practical protein sequence alignment with algebraic dynamic programming
DESCRIPTION
Practical Protein Sequence Alignment With Algebraic Dynamic Programming. Lyle Kopnicky PacSoft Research Group Tim Sheard, Adviser. Bioinformatics. DNA, RNA and proteins are strings Strings contain information Some problems Determine relatedness of strands of DNA - PowerPoint PPT PresentationTRANSCRIPT
Practical Protein Sequence Alignment With Algebraic Dynamic Programming
Lyle Kopnicky
PacSoft Research Group
Tim Sheard, Adviser

Bioinformatics
• DNA, RNA and proteins are strings• Strings contain information• Some problems
• Determine relatedness of strands of DNA• Figure out how RNA folds on itself• Identify proteins in a sample
GTTAGCGTGAATCTGTACTGAG

Tools for bioinformatics• Written in a general-purpose
programming language such as C• Designed to solve a narrow range of
problems• When problem doesn’t fit tool:
• Tweak data to fit tool – awkward, inefficient, may not fully solve problem
• Write new tools – time consuming, error-prone, require maintenance

The disconnect
#ifndef SS strncpy(pgm_name, "gsw", MAX_FN);#else strncpy(pgm_name, "ssw", MAX_FN);#endif standard_pam("BL50",ppst);
ppst->nsq = naa; ppst->nsqx = naax; for (i=0; i<=ppst->nsqx; i++) { ppst->sq[i]=aa[i]; /* sq = aa */ ppst->hsq[i]=haa[i]; /* hsq = haa */ ppst->sqx[i]=aax[i]; /* sq = aax */ ppst->hsqx[i]=haax[i]; /* hsq = haax */ } ppst->sq[ppst->nsqx+1] = ppst->sqx[ppst->nsqx+1] = '\0'; memcpy(qascii,aascii,sizeof(qascii)); /* set up the c_nt[] mapping */ ppst->c_nt[0]=0; for (i=1; i<=nnt; i++) { ppst->c_nt[i]=gc_nt[i]; ppst->c_nt[i+nnt]=gc_nt[i]+nnt; }}

The disconnect
• General purpose languages are easily available, efficient, BUT
• Biologists think: amino acids, matching, classification
• General-purpose languages provide characters, procedures, objects
• Domain experts may not be expert programmers
• A lot of design, development and maintenance time

Domains and tools
DomainDomain
Domain
Domain
Bioinformatics
Tool
Tool
Tool
Architecture
Physics
Finance
ChemistryMathematics
General-purpose languages

Libraries

Domain-specific languages• Represent a problem in the way domain
experts think of it• Examples: Excel, HTML, Matlab• General enough to capture a domain, specific
enough to reduce design time• Small change in requirements = small change
in program• Easy to answer “what-if” questions• Can be implemented efficiently using known
techniques

Collaboration with OHSU lab• Dr. Srinivasa Nagalla’s laboratory• Goals
• Gain domain knowledge• Discover goals of biologists• Potential users of DSL
• Began with protein identification problem

Protein identification problem
?
?
?
?
SD gel
breakup into peptideschromatography

Protein identification problem
tandem mass spectrometry
RPRR
AVA
A
AVAQFQFPRR
AVAQFPRR
AV
AQ
FP
RR
AVAQFPRR
de novo sequencing
[Po]QFPVGR
A[AV]QFPVGR
[Po]QFPRR
[241.10]QFPVGR
A[AV]QFPRR
database search
?
…TRSSRAGLQFPVGRVHRLLR…
[241.10]QFPVGR
AVAQFPRR
AVAQFPRR

Database search• Not an exact match• Mutations – substitutions, insertions,
deletions• Modifications – amino acids altered by
another molecule• De novo sequencer outputs a list of
ambiguous queries, e.g.
[229.07]HhNyG[PS][198.1]QHADD[ep]VD[Rz]R
unidentified mass unordered pair

