phycotoxins methods for detection
TRANSCRIPT

REVIEW
Determination of marine biotoxins relevant for regulations:from the mouse bioassay to coupled LC-MS methods
Bernd Christian & Bernd Luckas
Received: 18 September 2007 /Revised: 23 November 2007 /Accepted: 27 November 2007 /Published online: 15 December 2007# Springer-Verlag 2007
Abstract The frequency of occurrence and intensity ofharmful algal blooms (HABs) appear to be increasing on aglobal scale. Consequently, methods were established forthe evaluation of possible hazards caused by the enrichmentof algal toxins in the marine food chain. Different clinicaltypes of algae-related poisoning have attracted scientificattention: paralytic shellfish poisoning (PSP), diarrheticshellfish poisoning (DSP), and amnesic shellfish poisoning(ASP). In several countries fish specialties are consumedwhich may be contaminated with algal toxins typical for therespective region (e.g., ciguatera and tetrodotoxins). Bio-assays are common methods for the determination ofmarine biotoxins. However, biological tests are not com-pletely satisfactory, due to the low sensitivity and theabsence of specialized variations. Moreover, there isgrowing resistance against the use of animal experiments.Therefore, many efforts have been made to determine algaltoxins with chemical methods. In this context LC-MSmethods replaced HPLC methods with optical detectors,allowing both effective seafood control and monitoring ofphytoplankton in terms of the different groups of marinebiotoxins.
Keywords Marine biotoxins . LC-MS/MS . PSP toxins .
DSP toxins . Domoic acid . Tetrodotoxins . Ciguatera
Harmful algal blooms (HABs) and marine biotoxins
Anthropogenic contaminants such as polychlorinatedhydrocarbons accumulate in the tissue of warm-bloodedanimals. It is less known, however, that natural contami-nants may also accumulate along food chains. An examplefor this phenomenon is the ingestion and storage of algaltoxins in mussels. Mussels filter approximately 20 L h−1
water. During algal blooms water contains several millionalgae per liter. Although not all algae produce toxins, it isplausible that a significant accumulation of toxins willoccur in mussels.
Monitoring of toxins in seafood and risk assessment forhuman exposure is the main task of food control.Consumption of seafood contaminated with marine biotox-ins may cause serious diseases. Damage to the nervoussystem (paralytic shellfish poisoning, PSP), the intestinalsystem (diarrhetic shellfish poisoning, DSP), and loss ofmemory (amnesic shellfish poisoning, ASP) have beenobserved subject to the type of algal bloom. In addition,regionally specific algal toxins (e.g. ciguatera and tetrodo-toxins) have been identified in seafood specialties fromseveral countries.
The worldwide harvesting and transport of marineorganisms, in order to maintain the non-seasonal productionand import of seafood, has caused a potentially seriousthreat for the consumer. Therefore, the analysis of marinefood for marine biotoxins is an important task that must beconducted according to international regulations and instrict compliance with the respective restrictions.
The residue analyst specialized in the determination ofmarine biotoxins by chemical methods has to determinerapidly the toxins in complex biomatrices with high
Anal Bioanal Chem (2008) 391:117–134DOI 10.1007/s00216-007-1778-x
B. Christian (*) : B. LuckasInstitute for Nutrition, Friedrich-Schiller-University of Jena,Dornburger Str. 25,07743 Jena, Germanye-mail: [email protected]

sensitivity and, most importantly, unambiguously. Thesimultaneous realization of those demands was oftenaccompanied with difficulties. However, the introductionof LC-MS-based methods during the last decade hasallowed the analysis of algal toxins, especially thequantitative determination of those relevant for regulations,to be established in routine analysis.
Occurrence, chemical structure, toxicity, and analysisof marine biotoxins
Empirical scientific observations of harmful algal bloomsand toxic mussels were reported as early as 1937. At thattime, some algae were described which seemed to beresponsible for the production of toxins and a biologicaltest, the mouse bioassay, was developed to identify toxinsproduced by those algae. Up to now, the mouse bioassayhas been the most important international method for thedetection of algal toxins, and all analytical methodsdescribed below have to be evaluated against bioassays.However, such biological tests reveal only the total toxicityof a sample. The limited validity resulted in the need forphysicochemical methods for the detection of each individ-ual algal toxin. A further important argument against themouse bioassay is the growing resistance to the use ofanimal experiments [1].
Paralytic shellfish poisoning (PSP) toxins
The main producers of paralytic shellfish poisoning (PSP)toxins are dinoflagellates of the genus Alexandrium.Alexandrium occurs along the Atlantic and Pacific coast-lines where it may grow in great quantities, particularlyduring the summer period. Toxins produced by Alexandriumwere named PSP toxins due to the observation that theirconsumption caused symptoms of poisoning in warm-blooded species similar to paralytic phenomena such ascramp, signs of paralysis, and blocking of respiration. PSPtoxins are potential neurotoxins which specifically block the
excitation current in nerve and muscle cells resulting in signsof paralysis.
Consequently, the development of analytical methods forthe determination of poisoning caused by PSP toxins wasan important task. The mouse bioassay unambiguouslygives evidence of the toxic potential of a sample, since theapplication of higher toxin concentrations yields a shorten-ing of the time until death of the animals. However, thosebiological tests only reveal the total PSP toxicity of asample expressed in MU (mouse units) kg−1 or in PSP kg−1.Thus, the determination of individual PSP toxins was onlypossible after isolation and structure elucidation [2].
In 1957, a PSP toxin was isolated from Saxidomusgiganteus (clams) from Alaska, and in 1975 the chemicalstructure was assigned to the so-called saxitoxin (STX).Later on, more PSP toxins were identified which were allrelated to saxitoxin (STX) or N-1-hydroxy-saxitoxin (neo-saxitoxin, NEO). Today, it is convenient to distinguishbetween three groups of PSP toxins: carbamoyl toxins,N-sulfocarbamoyl toxins, and decarbamoyl toxins.
N-Sulfocarbamoyl toxins exhibit only low toxicity,whereas the toxicity of the carbamoyl and decarbamoyltoxins is significantly higher. During the manufacturing ofcanned seafood contaminated with PSP toxins, theN-sulfocarbamoyl toxins may be hydrolyzed to the moretoxic carbamoyl or decarbamoyl toxins, thus resulting in anincrease of the total PSP toxicity (Fig. 1).
In 1975, a fluorometric method was recommended forPSP determination in samples in addition to the mousebioassay [4]. PSP toxins which exhibit neither UVabsorption nor fluorescence were oxidized in alkalinesolution to fluorescent purine derivatives. After acidifica-tion the oxidation products′ intensity of fluorescence wasmeasured in solution. However, each PSP toxin differs bothin toxicity and fluorescence intensity after oxidation.Therefore, a chromatographic separation of the PSP toxinswas suggested prior to determination of the fluorescenceactivity, since the total PSP toxicity has to be calculatedfrom the individual toxin concentration and the absolutetoxicities of each PSP toxin. Hence, the unambiguous
Fig. 1 Chemical structure,toxicity, and molecular weightof PSP toxins [3]
118 Anal Bioanal Chem (2008) 391:117–134

assignment of the peaks in the chromatograms to definitePSP toxins is necessary for a proper quantification of PSPtoxins [3].
A breakthrough in the use of HPLC methods for PSPdetermination took place in 1984 when Sullivan et al. [5]succeeded in the separation of underivatized PSP toxins byion-pair chromatography with addition of alkylsulfonicacids followed by a post-column oxidation with periodicacid. A disadvantage of that method was the co-elution ofSTX and dc-STX resulting in a wrong total PSP toxicity,since the toxicity of STX is double that of dc-STX.Therefore, a separate determination of STX/dc-STX isessential for a comparison of the HPLC results with theresults from the mouse bioassay [6].
Oshima et al. [7] described an entire separation of PSPtoxins. However, that method requires three separateisocratic runs to analyze most of the PSP toxins and isslow and labor-intensive. As a consequence, the develop-ment of a new method allowing complete separation of allrelevant PSP toxins by a single chromatographic runfollowed by exact quantification with high sensitivity wasnecessary.
Recently, an HPLC method for PSP determination wasdeveloped based on ion-pair chromatography with post-column oxidation and fluorescence detection. The resultingchromatograms clearly showed a complete chromatograph-ic separation of all relevant PSP toxins such as the GTXsand dc-GTXs [8].
On the other hand, it is recommended that positivefindings of PSP toxins should be confirmed by applicationof mass spectrometry. However, eluents containing phos-phate and ion-pair-forming reagents prevented an efficientapplication of the LC-MS technique. Therefore, Jaime et al.[9] proposed the application of ion-exchange chromatogra-phy with eluents containing only volatile compounds todetermine PSP toxins with both fluorescence and MSdetection.
Electrospray ionization mass spectrometry (ESI-MS) iseffective for detection of the polar PSP toxins, which arequite basic and therefore form stable [M+H]+ ions. Thus,the direct detection of underivatized PSP toxins is possibleusing a mass spectrometer as detector [10].
Aversano et al. [11] examined hydrophilic interactionliquid chromatography (HILIC) mass spectrometry for theanalysis of PSP toxins. However, the GTXs were notseparated completely using a TSK-gel Amide-80 column,e.g., GTX-1 co-elutes with GTX-2. For that reason themethod can be applied only in combination with MS, butlacks in the combination with a fluorescence detector.Furthermore, inconstant retention times for PSP toxins indifferent seafood matrices were observed. As a conse-quence, the development of a new method allowing acomplete separation of all PSP toxins relevant for regu-
lations using one chromatographic run followed by selec-tive and sensitive quantification by a mass spectrometric orfluorimetric detection was required.
This challenge was solved by using a zwitterionic (ZIC)hydrophilic interaction chromatography (HILIC) column.In 2007, Diener et al. [12] published the application of aZIC-HILIC column (SeQuant, Haltern, Germany) for theseparation of underivatized PSP toxins including a new LC-MS/MS method which enabled the separation of all threegroups of PSP toxins in a single chromatographic run (Fig. 2).
In order to prove the applicability of this method forvarious sample materials, mussel extracts and the marinedinoflagellate Gymnodinium catenatum were analyzed byapplication of the new ZIC-HILIC column in combinationwith MS/MS detection (SRM mode). Independent of thesample matrix, mussels or algae, all present PSP toxinscould be determined (Fig. 3).
Although the limits of detection (LODs) are sometimeshigher with MS detection than with fluorescence detection
Time (min)
010 20 30 40
493/298
493/316
353/255
396/298
412/314
353/273
396/316
412/332
316/298
300/282
257/126
GT
X 1
GT
X 2
GT
X 4
dcG
TX
2
GT
X 3
dcG
TX
3
C 2
C 1
/
NE
OS
TX dc
ST
X
380/300
B 1
TIC
Fig. 2 HILIC-MS/MS analysis of a PSP standard mixture [12]
Anal Bioanal Chem (2008) 391:117–134 119

