ssuummmmeerr sscchhooooll: · certain, “phase field crystal modelling of liquid-crystal and...
TRANSCRIPT

1
SSSuuummmmmmeeerrr SSSccchhhoooooolll:::
CCCooommmpppuuutttaaatttiiiooonnnaaalll
MMMaaattteeerrriiiaaalllsss SSSccciiieeennnccceee
Miramar Palace, San Sebastian, Spain
June 28th – July 3th, 2010
CONTRIBUTIONS

2
SUMMER SCHOOL ORGANIZED BY
IGOR ABRIKOSOV, [email protected] JÖRG NEUGEBAUER, [email protected] ANDRÉS AYUELA, [email protected] PEPA CABRERA-SANFELIX, [email protected]
INVITED SPEAKERS
ABRIKOSOV IGOR
ALFE DARIO
ALONSO MARTIN JULIO ALFONSO
ASTA MARK
AYUELA ANDRES
BURKE KIERON
DIEZ-MUIÑO RICARDO
DRAUTZ RALF
DUDAREV SERGEI L.
GRABOWSKI BLAZEJ
HICKEL TILMANN
KORZHAVYI PAVEL
LECHERMANN FRANK
NEUGEBAUER JÖRG
ORDEJON PABLO
OZOLINS VIDVUDS
RAEBIGER HANNES
RINKE PATRICK
RUBAN ANDREI V.
SANCHEZ-PORTAL DANIEL
SIMAK SEGEI
VAN DE WALLE CHRIS
ORGANIZING COMMITTEE
PEDRO MIGUEL ECHENIQUE IGOR ABRIKOSOV
JÖRG NEUGEBAUER ANDRÉS AYUELA
PEPA CABRERA-SANFELIX

3
PREFACE
We would like to welcome all participants to the Summer School: Computational Materials
Science, being held in San Sebastian, from June 28 to July third, 2010. This Summer School
aims at the identification and promotion of the common elements developed in theoretical and
computational studies of materials properties across materials types, metals, ceramics,
materials for new energy technologies, electronic materials and minerals. To accomplish this
goal, the School brings together leading experts from a wide spectrum of materials
simulations including theory, modeling, and computation, engaged in the study of a broad
range of materials properties.
Therefore, this School provides a forum for exposing young researchers and students to most
recent state-of-the-art theoretical and computational developments in studying, understanding,
and predicting the properties of materials. Also, the School encourages interdisciplinary
contributions, such as between the fields of condensed matter physics and applied materials
sciences, chemistry, metallurgy, etc.
The attendance of the school has been kept below 100 participants so that active
communication between all members could be guaranteed. We are confident that the activity
of all the participants will result in a lively and successful school and, in addition, we hope
you will find the time to enjoy the surroundings and gastronomy of San Sebastian.
Igor Abrikosov Andrés Ayuela Jörg Neugebauer Pepa Cabrera-Sanfelix

4
Time Schedule
Monday, June 28th
(2010)
8:30 – 9:00 Registration
9:00 – 9:40 Basic class: J. Neugebauer, “Introduction to DFT”
9:50 – 10:30 Basic class: J. Neugebauer, “Introduction to DFT ”. Second part
10:30 – 11:00 Coffee break
11:00 – 11:40 Basic class: I. Abrikosov, “Ab initio theory of substitutional disorder: the
coherent potential approximation and beyond”
11:50 – 12:30 Basic class: I. Abrikosov, “Ab initio theory of substitutional disorder: the
coherent potential approximation and beyond”. Second part
12:30 – 15:00 Lunch
15:00 – 16:30 Poster Introduction Section (3 – 5 minutes talks)
1. A. Allard, “Tuning The Kohn Anomaly In The Phonon Dispersion Of Graphene By
Interaction With A Metallic Substrate.”
2. A. Glensk, “Ab initio prediction of thermodynamic data for selected phases of the Al-Mg-Si-
Cu system”.
3. A. Stroppa, “HYBRID FUNCTIONAL STUDIES OF MULTIFERROICS”.
4. M.S. Rakitin, “Effect of palladium and titanium impurities on hydrogen solubility in bcc
iron”.
5. G. M. Thomas, “SELF-ASSEMBLY OF DNA BASE THYMINE ON Cu(110) STUDY”.
6. E. Kabliman, “Mechanism Of Stabilization Of The Misfit Layer Compound (PbS)1.13TaS2”.
7. M. Rohrmüller, “Analysis of paramagnetic and ferromagnetic surface states in µc-Si:H via
the orbital magnetization”.
8. I. Errea, “Superconductivity and Novel Structures of Calcium Under Pressure from ab intio
Calculations”.
9. J. Ibañez, “Spin-Orbit Coupling in Tl/Si(1,1,1) surface”.
10. Z. Wang, “Thermoelectric transport properties of silicon from first principles”.
11. M. Certain, “Phase field crystal modelling of liquid-crystal and reconstructive phase
transitions with FCC and BCC lattices”.

5
12. A. Maître, “MODELING OF OF Co/IrMn BILAYER : PHASE TRANSITIONS AND
HYSTERESIS LOOPS”.
13. F. Gallino, “Characterization of nitrogen defective centers in zinc oxide”.
14. C. Motta, “Study of electronic transport in metal-metal junctions within the embedding
approach”.
15. L. Bläckberg, “Noble gas diffusion barriers for improved Nuclear-Test-Ban treaty
verification detection systems”.
16. G. Hautier, “A High-Throughput Computational Search for New Lithium-Ion Battery
Cathode Materials”.
17. M. Dahlqvist, “Stability trends of Mn+1AXn phases”.
18. L. Bjerg, “The Hunt For N-type ZnSb”.
19. S.S. Tsirkin, “Scattering of electrons and holes in surface states on Cu(110) and Ag(110)”.
20. Hemant Dixit, “Quasiparticle bandstructure of zincblende and rocksalt ZnO”.
16:30 – 17:00 Coffee break
17:00 – 18:30 Master Class: S. Simak, “Using VASP for materials simulations”.
18:30 – 19:00 Poster Introduction Section (3 – 5 minutes talks)
1. M. Amini, “Hydrogen impurities and oxygen vacancies in CdO”.
2. M.G. Vergniory, “Calculation Of Complex Band Structure Fro Plane-wave Pseudopotential
Hamiltonian”.
3. I. Etxebarria, “Condensation Of Several Structural Instabilities Via Secondary Distortions:
The Case Of Sr2MWO6 (M= Zn, Mg And Ni) Double Perovskites”.
4. D. Costa, “Towards an Atomistic kinetic Monte Carlo simulation of Iron Chromium
phasedecomposition based on an ab initio parameterisation”.
5. N. Tillack, “Ab initio study of nano-precipitate nucleation and growth in ferritic steels”.
6. E. Wachowicz, “Effect of impurities on structural, cohesive, and magnetic properties of grain
boundaries in α-Fe”.
7. B. Lange, “Constructing optimized atomic basis-sets with PW accuracy”.
8. D. Ma, “Solid solution strengthening investigated by first principles”.
9. I. Ulfat, “Annealing Induced Modifications in (GaMn)As: Electron Spectroscopic Studies”
10. A. Cammarata, “Local Structure Of Octahedral Site In Y-doped BaCeO3 And BaZrO3
Perovskite Compounds: A New Tetravalent Cation Substitution Model”
11. T. Pabisiak, “DFT study of formation and stability of Au nanostructure on rutile TiO2(110)”
19:00 – 21:00 Poster Session (and Refreshments)

6
Tuesday, June 29th
(2010)
9:00 – 9:40 Basic class: A. Ruban, “Alloys and magnetism”
9:50 – 10:30 Basic class: A. Ruban, “Alloys and magnetism”. Second Part
10:30 – 11:00 Coffee break
11:00 – 11:40 Basic class: M. Asta, “First-Principles Modeling of Configurational
Thermodynamics in Crystalline Alloys and Compounds”
11:50 – 12:30 Basic class: M. Asta, “First-Principles Modeling of Configurational
Thermodynamics in Crystalline Alloys and Compounds”. Second Part
12:30 – 15:00 Lunch
15:00 – 15:45 Special Class: A. Ayuela, “Magnetically driven shape memory alloys”
15:45– 16:30 Special Class: L-W.Wang, “Ab initio simulations for semiconductor
nanostructures and alloys for solar cell applications”
16:30 – 17:00 Coffee break
17:00 – 18:30 Master Class: A. V. Ruban, “Effective cluster interactions”
18:30 – 19:00 Poster Introduction Section (3 – 5 minutes talks)
1. K. Özdoğan, “The overview of the half-metallic Heusler alloys”.
2. P. Elstnerová, “Ab initio study of thermodynamic, structural, and elastic properties of Mg-
substituted crystalline calcite”.
3. M. Colakogullari, “An Investigation On The Covalent-like Transformation Of Undercooled
Liquid Silicon Using Orbital-Free Ab-initio Molecular Dynamics Method”.
4. U. Aydin, “Ab initio investigation of hydrogen solubility in 3d metals”.
5. A. Saracibar, “Molecular Dynamics In Zirconium Phosphates Systems”.
6. P. Rejmak, “QM/MM Studies On Ethene Adsorption On Cu(I) Exchanged Zeolites”.
7. J-B. Piochaud, “Microstructural evolution of austenitic alloys under irradiation modelled by
an ab initio based Atomic Kinetic Monte Carlo (AKMC) model”.
8. S. Rigamonte, “√7x√3 Indium on Si(111): one or two In layers?”
9. B. Grabowski, “Ab initio concepts for an efficient and accurate determination of
thermodynamic properties up to the melting point”
10. H. Sener Sen, “ELECTRONIC PROPERTIES OF GRAPHENE NANO-RIBBONS”.
11. C. M. Ulrich, “Modeling Diffusion in the Al-Au System with Parameters from Ab-Initio
Calculations”
19:00 – 21:00 Poster Session (and Refreshments)

7
Wednesday, June 30th
(2010)
9:00 – 9:40 Basic class: A. Drautz, “Bond-Order Potentials: from the electronic
structure to interatomic potentials”
9:50 – 10:30 Basic class: A. Drautz, “Bond-Order Potentials: from the electronic
structure to interatomic potentials”. Second Part
10:30 – 11:00 Coffee break
11:00 – 11:40 Basic class: S. Dudarev, “structure and dynamics of nano-defects in IRON
and OTHER BCC Metals”
11:50 – 12:30 Basic class: S. Dudarev, “structure and dynamics of nano-defects in IRON
and OTHER BCC Metals”. Second Part
12:30 – 15:00 Lunch
15:00 – … Guided tour or free afternoon
Thursday, July 1st (2010)
9:00 – 9:40 Basic class: K. Burke, “New functionals”
9:50 – 10:30 Basic class: K. Burke, “New functionals”. Second Part
10:30 – 11:00 Coffee break
11:00 – 11:40 Basic class: F. Lechermann, “An introduction to Dynamical Mean-Field
Theory and its application to strongly correlated materials”
11:50 – 12:30 Basic class: F. Lechermann, “An introduction to Dynamical Mean-Field
Theory and its application to strongly correlated materials”. Second Part
12:30 – 15:00 Lunch
15:00 – 16:30 Basic Class: C. van de Walle, “First-principles Investigations of Point
Defects”
16:30 – 17:00 Coffee break
17:00 – 18:30 Master Class: K. Burke, “Time-dependent DFT”.
18:30 – 19:15 Master Class: H. Raebiger, “Impurity States”.

8
Friday, July 2st (2010)
9:00 – 9:40 Basic class: V. Ozolins, “First-principles calculations of vibrational
thermodynamics at high temperatures”
9:50 – 10:30 Basic class: V. Ozolins, “First-principles calculations of vibrational
thermodynamics at high temperatures”. Second Part
10:30 – 11:00 Coffee break
11:00 – 11:40 Basic class: P. Ordejon, “Linear scaling methods in electronic structure
calculations”.
11:50 – 12:30 Basic class: D. Sánchez-Portal, “The SIESTA method: an efficient tool in
materials science”.
12:30 – 15:00 Lunch
15:00 – 15:45 Special Class: J. A. Alonso, “Hydrogen Storage in Nanoporous Carbons”
15:45 – 16:30 Special Class: P. Rinke, “GW Quasiparticle Calculations”
16:30 – 17:00 Coffee break
17:00 – 17:45 Master Class: P. Ordejon, “Advanced concepts in SIESTA”
17:45 – 18:30 Master Class: R. Diez-Muiño, “Theory of gas/surface dynamics”
21:00 Conference dinner

9
Saturday, July 3st (2010)
9:00 – 9:40 Basic class: P. Korzhavyi, “Interaction between ab initio and Calphad
approaches”
9:50 – 10:30 Basic class: P. Korzhavyi, “Interaction between ab initio and Calphad
approaches”. Second Part
10:30 – 11:00 Coffee break
11:00 – 11:40 Basic class: D. Alfe, “Liquids”.
11:50 – 12:30 Basic class: D. Alfe, “Liquids”. Second part
12:30 – 15:45 Lunch
15:45 – 16:30 Special Class: T. Hickel, “Using ab initio methods to predict thermodynamic
properties of metals”
16:30 – 17:00 Coffee break
17:00 – 18:30 Master Class: B. Grabowski, “Master Class on thermodynamics: How to
derive electronic and vibronic free energy surfaces from ab initio?”
(Close)

10
MONDAY
June 28th

11
Introduction into Density Functional Theory
Jörg Neugebauer
Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany
In this lecture a brief introduction into key concepts of various popular ab initio methods will be given,
advantages and present limitations will be discussed, and examples with respect to applications in
material science will be given. An important aspect to address real world problems is the limitation of
ab initio techniques to rather small system sizes of typically a few hundred atoms, which makes it
necessary to combine them with simulation techniques on the meso- and/or macroscale. Recent
developments and perspectives of such multiscale approaches will be given with specific focus on the
requirements on the ab initio side such as accuracy, efficiency etc.

12
Ab initio theory of substitutional disorder: the coherent potential
approximation and beyond
I. A. Abrikosov Department of Physics, Chemistry and Biology, Linköping University, Sweden
We review recent developments in the field of ab initio electronic structure theory and its application for studies of complex alloy systems. Basic ideas behind state-of-the-art techniques for first-principles theoretical calculations of the electronic structure and properties of intermetallic compounds and alloys based on the density functional theory are outlined. We concentrate on methods that allow for an efficient treatment of disorder effects [1,2], and illustrate their predictive power by numerous examples.
In general, a solution to the quantum mechanical problem for a solid is provided by the electronic structure theory [3]. However, a serious problem with its application occurs if a configuration of the solution phase, say A1-xBx, does not have any translational periodicity, which is the most common situation in practice. In this case the group theory, which is the corner stone of the modern electronic structure calculations, cannot be used directly. We will present modern approaches for solving the electronic structure problem and calculating the total energy for systems with substitutional disorder.
One obvious way to deal with a disordered system is to consider its fragment(s), to impose periodic boundary conditions, and to solve the KS problem for such “supercells”. As an alternative to the supercell approach one can reconstruct three-dimensional periodicity of the substitutionally disordered system by mapping the latter onto a suitably chosen ordered lattice of “effective” atoms, which describe the original system on the average. In terms of the electronic structure problem, one is ultimately interested in processes of electron scattering off the atoms in the system, the so-called multiple-scattering. The simplest mean field method, the coherent potential approximation (CPA), is constructed by placing effective scatterers at the sites of the original system. Scattering properties of these effective atoms have to be determined self-consistently from the condition that the scattering of electrons off the alloy components, embedded in the effective medium as impurities, vanishes on the average. The CPA is currently one of the most popular techniques to deal with substitutional disorder. However, CPA gives an approximate, mean-field description of the scattering properties of a system. A way beyond the mean-field theory in electronic structure calculations for solution phases is associated with the development of so-called O(N) methods [3]. Unfortunately, most of them can not deal with metallic systems at present. However, the locally self-consistent Green’s function method (LSGF) based on a concept of local self-consistency within the multiple scattering theory, has been applied for metals with considerable success. The basic ideas behind the LSGF method will be discussed. [1] I A. V. Ruban and I. A. Abrikosov, Rep. Prog. Phys. 71, 046501 (2008). [2] P. E. A. Turchi, I. A. Abrikosov, B. Burton, S. G. Fries, G. Grimvall, L. Kaufman, P. A. Korzhavyi, V. Rao Manga, M. Ohno, A. Pisch, A. Scott, and W. Zhang, CALPHAD 31, 4 (2007). [3] R. M. Martin, Electronic structure. Basic Theory and Practical Methods (Cambridge University Press, Cambridge, 2004)

13
Using VASP for materials simulations
S. I. Simak Theoretical Physics, IFM, Linköping University, SE-58183 Linköping (Sweden)
Practical aspects of materials simulation employing Vienna Ab Initio Simulation Package (VASP)1 will be addressed. VASP is a package for performing ab-initio quantum-mechanical simulations using pseudopotentials or the projector-augmented wave (PAW)2 method and a plane wave basis set. Basic techniques and choice of input parameters including potentials, density functional approximations, basis sets, k-point sampling etc. will be discussed in more detail, with a number of corresponding examples. References
1 G. Kresse and J. Hafner. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558, 1993; G. Kresse and J. Hafner. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251, 1994; G. Kresse and J. Furthmüller. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15, 1996; G. Kresse and J. Furthmüller. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169, 1996. 2 P. E. Blochl. Projector augmented-wave method. Phys. Rev. B 50, 17953, 1994; G. Kresse and D. Joubert. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758, 1999.

14
TUESDAY
June 29th

15
“Alloys and magnetism”
A.V. Ruban Materials Science and Engineering Department, KTH, Stockholm
(basic class)
Magnetism and bonding have the same origin in the case of 3d-metals and their alloys: valence
d-electrons. This determines their complex interplay, when, on one hand, magnetic interactions
can be extremely sensitive as to the local atomic structure and local chemical environment, and,
on the other hand, magnetic exchange interactions and local and global magnetic structure can
modify substantially chemical interactions between alloy components. This is also a
consequence of the fact that effective chemical and magnetic exchange interactions are on the
same energy scale.
This lecture is an introduction to the first-principles description of these phenomena and their
modeling on the atomic scale, which can be used in the statistical simulations of the phase
equilibria as magnetic as well as chemical. The ab initio methods for calculating effective
magnetic and chemical interactions, such as magnetic force theorem and generalized
perturbation methods will be introduced. Their application to the different alloy system
illustrating the above mention phenomena will be given.

16
First-Principles Modeling of Configurational Thermodynamics in
Crystalline Alloys and Compounds
Cluster Expansions and Related Approaches
Mark Asta1,2, Vitaly Alexandrov2, and Axel van de Walle3
1Department of Materials Science and Engineering, University of California, and Materials
Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
2NEAT ORU, University of California, Davis, CA, USA
3Materials Science and Engineering, California Institute of Technology, Pasadena, CA, USA
This lecture will cover first-principles methods for modeling the structure and thermodynamic
properties of substitutionally disordered crystalline materials, including alloys and ionic
compounds, within the framework of the so-called cluster expansion formalism.
Technologically-relevant materials are typically multicomponent systems, with solute species
added intentionally to enhance properties, or present as impurities originating from processing
or service. To impact the design of such materials, and to optimize their properties for
technological applications, it is generally critical to understand the thermodynamic stability of
competing phases as well as the ways in which aging of the materials can lead to changes in
atomic structure that can ultimately affect properties and performance. First-principles
computational methods, based on electronic density-functional theory, have found growing
applications in this area over the past decade owing to advances in methods, algorithms and
computational power. The lecture will begin with an introduction to the class of problems
that can be modeled by cluster expansion techniques, and will go on to describe the statistical-
mechanical foundation underlying the approach. After a review of the mathematical
formalism of cluster expansions, we will describe approaches used to fit the expansion

coefficients in this approach, including strategies used to account for long
interactions arising from, e.g., size mismatch between constituent species. N
the use of cluster-expansion models in Monte
of short and long-range order, and associated thermodynamic properties. The lecture will end
with a discussion of a few case studies taken from a
structured oxide compounds.
Figure 1: The figure on the right shows a schematic of the coupleddisorder present on an aliovalently doped fluorite crystal structsnapshot of a cluster-expansion based Monteright) and dopants (shown in red) display ordering along 110 planes in the fluorite structure.
coefficients in this approach, including strategies used to account for long
interactions arising from, e.g., size mismatch between constituent species. N
expansion models in Monte-Carlo simulations to derive equilibrium states
range order, and associated thermodynamic properties. The lecture will end
with a discussion of a few case studies taken from applications in Al alloys and fluorite
structured oxide compounds.
: The figure on the right shows a schematic of the coupled-sublattice configurational disorder present on an aliovalently doped fluorite crystal structure. The figure on the left is a
expansion based Monte-Carlo simulation where the vacancies (shown in right) and dopants (shown in red) display ordering along 110 planes in the fluorite structure.
17
coefficients in this approach, including strategies used to account for long-ranged interatomic
interactions arising from, e.g., size mismatch between constituent species. Next we will cover
Carlo simulations to derive equilibrium states
range order, and associated thermodynamic properties. The lecture will end
pplications in Al alloys and fluorite-
sublattice configurational ure. The figure on the left is a
Carlo simulation where the vacancies (shown in right) and dopants (shown in red) display ordering along 110 planes in the fluorite structure.

18
Magnetically driven shape memory alloys
A. Ayuela*
Centro de Física de Materiales CSIC UPV EHU, MPC, San Sebastian 20080, Spain
DIPC, San Sebastian 20018, Spain
* Work done in collaboration with Enkovaara J, Zayak AT, Entel P, Nordstrom L, Dube M,
Jalkanen J, Impola J, Nieminen RM.
Significant progress has been made both in experimentation and theoretical modelling of the
magnetic shape memory (MSM) effect, where magnetic field can induce strains of 10%. The
theoretical models used to analyze and interpret the different experiments provide reliable
information and insight into the physical changes involved in the magnetically driven shape
memory alloys. The aim of this talk is to discuss the present status of the computational
modelling we have done. First, the basic MSM requirements [1-2] and a brief summary of the
experimental results for the prototype material Ni-Mn-Ga are given. Then, in the context of
atomic-scale calculations, we focus primarily on the understanding of the structural variants
[1], magnetic anisotropy [2], and Curie temperatures [3]. Also we see an approach to describe
doping out of the stoichiometric alloys where we see the antiferromagnetic coupling of extra
Mn [4]. Finally, we bring into contact our modelling with recent results [5].
[1] Ayuela A, Enkovaara J, Ullakko K, et al. J. Phys-Condens Matter 11, 2017 (1999); Ayuela
A, Enkovaara J, Nieminen RM J. Phys-Condens Matter 14, 5325 (2002); Zayak AT, Entel P,
Enkovaara J, et al. J Phys-Condens Matter 15, 159 (2003); Zayak AT, Entel P, Enkovaara J,
et al. Phys. Rev. B 68, 132402 (2003).
[2] Enkovaara J, Ayuela A, Nordstrom L, et al Phys. Rev B 65, 134422 (2002); Enkovaara J,
Ayuela A, Nordstrom L, et al. J. App. Phys. 91, 7798 (2002).
[3] Enkovaara J, Ayuela A, Jalkanen J, et al. Phys Rev B 67, 054417 (2003).
[4] Enkovaara J, Heczko O, Ayuela A, et al.Phys. Rev. B 67, 212405 (2003).
[5] Ye, M. et al Phys. Rev. Lett. 104, 176401 (2010); Uijttewaal MA, Hickel T, Neugebauer
J, et al Phys. Rev. Lett. 102, 035702 (2009).

