the genetic composition and variability of …140351/fulltext01.pdf · the genetic composition and...
TRANSCRIPT

The Genetic Composition and Diversity of Francisella tularensis
Pär Larsson
Akademisk avhandling
som med vederbörligt tillstånd av rektorsämbetet vid Umeå Universitet för avläggande av medicine doktorsexamen i klinisk mikrobiologi med inriktning mot bakteriologi vid Medicinska fakulteten, framlägges till offentligt försvar vid Institutionen för Klinisk Mikrobiologi, sal E04 byggnad 6, torsdagen den 31 maj 2007, klockan 09.00. Avhandlingen kommer att försvaras på engelska. Fakultetsopponent: Dr. Andrew K Benson Department of Food Science & Technology University of Nebraska–Lincoln Lincoln, Nebraska USA
Department of Clinical Microbiology, Clinical Bacteriology Umeå University
Umeå 2007

Organization UMEÅ UNIVERSITY Department of Clinical Microbiology SE-901 87 Umeå, Sweden
Document type DOCTORAL DISSERTATION Date of publication May 2007
Author Pär Larsson Title The Genetic Composition and Diversity of Francisella tularensis Abstract Francisella tularensis is the causative agent of the debilitating, sometimes fatal zoonotic disease tularemia. Despite all F. tularensis bacteria having very similar genotypes and phenotypes, the disease varies significantly in severity depending on the subspecies of the infectious strain. To date, little information has been available on the genetic makeup of this pathogen, its evolution, and the genetic differences which characterize subspecific lineages. These are the main areas addressed in this thesis.
Using the F. tularensis subsp. tularensis SCHU S4 strain as a genetic reference, microarray-based comparative genomic hybridisations were used to investigate the differences in genomic composition of F. tularensis isolates. Overall, the strains analysed were very similar, matching the high degree of conservation previously observed at the sequence level. One striking finding was that subsp. mediasiatica was most similar to subsp. tularensis, despite their natural confinement to Central Asia and North America, respectively. Eight regions of difference (RDs) were found to characterize all European and American isolates of subsp. holarctica. One RD was highly polymorphic and, for the first time, allowed the development of an F. tularensis-specific PCR assay that discriminates between the four subspecies.
All RDs were found to have been the result of repeat-mediated excision. Therefore, a screening of loci flanked by direct repeats was undertaken, resulting in the identification of eight additional RDs. Data for the RDs along with a multiple locus sequence analysis suggested an evolutionary scenario for F. tularensis. This indicated a deep divergence of subsp. novicida and a deep divergence of the Japanese strains within subsp. holarctica. Since all virulence-attenuated strains investigated had deletions at two RDs, repeat-mediated excision may represent an important natural attenuating mechanism in F. tularensis.
A novel typing scheme for F. tularensis was evaluated, aiming to provide both a high resolution and support robust inference at deeper phylogenetic levels. The concept was based on a combination of MLVA (providing high resolution) and a class of insertion-deletion polymorphisms thought to evolve slowly (providing in-depth structural data). The data indicated that the insertion-deletion markers tested are relatively resistant to homoplasy, and that the statistical robustness could be significantly increased compared to the use of MLVA alone.
Finally, the genomic sequence of the highly virulent F. tularensis strain SCHU S4 was determined and analysed. Evidenced by numerous pseudogenes and disrupted metabolic pathways, the bacterium appears to be undergoing a genome reduction process whereby a large proportion of the genetic capacity gradually is lost. It is likely that F. tularensis has irreversibly has evolved into an obligate host-dependent bacterium, incapable of a free-living existence. Unexpectedly, the bacterium was found to be devoid of common virulence mechanisms such as classic toxins, or type III and IV secretion systems. Instead, the virulence of this bacterium is probably largely the result of specific and unusual mechanisms. For example, genes found in the genome sequence suggest that it might possess a poly-γ-glutamate capsule similar to the gram-positive anthrax bacterium Bacillus anthracis. Two perfect copies of a virulence-associated locus, the Francisella pathogenicity island, were also found.
Key words tularemia, Francisella tularensis, genotyping, evolution, microarray, genome sequencing, virulence, genome reduction Language: English ISBN: 978-91-7264-288-1-X ISSN: 0346-6612 Number of pages: 51 + 4 papers


UMEÅ UNIVERSITY MEDICAL DISSERTATIONS
New Series No 1094 ISSN 0346-6612 ISBN 978-91-7264-288-1 From the Department of Clinical Microbiology,
Clinical Bacteriology, Umeå University, Umeå Sweden
The Genetic Composition and Diversity of Francisella tularensis
Pär Larsson
Umeå 2007

Copyright © 2007 Pär Larsson
ISBN 978-91-7264-288-1
Printed in Sweden by Solfjädern Offset AB
2007

I love deadlines. I like the whooshing sound they make as they fly by.
Douglas Adams



TABLE OF CONTENTS
ORIGINAL PAPERS IN THESIS ii SAMMANFATTNING iii ABBREVIATIONS v INTRODUCTION 1 BACKGROUND 2
Historical account 2 Tularemia 5 Taxonomy and geographical distribution 6 Evolution and population structure 10 Genomic typing 11 Genome analysis 13
AIMS OF THE THESIS 14 METHODOLOGICAL CONSIDERATIONS 15
Genome sequencing 15 Comparative genomic hybridisation 16
RESULTS AND DISCUSSION 18 Comparative genomic hybridisations reveals strong compositional similarity within F. tularensis (paper I) 18 Phylogeny and direct repeat-mediated deletions among F. tularensis isolates (paper II and unpublished work) 23 Indel markers for low-resolution genetic typing (paper III) 28 The F. tularensis SCHU S4 genome sequence (paper IV) 32
Reductive evolution 33 Insertion sequence elements 34 IS elements and evolution 35 Virulence mechanisms 37 Whole-genome phylogeny 39
CONCLUSIONS 41 ACKNOWLEDGEMENTS 43 REFERENCES 44
i

ORIGINAL PAPERS IN THESIS This thesis is based on the following papers, which will be referred to by their Roman numerals (I-IV). I. Broekhuijsen M*, Larsson P*, Johansson A, Byström M,
Eriksson U, Larsson E, Prior RG, Sjöstedt A, Titball RW, Forsman M. Genome-wide DNA microarray analysis of Francisella tularensis strains demonstrates extensive genetic conservation within the species but identifies regions that are unique to the highly virulent F. tularensis subsp. tularensis. Journal of Clinical Microbiology, 2003; 41: 2924-31.
II. Svensson K, Larsson P, Johansson D, Byström M,
Forsman M, Johansson A. Evolution of subspecies of Francisella tularensis. Journal of Bacteriology, 2005; 187:3903-8.
III. Larsson P, Svensson K, Karlsson L, Guala D, Granberg M,
Forsman M, Johansson A Analysis of insertion-deletion markers with canonical properties for safe and rapid DNA-based typing of Francisella tularensis. Submitted.
IV. Larsson P, Oyston PC, Chain P, Chu MC, Duffield M,
Fuxelius HH, Garcia E, Halltorp G, Johansson D, Isherwood KE, Karp PD, Larsson E, Liu Y, Michell S, Prior J, Prior R, Malfatti S, Sjöstedt A, Svensson K, Thompson N, Vergez L, Wagg JK, Wren BW, Lindler LE, Andersson SG, Forsman M, Titball RW. The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nature Genetics, 2005; 37: 153-9.
Reprints were made with the permission of the publishers. *Shared first author
ii

SAMMANFATTNING Francisella tularensis orsakar den zoonotiska sjukdomen harpest, också kallad tularemi. Alla harpestbakterier visar stora genotypiska och fenotypiska likheter men trots detta skiljer sig bakteriens underarter avsevärt i virulens. Om man smittats av en stam som tillhör underarten tularensis (även kallad typ A), kan utgången i värsta fall bli dödlig. Avhandlingen syftade till att öka vår förståelse för hur harpestbakterier är besläktade, vad som skiljer olika evolutionära grenar av bakterien, och till en bestämning av det genetiska innehållet i en av de mest virulenta stammarna av harpestbakterien. Ett urval av harpestisolat, där alla underarter fanns representerade, undersöktes med mikromatris-baserad hybridisering (eng. ”array-based comparative genomic hybridisations”). Resultaten visade en hög homogenitet vad gäller genetiska sammansättningen hos isolaten. Underarterna tularensis och mediasiatica visade störst likhet, trots att dessa organismers geografiska utbredningsområden är åtskilda och begränsade till Nordamerika och Centralasien. Underarten holarctica (även kallad typ B), som finns spridd över stora delar av norra halvklotet och även i Sverige, var genetiskt mer avvikande. De genetiska regioner som innehöll skillnader, eng. regions of difference (RD), befanns ha uppkommit genom en speciell mutationsmekanism, sk. homolog rekombination. Detta faktum utnyttjades för att identifiera ytterligare mutationsbenägna genetiska regioner. I alla virulens-försvagade stammar upptäcktes att gener tappats i två RD. En hypotes är därför att homolog rekombination kan vara en viktig mekanism för naturlig försvagning av harpeststammar. En modell för hur harpestbakterien kan ha evolverat utarbetades också. Flera analyser indikerade att underarten novicida var den tidigast avvikande evolutionära grenen och på motsvarande sätt indikerades även inom underarten holarctica att japanska stammar avvikit tidigt. Resultaten gav stöd åt tidigare indikationer på ett nära släktskap mellan underarterna mediasiatica och tularensis. Dessutom utvecklades inom avhandlingsarbetet en ny typningsmetod för bestämning av hur harpestisolat är besläktade. Konceptet baserades på sn samtidig analys av två olika markörtyper, vilket medgav både en god statistisk robusthet och hög en upplösning. Metoden möjliggör på så sätt
iii

en enkel och säker bestämining av både underartstillhörighet och stamidentitet i samma anlays. Slutligen bestämdes och analyserades genomsekvensen för den högvirulenta harpeststammen SCHU S4. Ett stort antal icke-funktionella gener hittades i arvsmassan liksom ofullständinga reaktionsvägar i bakteriens predikterade metabolism. Sannolikt pågår en evolutionär process hos F. tularensis där gener succesivt förloras. Som en konsekvens kan bakterien sannolikt inte längre leva fritt utan har blivit obligat värd-beroende i dess naturliga livscykel. Harpestbakterien upptäcktes dessutom sakna vanligt förekommande virulensmekanismer, till exempel kända toxiner samt typ III och typ IV sekretionssystem. Istället indikerade analyserna att ovanliga eller unika mekanismer orsakar bakteriens virulens. Gener hittades som indikerar att harpestbakterien kan ha en kapsel som liknar den som finns hos antraxbakterien Bacillus anthracis.
iv

ABBREVIATIONS bp base pair DNA deoxyribonucleic acid IS insertion sequence MLST multilocus sequence typing MLVA multilocus variable-number tandem-repeat analysis PCR polymerase chain reaction PFGE pulsed-field gel electrophoresis RD region of difference RNA ribonucleic acid sp. species (plural: spp.) ssp. subspecies (also subspp.)
v


INTRODUCTION Francisella tularensis is a highly infectious facultative intracellular pathogen and the causative agent of the zoonotic disease tularemia. Four subspecies of F. tularensis are currently recognized: subspp. tularensis (type A), holarctica (type B), mediasiatica and novicida. The species is known to cause infections in a remarkable range of vertebrate (>100) and invertebrate (>150) species and can be transmitted via arthropods like ticks and mosquitoes. The resulting disease in humans is often severely debilitating and, if the infective strain belongs to the most virulent subspecies, tularensis, it can be fatal. Despite the potency of F. tularensis as a human pathogen and its potential for misuse as a biothreat agent, little information has existed on its genetic makeup and genomic variability among different strains. The present thesis seeks to expand our knowledge in these areas. The organization of this work follows two themes: The first three original papers (Paper I-III) cover interstrain variability of F. tularensis isolates as explored by several different methods. These include comparative genomic hybridisations, multilocus sequence typing, multilocus variable-number tandem-repeat analysis, and the exploration of two different categories of insertions and deletions mutations. By the use of these methods new theories were advanced on the interrelations and evolution of strains. In the last original paper (Paper IV) findings from analyses of the genomic sequence of the highly virulent strain SCHU S4 of F. tularensis are discussed. In this thesis I attempt to provide also a background with important aspects of the bacterium to put into perspective findings here presented. The significance of these papers is discussed in a broad context, sometimes complemented by unpublished data.
1

