september 2016 - university of sheffield
TRANSCRIPT

EVALUATE THE ABILITY OF MOLECULAR DESCRIPTORS FOR PREDICTING
BIOACTIVITY PROFILES OF COMPOUNDS WITH MACHINE LEARNING METHOD
A study submitted in partial fulfilment
of the requirements for the degree of
Master of Science in Data Science
at
THE UNIVERSITY OF SHEFFIELD
by
JINGYAN SUI
September 2016

Abstract
Research context
Drug discovery is a complex process for finding new drug candidates, and it is time-
consuming and high cost (Taylor, 2015). With the rapid development of human
genome technology and pharmacology, huge amounts of potential targets and
biological activity data are producing. With the accumulation of data redundancy
and complexity, simple analysis methods have been unable to meet the demand of
large data analysis. In this situation, as a kind of fast and low cost way,
computational methods in chemoinformatics has great significance in early drug
discovery process. Computer-aided drug discovery or design methods can be used
to improve the efficiency of drug discovery (Sliwoski, Kothiwale, Meiler, & Lowe,
2014).
Bioactivity profiles for compounds are generated from their bioactivity data. Insight
of mode of actions for compounds can be got from comparison of bioactivity
profiles, beside, compounds bioactivities are strongly correlated with their chemical
structures (Cheng, Wang, &Bryant, 2010).
Aim
The aim of this dissertation is to evaluate the effectiveness of different molecular
descriptors on their ability to predict the bioactivity profiles of compounds extracted
from the open ChEMBL database.
Methodology
Bioactivity data for compounds and targets were extracted from ChEMBL database,
and transformed into bioactivity profiles for compounds by EXCEL and Rstudio.
Two kinds of 2D fingerprints (Morgan, MACCS) and physiochemical properties of
compounds were calculated by KNIME and RDkit.
One of machine learning method named clustering was used in this dissertation. One
of clustering algorithms named k-means was implemented in WEKA to cluster
compounds into subgroups based on their bioactivity profiles, 2D fingerprints
(Morgan, MACCS) ,and physiochemical properties respectively.

4
Cluster purity was applied to evaluate the ability of molecular descriptors for
predicting bioactivity profiles of compounds.
Results
Bioactivity profiles, 2D fingerprints, and physiochemical properties of compounds
was extracted or calculated from data of ChEMBL. Compounds and targets were
analysed based on bioactivity profiles extracted from ChEMBL. For most
compounds in dataset, they were tested and bound with sporadic targets. For a very
few compounds, they were tested and bound with many targets. For targets, the
situation was similar. Clustering results were got by k-means algorithm and
evaluated by cluster purity.
Conclusion
Comparison of clustering results showed that both 2D fingerprints and
Physiochemical properties could predict bioactivity profiles of compounds to some
extent. In some cases, prediction of 2D fingerprints was better than that of
Physiochemical properties, sometimes the contrary. In order to find out which
method is better, more rigorous researches need to be done.

5
Acknowledgements
First and foremost, I would like to express my sincere thanks to my supervisor
Professor Valerie J. Gillet, for her excellent support and guidance throughout this
project.
I would like to thank Dr Christina Maria Founti for helping me to calculate
molecular descriptors, and Dr Gerard JP van Westen for contributing his data for
my dissertation.
I would like to express deep gratitude to my parents and my brother, for their
continuous love and support all the time.
I would also give a special thanks to my boyfriend Pengfei Yue, for his unremitting
encouragement throughout my study in the United Kingdom.

List of Figures
Figure 1 Partial presentation of downloaded dataset…………………………….. 22
Figure 2 Confidence score………………………………………………………..25
Figure 3 Number of target that each compound tested………………….………...44
Figure 4 Percent of compounds tested with targets……………………………….45
Figure 5 Number of Target that top 100 compound tested………………………45
Figure 6 Number of target that each compound hit………………………………46
Figure 7 Percent of compounds hit targets……………………………………….47
Figure 8 Number of target that top 100 compound hit……………………………47
Figure 9 Number of compounds that each target tested………………………….48
Figure 10 Percent of targets tested with compounds……………………………..49
Figure 11 Number of Compound that top 50 targets tested………………………49
Figure 12 Number of compounds that each target hit……………………………50
Figure 13 Percent of targets hit compounds………………………………………50
Figure 14 Number of compounds that each target hit…………………………….51
Figure 15 Selectivity of compounds……………………………………………...53
Figure 16 Selectivity of targets…………………………………………………...54
Figure 17 Distribution of targets…………………………………………………57
Figure 18 Cluster dendrogram for 6μM matrix…………………………………..58
Figure 19 Clustering result for 6μM matrix (Number= 4)……………………….59
Figure 20 Clustering result for 6μM matrix (Number= 8)………………………..59
Figure 21 Cluster dendrogram for 10μM matrix…………………………………60
Figure 22 Clustering result for 10μM matrix (Number= 4)………………………60
Figure 23 Clustering result for 10μM matrix (Number=8)………………………61
Figure 24 Cluster dendrogram for van Westen matrix……………………………61
Figure 25 Clustering result for van Westen matrix (Number=4)…………………62
Figure 26 Clustering result for van Westen matrix (Number=8)…………………62
Figure 27 Purity comparison for 6μM matrix……………………………………70
Figure 28 Purity comparison for 10μM matrix…………………………………..71
Figure 29 Purity comparison for van Westen matrix…………………………….73
Figure 30 Trend of ‘1’rate………………………………………………………..74
Figure 31 Purity comparison for four matrices…………………………………..76
Figure 32 Purity comparison for four matrices…………………………………...79

7
List of Tables
Table 1 Two bioactivity profiles & near complete bioactivity profiles…………...33
Table 2 Three bioactivity profiles & near complete bioactivity profiles…………35
Table 3 Example for calculating cluster purity…………………………………..40
Table 4 Summary of three bioactivity profiles……………………………….…..42
Table 5 Summary of three complete bioactivity profiles…………………….…..51
Table 6 Compounds in three complete matrices…………………………………55
Table 7 Targets in three complete matrices………………………………………57
Table 8 Clustering results for 6μM matrix by molecular descriptors (N= 4)…… 65
Table 9 Clustering results for 6μM matrix by molecular descriptors (N= 8)…….65
Table 10 Clustering results for 10μM matrix by molecular descriptors (N= 4).......66
Table 11 Clustering results for 10μM matrix by molecular descriptors (N= 8)…….67
Table 12 Clustering results for van Westen matrix by molecular descriptors (N= 4)
.…………………………………………………………………………………..…..68
Table 13 Clustering results for van Westen matrix by molecular descriptors (N= 8)
……………………………………………………………………………………….69
Table 14 Cluster purity summary for 6μM matrix (N=4)….....................................70
Table 15 Cluster purity summary for 6μM matrix (N=8)………..……….….…….70
Table 16 Cluster purity summary for 10μM matrix (N=4)……………….………..71
Table 17 Cluster purity summary for 10μM matrix (N=8)……………….………..71
Table 18 Cluster purity summary for van Westen matrix (N=4)……………….….72
Table 19 Cluster purity summary for van Westen matrix (N=8)……….……….…72
Table 20 Summary for four matrices.………………………………….….…….…74
Table 21 Clustering results for four matrices………………………….….…….….76
Table 22 Cluster purity for four matrices……………………………….…….……76
Table 23 Clustering results for four new matrices……………….……….……..…78

Table of Contents Abstract ........................................................................................................................ 3
Acknowledgements ...................................................................................................... 5
List of Figures .............................................................................................................. 6
List of Tables................................................................................................................ 7
Chapter 1 Introduction ............................................................................................... 11
1.1 Research context .......................................................................................... 11
1.2 Aim and objectives ....................................................................................... 13
1.3 Structure of dissertation ............................................................................... 13
Chapter 2 Literature Review ...................................................................................... 14
2.1 Drug discovery ............................................................................................. 14
2.2 Data-driven medicinal chemistry ................................................................. 14
2.3 Structure-activity relationship ...................................................................... 15
2.4 Bioactivity profile ........................................................................................ 16
2.5 Molecular descriptors ................................................................................... 18
2.6 Machine learning algorithms ....................................................................... 18
2.7 Conclusion ................................................................................................... 19
Chapter 3 Methodology and implementation ............................................................. 20
3.1 Data collection ............................................................................................. 20
3.1.1 Experimental parameters and thresholds .......................................... 20
3.1.2 Data acquisition from ChEMBL ....................................................... 21
3.1.3 Data pre-processing ........................................................................... 23
3.1.3.1 Data filtering .......................................................................... 26
3.1.3.2 Data transformation ................................................................ 27
3.1.3.3 Extracting a complete bioactivity matrix ............................... 29
3.1.3.4 A bioactivity matrix generated from van Westen’s dataset. .. 34
3.2 Calculation of molecular descriptors ........................................................... 35
3.2.1 2D fingerprint .................................................................................... 35
3.2.2 Physiochemical properties ................................................................ 36
3.3 Machine learning method ............................................................................. 36
3.3.1 Concept and principle ....................................................................... 36
3.3.2 Choosing appropriate number of clusters ......................................... 37

9
3.4 Evaluation method ....................................................................................... 38
3.5 Limitations/Constraints with methodology .................................................. 40
3.6 Ethical statement .......................................................................................... 41
Chapter 4 Results and discussion ............................................................................... 42
4.1 Bioactivity profiles ....................................................................................... 42
4.1.1 Results and descriptive analysis of bioactivity profiles .................... 42
4.1.1.1 Result and analysis for compounds ........................................ 43
4.1.1.2 Result and analysis for targets................................................ 47
4.1.2 Results and comparison of complete bioactivity profiles ................. 51
4.1.3 Analysis for selectivity of compounds and targets in complete matrices
.................................................................................................................... 52
4.1.3.1 Selectivity of compounds ....................................................... 52
4.1.3.2 Selectivity of targets ............................................................... 54
4.1.4 Analysis for compounds and targets in complete matrices ............... 55
4.1.4.1 Analysis for compounds in complete matrices ...................... 55
4.1.4.2 Analysis for targets in complete matrices .............................. 56
4.2 Clustering results by bioactivity profiles ..................................................... 58
4.2.1 Clustering results for compounds in 6μM matrix by bioactivity profiles
.................................................................................................................... 58
4.2.2 Clustering results for compounds in 10μM matrix by bioactivity
profiles........................................................................................................ 60
4.2.3 Clustering results for compounds in van Westen matrix by bioactivity
profiles........................................................................................................ 61
4.3 Molecular descriptors ................................................................................... 63
4.3.1 2D fingerprints .................................................................................. 63
4.3.1.1 Morgan fingerprints ............................................................... 63
4.3.1.2 MACCS fingerprints .............................................................. 63
4.3.2 Physiochemical properties ................................................................ 63
4.4 Clustering results by molecular descriptors ................................................. 64
4.4.1 Clustering results for compounds in 6μM matrix by molecular
descriptors .................................................................................................. 64
4.4.1.1 Cluster number = 4 ................................................................. 64
4.4.1.2 Cluster number = 8 ................................................................. 65

10
4.4.2 Clustering results for compounds in 10μM matrix by molecular
descriptors .................................................................................................. 66
4.4.2.1 Cluster number = 4 ................................................................. 66
4.4.2.2 Cluster number = 8 ................................................................. 67
4.4.3 Clustering results for compounds in van Westen matrix by molecular
descriptors .................................................................................................. 67
4.4.3.1 Cluster number = 4 ................................................................. 68
4.4.3.2 Cluster number = 8 ................................................................. 68
4.5 Comparison for different clustering results.................................................. 69
4.5.1 For compounds in 6μM matrix ......................................................... 69
4.5.2 For compounds in 10μM matrix ....................................................... 71
4.5.3 For compounds in van Westen matrix .............................................. 72
4.5.4 Further research for one group of compounds .................................. 74
4.5.4.1 Comparison for clustering results of three new matrices and van
Westen matrix .................................................................................... 74
4.5.4.2 Comparison for clustering results of adjusted matrices ......... 77
Chapter 5 Conclusion ................................................................................................. 80
References .................................................................................................................. 83
Appendix .................................................................................................................... 88
R code for iteration............................................................................................. 88

11
Chapter 1 Introduction
1.1 Research context
In the field of medicine, drug discovery is the process of finding new drug candidates,
which is not an easy process. Although it is just the beginning of the work of drug
research and development, it is time-consuming and high cost (Taylor, 2015). In
history of drug discovery, researchers found new drugs by the identification of
traditional drugs or by accidental discovery of active ingredients. In modern times, to
develop new drugs, researchers start from the confirmation of drug targets. Based on
confirming the targets, the follow-up research has the basis to continue. The following
step after confirming targets is to synthesize new compounds or to optimize the
structure of existing compounds. All the synthetic need to be tested experimentally to
find out their activity. Through these experiments, some compounds can be selected
to be candidates, which are called lead compounds. The activity data obtained from
the experiments combined with the structure of compounds can be used to make a
preliminary analysis for structure activity relationship. Structure activity relationship
can effectively guide the structure optimization of the following compounds. The
process of screening and optimization is often repeated many times until get rational
compounds with sufficient activity. However, a large number of compounds will be
excluded during the process of experimental testing. The whole process is expensive,
time consuming, and inefficient. Computer-aided drug discovery or design methods
can be used to predict lead compounds, reduce the number of compounds into the
experiments, and improve the efficiency of drug discovery (Sliwoski, Kothiwale,
Meiler, & Lowe, 2014).
With the rapid development of human genome technology and pharmacology, huge
amounts of potential targets and biological activity data are producing. With the
accumulation of data redundancy and complexity, simple analysis methods have been
unable to meet the demand of large data analysis. The growth of chemoinformatics can
meet the urgent need to solve the data processing and data analysis. The main research
of chemical information is how to properly select diverse subsets of compound, how
to characterize the drug molecular characteristics, how to identify molecular structure
and biological properties, and how to develop the corresponding computer software
and hardware (Fang, Liu, & Du, 2014).

12
Bioactivity profiles of compounds can be used to indicate bioactivity of compounds
about binding with different targets. If a compound can interact with a target, it means
the compound is active to the target. An increasing number of evidences that many
compounds can interact with a set of targets are changing drug discovery methods from
a single target to multi-target paradigm (Medina-Franco et al., 2013). Meanwhile,
Chemoinformatics techniques are being developed to predict compounds that relevant
to multi-target, although they were initially design to identify compounds matched a
single target.
Generally, experimental methods is difficult to widely carry out because of the
accuracy and cost constraints. In this situation, as a kind of fast and low cost way,
computational methods in chemoinformatics has great significance in early drug
discovery process. Chemoinformatics method in the post genome era is an important
application for predicting small molecule compounds and potential targets,
accelerating the drug development process.
Machine learning methods use advanced search techniques and algorithms to identify
effective and potential patterns from data sets (Lavecchia, 2015). It can find useful
information from a large number of data, improving the utilization of information.
Several machine learning methods have been used in drug discovery process
(Lavecchia, 2015). Machine learning methods can produce models with training set to
predict biological attributes, such as efficacy or absorption, distribution, metabolism,
and excretion (ADMET) properties. Researchers can use models to predict and analyse
properties of new compounds, to sort them for the following research, and to explore
their structure–activity relations (SARs) (Lavecchia, 2015). Machine learning
approaches can also be used in high-throughput screening to predict potential
compound and target pairs. Based on the generally valid assumption that “structurally
similar molecules exhibit similar biological activity compared dissimilar or less
similar molecules” (Lavecchia, 2015), machine learning methods can also be applied
to analyse chemical structural properties of compounds to predict their bioactivity.
Machine learning techniques can improve the collection, acquisition and use of
information that submerged in a large number of data, and extract insights from

13
information to help drug researchers to make more effective decision. Machine
learning methods can also improve the level of drug discovery and accelerate the speed
of drug development.
1.2 Aim and objectives
The aim of this dissertation is to compare the effectiveness of different molecular
descriptors on their ability to predict the bioactivity profiles of compounds extracted
from the ChEMBL database. The ChEMBL database is an open large bioactivity
database of molecules for drug discovery (Gaulton et al., 2012). It is useful to find out
effectiveness of different molecular descriptors. Because, in some cases, researchers
may be interested in finding compounds that are active against multiple targets as this
will increase the chance of a compound affecting multiple pathways in the body,
alternatively they may be interested in find compounds that hit some but not other
targets as this will make the compounds more selective.
Objective 1: collect useful data from ChEMBL to generate bioactivity profiles.
Objective 2: use machine learning method to divide compounds into clusters based on
their similarity of bioactivity.
Objective 3: calculate molecular descriptors by different methods.
Objective 4: use machine learning method to divide compounds into clusters based on
molecular descriptors, getting one clustering result for each molecular descriptor.
Objective 5: compare these clustering results to evaluate the ability of molecular
descriptors for predicting bioactivity of compounds, and get the information about
which calculate method is most useful.
1.3 Structure of dissertation
This dissertation is structured as follows:
Chapter 2 discusses the theoretical basis of this dissertation, and reviews the literature
of the application of chemoinformatics and machine learning in drug discovery field.

14
Chapter 3 describes the methodology and implementation of research, such as how to
extract data from ChEMBL, which thresholds or criteria applied in extraction, how to
implement clustering, how to calculate molecular descriptors with different software,
the criterion for evaluation of clusters, and how to implement evaluation. In addition,
this chapter talks about research limitations.
Chapter 4 presents and analyses extraction results, clustering results, and evaluation
results.
Chapter 5 presents conclusion, research limitation and possible suggestion in future
research.
Chapter 2 Literature Review
2.1 Drug discovery
The development of drug discovery goes through three main periods. The first period
was nineteenth century when medicinal chemists found out drug by chance. The
second period was from early twentieth Century to late stage. During this period, new
drug structures was found and many new techniques was development, such as
molecular modelling, combinatorial chemistry, automated high-throughput screening.
Based on these new discoveries, drug discovery was developed rapidly in the late
twentieth Century. The third period is the twenty-first century. In this period, new
technologies expanded and more biopharmaceutical drugs was approved for
therapeutic use (Pina, Hussain, & Roque, 2009).
Drug discovery pipeline usually contains target identification and selection, assay
development, generation of lead compound, optimisation of lead compound, and
clinical development (Hughes, Rees, Kalindjian, & Philpott, 2011).
2.2 Data-driven medicinal chemistry

15
With the development of computer and network technology, big data era has begun.
In big data era, the way medicinal chemists undertake research, is changing (Lusher et
al., 2014). The huge amount of data is providing many new opportunities for data-
driven research and change of current practices. Besides, big data brings some
challenges in medicinal chemistry. Modern research projects are becoming more
complexity than before and researchers need to work together with scientist from
different disciplines. Furthermore, team members maybe are from different sites or
different continents. How to share, manage, and use information from different
members is a challenge. Modern research needs to access and manage huge amounts
of data, which require all researchers to have the ability as data scientists. Researcher
need to collect relevant data, use machine learning tools to extract meaningful
information, and analyse results and patterns. With the use of modern technologies,
researcher can make better decisions based on the data.
At present, several open database in chemical field are available for researchers, such
as ChEMBL and PubChem. The huge data of compounds, targets, and their
interactions could be used by researchers for investigating associations between small
molecules and targets (Cheng, Wang, & Bryant, 2010).
2.3 Structure-activity relationship
Structure-activity relationship refers to the relationship between the chemical structure
of the drug or other physiological active substance and its physiological activity, and
it is one of the main research contents of the drug chemistry. The earliest researches
about structure-activity relationship use intuitive qualitative way to speculate the
relationship between physiologically active substance structure and its activity, and
then infer the target structure and structure of the active substance.
At present, computers are used for both qualitative and quantitative structure-activity
relationship modelling. Qualitative methods are usually classification methods, for
example, predict active or inactive; whereas quantitative methods predict quantitative
values such as IC50.