19
Ab initio simulations for semiconductor nanostructures and alloys
for solar cell applications
Lin-Wang Wang
Lawrence Berkeley National Laboratory
Inorganic semiconductor nanocrystals and alloys are often proposed to be used as active solar
cell materials. In this talk, I will present our recent ab initio simulations on such systems.
First, the binding
energy of the charge transfer exciton at CdTe/CdSe nanowire interface has been calculated. It
is found that the exciton binding energy can be as high as 0.4 eV, which poses a problem for
exciton dissociation. Second the electronic structure of GaN/ZnO alloy has been studied using
the ab initio method. A Hamiltonian model has been developed to describe the configuration
energy of this system. Monte Carlo simulation based on this model Hamiltonian is used to
study the atomic structures of the alloy. The electronic structure is then calculated after the
density functional theory band gap error has been corrected. Third, the electronic structures of
ZnTe:O has been calculated using the linear scaling three dimensional fragment method. This
material has been proposed to be used as an band gap intermediate state solar cell. The solar
cell efficiency has been calculated based on the absorption spectrum. Theoretical solar cell
efficiency as high as 60% has been predicted.

20
“Effective cluster interactions”
A.V. Ruban
Materials Science and Engineering Department, KTH, Stockholm.
(master class)
This class is intended as a guide to the practical use of two methods: screened generalized
perturbation method (SGPM) for calculating effective chemical interactions in metallic alloys
and magnetic force theorem method for magnetic exchange interaction parameters of the
Heisenberg magnetic Hamiltonian. The practical examples will be given showing how to
apply this method and check the consistency of the calculated interactions with the
configurational energetics of the corresponding systems. The calculated interactions will be
consequently used in the Monte Carlo simulations of the chemical order-disorder transition as
well as magnetic transition in real systems.

21
WEDNESDAY June 30th

22
Bond-Order Potentials: from the electronic structure to interatomic
potentials
Ralf Drautz¹, Bernhard Seiser², Thomas Hammerschmidt¹, David G. Pettifor²
¹ ICAMS, Ruhr-Universität Bochum, Germany
² Department of Materials, University of Oxford
Although quantum mechanics provides a fundamental and highly accurate basis for materials
science, it is rarely used as the basis for the development of a new material. This is due to the
hierarchical structure of materials, where several orders in length and time need to be bridged
between the electronic structure and the mechanical behaviour on the component scale. A
critical step for a systematic modelling of materials is the derivation of classical, effective
interatomic interactions from a quantum mechanical treatment of bond formation. For the
derivation of robust interatomic potentials we coarse grain the electronic structure at two
levels of approximation. In a first step, the description of the electronic structure is simplified
to the tight-binding approximation.
In a second step, a moments expansion of the tight-binding Hamiltonian results in a classical,
effective interatomic interaction, the analytic bond-order potential (BOP). The format of the
potential includes charge transfer and magnetic contributions to the binding energy. The BOP
reproduces the structural trend across the non-magnetic 4d and 5d TM series and displays the
experimental trend from anti-ferromagnetic order to ferromagnetic order across the 3d
transition metal series. For iron, the potential correctly predicts a large magnetic energy for
the alpha phase whereas the close-packed gamma and epsilon phases exhibit only a small
magnetic contribution to the binding energy. We will discuss the application of the bond-
order potentials to the prediction of phase stability in complex phases.

23
structure and dynamics of nano-defects in IRON and OTHER
BCC Metals1
S.L. Dudarev EURATOM/CCFE Fusion Association, Culham Centre for Fusion Energy,
Oxfordshire OX14 3DB, UK, [email protected]
The need to formulate a rational knowledge-based strategy for the development of fusion and fission materials has stimulated a renewed interest in fundamental materials modelling methodology. Recent developments show that modelling can indeed provide quantitative interpretation of experimentally observed changes in physical and mechanical properties of materials occurring under irradiation, make predictions needed for optimizing experimental tests, and even suggest routes for designing new alloys.
This presentation gives examples showing how multiscale mathematical modelling was successfully applied to solving practical problems of nuclear materials science. The examples include an ab initio study of small radiation defects in bcc transition metals [1], which for the first time explained the origin of low-temperature mobility of defects in tungsten, and highlighted the unusual effect of atomic magnetism on the structure of defects in iron; the development of a ‘magnetic’ interatomic potential for molecular dynamics simulations [2,3,4], its generalization to spin-lattice dynamics, where equations of motion follow the evolution of atomic coordinates and directions of magnetic moments [5], and the application of Magnetic Cluster Expansion (MCE) to the treatment of magnetic thermal fluctuations, and bcc-fcc α-γ-δ phase transitions in iron-based alloys [6].
Figure 1: Left: the magnetic (violet) and phonon (orange) fcc-bcc free energy differences for pure iron calculated using the Magnetic Cluster Expansion model [6]. Centre: the trajectory of a magnetic moment of an iron atom simulated using the spin-lattice dynamics algorithm [5]. Right: the real-space trajectory of the same atom simulated using spin-lattice dynamics [5].
Interpreting experimental observations requires algorithms for treating macroscopic time and length scales. The two papers [7,8], addressing the dynamical behaviour of ensembles of interacting defects, illustrate the complexity of the problem. To interpret in-situ electron microscope observations of migrating defects, it is necessary to formulate an alternative (to the well-established kinetic Monte Carlo) model for simulating the long-time-scale evolution of radiation-induced microstructures. The need for a new algorithm stems from the fact that understanding microscopic processes driving microstructural evolution, and matching simulations to experiment, requires modelling particular configurations of evolving defect
1This work was funded partly by the United Kingdom Engineering and Physical Sciences Research Council
under grant EP/G003955 and the European Communities under the contract of Association between EURATOM and CCFE.

24
structures. Some properties of irradiated materials depend on the statistics of their microstructure, formed by many defects and dislocations, and representing self-averaging quantities. At the same time, a microstructural evolution model requires understanding the dynamics of particular configurations of interacting defects, and comparing the results of simulations with local experimental observations, involving only a few defects, where no statistical averaging is possible. Langevin dynamics [8] is a new method for treating long-time-scale evolution of ensembles of interacting defects, which provides a conceptually consistent and computationally efficient framework for matching simulations to real-time experimental observations.
Figure 2: In-situ electron microscope observation (left) and a matching real-time Langevin dynamics simulation (right) showing correlated stochastic motion of two interacting nano-scale dislocation loops formed in pure iron under irradiation [8].
REFERENCES
1. D. Nguyen-Manh, A.P. Horsfield, and S.L. Dudarev, Self-interstitial atom defects in bcc transition metals: group-specific trends. Phys. Rev. B73 (2006) 020101.
2. S.L. Dudarev and P.M. Derlet, A ‘magnetic’ interatomic potential for molecular dynamics simulations. J. Phys.: Cond. Mat. 17 (2005) 7097; ibid. 19 (2007) 239001.
3. P.M. Derlet and S.L. Dudarev, Million-atom molecular dynamics simulations of magnetic iron. Progress in Materials Science 52 (2007) 299.
4. S. Chiesa, P.M. Derlet and S.L. Dudarev, Free energy of a 110 dumbbell interstitial defect in bcc Fe: harmonic and anharmonic contributions. Phys. Rev. B79 (2009) 214109
5. P.-W. Ma, C.H. Woo, and S.L. Dudarev, Large-scale simulations of the spin-lattice dynamics in ferromagnetic iron. Phys. Rev. B78 (2008) 024434.
6. M.Y. Lavrentiev, D. Nguyen-Manh, and S.L. Dudarev, Magnetic Cluster Expansion model for bcc-fcc transitions in Fe and Fe-Cr alloys. Phys. Rev. B81 (2010) 184202.
7. T.S. Hudson, S.L. Dudarev, M.J. Caturla, and A.P. Sutton, Effects of elastic interactions on post-cascade radiation damage evolution in kinetic Monte Carlo simulations. Philosophical Magazine 85 (2005) 661.
8. S.L. Dudarev, M.R. Gilbert, K. Arakawa, H. Mori, Z. Yao, M.L. Jenkins, and P.M. Derlet, A Langevin model for real-time Brownian dynamics of interacting nano-defects in irradiated metals. Phys. Rev. B81 (2010) in press, June 2010 issue of the journal

25
THURSDAY
July 1st

26
"New functionals"
Kieron Burke
UC Irvine, Depts of Physics and of Chemistry
In this talk, I will review basics of DFT, using material from my online book, ABC of DFT,
and elsewhere. I will discuss standard functionals, including both successes and failures, and
the current state-of-the-art. There will be a quiz at the end, including prizes.

27
An introduction to Dynamical Mean-Field Theory and its application to
strongly correlated materials
Frank Lechermann I. Institut für Theoretische Physik, Universität Hamburg, 22303 Hamburg, Germany
The celebrated density functional theory (DFT), e.g., within the local-density approximation (LDA), achieves for many weakly correlated materials excellent agreement between theory and experiment regarding the band structure, phase stability, static relaxation, phonon spectrum or ordered magnetic moments. However, materials with strong local electronic correlations cannot be described in this way. Focusing on the energy-band aspect, the LDA provides a conceptually invalid description in that case. For instance, the correlation induced Mott insulating state amounts to an electron localization in real space, which implies the overall breakdown of band theory. Thus strongly correlated systems require an explicit inclusion of many-body effects in order to capture the coherent quasiparticle excitations at low energy (i.e., close to the Fermi level) and the incoherent atomic-like excitations at higher energies. Inherent features like e.g, band-narrowing, spectral-weight transfer or the loss of coherency due to finite temperature are important results of such a manifest many-body nature. The dynamical mean-field theory (DMFT), developed 20 years ago, is the most comprehensive approach to describe such strongly correlated systems. Initially constructed for Hubbard-like model systems, DMFT becomes exact in the limit of high spatial dimensions or lattice coordination number. It corresponds to a mapping of the interacting lattice problem to the problem of a single quantum impurity subject to a mean-field given by an effective bath. In contrast to Hartree-Fock-type theories the mean field here is energy dependent and can thus cope with retardation effects, whereby the local quantum fluctuations on the impurity are fully taken into account. The self-consistent determination of the dynamical mean-field corresponds then to the solution of the original lattice problem with a local self energy. Hence the DMFT approximation amounts to the neglect of explicit spatial correlations. The actual numerical solution of the DMFT problem implies the use of quantum-impurity solvers, such as Quantum Monte-Carlo methods, exact diagonalization, slave-particle representations, etc…
Figure 1: DMFT construction. Lattice problem with on-site Coulomb interaction U is mapped
onto the problem of a single interacting site in an effective mean field ζ0(τ− τ ′).

28
Figure 2: Possible scheme for an LDA+DMFT calculation.
By combining DFT with DMFT within the so-called “LDA+DMFT” approach one ends up with a powerful approach to tackle strongly correlated materials on a realistic level. Several physical as well as technical aspects need however to be clarified and worked out in order to successfully merge electronic band-structure calculations with the DMFT method. For instance, the interacting Hamiltonian has to be defined and a suitable local-basis representation for the description of the local self-energy has to be introduced. Concerning the latter, Wannier-like techniques and projection methods have been applied, reinforcing the chemistry viewpoint in modern electronic structure calculations. In this talk, we will provide an introduction to the DMFT method as well as the relevant phenomenology of realistic many-body systems. Starting from the fundamental single-orbital Hubbard model Hamiltonian, we will elaborate on the DMFT description towards the application to demanding strong correlation problems in realistic materials systems. Recent developments in this very active field of research in theoretical condensed matter physics will be presented.

29
First-principles Investigations of Point Defects
Chris G. Van de Walle
Materials Department, University of California, Santa Barbara, CA 93103, USA
[email protected] Defects and impurities are often decisive in determining the physical properties of most materials. They control the conductivity of the material, and point defects also mediate diffusion. Experimental defect identification and characterization is typically difficult and indirect, usually requiring an ingenious combination of different techniques. First-principles calculations have emerged as a powerful approach that complements experiments and can even serve as a predictive tool. In these lectures I will describe the general methodology for performing defect calculations [1], addressing issues of geometry (supercells versus clusters), choice of first-principles technique, and computational issues such as convergence with respect to various parameters. Density functional theory (DFT), usually in conjunction with pseudopotential or projector augmented wave potentials, has emerged as the most commonly used first-principles approach for defects. Figure 1 shows the charge density of the defect state for an oxygen vacancy in ZnO.
Figure 1: Charge density of the defect-induced gap state for an oxygen vacancy in ZnO. In the neutral charge state, the defect state is occupied with two electrons. The isosurface corresponds to 10% of the maximum. The background shows the ZnO wurtzite lattice.

30
This approach has proven its value as an immensely powerful technique for assessing the structural properties of defect. Minimization of the total energy as a function of atomic positions yields the stable structure, including all relaxations of the host atoms, and most functionals [including the still most widely used local density approximation (LDA)] all yield results within reasonable error bars. Quite frequently, however, information about electronic structure is required, i.e., the position of defect levels that are introduced in the band gap of semiconductors or insulators. Since DFT in the LDA or generalized gradient approximation (GGA) severely underestimates the gap, the position of defect levels is subject to large error bars and cannot be directly compared with experiment. In turn, this affects the calculated formation energy of the defect, which determines its concentration. I will discuss particular methods that improve the description of band gaps, leading to results that can be directly compared to experiments on a quantitative level. These include LDA+U, screened hybrid functionals, the quasiparticle GW method, and the use of modified pseudopotentials. Advantages and limitations of these methods will be illustrated with examples and comparisons with experiment. Another issue that may occur in defect calculations is related to the geometry in which the calculations are performed. Typically, one wishes to address the dilute limit in which the defect concentration is low and defect-defect interactions are negligible. When performing calculations for charged defects in the supercell approach, long-range interactions may affect the calculated formation energies and transition levels. We have recently developed an approach based on a rigorous treatment of the electrostatic problem that outlines the conditions of validity of certain approximations and provides explicit expression for the quantities to be evaluated [2]. Work performed in collaboration with C. Freysoldt, A. Janotti, G. Kresse, J. Lyons, J. Neugebauer, P. Rinke, M. Scheffler, A. Singh, J. Varley, and J. Weber. [1] C. G. Van de Walle and J. Neugebauer, J. Appl. Phys. 95, 3851 (2004). [2] C. Freysoldt, J. Neugebauer, and C.G. Van de Walle, Phys. Rev. Lett. 102, 016402 (2009).

31
"Time-dependent DFT"
Kieron Burke
UC Irvine, Depts of Physics and of Chemistry
In this short class, I will quickly review basics of TDDFT, with an emphasis on excitations.

32
Impurity States
Hannes Raebiger Yokohama National University, Yokohama, Japan
[email protected] While defects and impurities semantically convey a somewhat negative meaning of ‘dirty’ or ‘imperfect’, semiconductor applications, including computer processors, photovoltaic cells, and various sensor devices etc. would be completely useless without such ‘dirt’ and ‘imperfections’. What makes semiconductors unique compared with metals or insulators is that tiny amounts of imperfections (around 1 ppm) can dramatically alter their conductive, optical, and even magnetic properties. Moreover, the abundance of such defects and impurities is rather simple to control via tuning the chemical environment during growth, and the thermochemical formation of various defects can be accurately predicted from quantum mechanical calculations. To asses the relevance of certain defect types, I will give an overview of density-functional theories to calculate defect formation enthalpies. Ideally, it would be desirable to carry out such calculations within self-interaction free total energy functionals, but in practice this is rarely possible. To this end, I will discuss both ‘post-processor’ corrections and simple nonlocal corrections to local-density approximations (LDA), which yield accurate thermochemical quantities for a similar CPU cost as an LDA-calculation. This is followed by a description of the relevant thermodynamics in order to actually assess the defect abundancies together with induced carrier concentrations. The main focus of this lecture is in understanding the electronic spectra of various impurity configurations, and how these impurity states ultimately affect the electronic, optical, and magnetic properties of the samples. While first principles calculations can be used to accurately predict these spectra, this does not entail understanding the physical mechanisms that lead to the specific electronic configurations. Here, symmetry considerations, together with simple tight binding theories are a useful tool – in parallel to ab initio calculations – to provide an in-depth understanding of the origin of specific impurity states. Here, I give an overview of the different types of defects one encounters. First of all, it is customary to distinguish between intrinsic and extrinsic defects, which further can be classified as shallow or deep. I will discuss in terms of simple LCAO models under what conditions intrinsic defects are shallow or deep [1], and show examples of oxide materials relevant in the design of photovoltaic cells. Finally, I will discuss the microscopic theory of charge regulation [2], which enables deep defects to exhibit multiple ‘charged states’, thus providing e.g. transparent materials with new catalytic functionalities relevant for photoelectrochemical water splitting for hydrogen fuel production, and is a driving force for microscopic phase separation (see e.g. Ref. 3) and defect gettering. [1] H. Raebiger, S. Lany, and A. Zunger, Phys. Rev. B 76, 045209 (2007). [2] H. Raebiger, S. Lany, and A. Zunger, Nature 453, 763 (2008). [3] H. Raebiger, A. Ayuela, and R. M. Nieminen, J. Phys.: Condens. matter 16, L457 (2004).

33
FRIDAY
July 2st

34
First-principles calculations of vibrational thermodynamics at high
temperatures
Vidvuds Ozolins
Department of Materials Science and Engineering,
University of California, Los Angeles, California 90095-1595, USA
E-mail: [email protected]
We will review the basic principles and methods of calculating vibrational contributions to the
free energies of solids at high temperatures. The harmonic and quasiharmonic phonon
theories, both within the linear response and supercell force constant method, will be
introduced together with the key applications from the literature. In the strongly anharmonic
limit where the quasiharmonic theory becomes inadequate, we will explain the most popular
state-of-the-art methods based on thermodynamic integration techniques. The case of high-
temperature metastable phases and relation to the CALPHAD calculations of phase diagrams
will be covered in detail. The lecture is expected to be useful for those who are interested in
modeling the thermodynamic properties of solid phases and thermodynamic driving forces for
structural transformations in pure elements and alloys using first-principles density-functional
theory techniques.

35
Linear scaling methods in electronic structure calculations
Pablo Ordejón
Centre d’Investigació en Nanociència i Nanotecnologia-CIN2 (CSIC-ICN), Campus UAB,
08193 Bellaterra, Spain
I will review the basic ideas behind the so-called "linear scaling" or Order-N methods for
electronic structures [1]. These methods were developed to speed-up the calculation of
electronic properties of large systems, overcoming the superlinear scaling imposed by the
standard diagonalization methods in solving a one-electron Hamiltonian. These linear scaling
methods always involve physically motivated approximations (mainly, localization ideas),
which imply that the solutions are approximate and that, therefore, errors must be carefully
tested and controlled. Linear scaling methods have also been developed to compute the
Hamiltonian matrix in Density Functional methods, and these will also be presented in this
talk.
[1] A review of some of these methodologies can be found, for example, in the articles: P. Ordejón, “Order-N tight-binding methods for electronic-structure and molecular dynamics”, Comp. Mat. Sci. 12, 157 (1998); and S. Goedecker,“Linear scaling electronic structure methods”, Rev. Mod. Phys. 71, 1085 (1999)

36
The SIESTA method: an efficient tool in materials science
Daniel Sánchez-Portal
Centro de Física de Materiales CSIC-UPV/EHU, Materials Physics Center (MPC, and
Donostia International Physics Center (DIPC),
Paseo Manuel de Lardizabal 4-5, 20018 Donostia, Spain
Email: [email protected]
The SIESTA method is an approach to compute the electronic properties and perform atomistic simulations of complex materials from first principles [1-7]. Large systems, with an unprecedented number of atoms, can be studied while keeping the computational cost at a reasonable level. The SIESTA code is freely available for the academic community (visit http://www.icmab.es/siesta), and this has made it a widely used tool for the study of materials. It has been applied to a large variety of systems including surfaces, adsorbates, nanotubes, nanoclusters, biological molecules, amorphous semiconductors, ferroelectric films, low-dimensional metals, etc. Here I will briefly present some of the ideas and algorithms behind the capabilities of the code, and present some recent applications in materials science. The capabilities of SIESTA are exemplified in Figure 1, where the distribution of the highest occupied molecular orbital (HOMO) of a short DNA chain (polyA-polyT) coupled to two carbon nanotubes has been plotted. The simulation cell contains 580 atoms. The output of this calculation will be used as an input to calculate transport using the TranSIESTA utility as will be explained by P. Ordejón in his talk. [1] The main team of SIESTA developers is composed by E. Artacho, J. M. Cela, J. Gale, A. García, J. Junquera, R. M. Martin, P. Ordejón, D. Sánchez-Portal and J. M. Soler. It is also necessary to acknowledge the generous work of many people who have contributed to the development of the code over the years with their implementations, ideas, suggestions and bug reports. [2] P. Ordejón, E. Artacho, and J. M. Soler, Self-consistent order-N density-functional calculations for very large systems, Phys. Rev. B 53, R10441 (1996) [3] D. Sánchez Portal, P. Ordejón, E. Artacho, J. M. Soler, Density-functional method for very large systems with LCAO basis sets, Int. J. Quantum Chem. 63, 453 (1997) [4] J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón, D. Sánchez-Portal, The SIESTA method for ab initio order-N materials simulations, Journal of Physics: Condensed Matter 14, 2745 (2002) [5] J. Junquera, O. Paz, D. Sánchez-Portal, E. Artacho, Numerical orbitals for linear-scaling calculations, Physical Review B 64, 235111 (2001) [6] D. Sánchez-Portal, P. Ordejón, E. Canadell, Computing the properties of materials from first principles with SIESTA, Bonding and Structure 113, 103 (2004) [7] E. Artacho , E Anglada , O Diéguez , J D Gale , A García , J Junquera , R M Martin , P Ordejón , J M Pruneda , D Sánchez-Portal and J M Soler, The SIESTA method; developments and applicability, J. Phys.: Condens. Matter 20 064208 (2008)

