toll-likereceptor9agonistimocooperateswithcetuximab in ... · sudhir agrawal8, federica di...
TRANSCRIPT

Cancer Therapy: Preclinical
Toll-like Receptor 9 Agonist IMOCooperateswithCetuximabin K-Ras Mutant Colorectal and Pancreatic Cancers
Roberta Rosa1,3, Davide Melisi2,4, Vincenzo Damiano1, Roberto Bianco1, Sonia Garofalo1, Teresa Gelardi5,Sudhir Agrawal8, Federica Di Nicolantonio9,10, Aldo Scarpa6,7, Alberto Bardelli9,10, and Giampaolo Tortora5
AbstractPurpose: K-Ras somatic mutations are a strong predictive biomarker for resistance to epidermal growth
factor receptor (EGFR) inhibitors in patients with colorectal and pancreatic cancer. We previously showed
that the novel Toll-like receptor 9 (TLR9) agonist immunomodulatory oligonucleotide (IMO) has a strong
in vivo activity in colorectal cancer models by interfering with EGFR-related signaling and synergizing with
the anti-EGFR monoclonal antibody cetuximab.
Experimental Design: In the present study, we investigated, both in vitro and in vivo, the antitumor effect
of IMO alone or in combination with cetuximab in subcutaneous colon and orthotopic pancreatic cancer
models harboring K-Ras mutations and resistance to EGFR inhibitors.
Results:We showed that IMOwas able to significantly restore the sensitivity of K-Rasmutant cancer cells
to cetuximab, producing a marked inhibition of cell survival and a complete suppression of mitogen—
activated protein kinase phosphorylation, when used in combination with cetuximab. IMO interfered with
EGFR-dependent signaling, modulating the functional interaction between TLR9 and EGFR. In vivo, IMO
plus cetuximab combination caused a potent and long-lasting cooperative antitumor activity in LS174T
colorectal cancer and in orthotopic AsPC1 pancreatic cancer. The capability of IMO to restore cetuximab
sensitivity was further confirmed by using K-Ras mutant colorectal cancer cell models obtained through
homologous recombination technology.
Conclusions: We showed that IMO markedly inhibits growth of K-Ras mutant colon and pancreatic
cancers in vitroand innudemice andcooperateswith cetuximabviamultiplemechanismsofaction. Therefore,
we propose IMO plus cetuximab as a therapeutic strategy for K-Ras wild-type as well for K-Ras mutant,
cetuximab-resistant colorectal and pancreatic cancers. Clin Cancer Res; 17(20); 6531–41. �2011 AACR.
Introduction
The epidermal growth factor receptor (EGFR) is a tyrosinekinase receptor (TKR) playing a key role in the developmentand progression of several human epithelial cancers. There-
fore, EGFR targeting has become a successful approach forcancer treatment (1). Two anti-EGFR monoclonal antibo-dies (mAb), cetuximab and panitumumab, and 2 small-molecule selective tyrosine kinase inhibitors (TKI), gefitiniband erlotinib, have been approved for the treatment ofseveral tumors (2–5). However, an emerging issue in cancerpatients is the occurrence of constitutive resistance or thedevelopment of acquired refractoriness to anti-EGFR drugsby several different mechanisms (6). These include specificmutations or loss of EGFR, alternative signaling by differentTKRs, constitutive activation of downstream signal trans-ducers, such as the serine/threonine kinase Akt or the smallGTPase Ras, and induction of angiogenesis by tumor-derived factors such as the VEGF (1, 7). Particularly, muta-tions of theK-Ras gene,most frequently in codon 12of exon2, produce a single amino acid change resulting in mutantRas proteins that are insensitive to the function of GTPase—activating protein and are constitutively active. These muta-tions are among the most common genetic alterations inhuman cancers, occurring in30%of colorectal cancers, 75%to 90%of pancreatic cancers, 50%of cholangiocarcinomas,and 20% of lung carcinomas (1, 8, 9). K-Ras mutationsleading to constitutive activation of the Ras/MAPK signaling
Authors' Affiliations: 1Dipartmento di Endocrinologia ed Oncologia Mole-colare e Clinica, Universit�a di Napoli "Federico II"; 2Unit�a di FarmacologiaSperimentale, Istituto Nazionale per lo Studio e la Cura dei Tumori-Fonda-zione "G. Pascale," Naples, Italy; Departments of 3Surgical Oncology and4Gastrointestinal Medical Oncology, The University of TexasMDAndersonCancer Center, Houston, Texas; 5Oncologia Medica, 6Dipartimento diPatologia e Diagnostica, Universit�a di Verona; 7ARC-NET, Center forApplied Research in Cancer, Verona, Italy; 8Idera Pharmaceuticals, Cam-bridge, Massachusetts; 9Institute for Cancer Research and Treatment,Universit�a di Torino, Torino; and 10FIRC Institute of Molecular Oncology,Milan, Italy
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Author:Giampaolo Tortora, OncologiaMedica, Universityof Verona, Policlinico BorgoRoma, Piazzale Scuro, 10, Verona 37134, Italy.Phone: 39-045-8128519; Fax: 39-045-8128519; E-mail:[email protected]
doi: 10.1158/1078-0432.CCR-10-3376
�2011 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 6531
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

pathway represent a strong predictive biomarker for resis-tance to EGFR inhibitors. Several studies reported thatmutations in the K-Ras gene are associated with lack ofresponse to anti-EGFR mAbs in advanced colorectal cancerand to EGFR TKIs in patients with non–small-cell lungcancer (10–12). It has been reported that K-Ras mutationscould be a predictive marker for erlotinib resistance also inpancreatic cancer (13, 14).
Toll-like receptor 9 (TLR9) participates in vertebrateimmune system recognizing unmethylated CpG dinucleo-tides, common in bacterial and viral DNA but not invertebrate DNA, and initiating a cascade of potent Th1-typeinnate and adaptive immune responses (15, 16). TLR9agonists are synthetic oligodeoxynucleotides containingCpGmotifs, developed as immunoprotective or antiallergicagents, vaccine adjuvants, or antitumor agents (16). Theseagents are able to potentiate the antitumor immuneresponses through the activation of natural killer, dendritic,and cytotoxic T cells, increased production of antitumorcytokines, and enhanced antibody-dependent cell-mediat-ed cytotoxicity (ADCC). Second-generation synthetic TLR9agonists, referred to as immunomodulatory oligonucleo-tides (IMO), contain a novel structure with 2 accessible 50-ends critical for TLR9 recognition and exhibit higher met-abolic stability than conventional TLR9 agonists (17). TLR9agonists, such as IMOs, are showing promising results asanticancer agents in clinical studies; however, themolecularmechanisms by which they affect tumor growth and angio-genesis have not been fully elucidated.
We have previously contributed to clarify some of thesemechanisms showing that a TLR9 agonist, IMO, potenti-ates the ADCC activity of anti-EGFR mAb cetuximab (18)and anti-HER2 mAb trastuzumab (19) in in vivo modelsof colorectal and breast cancers, respectively. In addition
to this immunomodulating function, we showed thatIMO acts also by impairing EGFR signaling and potentlysynergizes in vivo both with cetuximab or EGFR TKIgefitinib in EGFR-addicted colorectal cancer models(20). Finally, we showed that IMO cooperates in vivowith anti-VEGF mAb bevacizumab in EGFR-independentcolorectal cancer models by affecting function of endo-thelial cells (18). These findings opened the path to theongoing clinical studies combining TLR9 agonists withEGFR inhibitors in cancer patients (http://clinicaltrials.gov/ct2/show/NCT00719199).
