the american journal of human genetics supplemental …€¦ · · 2016-03-24she continued to...
TRANSCRIPT

The American Journal of Human Genetics
Supplemental Data
Recurrent Muscle Weakness with Rhabdomyolysis,
Metabolic Crises, and Cardiac Arrhythmia
due to Bi-allelic TANGO2 Mutations
Seema R. Lalani, Pengfei Liu, Jill A. Rosenfeld, Levi B. Watkin, Theodore Chiang,
Magalie Leduc, Wenmiao Zhu, Yan Ding, Shujuan Pan, Francesco Vetrini, Christina
Miyake, Marwan Shinawi, Tomasz Gambin, Mohammad K. Eldomery, Zeynep Hande
Coban Akdemir, Lisa Emrick, Yael Wilnai, Susan Schelley, Mary Kay Koenig, Nada
Memon, Laura S. Farach, Bradley P. Coe, Mahshid Azamian, Patricia Hernandez, Gladys
Zapata, Shalini N. Jhangiani, Donna M. Muzny, Timothy Lotze, Gary Clark, Angus
Wilfong, Hope Northrup, Adekunle Adesina, Carlos A. Bacino, Fernando Scaglia,
Penelope E. Bonnen, Jane Crosson, Jessica Duis, Gustavo H.B. Maegawa, David
Coman, Anita Inwood, Jim McGill, Eric Boerwinkle, Brett Graham, Art Beaudet,
Christine M. Eng, Neil Hanchard, Fan Xia, Jordan S. Orange, Richard A. Gibbs, James
R. Lupski, and Yaping Yang

Supplemental Data
Supplemental Case Reports
In the fifth family of Hispanic/Latino origin, subject 5 was diagnosed with speech and motor
delay, prolonged QTc interval and seizures, prior to her acute presentation at 7 years of age with
rhabdomyolysis. She had a concurrent mycoplasma infection at that time. Her CPK was reported
to be 60,000 U/L accompanied by myoglobinuria. She had another such episode preceded by
muscle weakness at 13 years of age, with acute rhabdomyolysis and cardiac arrest. She was off
nodalol for about a month prior to her presentation. Her CPK was 33,227 U/L. She was found to
have torsade de pointes from prolonged QTc interval and required defibrillation. Her highest
AST was 953 U/L, ALT was 540 U/L, repeat CPK was 16,790 U/L, with CPK-MB of 291 U/L.
She continued to have brief episodes of ventricular tachycardia (VT) with recurrent pulseless
arrest. Her serum potassium was normal during this time, ranging from 3.8-5.0 mmol/l. She had
a prolonged hospitalization course due to cardiac arrhythmia and subsequently had placement of
dual chamber implantable cardioverter defibrillator (ICD). Prior to discharge from the hospital,
she was ambulating with assistance, and her CPK was mildly elevated to 383 U/L.
Subject 6, a Hispanic male, was born to consanguineous parents who were second cousins once
removed. He was healthy until 1 year of age, albeit with developmental delay. Around one year
of age, he developed emesis and difficulty breathing, and was found to have hypoglycemia of 16
mg/dL, metabolic acidosis with blood gas showing pH of 7.1, bicarbonate of 16 mEq/L (normal
range 22-28 mEq/L), and a base deficit of 24.2 mmol/L. Lactate was elevated to 11.7 mmol/L.
Plasma ammonia level was mildly elevated to 102 μmol/L. CPK was not done during the acute
presentation. An acylcarnitine profile showed elevation of long and very long chain acylcarnitine