37
Figure 1. Distribution of the highest occupied molecular orbital (HOMO) of a short DNA chain (polyA-polyT) coupled to two carbon nanotubes as calculated with SIESTA. The simulation cell contains 580 atoms. Later on, the output of this calculation will be used as the input to calculate electronic transport in this system using the TranSIESTA utility. (Courtesy of Prof. P. Ordejón)

38
Hydrogen Storage in Nanoporous Carbons
Julio A. Alonso Departamento de Física Teórica, Atómica y Optica
University of Valladolid 47011 Valladolid, Spain
Hydrogen is a candidate to replace gasoline as a fuel in cars. Prototypes of electric cars in which the electric power is generated by the reaction of hydrogen with atmospheric oxygen in a fuel cell have already been built. The process is noncontaminant and only produces water. Hydrogen has a high energy density by mass, 120 MegaJoules/gram, nearly three times that of gasoline. However, hydrogen is a gas, and consequently its energy density by volume is much smaller than the value of 35 MegaJoules/liter of gasoline. Even liquid hydrogen has a volumetric energy density that is only about one fourth of that value. In the present prototype cars hydrogen is stored as a compressed gas in the tank of the car. This storage method is not very efficient. A few years ago the U.S. Department of Energy (DOE) established a hydrogen storage target for the year 2010 of 6 per cent of the storage system weight for onboard automotive applications. This has motivated a lot of research on light materials that could store enough hydrogen to fulfill the target. Computational materials science can help experimentalists in searching for materials with promising storage properties. One type of materials that is currently investigated, and which is the focus here, is the class of porous carbons. These light materials, cheap and easy to produce, contain a network of interconnected pores of nanometric size. Molecular dynamics simulations using effective many-atom potentials indicate that the walls of the pores have the form of planar and curved graphitized ribbons with many defects and open terminations [1]. Nanoporous carbons have large specific surface areas, several 100 m2 per gram. The potential of nanoporous carbons to store hydrogen is based on the physisorption of molecular hydrogen on the graphitic surface. The conditions for automotive applications require reversible adsorption/desorption cycles, and this establishes strict conditions on the adsorption enthalpies.
Figure 1. Potential energy curves for the interaction of a hydrogen molecule with a graphene layer, clean and
doped with lithium [2].
A graphene slitpore, consisting in two parallel graphene layers separated a certain distance d, gives a simple model for the pores existing in nanoporous carbons. The measured storage capacities of nanoporous carbons are often analyzed by assuming the pores in the material to have the form of slitpores. The adsorption of H2 on a planar graphene layer has been studied

39
using DFT. Figure 1 shows the interaction potential calculated using the LDA approximation. The binding energy is 70 - 90 meV/molecule. The interaction energy arises from two main contributions, one attractive and one repulsive. The sharp repulsive wall is due to the short-range repulsion that develops when the closed electronic shell of the hydrogen molecule overlaps substantially with the electron gas of the substrate. This contribution is sensitive to the local electron density sampled by the hydrogen molecule in its approach to the graphene layer, and consequently, it is sensitive to the adsorption site. The attractive contribution is mainly due to electronic exchange and correlation effects. The minimum of the interaction
potential energy in Fig. 1 occurs at 2.75o
A ; then, in a slitpore of width about 6o
A the hydrogen molecule interacts optimally with the two parallel surfaces, increasing the binding energy by a factor of two. That is, the binding energy of the molecule in a slitpore of that size will be Eb = 150 - 200 meV. Cylindrical nanopores can be modelled as the inner channel of a single wall carbon nanotube (SWCNT). The interaction potential between a hydrogen molecule and a (5, 5) nanobube,
whose radius is 6.44 a.u. (1o
A = 1.88 a.u.), is plotted in Figure 2. The different curves correspond to several orientations of the molecule relative to the nanotube. The largest binding energy, 170 meV, occurs for the molecule inside the SWNT, and this is due to the curvature effect. The binding for the molecule inside is sensitive to the radius. For instance, Eb = 120 meV for a (6, 6) nanotube. The difference gives a hint that narrow cylindrical pores may enhance the storage of hydrogen.
Figure 2. Interaction energy of a hydrogen molecule and a (5, 5) single wall carbon nanotube as a function of the
distance to the nanotube axis. The vertical line represents the nanotube wall. The left curves correspond to the molecule inside the nanotube, and the right curves to the molecule outside. Different curves indicate different
orientations and sites of approach.
The storage capacity of planar slitpores can be calculated using a quantum thermodynamical model [3, 4]. The model takes into account the quantum behavior of the hydrogen molecules confined in the volume of the slitpore and uses the experimental equation of state of hydrogen. The results of the model for storage at 298 K are in good agreement with measurements for activated carbons [5]. The conclusion from the theoretical calculations is that the DOE goal for the gravimetric storage capacity appears to be accessible for nanoporous carbons at 77 K, that is, at low temperatures. Of course, those temperatures prevent applications in the car industry, although other applications requiring low temperatures may be possible. At room temperature and for optimized slitpore sizes, a gravimetric storage capacity of 3.1 % is predicted at pressures of 10 MPa (see Fig. 3). The curves in this Figure indicate that the DOE target is not reached at room

40
temperature. However, the storage capacity increases with the external pressure and this indicates a possible path to enhance the storage.
Figure 3. Calculated gravimetric capacities of graphene slitpores as a function of the pore width at 300 K, and different pressures: 0.1, 1, 5 and 10 MPa [4]. The DOE target is indicated by he continuous horizontal line. The
dashed horizontal line represents a coverage of one H2 molecule per two hexagons.
Doping with some impurities represents a promising strategy to enhance the adsorption binding energies of molecular hydrogen to graphitized surfaces. Cabria et al. [2] have performed calculations comparing the physisorption of H2 on pure and Li-doped planar and curved graphitic surfaces. The Li atom transfers electronic charge to the carbon layer, and the presence of the partially charged Li atom polarizes the nearby H2 molecules, enhancing their adsorption binding energies in a factor of 2. This is shown in Figure 1 for the case of a planar graphene layer. The combination of the confinement effect of the pore and the polarization effect due to impurities increases the adsorption binding energies, which reach values of more than 300 meV per molecule [6], already approaching the magnitude required for efficient cyclic adsorption-desorption under normal operating conditions. This may be a promising line of research. Acknowledgements: This work was supported by MICINN (Grant MAT2008-06483-C03-01) and Junta de Castilla y León (Grants VA017A08 and GR23). References 1. M. J. López, I. Cabria and J. A. Alonso, manuscript in preparation. 2. I. Cabria, M J. López and J. A. Alonso, J. Chem. Phys. 123, 204721 (2005) 3. S. Patchkovskii, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Hrine and G. Seifert, Proc. Nat. Acad. Sci. USA 102, 10439 (2005). 4. I. Cabria, M. J. López and J. A. Alonso, Carbon 45, 2649 (2007). 5. M. Jorda-Beneyto, F. Suarez-García, D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon 45, 293 (2007). 6. I. Cabria, M J. López and J. A. Alonso, J. Chem. Phys. 128, 144704 (2008).

41
GW Quasiparticle Calculations
Patrick Rinke
Fritz-Haber-Institut der Max-Planck-Gesellschaft
Faradayweg 4–6, 14195 Berlin
In material science excited states are ubiquitous. For certain applications such as
optoelectronic devices the excitation process is of primary concern. More generally, our
understanding of the response of a material to an external perturbation such as excitation by
light or electrons is fundamental for characterizing its properties and utilizing its potential for
new applications.
In this lecture I will focus on photoemission spectroscopy (PES) and its inverse counterpart
(IPES). The success of PES and IPES owes much to the interpretation of the photo-electron
spectra in terms of single-particle-like excitations or quasiparticles. I will introduce the
connection between photoemission spectroscopy and many-body perturbation theory
(MBPT). For solids MBPT in Hedin’s GW approximation, where G refers to the Green’s
function and W to the dynamically screened Coulomb interaction, has become the method of
choice for an ab initio calculation of the quasiparticle energy spectrum. The GW approach is
best know for ameliorating the band-gap problem of Kohn-Sham density-functional theory,
but is of course not restricted to the calculation of quasiparticle band gaps. I will review the
current state of the art and illustrate practical aspects of GW calculations before discussing
open questions and future developments.

42
Advanced concepts in SIESTA
Pablo Ordejón
Centre d’Investigació en Nanociència i Nanotecnologia-CIN2 (CSIC-ICN), Campus UAB,
08193 Bellaterra, Spain
In this talk, I will describe some of the less standard calculations which are available in the
SIESTA package [1]. In particular, I will describe the TranSIESTA utility [2], which allows
the calculation of non-equilibrium transport properties in nanoscale devices. I will also
present a QM/MM methodology [3,4] that has been recently implemented in SIESTA, in
which the full DFT description of a part of the system is combined with classical potentials
for the rest of the atoms. This technique is useful for the study of very large systems in which
chemical reactions only take place in a small region, which must be treated at the QM level.
[1] J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón, D. Sánchez-Portal, The
SIESTA method for ab initio order-N materials simulations, Journal of Physics: Condensed Matter 14,
2745 (2002)
[2] Mads Brandbyge, José-Luis Mozos, Pablo Ordejón, Jeremy Taylor, and Kurt Stokbro,
Density-functional method for non-equilibrium electron transport, Phys. Rev. B 65, 165401 (2002)
[3] A. Crespo, D. A. Scherlis, M. A. Marti, P. Ordejón, A. E. Roitberg and D. A. Estrin, A DFT based
QM-MM Approach Designed for the Treatment of Large Molecular Systems: Application to
Chorismate Mutase, J. Phys. Chem. B 107, 13728 (2003)
[4] C. Sanz, A. García, P. Ordejón, to be published.

43
Theory of gas/surface dynamics
Ricardo Díez Muiño
Centro de Física de Materiales, Centro Mixto CSIC-UPV/EHU, San Sebastián, Spain, and
Donostia International Physics Center DIPC, San Sebastián, Spain
Understanding and mastering the dynamics of elementary reactive processes at surfaces is a
basic ingredient to control many physical and chemical processes. In particular, metal
surfaces are effective chemical agents capable of adsorbing and/or dissociating molecules
impinging from the gas phase. When the molecules approach the surface, intramolecular
chemical bonds can break down and new ones be formed with the surface. Over the last years,
the combination of experimental molecular-beam techniques and refined theoretical
calculations based on ab-initio methods have led research on the field to a new stage, in which
detailed investigations of the kinetics and dynamics of molecular reactivity at surfaces are
possible.
In this talk, we will describe the use of first-principles electronic structure calculations to
describe the details of the interaction between small diatomic molecules and metal surfaces.
We will cover different methodologies to build accurate multidimensional potential energy
surfaces (PESs), based on the adiabatic approximation. Once the PES of the system is known,
we will show how the dynamics of several processes can be analyzed by solving classical or
quantum equations of motion. Finally, we will discuss the possible relevance of non-adiabatic
effects in elementary reactive processes at surfaces, with special emphasis on the excitation of
electron-hole pairs in the metal surface.
.
References
[1] M. Alducin, R. Díez Muiño, H.F. Busnengo and A. Salin, Phys. Rev. Lett. 97 056102
(2006).
[2] J.I. Juaristi, M. Alducin, R. Díez Muiño, H.F. Busnengo and A. Salin, Phys. Rev. Lett.
100 116102 (2008).

44
SATURDAY
July 3st

45
Interaction between ab initio and Calphad approaches
Pavel A. Korzhavyi
Department of Materials Science and Engineering
Royal Institute of Technology (KTH)
100 44 Stockholm, SWEDEN
Abstract
Density functional theory enables one to obtain the ground-state energy of any given atomic
configuration (static as well as dynamic) through self-consistent ab initio calculations. This
possibility is of direct interest for the scientific community working with CALculations of
PHAse Diagrams (CALPHAD) and other types of empirically-based thermodynamic
modeling. However, the interaction between the two communities, one doing ab initio
calculations and the other performing Calphad modeling is not limited to a straightforward
use of ab initio-calculated total energies in place of experimental data. A mutually enriching
information exchange is actually taking place, and that involves several levels of abstraction:
Plain numerical data, their interpretation in the form of physical and mathematical models, as
well as general concepts and approaches in modeling are being exchanged, discussed and
developed jointly. Some examples of such collaborative projects, dealing with modeling of
industrially-relevant materials, will be discussed.

46
Liquids
Dario Alfè
This lecture will focus on how to obtain various properties of liquids using computer simulations, and in particular how to calculate melting properties. The initial part of the lecture will introduce the main concepts that help to characterise liquids, and how to distinguish liquids from solids and gases. These will include importance of collisional processes and short range correlations, as well as lack of long range order. It will be mentioned that the strong repulsions at short distances, caused by the overlap of the valence electronic charges, is the main ingredient in the determining the structure of liquids. Long range, smoother, attractive forces play a minor role for the structure, although they of course provide cohesive energy. I will briefly mention the source of these long range interactions, which in some cases are due to dispersive multipole-multipole interactions caused by spontaneous fluctuations in the electron densities. Combining these concepts I will introduce the Lennard-Jones potential. I will introduce the concepts of reduced density and reduced temperature, in terms of typical internuclear distances and internuclear energies, and mention that in liquids these two quantities are both of order ~ 1. Many properties of liquids can be obtained by a detailed microscopic analysis of the motion of the constituents atoms. The computer simulation of liquids goes back to the pioneering molecular dynamics (MD) work of Alder and Wainwright (1959). I will recall the basis concepts of molecular dynamics, including the solution of the Newton’s equation of motion, and how to solve them numerically on a computer: the Verlet algorithm. Simulation boxes, periodic boundary conditions to reduce surface effects. Thermodynamic properties of a system can be expressed as averages of appropriate functions of the coordinates and momenta of the constituent particles (with some important exceptions). Averages can be taken along MD trajectories. For example, the temperature of the system is proportional to the time average of the kinetic energy of the constituent particles, averaged over the total number of particles. Alternatively, ensemble averages are taken by generating a collection of imaginary systems, all representative of the system of interest in the sense that they have the same macroscopic properties, in which the position and the momenta of the particles are distributed according to some appropriate probability density. Recall the most important ensembles. Microcanonical (NVE), constant number of particles N, volume V and energy E. This ensemble is the natural outcome of a MD simulation for an isolated system, where the Hamiltonian is a constant of the motion. It is defined by the hypersurface in phase space of constant energy E. Canonical (NVT), constant N, V and temperature T. Isothermal-isobaric (NpT), constant N, pressure p and T. All these ensembles can be generated using Monte Carlo methods, or MD with appropriately modified Lagrangians. Introduce the concept of extended systems to include barostats and thermostats in MD. Under the hypothesis of ergodicity (after a long enough time a trajectory will have visited an equal number of times all points on the appropriate hyper-surface in phase space), time averages are equivalent to ensemble averages, and therefore it is possible to use MD to

47
evaluate thermodynamic properties in each of the ensembles mentioned above. The advantage of MD over the Monte Carlo method is that with MD one has access also to dynamical properties, like diffusion and correlation functions. Main ingredients in MD are forces on the atoms and energy of the system. Classical potentials versus ab-initio methods (density functional theory, quantum Monte Carlo). Brief mention of the Car-Parrinello method (1985), extended system to include the electronic degrees of freedom in the Lagrangian and perform ab-initio molecular dynamics (AIMD) simulations. Born-Oppenheimer AIMD. Example of static and dynamical properties of liquids calculated with AIMD: radial distribution functions, structure factors, diffusion coefficients, viscosity. Introduction to melting. Methods to calculate melting curves, including free energy calculations and coexistence of phases. Discussion of advantages and disadvantages of the methods: coexistence is fairly straightforward, but it needs large simulation cells (~ 1000 atoms), only recently become possible with AIMD. Show examples: Al, MgO (important for the mantle of the Earth), Fe (important for the Earth’s core) and Li (peculiar behaviour of the melting curve which displays a maximum). The free energy method is more involved and intricate, but has the advantage that can be used on fairly small systems (64 atoms) if the reference systems are good. Moreover, free energies also give access to a wealth of thermodynamic properties. I will discuss the method of thermodynamic integration to calculate free energies, and the importance of building a hierarchy of good reference systems, which are at the base for the success of the method. Methods to construct good reference systems. Free energies can be calculated for a range of pressures and temperatures, and therefore give access to whole melting curves, rather than to a set of melting points obtained in each coexistence simulation. In the spirit of building a hierarchy of reference systems, I will discuss recent applications of the quantum Monte Carlo method to the calculation of free energies, and in particular to the calculation of the melting temperature of iron at Earth’s core conditions.

48
Using ab initio methods to predict thermodynamic properties of
metals
T. Hickel, B. Grabowski, F, Körmann, A. Dick, J. Neugebauer
Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Str. 1, 40237 Düsseldorf, Germany
Although density functional theory (DFT) was originally developed as a ground state theory, recent
methodological developments extend it to predict thermodynamic properties of materials at
temperatures T > 0K. The key quantity for this purpose is the Helmholtz free energy, since its
knowledge provides not only access to derived properties like the heat capacity, but also to phase
transition temperatures. Within this presentation we will therefore discuss the capabilities and
accuracy of present day implementations (xc-functionals) of DFT in determining all relevant
contributions to free energies of metals. We will repeat the concepts of the quasiharmonic
approximation [1], which yield the dominant contribution to the free energy, and of extensions to
include anharmonic lattice vibrations [2]. For magnetic materials such as iron proper quantum-
mechanical treatments of magnetic excitation will be presented, pointing also out that classical
simulations are often not sufficient [3]. The focus of the talk will be on realistic material systems, for
which we will show that an integrated approach, combining these effects, leads to highly accurate free
energies. It will be shown that the predictive power of the approach can be estimated by comparing the
deviations between different xc-functionals. For the examples aluminum, cementite and a shape
memory alloy [4] we will demonstrate that the introduced ab initio methods can be extremely helpful
to identify the relevance of the individual free energy contributions for thermodynamic trends and
phase transitions.
[1] B. Grabowski, T. Hickel and J. Neugebauer, Phys. Rev. B 76, 024309 (2007).
[2] B. Grabowski, L. Ismer, T. Hickel and J. Neugebauer, Phys. Rev. B 79, 134106 (2009).
[3] F. Körmann, A. Dick, B. Grabowski, B. Hallstedt, T. Hickel, J. Neugebauer, Phys. Rev. B
78, 235302 (2008).
[4] M.A. Uijttewaal, T. Hickel, J. Neugebauer, M.E. Gruner, P. Entel, Phys. Rev. Lett. 102,
035702 (2009).

49
Master Class on thermodynamics: How to derive electronic and vibronic
free energy surfaces from ab initio?
Blazej Grabowski
Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany
An accurate description of thermodynamic quantities at finite temperatures is a key ingredient in designing and processing new materials with optimized properties. Traditional approaches to describe thermodynamic quantities have been based on physical understanding derived from the meso- and macroscopic scale. Due to the high complexity real materials can exhibit at microscopic scales, it became clear that such approaches, although extremely valuable, are facing fundamental limits and that further progress is only possible by a combination with ab
initio based methods. One such ab initio method, the density functional theory (DFT), has turned to be very accurate yet computationally efficient in describing material properties at T=0 K. The extension to finite temperatures is however accompanied by conceptual and computational challenges. The conceptual difficulty arises from the fact that classical DFT calculations provide only ground-state properties and neglect thermodynamic excitations. The additional computational effort to treat these excitations is significant due to cumbersome calculations required to sample the enlarged phase space with high demands on the accuracy. It is therefore of crucial importance to provide and advance efficient statistical concepts that allow to obtain DFT based finite temperature properties efficiently but still with a high numerical accuracy. In this master class, we will provide an interactive introduction to the current state-of-the-art methods to calculate finite temperature material properties based on DFT. We will focus on the calculation of the central thermodynamic quantity, the free energy surface, which contains all thermodynamic information needed to derive any other quantity. In particular, we will discuss the practical procedures developed to include electronic and vibronic excitations and show that these contributions constitute a dominant part of the free energy. For the latter, the quantized harmonic lattice vibrations (phonons) play a decisive role and their computation will be therefore discussed intensively. We will also show how one can go beyond the usually applied non-interacting phonon description by the inclusion of so called anharmonic effects. It is particularly in this domain where efficient statistical methods are crucial and where still a lot of space for improvement exists. Based on a flexible interactive format of the master class, practical knowledge will be provided to the participants allowing them to make first steps in the quickly developing yet challenging field of ab initio finite temperature calculations.

50
Posters

51
Tuning The Kohn Anomaly In The Phonon Dispersion Of Graphene By
Interaction With A Metallic Substrate.
Adrien ALLARD1, Ludger Wirtz1
1 Institute for Electronics, Microelectronics, and Nanotechnology // CNRS-UMR 8520 // Lille,
France
The phonon dispersion of graphene is known to display two strong Kohn Anomalies (kinks) in the highest optical branch (HOB) at the high-symmetry points Gamma and K. The phonon slope around the Kohn anomalies is related to the electron-phonon-coupling (EPC) with the graphene Pi bands. We show that this EPC which has strong impact, e.g., on Raman scattering and electron transport can be strongly modified due to interaction with a metallic substrate. For graphene grown on a Ni(111) surface, a total suppression of the Kohn anomaly occurs: the HOB around Gamma and K becomes completely flat. This is due to the strong hybridization of the graphene Pi bands with the Nickel d bands which lifts the linear crossing of the Pi bands at K. For other metallic substrates, where the distance between the graphene sheet and the substrate is larger, hybridization is much less pronounced and the Kohn anomaly is only weakly perturbed. From experimental phonon dispersions one can therefore draw conclusions about the interaction strength between graphene and its different substrates.