the sensitivity of the MS detection is sufficient to controlseafood at the regulatory limit for a PSP toxin content at800 μg STX equivalents kg−1 wet weight of soft tissues.
Diarrhetic shellfish poisoning (DSP) toxins
Since the first reports of diarrheic shellfish poisoning (DSP)in Japan, 1978, the illness is now recognized as a threat topublic health throughout the world. All DSP toxins arerelatively nonpolar, with molecular weights higher than500, and easily extractable by organic solvents such asmethanol and chloroform. Most DSP toxins are polyethercompounds with distinctive chemical structures and widelyvarying functional groups resulting in different toxicolog-ical and chemical characteristics [13].
Marine biotoxins in shellfish have traditionally beencategorized and regulated according to the poisoning syn-dromes that they evoke in human beings or animals. Forexample, okadaic acid (OA) and dinophysistoxins (DTXs)have been classified as diarrhetic shellfish poisoning (DSP)toxins due to their production of symptoms of diarrhea.Unfortunately, when pectenotoxins (PTXs) and yessotoxins(YTXs) were discovered and before their toxicity was
understood, they were placed into the DSP category becausethey often occur together with OA and DTXs and weredetected by the mouse bioassay procedure first used generallyto detect DSP toxins in lipophilic extracts.
With the continuing discovery of a series of newlipophilic toxins, such as azaspiracids (AZAs), gymnodi-mines, and spirolides, it became clear that a better approachwould be to categorize the toxins strictly according to theirchemical classes rather than their toxic symptoms. Thiswould allow seafood safety to be regulated according toallowable levels of specific toxins rather than to the resultof a specific assay. This is the path on which all othercontaminants in food, such as mycotoxins and pesticides,are controlled [14].
Okadaic acid and dinophysistoxins (OA and DTXs)
Dinophysistoxin-1 (DTX-1) was identified as the causativetoxin for incidents of diarrhetic shellfish poisoning in Japan,and it originates from the dinoflagellate Dinophysis fortii.Although DTX-1 is found in Europe to a limited extent, itwas identified as the major toxin in shellfish from Norway.
Most studies have implicated okadaic acid (OA) as theresponsible toxin arising from a variety of Dinophysisspecies. DTX-2, an isomer of OA, was isolated from Irishmussels. In addition, DTX-3 has been observed in planktonand shellfish (Fig. 4).
The term DTX-3 was originally coined to describe agroup of compounds in which saturated or unsaturatedC14–C18 fatty acid moieties are attached through the 7-hydroxyl group of DTX-1. Subsequently, it was shown thatany of the parent toxins, OA, DTX-1, and DTX-2, can beacylated and are shellfish metabolites [15].
All lipophilic toxins can be extracted from plankton orshellfish tissues by organic solvents such as acetone,methanol, and acetonitrile. The use of acetone for extractionis required if the mouse bioassay is applied for thedetermination of OA, DTXs, PTXs, and YTXs [16].However, 80% methanol in water is recommended as aneffective extraction solvent for application of HPLCmethods with fluorescence detection or the LC-MS/MStechnique [17]. After addition of more water to reduce thepercentage of methanol to approximately 50%, toxins canbe partitioned between chloroform or dichloromethanewhich are then evaporated to dryness and the residue canbe redissolved in 0.5–1.0 mL methanol.
In addition, the different ester derivatives, e.g., DTX-3,can be hydrolyzed quantitatively to their parent toxins bytreatment of the aqueous methanol extract with 0.25 MNaOH [18]. This procedure enables the unambiguousdetermination of both the DSP content of a sample materialas OA equivalents and the percentage of DTX-3, indepen-dent of the method used for analysis [19].
Time (min)
010 20 30 40
353/255
396/298
353/273
396/316257/126
GT
X 2
dcG
TX
2
GT
X 3 dc
GT
X 3
dcS
TX
TIC
C 1
/
C 2
493/316
493/298
Fig. 3 HILIC-MS/MS analysis of a Gymnodinium catenatum extract(0.03 M HAc), Baja California, Mexico [12]
120 Anal Bioanal Chem (2008) 391:117–134

In this context it is important that the DSP profile ofmussels can consist of a higher percentage of OA esters andtheir analogs than of other DSP toxins (see Fig. 5).
Pectenotoxins (PTXs)
The pectenotoxins (PTXs), named after the genus of scallopfrom which they were first isolated, Patinopecten yessoensis,are a group of DSP toxins originating from Dinophysisspecies throughout the world (Fig. 6).
PTX-2 is the main toxin in this group. When accumu-lated in shellfish, the methyl group at position C-43 isoxidized to the corresponding alcohol (PTX-1), aldehyde(PTX-3), and carboxylate (PTX-6) forms. The spiroketalsystem in rings A and B can also undergo rearrangementand/or epimerization under acidic conditions to produce
PTX-4 and PTX-7 through PTX-9. The lactone ring inPTX-2 may be opened to yield pectenotoxin seco acid(PTX-2sa), epimerization yields 7-epi-PTX-2sa [20].
PTXs appear to be highly toxic by intraperitonealinjection, leading to positive responses in the mousebioassay for lipophilic marine biotoxins. Because of that,the maximum level of PTXs (PTX-1 and PTX-2) permittedin European shellfish has been set at 160 μg kg−1 OAequivalents [16, 21]. However, PTXs appear to be of lowtoxicity orally and, unlike OA, they do not cause diarrhoea[22, 23].
All pectenotoxins absorb between 235 nm and 239 nm(UV). Detailed stability studies have not been performed,but it is known that rearrangements can occur under acidicconditions [24]. In addition, PTXs are easily destroyedunder strongly basic conditions. This fact has to be
Fig. 5 SIM chromatograms of edible parts of razor clams: a before NaOH hydrolysis, b after hydrolysis; OA=0.9 and 50.1 μg OA/100 g,respectively [18]
O
O
O
OOOH
O
Me O
OH Me
OHO
OHMeOR3
Me
H HR2
R1
okadaic acid (OA) R1 = CH3 R2 = H R3 = H
dinophysistoxin-1 (DTX-1) R1 = CH3 R2 = CH3 R3 = H
dinophysistoxin-2 (DTX-2) R1 = H R2 = CH3 R3 = H
dinophysistoxin-3 (DTX-3) R1, R2 = H or CH3 R3 = acyl
Fig. 4 Chemical structures ofthe DSP toxins OA and DTXs
Anal Bioanal Chem (2008) 391:117–134 121