Because K-Rasmutations are a major limitation for usingEGFR inhibitors in patients with colorectal and pancreaticcancer, we investigated the effects of IMO alone and incombination with cetuximab in colon and pancreatic can-cer models harboring a mutant K-Ras.
Methods
CompoundsThe anti-EGFR mAb cetuximab was kindly provided
by ImClone Systems. IMO, 50-TCTGACRTTCT-X-TCTTRCAGTCT-50 (X and R are glycerol linker and 20-deoxy-7-deazaguanosine, respectively), was synthesizedwith phosphorothioate backbone, purified and analyzedas described previously (21).
Cell culturesHuman GEO, SW48, and LS174T colon carcinoma cell
lines were obtained from the American Type Culture Col-lection. Human AsPC1/GLT pancreatic cancer cells werepreviously described (22). G12V K-Ras mutant SW48 cellswere generated through the homologous recombinationtechnology as previously described (23). Human T3M4 andMIA PaCa-2 pancreatic carcinoma cell lines were kindlyprovided by Prof. A. Scarpa (Departments of Pathology,University of Verona). GEO-CR (cetuximab resistant) cellswere established as previously described (24). Cells weremaintained in McCoy’s media or Dulbecco’s ModifiedEagle’s Media supplemented with 10% heat-inactivatedFBS, 20 mmol/L HEPES (pH ¼ 7.4), penicillin (100 UI/mL), streptomycin (100mg/mL), and 4mmol/L glutamine(ICN Biomedicals Ltd.) in a humidified atmosphere of 95%air and 5% CO2 at 37
�C.
Soft agar growth assayCells (104 cells per well) were suspended in 0.3% Difco
Noble agar (Difco) supplemented with complete medium,layered over 0.8% agar-medium base layer, and treated withdifferent concentrations of cetuximab or IMO. After 10 to 14days, cells were stained with nitroblue tetrazolium (SigmaChemical Co.) and colonies greater than 0.05 mm werecountedusing theQuantityOneSoftwarePackage (Bio-Rad).
MTT survival assayCells (104 cells perwell) were grown in 24-well plates and
exposed to increasing doses of cetuximab or IMO, alone orin combination. The percentage of cell survival was
Translational Relevance
In this study, we show the antitumor effect of thecombination of immunomodulatory oligonucleotide(IMO) and cetuximab in colon and pancreatic cancermodels harboring K-Ras mutations and resistance toEGFR inhibitors, both in vitro and in vivo. We show thatIMO significantly restores cetuximab sensitivity in bothintrinsically mutated and knocked-in cells for mutant K-Ras gene, suppressing activated expression of mitogen—activated protein kinase when used in combination withcetuximab. A possible mechanism for this effect is thecapability of IMO to modulate a structural/functionalinteraction between TLR9 and EGFR. The antitumoreffect of the combination of IMO and cetuximab wasconfirmedalso in vivo in clinically relevant tumormodelsharboring mutant K-Ras: a subcutaneous colorectal andanorthotopic pancreatic cancermodel.Ourdata supportIMO plus cetuximab as a novel therapeutic strategy to betested in a clinical setting for K-Ras mutant, cetuximab-resistant colon and pancreatic cancer patients.
Rosa et al.
Clin Cancer Res; 17(20) October 15, 2011 Clinical Cancer Research6532
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

determined using the MTT assay according to manufac-turer’s instructions.
Immunoprecipitation and Western blot analysisTotal cell lysates were obtained from cell cultures. The
protein extracts were resolved by 4% to 15%SDS-PAGE andprobed with anti-human, polyclonal pEGFR, polyclonalEGFR, monoclonal pMAPK, monoclonal MAPK (SantaCruz Biotechnology), monoclonal pAkt, polyclonal Akt(Cell Signaling Technologies), and monoclonal actin (Sig-ma-Aldrich). Immunoreactive proteins were visualized byenhanced chemiluminescence (Pierce). Immunoprecipita-tions were conducted using monoclonal anti-TLR9 anti-body (Calbiochem/EMDBiosciences) and blottingwith thesame antibody or polyclonal anti-EGFR antibody, follow-ing the procedures described above. As negative control,lysis buffer was mixed with anti-TLR9 antibody.
Ras activation assayThe analysis of Ras activation was conducted by an
immunoprecipitation assay with GST-Raf1-RBD (Ras-bind-ing domain), as previously described (23). The K-Ras pro-tein was detected with Anti-K-Ras (F234) mAb (Santa CruzBiotechnology).
Nude mice cancer xenograft modelsFive-week-old Balb/cAnNCrlBR athymic (nuþ/nuþ) mice
(Charles River Laboratories)maintained in accordancewithinstitutional guidelines of the University of Naples AnimalCare Committee and in accordance with the Declaration ofHelsinki were injected subcutaneously with LS174T humancolon cancer cells (107 cells per mice) resuspended in 200mL of Matrigel (Collaborative Biomedical Products). After 7days, tumors were detected and groups of 10 mice wererandomized to receive the following treatments: IMO1mg/kg intraperitoneally 3 times a week for 4 weeks; cetuximab10 mg/kg intraperitoneally twice a week for 3 weeks; thecombination of these agents on days 7 to 11, 14 to 18, and21 to 25, continuing only IMO on days 28 to 32, or vehiclesonly as control. Tumor diameter was assessedwith a Verniercaliper, and tumor volume (cm3) was measured using thefollowing formula: p/6� larger diameter � (smaller diam-eter)2 as previously reported (19).To produce pancreatic cancer orthotopic xenograft mod-
el, 6- to 8-week-old female athymic nude mice (NCI-nu)were purchased from the Animal Production Area of theNational Cancer Institute-Frederick Cancer Research Facil-ity (Frederick, MD). All mice were housed and treated inaccordance with the guidelines of Animal Care and UseCommittee of TheUniversity of TexasMDAndersonCancerCenter, and themice weremaintained in specific pathogen-free conditions. The facilities were approved by the Associ-ation for Assessment and Accreditation of Laboratory Ani-mal Care and met all current regulations and standards oftheU.S.Departments of Agriculture andHealth andHumanServices and theNIH. The orthotopic injection of pancreaticcancer cells was conducted as described previously (25).Briefly, the mice were anesthetized with a 1.5% isoflurane–
air mixture. A small incision in the left abdominal flankwasmade, and the spleen was exteriorized. Tumor cells (1.0 �106 cells in 50 mL of HBSS) were injected subcapsularly in aregion of the pancreas just beneath the spleen. A 30-gaugeneedle, 1-mL disposable syringe, and calibrated, push but-ton–controlled dispensing device (Hamilton Syringe) wasused to inject the tumor cell suspension. A successfulsubcapsular intrapancreatic injection of tumor cells wasidentified by the appearance of a fluid bleb without intra-peritoneal leakage. To prevent such leakage, a cotton swabwas held over the injection site for 1 minute. One layer ofthe abdominal wound was closed with wound clips (Auto-clip; Clay Adams). The animals tolerated the surgical pro-cedure well and no anesthesia-related deaths occurred. Allmice were weighed weekly and observed for tumor growth.Seven days after injection of tumor cells, tumor-bearingmice were randomly assigned (n ¼ 5 per group) to receivethe following: IMO 1 mg/kg intraperitoneally 3 times aweek for 4weeks; cetuximab 10mg/kg intraperitoneal twicea week for 3weeks; the combination of these agents on days7 to11, 14 to 18, and21 to 25, continuingonly IMOondays28 to 32, or vehicles only as control. Diseasewas consideredbulky when the tumor burden was prominent in themouseabdomen (tumor volume �2.0 cm3). When at least 3 of5 mice per treatment group presented a bulky disease, themedian survival duration was considered achieved for thatgroup. At themedian survival duration of the control group,the tumor growth in mice of all groups was evaluated usingthe bioluminescence emitted by the tumor cells. Biolumi-nescence imaging was conducted using a cryogenicallycooled IVIS 100 imaging system coupled to a data acqui-sition computer running Living Image software (Xenogen).The mice were euthanized by CO2 inhalation when evi-dence of advanced bulky disease was present, which wasconsidered the day of death for survival evaluation.
