente biológico (enzima) se hace necesario el uso de un mediador que permita el transporte de electrones desde y hacia los centros de oxido-reducción de la enzima, los cuales la mayoría de las veces se encuentran envueltos entre gruesas capas de proteínas.
Hoy en día los componentes biológicos más usados en los biosensores son las enzimas, esto debido a la alta especificidad y diversidad de las reacciones mediadas por ellas median. Los sensores basados en enzimas “puras” son en principio, muy selectivos y con tiempos de respuesta relativamente rápidos. Sin embargo, la extracción y sobre todo la purificación de la enzima puede ser un proceso largo y costoso, por lo cual se hace de imperiosa necesidad investigar si es posible el desarrollo de biosensores de alta especificidad y bajo costo, mediante el uso de extractos celulares que contienen el acerbo enzimático. Además, el aislamiento de la enzima de su entorno y condición natural puede conllevar a su desnaturalización y por ende a la pérdida de actividad enzimática
3.11 APLICACIÓN DE BIOSENSORES
3.11.1 SEGURIDAD ALIMENTARIA
El concepto de seguridad alimentaria implica garantizar la producción y comercialización de alimentos que no supongan un riesgo potencial para la salud del consumidor. En este campo los biosensores se utilizan para detectar:
Compuestos xenobióticos, es decir, sustancias externas al producto alimenticio que no han sintetizado los seres vivos (aditivos, fármacos, plaguicidas, etc).
Ciertos componentes del alimento (alérgenos y antinutrientes).
Toxinas de diversos orígenes (toxinas bacterianas, micotoxinas y toxinas marinas).
Microorganismos patógenos que afectan al hombre, al ganado y a los cultivos.
3.11.2 ADITIVOS
Es de gran importancia conocer en un alimento la cantidad y tipo de aditivos incorporados ya que estos se encuentran estrechamente regulados por la legislación comunitaria. Su detección y cuantificación son importantes para evitar posibles usos fraudulentos o malas prácticas de fabricación. Además, estos compuestos pueden provocar problemas de intolerancias, alergias u otras reacciones adversas a determinados grupos de la población. De momento, son pocos los ejemplos de biosensores aplicados a este campo. Se han identificado dispositivos para el análisis de edulcorantes artificiales, aspartame y sorbitol, y de conservantes, ácido benzoico y sulfitos.

16
3.11.3 FARMACOS
El uso de fármacos para el tratamiento de animales, tanto con fines terapéuticos como con fines de promoción del crecimiento está muy extendido en ganadería. Como consecuencia de este uso, pueden aparecer residuos en los alimentos de origen animal, de ahí el interés en su detección, tanto en los animales vivos, como en las canales o en los productos derivados de los mismos (leche, huevos, miel, etc.). Los métodos tradicionales de cribado de muestras para detectar estos fármacos en ocasiones no tienen suficiente sensibilidad, y los métodos más actuales como cromatografía y espectrometría de masas requieren personal cualificado.
3.11.4 RESIDUOS DE PLAGUICIDAS Y FERTILIZANTES
La presencia de residuos de plaguicidas y fertilizantes en productos destinados al consumo humano es indeseable porque muchos de ellos cuentan con una toxicidad elevada. Algunos plaguicidas tienen la capacidad de acumularse en el tejido graso animal mientras que los nitratos, nitritos y fosfatos procedentes del empleo abusivo de fertilizantes contaminan el medio.
3.11.5 COMPONENTES DEL ALIMENTO
Los factores antinutricionales y los alérgenos son compuestos presentes de forma natural en el alimento que pueden ocasionar trastornos en el organismo. Los primeros dificultan o impiden la absorción y metabolización de distintos nutrientes produciendo un déficit de los mismos. En cambio, los alérgenos desencadenan una respuesta inmune en personas hipersensibles.
3.11.6 BIOTOXINAS
En los alimentos pueden aparecer toxinas que pueden relacionarse con cuadros de intoxicación alimentaria. Estas toxinas pueden proceder del crecimiento de bacterias, de hongos (micotoxinas) o por contaminación de productos de origen marino (ficotoxinas). Al igual que ocurre con los patógenos de alimentos es necesario un conocimiento rápido de su presencia en los alimentos con el fin de conseguir una protección del consumidor adecuada.
La mayor parte de estas toxinas son compuestas de naturaleza proteica y su análisis suele ser complejo, ya que su detección y caracterización suele implicar procesos de extracción y purificación que requieren tiempos relativamente largos e incluso ensayos en animales, como el ensayo de ratón. Los biosensores podrían ser una alternativa ya que no es necesaria la purificación de los compuestos.
3.11.7 MICROORGANISMOS PATOGENOS
El aumento en los últimos años de toxiinfecciones producidas por bacterias patógenas ha incrementado la necesidad de métodos de detección más rápidos, sensibles y específicos. En la actualidad se utilizan métodos basados en anticuerpos y en ADN, que han permitido acortar el tiempo de análisis pero no una detección en tiempo real.

17
Frente a estos métodos de análisis, los biosensores tienen el potencial de que permiten la detección en tiempo real, pero presentan como inconveniente que, en caso de un número bajo de microorganismos, es necesario hacer un pre- enriquecimiento o una concentración de la muestra, con el consiguiente aumento del tiempo de análisis.
3.11.8 CALIDAD ALIMENTARIA
La calidad alimentaria se puede entender como aquellos factores que diferencian los productos de acuerdo con sus características organolépticas, de composición o con sus propiedades funcionales.
Los biosensores intervienen en el análisis de la composición de los alimentos permiten caracterizarlos y comprobar si contienen las cantidades que se requieren de los distintos componentes, tanto para aquellos que se encuentran de manera natural en los alimentos como para otros compuestos que se añaden para su enriquecimiento, como es el caso de algunas vitaminas o minerales.
3.11.9 CONTROL DE PROCESOS
Los sistemas de monitorización continua de un proceso industrial permiten detectar en tiempo real los posibles errores de la cadena de producción para subsanarlos de manera inmediata. Hasta el momento se han utilizado en el control de parámetros como el pH, la temperatura, la presión, etc. Gracias a las tecnologías de biosensores ahora también pueden determinarse y cuantificarse on line diversos compuestos de gran importancia.
3.11.10 SENSOR PARA BIOXIDO DE CARBONO.
El CO2 es un gas contaminante atmosférico proveniente de la combustión de materiales orgánicos y es el resultado del proceso de respiración de tejidos vivos. Este CO2 inhibe la capacidad de la sangre para transportar oxigeno, puesto que en los pulmones, el gas pasa rápidamente al torrente sanguíneo uniéndose a la célula encargada de transportar el oxígeno y desplazando a las moléculas de oxigeno. Cabe anotar que la afinidad de la Hemoglobina por el dióxido o el monóxido de carbono son 200 veces mayor que la del oxígeno. Además, es importante determinar la concentración de este gas en ambientes donde hay tejidos vivos, como bodegas donde se almacenan frutas, invernaderos de plantas, entre otras, puesto que este gas puede alterar la fotosíntesis en las plantas y la duración en buen estado del tejido.
En las ciudades, los automóviles y las industrias, son uno de los mayores contribuyentes a la contaminación de la atmósfera por CO y CO2. Estos gases tienden a concentrarse en lugares poco ventilados, interiores de edificios, estacionamiento de vehículos, estufas, hornos, humos de cigarrillos, bodegas de almacenamiento, entre otros. Así la detección y el monitoreo de estos dos gases se convierte en un objetivo de la política gubernamental.
En este orden de ideas se considera que el bióxido de carbono es uno de los gases más importantes que se encuentran en el medio ambiente, puede ser

18
encontrado como reactivo o como producto en un gran número de reacciones químicas, el esfuerzo que se ha venido dando para el estudio de este gas a sido considerable, dedicado al desarrollo de técnicas analíticas para la determinación cuantitativa y cualitativa de este gas.
Los sensores de CO2 sirven en la industria en monitoreo de la calidad del aire ambiente, emisiones por vehículos automotores, bodegas de almacenamiento de tejidos (frutas, verduras, etc), viviendas y edificios de trabajo.
Las técnicas tradicionales están basadas en la estimación indirecta de concentraciones de CO2, como lo son muestras del pH y la alcalinidad. Uno de los dispositivos más comunes empleados para la detección del CO2, es el detector infrarrojo (IR), usado casi exclusivamente para detectar CO2 gaseoso, no siendo una técnica muy confiable por la facilidad de interferencia que se puede producir por alguna clase de absorbente presente en la muestra, como es el caso del monóxido de carbono.
Muchos sensores químicos ópticos han sido desarrollados para la detección de CO2. Este tipo de sensores se caracterizan por poseer una alta estabilidad química y mecánica, además de ser económicos y fáciles de fabricar. Siendo un inconveniente de este tipo se sensores en algunos casos la mala sensibilidad y precisión, y el Hecho de que su respuesta es lenta especialmente en medio acuoso.
Teniendo en cuenta estas consideraciones los sensores electroquímicos son hasta ahora los más adecuados para mediciones de CO2, en la actualidad para mediciones continuas de bióxido de carbono se usa el electrodo tipo Severinghaus, para esta prueba un electrodo de vidrio detecta el cambio en el pH en una solución buffer inducida por la difusión del CO2 a través de una membrana hidrofóbica, aunque este tipo de sensor es uno de los más conocidos y empleado es importante avanzar en estudios de nuevas técnicas y más aun técnicas que involucren extractos enzimáticos purificados debido a su alta especificad por la actividad catalítica que presentan.
La investigación en sensores electroquímicos para CO2 y otros gases de interés ambiental es activa. El método de detección de CO2 propuesto en este trabajo está basado en la reacción de oxidorreducción que lleva a cabo la bacteria Methanococcus deltae, bacteria metanogénica de las aguas residuales, por medio de la cual el CO2 en presencia de H2 se reduce a formaldehído y posteriormente a metano (CH4).
3.12 INMOVILIZACION DE ENZIMAS
3.12.1 MÉTODOS DE INMOVILIZACIÓN DE ENZIMAS POR RETENCIÓN FÍSICA 3.12.2 ATRAPAMIENTO Consiste en la retención física de la enzima en las cavidades interiores de una matriz sólida porosa constituida generalmente por prepolímeros fotoentrucruzables o polímeros del tipo poliacrilamida, colágeno, alginato, carraginato o resinas de poliuretano. El proceso de inmovilización se lleva a