considered in analyses of samples containing PTXs and,consequently, sample preparation has to be performed at apH value near 7.
On the other hand, comparison of the PTX profiles of thetoxic dinoflagellate Dinophysis acuta, of Greenshell mussels(Perna canalicus), and Blue mussels (Mytilus galloprovin-cialis) from New Zealand by application of an LC-MStechnique revealed that the major PTX homolog in D. acutawas PTX-2, whereas both Greenshell and Blue musselscontained PTX-2sa as the predominant toxin. More than90% of PTX-2 isolated from D. acuta was rapidly convertedto PTX-2sa and its epimer 7-epi-PTX-2sa in the Greenshellmussel extract (Fig. 7). The conversion from PTX-2 to PTX-2sa and 7-epi-PTX-2sa was not observed in phosphatebuffers at various pH ranging from 4.1 to 9.1. These findingsindicate that PTX-2sa and 7-epi-PTX-2sa are not artifacttoxins resulting from hydrolysis of PTX-2, but they arisefrom the conversion of PTX-2 by mussel tissues [25, 26].
Yessotoxins (YTXs)
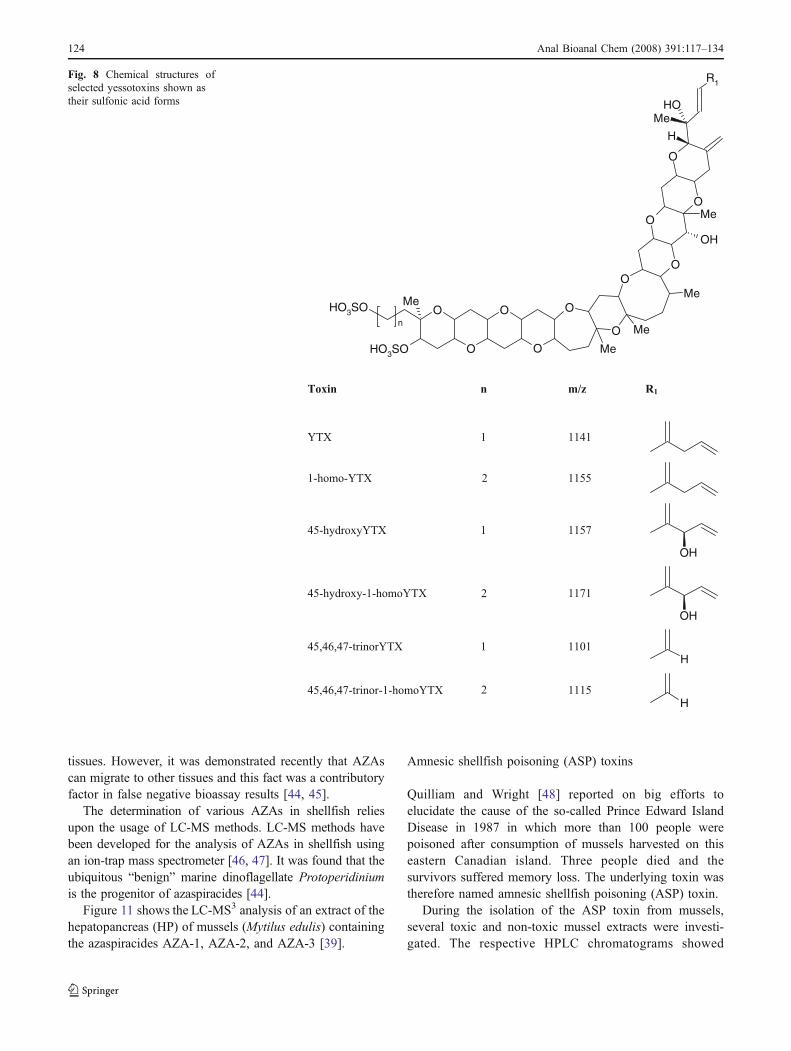
Yessotoxins (YTXs) are sulfated polyethers (Fig. 8) pro-duced by the dinoflagellates Protoceratium reticulatum andGonyaulax polyedrum in many parts of the world [27–30].YTXs are toxic by intraperitoneal injection in mice andtherefore give an increase to positive results in thetraditional mouse bioassay for DSP toxins [31].
A number of oxidized YTXs, apparently arising frommetabolism of YTX and 1-homo-YTX present in ingestedalgae, were subsequently isolated from shellfish by mousebioassay guided fractionation [32].
Antibodies to yessotoxin (YTX) have been produced andused to develop an ELISA. Its application for analyses ofalgal samples gave results two to three times higher thanwith LC-MS for known YTX, and for shellfish the resultsby ELISA were three to nine times higher than the resultsobtained from LC-MS. Those results suggested that algaecould produce a series of YTXs which were recognized bythe antibody and that those analogs had been metabolizedin shellfish [33].
In spite of recent evidence that YTXs may have nosignificantly acute toxicity when administered orally tomice [30], the actual quarantine level of YTX, 45-hydroxyYTX, 1-homo-YTX, and 45-hydroxy-1-homoYTXin shellfish is 1 mg kg−1 in the European Union [16, 21].
Recently, a number of LC-MS methods have beendemonstrated as powerful techniques for the quantification ofYTXs analogs and structure elucidation of novel toxins inplankton and shellfish [34–37]. Figure 9 shows the determi-nation of YTXs in mussel tissue obtained using LC-MS3 [37].
Azaspiracid poisoning (AZP) toxins
A shellfish poisoning event occurred in the Netherlands in1995 when at least eight severe gastrointestinal illnesses inhumans were reported after the consumption of mussels(Mytilus edulis). These shellfish were traced to KillaryHarbor, Ireland, where subsequent investigations revealedthat a number of local intoxications had also occurred.Unfortunately, this toxicity was not covered within the DSPtoxin-monitoring program in Ireland, which involvedbiweekly sampling in this region and utilized a recently
O
O
O
33
O
O
O
O
O
R43
OMe
OH
Me
1
O
O
Me
MeMe
OH
MeOH
R C1-C33
Pectenotoxin-1 CH2OH -O-
Pectenotoxin-2 CH3 -O-
Pectenotoxin-3 CHO -O-
Pectenotoxin-6 COOH -O-
Pectenotoxin-2 seco acid CH3 -OH HO-
Fig. 6 Chemical structures ofPTX homologs
122 Anal Bioanal Chem (2008) 391:117–134

modified rat bioassay. In subsequent chemical analyses ofthose shellfish, only low levels of OA and DTX-2 weredetected. In the DSP mouse test, a slow progressiveparalysis was observed using extracts of mussels, and theseneurotoxic symptoms were quite different from thosetypical for DSP toxins. In addition, the extracts weresubjected ESI-MS and 1H NMR analysis. The skeletalstructures of a toxin group named azaspiracids (AZAs)were elucidated [38]. It was apparent that they containednovel spiro-linked five- and six-membered rings, with oneof those containing nitrogen (Fig. 10).
In toxicity studies using azaspiracids, mice becameprogressively paralyzed with labored breathing. Diarrheawas not observed and, at low doses, mice died within 2–3 days. Histopathological changes were observed in micewhich were induced by both intraperitoneal and oraladministration of azaspiracids. Subsequently, azaspiracid
analogs have been isolated from Irish mussels, of whichAZA-1, AZA-2, and AZA-3 seem to be the maincompounds responsible for contamination. Methylazaspir-acid (AZA-2) and desmethylazaspirazid (AZA-3) occur inshellfish at lower concentrations than azaspiracid (AZA-1),but they are more toxic in the mouse bioassay [39]. Thechemical structure of AZA-4 and AZA-5 suggests that theyare oxidized metabolites of AZA-3 [40, 41].
Although AZAs were first identified in mussels thatwere cultivated in Ireland, a widespread European distri-bution of these toxins has recently been confirmed [42, 43],and the EU regulatory limit for the combined AZA-1,AZA-2, and AZA-3 content has been set at 0.16 μg g−1
total shellfish tissue [16, 21].Most regulatory testing only examines the digestive
glands (DG). That practice was a consequence of an earlyreport mentioning that DSP toxins are concentrated in these
Fig. 7 LC-MS chromatogramof PTX homologs obtainedfrom Greenshell mussels col-lected from Wedge Point, QueenCharlotte Sound, New Zealandin 1998 [24]: a TIC obtained bynegative mode with full-scanmonitoring (m/z 850–910), bextracted ion mass chromato-gram (m/z 857–858) (PTX-2), cextracted ion mass chromato-gram (m/z 875–876) (PTX-2saand 7-epi-PTX-2sa)
Anal Bioanal Chem (2008) 391:117–134 123
tissues. However, it was demonstrated recently that AZAscan migrate to other tissues and this fact was a contributoryfactor in false negative bioassay results [44, 45].
The determination of various AZAs in shellfish reliesupon the usage of LC-MS methods. LC-MS methods havebeen developed for the analysis of AZAs in shellfish usingan ion-trap mass spectrometer [46, 47]. It was found that theubiquitous “benign” marine dinoflagellate Protoperidiniumis the progenitor of azaspiracides [44].
Figure 11 shows the LC-MS3 analysis of an extract of thehepatopancreas (HP) of mussels (Mytilus edulis) containingthe azaspiracides AZA-1, AZA-2, and AZA-3 [39].
Amnesic shellfish poisoning (ASP) toxins
Quilliam and Wright [48] reported on big efforts toelucidate the cause of the so-called Prince Edward IslandDisease in 1987 in which more than 100 people werepoisoned after consumption of mussels harvested on thiseastern Canadian island. Three people died and thesurvivors suffered memory loss. The underlying toxin wastherefore named amnesic shellfish poisoning (ASP) toxin.
During the isolation of the ASP toxin from mussels,several toxic and non-toxic mussel extracts were investi-gated. The respective HPLC chromatograms showed
O
O
O
O
O
O
OO
O
O
O
HO3SOn Me
HO3SO Me
Me
Me
OH
Me
R1
H
MeOH
Toxin n m/z R1
YTX 1 1141
1-homo-YTX 2 1155
45-hydroxyYTX 1 1157
OH
45-hydroxy-1-homoYTX 1171
OH
45,46,47-trinorYTX 1 1101
H
45,46,47-trinor-1-homoYTX 1115
H
2
2
Fig. 8 Chemical structures ofselected yessotoxins shown astheir sulfonic acid forms
124 Anal Bioanal Chem (2008) 391:117–134

