porting and optimizing gpaw - prace training … · porting and optimizing gpaw jussi enkovaara...
TRANSCRIPT

Porting and optimizing GPAW
Jussi EnkovaaraComputing Environment and Applications
CSC – the finnish IT center for science

OutlineOverview of GPAW
• Features• Programming model
Porting• Power 6, Cray XT4/5, Blue Gene P
Performance and optimization for standard density functional theory
• Theoretical background• Implementation details• Serial performance• Parallel performance and enhancements
Performance for time-dependent density functional theorySummary

Acknowledgements All the development is done in close collaboration with the
whole GPAW development team:• Argonne National Laboratory, USA• CAMd, Technical University of Denmark• Department of Physics, Helsinki University of Technology, Finland• Department of Physics, Jyväskylä University, Finland• Freiburg Materials Research Center, Germany• Institute of Physics, Tampere University of Technology, Finland
Key developers in optimization efforts• Jens Jörgen Mortensen, DTU• Marcin Dulak, DTU• Nichols Romero, Argonne

GPAW Software package for quantum mechanical calculations
of nanostructures Ground state properties
• Total energies, structural optimizations, ...• Density functional theory
Excited state properties• Optical spectra, ...• Time-dependent density functional theory
System sizes up to few nm, few thousand electrons Open source software licensed under GPL
wiki.fysik.dtu.dk/gpaw

Programming model: Python + C
Python• Modern, general purpose object oriented
programming language• Interpreted language• Rapid development• Easy to extend with C (and Fortran) routines
Message passing interface (MPI) Core low level routines implemented in C
• MPI-calls directly from C
High level algorithms implemented in Python
• Python interfaces to BLAS and LAPACK• Python interfaces to MPI
Execution time:
Lines of code:
Python C
C
BLAS, LAPACK, MPI, numpy

Programming model Python makes it easy to focus on efficient high level algorithms
Installation can be be intricate in special operating systems
• Catamount, CLE• BlueGene
Debugging and profiling tools are often only for C or Fortran programs
• C-extensions can be profiled easily• Possible to have Python-interfaces to profiling
API such as CrayPAT
# Calculate the residual of pR_G # dR_G = (H - e S) pR_G hamiltonian.apply(pR_G, dR_G, kpt) overlap.apply(pR_G, self.work[1], kpt) axpy(-kpt.eps_n[n], self.work[1], dR_G)
RdR = self.comm.sum(real(npy.vdot(R_G, dR_G)))
Python code sniplet from iterative eigensolver

Porting Software requirements for GPAW
• Python• Numpy (Python package for numerical computations)• LAPACK, BLAS (SCALAPACK)• MPI
Python distutils• “makefile” for numpy and GPAW• Use same compiler and options as when building Python• Straightforward installation in standard Unix-systems• Cross compilation can be more difficult
For parallel calculations GPAW uses special Python interpreter:
int main(int argc, char **argv){ int status; MPI_Init(&argc, &argv); status = Py_Main(argc, argv); MPI_Finalize(); return status;}

Porting to Power6 on Linux
Standard Unix system Current porting with gcc Straightforward porting wiki.fysik.dtu.dk/gpaw/install/Linux/huygens.htm

Porting to Cray XT
Python is not supported in Catamount or CNL operating systems No shared libraries
• Normally, the python standard library consists of pure Python modules and of C-extensions
• C-extensions are dynamically loaded libraries
All the needed C-extensions have to build statically into libpython.a
Custom dynamic Python module loader Python is needed on front end node for build process Python is need on compute nodes for calculations Once Python is ported, building Numpy and GPAW is relatively
straigthforward wiki.fysik.dtu.dk/gpaw/install/Cray/louhi.html

Porting to Blue Gene P
Python is supported on Blue Gene P Shared libraries are supported on Blue Gene P Python is build with gcc, using xlc for GPAW requires
conversion of gcc options to xlc options Cross compilation can be tricky:
• using accidently front-end libraries produces code which works 95 % of test cases
• it can be difficult to locate the errors to wrong libraries
wiki.fysik.dtu.dk/gpaw/install/BGP/jugene.html