Sequence alignment• Find the best match between query
string and target (database) string• Each match is also called an alignment• Alignments are scored
WTABRRFCWGYPD KWGGSCASPNE F WT PDPYK
Target string
Query string

Alignment tools• Smith-Waterman dynamic programming
algorithm most common• O((n+m)2), where n and m are length of
strings• Run on every query-target pair, and data
sets are large• Requires precise query string
• FASTA, BLAST address speed problem• First look for runs of exact matches• Align on localized area

Fitting queries into FASTA• Generate all possible exact queries• Could be dozens of possibilities
NQQNGGGANGNAGQGGGQAGGG
[241.10]NEM[NP]YR
NP
PN
• Each one must be aligned – takes time• Output must be converted back to
original query string

Algebraic Dynamic Programming• Robert Giegerich, 2000• Generic approach to solving dynamic
programming problems using parsing• Domain-specific embedded language in
Haskell• Ambiguous queries can be represented
directly as a grammar• Searching with an ambiguous query slower,
but only one search instead of dozens• Still takes O((n+m)2) time
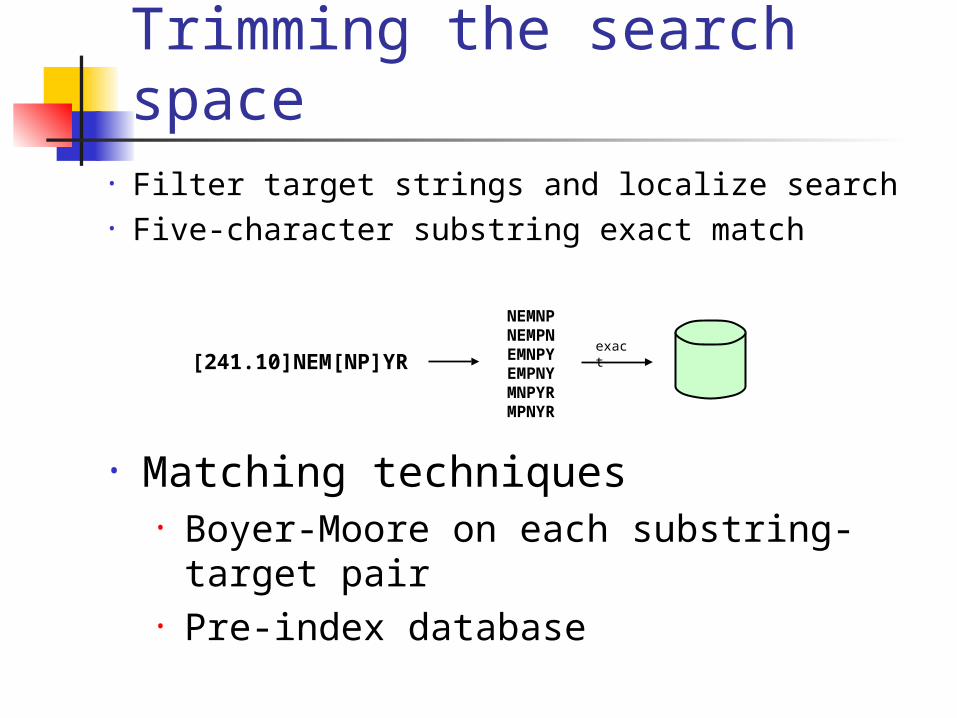
Trimming the search space• Filter target strings and localize search• Five-character substring exact match
NEMNPNEMPNEMNPYEMPNYMNPYRMPNYR
[241.10]NEM[NP]YRexact
• Matching techniques• Boyer-Moore on each substring-target pair• Pre-index database

Boyer-Moore• Finds an exact occurrence of one string
inside another• Doesn’t check every position – knows
when to skip ahead• Can run in sublinear time
SYNSNTLNNDIMLIKLKSAASLN xSAASL
SYNSNTLNNDIMLIKLKSAASLN x SAASL