16
Furthermore, quantitative structure-activity relationship that uses computer as an
auxiliary tool has become the main direction of this field. Accordingly, quantitative
structure-activity relationship has become one of the important methods for rational
drug design.
The relationship between molecular structure and biological activity across multiple
targets is important in hit selection and hit-to-lead projects (Wawer et al., 2010). Hit
selection is the process of selecting hits, and a hit is a compound with some desired
effects in a high throughput screening. Selecting compounds with desired effects is
one of the major goals for high throughput screening. After limited optimization, some
hits are identify as lead compounds. This process is called hit-to-lead process (Deprez-
Poulain & Deprez, 2004).
Cheng, Wang, &Bryant (2010) and Petrone et al. (2012) respectively compared
compounds based on their bioactivity and found that compounds with similar
bioactivity tend to hit similar targets.
2.4 Bioactivity profile
Selectivity trends have extensive implications in various field of drug discovery, such
as target selection, compound development prioritization, patient tailoring, mechanism
of action, and toxicity (Sutherland et al., 2013). Selectivity trends means the selectivity
pattern of compounds against targets, for example, what kind of compound are more
selective than other kinds or which compounds have similar selectivity. Many research
have been conducted to explore and analyse compound selectivity trends.
Davis et al. (2011) tested the interaction of 72 kinase inhibitors with 442 kinases and
the results showed interaction patterns and selectivity characteristics. From the
interaction patterns, a class of group-selective inhibitors showed similar selectivity
against a single subfamily of kinases, but dissimilar selectivity against kinases outside
the subfamily. The research also illustrated that, generally, type I inhibitors are less
selective than type II inhibitors. In this research, most type II inhibitors prefer a "DFG-
out" conformation of activation loop, while type I inhibitors do not require a "DFG-

17
out". The reason why some inhibitors show similar selectivity may be explained by
the structure-activity relationship. That is, compounds with similar chemical structure
show similar bioactivity.
Selectivity trend could be represented by bioactivity profiles for compounds and
targets. Bioactivity profiles could be represented by matrix of binary (active, inactive)
values or other values, which is used in this dissertaion. Besides, there are other kind
of bioactivity profiles. Backman & Girke (2016) introduced a ternary representation:
0 for missing or untested values, 1 for inactive values, and 2 for active values. Helal et
al. (2016) designed and evaluated a kind of bioactivity profiles in Z-score matrix. In
terms of how to generate bioactivity profiles from bioactivity values, different
researchers applied different threshold based on their need. For many research, it is
sensible default for threshold IC50 = 6μM, but it is appropriate to adjust the threshold
according to the quantity of active compounds, that is, a higher threshold is suitable
when large number of compounds would be identified as active, and a lower threshold
is suitable when small number of compounds would be identified as active (Clark &
Ekins, 2015). Paolini et al. (2006) used 10μM as activity threshold in their research,
and Bender et al. (2007) also used 10μM for IC50 or Ki in research. Martı´nez-Jime nez
et al. (2015) used 10μM for IC50, Ki or EC50 to extract bioactivity data form ChEMBL.
Insight of mode of actions for molecules could be got from comparison of bioactivity
profiles (Cheng, Wang, &Bryant, 2010). Cheng, Wang, &Bryant (2010) investigated
“correlations among chemical structures, bioactivity profiles and molecular targets of
small molecules”. They did hierarchical clustering of compounds according to their
bioactivity profiles and found that compounds were divided into clusters with similar
bioactivity. They also found that compounds bioactivities were strongly correlated
with chemical structures.
From bioactivity profiles, some properties of compounds and targets can be
statistically analysed. For example, selectivity of compounds and targets can be
analysed. Karaman et al. (2008) introduced a concept of selectivity score to do it.
Selectivity score represents the ability of a compound binding with a group of targets.
Similarly, it represents the ability of targets binding with a group of targets.

18
From bioactivity profiles, compounds with similar bioactivity can be approximately
clustered into same group by machine learning methods. Cheng, Wang, &Bryant (2010)
used hierarchical clustering to cluster compounds into groups with similar mode of
actions.
2.5 Molecular descriptors
Molecular descriptors are numerical values that represent molecules properties. They
can be used to analyse chemical structural information of molecules. Many different
molecular descriptors have been created and they can be calculated for different
purposes. There are two main descriptors: descriptors calculated from the 2D structure
and descriptors based on 3D representations. In this dissertation, two kinds of
descriptors calculated form 2D structure will be used to predict compounds selectivity,
which are physicochemical properties and 2D fingerprints. There are many kinds of
physicochemical properties such as hydrophobicity, lipophilicity, and so on.
Hydrophobicity (logP) is commonly used descriptor in drug discovery. It is an
important physicochemical property for representing the activity and transport of
compounds, which is commonly used for relatively large data sets. 2D fingerprints are
also frequently used descriptors, which are a kind of binary fragment descriptors. They
are "concerned with the chemical bonding between atoms rather than their 3D
structures", and there are two different kinds of 2D fingerprints: one kind "based on
the use of a fragment dictionary", and the other kind "based on hashed methods"
(Leach & Gillet, 2003). The good ability of 2D fingerprints for similarity searching
have been proved (Leach & Gillet, 2003).
2.6 Machine learning algorithms
Machine learning usually contains two kind of tasks: supervised leaning and
unsupervised learning. For supervised learning, training data (the input and desired
results) is usually given to build model, while for unsupervised learning, the model is
built without knowing correct labels, and it is used to divide the input data into clusters
according to their statistical properties. Classification, regression, and causal
modelling are typically supervised learning, while clustering, co-occurrence grouping,

19
and behaviour profiling are typically unsupervised learning, in addition, similarity
matching and link prediction are supervised or unsupervised learning (Provost &
Fawcett, 2013).
Lavecchia (2015) compared five kind of machine learning algorithms and their
applications in drug discovery: support vector machines, decision tree, naïve bayesian
classifier, k-nearest neighbours, and artificial neural networks. These methods are
widely used in chemoinformatics and in drug discovery. The relative software are
easily accessible and simple to implement, therefore these tools have become popular.
It is important for researchers to know how to use these methods properly to generate
useful models.
Besides, another machine learning approach - cluster method plays a wide role in many
fields such as medicine, social sciences, engineering and astronomy (Leach & Gillet,
2003). There are a large number of algorithms in this method such as hierarchical
algorithm and k-means algorithm (Witten, Frank & Hall, 2011). Most clustering
algorithm are non-overlapping, while some clustering algorithms are overlapping, that
is, one object belongs to more than one cluster (Witten, Frank & Hall, 2011). Cheng,
Wang, &Bryant (2010) applied hierarchical clustering to investigate “correlations
among chemical structures, bioactivity profiles and molecular targets of small
molecules”.
2.7 Conclusion
The continual growth of data in amount and complexity bring opportunities to drug
discovery, at the meantime, it also bring many challenges in collecting, managing, and
using big data. Chemoinformatics and machine learning methods can help to meet the
need to solve the data processing and information extraction tasks. Structure-activity
relationship is useful in hit selection and hit-to-lead projects. Based on the theory that
structurally similar molecules are likely to exhibits similar biological activity, proper
molecular descriptors that representing molecules structure properties can be used to
analyse the similarity of compounds and predict their biological activity.

20
Chapter 3 Methodology and implementation
3.1 Data collection
Experimental data and chemical structural information used in this dissertation are
collected from ChEMBL database, which is developed by European Bioinformatics
Institute. There are 11,019 targets, 1,928,903 compound records and 1,592,191 distinct
compounds in the database as well as relative bioactivity data and chemical structural
information.
ChEMBL database is an open large database of “bioactive drug-like small molecules”
(ChEMBL FAQ, 2014). There are “2-D structures”, “calculated properties” and
“abstracted bioactivities”, such as “binding constants”, “pharmacology and ADMET
data” about molecules, and these data are manually extracted from primary scientific
literature (ChEMBL FAQ, 2014). Usually, it is updated every three or four months
(ChEMBL FAQ, 2014). At present, ChEMBL database has been updated to the
ChEMBLdb21.
ChEMBL database can be used to deal with a wide range of drug discovery problems.
Data can be applied to identify “suitable chemical tools for a target”, investigate
“selectivity and off-targets effects of drugs”, and mine large-scale data (Bento et al.,
2014). Researchers can download data or software from ChEMBL to do their
research.
ChEMBL also provides users with the function of filtering data, therefore bioactivity
data and structural information used in this dissertation are filtered by ChEMBL and
downloaded in EXCEL format.
3.1.1 Experimental parameters and thresholds
There are various types of activity information in ChEMBL database, such as IC50, Ki,
EC50, and so on, with total 13,967,816 activity records. IC50 means half maximal

21
inhibitory concentration, which represents the concentration of a compound that is
needed for 50% inhibition in experiments, and EC50 means the concentration giving
half maximal effective response of a compound (Beck et al., 2012). Ki is inhibition
constant, which can be calculated from IC50 (Burlingham & Widlanski, 2003). Both of
them are measures of the effectiveness of compound.
Throughout the process of collecting data, experimental parameters and thresholds
used in this dissertation were adjusted several times, based on literatures and obtained
results of bioactivity profiles. In the beginning of collecting bioactivity data from
ChEMBL, three types of experimental parameters are considered to generate
bioactivity profiles, which are IC50, Ki, and EC50. However, in the later attempt, IC50
became the only filter parameter. The reason why changed experimental parameters is
explained in details in section of data pre-procession.
For many research, it is sensible default for threshold IC50 = 6μM, but it is appropriate
to adjust the threshold according to the quantity of active compounds, that is, a higher
threshold is suitable when large number of compounds would be identified as active,
and a lower threshold is suitable when small number of compounds would be identified
as active (Clark & Ekins, 2015). Paolini et al. (2006) used 10μM as activity threshold
in their research, and Bender et al. (2007) also used 10μM for IC50 or Ki in research.
Martı´nez-Jime´nez et al. (2015) used 10μM for IC50, Ki or EC50 to extract bioactivity
data form ChEMBL.
In this dissertation, three values (1μM, 6μM, and 10μM) of threshold were tried to
identify activity. Eventually, 6μM and 10μM were selected to filter IC50 data in order
to create several different bioactivity profiles for the later clustering and comparison.
3.1.2 Data acquisition from ChEMBL
On the home page of ChEMBL, there is a button named “browse targets” under search
box. When click this button, a new page can open and present a target tree, which show
names and quantity of various targets. When click protein kinases, another new page
can open and show search result, which contains 628 protein kinases and a summary
22
of their relative information. In order to get bioactivity data of these protein kinases,
users can choose required data by the function of “filter bioactivities”. When filter
bioactivities by IC50, Ki, and EC50, a dataset that contained interaction records of
compounds and protein kinases were created by ChEMBL. This dataset can be
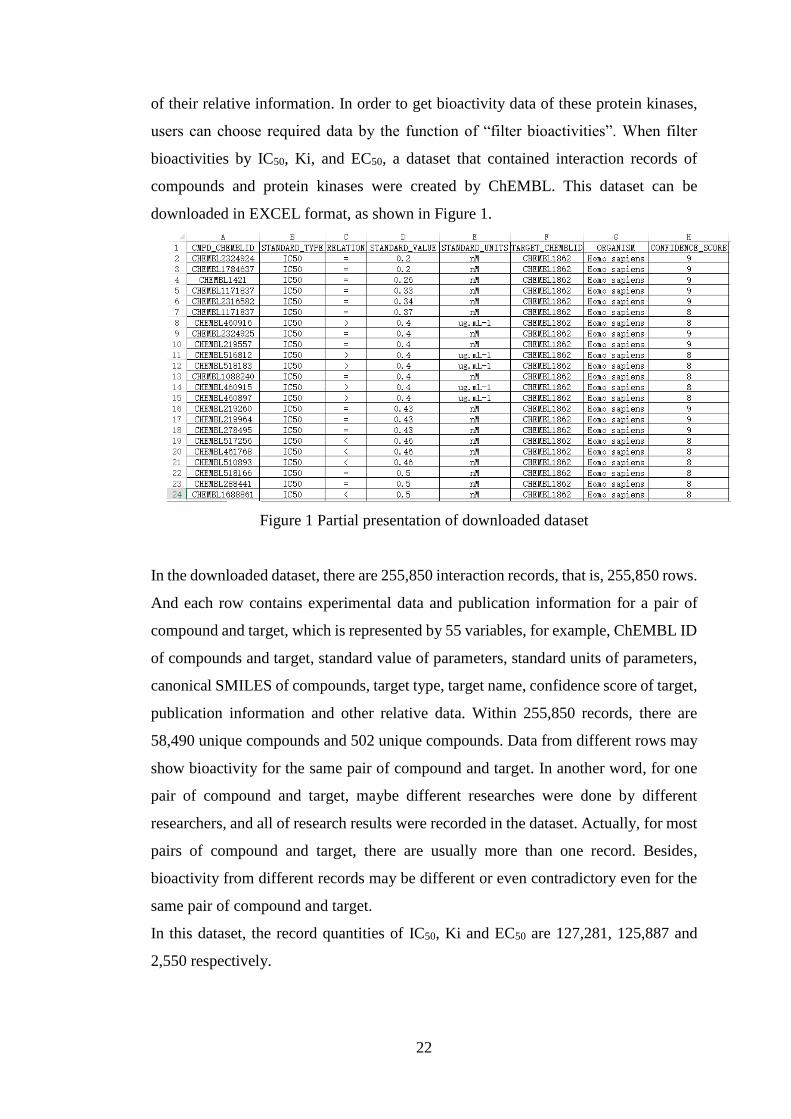
downloaded in EXCEL format, as shown in Figure 1.
Figure 1 Partial presentation of downloaded dataset
In the downloaded dataset, there are 255,850 interaction records, that is, 255,850 rows.
And each row contains experimental data and publication information for a pair of
compound and target, which is represented by 55 variables, for example, ChEMBL ID
of compounds and target, standard value of parameters, standard units of parameters,
canonical SMILES of compounds, target type, target name, confidence score of target,
publication information and other relative data. Within 255,850 records, there are
58,490 unique compounds and 502 unique compounds. Data from different rows may
show bioactivity for the same pair of compound and target. In another word, for one
pair of compound and target, maybe different researches were done by different
researchers, and all of research results were recorded in the dataset. Actually, for most
pairs of compound and target, there are usually more than one record. Besides,
bioactivity from different records may be different or even contradictory even for the
same pair of compound and target.
In this dataset, the record quantities of IC50, Ki and EC50 are 127,281, 125,887 and
2,550 respectively.

23
It should be note that these 628 protein kinases in this dataset come from 17 different
species, such as Homo sapiens, bacillus subtilis, eimeria tenella, and so on. This
dissertation focuses on protein kinases of Homo sapiens, therefore, further filtering
work need to be carried out in data pre-processing. For Homo sapiens, quantities of
IC50, Ki and EC50 in this dataset are 122,285, 124,535 and 2,438 respectively. In
another word, most records in the dataset are about Homo sapiens
3.1.3 Data pre-processing
As mentioned above, there are 255,850 interaction records in the dataset downloaded
from ChEMBL, with 55 variables. Most variables in this dataset are not needed for
creating bioactivity profiles. Therefore, delete other variables, except
CMPD_CHEMBLID, STANDARD_TYPE, RELATION, STANDARD_VALUE,
STANDARD_UNITS, TARGET_CHEMBLID, ORGANISM, and
CONFIDENCE_SCORE.
In the early stage of data pre-processing, confidence score was not considered.
Meanwhile, three experimental parameters (IC50, Ki and EC50) were used to filter
interaction records. Initially, the threshold of compound activity was set to 1μM.
When value of experimental parameters in a record was less than 1μM, the
compound recorded in this record was considered to be active to the target in the
same record. Otherwise, when the value was equal to or more than 1μM (1000nM),
the compound was considered to be inactive. On this basis, the original dataset was
transform to bioactivity matrix, the rows of which contained ChEMBL IDs of
targets and the columns of which contained ChEMBL IDs of compounds. When a
compound was considered to be active to a target, the value in the corresponding
position of the matrix was set to 1. Accordingly, when a compound was considered
to be inactive to a target, the value was set to 0.
Actually, there were usually more than one records for a pair of compound and
target, and some records are duplicate and contradictory for experiment result in the
original dataset that downloaded from ChEMBL. So a more complicated method
was used to assign values to a matrix. Taking threshold < 1000nM as an example,

24
value 1 was assigned to records whose IC50/Ki/EC50 < 1000nM, and assigned 0 to
records whose IC50/Ki/EC50 ≥ 1000nM, then the mean value of all records for the
same pair of compound and target is used as the bioactivity value of this pair of
compound and target. That is, when all records of a pair of compound and target
have value=1, their mean value is 1, then this compound is identified as active to
this target. Similarly, when all records of a pair of compound and target have
value=0, their mean value is 0, then this compound is identified as inactive to this
target. When some records for a pair of compound and target have value=1 and
some records for the same pair of compound and target have value=0, their mean
value is decimal. Therefore, there are three kind of values (1, 0, and decimals) in the
bioactivity profiles during extraction process. Then decimals were deleted from
bioactivity profiles, getting binary tables. Actually, deleting decimals does not mean
deleting compounds, but it means deleting decimal values in the matrix. In the
obtained matrix, the ratio of decimal to integer (0 and 1) is around 0.01, that is, the
ratio of deleted values is very small. In summary, filtering criteria in this dissertation
were more stringent. A compound was identified as active to a target when all
records about it show a consistent result. Contradictory results was treated as
missing value for a pair of compounds and target.
When threshold <1μM, a bioactivity matrix was created through the above method,
which has 54,397 unique compounds and 398 unique targets, with binary values 1
and 0. The reason why quantities of compounds and targets were less than original
dataset is that some were deleted during filtering process, which is described in
detail in section of data filtering. In the obtained matrix, there are a large number of
missing values. Because, for most pairs of compound and target, no experiments
had been done to provide their activity information. Then, by using a mathematical
idea and Rstudio described later, a complete bioactivity matrix was finally extracted
from this 54,397*398 matrix. In the complete matrix, there are 256 compounds and
99 targets.
However, there were two problems in this stage of processing. One problem was to
set the same threshold value for the three parameters. The other was not to consider
confidence score. From Cheng-Prusoff relationship (Burlingham & Widlanski,

25
2003), relationship between IC50 and Ki is described as IC50=Ki (1+[S]/Km) for
competitive inhibitors and IC50=Ki (1+Km/[S]) for uncompetitive inhibitors. This
means IC50 is normally bigger than Ki, unless the following situations: IC50≈Ki for
competitive inhibitors when [S]≈0, and IC50≈Ki for uncompetitive
inhibitors when [S]≈+∞ (Burlingham & Widlanski, 2003). Based on this theory, it
is not appreciate to set a same threshold value for both IC50 and Ki. In order to get
a consistent standard for IC50 and Ki, concentration of substrate and Km for each
assay are needed to transform Ki data into IC50 data, but this work is not easy to
achieve. Besides, for most compounds, they had both IC50 and Ki values. In order
to ensure the strict filtering, only using one of them is feasible. Therefore, IC50
was used to identify activity for compounds in the following work, instead of using
both IC50 and Ki at the same time. In addition, the quantity of EC50 in original
dataset is a very small number, with only 2,550 records, accounting for 2% of the
number of IC50 records. Based on the above reasons, only IC50 was used in the
following work.
Confidence score was assigned to the assay-to-target relationships by ChEMBL. It
represented “both the type of target assigned to a particular assay and the confidence
that the target assigned is the correct target for that assay”, and details about
confidence score was displayed in Figure 2 (ChEMBL FAQ, 2014).
Figure 2 Confidence score (ChEMBL FAQ, 2014)
In the second stage of data pre-processing, only IC50 was used as the parameter for
distinguishing activity. Meanwhile, confidence score was used to filter interaction
records. When adding confidence score=9 as a filter limitation, a new bioactivity

26
matrix was produced, which contained 25,194 unique compounds and 289 unique
targets, with binary values and missing values. Then a complete bioactivity matrix
was extracted from the 25,194*289 matrix, with 17 unique compounds and 11
unique targets. However, there were some rows with all “0” values, which meant
some compounds were inactive to each targets in the final matrix. Such rows in
complete matrix were not helpful for the following clustering part. After deleting
these rows, the size of the complete matrix became less, which was not big enough
for the following work.
Then, both confidence score 8 and 9 were considered to be filter limitations in the
next attempt. In this attempt, a bioactivity matrix with 44,254 unique compounds
and 330 unique targets was produced. Then a complete bioactivity matrix was
extracted from the 44,254*330 matrix, which consisted of 20 unique compounds
and 16 unique targets. But after deleting rows with all “0” values, its size reduced
to 12*16. Besides, the number of “1” value in the matrix was too small, which meant
the quantity of pairs of compound and target that could be defined as active was too
few. In another word, the threshold of IC50 needs to be larger, so that to increase the
number of pairs of active compound and target.
In the third stage of data pre-processing, two bigger values of threshold were used
to generate bioactivity matrix. They were 6μM and 10μM. At the same time,
confidence score 8 and 9 were considered to filter data. Next, taking 6μM as an
example, the method of data pre-process is explained in details. In the ChEMBL
dataset, 6μM is expressed in 6000nM.
3.1.3.1 Data filtering
On the basis of the downloaded dataset with 255,850 interaction records, filtering steps
are as following:
a. Delete other variables, except “CMPD_CHEMBLID”,
“STANDARD_TYPE”, “RELATION”, “STANDARD_VALUE”,

27
“STANDARD_UNITS”, “TARGET_CHEMBLID”, “ORGANISM”, and
“CONFIDENCE_SCORE”.
b. Filter data about “IC50”, “nM”, “Homo sapiens”, and “confidence score=9
and 8” in columns of STANDARD_TYPE, STANDARD_UNITS,
ORGANISM, and CONFIDENCE_SCORE respectively. After filtering, the
quantity of interaction records reduced to 94,600. Actually, data in ChEMBL
were manually extracted from different literatures, therefore many different
types of units are contained in dataset, such as nM, ug.mL-1, %, ucm, uM-1,
umol/dm3. The number of nM accounted for more than 90% in the
downloaded dataset.
c. There were six relations in the column “RELATION”, including “<”, “≤”,
“=”, “>”, “≥”, and “>>”. In next step, different relations were filtered
according to different thresholds. Meanwhile, uncertain records were deleted.
For example, when threshold IC50 = 6μM (6000nM), a record that IC50 > 5000
was confused to identify the corresponding compound as active or inactive
to its target, because this IC50 may be 5500 or 7000, that is, it was hard to
identified as less or more than threshold. In this case, the record need to be
deleted. The method of how to delete such records are described in the
following section.
3.1.3.2 Data transformation
a. During data preparation, each record was set a value of 1 or 0 according to
their IC50 less or more than threshold. There are two advantages to do so. One
advantage was to help to delete uncertain records, which was described in step
c. The other advantage was to help to identify and delete contradictory records,
which was described in step f. On the basis of the dataset after filtering, sort
dataset from small to large according to the value of “STANDARD_VALUE”.
b. In the column of “STANDARD_VALUE”, replace values of less than 6000
with “1”, and replace values of equal to or more than 6000 with “0”.

