struktura nanomateriálů 04...
TRANSCRIPT

Struktura nanomateriálů
Jindřich Leitner
Ústav inženýrství pevných látek VŠCHT Praha
(var_04, únor 2015 ‐ pracovní)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
2
… „One nanometer is one billionth of a meter. It is a magical point on the scale of length, for this is the point where the smallest man‐made devices meet the atoms and molecules of the natural world.“
Professor Eugen Wong Assistant Director of the National Science Foundation
1999

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
3
OBSAH 1. Úvod .......... ... 5 2. Povrch pevných látek .......... ... 7
2.1 Povrchová práce, povrchová energie, povrchové napětí:
definice a základní termodynamické vztahy .......... ... 7 2.2 Povrchová energie: eperimentální stanovení, výpočty a korelace .......... ... 10 2.3 Povrchové napětí: eperimentální stanovení, výpočty a korelace .......... ... 16 2.4 Relaxace a rekonstrukce povrchu pevných látek .......... ... 17 2.5 Zakřivená fázová rozhraní: Youngova-Laplaceova rovnice, závislost
povrchové energie na křivosti fázového rozhraní .......... ... 19 3. Struktura nanočástic a atomárních klastrů .......... ... 29
3.1 Geometrie polyedrů .......... ... 31 3.2 Nanočástice tvořené řádově 103 a více atomy: Wulffova konstrukce .......... ... 33 3.3 Klastry tvořené 101–103 atomů: pseudokrystalické struktury a struktury s nízkou mírou uspořádání .......... ... 44 3.4 Hustota nanočástic: Youngův-Laplaceův efekt a core-shell model .......... ... 49 3.5 Závislost hustoty nanočástic na teplotě a tlaku .......... ... 61
Seznam symbolů a zkratek

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
4
1. Úvod Nanoobjekty (nanočástice/nanoklastry, nanovlákna a nanovrstvy) a nanostrukturované materiály (polykrystalické materiály s velikostí krystalitů a kompozitní materiály s velikostí částic 1-100 nm) vykazují řadu kvantitativně i kvalitativně odlišných vlastností ve srovnání s materiály s charakteristickými rozměry nad 1 mikrometr. V mnoha případech se od objemových materiálů liší též svojí strukturou, a to jak na atomární úrovni (v důsledku povrchových efektů jsou stabilní jiné strukturní modifikace), tak z hlediska formálně makroskopického (rozdílná hustota nano- a objemových materiálů). Jedním z důvodů proč tomu tak je, je vzrůstající podíl povrchových atomů se zmenšujícími se rozměry objektu. Povrchové atomy mají nižší počet sousedů (vytvářejí menší počet vazeb) než atomy v objemu materiály. To podstatným způsobem ovlivňuje vazebné energie těchto atomů a rovněž jejich prostorové uspořádání – meziatomové vzdálenost ve směru kolmém na povrch i v rovině povrchu (relaxace a rekonstrukce povrchu vedoucí ke snížení povrchové energie). V případě nanočástic a nanovláken jejich zakřivený povrch dále ovlivňuje strukturu těchto objektu v důsledku Youngova-Laplaceova efektu. Tlak indukovaný v důsledku povrchového napětí na konkávní straně rozhraní vede ke kontrakci meziatomových vzdáleností (zvýšení hustoty) a v řadě případů, spolu s povrchovým efektem (povrchová energie), může vést k zvýšení termodynamické stability v objemu nestabilních strukturních modifikací. Tento text pojednává o struktuře nanomateriálů. V první části jsou shrnuty základní poznatky o povrchových vlastnostech pevných látek (zejména kovů) relevantní pro popis struktury nanoobjektů (povrchová práce, povrchová energie, povrchové napětí, Gibbsova termodynamika fázových rozhraní, zakřivená fázová rozhraní, relaxace a rekonstrukce povrchů pevných látek). Druhá část je věnována struktuře individuálních nanočástic a atomárních klastrů. Jsou zde uvedeny obecné principy, které ovlivňují rovnovážná uspořádání těchto objektů a shrnuty vztahy popisující jejich stavové chování. Z důvodu přiměřeného rozsahu této práce není většina zde uvedených matematických vztahů podrobně odvozena. Rovněž zde nejsou diskutovány všechny publikované práce věnované dané problematice ale pouze ty, dle mínění autorů, nejvýznamnější, přinášející nové a zásadní teoretické či experimentální poznatky. Zdrojem dalších informací v této oblasti mohou být vybrané partie řady monografií o nanomateriálech, např. 1-6 nebo přehledové práce podrobně pojednávající o dílčích aspektech zde předložené problematiky: termodynamika povrchů a fázových rozhraní (Ref.7, 8), povrchová a mezifázová energie a napětí/stres (Ref.9-11), struktura nanočástic (Ref.12-14), struktura nanomateriálů (Ref.15) nebo fázová stabilita nanostruktur (Ref.16-18). Mnoho další informací o vlivu velikosti a tvaru nanobjektů na jejich fyzikální a chemické vlastnosti lze nalézt v desítkách dalších souhrnných publikací např.19-23 a přehledných pracích odkazovaných v následujících kapitolách. Literatura: 1. G. Cao: Nanostructures & Nanomaterials: Synthesis, Properties & Applications. Imperial College Press,
London 2004. 2. E. Roduner: Nanoscopic Materials. Size-dependent Phenomena. RSC, Cambridge 2006. 3. C.Bréchignac, P. Houdy, M. Lahmani: Nanomaterials and Namochemistry. Springer, 2007. 4. G.L. Hornyak, J. Dutta, H.F. Tibbals, A.K. Rao: Introduction to Nanoscience. CRC Press, Boca Raton 2008.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
5
5. D. Vollath: Nanomaterials: An Introduction to Synthesis, Properties and Applications, Wiley-VCH, 2008. 6. K.J. Klabunde, R.M. Richards (Eds.): Nanoscale Materials in Chemistry. 2nd.Ed., Wiley, Hoboken 2009. 7. A.I. Rusanov: Surface thermodynamics revisited, Surf. Sci. Report 58 (2005) 111-239. 8. R.C. Cammarata: Generalized thermodynamics of surfaces with applications to small solid systems, in Solid
State Physics, Vol. 61 (H. Ehrenreich, F. Spaepen, Eds.), Academic Press, 2009. 9. W. Haiss: Surface stress of clean and adsorbate-covered solids, Rep. Prog. Phys. 64 (2001) 591-648. 10. P. Müller, A. Saúl: Elastic effects on surface physics, Surf. Sci. Rep. 54 (2004) 157-258. 11. Q. Jiang, H.M. Lu: Size dependent interface energy and its applications, Surf. Sci. Rep. 63 (2008) 427-464. 12. T.P. Martin: Shells of atoms, Phys. Rep. 273 (1996) 199-241. 13. Z.L. Wang: Transmission Electron microscopy of shape-controlled nanocrystals and their assemblies, J.
Phys. Chem. B 104 (2000) 1153-1175. 14. F. Baletto, R. Ferrando, Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic
effects, Rev. Modern Phys. 77 (2005) 371-423. 15. H. Gleiter: Nanostructured Materials: Basic consepts and microstructure, Acta Mater. 48 (2000) 1-29. 16. F.D. Fischer, T. Waitz, D. Vollath, N.K. Simha: On the role of surface energy and surface stress in phase-
transforming nanoparticles, Prog. Mater. Sci. 53 (2008) 481-527. 17. Q. Jiang, S. Li: Thermodynamic considerations on solid structural transition temperatures of nanocrystals,
J. Comput. Theor. Nanosci. 5 (2008) 2346-2364. 18. Q. Jiang, C.C. Yang: Size effect on the phase stabiliy of nanostructures, Current Nanosci. 4 (2008) 179-200. 19. C.X. Wang, G.W. Yang: Thermodynamics of metastable phase nucleation at the nanoscale, Mater. Sci. Eng.
R 49 (2005) 157-202. 20. Q.S. Mei, K. Lu: Melting and superheating of crystalline solids: From bulk to nanocrystals, Prog. Mater.
Sci. 52 (2007) 1175-1262. 21. R.A. Andrievsky: Size-dependent effects in properties of nanostructured materials, Rev. Adv. Mater. Sci. 21
(2009) 107-133. 22. G. Guisbiers, D. Ganguli (Eds.): Size effect in metals, semiconductors and inorganic compounds, Key Eng.
Mater. Vol. 444, Trans Tech Publ. 2010. 23. S.C. Parida (Ed.): Thermal and thermodynamic stability of nanomaterials, Mater. Sci. Forum Vol. 653,
Trans Tech Publ. 2010.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
6
2. Povrch pevných látek Atomy na povrchu pevných látek jsou odlišné od atomů objemových. Tato různost je dobře popsatelná na úrovni nanosvěta, avšak v mnoha případech naše makroskopické pozorování přináší pouze „průměrnou“ informaci o chování nanoobjektů a výjimečnost povrchových atomů lze dokladovat pouze teoretickými výpočty. Ztráta sousedů povrchových atomů při vzniku nového povrchu pevné látky (např. dělením původního tělesa) vede ke změnám vazebných poměrů těchto atomů, což se projeví jak změnou energie jednotlivých vazeb, tak rovnovážnou vzdáleností vázaných atomů. Snížení koordinačního čísla u kovových prvků vede ke zvýšení vazebné energie a současně ke zkrácení vazby mezi atomy. Kohezní energie vztažená na jeden atom je u povrchových atomů nižší než atomů objemových, a tedy celková energie objektů s vysokým podílem povrchových atomů je vyšší než energie stejného počtu atomů v objemu daného materiálu. 2.1 Povrchová práce, povrchová energie, povrchové napětí:
definice a základní termodynamické vztahy Vytvoření nového povrchu pevné látky je spojeno s povrchovou prací, kterou systém musí vykonat, čímž vzrůstá jeho vnitřní energie (v opačném případě byl se systém samovolně dělil na menší části s větším celkovým povrchem). Označme symbolem γsurf vratně vykonanou práci (specific surface work), kterou musí uzavřený jednosložkový systém vykonat při vytvoření nového povrchu jednotkové plochy (jsou přerušeny vazby mezi atomy, na novém povrchu se objeví nové atomy, jsou zachovány délky vazeb, nemění se atomová hustota povrchu). Platí
surf surf dw Aδ γ= (2-1) Tato práce, někdy označovaná jako (specifická) povrchová energie, je skalární veličinou. Její hodnota pro danou pevnou látku závisí na charakteru povrchu. U monokrystalických fází závisí na krystalografické orientaci povrchu (roviny určené Millerovými indexy (hkl)), neboť každá rovina má jiný počet sousedních atomů, mezi kterými se při vzniku nového povrchu přeruší vazby. Např. v případě ideální fcc struktury má každý atom roviny (111) v sousední rovině tři nejbližší sousedy, v rovině (100) čtyři a v rovině (110) šest a empirická relace γ(111) < γ(100) < γ(110) platí pro většinu kovů v této struktuře. V rámci Gibbsovy termodynamiky povrchů lze vnitřní energii heterogenního systému tvořeného objemovými fázemi α a β a fázovým rozhraním (symbol σ) o ploše A vyjádřit jako U U U u Aα β σ= + + (2-2) kde uσ je vnitřní energie jednotkové plochy fázového rozhraní. Pro diferenciál dU pak platí d d d dU U U u Aα β σ= + + (2-3)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
7
Vyjádříme-li diferenciál dUφ (φ = α, β) ze spojených formulací první a druhé věty termodynamické ve tvaru
1 1d d d d , ,U T S p V nϕ ϕ ϕ ϕµ ϕ α β= − + = (2-4) lze spojením rovnic (2-3) a (2-4) odvodit vztah (dle Gibbsova konceptu platí Vσ = 0)
surfd d d d d d dnU T S p V u Ts A T S p V AA
σσ σ µ γ
⎛ ⎞= − + − − = − +⎜ ⎟⎜ ⎟
⎝ ⎠ (2-5)
Jelikož v jednosložkovém systému lze vhodnou volbou matematického ekvivalentu fázového rozhraní (tzv. dividing surface) zajistit platnost podmínky nσ = 0, pro povrchovou práci spojenou s vytvořením jednotky nového povrchu platí vztah
surf u Ts f gσ σ σ σγ = − = = (2-6) kde f σ a g σ jsou Helmholtzova resp. Gibbsova energie jednotkové plochy fázového rozhraní. Již ve své klasické práci o rovnováze heterogenních systémů1 Gibbs připomíná, že vedle zvětšení plochy povrchu pevných látek vytvořením povrchu nového (např. dělením původního tělesa nebo depozicí částic/vrstev pevné látky z plynné fáze) existuje další možnost, a to elastická deformace (roztažení) povrchu stávajícího. Reversibilně vykonaná práce při vzniku jednotkové plochy elastickou deformací již existujícího povrchu tělesa (nejsou přerušeny vazby mezi atomy, na novém deformovaném povrchu se neobjeví nové atomy, mění se atomová hustota) se nazývá povrchové napětí (surface stress)a. Pro diferenciál této práce v případě izotropního prostředí platí vztah
( )elast surfd d dw A fAγ ε= = (2-7) kde f je izotropní povrchové napětí a ε je elastická deformace povrchu. V obecném případě je povrchové napětí fij tenzorem (pro izotropní prostředí je to skalár). Vztah mezi složkami tenzoru napětí fij a povrchovou energií γsurf lze vyjádřit jako
surfij ij
ijf γδ γ
ε∂
= +∂
(2-8)
kde δij je Kroneckerův symbol a εij je složka tenzoru elastické deformace. Jelikož při deformaci povrchu se uplatní pouze tažná/tlaková napětí (i = j), lze deformaci εii definovat jako
a Názvosloví a symbolika energetických veličin souvisejících s povrchem jsou v české i anglicky psané literatuře nejednotné, což vede k častým nedorozuměním i nesprávné interpretaci jednotlivých veličin a jejich hodnot (viz např. R.G. Linford: Symbols and nomenclature in solid surface thermodynamics, Electroanal. Chem. Interfac. Electrochem. 47 (1973) 155-157, P.R. Couchman, D.H. Everet: A comment on certain parameters and equations in surface thermodynamics, J. Electroanal. Chem. 67 (1976) 382-386).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
8
dd , , ,iii
i
l i x y zl
ε = = (2-9)
Deformace εii stejně jako povrchová energie γsurf jsou zde vyjádřeny vzhledem k aktuální (tj. deformované) ploše povrchu o velikosti A (tzv. Eulerův souřadný systém). Obě veličiny lze vyjádřit vzhledem k původní (tj. nedeformované) ploše o velikosti A0 (tzv. Lagrangeův souřadný systém), a pak platí
L0
dd , , ,iii
i
l i x y zl
ε = = (2-10)
a
LL
Lijij
f γε∂
=∂
(2-11)
Z hlediska mikroskopického (atomární stavby hmoty) lze povrchové napětí interpretovat jako síly (působící na jednotku délky v rovině povrchu), které brání atomům povrchové vrstvy změnit své polohy a dosáhnout tak aktuálních rovnovážných vzdáleností mezi sousedními atomy po vzniku nového povrchu. Zatímco pohyb atomů ve směru kolmém na povrch (směr osy z) je relativně snadný a bezprostředně po vzniku nového povrchu dochází k jeho relaxaci a fzz = 0 (viz část 2.4), je rekonstrukce povrchu, tj. změna rozložení atomů v rovině povrchu, obtížná, neboť jí brání snaha podpovrchových vrstev o zachování původní (objemové) geometrie. Stres v rovině x-y není přítomen pouze v povrchové vrstvě, ale projevuje se i v několika podpovrchových vrstvách atomů. Z rovnice (2-11) je zřejmé, že hodnota povrchového napětí je kladná (tahové napětí), pokud ke snížení povrchové energie dochází resp. by docházelo zmenšením plochy povrchu (∂ε < 0) a naopak záporná (tlakové napětí), pokud snížení povrchové energie je spojeno se zvětšením plochy povrchu (∂ε > 0).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
9
2.2 Povrchová energie: experimentální stanovení, výpočty a korelace
Povrchová energie (vratně vykonaná práce při vytvoření jednotkové plochy nového povrchu jednosložkového systému) je základní energetická veličina charakterizující povrch pevných látek a významně ovlivňující energetiku (stabilitu) nanoobjektů. Jelikož hodnota povrchové energie pevných látek je velmi citlivá na stav povrchu (relaxace, rekonstrukce, adsorbované molekuly na povrchu aj.) je poměrně obtížné hodnoty γsurf stanovit experimentálně. Kromě značné technické náročnosti je nevýhodou experimentálních metod rovněž praktická nemožnost stanovit hodnoty povrchové energie pro různé krystalografické roviny, tedy získáváme jednu, průměrnou hodnotu γsurf. Jinou možností, jak získat hodnoty γsurf pro danou pevnou látku jsou různé empirické, semiempirické či zcela teoretické výpočty. Pro experimentální stanovení γsurf čistých kovů (kovových prvků) jsou nejčastěji užívány metody Zero creep (např. Ref.14-18) a Multiphase equilibrium (např. Ref.19-22). V prvém případě je sledována mechanická rovnováha na tenkých vláknech (drátech) nebo fóliích polykrystalického materiálu, které jsou zatíženy závažími o různé hmotnosti. Měření se obvykle provádí při teplotách cca 90–95 % teploty tání, kdy se již projevuje v důsledku zvýšené pohyblivosti atomů vysokoteplotní creep a materiál má snahu se smršťovat (plastickou deformací zmenšovat plochu svého povrchu). Z interpolované hmotnosti závaží, která právě eliminuje spontánní kontrakci a z rozměrů vzorku lze stanovit hodnotu γsurf. Nevýhodou této metody je nezbytnost pracovat při vysokých teplotách, kdy stav povrchu je významně ovlivněn přítomností krystalových poruch a může docházet k jeho rekonstrukci (viz dále). V případě výskytu více alotropických modifikací lze získat hodnoty γsurf jen pro fáze stabilní při nejvyšších teplotách. V druhém případě je hodnota γsurf(s/g) počítána ze známé hodnoty γsurf(l/v) pro pracovní kapalinu (taveninu) a experimentálně stanovených hodnot jednoho kontaktního a tří dihedrálních úhlů. Kontaktní úhel je změřen pro rozhraní studovaná pevná látka/pracovní kapalina, dyhedrální úhly mezi hranicemi zrn u povrchu jsou měřeny za přítomnosti pracovní kapaliny, par pracovní kapaliny a inertní atmosféry (obsahující též páry studované pevné látky). Měření se provádí obvykle provádí při teplotách cca 40–60 % teploty tání. Jako pracovní kapalinu, která vytváří kapku na povrchu studovaného materiálu, lze v případě kovů užít jiný, níže tající kov, který se ve studovaném kovu prakticky nerozpouští (např. Cr(s)/Ag(l), Cu(s)/Pb(l), Mo(s)/Sn(l) neboTa(s)/Cu(l)). Experimentální hodnoty γsurf pro kovové prvky získané uvedenými, ale i dalšími, méně často užívanými metodami byly souhrnně publikovány Kumikovem a Khokonovem23 (data do r.1980) a Luikkonenem et al.24. Tyson a Miller25 navrhli empirický postup výpočtu hodnoty γsurf(s/g) pro kovy z hodnot γsurf(l/g), které jsou snáze experimentálně dostupné. Při teplotě tání platí (Ref.25): γsurf(s/g)/γsurf(l/g) = 1,18 ± 0,03. Pro přepočet na jinou teplotu užili zobecněnou hodnotu
surf(s/g) s RT
σγ∂= − ≈
∂ (2-12)
a tedy γsurf(s/g)(TF) – γsurf(s/g)(0) ≈ RTF/A, kde A je plocha zaplněná jedním molem povrchových atomů. Také tato metoda neumožňuje postihnout povrchovou anizotropii a získat různé hodnoty γsurf pro různé krystalografické roviny.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
10
Jinou metodou pro přímé měření povrchové energie je rozpouštěcí kalorimetrie, která je užívána zejména pro oxidy (Ref.26-28). Principem je stanovení rozpouštěcího tepla studovaného materiálu v závislosti na velikosti jeho měrného povrchu. Hodnotu průměrné povrchové energie pak získáme jako směrnici lineární závislosti entalpie vztažené k hodnotě objemového materiálu (viz obr. 1).
Obrázek 1 Závislost entalpie TiO2(rutil) na velikosti měrného povrchu získaná z rozpouštěcích tepel
práškových vzorků v tavenině 3Na2O.4MoO3 při teplotě 975 K (Ref.28) Hodnoty povrchové energie závisí na teplotě. Obecně platí, že hodnota γsurf s rostoucí teplotou klesá. Teplotní závislost bývá obvykle vyjádřena empirickým vztahem ve tvaru
( )Fsurf A B T Tγ = − − (2-13)
kde parametry A, B > 0. Parametr A odpovídá hodnotě γsurf při teplotě tání, parametr B záporně vzaté derivaci γsurf podle teploty. V další části se budeme věnovat možnostem výpočtu hodnot γsurf. Hned v úvodu této partie je třeba zdůraznit, že přímé porovnání experimentálních a vypočtených hodnot je zavádějící, neboť slovy prof. Navrotské28 „Thus comparing theory and experiment is indeed comparing apples and oranges, ...“ Jako zcela empirický (a také nejjednodušší) postup lze označit Broken-bond model (BBM) Ref.29-31, na základě kterého je hodnota povrchové energie vypočtena z počtu přerušených vazeb při vzniku povrchu a vazebných energií. V nejjednodušší variantě uvažujeme pouze vazby mezi nejbližšími sousedy v příslušné struktuře. Označme jako ∆Z(hkl) = Zbulk – Zsurf(hkl) rozdíl koordinačního čísla (počtu nejbližších sousedů) objemových atomů a koordinačního čísla atomů na povrchu v krystalografické rovině určené Millerovými indexy (hkl). Pak platí
( ) coh surf ( ) ( ) cohsurf ( ) ( )
A bulk bulk A1hkl hkl hkl
hkl hklE Z E
ZN Z Z Nρ ρ
γ⎛ ⎞
= ∆ = −⎜ ⎟⎝ ⎠
(2-14)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
11
Podíl Ecoh/(NA×Zbulk) představuje energii jedné vazby, která nezávisí na poloze atomu (objem resp. povrch s různou krystalografickou orientací). Parametr ρ(hkl) představuje atomární hustotu (počet atomů na jednotku plochy) v dané krystalografické rovině. Hodnoty ∆Z(hkl) a ρ(hkl) lze určit z geometrie dané struktury. Např. pro strukturu fcc platí31 (h ≥ k ≥ l)
( )
( )
2 , , jsou lichá čísla
4 2 ostatní variantyhkl
hkl
Z h k h k l
Z h k
∆ = +
∆ = + (2-15)
a tedy ∆Z(111) = 3, ∆Z(110) = 6 a ∆Z(100) = 4. Dále lze vypočítat atomární hustoty ρ(111) = 4/(√3a2), ρ(110) = 2/(√2a2) a ρ(100) = 2/(a2) atomů na jednotku plochy (konkrétní rozměr závisí na jednotkách, v jakých je vyjádřen mřížkový parametr a). I v této nejjednodušší variantě lze získat různé hodnoty γsurf(hkl) pro různé krystalografické roviny, avšak tato anizotropie je pro danou strukturu generalizovaná (nezáleží na tom, o jaký prvek se jedná). Tak např. pro strukturu fcc vždy získáme hodnoty γ(111) < γ(100) < γ(110) a platí γ(100)/γ(111) = 1,15 a γ(110)/γ(111) = 1,22. Anizotropie povrchové energie určená na základě této metody je v poměrně dobré shodě s výsledky teoretických výpočtů (viz dále). Jiang et al.31 modifikovali rovnici (2-14) a pro kovy v netěsnějším uspořádání (struktura fcc resp. hcp) vztah
surf ( ) surf ( ) ( ) cohsurf ( )
bulk bulk A2
2hkl hkl hkl
hklZ Z E
Z Z Nρ
γ⎛ ⎞⎜ ⎟= − −⎜ ⎟⎝ ⎠
(2-16)
Pro prvky v jiných strukturních modifikacích (bcc a struktura diamantu) při výpočtu γsurf zohlednili i vazby mezi dalšími nejbližšími atomy. Později byl jejich původní přístup pro některé struktury a krystalografické roviny upřesněn zohledněním rozdílné koordinace atomů vystavených okolnímu prostředí v povrchové a první podpovrchové vrstvě32-33. Pro výpočet povrchové energie (i dalších parametrů povrchu jako povrchového napětí a mezirovinných vzdáleností relaxovaných povrchů) čistých kovů jsou nejčastěji užívány semiempirické postupy. Zjednodušeně vyjádřeno, semiempirické postupy vycházejí z určitého fyzikálního modelu, na základě kterého je vytvořen předpis pro kohezní (potenciální, konfigurační) energii dané pevné látky. Tento předpis obsahuje určité parametry, jejichž hodnoty je třeba nastavit tak, aby bylo dosaženo shody s příslušnými experimentálními údaji. V některých případech mohou být pro parametrizaci modelu využita i data získaná teoretickými ab-initio výpočty (viz dále). Z literární rešerše je zřejmé, že z celé řady vhodných semiempirických postupů jsou pro výpočet γsurf užívány zejména dva: Embeded atom method (EAM) a Equivalent crystal theory (ECT). Oba postupy umožňují výpočet povrchové energie jak nerelaxovaných, tak relaxovaných povrchů (viz část 2.4). Pro výpočty jsou rovněž využívána další empirické postupy, jako např. metoda Tight-binding second moment nebo empirické potenciály Finnis-Sinclair a Sutton-Chen (Ref.34-37). V rámci metody EAM, resp. její modifikované podoby MEAM38-42, je celková energie systému tvořeného N atomy vyjádřena jako suma příspěvků těchto atomů (i) skládajících se z potenciální energie párové interakce dvou sousedních atomů i-j jejichž vzdálenost je Rij, φij(Rij), a potenciální energie atomu i v silovém poli elektronů všech ostatních atomů, jehož lokální elektronová hustota v poloze atomu i je ρi (tzv. embedding energy):