BACKGROUND
Historical account In 1911, George Walter McCoy described a plague-like illness of ground squirrels in Tulare County, California [1]; this was the first definitive report of the existence of tularemia. The disease no doubt existed before this date, but interpretations of earlier records have been equivocal. For example, Homma Soken, a Japanese physician, reported poisoning or an illness resembling the symptoms of tularaemia as early as 1837 [2]. It has even been speculated that an epidemic of tularemia was the trigger that ultimately led to the exodus of the Hebrew community from Egypt in biblical times during the rule of Ramses II [3]. In 1912, McCoy and Charles W. Chapin isolated the causative agent, naming it Bacterium tularense after Tulare County, California, where the work was conducted [4]. Two years later, the bacterium was isolated from man Extensive research was conducted during subsequent years, and Edward Francis linked several clinical syndromes to the pathogen, and coined the following designations for the disease; “rabbit fever”, “market’s men disease” and “deer-fly fever” [5]. In 1925 it was also established that the Japanese disease Yato-byo (wild hare disease) was tularemia, after Francis had received and examined the causative agent sent from the Japanese physician Hachiro O’Hara [2]. Tularemia was later discovered to be endemic in parts of Asia and in Europe. In tribute to Edward Francis, who also coined the name of the disease, the pathogen was later renamed Francisella tularensis. F. tularensis was quickly recognized as a significant cause of disease, but also as a potent biothreat agent. At Unit 731, an infamous covert medical experimental facility of the Imperial Japanese Army in Manchuria, experiments on humans were performed between 1932 and 1945 [6]. The aim was to investigate the effects of exposure to F. tularensis and other pathogens. Even today anti-Japanese sentiments flourish in China, at least partly because of the atrocities perpetrated at Unit 731. Allegations of illicit use have also been made by the defector Kanathan Alibekov (now known as Ken Alibek), who served as deputy director of the Soviet biological weapons program organization Biopreparat. He
2

contends that tularemia was used by the Soviet Red Army to hinder advances of German panzer troops shortly before the Battle of Stalingrad in 1942 [7]. The high incidence of pneumonic tularemia (70 %) was one indication, according to Alibek, that deliberate air-borne dissemination was the cause of the outbreak. His claims have largely been refuted, however [8]. Biological weapons programs were initiated in several countries as well as the Soviet Union before and during the Second World War, but few lasted much beyond its end [9]. However, during the 1950s and 1960s the US military accelerated research into biological weapons, particularly F. tularensis, which replaced Bacillus anthracis (anthrax) as the agent of choice. Reportedly, these efforts were also paralleled in the Soviet Union. While stockpiles held by the United States were destroyed by 1973, it is believed that the Soviet Union continued weapons production using antibiotic- and possibly also vaccine-resistant strains into the early 1990s [7,9]. In September 2001, B. anthracis spores were sent a number of times via the U.S. Mail, causing twenty-two suspected or confirmed cases of anthrax, including five deaths. These events spurred an international anthrax scare and a global wave of anthrax hoax letters, but also raised issues about the preparedness of many countries to counter acts of bioterrorism. Owing to concerns about bioterrorism, the interest in this F. tularensis has also been renewed as reflected in an increased number of scientific publications on the pathogen during recent years (Fig. 1).
3

Figure 1. Frequency of scientific publications relating to tularemia. Frequencies were determined by searching the PubMed bibliographic database, and retrieving all records of all publications between 1980 and February 2007 that contained any of the terms “Francisella”, “tularensis”, “tularense”, “tularaemia”, “tularemia”.
4

Tularemia Two clinically important subtypes of F. tularensis have been recognized; they are denoted type A and type B, and both cause tularemia. Type A is known to be more pathogenic and results in more fulminant diseases, but otherwise infections by both types of bacteria result in clinically similar manifestations. Recent evidence suggests the high virulence of type A may not be valid for all bacteria, but restricted to one subpopulation [10] (this is discussed further in the following section). The different manifestations of tularemia depend on the entry route. Ulceroglandular tularemia is the most common manifestation; this is usually the result of being bitten by an infected arthropod (such as a mosquito or a tick). A primary ulcer on the skin can develop at the site of infection and nearby lymph nodes subsequently become swollen and painful. Other symptoms include fever, chills, headache and exhaustion. If the primary site of infection cannot be found, the disease is instead classified as glandular tularemia, but is conceptually identical to ulceroglandular tularemia otherwise. More than 90% of all cases of tularemia in Scandinavia are ulceroglandular or glandular [11]. Other manifestations include oropharyngeal tularemia (affecting the mouth and throat), oculoglandular tularemia (affecting the eyes) and typhoidal tularemia, where the patient suffers from fever and general symptoms but there is no evidence of the route of inoculation. The most serious manifestation is respiratory tularemia, where symptoms typical of pneumonia develop: coughing, chest pains and difficult breathing. A life-threatening condition can develop if the infectious strain is of type A. The bacterium can also be spread to the lungs from other parts of the body and cause secondary pneumonia.
5

Taxonomy and geographical distribution The γ-proteobacterial family Francisellaceae contains only the genus Francisella, and lacks close relatives that are pathogenic to humans. Instead, 16S rRNA data suggest that the ciliate endosymbiont Caedibacter taeniospiralis [12] (87% sequence similarity of the 16S rRNA molecule to that in F. tularensis) is a member of a sister clade. The fish pathogen Piscirickettsia salmonis [13] is also a phylogenetic relative, although more distant. Two species of the genus Francisella are accepted according to current taxonomy. The second species is F. philomiragia, which rarely causes disease and only in immuno-compromised individuals or near-drowning victims [14]. It is becoming increasingly clear that this genus will gain additional taxonomic members in the near future. Different genetic clades of Francisella-like bacteria have emerged recently: pathogens capable of enzootic disease in several species of fish [15]; tick endosymbionts; and bacteria in soils and sediment. To visualize relationships between these bacteria, a phylogenetic analysis based on reported sequences for small-subunit ribosomal RNA molecules (16S) was performed (Fig. 2). Details are provided in the legend.
Figure 2. (next page) Phylogeny of Francisella and representative relatives based on 1070 bp of the 16S ribosomal gene sequences (unpubl.). This slowly evolving gene identifies the closest bacterial relatives and places them in the broad context of other Gram-negative bacteria. However, the lack of variation among Francisella subspecies provides little resolution between them. Neighbour–Joining analysis was used to construct the tree and bootstrap values based on 1,000 replications are indicated at the branching points. Tree topologies and bootstrap supports using minimal evolution or parsimony algorithms were highly similar (not shown). Sequences in groups IV, VII and VIII beginning with “clone” are from the environmental sampling by Barns et al (2005) in Houston Texas. The scale bar indicates the expected genetic distance of 0.02 nucleotide changes per site.
6

AF-01-6 AY928389AF-03-27 AY928393AF-01-23 AY928390
Tilapia parasite TPT-541 AF206675 Ehime-1 AB194068cf. CYH-2002 AF385857
piscicida 2006-Chile AM403242 2005 50 F292 6C DQ295795piscicida GM2212-2005 DQ309246
F. philomiragia ATCC 25017 AY928395F. philomiragia ATCC 25015 AY928394 clone 034b AY968301clone 034c AY968302
clone 039d AY968300clone 005b AY968294clone 005c AY968297clone 013b AY968299clone 015d AY968298
clone 027c AY968289clone 027d AY968290
clone 027a AY968287D. variabilis symbiont AY805305D. variabilis symbiont AY805306D. variabilis symbiont AY805307
O. moubata symbiont B AB001522Wolbachia persica M21292
novicida-like 3523 AY243028 clone 034a AY968283 clone 039b AY968285clone 039a AY968284novicida U112 CP000439tularensis FSC054 AY968224tularensis SCHU S4 AJ749949holarctica FSC090 AJ698864holarctica LVS AM233362mediasiatica FSC147 AY968234
clone 045a AY968303clone 045b AY968304
Thiotrichales bacterium CML28 AB176554Caedibacter taeniospiralis AY102612
Thiotrichales bacterium EC7 DQ889939Piscirickettsia salmonis LF-89 U36941Piscirickettsia salmonis ATL-4-91 U36915Piscirickettsia salmonis NOR-92 U36942
100/100/100
100/100/99
100/100/100
100/100/99
81/91/78
96/93/77
100/100/99
76/80/74
73/77/79
0.02
IXThiotrichales spp.
XPiscirickettsiasalmonis
IFish parasites
IIIFrancisellaphilomiragiagroup
IVSoil bacteria
VSoil bacteria
VITickendosymbionts
VIIFrancisellatularensisgroup
VIIISoil bacteria
IIFish parasites
Topology Genetic cladeStrain
7

The phylogenetic analysis indicates a major split deep within the Francisellaceae, dividing the lineages that contain the species F. tularensis and F. philomiragia. Thus, the taxonomic division between these two accepted species is supported by phylogeny. As previously reported [16], the strains of F. philomiragia appear to closely resemble isolates that have been found to cause disease in several species of fish. Species reported to be affected by Francisella infections include tilapia (Orechromis spp.) [17,18], hybrid striped bass (Morone saxatilis) [15], three-line grunt (Parapristipoma trilineatum) [19], and Atlantic cod (Gadus morhua) [16,20]. Thus, both freshwater and saltwater fish can be affected, and these infections may be of considerable economic importance. Taxonomic assignments at the species level have been proposed for several of these pathogens. Given their phylogenetic relatedness, however, subspecies assignments may be more appropriate. The first isolation of a Francisella-like endosymbiont of ticks occurred in 1961 in Egypt. It was initially described as a Wolbachia-like bacterium, and became classified as Wolbachia persica. Based on molecular sequence data, it was later found to be a close relative of F. tularensis [21]. Several other Francisella-like endosymbionts have since been found in at least five different species of ticks, including two common North American Dermacentor species: the Rocky Mountain wood tick (D. andersoni) and the American dog tick (D. variabilis) [22]. The phylogenetic analysis demonstrates that the Francisella-like tick endosymbionts, for which reliable 16S molecular sequence data were available, appear to comprise a clade with a common ancestor. Although supported by bootstrap values of less than 90%, data suggest that this clade could be phylogenetically more related to F. tularensis than to strains of F. philomiragia. Possibly, these bacteria may receive classification as new Francisella subspecies in the future. An indeterminate, but possibly substantial, number of Francisella-like bacteria may also exist in the environment. This is suggested by one study where PCR amplification was performed on DNA extracts from environmental soil and sediment samples, specifically targeting Francisella 16S rRNA genes [23]. Sequence and phylogenetic analysis of the amplicons obtained indicated the existence of five new genetic clades, two of which were closely related to F. tularensis and F. philomiragia, while the others exhibited more intermediate phylogenetic positions. These sequences were also included in the phylogenetic analysis performed herein (see “clones”, Figure 2).
8