28
c. Filter “1” values in the column “STANDARD_VALUE”. At the same time,
filter “<”, “≤” and “=” in the column of “RELATION”, excluding “>” and “≥”.
Then get 67,294 interaction records, which are considered “active” records for
compounds. In some records, IC50 > 4000nM, which meant IC50 might be
5000nM or 7000nM. In this situation, it was hard to say the compound was
active or not. Through this step, such uncertain records were excluded.
Filter “0” values in the column “STANDARD_VALUE”. At the same time,
filter “=”, “>”, “≥” and “>>” in the column of “RELATION”, excluding “<”
and “≤”. Then get 24,801 interaction records, which are considered “inactive”
records for compounds. Similarly, in some records, IC50 < 8000nM, which
meant IC50 might be 5000nM or 7000nM. In this situation, it was also hard to
say the compound was inactive or not. Through this step, such uncertain
records were excluded.
d. Combine “active” records and “inactive” records in one sheet of EXCEL,
getting a dataset with 92,095 interaction records.,After step c, uncertain
records were deleted, then remaining records were less than records got from
section of data filtering. Through step c and d, for different threshold, different
records could be excluded. That is, remaining compounds and targets might
be different for different thresholds. Therefore, obtained bioactivity profiles
with threshold < 6μM and threshold < 10μM might be different in size,
compounds and targets.
e. With the function of pivot table in EXCEL, assign “CMPD_CHEMBLID” to
“column” and “TARGET_CHEMBLID” to “row”, getting a bioactivity matrix
with 43,866 compounds in the first column and 329 targets in the first row.
f. In order to calculate values for the bioactivity matrix, assign “mean value” to
“value” in the pivot table, getting a matrix with values of “0”, “1”, and some
decimals. “1” value in a cell meant the corresponding compound was active to
the corresponding target. “0” value in a cell meant the corresponding
compound was inactive to the corresponding target. The reason why some

29
decimals were generated in the pivot table was that some pairs of compound
and target have contradictory records as described in the early stage of data
pre-process. Decimals were helpful to identified contradictory records for
these pairs of compounds and targets that showed contradictory activity in
different experiments. The ratio of decimal to integer (0 and 1) is 0.013.
Decimal were deleted in this dissertation with IF function in EXCEL. Then a
binary bioactivity profiles were generated.
3.1.3.3 Extracting a complete bioactivity matrix
Through the above steps, a bioactivity matrix was generated, with 43,866
compounds and 329 targets (Supplementary Table 1). Actually, there are many
missing values in this table, because many pairs of compound and target were not
verified by experiment. The proportion of cells with values in the table was
represented by density rate. Density rate of this matrix was 0.528%. The
proportion of cells with value 1 in the table was represented by “1” rate. It meant
percentage of compounds binding with targets. “1” rate of this matrix was 0.372%.
Then complete bioactivity matrix could be extracted from this matrix. However,
different complete bioactivity matrices in different size could be extracted with
different methods. Even in the same way, different attempts generated different
complete matrices.
A mathematical idea was used to extract a complete bioactivity matrix from
bioactivity profiles in this dissertation. The methodology of this idea is based on the
optimization theory in mathematics. Mathematical optimization is the theory of
choosing a best solution from some available alternatives according to some criteria
(Boyd & Vandenberghe, 2004). In each iteration, rows and columns were
respectively sorted according their number of values from high to low, and some
rows and columns with least quantity of values are deleted from the inputted
matrix. That is, the complete matrix outputted from iterations is the best one for
current inputted matrix. The method is shown as following:

30
a. Sort the entire matrix by the number of values in per row and per column in
order from large to small. Then the most dense part of values appeared in the
top left of the matrix.
b. Delete some rows and columns that contained the least amount of values,
getting a new matrix with less size.
c. Resort the obtained matrix by the number of values in per row and per column,
which meant repeat the first step.
d. Repeat the second step.
e. Repeat the sorting and deleting work for many times, until get a complete
matrix.
The above iteration could be done by writing and running several functions in
Rstudio or by manually. However, the size of obtained complete matrix was not big
enough. Then, the iteration was stopped at some level before the complete matrix
was obtained, in order to retain enough compounds and targets in the final matrix to
do further analysis. This meant there were some blank space, ie missing values, in
the final matrix. With Rstudio, the density rate of each matrix that generated during
the iteration was calculated, helping users to decide which level is the appropriate
time to stop the iterative process. This could ensure the balance between the size of
final matrix and density rate of values in final matrix.
In this dissertation, an extra row named “count1” and an extra column named
“count2” were added at the end of the 43,866*329 matrix. Count1 and count2 were
used to calculate and sort the number of values in each columns and each rows.
Then, manual method and Rstudio were combined to do the iteration. Due to the
size of the matrix before iteration was relatively large, only using the manual method
was ineffective and time consuming. Meanwhile, for most compounds and targets,
only a few of them could react with each other, in another word, most of cells in the
lower right part of matrix were blank. It was more efficient for Rstudio to do
iteration after deleting these rows and columns with large range of blank cells.
The manual part follow these steps:
a. Sort the entire matrix by count1 and count2 from large to small, thus the most
dense part of values appeared in the top left of the matrix,

31
b. Delete compounds whose count2 value ≤ 3, thus the number of compound
reduced from 43,866 to 3,101, then resort matrix by count1,
c. Delete targets whose count1 value ≤ 3, then the number of targets reduced
from 329 to 244,resort matrix by count2,
d. Delete compounds whose count2 value ≤ 5, then the number of compound
reduced to 1363, resort matrix by count1,
e. Delete targets whose count1 value ≤ 5, then the number of targets reduced to
201,resort matrix by count2,
f. Delete compounds whose count2 value ≤ 6,then the number of compound
reduced to 1001, resort matrix by count1,
g. Delete targets whose count1 value ≤ 6, then the number of targets reduced to
180, resort matrix by count2.
After the manually iterative process, the size of bioactivity matrix reduced to
1001*180, which meant there were 1001 compounds and 180 targets in the matrix.
Then the matrix was inputted into Rstudio to do iterations. R code is placed in the
appendix. There are two FOR functions in the code, whose iterative parameters need
to try many times to find a best option. For example, set iterative parameters to
2:13, and run FOR functions repeatedly, after many times iterations, a 39*32 matrix
could be generated, with density rate of 74.6%. Then run more iterations, smaller
matrices could be got. The meaning of iterative parameters 2:13 is that 2 columns
and 13 rows were deleted per time.
There are some rows with all “0” values in the obtained matrix. After deleting these
rows, the size of the final matrix reduced to 28*32, with density rate of 72.54% and
“1” rate of 20.60%. “1” rate equals the number of “1” values/the number of all cells
in the matrix.
In the view of activity values were rarer than inactivity values, missing values in the
final matrix were replaced by “0”. Backman and Girke (2016) handled missing
activity values by assigning inactivity values to them, that was to say, setting them
“0” values. They also introduced another method to deal with missing values, which
was to use three values: 0 for missing or untested values, 1 for inactive values, and
2 for active values (Backman & Girke, 2016). Both methods were reasonable. The

32
first method made bioactivity profiles more clear, so it was adopted in this
dissertation.
After all the steps above, a complete bioactivity profiles was generated, which
contained 28 compounds and 32 targets, with binary values. This matrix was
obtained based on the threshold that IC50 < 6μM. In the later part, this matrix was
called “6μM matrix” (Supplementary Table 4). It’s density rate and “1” rate were
72.54% and 20.6% respectively.
Similarly, a complete bioactivity profiles could be generated based on a threshold
IC50 < 10μM. When threshold < 10μM, after data filtering, data transformation, data
cleaning, a 43856*329 matrix (Supplementary Table 2) was obtained from original
records. It’s density rate and “1” rate were 0.527% and 0.39% respectively. During
data transformation, the ratio of decimal to integer (0 and 1) is 0.01.
A question may be asked why size of matrix decreased from 43866*329 to 43856*329
with threshold increased from 6μM to 10μM. Because there were some records with
IC50 > X, where X ∈(6μM, 10μM). When filter records with threshold < 6μM,
compounds in these records were identified as inactive, while when filter records with
threshold < 10μM, these records were identified as confused records and should be
deleted, as described in step c of section data transformation. This led to less records
remaining in the second matrix, thereby the second matrix had less size.
Next, extract complete matrix from the second matrix. Delete some rows and columns
manually, following these steps using the same process as above however the number
of compounds remaining after each step is different:
a. Sort the entire matrix by count1 and count2 from large to small, thus the most
dense part of values appeared in the top left of the matrix,
b. Delete compounds whose count2 value ≤ 3, thus the number of compound
reduced from 43,856 to 3100, then resort matrix by count1,
c. Delete targets whose count1 value ≤ 3, then the number of targets reduced
from 329 to 245,resort matrix by count2,
33
d. Delete compounds whose count2 value ≤ 5, then the number of compound
reduced to 1360, resort matrix by count1,
e. Delete targets whose count1 value ≤ 5, then the number of targets reduced to
201,resort matrix by count2,
f. Delete compounds whose count2 value ≤ 6,then the number of compound
reduced to 997, resort matrix by count1,
g. Delete targets whose count1 value ≤ 6, then the number of targets reduced to
180, resort matrix by count2.
During iteration with Rstudio, iterative parameter was set to 2:13, which meant 2
columns and 13 rows were deleted per time. After iteration, a 35*32 matrix was
generated. After deleting rows with all 0 values and dealing with missing values, a
complete 27*32matrix was obtained, with binary values. In the later study, this matrix
was called “10μM matrix” (Supplementary Table 5). It’s density rate and “1” rate were
74.77% and 22.22% respectively.
In summary, two different bioactivity profiles and near complete matrix were
extracted from the downloaded dataset.
Bioactivity profiles Near complete bioactivity profiles
Number of
compound
Number
of target
Density
Rate “1” Rate
Number of
compound
Number
of target
Densit
y Rate
“1”
Rate
Threshold
IC50<6μM 43866 329 0.528% 0.372% 28 32 72.54% 20.60%
Threshold
IC50<10μM 43856 329 0.527% 0.390% 27 32 74.77% 22.22%
Table 1 Two bioactivity profiles & near complete bioactivity profiles
Can be seen from the table, sizes of bioactivity profiles are different between using
different thresholds, so is complete bioactivity profiles. The reason is that valid
records extracted from the downloaded dataset are different with different
thresholds, as described in details in section of data transformation. That is, different
compounds and targets are obtained between two extractions. Therefore, sizes of
two bioactivity profiles are different. The methodology of extracting complete

34
matrix from bioactivity profiles is based on the idea of optimization in mathematics.
In each iteration, rows and columns with least quantity of data are deleted from the
inputted matrix. That is, the complete matrix outputted from iteration is the best one
for current inputted matrix. Therefore, the two complete matrices are the best to
their original matrices respectively. They do not necessarily have the same
compounds and targets, because the original matrices are different. Besides, it may
be good for clustering repeatedly with different datasets. This may lead to a more
objective result. Of course, another method can be used to extract complete matrix
for different thresholds. It is choosing the same compounds and targets for 10μM
matrix as that in 6μM matrix. This method is easy to implement. However, too single
sample may bias experiment result. Therefore, in this dissertation, different samples
are used to avoid bias result. Actually, the two complete matrices are not significant
different because of the similar thresholds. However, if thresholds are significant
different, there may be much difference between complete matrices.
3.1.3.4 A bioactivity matrix generated from van Westen’s dataset.
A researcher named van Westen (Personal communication, July 2016) also
extracted interaction records from ChEMBL for some compounds and targets. In
order to do more comparison and analysis for different bioactivity profiles, an extra
bioactivity matrix was extracted from his dataset. His dataset was in SD format, and
an Open Source data mining Platform named KNIME was used to read and rewrite
the SD file into EXCEL format (Mazanetz, Marmon, Reisser, & Morao, 2012).
In this dataset, there were 237,081 interaction records and more than one type of targets,
including protein kinases of Homo sapiens. van Westen applied 6.5 log units (~300
nM) as his threshold to identify compounds as active or inactive. After screening
protein kinases of Homo sapiens from the dataset, 30,335 records were obtained. Then
these interaction records were transformed into bioactivity matrix for corresponding
20188 compounds and 181 targets with the function pivot table in EXCEL
(Supplementary Table 3). It’s density rate and “1” rate were 0.83% and 0.476%
respectively.

35
An extra row named “count1” and an extra column named “count2” were added at
the end of the 20188*181 matrix. Count1 and count2 were used to calculate the number
of values in each columns and each rows. Then the 20188*181 matrix was sorted by
count1 and count2 from large to small, thus the most dense part of values appeared in
the top left of the matrix. After deleting compounds whose count2 value ≤ 8, the
number of compound reduced to 105, then resorted matrix by count1. After deleting
targets whose count1 value ≤ 11, the number of targets reduced to 108. During iteration
with Rstudio, iterative parameter was set to 1:1, which meant 1 columns and 1 rows
were deleted per time. After iteration, a 27*30 matrix was generated. After deleting
rows with all 0 values and dealing with missing values, a complete 26*30 matrix was
obtained, with binary values. In the later part, this matrix was called “van Westen
matrix” (Supplementary Table 6). It’s density rate and “1” rate were 75% and 35.9%
respectively.
In summary, three near complete matrices were generated at last.
Bioactivity profiles Near complete bioactivity profiles
Number of
compound
Number
of target
Density
Rate “1” Rate
Number of
compound
Number
of target
Density
Rate “1” Rate
Threshold
IC50<6μM 43866 329 0.528% 0.372% 28 32 72.54% 20.60%
Threshold
IC50<10μM 43856 329 0.527% 0.390% 27 32 74.77% 22.22%
van Westen’s
data
IC50<300nM
20189 181 0.830% 0.476% 26 30 75.00% 35.90%
Table 2 Three bioactivity profiles & near complete bioactivity profiles
3.2 Calculation of molecular descriptors
3.2.1 2D fingerprint
There are many different types of fingerprints for compounds, for example ECPF,

36
Morgan, and MACCS. ECFP algorithm was originated from a variant of Morgan
algorithm, and it made some changes to Morgan (Rogers & Hahn, 2010). Rogers
and Hahn (2010) described that “an iterative process assigned numeric identifiers to
each atom” from which a 2D fingerprint is generated. MACCS algorithm generated
MACCS keys for a molecule, and the result is a 167-bit vector. The Morgan
fingerprint, which is very similar to ECFP, and MACCS fingerprints were used as
calculated molecular descriptors in this dissertation.
RDKit is integrated into KNIME and was used to calculate 2D descriptors from
SMILES strings for compounds. 2D fingerprints calculated by Morgan and MACCS
algorithms for compounds were presented in the form of string values, which need
to be split into single digits like 0 and 1, generating a matrix with binary values. An
easy method for the transformation was to copy and paste these string values into a
word file, then add commas behind each character and save the word file as notepad
format. After that, opened a new EXCEL file, and imported data through the
function “DATA” & “FROM NOTEPAD”, splitting string value into single digit.
Eventually, a binary matrix was generated in EXCEL format.
3.2.2 Physiochemical properties
Five kinds of physiochemical properties were contained in matrix, which were
SlogP, SMR, LabuteASA, TPSA, and ExactMW. Actually, this is a small number
of physiochemical properties. This is because of the limitation of software. RDKit
was integrated into KNIME to calculate physiochemical properties from SMILES
for compounds. The obtained matrix was standardized with the formula Z=(x-
mean(x))/STD(x) (Larsen & Marx, 1986). The standardization was done in EXCEL
with the formulas AVERAGE and STDEV.S.
3.3 Machine learning method
3.3.1 Concept and principle

37
In this dissertation, k-means algorithm in WEKA was used more than once to divide
compounds into clusters based on their bioactivity profiles or molecular descriptors.
K-means method is a typical clustering algorithm based on Euclidean distance, using
Euclidean distance as the similarity evaluation index, that is, when the Euclidean
distance between two objects is less, their similarity is greater. The principle of k-
means method is following this:
Firstly, choose K objects from data set as the initial cluster centres, and the remaining
other objects are assigned to its nearest clusters respectively according to Euclidean
distance (cluster similarity) between each object and each cluster centre.
Secondly, calculate each new cluster centre for the received clusters, then objects are
assigned to a nearest clusters based on each new cluster centre.
Thirdly, keep repeating the process of calculating and assigning, until no change
occurs.
The compounds within the same cluster have similar properties, while compounds
from different clusters have dissimilar properties.
3.3.2 Choosing appropriate number of clusters
Leskovec, Rajaraman & Ullman (2014) described a method to decide the appropriate
number of clusters in their book. They suggested to start clustering dataset with N=2.
N represented the number of clusters in this dissertation. When average diameter of
clusters did not change significantly with the increase of N, this value of N was the
appropriate number. Here, the diameter of a cluster was the maximum distance
between any two points of the cluster.
In clustering result of WEKA, a parameter named “within cluster sum of squared errors”
showed the discrete degree of points within cluster. When this value is greater, distance
between points is greater, which means the diameter of a cluster is greater. Thereby,
this parameter can also reflect the diameter of a cluster.