19
cabo mediante la suspensión de la enzima en una solución del monómero. Seguidamente se inicia la polimerización por un cambio de temperatura o mediante la adición de un reactivo químico. El atrapamiento puede ser en geles o en fibras, que suelen ser más resistentes que los geles. En el primer caso, la enzima queda atrapada en el interior de un gel, mientras que en el segundo caso la enzima se encuentra ocluida dentro de las micro cavidades de una fibra sintética. El atrapamiento, de gran sencillez desde el punto de vista experimental, requiere poca cantidad de enzima para obtener derivados activos. Como ventaja adicional, la enzima no sufre ninguna alteración en su estructura. De todas formas, el atrapamiento requiere un control riguroso de las condiciones de polimerización, así como la comprobación de que la naturaleza química del proceso no altera los grupos reactivos de la proteína.
Inclusión en membranas: Dividida en dos tipos: 1) Microencapsulación: En esta técnica, las enzimas están rodeadas de membranas semipermeables que permiten el paso de moléculas de sustrato y producto, pero no de enzima. Estas membranas semipermeables pueden ser permanentes (originadas por polimerización interfacial) o no permanentes (generadas por surfactantes, también llamadas “micelas reversas”). Las cápsulas obtenidas son de forma esférica, con tamaños comprendidos entre 1 y 100 nm de diámetro. Mediante este método se pueden encapsular simultáneamente una gran variedad de enzimas, células o biomoléculas, permitiendo que se lleven a cabo determinadas reacciones que suceden en múltiples pasos. 2) Reactores de membrana: El desarrollo de reactores o sistemas que contengan enzimas atrapadas ha despertado gran interés en la industria. Estos reactores emplean membranas permeables al producto final, permeables o no al sustrato inicial y obviamente impermeables a la enzima. Mediante una bomba se establece un flujo líquido de sustrato que atraviesa el reactor.

20
En general, en esta metodología, se procede inicialmente a la adsorción de la enzima sobre la membrana que formará el reactor. Esta adsorción se puede realizar de dos formas:
1. mediante el paso de una solución tamponada de enzima a través de la membrana.
2. por contacto continuo de una solución de enzima con la membrana. 3.12.3 MÉTODOS DE INMOVILIZACIÓN DE ENZIMAS POR UNIÓN QUÍMICA 3.12.4 UNION A SOPORTES Son los métodos de inmovilización más utilizados y de los que se dispone de una mayor información. La elección del soporte y del tipo de enlace resultan determinantes en el comportamiento posterior del biocatalizador. Se debe procurar que la inmovilización incremente la afinidad por el sustrato, disminuya la inhibición, amplié el intervalo de pH óptimo y reduzca las posibles contaminaciones microbianas. Además el soporte debe tener resistencia mecánica adecuada a las condiciones de operación del reactor y ser fácilmente separable del medio líquido para que pueda ser reutilizado. Se han utilizado una gran variedad de materiales como soportes para la inmovilización de numerosas enzimas. Estos materiales difieren en tamaño, densidad, porosidad y forma, aunque generalmente nos los encontramos en forma de cilindro, hojas, fibras y más corrientemente en forma de esferas. Los soportes pueden clasificarse en dos grandes grupos: 1. Soportes inorgánicos: Dentro de este grupo tenemos una gran variedad de soportes, que pueden ser naturales (arcillas como la bentonita, piedra pómez, sílice, etc.) o materiales manufacturados (óxidos de metales y vidrio de tamaño de poro controlado, vidrio no poroso, alúmina, cerámicas, gel de sílice, etc.) 2. Soportes orgánicos: Se pueden clasificar en:
Polímeros naturales: a su vez divididos en: Polisacáridos (celulosa, almidón, dextranos, agar-agar, agarosa, alginatos, quitina, chitosan, etc). Proteínas fibrosas (colágeno, queratina, etc.)
Polímeros sintéticos: divididos en: Poliolefinas (como el poliestireno) Polímeros acrílicos (poliacrilatos, poliacrilamidas, polimetacrilatos, etc.)
Las enzimas se pueden unir a estos soportes mediante adsorción o por unión covalente.

21
3.12.5 ADSORCION En la adsorción, la enzima se une a un soporte sin funcionalizar mediante interacciones iónicas, fuerzas de Van der Waals y por puentes de hidrógeno. Los principales factores que influyen en la adsorción, son:
1. el pH del medio: controla el número y la naturaleza de las cargas que presenta la superficie de la proteína y del sólido.
2. la fuerza iónica: al aumentar la fuerza iónica se produce la desorción de la enzima, ya que los iones inorgánicos se unen con más fuerza al soporte que la proteína.
3. el diámetro de poro: debe ser aproximadamente dos veces el tamaño del eje mayor de la enzima.
4. la presencia de iones que actúen como cofactores de la enzima, ya que pueden incrementar la carga enzimática del derivado.
Como principales ventajas de este método destacan:
su preparación sencilla.
su bajo coste.
no hay cambios de especificidad enzimática.
los derivados son estables en medios de trabajo con bajo contenido en agua.
Los inconvenientes de la adsorción son principalmente:
la optimización de las variables que controlan la adsorción.
los derivados obtenidos son poco estables desde el punto de vista mecánico.
la unión al soporte es débil. Una variante dentro de la técnica de la adsorción consiste en emplear resinas de intercambio iónico, las cuales contienen grupos funcionales y contraiones

22
móviles. Estos contraiones se pueden intercambiar reversiblemente por otros iones de la misma carga, sin que se produzcan cambios en la matriz insoluble. 3.12.6 UNION COVALENTE La unión covalente de una enzima a un soporte es quizá el método de inmovilización más interesante desde el punto de vista industrial. La metodología de la unión covalente se basa en la activación de grupos químicos del soporte para que reaccionen con nucleófilos de las proteínas. De entre los 20 aminoácidos diferentes que se encuentran en la estructura de las enzimas, los más empleados para la formación de enlaces con el soporte son principalmente la lisina, la cisteína, la tirosina y la histidina, y en menor medida la metionina, el triptófano, la arginina y el ácido aspártico y glutámico. El resto de aminoácidos, debido a su carácter hidrófobo, no se encuentran expuestos hacia el exterior de la superficie proteica, y no pueden intervenir en la unión covalente. Este método presenta las siguientes ventajas: La manipulación de los derivados inmovilizados es sencilla. La carga de enzima permanece constante después de la inmovilización. Los derivados pueden utilizarse en reactores en continuo,
empaquetados, de lecho fluidizado o tanque agitado. Una mayor resistencia a la desactivación por el efecto de la temperatura,
de los disolventes orgánicos o del pH, al tener estabilizada su estructura terciaria.
En cambio la inmovilización por enlace covalente también presenta una serie de inconvenientes: Es necesario conocer la densidad de grupos activos por unidad de
superficie, ya que condiciona el número de uniones enzima-soporte y su geometría, que puede ser distorsionante y conducir a derivados inactivos.
El proceso de inmovilización puede alterar la estructura del centro activo. Para evitar esta posible alteración, se realiza la inmovilización en presencia de un inhibidor que bloquee el centro activo.
La inmovilización covalente no es aconsejable en aquellas enzimas muy sensibles a cambios de pH, fuerza iónica, etc.
3.12.7 RETICULADO También denominado entrecruzamiento o cross-linking, es una técnica que ha sido ampliamente utilizada en la estabilización de muchas enzimas. El método del reticulado consiste en uso de reactivos bifuncionales que originan uniones intermoleculares entre las moléculas de enzima. Como reactivos bifuncionales se pueden emplear dialdehídos, diiminoésteres, diisocianatos, sales de bisdiazonio e, incluso, diaminas si están activadas con carbodiimida. El resultado del reticulado son enzimas con enlaces intermoleculares irreversibles capaces de resistir condiciones extremas de pH y temperatura.

23
El co-reticulado, permite eliminar las pérdidas de actividad enzimática debidas a efectos difusionales, mediante el entrecruzamiento de las enzimas con una proteína sin actividad enzimática y rica en residuos de lisina (por ejemplo, la albúmina bovina). Un procedimiento mixto de inmovilización muy común consiste en inmovilizar la enzima por adsorción sobre una resina de intercambio iónico o un soporte polimérico (con lo que se consigue una elevada carga enzimática) y posteriormente añadir el reactivo bifuncional. Actualmente el método más novedoso de cross-linking consiste en la cristalización de enzimas y su posterior reticulado con glutaraldehído (Cross-Linked Enzyme Crystals o CLECs). El aumento de estabilidad se basa en la obtención de un entramado cristalino, donde las moléculas de enzima están rodeadas exclusivamente por otras moléculas de proteína. De esta manera la propia enzima actúa como soporte, y su estructura terciaria está estabilizada por las uniones covalentes intermoleculares. La estructura cristalina posee canales microscópicos que permiten el paso de sustratos hasta el centro activo de la enzima donde se cataliza la reacción. Estos cristales pueden soportar temperaturas elevadas y pH extremos, así como la acción de las proteasas. Esta tecnología se ha aplicado a enzimas muy diferentes, las cuales se han utilizado en la obtención de compuestos enatioméricamente puros y en la síntesis de péptidos. 3.12.8 EFECTOS DE LA INMOVILIZACIÓN A menudo, la inmovilización altera significativamente el comportamiento de las enzimas. En primer lugar se producen cambios en su estabilidad. En segundo lugar, la enzima inmovilizada es un sistema heterogéneo en el cual todos los componentes que intervienen en el proceso catalítico (pH, sustratos, productos, inhibidores, cofactores, activadores, etc.) se encuentran en interface: en el medio de reacción y en la fase constituida por el soporte con la enzima. Como consecuencia, la actividad enzimática se ve afectada por efectos de tipo difusional, estérico y del microentorno. 3.12.9 EFECTOS EN LA ESTABILIDAD Generalmente se observa un incremento en la estabilidad de las enzimas después de su inmovilización, que se debe principalmente a las siguientes razones: Una estabilización conformacional de la enzima debido a la existencia de
uniones multipuntuales enzima-soporte. La estructura terciaria de la enzima adquiere una mayor rigidez y se hace más resistente a la desactivación térmica o química. Este tipo de estabilización se obtiene únicamente en aquellos métodos en los que intervienen enlaces de tipo covalente, como el reticulado o la unión a soportes activados (Figura4).
La inmovilización covalente multipuntual se puede combinar con la adición de reactivos bifuncionales que den mayor rigidez a la estructura de la enzima.