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
12
tot1 ( ) ( )2 ij ij i i
i j iE R Fφ ρ
≠
⎛ ⎞⎜ ⎟= +⎜ ⎟⎝ ⎠
∑ ∑ (2-17)
Modifikace modelu spočívá v přesnějším vyjádření druhého členu, kdy prostorové rozložení hustoty elektronů je získáno superpozicí sféricky symetrického příspěvku (typického pro kovy) a směrově (úhlově) závislých příspěvků (typických pro prvky s kovalentní vazbou). Model obsahuje řadu adjustabilních parametrů (8 až 15 podle verze), přičemž hodnoty některých z nich úzce souvisejí s experimentálně dostupnými vlastnostmi dané látky (atomární objem, mřížkový parametr, kohezní energie, energie vzniku vakancí, elastické konstanty, objemový modul pružnosti). Pro výpočet povrchové energie při teplotě 0 K krystalu, který je ve dvou směrech (x-y) nekonečný a ve třetím směru (z) polonekonečný lze zapsat zcela obecný vztah ve tvaru
( )surf ( ) ( ) pot,surf( )/at pot,bulk/atk
hkl hkl hklk
E Eγ ρ= −∑ (2-18)
kde Ek
pot,surf(hkl)/at a Epot,bulk/at jsou vypočtené potenciální energie atomů v k-té povrchové/podpovrchové vrstvě (k = 1 je povrchová vrstva atomů), které k hodnotě γsurf(hkl) přispívají, a v objemu materiálu. EAM/MAEM model byl využit k výpočtu povrchové energie kovových prvků ve struktuře fcc (Ref.39, 47, 50-54), bcc (Ref.46, 55-56) i hcp (Ref.43, 57). Model je rovněž užíván k výpočtům povrchového napětí a relaxace povrchu (viz dále). Metoda ECT (Ref.58-62) vychází z předpokladu, že energie každého jednotlivého atomu je dána počtem jeho sousedů (NN-nejbližších a NNN-dalších nejbližších) a vzdálenostmi mezi nimi. Závislost kohezní energie na meziatomové vzdálenosti charakterizované např. rozměry Wignerovy-Seitzovy buňky lze vyjádřit univerzálním vztahem ve tvaru59, 60, 63
( )eq *coh WS coh WS( ) ( ) 1 * e rE r E r r −= − + (2-19)
kde Ecoh je kohezní energie, rWS je poloměr Wignerovy-Seitzovy buňky a r* je redukovaná délka vyjádřena jako
( )eq
WS WS1 2eq eq
coh WS WS
*( ) 12
r rr
E r B rπ
−= (2-20)
B je objemový modul pružnosti. Povrchové atomy mají menší počet sousedů než atomy objemové, což vede ke snížení jejich kohezní energie (vzhledem k energii atomů objemových). V rámci metody ECT postulujeme existenci tzv. ekvivalentního krystalu, který má stejnou strukturu jako reálný krystal, avšak jiné meziatomové vzdálenosti. Rozdíl kohezní energie v tomto nerovnovážném stavu (charakterizovaném parametrem rWS) a rovnovážném stavu (req
WS) je pak roven změně kohezní energie povrchových atomů reálného krystalu v důsledku snížení počtu vazeb. Jinými slovy: snížení počtu vazeb povrchových atomů v reálném krystalu (při zachování jejich délky) je modelováno jako prodloužení vazeb objemových atomů v ekvivalentním krystalu (při zachování jejich počtu). Z rovnosti těchto energií jsou vypočteny meziatomové vzdálenosti RNN a RNNN objemových atomů a pro tyto

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
13
vzdálenosti pak dosazením do vztahů (2-19) a (2-20) povrchová energie (a je mřížkový parametr)
( )eq *surf ( ) coh WS( ) 1 1 * e a
hkl E r aγ −⎡ ⎤= − +⎣ ⎦ (2-21)
( )
( )eq
NN WS1 2eq eq
coh WS WS
*( ) 12
R c ra
E r Brπ
−= (2-22)
Pro výpočet povrchové energie relaxovaných povrchů je zohledněn další člen popisující anizotropní deformaci (změny délky vazeb) mezi povrchovou a podpovrchovými vrstvami atomů. ECT model byl využit k výpočtu povrchové energie kovových prvků ve struktuře fcc, bcc i hcp (Ref.64-65). Model je rovněž užíván k výpočtům povrchového napětí a relaxace povrchu (viz dále). Přesné hodnoty povrchové energie lze získat na základě teoretických ab-initio výpočtů. Základem je výpočet celkové energie tzv. superbuňky (supercell), ve které se střídavě opakují bloky (slab) tvořené několika atomovými vrstvami daného kovu s příslušnou povrchovou orientací a vrstva vakua. Povrchová energie je pak určena jako rozdíl celkové energie bloku (Eslab) a energií stejného počtu bulkových atomů
( )surf ( ) slab bulk1lim2
Nhkl N
E N Eγ→∞
= − ⋅ (2-23)
přičemž v případě kovů je obvykle uvažováno 8-15 vrstev atomů. Jistým problémem je konvergence vypočtených hodnot γsurf pro rostoucí počet vrstev atomů uvažovaných při výpočtu pokud je hodnota Ebulk získána z přímého výpočtu pro objemový krystal (Ref.66-67). Pro výpočet celkové energie je obvykle využíván koncept DFT (Density functional theory) v aproximaci LDA (Local density approximation) nebo GGA (Generalized gradient approximation) pro výměnný/korelační potenciál. Při výpočtech elektronové hustoty jsou užívány různé báze, ovykle LAPW (Linear augmented plane waves), LMTO (Linear muffin tin orbitals) nebo PP (Pseudopotentials). Na základě teoretických výpočtů byly získány hodnoty povrchové energie různých povrchů (definovaných indexy (hkl)) pro téměř všechny prvky v pevném stavu. Z obrovského počtu prací zde citujme alespoň obsáhlejší studie zaměřené na kovy (Ref.68-74). Z vypočtených hodnot povrchových energií kovových prvků je patrná korelace s hodnotami příslušných kohezních energií. Na obr. 2 jsou vyneseny hodnoty γsurf pro vždy pro nejhustěji obsazenou krystalografickou rovinu proti hodnotám kohezní energie Ecoh (Ref.75). Hodnoty povrchové energie byly získány ab-initio výpočtem70 (metoda full charge density LMTO). Body jsou proloženy lineární závislostí s koeficientem regrese R2 = 0,86.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
14
0 200 400 600 8000
1000
2000
3000
4000
5000
γsurf = 4,62 Ecoh
γ surf (m
J m
-2)
Ecoh (kJ mol-1)
Obrázek 2
Korelace mezi povrchovou a kohezní energii pro kovové prvky Korelace mezi povrchovou a kohezní energií byla zmíněna řadou autorů63, 76-77. Jelikož kohezní energie koreluje s teplotou tání, je možné rovněž povrchovou energii korelovat s teplotou tání resp. s podílem teploty tání a druhé mocniny meziatomové vzdálenosti d, dNN(fcc) = √2a/2, dNN(bcc) = √3a/2 (Ref.78). Tato korelace je ukázána na obr. 3. Body jsou proloženy lineární závislostí s koeficientem regrese R2 = 0,94.
0 10 20 30 40 500
1000
2000
3000
4000
5000
γsurf = 89,1x10-3T F/d 2
γ surf (m
J m
-2)
10-3T F/d 2 (K nm-2)
Obrázek 3
Korelace mezi povrchovou energii a podílem T F/d 2 pro kovové prvky

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
15
2.3 Povrchové napětí: experimentální stanovení, výpočty a korelace
Experimentální stanovení hodnot povrchového napětí (resp. složek tenzoru povrchového napětí) čistých povrchů kovů je velice komplikované79-80 a neexistuje žádná přímá metoda pro tato měření. Obecně lze povrchové napětí stanovit z deformace, která je s ním spojená. Této souvislosti využívá metoda stanovení izotropního povrchového napětí ze změn mřížkových parametrů u částic malých rozměrů (nanočástic) (podrobný výklad a odvození rovnice viz část 2.5 a 3.3). Pro výpočet povrchového napětí f kubických krystalů je užíván vztah81-84
3 32 2 T
a a rf rBa a κ∆ ∆
= − = − (2-24)
kde ∆a/a je relativní kontrakce mřížkového parametru, B (B = 1/κT) je objemový modul pružnosti, κT je koeficient izotermní stlačitelnosti a r je poloměr částice, u které předpokládáme tvar koule. Změna mřížkového parametru je obvykle stanovena metodou elektronové difrakce. Bylo zjištěno, že hodnoty ∆a/a, a tedy i povrchové napětí f, se mírně liší podle toho, z jaké roviny (hkl) byly stanoveny. Tímto způsobem byly zjištěny hodnoty průměrného (izotropního) povrchového napětí např. pro Au, Ag, Pt nebo Cu (viz tabulka 1) S ohledem na experimentální náročnost stanovení hodnot povrchového napětí a prakticky nemožnost získat tyto hodnoty pro různé krystalografické roviny (hkl) jsou hodnoty f(hkl) obvykle počítány, a to jak na základě semiempirických postupů, tak teoretických ab-initio výpočtů.
Tabulka 1 Izotropní povrchové napětí vybraných vzácných kovů ve struktuře fcc
stanovené z kontrakce mřížkového parametru u nanočástic
Prvek Velikost částic d (nm) Rovina (hkl) κT
(Pa–1) Povrchové napětí
(J m–2) (Ref.)
Au 3,5–12,5 průměr (220) a (311) 5,99×10–12 1,18 ± 0,2 (81)
Au 2,5–14 5,99×10–12 3,83 (85) Au 3–40 (220) 3,08 ± 0,7 (86) Au 3–40 (422) 3,19 ± 1,0 (86) Au 1–10 (111) 5,85×10–12 3,88 ± 1,45 (87)
Ag 1–6 (111) 9,65×10–12 2,55 ± 1,38 (87) Ag 3–17,8 (220) 1,42 ± 0,3 (88)
Pt 3–40 (220) 3,86 ± 0,7 (86) Pt 3–40 (422) 4,44 ± 1,0 (86) Pt 3,8–24,4 (220) 2,57 ± 0,4 (89) Pt 3,8–24,4 (111) 2,27 ± 0,6 (89)
Cu 2,4–18,4 (220) 0,0 ± 0,45 (89)
Pd 1,4–5 (111) 5,29×10–12 6,0 ± 0,45 ()

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
16
2.4 Relaxace a rekonstrukce povrchu pevných látek Napětí v povrchové vrstvě atomů (a v několika málo dalších podpovrchových), které je důsledkem snahy o změnu prostorového rozložení atomů v reakci na změny silových interakcí na nově vzniklém povrchu vede k procesům relaxace a rekonstrukce povrchu. Jako relaxace povrchu jsou označovány změny v meziatomových vzdálenostech ve směru kolmém na povrch (směr osy z). Tyto změny mohou být kladné (dilatace) i záporné (kontrakce) a jsou nejvýraznější pro vzdálenosti mezi prvními dvěma atomárními vrstvami (viz obr. 4). Míra relaxace závisí na krystalografické orientaci povrchových ploch (plošné atomární hustotě): čím je atomární hustota vyšší, tím je relaxace méně výrazná. Např. u fcc struktury tedy roste v pořadí (111) → (100) → (110). U nejtěsněji uspořádaných struktur kovů odeznívá ve vzdálenosti 4 až 5 atomárních vrstev.
Obrázek 4 Relaxace povrchové a podpovrchové vrstvy atomů (dB je rovnovážná meziatomová vzdálenost v objemu
materiálu, ∆d12 < 0, ∆d23 > 0) Relaxaci na rovinných rozhraních lze sledovat experimentálně (obvykle pomocí difrakce elektronů s nízkou energií, metoda LEED) (Ref.90-92) nebo jí lze mapovat na základě teoretických ab-initio výpočtů (Ref.72-74, 93) popř. semiempirických postupů (Embedded Atom Method, Equivalent Crystal Theory aj.) (Ref.50, 54, 64, 94-95). Relaxaci povrchu Ni(111) pozorovali metodou elektronové difrakce již v roce 1927 C.J. Davisson a L.H. Germer (Ref.96). První výpočty, a to pro (100) povrch iontových krystalů typu NaCl provedli o rok později J.E. Lennard-Jones a B.M. Dent (Ref.97). Rekonstrukcí povrchu jsou míněny změny v uspořádání atomů v rovině povrchu (obvykle x-y), při kterých dochází ke ztrátě strukturní koherence s podpovrchovými vrstvami a objemem materiálů. Rekonstrukce povrchu není tak snadná jako jeho relaxace a dochází k ní většinou za zvýšených teplot (blízko teploty tání). Při rekonstrukci může docházet buď pouze ke změnám poloh atomů, nebo ke změnám plošné hustoty atomů v povrchové vrstvě (zvýšení resp. snížení). Pro posouzení schopnosti daného povrchu spontánně se rekonstruovat a snížit
dbulk
(1+∆d12)dbulk
(1+∆d23)dbulk

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
17
tak energii povrchové vrstvy atomů bylo navrženo několik kriterií98-100. Na základě porovnání experimentálních (zejména LEED a STM) výsledků a výsledků teoretických a semiempirických výpočtů bylo zjištěno, že u kovů s fcc strukturou není rekonstrukce vždy spojena se snížením povrchového napětí v povrchové (x-y) vrstvě atomů. Významným parametrem pro posouzení stability/nestability daného povrchu je pak derivace povrchové energie podle deformace (∂γ/∂εij), která je dle rovnice (2-7) rovna rozdílu povrchového stresu a povrchové energie fij – γ. Jako příklad je na obr. 4a ukázána rekonstrukce povrchu Au(100)-(1×2) missing-row (Ref. 100a).
Obrázek 4a Rekonstrukce povrchu Au(100)-(1×2) missing-row (Ref. 100a)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
18
2.5 Zakřivená fázová rozhraní: YoungovaLaplaceova rovnice, závislost povrchové energie na křivosti fázového rozhraní V případě zakřivených fázových rozhraní dochází na konkávní straně rozhraní ke kompresi hmoty v důsledku existence povrchového napětí. Je-li poloměr křivosti velmi malý (nanočástice, nanovlákna), zvyšuje se tak „průměrná“ hustota nanobjektu oproti hustotě objemového materiálu. Ačkoliv bylo teoretickými výpočty i experimentálně prokázáno, že v případě nanočástic dochází ke zkrácení meziatomových vzdáleností (a tedy ke zvýšení hustoty) jen těsně pod povrchem a uvnitř částice meziatomové vzdálenosti zůstávají zachovány jako v objemovém materiálu, lze v prvním přiblížení reálnou nanočástici popsat jako izotropní elastické kontinuum s dostatečně vysokou pohyblivostí atomů chovající se jako kulatá kapka kapaliny o poloměru r. Rozdíl tlaků ∆p na konvexní (pout) a konkávní (pin) straně fázového rozhraní pak vyjádříme pomocí Youngovy-Laplaceovy rovnice101, 102 ve tvaru
in out 2 fp p pr
∆ = − = (2-25)
kde f je průměrné izotropní povrchové napětí daného materiálu. Youngova-Laplaceova rovnice je vyjádřením mechanické rovnováhy sil na zakřiveném fázovém rozhraní. Uvažujme nejprve případ rovinného rozhraní oddělujícího fáze α a β – 2D průmět (obr. 5). Rozhraní chápejme jako nekonečně tenkou pružnou membránu. Toto rozhraní je izotropně napínáno silou, která působí v ploše rozhraní a jejíž působiště je umístěno podél obvodu rozhraní. Jako povrchové/mezifázové napětí f označme sílu vztaženou na jednotku délky obvodu rozhraní. Bilanci sil proveďme na elementu plochy rozhraní dA = da × da. Ve směru osy z kolmé na rozhraní působí proti sobě síly Fα a Fβ v důsledku hydrostatických tlaků pα ve fázi α a pβ ve fázi β. V rovině x-y působí izotropní povrchové napětí f, přičemž celková síla působící po obvodu je F = 4fda. Jelikož síla F neovlivňuje bilanci sil ve směru osy z, platí pα = pβ.
z
x
plocha dA = (da)2
Fα = pαdA
Fβ = pβdA
Fx = f da
A B
z
x
plocha dA = (da)2
Fα = pαdA
Fβ = pβdA
Fx = f da
z
x
plocha dA = (da)2
Fα = pαdA
Fβ = pβdA
Fx = f da
z
x
plocha dA = (da)2
Fα = pαdA
Fβ = pβdA
Fx = f da
z
x
plocha dA = (da)2
Fα = pαdA
Fβ = pβdA
Fx = f da
z
x
z
x
z
x
z
x
z
x
plocha dA = (da)2
Fα = pαdA
Fβ = pβdA
Fx = f da
A B
Obrázek 5 Bilance sil (izotropní tlak a povrchové napětí) na rovinném rozhraní

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
19
V případě zakřiveného rozhraní působí povrchové síly v tečné rovině k rovině rozhraní, která není totožná s x-y rovinou a její sklon vůči osy z se mění. Je-li působiště sil umístěno po obvodu rozraní, pak síla Ff má nenulový průmět do směru osy z a ovlivňuje bilanci sil v tomto směru. Rovnost tlaků pα = pβ na zakřiveném rozhraní tak neplatí. Situace pro rozhraní ve tvaru kulové plochy je znázorněna na obr. 6.
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
xBA
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
z
x
z
x
z
x
z
x
z
x
z
x
z
xBA
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
(a) (b)
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
xBA
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
z
x
z
x
z
x
z
x
z
x
z
x
z
xBA
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
xBA
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
plocha dA = (rdφ)2
Fα = pαdA
Fβ = pβdA
F = f rdφ
úhel dφpoloměr r
z
x
z
x
z
x
z
x
z
x
z
x
z
x
z
xBA
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
A
F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)F = f rdφ
z
x
úhel ½dφ
Fz = Fsin(½dφ)
(a) (b)
Obrázek 6 Bilance sil (izotropní tlak a povrchové napětí) na zakřiveném rozhraní (fáze α je uvnitř kapky, fáze β vně)
Bilanci sil nyní provedeme na elementu plochy rozhraní dA = rdφ × rdφ (r je poloměr příslušné koule, dφ je úhel, který ve směru os x a y vymezuje plochu dA). V bodě A působí na hranu elementu síla F = frdφ. Stejně velká síla působí i v bodě B na protilehlou hranu elementu rovnoběžnou s osou y a také na obě hrany rovnoběžné s osou x. Na obr. 6b je znázorněn rozklad síly, působící v bodě A do směrů os x a z. Platí:
( ) ( )2sin 1 2d 1 2d 1 2 dzF F F f rϕ ϕ ϕ= = (2.26) V případě kulové plochy a stejného úhlu dφ ve směru x a y jsou všechny průměty do osy z tečných sil působících po obvodě elementu stejné, a tedy
( )24 2 dzF F f r ϕ= = (2.26a) Bilance sil ve směru osy z (obr. 6b) má nyní tvar
( ) ( ) ( )2 2 2α βd d 2 dp r p r f rϕ ϕ ϕ= + (2.26b)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
20
odkud plyne α β 2 fp p
r− = (2.26c)
V případě kapalin je f = γ, a tedy platí α β 2p p
rγ
− = (2.26d)
V případě obecně zakřivené plochy s křivostmi cx = 1/rx a cy = 1/ry a obdélníkový element plochy dA = rxdφx × rydφy má bilance sil následující podobu:
d d d dx x y y x yF f r f rϕ ϕ ϕ ϕ= + (2.26e)
( )α βd d d d d dx x y y x x y y x y x yp r r p r r f r rϕ ϕ ϕ ϕ ϕ ϕ= + + (2.26f)
α β 1 1 2x y
x y x y
r rp p f f Hf
r r r r⎛ ⎞+
− = = + =⎜ ⎟⎜ ⎟⎝ ⎠
(2.26g)
kde H je tzv. střední křivost elementu dA. Dále je ukázáno alternativní (termodynamické) odvození Youngovy-Laplaceovy rovnice pro fázové rozhraní (l)-(g). Uvažujme izolovaný jednosložkový systém při stálé teplotě T a stálém objemu V tvořený kulatou kapkou (fáze α) o poloměru r a plynnou fází (fáze β) (viz obr. 7)
Vα, pα
Vβ, pβ
r
Vα, pα
Vβ, pβ
r
Obrázek 7 Izolovaný jednosložkový systém tvořený kulatou kapkou (fáze α) spojitě obklopenou plynnou fází (fáze β)
Tlak v kapce je pα a v okolní plynné atmosféře pβ. Kapka může vratně měnit svoji velikost (objem Vα), čímž se mění i objem plynné fáze Vβ a velikost plochy fázového rozhraní A (povrchu kapky). Platí:
α α β βvol surfd d d d 0U w w p V p V Aδ δ γ= + = − − + = (2.27)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
21
Jelikož celkový objem systému je konstantní (dV = 0), platí dVα = –dVβ, a tedy
( )α β d dp p V Aα γ− = (2.27a)
( )α β d 2d
Ap prVαγγ− = = (2.27b)
Zakřivenost fázového rozhraní a obecně limitované rozměry nanobjeků mají vliv na hodnotu povrchové energie γsurf. Tato závislost byla pro případ rozhraní l-g (γsurf(l/g)) postulována již v roce 1949 Tolmanem (Ref.123-126). Pro závislost γsurf(l/g) na poloměru kapky r odvodil Tolman vztah
surf(l/g)
surf(l/g),
( ) 1 211 2
rr r
γ δγ δ∞
= ≈ −+
(2-29)
kde parametr δ je tzv. Tolmanova délka, která má význam vzdálenosti mezi Gibbsem definovaným fázovým rozhraním (equimolar dividing surface) a rozhraním, kde působí povrchové napětí (surface of tension) zvyšující dle Youngovy- Laplaceovy rovnice (2-25) tlak uvnitř kapky. Přibližné vyjádření ve vztahu (2-29) bylo získáno rozvojem v Taylorovu řadu v bodě δ/r = 0, a vztah tak platí pouze pro „velké“ kapky r>> δ. Závislost γsurf(l/g) = f(r) je předmětem řady teoretických prací (přímé experimentální stanovení je velmi obtížné a analýza nepřímých experimentálních dat je problematická), jejichž výsledky však nejsou zcela ve shodě (kladné resp. záporné hodnoty parametru δ, nezávislost resp. závislost δ na velikosti kapky, platnost rovnice (2-29) vzhledem k velikosti kapky atd.). Pro kladné hodnoty parametru δ tak podle vztahu (2-29) povrchová energie γsurf(l/g) klesá s klesajícím rozměrem kapky, pro záporné hodnoty naopak roste. Pro závislost povrchové energie γsurf(s/g) na poloměru pevné kulaté částice r bylo navrženo několik různých vztahů (Ref.127-132). Většina z nich je založena na empiricky postulované relaci
surf(s/g) coh
surf(s/g), coh,
( ) ( )r E rE
γγ ∞ ∞
= (2-30)
která vychází z úměry mezi povrchovou a kohezní energií (např. rovnice (2-14) a (2-16)) a předpokladu, že příslušná konstanta úměrnosti je nezávislá na rozměru částice (stejná hodnota pro rovinné i zakřivené rozhraní). Jiang a spol. (Ref.129-131) navrhli vztah
surf(s/g) subl m at
surf(s/g),
( )1
3r S r
R rγγ ∞
∆= − (2-31)
ve kterém ∆sublSmje teplotně nezávislá změna molární entropie při sublimaci dané látky. Stejně jako vztah (2-29) pro kladné hodnoty parametru δ i rovnice (3-31) predikuje pokles hodnoty povrchové energie se zmenšujícími se rozměry částice. Kvalitativně shodný průběh závislosti γsurf(s/g) = f(r) prezentoval Attarian Shandiz (Ref.132), který závislost kohezní energie nanočástice Ecoh = f(r) odvodil na základě konceptu efektivního koordinačního čísla.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
22
Výše zmíněný trend, tedy pokles hodnoty povrchové energie se zmenšujícími se rozměry částice, není v relevantní literatuře akceptován bez výhrad a byly publikovány teoretické i experimentální práce, které předkládají zcela opačné argumenty pro vyšší hodnoty γsurf(s/g) u nanočástic ve srovnání s objemovým materiálem. Tyto rozpory lze v některých případech vysvětlit různou interpretací experimentálních dat (hodnoty povrchové energie získané na základě stanovení teploty sublimace u nanočástic různé velikosti, Ref.133-137). Experimentálně získané hodnoty γsurf(s/g) pro anatas (částice o velikosti 4-34 nm, Ref.138) ukazují na složitější závislost γsurf(s/g) = f(r) s maximem pro částice ≈14 nm a s poklesem pro částice menších velikostí. Závislost povrchové energie na rozměrech částic v subnanometrové oblasti (klastrů) lze studovat pouze na základě modelových výpočtů. Ab-initio výpočty pro částice Al 0,3-1 nm a empirické výpočty (EAM) pro částice 0,3-3,5 nm ukazují, že hodnota γsurf(s/g) se zmenšující se hodnotou r mírně roste: r = 30,2 nm, γsurf(s/g) = 960 mJ m–2, r = 9,8 nm, γsurf(s/g) = 1090 mJ m–2, r = 2,7 nm, γsurf(s/g) = 2050 mJ m–2 (Ref.139). Určitým problémem při interpretaci vypočtených hodnot je nejistota v určení rozměrů částice tvořené definovaným počtem atomů (electronic spilout factor), a tedy určení velikosti povrchu, ke kterému je hodnota povrchové energie vztažena. Podrobně byla studována závislost γsurf(s/g) = f(Nat) pro nanočástice Ag (Ref.140-142) (viz obr. 8).
0 200 400 600 800 10000.0
0.5
1.0
1.5
2.0
2.5
3.0
N = 429r = 1,2 nm
DFT (Ref.140) EAM (Ref.140) DFT (Ref.141) DFT (Ref.142)
γ surf (J
m-2)
NAg
N = 887r = 1,5 nm
Obrázek 8 Závislost povrchové energie nanočástic Ag na počtu atomů částici tvořících (Ref.140-142)
Závěrem lze konstatovat, že jistá neurčitost/nejasnost publikovaných výsledků souvisí zejména: i) s náročností příslušných experimentů, ii) s rozdílnou interpretací experimentálních dat včetně záměny veličin povrchová energie/povrchové napětí, iii) s tvarem nanočástic (kvazisféricka částice s fcc strukturou, ikosaedr, komolý dekaedr, komolý oktaedr – viz část 3.1), pro který jsou výpočty prováděny,