38
In the clustering process, value of this parameter continued to become smaller as N
increased. This means there is no suitable K for datasets in this dissertation. The reason
for this situation may be the size of dataset in this dissertation is not big enough, so
that distances between points are relatively large. In this case, the appropriate N can
be set according to the need of research.
3.4 Evaluation method
In this dissertation, clusters generated using 2D fingerprints and physiochemical
properties were compared with clusters based on the bioactivity profiles
respectively. In another word, clustering result of bioactivity profiles was treated as
standard class information to evaluate cluster quality of molecular descriptors,
which was evaluating the ability of molecular descriptors for predicting bioactivity
profiles. In this case, several methods can be used to evaluate the quality of clusters,
such as measuring cluster purity, running classification algorithms, and measuring
precision, recall or F-measure.
In this dissertation, cluster purity was used to do evaluation. Manning, Raghavan,
& Schutze (2008) described a formula to calculate cluster purity as following:
Purity(Ω, C) = 1
𝑁∑ max
𝑗|𝜔𝑘
𝑘
∩ 𝑐𝑗|
In this formula, Ω is clusters, C is classes, and N is the number of all objects. Based
on the formula, Objects in cluster k were compared with each class, then each object
in cluster k was labelled by class number, that is, after comparison, it can be got that
how many objects in cluster k belonged class1, class2, class 3…, for example, the
corresponding quantity was k1, k2, k3…, then chose the biggest one of these numbers.
With the same steps, objects in each cluster were compared with each class, then the
biggest number for each cluster was got. At last, these biggest numbers for each
cluster were added up to get a total number, and the total number was divided by N
to get a result, which was purity (Ω, C).

39
In this dissertation, cluster of bioactivity profiles was standard class for compounds,
and clusters of molecular descriptors were evaluated to see how similar they were
to the standard cluster. When the purity was higher, the similarity was higher.
The calculation for purity could be done in EXCEL. For example, as shown in Table
3, a group of compounds (28) were clustered into 4 subgroups by bioactivity profiles
and Morgan fingerprints respectively. The results for these two methods were shown
in columns “Experimental cluster” and “Morgan cluster” respectively. Experimental
cluster was treated as standard class. And Morgan cluster was compared with
standard class to get the conclusion that how similarity between these two clusters.
For easy of counting, compounds in different Morgan clusters were distinguished
by colours. In the right part of the following table, the “YES” in the first yellow
row means that CHEMBL1094408 was in both Morgan cluster1 and Class0.
Similarly, position of each compound was shown in the table. “YES” could be
calculated by IF function in EXCEL, for example, IF(COUNTIF($A$2:$A$10,
C2)>0, "YES", "").
Apparently, among four classes, compounds in Class0 appeared most frequently in
Morgan cluster0, and other cluster also could got the class that compounds appeared
most frequently. Then count the quantity of compounds in the obtained pairs of
cluster and class.
Purity (Morgan, Class)
= 1
28 (Morgan0∩Class0+ Morgan1∩Class1+ Morgan2∩Class2+ Morgan3∩Class3)
= 1
28 (4+1+11+1)
= 0.68
On a special note, in the obtained results, the same serial number of each cluster and
class in this example was just a coincidence.
CMPD_CHEMBLID Experimental
cluster CMPD_CHEMBLID
Morgan
cluster Class0 Class1 Class2 Class3
CHEMBL103667 0 CHEMBL1094408 0 YES

40
CHEMBL1094408 0 CHEMBL1738758 0 YES
CHEMBL1738758 0 CHEMBL1933576 0 YES
CHEMBL1929238 0 CHEMBL1933582 0 YES
CHEMBL1933576 0 CHEMBL564829 0 YES
CHEMBL1933582 0 CHEMBL1834657 1 YES
CHEMBL2148053 0 CHEMBL2148053 1 YES
CHEMBL2312303 0 CHEMBL103667 2 YES
CHEMBL2312304 0 CHEMBL1230607 2 YES
CHEMBL1258913 1 CHEMBL1258913 2 YES
CHEMBL1834657 1 CHEMBL1270230 2 YES
CHEMBL388978 1 CHEMBL1929238 2 YES
CHEMBL402548 1 CHEMBL225519 2 YES
CHEMBL564829 1 CHEMBL2312303 2 YES
CHEMBL1230607 2 CHEMBL2312304 2 YES
CHEMBL1270230 2 CHEMBL2403108 2 YES
CHEMBL225519 2 CHEMBL388978 2 YES
CHEMBL461139 2 CHEMBL402548 2 YES
CHEMBL461140 2 CHEMBL460472 2 YES
CHEMBL565612 2 CHEMBL461139 2 YES
CHEMBL590109 2 CHEMBL461140 2 YES
CHEMBL597754 2 CHEMBL565612 2 YES
CHEMBL599224 2 CHEMBL590109 2 YES
CHEMBL599428 2 CHEMBL597754 2 YES
CHEMBL604712 2 CHEMBL599224 2 YES
CHEMBL2403108 3 CHEMBL599428 2 YES
CHEMBL373882 3 CHEMBL604712 2 YES
CHEMBL460472 3 CHEMBL373882 3 YES
Table 3 Example for calculating cluster purity
3.5 Limitations/Constraints with methodology
a. In this dissertation, only IC50 was used to identify activity for compounds,
which did not make full use of various activity data in ChEMBL, hoping that
a better way could be found to identify bioactivity for compounds.

41
b. A small number of physicochemical properties was used for this descriptor,
because of the limitation of software. Actually, there are many others that
could have been used.
c. When a dataset is huge, it is inefficiency and time consuming to pre-process
the dataset in EXCEL.
d. In the process of extracting complete bioactivity profiles, the written code for
iteration was not the best. The final matrix got after iteration was just one of
many possibilities, and may not be the best one. This was actually a waste of
data resources.
e. Because of the theory described by Leskovec, Rajaraman & Ullman (2014)
for choosing the number of clusters was not suitable for datasets in this
dissertation, different values of N were tried for the following study. Actually,
this tentative approach of determination for N was not rigorous.
f. The efficiency for calculating cluster purity in EXCEL was not high, hoping
to find a better way to do calculations.
g. Clustering was done by a single way with WEKA and K-means algorithm,
which may be not enough to eliminate the errors caused by software and
algorithms themselves. A comprehensive comparison may be necessary to
apply a variety of soft wares and algorithms in future research.
h. In this research, not all the compounds and target data contained in ChEMBL
could be used to study the selectivity for compounds. Activity data for
selected sets of compounds and targets were required to be verified by
previous experiments. However, a considerable portion of compounds and
targets did not have corresponding experimental data. Based on this limitation,
the data collected from ChEMBL was not sufficient for the research, which
led to the analysis results was not perfect as predicted.
3.6 Ethical statement
This dissertation has no risk. The topic of this dissertation is a normal question in
drug discovery field, not a sensitive topic. There is no need for this dissertation to
choose participants for interviews or surveys, so there is no ethical issues for
considering sample or privacy.

42
Most data used in this dissertation were extracted from the online open ChEMBL
database. Some data were from another researcher named Gerard van Westen, and
the using of his data was permitted by him.
Chapter 4 Results and discussion
4.1 Bioactivity profiles
Three bioactivity profiles were created from ChEMBL database with different
thresholds. Two of them were created directly from dataset that downloaded from
ChEMBL, and one was created from van Westen’s dataset that also came from
ChEMBL. Accordingly, three complete bioactivity profiles were extracted from
these three bioactivity profiles.
4.1.1 Results and descriptive analysis of bioactivity profiles
Bioactivity profiles Number of compound Number of target Density
Rate “1” Rate
Threshold IC50<6μM 43866 329 0.528% 0.372%
Threshold IC50<10μM 43856 329 0.527% 0.390%
van Westen’s data
IC50 < 300nM 20189 181 0.830% 0.476%
Table 4 Summary of three bioactivity profiles
Although the first two matrix were extracted from ChEMBL with same
methodology, their sizes were slightly different. The reason why this happened were
explained in extraction section.
Can be seen from the first rows of Table 4, “1”rate increased with the increased
value of threshold. That is, the number of binding pairs of compounds and targets
increased with the increased thresholds. Actually, based on the different compounds
in these two matrices, “1” rate should not be compared directly between two

43
matrices. It is reasonable to compare “1” rate between two matrices with same
compounds and targets. Therefore, different compounds in that two matrices were
deleted, getting two new matrices with same compounds. Meanwhile, targets in two
matrices were same, then no need to change them. Size of new matrices was
43856*329, and their “1” rate were 0.3716% (6μM) and 0.3896% (10μM).
Obviously, it was true that “1” rate was increased with threshold increasing.
It is interesting that the third matrix in Table 4 had the highest “1” rate, while it
applied the lowest threshold. After researching values of IC50 for some pairs of
compounds and targets, one reason for this might be the difference between criteria
applied in different extraction. For example, for compound CHEMBL535 and target
CHEMBL279, there are 40 interaction records of IC50 in ChEMBL. Some values
are less than 300nM, while some values are far greater than 300nM. In this situation,
compound CHEMBL535 was identified as active to target CHEMBL279 by van
Westen, but, based on extraction criteria in this dissertation, compound
CHEMBL535 could not be identified as active to target CHEMBL279, because their
records did not show a consistent result. In this dissertation, the value in the
corresponding cell associated with the compound and target was assigned to a
missing value. However, the number of such missing values was not so much as to
cause obvious gap as shown in the table. Therefore, there might be other reasons.
4.1.1.1 Result and analysis for compounds
In this part, the first bioactivity profiles is analysed in details as an example. There
were 43866 compounds and 329 targets in the result.
a. Test situation
Compounds in the matrix were sorted from large to small by the number of targets
that each compound tested with. From the left part of Figure 1, difference in the
quantity of tested targets was significant for small portion of compounds. For these
compounds, the number of targets tested with them was relatively large. Form the

44
bottom of Figure 1, the curve was smooth, indicating that difference in the quantity
of tested targets was very small for most compounds. For most compounds, they
were tested with sporadic targets.
From Figure 3, compounds that were tested with more than 10 targets accounted for
less than 1%. Can be seen from Figure 4, more than 99% of compounds were tested
with less than 9 targets.
According to the quantity of targets that each compound tested with, top 100
compounds were ranked and showed in Figure 5. There are only 8 compounds that
were tested with more than 60 targets. Only top 20 compounds were tested with
more than 40 targets. For the other 80 compounds, the number of targets that each
compound tested varied from 40 to 20.
Figure 3 Number of target that each compound tested
0
20
40
60
80
100
120
140
160
0 10000 20000 30000 40000 50000
Nu
mb
er o
f Ta
rget
s
Number of Compounds

45
Figure 4 Percent of compounds tested with targets
Figure 5 Number of Target that top 100 compound tested
b. Hit situation
Compounds in the first matrix were sorted from large to small by the number of
targets that each compound hit. From the left part of Figure 6, difference in the
quantity of targets that each compound hit was significant for small portion of
compounds. For these compounds, the number of targets that they hit was relatively
large. Form the bottom of Figure 6, the curve was smooth, indicating that difference
99.046%
0.678%
0.165%
0.052%
0.032%
0.009%
0.002%
0.005%
0.000%
0.002%
0.009%0.000%
10.000%
20.000%
30.000%
40.000%
50.000%
60.000%
70.000%
80.000%
90.000%
100.000%
0-9 10-19 20-29 30-39 40-49 50-59 60-69 70-79 80-89 90-99 100and
more
Perc
ent
of
com
po
un
ds
Number of targets that tested with compounds
0
20
40
60
80
100
120
140
160
0 20 40 60 80 100
Nu
mb
er o
f ta
rget
s
Number of compounds

46
in the quantity of hit targets was very small for most compounds. For most
compounds, they were active to sporadic targets.
From Figure 7, compounds that were active to more than 10 targets accounted for
less than 0.3%. More than 99.7% of compounds were active to less than 9 targets.
According to the quantity of targets that each compound hit, top 100 compounds
were ranked and showed in Figure 8. There are only 2 compounds that were active
to more than 40 targets. Only top 20 compounds are active to more than 19 targets.
For the other 80 compounds, the number of targets that each compound hit varied
from 19 to 10.
Figure 6 Number of target that each compound hit
0
10
20
30
40
50
60
70
80
90
100
0 10000 20000 30000 40000 50000
Nu
mb
er o
f Ta
rget
s
Number of Compounds

47
Figure 7 Percent of compounds hit targets
Figure 8 Number of target that top 100 compound hit
4.1.1.2 Result and analysis for targets
In this part, the first bioactivity profiles was analysed in details as an example too.
a. Test situation
Targets in the matrix were sorted from large to small by the number of compounds that
each target tested with. From the left part of Figure 9, difference in the quantity of
99.761%
0.196%
0.036%
0.002%
0.002%
0.000%
0.000%
0.000%
0.000%
0.002%
0.000%
10.000%
20.000%
30.000%
40.000%
50.000%
60.000%
70.000%
80.000%
90.000%
100.000%
0-9 10-19 20-29 30-39 40-49 50-59 60-69 70-79 80-89 90-99
Perc
ent
of
com
po
un
ds
Number of targets that hit compounds
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100
Nu
mb
er o
f Ta
rget
s
Number of Compounds
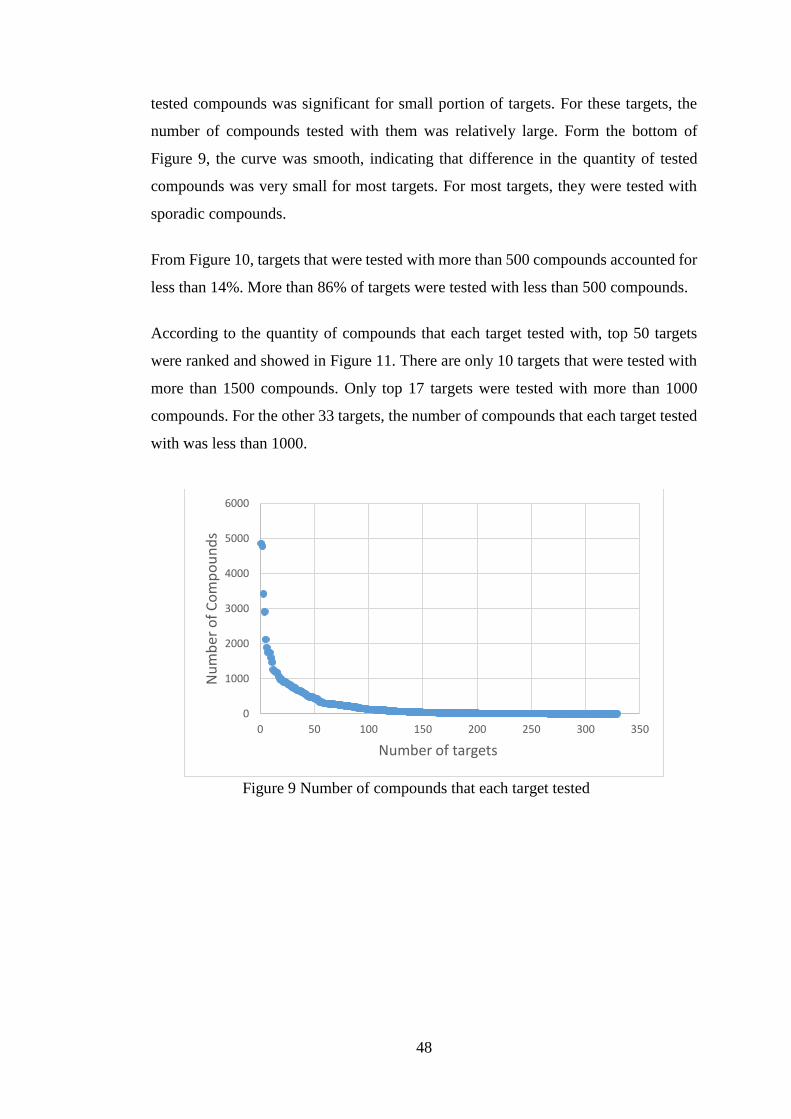
48
tested compounds was significant for small portion of targets. For these targets, the
number of compounds tested with them was relatively large. Form the bottom of
Figure 9, the curve was smooth, indicating that difference in the quantity of tested
compounds was very small for most targets. For most targets, they were tested with
sporadic compounds.
From Figure 10, targets that were tested with more than 500 compounds accounted for
less than 14%. More than 86% of targets were tested with less than 500 compounds.
According to the quantity of compounds that each target tested with, top 50 targets
were ranked and showed in Figure 11. There are only 10 targets that were tested with
more than 1500 compounds. Only top 17 targets were tested with more than 1000
compounds. For the other 33 targets, the number of compounds that each target tested
with was less than 1000.
Figure 9 Number of compounds that each target tested
0
1000
2000
3000
4000
5000
6000
0 50 100 150 200 250 300 350
Nu
mb
er o
f C
om
po
un
ds
Number of targets
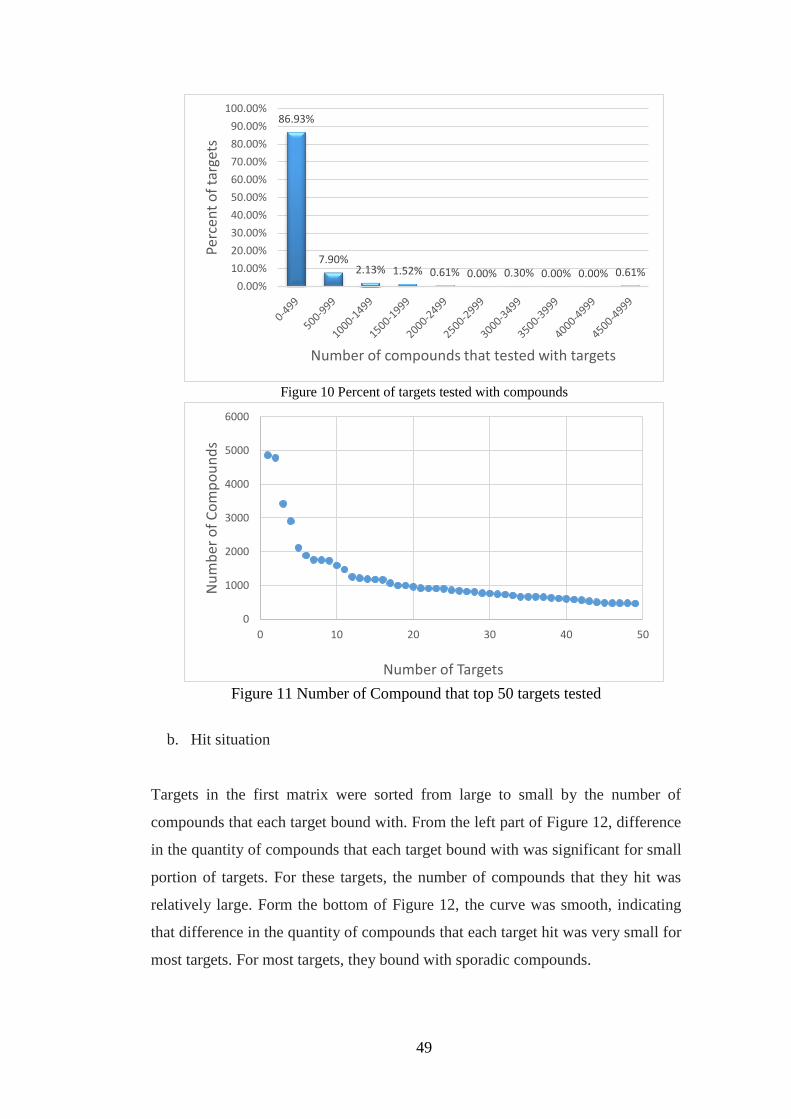
49
Figure 10 Percent of targets tested with compounds
Figure 11 Number of Compound that top 50 targets tested
b. Hit situation
Targets in the first matrix were sorted from large to small by the number of
compounds that each target bound with. From the left part of Figure 12, difference
in the quantity of compounds that each target bound with was significant for small
portion of targets. For these targets, the number of compounds that they hit was
relatively large. Form the bottom of Figure 12, the curve was smooth, indicating
that difference in the quantity of compounds that each target hit was very small for
most targets. For most targets, they bound with sporadic compounds.
86.93%
7.90%2.13% 1.52% 0.61% 0.00% 0.30% 0.00% 0.00% 0.61%
0.00%
10.00%
20.00%
30.00%
40.00%
50.00%
60.00%
70.00%
80.00%
90.00%
100.00%
Perc
ent
of
targ
ets
Number of compounds that tested with targets
0
1000
2000
3000
4000
5000
6000
0 10 20 30 40 50
Nu
mb
er o
f C
om
po
un
ds
Number of Targets