Density functional theory
Theoretical background Numerical methods and algorithms
• Uniform real-space grids• Multigrid method
Serial performance Parallel enhancements and performance
• mostly for Cray XT

Density functional theory
Many-body Schrödinger equation
Can be solved exactly for single electron 3N degrees of freedom, practical systems can
contain thousands of electrons Computational and storage requirements scale as
O(exp(N)) Hohenberg and Kohn: ground state properties are
unique functionals of the electron density

Density functional theory Kohn-Sham equations
Self-consistent set of single particle equations for N electrons First-principles calculations, atomic numbers are the only input
parameters Currently, maximum system sizes are typically few hundred atoms
or 2-3 nm

Projector augmented wave (PAW) method
Only valence electrons are chemically active Wave functions vary rapidly near atomic nuclei In projector augmented wave (PAW) method core electrons
are frozen and one works with smooth pseudo wave functions Wave functions and expectation values can be represented as
= + -
Smooth part Atomic corrections
PAW method introduces non-local term in effective potential

Real space grids Wave functions, electron densities, and potentials are represented
on uniform grids. Single parameter, grid spacing h
h
Accuracy of calculation can be improved systematically by decreasing the grid spacing

Finite differences
Both the Poisson equation and kinetic energy contain the Laplacian which can be approximated with finite differences
Accuracy depends on the order of the stencil N Sparse matrix, storage not needed Cost in operating to wave function is proportional to the number of
grid points

Multigrid method General framework for solving differential equations using a
hierarchy of discretizations
Recursive V-cycle Transform the original equation to a coarser
discretization• restriction operation
Correct the solution with results from coarser level
• interpolation operation

Self-consistency cycle0)Initial guess for the density and wave functions
1)Calculate HamiltonianPoisson equation is solved with multigridO(N) scaling with system size
2)Subspace diagonalization of wave functionsConstruct Hamiltonian matrixDiagonalize matrixRotate wave functionsO(N3) scaling
3)Iterative refinement of wave functionsPreconditioning with multigridO(N2) scaling
4)Orthonormalization of wave functionsCalculate overlap matrixCholesky decompositionRotate wave functionsO(N3) scaling
5)Calculate new charge density
6)Return to 1 until converged
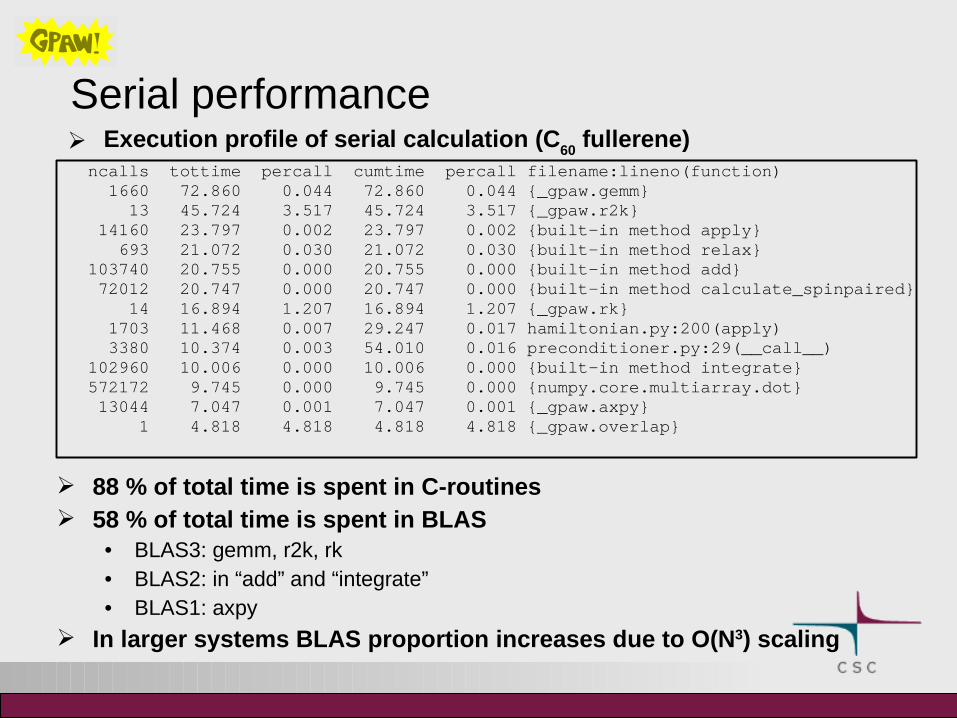
Serial performance Execution profile of serial calculation (C
60 fullerene)
ncalls tottime percall cumtime percall filename:lineno(function) 1660 72.860 0.044 72.860 0.044 {_gpaw.gemm} 13 45.724 3.517 45.724 3.517 {_gpaw.r2k} 14160 23.797 0.002 23.797 0.002 {built-in method apply} 693 21.072 0.030 21.072 0.030 {built-in method relax} 103740 20.755 0.000 20.755 0.000 {built-in method add} 72012 20.747 0.000 20.747 0.000 {built-in method calculate_spinpaired} 14 16.894 1.207 16.894 1.207 {_gpaw.rk} 1703 11.468 0.007 29.247 0.017 hamiltonian.py:200(apply) 3380 10.374 0.003 54.010 0.016 preconditioner.py:29(__call__) 102960 10.006 0.000 10.006 0.000 {built-in method integrate} 572172 9.745 0.000 9.745 0.000 {numpy.core.multiarray.dot} 13044 7.047 0.001 7.047 0.001 {_gpaw.axpy} 1 4.818 4.818 4.818 4.818 {_gpaw.overlap}
88 % of total time is spent in C-routines 58 % of total time is spent in BLAS
• BLAS3: gemm, r2k, rk• BLAS2: in “add” and “integrate”• BLAS1: axpy
In larger systems BLAS proportion increases due to O(N3) scaling