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
23
iv) s jemností klasifikací atomových poloh (plocha povrchu, hrana, vrchol, podpovrchová vrstva, jádro částice), pro které jsou kohezní energie počítány. Literatura: 1. J.W. Gibbs: On the equilibrium of heterogeneous substances, in: The Scientific Papers of J. Williard Gibbs,
Vol. One Thermodynamics, pp. 55-353, OX Bow Press, Woodbridge 1993. 2. R. Shuttleworth: The surface tension of solids, Proc. Phys. Soc. A63 (1950) 444-457. 3. J.S. Vermaak, C.W. Mays, D. Kuhlmann-Wilsdorf: On surface stress and surface tension. I. Theoretical
consideration, Surf. Sci. 12 (1968) 128-133. 4. P.R. Couchman, W.A. Jesser: On the thermodynamic of surfaces, Surf. Sci. 34 (1973) 212-224. 5. R.G. Linford: Surface Thermodynamics of solids, Solid State Surf. Sci. 2 (1973) 1-152. 6. R.C. Cammarata: Surface and interface stress effects in thin films, Prog. Surf. Sci. 46 (1994) 1-38. 7. E.M. Gutman: On the thermodynamic definition of surface stress, J. Phys.: Condens. Matter 7 (1995) L663-
L667. 8. R.C. Cammarata: Surface and interface stress effects of interfacial and nanostructured materials, Mat. Sci.
Eng. A 237 (1997) 180-184. 9. W.D. Nix, H. Gao: An atomistic interpretation of interface stress, Script. Mater. 12 (1998) 1653-1661. 10. W. Haiss: Surface stress of clean and adsorbate-covered solids, Rep. Prog. Phys. 64 (2001) 591-648. 11. D. Kramer, J. Weissmüller: A note on surface stress and surface tension and their interrelation via
Shuttleworth’s equation and Lippmann equation, Surf. Sci. 601 (2007) 3042-3051. 12. F.D. Fischer, T. Waitz, D. Vollath, N.K. Simha: On the role of surface energy and surface stress in phase-
transforming nanoparticles, Prog. mater. Sci. 53 (2008) 481-527. 13. R.C. Cammarata: Generalized thermodynamics of surfaces with applications to small solid systems, in:
Solid State Physics, Vol. 61 (H. Ehrenreich, F. Spaepen, Eds.), pp. 1-75, Academic Press 2009. 14. H. Udin, A.J. Shaler, J. Wulff: The surface tension of solid copper, Trans. Am. Inst. Mining, Metall. Petrol.
Engnrs. 1 (1949) 186-190. 15. V.K. Kumikov: The measurement of the surface tension of some pure metals in the solid state, Mater. Sci.
Eng. 60 (1983) L23-L24. 16. D. Josell: Exact solution for the zero creep load of a wire, Acta Metall. Mater. 41 (1993) 2179-2183. 17. V.K. Kumikov, Kh.M. Guketlov, M.V. Gedgagova: Measurement of surface tension of electronic materials,
Izv. Roc. Akad. Nauk, Ser. Fiz. 70 (2006) 588-590. 18. S.N. Zhevnenko: Isotherms of surface tension in copper-based systems, Fiz. Metall. Metalloved. 106 (2008)
286-293. 19. E.N. Hodkin, M.G. Nicholas, D.M. Poole: The surface energies of solid molybdenum, niobium, tantalum
and tungsten, J. Less-Common Met. 20 (1970) 93-103. 20. B.C. Allen: The iterfacial free energies of solid chromium, molybdenum and tungsten, J. Less-Common
Met. 29 (1972) 263-281. 21. M. Nakamoto, M. Liukkonen, M. Friman, E. Heikinheimo, M. Hämäläinen, L.Holappa: Measurement of
surface tension of solid Cu by improved multiphase equilibrium, Metall. Mater. Trans. B 39B (2008) 570-580.
22. M. Nakamoto, M. Liukkonen, M. Friman, E. Heikinheimo, M. Hämäläinen, L.Holappa, T. Yamamoto: Novel multiphase equilibrium metod using a Na2O-SiO2 droplet, Scripta Mater. 62 (2010) 871-874.
23. V.K. Kumikov, Kh.B. Khokonov: On the measurement of surface free energy and surface tension of solid metals, J. Appl. Phys. 54 (1983) 1346-1350.
24. M. Liukkonen, M. Friman, M. Nakamoto, M. Hamalainen, L. Holappa: Assessment of surface energy functions for solid elements, Report TKK-MT-191, 56 pp. Helsinki University of Technology, 2007.
25. W.R. Tyson, W.A. Miller: Surface free energies of solid metals estimation from liquid surface tension measurement, Surf. Sci. 62 (1977) 267-276.
26. A. Navrotsky: Calorimetry of nanoparticles, surfaces, interfaces, thin films, and multilayers,J. Chem. Thermodyn. 39 (2007) 2-9.
27. A. Navrotsky, L. Mazeina, J. Majzlan: Size-driven structural and thermodynamic complexity in iron oxides, Science 319 (2008) 1635-1638.
28. A. Navrotsky: Energetics of oxide nanoparticles, Int. J. Quantum Chem. 209 (2009) 2647-2657. 29. Z.A. Matysina: The relative surface energy of hexagonal close-packed crystals, Mater. Chem. Phys. 60
(1999) 70-78.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
24
30. S.G. Wang, E.K. Tian, C.W. Lung: Surface energy of arbitrary crystal plane of bcc and fcc metals, J. Phys. Chem. Solids 61 (2000) 1295-1300.
31. Q. Jiang, H.M. Lu, M. Zhao: Modelling of surface energies of elemental crystals, J. Phys.: Condens. Matter 16 (2004) 521-530.
32. J. Li, X. Luo, P. Hu, S. Dong: Comment on “Modelling of surface energies of elemental crystals“, J. Phys.: Condens. matter 21 (2009) 198001.
33. D. Liu, H.M. Lu, Q. Jiang: Reply to comment on “Modelling of surface energies of elemental crystals“, J. Phys.: Condens. Matter 21 (2009) 198002.
34. M.W. Finnis, J.E. Sinclair: A simple empirical N-body potential for transition metals, Phil. Mag. A 50 (1984) 139-146.
35. A.P. Sutton, J. Chen: Long-range Finnis-Sinclair potentials, Phil. Mag. Lett. 61 (1990) 139-146. 36. B.D. Todd, R.M. Lynden-Bell: Surface and bulk properties of metals modelled with Sutton-Chen potentials,
Surf. Sci. 281 (1993) 191-206. 37. J.H. Li, X.D. Dai, S.H. Liang, K.P. Tai, Y. Kong, B.X. Liu: Interatomic potentials of the binary transition
metal systems and some applications in materials physics, Phys. Rep. 455 (2008) 1–134. 38. M.S. Daw, M.I. Baskes: Embedded-atom method: Derivation and application to impurities, surfaces, and
other defects in metals, Phys. Rev. B 29 (1984) 6443-6453. 39. S.M. Foiles, M.I. Baskes, M.S. Daw: Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni,
Pd, Pt, and their alloys, Phys. Rev. B 33 (1986) 7983-7991. 40. M.I. Baskes, J.S. Nelson, A.F. Wright: Semiempirical modified embedded-atom potentials for silicon and
germanium, Phys. Rev. B 40 (1989) 6085-6100. 41. M.I. Baskes: Modified embedded-atom potentials for cubic materials and impurities, Phys. Rev. B 46
(1992) 2727-2742. 42. M.S. Daw, S.M. Foiles, M.I. Baskes: The embedded-atom method: a review of theory and applications,
Mater. Sci. Rep. 9 (1993) 251-310. 43. M.I. Baskes, R.A. Johnson: Modified embedded atom potentials for HCP metals, Modelling Simul. Mater.
Sci. Eng. 2 (1994) 147-163. 44. M.I. Baskes: Determination of modified embedded atom method parameters for nickel, Mater. Chem. Phys.
50 (1997) 152-158. 45. B.J. Lee, M.I. Baskes: Second nearest-neighbor modified embedded-atom-method potential, Phys. Rev. B
62 (2000) 8564-8567. 46. B.J. Lee, M.I. Baskes H. Kim, Y.K. Cho: Second nearest-neighbor modified embedded-atom-method
potential for bcc transition metals, Phys. Rev. B 64 (2001) 184102. 47. B.J. Lee, J.H. Shim, M.I. Baskes: Semiempirical atomic potentials for the fcc. metals Cu, Ag, Au, Ni, Pd, Pt,
Al, and Pb based on first and second nearest-neighbor modified embedded atom method, Phys. Rev. B 68 (2003) 144112.
48. S. Ryu, C.R. Weinberger, M.I. Baskes, W. Cai: Improved modified embedded-atom method potentials for gold and silicon, Modelling Simul. Mater. Sci. Eng. 17 (2009) 075008.
49. F. Yang, Y.W. Liu, L.H. Ou, X. Wang, S.L. Chen: Density functional theory (DFT)-based modified embedded atom method potentials: Bridging the gap between nanoscale theoretical simulations and DFT calculations, Sci. China Chem. 53 (2010) 411-418.
50. J. Wan, Y.L. Fan, D.W. Gong, S.G. Shen, X.Q. Fan: Surface relaxation and stress of fcc metals: Cu, Ag, Au, Ni, Pd, Pt, Al and Pb, Modelling Simul. Mater. Sci. Eng. 7 (1999) 189-206.
51. J.M. Zhang, F. Ma, K.W. Xu: Calculation of the surface energy of fcc metals with modified embedde-atom method, Chin. Phys. 13 (2004) 1082-1090.
52. J.M. Zhang, F. Ma, K.W. Xu: Calculation of the surface energy of fcc metals with modified embedde-atom method, Appl. Surf. Sci. 229 (2004) 34-42.
53. Y.N. Wen, J.M. Zhang: Surface energy calculation of the fcc metals by using the MAEAM, Solid State Commun. 144 (2007) 163-167.
54. T.M. Trimble, R.C. Cammarata: Many-body effects on surface stress, surface energy and surface relaxation of fcc metals, Surf. Sci. 602 (2008) 2339-2347.
55. J.M. Zhang, D.D. Wang, K.W. Xu: Calculation of the surface energy of bcc transition metals by using the second nearest–neighbor modified embedded atom method, Appl. Surf. Sci. 252 (2006) 8217-8222.
56. Y.N. Wen, J.M. Zhang: Surface energy calculation of the bcc metals by using the MAEAM, Comput. Mater. Sci. 42 (2008) 281-285.
57. J.M. Zhang, D.D. Wang, K.W. Xu: Calculation of the surface energy of hcp metals by using the modified embedded atom method, Appl. Surf. Sci. 253 (2006) 2018–2024.
58. J.R Smith, A. Banerjea: New approach to calculation of total energies of solids with defects: surface-energy anisotropies, Phys. Rev. Lett. 59 (1987) 2451–2454.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
25
59. J.R Smith, T. Perry, A. Banerjea, J. Ferrante, G. Bozzolo: Equivalent-crystal theory of metal and semiconductor surfaces and defects, Phys. Rev. B 44 (1991) 6444–6465.
60. G. Bozzolo, J. Ferrante, A.M. Rodríguez: Modelling of surfaces. 1. Monoatomic metallic surfaces using equivalen crystal theory, J. Comput.-Aided Mater. Design 1 (1993) 285-304.
61. J. Ferrante, F.R. Zypman: Generalization of equivalent crystal theory to include angular dependence, Comput. Mater. Sci. 36 (2006) 425-431.
62. F.R. Zypman, J. Ferrante: Analytical algorithm for equivalent crystal theory, Comput. Mater. Sci. 42 (2008) 659-663.
63. J.H. Rose, J.R. Smith, J. Ferrante: Universal features of bonding in metals, Phys. Rev. B 28 (1983) 1835-1845.
64. A.M. Rodríguez, G. Bozzolo, J. Ferrante: Multilayer relaxation and surface energies of fcc and bcc metals using equivalent crystal theory, Surf. Sci. 289 (1993) 100-126.
65. E. Aghemenloh, J.O.A. Idiodi, S.O. Azi: Surface energies of hcp metals using equivalen crystal theory, Comput. Mater. Sci. 46 (2009) 524-530.
66. J.C. Boettger: Nonconvergence of surface energies obtained from thin-film calculations, Phys. Rew. B 49 (1994) 16798-16800.
67. V. Fiorentini, M. Methfessel: Extracting convergent surface energfies from slab calculations, J. Phys.: Condens. Matter 8 (1996) 6525-6529.
68. M. Methfessel, D. Hennig, M. Scheffler: Trends of the surface relaxation, surface energies, and work functions of the 4d transition metals, Phys. Rev. B 46 (1992) 4816-4829.
69. H.L. Skriver, N.M. Rosengaard: Surface energy and work function of elemental metals, Phys. Rev. B 46 (1992) 7157-7168.
70. L. Vitos, A.V. Ruban, H.L. Skriver, J. Kollár: The surface energy of metals, Surf. Sci. 411 (1998) 186-202. 71. I. Galanakis, N. Papanikolaou, P.H. Dederichs: Applicability of the broken-bond rule to the surface energy
of the fcc metals, Surf. Sci. 511 (2002) 1-12. 72. J. Kollár, L. Vitos, J.M. Osorio-Guillén, R. Ahuja: Calculation of surface stress for fcc transition metals,
Phys. Rev. B 62 (2003) 245417 (5 pp). 73. V. Zólyomi, L. Vitos, S.K. Kwon, J. Kollár: Surface relaxation and stress for 5d transition metals, J. Phys.:
Condens. Matter 21 (2009) 095007 (5 pp). 74. N.E. Singh-Miller, N. Marzari: Surface energies, work functions, and surface relaxations of low-index
metallic surfaces from first principles, Phys. Rev. B 80 (2009) 235407 (9 pp). 75. D.A. Young: Phase diagrams of the elements. University of California Press, Berkeley 1991. 76. A.I. Rusanov: Surface and cohesive energy of condensed bodies, J. Colloid Interface Sci. 90 (1982) 143-
147. 77. S.C. Vanithakumari, K.K. Nanda: A universal relation for the cohesive energy of nanoparticles, Phys. Lett.
A 372 (2008) 6930-6934. 78. C.L. Reynolds, Jr., P.R. Couchman, F.E. Karasz: On the relation between surface energy, melting
temperature and interatomic separation for metals, Phil. mag. 34 (1976) 659-661. 79. W. Haiss: Surface stress of clean and adsorbate-covered solids, Rep. Prog. Phys. 64 (2001) 591-648. 80. D. Sander: Surface stress: implications and measurements, Current Opinion Solid State Mater. Sci. (2003)
51–57. 81. C.W. Mayes, J.S. Vermaak, D. Kuhlmann-Wilsdorf: On surface stress and surface tension. II.
Determination of the surface stress og gold, Surf. Sci. 12 (1968) 134-140. 82. A.M. Stoneham: Measurement of surface tension by lattice parameter changes: theory for facetted
microcrystals, J. Phys. C: Solid State Phys. 10 (1977) 1175-1179. 83. K.K. Nanda, S.N. Behera, S.N. Sahu: The lattice contraction of nanometre-sized Sn and Bi particles
produced by an electrohydrodynamic technique, J. Phys. C: Solid State Phys. 13 (2001) 2861-2864. 84. Q. Jiang, L.H. Liang, D.S. Zhao: Lattice contraction and surface stress of fcc nanocrystals, J. Phys. Chem.
B 105 (2001) 6275-6277. 85. C. Solliard, P. Buffat: Crystal size variation of small gold crystals due to size effect, J. Phys. 38 (1977) C2-
167-C2-170. 86. C. Solliard, M. Flueli: Surface stress and size effect on the lattice parameter in small particles of gold and
platinum, Surf. Sci. 156 (1985) 487-494. 87. H. Hofmeister: High-resolution electron microscopy studies of metal nanoparticles: shape and twin defects,
and surface stress effects, J. Optoelectron. Advanced Mater. 9 (2007) 99-105. 88. H.J. Wasserman, J.S. Vermaak: Determination of a lattice contraction in very small silver particles, Surf.
Sci. 22 (1970) 164-172. 89. H.J. Wasserman, J.S. Vermaak: Determination of the surface stress of copper and platinum, Surf. Sci.32
(1972) 168-174.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
26
90. R.D. Diehl, J. Ledieu, N. Ferralis, A.W. Szmodis, R. McGrath: Low-energy electron diffraction from quasicrystal surfaces, J. Phys. Condens. Matter 15 (2003) R63-R81.
91. C.A. Gerken, G.A. Somorjai: Low-energy electron diffraction, in: Characterization of Materials (E.N. Kaufmann, Ed.), pp. 1120-1134, Wiley-Interscience, 2003.
92. W. Moritz, J. Landskron, M. Deschauer: Perspectives for surface structure analysis with low energy electron diffraction, Surf. Sci. 603 (2009) 1306-1314.
93. K. Kadas, Z. Nabi, S.K. Kwon, L. Vitos, R. Ahuja, B. Johansson, J. Kollar: Surface relaxation and surface stress of 4d transition metals, Surf. Sci. 600 (2006) 395–402.
94. G. Bozzolo, A.M. Rodríguez, J. Ferrante: Multilayer relaxation and surface energies of metallic surfaces, Surf. Sci. 315 (1994) 204-214.
95. J.M. Zhang, Y. Shu, K.W. Xu: Multilayer relaxation of fcc metals (001) surfaces: A modified embedded atom method study, Solid State Commun. 137 (2006) 441-445.
96. C.J. Davisson, L.H. Germer: Diffraction of electrons by a single crystal of nickel, Phys. Rev. 30 (1927) 705-740.
97. J.E. Lennard-Jones, B.M. Dent: The change in lattice spacing at a crystal boundary, Proc. Royal Soc. London. Series A, 121 (1928) 247-259.
98. R.J. Needs, M.J. Godfrey, M. Mansfield: Theory of surface stress and surface reconstruction, Surf. Sci. 242 (1991) 215-221.
99. R.C. Cammarata: Continuum odel for surface reconstructions in (111) and (100) oriented surfaces of fcc metals, Surf. Sci. 279 (1992) 341-348.
100. S. Olivier, G. Tréglia, A. Saúl, F. Willaime: Influence of surface stress in the missing row reconstruction of fcc transition metals, Surf. Sci. 600 (2006) 5131-5135.
100a D.D. dos Reis, F.R. Negreiros, V.E. de Carvalho, E.A. Soares: Geometry of the Au(110)-(1 × 2) missing-row clean surface: A New LEED and DFT study, Surf. Sci. 604 (2010) 568–573.
101. S.W. Ip, J.M. Toguri: The equivalence of surface tension, surface energy and surface free energy, J. Mater. Sci. 29 (1994) 688-692.
102. T. Chen, M.S. Chiu, C.N. Weng: Derivation of the generalized Young-Laplace equation of curved interfaces in nanoscaled solids, J. Appl. Phys. 100 (2006) 074308 (5 pp).
POZOR: tady je mezera !!! 123. R.C. Tolman: The effect of droplet size on surface tension, J. Chem. Phys. 17 (1949) 333-337. 124. H.M. Lu, Q. Jiang: Size-dependent surface tension and Tolman’s length of droplets, Langmuir 21 (2005)
779-781. 125. E.M. Blokhius J. Kuipers: Thermodynamic expression for the Tolman length, J. Chem. Phys. 124 (2006)
074701 (8 pp). 126. R.Z. Zhu, X.S. Wang: Thermodynamic theory of the Tolman’s length, Chin. Phys. B 19 (2010) 076801 (4
pp). 127. R.A. Andrievski: Size-dependent effects in properties of nanostructured materials, Rev. Adv. Mater. 21
(2009) 107-133. 128. R.A. Andrievski, A.V. Khachoyan: Role of size-dependent effects and interfaces in physicochemical
properties of consolidated nanomaterials, Russ. J. Gen. Chem. 80 (2010) 555-566. 129. Q. Jiang, J.C. Li, B.Q. Chi: Size-dependent cohesive energy of nanocrystals, Chem. Phys. Lett. 366 (2002)
551-554. 130. H.M. Lu, Q. Jiang: Size-dependent surface energies of nanocrystals, J. Phys. Chem. B 108 (2004) 5617-
5619. 131. Q. Jiang, H.M. Lu: Size dependent interface energy and its applications, Surf. Sci. Rep. 63 (2008) 427-464. 132. M. Attarian Shandiz: Effective coordination number model for size dependency of physical properties of
nanocrystals, J. Phys.: Condens. Mater 20 (2008) 325237 (9 pp). 133. K.K. Nanda, A. Maisels, F.E. Kruis, H. Fissan, S. Stappert: Higher surface energy of free nanopaerticles,
Phys. Rev. Lett. 91 (2003) 106102 (4 pp). 134. K.K. Nanda: Bulk cohesive energy and surface tension from the size-dependent evaporation study of
nanoparticles, Appl. Phys. Lett. 87 (2005) 021909 (3 pp). 135. S.C. Vanithakumari, K.K. Nanda: Phenomenological predictions of cohesive energy and structural
transition of nanoparticles, J. Phys. Chem. B 110 (2006) 1033-1037. 136. D. Xie, M. Wang, L. Cao:A simplified model to calculate the higher surface energy of free-standing
nanocrystals, phys. stat. sol. (b) 242 (2005) R76-R78. 137. G. Ouyang, X. Tan, G. Yang: Thermodynamic model of the surface energy of nanocrystals, Phys. Rev. B 74
(2006) 195408 (5 pp).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
27
138. H. Zhang, B. Chen, J.F. Banfield: The size dependent of the surface energy of titania nanocrystals, Phys. Chem. Chem. Phys. 11 (2009) 2553-2558.
139. B. Madasani, I. Vasiliev: Computational study of the surface properties of aluminum nanoparticles, Surf. Sci. 603 (2009) 2042-2046.
140. B. Medasani, Y.H. Park, I. Vasiliev: Theoretical study of the surface energy, stress, and lattice contraction of silver nanoparticles, Phys. Rev. B 75 (2007) 235436 (6 pp).
141. D. Liu, J.S. Lian, Q. Jiang: Surface energy and electronic structures of Ag quasicrystal clusters, J. Phys. Chem. C Lett. 113 (2009) 1168-1170.
142. D. Liu, Y.F. Zhu, Q. Jiang: Site and structure dependent cohesive energy in several Ag clusters, J. Phys. Chem. C 113 (2009) 10907-10912.
143. R. Lamber, S. Wetjen, N.I. Jaeger: Size dependence of the lattice parameter of small palladium particles, Phys. Rev. B 51 (1995) 10968-10971.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
28
3. Struktura nanočástic a atomárních klastrů Nanočástice nelze obecně chápat jako „velmi malé“ monokrystaly. Vlivem povrchových jevů (povrchová energie/povrchové napětí) mohou být atomární struktura nanočástic a vnější geometrický tvar odlišné od objemového materiálu a i v případě, že nanočástice vykazují atomové uspořádání (např. struktura fcc) stejné jako bulk, nemusí být meziatomové vzdálenosti v celém objemu nanočástice stejné, shodné s bulkem. Podle obecně akceptovaného modelu core-shell existuje tenká povrchová vrstva, ve které jsou v důsledku snížení počtu svých sousedů atomy vázány menším počtem vazeb. U většiny pevných látek (zejména kovů) jsou tyto vazby kratší, což vede ke zvýšení „průměrné“ hustoty nanočástice, ale byly pozorovány i opačné trendy – prodloužené vazby a nižší hustota. Tento jev je významný u částic s rozměry pod cca 10 nm a s klesajícími rozměry nanočástic se progresivně zvětšuje. Pokusíme-li se odhadnout velikost „ideální“ nanočástice tvaru koule, pak z jednoduché geometrické představy plyne, že pro nejtěsnější uspořádání stejně velkých Nat atomů o poloměru rat je průměr nanočástice dnp dán vztahem
( )3np at at0,74 2d N r= (3-1)
Parametr 0,74 v rovnici (3-1) představuje podíl zaplnění prostoru pro nejtěsnější uspořádání atomů v fcc a hcp struktuře. Závislost dnp = f(Nat) pro Au (rAu = 0,144 nm) je ukázána na obr. 9. Ze vztahu (3-1) vypočteme, že nanočástice Au o průměru 2 nm je tvořena cca 250 atomy, částice o průměru 10 nm již 31000 atomy.
0 10000 20000 30000 40000 500000
2
4
6
8
10
12
rAu = 0,144 nmffcc = 0,74
d np (n
m)
NAu
Obrázek 9 Závislost průměru kulaté nanočástice Au ve struktuře fcc na počtu atomů částici tvořících (rovnice (3.1))
Koule je geometrický prostorový útvar, který vykazuje nejmenší plochu povrchu na jednotku objemu. Reálné nanočástice nejsou ideálně kulaté, ale mají tvar polyedrů s různým počtem a