Tularemia in humans is exclusively caused by subspecies of F. tularensis, which form a monophyletic group (Figure 2). For reasons unknown, tularemia exists only between 30 degrees and 71 degrees north latitude in continental Europe (but not the United Kingdom), North America, Mexico, the Middle East, Russia, Mediterranean Africa, Japan, Korea, and China [24]. One atypical case of tularemia was recently reported from Australia [25], but there have been no official cases in South America, thus rendering it the only human populated tularemia-free continent F. tularensis bacteria that cause human disease belong predominantly to the subspecies tularensis (type A) and holarctica (type B), of which the former is considered more virulent for humans [11]. New findings may however require a more detailed systematic classification to differentiate strains that exhibit different levels of virulence. Previous studies by Johansson and colleagues [26] and Farlow and colleagues [27] demonstrated that the subspecies tularensis could be further divided into two distinct subpopulations, A.I and A.II., and that these also were associated with distinct geographical distributions. Recently, this division was identified also in a study by Staples and colleagues [10]. This study was performed using a different method (PFGE instead of MLVA), but results for overlapping parts of the strain collections suggest that an exact correspondence between the subpopulations. A very interesting additional finding in the work by Staples was however that strains belonging to the subpopulation A.II (denoted A-west by Staples) seemed less pathogenic. The case-fatality rate for type A.I (A-east) infections was 14%, compared with 0% for type A.II (A-west) infections. It is possible therefore that only strains belonging to the subpopulation A.I cause the severe form of tularemia, which has been previously associated with all strains of F. tularensis subsp. tularensis (type A). In addition to the F. tularensis subsp. tularensis and holarctica, two clinically less important subspecies are also recognized. The subspecies mediasiatica has been collected only in central Asia. This subspecies has been found to display a level of virulence comparable to strains of subsp. holarctica [28,29]. Members of subsp. mediasiatica are human pathogens but human infections occur very rarely (pers. comm. T. Kunitsa). F. tularensis subsp. novicida is also a rare human pathogen, and then only in immuno-compromised individuals [30].. Because they are less virulent and easier to manipulate genetically, strains of the subsp. novicida have been used extensively as a model for the more virulent F. tularensis subspecies. The discovery of a novicida-like isolate in Australia was
9

recently reported [25], thus being the only subspecies of F. tularensis not confined to the Northern Hemisphere
Evolution and population structure A bacterium can principally evolve by three routes, either alone or in combination: either by the acquisition of genes that previously evolved elsewhere, by mutation of genes that already exist or by loss of genetic material. An important consequence of transferred genetic material can be the acquisition of resistance to certain antibiotics, resulting from conjugal transfer of plasmid DNA. It has been found also that genetic material of distant foreign origin can be integrated into the chromosome. Such laterally transferred genomic regions that have been found important for virulence have been termed pathogenicity islands (PAI). Many pathogens appear to have emerged after such events (see [31] for a review). Evolution by loss of genetic capacity has its most extreme representation among intracellular symbionts. An example is provided by the aphid endosymboint Buchnera aphidicola BCc, which carries a genome with the size of only 422,434-base pairs [32], the smallest bacterial genome known to date. It has become evident that chromosomal material also readily may be exchanged between closely related bacteria within a population. This can happen to such an extent that it affects the population structure. At the extreme “panmictic” end of this spectrum is Neisseria meningitidis, a causative agent of meningitis, for which recombination has been determined to be the predominant mode of evolution. In this bacterium, recombination occurs, on average, at a higher rate than nucleotide substitutions [33]. At the other “clonal” end of the spectrum are vertically inherited endosymbionts [34], as well as common pathogens such as Salmonella spp. An intracellular lifestyle, however, does not preclude recombination, as evident from bacteria such as Chlamydia trachomatis [35], Wolbachia spp. [35] and Leginella pneumophila [36]. The extent of such homologous recombination within species appears to be closely regulated by the degree of similarity of the recombining DNA, where the relative recombination rate is inversely proportional to the sequence divergence [37].
10

Genomic typing Traditionally, phenotypic characteristics of bacterial isolates have been used for their identification, such as ability to grow on specific media, capacity for various fermentation reactions, or serotype. These methods represent classical bacteriology and are routinely performed in bacteriological laboratories. More recently, methods have been developed that make use of differences among strains at the genetic level. A plethora of different genetic typing methods have been developed during the last decades (see reference [38] for a review). The choice of typing method depends ultimately on the purpose for which the data will be used. Some investigations may be addressed adequately by classical phenotypic classifications (for example, which pathogen is it?), while others may require a level of resolution not provided by these methods (such as epidemiological tracking of a strain). An ultimate method should be simple, fast, inexpensive, safe, reproducible, and enable robust and accurate estimates of relationships between both closely related microbes as well as distantly related. The different existing methods often represent compromises between these criteria. Ideally, to determine the identity of a bacterium, the optimal level of information is obtained from determining its entire genomic sequence. Although this level of information is useful for various purposes (see paper IV), it is, usually, an unrealistic method for typing. While it is falling rapidly, the cost of genome sequencing is still high (~10 000 Euro, by pyrosequencing). For some particular isolates, however, costs of that magnitude may not be preventive. An example is provided by a recent investigation into the origins of the idiosyncratic Slovakian F. tularensis isolate FSC198 [39]. This strain represents one of a handful of European F. tularensis subsp. tularensis (type A) organisms, all isolated from a geographically distinct area. In the analysis, extremely few genomic loci were found that contained polymorphic sites differentiating the FSC198 from the strain SCHU S4, thus indicating a close connection between these strains. As a typing method, genome sequencing represents an exception and is practically never used or considered for this purpose. Instead, commonly used methods specifically target genomic variability. Some methods with current relevance to F. tularensis typing are introduced below.
11

Macro-restriction analysis resolved by gel electrophoresis (PFGE) is, as the name suggests, based on digestion of the bacterial genome using rare-cutting restriction enzymes. In order to visualize the resulting fragments of DNA, these are separated on agarose gels by alternating electric fields. Rearrangements or polymorphisms at the cleavage sites result in different banding patterns. This represents a “gold standard” for typing of many pathogens, including Salmonella enteritidis [40] and methicillin-resistant Staphylococcus aureus [41]. PFGE has been standardized and become a prioritized tool for use in large networks of laboratories in both the USA (PulseNet) and Europe (PulseNet Europe). Its use has also been advocated for F. tularensis [10]. This method is, however, inherently dependent on interpretation of gel-images by analysis software, and requires stringent adherence to common protocols to allow inter-laboratory comparisons. It is also quite labour-intensive and requires prolonged handling of the strains to be typed, and provides a limited resolution [42]. For analysis of F. tularensis, because of its extremely contagious nature [43], the use of PFGE creates a risk of laboratory-acquired infections. Approaches that target specific, defined loci usually offer improved reproducibility, since they have built-in mechanisms to detect and discard unsuccessful assays. For example, a PCR assay has been developed to distinguish type B F. tularensis strains from type A strains by analysis of a 30-bp polymorphism [42]. If the fragment size obtained from this assay does not fall within an expected range of sizes, the result is considered invalid. Similarly, DNA sequencing provides quality scores that enable discrimination of good sequences from bad. One method which has attracted increasing interest in recent years, making use of defined genomic targets, is multilocus variable-number tandem-repeat analysis (MLVA). This method is conceptually based on analysis of variations in copy number of tandem repeats at several loci in the genome. These loci are typically highly variable and are considered to be the fastest mutating parts of the DNA. For F. tularensis, this is the only method which has enabled discrimination between individual isolates [26,27]. MLVA has also an advantage since it makes use of several genomic targets. Methods that characterize a single locus may be easier but more risky to use of if it is possible that the bacterium being typed recombines. Thus, knowledge about the bacterial population structure can be important in epidemiological investigations and have impact on the choice of typing method. If an investigation is performed in a forensic
12

setting where not only probable links but statistically valid evidence for attributional purposes is sought, a complete understanding of the population of a bacterium is critical.
Genome analysis The first bacterial genome sequence to be completely determined was Haemophilus influenzae, reported in 1995 [44]. Since then, the genomes of more than 500 microbial species and strains been determined and almost 1100 genome projects are currently in progress, as reported on ‘genomes online’ [45]. The availability of genome sequence information has profoundly changed many aspects of microbiology research. Commonly used experimental techniques, such as proteomics, are today heavily dependent on genomic information. Completely new research areas have emerged also, such as computational biology, bioinformatics, and systems biology, where a corner stone is provided by availability of genomic sequences. We have learnt from genomic information how bacterial evolution occurs by different mechanisms and have exploited such information for diagnostics, but it has also enabled us to fundamentally understand inner workings of bacteria. We can today predict both metabolic and other functional capacities of bacteria by comparisons with other better understood bacteria. Such information can provide a fundament for rational design of antimicrobials [46] and in vaccine development (so called Reverse Vaccinology) [47].
13

AIMS OF THE THESIS The aims of this thesis are: I. To identify genetic differences between subspecies,
subpopulations and strains of Francisella tularensis in order to: (a) identify putative virulence determinants and functional differences (b) provide improved means to better distinguish between evolutionary lineages (c) delineate interrelations between lineages
II. To analyse the genomic content of Francisella tularensis subsp.
tularensis strain SCHU S4 in order to increase our understanding of its biology, pathogenic mechanisms, and evolutionary adaptations.
14

METHODOLOGICAL CONSIDERATIONS
Genome sequencing The basic technology used in most genome projects today is still the same as what was used in 1995, although it has been greatly refined and the cost per sequenced base significantly reduced. The strategy used in genomic sequencing used is termed “shotgun” sequencing. It entails fragmenting of the chromosome, cloning the pieces into plasmids, and determination their molecular sequences by classical Sanger sequencing. All sequence fragments are then collected and assembled by computers into contiguous DNA sequences. Ideally, one contiguous sequence should result for every chromosome present in the bacterium after the assembly process. This does not always happen, despite that theoretical requirements on a sufficient number of sequence reads have been met. Since every shotgun fragment must be cloned and replicated by culturing in Escherichia coli bacteria, expression of gene products from the inserts can occur. Sometimes such gene products are toxic to the bacterium and will be selected against, leading to the presence of gaps at such positions in the sequence. A different problem concerns the presence of multiple copies of near-identical genomic regions, such as genomic duplications and the presence of insertion sequence (IS) elements. At such positions, the assembly program most often fails to correctly resolve the sequence since the reads originating from different copies of a repeat are identical. During the finishing phase of the SCHU S4 genome sequencing project (paper IV), the presence of repetitive DNA caused significant problems. The finished genomic sequence was found to contain five types of insertion sequence elements. Two were present in single copies, but tree IS elements where present in 50, 16, and 3 copies each. Moreover, one large genomic region was duplicated, corresponding to the Francisella pathogenicity island (FPI) [48], and one genetic region was triplicated. To solve the puzzle, different strategies were used. One method used was to identify clones with inserts that spanned IS elements in the genome. To the fullest extent possible, sequence reads were linked together in the
15

database according to the clones they had been sequenced from (scaffolding). Since clone information for each read was not entirely reliable significant portions of this work had was done manually. Also, for many clones, good quality sequence existed only from one direction. Resequencing of such clones was therefore necessary to obtain sequence information from both ends. A combinatorial PCR approach was also employed where pools of primers where combined and tested to identify primer pairs capable of amplification and, thus, matching ends for IS elements. Finally, to solve the last pieces of the puzzle, macrorestriction digestion and pulsed-field gel electrophoresis was used to correctly match the ends of the large duplications and triplications.
Comparative genomic hybridisation Array-based comparative genomic hybridisation (aCGH) is a tool to that enables interrogation of the genomic compositional diversity among bacterial strains. In this method, one bacterial strain usually serves as a reference, or index, to which other strains are compared. The index strain provides a template for generating DNA sequence probes that are attached onto the microarray. From other strains that are tested, DNA is extracted and hybridized to the probes on the microarray. In theory, probes which contain DNA sequences that are not represented in a tested strain will produce low signals and those present will produce high signals. The aCGH study (paper I) in the present thesis made use of a genomic library that already was existing, constructed for the F. tularensis SCHU S4 genome sequencing project (previous section). Use of this library, and universal primers for probe amplification by PCR, provided a reasonable cost for obtaining the microarray probes. While this strategy provides convenience and power, it is important to note that limitations of the technology exist. It follows from the use of large microarray probes (mean size 1405 bp) that detection of small deletions relative the reference strain can be impossible. Due to a variety of reasons [49], a problem concerns also the fact that microarray data may suffer from biases and noise. It has for example been recognized that microarray hybridisation under cover slips can result in uneven
16
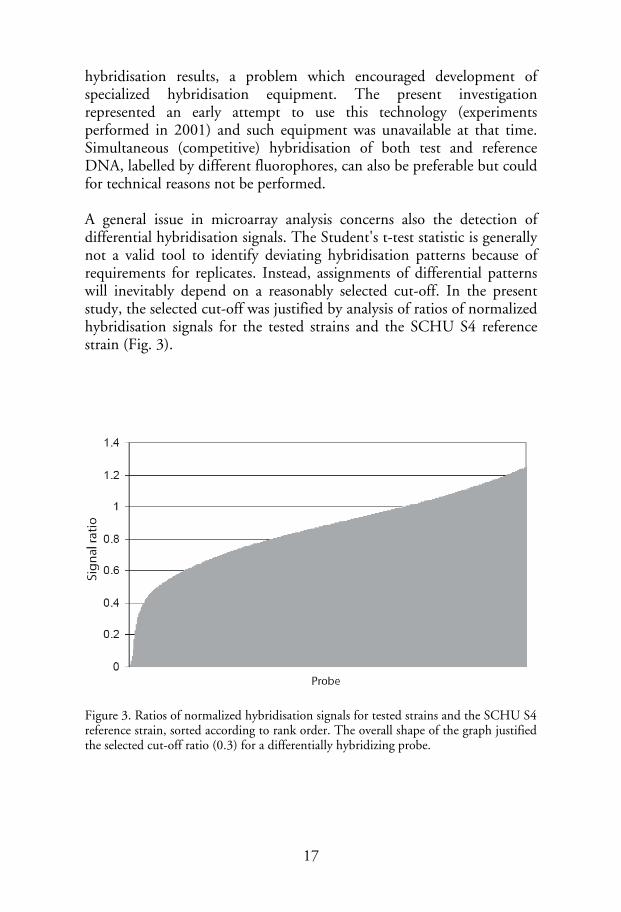
hybridisation results, a problem which encouraged development of specialized hybridisation equipment. The present investigation represented an early attempt to use this technology (experiments performed in 2001) and such equipment was unavailable at that time. Simultaneous (competitive) hybridisation of both test and reference DNA, labelled by different fluorophores, can also be preferable but could for technical reasons not be performed. A general issue in microarray analysis concerns also the detection of differential hybridisation signals. The Student's t-test statistic is generally not a valid tool to identify deviating hybridisation patterns because of requirements for replicates. Instead, assignments of differential patterns will inevitably depend on a reasonably selected cut-off. In the present study, the selected cut-off was justified by analysis of ratios of normalized hybridisation signals for the tested strains and the SCHU S4 reference strain (Fig. 3).
Figure 3. Ratios of normalized hybridisation signals for tested strains and the SCHU S4 reference strain, sorted according to rank order. The overall shape of the graph justified the selected cut-off ratio (0.3) for a differentially hybridizing probe.
17