Statistical analysisThe results of in vitro experiments were analyzed for
statistical significance of differences by the Student t testor nonlinear regression analysis and were expressed asmeans, SDs, and 95% CIs for at least 3 independent experi-ments conducted in triplicates. The statistical significance ofdifferences in tumor growth was determined by 1-wayANOVA and the Dunnett multiple comparison posttest,whereas the significance of the differences in survival wasevaluated by a log-rank test. All reported P values were 2sided. All analyses were conducted with the BMDP NewSystem Statistical Package version 1.0 for Microsoft Win-dows (BMDP Statistical Software).
Results
Characterizationofapanelof colorectal andpancreaticcancer cell lines
For this study, we selected a panel of colorectal andpancreatic human cancer cell lines, whose K-Ras status isreported in the catalogue of somatic mutations in cancerprovided by Sanger Institute (�COSMIC database,
TLR9 Agonist in K-Ras Mutant Colon and Pancreatic Cancers
www.aacrjournals.org Clin Cancer Res; 17(20) October 15, 2011 6533
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

Catalogue of Somatic Mutations In Cancer, http://www.sanger.ac.uk/) and in previous studies (26–28). Particular-ly, SW48 and T3M4 cells have a wild-type K-Ras gene. BothLS174T and MIA PaCa-2 exhibit K-Ras mutations(Gly12Asp for LS174T cells; Gly12Cys for MIA PaCa-2cells). GEO cells were previously reported as positive fora K-Ras mutation (Gly12Ala; ref. 27), as we verified (datanot shown).
The expression of EGFR and TLR9 proteins in colorectaland pancreatic cell lines was evaluated through Westernblot analysis (Supplementary Fig. S1A). GEO, SW48, andLS174T human colon cancer cells exhibit a differentialexpression of EGFR, higher in SW48 than in GEO andLS174T cells. Among the pancreatic cancer cells, T3M4showed higher EGFR levels than MIA PaCa-2 cells. Colo-rectal and pancreatic cancer cells expressed different levelsof TLR9, resulting higher in GEO than in SW48 and LS174Tcells, and similar in T3M4 and MIA PaCa-2 cells.
Then, we characterized the sensitivity of these differentcell lines to the anti-EGFR drugs cetuximab and erlotinib orthe TLR9 agonist IMO (Supplementary Fig. S1B), as mea-sured through soft agar assays. GEO and SW48 colon cancercells were responsive to both cetuximab (IC50 ¼ 7 and35 nmol/L, respectively) and erlotinib (IC50 ¼ 0.5 and 1mmol/L, respectively). Conversely, K-Ras mutant LS174Tcolon cancer cells were resistant to these agents (IC50 >
140 nmol/L for cetuximab and >5 mmol/L for erlotinib).Whereas K-Ras wild-type T3M4 pancreatic cancer cellsshowed a marked sensitivity to both cetuximab (IC50 ¼7 nmol/L) and erlotinib (IC50 ¼ 1 mmol/L), K-Ras mutantMIA PaCa-2 cells were only modestly inhibited even whenhighdoseswere used (IC50¼140nmol/L for cetuximaband>5mmol/L for erlotinib). IMOmoderately inhibited the softagar growth of all the cell lines, independently of TLR9expression levels andK-Ras status (Supplementary Fig. S1B).
IMO restores sensitivity of K-Rasmutant colorectal andpancreatic cancer cell lines to cetuximab
Because we have previously shown that IMO is able tointerfere with EGFR-related signaling and has a synergisticantitumor effectwith EGFR inhibitors (20), in this study,weevaluated the ability of IMO to restore cetuximab sensitivityin K-Ras mutant, cetuximab-resistant cancer cells. We firsttested the combination of IMO and cetuximab on soft agargrowth of colorectal and pancreatic cancer cells. As shownin Fig. 1, IMO significantly potentiated the activity ofcetuximab in all of the cell lines studied (cetuximab IC50:GEO plus IMO, 8.21E-10, 95% CI ¼ 3.3038e-010 to2.0416e-009 vs. GEO control, 8.93E-09, 95% CI ¼6.1600e-009 to 1.2948e-008; SW48 plus IMO, 3.24E-09,95% CI ¼ 1.4267e-009 to 7.3485e-009 vs. SW48 control,3.29E-08, 95% CI ¼ 2.4814e-008 to 4.3536e-008; LS174T
GEO Soft agar growth assaySW48
LS174T
Cetuximab (log mol/L)
Per
cen
tag
e o
f g
row
th
Per
cen
tag
e o
f g
row
th
Per
cen
tag
e o
f g
row
th
Control
Control
100
50
0
100
50
0
100
50
0–9 –8 –7 –6
Cetuximab (log mol/L)
Control –9 –8 –7 –6
Cetuximab (log mol/L)
Control –9 –8 –7 –6
IMO
T3M4 MIA PaCa-2
Cetuximab (log mol/L)
Per
cen
tag
e o
f g
row
th
Control
Control
100
50
0–9 –8 –7 –6
IMO
Cetuximab (log mol/L)
Per
cen
tag
e o
f g
row
th
Control
Control
100
50
0–9 –8 –7 –6
IMO
ControlIMO
ControlIMO
Figure 1. Effect of IMO on soft agar growth of colorectal and pancreatic cancer cell lines treated with cetuximab. Percentage of growth of cells treated withincreasing doses of cetuximab (log scale) in presence or absence of IMO (1 mmol/L), asmeasured by the soft agar assay. Cells were treated with the indicatedconcentrations of the drugs each day for 3 consecutive days. Colonies were counted after 10 to 14 days using the Quantity One Software Package (Bio-Rad).Data representmeans and 95%CIs of 3 independent experiments conducted in triplicate. Curveswere fitted by nonlinear regression analysis. Bars, 95%CIs.
Rosa et al.