characteristic differences compared with non-toxic frac-tions. On the basis of the UV spectrum the correspondingcompound was identified as domoic acid. The amino aciddomoic acid (DA) has an effect on the nervous system andacts as a glutamic acid agonist, which explains the amnesiasince glutamic acid plays an important role for the storageof information [49]. In the case of the DA, it was surprisingthat a diatom (Pseudonitzschia spp.) widely spread alongthe Canadian coast and in European waters was theproducer of DA. Therefore, mussels harvested in Europemay be also contaminated with DA [50].
Domoic acid became a subject of interest for food controllaboratories after introduction of a limit of 20 mg DA kg−1
flesh of mussels [21]. HPLC separation of the underivatizeddomoic acid on reversed-phase (RP) columns followed byUV detection at 242 nm was suggested. In addition, severalmethods involving MS have also been used for thedetermination of DA and its analogs in shellfish, followingthe solid-phase extraction (SPE) cleanup of extracts [51].Hummert et al. [52] succeeded in the determination of DA in
algae and mussels at concentrations of 1.0 mg DA kg−1 byapplication of an automated HPLC system with column-switching system and UV detection. The mass spectrometricdetermination is another sensitive and selective method fordomoic acid analysis [53]. However, a thorough purificationof the sample extracts is necessary for an unambiguousdetermination of DA [54].
Both LC-UV and LC-MS methods were applied for thedetermination ofDA concerning EC legislation [55]. Figure 12shows an ESI mass spectrum (positive mode) of DA [56].
Tetrodotoxins (TTXs)
Puffer fish (Takifugu spp.) is a delicacy in Japan and someother Asiatic countries. Unfortunately, every now and thenserious poisonings have been reported after consumption ofthat fish species [57]. Consequently, a so-called tetrodotox-in (TTX) was isolated from puffer fish followed bystructure elucidation [58–62]. Later on, it was discoveredthat TTX and its analogs (Fig. 13) also occur in othermarine organisms and some terrestrial vertebrates [63, 64].
TTX and its derivatives are neither produced by thesefish species nor by algae but from bacteria [66–68].However, several serious intoxications after consumption
NH39
37
O
30
O
24
23
22
14
O10
OO
8
OH 1
O
3
R1
H
R2
OCH3
H
H
20 OHH
OH
26H
R3
R4
CH3
OO
H
H
CH3
CH3
CH3
D
I
A BC
E
F
GH
R1 R2 R3 R4 MW
Azaspirazid (AZA-1) H H CH3 H 841.5
Azaspirazid-2 (AZA-2) H CH3 CH3 H 855.5
Azaspirazid-3 (AZA-3) H H H H 827.5
Azaspirazid-4 (AZA-4) OH H H H 843.5
Azaspirazid-5 (AZA-5) H H H OH 843.5
Fig. 10 Chemical structures and molecular weights of variousazaspiracid toxins
Fig. 9 Determination of YTX (0.20 μg g−1) and 45-OH-YTX(0.11 μg g−1) in mussel tissue using LC-MS3 [37]
Anal Bioanal Chem (2008) 391:117–134 125

of puffer fish and further species led to the result thatanalysis of tetrodotoxins is a task similar to the determina-tion of algal toxins [69]. The toxic effect of tetrodotoxins isbased on blocking of the sodium channels in nerve cellsand, consequently, the mouse bioassay is suitable fordetermination of those potential neurotoxins [70].
Moreover, it was discovered that puffer fish may containboth TTXs and PSP toxins [71–73]. Therefore, specificmethods were required for the determination of TTXs and
PSP toxins and, consequently, the application of chromato-graphic methods was suggested [74].
The detection of fugu toxin was a major problem sinceTTXs neither show UV absorption nor fluorescence activity[75]. Therefore, the HPLC system was equipped with areaction unit for post-column derivatization, enabling aselective and sensitive determination of TTX and its deriva-tives. After chromatographic separation TTX is converted intoa fluorescent derivative by addition of NaOH [76]. Further-
Fig. 12 Positive ion ESI massspectrum of domoic acid,MW 311.3 [56]
Fig. 11 Chromatogram obtainedfrom the LC-MS3 analysis ofmussels from Sognefjord:a m/z 828.5→810.5→792.5;b m/z 842.5→824.5→806.5;c m/z 856.5→838.5→820.5corresponding to AZA-1,AZA-2, and AZA-3,respectively [39]
126 Anal Bioanal Chem (2008) 391:117–134

more, it is possible to use such an HPLC system for thesimultaneous determination of TTX and PSP toxins [77].
Since ESI-MS was introduced for analysis of marinebiotoxins [78], this technique also proved to be a veryeffective tool in the analysis of TTXs. Reliable methodsusing LC-ESI-MS for the determination of tetrodotoxin inpuffer fish have been developed, too [61, 79].
TTXs are generally extracted with an aqueous ormethanolic solution of acetic acid because TTXs are stablein weakly acidic aqueous conditions and under heating.TTX is present at higher concentrations than its derivativesin samples of puffer fish and newt, such that 5-deoxyTTX,5,6,11-trideoxyTTX, and 4,9-anhydroTTX are consideredto be almost non-toxic analogs [80].
Recently, the HILIC column TSKgel Amide-80 wasapplied successfully for ESI-LC-MS determination ofTTXs. It was found that in eggs of the puffer fish Fugupoecilonotus the concentration of 5,6,11-trideoxyTTX washigher than that of TTX, whereas that analog was notdetected in the skin of the newt Cynops ensicauda [81].
Diener et al. [65] used a ZIC-HILIC column for thedetermination of TTXs in separated tissues of Bangladeshimarine puffer, Takifugu oblongus (Fig. 14). TTX waspredominant in skin, muscle, and liver, whereas 5,6,11-trideoxyTTX was preponderant in the ovary.
In the Annex III, Section VII, Chapter V of the Regulation(EC) No 853/2004 of the European Parliament and of theCouncil of 29 April 2004 [21], health standards for livebivalve mollusks concerning the marine biotoxins wereestablished. This regulation also contains (in Section VIII,Chapter V) health standards for fishery products in which thefollowing specific requirements are defined with regard totoxins harmful to human health: fishery products derived frompoisonous fish of the families Tetraodontidae, Molidae,Diodontidae, and Canthigasteridae as well as fishery productscontaining biotoxins such as ciguatera or muscle-paralyzingtoxins must not to be brought onto the market [82].
Ciguatera (CTXs)
The term ciguatera fish poisoning was first used in theCaribbean to describe an intoxication induced by ingestionof a marine snail, Turbo pica (called cigua by the Cubannatives). Now the world is used this term to describe theintoxication caused by consumption of certain fish, primar-ily reef fish from the tropical and subtropical areas of theCaribbean Sea and the Pacific Ocean, which have accumu-lated specific toxins (CTXs) via their diet.
The more than 175 ciguateric symptoms that have beenreported can be classified into four categories: gastrointes-
OR4
O
R3
OH
NH
HO
R2
OH
+H2N
HN
R1
OR2
O
R1
OH
NHH
OH
+H2N
HN
O
HR2
O
R1
OH
NH
HO
HO
O
+H2N
HN
H
R1
R2
R3 R
4 [M+H]
+
TTX OH OH CH2OH 320
4-epiTTX OH H OH CH2OH 320
6-epiTTX H OH CH2OH OH 320
11-deoxyTTX H OH OH CH3 304
norTTX-6(S)-ol H OH OH H 290
norTTX-6(R)-ol H OH H OH 290
norTTX-6, 6-diol H OH OH OH 306
R1 R
2 [M+H]
+
anhydroTTX OH CH2OH 302
6-epianhydroTTX CH2OH OH 302
R1 R
2 [M+H]
+
5-deoxyTTX OH CH2OH 304
trideoxyTTX H CH3 272
H
Fig. 13 Chemical structures ofTTXs [65]
Anal Bioanal Chem (2008) 391:117–134 127

