rom j morphol embryol 2014, 55(2 suppl):649–653 r j m e ... · rom j morphol embryol 2014, 55(2...
TRANSCRIPT

Rom J Morphol Embryol 2014, 55(2 Suppl):649–653
ISSN (print) 1220–0522 ISSN (on-line) 2066–8279
CCAASSEE RREEPPOORRTTSS
Histopathological findings concerning ocular melanomas
MARIANA COSTACHE1), OANA MARIA PĂTRAŞCU2), ADRIAN DUMITRU2), DIANA COSTACHE3), LILIANA MARY VOINEA4), OLGA SIMIONESCU5), MARIA SAJIN1)
1)Department of Pathology, “Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania 2)Department of Pathology, Emergency University Hospital, Bucharest, Romania 3)Medical student, “Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania 4)Department of Ophthalmology, “Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania 5)Department of Dermatology, “Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania
Abstract Ocular melanoma is rare in clinical practice. In this study, we present three cases of ocular melanoma surgically removed in the Department of Ophthalmology of the Emergency University Hospital of Bucharest, Romania, and diagnosed in the Department of Pathology of the same hospital using conventional histopathological techniques and immunohistochemical tests.
Keywords: ocular melanoma, uvea, immunohistochemical tests, genetic predisposition.
Introduction
Melanoma is a malignant proliferation of melanocytes, cells that are found mainly in the structure of the epidermis but also in certain structures of the eye, and is considered one of the most aggressive cancers. These cells are also responsible for skin, iris and hair color.
Tumors identified in the complex structure of the eye, benign or malignant, cause irreversible functional changes sometimes with great clinical impact. Melanoma is the most common primary malignant neoplasm of the eye identified in adult person, the second being the intraocular lymphomas [1]. However, primary ocular melanoma is rare.
Despite progress in terms of local and systemic therapy of melanoma, the survival rate of patients has not changed significantly in the last three decades; the prognosis is reserved, even for patients with early-stage tumors, being well known the evolution of this aggressive tumor type.
Epidemiology and risk factors
Despite being the most common malignant tumor of the eye, this potentially fatal type of cancer is rare in the general population, some studies reporting an incidence between 5.3 and 10.9 cases per one million people [2], compared with cutaneous melanoma whose incidence is ten times higher. Like cutaneous melanoma, this type of cancer is particularly common among Caucasians, north-western European populations with light phototype, in contrast with the black population of Africa, North America and the Asian population [3]. This suggests the etiologic role of UV rays as well as the protective role of iris pigmentation. It appears to be a slight predomi-nance among males in terms of gender ratio.
The iris nevi, common in the general population, has not been unequivocally associated with the occurrence
of ocular melanoma [4], although it is possible to consider their degeneration into malignant tissue structure such as melanoma, but such a development is rare or unlikely.
Finally, risk factors that should be looked for, besides pigment phenotype and exposure to ultraviolet radiation, include personal and family history, for example con-genital ocular melanosis history, xeroderma pigmentosum, dysplastic nevi syndrome, history of uveal nevus or uveal melanoma. Also, living environment and working conditions are often key elements in tracking risk factors [5, 6].
Materials and Methods
In 2012, three cases of ocular melanoma were identified in the Emergency University Hospital of Bucharest, Romania. Patients with ages between 60 and 77 years presented mainly with blurred vision in the Department of Ophthalmology of the hospital. They also experienced visual field defect, photopsia, irritation, mild pain, metamorphopsia, floaters, redness and pressure of the affected eyeballs. Here, clinical and laboratory examinations were performed raising suspicion of ocular melanoma and the diagnosis was further confirmed by the Department of Pathology of the same hospital. After surgical enucleation of the affected eyeball, pieces of its tissue were sent for histopathological examination and diagnosis.
Samples of tissue were fixed with 10% buffered formalin and were processed by conventional histo-pathological method using inclusion in paraffin and Hematoxylin–Eosin (HE) staining and also immuno-histochemical tests were performed. The paraffin blocks acquired by histopathological processing were section at microtome resulting sections with 3-μm thickness mounted on slides cover with poly-L-Lysine. After that, the sections were deparaffinized in toluene and alcohol
R J M ERomanian Journal of
Morphology & Embryologyhttp://www.rjme.ro/
Mariana Costache et al.