Clin Cancer Res; 17(20) October 15, 2011 Clinical Cancer Research6534
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

plus IMO, 4.08E-08, 95%CI¼ 2.6385e-008 to 6.2935e-008vs. LS174T control, 6.77E-07, 95% CI ¼ 2.0322e-007 to2.2525e-006; T3M4 plus IMO, 9.91E-10, 95% CI ¼5.3450e-010 to 1.8375e-009 vs. T3M4 control, 1.58E-08,95% CI ¼ 9.7619e-009 to 2.5507e-008; MIA PaCa-2 plusIMO, 3.07E-08, 95% CI ¼ 1.7744e-008 to 5.2965e-008 vs.MIA PaCa-2 control, 2.45E-07, 95% CI ¼ 1.1057e-007 to5.4341e-007; all P < 0.0001). In particular, at higher andmore clinically relevant doses of cetuximab, this effect wasmore relevant in K-Ras mutant, cetuximab-resistant cells(percentage of cell growth after treatment with cetuximab140 nmol/L: GEO plus IMO, mean ¼ 11.03%, 95% CI ¼3.083–18.98 vs. GEO control, mean ¼ 22.03%, 95% CI ¼15.94–28.13, P ¼ 0.0091; SW48 plus IMO, mean ¼23.03%, 95% CI ¼ 17.14–28.92 vs. SW48 control, mean¼ 33.03%, 95% CI ¼ 25.76–40.30, P ¼ 0.01; LS174T plusIMO, mean ¼ 38.97%, 95% CI ¼ 30.22–47.72 vs. LS174Tcontrol, mean ¼ 64.97%, 95% CI ¼ 53.75–76.19, P ¼0.0014; T3M4 plus IMO, mean ¼ 15.97%, 95% CI ¼8.538–23.40 vs. T3M4 control, mean ¼ 31.00%, 95% CI¼ 24.79–37.21, P¼ 0.0026;MIA PaCa-2 plus IMO,mean¼30.00%, 95% CI ¼ 19.49–40.51 vs. MIA PaCa-2 control,mean¼ 54.97%, 95%CI¼ 42.49–67.44, P¼ 0.0028; Fig. 1and Supplementary Fig. S2).Moreover, as shown in Supplementary Fig. S3, results of
MTT assays showed that the combined treatment with IMOand cetuximab efficiently inhibited survival of all the colonand pancreatic cancer cells, producing an additive effect.More importantly, IMO partially recovered (by about 25%)the antiproliferative effect of cetuximab in both K-Rasmutant LS174T and MIA PaCa-2 cells.We then analyzed the effect of those treatments on the
activation of EGFR-dependent signal transduction. IMOwas able to reduce EGFR phosphorylation, with down-stream inhibition of Akt and mitogen—activated proteinkinase (MAPK), in most cell lines. In the cetuximab-sensi-tive cells, cetuximabwas able to counteract the effects of EGFstimulation, reducing phosphorylation/activation of EGFR,Akt, and MAPK. Conversely, in the cetuximab-resistant
LS174T and MIA PaCa-2 cells, cetuximab reduced theEGF-induced EGFR phosphorylation but was unable tointerfere with phosphorylation of MAPK, the main Rasdownstream signal transducer. A variable effect of cetux-imab on Akt phosphorylation was detected on these celllines. The combination IMO plus cetuximab inhibitedEGFR-dependent signaling in the K-Rasmutant LS174T andMIA PaCa-2 cells, producing a total suppression of MAPKphosphorylation (Fig. 2).
These data show that IMO partly restores cetuximabsensitivity of K-Rasmutant colorectal and pancreatic cancercell lines, producing a significant inhibition of cell growthand survival and a strong interference with EGFR-depen-dent signal transduction when used in combination withcetuximab.
IMO modulates a functional interaction between TLR9and EGFR in colorectal and pancreatic cancer cells
We investigatedwhether the inhibition of EGFR signalingby IMO could be related to a modulation of the interactionbetween TLR9 and EGFR, similar to what we previouslydescribed in breast cancer cells (19). To this aim, we con-ducted a time-course experiment over a 25-hour window.Cells cultured in complete medium were treated with IMO(1 mmol/L) at 0 and 24 hours, and cell lysates were obtainedat 1, 24, and 25 hours. We immunoprecipitated GEO,SW48, LS174T, and MIA PaCa-2 cell lysates with an anti-TLR9 antibody and blotted with an anti-EGFR antibody. Asnegative control, lysis buffer was mixed with monoclonalanti-TLR9 antibody. In both cetuximab-sensitive GEO andSW48 and cetuximab-resistant LS174T and MIA PaCa-2cells, TLR9 coimmunoprecipitated with EGFR. IMO mod-ulated this interaction, which was reduced 1 hour after eachIMO treatment, and completely restored within 24 hoursfrom IMO treatment (Fig. 3). The kinetics of this modula-tion is consistent with the EGFR signaling inhibitioninduced by IMO, suggesting that the disruption of TLR9/EGFR interaction may play a critical role in the ability ofIMO to interfere with EGFR-dependent signal transduction.
Figure 2. Effect of IMO on signaltransduction of colorectal andpancreatic cancer cell lines treatedwith cetuximab. Western blottingon total cell lysates from cellscultured in serum-free medium,treated for 1 hour with IMO(1 mmol/L), cetuximab (140 nmol/L),or their combination and stimulatedfor 15 minutes with EGF (50 ng/mL)before protein extraction.
TLR9 Agonist in K-Ras Mutant Colon and Pancreatic Cancers
www.aacrjournals.org Clin Cancer Res; 17(20) October 15, 2011 6535
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

Combination of cetuximab with IMO inhibits tumorgrowth in athymic mice bearing colon or pancreaticcancer xenografts
To confirm the antitumor effect of the combination IMOplus cetuximab also in clinically relevant in vivomodels ofK-Ras mutant tumors, we used both subcutaneous LS174Tcolorectal cancer and AsPC1/GLT pancreatic cancer ortho-topic xenograft models.
In LS174T colorectal cancer model, on day 42—6 weeksafter tumor cell injection—all of the mice in the controlgroup reached themaximum allowed tumor size of about 2cm3. Tumors treated with cetuximab initially responded tothis agent but then resumed an exponential growth rate. Theaddition of IMO to cetuximab caused a potent and long-lasting cooperative antitumor activity with a significanttumor growth inhibition (median tumor size < 0.7 cm3)at the end of the experiment on day 105 (Fig. 4A). Accord-ingly, the mice treated with the combination of IMO andcetuximab showed a statistically significantly prolongedmedian survival duration compared with the mice treatedwith cetuximab as single agent (IMO plus cetuximab vs.cetuximab, median survival: 107 vs. 66.5 days, HR ¼0.3054, 95% CI ¼ 0.08261–0.8024, P ¼ 0.0193) or withthe controls (IMO plus cetuximab vs. control, mediansurvival: 107 vs. 31 days, HR ¼ 0.1459, 95% CI ¼0.01068–0.1619, P < 0.0001; Fig. 4B).
In theAsPC1/GLTpancreatic cancermodel, at themediansurvival of mice in the control group (49 days), tumor
growth was evaluated in all groups of mice on the basis ofthe bioluminescence emitted by tumor cells. Cetuximabstill produced a 50% tumor growth inhibition, likely due toits ADCC activity, whereas IMO was much more effective,reducing the tumor burden by about 85%. This antitumoreffect was further increased by the combined treatment ofIMO and cetuximab (Fig. 4C and D). Accordingly, thepancreatic cancer–bearing mice treated with the combinedtreatment showed a statistically significantly prolongedmedian survival duration compared with the mice treatedwith cetuximab as single agent (IMO plus cetuximab vs.cetuximab, median survival: 70 vs. 55 days, HR ¼ 0.3372,95% CI ¼ 0.04299–0.9846, P ¼ 0.0478) or with the con-trols (IMO plus cetuximab vs. control, median survival: 70vs. 49 days, HR¼ 0.2287, 95%CI¼ 0.007088–0.3325, P¼0.0021; Fig. 4E). The treatments were well tolerated, and noweight loss or other signs of acute or delayed toxicity wereobserved.