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
29
tvarem stěn (viz obr. 10). Proto je následující část věnována obecné geometrii těchto prostorových útvarů a jejich analýze. V dalších částech jsou pak shrnuty poznatky týkající se struktury nanočástic (zejména kovových prvků), a to nejprve pro nanočástice tvořené řádově 103-104 atomy, u kterých lze aplikovat top-down přístup a dále pro nanočástice tvořené 101-103 atomy (atomární klastry), jejichž strukturu je výhodné sledovat naopak v režimu bottom-up.
Obrázek 10 Nanočástice Ag ve tvaru krychle (A), kubooktaedru (B) a oktaedru (C) (Ref.7)
V dalším textu jsou diskutovány pouze energetické/termodynamické faktory mající vliv na atomární strukturu a tvar nanočástic. Kromě nich mají na strukturu a tvar nanočástic vliv i faktory kinetické, podmíněné zejména způsobem přípravy a dalšího zpracování. V mnoha případech jsou v dané populaci nanočástic identifikovány různě velké částice s různým tvarem i strukturou. Z hlediska termodynamického jsou některé z nich nestabilní, avšak v určitém časovém intervalu stálé. Obecně platí, že malé částice jsou v důsledku povrchové energie méně stabilní než částice velké (bulk), a tedy může spontánně docházet k jejich koalescenci. V souborech zejména velmi malých částic (pod 3 nm) byla opakovaně pozorována proměnná morfologie částic, tedy fluktuace velikost-tvar-struktura. Tato problematika je předmětem značného badatelského zájmu jak ze strany experimentálního sledování morfologie nanočástic – téměř volných, na podložce, suspendovaných v různých kapalinách nebo izolovaných v polymerních či skelných matricích (XRD, SEM, TEM, ...), tak v rámci počítačového modelování struktur a jejich stability (teoretické ab-initio výpočty, semiempirické výpočty, minimalizace Gibbsovy energie aj.) a byla zpracována v řadě přehledných prací, např. Ref.1-10).
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
30
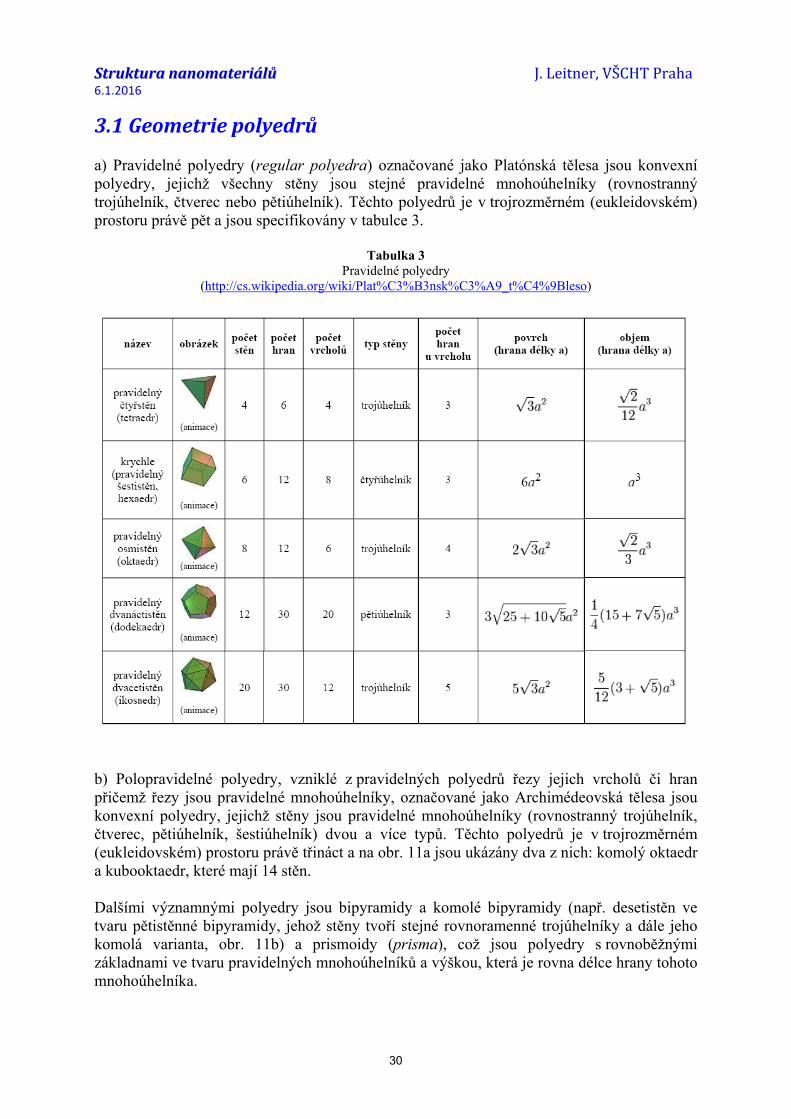
3.1 Geometrie polyedrů a) Pravidelné polyedry (regular polyedra) označované jako Platónská tělesa jsou konvexní polyedry, jejichž všechny stěny jsou stejné pravidelné mnohoúhelníky (rovnostranný trojúhelník, čtverec nebo pětiúhelník). Těchto polyedrů je v trojrozměrném (eukleidovském) prostoru právě pět a jsou specifikovány v tabulce 3.
Tabulka 3 Pravidelné polyedry
(http://cs.wikipedia.org/wiki/Plat%C3%B3nsk%C3%A9_t%C4%9Bleso)
b) Polopravidelné polyedry, vzniklé z pravidelných polyedrů řezy jejich vrcholů či hran přičemž řezy jsou pravidelné mnohoúhelníky, označované jako Archimédeovská tělesa jsou konvexní polyedry, jejichž stěny jsou pravidelné mnohoúhelníky (rovnostranný trojúhelník, čtverec, pětiúhelník, šestiúhelník) dvou a více typů. Těchto polyedrů je v trojrozměrném (eukleidovském) prostoru právě třináct a na obr. 11a jsou ukázány dva z nich: komolý oktaedr a kubooktaedr, které mají 14 stěn. Dalšími významnými polyedry jsou bipyramidy a komolé bipyramidy (např. desetistěn ve tvaru pětistěnné bipyramidy, jehož stěny tvoří stejné rovnoramenné trojúhelníky a dále jeho komolá varianta, obr. 11b) a prismoidy (prisma), což jsou polyedry s rovnoběžnými základnami ve tvaru pravidelných mnohoúhelníků a výškou, která je rovna délce hrany tohoto mnohoúhelníka.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
31
Oktaedr Komolý oktaedr KubooktaedrOktaedr Komolý oktaedr Kubooktaedr
Obrázek 11a Vybrané polyedry: pravidelný oktaedr, komolý oktaedr a kubooktaedr
Dekaedr Komolý dekaedr IkosaedrDekaedr Komolý dekaedr Ikosaedr
Obrázek 11b Vybrané polyedry: dekaedr (pentagonální bipyramida), komolý dekaedr a ikosaedr

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
32
3.2 Nanočástice tvořené řádově 103 a více atomy: Wulffova konstrukce
Snižující se počet atomů tvořících částici zvyšuje relativní význam energetických charakteristik povrchu (vzhledem k vlastnostem objemovým), což ve svých důsledcích určuje jak atomární strukturu částice, tak její vnější tvar. Uvažujme nejprve částici tvořenou pevně daným počtem atomů uspořádaných v určité krystalické struktuře (např. fcc). Zanedbáme-li okrajové efekty, pak celkový objem částice je dán počtem atomů a koeficientem zaplnění prostoru (např. 0,74 pro fcc strukturu). Daný objem lze realizovat různými geometrickými tvary částice, přičemž tyto tvary se liší velikostí plochy povrchu vztažené na jednotku objemu anp = Anp/Vnp. Jelikož povrch je nositelem energie (povrchová energie γsurf), která zvyšuje celkovou energii částice (Etot = Ebulk + Esurf), a tedy snižuje její stabilitu, je z hlediska čistě geometrického nejstabilnějším tvarem koule s minimální hodnotou parametru anp. Reálné nanočástice mají obvykle tvar různých polyedrů ohraničených různými krystalografickými rovinami (hkl). Daný polyedr lze charakterizovat tzv. tvarovým faktorem α, který je definován jako podíl plochy povrchu polyedru a povrchu koule (Asph) se stejným objemem: α = Anp/Asph = anp/asph. Výpočtem získáme hodnoty α20 = 1,06 (ikosaedr), α12 = 1,10 (dodekaedr), α8 = 1,18 (oktaedr), α6 = 1,24 (hexaedr-krychle) a α4 = 1,49 (tetraedr). Výsledný tvar částice tak určují dva faktory: (i) hodnota parametru anp a (ii) anizotropie povrchové energie, tedy různé hodnoty γsurf(hkl) pro různé roviny (hkl) ohraničující daný polyedr. Optimální tvar částice lze za jistých předpokladů získat postupem, který je v literatuře označován jako Wulffova konstrukce (Ref.11, 12). Matematicky se jedná o minimalizaci povrchové/mezifázové Helmholtzovy energie Fσ nanočástice o daném objemu při stálé teplotě:
[ ]σsurf,1 min, ,N
j jjF A T Vγ=
= →∑ (3-2)
Suma je provedena přes všechny povrchové plochy Aj. Pro řešení aplikujeme metodu Lagrangeových multiplikátorů. Jako proměnnou, která charakterizuje změnu hodnoty F s měnící se velikostí povrchových ploch Aj použijeme kolmé vzdálenosti od těžiště polyedru k různým povrchovým plochám hj. Vaznou podmínkou je konstantní objem, tedy dV = 0. Platí
0, 1,...,j j
F V j Nh h
λ∂ ∂− = =
∂ ∂ (3-3)
Celkový objem polyedru lze vyjádřit vztahem (objem polyedru V je homogenní funkcí 3. řádu proměnných h1, …, hN)
1 1 11 13 3
N N Nj j j jj j j
j
VV V h A hh= = =∂
= = =∂∑ ∑ ∑ (3-4)
kde Vj jsou objemy dílčích jehlanů majících základnu o ploše Aj a vrchol v těžišti polyedru. Pro dílčí povrchové plochy Aj dále platí (každá povrchová plocha Aj je homogenní funkcí 2. řádu proměnných h1, …, hN)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
33
112
N jj jj
j
AA h
h=
∂=
∂∑ (3-5)
Spojením vztahů (3-2) až (3-5) získáme soustavu rovnic
surf,1 1 0, 1,...,2
N Ni ii ii i
j j
A Ah j Nh h
λγ= =∂ ∂
− = =∂ ∂∑ ∑ (3-6)
kterou upravíme ve tvar
surf,1 0, 1,...,2
N ii ii
j
A h j Nh
λγ=∂ ⎛ ⎞− = =⎜ ⎟∂ ⎝ ⎠
∑ (3-7)
Řešení této soustavy, které vede k minimální hodnotě Fσ při [T,V] má tvar
surf, 0, 1,...,2i ih i Nλγ − = = (3-8)
a odtud
surf,1 surf,2 surf,
1 2...
2N
Nh h hγ γ γ λ
= = = = (3-9)
Parametry h1, h2, ..., hN tak definují výsledný tvar polyedru (částice), přičemž platí čím větší hodnota povrchové energie, tím větší vzdálenost (hi) dané plochy od těžiště a tím menší velikost této plochy Ai. Tímto postupem byl jako optimální tvar částice se strukturou fcc určen komolý oktaedr jehož povrch tvoří 8 šestiúhelníkových a 6 čtvercových ploch (viz obr. 11). Šestiúhelníkové plochy odpovídají rovinám (111) a čtvercové rovinám (100) a protože platí γsurf(111) < γsurf(100) je poměr ploch A(111)/A(100) > 1. Pro částice se strukturou bcc je optimálním tvarem rombický dodekaedr ohraničený 12 kosočtvercovými plochami, které odpovídají rovinám (110). Rovnovážný tvar krystalů lze vizualizovat v tzv. Wulffově diagramu, ve kterém je v polárních souřadnicích vynesena hodnota povrchové energie γsurf(hkl) v jednotlivých směrech [hkl]. V případě, že povrchová energie je izotropní (hodnoty γsurf(hkl) nezávisí na (hkl)), odpovídá Wulffův diagram kulové ploše. Wulffova konstrukce v 2D přiblížení a odvozený optimální tvar „krystalu“ je ukázán na obr. 12a. Na obr. 12b je překreslen výsledný tvar 2D krystalu.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
34
0
45
90
135
180
225
270
315
(10)γ(11)γ
0
45
90
135
180
225
270
315
(10)γ(11)γ
(10)γ(11)γ
[ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1− [ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1− [ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1−
Optimální tvar krystalu
0
45
90
135
180
225
270
315
(10)γ(11)γ
0
45
90
135
180
225
270
315
(10)γ(11)γ
(10)γ(11)γ
[ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1− [ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1− [ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1−
Optimální tvar krystalu
0
45
90
135
180
225
270
315
(10)γ(11)γ
0
45
90
135
180
225
270
315
(10)γ(11)γ
(10)γ(11)γ
0
45
90
135
180
225
270
315
(10)γ(11)γ
0
45
90
135
180
225
270
315
(10)γ(11)γ
(10)γ(11)γ
[ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1− [ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1− [ ]1,1
[ ]0,1
[ ]1,0
[ ]0, 1−
[ ]1,0−
[ ]1, 1−[ ]1, 1− −
[ ]1,1−
Optimální tvar krystalu
Obrázek 12a
Diagram závislosti γsurf(hkl) na krystalografických směrech a Wulffova konstrukce optimálního tvaru krystalu v 2D přiblížení
(10)h(11)h
(10)a
(11)a
(10)h(11)h
(10)h(11)h
(10)a
(11)a
Obrázek 12b Optimální tvaru krystalu (Wulffova konstrukce) v 2D přiblížení
Podle rovnice (3-9) platí pro 2D krystal na obr. 12b relace h(10)/h(11) = γ(10)/γ(11), kde γ(10) a γ(11) jsou energie jednotky obvodu s orientací (10) a (11). Na obr. 13 je vypočtená závislost celkové „obvodové“ energie E = 4a(10)γ(10) + 4a(11)γ(11) na podílu h(10)/h(11). Tento podíl je nejmenší pro čtverec se stranami kolmými na směry [1,0], [0,1], [–1,0] a [0,–1] (a(11) = 0, h(10)/h(11) = 1/√2) a největší pro čtverec se stranami kolmými na směry [1,1], [–1,1], [1,–1] a [–1,–1] ((a(10) = 0, h(10)/h(11) = √2). Výpočet byl proveden pro konstantní jednotkovou plochu 2D krystalu. Obvod obou krajních tvarů je stejný (o = 4), nejmenší hodnota obvodu odpovídá stejně velkým stranám a(10) = a(11) (h(10)/h(11) = 1). Takový tvar je z hlediska „obvodové“ energie tvarem optimálním pokud platí γ(10) = γ(11). V případě anizotropie (γ(10) ≠ γ(11)) minimum na křivce E = f(h(10)/h(11)) neodpovídá minimu závislosti o = f(h(10)/h(11)) (viz obr. 13).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
35
0.7 0.8 0.9 1.0 1.1 1.2 1.3 1.43.4
3.6
3.8
4.0
4.2
4.4
4.6
4.8
5.0
γ(10)/γ(11) = 1,25a(10) = 17,6 %
γ(10)/γ(11) = 1,15a(10) = 29,7 %
E =
4a (1
0)γ (1
0) +
4a (1
1)γ (1
1)
h(10)/h(11)
γ(10)/γ(11) = 1a(10) = 50,0 %
Obrázek 13 Závislost „obvodové“ energie E = 4a(10)γ(10) + 4a(11)γ(11) na podílu vzdáleností h(10)/h(11), viz obr. 12
(křivka pro poměr γ(10)/γ(11) = 1 současně představuje závislost obvodu)
Výše popsaná Wulffova konstrukce pro stanovení rovnovážného tvaru nanočástic má určitá omezení: (i) Nepredikuje optimální (rovnovážný) tvar částice, nýbrž optimalizuje geometrii tvaru uvažovaného. V případě nanočástic však bylo experimentálně i teoreticky prokázáno, že jejich tvar závisí na velikosti a vedle polyedrů odvozených z bulkové krystalové struktury (např. fcc) mohou nanočástice vykazovat i jiné tvary, např. dekaedr nebo ikosaedr, odvozené z tzv. pseudokrystalické struktury. (ii) Byla navržena pro „makrokrystaly“, u kterých lze zanedbat výjimečné vlastnosti atomů umístěných na hranách a ve vrcholech příslušného polyedru. U nanočástic tvořených řádově 102 atomy je však podíl těchto atomů významný, a protože povrchová (kohezní) energie těchto atomů se obecně liší od energie atomů uvnitř povrchových ploch mají tyto atomy vliv na rovnovážný tvar nanočástice (Ref.12b, 13). Termodynamický model pro stanovení rovnovážného tvaru nanočástic v závislosti na jejich atomární struktuře a velikosti prezentovala profesorka Barnard se svými spolupracovníky (Ref.14-19). Stejně jako v případě Wulffovy konstrukce model přímo nepredikuje nejstabilnější tvar nanočástice (s minimální hodnotou Gibbsovy energie), ale umožňuje vypočítat Gibbsovu energii v závislosti na velikosti (počtu atomů) pro různé zvolené tvary a z porovnání z nich určit ten stabilní. V rámci tohoto modelu je celková Gibbsova energie nanočástice vyjádřena jako suma čtyř příspěvků – objemového (bulk), povrchových ploch (surf), hran (edge) a vrcholů (corner):
np bulk surf edge cornerG G G G G= + + + (3-10)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
36
Příspěvky Gsurf, Gedge a Gcorner jsou vyjádřeny jako sumy přes jednotlivé položky. Pro povrchový term tak platí vztah
A Asurf surf, surf,i i i ii i
MG A q fγ γρ
= =∑ ∑ (3-11)
kde qA = Anp/Vnp a fi
A = Ai/Anp a suma je přes všechny povrchové plochy (hkl). Analogicky pro příspěvek hran délky Lnp = ∑Lj platí
L Ledge edge,j j j jj j
MG L q fγ λρ
= =∑ ∑ (3-12)
Veličina γedge,j má význam energie délkové jednotky hrany typu j, qL = Lnp/Vnp a fj
L = Lj/Lnp a suma je přes všechny povrchové hrany. Pro příspěvek vrcholů v počtu Nnp = ∑Nk pak platí
C Ccorner corner,k k k kk k
MG N q fγ ερ
= =∑ ∑ (3-13)
Veličina γcorner,k má význam energie jednoho vrcholu typu k, qC = Nnp/Vnp a fk
L = Nk/Nnp a suma je přes všechny vrcholy nanočástice. Provedené srovnávací výpočty (Ref.13) ukazují, že vliv hran a vrcholů je významný jen u velmi malých částic (pod cca 2 nm).V rámci modelu je rovněž uvažována objemová kontrakce nanočástic v důsledku zvýšeného vnitřního tlaku (viz část 3.4). Jako příklad výstupu takové termodynamické studie jsou na obr. 14 prezentovány výsledky pro nanočástice uhlíku v diamantové struktuře.
Obrázek 14
Závislost celkové Gibbsovy energie nanočástic C(dia) na počtu atomů částici tvořící (Ref.13)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
37
Experimentální pozorování i teoretické výpočty ukazují, že vedle monokrystalických nanočástic odvozených z bulkové struktury existují i jiné struktury a tvary nanočástic. Pro prvky krystalizující v fcc struktuře jsou typické částice s pětičetnou osou symetrie vykazující vícenásobné dvojčatění (multiply-twinned particles) ohraničené plochami s nízkou povrchovou energii (Ref.20-30). Výslednými tvary jsou pentagonální bipyramida (dekaedr) a ikosaedr (viz obr. 11b), popř. jejich komolé varianty, vzniklé skládáním tetraedrických bloků s (111) povrchovými plochami. Hraniční plochy tetraedrů jsou rovinami dvojčatění, které jsou, obdobně jako povrch krystalu, nositeli energie. Jelikož příslušně orientované pravidelné tetraedry zcela nezaplní prostor dekaedru resp. ikosaedru, dochází k jejich deformaci, která dále zvyšuje celkovou energie částice. Jelikož energie spojená s deformací je úměrná velkosti částice, jsou oba typy částic energeticky výhodné pouze v oblasti malých rozměrů (obvykle pod 1-2 nm). Výše popsaný dekaedr je tvořen pěti tetraedry s jednou společnou hranou, která je osou bipyramidy. Uvažujeme-li pravidelný dekaedr o jednotkové hraně, pak jeho objem je (5 + √5)/12. Objem pěti pravidelných tetraedrů o jednotkové hraně je 5×√2/12, a tedy vyplní prostor dekaedru z 97,72 %. Vyjádříme-li neúplnost zaplnění objemu dekaedru prostorovým úhlem u osy bipyramidy, pak volný prostor odpovídá úhlu přibližně 7,4° (zaplněný prostor je 5×70,5° = 352,6°). Tento volný prostor lze zaplnit pouze za cenu deformace původně pravidelných tetraedrických jednotek. Vzniklá struktura má nižší symetrii než původní fcc struktura a o cca 2 % nižší hustotu: atomy zaplňují objem z 72,36 % (v nejtěsnějším uspořádání fcc z 74,05 %). Dekaedr má velký poměr A/V = 7,18 (jednotková hrana), který lze snížit řezem obvodových hran rovinami (100) za vzniku komolého dekaedru (obr. 11b). Dalšího snížení energie lze docílit nahrazením kolmých hran mezi rovinami (100) zářezy ve tvaru písmene V, které zvýši podíl povrchových rovin (111) – tzv. Mark’s truncated decahedra (Ref.12, 25). Ikosaedr – tzv. Mackay icosahedron – (tvořený dvaceti identickými tetraedry) má výrazně nižší poměr A/V = 3,97 (jednotková hrana) než komolý dekaedr, avšak vyšší energetický příspěvek v důsledku objemové deformace. Z důvodu stérického omezení zbývá ve středu mezi pravidelnými tetraedry volný prostor, který odpovídá prostorovému úhlu Ω = 1,54 sr (vrcholový úhel Ωtetra = 3arccos(1/3) – π = 0,5513 sr, úhlový misfit ve středu ikosaedru Ω = 4π – 20⋅Ωtetra = 1,54 sr). Deformace původně pravidelných tetraedrů spočívá v prodloužení vnějších hran (povrchové hrany ikosaedru) o cca 5%, čímž se zvýší úhly mezi stranami stěn přiléhajících k vnitřním vrcholům (střed ikosaedru) ze 60° na cca 63,5°. Současně se sníží symetrie vzhledem k původní fcc struktuře a o cca 7 % se sníží hustota: atomy zaplňují objem z 68,82 % (v nejtěsnější uspořádání fcc z 74,05 %). Vnější tvar částice tvořené určitým počtem atomů je ovlivňován řadou dalších faktorů, zejména přítomností adsorbovaných atomů/molekul na povrchu, teplotou a v neposlední řadě charakterem nanočástic – volné resp. téměř volné nebo deponované na podložce (Ref.1, 6, 31). Vliv teploty na relativní zastoupení různých tvarů v populaci nanočástic Cu připravených kondenzací z plynné fáze na podložce SrTiO3 při různých teplotách depozice (Ref.32) je ukázán na obr. 15.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
38
Obrázek 15 Vliv teploty na relativní zastoupení různých tvarů v populaci nanočástic Cu
připravených kondenzací z plynné fáze na podložce SrTiO3 (Ref.32) Povrchové jevy (povrchová energie + povrchové napětí) ovlivňují u monokrystalických nanočástic rovněž atomární strukturu, která tak vykazuje závislost na velikosti. Vyjdeme-li z výše popsaného přístupu profesorky Barnard a v rovnici (3-10) budeme uvažovat pouze první dva příspěvky Gibbsovy energie, pak pro Gibbsovu energii nanočástice ve fázi φ (např. fcc, hcp nebo bcc) objemu Vnp při teplotě T a tlaku okolí p°platí
( ) ( )A
npnp np m surf,
m np
2, , 1 i ii
i ii
f AVG V T p G T A
V B rϕ ϕ γ
⎡ ⎤⎛ ⎞⎢ ⎥⎜ ⎟= + −
⎜ ⎟⎢ ⎥⎝ ⎠⎣ ⎦
∑ ∑ (3-14)
Člen v kulatých závorkách na pravé straně rovnice (3-14) představuje korekci na zvýšený tlak uvnitř částice. Porovnáním vypočtených hodnot molární Gibbsovy energie pro nanočástice tvořené určitým počtem atomů v různých strukturních modifikacích - fázích φ (lišící se parametry Gm
o, Vm, B, f a γsurf) určíme pro danou teplotu a velikost (počet atomů) stabilní strukturu (fázi φ) s minimální hodnotou Gnp. Tento přístup v komplexnějším zpracování (uvažují se nesférické tvary nanočástic získané optimalizací pro daný počet atomů a anizotropie hodnot f(hkl) a γsurf(hkl)) užili Barnard a spol. pro predikci relativní termodynamické stability různých strukturních modifikací TiO2(Ref.17) a ZrO2(Ref18). Na obr. 16 je ukázána závislost Gibbsovy energie tvarově optimalizovaných nanočástic ZrO2 v tetragonální (t) a monoklinické (m) struktuře při teplotě 298 K na velikosti částic (počtu vzorcových jednotek). Hodnoty G jsou vztaženy k hodnotě pro objemový m-ZrO2. Ačkoliv objemový ZrO2 je až do teploty ≈1170°C stabilní v monoklinické struktuře, částice m-ZrO2 o velikosti pod cca 13 nm jsou nestabilní vzhledem t-ZrO2 již při teplotě pokojové.
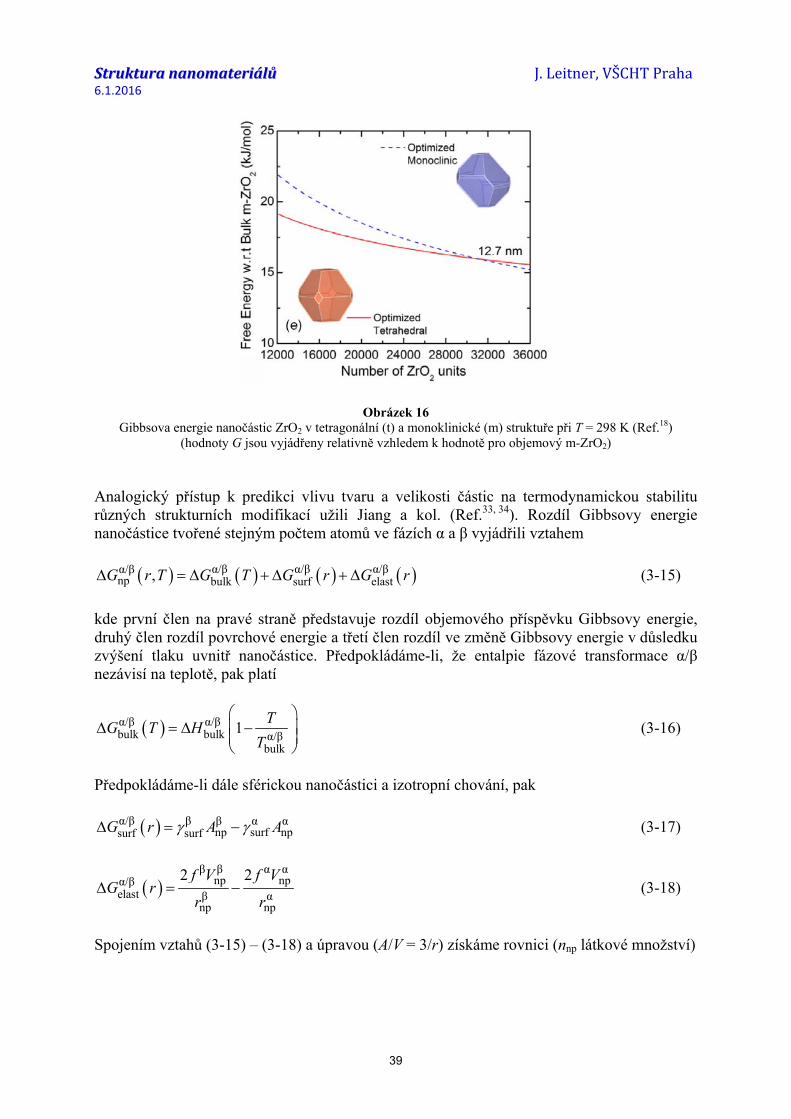
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
39
Obrázek 16 Gibbsova energie nanočástic ZrO2 v tetragonální (t) a monoklinické (m) struktuře při T = 298 K (Ref.18)
(hodnoty G jsou vyjádřeny relativně vzhledem k hodnotě pro objemový m-ZrO2) Analogický přístup k predikci vlivu tvaru a velikosti částic na termodynamickou stabilitu různých strukturních modifikací užili Jiang a kol. (Ref.33, 34). Rozdíl Gibbsovy energie nanočástice tvořené stejným počtem atomů ve fázích α a β vyjádřili vztahem
( ) ( ) ( ) ( )α/β α/β α/βα/βnp bulk surf elast,G r T G T G r G r∆ = ∆ + ∆ + ∆ (3-15)
kde první člen na pravé straně představuje rozdíl objemového příspěvku Gibbsovy energie, druhý člen rozdíl povrchové energie a třetí člen rozdíl ve změně Gibbsovy energie v důsledku zvýšení tlaku uvnitř nanočástice. Předpokládáme-li, že entalpie fázové transformace α/β nezávisí na teplotě, pak platí
( )α/β α/βbulk bulk α/β
bulk1 TG T H
T
⎛ ⎞∆ = ∆ −⎜ ⎟⎜ ⎟
⎝ ⎠ (3-16)
Předpokládáme-li dále sférickou nanočástici a izotropní chování, pak
( )α/β β β α αnp surf npsurf surfG r A Aγ γ∆ = − (3-17)
( )β β α α
np npα/βelast β α
np np
2 2f V f VG r
r r∆ = − (3-18)
Spojením vztahů (3-15) – (3-18) a úpravou (A/V = 3/r) získáme rovnici (nnp látkové množství)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
40
( )
( ) ( )
α/β β β α α β β α αnp α/β msurf surf m m
bulk α/β β α β αnp np np np npbulk
ββ β α α αm m surfsurfα/β
bulk α/β β αnp npbulk
, 3 2 21
3 2 3 21
G r T V A f V f VTHn r r r rT
V f V fTHr rT
γ γ
γ γ
⎛ ⎞∆= ∆ − + − + − =⎜ ⎟⎜ ⎟
⎝ ⎠
+ +⎛ ⎞= ∆ − + −⎜ ⎟⎜ ⎟
⎝ ⎠
(3-19)
V rámci Jiangova modelu je teplota transformace α ↔ β definována jako teplota, při které platí ∆Gα/β = 0. Platí:
( ) ( )ββ β α α αnp np surfsurfα/β
bulk α/β β αnp npbulk
3 2 3 21
V f V fTHr rT
γ γ+ +⎛ ⎞∆ − = − +⎜ ⎟⎜ ⎟
⎝ ⎠ (3-19a)
Předpokládáme-li ραnp = ρβnp = ρnp, je rαnp = rβnp = rnp a Vα
m = Vβm = Vm a
( ) ( )
( ) ( )
α/β β β α αmsurfbulk surfα/β
npbulk
β α β αm msurfsurf
np np
1 3 2 3 2
3 2
VTH f frT
V V f fr r
γ γ
γ γ
⎛ ⎞⎡ ⎤∆ − = − + − + =⎜ ⎟ ⎣ ⎦⎜ ⎟
⎝ ⎠
= − − − −
(3-19b)
Teplotu transformace lze z rovnic (3-19a) nebo (3-19b) explicitně vyjádřit jako funkci velikosti nanočástic a stejně tak lze vyjádřit velikost nanočástic jako funkci teploty transformace. Tento přístup byl užit při modelování závislostí Ttr-rnp pro C (Ref.35), ZrO2 (Ref.36), Al2O3, Fe2O3, TiO2 (Ref.37), ZnS (Ref.38), CdSe, ZnO (Ref.39) a jiných polovodičů typu AIIBVI (Ref.40). Výsledky výpočtů (Ref.40) pro fázovou transformaci nanočástic ZnS ve struktuře sfaleritu (kubické) a wurtzitu (hexagonální) jsou prezentovány na obr. 17.
Obrázek 17 Relace mezi rozměry nanočástic a teplotou transformace sfaleritové a wurtzitové struktury ZnS (Ref.40)
(předpokládán kulatý tvar nanočástic a izotropní chování povrchu) Experimentální data (Ref.41), (Ref. 42)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
41
Částečně odlišnou závislost mezi teplotou transformace a velikostí částice lze odvodit z podmínky rovnováhy pro systém znázorněný na obr. 18. Kulatá částice pevné látky ve struktuře α o poloměru rα je při teplotě T a tlaku pg v rovnováze se svými parami. Za stejných T a pg v rovnováze koexistuje i kulatá nanočástice téže látky ve struktuře β o poloměru rβ. Třífázový systém (α)-(β)-(g) má konstantní objem, „vnitřní“ tlak v částicích pα a pβ závisí na velikosti částic. Pro tuto rovnováhu (trojný bod) platí (Ref.43-45)
Vα, pαVβ, pβ
T, pgVα, pαVα, pα
Vβ, pβVβ, pβ
T, pg
Obrázek 18
Geometrické uspořádání systému pro odvození závislosti mezi teplotou fázové transformace a velikostí nanočástic
( ) ( )ββ β α α αm surf msurfg β α
g β αβ αnp np
2 2( , ) ( , ) ( , )
f V f VT p T p T p
r r
γ γµ µ µ
− −= − = − (3-20)
Chemický potenciál ve fázi φ (φ = α, β) při teplotě T a tlaku pφ vyjádříme jako
( )o
om m
o o om m m m
np
( , ) ( , ) d
2( , ) ( , )
p
pT p G T p V p
fG T p V p p G T p Vr
ϕϕ ϕ ϕ
ϕ
ϕϕ ϕ ϕ ϕ ϕ
ϕ
µ = + =
= + − = +
∫ (3-21)
Ve vztahu (3-21) předpokládáme nezávislost molárního objemu na tlaku a izotropní povrch s hodnotou povrchového napětí fφ. Spojením vztahů (3-20) a (3-21) pro φ = α, β získáme relaci
β β α αα/β α/βo o msurf surf m
m,β m,α bulk bulk α/β β αnp npbulk
2 2( , ) ( , ) 1V VTG T p G T p G H
r rTγ γ⎛ ⎞
− = ∆ = ∆ − = −⎜ ⎟⎜ ⎟⎝ ⎠
(3-22)
Při odvození předpokládáme teplotně nezávislou entalpii transformace objemového materiálu ∆Hα/β
bulk. Z porovnání se vztahem (3-19) je zřejmé, že rovnovážná podmínka odvozená tímto postupem explicitně neobsahuje povrchová napětí f α a f β a číselná konstanta u povrchové energie je 2 (3-22) namísto 3 (3-19). Tento rozdíl je dán výchozími předpoklady pro odvození uvedených vztahů, a to konstantním objemem resp. tlakem systému.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
42
Vztah (3-22) lze dále zjednodušit za předpokladu malých rozdílů v hustotě fází α a β. Pokud je zcela zanedbáme, platí: rαnp = rβnp = rnp a Vα
m = Vβm = Vm a z rovnice (2-22) získáme vztahy
( )β αm surfsurf
α/β α/βnpbulk bulk
2 11VT
rT H
γ γ−= −
∆ (3-23)
( )( )β α
m surfsurfnp α/β α/β
bulk bulk
2
1
Vr
H T T
γ γ−=∆ −
(3-24)
Budeme-li rozdíl hustot uvažovat, pak změnu teploty transformace v závislosti na velikosti částice ve struktuře α vyjádříme jako
( )2 3βα α βsurf surf
α/β α/β ααnpbulk bulk
211
MT
rT H
γ γ ρ ρ
ρ
⎡ ⎤−⎢ ⎥⎣ ⎦= −∆
(3-25)
a závislost velikosti částice ve struktuře α na teplotě jako
( )( )
2 3βα α βsurf surf
αnp α/β α/βα
bulk bulk
2
1
Mr
H T T
γ γ ρ ρ
ρ
⎡ ⎤−⎢ ⎥⎣ ⎦=∆ −
(3-25)
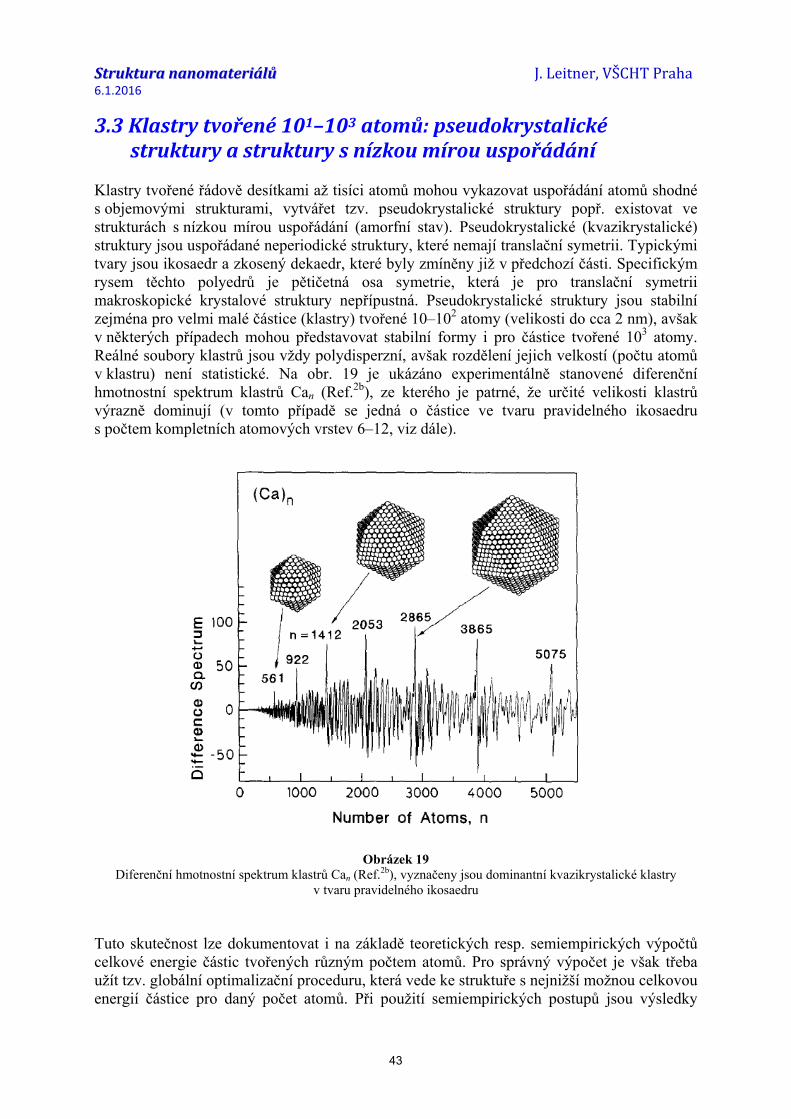
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
43
3.3 Klastry tvořené 101–103 atomů: pseudokrystalické struktury a struktury s nízkou mírou uspořádání
Klastry tvořené řádově desítkami až tisíci atomů mohou vykazovat uspořádání atomů shodné s objemovými strukturami, vytvářet tzv. pseudokrystalické struktury popř. existovat ve strukturách s nízkou mírou uspořádání (amorfní stav). Pseudokrystalické (kvazikrystalické) struktury jsou uspořádané neperiodické struktury, které nemají translační symetrii. Typickými tvary jsou ikosaedr a zkosený dekaedr, které byly zmíněny již v předchozí části. Specifickým rysem těchto polyedrů je pětičetná osa symetrie, která je pro translační symetrii makroskopické krystalové struktury nepřípustná. Pseudokrystalické struktury jsou stabilní zejména pro velmi malé částice (klastry) tvořené 10–102 atomy (velikosti do cca 2 nm), avšak v některých případech mohou představovat stabilní formy i pro částice tvořené 103 atomy. Reálné soubory klastrů jsou vždy polydisperzní, avšak rozdělení jejich velkostí (počtu atomů v klastru) není statistické. Na obr. 19 je ukázáno experimentálně stanovené diferenční hmotnostní spektrum klastrů Can (Ref.2b), ze kterého je patrné, že určité velikosti klastrů výrazně dominují (v tomto případě se jedná o částice ve tvaru pravidelného ikosaedru s počtem kompletních atomových vrstev 6–12, viz dále).
Obrázek 19 Diferenční hmotnostní spektrum klastrů Can (Ref.2b), vyznačeny jsou dominantní kvazikrystalické klastry
v tvaru pravidelného ikosaedru Tuto skutečnost lze dokumentovat i na základě teoretických resp. semiempirických výpočtů celkové energie částic tvořených různým počtem atomů. Pro správný výpočet je však třeba užít tzv. globální optimalizační proceduru, která vede ke struktuře s nejnižší možnou celkovou energií částice pro daný počet atomů. Při použití semiempirických postupů jsou výsledky

