shedding some light on the insights lurking in the pma database
TRANSCRIPT

Shedding Some Light
on the Insights Lurking
in the PMA Database
By Revital Hirsch

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 1
1 It’s All About the Context
May 28, 2016 marked forty years of modern day federal authority over medical devices in the U.S. Not
only is this period brief in and of itself, but it’s also shorter by half compared to the duration of federal authority
over pharmaceuticals, which began with the Food, Drug and Cosmetics Act of 1938. In the past several years
the FDA has been the target of much criticism with regards to the approval of high-risk medical devices. Some
of that critique is likely merited, but it is important to keep in mind that the medical device arm of the FDA
is a work-in-progress that has had considerably less time to invent itself in the larger context of history.
Since the beginning of 2009 the FDA has prepared and made available to the public comprehensive
quarterly performance reports. While valuable to promote standardized measurement and transparency,
analyses prepared by the Agency portray its performance vis-à-vis the metrics and goals by which it is
measured. In contrast, this article is anchored in the industry / business-oriented point of view. The difference
in perspectives permeates the underlying quantitative analytics that are the cornerstone of this paper:
Analyses prepared by the FDA are based upon its fiscal year, which begins October 1st of a calendar year
and ends September 30th of the following year.
Industry mostly favors the use of calendar years.
FDA analyses are based predominantly on filing cohorts in which PMA submissions are grouped according
to the date they are accepted for filing.
Filing cohorts are inarguably the most homogenous; but with some PMA reviews exceeding 24 months it
can take 2-3 years for a cohort to close and the final analysis of its data to become available. By then
circumstances in Industry and the Agency may have changed dramatically.
An alternative to filing cohorts are decision or approval cohorts that group PMA submissions according to
their MDUFA (Medical Device User Fee Amendment) decision or approval dates. Decision / approval
cohorts are not as homogenous but they paint the most up-to-date picture.
The FDA’s primary unit for measurement of time is net review days. This counts only calendar days in which
the Agency’s review clock was ticking.
While time may stop for the FDA it does not stop for the companies waiting on their submission
approvals. Therefore, an alternative metric reflecting that difference is the total elapsed time from filing date
to approval date.
FDA analyses encompass all PMA submissions that reached MDUFA decision1.
Analyses generated from the downloadable PMA database encompass only submissions for which the
final decision was ‘approval’. (Failed applications are excluded from the database.) Over the last decade
the subset of approved PMAs has accounted for 79% of all PMAs that were accepted for filing.
Without analyzing the failed submissions it is impossible to know how – if at all – their exclusion affects
the outcomes of the statistical analysis.

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 2
What We Currently See
As part of its MDUFA reporting the FDA provides a core set of graphs depicting different aspects of its
performance. Three of these charts were reproduced below and supplemented by comparable analyses origi-
nating in the PMA database and reflecting the industry / business perspective.
[1] PMA Review Time
A primary performance metric is the review time of PMA submissions, officially known as ‘Average Time
to MDUFA Decision’. This metric is defined as2:
the three-year rolling average of the annual total times to decision (i.e. the average shown for fiscal year 2012
is actually the average of fiscal years 2010 through 2012);
within a closed cohort (a cohort is closed when 95% of its submissions have reached a decision);
excluding the highest and lowest 5% of values.
In Exhibit 1 ‘time to MDUFA decision’ has been supplemented by ‘time to approval’, an alternative metric
that uses only the data from the fiscal year under discussion (no rolling average) and includes all data points
(no exclusion of outliers). Added to that is a modified ‘time to approval’ that is subjected to the same data
manipulations that are applied to the official metric.
Exhibit 1. Average Time to MDUFA Decision vs. Average Time to Approval
* Cohorts still open. Time to MDUFA Decision and/or Time to Approval will increase.
FY 2010 pending final decision for Guided Therapeutics’ PMA for its LuViva® Advanced Cervical Scan.
FY 2013 pending final decision for St. Jude Medical’s PMA for its Amplatzer™ PFO Occluder.
Sources: Time to MDUFA decision: FDA Quarterly Meeting on MDUFA III Performance, May 2, 2016.
Time to approval: analysis of the PMA database.
Time to MDUFA decision (measured in days) excludes periods of time during which the review clock is
stopped. It does not represent a continuous stretch of time. As a result, translating the days into a different unit
– months, for example – would be misleading as it would indicate a continuity that is simply not there. In
comparison, time to approval measures the total elapsed time from a PMA’s filing to its approval.
180
270
360
450
540
630
720
6
9
12
15
18
21
24
2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010* 2011 2012 2013* 2014* 2015*
Day
s
Mo
nth
s
FDA Fiscal Year Ending September 30 (Filing Cohort)
Approval
Approval (modified)
MDUFA Decision

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 3
As a metric that does not measure a continuous period of time, ‘time to MDUFA decision’ is all but
useless for the purpose of business forecasting. In contrast, ‘time to approval’ is a straightforward metric that
can be used to objectively estimate the timing of a submission’s approval – a cornerstone assumption for a
cascade of critical commercialization-related business planning and decision-making.
Continuity aside, the data manipulation that goes into calculating ‘time to MDUFA decision’ does more
to mask changes in Agency performance than it does to provide transparency. The three-year rolling average
levels out peaks and troughs (seen in the modified ‘time to approval’ as it relates to the straightforward version),
presenting lower-than-actual performance variability. For example, the data points from fiscal years 2009 and
2010 (two of the Agency’s worst performing years) skew the 2011 rolling average upwards, obscuring the
considerable decrease in the average review time that year.
But even ‘time to approval’ doesn’t tell the whole story because “the FDA review clock begins as of the
date of receipt of the most recent [emphasis added] submission or amendment that made the PMA complete
and on which the FDA based its “Accepted” and “Filed” decision.”3 Depending on the number of acceptance
and filing review cycles (see Appendix A) several weeks – or even a few months – may elapse between a
submission’s receipt date and its formal filing date.
This period of time – from receipt to filing – is outside the scope of the FDA’s review clock and is not
reflected in ‘time to MDUFA decision’. Nor is it conveyed in ‘time to approval’ because the original receipt
date is not included in the downloadable PMA database. If the receipt-to-filing period were included in the
metrics all three graphs in Exhibit 1 would shift upwards.
Similarly, at the tail end of the approval process, the FDA review clock stops once the MDUFA decision
is made; but that may not be the end of the PMA review process:
Nevro announced in January 2015 that it had received an ‘approvable’ letter from the FDA for its Senza®
spinal cord stimulation system. The final approval arrived 3½ months afterwards (and six weeks earlier
than expected4).
After a particularly tortuous regulatory review process (See Appendix B), Wright Medical Group
announced in October 2014 that it had received an ‘approvable’ letter from the FDA for its Augment®
Bone Graft. Another 10 months would pass before the product was finally approved.
In April 2015, as part of a larger strategic initiative to focus on its core business, Greatbatch announced
its plan to spin out its Algostim spinal cord stimulation business once the product obtained regulatory
approval. At that time FDA had already indicated to Greatbatch that the Algostim submission was
approvable subject to an inspection of the manufacturing facilities, methods and controls. The final
approval arrived 7 months later.
‘Time to MDUFA decision’ excludes this period at the end of the review process. ‘Time to approval’, on
the other hand, counts every single day that passes until the final approval decision has been reached.
Ideally, in addition to the filing and approval dates, the downloadable PMA database would disclose all
the major dates associated with each PMA. This includes the original receipt date, the acceptance date and the
MDUFA decision dates, which may number more than one as is the case with ‘approvable’ and ‘not approv-
able’ determinations that are eventually ‘approved’. This additional granularity would undoubtedly increase
transparency as well as assist filers in their forecasting and business decision-making.