IMO restores sensitivity to cetuximab in colorectalcancer cells knocked-in for mutant K-Ras gene
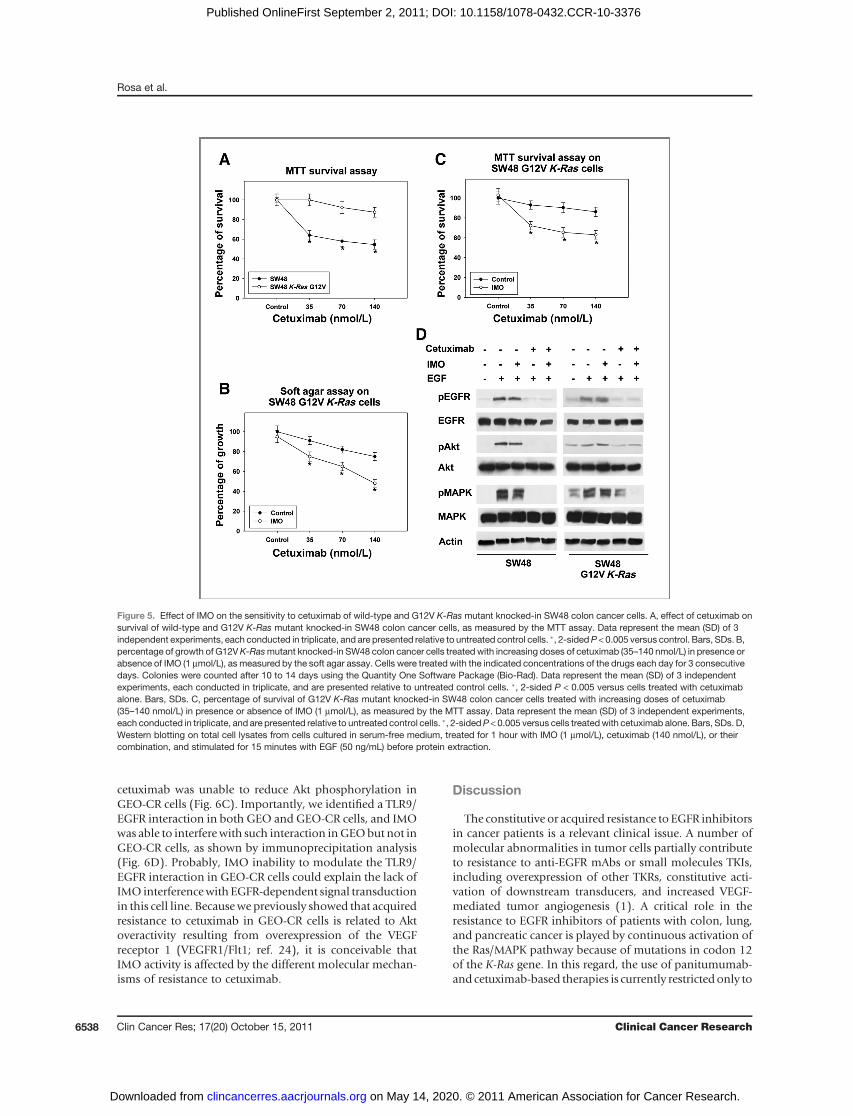
To further confirm the activity of IMO on K-Rasmutantcancer cells, we used isogenic wild-type and Gly12ValK-Ras mutant SW48 colon cancer cells obtained throughhomologous recombination. This technology allowsto introduce individual or multiple cancer mutationsinto the genome of cancer cells; as a result, the hetero-zygous-mutated genes are expressed under their endoge-nous promoters, thus closely recapitulating the lesionsobserved in human tumors (29). Accordingly to theirK-Ras status, the mutant SW48 cells were resistant tocetuximab compared with their wild-type control cells(Fig. 5A). However, combining IMO with increasingdoses of cetuximab significantly reversed this resistance,although not to the same level as that of the parental cellline, both in soft agar and MTT assays (Fig. 5B and C).Then, we compared the effects of IMO and cetuximab onthe activation of EGFR-dependent signal transduction inwild-type and K-Rasmutant SW48 cells. In the cetuximab-sensitive SW48 K-Ras wild-type parental cells, cetuximabefficiently inhibited EGFR signaling, suppressing phos-phorylation/activation of EGFR, Akt, and MAPK. Con-versely, in the cetuximab-resistant K-Ras mutant SW48cells, cetuximab reduced the EGF-induced EGFR phos-phorylation but was unable to inhibit MAPK phosphory-lation and slightly reduced Akt phosphorylation.Although IMO alone did not reduce pMAPK in bothwild-type and K-Ras mutant SW48 cells, a complete sup-pression of MAPK phosphorylation was achieved by thecombined treatment with IMO plus cetuximab in Gly12-Val K-Ras mutant SW48 cells (Fig. 5D). We analyzedwhether the combination of IMO and cetuximab couldinterfere with activation of K-Ras. The results (shown inSupplementary Fig. S3) seem to rule out this hypothesis.
These data confirm that IMO is able to restore cetuximabsensitivity in Gly12Val K-Rasmutant colorectal cancer cells,but further studies are required to fully elucidate the mech-anism by which this is achieved.
Figure 3. Interaction between TLR9 and EGFR in colorectal andpancreatic cancer cell lines. A, representative diagram of the time-courseexperiment, investigating the interaction between TLR9 and EGFR incolorectal and pancreatic cancer cell lines over a 25-hour window. Cellscultured in complete medium were treated with IMO at 0 and 24 hours.Cell lysateswere obtained at 1, 24, and 25 hours. B, immunoprecipitation(IP) using an anti-TLR9 antibody and blotting with an anti-EGFR antibodyon GEO, SW48, and LS174T colon and MIA PaCa-2 pancreatic cancercells cultured in complete medium and treated with IMO (1 mmol/L) atdifferent times. As negative control, lysis bufferwasmixedwith anti-TLR9antibody. WB, Western blotting.
Rosa et al.
Clin Cancer Res; 17(20) October 15, 2011 Clinical Cancer Research6536
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

IMO is ineffective in a colorectal cancer model ofacquired resistance to cetuximab due to AktoveractivityWe previously showed that IMO is unable to inhibit in
vivo growth in GEO-CR (cetuximab resistant) tumors, acolon cancer model of acquired resistance to cetuximab(20). In the present study, we found that combining IMOwith increasing doses of cetuximab did not reverse cetux-
imab resistance in GEO-CR cells, both in soft agar and MTTassays (Fig. 6A and B). Moreover, whereas IMO inhibitedEGFR-dependent signal transduction in GEO original cells,it was ineffective in reducing phosphorylation/activation ofEGFR signal transducers, such as Akt andMAPK, inGEO-CRcells. As previously showed (20), treatment of GEO-CR cellswith cetuximab reduced EGFR and MAPK phosphorylationbut not Akt phosphorylation. The combination IMO plus
Tum
or
volu
me
(ph
oto
n/s
ec)
Per
cen
tag
e o
f su
rviv
al
C
D
E
Days
8 × 106
7 × 106
6 × 106
5 × 106
4 × 106
3 × 106
2 × 106
1 × 106
0
100
50
0
Cetuximab
Control
IMO + cetuximab
IMO
Cetux
imab
Contro
l
IMO +
cetu
ximabIM
O
*
Cetuximab
Control
IMO + cetuximab
IMO
AsPC1 orthotopic
0 25 50 75
A
Tum
or
volu
me
(cm
3 )P
erce
nta
ge
of
surv
ival
B
LS174T s.c.
