research article error analysis of deep sequencing of
TRANSCRIPT

Hindawi Publishing CorporationComputational and Mathematical Methods in MedicineVolume 2013 Article ID 491612 13 pageshttpdxdoiorg1011552013491612
Research ArticleError Analysis of Deep Sequencing of Phage LibrariesPeptides Censored in Sequencing
Wadim L Matochko and Ratmir Derda
Department of Chemistry and Alberta Glycomics Centre University of Alberta Edmonton AB Canada T6G 2G2
Correspondence should be addressed to Ratmir Derda ratmirualbertaca
Received 6 May 2013 Accepted 30 July 2013
Academic Editor Yanxin Huang
Copyright copy 2013 W L Matochko and R Derda This is an open access article distributed under the Creative CommonsAttribution License which permits unrestricted use distribution and reproduction in any medium provided the original work isproperly cited
Next-generation sequencing techniques empower selection of ligands from phage-display libraries because they can detect lowabundant clones and quantify changes in the copy numbers of clones without excessive selection rounds Identification of errors indeep sequencing data is the most critical step in this process because these techniques have error rates gt1 Mechanisms that yielderrors in Illumina and other techniques have been proposed but no reports to date describe error analysis in phage libraries Ourpaper focuses on error analysis of 7-mer peptide libraries sequenced by Illumina method Low theoretical complexity of this phagelibrary as compared to complexity of long genetic reads and genomes allowed us to describe this library using convenient linearvector and operator framework We describe a phage library as Ntimes1 frequency vector 119899 = n
119894 where n
119894is the copy number of the
119894th sequence and N is the theoretical diversity that is the total number of all possible sequences Any manipulation to the library isan operator acting on 119899 Selection amplification or sequencing could be described as a product of a N timesNmatrix and a stochasticsampling operator (Sa) The latter is a random diagonal matrix that describes sampling of a library In this paper we focus on theproperties of Sa and use them to define the sequencing operator (Seq) Sequencingwithout any bias and errors is Seq = Sa IN whereIN is a N times N unity matrix Any bias in sequencing changes IN to a nonunity matrix We identified a diagonal censorship matrix(CEN) which describes elimination or statistically significant downsampling of specific reads during the sequencing process
1 Introduction
In vitro selection experimentsmdashsuch as phage display [1 2]RNA display SELEX and DNA aptamer selection [3 4]mdashemploy large libraries from which 102ndash106 active sequencesare identified through iterative rounds of selection andamplificationWith the recent emergence of deep sequencingit became possible to extract a large amount of informationfrom the libraries before and after selection [5ndash10] Deepexamination of the library is a promising technique for directevaluation of binding capacities of all binding sequences fromone panning experiment Deep sequencing also allows thecharacterization of unwanted phenomena in selection suchas amplification bias [6 11]
Analysis of 106 reads by deep sequencing gave rise to alarge number of errors that were not present in the analysisbased on the small number of sequences obtained usingthe Sanger method Analysis of errors in information-rich
datasets is a problem with over 50 years of history correctionof digital data made of bits or words is a topic of intenseresearch in communication theory [12] As phage displayoperates with limited digital sets data analysis techniquesfrom the communication theory could be applied to phagedisplay For example Rodi and coworkers used a positionalfrequency matrix to calculate the informational content orShannon entropy of each sequence [13] This approach couldbe used to distinguish potential fast growing sequencesfrom potential hits [14] With the introduction of deepsequencing the problem of error analysis in phage displaybecomes identical to a classical information theory problemldquoreproducing at one point either exactly or approximatelya message selected at another pointrdquo [15] The ldquomessagerdquo isthe sequence information stored in the library Sequencingprocess transmits this information andmakes either stochas-tic or predictable errors Understanding the sources of errorsduring sequencing could provide mechanisms for bypassing
2 Computational and Mathematical Methods in Medicine
them for correcting the errors and for maximizing theamount of useful information received from sequencing
There are over 10000 published literature reports thatcontain the terms ldquodeep sequencingrdquo or ldquonext generationsequencingrdquo or any of the trademark names such as ldquoIllu-minardquo (reference ISI database) Among these reports lessthan 10 published reports describe sequencing of phage-displayed libraries [5ndash7 9 10 16ndash19] Deep sequencing effortsin the literature are largely focused on genome assembly andmetagenomic analysesThe error analysis techniques tailoredfor genome assembly cannot be used directly for analysis ofphage libraries because the data output from phage librarysequencing is very different from the genome assembly Ingenome assembly genomic DNA is shredded into randomfragments and sequenced The genome is then assembledfrom these fragments in silico Although multiple fragmentscover each area of the genome the probability to observetwo identically shredded fragments is very small Two exactsequences thus could be considered amplification artifactsand removed by error analysis software On the contraryin phage-display sequencing the reads are exactly of thesame length Duplication of the same read is important forvalidation of the accuracy of this read Some researchers focusexclusively on reads that have been observed multiple timesand discard singleton reads as erroneous [5] Within eachlibrary the copy numbers of sequences range continuouslyby six or more orders of magnitude [5 6 9] Some phageclones are observed in the entire library only a few timesother clones could be present at copy number of 100000 persequencing run [5 6 9] Unlike multiple cells with identicalgenomes each screen is unique identical set of sequenceswith identical copy numbers cannot be obtained even if thescreen is repeated due to stochastic number of the screen thatcontains low copy number of binding clones [20]
Metagenomic analyses of microorganisms recoveredfrom environmental samples [21 22] also known as ldquomicro-biomerdquo [23] and ldquoviriomerdquo analyses [24] encountered similarproblems to those observed in phage library analysis theconcentration of species observed in a particular sampleis unequal [25] The abundance of species might range bya few orders of magnitude [26] It is possible that erroranalysis tools developed in the above areas could find usein phage display sequencing For example there are multiplepublished algorithms for removing errors from low copynumber reads to ascertain that low copy number sequencesare new species and not sequencing errors (eg see [27ndash29] and references within) Metagenomic analysis is usu-ally more complex than analysis of phage-display librariesFirst in metagenomics the bacterial or viral genes mustbe assembled from short reads de novo Second there isno simple relationship between phylogenetic classification ofldquospeciesrdquo and the observed DNA sequence Third the exactnumber of species in the environment is unknown On theother hand sequencing of phage-displayed peptide librarieshas none of these problems (i) it requires no assemblysteps because each sequence is covered by one read (ii) auniqueDNA sequence defines a unique ldquospeciesrdquo and (iii) thetheoretical complexity in synthetic libraries is known exactlyFor small libraries such as the library of 7-mer peptides
the complexity (20)7 is within the reach of next-generationsequencing We see phage-displayed peptide libraries as anideal model playground for the development of optimalerror analysis and error correction protocols It is possiblethat error analysis developed from phage libraries analysiscould then be used in other areas such as genomic andmetagenomic analyses
The errors in sequencing could be divided into ldquoanno-tatedrdquo and ldquoinvisiblerdquo The ldquoannotatedrdquo errors that originatefrom misincorporation of nucleotides are annotated usingPhred quality score [30]These annotated errors are removedduring the processing (see below) Examples of ldquoinvisiblerdquoerrors are sequence-specific frame shifts that lead to emer-gence of truncated reads during the Illumina sequencing [31]Invisible errors could also originate during the preparation ofthe libraries for sequencing Examples are removal of AT-richfragments during purification of dsDNA [32] and erroneousincorporation of nucleotides during PCR [33 34] Mutationshave the most significant impact on the observed diversity ofthe libraryThere are 63 ways to misspell a 21-mer-nucleotidesequence with a one-letter error (point mutation) The largedynamic range in concentrations of clones in the phagelibrary exacerbates the problem Clones that are present inhigh abundancemdash105 copies per readmdashare more prone toyield errors [6] For example we observed that random pointmutations convert several short sequencewith a copy numberof 105 to a library of sequences with copy numbers rangingfrom 1 to 102 [11] In attempt to unify error analysis into oneconvenient theoretical framework we generalized all errorsas follows All errors either lead to disappearance of particularsequence or its conversion to another sequence of the samelength Errors thus operate within a finite sequence spaceand it should be possible to use elementary linear algebra togeneralize most processes that lead to errors
2 Theoretical Description
See Table 1
21 Operator Description of the Phage-Display Library andSelection Process In our previous reports we described thephage library as a multiset or a set in which members canappear more than once [35] This description also simplifiesthe analysis of the errors in these libraries The multisetdescription represents a library with N theoretical membersas an ordered set of N sequences and N times 1 copy numbervector (119899) with positive integer copy numbers (Figure 1(a))Any manipulation of a phage librarymdashsuch as erroneousreading or selectionmdashchanges the numbers within the copynumber vector All manipulations to the multiset thus couldbe described by operators (Op) that convert vector 119899
1to
another vector 1198992as 1198992= Op 119899
1(Figure 1(c)) For an N times 1
vector the operator is NtimesNmatrix If elements are selected oreliminated independently of one another the NtimesNmatrix isdiagonal (Figure 1(d)) This approach is uniquely convenientfor libraries of short reads For example a library of 7-merscontains exactly 207 = 128 times 109 peptides and is described
Computational and Mathematical Methods in Medicine 3
Table 1 Symbols and definitions used in the theoretical description section
Symbols MeaningA a f m n k Unless specified otherwise normal font designates scalars119860 119886119873 119875 1n 13n Italic font designates vectors Different vectors can be distinguished by the left-superscript notationA a Abc Pan Sa Bold font designates operators or matrices (here all operators are matrices)1AfSa09Sa05Sa Operators can be distinguished by the left-superscript notation For sampling operator Sa this
notation specifies the sampling fraction of the Sa operatorA1 a2 A119894 a119895 Normal font with right subscript designates scalar values of the vectorA11 A21 Aij Aii Normal font with two right subscripts designates scalar values of the 2D matrix1003817100381710038171003817A1 A5
1003817100381710038171003817
Description of the scalar elements in the vector10038171003817100381710038171003817Aij Aii
10038171003817100381710038171003817
Description of the scalar elements in the matrixx isin [A B] Scalar x belongs to the inclusive scalar interval [A B] that is A le x le B119909 isin [119860119861] Vector x belongs to the ldquovector intervalrdquo [119860119861] that is for every element Ai le xi le Bi
ABC X Set where A B C X are the unique elements of the set
A(a) B(b) X(x) Multiset (2-tuple) where A B X are the unique elements and a b x are the scalars describing thecopy numbers of the A B X elements
IN Unity matrix of the Nth order that is N times Nmatrix 10038171003817100381710038171003817Aij10038171003817100381710038171003817 Aij = 120575ij (Kronecker delta)
completely using a 109-element vector This size is accessibleto the computational capacity of most desktop computers
In operator notation phage display can be described as
Sel = Pan Naive (1)
where Naive is the copy number vector for naıve librarySel is the copy number vector after panning and Pan isa panning operator In standard phage display the Panoperator is a complex product of all manipulation steps(binding amplification dilutions etc) If a screen uses noamplification and uses deep-sequencing [9 16] or large-scaleSanger sequencing [36 37] to analyze the enrichment itmight be possible to define the panning process as a simpleproduct of two operators as follows
Pan = fSa Ka (2)
Sel = fSa Ka Naive (3)
where Ka is a deterministic ldquoassociationrdquo operator whichcontains association constants for every phage clone presentin the library Description of such operator is beyond thescope of this paper and we recommend consulting otherreports that attempted to generalize the selection procedure[20] Another operator in (3) is a sampling operator ( fSa)which describes stochastic sampling of the library with msequences to yield a sublibrarywith flowastm-sequences where f isin[0 1] is a sampling fraction fSa operator has the followingproperties which emanate from physical properties of thesampling procedure
(I)
fSai 0 = 0 (sampling does not create new
sequences from nonexisting sequences) (4)
(II) fSa is a diagonal operator with diagonal scalar func-tions Sa
11Sa22sdot sdot sdot SaNN Sai(0) = 0
(III) In 119861 = fSa 119860 119861 is a vector of positive integersB119894ge 0 and sum(119861) = flowastsum(119860) Integer values
ensure that the observable values of the operator havephysical meaning The clone could be observed once(1) multiple times (2 3 etc) or not observed at all(0)
(IV) Sa is nondeterministic operator When applied to thesame vector the operator does not yield the sameresult but one of the possible vectors that satisfy rules(IndashIII) The majority of the solutions of the operatorhowever reside within a deterministic confidenceinterval fSa 119860 isin [ lo119862 hiC]
(V) As a consequence from (IV) operator Sa is nonlinearnoncommutative and nondistributive
(VI) Large sum of sampling operators with same f shouldldquoaverage outrdquo to yield IN unity matrix
(fSa1+
fSa2+
fSa3+ sdot sdot sdot
fSak)k
997888rarr flowastIN
as k 997888rarr infin
(5)
The Sa operator is simple to implement as a randomarray indexing function in any programming language (egsee Supplementary Schemes S1 and S2 available online athttpdxdoiorg1011552013491612) It might be possibleto express fSa analytically for any f as a diagonal matrix(Figure 1(d)) In this paper we use numerical treatment byan array sampling function because it is more convenient formultisets of general structure We tested the random index-ing implementation to show that the sampling algorithmyields a normal distribution for a large number of samples(Supplementary Figure S1) Despite the simplicity of fSa
4 Computational and Mathematical Methods in Medicine
Any phage library is a multiset defined by sequence set and copy number vector
Lib = [S n] middot middot middot middot middot middot
n1n2nN
N number of all possiblesequences in the libraryni copy number of theith sequence in the library