52
Ab initio prediction of thermodynamic data for selected phases of the Al-
Mg-Si-Cu system
Albert Glensk1, Blazej Grabowski1, Tilmann Hickel1, Jörg Neugebauer1
1Max-Planck Institut für Eisenforschung GmbH, Max-Planck-Strasse 1, D-40237 Düsseldorf, Germany
Al-Mg-Si-Cu alloys are widely used in engineering applications due to their excellent mechanical properties. The adjustment of processing routes is one of the most decisive steps for tailoring properties to specific needs and can e.g. significantly increase the strength of Al-Mg-Si-Cu alloys [1]. To address this challenge quantitative simulations (like the CALPHAD approach) need exact thermodynamic potentials, which are nowadays mostly gained from experiment. First principles theoretical calculations emerge as an alternative for reliable thermodynamic functions in cases of non-existent experimental data or in regions of phase boundaries where the reliability of experiments is limited due to the transient nature of metastable phases. The primary goal of this project is to provide highly accurate ab initio free energies as a function of temperature and molar volume for selected binary, ternary and quaternary phases of the Al-Mg-Si-Cu system. Based on this thermodynamic potential Helmholtz and Gibbs free energies, heat capacities, thermal expansion and vacancy concentrations will be derived. In particular finite temperature effects due to lattice vibrations will be considered. Our results show that converged phonon dispersions are indispensable in order to gain highly accurate free energies: Compared to total energies though, phonon energies respond much more sensitive to changes in relevant parameters. Systematic studies of these dependencies for hcp Mg will be presented. For complex phases, such as GP-zones, ß'' and ß phases , static total energy calculations and enthalpies of formation have been obtained and will be discussed.
[1] H. Zhang, Y. Wang, S.L. Shanbg, C.Ravi, C. Wolverton, L.Q. Chen, Z.K. Liu,
CALPHAD 34 (2010) 20 - 25

53
HYBRID FUNCTIONAL STUDIES OF MULTIFERROICS Alessandro Stroppa1*, Silvia Picozzi1
1 SPIN-CNR, University of l'Aquila, Italy *Email address: [email protected]
Multiferroics, i.e. materials where ferroelectricity and magnetism coexist, are considered as a "hot topic" in condensed matter physics (h-index for multiferroics: 49; almost 400 published articles in 2009). From the computational point of view, their description poses serious problems. These compounds are often (strongly) correlated materials, involving d- and f-electronic charge with significant spatial localization. First-principles density functional theory (DFT) calculations in the most commonly applied approximations (LDA, GGA) are not accurate enough to describe the complex electronic structure of these materials, where spin-charge-lattice degrees of freedom are strongly linked. Recently, hybrid Fock exchange/density functional theory functionals have received great attention in the solid state community. In particular, the HSE (Heyd-Scuseria-Ernzerhof) screened hybrid functional, widely applied in solid state calculations[1],is becoming a new computational paradigm for accurate electronic structure calculations of complex materials. In fact, HSE has been recently applied to solid states systems and has shown good results with respect to standard local DFT approaches, especially for insulating and correlated systems. Despite this, very few studies exist in the multiferroic field, and clearly much work is needed in order to assess the performance of HSE in this recent field of material research. In this framework, we focus on two prototypical systems such as BiFeO3 (BFO) and HoMnO3 (HMO). We perform an accurate study of the structural, electronic, magnetic and ferroelectric properties based on the comparison of the performance of standard DFT approaches, and more advanced and recent hybrid-DFT, such HSE. For BFO and HMO, by comparing our calculations with available experimental data, our study points towards a truly realistic description of multifunctional materials, such as multiferroics. For BFO, we also included state-of-the-art GW calculations, which, to the best of our knowledge, have not yet been done. ACKNOWLEDGEMENTS The research leading to these results has received funding from the European Research Council under the European-Community, 7th Framework Programme – FP7 (20072013)/ERC Grant Agreement n. 203523. Computational support by Caspur Supercomputing Center in Roma is gratefully acknowledged. REFERENCES [1] Heyd, J. Scuseria, G.E. and Ernzerhof, M. (2003) Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys.,118, 8207-8216 [2] Stroppa, A. and Picozzi, S (2010), Hybrid functional study of proper and improper multiferroics, Phys. Chem. Chem. Phys., 12, 5405-5416

54
Effect of palladium and titanium impurities
on hydrogen solubility in bcc iron
M.S. Rakitin, A.A. Mirzoev South Ural State University, Chelyabinsk, Russia
E-mail: [email protected] Hydrogen is known to cause some types of structural defects in iron and steels which can lead to cracks and embrittlement of the materials. Solubility of hydrogen decreases with cooling. As a result high pressure inside the materials can cause their failure. One of the ways to avoid such negative effect is to add impurities of Pd or Ti during steel production, so hydrogen atoms can be trapped by them. We have made an attempt to understand the nature of the phenomenon and eliminate disagreements about preferable position of the impurities to trap hydrogen. Several series of ab initio calculations have been performed for ferromagnetic bcc iron with hydrogen and small impurities of Pd and Ti with structural relaxation. We used WIEN2k [1] program realization of DFT method within the generalized gradient approximation of the PBE form for electron exchange and correlation. We employed LAPW method, which is an all-electron DFT technique. According to our convergence tests, we used a kinetic energy cutoff of 340 eV for all calculations. Structural relaxations were performed until forces on each atom were below 1 Ry/a.u. We used a k-point sampling of 24 for the Fe53MeH supercell (Me=Pd, Ti) and experimental lattice parameter of bcc Fe (5.4169 a.u.). In our earlier work we simulated dissolution of hydrogen in pure Fe54 with almost identical parameters and obtained results which are in good agreement with the results of Jiang and Carter [2]. Namely, we found the tetrahedral site of bcc iron to be more stable than the octahedral site for hydrogen dissolution in Fe54H supercell and dissolution energy of hydrogen in the tetrahedral site is 0.30 eV and 0.19 eV for the unrelaxed and relaxed structures respectively with the experimental value of 0.296 eV [3]. In the present work an atom of hydrogen was placed in the tetrahedral site. Case of hydrogen in the octahedral site hasn’t been studied due to instability of this site. Atoms of Pd and Ti have been inserted separately in iron lattice as substitutional impurities. We were varying position of each impurity relative to hydrogen atom. Figure 1 displays the results of the calculations for the unrelaxed structures. Both the impurities trap hydrogen atom in their third coordination spheres. Atom of Pd has the most effect on hydrogen trapping in bcc iron. For the relaxed structures of the Fe53PdH and Fe53TiH systems Figure 2 clearly shows that palladium traps hydrogen atom in its second coordination sphere, when Ti keeps on holding H in its third sphere. Pd cannot trap H in the first coordination sphere due to its instability. The work is supported by Federal Aim Program (State contract No. 1939 from October 29, 2009) and grant No. 2.1.1/1776.

55
Fig. 1. Dependence of dissolution energy of hydrogen on distance from Pd and Ti to H
for unrelaxed structures.
Fig. 2. Dependence of dissolution energy of hydrogen on distance from Pd and Ti to H
for relaxed structures. References 1. Schwarz, K. Electronic structure calculations of solids using the WIEN2k package for
material science / K. Schwarz, P. Blaha, G.K.H. Madsen, Computer Physics Communications 147, 71 (2002).
2. D. E. Jiang, E. A. Carter, Phys. Rev. В 70, 064102 (2004). 3. J. P. Hirth, Metall. Trans. A 11A, 861 (1980).

56
SELF-ASSEMBLY OF DNA BASE THYMINE ON Cu(110) STUDY
G. M. Thomas, S. Haq, I. Temprano, G. Darling, M. Dyer, R. Raval
Surface Science Research Centre, University of Liverpool (UK) The development of electronic devices like Organic Light-Emitting Diodes (OLEDs) has lead to an alternative technology to the silicon-based one. The emerging field of molecular electronics focuses on using organic molecules adsorbed onto surfaces as basic functional units to construct devices. The understanding and control of directed self-assembly techniques focuses on the notion that molecular devices can be synthesized from first principles [1]. Molecular devices require their physical properties to be tuned and molecules with several functional groups that adsorb on a surface through one group leaving the others to react with other molecules can be used to selectively modify the geometry and electronic properties of the system. To this end, the interaction of biologically-related molecules with surfaces is attracting increasing interest due to its potential relevance to areas such as biocompatibility, biosensors and the fabrication of novel biomaterials. It is now possible to model the complex self-assembled structures of DNA molecules on the surfaces of crystals. These are too complex to be investigated on the atomic scale, however, the interaction of these is governed by the building blocks of these molecules (the nucleic acids) and these have been modelled using various experimental and theoretical techniques [2]. In this case study the adsorption properties of thymine on Cu(110) was studied in some detail. This revealed that thymine spontaneously self-assembles into various different structures at different temperatures [3,4]. References:- [1] V. Caciuc, N. Atodiresei, P. Lazic, Y. Morikawa, S. Blugel, “Van der Waals Study of Thymine on Cu(110) Surface”, 2009. [2] Yamada, T.; Shirasaka, K.; Takano, A.; Kawai, M. Surf. Sci 2004, 561, 233. [3] Rocco, M. L. M.; Dudde, R.; Frank, K.-H.; Koch, E.-E. Chem. Phys. Lett. 1989, 160, 366. [4] Allegretti, F.; Polcik, M.; Woodruff, D. P. Surf Sci. 2007, 601, 3611.

57
Mechanism Of Stabilization Of The Misfit Layer Compound
(PbS)1.13TaS2
Evgeniya Kabliman1, Peter Blaha1
1 Institute of Materials Chemistry Vienna University of Technology Getreidemarkt 9/165-TC A-1060 Vienna, Austria
In the present work we perform ab initio electronic structure calculations of the (PbS)1.13TaS2 misfit layer compound in order to understand the basic mechanism of its stabilization. The so-called “misfit layer compounds” can be described by the general formula (MX)1+x(TX2)m (M = Sn,Pb, Bi, Sb or rare-earth metal; X = S or Se; T = Ti, V, Cr, Nb or Ta; 0.08 < x < 0.26; m = 1,2). They are built up of alternating MX double layers (originating from the NaCl structure of MX) and hexagonal TX2 sandwiches. The lattice parameters of these compounds do not match in one of the directions and therefore lack three-dimensional periodicity. The stability of the misfit layer compound, the interlayer chemical bonding and the effect of incommensurability on the physical properties are still not fully understood and despite extensive efforts the dominant interlayer bonding mechanism of these compounds has remained elusive. Two models have been put forward based on experiments: i) a systematic depletion of M atoms and a corresponding excess of T atoms were found in some experiments, suggesting that T atoms were substituted into the MX layers and the corresponding nonstoichiometry is essential for stability, and ii) a metal cross substitution mechanism, where M and T atoms are exchanged in the MX and TX2 layers, which alters the charge balance between the two layers in a way that strongly enhances the interlayer bonding.
The investigations of (PbS)1.13TaS2 are based on the density functional theory and the resulting Kohn-Sham equations are solved using the augmented plane wave + local orbitals (APW+lo) method as embodied in the WIEN2k code. The incommensurable structure is modeled by a unit cell consisting of seven TaS2 and four PbS units, each one stacked in double layers in a Bb2b space group structure with 74 atoms/unit cell. A full relaxation of this structure has been performed but an energy analysis reveals that the misfit layer compound is hardly more stable than the parent compounds and would not form as a stoichiometric compound. In the next step, the two models for stabilization, namely the metal cross-substitution (where some Ta atoms from the TaS2 layer are exchanged with Pb atoms in the PbS layer) and the nonstoichiometry mechanism (where a sizable amount of Ta atoms substitute Pb atoms in the PbS layer) have been investigated using four times larger supercells (296 atoms/cell). It was found that the metal (Pb,Ta) cross substitution cannot stabilize the (PbS)1.13TaS2 compound according to the calculated formation energies. On the contrary, the nonstoichiometry model, where Ta atoms substitute Pb in the PbS layer, has a strongly stabilizing effect. Therefore, this mechanism can be the origin of the stabilization of the misfit layer compounds. We find a minimum in the formation energy for an impurity concentration around n(Taimp) = 0.15 in very good agreement with experimental estimates. Of course, due to high temperature and non-equillibrium conditions when these materials are grown, also Pb impurities in the TaS2 layer are possible. The results are explained by analysis of partial densities of states and the calculated XPS core level shifts agree reasonably with experiment.

58
Analysis of paramagnetic and ferromagnetic surface states in µc-Si:H via
the orbital magnetization
M. Rohrmüller, U. Gerstmann, and W.G. Schmidt Theoretische Physik, Universität Paderborn, 33095 Paderborn, Germany
Electron paramagnetic resonance (EPR) provides a powerful tool to analyse the microscopic structure of paramagnetic systems. For defects in semiconductors, this is well known and frequently used since several decades. In this work, we show that the magnetic properties of surface structures are exceptionally sensitive quantities which help to elucidate the microscopic structures at the surfaces. Hydrogenated microcrystalline silicon (µc-Si:H , see figure 1)) provides a very interesting example: The material can be used for efficient and low-cost solar cells [1], which does not suffer too much from the notorious light-induced degradation, known as the Staebler-Wronski effect. Best cell performance, however is achieved for material grown close to the transition to amorphous growth [2]. The porosity of this material allows in-diffusion of atmospheric gases. The main effects to consider are, thus, oxidation and adsorption of hydrogen on surfaces.
Fig. 1: Material for solar cells from µc-Si:H (left) to a-Si:H (right).
In the framework of density functional theory, we use a recently developed Berry Phase formula for the orbital magnetization [3] to characterize the magnetic properties of the resulting surface states. For paramagnetic systems the approach allows to calculate the electronic g-tensor [4] providing similar results as obtained within linear response theory [5, 6].We analyze the influence of the H coverage and compare these results to the observed increase of the EPR-resonance at g = 2.0043 and g = 2.0052 [7]. The novel non-perturbative approach [5] requires that spin-orbit coupling is included explicitly in the Hamiltonian. In the present implementation, this is done in a collinear approximation. Steps beyond this approximation are discussed in connection with spin-orbit coupling of the Rashbah-type [8] that is obtained for a isolated dangling bond at Si(111) surface (see figure 2).

59
Fig. 2: Magnetization density of an isolated dangling bond at an otherwise hydrogenated Si(111) surface. The small arrows (right) indicate the direction of the hyperfine tensors of the Si nuclei. Furthermore, the method via the orbital magnetization allows even to treat metallic surfaces like the famous Si(111) 7×7 reconstruction [9]. Hence, the ab initio calculation of the orbital magnetization is shown to be a general key to a better understanding of surfaces structures, not only for paramagnetic systems, but also for ferromagnetic nanostructures. [1] E. Vallet-Sauvain, U. Kroll, J. Meier, A. Sah J. Pohl, J. Appl. Phys. 87, 3137(2000). [2] S. Klein, F. Finger, R. Carius, T.Dylla, B. Rech, M. Grimm, L. Houben, M. Stutzmann,Thin Solid Films 430, 202, (2003). [3] R. Resta, J. Phys. Cond. Matter 22, 123201 (2010). [4] D. Ceresoli, U. Gerstmann, A. P. Seitsonen, F. Mauri, Phys. Rev. B 81, 060409(R) (2010). [5] Ch. J. Pickard, F. Mauri, Phys. Rev. Lett. 88, 086403 (2002). [6] U. Gerstmann, M. Rohrm¨uller, F. Mauri, W. G. Schmidt, Phys. Status Solidi C7, 157 (2010). [7] J. Behrends, A. Schnegg, C. Boehme, S. Haas, H. Stiebig, F. Finger, B. Rech, K.Lips, Journal of Non-Crystalline Solids 354, 2411 (2008). [8] E.I. Rashba, Fiz. Tverdogo Tela 2, 1224 (1960); Sov. Phys. Solid State 2, 1109 (1960). [9] J. Ortega, F. Flores, A. L. Yeyati, Phys. Rev. B 58, 4584 (1998).

60
Superconductivity and Novel Structures of Calcium Under Pressure from
ab intio Calculations
Ion Errea1,2, Aitor Bergara1,2,3
1Materia Kondentsatuaren Fisika Saila, Zientzia eta Teknologia Fakultatea, Euskal Herriko Unibertsitatea, 644 Postakutxatila,
48080 Bilbao, Basque Country, Spain 2Donostia International Physics Center, Paseo de Manuel Lardizabal, 20018 Donostia,
Basque Country, Spain 3Centro de Física de Materiales (CFM), Materials Physics Center (MPC), Centro Mixto
CSIC-UPV/EHU, Edificio Korta, Avenida de Tolosa 72, Donostia, 20018 Basque Country, Spain
E-mail: [email protected]
Experimental studies at room temperature [1] suggest that calcium undergoes a series of phase transitions under pressure: fcc → bcc → sc → CaIV → CaV. Remarkably, Ca is a superconductor above 50 GPa and its Tc rapidly soars with pressure reaching 25 K at 161 Gpa [2], the largest Tc ever recorded in an element. Nevertheless, using evolutionary ab initio simulations at 0 K, we found a different phase diagram for Ca [3]. The fcc to bcc phase transition is properly predicted by our calculation, but between 33 and 134 GPa we find a series of simple-cubic-related structures (I41amd, C2/c-12, P43212-8, Pnma-4). Above 134 GPa an incommensurate phase becomes favorable. Although the predicted phase sequence and the one measured at room temperature differ, as can be seen in Fig. 1 the experimental values obtained for the superconducting critical temperature, Tc, agree well with our theoretical ab initio results. This result strongly suggest that at 0 K the phase sequence is the one presented by us. This work was performed in collaboration with Artem R. Oganov, Yanming Ma, Ying Xu and Andriy O. Lyakhov. Ion Errea would like to thank the Basque Department of Education and Research for financial help and SGI-IZO SGIker UPV-EHU for the allocation of computational resources. References
1. T. Yabuuchi, Y. Nakamoto, K. Simizu and T. Kikegawa, J. Phys. Soc. Japan 74, 2391 (2005) 2. T. Yabuuchi, T. Matsuoka, Y. Nakamoto and K. Simizu, J. Phys. Soc. Japan 75, 083703 (2006) 3. Artem R. Oganov, Yanming Ma, Ying Xu, Ion Errea, Aitor Bergara and Andriy O. Lyakhov, PNAS 107,
7646 (2010)
Fig. 1. Comparison between experimentally measured [2] and calculated Tc of Ca in different phases.

61
Spin-Orbit Coupling in Tl/Si(1,1,1) surface
Julen Ibañeza, Asier Eigurena,b, Aitor Bergaraa,b,c
aMateria Kondentsatuaren Fisika Saila, Zientzia eta Teknologia Fakultatea, Euskal Herriko
Unibertsitatea, Bilbo, Basque Country, Spain bDonostia International Physics Center (DIPC), Paseo de Manuel Lardizabal, Donostia,
Basque Country, Spain cCentro de F´ısica de Materiales (CFM), Materials Physics Center (MPC), Centro MixtCSIC-UPV/EHU, Edificio Korta, Avenida de Tolosa 72, Donostia, 20018 Basque Country, Spain
Since the rather unexpected discovery of spin-splitting of the Shockley type surface states [1] this phenomenon has attracted considerable inter-est due to its relevance in the emergent field of spintronics [2]. During the last decade, a great effort has been concentrated in order to produce and understand device functions based on the electron spin, where surface and interface studies play an outstanding role. We present an analysis of the effect of the spin-orbit coupling on the electronic surface states in the Tl/Si (1,1,1) surface, as a model system to understand the spin structure and spin-orbit coupling phenomena in surfaces and interfaces. Our calculations show the existence of two spin-split surface states crossing the Fermi level. These states form an effective two dimensional spin polar-ized electron gas due to the semiconductor nature of the Si substrate, the Fermi surface being completely defined by them. Among other quantities, we report the detailed electron/spin structure of surface electrons, providing the momentum dependent magnetization and energies of the relevant surface states.
References
[1] S. LaShell and B. A. McDougall and E. Jensen. Phys. Rev. Lett.77, 3419 (1996). [2] S. A. Wolf and D. D. Awschalom and R. A. Buhrman and J. M. Daughton and S. von Molmar and M. L. Roukes abd A. Y. Chtchelkanova and D. M. Treger. Science 294, 1488 (2001).