24
Una protección frente a las proteasas en el medio. Se ha visto que la
unión de proteasas a un soporte elimina su capacidad proteolítica, y evita su autolisis.
Se evita la agregación intermolecular al mantener las moléculas de
enzima retenidas en una determinada región del espacio.
Existe una alteración del microentorno del enzima debida a la interacción de la enzima con el soporte. Por ejemplo, si una enzima sensible al oxígeno (como las nitrogenasas, hidrogenasas, etc.) se sitúa en la superficie de un soporte cargado, la fuerza iónica efectiva en el microentorno de la enzima será muy alta y, como consecuencia, la concentración de oxígeno disuelto será mucho menor en esa zona que en el medio de reacción .
En otros casos el soporte tiene un efecto tamponador de tal manera que mantiene el pH óptimo de la enzima en su microentorno, aunque en la disolución se produzcan cambios importantes de pH. Por otra parte, en aquellas reacciones catalizadas por enzimas inmovilizadas en presencia de disolventes orgánicos, la “acuofilia” del soporte o su capacidad para retener agua, regula la actividad de la enzima. Cuanto mayor es la acuofilia del soporte, más agua adsorbe y la enzima poseerá la cantidad necesaria de agua en su microentorno para mantener su conformación activa.

25
3.12.10 Efectos en la actividad enzimática Tras una inmovilización, la actividad de la enzima puede disminuir e incluso perderse por diversas razones. Si pierde totalmente la actividad enzimática puede ser debido a que:
1. La unión al soporte se produce de tal forma que el paso del sustrato al centro activo está impedido.
2. Los grupos reactivos del soporte reaccionan con algún aminoácido que forme parte del centro activo o que sea esencial para la actividad catalítica de la enzima.
3. La inmovilización puede originar un cambio conformacional que da lugar a una forma inactiva.
4. Las condiciones experimentales del proceso causan la desnaturalización o desactivación de la enzima.
Si la pérdida de actividad no es total después de la inmovilización, los cambios (disminución o aumento de la actividad enzimática) se deberán principalmente a efectos difusionales, electrostáticos, estéricos y/o del microentorno. 1) Efectos difusionales: Como consecuencia de la inmovilización, la difusión de los sustratos hacia el centro activo de la enzima puede estar impedida por resistencias de tipo externo e interno.
A) resistencias difusionales externas: si el soporte es insoluble en el medio de reacción, el sustrato deberá atravesar la película líquida estacionaria (capa de Nernst o de disfusión) que rodea el soporte. En las proximidades de un soporte no cargado, la concentración de sustrato es menor que en el resto de la disolución, puesto que existe un gradiente de concentración a través de la zona de difusión. Por tanto, los valores de km para las enzimas inmovilizadas son siempre aparentes. B) resistencias difusionales internas: debida a que los sustratos tienen que atravesar el interior del gel, microcápsula, fibra o poro del soporte donde se encuentra la enzima inmovilizada. Existen diversas maneras de minimizar estos efectos difusionales como por ejemplo: disminuir el tamaño del biocatalizador, aumentar la concentración de sustrato, incrementar la agitación o el flujo en el reactor, etc. Con estas medidas se consigue reducir el grosor de la capa de Nernst, y como consecuencia, el valor de km’ disminuye. 2) Efectos electrostáticos entre el sustrato y el soporte, de tal manera que, si tienen la misma carga existe una repulsión mutua, mientras que si las cargas son opuestas hay atracción. Cuando el sustrato y el soporte tienen cargas opuestas, el valor de km’ aparente puede verse reducido hasta varias veces por debajo del obtenido en disolución. Hornby y cols. Desarrollaron una expresión matemática que permite calcular la actividad de las enzimas

26
inmovilizadas, teniendo en cuenta tanto los factores de difusión como los electrostáticos. La expresión de Michaelis-Menten en este caso es:
Donde x=espesor de la capa de Nernst; D=coeficiente de difusión; T=temperatura (ºK); z=valencia del sustrato; F=constante de Faraday; R=constante universal de los gases y V=gradiente de potencial en el soporte. Se puede disminuir el valor de km’, variando tanto el término de difusión como el término electrostático. En el primer caso, la disminución del valor de km’ se consigue al disminuir el tamaño del soporte (con lo que disminuye el término “x”) o al aumentar el flujo o la agitación. En el término electrostático, si “z” y “V” tienen el mismo signo, es decir si el soporte y el sustrato tienen la misma carga, el término electrostático es menor de 1 y km’ aumenta. Si tienen cargas opuestas, la km’ disminuye. Si no poseen carga, km’ sólo dependerá del término de disfusión.
3) Impedimentos estéricos o de tamaño de sustrato. En un principio, cualquier enzima puede ser inmovilizada sin que haya una pérdida apreciable de su actividad. Este hecho suele ser válido en el caso de que el sustrato sea de bajo peso molecular, pero si se trata de sustratos con pesos moleculares elevados, la actividad de la enzima inmovilizada disminuye drásticamente. Por ejemplo, muchas hidrolasas unidas covalentemente a soportes sólidos, a pesar de que son muy activas frente a sustratos pequeños, muestran una actividad muy baja hacia proteínas, polisacáridos y ácidos nucleicos. Este “efecto estérico” se puede evitar mediante una inmovilización covalente a través de un brazo espaciador enzima-soporte más largo. 4) Efectos en el microentorno: La enzima inmovilizada se encuentra en un entorno diferente al habitual, especialmente cuando el soporte tiene grupos cargados eléctricamente. El efecto observado suele ser un desplazamiento en el valor del pH óptimo de la catálisis enzimática y, muchas veces, un ensanchamiento en el intervalo de pH en el cual la enzima puede actuar. Por ejemplo, una enzima unida a un soporte cargado negativamente (v.g. CM-sephadex) tendrá en su microentorno una concentración mayor de hidrogeniones que en el medio de reacción. Como resultado, la enzima inmovilizada será más activa a un pH más alcalino. La enzima sería más activa a pH más ácidos si estuviera unida a un soporte cargado positivamente (v.g. DEAE-sephadex).
27
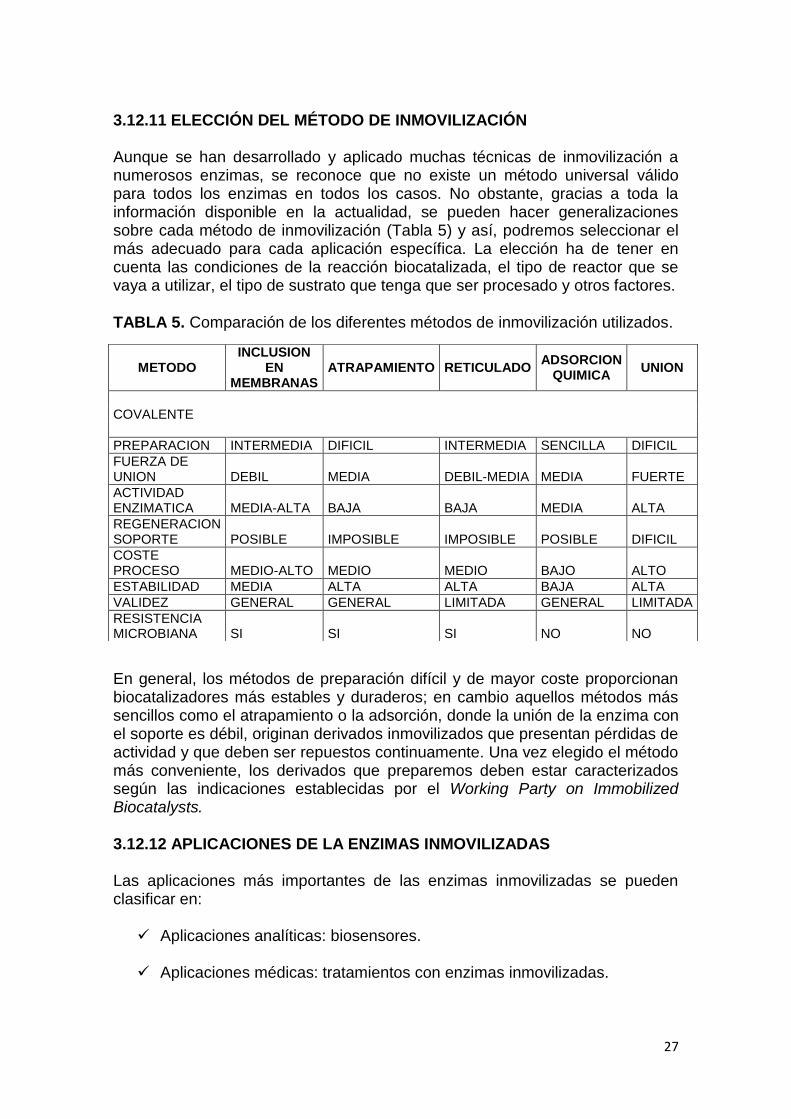
3.12.11 ELECCIÓN DEL MÉTODO DE INMOVILIZACIÓN Aunque se han desarrollado y aplicado muchas técnicas de inmovilización a numerosos enzimas, se reconoce que no existe un método universal válido para todos los enzimas en todos los casos. No obstante, gracias a toda la información disponible en la actualidad, se pueden hacer generalizaciones sobre cada método de inmovilización (Tabla 5) y así, podremos seleccionar el más adecuado para cada aplicación específica. La elección ha de tener en cuenta las condiciones de la reacción biocatalizada, el tipo de reactor que se vaya a utilizar, el tipo de sustrato que tenga que ser procesado y otros factores. TABLA 5. Comparación de los diferentes métodos de inmovilización utilizados.
En general, los métodos de preparación difícil y de mayor coste proporcionan biocatalizadores más estables y duraderos; en cambio aquellos métodos más sencillos como el atrapamiento o la adsorción, donde la unión de la enzima con el soporte es débil, originan derivados inmovilizados que presentan pérdidas de actividad y que deben ser repuestos continuamente. Una vez elegido el método más conveniente, los derivados que preparemos deben estar caracterizados según las indicaciones establecidas por el Working Party on Immobilized Biocatalysts. 3.12.12 APLICACIONES DE LA ENZIMAS INMOVILIZADAS Las aplicaciones más importantes de las enzimas inmovilizadas se pueden clasificar en: Aplicaciones analíticas: biosensores.
Aplicaciones médicas: tratamientos con enzimas inmovilizadas.
METODO INCLUSION
EN MEMBRANAS
ATRAPAMIENTO RETICULADO ADSORCION
QUIMICA UNION
COVALENTE
PREPARACION INTERMEDIA DIFICIL INTERMEDIA SENCILLA DIFICIL
FUERZA DE UNION DEBIL MEDIA DEBIL-MEDIA MEDIA FUERTE
ACTIVIDAD ENZIMATICA MEDIA-ALTA BAJA BAJA MEDIA ALTA
REGENERACION SOPORTE POSIBLE IMPOSIBLE IMPOSIBLE POSIBLE DIFICIL
COSTE PROCESO MEDIO-ALTO MEDIO MEDIO BAJO ALTO
ESTABILIDAD MEDIA ALTA ALTA BAJA ALTA
VALIDEZ GENERAL GENERAL LIMITADA GENERAL LIMITADA
RESISTENCIA MICROBIANA SI SI SI NO NO