Lib = [S = sequence1sequence2 n = ]sequenceN
(a)
Total (sum) and unique sequences (uni) 11
1middot middot middotsum(n) = n middot |1| =
|1| =Nsumi=1
ni
uni(n) =Nsumi=1
1 minus 120575[ni] 0 n ne 0 delta120575[n] = 1 n = 0 Kronecker
(b)
middot middot middot
Any manipulation of library is an operator acting on the ni vector (copy number vector)Example sampling of the library
1n =
1n11n2
1nN
middot middot middot
2n =
2n12n2
2nN1Lib = [S 1n]
2Lib = [S 2n]
T1 total sequencesT2 total sequences
Sum(1n) = T1 Sum(2n) = sum(Sa 1n) = T2
2n = Sa 1
n
Sa) acts on 1n vectorSampling operator (
Some elementspresent in 1st
library are ldquogonerdquofrom the 2nd library
2ni = 0
1ni 0ne
(c)
rnrn
rn
0 00 0
0 0
Example sampling operator
Properties of Sa
middot middot middotmiddot middot middot
middot middot middot
Sa(n) = round T1
T2
N Irndn where N Irnd =
⋱
k
Operators are N times N matrices Random N times Ndiagonal matrix
(1) Sa(middot middot middot(Sa(Sa(n)) )middot middot middot) asymp Sa(n)
sum Sa(n) rarr infinn as k rarr(2) 1
k
((
(d)
amplificationSynthesis ofnucleotides
Theoretical library
Example each random
Ligation
Sequencing
Panning
Naive librarySynthetic library Selected (panned) library
General problem of selection
Sequencing
Multiset description of the phage display selection
transformationgrowth
nucleotide is present once
T = [S 1]
The ith sequence Sihas copy number syn ni
enriched sequences x i gt 1 depleted sequences x i lt 1
P and N cannot be measured They have to be approximated from the ldquoobservablesrdquo
From P and N find enrichment E = [S x] x e R+obs
P = [S panobsn]
P = [S pann]N = [S naive
n]
S = [S synn]
obsN = [S naive
obsn]
(e)
Figure 1 (a) Phage library can be described by multisets made of S = sequence set and 119899 = vector of copy numbers Any change to thelibrary can be described as functionoperator acting on the 119899 (b) Relevant functions are calculations of total sequences (sum) and uniquesequences (uni) (c) Any transformation of library to another library is an operator acting on 119899 Sampling of libraries to yield a sublibrary isthe most important operator (d) It can be described as N times N matrix Specifically Sa is a diagonal matrix of values derived from randomdistribution Rounding function is necessary to ensure the physical meaning of the sampling results Sa acting on the same vector yieldsone of many vectors that have the same number of total elements As a consequence Sa is nonlinear nondistributive and noncommutativeoperator Average of many Sa operators is a scalar (dilution factor) (e) Any screen of any library can be described as operators acting onthe copy number vectors of the naıve (or theoretical) library Copy number vectors cannot be observed directly They have to be measuredthrough sequencing As sequencing contains sampling process (Sa operator) the result of sequencing is nondeterministic Sequencing yieldsone of many possible observed copy number vectors none of which are equal to the real copy number vector
implementationmdashthe entire code is lt30 lines in MatLabmdashthe script allows rapid calculation of the results of fSa for amultiset of reasonable size (severalmillion sequences Figures4 and 5) on a desktop computer
We evaluated the behaviors of 05Sa for several multisetsThe probability to observe a specific solution is describedin Figure 3(b) Individual solutions can be represented aslines with nodes on 119883119884-plane where each node represents
one element of the multiset (Figures 3(d) and 3(e)) Themost probable solutions reside near the ldquoexpected solutionrdquo(represented as dotted line) and the probability to observea solution where many elements deviate from the probablesolution is low (Figure 3(e)) Graphical representation of thesolutions highlights that sampling could lead to deviationof the frequency of the individual elements of the multisetfor example Figure 3(e) describes gt2 fold deviation from
Computational and Mathematical Methods in Medicine 5
Library of phage
Sampling
Growthof phage
Isolationof DNA
PCRof DNA
Sequencing Analysis of
Sa Gr
Operator description of each step
Sa Is Pc Sa Se AnSa
of DNA sequencing
(observed)Accepted
sequencing data
realN = [S real
n]
obs N = [S obs n]
(a)
Relation between observed copy numbers and ldquorealrdquo copy numbers
Analysisoperator
obs n =f 5Sa An f 4Sa Seq f 3Sa PCR f 2Sa Is f1Sa Gr real n
Growth bias betoassumedandneglected(usually IN )Isolation bias betoassumedandneglected(usually IN )
PCR bias betoassumedandneglected(usually IN )Sequencing bias betoassumedandneglected(usually IN )
(b)
10
0
0 00 0
0 0
0011
0
0
0
Analysis operator is a matrix that could compensate for problems and censorships caused by Se Pc Is Gr operators
f5Sa An (f4Sa Seq f3Sa PCR f2Sa Is f1Sa Gr real ni) sim Sa real ni
Seq 1 is accepted as isSeq 2 is considered a typoand renamed to seq 3Seq 3 is accepted as isSeq N is deleted (possible error)
middot middot middot
⋱
middot middot middot
middot middot middot
An =
(c)
Figure 2 Operator description of the deep sequencing process (a) A library of phage must be processed before deep sequencing Each stepinvolves sampling which is either a deliberate partitioning of the sample or random loss of the sample Each sample preparation state could(and does) introduce bias in sequence abundance Each step thus is an operator chat changeing the 119899 vector (b) If we ignore bias duringpreparation operators could be approximated as unity vectors and sequencing could be represented as a product of sampling and analysisoperators (c) Analysis operator (An) is a binary decision matrix which describes what sequences are and are not considered as errorsDecisions such as removal of sequences or correction of sequences are the most important because they decide which ldquoobservedrdquo sequencesare considered ldquorealrdquo To make the analysis of the selection process meaningful the same An operator should be used in all analyses
the expected value for one of the elements Figure 3(f) showsthat the solution in which two elements deviate by gt2 foldis improbable This observation is a simple consequence ofthe multiplicity of the probabilities (large deviation from theaverage has probability 119901 and the probability to observe thisdeviation twice is 1199012)
Even in small multisets such as A(1) B(2) C(3) D(4)made of four unique and 10 total elements05Sa A(1) B(2) C(3) D(4) operation yields large numberof solutions with equal probability termed as redundantsolutions (eg solutions that have equal probability inFigure 3(b)) Redundancy depends on the structure of themultiset (Figure S2)This redundancy makes the calculationsof all probable solutions of Sa impractical For sets even with5-6 unique elements identification of all vectors 119861 whichsatisfy equation 119861 = fSa 119860 and reside within a 95 intervalrequires hundreds of thousands of iterations (Figure S2and S3) On the other hand calculation of the confidenceinterval of each element 119861i of the vector 119861 converges rapidlyA multiset A
1000 = A
1(1) A2(2) sdot sdot sdotA
1000(1000) with
1000 unique elements and 1 + 2 + 3 + sdot sdot sdot + 1000 = 500 500total elements is similar to an average deep sequencingdata set (Figure 4) Calculation of all probable solutions of05Sa A
1000 is beyond the capabilities of most computers
However the 999 confidence interval of all elements ofvector 119861 = 05Sa A
1000 can be calculated in sim2 minutes on
an average desktop computer The red dots in Figure 4 arelo119862119894and hi119862119894or the 999 high and low confidence interval
of all elements Bi (Figure 4)
The sampling operator is critical in phage display becausesampling of libraries occurs in every step of the selection andthe preparation of libraries for sequencing The stochasticnature of sampling operators makes two identical screensldquosimilar within a confidence intervalrdquo Solving (1) exactly isnot possible but it should be possible to estimate the solutionwithin a confidence interval Consider
Sel isin [ loKa Naive hiKa Naive] (6)
where loKa and hiKa are diagonal matrices of the upperand lower confidence intervals for the association constantsA simulation of the behavior of the Sa operator (Figures3 and S3) suggests that the relative sizes of the confidenceintervalsmight be impractically largewhen the copy numbersof sequences are lt10
Multiple sampling events of the Sa operator yield anormal distribution for each element of the vector (Figure 3)Fitting this normal distribution could yield a ldquotruerdquo valueof the process This process is identical to the extrapolationof the average from the normal distribution of noisy dataMultiple algorithms for such extrapolation exist for one- andmultidimensional stochastic processes [38 39] We believethat Sa behaves as a one-dimensional stochastic process andit might be possible to extrapolate the true value of thesampling from 7 to 10 repeated instances of Sa (ie thenumber of data sufficient to fit an 1D normal distribution)The necessary practical steps towards solving (3) or (10)are the following (i) Eliminate or account for any bias notrelated to binding (eg growth bias) (ii) Repeat the screen
6 Computational and Mathematical Methods in Medicine
Probability to observe specific solution
BB C
C
C
AD
DD
D B CCD
D[0 1 2 2]A B C D
[1 2 3 4]A B C D Sa
95
confi
denc
e int
erva
l
0 2 3 01 0 0 40 1 0 41 1 3 00 0 1 41 2 2 00 2 0 31 0 3 10 0 3 21 2 0 20 1 3 11 1 0 31 2 1 10 0 2 31 0 1 30 2 2 11 0 2 20 2 1 21 1 2 10 1 1 30 1 2 21 1 1 2
0 002 004 006 008 01 012 014
Copy number before
Cop
y nu
mbe
r afte
r
0
1
2
3
4
(a)
(c) (d)
(e)
(h)(g) (i)
(f)(b)
1 2 3 4
Probability to observe specific solution0 002 004 006 008 01 012 014
100000 trials
Sa50
1
6385
5000 trials2957
7
42
1 2 3 4 5 6 7 8012345678
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
One of the solutions
[1 2 3 4 5 6 7 8]A B C D E F G H
1 2 3 4 5 6 7 8
012345678
Cop
y nu
mbe
r afte
r the
sam
plin
g
Number of data per point
Copy number before the sampling
50
BB
CCCADD
DD H HH
G
HHH
HHGG G
GGG
EE
EE
E
E
FF
FF
FF
B
CAD DH
G
HHH
GG
E EE
FF
F
[1 1 1 2 3 3 3 4]A B C D E F G H
Sa 50
Most probable solution
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
[0 2 3 0]A B C D
[1 1 1 2]A B C D
[0 2 1 2]A B C D
the s
ampl
ing
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
the sampling
Copy number before the sampling
Copy number before the sampling
Copy number before the sampling
gt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
solutions
P = 014
P lt 0001
P = 007
after 5000 trials
Figure 3 (a) Testing the sampling operator implemented as random indexing function using a model multiset (b) In 100000 trials weobserved 22 unique solutions from which 14 resided in a 95 confidence interval Solutions with 0 and 1 copies of element A were foundat equal abundances (ldquoredundant solutionsrdquo) (c) Representation of the most probable solution as a line with 4 nodes ldquo119901rdquo is a probabilityto find the solution dotted line is an expected ldquoaverage solutionrdquo for 50 sampling (d) The 5th most probable solution (e) least probablesolution deviates the most form the average (f) combination of all solutions Red thick lines describe the most probable solutions thin bluelines describe the least probable solutions (g) Sampling of larger multisets yields more possible solutions (here 2957 in 5000 trials) (h)All solutions of the sampling represented as lines (i) Probability to observe a particular copy number after sampling While (h) is the mostaccurate representations of the confidence intervals the thin blue lines describe solutions outside the confidence interval this representation isimpractical due to large number of redundant solutions in largermultisets In (i) confidence interval could be extrapolated from distributionsof individual copy numbers (e) red dots are on or outside the confidence interval
several times (iii) Measure all copy numbers of all sequencesincluding zero values with high confidence Requirement (i)has been an ongoing effort in our group [11 40] and othergroups [13 41ndash43] for review see [11 44] Deep sequencing
makes it simple to satisfy requirement (ii) and obtainmultipleinstances of the same experiment For example we describedthe Illumina sequencing method that allows using barcodedprimers to sequence 18 unrelated experiments in one deep
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

2 Computational and Mathematical Methods in Medicine
them for correcting the errors and for maximizing theamount of useful information received from sequencing
There are over 10000 published literature reports thatcontain the terms ldquodeep sequencingrdquo or ldquonext generationsequencingrdquo or any of the trademark names such as ldquoIllu-minardquo (reference ISI database) Among these reports lessthan 10 published reports describe sequencing of phage-displayed libraries [5ndash7 9 10 16ndash19] Deep sequencing effortsin the literature are largely focused on genome assembly andmetagenomic analysesThe error analysis techniques tailoredfor genome assembly cannot be used directly for analysis ofphage libraries because the data output from phage librarysequencing is very different from the genome assembly Ingenome assembly genomic DNA is shredded into randomfragments and sequenced The genome is then assembledfrom these fragments in silico Although multiple fragmentscover each area of the genome the probability to observetwo identically shredded fragments is very small Two exactsequences thus could be considered amplification artifactsand removed by error analysis software On the contraryin phage-display sequencing the reads are exactly of thesame length Duplication of the same read is important forvalidation of the accuracy of this read Some researchers focusexclusively on reads that have been observed multiple timesand discard singleton reads as erroneous [5] Within eachlibrary the copy numbers of sequences range continuouslyby six or more orders of magnitude [5 6 9] Some phageclones are observed in the entire library only a few timesother clones could be present at copy number of 100000 persequencing run [5 6 9] Unlike multiple cells with identicalgenomes each screen is unique identical set of sequenceswith identical copy numbers cannot be obtained even if thescreen is repeated due to stochastic number of the screen thatcontains low copy number of binding clones [20]
Metagenomic analyses of microorganisms recoveredfrom environmental samples [21 22] also known as ldquomicro-biomerdquo [23] and ldquoviriomerdquo analyses [24] encountered similarproblems to those observed in phage library analysis theconcentration of species observed in a particular sampleis unequal [25] The abundance of species might range bya few orders of magnitude [26] It is possible that erroranalysis tools developed in the above areas could find usein phage display sequencing For example there are multiplepublished algorithms for removing errors from low copynumber reads to ascertain that low copy number sequencesare new species and not sequencing errors (eg see [27ndash29] and references within) Metagenomic analysis is usu-ally more complex than analysis of phage-display librariesFirst in metagenomics the bacterial or viral genes mustbe assembled from short reads de novo Second there isno simple relationship between phylogenetic classification ofldquospeciesrdquo and the observed DNA sequence Third the exactnumber of species in the environment is unknown On theother hand sequencing of phage-displayed peptide librarieshas none of these problems (i) it requires no assemblysteps because each sequence is covered by one read (ii) auniqueDNA sequence defines a unique ldquospeciesrdquo and (iii) thetheoretical complexity in synthetic libraries is known exactlyFor small libraries such as the library of 7-mer peptides