650
successive baths, one hour, 15 minutes by bath, rehydration (three successive alcohol baths with decreased concen-tration: 96%, 80% and 70%, 10 minutes in each bath and followed by a bath with distillated water, where the sections were hold for 10 minutes). Washing in PBS (phosphate saline buffer), incubation with normal serum, for 20 minutes, incubation with primary antibody over-night, Dako LSAB kit, washing in carbonate buffer and development in 3,3’-diaminobenzidine hydrochloride/ hydrogen peroxide and nuclear counterstain with Mayer’s Hematoxylin. We used the following antibodies from NeoMarkers LabVision: Pancytokeratin, clone AE1/AE3 (Thermo Fisher Scientific Inc., USA, 1:100 dilution), Ki67, clone SP6 (Thermo Fisher Scientific Inc., USA, 1:200 dilution), S100 protein, clone 4C4.9 (Thermo Fisher Scientific Inc., USA, 1:100 dilution), Vimentin, clone V9 (Thermo Fisher Scientific Inc., USA, 1:200 dilution), MART-1/Melan-A, polyclonal (Thermo Fisher Scientific Inc., USA, 1:100 dilution), Melanoma, clone HMB-45 (Thermo Fisher Scientific Inc., USA, 1:80 dilution). The immunoreactive cells from each cases were semiquanti-tative evaluate as follow: diffuse positive, >75% positive cells; positive, 25–75% positive cells; focal positive, <25% positive cells and negative cells.
Results
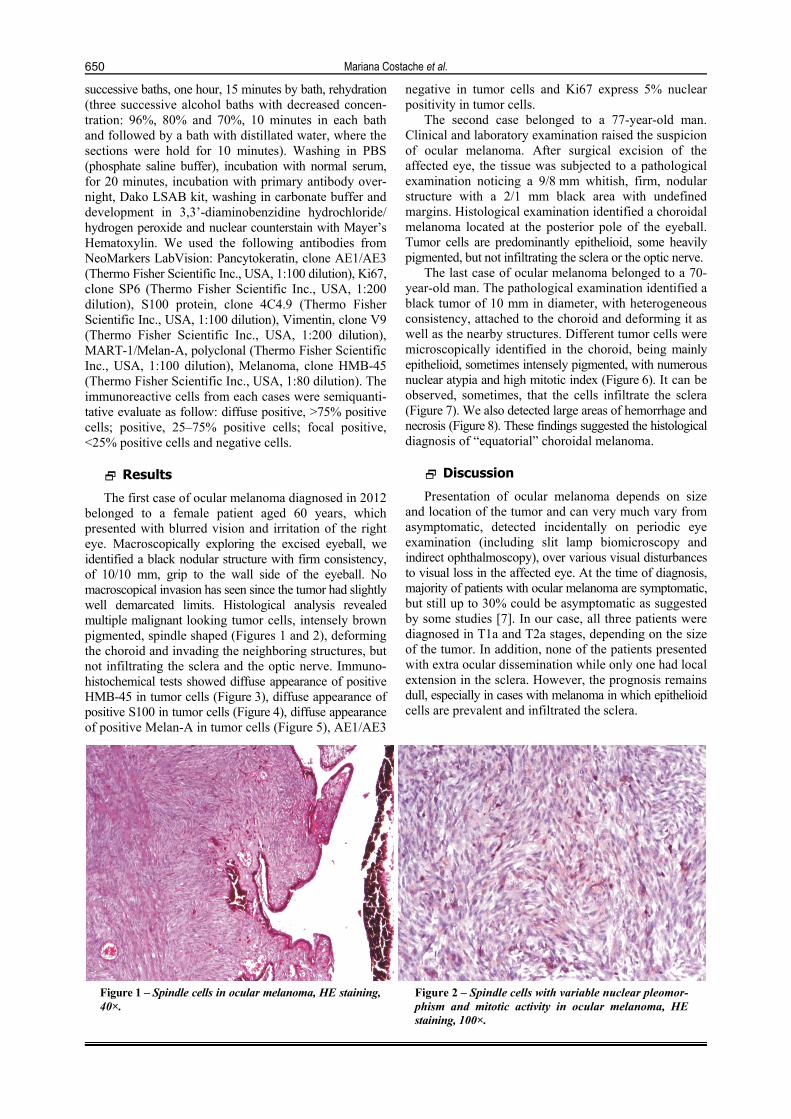
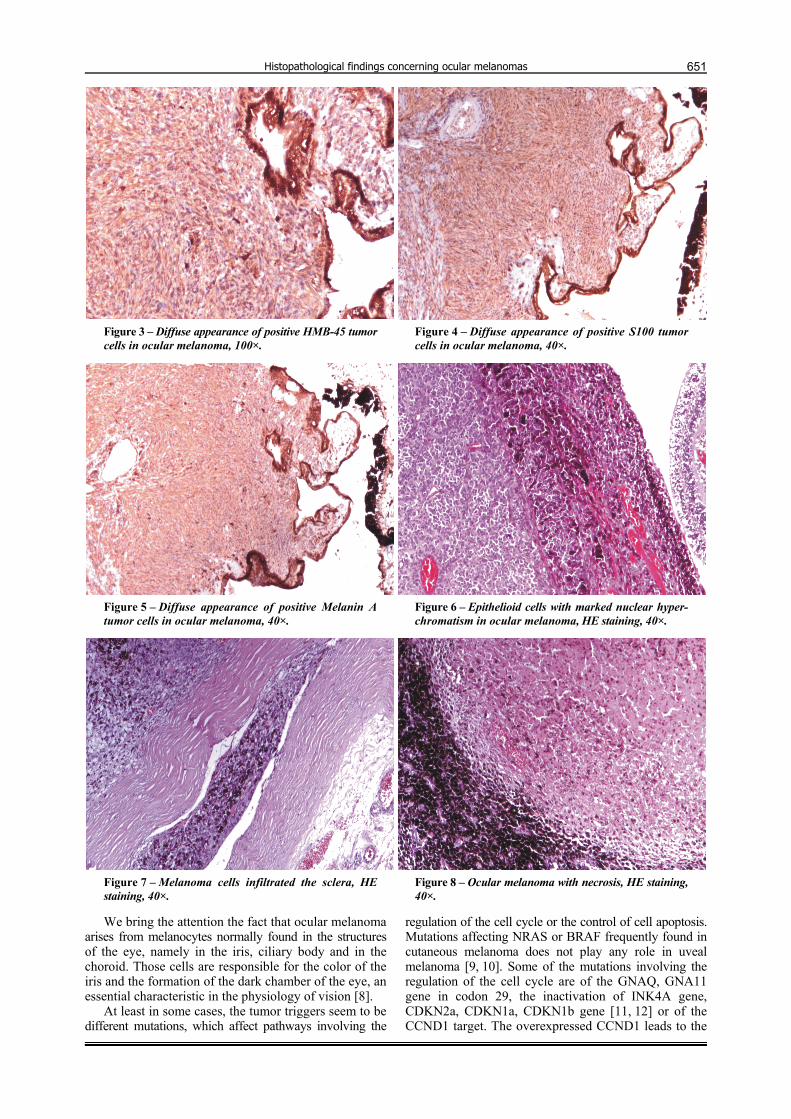