Parallelization strategies
Domain decomposition Parallelization over k-points and spin Parallelization over electronic states

Parallelization over k-points and spin
In (small) periodic systems parallelization over k-points• almost trivial parallelization• number of k-points decreases with increasing system size
Parallelization over spin in magnetic systems• almost trivial parallelization• only two spin degrees of freedom
Communication only when summing for charge density

Domain decomposition
Domain decomposition: real-space grid is divided to different processors
Only local communication Total amount of communication is
small Finite-difference:
• computation NG
/ P
• communication ( NG / P ) 2/3
Finite difference Laplacian
P1
P3 P4
P2
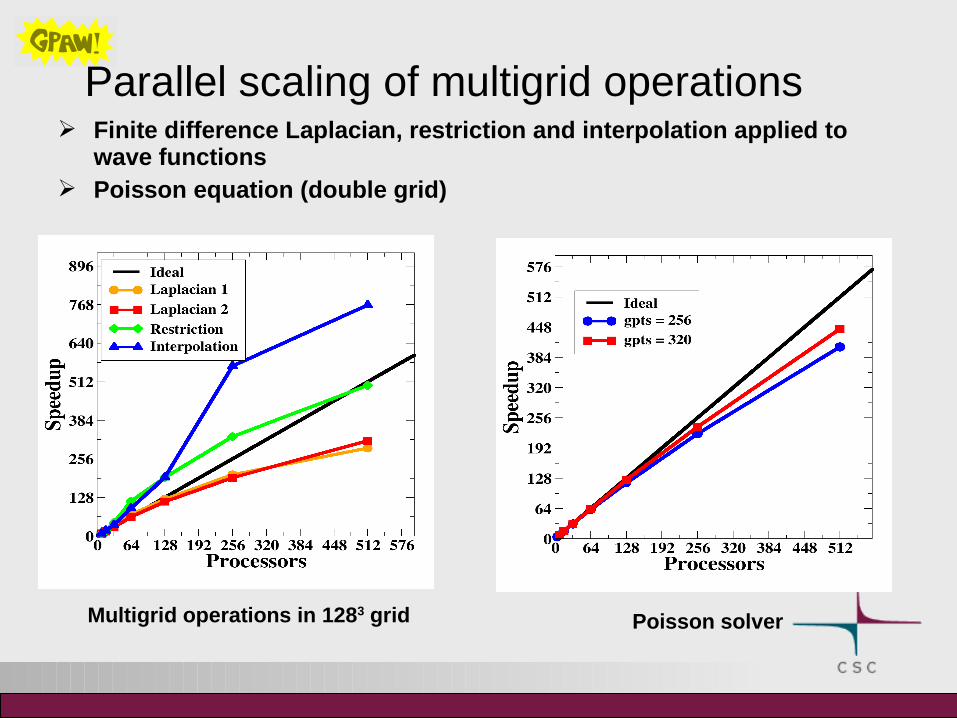
Parallel scaling of multigrid operations Finite difference Laplacian, restriction and interpolation applied to
wave functions Poisson equation (double grid)
Poisson solverMultigrid operations in 1283 grid