species, including C12, C14, C14-OH, C14-1-OH, C16, C16-1, C16-OH, and C16-1-OH. The
magnitude of the elevations was minor with the exception of C14, which was three times the
upper limit of normal. There was massive excretion of lactic acid on urine organic acids, as well
as marked elevations of 3-hydroxy-N-butyric and acetoacetic acids (ketone bodies), as well as
adipic, suberic, sebacic, and 3-hydroxysebacic (dicarboxylic) acids. He was stabilized with
normalization of biochemical studies except for lactate, which remained elevated to 4.1 mmol/L.
His CPK after stabilization was normal at 103 U/L. Chromosomal microarray and Fragile X
studies were performed due to his developmental delay and were normal. Due to failure to thrive,
a gastrostomy was placed to provide adequate nutrition. When evaluated at 16 months of age, he
was unable to sit without assistance and was non-verbal. Physical examination was pertinent for
head circumference at the 6th percentile for age (FOC 45.5 cm) and hypotonia.
Subject 7 was of European ancestry, born to non-consanguineous parents. He was diagnosed
with generalized myoclonic and atonic seizures at 18 months of age. Ketogenic diet was
instituted at 2-½ years of age, and seven days after commencing the diet, he developed lethargy
and hypoglycemia (blood glucose of 12 mg/dL). Dark urine prompted further studies and he was
found to have acute rhabdomyolysis with CPK of 258,000 U/L. Following this episode, he had
neuroregression with mild ataxia, spasticity of lower extremities, and global developmental
delay. Muscle biopsy studies showed normal electron transport chain and mitochondrial DNA
content. Acylcarnitine profile, when he was metabolically stable, showed no abnormalities. Brain
MRI was essentially normal. Echocardiogram and 24-hour holter monitoring were normal. He
had one more similar episode of rhabdomyolysis before 4 years of age. His physical exam at age
9 years demonstrated myopathic facies, with brisk deep tendon reflexes, bilateral clonus and
scissoring gait.

Subjects 8, 9 and 10 from family 8 were born to non-consanguineous parents of European origin.
Subjects 8 (II-1-female) and 9 (II-2-male) were a product of a dichorionic, diamniotic twin
pregnancy. Global developmental delay was noted in the twins by 9 months of age. Seizures
were apparent in the female by 11 months and male by 15 months. Brain MRI in both twins
showed generalized cerebral atrophy with Wallerian degeneration of the cerebral peduncles.
Barium swallow showed oropharyngeal dysphagia. The CPK in subject 9 was always slightly
elevated, but with an intercurrent illness at 15 months, he had rhabdomyolysis with a peak CPK
of 17,900 U/L. Ammonia was mildly elevated to 120 umol/L. The twin female also had
rhabdomyolysis with CPK peaking at 97,500 U/L. The physical examination of both individuals
was pertinent for deceleration of growth parameters, hypertonia, and hyperreflexia with clonus.
The echocardiogram in the male was normal, however hypertrophic cardiomyopathy was noted
in the female at 20 months of age, requiring treatment with frusemide and captopril.