The first case of ocular melanoma diagnosed in 2012 belonged to a female patient aged 60 years, which presented with blurred vision and irritation of the right eye. Macroscopically exploring the excised eyeball, we identified a black nodular structure with firm consistency, of 10/10 mm, grip to the wall side of the eyeball. No macroscopical invasion has seen since the tumor had slightly well demarcated limits. Histological analysis revealed multiple malignant looking tumor cells, intensely brown pigmented, spindle shaped (Figures 1 and 2), deforming the choroid and invading the neighboring structures, but not infiltrating the sclera and the optic nerve. Immuno-histochemical tests showed diffuse appearance of positive HMB-45 in tumor cells (Figure 3), diffuse appearance of positive S100 in tumor cells (Figure 4), diffuse appearance of positive Melan-A in tumor cells (Figure 5), AE1/AE3
negative in tumor cells and Ki67 express 5% nuclear positivity in tumor cells.
The second case belonged to a 77-year-old man. Clinical and laboratory examination raised the suspicion of ocular melanoma. After surgical excision of the affected eye, the tissue was subjected to a pathological examination noticing a 9/8 mm whitish, firm, nodular structure with a 2/1 mm black area with undefined margins. Histological examination identified a choroidal melanoma located at the posterior pole of the eyeball. Tumor cells are predominantly epithelioid, some heavily pigmented, but not infiltrating the sclera or the optic nerve.
