reaction path s0 ff oott qquuíímmiiccaa ii - uah.es · 5.2.1 región de intersecciones cónicas...
TRANSCRIPT

H H
MINIMUM
TS
S1/S0 CI
IRD
BP
S0 RELAXATION CHANNEL
PHOTOCHEMICAL REACTION PATH
THERMAL RECTION PATH
6
CIb 12.0
TS49.
st 8
CIst 17.9
H
H
H
DIR -27.2
RI -20.9
TS1VI
TS2VI
4.9
TSDBS -20.1
-5.4
TS
3
11.1
TSSBV
29.4
TSDIR'
12.7
S0
S1S0
5
S1
HH
H
Scheme 11
+
hν
7
2
2'
1 1'
1'DBS1DBS
1
5
1
5
21
21
21
2
1
-36.4-33.3
-26.9
-12.6
TS
0.0
RI-20.9
D8h
C2v
C v
CsD2d
C2
C2
Cs
D4h
Cs
CAPÍTULO
5
CsC2EEll eessppaacciioo ddee iinntteerrsseecccciióónn FFoottooqquuíímmiiccaa IIII
2
5.1 Introducción 159 5.2 Topología del Espacio de Intersección 160 5.2.1 Región de Intersecciones Cónicas 160 5.2.2 Región de Cruce de Sistemas 161 5.3 Espectro de absorción del 1,3,5,7-Ciclooctatetraeno 162 5.3.1 Métodos Teóricos y Detalles Computacionales 163 5.3.2 Espectro de Absorción Singlete-Singlete 166 5.3.3 Espectro de Absorción Singlete-Triplete 170 5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 173 5.4.1 Transición Franck-Condon S0→S2 y Población del Estado S1 en el COT 173 5.4.2 Región de Intersecciones Cónicas en el COT 177 5.4.3 Relajación en S0. Formación de Fotoproductos 195 5.5 El Espacio de Intersección (S1/S0) y (T1/S0) en el barreleno 223 5.5.1 Cicloadición [2+2] versus reagrupamiento di-π-metano singlete en el barreleno 224
5.5.2 Reacción triplete di-π-metano en el barreleno. Eficiencia de los canales de cruce de sistemas (T1/S0) 228
5.6 Conclusiones 232
-157-

5.1 Introducción 159
55..11 IInnttrroodduucccciióónn
Al igual que el análisis de la SEP del estado fundamental de un compuesto nos permite
conocer los procesos químicos activados térmicamente que puede sufrir, el análisis de la
SEP de los estados excitados nos permite conocer de la misma manera los procesos
fotoquímicos. Para las reacciones fotoquímicas que transcurren a través de una
transición diabática es interesante estudiar las propiedades de la región de cruce entre
los estados (excitado y fundamental) que participan en la reacción fotoquímica. El
análisis de este espacio de intersección nos permite establecer la relación que existe
entre distintas regiones de cruce (regiones de intersecciones cónicas) y las distintas
reacciones fotoquímicas que puede sufrir el sistema.
En primer lugar (Sección 5.2) se discutirán brevemente algunas de las propiedades
principales de los espacios de intersección tanto entre SEP de la misma multiplicidad
como de distinta.
Posteriormente se estudiará el proceso de excitación electrónica, tanto singlete como
triplete en el COT (Sección 5.3), fundamental en el entendimiento de su fotoquímica, ya
que nos permitirá analizar que tipo de transiciones diabáticas son posibles en este
compuesto.
La evolución tras la excitación del COT al estado S2 para poblar el estado S1 se analiza
en la Sección 5.4, en la cual se discute la accesibilidad de distintas regiones de
intersecciones cónicas en el decaimiento del COT al estado fundamental. Asimismo se
estudia gran parte del espacio de intersección S1/S0 a través de la localización de
diversas CoIn mínimas en energía, y se establecen relaciones entre las distintas familias
de intersecciones cónicas y las diversas reacciones fotoquímicas en las que pueden
existir cruces diabáticos.
Por último (Sección 5.5), se estudiará una de las reacciones (el reagrupamiento di-π
metano) más importantes que sufre un isómero de COT (el barreleno), la cual transcurre
tanto a través del estado triplete como del singlete, siendo fundamental el análisis del
espacio de cruce S1/S0 y T1/S0 para el correcto entendimiento del mecanismo de
reacción y su posible modulación.

160 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
55..22 TTooppoollooggííaa ddeell EEssppaacciioo ddee IInntteerrsseecccciióónn
En este punto se describirán muy brevemente algunas de las propiedades fundamentales
del espacio de intersección entre estados, tanto de la misma multiplicidad
(Intersecciones Cónicas) como de distinta multiplicidad, limitándonos en estos últimos
al cruce entre estados singlete y triplete (Cruce de Sistemas).
5.2.1 Región de Intersecciones Cónicas
Las Intersecciones Cónicas (CoIn) presentan un espacio de intersección de dimensión
3N-8 (ver punto 2.3.3). Esto es debido a que el acoplamiento entre estados electrónicos
de la misma multiplicidad es en general suficientemente grande como para que en la
coordenada correspondiente a dicho acoplamiento desaparezca la degeneración
energética. Por lo tanto esta coordenada junto con la correspondiente a la diferencia de
gradientes conforman el subespacio bidimensional en el que desaparece la degeneración
energética.
En muchas reacciones fotoquímicas diabáticas el cruce entre estados se realiza a través
de CoIn mínimas en energía, esto es, mínimos en el subespacio de intersección de
dimensión 3N-8. Sin embargo, en algunas de ellas están involucradas intersecciones
cónicas que no son mínimas, lo cual puede ocurrir en reacciones ultrarrápidas o en
reacciones en las que es necesario superar un TS en el estado excitado (un ejemplo es la
compleja fotoquímica del TOD en el estado singlete estudiada en el Capítulo cuarto).
Por lo tanto, puesto que en general son las CoIn mínimas en energía y en alguna ocasión
zonas cercanas a la misma las que determinan la fotoquímica de un sistema, es útil
estudiar este espacio de intersección, e intentar establecer relaciones (en base a
propiedades estructurales electrónicas y energéticas) entre las distintas CoIn.
Las CoIn mínimas en energía en hidrocarburos insaturados (como el COT y sus
isómeros de valencia) tienen un carácter tetrarradicálico, y en general el espacio de
intersección también posee este carácter. Desplazamientos en la localización de los
radicales en la CoIn pueden permitir establecer trayectorias continuas en el espacio de
configuraciones nucleares que “unan” CoIn mínimas en energía en el subespacio de

5.2 Topología del Espacio de Intersección 161
intersección. Como se verá es posible la existencia de estados de transición en este
subespacio que “unen” las distintas CoIn mínimas en energía.
Los métodos computacionales implementados en los paquetes de cálculo para
identificar las propiedades del espacio de intersección son escasos, por lo que el análisis
de estas propiedades es especialmente complejo, y el estudio que se ha llevado a cabo
tiene únicamente carácter cualitativo.
5.2.2 Región de Cruce de Sistemas
Como ya se indicó, por cruce de sistemas nos referimos generalmente a puntos de la
SEP en la que los estados Sn y Tm presentan degeneración energética, donde n y m
suelen ser números menores de dos (ver punto 2.3.2). También se explicó que debido al
débil acoplamiento entre los estados S y T (acoplamiento espín-órbita) la región de
cruce entre estos estados es de dimensión uno, por lo que en 3N-7 coordenadas la
degeneración energética de las SEP permanece y en una coordenada (la correspondiente
al vector diferencia de gradientes) la degeneración entre estados desaparece. Por lo
tanto, el análisis de esta región es mucho más simple que la que ocurre entre estados de
la misma multiplicidad.
Al contrario que las intersecciones cónicas, los cruces de sistemas no tienen por qué ser
canales efectivos para el salto electrónico, lo que se debe principalmente al ya
mencionado débil acoplamiento que suele existir entre estados tripletes y singletes. Por
lo tanto, el análisis de la región de cruce de sistemas debe ir acompañado de un análisis
en la magnitud del acoplamiento que permite la transición.
En el ejemplo que se ha estudiado de una región de cruce de sistemas (Sección 5.5),
existen distintas zonas de degeneración energética entre los estados T1 y S0 posibles
para el decaimiento al estado fundamental. Un análisis conjunto de las regiones de cruce
junto con el cálculo del acoplamiento espín-órbita nos permite establecer los caminos de
reacción favorecidos e impedidos, así como proponer posibles maneras de controlar
estos caminos de reacción.

162 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
55..33 EEssppeeccttrroo ddee aabbssoorrcciióónn ddeell 11,,33,,55,,77--CCiiccllooooccttaatteettrraaeennoo
La elucidación de los espectros de absorción (transiciones singlete y triplete) del
1,3,5,7-Cicloctatetraeno (COT) es fundamental para el correcto entendimiento de su
comportamiento fotoquímico y como quencher en los procesos de transferencia de
energía triplet-triplete como se verá más adelante.
A pesar de las interesantes propiedades del COT tanto en su comportamiento
fotoquímico como fotofísico, las cuales han sido ampliamente estudiadas, son pocos los
estudios experimentales (Langseth y Brodersen, 1949; Frueholz y Kuppermann, 1978;
Perkampus, 1992) y teóricos (Allinger et al., 1965; Van-Catledge, 1971; Palmer, 1988;
Hassenruck et al., 1989) que se han realizado para la determinación de las propiedades
de su espectro electrónico.
Estudios experimentales de difracción electrónica han determinado una estructura para
el estado fundamental del COT en forma de “bote” con una simetría D2d, presentando
alternancia de dobles enlaces completamente localizados (Traetteberg, 1966),
llegándose a la misma conclusión desde argumentos teóricos (Hassenruck et al., 1989).
El COT es uno de los hidrocarburos cíclicos insaturados miembro de la familia de
compuestos antiaromáticos, lo cual se ve reflejado en su estructura, muy lejana de la
planaridad, y en la localización de los dobles enlaces. Debido a esta especial estructura,
en el estudio teórico de sus propiedades electrónicas ha de tenerse en cuenta la
interacción entre dobles enlaces a través del espacio y a través de enlace.
El espectro de absorción singlete → singlete del COT en fase gaseosa presenta una
banda amplia de baja intensidad en la región de 4.00–4.77 eV, con un máximo
aproximadamente a 4.39 eV. Asimismo, presenta una banda intensa con un máximo a
6.42 eV y un hombro a 6.05 eV (Allinger et al., 1965; Hassenruck et al., 1989), energía
que supone el límite experimental del estudio.
Por otro lado, Frueholz y Kuppermann (Frueholz y Kuppermann, 1978) identificaron
tres bandas del espectro singlete → triplete con unos máximos a 3.05, 4.05 y 4.84 eV,
así como varias transiciones singlete → singlete hasta los 14 eV.
Por el contrario, la identificación de los estados Rydberg más bajos en energía no ha
sido posible experimentalmente, estando la asignación de las bandas experimentales
muy condicionada por los resultados semiempíricos ya existentes en los cuales sólo los
estados de valencia podían ser calculados (Frueholz y Kuppermann, 1978).

5.3 Espectro de Absorción del 1,3,5,7-Ciclooctatetraeno 163
En esta sección (5.3) se presenta un estudio de los espectros singlete → singlete y
singlete → triplete usando la teoría multiconfiguracional con corrección perturbacional
hasta segundo orden a través del método CASPT2 (Anderson et al., 1990; Anderson et
al., 1992) según ha sido implementado en el paquete de programas MOLCAS (Molcas
v.5, 1999), método que se ha demostrado muy útil en el estudio de las propiedades
espectroscópicas de muchos compuestos (Roos et al., 1995, 1996; Merchán et al.,
1998,1999).
5.3.1 Métodos Teóricos y Detalles Computacionales
Los cálculos fueron realizados con simetría C2v, imponiendo las restricciones
correspondientes a la simetría D2d tanto en los parámetros geométricos, como en la
función de onda CASSCF. La orientación así como la estructura del COT en el estado
fundamental se muestran en la figura 5.1.
Z
X
Y
Fig. 5.1. Orientación de la molécula COT (D2d) usada en el cálculo de sus propiedades espectroscópicas.
A partir de ahora se usará la notación (n(a1);n(e);n(b2);n(b1);n(a2)) para indicar el
número (n) de orbitales de la correspondiente simetría (a1,e,b2,b1 y a2) ocupados, en la
representación D2d. Así, la función de onda de capa cerrada obtenida aplicando la teoría
de Hartree-Fock restringida viene dada por la configuración (5;7;4;3;2), por lo tanto con

164 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
un total de 28 orbitales moleculares ocupados. Puesto que se ha usado simetría C2v,
dicha configuración equivale a (9;7;7;5) correspondiente a las simetrías a1,b1,b2 y a2 del
grupo C2v, teniendo presente la relación entre ambos grupos de simetría: D2d/C2v
a1+b2/a1, e/b1+b2, b1+a2/a2. (Fig. 5.2).
Fig. 5.2. Representación de los cinco OM canónicos ocupados más altos en energía, así como los 4 desocupados de simetría π, calculados para la geometría de equilibrio del COT con orbitales naturales atómicos (ver texto para detalles). Las energías de los mismos entre paréntesis también se incluyen.
Para la optimización geométrica del estado fundamental se usó como espacio activo el
formado por los orbitales π, lo cual comprende 8 electrones π distribuidos en los 8

5.3 Espectro de Absorción del 1,3,5,7-Ciclooctatetraeno 165
orbitales de valencia π: (2;2;2;2) para la representación C2v, siendo los restantes
orbitales inactivos: (7;6;6;5). Para realizar la optimización geométrica en el estado
fundamental se usaron funciones de base de orbitales atómicos naturales (ANO)§
obtenidas del conjunto de funciones primitivas C(14s9p4d)/H(8s4p) con el esquema de
contracción C[4s3p1d]/H[2s1p] (Widmark et al., 1990). Para el cálculo de las
propiedades espectroscópicas del COT a estas funciones de base se le añadieron dos
funciones difusas del tipo s, dos p y dos d cuyo origen se sitúa en el centro de la
molécula para poder estudiar los estados Rydberg.
Todas las funciones de onda CASSCF fueron usadas como funciones de referencia en el
tratamiento perturbacional de segundo orden a través del método CASPT2.
Por otro lado, el efecto de la presencia de estados intrusos débilmente actuantes, fue
minimizada a través del uso de la técnica denominada “imaginary level shift” (Forsberg
y Malmqvist,1997). Después del análisis de la influencia en las raíces (energías)
obtenidas al incrementar el denominador del cociente correspondiente a la corrección de
segundo orden de la energía en una cantidad, se determinó como óptimo el valor de 0.2
a.u.
Puesto que los cálculos se llevaron a cabo usando simetría C2v, la determinación de los
distintos estados estudiados se dividió en tres grupos: los estados de simetría A1, B1 y
A2. No se incluyen los estados B2 debido a que la suma B1+B2 conforma (como ya se ha
dicho antes) el estado E de simetría D2d, luego los estados determinados tanto dentro de
una simetría como de la otra son degenerados y poseen las mismas propiedades.
Los espacios activos usados han variado dependiendo de la simetría (una de las tres
simetrías C2v antes mencionada) de los estados calculados. Para cada una de las
simetrías estudiadas se han realizado cálculos “Multi-State” CASPT2 (Finley et al.,
1998) tomando como referencia también funciones “Multi-State” CASSCF en las que
todos los estados de la misma simetría poseen el mismo peso.
En concreto, para los estados de simetría 1A1 (C2v), se usó una función de onda de
referencia CASSCF (6;2;2;3) (es decir, con 13 orbitales) y un total de 10 electrones
activos, determinando 14 estados de esta simetría. La inclusión de un orbital ocupado
adicional en el espacio activo sirve para tener en cuenta el estado 11B2(3b1→3a2) que se
§ Acrónimo del inglés Atomic Natural Orbital (ANO)

166 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
posee una energía de 10.09eV a nivel CASSCF pero de tan solo 6.14eV tras la inclusión
de la correlación dinámica a través del método CASPT2.
Para los estados de simetría 1B1 (C2v) se usó una función de onda de referencia CASSCF
(3;4;2;3) con ocho electrones activos. De esta manera se determinaron nueve estados de
esta simetría.
Por último, dentro de la simetría 1A2 (C2v) se utilizó para los cálculos CASPT2 una
función de referencia CASSCF (2;2;2;3) para la determinación de cuatro estados de esta
simetría.
Todas las energías calculadas para los distintos estados (tanto singlete como triplete) se
refieren a la energía para la geometría optimizada usando el mismo espacio activo que
en el correspondiente cálculo del estado fundamental.
5.3.2 Espectro de Absorción Singlete-Singlete
Para el cálculo de todas las transiciones electrónicas verticales, se usó como geometría
de equilibrio del COT en el estado fundamental (S0) aquella optimizada a nivel
CASSCF con un espacio activo (2;2;2;2) y con las funciones de base antes descritas. La
geometría así obtenida presenta una gran similitud con la determinada mediante
difracción electrónica (Traetteberg, 1966) (Fig. 5.3).
Los resultados obtenidos en el estudio del espectro de absorción singlete → singlete y
que a continuación se pasarán a describir con detalles, están recogidos en la tabla 5.1.
El primer estado excitado del COT posee simetría A2. La función de onda de este estado
está básicamente dominada por una sola configuración correspondiente a la transición
HOMO → LUMO, que en términos de orbitales moleculares canónicos corresponde a la
transición 5a1 → 3a2. Esta transición es prohibida por dipolo eléctrico, lo cual ya fue
establecido con anterioridad (Frueholz y Kuppermann, 1978). Los valores
experimentales del máximo de esta primera banda están entorno a los 4.4 eV (Allinger
et al., 1965; Frueholz y Kuppermann, 1978; Hassenruck et al., 1989), lo cual está un
poco lejano a los 3.8 eV calculados (CASPT2).

5.3 Espectro de Absorción del 1,3,5,7-Ciclooctatetraeno 167
( )
( ) 1.3411.340
r C C= = A
( )
( ) 1.4771.476
r C C− = A
( )
( ) 127.1º126.1º
C C C∠ = − =
( )
( ) 117.6º117.6º
C C H∠ = − =
( ) 55.1ºC C C C∠ = − = =
( ) 1.078r C H− = Å
Fig. 5.3. Parámetros estructurales del COT calculados a nivel CASSCF (8 electrones π y 8 orbitales π) con funciones de base de “Orbitales Atómicos Naturales” (ver texto). Entre paréntesis se dan los valores experimetales obtenidos mediante difracción electrónica (Traetteberg, 1966).
Una explicación posible para esta desviación reside principalmente en dos factores. El
primero, la alta pendiente de la SEP en este estado excitado hace que pequeñas
desviaciones de la geometría usada para el cálculo de las propiedades espectroscópicas
correspondientes a las transiciones verticales provoquen un error significativo en la
determinación de la energía correspondiente a la transición electrónica. Sin embargo,
esta fuente de error debe ser menor, ya que no solamente la geometría usada es muy
cercana a la experimetal, sino que además otros estados excitados en los que existen
pendientes similares de la SEP y que han sido estudiados no presentan un error tan
grande. Por otro lado, debido a que la transición es prohibida (permitida sólo por
acoplamiento vibrónico) y puesto que la geometría relajada en el estado excitado difiere
mucho de la correspondiente a la transición vertical, la amplia progresión de bandas
vibracionales (factores de Franck-Condon) complica notablemente la asignación de la
energía de transición vertical de manera experimental.
No obstante, estudios teóricos empleando el método de interacción de configuraciones
multirreferencial (MRCI) (Palmer, 1988) predijeron una transición a 4.37 eV. Este
acuerdo con los datos experimentales es fortuito, y se debe al uso de unas funciones de
base limitadas (funciones “split-valence”). De hecho, resultados similares se pueden
obtener a nivel CASPT2 usando funciones de base similares, como se verá a
continuación.
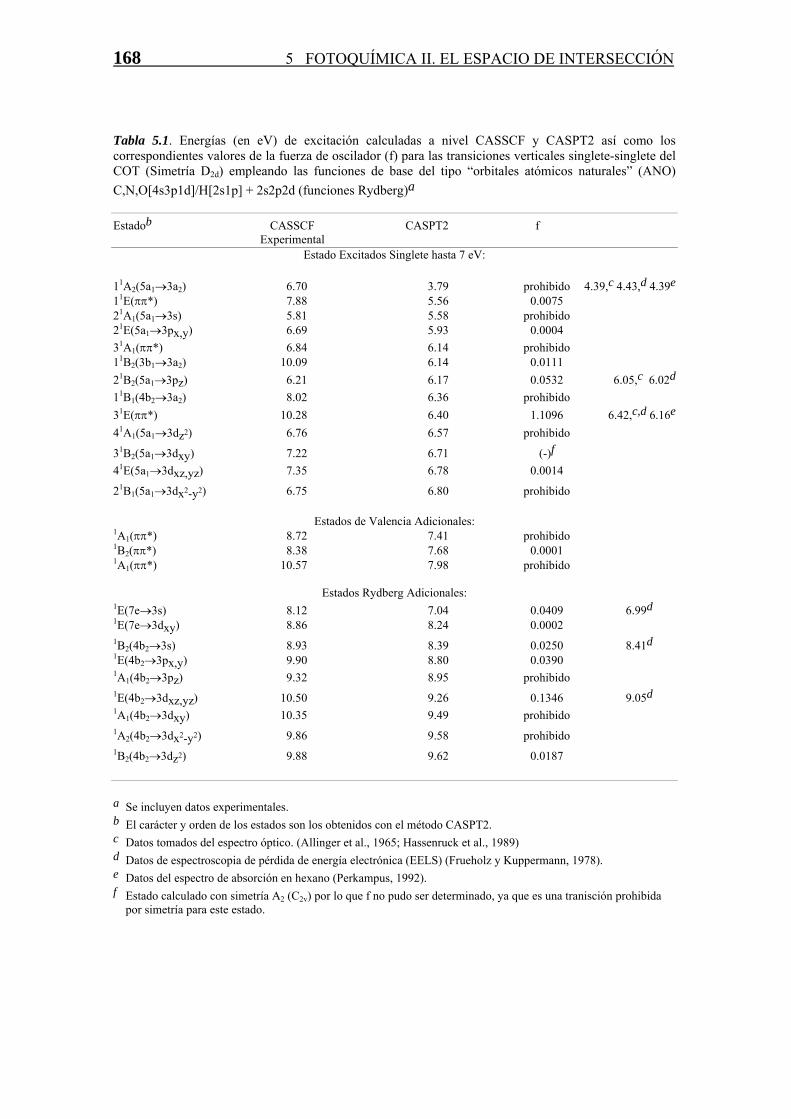
168 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Tabla 5.1. Energías (en eV) de excitación calculadas a nivel CASSCF y CASPT2 así como los correspondientes valores de la fuerza de oscilador (f) para las transiciones verticales singlete-singlete del COT (Simetría D2d) empleando las funciones de base del tipo “orbitales atómicos naturales” (ANO) C,N,O[4s3p1d]/H[2s1p] + 2s2p2d (funciones Rydberg)a Estadob CASSCF CASPT2 f Experimental
Estado Excitados Singlete hasta 7 eV: 11A2(5a1→3a2) 6.70 3.79 prohibido 4.39,c 4.43,d 4.39e 11E(ππ*) 7.88 5.56 0.0075 21A1(5a1→3s) 5.81 5.58 prohibido 21E(5a1→3px,y) 6.69 5.93 0.0004 31A1(ππ*) 6.84 6.14 prohibido 11B2(3b1→3a2) 10.09 6.14 0.0111 21B2(5a1→3pz) 6.21 6.17 0.0532 6.05,c 6.02d 11B1(4b2→3a2) 8.02 6.36 prohibido 31E(ππ*) 10.28 6.40 1.1096 6.42,c,d 6.16e 41A1(5a1→3dz2) 6.76 6.57 prohibido
31B2(5a1→3dxy) 7.22 6.71 (-)f 41E(5a1→3dxz,yz) 7.35 6.78 0.0014
21B1(5a1→3dx2-y2) 6.75 6.80 prohibido
Estados de Valencia Adicionales: 1A1(ππ*) 8.72 7.41 prohibido 1B2(ππ*) 8.38 7.68 0.0001 1A1(ππ*) 10.57 7.98 prohibido
Estados Rydberg Adicionales:
1E(7e→3s) 8.12 7.04 0.0409 6.99d 1E(7e→3dxy) 8.86 8.24 0.0002 1B2(4b2→3s) 8.93 8.39 0.0250 8.41d 1E(4b2→3px,y) 9.90 8.80 0.0390 1A1(4b2→3pz) 9.32 8.95 prohibido 1E(4b2→3dxz,yz) 10.50 9.26 0.1346 9.05d 1A1(4b2→3dxy) 10.35 9.49 prohibido 1A2(4b2→3dx2-y2) 9.86 9.58 prohibido 1B2(4b2→3dz2) 9.88 9.62 0.0187 a Se incluyen datos experimentales. b El carácter y orden de los estados son los obtenidos con el método CASPT2. c Datos tomados del espectro óptico. (Allinger et al., 1965; Hassenruck et al., 1989) d Datos de espectroscopia de pérdida de energía electrónica (EELS) (Frueholz y Kuppermann, 1978).e Datos del espectro de absorción en hexano (Perkampus, 1992). f Estado calculado con simetría A2 (C2v) por lo que f no pudo ser determinado, ya que es una tranisción prohibida
por simetría para este estado.