Acylcarnitine profile, mitochondrial studies, including POLG sequencing and respiratory chain
studies were normal. At 22 months of age, the affected male was found unconscious with blood
glucose of 21 mg/dL and lactate of 7.1 mmol/L. He was transferred to a pediatric intensive care
unit but unfortunately died the next day. The female twin passed away soon after her second
birthday due to worsening cardiomyopathy. The sibling of the twins (II-3-female; subject 10) is
currently living at one year of age, but has had seven episodes of severe acidosis usually
triggered by emesis, with pH going as low as 6.9, in association with lactic acidosis (peak lactate
17.1 mmol/L) and hypoglycemia. Ammonia was elevated during these episodes with highest
being 520 umol/L. Her CPK is usually normal and has been as high as 248 U/L. Urine organic
acids showed marked ketoacidosis, lactic acidosis and dicarboxylic aciduria. Plasma
acylcarnitine profile showed a pattern suggestive of VLCAD deficiency: C14:1 of 4.9 umol/L

(normal <0.73 umol/L), C14 of 1.5umol/L (normal <0.34 umol/L) and C10 of 0.6 umol/L
(normal <0.40 umol/L) with normal 3-hydroxybutyrylcarnitine and acetylcarnitine.
In family 9, subjects 11 and 12 were born to Middle Eastern consanguineous parents from Saudi
Arabia. Subject 11 (II-1) was a product of a monozygotic twin pair. The onset of first
neurological symptoms occurred by 2 years of age with recurrent jerky movements of upper and
lower limbs associated with recurrent falls. He was noted to have a progressively unsteady gait
and severe dysarthria with unintelligible speech. By 8 years, he was diagnosed with seizures,
dystonia, and global developmental delay. The brain MRI showed microcephalic appearance of
the brain but no structural abnormality. His TSH was reportedly high and T4 was normal.
During his hospitalization at age 8 years, he presented with generalized progressive weakness
and was found to have acute rhabdomyolysis. The initial evaluation showed elevated plasma
CPK >100,000 U/L and serum myoglobin of 8,366 ng/mL (normal <85 ng/mL). Aldolase was
elevated to 240 U/L, AST was 2,773 U/L and ALT was 1,305 U/L. Plasma acylcarnintine profile
showed elevations of propionyl-carnitine (C3; 1.01 umol/L: normal range <0.92 umol/L) and
decenoyl-carnitine (C10; 0.52 umol/L; normal range <0.40 umol/L) species. Cardiac assessment
showed normal ECG and echocardiogram. The twin monozygotic sibling of subject 11 shared a
similar clinical course and died at the age of 7 during an acute crisis of rhabdomyolysis. He had a
history of seizures with confirmatory atypical EEGs, progressive gait instability, and global
developmental delayed first noted by 2 years of age and reported dystonia versus spastic
diplegia. At 7 years of age, he presented with worsening of generalized weakness, inability to
walk, poor feeding, constant drooling and weight loss of about 3 kg during this time period.
Initial work-up showed a CPK of 33,048 U/L, elevated transaminases, with AST of 1,196 U/L,
an ALT of 423 U/L, and a serum myoglobin of 9,080 ng/mL. The highest CPK was 37,000 U/L