RESULTS AND DISCUSSION
Comparative genomic hybridisations reveals strong compositional similarity within F. tularensis (paper I) F. tularensis is recognized as having strong sequence level genetic conservation [50]. Analysis of small subunit (SSU) ribosomal RNA sequences demonstrates that representatives from each of the subspecies tularensis, holarctica, mediasiatica and novicida, are distinguished from one another only one of 1450 aligned nucleotides, despite their marked differences in virulence (unpubl.). To investigate the genomic content of different bacterial strains, we developed a DNA microarray, based on a set of 1,832 duplicate probes. The probes were obtained from a shotgun library used for genome sequencing of the F. tularensis subsp. tularensis strain SCHU S4. Based on the available data, the clones containing the inserts that we wanted to amplify and spot onto the microarray, were selected to achieve maximum genomic coverage and minimal redundancy. The total insert size of the clone set was estimated to cover 95% of the F. tularensis genome. Twenty-seven strains of F. tularensis, representing each of the four subspecies, were ultimately analyzed using the microarray in the study. Overall, few probes were found to display differential hybridisation signals that could be used to distinguish between the strains. The two most divergent strains analyzed were FSC040 (subsp. novicida) and ATCC 6223 (subsp. tularensis, avirulent). Differential hybridisation patterns (determined using the selected cut off) were detected for 3.7% and 2.6% of the probes for the ATCC 6223 and FSC040 strains respectively; many of these patterns were unique for each strain. The most striking finding from the hybridisation signal analysis was that few probes (0.7-1.2%) differentiated strains of the subsp. mediasiatica from the reference strain. Strains of this subspecies have only been isolated in central Asia, and exhibit a level of virulence similar to strains of the subsp. holarctica [28,29]. In contrast, the highly virulent subsp. tularensis, to which the reference strain belongs, is believed to be
18

Strain Subspecies
FSC230 tularensis (ATCC 6223, avirulent)FSC040 novicida (ATCC 15482)FSC148 mediasiaticaFSC054 tularensisFSC053 tularensisFSC046 tularensisFSC041 tularensisFSC042 tularensisFSC237 tularensis, SCHU S4FSC198 tularensisFSC043 tularensisFSC149 mediasiaticaFSC147 mediasiaticaFSC199 tualrensisFSC075 holarctica (Japan)FSC022 holarctica (Japan)FSC021 holarctica (Japan)FSC035 holarcticaFSC076 holarcticaFSC200 holarcticaFSC157 holarcticaFSC150 holarcticaFSC155 holarctica (Live Vaccine Strain)FSC124 holarcticaFSC080 holarcticaFSC089 holarcticaFSC032 holarctica
Figure 4. Hierarchical cluster analysis of F. tularensis DNA microarray hybridisations. Strain identity is indicated by FSC number and subspecies. naturally confined to North America. Despite differences in geographical distribution, and different levels of virulence, these two subspecies appear closely related. With the exception of the FSC040 strain, which belongs to the subsp. novicida, and the atypical type strain of F. tularensis ATCC 6223, European and American F. tularensis subsp. holarctica strains were, on average, the most divergent, displaying differential hybridizing signals for 1-3% of the probes. The Japanese F. tularensis subsp. holarctica strains displayed differential patterns for 1-2% of the probes and appeared to represent an intermediate between the subsp. tularensis (or mediasiatica) and the European and American subsp. holarctica strains. This intermittent status was also demonstrated by a deep and divergence of
19

this lineage in a hierarchical cluster analysis (Fig. 4). Previously, Japanese subsp. holarctica strains have been found to exhibit phenotypic differences to other strains of the same subspecies. Unlike other subsp. holarctica strains, Japanese subsp. holarctica can ferment glycerol into acid [28,51], but, in common with other members of the subsp. holarctica, the Japanese strains lack citrulline ureidase activity [28], which is a typical phenotypic marker for the subspp. tularensis and mediasiatica. Many differentially hybridizing probes were found to cluster in contiguous genomic regions; we designated these F. tularensis ‘regions of difference’ (RDs). It was apparent that the ‘presence’ or ‘absence’ of genes within F. tularensis RDs reflected the different subspecies of the strains. We focused further studies on eight such regions which had lengths between 0.6 kb and 11.5 kb, and which differentiated strains of subsp. tularensis (type A) from subsp. holarctica (type B, non-Japanese). PCR and sequencing revealed that repeat motifs 9- to 38-bp long, were present in the corresponding SCHU S4 sequence for all RDs, except RD8. Instead, this region was flanked by insertion sequence elements. Analysis of F. tularensis RD1 to RD8 identified a total of 21 open reading frames in the sequences, which were absent from F. tularensis subsp. holarctica strains, with several open reading frames homologous to known bacterial genes. RD1 was highly polymorphic (Fig. 5); by targeting this region it was possible to develop a single gel-based PCR assay that distinguished all four subsp. of F. tularensis, as well as Japanese strains of the subsp. holarctica. Potential explanations for the different levels of virulence exhibited by type A and type B strains of F. tularensis were considered, and several potential mechanisms were proposed. However, none of these have yet been demonstrated as important for virulence in other studies. RD6 was not discussed with respect to virulence in paper I, but was later found to correspond to a genetic deletion within the pdpD gene. This gene is positioned in a genetic region intimately associated with virulence, the Francisella Pathogenicity Island (FPI), and was later proposed as a potential explanation for the differences in virulence observed between type A and B F. tularensis bacteria [48]. Recent work by Francis Nano suggests however that this gene is not essential for virulence [52], at least not in the models that have been investigated to date.
20

Figure 5. Schematic depiction of the variable chromosomal region, RD1, identified among the different F. tularensis subspecies with insertions and deletions. A further objective of this study was to identify possible sources of the attenuation demonstrated by the live vaccine strain (LVS) of F. tularensis. Therefore, the hybridisation pattern for the LVS strain was meticulously compared to patterns obtained for European and North American strains of the subsp. holarctica, the group of strains to which the LVS should belong. However, we were unable to identify any deleted genomic regions in the LVS that were present in a virulent strain. Most RDs contain pseudogenes of the reference strain SCHU S4, or pseudogenes that are present elsewhere in the operons and which can be disrupted by deletions. This might suggest that most of these genes are not used by either subspecies, but have not yet been lost from subsp. tularensis.
21

In a recent study by Samrakandi et al. [53], differences between F. tularensis isolates were also investigated using comparative genomic hybridisations by microarray. Their results were broadly similar to ours, and they obtained similar percentages of probes displaying differential hybridisation signals. Differences between the results exist nonetheless; the study by Samrakandi et al. identified six polymorphic regions not detected in our microarray study, but failed to identify three regions that we detected. As also noted by Samrakandi et al., neither microarray study was comprehensive, which might account for these differences. Other explanations may also include the noisy nature of microarray data, or a technical inability to identify existing differential patterns. Genome sequencing of various bacteria has revealed that the similarity in genomic content is not always highly correlated to sequence similarity [54]. For example, up to more than 36% of the total gene content was found to be specific to individual lineages of Escherichia coli [55]. However, in F. tularensis, the observed limited sequence diversity appears to closely correlate with limited compositional diversity.
22

Phylogeny and direct repeat-mediated deletions among F. tularensis isolates (paper II and unpublished work) To further delineate the evolutionary relationships between different strains and subspecies of F. tularensis, and also to identify genomic differences that might correspond to functional correlates, two sets of analyses were performed that aimed to identify single nucleotide variations (SNPs) and genomic deletions across strains and subspecies. We found that all the deletions previously identified (paper I) resulted from repeat-mediated excision; RDs1-7 were associated with short direct repeats and RD8 was flanked by insertion sequence elements. We hypothesized that this might represent a significant propensity, such that deletion polymorphisms could also be found in regions containing direct repeats. The genome sequence of strain SCHU S4 was, therefore, scrutinized for direct repetitive motifs, and over 70 identified genomic regions were tested for size polymorphisms in a panel of five isolates. Regions that exhibited size polymorphisms on agarose gels were further analyzed in 45 strains. In a parallel study, single nucleotide polymorphisms (SNPs) were identified by sequencing seven genes in different strains of F. tularensis. By using the same set of PCR primers it was also possible to amplify four of the seven genes in Wolbachia persica, a bacterium previously recognized as a relative of F. tularensis [21]. By using the molecular data we collected, three phylogenetic analyses were conducted that mutually supported each other: (i) a ‘manual’ parsimony analysis was performed using the deletion polymorphisms; (ii) a multi-protein phylogeny was inferred using protein translations of a common set of four genes available for W. persica and F. tularensis strains. Two outgroup species were also included (Yersinia pestis and Agrobacterium tumefaciens) and their sequence information was obtained from GenBank; and (iii) a multi-gene phylogeny was performed which only included F. tularensis strains, using the more informative set of seven genes. The deletion markers analyzed in this study provided information that cannot be extracted from reversible characters at equilibrium (such as SNPs). Although they may arise independently, deletions cannot be
23