5.3 Espectro de Absorción del 1,3,5,7-Ciclooctatetraeno 169
Empleando funciones ANO, con un esquema de contracción C[3s2p]/H[2s], el cálculo
CASPT2 ofrece un valor para la transición electrónica de 4.45 eV, consistente con los
resultados MRCI. Añadiendo funciones polarizadas al carbono, C[3s2p1d]/H[2s], la
energía de excitación cae a 3.92 eV, a su vez consistente con la energía calculada a nivel
CASPT2/6-31G(d) igual a 4.00 eV. Usando el esquema de contracción mayor de los
puestos a prueba con este tipo de funciones de base (ANO), se obtiene un valor de 3.80
eV muy parecido al presentado en la tabla 5.1. Por lo tanto, los mejores resultados
teóricos predicen una energía de excitación vertical alrededor de 3.80 – 4.00 eV. Por lo
tanto, sin considerar el posible error de cálculo debido a ligeras desviaciones en la
geometría usada, y que ya ha sido comentado anteriormente, la diferencia entre el
máximo de absorción de dicha banda experimental y la energía correspondiente a la
transición vertical distan aproximadamente medio electronvoltio.
Las bandas de simetría E, esto es, 11E(ππ*) y 31E(ππ*) están dominadas principalmente
por dos configuraciones, correspondientes a las monoexcitaciones HOMO-1 → LUMO,
y HOMO → LUMO+1. De hecho, la diferencia de energías entre HOMO-1 y LUMO, y
HOMO y LUMO+1 son muy similares, 12.52 y 12.39 eV, lo que hace que puedan
interaccionar dando lugar a una combinación lineal positiva y otra negativa, como de
hecho ocurre. Desde esta perspectiva es posible racionalizar también el valor de los
momentos de transición. La correspondiente combinación lineal negativa, que da lugar
al estado 11E(ππ*), presenta el menor momento de transición, ya que los
correspondientes a las monoexcitaciones se cancelan (f(11A1→11E(ππ*)) =0.0075), mientras
que en la combinación lineal positiva se suman (f(11A1→31E(ππ*)) =1.1096).
Por otro lado, entre los estados de valencia 11E(ππ*) y 31E(ππ*), existen otros tres
estados más de la misma naturaleza. Los estados 31A1(ππ*) y 11B2(3b1→3a2) a nivel
CASPT2 son degenerados en energía (6.14 eV), mientras que el estado 11B1(4b2→3a2)
se encuentra a 6.36 eV, y por lo tanto muy próximo al 31E(ππ*). El estado 31A1(ππ*)
tiene un carácter de doble excitación electrónica, con una contribución alta (33.2%) de
la configuración correspondiente a la doble transición (HOMO→LUMO)2. El estado
11B2(3b1→3a2) está descrito principalmente por la configuración correspondiente a la
excitación simple de un electrón desde el orbital σ 3b1, al orbital π 3a2 (LUMO). La
fuerza del oscilador correspondiente a la transición 11A1→11B2 es de 0.011, y puede
contribuir en alguna medida al hombro que aparece en el espectro de absorción a

170 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
aproximadamente 6 eV (Allinger et al., 1965; Frueholz y Kuppermann, 1978;
Hassenruck et al., 1989). La asignación que se ha hecho de este hombro a la transición
11A1→11E no puede ser apoyada en base a los resultados obtenidos. Por el contrario, la
transición al estado Rydberg 21B2(5a1→3pz) que aparece a nivel CASPT2 a 6.17 eV y
que tiene una fuerza de oscilador de 0.05 parece la mejor candidata para explicar la
existencia de dicho hombro. Esta asignación viene apoyada por el hecho de que en
disolución (hexano) ya no aparece tal hombro en el espectro de absorción (Perkampus,
1992), como cabe esperar en base a la propiedad de los estados Rydberg, que
desaparecen en fase condensada y en disolución (Robin, 1975).
5.3.3 Espectro de Absorción Singlete-Triplete
Los resultados obtenidos del análisis de distintas transiciones en las que están
involucrados estados triplete y singlete a nivel CASSCF y CASPT2, se muestran en la
tabla 5.2.
Tabla 5.2. Energías (eV) de excitación calculadas con los métodos CASSCF y CASPT2 para los estados triplete más bajos en energía del COT, empleando las funciones de base del tipo orbitales atómicos naturales (ANO): C,N,O[4s3p1d]/H[2s1p] + 2s2p2d (funciones Rydberg)a Estado Transición CASSCF CASPT2 Experimental 13A2(5a1→3a2) S-T vertical 3.66 2.82 3.05,b 2.89c S-T 0-0 1.42 0.78 emisión max. 0.66 0.22 13E(ππ*) S-T vertical 4.31 3.84 4.05b 13B1(ππ*) S-T vertical 5.18 4.69 4.84b 13A1(5a1→3s) S-T vertical 6.04 5.54 a Se incluyen datos experimentales. b Datos de espectroscopia de pérdida de energía electrónica (EELS) (Frueholz y Kuppermann, 1978).c Dato tomado de análisis cinéticos (ver Capítulo sexto) empleando datos experimentales de constantes de
transferencia de energía triplete-triplete de diversos dadores al COT.( Forward et al., 1993; Das y Priyadarsini, 1994)
El primer estado Rydberg, 13A1(5a1→3s), aparece a 6.04 eV, y por debajo de él se han
determinado tres estados de valencia, de simetrías: 13A2(5a1→3a2), 13E(ππ*) y
13B1(ππ*). Estos tres estados triplete de valencia están localizados a 3.66, 4.31 y 5.18

5.3 Espectro de Absorción del 1,3,5,7-Ciclooctatetraeno 171
eV, y pueden ser relacionados con los hallados experimentalmente (Frueholz y
Kuppermann, 1978) situados a 3.05, 4.05 y 4.84 eV.
La asignación de las dos primeras bandas corresponde con la que hiciera Palmer
(Palmer, 1988) en base a cálculos de interacción de configuraciones (CI) con un
pequeño conjunto de funciones de base. Sin embargo surgen unas cuantas discrepancias
con respecto a otras bandas. En primer lugar, la transición 11A1→13E(ππ*) la sitúa 1 eV
por encima de la energía CASPT2 presentada aquí, lo que implica una asignación
incorrecta a dicha banda. Por otro lado, el mismo autor, sitúa al estado 23A2 por debajo
del 13E(ππ*), y lo propone como la tercera banda de valencia para el estado triplete,
mientras que esta transición está localizada, a nivel CASPT2, muy por encima de la
banda 13B1(ππ*), a unos 2.5 eV.
Hay que hacer notar que existe un error sistemático en los resultados obtenidos
mediante los cálculos CASPT2 y aquellos obtenidos mediante espectroscopia de
pérdida de energía electrónica (Frueholz y Kuppermann, 1978). En general, los cálculos
subestiman la energía en todas las transiciones electrónicas entorno a 0.2 eV. Este hecho
puede ser atribuido en parte al hamiltoniano de orden cero usado (del tipo Møller-
Plesset) que favorece sistemas de capa abierta y puede dar lugar a tripletes de energía
ligeramente baja. Este hecho está bien documentado y se pueden encontrar numerosas
referencias que lo describen, por ejemplo en (Roos et al., 1996). A pesar de esta
desviación sistemática, las primeras transiciones a estados triplete se han identificado
correctamente por lo que no es necesario recurrir a métodos más sofisticados.
Experimentos de transferencia de energía han permitido (como se verá con mucho más
detalle en el capítulo sexto) determinar la energía triplete del COT (Forward et al.,
1993; Das y Priyadarsini, 1994), aunque existe una discrepancia considerable entre
ellos. Sin embargo a partir de estos experimentos de determinación de constantes de
transferencia de energía, y aplicando un modelo teórico que será descrito en detalle en
el sexto capítulo, se ha determinado una energía triplete de 2.89eV, muy cercana a la
calculada, 2.82 eV, con la metodología CASPT2.
El peso de las distintas configuraciones en la función de onda varía de la siguiente
manera. Para la primera transición, la 11A1→13A2, la configuración predominante es la
debida a la transición HOMO → LUMO. Para la segunda transición (11A1→13E), la
función de onda está compuesta principalmente por una combinación lineal de dos
configuraciones correspondientes a monoexcitaciones, las cuales son: HOMO-1 →

172 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
LUMO (49.6%) y HOMO → LUMO+1 (37.4%). Por último, la tercera banda de
valencia posee un carácter multiconfiguracional donde existen numerosas
contribuciones de distintas configuraciones con pesos en la función de onda similares.
También se estudió, debido a su importancia en la explicación del comportamiento no-
vertical en el proceso de transferencia de energía triplete-triplete en el que participa el
COT como aceptor (ver Capítulo 6), la transición 0-0, es decir, la transición entre el
estado fundamental vibracional y electrónico del COT (11A1 y simetría molecular D2d)
al primer estado excitado triplete (13A2g y simetría molecular D8h). En primer lugar se
optimizó la estructura molecular en el estado triplete a nivel CASSCF (8 electrones, 8
orbitales) con funciones de base del tipo ANO C[4s3p1d]/H[2s1p] dando lugar a una
geometría plana de simetría D8h en la que los dos únicos parámetros geométricos de la
molécula son la distancia entre carbonos adyacentes (r(C-C)=1.404 Å) y la distancia
carbono-hidrógeno (r(C-H)=1.077 Å). A partir de esta estructura, se realizaron los
correspondientes cálculos a nivel CASPT2, obteniéndose una diferencia de energías
entre los mínimos de la superficie de energía potencial (D2d en 11A1 y D8h en 3A2g) de
0.78 eV. No se realizaron correcciones a la energía vibracional residual, las cuales no
suelen ser mayores de 0.1 eV, variación que está dentro del error intrínseco del método
CASPT2. Como ha sido apuntado por otros autores (ver capítulo sexto para detalles), la
gran diferencia entre la energía de transición vertical y la 0-0 (de unos 2 eV) está
íntimamente relacionada con el comportamiento no-vertical del COT como aceptor en
el proceso de transferencia de energía triplete-triplete.
Por último, se ha estudiado también la longitud de onda a la que emite el COT en el
primer estado excitado triplete, es decir, el máximo del espectro de fosforescencia. La
transición que da lugar a este espectro de emisión es la siguiente: 3A2g→1B2g. La energía
calculada correspondiente a esta transición es de tan sólo 0.22 eV, y por lo tanto cae en
el rango de la radiación infrarroja. Esta estimación de la energía está en concordancia
con los resultados ab initio ya existentes (Hrovat y Borden, 1997). También existen
estudios experimentales en los que se pone de manifiesto la menor energía del estado
singlete que del triplete en esta molécula antiaromática (Wenthold et al., 1996), tal y
como los cálculos CASPT2 demuestran, lo que implica la violación de la regla de Hund.
A propósito de la violación de esta regla, se ha sugerido que las moléculas
antiaromáticas no deberían satisfacerla (Zilberg y Haas, 1998), lo cual está en acuerdo
con los resultados obtenidos en el COT.

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 173
55..44 EEssppaacciioo ddee IInntteerrsseecccciióónn ((SS11//SS00)) eenn eell 11,,33,,55,,77--CCiiccllooooccttaatteettrraaeennoo
En este punto se va a estudiar parte de la compleja fotoquímica del 1,3,5,7-
Ciclooctatetraeno (COT) en base a las propiedades de las PES de los estados
electrónicos involucrados (S1 y S0 principalmente) en dicha reactividad.
En primer lugar se tratará el proceso de relajación tras la primera transición permitida:
S0 → S2. Este proceso provoca la población del estado S1 para el cual la estructura de
equilibrio es plana octogonal (simetría D8h), que sirve al sistema de intermedio para su
posterior decaimiento al estado fundamental a través de una transición diabática.
Posteriormente se analizará el espacio de intersección o región de intersecciones cónicas
(CoInR) que son accesibles para el COT así como otras regiones del mismo. Se
establece una conexión entre todas las CoIn mínimas en energía encontradas y se
conforma un panorama de reactividad no solamente descriptivo sino también predictivo.
Por último, se describen los procesos de relajación vibracional desde las intersecciones
cónicas asociadas a las transiciones diabáticas antes descritas.
5.4.1 Transición Franck-Condon S0→S2 y Población del Estado S1 en el
COT
A la luz de los resultados obtenidos del estudio del espectro de absorción del COT
(sección 5.3), la primera de las transiciones electrónicas permitidas desde el estado
fundamental es al estado 11E (S2), ya que las correspondientes a los estados 11A2 (S1) y
21A1(S3) son prohibidas por simetría, debido a el producto de las representaciones
irreducibles de simetría del estado inicial 11A1 (S0) de la componente (x,y,z) del
momento dipolar y del estado final no contienen la representación totalmente simétrica,
como ya se vio en la sección 5.3. La naturaleza de la transición 11A1 → 11E (S0 → S2)
está marcada principalmente por la contribución de dos configuraciones
correspondientes a las mono-excitaciones: HOMO-1 → LUMO, y HOMO →
LUMO+1, con una fuerza de oscilador de f=0.0075. Por lo tanto, esta transición
S0(11A1) → S2(11E) es la primera permitida por dipolo eléctrico, y ha sido el punto de
partida en el estudio de la relajación tras la transición FC.
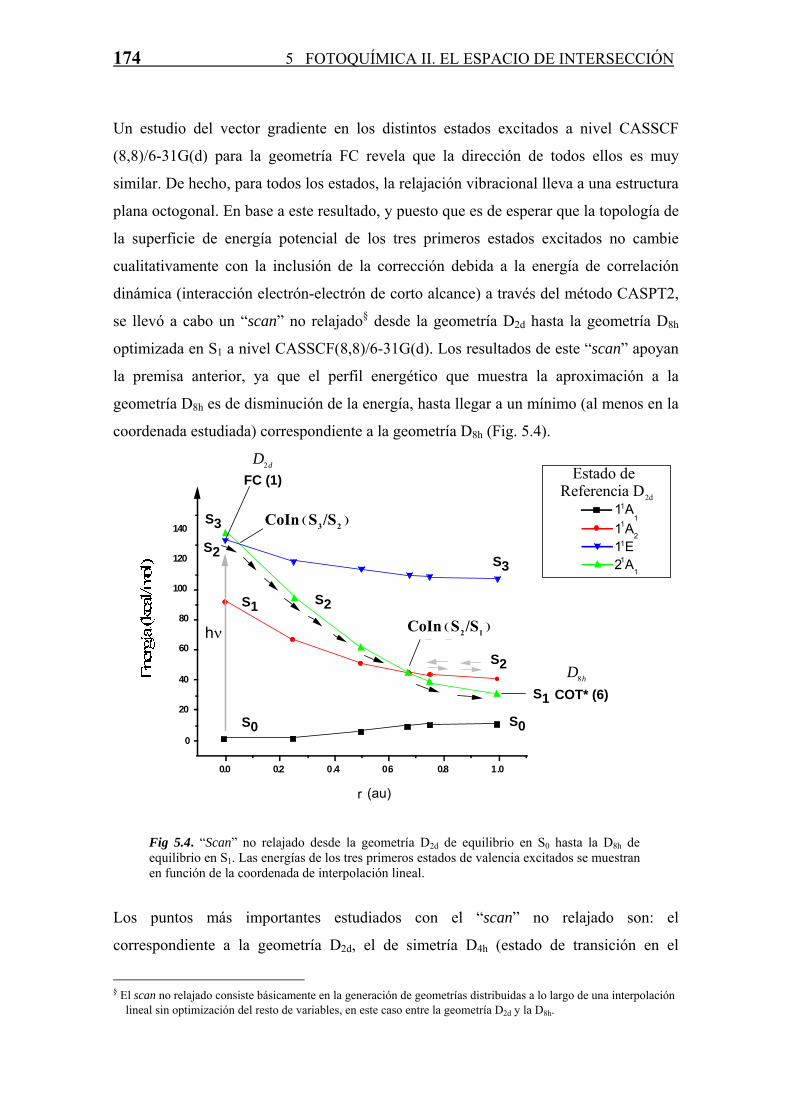
174 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Un estudio del vector gradiente en los distintos estados excitados a nivel CASSCF
(8,8)/6-31G(d) para la geometría FC revela que la dirección de todos ellos es muy
similar. De hecho, para todos los estados, la relajación vibracional lleva a una estructura
plana octogonal. En base a este resultado, y puesto que es de esperar que la topología de
la superficie de energía potencial de los tres primeros estados excitados no cambie
cualitativamente con la inclusión de la corrección debida a la energía de correlación
dinámica (interacción electrón-electrón de corto alcance) a través del método CASPT2,
se llevó a cabo un “scan” no relajado§ desde la geometría D2d hasta la geometría D8h
optimizada en S1 a nivel CASSCF(8,8)/6-31G(d). Los resultados de este “scan” apoyan
la premisa anterior, ya que el perfil energético que muestra la aproximación a la
geometría D8h es de disminución de la energía, hasta llegar a un mínimo (al menos en la
coordenada estudiada) correspondiente a la geometría D8h (Fig. 5.4).
0.0 0.2 0.4 0.6 0.8 1.0
0
20
40
60
80
100
120
140
Ene
rgy
(kca
l/mol
)
D2d
reference state
r (au)
FC (1)
COT* (6)
S3/S2 CI
S2/S1 CI
S1
S2
S3
S0
S1
S2
S3
S0
hν
11A1
11A2
11E21A
1
S2
2dD
8hD
2d
Estado de Referencia D
( )3 2CoIn S /S
( )2 1CoIn S /S
8hD
2dD
Fig 5.4. “Scan” no relajado desde la geometría D2d de equilibrio en S0 hasta la D8h de equilibrio en S1. Las energías de los tres primeros estados de valencia excitados se muestran en función de la coordenada de interpolación lineal.
Los puntos más importantes estudiados con el “scan” no relajado son: el
correspondiente a la geometría D2d, el de simetría D4h (estado de transición en el
§ El scan no relajado consiste básicamente en la generación de geometrías distribuidas a lo largo de una interpolación
lineal sin optimización del resto de variables, en este caso entre la geometría D2d y la D8h.
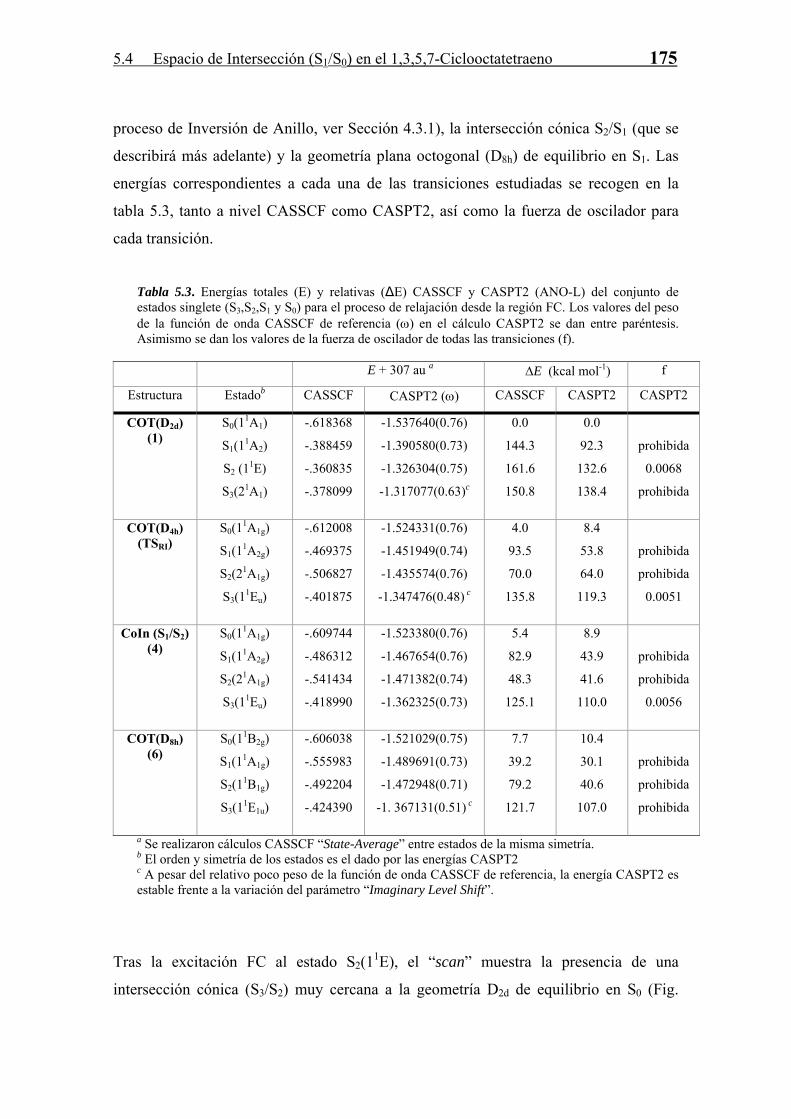
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 175
proceso de Inversión de Anillo, ver Sección 4.3.1), la intersección cónica S2/S1 (que se
describirá más adelante) y la geometría plana octogonal (D8h) de equilibrio en S1. Las
energías correspondientes a cada una de las transiciones estudiadas se recogen en la
tabla 5.3, tanto a nivel CASSCF como CASPT2, así como la fuerza de oscilador para
cada transición.
Tabla 5.3. Energías totales (E) y relativas (∆E) CASSCF y CASPT2 (ANO-L) del conjunto de estados singlete (S3,S2,S1 y S0) para el proceso de relajación desde la región FC. Los valores del peso de la función de onda CASSCF de referencia (ω) en el cálculo CASPT2 se dan entre paréntesis. Asimismo se dan los valores de la fuerza de oscilador de todas las transiciones (f).
E + 307 au a ∆E (kcal mol-1) f
Estructura Estadob CASSCF CASPT2 (ω) CASSCF CASPT2 CASPT2
COT(D2d) (1)
S0(11A1)
S1(11A2)
S2 (11E)
S3(21A1)
-.618368
-.388459
-.360835
-.378099
-1.537640(0.76)
-1.390580(0.73)
-1.326304(0.75)
-1.317077(0.63)c
0.0
144.3
161.6
150.8
0.0
92.3
132.6
138.4
prohibida
0.0068
prohibida
COT(D4h) (TSRI)
S0(11A1g)
S1(11A2g)
S2(21A1g)
S3(11Eu)
-.612008
-.469375
-.506827
-.401875
-1.524331(0.76)
-1.451949(0.74)
-1.435574(0.76)
-1.347476(0.48) c
4.0
93.5
70.0
135.8
8.4
53.8
64.0
119.3
prohibida
prohibida
0.0051
CoIn (S1/S2) (4)
S0(11A1g)
S1(11A2g)
S2(21A1g)
S3(11Eu)
-.609744
-.486312
-.541434
-.418990
-1.523380(0.76)
-1.467654(0.76)
-1.471382(0.74)
-1.362325(0.73)
5.4
82.9
48.3
125.1
8.9
43.9
41.6
110.0
prohibida
prohibida
0.0056
COT(D8h) (6)
S0(11B2g)
S1(11A1g)
S2(11B1g)
S3(11E1u)
-.606038
-.555983
-.492204
-.424390
-1.521029(0.75)
-1.489691(0.73)
-1.472948(0.71)
-1. 367131(0.51) c
7.7
39.2
79.2
121.7
10.4
30.1
40.6
107.0
prohibida
prohibida
prohibida
a Se realizaron cálculos CASSCF “State-Average” entre estados de la misma simetría. b El orden y simetría de los estados es el dado por las energías CASPT2 c A pesar del relativo poco peso de la función de onda CASSCF de referencia, la energía CASPT2 es estable frente a la variación del parámetro “Imaginary Level Shift”.
Tras la excitación FC al estado S2(11E), el “scan” muestra la presencia de una
intersección cónica (S3/S2) muy cercana a la geometría D2d de equilibrio en S0 (Fig.

176 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
5.4), y que es del tipo “inclinada” (ver Sección 2.3). Esta CoIn puede ser evitada por el
COT en el proceso de relajación ya que la otra coordenada (no representada en la figura
5.4) para la que desaparece la degeneración energética, posibilita un camino que evita la
población a través de esta CoIn del estado S3. Por lo tanto, tras la transición FC la
molécula permanece en el estado S2.
Siguiendo el camino de relajación en S2, se llega a otro cruce diabático (S2/S1) (Fig.
5.5). Esta segunda intersección cónica es también del tipo “inclinada” lo que hace
posible que se pueble el estado S2, aunque la energía interna adquirida en el proceso de
relajación vibracional es suficiente para que el sistema supere con gran facilidad la
pequeña energía de activación (unas 4 kcal/mol como límite superior) necesaria para
acceder desde la geometría D8h en S2 a la intersección cónica S2/S1 (Fig. 5.4).
a b
Fig. 5.5. Vectores “diferencia de gradientes” (a) y “acoplamiento derivativo” (b), para la intersección cónica S2/S1 de simetría D2d.
El análisis de los vectores “diferencia de gradientes” y “acoplamiento derivativo” (Fig.
5.5) nos permite predecir qué tipo de relajación ocurre tras la transición S2/S1. Al
alcanzar el sistema esta CoIn, existen dos componentes donde la degeneración
energética desparece, dados como ya se ha visto por los vectores antes mencionados que
conforman el espacio de ramificación. Estos dos vectores corresponden principalmente
a dos movimientos moleculares, que son el de redistribución de carga π hacia una
distribución equitativa de la misma en los 8 enlaces C-C, y el de aplanamiento del
sistema. Amabas distorsiones llevan consigo la formación de una estructura plana y
octogonal (D8h) que es la que corresponde a la geometría de equilibrio en S1. Por lo
tanto, esta CoIn S2/S1 es una vía efectiva de población del estado S1. Por lo tanto, se

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 177
comprueba cómo todas las posibles relajaciones tras la excitación FC llevan a la
población del estado S1 (geometría plana octogonal), ya sea directamente a través del
cruce diabático S2/S1 o a través del intermedio plano en S2 usando el mismo cruce para
poblar S1. Este proceso de relajación marca la fotoquímica del COT. Algunos de cuyos
aspectos se estudian en los siguientes puntos.
5.4.2 Región de Intersecciones Cónicas en el COT
Tras la población del estado S1, el COT puede evolucionar por distintos caminos para
decaer al estado fundamental. Debido a la pequeña diferencia de energía entre S1 y S0
para la geometría de equilibrio en S1, es posible que el sistema decaiga al estado
fundamental radiativamente a través de la emisión de fluorescencia. Este decaimiento
provoca la recuperación del COT a través de los procesos de “desplazamiento de
enlace” e “inversión de anillo” que ya se describieron detalladamente en la Sección 4.3,
ya que la estructura de equilibrio en S1 es muy similar al TS para el proceso de
“desplazamiento de enlace”. Sin embargo, existen canales de desactivación no radiante
que dan lugar a la formación de distintos productos en el estado fundamental.
La vía de desactivación más baja en energía corresponde con la transición S1/S0 a través
de una CoIn de simetría C2v (Garavelli et al., 2001). Este cruce diabático es el
responsable no solamente del proceso de “desplazamiento de enlace” sino también de la
formación de Semibulvaleno (SBV) (Esquema 5.1).
Sin embargo existen otros cruces diabáticos de mayor energía que si bien no son
alcanzables directamente por el COT en este proceso fotoquímico, si lo son en otros
procesos fotoquímicos en los que intervienen isómeros de valencia del COT (algunos
ejemplos ya se han tratado a lo largo de la Tesis y otro se estudiarán más adelante). Por
lo tanto tiene un interés especial el poder establecer la topología de la región de cruce en
el COT y sus isómeros de valencia.
El conocimiento de las características de este subespacio de cruce S1/S0 es muy
importante en un compuesto como el COT, que puede servir de modelo para numerosos
compuestos cíclicos insaturados, ya que las características de muchas intersecciones
cónicas parecen ser comunes en un mismo tipo de compuestos, independientemente de
su tamaño o composición concreta (por ejemplo las intersecciones cónicas encontradas
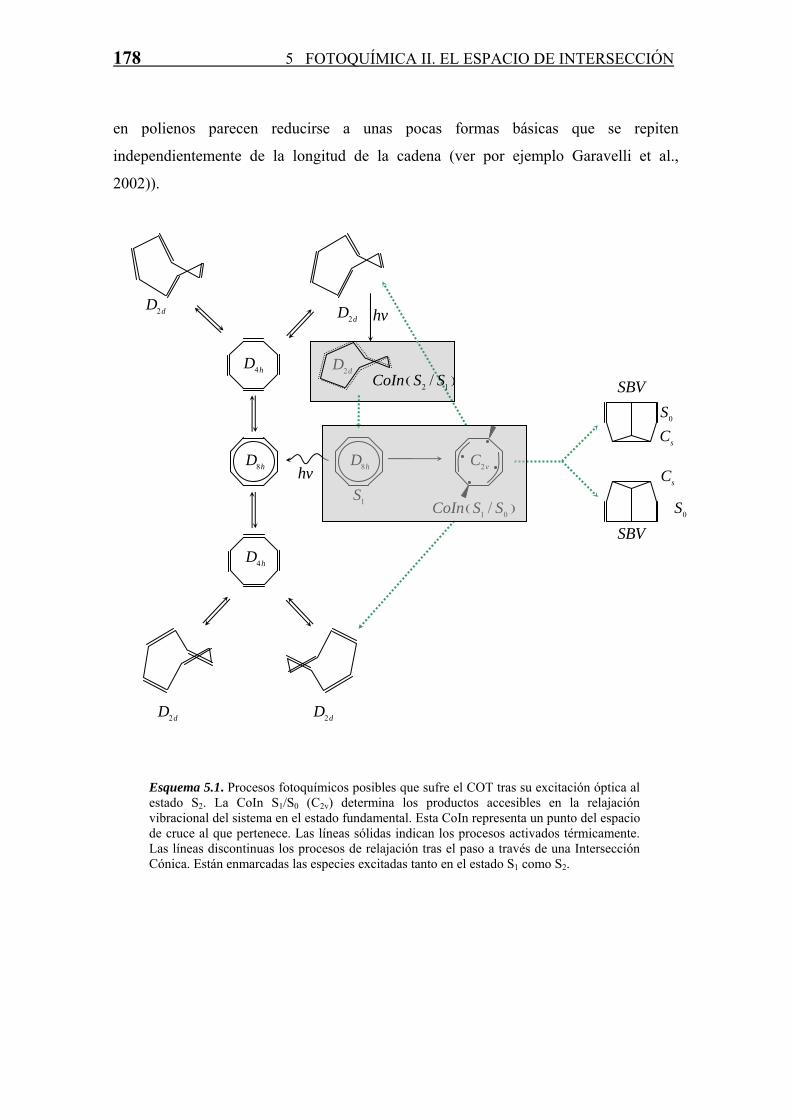
178 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
en polienos parecen reducirse a unas pocas formas básicas que se repiten
independientemente de la longitud de la cadena (ver por ejemplo Garavelli et al.,
2002)).
0S
2dD
1S
8hD
hv
hv
( )1 0/CoIn S S
2vC
( )2 1/CoIn S S
sC
sC
SBV
SBV
0S
8hD
4hD
4hD
2dD2dD
2dD2dD
Esquema 5.1. Procesos fotoquímicos posibles que sufre el COT tras su excitación óptica al estado S2. La CoIn S1/S0 (C2v) determina los productos accesibles en la relajación vibracional del sistema en el estado fundamental. Esta CoIn representa un punto del espacio de cruce al que pertenece. Las líneas sólidas indican los procesos activados térmicamente. Las líneas discontinuas los procesos de relajación tras el paso a través de una Intersección Cónica. Están enmarcadas las especies excitadas tanto en el estado S1 como S2.