during the acute presentation. He ultimately died in the hospital after a cardiac arrest on the
ventilator.
Subject 12, the younger sibling of the twins in family 9 (II-3), was similarly affected. He was
diagnosed with developmental delay, seizures, hypothyroidism, microcephaly, progressive
spastic diplegia and episodes of poor balance by 4 years of age. He had multiple hospital
admissions for acute episodes of rhabdomyolysis with a peak CPK of 54,258 U/L and maximum
elevated lactate of 8.5 mmol/L. During one these admissions, ECG was characterized by QTc
prolongation. During an acute rhabdomyolysis episode, he developed torsade de pointes
progressing to VT and pulseless electrical activity (PEA) requiring resuscitation. Laboratory
studies at this time showed potassium of 2.4 meq/L (normal range 3.5-5.1 meq/L), magnesium of
2 meq/L (normal range 1.3-2 meq/L), and ionized calcium 1.07 mmol/L (normal range 1.13-1.32
mmol/L). During this admission, the CPK levels reached 38,975 U/L, and serum myoglobin was
elevated to 5,499 ng/mL. Given the clinical improvement, the uncertainty of the etiology and
potential risk of recurrent VT and PEA, an ICD was placed. He later developed involuntary
choreiform movements of the upper extremities. The brain MRI demonstrated subtle
abnormalities in the right lentiform nucleus. A muscle biopsy was performed and quantitative
PCR assay showed no evidence of mitochondrial DNA depletion. Respiratory mitochondrial
complexes enzymatic activity was within normal range. The biopsy revealed mild acute
neurogenic atrophy with no ragged red fibers or COX negative fibers. During the diagnosis
process, a mitochondrial disorder was suspected and vitamins B1, B6, C, E, riboflavin,
levocarnitine and co-enzyme Q-10 were introduced. Interestingly, since starting on these
medications no further episodes of acute rhabdomyolysis and metabolic decompensation were
noted.
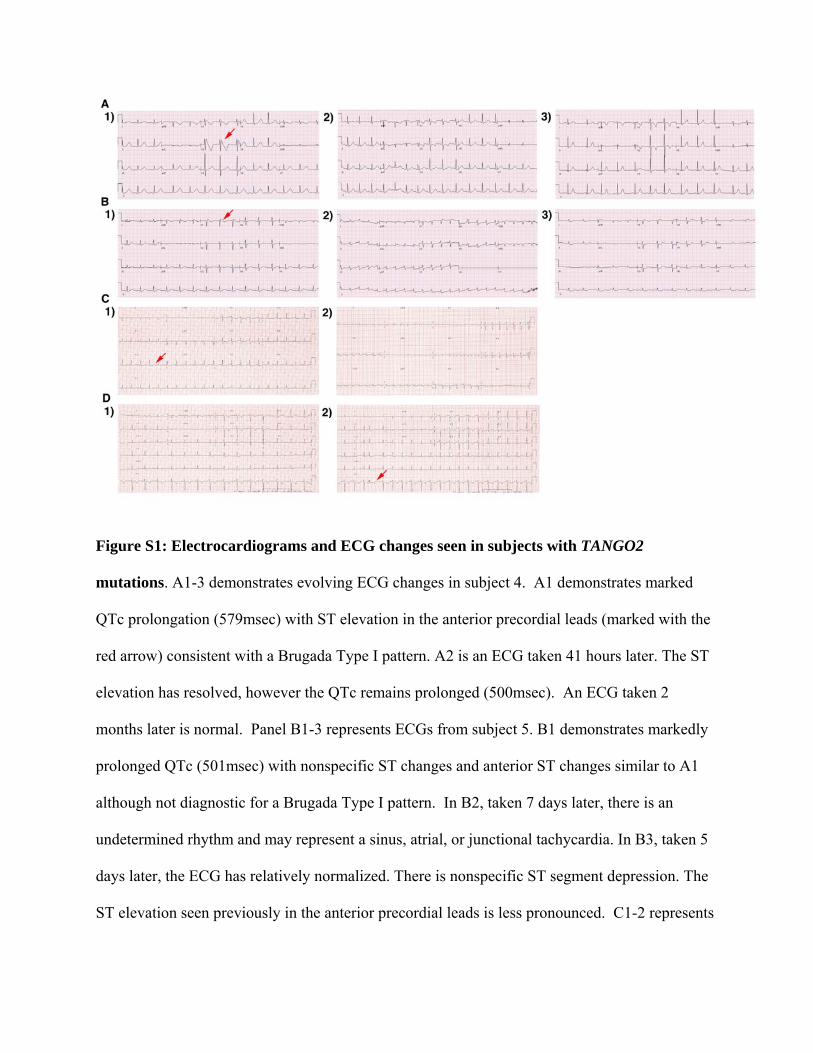
Figure S1: Electrocardiograms and ECG changes seen in subjects with TANGO2
mutations. A1-3 demonstrates evolving ECG changes in subject 4. A1 demonstrates marked
QTc prolongation (579msec) with ST elevation in the anterior precordial leads (marked with the
red arrow) consistent with a Brugada Type I pattern. A2 is an ECG taken 41 hours later. The ST
elevation has resolved, however the QTc remains prolonged (500msec). An ECG taken 2
months later is normal. Panel B1-3 represents ECGs from subject 5. B1 demonstrates markedly
prolonged QTc (501msec) with nonspecific ST changes and anterior ST changes similar to A1
although not diagnostic for a Brugada Type I pattern. In B2, taken 7 days later, there is an
undetermined rhythm and may represent a sinus, atrial, or junctional tachycardia. In B3, taken 5
days later, the ECG has relatively normalized. There is nonspecific ST segment depression. The
ST elevation seen previously in the anterior precordial leads is less pronounced. C1-2 represents