50
From Figure 13, targets that bound with more than 500 compounds accounted for
less than 11%. More than 89% of targets bound with less than 500 compounds.
According to the quantity of compounds that each target hit, top 50 targets were
ranked and showed in Figure 14. Only top 11 targets bound with more than 1000
compounds. For the other 39 targets, the number of compounds that each target
bound with less than 1000.
Figure 12 Number of compounds that each target hit
Figure 13 Percent of targets hit compounds
0
500
1000
1500
2000
2500
3000
3500
4000
0 50 100 150 200 250 300 350
Nu
mb
er o
f C
om
po
un
ds
Number of targets
89.36%
7.29%1.82% 0.61% 0.00% 0.61% 0.00% 0.30%
0.00%
10.00%
20.00%
30.00%
40.00%
50.00%
60.00%
70.00%
80.00%
90.00%
100.00%
Perc
ent
of
targ
ets
Number of compounds that hit targets

51
Figure 14 Number of compounds that each target hit
For compounds and targets in the other two bioactivity profiles, their tested trend
and hit trend were similar to that of the first matrix. That is, for small portion of
compounds, they were tested/bound with relatively large quantity of targets, while
vast majority of compounds were tested/bound with sporadic targets. For targets, it
was similar trend.
4.1.2 Results and comparison of complete bioactivity profiles
According to methods of extraction and dealing with missing value described in the
Chapter3, three complete bioactivity profiles were generated from those three
bioactivity profiles described above. Comparison for their size and “1” rate are in
Table 5.
Complete bioactivity profiles Number of
compound
Number of
target “1” Rate
6μM matrix 28 32 20.60%
10μM matrix 27 32 22.22%
van Westen matrix 26 30 35.90%
Table 5 Summary of three complete bioactivity profiles
“1” rate = (number of “1”) / (number of all cells in a matrix)
0
500
1000
1500
2000
2500
3000
3500
4000
0 10 20 30 40 50
Nu
mb
er o
f C
om
po
un
ds
Number of targets

52
Can be seen from Table 5, “1” rate of 10μM matrix was a little higher than that of
6μM matrix, while “1” rate of van Westen matrix was significantly higher than
others. This meant binding rate of compound and target in van Westen matrix are
apparently higher than that in other matrices.
Theoretically, binding rate should increase with increasing value of threshold.
Binding rate in 10μM matrix was higher than that of 6μM matrix. However, binding
rate of van Westen matrix should be lower than that of other matrices, because it
used a lower threshold 300nM.
4.1.3 Analysis for selectivity of compounds and targets in complete
matrices
There are three groups of compounds/targets in three matrix of Table 5. Analysis
and comparison for selectivity of compounds/targets were displayed in this part.
Karaman et al. (2008) introduced the concept of selectivity score to analyse kinases
and compounds.
Selectivity score of a compound
= (the number of targets that the compound hit)
(the number of all targets)
Selectivity score of a target
= (the number of compounds that the target hit)
(the number of all compounds)
4.1.3.1 Selectivity of compounds
Can be seen from Figure 15, the difference of Selectivity score among different
groups of compounds was significant.
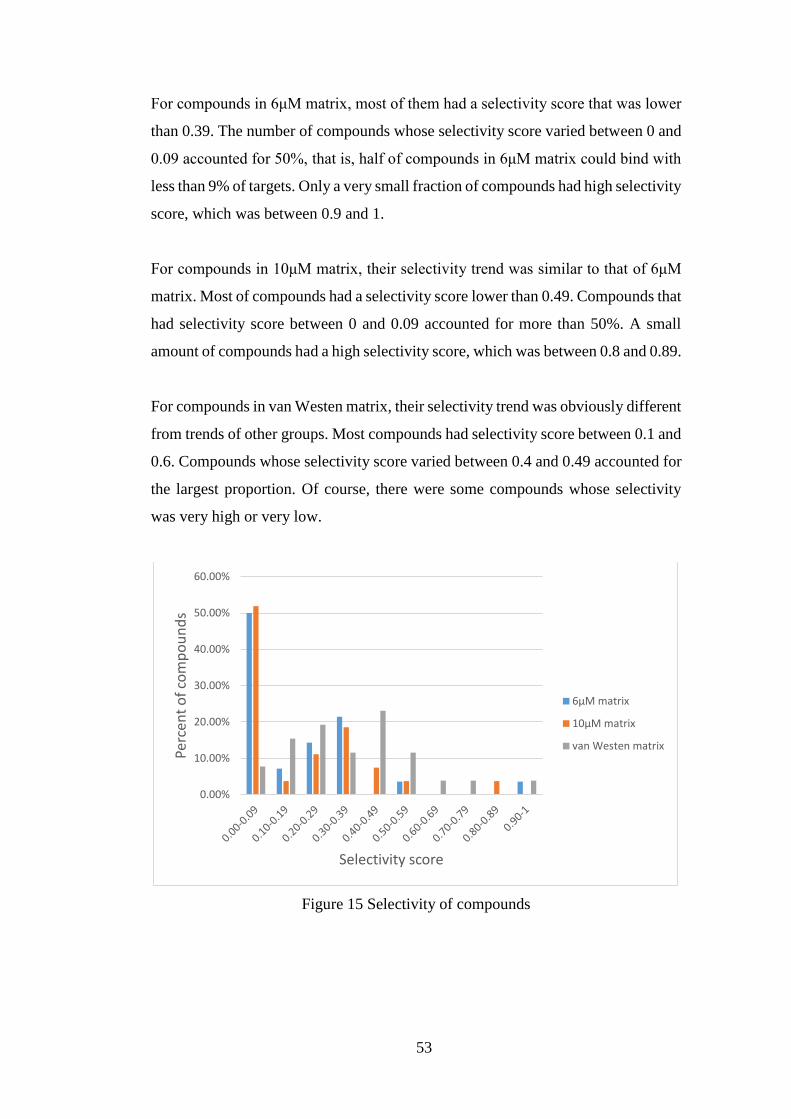
53
For compounds in 6μM matrix, most of them had a selectivity score that was lower
than 0.39. The number of compounds whose selectivity score varied between 0 and
0.09 accounted for 50%, that is, half of compounds in 6μM matrix could bind with
less than 9% of targets. Only a very small fraction of compounds had high selectivity
score, which was between 0.9 and 1.
For compounds in 10μM matrix, their selectivity trend was similar to that of 6μM
matrix. Most of compounds had a selectivity score lower than 0.49. Compounds that
had selectivity score between 0 and 0.09 accounted for more than 50%. A small
amount of compounds had a high selectivity score, which was between 0.8 and 0.89.
For compounds in van Westen matrix, their selectivity trend was obviously different
from trends of other groups. Most compounds had selectivity score between 0.1 and
0.6. Compounds whose selectivity score varied between 0.4 and 0.49 accounted for
the largest proportion. Of course, there were some compounds whose selectivity
was very high or very low.
Figure 15 Selectivity of compounds
0.00%
10.00%
20.00%
30.00%
40.00%
50.00%
60.00%
Perc
ent
of
com
po
un
ds
Selectivity score
6μM matrix
10μM matrix
van Westen matrix
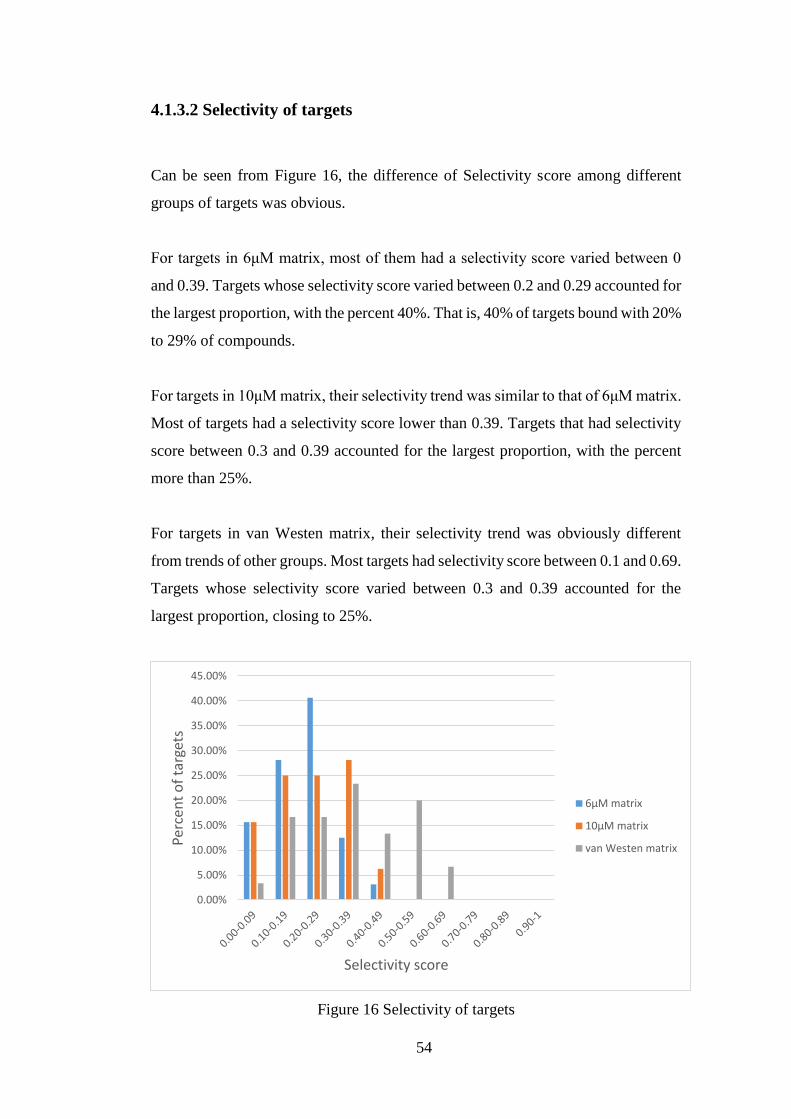
54
4.1.3.2 Selectivity of targets
Can be seen from Figure 16, the difference of Selectivity score among different
groups of targets was obvious.
For targets in 6μM matrix, most of them had a selectivity score varied between 0
and 0.39. Targets whose selectivity score varied between 0.2 and 0.29 accounted for
the largest proportion, with the percent 40%. That is, 40% of targets bound with 20%
to 29% of compounds.
For targets in 10μM matrix, their selectivity trend was similar to that of 6μM matrix.
Most of targets had a selectivity score lower than 0.39. Targets that had selectivity
score between 0.3 and 0.39 accounted for the largest proportion, with the percent
more than 25%.
For targets in van Westen matrix, their selectivity trend was obviously different
from trends of other groups. Most targets had selectivity score between 0.1 and 0.69.
Targets whose selectivity score varied between 0.3 and 0.39 accounted for the
largest proportion, closing to 25%.
Figure 16 Selectivity of targets
0.00%
5.00%
10.00%
15.00%
20.00%
25.00%
30.00%
35.00%
40.00%
45.00%
Perc
ent
of
targ
ets
Selectivity score
6μM matrix
10μM matrix
van Westen matrix
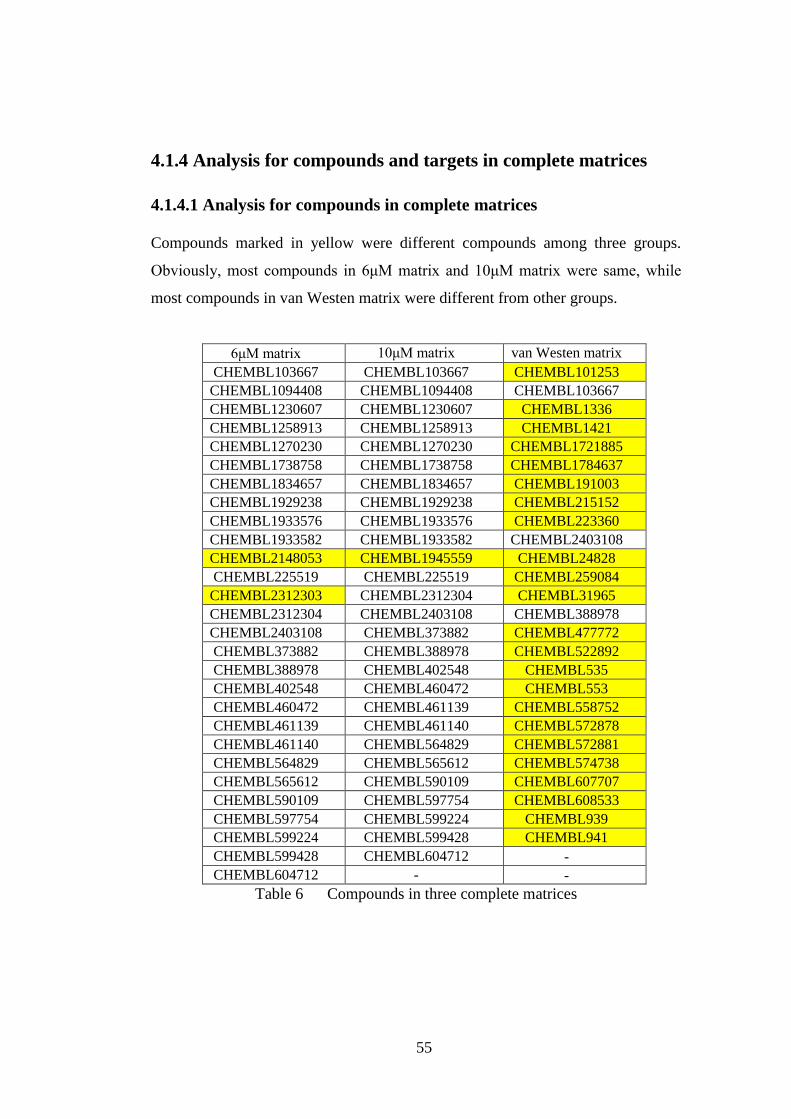
55
4.1.4 Analysis for compounds and targets in complete matrices
4.1.4.1 Analysis for compounds in complete matrices
Compounds marked in yellow were different compounds among three groups.
Obviously, most compounds in 6μM matrix and 10μM matrix were same, while
most compounds in van Westen matrix were different from other groups.
6μM matrix 10μM matrix van Westen matrix
CHEMBL103667 CHEMBL103667 CHEMBL101253
CHEMBL1094408 CHEMBL1094408 CHEMBL103667
CHEMBL1230607 CHEMBL1230607 CHEMBL1336
CHEMBL1258913 CHEMBL1258913 CHEMBL1421
CHEMBL1270230 CHEMBL1270230 CHEMBL1721885
CHEMBL1738758 CHEMBL1738758 CHEMBL1784637
CHEMBL1834657 CHEMBL1834657 CHEMBL191003
CHEMBL1929238 CHEMBL1929238 CHEMBL215152
CHEMBL1933576 CHEMBL1933576 CHEMBL223360
CHEMBL1933582 CHEMBL1933582 CHEMBL2403108
CHEMBL2148053 CHEMBL1945559 CHEMBL24828
CHEMBL225519 CHEMBL225519 CHEMBL259084
CHEMBL2312303 CHEMBL2312304 CHEMBL31965
CHEMBL2312304 CHEMBL2403108 CHEMBL388978
CHEMBL2403108 CHEMBL373882 CHEMBL477772
CHEMBL373882 CHEMBL388978 CHEMBL522892
CHEMBL388978 CHEMBL402548 CHEMBL535
CHEMBL402548 CHEMBL460472 CHEMBL553
CHEMBL460472 CHEMBL461139 CHEMBL558752
CHEMBL461139 CHEMBL461140 CHEMBL572878
CHEMBL461140 CHEMBL564829 CHEMBL572881
CHEMBL564829 CHEMBL565612 CHEMBL574738
CHEMBL565612 CHEMBL590109 CHEMBL607707
CHEMBL590109 CHEMBL597754 CHEMBL608533
CHEMBL597754 CHEMBL599224 CHEMBL939
CHEMBL599224 CHEMBL599428 CHEMBL941
CHEMBL599428 CHEMBL604712 -
CHEMBL604712 - -
Table 6 Compounds in three complete matrices
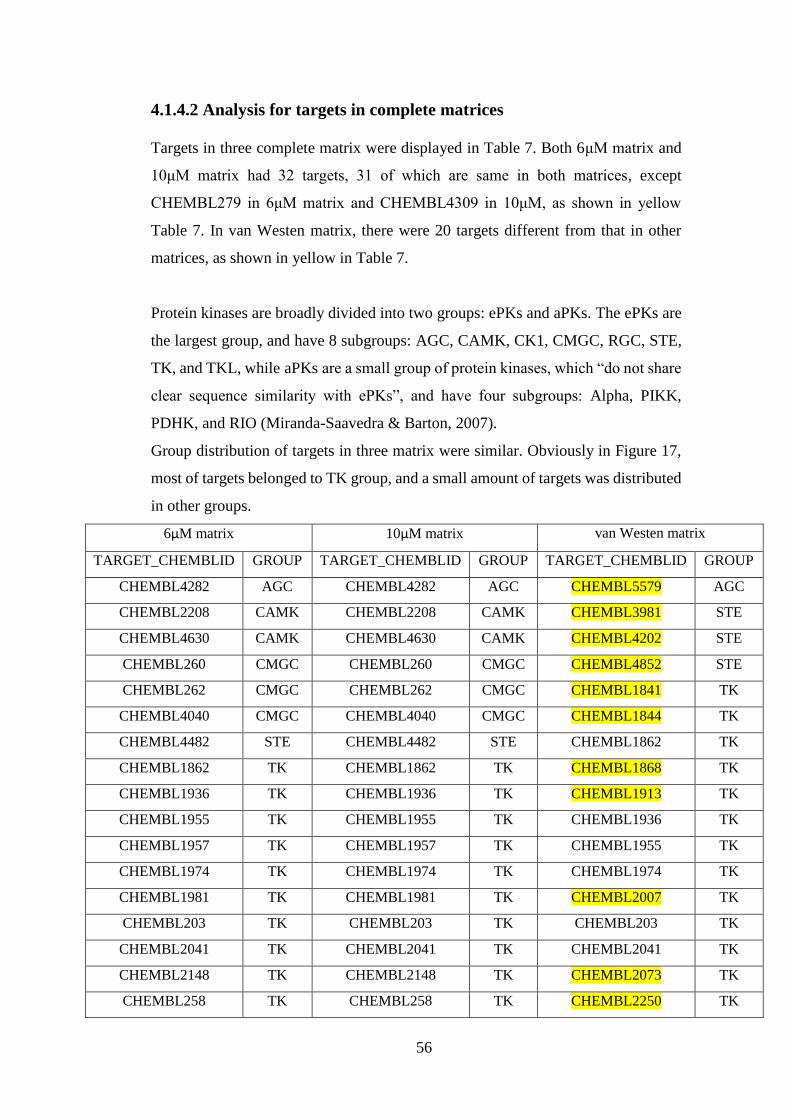
56
4.1.4.2 Analysis for targets in complete matrices
Targets in three complete matrix were displayed in Table 7. Both 6μM matrix and
10μM matrix had 32 targets, 31 of which are same in both matrices, except
CHEMBL279 in 6μM matrix and CHEMBL4309 in 10μM, as shown in yellow
Table 7. In van Westen matrix, there were 20 targets different from that in other
matrices, as shown in yellow in Table 7.
Protein kinases are broadly divided into two groups: ePKs and aPKs. The ePKs are
the largest group, and have 8 subgroups: AGC, CAMK, CK1, CMGC, RGC, STE,
TK, and TKL, while aPKs are a small group of protein kinases, which “do not share
clear sequence similarity with ePKs”, and have four subgroups: Alpha, PIKK,
PDHK, and RIO (Miranda-Saavedra & Barton, 2007).
Group distribution of targets in three matrix were similar. Obviously in Figure 17,
most of targets belonged to TK group, and a small amount of targets was distributed
in other groups.
6μM matrix 10μM matrix van Westen matrix
TARGET_CHEMBLID GROUP TARGET_CHEMBLID GROUP TARGET_CHEMBLID GROUP
CHEMBL4282 AGC CHEMBL4282 AGC CHEMBL5579 AGC
CHEMBL2208 CAMK CHEMBL2208 CAMK CHEMBL3981 STE
CHEMBL4630 CAMK CHEMBL4630 CAMK CHEMBL4202 STE
CHEMBL260 CMGC CHEMBL260 CMGC CHEMBL4852 STE
CHEMBL262 CMGC CHEMBL262 CMGC CHEMBL1841 TK
CHEMBL4040 CMGC CHEMBL4040 CMGC CHEMBL1844 TK
CHEMBL4482 STE CHEMBL4482 STE CHEMBL1862 TK
CHEMBL1862 TK CHEMBL1862 TK CHEMBL1868 TK
CHEMBL1936 TK CHEMBL1936 TK CHEMBL1913 TK
CHEMBL1955 TK CHEMBL1955 TK CHEMBL1936 TK
CHEMBL1957 TK CHEMBL1957 TK CHEMBL1955 TK
CHEMBL1974 TK CHEMBL1974 TK CHEMBL1974 TK
CHEMBL1981 TK CHEMBL1981 TK CHEMBL2007 TK
CHEMBL203 TK CHEMBL203 TK CHEMBL203 TK
CHEMBL2041 TK CHEMBL2041 TK CHEMBL2041 TK
CHEMBL2148 TK CHEMBL2148 TK CHEMBL2073 TK
CHEMBL258 TK CHEMBL258 TK CHEMBL2250 TK