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 179
Este estudio pormenorizado del espacio de intersección se ha realizado pretendiendo
describirlo completamente, para lo cual se han determinando las propiedades
electrónicas y estructurales de una gran cantidad de intersecciones cónicas mínimas en
energía, estableciendo además en algunos casos la relación existente entre estos
mínimos.
El estudio de estas CoIn se dividirá en función de los enlaces sigma intra-anulares que
presenta la estructura. Para ello, y al igual que en los cálculos anteriores, se ha usado el
método multiconfiguracional CASSCF, empleando como espacio activo el compuesto
por los electrones π, por lo que las CoIn cíclicas sin enlaces σ se han tratado con el
método CASSCF(8,8), las que presentan un enlace intra-anular a nivel CASSCF(6,6) y
las que presentan dos a nivel CASSCF(4,4). Siempre se ha usado una base de cálculo
6-31G(d). En primer lugar se estudiarán las características electrónicas y estructurales
de las CoIn que no presentan ningún enlace sigma.
1. CoIn Cíclicas Abiertas (sin enlaces σ intra-anulares).
Todas las Intersecciones Cónicas mínimas en energía encontradas poseen una estructura
tetrarradicálica, en concordancia con los numerosos ejemplos descritos para
hidrocarburos insaturados (Bernardi et. al, 1990; Bernardi et. al, 1990b; Olivucci et al.,
1993; Olivucci et al., 1994).
En base a esta distribución tetrarradicálica es posible postular distintas distribuciones
electrónicas (Esquema 5.2).
Esquema 5.2. Posibles distribuciones tetrarradicálicas en un anillo de ocho eslabones (derivadas estructuralmente del COT). Enmarcadas están las distribuciones electrónicas encontradas en la SEP que se corresponden con intersecciones cónicas mínimas (en el espacio de intersección) en energía.
Se ha llevado a cabo el estudio de las intersecciones cónicas de ciclo abierto (sin enlaces
sigma aparte de los correspondientes al anillo) a partir de las posibilidades presentadas
en el esquema 5.2. Se han identificado ocho CoIn, cada una de las cuales se relaciona
con alguna de las estructuras tetrarradicálicas propuestas. Sin embargo, estas ocho CoIn
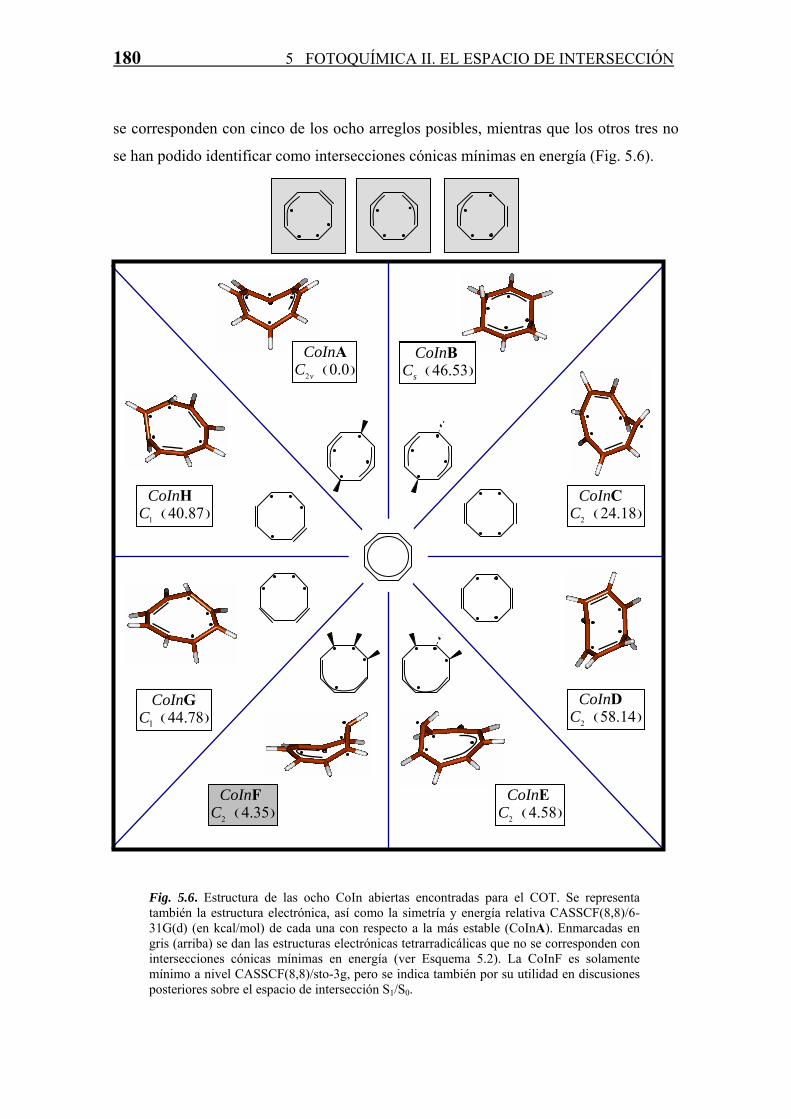
180 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
se corresponden con cinco de los ocho arreglos posibles, mientras que los otros tres no
se han podido identificar como intersecciones cónicas mínimas en energía (Fig. 5.6).
( )1 40.87CoIn
CH
( )2 0.0v
CoInC
A( )37.13s
CoInC
B
( )2 24.18CoIn
CC
( )2 58.14CoIn
CD
( )2 4.58CoIn
CE
( )2 4.35CoIn
CF
( )1 38.48CoIn
CG
( )1
44.78CoIn
CG
( )
46.53s
CoInC
B
Fig. 5.6. Estructura de las ocho CoIn abiertas encontradas para el COT. Se representa también la estructura electrónica, así como la simetría y energía relativa CASSCF(8,8)/6-31G(d) (en kcal/mol) de cada una con respecto a la más estable (CoInA). Enmarcadas en gris (arriba) se dan las estructuras electrónicas tetrarradicálicas que no se corresponden con intersecciones cónicas mínimas en energía (ver Esquema 5.2). La CoInF es solamente mínimo a nivel CASSCF(8,8)/sto-3g, pero se indica también por su utilidad en discusiones posteriores sobre el espacio de intersección S1/S0.
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 181
Las estructuras y datos energéticos de las CoIn encontradas se recogen en la figura 5.6 y
en la tabla 5.4. Pueden existir grandes diferencias energéticas y estructurales entre CoIn
con un mismo arreglo electrónico por lo que se requieren tanto criterios estructurales
como electrónicos para determinar la estabilidad relativa de una determinada
intersección cónica. Por lo tanto, para un mismo esquema de localización de los
radicales (uno de los ocho arreglos posibles del esquema 5.2) pueden existir varias CoIn
que poseen energías que pueden diferir notablemente.
Estas diferencias las encontramos por ejemplo entre las CoInA y B, para las cuales la
orientación de dos de los hidrógenos opuestos en el anillo provoca una diferencia de
energías entre ambas de ~46 kcal/mol, a pesar de tener una distribución electrónica
similar. Por lo tanto, recurrir únicamente a la distribución electrónica en la molécula de
los cuatro radicales para racionalizar la estabilidad de las intersecciones cónicas puede
ser en algunos casos insuficiente.
Tabla 5.4. Energías electrónicas (absolutas y relativas) de las 8 intersecciones cónicas de ciclo abierto encontradas a nivel CASSCF(8,8)/6-31G(d).
CoIn E(S0)a
E(S1)a
E(S0)b E(S1)b ∆E(S1/S0)b
A -307.51408 -307.51400 0.00 0.05 0.05
B -307.44016 -307.42993 46.38 46.53 0.14
C -307.48049 -307.48019 21.08 21.27 0.19
D -307.42652 -307.42607 54.94 55.23 0.28
E -307.51155 -307.51143 1.59 1.66 0.07
Fc - - 4.35 4.42 0.07
G -307.44275 -307.44271 44.76 44.78 0.03
H -307.45365 -307.45360 37.92 37.95 0.03
a Energía electrónica en hartrees b Energía electrónica relativa a la CoInA (la más estable) en kcal/mol. c Energía relativa CASSCF(8,8)/sto-3g con respecto a la CoInA al mismo nivel de cálculo
Es posible no obstante tomar las estructuras más estables de cada arreglo electrónico,
para las cuales es de suponer que la desestabilización por motivos estructurales está
182 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
minimizada, e intentar estudiar la estabilidad de éstos en cada CoIn en base a
argumentos sencillos de estabilización de radicales. Un método muy sencillo e intuitivo
desde el punto de vista químico (ya que hace uso del concepto de deslocalización
electrónica de una manera muy simple) es el método de Hückel (Hückel, 1931). Con
este método se pueden comparar estabilidades de distintos arreglos electrónicos en
función de la integral de intercambio β, que depende del apareamiento de los electrones
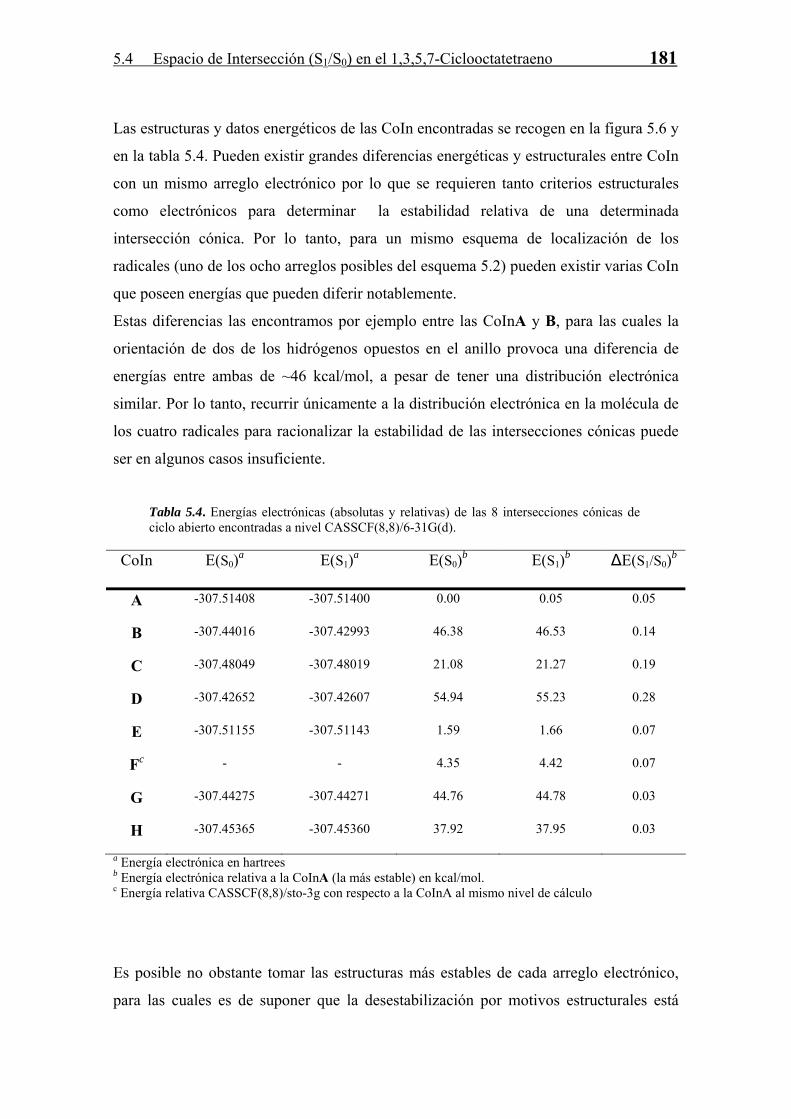
π y de su deslocalización a través del anillo. Tomando pues las CoIn más estables de
cada arreglo electrónico correspondiente a una energía distinta en el método de Hückel,
observamos que existe una relación cualitativa (la asombrosa buena correlación
cuantitativa entre ambos métodos es fortuita) entre la energía CASSCF y la calculada
con el método semiempírico de Hückel (Fig. 5.7).
3.8 4.0 4.2 4.4 4.6 4.8 5.0 5.2 5.4 5.6 5.8
-0.515
-0.510
-0.505
-0.500
-0.495
-0.490
-0.485
-0.480
-0.475
E(C
ASS
CF(
8,8)
/6-3
1G(d
)+30
7) /
a.u.
Erel(Hückel) /β
Fig. 5.7. Energía de las intersecciones cónicas más estables para cada arreglo de Hückel distinto a nivel CASSCF(8,8)/6-31g* y con el método de Hückel (en unidades de la integral de intercambio β).
Esta correlación pone de manifiesto el importante papel que juega la estructura
electrónica en la estabilización de las intersecciones cónicas, de tal manera que a mayor
deslocalización de los radicales (cuatro) en el anillo, mayor es la estabilidad de la
misma. Por lo tanto, la inclusión de sustituyentes en la molécula que estabilizan una

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 183
determinada disposición de los radicales correspondiente a una CoIn con una estructura
electrónica concreta, debe hacer posible la estabilización energética de la misma, y
eventualmente la activación del canal fotoquímico correspondiente a dicha intersección
cónica.
Además, se ha abordado el estudio de la continuidad del espacio de intersección de
manera cualitativa, es decir, se ha estudiado qué intersecciones cónicas (mínimas en
energía) pertenecen al mismo subespacio continuo de cruce o degeneración energética
(S1/S0).
Este estudio es complejo ya que no existen métodos implementados en paquetes de
cálculo que permitan el estudio de estas zonas de la SEP. Sin embargo, es posible
estudiar cualitativamente este problema recurriendo al análisis del espacio en el que
desaparece la degeneración energética (espacio de ramificación) de las CoIn entre las
que se pretende estudiar si existe o no esta relación. El planteamiento para llevar a cabo
este estudio es muy sencillo. Para que exista un camino (estamos interesados en el de
mínima energía) dentro del espacio de intersección que una dos CoIn, éste debe ser
perpendicular al espacio de ramificación. Por lo tanto, la dirección inicial de esta
trayectoria que une dos CoIn es conocida en tanto que debe ser ortogonal al espacio de
ramificación de la primera intersección cónica. Asimismo ocurre con la dirección de
llegada de esta trayectoria a la segunda CoIn. Sabiendo esto podemos preguntarnos en
qué medida es viable una trayectoria que una ambas CoIn y que cumpla las restricciones
descritas. Si no tenemos más información que la estructura de las intersecciones cónicas
que queremos relacionar y los vectores que definen el espacio de ramificación de cada
una de ellas, podemos proceder al estudio del problema como se explicará a
continuación. Para ello se usará el esquema 5.3 como ejemplo ilustrativo.
En este esquema se ha representado la posición de las CoIn en el espacio de
coordenadas internas de la molécula mediante dos puntos azules. Las flechas negras
representan la dirección del espacio de ramificación (por sencillez se ha representado
por un vector solamente y de esta manera no es necesario recurrir a representaciones
tridimensionales) tanto en CoIn1 como en CoIn2. Las flechas grises representan la
dirección de este mismo subespacio en el espacio de coordenadas. Una curva que
pertenece al espacio de intersección y que une dos CoIn mínimas (1 y 2) debe ser
perpendicular en todo momento, como ya se ha indicado, al espacio de ramificación, por
lo tanto, si como ocurre en el primer caso (caso (a) en el esquema 5.3) la trayectoria
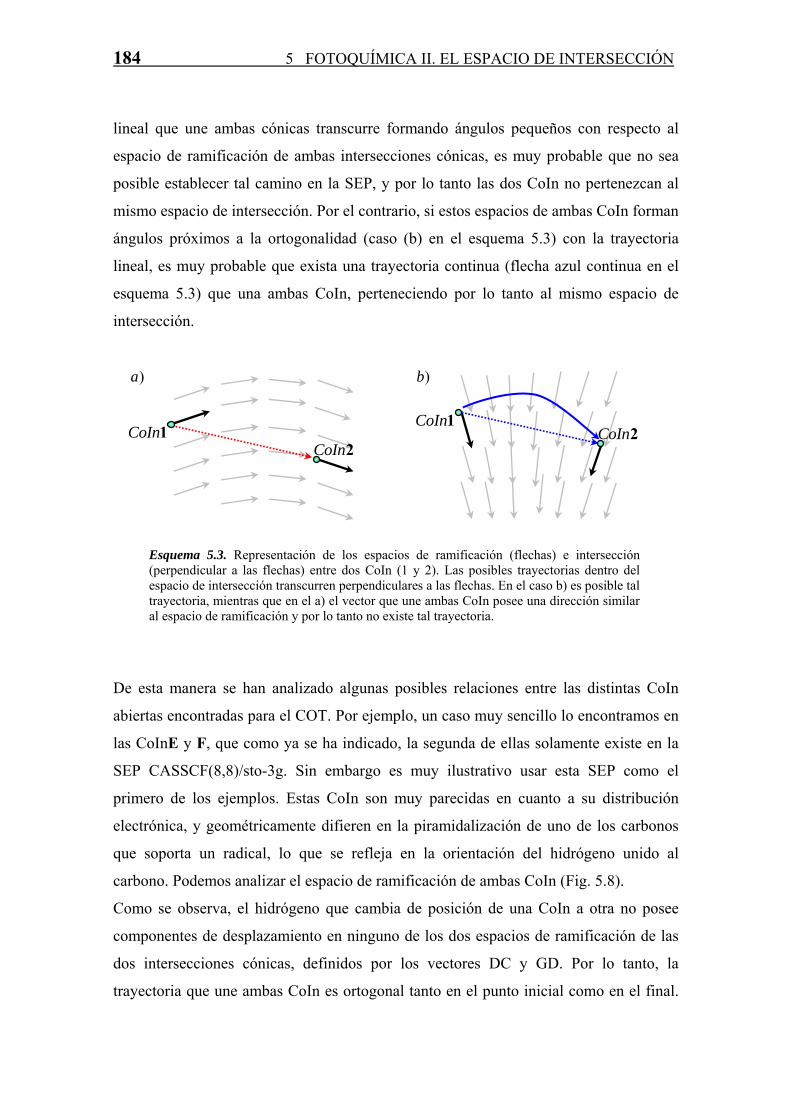
184 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
lineal que une ambas cónicas transcurre formando ángulos pequeños con respecto al
espacio de ramificación de ambas intersecciones cónicas, es muy probable que no sea
posible establecer tal camino en la SEP, y por lo tanto las dos CoIn no pertenezcan al
mismo espacio de intersección. Por el contrario, si estos espacios de ambas CoIn forman
ángulos próximos a la ortogonalidad (caso (b) en el esquema 5.3) con la trayectoria
lineal, es muy probable que exista una trayectoria continua (flecha azul continua en el
esquema 5.3) que una ambas CoIn, perteneciendo por lo tanto al mismo espacio de
intersección.
CoIn1CoIn2
CoIn1CoIn2
)a )b
Esquema 5.3. Representación de los espacios de ramificación (flechas) e intersección (perpendicular a las flechas) entre dos CoIn (1 y 2). Las posibles trayectorias dentro del espacio de intersección transcurren perpendiculares a las flechas. En el caso b) es posible tal trayectoria, mientras que en el a) el vector que une ambas CoIn posee una dirección similar al espacio de ramificación y por lo tanto no existe tal trayectoria.
De esta manera se han analizado algunas posibles relaciones entre las distintas CoIn
abiertas encontradas para el COT. Por ejemplo, un caso muy sencillo lo encontramos en
las CoInE y F, que como ya se ha indicado, la segunda de ellas solamente existe en la
SEP CASSCF(8,8)/sto-3g. Sin embargo es muy ilustrativo usar esta SEP como el
primero de los ejemplos. Estas CoIn son muy parecidas en cuanto a su distribución
electrónica, y geométricamente difieren en la piramidalización de uno de los carbonos
que soporta un radical, lo que se refleja en la orientación del hidrógeno unido al
carbono. Podemos analizar el espacio de ramificación de ambas CoIn (Fig. 5.8).
Como se observa, el hidrógeno que cambia de posición de una CoIn a otra no posee
componentes de desplazamiento en ninguno de los dos espacios de ramificación de las
dos intersecciones cónicas, definidos por los vectores DC y GD. Por lo tanto, la
trayectoria que une ambas CoIn es ortogonal tanto en el punto inicial como en el final.

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 185
Si no ocurren grandes cambios en el espacio de ramificación a lo largo de la trayectoria
que debe “unir” ambas CoIn, es muy probable que ambas pertenezcan a la misma región
de intersecciones cónicas y por lo tanto al mismo espacio de intersección. De hecho, se
puede ver cómo los vectores DC y GD en las dos CoIn son muy parecidos, por lo que es
muy improbable que no exista una trayectoria (en el espacio de intersección) que una
ambas CoIn.
DC GD DC GD
Fig. 5.8. Vectores DC (izquierda) y GD (derecha) para la CoinF (izquierda) y E (derecha). Como se puede observar, el hidrógeno que cambia de posición en una intersección cónica respecto a la otra (señalado con una flecha granate) no posee componente alguna en el “espacio de ramificación”, definido por los vectores DC y GD, para ninguna de las dos CoIn.
De hecho, realizando un “scan” relajado entre las dos estructuras, se puede comprobar
que existe dicho espacio de intersección continuo, y que además existe un máximo,
correspondiente al estado de transición entre ambos (Fig. 5.9).
3.30
1.45 1.39
1.441.47 3.541.43
1.401.44
1.463.421.44
1.391.46
1.47
CoInECoInF CoIn E-FTS
Fig. 5.9. Intersecciones cónicas E y F así como la TS entre ambas (CoInTSE-F) dentro del espacio de intersección (S1/S0). Las distancias se dan en Angstroms.
Más interesante es establecer este tipo de relación entre dos CoIn que tienen un arreglo
electrónico distinto, es el caso por ejemplo de las CoInA y C.

186 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
El procedimiento general en todos los cálculos ha sido el siguiente: primero se orientan
las geometrías moleculares correspondientes a las CoIn optimizadas en base a sus ejes
de inercia con el programa (ver Apéndice B). Posteriormente, se genera el vector de
posición desde la primera a la segunda intersección cónica, en coordenadas cartesianas.
Este vector da la dirección de la trayectoria más corta (en estas coordenadas) que une
ambas CoIn. Puesto que se conocen los vectores “acoplamiento derivativo” (DC) y
“diferencia de gradientes” (GD) del cálculo de optimización de ambas intersecciones
cónicas, es posible calcular el ángulo que forman éstos (tanto para la primera como para
la segunda CoIn) con el primero de los vectores (el correspondiente a la trayectoria
lineal entre las dos CoIn). De esta manera se obtiene el ángulo formado entre la
trayectoria recta que une ambas CoIn y el espacio de ramificación. Si este ángulo está
próximo a la ortogonalidad, muy probablemente exista una trayectoria continua en el
espacio de intersección que una ambas CoIn, por el contrario, si esto no ocurre,
probablemente pertenezcan a espacios de intersección distintos.
Para las CoInA y C, se ha procedido como se acaba de describir. Es fácil comprobar por
inspección directa que los vectores DC y GD en ambas CoIn son muy parecidos (Fig.
5.10). El vector en el espacio de configuraciones nucleares que une ambas CoIn forma
un ángulo de 80.1º y 88.1º con los vectores DC y GD de la CoInA, mientras que con los
mismos de la CoInC forma 91.1º y 88.3º respectivamente.
DC GD DC GD
Fig. 5.10. Vectores DC y GD de las CoInA (izquierda) y C (derecha).
Estos resultados nos hacen postular la existencia de un camino de mínima energía que
une ambas CoIn en el espacio de intersección. Para este caso, se ha realizado un scan
relajado en el subespacio de intersección, encontrando un máximo a lo largo de este
espacio continuo de degeneración energética, que corresponde al estado de transición

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 187
en dicho subespacio. Este estado de transición dentro del espacio de intersección se
halla muy próximo a la CoInC, por lo que posee una similitud (estructural y electrónica)
mucho mayor con esta CoIn que con la CoInA (Fig. 5.11).
CoInACoInC CoIn A-CTS
3.50
2.51
3.03
2.42
3.09
2.39
Fig. 5.11. Estructura de las CoIn C y A, así como de la intersección cónica (aproximada) estado de transición entre ambas en el espacio de intersección, obtenida a partir de un “scan” relajado en dicho espacio. Las distancias se dan en angstroms
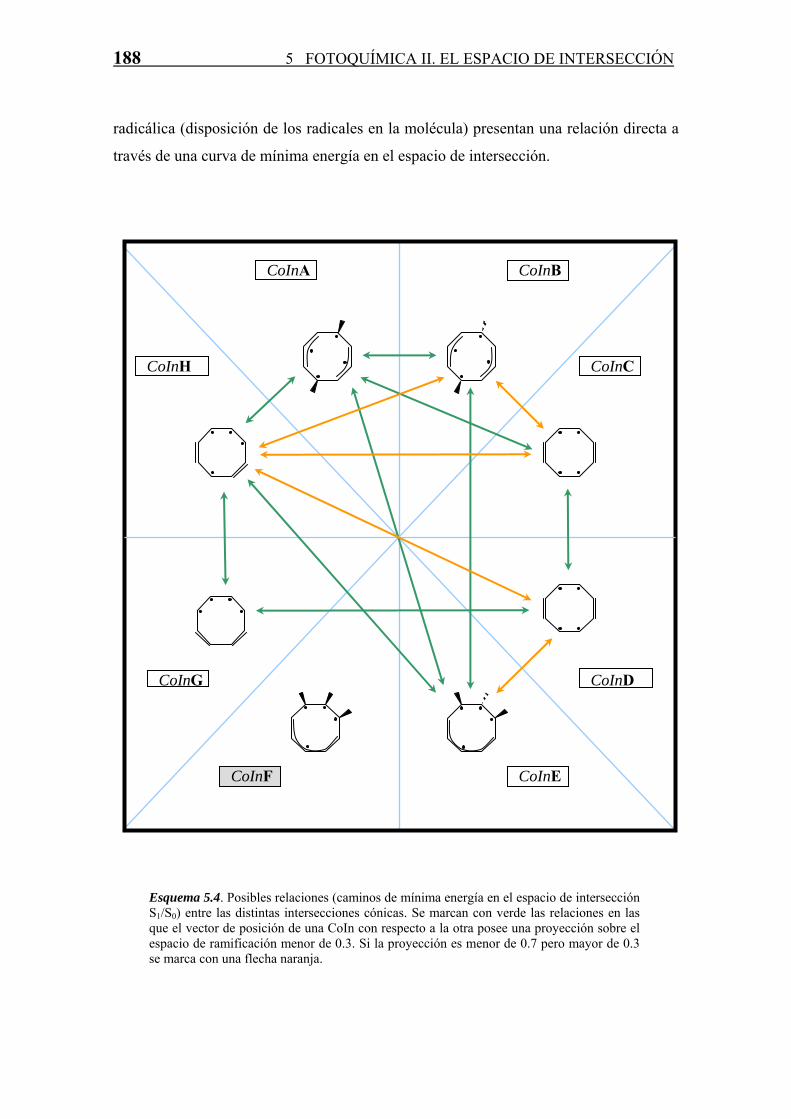
El estudio de las posibles uniones entre las distintas CoIn(A-H) por caminos de mínima
energía en el espacio de intersección se ha llevado a cabo usando la metodología antes
descrita. Así para los casos en que las dos proyecciones del vector que une ambas CoIn
en el espacio de ramificación es menor de 0.3 (30%) se ha considerado como probable
la existencia de dicho camino. Por el contrario si la proyección sobre dicho espacio es
menor que 0.7 pero mayor que 0.3 no se puede asegurar claramente nada acerca de la
existencia de tal camino. Por último, si la proyección es mayor de 0.7 (70%) muy
probablemente no existe un camino de mínima energía en el espacio de intersección que
una ambas intersecciones cónicas. Los resultados se resumen en el esquema 5.4. En ella,
los caminos establecidos claramente (proyección menor de 0.3) se marcan con una
flecha verde, mientras que los caminos que presentan dudas (proyección entre 0.3 y 0.7)
se marcan con una flecha naranja. Es importante destacar que entre todas las
intersecciones cónicas encontradas que pertenecen a este primer tipo (sin enlaces σ
intra-anulares) existe una relación directa o indirecta, por lo que es de esperar que todas
estas CoIn pertenezcan a un mismo espacio de intersección. Además, otra conclusión
importante a la que se llega es que intersecciones cónicas con una misma estructura
188 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
radicálica (disposición de los radicales en la molécula) presentan una relación directa a
través de una curva de mínima energía en el espacio de intersección.
CoInH
CoInA CoInB
CoInC
CoInD
CoInECoInF
CoInG
Esquema 5.4. Posibles relaciones (caminos de mínima energía en el espacio de intersección S1/S0) entre las distintas intersecciones cónicas. Se marcan con verde las relaciones en las que el vector de posición de una CoIn con respecto a la otra posee una proyección sobre el espacio de ramificación menor de 0.3. Si la proyección es menor de 0.7 pero mayor de 0.3 se marca con una flecha naranja.