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 4
[2] PMA Approval Rate
A second performance metric is the approval rate of PMAs from the total number of submissions that
are accepted for filing. The formal metric, which is based upon a decision cohort, has been supplemented by
a comparable graph using a filing cohort. (See Exhibit 2.)
Anecdotally, this is the only PMA-related graph prepared by the FDA that uses a decision cohort. The
exhibit illustrates the different perspectives inherent to the two types of cohorts and it highlights the tradeoff
between them: the less homogenous decision cohort provides the most up-to-date information while the
homogenous filing cohort is either unavailable or incomplete for the last three or more fiscal years.
Exhibit 2. PMA Approval Rates, Decision vs. Filing Cohorts
* Filing cohorts still open. PMA approval rates may increase. 2015 cohort percentage is preliminary.
Sources: Decision Cohort – FDA Quarterly Meeting on MDUFA III Performance, May 2, 2016.
Filing Cohort – FDA materials and analysis of the PMA database.
PMA approval rates have fluctuated dramatically – bottoming out in the 2009 filing cohort at 56% and
peaking two years later at 91%. But perhaps more interesting than the approval rate is the failure rate. In the
2012-2014 filing cohort a total of 69 PMAs were approved whereas 9 were withdrawn or denied. These
numbers reflect a ratio of 1 failed submission for every 8 successful ones – an unacceptably high rate of failure.
Looking back a full decade paints an even more dismal picture of a 20% failure rate.
Analyzing failed submissions could pinpoint causes for failure, some of which may be addressable by
either filers or the Agency to reduce failure rates. Unfortunately, data regarding failed PMA submissions are
not made available to the public, effectively curtailing such research.
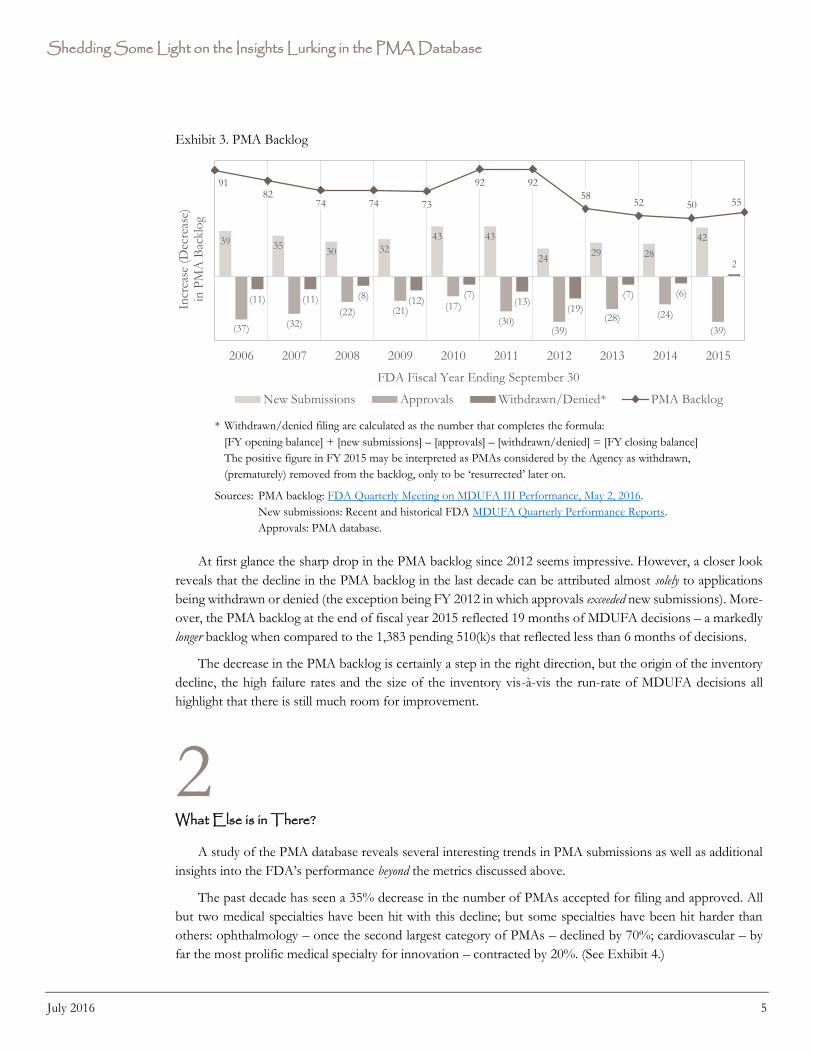
[3] PMA Backlog
A third performance metric is the inventory of pending PMAs, which is defined as original PMAs under
review or on hold. The FDA-generated graph has been supplemented by the underlying movements in
inventory – new submissions accepted for filing, approvals and withdrawals – that explain the changes in the
PMA backlog. (See Exhibit 3.)
50%
60%
70%
80%
90%
100%
2001 2002 2003 2004 2005 2006 2007 2008 2009 2010* 2011 2012 2013* 2014* 2015*
PM
A A
pp
roval
Rat
e
FDA Fiscal Year Ending September 30
Decision Cohort
Filing Cohort
what percentage of MDUFA decisions this year were 'approval'?
what percentage of PMAs accepted forfiling this year were eventually approved?
Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 5
Exhibit 3. PMA Backlog
* Withdrawn/denied filing are calculated as the number that completes the formula:
[FY opening balance] + [new submissions] – [approvals] – [withdrawn/denied] = [FY closing balance]
The positive figure in FY 2015 may be interpreted as PMAs considered by the Agency as withdrawn,
(prematurely) removed from the backlog, only to be ‘resurrected’ later on.
Sources: PMA backlog: FDA Quarterly Meeting on MDUFA III Performance, May 2, 2016.
New submissions: Recent and historical FDA MDUFA Quarterly Performance Reports.
Approvals: PMA database.
At first glance the sharp drop in the PMA backlog since 2012 seems impressive. However, a closer look
reveals that the decline in the PMA backlog in the last decade can be attributed almost solely to applications
being withdrawn or denied (the exception being FY 2012 in which approvals exceeded new submissions). More-
over, the PMA backlog at the end of fiscal year 2015 reflected 19 months of MDUFA decisions – a markedly
longer backlog when compared to the 1,383 pending 510(k)s that reflected less than 6 months of decisions.
The decrease in the PMA backlog is certainly a step in the right direction, but the origin of the inventory
decline, the high failure rates and the size of the inventory vis-à-vis the run-rate of MDUFA decisions all
highlight that there is still much room for improvement.
2 What Else is in There?
A study of the PMA database reveals several interesting trends in PMA submissions as well as additional
insights into the FDA’s performance beyond the metrics discussed above.
The past decade has seen a 35% decrease in the number of PMAs accepted for filing and approved. All
but two medical specialties have been hit with this decline; but some specialties have been hit harder than
others: ophthalmology – once the second largest category of PMAs – declined by 70%; cardiovascular – by
far the most prolific medical specialty for innovation – contracted by 20%. (See Exhibit 4.)
39 35 30 32
43 43
24 29 28
42
(37) (32)(22) (21) (17)
(30)(39)
(28) (24)
(39)
(11) (11) (8) (12)(7)
(13)(19)
(7) (6)
2
91 82
74 74 73
92 92
58 52 50 55
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
Incr
ease
(D
ecre
ase)
in
PM
A B
acklo
g
FDA Fiscal Year Ending September 30
New Submissions Approvals Withdrawn/Denied* PMA Backlog