57
CHEMBL2599 TK CHEMBL2599 TK CHEMBL258 TK
CHEMBL267 TK CHEMBL2695 TK CHEMBL267 TK
CHEMBL2695 TK CHEMBL279 TK CHEMBL279 TK
CHEMBL279 TK CHEMBL2803 TK CHEMBL3234 TK
CHEMBL2803 TK CHEMBL2815 TK CHEMBL3650 TK
CHEMBL2815 TK CHEMBL2971 TK CHEMBL3905 TK
CHEMBL2971 TK CHEMBL3650 TK CHEMBL4142 TK
CHEMBL3650 TK CHEMBL3717 TK CHEMBL4223 TK
CHEMBL3717 TK CHEMBL3905 TK CHEMBL4454 TK
CHEMBL3905 TK CHEMBL4247 TKL CHEMBL5274 TK
CHEMBL4247 TKL CHEMBL1991 Other CHEMBL5319 TK
CHEMBL1991 Other CHEMBL2185 Other CHEMBL3935 Other
CHEMBL2185 Other CHEMBL3024 Other CHEMBL4355 Other
CHEMBL3024 Other CHEMBL4309 Other - -
CHEMBL4722 Other CHEMBL4722 Other - -
Table 7 Targets in three complete matrices
Figure 17 Distribution of targets
0.00%
10.00%
20.00%
30.00%
40.00%
50.00%
60.00%
70.00%
80.00%
90.00%
AGC CAMK CMGC STE TK TKL Other
Perc
ent
of
each
gru
op
Protein kinases groups
6μM matrix
10μM matrix
van Westen matrix

58
4.2 Clustering results by bioactivity profiles
All datasets of bioactivity profiles in EXCEL format need to be changed into csv
format for clustering in WEKA. The value of seed for each clustering operation used
the default value 10.
As explained in methodology section, there is no appropriate value for the number
of clusters in this project. Then the number of clusters can be determined according
to research need.
4.2.1 Clustering results for compounds in 6μM matrix by bioactivity
profiles
The cluster dendrogram in Figure 18 was draw in Rstudio. It was used to directly
show distance between different compounds in 6μM matrix.
Based on the cluster dendrogram, two levels were chosen to implement clustering
by K-means algorithm, which were number of clusters equalled to 4 and 8
respectively. Then compounds were clustered into 4 and 8 clusters respectively in
WEKA. Clustering results were shown in Figure 19 and 20.

59
Figure 18 Cluster dendrogram for 6μM matrix
Figure 19 Clustering result for 6μM matrix (Number= 4)

60
Figure 20 Clustering result for 6μM matrix (Number= 8)
4.2.2 Clustering results for compounds in 10μM matrix by
bioactivity profiles
Similarly, cluster dendrogram and clustering results for compounds in 10μM matrix
were shown in the following three figures.
Figure 21 Cluster dendrogram for 10μM matrix
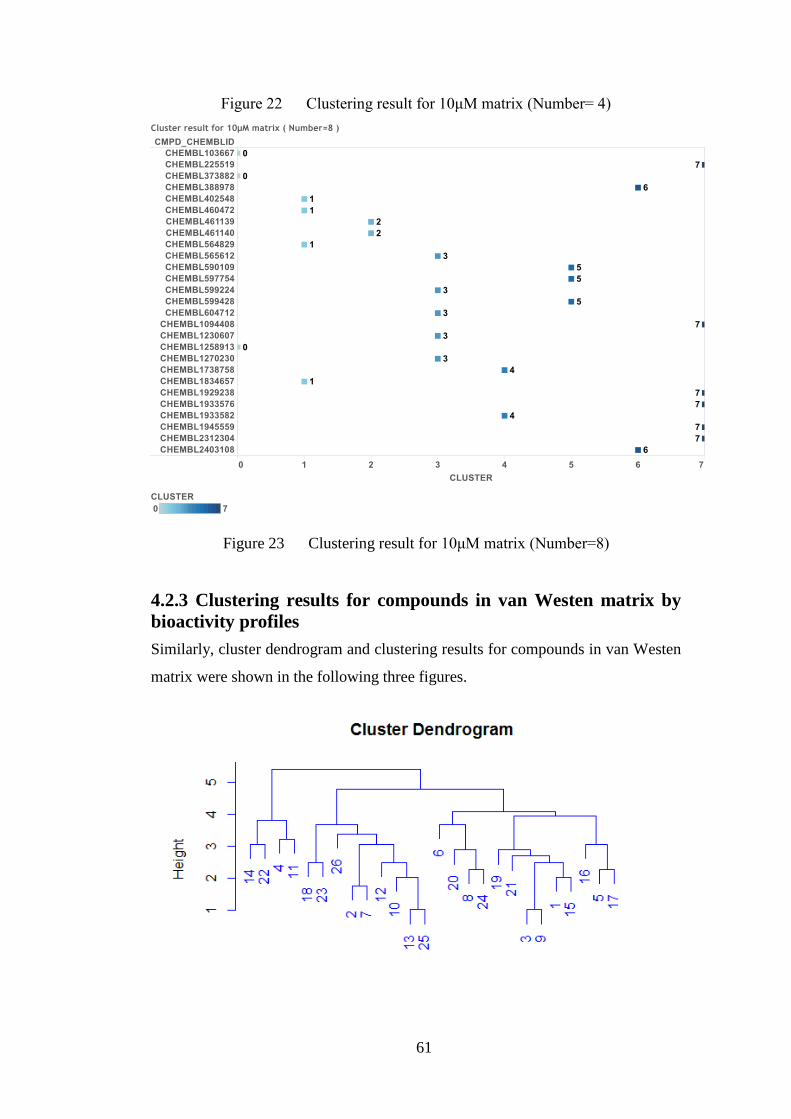
61
Figure 22 Clustering result for 10μM matrix (Number= 4)
Figure 23 Clustering result for 10μM matrix (Number=8)
4.2.3 Clustering results for compounds in van Westen matrix by
bioactivity profiles
Similarly, cluster dendrogram and clustering results for compounds in van Westen
matrix were shown in the following three figures.

62
Figure 24 Cluster dendrogram for van Westen matrix
Figure 25 Clustering result for van Westen matrix (Number=4)
Figure 26 Clustering result for van Westen matrix (Number=8)

63
4.3 Molecular descriptors
Two kind of molecular descriptors were calculated for the three group of compounds.
Furthermore, both Morgan and MACCS were used to calculate for 2D fingerprints.
4.3.1 2D fingerprints
4.3.1.1 Morgan fingerprints
Morgan fingerprints were calculated from SMILES for compounds in 6μM matrix
(Supplementary Table 7).
Morgan fingerprints were calculated from SMILES for compounds in 10μM matrix
(Supplementary Table 8).
Morgan fingerprints were calculated from SMILES for compounds in van Westen
matrix (Supplementary Table 9).
4.3.1.2 MACCS fingerprints
MACCS fingerprints were calculated from SMILES for compounds in 6μM matrix
(Supplementary Table 10).
MACCS fingerprints were calculated from SMILES for compounds in 10μM matrix
(Supplementary Table 11).
MACCS fingerprints were calculated from SMILES for compounds in van Westen
matrix (Supplementary Table 12).
4.3.2 Physiochemical properties
Physiochemical properties were calculated from SMILES for compounds in 6μM
matrix (Supplementary Table 13).
Physiochemical properties were calculated from SMILES for compounds in 10μM
matrix (Supplementary Table 14).

64
Physiochemical properties were calculated from SMILES for compounds in van
Westen matrix (Supplementary Table 15).
4.4 Clustering results by molecular descriptors
Similar to the clustering results by bioactivity profiles, the three group of
compounds were clustered respectively by their 2D fingerprints and physiochemical
properties. Besides, each group were clustered two times based on two options for
the number of clusters (4 and 8).
4.4.1 Clustering results for compounds in 6μM matrix by molecular
descriptors
There are 28 compounds in 6μM matrix. According to their 2D fingerprints and
physiochemical properties, they were clustered into different results respectively, as
shown in Table 8 and Table 9.
4.4.1.1 Cluster number = 4
CMPD_CHEMBLID Morgan
CLUSTER
MACCS
CLUSTER
PCP
CLUSTER
CHEMBL103667 2 0 3
CHEMBL1094408 0 0 0
CHEMBL1230607 2 0 0
CHEMBL1258913 2 2 3
CHEMBL1270230 2 2 2
CHEMBL1738758 0 0 0
CHEMBL1834657 1 1 1
CHEMBL1929238 2 3 3
CHEMBL1933576 0 0 0
CHEMBL1933582 0 0 0
CHEMBL2148053 1 1 1
CHEMBL225519 2 2 2
CHEMBL2312303 2 0 3
CHEMBL2312304 2 0 3
CHEMBL2403108 2 3 3
CHEMBL373882 3 3 3
CHEMBL388978 2 0 3
CHEMBL402548 2 0 0
65
CHEMBL460472 2 0 3
CHEMBL461139 2 2 0
CHEMBL461140 2 3 0
CHEMBL564829 0 0 0
CHEMBL565612 2 0 0
CHEMBL590109 2 2 0
CHEMBL597754 2 0 0
CHEMBL599224 2 0 0
CHEMBL599428 2 0 0
CHEMBL604712 2 3 0
Table 8 Clustering results for 6μM matrix by molecular descriptors ( Number= 4 )
4.4.1.2 Cluster number = 8
CMPD_CHEMBLID Morgan
CLUSTER
MACCS
CLUSTER
PCP
CLUSTER
CHEMBL103667 6 6 3
CHEMBL1094408 0 0 4
CHEMBL1230607 2 6 4
CHEMBL1258913 6 6 3
CHEMBL1270230 2 2 2
CHEMBL1738758 0 0 6
CHEMBL1834657 1 1 1
CHEMBL1929238 2 3 3
CHEMBL1933576 0 0 6
CHEMBL1933582 0 0 0
CHEMBL2148053 7 7 7
CHEMBL225519 2 2 2
CHEMBL2312303 2 0 5
CHEMBL2312304 2 0 5
CHEMBL2403108 2 7 3
CHEMBL373882 3 3 3
CHEMBL388978 2 0 0
CHEMBL402548 6 6 6
CHEMBL460472 2 6 3
CHEMBL461139 2 4 4
CHEMBL461140 2 3 4
CHEMBL564829 4 6 6
CHEMBL565612 4 4 4
CHEMBL590109 5 5 5
CHEMBL597754 4 6 6
CHEMBL599224 4 6 4
CHEMBL599428 4 6 6
CHEMBL604712 2 6 4
Table 9 Clustering results for 6μM matrix by molecular descriptors (Number= 8)

66
4.4.2 Clustering results for compounds in 10μM matrix by
molecular descriptors
There are 27 compounds in 10μM matrix. According to their 2D fingerprints and
physiochemical properties, they were clustered into different results respectively, as
shown in Table 10 and Table 11.
4.4.2.1 Cluster number = 4
CMPD_CHEMBLID Morgan
CLUSTER
MACCS
CLUSER PCP CLUSTER
CHEMBL103667 0 0 0
CHEMBL1094408 3 3 2
CHEMBL1230607 3 3 3
CHEMBL1258913 0 2 0
CHEMBL1270230 3 3 3
CHEMBL1738758 3 0 2
CHEMBL1834657 1 1 1
CHEMBL1929238 0 0 0
CHEMBL1933576 3 3 2
CHEMBL1933582 3 3 2
CHEMBL1945559 3 3 3
CHEMBL225519 2 2 3
CHEMBL2312304 3 2 2
CHEMBL2403108 3 1 0
CHEMBL373882 1 1 0
CHEMBL388978 3 0 2
CHEMBL402548 1 3 2
CHEMBL460472 0 0 0
CHEMBL461139 2 2 2
CHEMBL461140 2 2 2
CHEMBL564829 3 3 2
CHEMBL565612 3 3 2
CHEMBL590109 3 3 2
CHEMBL597754 3 3 2
CHEMBL599224 3 3 3
CHEMBL599428 3 3 2
CHEMBL604712 3 3 3
Table 10 Clustering results for 10μM matrix by molecular descriptors
(Number= 4)
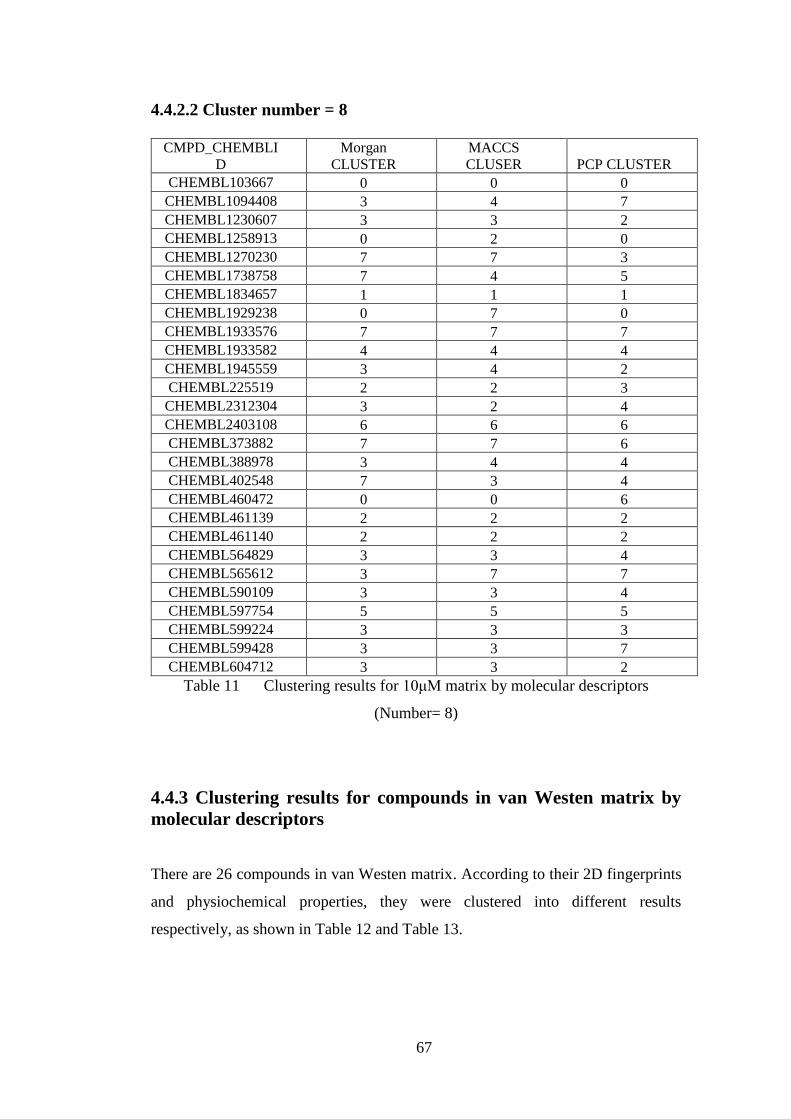
67
4.4.2.2 Cluster number = 8
CMPD_CHEMBLI
D
Morgan
CLUSTER
MACCS
CLUSER PCP CLUSTER
CHEMBL103667 0 0 0
CHEMBL1094408 3 4 7
CHEMBL1230607 3 3 2
CHEMBL1258913 0 2 0
CHEMBL1270230 7 7 3
CHEMBL1738758 7 4 5
CHEMBL1834657 1 1 1
CHEMBL1929238 0 7 0
CHEMBL1933576 7 7 7
CHEMBL1933582 4 4 4
CHEMBL1945559 3 4 2
CHEMBL225519 2 2 3
CHEMBL2312304 3 2 4
CHEMBL2403108 6 6 6
CHEMBL373882 7 7 6
CHEMBL388978 3 4 4
CHEMBL402548 7 3 4
CHEMBL460472 0 0 6
CHEMBL461139 2 2 2
CHEMBL461140 2 2 2
CHEMBL564829 3 3 4
CHEMBL565612 3 7 7
CHEMBL590109 3 3 4
CHEMBL597754 5 5 5
CHEMBL599224 3 3 3
CHEMBL599428 3 3 7
CHEMBL604712 3 3 2
Table 11 Clustering results for 10μM matrix by molecular descriptors
(Number= 8)
4.4.3 Clustering results for compounds in van Westen matrix by
molecular descriptors
There are 26 compounds in van Westen matrix. According to their 2D fingerprints
and physiochemical properties, they were clustered into different results
respectively, as shown in Table 12 and Table 13.

68
4.4.3.1 Cluster number = 4
CMPD_CHEMBLI
D
Morgan
CLUSTER
MACCS
CLUSER PCP CLUSTER
CHEMBL101253 3 0 0
CHEMBL103667 3 2 2
CHEMBL1336 2 2 3
CHEMBL1421 1 1 3
CHEMBL1721885 3 3 0
CHEMBL1784637 1 1 1
CHEMBL191003 0 0 0
CHEMBL215152 2 3 2
CHEMBL223360 0 0 0
CHEMBL2403108 0 2 2
CHEMBL24828 3 3 3
CHEMBL259084 0 0 3
CHEMBL31965 3 3 3
CHEMBL388978 0 1 3
CHEMBL477772 0 0 0
CHEMBL522892 0 0 0
CHEMBL535 0 3 0
CHEMBL553 3 3 0
CHEMBL558752 2 0 3
CHEMBL572878 0 0 3
CHEMBL572881 1 0 0
CHEMBL574738 2 2 2
CHEMBL607707 3 3 3
CHEMBL608533 0 1 2
CHEMBL939 3 3 3
CHEMBL941 1 0 2
Table 12 Clustering results for van Westen matrix by molecular descriptors
(Number= 4)
4.4.3.2 Cluster number = 8
CMPD_CHEMBLID Morgan
CLUSTER MACCS CLUSER PCP CLUSTER
CHEMBL101253 7 4 7
CHEMBL103667 3 2 2
CHEMBL1336 2 4 3
CHEMBL1421 1 1 3
CHEMBL1721885 7 3 0
CHEMBL1784637 1 1 1
CHEMBL191003 0 4 0
CHEMBL215152 6 3 3

69
CHEMBL223360 0 4 7
CHEMBL2403108 6 2 2
CHEMBL24828 6 6 5
CHEMBL259084 6 4 4
CHEMBL31965 3 3 3
CHEMBL388978 5 5 5
CHEMBL477772 4 4 0
CHEMBL522892 0 0 6
CHEMBL535 7 0 6
CHEMBL553 6 6 6
CHEMBL558752 4 4 4
CHEMBL572878 0 0 3
CHEMBL572881 7 7 7
CHEMBL574738 2 2 2
CHEMBL607707 3 3 3
CHEMBL608533 5 5 2
CHEMBL939 3 3 5
CHEMBL941 1 0 2
Table 13 Clustering results for van Westen matrix by molecular descriptors
(Number= 8)
4.5 Comparison for different clustering results
4.5.1 For compounds in 6μM matrix
In this dissertation, clusters of bioactivity profiles were standard classes for
compounds, and clusters of molecular descriptors were compared with standard
classes to see how similar they were to the standard classes. When cluster purity
was higher, the similarity was higher.
As shown in Table 14, when N = 4, cluster purity of Morgan algorithm is the higher
than physiochemical properties method, which means clustering result of Morgan
algorithm is closer to experimental result than that of physiochemical properties
method. If only from this view, it can be said that 2D fingerprints are better than
physiochemical properties in predicting activity of compounds. However, at the
same time, cluster purity of MACCS algorithm is less than that of physiochemical
properties method. That is to say, from MACCS perspective, 2D fingerprints are
worse than physiochemical properties in predicting activity of compounds.