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 189
2. CoIn Bicíclicas (con un enlace σ intra-anular).
Se han estudiado los correspondientes biciclos (un solo enlace sigma intra-anular). Para
ello, se han establecido los arreglos tetrarradicálicos posibles que se pueden plantear
con un enlace intra-anular (Esquema 5.5), para posteriormente localizar aquellos que se
corresponden con intersecciones cónicas S1/S0.
Esquema 5.5. Posibles estructuras electrónicas tetrarradicálicas distintas en biciclos (un solo enlace sigma intra-anular) derivados del COT. Está enmarcada la estructura tetrarradicálica que no se corresponde con una CoIn mínima en energía.
Al igual que para las intersecciones cónicas cíclicas sin enlaces intranulares, para las
que poseen un solo enlace sigma intra-anular se han estudiado todos los posibles
arreglos tetrarradicálicos (Esquema 5.5). Un total de diez posibles arreglos distintos se
han encontrado, los cuales se pueden dividir en dos tipos: los tetrarradicales localizados
sobre cuatro centros, y aquellos en los que uno de los radicales es alílico. De las diez
estructuras tetrarradicálicas formalmente distintas, se han encontrado nueve CoIn que se
corresponden con otras tantas de estas estructuras. Por el contrario, una de ellas no se ha
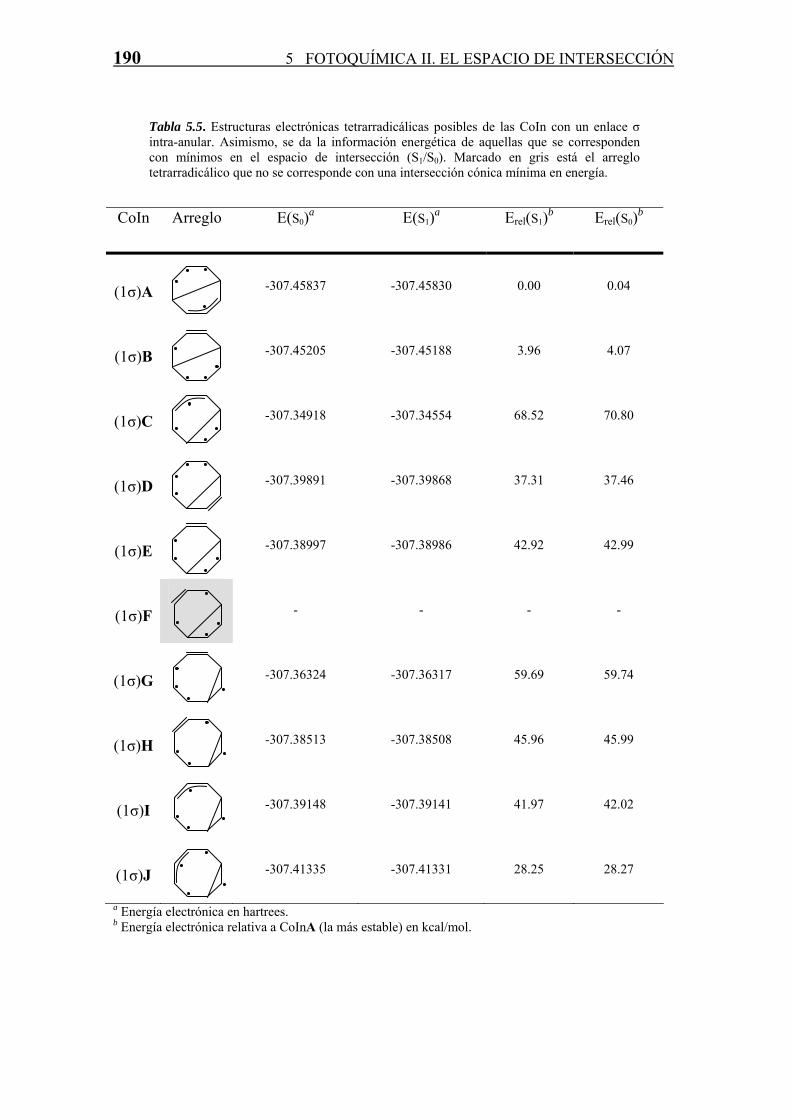
podido identificar como mínimo en el espacio de intersección (S1/S0) (Tabla 5.5). Esta
estructura, que no se corresponde con una CoIn de mínima energía ((1σ)F) posee un
doble enlace C=C flanqueado por dos birradicales aislados. Probablemente la escasa
interacción de los radicales aislados junto con la también casi nula interacción con los
dos radicales localizados sobre el etileno, ya sea a través de enlace o a través del
espacio, hacen que no exista la correspondiente intersección cónica mínima en energía.
190 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Tabla 5.5. Estructuras electrónicas tetrarradicálicas posibles de las CoIn con un enlace σ intra-anular. Asimismo, se da la información energética de aquellas que se corresponden con mínimos en el espacio de intersección (S1/S0). Marcado en gris está el arreglo tetrarradicálico que no se corresponde con una intersección cónica mínima en energía.
CoIn Arreglo E(S0)a E(S1)a Erel(S1)b Erel(S0)b
(1σ)A
-307.45837
-307.45830
0.00
0.04
(1σ)B
-307.45205
-307.45188
3.96
4.07
(1σ)C
-307.34918
-307.34554
68.52
70.80
(1σ)D
-307.39891
-307.39868
37.31
37.46
(1σ)E
-307.38997
-307.38986
42.92
42.99
(1σ)F
-
-
-
-
(1σ)G
-307.36324
-307.36317
59.69
59.74
(1σ)H
-307.38513
-307.38508
45.96
45.99
(1σ)I
-307.39148
-307.39141
41.97
42.02
(1σ)J
-307.41335
-307.41331
28.25
28.27
a Energía electrónica en hartrees. b Energía electrónica relativa a CoInA (la más estable) en kcal/mol.

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 191
Las estructuras de las intersecciones cónicas encontradas son muy diversas. La más
estable de todas ellas (la CoIn(1σ)A) posee por un lado la menor tensión de anillo debido
a que los dos anillos de la molécula son de cinco eslabones, y por otro lado, uno de los
radicales está deslocalizado en un alilo (lo cual estabiliza la CoIn), mientras que los
otros tres se encuentran formando la estructura -(CH)3- “retorcido” ya descrita en
intersecciones cónicas en polienos (ver por ejemplo Celani et al., 1995b). La segunda
más estable, a tan solo unas ~3 kcal/mol de la primera es la CoIn(1σ)B, la cual no posee
la estabilidad procurada por la deslocalización del radical alílico que presenta la
anterior, pero que se estabiliza debido a la interacción que existe entre el radical
solitario situado en uno de los dos anillos de cinco eslabones con uno de los radicales de
la estructura -(CH)3- “retorcida” del otro.
El resto de CoIn poseen una energía claramente mayor (desde ~28 hasta ~70 kcal/mol
más inestables que la CoInA) y sus estructuras se presentan en la figura 5.12.
En general, las CoIn que presentan un ciclo de cuatro eslabones (y por lo tanto otro de
seis) son más estables que las que presentan uno de tres (y otro de siete), debido a la
tensión mayor del anillo. Sin embargo esta comparación no se puede establecer
claramente ya que no existen arreglos electrónicos comparables para una y otra
disposición. Curiosamente, una de las CoIn más bajas ((1σ)J) en energía posee un anillo
ciclopropano (de tres eslabones). La alta estabilización de esta estructura (CoIn(1σ)J) es
debida no solamente a la deslocalización en un grupo alílico de uno de los radicales,
sino también a la interacción espacial entre dos de los birradicales solitarios que
presentan una disposición análoga a la CoInA en los ciclos sin enlaces intra-anulares, y
que a su vez es la CoIn de más baja energía de todas las encontradas.
Las propiedades estructurales y electrónicas de cada intersección cónica condicionan los
fotoproductos de la reacción diabática que transcurre a través de estos cruces diabáticos
entre los estados S1 y S0. En cualquier caso, la formación de fotoproductos se discutirá
en detalle más adelante (ver punto 5.4.3).
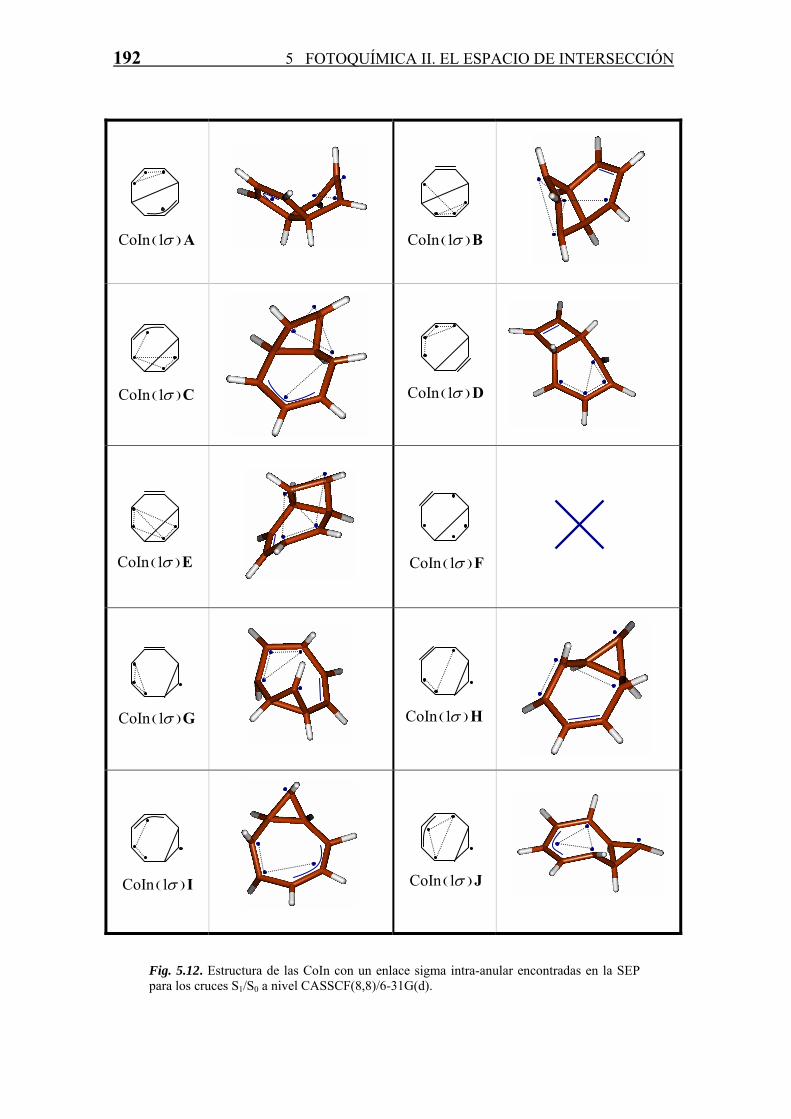
192 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
( )CoIn 1σ A ( )CoIn 1σ B
( )CoIn 1σ D( )CoIn 1σ C
( )CoIn 1σ E ( )CoIn 1σ F
( )CoIn 1σ H( )CoIn 1σ G
( )CoIn 1σ J( )CoIn 1σ I
Fig. 5.12. Estructura de las CoIn con un enlace sigma intra-anular encontradas en la SEP para los cruces S1/S0 a nivel CASSCF(8,8)/6-31G(d).

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 193
3. CoIn Tricíclicas (con dos enlaces σ intra-anulares)
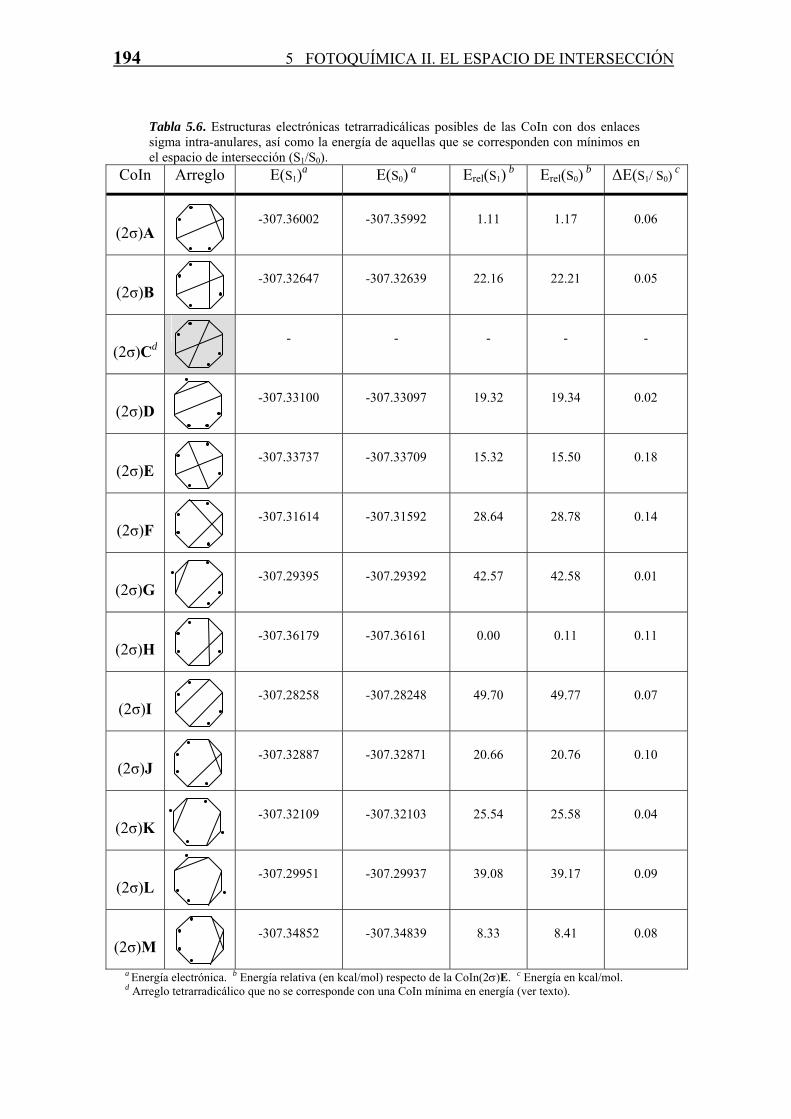
En total existen trece estructuras tetrarradicálicas distintas formalmente (Esquema 5.6).
De las trece estructuras solamente una de ellas no se corresponde con una CoIn de
mínima energía.
Esquema 5.6. Posibles estructuras electrónicas tetrarradicálicas distintas derivadas del COT y que posee dos enlaces sigma intra-anulares.
Al contrario que en las situaciones anteriores (uno o ningún enlace sigma intra-anular),
para este caso no puede darse deslocalización π-electrónica de los radicales en ninguna
agrupación posible, ya que una vez formados los dos enlaces sigma, únicamente quedan
cuatro centros libres para albergar los correspondientes radicales.
Por lo tanto, dos son los factores que deben marcar la estabilidad de las distintas
intersecciones cónicas, estos son, los factores estéricos derivados fundamentalmente de
la tensión de enlace de los ciclos formados, y la estabilización de los radicales debida a
la interacción entre ellos fundamentalmente a través del espacio (no a través de enlace).
Así, la CoIn(2σ)E es la más estable (Tabla 5.6) , ya que los cuatro ciclos que posee son
de cinco eslabones, y por lo tanto la tensión estérica debido a la tensión anular está
minimizada, ya que los anillos de cinco eslabones son los más estables en cuanto a su
tensión anular para hidrocarburos saturados. Por otro lado, los cuatro carbonos que
soportan los radicales están situados en el mismo plano a una distancia de 2.29 Å, por lo
que presentan una cierta estabilización debida a dicha interacción.
El arreglo electrónico (2σ)C que no se corresponde con una intersección cónica mínima
en energía, presenta los cuatro radicales agrupados de dos en dos, y muy separados en el
espacio, lo que hace a esta estructura muy inestable.
194 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Tabla 5.6. Estructuras electrónicas tetrarradicálicas posibles de las CoIn con dos enlaces sigma intra-anulares, así como la energía de aquellas que se corresponden con mínimos en el espacio de intersección (S1/S0).
CoIn Arreglo E(S1)a E(S0) a Erel(S1) b Erel(S0) b ∆E(S1/ S0) c
(2σ)A
-307.36002
-307.35992
1.11
1.17
0.06
(2σ)B
-307.32647
-307.32639
22.16
22.21
0.05
(2σ)Cd
-
-
-
-
-
(2σ)D
-307.33100
-307.33097
19.32
19.34
0.02
(2σ)E
-307.33737
-307.33709
15.32
15.50
0.18
(2σ)F
-307.31614
-307.31592
28.64
28.78
0.14
(2σ)G
-307.29395
-307.29392
42.57
42.58
0.01
(2σ)H
-307.36179
-307.36161
0.00
0.11
0.11
(2σ)I
-307.28258
-307.28248
49.70
49.77
0.07
(2σ)J
-307.32887
-307.32871
20.66
20.76
0.10
(2σ)K
-307.32109
-307.32103
25.54
25.58
0.04
(2σ)L
-307.29951
-307.29937
39.08
39.17
0.09
(2σ)M
-307.34852
-307.34839
8.33
8.41
0.08
a Energía electrónica. b Energía relativa (en kcal/mol) respecto de la CoIn(2σ)E. c Energía en kcal/mol. d Arreglo tetrarradicálico que no se corresponde con una CoIn mínima en energía (ver texto).

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 195
La estructura detallada de las CoIn y su relación con la reactividad fotoquímica se
abordan con más profundidad en el próximo punto.
5.4.3 Relajación en S0. Formación de Fotoproductos
Cada CoIn, como ya se ha comentado a lo largo de la Tesis, es una estructura que sirve
de “paso” en una reacción diabática entre dos estados electrónicos. La estructura y
propiedades de estas CoIn marcan la naturaleza de los fotoproductos formados en la
reacción. Para todas las CoIn (S1/S0) derivadas del COT se han estudiado, en base a las
propiedades del espacio de ramificación definido por los vectores acoplamiento
derivativo (DC) y diferencia de gradientes (GD), los posibles productos formados a
partir de las mismas en el proceso de relajación vibracional en S0. Este estudio se divide
según la clasificación de número del enlaces σ intra-anular que posee la intersección
cónica.
· Intersecciones Cónicas sin enlaces σ intra-anulares.
Algunas de estas CoIn están involucradas en reacciones fotoquímicas diabáticas que ya
han sido estudiadas teóricamente (ver Capítulo cuarto de esta Tesis y (Garavelli et al.,
2001;2002)), sin embargo la mayoría de ellas no han sido objeto de estudio.
A continuación se presenta un estudio en base a las propiedades del espacio de
ramificación tal y como ha sido descrito en el segundo capítulo. Para llevar a cabo tal
estudio se ha usado el programa RELAXATION (ver Apéndice D) gracias al cual se
han podido estudiar los posibles caminos de relajación vibracional a través de cada CoIn
en base a las propiedades de los vectores DC y GD así como de la proyección sobre
dicho espacio de distintos vectores que definen direcciones iniciales de relajación.
La CoInA es la más estable de todas las intersecciones cónicas de fórmula (CH)8
encontradas, y de hecho, como ya se ha visto en esta misma Sección 5.4, es la que
emplea el COT en el proceso de decaimiento al estado fundamental (S0) tras la
excitación al estado S2 y posterior decaimiento a través de una CoIn S2/S1 al estado
excitado S1 (geometría de equilibrio plana D8h).
La CoInB posee una estructura electrónica similar a la A, sin embargo, tiene una energía
considerablemente mayor (~46 kcal/mol) debido en gran medida a la estructura más
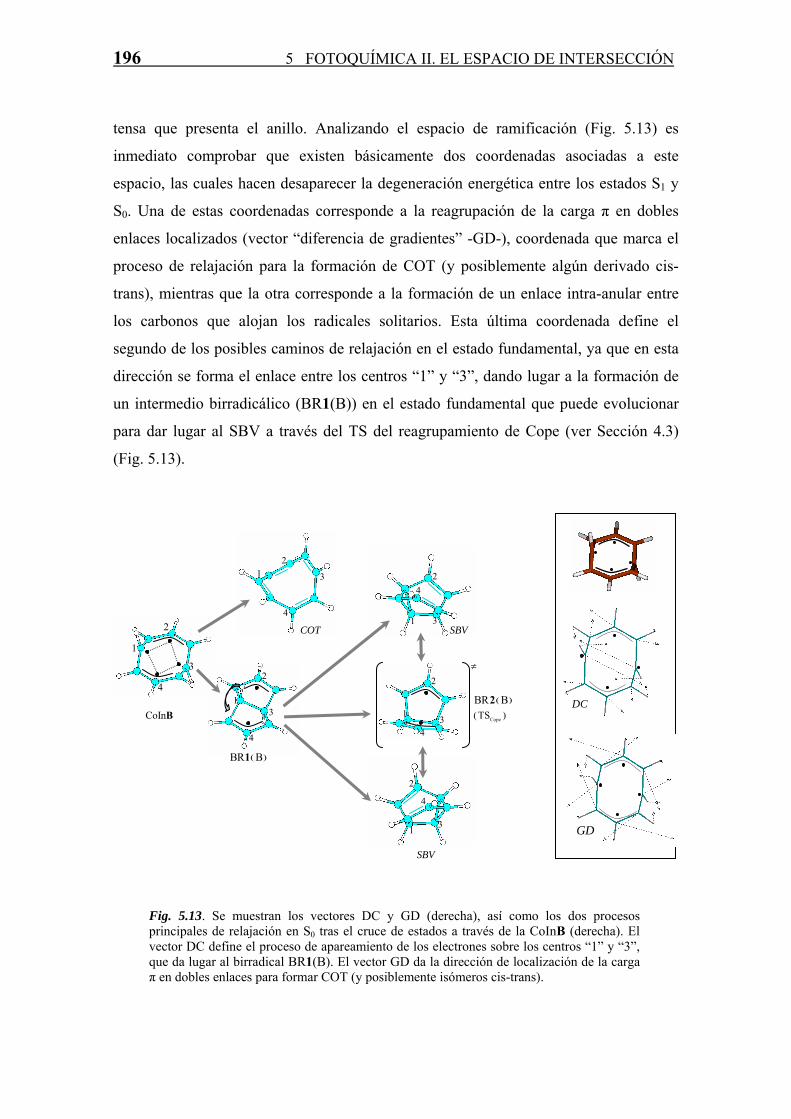
196 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
tensa que presenta el anillo. Analizando el espacio de ramificación (Fig. 5.13) es
inmediato comprobar que existen básicamente dos coordenadas asociadas a este
espacio, las cuales hacen desaparecer la degeneración energética entre los estados S1 y
S0. Una de estas coordenadas corresponde a la reagrupación de la carga π en dobles
enlaces localizados (vector “diferencia de gradientes” -GD-), coordenada que marca el
proceso de relajación para la formación de COT (y posiblemente algún derivado cis-
trans), mientras que la otra corresponde a la formación de un enlace intra-anular entre
los carbonos que alojan los radicales solitarios. Esta última coordenada define el
segundo de los posibles caminos de relajación en el estado fundamental, ya que en esta
dirección se forma el enlace entre los centros “1” y “3”, dando lugar a la formación de
un intermedio birradicálico (BR1(B)) en el estado fundamental que puede evolucionar
para dar lugar al SBV a través del TS del reagrupamiento de Cope (ver Sección 4.3)
(Fig. 5.13).
CoInB
BR1( )B
( )Cope
BR2( )TS
B
≠
COT
SBV
SBV
DC
GD
1
2
3
4
12
3
4
1
2
34
1
2
3
4
1
2
3
4
( )BR B1
( )BR B21
2
3
4
Fig. 5.13. Se muestran los vectores DC y GD (derecha), así como los dos procesos principales de relajación en S0 tras el cruce de estados a través de la CoInB (derecha). El vector DC define el proceso de apareamiento de los electrones sobre los centros “1” y “3”, que da lugar al birradical BR1(B). El vector GD da la dirección de localización de la carga π en dobles enlaces para formar COT (y posiblemente isómeros cis-trans).
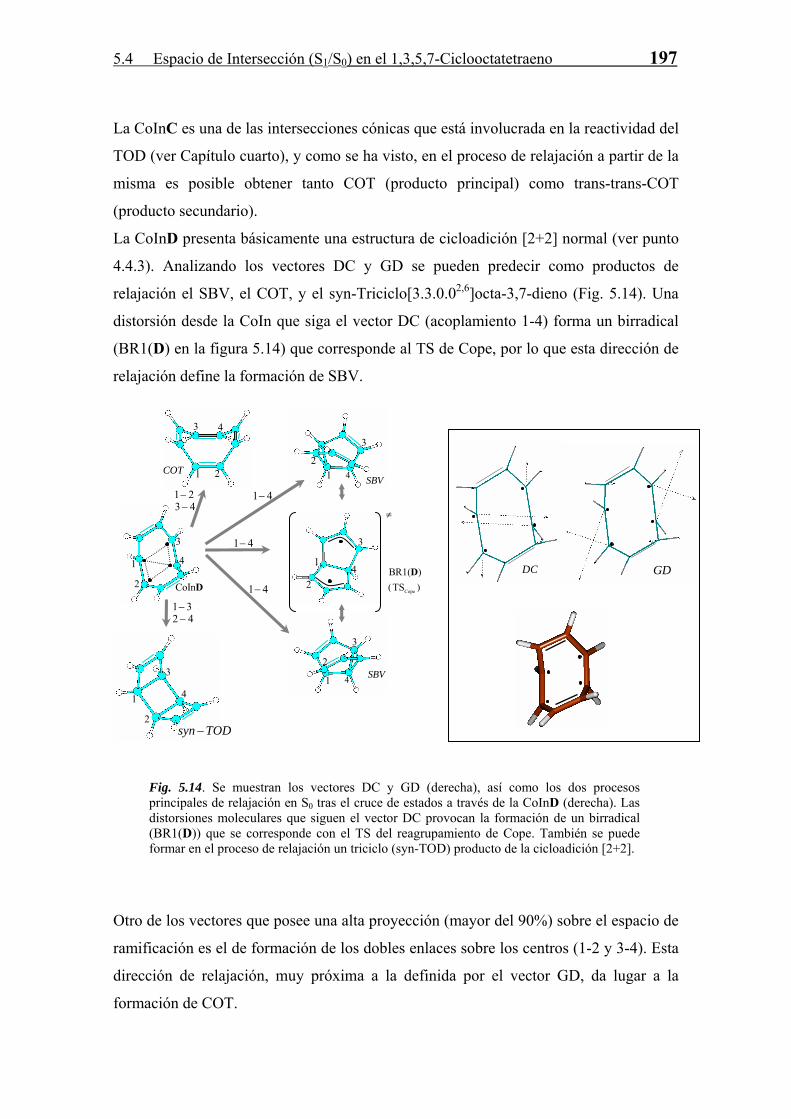
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 197
La CoInC es una de las intersecciones cónicas que está involucrada en la reactividad del
TOD (ver Capítulo cuarto), y como se ha visto, en el proceso de relajación a partir de la
misma es posible obtener tanto COT (producto principal) como trans-trans-COT
(producto secundario).
La CoInD presenta básicamente una estructura de cicloadición [2+2] normal (ver punto
4.4.3). Analizando los vectores DC y GD se pueden predecir como productos de
relajación el SBV, el COT, y el syn-Triciclo[3.3.0.02,6]octa-3,7-dieno (Fig. 5.14). Una
distorsión desde la CoIn que siga el vector DC (acoplamiento 1-4) forma un birradical
(BR1(D) en la figura 5.14) que corresponde al TS de Cope, por lo que esta dirección de
relajación define la formación de SBV.
≠
SBV
CoInD ( )Cope
BR1( )TS
D
SBV
DC GD
COT
syn TOD−
1 23 4−−
1
2
3
4
1 2
3 4
1
2
3
4
12
3
4
1
2
3
4
1 32 4−−
1 4−
1 4−
1 4−
1
2
3
4
Fig. 5.14. Se muestran los vectores DC y GD (derecha), así como los dos procesos principales de relajación en S0 tras el cruce de estados a través de la CoInD (derecha). Las distorsiones moleculares que siguen el vector DC provocan la formación de un birradical (BR1(D)) que se corresponde con el TS del reagrupamiento de Cope. También se puede formar en el proceso de relajación un triciclo (syn-TOD) producto de la cicloadición [2+2].
Otro de los vectores que posee una alta proyección (mayor del 90%) sobre el espacio de
ramificación es el de formación de los dobles enlaces sobre los centros (1-2 y 3-4). Esta
dirección de relajación, muy próxima a la definida por el vector GD, da lugar a la
formación de COT.