The last case of ocular melanoma belonged to a 70-year-old man. The pathological examination identified a black tumor of 10 mm in diameter, with heterogeneous consistency, attached to the choroid and deforming it as well as the nearby structures. Different tumor cells were microscopically identified in the choroid, being mainly epithelioid, sometimes intensely pigmented, with numerous nuclear atypia and high mitotic index (Figure 6). It can be observed, sometimes, that the cells infiltrate the sclera (Figure 7). We also detected large areas of hemorrhage and necrosis (Figure 8). These findings suggested the histological diagnosis of “equatorial” choroidal melanoma.
Discussion
Presentation of ocular melanoma depends on size and location of the tumor and can very much vary from asymptomatic, detected incidentally on periodic eye examination (including slit lamp biomicroscopy and indirect ophthalmoscopy), over various visual disturbances to visual loss in the affected eye. At the time of diagnosis, majority of patients with ocular melanoma are symptomatic, but still up to 30% could be asymptomatic as suggested by some studies [7]. In our case, all three patients were diagnosed in T1a and T2a stages, depending on the size of the tumor. In addition, none of the patients presented with extra ocular dissemination while only one had local extension in the sclera. However, the prognosis remains dull, especially in cases with melanoma in which epithelioid cells are prevalent and infiltrated the sclera.
Figure 1 – Spindle cells in ocular melanoma, HE staining, 40×.
Figure 2 – Spindle cells with variable nuclear pleomor-phism and mitotic activity in ocular melanoma, HE staining, 100×.
Histopathological findings concerning ocular melanomas
651
Figure 3 – Diffuse appearance of positive HMB-45 tumor cells in ocular melanoma, 100×.
Figure 4 – Diffuse appearance of positive S100 tumor cells in ocular melanoma, 40×.
Figure 5 – Diffuse appearance of positive Melanin A tumor cells in ocular melanoma, 40×.
Figure 6 – Epithelioid cells with marked nuclear hyper-chromatism in ocular melanoma, HE staining, 40×.
Figure 7 – Melanoma cells infiltrated the sclera, HE staining, 40×.
Figure 8 – Ocular melanoma with necrosis, HE staining, 40×.
We bring the attention the fact that ocular melanoma arises from melanocytes normally found in the structures of the eye, namely in the iris, ciliary body and in the choroid. Those cells are responsible for the color of the iris and the formation of the dark chamber of the eye, an essential characteristic in the physiology of vision [8].
At least in some cases, the tumor triggers seem to be different mutations, which affect pathways involving the
regulation of the cell cycle or the control of cell apoptosis. Mutations affecting NRAS or BRAF frequently found in cutaneous melanoma does not play any role in uveal melanoma [9, 10]. Some of the mutations involving the regulation of the cell cycle are of the GNAQ, GNA11 gene in codon 29, the inactivation of INK4A gene, CDKN2a, CDKN1a, CDKN1b gene [11, 12] or of the CCND1 target. The overexpressed CCND1 leads to the

Mariana Costache et al.
652
inactivation of the retinoblastoma tumor suppressor gene and appear to involve the RAF/MEK/ERK pathway [13]. On the other hand, the inactivation of the p53 pathway, defects in the Bcl-2 pathway and activation of the pro-survival PI3K-AKT pathway have been associated with inhibition of apoptosis in uveal melanoma [14]. Other important triggers of the ocular melanoma, especially in young adults, are mutations of the BAP1 gene (BRCA1-associated protein 1 or ubiquitin carboxy-terminal hidrolase) – a tumor-suppressor gene found on the 3rd chromosome (3p21-p21.2). Also, monosomy 3 is a frequently discovered genetic abnormality of the ocular melanoma [13] closely associated with metastasis [15].
One classification of the malignant melanocytic tumors regards the affected ocular structure. Hence, the most common type of ocular melanoma is represented by the uveal melanoma, which affects the iris, ciliary body or the choroid. This type of tumor can involve one, two or all three structures. This type of neoplasia can also affect the conjunctiva or the eyelid, but those are extremely rare cases [16, 17].