28
Aplicaciones industriales: en la industria química, farmacéutica, alimentaria y de tratamiento de residuos.

29
4. METODOLOGÍA
4.1. Lugar de trabajo.
La bacteria Methanococcus deltae y las demás cepas bacterianas aisladas
carboxidótrofas se cultivaron y crecieron en el laboratorio de microbiología de la
Universidad de San Buenaventura-Sede Cali.
Las pruebas de tinción microbiológicas (Tinción de Gram, esporas y cápsula)
para corroborar la identificación del género Methanococcus sp fueron
realizados en el Laboratorio de Microbiología de la Universidad de San
Buenaventura.
Las pruebas Bioquímicas de identificación fueron realizadas en el laboratorio
de Microbiología de la Universidad de San Buenaventura.
Las pruebas para la determinación de la actividad enzimática del extracto
bacteriano obtenido, así como la inmovilización enzimática fueron realizadas en
el laboratorio de Biología Molecular de la Universidad del Valle.
4.2 Material biológico.
La bacteria Methanococcus deltae se compró a la American Type Culture
Collection, (ATCC # 35294), además se tomaron varias muestras del lago
principal de la Universidad de San Buenaventura-Seccional Cali, con el objetivo
de aislar y cultivar cepas nativas de Methanococcus sp.
4.3 Métodos
4.3.1 Cultivo de la bacterias del género Methanococcus sp.
Para los diferentes cultivos realizados de la bacteria Methanococcus deltae, así
como de las bacterias que utilizan bióxido de carbono aisladas del lago
principal de la Universidad de San Buenaventura, Sede Cali, Valle del Cauca,
se utilizaron los medios descritos a continuación::
Medio de Crecimiento: Para el crecimiento autotrófico de la bacteria se utilizó el
medio sugerido por la ATCC (1343) para estuarios marinos. Este medio
contenía los siguientes reactivos:
Solución Salina Completa ……….............................200.0 ml
Wolfe's Mineral Solution ……………...............................4.0 ml
Wolfe's Vitamin Solution………………............................4.0 ml
20% Extracto de Levadura- Solución de Trypticasa…....4.0 ml
0.1% Resazurin...............................................................0.4 ml
25% Solución de Acetato de Sodio.................................2.0 ml

30
0.2% Solución de Fe(NH)…............................................0.4 ml NaCl..................................................................................3.6 g
Cysteine-Sulfide Reducing Agent………………............16.0 ml
Agar (Para medios sólidos)............................................8.0 g
Agua Destilada estéril……........................................180.0 ml
Se ajusta el medio a un pH final de 7.0.se calienta hasta ebullición y se somete
a una atmosfera de 80% N, 20% CO y se agrega el agente reductor.
La solución salina estaba compuesta de la siguiente forma:
KCl......................................0.67 g
MgCl2 6H O...........................5.5 g
MgSO4 7H O.........................6.9 g
NH4CL...................................0.5 g
CaCl2 2H2 O........................0.28 g
K 2HPO4 .............................0.28 g
Agua Destilada.......................1.0 L
La solución Vitamínica de Wolf tenía la siguiente composición:
Biotina………...............................2.0 mg
Äcido Fólico……..........................2.0 mg
Hidroclururo de Piridoxina ........10.0 mg
Thiamina . HCl..............................5.0 mg
Riboflavina……….........................5.0 mg
Acido Nicotinico…….....................5.0 mg
Pantotenato de Calcio………….....5.0 mg
Vitamina B12.................................0.1 mg
Äcido p-Aminobenzoico ...............5.0 mg
Agua Destilada …………..................1.0 L
La Solución Mineral de Wolf estaba compuesta por:
Acido Nitrilo acético.....................1.5 g
MgSO4 7H O...............................3.0 g
MnSO4. H2 O...............................0.5 g

31
NaCl............................................1.0 g
FeSO4 7H2 O..............................0.1 g
CoCl2 6H2 O................................0.1 g
CaCl2..........................................0.1 g
ZnSO4 7H2 O ............................0.1 g
CuSO4 5H2 O............................0.01 g
AlK(SO4)2 . 12H2 O...................0.01 g
H3 BO 3 ....................................0.01 g
Na2 MoO4 . 2H2 O....................0.01 g
Agua Destilada...........................1.0 L
El crecimiento autotrófico fue realizado mediante la inoculación de
Methanococcus deltae o de las bacterias Metanogénicas aisladas en un medio
mineral líquido, en el que la única fuente de carbono disponible para la bacteria
provenía del bióxido de carbono. Methanococcus deltae fue crecida del
liofilizado original adquirido a la ATCC, en el medio de cultivo descrito
anteriormente, del cual se realizaron cultivos sucesivos hasta obtener 200 ml.
Las bacterias degradadoras de Bióxido de carbono se crecieron medio de
cultivo sugerido por la ATCC sólido el cual fue sembrado por el método de
estrías y colocado en un que contenía una papeleta de bióxido de carbón y
por lo tanto el ambiente estaba saturado de este gas. Se dejó crecer durante el
tiempo necesario para que las células formaran colonias. A partir de los cultivos
en medio sólido se procedió a repetir el crecimiento en medio líquido bajo las
mismas condiciones. Una vez la población bacteriana llegó a la fase de
crecimiento exponencial se procedió a la lisis bacteriana por diferentes
métodos.

32
Figura 5. Crecimiento anaeróbico de la bacteria Methanococcus deltae en
cajas de petri con ambiente rico en Bióxido de carbono.
Fuente: Duran, C.A; HETZ, J; PARDO, J.F, 2009
Una vez se observó el crecimiento bacteriano debido a la turbidez del medio,
este fue rectificado por recuento celular en la cámara de Newbauer,
posteriormente se realizaron resiembras en medio sólido para la
caracterización de las colonias. Cabe anotar que el medio de crecimiento sólido
poseía la misma composición que el medio líquido solo que en el primero se
utilizó como agente solidificante agar (15 g/L). La pureza de los cultivos se
aseguró por réplicas sucesivas y verificación por microscopía después de la
coloración de Gram.
4.3.2 Aislamiento e identificación bacteriana.
A 100 ml del medio descrito anteriormente se le inocularon 10 ml del agua
proveniente del lago principal de la Universidad de San Buenaventura-sede
Cali. Este cultivo se creció en una cámara de anaerobiosis que contenía un
medio saturado de bióxido de carbono. Posteriormente, se procedió a realizar
un recuento microbiológico directo por cámara de Newbauer, Posteriormente,
se procedió a realizar una resiembra en 100 ml de medio mineral líquido a
partir de 10 ml de inoculos tomados de la muestra anterior. A partir de esta
muestra líquida se obtuvo una muestra para sembrarla en un cultivo sólido. A
cada colonia se le realizó coloración de Gram, y caracterización teniendo en
cuenta la forma, tamaño, color, elevación y tipo de borde de la colonia
(Caracterización morfológica).