Pre-indexing the database• Build a tree in which to look up
substrings, find positions in database• Substring tree: A substring at each node• Suffix tree: Path along labeled edges
describes substring
substring tree suffix tree
TADTA
ATD AD
TA
TAD, 2ADT, 2
DTA, 3
32
1

Sample output
Query string:[229.07]HhNyG[PS][198.1]QHADD[EP]VD[Rz]R:{21}
Target string:>MK14_HUMAN (Q16539) Mitogen-activated protein kinase 14MSQERPTFYRQELNKTIWEVPERYQNLSPVGSGAYGSVCAAFDTKTGLRVAVKKLSRPFQSIIHAKRTYRELRLLKHMKHENVIGLLDVFTPARSLEEFNDVYLVTHLMGADLNNIVKCQKLTDDHVQFLIYQILRGLKYIHSADIIHRDLKPSNLAVNEDCELKILDFGLARHTDDEMTGYVATRWYRAPEIMLNWMHYNQTVDIWSVGCIMAELLTGRTLFPGTDHIDQLKLILRLVGTPGAELLKKISSESARNYIQSLTQMPKMNFANVFIGANPLAVDLLEKMLVLDSDKRITAAQALAHAYFAQYHDPDDEPVADPYDQSFESRDLLIDEWKSLTYDEVISFVPPPLDQEEMES:{360}
Target range: 290-326LDSDKRITAAQALAHA YFAQYHDPDDEPVADPYDQSF :|XvvvXX:|^|X^||//|^|XXX -HhNyGPS-Q HA DDEPV DRzR Score: 31Time: 0.045 secs

Time and space usage
Small set Full set
Number of queries 5 4800
Query length 7–17 up to 21
Number of target strings 179 8500
Target string length up to 700 up to 7000
Small set Full set
time time space
No heuristic 2m35s — —
Boyer-Moore 3s — —
Substring tree pre-indexing 1s 5m 750MB
lookup 1s 1h49m 1GB

Smith-Waterman (1981)
• Local alignment problem• Dynamic programming algorithm• Pathways through table represent
alignments• Entry represents best score of an
alignment starting here, ending anywhere

S.-W. alignment scoring
WTABRRFCWTYPDG WKGGSCASPNE
GE F WT PDPYDAW QAPT
match/substitution: s(a1,a2)
gap: -w x length
start/end = 0
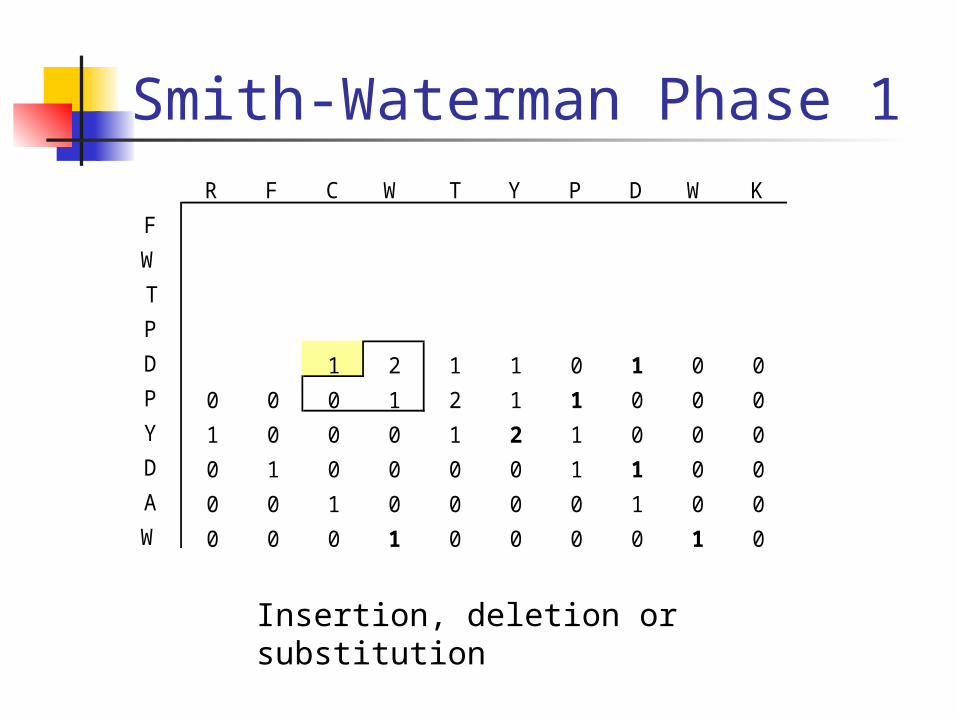
Smith-Waterman Phase 1R F C W T Y P D W K
F
W
T
P
D 1 2 1 1 0 1 0 0
P 0 0 0 1 2 1 1 0 0 0
Y 1 0 0 0 1 2 1 0 0 0
D 0 1 0 0 0 0 1 1 0 0
A 0 0 1 0 0 0 0 1 0 0
W 0 0 0 1 0 0 0 0 1 0
Insertion, deletion or substitution