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
44
výpočtů významně ovlivněny použitým meziatomovým potenciálem. Na obr. 20 je ukázána závislost redukované energie nanočástic Ni na počtu atomů částici tvořících (Ref.51). Energie nanočástic byla vypočtena na základě Sutton-Chenova (9-6) potenciálu a je vztažena k hodnotě ENi získané z proložení individuálních hodnot EN (N je počet atomů). Klastry tvořené 13 a 55 atomy mají tvar pravidelného ikosaedru, morfologie 38-atomového klastru je odvozena z fcc struktury (zkosený oktaedr) a 75-atomový klastr má tvar zkoseného dekaedru (viz dále).
Obrázek 20 Energie nanočástic Ni v závislosti na počtu atomů N částici tvořících
(energie E je vztažena k „průměrné“ hodnotě ENi = f(NNi) V rámci dané velikosti především malých klastrů existuje řada tvarů, které se liší svým prostorovým uspořádáním atomů (lze je označit za izomery), ale mají velmi blízké hodnoty celkové energie, a mohou tedy v dané populaci částic koexistovat. Stabilní uspořádané atomové klastry lze v prvním přiblížení popsat jako koncentrické útvary s pravidelnými vrstvami/slupkami atomů (shell-structure) (Ref.2, 46-52)b. Tato jednoduchá definice by však pomíjela řadu významných geometrických tvarů. Uvažujme prostou krychlovou strukturu (viz obr. 21a), kde atomy jsou umístěny pouze v rozích krychle. 27 atomů (3×3×3) vytváří strukturu s centrálním atomem (viz obr. 21b), který má ve vzdálenosti a (hrana krychle) 6 sousedních atomů, ve vzdálenosti √2⋅a (stěnová úhlopříčka) 12 atomů a ve vzdálenosti √3⋅a (tělesová úhlopříčka) 8 atomů. Z výše uvedené definice plyne, že další klastr s centrálním atomem je tvořen 125 atomy (5×5×5) (viz obr. 21d). Pokud ale uvažujeme i krychli tvořenou 64 atomy (4×4×4) (viz obr. 21c), která nemá centrální atom nýbrž centrální krychli tvořenou osmi atomy, musíme výše uvedenou definici klastru modifikovat (rozšířit).
b V dalším textu nebudeme uvažovat klastry tvořené velmi malým počtem atomů (méně než cca 10), jejichž stabilní tvary mohou být lineární řetězce nebo 2D struktury – nelineární řetězce a cykly.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
45
Vrstevnaté klastry tedy nemusí být útvary s centrálním atomem, další vrstvy atomů nemusí překrývat všechny vrstvy předchozí, avšak symetrie vnějšího tvaru musí být s každou další vrstvou zachována.
a b c da b c da b c d
Obrázek 21 Uspořádání atomů v prosté kubické struktuře
(počet atomů na hranách krychle je 2(a), 3(b), 4(c) a 5(d), klastr na obr. (b) a (d) má ve středu právě jeden atom, klastr na obr (c) má ve středu krychli tvořenou osmi atomy, viz obr. (a))
Dominantní velikost klastrů odpovídá určitým počtům atomů (tzv. magic number), které, dle výše uvedených pravidel, vytvářejí vrstvu po vrstvě klastr dané geometrie. Jako příklad uvažujme genezi atomových klastrů postupným zaplňováním následných koncentrických atomových vrstev. Zvolený středový atom klastru má v první koordinační sféře celkem 12 sousedů. Způsobů, jak pravidelně umístit 12 atomů první vrstvy kolem centrálního atomu je více. V uspořádání bulkové fcc struktury (nejtěsnější uspořádání, zaplnění prostoru 74,05 %) leží 6 atomů ve stejné rovině jako centrální atom, tři v rovině nad a tři v rovině pod, přičemž všechny atomy první sféry se dotýkají centrálního atomu. Výsledným tvarem je kubooktaedr, čtrnáctistěn s osmi stěnami ve tvaru rovnostranného trojúhelníku a šesti stěnami ve tvaru čtverce (viz obr. 22a). Jinou možností je umístit atomy první vrstvy do vrcholů pravidelného ikosaedru (deformace fcc struktury, zaplnění prostoru 68,82 %) (viz obr. 22b, Ref.4).
Obrázek 22
Nanočástice ve tvaru komolého oktaedru (a) a pravidelného ikosaedru (b) (Ref.4)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
46
Z geometrie daného tvaru lze odvodit vztahy mezi počtem vrstev ν a počtem atomů Nat částici tvořících a rovněž podíly povrchových atomů ve specifických polohách (plocha-hrana-vrchol) (Ref.2, 46-52). Tak např. pro výše zmíněné kubooktaedr a ikosaedr platí (centrální atomν = 1)
3 2at
10 115 13 3
N ν ν ν= − + − (3-26)
resp. (centrální atomν = 0)
3 2at
10 115 13 3
N ν ν ν= + + + (3-27)
Počet atomů v ν-té vrstvě kubooktaedru resp. ikosaedru je dán vztahem (centrální atomν = 0)
σ 2at 10 2N ν= + (3-28)
Podíl povrchových atomů z celkového počtu atomů částici tvořících (někdy označovaný jako disperze D = Nσ
at/Nat) je pro uvažované geometrické tvary v závislosti na počtu vrstev ν ukázán na obr. 23 (Ref.48).
Obrázek 23 Disperze (podíl povrchových atomů z celkového počtu atomů částici tvořících, D = Nσ
at/Nat) v závislosti na počtu vrstev (cluster order ν ) nanočástice ve tvaru ikosaedru nebo kubooktaedru (Ref.48)
V tabulce 4 jsou pro vybrané tvary nanočástic (pravidelný tetraedr, pravidelný oktaedr, kubooktaedr/ikosaedr a dekaedr-pentagonální bipyramida) uvedeny celkové počty atomů a podíly povrchových atomů pro počet vrstev klastru 1-6 (Ref.48, 49).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
47
Tabulka 4 Počty atomů Nat tvořících klastry různých tvarů s plně obsazenými vrstvami ν = 1–6
a podíly povrchový atomů D = Nσat/Nat (Ref.2b, 47–49)
(Kurzivou uvedené údaje odpovídají klastrům, které nemají centrální atom)
Tetraedr Oktaedr Kubooktaedr Ikosaedr Dekaedr
ν Nat D Nat D Nat D Nat D
0 1 - 1 - 1 - 1 - 1 4 1 6 1 13 0,923 7 1 2 10 1 19 0,947 55 0,764 23 0,957 3 20 1 44 0,864 147 0,626 54 0,870 4 35 0,971 85 0,776 309 0,524 105 0,781 5 56 0,929 146 0,699 561 0,449 181 0,702 6 84 0,881 231 0,632 923 0,392 287 0,634
Kromě výše popsaného geometrického principu (kompletní atomové vrstvy/slupky klastrů) existuje u některých látek další faktor, který určuje preferovanou konfiguraci zejména velmi malých klastrů. Jedná se o klastry s uzavřenými elektronovými slupkami, které jsou typické zejména pro alkalické kovy. Počty atomů ve stabilních klastrech lze určit na základě jednoduchého kvantově mechanického modelu (tzv. spherical jellium model), v rámci kterého se delokalizované valenční elektrony jednotlivých atomů volně pohybují v homogenním, sféricky symetrickém potenciálovém poli kladného náboje („positive jelly“) vytvořeném zbytky atomů (ion core = nuclei + core electrones) (Ref.2, 5, 53-58). Řešením příslušné Schrödingerovy rovnice získáme diskrétní energetické hladiny elektronů charakterizované, analogicky jako u individuálních atomů, hlavním kvantovým číslem n a vedlejším (orbitalovým, angular momentum) kvantovým číslem l s degenerací 2×(2l + 1). Tyto hladiny jsou postupně (od nejnižší) obsazovány valenčními elektrony jednotlivých atomů, přičemž stabilní klastry odpovídají plnému obsazení orbitalů elektrony. Tímto způsobem lze generovat číselnou řadu počtu valenčních elektronů 2, 8, 18, 20, 34, 40, 58, 90, 92, 138, 198, 268, 338, 440, 562, 704, 852, 1032, ..., která v případě alkalických kovů s jedním valenčním elektronem přímo odpovídá počtu atomů ve stabilních klastrech. Stabilní klastry tvořené „magickým“ počtem atomů (na základě geometrického či elektronového principu) vykazují ve srovnání s klastry tvořenými jiným počtem atomů výjimečné vlastnosti. Se zvýšenou stabilitou souvisí např. vyšší hodnoty kohezní a disociační energie nebo teploty tání a entalpie tání, které tak vykazují výrazné fluktuace v závislosti na počtu atomů klastr tvořících. ... (doplnit Obr. 24, Ref, 59)
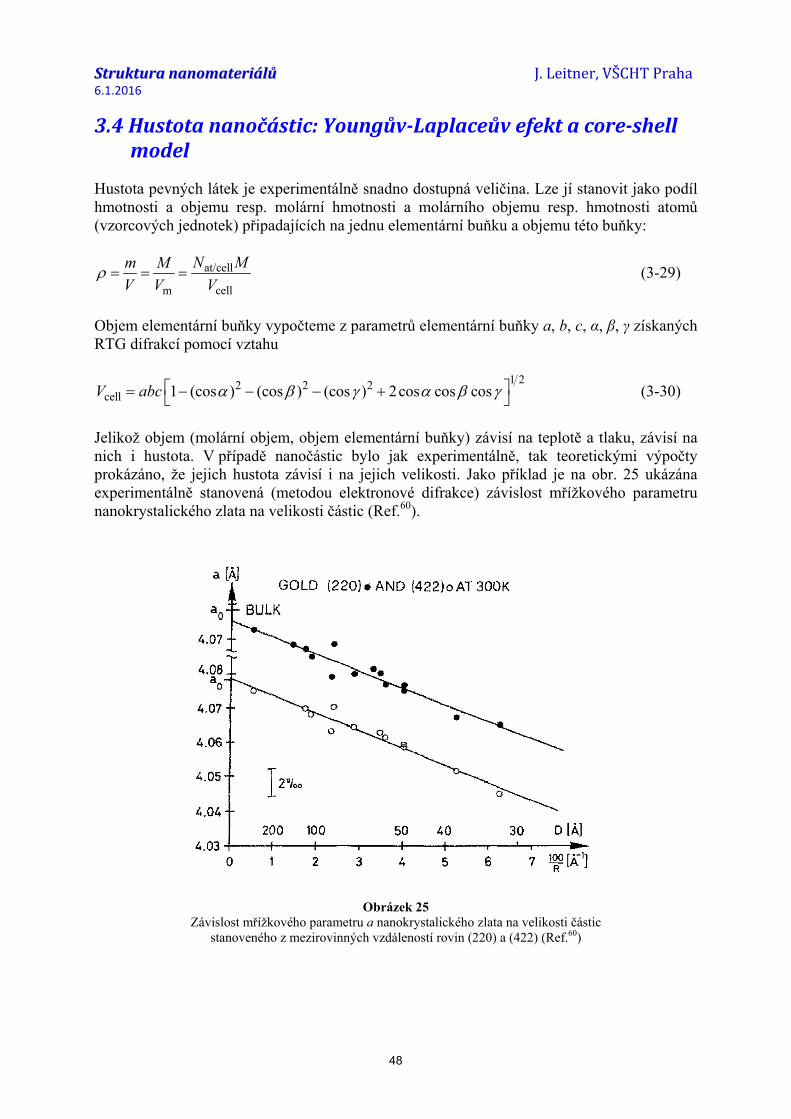
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
48
3.4 Hustota nanočástic: YoungůvLaplaceův efekt a coreshell model
Hustota pevných látek je experimentálně snadno dostupná veličina. Lze jí stanovit jako podíl hmotnosti a objemu resp. molární hmotnosti a molárního objemu resp. hmotnosti atomů (vzorcových jednotek) připadajících na jednu elementární buňku a objemu této buňky:
at/cell
m cell
N Mm MV V V
ρ = = = (3-29)
Objem elementární buňky vypočteme z parametrů elementární buňky a, b, c, α, β, γ získaných RTG difrakcí pomocí vztahu
1 22 2 2cell 1 (cos ) (cos ) (cos ) 2cos cos cosV abc α β γ α β γ⎡ ⎤= − − − +⎣ ⎦ (3-30)
Jelikož objem (molární objem, objem elementární buňky) závisí na teplotě a tlaku, závisí na nich i hustota. V případě nanočástic bylo jak experimentálně, tak teoretickými výpočty prokázáno, že jejich hustota závisí i na jejich velikosti. Jako příklad je na obr. 25 ukázána experimentálně stanovená (metodou elektronové difrakce) závislost mřížkového parametru nanokrystalického zlata na velikosti částic (Ref.60).
Obrázek 25 Závislost mřížkového parametru a nanokrystalického zlata na velikosti částic
stanoveného z mezirovinných vzdáleností rovin (220) a (422) (Ref.60)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
49
Na obr. 26 je ukázána závislost relativní kontrakce mřížkového parametru Al(fcc) se zmenšujícím se rozměrem nanočástice získaná semiempirickým výpočtem – Embedded atom method, viz část 2.2 (Ref.61).
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5-7
-6
-5
-4
-3
-2
-1
0
∆a
/abu
lk (%
)
Nanoparticle radius r (nm)
0
2000
4000
6000
8000
10000
12000
Num
ber o
f ato
ms
Obrázek 26 Závislost relativní změny mřížkového parametr a nanokrystalického Al na velikosti částic (Ref.61)
Pro interpretaci závislosti mřížkových parametrů (objemu, hustoty) volných nanočástic na jejich velikosti byla navržena řada teoretických modelů. Nejjednodušší model (Ref.62-65) chápe nanočástici jako elastické kontinuum jehož objem se vlivem zvýšeného vnitřního tlaku (Youngův-Lapalceův efekt) zmenšuje, přičemž tlaková závislost objemu je vyjádřena z definice koeficientu izotermní stlačitelnosti κT
1 1T
T
V VV p V p
κ⎛ ⎞∂ ∆
= − ≅ −⎜ ⎟∂ ∆⎝ ⎠ (3-31)
Objem V vystupující v rovnici (3-31) je obvykle molární objem dané látky, avšak může být nahrazen objemem elementární buňky. Pro krychlové soustavy platí V = a3 a dV/V = 3da/a (a je mřížkový parametr), a tak spojením vztahu (3-31) a Youngovy-Laplaceovy rovnice (2-25) získáme vztah (viz část 2.3, rovnice (2-24))
3 2 2T T
V a f fpV a r B r
κ κ∆ ∆= = − ∆ = − = − (3-32)
Z tvaru rovnice (3-32) je zřejmé, že kontrakce mřížkových parametrů resp. objemu (předpokládáme kladnou hodnotu povrchového napětí f) je lineární funkcí 1/r. Na obr. 27 je ukázáno porovnání modelové predikce dle rovnice (3-32) a experimentálních dat pro nanočástice Bi (Ref.66).
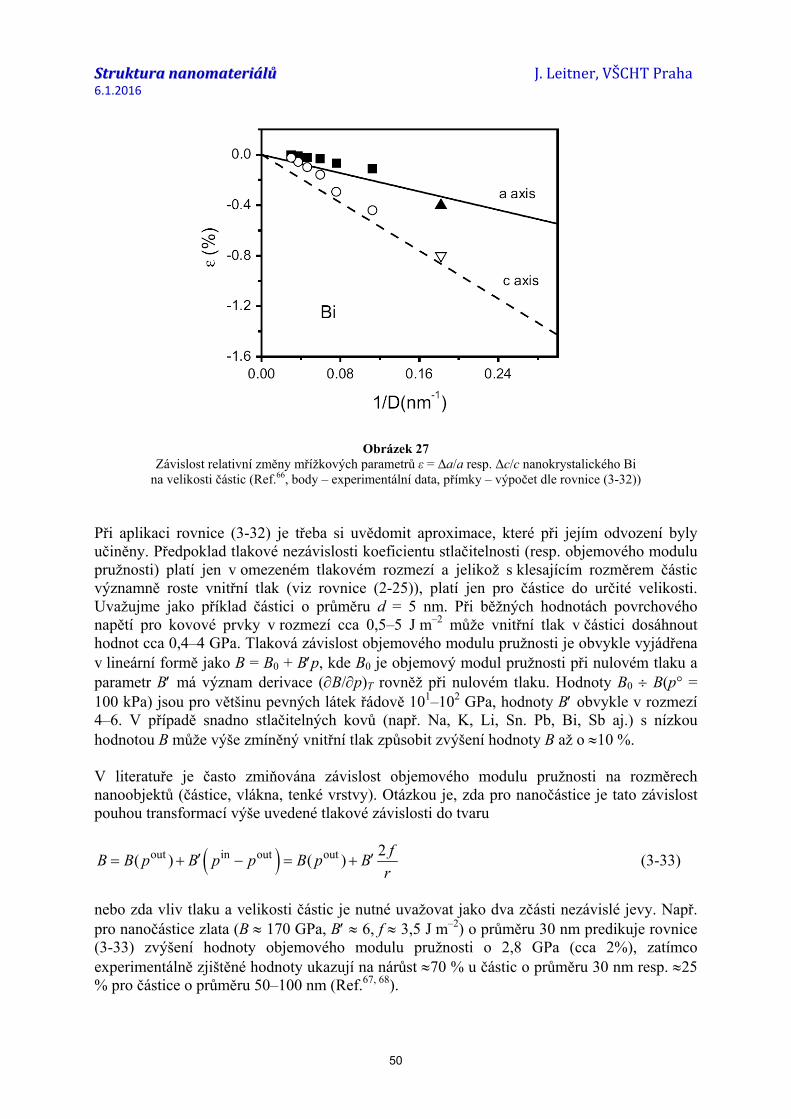
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
50
Obrázek 27 Závislost relativní změny mřížkových parametrů ε = ∆a/a resp. ∆c/c nanokrystalického Bi
na velikosti částic (Ref.66, body – experimentální data, přímky – výpočet dle rovnice (3-32)) Při aplikaci rovnice (3-32) je třeba si uvědomit aproximace, které při jejím odvození byly učiněny. Předpoklad tlakové nezávislosti koeficientu stlačitelnosti (resp. objemového modulu pružnosti) platí jen v omezeném tlakovém rozmezí a jelikož s klesajícím rozměrem částic významně roste vnitřní tlak (viz rovnice (2-25)), platí jen pro částice do určité velikosti. Uvažujme jako příklad částici o průměru d = 5 nm. Při běžných hodnotách povrchového napětí pro kovové prvky v rozmezí cca 0,5–5 J m–2 může vnitřní tlak v částici dosáhnout hodnot cca 0,4–4 GPa. Tlaková závislost objemového modulu pružnosti je obvykle vyjádřena v lineární formě jako B = B0 + B′p, kde B0 je objemový modul pružnosti při nulovém tlaku a parametr B′ má význam derivace (∂B/∂p)T rovněž při nulovém tlaku. Hodnoty B0 ÷ B(p° = 100 kPa) jsou pro většinu pevných látek řádově 101–102 GPa, hodnoty B′ obvykle v rozmezí 4–6. V případě snadno stlačitelných kovů (např. Na, K, Li, Sn. Pb, Bi, Sb aj.) s nízkou hodnotou B může výše zmíněný vnitřní tlak způsobit zvýšení hodnoty B až o ≈10 %. V literatuře je často zmiňována závislost objemového modulu pružnosti na rozměrech nanoobjektů (částice, vlákna, tenké vrstvy). Otázkou je, zda pro nanočástice je tato závislost pouhou transformací výše uvedené tlakové závislosti do tvaru
( )out in out out 2( ) ( ) fB B p B p p B p Br
′ ′= + − = + (3-33)
nebo zda vliv tlaku a velikosti částic je nutné uvažovat jako dva zčásti nezávislé jevy. Např. pro nanočástice zlata (B ≈ 170 GPa, B′ ≈ 6, f ≈ 3,5 J m–2) o průměru 30 nm predikuje rovnice (3-33) zvýšení hodnoty objemového modulu pružnosti o 2,8 GPa (cca 2%), zatímco experimentálně zjištěné hodnoty ukazují na nárůst ≈70 % u částic o průměru 30 nm resp. ≈25 % pro částice o průměru 50–100 nm (Ref.67, 68).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
51
Růst hodnoty objemového modulu pružnosti s klesajícím rozměrem ideálně krystalické nanočástice předpovídá model navržený Ouyangem et al.69 V rámci tohoto modelu je uvažována core-shell struktura částice (Ref.70, 71). V povrchové vrstvě nanočástice dochází vlivem sníženého počtu sousedních atomů (koordinačního čísla) k zesílení a zkrácení vazeb. Tím dochází k zpevnění této oblasti, které se projeví zvýšením průměrné hodnoty B celé částice. Modelová predikce závislosti relativní změny objemového modulu pružnosti Ag na velikosti nanočástic je ukázána na obr. 28.
Obrázek 28 Závislost relativní změny objemového modulu pružnosti Ag na velikosti částic (Ref.69)
- experimentální hodnota B0 = 121 GPa pro částice o průměru 10 nm (Ref.67) U některých materiálů (např. γ-Al2O3 (viz obr. 29, Ref.72), SnO2 (Ref.73), PbS (Ref.74) nebo ZnS (Ref.75)), dochází naopak ke snižování hodnoty B s klesající velikostí částic.
Obrázek 29 Závislost objemového modulu pružnosti γ-Al2O3 na velikosti částic (Ref.72)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
52
To může být vysvětleno buď prodloužením meziatomových vzdáleností v povrchové vrstvě částice (záporná hodnota povrchového napětí f), nebo skutečností, že za zvýšených tlaků (řádově 101 GPa) dochází k podstatnému zhutnění materiálu a jedná se tedy spíše o kompaktní nanokrystalický materiál než o volné shluky individuálních nanočástic. Nanokrystalický materiál je tvořen náhodně orientovanými monokrystalickými zrny (původními nanočásticemi) a hranicemi těchto krystalitů s nižší hustotou a vyšší stlačitelností (Ref.76-78). Průměrná hodnota objemového modulu pružnosti tak může být u nanokrystalických materiálů nižší než u materiálů objemových, přičemž s klesající velikostí krystalitů roste objemový podíl hranice zrn a hodnota B tak se zmenšujícími se rozměry částic klesá. U řady jiných materiálů nebyl vliv velikosti částic na hodnotu objemového modulu pružnosti prokázán. Údaje o vlivu velikosti částic na hodnoty objemového modulu pružnosti uváděné v literatuře nejsou v mnoha případech ve shodě. Tak např. pro Ni bylo pozorováno jak zpevnění, tedy zvýšení hodnot B, tak naopak snížení hodnot B v závislosti na rozměru částic (Ref.79-84, viz tabulka 5). Pro objemový materiál jsou udávány hodnoty B0 v rozmezí 180-186 GPa.
Tabulka 5 Experimentálně stanovené hodnoty objemového modulu pružnosti při nulovém tlaku B0
a derivace B′ = (∂B/∂p)p = 0 pro Ni
Materiál pmax (GPa) Způsob komprese B0 (GPa) B′ (Ref.)
Nanočástice 20 nm
55 quasi-hydrostatic non-hydrostatic
217,5 ± 14 185,4 ± 10
4 (fixní) 4 (fixní)
(79)
Nanočástice 20 nm
61,5 non-hydrostatic non-hydrostatic
229 ± 4
228 ± 15
4 (fixní)
4,02 ± 0,51 (80)
Nanočástice 8-10 nm
7,4 non-hydrostatic
161 ± 3
4 (fixní) (81, 82)
Nanočástice 150-250 nm
44,7 quasi-hydrostatic
232 ± 11
2,5 ± 0,5 (83)
Nanočástice 62 nm
56 non-hydrostatic
216
4 (fixní) (84)
Hlavní důvody značného rozptylu literárních údajů spočívají v metodice měření a zpracování získaných dat. Obvyklý experimentální postup určení hodnoty objemového modulu pružnosti spočívá ve stanovení závislosti molárního objemu resp. objemu elementární buňky na tlaku a analýze této závislosti na základě vhodné stavové rovnice. Příslušný objem je vypočten z parametrů elementární buňky získaných z vysokotlakých XRD měření, přičemž výsledek je ovlivněn jednak způsobem dosažení vysokých tlaků (kvazihydrostatická resp. anizotropní komprese), jednak použitým standardem pro určení přesné hodnoty tlaku. Pro analýzu dat je obvykle užívána Birchova-Murnaghanova stavová rovnice85, jejímiž parametry jsou hodnoty B0 a B′. V některých případech však byla užita pevná hodnota B′ = 4 a optimalizován pouze parametr B0. Stejný koncept (nanočástice jako elastické kontinuum), ale jiný přístup k vysvětlení objemové kontrakce (kontrakce parametrů elementární buňky) užili Qi a spol. (Ref.86-88). V rámci jejich