Clinically, melanoma of the iris is frequently found on the lower part of the iris. The tumor presents itself as an elevated or flat mass with various degrees of pigmentation [18], it can grow in volume or it can expand in a ring configuration, leading to iris dislocations and dysfunctions. Usually, the patient observes an abnormality of the iris, thus presenting sooner to the doctor unlike other types of uveal melanoma. Therefore, melanoma of the iris is considered to be less aggressive, being the most rapidly diagnosed tumor of the uveal tract [19].
Melanoma of the ciliary body rarely affects only this structure. More commonly, it is expanded in the iris or the choroid [20]. In most of the cases, the patient has no symptoms; hence, the tumor is later diagnosed and can grow bigger in size. The main complications – astigmatism, dislocation of the lens, acute glaucoma or cataract – can also appear as first clinical signs of the tumor.
Finally, choroidal melanoma presents itself as an irregular, slate-gray, solid [21] mass associated with decreased vision, retinal detachment or scleral invasion.
Accuracy of diagnosis established by clinical examination is nowadays very high, over 99%.
Ocular melanoma is diagnosed in the adults from the fifth decade, reaching a maximum at about 70-year-old. It is exceptionally identified in young children [22].
Usually, the diagnostic of uveal melanoma is raised by common ophthalmological investigations such as slit lamp biomicroscopy or eyeground examination. Ultra-sonography, oculo-CT or MRI are useful for diagnosis of the extra scleral invasion. However, results of at least one study showed that ocular melanomas were initially missed or misdiagnosed in 23% [23] of patients, which resulted in more advanced tumor and higher rate of primary enucleation among those patients; still, the accuracy of the diagnosis is given after the histopathological examination of the tumor.
The histological classification of the uveal melanoma is the modified Callender classification, in which the tumors are divided in four major histopathological types: spindle A and B type melanoma, out of which 45% with good prognosis, mixed cells melanoma with less good prognosis, epithelioid cells melanoma and necrotic melanoma. The last two have an unfavorable prognosis [18].
Type A spindle cells present fusiform, small nuclei, no nucleoli, their chromatin is frequently arranged in a linear fashion along the central axis of the nucleus [16, 18] and they have few mitotic figures. Type B cells are larger, more pleomorphic cells, with bigger ovoid nuclei and bulging nucleoli. They have mitotic activity. The most aggressive type of cells is the epithelioid cells. Those cells are irregular shape cells, larger than the other two types, even gigantic, with big, anaplastic nuclei and bulging nucleoli. Epithelioid cells are characteristically less cohesive than the spindle cells.
Often, this type of melanoma is formed of a mixture of the three types of cells. The presence of the epithelioid cells associated with various mitosis and anaplasia raises a poor prognosis. Also, the presence of inflammatory infiltrate, especially of mast cells, was correlated with a poor survival rate [24]. The mast cells contain lipofuscin and melanin because of their phagocytic activity. In addition, they produce VEGF, vascular endothelial factor, which plays an important role in formation of new blood vessels [25, 26].
Immunohistochemically, melanomas are reactive for S100 protein, HMB-45, Melan-A [27], vimentin and some low molecular weight keratins. It has been suggested that the melanoma cells exhibiting keratin immunostaining have a greater metastatic potential [28].
Local complications are various and important, some of what represent first clinical signs. We recall that the dysfunctions of the iris and the lens appear mostly in cases of iris and ciliary body melanoma. Necrosis, end-ophthalmitis, secondary glaucoma, retinal detachment, intraocular hemorrhage or fibrosis are the common local complications of the extensive choroidal melanoma [29]. General complications are represented preferentially by liver metastasis [30], but also lung, skin or central nervous system metastasis. They appear only after blood stream dissemination. Shields and colleagues showed that risk for metastases is greatly increasing with tumor thickness, and each millimeter increase in tumor thickness showed a 1.06 hazard ratio with increasing tumor thickness risk for metastases at 10 years showed increase from 6% for tumors 0–1 mm in thickness up to 51% for tumors over 10 mm in thickness [19] as for our three patients. The mortality is, unfortunately, constant and quite big, reaching about 40–45% at 10 years. Poor prognostic factors also include the age of the patient, the site and size of the tumor [31], trans-scleral infiltration or the invasion of the Bruch membrane and of the optic nerve. Better under-standing of underlying histological, genetic and molecular abnormalities implicated in development, progression and metastasis of ocular melanomas provides a great opportunity for development of targeted therapy.