198 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Por último, el vector GD (en el sentido opuesto al representado en la figura 5.14), define
una distorsión molecular que es la responsable de la formación del producto de
cicloadición [2+2], el syn-Triciclo[3.3.0.02,6]octa-3,7-dieno (syn-TOD en la figura
5.14).
Las CoInE ha sido estudiada teóricamente en el marco de la reactividad del COT,
proponiéndose como cruce diabático en una ruta impedida en la fotoquímica del COT
(Garavelli et al., 2002). En base al estudio del espacio de ramificación se predice la
formación de dos productos mayoritarios (Fig. 5.15) como se verá a continuación.
DC GD
COT
CoInE
1
23
4
1
234
1
2 3
4
( )BR E11 3−
( )1 3− −
Fig. 5.15. Se muestran los vectores “acoplamiento derivativo” (DC) y “diferencia de gradientes” (GD) (derecha), así como los dos procesos principales de relajación en S0 tras el cruce de estados a través de la CoInE (derecha).
Es el vector GD en este caso el que define los dos principales caminos de relajación en
S0, ya que lleva asociado el acoplamiento entre los electrones “1” y “3”. Por lo tanto,
direcciones iniciales de relajación próximas al vector GD (Fig. 5.15) dan lugar al
alejamiento de los carbonos “1” y “3”, acoplando progresivamente electrones contiguos
en el anillo para dar dobles enlaces localizados y formar COT, en analogía con los
productos formados desde CoIn de hidrocarburos cíclicos insaturados que presentan una
estructura similar a ésta con un grupo -(CH)3- “retorcido” (Palmer, 1993). Por otro lado,
en la dirección opuesta a la representada en la figura 5.15 por el vector GD, los

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 199
electrones “1” y “3” se acoplan, dando lugar a la formación del intermedio birradical
ciclopropano denominado BR1(E) en la figura 5.15.
Usando el método IRD (Celani et al., 1995) es posible localizar otros productos
secundarios de relajación vibracional en S0 (Garavelli et al., 2002) aparte de los
determinados cualitativamente con este análisis.
La CoInG presenta una estructura con un grupo trirradicálico -(CH)3- “retorcido”
análogo a la que presenta la correspondiente intersección cónica (S1/S0) del benceno
(Palmer, 1993). Sin embargo, para la CoInG, un cuarto centro radicálico se halla
contiguo a esta estructura, en lugar de deslocalizado en un grupo alílico. La estructura
-(CH)3- la conforman los centros 2-3-4 (Fig. 5.16), siendo el carbono “1” el cuarto
centro radicálico. El análisis del espacio de ramificación es más complejo que en los
casos anteriores, y la simple inspección de los vectores DC y GD no esclarece los
posibles productos de relajación vibracional en el estado fundamental.
DC GD12
34
CoInG
COTt −
1 23
4
1
2
34
2 4−
1 2−
( )BR G1
( )Proy 1-2( )Proy 2-4
Fig. 5.16. Se muestran los vectores DC y GD y las proyecciones de los vectores de formación de los enlaces “2-4” (Proy(2-4)) y “1-2” (Proy(1-2)) (derecha), así como los dos procesos principales de relajación en S0 tras el cruce de estados a través de la CoInE que corresponden a la formación del COT y del birradical BR1(G) (izquierda).
Proyectando sobre el espacio de ramificación el vector de formación del enlace 2-4 se
obtiene el vector Proy(2-4) (Fig. 5.16). La proyección de este vector es muy alta (~80%)
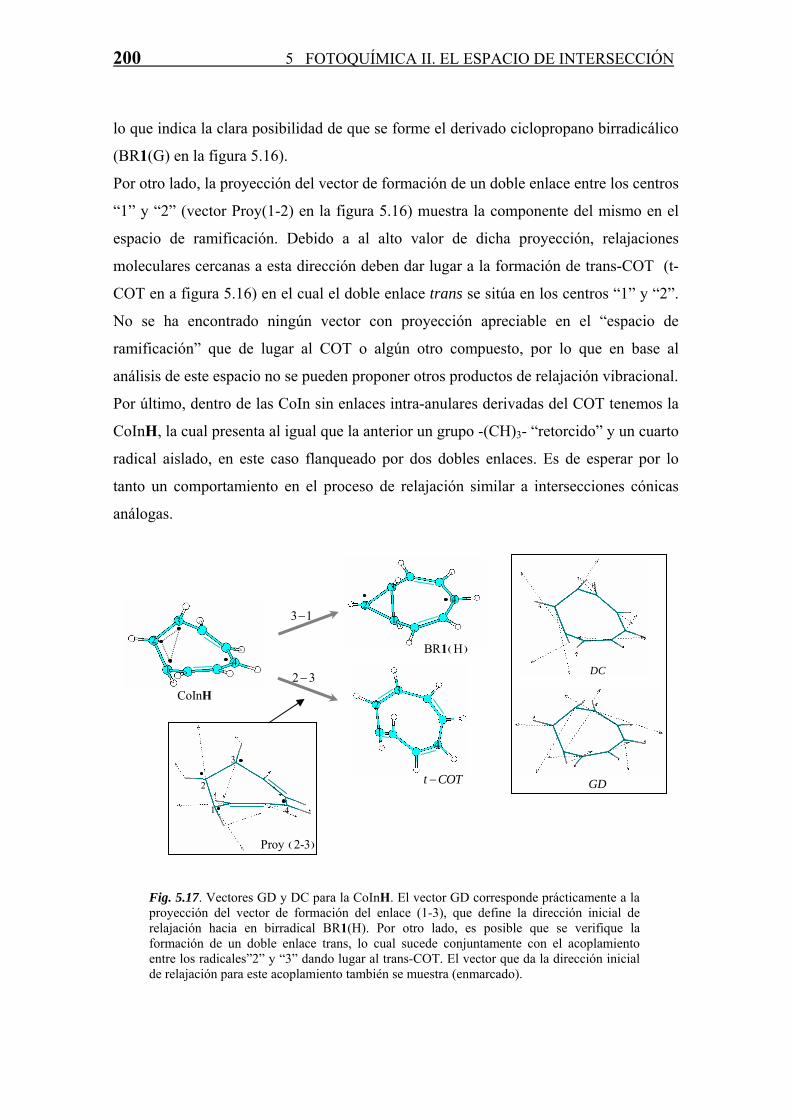
200 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
lo que indica la clara posibilidad de que se forme el derivado ciclopropano birradicálico
(BR1(G) en la figura 5.16).
Por otro lado, la proyección del vector de formación de un doble enlace entre los centros
“1” y “2” (vector Proy(1-2) en la figura 5.16) muestra la componente del mismo en el
espacio de ramificación. Debido a al alto valor de dicha proyección, relajaciones
moleculares cercanas a esta dirección deben dar lugar a la formación de trans-COT (t-
COT en a figura 5.16) en el cual el doble enlace trans se sitúa en los centros “1” y “2”.
No se ha encontrado ningún vector con proyección apreciable en el “espacio de
ramificación” que de lugar al COT o algún otro compuesto, por lo que en base al
análisis de este espacio no se pueden proponer otros productos de relajación vibracional.
Por último, dentro de las CoIn sin enlaces intra-anulares derivadas del COT tenemos la
CoInH, la cual presenta al igual que la anterior un grupo -(CH)3- “retorcido” y un cuarto
radical aislado, en este caso flanqueado por dos dobles enlaces. Es de esperar por lo
tanto un comportamiento en el proceso de relajación similar a intersecciones cónicas
análogas.
1
2
3
4
1
23
4
3 1−
2 3−
12
34
CoInH
t COT−
( )BR H1
DC
GD
1
2
3
4
( )Proy 2-3
Fig. 5.17. Vectores GD y DC para la CoInH. El vector GD corresponde prácticamente a la proyección del vector de formación del enlace (1-3), que define la dirección inicial de relajación hacia en birradical BR1(H). Por otro lado, es posible que se verifique la formación de un doble enlace trans, lo cual sucede conjuntamente con el acoplamiento entre los radicales”2” y “3” dando lugar al trans-COT. El vector que da la dirección inicial de relajación para este acoplamiento también se muestra (enmarcado).

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 201
De esta manera, para la CoInH, existen dos coordenadas (vectores) asociadas con
sendas direcciones iniciales de relajación que poseen una alta proyección en el espacio
de ramificación. Por un lado el acoplamiento de los radicales “1” y “3”, cuya
coordenada coincide prácticamente con la determinada por el vector GD (Fig. 5.17), es
responsable de la formación de un birradical ciclopropano BR1(H).
Por otro lado, el vector de acoplamiento entre los radicales “2” y “3”, que también
posee una alta proyección sobre el espacio de ramificación, lleva asociado una gran
contribución en la formación de un enlace doble trans entre el carbono “1” y el carbono
adyacente al mismo no denotado con ningún número (Fig. 5.17). Esta contribución a la
formación del doble enlace permite establecer un segundo producto de relajación, el
trans-COT (t-COT). Además, los desplazamientos moleculares asociados a esta
coordenada se pueden apreciar en el vector Proy (2-3) de la figura 5.17, que
corresponde a la proyección del vector de formación del doble enlace (2-3) y de torsión
del diedro correspondiente para la formación del doble enlace trans sobre el espacio de
ramificación definido por los vectores DC y GD. Ningún otro producto de relajación
(por ejemplo el COT) posee un vector de dirección de relajación inicial con una
proyección grande sobre los vectores DC y GD, aunque la formación de otros productos
(por ejemplo de isomería de doble enlace) minoritarios no se puede excluir.
· Intersecciones Cónicas bicíclicas con un enlace σ intra-anular.
Para las intersecciones cónicas que presentan un enlace sigma intra-anular se ha llevado
a cabo el estudio sistemático, en base a las propiedades del espacio de ramificación, de
los posibles productos de relajación tras el cruce diabático a través de dichas CoIn.
La CoIn(1σ)A pertenece al tipo de las que poseen un grupo -(CH)3- “retorcido”, grupo
que se localiza en uno de los dos anillo (cuyos centros están etiquetados como “1” “2” y
“3” en la figura 5.18)
El cuarto radical se halla deslocalizado en un grupo alilo, lo que proporciona una mayor
estabilidad al cuarto radical, el cual no interviene en los procesos de relajación
vibracional, ya que en ninguno de los vectores del espacio de ramificación (DC y GD)
los carbonos que lo alojan poseen contribución alguna (Fig. 5.18).
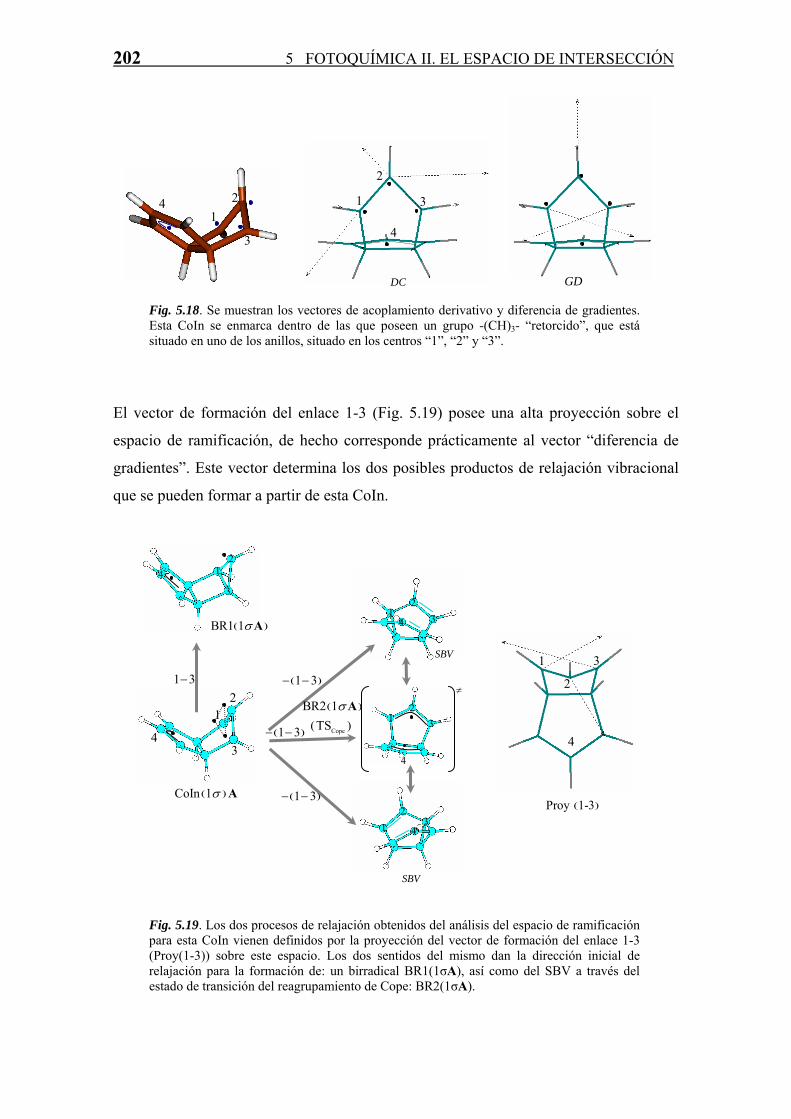
202 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
DC GD
1
2
3
41
2
3
4
Fig. 5.18. Se muestran los vectores de acoplamiento derivativo y diferencia de gradientes. Esta CoIn se enmarca dentro de las que poseen un grupo -(CH)3- “retorcido”, que está situado en uno de los anillos, situado en los centros “1”, “2” y “3”.
El vector de formación del enlace 1-3 (Fig. 5.19) posee una alta proyección sobre el
espacio de ramificación, de hecho corresponde prácticamente al vector “diferencia de
gradientes”. Este vector determina los dos posibles productos de relajación vibracional
que se pueden formar a partir de esta CoIn.
≠
SBV
SBV 1
2
3
4
12
34
( )BR1 1σA
( )CoIn 1σ A( )Proy 1-3
( )
( )Cope
BR2 1 TS
σA
12
34
12
34
12
3
4
12
34
1 3− ( )1 3− −
( )1 3− −
( )1 3− −
Fig. 5.19. Los dos procesos de relajación obtenidos del análisis del espacio de ramificación para esta CoIn vienen definidos por la proyección del vector de formación del enlace 1-3 (Proy(1-3)) sobre este espacio. Los dos sentidos del mismo dan la dirección inicial de relajación para la formación de: un birradical BR1(1σA), así como del SBV a través del estado de transición del reagrupamiento de Cope: BR2(1σA).

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 203
A través de la formación de un enlace σ (1-3) se obtiene un compuesto birradicálico
(BR1(1σA) en la figura 5.19), mientras que el sentido contrario de este mismo vector
indica la dirección inicial de relajación para la formación de un segundo birradical que
corresponde, como en ocasiones anteriores, al estado de transición del reagrupamiento
de Cope entre SBV equivalentes (Fig. 5.19).
La CoIn(1σ)B presenta una estructura parecida a la anterior, solo que en este caso el
cuarto radical no está deslocalizado en un alilo, sino que está localizado en un carbono.
Además presenta una interacción a través del espacio con el grupo -(CH)3- “retorcido”,
de tal manera que los productos de relajación ya no son los característicos de este grupo,
sino que están marcados por la interacción entre el radical solitario y este grupo. De
hecho, la CoIn presenta una estructura parecida a la que se ha propuesto para reacciones
di-π-metano que transcurren por reacción fotoquímica directa (Reguero et al., 1993). El
proceso de relajación está determinado fundamentalmente por el vector de formación de
enlace sigma entre los centros “4” y “2” (Fig. 5.20).
≠
SBV
SBVDC GD
1
2
34
12
3
4
( )CoIn 1σ B( )BR1 1σB
( )
( )Cope
BR2 1 TS
σB
( )2 4 GD− = −
GD
GD
GD
1
2
34
12 3
4
1 23
4
1 23
4
SBV
Fig. 5.20. Se muestran los vectores DC y GD (arriba). El vector de formación del enlace 2-4 (que se corresponde prácticamente con el vector “-GD”) define los dos productos de relajación, por un lado la formación del birradical BR1(1σB) por formación de dicho enlace (2-4), y por otro la formación del birradical BR2(1σB) (TS de Cope en el SBV) que da lugar a la formación de SBV.
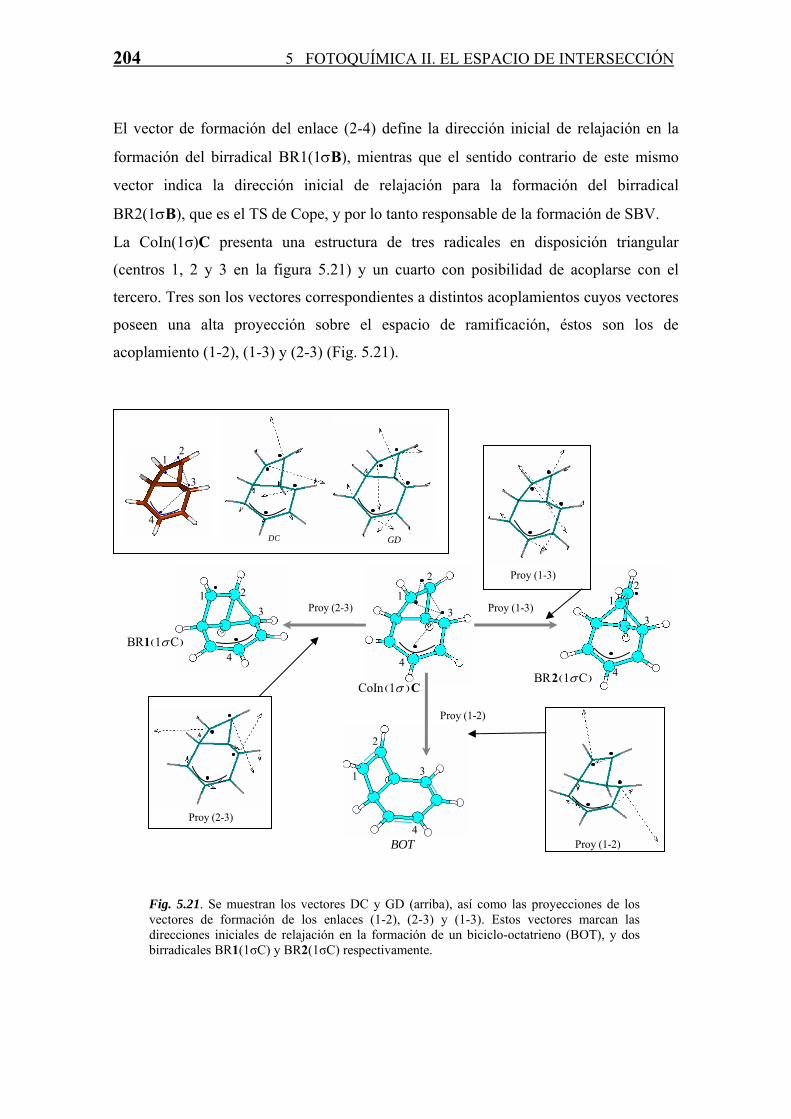
204 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
El vector de formación del enlace (2-4) define la dirección inicial de relajación en la
formación del birradical BR1(1σB), mientras que el sentido contrario de este mismo
vector indica la dirección inicial de relajación para la formación del birradical
BR2(1σB), que es el TS de Cope, y por lo tanto responsable de la formación de SBV.
La CoIn(1σ)C presenta una estructura de tres radicales en disposición triangular
(centros 1, 2 y 3 en la figura 5.21) y un cuarto con posibilidad de acoplarse con el
tercero. Tres son los vectores correspondientes a distintos acoplamientos cuyos vectores
poseen una alta proyección sobre el espacio de ramificación, éstos son los de
acoplamiento (1-2), (1-3) y (2-3) (Fig. 5.21).
1
2
3
4
1 2
3
4
12
3
4
1
2
3
4
Proy (2-3) Proy (1-3)
Proy (1-2)
Proy (2-3)
Proy (1-2)
Proy (1-3)
12
3
4DC GD
( )CoIn 1σ C
( )BR 1 Cσ1
( )BR 1 Cσ2
BOT
Fig. 5.21. Se muestran los vectores DC y GD (arriba), así como las proyecciones de los vectores de formación de los enlaces (1-2), (2-3) y (1-3). Estos vectores marcan las direcciones iniciales de relajación en la formación de un biciclo-octatrieno (BOT), y dos birradicales BR1(1σC) y BR2(1σC) respectivamente.

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 205
Estos tres vectores corresponden al acoplamiento de distintos electrones. En primer
lugar, el acoplamiento de los electrones “1” y “2” da lugar a la formación de un doble
enlace entre estos centros, y un reagrupamiento de carga en el otro anillo de tal manera
que se forma un biciclo-octatrieno (BOT).
Los vectores de los dos acoplamientos restantes: (2-3) y (1-3) proyectados sobre el
espacio de ramificación (Fig.5.21) dan las direcciones iniciales de relajación para la
formación de sendos birradicales: BR1(1σC) y BR2(1σC) respectivamente.
La CoIn(1σ)D presenta una estructura similar a otras intersecciones cónicas ya
estudiadas. Posee un grupo -(CH)3- “retorcido” y un cuarto centro radicálico contiguo al
mismo. Los posibles productos de relajación son por lo tanto los característicos de este
tipo de CoIn, es decir, la formación de un ciclopropano derivado así como la apertura de
este ciclo dando lugar a un bicilo-octatrieno (BOT) (Fig. 5.22).
DC GD
( )BR1 1σD
1
2
34
12
34
BOT
12
34
1 3− 1 2 , 3 4− −
( )CoIn 1σ D
Fig. 5.22. Se muestran los vectores DC y GD (arriba). Existen dos posibles direcciones iniciales de relajación vibracional a partir de esta CoIn. En primer lugar, la formación del enlace 1-3 (cuya dirección inicial en el espacio de ramificación viene dada por el vector DC) da lugar al birradical BR1(1σD), mientras que por otro lado, el acoplamiento de los radicales 1-2 y 3-4 da lugar a la formación de un biciclo-octatrieno (BOT).
La CoIn(1σ)E corresponde claramente a una intersección cónica de cicloadición [2+2].
Presenta cuatro radicales situados sobre cuatro carbonos contiguos dos a dos. Estos
cuatro carbonos están situados prácticamente en el mismo plano, y la disposición de los
orbitales semiocupados es la que presentan normalmente este tipo de CoIn (Fig. 5.23).
Los vectores DC y GD son también análogos a los que poseen los de las