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
53
modelu je uvažován vznik N kulatých nanočástic o průměru d1 z relativně velké (bulkové) kulaté částice o průměru d0. Platí (přibližně):
30 0
1 1
V dNV d
⎛ ⎞= = ⎜ ⎟
⎝ ⎠ (3-34)
Tento proces je doprovázen: 1. Zvětšením celkového povrchu souboru nanočástic a zvýšením povrchové energie o ∆Esurf. 2. Elastickou deformací každé jednotlivé nanočástice v důsledku povrchového napětí, čímž se zmenší její průměr na hodnotu d(1 – ε). Pro celkovou změnu energie při tomto procesu platí:
( )
surf elast22
01surf surf
321
elast
4 12 2
42
E E E
ddE N
dE N G
γ π ε
π ε
∆ = ∆ + ∆
⎧ ⎫⎪ ⎪⎡ ⎤⎡ ⎤∆ = − −⎨ ⎬⎢ ⎥⎢ ⎥⎣ ⎦ ⎣ ⎦⎪ ⎪⎩ ⎭
⎛ ⎞∆ = ⎜ ⎟⎝ ⎠
(3-35)
Ve vztahu pro energii elastické deformace je symbolem G označen modul pružnosti ve smyku/střihu (shear modulus). Ze vztahu (3-35) je zřejmé, že kladná hodnota ε snižuje hodnotu povrchové energie a naopak zvyšuje hodnotu elastické energie. Minimalizací celkové energie vzhledem k deformaci ε (dE/dε = 0) získáme rovnovážnou hodnotu εeq
( )eqsurf
11 G d
επ γ
=+
(3-36)
a odtud relativní změnu (kontrakci) mřížkového parametru ve tvaru
11
aa K d∆
=+
(3-37)
kde K = πG/γsurf. Vztah (3-36) byl později (Ref.87, 88) rozšířen zavedením tzv. tvarového faktoru na nesférické částice. Další modifikaci a rozšíření modelu nanočástice jako elastického kontinua komprimovaného v důsledku povrchového napětí navrhli Huang a spol. (Ref.89). Modifikace spočívá ve vyjádření kontrakce vzhledem k rovnovážným (deformovaným) rozměrům částice a rozšíření v zahrnutí závislosti povrchového napětí (energie) na velikosti částice, což umožňuje použít model i pro velmi malé částice. Modelové rovnice jsou odvozeny pro kulatou nanočástici o původním (nedeformovaném) poloměru r0, který se vlivem povrchového napětí zmenší na hodnotu r. Pro závislost povrchové energie na poloměru částice je užita Tolmanova rovnice (2-29). Pro relativní kontrakci mřížkového parametru vzhledem k rovnovážné (deformované) hodnotě a tak odvodili rovnici

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
54
( )2
2 3
2 4 4 3
a f Ba r r r f Bδ δ δ
∆= −
+ + + + − (3-38)
ve které f je povrchové napětí na rovinném povrchu a δ je parametr (Tolmanova délka). Výsledky řady experimentálních prací (např. Ref.90-98) i teoretických výpočtů (např. Ref.99-102) ukazují, že vzdálenosti (délky vazeb) mezi atomy nejsou v celém objemu nanočástice stejné. V povrchové a několika málo (1–3) podpovrchových vrstvách s nižší koordinací atomů jsou vzdálenosti kratší než meziatomové vzdálenosti v jádru nanočástice, které se příliš neliší od meziatomových vzdáleností v bulku (tzv. core-shell modelc). Takový případ je pro nanočástice Au ukázán na obr. 30 (Ref.99)
Obrázek 30 Závislost průměrné meziatomové vzdálenosti dAu–Au v kulaté nanočástici tvořené 1830 atomy Au
na vzdálenosti od středu částice (Ref.99) + - experimentální body (Ref94) získané elektronovou difrakcí
- semiempirický výpočet (MD, potenciál Cleri-Rosato) Strukturní model core-shell byl podrobně rozpracován Paloszem a spol. (Ref.94-98) na základě RTG difrakce nanočástic různých pevných látek (zejména C(dia) a SiC). V rámci tohoto modelu byl formulován koncept „zdánlivého mřížkového parametru“ (apparent lattice parameter – alp), kde mřížkový parametr je stanoven v závislosti na hodnotě difrakčního vektoru Q = 2π/dhkl (viz obr. 31). Zatímco RTG difrakční reflexe pro velké hodnoty Q poskytují informace o struktuře jádra, reflexe pro malé hodnoty Q jsou citlivé na strukturu povrchové vrstvy nanočástice. c Termín core-shell se užívá i pro vícesložkové nanočástice (slitiny, kov/oxid aj.), jejichž jádro se svým složením liší od povrchové vrstvy.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
55
Obrázek 31 Závislost „zdánlivého mřížkového parametru“ (alp) na hodnotě difrakčního vektoru Q pro přibližně sférické
nanočástice C(dia) o průměru 3, 4 a 8 nm (Ref.94). RTG difrakční reflexe pro velké hodnoty Q poskytují informace o struktuře jádra, reflexe pro malé hodnoty Q jsou citlivé na strukturu povrchové vrstvy nanočástice.
Povrchové vrstvy nanočástice v tomto případě vykazují mírnou expanzi struktury. Geometrie core-shell je základním principem modelu BOLS (Bond-order-length-strength) navrženého a podrobně rozpracovaného profesorem Sunem a spol. (Ref.71, 103-105). Uvažujme kulatou nanočástici o poloměru R (viz obr. 32) tvořenou N atomy o průměru d.
1R r=
1 1d c d=
2 2d c d=
1R r=
1 1d c d=
1R r=
1 1d c d=
2 2d c d=
Obrázek 32 Geometrie kulaté nanočástice v rámci modelu BOLS (Ref.71)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
56
V případě nejtěsnějšího uspořádání atomů odpovídá hodnota d také délce vazby mezi nimi. Délka vazby je významně závislá na koordinačním čísle z, tedy počtu vazeb, které daný atom se svými sousedy vytváří. Pro tuto závislost navrhli Sun a spol. vztah
( )0
21 exp 12 8
ii
i i
dcd z z
= =⎡ ⎤+ −⎣ ⎦
(3-39)
ve kterém je d0 bulková hodnota délky vazby (pro nejtěsnější uspořádání atomů odpovídá koordinačnímu číslu z = 12) a di redukovaná délka vazby mezi atomy s koordinačním číslem zi (zi < 12). Pro popis kontrakce nanočástic je uvažována redukce vazeb pouze mezi první a druhou (d1) a druhou a třetí (d2) povrchovou/podpovrchovou vrstvou atomů, přičemž pro příslušná koordinační čísla z1 a z2 platí
( )1
2 1
4 1 0,752
z Kz z= −
= + (3-40)
Parametr K = R/d0 je bezrozměrným vyjádřením velikosti částice. Pro rovinné rozhraní K → ∞ a z1 = 4, pro nejmenší nanočástici R = 1,5d0, K = 1,5 a z1 = 2. Průměrnou relativní délkovou kontrakci (průměrná délka vazby mezi atomy, průměrné mřížkové parametry) pak vypočteme jako
( ) ( )av 0 1 21 2
0 0
1 1d d N Nd c cd d N N
−∆= = − + − (3-41)
Podíly atomů v první (N1/N) a druhé (N2/N) vrstvě atomů částice určíme ze vztahů
( )3 331 1 11 1 1
31
1 1r r dN V c
N V r K− − ⎛ ⎞= = = − −⎜ ⎟
⎝ ⎠ (3-42a)
( )3 3 33
2 2 22 2 1 1 23
1
1 1r r dN V c c c
N V r K K K− − ⎛ ⎞ ⎛ ⎞= = = − − − −⎜ ⎟ ⎜ ⎟
⎝ ⎠ ⎝ ⎠ (3-42b)
kde r1 = R a r2 = r1 – d1. BOLS model tak predikuje průměrnou kontrakci mřížkových parametrů nanočástice resp. průměrnou meziatomovou vzdálenost bez využití Youngovy-Laplaceovy rovnice a v modelových vztazích nevystupují žádné veličiny charakterizující povrch (povrchová energie, povrchové napětí) ani objem (objemová stlačitelnost, objemový modul pružnosti) daného materiálu. Výsledky modelové predikce pro řadu kovových prvků a porovnání s výsledky experimentálními jsou ukázány na obr. 33 (Ref.71).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
57
Obrázek 33 Kontrakce mřížkových parametrů nanočástic a nanovrstev kovů v fcc struktuře v závislosti na parametru
K = R/dat (nanočástice poloměru R) resp. K = H/dat (nanovrstva tloušťky H) predikovaná na základě modelu BOLS (Ref.71) a porovnání s údaji experimentálními (při výpočtu kontrakce v nanovrstvách byly užity rovnice
(3-42) v modifikované podobě) Všechny výše uvedené modelové vztahy predikují kontrakci mřížkových parametrů (zvýšení hustoty) nanočástic vzhledem k bulkovým hodnotám (ve vtahu (3-32) předpokládáme kladnou hodnotu povrchového napětí f). V některých případech však byla experimentálně pozorována naopak expanze mřížkových parametrů, a to jak u kovových, tak nekovových (např. oxidy, karbidy aj.) nanočástic. V případě kovových nanočástic je při interpretaci výsledků zcela zásadní rozlišovat mezi volnými nanočásticemi v plynné atmosféře s původním („čistým“) povrchem, s adsorbáty či s oxidickou vrstvou, nanočásticemi stabilizovanými v různých suspenzích a nanočásticemi fixovanými v různých matricích (nejčastěji amorfní oxidy nebo organické polymerní materiály). Opakovaně bylo prokázáno (Ref.30, 106-109), že i velmi tenká vrstva amorfního nebo krystalického oxidu na povrchu původní částice vytváří na rozhraní kov-(core)/(shell)-oxid kompresivní mezifázový stress (záporná hodnota f), což vede (viz rovnice (3-32)) k expanzi průměrného mřížkového parametru jádra částice. Jako příklad je na obr. 34 ukázána závislost relativní změny mřížkového parametru Ni na velikosti nanočástic, ze které byla určena hodnota mezifázového napětí na rozhraní Ni/NiO: f = –17,5 N m–1 (Ref.106).
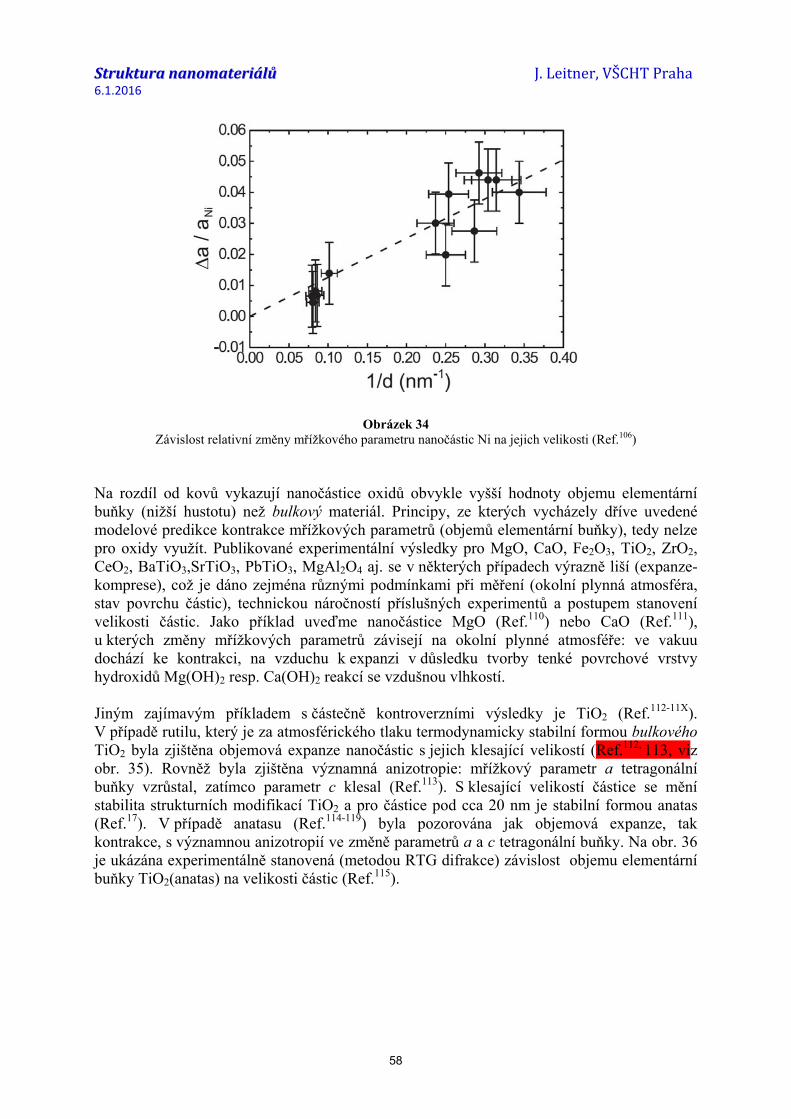
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
58
Obrázek 34 Závislost relativní změny mřížkového parametru nanočástic Ni na jejich velikosti (Ref.106)
Na rozdíl od kovů vykazují nanočástice oxidů obvykle vyšší hodnoty objemu elementární buňky (nižší hustotu) než bulkový materiál. Principy, ze kterých vycházely dříve uvedené modelové predikce kontrakce mřížkových parametrů (objemů elementární buňky), tedy nelze pro oxidy využít. Publikované experimentální výsledky pro MgO, CaO, Fe2O3, TiO2, ZrO2, CeO2, BaTiO3,SrTiO3, PbTiO3, MgAl2O4 aj. se v některých případech výrazně liší (expanze-komprese), což je dáno zejména různými podmínkami při měření (okolní plynná atmosféra, stav povrchu částic), technickou náročností příslušných experimentů a postupem stanovení velikosti částic. Jako příklad uveďme nanočástice MgO (Ref.110) nebo CaO (Ref.111), u kterých změny mřížkových parametrů závisejí na okolní plynné atmosféře: ve vakuu dochází ke kontrakci, na vzduchu k expanzi v důsledku tvorby tenké povrchové vrstvy hydroxidů Mg(OH)2 resp. Ca(OH)2 reakcí se vzdušnou vlhkostí. Jiným zajímavým příkladem s částečně kontroverzními výsledky je TiO2 (Ref.112-11X). V případě rutilu, který je za atmosférického tlaku termodynamicky stabilní formou bulkového TiO2 byla zjištěna objemová expanze nanočástic s jejich klesající velikostí (Ref.112, 113, viz obr. 35). Rovněž byla zjištěna významná anizotropie: mřížkový parametr a tetragonální buňky vzrůstal, zatímco parametr c klesal (Ref.113). S klesající velikostí částice se mění stabilita strukturních modifikací TiO2 a pro částice pod cca 20 nm je stabilní formou anatas (Ref.17). V případě anatasu (Ref.114-119) byla pozorována jak objemová expanze, tak kontrakce, s významnou anizotropií ve změně parametrů a a c tetragonální buňky. Na obr. 36 je ukázána experimentálně stanovená (metodou RTG difrakce) závislost objemu elementární buňky TiO2(anatas) na velikosti částic (Ref.115).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
59
Obrázek 35 Závislost objemu elementární buňky TiO2(rutil) na velikosti částic (Ref.113)
- experimentální data (Ref.112), - experimentální data (Ref.113)
Obrázek 35 Závislost objemu elementární buňky TiO2(anatas) na velikosti částic (Ref.115)
Ačkoliv bylo navrženo několik modelů interpretujících experimentálně pozorované změny mřížkových parametrů resp. objemu elementární buňky nanočástic různých oxidů, jejich použití je značně omezené a rovněž problematické v případech, kdy experimentální data jsou protichůdná. Model navržený Andersonem a Scholzem (Ref.120) pro iontové sloučeniny s halitovou strukturou (MgO, CaO aj.) vychází ze závislosti mřížkové energie na velikosti částice a predikuje kontrakci parametrů elementární buňky. Rovněž v rámci modelu navrženého Perebeinosem a spol. (Ref.121) se předpokládá, že v nanočásticích oxidů dochází