At the same time, the lack of pigmentation, the presence of inflammatory infiltrate and the presence of a particular type of cells are essential characteristics of the treatment and further follow-up of the patients as well as important prognosis factors [32]. Moreover, new potential target therapeutic agents based of molecular/genetic profiles are currently being researched [33].
Conclusions
Even if ocular melanomas are more frequently diagnosed in the first and second stages, clarifying the

Histopathological findings concerning ocular melanomas
653
risk of metastasis remains a real challenge both clinically and therapeutically. The changeless prognosis as well as the multitude of treatments had not increase the survival, nor the quality of life, requesting more investigations on this matter. Hopefully, in near future emerging knowledge of the pathogenesis of ocular melanomas will translate into a novel and more effective systemic therapeutic agents which will increase, currently poor, prognosis of patients with metastatic disease.
References [1] Striker PT, Kumar V, Neoplasia. In: Kumar V, Abbas AK,
Fausto N, Aster J, Robbins and Cotran basis pathology, 8th edition, Elsevier–Saunders, Philadelphia, 2009, 192–223.
[2] Virgili G, Gatta G, Ciccolallo L, Capocaccia R, Biggeri A, Crocetti E, Lutz JM, Paci E; EUROCARE Working Group, Incidence of uveal melanoma in Europe, Ophthalmology, 2007, 114(12):2309–2315.
[3] Inskip PD, Devesa SS, Fraumeni JF Jr, Trends in the incidence of ocular melanoma in the United States, 1974–1998, Cancer Causes Control, 2003, 14(3):251–257.
[4] Baderca F, Solovan C, Boghian L, Epidemiological and morphological data of ocular melanocytic lesions, Rom J Morphol Embryol, 2013, 54(1):77–83.
[5] Holly EA, Aston DA, Char DH, Kristiansen JJ, Ahn DK, Uveal melanoma in relation to ultraviolet light exposure and host factors, Cancer Res, 1990, 50(18):5773–5777.
[6] Guénel P, Laforest L, Cyr D, Févotte J, Sabroe S, Dufour C, Lutz JM, Lynge E, Occupational risk factors, ultraviolet radiation, and ocular melanoma: a case-control study in France, Cancer Causes Control, 2001, 12(5):451–459.
[7] Indrei A, Cianga P, Florea ID, Haba D, Foia L, Cianga CM, A rare case of double recurrent choroidal melanoma, with distinctive immunohistochemical features, Rom J Morphol Embryol, 2010, 51(1):187–193.
[8] Harbour JW, Brantley MA Jr, Hollingsworth H, Gordon M, Association between choroidal pigmentation and posterior uveal melanoma in a white population, Br J Ophthalmol, 2004, 88(1):39–43.
[9] Saldanha G, Purnell D, Fletcher A, Potter L, Gillies A, Pringle JH, High BRAF mutation frequency does not characterize all melanocytic tumor types, Int J Cancer, 2004, 111(5):705–710.
[10] Verdijk RM, van den Bosch T, Naus NC, Paridaens D, Mooy CM, de Klein A, Histopathologic, immunohistochemical, ultrastructural, and cytogenetic analysis of oncocytic uveal melanoma, Arch Ophthalmol, 2011, 129(11):1501–1502.
[11] Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, Ishov AM, Tommerup N, Vissing H, Sekido Y, Minna J, Borodovsky A, Schultz DC, Wilkinson KD, Maul GG, Barlev N, Berger SL, Prendergast GC, Rauscher FJ 3rd, BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression, Oncogene, 1998, 16(9):1097–1112.
[12] Onken MD, Worley LA, Tuscan MD, Harbour JW, An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma, J Mol Diagn, 2010, 12(4):461–468.
[13] Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW, Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to mono-somy 3 for predicting metastasis in uveal melanoma, Clin Cancer Res, 2007, 13(10):2923–2927.
[14] An J, Wan H, Zhou X, Hu DN, Wang L, Hao L, Yan D, Shi F, Zhou Z, Wang J, Hu S, Yu J, Qu J, A comparative trans-criptomic analysis of uveal melanoma and normal uveal melanocyte, PLoS One, 2011, 6(1):e16516.