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 6
Exhibit 4. PMA Approvals by Medical Specialty
Source: PMA database.
A variety of causes could explain the decline in the number of PMAs. Frequently cited are the economic
downturn, scarcity of venture funding, stagnating government budgets for basic scientific research and the
increasing regulatory burden that may be pushing innovation out of the U.S. and into more regulatory-friendly
geographies. Somewhat overlooked is a ‘technical’ explanation in the form of product downgrades (from Class
III to II) that have taken place in recent years, stemming the flow of PMAs in particular medical specialties:
There were two major product downgrades in radiology: bone sonometers5 in 2008 and full field digital
mammography systems6 in 2010. From 1996 to 2005 eight PMAs for bone sonometers and four for
mammography systems were approved. After the downgrade, twenty three 510(k)s for mammography
systems were cleared, seven of which were new entrants that would have required a PMA be filed had the
downgrade not occurred.
Excluding the two reclassified product codes (or, conversely, including the post-downgrade new entrants),
the number of PMAs in radiology still declined, but not as severely as Exhibit 4 indicates.
There was one significant product code downgrade in orthopedics in 2007 of intervertebral body fusion
devices that contain bone grafting material7. Between 1996 and 2002 six PMAs for such devices were
submitted and approved. After the downgrade and until the end of 2015, this product code was flooded
by more than four hundred 510(k)s submitted by 135 different companies.
Excluding the downgraded products from the two decades’ comparison lessens the severity of the decline
in the number of orthopedic PMAs. Comparing the two decades including the post-downgrade new
entrants would be misleading because the vast majority of them would probably not have entered the
market had the downgrade not occurred.
This ‘technical’ explanation only goes so far, however. No examples of similar significance can be found
to make headway towards explaining the last decade’s decline in any other medical specialty.
37%
16%
9%
6%
5%
4%
4%
4%
4%
11%
29%
9%
8%
3%
11%
9%
6%
3%
6%
15%
Cardiovascular
Microbiology
Orthopedics
Pathology
Ophthalmology
Surgery
Gastroenterolo…
Immunology
Radiology
Other
Percentage of Approved PMAs
2006-2015
1996-2005
95
40
22
16
14
11
10
10
9
28
120
38
33
11
47
36
24
14
24
63
Cardiovascular
Microbiology
Orthopedics
Pathology
Ophthalmology
Surgery
Gastroenterology…
Immunology
Radiology
Other
Number of Approved PMAs
Cardiovascular
Microbiology
Orthopedics
Pathology
Ophthalmology
Surgery
Gastro/Urology
Immunology
Radiology
All Other
38%
16%
8%
6%
5%
4%
4%
4%
4%
10%
29%
9%
8%
3%
11%
9%
3%
6%
6%
15%
Cardiovascular
Microbiology
Orthopedics
Pathology
Ophthalmology
Surgery
Immunology
Radiology
Gastroenterolo…
Other
Percentage of Approved PMAs
2006-2015
1996-2005
38%
16%
8%
6%
5%
4%
4%
4%
4%
10%
29%
9%
8%
3%
11%
9%
3%
6%
6%
15%
Cardiovascular
Microbiology
Orthopedics
Pathology
Ophthalmology
Surgery
Immunology
Radiology
Gastroenterolo…
Other
Percentage of Approved PMAs
2006-2015
1996-2005
Calendar Years
(Filing Cohort)

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 7
All PMAs are Not Created Equal
Original PMAs are a small group to begin with – averaging 35 submissions per year. One might expect
this group of products to be cohesive and behave similarly in their passage through the regulatory process,
but this could not be further from the truth. Analysis of the PMA database shows that there is quite a
divergence from the mean approval time among medical specialties. (See Exhibit 5.)
Microbiology and pathology – both ex
vivo devices – are typically characterized by
shorter-than-average review times.
Similarly, the mean approval time for
cardiovascular and ophthalmic PMAs is 2-
3 months shorter than the overall average.
At the other end of the spectrum are
surgery and orthopedics – two specialties
that are characterized by extremely long
submission review times. Several PMAs in
these two specialties have required four,
five or more years to obtain FDA approval.
These wide deviations from the mean
are not limited to a certain period in the
Agency’s recent history. In fact, they are
almost a constant. The statistical standard
deviation from the mean (depicted in Exhibit 6) has remained consistently high.
Most years in the graph below were characterized by a relative standard deviation that exceeded 70% of
the mean. Calendar year 2013 is the first cohort since 2000 to show not only shorter average PMA approval
time but also a significantly smaller relative standard deviation.
Exhibit 6. Average Time to PMA Approval Plus/Minus One Standard Deviation
* Cohorts still open. Average time to approval and the standard deviation may change.
Source: PMA database.
0
6
12
18
24
30
36
2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010* 2011 2012* 2013 2014* 2015*
Aver
age
Tim
e to
Ap
pro
val
(M
on
ths)
Calendar Year (Filing Cohort)
Exhibit 5. Mean PMA Approval Time by Medical Specialty
and their Deviation from the Overall Average
Microbiology
Pathology
Cardiovascular
Ophthalmology
Neurology
Radiology
Orthopedic
Surgery
All
Calendar Years 2006-2015 (Filing Cohort)
Source: PMA Database.
36.2
28.1
20.4
17.4
14.0
13.8
11.9
11.4
Surgery
Orthopedic
Radiology
Neurology
Ophthalmology
Cardiovascular
Pathology
Microbiology
mean: 16.5 months

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 8
The decline in the annual average time to approval for a PMA in the last five years has been accompanied
by a notable increase in the percentage of PMAs gaining approval within 12 months of filing. (See Exhibit 7.)
Exhibit 7. PMA Approvals by Review Time
* Cohorts still open. Breakdown by review time may change. 2015 cohort breakdown is preliminary.
Source: PMA Database.
During the Agency’s worst performing years (2005-2009) only one third of PMAs was gaining approval
within 12 months of filing. Calendar years 2010-2014 saw a return to the more normalized 50%-60% rates.
Multiple factors are likely contributing to these changes. One of these factors is the divergence of review
times across medical specialties, which suggests that the mix of PMA submissions across medical specialties
in any given year may affect the overall time to MDUFA decision for that specific year.
Additional factors that likely affect the annual statistics include:
Submission Risk Profiles. The inclusion and exclusion criteria for Class III medical devices that are
subject to the PMA regulatory process fluctuate over time. As discussed earlier, several product codes
originally classified as Class III have been downgraded to Class II; but at the same time products that were
previously governed by the 510(k) process have been upgraded to PMAs.
July 2012 marked the upgrade of permanent pacemaker electrodes8 from Class II to Class III. Pursuant
to this change two PMAs for pacing leads were approved in April 2015 – both were products that had
been sold commercially in the U.S. since the early 1990s.
In February 2016 the FDA issued a final order9 requiring metal-on-metal hip manufacturers to file PMAs
for their devices within three months if they want to keep their products on the market. As a result, we
can expect to see an uptick in orthopedic PMA submissions in 2016.
Product Type (Branded vs. Generic). With the regulatory pathway already in place, Class III medical
devices may begin seeing increased generic competition in upcoming years.
September 2015 marked the approval of a generic intraarticular injection of hyaluronic acid for osteo-
arthritis of the knee. The PMA submission was approved within 17 months – 43% faster than the average
of the nine other products previously approved within the same product code.
55%67%
58% 54% 55%
29%40%
33% 33% 35%46%
58% 53%44%
52%
80%
40% 14% 31%33%
38%
43%33% 47%
33%24%
32%21%
21%48%
48%
20%12%10%
14%20% 7%
13% 29%
11% 13% 21%
7% 11% 14%7%
13%21%
12% 11% 8%
2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010* 2011 2012* 2013 2014* 2015*
Per
cen
tage
of
Ap
pro
ved
PM
As
Calendar Year (Filing Cohort)
≤ 12 months 12-24 months 24-36 months > 36 months