Ancestor ofF. tularensis
subsp. mediasiatic
subsp. holarcticafrom Japan
subsp. holarcticafrom Eurasia andN. America
subsp. novicida
subsp. tularensis
Deletion of: RD1c
Extensive singlenucleotide variationin seven genes
Presence of: RD1c RD6 RD2 RD7 RD3 RD11 RD4 RD16 RD5
Deletion of: RD2 RD6 RD3 RD11 RD5
Deletion of: RD4 RD16 RD7
(a)
(b) (c)
Figure 6. (a) Evolutionary scenario of F. tularensis based on deletion characters. The scheme reads in evolutionary time from left to right. (b) Four-gene tree constructed using amino acid sequences (631 amino acids). (c) Seven-gene analysis using nucleotide sequences (3,135 bp).
reversed. The pattern of deletions dictates that the subsp. tularensis lineage must have diverged earlier in evolutionary time than the formation of the subsp. holarctica. The complete lack of deletions shared between the subspp. holarctica, mediasiatica and novicida further suggest, by the principle of parsimony, that these strains do not belong to the same lineage. Thus, this information and the divergent nature of the subsp. novicida suggested an evolutionary scenario that is described in Fig. 6a. This scenario was entirely congruent with phylogenetic scenarios that were inferred using only SNP-based evidence (Fig. 6b, 6c). The results from the protein phylogeny indicated that the inclusion of representatives of A. tumefaciens and Y. pestis, which belong to the α-subgroup of the proteobacteria and a different clade within the γ-subgroup, further supported a deep divergence in the subsp. novicida lineage (Fig. 6b). Using the seven-gene dataset, which had greater
24

resolution, the general order in which the subspecies and strains are likely to have diverged could be delineated (Fig. 6c). The seven-gene analysis suggested further monophyly of the subspp. mediasiatica and tularensis, and indicated a split within the subsp. tularensis. This division corresponds to the subclades A.I and A.II of the subsp. tularensis, which have been previously recognized and supported by both multilocus variable number tandem repeat analysis (MLVA) [26,27] and more recently by macrorestriction analysis and pulsed-field gel electrophoresis [10]. In agreement with results obtained by comparative genomic hybridisations (paper I), the inferred topology indicated also the monophyly of the subspp. mediasiatica and tularensis. However, only one nucleotide character was found to be responsible for the formation of this clade. Thus, the possibility exists that this association might have been the result of a homoplastic character sampled by chance and not the result of common ancestry. The possibility that recombination between different F. tularensis lineages has occurred was also addressed in paper II, but no evidence to support such events was found. The sequence data provided a perfect phylogeny for the included taxa. Evidence of recombination was reported previously for F. tularensis [56], but an analysis of the data has indicated that these conclusions may have been inaccurate (pers. comm. A. Johansson). Nonetheless, it cannot be ruled out that this species could recombine. In the studies performed so far, only few genes have been sampled, and the odds of detecting recombination are also limited due to the extensive sequence identity within F. tularensis. It may be also that recombination could be restricted and only occur within subpopulations of F. tularensis if these correspond to separate ecotypes [57]. The availability of more genomic sequences for F. tularensis isolates will provide an opportunity to better investigate inter-subspecies recombination. The existence of multiple genome sequences has also created an opportunity to resolve uncertainties about the phylolgenetic structure. In recent unpublished work using genomic data, the phylogenetic position was inferred for the F. tularensis subsp. mediasiatica, confirming the monophyly of subspp. mediasiatica and tularensis and verifying also the phylogenetic positions of other described subclades (Fig. 7).
25

Schu S4
WY96
FSC 147
LVS
OSU18
U112
ATCC 25017
100
100
100
100
0.1
100
100
100
1000.01
Schu S4
WY96
FSC 147
LVS
OSU18
U112
100
tularensis A.I
tularensis A.IImediasiatica
holarctica B.I
holarctica B.II
novicida
philomiragia
tularensis A.I
tularensis A.II
mediasiatica
holarctica B.I
holarctica B.II
novicida
Topology Genetic cladeStrain
(a)
(b)
Figure 7. Whole-genome phylogeny inferred using Neighbours-Joining. The analysis was based on 44,244 variable nucleotide sites, extracted from whole-genome alignments for genomic sequences of the included strains, and the p-distance metric. The support values indicate percent bootstrap support for each node, and was calculated using 500 pseudo-replicates. (a) In the tree depicted in the upper part of the figure, all strains used for inference are included, whereas (b) the tree in the lower part of the figure corresponds to a subtree of the same inferred topology. The scale bars indicate lengths corresponding to the number of nucleotide substitutions per site, given below the bars. All sites were selected on the criterion that they are variable, branch lengths should be therefore interpreted as a relative measure and not as absolute genetic distances between genome sequences. Among genes mutated by repeat-mediated excision, some require discussion with regard to virulence properties. Genes discovered in the polymorphic RD18 and RD19, also independently identified by Samrakandi et al. [53], were found to discriminate among isolates of individual subspecies. Interestingly, the genes present in RD19 represent type IV pili building block proteins, which are associated with virulence in several pathogens including Neisseria spp., Pseudomonas aeruginosa and Vibrio cholerae (see reference [58] for a review). Recombination and
26

deletion of these genes have occurred in the LVS of F. tularensis. Although evidence suggests the possibility of a different functional role of this protein in F. tularensis, mutation of this gene was recently found to result in an attenuated phenotype [59]. Genes discovered in the polymorphic genomic region of RD18 represent genes of a novel protein family unique to F. tularensis. A targeted mutation of one gene (FTT0918) in this region of the highly virulent SCHU S4 strain caused significant attenuation of virulence in mice [60]. The level of conservation among F. tularensis subsp. holarctica strains is extreme; a level of sequence similarity of >99.92% was recently found for the Live Vaccine Strain (LVS) of F. tularensis and the strain FSC200, a virulent counterpart [61]. As both mutations mentioned above were previously found to be attenuating in F. tularensis, it is possible that the attenuation of LVS may largely be explained by these two mutations. Several studies have shown that the vaccination using LVS can provide protective immunity against tularemia [62], but the uncertain nature of the attenuation of this strain has restricted its use because of a reversal risk. However, if it could be shown that the source and origin of the F. tularensis LVS attenuation is confined to these mutations, restrictions on use of the LVS as a vaccine could be lifted. A typical example of direct repeat-mediated excisions is provided in Fig. 8, illustrating the deletion and typical formation of a composite repeat motif.
12 bp composite repeat
3'subsp. tularensis
subsp. holarctica from Japansubsp. mediasiatica
subsp. novicida
subsp. holarctica fromEurasia and N. America
5'∆ (1323 bp)
Deletion of RD 4
3'5'
12 bp direct repeatsGGAGTTATCTCGGCTTTTAACTTTCCTGTAGC
ATTTATTAGTCAGCTTTTAACAAATGAATTCT
DR1 DR2DR1
DR2
GGAGTTATCTCGGCTTTTAACAAATGAATTCT
composite repeat Figure 8. Unidirectional repeat-mediated deletion mechanisms exemplified by RD4. Flanking direct repeats (DR1 and DR2) and the resulting composite repeat are depicted as hatched bars. Open bars represent open reading frames that were truncated by the deletion. Partial nucleotide sequences are shown before (DR1 and DR2) and after (composite repeat) the deletion event.
27

Indel markers for low-resolution genetic typing (paper III) During the past decade, the genetic typing method, multilocus variable number tandem repeat analysis (MLVA), has received increasing interest and has been developed for a large number of pathogens [63]. For F. tularensis, MLVA is the only method that can currently distinguish isolates at the strain-level. However, while necessary for MLVA to be useful, the high mutation rate of these markers represents a risk for spurious phylogenetic estimates, especially at larger genetic distances. Moreover, it has been recognized that simple sequence repeats, which constitute MLVA markers, may experience functional constraints. These sequences, called contingency loci, are sometimes exploited as genotypic switches to generate phenotypic variation and enhance fitness [64,65]. Thus, the risks of homoplasy are evident, due to chance and convergent selection forces, which have been previously noted by several authors [65-68]. One solution to this problem is to combine MLVA with a marker analysis that has an inherently lower mutation rate. This way, MLVA can provide fine-grained resolution, while slowly evolving markers afford a supportive ‘backbone’. Single nucleotide polymorphisms (SNPs) have been used for this purpose in previous studies of B. anthracis [67,69]. Apart from a low mutation rate, SNPs offer several other benefits, including well-established models to support their analysis [70]. However, one limitation, which may preclude their use as a widespread clinical tool, is that the analysis of SNP and MLVA markers require separate assays, thereby increasing complexity, time and cost of their use. Therefore a stable class of insertion-deletion (Indel) polymorphisms were sought in paper III (rather than SNPs) to complement the MLVA markers. To identify Indels present in F. tularensis, five genome sequences were used, representing each of the currently recognized subspecies of F. tularensis from at least one genomic sequence. The method to identify polymorphisms is simple, but has only recently become possible with the availability of genomic sequences. By applying a strategy that involved automated multiple alignment of genomic sequences and searching, 280 loci across lineages were identified as containing size polymorphisms.
28

By definition, all size polymorphisms are insertions or deletions (Indels) in DNA, but their evolutionary rates diverge widely, as a result of different mechanisms of formation. MLVA markers are formed through a slipped-strand mechanism and are highly variable [63]. We previously found (papers I-II) that F. tularensis Indels also commonly form by recombination of repeated DNA sequence motifs. However, the fact that direct sequence motifs can be used to predict Indel loci (paper II) suggests that there is a risk that such loci may exhibit significant homoplasy. Using the same argument, we hypothesized that loci lacking direct repeats (if they exist) might provide markers that are less prone to homoplasy. Analysis of the 280 Indel loci that we identified revealed that direct repeat motifs commonly existed in the longer alleles, and such repeats were very short or did not exist at all. To get an overview of the data, the frequency of the loci was analyzed, as a function of repeat length, and was found to conform to a bimodal distribution (Fig. 9). This shape commonly arises when two separate unimodal distributions are combined, which supports the possibility that separate molecular mechanisms could be responsible for their mutation. One mechanism might be dependent on the presence of direct DNA repeats, while at least one other ‘illegitimate’ mechanism might exist that is independent or less dependent on such repeats.
Direct repeat length (bp)
Freq
uenc
y
0 5 10 15 20 25 30
010
2030
4050
60
Figure 9. Comparative genomics of five genomes of F. tularensis isolates revealed a bimodal frequency distribution of lengths of direct repeats, associated with Indel
29

mutations. This fact is suggestive of separate underlying mechanisms responsible for Indel formation. The frequency of homoplasy in the data was then estimated and compared to the overall Indel-based topology (congruent with the topology in Fig. 7). Among the 158 loci present in the left mode of the distribution (short or no repeats), none were found to be incongruent. Conversely, among the 122 loci found in the right mode of the distribution (longer repeats), four incongruent loci were found. This analysis supports the possibility of lower homoplasy frequency for loci with or without only short repeat motifs. Thirty-eight Indel loci were selected to further evaluate their potential as genetic typing markers. The selected loci encompassed the complete range of evolutionary patterns and were physically well separated; this was determined from their locations on the F. tularensis strain SCHU S4 chromosome. The Indel loci were assayed together with a set of 25 previously used [26,27] MLVA markers, in 23 F. tularensis strains, which comprised representatives from all the major accepted genetic clades. Analysis of the Indel loci demonstrated the extent that Indels can provide a similar functionality to SNPs [67,69] in providing the canonical structure (Fig. 10). Bootstrap analysis demonstrated that the topology inferred using MLVA data was weakly supported at most nodes, but we found significantly increased statistical support for the combined sets of MLVA and Indel data. For strains belonging to the subsp. holarctica, a combined analysis also provided higher resolution than obtained for any of the individual datasets. This approach, using the two markers, provided increased support for the basic structure in the data, as well as higher resolution. In this study, alleles were tested individually, but future optimizations may include multiplexing of assays and a reduction in the number of specific markers for each clade; the number of MLVA markers may also be reduced. Further future optimization might also include selecting markers that are specific for currently unidentified clades. Clearly, the ability to identify clade-specific Indels will improve in the future as the number of available genomic sequences for F. tularensis (and other bacteria) rapidly increases. This method represents a significant improvement to using only MLVA markers. It should be noted that the combination of MLVA and Indel loci offers obvious advantages compared with using macrorestriction analysis resolved by pulsed-field gel electrophoresis (PFGE), that has
30
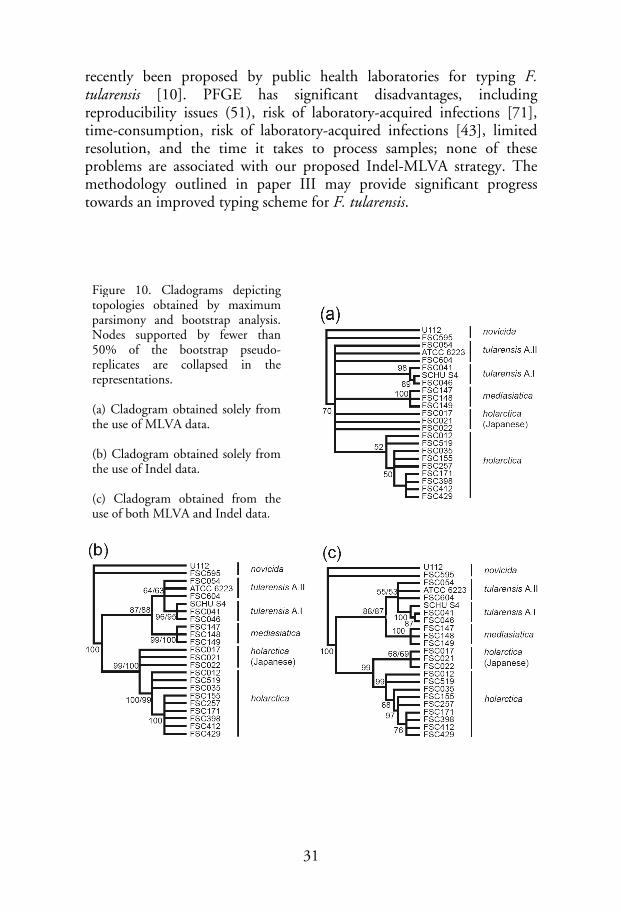
recently been proposed by public health laboratories for typing F. tularensis [10]. PFGE has significant disadvantages, including reproducibility issues (51), risk of laboratory-acquired infections [71], time-consumption, risk of laboratory-acquired infections [43], limited resolution, and the time it takes to process samples; none of these problems are associated with our proposed Indel-MLVA strategy. The methodology outlined in paper III may provide significant progress towards an improved typing scheme for F. tularensis.
Figure 10. Cladograms depicting topologies obtained by maximum parsimony and bootstrap analysis. Nodes supported by fewer than 50% of the bootstrap pseudo-replicates are collapsed in the representations. (a) Cladogram obtained solely from the use of MLVA data. (b) Cladogram obtained solely from the use of Indel data. (c) Cladogram obtained from the use of both MLVA and Indel data.
31