ECGs taken from subject 8. The initial ECG (C1) demonstrates QTc prolongation (QTc
490msec, marked by red arrow). C2 is taken few days later and demonstrates low voltages QRS
with nonspecific ST changes. D1-2. The baseline ECG (D1) in subject 9 does not demonstrate
QTc prolongation. Multiple subsequent ECGs demonstrate frequent isolated premature
ventricular contractions (marked by red arrow in D2).

Chr22:20028913 GCAAACTCTGCCTCCCGGGTTCACACCATTGTCCTGCCTCAGCCTCCCGAGTAGCTGGGACTACAGGCGCCCGCCACCACACCTGGCTA Exon3-9del GCAAGCTCCGCCTCCCGGGTTCACGCCATTCTCCTGCCTCAGCCTCCCGAGTAGCTGGGACTACAGGCGCCCCCTACCACGCCCGGCTA Chr22:20062731 ACAGAGTGAGACTCCGTCTCAAAAATAAAAAGTAAAATAATAATAATAATAATAAAAGGTAAACAGAAATCTTCATAATCTCAAATACT Chr22:20029002 ATTTTTT--GTATTTTTAGTAGAGACGGGGTTTTACCGTGTTAGCCAGGATGGTCTCGATCTCTTGACCTCGTGATCCTCCCACCTTGG Exon3-9del ATTTTTTTTGTATTTTTAGTAGAGACGGGGTTTCACCGTGTTAGCCAGGATGGTCTCGATCTCCTGACCTCGTGATCCGCCCGCCTCGG Chr22:20062820 GTAAGAAAACTTTGTCATTTCAACAGAGAAGATCAAGTTAAAGTTCTGCATCATAGCACTACTAATAAAGCTAATTTTAACAAAACCTT Chr22:20029089 CCTCTCAAAGTGCTGGGATTACAGGCGTGAGCCACCGGGCCCGGCCTAATTTTTGTATTTTTAATAGAGATGGGGTTTCACCATCTTGG Exon3-9del CCTCCCAAAGTGCTGGGATTACAGGCGTGAGCCACCGCGCCCGGCCAGATTTCTTTTCTATAGATGTTTTACAACTTAAAAAAAATATC Chr22:20062909 ATAAATGAATCCATCCAATCTCATGCAAGATAATATTTCTTTTCCAAGATTTCTTTTCTATAGATCTTTTACAACTTAAAAAAAATATC Chr22:20029178 CCAGGCTGGTCTTGAACTCCTGACCTCATGATCCACCCACCTCGGCCTCGCAAAATGCTGGGATTACAGGCATGAGCCACCGTACCCAG Exon3-9del TGGCCGGGCACCGTGGCTCATGCCTGTAATCCCAGCACTTTGGGAGGCCGAGGCGGGTGGATCATGAGGGCAGGAGATTGAGACCAGCC Chr22:20062998 TGGCCGGGCACGGTGGCTCATGCCTGTAATCCCAGCACTTTGGGAGGCCGAGGCGGGTGGATCATGAGGTGAGGAGATCGAGACCAGCC
rs573015985 A: 99.980% (5007 / 5008); G: 0.020% (1 / 5008)
rs544842733 No data available
rs193263084 No data available
rs530937322 No data available
rs174881 C: 50.000% (4 / 8); G: 50.000% (4 / 8)
rs73150865 C: 66.667% (4 / 6); G: 33.333% (2 / 6)
rs695492 G: 50.000% (3 / 6); T: 50.000% (3 / 6)
rs695519 C: 50.000% (2 / 4); G: 50.000% (2 / 4)
rs174883 C: 25.000% (1 / 4); T: 75.000% (3 / 4)
Figure S2: Tag SNPs around the breakpoint for the TANGO2 exons 3-9 deletion in families 2, 7
and 8. The breakpoint junction sequence is aligned with reference sequences to illustrate the transition
from the proximal (red) to the distal (blue) breakpoints. Nucleotides that do not match perfectly to
reference sequences are highlighted in yellow or green. Those in yellow are variants present in
dbSNP142. Their rs numbers and allele frequencies, if available, are listed in the order they appear. The
12 nucleotides in green are absent in dbSNP142, potentially reflecting novel private mutations.

Figure S3: TANGO2 p.Gly154Arg haplotypes. The figure shows 23 SNPs (columns) with
minor allele frequency (MAF) > 1% (dbSNP142) in the ~1.4-Mb region surrounding TANGO2
on the cSNP array. The rs numbers and associated genomic position (hg19) are given,
respectively, below and above each SNP position. Each row represents a unique subject. At each
SNP position dark blue shaded squares represent the homozygous genotype of one allele and
light blue the homozygous genotype of the alternate allele; heterozygous genotypes are
represented in mid-blue. Genotypes for subjects 1, 3, and 4 have the same homozygous genotype
at each position across the ~1.4 Mb region interrogated, suggesting a shared ancestry; subject 6
(consanguineous) shows a similar pattern of homozygosity across the region, but with different
genotype states at most SNP positions. Subjects with exons 3-9 deletion share a smaller region
of homozygosity (~825 kb).
*SNPs with MAF <5% but >1%

Figure S4: Multiple protein sequence alignment using ClustalW. Multiple protein sequence
alignment using ClustalW shows evolutionary conservation of Gly154 residues in TANGO2
orthologs. The Gly154 is conserved from D. melanogaster to Human, but not in C. elegans. The
change was predicted to be probably damaging and damaging by PolyPhen 2 and SIFT programs
respectively.

Figure S5: Tertiary structures of the wild-type (WT) and the p.Gly154Arg mutant
TANGO2 proteins. Images are predicted by Phyre2 web server
(http://www.sbg.bio.ic.ac.uk/phyre2) based on sequence homology with proteins with known 3D
structures. Arrows show the Gly or Arg with side chains in spheres at position 154. Arrowheads
show the structural differences between the WT and the p.Gly154Arg mutant.