Days
Days
Treatment
2.5
2.0
1.5
1.0
0.5
0.0
100
50
0
Cetuximab
Control
IMO + cetuximab
IMO
Cetuximab
Control
IMO + cetuximab
IMO
0
0 25 50 75 100 125
7 14 21 28 35 42 49 56 63 70 77 84 91 98105112
Figure 4. Effect of IMOandcetuximab in combination on tumor growth and survival of athymicmice bearing subcutaneous or orthotopic tumors. A, after 7daysfollowing subcutaneous injection of LS174T colon cancer cell, mice were randomized (n¼ 10 per group) to receive IMO, cetuximab, or their combination, asdescribed in the Methods section. The Student t test was used to compare tumor sizes among different treatment groups at the median survival time of thecontrol group (31 days). They resulted statistically significant for the combination versus single agents (P < 0.0001). Bars, SDs. B, median survival rateresulted statistically significant for IMOandcetuximab versus cetuximab and versus control (log-rank test). C,mice bearingorthotopicAsPC1/GLTpancreatictumors were randomly allocated to be treated with IMO, cetuximab, or their combination, according to the schedule described in the Methods section. At themedian survival time of the control group (49 days), tumor growth in mice of all groups was evaluated on the basis of the bioluminescence emitted bytumor cells, as described in theMethods section. D, tumor volumewasquantified as the sumof all detected photonswithin the region of the tumor per second.�, P < 0.05 combination versus control by 1-way ANOVA and the Dunnett multiple comparison posttest. Bars, SDs. E, mice were sacrificed by CO2 inhalationwhen evidence of advanced bulky disease was present. The day of sacrifice was considered the day of death for survival evaluation. Median survivalrate resulted statistically significant for IMO and cetuximab versus cetuximab and versus control (log-rank test).
TLR9 Agonist in K-Ras Mutant Colon and Pancreatic Cancers
www.aacrjournals.org Clin Cancer Res; 17(20) October 15, 2011 6537
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376
cetuximab was unable to reduce Akt phosphorylation inGEO-CR cells (Fig. 6C). Importantly, we identified a TLR9/EGFR interaction in both GEO and GEO-CR cells, and IMOwas able to interferewith such interaction inGEObut not inGEO-CR cells, as shown by immunoprecipitation analysis(Fig. 6D). Probably, IMO inability to modulate the TLR9/EGFR interaction in GEO-CR cells could explain the lack ofIMO interferencewith EGFR-dependent signal transductionin this cell line. Becausewe previously showed that acquiredresistance to cetuximab in GEO-CR cells is related to Aktoveractivity resulting from overexpression of the VEGFreceptor 1 (VEGFR1/Flt1; ref. 24), it is conceivable thatIMO activity is affected by the different molecular mechan-isms of resistance to cetuximab.
Discussion
The constitutive or acquired resistance to EGFR inhibitorsin cancer patients is a relevant clinical issue. A number ofmolecular abnormalities in tumor cells partially contributeto resistance to anti-EGFR mAbs or small molecules TKIs,including overexpression of other TKRs, constitutive acti-vation of downstream transducers, and increased VEGF-mediated tumor angiogenesis (1). A critical role in theresistance to EGFR inhibitors of patients with colon, lung,and pancreatic cancer is played by continuous activation ofthe Ras/MAPK pathway because of mutations in codon 12of the K-Ras gene. In this regard, the use of panitumumab-and cetuximab-based therapies is currently restrictedonly to
Figure 5. Effect of IMO on the sensitivity to cetuximab of wild-type and G12V K-Ras mutant knocked-in SW48 colon cancer cells. A, effect of cetuximab onsurvival of wild-type and G12V K-Ras mutant knocked-in SW48 colon cancer cells, as measured by the MTT assay. Data represent the mean (SD) of 3independent experiments, each conducted in triplicate, and are presented relative to untreated control cells. �, 2-sidedP < 0.005 versus control. Bars, SDs. B,percentage of growth of G12VK-Rasmutant knocked-in SW48 colon cancer cells treated with increasing doses of cetuximab (35–140 nmol/L) in presence orabsence of IMO (1 mmol/L), as measured by the soft agar assay. Cells were treated with the indicated concentrations of the drugs each day for 3 consecutivedays. Colonies were counted after 10 to 14 days using the Quantity One Software Package (Bio-Rad). Data represent the mean (SD) of 3 independentexperiments, each conducted in triplicate, and are presented relative to untreated control cells. �, 2-sided P < 0.005 versus cells treated with cetuximabalone. Bars, SDs. C, percentage of survival of G12V K-Ras mutant knocked-in SW48 colon cancer cells treated with increasing doses of cetuximab(35–140 nmol/L) in presence or absence of IMO (1 mmol/L), as measured by the MTT assay. Data represent the mean (SD) of 3 independent experiments,each conducted in triplicate, and are presented relative to untreated control cells. �, 2-sidedP < 0.005 versus cells treatedwith cetuximab alone. Bars, SDs. D,Western blotting on total cell lysates from cells cultured in serum-free medium, treated for 1 hour with IMO (1 mmol/L), cetuximab (140 nmol/L), or theircombination, and stimulated for 15 minutes with EGF (50 ng/mL) before protein extraction.
Rosa et al.
Clin Cancer Res; 17(20) October 15, 2011 Clinical Cancer Research6538
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

patients with wild-type K-Ras metastatic colorectal cancer(7). Therefore, there is an unmet need to identify andclinically validate novel therapeutic strategies based oncombination of different targeted agents able to hinder theoccurrence of resistance to EGFR antagonists.In this study, we investigated the antitumor effect of the
TLR9 agonist IMO as single agent or in combination withcetuximab in colorectal and pancreatic cancer models har-boring the oncogenic K-Rasmutations conferring resistanceto EGFR inhibitors.We found a correlation between K-Ras status and sensi-
tivity to EGFR antagonists in all the cell lines tested, with theexception of the human GEO colorectal cancer cells. Eventhough these cells bear a low-frequency K-Ras mutation(Gly12Ala), they maintain a measurable sensitivity to bothcetuximab and erlotinib. This is in agreement with theevidence that different K-Ras mutations have different rel-evance in mediating resistance to EGFR inhibitors. In thisregard, a small number of patients carrying K-Ras mutanttumors have been reported to respond to either cetuximabor panitumumab (23, 30–32).
We showed that IMO is able to restore sensitivity tocetuximab in K-Ras mutant cell lines. The combination ofIMO plus cetuximab produced a significant inhibition ofcell growth and survival and a strong interference withEGFR-dependent signal transduction in K-Rasmutant colo-rectal and pancreatic cancer cells, totally suppressing phos-phorylation/activation ofMAPK, themain signal transducerdownstream to Ras. We have reported that the ability ofIMO to interfere with EGFR-dependent signaling is associ-ated with themodulation of the structural/functional inter-action between TLR9 and EGFR (19). In this study, wedocument that the kinetics of this modulation is consistentwith the ability of IMO to inhibit EGFR signaling anddisrupt the interaction between TLR9 and EGFR. Cross-talksbetween EGFR and TLR signaling pathways were described(33–35), and other studies hypothesized the existence of apotential TLR9 coreceptor at the plasma membrane able tointeract withCpGs before the activation of endosomal TLR9(36).Our resultsmay suggest a role for EGFR in this process,but further investigations dissecting the relationshipsbetween EGFR and TLR9 are needed.