70
From Table 15, when N = 8, cluster purities of both Morgan and MACCS algorithm
are higher than that of physiochemical properties method. In this case, it is clear that
clustering result of 2D fingerprints is closer to experimental result. In another word,
this moment, 2D fingerprints are better than physiochemical properties in predicting
activity of compounds.
Cluster number = 4 Cluster purity
Bioactivity profiles & Morgan fingerprints 0.61
Bioactivity profiles & MACCS fingerprints 0.50
Bioactivity profiles & Physiochemical properties 0.57
Table 14 Cluster purity summary for 6μM matrix (N=4)
Cluster number = 8 Cluster purity
Bioactivity profiles & Morgan fingerprints 0.68
Bioactivity profiles & MACCS fingerprints 0.61
Bioactivity profiles & Physiochemical properties 0.57
Table 15 Cluster purity summary for 6μM matrix (N=8)
Besides, no matter to Morgan or MACCS algorithm, cluster purity is increased with
the increase of N, as shown in Figure 27. This means, the number of clusters might
affect evaluation result. In another word, for a group of compounds and a kind of
fingerprints, when choosing different number of clusters to implement clustering,
maybe getting different evaluation result. However, cluster purity of
physiochemical properties method did not change with the increase of cluster
numbers.
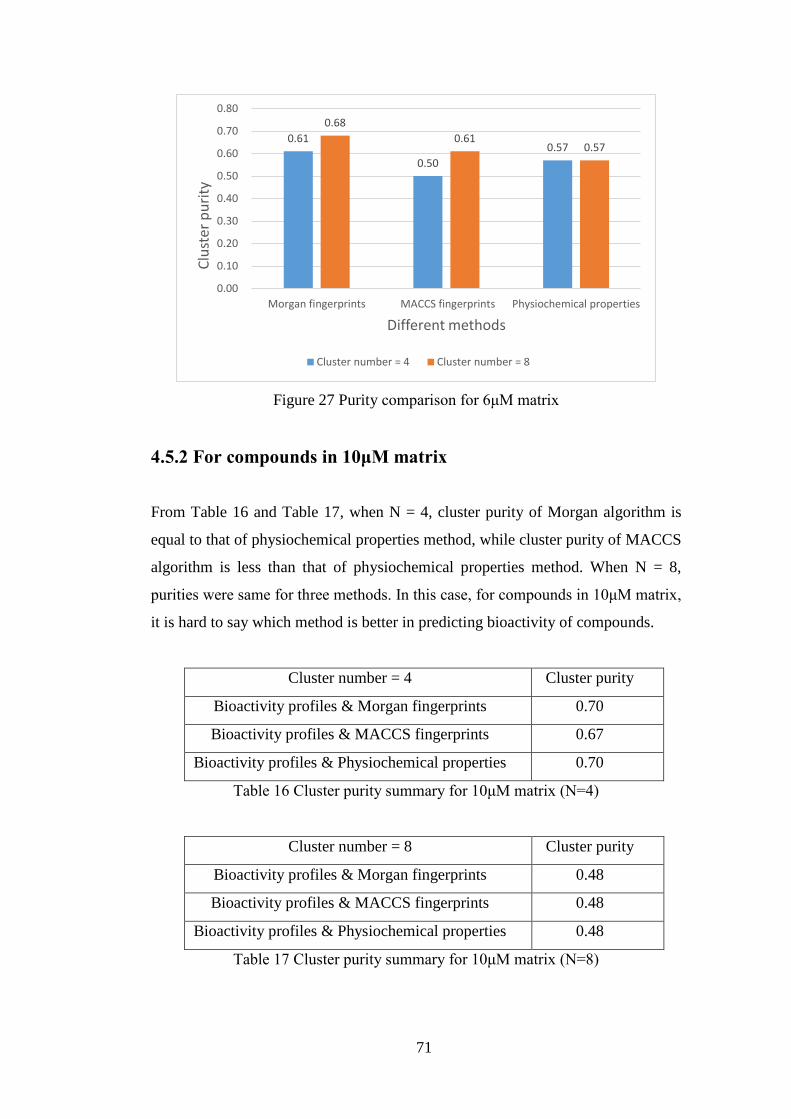
71
Figure 27 Purity comparison for 6μM matrix
4.5.2 For compounds in 10μM matrix
From Table 16 and Table 17, when N = 4, cluster purity of Morgan algorithm is
equal to that of physiochemical properties method, while cluster purity of MACCS
algorithm is less than that of physiochemical properties method. When N = 8,
purities were same for three methods. In this case, for compounds in 10μM matrix,
it is hard to say which method is better in predicting bioactivity of compounds.
Cluster number = 4 Cluster purity
Bioactivity profiles & Morgan fingerprints 0.70
Bioactivity profiles & MACCS fingerprints 0.67
Bioactivity profiles & Physiochemical properties 0.70
Table 16 Cluster purity summary for 10μM matrix (N=4)
Cluster number = 8 Cluster purity
Bioactivity profiles & Morgan fingerprints 0.48
Bioactivity profiles & MACCS fingerprints 0.48
Bioactivity profiles & Physiochemical properties 0.48
Table 17 Cluster purity summary for 10μM matrix (N=8)
0.61
0.50
0.57
0.68
0.61 0.57
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
Morgan fingerprints MACCS fingerprints Physiochemical properties
Clu
ster
pu
rity
Different methods
Cluster number = 4 Cluster number = 8

72
Figure 28 Purity comparison for 10μM matrix
Besides, cluster purity of each method is decreased with the increase of cluster
number, as shown in Figure 28. That is, evaluation result was affected by number
of clusters.
4.5.3 For compounds in van Westen matrix
Contrary to the results of the other matrices, from Table 18 and Table 19, cluster
purity of both Morgan and MACCS algorithm is lower than that of physiochemical
properties method. That is, in this case, clustering result of physiochemical
properties method is closer to experimental result than that of 2D fingerprints. In
another word, for compounds in this matrix, 2D fingerprints are worse than
physiochemical properties in predicting activity of compounds.
Cluster number = 4 Cluster purity
Bioactivity profiles & Morgan fingerprints 0.46
Bioactivity profiles & MACCS fingerprints 0.46
Bioactivity profiles & Physiochemical properties 0.58
Table 18 Cluster purity summary for van Westen matrix (N=4)
Cluster number = 8 Cluster purity
Bioactivity profiles & Morgan fingerprints 0.46
0.70 0.67
0.70
0.48 0.48 0.48
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
Morgan fingerprints MACCS fingerprints Physiochemical properties
Clu
ster
pu
rity
Different methods
Cluster number = 4 Cluster number = 8

73
Bioactivity profiles & MACCS fingerprints 0.50
Bioactivity profiles & Physiochemical properties 0.54
Table 19 Cluster purity summary for van Westen matrix (N=8)
Besides, cluster purity of Morgan algorithm does not change with the increase of
cluster number, as shown in Figure 29, while cluster purity of MACCS algorithm is
increased. In addition, cluster purity of physiochemical properties method is
decreased with the increase of cluster number.
Figure 29 Purity comparison for van Westen matrix
In summary, for different groups of compounds with different thresholds, both 2D
fingerprints and Physiochemical properties can predict bioactivity of compounds to
some extent. However, results of three groups did not show a consistent result about
which method had the highest ability of prediction. One set of data in three sets
shows that 2D fingerprints are better than physiochemical properties in predicting
bioactivity of compounds. One showed an opposite result that physiochemical
properties are better than 2D fingerprints in predicting bioactivity of compounds.
The other shows there is no significant difference in prediction ability between 2D
fingerprints and physiochemical properties.
Then further research was done on one group of compounds with different threshold.
0.46 0.46
0.58
0.46 0.50
0.54
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
Morgan fingerprints MACCS fingerprints Physiochemical properties
Clu
ster
pu
rity
Different methods
Cluster number = 4 Cluster number = 8
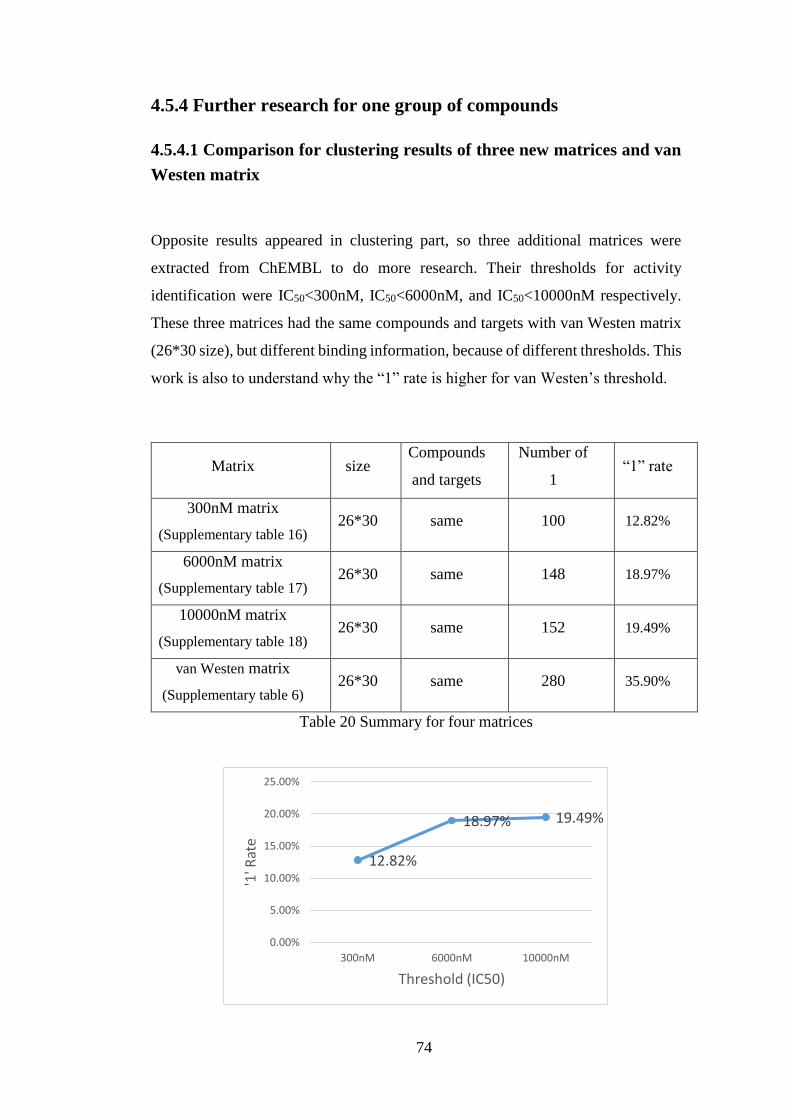
74
4.5.4 Further research for one group of compounds
4.5.4.1 Comparison for clustering results of three new matrices and van
Westen matrix
Opposite results appeared in clustering part, so three additional matrices were
extracted from ChEMBL to do more research. Their thresholds for activity
identification were IC50<300nM, IC50<6000nM, and IC50<10000nM respectively.
These three matrices had the same compounds and targets with van Westen matrix
(26*30 size), but different binding information, because of different thresholds. This
work is also to understand why the “1” rate is higher for van Westen’s threshold.
Matrix size Compounds
and targets
Number of
1 “1” rate
300nM matrix
(Supplementary table 16) 26*30 same 100 12.82%
6000nM matrix
(Supplementary table 17) 26*30 same 148 18.97%
10000nM matrix
(Supplementary table 18) 26*30 same 152 19.49%
van Westen matrix
(Supplementary table 6) 26*30 same 280 35.90%
Table 20 Summary for four matrices
12.82%
18.97% 19.49%
0.00%
5.00%
10.00%
15.00%
20.00%
25.00%
300nM 6000nM 10000nM
'1' R
ate
Threshold (IC50)
75
Figure 30 Trend of ‘1’rate
As shown in Figure 30, “1” rate increases with increasing of threshold values. This
means, the more inexactitude the standard is, the more compounds and targets are
thought to be able to bind with each other. For matrices with different sizes, different
compounds and different targets, their ‘1’ rate may does not follow this trend with
the increasing of threshold.
For 300nM matrix and van Westen matrix, they applied the same threshold, but had
different “1” rate. As analysed before, some reason might lead to this confused result.
In Table 21, clustering results of three new matrices and van Westen matrix (section
4.5.3) were put together. Compounds in each matrix were clustered with number of
clusters 4 or 8, so there were 8 sets of clusters for bioactivity profiles. They were
treated as 8 sets of standard classes for calculating cluster purity. Then,
corresponding clustering results of Morgan fingerprints, MACCS fingerprints, and
Physiochemical properties were compared with each set of classes respectively.
Cluster purities for them were placed in Table 22 and Figure 31.
Matrix size
26*30
Bioactivity profiles 2D fingerprints Physiochemi
cal
properties 300 nM 6000 nM
10000
nM
van
Westen Morgan MACCS
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8 N=4 N=8
CHEMBL101253 2 6 2 7 0 7 0 7 3 7 0 4 0 7
CHEMBL103667 2 7 2 7 0 7 3 4 3 3 2 2 2 2
CHEMBL1336 0 0 0 0 0 0 0 7 2 2 2 4 3 3
CHEMBL1421 0 4 0 5 0 5 2 5 1 1 1 1 3 3
CHEMBL1721885 2 6 2 2 2 2 0 7 3 7 3 3 0 0
CHEMBL1784637 1 1 1 1 1 1 1 1 1 1 1 1 1 1
CHEMBL191003 2 6 2 4 2 4 3 6 0 0 0 4 0 0
CHEMBL215152 2 2 2 2 2 2 0 0 2 6 3 3 2 3
CHEMBL223360 0 0 0 0 0 0 0 7 0 0 0 4 0 7
CHEMBL2403108 2 6 1 5 1 5 3 3 0 6 2 2 2 2
CHEMBL24828 2 6 2 7 2 7 2 2 3 6 3 6 3 5
76
CHEMBL259084 2 6 2 2 2 2 3 3 0 6 0 4 3 4
CHEMBL31965 3 3 3 3 3 3 3 3 3 3 3 3 3 3
CHEMBL388978 1 5 1 5 1 5 2 5 0 5 1 5 3 5
CHEMBL477772 0 0 0 0 0 0 1 7 0 4 0 4 0 0
CHEMBL522892 0 0 0 0 0 0 0 0 0 0 0 0 0 6
CHEMBL535 0 4 0 0 0 0 0 0 0 7 3 0 0 6
CHEMBL553 2 6 3 6 3 6 3 6 3 6 3 6 0 6
CHEMBL558752 2 6 2 4 2 4 3 4 2 4 0 4 3 4
CHEMBL572878 2 2 2 2 2 2 1 4 0 0 0 0 3 3
CHEMBL572881 2 7 2 7 2 7 0 7 1 7 0 7 0 7
CHEMBL574738 2 2 2 2 2 2 2 2 2 2 2 2 2 2
CHEMBL607707 2 6 2 2 2 2 3 6 3 3 3 3 3 3
CHEMBL608533 2 2 2 2 2 2 0 4 0 5 1 5 2 2
CHEMBL939 2 6 3 6 3 6 3 3 3 3 3 3 3 5
CHEMBL941 2 6 2 6 2 6 3 4 1 1 0 0 2 2
Table 21 Clustering results for four matrices
Matrix
size
26*30
300nM
matrix
6000nM
matrix
10000nM
matrix
van Westen
matrix
N=4 N=8 N=4 N=8 N=4 N=8 N=4 N=8
Morgan 0.65 0.52 0.52 0.42 0.46 0.42 0.46 0.46
MACCS 0.69 0.52 0.58 0.50 0.54 0.50 0.46 0.50
PCP 0.69 0.50 0.58 0.50 0.58 0.50 0.58 0.54
Table 22 Cluster purity for four matrices
PCP means Physiochemical properties

77
Figure 31 Purity comparison for four matrices
PCP means Physiochemical properties
Can be seen from Figure 31, for van Westen classes, purities of physiochemical
properties method were higher than Morgan and MACCS algorithms, no matter
number of clusters is 4 or 8. That is, in this situation, physiochemical properties
method was better than 2D fingerprints in predicting bioactivity of compounds.
However, for other classes, sometimes prediction of 2D fingerprints was better than
that of physiochemical properties, sometimes the contrary.
4.5.4.2 Comparison for clustering results of adjusted matrices
In last section, clustering results of three new matrices were compared with that of
van Westen matrix. Actually, there were some rows with all “0” values in these
three new matrices. The reason why these rows were not deleted is that to ensure
these matrices had same compounds and targets with van Westen matrix, and ensure
fairness of comparison. However, these rows might affect the accuracy of clustering,
so they were deleted in the following analysis and comparison.
In 300nM matrix, the number of rows with all”0”value was the most. Five
compounds of this matrix were active to none of targets, and only one or two of
these compounds in other matrices were active to none of targets. So these five
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
N=4 N=8 N=4 N=8 N=4 N=8 N=4 N=8
300nM 6000nM 10000nM Gerard
Cu
ster
Pu
rity
Diffrent sets of classes
Morgan MACCS PCP