the complexity (20)7 is within the reach of next-generationsequencing We see phage-displayed peptide libraries as anideal model playground for the development of optimalerror analysis and error correction protocols It is possiblethat error analysis developed from phage libraries analysiscould then be used in other areas such as genomic andmetagenomic analyses
The errors in sequencing could be divided into ldquoanno-tatedrdquo and ldquoinvisiblerdquo The ldquoannotatedrdquo errors that originatefrom misincorporation of nucleotides are annotated usingPhred quality score [30]These annotated errors are removedduring the processing (see below) Examples of ldquoinvisiblerdquoerrors are sequence-specific frame shifts that lead to emer-gence of truncated reads during the Illumina sequencing [31]Invisible errors could also originate during the preparation ofthe libraries for sequencing Examples are removal of AT-richfragments during purification of dsDNA [32] and erroneousincorporation of nucleotides during PCR [33 34] Mutationshave the most significant impact on the observed diversity ofthe libraryThere are 63 ways to misspell a 21-mer-nucleotidesequence with a one-letter error (point mutation) The largedynamic range in concentrations of clones in the phagelibrary exacerbates the problem Clones that are present inhigh abundancemdash105 copies per readmdashare more prone toyield errors [6] For example we observed that random pointmutations convert several short sequencewith a copy numberof 105 to a library of sequences with copy numbers rangingfrom 1 to 102 [11] In attempt to unify error analysis into oneconvenient theoretical framework we generalized all errorsas follows All errors either lead to disappearance of particularsequence or its conversion to another sequence of the samelength Errors thus operate within a finite sequence spaceand it should be possible to use elementary linear algebra togeneralize most processes that lead to errors
2 Theoretical Description
See Table 1
21 Operator Description of the Phage-Display Library andSelection Process In our previous reports we described thephage library as a multiset or a set in which members canappear more than once [35] This description also simplifiesthe analysis of the errors in these libraries The multisetdescription represents a library with N theoretical membersas an ordered set of N sequences and N times 1 copy numbervector (119899) with positive integer copy numbers (Figure 1(a))Any manipulation of a phage librarymdashsuch as erroneousreading or selectionmdashchanges the numbers within the copynumber vector All manipulations to the multiset thus couldbe described by operators (Op) that convert vector 119899
1to
another vector 1198992as 1198992= Op 119899
1(Figure 1(c)) For an N times 1
vector the operator is NtimesNmatrix If elements are selected oreliminated independently of one another the NtimesNmatrix isdiagonal (Figure 1(d)) This approach is uniquely convenientfor libraries of short reads For example a library of 7-merscontains exactly 207 = 128 times 109 peptides and is described
Computational and Mathematical Methods in Medicine 3
Table 1 Symbols and definitions used in the theoretical description section
Symbols MeaningA a f m n k Unless specified otherwise normal font designates scalars119860 119886119873 119875 1n 13n Italic font designates vectors Different vectors can be distinguished by the left-superscript notationA a Abc Pan Sa Bold font designates operators or matrices (here all operators are matrices)1AfSa09Sa05Sa Operators can be distinguished by the left-superscript notation For sampling operator Sa this
notation specifies the sampling fraction of the Sa operatorA1 a2 A119894 a119895 Normal font with right subscript designates scalar values of the vectorA11 A21 Aij Aii Normal font with two right subscripts designates scalar values of the 2D matrix1003817100381710038171003817A1 A5
1003817100381710038171003817
Description of the scalar elements in the vector10038171003817100381710038171003817Aij Aii
10038171003817100381710038171003817
Description of the scalar elements in the matrixx isin [A B] Scalar x belongs to the inclusive scalar interval [A B] that is A le x le B119909 isin [119860119861] Vector x belongs to the ldquovector intervalrdquo [119860119861] that is for every element Ai le xi le Bi
ABC X Set where A B C X are the unique elements of the set
A(a) B(b) X(x) Multiset (2-tuple) where A B X are the unique elements and a b x are the scalars describing thecopy numbers of the A B X elements
IN Unity matrix of the Nth order that is N times Nmatrix 10038171003817100381710038171003817Aij10038171003817100381710038171003817 Aij = 120575ij (Kronecker delta)
completely using a 109-element vector This size is accessibleto the computational capacity of most desktop computers
In operator notation phage display can be described as
Sel = Pan Naive (1)
where Naive is the copy number vector for naıve librarySel is the copy number vector after panning and Pan isa panning operator In standard phage display the Panoperator is a complex product of all manipulation steps(binding amplification dilutions etc) If a screen uses noamplification and uses deep-sequencing [9 16] or large-scaleSanger sequencing [36 37] to analyze the enrichment itmight be possible to define the panning process as a simpleproduct of two operators as follows
Pan = fSa Ka (2)
Sel = fSa Ka Naive (3)
where Ka is a deterministic ldquoassociationrdquo operator whichcontains association constants for every phage clone presentin the library Description of such operator is beyond thescope of this paper and we recommend consulting otherreports that attempted to generalize the selection procedure[20] Another operator in (3) is a sampling operator ( fSa)which describes stochastic sampling of the library with msequences to yield a sublibrarywith flowastm-sequences where f isin[0 1] is a sampling fraction fSa operator has the followingproperties which emanate from physical properties of thesampling procedure
(I)
fSai 0 = 0 (sampling does not create new
sequences from nonexisting sequences) (4)
(II) fSa is a diagonal operator with diagonal scalar func-tions Sa
11Sa22sdot sdot sdot SaNN Sai(0) = 0
(III) In 119861 = fSa 119860 119861 is a vector of positive integersB119894ge 0 and sum(119861) = flowastsum(119860) Integer values
ensure that the observable values of the operator havephysical meaning The clone could be observed once(1) multiple times (2 3 etc) or not observed at all(0)
(IV) Sa is nondeterministic operator When applied to thesame vector the operator does not yield the sameresult but one of the possible vectors that satisfy rules(IndashIII) The majority of the solutions of the operatorhowever reside within a deterministic confidenceinterval fSa 119860 isin [ lo119862 hiC]
(V) As a consequence from (IV) operator Sa is nonlinearnoncommutative and nondistributive
(VI) Large sum of sampling operators with same f shouldldquoaverage outrdquo to yield IN unity matrix
(fSa1+
fSa2+
fSa3+ sdot sdot sdot
fSak)k
997888rarr flowastIN
as k 997888rarr infin
(5)
The Sa operator is simple to implement as a randomarray indexing function in any programming language (egsee Supplementary Schemes S1 and S2 available online athttpdxdoiorg1011552013491612) It might be possibleto express fSa analytically for any f as a diagonal matrix(Figure 1(d)) In this paper we use numerical treatment byan array sampling function because it is more convenient formultisets of general structure We tested the random index-ing implementation to show that the sampling algorithmyields a normal distribution for a large number of samples(Supplementary Figure S1) Despite the simplicity of fSa
4 Computational and Mathematical Methods in Medicine
Any phage library is a multiset defined by sequence set and copy number vector
Lib = [S n] middot middot middot middot middot middot
n1n2nN
N number of all possiblesequences in the libraryni copy number of theith sequence in the library
Lib = [S = sequence1sequence2 n = ]sequenceN
(a)
Total (sum) and unique sequences (uni) 11
1middot middot middotsum(n) = n middot |1| =
|1| =Nsumi=1
ni
uni(n) =Nsumi=1
1 minus 120575[ni] 0 n ne 0 delta120575[n] = 1 n = 0 Kronecker
(b)
middot middot middot
Any manipulation of library is an operator acting on the ni vector (copy number vector)Example sampling of the library
1n =
1n11n2
1nN
middot middot middot
2n =
2n12n2
2nN1Lib = [S 1n]
2Lib = [S 2n]
T1 total sequencesT2 total sequences
Sum(1n) = T1 Sum(2n) = sum(Sa 1n) = T2
2n = Sa 1
n
Sa) acts on 1n vectorSampling operator (
Some elementspresent in 1st
library are ldquogonerdquofrom the 2nd library
2ni = 0
1ni 0ne
(c)
rnrn
rn
0 00 0
0 0
Example sampling operator
Properties of Sa
middot middot middotmiddot middot middot
middot middot middot
Sa(n) = round T1
T2
N Irndn where N Irnd =
⋱
k
Operators are N times N matrices Random N times Ndiagonal matrix
(1) Sa(middot middot middot(Sa(Sa(n)) )middot middot middot) asymp Sa(n)
sum Sa(n) rarr infinn as k rarr(2) 1
k
((
(d)
amplificationSynthesis ofnucleotides
Theoretical library
Example each random
Ligation
Sequencing
Panning
Naive librarySynthetic library Selected (panned) library
General problem of selection
Sequencing
Multiset description of the phage display selection
transformationgrowth
nucleotide is present once
T = [S 1]
The ith sequence Sihas copy number syn ni
enriched sequences x i gt 1 depleted sequences x i lt 1
P and N cannot be measured They have to be approximated from the ldquoobservablesrdquo
From P and N find enrichment E = [S x] x e R+obs
P = [S panobsn]
P = [S pann]N = [S naive
n]
S = [S synn]
obsN = [S naive
obsn]
(e)
Figure 1 (a) Phage library can be described by multisets made of S = sequence set and 119899 = vector of copy numbers Any change to thelibrary can be described as functionoperator acting on the 119899 (b) Relevant functions are calculations of total sequences (sum) and uniquesequences (uni) (c) Any transformation of library to another library is an operator acting on 119899 Sampling of libraries to yield a sublibrary isthe most important operator (d) It can be described as N times N matrix Specifically Sa is a diagonal matrix of values derived from randomdistribution Rounding function is necessary to ensure the physical meaning of the sampling results Sa acting on the same vector yieldsone of many vectors that have the same number of total elements As a consequence Sa is nonlinear nondistributive and noncommutativeoperator Average of many Sa operators is a scalar (dilution factor) (e) Any screen of any library can be described as operators acting onthe copy number vectors of the naıve (or theoretical) library Copy number vectors cannot be observed directly They have to be measuredthrough sequencing As sequencing contains sampling process (Sa operator) the result of sequencing is nondeterministic Sequencing yieldsone of many possible observed copy number vectors none of which are equal to the real copy number vector
implementationmdashthe entire code is lt30 lines in MatLabmdashthe script allows rapid calculation of the results of fSa for amultiset of reasonable size (severalmillion sequences Figures4 and 5) on a desktop computer
We evaluated the behaviors of 05Sa for several multisetsThe probability to observe a specific solution is describedin Figure 3(b) Individual solutions can be represented aslines with nodes on 119883119884-plane where each node represents
one element of the multiset (Figures 3(d) and 3(e)) Themost probable solutions reside near the ldquoexpected solutionrdquo(represented as dotted line) and the probability to observea solution where many elements deviate from the probablesolution is low (Figure 3(e)) Graphical representation of thesolutions highlights that sampling could lead to deviationof the frequency of the individual elements of the multisetfor example Figure 3(e) describes gt2 fold deviation from
Computational and Mathematical Methods in Medicine 5
Library of phage
Sampling
Growthof phage
Isolationof DNA
PCRof DNA
Sequencing Analysis of
Sa Gr
Operator description of each step
Sa Is Pc Sa Se AnSa
of DNA sequencing
(observed)Accepted
sequencing data
realN = [S real
n]
obs N = [S obs n]
(a)
Relation between observed copy numbers and ldquorealrdquo copy numbers
Analysisoperator
obs n =f 5Sa An f 4Sa Seq f 3Sa PCR f 2Sa Is f1Sa Gr real n
Growth bias betoassumedandneglected(usually IN )Isolation bias betoassumedandneglected(usually IN )
PCR bias betoassumedandneglected(usually IN )Sequencing bias betoassumedandneglected(usually IN )
(b)
10
0
0 00 0
0 0
0011
0
0
0
Analysis operator is a matrix that could compensate for problems and censorships caused by Se Pc Is Gr operators
f5Sa An (f4Sa Seq f3Sa PCR f2Sa Is f1Sa Gr real ni) sim Sa real ni
Seq 1 is accepted as isSeq 2 is considered a typoand renamed to seq 3Seq 3 is accepted as isSeq N is deleted (possible error)
middot middot middot
⋱
middot middot middot
middot middot middot
An =
(c)
Figure 2 Operator description of the deep sequencing process (a) A library of phage must be processed before deep sequencing Each stepinvolves sampling which is either a deliberate partitioning of the sample or random loss of the sample Each sample preparation state could(and does) introduce bias in sequence abundance Each step thus is an operator chat changeing the 119899 vector (b) If we ignore bias duringpreparation operators could be approximated as unity vectors and sequencing could be represented as a product of sampling and analysisoperators (c) Analysis operator (An) is a binary decision matrix which describes what sequences are and are not considered as errorsDecisions such as removal of sequences or correction of sequences are the most important because they decide which ldquoobservedrdquo sequencesare considered ldquorealrdquo To make the analysis of the selection process meaningful the same An operator should be used in all analyses
the expected value for one of the elements Figure 3(f) showsthat the solution in which two elements deviate by gt2 foldis improbable This observation is a simple consequence ofthe multiplicity of the probabilities (large deviation from theaverage has probability 119901 and the probability to observe thisdeviation twice is 1199012)
Even in small multisets such as A(1) B(2) C(3) D(4)made of four unique and 10 total elements05Sa A(1) B(2) C(3) D(4) operation yields large numberof solutions with equal probability termed as redundantsolutions (eg solutions that have equal probability inFigure 3(b)) Redundancy depends on the structure of themultiset (Figure S2)This redundancy makes the calculationsof all probable solutions of Sa impractical For sets even with5-6 unique elements identification of all vectors 119861 whichsatisfy equation 119861 = fSa 119860 and reside within a 95 intervalrequires hundreds of thousands of iterations (Figure S2and S3) On the other hand calculation of the confidenceinterval of each element 119861i of the vector 119861 converges rapidlyA multiset A
1000 = A
1(1) A2(2) sdot sdot sdotA
1000(1000) with
1000 unique elements and 1 + 2 + 3 + sdot sdot sdot + 1000 = 500 500total elements is similar to an average deep sequencingdata set (Figure 4) Calculation of all probable solutions of05Sa A
1000 is beyond the capabilities of most computers
However the 999 confidence interval of all elements ofvector 119861 = 05Sa A
1000 can be calculated in sim2 minutes on
an average desktop computer The red dots in Figure 4 arelo119862119894and hi119862119894or the 999 high and low confidence interval
of all elements Bi (Figure 4)
The sampling operator is critical in phage display becausesampling of libraries occurs in every step of the selection andthe preparation of libraries for sequencing The stochasticnature of sampling operators makes two identical screensldquosimilar within a confidence intervalrdquo Solving (1) exactly isnot possible but it should be possible to estimate the solutionwithin a confidence interval Consider