33
A las colonias bacterianas aisladas puras se les realizaron pruebas para
determinar la presencia de esporas y pruebas bioquímicas. Estas pruebas
fueron realizadas 3 veces.
Para la realización de las pruebas bioquímicas se comenzó con un sembrado
de las bacterias en agar MacConkey, el cual es un medio diferencial que
permite diferencias las bacterias que hidrolizan lactosa de las que no lo hacen.
Esta hidrólisis produce ácidos orgánicos y las colonias que forman adquieren
un color rojizo. Además, se continuaron con las siguientes pruebas
bioquímicas:
a) Catalasa. Se sembraron bacterias por estrías para determinar si poseían la enzima
que descompone el peróxido de hidrogeno.
b) Citrato, Se sembró en agar citrato de Simmons y se determinó si las bacterias
eran capaces de utilizar el citrato como única fuente de carbono.
c) Descarboxilasa En este medio se prueba la descarboxilación de los aminoácidos.
Contiene púrpura de bromocresol el cual toma un color de púrpura
intenso a pH alcalino.
d) Licuefacción de la gelatina. Esta prueba ayuda a la identificación de los géneros Serratia,
Pseudomonas; Clostridium y Flavobacterium, que son capaces de
hidrolizar la gelatina destruyendo el medio.
e) Hidrólisis de Almidón. Las bacterias se sembraron por estrías en un medio que contenía
almidón. Después de crecidas las colonias se agrega lugol para
identificar si el almidón permanece intacto o no. Las bacterias que
hidrolizan el almidón poseen las amilasas necesarias para su
degradación.
f) Hidrólisis de Caseína. Las bacterias fueron sembradas en un medio de cultivo cuyo
componente más abundante era caseína. Esta prueba permite
reconocer si las bacterias son capaces de degradar la caseína o no.
g) Producción de sulfuro de hidrógeno. Se utilizó el medio agar hierro triple azúcar, el cuál fue sembrado en
profundidad y donde se pudo determinar la producción de sulfuro de
hidrógeno debido a la ruptura de tiosulfato. Esta prueba permite
identificar los géneros Proteus y Salmonella, los cuales dan positivos.

34
h) Prueba de Indol. Las bacterias fueron sembradas en medio SIM. Esta prueba permite
identificar si las bacterias inoculadas realizan la conversión del triptófano
de las proteínas.
i) Prueba de Rojo de Metilo. Se realizó la inoculación bacteriana en un caldo MRVP con ácidos
orgánicos. Algunos géneros bacterianos como E.coli y Edwarsiella sp
son capaces de fermentar los ácidos y disminuir el pH por debajo de 4.3.
j) Prueba de Voges- Proskauer. En esta prueba la acetoína es producida a partir del caldo MRVP. En
esta prueba se diferencian los géneros como Klebsiella sp, Enterobacter
sp y E.coli.
k) Prueba de Oxidasa Positiva.
En esta prueba se realizó un sembrado por estrías en agar. Esta prueba
permite detectar las colonias oxidasas positivas adicionando el reactivo
de oxidasa sobre las colonias e identificando la coloración que toma.
l) Prueba del Nitrato.
Se inoculan las bacterias escogidas en un caldo con nitrato, después de
la incubación se determina la producción de nitrito a partir de nitrato,
mediante el ácido naftilamina sulfanílico. Esta reacción es típica de
enterobacterias.
m) Agar cetrimide.
Este agar se inocula mediante el método de estrías y se observa la
coloración tomada por las colonias después de la incubación. Esta
prueba nos determinó si algunas de las cepas aisladas poseen el
pigmento piocianina, típica del género Pseudomonas.
4.3.3 Preparación de los extractos enzimáticos obtenidos por tres
métodos de lisis celular diferente.
4.3.3.1 Lisis celular por Sonicación.
La obtención de la lisis celular por medio de la sonicación es un procedimiento
muy utilizado en la biología. Para esto se cultivarán las células en 200 ml de
medio líquido y se esperará hasta que la población bacteriana llegue a la fase
máxima de crecimiento (Fase exponencial). Esta fase exponencial fue
alcanzada a las 2 horas aproximadamente.

35
Posteriormente se completó el rompimiento celular mediante el sometimiento a
sonicación en un Equipo Branson Sonifier (VWR Company), con las siguientes
indicaciones:
Output Control: 7
Duty Cycle: 60.
Time: 10 Minutos.
La muestra fue colocada en hielo mientras se sometía a sonicación, con el
objetivo de evitar su calentamiento.
Figura 6. Rompimiento celular mediante sonicación. Equipo Branson.-Sonifier.
Universidad del Valle. Departamento de Biología. Laboratorio de Biología
Molecular de Levaduras.
Fuente: Duran, C.A; HETZ, J; PARDO, J.F, 2009
El rompimiento celular fue verificado por observación al microscopio de luz. El
extracto crudo resultante fue fraccionado en muestras de 5 ml y almacenado a
-20 C para la realización de los demás procedimientos. Sin embargo, para
determinar la eficiencia del proceso se procedió a realizar un conteo celular por
cámara de Newbauer antes y después del proceso.
4.3.3.2 Lisis celular por Fricción mecánica.
Se realiza una inoculación de 200 ml del medio de cultivo con la bacteria
Methanococcus deltae. Se incuba a 37 C hasta alcanzar la fase de crecimiento
exponencial la cual se alcanza aproximadamente a las 2 horas de inoculado el
medio. En esta fase de crecimiento exponencial se hace un recuento utilizando
la cámara de Newbauer alcanzándose una población mayor a 18 millones de
bacterias/ cc.

36
Se realizaron dos rompimientos por fricción mecánica para lo cual se empleo 2
gr de arena fina estéril y mediante un mortero de porcelana se procedió a
aplicar la fuerza necesaria para llegar a la lisis bacteriana. El proceso de
maceramiento tuvo una duración de 25 minutos aproximadamente. Una vez
completada la maceración manual se procedió a realizar una centrifugación a
6 000 rpm para eliminar los residuos de arena y proceder al conteo celular.
Figura 7. Proceso de ruptura mecánica mediante el uso de arena estéril y
posterior centrifugación a 6000 rpm.
Fuente: Duran, C.A; HETZ, J; PARDO, J.F, 2009
Para determinar la efectividad de este método se realizó un conteo de células
antes y después del proceso.
4.3.3.3 Lisis celular por Agentes Químicos.
La adición de enzimas para romper la pared celular bacteriana compuesta por
el polisacárido conocido con el nombre de peptidoglucano es una práctica
habitual en los laboratorios de Biología y Microbiología.
En esta ocasión se utilizó la enzima liticasa, la cual digiere parte de la pared
celular bacteriana provocando la ruptura o lisis de esta. Una vez digerida la
pared celular se procede a una centrifugación a 6000 rpm para eliminar los
restos de pared bacteriana y obtener el sobrenadante que contenía nuestra
enzima de interés.
Para comenzar con el procedimiento se agregó a un cultivo de liticasa con las
poblaciones bacterianas de M. deltae en fase exponencial la enzima liticasa (2
ml por cada 25 ml de suspensión bacteriana), preparada en 0,89 g de NaCl y
100 ml de agua destilada. Las Bacterias con liticasa se colocaron en un baño
maría durante al menos 30 min-1 hora y 40 C.
Para determinar la efectividad de este método se realizó un conteo de células
antes y después del proceso.

37
4.3.4. Semipurificación de los extractos enzimáticos obtenidos a partir de
la lisis celular.
Columna de DEAE.
El extracto crudo proveniente de la lisis celular de mayor eficiencia (Liticasa)
fue sometida a dos pasos de purificación con el propósito de mejorar la
actividad específica enzimática y desechar proteínas que no eran de nuestro
interés.
Para esto se sometió el extracto crudo primero a una columna de DEAE, la cual
fue armada en el laboratorio de biología de la Universidad de San
Buenaventura, Cali.
Figura 8. Columna de DEAE utilizada en la semipurificación enzimática.
Fuente: Duran, C.A; HETZ, J; PARDO, J.F, 2009
La columna de DEAE fue lavada 2 veces con buffer fosfato de sodio, 0,2 M, pH
7. Luego de lavada la columna se tapó y se dejó con el buffer 24 horas antes
de su utilización.
Se tomó la muestra de lisis celular y se eluyeron en 18 ml del buffer fosfato de
sodio, 0,2 M, pH 7se colocaron en la columna armada a presión atmosférica
constante y se recolectaron las fracciones en tubos de 1,5 ml a las cuales se
les realizaron las pruebas de actividad enzimática. Solo las fracciones que
poseían actividad enzimática fueron sometidas al siguiente paso de purificación
con Sephadex g-100.

38
Columna de Sephadex.
Las fracciones que pasaron por la columna de DEAE, y las cuales poseían
actividad enzimática se pasaron por una columna de exclusión por tamaño
preparada en el laboratorio de Biología Molecular de levaduras (Universidad del
Valle) y según el protocolo de Cooper & Scopes (1982)
El gel utilizado fue Sephadex g-100, capaz de separar proteínas que posean
tamaños entre 40 000 y 150 000.
Un gramo de Sephadex fue solubilizado en 10 ml de alcohol etílico al 10% y
posteriormente colocado en una pipeta, obteniendo una columna de 1,3*10 cm
de alto.
La columna de Sephadex fue lavada 4 veces con buffer fosfato de sodio, 0,2 M,
pH 7. Luego de lavada la columna se tapó y se dejó con el buffer 3 días antes
de usarla.
En la siguiente tabla se puede observar algunas de las características del
Sephadex empleado en la semipurificación del extracto crudo.
Tabla 6: Características del gel de Sephadex G-100 empleado en la
semipurificación del extracto enzimático.
Descripción Característica
1. Tipo de Gel Sephadex g-100
2. Diámetro de la partícula seca. 40-120 micrómetros.
3. Solvente utilizado. Etanol al 10%
4. Tiempo mínimo requerido antes de su utilización.
3 días.
5. Tasa de elusión. 1.0 ml/h
6. Presión Atmosférica.
7. Buffer de muestra usado. Buffer fosfato de Sodio, 0.2 M, pH 7.0
Fuente: Duran, C.A; HETZ, J; PARDO, J.F, 2009
4.3.5 Medición de la actividad enzimática de los extractos celulares
obtenidos a partir de los tres métodos de lisis bacteriana.
La actividad enzimática fue estimada espectrofotométricamente mediante la
reducción del azul de metileno en bióxido de carbono.