Figure 11. (unpubl.) Comparison of the composition, in mole percent of amino acid, between different bacterial species. The comparison was based on analysis of XXX orthologous genes, determined by reciprocal best-hit blast searches for all translated open reading frames for the genomic sequences of strains shown in the legend. The included strains were ranked in descending order of AT content. In the histogram species are arranged likewise in order of AT content; the bacterium with the highest AT content (F. tularensis) is present to the right for each amino acid, and the bacterium with the lowest AT content (Burkholderia pseudomallei) to the left. Similarly, the amino acids are arranged according to AT content of their corresponding encoding codon families; on the right hand side of the histogram are amino acids encoded by AT-rich codon families and amino acids encoded by GC-rich codon families are presented to the left.
The F. tularensis SCHU S4 genome sequence (paper IV) In paper IV, the complete genome sequence of the strain SCHU S4 of F. tularensis was determined and analyzed. This strain provides a fully virulent prototypic representative of the highly virulent subsp. tularensis. The reasons for the high virulence of this subspecies are contained in its genomic sequence, along with clues to other aspects of its biology and the evolutionary processes that affected it. The availability of the genome sequence has also provided a foundation for future experimental and theoretical work on this species. A prominent feature of the F. tularensis genome sequence was the high AT content – 67.1%. It has been shown that such biases affect amino acid composition [72] (69), and this was the case for F. tularensis. An analysis of its amino acid content in comparison to other bacteria demonstrates an excessive representation of amino acids encoded by AT-
32

rich codon families, such as isoleucine; amino acids encoded by GC-rich codon families, such as alanine, are disproportionately few (unpubl. Fig. 11).
Reductive evolution The F. tularensis genomic sequence was also found to contain an abundance of pseudogenes and disrupted metabolic pathways. This signifies an ongoing genome reduction process; a feature commonly found in bacteria with obligate intracellular associations. Because of a nutrient-rich and invariant milieu, such bacteria are believed to experience relaxed selectional constraints and consequent loss of functions that are dispensable (i.e. “use it or lose it”). It has also been argued that population “bottlenecks” (reduction in effective population size) may accelerate propagation and fixation of also slightly deleterious mutations. Repeated bottlenecks, affecting vertical transition, are thought to play a role in the evolution of some endosymbionts [32,73]. Bottlenecks may also arise during the colonisation of a new niche, and have been suggested as an explanation for the high number of pseudogenes observed in some pathogens, such as Salmonella enterica serovar Typhi [74], and Y. pestis [75]. Some reports also suggest that adaptive selectional forces may cause genomic reduction [76,77]. It is not yet clear which of these models best describes the reductive process in F. tularensis. The bacterium appears to be adapted to a rich supply of nutrients and an invariable lifestyle. It has lost the ability to synthesize several amino acids, an observation supported by the fastidious nutritional requirements exhibited by F. tularensis [78]. Also, few transcriptional regulators were found in the genome. Using the annotation model provided by the Institute for Genomic Research (TIGR), the strain SCHU S4 is found to contain a total of 47 regulatory proteins, a number broadly similar to that for other intracellular endosymbionts and parasites, such as Buchnera aphidicola Sg (9), Coxiella burnetii RSA 493 (53), Rickettsia prowazekii Madrid E (26). This number is very different from E. coli CFT073 (417) and Bacillus cereus 10987 (386), which are free-living. An in-depth analysis of transcriptional regulators in F. tularensis indicates that that many are orphaned components of two-component systems; these are
33

probably non-functional. F. tularensis is considered to be a facultative intracellular bacterium because of its ability to grow inside cells as well as its capacity for extracellular culturing. It is possible that the bacterium, in its natural lifecycle, is confined to an intrabiotic or intracellular environment.
Insertion sequence elements Two types of insertion sequences (ISFtu1 and ISFtu2) were previously identified in F. tularensis. The analysis performed in paper IV revealed that these are the predominant types in the genome, with 50 copies of ISFtu1, a transposon belonging to the IS630 family, and sixteen copies of ISFtu2, an IS5-family element. In addition, novel IS elements were discovered: ISFtu3 to 5, although there were only a few copies of these. While most members of the IS630 family of insertion sequences possess a single open reading frame (32) it was found that translation of the ISFtu1 CDS requires ribosomal frameshifting (or transcriptional slippage). In ISFtu1, the first aspartic acid residue of the DDE triad, which is essential for transposition, is generated only following a frameshift. This could be a mechanism for controlling the transposition rate of this element. The recent availability of additional F. tularensis genomic sequences has allowed comparisons to be made between ISFtu1 elements in different lineages. From such comparisons (unpubl.), ISFtu1 appears to be present in all lineages, albeit with different numbers of copies (Table 1). Such comparisons also revealed that the frameshift mutation was present in all lineages for which genomic sequence data exist, further supporting the suggestion that there is a universal frameshifting mechanism.
34

Table 1. Copies of ISFtu1 in F. tularensis genome sequences
Genetic clade Strain Copies Ftu1 (no) GenBank acc. Reference
subsp. tularensis (A.I) SCHU S4 50 AJ749949 Paper IV (A.II) WY96-3418 52 CP000608 Unpubl.
subsp. mediasiatica FSC 147 many* NA [79] FSC 148 many* NA [79]
subsp. holarctica LVS 59 AM233362 Unpubl. OSU18 59 CP000437 [80]
subsp. novicida U112 1 CP000439 Unpubl. *Expansion indicated by restriction fragment length polymorphisms using an ISFtu1-specific probe.
IS elements and evolution It is interesting to note that, previously, expansions of insertion sequences have been found to accompany the evolution of several pathogenic bacterial species. For example, genomic comparisons of the species Bordetella bronchiseptica, Bordetella parapertussis and Bordetella pertussis, indicate such expansions in the two latter species, which are capable of causing whooping cough in humans. In contrast IS elements were not found in B. bronchiseptica, which causes a mild and often undetected respiratory disease [81]. Genomic data suggests that B. pertussis and B. parapertussis evolved from an ancestral strain similar in genetic composition to B. bronchiseptica. An example of this mode of evolution is also provided by the yersiniae, where the evolution of Yersinia pestis from an ancestral strain resembling Yersinia pseudotuberculosis was accompanied by a amplification of IS elements [82].
35

In F. tularensis, the numbers of pseudogenes present in the genomic sequences are high in strains that belong to subspp. holarctica (LVS, OSU18) and tularensis (SCHU S4), while the annotated genome sequence of the only representative available for subsp. novicida demonstrates only 14 pseudogenes. Future analysis will demonstrate how many genes are uniquely present in the different subspecies of F. tularensis, but considering data currently available, it appears likely that F. tularensis subspp. tularensis, holarctica and mediasiatica, evolved from an ancestral strain that was more similar in composition to F. tularensis subsp. novicida. The presence of only one copy of ISFtu1 in the subsp. novicida strain (Table 1) suggests that an amplification of this IS element occurred in the other F. tularensis lineages after diverging from the subsp. novicida lineage. It is interesting that this correlates to an increase in virulence, as observed in yersiniae and bordetellae. Since ISFtu1 was probably already present in the progenitor of both subsp. novicida and the other lineages, it is likely that the observed amplification of this IS element did not occur primarily as a result of a high infectivity. Rather, it may indicate that the bacteria experienced a population bottleneck - slightly deleterious mutations permitted by a reduced purifying selection - or that they invaded a new niche in which amplification was facilitated by loss of selectional restriction on particular superfluous loci. Both mechanisms could have contributed. It is interesting to note that the amplification of ISFtu1 permitted the duplication of the FPI as a result of the insertion of a copy of the IS element on the flank of this region. The FPI represents a locus that is central to the special mode of virulence exhibited by this bacterium. Within the conceptual framework of source–sink dynamics [83], it is tempting to suggest a model where the bacterium inefficiently invaded a new niche, which allowed amplification of the IS element, after which duplication of the FPI followed as a gene dosage adaptation to its new environment.
36

1350
5SrRNA
23SrRNA
16SrRNA
pdpA1 pdpB1 1346
1347
1348
1349 1351
1352
1353
pdpC1
1355
iglD1
iglC1
iglB1
iglA1
pdpD1
1361
1362'
ISFtu1
1705
5SrRNA
23SrRNA
16SrRNA
pdpA2 pdpB2 1701
1702
1703
1704 1706
1707
1708
pdpC2
1710
iglD2
iglC2
iglB2
iglA2
pdpD2
1716
1717'
ISFtu1
55.23
32.26
19.89
10 kb
1,374,371
1,767,715
1,408,281
1,801,625
10 kb
Figure 12. The organization of the duplicated region in F. tularensis strain SCHU S4. The left scale indicates G+C content. Blue indicates RNA coding regions; green indicates open reading frames encoding hypothetical proteins; brown indicates pseudogenes; and pink indicates IS elements. Open reading frame labels refer to the corresponding annotated gene or FTT number in the genome sequence of SCHU S4.
Virulence mechanisms Our understanding of the virulence mechanisms effective in F. tularensis is limited. The genome sequence failed to reveal the existence of common virulence mechanisms such as toxins or machineries for type III or IV secretion, which commonly constitute virulence determinants in many pathogens. It was intriguing that F. tularensis appeared to contain a locus for the production of siderophores, while being devoid of classical uptake systems for iron acquisition, commonly important for virulence. Analysis of the genome sequence suggests that the virulence of F. tularensis is based on unusual mechanisms. As with subsp. holarctica LVS strain [48], but unlike subsp. novicida, the SCHU S4 strain was found to contain a perfect duplication of a region of significant importance for F. tularensis virulence (Fig. 12). The analysis of genomic sequences demonstrated that this region is also
37

