passive air samplers for semivolatile organic …...ii passive air samplers for semivolatile organic...
TRANSCRIPT

Passive Air Samplers for Semivolatile Organic Compounds: Experiments, Modeling, and Field Application
by
Xianming Zhang
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Department of Chemistry University of Toronto
© Copyright by Xianming Zhang 2012

ii
Passive Air Samplers for Semivolatile Organic Compounds:
Experiments, Modeling, and Field Application
Xianming Zhang
Doctor of Philosophy
Department of Chemistry
University of Toronto
2012
Abstract
Knowledge gaps related to mass transfer processes involved in passive air sampling of
semivolatile organic compounds and factors potentially influencing passive sampling rates
(PSRs) were addressed with controlled laboratory experiments, mass transfer modeling, and a
field sampling campaign. The observed non-uniform SVOC distributions within porous passive
sampling media (PSMs) contradict an assumption in an earlier passive air sampling theory and
proved the existence of a kinetic resistance on the PSM side. This resistance can affect PSRs as
revealed by a new PAS model which is based on fundamental laws of mass transfer in air and
porous media. By considering mass transfer processes within the PSM, the model is able to
explain the large variations of field calibrated PSRs with temperature and between SVOC species
and the two-stage uptake process, which cannot be addressed by the earlier PAS theory. Because
the PSM side kinetic resistance invalidates the assumption that depuration compounds added to
the PSM prior to deployment are subject to the same kinetic resistance as the sampled SVOCs,
PSRs derived from the loss rates of depuration compounds can differ from the actual PSRs of the
sampled SVOCs. Using such PSRs could thus introduce additional uncertainty to PAS-derived
air concentrations.

iii
Experiments using XAD-resin and silica-gel filled mesh cylinder as PSMs for the uptake of
SVOCs and water vapor respectively revealed that sorbent in the inner portion of the PSM does
not take part in chemical uptake; PSRs are thus proportional to the interfacial transfer area but
not the amount of the sorbent. Accordingly, thinner PSM can be used to reduce the amount of
sorbent while keeping or even increasing the PSRs. Optimized designs of PASs could be tested
time efficiently using the gravimetrical approach based on water vapor uptake by silica gel.

iv
Acknowledgments
First, I would like to express my sincere gratitude to my supervisor, Prof. Frank Wania, for his
continuous support during my PhD study. His deep insight and innovative ideas in the field of
environmental chemistry have been guiding me throughout my PhD. I also thank Ying Lei for
the guidance and assistance in the lab. I would like to thank my supervisory/exam committee
members, Profs. Terry Bidleman, Miriam Diamond, Jennifer Murphy and Eric Reiner for their
guidance during my PhD study, and Prof. Thomas Holsen (Clarkson University) for being part of
my defense committee.
Thanks also go to the collaborators in my PhD research projects: Dr. Takeshi Nakano and
Masahiro Tsurukawa (Hyogo Prefecture Institute of Environmental Sciences, Japan), Prof. Akira
Kondo (Osaka University, Japan), and Dr. John Barnes (Mauna Loa Observatory, USA). I
appreciate the collaborations with you. Without these collaborations, the accomplishments I’ve
made during my PhD study would not have been possible. I also thank Prof. Kai-Uwe Goss
(Helmholtz-Centre for Environmental Research–UFZ, Germany), Dr. Eldbjørg Heimstad
(Norwegian Institute for Air Research–NILU, Norway), Dr. Li Shen, Dr. Satyendra Bhavsar
(Ontario Ministry of Environment), and Ingjerd Krogseth (NILU) for the opportunities to work
together on some projects beyond this thesis.
I would like to thank my colleagues in the Wania Group–Dr. Jon Arnot, Dr. James Armitage, Dr.
Trevor Brown, Anya Gawor, Johnny Westgate, Cristina Quinn, Dr. Hang Xiao, Dr. Chuba
Shunthirasingham, Dr. Steve Hayward and summer students – Cindy Wong and Xiaoshu Cao for
different types of assistance during my PhD.
I am gratitude to the graduate student advisors Ms. Anna Liza Villavelez and Ms. Denise Ing at
the Department of Chemistry and Mr. Pavel Pripa at the Centre for Environment, for their help
during my PhD.
I would like to acknowledge the Graduate Student Award from the Centre for Global Change
Sciences (University of Toronto) for supporting my field work; the Ontario Graduate Scholarship
for financial support; travel fellowships for me to attend conferences by the Department of
Chemistry, Faculty of Arts and Sciences and School of Graduate Studies, University of Toronto
Finally, I extend my thanks to my family and friends, who have always been there providing
continuous support and encouragement.

v
Table of Contents
Acknowledgments .......................................................................................................................... iv
Table of Contents ............................................................................................................................ v
List of Tables ................................................................................................................................ xi
List of Figures .............................................................................................................................. xiii
List of Acronyms .......................................................................................................................... xx
Chapter 1. Passive Air Samplers for Semivolatile Organic Compounds: An Overview ................ 1
1.1 A Historical Perspective on the Development of Passive Air Sampling Techniques ......... 1
1.2 Applications of Passive Air Samplers for SVOCs .............................................................. 5
1.3 Mechanism and Theory of Passive Air Sampling ............................................................... 7
1.4 Factors Influencing Passive Air Sampling Rates .............................................................. 11
1.5 Objective and Structure of the Thesis ............................................................................... 14
Chapter 2. Sampling Medium Side Resistance to Uptake of Semi-volatile Organic Compounds
in Passive Air Samplers ........................................................................................... 16
2.1 Abstract ............................................................................................................................. 17
2.2 Introduction ....................................................................................................................... 17
2.3 Materials and Methods ...................................................................................................... 19
2.3.1 Passive Sampling Media. ...................................................................................... 19
2.3.2 Chemicals. ............................................................................................................. 20
2.3.3 Sampling Design. .................................................................................................. 20
2.3.4 Sample Extraction and Analysis. .......................................................................... 21
2.3.5 QA/QC. ................................................................................................................. 22
2.3.6 Derivation of passive air sampling rates. .............................................................. 22
2.3.7 Derivation of the effective diffusivities on the PSM side. .................................... 22
2.3.8 Mechanistic model of effective diffusivity in porous media. ............................... 23
2.4 Results and Discussion ..................................................................................................... 23

vi
2.4.1 Passive Air Sampling Rates. ................................................................................. 23
2.4.2 Evidence of kinetic resistance on chemical transfer within PSM. ........................ 25
2.4.2.1 PCB Uptake from Air. ............................................................................ 25
2.4.2.2 Depuration Compounds. ......................................................................... 27
2.4.3 Mass transfer coefficient for chemical diffusion between the two PUF layers
(kPUF12). .................................................................................................................. 28
2.4.4 Effective PSM-side diffusivities (DE,PUF). ............................................................ 29
2.4.5 Further Comments on the PSM-Side Kinetic Resistance and Its Implications. ... 30
2.5 Acknowledgments ............................................................................................................. 32
Supporting Information of Chapter 2 ....................................................................................... 33
Determination of PSM-air partition coefficients and sorption enthalpies of PCB
congeners using poly-parameter linear free energy relationships ......................... 33
Detailed information on the depuration compounds and spiking procedures ................... 40
Detailed information on the depuration compounds and spiking procedures ................... 40
Description of the two-layer mass balance model used to derive effective diffusivities
of PCBs through the passive sampling medium ................................................... 43
Transfer kinetics of the depuration compounds ................................................................ 50
Chapter 3. Modeling the uptake of semi-volatile organic compounds by passive air samplers:
Importance of mass transfer processes within the porous sampling media ............. 54
3.1 Abstract ............................................................................................................................. 55
3.2 Introduction ....................................................................................................................... 55
3.3 Methods ............................................................................................................................. 57
3.3.1 Conceptual Model of Chemical Mass Transfer during Passive Air Sampling. .... 57
3.3.2 Mathematical Model of Chemical Mass Transfer during Passive Air Sampling. 58
3.3.2.1 Diffusion Across the Stagnant Air Layer. .............................................. 59
3.3.2.2 Diffusion within the Porous PSM. .......................................................... 59
3.3.2.3 Chemical Exchange between Air-filled Macro-pores and XAD Pellets 60
3.3.2.4 Model Solution ....................................................................................... 61

vii
3.3.3 Sensitivity Analysis .............................................................................................. 61
3.3.4 Model Application ................................................................................................ 62
3.4 Results and Discussion ..................................................................................................... 63
3.4.1 Influence of Mass Transfer Processes and Associated Parameters on the
Passive Air Sampling Rate. ................................................................................... 63
3.4.2 Influence of Chemical Properties and Temperatures on Passive Air Sampling
Rates. ..................................................................................................................... 67
3.4.3 Two-Stage Uptake Process. .................................................................................. 70
3.4.4 Non-Uniform Chemical Distribution within Passive Sampling Media. ............... 70
3.4.5 Knowledge Gap and Implications. ........................................................................ 71
3.5 Acknowledgments ............................................................................................................. 72
Supporting Information of Chapter 3 ....................................................................................... 73
Mathematical Model of Chemical Uptake by XAD-PAS. ................................................ 73
Mathematical Model of Chemical Uptake by PUF-PAS. ................................................. 76
Chapter 4. Influence of Sampler Configuration on the Uptake Kinetics of a Passive Air
Sampler .................................................................................................................... 88
4.1 Abstract ............................................................................................................................. 89
4.2 Introduction ....................................................................................................................... 89
4.3 Materials and Methods ...................................................................................................... 91
4.3.1 Setup for Water Uptake Experiments ................................................................... 91
4.3.2 Characterizing Water Uptake by Silica-gel .......................................................... 92
4.3.3 Assessment of Different Sampler Configurations ................................................ 92
4.3.4 Indoor Calibration of XAD-based Passive Air Samplers Using Sampling
Media of Different Diameters ............................................................................... 93
4.3.5 Sample Extraction and Preparation ....................................................................... 94
4.3.6 PCB Analysis ........................................................................................................ 94
4.3.7 QA/QC .................................................................................................................. 94
4.4 Results and Discussion ..................................................................................................... 95

viii
4.4.1 Characteristics of Water Uptake by Silica Gel ..................................................... 95
4.4.2 Effect of Interfacial Transfer Area and Sorbent Amount on Uptake .................... 96
4.4.3 Effect of the Position of the PSM within the Sampler Housing On Uptake ......... 98
4.4.4 Effect of Dimensions of the Sampling Medium and Sampler Housing on
Uptake ................................................................................................................. 100
4.4.5 Uptake of PCBs by XAD-filled Mesh Cylinder of Different Diameters ............ 101
4.4.6 Water Uptake by Silica Gel vs. SVOC Uptake by XAD .................................... 102
4.4.7 Implications ......................................................................................................... 103
4.5 Acknowledgments ........................................................................................................... 104
Supporting Information of Chapter 4 ..................................................................................... 105
Derivation of KSA and kO from curve ftting on the experimental data. ........................... 109
Chapter 5. Wind Effect on Chemical Uptake and Axial Distribution in the Sampling Medium
of a Passive Air Sampler ........................................................................................ 114
5.1 Abstract ........................................................................................................................... 115
5.2 Introduction ..................................................................................................................... 115
5.3 Materials and Methods .................................................................................................... 117
5.3.1 Experimental Setup ............................................................................................. 117
5.3.1.1 Axial Distribution of Chemicals in the Sampling Medium .................. 117
5.3.1.2 Wind Effect on Passive Air Sampling Kinetics.................................... 118
5.3.2 Sample Preparation and Extraction ..................................................................... 119
5.3.3 Chemical Analysis .............................................................................................. 119
5.3.4 QA/QC ................................................................................................................ 119
5.3.5 Computational Fluid Dynamics Simulation ........................................................ 120
5.4 Results and Discussion ................................................................................................... 121
5.4.1 Indoor Experiment on Axial Distributions of PCBs in the XAD-filled Mesh
Cylinder ............................................................................................................... 121
5.4.2 Outdoor Experiment on Axial Distributions of PCBs in the XAD mesh
cylinder ............................................................................................................... 124

ix
5.4.3 Wind Effect on Passive Sampling Kinetics ........................................................ 126
5.4.4 Simulated Wind Conditions in the Sampler ........................................................ 127
5.4.5 Implications and Further Research Questions Originating From This Study ..... 128
5.5 Acknowledgments ........................................................................................................... 129
Supporting Information of Chapter 5 ..................................................................................... 130
Testing the slopes of two linear regressions using analysis of covariance (ANCOVA). 137
Chapter 6. Application of passive air samplers and flow-through air samplers to assess semi-
volatile organic contaminants in the atmosphere of Hawaii .................................. 146
6.1 Abstract ........................................................................................................................... 147
6.2 Introduction ..................................................................................................................... 147
6.3 Materials and Methods .................................................................................................... 149
6.3.1 Sampling Sites .................................................................................................... 149
6.3.2 Sampling Campaign ............................................................................................ 150
6.3.3 Sample Extraction ............................................................................................... 151
6.3.4 Sample Analysis .................................................................................................. 151
6.3.5 QA/QC ................................................................................................................ 152
6.3.6 Air Mass Back Trajectory Analysis .................................................................... 152
6.4 Results and Discussion ................................................................................................... 152
6.4.1 PAHs and PBDEs Accumulated in PASs of Different Configuration ................ 152
6.4.2 Passive Air Sampler Derived Spatial Variations of PAHs and PBDEs .............. 154
6.4.3 Monthly Variations of PAHs and PBDEs ........................................................... 157
6.4.4 Global Background Levels of Atmospheric PAHs and PBDEs ......................... 158
6.4.5 Origin of SVOCs in Hawaii: Long Range Atmospheric Transport vs. Material
Flows ................................................................................................................... 160
6.5 Acknowledgments ........................................................................................................... 161
Supporting Information of Chapter 6 ..................................................................................... 162
Chapter 7. Conclusions and Outlook .......................................................................................... 169

x
7.1 Conclusions ..................................................................................................................... 169
7.2 Overall Implications ........................................................................................................ 171
7.2.1 Uncertainty associated with passive air sampling derived air concentrations .... 171
7.2.2 Problems involved in deriving passive sampling rates from the loss of
depuration compounds from porous sampling media. ........................................ 173
7.2.3 Insights into the optimization of passive air sampler designs ............................. 174
7.3 Further Research Needs and Recommendations ............................................................. 176
References 178

xi
List of Tables
Table S2.1 XAD-air partition coefficients (KXAD/A) and sorption enthalpies (ΔHS, XAD, J/mol)
for PCBs................................................................................................................... 34
Table S2.1 (continued) ............................................................................................................... 35
Table S2.1 (continued) ............................................................................................................... 36
Table S2.2 PUF-air partition coefficients (KPUF/A) and sorption enthalpies (ΔHS, PUF, J/mol)
for PCBs................................................................................................................... 37
Table S2.2 (continued) ............................................................................................................... 38
Table S2.2 (continued) ............................................................................................................... 39
Table S2.3 Limit of detection a (LOD) of PCBs analyzed using HRGC/MS ............................ 42
Table S2.4 Congener-specific passive air sampling rates of PCBs derived using linear least
squares fitting........................................................................................................... 46
Table S2.4 (continued) ............................................................................................................... 47
Table S2.4 (continued) ............................................................................................................... 48
Table S2.5 Passive air sampling rates determined in different studies using XAD and PUF as
PSM. ........................................................................................................................ 49
Table S3.1 Properties of the modeled passive air sampling media ............................................ 80
Table S4.1 Target ions, quanlify ions and limit of detection (LOD) of the chemicals analyzed
using GC-MS selected ion monitoring mode. ....................................................... 108
Table S4.1 (continued) ............................................................................................................. 109
Table S4.2 Parameters derived from the fitting of the water uptake kinetics .......................... 110
Table S4.3 Overall mass transfer coefficient from the air to the sampling medium for
selected SVOCs derived based on the water uptake kinetics a .............................. 113
Table S5.1 Target ions, quanlify ions and limit of detection (LOD) of the PCB homolog
groups analyzed using GC-MS selected ion monitoring mode. ............................ 133
Table S5.2 Two-factorial ANOVA and Scheffé's post hoc test on the PCB congeners
accumulated at the three axially segmented PSM ................................................. 136
Table S5.3 Descriptive statistics on the temperature (°C) recorded by the temperature logger
in the passive air samplers deployed outdoors ...................................................... 140

xii
Table S5.4 Passive sampling rates (PSRs) derived as the slopes of the regressiona between
the deployment time and equivalent sampling volume. ........................................ 144
Table S6.1 Geographic coordinates and elevations of the sampling sites ............................... 162
Table S6.2 Information on the 100 μL surrogate standards spiked prior to sample
extractions .............................................................................................................. 164
Table S6.3 Precursor ions, product ions and collision energies for the multiple reaction
monitoring mode for PAH analysis ....................................................................... 165
Table S6.4 Precursor ions, product ions and collision energies for the multiple reaction
monitoring mode for PBDE analysis ..................................................................... 166
Table S6.5 APOWin (v1.92) estimated half life of reaction with hydroxyl radicals in the
atmosphere ............................................................................................................. 168

xiii
List of Figures
Figure 1.1 Twenty-year trends of the number of studies published on the topic of “passive
air sampler/sampling”. Data retrieved from the Web of Knowledge Results
Analysis Tool. ............................................................................................................ 2
Figure 1.2 Schematic of (a) the polyurethane foam (PUF) based passive air sampler and (b)
the cylindrical XAD-resin based passive air sampler. ............................................... 3
Figure 1.3 Structure of this thesis and task involved/skills developed from the studies. ......... 13
Figure 2.1 Design of the layered passive air sampling media (XAD and PUF) used to study
the distribution of PCBs within the passive sampling medium. .............................. 20
Figure 2.2 Comparison of the passive air sampling rates of PCB homologs between the
passive sampling media of XAD and PUF positioned in the same type of
cylindrical sampling housing. .................................................................................. 24
Figure 2.3 PCB accumulation and distribution in the outer, middle and inner layers of the
passive sampling media (PUF and XAD). Plots are based on duplicated
measurements. Mono-PCB (PCB-1) and Penta-PCB (PCB-98/95) are used to
illustrate the differences between PCBs of different chlorination or
physicochemical properties. .................................................................................... 27
Figure 2.4 The relationship between the PUF-air partition coefficients (KPUF/A at 20°C) and
the mass transfer coefficients for chemical diffusion between the two PUF layers
(kPUF12, m/h). The data points represent selected mono-, di-, and tri-CB congeners
that penetrated into the inner PUF with detectable amounts. The dash lines
indicate 95% confidence interval of the regression model. ..................................... 28
Figure 2.5 Relationship between the effective diffusivity in PUF (DE,PUF, m2/h) and the
PUF/air partition coefficient (KPUF/A) for PCBs. The upper- and lower-bound
experimentally derived DE,PUF were based on a diffusion length of 1 and 2.5 cm,
respectively. The upper- and lower-bound modeled DE,PUF were based on a f /rSA
value of 0.14 and 0.53, respectively. ....................................................................... 30
Figure S2.1 Illustration of the sampling scheme in this study. ................................................... 41
Figure S2.2 Reproducibility of the duplicated samples as represented by the relative
difference of the sampling rate R (m3/h) between duplicates. The relative
difference is defined as 1 2
1 20.5( )
R R
R R ..................................................................... 41
Figure S2.3 Analytical procedure recovery of the surrogate standards spiked prior to sample
extraction. ................................................................................................................ 42
Figure S2.4 Illustration of the two-layer mass balance model used to derive effective
diffusivities of PCBs through the passive sampling medium. ................................. 43

xiv
Figure S2.5 Relationship between homolog-specific molecular diffusivities in air and passive
air sampling rates. The molecular diffusivities in air are derived from the Fuller-
Schettler-Giddings equation109
; the passive air sampling rate is based on the
median of the congener-specific sampling rates in each homolog group. ............... 50
Figure S2.6 Changes of the amounts of depuration compounds (tri- and hepta-CBs) spiked to
the inner, middle, and outer layer of PUF. The amount of chemicals present in
each layer (Mi) was normalized to the amount (M0) in the field blanks (samples
retrieved at t=0). ...................................................................................................... 52
Figure S2.7 Changes of the amounts of depuration compounds (mono-/di- and tri-CBs)
spiked to the inner, middle, and outer layer of XAD. The amount of chemicals
present in each layer (Mi) was normalized to the amount (M0) in the field blanks
(samples retrieved at t=0). ....................................................................................... 53
Figure S2.8 Illustration of the sensitivity of DEPUF to the variations of DA and KPUF/A. (a)
based on f/rSA value of 0.18; (B) based on f/rSA value of 0.45. ............................... 53
Figure 3.1 Conceptual diagram of the chemical mass transfer processes between air and the
passive sampling media (PSMs) in the (a) XAD-resin based passive air sampler
and (b) polyurethane foam based passive air sampler. The mass transfer
processes include: (1) diffusion through the stagnant air layer surrounding the
PSM; (2) diffusion through macro-pores within the PSM; (3) sorption/desorption
between porous air and solid PSM material. The microstructure of polyurethane
foam was taken from a micrograph contributed by JA Elliott to the DoITPoMS
Micrograph Library, University of Cambridge under the Creative Commons
Attribution Non-Commercial Share Alike license. ................................................. 58
Figure 3.2 Sensitivity (SC) of the sampling rate (PSR, m3/d) of the XAD-based passive air
sampler for compounds with different equilibrium partition coefficients between
XAD and air (KXAD/A) and different sorption rate constants (ksorb) to changes in
(a) the thickness of the stagnant air layer (δBL), (b) the molecular diffusivity in
bulk air (DA), (c) the molecular diffusivity in the macroporous fraction within the
XAD (DPA), (d) KXAD/A, and (e) ksorb. δBL = 0.01 cm was used as the baseline for
the SC calculations. Based on the other five panels, panel (f) identifies four
regions, in which the PSR is predominantly influenced by a particular mass
transfer process. ....................................................................................................... 64
Figure 3.3 Illustration of the dependence of passive sampling rates (PSRs) on chemical
properties and temperature. Molecular size: M1 > M2; temperature T1 < T2. The
map depicting PSRs in the KSA-ksorb chemical space was constructed based on the
model for a XAD-passive air sampler deployed for 360 d assuming a stagnant air
boundary layer thickness δBL of 0.01 cm. PSRs exceeding 5 m3/d were calculated
for the combination of large KSA and large ksorb (hatched area), which is unlikely
to exist among real chemicals. ................................................................................. 67
Figure S3.1 Illustration showing the discretization of the PSM of the XAD-PAS to solve the
diffusion equations. m = 200 and n = 50 were used in this study. .......................... 75

xv
Figure S3.2 Illustration showing the discretization of the PSM of the PUF-PAS to solve the
diffusion equations. m = 200 and n = 50 were used in this study. .......................... 79
Figure S3.3 Illustration of how passive air sampling rates (PSRs) were derived from a linear
fit on six discrete data points placed equidistantly on the uptake curve generated
by the model. ........................................................................................................... 81
Figure S3.4 Distribution of the difference between KXAD/A and KPUF/A for chlorothalonil,
endosulfan I, endosulfan II, atrazine, alachlor, metolachlor, trifluralin, HCB, α-
HCH, γ-HCH and 209 PCB congeners based on calculations using polyparameter
linear free energy relationships (ppLFERs).38,111,119
................................................ 81
Figure S3.5 Empirical relationships of XAD/air partition coefficient (KXAD/A) and PUF/air
partition coefficient (KPUF/A)with the diffusivity of chemicals in air (DA, cm2/s)
based on 209 polychlorinated biphenyl congeners and 10 organochlorinated
pesticides (namely, chlorothalonil, endosulfan I, endosulfan II, atrazine, alachlor,
metolachlor, trifluralin, HCB, α-HCH, and γ-HCH). KXAD/A and KPUF/A of the
chemicals were calculated using polyparameter linear free energy relationships
(ppLFERs).38,111,119
DA was calculated using the Fuller-Schettler-Giddings
equation with La Bas molar volumes.109
................................................................. 82
Figure S3.6 Illustration of the relationship between the change of internal energy (ΔUSA,
from air phase M to sorbed phase M···S) and the activation energies of sorption
(Ea+) and desorption (Ea–). ....................................................................................... 83
Figure S3.7 Sensitivities of passive air sampling rate (m3/d) of XAD-PAS (left) and PUF-
PAS (right) (deployed for 90 d) to changes of molecular diffusivity in bulk air
(DA), molecular diffusivity in the macroporous fraction within the PSM (DPA),
equilibrium partition coefficient between the sorbent and air (KSA), and the
sorption rate constant (ksorb) based on stagnant boundary layer thickness δBL of
0.001 cm (top), 0.01 cm (centre), and 0.1 cm (bottom). .......................................... 84
Figure S3.8 Comparison between cylindrical and disk-like PSM configurations for the
sensitivities of passive air sampling rate (m3/d) to the changes of in bulk air (DA),
molecular diffusivity in the macroporous fraction within the media PSM (DPA),
equilibrium partition coefficient between the sorbent and air (KSA), and the
sorption rate constant (ksorb) at a stagnant boundary layer thickness δBL of
0.01cm. .................................................................................................................... 85
Figure S3.9 Modeled passive air sampling rates as a function of equilibrium partition
coefficient between the XAD and air KXAD/A and the sorption rate constant ksorb
with stagnant air layers of 0.1, 0.01, and 0.001 cm thickness. ................................ 86
Figure S3.10 Modeled chemical uptake curve in passive air sampling of chemicals with
different combinations of KPUF/A and ksorb. .............................................................. 86
Figure S3.11 Penetration depth (defined as the thickness of outer sampling medium layer
which accumulates 90% of the sampled chemical amount) of chemicals in XAD
and PUF, both in cylindrical and in disk configuration. .......................................... 87

xvi
Figure 4.1 Measured and model-fitted equivalent air volume derived from passive sampling
of water vapor from air using silica gel filled mesh cylinder as a sampling
medium. Data were recorded every 1 min for the first 30 min and every 5 min
afterwards. ............................................................................................................... 95
Figure 4.2 Effect of interfacial transfer area and sorbent amount on the uptake of water
vapor from air by silica gel. I and II: short and long silica gel filled mesh cylinder
in short and long housing; III: long mesh cylinder with a metal rod positioned at
the center with silica gel surrounding it. Ratios of the interfacial transfer area to
bulk XAD volume for I, II and III are 1, 1 and 1.25 cm-1
respectively. .................. 97
Figure 4.3 Effect of the distance of the silica gel filled mesh cylinder to the opening of the
sampler housing on the uptake of water vapor from air by silica gel. I and II: long
mesh cylinder at different positions within long housing; III-V: short mesh
cylinder at different positions within long housing. ................................................ 98
Figure 4.4 Effect of dimensions of the sampling medium and sampler housing on the uptake
of water vapor from air by silica gel. I-III: silica-gel filled mesh cylinder (lC=10
cm, dC=2cm) without housing, in a housing with dH=6 cm, and in a housing with
dH=10.5 cm; IV and V: silica-gel filled mesh cylinder (lC=10 cm, dC=1.2cm and
3 cm) in a housing with dH=10.5 cm. ...................................................................... 99
Figure 4.5 Comparison of passive sampling rates of PCBs between passive sampling
medium of different diameters. Data of 1.2-cm and 2-cm mesh cylinder were
obtained in this study; data of the 3-cm mesh cylinder were based on the sum of
three concentric layers in a previous study.126
....................................................... 102
Figure S4.1 Schematic of the cylindrical passive air samplers. (a) long version with 20 cm-
long mesh cylinder; (b) short version with 10 cm-long mesh cylinder. ................ 105
Figure S4.2 Illustration of gravimetrical experiment for passive air sampling of water using
silica gel filled mesh cylinder as the passive sampling medium. .......................... 105
Figure S4.3 Experiment setup to investigate the effect of interfacial transfer area and sorbent
amount on uptake of water vapor from air by silica gel. ....................................... 106
Figure S4.4 Experiment setup to investigate the effect of the distance of the silica gel filled
mesh cylinder to the opening of the sampler housing on uptake of water vapor
from air by silica gel. ............................................................................................. 106
Figure S4.5 Experiment setup to investigate the effect of Dimensions of the sampling
medium and sampler housing on uptake of water vapor from air by silica gel. .... 106
Figure S4.6 Schematics of the passive air sampler calibration for indoor PCBs. ..................... 107
Figure S4.7 Measured and model-fit equivalent air volume derived from the duplicated water
uptake experiment .................................................................................................. 110
Figure S4.8 Reproducibility of water uptake experiment on different sampler configurations.

xvii
The coefficient of variance is based on 6 replicated experiments ......................... 111
Figure S4.9 Method recovery of PCB analysis based on 13
C-PCB surrogate standards spiked
into the samples before extraction ......................................................................... 111
Figure S4.10 Congener specific PCB sampling rates (R) and interfacial transfer area
normalized sampling rate (SR) of XAD-PAS indoors. Sampling rates of the 1.2-
cm and 2-cm mesh cylinder were obtained from calibrations in this study;
sampling rates of the 3-cm were retrieved from a previous study126
based on the
sum of three concentric layers. .............................................................................. 112
Figure 5.1 Spatial distribution of speed (m/s) of the lab generated wind. Wind speeds were
measured with a hot-wire anemometer at a resolution of 2 cm. The round and
elliptical rings represent the position (projective planes of the opening) of the
straight and 45° slanted passive air samplers, respectively ................................... 121
Figure 5.2 Amounts of PCBs accumulated in the three axial segments of XAD-resin based
passive air samplers deployed indoors under windy condition generated using
electric fans (L1W1 and L1W2), wind still condition (L1-L4) and deployed
outdoors with normal sampler configuration (ODN), with black painted housings
(ODB) and with housings shaded from sunlight (ODC). The sum of the amounts
in the three segments is compared with the amount in a non-segmented sampler
deployed simultaneously at the same location. The whiskers indicate the root
mean square of the distances of the two points to the average. ............................. 122
Figure 5.3 Masses of PCBs accumulated in the three axial segments of two XAD-resin
based passive air samplers deployed under wind still and lab generated windy
conditions ............................................................................................................... 123
Figure 5.4 Passive air sampling kinetics (Penta-CB110 as an example) for samplers under
windy (lab generated wind blowing at 45° slanted angle and at straight angle
towards the cylindrical passive air samplers) and wind still conditions ................ 126
Figure 5.5 Computational fluid dynamic simulations of wind field on the cross sections at
the top (a and d), middle (b and e) and bottom (c and f) of the XAD mesh
cylinders within the housing of the passive air samplers subject to wind blowing
at straight (a-c) and at 45° slanted angles (e-f) towards the sampler ..................... 128
Figure S5.1 Passive air samplers with axially segmented XAD-filled mesh cylinder to study
the axial chemical distribution within the sampling medium. ............................... 130
Figure S5.2 Experiment setup to study chemical distributions in the axially segmented
passive sampling medium (XAD mesh cylinder) under wind and wind still
conditions. .............................................................................................................. 130
Figure S5.3 Experiment setup to study potential effect of solar radiation on chemical uptake
and axial distribution within the XAD mesh cylinder. .......................................... 131
Figure S5.4 Experiment setup to study potential wind effects on chemical uptake by the XAD

xviii
passive air sampler. ................................................................................................ 132
Figure S5.5 Variations of wind speed measured at the mouth of the fans (point A of Figure 1)
and at the openings of the sampler housings (point B of Figure 1) for the 24
passive air samplers subjected under lab generated windy conditions. ................. 132
Figure S5.6 Amounts of PCBs accumulated in the three axially segment3ed passive air
sampling medium (XAD mesh cylinder) of passive air samplers deployed in the
four indoor locations (L1-4), passive air samplers with lab generated wind (L1W),
and at outdoor location (OD) ................................................................................. 134
Figure S5.7 Distribution of PCBs in the three axially segmented XAD mesh cylinders in the
duplicated PASs blown with lab generated wind. ................................................. 135
Figure S5.8 Distribution of PCBs in the three axially segmented XAD mesh cylinders in the
duplicated PASs (a) under the quasi wind still condition; (b) under the lab
generated windy condition; (c) in outdoor environment ....................................... 135
Figure S5.9 Mass of PCBs accumulated in the three axially segmented passive air sampling
medium (XAD mesh cylinder) of passive air samplers deployed outdoors (a)
under normal condition (b) with black sampler housing and (c) with black
sampler housing shaded from direct sunshine. ...................................................... 138
Figure S5.10 Distribution of PCBs in the three axially segmented XAD mesh cylinders in the
normal housings, back housings and housings shaded from sunshine. ................. 139
Figure S5.11 Temperature differences in the normal, black, and shaded passive sampler
housing. .................................................................................................................. 141
Figure S5.12 Comparison of temperatures (°C) at different positions within the passive air
sampling housing. .................................................................................................. 142
Figure S5.13 Passive air sampling kinetics for samplers under windy (lab generated wind
blowing at 45° slanted angle and at straight angle towards the cylindrical passive
air samplers) and wind still conditions. ................................................................. 143
Figure S5.14 (a) Passive air sampling rates of PCBs under quasi wind still condition and with
lab generated wind blowing at straight and 45° slanted angles towards the passive
air samplers; (b) statistical test on the difference of passive air sampling rates
between the two windy conditions. ....................................................................... 145
Figure 6.1 Locations of the sampling sites on the Big Island of Hawaii. A-I: passive air
samplers; A and F: flow-through air samplers. ..................................................... 149
Figure 6.4 Flow-through sampler derived air concentrations of fluorene (Fluo),
phenanthrene (Phe), fluoranthene (Flu), pyrene (Pyr), BDE47and BDE99 during
the five sampling months. ...................................................................................... 157
Figure 6.5 Comparison of the PAH air concentrations measured at Mauna Loa in this study

xix
using flow-through samplers (based on data from five sampling months) and
passive air samplers (based on passive sampling rate range of 0.5-5.5 m3/d from
previous calibrations20,89,106
) with those at Arctic background sites.180
................ 159
Figure 6.6 Comparison of the PBDE air concentrations measured at Mauna Loa in this study
using flow-through samplers (based on data from five sampling months) and
passive air samplers (based on passive sampling rate range of 0.5-5.5 m3/d from
previous calibrations) with those at other global background sites. ...................... 160
Figure S6.1 Illustration of the three configurations of passive air samplers used in this study 163
Figure S6.2 Daily averaged temperature profiles at the sampling sites. ................................... 163
Figure S6.3 Decreasing trend of SVOC levels along the transect A to F. ................................ 167
Figure S6.4 Endpoint density of trajectories arriving at site A and F during the five sampling
months based on 14 d back trajectory calculated using HYSPLIT model at every
6 h interval. ............................................................................................................ 168
Figure 7.1 Illustration of factors potentially contributing to the uncertainty of passive air
sampling derived air concentration (CA). .............................................................. 173
Figure 7.2 Illustrations of suggested approaches to optimize the design of passive air
samplers using XAD resin as the sampling medium. (a) Using mesh cylinder of
smaller diameter. (b) Using disk-shaped mesh container. ..................................... 175

xx
List of Acronyms
AAS active air sampler
ANCOVA analysis of covariance
ANOVA analysis of variance
CFD computational fluid dynamics
CI confidence interval
DC depuration compound
Flu fluoranthene
Fluo fluorene
FT free troposphere
FTS flow-through sampler
GC/MS gas chromatography mass spectrometry
GC/MS/MS gas chromatography tandem mass spectrometry
GDAS global data assimilation system
HRGC/MS high resolution gas chromatography mass spectrometry
HVAAS high-volume active air sampler
HYSPLIT hybrid single-particle Lagrangian integrated trajectory
ID inner diameter
LLSF linear least squares fitting
LOD limit of detection
MRM multiple reaction monitoring
PAH polycyclic aromatic hydrocarbon
PAS passive air sampler
PBDE polybrominated biphenyl ether
PCB polychlorinated biphenyl
Phe phenanthrene
POP persistent organic pollutant
PSM passive sampling medium
PSR (or R) passive sampling rate
PUF polyurethane foam
Pyr pyrene

xxi
QA/QC quality assurance/quality control
RSD relative standard deviation
SA surface area
SI supporting information
SIM selective ion monitoring
SIP sorbent impregnated polyurethane foam
SPMD semi-permeable membrane devices
SR surface area normalized passive sampling rate
SVOC semivolatile organic compound
VMS volatile methyl siloxane
VOC volatile organic compound
XAD styrene-divinylbenzene copolymer

1
Chapter 1. Passive Air Samplers for Semivolatile Organic Compounds: An
Overview
1.1 A Historical Perspective on the Development of Passive Air Sampling Techniques
Passive air sampling techniques based on molecular diffusion and sorption to various sorbents as
sampling media have been developed to sample and monitor gaseous contaminants in the air as
early as the 1970s.1,2
Early passive air samplers (PASs), which were more often referred to as
diffusive samplers at that time, had been widely used to sample inorganic atmospheric
contaminants or volatile organic contaminants (VOCs) in order to assess occupational exposures
to these contaminants.1-5
Statistics on the number of publications on the topic of “passive air
sampler/sampling” over the past 20 years (Figure 1.1) shows that the application of PASs was
quite limited before the 1990s. Since the 1990s, the applications of PASs have increased rapidly.
Apart from sampling VOCs, the application of PASs was expanded to semivolatile organic
compounds (SVOCs) such as polychlorinated biphenyls (PCBs) in the early 1990s.6-8
Triolein-
containing semi-permeable membrane devices (SPMDs) were used as the passive sampling
medium (PSM), which could assure PCBs with more than four chlorines experience a linear
uptake of the from the air to the PSM for ~a month.9 Later on, SPMDs became widely used for
monitoring SVOCs in air.10-15
Along with SPMDs, other devices have also been developed and used as PSMs in PASs for
SVOCs. These PSMs include polymer-coated fibers,16
glass disks,17
stir bars and silicone
tubing.18
However, these PSMs have not been as widely used as SPMDs in air monitoring
campaign for SVOCs. Despite of the wide use of SPMDs as PASs for SVOCs, there are some
disadvantages associated with SPMDs:19,20
for some SVOCs with relatively high volatility, the
linear uptake stage could be shorter than the PAS deployment time; chemicals accumulated in
SPMDs have to permeate through the polyethylene film into the triolein solvent system and these
processes result in complex uptake kinetics.
In the early 2000s, polyurethane foam (PUF) disk were first employed as PSM for PASs (Figure
1.2a).9 PUF-PASs have longer linear uptake ranges than SPMDs
9 and are relatively easy to

2
deploy, retrieve, and extract. Analytical chemists involved in taking air samples for SVOC
analysis were also familiar with using PUF. PUF-PASs thus overcame some of the disadvantage
of the SPMDs and have been widely used to monitor SVOCs in air since.21-30
At the same time, a
PAS using XAD-2 resin (styrene-divinylbenzene copolymer) as the PSM (Figure 1.2b) had also
been developed and gained wide use.20,31-37
Because the capacity of XAD for SVOCs is
generally higher than that of PUF,38
the XAD-PAS is more suitable for longer deployment
periods and/or for organic chemicals with higher volatilities such as fluorinated telomer alcohols
and volatile methyl siloxanes. Because of the lower capacities of PUF for these chemicals, the
passive sampling rates (PSRs) would decrease over the passive sampling period, which make
PUF-PAS unsuitable for quantifying the air concentrations accurately. Because of this drawback,
PUF disks have recently been impregnated with XAD powder and the resultant sorbent
impregnated PUF (SIP) has also been used as PSM for sampling those volatile compounds.39-42
Although impregnating PUF with XAD can somewhat increase the capacity of the PSM for
SVOCs, the preparation procedure is deemed labor intensive and sometimes XAD can get
dislodged from the PUF during handling exposed samplers, possibly causing some of the
sampled chemicals to get lost.
Figure 1.1 Twenty-year trends of the number of studies published on the topic of “passive air
sampler/sampling”. Data retrieved from the Web of Knowledge Results Analysis Tool.
0
20
40
60
80
100
120
140
160
180
1982
1983
1984
1985
1986
1989
1990
1991
1992
1993
1994
1995
1996
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
2010
2011
Nu
mb
er
of
Pu
blic
aito
ns
Year
Stockholm Convention
becomes effective

3
Figure 1.2 Schematic of (a) the polyurethane foam (PUF) based passive air sampler and (b)
the cylindrical XAD-resin based passive air sampler.
Since 2004 when the Stockholm Convention on persistent organic pollutants (POPs) came into
force,43
applications of PASs to monitor POPs (including POP-like SVOCs) have seen a
dramatic increase (Figure 1.1). The Convention introduced international controls on the
production and uses of POPs. Because the atmosphere is an important medium in the global
cycling of POPs and supplies of POPs to terrestrial and aquatic food webs, reducing emissions of
POPs to the atmosphere is the main focus of international regulations including the Stockholm
Convention. Under the Stockholm Convention, participating countries are required to conduct
source inventories and provide environmental monitoring evidence that the Convention is
effective in reducing the ambient levels of POPs.44
PASs provide a cost-effective approach to
complete the tasks because PASs are relatively inexpensive to produce and operate and they do
not require electricity supply and regular maintenance by skilled workers.20
Because of these
advantages, PASs have been widely used for routinely sampling air for POPs at a number of
locations, especially at remote sites where conditions do not support conventional active air
samplers.
(a)
(b)
2 cm
10
cm
2 cm
20
cm
stagnant air layer
mesoporousXAD pellet
macroporesbetween
XAD pellets
XAD resin filled mesh
cylinder
polyurethane foam (PUF) disk
14 cm
1.4 cm
stagnant air layer
macroporeswithin PUF
cross-linked PUF material

4
Sampling rates (PSRs) of PAS are generally low (< 5 m3/d) which limit them from providing
information on SVOCs in air at a high temporal resolution. To overcome the limitation while
retaining other advantages, in 2006, a flow-through sampler (FTS) was developed to sample
SVOCs from air.45
The FTS consists of a horizontally oriented flow tube, which turns into the
wind with the help of vanes. It relies on the wind to pass air through a plug of polyurethane foam
that serves as the sampling medium. Such a design can increase the sampling rate (> 15 m m3/d)
and has proven useful for monitoring SVOCs in remote areas with a much higher temporal
resolution and/or lower detection limits than PAS.46
As PASs became more and more popular for sampling SVOCs in air, attempts have been made
to modify existing PASs in order to add more functionality. In 2007, Tao et al.47
modified the
existing PUF-PAS design to allow it to sample SVOCs in both gas and particle phases. The idea
of using a modified PUF-PASs to sample both gas and particle phase SVOCs was later adopted
by Abdallah and Harrad to sample PBDEs indoors.48
A PAS that is able to sample both gas and
particle phase SVOCs at sampling rates that can be quantified well would be useful. However,
PSRs for particle phase SVOC can be influenced by many factors and are thus subject to large
variations,47
which limits the applicability of PASs to sample SVOCs in the particle phase. In
2008, Tao et al.49
developed a PAS that positioned a PUF in a flow duct with one-way valves,
which enabled the collection of SVOCs in air masses arriving from specific directions. However,
the wind direction at the scale of the sampler could be affected by many factors, such as the
characteristics of the local terrain and thus not necessarily reflect the wind direction or air mass
origin at the atmospheric scale. This drawback limited the adoption of this PAS in further field
campaigns.
Apart from synthetic sorbents, natural organic phases with relatively high capacity for SVOCs
have also been used as PSM for SOVCs in air. For example, organic film on impervious
surfaces, leaves, bark, lichen, pine needles, soil, snowpack and even butter have been used as
natural PASs to assess SVOCs in air.50-55
Although these natural organic phases can somewhat
reflect the atmospheric concentrations of SVOCs, the chemical uptake capacity and kinetics can
be influenced by many factors,56
which would cause large variations in chemical uptake rates
and thus the derived air concentrations. This disadvantage limits the potential application of
these natural organic phases as PSM.

5
1.2 Applications of Passive Air Samplers for SVOCs
PASs have been applied in diverse studies on SVOCs in air. The applications include monitor
SVOCs in the atmosphere at various scales for long term average air concentrations and to study
emission sources of SVOCs and the processes governing their fate in the environment based on
the spatial distributions.
On the global scale, with the purpose to provide comparable monitoring data on the presence of
POPs and thus to evaluate the effectiveness of the Stockholm Convention, a Global Atmospheric
Passive Sampling (GAPS) network was initialized in 2004 and is still ongoing today.57,58
This
network uses PUF-PASs and XAD-PASs at over 40 sites on the seven continents. Seasonal and
yearly averaged air concentrations of SVOCs at these sites provided by GAPS are useful to
assess the spatial and temporal distributions of legacy and emerging SVOCs globally58-60
and
supply data for the evaluation of global chemical fate models.61
There have been a number of PAS networks/campaigns at the regional scales. Since 1994, a
network using SPMDs has been setup on a latitudinal transect from southern England to northern
Norway with the purpose of studying the sources, long range atmospheric transport,
fractionation, and global clearance processes controlling ambient levels of SVOCs62-64
From
2000-2001, a campaign using XAD-PASs at 40 sites was conducted to discern the large-scale
variability of SVOCs in the North American atmosphere.31,32,65
This was the largest and most
extensive PAS network for SVOCs at that time.31
In this study, regions dominated by
primary/secondary sources were identified with chiral signatures of hexachlorocyclohexane and
DDT. In the summer of 2002, PUF-PASs were deployed at remote/rural/urban locations in 22
countries in Europe. The PAS monitored SVOCs across Europe reflected suspected regional
emission patterns and highlighted localized hotspots.66,67
From 2002-2003, PUF-PASs were
deployed on a seasonal basis to study seasonal and spatial distribution of various POPs in the
Laurentian Great Lakes region.68
The sampling sites overlapped with the Integrated Atmospheric
Deposition Network (IADN), which monitors POPs with high-volume active air samplers
(HVAAS). The comparison of PAS-derived air concentrations with the measurements relying on
HVAAS indicated the feasibility of PAS as an effective tool for monitoring POPs in air. In the
autumn of 2004, the first PAS campaign in Asia deployed PUF-PASs at 77 sites to study the
occurrence and spatial distributions of POPs in four countries69
. In the summer of 2006, PUF-

6
PASs were deployed at 86 European background sites to gain insight in the spatial patterns of
SVOCs in background air across Europe.70
With comparisons to HVAAS derived air
concentrations, it was illustrated that PAS campaigns can serve as useful inter-comparison within
and across existing monitoring networks.
PASs have also been widely used on an urban scale. The occurrence and distributions of SVOCs
in urban areas have been attracting attention because of the high population density and emission
sources in urban areas. In the urban air, some SVOCs such as agrochemicals are mainly used
outside of urban area while other SVOCs such as flame retardants are mainly found in consumer
products, which make urban areas hotspots. PAS provide an easy and effective tool to assess the
spatial distribution of SVOCs along an urban-rural transect. The urban-rural distribution patterns
enable the identification of predominant sources/source regions of SVOCs in urban areas.
Studies deploying PASs along urban-rural transects in Toronto, Canada and Birmingham, UK
revealed different spatial distribution patterns for different SVOCs.21,25,27,71,72
For
polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs), which had been
used widely in building materials and consumer products, higher concentrations were observed
in urban than rural regions.21,27,72
For organochlorinated pesticides such as dieldrin, DDT and
endosulfan, elevated concentrations were observed in rural areas where the chemicals might
enter the air from previously treated agricultural soils or from past uses.25
As people generally spend most of their time indoors, levels of SVOCs present in indoor air are
essential for assessing inhalation and dermal exposure. With the development of PASs for
SVOCs, they have also been widely used in indoor environments. Because of noise free
operating conditions, PASs can be used indoors without disturbing normal activities of the
occupants. So far, PASs have seen been used in various types of indoor or micro-environments,
including cars, homes, offices, classrooms, daycare centres, gymnasiums, etc.30,48,73-75
By
simultaneously sampling indoor and outdoor air, the indoor environment has been identified as a
source of some SVOCs (e.g. PBDEs) associated with consumer products.23,76,77
Based on the spatial distributions of SVOCs in the air, PASs is a useful tool to assess emission
sources, environmental processes and fate of SVOCs. For example, by comparing congener
profiles of PBDEs sampled by PASs in different indoor locations with congener profiles of
technical PBDE mixtures and potential emission sources, sources of PBDEs in each indoor

7
location were identified and related to different consumer products.30
By deploying PASs at
different sections of a waste water treatment plant and a land fill, the emission rates of volatile
methyl siloxanes (VMSs) and polyfluoroalkyl compounds from the sites were estimated.78,79
Using PAS at different heights above the soil surface, the air-soil exchange flux of gaseous
SVOCs was quantified based on the vertical concentration profiles.80
By analyzing air
concentrations of SVOCs using PASs along elevation transects in mountains and comparing air
with soil concentrations, the mechanism of cold trapping for SVOCs in mountain regions was
evaluated.36,37,81,82
1.3 Mechanism and Theory of Passive Air Sampling
Passive air sampling is based on the sorption of target chemical from ambient air to the PSM.
When a PAS is deployed, the target chemicals have higher fugacities in the air than in the PSM,
and the chemicals diffuse from the ambient air to the PSM until equilibrium is reached (i.e. the
fugacity in the PSM equals that in the ambient air). In order to derive ambient air concentrations,
two possible approaches can be applied. The first is the equilibrium approach, which derives the
air concentrations based on the amounts (concentrations) of chemicals accumulated in the PSM
at equilibrium and the equilibrium partition coefficients between PSM and air. In order to use
this approach, equilibrium between PSM and air must be ensured. However, equilibrium time for
chemicals of different physicochemical properties are different and the amounts of chemicals
accumulated in the PSM at equilibrium varies highly with fluctuations in temperatures and
ambient air concentrations. Depending on the time required for a chemical to reach equilibrium
between the PSM and air, the air concentrations derived using this approach may only be
representative of a short period prior to the retrieval of the PSM. These disadvantages of the
equilibrium approach limit the applications of equilibrium PAS to monitor air concentrations.
The second approach to determine ambient air concentrations from the amount of chemicals
sorbed to the PSM is based on the uptake kinetics and is normally adopted in passive air
sampling for SVOCs. At the initial uptake stage (referred as the linear uptake range) when the
PSM is far from equilibrium, the passive air sampling rate (PSR, m3/d) is approximately
constant. The linear uptake range is operationally defined as the time until the PSM has
accumulated 25% of the equilibrium amount.9,38
Normalizing the chemical uptake rate with the
corresponding air concentration gives the passive sampling rate. In theory, PSRs depend on the

8
overall mass transfer coefficients of the chemicals from ambient air to the PSM and the
interfacial transfer area between the PSM and surrounding air. Prior to the work described in this
thesis, the overall chemical mass transfer involved in passive air sampling was viewed as a three-
step process:83
mass transfer from ambient air to the interior of the passive sampler housing,
diffusions from air inside the sampler housing to the PSM-air interface and from the interface
into the PSM phase. To describe the processes mathematically, the Whitman two-film theory84
was adopted and a mass balance equation for the target chemicals in the PSM can be constructed
as:
( / )
( / )
S S
S
O S A S SA
O S A S S SA
O S A
dm dCV
dt dt
k A C C K
k A C m V K
k A C
(Equation 1.1)
where mS is the amount of a chemical accumulated in the PSM; t is PAS deployment time; CA is
air concentration of the chemical; CS is the concentration in the bulk PSM when assuming
uniform chemical distribution within the PSM; KSA is the PSM-air equilibrium partition
coefficient; VS and AS are the volume and surface area of the bulk PSM; kO is the overall mass
transfer coefficient, which can be derived from the coefficients of mass transfer from ambient air
to the interior of the passive sampler housing (kA, H), diffusions across the air side interfacial
boundary layer (kA, BL) and diffusion through interfacial boundary layer on the PSM side (kS):83
, ,
1 1 1 1
O A H A BL S SAk k k k K
(Equation 1.2)
During the initial uptake stage, when the chemical concentrations in the PSM are low, and KSA
values for SVOCs are normally large, so that the term CS/KSA, representing re-evaporation of
chemicals from the surface of PSM to the air, can be eliminated in Equation 1.1. The mass
balance equation (Equation 1.1) becomes:
S S
S O S A
dm dCV k A C
dt dt (Equation 1.3)
kO AS here is the defined PSR:

9
O SPSR k A (Equation 1.4)
According to the PAS theory9,83
based on the two film model,84
transfer from ambient air to the
interior of the PAS housing and diffusion within the PSM do not kinetically limit the overall
mass transfer coefficient. So that
/O S A S S A BL
PSR k A k A A D (Equation 1.5)
where DA is the chemical’s molecular diffusivity in air and δBL is the thickness of the stagnant air
boundary layer. According to the Fuller, Schettler and Giddings (FSG) equation for air-phase
diffusivity (where T is absolute temperature in K, MA and VA are the molecular mass and
diffusion volume of air, M and V are the molecular mass and diffusion volume of the target
chemical, P is the atmospheric pressure in atm): 85
3 1.75 0.5
1/ 3 1/ 3 2
10 (1 / 1 / )
( )
A
A
A
T M MD
P V V
(Equation 1.6)
DA is not particularly sensitive to variations in temperatures or the molecular size of the
chemicals. Therefore, little variation of PSRs was expected among different chemicals and at
different temperatures. This would be ideal for passive air sampling, as a single PSR would
suffice to derive air concentrations for all chemicals at different temperatures.
Based on Equation 1.5, PSRs can be calculated from AS, DA, and δBL. Although the former two
parameters can be determined easily, it is difficult to get δBL. As such, it is not a common
practice to determine PSR using a theoretical approach.86
Instead, empirical PSRs are mainly
determined from calibrations against active air samplers (AASs).20,47,87,88
In PAS calibrations, the
amount of a chemical accumulated in the PSM at different time points are divided by an AAS
derived air concentration to get an equilibrium sampling volume (VEq). When plotting VEq against
the sampling time and applying a linear least squared fit, the slope of the fitted line is the PSR.
Because the wind conditions at the sampling sites can be different from that at the site where a
PAS had been calibrated, PSRs derived from the loss of performance reference compounds
(PRCs) or depuration compounds (DCs) added to the PSM prior to deployment of the PASs in
the field22
are gaining in popularity. This approach to obtaining the PSRs of the target chemicals

10
somewhat accounts for the effect of wind at different sampling sites. Chemicals used as PRCs
cannot be present in the sampled air, i.e. CA = 0. Applying Equation 1.1 to the DCs we obtain:
,
, , , , ,/ / ( )
S DC
O DC S S DC SA DC S DC S SA DC
dmk A C K PSR m V K
dt (Equation 1.7)
By assuming that the overall mass transfer coefficients for the target chemicals sampled by PASs
from air and for the loss of DCs from the PSM to the air are only kinetically limited by diffusion
through the stagnant air boundary layer surrounding the bulk PSM, we obtain:
,
, ,
A DC A
O DC A DC A O
BL BL
D Dk k k k
(Equation 1.8)
So Equation 1.7 becomes
,
, ,/ ( )
S DC
S DC S SA DC
dmPSR m V K
dt (Equation 1.9)
Solving Equation 1.9 makes it possible to calculate a PSR based on the amount of a DC initially
spiked to the PSM (mS,DC.(0)), the length of time the PAS was deployed in the field (t), the
amount of DC left at the end of deployment (mS,DC.(t)), the volume of the PSM (VS) and the
partition coefficient of the DC between the PSM and air (KSA, DC) at the average temperature of
the sampling period:
, ,
,
ln[ (0) / ( )]S DC S DC
S SA DC
m m tPSR V K
t (Equation 1.10)
The loss rates of DCs from the PSM are related to wind speed. This approach using DCs to
derive PSRs relies on the PAS theory and the associated assumptions stated above.
Although PAS theory based on the two-film model has been widely applied to describe the
kinetics of passive air sampling for SVOCs, this theory has not been able to explain some field
observations. According to this theory, temperature and molecular properties only affect the
uptake kinetics via the molecular diffusivity in air (DA). However, this cannot explain the large
variations of PSRs among different chemicals or at different temperatures observed in field
calibrations of PASs for SVOCS.28,87,89,90

11
Not only do the PSRs observed in the fields vary more than can be explained by the current
theory, but also a key assumption of the two film model raises questions. The two-film model
was originally developed by Lewis and Whitman84
to describe mass transfer between air and
water. The two-film model requires that “in the main body of either liquid or gas […] the
concentration of solute in the fluid is essentially uniform at all points”. Besides, in Equation 1.1,
the chemical concentration within the PSM is also assumed to be uniform. Nevertheless, when
the two-film was applied to PASs, this assumption has been ignored and its validity not tested. If
this assumption is not fulfilled, it would not be appropriate to conclude that the kinetic resistance
on the PSM side can be neglected based on Equation 1.1 derived from the two-film model.
1.4 Factors Influencing Passive Air Sampling Rates
As PASs have been applied widely to monitor SVOCs in air, factors potentially influencing
PSRs have also been identified and studied. One influential factor is wind, which could affect the
thickness of the stagnant air boundary layer and therefore the mass transfer coefficient. Although
the housing of PASs can dampen the effect of wind on the thickness of the stagnant air boundary
layer surrounding the PSM and thus on the chemical uptake kinetics, PSRs have been found to
be dependent on wind speed.90-94
Studies suggested at wind speeds over 1 m/s (3.6 km/h) the
PSRs of PUF-PAS increases exponentially with wind speed. This factor could cause variations in
the calibrated PSRs of the PUF-disk PASs by as much as an order of magnitude.87,92,95
The
influence of wind on PSRs appears to be associated with the design of the PAS housing.
Comparing chemical uptake by the double-bowl PUF-PASs and the cylindrical XAD-PASs
deployed side-by-side at over 30 sites of the Global Atmosperic Passive Sampling network,57,58
the XAD-PASs appeared less influenced by wind.96
A wind-tunnel study suggested little wind
effect on the water uptake by silica-gel filled mesh cylinders at wind speed of 5-15 m/s,20
but
field deployments of XAD-PAS noted higher PSRs at sites exposed to strong winds.36,37
Besides wind speed, wind direction towards the PAS housing can also influence the PSRs by
varying the wind speed within the PAS housing relative to the ambient wind speed.97,98
The
direction at which the wind is blowing at a PAS may be affected by the local terrain of the
deployment site. For example, PASs deployed along a slope may have valley to mountain winds
preferentially blowing at an angle towards it.34
The effect of wind directions on the PUF-PAS
was studied by measuring the rate of water evaporation from a PUF disk in a wind channel.

12
Results suggested wind of the same speed blowing at different directions towards the double-
bowl PAS could vary the water evaporation rate by as much as 40%. Based on this result, a
similar effect may be infered for the PSRs of SVOCs. However, no study based on uptake of
SVOCs by PAS under different angles of wind incidence has so far been conducted.
Because the PAS housing affects the air movement or wind conditions around the PSM, PSRs
can also depend on the PAS configurations (or designs). In a previous indoor calibration study
using a PAS with the PUF disk positioned in a housing that was more confined than the typically
used double-bowl PAS,87
Tao et al.99
observed a lower PSR (and a lower surface area normalized
PSR). In a study using a modified double-bowl PUF-PAS, in which PUF was moved further
from the opening of the housing compared to the original PUF-PAS design, 28
Abdallah and
Harrad48
noted a decreased rate of chemical uptake by PUF. The influence of sampler
configuration on PSR can be rationalized in two ways. Different configurations could result in a
different thickness of the stagnant boundary layer surrounding the PSM48
or they could affect the
mass transfer of chemicals from ambient air into the PAS housing. Once this process becomes
slower than the rate of chemical uptake by the PSM, the chemical concentration within the
sampler housing would be lower than the ambient air concentration and cause a so-called
starvation effect,100
which could also affect the PSR. Although for a given type of PAS, the
configuration is fixed and will not cause variations among PASs of the same design,
understanding the influence of sampler configurations on the PSRs would provide useful
information to optimize the design of PASs.
Depending on wind conditions and PAS configuration, particles in ambient air could possibly
enter the PAS and be trapped by the PSM. A number of studies using the double-bowl PAS
indicated the presence of particles on the PUF.28,48,74,87,90,98
If the fraction of particles in the air
been trapped by the PSM would be consistent under different environmental conditions, it would
be feasible to quantify SVOC in both gas and particle phase in the air. However, different studies
do not agree on the fraction of ambient particles that is sampled by the PUF-PAS and thus on the
influence of particle trapping on PSRs74,90,98
This is likely because of the strong influence of
wind conditions on particle sampling rates. Therefore, unless the fraction of particles sampled by
a PAS can be well controlled and quantified, PASs should mainly target SVOCs in the gas phase
and avoid the uncertainty introduced by unpredictable particle trapping. SVOCs largely bound to

13
particles have not been detected in the PSM of the cylindrical XAD-PAS, suggesting that this
sampler configuration may be superior to the double-bowl PAS housing design.35,101,102
According to the current PAS theory,83,103
PSRs should vary little with temperature and among
different SVOCs. However, field studies indicate that the variation of PSRs with temperatures
and SVOC species is much larger than the theory predicts. PSRs of the double-bowl PUF-PAS
for gas-phase-associated SVOCs decreased with temperatures while those for particle-associated
SVOCs increased with temperatures.87,90
Yet, in another study also using PUF as the PSM,
higher PSRs of gas-phase PAHs were found at lower temperatures.49
The variation of PSRs for
different SVOC species is also larger than can be explained by PAS theory. In calibrations of
PUF-PAS for PCBs, the PSR for hepta-CBs could be six times higher than that of tri-CBs.87
Such trends of increasing PSRs with increasing molecular weight of the sampled chemicals have
been observed by many studies using the PUF-PASs.28,88,104,105
Large variations of PSRs with
temperature and between SVOC species have also been observed for the XAD-PAS. However,
the observed trend is different from that in most studies on the PUF-PAS. Lower PSRs for the
XAD-PAS were observed at low temperatures and chemicals with lower molecular weight were
found with higher PSRs.20,89,106
Based on these field observations on the variation of PSRs, it is
inferred that factors other than those included in the current PAS theory may greatly influence
PSRs.
Figure 1.3 Structure of this thesis and task involved/skills developed from the studies.
Passive Air
Sampling
Mechanism
Influence
Precision Balance
Passive Sampler housing
Silica gel Packed Mesh
Cylinder
Ch
apte
r 4
1
2 3
log KSA
log
(k s
orb
/ d–
1)
6 7 8 9 10
9
8
7
6
5
4
KSADA
ksorb
DPA
Ch
apte
r3
kPSMkAir
DE
Passive Sampling Media (PSM)
Ch
apte
r2C
hap
ter5
Ch
apte
r6
Application
Model Development
Soxhlet Extraction
Numerical Computation
GC/MS(/MS) Analysis
Field Sampling
Data Analysis
Trajectory Analysis
GC/MS(/MS) Method Development
Accelerated Solvent Extraction
Experiment Design

14
1.5 Objective and Structure of the Thesis
Starting from the current understanding and knowledge gaps of PASs, a combination of
controlled laboratory experiments, mathematical modeling and field work was applied to gain
further insight into the mechanism and processes involved in passive air sampling of SVOCs, to
investigate the factors influencing PSRs, and to understand the fate and behavior of SVOCs in
the environment using PASs in the field (Figure 1.3). These objectives were addressed in the
following five chapters of the thesis:
Chapter 2 describes a kinetic uptake experiment using cylindrical PSMs that had been
concentrically segmented into concentric layers to test whether SVOCs distribute uniformly
along the radial direction of the PSMs and whether a kinetic resistance to chemical transfer
within the PSMs exists. Both XAD and PUF were positioned in the same type of sampler
housing to eliminate the variation caused by the different housing designs.
Chapter 3 is based on the results of Chapter 2, which suggested chemical transfer within the
PSM is not properly described by current PAS theory. This chapter describes the development
and applications of a new PAS model. This model relies on the fundamental laws of mass
transfer in the gas phase and in porous media and of chemical exchange between air and
sorbents. This model is independent of the assumption of uniform chemical distribution within
the PSM. The model was used to illustrate the kinetic resistance within the PSM and to explain
the field observation of the dependence of PSRs on temperatures and SVOC species as well as
the two-stage uptake processes observed in some studies, which cannot be explained by current
PAS theory.
Chapter 4 focuses on the influence of sampler configurations on chemical uptake by the
cylindrical PAS. In this study, PSRs of various PAS configurations were tested using a
gravimetrical approach developed to study the kinetics of water vapor uptake from indoor air by
silica gel placed inside cylindrical PAS as a surrogate of SVOC uptake by the XAD-PASs.
Chapter 5 assesses the effect of wind on the uptake in cylindrical XAD-PASs. The distributions
of the sampled chemicals along the axial direction of the XAD-filled mesh cylinders were

15
studied in PASs deployed under quasi wind still and lab generated windy conditions indoors as
well as under normal outdoor conditions. The kinetics of chemical uptake by the PASs was
investigated under indoor quasi wind still condition and with lab generated wind blowing at
straight and 45° slanted angles towards the PASs. Computational fluid dynamic simulations were
also conducted to investigate wind patterns within the PAS housings under the two different
windy conditions.
Chapter 6 describes a field study using XAD-PASs and FTSs on the Big Island of Hawaii with
the purpose to test the potential starvation effect of PASs in the field, to explore the vertical
distribution of SVOCs along an altitudinal transect, and to assess global SVOC background
concentrations over the Central Northern Pacific.
Chapter 7 integrates the study presented in Chapter 2 to Chapter 6 and gives the conclusions of
the thesis and identifies further research needs in order to gain further insight on the mechanism
and influential factors of passive air sampling.

16
Chapter 2. Sampling Medium Side Resistance to Uptake of Semi-volatile
Organic Compounds in Passive Air Samplers
Xianming Zhang, Masahiro Tsurukawa, Takeshi Nakano, Ying D. Lei, Frank Wania
Environmental Science & Technology 2011, 45, 10509-10515.
Contributions: X. Zhang designed the experiment under the guidance of F. Wania and Y.D. Lei.
Y.D. Lei assisted in designing the layered mesh cylinders. X. Zhang deployed the samplers and
extracted the samples. M. Tsurukawa and T. Nakano offered the opportunity to use a high-
resolution GC-MS to analyze the samples and provided assistance in analyzing the samples. X.
Zhang processed the chromatograms and interpreted the data. Under the guidance of F. Wania,
X. Zhang wrote the manuscript, revised it and responded to reviewers’ comments.
Reproduced with permission from Environmental Science and Technology
Copyright 2011 American Chemical Society
kPSMkAir
DE
Passive Sampling Media (PSM)

17
2.1 Abstract
Current theory of the uptake of semi-volatile organic compounds in passive air samplers (PAS)
assumes uniform chemical distribution and no kinetic resistance within the passive sampling
media (PSM) such as polystyrene-divinylbenzene resin (XAD) and polyurethane foam (PUF).
However, these assumptions have not been tested experimentally and are challenged by some
recently reported observations. In order to test the assumptions, we performed kinetic uptake
experiments indoors using cylindrical PSM that had been concentrically segmented into three
layers. Both, XAD and PUF, were positioned in the same type of sampler housing to eliminate
the variation caused by the different housing designs, which enabled us to quantify differences in
uptake caused by the properties of the PSM. Duplicated XAD (PUF) samples were retrieved
after being deployed for 0, 1 (0.5), 2 (1), 4 (2), 8 (4), 12 (8) and 24 (12) weeks. Upon retrieval,
the PSM layers were separated and analyzed individually for PCBs. Passive sampling rates (R)
were lower for heavier PCB homologs. Within a homolog group, R for XAD was higher than
that for PUF, from which we infer that the design of the “cylindrical can” housing typically used
for XAD PAS lowers the R compared to the “double bowl” shelter commonly used for PUF-disk
PAS. Outer layers of the PSM sequestered much higher levels of PCBs than inner layers,
indicative of a kinetic resistance to chemical transfer within the PSM. The effective diffusivities
for chemical transfer within PSM were derived and were found negatively correlated with the
partition coefficients between the PSM and air. Based on the results, we conclude that the PSM-
side kinetic resistance should be considered when investigating factors influencing R and when
deriving R based on the loss of depuration compounds.
2.2 Introduction
Dynamic-uptake based passive air samplers (PAS) such as those based on polystyrene
divinylbenzene (XAD)20
and polyurethane foam (PUF)9 are increasingly used to study persistent
semi-volatile organic compounds (SVOCs) in the atmosphere. Such PAS are capable of time-
integrated sampling with relatively low cost and simple operation, which is independent from
power supply and free of noise.9,20,83
Because of these advantages over the traditional high
volume air sampler PAS are widely applied to understand spatial and long term temporal trends,
identify sources, and assess human exposure to SVOCs in various types of environment.30,58,74

18
The mechanism of uptake in PASs is based on the molecular diffusion from air to passive
sampling medium (PSM). Conceptually, the process of SVOC uptake in PAS has been described
using the two-film diffusion model,9,84
which was originally proposed to describe mass transfer
across gas-liquid interfaces.84
According to the two-film model, “in the main body of either
liquid and gas, […] the concentration of solute in the fluid is essentially uniform at all points”.84
As indicated by the current “theory”,9,83
the kinetic resistance within the PSM is inversely related
to a chemical’s PSM/air partition coefficient and thus negligible for SVOCs due to their large
PSM/air partition coefficients. Therefore, the resistance posed by the air boundary layer is
regarded as controlling the rate of SVOC uptake in PAS. During the initial uptake stage
(operationally defined as the linear uptake range), chemical concentrations on the PSM are so
low that surface evaporation is neglible. As such, chemical uptake in PAS can be quantified with
a simple linear equation involving a sampling rate (R, m3/d) that only depends on the surface
area of the PSM, the chemical’s molecular diffusivity in air (DA) and the boundary layer
thickness.83
Because the boundary layer thickness is difficult to quantify directly, in practice, R is
usually determined by calibrations against air concentrations determined using active samplers.
A number of PAS calibration studies have determined R for both XAD-PAS and PUF-PAS under
different environmental conditions.20,28,87,89,106,107
Based on these studies, XAD-PAS have a
higher sampling capacity or longer linear uptake range than PUF-PAS.38
The high capacity
makes XAD-PAS superior for integrated sampling over long time periods, especially for
relatively more volatile compounds such as the fluorotelomer alcohols.108
However, XAD-PAS
generally have a two- to five-fold lower R than PUF-PAS. So far, it is unclear whether the
different R is caused by differences in the properties of XAD and PUF or by differences between
the housing configurations typically employed with the two PAS.
R for both of the PAS varies among different chemicals or at different temperatures.28,87,89,90
Such variations are larger than can be explained by the dependence of DA on chemical properties
or temperature (Fuller-Schettler-Giddings equation),109
indicating some other influential factors
may exist. For the PUF-PAS, some studies observed higher R for chemicals with low
volatility,87,104
an observation attributed to the binding of such chemicals to particles, which are
trapped by the PUF. Conflicting results showing lower R for particle-bound chemicals have also
been found.110
Previous studies on the temperature dependence of the R for PUF-PAS also
yielded inconsistent results. Increased R for some particle-bound PAHs was observed as

19
temperature increases, which was explained with a shifting from particle to gas phase at higher
temperatures.87,110
However, a negative correlation was found for BDE-99, which is also likely to
undergo gas-particle phase exchange.104
Calibrations for selected pesticides conducted at
different latitudes yielded R for XAD-PAS that are higher at higher temperatures.20,89
However,
this cannot be due to shifts in the atmospheric phase distribution because the gas-particle
exchange behavior of these pesticides is not sensitive to temperature in the environmental
temperature range. Here, we hypothesize that SVOCs distribute non-uniformly within the PSM
and the PSM-side kinetic resistance could also affect R. This resistance might help explain the
variation of R between chemicals and with temperatures. In order to explain the variation of R
with sampling time, Chaemfa et al.107
postulated a two-phase uptake processes: chemicals first
sorb to the surface of PUF and then penetrate into the PUF at a slower rate. This is essentially
similar to our PSM-side kinetic resistance hypothesis. However, no further investigation has
sought to confirm this hypothesis that challenges the current PAS uptake theory.
In this study, we aim to (1) investigate whether PSM or housing differences cause the different
sampling rate between XAD-PAS and PUF-PAS, (2) test our hypothesis on chemical distribution
and kinetic resistance within PSM, and (3) quantify the effective diffusivity of chemical transfer
within the PSM. To achieve these objectives, we performed a kinetic uptake experiment using
concentrically segmented XAD and PUF positioned in the same type of sampler housing.
2.3 Materials and Methods
2.3.1 Passive Sampling Media.
XAD packed in mesh cylinders and PUF were selected for this study because they are the most
widely used passive sampling media (PSM) for SVOCs in air. Instead of the PUF disk
commonly used in the “double-bowl” type PAS,9 a cylindrical PUF plug (8 cm diameter, 8 cm
high) was made from 1-cm-thick PUF sheets (Pacwill Environmental, density ~0.02 g/cm3) and
placed in the “cylindrical can” housing commonly used with XAD-PAS (Figure 2.1) to eliminate
the influence of sampler housing design when comparing the uptake characteristics of the two
PSM. The XAD-filled mesh cylinder and cylindrical PUF were concentrically segmented into
three layers (outer, mid, and inner). The PSM layers can be separated upon sample retrieval.
Detailed dimensions of the PSM are given in Figure 2.1. Before sampling, the segmented PUF
components were sequentially cleaned with soap water, deionized water and Soxhlet-extracted

20
with acetone for 24 h and with petroleum ether for another 24 h. The XAD-2 resin was
purchased pre-cleaned (Sigma-Aldrich).
Figure 2.1 Design of the layered passive air sampling media (XAD and PUF) used to study
the distribution of PCBs within the passive sampling medium.
2.3.2 Chemicals.
Polychlorinated biphenyls (PCBs) were selected as the target chemicals for this study because
the PCB congeners cover a wide range of partitioning properties (e.g. PSM/air partition
coefficient), which also partially overlaps with other SVOCs of environmental interest such as
organochlorine pesticides, polycyclic aromatic compounds and brominated flame retardants.
PSM (XAD and PUF)/air partition coefficients for individual PCB congeners, estimated using
poly-parameter linear free energy relationships38,111
and recently updated PCB solute
descriptors,111
were compiled in Table S2.1 and S2.2 in the supporting information (SI) and were
used for further data analysis.
2.3.3 Sampling Design.
Before deployment, the three layers of PSM were spiked with three different groups of
depuration compounds (DCs) comprised of 13
C-labeled PCB congeners or non-labeled PCB
8cm
8cm21cm 8cm
15cm 6cm
17cm 4cm
2cm
1cm
XAD
resin
PUF

21
congeners that are not present in ambient air. Different groups of DCs were applied to different
PSM layers. Detailed information on DCs and spiking procedure is provided in the SI. An
unoccupied office previously identified as being contaminated with PCBs was selected as the
sampling site. Duplicated XAD (PUF) samples were retrieved after been deployed for 0, 1 (0.5),
2 (1), 4 (2), 8 (4), 12 (8) and 24 (12) weeks. Deployment lengths for PUF-PAS were shorter,
because we had anticipated faster uptake than for XAD-PAS. Upon retrieval, the layered PSM
were separated, individually sealed in pre-cleaned aluminum foil and Ziploc bags, and stored at -
20 °C before extraction within two weeks. Along with the PAS, a low-volume active sampler
(BGI Inc., 2.9 ± 0.2 m3/d) with a PUF-XAD-PUF sandwich (5 g of XAD between two 2 cm i.d.
× 3 cm PUF plugs) as the sampling medium was used to measure the PCB air concentrations
with monthly resolution. The sampling scheme is illustrated in Figure S2.1.
2.3.4 Sample Extraction and Analysis.
Each sample was Soxhlet extracted for 24 h in ~500 ml petroleum ether (PUF) or 1:1
acetone:hexane (XAD and PUF-XAD-PUF sandwiches). The extract was roto-evaporated to ~2
mL and eluted through a disposable pasteur pipet packed with dehydrated sodium sulphate to
remove moisture. The eluent was blown down with high purity N2, solvent exchanged to iso-
octane and reduced to ~0.5 mL in a GC vial, to which 100 ng mirex was added for volume
correction and as internal standard for PCB quantification.
PCBs in the samples were analyzed with an Agilent 5890 gas chromatograph (GC) coupled with
a JMS-800 double focusing high resolution mass spectrometer (HRMS, resolution ≥ 60 000).
The detailed method for instrumental analysis is described by Matsumura et al..112
Briefly, 1.0
μL of the sample was injected in splitless mode with the injector temperature at 280 °C. PCBs in
the sample were separated using an HT8-PCB column (0.25 mm i.d. × 60 m, SGE Analytical
Science) with helium (1 mL/min) as the carrier gas. The GC oven was programmed from 120 to
180 °C at 20 °C/min, to 260 °C at 2 °C/min, to 300 °C at 5 °C/min, and then held isothermal for
4 min. The HRMS was operated under EI and SIM mode with the interface and chamber
operated at 260 °C.

22
2.3.5 QA/QC.
All samples were duplicated to quantify reproducibility. Data analysis for all samples was based
on both duplicates except for the XAD 12-month inner layer, of which one duplicate was lost
during sample preparation. The relative difference between the passive sampling rates derived
from duplicated samples was generally less than 10% (Figure S2.2). Duplicated field blanks for
both XAD- and PUF-PAS were treated as time zero values in the analysis of chemical uptake
kinetics. Prior to extraction, each sample was spiked with 100 μL 250 pg/μL 13
C12-labeled PCB-
77, -101, -141 and -178 (Cambridge Isotope) as surrogate standards. Recoveries of the four
surrogate standards ranged between 74 and 131% with an interquatile range <15% (Figure S2.3).
2.3.6 Derivation of passive air sampling rates.
Passive air sampling rates (R, m3/d) and the PSM-side effective diffusivities (DE, m
2/h) were
obtained by linear least squares fitting (LLSF) to all duplicated data points. For data below the
LOD, random numbers between 0 and LOD were assigned.113,114
The method of using LLSF to
derive R has been applied in other studies.9,107
Briefly, R equals the slope of the linear least
square fitted line of the equivalent sampling volume (Veq) over the PAS deployment time; Veq is
calculated as the amount of a chemical sequestered in the PSM (sum of the three layers) divided
by the ambient air concentration measured using the active air sampler.
2.3.7 Derivation of the effective diffusivities on the PSM side.
To derive DE on the PSM side, a two-layered PSM mass balance model was developed (Figure
S2.4, Equation S2.6-Equation S2.14 and the relevant text in SI). The outer layer in the
abovementioned experiments is referred to as Layer 1; since few PCB congeners were detected
in the inner layer, the inner and mid layers in the experiment were combined and are referred to
as Layer 2 hereafter. Starting from the chemical mass balance equations (Equation S2.6 and
Equation S2.7) for the two layers, a relationship between the amounts of chemical sequestered in
Layers 1 and 2 was derived:
2
2 12 1 1 2
1
1( ) [ (0) ( )] (0)
2
PSM
Am t k m m t t m
V
(Equation 2.1)
where m1(t) and m2(t) [dimension: M] are the amount of the chemical sequestered in Layer 1 and
2 at time t [T]; kPSM12 [LT-1
] is the mass transfer coefficient for chemical diffusion between the

23
two layers of the PSM and kPSM12 = DE, PSM / δ, where DE, PSM [L2T
-1] is the effective diffusivity
of the chemical in the PSM and δ [L] is the diffusion length; A2 [L2] is the area between Layer 1
and 2; V1 [L3] is the Layer 1 volume of the PSM. Let Xt = [m1(0) + m1(t)]·t, Yt = m2(t), and apply
LLSF to Xt and Yt, the slope of the fitted line equals to kPSM12 A2 / (2V1), from which kPSM12 can be
determined. Further, if δ is known, DE, PSM can be determined.
2.3.8 Mechanistic model of effective diffusivity in porous media.
A previously developed modeling approach for the effective diffusivity of chemicals in porous
media, such as soil and sediment, which considers sorption and tortuosity109,115
was applied to fit
the effective diffusivity in PUF (DE, PUF ) derived in this study:
A
E ,PUF A A A
SA PUF/A SA PUF/A
1
1
DfD f D f D
r K r K
(Equation 2.2)
where DE, PUF [L2T
-1] is the effective diffusivity in PUF, DA [L
2T
-1] is the molecular diffusivity in
bulk air, ΦA [dimensionless] is the fraction of the chemical in the air-filled PUF pore space, f
[dimensionless] is a correction factor related to intra-aggregate porosity and tortuosity,115
rSA
[L3
(PUF)L-3
(A)] is the volume ratio between the solid PUF material and the porous air space in
PUF, and KPUF/A [L3
(A)L-3
(PUF)] is the chemical partition coefficient between PUF and air. The
ratio f/rSA is a property of the porous medium that decreases with increasing density and
tortuosity of the PUF.
2.4 Results and Discussion
2.4.1 Passive Air Sampling Rates.
To compare the performance of XAD and PUF, we studied the PCB uptake kinetics on the two
PSM placed in housings of the same design (Figure 2.1). The median of the R for the PCB
congeners in each homolog group ranged 0.12-0.23 m3/d and 0.08-0.16 m
3/d for an XAD and
PUF-based PAS, respectively (Figure 2.2). R derived for the individual PCB congeners are
reported in Table S2.3. Because the configurations of the PSM used in this study were different
from those used previously, it is not feasible to directly compare R with those reported in other
studies. Therefore, R (m3/d) was normalized to the PSM surface area (dm
2) and the normalized
sampling rate (SR, m3/d/dm
2) was used for comparison (Table S2.4). XAD-based SR ranged
0.11-0.32 m3/d/dm
2, which is approximately 5 to 10-fold lower than SR from previous outdoor

24
calibrations for XAD-PAS.20,89,106
This is in agreement with previous studies on the PUF-PAS,
which indicate that outdoor SR can be as much as ~50-fold higher than indoor SR.28,87
The lower
SR observed indoors by this and other studies can be attributed to the different extent of air
movement indoors and outdoors. Relatively wind-still indoor conditions tend to increase the
thickness of the air boundary layer surrounding the PSM and reduce R. The low air movement
indoors could also increase the resistance to chemical transfer from ambient air into the PAS
housing, which could possibly cause a “starvation” effect100
and make the air concentration
within the housing lower than the ambient air. However, such an effect would exist and lower the
passive sampling rate only if the resistance for a chemical to diffuse into the housing from
ambient air is higher than that for chemical uptake by the PSM. Because it is difficult to measure
the actual air concentration of SVOC within the PAS housing without disturbing its normal
operational conditions, such a “starvation” effect on PAS for SVOC has so far not been
confirmed experimentally.
Figure 2.2 Comparison of the passive air sampling rates of PCB homologs between the
passive sampling media of XAD and PUF positioned in the same type of cylindrical sampling
housing.
PUF-based SR of this study ranged 0.02-0.07 m3/d/dm
2, which is ~5- and ~30-fold lower than the
calibrated indoor SR by Hazrati and Harrad28
and Shoeib and Harner9. Apart from inter-study
variations (~5 times difference for the same type of PAS between ref.9 and
28), different sampler
configurations could provide a possible explanation for the lower PUF-based SR observed here.
median25%ile
75%ile
non-outlier min
non-outlier max
XAD
PUF
1Cl 2Cl 4Cl 5Cl 6Cl 7Cl3Cl
0.05
0.15
0.20
0.30
0.25
0.10Sam
plin
g R
ate
R (
m3/d
)
PCB Homolog Groups

25
In this study, a PUF-cylinder was positioned in a “cylindrical can” rather than the more
commonly used arrangement of a disk in a “double bowl” housing.9,28,87,107
This different
configuration could increase the thickness of stagnant air around the PUF, increase the kinetic
resistance for a chemical to diffuse into the housing from ambient air, and thus lower the SR.
Evidence of the effect of sampler configuration on passive sampling rates can also be found in
studies by Tao et al.47,116
: PUF-disks positioned in a more confined housing had ~10-fold lower
SR than PUF disks in a “double bowl” shelter. Such evidence of the effect of sampler
configuration on passive sampling rate indicates that the housing design may also contribute to
the kinetic resistance to chemical uptake.
The homolog-specific R decreases from the lighter to the heavier PCBs for both PSM (Figure
2.2). This is in contrast with previous studies on the PUF-PAS, which found higher R for heavier
congeners.28,87,107
A higher fraction of heavier congeners is particle-bound in air. The higher R
for heavier congeners was attributed to particles being captured by the PUF-disk.28,87,107
Unlike
the PUF-PAS, in which particle-bound chemicals were often detected,98,117
the XAD-PAS is
unlikely to trap atmospheric particles since few particle-bound chemicals have ever been
detected in XAD-filled mesh cylinders positioned in a cylindrical housing.58
The semi-enclosed
configuration of the “cylindrical can” shelter greatly limits advective air flow into the housing
and thus few particles may enter the housing and get trapped on the PSM. Excluding the effect of
particle-bound chemicals, chemical sequestration on the PSM is mainly determined by chemical
transport from the ambient air to the PSM via diffusion in the gas phase. This is supported by the
positive correlations between the homolog-specific passive sampling rate and the gaseous
molecular diffusivity of the chemicals (Figure S2.5).
2.4.2 Evidence of kinetic resistance on chemical transfer within PSM.
The kinetics of PSM-side mass transfer of the DCs and PCBs from air was investigated by
analyzing the amount sequestered in each layer after different deployment times.
2.4.2.1 PCB Uptake from Air.
Higher PCB levels were found in the outer layer than in the middle and inner layer over the
whole sampling period (Figure 2.3 and Table S2.5 and S2.6). Within the first month of PAS
deployment, the PCBs were either not detected in the middle or inner layers of PUF or at levels

26
no different from the blanks. Mono-CBs could be detected in the middle and inner PUF layer
after 4 and 8 weeks of deployment, respectively. Nevertheless, even after 12 weeks of
deployment, the amount of mono-CBs in the middle and inner PUF layers was only ~20% and
~5% of that in the outer layer (Figure 2.3). Heavier PCBs could hardly be detected in the inner
PUF layer, even after 12 weeks. For the mono- to tetra-PCBs detected in the middle PUF layer,
the ratio of the amount in middle and outer layer was generally lower for the heavier congeners.
No detectable amounts of penta-CBs and higher chlorinated PCBs could be found penetrating to
the middle layer even after 12 weeks (Figure 2.3). Lighter PCBs appeared to diffuse more easily
to the inner PUF layer: mono-CBs could diffuse through the 2-cm outer and middle PUF layer
into the inner layer. This is because lighter PCBs have lower sorption affinity to PUF (i.e. a
lower KPUF/A), allowing for a higher fraction to be in the gas phase of the PUF pores. The non-
uniform PCB distribution within the PSM contradicts the assumption in the current passive air
sampling theory84
describing chemical uptake in PAS.9,83
Compared to the PUF, less of the Mono-CBs were found penetrating into the XAD (Figure 2.3).
Even after 24 weeks, the amount sequestered in the middle layer was only ~1% of that in the
outer layer and no PCBs could be detected in the inner layer. This is in line with KXAD/A being
higher than KPUF/A for individual PCB congeners (Table S2.1 and S2.2), which make them less
likely to be in the porous air phase and available for diffusion through the XAD-PSM. However,
despite different KXAD/A values, the amount of PCBs sequestered in the middle layer relative to
that in the outer layer was very similar for different PCB homologs; even for the heavier PCB
homologs such as hepta-CBs the middle layer contained approximately ~1% of the amount in the
outer layer. We attribute this to the incomplete shielding of the middle XAD layer from ambient
air. The XAD resin may have settled during the deployment period and left the upper part of the
XAD in the inner mesh cylinders partially exposed to ambient air. Therefore, we can only infer
that less than 1% of the PCBs in the outer XAD layer would penetrate to the middle layer by
diffusion through the pores. This low diffusion rate also indicates that even if only 1% of the
middle XAD layer was exposed to ambient air, the amount detected in the middle layer can not
reflect the diffusion across the outer layer. Therefore, we do not further interpret the data for the
layered XAD-PSM but focus on the layered PUF-PSM, of which the inside layer was completely
covered by the outer one.

27
Figure 2.3 PCB accumulation and distribution in the outer, middle and inner layers of the
passive sampling media (PUF and XAD). Plots are based on duplicated measurements. Mono-
PCB (PCB-1) and Penta-PCB (PCB-98/95) are used to illustrate the differences between PCBs
of different chlorination or physicochemical properties.
2.4.2.2 Depuration Compounds.
Transport of the spiked DCs between the PSM layers was observed. The data for the DCs are
presented (Figure S2.6 and S2.7) and discussed in the SI.
0
5
10
15
20
25
30
35
40
45
0 20 40 60 80 100 120 140 160 180
0
2
4
6
8
10
12
0 20 40 60 80 100
out
mid
in
0
1
2
3
4
5
6
7
0 20 40 60 80 100
PUF Mono-CB
Eq
uiv
ale
nt A
ir V
olu
me (m
3)
Deployment Time (d)
PUF Penta-CB
0
5
10
15
20
25
30
35
40
0 20 40 60 80 100 120 140 160 180
XAD Mono-CB XAD Penta-CB

28
Figure 2.4 The relationship between the PUF-air partition coefficients (KPUF/A at 20°C) and
the mass transfer coefficients for chemical diffusion between the two PUF layers (kPUF12, m/h).
The data points represent selected mono-, di-, and tri-CB congeners that penetrated into the inner
PUF with detectable amounts. The dash lines indicate 95% confidence interval of the regression
model.
2.4.3 Mass transfer coefficient for chemical diffusion between the two PUF layers (kPUF12).
kPUF12 was derived by fitting the amount of chemical accumulated in each PUF layer to the two-
layered mass balance model (Equation 2.1). kPUF12 was calculated only if the coefficient of
determination of the LLSF was over 0.7. The kPUF12 could only be derived for mono-, di- and tri-
CBs because heavier PCBs could not be detected in Layer 2. The derived kPUF12 ranged from
4.0×10-4
m/h for PCB-28 (tri-CB) to 1.1×10-2
m/h for PCB-1 (mono-CB) (Figure 2.4). A
negative correlation (Spearman’s ρ=0.91, p<10-4
) was found between kPUF12 and the PUF-air
partition coefficients (KPUF/A). A simple regression model to predict kPUF12 from KPUF/A (Figure
2.4) shows that 81% of the variation in the experimentally derived kPUF12 can be accounted for by
the variation in KPUF/A. The kPUF12 is related to the diffusion distance within the PUF and thus
5 5.5 6 6.5
-4.5
-4
-3.5
-3
-2.5
-2
-1.5
X = log KPUF/A
Y = -1.20( 0.16)·X+3.99( 0.93)
R2 = 0.81, p < 10-5
95% C.I.
Regression
line
Y =
lo
g k
PU
F1
2

29
affected by the dimensions of the PUF. To exclude this factor, we derived the effective
diffusivity (DE, PUF).
2.4.4 Effective PSM-side diffusivities (DE,PUF).
As the product of kPUF12 and diffusion length, DE,PUF excludes the effect of PUF dimensions and
should only depend on the properties of the PUF and chemical. Because kPUF12 was derived from
chemical concentrations in two discrete PUF layers of finite thickness, we do not have
information on the diffusion length within the PUF. Therefore, a range between 1 cm (thickness
of Layer 1) and 2.5 cm (thickness of Layer 1 plus half the thickness of Layer 2) was used to
represent the potential distance that chemicals diffusing from Layer 1 to Layer 2 are traversing.
The magnitude of DE,PUF ranged from 10-9
m2/h for tri-CBs to 10
-7 m
2/h for mono-CBs (Figure
2.5). Although chemical diffusion in PUF occurs in the air-filled pore space, the effective
diffusivity in PUF is lower than the diffusivity in air by a factor of 105-10
7. The low diffusivity
in PUF is mainly attributed to the relatively large KPUF/A and thus a low fraction of the chemical
in the porous air phase, where chemical diffusion within PUF occurs. Another factor lowering
the chemical diffusivity in PUF is the tortuous diffusion pathway within the PUF, which
increases the diffusion length and decreases DE,PUF. The influence of these factors on DE, PUF is
also illustrated by the mechanistic model of chemical diffusion in porous media (Equation 2.2).
Fitting the DE,PUF (upper- and lower-bound value) derived in this study, we estimated f/rSA ranges
between 0.18 (95% CI: 0.14-0.21) and 0.45 (95% CI: 0.35-0.53). Based on the model, DE, PUF
were calculated for all PCB congeners (Figure 2.5). DE,PUF decreases by over 5 orders of
magnitude from mono- to deca-CB. This variation in DE, PUF is mainly due to the variation in
KPUF/A, because DA varies by less than 50% among different PCB congeners (Figure S2.8). The
upper- and lower-bound DE, PUF from the model differ by ~0.6 log-unit, which represents the
range of f/rSA caused by potential variations of physical PUF properties. Using PUF with
densities of 0.021 and 0.035 g/cm3, Chaemfa et al.
118 found no significant difference in sampling
rates during 12 weeks of uptake. Based on our hypothesis, slightly higher uptake rates would be
expected in low density PUF. This finding suggests that in the currently used PUF, f/rSA varies
less than the difference between our upper- and lower-bound values. Interestingly, although
overall uptake rates were not significantly different, Chaemfa et al. noted a faster uptake of some
PCBs in the low density PUF during early uptake.118

30
Figure 2.5 Relationship between the effective diffusivity in PUF (DE,PUF, m2/h) and the
PUF/air partition coefficient (KPUF/A) for PCBs. The upper- and lower-bound experimentally
derived DE,PUF were based on a diffusion length of 1 and 2.5 cm, respectively. The upper- and
lower-bound modeled DE,PUF were based on a f /rSA value of 0.14 and 0.53, respectively.
2.4.5 Further Comments on the PSM-Side Kinetic Resistance and Its Implications.
Based on our experimental results and evidence from previous studies,89,107,118
we conclude that a
kinetic resistance to chemical transfer exists within the PSM (PUF and XAD).
The PSM in this study was a cylindrical PUF plug of 8 cm diameter. However, because DE, PUF of
a chemical only depends on the properties of the PUF material but not on its shape, it should be
possible to extrapolate the results of this study to the widely used 1 cm PUF disk. Because the
experiment was conducted indoors and the PSM were positioned in a housing that effectively
shields the wind, advective transport of chemicals within the PSM did likely not occur. This
agrees with Bohlin et al.117
, who observed only a minor influence of wind on PUF-PAS deployed
indoors. In PAS campaigns conducted outdoors, however, wind is likely to pass through the
”double bowl”-type housing, resulting in increasing sampling rates with increasing wind speed.95
-13
-12
-11
-10
-9
-8
-7
-6
4.5 5.5 6.5 7.5 8.5 9.5 10.5
log
DE,
PU
F
log KPUF/A
1Cl
10ClExp. derived
Upper bound
Lower bound
Modeled

31
Such a wind effect on the sampling rate can be caused by a decrease in the thickness of the air
boundary layer and/or an increased effective diffusivity within the PSM. According to CFD
simulations on the PUF-PAS,91
the wind velocity approaches zero at the PUF surface. Therefore,
if the wind does not blow directly toward the PUF, wind should have little, if any, effect on DE,
PUF. However, the CFD simulations rely on assumed scenarios of wind and other conditions.
Based on the existing information on PAS under environmental conditions outdoors, we cannot
exclude the possibility of advective chemical transport within the PSM. Further studies are
needed to understand the potential advective transport within PSM and its effect on the PSM-side
kinetic resistance under various wind conditions.
The non-uniform chemical distribution within the PSM affects the calculation of the maximum
linear uptake capacity of a PAS and the characteristic times of linear uptake or equilibration.
Assuming a uniform chemical distribution within the PSM83
will lead to an overestimation of
both the uptake capacity and the characteristic times because only the outer layer of the PSM is
available for the sampled chemicals. Knowledge of the non-uniform chemical distribution can
also help optimization of PAS design. Thinner PSM with a high surface area increase the
sampling rate R without a significant loss in uptake capacity.
The non-uniform chemical distribution within PSM also challenges the current passive air
sampling theory.83
Based on the two-film model,84
it assumes the sampled chemical is uniformly
distributed within the PSM and a kinetic resistance to chemical uptake and loss only arises from
the air boundary layer. This conceptual approach failed to explain chemical- and temperature-
specific passive sampling rates,87,89
because the experimentally observed variation of R between
chemicals and with temperatures was much larger than that can be explained by the compound-
specificity and temperature dependence of DA.20,89
In this study, we found that the PSM-side
kinetic resistance correlates with KPUF/A, which varies more among different chemicals and at
different temperatures than DA. Qualitatively, this agrees with the experimental observations. It
would be desirable to quantitatively compare the kinetic resistance (i.e. reciprocal of the mass
transfer coefficients) introduced by air boundary layer and PSM. However, we currently do not know
the thickness of the boundary layer or the average diffusion length within the PSM, which are
necessary to convert the diffusivities to mass transfer coefficients. Because DE, PUF are more than 7
orders of magnitude lower than DA, the PSM side resistance will play a role in the overall uptake as
long as the average diffusion length within the PSM exceeds 1/107 of the boundary layer thickness. A

32
model that does not rely on the assumption of a uniform chemical distribution within the PSM
will be required to quantitatively understand the PSM-side kinetic resistance and its influence on
sampling rates.
The current passive air sampling theory has also been used to describe the loss of DCs from the
PSM and to derive sampler-specific sampling rates.83,95
This approach relies on the assumption
that the uptake of the sampled chemicals and the loss of the DCs are subjected to the same
kinetic resistances.95
This assumption would likely be true if the kinetic resistance of the air
boundary layer were rate-limiting. However, because the kinetic resistance on the PSM side is
not negligible, the kinetic resistance to uptake and loss would only be identical, if the distribution
of DCs and sampled chemicals within the PSM were the same. Such rigid conditions are
impossible to meet because the distribution of the sampled chemicals within the PSM is
unknown beforehand. Therefore, such uncertainty should be considered when interpreting PAS-
based air concentrations calculated using R derived from the loss of DCs. Further efforts are
necessary to quantify and correct the uncertainty of R derived from the loss of DCs.
2.5 Acknowledgments
The authors are grateful to James Armitage for sharing the idea for the design of the described
experiments and to the Canadian Foundation for Climate and Atmospheric Sciences and the
Natural Sciences and Engineering Research Council of Canada for funding. XZ acknowledges
the Centre for Global Change Science at the University of Toronto for supporting the visit to
HIES.

33
Supporting Information of Chapter 2
Determination of PSM-air partition coefficients and sorption enthalpies of PCB congeners using poly-parameter linear free energy relationships
Poly-parameter linear free energy relationships (pp-LFERs) are available for XAD-air and PUF-
air partition coefficients (KXAD/A and KPUF/A).38,119
Hayward et al. (1) estimated KXAD/A and
KPUF/A for individual PCB congeners using these pp-LFERs and solute descriptors reported by
Abraham et al..120
Recently, van Noort et al.111
showed the PCB solute descriptors from ref. (3)
work poorly for highly ortho-chlorinated PCBs, making it is necessary to update the estimation
of partition coefficients using the pp-LFERs and the new solute descriptors.111
To calculate
KXAD/A at 20 °C, the pp-LFER:
log KXAD/A (20 °C) = 0.45A + 0.78L -0.37E + 1.96
(Equation S2.1)
by Hayward et al.38
was used. From KXAD/A at 20 °C, KXAD/A at other temperatures can also be
derived using the van’t Hoff equation:
S1
2 1 2
( ) 1 1log ( )
( ) 2.303
HK T
K T R T T (Equation S2.2)
The sorption enthalpies (ΔHS,XAD in J/mol) can be estimated by another pp-LFER 38
:
S, X AD (J/m ol) ( 17.5 2.36 2.44 27.3) 1000 H A L E
(Equation S2.3)
The K’PUF/A was first calculated using the pp-LFERs reported by Kamprad and Goss 119
:
K’PUF/A (15°C) = 3.66A + 1.69S + 0.71L16 + 0.36V - 0.15 (Equation S2.4)
Note that K’PUF/A has units of cm3/g. K’PUF/A (15 °C) was adjusted to 20 °C using the van’t Hoff
equation and sorption enthalpies (ΔHS, PUF in J/mol) calculated using 119
:
S, PU F (J/m ol) ( 46.6 4.3 12.8 17.6 2.7) 1000 H A L V S (Equation S2.5)
Then, K’PUF/A (20 °C) was converted to unitless KPUF/A (20 °C) using a PUF density of 0.02
g/cm3. KXAD/A and KPUF/A at 20 °C are listed in Table S2.1 and Table S2.2.

34
Table S2.1 XAD-air partition coefficients (KXAD/A) and sorption enthalpies (ΔHS, XAD, J/mol)
for PCBs
PCB Congener ΔHS, XAD log KXAD/A (20°C)
PCB Congener ΔHS, XAD log KXAD/A (20°C) IUPAC# J/mol (-)
IUPAC# J/mol (-)
1 -39289 6.57
43 -42405 7.79 2 -40313 6.89
44 -42405 7.79
3 -40313 6.89
45 -41475 7.50 4 -39682 6.77
46 -41475 7.50
5 -40691 7.09
47 -42405 7.79 6 -40691 7.09
48 -42405 7.79
7 -40691 7.09
49 -42405 7.79 8 -40691 7.09
50 -41475 7.50
9 -40691 7.09
51 -41475 7.50 10 -39682 6.77
52 -42405 7.79
11 -41738 7.42
53 -41475 7.50 12 -41738 7.42
54 -40529 7.20
13 -41738 7.42
55 -43496 8.13 14 -41738 7.42
56 -43496 8.13
15 -41738 7.42
57 -43496 8.13 16 -41043 7.28
58 -43496 8.13
17 -41043 7.28
59 -42405 7.79 18 -41043 7.28
60 -43496 8.13
19 -40132 7.00
61 -43496 8.13 20 -42093 7.61
62 -42405 7.79
21 -42093 7.61
63 -43496 8.13 22 -42093 7.61
64 -42405 7.79
23 -42093 7.61
65 -42405 7.79 24 -41043 7.28
66 -43496 8.13
25 -42093 7.61
67 -43496 8.13 26 -42093 7.61
68 -43496 8.13
27 -41043 7.28
69 -42405 7.79 28 -42093 7.61
70 -43496 8.13
29 -42093 7.61
71 -42405 7.79 30 -41043 7.28
72 -43496 8.13
31 -42093 7.61
73 -42405 7.79 32 -41043 7.28
74 -43496 8.13
33 -42093 7.61
75 -42405 7.79 34 -42093 7.61
76 -43496 8.13
35 -43162 7.95
77 -44587 8.48 36 -43162 7.95
78 -44587 8.48
37 -43162 7.95
79 -44587 8.48 38 -43162 7.95
80 -44587 8.48
39 -43162 7.95
81 -44587 8.48 40 -42405 7.79
82 -43766 8.30
41 -42405 7.79
83 -43766 8.30 42 -42405 7.79
84 -42817 8.01

35
Table S2.1 (continued)
PCB Congener ΔHS, XAD log KXAD/A (20°C)
PCB Congener ΔHS, XAD log KXAD/A (20°C) IUPAC# J/mol (-)
IUPAC# J/mol (-)
85 -43766 8.30
127 -46011 9.00 86 -43766 8.30
128 -45127 8.81
87 -43766 8.30
129 -45127 8.81 88 -42817 8.01
130 -45127 8.81
89 -42817 8.01
131 -44160 8.52 90 -43766 8.30
132 -44160 8.52
91 -42817 8.01
133 -45127 8.81 92 -43766 8.30
134 -44160 8.52
93 -42817 8.01
135 -44160 8.52 94 -42817 8.01
136 -43133 8.20
95 -42817 8.01
137 -45127 8.81 96 -41831 7.70
138 -45127 8.81
97 -43766 8.30
139 -44160 8.52 98 -42817 8.01
140 -44160 8.52
99 -43766 8.30
141 -45127 8.81 100 -42817 8.01
142 -44160 8.52
101 -43766 8.30
143 -44160 8.52 102 -42817 8.01
144 -44160 8.52
103 -42817 8.01
145 -43133 8.20 104 -41831 7.70
146 -45127 8.81
105 -44898 8.65
147 -44160 8.52 106 -44898 8.65
148 -44160 8.52
107 -44898 8.65
149 -44160 8.52 108 -44898 8.65
150 -43133 8.20
109 -43766 8.30
151 -44160 8.52 110 -43766 8.30
152 -43133 8.20
111 -44898 8.65
153 -45127 8.81 112 -43766 8.30
154 -44160 8.52
113 -43766 8.30
155 -43133 8.20 114 -44898 8.65
156 -46300 9.17
115 -43766 8.30
157 -46300 9.17 116 -43766 8.30
158 -45127 8.81
117 -43766 8.30
159 -46300 9.17 118 -44898 8.65
160 -45127 8.81
119 -43766 8.30
161 -45127 8.81 120 -44898 8.65
162 -46300 9.17
121 -43766 8.30
163 -45127 8.81 122 -44898 8.65
164 -45127 8.81
123 -44898 8.65
165 -45127 8.81 124 -44898 8.65
166 -45127 8.81
125 -43766 8.30
167 -46300 9.17 126 -46011 9.00
168 -44160 8.52

36
Table S2.1 (continued)
PCB Congener ΔHS, XAD log KXAD/A (20°C)
PCB Congener ΔHS, XAD log KXAD/A (20°C) IUPAC# J/mol (-)
IUPAC# J/mol (-)
169 -47436 9.53
190 -46488 9.32 170 -46488 9.32
191 -46488 9.32
171 -46488 9.32
192 -46488 9.32 172 -46488 9.32
193 -46488 9.32
173 -46488 9.32
194 -47850 9.83 174 -45503 9.03
195 -46846 9.54
175 -45503 9.03
196 -46846 9.54 176 -44435 8.70
197 -45737 9.20
177 -45503 9.03
198 -46846 9.54 178 -45503 9.03
201 -46846 9.54
179 -44435 8.70
199 -45737 9.20 180 -46488 9.32
200 -45737 9.20
181 -45503 9.03
202 -45737 9.20 182 -45503 9.03
203 -46846 9.54
183 -45503 9.03
204 -45737 9.20 184 -44435 8.70
205 -47850 9.83
185 -45503 9.03
206 -48189 10.05 186 -44435 8.70
207 -47039 9.70
187 -45503 9.03
208 -47039 9.70 188 -44435 8.70
209 -48341 10.20
189 -47702 9.69

37
Table S2.2 PUF-air partition coefficients (KPUF/A) and sorption enthalpies (ΔHS, PUF, J/mol)
for PCBs
PCB Congener ΔHS, PUF log KPUF/A (20°C)
PCB Congener ΔHS, PUF log KPUF/A (20°C) IUPAC# J/mol (-)
IUPAC# J/mol (-)
1 -61893 4.81
43 -81562 6.86 2 -62965 5.01
44 -82306 6.98
3 -63184 5.05
45 -80818 6.74 4 -68160 5.43
46 -81463 6.84
5 -69326 5.65
47 -81941 6.92 6 -69231 5.63
48 -81829 6.90
7 -68784 5.56
49 -81764 6.89 8 -69450 5.67
50 -80337 6.66
9 -68608 5.53
51 -80922 6.75 10 -67945 5.40
52 -81584 6.86
11 -70302 5.83
53 -80745 6.73 12 -70535 5.87
54 -79903 6.59
13 -70522 5.86
55 -83696 7.23 14 -69821 5.75
56 -84233 7.32
15 -70745 5.90
57 -82634 7.06 16 -75592 6.26
58 -83150 7.14
17 -75050 6.18
59 -82212 6.96 18 -74874 6.15
60 -83919 7.26
19 -74031 6.01
61 -83567 7.21 20 -76663 6.46
62 -81945 6.92
21 -76358 6.41
63 -82857 7.09 22 -76887 6.50
64 -82461 7.00
23 -75296 6.24
65 -82203 6.96 24 -74904 6.15
66 -83691 7.23
25 -76122 6.37
67 -82900 7.10 26 -75941 6.34
68 -82608 7.05
27 -75424 6.24
69 -81377 6.83 28 -76345 6.41
70 -83597 7.21
29 -75563 6.28
71 -83024 7.10 30 -74487 6.08
72 -82427 7.02
31 -76165 6.38
73 -81941 6.92 32 -75721 6.28
74 -83120 7.13
33 -76801 6.48
75 -81597 6.86 34 -75717 6.31
76 -83661 7.22
35 -77872 6.68
77 -85442 7.54 36 -76845 6.52
78 -84728 7.42
37 -78092 6.72
79 -84354 7.36 38 -77365 6.60
80 -83387 7.20
39 -77008 6.54
81 -84922 7.45 40 -83029 7.10
82 -84190 7.13
41 -82624 7.03
83 -87428 7.65 42 -82487 7.01
84 -86684 7.53

38
Table S2.2 (continued)
PCB Congener ΔHS, PUF log KPUF/A (20°C)
PCB Congener ΔHS, PUF log KPUF/A (20°C) IUPAC# J/mol (-)
IUPAC# J/mol (-)
85 -87948 7.73
127 -89622 8.05 86 -87578 7.67
128 -97089 8.63
87 -87772 7.71
129 -96577 8.55 88 -85226 7.29
130 -96027 8.46
89 -86929 7.57
131 -94230 8.17 90 -86886 7.56
132 -95283 8.34
91 -86142 7.44
133 -94965 8.28 92 -86710 7.53
134 -94307 8.18
93 -85308 7.31
135 -94221 8.16 94 -85867 7.40
136 -93477 8.04
95 -85966 7.41
137 -96036 8.46 96 -85123 7.28
138 -96294 8.50
97 -87695 7.69
139 -94337 8.18 98 -85820 7.39
140 -94333 8.18
99 -87166 7.61
141 -94372 8.19 100 -85278 7.30
142 -94836 8.26
101 -86985 7.58
143 -95017 8.29 102 -86134 7.44
144 -93507 8.05
103 -85097 7.27
145 -92664 7.91 104 -84302 7.14
146 -95240 8.33
105 -89755 8.05
147 -93757 8.09 106 -88615 7.87
148 -93357 8.02
107 -88637 7.87
149 -94488 8.21 108 -88701 7.88
150 -92613 7.90
109 -87720 7.70
151 -93585 8.06 110 -88245 7.78
152 -92746 7.92
111 -87553 7.70
153 -95498 8.37 112 -86731 7.54
154 -93623 8.07
113 -87161 7.61
155 -91749 7.76 114 -88869 7.91
156 -97782 8.77
115 -86873 7.56
157 -98096 8.82 116 -87544 7.67
158 -95786 8.42
117 -87677 7.69
159 -97111 8.63 118 -88903 7.91
160 -95511 8.37
119 -87381 7.64
161 -94703 8.24 120 -87816 7.74
162 -97090 8.65
121 -86306 7.47
163 -95868 8.43 122 -89497 8.01
164 -96642 8.56
123 -88955 7.92
165 -95090 8.30 124 -88779 7.89
166 -96530 8.54
125 -88288 7.79
167 -97300 8.69 126 -90650 8.22
168 -95778 8.42

39
Table S2.2 (continued)
PCB Congener ΔHS, PUF log KPUF/A (20°C)
PCB Congener ΔHS, PUF log KPUF/A (20°C)
IUPAC# J/mol (-)
IUPAC# J/mol (-)
169 -99189 9.02
190 -103081 9.23
170 -103610 9.31
191 -102617 9.15
171 -101262 8.93
192 -102006 9.05
172 -102548 9.14
193 -102698 9.16
173 -101524 8.97
194 -110083 9.99
174 -101813 9.02
195 -108578 9.74
175 -100196 8.76
196 -107779 9.62
176 -99366 8.62
197 -105431 9.23
177 -101426 8.96
198 -107490 9.57
178 -100303 8.78
201 -107856 9.63
179 -99448 8.64
199 -106747 9.45
180 -102913 9.20
200 -106643 9.43
181 -100978 8.89
202 -105590 9.26
182 -100948 8.88
203 -107671 9.60
183 -100475 8.80
204 -105598 9.26
184 -98588 8.50
205 -109911 9.96
185 -100802 8.86
206 -113507 10.39
186 -99959 8.72
207 -111159 10.00
187 -100544 8.82
208 -111236 10.02
188 -98669 8.51
209 -116882 10.77
189 -104617 9.50

40
Detailed information on the depuration compounds and spiking procedures
PUF. Before sampling and after cleaning, the three cylindrical PUF layers were fortified with
three different groups of depuration compounds (DCs): 15 mL of 1.4 ng/mL PCB-36 and PCB-
186 in hexane were applied to the outer layer, 15 mL of 1.4 ng/mL PCB-38 and PCB-188 in
hexane were applied to the middle layer, and 15 mL of 1.4 ng/mL PCB-39 and PCB-190 in
hexane were applied to the inner layer. Based on a pre-test, 15 mL of solvent was sufficient to
fully wet the PUF sheets, allowing the DCs to achieve a relatively uniform distribution in the
PUF. When applying the DCs, each PUF sheet was placed on a piece of aluminum foil (baked
for >4 hr at 450 °C), the 15 mL DC solution was spiked evenly onto the PUF using a pipette. The
spiked PUF sheets were placed in a fume hood for ~ 1 h to let the solvent evaporate before
assembling the sheets into the concentrically layered PUF cylinder.
XAD. 200 g, 250 g, and 450 g clean XAD-2 resin were transferred into three glass jars in order to
be used to fill the inner, middle and outer layer of the mesh cylinders, respectively. To the jar
containing 200 g XAD (to be used to fill the inner layer) 4000 ng 13
C-PCB-1 and 3500 ng PCB-
36 in 10 ml hexane was added; to the jar containing 250 g XAD (to be used to fill the middle
layer) 4000 ng 13
C-PCB-4 and 3500 ng PCB-38 in 10 ml hexane was added; to the jar containing
450 g XAD (to be used to fill the outer layer) 8000 ng 13
C-PCB-8 and 7000 ng PCB-39 in 10ml
hexane was added. The spiked XAD was shaken in the jars to uniformly distribute the spiked
DCs. Since not all the prepared XAD was used, the initial DC levels in each of the XAD layers
was determined from the duplicated blanks we prepared assuming that the DCs are uniformly
distributed in the XAD in the glass jars.

41
Figure S2.1 Illustration of the sampling scheme in this study.
Figure S2.2 Reproducibility of the duplicated samples as represented by the relative difference
of the sampling rate R (m3/h) between duplicates. The relative difference is defined as
1 2
1 20.5( )
R R
R R
t=0 (Field Blank)
0.5w/1w
1w/2w
2w/4w
4w/8w
8w/12w
12w/24w
PUF/XAD
4w 8w 12wLowVol Sampler 2.9 m3/day
24w
PUF/XAD/PUF
Layered PSM
16w 20w
-20%
-10%
0%
10%
20%PUFXAD
Rel
ativ
e D
iffe
ren
ce
1Cl 2Cl 4Cl 5Cl 6Cl 7Cl3ClPCB Homolog Groups

42
Figure S2.3 Analytical procedure recovery of the surrogate standards spiked prior to sample
extraction.
Table S2.3 Limit of detection a (LOD) of PCBs analyzed using HRGC/MS
PCB Homolog Mono- Di- Tri- Tetra- Penta- Hexa- Hepta-
LOD (pg/sample) 10 20 5 5 5 5 7
a
defined as the chemical amount corresponding to the signal-to-noise ratio of 3 LOD of each
PCB homolog is average of the LOD of each congener in the homolog group.
50
60
70
80
90
100
110
120
130
Recovery
(%
)

43
Description of the two-layer mass balance model used to derive effective diffusivities of PCBs through the passive sampling medium
The mass balance of the chemical in the outer (Layer 1) and inner layer (Layer 2) can be
expressed as:
1
1 1 2 1 2( / ) ( )
E
A A SA
dm Dk A C C K A C C
dt (Equation S2.6)
2
2 1 2( )
E
dm DA C C
dt (Equation S2.7)
where m1 and m2 [dimension: M] are the amounts of the chemical sequestered in Layer 1 and 2; t
[T] is time; kA[LT-1
] is the mass transfer coefficient for chemical crossing the air-side boundary
layer; A1 [L2] is the surface area between air and Layer 1; CA [ML
-3] is the chemical
concentration in air; C1 and C2 [ML-3
] are the concentrations of the chemical sequestered in
Layer 1 and 2; KSA [dimensionless] is the partition coefficient between the passive sampling
medium (PSM) and air; DE [L2T
-1] is the effective diffusion coefficient of the chemical in the
PSM; δ [L] is the diffusion length of the chemical within the PSM.
Figure S2.4 Illustration of the two-layer mass balance model used to derive effective
diffusivities of PCBs through the passive sampling medium.
The measured data indicate that the amount of chemical penetrating to Layer 2 is less than 1% of
that staying in Layer 1, i.e. the chemical exchange between outer and inner layer is negligible
δ
m1(t), C1(t)
m2(t), C2(t)
DE
CA
A1
kA
A2
boundary layer
δbl

44
compared to the chemical transfer from air to Layer 1. Thus, Equation S2.6 and Equation S2.7
can be simplified to:
1
1 1( / )
A A SA
dmk A C C K
dt
(Equation S2.8)
2
2 1
Edm D
A Cdt
(Equation S2.9)
Further, the uptake kinetics of the chemical in the first layer was generally linear, thus Equation
S2.8 can be further simplified to:
1
1A A
dmk A C
dt
(Equation S2.10)
Integrated from 0 to t, Equation S2.10 becomes:
1 1 1 1 1( ) ( ) (0)
A AV C t m t k A C t m
(Equation S2.11)
From Equation S2.9 and Equation S2.11,
2 2
1 1
1
[ (0)]
E
A A
dm D Ak A C t m
dt V (Equation S2.12)
Integrated from 0 to t, Equation S2.12 becomes:
22 2
2 1 1 2
1 1
1( ) (0) (0)
2 E E
A A
D A D Am t k A C t m t m
V V (Equation S2.13)
Because kA depends on the boundary layer thickness, which varies by the air conditions around
the PSM and is highly uncertain, the term 1A A
k A C t in Equation S2.13 can be replaced with that
in Equation S2.11:

45
2
2 1 1 2
1
1( ) [ (0) ( )] (0)
2
ED A
m t m m t t mV
(Equation S2.14)
In the experiment, different PCB congeners sequestered in Layer 1 and 2 (m1, m2) were measured
at seven time points. Let 1 1
[ (0) ( )]t
X m m t t and 2( )
tY m t .
tX and
tY can be plotted against
each other and subjected to linear least squares fitting. The slope of the fitted line is equal to
2 1/ (2 )
ED A V . From the slope, the mass transfer coefficient from Layer 1 to Layer 2 (defined as
kPSM12 = DE/δ) can be calculated, because the dimensional parameters A2 and V1 are known.
Furthermore, if the diffusion length within the PSM is known, the effective diffusion coefficient
in the PSM (E
D ) can be derived.

46
Table S2.4 Congener-specific passive air sampling rates of PCBs derived using linear least
squares fitting
PCB Homolog
PCB Congener IUPAC #
PUF XAD Sampling Rate
(m3/d) R2
Sampling Rate (m3/d)
R2
Mono- #1 0.16 0.98 0.23 0.96 Mono- #3 0.19 0.99 0.27 0.98 Mono- #2 0.13 0.97 0.20 0.98
Di- #10 0.10 0.95 0.17 0.95 Di- #4 0.11 0.96 0.19 0.97 Di- #9 0.08 0.96 0.12 0.91 Di- #7 0.14 0.96 0.18 0.97 Di- #6 0.14 0.98 0.19 0.98 Di- #8#5 0.14 0.97 0.18 0.98 Di- #11 0.12 0.96 0.17 0.97 Di- #13#12 0.12 0.97 0.17 0.91 Di- #15 0.14 0.96 0.20 0.97 Tri- #19 0.14 0.99 0.21 0.99 Tri- #18 0.15 0.98 0.20 0.99 Tri- #17 0.15 0.99 0.20 0.99 Tri- #24 0.14 0.97 0.21 0.98 Tri- #27 0.15 0.99 0.20 0.99 Tri- #32 0.15 0.99 0.21 0.98 Tri- #16 0.15 0.99 0.22 0.98 Tri- #34 0.11 0.88 0.20 0.88 Tri- #29 0.15 0.90 0.23 0.97 Tri- #26 0.15 0.99 0.20 0.98 Tri- #25 0.15 0.98 0.21 0.98 Tri- #31 0.14 0.99 0.20 0.98 Tri- #28 0.15 0.99 0.20 0.99 Tri- #22 0.15 0.99 0.19 0.98 Tri- #35 0.17 0.97 0.26 0.78 Tri- #37 0.15 0.99 0.19 0.98

47
Table S2.4 (continued)
PCB Homolog
PCB Congener IUPAC #
PUF XAD Sampling Rate
(m3/d) R2
Sampling Rate (m3/d)
R2
Tetra- #53 0.15 0.99 0.18 0.98 Tetra- #51 0.15 0.99 0.20 0.98 Tetra- #45 0.16 0.99 0.18 0.98 Tetra- #46 0.15 0.99 0.18 0.98 Tetra- #52#69 0.16 0.99 0.22 0.98 Tetra- #43#49 0.15 0.99 0.18 0.99 Tetra- #48#47 0.16 0.99 0.18 0.98 Tetra- #44 0.15 0.99 0.14 0.97 Tetra- #42 0.16 0.98 0.19 0.99 Tetra- #64 0.16 0.98 0.18 0.99 Tetra- #71 0.15 0.99 0.18 0.98 Tetra- #40 0.15 0.99 0.18 0.98 Tetra- #67 0.15 0.94 0.17 0.97 Tetra- #63 0.16 0.98 0.18 0.98 Tetra- #74 0.16 0.99 0.18 0.98 Tetra- #70 0.15 0.98 0.17 0.97 Tetra- #66 0.16 0.98 0.18 0.98 Tetra- #55 0.06 0.99 0.22 0.82 Tetra- #60 0.15 0.96 0.17 0.99 Tetra- #56 0.15 0.98 0.18 0.98 Tetra- #78 0.16 0.96 0.14 0.99 Tetra- #81 0.15 0.95 0.15 0.94 Tetra- #77 0.11 0.90 0.15 0.92 Tetra- #96 0.08 0.98 0.20 0.94 Tetra- #103 0.08 0.97 0.20 0.96 Penta- #100 0.12 0.98 0.20 0.96 Penta- #94 0.14 0.98 0.24 0.97 Penta- #102#93 0.17 0.99 0.15 0.96 Penta- #98#95 0.08 0.96 0.20 0.98 Penta- #91 0.11 0.99 0.18 0.97 Penta- #92 0.12 0.99 0.19 0.98 Penta- #84 0.09 0.99 0.17 0.97 Penta- #89 0.08 0.95 0.17 0.89 Penta- #90#101 0.07 0.96 0.19 0.98 Penta- #99 0.11 0.99 0.17 0.97 Penta- #112#119 0.11 0.99 0.17 0.97 Penta- #83 0.10 0.98 0.18 0.97 Penta- #86#117#97 0.11 0.98 0.18 0.97 Penta- #85 0.09 0.98 0.16 0.97 Penta- #87#115 0.10 0.98 0.17 0.93 Penta- #120#110 0.07 0.97 0.16 0.86 Penta- #82 0.08 0.96 0.16 0.96 Penta- #124 0.09 0.98 0.16 0.97 Penta- #109#107 0.10 0.98 0.16 0.97 Penta- #118 0.09 0.98 0.15 0.97 Penta- #114 0.10 0.94 0.15 0.93 Penta- #122 0.10 0.87 0.19 0.96

48
Table S2.4 (continued)
PCB Homolog
PCB Congener IUPAC #
PUF XAD Sampling Rate
(m3/d) R2
Sampling Rate (m3/d)
R2
Hexa- #150 0.11 0.92 0.18 0.96 Hexa- #152 0.10 0.94 0.17 0.97 Hexa- #145 0.09 0.85 0.17 0.97 Hexa- #136 0.08 0.98 0.14 0.98 Hexa- #154 0.10 0.97 0.16 0.98 Hexa- #151 0.10 0.98 0.14 0.99 Hexa- #135 0.10 0.98 0.14 0.99 Hexa- #144 0.10 0.98 0.13 0.99 Hexa- #147 0.09 0.97 0.15 0.98 Hexa- #149#139 0.10 0.98 0.14 0.99 Hexa- #143 0.10 0.83 0.14 0.94 Hexa- #134 0.10 0.98 0.14 0.99 Hexa- #131 0.09 0.97 0.14 0.99 Hexa- #146 0.09 0.98 0.14 0.98 Hexa- #132 0.08 0.97 0.13 0.98 Hexa- #153 0.08 0.98 0.12 0.98 Hexa- #141 0.07 0.98 0.13 0.98 Hexa- #137 0.09 0.98 0.13 0.99 Hexa- #130 0.08 0.98 0.12 0.99 Hexa- #164#163 0.09 0.97 0.13 0.99 Hexa- #138 0.08 0.85 0.12 0.99 Hexa- #158 0.09 0.98 0.12 0.99 Hexa- #129 0.09 0.97 0.12 0.99 Hexa- #166 0.10 0.87 0.13 0.98 Hexa- #128 0.08 0.95 0.13 0.98 Hexa- #167 0.12 0.95 0.15 0.99 Hexa- #156 0.13 0.91 0.15 0.99 Hepta- #179 0.09 0.98 0.13 0.98 Hepta- #176 0.09 0.98 0.13 0.98 Hepta- #178 0.08 0.89 0.13 0.98 Hepta- #175 0.09 0.89 0.10 0.96 Hepta- #182#187 0.08 0.96 0.12 0.98 Hepta- #183 0.08 0.94 0.12 0.98 Hepta- #185 0.08 0.96 0.12 0.98 Hepta- #174 0.08 0.94 0.12 0.98 Hepta- #177 0.08 0.93 0.12 0.97 Hepta- #171 0.08 0.93 0.12 0.94 Hepta- #172 0.09 0.81 0.11 0.99 Hepta- #180 0.12 0.85 0.11 0.98 Hepta- #170 0.12 0.83 0.10 0.97

49
Table S2.5 Passive air sampling rates determined in different studies using XAD and
PUF as PSM.
PSM R
(m3/d) SAa
(dm2) SRb
(m3/d/dm2) Environmen
t Type Chemical Studyc
XAD
0.1-0.3 0.94 0.11-0.32 indoor PCBs this study
0.4-2.3 0.63 0.63-3.7 outdoor pesticide
s Hayward, et al. (2010)
0.8-5.4 1.26 0.63-4.3 outdoor pesticide
s Gouin, et al. (2008)
0.4-2.2 1.26 0.32-1.7 outdoor pesticide
s Wania, et al. (2003)
PUF
0.06-0.2 3.02 0.02-0.07 indoor PCBs this study
0.57-1.55 3.6 0.16-0.43 indoor PCBs Hazrati and Harrad (2007)
2.0-8.3 3.65 0.55-2.27 indoor PCBs Shoeib and Harner (2002)
0.66-24 3.6 0.18-6.7 outdoor PCBs Melymuk, et al. (2010)
2.9-7.3 3.6 0.81-2.03 outdoor PCBs Chaemfa, et al. (2008)
0.10 ± 0.01
1.88 0.053 ± 0.005 indoor PAHs Tao, et al. (2007)
0.38 ± 0.51
2.42 0.16 ± 0.21 outdoor PAHs Tao, et al. (2009)
a surface area between PSM and air; b surface area normalized sampling rate; c reference 9,20,47,87,89,106,107,116

50
Figure S2.5 Relationship between homolog-specific molecular diffusivities in air and passive
air sampling rates. The molecular diffusivities in air are derived from the Fuller-Schettler-
Giddings equation109
; the passive air sampling rate is based on the median of the congener-
specific sampling rates in each homolog group.
Transfer kinetics of the depuration compounds
The depuration compounds spiked to the inner PUF layer (PCB-39 and PCB-190) gradually
migrated outward during deployment (Figure S2.6). PCB-39 and PCB-190 decreased in the inner
layer and increased in the middle and outer layer. The amount accumulated in the outer layer was
lower than that in the middle layer. The mass balance of the DCs was checked by the sum of the
DCs in the three layers. The sum of PCB-39 and PCB-190 ranged from 80 % to 100 % of the
initially spiked amount. After 84 d, ~20 % and ~10 % of the initially spiked PCB-39 of PCB-190
had move into the middle and inner layers.
The depuration compounds spiked to the middle PUF layer (PCB-38 and PCB-188) migrated
both to the inner and outer layer during deployment (Figure S2.6). No difference was observed
between the amount in the inner and outer layer. The sum of PCB-38 and PCB-188 in the three
0.05
0.10
0.15
0.20
0.25
0.014 0.016 0.018 0.02 0.022
R(m
3/d
)
DA (m2/h)
XAD
PUF

51
layers ranged from 90 % to 110 % of the initially spiked amount. After 84 d, ~30 % and ~20 %
of the initially spiked PCB-38 of PCB-180 had moved into the inner and outer layers.
The depuration compounds spiked to the outer PUF layer (PCB-36 and PCB-186) migrated
inward to the middle and inner layer during deployment (Figure S2.6). Similar to the DCs spiked
to the inner layer, higher amounts were detected in the adjacent layer. The sum of PCB-186 in
the three layers ranged from 60 % to 100 % of the initially spiked amount. For PCB-36, the sum
of the three layers ranged from 60 % to 140 % of the initially spiked amount; the amount in the
outer layer appeared to increase gradually. Although PCB-36 is a non-Aroclor PCB121
and had
not been reported in PCB air profiles, from our low volume sampler analysis, we found PCB-36
had a level of ~0.7 ng/m3 in the indoor air we sampled. This explains the increasing levels of
PCB-36 in the outer layer of both PUF and XAD. Up to now, except for some studies on PCB-
11,122,123
few non-Aroclor PCBs have been analyzed and reported. Considering the likelihood of
occurrence and toxicity of these congeners, further studies on these non-Aroclor PCBs are
warranted.
PCBs sorb more strongly to XAD than to PUF.38
Therefore, more volatile PCB congeners
(mono-/di- and tri-CBs) were spiked onto XAD to increase the likelihood of observing a transfer
between the XAD layers. Nevertheless, even for the most volatile congeners (13
C PCB-1) spiked
to the inner XAD layer, no significant transfer to the other layers was observed. This is also the
same for all the other di- and tri-CBs spiked as DCs to the inner and middle XAD layer (Figure
S2.7).
Overall, the results for the DCs serve as further evidence of the existence of kinetic resistance to
chemical transfer within the PSM. They also provide further evidence of a relationship between a
chemical’s mobility within a PSM and the KPSM/A. We can also conclude that DCs initially
present in the inner part of PSM are less likely to evaporate to the ambient air than those closer
to the surface.

52
Figure S2.6 Changes of the amounts of depuration compounds (tri- and hepta-CBs) spiked to
the inner, middle, and outer layer of PUF. The amount of chemicals present in each layer (Mi)
was normalized to the amount (M0) in the field blanks (samples retrieved at t=0).
0
20
40
60
80
100
120
0 20 40 60 80 100
0
20
40
60
80
100
120
0 20 40 60 80 100
0
20
40
60
80
100
120
140
0 20 40 60 80 100
0
20
40
60
80
100
120
0 20 40 60 80 100
0
20
40
60
80
100
120
0 20 40 60 80 100
0
20
40
60
80
100
120
0 20 40 60 80 100
PCB-39 spiked to inner PUF
0
20
40
60
80
100
120
0 20 40 60 80 100
in
mid
out
PCB-38 spiked to middle PUF PCB-36 spiked to outer PUF
PCB-190 spiked to inner PUF PCB-188 spiked to middle PUF PCB-186 spiked to outer PUF
Deployment Time (d)
( M
i/
M0
) 1
00
%
in+mid+out
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180
0
20
40
60
80
100
120
0 30 60 90 120 150 180
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180
0
20
40
60
80
100
120
0 30 60 90 120 150 180
0
20
40
60
80
100
120
0 30 60 90 120 150 180
0
20
40
60
80
100
120
0 30 60 90 120 150 180
Deployment Time (d)
( M
i/
M0
) 1
00
%
13C PCB-1spiked to inner XAD 13C PCB-4 spiked to middle XAD 13C PCB-8 spiked to outer XAD
PCB-36 spiked to inner XAD PCB-38 spiked to middle XAD PCB-39 spiked to outer XAD
0
20
40
60
80
100
120
0 20 40 60 80 100
in
mid
out
in+mid+out

53
Figure S2.7 Changes of the amounts of depuration compounds (mono-/di- and tri-CBs) spiked
to the inner, middle, and outer layer of XAD. The amount of chemicals present in each layer (Mi)
was normalized to the amount (M0) in the field blanks (samples retrieved at t=0).
Figure S2.8 Illustration of the sensitivity of DEPUF to the variations of DA and KPUF/A. (a) based
on f/rSA value of 0.18; (B) based on f/rSA value of 0.45.
0.012 0.014 0.016 0.018 0.0204
5
6
7
8
9
10
11
log
KP
UF
/A
DA
-14
-13
-12
-11
-10
-9
-8
-7
-6
0.012 0.014 0.016 0.018 0.0204
5
6
7
8
9
10
11
log
KP
UF
/A
DA
-14-13-12-11-10-9-8-7-6
DA (m2/h)
log
KP
UF/
A
1Cl
10Cl
1Cl
10Cl
(a) (b)
0.012 0.014 0.016 0.018 0.0204
5
6
7
8
9
10
11
log
KP
UF
/A
DA
-14-13-12-11-10-9-8-7-6
DE,PUF
(m2/h)

54
Chapter 3. Modeling the uptake of semi-volatile organic compounds by
passive air samplers: Importance of mass transfer processes within the porous sampling media
Xianming Zhang, Frank Wania
Contributions: X. Zhang developed the model, programmed to solve the model under different
scenarios. X. Zhang interpreted the model output, wrote the manuscript, revised it and responded
to reviewers’ comments under the guidance of F. Wania.
1
2 3
log KSA
log
(k
sorb
/ d–
1)
6 7 8 9 10
9
8
7
6
5
4
KSADA
ksorb
DPA

55
3.1 Abstract
Air sampling based on diffusion of target molecules from the atmospheric gas phase to passive
sampling media (PSMs) is currently modeled using the two-film approach. Originally developed
to describe chemical exchange between air and water, it assumes a uniform chemical distribution
in the bulk phases on either side of the interfacial films. Although such an assumption may be
satisfied when modeling uptake in PSMs in which chemicals have high mobility, its validity is
questionable for PSMs such as polyurethane foam disks and XAD-resin packed mesh cylinders.
Mass transfer of chemicals through the PSMs may be subject to a large resistance because of the
low mass fraction of gas-phase chemicals in the pores, where diffusion occurs. Here we present a
model that does not assume that chemicals distribute uniformly in the PSMs. It describes the
sequential diffusion of vapors through a stagnant air-side boundary layer and the PSM pores, and
the reversible sorption onto the PSM. Sensitivity analyses reveal the potential influence of the
latter two processes on passive sampling rates (PSRs) unless the air-side boundary layer is
assumed to be extremely thick (i.e. representative of negligible wind speeds). The model also
reveals that the temperature dependence of PSRs, differences in PSRs between different
compounds, and a two-stage uptake, all observed in field calibrations, can be attributed to those
mass transfer processes within the PSM. The kinetics of chemical sorption to the PSM from the
gas phase in the macro-pores is a knowledge gap that needs to be addressed before the model can
be applied to specific compounds.
3.2 Introduction
Over the past decades, various types of passive air samplers (PASs) have been developed to
monitor semivolatile organic compounds (SVOCs) in air.9,13,20,124
Due to the advantages of low
cost, simple and noise-free operation and no power requirement, applications of PASs range
widely from investigating spatial and long term temporal trends of SVOCs at local, regional and
global scales42,58,64,65
to identifying sources and assessing exposures of SVOCs in the air of
various environments.30,74,79
Passive air sampling is based on molecular diffusion from the atmospheric gas phase to a passive
sampling medium (PSM) such as polyethylene,13
polymer-coated glass,125
polyurethane foam
(PUF),9 and XAD-resin.
20 Unlike polyethylene or polymer-coated glass-based PAS, where
SVOCs accumulate in thin layers in contact with air, the PSM in PUF and XAD-based PAS is

56
relatively thick and porous. Chemical uptake by PSMs has been described using the two-film
model84
, which is often referred to as passive air sampling theory.9,83,86
The two-film model was
originally developed by Lewis and Whitman84
to describe mass transfer between air and water.
By replacing the water compartment with the PSM, the two-film model approach is applied to
describe chemical transfer from air to the PSM. However, the two-film model requires that “in
the main body of either liquid or gas […] the concentration of solute in the fluid is essentially
uniform at all points.”84
While this assumption may be satisfied when modeling uptake in PSMs
in which chemicals have high mobility, its validity is questionable for thick, porous PSMs such
as PUF and XAD. Chemical transfer within such porous PSMs primarily occurs in the gas-filled
pores, which limits the transfer kinetics because only a small mass fraction of the SVOCs may be
in the porous gas phase within the PSM. Recently, a passive sampling experiment conducted
using concentrically layered XAD and PUF indicated that SVOCs did not distribute uniformly
within these PSMs over the exposure period (168 d for XAD or 84 d for PUF) but remained
predominantly in outer layers in close contact with air (Chapter 2 and Chapter 4).126,127
Therefore, using the two-film approach and assuming uniform chemical distributions within the
PSMs, the current PAS theory may not be able to fully describe the uptake of SVOCs from air to
these bulk PSMs.
If uptake in a PAS were indeed limited by the air side resistance only,9,83,86
under a given wind
condition or thickness of the stagnant air layer surrounding the PSM, a chemical’s passive
sampling rate (PSR) should be proportional to its diffusivity in air (DA). According to the Fuller-
Schettler-Giddings Equation,85
DA is a function of atmospheric pressure, temperature, and
molecular size.87,89
However, DA is not sufficiently sensitive to these parameters to explain
variations of PSR with temperature and differences in PSR between compounds/congeners
observed in PAS calibration studies.20,28,87,89,90
Whereas shifts in gas-particle partitioning87
can to
some extent explain PSR variations for SVOCs of very low volatility, they cannot serve as an
explanation for the observed variations in the PSRs of most SVOCs, indicating that other factors
must play a role. Furthermore, based on the two-film PAS theory,9,83,86
the sampled amount (or
equivalent air volume) increases linearly with time during the initial stage of chemical uptake by
a PAS until uptake gradually slows due to re-evaporation from the PSM back to air. Deviating
from this pattern, some recent calibration studies for PUF-PAS observed high PSRs initially
followed by lower, yet relatively constant PSRs during later uptake.88,128
Although these

57
observations were conceptually described as a two-stage uptake mechanism88
, no attempt was
made to reconcile them with two-film PAS theory which fails to explain such behavior.
The objective of this study was to develop a model that does not require the assumption that
chemicals distribute uniformly in the PSM but considers chemical diffusion through the stagnant
air layer surrounding the PSM, diffusion through the air-filled pores within the PSM and
sorption/desorption between the gas phase and the PSM material. The model is then applied to
illustrate how the mass transfer processes and associated parameters affect the PSR.
3.3 Methods
3.3.1 Conceptual Model of Chemical Mass Transfer during Passive Air Sampling.
Currently, when describing the kinetics of SVOC uptake from air to PAS or the depuration from
PSM to air, the PSM is treated as a bulk phase, in which chemical distribution is uniform and
therefore no chemical mass transfer processes need to be considered.9,20,83
Based on our previous
experiments indicating the existence of a kinetic resistance to SVOC mass transfer within porous
PSMs such as XAD resin and PUF, (Chapter 2)126
we propose an alternative conceptual
framework of a three-stage mass transfer process to describe sampling of SVOCs in PAS, which
is illustrated in Figure 3.1. The first stage is the mass transfer of SVOCs through the stagnant air
layer surrounding the bulk PSM (process 1 in Figure 3.1). This process is the same as that
described by the two-film PAS theory.9,83
After crossing the stagnant air layer, molecules can
diffuse deeper into the PSM through macro-pores (process 2 in Figure 3.1). The terms “macro-
pore” and “meso-pore” refer to pores with diameters of >50 nm and 2-50 nm, respectively.129
Simultaneously, molecules can sorb to the solid PSM material (process 3 in Figure 3.1). As XAD
pellets have meso-pores ~9 nm in diameter,130
the molecules will not only adsorb to the outer
pellet surface, but will also diffuse through the meso-pores and sorb to sites deeper within the
XAD pellets. Therefore, process 3 for the XAD-PAS involves both meso-pore diffusion and
sorption/desorption.

58
Figure 3.1 Conceptual diagram of the chemical mass transfer processes between air and the
passive sampling media (PSMs) in the (a) XAD-resin based passive air sampler and (b)
polyurethane foam based passive air sampler. The mass transfer processes include: (1) diffusion
through the stagnant air layer surrounding the PSM; (2) diffusion through macro-pores within the
PSM; (3) sorption/desorption between porous air and solid PSM material. The microstructure of
polyurethane foam was taken from a micrograph contributed by JA Elliott to the DoITPoMS
Micrograph Library, University of Cambridge under the Creative Commons Attribution Non-
Commercial Share Alike license.
3.3.2 Mathematical Model of Chemical Mass Transfer during Passive Air Sampling.
To quantitatively describe (i) molecular diffusion through the stagnant air layer surrounding the
PSM, (ii) diffusion though the macro-pores within the PSM and (iii) sorption/desorption between
gas-filled macro-pores and solid PSM material, we applied (i) Fick’s law, (ii) the diffusion-
reaction equation based on Fick’s law and the principles of mass conservation, and (iii) the law
of mass action. Due to the different geometries of XAD-filled mesh cylinders and PUF disks
(Figure 3.1), Fick’s law of diffusion in cylindrical coordinates and in a plane was applied to
1
23
stagnant air layer
mesoporousXAD pellet
macroporesbetween
XAD pellets
1
2
3
stagnant air layer
macroporeswithin PUF
cross-linked PUF material
(a)
(b)
XAD resin filled mesh
cylinder
polyurethane foam (PUF) disk

59
XAD-PAS and PUF-PAS, respectively. The model for the XAD-PAS is presented below; the
one for the PUF-PAS is described in the Supporting Information (SI).
3.3.2.1 Diffusion Across the Stagnant Air Layer.
Close to the interface between air and the bulk PSM, eddies become diminished owing to the
viscous nature of air and the air flow rate decreases drastically because of frictional forces.109
As
a result, a stagnant air layer (air-side boundary layer) is formed at the interface. Chemical mass
transfer through this layer is attributed to molecular diffusion, which can be described by Fick’s
law:131
2
A A A
A A S S BL2
( , ) 1,
C r t C CD D r r r
t r r r
(Equation 3.1)
where CA (ng/cm3) is the vapor concentration in air at position r (cm) at PAS deployment time t
(d); r is the position on the radial coordinate originating in the center of the XAD-filled mesh
cylinder (Equation S3.1); rS (cm) is the radius of this cylinder and δBL (cm) is the thickness of the
stagnant air layer. DA (cm2/d) is the molecular diffusivity in bulk air.
3.3.2.2 Diffusion within the Porous PSM.
Within the PSM, it is assumed diffusion along the radial coordinate only occurs in the air-filled
macro-pores (i.e. negligible diffusion through solid phase). Different from diffusion in bulk air,
diffusion in porous media is retarded due to the more tortuous path and reduced area for
diffusion.132
Thus, diffusivity in the porous air phase (DPA, cm2/d) is related to the diffusivity in
bulk air and the void fraction (ε, unitless) of the PSM:132
DPA = DA·ε4/3
(Equation 3.2)
The behavior of molecules in the macro-pores is not only related to diffusion, but also affected
by the kinetics of reversible sorption of the vapor in the macro-pores to the XAD pellets. A mass
balance equation for molecules in the macro-pores subject to these two processes is:
2
SA A A
PA PA S2
( , ) 1, 0
CC r t C CD D r r
t r r r t
(Equation 3.3)
where CA (ng/cm3) is the concentration in the air-filled macro-pores, ρ (g/cm
3) is the density of
bulk XAD, and CS (ng/g) is the mass concentration in the XAD pellets.

60
3.3.2.3 Chemical Exchange between Air-filled Macro-pores and XAD Pellets
This process can be represented by the chemical equation109
:
(Equation 3.4)
where M represents the gas phase molecule in the macro-pores, S represents the polymeric
sorbent, and M···S represents the sorbed molecule. Due to the large amount of meso-pores within
XAD, the sites available for sorption can be assumed to be constant and Equation 3.4 can be
simplified to:133
(Equation 3.5)
where ksorb (d-1
) and kdes (d-1
) are the sorption and desorption rate constants, respectively. ksorb and
kdes are related to the equilibrium partition coefficient between the sorbent and air (KSA = ksorb /
kdes). Note that sorption/desorption as used here comprises also molecular diffusion through the
meso-pores within the XAD pellets. In principle, such meso-pore diffusion could be described
with an additional diffusion equation of spherical coordinates.134,135
The size of individual XAD
pellets is much smaller than that of the XAD-filled mesh cylinder. During passive air sampling,
the diffusion path through the bulk XAD-filled mesh cylinder is much longer than the
intraparticle diffusion path. Thus, intraparticle diffusion should have a trivial effect on the
overall mass transfer kinetics. Furthermore, due to the lack of information on chemical transfer
within XAD pellets, for the purpose of this study, the kinetics of mass transfer processes within
XAD pellets were integrated to ksorb and kdes. Similar approaches have been adopted to model
sorption of chemicals to sorbents such as activated carbons and sediments.133,136
Applying the
law of mass action to the pseudo first order reaction (Equation 3.5),133,137
the mass balance of
chemical sorbed to the PSM can be quantified by:
S A
S S
( , ), 0
sorb
des
C r t k Ck C r r
t
(Equation 3.6)

61
3.3.2.4 Model Solution
By replacing the spatial derivatives with finite differences (200 nodes in the PSM and 50 nodes
in the stagnant air layer, which is illustrated in Equation S3.1 and Equation S3.2), the partial
differential equations (Equation 3.1, 3.3, 3.6) become a system of ordinary differential equations
(details in the SI), which can be solved numerically under the initial (t = 0) and
boundary/interfacial (r = 0; r = rS; r = rS + δBL) conditions described in the SI. The model outputs
chemical concentrations in the stagnant air layer, in the macro-porous air phase and in the PSM
as a function of space and time. By spatially integrating the concentrations, the amount
accumulated in the PSM at any time point can be derived. Following the practice of PAS field
calibrations,87
in which a PSR is often derived as the slope of the linear regression between the
equivalent air volume and the length of deployment, we calculated model-derived PSRs by
selecting six equally spaced time points from zero to the maximum deployment time and
applying a linear fit (forced through the origin) with the corresponding equivalent sampling
volumes retrieved from the model output (example in Equation S3.3). Note that these model-
derived PSRs are not the same as the theoretical ('intrinsic') PSRs defined in Bartkow et al.,83
which are presumed constant for a given set of conditions (e.g. diffusivity in air, boundary layer
thickness, temperature). In fact, the instantaneous PSR is always changing, even during the so-
called linear uptake phase.
3.3.3 Sensitivity Analysis
To investigate which of the three chemical mass transfer processes involved in passive air
sampling is more influential on the PSR (m3/d), we performed a sensitivity analysis on the PSR
(90 d deployment time) by varying by ±10% one of the parameters (δBL, DA, DPA, KSA and ksorb)
governing the three mass transfer processes. Note that although DA, DPA, and KSA are correlated
(Figure S3.5), we only varied one parameter at a time in order to reveal the influential processes
and parameters. Sensitivity coefficients (SC) were calculated as SC = (Δy / y) / (Δx / x) = [(y+ –
y–) / (y+ + y–)] / [(x+ – x–) / (x+ + x–)], in which x and y are the model input and output,
respectively and the subscripts + and – designate values with the model input parameters
increased and decreased by 10 %, respectively. Because the influence of a model parameter on
model results is often dependent on the value of that and other parameters, we conducted a
global sensitivity analysis considering combinations of the parameters varying over a wide, but
reasonable range. KXAD/A and KPUF/A for specific chemicals have been well established.38,119
The

62
ranges selected for KXAD/A (106 ≤ KXAD/A ≤ 10
10) and KPUF/A (10
5 ≤ KPUF/A ≤ 10
9) cover most
SVOCs (on average, KXAD/A are larger than KPUF/A by ~100.8
; Figure S3.4).38,126
The ranges of DA
and DPA are determined by the KSA range due to the correlation between these parameters
(Equation S3.5 and Equation 3.2). A previous study established that the thickness of the air
boundary layer surrounding a cylinder of 2 cm diameter ranged between 0.1 and 0.01 cm under
wind speeds between 1 and 10 m/s.138
Since wind speeds can exceed 10 m/s, we selected 0.01 cm
as the base case for δBL and investigated the δBL range between 0.001 and 0.1 cm. Because no
empirical information on likely values of ksorb for sorption of SVOCs onto XAD or PUF existed,
we chose a range of ksorb values (104 d
-1 ≤ ksorb ≤ 10
9 d
-1) for which the model yields PSRs that
match the range of those measured in calibration studies. ksorb values reported for the sorption of
VOCs onto activated carbon also fall within this range.136
Adsorption is generally believed to be the primary process for the retention of chemicals in
polymers at temperatures below their glassy state transition temperature (Tg).139
The Tg of XAD
is above 100 °C,140
which suggests that adsorption is dominant at environmentally relevant
temperatures. However, according to the dual-mode sorption theory, dissolution (partition) of the
chemical into the polymer could also occur at temperatures below Tg.141
Even though the
contribution of partition/absorption relative to adsorption is likely to be very small, we do not
distinguish “adsorption” and “absorption” but describe the uptake of chemical from air to XAD
using the general term“sorption”.
3.3.4 Model Application
The model described above was applied to investigate the variation of PSRs with chemical
properties and temperatures. Such variations observed in field PAS calibrations20,87,89
are larger
than that can be explained by the two-film PAS model,9,83
which presumes that temperature
influences PSRs only via the influence on DA. In the model presented, PSR is a function of DA,
DPA, KSA, ksorb and kdes. These parameters vary between different chemicals and are temperature
dependent. The temperature dependence of DA and DPA is quantified by the Fuller-Schettler-
Giddings equation85
and the dependence of KSA can be quantified with the van’t Hoff equation
using a measured or predicted internal energy of sorption (ΔUSA).38,119
The temperature
dependence of ksorb and kdes can be described with the Arrhenius equation:
ksorb = A1 exp(–Ea+ / RT) (Equation 3.7)

63
kdes = A2 exp(–Ea– / RT) (Equation 3.7’)
where A1 and A2 are pre-exponential factors, Ea+ and Ea– are the activation energies of the
forward and backward reactions in Equation 3.5, R is the ideal gas constant, and T is absolute
temperature (K). E+, E– and ΔUSA are interrelated (illustrated in Figure S3.6) through:
ΔUSA = Ea+ – Ea–. (Equation 3.8)
Using the model, we also investigated the chemical uptake curve with the intention of explaining
a rapid decrease in the PSR of the PUF-PAS after the first few weeks of sampling.88,104
Lastly,
with the model, we calculated the penetration depths of chemicals in the PSM. Because there is a
lack of information on ksorb or kdes (and the parameters in Equation 3.7 and Equation 3.8) for
specific chemicals, model calculations were performed on a range of values.
3.4 Results and Discussion
3.4.1 Influence of Mass Transfer Processes and Associated Parameters on the Passive Air Sampling Rate.
The sensitivity analysis reveals how the chemical mass transfer through the stagnant air layer
surrounding the PSM (δBL, DA), the diffusive mass transfer in the macro-pores within the PSM
(DPA), and the reversible sorption from the gas phase to the PSM (KSA and ksorb) influence the
PSR. The sensitivity of the PSR to model parameters was calculated and displayed in the
coordinate system defined by KSA and ksorb (referred to as sensitivity map hereafter) at different
δBL (Figure 3.2 and Figure S3.7). Based on the sensitivity (SC > 0.5) of the parameters, the
sensitivity map could generally be divided into four regions (shown in Figure 3.2f) for both
XAD-PAS and PUF-PAS for different assumed values of δBL.

64
Figure 3.2 Sensitivity (SC) of the sampling rate (PSR, m3/d) of the XAD-based passive air
sampler for compounds with different equilibrium partition coefficients between XAD and air
(KXAD/A) and different sorption rate constants (ksorb) to changes in (a) the thickness of the
stagnant air layer (δBL), (b) the molecular diffusivity in bulk air (DA), (c) the molecular
diffusivity in the macroporous fraction within the XAD (DPA), (d) KXAD/A, and (e) ksorb. δBL =
0.01 cm was used as the baseline for the SC calculations. Based on the other five panels, panel
(f) identifies four regions, in which the PSR is predominantly influenced by a particular mass
transfer process.
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
log KXAD/A
9
8
7
6
5
4
log
(k
so
rb/
d–
1)
9
8
7
6
5
4
9
8
7
6
5
4
6 7 8 9 10
6 7 8 9 10
6 7 8 9 10
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1
-0.8
-0.6
-0.4
-0.2
9
8
7
6
5
4
SC
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
9
8
7
6
5
4
9
8
7
6
5
46 7 8 9 10
I
IIIII
IV

65
In region I, PSRs are most sensitive to δBL and DA (Figure 3.2a and b). δBL and DA together
determine the mass transfer coefficient in the stagnant air layer (kA= DA / δBL), which explains
why the effects of δBL and DA on PSR are equal in magnitude but reverse in direction. In other
words, PSR would increase (decrease) equally either by increasing (decreasing) DA or decreasing
(increasing) δBL to the same extent. Therefore, hereafter we focus only on DA in our analysis of
mass transfer within the stagnant air layer. In this region, the kinetics of the overall mass transfer
is limited by the chemical diffusion through the stagnant air layer surrounding the PSM. Moving
from region I to region II, PSRs become less sensitive to DA and more sensitive to DPA (Figure
3.2c), indicating that the chemical mass transfer through the macro-pores within the PSM
becomes more influential on the PSRs. Chemicals in region I have high KSA and ksorb relative to
those in region II. The PSM has a high capacity for such chemicals, which therefore are more
likely to sorb to the outer layer of the PSM than to penetrate to the inside. Thus, DPA is less
influential on the PSR of chemicals in region I than of those in region II.
In region III, PSRs are most sensitive to a chemical’s KSA or the uptake capacity of the PSM
(Figure 3.2d). Chemicals in this region have relatively low KSA and high ksorb. The low PSM
capacities for the chemicals and the fast rates of sorption/desorption to the PSM allow surface
evaporation to play an important role in chemical mass transfer between air and the PSM.
Lowering the KSA increases the rate of chemical evaporation from the PSM and thus reduces the
PSR during the deployment period. In region IV, PSRs are most sensitive to ksorb. The reason is
that in this region, ksorb is low and the overall rate of mass transfer from ambient air to the PSM
is kinetically limited by the rate of sorption from the gas phase in the macro-pores to the solid
PSM material. In both regions III and IV, PSRs are also sensitive to DPA (Figure 3.2c). In region
III, an increased DPA facilitates penetration into the PSM, which competes with the surface
evaporation process that cause chemical loss from the PSM. In region IV, while the PSRs are
limited by the sorption kinetics, an increased DPA makes more of the sorbent deep inside the
PSM accessible for sorption, and thus increases the PSRs.
The boundaries between the four regions shift on the sensitivity map when the thickness of the
stagnant air layer (δBL) is changed due to, for example, a change in wind conditions (Figure
S3.7). The thicker the stagnant air layer, the larger the number of chemicals whose overall mass
transfer is controlled by the diffusion across the stagnant air layer. Thus, when δBL increases, the
sensitivity of PSRs to DA or δBL increases and the boundary between region I and II shifts

66
towards the centre of map. As the kinetic resistance to diffusion through the stagnant air layer
increases with increasing δBL, so does the kinetic resistance to evaporation from the PSM. This
decreases the influence of surface evaporation on the PSRs for chemicals for which this process
is important (i.e., chemicals with low KSA). Therefore, region III shifts towards the lower KSA
with increased δBL. As the kinetics for chemical crossing the stagnant air layer becomes more
influential to the overall uptake, the sorption rate would have to be lower in order to kinetically
limit the overall uptake process. Thus, region IV shifts towards lower ksorb as δBL increases.
Comparing the sensitivity maps based on the models for XAD-PAS and PUF-PAS (Figure S3.7),
region I for the PUF-PAS extends to lower KSA and lower ksorb than for the XAD-PAS, indicating
that the stagnant air layer resistance (DA/δBL) is more important in determining the PSR in PUF-
PAS than in XAD-PAS. This difference could be due to differences in the configuration
(cylindrical XAD-resin filled mesh cylinder vs. planar PUF sheet) and/or physical properties
(density and macro-pore fraction) of the PSM. To investigate the contributions of these two
factors, we conducted a sensitivity analysis using models in which the cylindrical XAD-filled
mesh cylinder in the XAD-PAS model was replaced by a cylindrical PUF of the same dimension
or the PUF disk in the PUF-PAS model was changed to an XAD disk. The sensitivity maps
based on the modified models (Figure S3.8) indicate that it is the density and porosity (Table
S3.1) of the PSMs rather than their geometrical arrangement that explains the difference in the
importance of the stagnant boundary layer.
A specific chemical with known KSA and ksorb is represented by a point on the sensitivity maps.
KSAs for both XAD and PUF have been well characterized and are generally positively correlated
with molecular size. So far, information on the rates of sorption and desorption (i.e., ksorb and
kdes) to PSM is lacking. However, based on the theory of mass transfer between a fluid and a
single spherical particle,142
ksorb is positively correlated with the molecular diffusivity in the fluid
and thus negatively correlated with the molecular volume. Therefore, KSA is presumably
negatively correlated with ksorb. As such, points representing real chemicals on the sensitivity
map are more likely to distribute within a belt from the top left to the bottom right and the
likelihoods for a chemical to have both high (low) KSA and high (low) ksorb (i.e. distributed in the
upper right and lower left region of the map) are low.

67
3.4.2 Influence of Chemical Properties and Temperatures on Passive Air Sampling Rates.
Figure 3.3 Illustration of the dependence of passive sampling rates (PSRs) on chemical
properties and temperature. Molecular size: M1 > M2; temperature T1 < T2. The map depicting
PSRs in the KSA-ksorb chemical space was constructed based on the model for a XAD-passive air
sampler deployed for 360 d assuming a stagnant air boundary layer thickness δBL of 0.01 cm.
PSRs exceeding 5 m3/d were calculated for the combination of large KSA and large ksorb (hatched
area), which is unlikely to exist among real chemicals.
log KSA
log
(kSo
rb/
d–1
)
6 7 8 9 1010
4
105
106
107
108
109
ksorb(d
-1)
log KSA
0.250
1.25
2.25
3.25
4.25
5.00
6 7 8 9 1010
4
105
106
107
108
109
k sorb(d
-1)
log KSA
0.2500.5000.7501.001.251.501.752.002.252.502.753.003.253.503.754.004.254.504.755.00
0
1
5
0.5
1.52
2.5
3
3.5
44.5
PSR (m3/d)
6 7 8 9 10
9
8
7
6
5
4
T1
T2
T1
T2
M1
M2
M1
M2

68
In order to explain the variation of PSRs with temperature and between different compounds, we
constructed a chemical space map displaying the PSR calculated for different combinations of
KSA and ksorb. As an illustration, the chemical space map showing the PSR for uptake in an XAD-
PAS deployed for 360 d assuming δBL= 0.01 cm is shown in Figure 3.3. A color scale represents
the PSR with changes in KSA and ksorb. Maps with lower and higher wind exposure of the XAD-
PAS (δBL= 0.1 and 0.001cm) are presented in Figure S3.9. Recall that KSA is correlated with DA
and DPA (Figure S3.5) and thus the maps integrate the variation of DA and DPA with KSA. The
maps display L-shaped strips of different colors, each representing a range of PSRs. The dashed
line connecting the inflection points of all the L-shaped strips divides the map into two regions.
On the upper-left (lower-right) region, the strips are generally parallel with the y-axis (x-axis),
indicating PSRs for chemicals in this region are sensitive to changes in KSA (ksorb) but not
sensitive to the changes in ksorb (KSA). This pattern of PSRs in the chemical space map agrees
with the results of the sensitivity analysis (Figure 3.2d and e).
The hatched area on the top right of the map represents a PSR above 5 m3/d, which has rarely
been observed in a field calibration using the XAD-PAS. As mentioned before, KSA is generally
negatively correlated with ksorb; smaller chemicals in a homologous series tend to have a low KSA
and a high ksorb. Therefore, points representing a homologous series on the map would distribute
along a line from the bottom right to the top left and a point representing a chemical on the
chemical space map would shift towards the upper left (lower right) at higher (lower)
temperatures. As indicated by the model results, different chemicals or a chemical at different
temperatures would be expected to have different PSRs. Depending on the chemical properties
(KSA and ksorb), the direction in which the PSR changes with chemical properties and
temperatures can be different. For chemicals in the lower right of the map (high KSA and low
ksorb), the PSR for a homologous series decreases with increasing molecular size (M1 > M2 in
Figure 3.3). In contrast, if the sorption of the chemicals to the sorbent is fast (ksorb is large) so that
the chemicals are positioned on the upper left, a chemical with large molecular size (M1) would
have a higher PSR than a smaller chemical (M2). Similarly, the variation of the PSR with
temperature can be different depending on a chemical’s KSA and ksorb, i.e. its position in the map.
PSRs for chemicals positioned in the lower right (upper left) increase (decrease) as temperature
increases (from T1 to T2 as illustrated in Figure 3.3).

69
The model facilitates a mechanistic explanation of this seemingly contradictory behavior. The
PSR of chemicals in the lower right is kinetically controlled by the sorption process and the
diffusion within the PSM. As KSA decreases and ksorb increases with a decrease in molecular size
or an increase in temperature, the fraction in the air-filled pores increases, facilitating the
penetration into the PSM. The increased diffusivity within the PSM and the increased sorption
rate (ksorb) will increase the PSR. For chemicals in the upper left region, ksorb is large so that
chemicals accumulate rapidly at the PSM surface. Increased chemical accumulation at the
surface with increased ksorb and the decreased sorbent capacity (KSA) would enhance the role that
surface evaporation plays in decreasing the PSR.
Many studies on passive air sampling have observed the variation of PSRs among different
chemicals or for a chemical at different temperatures.20,87,89,90
For the XAD-PAS, PSRs were
found to be positively correlated with DA but the variation in PSR was much larger than that of
DA.20,89
For the PUF-PAS, the relationship between PSRs and DA seems more complicated. Some
studies observed a negative correlation of PSRs with DA: higher PSRs for chemicals at lower
temperatures and for highly chlorinated biphenyls.87,90
On the contrary, lower PSRs for highly
brominated diphenyl ethers have also been observed.104
One explanation for the variation of
PSRs in PUF-PAS is a shift in the gas-particle distribution of the target SVOCs:87
at lower
(higher) temperature or for a heavier (lighter) congener, a higher (lower) fraction of the
chemicals would be in the particle phase so the amount of chemicals in the gas phase available
for uptake decreases (increases). Considering a lower sampling efficiency for particle-bound
chemicals than for chemicals in the gas phase, PSR calibrated against bulk air concentrations
becomes lower (higher). Although this process could possibly affect the PSR of SVOCs with
very low volatility (e.g. highly brominated diphenyl ethers), it cannot explain the PSR variation
for chemicals predominantly in gas phase (e.g. tri- and tetra-chlorinated biphenyls). Comparing
the field observations with the map in Figure 3.3, it seems that a model that considers the kinetic
resistance within the PSM can explain the observed PSR variations for both XAD-PAS and PUF-
PAS. The observed behavior of the XAD-PAS agrees with the case on the bottom right of the
map (higher KSA and lower ksorb) and that of the PUF-PAS follows that in the upper left region
(lower KSA and higher ksorb). While KSA for XAD has been found to be generally higher than that
for PUF, no information on ksorb for XAD or PUF is currently available. Because sorption to

70
XAD involves diffusion through the meso-pores within each pellet, which may limit the sorption
kinetics, ksorb for XAD is presumably lower than for PUF.
3.4.3 Two-Stage Uptake Process.
In the PUF-PAS calibration studies by Chaemfa et al.88
and by Tsurukawa et al.,128
some
chemicals are observed to have a high PSR initially (~1-2 weeks) after which the PSR drops and
stays relatively constant. A two-stage uptake was hypothesized to explain such an observation
but no quantitative studies were conducted and no mechanistic explanation was formulated. We
used the model to construct the uptake curves for chemicals with different combinations of KSA
and ksorb (Figure S3.10). Two-stage uptake was predicted for chemicals with high ksorb. Sorption
of such chemicals to the surface of the PSM is faster than diffusive penetration into the PSM.
Initially, sorption occurs at the surface of the PSM, the resistance to diffusion in the PSM pores
is not rate-controlling, and the PSR is determined by the fast sorption rate. As the PSM surface
becomes saturated, the chemical either evaporates or diffuses into the PSM. The overall uptake
kinetics is then determined by the rate of diffusion into the PSM, which leads to a decreased PSR
compared with the initial uptake phase. Although the model can mechanistically explain a two-
stage uptake, we are unable to use the model to predict the uptake for specific compounds due to
the of lack information on ksorb.
3.4.4 Non-Uniform Chemical Distribution within Passive Sampling Media.
The model describing the mass transfer processes within PSMs is capable of calculating the
radial distribution of chemicals within the PSM. Agreeing with experimental evidence,(Chapter
2)126
the model calculations for the KSA-ksorb chemical space indicate a non-uniform chemical
distribution within the PSMs. For the majority of KSA and ksorb combinations, >90% of the
amount of chemical accumulated in the PSM of both XAD- and PUF-PAS are constrained within
a surface layer of less than 0.4 cm thickness after 90 d of deployment (Figure S3.11). Our
experiments investigating chemical distribution within the PSMs were based on cylindrical PSM
configurations (Chapter 2)126
. The mass transfer of chemicals within such cylindrical PSMs
might be retarded as the cross-sectional area for diffusion decreases from the outer to the inner
part of the PSM. This could possibly add some uncertainty when extrapolating the experimental
results based on PSMs of cylindrical configuration to those of planar configuration (disk). Model

71
calculations for PSMs in both cylindrical and planar configurations (Figure S3.11) revealed no
obvious differences in the penetration depth (defined as the thickness of the outer PSM layer
which accumulates 90% of the sampled amount). This indicates that the non-uniform distribution
within the PSM is mainly determined by the competition between sorption and diffusion deeper
into the PSM rather than the PSM configuration.
3.4.5 Knowledge Gap and Implications.
In this study, the new model was primarily used to provide mechanism insight into some field
observations that could not be explained with the two-film PAS theory. The new model’s
capability to describe the behavior of specific chemicals is mainly limited by the lack of
quantitative information on ksorb, either from measurements or predictions. Unlike other model
parameters such as DA, DPA and KSA, which either have been measured or can be predicted with
established theories for SVOCs,38,109,119
ksorb has only been studied for the sorption of some
VOCs (e.g. benzene, carbon tetrachloride) on a few sorbents other than XAD or PUF.136,143
In
order to quantitatively describe specific chemicals and to expand the application of the model,
ksorb and its temperature dependence need to be quantified for SVOCs and the PSMs commonly
used in PASs. Once ksorb is available for specific chemicals, this model could be used to predict
chemical specific PSRs at different temperatures. The model results on the chemical distribution
within the PSMs could also be useful when optimizing the design of PASs with the intention of
improving the sampling efficiency. For example, increasing the interfacial area/volume ratio of a
PSM would prevent the sorbent in the inner part of the PSM from being wasted.
In field applications of PASs, PSRs are often determined from calibrations against active air
samplers instead of being calculated from PAS theory. Therefore, although both experimental
(Chapter 2) 126
and modeling evidence (this study) indicates non-uniform chemical distribution
within porous PSMs, which contradicts the assumption in the two-film approach, interpretation
of PAS data using empirically derived PSRs will not be affected, as long as PSRs are used that
are compound- and sampling site specific. The two-film PAS theory has previously been used to
estimate linear uptake ranges9,38,87
or to calculate PSRs from the observed loss of depuration
compounds.95
By neglecting to consider the kinetic resistance within the PSM, linear uptake
ranges tend to be overestimated because deeper parts of the PSM are not readily accessible. This
agrees with observed linear uptake ranges that are shorter than estimated for heavier compounds

72
that do not penetrate readily into the PSMs.95
PSRs, through their dependence on KSA and ksorb,
are clearly compound-specific, and the applicability of a PSR obtained for one type of
(depuration) compound to another cannot be assumed but would need to be demonstrated. Even
if depuration compounds are isotopically labeled analogs of the target compounds, PSRs derived
from the loss of depuration compounds (without accounting for a kinetic resistance within the
PSM) may deviate from the PSRs of chemicals sampled from air because their distributions
within the PSM are different and thus result in different kinetic resistances within the PSM.
3.5 Acknowledgments
We acknowledge research funding from the Canadian Foundation for Climate and Atmospheric
Sciences and the Natural Sciences and Engineering Research Council of Canada. X. Zhang
acknowledges financial support from an Ontario Graduate Scholarship.

73
Supporting Information of Chapter 3
Mathematical Model of Chemical Uptake by XAD-PAS.
Discretization of Partial Differential Equations. As illustrated in Figure S3.1, the governing
partial differential equations (Equation 3.1, 3.3, 3.6) of the model can be discretized on the
spatial derivative with finite differences into a system of ordinary differential equations:
1 1 1 1
A A A A A A
A A2
A S A A
2 1, 1 1
( ) 2
i i i i i idC C C C C C
D D m i m ndt r i m
(Equation S3.1)
1 1 1 1
sorbA A A A A A
PA PA A des S2
S S S
2 1, 1 1
2
i i i i i i
i ikdC C C C C CD D C k C i m
dt i(Equation S3.2)
S sorb
A des S , 1 1
i
i idC kC k C i m
dt (Equation S3.3)
The system of ordinary differential equations can be solved with information on initial
conditions and boundary/interfacial conditions.
Initial Conditions. Initially (t = 0), the chemical concentration in the air surrounding the PSM
(XAD mesh cylinder) equals the ambient air concentration (CAA, ng/cm3):
A AA S S( , 0) ,
BLC r C r r r (Equation S3.4)
or A AA
(0) , 1 i
C C m i m n (Equation S3.4’)
Chemicals initially in the PSM (e.g. deliberately spiked depuration compounds) are assumed to
be uniformly distributed within the PSM and to have reached equilibrium between PSM and
macroporous air:
0
A S
S SA
( , 0) , 0(1 - )
nC r r r
V K (Equation S3.5)
or 0
A
S SA
(0) , 0(1 - )
i nC i m
V K (Equation S3.5’)

74
0 SA
S S
S SA
(1 - )( , 0) , 0
(1 - )
n KC r r r
V K (Equation S3.6)
or 0 SA
S
S SA
(1 - )(0) , 0
(1 - )
i n KC i m
V K (Equation S3.6)
where VS (cm3) is the volume of the PSM. For depuration compounds spiked to the PSM at the
beginning of a passive air sampling campaign, n0 (ng) is the initial amount within the PSM. For
the target chemicals sampled by the PAS, n0 equals the blank level of the chemicals in the PSM.
Assuming blank levels are negligible, Equation S3.5and Equation S3.6 become:
A S S( , 0) ( , 0) 0 , 0 C r C r r r (Equation S3.7)
or A S
(0) (0) 0 , 0 i i
C C i m (Equation S3.7’)
Boundary/Interfacial Conditions. At the cylindrical axis of the XAD mesh cylinder (r = 0 or i =
0), because of symmetry, there is no radial flux, therefore:
A(0, )
0 ,
C tt
r (Equation S3.8)
From Equation 3.3 and Equation S3.6,
SA(0, )(0, )
C tC t
t t (Equation S3.9)
or 00
0 0SA
sorb A des S
dCdCk C k C
dt dt (Equation S3.9’)
At the interface between the PSM and stagnant air (r = rS or i = m), applying a mass balance
equation to the macroporous air phase of an imaginary layer between rS – 0.5δS and rS + 0.5δA,
we obtain:
1 1
A A A A A
A PA sorb A des S
A A
( )2
m m m m m
m mS A
S
dC C C C CD D k C k C
dt (Equation S3.10)

75
S sorb
A des S
m
m mdC kC k C
dt (Equation S3.11)
At the boundary of the stagnant air layer (r = rS + δBL or i = m + n), applying a mass balance
equation to an imaginary layer between rS + δBL – 0.5δA and rS + δBL + 0.5δA, we get:
1
A AA A A
A 2
A
2
m n m n m ndC C C C
Ddt
(Equation S3.12)
An ordinary differential equation system (A
iC and
S
jC as dependents of t, where i = 0…m + n; j =
0…m) composed by Equation S3.1 to Equation S3.8 can be solved numerically to get A
iC and
S
jC at a given time t.
Figure S3.1 Illustration showing the discretization of the PSM of the XAD-PAS to solve the
diffusion equations. m = 200 and n = 50 were used in this study.
01
i=-1
m-1
m+1m
2
m+n-1m+n
rS
δBL
2 cm
10
cm

76
Mathematical Model of Chemical Uptake by PUF-PAS.
Diffusion and Sorption/Desorption Equations. Different from the XAD-PAS, whose PSM is
cylindrical, the widely used PUF-PAS has a PUF disk as the PSM. Chemical mass transfer
within the PUF disk can be modeled using the diffusion equations for a planar sheet. The
equations corresponding to Equation 3.1, 3.3, 3.6 are:
2
A A
A BL2
( , ),
C z t CD L z L
t z (Equation S3.13)
2
SA A
PA 2
( , ), 0
CC z t CD z L
t z t (Equation S3.14)
S A
S
( , ), 0
sorb
des
C z t k Ck C z L
t (Equation S3.15)
where L (cm) is half of the thickness of the PUF disk; z (cm) is the position at the coordinate
originated from the half thickness of the PUF disk (Figure S3.2).
Equation S3.13, Equation S3.14, and Equation S3.15 can be discretized as:
1 1
A A A A
A 2
A
2, 1 1
i i i idC C C C
D m i m ndt
(Equation S3.16)
1 1
sorbA A A A
PA A des S2
S
2, 1 1
i i i i
i ikdC C C CD C k C i m
dt (Equation S3.17)
S sorb
A des S , 1 1
i
i idC kC k C i m
dt (Equation S3.18)
Initial Conditions. Initially (t = 0), chemical concentration in the air surrounding the PUF disk
equals the ambient air concentration (CAA, ng/cm3):
A AA( , 0) ,
BLC z C L z L (Equation S3.19)
or A AA
(0) , 1 i
C C m i m n (Equation S3.19’)

77
Chemicals initially in the PUF are assumed uniformly distributed and reach equilibrium between
PUF and macroporous air:
0
A
S SA
( , 0) , 0(1 - )
nC z z L
V K (Equation S3.20)
or 0
A
S SA
(0) , 0(1 - )
i nC i m
V K (Equation S3.20’)
0 SA
S
S SA
(1 - )( , 0) , 0
(1 - )
n KC z z L
V K (Equation S3.21)
0 SA
S
S SA
(1 - )(0) , 0
(1 - )
i n KC i m
V K (Equation S3.21’)
where VS (cm3) is the volume of the PUF disk; For depuration compounds spiked to the PUF at
the beginning of passive air sampling campaign, n0 (ng) is the amount of depuration compounds
within the PUF initially. For the target chemicals sampled by the PAS, n0 equals the blank level
of the chemicals on the PUF. Assuming blank levels are negligible, Equation S3.20 and Equation
S3.21 become:
A S S( , 0) ( , 0) 0 , 0 C r C r r r (Equation S3.22)
or A S
(0) (0) 0 , 0 i i
C C i m (Equation S3.22’)
Boundary/Interfacial Conditions. At the half depth of the PUF disk (z = 0 or i = 0), because of
symmetry, there is no radial flux, therefore:
A(0, )
0 ,
C tt
z (Equation S3.23)
From Equation S3.14 and Equation S3.23,
SA(0, )(0, )
C tC t
t t (Equation S3.24)
or 00
0 0SA
sorb A des S
dCdCk C k C
dt dt (Equation S3.24’)

78
At the interface between the PUF and stagnant air (z = L or i = m), applying a mass balance
equation to the macroporous air phase of an imaginary layers between L – 0.5δS and L + 0.5δA,
we have:
1 1
A A A A A
A PA sorb A des S
A A
( )2
m m m m m
m mS A
S
dC C C C CD D k C k C
dt (Equation S3.25)
S sorb
A des S
m
m mdC kC k C
dt (Equation S3.26)
At the boundary of the stagnant air layer (z = L + δBL or i = m + n), applying a mass balance
equation to an imaginary layers between L + δBL – 0.5δA and L + δBL + 0.5δA, we get:
1
A AA A A
A 2
A
2
m n m n m ndC C C C
Ddt
(Equation S3.27)
An ordinary differential equation system (A
iC and
S
jC as dependents of t, where i = 0…m + n; j =
0…m) composed by Equation S3.16, Equation S3.17, Equation S3.18, Equation S3.22, Equation
S3.20, Equation S3.21, Equation S3.24, Equation S3.25, Equation S3.26, and Equation S3.27 can
be solved numerically to get A
iC and
S
jC at a given time t.

79
Figure S3.2 Illustration showing the discretization of the PSM of the PUF-PAS to solve the
diffusion equations. m = 200 and n = 50 were used in this study.
14 cm
1.5 cm
L
L
i=0…
…
i=1
i=-1
i=m-1
i=m+1i=m
zi
…
i=m+n-1i=m+n
δS = rS /m
δA = δBL /n

80
Table S3.1 Properties of the modeled passive air sampling media
PAS
PSM Radius
(cm)
PSM
Height/Thickness
(cm)
Density
(g/cm3)
Void
fraction, ε
1 10 1.08 ref.130
0.45 ref.130
14 1.5 0.0213 ref.9
0.97 ref.119

81
Figure S3.3 Illustration of how passive air sampling rates (PSRs) were derived from a linear
fit on six discrete data points placed equidistantly on the uptake curve generated by the model.
Figure S3.4 Distribution of the difference between KXAD/A and KPUF/A for chlorothalonil,
endosulfan I, endosulfan II, atrazine, alachlor, metolachlor, trifluralin, HCB, α-HCH, γ-HCH and
209 PCB congeners based on calculations using polyparameter linear free energy relationships
(ppLFERs).38,111,119
0 10 20 30 40 50 60 70 80 90
0
20
40
60
80
100
120
140
160
180
200
PAS deployment time (d)
Equiv
ale
nt
sa
mplin
g vo
lum
e (
m3) Fit line based on the data points
PAS sampling rate = slope =2.3 m3/d
-0.4 0.0 0.4 0.8 1.2 1.6 2.0 2.40
10
20
30
40
50
Num
be
r o
f o
bse
rva
tio
n
μ=0.800, σ=0.386
log KXAD/A - log KPUF/A

82
Figure S3.5 Empirical relationships of XAD/air partition coefficient (KXAD/A) and PUF/air
partition coefficient (KPUF/A)with the diffusivity of chemicals in air (DA, cm2/s) based on 209
polychlorinated biphenyl congeners and 10 organochlorinated pesticides (namely, chlorothalonil,
endosulfan I, endosulfan II, atrazine, alachlor, metolachlor, trifluralin, HCB, α-HCH, and γ-
HCH). KXAD/A and KPUF/A of the chemicals were calculated using polyparameter linear free
energy relationships (ppLFERs).38,111,119
DA was calculated using the Fuller-Schettler-Giddings
equation with La Bas molar volumes.109
7.0 8.0 9.0 10.0 11.00.030
0.035
0.040
0.045
0.050
0.055
log KXAD/A
DA
/ (
cm2 s
-1)
DA /(cm2s-1)= -0.0041logKXAD/A+0.0781
R2 = 0.821
T = 281K
6.0 7.0 8.0 9.0 10.0
0.038
0.042
0.046
0.050
0.054
0.058DA /(cm2s-1)= -0.0044logKXAD/A+0.0822
R2 = 0.825
T = 293K
5 6 7 8 9 10 11 120.034
0.038
0.042
0.046
0.050
0.054
4 5 6 7 8 9 10 11
0.038
0.042
0.046
0.050
0.054
0.058
T = 281K T = 293K
DA /(cm2s-1)= -0.0024logKPUF/A+0.0623
R2 = 0.762
DA /(cm2s-1)= -0.0028logKPUF/A+0.0667
R2 = 0.763
log KPUF/A

83
Figure S3.6 Illustration of the relationship between the change of internal energy (ΔUSA,
from air phase M to sorbed phase M···S) and the activation energies of sorption (Ea+) and
desorption (Ea–).
M
M···S
Ea+
Ea–
ΔUSAPo
ten
tial
En
ergy

84
Figure S3.7 Sensitivities of passive air sampling rate (m3/d) of XAD-PAS (left) and PUF-
PAS (right) (deployed for 90 d) to changes of molecular diffusivity in bulk air (DA), molecular
diffusivity in the macroporous fraction within the PSM (DPA), equilibrium partition coefficient
between the sorbent and air (KSA), and the sorption rate constant (ksorb) based on stagnant
boundary layer thickness δBL of 0.001 cm (top), 0.01 cm (centre), and 0.1 cm (bottom).
δB
L=
0.0
1 c
mδ
BL
= 0
.00
1 c
mδ
BL
= 0
.1 c
m
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
log KXAD/A
9
8
7
6
5
4
log
(kSo
rb/
d–1
)
9
8
7
6
5
4
9
8
7
6
5
4
6 7 8 9 10
6 7 8 9 10 6 7 8 9 10
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
log KXAD/A
9
8
7
6
5
4
log
(kSo
rb/
d–1
)
9
8
7
6
5
4
9
8
7
6
5
4
6 7 8 9 10
6 7 8 9 10 6 7 8 9 10
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
log KXAD/A
9
8
7
6
5
4
log
(kSo
rb/
d–1
)
9
8
7
6
5
4
9
8
7
6
5
4
6 7 8 9 10
6 7 8 9 10 6 7 8 9 10
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
XAD-PAS PUF-PAS
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
5 6 7 8 9
5 6 7 8 9 5 6 7 8 9
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
log
(kSo
rb/
d–1
)5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
DB
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
5 6 7 8 9
5 6 7 8 9 5 6 7 8 9
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
log KPUF/A
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
5 6 7 8 9
5 6 7 8 9 5 6 7 8 9
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
log KPUF/A
log KPUF/A
log
(kSo
rb/
d–1
)lo
g (k
Sorb
/ d–1
)

85
Figure S3.8 Comparison between cylindrical and disk-like PSM configurations for the
sensitivities of passive air sampling rate (m3/d) to the changes of in bulk air (DA), molecular
diffusivity in the macroporous fraction within the media PSM (DPA), equilibrium partition
coefficient between the sorbent and air (KSA), and the sorption rate constant (ksorb) at a stagnant
boundary layer thickness δBL of 0.01cm.
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
δBL = 0.01 cm
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
6 7 8 9 10
log KXAD/A
9
8
7
6
5
4
log
(kSo
rb/
d–1
)
9
8
7
6
5
4
9
8
7
6
5
4
6 7 8 9 10
6 7 8 9 10 6 7 8 9 10
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
log
(kSo
rb/
d–1
)
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
5 6 7 8 9
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
5 6 7 8 9
5 6 7 8 9 5 6 7 8 9
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
log KPUF/A
log
(kSo
rb/
s–1
)
5 6 7 8 9
log KPUF/A
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
5 6 7 8 9
5 6 7 8 9 5 6 7 8 9
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1
-0.8
-0.6
-0.4
-0.2
9
8
7
6
5
4
6 7 8 9 10
log KXAD/A
9
8
7
6
5
4
log
(kSo
rb/
d–1
)
9
8
7
6
5
4
9
8
7
6
5
4
6 7 8 9 10
6 7 8 9 10 6 7 8 9 10
5 6 7 8 9
10000
100000
1000000
1E7
1E8
1E9
D
B
-1.000-0.9000-0.8000-0.7000-0.6000-0.5000-0.4000-0.3000-0.2000-0.10000.0000.10000.20000.30000.40000.50000.60000.70000.80000.90001.000
0
0.2
1.0
0.4
0.6
0.8
-1.0
-0.8
-0.6
-0.4
-0.2
9
8
7
6
5
4
9
8
7
6
5
4
9
8
7
6
5
4
XAD
XAD
PUF
PUF

86
Figure S3.9 Modeled passive air sampling rates as a function of equilibrium partition
coefficient between the XAD and air KXAD/A and the sorption rate constant ksorb with stagnant air
layers of 0.1, 0.01, and 0.001 cm thickness.
Figure S3.10 Modeled chemical uptake curve in passive air sampling of chemicals with
different combinations of KPUF/A and ksorb.
6 7 8 9 1010
4
105
106
107
108
109
k sorb(d
-1)
log KSA
0.2500.5000.7501.001.251.501.752.002.252.502.753.003.253.503.754.004.254.504.755.00
6 7 8 9 100
1
5
0.5
1.52
2.5
3
3.5
44.5
log KXAD/A
log
(kSo
rb/
d–1
)
δBL = 0.01 cm
6 7 8 9 10
δBL = 0.001 cm9
8
7
6
5
46 7 8 9 10
δBL = 0.1 cm R (m3/d) 9
8
7
6
5
4
9
8
7
6
5
46 7 8 9 10
104
105
106
107
108
109
ksorb(d
-1)
log KSA
0.2500.5000.7501.001.251.501.752.002.252.502.753.003.253.503.754.004.254.504.755.00
6 7 8 9 1010
4
105
106
107
108
109
kso
rb(d
-1)
log KSA
0.250
1.25
2.25
3.25
4.25
5.00
6 7 8 9 1010
4
105
106
107
108
109
kso
rb(d
-1)
log KSA
0.2500.5000.7501.001.251.501.752.002.252.502.753.003.253.503.754.004.254.504.755.00
0
20
40
60
80
100
120
140
160
180
0 30 60 90Deployment time (d)
Eq
uiv
ale
ntsam
plin
g v
olu
me (
m3)
(8, 7)(8, 8)
(7, 6)
(8, 6)(9, 6)(10, 6)
(8, 9)
(logKSA, logksorb)

87
Figure S3.11 Penetration depth (defined as the thickness of outer sampling medium layer
which accumulates 90% of the sampled chemical amount) of chemicals in XAD and PUF, both
in cylindrical and in disk configuration.
t = 90 d
XAD
XAD
PUF
PUF
9
8
7
6
5
46 7 8 9 106 7 8 9 10
4
5
6
7
8
9
B
A
0.00.0500.100.150.200.250.300.350.400.450.500.550.600.650.700.750.800.850.900.951.0
5 6 7 8 9
4
5
6
7
8
9
B
A
0.00.0500.100.150.200.250.300.350.400.450.500.550.600.650.700.750.800.850.900.951.0
6 7 8 9 10
4
5
6
7
8
9
B
A
0.00.0500.100.150.200.250.300.350.400.450.500.550.600.650.700.750.800.850.900.951.0
9
8
7
6
5
45 6 7 8 9
9
8
7
6
5
46 7 8 9 10
9
8
7
6
5
45 6 7 8 9
6 7 8 9 10
4
5
6
7
8
9
B
A
0.0
0.050
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
0.75
0.80
0.85
0.90
0.95
1.0
0
0.1
0.4
0.2
1.0
0.7
0.6
0.5
0.3
0.8
0.9
Penetration depth (cm)
5 6 7 8 9
4
5
6
7
8
9
B
A
0.00.0500.100.150.200.250.300.350.400.450.500.550.600.650.700.750.800.850.900.951.0
log KXAD/A
log
(kSo
rb/
d–1
)
log KPUF/A
log
(kSo
rb/
d–1
)
log KXAD/A
log
(kSo
rb/
d–1
)
log KPUF/A
log
(kSo
rb/
d–1
)

88
Chapter 4. Influence of Sampler Configuration on the Uptake Kinetics of a
Passive Air Sampler
Xianming Zhang, Cindy Wong, Ying D. Lei, Frank Wania
Environmental Science & Technology 2012, 46, 397-403.
Contributions: X. Zhang designed the experiments under the guidance of F. Wania. X. Zhang
conducted the water uptake experiment. X. Zhang supervised C. Wong to conduct the calibration
experiment. Y.D. Lei provided assistance during instrumental analysis and in making the mesh
cylinders of different diameters. X. Zhang processed the chromatograms, interpreted the data.
Under the guidance of F. Wania, X. Zhang wrote the manuscript, revised it and responded to
reviewers’ comments.
Reproduced with permission from Environmental Science and Technology
Copyright 2012 American Chemical Society
Precision Balance
Passive Sampler housing
Silica gel Packed Mesh
Cylinder

89
4.1 Abstract
Passive air samplers (PAS) are simple and cost-effective tools to monitor semi-volatile organic
compounds in air. Chemical uptake occurs by molecular diffusion from ambient air to a passive
sampling medium (PSM). Previous calibration studies indicate that even for the same type of
PAS, passive air sampling rates (R, m3
air/d) can be highly variable due to the influence of a
number of factors. Earlier studies mainly focused on factors (e.g. wind speed and temperature)
influencing R via the kinetic resistance posed by the air boundary layer surrounding the PSM
because that layer was deemed to be the main factor determining the uptake kinetics. Whereas
recent calibration studies suggest that the PAS configuration can influence R, so far few studies
have specifically focused on this factor. In this study, with the objective to understand the effect
of PAS configurations on R, we applied a gravimetrical approach to study the uptake kinetics of
water vapor from indoor air in silica gel placed inside cylindrical PAS of various configurations.
We also conducted an indoor calibration for polychlorinated biphenyls on the same type of PAS
using XAD-resin as the PSM. R was found to be proportional to the interfacial transfer area of
the PSM but not the amount of the PSM because chemicals mainly accumulated in the outer
layer of the PSM during the deployment time of the PAS. The sampler housing and the PSM can
introduce kinetic resistance to chemical uptake as indicated by changes in R caused by
positioning the PSM at different distances from the opening of the sampler housing and by using
PSM of different diameters. Information gained from this study is useful for optimizing the PAS
design with the objective to reduce the material and shipping costs without sacrificing sampling
efficiency.
4.2 Introduction
Passive air samplers (PAS) are finding widespread and increasing use in monitoring semi-
volatile organic contaminants (SVOC) in the air due to a number of advantages, which include
(i) the capability of extended time-integrated sampling; (ii) the independence from power
supplies and regular maintenance and (iii) the relatively low production and operational cost. As
a result, PAS have been widely applied in studies on SVOC in both outdoor31,144
and
indoor{Bohlin, 2010 #256;Zhang, 2011 #249} environments and proved effective in
characterizing concentrations, temporal and spatial trends, and potential human exposure to
SVOC in air.27,145,146

90
Passive air sampling is based on molecular diffusion of the SVOC from ambient air into the
passive sampling medium (PSM). This uptake process has been described with the two-film
diffusion theory which assumes uniform chemical distribution within both air and the PSM.
Applying a mass balance to the SVOC in the PSM, the amount of SVOC accumulated in the
PSM (mS) can be derived as a function of PAS deployment time (t), SVOC air concentration
(CA), PSM-air equilibrium partition or sorption coefficient (KSA), the volume (VS) and surface
area (A) of the PSM, and the overall mass transfer coefficient (kO) for the uptake of the SVOC:
O
SA S
S A S SA(1 )
k At
K Vm C V K e (Equation 4.1)
The uptake can be approximated as a linear function of t when the surface evaporation of the
SVOC from the PSM to the air is negligible during the initial uptake stage, which is referred as
the quasi-linear range and operationally defined as the period when the amount of chemical in
the PSM is less than 25% of the equilibration amount.9,38
During the quasi-linear range, the
amount of SVOCs accumulated in the PSM as a function of PAS deployment time can be
simplified from Equation 4.1:
mS = kO·A·CA·t = R·CA·t (Equation 4.2)
where the passive sampling rate R equals kO∙A. In order to derive CA from mS and t, using
Equation 4.2, PAS for SVOCs should be deployed within the quasi-linear range, during which R
does not vary with t. The overall mass transfer coefficient for chemical uptake from ambient air
to PSM (kO) is inversely proportional to the overall kinetic resistance (rO) which is the sum of the
kinetic resistances posed by the sampler housing (rH), by the air boundary layer surrounding the
PSM (rBL), and by the PSM (rPSM).83
These individual kinetic resistance terms depend on
boundary layer thickness and diffusion length, which are difficult to measure directly. Thus, it is
not practical to calculate R from the individual kinetic resistance terms. Instead, R is typically
acquired from a calibration of the PAS against an active air sampler.
PAS calibrations have been conducted under various environmental conditions using
polyurethane foam (PUF) disk or XAD resin as the PSM.9,20,47,87,88
Even for the same type of
PUF-disk PAS, R can vary by as much as an order of magnitude between different studies.87
Such a large variation in R can introduce uncertainty to PAS-derived air concentrations.

91
Therefore, it is important to understand the factors influencing R. So far, several studies have
attempted to understand the effect of temperature and wind speed on R.20,90,92
Besides the
temperature and wind effect, there is evidence showing PAS configuration could also affect R. In
a previous indoor calibration study, Tao et al.47
observed a lower R (and a lower surface area
normalized R) for a PAS with the PUF disk positioned in a housing that was more confined than
the typically used double-bowl PAS.87
Abdallah and Harrad48
noted a decreased rate of chemical
uptake by PUF when it was moved further from the opening of the housing compared to the
original PUF-PAS design.28
While these studies clearly indicate that the PAS configurations can
affect R, no studies have systematically focused on this effect so far.
With the objective to understand the effect of PAS configurations on R, we applied a
gravimetrical approach to study the uptake of water vapor from indoor air in silica gel placed
inside cylindrical PAS of variable configuration (Schematic of the PAS are shown in Figure
S4.1). These studies were supplemented with an indoor calibration for polychlorinated biphenyl
(PCB) uptake in the same type of PAS with XAD-resin as the PSM.
4.3 Materials and Methods
4.3.1 Setup for Water Uptake Experiments
The experiments were performed using the PAS design by Wania et al.14
This sampler consists
of a stainless steel mesh cylinder (cylinder diameter dC=2 cm) filled with PSM XAD and hung
into an inverted cylindrical steel can (housing diameter dH=10.5 cm). Both long (cylinder length
lC=20 cm, housing length lH=30 cm) and short versions (lC=10 cm, lH=18 cm) of the sampler
have been used. Using water as a surrogate for SVOCs in PAS experiments has proven to be an
effective approach to studying the influence of factors that are largely independent of the
chemical nature of the sampling medium and the sampled chemicals.20,97
An earlier experimental
setup20
required taking the PSM out of the PAS housing to gravimetrically measure the amount
of water taken up. In this study, we hung mesh cylinders filled with silica-gel (Aldrich, 35-60
mesh, pore size 150 Å, bulk density 0.33g/cm3, conditioned overnight at 120 °C before use) from
a precision balance (Sartorius ED423S, Readability 0.001g) placed on a table with a hole (Figure
S4.2). The cylinder can still be placed into a housing, if its lid has a small opening. The balance
reading (gram of water accumulated, mS), ambient temperature and relative humidity, measured
with a digital psychrometer (Reuter-Stokes RSS230), were recorded at preset time intervals

92
using a data logger. The sensor of the psychrometer was placed outside of the housing with a
horizontal distance of 2 cm away from the opening of the housing. From the temperature and
relative humidity, the water concentration in air (CA, gwater/m3
air) could be derived,147
and the
equivalent volume (m3) of air sampled was calculated as VA, eqv = mS /CA.
9
4.3.2 Characterizing Water Uptake by Silica-gel
With the experimental setup, we tested the characteristics of silica gel using a long PAS (lC=20
cm, dC=2 cm) placed in a long housing (lH=30 cm, dH=10.5 cm) (Figure S4.1a). Duplicate
experiments on water uptake for 3 h were conducted and the VA, eqv was plotted against time and
fitted with:
A,eqv(1 )
b tV a e
(Equation 4.3)
where a = KSA·VS and b = A·kO/(KSA·VS). KSA [dimension: unitless] is the partition or sorption
coefficient between the sorbent (silica-gel) and air; VS [M3] is the volume of the PSM; A [M
2] is
the interfacial transfer area between the PSM and air (the lateral area of the mesh cylinder); kO
[M·T-1
] is the overall mass transfer coefficient. In other studies,9 kA (the mass transfer coefficient
on the air side) may have been used instead of kO because the uptake was thought kinetically
limited by the air-side boundary layer. However, kinetic resistance could also be introduced from
the sampler housing and the PSM.83,148
Thus, we use the overall mass transfer coefficient here to
represent all possible kinetic resistances. KSA and kO were derived from the fitting parameters a
and b (see SI for detail).
4.3.3 Assessment of Different Sampler Configurations
In order to test the hypothesis that R during the linear uptake stage is governed by the interfacial
transfer area rather than the amount of sorbent, we performed a water uptake experiment with a
short and long cylinders (lC=10 or 20 cm, dC=2 cm) filled completely with silica gel (~9.5 g and
19 g), and a long mesh cylinder with a metal rod (20 cm long, 0.9 cm in diameter) placed at the
center with silica gel (~15 g) surrounding it (Figure S4.3). We further tested whether R is
affected by the distance of the PSM cylinder from the opening of the PAS housing. R was
measured for a 20 cm cylinder positioned at two different positions and a 10 cm cylinder (both
with dC=2 cm) positioned at three different positions within the long PAS housing (lH=30 cm,

93
dH=10.5 cm) (Figure S4.4). In addition, we investigated how the configurations of the PAS
housing and PSM affect R. Uptake experiments were performed using the regular PAS housing
(dH=10.5 cm) with thin (dC=1.2 cm), regular (dC=2 cm) and thick (dC=3 cm) mesh cylinders,
and using the 2 cm diameter mesh cylinder without PAS housing, with thin (dH=6 cm) and
regular PAS housing (Figure S4.5).
4.3.4 Indoor Calibration of XAD-based Passive Air Samplers Using Sampling Media of Different Diameters
An experiment on the uptake kinetics of PCBs in the XAD-PAS (Figure S4.1b) was conducted in
an unoccupied office. The office had previously been identified as being heavily contaminated
with PCBs (air concentration of ∑PCB = 200 ± 40 ng/m3). Previously extracted XAD-2 resin
(20–60 mesh) was cleaned by Soxhlet extraction with acetone for 24 h and hexane for 24 h. PAS
with XAD-filled mesh cylinders (lC=10 cm, dC=1.2 cm or 2 cm) were deployed in the office for
0 (as the field blanks), 2, 4, 6, 8, 10, and 12 weeks between July and September, 2010. PCBs in
the air of the office had been continuously monitored at monthly resolution since April 2010
using a low volume air sampler (BGI Inc., 2.9 ± 0.2 m3/d) with a PUF-XAD-PUF sandwich (5 g
of XAD between two 2 cm i.d. × 3 cm PUF plugs) as the sampling medium.126
The passive air
sampling rate was calibrated based on the PCB concentrations in the bulk air (gas and particle
phase not separated) monitored with the low volume air sampler. The resolution of the active
sampling was half of the retrieval frequency of the PAS, which could potentially introduce some
uncertainty to the calibration. To evaluate this uncertainty, another set of seven PAS with XAD-
filled mesh cylinders (dC=2 cm) were deployed two weeks after the first set and retrieved at the
same frequency (sampling scheme illustrated in Figure S4.6). Upon retrieval, the PSM were
individually sealed in pre-cleaned aluminum foil and Ziploc bags, and stored at –20°C (storage
time < 1 month) until extraction.
Unlike the water uptake experiment, which investigated uptake using mesh cylinders of three
diameters (1.2, 2 and 3 cm), we did not include XAD-filled mesh cylinders of dC=3 cm in the
PCB uptake experiment of this study because we had gained such information in a previous
study.126
In that study, mesh cylinders of dC=3 cm were concentrically separated into three
layers, and PCB uptake by the XAD within each of the three layers was analyzed. Herein, we
used the sum of the amount of PCB accumulated in the three concentric layers to represent
uptake to mesh cylinders of dC = 3 cm.

94
4.3.5 Sample Extraction and Preparation
The XAD resin of each sample was extracted using a Dionex ASE-350 system with 33 ml
extraction cells. Before use, the extraction cells had been ultrasonically cleaned sequentially with
deionized water, acetone, and hexane. Prior to extraction, each sample was spiked with 100 μL
0.25 ng∙μL-1
of 13
C-labeled PCB-77, -101, -141, and -178 as surrogate standards. The ASE
conditions followed that by Primbs et al.149
: solvent 50:50 hexane:acetone; temperature 75°C;
pressure 1500 psi; static time 5 min; static cycles 3; flush volume 100%; purge time 240 s. Each
extract was roto-evaporated to ~2 mL and filtered through ~1 g of anhydrous sodium sulfate
packed in a disposable pasteur pipet to remove moisture. The eluent was solvent exchanged to
isooctane, blown down with high purity nitrogen, transferred to a GC vial, and further reduced to
0.5 mL. To the GC vial, 10 μL of 10 ng∙μL-1
mirex was added as the internal standard for PCB
quantification.
4.3.6 PCB Analysis
PCBs in the samples were analyzed using an Agilent 6890 gas chromatograph coupled with an
Agilent 7683 auto-sampler and an Agilent 5973 mass spectrometric detector. 1.0 μL of the
sample was injected in splitless mode with the injector temperature at 250 °C. PCBs in the
sample were separated using a DB5-MS capillary column (60 m length × 0.25 mm i.d., 0.25 μm
film thickness, J&W Scientific) with helium (25 psi, 1.4 mL/min) as the carrier gas. The column
temperature program started from 80 °C for 1 min, to 160 °C at 10 °C·min-1
, to 280 °C at
3 °C·min-1
, and held for 6 min. The mass spectrometric detector was operated in electron impact
ionization (70 eV) and selective ion monitoring mode. Temperatures for the ion source and
quadrupole were 230 °C and 150 °C. The targeted PCB congeners and the monitored ions
analyzed are listed in Table S4.1.
4.3.7 QA/QC
The relative difference of the KSA and kO derived from the duplicates of 3-hour water uptake
experiments were ~5% (Figure 4.1 and Figure S4.7). The coefficients of variation of water
uptake rates derived from 6 replicates for each PAS configuration were <10% (Figure S4.8). In
the PAS indoor calibration experiment, the differences between the sampling rates (R) derived
from the two sets of PAS deployed with two-week lag time were ~30%. This represents the
uncertainty of the R-values derived from the low volume air sampler with a resolution at half of

95
the retrieval frequency of the PAS. Method recoveries derived from the labeled PCB congeners
were 76-120% with an interquartile range of 21% (Figure S4.9). Three solvent blanks and three
field blanks were analyzed. No target compounds were observed in the solvent and field blanks
except for PCB-44 in the field blanks. The PCB-44 field blank levels were ~20% of the sample
with the lowest concentration. The blanks were considered as time zero levels in the linear fitting
to derive R.
Figure 4.1 Measured and model-fitted equivalent air volume derived from passive sampling
of water vapor from air using silica gel filled mesh cylinder as a sampling medium. Data were
recorded every 1 min for the first 30 min and every 5 min afterwards.
4.4 Results and Discussion
4.4.1 Characteristics of Water Uptake by Silica Gel
Water uptake by silica gel filled mesh cylinders placed in a housing was continuously monitored
for 3 h using the gravimetric method (Figure S4.2). The plot between the equivalent volume of
air sampled and sampling time (Figure 4.1) reveals that the sampling rate (slope of the plot)
gradually decreased over the 3 h deployment time. This is due to the evaporation of the
accumulated water vapor from silica gel to air. Similar to the uptake of SVOC by an XAD-filled
0 50 100 150 200 250 300
0.00
0.02
0.04
0.06
0.08
0.10
Eq
uiv
ale
nt a
ir v
olu
me
(m
3)
Deployment time (min)
Measured
Model Fit
Model Fit Result:
y = a*(1 - exp(-b*x))
a = 0.1104±0.0004
b = 0.0099±0.0001
R2 = 0.999

96
mesh cylinder,20
the initial uptake stage is quasi-linear. The slope of the uptake curve changes
little within this quasi-linear range. Based on this slope, the air concentration of a chemical can
be calculated from the amount accumulated in the PSM.
The quasi-linear range is determined by the kinetic (kO) and thermodynamic (KSA) properties of
the sampled chemicals. Fitting the uptake curve to the theoretical equation (Equation 4.1 and
Table S4.2), we derived a kO for the water uptake by a silica gel filled mesh cylinder of 127 m/d;
the equilibrium sorption coefficient of water vapor to silica gel (KSA) was 1.8×103. Applying
these two parameters in Equation 4.3, the quasi-linear range for water uptake by silica gel is 27
min. Note this quasi-linear range is based on an experiment using the long mesh cylinder. To
ensure uptake is within the quasi-linear range during all experiments, the first 10 min were
selected to derive the R.
4.4.2 Effect of Interfacial Transfer Area and Sorbent Amount on Uptake
Water uptake experiments were conducted on the regular short and long PAS (Figure S4.3). The
short PAS gave an R of 0.87 ± 0.02 m3/d, which is about half of the R for the long PAS (1.58 ±
0.08 m3/d). This difference between short and long PAS agrees with the field-calibrated
sampling rates of SVOCs using XAD-resin as the PSM.108
The reduced sampling rate for the
short PAS could be due to the reduced interfacial transfer area between air and PSM and/or the
reduced PSM amount.
Water uptake was also measured in a long PAS with silica gel in the outer part of the mesh
cylinder and a metal rod at the center. The interfacial transfer area is the same as for the regular
long PAS while the amount of sorbent is reduced by ~25%. Despite the reduced sorbent amount,
R was not statistically different (Mann–Whitney U test, p=0.8) from that of the regular long PAS
(Figure 4.2a). This indicates water vapor penetrates into the inside of the silica gel filled mesh
cylinder more slowly than uptake from the air occurs; i.e. most of the water molecules sorb to the
outer layer of the silica gel and the inner portion of the silica gel is not participating in the
accumulation of water molecules, at least during the initial 1/3 of the quasi-linear uptake range,
during which the experiments were conducted. Such non-uniform distribution of the sorbate
within the PSM has also been observed for SVOCs in the PSMs PUF and XAD.126
This
observation confirms that passive sampling efficiency could be improved by maximizing the
surface area/volume ratio A/VS of the PSM.83
For example, with the same amount (volume) of

97
sorbent, the normal cylindrical mesh cylinders can be replaced with several slim ones, which
would yield an increased A/VS and thus sampling rate.
Figure 4.2 Effect of interfacial transfer area and sorbent amount on the uptake of water
vapor from air by silica gel. I and II: short and long silica gel filled mesh cylinder in short and
long housing; III: long mesh cylinder with a metal rod positioned at the center with silica gel
surrounding it. Ratios of the interfacial transfer area to bulk XAD volume for I, II and III are 1, 1
and 1.25 cm-1
respectively.
Because interfacial transfer area is a key factor in determining R, we also compared sampling
rates normalized to the interfacial transfer area (SR, m3/d/m
2). SR of the long PAS with the metal
rod in the centre and of the regular long PAS are not statistically different (Mann–Whitney U
test, p = 0.8) but are lower (Mann–Whitney U test, p = 10–4
) than that of the short PAS (Figure
4.2b).
I II III0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
Median
Interquartile Range
Non-Outlier Range
Outliers
R(m
3a
ir/ day /
sam
ple
r)
0
20
40
60
80
100
120
140
SR
(m3
air
/ day /
m2
PS
M a
rea)
A
B
10
cm
10
cm
13 c
m
23 c
m

98
Figure 4.3 Effect of the distance of the silica gel filled mesh cylinder to the opening of the
sampler housing on the uptake of water vapor from air by silica gel. I and II: long mesh cylinder
at different positions within long housing; III-V: short mesh cylinder at different positions within
long housing.
4.4.3 Effect of the Position of the PSM within the Sampler Housing On Uptake
We hypothesized that the average distance between the PSM and the opening of the sampler
housing could affect the uptake rate. To test this hypothesis, water uptake experiments were
conducted by positioning the silica gel filled mesh cylinder at different positions within the
sampler housing (Figure S4.4). We could vary the distance of the 20-cm mesh cylinder to the
opening of the long housing by 2 cm (Figure 4.3-I and II). This small difference had no
statistically significant effect on R (Mann–Whitney U test, p = 0.7). Thus, we set up three
configurations (Figure 4.3-III to V) using the 10-cm mesh cylinder in the long housing. The
R(m
3a
ir/
da
y /
sam
ple
r)
0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Median
Interquartile Range
Non-Outlier Range
Outliers
90
110
130
150
170
190
SR
(m3
air
/ d
ay /
m2
PS
M a
rea)
A
B
I II III IV V
20
cm
23
cm
10
cm
6.5
6.5 1
1cm
21
cm

99
distance of the mesh cylinder to the opening of the housing varied by 6.5 cm between the three
configurations. Statistically significant different R between each of the three configurations were
observed (Mann–Whitney U test, p < 10–3
). The closer the PSM to the opening, the higher was
R. This also explained why the short cylindrical PAS (Figure 4.2-I) had a higher SR than the long
one (Figure 4.2-II). This results is also in line with the studies using PUF-disk PAS to sample
SVOCs. When Abdallah and Harrad48
mounted the PUF-disk further from the opening of the
housing than in the regular configuration of the PUF-disk PAS, lower uptake rates were
observed.28,48
Figure 4.4 Effect of dimensions of the sampling medium and sampler housing on the uptake
of water vapor from air by silica gel. I-III: silica-gel filled mesh cylinder (lC=10 cm, dC=2cm)
without housing, in a housing with dH=6 cm, and in a housing with dH=10.5 cm; IV and V:
silica-gel filled mesh cylinder (lC=10 cm, dC=1.2cm and 3 cm) in a housing with dH=10.5 cm.
I II III IV V0
0.2
0.4
0.6
0.8
1.0
1.2
R(m
3a
ir/ day /
sam
ple
r)
10.5 cm6 cm2 cm
10
cm
1.2 cm 3 cm
100
120
140
160
180
SR
(m3
air
/ day /
m2
PS
M a
rea)
Median
Interquartile RangeNon-Outlier Range
OutliersA
B

100
The different sampling rates for PSM-filled cylinders positioned at different positions of the
housing can be explained by a different air boundary layer thickness and/or housing resistance
rH. The thickness of the air boundary layer is negatively correlated with the strength of air
turbulence.109
The closer the sampling medium is to the opening of the housing, the more
susceptible it is to turbulence in ambient air. Thus, PSM placed closer to the opening will
presumably be surrounded by a thinner boundary layer, which leads to a higher uptake rate.
Besides the boundary layer thickness, the housing resistance rH could also contribute to different
sampling rates. In a previous definition83, rH is only related to the rate of air entering the PAS
housing via advection. When air around the PAS is turbulent, chemical may indeed enter the
housing via advection and rH is unlikely to affect the overall sampling rate. However, under wind
still conditions chemical is more likely to enter the housing via diffusion. The diffusion length
for molecules transferring from ambient air to the boundary layer is different for PSM mounted
at different positions within the housing, which lead to different rH. If the overall mass transfer
coefficient for this transfer through the housing is lower than that that for transfer through the
boundary layer, then rH could also explain different uptake rates. Although both the boundary
layer thickness and rH could play a role, we cannot presently distinguish the two or tell which is
more influential.
This study was conducted indoors. In outdoor environments, stronger air turbulence reduces the
boundary layer thickness and, in addition to diffusion, advection can contribute to chemical
transport from ambient air to the PSM-air boundary layer. This advection could reduce rH. Thus,
whether similar results would be observed for the PAS deployed outdoors merits further study.
4.4.4 Effect of Dimensions of the Sampling Medium and Sampler Housing on Uptake
Water uptake experiments were conducted using the silica gel filled mesh cylinders directly
exposed to ambient air, and mounted in a housing with a narrow diameter (dH=6 cm) and in a
regular housing (Figure S4.5). When the PSM cylinder was directly exposed to ambient air, R
was ~1.5 times higher than when it was positioned in the housing (Figure 4.4-I to III). This is
expected because there is no housing resistance rH if the PSM is directly exposed to ambient air.
Besides, stronger air turbulence and thus a thinner air boundary layer around a PSM directly
exposed to ambient air would also explain an increase in R. This observation is consistent with a
study using semipermeable membrane devices as PSM: sampling rate decreased when the PSM

101
was protected within a shelter.93
The sampling rate for the PSM cylinder positioned in the narrow
housing (Figure 4.4-II) was ~20% lower than that of the regular configuration (Figure 4.4-III).
This agrees with the lower indoor passive air sampling rates of PAHs determined by Tao et al.,47
who mounted PUF disks in a more confined housing than the regular double-bowl PAS.9 A
lower R for a PSM in more confined housing can be explained by the limited air turbulence and
thicker boundary layer around the PSM.
Water uptake experiments were also conducted using silica gel filled mesh cylinders of different
diameters dC mounted in regular housings (dH=10.5 cm) (Figure 4.4-III to V). Since the
interfacial transfer area determines the sampling rate, R increased with increasing dC. When
normalizing the sampling rate to the interfacial transfer area, we noted that SR decreased with
increasing dC. One possible cause for the reduced SR with PSM of larger dC is the reduced space
between the PSM and the inner wall of the housing (dH - dC)/2, which may increase rH or the
boundary layer thickness and thus rBL. Comparing Figure 4.4-II and -III, SR decreased ~20%
upon a reduction of (dH - dC)/2 by ~50% (from 4.25 cm to 2 cm). The SR differences between the
set-ups shown in Figure 4.4-III, -IV and –V are of the same magnitude (~20%), even though (dH
- dC)/2 changed much less. This indicates that this distance plays a minor role and the different
observed SR can be attributed to the PSM of different dC.
4.4.5 Uptake of PCBs by XAD-filled Mesh Cylinder of Different Diameters
An indoor calibration of PCBs uptake in XAD-PAS was conducted using PSM cylinders with dC
of 1.2 and 2 cm. The sampling rates for XAD-PAS using a PSM with dC of 3 cm were retrieved
from an earlier study126
based on the sum of the amount of chemical accumulated in three
concentric layers. Similar to the water uptake by silica gel, the R for PCB uptake in XAD-PAS
increases with dC and thus with the interfacial transfer area (Figure 4.5). The interfacial transfer
area normalized sampling rates SR for the PAS with dC=2 cm was slightly, but significantly
lower (p<10–5
, Wilcoxon signed-rank test for the PCB congeners in Figure S4.10) than the SR for
the PAS with dC=1.2 cm. This is similar to the water uptake by silica gel-filled mesh cylinder of
different dC. Contrary to expectations based on this trend, SR of the PAS with a dC of 3 cm was
higher than the SR for the PAS with a dC of 2 and 1.2 cm (Figure 4.5), except for the penta-CBs.
The explanation is likely to be found in the chemical analysis. The SR for the wide cylinders is
derived from the sum of the amounts in three layers, which are therefore subject to a higher

102
uncertainty, illustrated by the longer whiskers in Figure 4.5. Furthermore, the samples for the
PAS with a dC of 3 cm were analyzed at a different laboratory with a different method126
and the
inter-lab variation of SVOC analyses (RSD 10-150% with an average of 70% for PCBs in air
extract150
) could easily be larger than the differences between the SR of a set up with dC of 1.2
and 3 cm.
Figure 4.5 Comparison of passive sampling rates of PCBs between passive sampling
medium of different diameters. Data of 1.2-cm and 2-cm mesh cylinder were obtained in this
study; data of the 3-cm mesh cylinder were based on the sum of three concentric layers in a
previous study.126
4.4.6 Water Uptake by Silica Gel vs. SVOC Uptake by XAD
From the above experiments and previous studies on SVOC uptake by XAD-based PAS,20,108
we
can conclude that water uptake by silica gel (Figure 4.1) and SVOC uptake by XAD20
follow the
same pattern: an initial quasi-linear uptake phase is followed by a gradually decreasing rate of
uptake until eventually equilibrium is reached. Because of such similarity, water uptake and loss
kinetics have proven useful in evaluating the kinetics of SVOC uptake in both XAD-based and
PUF-based PAS.20,97
The time scale of uptake is of course widely different: the quasi-linear
0.00
0.05
0.10
0.15
0.20
0
5
10
15
20
Tri-CB Tetra-CB Penta-CB Hexa-CB
Diameter of mesh cylinder
(cm)
1.2 cm
2 cm
3 cm
R(m
3a
ir/
da
y /
sa
mp
ler)
SR
(m3
air
/ d
ay /
m2
PS
M a
rea)

103
range for water uptake by silica gel (< 30 min) is much shorter than that of SVOC uptake (a few
months108
). This is because of the higher kO for water uptake by silica gel and the lower holding
capacity of silica gel for water (KSA). The short time scale for water uptake makes it feasible to
conduct a number of experiments quickly and at low cost to investigate numerous factors. An
example of such a factor is the potential resistances posed by housing rH and boundary layer rBL.
These two contributions to the overall kinetic resistance rO could be further affected by wind
conditions20
and passive sampler configuration. Therefore, uptake of water vapor by silica gel
can be used for a preliminary assessment of the influence of various factors on chemical uptake
of SVOCs by XAD.
If we assume that only the resistance at the air-boundary layer affects kO (kO = kA), kO would be
proportional to the chemical’s molecular diffusivity in air (DA), which can be estimated using the
Fuller-Schettler-Giddings equation.85
Based on kO for water uptake (139 m/d based on the
configuration of Figure 4.2-I) and DA of water (0.0015 m2/min) and SVOCs (Table S4.3), kO for
the SVOCs (kO,SVOC = kO,Water·DA,SVOC / DA,Water) are estimated to range from 23 to 29 m/d (Table
S4.3). The kO of tri- to hexa-CBs estimated this way (24–28 m/d) are ~2 times higher than the kO
(9–12 m/d, equivalent to SR in Figure S4.10) calculated from the indoor calibration of the XAD-
PAS (dC=2 cm) for PCBs. Because both experiments were conducted indoors with the same PAS
configuration and because the sorbents used as the PSM (silica gel and XAD) have a similar
particle size, the thickness of the boundary layer surrounding the PSM is presumably identical in
the two experiments. Therefore, if the uptake kinetics were only affected by the resistance from
the boundary layer, kO for SVOC derived from the water uptake experiment should have matched
that derived from the calibration experiment. However, because the observed kO from the
calibration were 2-fold lower, we can infer that the uptake of SVOCs by PSM is kinetically
limited not only by the boundary layer, but likely is also affected by a resistance within the PSM.
This is in line with our previous study indicating that SVOCs do not uniformly distribute within
the PSM.126
4.4.7 Implications
Using silica gel as a PSM to sample water vapor from air is an effective approach to study
factors that influence uptake in PAS and are independent of the sampling media and target
chemicals. The short time scale of the water uptake makes it time-efficient to conduct numerous

104
passive sampling experiments, increasing precision through sufficient replication and allowing
for a variety of experimental conditions. Besides the air boundary layer surrounding the sampling
medium, the sampler housing and the sampling medium appear to contribute kinetic resistance to
chemical uptake, especially in the indoor environment where the air turbulence is relatively
limited. Based on the information gained from this study, a smaller housing with one or multiple
cylinders of smaller diameter could be used as an alternative to the current PAS design (Figure
S4.11). A smaller housing would reduce the cost for material and for shipping to sampling sites,
although the confined configuration would introduce more kinetic resistance causing the
sampling rate to decrease slightly. However, for a given amount of PSM, multiple mesh
cylinders with smaller diameter could increase the interfacial transfer area, which would
compensate for the increased kinetic resistance introduced by a smaller housing.
4.5 Acknowledgments
We acknowledge research funding from the Canadian Foundation for Climate and Atmospheric
Sciences and the Natural Sciences and Engineering Research Council of Canada. X. Zhang also
acknowledges financial support through the Ontario Graduate Scholarship.

105
Supporting Information of Chapter 4
Figure S4.1 Schematic of the cylindrical passive air samplers. (a) long version with 20 cm-
long mesh cylinder; (b) short version with 10 cm-long mesh cylinder.
Figure S4.2 Illustration of gravimetrical experiment for passive air sampling of water using
silica gel filled mesh cylinder as the passive sampling medium.
20
cm
2 cm
sorbent-filled stainless steel mesh cylinder
sampler housing
10.5 cm
10 c
m
2 cm
10.5 cm
(a) (b)
3 cm
3 cm
Precision Balance
Passive Sampler housing
Silica gel Packed Mesh
Cylinder

106
Figure S4.3 Experiment setup to investigate the effect of interfacial transfer area and sorbent
amount on uptake of water vapor from air by silica gel.
Figure S4.4 Experiment setup to investigate the effect of the distance of the silica gel filled
mesh cylinder to the opening of the sampler housing on uptake of water vapor from air by silica
gel.
Figure S4.5 Experiment setup to investigate the effect of Dimensions of the sampling medium
and sampler housing on uptake of water vapor from air by silica gel.
3
metal rod to take up
inner space of
mesh cylinder
10
cm
10
cm
3
20
cm
2 cm
10cm
6.5 cm
6.5 cm
11cm
21cm
3 cm10.5 cm6 cm
10
cm
2 cm 1.2 cm
3

107
Figure S4.6 Schematics of the passive air sampler calibration for indoor PCBs.
2 w
4 w
6 w
8 w
10 w
12 w
t = 0 (field blank)
2 w
4 w
6 w
8 w
10 w
12 w
t = 0 (field blank)
LowVol Sampler 2.9 m3/day
PUF/XAD/PUF
10.5 cm1.2 cm2 cm

108
Table S4.1 Target ions, quanlify ions and limit of detection (LOD) of the chemicals analyzed
using GC-MS selected ion monitoring mode.
Class PCB Homolog
Chemical Target
Ion Qualify
Ion (Qual. /Targ.)
*100% LOD a
(ng/sample)
Internal Standard
Mirex 272 274 81.1 n/a Surrogate Standard Tetra- 13CPCB77 304 302 77.2 n/a Surrogate Standard Penta- 13CPCB101 338 340 64.8 n/a Surrogate Standard Hexa- 13CPCB141 372 374 81 n/a Surrogate Standard Hepta- 13CPCB178 406 408 97.2 n/a Target Analyte Di- PCB8 222 224 65.6 0.5 Target Analyte Di- PCB15 222 224 65.6 0.6 Target Analyte Tri- PCB18 256 258 98 0.2 Target Analyte Tri- PCB17 256 258 98 0.2 Target Analyte Tri- PCB16/32 256 258 98 0.5 Target Analyte Tri- PCB31/28 256 258 98 0.5 Target Analyte Tri- PCB33 256 258 98 0.9 Target Analyte Tri- PCB37 256 258 98 0.9 Target Analyte Tetra- PCB52 292 290 76.7 0.1 Target Analyte Tetra- PCB49 292 290 76.7 1.0 Target Analyte Tetra- PCB44 292 290 76.7 1.7 Target Analyte Tetra- PCB42 292 290 76.7 0.9 Target Analyte Tetra- PCB74 292 290 76.7 1.0 Target Analyte Tetra- PCB66 292 290 76.7 1.8 Target Analyte Tetra- PCB56/60 292 290 76.7 1.8 Target Analyte Tetra- PCB81 292 290 76.7 0.3 Target Analyte Tetra- PCB77 292 290 76.7 0.6 Target Analyte Penta- PCB95 326 328 65.3 0.2 Target Analyte Penta- PCB101 326 328 65.3 1.0 Target Analyte Penta- PCB99 326 328 65.3 1.4 Target Analyte Penta- PCB87 326 328 65.3 0.6 Target Analyte Penta- PCB110 326 328 65.3 1.3 Target Analyte Penta- PCB123 326 328 65.3 0.2 Target Analyte Penta- PCB118 326 328 65.3 0.2 Target Analyte Penta- PCB114 326 328 65.3 0.1 Target Analyte Penta- PCB105 326 328 65.3 1.7 Target Analyte Penta- PCB126 326 328 65.3 2.8 Target Analyte Hexa- PCB151 360 362 81.4 0.1 Target Analyte Hexa- PCB149 360 362 81.4 1.3 Target Analyte Hexa- PCB153 360 362 81.4 1.1 Target Analyte Hexa- PCB137 360 362 81.4 0.1 Target Analyte Hexa- PCB138 360 362 81.4 1.0 Target Analyte Hexa- PCB128 360 362 81.4 3.1 Target Analyte Hexa- PCB156 360 362 81.4 0.2

109
Table S4.1 (continued)
Class PCB Homolog
Chemical Target
Ion Qualify
Ion (Qual. /Targ.)
*100% LOD a
(ng/sample)
Target Analyte Hexa- PCB157 360 362 81.4 1.4
Target Analyte Hepta- PCB187 394 396 97.6 0.3
Target Analyte Hepta- PCB183 394 396 97.6 0.3
Target Analyte Hepta- PCB185 394 396 97.6 1.0
Target Analyte Hepta- PCB174 394 396 97.6 0.6
Target Analyte Hepta- PCB177 394 396 97.6 1.1
Target Analyte Hepta- PCB171 394 396 97.6 1.7
Target Analyte Hepta- PCB180 394 396 97.6 0.2
Target Analyte Hepta- PCB170 394 396 97.6 1.1
Target Analyte Octa- PCB199 430 428 87.9 0.3
Target Analyte Octa- PCB200 430 428 87.9 0.3
Target Analyte Octa- PCB203 430 428 87.9 0.3
Target Analyte Octa- PCB195 430 428 87.9 0.4
Target Analyte Octa- PCB194 430 428 87.9 0.2
Target Analyte Octa- PCB205 430 428 87.9 0.7
Target Analyte Nona- PCB207 464 462 76.9 0.2
Target Analyte Nona- PCB206 464 462 76.9 0.5
Target Analyte Deca- PCB209 498 500 86.7 0.3 a LOD calculated as the chemical amount of which the instrument detects a signal corresponding
to three times of the noise level.
Derivation of KSA and kO from curve ftting on the experimental data.
A
SA S
A,eqv SA S(1 )
k At
K VV K V e
where a = KSA·VS and b = A·kO/(KSA·VS). KSA [unitless] is the partition coefficient between the
sorbent (silica-gel) and air; VS [M3] is the volume of PSM; A [M2] is the interfacial transfer area
between the PSM and air (the lateral area of the mesh cylinder); kO [M·T-1
] is the overall mass
transfer coefficient.
Fitting Equation: y = a(1– e–bx
) where a = KSA·VS, b = kA·A/(KSA·VS)
VS = πr2h=π·0.01
2·0.2=6.28×10
–5 m
3
A / VS = 2πrh / πr2h = 2 / r
KSA= a/VS

110
kO = b·KSA·VS /A= b·KSA·r /2
Figure S4.7 Measured and model-fit equivalent air volume derived from the duplicated water
uptake experiment
Table S4.2 Parameters derived from the fitting of the water uptake kinetics
Replicate 1 Replicate 2 Average
a 0.1104 0.1190 0.1147
b (min-1
) 0.0099 0.0094 0.0097
KSA= a/VS 1758 1894 1826
kO = b·KSA·r /2 (m·min-1
) 0.087 0.089 0.088
log KSA 3.24 3.28 3.26
kO (m/d) 125.2 128.2 126.7
0 50 100 150 200 250 300 3500.00
0.02
0.04
0.06
0.08
0.10
0.12
Eq
uiv
ale
nt A
ir V
olu
me
Sa
mp
led
(m
3)
Time (min)
Model Fit Result:
y = a*(1 - exp(-b*x))
a = 0.1190±0.0003
b = 0.0094±0.0001
R2 = 0.999
Measured
Model Fit

111
Figure S4.8 Reproducibility of water uptake experiment on different sampler configurations.
The coefficient of variance is based on 6 replicated experiments
Figure S4.9 Method recovery of PCB analysis based on 13
C-PCB surrogate standards spiked
into the samples before extraction
0
5
10
15
20C
oe
ffic
ien
t o
f Var
ian
ce (%
)
70
80
90
100
110
120
Reco
ve
ry (
%)
max
75%ile
mean
median
25%ile
min
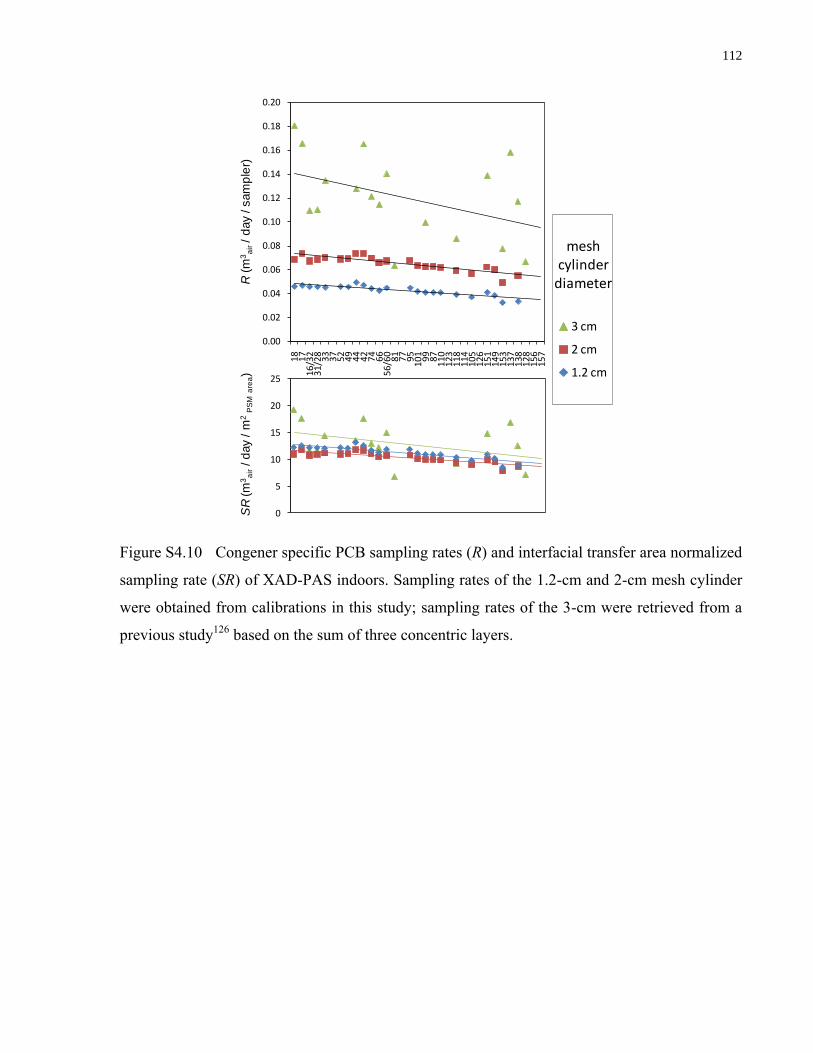
112
Figure S4.10 Congener specific PCB sampling rates (R) and interfacial transfer area normalized
sampling rate (SR) of XAD-PAS indoors. Sampling rates of the 1.2-cm and 2-cm mesh cylinder
were obtained from calibrations in this study; sampling rates of the 3-cm were retrieved from a
previous study126
based on the sum of three concentric layers.
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
18 17
16
/32
31
/28 33 37 52 49 44 42 74 66
56
/60 81 77 95
101 99 87
110
123
118
114
105
126
151
149
153
137
138
128
156
157
3 cm
2 cm
1.2 cm
0
5
10
15
20
25
R(m
3a
ir/
day / s
am
ple
r)S
R(m
3a
ir/ d
ay / m
2P
SM
a
rea)
mesh cylinder
diameter
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
18
17
16
/32
31
/28 33
37
52
49
44
42
74
66
56
/60 81
77
95
10
19
98
71
10
12
31
18
11
41
05
12
61
51
14
91
53
13
71
38
12
81
56
15
7
3 cm
2 cm
1.2 cm

113
Table S4.3 Overall mass transfer coefficient from the air to the sampling medium for selected
SVOCs derived based on the water uptake kinetics a
Chemical
Molar
Volume b
cm3/mol
MW
g/mol
DA c
cm2/s
DA
m2/min
kO
m/min
kO
m/d
H2O (g) 15 18 0.250 0.00150 0.088 139
HCB 236 285 0.052 0.00031 0.018 29
HCH 259 291 0.050 0.00030 0.018 28
endosulfan 330 407 0.044 0.00026 0.015 24
dieldrin 315 381 0.045 0.00027 0.016 25
trans-nonachlor 361 444 0.042 0.00025 0.015 23
tri-Cl biphenyl 255 257 0.050 0.00030 0.018 28
tetra-Cl biphenyl 279 291 0.048 0.00028 0.017 26
penta-Cl biphenyl 304 327 0.046 0.00027 0.016 25
hexa-Cl biphenyl 329 361 0.044 0.00026 0.015 24
hepta-Cl biphenyl 353 395 0.042 0.00025 0.015 23
a Assuming uptake kinetic resistance only from the air boundary layer.
b Based on the Le Bas molar volume estimation method.
151
c Calculated using Fuller-Schettler-Giddins equation at 295K.
85
Figure S4.11 Currently used long (a) and short (b) version of the cylindrical passive air sampler
and a modified design (c) proposed according to the information gained from this study.
(b) (c)(a)

114
Chapter 5. Wind Effect on Chemical Uptake and Axial Distribution in the
Sampling Medium of a Passive Air Sampler
Xianming Zhang, Trevor N. Brown, Akira Kondo, Ying D. Lei, Frank Wania
Contributions: X. Zhang designed the experiment on the axial distribution of chemicals in the
sampling medium under the guidance of F. Wania. X. Zhang performed the experiment,
extracted the samples, performed the analysis using GC-MS with the assistance of Y.D. Lei, and
processed the chromatograms. T. N. Brown and F. Wania designed the experiment studying the
wind effect on passive air sampling kinetics. T. Brown conducted the experiment, extracted the
samples, and performed GC-MS analysis with the assistance of Y.D. Lei. X. Zhang processed
the chromatograms of those analyses. A. Kondo conducted the computational fluid dynamic
simulations. X. Zhang interpreted all the data and wrote the manuscript with the guidance of F.
Wania,

115
5.1 Abstract
Passive air samplers (PASs) deployed in different types of environment operate under different
wind conditions, which may affect sampling rates. To investigate the effect of wind on the
uptake in cylindrical PASs using XAD resin as the sampling medium, we conducted two sets of
experiments. The first set of experiments focused on the distribution of the sampled chemicals
along the axial direction of the XAD-filled mesh cylinders. Axially segmented PASs were
deployed under quasi wind still and lab generated windy conditions indoors as well as under
normal outdoor conditions. Whereas under windy condition in the lab, the sampled chemicals
were uniformly distributed within the XAD under the quasi wind still and outdoor conditions, the
segment of the XAD-filled cylinder closer to the opening of the PAS housing had higher
chemical uptake rates. The differences between the segments were smaller for the outdoor PASs.
In the second set of experiments, the kinetics of chemical uptake by the PASs was investigated
under indoor quasi wind still condition and with lab generated wind blowing at straight and 45°
slanted angles towards the PASs. Passive sampling rates under the two windy conditions were
similar and were ~4 times higher than under quasi wind still condition. Computational fluid
dynamic simulations indicated similar wind patterns within the PAS housings under the two
windy conditions. Wind mainly affected the part of XAD mesh cylinders closer to the opening of
the housing.
5.2 Introduction
Allowing for time-integrated monitoring of the air concentrations of semivolatile organic
compounds (SVOCs) at low cost and without power requirement, passive air samplers (PASs)
have seen increasing use over the past decades.21,22,31,61,81,152
PASs gained more popularity
especially since 2004 when the Stockholm Convention153
came into force and various
stakeholders became interested in evaluating its effectiveness in reducing levels of persistent
organic pollutants in the global atmosphere. Having been used in several long term monitoring
campaigns,42,57,144
PASs have proven effective in studying the interannual time trends of SVOCs
in air. PASs have also found use in assessing spatial distributions of SVOCs at different
scales25,32,33,154
and identifying potential sources of, and human exposure to, SVOCs in various
types of environments.30,48,155

116
Environmental conditions under which PASs are deployed can vary tremendously. For example,
wind at sampling sites outdoors is generally much stronger than during indoor deployments156
and due to differences in surface roughness, sites in urban or forested areas tend to be less windy
than those in unobstructed locations.157
Different wind conditions may impact passive sampling
rates (PSR). Studies on the “double-bowl” PUF-disk PAS suggested that the housing of PAS can
somewhat dampen the effect of wind on PSRs at ambient wind speed below 1 m/s.91,92,95
However, under wind speeds in excess of 1m/s (3.6 km/h), which is quite commonly observed in
outdoor settings,87,90,92
the PSRs increased exponentially with wind speed.92,95
Such effects could
possibly cause variations in the calibrated PSRs of the PUF-disk PASs by as much as an order of
magnitude.87
In order to account for wind effects on PSRs, depuration compouds (DCs) have commonly been
added to sampling media prior to deploying a PASs; site-specific PSRs are then derived from the
loss kinetics of the DCs.95
The loss rates of DCs and thus the DC-derived PSRs were found to
correlate with wind speed but air concentrations derived with PSRs based on DC-loss rates are
biased towards windy days.92,95
The use of DCs rests on the assumption that “uptake and loss
mass transfer directions are opposite to each other”.95
Recent studies have shown this
requirement hardly be satisfied because of the existence of a mass transfer resistance within
porous passive sampling media.(Chapter 3) 126
An alternative approach to address the wind effect
is to have the sampling medium better sheltered from the wind. For example, PUF-disks
mounted at the ceiling of cylindrical housings appeared to be less effected by ambient wind but
the PSRs were lower than for the “double-bowl” PASs.47,116
Comparing chemical uptake by the
“double-bowl” PUF-PASs and the “cylindrical can” XAD-PASs deployed side by side at over 30
sites of the Global Atmosperic Passive Sampling network,57,144
the XAD-PASs appeared less
influenced by wind.96
While some studies have focused on the wind effect on the PUF-
PAS,90,92,95,97
studies quantitatively investigating the effect of wind on PSRs of the XAD-PAS
are still very limited. A wind-tunnel study suggested little wind effect on the water uptake by
silica-gel filled mesh cylinders at wind speed of 5-15 m/s,20
but field deployments of XAD-PAS
noted higher PSRs at sites exposed to strong winds.36,37
Besides wind speed, the angle at which the wind is blowing at a PAS may also affect the PSR.
This angle may be affected by the local terrain of the deployment site. For example, PASs
deployed along a slope may have valley to mountain winds preferentially blowing at an angle

117
towards it.34
As such, studies on how PSRs are influencd by the angle of wind incidence would
be useful. Although a recent study investigated the influence of the wind angle on the rate of
water evaporation from a PUF-disk placed in a double-bowl housing,97
no studies have tested the
effect of wind angle on chemical uptake by PASs.
PASs deployed at different sites of the world also differ in terms of insolation. Intense solar
radiation may generate a temperature gradient within a PAS housing and thus heat-induced air
movement (or heat convection) within the housing. So far, heat convection has only been
hypothesized as a potential factors influencing PSRs89
but no experiment has been performed to
test the hypothesis.
With the objective of filling the knowledge gaps identified above, we conducted experiments to
test (i) whether the sampling efficiency of the XAD-PAS varies along the axial direction of the
resin-filled mesh cylinder in both indoor and outdoor environments, (ii) whether wind and its
angle of incidence will affect chemical uptake by the XAD-PAS, and (iii) whether heat
convection will affect the uptake rate. Besides the experiments, we also conducted a
computational fluid dynamics (CFD) simulation to study the wind field within the housing of the
XAD-PAS.
5.3 Materials and Methods
5.3.1 Experimental Setup
5.3.1.1 Axial Distribution of Chemicals in the Sampling Medium
The PAS developed by Wania et al.20
was used in this study. This PAS consists of a stainless
steel mesh cylinder (length 20 cm, diameter 2 cm) filled with XAD-2 resin (Sigma-Aldrich, pre-
cleaned) and hung within a cylindrical steel housing (length 30 cm, diameter 10.5 cm). In order
to study the axial distributions of the sampled chemicals, the normal mesh cylinder (20 cm in
length) was divided into three segments of equal length (based on their positions in the sampler
housing, they are hereafter referred to as bottom, middle and top, Figure S5.1). The three
cylinder segments are connected with screws on the caps, which allows for easy assembly and
disassembly.

118
Two PASs with axially segmented cylinders and one PAS with a normal 20-cm-long cylinder
were each deployed at four indoor locations (referred as L1–4 hereafter) for six months (Figure
S5.2). In L1, we also studied preliminarily the wind effect on the axial distribution of the
sampled chemicals. Two fans (Delta Electronics Inc. BFC1212B, 12V, 1.1A, 2800 rpm) were set
up to blow at a 45° angle towards the opening of a PAS with an axially segmented cylinder
(Figure S5.2). In this preliminary study, wind speeds were not measured because the aim was to
test whether wind has any effect on uptake and axial distribution of chemicals in the XAD-PAS.
PASs with axially segmented cylinders were also deployed for three months on the roof of the
building in which L1 is located (referred as outdoor location or OD hereafter). In addition to
allowing for a comparison with the indoor experiment, this roof-top experiment also served as a
preliminary test of the potential effect of sunshine-induced heat convection on chemical uptake
and axial distribution in XAD-filled mesh cylinders. Along with two regular PASs with axially
segmented cylinders (identical to those used in L1–4), two PASs had housings that were painted
black on the outside to increase solar heat absorption, and another two were shaded from
sunshine by steel covers (Figure S5.3). By thus varying the amount of solar radiation absorbed
by the PAS housings we hoped to represent PASs deployed at sampling sites with different
insolation, and thus different potential to generate heat convection and affect PSRs. Three Smart
Button temperature loggers (ACR System Inc.) were placed within the PAS housings at the
levels corresponding to the three PSM segments (Figure S5.3) to record temperature gradients
within the housing with a one-hour frequency during the sampling period.
5.3.1.2 Wind Effect on Passive Air Sampling Kinetics
Based on the preliminary experiment indicating that windy conditions would increase chemical
uptake by PASs, calibrations were conducted using the short XAD-PASs under wind still indoor
condition, and under lab generated wind blowing at 45° and 90° towards the opening of the
housing (referred to as slanted angle and straight angle, respectively, hereafter, Figure S5.4). We
hypothesize wind blowing with a slanted angle at the PAS would result in a higher PSR. The
generated wind condition measured on the horizontal plain parallel to the fans is shown in Figure
5.1. Based on the spatial distribution of wind generated by the fans and the average outdoor wind
speed of ~4 m/s,92
the center at the opening of PAS was set up at point B (Figure 5.1). Although
wind speeds measured at point A had no significant difference (p = 0.29, Wilcoxon rank-sum test)

119
between the straight PASs and the slanted PASs, the average wind speed measured at point B for
the straight PASs was 4.3 ± 0.2 m/s, which was significant higher (p < 0.001) than the 3.6 ± 0.3
m/s for slanted PASs (Figure S5.5).
5.3.2 Sample Preparation and Extraction
Upon retrieval, the XAD-filled mesh cylinders were stored in air-tight metal tubes and placed in
a -20°C freezer until extraction. Segmented mesh cylinders were disassembled, and stored and
analyzed individually. Before extraction, each sample was spiked with 100 μL of a solution with
0.25 ng/μL 13
C12-labeled polychlorinated biphenyl congeners PCB-77, -101, -141 and -178
(Cambridge Isotope Labs) as surrogate standards. Each sample was Soxhlet extracted for 24 h
with ~500 ml dichloromethane. Extracts were roto-evaporated to ~2 mL and eluted through
dehydrated sodium sulphate packed in a disposable pasteur pipet to remove moisture. The eluent
was blown down with high purity (5.0) nitrogen, solvent exchanged to iso-octane and reduced to
~0.5 mL in a vial, to which 100 ng mirex was added as internal standard for quantification.
5.3.3 Chemical Analysis
PCBs, whose partition properties overlap with many SVOCs of environmental interest, were
selected as the target chemicals. An Agilent 6890 gas chromatograph coupled with an Agilent
7683 auto-sampler and an Agilent 5973 mass spectrometric detector were used for the analysis.
PCBs in 1.0 μL of extract were injected in splitless mode (injector temperature 250 °C) and
separated using a DB5-MS capillary column (60 m length × 0.25 mm i.d., 0.25 μm film
thickness, J&W Scientific) with helium (1.4 mL/min) as carrier gas. The chromatograph’s oven
temperature was programmed as 80 °C for 1 min, to 160 °C at 10 °C·min-1
, to 280 °C at
3 °C·min-1
, and held for 6 min. Temperatures for the ion source and quadrupole of the mass
spectrometer were 230 °C and 150 °C. The mass spectrometer was operated in electron impact
ionization (70 eV) and selective ion monitoring mode. The quantitative and qualitative ions
monitored are listed in Table S5.1.
5.3.4 QA/QC
Recoveries of the PCBs as indicated by the four surrogate standards ranged 73–144%
(interquartile range < 15%) for samples in the axial distribution experiment and 67-156%
(interquartile range < 20%) for samples in the uptake kinetics experiment. One field blank was

120
included at each of the sampling locations for the axial distribution experiment and four field
blanks were included for the uptake kinetics experiment. A solvent blank was included in every
batch (every 5 samples) of Soxhlet extractions. No PCBs were detected in the solvent blanks.
One field blank for the uptake kinetics experiment had levels of PCB-52, -49, -74, -99, -101, -
110, -153 at 5–15 % of the samples. Apart from that blank, all the field blanks for the axial
distribution experiment and three of the four field blanks for the uptake kinetics experiment
contained less than 5% of the PCB amounts in the samples. Because the recoveries and blank
levels were smaller than the variability of trace organic contaminants analysis, the reported
values were not recovery or blank corrected. At site L4, interference appeared to affect the
analysis of PCB-31/28, -49, -44 because the abundance ratios between the qualifying and
quantifying ions peaks deviated by more than 30% from the theoretical values, while for samples
from other locations, the differences were < 15%. As such, PCB-31/28, -49, -44 in samples from
L4 were not included in the data analysis.
In the preliminary experiment studying the effect of wind on chemical uptake and axial
distribution (Figure S5.2), the sum of the amounts of PCBs in the three segments matched well
the amount of PCBs accumulated in the long PSM (relative difference 11% ± 8%), indicating
that segmentation did not affect overall chemical uptake. Excluding the two indoor PASs with
blowing fans, the relative difference between duplicate PASs with axially segmented cylinders
was 17% ± 14% for the targeted PCB congeners. The amounts of PCBs sampled by the two
indoor PASs with blowing fans had a relative difference of 58% ± 7% (discussed below). In the
experiment on the wind effect on PAS uptake kinetics, the CV of the wind speeds in the
modified setup of fans and PASs was 4% at point A of Figure 5.1 for all the PASs, 4% and 8% at
point B for the straight and slanted PASs, respectively. The relative differences in the PCBs
analyzed between duplicates were 42% ± 2%, 17% ± 11%, and 22% ± 17% for the wind still,
straight wind and slanted wind conditons, respectively. The wind speeds (measured at position A,
Figure 5.1) for the 24 fans after running for 100 d were 87-103% of that measured at the
beginning of the experiment.
5.3.5 Computational Fluid Dynamics Simulation
Wind fields within the sampler housing under the two ambient wind conditions corresponding to
the experiment were assessed via computational fluid dynamics (CFD) simulations. The CFD

121
simulations were based on the continuity equation and the equations on the conservation of mass
and momentum. The standard k-ε turbulent model for high Reynolds number was adopted. The
wall function was applied to the surface of the PAS housing. All equations were solved by the
Semi-Implicit Method for Pressure Linked Equations (SIMPLE Algorithm). The selected domain
for the calculations was large enough so that the ambient winds were not influenced by the PAS.
Figure 5.1 Spatial distribution of speed (m/s) of the lab generated wind. Wind speeds were
measured with a hot-wire anemometer at a resolution of 2 cm. The round and elliptical rings
represent the position (projective planes of the opening) of the straight and 45° slanted passive
air samplers, respectively
5.4 Results and Discussion
5.4.1 Indoor Experiment on Axial Distributions of PCBs in the XAD-filled Mesh Cylinder
The axial distribution of PCBs in the passive sampling medium was investigated by analyzing
PCBs in each segment of axially segmented XAD-filled mesh cylinders. In all four indoor
locations (L1-L4) the sum of the amounts of a PCB congener in the three segments was not
significantly different (p = 0.53, Wilcoxon signed-rank test) from the amount in the non-
segmented mesh cylinder deployed at the same location (Figure 5.2 and Figure S5.6), indicating
that segmentation did not change uptake characteristics. The amount of PCBs accumulated in the
three segments appeared to be different (Figure S5.6). Analysis of variance (ANOVA) and
multiple comparisons on log-transformed data (Table S5.2) indicated that at L1, L2 and L4, the
amounts of PCBs in the bottom segments was significantly higher (p < 0.05) than that in the
0 4 8 12 16 20 24 28 32 36 40
x (cm)
0123456789101112
0
4
8
12
16
wind speed(m/s)
y (c
m)
A
B
B B

122
middle and top segments. This agrees with a previous study showing a higher PSR of water
when a silica gel filled mesh cylinder was positioned closer to the opening of the sampler
housing.127
L1, L2, and L4 are offices or storage rooms with little activity and thus air turbulence.
Under such wind still condition, the stagnant air layer surrounding the XAD-filled mesh cylinder
farther from the opening of the PAS housing is presumably thicker. Thus a higher chemical
uptake rate applies to the bottom segments of the cylinders than the middle and top segments.
Another possible explanation for the higher uptake rate for the bottom segment is the so-called
“starvation effect”: if the rate of gas diffusion within the housing is smaller than the chemical
uptake rate by the sampling medium, the concentrations of the chemicals in the air surrounding
the middle and top segments could be lower than that in the air surrounding the bottom segment.
A study48
using PUF as the sampling medium observed a decreased PSR when the PUF was
moved further from the opening of the sampler housing. The decreased PSR was partially
attributed to less particles being trapped by a PUF placed further from the opening.48
In the
present study, the congeners with less than 5 chlorines that are predominantly (>95%) in the gas
phase also showed decreased uptake in the middle and top cylinder segments, indicating that
chemical uptake could be limited by gas diffusion within the housing.
Figure 5.2 Amounts of PCBs accumulated in the three axial segments of XAD-resin based
passive air samplers deployed indoors under windy condition generated using electric fans
(L1W1 and L1W2), wind still condition (L1-L4) and deployed outdoors with normal sampler
configuration (ODN), with black painted housings (ODB) and with housings shaded from
sunlight (ODC). The sum of the amounts in the three segments is compared with the amount in a
non-segmented sampler deployed simultaneously at the same location. The whiskers indicate the
root mean square of the distances of the two points to the average.
The amounts of PCBs accumulated in the three segments of the PASs deployed at L3 were not
significantly different (Table S5.2). L3 is an underground cargo loading area with truck traffic
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
L1SW0
10000
20000
30000
40000
50000
60000
70000
80000
90000
L1SW
29%
38%
33%
29%
36%
35%
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
L1SW
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10
200
400
600
800
1000
L1SW
0
200
400
600
800
1000
1
0
200
400
600
800
1000
L1SW0
200
400
600
800
1000
1
0
200
400
600
800
1000
L1SW
0
200
400
600
800
1000
1
0
200
400
600
800
1000
1200
1400
L1SW
0
200
400
600
800
1000
1200
1400
L1SW
0
200
400
600
800
1000
1200
1400
L1SW
23%
23%
47%
24%
30%
46%
34%
28%
37%
28%29%
43%
30%
34%
36%
33%
31%
37%
27%
33%
41%
L2 L3 L4L1 ODCODN ODB
Σ 15P
CB
am
ou
nt
(μg)
L1W1 L1W2
908070605040302010
0
9876543210
1
0.8
0.6
0.4
0.2
0
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0

123
and other activities; thus air turbulence is expected to be stronger here than the other indoor
locations. Stronger air turbulence may expose the upper segments of the mesh cylinder to wind
to a similar extent as the bottom segments, resulting in similar uptake rates for the three
segments.
Figure 5.3 Masses of PCBs accumulated in the three axial segments of two XAD-resin based
passive air samplers deployed under wind still and lab generated windy conditions
To test whether air turbulence could affect the axial distribution of chemicals in the XAD-filled
mesh cylinder, we set up two electric fans blowing at 45° angle towards the openings of two
PASs with segmented cylinders (Figure S5.2). While the PCB congener profile under these
conditions was similar to that under wind still condition (Figure S5.6), exposure to constant wind
increased the amounts of PCBs accumulated in the PASs ~8 times. The relative difference in the
amounts of PCBs sampled between the duplicates under the windy condition was over 50%,
which was larger than that under wind still conditions. Such large variation is probably caused by
the difficulty of precisely replicating wind patterns; both speed and angle of incidence of the
wind are potential factors varying the uptake rate. In a previous study,20
no differences in the
uptake rate was observed for the same type of PAS with wind blowing at speeds between 5 and
15 m/s. However, the wind was blowing at a straight angle towards the PAS, in contrast to this
preliminary experiment, in which the fans were set up to generate wind at a 45° angle towards
the openings of the PASs.
y = 3.93xR² = 0.992
y = 7.32xR² = 0.995
y = 6.83xR² = 0.995
0
500
1000
1500
2000
2500
3000
3500
0 200 400 600 800
y = 7.63x
R² = 0.987
y = 12.74x
R² = 0.992
y = 13.01xR² = 0.992
0
1000
2000
3000
4000
5000
6000
0 200 400 600 800
PCB mass without wind (ng)
PC
B m
ass
wit
h w
ind
(n
g)
(a) (b)1:1 line
1:1 line

124
Despite of the variations between the duplicates in the absolute PCB amounts accumulated under
lab-generated windy conditions, the relative distribution of PCBs among the three segments was
quite consistent (p = 0.9) (Figure S5.7): No difference in the distributions of PCBs among the
three segments was observed under the lab generated windy condition (Figure S5.8and Table
S5.2).
Comparing the amounts of PCBs accumulated in the segmented cylinders of PASs deployed in
L1 under wind still and windy conditions (Figure 5.3), we note that wind increased the uptake
rates for all three segments of the mesh cylinder (all the points in Figure 5.3 fall on the upper left
side of the 1:1 line). In each segment of the XAD mesh cylinder no statistical difference in the
extent of increase (relative to wind still condition) was found among different PCB congeners
(the points representing different PCB congeners in a segment fall on a line through the origin in
Figure 5.3). Wind increased the uptake rate of the top and middle segments to the same extent
(no significant difference was found between the slopes of the corresponding red and green
regression lines in Figure 5.3; p > 0.4 for the interaction factor in the analysis of covariance;
more detail on the statistical test of the slopes are presented in SI). In both duplicates, the
increase in the uptake rate (the slopes of the lines in Figure 5.3a and b) for the top and middle
segments was ~1.7 times higher than the increase of the uptake in the bottom segments. The
smaller increase of uptake rate for the bottom segments is likely due to them being already
influenced by the air movement in a normal ventilated indoor environment with limited activities.
5.4.2 Outdoor Experiment on Axial Distributions of PCBs in the XAD mesh cylinder
Passive air samplers with axially segmented XAD-filled mesh cylinders in normal housings,
black painted housings and housings shaded from the sunlight were deployed outdoors to test
whether solar radiation would affect chemical uptake and axial distribution in PASs (Figure
S5.3). PCBs accumulating in PASs deployed within and on top of the same building (Figure S5.6)
had similar congener profiles. Even though PSRs tend to be higher outdoors than indoors,127
the
amounts of PCBs accumulated in the PASs deployed at L1 were ~10 times higher than in the
outdoor PASs. This suggests that the building was the source of the PCBs measured on the
building rooftop. This is consistent with previous studies,30,158
which suggested that PCBs are
still continuously emitted from indoor sources.

125
The PASs deployed outdoors showed non-uniform axial distributions of PCBs in the XAD mesh
cylinder (Figure S5.10). This is in contrast with the PASs under the lab generated windy
conditions, in which no statistical differences were observed for the masses of PCBs
accumulated in the three segments of XAD mesh cylinders but similar to the indoor PASs under
wind still condition. During the outdoor deployment segments that are closer to the opening of
the PAS housing generally accumulated more PCBs. The only exception was the middle
segments of the two cylinders deployed in black housings, which both accumulated less PCBs
than the top segment. Different from the wind still indoor conditions where the PCB amounts in
the top segments were 51 ± 2% of that in the bottom segments, the outdoor PASs (normal
housing) showed only a 17 ± 3% decline in the accumulated PCB amount from the bottom to the
top segment. With respect to the axial chemical distributions within the XAD-filled mesh
cylinder, the outdoor deployment falls in between the wind still and artificially windy indoor
deployment.
The total PCB amounts (i.e. sum of amounts in three segments) taken up by the PAS placed in
black housings were no different (Scheffe multiple comparison p = 0.17) from those in shaded
housings, but higher (p < 0.001) than those in normal housings. The shaded housing had more
PCBs (41±1)% in the bottom segments than the normal and black housing, which had (36±1)%
and (37±2)% of PCBs accumulated in the bottom segments. Because all the PASs were deployed
at the same location, they were exposed to the same wind. The lower portion of PCBs
accumulating in the middle and top segments could be caused by less heat convection in the
housing shaded from sunlight. In accordance with expectations, the records of the temperature
loggers (Figure S5.11 and Table S5.3) showed that the black housing experienced the highest
maximum temperatures, followed by the normal housing and the shaded housing. However,
differences were mostly less than 2 °C and occurred only during the day when the samplers were
exposed to direct sunshine. For most of the sampling period, there was no difference in the
temperatures measured in different housings and thus the temperature differences averaged over
the whole sampling period were small (<1°C). No temperature gradients were observed within
any of the sampler housings (Figure S5.12). We conclude that any variation in the PSRs
potentially caused by heat convections is likely so small to be dwarfed by other factors with
greater influence on the kinetics of uptake.

126
5.4.3 Wind Effect on Passive Sampling Kinetics
Calibrations of PASs under lab generated windy and wind still conditions were conducted
indoors to investigate the wind effect on chemical uptake. Similar to the preliminary experiment
(Figure 5.3), uptake of PCBs was faster under windy conditions; wind at a speed of ~4 m/s
increased PSRs ~4-fold (Figure 5.4 and Figure S5.13). This increase in PSRs was smaller than
the 10-fold increase in PSRs observed for the PUF-disk PAS at a wind speed of ~2 m/s
compared to wind still conditions.92
Windy conditions reduce the thickness of the stagnant air
layer, which results in less kinetic resistance to chemical mass transfer and thus elevated PSRs.
The study by Tuduri et al.92
identified a threshold of wind speed (1 m/s) below which wind had
no significant effect on the PSR in PUF-disk PAS. In an earlier experiment measuring water
uptake in a wind tunnel the PSR of the mesh cylinder-PAS did not vary within the wind speed
range from 5 to 15 m/s.20
Based on this, we hypothesize that wind only affect PSRs within
certain ranges of wind speeds. Further studies would be necessary to test the effect of wind on
PSRs at different wind speeds.
Figure 5.4 Passive air sampling kinetics (Penta-CB110 as an example) for samplers under
windy (lab generated wind blowing at 45° slanted angle and at straight angle towards the
cylindrical passive air samplers) and wind still conditions
y = (0.97±0.03)xR2=0.99
y = (0.84±0.03)xR2=0.98
y = (0.25±0.02)xR2=0.93
0
20
40
60
80
100
120
140
160
180
0 40 80 120 160
Windy, Slanted Angle
Windy, Straight Angle
No Wind
Deployment time (d)
Equ
ival
ent
Air
Vo
lum
e (m
3)

127
The ratio between the uptake rates under windy and wind still condition is a measure of the wind
effect. These ratios were 3.8±0.2 and 3.3±0.2 with the wind blowing at a slanted angle and at a
straight angle, respectively. These ratios correlated (r = 0.71, p = 0.003) with the particle bound
fractions if the wind hit the PAS at a slanted angle, but not if the angle of incidence was 90° (p =
0.2). This observation may suggest that a slanted angle of incidence causes more particles to be
trapped in the XAD-filled mesh cylinders. This would be consistent with previous studies on the
PUF-PAS which indicated that wind accelerates particle transfer from air to PUF.87
The PSRs for PCBs under slanted wind conditions were higher than when the wind blew the
PAS at a straight angle (Table S5.4), which agrees with our hypothesis. However, ANCOVA
(see SI) indicated no significant difference in the slopes of the uptake curve (i.e. PSRs) between
the straight and slanted windy conditions for the majority of PCBs congeners (Figure S5.14). In
contrast to a previous study with the PUF-PAS showing a significant influence of the angle of
wind incidence on PSRs,97
observations in this study suggest that this angle has little effect on
the chemical uptake kinetics of the cylindrical XAD-PAS. In the field PASs oftentimes are
subject to highly variable wind conditions due to the effect of local terrain such as a mountain
slope and lack of influence of such variations on PSRs would assist in increasing the precision of
the measurement. Nevertheless, this study only tested two wind directions relative to the PASs,
and further study on wind blowing at other angles is necessary in order to provide more solid
evidence to support this conclusion.
5.4.4 Simulated Wind Conditions in the Sampler
CFD simulations were performed to investigate wind fields in the PASs subject to wind blowing
at straight and at 45° slanted angles towards the PAS. From the simulated wind field at the cross
sections at the bottom, middle and top of the PAS (Figure 5.5), we note the wind speeds were
over 70% lower inside than outside the housing, suggesting the housing largely shields the
sampling medium from the wind. This is consistent with a previous study showing that the
sampler housing dampened the wind and PSR variability in PUF-disk PAS.92
In the PAS with
wind blowing at a straight angle, wind speeds at the top and middle cross sections were similar
but lower than that at the bottom. Wind blowing at 45° angle seems to have more influence on
the wind exposure of the sampling medium within the housings; in particular, the middle and top
cross sections are predicted to have a higher wind exposure than if the wind is blowing at the

128
PAS at a straight angle. Nevertheless, the differences in the wind speeds within the housing of
PAS subjected to straight and slanted wind are predicted to be small, which probably explains
the non-significant difference in the PSRs between the two wind conditions.
Figure 5.5 Computational fluid dynamic simulations of wind field on the cross sections at the
top (a and d), middle (b and e) and bottom (c and f) of the XAD mesh cylinders within the
housing of the passive air samplers subject to wind blowing at straight (a-c) and at 45° slanted
angles (e-f) towards the sampler
5.4.5 Implications and Further Research Questions Originating From This Study
The present study shows that wind would increase the PSRs and reduce the non-uniform axial
distributions of chemicals in the cylindrical sampling medium of the XAD-PAS. When applying
PASs under less windy conditions such as indoors, PSRs obtained in outdoor calibration studies
are unlikely to be valid; PAS will need to be recalibrated for use under conditions with limited
air turbulence. Higher PSR induced by wind allow for shorter PAS deployment times, if the
sampled chemicals are close to the detection limit. Windy conditions also tend to reduce the non-
uniform uptake by the sampling medium at different distances from the opening of the housing.
0 1 2 3 4 5Wind speed (m/s)
(a)
(b)
(c)
(d)
(e)
(f)

129
However, the influence of wind on PSRs can introduce large uncertainty to PAS-derived air
concentrations. Therefore, optimization of PAS design would seek to minimize the influence of
wind (e.g. by covering the opening of the housing with a fine mesh screen) while maximizing the
PSR by varying other factors that are independent of environmental conditions (e.g. the
configuration of the sampling medium). The results of this study show a significant increase in
PSRs between wind-still conditions and wind speeds of ~4m/s while an earlier study indicated no
significant change of PSRs when ambient wind speeds increased from 5 to 15 m/s. This suggests
that the wind effect on PSRs may depend on the wind speed range. As such, further studies on
the wind effect on the PSRs at different wind speeds are worthwhile to fill this knowledge gap.
5.5 Acknowledgments
We acknowledge research funding from the Canadian Foundation for Climate and Atmospheric
Sciences and the Natural Sciences and Engineering Research Council of Canada. XZ also
acknowledges financial support by an Ontario Graduate Scholarship.

130
Supporting Information of Chapter 5
Figure S5.1 Passive air samplers with axially segmented XAD-filled mesh cylinder to study
the axial chemical distribution within the sampling medium.
Figure S5.2 Experiment setup to study chemical distributions in the axially segmented passive
sampling medium (XAD mesh cylinder) under wind and wind still conditions.

131
Figure S5.3 Experiment setup to study potential effect of solar radiation on chemical uptake
and axial distribution within the XAD mesh cylinder.

132
Figure S5.4 Experiment setup to study potential wind effects on chemical uptake by the XAD
passive air sampler.
Figure S5.5 Variations of wind speed measured at the mouth of the fans (point A of Figure 1)
and at the openings of the sampler housings (point B of Figure 1) for the 24 passive air samplers
subjected under lab generated windy conditions.
2.5
3
3.5
4
4.5
5
5.5
6
11.0 11.5 12.0 12.5 13.0 13.5
12.0 ± 0.4
4.3 ± 0.2
3.6 ± 0.3
11.8 ± 0.4
Wind speed at the mouth of the fan (m/s)
Win
d s
pee
d a
t th
e m
ou
th o
f th
e PA
S (m
/s)

133
Table S5.1 Target ions, quanlify ions and limit of detection (LOD) of the PCB homolog
groups analyzed using GC-MS selected ion monitoring mode.
a LOD calculated as the chemical amount of which the instrument detects a signal corresponding
to three times of the noise level.
Class Chemical Target
Ion
Qualify
Ion
(Qual. /Targ.)
*100%
LOD a
(ng/sample)
Internal Standard Mirex 272 274 81.1 n/a
Surrogate Standard 13CPCB77 304 302 77.2 n/a
Surrogate Standard 13CPCB101 338 340 64.8 n/a
Surrogate Standard 13CPCB141 372 374 81 n/a
Surrogate Standard 13CPCB178 406 408 97.2 n/a
Target Analyte Tri-CB 256 258 98 0.5
Target Analyte Tetra-CB 292 290 76.7 1
Target Analyte Penta-CB 326 328 65.3 0.2
Target Analyte Hexa-CB 360 362 81.4 1.5

134
Figure S5.6 Amounts of PCBs accumulated in the three axially segment3ed passive air
sampling medium (XAD mesh cylinder) of passive air samplers deployed in the four indoor
locations (L1-4), passive air samplers with lab generated wind (L1W), and at outdoor location
(OD)
0
400
800
1200
1600
0
20
40
60
80
100
120
0
50
100
150
200
0
20
40
60
80
100
0
50
100
150
200
250
L1
L2
OD
L3
L4
Am
ou
nt o
f P
CB
s a
ccu
mu
late
d in
th
e p
ass
ive
sam
pli
ng
med
ium
(ng)
PCB congener
(a)
(f)
(d)
(c)
(b)
0
2000
4000
6000
8000
10000
12000
14000(e) L1W

135
Figure S5.7 Distribution of PCBs in the three axially segmented XAD mesh cylinders in the
duplicated PASs blown with lab generated wind.
Figure S5.8 Distribution of PCBs in the three axially segmented XAD mesh cylinders in the
duplicated PASs (a) under the quasi wind still condition; (b) under the lab generated windy
condition; (c) in outdoor environment
0%
20%
40%
60%
80%
100%
0%
20%
40%
60%
80%
100%
Per
cen
t of
PC
Bs
acc
um
ula
ted
PCB congener
Perc
enta
ge o
f PC
Bs
accu
mu
late
d in
eac
h s
egm
ent
PCB congener
(a)
(b)
(c)
0%
20%
40%
60%
80%
100%
0%
20%
40%
60%
80%
100%
31
/28
52
49
44
74
66
95
10
1
99
87
11
0
11
8
14
9
15
3
13
8
0%
20%
40%
60%
80%
100%
31
/28
52
49
44
74
66
95
10
1
99
87
11
0
11
8
14
9
15
3
13
8

136
Table S5.2 Two-factorial ANOVA and Scheffé's post hoc test on the PCB congeners
accumulated at the three axially segmented PSM
Sample
ANOVA on ln-transformed PCB amount
Scheffé's Post Hoc Test
SS df MS F p
B M T
L1
PSM Segment 8.0 2.0 4.0 148.8 0.000
B
<0.001 <0.001
PCB Congener 63.6 14.0 4.5 169.2 0.000
M <0.001
<0.001
Segment * Congener 0.0 28.0 0.0 0.0 1.000
T <0.001 <0.001
B M T
L1_Wind
PSM Segment 0.9 2.0 0.5 2.5 0.092
B
0.774 0.342
PCB Congener 64.4 14.0 4.6 24.9 0.000
M 0.774
0.1
Segment * Congener 0.0 28.0 0.0 0.0 1.000
T 0.342 0.1
B M T
L2
PSM Segment 7.0 2.0 3.5 70.9 0.000
B
<0.001 <0.001
PCB Congener 48.7 14.0 3.5 70.6 0.000
M <0.001
<0.001
Segment * Congener 0.3 28.0 0.0 0.2 1.000
T <0.001 <0.001
B M T
L3
PSM Segment 1.2 2.0 0.6 9.9 0.000
B
<0.001 0.209
PCB Congener 42.7 14.0 3.1 49.1 0.000
M <0.001
0.04
Segment * Congener 0.1 28.0 0.0 0.1 1.000
T 0.209 0.04
B M T
L4
PSM Segment 2.4 2.0 1.2 36.6 0.000
B
<0.001 <0.001
PCB Congener 39.9 11.0 3.6 111.4 0.000
M <0.001
0.787
Segment * Congener 0.1 22.0 0.0 0.2 1.000
T <0.001 0.787
B M T
OD
PSM Segment 0.5 2.0 0.2 79.6 0.000
B
<0.001 <0.001
PCB Congener 42.5 14.0 3.0 1034.8 0.000
M <0.001
<0.001
Segment * Congener 0.0 28.0 0.0 0.3 0.999
T <0.001 <0.001
B M T
OD_Black
PSM Segment 2.7 2.0 1.4 197.1 0.000
B
<0.001 <0.001
PCB Congener 42.2 14.0 3.0 432.4 0.000
M <0.001
<0.001
Segment * Congener 0.0 28.0 0.0 0.2 1.000
T <0.001 <0.001
B M T
OD_Covered
PSM Segment 0.5 2.0 0.2 177.9 0.000
B
<0.001 <0.001
PCB Congener 44.2 14.0 3.2 2428.6 0.000
M <0.001
<0.001
Segment * Congener 0.0 28.0 0.0 0.3 0.998
T <0.001 <0.001

137
Testing the slopes of two linear regressions using analysis of covariance (ANCOVA).
ANCOVA Model: ( ) i j i ij i j
Y A X X
i jY : The value of the response variable for the jth observation in the ith treatment of factor A
: The overall mean value of the response variable
iA : The effect of the ith treatment of factor A, defined as the difference of the mean of each A
and the overall mean ( i i
A )
: A combined regression coefficient representing the pooling of the regression slopes of Y on
X within each group.
ijX : Covariate value for the jth replicated observation from the ith level of factor A
X : mean value of covariate
i j
: Unexplained error associated with jth replicate observation from the ith level of factor A
Null hypothesis to test the slopes of regression lines (H0): no difference between the regression
coefficients under treatment of A1, A2…Ai (i.e.1 2
... i)
When the interaction effect between the treatment factor A and the covariate in the ANCOVA
model is significant, the effect the covariate on the response depends on the treatment factor,
which means the slopes of regressions for each treatment factor are not statistically the same and
H0 is rejected.

138
Figure S5.9 Mass of PCBs accumulated in the three axially segmented passive air sampling
medium (XAD mesh cylinder) of passive air samplers deployed outdoors (a) under normal
condition (b) with black sampler housing and (c) with black sampler housing shaded from direct
sunshine.
0
50
100
150
200
250
0
50
100
150
200
250
0
50
100
150
200
250
PCB
am
ou
nt a
ccu
mu
late
d (n
g)
PCB congener
(a)
(b)
(c)

139
Figure S5.10 Distribution of PCBs in the three axially segmented XAD mesh cylinders in the
normal housings, back housings and housings shaded from sunshine.
0%
20%
40%
60%
80%
100%
0%
20%
40%
60%
80%
100%
0%
20%
40%
60%
80%
100%Perc
ent o
f PC
Bs
accu
mu
late
d
PCB congener

140
Table S5.3 Descriptive statistics on the temperature (°C) recorded by the temperature logger
in the passive air samplers deployed outdoors
Logger position in the
passive air sampler
Logger recorded temperature (°C)
A B C D E F G H I
Max 49.0 50.5 49.5 43.0 45.5 45.0 37.0 38.5 39.0
75%ile 22.0 22.0 21.5 21.5 21.5 21.5 20.5 20.5 21.0
Mean 16.8 16.9 16.9 16.8 16.8 16.4 15.7 16.0 16.4
Median 15.5 15.5 15.5 15.5 15.5 15.5 15.0 15.5 15.5
25%ile 10.5 10.5 11.0 11.5 11.0 11.0 11.0 11.0 11.5
Min -2.5 -2.5 -1.5 -1.5 -1.5 -1.5 -1.5 -1.0 -1.0
Outer wall
of PAS housing painted
black
Sun shelter
Temperature
logger
I
H
G
F
E
D
C
B
A

141
Figure S5.11 Temperature differences in the normal, black, and shaded passive sampler
housing.
-2 0 2 4 6 8 10 12 140
200
400
600
Y A
xis
Title
X Axis Title
BlackT-CovT
-2 0 2 4 6 8 10 12 140
200
400
600
Y A
xis
Title
X Axis Title
BlackM-CovM
-2 0 2 4 6 8 10 12 140
200
400
600
Y A
xis
Title
X Axis Title
BlackB-CovB
-2 0 2 4 60
200
400
600
800
Y A
xis
Title
X Axis Title
BlackT-NormT
-2 0 2 4 60
200
400
600
800
Y A
xis
Title
X Axis Title
BlackM-NormM
-2 0 2 4 60
200
400
600
800
Y A
xis
Title
X Axis Title
BlackB-NormB
-2 0 2 4 6 8 100
200
400
600
800
1000
Y A
xis
Title
X Axis Title
NormT-CovT
-2 0 2 4 6 8 100
200
400
600
800
1000
Y A
xis
Title
X Axis Title
NormM-CovM
-2 0 2 4 6 8 100
200
400
600
800
1000
Y A
xis
Title
X Axis Title
NormB-CovB
Black - Normal Normal-Covered Black - Covered
Top
Middle
Bottom
Top
Middle
Bottom
Top
Middle
Bottom
Temperature Difference (°C)
Nu
mb
er o
f O
bse
rvat
ion
Bottom
Middle
Top

142
Figure S5.12 Comparison of temperatures (°C) at different positions within the passive air
sampling housing.
y = 0.97x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 1.00x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 0.97x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 1.02x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 1.04x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 1.02x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 1.01x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 0.99x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
y = 0.98x
-5
5
15
25
35
45
-5 0 5 10 15 20 25 30 35 40 45
Sun shelter
I
H
G
F
E
D
C
B
A
A
B
A
C
B
C
D
E
D
F
E
F
G
H
G
I
H
I

143
Figure S5.13 Passive air sampling kinetics for samplers under windy (lab generated wind
blowing at 45° slanted angle and at straight angle towards the cylindrical passive air samplers)
and wind still conditions.
y = 0.49xy = 0.44x
y = 0.13x0
10
20
30
40
50
60
70
80
90
0 40 80 120 160
TriCB31/28
y = 0.64xy = 0.58x
y = 0.17x0
20
40
60
80
100
120
0 40 80 120 160
TetraCB52
y = 0.75xy = 0.65x
y = 0.20x0
20
40
60
80
100
120
140
0 40 80 120 160
TetraCB49
y = 0.58xy = 0.50x
y = 0.16x0
20
40
60
80
100
120
0 40 80 120 160
TetraCB44
y = 0.74xy = 0.65x
y = 0.19x0
20
40
60
80
100
120
140
0 40 80 120 160
PentaCB95
y = 0.92xy = 0.81x
y = 0.24x0
20
40
60
80
100
120
140
160
180
0 40 80 120 160
TetraCB66
y = 0.81xy = 0.70x
y = 0.21x0
20
40
60
80
100
120
140
160
0 40 80 120 160
PentaCB101
y = 0.67xy = 0.58x
y = 0.17x0
20
40
60
80
100
120
0 40 80 120 160
PentaCB99
y = 0.85xy = 0.74x
y = 0.22x0
20
40
60
80
100
120
140
160
0 40 80 120 160
PentaCB87
y = 0.97xy = 0.84x
y = 0.25x0
20
40
60
80
100
120
140
160
180
0 40 80 120 160
PentaCB110
y = 0.94xy = 0.81x
y = 0.24x0
20
40
60
80
100
120
140
160
180
0 40 80 120 160
HexaCB149
y = 1.21xy = 1.05x
y = 0.32x0
50
100
150
200
250
0 40 80 120 160
HexaCB118
y 1.00xy = 0.82x
y = 0.26x0
20
40
60
80
100
120
140
160
180
0 40 80 120 160
TetraCB74
y = 1.21x
y = 1.00x
y = 0.29x0
50
100
150
200
250
0 40 80 120 160
HexaCB153
y = 1.35xy = 1.16x
y = 0.33x0
50
100
150
200
250
0 40 80 120 160
HexaCB138
Deployment time (d)
Equ
ival
ent
Air
Vo
lum
e (m
3)

144
Table S5.4 Passive sampling rates (PSRs) derived as the slopes of the regressiona between
the deployment time and equivalent sampling volume.
Windy, Slanted Angle
Windy, straight
no wind
PCB congener PSR (m3/d) SE b R2 c
PSR (m3/d) SE R2
PSR (m3/d) SE R2
TriCB31/28 0.49 0.02 0.98
0.44 0.02 0.99
0.13 0.01 0.93
TetraCB52 0.64 0.02 0.99
0.58 0.02 0.98
0.17 0.01 0.94
TetraCB49 0.75 0.03 0.98
0.65 0.02 0.99
0.20 0.02 0.93
TetraCB44 0.58 0.02 0.98
0.50 0.02 0.98
0.16 0.01 0.93
TetraCB74 1.00 0.04 0.99
0.89 0.04 0.98
0.26 0.02 0.95
TetraCB66 0.92 0.03 0.99
0.81 0.03 0.99
0.24 0.02 0.92
PentaCB95 0.74 0.03 0.99
0.65 0.02 0.98
0.19 0.02 0.94
PentaCB101 0.81 0.03 0.99
0.70 0.03 0.99
0.21 0.02 0.94
PentaCB99 0.67 0.03 0.98
0.58 0.02 0.98
0.17 0.01 0.93
PentaCB87 0.85 0.03 0.99
0.74 0.03 0.98
0.22 0.02 0.93
PentaCB110 0.97 0.03 0.99
0.84 0.03 0.98
0.25 0.02 0.93
HexaCB149 0.94 0.03 0.99
0.81 0.03 0.98
0.24 0.02 0.93
PentaCB118 1.21 0.05 0.98
1.05 0.03 0.99
0.32 0.03 0.93
HexaCB153 1.21 0.05 0.98
1.00 0.04 0.98
0.29 0.03 0.92
HexaCB138 1.35 0.04 0.99
1.16 0.05 0.98
0.33 0.03 0.93
a regression forced through the origin
b Standard error of the regression coefficients (PSRs)
c For regression through the origin (the no-intercept model), R
2 measures the proportion of the
variability in the dependent variable explained by regression. This cannot be compared to R2 for
models which include an intercept

145
Figure S5.14 (a) Passive air sampling rates of PCBs under quasi wind still condition and with
lab generated wind blowing at straight and 45° slanted angles towards the passive air samplers;
(b) statistical test on the difference of passive air sampling rates between the two windy
conditions.
0.00.20.40.60.81.01.21.4
0
0.05
0.1
0.15
0.2
PCB congener
PSR
(m3/d
)P
valu
e
(a)
(b) PSR difference betweentwo windy conditions
significant
non-significant

146
Chapter 6. Application of passive air samplers and flow-through air samplers to assess semi-volatile organic contaminants in the atmosphere of
Hawaii
Xianming Zhang, John Barnes, Ying D. Lei, Frank Wania
Contributions: F. Wania and X. Zhang planned the sampling campaign. X. Zhang did the field
work with the assistance of J. Barnes, J. Armitage, and A. Gawor. X. Zhang extracted the
samples, performed analysis using GC/MS/MS under the guidance of Y.D. Lei, and processed
the chromatograms. X. Zhang interpreted the data and wrote the manuscript with the guidance of
F. Wania.

147
6.1 Abstract
An air sampling campaign using passive air samplers (PASs) and flow-through samplers (FTSs)
for semivolatile organic compounds (SVOCs) was conducted on the Big Island of Hawaii with
the purpose to (1) test the potential starvation effect of PASs in the field, (2) explore the vertical
distribution of SVOCs along an altitudinal transect, and (3) assess global SVOC background
concentrations over the Central Northern Pacific. XAD-resin based PASs were deployed from
May to September 2011 at six sites along a transect from the northeastern coast to the Mauna
Loa Observatory 3400 m above sea level and at three control sites on the island. By crossing the
trade wind inversion layer the transect ranged from the marine boundary layer to the free
troposphere. At the two ends of the transect, FTSs were deployed to simultaneously sample air at
monthly resolution. Based on a comparison of the amounts of polycyclic aromatic hydrocarbons
(PAHs) and polybrominated diphenyl ethers (PBDEs) accumulated in differently configured
PAS deployed side-by-side, the starvation effect was judged insignificant, i.e. the kinetic
resistance for chemical transfer from ambient air into the sampler housing had no significant
influence on overall chemical uptake. Elevated PAHs and PBDEs levels at two sites close to Hilo
indicated contributions of local sources to the SVOCs in air. SVOC concentrations decreased
with increasing elevation. Higher rates of decrease for PAHs than for PBDEs correspond to
higher atmospheric degradation rates of PAHs than PBDEs. Levels of PAHs and PBDEs at the
Mauna Loa Observatory were generally at the lower end of the range of concentrations reported
at other remote sites, including the Arctic. However, in contrast to the Arctic, long range
atmospheric transport is deemed less important than human-induced material flow as the source
of the SVOCs to the island’s atmosphere. The latter process would be important in the chemical
life cycle impact assessment for environments such as isolated tropical islands.
6.2 Introduction
Semivolatile organic chemicals (SVOCs) such as polybrominated diphenyl ethers (PBDEs),
polycyclic aromatic hydrocarbons (PAHs) have attracted great concerns because of their
potential hazard to environment and humans.30,66,159,160
Some SVOCs (e.g. PBDEs) were
produced intentionally for enhancing the function of commercial products while others (e.g.
PAHs) are generated from natural or anthropogenic processes such as combustion. Due to
national and international regulations on some SVOCs, re-volatilization from soils and oceans is

148
gaining in importance relative to primary emissions to the atmosphere.161
Upon entering the
atmosphere, SVOCs are prone to undergo long range atmospheric transport to remote regions
where local emissions are low. Global atmospheric transport of SVOCs mainly occurs in the free
troposphere (FT) due to higher wind speeds and limited exchange with the earth’s surface.162,163
As such, investigating the occurrence of SVOCs in the FT is important for understanding
atmospheric transport of SVOCs to remote regions.163
Due to the difficulty of accessing
sampling sites, much fewer measurements of SVOCs have been conducted in the FT than the
planetary boundary layer. Such measurements were mainly conducted from aircraft164,165
or at
high mountain sites,160,163,166
and most have been conducted over the continents. Only a few have
focused on SVOCs in the FT over the oceans by sampling high altitude sites on an island.163
SVOCs in air are conventionally sampled using high-volume active air samplers
(HVAASs),167,168
which are able to provide high temporal resolution and information on
gas/particle partitioning.108,163,169
However, HVAASs require electricity and frequent operator
visits and are difficult and expensive to deploy at multiple sites to study the spatial variations of
SVOCs in the air of remote regions.108
Overcoming these disadvantages, passive air samplers
(PASs) are increasingly used to obtain time-integrated SVOC air concentrations in various types
of environment.42,72,154
PASs are especially useful in providing high spatial resolution data to
elucidate the fate of SVOCs in remote regions such as mountains.34,37,166,170-172
Although PASs
have many advantages and have been widely used, passive sampling rates (PSRs) are potentially
influenced by many factors. Understanding these factors is necessary in order to properly
interpret and compare PAS-derived air concentrations. A few studies have investigated
potentially influential factors such as wind, temperature, properties of the target chemicals, and
sampler configuration.89,90,97,127
A question arose from our previous study:126
Does a starvation
effect exist and does it affect PSRs of the XAD-resin based PASs? This effect refers to lower
concentrations of target chemicals within the PAS housing than in ambient air, which would
occur if the uptake of chemicals by the sampling medium is faster than the rate of transfer from
ambient air into the PAS housing. So far, the existence of such an effect has not been tested in
the field.
While time-integrated sampling is an advantage of PASs, the PSRs are generally low (< 5 m3/d)
so that PASs have to be deployed from several months to a year in order to allow for the
detection of the accumulated chemicals. To overcome the limitation of low temporal resolution

149
while keeping other advantages of PASs, a flow-through sampler (FTS) was developed to sample
SVOCs in air.45
The FTS consists of a horizontally oriented flow tube, which turns into the wind
with the help of vanes. It relies on the wind to pass air through a plug of polyurethane foam that
serves as the sampling medium. Such a design can increase the sampling rate (> 15 m m3/d) and
has proven useful for monitoring SVOCs in remote areas with a much higher temporal resolution
than PAS.46
In this study, an air sampling campaign using PASs and FTSs was conducted on the Big Island
of Hawaii with the aim to (1) test the potential starvation effect on PASs ; (2) explore the vertical
variations of SVOCs from sea level to the top of the Mauna Loa volcano, (3) assess the
occurrence of several groups of SVOCs in the FT over the central northern Pacific and compare
their concentrations with those in the marine boundary layer.
Figure 6.1 Locations of the sampling sites on the Big Island of Hawaii. A-I: passive air
samplers; A and F: flow-through air samplers.
6.3 Materials and Methods
6.3.1 Sampling Sites
Located in the Central Northern Pacific, the archipelago of Hawaii is a relatively easily accessed
place far from the continents (>3000 km). The air sampling campaign was conducted on the Big
Island of Hawaii, which is a volcanic island covering 10,432 km2.173,174
Less than 1.5 million

150
people live within a 3000 km radius of the island, which itself has a population of 185,000.29
The
elevation on the island rises by more than 3000 m over a horizontal distance of less than 60 km.
Prevailing northeasterly trade wind bring marine air to the island. A persistent trade wind
inversion caps the atmospheric planetary boundary layer at approximately 2000 m above sea
level.175
6.3.2 Sampling Campaign
Cylindrical PASs using XAD-resin filled mesh cylinder as the passive sampling medium (PSM)
were deployed to conduct time-integrated sampling at nine sites (labeled A-I in Figure 6.1) from
May to September, 2011. Six of these sites form a transect from the northeastern coast (Site A,
elevation: 0 m) to the Mauna Loa Observatory (Site F, elevation: 3400 m) and extend from the
planetary boundary layer (sites A-D) to the FT (sites E and F). For comparison PAS were also
deployed at sites on the east end of the island (G), in Volcano Village (H), and in the northeast of
the island (I). Two FTSs were deployed at sites A and F to sample air at a monthly temporal
resolution during the PAS deployment period. Detailed information on the location and elevation
of the sampling sites is given in Table S6.1 of the Supporting Information (SI).
At each of the sites (except A and H) three XAD-PASs (Figure S6.1) were deployed: a long one,
a short one and a short one with two XAD-resin filled mesh cylinders in one housing. By
comparing the amounts of SVOCs accumulated in three differently configured PASs deployed
side-by-side, we aimed to probe the existence of a starvation effect. At each site, a temperature
logger (ACR System Inc.) was mounted in a PAS housing to record the temperatures during the
sampling period (Figure S6.2).
Pre-extracted XAD resin used in the PASs was cleaned by Soxhlet extractions with acetone for
24 h and with hexane for another 24h. Polyurethane foam (PUF) plugs used in the FTSs were
cleaned up by tap water and deionized water and then Soxhlet extracted with acetone for 24 h
and with petroleum ether for another 24h. XAD-resin filled mesh cylinders were sealed in pre-
cleaned stainless steel tubes and PUF plugs were stored in pre-cleaned air tight glass jars before
being used in the field. Upon retrieval, each XAD-filled mesh cylinder and each PUF plug was
separately placed in their original containers and stored in a freezer in Hilo, Hawaii before being
transported to a freezer in the lab in Toronto at the end of the sampling campaign. The samples

151
from the FTSs and from the PASs were extracted within one month and three months of retrieval,
respectively.
6.3.3 Sample Extraction
Prior to extraction, each sample was spiked with 100 μL of isotope-labeled standards. Identities
and concentrations of those standards are listed in Table S6.2. The XAD-resin was extracted by
accelerated solvent extraction (Dionex ASE-350) using 33 or 66 ml cells for the short and long
mesh cylinders, respectively. The ASE conditions followed a methods previously used in our
lab127,149
: solvent 50:50 hexane:acetone; temperature 75°C; pressure 1500 psi; static time 5 min;
static cycles 3; flush volume 100%; purge time 240 s. The PUF plugs were Sohlet extracted with
petroleum ether for 24 h. After extraction, each extract was roto-evaporated to ~2 mL and
filtered through ~1 g of anhydrous sodium sulfate packed in a disposable pasteur pipet to remove
moisture. The eluent was solvent exchanged to isooctane, blown down with high purity nitrogen,
transferred to a GC vial, and further reduced to 0.5 mL. To the GC vial, 10 μL of 10 ng∙μL-1
mirex and 20 μL of 1 ng∙μL-1
each of BDE-75, 116, 205 were added to quantify the recovery of
the surrogates.
6.3.4 Sample Analysis
Both PAHs and PBDEs were analyzed using an Agilent 7890A gas chromatography (GC)
coupled to an Agilent 7000A triple quadrupole mass spectrometry (MS/MS) with EI source. For
PAH analysis, 1.0 μL of the sample was injected in splitless mode with the injector temperature
at 250 °C. PAHs in the sample were separated using a J&W HP-5MS column (30m × 250 μm ID
× 0.25 μm film thickness) with helium (1.2 mL/min) as the carrier gas. The column temperature
program started from 90 °C for 1 min, to 250 °C at 10 °C·min-1
, to 300 °C at 5 °C·min-1
, and held
for 3 min. The interface, source and quadrupole temperatures were set at 250 °C, 230 °C and
150°C, respectively. For PBDE analysis, 2.0 μL of the sample was injected in splitless mode
with the injector temperature at 285 °C. PBDEs in the sample were separated using a J&W HP-
5MS column (15m × 250 μm ID × 0.25 μm film thickness) with helium (1.8 mL/min) as the
carrier gas. The column temperature program started from 100 °C, to 185 °C at 25 °C·min-1
, to
275 °C at 15 °C·min-1
, to 315 °C at 45 °C·min-1
, and held for 6 min. The interface, source and
quadrupole temperatures were set at 300 °C, 230 °C and 150 °C, respectively. Both PAHs and
PBDEs were detected using multiple reaction monitoring (MRM) mode with He (2.25 mL/min)

152
as the quench gas and N2 (1.5 mL/min) as the collision gas. The precursor ions, product ions and
collision energies selected and monitored for PAH analysis are listed in Table S6.3. Those for
PBDE analysis are listed in Table S6.4.
6.3.5 QA/QC
Field blanks were collected by exposing the sampling medium to the air at the sampling sites for
1 min and by storing and transporting them the same way as the samples until analysis. 8 field
blanks of the XAD-filled mesh cylinder were collected. 10 field blanks (1 for each site every
month) were originally planned for the FTSs during the whole sampling campaign. However,
two glass jars were broken while being transported to the field so the number of field blanks for
the FTSs was reduced to 8. Of the PAHs and PBDEs analyzed, only fluorene and phenanthrene
can be detected in the blanks for the FTSs. The blank levels (1-10 ng/sample) were <5% for 90%
of the samples and <10% for all the samples. The reported data were not blank corrected.
Recoveries of the isotope labeled standards spiked prior to extraction were 60-148% PAHs and
56-121% for PBDEs. The reported data were recovery corrected.
6.3.6 Air Mass Back Trajectory Analysis
The origins of air masses arriving at sampling sites A and F were assessed via back trajectories
calculated using the hybrid single-particle Lagrangian integrated trajectory (HYSPLIT) model.176
Based on the Global Data Assimilation System (GDAS) 1 degree latitude longitude
meteorological dataset, 14 d back trajectories for air masses arriving at 50 m above ground of the
two sites were calculated every 6 h for the entire sampling period. The point densities of
endpoints of the trajectories were derived and mapped using the spatial analysis tool of ArcGIS
10.0.
6.4 Results and Discussion
6.4.1 PAHs and PBDEs Accumulated in PASs of Different Configuration
Deploying side-by-side a short PAS with two 10-cm-long XAD filled mesh cylinders, a short
PAS with one 10-cm-long XAD filled mesh cylinder and a long PAS with one 20-cm-long XAD
filled mesh cylinder (Figure S6.2), we intended to test the existence of starvation effect of the
XAD-PAS, i.e. whether the rate of uptake by the PSM is faster than the rate of chemical transfer
from ambient air into the sampler housing.

153
Figure 6.2 Comparison of the amounts (ng) of chemicals (fluorene, phenanthrene,
fluoranthene, pyrene, BDE47and BDE99) sampled by PASs of different configurations.
Had the starvation existed, the amounts of SVOCs accumulated in each of the two short XAD
filled mesh cylinders placed in a single housing of PAS would be less than the SVOC amounts
sampled by the PAS with only one short XAD-filled mesh cylinder. No such difference was
observed (Figure 6.2a, Wilcoxon signed ranks test, p = 0.17), indicating that chemical transfer
from ambient air to housing (i.e. the resistance posed by the sampler housing) does not
kinetically limit the PSRs of PASs deployed under general outdoor conditions.
Previous studies had indicated that PSRs for the cylindrical XAD-PAS deployed outdoors are
lower than those of the double-bowl shaped PAS using PUF as the PSM.108
The different design
of the sampling housings is one possible cause for different PSRs between the two PASs. The
PSM is more confined in the cylindrical sampler housing of the XAD-PAS than in the double-
bowl housing of the PUF-PAS, which could limit chemical uptake kinetics. Our results indicate
that the cylindrical sampling housing poses no significant kinetic limit on the outdoor PSRs. This
adds merit to the cylindrical PAS housing design because the PSM in such a housing is less
susceptible to the influence of wind than in the double-bowl housing.96
Since a higher influence
of wind on chemical uptake by the PSM adds uncertainty to PSRs,93,118
an ideal PAS housing
design would seek to minimize the wind exposure of the PSM without adding to the overall
kinetic resistance to chemical transfer from ambient air to PSM. Even if we found that outdoors
the PSR is not limited by the cylindrical sampling housing, we cannot infer that this is also the
case in an indoor environment. Lower air turbulence is likely to increase the kinetic resistance
for chemical transfer from ambient air to the inside of the sampler housing. Therefore, similar
0
10
20
30
0 10 20 30
0
5
10
15
0 5 10 15
p=0.17
(a)
0
10
20
30
0 10 20 30
p=0.51
(c)
2 ×
(b)
p=0.64

154
experiments should be conducted indoors to test whether kinetic resistance from the sampler
housing is important under conditions of low air turbulence.
The amounts of SVOCs sampled by the long XAD-PASs were double of those sampled by the
short ones (Figure 6.2b). This implies that under outdoor conditions, no additional uncertainty is
introduced when applying PSRs obtained from calibrations with one type of XAD-PAS to the
other by simply dividing or multiplying the PSRs by two. Because no starvation effect existed,
the amounts of SVOCs sampled by the long PAS did not differ (p = 0.51) from the sum of the
two short XAD-filled mesh cylinders put in one sampler housing (Figure 6.2c). This was
expected as the same interfacial transfer area between the PSM and surrounding air, which
determines the PSR, was the same.127
This result validates our proposed approach for improving
the PAS design:127
instead of using a single piece of PSM in a large PAS housing, several
smaller pieces can be positioned in a smaller PAS housing, which would reduce the expense of
making and shipping large PAS housings without reducing the interfacial transfer area between
the PSM and the surrounding air, and thus also the PSR.
This result also implies that air samples can be duplicated (or triplicated) by putting two (or three)
XAD-filled mesh cylinders in one sampler housing. Again, this could reduce the expenses of
shipping PAS housings to remote field sites. Note that with a larger amount of additional PSM in
a sampler housing the rate of chemical uptake by the PSM may increase and eventually exceed
the rates of chemical transfer from ambient air to the inside of the sampler housing, resulting in a
starvation effect. The maximum amount of PSM that can be used in a single sampler housing
under different environmental conditions without causing a starvation effect remains to be
established.
6.4.2 Passive Air Sampler Derived Spatial Variations of PAHs and PBDEs
Large spatial variations of the XAD-PAS-derived levels (ng/PAS) of PAHs and PBDEs were
observed. The highest PAH levels were found at sampling site A (Figure 6.3a), which is at the
northeastern coast of the island and faces the northwestern Pacific Ocean. Although the
population density around site A is lower than that around site B located in the urban area of Hilo,
Hilo airport is ~1000 m south, a wharf with petroleum refinery facilities is ~600 m west and a
beach park is ~200 m northeast of site A. As petroleum and other fuel combustion could be

155
sources of PAHs, these facilities around site A could possibly contribute to the ~50% higher
PAHs levels at site A than at site B. PAH levels at site A were ~30 times higher than that at site
G, another coastal site ~40 km southeast of site A. The difference between the two coastal sites
indicates PAHs measured at site A were mainly from local sources around site A, instead of from
the marine boundary layer.
Figure 6.3 Spatial distributions of (a) polyaromatic hydrocarbons (PAHs) and (b)
polybrominated diphenyl ethers (PBDEs) sampled by passive air samplers (20 cm long XAD
filled mesh cylinder) on the Big Island of Hawaii. Dash lines indicate the altitude of the sampling
sites.
0
500
1000
1500
2000
2500
3000
3500
Alt
itu
de(m
)0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0
20
40
60
80
PAHs
(ng/PAS)
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
Pyrene
Fluoranthene
Phenanthrene
Fluorene
0.0
50.0
100.0
150.0
200.0
250.0
A B C D E F G H I
Pyrene
Fluoranthene
Phenanthrene
Fluorene
(a)
A
H
B
G
C
I
F
D
E
BDE99
BDE47
PBDEs
(ng/PAS)
0
0.5
1.5
2
2.5
0
500
1000
1500
2000
2500
3000
3500
Alt
itu
de(m
)
1 (b)A
H
B
G
C
I
F
D
E

156
Comparing PAH levels along the transect A to F, the impact of PAH sources in Hilo, which
caused the higher PAH levels at site A and B, is limited to a small area. At site C, ~15 km
southwest of Hilo, PAH levels decreased by an order of magnitude compared to site B. From site
C to site D, PAHs levels decreased by a further 50%. PAHs levels at site E and F were quite
close to those at site D. Comparing the PAH levels at the three control sites (G, H, I) to those
along the transect, PAHs levels at site G and H were close to those at D to F. These sites reflect
the background levels of PAHs on the island and perhaps around the world. At site I close to
Waimea, a small town with a population of ~10 000173
at an elevation of ~800 m above sea level,
PAH levels were between those of sites B and C. The distribution of PAHs across the island
indicates that local sources close to the sampling sites are the main contributor to the variability
in the measured PAH levels.
Different from the PAH spatial distribution, highest PBDEs (BDE47 and 99) levels were
observed in Hilo (site B, Figure 6.3b). This agree with other studies on the spatial distribution of
PBDEs along urban-rural transects and indicates that urban area have strong sources of
PBDEs.25,72
At site A, ~4 km northeast from site B, PBDE levels were ~40% lower. At site G,
another coastal site on the far east of the island with few human inhabitants within a 5 km radius,
PBDE levels were ~30% of that at site A. From this, we can attribute PBDEs measured at site A
mainly to local sources., PBDE levels at site C and D, 10 km and 30 km southwest of site B,
were 35% and 20% of that at site B. Above the trade wind inversion layer (sites E and F), PBDE
in the FT were at similar level. The PBDE levels were also quite similar at the three control sites
(G, H, I) and comparable to the levels in the FT.
The rates of decrease along the A-F transect were lower for the PBDEs than for the PAHs
(Figure S6.3). When the source of an SVOC is at one end of a transect, the concentrations of
SVOCs in air decrease with distance from that end because of dilution, dry and wet deposition,
and degradation.177
The effect of dilution should be identical for different chemicals. Because
BDE47 and 99 have higher octanol/air and lower air/water partition coefficients than the three-
and four-ring PAHs we quantified, the deposition rates for the PBDEs should be higher than for
the PAHs. Therefore, if deposition were to control the rate of concentration decrease, levels of
PBDEs would decrease faster than those of the PAHs along the transect. Because this was not
the case, we postulate that the faster decline in the PAH concentration is due to their faster
atmospheric degradation. This is supported by the AOPWin (v1.92)178
estimated atmospheric

157
half-lives due to reaction with the hydroxyl radical (Table S6.5), which are ~1 d for the PAHs
and > 15 d for the PBDEs.
6.4.3 Monthly Variations of PAHs and PBDEs
Figure 6.4 Flow-through sampler derived air concentrations of fluorene (Fluo), phenanthrene
(Phe), fluoranthene (Flu), pyrene (Pyr), BDE47and BDE99 during the five sampling months.
FTS-derived air concentrations of PAHs and PBDEs at site A and F in each of the five sampling
months are shown in Figure 6.4. Concentrations of the four PAHs (fluorene, phenanthrene,
fluoranthene, pyrene) at site A were ~50-70% lower in August than during the other sampling
0
1000
2000
3000
4000
5000
May Jun Jul Aug Sept0
30
60
90
120
150
May Jun Jul Aug Sept
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
May
Ju
n
Ju
l
Au
g
Sep
t
Site A
BDE99
BDE47
Co
nce
ntr
atio
n (
pg
/m3)
0
1
2
3
4
5
6
7
8
May
Ju
n
Ju
l
Au
g
Sep
t
24 88
Site F
Fluo Phe Flu Pyr
283 282
(a) (b)
(c) (d)

158
months. At site F, the PAH concentrations in August and September were ~3 fold of those in
May to July. Based on the endpoint density of 14 d back trajectories during the sampling month
(Figure S6.4), it seems that differences in the origin of the air mass cannot explain the observed
variations. The back trajectories indicate that there is little change in the air mass origin over the
five sampling month: the air always originated from above the Pacific Ocean to the NE of
Hawaii. The elevated PAHs in August and September at site F were therefore presumably due to
local sources. BDE47 and 99 showed little variations at site A from May to August, but in
September levels were ~ 5 and 30 times higher. Again we cannot attribute this to the origin of
the air mass. In the September sample, BDE99 was higher than BDE47 while in previous months,
BDE47 was dominant. This could indicate a different source of PBDEs to the September sample.
Since we don’t have replicates for the FTS, we cannot exclude the possibility of that specific
sample having become contaminated during the transport to/from the field, although the field
blanks indicate little sample contamination. The monthly variations of PBDEs at site F were
different: concentrations of BDE47 and 99 in May and August were ~4 times higher than in June
and July and ~2 times higher than in September. It is interesting to compare the relative
abundance of BDE47 and 99 at the two sites. At site A, except the September sample,
concentrations of BDE47 are higher than BDE99 while at site F, the two congeners have about
equal abundance. This is consistent with a higher estimated atmospheric transport potential for
BDE47 compared to 99.179
Note that no such change between sites A and F is apparent in the
PASs, because they mainly sample the gas phase while FTSs sample both gas and particle phase
SVOCs from air.
6.4.4 Global Background Levels of Atmospheric PAHs and PBDEs
Being more than 3000 km from the nearest continent, Hawaii can be viewed as a tropical region
remote from global SVOC emissions. The Mauna Loa sampling site (site F) in particular is in the
FT above the trade wind inversion layer and should reflect global background concentrations
over the Central North Pacific Ocean. Concentrations of fluorene, phenanthrene, fluoranthene,
pyrene, BDE47 and BDE99 at Mauna Loa were compared with those reported for other remote
sites. Because the XAD-PAS samples only gas phase SVOCs and the conversion to volumetric
concentrations depends on uncertain PSRs, FTS-derived air concentrations were used for the
comparison. Fluorene, fluoranthene and pyrene concentrations at Mauna Loa are comparable to
HVAAS-derived concentrations reported for Kinngait, Nunavut, and lower than those reported

159
for Alert, Nunavut, and Ny Ålesund, Spitsbergen 180
(Figure 6.5). Phenanthrene concentrations at
Mauna Loa, however, were more comparable to those in Ny Ålesund and higher than those in the
Canadian High Arctic. FTS-measured concentrations of BDE47 and 99 were at the lower bound
of the PAS derived concentrations. The FTS-derived concentrations of BDE47 and BDE99 on
Mauna Loa were close to the lower end of the concentration ranges reported for Nam Co on the
Tibet Plateau,46
, Alert in the Canadian High Arctic46,181
and Nuuk in Greenland,46,181
, except that
concentrations of BDE99 in Nam Co and on Mauna Loa were similar (Figure 6.5). These
comparisons indicate that PAHs and PBDEs measured at Mauna Loa reflect the global
background levels of these SVOCs.
Figure 6.5 Comparison of the PAH air concentrations measured at Mauna Loa in this study
using flow-through samplers (based on data from five sampling months) and passive air samplers
(based on passive sampling rate range of 0.5-5.5 m3/d from previous calibrations
20,89,106) with
those at Arctic background sites.180
0
50
100
150
200
250
0
50
100
150
200
250
0 1 2
0
50
100
150
200
250
0 1 20
50
100
150
200
0
50
100
150
200
0 1 2
0
50
100
150
200
0 1 2
525
0
10
20
30
40
50
0
10
20
30
40
50
0 1 2
0
10
20
30
40
50
0 1 2
120
0
10
20
30
0
10
20
30
0 1 2
0
10
20
30
0 1 2
75
FTS PAS FTS PAS
Fluo Phe
Flu Pyr
Co
nce
ntr
atio
n (p
g/m
3)
max
mean
median
min

160
Figure 6.6 Comparison of the PBDE air concentrations measured at Mauna Loa in this study
using flow-through samplers (based on data from five sampling months) and passive air samplers
(based on passive sampling rate range of 0.5-5.5 m3/d from previous calibrations) with those at
other global background sites.
6.4.5 Origin of SVOCs in Hawaii: Long Range Atmospheric Transport vs. Material Flows
Based on the back trajectory analysis and the spatial distributions of PAHs and PBDEs on the
Big Island of Hawaii, we conclude that SVOCs on the tropical island largely originate locally.
This is in contrast to the Arctic regions, where SVOCs mainly originated from long range
atmospheric transport from continental source regions rather than from local sources, which are
very limited because of the low population density. While PAHs are emitted from combustion
sources, PBDEs emissions are, in the absence of production facilities, mainly associated with
consumer products, to which the chemicals had been added as a flame retardant. Five years after
the ban on penta-BDEs,182
elevated penta-BDE concentrations can still be observed in air close
to the urban area of the island, indicative of emissions from consumer products and the long life
time of PBDEs in those products. As, to our knowledge, no PBDEs have been produced on the
island, the PBDE stock on the island is mainly associated with material flows associated with
human activities. Such chemical transport via material flow to a “remote” island could be a more
important process than the transport of chemical in the natural environment and deserves
attention during environmental impact assessments of chemicals in such ecosystems.
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2
0
0.5
1
1.5
2
2.5
3
0 1 2
0
0.5
1
1.5
2
2.5
3
0 1 20
0.5
1
1.5
2
2.5
3
0 1 2
0
0.5
1
1.5
2
2.5
3
0 1 2
0
0.5
1
1.5
2
2.5
3
0 1 2FTS PAS
12
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2
5.3
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2FTS PAS
BDE47 BDE99C
on
cen
trat
ion
(pg
/m3 )
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2
max
mean
median
min

161
6.5 Acknowledgments
We appreciate assistance provided by A. Gawor, J. Armitage, H. Xiao, P. Suganuma, B.
Wiecking, and K. Hopkins. We acknowledge funding from a Graduate Student Award from the
Centre for Global Change Science at the University of Toronto, an Ontario Graduate Scholarship
and the Natural Sciences and Engineering Research Council of Canada.

162
Supporting Information of Chapter 6
Table S6.1 Geographic coordinates and elevations of the sampling sites
SiteCode Lattitude Longitude Elevation (m) Scene
A 19°43'53.38"N 155°2'51.89"W 0
B 19°42'28.67"N 155° 4'29.04"W 11
C 19°40'49.88"N 155°10'48.00"W 587
D 19°40'23.00"N 155°22'12.00"W 1699
E 19°37'15.42"N 155°28'25.20"W 2240
F 19°32'9.03"N 155°34'30.97"W 3400
G 19°30'58.22"N 154°48'38.83"W 2
H 19°25'46.36"N 155°13'41.46"W 1123
I 20° 1'53.58"N 155°41'42.72"W 784

163
Figure S6.1 Illustration of the three configurations of passive air samplers used in this study
Figure S6.2 Daily averaged temperature profiles at the sampling sites.
10 cm10 cm
(a) (b) (c)
0
5
10
15
20
25
30
25 28 1 4 7 10 13 16 19 22 25 28 31 3 6 9 12 15 18 21 24 27 30 3 6 9 12 15 18 21 24 27 30 2 5 8 11 14 17 20 23 26 29 1 4 7 10 13 16 19 22 25 28
A
B
C
D
E
F
0
5
10
15
20
25
30
25 28 1 4 7 10 13 16 19 22 25 28 31 3 6 9 12 15 18 21 24 27 30 3 6 9 12 15 18 21 24 27 30 2 5 8 11 14 17 20 23 26 29 1 4 7 10 13 16 19 22 25 28
G
H
I
Dai
ly a
vera
ged
tem
per
atu
re (°
C)
05/0
1/20
11
09/0
1/20
11
08/1
5/20
11
07/1
5/20
11
06/1
5/20
11
05/1
5/20
11
06/0
1/20
11
07/0
1/20
11
08/0
1/20
11
08/1
5/20
11
10/0
1/20
11
0
11
587
1699
2240
3400
SiteElev. (m)
2
1123
784

164
Table S6.2 Information on the 100 μL surrogate standards spiked prior to sample extractions
Chemical Concentration (ng/uL)
Chemical Concentration (ng/uL)
13C12 BDE28 0.2
D10 Acenaphthene 0.25
13C12 BDE47 0.19
D8 Acenaphthylene 0.25
13C12 BDE153 0.19
D10 Anthracene 0.25
13C12 BDE209 0.84
D12 Benz[a]anthracene 0.25
D12 Benzo[b]fluoranthene 0.25
13C12 PCB77 0.2
D12 Benzo[k]fluoranthene 0.25
13C12 PCB101 0.2
D12 Benzo[g,h,i]perylene 0.25
13C12 PCB141 0.2
D12 Benzo[a]pyrene 0.25
13C12 PCB178 0.2
D12 Chrysene 0.25
D14 Dibenz[a,h]anthracene 0.25
D4 endosulfan 0.25
D10 Fluoranthene 0.25
D5 atrazine 0.25
D10 Fluorene 0.25
D10 chlorpyrifos 0.25
D12 Indeno[1,2,3-cd]pyrene 0.25
D14 trifluralin 0.25
D8 Naphthalene 0.25
13C6 HCB 0.25
D10 Phenathrene 0.25
13C6 aHCH 0.25
D10 Pyrene 0.25
13C6 gHCH 0.25
13C6 PeCB 0.25
13C4 dieldrin 0.25
13C10 trans chlordane 0.25
13C12 4,4 DDT 0.25

165
Table S6.3 Precursor ions, product ions and collision energies for the multiple reaction
monitoring mode for PAH analysis
Chemica
l
Precursor
Ion
Product
Ion
Collision
Energy
Chemical Precursor
Ion
Product
Ion
Collision
Energy
Fluo 166.0 165.0 30
D10-Fluo 176.0 174.0 30
Phe 178.0 152.0 20
D10-Phe 188.0 160.0 34
Ant 178.0 152.0 20
D10-Ant 188.0 184.0 34
Flu 202.0 201.0 30
D10-Flu 212.0 210.0 30
Pyr 202.0 201.0 30
D10-Pyr 212.0 210.0 30
Chry 228.0 226.0 38
D12-Chry 240.0 236.0 38
BaA 228.0 226.0 38
D12-BaA 240.0 236.0 38
BbF 252.0 250.0 42
D12-BbF 264.0 260.0 42
BkF 252.0 250.0 42
D12-BkF 264.0 260.0 42
BeP 252.0 250.0 42
BaP 252.0 250.0 42
D12-BaP 264.0 260.3 42
IP 276.0 274.0 42
D12-IP 288.0 284.0 42
DBA 278.0 276.0 38
D14-DBA 292.0 284.0 40
BghiP 276.0 274.0 30
D12-BghiP 288.0 284.0 38
Mirex 274.0 274.0 0
Fluo: fluorene; Phe: phenanthrene; Ant: anthrancene; Flu: fluoranthene; Pyr: pyrene; Chry: chrysene; BaA:
benzo(a)pyrene; BbF: benzo(b)fluoranthene; BkF: benzo(k)fluoranthene; BeP: benzo(e)pyrene; BaP:
benzo(a)pyrene; IP: Indeno(1,2,3-c,d)pyrene; DBA: Dibenzo(a,b)anthracene; BghiP: Benzo(g,h,i)perylene

166
Table S6.4 Precursor ions, product ions and collision energies for the multiple reaction
monitoring mode for PBDE analysis
Chemical Precursor
Ion
Product
Ion
Collision
Energy
Chemical Precursor
Ion
Product
Ion
Collision
Energy
BDE-17 247.9 139.0 30
13C-BDE-28 259.9 150.1 30
BDE-28 247.9 139.0 30
13C -BDE-47 497.7 337.9 25
BDE-47 485.7 325.8 55
13C -BDE153 655.7 495.7 25
BDE-66 325.9 138.0 55
13C -BDE209 811.4 651.1 55
BDE-71 325.9 138.0 55
BDE-100 565.7 405.7 55
BDE-75 325.9 138.0 55
BDE-99 565.7 405.7 55
BDE-116 403.7 137.1 25
BDE-138 643.6 483.6 25
BDE-205 801.5 641.6 25
BDE-153 643.6 483.6 25
BDE-154 643.6 483.6 25
BDE-181 561.6 454.6 30
BDE-183 561.6 454.6 30
BDE-190 561.6 454.6 30
BDE-209 799.7 639.6 55

167
Figure S6.3 Decreasing trend of SVOC levels along the transect A to F.
y = -0.06x + 2.28R² = 0.75
-4-3-2-101234
0 20 40 60
y = -0.06x + 3.20R² = 0.80
-4-3-2-101234
0 20 40 60
y = -0.07x + 1.14R² = 0.78
-4-3-2-101234
0 20 40 60
y = -0.05x + 0.18R² = 0.68
-4-3-2-101234
0 20 40 60
y = -0.04x + 0.19R² = 0.88
-4-3-2-101234
0 20 40 60
y = -0.02x - 1.38R² = 0.79
-4-3-2-101234
0 20 40 60
Horizontal distance relative to site A (km)
Fluorene Phenanthrene
Fluoranthene Pyrene
BDE47 BDE99
ln(n
g/P
AS)

168
Table S6.5 APOWin (v1.92) estimated half-life of reaction with hydroxyl radicals in the
atmosphere
Chemical Fluorene Phenanthrene Fluoranthene Pyrene BDE47 BDE99
tOH, 1/2 (d) 1.2 0.8 0.4 0.2 16.4 33.6
Figure S6.4 Endpoint density of trajectories arriving at site A and F during the five sampling
months based on 14 d back trajectory calculated using HYSPLIT model at every 6 h interval.
Site A Site F
Ap
r 2
6-M
ay 2
5, 2
01
1M
ay 2
6-J
un
25
, 20
11
Jun
26
-Ju
l25
, 20
11
Jul2
6-A
ug
25
, 20
11
Au
g26
-Sep
t 2
5, 2
01
1

169
Chapter 7. Conclusions and Outlook
7.1 Conclusions
Although passive air samplers (PASs) have been widely used for monitoring semivolatile
organic compounds (SVOCs) in air, chemical mass transfer processes involved in passive air
sampling had not been fully understood prior to this thesis. Whilst many studies have
investigated factors potentially influencing passive sampling rate (PSRs), many of those
influences could not be explained or predicted with the understanding of the mass transfer
processes that was prevalent prior to the research described in this thesis. In order to fill these
knowledge gaps and to gain further insight into the mechanism of passive air sampling and into
the factors that may influence passive air sampling rates, a series of studies combining controlled
laboratory experiments, mass transfer process modeling, with a field sampling campaign was
conducted. Major conclusions drawn from the results of these studies presented in Chapter 2
through Chapter 6 of this thesis include:
(1) PSRs for the cylindrical XAD-PAS that are reported to be lower than for the double-bowl
polyurethane foam (PUF)-PAS are likely caused by the different configuration of the
sampler housing, and not by the different properties of XAD and PUF.
(2) During the deployment time period of PASs, the sampled SVOCs are unlikely to become
uniformly distributed within the porous passive sampling media (PSMs) of both XAD-
filled mesh cylinders and PUF disks.
(3) SVOCs with higher fractions in the air phase of a porous PSM penetrate deeper into the
PSM. The mass transfer coefficients and the effective diffusivities of SVOCs transfer
within the PSM are negatively correlated with a SVOC’s partition coefficient between
PSM and air.
(4) Because it assumes uniform chemical distribution within the PSM, the widely adopted two-
film model for SVOC uptake in PAS fails to properly describe the mass transfer processes
involved in, and thus the kinetics of, passive air sampling using porous PSM. Neglecting
the influence of the kinetic resistance within the PSM on the overall PSR, as is done in the
two-film model, is therefore not justified.
(5) The kinetic resistance within the porous PSM can have a strong influence on PSRs as
indicated by a model that is based on fundamental laws of mass transfer in air and in

170
porous media and of exchange between air and sorbent, but does not require a uniform
chemical distribution within the PSM.
(6) The kinetic resistance within the porous PSM is negatively correlated with the chemical’s
diffusivity in the air-filled pore space within the PSM, the chemical partition coefficient
and the rate of exchange between the air in the pores and the sorbent. The latter two
parameters vary more with temperature and between chemical species than the
diffusivities.
(7) The large variations of field-calibrated PSRs with temperature and between chemical
species can be explained by the influence of the chemical partition coefficient and the rate
of exchange between gas phase and sorbent on the kinetic resistance within the PSM.
(8) The two-stage uptake process observed for some chemicals in PAS calibration studies is
the result of the kinetic resistance within the PSM. During the initial uptake stage,
chemicals mainly sorb to the surface of the bulk PSM; thus the kinetic resistance within the
PSM is not relevant, and fast chemical uptake is observed. As the surface get saturated,
chemical uptake by the PSM requires diffusion through the porous PSM and the kinetic
resistance within the PSM decreases the overall PSRs.
(9) Because of the evidence that the kinetic resistance within the PSM influences the kinetics
of chemical exchange between ambient air and a porous PSM, the overall kinetic resistance
on the loss of depurations compounds from the PSM to air would be different from the that
on the uptake of target chemicals from air to the PSM. Thus, PSRs derived from the loss
rates of depuration compounds may deviate from the true PSRs.
(10) Water uptake by silica gel follows the same pattern as SVOC uptake by XAD: an initial
quasi-linear uptake phase is followed by a gradually decreasing rate of uptake until
eventually equilibrium is reached. Using water vapor uptake from air by silica-gel filled
mesh cylinders as a surrogate for SVOC uptake by XAD filled mesh cylinders is an
efficient approach to assess the role of those factors that influence PSRs in PAS but are
independent of chemicals and PSM (e.g. sampler configurations).
(11) PSRs are proportional to the interfacial transfer area but not the amount of the PSM
because chemicals mainly accumulate in the outer layer of the PSM during the deployment
time of PASs. With a given amount of sorbent used as PSM, increasing the ratio of
interfacial transfer area to volume can improve the use efficiency of the PSM and also the
PSRs.

171
(12) Under wind still indoor conditions, PSM placed closer to the opening of the PAS housing
tends to have a higher PSR than PSM further away from that opening. This is likely caused
by a different thickness of the stagnant air boundary layer surrounding the PSM at different
positions within the sampling housing. Such non-uniform distributions of SVOCs along the
axial direction of the XAD filled mesh cylinder can be eliminated by wind either
artificially generated in the lab or typically being present under normal outdoor conditions.
(13) Even for the cylindrical XAD-PAS in which the PSM is positioned in a semi-enclosed
sampler housing, wind can have a strong influence on the PSRs. PSRs can increase as
much as 5 fold from wind still to 4 m/s wind speed.
(14) The potential starvation effect on the PSR is insignificant for the cylindrical XAD-PAS
deployed outdoors, i.e. the rate of air exchange between the outside and inside of the PAS
sampler housing is not rate-limiting. Multiple XAD-filled mesh cylinders can be put in one
sampler housing to increase the amount of chemical sampled or to serve as replicates.
(15) The atmospheric distribution patterns of PBDEs and PAHs measured with PASs along an
elevation transect from Hilo to Mauna Loa Observatory on Hawaii suggested faster rates of
decrease for PAHs than for PBDEs, which corresponds to higher atmospheric degradation
rates of PAHs than PBDEs.
(16) In contrast to the Arctic, where long range atmospheric transport is deemed the
predominant input pathways for SVOCs, on isolated tropical islands, human-induced
material flow can be the dominant source of the SVOCs, which could be important in the
chemical life cycle impact assessment for such environments.
7.2 Overall Implications
7.2.1 Uncertainty associated with passive air sampling derived air concentrations
The air concentration (CA) of a SVOC derived using passive air samplers are based on the
amount (mchem) of the SVOC detected in the passive sampling medium and the passive air
sampling rate (PSR). The uncertainly associated with the PAS-derived air concentrations can
thus be attributed to both mchem and PSR (Figure 7.1). mchem is obtained by solvent extraction of
the sampling medium and subsequent instrumental analysis of the sample extract. The variations
of replicated PASs deployed side by side can reveal the uncertainty from sample extraction and
analysis. The coefficients of variation of mchem between replicated PASs are generally <30%.

172
This uncertainty can be characterized through sample replication and generally is smaller than
the uncertainty originating from the PSR. PSRs are subject to the influence by temperatures,
chemical species and wind conditions. Previous PAS calibrations20,28,87,88,104,183
indicated that
such factors can results in variations in PSRs as large as one order of magnitude. The uncertainty
in PSR thus contributes most to the uncertainty in PAS derived air concentrations.
With this study, the current understanding of how temperature and chemical species affect the
PSRs has been advanced. With sampling medium side kinetic resistance influencing the overall
chemical uptake kinetics (Chapter 2 and 3), the influence of temperatures and chemical species
on PSR can be much larger than characterized by a previous passive sampling model9,89,184
. Even
using a semi-enclosed cylindrical housing design, the PSRs under windy condition could be
more than 5 times higher than those under relative wind still indoor conditions (Chapter 5),
Therefore, PAS-derived air concentration is likely to have large uncertainty when it was
calculated with a PSR calibrated at a different temperature, under different wind conditions,
and/or for a different SVOC.
In this study (Chapter 3), a model was developed to semi-quantitatively understand the influence
of temperature and chemical species on PSRs. However, because of the lack of information on
the sorption rate constant, the model can still not be used to quantitatively predict those
influences and thus the uncertainty associated with the PSR. Therefore, when a question can be
addressed using mchem, i.e. the amount of chemical accumulated in a PAS (ng/sampler), it is
recommended to avoid converting mchem to the volumetric air concentrations CA (ng/m3). It is
also preferable to rely on concentration ratios of two chemicals than on the absolute
concentrations to derive information because by taking the ratio, the influence of wind and
temperature on PSRs and thus the associated uncertainty can be eliminated. When it is necessary
to obtain CA in order to address a study’s questions, it is recommended to use PSRs calibrated for
the same type of chemicals under similar climate and wind conditions and to explicitly take the
uncertainty into account when comparing concentrations derived by passive air samplers.

173
Figure 7.1 Illustration of factors potentially contributing to the uncertainty of passive air
sampling derived air concentration (CA).
7.2.2 Problems involved in deriving passive sampling rates from the loss of depuration compounds from porous sampling media.
Different temperature and wind conditions between the site where a PAS calibration was
conducted and the actual sampling site can introduce uncertainty to the PAS derived air
concentrations. An approach based on the loss rate of depuration compounds (DCs) has been
applied to derive sampling site specific PSRs, which was believed to correct for the influence of
wind and temperature on the uptake of the target SVOCs.95
A key assumption involved in
converting the loss rates of DCs to the uptake rates of the target SVOCs is that the overall mass
transfer coefficient equals the mass transfer coefficient across the stagnant air boundary layer
surrounding the bulk sampling medium (Equation 1.5) so that the kinetic resistance for the DCs
equals that for the target SVOCs (Equation 1.8). However, evidence from this study (Chapter 2,
3, 4) indicates such assumption is not valid for thick porous sampling media such as a PUF-disk
and XAD-filled mesh cylinder. Because of the existence of a kinetic resistance residing within
the PSM, DCs and the target SVOCs would only be subject to identical kinetic resistances if the
distributions of the DCs and the sampled SVOCs in the PSM were identical. Because the
distribution of the target SVOCs is unknown beforehand, it is impossible for the DCs and target
SVOCs to be distributed identically within PSM and thus be subject to identical kinetic
resistances.
CA = mchem / PSR
mchem
WindTemp.Sample analysis Chem.
PSR

174
A DC-derived PSR is based on the fraction of a DC lost from the PSM after the sampling period
(Equation 1.10). The common approach of applying DCs is to soak the whole PSM in DC
containing solvent and let the solvent evaporate before sampling.95
Presumably, this approach
leads to a uniform distribution of DCs in the PSM. If that is the case, DCs, on average, would
have a longer diffusion pathway within the PSM before evaporating to the air than the target
SVOCs sampled from the air. Thus, PSRs derived based on the loss of DCs (Equation 1.10) tend
to be underestimated and a greater extent of such bias is expected for compounds subject to more
kinetic resistance within the PSM (compounds with high KPSM/A).
Loss rates of DCs are somewhat correlated with sampling site characteristics such as wind speed
and temperature and can therefore be used to assess semi-quantitatively the influence of site
conditions on PSRs. However, because of the kinetic resistance within the PSM, it is not feasible
to quantitatively derive PSRs based on the loss of DCs. The uncertainty of DC-derived PSRs
introduced due to the failure to consider the PSM side resistance could possibly exceed the
uncertainty introduced by using a PSR from a calibration at a different site. Therefore, for the
PUF- and XAD-PASs with thick porous sampling media, using DCs cannot be recommended as
an approach to reducing the uncertainty of PSRs introduced by temperature and wind. The
derivation of air concentrations from PUF and/or XAD-based PAS will still need to rely on
calibrated PSRs, so long as the calibration site and the sampling site have similar characteristics
in terms of temperature and wind exposure.
7.2.3 Insights into the optimization of passive air sampler designs
As a dynamic uptake PAS should remain in the linear uptake range for as long as possible, PSMs
for such PAS should have a high sorption capacity (PSM/air partition coefficient) for SVOCs. In
particular, a higher sorption capacity will enable the PAS to be used for a broader range of
chemicals (more volatile compounds) and/or for a longer period of time. Comparing PUF and
XAD, the two PSMs most commonly used for dynamic passive air sampling of SVOCs, PUF/air
partition coefficients are generally lower than XAD/air partition coefficients.38
Therefore, a
dynamic uptake PAS using XAD as the PSM can be applied to more volatile compounds, which
might not be within the linear uptake range when using PUF as the PSM. Because of this
advantage, the suggested approaches to optimize the design of PAS are based on using XAD as
the PSM.

175
Based on the findings of this study (Chapter 2, 3, 4), during the typical deployment time of
PASs, the sampled chemicals do not penetrate deeply into the PSM and thus the sorbent deeper
in the PSM is not efficiently used for chemical uptake. It is the surface area between the bulk
PSM and air (interfacial transfer area) rather than the total amount of sorbent that determines the
PSR. So an optimized design would seek to minimize the amount of sorbent while maximizing
the interfacial transfer area. One approach to achieve this is to use multiple XAD-filled mesh
cylinders of small diameters. Such an approach would greatly improve the use efficiency of the
XAD resin. As illustrated in Figure 7.2(a), using two XAD-filled mesh cylinders of 1 cm in
diameter instead of one of 2 cm in diameter, the interfacial transfer area remains unchanged
while the amount of XAD is reduced by half. Since XAD resin (pre-cleaned or cleaned with
solvent extraction) is the most expensive component of the PAS, this approach could
significantly reduce the cost of using PAS.
Figure 7.2 Illustrations of suggested approaches to optimize the design of passive air
samplers using XAD resin as the sampling medium. (a) Using mesh cylinder of smaller diameter.
(b) Using disk-shaped mesh container.
d=1 cm h=10 cm
d=2 cm h=10 cm
Same surface area
Half amount of XAD
d=2 cm h=10cm
d=5 cm h= 0.4 cm
Same amount of XAD 2.5 x surface area
(a)
(b)

176
An alternative approach is to use a disk shaped PSM instead of a cylindrical PSM as illustrated
in Figure 7.2(b). With the amount of XAD in a 2-cm diameter mesh cylinder, a disk shaped
container with a diameter of 5 cm and a thickness of 0.4 cm can be filled. Such a disk shaped
PSM has an interfacial transfer area 2.5 times higher. If no other factors affecting the PSR
changes, the PSR is expected to increase by 2.5 times, which would enable the detection of
chemicals with lower air concentrations and/or the reduction of sampling times. A disk shaped
PSM could also be mounted further from the opening of the PAS housing, which could buffer
the effect of wind on the PSR. Another way to reduce the effect of wind could be to cover the
opening of the PAS housing with a fine mesh screen. Presumably such a screen would buffer the
wind within the PAS housing.
The time-efficient gravimetrical approach of measuring water uptake in silica gel developed in
this study (Chapter 4) could be used as an initial test of these proposed PAS designs. The short
experimental time frame of this approach allows the screening for a number of different PAS
designs to select a few optimized designs for further testing using XAD and SVOCs as the PSM
and target chemicals.
7.3 Further Research Needs and Recommendations
While some knowledge gaps on the mechanism of and influential factors on passive air sampling
have been addressed with this study, it also identified some new knowledge gaps.
In Chapter 3, the kinetics of chemical exchange between gas phase and sorbent was identified as
an important parameters affecting the overall PSRs. However, sorption rate constants are not
available for SVOCs and the sorbents commonly used in PASs. Such kinetic rate constants are a
knowledge gap that prevents the use of the newly developed model in the prediction of the
variations of PSRs with temperature and between different SVOCs. Further research quantifying
these sorption rate constants would be worthwhile.
In Chapter 5, PSRs for XAD-PAS were observed to be higher at windy conditions than under
wind still conditions. However, a previous wind-tunnel study suggested little wind effect on the
water uptake by silica-gel filled mesh cylinders at wind speed between 5 and 15 m/s.20
The
model presented in Chapter 3 suggests that when the thickness of the stagnant air boundary layer
is smaller than 0.01 cm, PSRs are not so sensitive to the boundary layer thickness, which is

177
affected by wind speed. So it seems that there may exist a threshold in wind speed above which
PSRs are not sensitive to wind. Identifying such a threshold would be important for
understanding and quantifying the impact of wind on PSRs. This could be done by measuring
PSRs at different wind speeds starting from 0 m/s.
Chapter 6 suggests with one XAD-filled mesh cylinder in a sampling housing deployed outdoors,
the starvation effect was insignificant and multiple XAD-filled mesh cylinders can be put in one
housing to increase the amount of chemicals sampled or the number of replicates. With
additional PSM in a sampler housing, the rate of chemical uptake by the PSM may increase and
eventually exceed the rates of chemical transfer from ambient air to the inside of the sampler
housing, resulting in a starvation effect. Therefore, it would be useful to establish the maximum
amount of PSM that can be used in a single sampler housing under different environmental
conditions (indoors, outdoors with different exposure to air turbulence) without causing a
starvation effect.
Although they have many advantages compared to conventional active air samplers, PASs are
also influenced by many factors. Variations in the PSRs as large as one order of magnitude due
to the influence of these factors should be expected. When comparing PAS derived air
concentrations measured at different sites and under different conditions, the uncertainty
originating from these different factors of influence should be considered. The measurement
uncertainties that can be tolerated should be considered when deciding whether to use PASs in a
sampling campaign. With more and more quantitative information on the factors influencing the
PSRs becoming available (including as a result of this thesis), further effort would be worthwhile
to modify the current PAS designs with an aim to minimize the uncertainties in the PSRs
introduced by these factors, while maximizing the PSRs and the use efficiency of the PSM.

178
References
(1) Campbell, J. E.; Konzen, R. B. The development of a passive dosimeter for airborne
aniline vapors. Am. Ind. Hyg. Assoc. J. 1980, 41, 180-184.
(2) Mazur, J. F.; Podolak, G. E.; Esposito, G. G.; Rinehart, D. S.; Glenn, R. E. Evaluation of
a passive dosimeter for collection of 2-bromo-2-chloro-1,1,1-trifluoroethane and 2-chloro-1,1,2-
trifluoroethyl difluoromethyl ether in hospital operating-rooms. Am. Ind. Hyg. Assoc. J. 1980, 41,
317-321.
(3) Mehta, S.; Burton, P.; Simms, J. S. Monitoring of occupational exposure to nitrous-oxide.
Can. Anaesth. Soc. J. 1978, 25, 419-423.
(4) Ohmori, K.; Ikemi, Y.; Watanabe, S.; Kitazume, M. A simple method for monitoring
ozone exposure. Jpn. J. Ind. Health 1980, 22, 249-255.
(5) McCammon, C. S.; Edwards, S. L.; Hull, R. D.; Woodfin, W. J. A comparison of 4
personal sampling methods for the determination of mercury vapor. Am. Ind. Hyg. Assoc. J.
1980, 41, 528-531.
(6) Petty, J. D.; Huckins, J. N.; Zajicek, J. L. Application of semipermeable-membrane
devices (SPMDs) as passive air samplers. Chemosphere 1993, 27, 1609-1624.
(7) Prest, H. F.; Huckins, J. N.; Petty, J. D.; Herve, S.; Paasivirta, J.; Heinonen, P. A survey
of recent results in passive sampling of water and air by semipermeable membrane devices. Mar.
Pollut. Bull. 1995, 31, 306-312.
(8) Prest, H. F.; Jacobsen, L. A.; Huckins, J. N. Passive sampling of water and coastal air via
semipermeable-membrane devices. Chemosphere 1995, 30, 1351-1361.
(9) Shoeib, M.; Harner, T. Characterization and comparison of three passive air samplers for
persistent organic pollutants. Environ. Sci. Technol. 2002, 36, 4142-4151.
(10) Ockenden, W. A.; Corrigan, B. P.; Howsam, M.; Jones, K. C. Further developments in
the use of semipermeable membrane devices as passive air samplers: Application to PCBs.
Environ. Sci. Technol. 2001, 35, 4536-4543.
(11) Lohmann, R.; Corrigan, B. P.; Howsam, M.; Jones, K. C.; Ockenden, W. A. Further
developments in the use of semipermeable membrane devices (SPMDs) as passive air samplers
for persistent organic pollutants: Field application in a spatial survey of PCDD/Fs and PAHs.
Environ. Sci. Technol. 2001, 35, 2576-2582.
(12) Ockenden, W. A.; Prest, H. F.; Thomas, G. O.; Sweetman, A.; Jones, K. C. Passive air
sampling of PCBs: Field calculation of atmospheric sampling rates by triolein-containing
semipermeable membrane devices. Environ. Sci. Technol. 1998, 32, 1538-1543.

179
(13) Bartkow, M. E.; Hawker, D. W.; Kennedy, K. E.; Müller, J. F. Characterizing uptake
kinetics of PAHs from the air using polyethylene-based passive air samplers of multiple surface
area-to-volume ratios. Environ. Sci. Technol. 2004, 38, 2701-2706.
(14) Söderström, H.; Hajslova, J.; Kocourek, V.; Siegmund, B.; Kocan, A.; Obiedzinski, W.;
Tysklind, M.; Bergqvist, P. A. PAHs and nitrated PAHs in air of five European countries
determined using SPMDs as passive samplers. Atmos. Environ. 2005, 39, 1627-1640.
(15) Strandberg, B.; Gustafson, P.; Söderström, H.; Barregard, L.; Bergqvist, P. A.; Sallsten,
G. The use of semipermeable membrane devices as passive samplers to determine persistent
organic compounds in indoor air. J. Environ. Monit. 2006, 8, 257-262.
(16) Khaled, A.; Pawliszyn, J. Time-weighted average sampling of volatile and semi-volatile
airborne organic compounds by the solid-phase microextraction device. J. Chromatogr. A 2000,
892, 455-467.
(17) Wilcockson, J. B.; Gobas, F. A. P. Thin-film solid-phase extraction to measure fugacities
of organic chemicals with low volatility in biological samples. Environ. Sci. Technol. 2001, 35,
1425-1431.
(18) Wennrich, L.; Popp, P.; Hafner, C. Novel integrative passive samplers for the long-term
monitoring of semivolatile organic air pollutants. J. Environ. Monit. 2002, 4, 371-376.
(19) Ockenden, W. A.; Jaward, F. M.; Jones, K. C. Atmospheric sampling of persistent
organic pollutants: Needs, applications and advances in passive air sampling techniques. The
Scientific World 2001, 1, 557-575.
(20) Wania, F.; Shen, L.; Lei, Y. D.; Teixeira, C.; Muir, D. C. G. Development and calibration
of a resin-based passive sampling system for monitoring persistent organic pollutants in the
atmosphere. Environ. Sci. Technol. 2003, 37, 1352-1359.
(21) Harner, T.; Shoeib, M.; Diamond, M.; Stern, G.; Rosenberg, B. Using passive air
samplers to assess urban-Rural trends for persistent organic pollutants. 1. Polychlorinated
biphenyls and organochlorine pesticides. Environ. Sci. Technol. 2004, 38, 4474-4483.
(22) Pozo, K.; Harner, T.; Shoeib, M.; Urrutia, R.; Barra, R.; Parra, O.; Focardi, S. Passive-
sampler derived air concentrations of persistent organic pollutants on a north-south transect in
Chile. Environ. Sci. Technol. 2004, 38, 6529-6537.
(23) Wilford, B. H.; Harner, T.; Zhu, J. P.; Shoeib, M.; Jones, K. C. Passive sampling survey
of polybrominated diphenyl ether flame retardants in indoor and outdoor air in Ottawa, Canada:
Implications for sources and exposure. Environ. Sci. Technol. 2004, 38, 5312-5318.
(24) Harner, T.; Pozo, K.; Gouin, T.; Macdonald, A. M.; Hung, H.; Cainey, J.; Peters, A.
Global pilot study for persistent organic pollutants (POPs) using PUF disk passive air samplers.
Environ. Pollut. 2006, 144, 445-452.

180
(25) Harner, T.; Shoeib, M.; Diamond, M.; Ikonomou, M.; Stern, G. Passive sampler derived
air concentrations of PBDEs along an urban-rural transect: Spatial and temporal trends.
Chemosphere 2006, 64, 262-267.
(26) Harrad, S.; Hazrati, S.; Ibarra, C. Concentrations of polychlorinated biphenyls in indoor
air and polybrominated diphenyl ethers in indoor air and dust in Birmingham, United Kingdom:
Implications for human exposure. Environ. Sci. Technol. 2006, 40, 4633-4638.
(27) Harrad, S.; Hunter, S. Concentrations of polybrominated diphenyl ethers in air and soil
on a rural-urban transect across a major UK conurbation. Environ. Sci. Technol. 2006, 40, 4548-
4553.
(28) Hazrati, S.; Harrad, S. Calibration of polyurethane foam (PUF) disk passive air samplers
for quantitative measurement of polychlorinated biphenyls (PCBs) and polybrominated diphenyl
ethers (PBDEs): Factors influencing sampling rates. Chemosphere 2007, 67, 448-455.
(29) Jamshidi, A.; Hunter, S.; Hazrati, S.; Harrad, S. Concentrations and chiral signatures of
polychlorinated biphenyls in outdoor and indoor air and soil in a major UK conurbation.
Environ. Sci. Technol. 2007, 41, 2153-2158.
(30) Zhang, X. M.; Diamond, M. L.; Robson, M.; Harrad, S. Sources, emissions, and fate of
polybrominated diphenyl ethers and polychlorinated biphenyls indoors in Toronto, Canada.
Environ. Sci. Technol. 2011, 45, 3268-3274.
(31) Shen, L.; Wania, F.; Lei, Y. D.; Teixeira, C.; Muir, D. C. G.; Bidleman, T. F.
Hexachlorocyclohexanes in the north American atmosphere. Environ. Sci. Technol. 2004, 38,
965-975.
(32) Shen, L.; Wania, F.; Lei, Y. D.; Teixeira, C.; Muir, D. C. G.; Xiao, H. Polychlorinated
biphenyls and polybrominated diphenyl ethers in the North American atmosphere. Environ.
Pollut. 2006, 144, 434-444.
(33) Daly, G. L.; Lei, Y. D.; Teixeira, C.; Muir, D. C. G.; Castillo, L. E.; Jantunen, L. M. M.;
Wania, F. Organochlorine pesticides in the soils and atmosphere of Costa Rica. Environ. Sci.
Technol. 2007, 41, 1124-1130.
(34) Daly, G. L.; Lei, Y. D.; Teixeira, C.; Muir, D. C. G.; Wania, F. Pesticides in western
Canadian mountain air and soil. Environ. Sci. Technol. 2007, 41, 6020-6025.
(35) Choi, S. D.; Shunthirasingham, C.; Daly, G. L.; Xiao, H.; Lei, Y. D.; Wania, F. Levels of
polycyclic aromatic hydrocarbons in Canadian mountain air and soil are controlled by proximity
to roads. Environ. Pollut. 2009, 157, 3199-3206.
(36) Liu, W. J.; Chen, D. Z.; Liu, X. D.; Zheng, X. Y.; Yang, W.; Westgate, J. N.; Wania, F.
Transport of Semivolatile Organic Compounds to the Tibetan Plateau: Spatial and Temporal
Variation in Air Concentrations in Mountainous Western Sichuan, China. Environ. Sci. Technol.
2010, 44, 1559-1565.

181
(37) Shunthirasingham, C.; Barra, R.; Mendoza, G.; Montory, M.; Oyiliagu, C. E.; Lei, Y. D.;
Wania, F. Spatial variability of atmospheric semivolatile organic compounds in Chile. Atmos.
Environ. 2011, 45, 303-309.
(38) Hayward, S. J.; Lei, Y. D.; Wania, F. Sorption of a diverse set of organic chemical vapors
onto XAD-2 resin: Measurement, prediction and implications for air sampling. Atmos. Environ.
2011, 45, 296-302.
(39) Genualdi, S.; Harner, T.; Cheng, Y.; MacLeod, M.; Hansen, K. M.; van Egmond, R.;
Shoeib, M.; Lee, S. C. Global distribution of linear and cyclic volatile methyl siloxanes in air.
Environ. Sci. Technol. 2011, 45, 3349-3354.
(40) Vierke, L.; Ahrens, L.; Shoeib, M.; Reiner, E. J.; Guo, R.; Palm, W. U.; Ebinghaus, R.;
Harner, T. Air concentrations and particle-gas partitioning of polyfluoroalkyl compounds at a
wastewater treatment plant. Environmental Chemistry 2011, 8, 363-371.
(41) Koblizkova, M.; Genualdi, S.; Lee, S. C.; Harner, T. Application of sorbent impregnated
polyurethane foam (SIP) disk passive air samplers for investigating organochlorine pesticides
and polybrominated diphenyl ethers at the global scale. Environ. Sci. Technol. 2012, 46, 391-
396.
(42) Schuster, J. K.; Gioia, R.; Harner, T.; Lee, S. C.; Breivik, K.; Jones, K. C. Assessment of
sorbent impregnated PUF disks (SIPs) for long-term sampling of legacy POPs. J. Environ.
Monit. 2012, 14, 71-78.
(43) United Nations Environment Programme. Stockholm Convention on persistent organic
pollutants (POPs). Available at: http://chm.pops.int/ (accessed Apr 2012).
(44) Harner, T.; Bartkow, M.; Holoubek, I.; Klanova, J.; Wania, F.; Gioia, R.; Moeckel, C.;
Sweetman, A. J.; Jones, K. C. Passive air sampling for persistent organic pollutants: Introductory
remarks to the special issue. Environ. Pollut. 2006, 144, 361-364.
(45) Xiao, H.; Hung, H.; Harner, T.; Lei, Y. D.; Johnston, G. W.; Wania, F. A flow-through
sampler for semivolatile organic compounds in air. Environ. Sci. Technol. 2007, 41, 250-256.
(46) Xiao, H.; Shen, L.; Su, Y. S.; Barresi, E.; DeJong, M.; Hung, H. L.; Lei, Y. D.; Wania,
F.; Reiner, E. J.; Sverko, E.; Kang, S. C. Atmospheric concentrations of halogenated flame
retardants at two remote locations: The Canadian High Arctic and the Tibetan Plateau. Environ.
Pollut. 2012, 161, 154-161.
(47) Tao, S.; Liu, Y. N.; Xu, W.; Lang, C.; Liu, S. Z.; Dou, H.; Liu, W. X. Calibration of a
passive sampler for both gaseous and particulate phase polycyclic aromatic hydrocarbons.
Environ. Sci. Technol. 2007, 41, 568-573.
(48) Abdallah, M. A. E.; Harrad, S. Modification and calibration of a passive air sampler for
monitoring vapor and particulate phase brominated flame retardants in indoor air: Application to
car interiors. Environ. Sci. Technol. 2010, 44, 3059-3065.

182
(49) Tao, S.; Liu, Y. N.; Lang, C.; Wang, W. T.; Yuan, H. S.; Zhang, D. Y.; Qiu, W. X.; Liu,
J. M.; Liu, Z. G.; Liu, S. Z.; Yi, R.; Ji, M.; Liu, X. X. A directional passive air sampler for
monitoring polycyclic aromatic hydrocarbons (PAHs) in air mass. Environ. Pollut. 2008, 156,
435-441.
(50) Schrlau, J. E.; Geiser, L.; Hageman, K. J.; Landers, D. H.; Simonich, S. M. Comparison
of lichen, conifer needles, passive air sampling devices, and snowpack as passive sampling
media to measure semi-volatile organic compounds in remote atmospheres. Environ. Sci.
Technol. 2011, 45, 10354-10361.
(51) Simonich, S. L.; Hites, R. A. Vegetation-atmosphere partitioning of polycyclic aromatic
hydrocarbons. Environ. Sci. Technol. 1994, 28, 939-943.
(52) Meijer, S. N.; Steinnes, E.; Ockenden, W. A.; Jones, K. C. Influence of environmental
variables on the spatial distribution of PCBs in Norwegian and UK soils: Implications for global
cycling. Environ. Sci. Technol. 2002, 36, 2146-2153.
(53) Wong, F.; Harner, T.; Liu, Q. T.; Diamond, M. L. Using experimental and forest soils to
investigate the uptake of polycyclic aromatic hydrocarbons (PAHs) along an urban-rural
gradient. Environ. Pollut. 2004, 129, 387-398.
(54) Kalantzi, O. I.; Alcock, R. E.; Johnston, P. A.; Santillo, D.; Stringer, R. L.; Thomas, G.
O.; Jones, K. C. The global distribution of PCBs and organochlorine pesticides in butter.
Environ. Sci. Technol. 2001, 35, 1013-1018.
(55) Liu, Q. T.; Chen, R.; McCarry, B. E.; Diamond, M. L.; Bahavar, B. Characterization of
polar organic compounds in the organic film on indoor and outdoor glass windows. Environ. Sci.
Technol. 2003, 37, 2340-2349.
(56) Müller, J. F.; Hawker, D. W.; Connell, D. W.; Kömp, P.; McLachlan, M. S. Passive
sampling of atmospheric SOCs using tristearin-coated fibreglass sheets. Atmos. Environ. 2000,
34, 3525-3534.
(57) Pozo, K.; Harner, T.; Wania, F.; Muir, D. C. G.; Jones, K. C.; Barrie, L. A. Toward a
global network for persistent organic pollutants in air: Results from the GAPS study. Environ.
Sci. Technol. 2006, 40, 4867-4873.
(58) Shunthirasingham, C.; Oyiliagu, C. E.; Cao, X. S.; Gouin, T.; Wania, F.; Lee, S. C.;
Pozo, K.; Harner, T.; Muir, D. C. G. Spatial and temporal pattern of pesticides in the global
atmosphere. J. Environ. Monit. 2010, 12, 1650-1657.
(59) Lee, S. C.; Harner, T.; Pozo, K.; Shoeib, M.; Wania, F.; Muir, D. C. G.; Barrie, L. A.;
Jones, K. C. Polychlorinated naphthalenes in the Global Atmospheric Passive Sampling (GAPS)
study. Environ. Sci. Technol. 2007, 41, 2680-2687.
(60) Pozo, K.; Harner, T.; Lee, S. C.; Wania, F.; Muir, D. C. G.; Jones, K. C. Seasonally
resolved concentrations of persistent organic pollutants in the global atmosphere from the first
year of the GAPS study. Environ. Sci. Technol. 2009, 43, 796-803.

183
(61) Genualdi, S.; Lee, S. C.; Shoeib, M.; Gawor, A.; Ahrens, L.; Harner, T. Global pilot
study of legacy and emerging persistent organic pollutants using sorbent-impregnated
polyurethane foam disk passive air samplers. Environ. Sci. Technol. 2010, 44, 5534-5539.
(62) Ockenden, W. A.; Sweetman, A. J.; Prest, H. F.; Steinnes, E.; Jones, K. C. Toward an
understanding of the global atmospheric distribution of persistent organic pollutants: The use of
semipermeable membrane devices as time-integrated passive samplers. Environ. Sci. Technol.
1998, 32, 2795-2803.
(63) Meijer, S. N.; Ockenden, W. A.; Steinnes, E.; Corrigan, B. P.; Jones, K. C. Spatial and
temporal trends of POPs in Norwegian and UK background air: Implications for global cycling.
Environ. Sci. Technol. 2003, 37, 454-461.
(64) Schuster, J. K.; Gioia, R.; Breivik, K.; Steinnes, E.; Scheringer, M.; Jones, K. C. Trends
in European background air reflect reductions in primary emissions of PCBs and PBDEs. .
Environ. Sci. Technol. 2010, 44, 6760-6766.
(65) Shen, L.; Wania, F.; Lei, Y. D.; Teixeira, C.; Muir, D. C. G.; Bidleman, T. F.
Atmospheric distribution and long-range transport behavior of organochlorine pesticides in north
America. Environ. Sci. Technol. 2005, 39, 409-420.
(66) Jaward, F. M.; Farrar, N. J.; Harner, T.; Sweetman, A. J.; Jones, K. C. Passive air
sampling of polycyclic aromatic hydrocarbons and polychlorinated naphthalenes across Europe.
Environ. Toxicol. Chem. 2004, 23, 1355-1364.
(67) Jaward, F. M.; Farrar, N. J.; Harner, T.; Sweetman, A. J.; Jones, K. C. Passive air
sampling of PCBs, PBDEs, and organochlorine pesticides across Europe. Environ. Sci. Technol.
2004, 38, 34-41.
(68) Gouin, T.; Harner, T.; Blanchard, P.; Mackay, D. Passive and active air samplers as
complementary methods for investigating persistent organic pollutants in the Great Lakes basin.
Environ. Sci. Technol. 2005, 39, 9115-9122.
(69) Jaward, T. M.; Zhang, G.; Nam, J. J.; Sweetman, A. J.; Obbard, J. P.; Kobara, Y.; Jones,
K. C. Passive air sampling of polychlorinated biphenyls, organochlorine compounds, and
polybrominated diphenyl ethers across Asia. Environ. Sci. Technol. 2005, 39, 8638-8645.
(70) Halse, A. K.; Schlabach, M.; Eckhardt, S.; Sweetman, A.; Jones, K. C.; Breivik, K.
Spatial variability of POPs in European background air. Atmos. Chem. Phys. 2011, 11, 1549-
1564.
(71) Motelay-Massei, A.; Harner, T.; Shoeib, M.; Diamond, M.; Stern, G.; Rosenberg, B.
Using passive air samplers to assess urban-rural trends for persistent organic pollutants and
polycyclic aromatic hydrocarbons. 2. Seasonal trends for PAHs, PCBs, and organochlorine
pesticides. Environ. Sci. Technol. 2005, 39, 5763-5773.
(72) Melymuk, L.; Robson, M.; Helm, P. A.; Diamond, M. L. PCBs, PBDEs, and PAHs in
Toronto air: Spatial and seasonal trends and implications for contaminant transport. Sci. Total
Environ. 2012, 429, 272-280.

184
(73) Harrad, S.; Goosey, E.; Desborough, J.; Abdallah, M. A. E.; Roosens, L.; Covaci, A. Dust
from UK primary school classrooms and daycare centers: The significance of dust ss a pathway
of exposure of young UK children to brominated flame retardants and polychlorinated biphenyls.
Environ. Sci. Technol. 2010, 44, 4198-4202.
(74) Bohlin, P.; Jones, K. C.; Levin, J. O.; Lindahl, R.; Strandberg, B. Field evaluation of a
passive personal air sampler for screening of PAH exposure in workplaces. J. Environ. Monit.
2010, 12, 1437-1444.
(75) Harrad, S.; Ibarra, C.; Abdallah, M. A. E.; Boon, R.; Neels, H.; Covaci, A.
Concentrations of brominated flame retardants in dust from United Kingdom cars, homes, and
offices: Causes of variability and implications for human exposure. Environ. Int. 2008, 34, 1170-
1175.
(76) Shoeib, M.; Harner, T.; Wilford, B. H.; Jones, K. C.; Zhu, J. Perfluorinated Sulfonamides
in Indoor and Outdoor Air and Indoor Dust:鈥?Occurrence, Partitioning, and Human Exposure.
Environ. Sci. Technol. 2005, 39, 6599-6606.
(77) Bohlin, P.; Jones, K. C.; Tovalin, H.; Strandberg, B. Observations on persistent organic
pollutants in indoor and outdoor air using passive polyurethane foam samplers. Atmos. Environ.
2008, 42, 7234-7241.
(78) Ahrens, L.; Shoeib, M.; Harner, T.; Lee, S. C.; Guo, R.; Reiner, E. J. Wastewater
treatment plant and landfills as sources of polyfluoroalkyl compounds to the atmosphere.
Environ. Sci. Technol. 2011, 45, 8098-8105.
(79) Cheng, Y.; Shoeib, M.; Ahrens, L.; Harner, T.; Ma, J. M. Wastewater treatment plants
and landfills emit volatile methyl siloxanes (VMSs) to the atmosphere: Investigations using a
new passive air sampler. Environ. Pollut. 2011, 159, 2380-2386.
(80) Zhang, Y. Z.; Deng, S. X.; Liu, Y. A.; Shen, G. F.; Li, X. Q.; Cao, J.; Wang, X. L.; Reid,
B.; Tao, S. A passive air sampler for characterizing the vertical concentration profile of gaseous
phase polycyclic aromatic hydrocarbons in near soil surface air. Environ. Pollut. 2011, 159, 694-
699.
(81) Daly, G. L.; Lei, Y. D.; Castillo, L. E.; Muir, D. C. G.; Wania, F. Polycyclic aromatic
hydrocarbons in Costa Rican air and soil: A tropical/temperate comparison. Atmos. Environ.
2007, 41, 7339-7350.
(82) Loewen, M.; Wania, F.; Wang, F. Y.; Tomy, G. Altitudinal transect of atmospheric and
aqueous fluorinated organic compounds in western Canada. Environ. Sci. Technol. 2008, 42,
2374-2379.
(83) Bartkow, M. E.; Booij, K.; Kennedy, K. E.; Müller, J. F.; Hawker, D. W. Passive air
sampling theory for semivolatile organic compounds. Chemosphere 2005, 60, 170-176.
(84) Lewis, W. K.; Whitman, W. G. Principles of gas absorption. Ind. Eng. Chem. 1924, 16,
1215-1220.

185
(85) Fuller, E. N.; Schettler, P. D.; Giddings, J. C. A new method for prediction of binary gas-
phase diffusion coefficients. Ind. Eng. Chem. 1966, 58, 18-27.
(86) Tuduri, L.; Millet, M.; Briand, O.; Montury, M. Passive air sampling of semi-volatile
organic compounds. Trac-Trends in Analytical Chemistry 2012, 31, 38-49.
(87) Melymuk, L.; Robson, M.; Helm, P. A.; Diamond, M. L. Evaluation of passive air
sampler calibrations: Selection of sampling rates and implications for the measurement of
persistent organic pollutants in air. Atmos. Environ. 2011, 45, 1867-1875.
(88) Chaemfa, C.; Barber, J. L.; Gocht, T.; Harner, T.; Holoubek, I.; Klanova, J.; Jones, K. C.
Field calibration of polyurethane foam (PUF) disk passive air samplers for PCBs and OC
pesticides. Environ. Pollut. 2008, 156, 1290-1297.
(89) Gouin, T.; Wania, F.; Ruepert, C.; Castillo, L. E. Field testing passive air samplers for
current use pesticides in a tropical environment. Environ. Sci. Technol. 2008, 42, 6625-6630.
(90) Klánová, J.; Èupr, P.; Kohoutek, J.; Harner, T. Assessing the influence of meteorological
parameters on the performance of polyurethane foam-based passive air samplers. Environ. Sci.
Technol. 2008, 42, 550-555.
(91) Thomas, J.; Holsen, T. M.; Dhaniyala, S. Computational fluid dynamic modeling of two
passive samplers. Environ. Pollut. 2006, 144, 384-392.
(92) Tuduri, L.; Harner, T.; Hung, H. Polyurethane foam (PUF) disks passive air samplers:
Wind effect on sampling rates. Environ. Pollut. 2006, 144, 377-383.
(93) Söderström, H. S.; Bergqvist, P. A. Passive air sampling using semipermeable membrane
devices at different wind-speeds in situ calibrated by performance reference compounds.
Environ. Sci. Technol. 2004, 38, 4828-4834.
(94) Söderström, H. S.; Bergqvist, P. A. Wind effects on passive air sampling of PAHs and
PCBs. Bull. Environ. Contam. Toxicol. 2005, 74, 429-436.
(95) Moeckel, C.; Harner, T.; Nizzetto, L.; Strandberg, B.; Lindroth, A.; Jones, K. C. Use of
depuration compounds in passive air samplers: results from active sampling-supported field
deployment, potential uses, and recommendations. Environ. Sci. Technol. 2009, 43, 3227-3232.
(96) Shunthirasingham, C. Pesticide fate in different climates. University of Toronto PhD
thesis, Toronto, 2010.
(97) May, A. A.; Ashman, P.; Huang, J.; Dhaniyala, S.; Holsen, T. M. Evaluation of the
polyurethane foam (PUF) disk passive air sampler: Computational modeling and experimental
measurements. Atmos. Environ. 2011, 45, 4354-4359.
(98) Chaemfa, C.; Wild, E.; Davison, B.; Barber, J. L.; Jones, K. C. A study of aerosol
entrapment and the influence of wind speed, chamber design and foam density on polyurethane
foam passive air samplers used for persistent organic pollutants. J. Environ. Monit. 2009, 11,
1135-1139.

186
(99) Tao, S.; Liu, Y.; Xu, W.; Lang, C.; Liu, S.; Dou, H.; Liu, W. Calibration of a passive
sampler for both gaseous and particulate phase polycyclic aromatic hydrocarbons. Environ. Sci.
Technol. 2007, 41, 568-73.
(100) Brown, R. H. The use of diffusive samplers for monitoring of ambient air. Pure Appl.
Chem. 1993, 65, 1859-1874.
(101) Barthel, P.; Thuens, S.; Shunthirasingham, C.; Westgate, J. N.; Wania, F.; Radke, M.
Application of XAD-resin based passive air samplers to assess local (roadside) and regional
patterns of persistent organic pollutants. Environ. Pollut. 2012, 166, 218-225.
(102) Westgate, J. N.; Shunthirasingham, C.; Oyiliagu, C. E.; von Waldow, H.; Wania, F.
Three methods for quantifying proximity of air sampling sites to spatially resolved emissions of
semi-volatile organic contaminants. Atmos. Environ. 2010, 44, 4380-4387.
(103) Shoeib, M.; Harner, T. Using measured octanol-air partition coefficients to explain
environmental partitioning of organochlorine pesticides. Environ. Toxicol. Chem. 2002, 21, 984-
990.
(104) Chaemfa, C.; Barber, J. L.; Moeckel, C.; Gocht, T.; Harner, T.; Holoubek, I.; Klanova, J.;
Jones, K. C. Field calibration of polyurethane foam disk passive air samplers for PBDEs. J.
Environ. Monit. 2009, 11, 1859-1865.
(105) Du, S.; Wall, S. I.; Cacia, D.; Rodenburg, L. A. Passive air sampling for polychlorinated
biphenyls in the Philadelphia metropolitan area. Environ. Sci. Technol. 2009, 43, 1287-1292.
(106) Hayward, S. J. Fate of current-use pesticides in the Canadian atmosphere. Ph.D.Thesis.
University of Toronto, Toronto, 2010.
(107) Chaemfa, C.; Barber, J. L.; Gocht, T.; Harner, T.; Holoubek, I.; Klanova, J.; Jones, K. C.
Field calibration of polyurethane foam (PUF) disk passive air samplers for PCBs and OC
pesticides. Environ. Pollut. 2008, 156, 1290-7.
(108) Hayward, S. J.; Gouin, T.; Wania, F. Comparison of four active and passive sampling
techniques for pesticides in air. Environ. Sci. Technol. 2010, 44, 3410-3416.
(109) Schwarzenbach, R. P.; Gschwend, P. M.; Imboden, D. M. Environmental Organic
Chemistry. 2nd ed.; Wiley: Hoboken, N.J., 2003.
(110) Klanova, J.; Eupr, P.; Kohoutek, J.; Harner, T. Assessing the influence of meteorological
parameters on the performance of polyurethane foam-based passive air samplers. Environ. Sci.
Technol. 2008, 42, 550-555.
(111) van Noort, P. C. M.; Haftka, J. J. H.; Parsons, J. R. Updated Abraham solvation
parameters for polychlorinated biphenyls. Environ. Sci. Technol. 2010, 44, 7037-7042.
(112) Matsumura, C.; Tsurukawa, M.; Nakano, T.; Ezaki, T.; Ohashi, M. Elution orders of all
209 PCBs congeners on capillary column HT8-PCB. J. Environ. Chem. 2002, 12, 855-866.

187
(113) Antweiler, R. C.; Taylor, H. E. Evaluation of statistical treatments of left-censored
environmental data using coincident uncensored data sets: I. Summary statistics. Environ. Sci.
Technol. 2008, 42, 3732-3738.
(114) Aruga, R. Treatment of responses below the detection limit: some current techniques
compared by factor analysis on environmental data. Anal. Chim. Acta 1997, 354, 255-262.
(115) Wu, S. C.; Gschwend, P. M. Sorption kinetics of hydrophobic organic-compounds to
natural sediments and soils. Environ. Sci. Technol. 1986, 20, 717-725.
(116) Tao, S.; Cao, J.; Wang, W. T.; Zhao, J. Y.; Wang, W.; Wang, Z. H.; Cao, H. Y.; Xing, B.
S. A passive sampler with improved performance for collecting gaseous and particulate phase
polycyclic aromatic hydrocarbons in air. Environ. Sci. Technol. 2009, 43, 4124-4129.
(117) Bohlin, P.; Jones, K. C.; Strandberg, B. Field evaluation of polyurethane foam passive air
samplers to assess airborne PAHs in occupational environments. Environ. Sci. Technol. 2010, 44,
749-754.
(118) Chaemfa, C.; Barber, J. L.; Kim, K. S.; Harner, T.; Jones, K. C. Further studies on the
uptake of persistent organic pollutants (POPs) by polyurethane foam disk passive air samplers.
Atmos. Environ. 2009, 43, 3843-3849.
(119) Kamprad, I.; Goss, K. U. Systematic investigation of the sorption properties of
polyurethane foams for organic vapors. Anal. Chem. 2007, 79, 4222-4227.
(120) Abraham, M. H.; Al-Hussaini, A. J. M. Solvation parameters for the 209 PCBs:
calculation of physicochemical properties. J. Environ. Monit. 2005, 7, 295-301.
(121) Frame, G. M.; Cochran, J. W.; Bowadt, S. S. Complete PCB congener distributions for
17 aroclor mixtures determined by 3 HRGC systems optimized for comprehensive, quantitative,
congener-specific analysis. HRC-J High Res. Chrom. 1996, 19, 657-668.
(122) Hu, D. F.; Martinez, A.; Hornbuckle, K. C. Discovery of non-Aroclor PCB (3,3 '-
dichlorobiphenyl) in Chicago air. Environ. Sci. Technol. 2008, 42, 7873-7877.
(123) Rodenburg, L. A.; Guo, J.; Du, S. Y.; Cavallo, G. J. Evidence for unique and ubiquitous
environmental sources of 3,3 '-dichlorobiphenyl (PCB 11). Environ. Sci. Technol. 2010, 44,
2816-2821.
(124) Farrar, N. J.; Harner, T.; Shoeib, M.; Sweetman, A.; Jones, K. C. Field deployment of
thin film passive air samplers for persistent organic pollutants: A study in the urban atmospheric
boundary layer. Environ. Sci. Technol. 2005, 39, 42-48.
(125) Farrar, N. J.; Harner, T. J.; Sweetman, A. J.; Jones, K. C. Field calibration of rapidly
equilibrating thin-film passive air samplers and their potential application for low-volume air
sampling studies. Environ. Sci. Technol. 2005, 39, 261-267.

188
(126) Zhang, X. M.; Tsurukawa, M.; Nakano, T.; Lei, Y. D.; Wania, F. Sampling medium side
resistance to uptake of semivolatile organic compounds in passive air samplers. Environ. Sci.
Technol. 2011, 45, 10509-10515.
(127) Zhang, X. M.; Wong, C.; Lei, Y. D.; Wania, F. Influence of sampler configuration on the
uptake kinetics of a passive air sampler. Environ. Sci. Technol. 2012, 46, 397-403.
(128) Tsurukawa, M.; Suzuki, M.; Okuno, T.; Takemine, S.; Okada, Y.; Matsumura, C.;
Nakano, T. Calibration and field survey of passive air samplers for persistent organic pollutants.
Organohalogen Compd. 2010, 72, 884-887.
(129) Sing, K. S. W.; Everett, D. H.; Haul, R. A. W.; Moscou, L.; Pierotti, R. A.; Rouquerol, J.;
Siemieniewska, T. Physisorption data for gas solid systems with special reference to the
determination of surface-area and porosity. Pure Appl. Chem. 1985, 57, 603-619.
(130) Sigma-Aldrich. Amberlite XAD-2 polymeric adsorbent product specification. Available
online:
http://www.sigmaaldrich.com/etc/medialib/docs/supelco/product_information_sheet/4802.par
(accessed Jan 2012).
(131) Louch, D.; Motlagh, S.; Pawliszyn, J. Organic-compound extraction from water using
liquid-coated fused-silica fibers. Anal. Chem. 1992, 64, 1187-1199.
(132) Mackay, D. Multimedia environmental models: the fugacity approach. 2nd ed.; Lewis
Publishers: Boca Raton, 2001.
(133) Harper, M. P.; Davison, W.; Tych, W. DIFS - a modelling and simulation tool for DGT
induced trace metal remobilisation in sediments and soils. Environ. Model. Soft. 2000, 15, 55-66.
(134) Ruthven, D. M. Principles of adsorption and adsorption processes. Wiley: New York,
1984.
(135) Katsanos, N. A.; Bakaoukas, N.; Koliadima, A.; Karaiskakis, G.; Jannussis, A. Diffusion
and adsorption measurements in porous solids by inverse gas chromatography. J. Phys. Chem. B
2005, 109, 11240-11246.
(136) Chuang, C. L.; Chiang, P. C.; Chang, E. E. Modeling VOCs adsorption onto activated
carbon. Chemosphere 2003, 53, 17-27.
(137) Tovbin, Y. K. Chapter 4. Theory of adsorption-desorption kinetics on flat heterogeneous
surfaces. In Studies in Surface Science and Catalysis, Rudziński, W.; Steele, W. A.; Zgrablich,
G., Eds. Elsevier: 1997; Vol. 104, pp 201-284.
(138) Nobel, P. S. Boundary-layers of air adjacent to cylinders - estimation of effective
thickness and measurements on plant material. Plant Physiol. 1974, 54, 177-181.
(139) Courval, G.; Gray, D. G. Effect of surface adsorption on gas-chromatographic
measurements near polymer melting transitions. Macromolecules 1975, 8, 326-331.

189
(140) Nakagawa, H.; Tsuge, S. Characterization of styrene divinylbenzene copolymers by high-
resolution pyrolysis-gas chromatography. Macromolecules 1985, 18, 2068-2072.
(141) Panda, S.; Bu, Q.; Huang, B.; Edwards, R. R.; Liao, Q.; Yun, K. S.; Parcher, J. F. Mass
spectrometric inverse gas chromatography: Investigation of polymeric phase transitions. Anal.
Chem. 1997, 69, 2485-2495.
(142) Suzuki, M. Adsorption Engineering. Kodansha; Elsevier: Tokyo, 1990.
(143) Ribeiro, A. M.; Sauer, T. P.; Grande, C. A.; Moreira, R. F. P. M.; Loureiro, J. M.;
Rodrigues, A. E. Adsorption equilibrium and kinetics of water vapor on different adsorbents.
Ind. Eng. Chem. Res. 2008, 47, 7019-7026.
(144) Shunthirasingham, C.; Oyiliagu, C. E.; Cao, X.; Gouin, T.; Wania, F.; Lee, S. C.; Pozo,
K.; Harner, T.; Muir, D. C. G. Spatial and temporal pattern of pesticides in the global
atmosphere. J. Environ. Monit. 2010, 12, 1650-1657.
(145) Gevao, B.; Al-Bahloul, M.; Al-Ghadban, A. N.; Ali, L.; Al-Omair, A.; Helaleh, M.; Al-
Matrouk, K.; Zafar, J. Polybrominated diphenyl ethers in indoor air in Kuwait: Implications for
human exposure. Atmos. Environ. 2006, 40, 1419-1426.
(146) Klánová, J.; Čupr, P.; Holoubek, I.; Borůvková, J.; Přibylová, P.; Kareš, R.; Tomšej, T.;
Ocelka, T. Monitoring of persistent organic pollutants in Africa. Part 1: Passive air sampling
across the continent in 2008. J. Environ. Monit. 2009, 11, 1952-1963.
(147) Lide, D. R., CRC handbook of chemistry and physics: a ready-reference book of
chemical and physical data. 87th ed.; Taylor and Francis: New York, 2006.
(148) Zhang, X. M.; Wania, F. Modeling the uptake of semi-volatile organic compounds by
passive air samplers: Importance of mass transfer processes within the porous sampling media.
Submitted for publication.
(149) Primbs, T.; Genualdi, S.; Simonich, S. M. Solvent selection for pressurized liquid
extraction of polymeric sorbents used in air sampling. Environ. Toxicol. Chem. 2008, 27, 1267-
1272.
(150) Su, Y.; Hung, H. Inter-laboratory comparison study on measuring semi-volatile organic
chemicals in standards and air samples. Environ. Pollut. 2010, 158, 3365-3371.
(151) Gulliver, J. S. Introduction to chemical transport in the environment. Cambridge
University Press: New York, 2007.
(152) Choi, S. D.; Baek, S. Y.; Chang, Y. S.; Wania, F.; Ikonomou, M. G.; Yoon, Y. J.; Park,
B. K.; Hong, S. Passive air sampling of polychlorinated biphenyls and organochlorine pesticides
at the Korean Arctic and Antarctic research stations: Implications for long-range transport and
local pollution. Environ. Sci. Technol. 2008, 42, 7125-7131.
(153) United Nations Environment Programme. Stockholm Convention on persistent organic
pollutants (POPs). Available at: http://chm.pops.int/Convention/ (accessed Apr 2012).

190
(154) Liu, S. Z.; Tao, S.; Liu, W. X.; Dou, H.; Liu, Y. N.; Zhao, J. Y.; Little, M. G.; Tian, Z. F.;
Wang, J. F.; Wang, L. G.; Gao, Y. Seasonal and spatial occurrence and distribution of
atmospheric polycyclic aromatic hydrocarbons (PAHs) in rural and urban areas of the North
Chinese Plain. Environ. Pollut. 2008, 156, 651-656.
(155) Bohlin, P.; Jones, K. C.; Strandberg, B. Field evaluation of polyurethane foam passive air
samplers to assess airborne PAHs in occupational environments. Environ. Sci. Technol. 2009, 44,
749-754.
(156) Baldwin, P. E. J.; Maynard, A. D. A survey of wind speeds in indoor workplaces. Ann.
Occup. Hyg. 1998, 42, 303-313.
(157) Bornstein, R. D.; Johnson, D. S. Urban-rural wind velocity differences. Atmos. Environ.
1977, 11, 597-604.
(158) Diamond, M. L.; Melymuk, L.; Csiszar, S. A.; Robson, M. Estimation of PCB stocks,
emissions, and urban fate: will our policies reduce concentrations and exposure? Environ. Sci.
Technol. 2010, 44, 2777-2783.
(159) Wang, W. T.; Simonich, S. L. M.; Wang, W.; Giri, B.; Zhao, J. Y.; Xue, M. A.; Cao, J.;
Lu, X. X.; Tao, S. Atmospheric polycyclic aromatic hydrocarbon concentrations and gas/particle
partitioning at background, rural village and urban sites in the North China Plain. Atmospheric
Research 2011, 99, 197-206.
(160) Lammel, G.; Klanova, J.; Kohoutek, J.; Prokes, R.; Ries, L.; Stohl, A. Observation and
origin of organochlorine compounds and polycyclic aromatic hydrocarbons in the free
troposphere over central Europe. Environ. Pollut. 2009, 157, 3264-3271.
(161) Nizzetto, L.; Lohmann, R.; Gioia, R.; Dachs, J.; Jones, K. C. Atlantic Ocean surface
waters buffer declining atmospheric concentrations of persistent organic pollutants. Environ. Sci.
Technol. 2010, 44, 6978-6984.
(162) Zarfl, C.; Scheringer, M.; Matthies, M. Screening criteria for long-range transport
potential of organic substances in water. Environ. Sci. Technol. 2011, 45, 10075-10081.
(163) Van Drooge, B. L.; Grimalt, J. O.; Garcia, C. J. T.; Cuevas, E. Semivolatile
organochlorine compounds in the free troposphere of the northeastern Atlantic. Environ. Sci.
Technol. 2002, 36, 1155-1161.
(164) Knap, A. H.; Binkley, K. S. Chlorinated organic-compounds in the troposphere over the
western north-atlantic ocean measured by aircraft. Atmos. Environ. 1991, 25A, 1507-1516.
(165) Harner, T.; Shoeib, M.; Kozma, M.; Gobas, F. A. P. C.; Li, S. M.
Hexachlorocyclohexanes and endosulfans in urban, rural, and high altitude air samples in the
Fraser Valley, British Columbia: Evidence for trans-Pacific transport. Environ. Sci. Technol.
2005, 39, 724-731.

191
(166) Van Drooge, B. L.; Grimalt, J. O.; Booij, K.; Camarero, L.; Catalan, J. Passive sampling
of atmospheric organochlorine compounds by SPMDs in a remote high mountain area. Atmos.
Environ. 2005, 39, 5195-5204.
(167) Burgoyne, T. W.; Hites, R. A. Effects of temperature and wind direction on the
atmospheric concentrations of -endosulfan. Environ. Sci. Technol. 1993, 27, 910-914.
(168) Halsall, C. J.; Coleman, P. J.; Davis, B. J.; Burnett, V.; Waterhouse, K. S.; Hardingjones,
P.; Jones, K. C. Polycyclic aromatic-hydrocarbons in UK urban air. Environ. Sci. Technol. 1994,
28, 2380-2386.
(169) Gasic, B.; Moeckel, C.; Macleod, M.; Brunner, J.; Scheringer, M.; Jones, K. C.;
Hungerbühler, K. Measuring and modeling short-term variability of PCBs in air and
characterization of urban source strength in Zurich, Switzerland. Environ. Sci. Technol. 2009, 43,
769-776.
(170) Estellano, V. H.; Pozo, K.; Harner, T.; Franken, M.; Zaballa, M. Altitudinal and seasonal
variations of persistent organic pollutants in the Bolivian Andes Mountains. Environ. Sci.
Technol. 2008, 42, 2528-2534.
(171) Jaward, F. M.; Di Guardo, A.; Nizzetto, L.; Cassani, C.; Raffaele, F.; Ferretti, R.; Jones,
K. C. PCBs and selected organochlorine compounds in Italian mountain air: the influence of
altitude and forest ecosystem type. Environ. Sci. Technol. 2005, 39, 3455-3463.
(172) Liu, X.; Zhang, G.; Jones, K. C.; Li, X. D.; Peng, X. Z.; Qi, S. H. Compositional
fractionation of polycyclic aromatic hydrocarbons (PAHs) in mosses (Hypnum plumaeformae
WILS.) from the northern slope of Nanling Mountains, South China. Atmos. Environ. 2005, 39,
5490-5499.
(173) U.S. Cencus Bareau. 2010 U.S. Census. http://2010.census.gov/2010census/ (accessed
Jun. 2012).
(174) Juvik, S. P.; Juvik, J. O. Atlas of Hawaii. Univ of Hawaii Pr: 1998.
(175) Sanderson, M. Prevailing trade winds : climate and weather in Hawaii. University of
Hawaii Press: Honolulu, 1993.
(176) Draxler, R. R.; Rolph, G. D. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated
Trajectory) Model access via NOAA ARL READY Website
(http://ready.arl.noaa.gov/HYSPLIT.php). NOAA Air Resources Laboratory, Silver Spring, MD.,
2012.
(177) Wania, F.; Westgate, J. N. On the mechanism of mountain cold-trapping of organic
chemicals. Environ. Sci. Technol. 2008, 42, 9092-9098.
(178) U.S. EPA Estimation Programs Interface Suite™ for Microsoft Windows, v 4.00. United
States Environmental Protection Agency, Washington, DC, USA.

192
(179) Wania, F.; Dugani, C. B. Assessing the long-range transport potential of polybrominated
diphenyl ethers: a comparison of four multimedia models. Environ. Toxicol. Chem. 2003, 22,
1252-1261.
(180) Hung, H.; Blanchard, P.; Halsall, C. J.; Bidleman, T. F.; Stern, G. A.; Fellin, P.; Muir, D.
C. G.; Barrie, L. A.; Jantunen, L. M.; Helm, P. A.; Ma, J.; Konoplev, A. Temporal and spatial
variabilities of atmospheric polychlorinated biphenyls (PCBs), organochlorine (OC) pesticides
and polycyclic aromatic hydrocarbons (PAHs) in the Canadian Arctic: Results from a decade of
monitoring. Sci. Total Environ. 2005, 342, 119-144.
(181) Bossi, R.; Skov, H.; Vorkamp, K.; Christensen, J.; Rastogi, S. C.; Egelov, A.; Petersen,
D. Atmospheric concentrations of organochlorine pesticides, polybrominated diphenyl ethers and
polychloronaphthalenes in Nuuk, South-West Greenland. Atmos. Environ. 2008, 42, 7293-7303.
(182) La Guardia, M. J.; Hale, R. C.; Harvey, E. Detailed polybrominated diphenyl ether
(PBDE) congener composition of the widely used penta-, octa-, and deca-PBDE technical flame-
retardant mixtures. Environ. Sci. Technol. 2006, 40, 6247-6254.
(183) Harrad, S.; Abdallah, M. A. E. Calibration of two passive air sampler configurations for
monitoring concentrations of hexabromocyclododecanes in indoor air. J. Environ. Monit. 2008,
10, 527-531.
(184) Wang, X. P.; Gong, P.; Yao, T. D.; Jones, K. C. Passive air sampling of organochlorine
pesticides, polychlorinated biphenyls, and polybrominated diphenyl ethers across the Tibetan
Plateau. Environ. Sci. Technol. 2010, 44, 2988-2993.