Sel isin [ loKa Naive hiKa Naive] (6)
where loKa and hiKa are diagonal matrices of the upperand lower confidence intervals for the association constantsA simulation of the behavior of the Sa operator (Figures3 and S3) suggests that the relative sizes of the confidenceintervalsmight be impractically largewhen the copy numbersof sequences are lt10
Multiple sampling events of the Sa operator yield anormal distribution for each element of the vector (Figure 3)Fitting this normal distribution could yield a ldquotruerdquo valueof the process This process is identical to the extrapolationof the average from the normal distribution of noisy dataMultiple algorithms for such extrapolation exist for one- andmultidimensional stochastic processes [38 39] We believethat Sa behaves as a one-dimensional stochastic process andit might be possible to extrapolate the true value of thesampling from 7 to 10 repeated instances of Sa (ie thenumber of data sufficient to fit an 1D normal distribution)The necessary practical steps towards solving (3) or (10)are the following (i) Eliminate or account for any bias notrelated to binding (eg growth bias) (ii) Repeat the screen
6 Computational and Mathematical Methods in Medicine
Probability to observe specific solution
BB C
C
C
AD
DD
D B CCD
D[0 1 2 2]A B C D
[1 2 3 4]A B C D Sa
95
confi
denc
e int
erva
l
0 2 3 01 0 0 40 1 0 41 1 3 00 0 1 41 2 2 00 2 0 31 0 3 10 0 3 21 2 0 20 1 3 11 1 0 31 2 1 10 0 2 31 0 1 30 2 2 11 0 2 20 2 1 21 1 2 10 1 1 30 1 2 21 1 1 2
0 002 004 006 008 01 012 014
Copy number before
Cop
y nu
mbe
r afte
r
0
1
2
3
4
(a)
(c) (d)
(e)
(h)(g) (i)
(f)(b)
1 2 3 4
Probability to observe specific solution0 002 004 006 008 01 012 014
100000 trials
Sa50
1
6385
5000 trials2957
7
42
1 2 3 4 5 6 7 8012345678
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
One of the solutions
[1 2 3 4 5 6 7 8]A B C D E F G H
1 2 3 4 5 6 7 8
012345678
Cop
y nu
mbe
r afte
r the
sam
plin
g
Number of data per point
Copy number before the sampling
50
BB
CCCADD
DD H HH
G
HHH
HHGG G
GGG
EE
EE
E
E
FF
FF
FF
B
CAD DH
G
HHH
GG
E EE
FF
F
[1 1 1 2 3 3 3 4]A B C D E F G H
Sa 50
Most probable solution
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
[0 2 3 0]A B C D
[1 1 1 2]A B C D
[0 2 1 2]A B C D
the s
ampl
ing
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
the sampling
Copy number before the sampling
Copy number before the sampling
Copy number before the sampling
gt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
solutions
P = 014
P lt 0001
P = 007
after 5000 trials
Figure 3 (a) Testing the sampling operator implemented as random indexing function using a model multiset (b) In 100000 trials weobserved 22 unique solutions from which 14 resided in a 95 confidence interval Solutions with 0 and 1 copies of element A were foundat equal abundances (ldquoredundant solutionsrdquo) (c) Representation of the most probable solution as a line with 4 nodes ldquo119901rdquo is a probabilityto find the solution dotted line is an expected ldquoaverage solutionrdquo for 50 sampling (d) The 5th most probable solution (e) least probablesolution deviates the most form the average (f) combination of all solutions Red thick lines describe the most probable solutions thin bluelines describe the least probable solutions (g) Sampling of larger multisets yields more possible solutions (here 2957 in 5000 trials) (h)All solutions of the sampling represented as lines (i) Probability to observe a particular copy number after sampling While (h) is the mostaccurate representations of the confidence intervals the thin blue lines describe solutions outside the confidence interval this representation isimpractical due to large number of redundant solutions in largermultisets In (i) confidence interval could be extrapolated from distributionsof individual copy numbers (e) red dots are on or outside the confidence interval
several times (iii) Measure all copy numbers of all sequencesincluding zero values with high confidence Requirement (i)has been an ongoing effort in our group [11 40] and othergroups [13 41ndash43] for review see [11 44] Deep sequencing
makes it simple to satisfy requirement (ii) and obtainmultipleinstances of the same experiment For example we describedthe Illumina sequencing method that allows using barcodedprimers to sequence 18 unrelated experiments in one deep
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

Computational and Mathematical Methods in Medicine 3
Table 1 Symbols and definitions used in the theoretical description section
Symbols MeaningA a f m n k Unless specified otherwise normal font designates scalars119860 119886119873 119875 1n 13n Italic font designates vectors Different vectors can be distinguished by the left-superscript notationA a Abc Pan Sa Bold font designates operators or matrices (here all operators are matrices)1AfSa09Sa05Sa Operators can be distinguished by the left-superscript notation For sampling operator Sa this
notation specifies the sampling fraction of the Sa operatorA1 a2 A119894 a119895 Normal font with right subscript designates scalar values of the vectorA11 A21 Aij Aii Normal font with two right subscripts designates scalar values of the 2D matrix1003817100381710038171003817A1 A5
1003817100381710038171003817
Description of the scalar elements in the vector10038171003817100381710038171003817Aij Aii
10038171003817100381710038171003817
Description of the scalar elements in the matrixx isin [A B] Scalar x belongs to the inclusive scalar interval [A B] that is A le x le B119909 isin [119860119861] Vector x belongs to the ldquovector intervalrdquo [119860119861] that is for every element Ai le xi le Bi
ABC X Set where A B C X are the unique elements of the set
A(a) B(b) X(x) Multiset (2-tuple) where A B X are the unique elements and a b x are the scalars describing thecopy numbers of the A B X elements
IN Unity matrix of the Nth order that is N times Nmatrix 10038171003817100381710038171003817Aij10038171003817100381710038171003817 Aij = 120575ij (Kronecker delta)
completely using a 109-element vector This size is accessibleto the computational capacity of most desktop computers
In operator notation phage display can be described as
Sel = Pan Naive (1)
where Naive is the copy number vector for naıve librarySel is the copy number vector after panning and Pan isa panning operator In standard phage display the Panoperator is a complex product of all manipulation steps(binding amplification dilutions etc) If a screen uses noamplification and uses deep-sequencing [9 16] or large-scaleSanger sequencing [36 37] to analyze the enrichment itmight be possible to define the panning process as a simpleproduct of two operators as follows
Pan = fSa Ka (2)
Sel = fSa Ka Naive (3)
where Ka is a deterministic ldquoassociationrdquo operator whichcontains association constants for every phage clone presentin the library Description of such operator is beyond thescope of this paper and we recommend consulting otherreports that attempted to generalize the selection procedure[20] Another operator in (3) is a sampling operator ( fSa)which describes stochastic sampling of the library with msequences to yield a sublibrarywith flowastm-sequences where f isin[0 1] is a sampling fraction fSa operator has the followingproperties which emanate from physical properties of thesampling procedure
(I)
fSai 0 = 0 (sampling does not create new
sequences from nonexisting sequences) (4)
(II) fSa is a diagonal operator with diagonal scalar func-tions Sa
11Sa22sdot sdot sdot SaNN Sai(0) = 0
(III) In 119861 = fSa 119860 119861 is a vector of positive integersB119894ge 0 and sum(119861) = flowastsum(119860) Integer values
ensure that the observable values of the operator havephysical meaning The clone could be observed once(1) multiple times (2 3 etc) or not observed at all(0)
(IV) Sa is nondeterministic operator When applied to thesame vector the operator does not yield the sameresult but one of the possible vectors that satisfy rules(IndashIII) The majority of the solutions of the operatorhowever reside within a deterministic confidenceinterval fSa 119860 isin [ lo119862 hiC]
(V) As a consequence from (IV) operator Sa is nonlinearnoncommutative and nondistributive
(VI) Large sum of sampling operators with same f shouldldquoaverage outrdquo to yield IN unity matrix
(fSa1+
fSa2+
fSa3+ sdot sdot sdot
fSak)k
997888rarr flowastIN
as k 997888rarr infin
(5)
The Sa operator is simple to implement as a randomarray indexing function in any programming language (egsee Supplementary Schemes S1 and S2 available online athttpdxdoiorg1011552013491612) It might be possibleto express fSa analytically for any f as a diagonal matrix(Figure 1(d)) In this paper we use numerical treatment byan array sampling function because it is more convenient formultisets of general structure We tested the random index-ing implementation to show that the sampling algorithmyields a normal distribution for a large number of samples(Supplementary Figure S1) Despite the simplicity of fSa
4 Computational and Mathematical Methods in Medicine
Any phage library is a multiset defined by sequence set and copy number vector
Lib = [S n] middot middot middot middot middot middot
n1n2nN
N number of all possiblesequences in the libraryni copy number of theith sequence in the library
Lib = [S = sequence1sequence2 n = ]sequenceN
(a)
Total (sum) and unique sequences (uni) 11
1middot middot middotsum(n) = n middot |1| =
|1| =Nsumi=1
ni
uni(n) =Nsumi=1
1 minus 120575[ni] 0 n ne 0 delta120575[n] = 1 n = 0 Kronecker
(b)
middot middot middot
Any manipulation of library is an operator acting on the ni vector (copy number vector)Example sampling of the library
1n =
1n11n2
1nN
middot middot middot
2n =
2n12n2
2nN1Lib = [S 1n]
2Lib = [S 2n]
T1 total sequencesT2 total sequences
Sum(1n) = T1 Sum(2n) = sum(Sa 1n) = T2
2n = Sa 1
n
Sa) acts on 1n vectorSampling operator (
Some elementspresent in 1st
library are ldquogonerdquofrom the 2nd library
2ni = 0
1ni 0ne
(c)
rnrn
rn
0 00 0
0 0
Example sampling operator
Properties of Sa
middot middot middotmiddot middot middot
middot middot middot
Sa(n) = round T1
T2
N Irndn where N Irnd =
⋱
k
Operators are N times N matrices Random N times Ndiagonal matrix
(1) Sa(middot middot middot(Sa(Sa(n)) )middot middot middot) asymp Sa(n)
sum Sa(n) rarr infinn as k rarr(2) 1
k
((
(d)
amplificationSynthesis ofnucleotides
Theoretical library
Example each random
Ligation
Sequencing
Panning
Naive librarySynthetic library Selected (panned) library
General problem of selection
Sequencing
Multiset description of the phage display selection
transformationgrowth
nucleotide is present once
T = [S 1]
The ith sequence Sihas copy number syn ni
enriched sequences x i gt 1 depleted sequences x i lt 1
P and N cannot be measured They have to be approximated from the ldquoobservablesrdquo
From P and N find enrichment E = [S x] x e R+obs
P = [S panobsn]
P = [S pann]N = [S naive
n]
S = [S synn]
obsN = [S naive
obsn]
(e)
Figure 1 (a) Phage library can be described by multisets made of S = sequence set and 119899 = vector of copy numbers Any change to thelibrary can be described as functionoperator acting on the 119899 (b) Relevant functions are calculations of total sequences (sum) and uniquesequences (uni) (c) Any transformation of library to another library is an operator acting on 119899 Sampling of libraries to yield a sublibrary isthe most important operator (d) It can be described as N times N matrix Specifically Sa is a diagonal matrix of values derived from randomdistribution Rounding function is necessary to ensure the physical meaning of the sampling results Sa acting on the same vector yieldsone of many vectors that have the same number of total elements As a consequence Sa is nonlinear nondistributive and noncommutativeoperator Average of many Sa operators is a scalar (dilution factor) (e) Any screen of any library can be described as operators acting onthe copy number vectors of the naıve (or theoretical) library Copy number vectors cannot be observed directly They have to be measuredthrough sequencing As sequencing contains sampling process (Sa operator) the result of sequencing is nondeterministic Sequencing yieldsone of many possible observed copy number vectors none of which are equal to the real copy number vector
implementationmdashthe entire code is lt30 lines in MatLabmdashthe script allows rapid calculation of the results of fSa for amultiset of reasonable size (severalmillion sequences Figures4 and 5) on a desktop computer
We evaluated the behaviors of 05Sa for several multisetsThe probability to observe a specific solution is describedin Figure 3(b) Individual solutions can be represented aslines with nodes on 119883119884-plane where each node represents
one element of the multiset (Figures 3(d) and 3(e)) Themost probable solutions reside near the ldquoexpected solutionrdquo(represented as dotted line) and the probability to observea solution where many elements deviate from the probablesolution is low (Figure 3(e)) Graphical representation of thesolutions highlights that sampling could lead to deviationof the frequency of the individual elements of the multisetfor example Figure 3(e) describes gt2 fold deviation from
Computational and Mathematical Methods in Medicine 5
Library of phage
Sampling
Growthof phage
Isolationof DNA
PCRof DNA
Sequencing Analysis of
Sa Gr
Operator description of each step
Sa Is Pc Sa Se AnSa
of DNA sequencing
(observed)Accepted
sequencing data
realN = [S real
n]
obs N = [S obs n]
(a)
Relation between observed copy numbers and ldquorealrdquo copy numbers
Analysisoperator
obs n =f 5Sa An f 4Sa Seq f 3Sa PCR f 2Sa Is f1Sa Gr real n
Growth bias betoassumedandneglected(usually IN )Isolation bias betoassumedandneglected(usually IN )
PCR bias betoassumedandneglected(usually IN )Sequencing bias betoassumedandneglected(usually IN )
(b)
10
0
0 00 0
0 0
0011
0
0
0
Analysis operator is a matrix that could compensate for problems and censorships caused by Se Pc Is Gr operators
f5Sa An (f4Sa Seq f3Sa PCR f2Sa Is f1Sa Gr real ni) sim Sa real ni
Seq 1 is accepted as isSeq 2 is considered a typoand renamed to seq 3Seq 3 is accepted as isSeq N is deleted (possible error)
middot middot middot
⋱
middot middot middot
middot middot middot
An =
(c)
Figure 2 Operator description of the deep sequencing process (a) A library of phage must be processed before deep sequencing Each stepinvolves sampling which is either a deliberate partitioning of the sample or random loss of the sample Each sample preparation state could(and does) introduce bias in sequence abundance Each step thus is an operator chat changeing the 119899 vector (b) If we ignore bias duringpreparation operators could be approximated as unity vectors and sequencing could be represented as a product of sampling and analysisoperators (c) Analysis operator (An) is a binary decision matrix which describes what sequences are and are not considered as errorsDecisions such as removal of sequences or correction of sequences are the most important because they decide which ldquoobservedrdquo sequencesare considered ldquorealrdquo To make the analysis of the selection process meaningful the same An operator should be used in all analyses
the expected value for one of the elements Figure 3(f) showsthat the solution in which two elements deviate by gt2 foldis improbable This observation is a simple consequence ofthe multiplicity of the probabilities (large deviation from theaverage has probability 119901 and the probability to observe thisdeviation twice is 1199012)