78
compounds were deleted from the four matrices: 300nM matrix, 6000nM matrix,
10000nM matrix, and van Westen matrix, in order to ensure no rows with all “0”
values in matrices, so as to ensure accuracy of clustering. Then, four new matrices
were generated. In this section, they were named 300nM_new matrix (Supplementary
table 19), 6000nM_new matrix (Supplementary table 20), 10000nM_new matrix
(Supplementary table 21), and van Westen_new matrix (Supplementary table 22)
respectively.
The size of each matrix was 21*30, with same compounds and targets in each matrix.
Morgan fingerprints (Supplementary table 23), MACCS fingerprints (Supplementary
table 24), and Physiochemical properties (Supplementary table 25) of this group
compounds were calculated respectively.
Clustering results were placed in Table 23. Similarly, after calculation, cluster
purities of each methods were shown in Table 24 and Figure 32.
Matrix size
21*30
Bioactivity profiles 2D fingerprints Physiochem
ical
properties
300nM_n
ew
6000nM_
new
10000nM
_new
van
Westen_
new
Morgan MACCS
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8
N=
4
N=
8 N=4 N=8
CHEMBL101253 1 1 1 1 1 1 1 1 1 0 1 1 1 1
CHEMBL103667 1 7 0 7 1 7 3 3 3 1 3 5 2 5
CHEMBL1336 2 6 2 6 2 6 1 6 1 3 3 6 3 3
CHEMBL1421 0 0 3 3 3 3 0 0 1 1 3 5 0 4
CHEMBL1784637 2 2 2 2 2 2 1 1 1 4 3 5 2 5
CHEMBL191003 1 1 0 4 0 4 3 3 1 7 1 6 3 6
CHEMBL215152 1 4 1 4 0 4 1 4 3 1 3 5 2 4
CHEMBL223360 1 6 1 6 1 6 1 6 1 3 1 6 3 7
CHEMBL2403108 1 1 2 2 2 2 3 3 1 6 3 3 2 5
CHEMBL24828 0 0 0 0 0 0 0 0 0 1 0 0 0 0
CHEMBL31965 1 5 3 5 0 5 3 5 3 5 3 5 0 2
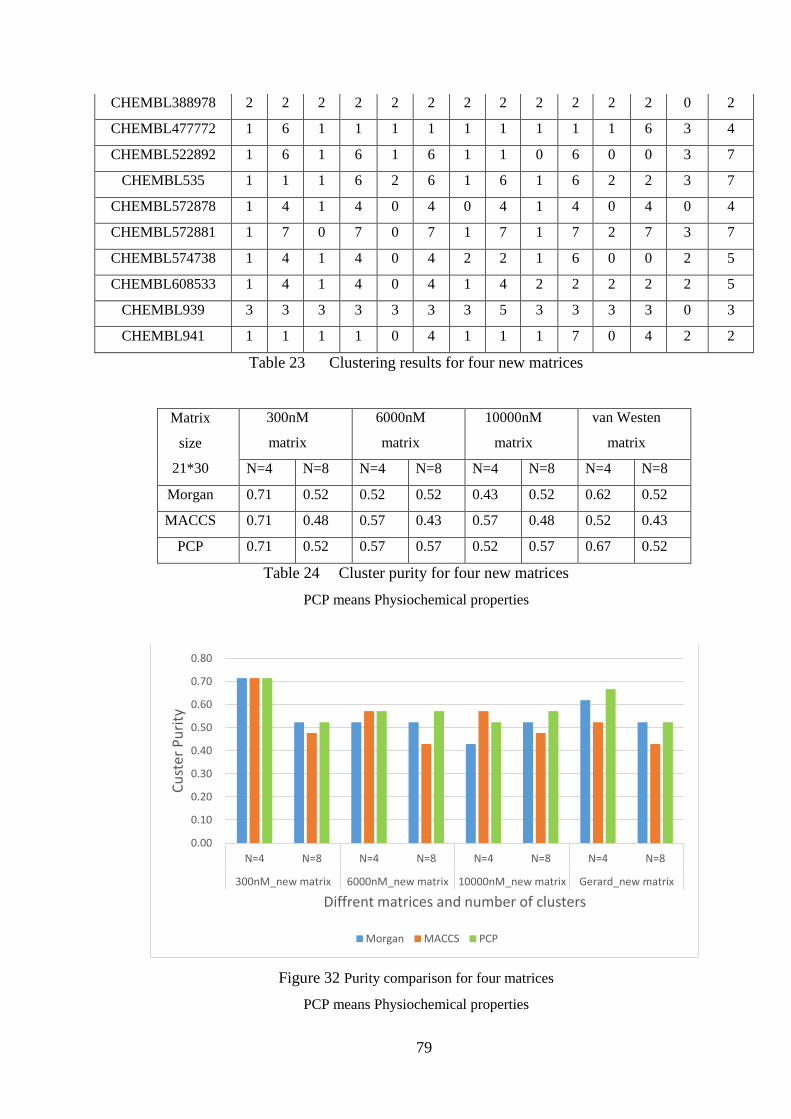
79
CHEMBL388978 2 2 2 2 2 2 2 2 2 2 2 2 0 2
CHEMBL477772 1 6 1 1 1 1 1 1 1 1 1 6 3 4
CHEMBL522892 1 6 1 6 1 6 1 1 0 6 0 0 3 7
CHEMBL535 1 1 1 6 2 6 1 6 1 6 2 2 3 7
CHEMBL572878 1 4 1 4 0 4 0 4 1 4 0 4 0 4
CHEMBL572881 1 7 0 7 0 7 1 7 1 7 2 7 3 7
CHEMBL574738 1 4 1 4 0 4 2 2 1 6 0 0 2 5
CHEMBL608533 1 4 1 4 0 4 1 4 2 2 2 2 2 5
CHEMBL939 3 3 3 3 3 3 3 5 3 3 3 3 0 3
CHEMBL941 1 1 1 1 0 4 1 1 1 7 0 4 2 2
Table 23 Clustering results for four new matrices
Matrix
size
21*30
300nM
matrix
6000nM
matrix
10000nM
matrix
van Westen
matrix
N=4 N=8 N=4 N=8 N=4 N=8 N=4 N=8
Morgan 0.71 0.52 0.52 0.52 0.43 0.52 0.62 0.52
MACCS 0.71 0.48 0.57 0.43 0.57 0.48 0.52 0.43
PCP 0.71 0.52 0.57 0.57 0.52 0.57 0.67 0.52
Table 24 Cluster purity for four new matrices
PCP means Physiochemical properties
Figure 32 Purity comparison for four matrices
PCP means Physiochemical properties
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
N=4 N=8 N=4 N=8 N=4 N=8 N=4 N=8
300nM_new matrix 6000nM_new matrix 10000nM_new matrix Gerard_new matrix
Cu
ster
Pu
rity
Diffrent matrices and number of clusters
Morgan MACCS PCP

80
Can be seen from Figure 32, for van Westen classes, only when N=4, purity of
physiochemical properties method was higher than Morgan and MACCS algorithms.
When n=8, purity of Morgan algorithm was as same as that of physiochemical
properties method. That is, for van Westen_new matrix, physiochemical properties
method was not always better than 2D fingerprints in predicting bioactivity of
compounds. Besides, for other classes, sometimes prediction of 2D fingerprints was
better than that of Physiochemical properties, sometimes the contrary.
In summary, no matter for different groups or the same group of compounds, both 2D
fingerprints and Physiochemical properties could predict bioactivity of compounds to
some extent, while which method was definitely better than the other one, more
rigorous researches need to be done. Actually, many factors may affect results. For
example, criteria for data extraction, size of complete matrix, cluster algorithms and
software, number of clusters, or other factors. In this dissertation, contrary records
were deleted at the step of data extraction, while if remain these records and identify
corresponding compounds as active or inactive according to the number of active
records or inactive records, result may be affected. Besides, if expand size of complete
matrix, result may be affected too.
Chapter 5 Conclusion
In this dissertation, bioactivity data and SMILES strings of compounds were extracted
from ChEMBL database respectively with different bioactivity thresholds (6μM,
10μM), then bioactivity data were transformed into two bioactivity profiles with
different compounds, and bioactivity profiles in this dissertation were represented by
binary values (1 meant active, 0 meant in active). “1” rate in dataset were increased
with threshold value increasing.
By using a mathematical idea and Rstudio, two complete bioactivity profiles were
extracted from obtained bioactivity profiles respectively. The methodology of
extracting complete matrix from bioactivity profiles is based on the idea of
optimization in mathematics. In each iteration, rows and columns were respectively

81
sorted according their number of values from high to low, and some rows and columns
with least quantity of values are deleted from the inputted matrix. That is, the complete
matrix outputted by iterations is the best one for current inputted matrix. Therefore,
the two complete matrices are the best to their original matrices respectively. Based
on their original inputted matrices were different, two outputted matrices did not
necessarily have same compounds and targets. Actually, compounds, targets and size
were different for two complete matrices. It was good for clustering repeatedly with
different matrices. Because this led to a more objective result, with less bias.
Besides, a complete bioactivity profiles was extracted from dataset of a researcher
named van Westen. This complete matrix also had different compounds and target
from other two complete matrices.
Bioactivity profiles were analysed from different angles. For most compounds in
dataset, they were tested and bound with sporadic targets. For a very few compounds,
they were tested and bound with many targets. For targets, the situation was similar.
For most compounds and targets in complete matrices, their selectivity score were
lower than 0.49.
Molecular descriptors such as 2D fingerprints and physiochemical properties for
compounds were calculated from their SMILES strings by KNIME and RDkit, and
two kinds of fingerprints were calculated by Morgan and MACCS algorithm
respectively.
One of machine learning methods named k-means was applied in WEKA to cluster
each group of compounds into subgroups, based on their bioactivity profiles, 2D
fingerprints, and physiochemical properties respectively.
Eventually, this dissertation compared and evaluated the ability of each molecular
descriptor for predicting bioactivity profiles of compounds. Evaluation of clusters was
achieved by using cluster purity. Clustering result of bioactivity profiles was treated
as standard class information to evaluate cluster quality of molecular descriptors.
Clusters of 2D fingerprints and physiochemical properties were compared with

82
standard class respectively to calculate cluster purity, then cluster purity were
compared to get result.
For three different groups of compounds with different thresholds, both 2D
fingerprints and Physiochemical properties can predict bioactivity of compounds to
some extent. In some cases, cluster purity could reach up to 0.7, that is, the similarity
between predictions and bioactivity profiles was as high as 70% for some cases. Beside,
average purity for each method were around 0.5. Overall speaking, it is helpful to use
these methods to predict bioactivity of compounds. There is certain possibility to partly
know bioactivity of compounds. It is better than there is no such methods. However,
all comparisons did not show a consistent result about which method had the highest
ability of predicting bioactivity of compounds. For one group of compounds, 2D
fingerprints were better than physiochemical properties in prediction. For one group,
physiochemical properties were better than 2D fingerprints. And the other group, there
was no significant difference in prediction ability between 2D fingerprints and
physiochemical properties. Then further research was done on one group of
compounds with different threshold. The similar result appeared. In summary, both 2D
fingerprints and Physiochemical properties could predict bioactivity of compounds to
some extent, while which method was definitely better than the other one, more
rigorous researches need to be done.
Actually, many factors may affect results. For example, criteria for data extraction,
size of complete matrix, cluster algorithms and software, number of clusters, or other
factors. In this dissertation, contrary records were deleted at the step of data extraction,
while if remain these records and identify corresponding compounds as active or
inactive according to the number of active records or inactive records, result may be
affected. Besides, if expand size of complete matrix, result may be affected too.
In this dissertation, only IC50 was used to identify activity for compounds, which did
not make full use of various activity data in ChEMBL. In the future, a better way could
be tried to identify bioactivity for compounds, and bigger size of complete matrix
could be extracted. Therefore, maybe better clusters could be got based on more
compounds in matrix. Besides, a comprehensive comparison may be necessary to
apply a variety of soft wares and algorithms in future research.

83
References
Anastassiadis, T., Deacon, S. W., Devarajan, K., Ma, H., & Peterson, J. R. (2011).
Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor
selectivity. Nature biotechnology, 29(11), 1039-1045.
Backman, T. W. H., & Girke, T. (2016). bioassayR: Cross-Target Analysis of Small
Molecule Bioactivity. Journal of Chemical Information and Modeling,56(7), 1237-
1242.
Beck, B., Chen, Y. F., Dere, W., Devanarayan, V., Eastwood, B. J., Farmen, M.
W., ... & Sawyer, B. D. (2012). Assay Operations for SAR Support.
Bender, A., Scheiber, J., Glick, M., Davies, J. W., Azzaoui, K., Hamon, J., ... &
Jenkins, J. L. (2007). Analysis of pharmacology data and the prediction of adverse
drug reactions and off ‐ target effects from chemical structure.
ChemMedChem, 2(6), 861-873.
Bento, A. P., Gaulton, A., Hersey, A., Bellis, L. J., Chambers, J., Davies, M., Krüger,
F. A., Light, Y., Mak, L., McGlinchey, S., Nowotka, M., Papadatos, G., Santos, R.,
& Overington, J. P. (2014) “The ChEMBL bioactivity database: an update.”
Nucleic Acids Res., 42 1083-1090.
DOI: 10.1093/nar/gkt1031
PMID: 24214965
Boyd, S., & Vandenberghe, L. (2004). Convex optimization. Cambridge university
press.
Burlingham, B. T., & Widlanski, T. S. (2003). An intuitive look at the relationship
of Ki and IC50: A more general use for the Dixon plot. J. Chem. Educ, 80(2), 214.
ChEMBL FAQ, (2014). Retrieved from https://www.ebi.ac.uk/ChEMBL/faq

84
Cheng, T., Wang, Y., & Bryant, S. H. (2010). Investigating the correlations among
the chemical structures, bioactivity profiles and molecular targets of small
molecules. Bioinformatics, 26(22), 2881-2888.
Davis, M. I., Hunt, J. P., Herrgard, S., Ciceri, P., Wodicka, L. M., Pallares, G., ... &
Zarrinkar, P. P. (2011). Comprehensive analysis of kinase inhibitor
selectivity. Nature biotechnology, 29(11), 1046-1051.
Deprez-Poulain, R., & Deprez, B. (2004). Facts, figures and trends in lead
generation. Current topics in medicinal chemistry, 4(6), 569-580.
Fang, J. S., Liu, A. L., & Du, G. H. (2014). Research advance in the drug target
prediction based on chemoinformatics. Acta pharmaceutica Sinica, 49(10), 1357-
1364.
Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., ... &
Overington, J. P. (2012). ChEMBL: a large-scale bioactivity database for drug
discovery. Nucleic acids research, 40(D1), D1100-D1107.
Helal, K. Y., Maciejewski, M., Gregori-Puigjane, E., Glick, M., & Wassermann, A.
M. (2016). Public domain HTS fingerprints: design and evaluation of compound
bioactivity profiles from PubChem”s bioassay repository. Journal of chemical
information and modeling, 56(2), 390-398.
Hughes, J. P., Rees, S., Kalindjian, S. B., & Philpott, K. L. (2011). Principles of
early drug discovery. British journal of pharmacology, 162(6), 1239-1249.
Karaman, M. W., Herrgard, S., Treiber, D. K., Gallant, P., Atteridge, C. E.,
Campbell, B. T., ... & Faraoni, R. (2008). A quantitative analysis of kinase inhibitor
selectivity. Nature biotechnology, 26(1), 127-132.
Larsen, R. J., & Marx, M. L. (1986). An introduction to mathematical statistics and
its applications.

85
Lavecchia, A. (2015). Machine-learning approaches in drug discovery: methods and
applications. Drug discovery today, 20(3), 318-331.
Leach, A. R., & Gillet, V. J. (2003). An introduction to chemoinformatics. Kluwer
Academic Publishers.
Leskovec, J., Rajaraman, A., & Ullman, J. D. (2014). Mining of massive datasets.
Cambridge University Press
Lusher, S. J., McGuire, R., van Schaik, R. C., Nicholson, C. D., & de Vlieg, J. (2014).
Data-driven medicinal chemistry in the era of big data. Drug discovery today, 19(7),
859-868.
Manning, C. D., Raghavan, P., & Schutze, H. (2008) Introduction to Information
Retrieval. Cambridge University Press
Martínez-Jiménez, F., Papadatos, G., Yang, L., Wallace, I. M., Kumar, V., Pieper,
U., ... & Marti-Renom, M. A. (2013). Target prediction for an open access set of
compounds active against Mycobacterium tuberculosis. PLoS Comput Biol, 9(10),
e1003253.
Mazanetz, M. P., Marmon R. J., Reisser, C. BT., & Morao, I. (2012). Drug discovery
applications for KNIME: an open source data mining platform.Current topics in
medicinal chemistry, 12(18), 1965-1979.
Medina-Franco, J. L., Giulianotti, M. A., Welmaker, G. S., & Houghten, R. A.
(2013). Shifting from the single to the multitarget paradigm in drug discovery. Drug
discovery today, 18(9), 495-501.
Miranda-Saavedra, D., & Barton, G.J. (2007) Classification and functional
annotation of eukaryotic protein kinases. Proteins 68, 893-914.

86
Paolini, G. V., Shapland, R. H., van Hoorn, W. P., Mason, J. S., & Hopkins, A. L.
(2006). Global mapping of pharmacological space. Nature biotechnology, 24(7),
805-815.
Petrone, P. M., Simms, B., Nigsch, F., Lounkine, E., Kutchukian, P., Cornett, A., ...
& Glick, M. (2012). Rethinking molecular similarity: comparing compounds on the
basis of biological activity. ACS chemical biology, 7(8), 1399-1409.
Personal communication, (July, 2016). van Westen, G.
Pina, A. S., Hussain, A., & Roque, A. C. A. (2010). An historical overview of drug
discovery. Ligand-Macromolecular Interactions in Drug Discovery: Methods and
Protocols, 3-12.
Provost, F., & Fawcett, T. (2013). Data Science for Business: What you need to
know about data mining and data-analytic thinking. "O'Reilly Media, Inc.".
Rogers, D., & Hahn, M. (2010). Extended-connectivity fingerprints. Journal of
chemical information and modeling, 50(5), 742-754.
Sliwoski, G., Kothiwale, S., Meiler, J., & Lowe, E. W. (2014). Computational
methods in drug discovery. Pharmacological reviews, 66(1), 334-395.
Sutherland, J. J., Gao, C., Cahya, S., & Vieth, M. (2013). What general conclusions
can we draw from kinase profiling data sets?. Biochimica et Biophysica Acta (BBA)-
Proteins and Proteomics, 1834(7), 1425-1433.
Taylor, D. (2015). The Pharmaceutical Industry and the Future of Drug
Development, Pharmaceuticals in the Environment, pp. 1-33
Wawer, M., Lounkine, E., Wassermann, A. M., & Bajorath, J. (2010). Data
structures and computational tools for the extraction of SAR information from large
compound sets. Drug discovery today, 15(15), 630-639.

87
Witten, I. H., Frank, E., & Hall, M. A. (2011). Data Mining: Practical machine
learning tools and techniques (3rd edition.). Morgan Kaufmann Publishers

88
Appendix
R code for iteration
setwd("d:/data")
raw_data<-read.csv("data.csv",header=T,encoding=“UTF-8”,as.is=TRUE)
data<-raw_data[1:(nrow(raw_data)-1),]
data <- subset( data, select = -count2 )
row.names(data)<-data[,1]
data <- subset( data, select = -c(1))
row<-c(0)
col<-c(0)
count_col<-function(data)
{
for (i in 1 : ncol(data))
{
col[i]<-sum(is.na(data[,i]))
}
return(col)
}
count_row<-function(data)
{
for (i in 1 : nrow(data))
{
row[i]<-sum(is.na(data[i,]))
}
return(row)
}
delet_col<-function(data)
{

89
j<-which.max(col)
data <- subset( data, select = -c(j))
return(data)
}
delet_row<-function(data)
{
j<-which.max(row)
data<-data[-j,]
return(data)
}
result<-function(data)
{
num_na<-0
for(i in 1:nrow(data))
num_na<-num_na+sum(is.na(data[i,]))
return(1-num_na/(nrow(data)*ncol(data)))
}
for(i in 1:2)
{
col<-c(0)
col<-count_col(data)
data<-delet_col(data)
}
result(data)
for(i in 1:13)
{
row<-c(0)
row<-count_row(data)
data<-delet_row(data)
print(i)

90
}
result(data)
write.csv(data, file="result.csv",row.names=T,quote=F)

Access to Dissertation
A Dissertation submitted to the University may be held by the Department (or School) within which the Dissertation was
undertaken and made available for borrowing or consultation in accordance with University Regulations.
Requests for the loan of dissertations may be received from libraries in the UK and overseas. The Department may also receive
requests from other organisations, as well as individuals. The conservation of the original dissertation is better assured if the
Department and/or Library can fulfill such requests by sending a copy. The Department may also make your dissertation
available via its web pages.
In certain cases where confidentiality of information is concerned, if either the author or the supervisor so requests, the
Department will withhold the dissertation from loan or consultation for the period specified below. Where no such
restriction is in force, the Department may also deposit the Dissertation in the University of Sheffield Library.
To be completed by the Author – Select (a) or (b) by placing a tick in the appropriate box
If you are willing to give permission for the Information School to make your dissertation available in these ways, please
complete the following:
(a) Subject to the General Regulation on Intellectual Property, I, the author, agree to this dissertation being made
immediately available through the Department and/or University Library for consultation, and for the Department
and/or Library to reproduce this dissertation in whole or part in order to supply single copies for the purpose of
research or private study
(b) Subject to the General Regulation on Intellectual Property, I, the author, request that this dissertation be withheld
from loan, consultation or reproduction for a period of [ ] years from the date of its submission. Subsequent to
this period, I agree to this dissertation being made available through the Department and/or University Library for
consultation, and for the Department and/or Library to reproduce this dissertation in whole or part in order to supply
single copies for the purpose of research or private study
Name Jingyan Sui
Department Information School
Signed Jingyan Sui Date September 1, 2016
To be completed by the Supervisor – Select (a) or (b) by placing a tick in the appropriate box
(a) I, the supervisor, agree to this dissertation being made immediately available through the Department and/or
University Library for loan or consultation, subject to any special restrictions (*) agreed with external organisations
as part of a collaborative project.
*Special
restrictions
(b) I, the supervisor, request that this dissertation be withheld from loan, consultation or reproduction for a period of
[ ] years from the date of its submission. Subsequent to this period, I, agree to this dissertation being made
available through the Department and/or University Library for loan or consultation, subject to any special
restrictions (*) agreed with external organisations as part of a collaborative project
Name
Department
Signed Date
THIS SHEET MUST BE SUBMITTED WITH DISSERTATIONS BY DEPARTMENTAL REQUIREMENTS.