62
Thermoelectric transport properties of silicon from first principles
Z. Wang.1, S. Wang1, N Vast2, J. Sjakste2, V. Tuyterev2, N. Mingo1
1LITEN, CEA-Grenoble, 17 rue des Martyrs, 38054 Grenoble Cedex 9. 2 Laboratoire des Solides Irradiés, Ecole Polytechnique, CEA, CNRS UMR 7642, 91128
Palaiseau.
We have implemented an ab-initio approach to compute the linear response thermoelectric coefficients of semiconductors. The ab-initio band structure and electron-phonon scattering rates are obtained from density functional theory using the Quantum Espresso package. Ionized impurity and electron-plasmon interactions are approximated via standard models. The linearized Boltzmann equation is then solved exactly without using the iterative method. The approach is applied to crystalline silicon. We present results for mobility, Seebeck coefficient, and electronic contribution to the thermal conductivity, as a function of carrier concentration and temperature. The calculated coefficients are in good agreement with experimental results. Author Email : [email protected]

63
Phase field crystal modelling of liquid-crystal and reconstructive
phase transitions with FCC and BCC lattices
M. Certain and H. Zapolsky
Groupe de Physique des Matériaux (GPM) – UMR 6634 - Avenue de l'Université BP 12
76801 Saint Etienne du Rouvray, CEDEX, France
The phase-field method is a very powerful tool in modelling phase transformation and microstructural evolution in solids at mesoscopic scale. However, some important phenomenons like defects formation, grain boundaries motion or reconstructive phase transitions require atomic scale study. Recently an approach called phase-field-crystal (PFC) modelling has been developed to incorporate atomic-level crystalline structures into standard continuum theory for pure and binary systems [Elder, Khachaturyan]. The PFC model describes the diffusive, large-time-scale dynamics of the atomic density field ρ, which is spatially periodic on atomic length scales. In spite of the PFC method has been successfully applied to describe qualitatively a wide range of phenomena, its predictive capability and quantitative description of phase transition remains largely unexplored. One of the main questions is how to evaluate the non-local party of the free energy for different crystal symmetries. Until now only triangular lattice in two dimensions and the body centred cubic (BCC) structure in three dimensions were reproduced by PFC method. In our work we investigated the stability of BCC, face centred cubic (FCC) and simple cubic (CS) lattices. We show that using diffraction patterns we can construct a spherically symmetric pair interaction potential that yields the CS, BCC or FCC lattice as its ground state. As example, the potential that stabilise CS structure is shown in Fig.1. The form of this potential is such that the first neighbour, and the second neighbour lie at a distance at which the pair potential is negative. Using this potential we reproduced the liquid-CS phase transition in 2D. In our simulation we track not only the crystal structure of nucleus but also the facet form of interface solid/liquid, as shown in Fig.2. We have optimised other isotropic interaction potentials to reproduce the liquid-fcc and liquid-bcc phase transition. We discuss also the influence of the shape of the interaction potentials on the stability of phases with different symmetry.
Figure 1: Double-well potential used to stabilise Figure 2: Simulation of quadratic nucleus the quadratic structure in two dimensions. from liquid state at reduced time t* = 10000

64
Using these optimised potentials, it is now possible to model reconstructive FCC/BCC phase transition. Indeed, although reconstructive transformations have been studied for more than a century, the atomistic processes leading to this structural transition are still a subject of debate. The phase field crystal (PFC) modelling can give an important insight on the dynamic of such transition and even describe the formation of dislocations at the FCC/BCC interface. [1] K.R. Elder, M. Katakowski, M. Haataja and M. Grant « Modeling Elasticity in Crystal Growth » Phys.Rev. Lett. 24, 2002 [2] K.R. Elder and M. Grant « Modeling elastic and plastic deformations in nonequilibrium processing unsing phase field crystals » Phys. Rev. E 70, 2004 [3] Y.M. Jin and A.G. Khachaturyan « Atomic density function theory and modelling of microstructure evolution at the atomic scale » J. App. Phys. 100, 2006

65
MODELING OF OF Co/IrMn BILAYER :
PHASE TRANSITIONS AND HYSTERESIS LOOPS
A. Maître1, D. Ledue
1, R. Patte
1, L. Lechevallier
1, P. E. Berche
1
1Groupe de Physique des Matériaux UMR 6634 CNRS - Université de Rouen
Co/IrMn bilayers exhibit exchange anisotropy. This anisotropy which was discovered in 1956 by Meiklejohn and Bean [1], is unidirectional, and is evidenced by a shift in magnetic field (HE) of the hysteresis loop (Fig. 1). It appears for systems with a ferromagnet (FM)/ antiferromagnet (AFM) interface which are field-cooled from a temperature above the Neel temperature of the AFM (TN) and below the Curie temperature of the FM layer (TC), until a temperature below TN.
Since, the exchange anisotropy is strongly related to the type of the FM/AFM
interface, we want to focus here on the effect of the FM/AFM interface roughness. We wish to study the impact of this latter on the phase transition of Co/IrMn bilayer, and also on the hysteresis loops, in particular on the exchange field HE, We also investigate the temperature dependence of the exchange and coercive fields. The study of phase transitions will be done using the standard Monte Carlo (MC) method based on the Metropolis algorithm [2]. For the simulation of hysteresis loops, the method used is the time step quantified MC method [3].
M
HE
H
FIG. 1. Schematic représentation of a hysteresis loop « shift » (HE is called exchange field)
FIG. 2. Schematic principle of the time step quantified Monte Carlo method

66
In this algorithm, a site i is randomly selected and the new orientation of the spin of
this site within a cone with a given size is proposed. To achieve this procedure, a random vector u with uniform probability distribution within a sphere of radius R <1 is added to the initial normalized spin si=Si/||Si||. The new orientation of the spin is then given by the unit vector si'=si+u/||si+u|| (Fig. 2). If the energy difference ∆E between the new direction (si') and the initial orientation (si) is negative, the transition is accepted because it lowers the energy. Otherwise, the transition is accepted with a probability rate proportional to
exp(−∆E/kBT). The variation in R is used to vary the acceptance rate in order to optimize the efficiency of the algorithm. Consequently, this technique allows to simulate the temperature effect on hysteresis loops with reasonable computer time [4]. Our first results seem to indicate that the roughness increases the exchange bias field.
1. W. H. Meiklejohn and C. P. Bean, Phys. Rev. 102 (1956), 1413 2. N. Metropolis, A. Rosenbluth, M. Rosenbluth, A. Teller and E. Teller, J. Chem. Phys.
21 (1953), 1087 3. U. Nowak, R. W. Chantrell and E. C. Kennedy, Phys. Rev. Lett. 84 (2000), 163 4. E. Talbot, D. Ledue and P.E. Berche, J. Appl. Phys. 106 (2009), 023901

67
“Characterization of nitrogen defective centers in zinc oxide”
F. Gallino, G. Pacchioni, C. Di Valentin
Dipartimento di Scienza dei Materiali, Università di Milano-Bicocca,
via Cozzi 53, 20125, Milano, Italy
M. Chiesa, E. Giamello
Dipartimento di Chimica IFM, Università di Torino and NIS Nanostructured Interfaces and
Surfaces, Centre of Excellence, via Giuria 7, 10125, Torino, Italy
Zinc oxide is a wide gap semiconductor (3.4 eV) that has recently captured a remarkable
interest of the scientific community because of the large variety of applications that it offers.
The two main promising application fields are optoelectronics and spintronics that, however,
are severely hampered by the native n-type conductivity of ZnO. A stable, efficient and
reproducible p-type doping represents the current challenge and nitrogen is the most
promising candidate as p-dopant. Even though different groups have reported p-type
conductivity for N-doped thin films and bulk crystals, the matter is still controversial and it
remains an open issue until an atomistic description of nitrogen incorporation and of its bulk
species is achieved.
In this context, we faced a combined experimental and theoretical studyi with the aim of
understanding the defective centres produced upon nitrogen doping of polycrystalline ZnO.
We obtained two nitrogen paramagnetic species by ammonia annealing, a prevailing
monomeric species and a secondary dimeric defect. Electron spin echo (ESE) detected EPR
and HYSCORE experiments provided a detailed characterization of the hyperfine and
quadrupolar tensors of the first species. Theoretical spin-polarized calculations, performed
using the hybrid B3LYP exchange-correlation functional as implemented in the periodic
CRYSTAL06 code based on atomic Gaussian basis-sets, showed unambiguously that the
defect consists of a N atom replacing a lattice O, NO. By means of a theoretical approachii that
enables one to calculate thermodynamic (εtherm) and optical (εopt) transition levels from single
particle Kohn-Sham eigenvalues, we estimated εtherm(0/-1)=1.47 eV and εopt(0/-1)=2.16 eV for
this acceptor species. These results contribute to exclude that N-doping is a viable route for p-
type doping of ZnO.

i F. Gallino, C. Di Valentin, G. Pacchioni, M. Chiesa, E. Giamello, J. Mater. Chem. 20, 689 (2010).
ii F. Gallino, C. Di Valentin, G. Pacchioni, J. Chem. Phys., submitted.
F. Gallino, C. Di Valentin, G. Pacchioni, M. Chiesa, E. Giamello, J. Mater. Chem. 20, 689 (2010).
F. Gallino, C. Di Valentin, G. Pacchioni, J. Chem. Phys., submitted.
68
F. Gallino, C. Di Valentin, G. Pacchioni, M. Chiesa, E. Giamello, J. Mater. Chem. 20, 689 (2010).

69
Study of electronic transport in metal-metal junctions within the
embedding approach
C. Motta, M.I. Trioni, G. Fratesi and G.P. Brivio
Dipartimento di Scienza dei Materiali,
Universit`a di Milano-Bicocca, via Cozzi 53, 20125 Milano, Italy
The calculation of electronic transport through a system is a subject of great interest, in particular in the field of molecular electronics. The most common approach to study quantum transport is the Landauer scattering formalism, often formulated in terms of the Green’s function. This theory can be cast within the framework of the embedding method of Inglesfield [1], which describes the infinitely extended substrates. The embedding technique is a theory that allows one to calculate the Green’s function in a limited region of space, taking into account the effect of the rest of the space [2]. This is done by adding an embedding potential to the Hamiltonian, which includes the effect of a infinite substrate. This potential ensures the wavefunctions to have the correct boundary conditions at the interface between each subvolume, allowing them to match with those of the substrate. It is thus a suitable method to investigate the properties of the scattering region in a lead-interface-lead system. In our work we study the ballistic conductance at the �� point of a system formed by two metallic electrodes and a resonance in the interface. In order to reproduce and to tune an ad-hoc resonant state in the interface, we add a gaussian-well contribution to the potential. In particular, we consider two interfaces formed by two different Cu surfaces, Cu(111) and Cu(100), reproduced by using a one-dimensional modulated potential [3]. We present the calculated Landauer conductance with and without the ad-hoc interface resonant state, and the current-voltage characteristics. As an example, in Fig. 1 we show the DOS and the conductance of the Cu(111) interface for different depths (V0) of the gaussian. As V0 increases, the resonant state (represented by dotted vertical lines in the gap) shifts to lower energies and it hybridizes with the surface state, pushing it into the conduction band. When it occurs, this state acts as a bridge between the two leads, and a strong peak appears in the conductance Γ(E). For what concerns the I-V characteristic of the interface, it is affected by the presence of resonances, which shift with the applied field. These simple model systems allow us to gain insight into the behaviour of a real molecular junction, where the presence of molecular resonances play a central role in the charge transport between two electrodes.
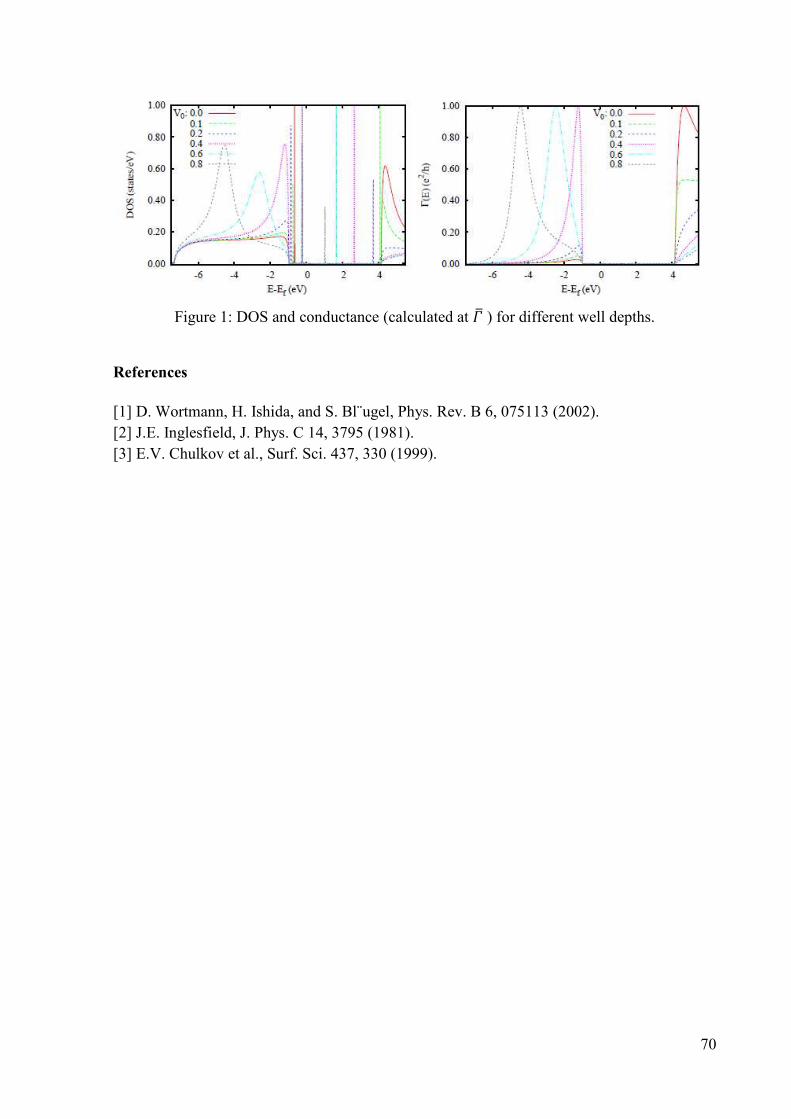
70
Figure 1: DOS and conductance (calculated at �� ) for different well depths.
References
[1] D. Wortmann, H. Ishida, and S. Bl¨ugel, Phys. Rev. B 6, 075113 (2002). [2] J.E. Inglesfield, J. Phys. C 14, 3795 (1981). [3] E.V. Chulkov et al., Surf. Sci. 437, 330 (1999).

71
Noble gas diffusion barriers for improved Nuclear-Test-Ban treaty
verification detection systems.
L. Bläckberg1*, A. Ringbom2, H. Sjöstrand3 and M. Klintenberg1 1Materials Theory, Department of Physics and Astronomy, Uppsala University
2Swedish Defence Research Agency 3Applied Nuclear Physics, Department of Physics and Astronomy, Uppsala University
*[email protected] The Comprehensive Nuclear‐Test‐Ban Treaty (CTBT) bans all nuclear explosions1. In order to verify its compliance a worldwide network of sensors, the international monitoring system (IMS), is now being constructed. The network is designed to detect the two basic phenomena caused by a nuclear explosion: energy release and radionuclide production. An important technology used to verify the nuclear nature of an underground explosion is the detection of radioactive noble gases, in particular xenon. In an underground nuclear explosion most of the produced radioactivity will remain in the cavity. However about 15% of the fission products come in the form of noble gases, which due to their inert chemical properties have a good chance of reaching the surface, and allow for detection. One system used within the IMS is the Swedish Automatic Unit for Noble Gas Acquisition (SAUNA)2. This system collects air, extracts a xenon sample and measures decay modes from interesting radioxenon isotopes (131mXe, 133mXe, 133Xe and 135Xe) using a NaI(Tl)‐ plastic scintillator gamma‐beta coincidence detector. The main drawback with the current setup of the detector is that during the measurements radioxenon diffuses into the plastic scintillator causing an unwanted memory effect. One approach to remove or reduce this effect is to coat the surfaces exposed to the radioactive gas. The coating material should stop the xenon diffusion without impairing detector properties such as resolution and efficiency. A thin coating is advantageous in this regard since the particles being detected retain most of their energy passing through the coating and in to the active volume of the detector. In this work possible candidate materials are presented, and special emphasis is given to the possibility of using graphene as a coating material for this application. A mono‐layer of sp2
‐bonded carbon (graphene) is a material with great technological promise because of, for example, its transport‐, electrical‐, optical‐ and mechanical properties. It has the potential of being a possible candidate for this application since it is known to efficiently block diffusion of helium3 and since it is by definition thin. However, up‐to‐date fabrication of large area graphene sheets is difficult. Therefore the ability of various small graphene flakes on a porous substrate (i.e. polyvinyltoluene, which is constituting the base of the plastic scintillator) to act as a diffusion barrier for noble gases is here explored.

72
This has been done by studying the diffusion barrier experienced by a xenon atom approaching a ripped graphene sheet. Rips of varying widths have been constructed with Stone‐Wales edges (see Figure 1), since this is the lowest energy configuration4.
The electronic structure for the system has been computed using VASP. The total energy of the relaxed structure has been calculated for different rip widths (rw) and different Xe‐graphene distances (h), see Figure 2.
The obtained results show that even a ripped graphene sheet would reduce xenon diffusion at ambient conditions, and graphene could thus be a suitable coating material for this application. It is further observed that narrow rips self heal and that the xenon atom can assist in the healing of wider rips. References: 1http://www.ctbto.org 2 ”SAUNA – a system for automatic sampling, processing and analysis of radioactive xenon”, A. Ringbom et al. Nucl. Instr. Meth. Phys. Res. A 508, 542 (2003) 3 “Graphene: A perfect nanoballoon”, O. Leenaerts, B. Partoens and F. M. Peeters. Appl. Phys. Lett. 93, 193107 (2008) 4“Self‐passivating edge reconstructions of graphene”, P. Koskinen, S. Malola and H.
Häkkinen. Phys. Rev.Lett. 101, 115502 (2008).

73
A High-Throughput Computational Search for New Lithium-Ion Battery Cathode
Materials
Geoffroy Hautier, Anubhav Jain, Charles Moore, Hailong Chen, Robert Doe, Christopher Fischer,
Byoungwoo Kang, Jae Chul Kim, Xiaohua Ma, Tim Mueller, Kristin Persson, Gerbrand Ceder
Massachusetts Institute of Technology, United States
The current drive to replace CO2 emitting technologies based on fossil fuel combustion by cleaner technologies such as electric or hybrid vehicles requires the development of high performances energy storage. Lithium-ion batteries are widely recognized as one of the most promising energy storage technology. Large efforts are currently spent by the lithium-ion battery community to discover new cathode materials that will outperform current materials (e.g. LiCoO2, LiMn2O4 or LiFePO4) in terms of energy density, safety, rate capability and cost. Nowadays, ab initio computational methods are capable to predict essential lithium-ion electrode properties such as voltage, diffusivity, or thermodynamic stability. When coupled with the exponential rises in computational power available to research groups, this predictive power provides the opportunity for a large-scale computational search for new lithium-ion battery cathode materials. Tens of thousands of novel materials can be generated and computationally screened for battery performance, focusing experiments on the most promising candidates and expanding the coverage of new chemical spaces. In this poster, we will present how such a project can be set up, highlighting the challenges of automatic data generation, management and analysis as necessitated by the scale of a high-throughput computational project. Preliminary results on experimentally confirmed novel Li-ion battery materials discovered by our high-throughput screening approach will be presented. In addition, we will show how such a large pool of computational data can also be used to achieve a better understanding of the chemical factors determining important battery properties such as voltage or safety.

74
Stability trends of Mn+1AXn phases
Martin Dahlqvist1,*, Björn Alling1, Johanna Rosén1 1 Department of Physics, Chemistry, and Biology (IFM), Linköping University, SE-581
83 Linköping, Sweden
* Electronic mail: [email protected]
Mn+1AXn (n = 1-3), or MAX phases, are hexagonal nanolaminated carbides or nitrides
where M is an early transition metal, A is an A-group element and X is either carbon or
nitrogen. These materials are very interesting as their structure and combination of elements
results in a combination of both ceramic and metallic properties. From a vast number of
combinatorial possibilities of three MAX phase elements, approximately 60 phases have been
synthesized to date. A majority are 211 phases (n = 1). The question rises why only ~60 out
of approximately 650 Mn+1AXn combinations are known?
Furthermore, much experimental work has been attempted on phases not possible to
synthesize. We have therefore, based on developed a systematic method, investigated the
phase stability of an important subset of the Mn+1AXn phases: M = Sc, Ti, V, Cr, or Mn, A =
Al, and X = C or N. Through a combination of Density Functional Theory and a linear
optimization procedure including all known competing phases, we identify the set of most
competitive phases for n = 1-3 in each system. The approach has been used to avoid ad hoc
choices of competing phases, which may lead to incorrect indication of phase stability. Our
calculations completely reproduce experimental occurrences of stable MAX phases, see Table
I. We also identify and suggest an explanation for trends in stability for both carbon and
nitrogen based Mn+1AXn systems as the transition metal is changed across the 3d-series, see
Figure 1. Furthermore, we suggest that the high stability of the competing AlN (in comparison
to the lower stability of Al4C3) may explain why so few nitride Mn+1AlNn phases have been
synthesized as compared to corresponding carbides.
Based on here presented results, our method seems to be a reliable tool that can be used as
guidance for further search of new Mn+1AXn phases, as well as other multinary compounds,
before time consuming and expensive experimental investigations are attempted.
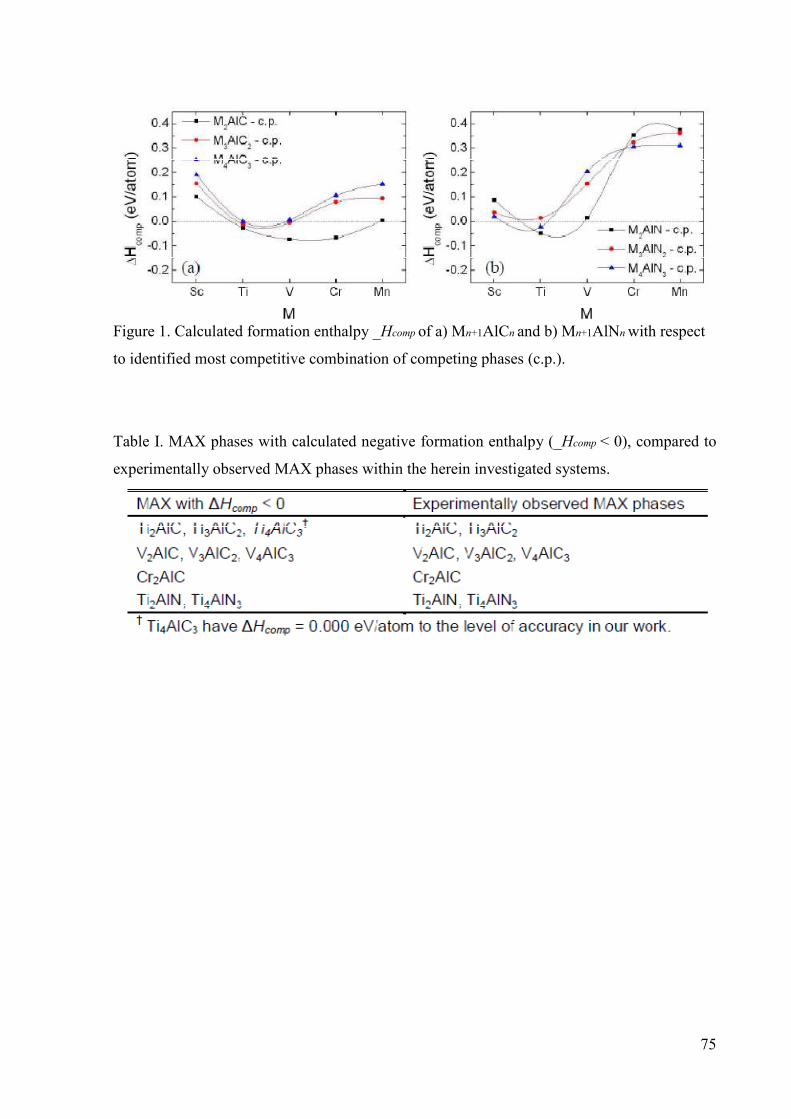
75
Figure 1. Calculated formation enthalpy _Hcomp of a) Mn+1AlCn and b) Mn+1AlNn with respect
to identified most competitive combination of competing phases (c.p.).
Table I. MAX phases with calculated negative formation enthalpy (_Hcomp < 0), compared to
experimentally observed MAX phases within the herein investigated systems.