Even in small multisets such as A(1) B(2) C(3) D(4)made of four unique and 10 total elements05Sa A(1) B(2) C(3) D(4) operation yields large numberof solutions with equal probability termed as redundantsolutions (eg solutions that have equal probability inFigure 3(b)) Redundancy depends on the structure of themultiset (Figure S2)This redundancy makes the calculationsof all probable solutions of Sa impractical For sets even with5-6 unique elements identification of all vectors 119861 whichsatisfy equation 119861 = fSa 119860 and reside within a 95 intervalrequires hundreds of thousands of iterations (Figure S2and S3) On the other hand calculation of the confidenceinterval of each element 119861i of the vector 119861 converges rapidlyA multiset A
1000 = A
1(1) A2(2) sdot sdot sdotA
1000(1000) with
1000 unique elements and 1 + 2 + 3 + sdot sdot sdot + 1000 = 500 500total elements is similar to an average deep sequencingdata set (Figure 4) Calculation of all probable solutions of05Sa A
1000 is beyond the capabilities of most computers
However the 999 confidence interval of all elements ofvector 119861 = 05Sa A
1000 can be calculated in sim2 minutes on
an average desktop computer The red dots in Figure 4 arelo119862119894and hi119862119894or the 999 high and low confidence interval
of all elements Bi (Figure 4)
The sampling operator is critical in phage display becausesampling of libraries occurs in every step of the selection andthe preparation of libraries for sequencing The stochasticnature of sampling operators makes two identical screensldquosimilar within a confidence intervalrdquo Solving (1) exactly isnot possible but it should be possible to estimate the solutionwithin a confidence interval Consider
Sel isin [ loKa Naive hiKa Naive] (6)
where loKa and hiKa are diagonal matrices of the upperand lower confidence intervals for the association constantsA simulation of the behavior of the Sa operator (Figures3 and S3) suggests that the relative sizes of the confidenceintervalsmight be impractically largewhen the copy numbersof sequences are lt10
Multiple sampling events of the Sa operator yield anormal distribution for each element of the vector (Figure 3)Fitting this normal distribution could yield a ldquotruerdquo valueof the process This process is identical to the extrapolationof the average from the normal distribution of noisy dataMultiple algorithms for such extrapolation exist for one- andmultidimensional stochastic processes [38 39] We believethat Sa behaves as a one-dimensional stochastic process andit might be possible to extrapolate the true value of thesampling from 7 to 10 repeated instances of Sa (ie thenumber of data sufficient to fit an 1D normal distribution)The necessary practical steps towards solving (3) or (10)are the following (i) Eliminate or account for any bias notrelated to binding (eg growth bias) (ii) Repeat the screen
6 Computational and Mathematical Methods in Medicine
Probability to observe specific solution
BB C
C
C
AD
DD
D B CCD
D[0 1 2 2]A B C D
[1 2 3 4]A B C D Sa
95
confi
denc
e int
erva
l
0 2 3 01 0 0 40 1 0 41 1 3 00 0 1 41 2 2 00 2 0 31 0 3 10 0 3 21 2 0 20 1 3 11 1 0 31 2 1 10 0 2 31 0 1 30 2 2 11 0 2 20 2 1 21 1 2 10 1 1 30 1 2 21 1 1 2
0 002 004 006 008 01 012 014
Copy number before
Cop
y nu
mbe
r afte
r
0
1
2
3
4
(a)
(c) (d)
(e)
(h)(g) (i)
(f)(b)
1 2 3 4
Probability to observe specific solution0 002 004 006 008 01 012 014
100000 trials
Sa50
1
6385
5000 trials2957
7
42
1 2 3 4 5 6 7 8012345678
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
One of the solutions
[1 2 3 4 5 6 7 8]A B C D E F G H
1 2 3 4 5 6 7 8
012345678
Cop
y nu
mbe
r afte
r the
sam
plin
g
Number of data per point
Copy number before the sampling
50
BB
CCCADD
DD H HH
G
HHH
HHGG G
GGG
EE
EE
E
E
FF
FF
FF
B
CAD DH
G
HHH
GG
E EE
FF
F
[1 1 1 2 3 3 3 4]A B C D E F G H
Sa 50
Most probable solution
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
[0 2 3 0]A B C D
[1 1 1 2]A B C D
[0 2 1 2]A B C D
the s
ampl
ing
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
the sampling
Copy number before the sampling
Copy number before the sampling
Copy number before the sampling
gt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
solutions
P = 014
P lt 0001
P = 007
after 5000 trials
Figure 3 (a) Testing the sampling operator implemented as random indexing function using a model multiset (b) In 100000 trials weobserved 22 unique solutions from which 14 resided in a 95 confidence interval Solutions with 0 and 1 copies of element A were foundat equal abundances (ldquoredundant solutionsrdquo) (c) Representation of the most probable solution as a line with 4 nodes ldquo119901rdquo is a probabilityto find the solution dotted line is an expected ldquoaverage solutionrdquo for 50 sampling (d) The 5th most probable solution (e) least probablesolution deviates the most form the average (f) combination of all solutions Red thick lines describe the most probable solutions thin bluelines describe the least probable solutions (g) Sampling of larger multisets yields more possible solutions (here 2957 in 5000 trials) (h)All solutions of the sampling represented as lines (i) Probability to observe a particular copy number after sampling While (h) is the mostaccurate representations of the confidence intervals the thin blue lines describe solutions outside the confidence interval this representation isimpractical due to large number of redundant solutions in largermultisets In (i) confidence interval could be extrapolated from distributionsof individual copy numbers (e) red dots are on or outside the confidence interval
several times (iii) Measure all copy numbers of all sequencesincluding zero values with high confidence Requirement (i)has been an ongoing effort in our group [11 40] and othergroups [13 41ndash43] for review see [11 44] Deep sequencing
makes it simple to satisfy requirement (ii) and obtainmultipleinstances of the same experiment For example we describedthe Illumina sequencing method that allows using barcodedprimers to sequence 18 unrelated experiments in one deep
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

4 Computational and Mathematical Methods in Medicine
Any phage library is a multiset defined by sequence set and copy number vector
Lib = [S n] middot middot middot middot middot middot
n1n2nN
N number of all possiblesequences in the libraryni copy number of theith sequence in the library
Lib = [S = sequence1sequence2 n = ]sequenceN
(a)
Total (sum) and unique sequences (uni) 11
1middot middot middotsum(n) = n middot |1| =
|1| =Nsumi=1
ni
uni(n) =Nsumi=1
1 minus 120575[ni] 0 n ne 0 delta120575[n] = 1 n = 0 Kronecker
(b)
middot middot middot
Any manipulation of library is an operator acting on the ni vector (copy number vector)Example sampling of the library
1n =
1n11n2
1nN
middot middot middot
2n =
2n12n2
2nN1Lib = [S 1n]
2Lib = [S 2n]
T1 total sequencesT2 total sequences
Sum(1n) = T1 Sum(2n) = sum(Sa 1n) = T2
2n = Sa 1
n
Sa) acts on 1n vectorSampling operator (
Some elementspresent in 1st
library are ldquogonerdquofrom the 2nd library
2ni = 0
1ni 0ne
(c)
rnrn
rn
0 00 0
0 0
Example sampling operator
Properties of Sa
middot middot middotmiddot middot middot
middot middot middot
Sa(n) = round T1
T2
N Irndn where N Irnd =
⋱
k
Operators are N times N matrices Random N times Ndiagonal matrix
(1) Sa(middot middot middot(Sa(Sa(n)) )middot middot middot) asymp Sa(n)
sum Sa(n) rarr infinn as k rarr(2) 1
k
((
(d)
amplificationSynthesis ofnucleotides
Theoretical library
Example each random
Ligation
Sequencing
Panning
Naive librarySynthetic library Selected (panned) library
General problem of selection
Sequencing
Multiset description of the phage display selection
transformationgrowth
nucleotide is present once
T = [S 1]
The ith sequence Sihas copy number syn ni
enriched sequences x i gt 1 depleted sequences x i lt 1
P and N cannot be measured They have to be approximated from the ldquoobservablesrdquo
From P and N find enrichment E = [S x] x e R+obs
P = [S panobsn]
P = [S pann]N = [S naive
n]
S = [S synn]
obsN = [S naive
obsn]
(e)
Figure 1 (a) Phage library can be described by multisets made of S = sequence set and 119899 = vector of copy numbers Any change to thelibrary can be described as functionoperator acting on the 119899 (b) Relevant functions are calculations of total sequences (sum) and uniquesequences (uni) (c) Any transformation of library to another library is an operator acting on 119899 Sampling of libraries to yield a sublibrary isthe most important operator (d) It can be described as N times N matrix Specifically Sa is a diagonal matrix of values derived from randomdistribution Rounding function is necessary to ensure the physical meaning of the sampling results Sa acting on the same vector yieldsone of many vectors that have the same number of total elements As a consequence Sa is nonlinear nondistributive and noncommutativeoperator Average of many Sa operators is a scalar (dilution factor) (e) Any screen of any library can be described as operators acting onthe copy number vectors of the naıve (or theoretical) library Copy number vectors cannot be observed directly They have to be measuredthrough sequencing As sequencing contains sampling process (Sa operator) the result of sequencing is nondeterministic Sequencing yieldsone of many possible observed copy number vectors none of which are equal to the real copy number vector
implementationmdashthe entire code is lt30 lines in MatLabmdashthe script allows rapid calculation of the results of fSa for amultiset of reasonable size (severalmillion sequences Figures4 and 5) on a desktop computer
We evaluated the behaviors of 05Sa for several multisetsThe probability to observe a specific solution is describedin Figure 3(b) Individual solutions can be represented aslines with nodes on 119883119884-plane where each node represents
one element of the multiset (Figures 3(d) and 3(e)) Themost probable solutions reside near the ldquoexpected solutionrdquo(represented as dotted line) and the probability to observea solution where many elements deviate from the probablesolution is low (Figure 3(e)) Graphical representation of thesolutions highlights that sampling could lead to deviationof the frequency of the individual elements of the multisetfor example Figure 3(e) describes gt2 fold deviation from
Computational and Mathematical Methods in Medicine 5
Library of phage
Sampling
Growthof phage
Isolationof DNA
PCRof DNA
Sequencing Analysis of
Sa Gr
Operator description of each step
Sa Is Pc Sa Se AnSa
of DNA sequencing
(observed)Accepted
sequencing data
realN = [S real
n]
obs N = [S obs n]
(a)
Relation between observed copy numbers and ldquorealrdquo copy numbers
Analysisoperator
obs n =f 5Sa An f 4Sa Seq f 3Sa PCR f 2Sa Is f1Sa Gr real n
Growth bias betoassumedandneglected(usually IN )Isolation bias betoassumedandneglected(usually IN )
PCR bias betoassumedandneglected(usually IN )Sequencing bias betoassumedandneglected(usually IN )
(b)
10
0
0 00 0
0 0
0011
0
0
0
Analysis operator is a matrix that could compensate for problems and censorships caused by Se Pc Is Gr operators
f5Sa An (f4Sa Seq f3Sa PCR f2Sa Is f1Sa Gr real ni) sim Sa real ni
Seq 1 is accepted as isSeq 2 is considered a typoand renamed to seq 3Seq 3 is accepted as isSeq N is deleted (possible error)
middot middot middot
⋱
middot middot middot
middot middot middot
An =
(c)
Figure 2 Operator description of the deep sequencing process (a) A library of phage must be processed before deep sequencing Each stepinvolves sampling which is either a deliberate partitioning of the sample or random loss of the sample Each sample preparation state could(and does) introduce bias in sequence abundance Each step thus is an operator chat changeing the 119899 vector (b) If we ignore bias duringpreparation operators could be approximated as unity vectors and sequencing could be represented as a product of sampling and analysisoperators (c) Analysis operator (An) is a binary decision matrix which describes what sequences are and are not considered as errorsDecisions such as removal of sequences or correction of sequences are the most important because they decide which ldquoobservedrdquo sequencesare considered ldquorealrdquo To make the analysis of the selection process meaningful the same An operator should be used in all analyses
the expected value for one of the elements Figure 3(f) showsthat the solution in which two elements deviate by gt2 foldis improbable This observation is a simple consequence ofthe multiplicity of the probabilities (large deviation from theaverage has probability 119901 and the probability to observe thisdeviation twice is 1199012)
Even in small multisets such as A(1) B(2) C(3) D(4)made of four unique and 10 total elements05Sa A(1) B(2) C(3) D(4) operation yields large numberof solutions with equal probability termed as redundantsolutions (eg solutions that have equal probability inFigure 3(b)) Redundancy depends on the structure of themultiset (Figure S2)This redundancy makes the calculationsof all probable solutions of Sa impractical For sets even with5-6 unique elements identification of all vectors 119861 whichsatisfy equation 119861 = fSa 119860 and reside within a 95 intervalrequires hundreds of thousands of iterations (Figure S2and S3) On the other hand calculation of the confidenceinterval of each element 119861i of the vector 119861 converges rapidlyA multiset A
1000 = A
1(1) A2(2) sdot sdot sdotA
1000(1000) with
1000 unique elements and 1 + 2 + 3 + sdot sdot sdot + 1000 = 500 500total elements is similar to an average deep sequencingdata set (Figure 4) Calculation of all probable solutions of05Sa A
1000 is beyond the capabilities of most computers
However the 999 confidence interval of all elements ofvector 119861 = 05Sa A
1000 can be calculated in sim2 minutes on
an average desktop computer The red dots in Figure 4 arelo119862119894and hi119862119894or the 999 high and low confidence interval
of all elements Bi (Figure 4)
The sampling operator is critical in phage display becausesampling of libraries occurs in every step of the selection andthe preparation of libraries for sequencing The stochasticnature of sampling operators makes two identical screensldquosimilar within a confidence intervalrdquo Solving (1) exactly isnot possible but it should be possible to estimate the solutionwithin a confidence interval Consider
Sel isin [ loKa Naive hiKa Naive] (6)
where loKa and hiKa are diagonal matrices of the upperand lower confidence intervals for the association constantsA simulation of the behavior of the Sa operator (Figures3 and S3) suggests that the relative sizes of the confidenceintervalsmight be impractically largewhen the copy numbersof sequences are lt10
Multiple sampling events of the Sa operator yield anormal distribution for each element of the vector (Figure 3)Fitting this normal distribution could yield a ldquotruerdquo valueof the process This process is identical to the extrapolationof the average from the normal distribution of noisy dataMultiple algorithms for such extrapolation exist for one- andmultidimensional stochastic processes [38 39] We believethat Sa behaves as a one-dimensional stochastic process andit might be possible to extrapolate the true value of thesampling from 7 to 10 repeated instances of Sa (ie thenumber of data sufficient to fit an 1D normal distribution)The necessary practical steps towards solving (3) or (10)are the following (i) Eliminate or account for any bias notrelated to binding (eg growth bias) (ii) Repeat the screen
6 Computational and Mathematical Methods in Medicine
Probability to observe specific solution
BB C
C
C
AD
DD
D B CCD
D[0 1 2 2]A B C D
[1 2 3 4]A B C D Sa
95
confi
denc
e int
erva
l
0 2 3 01 0 0 40 1 0 41 1 3 00 0 1 41 2 2 00 2 0 31 0 3 10 0 3 21 2 0 20 1 3 11 1 0 31 2 1 10 0 2 31 0 1 30 2 2 11 0 2 20 2 1 21 1 2 10 1 1 30 1 2 21 1 1 2
0 002 004 006 008 01 012 014
Copy number before
Cop
y nu
mbe
r afte
r
0
1
2
3
4
(a)
(c) (d)
(e)
(h)(g) (i)
(f)(b)
1 2 3 4
Probability to observe specific solution0 002 004 006 008 01 012 014
100000 trials
Sa50
1
6385
5000 trials2957
7
42