206 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
correspondientes CoIn de cicloadición [2+2] en sistemas sencillos como puede ser el
formado por dos etilenos.
En este tipo de intersecciones cónicas son posibles en general tres caminos distintos de
relajación en el estado fundamental, lo cual viene refrendado para esta CoIn concreta
por la alta proyección de tres direcciones iniciales de relajación sobre el espacio de
ramificación correspondientes a tres acoplamientos distintos de los cuatro radicales en
el estado fundamental. En primer lugar, es posible la evolución del sistema en la
dirección de formación del enlace 1-4 (Fig. 5.23). En esta dirección de relajación se
forma un birradical (BR1(1σE) en la figura 5.23)
DC
12
3 4
GD
1 2
3 4
1 2
34
1 2
3 4
1 2
3 4
1 2
3 4
1 2
3 4
( )Proy 1-3,2-4
( )Proy 1-4
1 2
34
12
3
4
12
34
12
34
( )BR1 1σE
BOT
( )CoIn 1σ E
TCO
Fig. 5.23. Vectores DC y GD así como las posibles relajaciones obtenidas del análisis de estos vectores. El acoplamiento entre los electrones “1” y “4” da lugar a la formación de un birradical (BR1(1σE)). El sentido opuesto de este vector define la dirección inicial de relajación correspondiente al apareamiento entre los electrones (1-2) y (3-4) y por lo tanto a la formación de un biciclo-octatrieno (BOT). Por otro lado, el apareamiento entre los electrones (1-3) y (2-4) que viene dado aproximadamente por el vector DC da lugar a la formación del producto de cicloadición [2+2] (tetraciclo[ 3.3.0.02,4.03,6]oct-7-eno=TCO).
En el sentido opuesto de este mismo vector, se alarga la distancia entre los centros 1 y 4,
por lo que se reagrupa la carga π, formándose un biciclo-octa-trieno (BOT). Por otro
lado, la proyección del vector de formación de los enlaces 1-3 y 2-4 sobre el espacio de
ramificación es considerablemente alta, ya que prácticamente coincide con el vector
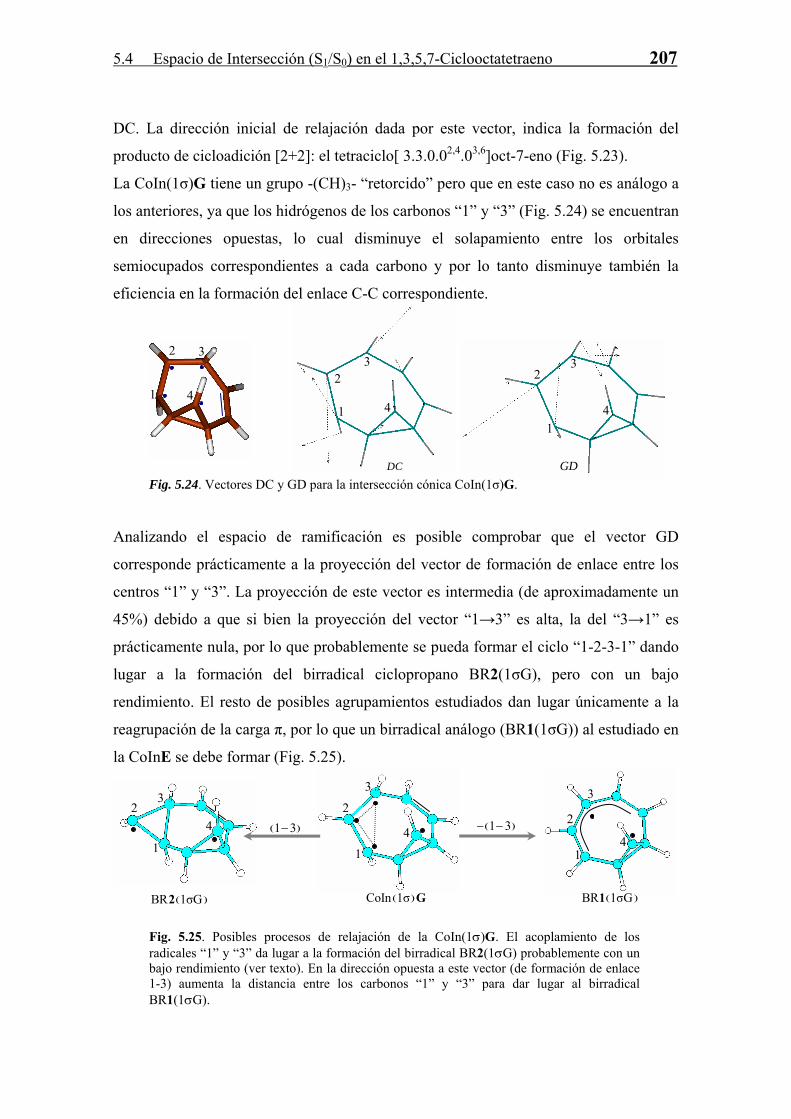
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 207
DC. La dirección inicial de relajación dada por este vector, indica la formación del
producto de cicloadición [2+2]: el tetraciclo[ 3.3.0.02,4.03,6]oct-7-eno (Fig. 5.23).
La CoIn(1σ)G tiene un grupo -(CH)3- “retorcido” pero que en este caso no es análogo a
los anteriores, ya que los hidrógenos de los carbonos “1” y “3” (Fig. 5.24) se encuentran
en direcciones opuestas, lo cual disminuye el solapamiento entre los orbitales
semiocupados correspondientes a cada carbono y por lo tanto disminuye también la
eficiencia en la formación del enlace C-C correspondiente.
DC
1
23
4
Fig. 5.24. Vectores DC y GD para la intersección cónica CoIn(1σ)G.
GD
1
23
41
2 3
4
Analizando el espacio de ramificación es posible comprobar que el vector GD
corresponde prácticamente a la proyección del vector de formación de enlace entre los
centros “1” y “3”. La proyección de este vector es intermedia (de aproximadamente un
45%) debido a que si bien la proyección del vector “1→3” es alta, la del “3→1” es
prácticamente nula, por lo que probablemente se pueda formar el ciclo “1-2-3-1” dando
lugar a la formación del birradical ciclopropano BR2(1σG), pero con un bajo
rendimiento. El resto de posibles agrupamientos estudiados dan lugar únicamente a la
reagrupación de la carga π, por lo que un birradical análogo (BR1(1σG)) al estudiado en
la CoInE se debe formar (Fig. 5.25).
1
2
3
41
23
4 ( )1 3−
1
2
3
4 ( )1 3− −
( )CoIn 1σ G ( )BR 1σG1( )BR 1σG2
Fig. 5.25. Posibles procesos de relajación de la CoIn(1σ)G. El acoplamiento de los radicales “1” y “3” da lugar a la formación del birradical BR2(1σG) probablemente con un bajo rendimiento (ver texto). En la dirección opuesta a este vector (de formación de enlace 1-3) aumenta la distancia entre los carbonos “1” y “3” para dar lugar al birradical BR1(1σG).
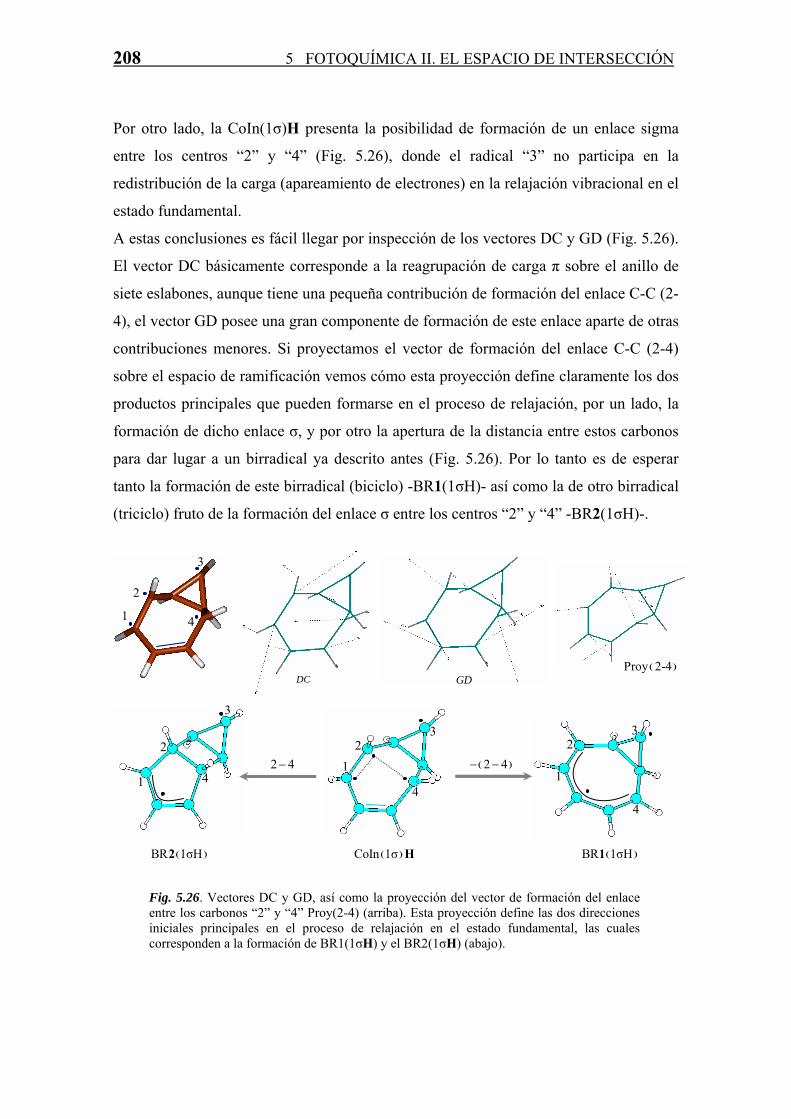
208 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Por otro lado, la CoIn(1σ)H presenta la posibilidad de formación de un enlace sigma
entre los centros “2” y “4” (Fig. 5.26), donde el radical “3” no participa en la
redistribución de la carga (apareamiento de electrones) en la relajación vibracional en el
estado fundamental.
A estas conclusiones es fácil llegar por inspección de los vectores DC y GD (Fig. 5.26).
El vector DC básicamente corresponde a la reagrupación de carga π sobre el anillo de
siete eslabones, aunque tiene una pequeña contribución de formación del enlace C-C (2-
4), el vector GD posee una gran componente de formación de este enlace aparte de otras
contribuciones menores. Si proyectamos el vector de formación del enlace C-C (2-4)
sobre el espacio de ramificación vemos cómo esta proyección define claramente los dos
productos principales que pueden formarse en el proceso de relajación, por un lado, la
formación de dicho enlace σ, y por otro la apertura de la distancia entre estos carbonos
para dar lugar a un birradical ya descrito antes (Fig. 5.26). Por lo tanto es de esperar
tanto la formación de este birradical (biciclo) -BR1(1σH)- así como la de otro birradical
(triciclo) fruto de la formación del enlace σ entre los centros “2” y “4” -BR2(1σH)-.
DC GD
1
2
3
4
( )Proy 2-4
12
3
41
23
4
1
2
3
42 4− ( )2 4− −
( )CoIn 1σ H ( )BR 1σH1( )BR 1σH2
Fig. 5.26. Vectores DC y GD, así como la proyección del vector de formación del enlace entre los carbonos “2” y “4” Proy(2-4) (arriba). Esta proyección define las dos direcciones iniciales principales en el proceso de relajación en el estado fundamental, las cuales corresponden a la formación de BR1(1σH) y el BR2(1σH) (abajo).
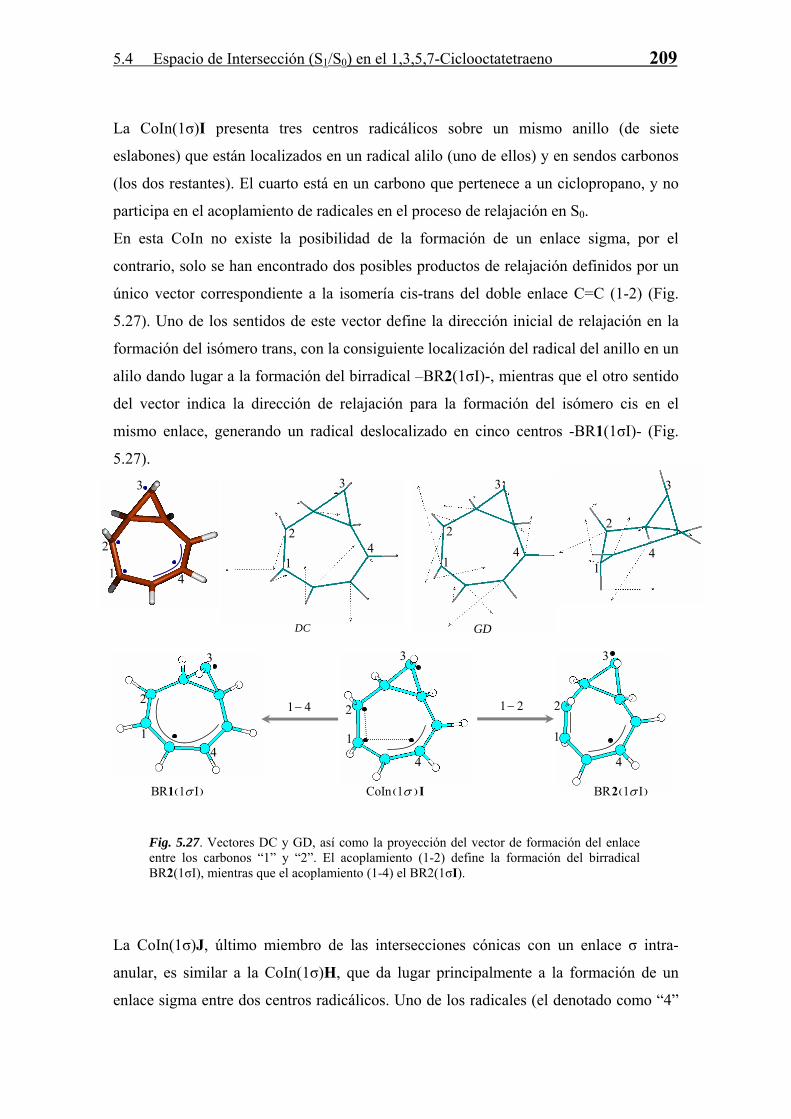
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 209
La CoIn(1σ)I presenta tres centros radicálicos sobre un mismo anillo (de siete
eslabones) que están localizados en un radical alilo (uno de ellos) y en sendos carbonos
(los dos restantes). El cuarto está en un carbono que pertenece a un ciclopropano, y no
participa en el acoplamiento de radicales en el proceso de relajación en S0.
En esta CoIn no existe la posibilidad de la formación de un enlace sigma, por el
contrario, solo se han encontrado dos posibles productos de relajación definidos por un
único vector correspondiente a la isomería cis-trans del doble enlace C=C (1-2) (Fig.
5.27). Uno de los sentidos de este vector define la dirección inicial de relajación en la
formación del isómero trans, con la consiguiente localización del radical del anillo en un
alilo dando lugar a la formación del birradical –BR2(1σI)-, mientras que el otro sentido
del vector indica la dirección de relajación para la formación del isómero cis en el
mismo enlace, generando un radical deslocalizado en cinco centros -BR1(1σI)- (Fig.
5.27).
DC GD
1
2
3
41
2
3
41
2
3
41
2
3
4
1
2
3
4
1
2
3
4
1
2
3
4
( )CoIn 1σ I( )BR 1 Iσ1 ( )BR 1 Iσ2
1 4− 1 2−
Fig. 5.27. Vectores DC y GD, así como la proyección del vector de formación del enlace entre los carbonos “1” y “2”. El acoplamiento (1-2) define la formación del birradical BR2(1σI), mientras que el acoplamiento (1-4) el BR2(1σI).
La CoIn(1σ)J, último miembro de las intersecciones cónicas con un enlace σ intra-
anular, es similar a la CoIn(1σ)H, que da lugar principalmente a la formación de un
enlace sigma entre dos centros radicálicos. Uno de los radicales (el denotado como “4”
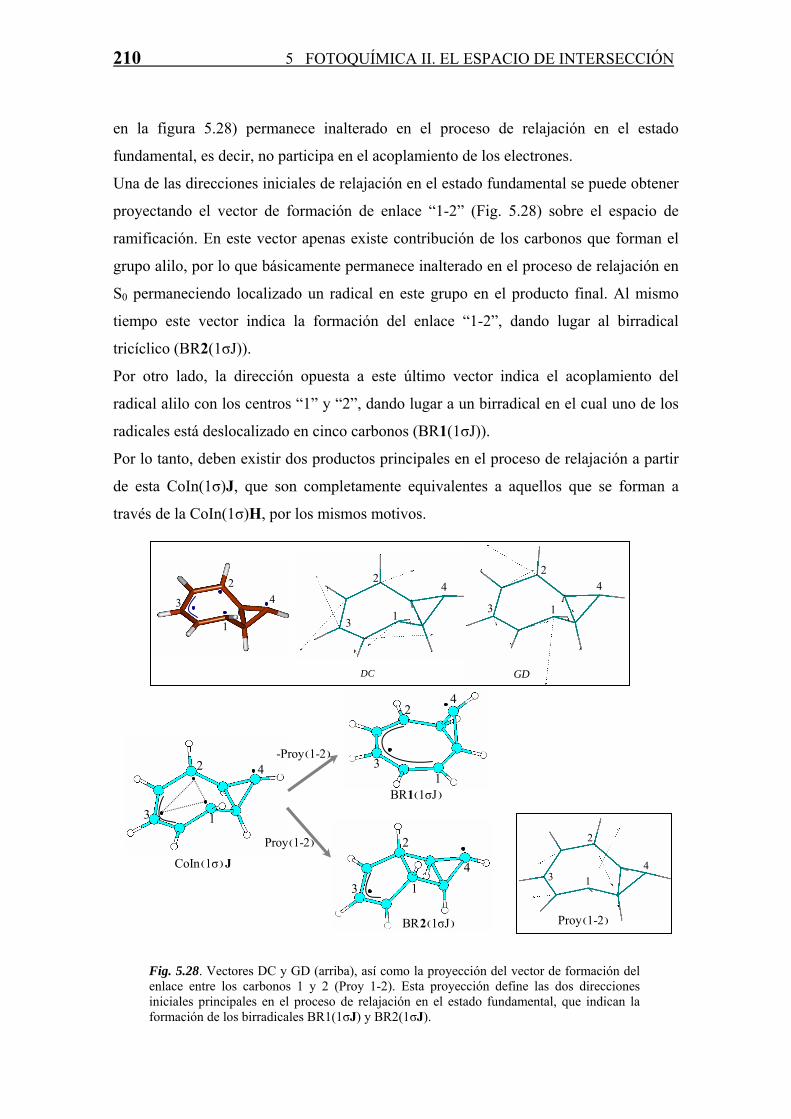
210 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
en la figura 5.28) permanece inalterado en el proceso de relajación en el estado
fundamental, es decir, no participa en el acoplamiento de los electrones.
Una de las direcciones iniciales de relajación en el estado fundamental se puede obtener
proyectando el vector de formación de enlace “1-2” (Fig. 5.28) sobre el espacio de
ramificación. En este vector apenas existe contribución de los carbonos que forman el
grupo alilo, por lo que básicamente permanece inalterado en el proceso de relajación en
S0 permaneciendo localizado un radical en este grupo en el producto final. Al mismo
tiempo este vector indica la formación del enlace “1-2”, dando lugar al birradical
tricíclico (BR2(1σJ)).
Por otro lado, la dirección opuesta a este último vector indica el acoplamiento del
radical alilo con los centros “1” y “2”, dando lugar a un birradical en el cual uno de los
radicales está deslocalizado en cinco carbonos (BR1(1σJ)).
Por lo tanto, deben existir dos productos principales en el proceso de relajación a partir
de esta CoIn(1σ)J, que son completamente equivalentes a aquellos que se forman a
través de la CoIn(1σ)H, por los mismos motivos.
DC
1
2
3
4
GD
1
2
3
4
1
2
3 4
1
2
3
4
1
2
3
4
1
2
3
4
1
2
34
( )Proy 1-2
( )Proy 1-2
( )-Proy 1-2
( )CoIn 1σ J
( )BR 1σJ1
( )BR 1σJ2
Fig. 5.28. Vectores DC y GD (arriba), así como la proyección del vector de formación del enlace entre los carbonos 1 y 2 (Proy 1-2). Esta proyección define las dos direcciones iniciales principales en el proceso de relajación en el estado fundamental, que indican la formación de los birradicales BR1(1σJ) y BR2(1σJ).
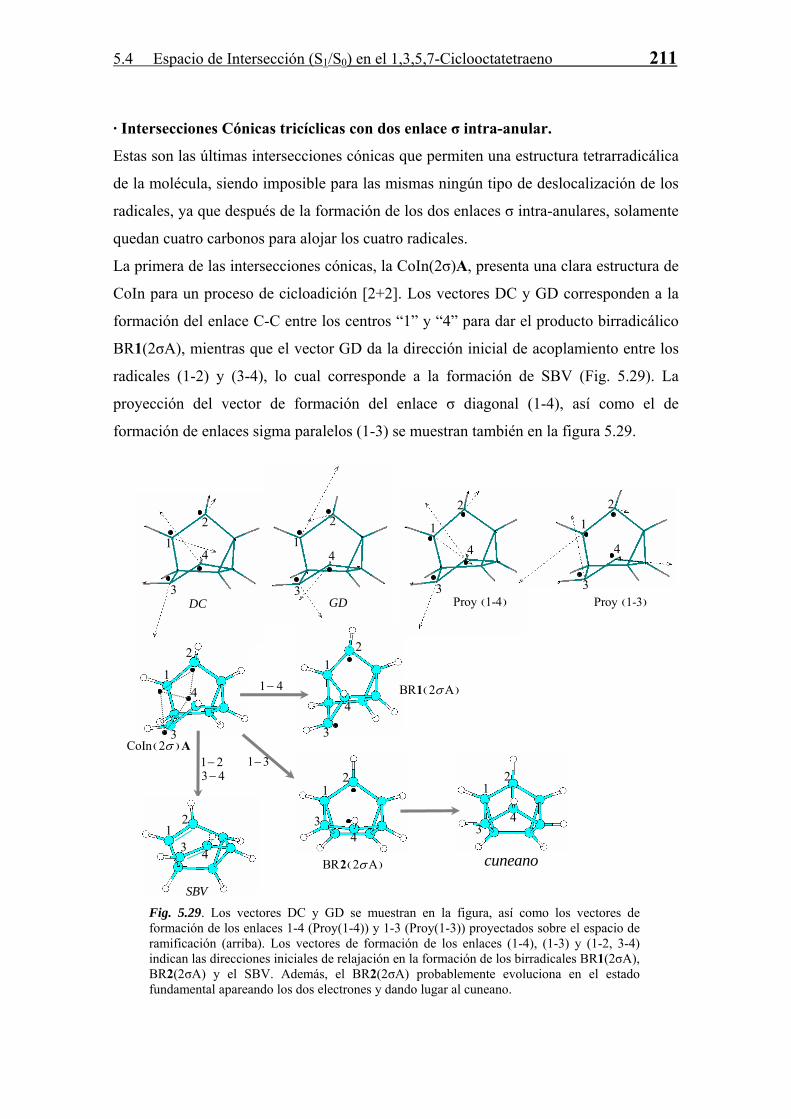
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 211
· Intersecciones Cónicas tricíclicas con dos enlace σ intra-anular.
Estas son las últimas intersecciones cónicas que permiten una estructura tetrarradicálica
de la molécula, siendo imposible para las mismas ningún tipo de deslocalización de los
radicales, ya que después de la formación de los dos enlaces σ intra-anulares, solamente
quedan cuatro carbonos para alojar los cuatro radicales.
La primera de las intersecciones cónicas, la CoIn(2σ)A, presenta una clara estructura de
CoIn para un proceso de cicloadición [2+2]. Los vectores DC y GD corresponden a la
formación del enlace C-C entre los centros “1” y “4” para dar el producto birradicálico
BR1(2σA), mientras que el vector GD da la dirección inicial de acoplamiento entre los
radicales (1-2) y (3-4), lo cual corresponde a la formación de SBV (Fig. 5.29). La
proyección del vector de formación del enlace σ diagonal (1-4), así como el de
formación de enlaces sigma paralelos (1-3) se muestran también en la figura 5.29.
12
3
41
2
3
4
1
2
3
4
12
3
4
DC GD ( )Proy 1-3( )Proy 1-4
1
2
3
4
12
3
4
31
2
4
3
12
4 3
12
4
( )CoIn 2σ A
1 4−
1 23 4−−
1 3−
( )BR 2 Aσ1
( )BR 2 Aσ2
SBV
cuneano
Fig. 5.29. Los vectores DC y GD se muestran en la figura, así como los vectores de formación de los enlaces 1-4 (Proy(1-4)) y 1-3 (Proy(1-3)) proyectados sobre el espacio de ramificación (arriba). Los vectores de formación de los enlaces (1-4), (1-3) y (1-2, 3-4) indican las direcciones iniciales de relajación en la formación de los birradicales BR1(2σA), BR2(2σA) y el SBV. Además, el BR2(2σA) probablemente evoluciona en el estado fundamental apareando los dos electrones y dando lugar al cuneano.

212 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Los productos que se pueden formar a partir de esta CoIn vienen dados por los vectores
de formación de enlace (1-4), (1-3) y (1-2, 3-4). Estos posibles acoplamientos de
electrones dan lugar en principio a tres posible productos, un birradical BR1(2σA), un
segundo birradical BR2(2σA) que puede evolucionar para dar el cuneano, y el SBV
respectivamente.
La segunda de estas intersecciones cónicas, la CoIn(2σ)B, es más compleja que las
anteriores, y no se puede enmarcar claramente en ninguno de los tipos de CoIn ya
descritos. Los únicos vectores que conllevan un acoplamiento de electrones con una
proyección alta en el espacio de ramificación son los de formación de los enlaces (1-2)
y (3-4) que corresponden prácticamente al mismo vector (Fig. 5.30).
1
24
3
DC
1
2
4
3
GD 1
2
43
( )Proy 3-4
1 2
4
3
( )Proy 1-2
1
23
4
12
43
( )CoIn 2σ B TCO
Fig. 5.30. Vectores DC y GD así como las proyecciones de los dos vectores de formación de los enlaces (3-4) y (1-2) sobre el espacio de ramificación (arriba). La única posible evolución encontrada a partir del análisis de este espacio corresponde a la formación de un tetraciclo (tetraciclo[ 3.3.0.02,4.03,6]oct-7-eno=TCO).
Por lo tanto, el único producto posible de relajación en el estado fundamental debería
corresponder al tetraciclo[3.3.0.02,4.03,6]oct-7-eno (=TCO en la figura 5.30).
La CoIn(2σ)D, corresponde al tipo de intersecciones cónicas que presenta un grupo
-(CH)3- “retorcido”. Los centros “1”,”2” y “3” (Fig. 5.31) están contiguos y alojan tres
radicales que intervienen en el proceso de relajación en el estado fundamental. Por el
contrario, el radical albergado en el carbono “4” no da lugar a acoplamiento con otros