Smith-Waterman Phase 2R F C W T Y P D W K
F 2 3 3 2 1 0 0 1 0 0
W 2 1 2 3 2 1 0 0 1 0
T 1 2 1 1 2 2 1 0 0 0
P 0 1 2 1 1 1 2 0 0 0
D 0 0 1 2 1 1 0 1 0 0
P 0 0 0 1 2 1 1 0 0 0
Y 1 0 0 0 1 2 1 0 0 0
D 0 1 0 0 0 0 1 1 0 0
A 0 0 1 0 0 0 0 1 0 0
W 0 0 0 1 0 0 0 0 1 0
a
Trace back maximal pathways CWTYPD FWT PD
a

Today’s problems
transpositions
AN NA
one-to-many
W NAC
endpoint scoring
FGAK +5 AGNCF... 85 116 39 85 100...
dual representations
We need a way to model new problems quickly
Trying to fit new data into old tools…

Recurrence relations• Basis of traditional DP• Hard to design and
understand• Mixes together search space,
scoring, and order of evaluation
• Subscript errors are common in implementation
}0,,)),(),((max{ ,11,211,1, wHwHjSiSsHH jijijiji
Smith-Waterman recurrence relation

Algebraic Dynamic Programming• Giegerich, 2000• Grammars describe search space• Set of functions (evaluation algebra)
specifies scoring• Solution space is pared down by an
objective function
first string $ gnirts dnoces

Grammar & eval. algebra
localign = start <<< string ~~~ internal ~~~ string
... h
internal = subst <<< aa ~~~ internal ~~~ aa |||delete <<< aa ~~~ internal |||insert <<< internal ~~~ aa |||end <<< string ~~~ ’$’ ~~~ string... h
subst(a1,score,a2) = score + s(a1,a2)insert(score,a) = score – wdelete(a,score) = score – wend(str1,$,str2) = 0h[score1,...,scorek] = max[score1,...,scorek]

Traditional DP vs. ADP
Traditional DP solutions ADPTricky to design & implement recurrence relations
Grammar and algebra like you think, no subscripts
Difficult to extend to alternate problem descriptions
Just change grammar and evaluation algebra
Careful about order of evaluation
Order of evaluation automatic
Time complexity depends on recurrence relation
Time complexity depends on form of grammar
Very fast in C Haskell can be slow

Design of a DSL• Implement sample applications
• Use a flexible, higher-order language• Abstract out common themes
• Data structures• Operations
• Decide how to handle errors• Run-time errors• Type system
• Speed up implementation by generating C• From embedded language, like PAN• Using standalone compiler

The next steps• Compare our alignments with biologists’• Accumulate protein scores• Try 2-3 character statistical matches• Pre-index using suffix trees• Reduce memory usage• Look for runs of exact matches as in
FASTA

Our contributions• Built a working relationship with bio lab
to understand domain• Demonstrated feasibility of aligning
large data sets in Haskell• Constructed a framework for
experimenting with representations for alignments, and heuristics for filtering target strings, localizing searches