[15] Harbour JW, Genomic research in uveal melanoma, Retina Today, December 2010.
[16] Collaborative Ocular Melanoma Group, Accuracy of diagnosis of choroidal melanomas in the Collaborative Ocular Melanoma Study. COMS report no. 1, Arch Ophthalmol, 1990, 108(9): 1268–1273.
[17] Papastefanou VP, Cohen VML, Uveal melanoma, J Skin Cancer, 2011, 2011:573974.
[18] Yanoff M, Sassani JW, Ocular melanocytic tumors in ocular pathology, 6th edition, Mosby–Elsevier, 2009, 667–732.
[19] Shields CL, Furuta M, Thangappan A, Nagori S, Mashayekhi A, Lally DR, Kelly CC, Rudich DS, Nagori AV, Wakade OA, Mehta S, Forte L, Long A, Dellacava EF, Kaplan B, Shields JA, Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes, Arch Ophthalmol, 2009, 127(8):989–998.
[20] Patel M, Smyth E, Chapman PB, Wolchok JD, Schwartz GK, Abramson DH, Carvajal RD, Therapeutic implications of the emerging molecular biology of uveal melanoma, Clin Cancer Res, 2011, 17(8):2087–2100.
[21] Coroi M, Muţiu G, Roşca E, Burtă L, Ilin R, Manole F, Choroidal melanocytes and associated pathology, Rom J Morphol Embryol, 2006, 47(3):269–272.
[22] Rubin R, Strayer DS, Rubin E, Rubin’s pathology: clinico-pathologic foundations of medicine, 6th edition, Lippincott Williams & Wilkins, 2011, 1393.
[23] Griewank KG, Murali R, Pathology and genetics of uveal melanoma, Pathology, 2013, 45(1):18–27.
[24] Ly LV, Bronkhorst IH, van Beelen E, Vrolijk J, Taylor AW, Versluis M, Luyten GP, Jager MJ, Inflammatory cytokines in eyes with uveal melanoma and relation with macrophage infiltration, Invest Ophthalmol Vis Sci, 2010, 51(11):5445–5451.
[25] Kenawy N, Lake SL, Coupland SE, Damato BE, Conjunctival melanoma and melanocytic intra-epithelial neoplasia, Cambridge Ophthalmological Symposium, Eye, 2013, 27(2):142–152.
[26] Kurli M, Finger PT, Melanocytic conjunctival tumors, Ophthalmol Clin North Am, 2005, 18(1):15–24, vii.
[27] Fernandes BF, Odashiro AN, Saraiva VS, Logan P, Antecka E, Burnier MN Jr, Immunohistochemical expression of melan-A and tyrosinase in uveal melanoma, J Carcinog, 2007, 6:6.
[28] Fuchs U, Kivelä T, Summanen P, Immonen I, Tarkkanen A, An immunohistochemical and prognostic analysis of cytokeratin expression in malignant uveal melanoma, Am J Pathol, 1992, 141(1):169–181.
[29] Damato EM, Damato BE, Detection and time to treatment of uveal melanoma in the United Kingdom: an evaluation of 2,384 patients, Ophthalmology, 2012, 119(8):1582–1589.
[30] Humphrey PA, Dehner LP, Pfeifer JD (eds), The Washington manual of surgical pathology, 1st edition, Lippincott Williams & Wilkins, 2008, 163–205.
[31] Damato B, Eleuteri A, Taktak AF, Coupland SE, Estimating prognosis for survival after treatment of choroidal melanoma, Progr Retin Eye Res, 2011, 30(5):285–295.
[32] Triozzi PL, Eng C, Singh AD, Targeted therapy for uveal melanoma, Cancer Treat Rev, 2008, 34(3):247–258.
[33] Albert DM, Kulkarni AD, Intraocular melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA (eds), DeVita, Hellman, and Rosenberg’s cancer: principles and practice of oncology, 9th edition, Lippincott Williams & Wilkins, Philadelphia, 2011, 2090–2098.
Corresponding author Mariana Costache, Associate Professor, MD, PhD, Department of Pathology, “Carol Davila” University of Medicine and Pharmacy, 8 Eroilor Sanitari Avenue, 050474 Bucharest, Romania; Phone/Fax +4021–318 07 62, e-mail: [email protected]
Received: January 10, 2014 Accepted: June 27, 2014