39
El ensayo de la actividad enzimática fue realizado de la siguiente manera: La
mezcla de reacción contenía 0,91 ml de buffer KH2PO4 (50 mM, pH 7), 0,05 ml
de glucosa en buffer 2 mM, 0,02 ml de azúl de metileno (2.5 mM), 0,01 ml de la
mezcla de (1 U de glucosa+ 1 U de catalasa en buffer). La reacción se inicia
aproximadamente con 200 microlitros del extracto enzimático, con un contenido
aproximado de 0,1 a 1.8 mg por ml.
Las mezclas de reacción, mantenidas en cubetas de 1 cm de diámetro fueron
burbujeadas durante 10 minutos con bióxido de carbono y posteriormente las
cubetas fueron cerradas para evitar el escape del gas y el contacto de los
reactantes con el oxígeno del aire.
El blanco poseía los mismos ingredientes que la mezcla de reacción con
excepción del bióxido de carbono y el extracto enzimático. El espectrofotómetro
utilizado era de doble haz, marca Shimadzu UV 160 A y los ensayos se
realizaron en un tiempo total de 1800 segundos, suficiente para que la reacción
se llevara a cabo completamente.
El azul de metileno en estado reducido absorbe a una longitud de onda de 664
nm. Cuando el azul de metileno pasa de un estado reducido a un estado
oxidado, se observa un cambio en la absorbancia a 664 nm la cual es
registrada por medio del espectrofotómetro. Este cambio de absorbancia es
proporcional a la cantidad de azul de metileno oxidado, el cual es
estequiométricamente equivalente al bióxido de carbono reducido en la
reacción:
CO2 +H20 + X (red) CO + X (ox), donde X es el azul de metileno.
La diferencia de la absorbancia se refiere al cambio de la absorbancia del azul
de metileno a 664 nm, mientras que el tiempo (segundos) es el requerido para
que la absorbancia se vuelva constante.
Estos valores de actividad enzimática fueron convertidos a unidades de
actividad enzimática, teniendo en cuenta el coeficiente de extinción del azúl de
metileno se calculó de forma experimental realizando una curva a diferentes
concentraciones de azul de metileno vs absorbancia, a una longitud de onda de
664 nm. El valor obtenido fue de 0,0708 cm2/nmol y su porcentaje de error con
respecto al reportado en literatura fue de 6,2%. (http://omlc.oqi.ed/spectra/mb)
4.3.6. Inmovilización enzimática.
El extracto enzimático obtenido a partir de la bacteria Methanococcus deltae se
inmovilizó con alginato de sodio. Este polisacárido producido por algas soluble en
agua, en una solución con iones divalentes como el Ca2+ polimeriza ya que los
iones producen entrecruzamiento.

40
Al polimerizar una solución de alginato conteniendo enzimas estas quedan
atrapadas dentro de las esferas que se generan.
Para la inmovilización se colocaron 10 ml del extracto enzimático en un vaso de
precipitado y se agregaron 100 mg de alginato de sodio (1.0 % final). Se
homogenizo el medio y se tomó con una jeringa de 10 ml. Se hizo gotear la
solución en un vaso de precipitado conteniendo solución de CaCl2 200 mM. Se
producen esferas de 3-5 mm de diámetro que polimerizan al entrar en contacto
con la solución de CaCl2. Las esferas se estabilizan durante 1 h en la solución de
CaCl2 a temperatura ambiente. Las esferas se lavaron con buffer acetato 0.1 M,
pH: 5.
La actividad de la enzima inmovilizada se determinó agregando 5 ml de
solución de sustrato e incubando 5 min a 37°. Luego se extrajo una alícuota de
0.5 ml y se agrega a cada tubo 4.5 ml de solución reveladora. Se midió la
absorbancia en espectrofotómetro a 640 nm.
Posteriormente la enzima inmovilizada se colocó en un electrodo de platino con el
objetivo de elaborar el Biosensor. Este electrodo transmitía la señal hacia un
detector el cual permitía determinar la concentración de bióxido de carbono en el
medio. El electrodo fue suministrado por el grupo de investigación en Biología
Molecular de la Universidad del Valle. (Figura 9).
Figura 9: Electrodo de Platino usado en la Investigación.
Fuente. Duran, C , 2010.

41
5. RESULTADOS Y DISCUSIÓN
5.1 Cultivo y crecimiento de la cepa bacteriana (M. deltae).
Aislamiento
Se obtuvieron al final de la experimentación dos cepas que utilizan el bióxido
de carbono como fuente de carbono a partir del lago principal de la Universidad
de San Buenaventura, Cali. Esta cepa fue crecida en un ambiente saturado de
Bióxido de carbono al igual que la cepa de methanococcus deltae comprada a
la ATCC.
La cepa aislada del lago fue almacenada a -20 C para posteriores estudios en
el laboratorio. A todas las cepas tanto las aisladas como a la cepa comprada a
la ATCC, se le realizó la coloración de gram, cápsula y espora, dando bacterias
bacilares, gram (-), sin esporas y sin cápsulas. (Figura 9 )
Figura 10: Coloración de Gram donde se observa un bacilo gran (-) y cloración
de cápsula donde se observa la ausencia de esta.
Cultivo.
Cultivo.
Los cultivos de todas las bacterias se realizaron en el medio anteriormente
mencionado y sugerido por la American Type Culture Collection (ATCC), en
una cámara de anaerobiosis saturada con bióxido de carbono, a 30 C. En estas
condiciones, las bacterias alcanzaron la fase de crecimiento exponencial en
aproximadamente 2 horas. En la Figura 10, se puede observar la curva de
crecimiento de la población de M. deltae donde se observa que la población
termina la fase de crecimiento exponencial al cabo de las dos horas.

42
Figura 11. Curva de crecimiento de la población de M. deltae
Fuente: DURAN, C.A; HETZ, J; PARDO, J.F, 2009
En medio sólido todas las bacterias mostraron características muy similares,
observándose colonias medianas y cremosas de borde regular.
5.2 PRUEBAS BIOQUÍMICAS REALIZADAS A LAS BACTERIAS AISLADAS
DEL LAGO.
Las pruebas bioquímicas fueron realizadas en el laboratorio de Microbiología
de la Universidad de San Buenaventura. Los resultados de estas pruebas se
muestran a continuación:
En la Tabla 7, se pueden observar los resultados de las pruebas bioquímicas
realizadas a las bacterias aisladas de lago de la Universidad de San
Buenaventura y la bacteria Methanococcus deltae comprada a la ATCC.

43
Tabla 7. Resultados de pruebas bioquímicas realizadas.
PRUEBA CEPAS AISLADAS Methanococcus deltae
(ATCC)
TSI
H2S - -
Lisina K/K K/K
Citrato + +
Urea - -
Indol - -
Motilidad + +
Rojo de Metilo - -
Voges Proskaver - -
Oxidasa - -
H. Gelatina - -
H. almidón - -
H. caseína - -
Agar cetrimida - -
Agar XLD - -
Agar Sangre - -
Fuente: HETZ, J; PARDO, J.F, 2009

44
Tal como se puede observar en la tabla anterior las bacterias aisladas
pertenecen al género Methanococcus, sin embargo, estas pruebas no son
concluyentes para determinar la especie bacteriana aislada. Más aún, se
sugiere que estas pruebas sean ratificadas por pruebas de análisis molecular
del ADN ribosomal 18S.
A partir de las pruebas bioquímicas anteriormente mencionadas podemos
inferir que las bacterias no pertenecen a los géneros Pseudomonas sp (Agar
cetrimide -), Estaphilococcus sp (Agar sangre -).
Esta bacteria según la literatura es flagelada polar lo cual se corrobora con la
prueba de motilidad positiva, no produce indol y es negativo para las pruebas
de citrato y oxidasa, lo cual descarta al género Pseudomonas. Sin embargo,
para las pruebas de las hidrólisis (H. almidón,. H. Caseína, H. gelatina), da
negativo y la prueba de lisina es K/K, estos resultados son típicos de bacterias
metanógenas,
5.3 LISIS BACTERIANA POR TRES DIFERENTES MÉTODOS.
Las bacterias metanógenas se sometieron a tres métodos diferentes de ruptura
para identificar cuál método era el más eficiente y fácil de realizar, con la
mayor cantidad de extracto proteico crudo obtenido. La tabla 8 muestran los
resultados de los procesos de lisis celular:
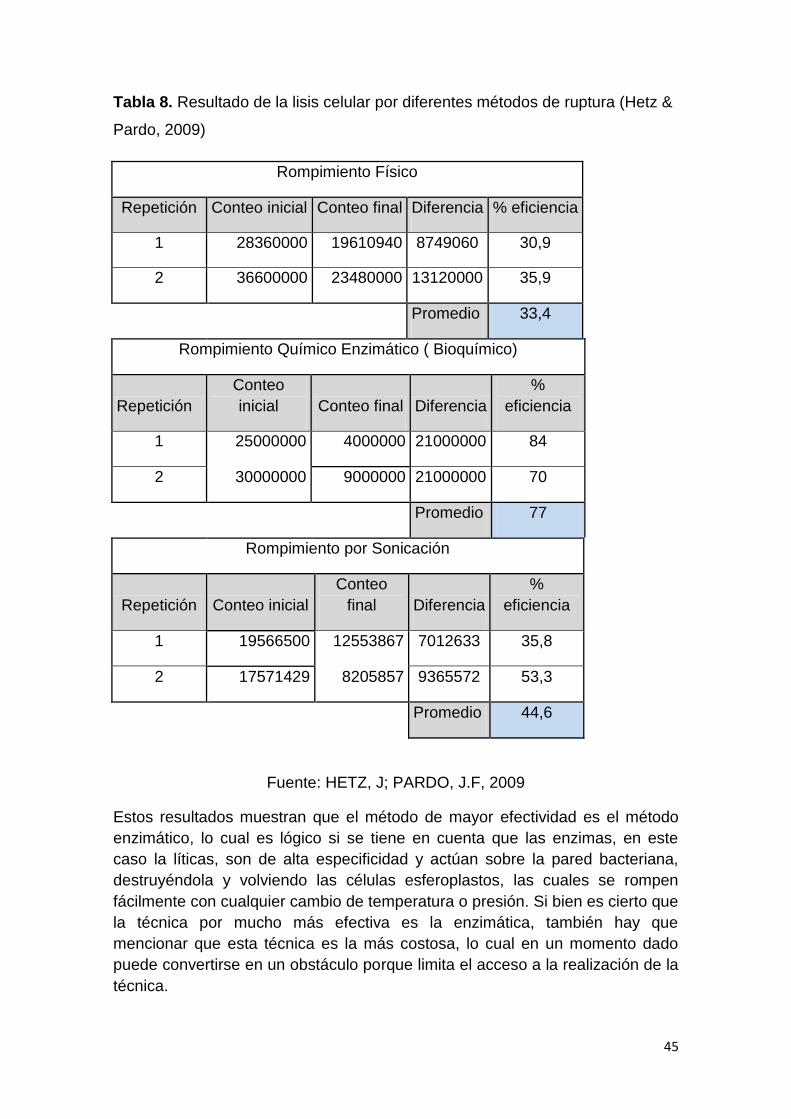
45
Tabla 8. Resultado de la lisis celular por diferentes métodos de ruptura (Hetz &
Pardo, 2009)
Rompimiento Físico
Repetición Conteo inicial Conteo final Diferencia % eficiencia
1 28360000 19610940 8749060 30,9
2 36600000 23480000 13120000 35,9
Promedio 33,4
Rompimiento Químico Enzimático ( Bioquímico)
Repetición
Conteo
inicial Conteo final Diferencia
%
eficiencia
1 25000000 4000000 21000000 84
2 30000000 9000000 21000000 70
Promedio 77
Rompimiento por Sonicación
Repetición Conteo inicial
Conteo
final Diferencia
%
eficiencia
1 19566500 12553867 7012633 35,8
2 17571429 8205857 9365572 53,3
Promedio 44,6
Fuente: HETZ, J; PARDO, J.F, 2009
Estos resultados muestran que el método de mayor efectividad es el método
enzimático, lo cual es lógico si se tiene en cuenta que las enzimas, en este
caso la líticas, son de alta especificidad y actúan sobre la pared bacteriana,
destruyéndola y volviendo las células esferoplastos, las cuales se rompen
fácilmente con cualquier cambio de temperatura o presión. Si bien es cierto que
la técnica por mucho más efectiva es la enzimática, también hay que
mencionar que esta técnica es la más costosa, lo cual en un momento dado
puede convertirse en un obstáculo porque limita el acceso a la realización de la
técnica.