Parallel performance with domain decomposition Test system: 256 water molecules, 768 atoms, 1056
electrons, 112 x 112 x 112 grid
Problem:• Matrix diagonalization in subspace rotation takes 270 s and it is
done serially!
Solution:• Use Scalapack for diagonalization
procs time (s) speedup
128 571
256 452 1.26

Domain decomposition and Scalapack Matrix diagonalization with Scalapack, 1056 x 1056 matrix
procs time (s) speedup
4 20 13
16 14 1.4
256 14 1
procs time (s) speedup
128 312
256 191 1.63
512 158 1.20
Whole calculation, 16 processors for Scalapack
Matrix diagonalization no longer bottleneck, however domain size of 14 x 14 x14 starts to be too small

Parallelization over electronic states
Non-trivial parallelization of eigenvalue problem• eigensolvers require orthonormalization• in practice, also subspace diagonalization is needed
Construction of matrices like
Matrix-vector products like
All-to-all communication of wave functions Large messages

Parallelization over electronic states 1D block cyclic distribution of wave functions Pipeline with overlapping computation and
communication• communication only between nearest neighbors
Example: 4 processors, 3 steps• step 0, the diagonal elements are calculated and
the same time the blocks needed for diagonal+1 elements are exchanged
• step 1, diagonal+1 elements are calculated while the next blocks are exchanged
• step 2, diagonal+2 elements are calculated while the next blocks are exchanged.
• step 3, diagonal+3 elements are calculated. For symmetric matrices step 3 is not needed
Generally, B / 2 + 1 pairs of send-receives (B=number of processors for state parallelization)
0 1 B-1...
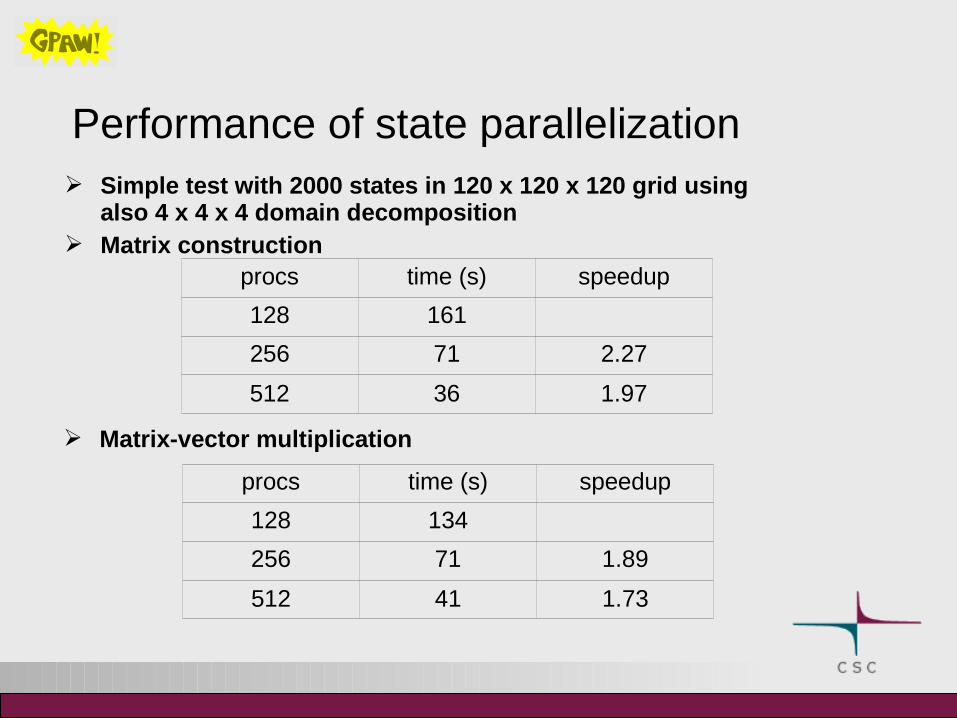
Performance of state parallelization Simple test with 2000 states in 120 x 120 x 120 grid using
also 4 x 4 x 4 domain decomposition Matrix construction
procs time (s) speedup
128 161
256 71 2.27
512 36 1.97
Matrix-vector multiplication
procs time (s) speedup
128 134
256 71 1.89
512 41 1.73