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
60
ke změně Madelungovy energie (mřížkové energie iontového krystalu), přičemž tato změna (pokles) vede k expanzi mřížkových parametrů. Model byl užit pro interpretaci experimentálních dat pro CeO2 a predikci BaTiO3. Jiný přístup k vysvětlení dilatace mřížkových parametrů v nanočásticích oxidů (MgO, Fe2O3) zvolil Fukuhara (Ref.122). Na základě Thomasovy-Fermiho aproximace vyjádřil elektronovou hustotu a Coulombický potenciál v nanočásticích jako funkci jejich velikosti a z této závislosti odvodil vztah pro dilataci parametrů elementární buňky. V řadě případů je však interpretace experimentálních dat specifická pro daný oxid. Např. expanze mřížkových parametrů CeO2 u malých nanočástic je vysvětlována povrchovou redukcí iontů Ce4+ → Ce3+ a současnou tvorbou kyslíkových vakancí. Expanze mřížkových parametrů TiO2(rutil) a podobně i SrTiO3 je vysvětlována silnými dipol-dipol interakcemi na povrchu částic vzniklých v důsledku deformace TiO6 oktaedrů. 3.5 Závislost hustoty nanočástic na teplotě a tlaku Hustota pevných látek závisí na teplotě a tlaku. Pro vyjádření této závislosti použijeme rovnici (3-29) ve které vyjádříme molární objem Vm jako funkci T a p ve tvaru (Ref.123)d
( )mln , ( , )d ( , )dV Td V T p T p T T p pα κ= − (3-43) kde αV(T, p) je koeficient izobarické objemové roztažnosti a κT(T, p) = 1/B(T, p) koeficient izotermní stlačitelnosti (B je modul objemové pružnosti). Uvažujme nejprve teplotní závislost molárního objemu při stálém tlaku p0 (p0 je obvykle tlak atmosférický). Z rovnice (3-43) plyne
( ) ( )ref
Tm 0 m ref 0 0, , exp ( , )dVT
V T p V T p T p Tα= ∫ (3-44)
Integrál na pravé straně rovnice (3-44) můžeme vyhodnotit ze známé závislosti αV = f(T, p). Na obr. 36 (Ref.124) jsou ukázány vypočtené závislosti koeficientu αV pro Pt na teplotě a tlaku. Obdobný průběh těchto závislostí vykazuje většina pevných látek – výjimkou jsou látky s negativním koeficientem objemové roztažnosti, např. Cu2O (0-100 K), Ag2O (0-300 K), ZrW2O8 (0-300 K) nebo PbTiO3 (300-750 K) (Ref.125). Závislost αV = f(T, p) lze vyjádřit obecným vtahem
( , ) ( , )d ( , ) d dV VV
p T
T p T pT p T pT p
α αα⎛ ⎞∂ ∂⎛ ⎞= + ⎜ ⎟⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
(3-45)
ve kterém obě parciální derivace závisí na teplotě a tlaku. Pro závislost αV na T a p byla v literatuře navržena řada vztahů.
d Rovnice (3-43) je jedním z možných vyjádření stavového (p-V-T) chování pevných látek. Existuje řada dalších stavových rovnic (EOS), jejichž výsledky se liší zejména v oblast velmi vysokých tlaků (O.L. Anderson: Equations of State of Solids for Geophysics and Ceramic Science, Oxford University Press, Oxford – New York, 1995).
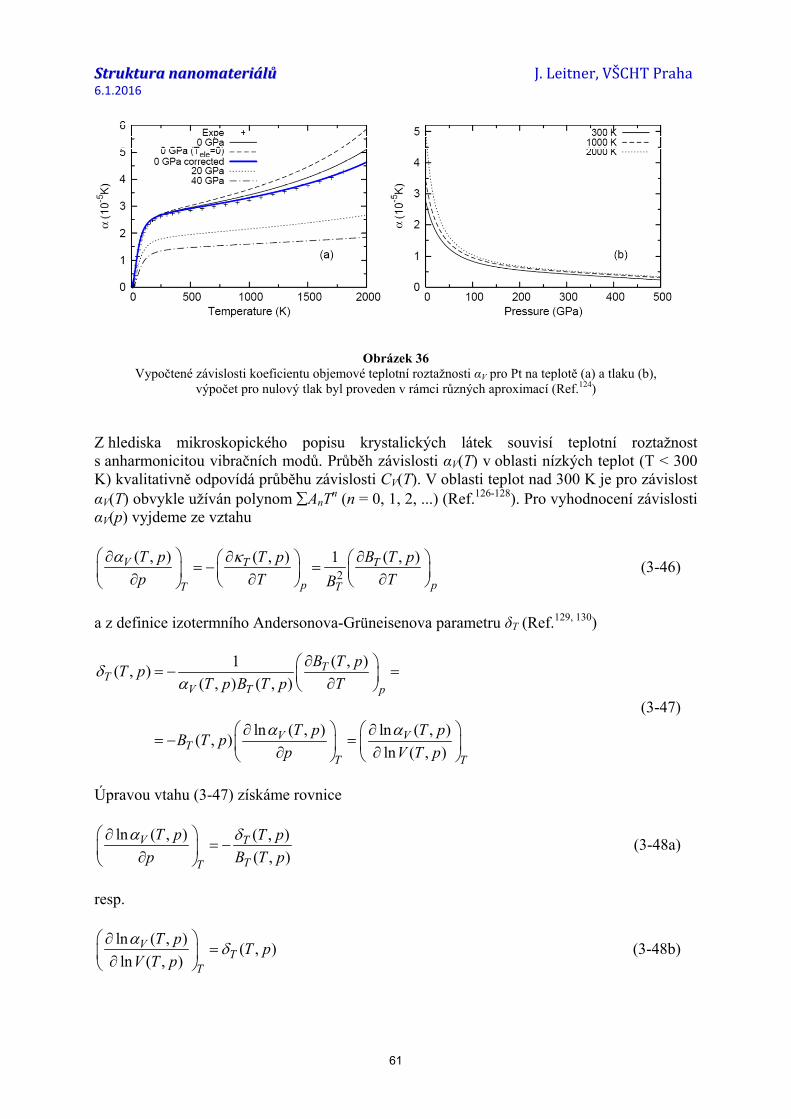
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
61
Obrázek 36 Vypočtené závislosti koeficientu objemové teplotní roztažnosti αV pro Pt na teplotě (a) a tlaku (b),
výpočet pro nulový tlak byl proveden v rámci různých aproximací (Ref.124) Z hlediska mikroskopického popisu krystalických látek souvisí teplotní roztažnost s anharmonicitou vibračních modů. Průběh závislosti αV(T) v oblasti nízkých teplot (T < 300 K) kvalitativně odpovídá průběhu závislosti CV(T). V oblasti teplot nad 300 K je pro závislost αV(T) obvykle užíván polynom ∑AnTn (n = 0, 1, 2, ...) (Ref.126-128). Pro vyhodnocení závislosti αV(p) vyjdeme ze vztahu
2( , ) ( , ) ( , )1V T T
p pTT
T p T p B T pp T TB
α κ⎛ ⎞∂ ∂ ∂⎛ ⎞ ⎛ ⎞= − =⎜ ⎟ ⎜ ⎟ ⎜ ⎟∂ ∂ ∂⎝ ⎠ ⎝ ⎠⎝ ⎠ (3-46)
a z definice izotermního Andersonova-Grüneisenova parametru δT (Ref.129, 130)
( , )1( , )( , ) ( , )
ln ( , ) ln ( , )( , )ln ( , )
TT
V T p
V VT
T T
B T pT pT p B T p T
T p T pB T pp V T p
δα
α α
∂⎛ ⎞= − =⎜ ⎟∂⎝ ⎠
⎛ ⎞ ⎛ ⎞∂ ∂= − =⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
(3-47)
Úpravou vtahu (3-47) získáme rovnice
ln ( , ) ( , )( , )
V T
TT
T p T pp B T p
α δ⎛ ⎞∂= −⎜ ⎟∂⎝ ⎠
(3-48a)
resp.
ln ( , ) ( , )ln ( , )
VT
T
T p T pV T pα δ
⎛ ⎞∂=⎜ ⎟∂⎝ ⎠
(3-48b)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
62
a jejich integrací
00
( , )( , ) ( , ) exp( , )
p TV V p T
T pT p T p dpB T pδα α
⎡ ⎤= −⎢ ⎥
⎣ ⎦∫ (3-49a)
resp.
00( , ) ( , ) exp ( , )d ln ( , )
VV V TV
T p T p T p V T pα α δ= ∫ (3-49b)
Zanedbáme-li závislost δT a BT na tlaku, lze tlakovou závislost αV(p) vyjádřit jako
0 0( , ) ( , ) exp ( )TV V
TT p T p p p
Bδα α
⎡ ⎤= − −⎢ ⎥
⎣ ⎦ (3-50a)
resp.
00
( , )( , ) ( , )( , )
T
V VV T pT p T pV T p
δ
α α⎛ ⎞
= ⎜ ⎟⎝ ⎠
(3-50b)
V případě nanočástic a jiných nanoobjektů byla pozorována závislost hodnoty koeficientu objemové/délkové teplotní roztažnosti na velikosti (Ref.131-137). Na obr. 37 je ukázána závislost mřížkového parametru Ag v bulku a ve formě nanočástic o průměru cca 5 nm na teplotě (Ref.133). Příslušný koeficient délkové teplotní roztažnosti (αL = (1/a)(∂a/∂T)p) je pro nanočástice menší než pro objemový materiál.
Obrázek 37 Závislost mřížkového parametru Ag v bulku a ve formě nanočástic o průměru cca 5 nm na teplotě (Ref.133)
(αV(bulk) = 6,6×10–5 K–1, αV(nano, air) = 5,1×10–5 K–1, αV(nano, vacuum) = 1,8×10–5 K–1)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
63
V případě selenu bylo pozorováno naopak zvýšení hodnoty koeficientu tepelné roztažnosti se zmenšující se velikostí nanokrystalů (Ref.130). Rovněž zvýšení hodnoty αL bylo na základě MD simulace predikováno pro nanočástice Fe (Ref.138). Změnu hodnoty αV v závislosti na velikosti nanočástice lze v prvním přiblížení vysvětlit jako důsledek Youngova-Laplaceova efektu. V částici o poloměru r je vyšší tlak než v jejím okolí o ∆p = 2f/r (viz rovnice (2-25)) a jelikož podle rovnice (3-50a) s rostoucím tlakem klesá hodnota αV (viz obr. 36) lze toto interpretovat jako důsledek zmenšujícího se rozměru částice. Platí:
0
0 0
( ) ( ) ( ) 2 ( / )exp 1( ) ( )
V V V T T
V V
p p p f Bp p r
α α α δα α− ∆ ⎡ ⎤= = − −⎢ ⎥⎣ ⎦
(3-51)
Pro Ag je B0 = 165,2 GPa, δT = 5,23 (Ref.127) a f = 1,8 J m–2 (viz tabulka 1) a pro nanočástice o průměru 2r = 5 nm rovnice (3-51) predikuje relativní snížení hodnoty αV o cca 5 %. Na základě analýzy dat z obr. 37 (Ref.133) lze odhadnout výrazně vyšší snížení hodnoty αV o cca 23 % resp. 73 % pro nanočástice na vzduchu resp. ve vakuu. Model, který predikuje vzrůst hodnoty αV s klesající velikostí částice navrhli Jiang a spol. (Ref.139-141). Pro poměr hodnot koeficientu teplotní roztažnosti částice o poloměru r αV(r) a objemového materiálu αV(∞) platí vztah
( )
2 FvibD
D at
( ) 2 / 3( ) exp( ) ( ) / 3 1
V
V
r S Rr r d
αα
⎡ ⎤⎛ ⎞ ∆Θ ∞= = ⎢ ⎥⎜ ⎟∞ Θ −⎢ ⎥⎝ ⎠ ⎣ ⎦
(3-52)
kde ΘD je charakteristická Debyeova teplota a ∆SF
vib je vibrační příspěvek molární entropie tání dané pevné látky. Na obr. 38 jsou ukázány vypočtené závislosti αV(r) pro nanočástice Se a Pb a porovnání s experimentálními daty (Ref.141).
Obrázek 38 Vypočtené závislosti αV(r) pro nanočástice Se a Pb a porovnání s experimentálními daty (Ref.141),
- exp. data pro Se, - exp. data pro Pb
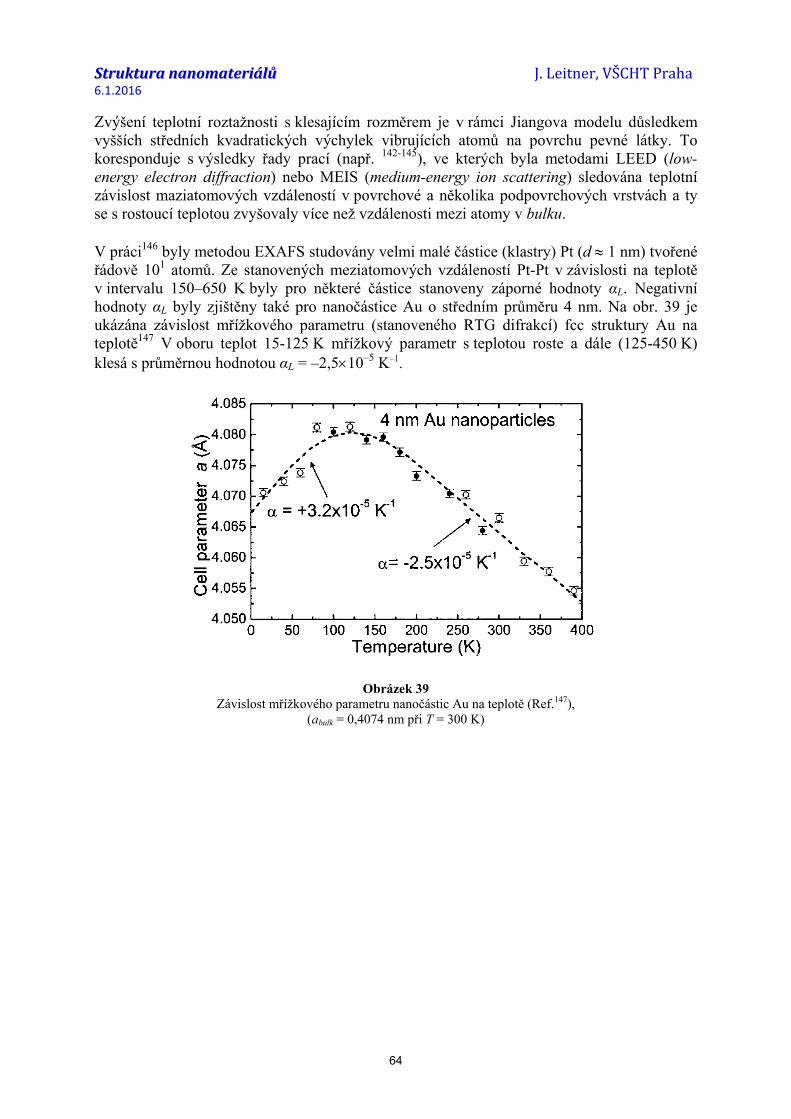
SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
64
Zvýšení teplotní roztažnosti s klesajícím rozměrem je v rámci Jiangova modelu důsledkem vyšších středních kvadratických výchylek vibrujících atomů na povrchu pevné látky. To koresponduje s výsledky řady prací (např. 142-145), ve kterých byla metodami LEED (low-energy electron diffraction) nebo MEIS (medium-energy ion scattering) sledována teplotní závislost maziatomových vzdáleností v povrchové a několika podpovrchových vrstvách a ty se s rostoucí teplotou zvyšovaly více než vzdálenosti mezi atomy v bulku. V práci146 byly metodou EXAFS studovány velmi malé částice (klastry) Pt (d ≈ 1 nm) tvořené řádově 101 atomů. Ze stanovených meziatomových vzdáleností Pt-Pt v závislosti na teplotě v intervalu 150–650 K byly pro některé částice stanoveny záporné hodnoty αL. Negativní hodnoty αL byly zjištěny také pro nanočástice Au o středním průměru 4 nm. Na obr. 39 je ukázána závislost mřížkového parametru (stanoveného RTG difrakcí) fcc struktury Au na teplotě147 V oboru teplot 15-125 K mřížkový parametr s teplotou roste a dále (125-450 K) klesá s průměrnou hodnotou αL = –2,5×10–5 K–1.
Obrázek 39 Závislost mřížkového parametru nanočástic Au na teplotě (Ref.147),
(abulk = 0,4074 nm při T = 300 K)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
65
Uvažujme nyní tlakovou závislost molárního objemu při stálé teplotě T0 (T0 je obvykle 298 K). Z rovnice (3-43) plyne
( ) ( )ref
pm 0 m 0 ref 0, , exp ( , )dTp
V T p V T p T p pκ⎡ ⎤= −⎢ ⎥⎣ ⎦∫ (3-53)
Integrál na pravé straně rovnice (3-53) můžeme vyhodnotit ze známé závislosti κT = 1/BT = f(T, p). Na obr. 40 (Ref.124) jsou ukázány vypočtené závislosti objemového modulu pružnosti K pro Pt na teplotě a tlaku. Pro vyjádření teplotní závislosti BT = f(T) je v oblasti teplot nad 300 K obvykle užíván polynom ∑AnTn (n = 0, 1, 2, ...) (Ref.148-150). O tlakové závislosti objemového modulu pružnosti a závislosti BT na velikosti částic bylo pojednáno již dříve (viz část 3.4).
Obrázek 40 Vypočtené závislosti objemových modulů pružnosti – izotermní BT (KT) a adiabatický BS (KS) pro Pt
na teplotě (a) a tlaku (b) (Ref.124)