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 9
Similarly, a handful of generic ceramic hip prostheses gained approval in less than half the time it took for
branded versions in the same product code; but a trio of generic spinal cord stimulators showed identical
average review time to branded products in the same product code.
There are too few examples of generic PMAs as of yet to indicate a trend, but it stands to reason that the
review times for generic Class III medical devices will be shorter than those of branded versions. Generic
PMAs enjoy several second-mover advantages such as using clinical data that has already been reviewed
by the Agency, leveraging the regulatory precedents created by earlier branded submissions, and avoiding
the mistakes that may have prolonged the review times of those products.
Filer Characteristics. A PMA sponsor’s maturity, regulatory experience and financial resources are all
likely to affect review time. (This is discussed and analyzed in detail in the next section.)
Review Types. A PMA may require a panel review and it may obtain expedited review status. Advisory
panels are convened for first-of-their-kind products that the Agency does not have prior experience
evaluating. Expedited status is given to products that demonstrate the potential to address unmet clinical
needs. Historically, both of these review types were guaranteed to prolong the review process.
In its May 2015 MDUFA performance update the FDA began breaking down its ‘time to MDUFA
decision’ chart into two separate graphs – for PMAs with and without a panel review. Those graphs
(reproduced in Exhibit 8) clearly depict the considerably longer review times for panel-reviewed PMAs.
Exhibit 8: Average Time to MDUFA Decision, PMAs With and Without a Panel Review
* Cohorts still open, average review time will increase.
Source: FDA Quarterly Meeting on MDUFA III Performance, May 2, 2016.
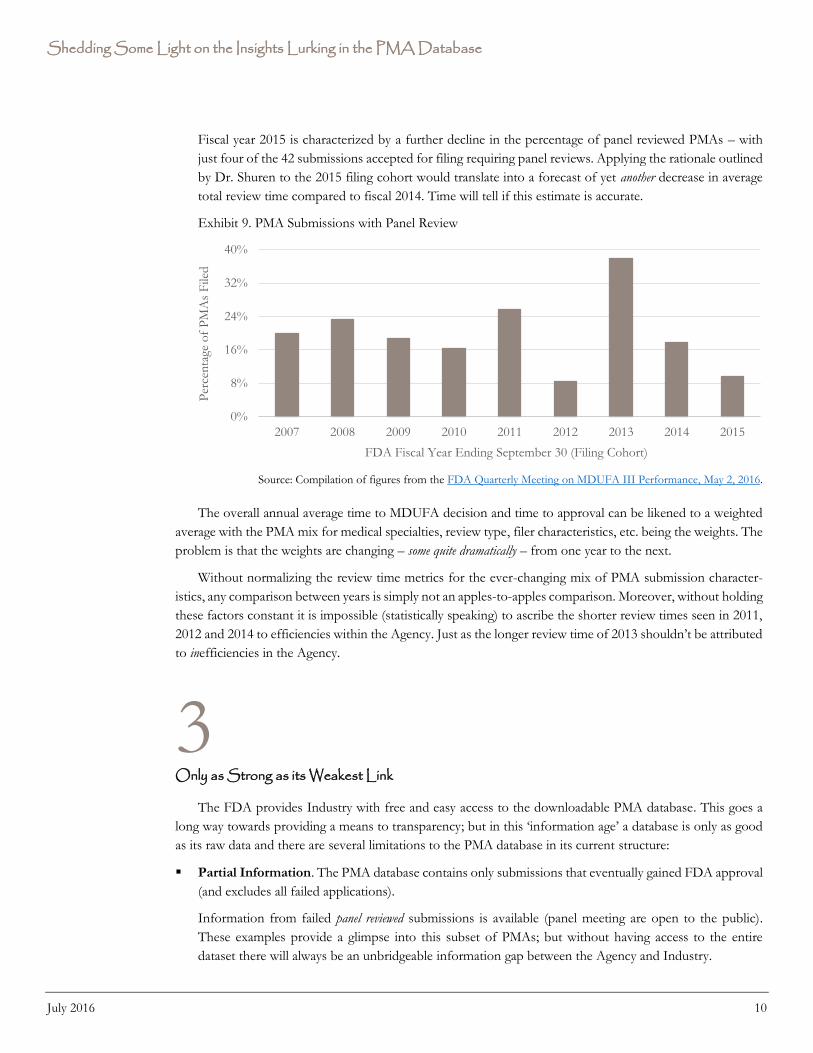
Overall time to MDUFA decision recorded a 40% increase in fiscal year 2013. This was driven by a surge
in the percentage of PMAs requiring a panel review. (See Exhibit 9.)
In his April 2015 statement to the U.S. Senate, CDRH Director, Dr. Jeffrey Shuren, stated that “A
decrease in the percent of PMAs which will go to an advisory panel in FY 2014 along with other program
improvements lead us to expect lower average total review times in FY 2014.”10
Indeed, average time to MDUFA decision for the 2014 filing cohort (which is 93% closed) declined by
40%. However, this improvement will erode somewhat as the review time for the last two PMAs awaiting
MDUFA decision has already crossed the two year mark.
180
270
360
450
540
630
720
2007 2008 2009 2010* 2011 2012 2013* 2014* 2015*
Day
s
FDA Fiscal Year Ending September 30 (Filing Cohort)
Panel Reviewed
All PMAs
Regular PMAs
Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 10
Fiscal year 2015 is characterized by a further decline in the percentage of panel reviewed PMAs – with
just four of the 42 submissions accepted for filing requiring panel reviews. Applying the rationale outlined
by Dr. Shuren to the 2015 filing cohort would translate into a forecast of yet another decrease in average
total review time compared to fiscal 2014. Time will tell if this estimate is accurate.
Exhibit 9. PMA Submissions with Panel Review
Source: Compilation of figures from the FDA Quarterly Meeting on MDUFA III Performance, May 2, 2016.
The overall annual average time to MDUFA decision and time to approval can be likened to a weighted
average with the PMA mix for medical specialties, review type, filer characteristics, etc. being the weights. The
problem is that the weights are changing – some quite dramatically – from one year to the next.
Without normalizing the review time metrics for the ever-changing mix of PMA submission character-
istics, any comparison between years is simply not an apples-to-apples comparison. Moreover, without holding
these factors constant it is impossible (statistically speaking) to ascribe the shorter review times seen in 2011,
2012 and 2014 to efficiencies within the Agency. Just as the longer review time of 2013 shouldn’t be attributed
to inefficiencies in the Agency.
3 Only as Strong as its Weakest Link
The FDA provides Industry with free and easy access to the downloadable PMA database. This goes a
long way towards providing a means to transparency; but in this ‘information age’ a database is only as good
as its raw data and there are several limitations to the PMA database in its current structure:
Partial Information. The PMA database contains only submissions that eventually gained FDA approval
(and excludes all failed applications).
Information from failed panel reviewed submissions is available (panel meeting are open to the public).
These examples provide a glimpse into this subset of PMAs; but without having access to the entire
dataset there will always be an unbridgeable information gap between the Agency and Industry.
0%
8%
16%
24%
32%
40%
2007 2008 2009 2010 2011 2012 2013 2014 2015
Per
cen
tage
of
PM
As
File
d
FDA Fiscal Year Ending September 30 (Filing Cohort)

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 11
Missing Fields. The database organically includes a ‘Yes/No’ field for PMA submissions that were
granted expedited review, yet it lacks comparable fields for panel reviewed applications and for
combination products. Similarly, the database contains the full street address, zip code and state of each
PMA’s sponsor yet lacks a field for the country.
These seemingly trivial pieces of missing information limit the axes by which the data can be stratified,
negatively affecting transparency; and for no good reason as this information is readily retrievable from
the public domain.
Lack of Standardization. The PMA database contains more than 80 PMAs that list ‘Medtronic’ as the
original sponsor. The problem is ‘Medtronic’ appears under 17 different name variations in the database.
This lack of standardization runs through the entire dataset, making it difficult to analyze the data using
commonly-available analytical software tools.
Retroactive Applicant Renaming. Once a PMA changes hands (which can happen multiple times in
chains of acquisitions or divestitures) the name of the original sponsor is overwritten with the name of the
most recent holder. For example, of the 82 PMAs that list Medtronic as the sponsor 15 submissions were
actually filed by 13 different companies that Medtronic acquired after the PMAs had been filed.
Retroactive applicant renaming results in the loss of invaluable historical information about the original
PMA sponsors. It impedes any attempt at accurately profiling the filers and irrevocably distorts the analysis
per the PMA sponsor characteristics:
- Were the original sponsors pre-revenue start-ups or multi-billion dollar corporations? Domestic or
foreign? Experienced filers or first-timers?
- Do review times vary among different stage filers? Do domestic companies have an advantage over
foreign ones? Does regulatory experience (or lack thereof) affect the speed of the review process?
A large portion of the time spent researching this paper was allocated to cleaning up the PMA database
to compensate for the shortfalls identified above. The cleanup included streamlining the data and
reconstructing the original filer identities. The database was then supplemented with additional layers of
information that provided new axes by which the data could be analyzed. These efforts were rewarded by
generating several surprising and powerful insights.
Streamlining the Data
The streamlining process began with creating a unique identifier for each sponsor. Two types of examples are:
all Medtronic name variations were unified to just ‘Medtronic’.
all the J&J medical device divisions (such as Animas, Biosense Webster, Depuy Synthes and Ethicon)
were consolidated into a single identifier under ‘Johnson & Johnson’.
Once the PMA holder data were streamlined, an image emerged of a highly centralized PMA holder
market. (See Exhibit 10.)
When Abbott closes its recently-announced acquisition of St. Jude Medical, it will become the single
largest PMA holder. Abbott and Medtronic (the previous largest PMA holder) each hold 1 out of every 10
active PMAs.