Rank placement
In Cray, large random variations in the execution time of matrix construction + matrix vector product
• with 512 cores, maximum time 140 s, minimum time 80 s
Ranks 0-63 contain first group of states, ranks 64-127 second group of states etc.
By default, processes are placed to nodes by increasing rank number
State parallelization involves large (10-40 MB) messages In realistic situation, bandwidth depends on physical
distance between the nodes Custom rank placement with MPICH_RANK_ORDER file:
• processes which exchange large messages are close to each other, i.e placement goes like 0,64,128,...
• coherent execution time of 75 s
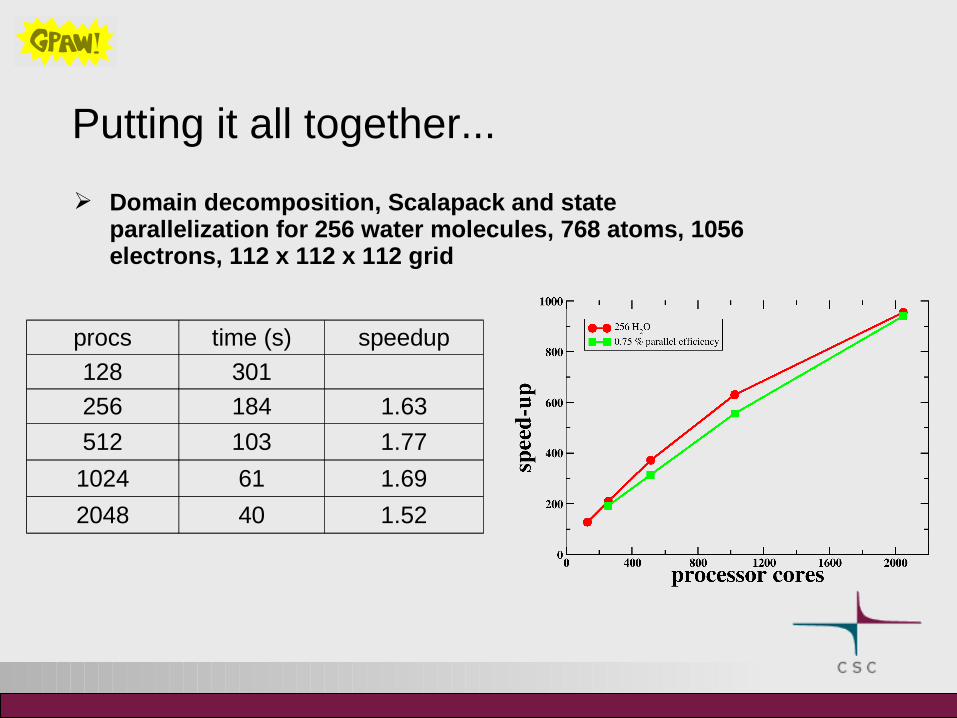
Putting it all together...
Domain decomposition, Scalapack and state parallelization for 256 water molecules, 768 atoms, 1056 electrons, 112 x 112 x 112 grid
procs time (s) speedup
128 301
256 184 1.63
512 103 1.77
1024 61 1.69
2048 40 1.52