Table S1: Clinical features of subjects with bi-allelic TANGO2 mutations
Family 1
Subject 1
II-1
Family 2
Subject 2
II-1
Family 3
Subject 3
II-1
Family 4
Subject 4
II-1
Family 5
Subject 5
II-1
Family 6
Subject 6
II-1
Family 7
Subject 7
III-2
Family 8
Subject 8
II-1
Family 8
Subject 9
II-2
Family 8
Subject 10
II-3
Family 9
Subject 11
II-2a
Family 9
Subject 12
II-3
Age at first
presentation
4 years
6 months 5 months 8 years 7 years 1 year 18 months 9 months 1 year 3.5 months 2 years 4 years
Current age 6 years 6 years 27 years 14 years 17 years 3 years 9 years Deceased
(2 years)
Deceased
(22 months)
11 months 11 years 8 years
Gender Female Female Female Male Female Male Male Female
Dizygotic twin
Male
Dizygotic twin
Female Male
Monozygotic
twin
Male
Ethnicity Hispanic Hispanic/
European
Hispanic Hispanic Hispanic Hispanic European European European European Arab Arab
Parental
relationship
Non-
consanguineous
Non-
consanguineous
Non-
consanguineous
Non-
consanguineous
Non-
consanguineous
Consanguineous Non-
consanguineous
Non-
consanguineous
Non-
consanguineous
Non-
consanguineous
Consanguineous Consanguineous
Nucleotide
change
c.460G>A c.460G>A
c.460G>A c.460G>A c.460G>A c.605+1G>A - - - - - -
Amino acid
change
p.Gly154Arg p.Gly154Arg p.Gly154Arg p.Gly154Arg p.Gly154Arg
Exonic deletion - Exons 3-9 del - - - - Exons 3-9 del Exons 3-9 del Exons 3-9 del Exons 3-9 del Exons 4-6 del
Exons 4-6 del
Mutation Homozygous Compound
heterozygous
Homozygous Homozygous Homozygous Homozygous Homozygous Homozygous Homozygous Homozygous Homozygous Homozygous
Highest CPK
205,000 U/L 287,230 U/L 22,000 U/L 16,674 U/L 60,000 U/L 103 U/L 258,000 u/L 97,500 U/L 17,900 U/L 248 U/L >100,000 U/L 54,258 U/L
Rhabdomyolysis
Yes Yes Yes Yes Yes No Yes Yes Yes No Yes Yes
Glucose
Hypoglycemia Hypoglycemia Hypoglycemia Normoglycemia Hypoglycemia Hypoglycemia Hypoglycemia Hypoglycemia Hypoglycemia Hypoglycemia Normoglycemia Normoglycemia
Ammonia
↑ ↑ Normal ↑ ND ↑ ↑ Normal ↑ ↑ ↑ ↑
Lactate ND ↑ ↑ Normal ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑
Brain MRI Mild diffuse
atrophy of left
cerebral
hemisphere,
mild optic
chiasm
hypoplasia
Asymmetric
and prominent
lateral
ventricles
No structural
abnormalities Mild cerebellar
volume loss
Prominent
lateral
ventricles,
likely due to
decrease in the
amount of
cerebral white
ND No structural
abnormalities
Generalized
cerebral
atrophy with
Wallerian
degeneration of
the cerebral
peduncles
Wallerian
degeneration of
the cerebral
peduncles
ND No structural
abnormalities
Subtle
abnormalities in
the right
lentiform
nucleus

matter
Seizures Yes No Yes Yes Yes No Yes Yes Yes No Yes Yes
Intellectual
disability
Yes Yes
Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes
Echocardiogram Normal Normal Normal Normal Normal ND Normal Hypertrophic
cardiomyopathy
Normal Normal Normal Normal
ECG/
Arrhythmia
Normal Prolonged QTc
interval,
ventricular
tachycardia
Ventricular
tachycardia
requiring ICD
placement
Prolonged QTc
interval,
Brugada pattern
Prolonged QTc
interval,
torsade de
pointes,
requiring ICD
placement
ND Normal Prolonged QTc
interval
Bradycardia
and premature
ventricular
contractions
and junctional
rhythm.
Prolonged QTc
interval
Normal Normal Prolonged QTc
interval, torsade
de pointes
progressing to
ventricular
tachycardia,
requiring ICD
placement
Other notable
problems
Acquired
microcephaly
Transient
hypothyroidism
considered to
be related to
amiodarone
Bilateral
hearing loss,
head
circumference
at 5%
Hypothyroidism Macrocephaly Failure to thrive Failure to
thrive
Failure to
thrive
responding to
nasogastric
feedings
Hypothyroidism Hypothyroidism
ND=Not determined
ICD= automatic implantable cardioverter-defibrillator
aMonozygotic male twin of Subject 11 had a very similar presentation, but passed away at 7 years of age before molecular confirmation could be obtained