76
The Hunt For N-type ZnSb
Lasse Bjerg1, Georg Madsen2
1 iNANO, Aarhus University 2 ICAMS, Ruhr-Universität Bochum
Thermoelectric materials have the ability to convert waste heat into usable electricity. Current state-of-the-
art thermoelectric materials are heavily doped semiconductors. n-type ZnSb is, by theoretical electron
density studies, predicted to be a good thermoelectric material. Unfortunately, only p-type ZnSb can readily
be synthesized.
One possible explanation for the preferred p-type conductivity of ZnSb may be that Zn vacancies form
easily in the material, thereby removing electrons from the valence band. The current poster focuses on the
thermodynamics of Zn vacancy formation.

77
Scattering of electrons and holes in surface states on Cu(110) and Ag(110)
S.S. Tsirkin1,2, S.V. Eremeev2,3, E.V. Chulkov 1Donostia International Physics Center (DIPC), 20018 San Sebastián/Donostia, Basque
Country, Spain 2Tomsk State University, Tomsk,Russia
3Institute of Strength Physics and Materials Science, Tomsk, Russia 4Departamento de Física de Materiales and Centro Mixto CSIC-UPV/EHU, Facultad de
Ciencias Químicas, UPV/EHU, Apdo. 1072,20080 San Sebastián/Donostia, Basque Country, Spain
E-mail: [email protected] We report a study of electron-phonon coupling and inelastic electron-electron scattering of electrons in occupied surface states (SS1) and holes in occupied surface states (SS2) on (110) surfaces of copper and silver at the Y point.
Electron-phonon contributions to the linewidth e ph−Γ and coupling parameters λ were calculated by use of the method elaborated in [1], which consists of three approximations: the electronic structure is described by a two-dimensional pseudopotential [2], phonon spectra are obtained on the basis of embedded-atom derived pseudopotentials [3] and the electron-ion potential is the Ashkroft pseudopotential with Thomas-Fermi screening. The unoccupied and the occupied surface states are localized in front of the atoms of the first and the second atomic layers respectively. We show that this difference in spatial distribution of surface states results scattering by different phonon modes. Thus, electronds in unoccupied states are mostly scattered by phonon modes, wich are localized in the first atomic layer and polarized in the [110] direction. Values of λ=0.08 were obtained for SS1 on both Cu and Ag. Electron-phonon coupling in SS2 is mainly driven by [110]-polarized vibrations of the second atomic layer and [001]-polarized vibrations of the first atomic layer, yielding greater values of the coupling parameter: λ=0.24 for Cu and λ=0.28 for Ag. The calculated coupling parameter of SS2 on Cu(110) is in excellent agreement with the photoemission value λ=0.23±0.02 [4]. The contribution of many-body electron-electron scattering to the linewidth e e−Γ is calculated within the GW approximation [5], the electronic structure being described by the two-dimensional pseudopotential model [2]. We show that e e−Γ of unoccupied states, calculated to be 39 and 58 meV for Cu and Ag respectively, is mostly contributed by transitions to bulk states, while equal contributions to linewidth from inter- and intraband transitions were obtained for occupied states. The value 21e e−Γ = meV for the occupied surface state on
Cu(110) combined with the calculated 17e ph−Γ = meV at 140T = K agrees with
photoemission result exp 48 6Γ = ± meV [4], while the difference ( )exp 10 6e ph e e− −Γ − Γ + Γ = ± meV may be attributed to scattering on defects. References 1. S.V. Eremeev, S.S. Tsirkin, E.V. Chulkov, JETP 137, 788 (2010) 2. S.S. Tsirkin, S.V. Eremeev, E.V. Chulkov, Surf. Sci. 604, 804 (2010) 3. M.S. Daw, M.I. Baskes, Phys. Rev. B 29, 6443 (1984) 4. P. Straube, F. Pforte, T. Michalke, K. Berge, A. Gerlach, and A. Goldmann, Phys. Rev. B
61, 14072 (2000). 5. L. Hedin, Phys. Rev. 139, A796 (1965).

78
Quasiparticle bandstructure of zincblende and rocksalt ZnO
Hemant Dixit, Rolando Saniz, Dirk Lamoen and Bart Partoens
CMT-group and EMAT, Departement Fysica Universiteit Antwerpen Groenenborgerlaan 171,
B-2020 Antwerpen, Belgium
The GW approximation [1] to many body perturbation theory represents the state of the art technique to calculate the quasiparticle correction to the band gap of solids and has been sucessfully applied to many materials. Although the GW approximation works well with pseudopotentials (PP’s) and plane wave basis sets used within Density Functional Theory Local density approximation (DFT-LDA), the II-VI materials are particularly challenging due to the strong p-d hybridization between the cation ’d’ and anion ’p’ states [2, 3]. The DFT-LDA resuslts in p-d overhybridization in the bandstructure which leads to underestimation of the calculated GWband gap. Recently, the self-consistent GW scheme [4] and quasiparticle correction based on the generalized Kohn-Sham schemes [5, 6] have been used to improve the GW band gap for II-VI semiconductors and insulators. Although these schemes make closer estimate of the band gap with the GW approximation, there is still need of an elaborate theory and it is a topic of current research. We present the GW bandgap calculated with the ABINIT [7] code, using the PP and plane wave basis set, for II-VI transparent oxides namely ZnO. The quasiparticle corrections are calculated systematically with different number of valence electrons in the cation-pseudopotential. We find that the 20-electron cation pseudopotential yields the best quasiparticle correction. The dependence of self-energy on the exchange interaction between the atomic orbitals is discussed. The postioning of the cation ’d’ energy level in the quasiparticle bandstructure is addressed. The correlation between the p-d hybridization and the underestimation of the non-self-consistent GW band gap is made through a comparison of zincblende (ZB) and rocksalt (RS) ZnO. The RS phase includes the inversion symmetry at the Gamma point in the Brillouin zone. The p and d states do not mix at the Gamma point and remain unhybridized, therefore the GW correction at the Gamma point is better for the RS-ZnO compared to the ZB counterpart. References [1] W. G. Aulbur, L. J¨onsson, and J. W. Wilkins, Solid State Phys. 54, 1 (2000). [2] M. Rohlfing, P. Kr¨uger, and J. Pollmann, Phys. Rev. Lett. 75, 3489 (1995). [3] M. Rohlfing, P. Kr¨uger, and J. Pollmann, Phys. Rev. B 57, 6485 (1998). [4] M. Shishkin and G. Kresse, Phys. Rev. B 75, 235102 (2007). [5] P. Rinke, A. Qteish, J. Neugebauer, C. Freysoldt, and M. Scheffler, New J. Phys. 7, 126 (2005). [6] F. Fuchs, J.Furthm¨uller, F. Bechstedt, M. Shishkin, and G. Kresse, Phys. Rev. B 76, 115109 (2007). [7] www.abinit.org

79
Hydrogen impurities and oxygen vacancies in CdO
Mozhgan Amini, Rolando Saniz, Hemant Dixit, Bart Partoens, Dirk Lamoen
Departement Fysica, Universiteit Antwerpen
Groenenborgerlaan 171, B-2020 Antwerpen, Belgium
The presence of even small concentrations of defects or impurities in semiconductors has a
major effect on their bulk electronic properties. CdO belongs to the transparent conductive
oxide (TCO) materials which have found many applications in optoelectronic devices. We
have used first principle calculations to study point defects in CdO based on density
functional theory within the local density approximation and beyond (LDA+U).
We have considered the hydrogen impurity atom in three charge states, once as an interstitial
atom and once as a substitutional atom for oxygen in order to find formation energies of these
different structures as a function of the fermi level, which determines the most stable state for
a given value of the fermi energy. We have also considered oxygen vacancies in three charge
states. Depending on the experimental conditions, we can have oxygen rich or poor
conditions. It is shown that under oxygen rich conditions, interstitial hydrogen with a positive
charge has the lowest formation energy, while under oxygen poor conditions the oxygen
vacancy in the 2+ charge state has the lowest formation energy. It is also remarkable that the
substitutional hydrogen defect, with a formation energy surprisingly close to the one of the
interstitional defect, is 6-fold coordinated. In contrast to conventional semiconductors, these
defects are all found to act as shallow donors. This explains the n-type conductivity found in
as-grown CdO.
Furthermore, the transition points between different charge states for the different studied
impurities are all close to each other, which supports the unification of the electrical behavior
of defects in CdO as recently shown experimentally.

80
Calculation Of Complex Band Structure Fro Plane-wave
Pseudopotential Hamiltonian
M. G. Vergniory1, C. Yang2, A. Garcia-Lekue1, L. W. Wang2 1 Donostia International Physics Center, Donostia, Spain
2 Lawrence Berkeley National Laboratory, Berkeley, CA, USA
We present a practical approach to calculate the complex band structure of an electrode for
quantum transport calculations. This method is designed for plane wave based Hamiltonian
with nonlocal pseudopotentials, although it can be used for any Hamiltonian with a large basis
set. Our method is particulary useful for the auxiliary periodic boundary condition transport
calculation. The complex band strcutures of copper and gold nanowires, and bulk gold
electrodes are presented.

81
Condensation Of Several Structural Instabilities Via Secondary Distortions: The
Case Of Sr2MWO6 (M= Zn, Mg And Ni) Double Perovskites
Iñigo Etxebarria1, Urko Petralanda1, Josu Mirena Igartua1 1 Euskal Herriko Unibertsitatea. Bilbao
Many perovskite and perovskite-like materials present several structural instabilities with
respect to a high-symmetry cubic phase. These instabilities correspond to tilts of the
constituent octahedral units that behave as rigid units and are associated to frozen phonons
with imaginary frequencies at different points of the Brillouin zone. Within the harmonic
approximation the ground state should correspond to the simultaneous condensation of all the
instabilities. Howevew, the strong positive biquadratic coupling between the distortion modes
is usually enough to penalize energetically the mixed ground state and a single distortion is
present at low temperatures. Compounds with double perovskite structure Sr2MWO6 (M=
Zn, Co, Ca, Cd, Mg, Ni) exhibit a second order structural phase transition from a cubic (Fm-
3m, #225) high-temperature phase to a tetragonal (I4/m, #87) one, and at lower temperatures
some of them (M= Zn, Co, Ca and Cd) present a first order phase transition to a monoclinic
phase (P21/n, #14) [1-3]. Both, the intermediate (tetragonal) and low symmetry (monoclinic)
structures, can be described as distorted high symmetry cubic phases, by the freezing of the
appropriate symmetry-breaking modes. The tetragonal phase can be achieved by the freezing
of a single primary mode of symmetry Γ4+, and the monoclinic structure can be explained by
the condensation of two modes of Γ4+ and X3+ symmetry. Alongside these primary
distortions that involve cooperative tilts of rigid octahedra, the condensation of ‘residual’
secondary modes compatible with the low-temperature symmetry provides the rest of degrees
of freedom present in the monoclinic phase. This description of distortions in terms of
symmetry-adapted modes is very convenient to constrain the configuration space to the
relevant degrees of freedom and to explore the role of the couplings among them. We have
performed ab initio energy calculations using the code WIEN2k to study the energy landscape
around the cubic phase of three compounds (M= Zn, Mg, Ni) of the family. In the three cases
the absolute minimum of the energy corresponds to the monoclinic structure. The compounds
with Ni and Mg present very shallow minima with respect to the tetragonal structure and
presumably quantum fluctuations should be responsible for the stability of the tetragonal
phase at low temperatures. The energy maps restricted to the space of the Γ4+ and X3+
distortions show that the stronger instability corresponds to the pure Γ4+ mode, and that there

82
is no minimum for the experimentally observed mixed configuration of the Γ4+ and X3+
modes due to the strong and positive biquadratic coupling between them. In fact, a secondary
hard mode of X5+ symmetry plays a crucial role to stabilize the low temperature phase; this
mode is trilinearly coupled with the two primary instabilities and its energetic contribution is
essential to compensate the effect of the biquadratic coupling and to stabilize the monoclinic
ground state. A similar scenario has been reported for SrBi2Ta2O9 [4], and it cannot be
discarded that the role of secondary distortions via trilinear couplings could be relevant in
other structural phase transitions where the condensation of several instabilities is present. [1]
M. Gateshki, J.M. Igartua and E. Herná�ndez-Bocanegra, J. Phys.: Condens.Matter 15, 6199
(2003) [2] B. Manoun, J.M. Igartua, M. Gateshki and S. K. Saxena, J. Phys.: Condens.Matter
16, 8367 (2004) [3] M. Gateshki, J.M. Igartua and A. Faik, J. Solid State Chem. 180, 2248
(2007). [4] J.M. Perez-Mato, M. Aroyo, Alberto García, P. Blaha, K. Schwarz, J. Schweifer
and K. Parlinski, Phys. Rev. B 70, 214111 (2004)

83
Towards an Atomistic kinetic Monte Carlo simulation of Iron
Chromium phasedecomposition based on an ab initio parameterisation
D. Costa1 2, G. Adjanor2, C. S. Becquart1, C. Domain1 2, P. Olsson2
1 Unité Matériaux et Transformation, CNRS UMR8207, Université de Lille 1, F-59655
Villeneuve d’Ascq Cédex, France
2 EDF-R&D Département MMC, Les Renardières, F-77818 Moret sur Loing Cédex, France
Thermal aging of iron chromium alloys is an important issue for the development of
generation IV structural materials as well as for duplex stainless steel components in current
generation nuclear power plants.
Chromium precipitation in the ferritic phase is observed in thermally aged materials and has
an embrittling effect on the alloy. The aim of this study is to provide a predictive tool for the
microstructure evolution of bcc iron chromium alloys under thermal aging using an atomistic
kinetic Monte Carlo approach. Several studies related to this issue have already been
performed, most of them with the common denominator of being based on semi-empirical
cohesive models. Here a study concerning the development of a kinetic Monte Carlo
parameterisation based on ab initio calculations is proposed. In particular, an investigation on
the influence of local ionic environment on the migration barriers is performed via an ab initio
approach.

84
Ab initio study of nano-precipitate nucleation and growth in ferritic steels
NATALIE TILLACK, TILMANN HICKEL, DIERK RAABE, and JÖRG NEUGEBAUER
MAX-PLANCK-INSTITUT FÜR EISENFORSCHUNG, DÜSSELDORF
The hardness and ductility of metallic alloys can be substantially improved by inducing nano-
precipitates having dimensions of only a few nm. To realize such nano-structures, chemical
compositions and process conditions have to be identified that form them in a spontaneous
(self-organized) manner.
For the example of ferritic alloys we have therefore studied nucleation and growth of nano-
particles combining ab initio calculations and kinetic Monte Carlo simulations. Using density
functional theory and two kinds of supercell approaches, we determine in a first step
formation and interaction energies of substitutional alloying elements in an FeMn or an Fe
matrix. In a second step the ab initio determined energies are used to construct the Master
equation, which is solved by a kinetic Monte Carlo approach. These simulations provide a
direct insight into the formation and size distribution of the nano-precipitates as function of
alloy composition, temperature and the calculated ab initio energies.

85
Effect of impurities on structural, cohesive, and magnetic properties of
grain boundaries in α-Fe
E. Wachowicz and A. Kiejna
Institute of Experimental Physics and Interdisciplinary Centre for Materials Modeling,
University of Wrocław, Plac M. Borna 9, PL-50-204 Wrocław
The effects of several metalloid (B), metalloid-like (C, P) and nonmetallic impurities (N, O
and S) on structure, energetics and mechanical properties of Σ3(111) and Σ 5(210) grain
boundaries (GBs) in ferromagnetic α-Fe have been studied from first principles. For two
di_erent concentrations and positions of impurity atoms the variations in GB properties have
been analyzed in terms of the structural, electronic, and magnetic properties of the system.
Most of the impurities enhance the relaxation of the interplanar spacing of the pure grains.
Interstitial impurities at both GBs are shown to increase separation of the grains while
substitutional ones in general either do not alter or decrease the grains' separation. It is shown
that at the Σ5 GB for all impurity atoms considered the positions in the boundary layer are
energetically favored independently of interstitial or substitutional site, wheras the enrichment
of the Σ 3 GB is favoured for the impurities of the interstitial sites as well as for a
substitutional P and C (Fig. 1).
Figure 1: Energy of segregation of various interstitial and substitutional impurities at the Σ3 and Σ5 GBs in iron.

86
We have found that most of the considered species placed in substitutional sites act as
embrittlers. Even boron which is known as a cohesion enhancer when placed in the interstice
of the Σ3(111) or Σ5(210) GB in iron, or in a substitutional position at the Σ5 GB, weakens
the cohesion when placed substitutionally at the Σ3 GB. Sulphur and oxygen are embrittlers in
all con_gurations and concentrations considered. Interstitial phosphorus and carbon in most
cases show strengthening effect. Nitrogen may act differently depending on the GB type and
concentration. An analysis of the components of the GB strengthening energy shows that the
GB embrittlement/strengthening results form a complicated interplay between chemical and
mechanical contributions, and the prevailing of one or the other component is both the GB
geometry and the impurity position and concentration specific.
In general, the impurity atom decrease the magnetic moments at the nearest neighbour Fe
atoms. Interstitial impurities modify magnetic moments on the GB atoms stronger than
substitutional ones. The moments on the impurities are much smaller in comparison to those
on iron and they do not exceed 0.15 µB. In most cases they are oriented antiparallel to the
moments on the neighbouring Fe atoms.

87
Constructing optimized atomic basis-sets with PW accuracy
Björn Lange, Christoph Freysoldt, and Jörg Neugebauer
Max-Planck Institut für Eisenforschung GmbH, 40237 Düsseldorf, Deutschland
Minimum atomic basis-sets as used e.g. for tight-binding calculations are commonly
constructed by fitting them or even only their matrix elements to experimental or theoretical
data. The resulting models are often optimized only for a specific environment, making a
systematic analysis of their transferability impossible. In order to achieve systematically
analyzable atom-centered basis-sets, a possible method is to start from a plane-wave(PW)
density-functional theory (DFT) calculation where the basis-set convergence is well
controlled [1,2]. The optimum shape for each orbital is determined by maximizing the overlap
of the orbital-set with the Bloch-states of the underlying plane-wave calculation. We keep the
spherical symmetry and freely optimize only the radial shape of the orbitals. The calculated
spillages of the Hilbert space norm are signi_cantly smaller (2-3 orders of magnitude) than
spillages obtained from free-atom LCAO basis-sets. An analysis of the Bloch state residues
(Fig. 1) shows which regions are not covered by the optimized basis-set, and thus reveals
improvement opportunities. To demonstrate the performance of this approach we provide
results of an extensive analysis of the transferability issues for prototype molecules and
semiconductors. Using the resulting optimized basis-sets in LCAO calculations for
semiconductors we achieve excellent agreement with PW calculations, e.g bandstructures
with meV accuracy (Fig. 2).
[1] D. Sanchez-Portal et al., Sol. Stat. Comm. 95, 685 (1995)
[2] W.C. Lu et al., J. Chem. Phys. 120, 2629 (2003)

88
Figure 1: State-summed residuum (isosurface: 4·10-5 ) for GaN. Ga orbitals as obtained from GaN optimization. Left: N orbitals from N2, showing optimization opportunities at N. Right: N orbitals from GaN, showing significant decrease of the state-summed residuum.
Figure 2: Comparison of di_erent bandstructure calculations. The lines represent a full DFT run, while the dotted bandstructures show the result of a self consistent LCAO bandstructure calculation with different basis sets.

89
Solid solution strengthening investigated by first principles
Duancheng Ma, Martin Friák, Johann von Pezold, Dierk Raabe, and Jörg Neugebauer
Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Strasse 1, D-402 37 Düsseldorf,
Germany
Solid solution strengthening is one of the most important strengthening mechanisms in
metallic systems. At the atomistic level, it is characterized by solute atoms interacting with
dislocations in the metallic matrix. Rather than investigating the dislocation-impurity
interaction explicitly, we pursue a multiscale approach, based on the combination of ab-initio
determined materials parameters with classical, linear elasticity-based solid-solution-
strengthening theories. In particular, by calculating the key parameters in the par- and di-
elastic models (lattice parameters and elastic properties) from first principles, the
strengthening capability of solute elements can be predicted.
In this work, the strengthening effect of a number of substituents (Cu, Mg, Li, Zn, Ca,
Sr, and Ir) in Al has been investigated. The parameters of interested, namely lattice
parameters and homogenized shear modulus are shown in Figure 1.
Figure 1 Ab initio determined compositional trends of the lattice parameters (a) and homogenized shear
modulus (b) of Al-X (X= Cu, Mg, Li, Zn, Ca, Sr, Ir) binary systems.
The predicted hardening effect of the different impurities is rationalized in terms of
their electronic structure. The change of the volume of the impurities and the matrix is
correlated with the charge transfer (explained in detail on the poster). Hence impurities
attracting electronic charge form the Al matrix generally give rise to reduced lattice
parameters, while substituents donating electronic charge to the host induce an increase in the
lattice parameter. The only exception is Li in Al. Li attracts the electrons from Al, which

90
makes Li smaller and the surrounding Al larger, but the overall volume is reduced. The
modulus change is attempted to be explained by ELF (electron localization function). Figure 2
shows four examples. In the case of Sr and Mg in Al, the impurities donate electrons to the
matrix. The electrons are localized near the 1st nearest Al, and between 1st and 3rd nearest
neighbor. When the impurities attract electrons from Al, such as Cu and Ir, the electrons tend
to localize between 1st and 2nd nearest neighbors.
Figure 2 Inter-connected maps of ELF in selected crystallographic planes calculated for Al31Sr1 (a) Al31Cu1(b)
Al31Mg1(c) and Al31Ir1(d).

91
Annealing Induced Modifications in (GaMn)As: Electron Spectroscopic Studies
Intikhab Ulfat
Department of Applied Physics
Chalmers University of Technology, SE-41296 Göteborg, Sweden
ABSTRACT
By incorporating magnetism into semiconductors, it may possibly be viable to enhance the
functionality of materials. An exceptionally important material in this context is GaAs, which
can be doped with Mn atoms. (GaMn)As has fascinated research community as a promising
candidate for spintronic application. It is quite appealing due to both its compatibility with
existing III-V technology and great progress in improving its magnetic properties. Being
fabricated by low temperature molecular beam epitaxy (LT-MBE), due to thermal instability
at elevated temperatures, the material contains a high density of various defects compensating
Mn acceptors. It is a well-established fact that the ferromagnetic state of (GaMn)As can be
stabilized via post growth annealing. Nevertheless, in general, the annealed (GaMn)As layers
do not remain useful for further epitaxial overgrowth that might be included in multilayer
structure.
In order to overcome this practical difficulty a procedure was devised in which annealing is
carried out under amorphous As. The focus of the present thesis is to study the MBE grown
(GaMn)As layers annealed under As capping using synchrotron based spectroscopy.
The formation of epitaxial MnAs with a 1x2 reconstruction resulting from the reaction
between out-diffusing Mn interstitials and As capping has been reported analysing the As3d
core levels. The use of topmost MnAs layer of (GaMn)As annealed under As capping has also
been described for depositing successive layers of Bi and Mn resulting in formation of
uniform surface layers. Moreover Mn2p emission from annealed and as-grown (GaMn)As
layers with varying GaAs overlayer thickness has been examined to describe the thickness
dependent mechanism that stops the diffusing of interstitial Mn from reaching the surface.