Things neglected by now
Initialization phase of the calculation does not scale so well
• initial guess for the wave functions• when python is started, lots of small files are read• in Cray, startup time increases rapidly with increasing processor
count• in production calculations initialization time should not be so bad,
however in small benchmarks it can be significant
GPAW (or DFT in general) is not very IO-intensive• in practice users might want restart files• for petascale production calculations parallel IO might be
necessary

Limiting factors in parallel scalability
Atomic spheres are not necessarily divided evenly to domains
• load imbalance which limits scalabilty of domain decomposition
Calculation of Hamiltonian• Can be parallelized only over domains
Matrix diagonalization• Scalapack scalability is limited due to modest matrix sizes
Communication patterns of finite-difference operations are not optimal, possibility for small enhancements

Time-dependent density-functional theory Generalization of density-functional theory also to time-
dependent cases Excited state properties
• excited state energies• optical absorption spectra• ...
Time-dependent Kohn-Sham equations

Parallelization schemes for TD-DFT
Similar parallelization possibilities than in DFT• domain decomposition• spins
Time-propagation schemes conserve orthonormality• parallelization over electronic states is almost trivial,
communication only when summing for charge density
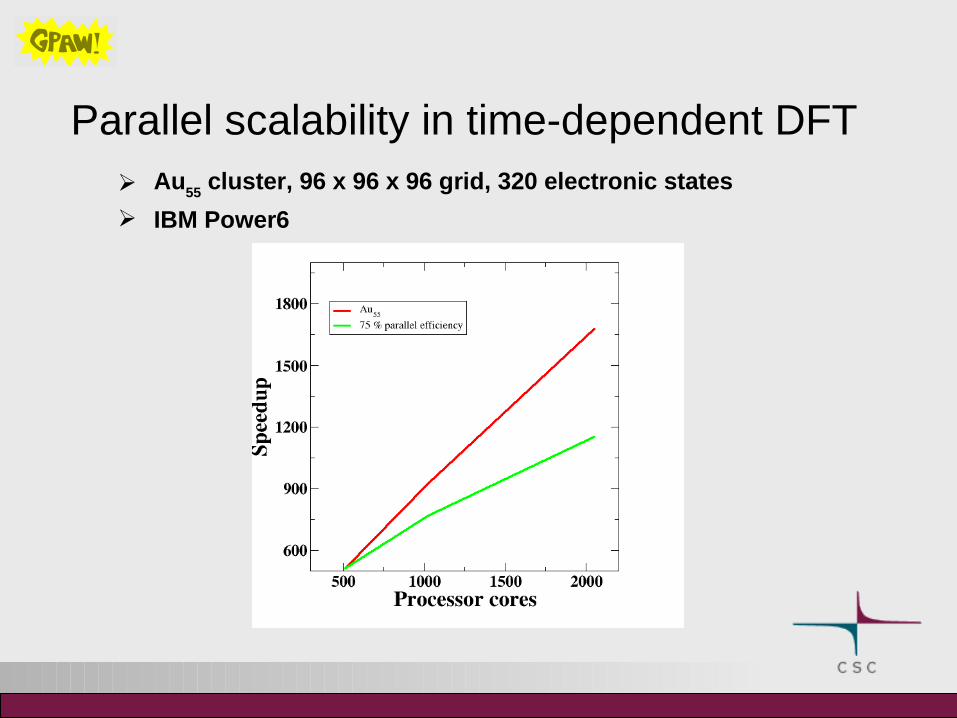
Parallel scalability in time-dependent DFT Au
55 cluster, 96 x 96 x 96 grid, 320 electronic states
IBM Power6

Possible enhancements for TDDFT
TD DFT uses only BLAS 1 routines, serial optimizations are more plausible than in DFT part
• Converting some Python routines to C
Optimizing communication of finite difference operations• probably larger benefits than in standard DFT
Algorithm is memory bandwidth limited, difficult to obtain very high FLOP rates

Summary and final thoughts Python + C programming model can be used for high
performance computing• porting especially in cross-compilation environments can be
tricky
Density functional theory code can be parallelized efficiently with multilevel parallelism
• rank placement can be important in petascale systems• large systems could scale to over 10 000 cores
Time-dependent density functional calculations scales extremely well
• 4000 cores already on relative small systems• large systems with 100 000 cores?
Good collaboration with developer community is very important