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 12
While ¾ of Abbott’s PMAs can be traced
back to acquisitions, Medtronic is characterized
by a higher proportion of internal development,
with ‘just’ half of its active Class III devices
originating in acquisitions.
Abbott’s acquisition of St. Jude Medical
may have boosted it to the overall largest PMA
holder, but Medtronic remains the largest
cardiovascular PMA holder with 21% of all
active Class III devices in the category.
Close behind Medtronic are Boston Scien-
tific and Abbott, each holding 17% of cardio-
vascular PMAs. In total, these three companies
hold 55% of all active Class III devices in the
segment making it exceptionally centralized.
In the ophthalmic space, Valeant Pharma-
ceuticals is the single largest PMA holder with
20% of active PMAs in the category. All but one
of Valeant’s Class III ophthalmic devices
originate in its acquisition of Bausch & Lomb in 2013. Bausch & Lomb itself was an active and dominant
acquirer of companies that had developed innovative ophthalmic PMA devices.
New Layers of Information
Once the original PMA sponsor identities were reconstructed, each of the PMA submissions in the data-
base was supplemented by a set of characteristics. These included:
Geographical location of the original filer (state and/or country, which was then attributed to world
regions and to a domestic vs. foreign designation);
Regulatory experience of the original filer (first time vs. experienced designation);
Stage of the original filer per one of four categories (giant, mature, early commercial and pre-revenue
based upon the trailing twelve month revenues at the time of the PMA filing).
These characteristics enabled stratification of PMAs and analysis of their review times across several
different axes, with regulatory experience and filer stage yielding the most interesting observations.
Historically, pre-revenue start-ups sponsored 10%-15% of PMAs and early-commercial companies
accounted for an additional 20%-25%. This all changed in 2006 when emerging companies disappeared
altogether from PMA sponsorship and early-commercial companies contracted by ⅔. (See Exhibit 11.)
Pre-revenue and early-commercial companies have since rebounded as a percent of PMA sponsors – with
the 2013 and 2014 filing cohorts completely back to ‘normal’. However, these companies’ resurgence came at
a price: their PMA review times increased considerably.
Exhibit 10. Snapshot of Active* PMA Holdings
* Excludes withdrawn, reclassified or converted PMAs.
Source: Enhanced PMA Database.
0%
20%
40%
60%
80%
100%
0% 20% 40% 60% 80% 100%
Per
cen
tage
of
PM
As
Percentage of Companies
All PMAs
Cardiovascular PMAs
Ophthalmic PMAs
749
248
101
N

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 13
Exhibit 11. PMA Approvals by Stage of Original Sponsor**
* Cohorts still open, percentages may change. 2015 cohort breakdown is preliminary.
** Filer stage is defined as one of four trailing-twelve-month (TTM) revenue categories at the time of the PMA filing:
Pre-revenue, Early commercial: <$100m, Mature: $100m - $1bn, and Giant: >$1bn.
TTM revenues could not be determined for all PMAs or PMA sponsors, hence not all filing cohorts sum to 100%.
Source: Enhanced PMA Database.
From 1996 to 2005 there was very little distinction in review times between different stage companies or
between experienced and first-time filers. The last decade (2006-2015) tells a different story altogether. (See
Exhibit 12.)
Early commercial companies and pre-revenue start-ups now require 4 and 8 more months, respectively,
to obtain PMA approval than they did a decade ago. The review times of mature and giant companies, on the
other hand, showed little-to-no change from the earlier to the later decade.
Similarly, inexperienced filers now require four more months to gain approval than they did in the earlier
decade whereas experienced filers’ timelines lengthened only marginally.
Across graphs, experienced filers’ review times closely resemble those of ‘giant’ corporations; and time-
lines for first-time sponsors seem to be in between those of pre-revenue and early commercial companies.
This is not a coincidence as there is significant (although not full) overlap of those cohorts.
Earlier stage companies and/or those that lack regulatory experience are at a clear disadvantage when it
comes to navigating the PMA regulatory process. Several things could explain this:
Inexperience. Younger companies, by their very nature, lack clinical and regulatory experience when
compared to mature companies. They have not yet transversed the regulatory learning curve as
experienced filers have.
Access to Resources. Start-ups often lack in-house clinical and regulatory infrastructure. Instead, they
rely on external consultants. In-house resources, by their sheer continuous presence, positively affect the
R&D process by developing a more ‘FDA ready’ product. This cannot be matched when using external
consultants, especially if these are brought in only when most of the R&D has already been completed.
7%
21%11% 8%
14% 12% 7%13% 11% 15%
9%
36%14%
19%20%
24% 24%
7%
13%13%
12%
16% 24% 26%
30%
26%
24%
12% 28%25%
18%
14%12%
30%13%
17%19%
19%
17%
16%
36% 21% 36%48% 38%
33% 60% 67% 50%
71%
46%61%
47%
37%48%
48%
2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010* 2011 2012* 2013 2014* 2015*
Per
cen
tage
of
Ap
pro
ved
PM
As
Calendar Year (Filing Cohort)
Pre-Revenue Early Commercial Mature Giant

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 14
Development Mindset. Mature companies developing innovative medical devices likely emphasize large
scale manufacturability, reimburse-ability and regulatory approvability from the earliest product ideation
phase and throughout the R&D process. This may not be the case in entrepreneurial venture backed start-
ups that may focus their efforts and limited financial resources on product engineering to the exclusion
of other aspects.
The ‘Technical’ Explanation. Start-ups may just be developing the most innovative of Class III
products. The first-of-their-kind highest-risk products that justifiably require lengthier regulatory reviews.
In the past decade 40% of all PMAs sponsored by pre-revenue and early commercial start-ups were for
first-in-product-code devices. The comparable statistic for PMAs sponsored by mature and giant com-
panies is a mere 17%.
Start-ups are in fact sponsoring PMAs higher in regulatory uncertainty and longer in review time (first-in-
code PMAs take 2 months longer than the average and 6 months longer than the median of subsequent
devices in the same product code).
Exhibit 12. PMA Review Time by Filer Stage and Experience
* n – data points for which filer stage can be determined.
N – all data points.
Source: Enhanced PMA Database.
PMAs sponsored by early-stage and/or inexperienced filers is an easily identifiable subset of submissions.
A deeper analysis of these filings may reveal where in the review process these PMAs are getting stuck, for how
long and – most importantly – why. These insights may open the door for regulatory process improvements
that are specific to this subset of filers. Unfortunately, the basic data necessary to conduct such research is not
available in the PMA database in its current form and cannot be sourced from the public domain.
Calendar Years
(Filing Cohort)
‘06-’15 ‘96-’05
Average
Median
First
Timers
Experienced
Filers
19.1
15.4 15.5
11.8
15.2
11.1
14.6
11.0
N*: 85 182 170 228
Pre-
Revenue
Early
Commercial
Mature Giant
PM
A R
evie
w T
ime
(Mo
nth
s)
22.7
16.314.6
10.8
15.1
11.6
16.2
12.3
16.2
11.0
14.1
11.0
13.7
11.0
n*: 18 48 49 103 38 78 134 124
19.2
14.2

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 15
4 How Does All of This Affect Patients?
From the outset this article has focused on the regulator and the industry, to the exclusion of a key group
of stakeholders – patients. All else aside, the bottom line is a 4-5 year average lag in often-times game-changing
medical devices reaching patients. (See Exhibit 13.)
Exhibit 13. The U.S.-EU PMA Device Lag*
* The device lag is measured as the total elapsed time between obtaining CE Mark and FDA Approval.
** PMAs for which CE Mark dates can be determined.
Devices for which only the year of CE Marking was available, a date of October 1st was assumed. There are
28 such occurrences in the dataset (11% of samples). These data points are spread across the sixteen year
period displayed in the graph such that a bias – if any – resulting from this assumption is negligible.
Sources: FDA Approval dates – the PMA database. CE Mark dates – the Summary of Safety and Effectiveness
Data documents of the PMAs, company press releases, quarterly or annual reports and investor presentations.
Some devices are reaching U.S. patients a decade or more later than they do European patients:
Medtronic’s DIAM spine stabilization system has been sold OUS since 2004 (a slightly modified design
has a marketing history dating as far back as 1997). DIAM has been used in more than 140,000 cases in
50+ countries and has not been withdrawn from marketing for any reason.
A PMA for the DIAM system was submitted in April 2014 and reviewed by the FDA’s Orthopaedic &
Rehabilitation Devices panel in February 2016. The panel voted against approval of the device on all fronts
– safety, effectiveness and risk-to-benefit ratio.
St. Jude Medical’s Amplatzer PFO Occluder gained CE Mark in February 1998. Four years later the device
obtained a Humanitarian Device Exemption (HDE) and began small-scale commercial sales in the U.S.
Those sales ceased in October 2006 when the FDA withdrew the HDE. (The Agency concluded that the
eligible patient population significantly exceeded the HDE regulatory limit of 4,000 patients per year.)
1
2
3
4
5
6
7
2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
U.S
. P
MA
Dev
ice
Lag
(Y
ears
)
Calendar Year (PMA Filing Cohort)
Average Lag
Median Lag
n**: 19 15 16 13 13 21 15 13 9 11 24 25 12 20 16 13