92
Local Structure Of Octahedral Site In Y-doped BaCeO3 And BaZrO3
Perovskite Compounds: A New Tetravalent Cation Substitution Model
Antonio Cammarata1,2, Pablo Ordejón3, Antonio Emanuele2, Antonino Martorana1, Dario Duca1
1 Dip. Chimica Inorganica e Analitica "S. Cannizzaro", Università degli Studi di Palermo,
Italy 2 Dip. Scienze Fisiche e Astronomiche, Università degli Studi di Palermo, Italy
3 Centre d’Investigació en Nanociencia i Nanotecnología - CIN2, CSIC-ICN, Barcelona, Spain
Local structure of octahedral site in Y-doped BaCeO3 and BaZrO3 perovskite
compounds: a new tetravalent cation substitution model
Proton conductivity in perovskite-type oxide materials has attracted great interest due to the wide range of applications such as membrane reactors, hydrogen sensors, batteries and fuel cells [H. Iwahara. Solid State Ionics, 77, 289–298, 1995; N. Bonanos, K. S. Knight, B. Ellis. Solid State Ionics, 79, 161–170, 1995; A. Tomita, T. Hibino, M. Suzuki, M. Sano. J. Mater. Sci., 39, 2493–2497, 2004.]. To improve the stability and efficiency of these materials, it is of fundamental importance to understand the elementary processes implied in protonic phenomena.
Hydrogen incorporation in the material is facilitated by the inclusion of trivalent species substituting the tetravalent cations. This leads to the formation of one oxygen vacancy for each pair of doping atoms. By steaming water vapor in the doped material, vacancies are transformed in hydroxyls groups, in this way introducing protons into the structure. Proton diffusion is mostly driven by phonon-assisted dynamics [K. D. Kreuer. Annu. Rev. Mater. Res., 33, 333–359, 2003; M. S. Islam. J. Mater. Chem., 10, 1027–1038, 2000.], as a consequence, the mechanism of protonic conduction is strongly influenced by local distortions affecting the proton environment and the doped sites. Hence, to characterize the local environments surrounding the substituted sites is of basic importance in rationalizing conduction details [M. S. Islam. J. Mater. Chem., 10, 1027–1038, 2000.].
In this work, we analyze the octahedral local environment of yttrium doped barium cerate and zirconate perovskite structures, being both among the most promising materials as solid oxide protonic conductors [K. D. Kreuer. Annu. Rev. Mater. Res., 33, 333–359, 2003.]. In the yttrium doped barium cerate derivatives, a characteristic bimodal Y−O distance distribution is found by EXAFS experiments; this was explained by the occurrence of peculiar axial distortions characterizing the octahedral site [A. Longo, F. Giannici, A. Balerna, C. Ingrao, F. Deganello, A. Martorana Chem. Mater. 18, 5782-5788, 2006.]. We already took into account the substitution of two nearest neighboring tetravalent atoms, obtaining an Y-O-Y configuration – see figure 1 and 2. By this model, it was possible to self-consistently analyze the charge and structure arrangement characterizing the Y-doped environment in the Y:BaCeO3 materials. In particular, we have found that the bimodal Y-O distance distribution could be attributed to local clustering of Y atoms; moreover, charge gradient considerations let us to infer i) that in the bulk of the BaCeO3 derivatives the protonic intra-octahedral diffusion is more likely to occur than the inter-octahedral one and ii) that the gradients in the local concentration of the charge density should play a minor role in the same protonic diffusion [A. Cammarata, A. Martorana, D. Duca, manuscript in preparation].

93
Starting from these results, we took into account also a double zirconium substitution in Y:BaZrO3, as we already did for the barium cerate perovskite. Since the structure of Y:BaZrO3 has cubic symmetry, all the oxygen atoms neighboring the first coordination shell of yttrium are equivalent. However, we have found that, when one proton is inserted in the Y-doped environment, the oxygen sites become distinct.
As a consequence, a study of the unprotonated doped environment could be not sufficient to perform predictions on its behavior after that proton defects are embedded in the structure. Furthermore, the presence of the proton generates, at least, two different transfer paths having different energy barriers, which are able i) to account for the cause of the underestimation of the activation energy of some computational works [M. E. Björketun, P. G. Sundell, G. Wahnström. Phys. Rev. B, 76, 54307–54315, 2007.] and ii) to explain the experimental values found for the proton transfer energy barrier.
These findings suggest to consider other protonated Y-doped systems that take into account structures different from those here discussed, in order to compare results and to improve models. Nevertheless, the outcomes here presented already allow one to infer that the Y local clustering could be considered as a characteristic feature of yttrium centers, when used as substituents of tetravalent cations in perovskite materials.

94
DFT study of formation and stability of Au nanostructures
on rutile TiO2(110) surface
Tomasz Pabisiak and Adam Kiejna
Institute of Experimental Physics, University of Wrocław, plac M. Borna 9,
PL- 50-204 Wrocław, Poland
Gold nanoparticles dispersed on rutile TiO2(110) surface are considered as a promising system for catalytic applications. The effect of size and shape of the gold aggregates, and the influence of surface reduction in determining the catalytic activity of Au/TiO2 system is still not completely explained. Experimentally, it has been proposed that one-dimensional Au structures can be formed in a controlled manner, using ordered oxygen vacancies created on the TiO2(110) surface during irradiation [1,2]. By applying gradient corrected density functional theory and the projector augmented wave method we examine the adsorption and formation of stable gold nanostructures on the oxygendeficient (110) surface of rutile TiO2 [3]. Starting from a monomer through the dimer and clusters consisting up to a dozen of Au atoms arranged in the elongated, one- or threedimensional structures (Fig.1), as well as the infinite Au rows are considered.
Fig.1. Side and top view on the 4ҳ2 cell of defected rutile TiO2(110) with six- and nine-atomic Au clusters adsorbed in (a) three-dimensional, and (b) elongated structures. Our research is focused on the role of oxygen vacancies and the influence of the amount of deposited gold on structural, electronic, and energetic properties of the system. The growth behavior of Au on TiO2(110) is discussed. A strong bonding (2 eV/atom) of the finite gold clusters to the O-deficient TiO2(110) is covalent. The bonding of elongated Au clusters is by 0.15 eV stronger than that of spatially more extended or three-dimensional clusters, and is strongest for the infinite Au rows. This points to the preference of a one-dimensional growth of Au on a highly O-deficient TiO2(110) surface. The electron charge transfer was examined and a negative charging of rows and clusters is discussed in the context of CO adsorption. References: [1] A. Locatelli, T. Pabisiak, A. Pavlovska, T. O. Mentes, L. Aballe, A. Kiejna, E. Bauer, J.Phys.: Condens. Matter 19 (2007) 082202. [2] T. O. Mentes, A. Locatelli, L. Aballe, A. Pavlovska, E. Bauer, T. Pabisiak, A. Kiejna,Phys. Rev. B 76 (2007) 155413. [3] T. Pabisiak, A. Kiejna, Phys. Rev. B 79 (2009) 085411.

95
The overview of the half-metallic Heusler alloys
Kemal Özdoğan*, Iosif Galanakis**,
*Gebze Institute of Technology, Department of Physics, Gebze-Kocaeli, Turkey
** University of Patras, Department of Materials Science, Patras-Greece
Email: [email protected]
Half-metallic Heusler alloys have attracted a great interest for spintronic applications due to
the high Curie temperatures and the structural similarity to the binary semiconductors during
the last decade. It is very important to control accurately the creation of the defects in these
alloys for the realistic application. In this work we present an overview of the electronic and
magnetic properties the half-metallic full-Heusler alloys. Using state-of-the-art electronic
structure calculations we show that the electronic and magnetic properties in these compounds
are related to the appearance of the minority-spin gap. In the half-metallic full Heusler alloys
the total spin magnetic moment in the unit cell follows the relation Zt-24, where Zt is the total
number of valence electrons. We present results for the following cases (i) the disorder in
Co2Cr(Mn)Al(Si) alloys, which is susceptible to destroy the perfect half-metallicity of the
bulk compounds and thus reduce the performance of devices. (ii) the appearance of the half-
metallic ferrimagnetism due to the creation of Cr(Mn) antisites in these compounds. (iii) Co
doping in Mn2VAl(Si) alloys leading to the fully-compensated half-metallic ferrimagnetism.
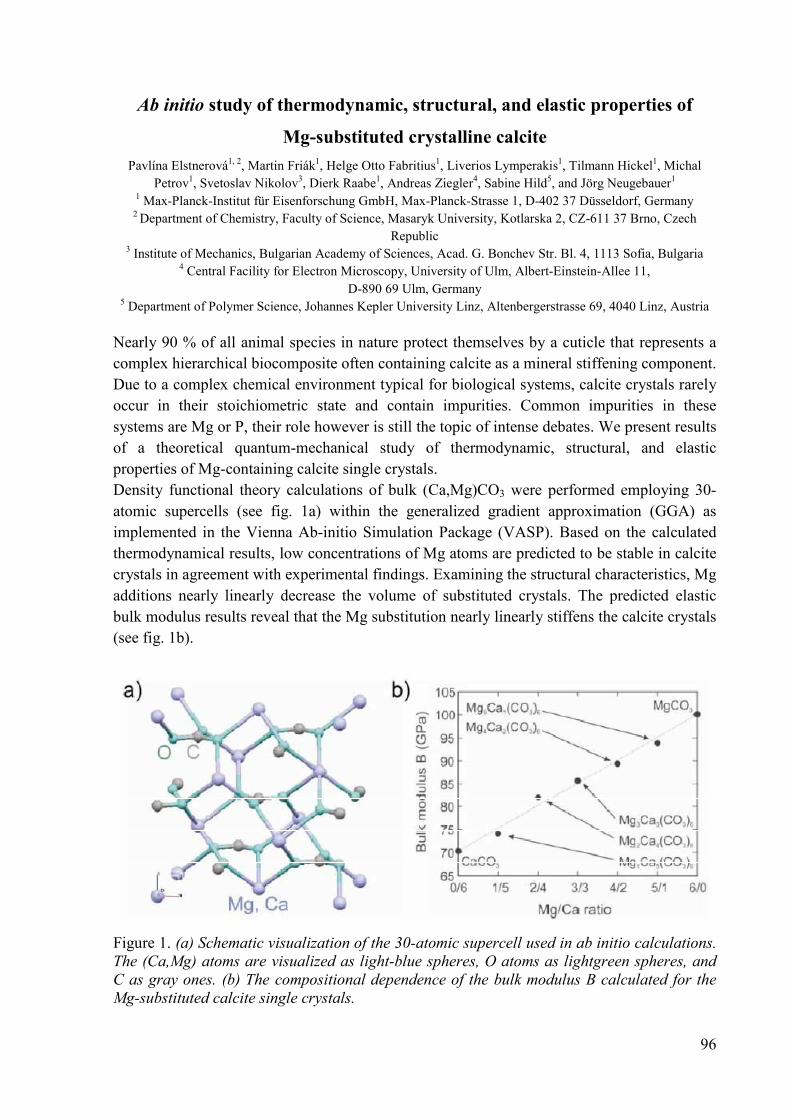
96
Ab initio study of thermodynamic, structural, and elastic properties of
Mg-substituted crystalline calcite
Pavlína Elstnerová1, 2, Martin Friák1, Helge Otto Fabritius1, Liverios Lymperakis1, Tilmann Hickel1, Michal Petrov1, Svetoslav Nikolov3, Dierk Raabe1, Andreas Ziegler4, Sabine Hild5, and Jörg Neugebauer1
1 Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Strasse 1, D-402 37 Düsseldorf, Germany 2 Department of Chemistry, Faculty of Science, Masaryk University, Kotlarska 2, CZ-611 37 Brno, Czech
Republic 3 Institute of Mechanics, Bulgarian Academy of Sciences, Acad. G. Bonchev Str. Bl. 4, 1113 Sofia, Bulgaria
4 Central Facility for Electron Microscopy, University of Ulm, Albert-Einstein-Allee 11, D-890 69 Ulm, Germany
5 Department of Polymer Science, Johannes Kepler University Linz, Altenbergerstrasse 69, 4040 Linz, Austria
Nearly 90 % of all animal species in nature protect themselves by a cuticle that represents a complex hierarchical biocomposite often containing calcite as a mineral stiffening component. Due to a complex chemical environment typical for biological systems, calcite crystals rarely occur in their stoichiometric state and contain impurities. Common impurities in these systems are Mg or P, their role however is still the topic of intense debates. We present results of a theoretical quantum-mechanical study of thermodynamic, structural, and elastic properties of Mg-containing calcite single crystals. Density functional theory calculations of bulk (Ca,Mg)CO3 were performed employing 30-atomic supercells (see fig. 1a) within the generalized gradient approximation (GGA) as implemented in the Vienna Ab-initio Simulation Package (VASP). Based on the calculated thermodynamical results, low concentrations of Mg atoms are predicted to be stable in calcite crystals in agreement with experimental findings. Examining the structural characteristics, Mg additions nearly linearly decrease the volume of substituted crystals. The predicted elastic bulk modulus results reveal that the Mg substitution nearly linearly stiffens the calcite crystals (see fig. 1b).
Figure 1. (a) Schematic visualization of the 30-atomic supercell used in ab initio calculations. The (Ca,Mg) atoms are visualized as light-blue spheres, O atoms as lightgreen spheres, and C as gray ones. (b) The compositional dependence of the bulk modulus B calculated for the Mg-substituted calcite single crystals.
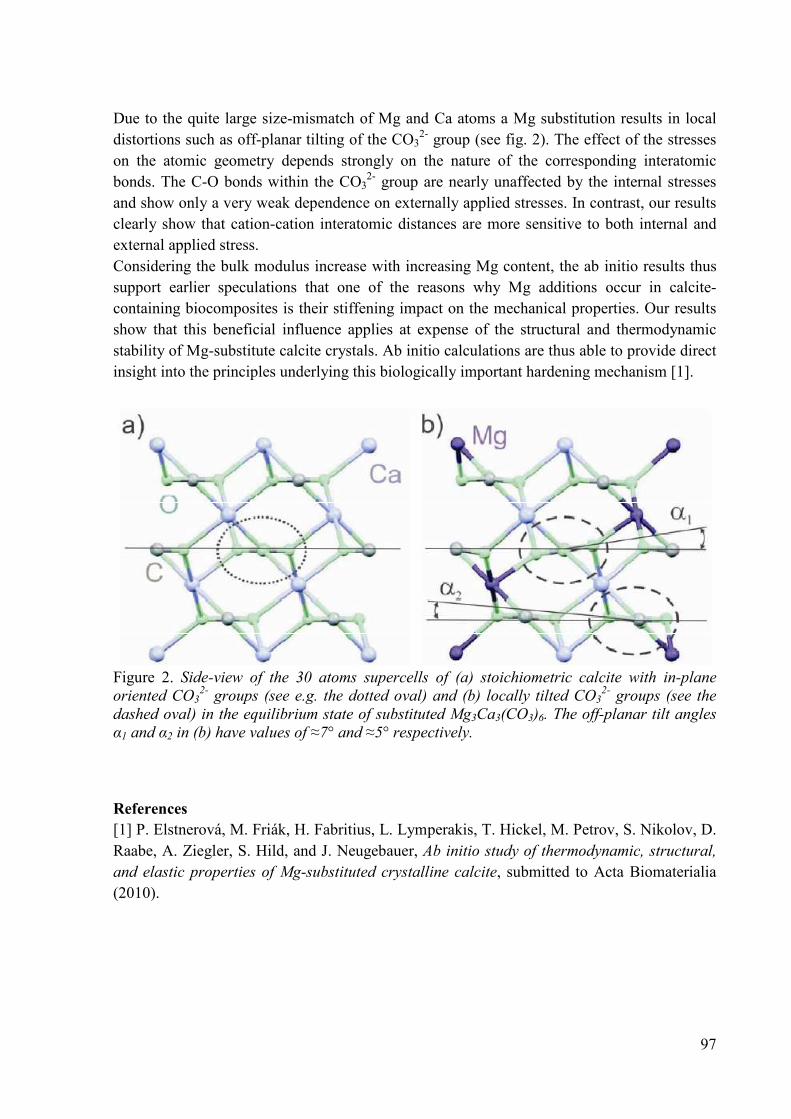
97
Due to the quite large size-mismatch of Mg and Ca atoms a Mg substitution results in local distortions such as off-planar tilting of the CO3
2- group (see fig. 2). The effect of the stresses on the atomic geometry depends strongly on the nature of the corresponding interatomic bonds. The C-O bonds within the CO3
2- group are nearly unaffected by the internal stresses and show only a very weak dependence on externally applied stresses. In contrast, our results clearly show that cation-cation interatomic distances are more sensitive to both internal and external applied stress. Considering the bulk modulus increase with increasing Mg content, the ab initio results thus support earlier speculations that one of the reasons why Mg additions occur in calcite-containing biocomposites is their stiffening impact on the mechanical properties. Our results show that this beneficial influence applies at expense of the structural and thermodynamic stability of Mg-substitute calcite crystals. Ab initio calculations are thus able to provide direct insight into the principles underlying this biologically important hardening mechanism [1].
Figure 2. Side-view of the 30 atoms supercells of (a) stoichiometric calcite with in-plane oriented CO3
2- groups (see e.g. the dotted oval) and (b) locally tilted CO32- groups (see the
dashed oval) in the equilibrium state of substituted Mg3Ca3(CO3)6. The off-planar tilt angles α1 and α2 in (b) have values of ≈7° and ≈5° respectively.
References
[1] P. Elstnerová, M. Friák, H. Fabritius, L. Lymperakis, T. Hickel, M. Petrov, S. Nikolov, D. Raabe, A. Ziegler, S. Hild, and J. Neugebauer, Ab initio study of thermodynamic, structural,
and elastic properties of Mg-substituted crystalline calcite, submitted to Acta Biomaterialia (2010).

98
An Investigation On The Covalent-like Transformation Of Undercooled
Liquid Silicon Using Orbital-Free Ab-initio Molecular Dynamics Method
Mutlu Colakogullari1, Luis E. Gonzalez2, Seyfettin Dalgic1, David J. Gonzalez2
1 Physics Department, Faculty of Sciences, Trakya University, 22100 Edirne, TURKEY
2 Departamento de Física Teórica, Universidad de Valladolid, 47011 Valladolid, SPAIN
In this work we show the results for static and dynamic properties of liquid silicon at several
temperatures between 1550 K and 1100 K, where is the undercooled region, using Orbital-
Free ab-initio Molecular Dynamics (OFAIMD) method. Local bond angle distributions and
average coordination numbers obtained by integrating the radial distribution function up to
first minimum indicate that the local order of liquid structure turns into to covalent-like
bounded open network structure from its metallic structure when the temperature is reduced.
Besides, single-particle properties such as mean square displacement and velocity
autocorrelation function also support the structure transformation. We also analyze the
variation of dynamic magnitudes and transport properties with temperatures. We have
compared our OFAIMD results with available experimental data and previous theoretical
studies.

99
Ab initio investigation of hydrogen solubility in 3d metals
U. Aydin, L. Ismer, T. Hickel, J. Neugebauer
Since the Mid-19th century it has been known, that transition metals (TM) can absorb
significant amounts of hydrogen, and that the presence of hydrogen can lead to serious
materials failures (so-called hydrogen embrittlement). Therefore, the energetics and dynamics
of hydrogen in TM are of critical importance in state-of-the-art materials design. Previously,
the hydrogen solubility has been studied both experimentally and theoretically. As far as
theoretical simulations are concerned, most of the studies rely on semi-empirical models that
had been developed to predict the solution enthalpy. Being based on empirical input, these
models unfortunately cannot provide a deeper understanding and/or insight into the decisive
underlying mechanisms involved in hydrogen-metal solid solutions. In the present work, we
employ ab initio calculations in order to shed more light on the mechanical and chemical
mechanisms governing hydrogen solution in a broad class of 3d TM under comparable
conditions. For this purpose, we have used PAW pseudopotentials implemented in VASP.
The electronic exchange and correlation has been described with GGA-PBE functionals. The
solution enthalpy for H in TM has been calculated by assuming a dilute limit for interstitial
hydrogen in high symmetry interstitial sites (octahedral or tetrahedral site) of 3d transition
metals in a nonmagnetic fcc structure. Furthermore, the effect of zero point vibrations on the
solution enthalpy has been studied. The analysis revealed an universal dependence on the
crystal lattice constant, with a material dependent interplay of chemical and strain
contributions. It turned out, that a hydrogen-metal distance of 2.3 Å is a characteristic value,
for which the H solubility has a maximum.