duplicated in subsp. mediasiatica strain FSC147 (unpubl.) as well as in strain WY96, which belongs to a separate clade (A.II) (unpubl.) of subsp. tularensis. It has been speculated that this region may represent horizontally transferred genetic material; it has therefore been named the “Francisella pathogenicity island” (FPI). Some proteins encoded by genes present in this region have recently been found to function collectively as a type VI secretion system, a conceptually new system described recently in Vibrio cholerae [84]. One highly speculative suggestion put forward in the paper was based on the observation that strains of F. tularensis characteristically produce ammonia and increase the pH if cultured in an acidified medium [78]. Interestingly, although the mechanism is uncertain, ammonium chloride can be used experimentally to prevent phagosome–lysosome fusion in eukaryotic cells [85]. Since F. tularensis also has the ability to prevent phagosome–lysosome fusion [86], we speculated that ammonia produced by this bacterium could play a role in virulence. Several genes were found that may directly account for ammonia production, although none these have yet been implicated in virulence. The hypothesis has gained partial, indirect support however as enzymes that degrade and synthesize cyanophycin have been implicated in virulence indicating the possibility of such a mechanism. Cyanophycin is an amino acid polymer composed of an aspartic acid backbone and arginine side groups, and its break-down is catalyzed by this enzyme, which might potentially produce a substrate for ammonia production. Although highly speculative, it is possible that the loss of individual enzymes that produce ammonia is compensated for by other enzymes, while the loss of the nitrogen source attenuates the bacterium. The existence of cyanophycin molecules is suggested by the observation of inclusion bodies in transmission electron micrograph (TEM) images of F. tularensis (not shown). Interesting genes that were identified further include capB and capC, which have homologs that are required for capsule biosynthesis and are crucial for virulence in the anthrax pathogen Bacillus anthracis. It was, therefore, hypothesised in paper IV that the capsule of F. tularensis, in common with B. anthracis, might contain poly-γ-glutamine. The capBC locus has recently been independently identified as a virulence determining region in two separate studies by Su [87] and Weiss [88]. A gene cluster was also highlighted in paper IV that could encode a polysaccharide in addition to the lipopolysaccharide O-antigen. These genes were also found in the study by Weiss to be virulence determinants [88]. All genes necessary for type IV pili biosynthesis were also identified,
38

components of which have been demonstrated to play key roles in virulence [59].
Whole-genome phylogeny F. tularensis is recognized as a γ-proteobacterial species [21], but its phylogenetic position within the subgroup has been uncertain. Based on analysis of 16S ribosomal RNA molecules, several phylogenetic investigations have suggested that this lineage could have a deep origin [89,90], maybe diverging even before the gamma/beta-proteobacterial division [91]. This pattern was corroborated by the phylogenetic analyses performed as part of the publication of the genome sequence, utilizing a concatenated set of 10 protein sequences for 15 γ-proteobacterial species, F. tularensis, and the Firmicute B. anthracis as an outgroup. The deep divergence was supported by strong bootstrap evidence (>75%) for more than 40% of individual phylogenetic gene trees, inferred from 200 sets of orthologous genes. Despite compelling evidence supporting a basal phylogenetic divergence of this lineage, a caveat should be noted. An issue concerning phylogenetic reconstructions is that compositionally heterogeneous taxa will tend to repel, and compositionally homogeneous taxa attract [92-94]. F. tularensis exhibits a significantly deviating amino acid composition (see Fig. 11) and a phylogenetic reconstruction that includes this bacterium exemplifies a situation where problems might be anticipated. Further studies on the impact of these biases on the phylogenetic position of F. tularensis are therefore warranted.
39

0.1100,100
100,100
100,100
100,100
100,100100,100
100,100
52,97
69,74
100,100
100,100
100,100100,100
100,94
Bacillus anthracis
Francisella tularensis
Legionella pneumophila
Coxiella burnetii
Xylella fastidiosa
Xanthomonas axonopodis
Xanthomonas campestris
Pseudomonas aeruginosa
Shewanella oneidensis
Vibrio cholerae
Haemophilus influenzae
Pasteurella multocida
Yersinia pestis
Salmonella typhimurium
Salmonella enterica
Shigella flexneri
Escherichia coli
Figure 13. Phylogenetic relationships of 16 γ-proteobacterial species inferred from a concatenated alignment of the proteins encoded by dnaA, ftsA, mfd, mraY, murB, murC, parC, recA, recG, and rpoC. Bacillus anthracis was used as the outgroup. The branch lengths are according to the reconstruction with the neighbour-joining method. Values at nodes are bootstrap support values for the neighbour-joining and maximum parsimony methods (in that order).
40

CONCLUSIONS
The species F. tularensis generally exhibits a strong genetic compositional homogeneity. The two subspp. mediasiatica and tularensis are highly similar in their composition, whereas strains of subsp. holarctica are more divergent. Japanese strains of subsp. holarctica are compositionally distinct from other strains of the subsp. holarctica. Size polymorphisms that distinguish F. tularensis lineages have commonly resulted from repeat-mediated excision. Such unidirectional events can provide evidence of phylogenetic rooting and evolutionary direction in phylogenetic inference. Phylogenetic analyses support a scenario where subsp. novicida diverged deeply within F. tularensis. Multilocus sequence typing and the presence of insertion-deletion polymorphisms corroborate the monophyly of subspp. tularensis and mediasiatica, and subsp. holarctica as a sister clade. This topology corresponds well to observed compositional similarities and differences between the subspecies. In F. tularensis, insertion-deletion polymorphisms exist which were probably formed by mechanisms other than repeat-mediated recombination. In the genetic typing of F. tularensis isolates, such polymorphisms may provide stable, canonical markers well-suited for combined analysis with multilocus variable-number tandem-repeat analysis markers. Direct-mediated excision is an important natural attenuating mechanism for F. tularensis strains: Genetic mutations at two loci identified in this work as polymorphic were later found to be effective in attenuating virulent F. tularensis isolates. The recent specialization to an intrabiotic or intracellular niche is suggested in F. tularensis by the obvious signs of an ongoing genome degradation process; many pseudogenes and disrupted metabolic pathways were found in its genomic sequence.
41

Unusual mechanisms contribute to the virulence of F. tularensis. For example, genes that encode type III and IV secretion systems are common virulence determinants in many pathogens, but are missing in F. tularensis. Also, the ability to acquire iron is likely crucial, and the bacterium has capacity to produce a siderphore, but lacks a classic uptake system for iron complexes. F. tularensis harbours a type IV pili system and a locus that putatively produce a poly-γ-glutamic acid capsule, similar to that found in B. anthracis. Both have been confirmed as virulence factors. Because of a phylogenetic and compositional resemblance, the reason for the high virulence of the subspecies tularensis subclade A.I may be better determined by genetic comparisons to the subsp. tularensis subclade A.II or mediasiatica than to subsp. holarctica.
42

ACKNOWLEDGEMENTS
It is a difficult task to list all the people who contributed, directly and indirectly, to this thesis. I therefore apologize in advance for anyone I have (inevitably…) forgotten to mention below. Firstly, I would like to thank my two excellent supervisors, Mats Forsman and Anders Sjöstedt for most things: support, guidance, common-sense, and deep knowledge of the subject. Especially during the last months, I have much appreciated Anders helpful attitude. Mats’ office has been located only one room away from my own, so I’ve had the opportunity to benefit from his clarity more often. Anders Johansson and Kerstin Svensson have also received continuous harassment, both at work and at home. They would all likely have asked for secret telephone numbers had I not finished this thesis soon. The rest of Mats’ FOI group is also just terrific bunch of people; Anki Daniel, Eva, Johanna, Linda, Malin, Mona, Per, Tina, Stina, Ulla, and the support I’ve received from the department has also been excellent, Britt and Ondina! And the university people; Margareta, Henrik, Igor, Helén, Helena, Konstantin, Linda, Malin, Mattias, Patrik, Carl, Blanka, Svenja, Stephan, what would tularemia be without you?! I would also like to thank the FOI and Försvarsmakten supporting this work, and all co-authors and collaborators, past and present; Martien Broekhuijsen at TNO, the Proteomics group at the Purkyne Military Medical Academy in Hradec Králové (Martin, Ivona, Juraj, Jana, Jirka, Ales) and the people at the DSTL, Porton Down (Stephen, Petra, Melanie, Rick). Finally, I need to apologize to my unscientific personal friends (Andreas and Maria, Håkan and Elin, Johan and Sara, Lars and Miriam Matts, Magnus and Miriam, Martin och Lena): I will make every effort to see you all a great deal more from now on. Mamma, Pappa, Edvin Ylva!
43

REFERENCES
[1] McCoy, G.W. (1911). Plague-like disease in rodents. Public
Health Bulletin, Washington DC 43, 53-71. [2] Ohara, S. (1954). Studies on yato-byo (Ohara's disease, tularemia
in Japan). I. Jpn J Exp Med 24, 69-79. [3] Trevisanato, S.I. (2004). Did an epidemic of tularemia in
Ancient Egypt affect the course of world history? Med Hypotheses 63, 905-10.
[4] McCoy, G.W. and Chapin, C.W. (1912). Further observations on a plague-like disease of rodents with a preliminary note on the causative agent, Bacterium tularense. The Journal of Infectious Diseases, Chicago 10
[5] Francis, E. (1928). A summary of the present knowledge of tularemia. . Medicine, Baltimore 7, 411–432.
[6] Riedel, S. (2004). Biological warfare and bioterrorism: a historical review. Proc (Bayl Univ Med Cent) 17, 400-6.
[7] Alibek, K. (1999) Biohazard, Random House Inc. New York. [8] Geissler, E. (2005). Alibek, Tularaemia and The Battle of
Stalingrad. THE CBW CONVENTIONS BULLETIN 69+70, 10-15.
[9] Wheelis, M., Rózsa, L. and Dando, M. (2006) Deadly Cultures: Biological Weapons Since 1945, Harvard University Press
[10] Staples, J.E., Kubota, K.A., Chalcraft, L.G., Mead, P.S. and Petersen, J.M. (2006). Epidemiologic and molecular analysis of human tularemia, United States, 1964-2004. Emerg Infect Dis 12, 1113-8.
[11] Tarnvik, A., Priebe, H.S. and Grunow, R. (2004). Tularaemia in Europe: an epidemiological overview. Scand J Infect Dis 36, 350-5.
[12] Beier, C.L., Horn, M., Michel, R., Schweikert, M., Gortz, H.D. and Wagner, M. (2002). The genus Caedibacter comprises endosymbionts of Paramecium spp. related to the Rickettsiales (Alphaproteobacteria) and to Francisella tularensis (Gammaproteobacteria). Appl Environ Microbiol 68, 6043-50.
[13] Fryer, J.L. and Hedrick, R.P. (2003). Piscirickettsia salmonis: a Gram-negative intracellular bacterial pathogen of fish. J Fish Dis 26, 251-62.
44

[14] Wenger, J.D., Hollis, D.G., Weaver, R.E., Baker, C.N., Brown, G.R., Brenner, D.J. and Broome, C.V. (1989). Infection caused by Francisella philomiragia (formerly Yersinia philomiragia). A newly recognized human pathogen. Ann Intern Med 110, 888-92.
[15] Ostland, V.E., Stannard, J.A., Creek, J.J., Hedrick, R.P., Ferguson, H.W., Carlberg, J.M. and Westerman, M.E. (2006). Aquatic Francisella-like bacterium associated with mortality of intensively cultured hybrid striped bass Morone chrysops x M. saxatilis. Dis Aquat Organ 72, 135-45.
[16] Olsen, A.B., Mikalsen, J., Rode, M., Alfjorden, A., Hoel, E., Straum-Lie, K., Haldorsen, R. and Colquhoun, D.J. (2006). A novel systemic granulomatous inflammatory disease in farmed Atlantic cod, Gadus morhua L., associated with a bacterium belonging to the genus Francisella. J Fish Dis 29, 307-11.
[17] Hsieh, C.Y., Tung, M.C., Tu, C., Chang, C. and Tsai, S. (2006). Enzootics of visceral granulomas associated with Francisella-like organism infection in tilapia (Oreochromis spp.). Aquaculture 254, 129-138.
[18] Kay, W., Petersen, B.O., Duus, J.O., Perry, M.B. and Vinogradov, E. (2006). Characterization of the lipopolysaccharide and beta-glucan of the fish pathogen Francisella victoria. Febs J 273, 3002-13.
[19] Kamaishi, T., Fukuda, Y., Nishiyama, M., Kawakami, H., Matsuyama, T., Yoshinaga, T. and Oseko, N. (2005). Identification and pathogenicity of intracellular Francisella bacterium in three-line grunt Parapristipoma trilineatum. Fish pathol. 40, 67–71.
[20] Nylund, A., Ottem, K.F., Watanabe, K., Karlsbakk, E. and Krossoy, B. (2006). Francisella sp. (Family Francisellaceae) causing mortality in Norwegian cod (Gadus morhua) farming. Arch Microbiol 185, 383-92.
[21] Forsman, M., Sandstrom, G. and Sjostedt, A. (1994). Analysis of 16S ribosomal DNA sequences of Francisella strains and utilization for determination of the phylogeny of the genus and for identification of strains by PCR. Int J Syst Bacteriol 44, 38-46.
[22] Scoles, G.A. (2004). Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J Med Entomol 41, 277-86.
45