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 16
Since 1998 more than 85,000 Amplatzer PFO devices have shipped worldwide, including approximately
9,700 units that were shipped domestically under the HDE.
A PMA for the device was submitted in November 2012 and reviewed by the Circulatory Systems Devices
panel in May 2016. The panel vote was mixed: 15-1 for safety, 9-7 for effectiveness and 11-5 that the
device’s benefits outweigh its risks. It remains to be seen whether the PMA is approved or not.
The Blame Game
How much of the PMA device lag time can be objectively ascribed to the U.S. regulatory process and the
FDA’s execution of that legal framework? The lag was tested for two major periods – the pre-submission
period and the filing review:
The pre-submission period spans the time required to conduct the pivotal trial that generates the safety
and effectiveness evidence used to support the PMA submission, to carry out the post-study statistical
analysis and to prepare the filing. Excluded from the pre-submission period is the time required to obtain
IDE approval (the inclusion of which would increase the burden even more).
For U.S. filings the length of the pre-submission period was determined using the start date of the pivotal
trial (sourced from the clinicaltrials.gov website) as the beginning of the period; and the filing date of the
PMA as the closing date of the period. PMAs approved in the 2006-2015 calendar year filing cohort
yielded an average pre-submission period of 40 months.
For European filings the pre-submission period was assumed to be 18-24 months.
Filing review time is a straightforward measurement starting at a product’s regulatory filing date and
ending at its approval.
FDA review time is easily quantified using the information found in the PMA database. For the same
cohort of PMAs used in the pre-submission period the average review time was 18 months.
CE review time cannot be measured in the same bottom-up approach because the CE database is not
open to the public. A CE review time of 6-9 months was assumed:
- The lower end of the range (6 months) reflects anecdotal evidence from a small number of PMAs for
which information was available in the public domain.
- The upper end of the range (9 months/270 days) reflects the MDUFA performance goal of 180 ‘FDA
days’ for the Agency to issue a MDUFA decision (for submissions that do not require an advisory
committee), to which 90 ‘industry days’ were added.
The results of this methodology11 are illustrated in Exhibit 14, which quantifies the temporal burden
associated with the U.S. PMA regulatory process in excess of obtaining CE Mark.
The pre-submission period associated with U.S. regulatory approval is 1.7x-2.2x the length of the Euro-
pean equivalent. This reflects the burden of significantly larger-scale clinical trials that are necessary to generate
the clinical efficacy data required by the U.S. regulatory system (in comparison to the safety and mechanical
performance data required in Europe). These larger trials and the demand for clinical efficacy have ripple
effects on the statistical analysis as well as filing preparation time – both of which are included in this period.

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 17
The PMA filing review is 2.0x-2.9x the length of the CE review. This reflects the burden of reviewing the
massively larger regulatory filings (yet another secondary effect of the requirement to prove clinical efficacy).
This longer domestic review may also be indicative of the bureaucracy and inefficiencies that characterize large
centralized government agencies in comparison to the decentralized and privatized structure of the European
notified bodies.
Exhibit 14: Temporal Burden of the U.S. PMA Regulatory Process in Excess of its European Equivalent
Source: Enhanced PMA Database.
For the last five years, the burden associated directly with the FDA and the higher standards of the U.S.
PMA regulatory process can be estimated at 23-32 months. This represents 40%-55% of the 4-5 year long
average U.S.-EU PMA device lag. (These figures would likely increase by several months and percentage points
if time-to-IDE approval were taken into account.)
During this time the PMA sponsor may be burning through $500k-$1m per month. A back-of-the-
envelope calculation that assumes an excess burden of 27 months and a monthly burn rate of $750k yields a
direct financial burden of $20M – per product – in reaching the U.S. market.
On average, 40 PMA submissions reach MDUFA decision every year. Multiplying this number by the
excess burden of $20M per product needed to obtain U.S. regulatory approval yields a direct burden on industry
of $800m per annum. Added to this are direct loss of revenues and profits as well as indirect costs such as loss
of patent protection earlier in the commercial lifetime of the product.
Seeing the Forrest for the Trees
Yes, the U.S. PMA device lag is considerable; and, yes, the domestic regulatory system is a primary
contributor to that lag. However, the U.S.-EU device lag is just one facet of a larger picture as it focuses only
on devices that were eventually approved in both regions. There are quite a few examples of devices that
gained CE Mark and never reached the U.S. market because they failed to meet the higher standards of its
regulatory system. (See Appendix C, which is based upon an FDA report on the topic12 and supplemented by
independent research.)
Excess2006-2010
Excess2011-2015
26.6
22.6
Pre-Submission
Period
Filing Review Total
Excess
Mo
nth
s
39.8
21.8
17.7 11.76.0
8.79.0
33.5
: n : 80 94174
18.0
24.0 15.8
24.5
35.6
31.6
FDA CE Excess
Calendar Years 2006-2015 (Filing Cohort)

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 18
Many – but not all – of these unsafe or ineffective devices were eventually pulled from international
markets. Viewed through this lens, the FDA was executing its mandate and protecting American patients from
potentially significant harm from devices that are ineffective or unsafe.
But neither the FDA nor the U.S. regulatory process are infallible. There are several examples of products
that were approved domestically and later recalled or pulled from the markets entirely:
AnCure, one of the first AAA stent grafts to be approved in September 1999, was withdrawn in March
2002 following malfunctions of the delivery system and failure to report adverse events.
The Sprint Fidelis family of four cardiac pacing leads, originally approved in June 2004 under supplements
S029 and S030 of P920015, were recalled in October 2007 due to a high rate of lead fractures. At the time,
approximately 268 thousand leads had been implanted worldwide of which 176 thousand had been
implanted in U.S. patients.
The HeartMate II left ventricular assist device has undergone multiple Class I recalls since it was first
approved in 2008. The recalls stemmed from hardware and software malfunctions, some of which caused
serious injuries and patient deaths.
There are also mirroring examples of devices that were initially rejected by the Agency and later gained
approval:
Prior to the September 2015 approval of Augment® Bone Graft the FDA had issued two ‘not approvable’
letters (the first of which came after an advisory committee voted in favor of the product’s approval). In
total, 5½ years elapsed from PMA submission to final approval.
A PMA for the Watchman Left Atrial Appendage Closure device was originally filed by start-up company
Atritech in August 2008. The submission was based upon the 800-patient PROTECT AF pivotal study.
In April 2009 the FDA’s Circulatory Systems panel voted narrowly in favor of a conditional approval citing
both safety and long-term efficacy concerns. In March 2010 the Agency issued a ‘not approvable’ letter.
A second study – the PREVAIL trial – encompassing 475 patients and designed to address the limitations
of the PROTECT AF data was launched in November 2010. Shortly afterwards Atritech was acquired by
Boston Scientific. In May 2013 Boston Scientific filed a new PMA for the Watchman device, which was
approved in March 2015.
Unfortunately, the broader perspective does not change the bottom line: safe and effective life-saving
medical devices are reaching American patients an average of 4-5 years after those same devices are approved
in Europe, and half of that lag can be directly attributed to the U.S. regulatory framework and the FDA.
Knowing the difficulties ahead of them, some companies may be deferring initiation of their domestic
regulatory activities, instead prioritizing European approval and commercialization. If true, this would lay an
even greater portion of the U.S.-EU device lag on the domestic regulatory system.
Worse yet, the increased standards of U.S. regulatory framework and the costs associated with it may
cause some companies to forego the domestic market altogether, effectively curtailing the variety of products
that become available to U.S. patients.
Recognizing the need for process improvements without compromising patient safety, the FDA launched
several initiatives in 2015 that aim to increase transparency, streamline the PMA process and expedite patient
access to new treatment options:

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 19
April 2015 marked the roll-out of the new Expedited Access Pathway13 Program. A key feature of the
EAP is the data development plan, which holds the potential to shift some pre-approval clinical data
collection to the post-market setting hence shortening time-to-market.
In the year since the program was rolled out 29 requests for EAP designation were submitted, 17 of which
were accepted. These include Cohera Medical’s Sylys GI surgical sealant, PathMaker Neurosystems’
MyoRegulator non-invasive neurostimulator, Blood Purification Technologies’ wearable artificial kidney
and ALung Technologies’ Hemolung Respiratory Assist System. These products will make for interesting
case studies as to the effectiveness of the EAP program and its ability to speed up the regulatory review
process for the benefit of PMA sponsors and patients alike.
Also in April 2015, the FDA released for comments a draft guidance regarding the acceptance of clinical
data from studies conducted outside the U.S.14 The guidance addresses differences in clinical conditions,
study populations and regulatory standards in an attempt to avoid duplication of studies and enable
companies to leverage OUS clinical data towards their domestic approval.
In May 2015 the Agency issued a draft guidance that outlines the principals for the incorporation of
adaptive design methodologies into medical device clinical trials.15 Adaptive design has the potential to
improve the cost-effectiveness of clinical trials and increase their chances of success.
In June 2015 the FDA issued a draft guidance clarifying the factors it considers when assessing the risks
and benefits in approving IDE studies.16 The clarifications may bring first-in-human and feasibility studies
back to the U.S. in an effort to strengthen the domestic industry.
On paper these reforms seem to hold much promise, but it may take several years before the outcomes of
these initiatives begin demonstrating a measurable effect in the quantitative metrics presented throughout this
article.
By comparing and contrasting different stakeholders’ perspectives and by taking a deeper dive into the
data, this paper has outlined practical changes and enhancements to the PMA database that can be carried out
in the immediate present to increase transparency between the FDA and Industry.
The deeper dive has also identified several potential avenues for follow-on research, including PMAs that
do not reach a positive conclusion and PMAs that are sponsored by early-stage and/or inexperienced filers.
Insights from such research may hold the key to longer-term regulatory process improvements within the
existing framework to promote high-risk medical device innovation and shorten these devices’ time-to-market
without compromising the higher standards of the domestic regulatory system or the safety of patients.

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 20
Appendix A: PMA Approval Process
The legal framework governing the PMA
approval process is the Food, Drug and
Cosmetic (FD&C) Act, Section 515 and
the Code of Federal Regulations (CFR),
Title 21, Part 814.
The purpose of the acceptance review
is to assess the completeness of a sub-
mission and it should be completed with-
in 15 calendar days after the receipt of
the application by the FDA’s document
control center.
The objective of the filing review is to
determine the basic adequacy of the tech-
nical elements of the submission.
The PMA applicant should be notified
within 45 calendar days of receipt
whether the PMA has been filed or not.
Neither the acceptance nor the filing
reviews should evaluate the submitted
information vis-à-vis the safety or
effectiveness of the device. This is the
purpose of the substantive review – to
evaluate the content of the filing as the
basis for the MDUFA decision.
In the FDA’s December 2012 guidance
to Industry and Agency staff, the PMA
Acceptance and Filing Review Policy
provides detailed checklists for the
acceptance and filing reviews.
The checklists are tools to ensure that the
submission contains the necessary infor-
mation in order to conduct a substantive
review.
Original PMA
Received
Acceptance
Review
Accepted
PMA
Amendment
Filing
Review
Additional
Review
Cycle(s)
Filed
PMA
Amendment
Additional
Review
Cycle(s)
Substantive
Review
by
FDA
MDUFA
Decision
Closed w/o
Substantive
Interaction
Not
Approvable
Voluntarily
Withdrawn
Approvable
by Advisory
Committee
Panel
Recomm-
endation
PMA
Amendment
Additional
Review
Cycle(s)
Refuse
to Accept
Refuse
to File
Major
Deficiency
Letter
Eligible for
MDUFA
Decision
PMA
Amendment
Additional
Review
Cycle
Acceptance
Review
Not
Completed
Filing
Review
Not
Completed
Public
Meeting
Approved

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 21
Appendix B: Augment® Bone Graft PMA Review Timeline
Date Milestone / Event Time (months)
Periodic Total
Feb 4, 2010 BioMimetic Therapeutics submits PMA P100006 for Augment Bone Graft. 0 0
May 7, 2010 P100006 is accepted for filing. 3 3
Sep 3, 2010 FDA issues a deficiency letter. 4 7
Nov 19, 2010 BioMimetic files a PMA amendment in response to the FDA’s deficiency letter. 3 9
May 12, 2011 FDA’s Orthopedics and Rehabilitation Devices Panel reviews the PMA and votes in favor
of Augment, although not unanimously. The advisory committee voted 12-6 in support of
safety, 10-8 in support of efficacy and 10-8 in support of a favorable benefit-to-risk profile.
5 15
Dec 20, 2011 FDA issues a post-panel ‘not approvable’ letter for P100006 requesting that BioMimetic
submit additional information regarding its pivotal trial and post-approval studies while
outlining a pathway to potential approval that did not require additional clinical trials.
7 22
Jun 15, 2012 BioMimetic submits a PMA amendment in response to FDA’s not approvable letter. 6 28
Sep 13, 2012 BioMimetic submits an additional amendment to its Augment PMA. 3 31
Nov 19, 2012 Wright Medical Group announces the acquisition of BioMimetic Therapeutics for up to
$380 million. Half of the amount is paid upfront and the remaining $190 million is to be
paid upon achievement of regulatory and commercial milestones.
- -
Jan 7, 2013 FDA sends BioMimetic an informal request for clarification. 4 35
Feb 5, 2013 BioMimetic provides a responses to FDA’s informal request. 1 36
Mar 1, 2013 Wright Medical Group completes acquisition of BioMimetic Therapeutics. - -
Aug 6, 2013 FDA issues a second ‘not approvable’ letter for the Augment Bone Graft PMA application. 6 42
Sep 5, 2013 Wright submits an appeal to the FDA requesting a convening of the Dispute Resolution
Panel to review the scientific and clinical issues under dispute, withdrawal of the ‘not
approvable’ determination and the issuance of an approvable letter identifying the minor
outstanding issues that need to be addressed for the Agency to approve Augment.
1 43
Oct 4, 2013 Wright Medical meets with CDRH to discuss the appeal. 1 44
Oct 31, 2013 CDRH agrees to convene a meeting of the Dispute Resolution Panel. The meeting was
later scheduled for May 19, 2014.
1 45
Mar 10, 2014 ODE reaches an agreement with Wright to accept a further amendment to Augment’s
PMA application in an attempt to circumvent the Dispute Resolution Panel.
4 49
Jun 24, 2014 Wright Medical submits the last amendment prior to being declared ‘approvable’. 3 53
Oct 27, 2014 FDA issues an ‘approvable letter’ for the Augment Bone Graft PMA submission. 4 57
Sep 1, 2015 P100006 for Augment Bone Graft is finally approved. 10 67