100
Molecular Dynamics In Zirconium Phosphates Systems
Amaia Saracibar1
1 Universitá di Perugia
Zirconium phosphates are extensively investigated members of the family of layered solid acids of tetravalent metals mainly because of their proton conductivity important for innovative technological applications [1,2,3]. The Zirconium phosphates Zr(HPO4)2 (shortly ZrP) can form different crystal structures. Our work has focused on alpha-ZrP for which structural, dynamics and conductivity data has been worked out. Using thermogravimetric analysis and differential thermal analysis monohydrated (alpha-ZrP*H2O) phosphates (form I) were found to form different phases [4,5] depending on the value of the temperature T when dehydrating (forms II and III). Proton conductivity was measured using the anhydrous pellicular zirconium phosphates [6]. To rationalize the experimental findings classical Molecular Dynamics (MD) calculations were performed using a suitable Force Field [7]. From the outcomes of such calculations carried out using the DL POLY [8] code, a wealth of information on the physical properties of the investigated system, such as the mean square displacement (MSD) from which the proton conductivity was estimated [9,10]. The comparison with the experiment shows that calculated conductivity is much larger than the one measured for forms I and II. By measuring the conductivity for pellets of microcrystalline alpha-ZrP [11] it was also found that a significant obstacle lowering the pellet conductivity is the integrating resistance. This suggests that experimental measurements might be largely in defect and that the agreement with theory could be much better if they could be carried out with a single crystal. References [1] A. Cleareld, in Design of new materials, D.L. Cocke, A. Cleareld Eds, Plenum Press, New York, p. 121 (1987) [2] G. Alberti, in Solid State Supramolecular Chemistry: Two and Three Dimensional Inorganic Networks, G. Alberti and T. Bein Eds, Comprehensive Supramolecular Chemistry Series, Pergamon, Oxford, vol. 7, chapt 5 (1996) [3] G. Alberti, M. Casciola, M. Pica, T. Tarpanelli and M. Sganappa, Fuel Cells, 5, 366 (2005) [4] U. Costantino, R. Vivani, V. Zima and E. Cernoskova, J. Solid State Chem. 132, 17 (1997) [5] G. Shuck, R. Melzer, R. Sonntag, R.E. Lechner, A. Bohn, K. Langer, M. Casciola, Solid State Ionics 77, 55 (1965) [6] G. Alberti, M. Casciola, U. Costantino and M. Leonardi, Solid State Ionics, 14, 289 (1984) [7] M. Porrini, PhD Thesis, University of Perugia, 2006; M. Porrini, A. Laganaà, Lecture Notes in Computer Science 4705, 295 (2007). [8] http://www.cse.clrc.ac.uk/msi/software/DL POLY/ [9] D.W.M. Hofmann, L.N. Kuleshova and B.D. Aguanno, J. Power Sources, doi:10.1016/j.jpowsour.2009.10.019 [10] J. Ennari, M. Elomaa, and F. Sundholm. Polymer, 40, 5035 (1999) [11] D. Bianchi, M. Casciola, Solid State Ionics, 17, 7 (1985).

101
QM/MM Studies On Ethene Adsorption On Cu(I) Exchanged Zeolites
Pawel Rejmak1,3, Mariusz Mitoraj2, Ewa Broclawik1
1 Institute of Catalysis and Surface Chemistry of Polish Academy of Sciences, Krakow,
Poland 2 Faculty of Chemistry, Jagiellonian University, Krakow, Poland
3 Donostia International Physics Center, Donostia-San Sebastian, Spain
Copper exchanged zeolites attract attention as the promising catalysts (removal od nitrogen
oxides, certain organic synthesis) or selective adsorbents. In present work the interaction of
various isolated Cu(I) sites in 2 types of zeolites (faujasite and MFI) with ethene molecule
(being employed both as reagent and as probe molecule in IR spectroscopy) is studied by
means of embedded cluster method. Structures, energetic stabilities and C=C stretching
vibrations in adsorption complexes are discussed. Furthermore, for interpretative purposes,
the interaction energies are decomposed, using new approach based on so called natural
orbitals for chemical valence.
It has been found that C2H4 always binds Cu(I) ion symmetrically by both C atoms. In
faujasite two local minima of similar stability on potential energy surface, differing by Cu(I)
site relaxation are found, that may be simultaneously populated in equilibrium. Binding
energies typically decrease with the degree of reconstruction of Cu(I) site after adsorption.
However, in certain cases, more distorted structure can be slightly more stable, if bonus from
favorable π* back donation overwhelms the distortion effects. Calculated values of binding
energies for Cu(I)-faujasite zeolite (about 80 kJ/mol) agree well with available
microcalorimetric data. Predicted ethene binding in MFI over two times stronger (none
experimental data known to authors).
On the basis of presented calculations it can be claimed that C=C stretching frequency is
neither site specific nor sensitive to the difference in local Al loading, but depends only on the
type of copper connectivity to oxygen nodes. The appearance of two C=C bands in
experimental IR spectra of Cu(I)-faujasite can be explained as the effect of coexistence of two
types of adsorption complexes, with Cu(I) coordinated to one or two framework tetrahedrons,
respectively. As in Cu(I)-MFI only one type of adsorption complexes with Cu(I) ion
coordinated to single tetrahedron exists, single C=C band is observed in IR spectra.

102

103
Microstructural evolution of austenitic alloys under irradiation modelled
by an ab initio based Atomic Kinetic Monte Carlo (AKMC) model
Jean-Baptiste Piochaud1, C. S. Becquart1, C. Domain1 2
1 Unité Matériaux et Transformation, CNRS UMR8207, Université de Lille 1, F-59655
Villeneuve d’Ascq Cédex, France
2 EDF-R&D Département MMC, Les Renardières, F-77818 Moret sur Loing Cédex, France
UNITE MATERIAUX ET TRANSFORMATIONS (CNRS UMR 8207), UNIVERSITE
LILLE1, BATIMENT C6, 59655 VILLENEUVE D 'ASCQ, FRANCE
In the nuclear industry, the limiting factor in plant life extension and new fission reactor
design is often the environmental degradation of the materials and particularly, the radiation
damage. Both the microstructural and microchemical changes are well known to degrade the
mechanical properties of materials. The PERFORM project aims at improving our knowledge
of the radiation induced-microstructural evolution of ferritic as well as austenitic steels which
are some of the most widely used materials in current light water reactors.
Atomic Kinetic Monte Carlo (AKMC) can provide an insight into the mechanisms by
which microstructure changes and segregations can occur in austentic alloys (FeNiCr).
However, one needs a suitable set of data to parameterize the AKMC code. Similarly to what
has been done for ferritic steels, we have undertaken to use DFT calculations to obtain
pertinent data for austenitic steels. The first ab initio calculations we performed on Fe-Ni-Cr
systems indicate that the magnetic ground state is not so easy to reach. A systematic study of
the magnetic behaviour of these alloys is thus under way which first results we present in this
poster.

104
√√√√7X√√√√3 Indium on Si(111): one or two In layers?
S. Rigamonti1, A. Arnau1, T. Nagao² and D. Sánchez-Portal¹ 1Donostia International Physics Center (DIPC)
and Centro-Mixto CSIC-UPV/EHU, San Sebastián, Spain 2National Institute for Materials Science, Tsukuba, Japan
E-mail: [email protected] Indium self-assembled overlayers on Si(111) are among the most studied metal-semiconductor surface structures. At least 10 different overlayer reconstructions have been reported for up to two In monolayers (ML). Different types of structural and electronic phase transitions (e.g. the temperature driven 8x2⇔4x1, the deposition driven √3x√3⇔2x2⇔√7x√3, the field induced √3x√3⇔2x2) have rised great interest in these surfaces.[1] At around one monolayer (ML) coverage, a commonly observed structure is the √7x√3 reconstruction. This structure has been the subject of many experimental studies, some of which point to a single In overlayer model (SL), with In atoms arranged in a quasi-rectangular (√7x√3-rec) or quasi-hexagonal (√7x√3-hex) fashion with coverages of 1.2ML and 1.0ML respectively [2], while others support a double-layer (DL) model instead, with 2~3ML coverage [3]. Recent angle-resolved photoemission (ARP) measurements shed light on the electronic structure of the √7x√3 phase, revealing a Fermi surface strikingly close to the ideal two dimensional electron gas.[4] Interpreted on the basis of the SL √7x√3-rec structural model proposed by Kraft et al. [2], the experimental results implied a charge transfer of ~0.45 el/In atom from about half of the Si dangling bonds to the two-dimensional electron gas formed at the In layer. Also, the conclusion was drawn that the dominant interaction determining the structure is metallic In-In. In this poster, we report ab-initio density functional theory (DFT) calculations for different √7x√3 In/Si(111) reconstructions.[5] Our calculations were performed with the SIESTA code within the local density approximation. We show that the Fermi surface (FS) and band-structure calculated for the SL √7x√3-rec model of Kraft et al. disagree with the experiments in Ref.[4]. We propose a DL structural model with 2.4ML coverage and find excelent agreement with experiments. For both the SL and the DL models the charge transfer from Si dangling bonds to the two dimensional gas is found to be negligible. We also show that the In-In interaction dominates over In-Si bonding. References 1. C. Kumpf et al., Phys. Rev. Lett 85 4916 (2000); A. Saranin, Surf. Sci. 388, 299 (1997);
A. A. Saranin, et al., Phys. Rev. B 56, 7449 (1997). 2. J. Kraft, M. G. Ramsey, and F. P. Netzer, Physical Review B 55, 5384 (1997); S.W.Cho,
et al., Physical Review B 67, 035414 (2003); A. A. Saranin, et al., Physical Review B 74, 035436 (2006).
3. S. Takeda, et al., Surface Science 415, 264 (1998); A. Pavlovska, E. Bauer, and M. Giessen, J. Vac. Sci. Technol. B 20, 2478 (2002).
4. E. Rotenberg, et al, Physical Review Letters 91, 246404 (2003). 5. S. Rigamonti, A. Arnau, T. Nagao and D. Sánchez-Portal, unpublished.

105
Ab initio concepts for an efficient and accurate determination of
thermodynamic properties up to the melting point
Blazej Grabowski, Tilmann Hickel, and Jörg Neugebauer
Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany
Phase diagrams play a crucial role in the thermodynamic evaluation and design of materials. In order to improve the existing description of phase diagrams, additional information on e.g. the volume/pressure dependence of thermodynamic properties, the detailed balance of the various physical excitation mechanisms, and the free energy of unstable phases is desirable. Since an experimental approach to this information is partly not even feasible, the interest in ab initio based methods, in particular density functional theory (DFT), has been recently re-intensified. While in principle ideally suited to tackle such basic questions, the actual application of DFT faces two key challenges: 1) Are the present days exchange-correlation functionals sufficient to guarantee the desired accuracy in the free energy? 2) Are the available statistical methods efficient enough to enable a DFT based calculation of all relevant free energy contributions up to the melting point? To address these two questions, we have performed a systematic DFT study of thermodynamic properties for a wide range of metals up to the melting point [1,2]. Two popular exchange-correlation functionals (LDA and GGA-PBE) have been considered and the quasiharmonic approximation (full volume/pressure dependence), including the electronic contribution as well as the first-order electron-phonon coupling, has been used. Besides the comparison of the thermal expansion, heat capacity, or the temperature dependent bulk modulus with experimental data, we also compared directly the constant pressure DFT free energies. The results showed over a large temperature range a very good agreement with CALPHAD data. For the specific case of aluminum, we extended the study beyond the quasiharmonic approximation including the explicitly anharmonic vibrations and the anharmonic vacancy contribution (both fully temperature and volume dependent). To master the challenge of calculating the computationally highly expensive anharmonic free energy, we have developed a hierarchical scheme to coarse grain the configurations space, which allows reducing the error in the ensemble average to < 1meV/atom [3]. Including all these excitation mechanisms, we were able to tackle the long standing debate about the dominating physical mechanisms determining the isobaric heat capacity of aluminum close to the melting point [3]. References: [1] B. Grabowski, T. Hickel, and J. Neugebauer, Phys. Rev. B 76, 024309 (2007). [2] B. Grabowski, PhD-Thesis, University Paderborn (2009). [3] B. Grabowski, L. Ismer, T. Hickel, and J. Neugebauer, Phys. Rev. B 79, 134106 (2009); see also the Viewpoint in Physics: G. Grimvall, Physics 2, 28 (2009).

106
ELECTRONIC PROPERTIES OF GRAPHENE NANO-RIBBONS
Huseyin Sener Sen1, Oguz Gulseren1
1 Bilkent University
Hüseyin Şener Şen1*, Oğuz Gülseren1
1Department of Phyics, Bilkent University, Ankara 06800, Turkey *[email protected]
Abstract- In this study we present a theoretical work on the electronic structure of several graphene nano-ribbons (up to the system with more than 1000 atoms) including all types (armchair, zigzag and chiral) using tight binding approach both in 1D and 0D. To handle the dangling bonds, we saturated the ribbon by the introduction of hydrogen atoms. We saw that the band gap of the ribbons depend both on the length of the ribbon and the angle of chirality. However, there is no simple function to fit the data as there is for Carbon Nanotubes. After the isolation of Graphene as a single layer in 2004 by Novoselov et al [1], the theoretical works became possible to be checked. Therefore, it became much more popular amongst both experimentalists and theorists. Having zero band gap, graphene cannot be used directly in applications as a semiconductor. However, Graphene Nano-Ribbons (GNRs), finite sized graphenes, can have a band gap different from zero. This band gap changes with the change in length and chirality angle of the ribbon. As the ribbon gets smaller, band gap increases.
Figure 1. Unit cell of 10-1CGNR with saturating hydrogen atoms.
We studied all types of GNRs (Armchair (AGNR), Zigzag (ZGNR), Chiral (CGNR)) in both 0D and 1D. Their names come from the shape of the long edge of the ribbon. We used tight binding approach in our study so that we were able to manage up to thousands of atoms easily. The parameters we used in our calculations are shown in table 1. Since the dangling bonds yields wrong and unrealistic band gap values,

107
Table 1. Tight binding parameters used in our calculations. [2] First nearest neighbor parameter | Value(eV) | Second nearest neighbor parameter | Value(eV) εs -7.30 Hss2 -0.18 εp 0.00 Hsp2 0.00 Hss -4.30 Hppσ2 0.35 Hsp 4.98 Hppπ2 -0.10 Hppσ 6.38 Hppπ -2.66 We saturated them with the introduction of hydrogen atoms. We generated the related parameters for hydrogen-carbon interaction using the parameters in table 1 so that they are compatible. There are some works on 1D AGNRs and ZGNRs in the literature showing great agreement with ours results which tells us that our parameters work fine [3,4]. 0D ZGNRs show zero HOMO-LUMO gap whatever its length is, whereas, HOMO-LUMO gap of 0D AGNRs show an exponential behavior increasing as the length of the ribbon decreases. Band gap of 1D CGNRs depend on the chirality angle and length of the ribbon. In determining the HOMO-LUMO gap of 0D CGNRs, however, in addition to those variables, width of the ribbon is also effective. Although there is a simple function, as indicated by Yorikawa and Muramatsu [5], for Carbon nanotubes showing how the band gap changes with chirality and length, we were not able to find such a function for CGNRs. However, in all GNRs, the band gap increases as the size of the ribbon decreases.
[1] Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos, S.V., Grigorieva, I.V., Firsov, A.A., Science 306, 666 (2004) [2] Tomanek, D., Louie, S.G., Phys. Rev. B 37, 14 (1987) [3] Pisani, L., Chan, J.A., Montanari, B., Harrison, N.M., Phys. Rev. B 75, 064418 (2007) [4] Son, Y-W., Cohen, M.L., Louie, S.G., Phys. Rev. Lett. 97, 216803 (2006) [5] Yorikawa, H., Muramatsu, S., Phys. Rev. B 52, 4 (1995)
Modeling Diffusion in the Al
Christian M. Ulrich
Elsässer
1Fraunhofer Institute for Mechanics of Materials IWM,Wöhlerstr. 11, 79108 Freiburg, Germany
2IZBS, Karlsruhe Institute of Technology, Kaiserstr. 12, 76131 3Institute of Physics of Materials, Academy of Sciences of the Czech Republic,
Žižkova 22, Cz*Email: [email protected]
The ongoing progress of miniaturization in microchip design and packaging technology has created steadily increasing demands to the integration density of thermosonic wire bonds, which are a common way to provide electrical contact to integrated circuits. wires on aluminium bond pads are of particular interest due to their industrial relevance. Microscopic defects in these contacts have been identified as one of the key causes for the failure of electronic components due to aging at elevateare frequently observed in micrographs. Their incidence is correlated with diffusion mediated phase transformation processes, which involve the formation and growth of stoichiometric compounds. Weak bonds commonly breDensity Functional Theory
mixed-basis pseudopotential code, was employed to obtain useful thermodynamic quantities that are necessary for modeling kinetics of the six ordAu system. In particular, activation energies and minimum energy trajectories for elementary jump processes in the compound AlAudiffusion coefficients for aluminium preexponential factors. In agreement with the empirical CuD*Au>>D*Al in AlAu4 at all temperatures. Results from phenomenological modeling using a thermodynamic extremal principle and the previously calculated parameters are presented, and qualitative agreement in the transformation kinetics of Al
Modeling Diffusion in the Al-Au System with Parameters from Ab
Calculations
hristian M. Ulrich1*, Adham Hashibon
1, Jiří Svoboda
Elsässer1,2
and Hermann Riedel1
Fraunhofer Institute for Mechanics of Materials IWM,
Wöhlerstr. 11, 79108 Freiburg, Germany IZBS, Karlsruhe Institute of Technology, Kaiserstr. 12, 76131 Karlsruhe,Germany
Institute of Physics of Materials, Academy of Sciences of the Czech Republic,Žižkova 22, Cz-616 62 Brno, Czech Republic *Email: [email protected]
The ongoing progress of miniaturization in microchip design and packaging technology has created steadily increasing demands to the integration density of thermosonic wire bonds, which are a common way to provide electrical contact to integrated circuits. wires on aluminium bond pads are of particular interest due to their industrial relevance. Microscopic defects in these contacts have been identified as one of the key causes for the failure of electronic components due to aging at elevated temperatures. Cracks and void layers are frequently observed in micrographs. Their incidence is correlated with diffusion mediated phase transformation processes, which involve the formation and growth of stoichiometric compounds. Weak bonds commonly break at phase boundaries. Density Functional Theory (DFT) in the Local Density Approximation
basis pseudopotential code, was employed to obtain useful thermodynamic quantities that are necessary for modeling kinetics of the six ordered low-temperature phases in the AlAu system. In particular, activation energies and minimum energy trajectories for elementary jump processes in the compound AlAu4 have been calculated and used to obtain the tracer diffusion coefficients for aluminium and gold as well as effective activation energies and preexponential factors. In agreement with the empirical Cu3Au rule, it is found that
at all temperatures. Results from phenomenological modeling using a thermodynamic extremal principle and the previously calculated parameters are presented, and qualitative agreement in the transformation kinetics of Al3Au8 into AlAu
108
Au System with Parameters from Ab-Initio
, Jiří Svoboda3, Christian
Fraunhofer Institute for Mechanics of Materials IWM,
Karlsruhe,Germany Institute of Physics of Materials, Academy of Sciences of the Czech Republic,
The ongoing progress of miniaturization in microchip design and packaging technology has created steadily increasing demands to the integration density of thermosonic wire bonds, which are a common way to provide electrical contact to integrated circuits. Contacts of gold wires on aluminium bond pads are of particular interest due to their industrial relevance. Microscopic defects in these contacts have been identified as one of the key causes for the
d temperatures. Cracks and void layers are frequently observed in micrographs. Their incidence is correlated with diffusion mediated phase transformation processes, which involve the formation and growth of stoichiometric
Density Approximation, implemented in a basis pseudopotential code, was employed to obtain useful thermodynamic quantities
temperature phases in the Al-Au system. In particular, activation energies and minimum energy trajectories for elementary
have been calculated and used to obtain the tracer and gold as well as effective activation energies and
Au rule, it is found that at all temperatures. Results from phenomenological modeling using a
thermodynamic extremal principle and the previously calculated parameters are presented, into AlAu4 is shown.

109
List of Participants ABRIKOSOV IGOR [email protected] LASSE BJERG [email protected]
ADJANOR GILLES gilles. [email protected] LEE JUNE GUNN [email protected]
ALLARD ADRIEN [email protected] LIND HANS [email protected]
ALONSO MARTIN JULIO ALFONSO [email protected] MAITRE ADELINE
ASTA M [email protected] MAURO PALUMBO [email protected]
AYUELA ANDRES [email protected] MORAES DE ALMEIDA JAMES [email protected]
BJORN LANGE [email protected] MOTTA CARLO [email protected]
BLACKBERG LISA [email protected] MOZHGAN AMINI [email protected]
BLAZEJ GRABOWSKI [email protected] MUTLU COLAKOGULLARI [email protected]
BURKE KIERON [email protected] NEUGEBAUER JORG [email protected]
CABRERA SANFELIX PEPA [email protected]
OKSANA SUKHOSTAVETS tiptop190 @mail.ru
CAMMARATA ANTONIO [email protected] ORDEJON PABLO [email protected]
CERTAIN MARILYNE [email protected] OZDOGAN KEMAL [email protected]
COSTA DAVIDE [email protected] OZOLINS VIDVUDS [email protected]
DAHLQVIST MARTIN [email protected] PAVLINA ELSTNEROVA [email protected]
DIEZ MUIÑO R. [email protected] PIOCHAUD JEAN BAPTISTE [email protected]
DMYTRO KANDASKALOV [email protected] PUKARI MERJA [email protected]
DRAIN JOHNNY [email protected] RAEBIGER H. [email protected]
DRAUTZ RALF [email protected] RAKITIN MAXIM [email protected]
DUANCHENG MA [email protected] RIGAMONT SANTIAGO [email protected]
DUDAREV L. [email protected] RINKE P. [email protected]
DUDEK MARTA [email protected] ROHRMULLER MARTIN
EKHOLM MARCUS [email protected] RUBAN A. [email protected]
ERREA ION [email protected] RUNEVALL ODD [email protected]
ETXEBARRIA IÑIGO [email protected] SANCHEZ PORTAL DANIEL [email protected]
EVGENY BLOKHIN [email protected] SARACIBAR AMAIA [email protected]
FREYSOLDT C. [email protected] SIMAK I. [email protected]
GALLINO FEDERICO [email protected] STASIEWICZ JULIUSZ [email protected]
GARCIA LEKUE ARANZAZU [email protected]
STROPPA ALESSANDRO [email protected]
GARCIA VERGNIORY MAIA [email protected] THOMAS GEOFFREY [email protected]
GLENSK ALBERT [email protected] TILLACK NATALIE [email protected]
GOYENOLA CECILIA [email protected] TOMASZ PABISIAK [email protected]
GRABOWSKI B. [email protected] TSIRKIN STEFAN [email protected]
HAUTIER GEOFFROY [email protected] UGUR AYDIN [email protected]
HEMANT DIXIT [email protected] ULRICH CHRISTIAN MARKUS
HICKEL T. [email protected] Ulfat Intikhab [email protected]
HUSEYIN SENER SEN [email protected] VAN DE WALLE CHRIS [email protected]
IBAÑEZ AZPÍROZ JULEN [email protected] VEKILOVA OLGA [email protected]
KABLIMAN EVGENIYA [email protected] VIMAL KISHORE [email protected]
KORZHAVYI PAVEL [email protected] WACHOWICZ ELWIRA [email protected]
LAFE DARIO [email protected] ZHAO QIAN [email protected]
LASA ANE [email protected] ZHAO WANG [email protected]

110