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 213
radicales, lo cual se manifiesta en la nula contribución del carbono “4” en los vectores
DC y GD (Fig. 5.31).
1
23
4
1
23
41
2
34
DC GD ( )Proy 1-3
( )BR 2 Dσ1 ( )BR 2 Dσ2( )CoIn 2σ D
1 2 3− −1 3−
Fig. 5.31. Se muestran los vectores DC y GD así como la proyección del vector de formación del enlace sigma (1-3) sobre el “espacio de ramificación”. Este último vector define las dos direcciones iniciales de relajación (dadas por los dos sentidos del vector), que dan lugar a la formación de los birradicales BR1(2σD) y BR2(2σD) respectivamente.
Al igual que en intersecciones cónicas análogas, básicamente existen dos posibles
evoluciones de la molécula en el estado fundamental, la primera correspondiente a la
formación del birradical ciclopropano (1-3) (BR1(2σD) en la figura 5.31) y la segunda,
la formación de un birradical alílico (BR2(2σD) en la misma figura) cuya dirección
inicial de relajación viene dada aproximadamente por el vector DC.
La CoIn(2σ)E, presenta unas curiosas propiedades, ya que si bien los radicales se hallan
localizados en carbonos no unidos directamente, los mismos presentan una estructura
cuadrada correspondiente básicamente a una cicloadición [2+2]. Al contrario que en las
cicloadiciones [2+2] anteriormente estudiadas, en ésta, debido a la poca movilidad que
presentan los cuatro carbonos en la estructura policíclica de la CoIn solamente una de
las relajaciones vibracionales es posible, que corresponde al apareamiento de los
electrones situados sobre los centros (1-2) y (3-4), o equivalentemente por simetría (2-3)
y (1-4). Así, si bien existe una importante proyección del vector de formación del enlace
(2-4) (Fig. 5.32) sobre el espacio de ramificación, no es posible la formación de este
enlace debido a la altísima tensión estérica que ello conllevaría. Por el contrario, la
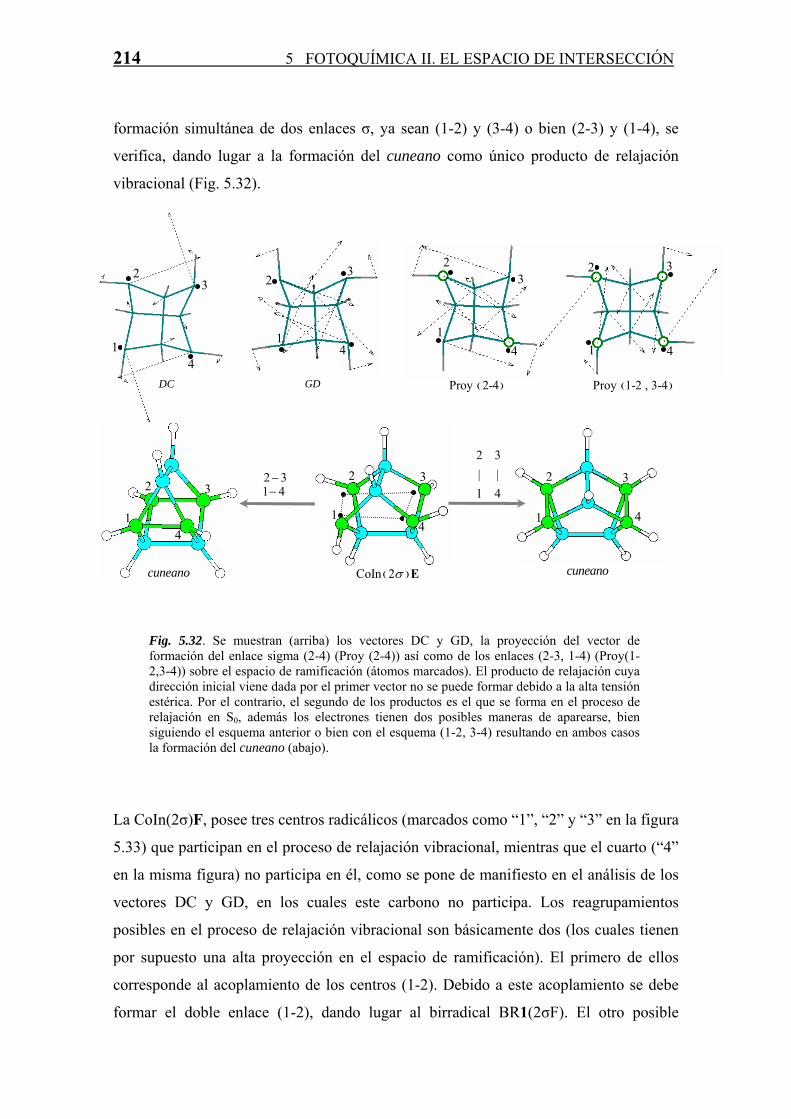
214 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
formación simultánea de dos enlaces σ, ya sean (1-2) y (3-4) o bien (2-3) y (1-4), se
verifica, dando lugar a la formación del cuneano como único producto de relajación
vibracional (Fig. 5.32).
( )CoIn 2σ E cuneano
1
23
4DC GD
1
23
41
23
4 1
2 3
4
1
2 3
41
2 3
4
2 31 4−−
2 3| |1 4
1
2 3
4
cuneano
( )Proy 2-4 ( )Proy 1-2 , 3-4
Fig. 5.32. Se muestran (arriba) los vectores DC y GD, la proyección del vector de formación del enlace sigma (2-4) (Proy (2-4)) así como de los enlaces (2-3, 1-4) (Proy(1-2,3-4)) sobre el espacio de ramificación (átomos marcados). El producto de relajación cuya dirección inicial viene dada por el primer vector no se puede formar debido a la alta tensión estérica. Por el contrario, el segundo de los productos es el que se forma en el proceso de relajación en S0, además los electrones tienen dos posibles maneras de aparearse, bien siguiendo el esquema anterior o bien con el esquema (1-2, 3-4) resultando en ambos casos la formación del cuneano (abajo).
La CoIn(2σ)F, posee tres centros radicálicos (marcados como “1”, “2” y “3” en la figura
5.33) que participan en el proceso de relajación vibracional, mientras que el cuarto (“4”
en la misma figura) no participa en él, como se pone de manifiesto en el análisis de los
vectores DC y GD, en los cuales este carbono no participa. Los reagrupamientos
posibles en el proceso de relajación vibracional son básicamente dos (los cuales tienen
por supuesto una alta proyección en el espacio de ramificación). El primero de ellos
corresponde al acoplamiento de los centros (1-2). Debido a este acoplamiento se debe
formar el doble enlace (1-2), dando lugar al birradical BR1(2σF). El otro posible
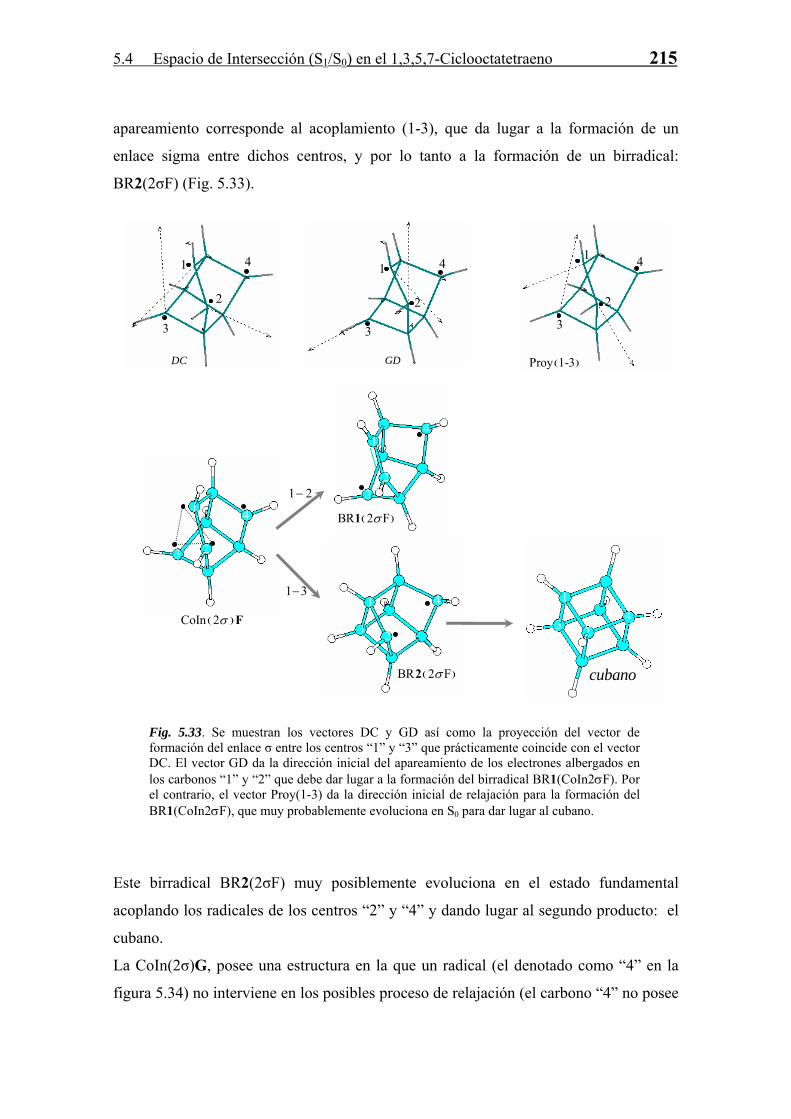
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 215
apareamiento corresponde al acoplamiento (1-3), que da lugar a la formación de un
enlace sigma entre dichos centros, y por lo tanto a la formación de un birradical:
BR2(2σF) (Fig. 5.33).
1
2
3
4 1
2
3
41
2
3
4
1
23
4
1
23
4
1
23
4 1
234
DC GD ( )Proy 1-3
cubano
( )CoIn 2σ F
( )BR 2 Fσ1
( )BR 2 Fσ2
1 2−
1 3−
Fig. 5.33. Se muestran los vectores DC y GD así como la proyección del vector de formación del enlace σ entre los centros “1” y “3” que prácticamente coincide con el vector DC. El vector GD da la dirección inicial del apareamiento de los electrones albergados en los carbonos “1” y “2” que debe dar lugar a la formación del birradical BR1(CoIn2σF). Por el contrario, el vector Proy(1-3) da la dirección inicial de relajación para la formación del BR1(CoIn2σF), que muy probablemente evoluciona en S0 para dar lugar al cubano.
Este birradical BR2(2σF) muy posiblemente evoluciona en el estado fundamental
acoplando los radicales de los centros “2” y “4” y dando lugar al segundo producto: el
cubano.
La CoIn(2σ)G, posee una estructura en la que un radical (el denotado como “4” en la
figura 5.34) no interviene en los posibles proceso de relajación (el carbono “4” no posee
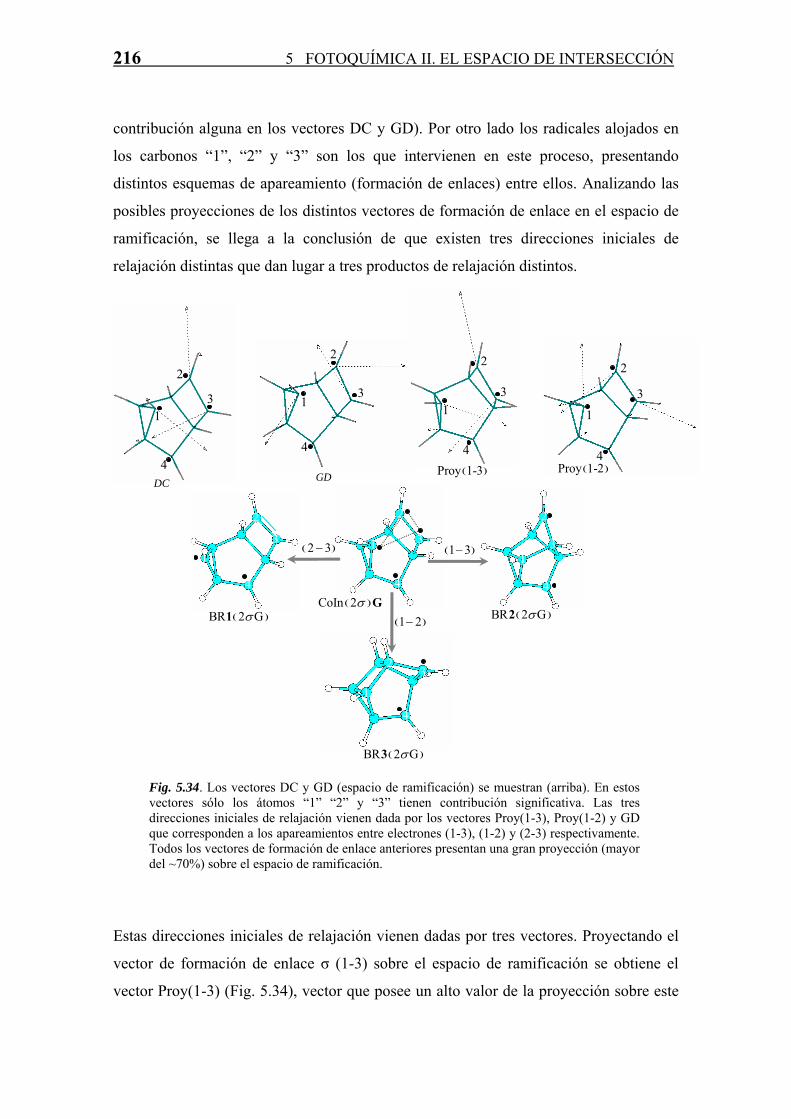
216 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
contribución alguna en los vectores DC y GD). Por otro lado los radicales alojados en
los carbonos “1”, “2” y “3” son los que intervienen en este proceso, presentando
distintos esquemas de apareamiento (formación de enlaces) entre ellos. Analizando las
posibles proyecciones de los distintos vectores de formación de enlace en el espacio de
ramificación, se llega a la conclusión de que existen tres direcciones iniciales de
relajación distintas que dan lugar a tres productos de relajación distintos.
2
3
4
1
2
3
4
1
2
3
4
1
2
3
4
1
DC GD( )Proy 1-3 ( )Proy 1-2
1
2
3
4
1
2
3
4
1
2
3
4
1
23
4
( )2 3− ( )1 3−
( )1 2−( )CoIn 2σ G
( )BR 2 Gσ1 ( )BR 2 Gσ2
( )BR 2 Gσ3
Fig. 5.34. Los vectores DC y GD (espacio de ramificación) se muestran (arriba). En estos vectores sólo los átomos “1” “2” y “3” tienen contribución significativa. Las tres direcciones iniciales de relajación vienen dada por los vectores Proy(1-3), Proy(1-2) y GD que corresponden a los apareamientos entre electrones (1-3), (1-2) y (2-3) respectivamente. Todos los vectores de formación de enlace anteriores presentan una gran proyección (mayor del ~70%) sobre el espacio de ramificación.
Estas direcciones iniciales de relajación vienen dadas por tres vectores. Proyectando el
vector de formación de enlace σ (1-3) sobre el espacio de ramificación se obtiene el
vector Proy(1-3) (Fig. 5.34), vector que posee un alto valor de la proyección sobre este

5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 217
espacio. Por otro lado, otro de los posibles apareamientos entre radicales que presenta
también una alta componente en el espacio de ramificación es el de formación del
enlace σ (1-2), cuya proyección en este espacio viene dada por el vector Proy(1-2) (Fig.
5.34). Por último, el apareamiento entre los electrones situados sobre los carbonos 2 y 3
se corresponde prácticamente con el vector GD. Cada uno de estos tres vectores definen
la dirección inicial de relajación en el estado fundamental para evolucionar hacia tres
birradicales respectivamente (Fig. 5.34).
La CoIn(2σ)H, posee una estructura ya vista antes en la que está presente un grupo
-(CH)3- “retorcido” definido por la secuencia de carbonos “1-2-3” (Fig. 5.35).
12
3
4
1
2
3
4
DC GD
1
2
34
12
3
412
3
4
( )CoIn 2σ H( )BR 2 Hσ1 ( )BR 2 Hσ2
1
2
3
41
23
4( )Proy 1-3
Fig. 5.35. Vectores DC y GD para esta CoIn. Además se representa el vector de formación del enlace (1-3) proyectado en el espacio de ramificación (dos vistas del mismo) (arriba). Este vector define las dos direcciones iniciales, dadas por los dos sentidos del vector, de relajación en el estado fundamental que dan lugar a la formación de los birradicales BR1(2σH) y BR2(2σH) (abajo).
El cuarto radical, situado sobre el carbono “4” no participa en el proceso de
reagrupamiento (apareamiento) de los electrones durante la relajación en S0. El vector
de formación del enlace (1-3), denotado en la figura 5.35 como Proy(1-3), posee una
alta proyección sobre el espacio de ramificación; además, ningún otro posee una
proyección significativa sobre dicho espacio. Este vector define las dos direcciones
iniciales de relajación posibles (dadas por los dos sentidos del vector). Por un lado la

218 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
formación del enlace (1-3) da lugar a la ciclación del anillo obteniéndose un birradical
que presenta un anillo ciclopropano BR2(2σH), mientras que la separación de estos dos
centros (1 y 3) da lugar a la formación de otro birradical para el cual uno de los
radicales es alílico BR1(2σH).
La CoIn(2σ)I, novena dentro de esta familia de intersecciones cónicas con dos enlaces
sigma intra-anulares, presenta una clara disposición de los cuatro radicales
correspondiente a una cicloadición [2+2] normal (Fig. 5.36).
12
34
1 2
3 4
1
34
1 2
3 4
( )CoIn 2σ I
( )BR 2 Iσ1
cubano
1 32 4−−
1 23 4−−
1 4−syn TOD−
2
12
3 4
12
3 4
DC
GD
Fig. 5.36. Se representan los vectores DC y GD (derecha). Los acoplamientos típicos en una intersección cónica correspondiente a una cicloadición [2+2] normal son posibles en ésta. Todos estos acoplamientos poseen una alta proyección sobre el espacio de ramificación. Por un lado el acoplamiento (1-4) da lugar a la formación del birradical BR1(2σI). Por otro lado, los acoplamientos (1-2,3-4) y (1-3,2-4) dan lugar a la formación de syn-TOD y cubano respectivamente.
Las posibles evoluciones en el proceso de relajación en el estado fundamental son las
mismas que en intersecciones cónicas análogas. Tres son los acoplamientos entre
electrones que pueden verificarse. Por un lado el acoplamiento entre los radicales
situados en los carbonos “1” y “4” da lugar a la formación del birradical BR1(2σI). Por
otro lado, la formación simultánea de los dobles enlaces entre los carbonos “1-2” y “3-
4” da lugar a la formación de syn-TOD (syn-Triciclo[4.2.0.02,5]octa-3,7-dieno). El
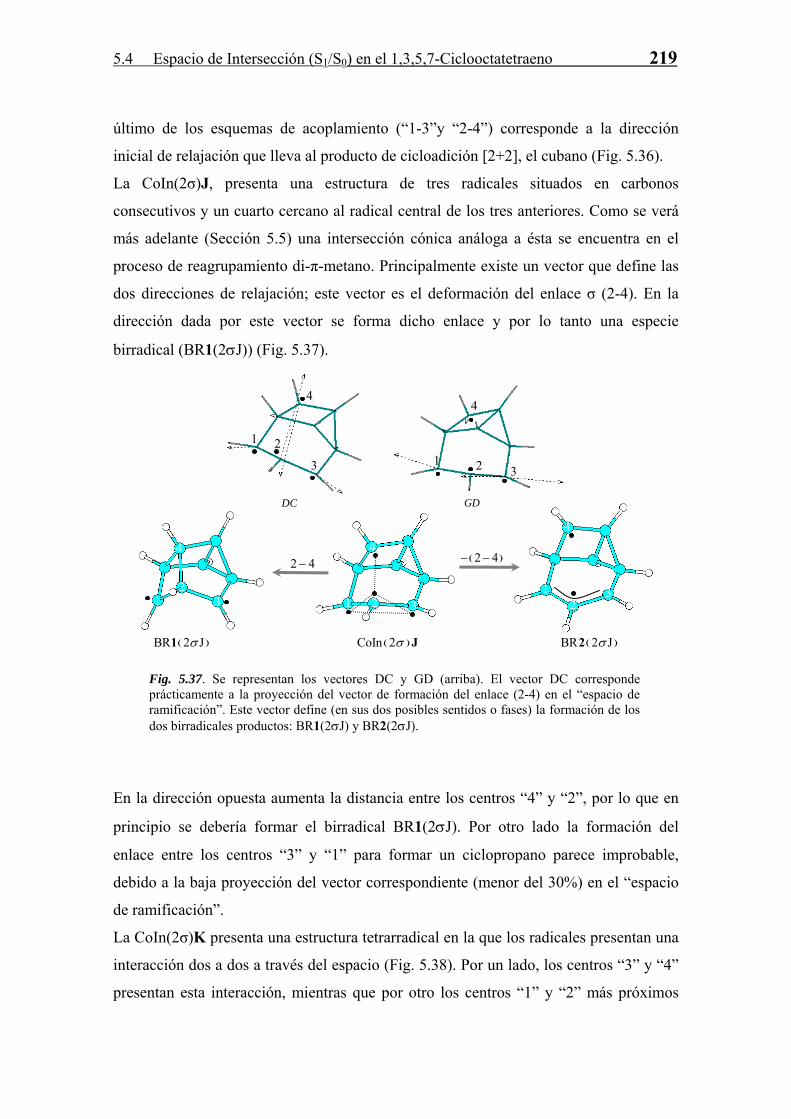
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 219
último de los esquemas de acoplamiento (“1-3”y “2-4”) corresponde a la dirección
inicial de relajación que lleva al producto de cicloadición [2+2], el cubano (Fig. 5.36).
La CoIn(2σ)J, presenta una estructura de tres radicales situados en carbonos
consecutivos y un cuarto cercano al radical central de los tres anteriores. Como se verá
más adelante (Sección 5.5) una intersección cónica análoga a ésta se encuentra en el
proceso de reagrupamiento di-π-metano. Principalmente existe un vector que define las
dos direcciones de relajación; este vector es el deformación del enlace σ (2-4). En la
dirección dada por este vector se forma dicho enlace y por lo tanto una especie
birradical (BR1(2σJ)) (Fig. 5.37).
1 2
3
4
1 2 3
4
DC GD
12
3
4
1 2 3
4
12
3
4
( )CoIn 2σ J( )BR 2 Jσ1 ( )BR 2 Jσ2
2 4− ( )2 4− −
Fig. 5.37. Se representan los vectores DC y GD (arriba). El vector DC corresponde prácticamente a la proyección del vector de formación del enlace (2-4) en el “espacio de ramificación”. Este vector define (en sus dos posibles sentidos o fases) la formación de los dos birradicales productos: BR1(2σJ) y BR2(2σJ).
En la dirección opuesta aumenta la distancia entre los centros “4” y “2”, por lo que en
principio se debería formar el birradical BR1(2σJ). Por otro lado la formación del
enlace entre los centros “3” y “1” para formar un ciclopropano parece improbable,
debido a la baja proyección del vector correspondiente (menor del 30%) en el “espacio
de ramificación”.
La CoIn(2σ)K presenta una estructura tetrarradical en la que los radicales presentan una
interacción dos a dos a través del espacio (Fig. 5.38). Por un lado, los centros “3” y “4”
presentan esta interacción, mientras que por otro los centros “1” y “2” más próximos
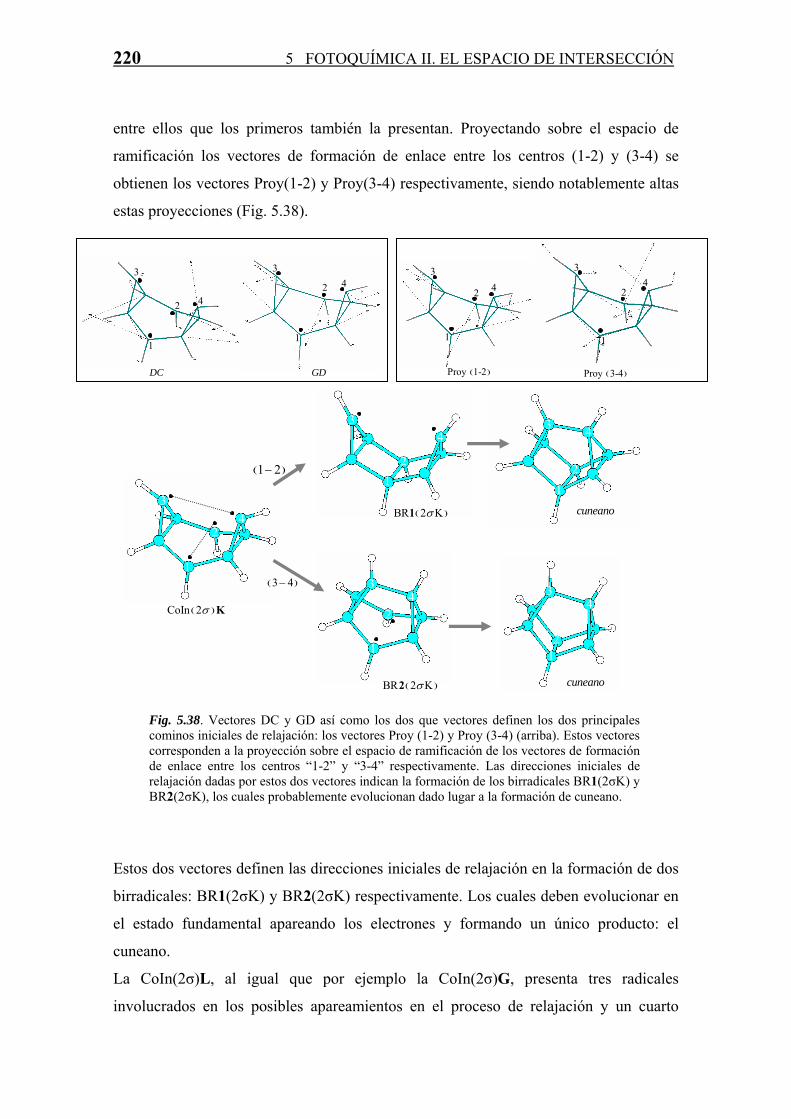
220 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
entre ellos que los primeros también la presentan. Proyectando sobre el espacio de
ramificación los vectores de formación de enlace entre los centros (1-2) y (3-4) se
obtienen los vectores Proy(1-2) y Proy(3-4) respectivamente, siendo notablemente altas
estas proyecciones (Fig. 5.38).
DC
1
2
3
4
GD
1
2
34
1
2
34
( )Proy 1-2
1
2
34
( )Proy 3-4
12
34
12
3 4
1
2
34
12
34
1
2
34
( )1 2−
( )CoIn 2σ K
( )BR 2 Kσ1
( )BR 2 Kσ2
cuneano
cuneano
( )3 4−
Fig. 5.38. Vectores DC y GD así como los dos que vectores definen los dos principales cominos iniciales de relajación: los vectores Proy (1-2) y Proy (3-4) (arriba). Estos vectores corresponden a la proyección sobre el espacio de ramificación de los vectores de formación de enlace entre los centros “1-2” y “3-4” respectivamente. Las direcciones iniciales de relajación dadas por estos dos vectores indican la formación de los birradicales BR1(2σK) y BR2(2σK), los cuales probablemente evolucionan dado lugar a la formación de cuneano.
Estos dos vectores definen las direcciones iniciales de relajación en la formación de dos
birradicales: BR1(2σK) y BR2(2σK) respectivamente. Los cuales deben evolucionar en
el estado fundamental apareando los electrones y formando un único producto: el
cuneano.
La CoIn(2σ)L, al igual que por ejemplo la CoIn(2σ)G, presenta tres radicales
involucrados en los posibles apareamientos en el proceso de relajación y un cuarto
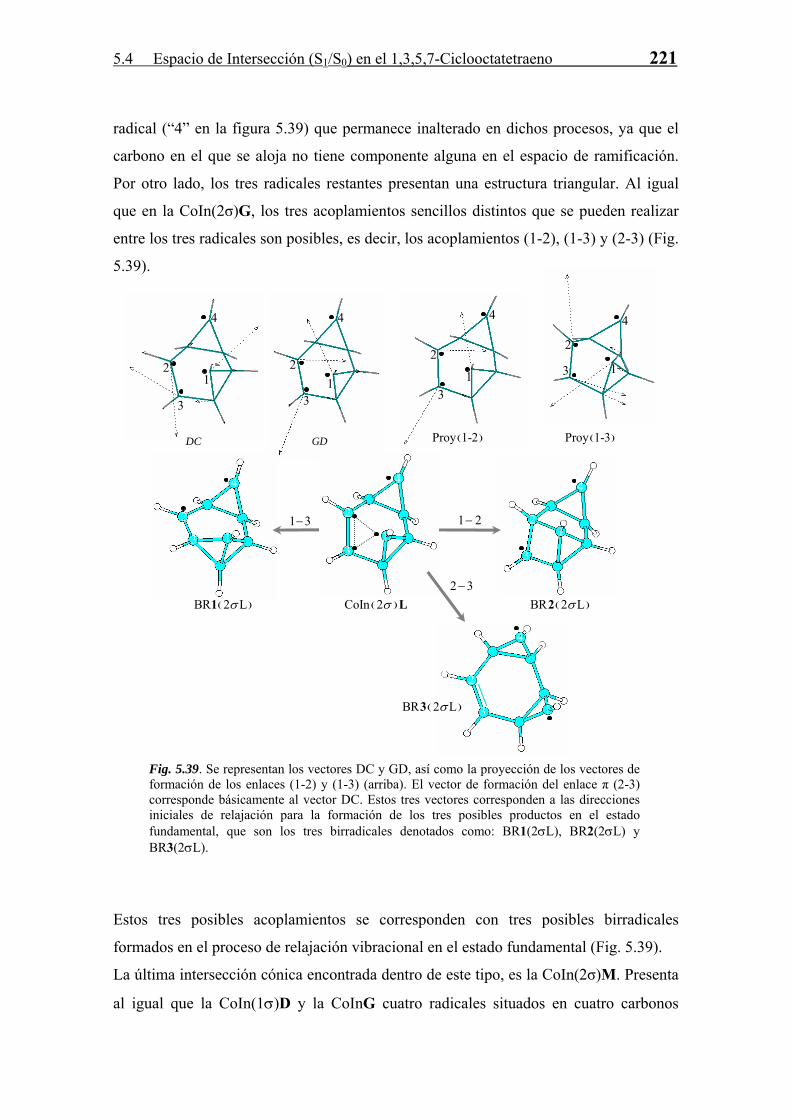
5.4 Espacio de Intersección (S1/S0) en el 1,3,5,7-Ciclooctatetraeno 221
radical (“4” en la figura 5.39) que permanece inalterado en dichos procesos, ya que el
carbono en el que se aloja no tiene componente alguna en el espacio de ramificación.
Por otro lado, los tres radicales restantes presentan una estructura triangular. Al igual
que en la CoIn(2σ)G, los tres acoplamientos sencillos distintos que se pueden realizar
entre los tres radicales son posibles, es decir, los acoplamientos (1-2), (1-3) y (2-3) (Fig.
5.39).
12
3
4
12
3
4
1
2
3
4
( )CoIn 2σ L( )BR 2 Lσ1 ( )BR 2 Lσ2
12
3
4
12
3
4
DC GD
1
2
3
4
1
2
3
4
( )Proy 1-2 ( )Proy 1-3
1
2
3
4
( )BR 2 Lσ3
1 3− 1 2−
2 3−
Fig. 5.39. Se representan los vectores DC y GD, así como la proyección de los vectores de formación de los enlaces (1-2) y (1-3) (arriba). El vector de formación del enlace π (2-3) corresponde básicamente al vector DC. Estos tres vectores corresponden a las direcciones iniciales de relajación para la formación de los tres posibles productos en el estado fundamental, que son los tres birradicales denotados como: BR1(2σL), BR2(2σL) y BR3(2σL).
Estos tres posibles acoplamientos se corresponden con tres posibles birradicales
formados en el proceso de relajación vibracional en el estado fundamental (Fig. 5.39).
La última intersección cónica encontrada dentro de este tipo, es la CoIn(2σ)M. Presenta
al igual que la CoIn(1σ)D y la CoInG cuatro radicales situados en cuatro carbonos

222 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
adyacentes. El esquema de acoplamiento es el mismo que en las anteriores, presentando
tres radicales (“1”, “2” y “3” en la figura 5.40) en disposición triangular y un cuarto que
puede acoplarse únicamente con el radical sobre el centro “3”. Son dos las posibles
direcciones iniciales de relajación correspondientes a distintos acoplamientos entre
electrones. La primera de ellas viene dada por la proyección del vector de formación de
los enlaces (1-2) y (3-4) sobre el espacio de ramificación (Fig. 5.40).
12
3
4DC
1 2
3
4
GD
1 23
4
( )Proy 1-2,3-4
1 2
3
4
1 2
3
4
12
34
( )CoIn 2σ M ( )BR 2 Mσ1
1 23 4−− 1 3−
2,8Triciclo[5.1.0.0 ]octa-3,5-dieno
Fig. 5.40. Vectores DC y GD para la CoIn(2σ)M. Además también se representa la proyección del vector de formación de los enlaces (1-2,3-4) sobre el espacio de ramificación. Este último vector da la dirección inicial de relajación para formar el Triciclo[5.1.0.02,8]octa-3,5-dieno. Por otro lado, el vector DC corresponde prácticamente a la proyección del vector de formación del enlace (1-3) sobre el espacio de ramificación, vector que da la dirección inicial para la formación del birradical BR1(2σM).
La menor flexibilidad del ciclo donde se encuentran los radicales (en comparación con
la intersección cónica abierta análoga CoInG) hace que no parezca posible la isomería
cis-trans en la formación del doble enlace entre los centro “1” y “2”, por lo que
probablemente sólo el isómero cis se pueda formar, que corresponde al
Triciclo[5.1.0.02,8]octa-3,5-dieno. Por otro lado, el vector de formación del enlace (1-3)
proyectado sobre el “espacio de ramificación” corresponde prácticamente al vector DC.
Siguiendo esta dirección inicial de relajación el sistema debería evolucionar para dar le
birradical BR1(2σM).