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 22
Appendix C: Examples of Class III Devices that Did Not Make the (U.S.) Cut
Med
ical
Sp
ecia
lty
Pro
du
ct
Cate
go
ry
N
Ex
am
ple
Co
mp
an
ies,
Pro
du
cts
L
ikely
/
Actu
al
Reg
. P
ath
way
CE
M
ark
D
ate
Ou
tco
me
An
esth
esio
logy
&
Res
pir
ato
ry
1
Em
ph
asys
Med
ical
, Z
eph
yr E
nd
ob
ron
chia
l V
alve
P070025
9/
03
PM
A n
ot
app
roved
, b
ut
pro
duct
no
t w
ith
dra
wn
fro
m
inte
rnat
ion
al m
arket
s. E
mp
has
ys’ as
sets
wer
e ac
quir
ed in
2009 b
y P
ulm
on
x, w
hic
h is
spo
nso
rin
g a
new
clin
ical
tri
al t
o s
tud
y th
e Z
eph
yr E
BV
.
Car
dio
logy
AA
A S
ten
t G
raft
s 6
Bo
sto
n S
cien
tifi
c (M
inT
ec),
Van
guar
d (
Ste
nto
r)
PM
A
7/
98
Wit
hd
raw
n.
Ed
war
ds
Lif
esci
ence
s, L
ifep
ath
6/
99
Sal
es w
ere
susp
end
ed in
Ap
ril 2000 a
nd
th
e p
rod
uct
w
as r
edes
ign
ed; b
ut
the
enti
re A
AA
pro
gram
was
d
isco
nti
nued
in
Ap
ril 2004.
Joh
nso
n &
Jo
hn
son
(T
ER
AM
ed),
Fo
rtro
n (
Ari
ba)
8/
01
Wit
hd
raw
n.
Car
dia
c A
bla
tio
n
Sys
tem
s 2
Car
dim
a, R
evel
atio
n T
x M
icro
cath
eter
Sys
tem
P
020039
11/
98
PM
A n
ot
app
roved
, b
ut
pro
duct
no
t w
ith
dra
wn
fro
m
intl
. m
arket
. C
om
pan
y w
ent
out
of
busi
nes
s in
2010.
Med
tro
nic
(A
bla
tio
n F
ron
tier
s), A
bla
tio
n S
yste
m
P100008
11/
06
PM
A n
ot
app
roved
. P
rod
uct
sti
ll s
ellin
g in
Euro
pe.
Car
dio
vas
cula
r Ste
nts
1
Joh
nso
n &
Jo
hn
son
(C
on
or
Med
syst
ems)
, C
oST
AR
Dru
g-E
luti
ng
Ste
nt
PM
A
2/
06
Pro
duct
fai
led
in
piv
ota
l tr
ial;
was
wit
hd
raw
n f
rom
E
uro
pea
n m
arket
in
May
2007.
Pas
sive
Co
nst
rain
ts
to T
reat
H
eart
Fai
lure
3
Aco
rn C
ard
iovas
cula
r, C
orC
ap
P040049
9/
00
PM
A n
ot
app
roved
, b
ut
pro
duct
no
t w
ith
dra
wn
fro
m
intl
. m
arket
s. C
om
pan
y w
ent
out
of
busi
nes
s in
2010.
Par
aco
r M
edic
al, P
arac
or
Hea
rtN
et
PM
A
6/
08
Pro
duct
no
t w
ith
dra
wn
fro
m in
tern
atio
nal
mar
ket
s,
but
com
pan
y w
ent
out
of
busi
nes
s in
2012.
PF
O
Occ
lud
ers
N
MT
Med
ical
, C
ard
ioSE
AL
/ S
TA
RF
lex
PM
A
6/
97
Pro
duct
s se
llin
g co
mm
erci
ally
in
Euro
pe.
Co
mp
any
wen
t o
ut
of
busi
nes
s. I
P
was
acq
uir
ed b
y W
.L. G
ore
in
2011.
A
GA
Med
ical
, A
mp
latz
er
P120021
2/
98
Acq
uir
ed b
y St.
Jud
e in
2010.
FD
A p
anel
rev
iew
ed t
he
PM
A in
M
ay 2
016. T
he
vo
te w
as m
ixed
.
10
Vel
oci
med
, P
rem
ere
1/
05
Acq
uir
ed b
y St.
Jud
e in
2005.
O
cclu
tech
, F
igulla
N
PM
A
10/
07
C
oh
erex
Med
ical
, F
latS
ten
t E
F
7/
09
Acq
uir
ed b
y J&
J in
2015.
P
FM
Med
ical
, N
it-O
cclu
d
7/
10
Clin
ical
C
hem
istr
y
No
nin
vas
ive
Blo
od
Glu
cose
M
on
ito
r 1
Pen
dra
gon
Med
ical
, P
end
ra
PM
A
5/
03
Wit
hd
raw
n.
Ob
stet
rics
&
Gyn
eco
logy
D
iagn
ost
ic
Sys
tem
s 2
Bio
fiel
d C
orp
., B
iofi
eld
Dia
gno
stic
Sys
tem
P
MA
6/
98
Wit
hd
raw
n.
Mir
abel
Med
ical
Sys
tem
s, T
-Sca
n 2
000 E
D
P050003
12/
02
PM
A n
ot
app
roved
. M
irab
el w
as p
lan
nin
g si
mult
a-n
eous
rollo
ut
in t
he
U.S
. an
d E
U, so
pro
duct
was
n
ever
so
ld in
Euro
pe.
Co
mp
any
wen
t o
ut
of
busi
nes
s.
Ort
ho
ped
ics
Ort
ho
ped
ic
Imp
lan
ts &
M
ater
ials
5
Q-M
ed, D
uro
lan
e K
nee
P
060013
5/
01
Nei
ther
PM
A w
as a
pp
roved
. B
oth
pro
duct
s ar
e se
llin
g co
mm
erci
ally
in
Euro
pe.
F
zio
Med
, O
xiP
lex/
SP
Gel
P
070023
7/
01
Gen
eral
&
Pla
stic
Surg
ery
Bre
ast
Imp
lan
ts
1
Lip
oM
atri
x, T
rilu
cen
t P
MA
12/
94
Wit
hd
raw
n f
rom
all
mar
ket
s in
Mar
ch 1
999.
Surg
ical
Sea
lan
ts
1
Co
vid
ien
(C
on
fluen
t Surg
ical
), P
leura
Sea
l P
MA
11/
07
Wit
hd
raw
n f
rom
all
mar
ket
s in
Oct
ob
er 2
010.

Shedding Some Light on the Insights Lurking in the PMA Database
July 2016 23
Endnotes & Sources
1 There are five possible FDA MDUFA decisions for PMA submissions: [1] Approval Order, which enables
the PMA sponsor to begin commercialization of the device in the U.S.; [2] Approvable Letter, indicating that
the FDA requires resolution of minor deficiencies and/or a GMP inspection prior to approval; [3] Not
Approvable Letter, indicating that FDA does not believe that the application can be approved due to
significant deficiencies; [4] Denial Order, indicating the FDA has denied approval of the application; [5]
Acknowledgement of Voluntary Withdrawal, indicating the FDA considers the PMA submission to have been
voluntarily withdrawn if an applicant has failed to respond to an approvable, major deficiency or not
approvable letter within 180 days.
2 FDA and Industry Actions on Premarket Approval Applications (PMAs): Effect on FDA Review Clock and
Goals. Guidance for Industry and FDA Staff. October 15, 2012.
3 Acceptance and Filing Reviews for Premarket Approval Applications. Guidance for Industry and FDA Staff.
December 31, 2012.
4 Inferred from a statement made by Andrew Galligan, Nevro CFO, regarding U.S. launch six weeks earlier
than expected, during the company’s Senza FDA Approval and First Quarter 2015 Earnings Conference Call
on May 11, 2015.
5 Final Rule issued July 17, 2008 in the Federal Register Volume 73, Docket No. FDA-2005-N-0346.
6 Final Rule issued November 5, 2010 in the Federal Register Volume 75, Docket No. FDA-2008-N-0273.
7 Final Rule issued June 12, 2007 in the Federal Register Volume 72, Docket No. 2006N-0019.
8 Final Rule issued July 6, 2012 in the Federal Register Volume 77, Docket No. FDA-2011-N-0505.
9 Final Rule issued February 18, 2016 in the Federal Register Volume 81, Docket No. FDA-2011-N-0661.
10 Continuing America’s Leadership: The Future of Medical Innovation for Patients. Statement of Jeffrey
Shuren, M.D., J.D., Director CDRH, before the U.S. Senate Committee on Health, Education, Labor and
Pensions. April 28, 2015.
11 This model was adapted from a train of thought presented in an analysis prepared by The Boston Consulting
Group in June 2012 titled Regulation and Access to Innovative Medical Technologies: A Comparison of the
FDA and EU Approval Processes and Their Impact on Patients and Industry.
12 U.S. Food and Drug Administration, Unsafe and Ineffective Devices Approved in the EU that were Not
Approved in the US. May 2012.
13 Expedited Access for Premarket Approval and De Novo Medical Devices Intended for Unmet Medical Need
for Life Threatening or Irreversibly Debilitating Diseases or Conditions. Guidance for Industry and FDA
Staff. April 13, 2015.
14 Acceptance of Medical Device Clinical Data from Studies Conducted Outside the United States. Draft
Guidance for Industry and FDA Staff. April 22, 2015.
15 Adaptive Designs for Medical Device Clinical Studies. Draft Guidance for Industry and FDA Staff. May 18,
2015.
16 Factors to Consider When Making Benefit-Risk Determinations for Medical Device Investigational Device
Exemptions (IDEs). Draft Guidance for IDE Sponsors, Sponsor-Investigators and FDA Staff. June 18, 2015.
Revital Hirsch is a seasoned professional in life sciences venture capital investing, specializing in medical devices .