1 2 3 4 5 6 7 8012345678
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
One of the solutions
[1 2 3 4 5 6 7 8]A B C D E F G H
1 2 3 4 5 6 7 8
012345678
Cop
y nu
mbe
r afte
r the
sam
plin
g
Number of data per point
Copy number before the sampling
50
BB
CCCADD
DD H HH
G
HHH
HHGG G
GGG
EE
EE
E
E
FF
FF
FF
B
CAD DH
G
HHH
GG
E EE
FF
F
[1 1 1 2 3 3 3 4]A B C D E F G H
Sa 50
Most probable solution
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
[0 2 3 0]A B C D
[1 1 1 2]A B C D
[0 2 1 2]A B C D
the s
ampl
ing
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
the sampling
Copy number before the sampling
Copy number before the sampling
Copy number before the sampling
gt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
solutions
P = 014
P lt 0001
P = 007
after 5000 trials
Figure 3 (a) Testing the sampling operator implemented as random indexing function using a model multiset (b) In 100000 trials weobserved 22 unique solutions from which 14 resided in a 95 confidence interval Solutions with 0 and 1 copies of element A were foundat equal abundances (ldquoredundant solutionsrdquo) (c) Representation of the most probable solution as a line with 4 nodes ldquo119901rdquo is a probabilityto find the solution dotted line is an expected ldquoaverage solutionrdquo for 50 sampling (d) The 5th most probable solution (e) least probablesolution deviates the most form the average (f) combination of all solutions Red thick lines describe the most probable solutions thin bluelines describe the least probable solutions (g) Sampling of larger multisets yields more possible solutions (here 2957 in 5000 trials) (h)All solutions of the sampling represented as lines (i) Probability to observe a particular copy number after sampling While (h) is the mostaccurate representations of the confidence intervals the thin blue lines describe solutions outside the confidence interval this representation isimpractical due to large number of redundant solutions in largermultisets In (i) confidence interval could be extrapolated from distributionsof individual copy numbers (e) red dots are on or outside the confidence interval
several times (iii) Measure all copy numbers of all sequencesincluding zero values with high confidence Requirement (i)has been an ongoing effort in our group [11 40] and othergroups [13 41ndash43] for review see [11 44] Deep sequencing
makes it simple to satisfy requirement (ii) and obtainmultipleinstances of the same experiment For example we describedthe Illumina sequencing method that allows using barcodedprimers to sequence 18 unrelated experiments in one deep
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

Computational and Mathematical Methods in Medicine 5
Library of phage
Sampling
Growthof phage
Isolationof DNA
PCRof DNA
Sequencing Analysis of
Sa Gr
Operator description of each step
Sa Is Pc Sa Se AnSa
of DNA sequencing
(observed)Accepted
sequencing data
realN = [S real
n]
obs N = [S obs n]
(a)
Relation between observed copy numbers and ldquorealrdquo copy numbers
Analysisoperator
obs n =f 5Sa An f 4Sa Seq f 3Sa PCR f 2Sa Is f1Sa Gr real n
Growth bias betoassumedandneglected(usually IN )Isolation bias betoassumedandneglected(usually IN )
PCR bias betoassumedandneglected(usually IN )Sequencing bias betoassumedandneglected(usually IN )
(b)
10
0
0 00 0
0 0
0011
0
0
0
Analysis operator is a matrix that could compensate for problems and censorships caused by Se Pc Is Gr operators
f5Sa An (f4Sa Seq f3Sa PCR f2Sa Is f1Sa Gr real ni) sim Sa real ni
Seq 1 is accepted as isSeq 2 is considered a typoand renamed to seq 3Seq 3 is accepted as isSeq N is deleted (possible error)
middot middot middot
⋱
middot middot middot
middot middot middot
An =
(c)
Figure 2 Operator description of the deep sequencing process (a) A library of phage must be processed before deep sequencing Each stepinvolves sampling which is either a deliberate partitioning of the sample or random loss of the sample Each sample preparation state could(and does) introduce bias in sequence abundance Each step thus is an operator chat changeing the 119899 vector (b) If we ignore bias duringpreparation operators could be approximated as unity vectors and sequencing could be represented as a product of sampling and analysisoperators (c) Analysis operator (An) is a binary decision matrix which describes what sequences are and are not considered as errorsDecisions such as removal of sequences or correction of sequences are the most important because they decide which ldquoobservedrdquo sequencesare considered ldquorealrdquo To make the analysis of the selection process meaningful the same An operator should be used in all analyses
the expected value for one of the elements Figure 3(f) showsthat the solution in which two elements deviate by gt2 foldis improbable This observation is a simple consequence ofthe multiplicity of the probabilities (large deviation from theaverage has probability 119901 and the probability to observe thisdeviation twice is 1199012)
Even in small multisets such as A(1) B(2) C(3) D(4)made of four unique and 10 total elements05Sa A(1) B(2) C(3) D(4) operation yields large numberof solutions with equal probability termed as redundantsolutions (eg solutions that have equal probability inFigure 3(b)) Redundancy depends on the structure of themultiset (Figure S2)This redundancy makes the calculationsof all probable solutions of Sa impractical For sets even with5-6 unique elements identification of all vectors 119861 whichsatisfy equation 119861 = fSa 119860 and reside within a 95 intervalrequires hundreds of thousands of iterations (Figure S2and S3) On the other hand calculation of the confidenceinterval of each element 119861i of the vector 119861 converges rapidlyA multiset A
1000 = A
1(1) A2(2) sdot sdot sdotA
1000(1000) with
1000 unique elements and 1 + 2 + 3 + sdot sdot sdot + 1000 = 500 500total elements is similar to an average deep sequencingdata set (Figure 4) Calculation of all probable solutions of05Sa A
1000 is beyond the capabilities of most computers
However the 999 confidence interval of all elements ofvector 119861 = 05Sa A
1000 can be calculated in sim2 minutes on
an average desktop computer The red dots in Figure 4 arelo119862119894and hi119862119894or the 999 high and low confidence interval
of all elements Bi (Figure 4)
The sampling operator is critical in phage display becausesampling of libraries occurs in every step of the selection andthe preparation of libraries for sequencing The stochasticnature of sampling operators makes two identical screensldquosimilar within a confidence intervalrdquo Solving (1) exactly isnot possible but it should be possible to estimate the solutionwithin a confidence interval Consider
Sel isin [ loKa Naive hiKa Naive] (6)
where loKa and hiKa are diagonal matrices of the upperand lower confidence intervals for the association constantsA simulation of the behavior of the Sa operator (Figures3 and S3) suggests that the relative sizes of the confidenceintervalsmight be impractically largewhen the copy numbersof sequences are lt10
Multiple sampling events of the Sa operator yield anormal distribution for each element of the vector (Figure 3)Fitting this normal distribution could yield a ldquotruerdquo valueof the process This process is identical to the extrapolationof the average from the normal distribution of noisy dataMultiple algorithms for such extrapolation exist for one- andmultidimensional stochastic processes [38 39] We believethat Sa behaves as a one-dimensional stochastic process andit might be possible to extrapolate the true value of thesampling from 7 to 10 repeated instances of Sa (ie thenumber of data sufficient to fit an 1D normal distribution)The necessary practical steps towards solving (3) or (10)are the following (i) Eliminate or account for any bias notrelated to binding (eg growth bias) (ii) Repeat the screen
6 Computational and Mathematical Methods in Medicine
Probability to observe specific solution
BB C
C
C
AD
DD
D B CCD
D[0 1 2 2]A B C D
[1 2 3 4]A B C D Sa
95
confi
denc
e int
erva
l
0 2 3 01 0 0 40 1 0 41 1 3 00 0 1 41 2 2 00 2 0 31 0 3 10 0 3 21 2 0 20 1 3 11 1 0 31 2 1 10 0 2 31 0 1 30 2 2 11 0 2 20 2 1 21 1 2 10 1 1 30 1 2 21 1 1 2
0 002 004 006 008 01 012 014
Copy number before
Cop
y nu
mbe
r afte
r
0
1
2
3
4
(a)
(c) (d)
(e)
(h)(g) (i)
(f)(b)
1 2 3 4
Probability to observe specific solution0 002 004 006 008 01 012 014
100000 trials
Sa50
1
6385
5000 trials2957
7
42
1 2 3 4 5 6 7 8012345678
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
One of the solutions
[1 2 3 4 5 6 7 8]A B C D E F G H
1 2 3 4 5 6 7 8
012345678
Cop
y nu
mbe
r afte
r the
sam
plin
g
Number of data per point
Copy number before the sampling
50
BB
CCCADD
DD H HH
G
HHH
HHGG G
GGG
EE
EE
E
E
FF
FF
FF
B
CAD DH
G
HHH
GG
E EE
FF
F
[1 1 1 2 3 3 3 4]A B C D E F G H
Sa 50
Most probable solution
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
[0 2 3 0]A B C D
[1 1 1 2]A B C D
[0 2 1 2]A B C D
the s
ampl
ing
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
the sampling
Copy number before the sampling
Copy number before the sampling
Copy number before the sampling
gt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
solutions
P = 014
P lt 0001
P = 007
after 5000 trials
Figure 3 (a) Testing the sampling operator implemented as random indexing function using a model multiset (b) In 100000 trials weobserved 22 unique solutions from which 14 resided in a 95 confidence interval Solutions with 0 and 1 copies of element A were foundat equal abundances (ldquoredundant solutionsrdquo) (c) Representation of the most probable solution as a line with 4 nodes ldquo119901rdquo is a probabilityto find the solution dotted line is an expected ldquoaverage solutionrdquo for 50 sampling (d) The 5th most probable solution (e) least probablesolution deviates the most form the average (f) combination of all solutions Red thick lines describe the most probable solutions thin bluelines describe the least probable solutions (g) Sampling of larger multisets yields more possible solutions (here 2957 in 5000 trials) (h)All solutions of the sampling represented as lines (i) Probability to observe a particular copy number after sampling While (h) is the mostaccurate representations of the confidence intervals the thin blue lines describe solutions outside the confidence interval this representation isimpractical due to large number of redundant solutions in largermultisets In (i) confidence interval could be extrapolated from distributionsof individual copy numbers (e) red dots are on or outside the confidence interval
several times (iii) Measure all copy numbers of all sequencesincluding zero values with high confidence Requirement (i)has been an ongoing effort in our group [11 40] and othergroups [13 41ndash43] for review see [11 44] Deep sequencing
makes it simple to satisfy requirement (ii) and obtainmultipleinstances of the same experiment For example we describedthe Illumina sequencing method that allows using barcodedprimers to sequence 18 unrelated experiments in one deep
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom
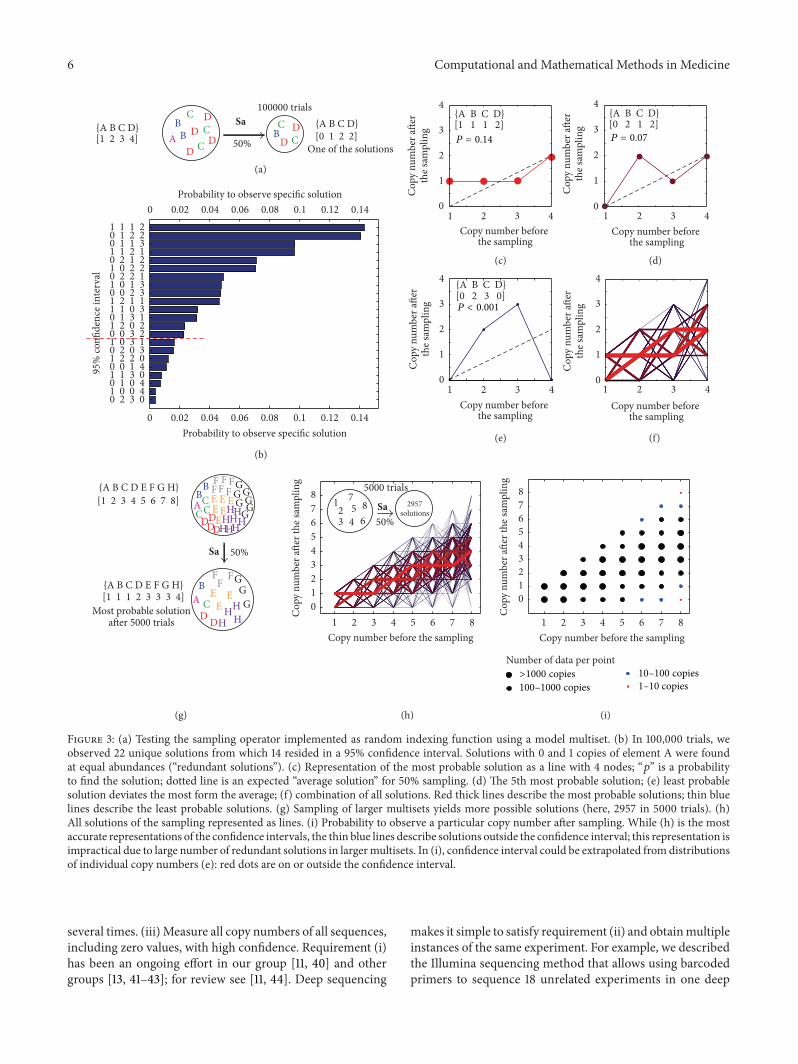
6 Computational and Mathematical Methods in Medicine
Probability to observe specific solution
BB C
C
C
AD
DD
D B CCD
D[0 1 2 2]A B C D
[1 2 3 4]A B C D Sa
95
confi
denc
e int
erva
l
0 2 3 01 0 0 40 1 0 41 1 3 00 0 1 41 2 2 00 2 0 31 0 3 10 0 3 21 2 0 20 1 3 11 1 0 31 2 1 10 0 2 31 0 1 30 2 2 11 0 2 20 2 1 21 1 2 10 1 1 30 1 2 21 1 1 2
0 002 004 006 008 01 012 014
Copy number before
Cop
y nu
mbe
r afte
r
0
1
2
3
4
(a)
(c) (d)
(e)
(h)(g) (i)
(f)(b)
1 2 3 4
Probability to observe specific solution0 002 004 006 008 01 012 014
100000 trials
Sa50
1
6385
5000 trials2957
7
42
1 2 3 4 5 6 7 8012345678
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
One of the solutions
[1 2 3 4 5 6 7 8]A B C D E F G H
1 2 3 4 5 6 7 8
012345678
Cop
y nu
mbe
r afte
r the
sam
plin
g
Number of data per point
Copy number before the sampling
50
BB
CCCADD
DD H HH
G
HHH
HHGG G
GGG
EE
EE
E
E
FF
FF
FF
B
CAD DH
G
HHH
GG
E EE
FF
F
[1 1 1 2 3 3 3 4]A B C D E F G H
Sa 50
Most probable solution
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
0
1
2
3
4
1 2 3 4
[0 2 3 0]A B C D
[1 1 1 2]A B C D
[0 2 1 2]A B C D
the s
ampl
ing
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
Cop
y nu
mbe
r afte
rth
e sam
plin
g
the sampling
Copy number before the sampling
Copy number before the sampling
Copy number before the sampling
gt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
solutions
P = 014
P lt 0001
P = 007
after 5000 trials
Figure 3 (a) Testing the sampling operator implemented as random indexing function using a model multiset (b) In 100000 trials weobserved 22 unique solutions from which 14 resided in a 95 confidence interval Solutions with 0 and 1 copies of element A were foundat equal abundances (ldquoredundant solutionsrdquo) (c) Representation of the most probable solution as a line with 4 nodes ldquo119901rdquo is a probabilityto find the solution dotted line is an expected ldquoaverage solutionrdquo for 50 sampling (d) The 5th most probable solution (e) least probablesolution deviates the most form the average (f) combination of all solutions Red thick lines describe the most probable solutions thin bluelines describe the least probable solutions (g) Sampling of larger multisets yields more possible solutions (here 2957 in 5000 trials) (h)All solutions of the sampling represented as lines (i) Probability to observe a particular copy number after sampling While (h) is the mostaccurate representations of the confidence intervals the thin blue lines describe solutions outside the confidence interval this representation isimpractical due to large number of redundant solutions in largermultisets In (i) confidence interval could be extrapolated from distributionsof individual copy numbers (e) red dots are on or outside the confidence interval
several times (iii) Measure all copy numbers of all sequencesincluding zero values with high confidence Requirement (i)has been an ongoing effort in our group [11 40] and othergroups [13 41ndash43] for review see [11 44] Deep sequencing
makes it simple to satisfy requirement (ii) and obtainmultipleinstances of the same experiment For example we describedthe Illumina sequencing method that allows using barcodedprimers to sequence 18 unrelated experiments in one deep
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom
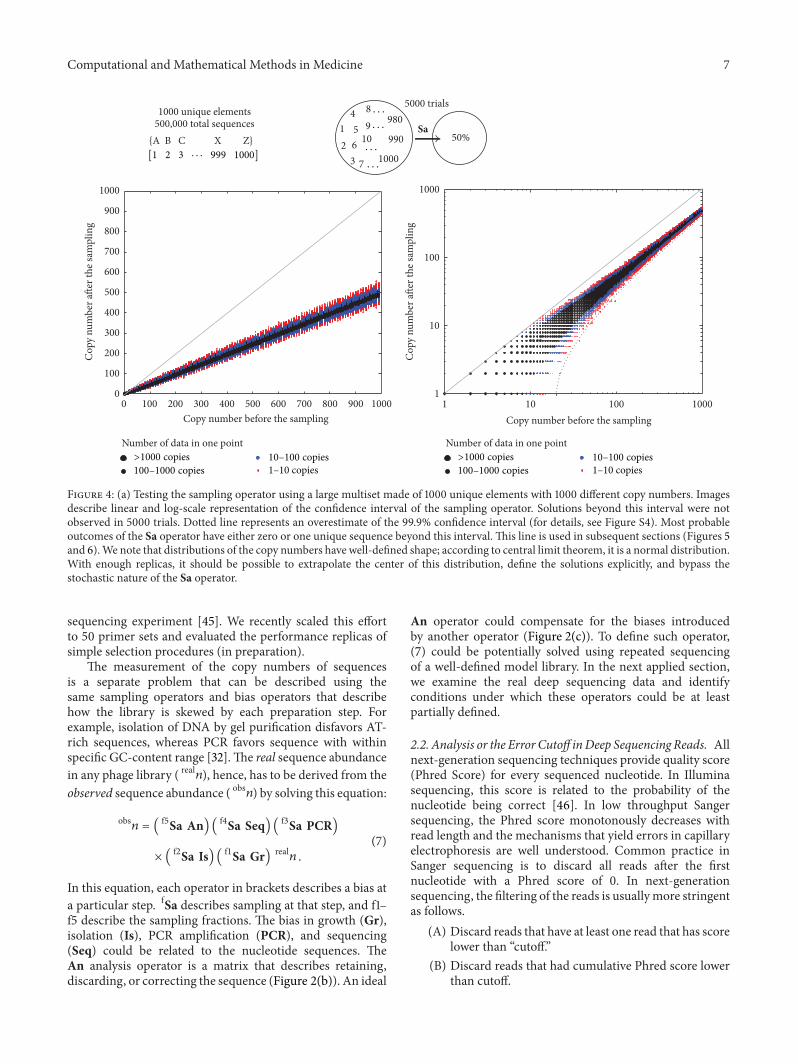
Computational and Mathematical Methods in Medicine 7
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
Number of data in one pointgt1000 copies100ndash1000 copies
10ndash100 copies1ndash10 copies
0 100 200 300 400 500 600 700 800 900 10000
100
200
300
400
500
600
700
800
900
1000
1 10 100 10001
10
100
1000
Copy number before the samplingC
opy
num
ber a
fter t
he sa
mpl
ing
A B C X Z
Copy number before the sampling
Cop
y nu
mbe
r afte
r the
sam
plin
g
1
1000
990
7
8
23
9 980
10Sa
456
50
5000 trials1000 unique elements
500000 total sequencesmiddot middot middot
middot middot middot
middot middot middot
middot middot middot[1 2 3 middot middot middot 999 1000]
Figure 4 (a) Testing the sampling operator using a large multiset made of 1000 unique elements with 1000 different copy numbers Imagesdescribe linear and log-scale representation of the confidence interval of the sampling operator Solutions beyond this interval were notobserved in 5000 trials Dotted line represents an overestimate of the 999 confidence interval (for details see Figure S4) Most probableoutcomes of the Sa operator have either zero or one unique sequence beyond this interval This line is used in subsequent sections (Figures 5and 6)We note that distributions of the copy numbers have well-defined shape according to central limit theorem it is a normal distributionWith enough replicas it should be possible to extrapolate the center of this distribution define the solutions explicitly and bypass thestochastic nature of the Sa operator