46
5.4 MEDICIÓN DE LA ACTIVIDAD ENZIMÁTICA.
El extracto crudo fue pasado por una columna de DEAE y posteriormente fue
pasado por una columna de exclusión por tamaño de Sephadex G-100,
resultando un total de 25 eppendorf de 1,0 ml. A cada uno de estos viales les
fue determinada la concentración proteica por el método de Folin Lowry y
aquellos en los cuales se reportaba la presencia de proteínas se les
determinaban la actividad enzimática.
La actividad enzimática se determinó por el cambio de observancia del azul de
metileno a 664 nm. (Fig 11). La actividad enzimática específica fue
determinada a partir de los valores del Δ de absorbancia, tiempo y
concentración de proteínas, mediante la fórmula:
Actividad enzimática específica= actividad enzimática/cantidad proteica
determinada.
Figura 12. Cambio en la absorbancia del azul de metileno vs tiempo, en
presencia de la enzima que degrada el CO2.
Fuente: Duran, C; HETZ, J; PARDO, J.F, 2009
En estas condiciones las unidades resultantes son micromoles de azul de
metileno reducido/tiempo * mg de proteína. Mediante el coeficiente de extinción
del azúl de metileno y la relación Beer-Lambert se puedo expresar la actividad
enzimática específica en unidades/mg de proteína.
A partir de estos datos se determinó la actividad enzimática específica la cual
se suponía iba incrementándose a medida que aumentaban los pasos de
purificación. Los datos de las fracciones, la concentración proteica y la
actividad específica de los 25 viales se pueden observar en la tabla siguiente:
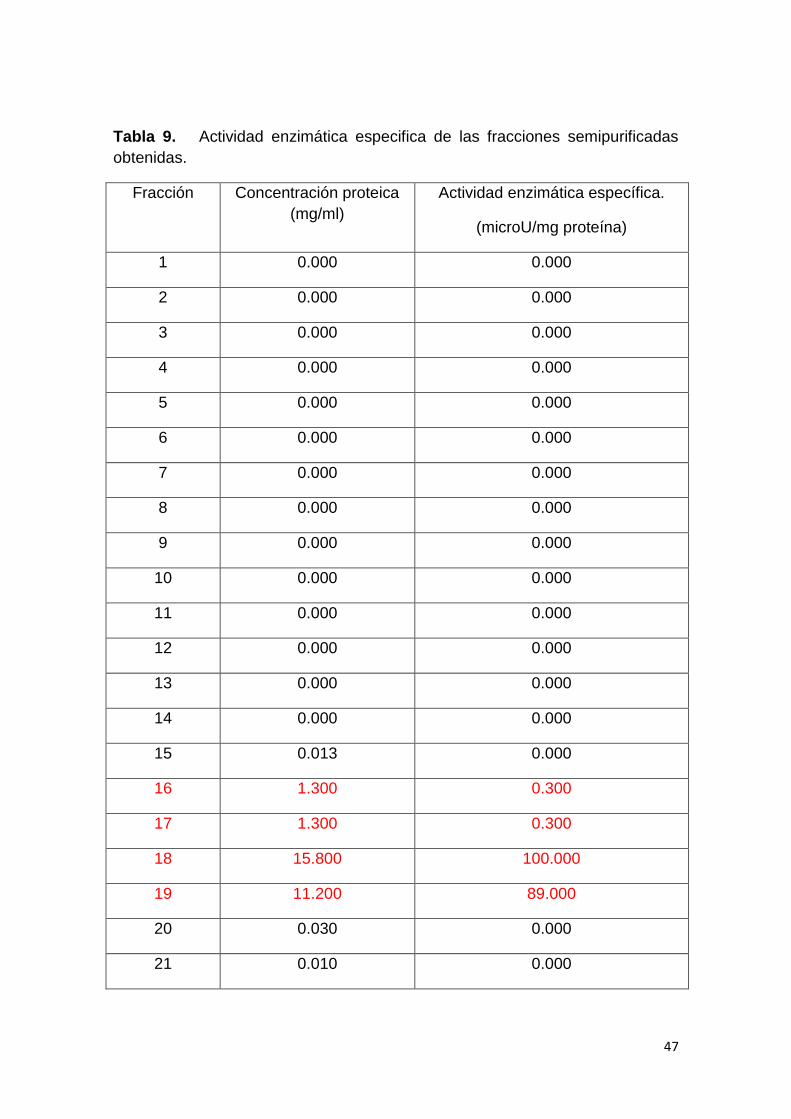
47
Tabla 9. Actividad enzimática especifica de las fracciones semipurificadas
obtenidas.
Fracción Concentración proteica
(mg/ml)
Actividad enzimática específica.
(microU/mg proteína)
1 0.000 0.000
2 0.000 0.000
3 0.000 0.000
4 0.000 0.000
5 0.000 0.000
6 0.000 0.000
7 0.000 0.000
8 0.000 0.000
9 0.000 0.000
10 0.000 0.000
11 0.000 0.000
12 0.000 0.000
13 0.000 0.000
14 0.000 0.000
15 0.013 0.000
16 1.300 0.300
17 1.300 0.300
18 15.800 100.000
19 11.200 89.000
20 0.030 0.000
21 0.010 0.000

48
22 0.000 0.000
23 0.000 0.000
24 0.000 0.000
25 0.000 0.000
Fuente: HETZ, J; PARDO, J.F, 2009
Las fracciones 16, 17, 18 y 19 que dieron tanto concentración de proteínas
como actividad enzimática se unieron en dos viales (M1 y M2
respectivamente), en M1 se unieron las muestras 16 y 17, mientras que en M2
se unieron las muestras 18 y 19. Las muestras 15, 19 y 21 se descartaron
puesto que si bien daba positivo para la concentración proteica no daba señal
de actividad enzimática, lo cual significaba que las proteínas encontradas en
esos viales no correspondían a la enzima de interés.
A partir de los datos anteriormente mostrados se puede deducir que se
obtuvieron extractos semipuros con actividad enzimática que pueden ser
utilizados para la elaboración del sensor bio-enzimático.
5.5 INMOVILIZACIÓN ENZIMÁTICA Y BIOSENSOR ENZIMÁTICO.
La inmovilización de la enzima se realizó de forma satisfactoria, por medio de
Alginato de Sodio, el cual a su vez se colocó en el electrodo de platino. Este
electrodo de platino festaba unido a un medidor de Bióxido de el cual media las
cantidades de este gas atrapado por la enzima.
La medición se realizo en el orden de los 5 ppm, lo cual hace esta técnica
altamente específica e indiscutiblemente interesante para medir la respiración
en frutas, vegetales y otras materias primas importantes a nivel agroindustrial.

49
6. CONCLUSIONES
1. Se logró una semipurificación de la enzima bióxido de carbono deshidrogenasa a partir de extractor celulares procedentes de la bacteria Methanococcus deltae.
2. El método de ruptura más eficiente fue el tratamiento con la enzima liticasa, puesto que ocurrió la lisis celular en menor tiempo y se obtuvo el mayor porcentaje de eficiencia medida como numero de células bacterianas lisadas.
3. La enzima semipurificada por la columna de DEAE y de exclusión por tamaño Sephadex G-100 poseía actividad enzimática, lo cual lo hace ideal para ser inmovilizada y utilizada en la fabricación del sensor enzimático.
4. Se logró el aislamiento de cepas nativas (1) de bacterias metanógenas a
partir del agua del lago principal de la Universidad de San Buenaventura-Cali.
5. No se observaron diferencias bioquímicas entre la cepa aislada y la cepa de Methanococcus deltae comprada a la ATCC.
6. Se logró inmovilizar la enzima procedenrte de la bacteria Metanococcus deltae, con actividad en<zimática para el bióxido de carbono por medio del alginato de Sodio.
7. Se obtuvo un prototipo experimental con un electrodo de platino que medía las concentraciones de bióxido de carbono en el orden de los 5 ppm.