5.5 El Espacio de Intersección (S1/S0) y (T1/S0) en el barreleno 223
55..55 EEll EEssppaacciioo ddee IInntteerrsseecccciióónn ((SS11//SS00)) yy ((TT11//SS00)) eenn eell bbaarrrreelleennoo
Existen numerosas reacciones, por ejemplo en química orgánica, en las que son posibles
varios caminos de reacción, y en las que además se verifican transiciones diabáticas. En
estos casos, el análisis de la región de cruce entre los estados implicados puede revelar
muchos detalles sobre el mecanismo de la reacción, ya que puede ser esta región de
cruce la que por su topología determine la accesibilidad de los distintos caminos. En
esta sección se aborda un ejemplo práctico de la importancia del espacio de intersección
en química orgánica. En concreto se estudiará la reacción di-π-metano en el
biciclo[2.2.2]octa-2,5,7-trieno (barreleno) a la luz de la influencia que tiene el espacio
de intersección (tanto el S1/S0 como el T1/S0) sobre los posibles mecanismos de la
reacción.
En 1966, Zimmerman y Grunewald (Zimmerman y Grunewald, 1966) realizaron por
primera vez la reacción di-π-metano (RDPM) en el barreleno, y desde entonces este tipo
de reacción ha sido motivo de estudio dentro de la fotoquímica orgánica (Turro, 1978;
para una revisión reciente ver: Zimmerman y Armesto, 1996).
La RDPM tiene lugar cuando una molécula con dos grupos vinílicos (o grupos
equivalentes que poseen un doble enlace π, por ejemplo grupos arilo) unidos a un
mismo carbono sp3 al ser irradiada reacciona para dar un vinilciclopropano (Fig. 5.41).
i
ii
hν
BR2 BR3
Fig. 5.41. (i) Mecanismo de Zimmerman, (Zimmerman y Grunewald, 1966; Zimmerman et al., 1967; Zimmerman y Mariano, 1969; Zimmerman, 1980) y (ii) Mecanismo de Bernardi y Robb (Reguero et al., 1993) para la reacción de reagrupamiento di-π-metano en el 1,4-pentadieno. Los birradicales 1,4 (BR2) y 1,3 (BR3) son intermedios de reacción en el mecanismo (i). BR3 es una especie inestable en el mecanismo (ii).
Se han propuesto dos mecanismos para esta reacción, por un lado, Zimermann y
colaboradores (Zimmerman y Grunewald, 1966; Zimmerman et al., 1967; Zimmerman
y Mariano, 1969; Zimmerman, 1980) han postulado la existencia de los birradicales
BR2 y BR3 en el camino de reacción, aunque pudiendo no ser dichos birradicales
especies estables (mínimos sobre la SEP). Por otro lado (Reguero et al., 1993), en base a

224 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
la teoría de las intersecciones cónicas, propusieron como estructura de transición
diabática en la reacción en estado singlete una CoIn con unas características que
posteriormente se describirán con detalle.
En primer lugar se discutirán los posibles mecanismos para la reacción fotoinducida en
ausencia de sensibilizador (vía singlete) en base al estudio del espacio de intersección
S1/S0. Por último se abordará desde el mismo punto de vista la reacción sensibilizada
(via triplete).
5.5.1 Cicloadición [2+2] versus reagrupamiento di-π-metano singlete en
el barreleno
El barreleno es una molécula que puede, en principio, dar por irradiación directa al
menos dos tipos de reacciones. Por poseer grupos vinílicos unidos a un carbono sp3
puede dar la RDPM, mientras que por tener dos grupos vinílicos próximos donde los
cuatro carbonos de los dos grupos están en el mismo plano es un sistema en el que
puede ocurrir una cicloadición [2+2]. Por lo tanto, surgen dos cuestiones: ¿Cuál de los
dos procesos está más favorecido? y ¿Por qué?.
Para ambas reacciones ha sido propuesto un mecanismo en el que se verifica una
transición diabática a través de sendas CoIn, cuyas propiedades estructurales y
electrónicas se discutirán más adelante (ver figura 5.41 para la estructura de las mismas
en el barreleno). En principio podríamos pensar que la estabilidad de estas estructuras
de cruce entre estados (S1/S0 en este caso) podría determinar la viabilidad de uno u otro
mecanismo, ya que como ha sido apuntado (Olivucci et al., 1994) existe una analogía
entre los estados de transición para reacciones activadas térmicamente y las
intersecciones cónicas en reacciones diabáticas fotoinducidas.
La diferencia de energía entre las CoIn mínimas en energía correspondientes a las
transiciones diabáticas antes descritas es de ~19 kcal/mol a nivel CASSCF(8,8)/6-
31G(d), siendo la correspondiente a la reacción DPM la más estable. Sin embargo, no es
esta reacción la que se verifica experimentalmente, sino la cicloadición [2+2].
Hay que cuidarse por lo tanto de atribuir en una reacción diabática a la CoIn más estable
la responsabilidad de la transición. En general se debe hacer un análisis más profundo
del problema.
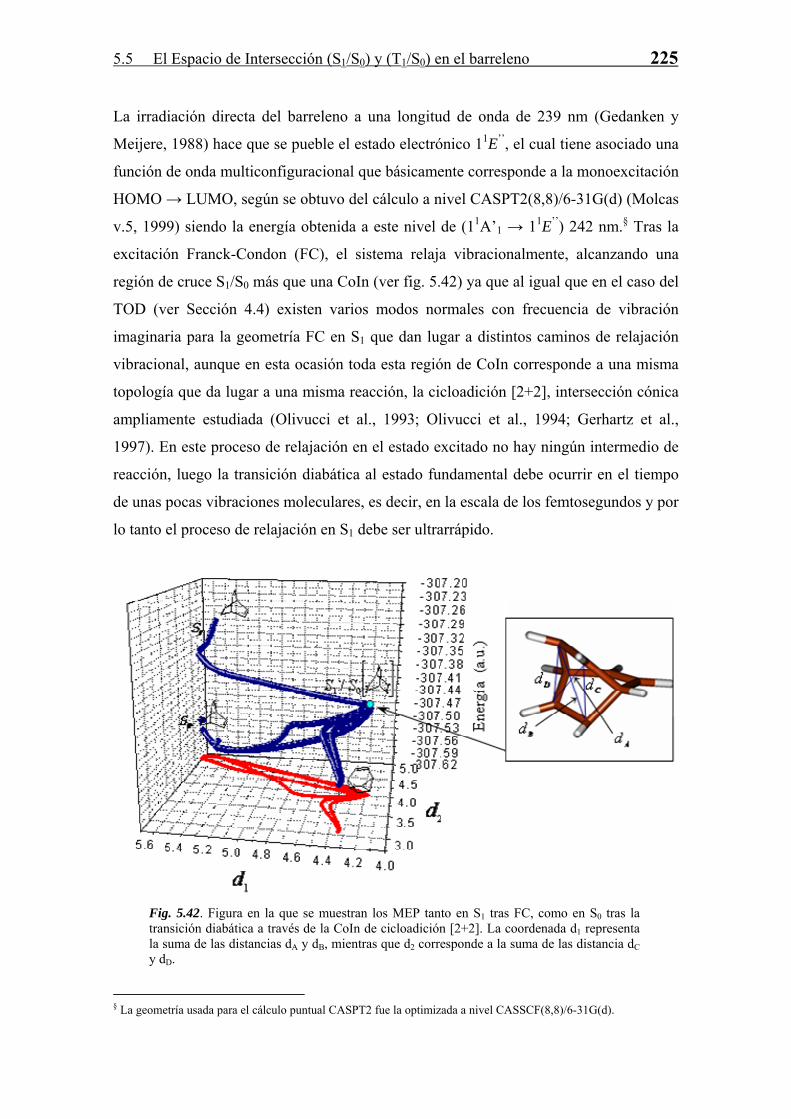
5.5 El Espacio de Intersección (S1/S0) y (T1/S0) en el barreleno 225
La irradiación directa del barreleno a una longitud de onda de 239 nm (Gedanken y
Meijere, 1988) hace que se pueble el estado electrónico 11E’’, el cual tiene asociado una
función de onda multiconfiguracional que básicamente corresponde a la monoexcitación
HOMO → LUMO, según se obtuvo del cálculo a nivel CASPT2(8,8)/6-31G(d) (Molcas
v.5, 1999) siendo la energía obtenida a este nivel de (11A’1 → 11E’’) 242 nm.§ Tras la
excitación Franck-Condon (FC), el sistema relaja vibracionalmente, alcanzando una
región de cruce S1/S0 más que una CoIn (ver fig. 5.42) ya que al igual que en el caso del
TOD (ver Sección 4.4) existen varios modos normales con frecuencia de vibración
imaginaria para la geometría FC en S1 que dan lugar a distintos caminos de relajación
vibracional, aunque en esta ocasión toda esta región de CoIn corresponde a una misma
topología que da lugar a una misma reacción, la cicloadición [2+2], intersección cónica
ampliamente estudiada (Olivucci et al., 1993; Olivucci et al., 1994; Gerhartz et al.,
1997). En este proceso de relajación en el estado excitado no hay ningún intermedio de
reacción, luego la transición diabática al estado fundamental debe ocurrir en el tiempo
de unas pocas vibraciones moleculares, es decir, en la escala de los femtosegundos y por
lo tanto el proceso de relajación en S1 debe ser ultrarrápido.
Fig. 5.42. Figura en la que se muestran los MEP tanto en S1 tras FC, como en S0 tras la transición diabática a través de la CoIn de cicloadición [2+2]. La coordenada d1 representa la suma de las distancias dA y dB, mientras que d2 corresponde a la suma de las distancia dC y dD.
§ La geometría usada para el cálculo puntual CASPT2 fue la optimizada a nivel CASSCF(8,8)/6-31G(d).

226 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Para las reacciones ultrarrápidas como es ésta, los argumentos energéticos antes
descritos en la predicción del camino de reacción en base a la estabilidad de las CoIn
(cuanto más estable es la CoIn más favorable es su accesibilidad) son del todo
inservibles, ya que no existe un equilibrio térmico de especies intermedias que permita
aplicar dicho razonamiento. Se puede hacer una analogía entre control cinético y
termodinámico de una reacción térmica con el control por estabilidad de la Intersección
Cónica o por accesibilidad de la misma. En este caso por lo tanto, es la accesibilidad de
la misma lo que prima por encima de la estabilidad.
Tras la transición diabática, a temperatura ambiente ocurre un fácil reagrupamiento en
el estado fundamental que lleva a la formación del COT, proceso que también ha sido
estudiado experimentalmente (Hassenruck et al., 1989) (Fig. 5.43).
La comparación de los resultados descritos aquí con los de (Reguero et al., 1993) pone
de manifiesto la estabilidad relativa similar entre los dos tipos de CoIn en el barreleno y
el 1,4-pentadieno respectivamente, debido a la similitud estructural y electrónica
existente entre las CoIn equivalentes de cada sistema. Sin embargo, como ya se apuntó,
la reactividad fotoquímica no puede ser comprendida en base a la diferencia de
estabilidad entre estas intersecciones cónicas.
hν
1 0/S S
∆ ∆1CI
BOT COT
hν
2CI SBV
∆ ∆
)a
)b
BARRELENO
BARRELENO
directa
directaultrarrápida
3 0BR en S
Fig. 5.43. Fotoquímica del barreleno tras irradiación directa a) Mecanismo permitido de cicloadición [2+2] b) Mecanismo de reagrupamiento di-π-metano prohibido. (las especies entre corchetes no son mínimos en la SEP). (BOT = Biciclo[4.2.0]octa-2,4,7-trieno,TCO = tetraciclo[4.2.0.02,8.05,7]octeno).

5.5 El Espacio de Intersección (S1/S0) y (T1/S0) en el barreleno 227
De hecho, ambas CoIn (tanto la correspondiente a la cicloadición [2+2] como la
relacionada con la reacción DPM) pertenecen al mismo subespacio de CoIn, es decir,
son mínimos conectados a través de un TS en el subespacio de degeneración energética
entre S0 y S1. La figura 5.44 muestra la estructura aproximada del estado de transición
en este subespacio.
1.84
1.99 2.31
2.01
( )19.34CoIn1
( )0.0CoIn2
( )39.29CoInTS
Fig. 5.44. Estructura de las dos CoIn mínimas en energía, así como la CoIn TS aproximada (perteneciente al subespacio continuo de CoIn) que une ambas CoIn mínimas. CoIn1 se corresponde con la CoIn que da lugar a la cicloadición [2+2], mientras que CoIn2 lo hace con la reacción de reagrupamiento di-π-metano. Las energías relativas en kcal/mol con respecto a la CoIn más estable (a nivel CASSCF(8,8)/6-31G(d)) se dan entre paréntesis.
Por el contrario, la reactividad está controlada por la región de CoIn a la que puede
acceder el sistema tras la transición FC. De esta manera, si la molécula en el estado
fundamental presenta dos grupos etilénicos cercanos y orientados apropiadamente (ver
arriba) el sistema alcanzará probablemente la región de CoIn correspondiente a la
cicloadición [2+2]. Por otro lado, si el sistema presenta flexibilidad conformacional de
tal manera que la molécula en el estado fundamental preserva los grupos etilénicos
separados, la región de CoIn correspondiente a la reacción DPM podrá ser visitada por
la molécula.
De esto concluimos que la reactividad fotoquímica en este tipo de reacciones debe estar
controlada principalmente por las propiedades del estado fundamental (S0), y no por
aquellas del estado excitado (S1), ya que la estabilidad conformacional en el estado
fundamental debe determinar la región de CoIn alcanzada tras la transición FC y por lo
tanto la naturaleza de los productos formados (Fig. 5.45).

228 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
Fig. 5.45. Representación esquemática de las SEP involucradas (S0 y S1) en la cicloadición [2+2] por irradiación del barreleno. La región de la SEP correspondiente al reagrupamiento di-π-metano no es accesible tras la transición FC.
Por lo tanto, debería ser posible controlar los fotoproductos diseñando, por ejemplo, los
sustituyentes de un sistema di-π-metano con el fin de optimizar, en el estado S0, la
conformación deseada.
5.5.2 Reacción di-π-metano vía triplete en el barreleno. Eficiencia de
los canales de cruce de sistemas (T1/S0)
La reacción sensibilizada (vía triplete) presenta una problemática distinta de la no
sensibilizada, casi opuesta. En general (salvo casos excepcionales) los cruces entre
estados de multiplicidad singlete y triplete en moléculas orgánicas que no contienen
átomos pesados son bastante ineficaces en el proceso de salto de estado electrónico. La
molécula requiere un gran número de “visitas” a la región de degeneración energética (o
región de Cruce de Sistemas) para conseguir que se produzca el cambio de estado,
debido a que es una transición prohibida por simetría (de espín). Solamente la inclusión

5.5 El Espacio de Intersección (S1/S0) y (T1/S0) en el barreleno 229
del acoplamiento espín-órbita en el hamiltoniano puede hacer dicha transición
débilmente permitida. Esto contrasta con la alta eficiencia (del 100% cuando existe
degeneración energética) de los cruces entre estados de la misma multiplicidad (CoIn).
Por lo tanto, como se verá a continuación para la reacción DPM sensibilizada, el control
de la reacción en general viene dado (aunque se hará una matización de este extremo)
por la estabilidad de las zonas de degeneración energética, al contrario de lo que ocurre
en la reacción DPM directa.
La irradiación del barreleno en presencia de un sensibilizador triplete, lleva a la
formación del producto de reagrupamiento DPM con un alto rendimiento (Zimmerman
et al., 1967). El mecanismo propuesto por Zimmerman y colaboradores (Zimmerman y
Grunewald, 1966; Zimmerman et al., 1967; Zimmerman y Mariano, 1969; Zimmerman,
1980) se ratifica en base al estudio detallado de las SEP involucradas en el proceso,
estudio realizado a nivel CASSCF(8,8)/6-31G(d) (Gaussian98, 1998).
La reacción DPM tiene lugar en cuatro etapas (Fig. 5.46).
hν3 sens
≠
≠
ISC
1BR 2BR1TS
2TSSBV ( )03BR S ( )13BR T
Fig. 5.46. Fotoquímica del barreleno por irradiación en presencia de sensibilizadores triplete, dando lugar al reagrupamiento di-π-metano. (las especies entre corchetes no son mínimos en la SEP).
En una primera etapa, tras la excitación en presencia de sensibilizador, el barreleno (en
T1) relaja hasta alcanzar un birradical-1,2 (BR1). Este BR1 (mínimo en T1) no presenta
los orbitales p semiocupados perpendiculares, como ocurre en compuestos análogos sin
las restricciones estructurales del barreleno. Existe un punto de cruce de sistemas (ISC)
mínimo en energía en el subespacio de cruce entre T1 y S0 que corresponde con esta
situación, pero se halla situado a 39 kcal/mol respecto de dicho mínimo, lo que hace que
esta vía de decaimiento a S0 esté impedida, cruce de sistemas que por otro lado, si fuese

230 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
un canal efectivo de decaimiento al estado fundamental, revertiría de nuevo al
barreleno.
El motor de la reacción es la minimización de las repulsiones electrónicas entre los dos
electrones de mismo espín. Posteriormente a la formación de BR1, se forma un
birradical-1,3 (BR2) y después un 1-alil,3-birradical (BR3), el cual es el más estable de
todos ellos. Estos dos birradicales ya habían sido identificados en la SEP de nafto-
barrelenos (Zimmerman et al., 1994). A medida que el sistema entra en la región de
BR2, las SEP singlete (S0) y triplete (T1) se aproximan, llegando a una situación de
cuasi-degeneración energética hasta BR3 (Fig. 5.47).
Fig. 5.47. Superficie de energía potencial esquemática de los estados T1 y S0, involucrados en la reacción vía triplete di-π-metano, cuyo camino de relajación en T1 se indica con flechas. La especie birradicálica más estable (BR3), la región de cruce de sistemas (ISC), así como el producto final de la fotorreacción, el semibulvaleno (SBV) se muestran en la figura.
Sin embargo, no solamente el birradical BR3 es el más estable de todos, sino que
además es el que presenta un mayor acoplamiento espín-órbita. Esto es debido a que los
orbitales semiocupados son paralelos (aproximadamente) en el BR2, mientras que son
prácticamente perpendiculares en BR3. Esto tiene un efecto claro en la constante de
velocidad de ISC, debido a la fuerte dependencia angular del término energético del
acoplamiento espín-órbita 1 SOT H S0 , como ya se pusiera de manifiesto en el
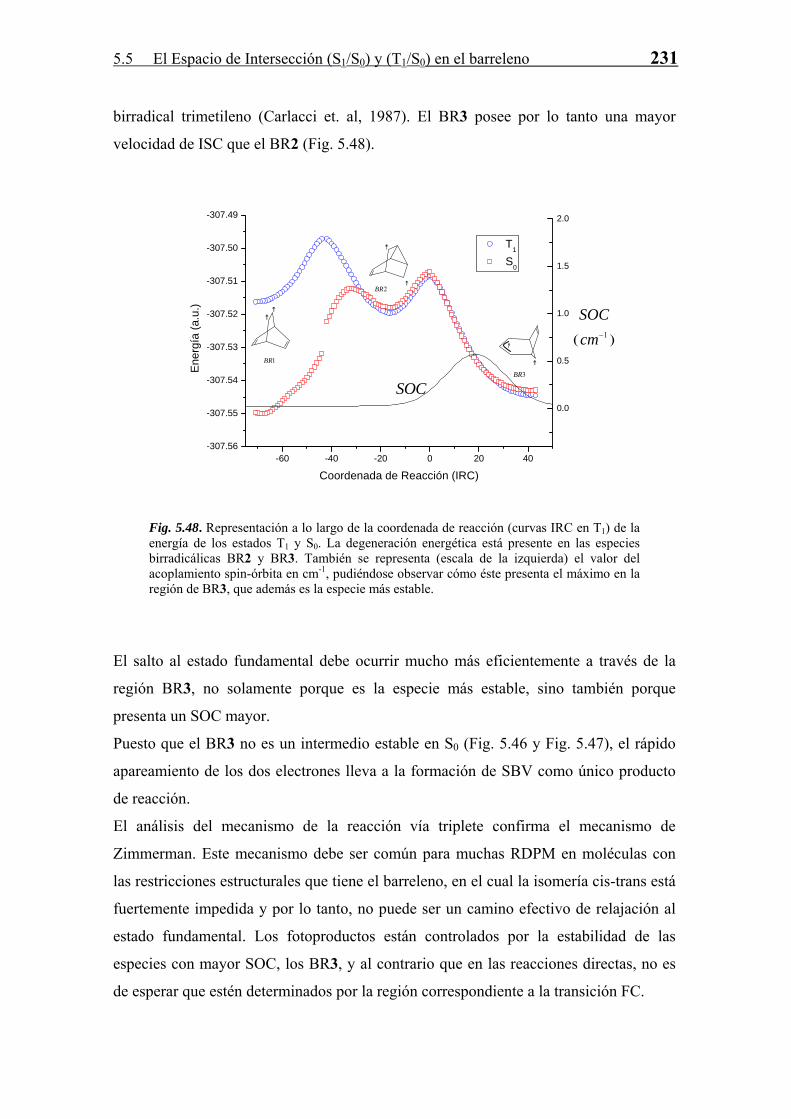
5.5 El Espacio de Intersección (S1/S0) y (T1/S0) en el barreleno 231
birradical trimetileno (Carlacci et. al, 1987). El BR3 posee por lo tanto una mayor
velocidad de ISC que el BR2 (Fig. 5.48).
-60 -40 -20 0 20 40-307.56
-307.55
-307.54
-307.53
-307.52
-307.51
-307.50
-307.49
Ene
rgía
(a.u
.)
Coordenada de Reacción (IRC)
T1
S0
0.0
0.5
1.0
1.5
2.0
1BR
2BR
3BR
SOC
( )1
SOCcm−
Fig. 5.48. Representación a lo largo de la coordenada de reacción (curvas IRC en T1) de la energía de los estados T1 y S0. La degeneración energética está presente en las especies birradicálicas BR2 y BR3. También se representa (escala de la izquierda) el valor del acoplamiento spin-órbita en cm-1, pudiéndose observar cómo éste presenta el máximo en la región de BR3, que además es la especie más estable.
El salto al estado fundamental debe ocurrir mucho más eficientemente a través de la
región BR3, no solamente porque es la especie más estable, sino también porque
presenta un SOC mayor.
Puesto que el BR3 no es un intermedio estable en S0 (Fig. 5.46 y Fig. 5.47), el rápido
apareamiento de los dos electrones lleva a la formación de SBV como único producto
de reacción.
El análisis del mecanismo de la reacción vía triplete confirma el mecanismo de
Zimmerman. Este mecanismo debe ser común para muchas RDPM en moléculas con
las restricciones estructurales que tiene el barreleno, en el cual la isomería cis-trans está
fuertemente impedida y por lo tanto, no puede ser un camino efectivo de relajación al
estado fundamental. Los fotoproductos están controlados por la estabilidad de las
especies con mayor SOC, los BR3, y al contrario que en las reacciones directas, no es
de esperar que estén determinados por la región correspondiente a la transición FC.

232 5 FOTOQUÍMICA II. EL ESPACIO DE INTERSECCIÓN
55..66 CCoonncclluussiioonneess
El espacio de intersección tiene una gran importancia en distintos procesos
fotoquímicos en los que están involucradas transiciones diabáticas. Distintos ejemplos
se han visto en este capítulo que ponen de manifiesto tal circunstancia. Por ejemplo, la
compleja evolución del COT tras la excitación óptica al estado S2 hasta el estado S1
involucra un cruce evitado entre estados y una intersección cónica S2/S1 a través de la
cual se puebla el estado S1. Posteriormente, en el proceso de decaimiento desde el
estado excitado S1 al estado fundamental, existen una serie de cruces diabáticos
(Intersecciones Cónicas) que pueden servir al sistema para decaer al estado
fundamental. Se ha visto como muchas de esas vías de decaimiento pertenecen al
mismo espacio de intersección, en el cual en gran medida se puede racionalizar la
estabilidad de las distintas intersecciones cónicas en base a sus propiedades electrónicas
y estructurales. Se ha estudiado sistemáticamente el espacio de intersección (S1/S0)
relacionado con el COT, incluyendo en este estudio intersecciones cónicas con distinto
número de instauraciones. Un estudio en base a las propiedades del espacio de
ramificación permite predecir el conjunto de productos de reacción (fotoproductos) que
pueden formarse a partir de cruces diabáticos que involucran a dichas intersecciones
cónicas.
Por último, se ha estudiado la importancia del espacio de intersección (tanto S1/S0 como
T1/S0) en una reacción muy importante que sufre un isómero de valencia del COT, el
barreleno. Las reacciones di-π-metano conforman un tipo de reacciones destacado en
química orgánica. Su mecanismo implica cruces diabáticos entre estados tanto de la
misma como de distinta multiplicidad de espín, por lo que el estudio de este espacio es
fundamental para ahondar en el mecanismo de la reacción y su modulación. En
concreto, se han presentado las características topológicas de este espacio así como
posibles formas de controlar la reacción en base a las propiedades del mismo. El
conocimiento de las propiedades del espacio de intersección puede ser de gran ayuda en
la comprensión de los mecanismos de reacción en numerosos sistemas, y su estudio
teórico siempre complicado, puede ser de gran utilidad en el plano experimental.