sequencing experiment [45] We recently scaled this effortto 50 primer sets and evaluated the performance replicas ofsimple selection procedures (in preparation)
The measurement of the copy numbers of sequencesis a separate problem that can be described using thesame sampling operators and bias operators that describehow the library is skewed by each preparation step Forexample isolation of DNA by gel purification disfavors AT-rich sequences whereas PCR favors sequence with withinspecific GC-content range [32]The real sequence abundancein any phage library ( real119899) hence has to be derived from theobserved sequence abundance ( obs119899) by solving this equation
obs119899 = (
f5Sa An) ( f4Sa Seq) ( f3Sa PCR)
times (f2Sa Is) ( f1Sa Gr) real119899
(7)
In this equation each operator in brackets describes a bias ata particular step fSa describes sampling at that step and f1ndashf5 describe the sampling fractions The bias in growth (Gr)isolation (Is) PCR amplification (PCR) and sequencing(Seq) could be related to the nucleotide sequences TheAn analysis operator is a matrix that describes retainingdiscarding or correcting the sequence (Figure 2(b)) An ideal
An operator could compensate for the biases introducedby another operator (Figure 2(c)) To define such operator(7) could be potentially solved using repeated sequencingof a well-defined model library In the next applied sectionwe examine the real deep sequencing data and identifyconditions under which these operators could be at leastpartially defined
22 Analysis or the Error Cutoff inDeep Sequencing Reads Allnext-generation sequencing techniques provide quality score(Phred Score) for every sequenced nucleotide In Illuminasequencing this score is related to the probability of thenucleotide being correct [46] In low throughput Sangersequencing the Phred score monotonously decreases withread length and the mechanisms that yield errors in capillaryelectrophoresis are well understood Common practice inSanger sequencing is to discard all reads after the firstnucleotide with a Phred score of 0 In next-generationsequencing the filtering of the reads is usuallymore stringentas follows
(A) Discard reads that have at least one read that has scorelower than ldquocutoffrdquo
(B) Discard reads that had cumulative Phred score lowerthan cutoff
8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

8 Computational and Mathematical Methods in Medicine
C7C
Tags NKKN Bar Adapter Sequence Quality string for sequenceEnd of
read
C7C
C7CC7CC7CC7C
Ph7
Ph7Ph7P12
Ph7
TGGG
GGGC
TGTG
ATGG
TTGG
CGGC
CGGG
CGGG
CGTC
TGTG
ATGC
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
TATTCTCACTCT
CGA CCCF
CCCF
CCCF
CCCF
CCCF
CCF
CCCF
F
CCCF
CCBF
CCCF
FFF
FFF
FFF
FFF
FDF
FFA
FFF
=A
FFF
FFF
FFF
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
DFFHBFFFGEFH
FHHHHHJJJJJJ
ADDDHFEFIEHG
FHGGHHJJJJJJ
FHHHHHJJJJJJ
FHHHHHJJJJJJ
TTG
GAC
GAC
GTA
TTG
CGA
TTG
TCA
TCA
CGA
JJJJJJJJJJJJJJJHHHGIJJJJJgtFHJJIJH simsimsimsimsimsimsim
JJJJJJJJJJHJJIJHIIJJJJBFHAHIHHHF simsimsimsimsimsimsim
JJJJJJJJJJJJGGHIJJJJJJJIJJIJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJIJJJJJJJJJJJJJIJJJ simsimsimsimsimsimsim
JJJJJJJJJJJIIJJJJJJIJIJJIEHHHFHFD simsimsimsimsimsimsim
JJJJJJJJJJJJJJJJJJJJJJJHIJJJJJJJJ simsimsimsimsimsimsim
JJJJJJJJJJJJIJJJJJJJJJJJJJJJJJJJH simsimsimsimsimsimsim
JJIJJIGIJIJEGIJGHFGIIJJFFGGIIGII simsimsimsimsimsimsim
JJJJJJJJJJJIJJJIGHGIHIIIGHGIJJJJH simsimsimsimsimsimsim
GAHGHFHGGDHEHBGGCEHIEGBFDHBDltA simsimsimsimsimsimsim
simsimsimsimsimsimsim
GCTTGTCCTCCGAAGGTGCATCTGCAGTGCGGT simsimsimsimsimsimsim
GCTTGTGCGGCGAATTCTTATCGGAATTGCGGT simsimsimsimsimsimsim
ACTCAGCCTCATCATACTCCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCGACGACTGTTCCAGCTTCGGGTGGAGGTTCG simsimsimsimsimsimsim
GCTTGTGATGCGCGTATGAATCAGCCTTGCGGT simsimsimsimsimsimsim
GCTTGTCAGGATAAGAATTCGCTTTTTTGCGGT simsimsimsimsimsimsim
GCTTGTAAGTCTCTTTCGAGTGCTCTGTGCGGT simsimsimsimsimsimsim
GCTTGTCCTCGTCTGTATCAGATGTCGTGCGGT simsimsimsimsimsimsim
GATGCGCATCGTACGTGTAATGGTGGAGGTTCG simsimsimsimsimsimsim
TGGCGTCATGATTCGTTTTAGACGGTTCAGCGT simsimsimsimsimsimsim
CGGGCTGCGTGTGCGAATCCTGGTGGAGGTTCG simsimsimsimsimsimsim
GEGIDFAEGIFCHC9FFAGHlowastDBAH
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
ltPERFgt
(a)
Sequencing resultsRemoval
All nucleotides
of errors
0L = [S 0n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph gt 30
Phred gt 30
(b)
Base call accuracy ()
Phred score cutoff Every nucleotide must have Phred score higher than
Mean 33-mer accuracy ()
1 5 8 13 20 30
210 680 840 950 990 999
00 00 01 18 717 967
950 975 987 992 995 996
(Base accuracy)33 ()
10cutoff10
33prodi=1
10Qi10
(c)
Cumulative base call accuracy or accuracy
100
10
1
01
001
0001
0000160 70 80 1009050403020100
100
10
01
1
001
0001
00001
of 33-mer sequence ()
Sequ
ence
s (
)
(d)
60
80
100
40
20
0
60
80
100
40
20
0gt90 gt95 gt98
Sequ
ence
s (
)
Sequ
ence
s (
)
Cumulative base call accuracy oraccuracy of 33-mer sequence ()
gt99
(e)
Figure 5 (a) Representative lines from the intermediate file from Illumina deep sequencing analysis (for more information seeERROR TAG data0001txt in Section 4 and our previous publication [6]) The reads have been parsed to identify adapters and barcodesEach read has been tagged according to the library type direction of the read and quality of the adapter regions We use this intermediatelibrary to identify reads that harbor erroneous nucleotides (b) Multiset view of the intermediate library The library contains subsets thathave low medium and high quality reads Error filtering of this intermediate library to eliminates any read with Phred score below 30 yieldsa high quality library of reads 30L (c) Mean accuracy of the reads in the library after error filtering ranges from 95 to 996 Even forvery low-quality cutoff Phred gt 1 the average read quality is 95 (d) Distribution of cumulative read accuracy in libraries processed usingdifferent cutoffs (e) Linear plot of the data presented in (d) with zoom in on the region with gt90 cumulative accuracy
(C) Use a combination of A and B (accept reads withminimal cutoff and minimal cumulative score)
Many of the error analyses in the area of deep sequencingare designed for genetic reads which have variable lengthsand unknown sequence throughout the whole read Analysisof the reads in a phage-display library is a simpler problembecause phage-derived constant adapter regions flank thevariable reads Identification of the adapter region is anecessary first step in the analysis Reads in which theadapters cannot be mapped cannot be used We designedalgorithms to recover reads even if adapters were hamperedby truncation deletion ormutation [6]We observed that the
reads flanked by the erroneous adapters had a significantlyhigher error rate than reads flanked by ldquoperfectrdquo adapters(unpublished) Example of mapping of flawed reads in Illu-mina sequencing is provided in ERROR TAG data0001txt(see Section 4) In the remaining sections we analyze thepopulation of the sequences preceded by a ldquoperfectrdquo adapterto identify possible sequence-specific biases
We analyzed a typical library sequenced by Illumina usingvarious cutoffs (Figure 5) We analyzed a 33-bp segment ofthe library that contained variable seven amino acids anda constant region and GGGS terminus A simple cutoffthat discards reads with Phred lt1 nucleotides yields library
Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

Computational and Mathematical Methods in Medicine 9
termed 1119899 which had an average 95 accuracy of the33-nucleotide read Reads that do not contain Phred = 0nucleotide rarely contain multiple low-quality reads The 1119899library was bimodal 80 of the reads had overall accuracyof 99 very few reads with accuracy 5ndash90 and significantnumber of reads with accuracy of 1 (Figures 5(d) and 5(e))These observations suggest that reads can be divided into (i)reads free of errors and (ii) reads with multiple errors
An example of a more stringent cutoff is eliminationof reads with Phred lt13 nucleotides this process yielded alibrary 13119899 in which every nucleotide had gt95 confidenceThe number of total reads in 13119899 was 10 less than numberof reads in 1119899 that is sum( 13119899) = 09sum( 1119899) The observedaverage read accuracy of the read in the 13119899 library was992 Theoretically the 095 confidence cutoff in a 33-mer nucleotide could yield reads with accuracy as low as(095)
33= 18 In practice the probability to find reads with
multiple nucleotides of 95 accuracy was vanishingly smallSpecifically among 500000 reads the lowest observed cumu-lative accuracy was 77 Such a result for example could beobtained in a sequence that has 27 ldquoperfectrdquo nucleotides and5 nucleotides with a Phred = 13 score (1)27(095)5 = 077Applying the most stringent cutoff to eliminate all reads witha Phred lt 30 yielded a library 30119899 in which every nucleotidehad 999 confidence The average confidence of the readsimproved subtly from 992 to 996 The number of totalreads in 30119899 was 30 less than number of reads in 13119899 thatis sum( 30119899) = 07sum( 13119899) It was not clear whether suchcutoff is an improvement or a detriment for analysis In thenext section we examined how frequency of the members ofthe library changed upon application of each error cutoff
23 Example of Error Analysis Sequence-Specific Censorshipduring Phred Quality Cutoff If errors occur by randomchance they should be uniformly distributed in all sequencesRemoval of erroneous read in that case should be identicalto sampling of the library by fSa operator where f is thesampling fraction For example consider the removal ofPhred lt 13 nucleotides from an unfiltered library (processdenoted as 1119899 rarr 13
119899) From the experiments we knowthat sum( 13119899) = 09sum( 1119899) if errors were distributedin sequences at random the 1119899 and 13119899 vectors should berelated as
13119899 =09Sa ( 1119899) (8)
The solutions should reside within a confidence interval
13119899 isin [
lo119862
hi119862] (9)
If errors occur preferentially in specific reads the frequencyof these reads should occur beyond the confidence intervalof the 09Sa This process could be described by a diagonalmatrix Bias as
13119899 =09Sa (Bias ( 1119899)) (10)
The elements of the diagonal matrix Bias = B119894119894 could be
estimated as follows
13119899119894isin [
lo119862119894
hi119862119894] B
119894119894= 1 (11)
13119899119894lt
lo119862119894 B
119894119894=
13119899119894
(091119899119894)
(12)
Figure 6(c) describes the representative solution of the09Sa( 1119899) (green dots) and the confidence interval (bluelines) Supplementary Scheme S3 describes the script thatcalculated this interval frommultiset 1119899 described as a plaintext file PhD7-Amp-0Ftxt using 10000 iterative calculationsof 09Sa( 1119899) This calculation required sim2 hours on a desktopcomputer Confidence interval was estimated as the mini-mum and maximum copy number found after 10000 iter-ations In this approximation of the confidence interval forsequences with the copy number lt10 before sampling it wasimpossible to determine whether the sequence disappeareddue to random sampling or due to bias The values of Biasoperator cannot be defined for these sequences and it couldbe assumed to be 1 (see (11)) For copy number gt10 howeversequence-specific bias can be readily detected We observedthat the removal of Phred lt13 reads yielded a multiset inwhich a large number of sequences deviated beyond theconfidence interval (Figure 6(d)) Their sequences could bereadily extracted by comparing the vector 13119899with the vectorof the lower confidence intervals lo
119862 (see (12)) The solutionof theBias can be illustrated graphically (Figure 6(e)) Top 30censored sequences are listed in Table S1 the other sequencescan be found in the supplementary information (file PhD7-Amp-0F-13F-CENtxt)
We performed similar calculations for 1119899 rarr 30119899 and13119899 rarr
30119899 processes The latter process is the most
interesting because 13119899 library has all nucleotides withinacceptable confidence range (gt95) and the distribution ofcumulative quality suggested that errors on average do notcluster in one read (Figure 5) The 13119899 rarr 30
119899 conversioneliminated 30 of the reads and copy numbers of manysequences deviated significantly from the random samplingthese sequences are represented by green dots outside the blueconfidence interval in Figure 6(h) Top 30 sequences are listedin Table S2 The censorship is not only sequence-specific butalso position-specific In sequences that had been censoredduring the 13119899 rarr 30119899 process lower quality reads clusteredaround 3-4 specific nucleotides (supplementary informationFigure S5)
The mechanism that leads to the disappearance of cen-sored sequences is not currently clear We attempted toidentify common motifs in censored sequences using twoapproaches (i) clustering and principal component analysesbased on Jukes-Cantor distance between sequences and(ii) identification of motifs using multiple unique sequenceidentifier software (MUSI) [17] These approaches couldnot detect any property common to censored reads whichwould make them significantly different from the othernoncensored reads Still we hypothesize that the observed
10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

10 Computational and Mathematical Methods in Medicine
Ph gt 30 Ph gt 30
Sequencing resultsRemovalof errors
30n = Bias Sa 0
n
All nucleotides
0L = [S
0n]
30L = [S 30n]
Phred gt 30
(a)
Sequencing results
Ph gt13
1T total sequences
13T total sequences
30T total sequences
30n = Bias Sa 13
n
13n = Bias Sa 1
n
30n = Bias Sa 1
n
13L = [S
13n]
0L = [S
0n]
1L = [S
1n]
30L = [S 30n]
Ph gt 30
Ph gt 13
Ph gt 0
Ph ge 1
Ph gt 30
(b)
38390
597122105
104
103
102
10
1
105
104
103
102
101
13n = Sa 1
n
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(c)
558480
77032
Inside the interval
Outsideintervalthe
of
105
104
103
102
10
1
105
104
103
102
101
13n = Bias 1
n
13n = Bias Sa 1
nC
opy
num
ber i
n Ph
redgt0
libra
ry
Copy number in Phred gt 30 library
(d)
1
01
001
Bias can be definedfor these sequences
Bias cannot be definedfor these sequences
105
104
103
102
101
Bias = 13n|I| |N||
1nminus1
Bias = |IN|
Ratio
of c
opy
num
ber i
nPh
redgt0
Phr
edgt13
Copy number in Phred gt 0 library
(e)
502655
132857
105
104
103
102
101
105
104
103
102
10
1
30n = Sa 1
n
Number of data points per pixelgt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
Copy number in Sa (Phred gt 0) library
(f)
438703
196809
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
Number of data points per pixel
30n = Bias Sa 1
n
30n = Bias 1
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt0
libra
ry
(g)
438703
119777
Inside the interval
Outside ofthe interval
105
104
103
102
10
1
105
104
103
102
101
30n = Bias 13
n
Number of data points per pixel
30n = Bias Sa 13
n
gt30000
gt10000
gt3000
gt1000 1ndash10
gt100
gt300
gt30
gt10
Copy number in Phred gt 30 library
Cop
y nu
mbe
r in
Phre
dgt13
libra
ry
(h)
Figure 6 (a) Operator and multiset description of the error filtering procedure Applying a Phred gt 30 cutoff to library filtered by Phredgt1cutoff ( 1n) yields a subpopulation of the library ( 30n) If errors are sequence-independent the 1n rarr 30n process should be identical torandom sampling ( 30n = Sa 1n) Any sequence-specific bias (Bias) should be detected as deviation from Sa 1n (b) Progressive samplingwith more stringent cutoff (c) Theoretical Sa 1n and theoretical 999 confidence interval (blue) (d) Observation of statistically significantdeviation from Sa operator dots beyond the blue line represent sequences prone to bias Red dots represent sequences that disappeared afterin 1n rarr 30n process or during Sa 1n sampling (e) Magnitude of the bias range from 5 to 100-fold (f) Bias in sampling of Phred gt 30 datafrom Phred gt 1 data ((f) is theory (g) is observed) (h) Bias upon sampling of Phred gt 30 data from Phred gt 13 Many sequences were lost inthis sampling and this loss was statistically significant beyond the 999 interval This result shows that some sequences have propensity toharbor low- and medium-quality reads Distribution of the errors is sequence specific