50
RECOMENDACIONES
A pesar de la gran cantidad de investigaciones sobre tecnologías de biosensores en agroindustria, y más en el sector de alimentos, los productos comercializados son escasos. Sería interesante que futuros proyectos del grupo de investigación del programa de ingeniería agroindustrial de la universidad de San Buenaventura se enfocaran en generar este tipo de tecnología cada vez más indispensables.
Se recomienda después de la lisis celular someter el extracto a un detergente tal como Tritón X-100, esto debido a que en reportes de literatura mencionan a esta enzima como ligada a membrana. Si se somete a tritón X-100, se aumentaría la concentración de enzima después de la lisis celular.
Se recomienda experimentar con otros métodos de inmovilización que puedan optimizar el proceso de fijación de la enzima al electrodo y de este modo obtener un mejor desempeño del biosensor desarrollado.

51
BIBLIOGRAFIA
BARD, A.J. Integrated Chemical Systems; Wiley & Sons: New York, 1994
BRAUN, R. D. Introduction to instrumental Analysis; Mc-Graw Hill: New York, 1987
CAMPÁS, M.; MIR, M.; O´SULLIVAN, C. Y KATAKIS, I. (2002) Biosensores al servicio de la industria alimentaria. 2º Congreso Español de Ingeniería de Alimentos, Lleida (España).
CHOI, M.F; Hawkins, P.; Novel dye-solvent solutions for the simultaneous detection of oxygen and carbon dioxide, Anal. Chem. 67 (1995) 3897–3902.
COMMERCIAL BIOSENSORS: Applications to Bioanalytical sensors (Techniques in Analytical Chemistry Series); Wiley & Sons: New York, 1998
COPELAN, R. A. 1993. Methods for Protein Analysis: A Practical Guide to Laboratory Protocols. Chapman & Hall, New York.
CUERVO, Raul; BRAVO, Enrique; BENÍTEZ, Neyla; TORRES, Walter; LARMAT, Fernando. Extracción y Purificación de la enzima monóxido de carbono deshidrogenasa a partir de la bacteria carboxidotrofa Oligotropha carboxidovorans. Revista Científica Guillermo de Ockham. Vol 3. No 2. Julio de 2005. 107-116
EGGINS, B. R. Biosensors: An Introduction; Wiley & Sons: New York, 1997.
ESPÍN, Neyda. La biotecnología en la valorización de desechos agroindustriales. Escuela Politécnica Nacional. Departamento de Bioprocesos. Ecuador. Disponible en Internet en: http://www.bvsde.paho.org/bvsaidis/ecuador10/biot.pdf
FRITZ, J.F. Anal. Chem., 1987, 59, 335A; P. K. DASGUPTA, Anal Chem., Cromatografía iónica moderna
GONZALEZ, Víctor; GARCÍA, Esther; RUIZ, Olga; GAGO, Lara; et al. Aplicaciones de Biosensores en la industria Agroalimentaria. Colección Vt. Dirección General de Universidades e Investigación. España 2005

52
GRATTAROLO, M.; MASSOBRIO, G. Bioelectronics Handbook: MOSFETs, Biosensors and Neurons; McGraw Hill: New York, 1998.
HALL, R. H. (2002) Biosensor technologies for detecting microbiological foodborne hazards Microbes and Infection, 4, 425–432.
HORTON H., Rober. Bioquímica, México : Prentice Hall Hispanoamericana, c1995
HUNT, B.J.; S.R.HOLDING; Size Exclusion Chomatography, Eds. New York: Chapman y Hall, 1998; Aqueus Size-exclusion Chromatography, P.L. Dubin, Ed. Amsterdam, Neth.: Elseiver, 1988, Handbook of Size- exclusion Chromatography, C. S. Wu, Ed. New York, Marcel Dekker, 1995.
IANNIELLO, R. M.; YACYNYCH, A. M. Anal. Chem. 1981, 53, 2090-2095.
JANATA, J.; JOSOWICZ, M.; DEVANEY, M. Analytical Chemestry 1994, 66, 207-228.
LEONARD, P.; HEARTY, S.; BRENNAN, J.; DUNNE, L.; QUINN, J.; CHAKRABORTY, T. Y O´KENNEDY, R. (2003). Advances in biosensors for detection of pathogens in food and water. Review. Enzyme and Microbial Technology, 32, 3-13.
LODISH, H., BERK. A,. ZIPURSKY. S. L,. MATSUDAIRA. P., BALTIMORE. D., J. DARNELL. 2002. Biologia Celular y Molecular. Editorial MedicaPanamericana. Mexico.
MELLO, L.D; KUBOTA, L.T. (2002). Review of the use of biosensors as analytical tools in the food and drink industries. Food Chemistry, nº 77, pág. 237-256.)
MEYER, O; SCHLEGEL, H. G. J. Bacteriol. 1980, 141, 74-80
MILLS, A. CHANG, Q. Fluorescence plastic thin-film sensor for carbon dioxide, Analyst 118 (1993) 839–843.
MONOGRAFÍA: Ciencia y tecnología para una sociedad abierta; COLCIENCIAS: Bogotá. 1990.
MORALES, M.D.; MORANTE, S.; ESCARPA, A.; GONZÁLEZ, M.C.; REVIEJO, A.J. Y PINGARRÓN, J.M. (2002) Design of a composite

53
amperometric enzyme electrode for the control of the benzoic acid content in food. Talanta, 57, 1189-1198.
ODACT, D.; TIMUR, S. Y TELEFONCU, A. (2004) Carboxyl esterase-alcohol oxidase based biosensor for aspartame determination. Food Chemistry, 84, 493-496.
PATEL P.D. Biosensors for measurement of analytes implicated in food safety: a review. Trends in Analytical Chemistry, vol. 21, nº 2, 96-115. (2002).
PERSON, D. M.; O’REILLY, C.; COLBY, J.; BLACK, G.W. Biochim, Biophys. Acta 1994, 1188(3), 432-438.
POCCIA, María Eugenia. Operaciones Biotecnológicas. Laboratorio de Fermentaciones. Facultad de Bioquímica y Ciencias Biológicas.[ citado en 2002] disponible en internet en: http://www.universia.com.ar/contenidos/investigacion/unl/C_BASICAS/biologia/1111.htm
SANZ GARCÍA, María Peña hora. Sistemas Multisensores para el Control de Calidad de Alimentos. España: Universidad Autónoma de Madrid, Facultad de Ciencias, 2004.
SEVERINGHAUS, J; BRADLEY, A.F. Electrodes for blood pO2 and pCO2 determination, Appl. Physiol. 13 (1958) 515–520.
SKOOG. HOLLER. NIEMAN. Principios DE ANALISIS INSTRUMENTAL quinta edición Mc Graw Hill
TIERNEY, M. J.; KIM; H. O. L.; MADOU, M.; OTAGAWA, T. Sensors and Actuators, B 1993, B13(1-3), 408-411
TREVAN, M. D., BOFFEY, S., GOULDING, K. H., STANBURY, P. 1990. Biotecnología: principios Biológicos. Ed. Acribia. Zaragosa. España.
VELASCO GARCÍA M. Y MOTTRAM T. (2003). Biosensor technology addressing agricultural problems. Review paper. Biosystems Engineering, vol. 84, nº 1, 1-12.
WALKER, J.M., GINGOLD, E.B. 1997 Biología molecular y

54
biotecnología. Editorial Acribia. Segunda Edición.
WEIGL, B.H; WOLFBEIS O.S. New hydrophobic materials for optical carbon dioxide sensors based on ion pairing, Anal. Chim. Acta 302 (1995) 249–254.)
Taylor, R.F. (1991) Protein Immobilization: fundamentals and applications, Marcel Dekker, New York.
Hartmeier, W. (1985) immobilized biocatalysts: from simple to complex systems. Trends Biotechnol. 3: 149-153.
Martinek, K.; Mozhaev, V.V. (1987) Immobilization of enzymes: an approach to fundamental studies in biochemistry.Adv. Enzymol. 57: 179-249.
Kennedy, J.F.; Cabral, J.M.S. (1983) Solid Phase Biochemistry. Schouten, W.H. (ed.) Wiley Pub., New York.
Klei, H.E.; Sundstrom, D.W.; Shim, D. (1985) Immobilization of enzymes by microencapsulation en Immobilized cells and enzymes: a practical approach J. Woodward, ed. IRL Press, pág. 49-54.
Katchalski-Katzir, E. (1993) Immobilized enzymes: learning from past successes and failures. Trends Biotechnol. 11: 471-478.
Chibata, I.; Tosa, T. (1980) Immobilized microbial cells and their applications. Trends Biochem. Sci. 5:88-90.
Honda, Y.; Kako, M.; Abiko, K.; Sogo, Y. (1993) Industrial Application of immobilized enzymes, Tanaka, A. et al. eds. Marcel Dekker, pág. 209-234.
Germain, P.; Crichton, R. (1988) Characterization of a chemically modified a-amilase immobilized on porous silica. J. Chem. Technol. Biotechnol. 41: 297 315.
Piffer, P.G.; Spagna, G.; Bustetto, L. (1987) Immobilization of endo-polygalacturonase on g-alumina for the treatment of fruit juices. J. Mol. Catal. 42: 137-149.
Magee, E.L.; Olson, N.F.; Linslay, R.C. (1981) Microencapsulation of cheese ripening systems: production of diacetyl and acetoin in cheese by encapsulated bacterial cell free extract. J. Diary Sci. 64: 616-621.

55