Figure 6. Effect of IMOon the sensitivity to cetuximabofGEO-CRcolon cancer cells. A, percentage of growth ofGEO-CR cells treatedwith increasing doses ofcetuximab (7–140 nmol/L) in presence or absence of IMO (1 mmol/L), asmeasured by the soft agar assay. Cells were treatedwith the indicated concentrationsof the drugs each day for 3 consecutive days. Colonies were counted after 10 to 14 days using the Quantity One Software Package (Bio-Rad). Data representthe mean (SD) of 3 independent experiments, each conducted in triplicate, and are presented relative to untreated control cells. Bars, SDs. B, percentage ofsurvival of GEO-CRcells treatedwith increasing doses of cetuximab (7–140nmol/L) in presence or absence of IMO (1 mmol/L), asmeasured by theMTT assay.Data represent the mean (SD) of 3 independent experiments, each conducted in triplicate, and are presented relative to untreated control cells. Bars, SDs. C,Western blotting on total cell lysates from GEO and GEO-CR cells cultured in serum-free medium, treated for 1 hour with IMO (1 mmol/L), cetuximab(140nmol/L), or their combination, andstimulated for 15minuteswithEGF (50ng/mL) beforeprotein extraction.D, immunoprecipitation (IP) usingan anti-TLR9antibody and blotting (WB) with an anti-EGFRantibody onGEOandGEO-CR cells cultured in completemedium and treatedwith IMO (1mmol/L) for 1 hour. Asnegative control, lysis buffer was mixed with anti-TLR9 antibody.
TLR9 Agonist in K-Ras Mutant Colon and Pancreatic Cancers
www.aacrjournals.org Clin Cancer Res; 17(20) October 15, 2011 6539
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

The antitumor effect of IMO on K-Ras mutant tumorswas confirmed also in vivo, in subcutaneous LS174Tcolorectal and orthotopic AsPC1 pancreatic cancermodels. In both models, cetuximab still produced asignificant tumor growth inhibition despite the intrinsicresistance to this agent in vitro. This effect is likely due toits ADCC activity, as we previously reported (19). Theantitumor activity of IMO was particularly evident inAsPC1 pancreatic cancer orthotopic model, with a reduc-tion of the tumor burden of about 85%. We hypo-thesized that this event may depend on the capabilityof IMO to interfere with different cell populations oftumor microenvironment, whose contribution to tumorgrowth could be higher in the orthotopic than in thesubcutaneous xenograft (37). The combination of IMOplus cetuximab caused a potent and long-lasting cooper-ative antitumor activity, with a significant inhibition oftumor growth and prolongation of mice survival.
Finally, we confirmed the ability of IMO to overcomecetuximab resistance associated with K-Ras mutations byusing targeted homologous recombination to introduce(knock-in) Gly12Val K-Ras–mutated allele in human coloncancer cells. This technology allows the construction ofmodels able to accurately recapitulate the genetic alterationspresent in human cancers, useful to identify genotype andtumor-specific pharmacologic responses (29, 38). WhileIMO alone did not produce any effect on EGFR signalingin this model, this agent was able to restore sensitivity tocetuximab, producing a significant inhibition of cell growthand survival and a strong interference with EGFR-depen-dent signal transduction when used in combination withcetuximab.
Taken together, our results show for the first time thatIMO is effective in restoring cetuximab antitumor activity inK-Ras mutant colon and pancreatic cancers. This effect islikely due to multiple mechanisms of action, includinginterference with EGFR signaling by modulation of the
TLR9/EGFR interaction, and enhancement of cetuximabADCC.
The combination of IMO plus cetuximab is currentlyunder clinical investigation, and the results of the presentstudy suggest that the combination with IMO could allowthe use of cetuximab also in a set of patients with K-Rasmutant tumors, where this agent would be otherwiseineffective.
Disclosure of Potential Conflicts of Interest
S. Agrawal has employment (other than primary affiliation, e.g., consult-ing) and G. Tortora has commercial research grant. No potential conflicts ofinterest were disclosed by the other authors.
Acknowledgments
Human T3M4 and MIA PaCa-2 pancreatic carcinoma cell lines werekindly provided by Prof. A. Scarpa (Department of Pathology and Diag-nostics, University of Verona).
Grant Support
This study was supported in part by Associazione Italiana per la Ricercasul Cancro (AIRC), Ministero della Salute, Ministero dell’Universit�a edella Ricerca (PRIN), and Regione Campania grants to G. Tortora. Thiswork was also supported in part by AIRC (Start-Up grant no. 10129),American-Italian Cancer Foundation, and The Sass Foundation for Med-ical Research grants to D. Melisi. V. Damiano was supported by afellowship from AIRC. This study was supported partly also by grantsfrom Italian AIRC IG 2009-2011 (A. Bardelli), AIRC "2010 SpecialProgram Molecular Clinical Oncology 5 � 1000" Project no 9970 (A.Bardelli), Regione Piemonte (F.D. Nicolantonio and A. Bardelli), ItalianMinistry of University and Research (A. Bardelli), European UnionSeventh Framework Programme grant agreement 259015 (A. Bardelli),FPRC, Fondazione Piemontese per la Ricerca sul Cancro Onlus (F.D.Nicolantonio and A. Bardelli).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received December 21, 2010; revised August 9, 2011; accepted August 11,2011; published OnlineFirst September 2, 2011.
References1. Ciardiello F, TortoraG. EGFR antagonists in cancer treatment. NEngl J
Med 2008;358:1160–74.2. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A,
et al. Cetuximab monotherapy and cetuximab plus irinotecan in irino-tecan-refractory metastatic colorectal cancer. N Engl J Med2004;351:337–45.
3. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al.Erlotinib plus gemcitabinecomparedwithgemcitabinealone in patientswith advancedpancreatic cancer: a phase III trial of theNationalCancerInstitute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
4. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR,Makhson A, et al. Cetuximab and chemotherapy as initial treatmentfor metastatic colorectal cancer. N Engl J Med 2009;360:1408–17.
5. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B,et al. Open-label phase III trial of panitumumab plus best supportivecare compared with best supportive care alone in patients withchemotherapy-refractory metastatic colorectal cancer. J Clin Oncol2007;25:1658–64.
6. Morgillo F, Bareschino MA, Bianco R, Tortora G, Ciardiello F. Primaryand acquired resistance to anti-EGFR targeted drugs in cancer ther-apy. Differentiation 2007;75:788–99.
7. Bardelli A, Siena S. Molecular mechanisms of resistance to cetux-imab and panitumumab in colorectal cancer. J Clin Oncol2010;28:1254–61.
8. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al.Core signaling pathways in human pancreatic cancers revealed byglobal genomic analyses. Science 2008;321:1801–6.
9. MalumbresM, BarbacidM. RASoncogenes: the first 30 years. Nat RevCancer 2003;3:459–65.
10. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, deBraud F, et al. Fluorouracil, leucovorin, and oxaliplatinwith andwithoutcetuximab in the first-line treatment of metastatic colorectal cancer. JClin Oncol 2009;27:663–71.
11. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, LiuDD, et al. KRAS mutation is an important predictor of resistance totherapy with epidermal growth factor receptor tyrosine kinaseinhibitors in non-small-cell lung cancer. Clin Cancer Res 2007;13:2890–6.