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
66
Literatura: 1. L.D. Marks: Experimental studies of small particle structures, Rep. Prog. Phys. 57 (1994) 603-649. 2. (a) T.P. Martin, T. Bergmann, H. Göhlich, T. Lange: Shell structure of clusters, J. Phys. Chem. 95 (1991)
6421-6429. (b) T.P. Martin: Shells of atoms, Phys. Rep. 273 (1996) 199-241. (c) T.P. Martin: From atoms to solids, Solid State Ionics 131 (2000) 3-12.
3. Z.L. Wang: Transmission Electron Microscopy of Shape-Controlled Nanocrystals and Their Assemblies, J. Phys. Chem. B 104 (2000) 1153-1175.
4. M.J. Yacamán, J.A. Ascencio, H.B. Liu, J. Gardea-Torresdey: Structure shape and stability of nanometric sized particles, J. Vac. Sci. Technol. B 19 (2001) 1091-1103.
5. F. Baletto, R. Ferrando, Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic effects, Rev. Modern Phys. 77 (2005) 371-423.
6. C.R. Henry: Morphology of supported nanoparticles, Prog. Surf. Sci. 80 (2005) 92-116. 7. A.R. Tao, S. Habas, P. Yang: Shape control of colloidal metal nanoparticles, Small 4 (2008) 310-325. 8. Y. Xia, Y. Xiong, B. Lim, S.E. Skrabalak: Shape-controlled synthesis of metal nanocrystals: Simple
chemistry meets complex physics?, Angew. Chem. Int. Ed. 48 (2009) 60-103. 9. A.S. Barnard: Modelling of nanoparticles: approaches to morphology and evolution, Rep. Prog. Phys. 73
(2010) 086502 (52pp). 10. A. Mayoral, H. Barron, R.E. Salas, A.V. Duran, M.J. Yacamán: Nanoparticle stability from the nano to the
meso interval, Nanoscale 2 (2010) 335–342. 11. C. Herring: Some Theorems on the Free Energies of Crystal Surfaces, Phys. Rev. 82 (1951) 87–93. 12. (a) L.D. Marks: Modified Wulff construction for twinned particles, J. Cryst. Growth 61 (1983) 556-566.
(b) L.D. Marks: Particle size effects on Wulff construction, Surf. Sci. 150 (1985) 358-366. 13. P. Müller, C. Mottet: Equilibrium nanoshapes: From thermodynamics to atomistic simulations, J. Comput.
Theor. Nanosci. 4 (2007) 316-325. 14. A.S. Barnard, P. Zapol: A model for the phase stability of arbitrary nanoparticles as a function of size and
shape, J. Chem. Phys. 121 (2004) 4276-4283. 15. A.S. Barnard, L.A. Curtiss: Computational nano-morphology: Modeling shape as well as size, Rev. Adv.
Mater. Sci. 10 (2005) 105-109. 16. A.S. Barnard, X.M. Lin, L.A. Curtiss: Equilibrium morphology of face-centered cubic gold nanoparticles >
3 nm and the shape changes induced by temperature, J. Phys. Chem. B 109 (2005) 24465-24472. 17. A.S. Barnard, L.A. Curtiss: Prediction of TiO2 nanoparticle phase and shape transitions controlled by
surface chemistry, Nano Lett. 5 (2005) 1261-1266. 18. A.S. Barnard, R.R. Yeredla, H. Xu: Modelling the effect of particle shape on the phase stability of ZrO2
nanoparticles, Nanotechnol. 17 (2006) 3039-3047. 19. A.S. Barnard: A thermodynamic model for the shape stability of twinned nanostructures, J. Phys.chem. B
110 (2006) 24498-24504. 20. S. Ino, S. Ogawa: Multiply twinned particles at earlier stage of gold film formation on alkalihalide crystals,
J. Phys. Soc. Jpn. 22 (1967) 1365-1374. 21. S. Ino: Stability of multiple-twinned particles, J. Phys. Soc. Jpn. 27 (1969) 941-953. 22. J.G. Allpress, J.V. Sanders:The structure and orientation of crystals in deposits of metals on mica, Surf. Sci.
7 (1967) 1-25. 23. J.G. Allpress, J.V. Sanders: The structure and stability of small clusters of atoms, Austr. J. Phys. 23 (1970)
23-36. 24. C.Y. Yang: Crystallography of decahedral and icosahedral particles I. Geometry of twinning, J. Cryst.
Growth 47 (1979) 274-282. 25. L.D. Marks: Surface structure and energetics of multiply twinned particles, Phil. Mag. A. 49 (1984) 81-93. 26. V.G. Gryaznov, J. Heydenreich, A.M. Kaprelov, S.A. Nepijko, A.E. Romanov, J. Urban: Pentagonal
symmetry and disclinations in small particles, Cryst. Res. Technol. 34 (1999) 1091-1119. 27. F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti, C. Mottet: Crossover among structural motifs in
transition and noble-metal clusters, J. Chem. Phys. 116 (2002) 3856-3863. 28. P.A. Buffat: Electron diffraction and HRTEM studies of multiple-twinned structures and dynamic events in
metal nanoparticles: facts and artefacts, Mater. Chem. Phys. 81 (2003) 368-375. 29. J.L. Elechiguerra, J.R. Gasga, M.J. Yacamán: The role of twinning in shape evolution of anisotropic noble
metal nanostructures, J. Mater. Chem. 16 (2006) 3906-3919. 30. H. Hofmeister: High-resolution electron microscopy studies of metal nanoparticles: shape and twin defects,
and surface stress effects, J. Optoelectron. Advanced Mater. 9 (2007) 99-105.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
67
31. W.L. Winterbottom: Equilibrium shape of a small particles in contact with a foreign substrate, Acta Metall. 15 (1967) 303-310.
32. F. Silly, M.R. Castell: Temperature-dependent stability of supported five-fold twinned copper nanocrystaůs, ACS Nano 3 (2009) 901-906.
33. Q. Jiang, S. Li: Thermodynamic considerations on solid structural transition temperatures of nanocrystals, J. Comput. Theor. Nanosci. 5 (2008) 2346-2364.
34. Q. Jiang, C.C. Yang: Size effect on the phase stabiliy of nanostructures, Current Nanosci. 4 (2008) 179-200. 35. Q. Jinag, Z.P. Chen: Thermodynamic phase stabilities of nanocrabon, Carbon 44 (2006) 79-83. 36. S. Li, W.T. Zheng, Q. Jiang: Size and pressure effects on solid transition temperature of ZrO2, Script. Mater.
54 (2006) 2091-2094. 37. S. Li, Z.H. Jiang, Q. Jiang: Thermodynamic phase stability of three nano-oxides, Mater. Res. Bul. 43 (2008)
3149-3154. 38. S. Li, J.S. Lian, Q. Jiang: Modeling size and surface effects on ZnS phase selection, Chem. Phys. Lett. 455
(2008) 202-206. 39. S. Li, Z. Wen, Q. Jiang: Pressure-induced phase transition of CdSe and ZnO nanocrystals, Sript. Mater. 59
(2008) 526-529. 40. S. Li, G.W. Yang: Phase transition of II-VI semiconductor nanocrystals, J. Phys. Chem. C 114 (2010)
15054-15060. 41. S.B. Qadri, E.F. Skelton, D. Hsu, A.D. Dinsmore, J. Yang, H.F. Gray, B.R. Ratan: Size-induced transition-
temperature reduction in nanoparticles of ZnS, Phys. Rev. B 60 (1999) 9191-9193. 42. H.Z. Zhang, F. Huang, B. Gilbert, J.F. Banfield: Molecular dynamics simulations, thermodynamic analysis,
and experimental study of phase stability of zinc sulfide nanoparticles, J. Phys. Chem. B 107 (2003) 13051-13060.
43. J.W. Cahn: Surface stress and the chemical equilibrium of small crystals – I. The case of the isotropic surface, Acta Metall. 28 (1980) 1333-1338.
44. R.C. Cammarata: Surface and interface stress effects in thin films, Prog. Surf. Sci. 46 (1994) 1-38. 45. R.C. Cammarata: Generalized thermodynamics of surfaces with applications to small solid systems, in Solid
State Physics, Vol. 61 (H. Ehrenreich, F. Spaepen, Eds.), Academic Press, 2009. 46. A.L. Mackay: A dense non-crystallographic packing of equal spheres, Acta Cryst. 15 (1962) 916-918. 47. B.K. Teo, N.J.A. Sloane: Magic numbers in polygonal and polyhedral clusters, Inorg. Chem. 24 (1985)
4545-4558. 48. J.M. Montejano-Carrizales, J.L. Morán-Lopéz: Geometrical characteristics of compact nanoclusters,
Nanostruct. Mater. 1 (1992) 397-409. 49. J.M. Montejano-Carrizales, F. Aguilera-Granja, J.L. Morán-Lopéz: Direct enumeration of the geometrical
characteristics of clusters, Nanostruct. Mater. 8 (1997) 269-287 50. J.P.K. Doye, D.J. Wales: Magic number and growth sequences of small face-centered-cubic and decahedral
clusters, Chem. Phys. Lett. 247 (1995) 339-347. 51. J.P.K. Doye, D.J. Wales: Global minima for transition metal clusters described by Sutton-Chen potentials,
New J. Chem. xxx (1998) 733-744. 52. V.Ya. Shevchenko, A.E. Madison: Structure of nanoparticles: I. Generalized crystallography of
nanoparticles and magic numbers, Glass Phys. Chem. 28 (2002) 40-43. 53. M.L. Cohen, M.Y Chou, W.D. Knight, W.A. de Heer: Physics of metal clusters, J. Phys. Chem. 91 (1987)
3141-3149. 54. W.A. de Heer, W.D. Knight, M.Y. Chou, M.L. Cohen: Electronic shell structure and metal clusters, Solid
State Phys. 40 (1987) 93-181. 55. T.P. Martin, T. Bergmann, H. Göhlich, T. Lange: Observation of electronic shells and shells of atoms in
large Na clusters, Chem. Phys. Lett. 172 (1990) 209-213. 56. Z. Lin, T. Slee, D.M.P. Mingos: A structural jellium model of cluster electronic structure, Chem. Phys. 142
(1990) 321-334. 57. L. Magaud, S.N. Khanna, P. Jena: Limitation on the success of the jellium model for metal clusters, Chem.
Phys. Lett. 183 (1991) 333-336. 58. W.A. de Heer: The physics of simple metal clusters: experimental aspects and simple models, Rev. Mod.
Phys. 65 (1993) 611-676. 59. ... 60. C. Solliard, M. Flueli: Surface stress and size effect on the lattice parameter in small particles of gold and
platinum, Surf. Sci. 156 (1985) 487-494. 61. B. Medasani, I. Vasiliev: Computational study of the surface properties of aluminum nanoparticles, Surf.
Sci. 603 (2009) 2042-2046.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
68
62. C.W. Mayes, J.S. Vermaak, D. Kuhlmann-Wilsdorf: On surface stress and surface tension. II. Determination of the surface stress og gold, Surf. Sci. 12 (1968) 134-140.
63. A.M. Stoneham: Measurement of surface tension by lattice parameter changes: theory for facetted microcrystals, J. Phys. C: Solid State Phys. 10 (1977) 1175-1179.
64. K.K. Nanda, S.N. Behera, S.N. Sahu: The lattice contraction of nanometre-sized Sn and Bi particles produced by an electrohydrodynamic technique, J. Phys. C: Solid State Phys. 13 (2001) 2861-2864.
65. Q. Jiang, L.H. Liang, D.S. Zhao: Lattice contraction and surface stress of fcc nanocrystals, J. Phys. Chem. B 105 (2001) 6275-6277.
66. L.H. Liang, J.C. Li, Q. Jiang: Size-dependent melting depression and lattice contraction of Bi nanocrystals, Physica B 334 (2003) 49–53.
67. G.F. Gu, G. Krauss, W. Steurer, F. Gramm, A. Cervellino: Unexpected high stiffness of Ag and Au nanoparticles, Phys. Rev. Lett. 100 (2008) 045502 (4 pp).
68. C.D. Martin, S.M. Antao, P.J. Chupas, P.L. Lee, S.D. Shastri, J.B. Parise: Quantitative high-pressure pair distribution function analysisi of nanocrystalline gold, Appl. Phys. Lett. 86 (2005) 061910 (3 pp).
69. G. Ouyang, W.G. Zhu, C.Q. Sun, Z.M. Zhu, S.Z. Liao: Atomistic origin of lattice strain on stiffness of nanoparticles, Phys. Chem. Chem. Phys. 12 (2010) 1543-1549.
70. G. Ouyang, X.L. Li, X. Tan, G.W. Yanga: Size-induced strain and stiffness of nanocrystals, Appl. Phys. Lett. 89 (2006) 031904 (3 pp).
71. C.Q. Sun: Size dependence of nanostructures: Impact of bond order deficiency, Prog. Solid State Chem. 35 (2007) 1-159.
72. B. Chen, D. Penwell, L.R. Benedetti, R. Jeanloz, M.B. Kruger: Particle-size effect on the compressibility of nanocrystalline alumina Phys. Rev. B 66 (2002) 144101 (4 pp).
73. Y. He, J.F. Liu, W. Chen, Y. Wang, H. Wang, Y.W. Zeng, G.Q. Zhang, L.N. Wang, J. Liu, T.D. Hu, H. Hahn, H. Gleiter, J.Z. Jiang: High-pressure behavior of SnO2 nanocrystals, Phys. Rev. B 72 (2005) 212102 (4 pp).
74. S.B. Qadri, J. Yang; B.R. Ratna, E.F. Skelton, J.Z. Hu: Pressure induced structural transitions in nanometer size particles of PbS, Appl. Phys. Lett. 69 (1996) 2205-2207.
75. Y. Pan, J. Yu, Z. Hu, H. Li, Q. Cui, G. Zou: Pressure-induced structural transitions of the zinc sulfiden nano-particles with different sizes, J. Mater. Sci. Technol. 23 (2007) 193-195.
76. H. Gleiter: Nanocrystalline materials, Prog. Mater. Sci. 33 (1989) 223-315. 77. C. Suryanarayana: Nanocrystalline materials, Int. Mater. Rev. 40 (1995) 41-64. 78. H. Gleiter: Nanostructured materials: Basic concenpts and microstructure, Acta Mater. 48 (2000) 1-29. 79. B. Chen, D. Penwell, M.B. Kruger: The compressibility of nanocrystalline nickel, Solid State Commun. 115
(2000) 191-194. 80. S. Rekhi, S.K. Saxena, R. Ahuja, B. Johansson, J. Hu: Experimental and theoretical investigations on the
compressibility of nanocrystalline nickel, J. Mater. Sci. 36 (2001) 4719-4721. 81. J. Zhang, Y. Zhao, B. Palosz: Comparative studies of compressibility between nanocrystalline and bulk
nickel, Appl. Phys. lett. 90 (2007) 043112 (3 pp). 82. Y. Zhao, T.D. Shen, J. Zhang: High p-T nano-mechanics of polycrystalline nickel, Nanoscale Res. Lett. 2
(2007) 476-491. 83. X.D. Li, L.Y. Tang, Y.C. Li, J. Liu: Compressibility of nickel nanoparticle chain, Chin. Phys. lett. 24 (2007)
1671-1673. 84. R. Selva Vennila, S.R. Kulkarni, S.K. Saxena, H.-P. Liermann, S.V. Sinogeikin: Compression behavior of
nanosized nickel and molybdenum, Appl. Phys. lett 89 (2006) 261901 (3 pp). 85. (a) P. Bose Roy, S. Bose Roy: Applicability of three-parameter equation of state of solids: compatibility
with first principle approaches and application to solid, J. Physi.: Condens Matter. 15 (2003) 1643-1663. (b) P. Bose Roy, S. Bose Roy: An isothermal equation of state for solids, Physica B 350 (2004) 375-388.
86. W.H. Qi, M.P. Wang, Y.C. Su: Size effect on the lattice parameters of nanoparticles, J. Mater.Sci. Lett. 21 (2002) 877-878.
87. W.H. Qi, M.P. Wang, Y.C. Su: Comment on: Size effect on the lattice parameters of nanoparticles, J. Mater.Sci. Lett. 22 (2003) 1333-1334.
88. W.H. Qi, M.P. Wang: Size and shape dependent lattice parameters of metallic nanoparticles, J. Nanopart. Res. 7 (2005) 51-57.
89. Z. Huang, P. Thomson, S. Di: Lattice contractions of a nanopartilce due to the surface tension: A model of elasticity, J. Phys. Chem. Solids 68 (2007) 530-535.
90. J. Woltersdorf, A.S. Nepijko, E. Pippel: Dependence of lattice parameters of small particles on the size of the nuclei, Surf. Sci. 106 (1981) 64-69.
91. ... 92. ...

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
69
93. W.J. Huang, R. Sun, J. Tao, L.D. Menard, R.G. Nuzzo, J.M. Zuo: Coordination-dependent surface atomic contraction in nanocrystals revealed by coherent diffraction, Nature Mater. 7 (2008) 308-313.
94. B. Palosz, E. Grzanka, S. Gierlotka, S. Stelmakh, R. Pielaszek, U. Bismayer, J. Neuefeind, H.P. Weber, W. Palosz: Diffraction studies of nanocrystals: Theory and experiment, Acta Phys. Pol. (A) 102 (2002) 57-82.
95. B. Palosz, E. Grzanka, S. Gierlotka, S. Stelmakh, R. Pielaszek, W. Lojkowski, U. Bismayer, J. Neuefeind, H.P. Weber, W. Palosz: Application of X-ray powder diffraction to nanomaterials – determination of the atomic structure of nanocrystals with relaxed and strained surfaces, Phase Transitions 76 (2003) 171-185.
96. S. Stalmakh, S. Gierlotka, E. Grzanka, H.P. Weber, B. Palosz: X-ray diffraction studies of thermal properties of the core and surface shell of isolated and sintered SiC nanocrystals, J. Alloys Compds. 382 (2004) 138-145.
97. B. Palosz, S. Stelmakh, E. Grzanka, S. Gierlotka, W. Palosz: Applicationof the apparent lattice parameter to determination of the core-shell structure of nanocrystals, Z. Kristallogr. 222 (2007) 580-594.
98. B. Palosz, E. Grzanka, S. Gierlotka, S. Stelmakh: Nanocrystals: breaking limitations of data analysis, Z. Kristallogr. 225 (2010) 588–598.
99. W. Qi, B. Huang, M. Wang: Bond-Length and -Energy Variation of Small Gold Nanoparticles, J. Comput. Theor. Nanosci. 6 (2009) 635-639.
100. W.H. Qi, B.Y. Huang, M.P. Wang, Z.M. Yin, J. Li: Molecular dynamic simulation of the size- and shape-dependent lattice parameter of small platinum nanoparticles, J. Nanopart. Res. 11 (2009) 575-580.
101. W.H. Qi, S.T. Lee: Core-shell structures of silicon nanoparticles and nanowires with free and hydrogenated surface, Phys. Chem. Lett. 483 (2009) 247-249.
102. W.H. Qi, B.Y. Huang, M.P. Wang: Structure of unsupported small palladium nanoparticles, Nanoscale Res. Lett. 4 (2009) 269-273.
103. C.Q. Sun: The lattice contraction of nanometre-sized Sn and Bi particles produced by an electrohydrodynamic technique, J. Phys.: Condens. Matter 11 (1999) 4801-4803.
104. C.Q. Sun, B.K. Tay, X.T. Zeng, S. Li, T.P. Chen, J. Zhou, H.L. Bai, E.Y. Jiang: Bond-order-bond-length-bond-strength (bond-OLS) correlation mechanism for shape and size dependence of a nanosolid, J. Phys.: Condens. Mater. 14 (2002) 7781-7795.
105. C.Q. Sun: Thermo-mechanical behavior of low-dimensional systems: The local bond average approach, Prog. Mater Sci. 54 (2009) 179-307.
106. (a) B. Rellinghaus, S. Stappert, E.F. Wassermann, H. Sauer, B. Spliethoff: The effect of oxidation on the structure of nickel nanoparticles, Eur. Phys. J. D 16 (2001) 249-252, (b) Y. Duan, J. Li: Structure study of nickel nanoparticles, Mater. Chem. Phys. 87 (2004) 452-454.
107. D. Shreiber, W.A. Jesser: Size dependence of lattice parameter for SixGe1–x nanoparticles, Surf. Sci. 600 (2006) 4584-4590.
108. B. Ingham, S.C. Hendy, D.D. Fong, P.H. Fuoss, J.A. Eastman, A. Lassesson, K.C. Tee, P.Y. Convers, S.A. Brown, M.P. Ryan, M.F. Toney: Synchrotron x-ray diffraction measurements of strain in metallic nanoparticles with oxide shells, J. Phys. D: Appl. Phys. 43 (2010) 072301 (6pp).
109. K. Du, F. Ernst, M.C. Pelsozy, J. Barthel, K. Tillmann: Expansion of interatomic distances in platinum catalyst nanoparticles, Acta Mater. 58 (2010) 836-845.
110. I.F. Guilliatt, N.H. Brett: Lattice constant variation in finely divided magnesium oxide, Trans. Faraday Soc. 65 (1969) 3328-3333.
111. I.F. Guilliatt, N.H. Brett: Variations in the lattice constant of calcium oxide, J. Mater. Sci. 5 (1970) 615-617.
112. G. Li, J. Boerio-Goates, B.F. Woodfield: Evidence of linear lattice expansion and covalency enhancement in rutile TiO2 nanocrystals, Appl. Phys. Lett. 85 (2004) 2059-20661.
113. A.Y. Kuznetsov, R. Machado, L.S. Gomes, C.A. Achete, V. Swamy, B.C. Muddle, V. Prakapenka: Size dependence of rutile TiO2 lattice parameters determined via simultaneous size, strain, and shape modeling, Appl. Phys. Lett. 94 (2009) 193117 (3 pp).
114. A.M. Tonejc, I. Djerdj, A. Tonejc: An analysis of evolution of grain size-lattice parameters dependence in nanocrystalline TiO2 anatase, Mater. Sci. Eng. C 19 (2002) 85-89.
115. G. Li, L. Li, J. Boerio-Goates, B.F. Woodfield: High purity anatase TiO2 nanocrystals: Near room-temperature synthesis, grain growth kinetics, and surface hydratation chemistry, J. Am. Chem. Soc. 127 (2005) 8659-8666.
116. V. Swamy, D. Manzies, B.C. Muddle, A. Kuznetsov, L.S. Dubrovinsky, Q. Dai, V. Dmitriev: Nonlinear size dependence of anatase TiO2 lattice parameters, Appl. Phys. Lett. 88 (2006) 243103 (3 pp).
117. I. Djerdj, A.M. Tonejc: Structural investigation of nanocrystalline TiO2 samples, J. Alloys Compnds. 413 (2006) 159-174.
118. M. Imteyaz Ahmad, S.S. Bhattacharya: Size effect on the lattice parameters of nanocrystalline anatas, Appl. Phys. lett. 95 (2009) 191906 (3 pp).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
70
119. H. Zhang, B. Chen, J.F. Banfield: The size dependence of the surface free energy of titania nanocrystals, Phys. Chem. Chem. Phys. 11 (2009) 2553-2558.
120. P.J. Anderson, A. Scholz: Lattice constants and energies of ionic microcrystals, Trans. Faraday Soc. 64 (1968) 2973-2987.
121. V. Perebeinos, S.W. Chan, F. Zhang: „Madelung model“ predictionof lattice parameter on nanocrystal size, Solid State Commun. 123 (2002) 295-297.
122. M. Fukuhara: Lattice expansion of nanoscale compound particles, Phys. Lett. A 313 (2003) 427-430. 123. R. Setton: An equation of state for condensed matter; application to solid chemical elements, J. Phys.
Chem. Solids 71 (2010) 776-783. 124. T. Sun, K. Umemoto, Z. Wu, J.C. Zheng, R.M. Wentzcovitch: Lattice dynamics and thermal equation of
state of platinum, Phys. Rev. B 78 (2008) 024304 (12 pp). 125. W. Miller, C.W. Smith, D.S. Mackenzie, K.E. Evans: Negative thermal expansion: a review, J. Mater. Sci.
44 (2009) 5441-5451. 126. (a) X.G. Lu, M. Selleby, B. Sundman: Assessment of molar volume and thermal expansion for selected bcc,
fcc and hcp metallic elements, CALPHAD 29 (2005) 68-89. (b) X.G. Lu, M. Selleby, B. Sundman: Theoretical modeling of molar volume and thermal expansion, Acta Mater. 53 (2005) 2259-2272.
127. J. Garai, A. Laugier: The temperature dependence of the isotherma bulk modulus at 1 bar pressure, J. Appl. Phys. 101 (2007) 023514 (4 pp).
128. M. Gu, Y. Zhou. C.Q. Sun: Local bond average for the thermally induced lattice expansion, J. Phys. Chem. B 112 (2008) 7992-7995.
129. Y.A., Chang: On the temperature dependence of the bulk modulus and the Anderson-Grüneisen parameter δ of oxide compounds, J. Phys. Chem. Solids 28 (1967) 697-701.
130. O.L. Anderson, D.G. Isaak: The dependence of the Anderson-Grüneisen parameter δT upon compression at extreme conditions, J. Phys. Chem. Solids 54 (1993) 221-227.
131. Y.H. Zhao, K. Lu: Grain-size dependence of thermal properties of nanocrystalline elemental selenium studied by x-ray diffraction, Phys. Rev. B 56 (1997) 14330-14337.
132. H. Zhang, B.S. Mitchell: Thermal expansion behavior and microstructure in bulk nanocrystalline selenium by thermoanalytical analysis, Mater. Sci. Eng. A 270 (1999) 237-243.
133. J. Hu, W. Cai, C. Li, Y. Gan, L. Chen: In-situ x-ray diffraction study of the thermal expansion of silver nanoparticles in ambient air and vacuum, Appl. Phys. Lett. 86 (2005) 151915 (3 pp).
134. L. Li, Y. Zhang, Y.W. Yang, X.H. Huang, G.H. Li, L.D. Zhang: Diameter-dependence thermal expansion properties of Bi nanowire arrays, Appl. Phys. Lett. 87 (2005) 031912 (3 pp).
135. X.J. Xu, G.T. Fei, W.H. Yu, L.D. Zhang, X. Ju, X.P. Hao, D.N. Wang, B.Y. Wang: In situ x-ray diffraction study of the size dependent thermal expansion of silver nanowires, Appl. Phys. Lett. 89 (2006) 181914 (3 pp).
136. Y. Wang, H. Zhao, Y. Hu, C. Ye, L. Zhang: Thermal expansion behavior of hexagonal Zn nanowires, J. Cryst. Growth 305 (2007) 8-11.
137. X.W. Wang, Z.H. Yuan, S.Q. Sun, Y.Q. Duan, L.J. Bie: Thermal expansion behaviors of hcp and fcc Co nanowires arrays, Phys. Lett. A 373 (2009) 2887-2889.
138. X.H. Li, M.H. Ma, J.F. Huang: Structure and properties of nanometer size materials III. Structures and physical properties of iron nanoparticles, Chin. J. Chem. 23 (2005) 693-702.
139. C.C. Yang, M.X. Xiao, W. Li, Q. Jiang: Size effects on Debye temperature, Einstein temperature, and volume thermal expansion coefficient of nanocrystals, Solid State Commun. 139 (2006) 148-152.
140. Y.F. Zhu, J.S. Lian, Q. Jiang: Modeling of the melting point, Debye temperature, thermal expansion coefficient, and specific heat of nanostructured materials, J. Phys. Chem. C 113 (2009) 16896-16900.
141. M. Zhao, Q. Jiang: Size effect on thermal properties in low-dimensional materials, Key Eng. Mater. 444 (2010) 189-217.
142. Y. Cao, E. Conrad: Anomalous thermal expansion of Ni(001), Phys. Rev. Lett. 65 (1990) 2808–2811. 143. K.H. Chae, H.C. Lu, T. Gustafsson: Medium-energy ion-scattering study of the temperature dependence of
the structure of Cu(111), Phys. Rev. B 54 (1996) 14082-14086. 144. E.A. Soares, G.S. Leatherman, R.D. Diehl, M.A. van Hove: Low-energy electron diffraction study of the
thermal expansion of Ag(111), Surf. Sci. 468 (2000) 129-136. 145. K. Pussi, J. Vuorinen, M. Lindroos, H.I. Li, J.D. Howe, K.J. Hanna, R.D. Diehl, P.K. Bandyopadhyay:
Temperature dependent LEED study of Pb111, Surf. Sci. 603 (2009) 2759-2763. 146. B. Roldan Cuenya, A.I. Frenkel, S. Mostafa, F. Behafarid, J.R. Croy, L.K. Ono, Q. Wang: Anomalous
lattice dynamics and thermal properties of supported size and shape-selected Pt nanoparticles, Phys. Rev. B 82 (2010) 155450 (8 pp).

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
71
147. W.H. Li, S.Y. Wu, C.C. Yang, S.K. Lai, K.C. Lee, H.L. Huang, H.D. Yang: Thermal contraction of Au nanoparticles, Phys. Rev. Lett. 89 (2002) 135504 (4 pp).
148. A. Fernández Guillermet, P. Gustafson, M. Hillert: The representation of thermodynamic properties at high pressures, J. Phys. Chem. Solids 46 (1985) 1427-1429.
149. S.K. Saxena, J. Zhang: Assessed high-temperature thermochemical data on some solids, J. Phys. Chem. Solids 50 (1989) 723-727.
150. N.A. Dubrovinskaya, L.S. Dubrovinsky, S.K. Saxena: Systematics of thermodynamic data on solids: Thermochemical and pressure-volume-temperature properties of some minerals, Geochim. Cosmochim. Acta 61 (1997) 4251-4158.

SSttrruukkttuurraa nnaannoommaatteerriiáállůů J. Leitner, VŠCHT Praha 6.1.2016
72
5. Seznam symbolů Proměnné A ... plocha a ... mřížkový parametr w ... práce U ... vnitřní energie (molární veličina) u ... specifická vnitřní energie povrchu (fázového rozhranní) vztažena na jednotku plochy T ... teplota (K) p ... tlak V ... objem S ... entropie (molární veličina) s ... specifická entropie povrchu (fázového rozhranní) vztažena na jednotku plochy f ... specifická Helmholtzova energie povrchu (fázového rozhranní) vztažena na jednotku plochy g ... specifická Gibbsova energie povrchu (fázového rozhranní) vztažena na jednotku plochy n ... látkové množství µ ... chemický potenciál f ... povrchové napětí (stres) ε ... deformace l ... délka R ... univerzální plynová konstanta E ... kohezní Z ... koordinační číslo (počet nejbližších sousedních atomů) NA ... Avogadrova konstanta ρ ... atomární hustota porchu Indexy surf ... povrchový bulk ... objemový α ... označení fáze β ... označení fáze σ ... vztahující se k povrchu (fázovému rozhraní) i ... označení složky j ... označení složky φ ... obecné označení fáze elast... elastický F ... tání coh ... kohezní (hkl) ... vztahující se k rovině s Millerovými indexy (hkl)