[23] Barns, S.M., Grow, C.C., Okinaka, R.T., Keim, P. and Kuske, C.R. (2005). Detection of diverse new Francisella-like bacteria in environmental samples. Appl Environ Microbiol 71, 5494-500.
[24] Sjöstedt, A. (2005) Bergey's manual of systematic bacteriology, Springer-Verlag. New York, N.Y.
[25] Whipp, M.J. et al. (2003). Characterization of a novicida-like subspecies of Francisella tularensis isolated in Australia. J Med Microbiol 52, 839-42.
[26] Johansson, A. et al. (2004). Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J Bacteriol 186, 5808-18.
[27] Farlow, J., Wagner, D.M., Dukerich, M., Stanley, M., Chu, M., Kubota, K., Petersen, J. and Keim, P. (2005). Francisella tularensis in the United States. Emerg Infect Dis 11, 1835-41.
[28] Sandstrom, G., Sjostedt, A., Forsman, M., Pavlovich, N.V. and Mishankin, B.N. (1992). Characterization and classification of strains of Francisella tularensis isolated in the central Asian focus of the Soviet Union and in Japan. J Clin Microbiol 30, 172-5.
[29] Olsufjev, N.G. and Meshcheryakova, I.S. (1982). Infraspecific taxonomy of tularemia agent Francisella tularensis McCoy et Chapin. J Hyg Epidemiol Microbiol Immunol 26, 291-9.
[30] Hollis, D.G., Weaver, R.E., Steigerwalt, A.G., Wenger, J.D., Moss, C.W. and Brenner, D.J. (1989). Francisella philomiragia comb. nov. (formerly Yersinia philomiragia) and Francisella tularensis biogroup novicida (formerly Francisella novicida) associated with human disease. J Clin Microbiol 27, 1601-8.
[31] Gal-Mor, O. and Finlay, B.B. (2006). Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol 8, 1707-19.
[32] Perez-Brocal, V. et al. (2006). A small microbial genome: the end of a long symbiotic relationship? Science 314, 312-3.
[33] Jolley, K.A., Wilson, D.J., Kriz, P., McVean, G. and Maiden, M.C. (2005). The influence of mutation, recombination, population history, and selection on patterns of genetic diversity in Neisseria meningitidis. Mol Biol Evol 22, 562-9.
[34] Moran, N.A. and Plague, G.R. (2004). Genomic changes following host restriction in bacteria. Curr Opin Genet Dev 14, 627-33.
[35] Baldo, L., Bordenstein, S., Wernegreen, J.J. and Werren, J.H. (2006). Widespread recombination throughout Wolbachia genomes. Mol Biol Evol 23, 437-49.
46

[36] Coscolla, M. and Gonzalez-Candelas, F. (2007). Population structure and recombination in environmental isolates of Legionella pneumophila. Environ Microbiol 9, 643-56.
[37] Fraser, C., Hanage, W.P. and Spratt, B.G. (2007). Recombination and the nature of bacterial speciation. Science 315, 476-80.
[38] van Belkum, A., Struelens, M., de Visser, A., Verbrugh, H. and Tibayrenc, M. (2001). Role of genomic typing in taxonomy, evolutionary genetics, and microbial epidemiology. Clin Microbiol Rev 14, 547-60.
[39] Chaudhuri, R.R. et al. (2007). Genome Sequencing Shows that European Isolates of Francisella tularensis Subspecies tularensis Are Almost Identical to US Laboratory Strain Schu S4. PLoS ONE 2, e352.
[40] Pang, J.C., Chiu, T.H., Helmuth, R., Schroeter, A., Guerra, B. and Tsen, H.Y. (2007). A pulsed field gel electrophoresis (PFGE) study that suggests a major world-wide clone of Salmonella enterica serovar Enteritidis. Int J Food Microbiol
[41] Strommenger, B., Kettlitz, C., Weniger, T., Harmsen, D., Friedrich, A.W. and Witte, W. (2006). Assignment of Staphylococcus isolates to groups by spa typing, SmaI macrorestriction analysis, and multilocus sequence typing. J Clin Microbiol 44, 2533-40.
[42] Johansson, A., Forsman, M. and Sjostedt, A. (2004). The development of tools for diagnosis of tularemia and typing of Francisella tularensis. Apmis 112, 898-907.
[43] Burke, D.S. (1977). Immunization against tularemia: analysis of the effectiveness of live Francisella tularensis vaccine in prevention of laboratory-acquired tularemia. J Infect Dis 135, 55-60.
[44] Fleischmann, R.D. et al. (1995). Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496-512.
[45] Liolios, K., Tavernarakis, N., Hugenholtz, P. and Kyrpides, N.C. (2006). The Genomes On Line Database (GOLD) v.2: a monitor of genome projects worldwide. Nucleic Acids Res 34, D332-4.
[46] Bailey, L. et al. (2007). Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett 581, 587-95.
47

[47] Scarselli, M., Giuliani, M.M., Adu-Bobie, J., Pizza, M. and Rappuoli, R. (2005). The impact of genomics on vaccine design. Trends Biotechnol 23, 84-91.
[48] Nano, F.E. et al. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol 186, 6430-6.
[49] Sievertzon, M., Nilsson, P. and Lundeberg, J. (2006). Improving reliability and performance of DNA microarrays. Expert Rev Mol Diagn 6, 481-92.
[50] Forsman, M., Sandstrom, G. and Jaurin, B. (1990). Identification of Francisella species and discrimination of type A and type B strains of F. tularensis by 16S rRNA analysis. Appl Environ Microbiol 56, 949-55.
[51] Jellison, W.L. (1974) Tularemia in North America, 1930-1974, University of Montana Foundation. Missoula.
[52] Nano, F. and Schmerk, C. (2007). The Francisella Pathogenicity Island. Ann N Y Acad Sci
[53] Samrakandi, M.M. et al. (2004). Genome diversity among regional populations of Francisella tularensis subspecies tularensis and Francisella tularensis subspecies holarctica isolated from the US. FEMS Microbiol Lett 237, 9-17.
[54] Konstantinidis, K.T. and Tiedje, J.M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102, 2567-72.
[55] Welch, R.A. et al. (2002). Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A 99, 17020-4.
[56] Nubel, U. et al. (2006). Population structure of Francisella tularensis. J Bacteriol 188, 5319-24.
[57] Cohan, F.M. (2001). Bacterial species and speciation. Syst Biol 50, 513-24.
[58] Burrows, L.L. (2005). Weapons of mass retraction. Mol Microbiol 57, 878-88.
[59] Forslund, A.L. et al. (2006). Direct repeat-mediated deletion of a type IV pilin gene results in major virulence attenuation of Francisella tularensis. Mol Microbiol 59, 1818-30.
[60] Twine, S. et al. (2005). A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect Immun 73, 8345-52.
48

[61] Rohmer, L. et al. (2006). Potential source of Francisella tularensis live vaccine strain attenuation determined by genome comparison. Infect Immun 74, 6895-906.
[62] Dennis, D.T. et al. (2001). Tularemia as a biological weapon: medical and public health management. Jama 285, 2763-73.
[63] van Belkum, A. (2007). Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol Med Microbiol 49, 22-7.
[64] Field, D., Magnasco, M.O., Moxon, E.R., Metzgar, D., Tanaka, M.M., Wills, C. and Thaler, D.S. (1999). Contingency loci, mutator alleles, and their interactions. Synergistic strategies for microbial evolution and adaptation in pathogenesis. Ann N Y Acad Sci 870, 378-82.
[65] van Belkum, A. (1999). The role of short sequence repeats in epidemiologic typing. Curr Opin Microbiol 2, 306-11.
[66] Achtman, M. et al. (2004). Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci U S A 101, 17837-42.
[67] Keim, P., Van Ert, M.N., Pearson, T., Vogler, A.J., Huynh, L.Y. and Wagner, D.M. (2004). Anthrax molecular epidemiology and forensics: using the appropriate marker for different evolutionary scales. Infect Genet Evol 4, 205-13.
[68] Arricau-Bouvery, N. et al. (2006). Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol 6, 38.
[69] Van Ert, M.N. et al. (2007). Strain-specific single-nucleotide polymorphism assays for the Bacillus anthracis Ames strain. J Clin Microbiol 45, 47-53.
[70] Nei, M. and Kumar, S. (2000) Molecular Evolution and Phylogenetics, Oxford University Press. New York.
[71] Peacock, S.J., de Silva, G.D., Justice, A., Cowland, A., Moore, C.E., Winearls, C.G. and Day, N.P. (2002). Comparison of multilocus sequence typing and pulsed-field gel electrophoresis as tools for typing Staphylococcus aureus isolates in a microepidemiological setting. J Clin Microbiol 40, 3764-70.
[72] Singer, G.A. and Hickey, D.A. (2000). Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol Biol Evol 17, 1581-8.
[73] Moran, N.A. (1996). Accelerated evolution and Muller's rachet in endosymbiotic bacteria. Proc Natl Acad Sci U S A 93, 2873-8.
49

[74] Parkhill, J. et al. (2001). Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413, 848-52.
[75] Parkhill, J. et al. (2001). Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413, 523-7.
[76] Dufresne, A., Garczarek, L. and Partensky, F. (2005). Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol 6, R14.
[77] Tsolaki, A.G. et al. (2004). Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc Natl Acad Sci U S A 101, 4865-70.
[78] Traub, A., Mager, J. and Grossowicz, N. (1955). Studies on the nutrition of Pasteurella tularensis. J Bacteriol 70, 60-9.
[79] Thomas, R., Johansson, A., Neeson, B., Isherwood, K., Sjostedt, A., Ellis, J. and Titball, R.W. (2003). Discrimination of human pathogenic subspecies of Francisella tularensis by using restriction fragment length polymorphism. J Clin Microbiol 41, 50-7.
[80] Petrosino, J.F. et al. (2006). Chromosome rearrangement and diversification of Francisella tularensis revealed by the type B (OSU18) genome sequence. J Bacteriol 188, 6977-85.
[81] Parkhill, J. et al. (2003). Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35, 32-40.
[82] Chain, P.S. et al. (2004). Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 101, 13826-31.
[83] Sokurenko, E.V., Gomulkiewicz, R. and Dykhuizen, D.E. (2006). Source-sink dynamics of virulence evolution. Nat Rev Microbiol 4, 548-55.
[84] Pukatzki, S., Ma, A.T., Sturtevant, D., Krastins, B., Sarracino, D., Nelson, W.C., Heidelberg, J.F. and Mekalanos, J.J. (2006). Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A 103, 1528-33.
[85] Gordon, A.H., Hart, P.D. and Young, M.R. (1980). Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature 286, 79-80.
[86] Anthony, L.D., Burke, R.D. and Nano, F.E. (1991). Growth of Francisella spp. in rodent macrophages. Infect Immun 59, 3291-6.
50

[87] Su, J., Yang, J., Zhao, D., Kawula, T.H., Banas, J.A. and Zhang, J.R. (2007). Genome-Wide Identification of Francisella tularensis Virulence Determinants. Infect Immun
[88] Weiss, D.S., Brotcke, A., Henry, T., Margolis, J.J., Chan, K. and Monack, D.M. (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A 104, 6037-42.
[89] Maurin, M. and Raoult, D. (1999). Q fever. Clin Microbiol Rev 12, 518-53.
[90] Roux, V., Bergoin, M., Lamaze, N. and Raoult, D. (1997). Reassessment of the taxonomic position of Rickettsiella grylli. Int J Syst Bacteriol 47, 1255-7.
[91] Noda, H., Munderloh, U.G. and Kurtti, T.J. (1997). Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol 63, 3926-32.
[92] Foster, P.G. and Hickey, D.A. (1999). Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J Mol Evol 48, 284-90.
[93] Foster, P.G. (2004). Modeling compositional heterogeneity. Syst Biol 53, 485-95.
[94] Bergsten, J. (2005). A review of long-branch attraction. Cladistics 21, 163–193.
51