tinal, neurological, cardiovascular, and general [83]. Thismultiphase intoxication is thought to be due to the presenceof different ciguatera-related compounds at different ratios[84, 85].
Although Scheuer et al. [86] isolated ciguatera in 1967,the structure was not elucidated until 1989 [87]. Furtherciguatoxins (CTXs) were later discovered in algae [88] andfish [89, 90].
CTXs are cyclic polyethers with similar structures to DSPtoxins and brevetoxins [91]. Therefore, a fast and unambig-uous determination of ciguatoxins with simple chemical testsis difficult [83].
The mouse bioassay was applied first to determine acontamination of fish with ciguatera [92]. Later, bioassayswith chicken [93] and mosquitos [94] and an enzymeimmunoassay, the so-called stick test, were applied fordetection of ciguatera [95].
Despite the further development of immunological andenzymatic methods, doubtful or false positive results wereobtained during the control of tropical fish on ciguatera. Itwas the introduction of a solid-phase immunobead assay(S-PIA) which, for the first time, resulted in ciguateravalues correlating with data obtained with other detectionmethods [96]. On the basis of this immunoassay a test kitnamed Ciguatect™ was introduced for the determination ofciguatoxins. However, Ciguatect™ can only be used for thedetermination of ciguatoxins in the absence of DSP toxinssince okadaic acid, for example, gives also positive reactionwith the kit. Therefore, application of Ciguatect™ has to beaccompanied with a second detection method for DSPtoxins [97]. This is of great importance, not only withrespect to the aforementioned food control regulation [82],but also with concern to the Regulation (EC) No. 854/2004of the European Parliament and the Council whichstipulates (in chapter II) that checks have to be done toensure that fishery products containing biotoxins such asciguatera are not placed on the market [98].
Intensive research was ongoing to develop LC-MS/MS-based methods for the determination of CTXs to overcome thelack of suitable physicochemical methods [99, 100]. It wasevident that differences exist between ciguatoxins from thePacific (P-CTXs) produced by certain strains of the dinofla-gellate Gambierdiscus toxicus as well as from the Caribbean(C-CTXs) and the Indian Ocean (I-CTXs) [101–103].Figure 15 shows the chemical structures of P-CTXs and C-CTXs, and Fig. 16 clearly demonstrates the formation of ionscharacteristic for CTXs. Interestingly, the retention time andmasses for I-CTX and C-CTX-1 were virtually indistinguish-able under the conditions applied for the ESI-LC-MSmeasurements [104].
Application of LC-MS/MS methods for determinationof assorted marine biotoxins in compliancewith legislation
Due to the cases of human intoxication frequently observedafter mussel consumption, governmental institutions andthe fishery industry have established control methods toensure that seafood contaminated by biotoxins does notreach the consumer [105]. The first step was to establishmonitoring programs for harmful algae blooms (HABs)followed by restrictions concerning the harvest of musselsin areas with an unacceptable amount of toxin-producingalgae per liter [106, 107]. Furthermore, efforts were made
Fig. 14 HILIC-MS analysis (SIM mode) of extracts of T. oblongus:a skin, b ovary [65]
128 Anal Bioanal Chem (2008) 391:117–134

to harmonize legal regulations and monitoring methods, toprovide a common basis for risk assessment [108], wherebyin 1958 USA and Canada were the first countries in theworld to establish the mouse bioassay and residue limits forPSP toxins with 400 MU and 80 μg PSP/100 g musseltissue, respectively [109, 110].
The European Union established reference laboratories tomonitor the shellfish production in the community [111].Moreover, specific rules for official controls concerning livebivalve molluscs from classified production areas wereestablished. Classified relaying and production areas have tobe periodically monitored to check the presence of toxin-producing plankton. The sampling frequency for toxin analysisin the molluscs is, as a general rule, to be weekly during theperiods at which harvesting is allowed. If any changes in toxinpopulations that may lead to toxin accumulation are detected,the sampling frequency of molluscs is to be increased orprecautionary closures of the areas are to be established untilthe results of toxin analysis are obtained [112].
To fulfill the tasks concerning the monitoring ofphytoplankton in classified production areas, LC-MS-basedmethods were developed for simultaneous determination ofvarious algal and cyanobacterial toxins extracted fromphytoplankton [113, 114], and, during recent years, espe-cially mussels from different marine regions have to becontrolled in compliance with the newsworthy legislation[115, 116]. The latter was focused on ASP and DSP toxinswhich should be determined simultaneously by applicationof a LC-MS/MS multiresidue method (Figs. 17 and 18).
A newly developed LC-MS/MS method allowing thedetermination of various marine biotoxins in shellfish wassubjected to a full single-laboratory validation and a limitedinterlaboratory study. The single-laboratory validationproved that the LC-MS/MS method is suitable for routinedetermination of ASP, DSP, and other lipophilic algal toxinsin shellfish with high specificity, good precision/accuracy,and low detection limits [117]. Furthermore, a study using
both mouse bioassay and an LC-MS-based method todetermine the lipophilic toxins associated with diarrheticshellfish poisoning in Japanese bivalves demonstrated thegood comparability of the results obtained with the mousebioassay and LC-MS [118].
Fig. 16 Mass spectra showing the different ratio of pseudomolecular([M+Na]+, [M+NH4]
+, [M+H]+) and product ([M+H-nH2O]+) ions
within the fragmentation patterns of a I-CTX and b C-CTX-1 [104]
Fig. 15 Structures of Pacificciguatoxin-1 (P-CTX-1) andCaribbean ciguatoxin-1(C-CTX-1) [104]
Anal Bioanal Chem (2008) 391:117–134 129

Conclusions
The safety of shellfish as food can be compromised bycontamination with marine biotoxins. This risk is managed bymonitoring programs that generally combine the identification
and enumeration of hazardous algal species in growing watersand the testing of flesh from shellfish samples [117].
There is currently a high degree of dependence onmouse-based bioassays but there is growing acceptance forthe need to develop and implement non-animal-based
Fig. 18 LC-MS/MS multiple reaction monitoring chromatogram with positive and negative ESI. Greenshell mussel flesh from site J02, collected7 February 2001. Each trace is normalized to the largest peak [115]
Fig. 17 Reversed-phase gradi-ent elution LC-MS analysis of arange of toxins in a blend ofcontaminated mussel tissueextracts. Selected ion monitor-ing was carried out on either[M+H]+ or [M+NH4]
+ ions,which are displayed as individualmass chromatograms. The toxinspresent include domoic acid(DA), spirolides (SpiroB/D),okadaic acid (OA), dinophysis-toxins (DTX1/2), pectenotoxins(PTX2 and PTX2sa), azapsirac-ids (AZA1, AZA2 and AZA3),and acyl esters of OA and DTX2(DTX3) [10]
130 Anal Bioanal Chem (2008) 391:117–134

methods. Recent advances in analytical instrumentationhave enabled the development of alternative methods suchas LC-MS [119].
Comparison of the quantitative results obtained forbivalve samples with the mouse bioassay and LC-MSindicates that LC-MS is suitable for routine monitoring ofmarine biotoxins and therefore this technology is becomingthe method of choice for the detection and quantification ofseveral marine biotoxins [120].
Moreover, further development and validation of analyt-ical methodology from algal toxins is highly desirable,because the enforcement of biotoxin legislation is ultimatelybased on the ability of analysts to identify and quantify thosetoxins accurately in seafood products.
References
1. Fernandez ML, Richard DJA, Cembella AD (2003) In vivoassays for phycotoxins. In: Hallegraeff G, Anderson DM,Cembella AD (eds) Manual on harmful marine microalgae.Intergovernmental Oceanographic Commission of UNESCO,Paris, ISBN 92-3-103871-0, p 349
2. Schantz EJ (1986) Chemistry and biology of saxitoxin andrelated toxins. Ann N Y Acad Sci 47:15–23
3. Luckas B, Hummert C, Oshima Y (2003) Analytical methods forparalytic shellfish poisons. In: Hallegraeff G, Anderson DM,Cembella AD (eds) Manual on harmful marine microalgae.Intergovernmental Oceanographic Commission of UNESCO, 7Place de Fontenoy, 75352 Paris 07 SP, ISBN 92-3-103871-0,pp 191–209
4. Bates HA, Rapoport H (1975) A chemical assay for saxitoxin,the paralytic shellfish poison. J Agric Food Chem 23:237–239
5. Sullivan JJ, Wekell MM (1984) Determination of paralyticshellfish poisoning toxins by high pressure liquid chromatogra-phy. In: Ragelis EP (ed) Seafood toxins, ACS Symposium Series262. American Chemical Society, Washington DC, pp 197–205
6. Luckas B (2000) Chemical analysis of PSP toxins. In: BotanaLM (ed) Seafood and freshwater toxins: pharmacology, physiol-ogy and detection. Marcel Dekker, New York, ISBN 8247-8956-3, pp 173–186
7. Oshima Y, Sugino K, Yasumoto T (1989) Latest advances inHPLC analysis of paralytic shellfish toxins. In: Natori S,Hashimoto K, Ueno Y (eds) Mycotoxins and phycotoxins ′88.Elsevier, Amsterdam, pp 319–328
8. Diener M, Erler K, Hiller S, Christian B, Luckas B (2006)Determination of paralytic shellfish poisoning (PSP) toxins indietary supplements by application of a new HPLC/FD method.Eur Food Res Technol 224:147–151
9. Jaime E, Hummert C, Hess P, Luckas B (2001) Ion-exchangeseparation of paralytic shellfish poisoning (PSP) toxins for high-performance liquid chromatography determination. J Chroma-togr A 929:43–49
10. Quilliam MA (2003) The role of chromatography in the hunt forred tide toxins. J Chromatogr A 1000:527–548
11. Aversano CD, Hess P, Quilliam MA (2005) Hydrophilicinteraction liquid chromatography-mass spectrometry for theanalysis of paralytic shellfish poisoning (PSP) toxins. J Chro-matogr A 1081:190–201
12. Diener M, Erler K, Christian B, Luckas B (2007) Application ofa new zwitterionic hydrophilic interaction chromatography
column for determination of paralytic shellfish poisoning toxins.J Sep Sci 30:1821–1826
13. Yasumoto T, Murata M (1985) Diarrhetic shellfish toxins.Tetrahedron 41:1019–1025
14. Quilliam MA (2003) Chemical methods for lipophilic shellfishtoxins. In: Hallegraeff G, Anderson DM, Cembella AD (eds)Manualon harmful marine microalgae. Intergovernmental oceanographiccommission of UNESCO, Paris, ISBN 92-3-103871-0, pp 211–245
15. Suzuki T, Ota H, Yamasaki M (1999) Direct evidence oftransformation of dinophysistoxin-1 to 7-O-acyl-dinophysis-toxin-1 (dinophysistoxin-3) in the scallop Patinopecten yessoen-sis. Toxicon 37:187–198
16. EC (2002) Commission decision 2002/225/EC of 15. March2002 laying down detailed rules for the implementation ofCouncil Directive 91/492/EEC as regards the maximum levelsand the methods of analysis of certain marine biotoxins inbivalve molluscs, echinoderms, tunicates and marine gastropods.Off J Eur Comm L75:62–64
17. James KJ, Bishop AG, Carmody EP, Kelly SS (2000) Detectionmethods for okadaic acid and analogues. In: Botana LM (ed)Seafood and freshwater toxins: pharmacology, physiology anddetection. Marcel Dekker, New York, ISBN 8247-8956-3,pp 217–238
18. Vale P, Sampayo MAM (2002) First confirmation of humandiarrhoeic poisoning by okadaic esters after ingestion of razorclams (Solen marginatus) and green crabs (Carcinus maenas) inAveiro lagoon, Portugal and detection of okadaic acid esters inphytoplankton. Toxicon 40:989–996
19. Suzuki T, Beuzenberg V, Mackenzie L, Quilliam MA (2004)Discovery of okadaic acid esters in the toxic dinoflagellateDinophysis acuta from New Zealand using liquid chromatogra-phy/tandem mass spectrometry. Rapid Commun Mass Spectrom18:1131–1138
20. Suzuki T, Beuzenberg V, Mackenzie L, Quilliam, MA (2003)Liquid chromatography-mass spectrometry of spiroketal stereo-isomers of pectenotoxins and the analysis of novel pectenotoxinsisomers in the toxic dinoflagellate Dinophysis acuta from NewZealand. J Chromatogr A 992:141–150
21. EC (2004) Regulation (EC) No. 853/2004 of the EuropeanParliament and of the Council of 29 April 2004 laying downspecific hygiene rules for food of animal origin. Off J EurCommun Annex III, Sect VII, Chap V, 2. L 226 pp 60–61
22. Miles CO, Wilkins AL, Samdal IA, Sandvik M, Petersen D,Quilliam MA, Naustvoll LJ, Rundberget T, Torgensen T,Hovgaard P, Jensen DJ, Cooney JM (2004) A novel pecteno-toxin, PTX-12, in Dinophysis spp. and shellfish from Norway.Chem Res Toxicol 17:1423–1433
23. Miles CO, Wilkins AL, Munday R, Dines MH, Hawkes AD,Briggs LR, Sandvik M, Jensen DJ, Cooney JM, Holland PT,Quilliam MA, MacKenzie AL, Beuzenberg V, Towers NR(2004) Isolation of pectenotoxin-2 seco acid, and preliminaryassessment of their acute toxicities. Toxicon 43:1–9
24. Suzuki T, Mackenzie L, Stirling D, Adamson J (2001)Pectenotoxin-2 seco acid: A toxin converted from pecteno-toxin-2 by the New Zealand Greenshell mussel Perna canalicus.Toxicon 39:507–514
25. Miles CO, Wilkins AL, Hawkes AD, Jensen DJ, Selwood AI,Beuzenberg V, Mackenzie AL, Cooney JM, Holland PT (2006)Isolation and identification of pectenotoxins-13 and -14 fromDinophysis acuta in New Zealand. Toxicon 48:152–159
26. Wilkins AL, Rehmann N, Torgersen T, Rundberget T, Keogh M,Petersen D, Hess P, Rise F, Miles CO (2006) Identification offatty acid esters of pectenotoxin-2 seco acid in blue mussels(Mytilus edulis) from Ireland. J Agric Food Chem 54:5672–5678
27. Miles CO, Wilkins AL, Hawkes AD, Selwood AI, Jensen DJ,Cooney JM, Beuzenberg V, MacKenzie AL (2006) Identification
Anal Bioanal Chem (2008) 391:117–134 131

of 45-hydroxy-46,47-dinoryessotoxin, 44-oxo-45,46,47-trinor-yessotoxin, and 9-methyl-42,43,45,46,47,55-heptanor-38-en-41-oxo-yessotoxin, and partial characterization of some minoryessotoxin, from Protoceratium reticulatum. Toxicon 47:229–240
28. Satake M, Eiki K, Ichimura T, Ota S, Sekiguchi K, Oshima Y(2006) Structure of 45,46,47-trinorhomoyessotoxin, a newyessotoxin analog, from Protoceratium reticulatum whichrepresents the first detection of a homoyessotoxin analog inJapan. Harmful Algae 5:731–735
29. Cañás IR, Hamilton B, Amandi MF, Furey A, James KJ (2004)Nano liquid chromatography with hybrid quadrupole time-of-flightmass spectrometry for the determination of yessotoxin in marinephytoplankton. J Chromatogr A 1056:253–256
30. Suzuki T, Horie Y, Koike K, Satake M, Oshima Y, Iwataki M,Yoshimatsu S (2007) Yessotoxin analogues in several strains ofProtoceratium reticulatum in Japan determined by liquidchromatography-hybrid triple quadrupole/linear ion trap massspectrometry. J Chromatogr A 1142:172–177
31. Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, MagnoS, Poletti R (2002) Direct detection of yessotoxin an itsanalogues by liquid chromatography coupled with electrosprayion trap mass spectrometry. J Chromatogr A 968:61–69
32. Finch SC, Wilkins AL, Hawkes AD, Jensen DJ, MacKenzie AL,Beuzenberg V, Quilliam MA, Olseng CD, Samdal IA, Aasen J,Selwood AI, Cooney JM, Sandvik M, Miles CO (2005) Isolationand identification of (44-R,S)-44,55-dihydroxy yessotoxin fromProtoceratium reticulatum, and its occurrence in extracts of shell-fish from New Zealand, Norway and Canada. Toxicon 46:160–170
33. Samdal IA, Aasen JAB, Briggs LR, Dahl E, Miles CO (2005)Comparison of ELISA and LC-MS analyses for yessotoxins inblue mussels (Mytilus edulis). Toxicon 46:7–15
34. Miles CO, Samdal IA, Aasen JAG, Jensen DJ, Quilliam MA,Petersen D, Briggs LM, Wilkins AL, Rise F, Cooney JM,MacKenzie AL (2005) Evidence for numerous analogs ofyessotoxin in Protoceratium reticulatum. Harmful Algae4:1075–1091
35. Rhodes L, McNabb P, de Salas M, Briggs L, Beuzenberg V,Gladstone M (2006) Yessotoxin production by Gonyaulaxspinifera. Harmful Algae 5:148–155
36. Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, MagnoS, Guerrini F, Pistocchi R, Boni L (2003) Complex yessotoxinsprofile in Protoceratium reticulatum from north-western Adriaticsea revealed by LC-MS analysis. Toxicon 42:7–14
37. Amandi MF, Furey A, Lehane M, Ramstad H, James KJ (2002)Liquid chromatography with electrospray ion-trap mass spec-trometry for the determination of yessotoxins in shellfish. JChromatogr A 976:329–334
38. James KJ, Bishop AG, Furey A (2000) New toxins on thehorizon. In: Botana LM (ed) Seafood and freshwater toxins:pharmacology, physiology and detection. Marcel Dekker, NewYork, ISBN 8247-8956-3, pp 693–714
39. James KJ, Furey A, Lehane M, Ramstad H, Aune T, Hovgaard P,Morris S, Higman W, Satake M, Yasumoto T (2002) First evidenceof an extensive northern European distribution of azaspiracidpoisoning (AZP) toxins in shellfish. Toxicon 40:909–915
40. Brombacher S, Edmonds S, Volmer DA (2002) Studies onazapsiracids biotoxins. II. Mass spectral behaviour and structuralelucidation of azaspiracid analogues. Rapid Commun MassSpectrom 16:2306–2316
41. James KJ, Sierra MD, Lehane M, Magdalena AB, Furey A(2003) Detection of five new hydroxyl analogues of azaspiracidsin shellfhish using multiple tandem mass spectrometry. Toxicon41:277–283
42. Vale P (2004) Is there a risk of human poisoning by azaspiracidsfrom shellfish harvested at the Portuguese coast? Toxicon44:943–947
43. Magdalena AB, Lehane M, Krys S, Fernández ML, Furey A,James KJ (2003) The first identification of azaspiracids inshellfish from France and Spain. Toxicon 42:105–108
44. James KJ, Moroney C, Roden C, Satake M, Yasumonto T,Lehane M, Furey A (2003) Ubiquitous ‘benign’ alga emerges asthe cause of shellfish contamination responsible for the humantoxic syndrome, azaspiracid poisoning. Toxicon 41:145–151
45. Hess P, Nguyen L, Aasen J, Keogh M, Kilcoyne J, McCarron P,Aune T (2005) Tissue distribution, effects of cooking andparameters affecting the extraction of azaspiracids from mussels,Mytilus edulis, prior to analysis by liquid chromatographycoupled to mass spectrometry. Toxicon 46:62–71
46. Draisci R, Palleschi L, Ferretti E, Furey A, James KJ, Satake M,Yasumoto T (2000) Development of a method for the identifi-cation of azaspiracid in shellfish by liquid chromatography-tandem mass spectrometry. J Chromatogr A 871:13–21
47. Lehane M, Sáez MJF, Magdalena AB, Cañás IR, Sierra MD,Hamilton B, Furey A, James KJ (2004) Liquid chromatography-multiple tandem mass spectrometry for the determination of tenazaspiracids, including hydroxyl analogues in shellfish. JChromatogr A 1024:63–70
48. Quilliam MA, Wright JLC (1989) The amnesic shellfishpoisoning mystery. Anal Chem 81:1053–1060
49. Todd ECD (1993) Domoic acid and amnesic shellfish poisoning-a review. J Food Prot 56:69–83
50. James KJ, Gillman M, Amandi MF, López-Rivera A, Puente PF,LehaneM,Mitrovic S, Furey A (2005) Amnesic shellfish poisoningtoxins in bivalve molluscs in Ireland. Toxicon 46:852–858
51. Lawrence JF, Lau VP, Cleroux C, Lewis D (1994) Comparison ofUV absorption and electrospray mass-spectrometry for the high-performance liquid chromatographic determination of domoic acidin shellfish and biological samples. J Chromatogr A 659:119–126
52. Hummert C, Reichelt M, Luckas B (1997) Automatic HPLC-UVDetermination of domoic acid in mussels and algae. Chroma-tographia 45:284–288
53. Furey A, Lehane M, Gillman M, Fernandez-Puente P, James KJ(2001) Determination of domoic acid in shellfish by liquidchromatography with electrospray ionization and multipletandem mass spectrometry. J Chromatogr A 938:167–174
54. López-Rivera A, Suárez-Isla BA, Eilers PP, Beaudry CG, Hall S,Amandi MF, Furey A, James KJ (2005) Improved high-performance liquid chromatographic method for the determina-tion of domoic acid and analogues in shellfish: effect of pH.Anal Bioanal Chem 381:1540–1545
55. Hess P, Morris S, Stobo LA, Brown NA, McEvoy JDG,Kennedy G, Young PB, Slattery D, McGovern E, McMahon T,Gallacher S (2005) LC-UV and LC-MS methods for thedetermination of domoic acid. Trends Anal Chem 24:358–367
56. Tor ER, Puschner B, Whitehead WE (2003) Rapid determination ofdomoic acid in serum and urine by liquid chromatography-electro-spray tandemmass spectrometry. J Agric Food Chem 51:1791–1796
57. Yokoo A (1950) Chemical studies on globefish poison. J ChemSoc Japan 71:590–593
58. Woodward RB (1964) The structure of tetrodotoxin. Pure ApplChem 9:49
59. Goto T, Kishi Y, Takahashi S, Hirata Y (1965) Tetrodotoxin.Tetrahedron 21:2059–2061
60. Yasumoto T, Murata M (1993) Marine Toxins Chem Rev93:1897–1909
61. Shoji YS, Yamashita MY, Miyazawa T, Yasumoto T (2001)Electrospray ionization mass spectrometry of tetrodotoxin and itsanalogs: liquid chromatography/mass spectrometry, tandem massspectrometry, and liqid chromatography/tandem mass spectrom-etry. Anal Biochem 290:10–17
62. Yotsu-Yamashita M (2001) Chemistry of puffer fish toxin. JToxicol Toxin Rev 20:51–66
132 Anal Bioanal Chem (2008) 391:117–134

63. Pires OR, Sebben A, Schwartz EF, Morales RAV, Bloch C,Schwartz CA (2005) Further report of the occurrence oftetrodotoxin and new analogues in the Anuran family Brachy-cephalidae. Toxicon 45:73–79
64. Yotsu-Yamashita M, Mebs D, Flachsenberger W (2007) Distri-bution of tetrodotoxin in the body of the blue-ringed octopus(Hapalochlaena maculosa). Toxicon 49:410–412
65. Diener M, Christian B, Ahmed S, Luckas B (2007) Determina-tion of tetrodotoxin and its analogs in the puffer fish Takifuguoblongus from Bangladesh by hydrophilic interaction chroma-tography and mass spectrometric detection. Anal Bioanal Chem.DOI 10.1007/100216-007-1602-7
66. Simidu U, Noguchi T, Hwang DF, Shida Y, Hashimoto K (1987)Marine bacteria which produce tetrodotoxin. Appl EnvironMicrobiol 53:1714–1715
67. Kodama M, Shimizu H, Sato S, Ogata T, Terao K (1995) Theinfection of bacteria in the liver cells of toxic puffer-a possiblecause for organisms to be made toxic by tetrodotoxin inassociation with bacteria. In: Lassus P, Arzul G, Erard-LeDennE, Gentien P, Marcaillou-LeBaut C (eds) Harmful marine algalblooms. Lavoisier, Paris, ISBN 1-898298-11-4, pp 457–462
68. Lee MJ, Jeong DY, Kim WS, Kim HD, Kim CH, Park WW, ParkYH, Kim KS, Kim HM, Kim DS (2000) A tetrodotoxin-producing Vibrio strain, LM-1, from the puffer fish Fuguvermicularis radiatus. Appl Environ Microbiol 66:1698–1701
69. Mosher HS, Fuhrman FA (1984) Occurrence and origin oftetrodotoxin. In: Ragelis EP (ed) Seafood toxins. ACS, Wash-ington DC, pp 333–334
70. Yasumoto T, Fukui M, Sasaki K, Sugiyama K (1995) Determi-nations of marine toxins in food. J Assoc Off Anal Chem Intern78:574–582
71. Nakamura M, Oshima Y, Yasumoto T (1984) Occurrence ofsaxitoxin in puffer fish. Toxicon 22:381–385
72. Yasumura D, Oshima Y, Yasumoto T, Alcala AC, Alcala LC(1986) Tetrodotoxin and paralytic shellfish toxins in Philippinecrabs. Agric Biol Chem 50:593–598
73. Nakashima K, Arakawa O, Taniyama S, Nonaka M, Takatani T,Yamamori K, Fuchi Y, Noguchi T (2004) Occurrence ofsaxitoxins as a major toxin in the ovary of a marine pufferArothron firmamentum. Toxicon 43:207–212
74. Onoue Y, Noguchi T, Nagashima Y, Hashimoto K, Kanoh S, ItoM, Tsukada K (1983) Separation of tetrodotoxin and paralyticshellfish poisons by high-performance liquid chromatographywith a fluorimetric detection using o-phthaldialdehyde. JChromatogr 257:373–379
75. Gawley RE, Shanmugasundaram M, Thorne JB, Tarkka RM(2005) Selective detection of saxitoxin over tetrodotoxin usingacridinylmethyl crown ether chemosensor. Toxicon 45:783–787
76. Yasumoto T, Michishita T (1985) Fluorimetric determination oftetrodotoxin by high-performance liquid chromatography. AgricBiol Chem 49:3077–3089
77. Nagashima Y, Maruyama J, Noguchi T, Hashimoto K (1987)Analysis of paralytic shellfish poison and tetrodotoxin by ion-pairing high-performance liquid chromatography. Nippon SuisanGakkaishi 53:819–823
78. Quilliam MA, Thomson BA, Scott GJ, Siu KWM (1989) Ion-spray mass spectrometry of marine neutrotoxins. Rapid CommMass Spectrom 3:145–150
79. Horie M, Kobayashi S, Shimizu N, Nakazawa H (2002) Determi-nation of tetrodotoxin in puffer-fish by liquid chromatography-electrospray ionization mass spectrometry. Analyst 127:755–759
80. Jang J, Yotsu-Yamashita M (2006) Distribution of tetrodotoxin,saxitoxin, and their analogs among tissues of the puffer fishFugu pardalis. Toxicon 48:980–987
81. Nakagawa T, Jang J, Yotsu-Yamashita M (2006) Hydrophilicinteraction liquid chromatography-electrospray ionization mass
spectrometry of tetrodotoxin and its analogs. Anal Biochem352:142–144
82. EC (2004) Regulation (EC) No. 853/2004 of the EuropeanParliament and of the Council of 29 April 2004 laying downspecific hygiene rules for food of animal origin. Off J EurCommun Annex III, Sect VII, Chap V, E L 226: p 68
83. Guzman-Perez SE, Park DL (2000) Ciguatera toxins: Chemistryand detection. In: Botana LM (ed) Seafood and freshwatertoxins: pharmacology, physiology and detection. Marcel Dekker,New York, ISBN 8247-8956-3, pp 401–418
84. Capra MF, Cameron J (1987) Ciguatera poisoning: pharmacol-ogy and pathology. Report to the Fishing Industries ResearchCommittee, Department of Primary Industries, Canberra, 1987
85. Lewis RJ, Sellin M (1992) Multiple ciguatoxins in the flesh offish. Toxicon 30:915–919
86. Scheuer PJ, Takahashi W, Tsutsumi J, Yoshida T (1967)Ciguatoxin: Isolation and chemical nature. Science 155:1267–1268
87. Murata M, Legrand AM, Ishibashi Y, Fukui M, Yasumoto T(1989) Structure of ciguatoxin and its congener. J Am Chem Soc111:8929–8931
88. Sperr AE, Doucette GJ (1996) Variation in growth rate andCiguatera toxin production among geographically distinct isolatesof Gambierdiscus toxicus. In: Yasumoto T, Oshima Y, Fukuyo Y(eds) Harmful and Toxic Algal Blooms. IntergovernmentalOceanographic Commission , UNESCO, Paris, pp 309–312
89. Lewis RJ, Sellin M, Poli MA, Norton RS, MacLeod JK, SheilMM (1991) Purification and characterization of ciguatoxins fromMoray Eel (Lycodontis javanicus, Muraenidae). Toxicon29:1115–1127
90. Gaboa PM, Park DL, Fremy JM (1992) Extraction andpurification of toxic fractions from barracuda (Sphyraenabarracuda) implicated in ciguatera poisoning. In: Tosteson TR(ed) Proceedings of the 3rd international conference on ciguaterafish poisoning. Polyscience, Quebec, pp 13–24
91. Lewis RJ, Molgo J, Adams DJ (2000) Ciguatera toxins:Pharmacology of toxins involved in ciguatera and related fishpoisoning. In: Botana LM (ed) Seafood and freshwater toxins:pharmacology, physiology and detection. Marcel Dekker, NewYork, ISBN 8247-8956-3 pp 419–447
92. Park DL (1994) Evolution of methods for assessing ciguateratoxins in fish. Rev Environ Contam Toxicol 136:1–20
93. Vernoux JP, Lahlou N, Magras LP, Greaux JB (1985) Chickfeeding test: a simple system to detect ciguatoxin. Acta tropica42:235–240
94. Chungue E, Bagnis RA, Parc E (1984) The use of the mosquito(Aedes aegypti) to detect ciguatoxin in surgeonfishes (Ctenochaetusstriatus). Toxicon 22:161–164
95. Hokama Y (1985) A rapid simplified enzyme immunoassay sticktest for the detection of ciguatoxin and related polyethers fromfish tissues. Toxicon 23:939–946
96. Hokama Y, Asahina AY, Hong TWP, Shang ES, Miyahara JT(1990) Evaluation of the stick enzyme immunoassay in Carnaxsp. and Seriola dumerili associated with ciguatera. J Clin LabAnal 4:363–366
97. Park DL (1995) Detection of ciguatera and diarrheic shellfishtoxins in finfish and shellfish with ciguatect kit. J Assoc OffAnal Chem Intern 78:533–537
98. EC (2004) Regulation (EC) No. 854/2004 of the EuropeanParliament and of the Council of 29. April 2004 laying downspecific rules for the organisation of official controls on productsof animal origin intended for human consumption. Off J EurCommun Annex III, Chap II, G L 226: p123
99. Pottier I, Hamilton B, Jones A, Lewis RJ, Vernoux JP (2003)Identification of slow and fast-acting toxins in a highlyciguatoxic barracuda (Sphyraena barracuda) by HPLC/MS andradiolabelled ligand binding. Toxicon 42:663–672
Anal Bioanal Chem (2008) 391:117–134 133

100. Bottein Dechraoui MY, Tiedeken JA, Persad R, Wang Z,Granade HR, Dickey RW, Ramsdell JS (2005) Use of twodetection methods to discriminate ciguatoxins from brevetoxins:application to great barracuda from Florida Keys. Toxicon46:261–270
101. Chateau-Degat M-L, Chinain M, Cerf N, Gingras S, Hubert B,Dewailly È (2005) Seawater temperature, Gambierdiscus spp.variability and incidence of ciguatera poisoning in FrenchPolynesia. Harmful Algae 4:1053–1062
102. Hamilton B, Hurbungs M, Jones A, Lewis RJ (2002) Multipleciguatoxins present in Indian ocean reef fish. Toxicon 40:1347–1353
103. Pottier I, Vernoux J-P, Jones A, Lewis RJ (2002) Character-isation of multiple Caribbean ciguatoxins and congeners inindividual specimens of horse-eye jack (Caranx latus) by high-performance liquid chromatography/mass spectrometry. Toxicon40:929–939
104. Hamilton B, Hurbungs M, Vernoux J-P, Jones A, Lewis RJ(2002) Isolation and characterisation of Indian ocean ciguatoxin.Toxicon 40:685–693
105. Shumway SE, von Egmond HP, Hurst JW, Bean LL (1995)Management of shellfish resources. In: Hallegraeff GM,Anderson DM, Cembella AD (eds) Manual on harmful marinemicroalgae. IOC Manuals and Guides 33, UNESCO, Paris, pp433–459
106. Reguera B, Campos MJ, Fraga S, Marino J, Bravo I (1991) Themonitoring of harmful algal blooms in Galicia (NW Spain). In:Fremy JM (ed) Proceedings of symposium on marine biotoxins.CNEVA, Maisons-Alfort, pp 217–223
107. Anderson DM (1989) Toxic algal blooms and red tides, a globalperspective. In: Okaichi T, Anderson DM, Nemoto T (eds) Redtides-biology environmental science and toxicology. Elsevier,New York, pp 11–16
108. van Egmond HP, Speijers GJA, van den Top HJ (1992) Currentsituation on worldwide regulations for marine phycotoxins. J NatToxins 1:67–85
109. Food and Drug Administration (1989) Compliance policy guide7108.20 on paralytic shellfish poison in clams, mussels, oysters-fresh, frozen or Canned. FDA, Washington DC, 30 Nov 1989
110. National Shellfish Sanitation Program (NSSP) (1990) Manual ofoperations, part I: Sanitation of shellfish growing areas, 1990revision. USDepartment of Health and Human Services, Public
Health Service, Food and Drug Administration (FDA), Wash-ington DC, C22–C24
111. Council of European Communities (1992) Council decision onreference laboratories for the monitoring of marine biotoxins.COM (92) 551 final. Brussels, 16 Dec 1992
112. EC (2004) Regulation (EC) No. 854/2004 of the EuropeanParliament and of the Council of 29. April 2004 laying downspecific rules for the organisation of official controls on productsof animal origin intended for human consumption. Off J EurCommun Annex II, Chap II, A-D L 226: pp 119–121
113. Hummert C, Rühl A, Reinhardt K, Gerdts G, Luckas B (2002)Simultaneous analysis of different algal toxins by LC-MS.Chromatographia 55:673–680
114. Dahlmann J, Budakowski WR, Luckas B (2003) Liquidchromatography-electrospray ionisation-mass spectrometrybased method for the simultaneous determination of algal andcyanobacterial toxins in phytoplankton from marine waters andlakes followed by tentative structural elucidation of micro-cystins. J Chromatogr A 994:45–57
115. MacKenzie L, Holland P, McNabb P, Beuzenberg V, Selwood A,Suzuki T (2002) Complex toxin profiles in phytoplankton andGreenshell mussels (Perna canaliculus), revealed by LC-MS/MSanalysis. Toxicon 40:1321–1330
116. Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, MagnoS, Santelia F, Tsoukatou M (2006) Investigation of the toxinprofile of Greek mussels Mytilus galloprovincialis by liquidchromatography-mass spectrometry. Toxicon 47:174–181
117. McNabb P, SelwoodAI, Holland PT (2005)Multiresidue method fordetermination of algal toxins in shellfish: single-laboratory validationand interlaboratory study. J Anal Off Anal Chem Int 88:761–772
118. Suzuki T, Jin T, Shirota Y, Mitsuya T, Okumura Y, Kamiyama T(2005) Quantification of lipophilic toxins associated withdiarrhetic shellfish poisoning in Japanese bivalves by liquidchromatography-mass spectrometry and comparison with mousebioassay. Fisheries Sci 71:1370–1378
119. Botana LM (ed) (2007) Phycotoxins: chemistry and biochemis-try. Blackwell, Ames, ISBN 978-0-8138-2700-1
120. Fux E, McMillan D, Bire R, Hess P (2007) Development of anultra-performance liquid chromatography-mass spectrometrymethod for the detection of lipophilic marine toxins. J ChromatogrA 1157:273–280
134 Anal Bioanal Chem (2008) 391:117–134