12. Van Cutsem E, Rougier P, K€ohne C, Stroh C, Schlichting M, Boke-meyer C. A meta-analysis of the CRYSTAL and OPUS studies com-bining cetuximab with chemotherapy (CT) as 1st-line treatment forpatients (pts) with metastatic colorectal cancer (mCRC): Results
Rosa et al.
Clin Cancer Res; 17(20) October 15, 2011 Clinical Cancer Research6540
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

according to KRAS and BRAF mutation status. Eur J Cancer 2009;7:S345, abstr 6077.
13. Laurent-Puig P, Taieb J. Lessons from Tarceva in pancreatic cancer:where are we now, and how should future trials be designed inpancreatic cancer? Curr Opin Oncol 2008;20:454–8.
14. Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D,Kosmidis P, et al. Assessment of somatic k-RAS mutations as amechanism associated with resistance to EGFR-targeted agents: asystematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol2008;9:962–72.
15. Agrawal S,KandimallaER. Synthetic agonists of Toll-like receptors 7, 8and 9. Biochem Soc Trans 2007;35:1461–7.
16. Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. NatRev Drug Discov 2006;5:471–84.
17. Yu D, Kandimalla ER, Bhagat L, Tang JY, Cong Y, Tang J, et al.`Immunomers'–novel 30-30-linked CpG oligodeoxyribonucleotides aspotent immunomodulatory agents. Nucleic Acids Res 2002;30:4460–9.
18. Damiano V, Caputo R, Garofalo S, Bianco R, Rosa R, Merola G, et al.TLR9 agonist acts by different mechanisms synergizing with bevaci-zumab in sensitive and cetuximab-resistant colon cancer xenografts.Proc Natl Acad Sci U S A 2007;104:12468–73.
19. Damiano V, Garofalo S, Rosa R, Bianco R, Caputo R, Gelardi T, et al. Anovel toll-like receptor 9 agonist cooperates with trastuzumab intrastuzumab-resistant breast tumors through multiple mechanisms ofaction. Clin Cancer Res 2009;15:6921–30.
20. Damiano V, Caputo R, Bianco R, D'Armiento FP, Leonardi A, DePlacido S, et al. Novel toll-like receptor 9 agonist induces epidermalgrowth factor receptor (EGFR) inhibition and synergistic antitumoractivity with EGFR inhibitors. Clin Cancer Res 2006;12:577–83.
21. Kandimalla ER, Bhagat L, Wang D, Yu D, Zhu FG, Tang J, et al.Divergent synthetic nucleotide motif recognition pattern: design anddevelopment of potent immunomodulatory oligodeoxyribonucleotideagents with distinct cytokine induction profiles. Nucleic Acids Res2003;31:2393–400.
22. Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G,et al. LY2109761, a novel transforming growth factor {beta} receptortype I and type II dual inhibitor, as a therapeutic approach tosuppressing pancreatic cancer metastasis. Mol Cancer Ther2008;7:829–40.
23. De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D,Siena S, et al. Association of KRAS p.G13D mutation with outcome inpatients with chemotherapy-refractory metastatic colorectal cancertreated with cetuximab. JAMA 2010;304:1812–20.
24. Bianco R, Rosa R, Damiano V, Daniele G, Gelardi T, Garofalo S, et al.Vascular endothelial growth factor receptor-1 contributes to resis-tance to anti-epidermal growth factor receptor drugs in human cancercells. Clin Cancer Res 2008;14:5069–80.
25. Melisi D, Ossovskaya V, Zhu C, Rosa R, Ling J, Dougherty PM, et al.Oral poly(ADP-ribose) polymerase-1 inhibitor BSI-401 has antitu-mor activity and synergizes with oxaliplatin against pancreatic
cancer, preventing acute neurotoxicity. Clin Cancer Res 2009;15:6367–77.
26. Hamada K, Monnai M, Kawai K, Nishime C, Kito C, Miyazaki N, et al.Livermetastasismodels of colon cancer for evaluation of drug efficacyusing NOD/Shi-scid IL2Rgammanull (NOG) mice. Int J Oncol2008;32:153–9.
27. Paranavitana CM. Non-radioactive detection of K-ras mutations bynested allele specific PCR and oligonucleotide hybridization. Mol CellProbes 1998;12:309–15.
28. Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, et al.KRAS/BRAF mutation status and ERK1/2 activation as biomarkers forMEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther2009;8:834–43.
29. Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macar-ulla T, et al. Deregulation of the PI3K and KRAS signaling pathways inhuman cancer cells determines their response to everolimus. J ClinInvest 2010;120:2858–66.
30. Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, ZanonC,MoroniM,Veronese S, et al. Oncogenic activation of the RAS/RAF signalingpathway impairs the response ofmetastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res2007;67:2643–8.
31. Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D,Tebbutt NC, et al. K-ras mutations and benefit from cetuximab inadvanced colorectal cancer. N Engl J Med 2008;359:1757–65.
32. Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, et al.PTEN expression and KRAS mutations on primary tumors andmetastases in the prediction of benefit from cetuximab plus irino-tecan for patients with metastatic colorectal cancer. J Clin Oncol2009;27:2622–9.
33. Koff JL, ShaoMX, Ueki IF, Nadel JA.Multiple TLRs activate EGFR via asignaling cascade to produce innate immune responses in airwayepithelium. Am J Physiol Lung Cell Mol Physiol 2008;294:L1068–75.
34. Liu K, AndersonGP, Bozinovski S. DNA vector augments inflammationin epithelial cells via EGFR-dependent regulation of TLR4 and TLR2.Am J Respir Cell Mol Biol 2008;39:305–11.
35. Mikami F, Gu H, Jono H, Andalibi A, Kai H, Li JD. Epidermal growthfactor receptor acts as a negative regulator for bacterium nontypeableHaemophilus influenzae-induced Toll-like receptor 2 expression via anSrc-dependent p38 mitogen-activated protein kinase signaling path-way. J Biol Chem 2005;280:36185–94.
36. Sanjuan MA, Rao N, Lai KT, Gu Y, Sun S, Fuchs A, et al. CpG-induced tyrosine phosphorylation occurs via a TLR9-independentmechanism and is required for cytokine secretion. J Cell Biol 2006;172:1057–68.
37. Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary topredict therapy of transplantable tumors in mice. Cancer MetastasisRev 1998;17:279–84.
38. Di Nicolantonio F, Arena S, Gallicchio M, Zecchin D, Martini M, FlontaSE, et al. Replacement of normal with mutant alleles in the genome ofnormal human cells unveils mutation-specific drug responses. ProcNatl Acad Sci U S A 2008;105:20864–9.
TLR9 Agonist in K-Ras Mutant Colon and Pancreatic Cancers
www.aacrjournals.org Clin Cancer Res; 17(20) October 15, 2011 6541
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376

2011;17:6531-6541. Published OnlineFirst September 2, 2011.Clin Cancer Res Roberta Rosa, Davide Melisi, Vincenzo Damiano, et al.
Mutant Colorectal and Pancreatic Cancers Ras-KToll-like Receptor 9 Agonist IMO Cooperates with Cetuximab in
Updated version
10.1158/1078-0432.CCR-10-3376doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2011/09/02/1078-0432.CCR-10-3376.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/17/20/6531.full#ref-list-1
This article cites 38 articles, 18 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/17/20/6531.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/17/20/6531To request permission to re-use all or part of this article, use this link
on May 14, 2020. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 2, 2011; DOI: 10.1158/1078-0432.CCR-10-3376