censorship represents sequence-specific errors which occurin every time such sequence passes though the Illuminaanalyzer For example the sequences listed in Tables S1 andS2 and supplementary files were censored in five independent
experiments which were pooled and processed simultane-ously in one Illumina run Analysis of other instances of Illu-mina sequencing performedby other groups could help prove(or disprove) that censorship is indeed sequence-specific
Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

Computational and Mathematical Methods in Medicine 11
and experiment-independent Sequence-specific censorshipduring Illumina analysis has been described in other publi-cations [46] The observations presented above suggest thatreading of some sequences in phage libraries does not yield anaccurate copy number Even if these sequences were enricheddue to binding their apparent copy number in sequencingwould be decreased due to sequencing bias If the magnitudeof bias is known however such error could be corrected Weanticipate that other biases could be calculated for these andother libraries in similar fashion Their calculation extendsbeyond the scope of this paper and it will be performed inour next publication
3 Discussion
31 Significance and Transformative Potential of Library-Wide Error Correction In the Medicinal Chemistry fieldstructure-activity relationships (SAR) and pharmacophoresare built using both positive and negative observations Itis the negative results that bear the most significance inthese studies because they allow mapping of the range ofconditions under which particular structure no longer worksFor example SAR of an R group of a ligand might be builton the following observations A ligand binds to the targetwhen the R group in the specific position is methyl or ethylchanging R to iso-propyl and tert-butyl ablates the bindingThis concludes that the R group must be a small alkyl groupAn analogous situation is found in SARof peptide ligands themost important information from alanine scan mutagenesisis loss of function because it helps identifying the importantresidues Interestingly loss-of-binding conclusions are neverapplied to phage-display The phage-display field is driven bypositive results Most publications report and follow up onlyon sequences enriched in the screen and consider only largecopy numbers interesting All papers focus on sequences thatwere found Very few papers in phage display ask why othersequences were not found
One of the reasons why phage display is not used forSAR-type analysis is because negative observations in phagelibrary cannot be determined with high confidence From apractical point of view measuring zero with high confidencerequires the largest number of observation (the highest depthof sequencing) The payoff however is immense one screenwith ldquoconfident zerosrdquo could potentially yield SAR for everypossible substitution of every possible amino acid We referto this (theoretical) possibility as ldquoInstant SARrdquo and itscondensed theoretical form is described in (3) or (9) and(10) This paper demonstrates that the depth of sequencing isnot the only problem towards this goal Accurate estimate ofnegative results requires complete characterization of the ori-gins of errors in sequencing which yield false negative valuesby censoring certain sequencing Other types of censorshipsuch as growth bias should be characterized and eliminatedas well As the phage display field is currently focused onpositive results the need for optimal error corrections andrecovery of erroneous reads is low With the rise of SAR-type applications in phage display error correction will berecognized as the most significant barrier because it could
lead to improper assignment of low frequencies and negativeresults Improved error correction strategies could assigna lower confidence to the sequence instead of eliminatingthe errors and labeling them as confident zero Propermathematical framework possibly similar to the one used inthis paper could be then used to carry all confidence intervalsthrough calculations to yield reliable SAR-type data
We note that the framework described in this paper issuitable for the analysis of the selection from libraries inwhich the diversity of the libraries before and after selectioncould be covered entirely by deep sequencing With thecurrent depth of sequencing it corresponds to medium-scale libraries of sim106 randommembers and affinity-maturedlibraries that containsim106 pointmutationsWe are in the pro-cess of generating these medium-scale libraries and runningselection procedures that will allow us to apply and refine ourframework In the future as technical capabilities and depthof sequencing increase the process would be applicable tolarger libraries as well
4 Methods
41 Generation of Z-Bars and Other Visualization TechniquesSequencing of the libraries has been described in our previouspublications [6 47] All data visualization in this paper wasdone by MATLAB scripts raw lowasteps output from MATLABscripts subject to minor postprocessing in Adobe Illustratorto adjust fonts relative dimensions of plots Core scripts aredescribed in the supplementary information Other scriptsare available in our previous publication [47] Illumina filesused for the analysis can be found in the directory athttpwwwchemualbertacasimderdamathbiology the fileERROR TAG data0001txt is an example of error-taggedreads PhD7-Amp-xxFtxt is the library filtered with xx Phredcutoff (xx = 1 13 and 30) file PhD7-Amp-13F-30F-COtxtdescribes confidence intervals for Phred(13) to Phred(30)filtering process other files with lowast-COtxt extension describeconfidence intervals of other processes Supplementary Fig-ures S1ndashS4 and Schemes S1 and S2 describe MATLAB imple-mentation of the Sa operator
Acknowledgments
This work was supported by funds from the University ofAlberta and Alberta Glycomics Centre
References
[1] J K Scott and G P Smith ldquoSearching for peptide ligands withan epitope libraryrdquo Science vol 249 no 4967 pp 386ndash390 1990
[2] G P Smith and V A Petrenko ldquoPhage displayrdquo ChemicalReviews vol 97 no 2 pp 391ndash410 1997
[3] A D Ellington and J W Szostak ldquoIn vitro selection of RNAmolecules that bind specific ligandsrdquo Nature vol 346 no 6287pp 818ndash822 1990
[4] C Tuerk and L Gold ldquoSystemic evolution of ligands byexponential enrichment RNA ligands to bacteriophage T4DNApolymeraserdquo Science vol 249 no 4968 pp 505ndash510 1990
12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

12 Computational and Mathematical Methods in Medicine
[5] E Dias-Neto D N Nunes R J Giordano et al ldquoNext-generation phage display integrating and comparing availablemolecular tools to enable costeffective high-throughput analy-sisrdquo PLoS ONE vol 4 no 12 Article ID e8338 2009
[6] W LMatochko K Chu B Jin SW Lee GMWhitesides andR Derda ldquoDeep sequencing analysis of phage libraries usingIllumina platformrdquoMethods vol 58 pp 47ndash55 2012
[7] A Ernst D Gfeller Z Kan et al ldquoCoevolution of PDZ domain-ligand interactions analyzed by high-throughput phage displayand deep sequencingrdquo Molecular BioSystems vol 6 no 10 pp1782ndash1790 2010
[8] G V Kupakuwana J E Crill M P McPike and P N BorerldquoAcyclic identification of aptamers for human alpha-thrombinusing over-represented libraries and deep sequencingrdquo PLoSONE vol 6 no 5 Article ID e19395 2011
[9] P A C rsquoT Hoen S M G Jirka B R Ten Broeke et alldquoPhage display screening without repetitious selection roundsrdquoAnalytical Biochemistry vol 421 no 2 pp 622ndash631 2012
[10] H Zhang A Torkamani T M Jones D I Ruiz J Ponsand R A Lerner ldquoPhenotype-information-phenotype cyclefor deconvolution of combinatorial antibody libraries selectedagainst complex systemsrdquo Proceedings of the National Academyof Sciences of the United States of America vol 108 no 33 pp13456ndash13461 2011
[11] R Derda S K Y Tang S C Li S Ng W Matochko and M RJafari ldquoDiversity of phage-displayed libraries of peptides duringpanning and amplificationrdquo Molecules vol 16 no 2 pp 1776ndash1803 2011
[12] R W Hamming ldquoError detecting and error correcting codesrdquoBell System Technical Journal vol 29 pp 147ndash160 1950
[13] D J Rodi A S Soares and L Makowski ldquoQuantitativeassessment of peptide sequence diversity in M13 combinatorialpeptide phage display librariesrdquo Journal of Molecular Biologyvol 322 no 5 pp 1039ndash1052 2002
[14] L Makowski ldquoQuantitative analysis of peptide librariesrdquo inPhage Nanobiotechnology chapter 3 2011
[15] C E Shannon ldquoAmathematical theory of communicationrdquo BellSystem Technical Journal vol 27 pp 379ndash423 1948
[16] U Ravn F Gueneau L Baerlocher et al ldquoBy-passing in vitroscreening next generation sequencing technologies applied toantibody display and in silico candidate selectionrdquoNucleic AcidsResearch vol 38 no 21 2010
[17] T Kim M S Tyndel H Huang et al ldquoMUSI an integratedsystem for identifying multiple specificity from very largepeptide or nucleic acid data setsrdquo Nucleic Acids Research vol40 no 6 article e47 2012
[18] J A Weinstein N Jiang R A White III D S Fisher and S RQuake ldquoHigh-throughput sequencing of the zebrafish antibodyrepertoirerdquo Science vol 324 no 5928 pp 807ndash810 2009
[19] B J DeKosky G C Ippolito R P Deschner et al et al ldquoHigh-throughput sequencing of the paired human immunoglobulinheavy and light chain repertoirerdquo Nature Biotechnology vol 31pp 166ndash169 2013
[20] B Levitan ldquoStochastic modeling and optimization of phagedisplayrdquo Journal of Molecular Biology vol 277 no 4 pp 893ndash916 1998
[21] C S Riesenfeld P D Schloss and J Handelsman ldquoMetage-nomics genomic analysis of microbial communitiesrdquo AnnualReview of Genetics vol 38 pp 525ndash552 2004
[22] M L Sogin H G Morrison J A Huber et al ldquoMicrobialdiversity in the deep sea and the underexplored lsquorare biospherersquordquo
Proceedings of the National Academy of Sciences of the UnitedStates of America vol 103 no 32 pp 12115ndash12120 2006
[23] F Backhed R E Ley J L Sonnenburg D A Peterson and JI Gordon ldquoHost-bacterial mutualism in the human intestinerdquoScience vol 307 no 5717 pp 1915ndash1920 2005
[24] N Beerenwinkel and O Zagordi ldquoUltra-deep sequencing forthe analysis of viral populationsrdquo Current Opinion in Virologyvol 1 no 5 pp 413ndash418 2011
[25] J A Huber D B MarkWelch H G Morrison et al ldquoMicrobialpopulation structures in the deep marine biosphererdquo Sciencevol 318 no 5847 pp 97ndash100 2007
[26] A Wilm P P K Aw D Bertrand et al ldquoLoFreq a sequence-quality aware ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencingdatasetsrdquo Nucleic Acids Research vol 40 pp 11189ndash11201 2012
[27] P J Turnbaugh C Quince J J Faith et al ldquoOrganismalgenetic and transcriptional variation in the deeply sequencedgut microbiomes of identical twinsrdquo Proceedings of the NationalAcademy of Sciences of the United States of America vol 107 no16 pp 7503ndash7508 2010
[28] CQuince A Lanzen T P Curtis et al ldquoAccurate determinationof microbial diversity from 454 pyrosequencing datardquo NatureMethods vol 6 no 9 pp 639ndash641 2009
[29] S J Watson M R A Welkers D P Depledge et al ldquoViralpopulation analysis and minority-variant detection using shortread next-generation sequencingrdquo Philosophical Transactions ofthe Royal Society B vol 368 no 1614 Article ID 20120205 2013
[30] D R Bentley S Balasubramanian H P Swerdlow et al et alldquoAccurate whole human genome sequencing using reversibleterminator chemistryrdquo Nature vol 456 pp 53ndash59 2008
[31] KNakamura TOshima TMorimoto et al ldquoSequence-specificerror profile of Illumina sequencersrdquo Nucleic Acids Researchvol 39 no 13 2011
[32] M A Quail I Kozarewa F Smith et al ldquoA large genomecenterrsquos improvements to the Illumina sequencing systemrdquoNature Methods vol 5 no 12 pp 1005ndash1010 2008
[33] O Zagordi R Klein M Daumer and N Beerenwinkel ldquoErrorcorrection of next-generation sequencing data and reliableestimation of HIV quasispeciesrdquoNucleic Acids Research vol 38no 21 pp 7400ndash7409 2010
[34] M W Schmitt S R Kennedy J J Salk E J Fox J B Hiattand L A Loeb ldquoDetection of ultra-rare mutations by next-generation sequencingrdquo Proceedings of the National Academyof Sciences of the United States of America vol 109 pp 14508ndash14513 2012
[35] A Syropoulos ldquoMathematics of multisetsrdquo in Proceedings of theWorkshop on Multiset Processing Multiset Processing Mathe-matical Computer Science and Molecular Computing Points ofView pp 347ndash358 Springer 2001
[36] W ArapM G KoloninM Trepel et al ldquoSteps towardmappingthe human vasculature by phage displayrdquo Nature Medicine vol8 no 2 pp 121ndash127 2002
[37] S Blond-Elguindi S E Cwirla W J Dower et al ldquoAffinitypanning of a library of peptides displayed on bacteriophagesreveals the binding specificity of BiPrdquoCell vol 75 no 4 pp 717ndash728 1993
[38] S Cox E Rosten J Monypenny et al ldquoBayesian localizationmicroscopy reveals nanoscale podosome dynamicsrdquo NatureMethods vol 9 no 2 pp 195ndash200 2012
[39] E Rosten G E Jones and S Cox ldquoImage plug-in for Bayesiananalysis of blinking and bleachingrdquoNature Methods vol 10 pp97ndash98 2013
Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

Computational and Mathematical Methods in Medicine 13
[40] W L Matochko S Ng M R Jafari J Romaniuk S K Y Tangand R Derda ldquoUniform amplification of phage display librariesin monodisperse emulsionsrdquoMethods vol 58 pp 18ndash27 2012
[41] E A Peters P J Schatz S S Johnson and W J DowerldquoMembrane insertion defects caused by positive charges in theearly mature region of protein pIII of filamentous phage fd canbe corrected by prlA suppressorsrdquo Journal of Bacteriology vol176 no 14 pp 4296ndash4305 1994
[42] L A Brammer B Bolduc J L Kass K M Felice C J Norenand M F Hall ldquoA target-unrelated peptide in an M13 phagedisplay library traced to an advantageous mutation in the geneII ribosome-binding siterdquo Analytical Biochemistry vol 373 no1 pp 88ndash98 2008
[43] G A Kuzmicheva P K Jayanna I B Sorokulova and VA Petrenko ldquoDiversity and censoring of landscape phagelibrariesrdquo Protein Engineering Design and Selection vol 22 no1 pp 9ndash18 2009
[44] D R Wilson and B B Finlay ldquoPhage display applicationsinnovations and issues in phage and host biologyrdquo CanadianJournal of Microbiology vol 44 no 4 pp 313ndash329 1998
[45] R Derda S K Y Tang and G M Whitesides ldquoUniformamplification of phage with different growth characteristics inindividual compartments consisting ofmonodisperse dropletsrdquoAngewandte Chemie vol 49 no 31 pp 5301ndash5304 2010
[46] J C Dohm C Lottaz T Borodina and H HimmelbauerldquoSubstantial biases in ultra-short read data sets from high-throughput DNA sequencingrdquo Nucleic Acids Research vol 36no 16 article e105 2008
[47] W L Matochko S C Li S K Y Tang and R DerdaldquoProspective identification of parasitic sequences in phagedisplay screensrdquo Nucleic Acids Research 2013
Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom

Submit your manuscripts athttpwwwhindawicom
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Disease Markers
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Immunology ResearchHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Parkinsonrsquos Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttpwwwhindawicom