papel funcional das isoformas gnnk+ e gnnk- de kit …
TRANSCRIPT

Fernanda de Paula Cury
PAPEL FUNCIONAL DAS ISOFORMAS GNNK+ E GNNK- DE KIT EM GLIOBLASTOMA
Dissertação apresentada ao Programa de Pós-Graduação da Fundação Pio XII – Hospital de Câncer de Barretos para obtenção do Título de Mestre em Ciências da Saúde. Área de Concentração: Oncologia Orientadora: Dra. Olga Catarina Lopes Martinho Co-orientador: Dr. Rui Manuel Vieira Reis
Barretos, SP
2016

Fernanda de Paula Cury
PAPEL FUNCIONAL DAS ISOFORMAS GNNK+ E GNNK- DE KIT EM GLIOBLASTOMA
Dissertação apresentada ao Programa de Pós-Graduação da Fundação Pio XII – Hospital de Câncer de Barretos para obtenção do Título de Mestre em Ciências da Saúde. Área de Concentração: Oncologia Orientadora: Dra. Olga Catarina Lopes Martinho Co-orientador: Dr. Rui Manuel Vieira Reis
Barretos, SP
2016

FICHA CATALOGRÁFICA
Preparada por Martins Fideles dos Santos Neto CRB 8/9570
Biblioteca da Fundação Pio XII – Hospital de Câncer de Barretos
C975p Cury, Fernanda de Paula. Papel funcional das isoformas GNNK+ e GNNK- de KIT em glioblastoma/
Fernanda de Paula Cury. - Barretos, SP 2016. 124 f. : il. Orientador: Drª. Olga Catarina Lopes Martinho Co-Orientador: Dr. Rui Manuel Vieira Reis Dissertação (Mestrado em Ciências da Saúde) – Fundação Pio XII – Hospital
de Câncer de Barretos, 2016. 1. Isoformas de RNA 2. Proteínas Proto-Oncogênicas c-KIT 3. Glioblastoma
4. Pacientes 5. Células 6. Terapêutica. I. Autor. II.Martinho, Olga Catarina Lopes. III. Reis, Rui Manuel Vieira. IV. Título
CDD 571.6

FOLHA DE APROVAÇÃO
Fernanda de Paula Cury
Papel funcional das isoformas GNNK+ e GNNK- de KIT em glioblastoma.
Dissertação apresentada ao Programa de Pós-Graduação da Fundação Pio XII – Hospital de
Câncer de Barretos para obtenção do Título de Mestre em Ciências da Saúde - Área de
Concentração: Oncologia
Data da aprovação: 02/03/2016
Banca Examinadora:
Prof.ª Dra. Glaucia Noeli Maroso Hajj
Instituição: Fundação Antônio Prudente, Hospital AC Camargo
Prof. Dr. Luciano Neder Serafini
Instituição: Faculdade de Medicina de Ribeirão Preto - USP
Prof.ª Dra. Olga Catarina Lopes Martinho
Orientadora
Prof. Dr. Rui Manuel Vieira Reis
Presidente da Banca

SUPORTE À PESQUISA POR AGÊNCIA DE FOMENTO
Este trabalho recebeu apoio da Fundação de Amparo a Pesquisa do Estado de São Paulo
(FAPESP) através de Bolsa de Mestrado - Regular (processo número 2014/03684-0) e Bolsa
Estágio de Pesquisa no Exterior – BEPE (processo número 2015/02691-6).
As opiniões, hipóteses e conclusões ou recomendações expressas neste material são de
responsabilidade dos autores e não necessariamente refletem a visão da FAPESP.

“Esta dissertação foi elaborada e está apresentada de acordo com as normas da Pós-
Graduação do Hospital de Câncer de Barretos – Fundação Pio XII, baseando-se no Regimento
do Programa de Pós-Graduação em Oncologia e no Manual de Apresentação de Dissertações
e Teses do Hospital de Câncer de Barretos. Os pesquisadores declaram ainda que este
trabalho foi realizado em concordância com o Código de Boas Práticas Científicas (FAPESP),
não havendo nada em seu conteúdo que possa ser considerado como plágio, fabricação ou
falsificação de dados. As opiniões, hipóteses e conclusões ou recomendações expressas
neste material são de responsabilidade dos autores e não necessariamente refletem a visão
da Fundação Pio XII – Hospital de Câncer de Barretos.”
“Embora o Núcleo de Apoio ao Pesquisador do Hospital de Câncer de Barretos tenha
realizado as análises estatísticas e orientado sua interpretação, a descrição da metodologia
estatística, a apresentação dos resultados e suas conclusões são de inteira responsabilidade
dos pesquisadores envolvidos.”

Aos meus pais, pelo amor incondicional,
carinho e preocupação. Pelo exemplo, apoio e
incentivo sempre.

AGRADECIMENTOS
À Profa. Dra. Olga Catarina Lopes Martinho, minha orientadora, por depositar tanta
confiança em mim, por todo ensinamento e ser tão presente apesar da distância. Por
compartilhar de tantas conquistas que, com certeza, foram nossas. Tenho uma imensa
admiração pela sua determinação, exemplo de pessoa e profissional. Além do mais, tenho
enorme orgulho em dizer que sou sua orientanda. Obrigada por tudo!
Ao Prof. Dr. Rui Manuel Vieira Reis, meu co-orientador, por toda ajuda e por estar sempre à
disposição. Apesar de alguns tropeços meus, agradeço pelas correções e elogios que tenho
certeza que me ajudaram amadurecer na pesquisa.
Ao Me. Renato Oliveira pela enorme paciência em ensinar, pela companhia nos almoços e
nos cafés, pelas risadas nos momentos mais críticos do mestrado, pelo abraço todos os dias
de manhã, por aguentar meus momentos de estresse, meus choros e, principalmente, pela
amizade.
Ao Dr. Matias Melendez, Dra. Viviane Aline, Me. André Lengert e Me. Adriana Cruvinel
pelas várias contribuições, pela disponibilidade e boa vontade em ajudar sempre. Obrigada,
também, pela amizade.
Ao Departamento de Patologia do Hospital de Câncer de Barretos, ao Dr. Cristovam
Scapulatempo e à Dra. Gisele Caravina de Almeida, pela revisão de lâminas e avaliação das
imunohistoquímicas, à Patrícia, Bia e Letícia pela eficiência na separação e cortes dos blocos
de parafina. Agradeço também ao Dr. José Manuel Lopes, do IPATIMUP, pela avaliação das
imunohistoquímicas.
Ao Biobanco do Hospital de Câncer de Barretos, pelo levantamento dos casos e extração de
RNA dos tecidos tumorais.
Aos médicos da Neurocirurgia, por sempre estarem dispostos a auxiliar nas dúvidas clínicas.

Ao NAP (Núcleo de Apoio ao Pesquisador) pelo auxílio na confecção do Banco de Dados e
análise estatística.
Ao SAME, em especial ao Dante e ao Kleber, pela eficiência em separar os prontuários.
Ao EPIT (Escritório de Projetos e Inovação Tecnológica) por toda eficiência nas compras de
reagentes e, em especial à Joyce, por todo auxílio com pedido de bolsa e relatórios FAPESP.
Aos membros das bancas de acompanhamento/qualificação e defesa(Dra. Gláucia Maroso
Hajj, Dr. Sérgio Vicente Serrano e Dr. Luciano Neder Serafini), pelas contribuições e
correções deste trabalho.
Ao Departamento de Pós-Graduação, essencialmente à Silvana e a Brenda, por toda
eficiência e organização, pela disposição em ajudar sempre e por lembrarem todos os
prazos.
À FAPESP pela concessão de bolsa de estudos e pela oportunidade da primeira experiência
em outro país.
Ao ICVS (Instituto de Investigação de Ciências da Vida e da Saúde – Universidade do Minho)
por me receber durante três meses e contribuir para esse trabalho.
Ao Hospital de Câncer de Barretos pelo apoio financeiro e toda infraestrutura que, com
certeza, é de uma qualidade imensurável.
Aos meus amigos de toda a vida, que acompanharam desde o início do mestrado, e mesmo
não entendendo muito sobre a pesquisa e biologia molecular, sempre compartilharam a
felicidade com as minhas conquistas, obrigada por compreenderem meus momentos de
ausência e, ainda assim, não desistirem de mim.

E finalmente, e extremamente importantes, à todas as amizades feitas no CPOM (Centro de
Pesquisa em Oncologia Molecular), que desde a iniciação científica, me receberam muito
bem. Em quatro anos, conheci praticamente todas as “gerações” e me orgulho muito de
fazer parte desse laboratório.
À Anna Luiza (Anninha) e Maraísa (Mara), simplesmente pela amizade. Encontrei em
vocês muita parceria, companheirismo, confiança e suporte. Com toda certeza se
tornaram essenciais e sei que posso contar com vocês em todos os momentos.
Obrigada.
Aos meus companheiros de bancada: Weder, pela amizade, pelas conversas durante
o café, por toda ajuda sempre, obrigada por ser essa pessoa tão fácil de dividir a
bancada (deve ser porque somos aquarianos!), à Karina, pela eficiência como
biologista e fazer de tudo pela organização do laboratório, obrigada pelo “bom dia”
de todas as manhãs, além de toda meiguice e alegria em pessoa e, à Izabela,
obrigada pelas conversas e por dividir conhecimento sobre a extração de novos
compostos.
À Paula Felício, Estela, Paula Pastrez, Aline Rocha, Tatiane Honda, Isana pela
amizade e por compartilhar os prazos do mestrado, as disciplinas da Pós-Graduação,
os trabalhos feitos em grupo, a espera de resposta das agências de fomento e a
alegria de aprovação nas bancas de acompanhamento/qualificação.
Ao Maicon (Irmão) pela amizade e pela alegria sempre, à Nathalia pela disposição em
ajudar sempre e à Carol Laus, Vânia, Lidia e Carol Carvalho, pelo auxílio em dúvidas
científicas e pelas experiências de vida trocadas.
Aos meus amigos residentes: Murilo por ter se tornado meu “best” em tão pouco
tempo, e Sâmia pelas conversas, risadas e por me entender tão bem.
A todos que direta ou indiretamente contribuíram para a realização deste trabalho.

“A grandeza de um ser humano não está no quanto ele sabe,
mas no quanto ele tem consciência que não sabe.”
(Augusto Cury)

ÍNDICE
1 INTRODUÇÃO 1
1.1 Câncer 2
1.2 Epidemiologia 3
1.3 Tumores Primários do Sistema Nervoso Central 6
1.4 Glioblastoma 7
1.5 Receptores Tirosina Quinase 7
1.6 KIT 9
1.7 KIT e Sistema Nervoso Central 11
1.8 Isoformas GNNK 13
1.9 Terapia-Alvo 15
2 JUSTIFICATIVA 18
3 OBJETIVOS 21
4 MATERIAL E MÉTODOS 23
4.1 Tecidos tumorais (Casos) 24
4.2 Linhagens celulares e cultura celular 25
4.3 Extração de RNA e RT-PCR (Reverse Transcription-Polymerase Chain Reaction) 25
4.4 Imunohistoquímica e Imunoflorescência para CD117 26
4.5 Transfecção estável de linhagens celulares 27
4.6 Ensaio de Viabilidade Celular 28
4.7 Ensaio de Clonogenicidade 28
4.8 Ensaio de Migração (Wound healing) 28
4.9 Ensaio de Invasão em Matrigel 29
4.10 Western blot 29
4.11 Ensaio in vivo em Membrana Corioalantóide (CAM) de embrião de galinha 30
4.12 Análise Estatística 30
4.13 Delineamento Experimental 31
4.13.1 Caracterizar a expressão de RNAm e proteína de KIT em glioblastoma 31

4.13.2 Impacto clínico das isoformas de KIT em pacientes com glioblastoma 31
4.13.3 Análise do papel tumorigênico das isoformas de KIT em glioblastomas 31
4.13.4 Análise das funções bioquímicas das isoformas de KIT em linhagens celulares de
glioblastoma 32
4.13.5 Ensaio in vivo em Membrana Corioalantóide (CAM) de embrião de galinha 32
5 RESULTADOS 33
5.1 Expressão de RNAm e proteína de KIT em glioblastoma 34
5.2 Impacto clínico das isoformas de KIT em pacientes com glioblastoma 38
5.3 Análise do papel tumorigênico das isoformas de KIT em glioblastomas 51
5.4 Análise das funções bioquímicas das isoformas de KIT em linhagens celulares de
glioblastoma 55
5.5 Ensaio in vivo em Membrana Corioalantóide (CAM) de embrião de galinha 57
6 DISCUSSÃO 60
7 CONCLUSÕES 67
8 PERSPECTIVAS 69
REFERÊNCIAS 71
ANEXOS 80

LISTA DE FIGURAS
Figura 1 - Estimativas para o ano de 2016 das taxas brutas de incidência por
100 habitantes e do número de casos novos de câncer, segundo
sexo (a: homens; b: mulheres) e localização primária para o Brasil. 5
Figura 2 - Esquema do desenvolvimento de células neuroectodermal
(neurônios e células da glia) e classificação dos tumores
neurológicos. No SNC, células neurais multipotentes originam três
tipos celulares principais no SNC – neurônios, oligodendrócitos e
astrócitos. A classificação dos tumores do SNC é baseada no seu
tipo celular predominante (Adaptado). 6
Figura 3 - Famílias dos Receptores Tirosina Quinase (RTK). Nesse esquema,
estão listados todos os membros de cada família. 9
Figura 4 - Análise de alterações em casos do TCGA no gene KIT em GBMs,
disponível no cBioPortal for Cancer Genomics. a: Avaliação de
alterações encontradas, mutação e amplificação, no gene KIT em
GBMs. b: Mutações encontradas no gene KIT em GBMs. 12
Figura 5 - Esquema das isoformas GNNK de KIT codificadas devido ao
splicing alternativo do RNAm que resultam na produção de duas
isoformas caracterizadas pela presença (+) ou ausência (-) de 12
nucleotídeos que codificam uma sequência tetrapeptídica GNNK. 13
Figura 6 - Caracterização das isoformas GNNK de KIT de um amplo painel
(73) de linhagens celulares tumorais. 15
Figura 7 - Alterações genéticas frequentes de GBMs em três vias de
sinalização críticas. Alterações nas sequências primárias e

mudanças significativas de copy number para os componentes das
vias de sinalização de (a) RTK/RAS/PI3K, (b) p53 e (c) RB. 16
Figura 8 - a: Análise por RT-PCR da expressão das isoformas GNNK de KIT de
linhagens estabelecidas de glioblastoma, linhagem estabelecida de
astrócitos humanos normais (NHA) e tecidos normais (NF: tecido
normal de cérebro fetal, NA e NB: tecidos normais de cérebro
adulto) em gel de agarose a 4%. b: Análise da expressão proteica
das linhagens estabelecidas de glioblastoma, NHA, NF e NA por
western blot utilizando o anticorpo de KIT (CD117 – DAKO A4502). 19
Figura 9 - Representação do número de amostras utilizadas para cada
técnica (RT-PCR e IHQ), número de prontuários revisados e as
respectivas instituições onde foram coletados. 24
Figura 10 - Análise da predominância das isoformas GNNK de KIT nos tecidos
congelados e parafinados de glioblastoma em cinco grupos:
negativo para RNAm, 100% GNNK+, 100% GNNK-, 75%
GNNK+/25% GNNK-, 75% GNNK-/25% GNNK+ e 50% GNNK+/50%
GNNK-. Foram avaliados 136 casos, sendo 120 de tecidos
congelados (Hospital de Câncer de Barretos - Brasil) e 19 de
tecidos parafinados (Hospital Pedro Hispano – Portugal). 34
Figura 11 - Análise da expressão da proteína KIT em cortes parafinados de
glioblastoma por Imunohistoquímica (IHC). a: GBM com baixa
expressão nas células tumorais; b: GBM com expressão moderada
nas células tumorais; c: expressão de CD117 nos microvasos
(tecido endotelial); d: expressão de CD117 na proliferação
microvascular (tecido endotelial). 35

Figura 12 - Análise da expressão das isoformas GNNK de KIT em 8 linhagens
celulares primárias de glioblastoma e análise dos respectivos
tecidos congelados dos pacientes. Análise da expressão proteica
de KIT das respectivas linhagens celulares. 36
Figura 13 - Análise dos níveis de expressão da proteína KIT, expressão do
ligante de KIT (SCF) e expressão de KIT fosforilado (resíduo de
tirosina 719) em células tumorais (A, B, C) e células endoteliais (D,
E, F). 37
Figura 14 - Curvas de sobrevivência de 136 pacientes, utilizando curvas de
Kaplan Meier e teste de log-rank. a: Curvas de sobrevivência com
amostras negativas pra KIT, com predominância da isoforma
GNNK+, com predominância da isoforma GNNK- e expressão igual
de ambas isoformas (p-valor 0,263); b: Curvas de sobrevivência de
amostras com KIT positivo e KIT negativo (p-valor 0,057); c: Curvas
de sobrevivência de amostras com a predominância de GNNK+ e
amostras com predominância de GNNK- (p-valor 0,291). 49
Figura 15 - Curvas de sobrevivência de 136 pacientes, utilizando Kaplan Meier
e teste de log-rank. a: Curvas de sobrevivência com amostras com
expressão positiva e negativa da proteína KIT nas células
neoplásicas (p-valor 0,517); b: Curvas de sobrevivência de
amostras com expressão positiva e negativa da proteína KIT no
tecido endotelial (p-valor 0,354). 50
Figura 16 - Confirmação da transfecção das isoformas GNNK+ e GNNK- de KIT
na linhagem GAMG através do RNAm, expressão da proteina KIT
(CD117) e confirmação da ativação através da expressão de KIT
fosforilado (p-KIT 719). 52

Figura 17 - Ensaios celulares realizados com a linhagem celular GAMG
transfectada estavelmente e selecionada com 300ug/ml de G418.
a: ensaio de viabilidade celular; b: ensaio de clonogenicidade; c:
ensaio de wound healing; d: ensaio de invasão em Matrigel. 53
Figura 18 - Ensaios celulares realizados com a linhagem celular U251
transfectada estavelmente e selecionada com 300ug/ml de G418.
a: Confirmação da transfecção das isoformas GNNK+ e GNNK- de
KIT na linhagem GAMG através do RNAm, expressão da proteina
KIT (CD117); b: ensaio de viabilidade celular; c: ensaio de
clonogenicidade; d: ensaio de wound healing. 54
Figura 19 - Efeito na ativação de KIT e vias intracelulares (MAPK e AKT) em
células transfectadas com as isoformas GNNK+ e GNNK- na
linhagem celular GAMG. Os gráficos apresentam a quantificação
das proteínas, sendo (proteína fosforilada/proteína total). 55
Figura 20 - Efeito na ativação de KIT e vias intracelulares em células
transfectadas com as isoformas GNNK+ e GNNK- na linhagem
celular U251. O gráfico apresenta a quantificação das proteínas,
sendo (proteína fosforilada/proteína total). 56
Figura 21 - Imagens de Imunofluorescência (IF) com 0, 15 e 30 min de
estimulação com SCF; utilizando anticorpo CD117 (DAKO A4502)
nas linhagens transfectadas e no pCDNA3 (vetor vazio). 57
Figura 22 - a: Análise da formação tumoral na CAM, nos dias 13 (in ovo) e 17
(ex ovo), com a linhagem GAMG; b: os tumores formados com as
linhagens transfectadas apresentaram maior média de perímetro
em comparação ao vetor vazio, com resultados estatisticamente
significativos (GNNK- p-valor <0,0001; GNNK+ p-valor 0,0154); c:

coloração de H&E e imunohistoquímica para CD117 (KIT) de cortes
feitos a partir da CAM incluída em parafina. 58
Figura 23 - a: Análise da formação tumoral na CAM, nos dias 13 (in ovo) e 17
(ex ovo), com a linhagem U251; b: os tumores formados com a
linhagem transfectada com a isoforma GNNK- apresentaram maior
média de perímetro em comparação ao vetor vazio, com
resultados estatisticamente significativos (p-valor 0,0311). 59

LISTA DE TABELAS
Tabela 1 - Casuística dos 21 casos que foram analisados de acordo com a
expressão da proteína KIT, expressão do ligante de KIT (SCF) e
expressão de KIT fosforilado (resíduo de tirosina 719) e suas
respectivas marcações. 38
Tabela 2 - Características e histórico da doença de 148 pacientes com GBM
do Hospital de Câncer de Barretos e Hospital Pedro Hispano
(Portugal). 40
Tabela 3 - Associação da expressão do RNA de KIT com as variáveis
demográficas e clínico-patológicas de pacientes com glioblastoma
(n=136). 41
Tabela 4 - Associação da predominância da isoforma GNNK- de KIT com as
variáveis demográficas e clínico-patológicas de pacientes com
glioblastoma (n=136). 42
Tabela 5 - Associação da predominância da isoforma GNNK+ de KIT com as
variáveis demográficas e clínico-patológicas de pacientes com
glioblastoma (n=136). 43
Tabela 6 - Associação da expressão da proteína KIT (células neoplásicas) com
as variáveis demográficas e clínico-patológicas de pacientes com
glioblastoma (n=70). 44
Tabela 7 - Associação da expressão da proteína KIT (tecido endotelial) com as
variáveis demográficas e clínico-patológicas de pacientes com
glioblastoma (n=70). 45

Tabela 8 - Associação da expressão da proteína KIT (células neoplásicas) com
a expressão do RNA e as isoformas de KIT de pacientes com
glioblastoma (n=63). 46
Tabela 9 - Associação da expressão da proteína KIT (tecido endotelial) com a
expressão do RNA e as isoformas de KIT de pacientes com
glioblastoma (n=63). 46
Tabela 10 - Associação entre os dados clínico-patológicos com a sobrevida de
pacientes com GBM. 47
Tabela 11 - Associação entre dados de expressão de KIT com a sobrevida de
pacientes com GBM. 48

LISTA DE ABREVIATURAS
CAM Ensaio em Membrana Corioalantóide
CD117 KIT; SCFR
cDNA DNA complementar
CEP Comitê de Ética em Pesquisa
DAB 3,3'-diaminobenzidine
DAPI 4',6-diamidino-2-phenylindole
DMEM Dulbecco's Modified Eagle Medium
DMSO Dimetilsulfóxido
DP Desvio padrão
FFPE Formalin-fixed paraffin-embedded
FLT3 Fms-Related Tyrosine Kinase
GBM Glioblastoma multiforme
GIST Gastrointestinal stromal tumors
GNNK Gly-Asn-Asn-Lys
GM-CSF Granulocyte-macrophage colony-stimulating factor
IC50 Half maximal inhibitory concentration
IF Imunofluorescência
IHQ Imunohistoquímica
INCA Instituto Nacional do Câncer
JAK/STAT Janus kinase/signal transducers and activators of transcription
kDa kilodalton
KPS Índice de Karnofsky
MAPK Mitogen-activated protein kinase
NHA Normal human astrocytes
PBS Phosphate-buffered saline
PDGFRA/B Platelet-derived growth fator receptor alpha/beta
PI3K Phosphoinositide 3-kinase
PTEN Phosphatase and tensin homolog
P/S Penicilina/Streptomicina

QT Quimioterapia
RB Proteína do Retinoblastoma
RNAm RNA mensageiro
RT Radioterapia
RTK Receptor Tirosina Quinase
RT-PCR Reverse Transcription Polymerase Chain Reaction
SCF Stem cell factor
SFB Soro fetal bovino
SNC Sistema Nervoso Central
TCG Tumores de Células Germinativas
TMZ Temozolamida
TCGA The Cancer Genome Atlas
TKI Inibidor Tirosina Quinase
VEGF Vascular endothelial growth factor
VEGFR Vascular endothelial growth factor receptor

LISTA DE SÍMBOLOS
% Porcentagem
˂ Menor
= Igual
˃ Maior
≤ Menor ou igual
≥ Maior ou igual
µ micro (106)

RESUMO
Cury FP. Papel funcional das isoformas GNNK + e GNNK- de KIT em glioblastoma. Dissertação
(Mestrado). Barretos: Hospital de Câncer de Barretos; 2016.
INTRODUÇÃO: O glioblastoma (GBM) é o tumor cerebral mais comum em adultos e uma das
doenças malignas mais mortais em humanos e essa situação não tem mudado
significativamente nas últimas décadas, devido às propriedades biológicas do GBM. A
proteína KIT, membro da família III dos receptores de tirosina quinase (RTK), está envolvida
com a tumorigênese de alguns tumores e devido à existência de uma pequena molécula
inibidora de KIT tem feito desta proteína um alvo molecular terapêutico para o câncer. Em
GBMs, tem sido observada a presença de amplificação gênica de KIT, em vez de mutações
ativantes neste gene. Devido ao splicing alternativo do RNAm, KIT é expressa em duas
diferentes isoformas, que são caracterizadas pela presença (+) ou ausência (-) da sequência
tetrapeptídica (GNNK) na região extracelular justamembranar. Estas isoformas demonstram
possuir características de sinalização intracelular distintas e também diferente atividade de
transformação tumorigênica em fibroblastos de camundongos. JUSTIFICATIVA: Resultados
preliminares tem mostrado que as isoformas GNNK são frequentemente co-expressas tanto
em linhagens celulares de GBMs quanto em tecidos normais, com GNNK- sendo a forma
predominante em linhagens celulares de GBM, enquanto que em tecidos normais cerebrais
é predominante a isoforma GNNK+, sugerindo que a isoforma GNNK- pode desempenhar um
papel na tumorigênese de GBM. Nosso trabalho é o primeiro a abordar o papel funcional das
isoformas GNNK de KIT em GBM. OBJETIVO: Dessa forma, neste projeto, temos o objetivo
de esclarecer o papel funcional e biológico das isoformas GNNK de KIT em GBMs a fim de
observar se as isoformas GNNK conferem sinalização celular e papel tumorigênico distintos,
além de observar o impacto clínico em pacientes com GBM. MATERIAL E MÉTODOS: Tecidos
congelados/parafinados de glioblastoma foram avaliados quanto à expressão das isoformas
GNNK por RT-PCR e quanto à expressão de KIT por imunohistoquímica. Além disso, foram
realizados ensaios celulares (in vitro e in vivo) com linhagens celulares de GBM transfectadas
com as isoformas GNNK. RESULTADOS: Podemos observar que o RNAm de KIT está presente
em 95% das amostras de tecidos tumorais congelados/parafinados, sendo predominante a

isoforma GNNK+, quanto à presença da proteína, expressa em uma frequência menor, está
presente em apenas 22,8% das células neoplásicas e 40,2% nas células endoteliais. Em uma
casuística de 21 casos, observamos que a expressão de p-KIT só é encontrada nos casos que
expressam SCF e KIT, indicando que KIT é ativado em GBM através de mecanismos
autócrinos/parácrinos. Em relação ao impacto clínico das isoformas, encontramos uma
tendência na sobrevida de pacientes que expressam o RNAm daqueles que não expressam,
estes por sua vez, apresentam uma melhor sobrevida. Nos ensaios celulares, notamos maior
viabilidade, potencial de invasão e proliferação tumoral das células transfectadas com a
isoforma GNNK-; também notamos maior crescimento tumoral nos ensaios in vivo.
CONCLUSÕES: Embora não conseguimos diferenças estatisticamente significativas entre a
expressão das isoformas e a sobrevida global, através dos ensaios celulares conseguimos
notar que há diferenças na ativação de KIT e no papel tumorigênico entre as isoformas,
sendo a GNNK- mais tumorigênica.
PALAVRAS-CHAVE: Isoformas; KIT; Glioblastoma; Terapia-Alvo; Tratamento; Biomarcador.

ABSTRACT
Cury FP. Functional role of KIT GNNK+ and GNNK- isoforms in glioblastoma. Dissertation
(Master’s degree). Barretos: Barretos Cancer Hospital; 2016.
INTRODUCTION: Glioblastoma is the most common adult brain tumor and one of the
deadliest human malignancies and this situation has not significantly changed in the last
decades, due to the biological properties of GBM. KIT, a member of the receptor tyrosine
kinase (RTK) family III, is involved with the tumorigenesis of some tumors and the existence
of specific small molecule inhibitors for KIT made this protein a key molecular therapeutic
target in cancer. In GBMs, it has been observed the presence of KIT gene amplification,
instead of activating mutations in this gene. Due to mRNA alternative splicing, KIT is
expressed by two different functional isoforms, which are characterized by the presence (+)
or absence (-) of a tetrapeptide sequence (GNNK) in the extracellular juxtamembrane region.
They were shown to display distinct intracellular signaling features and also different
tumorigenic transforming activities in mouse fibroblasts. JUSTIFICATIVE: Preliminary results
have showed that GNNK isoform are frequently co-expressed in both glioblastoma cell lines
and normal tissues, with GNNK- being the prevalent form in glioblastoma cell lines, whereas
in normal brain tissues there is predominance of GNNK+ isoform, suggesting that GNNK-
isoform could play a role in glioblastoma tumorigenesis. Hitherto, there are no reports
assessing KIT GNNK isoforms functional role of KIT GNNK isoforms in GBM. AIM: Thus, in this
project we aim to shed light on the functional and biological role of KIT GNNK isoforms in
GBMs to observed if the GNNK isoforms present cell signaling and distinct tumorigenic role,
in addition, observe the clinical impact in patients with GBM. MATERIAL AND METHODS:
Frozen/FFPE glioblastoma tissues was evaluated for expression of GNNK isoforms by RT-PCR
and also evaluated the KIT expression by immunohistochemistry. Additionally, cellular assays
were performed (in vitro and in vivo) in glioblastoma cell lines transfected with GNNK
isoforms. RESULTS: We observe that KIT mRNA is present in 95% of frozen/paraffin
embedded tumor tissues, with GNNK+ isoform predominance in tumor tissues, regarding the
protein presence, expressed in lower frequency, it is present in only 22,8% neoplastic cells
and 40,2% in endothelial cells. In 21 cases, we found that p-KIT expression is found only in

cases that express SCF and KIT, indicating that KIT is activated in GBM through
autocrine/paracrine mechanisms. Relative to isoforms clinical impact, we found differences
in survival between patients that express the mRNA of those who do not express, these in
turn have a better survival; in cellular assays, we noticed greater viability, potential invasion
and proliferation of tumor cells transfected with the isoform GNNK-; also noticed greater
tumor growth in in vivo assays. CONCLUSIONS: Although we cannot statistically significant
differences between the expression of isoforms and overall survival, through cellular assays
can notice that there are differences in the activation of KIT and tumorigenic role between
isoforms, and the GNNK- more tumorigenic.
KEYWORDS: Isoforms; KIT; Glioblastoma; Targeted Therapy; Treatment; Biomarker.

1
INTRODUÇÃO

2
1 INTRODUÇÃO
1.1 Câncer
O câncer é um termo genérico para várias doenças (mais de 100 tipos) que podem
afetar qualquer parte do corpo, é definido também como tumores malignos ou neoplasias.
Sua principal característica é o crescimento descontrolado de células anormais que crescem
além dos seus limites, e que podem invadir partes adjacentes e se espalhar para outros
órgãos, processo conhecido como metástase (OMS – Organização Mundial da Saúde). Essas
células em um determinado tecido já não são responsivas aos sinais específicos deste tecido
que regulam a diferenciação celular, sobrevivência, proliferação e morte.
A ideia de que o câncer é uma doença genética das células somáticas foi proposta em
1914 por Theodor Boveri e, desde então, outros estudos confirmam que o câncer é uma
doença genética, entretanto se diferencia por dois motivos do restante das outras doenças
genéticas: (1) é causado, na sua maioria, a partir de mutações somáticas, enquanto que
todas as outras doenças genéticas em mamíferos (exceto aquelas que envolvem genes
mitocondriais) são causadas unicamente por mutações nas linhagens germinativas; (2) cada
câncer se inicia não a partir de uma única mutação, mas a partir de um acúmulo de
mutações 1.
Como sabemos, em alguns tecidos adultos ocorre a proliferação celular contínua
como estratégia de renovação tecidual. Entretanto, em outros tecidos, essa proliferação
constante não acontece, exceto nos processos de cicatrização, como exemplo os
hepatócitos, células do músculo cardíaco e os neurônios, que permanecem funcionais por
longos períodos ou até mesmo durante toda a vida do organismo. Os danos genéticos em
duas principais classes de genes (Proto-oncogenes e Genes Supressores Tumorais) estão
envolvidos na perda dessa regulação celular, levando à falhas dos mecanismos que
controlam o crescimento e proliferação celular 2. Os proto-oncogenes são ativados por
alterações tornando-se oncogenes, que causa uma atividade excessiva na promoção do
crescimento. Os genes supressores tumorais normalmente reprimem o crescimento, então o
dano nesses genes permite o crescimento inapropriado.

3
Além disso, mecanismos epigenéticos, que são essenciais para o desenvolvimento
normal e manutenção do padrão de expressão gênica de tecidos específicos, podem
contribuir também para a perda da regulação celular, levando à alterações na função do
gene e transformação maligna celular. Mecanismos epigenéticos no câncer incluem
metilação do DNA, modificação de histonas e microRNAs 3.
Com a finalidade de compreender a diversidade das doenças neoplásicas foi proposto
por Hanahan e Weinberg (2000 e 2011) os Hallmarks of Cancer, que são capacidades
biológicas adquiridas durante o desenvolvimento dos tumores. Dessa forma, as células
normais precisariam adquirir uma sucessão dessas capacidades para o processo de
tumorigênese se iniciar. São os hallmarks: manutenção da sinalização proliferativa,
descontrole dos supressores de crescimento, resistência à morte celular, imortalidade,
indução de angiogênese, ativação de invasão e metástase, desregulação energética,
bloqueio da destruição do sistema imune, inflamação como indutor de tumores,
instabilidade genômica e mutações. A compreensão desses conceitos tende a afetar cada
vez mais o desenvolvimento de novos meios para tratar o câncer 4.
O processo de origem do câncer, é chamado de oncogênese ou tumorigênese, sendo
iniciado a partir de erros na duplicação ou reparo de genes que podem ocorrer
espontaneamente, ou pela alteração por carcinógenos. Dessa forma, esse processo é uma
interação entre a genética e o ambiente 2. Aproximadamente 5 – 10% de todos os cânceres
são herdados, sendo portanto, a maioria esporádicos 5. Além de causas genéticas, como as
síndromes hereditárias, já existe um conjunto de fatores ambientais e estilo de vida bem
definidos envolvidos na causa do câncer, são eles: tabagismo, álcool, dieta, atividade física,
fatores ocupacionais, fatores ambientais e agentes infecciosos 6.
1.2 Epidemiologia
Tumores primários do Sistema Nervoso Central (SNC) são representados por
neoplasias benignas e malignas que apresentam histologia e alterações moleculares
distintas. As neoplasias malignas constituem a principal causa de morte relacionada ao

4
câncer (sólido) na população pediátrica e a terceira em adultos de meia-idade, embora
representem menos de 2% de todas as neoplasias malignas 7-9.
Dentre os principais fatores de risco associados aos tumores do SNC estão as
síndromes genéticas raras, responsáveis por cerca de 5% dos gliomas malignos, são elas:
neurofibromatose do tipo 1 e 2, esclerose tuberosa, doença de Von Hippel-Lindau,
síndromes de Li-Fraumeni, Gorlin e Turcot. Em relação aos fatores ambientais, a exposição a
altas doses de radiação ionizante está associada ao aumento do risco, enquanto substâncias
químicas, exposição ocupacional, etilismo, tabagismo, radiação não-ionizante (telefone
celular), trauma craniano e infecções não apresentam resultados conclusivos 9-11.
Considerando os tumores do SNC malignos, os gliomas são responsáveis por 70% dos
casos, e os astrocitomas correspondem a 76% dos gliomas 12.
As taxas de incidência dos gliomas variam de acordo com o tipo histológico, idade de
diagnóstico, gênero, raça e país. Os gliomas são mais comuns em homens que em mulheres,
exceto os astrocitomas pilocíticos que ocorrem em taxas semelhantes 11. Os astrocitomas de
alto-grau (anaplásico e glioblastoma) aumentam sua incidência de acordo com a idade,
atingindo pacientes entre 75-84 anos, na sua maioria. Já os oligodendrogliomas e
oligoastrocitomas, são mais comuns em pacientes com idade entre 35-44 anos 13.
Para o Brasil, no ano de 2014, foram estimados 4.960 casos novos de câncer do SNC
em homens e 4.130 em mulheres, segundo o Instituto Nacional do Câncer (INCA). Mesmo
não sendo muito frequentes, os tumores de SNC contribuem significativamente para a
morbidade global. Durante as últimas décadas, a incidência e mortalidade dos tumores de
SNC têm aumentado na maioria dos países desenvolvidos e, isso se deve à melhoria de
novas tecnologias diagnósticas e também à maior expectativa de vida 14, 15. As estatísticas de
casos novos para 2016 no Brasil têm aumentado, cerca de 5.440 novos casos em homens e
4.830 novos casos em mulheres, como mostra a figura 1.

5
a.
b.
Figura 1 – Estimativas para o ano de 2016 das taxas brutas de incidência por 100 habitantes e do
número de casos novos de câncer, segundo sexo (a: homens; b: mulheres) e localização primária
para o Brasil 16.

6
1.3 Tumores Primários do Sistema Nervoso Central
A base para o conceito de classificação dos tumores do SNC foi lançada em 1856 por
Virchow, que descreveu a neuroglia. Atualmente, a classificação mais aceita e empregada
para tumores do SNC, é a classificação da OMS de 2007, que pode ser considerada como
uma escala de malignidade de acordo com o comportamento clínico e biológico, o perfil
genético e o prognóstico das neoplasias 17.
Os gliomas malignos são o grupo mais comum, representando aproximadamente 70%
de todos os tumores cerebrais, são denominados dessa forma devido à sua semelhança com
as células gliais do cérebro. São classificados de acordo com a diferenciação das células
progenitoras, apresentando propriedade de astrócitos, oligodendrócitos e células
ependimais 18.
Os gliomas são classificados histologicamente em astrocitomas, oligodendrogliomas,
oligoastrocitomas e ependimomas. Estes, por sua vez, são classificados de acordo com o
grau de malignidade: astrocitomas são graduados em astrocitoma pilocítico (grau I),
astrocitoma difuso (grau II), astrocitoma anaplásico (grau III) e glioblastoma (IV);
oligodendrogliomas e oligoastrocitomas são graduados em difuso (grau II) e anaplásico (grau
III) 19. Ependimomas são classificados em ependimoma mixopapilar (grau I), subependimoma
(grau I), ependimoma clássico (grau II) e ependimoma anaplásico (grau III) 17.
Diferente de outros tumores sólidos, nos gliomas raramente ocorre metástase para
outros locais do corpo 19.
Meduloblastoma
Oligodendroglioma
Oligoastrocitoma
Astrocitoma
Neurônios
Oligodendrócitos
Astrócitos
Células-tronco neurais
Células progenitoras
neurais
Células progenitoras gliais restritas
Células progenitoras
neurais restritas

7
Figura 2 – Esquema do desenvolvimento de células neuroectodermal (neurônios e células da glia) e
classificação dos tumores neurológicos. No SNC, células neurais multipotentes originam três tipos
celulares principais no SNC – neurônios, oligodendrócitos e astrócitos. A classificação dos tumores
do SNC é baseada no seu tipo celular predominante (Adaptado) 20.
1.4 Glioblastoma
A manifestação mais frequente e agressiva entre os gliomas, o GBM, apresenta uma
proliferação endotelial, necrose, alta densidade de células e atipias 18, é dividido em primário
e secundário, que afetam vias genéticas diferentes, afetam pacientes em idades diferentes
e, também apresentam prognósticos diferentes 21. O GBM, tipo mais agressivo entre os
astrocitomas, é responsável por 53,8% dos gliomas 12.
GBMs primários (de novo) acometem cerca de 80% dos casos e ocorrem em pacientes
mais idosos (média de idade: 62 anos), enquanto GBMs secundários desenvolvem-se a partir
de astrocitomas ou oligodendrogliomas de baixo-grau (média de idade: 45 anos); em
crianças, os GBMs são raros, acometendo apenas 3 % dos tumores cerebrais e do SNC em
pacientes de 0-19 anos de idades. O GBM possui um mau prognóstico com sobrevida relativa
bastante baixa, sendo a média de 16 meses; apenas alguns pacientes alcançam o tempo de
sobrevida de 2,5 anos e menos de 5 % dos pacientes alcançam uma sobrevida de 5 anos
após o diagnóstico 22.
Os glioblastomas apresentam a pior sobrevida global, onde os fatores prognósticos
mais importantes para a sobrevida após diagnóstico desses pacientes são: grau de ressecção
do tumor, idade do diagnóstico e índice de Karnofsky 23, 24.
O tratamento consiste na máxima ressecção cirúrgica, seguido de radioterapia (RT)
com temozolamida (TMZ) concomitante seguido de seis ciclos de manutenção de TMZ 25;
apesar de todos os avanços da neurocirurgia, radioterapia e quimioterapia – o fato
estatístico pouco mudou ao longo das últimas três décadas 7. Portanto, novas formas de
terapias são necessárias para melhoras significativas na sobrevivência desses pacientes.
1.5 Receptores Tirosina Quinase

8
Os Receptores Tirosina Quinase (RTK) são receptores de membrana celular que foram
descobertos há 25 anos atrás e, emergiram como chave regulatória de processos celulares
importantes, como proliferação, diferenciação, sobrevivência, metabolismo, migração
celular e controle do ciclo celular 26. São responsáveis por receber e transmitir os sinais
extracelulares, se ligando a fatores de crescimento e outras proteínas liberadas localmente
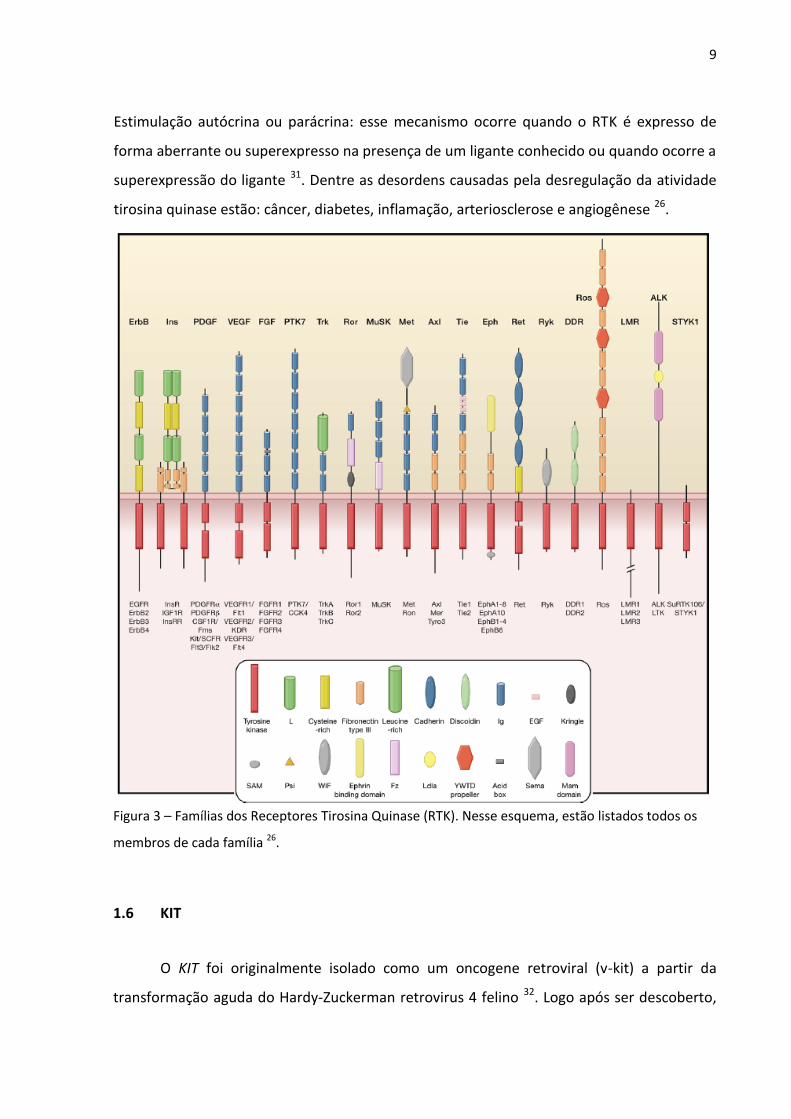
que estão presentes em baixas concentrações. Humanos possuem 58 RTKs conhecidos, que
são divididos em 20 subfamílias como ilustrado na figura 3. Todos os RTKs apresentam uma
estrutura bastante similar, são constituídos por uma porção extracelular com o sítio de
ligação para o fator de crescimento, uma região transmembrana e a região citoplasmática
que contém os domínios tirosina quinase (TK) além de uma terminação carboxila (C-) e
regiões regulatórias justamembrana 26, 27.
O ligante é responsável por induzir ou estabilizar a dimerização do receptor, levando
ao aumento da atividade quinase do RTK. O domínio intracelular catalítico deste receptor
apresenta sítios de ligação de ATP responsáveis por catalizar a autofosforilação de resíduos
tirosina citoplasmáticos, que servem como sítios de ligação para Src homology 2 (SH2) e
phosphotyrosine-binding (PTB) contendo proteínas como Shc, Grb2, Src, Cbl e PLC. Essas
proteínas, então, recrutam moléculas efetoras que ativam a cascata de sinalização
intracelular, sendo as principais: MAPK, PI3K/AKT e JAK/STAT 28.
Os RTKs apresentam uma atividade fundamental nas células normais, entretanto, a
desregulação da sua sinalização, seja por mecanismos autócrinos ou parácrinos dos fatores
de crescimento ou por alterações genéticas resultam em uma ativação da sinalização
constitutiva ou fortemente aumentada, levando à uma transformação maligna. São três os
principais mecanismos que levam à essa desregulação: (1) Amplificação gênica: ocorre a
superexpressão de um RTK na membrana celular aumentando a incidência da dimerização
do receptor mesmo na ausência do ligante, um exemplo importante é a superexpressão
e/ou amplificação de HER2 em casos de câncer de mama e ovário 29 e, também a
amplificação de EGFR em câncer de pulmão não-pequenas-células, bexiga, cervical, ovário,
rim, pâncreas e cabeça e pescoço; (2) Mutações gênicas: incluem deleções ou mutações no
domínio extracelular e alterações no domínio catalítico de ligação do ATP, um exemplo é a
forma mutada de EGFR, EGFRvIII, caracterizado pela deleção de 267 aminoácidos no
domínio extracelular levando a uma ativação constitutiva na ausência do ligante 30; e (3)
9
Estimulação autócrina ou parácrina: esse mecanismo ocorre quando o RTK é expresso de
forma aberrante ou superexpresso na presença de um ligante conhecido ou quando ocorre a
superexpressão do ligante 31. Dentre as desordens causadas pela desregulação da atividade
tirosina quinase estão: câncer, diabetes, inflamação, arteriosclerose e angiogênese 26.
Figura 3 – Famílias dos Receptores Tirosina Quinase (RTK). Nesse esquema, estão listados todos os
membros de cada família 26.
1.6 KIT
O KIT foi originalmente isolado como um oncogene retroviral (v-kit) a partir da
transformação aguda do Hardy-Zuckerman retrovirus 4 felino 32. Logo após ser descoberto,

10
KIT foi encontrado codificado pelo locus W que possui mais de 30 mutações conhecidas 33.
Além disso, a proteína KIT também denominada CD117, foi definida como um receptor
tirosina quinase (RTK) pertencente à subfamília III das RTKs, incluindo também PDGFRA/B
(Platelet-derived growth fator receptor alpha/beta), FLT3 (Fms-Related Tyrosine Kinase) e
GM-CSF (Granulocyte-macrophage colony-stimulating factor). Essa subfamília é composta
por um domínio extracelular e um domínio intracelular de tirosina quinase dividido em duas
partes. O domínio extracelular de KIT contém cinco imunoglobulinas (codificada pelo éxons
1-9), o domínio transmembrana (codificada pelo éxon 11) e o domínio tirosina quinase
(codificada pelos éxons 13-21) 34, e por fim, a extremidade terminal COOH. KIT é uma
proteína com 976 aminoácidos, fazendo com que o tamanho da proteína seja de
aproximadamente 110 kDa; ligações heterogêneas de glicosilação resultam na proteína
madura com 145 e 160 kDa 34, 35. Existem até nove sítios de N-glicosilação, a maioria
concentrada no domínio extracelular próximo à membrana plasmática. Outras modificações
pós-traducionais são conhecidas no KIT, incluindo a fosforilação nos resíduos tirosina e
serina 36 e também a ubiquitinação para regulação da sua internalização de degradação 37-39.
O ligante de KIT, também conhecido como Stem cell factor (SCF) ou Steel factor, é um
fator de crescimento que existe ligado à membrana ou então na forma solúvel. O gene que
codifica o ligante está localizado no cromossomo 10 em camundongos e no cromossomo 12
em humanos 40 e possui nove éxons 41. O SCF é expresso por fibroblastos e células
endoteliais em todo o corpo, promovendo a proliferação, migração e sobrevivência e
diferenciação de progenitores hematopoiéticos, de melanócitos e de células germinativas 40.
Sob circunstâncias normais, o ligante SCF se liga no KIT e ativa a sua função tirosina quinase
intrínseca formando um elo de ligação com duas moléculas de KIT a um dímero, levando a
uma fosforilação cruzada de dois resíduos de tirosina (Y568 e Y570) no domínio da
justamembranar autoinibitório e a transfosforilação do Y823 no local de ativação do domínio
tirosina quinase. A autofosforilação subsequente de outros resíduos de tirosina quinase
(Y703, Y721, Y730, Y736) permite a ativação de vias de cascatas de sinalização como MAPK,
PI3K/AKT e JAK/STAT. Essas cascatas de sinalização culminam na regulação gênica de vários
mecanismos celulares, incluindo apoptose, proliferação celular, diferenciação, adesão e
mobilidade 42. A sinalização de KIT é essencial para a manutenção de células-tronco durante
a hematopoiese, gametogênese, e melanogênese. Conforme estas observações, a falta de

11
KIT provoca múltiplos defeitos que incluem anemia, esterilidade, e despigmentação
(piebaldismo), e a ausência de KIT leva camundongos mutados à morte durante a primeira
semana de vida devido ao defeito hematopoiético 43.
Alterações em KIT têm sido implicadas no câncer. Diversas evidências, incluindo
algumas do nosso grupo 44-46, mostraram que as propriedades oncogênicas de KIT são devido
à mutações de ganho de função deste gene [por exemplo, tumores estromais
gastrointestinais (GISTs)], com a presença de mecanismos autócrinos/parácrinos com SCF
(por exemplo, carcinomas de pulmão e ginecológicos), e pela presença de amplificação
gênica (por exemplo gliomas e seminomas) 43, 47. Ressalta-se que o oncogene KIT representa
um alvo molecular terapêutico chave no câncer, com o desenvolvimento de inibidores
específicos, como o imatinib e sunitinib, levando à eficácia clínica sem precedentes no
tratamento de GIST com KIT (éxons 11 e 9 hotspots) mutado 48.
1.7 KIT e Sistema Nervoso Central
O papel do KIT no SNC está longe de ser bem compreendido. KIT está descrita como
sendo expressa em diferentes regiões do SNC durante o desenvolvimento normal do cérebro
49. Uma variedade de abordagens sugere que, no sistema nervoso de adultos, a sinalização
KIT pode exercer diversas funções como, o estabelecimento da conexão neuronal,
sobrevivência, e potenciar as atividades sinápticas. Além disso, o KIT exerce um papel na
diferenciação oligodendrocítica, na migração de células progenitoras à locais de danos no
cérebro e, é expresso em zonas neuroproliferativas do cérebro adulto 50-52.
Nos tumores do SNC, alguns estudos têm mostrado a superexpressão em
meduloblastoma, neuroblastoma e glioma 53. O nosso grupo analisou uma série de 185
gliomas, detectando a superexpressão de KIT em 16% dos casos e amplificação gênica de KIT
em 33% dos gliomas positivos para KIT, principalmente nos GBMs, sugerindo que a
amplificação gênica de KIT, em vez de mutações ativantes, pode ser o mecanismo molecular
subjacente à superexpressão da proteína em GBMs 46, 54. Estes resultados foram confirmados
por outros estudos 55-57, destacando o papel de KIT em um subconjunto de GBMs.
Interessantemente, um estudo relatou a superexpressão de KIT induz a proliferação de

12
astrócitos em camundongos, que é inibida pelo imatinib 58. Além disso, em outro estudo
onde verificaram a expressão de KIT em 345 tumores, sendo de 34 diferentes histologias,
observaram que somente os glioblastomas apresentam a expressão de KIT no tecido
endotelial em 59% dos casos de GBM (13/22), estes tumores são conhecidos por abrigar
proliferação microvascular com características morfológicas próprias 59.
Segundo dados do TCGA (The Cancer Genome Atlas) - TCGA, Nature 2008, TCGA, Cell
2013 e TCGA Provisório – disponível no cBioPortal for Cancer Genomics, foram avaliados
dados de mutação, deleção e amplificação de KIT em GBMs. No TCGA 2008, em 91 casos,
foram encontrados 11% (10 casos) de amplificação e 1,1% (1 caso) de mutação; no TCGA
2013, em 281 casos, foram encontrados 9,6% (27 casos) de amplificação e 1,1% (3 casos) de
mutação; e no TCGA Provisório, em 273 casos, foram encontrados 9,2% (25 casos) de
amplificação e 1,1% (3 casos) de mutação. As mutações encontradas foram V50M (2), D72E
(2), T132M (2) e A599T (1).
a.
Mutação
Deleção
Amplificação
Múltiplas alterações

13
b.
V5
0M
D7
2E
T13
2M
A5
99
T
Figura 4 – Análise de alterações em casos do TCGA no gene KIT em GBMs, disponível no cBioPortal
for Cancer Genomics. a: Avaliação de alterações encontradas, mutação e amplificação, no gene KIT
em GBMs. b: Mutações encontradas no gene KIT em GBMs.
1.8 Isoformas GNNK
O splicing alternativo do RNA mensageiro (RNAm), devido ao uso alternativo da
região de corte doadora 5’, resulta na produção de duas isoformas caracterizadas pela
presença (+) ou ausência (-) de 12 nucleotídeos que codificam uma sequência tetrapeptídica
Gly-Asn-Asn-Lys (GNNK) na parte extracelular da região justamembranar (éxon 9) como
ilustrado na figura 5. As isoformas GNNK+ e GNNK- também são denominadas de c-Kit e c-
KitA, respectivamente. São coexpressas na maioria dos tecidos, mas a isoforma GNNK-
geralmente é predominante 34, 60-62.
COOH
NH2
IG1
IG2
IG3
IG3
IG3
TM
TK1
TK2
GNNK
IG1
IG2
IG3
IG3
IG3
TM
TK1
TK2
NH2
COOH
G N N K
CO
OH
NH
2
IG1
IG2 IG
3
IG3
IG3
TM
TK1
TK2
…GCA TTT AAA GGT AAC AAC AAA GAG CAA
G N N K
…GCA TTT AAA GAG CAA
-
Reith et al, Embo J, 1991
Yarden at al, Embo J, 1987
cDNA
cDNA
+

14
Figura 5 – Esquema das isoformas GNNK de KIT codificadas devido ao splicing alternativo do RNA
mensageiro que resultam na produção de duas isoformas caracterizadas pela presença (+) ou
ausência (-) de 12 nucleotídeos que codificam uma sequência tetrapeptídica GNNK (Gly-Asn-Asn-Lys)
(Imagem adaptada).
Um estudo demonstrou que células NIH3T3 transfectadas com a isoforma GNNK- são
tumorigênicas in vitro e in vivo, enquanto células transfectadas com a isoforma GNNK+ não
são 63. Tem sido demonstrado também que essas isoformas diferem na intensidade e
duração de ativação de diferentes vias de sinalização, sendo a isoforma GNNK- tirosina
altamente fosforilada e rapidamente internalizada. Mais importante, o efeito de ambas
isoformas na cascata de vias de sinalização parece ser dependente do tipo celular 63-67. As
isoformas GNNK+ e GNNK- parecem ser coexpressas numa variedade de tipos celulares, com
a predominância da isoforma GNNK-. Em neoplasias hematológicas, os estudos sugerem
que não há diferenças entre a distribuição de células normais e neoplásicas em isoformas
GNNK. Em relação a tumores sólidos, os poucos relatos sobre diferentes neoplasias
apresentam resultados inconsistentes 61, 63-69. Um estudo mostrou que a co-expressão de
ambas isoformas resulta na diminuição da sobrevida global em pacientes com leucemia
mieloide aguda (LMA) comparado com pacientes que apresentam apenas a isoforma GNNK-
70.
Recentemente o nosso grupo realizou uma caracterização geral das isoformas de KIT
em um painel de 73 linhagens celulares comerciais provenientes de vários locais, como:
próstata, meduloblastoma, pâncreas, colo do útero, bexiga, cabeça e pescoço, mama,
esôfago, melanoma, cólon, glioma pediátrico, GBM, ovário e pulmão. Entre as diversas
linhagens celulares, podemos perceber, na maioria, a positividade de expressão de RNAm de
KIT com predominância da isoforma GNNK- (Martinho O et al, dados não publicados).

15
100% 100%
75%
50% 50%42,8%
33,3% 33,3%27,3%
20% 20%14,3%
25%
50%
25% 42,8% 66,6% 66,6%
54,5%
40% 40%
85,7%
100%
60%
20%10%
25%14,3% 18,2%
30%40%
20%
negativa 25% GNNK+; 75% GNNK- 50% GNNK+; 50% GNNK- 75% GNNK+; 25% GNNK- 100% GNNK+ 100% GNNK-
N=3 N=2 N=4 N=4 N=4 N=7 N=6 N=3 N=11 N=10 N=5 N=7 N=2 N=5
Figura 6 - Caracterização das isoformas GNNK de KIT de um amplo painel (73) de linhagens celulares
tumorais (Martinho O et al, dados não publicados).
1.9 Terapia-Alvo
Os GBMs são um dos maiores desafios para o tratamento do câncer, apresentam um
mau prognóstico devido à resistência terapêutica (heterogeneidade molecular intratumoral
e resistência às “glioma stem-cells”) e recorrência tumoral após a ressecção cirúrgica (alto
potencial invasivo tumoral) 71. Entretanto, a terapia-alvo têm sido uma promessa para o
tratamento de pacientes com gliomas malignos, devido ao sucesso alcançado em outros
tipos tumorais, como câncer de pulmão não-pequenas células 72, melanoma 73 e leucemia
mieloide aguda 74.
Em 2008, o TCGA (The Cancer Genome Atlas) identificou três vias de sinalização
principais na patogênese dos glioblastomas: receptores tirosina quinase (RTK)/RAS/PI3K, p53
e proteína do retinoblastoma (RB) 75. Além da via pró-angiogênica, importante para a
gliomagênese e manutenção do fenótipo dos gliomas 71.

16
Figura 7 – Alterações genéticas frequentes de GBMs em três vias de sinalização críticas. Alterações
nas sequências primárias e mudanças significativas de copy number para os componentes das vias de
sinalização de (a) RTK/RAS/PI3K, (b) p53 e (c) RB. Vermelho indica alterações genéticas ativantes,
com genes mais frequentemente alterados em tons de vermelho mais forte. Em azul, estão indicadas
alterações de inativação, quanto mais forte o azul, corresponde à maior porcentagem de alterações.
Para cada componente alterado de cada via de sinalização, o tipo da alteração e a porcentagem dos
tumores afetados estão indicados. Caixas azuis contêm as porcentagens finais de glioblastomas com
alterações em pelo menos um componente conhecido da via designada 75.
Terapia-alvo específicas para essas vias de sinalização tem sido a principal estratégia
de tratamento para a terapia molecular de gliomas. Entretanto, as vias de p53 e RB são
difíceis de tornar-se um alvo terapêutico, dessa forma, as vias dos RTK tem sido o principal
foco para potenciais estratégias terapêuticas. Receptores de fatores de crescimento (EGFR E
PDGFR, por exemplo) ou RTKs são proteínas que atravessam a membrana plasmática e
apresentam um domínio extracelular que se ligam aos seus respectivos ligantes (EGF e

17
PDGF, por exemplo) e domínios intracelulares associados à atividade tirosina quinase. Essas
alterações genéticas na via dos RTK/RAS/PI3K em GBM foi confirmada em mais de 88% dos
tumores 75. VEGFR (Vascular endothelial growth factor receptor) também é um RTK
responsável pela regulação da angiogênese, VEGF (Vascular endothelial growth factor) se
liga em VEGFR ativando cascatas de sinalização intracelular pelas vias de RAS/MAPK e
PI3K/AKT, promovendo a migração e proliferação de células endoteliais e estimulando a
formação de novos vasos sanguíneos, além disso, a expressão de VEGF tem sido relatada de
acordo com o grau de malignidade do tumor, por isso a proposta do alvo VEGF/VEGFR
parece efetiva para o controle do crescimento tumoral 76, 77. O Bevacizumab é um anticorpo
monoclonal contra o VEGF, aprovado pelo FDA em 2009 78. Entretanto, apesar da resposta e
aumento da sobrevida livre de progressão, o bevacizumab não beneficiou a sobrevida global
de pacientes com glioblastoma, tanto recorrentes quanto diagnosticados de novo 79-81.
Existem diversos inibidores de KIT, conhecidos como TKIs (tyrosine kinase inhibitor),
que atuam em outros receptores também, por exemplo: Imatinib/Gleevec (alvos: KIT e
PDGFRA), Sunitinib/Sutent (alvos: KIT, PDGFRA e VEGFR2), Sorafenib/Nexavar (alvos: KIT,
PDGFRA e VEGFR2), Dasatinib/Sprycel (alvos: Abl, Src e KIT), Nilotinib/Tasigna (alvos: Bcr-Abl
e KIT) e Cediranib/Recentin (alvos: Bcr-Abl, VEGFR e KIT). Todos já são aprovados pelo FDA
para outros tipos tumorais. É importante ressaltar que ensaios clínicos que avaliam a eficácia
de fármacos anti-KIT como agente único ou em combinação com a quimioterapia em
pacientes com GBM estão em andamento 82, 83, entretanto, é desconhecida sua resposta em
relação às isoformas GNNK de KIT em GBMs.

18
JUSTIFICATIVA

19
2 JUSTIFICATIVA
Como mencionado na revisão de literatura, KIT parece regular um importante papel
tanto no SNC normal quanto no patológico. Para completar o nosso conhecimento sobre o
impacto de alterações de KIT em GBMs usamos linhagens celulares de GBM como modelos
in vitro. Assim, preliminarmente caracterizamos um painel de 8 linhagens celulares de GBMs
para alterações moleculares de KIT. Nenhuma das linhagens celulares apresenta mutações
ativantes no gene KIT, e a metade delas apresentam amplificação gênica em KIT 84. Sete das
linhagens celulares são positivas para a expressão do RNAm de KIT, e apenas duas
expressam a proteína (Martinho O et al, dados pessoais). Portanto, outros mecanismos
podem regular a expressão gênica e função de KIT, como o splicing do RNAm.
Surpreendentemente, a avaliação da expressão das isoformas GNNK de KIT nunca foi
realizada em ambos os tecidos, normal e tumoral de cérebro. Assim, investigamos a
distribuição relativa das duas isoformas de KIT nas 8 linhagens celulares de GBM e em 3
amostras de tecido cerebral normal através RT-PCR utilizando primers específicos, como
mostrado na figura 8. Em 7 linhagens celulares que expressam KIT, nós encontramos a co-
expressão de ambas isoformas com a predominância da isoforma GNNK-. Em contraste, em
3 amostras de tecidos cerebrais normais encontramos uma baixa expressão da isoforma
GNNK- e predominância de GNNK+ (Martinho O et al, dados pessoais). Essa expressão
inversa dos níveis das isoformas GNNK de KIT entre células normais e tumorais nunca foi
descrito antes para outros tipos celulares. Então, os nossos resultados preliminares são
muito promissores, indicando que o aumento da expressão da isoforma GNNK- pode
desempenhar um papel importante na tumorigênese do GBM.
hGUSRN
Am
GNNK+GNNK-
Linhagens Celulares Tecido cerebral
SW
10
88
U2
51
SN
B-1
9
U3
73
U8
7-M
G
GA
MG
SW
17
83
A1
72
NH
A
NF
NA
NB
a.

20
Pro
teín
a
CD117
β-Actina
Linhagens Celulares Tecido cerebral
SW
10
88
U2
51
SN
B-1
9
U3
73
U8
7-M
G
GA
MG
SW
17
83
A1
72
NH
A
NF
NA
b.
Figura 8 – a: Análise da expressão das isoformas GNNK de KIT por RT-PCR em linhagens estabelecidas
de glioblastoma, linhagem estabelecida de astrócitos humanos normais (NHA) e tecidos normais (NF:
tecido normal de cérebro fetal, NA e NB: tecidos normais de cérebro adulto) em gel de agarose a 4%.
b: Análise da expressão proteica das linhagens estabelecidas de glioblastoma, NHA, NF e NA por
western blot utilizando o anticorpo de KIT (CD117 – DAKO A4502).

21
OBJETIVOS

22
3 OBJETIVOS
O principal objetivo deste projeto é, assim investigar o papel funcional e biológico das
isoformas GNNK de KIT em GBMs. Especificamente, pretendemos avaliar o papel das
isoformas GNNK na sinalização de KIT e analisar o papel tumorigênico das isoformas de KIT.
Finalmente, em uma série de GBMs queremos saber as implicações clínicas das isoformas de
KIT nestes pacientes. Assim, propõe-se dividir o presente projeto em cinco tarefas:
1. Caracterizar a expressão de RNAm e proteína de KIT em linhagens e tecidos tumorais
de glioblastoma;
2. Impacto clínico das isoformas de KIT em pacientes com glioblastoma;
3. Análise do papel tumorigênico das isoformas de KIT;
4. Análise das vias de sinalização das isoformas de KIT;
5. Análise do papel tumorigênico das isoformas de KIT utilizando ensaio in vivo em
Membrana Corioalantóide (CAM) de embrião de galinha.

23
MATERIAL E MÉTODOS

24
4 MATERIAL E MÉTODOS
4.1 Tecidos tumorais (Casos)
Cento e dezessete amostras de tecidos congelados, dezenove amostras de tecidos
parafinados (FFPE) e noventa e dois cortes de 4 µm de tecidos tumorais parafinados de GBM
foram recuperados do arquivo de Patologia do Hospital de Câncer de Barretos, Barretos, São
Paulo, Brasil, com a aprovação do Comitê de Ética em Pesquisa (CEP) nº 802/2014 (Anexo A,
B e C), do Hospital Pedro Hispano, Matosinhos, Portugal e do Hospital Santo António, Porto,
Portugal. Foram coletadas informações clinico-patológicas relevantes como: idade do
paciente, sexo, cor, tabagismo, etilismo, índice de Karnofsky (KPS), lateralidade,
radioterapia, quimioterapia e recidiva da doença. Informações de follow-up também foram
coletadas para todos os pacientes e sobrevida global foi definida como o tempo entre a data
do primeiro diagnóstico e a data da última informação ou morte do paciente (de acordo com
a ficha de coleta – Anexo D). Além disso, tecidos normais cerebrais foram utilizados, sendo
um tecido normal fetal (NF) e dois tecidos normais adultos (NA e NB).
Amostras de GBM Amostras de GBM Prontuários
71 tecidos parafinados de GBM – Hospital de Câncer
de Barretos
21 tecidos parafinados de GBM – Hospital Santo
António
129 prontuários – Hospital de Câncer de Barretos
19 prontuários – Hospital Pedro Hispano (PT)
RNA Proteína
117 tecidos congelados de GBM – Hospital de Câncer
de Barretos
19 tecidos parafinados –Hospital Pedro Hispano (PT)
Revisão
136 amostras para expressão do RNAm e
isoformas
92 amostras para expressão da proteína
148 prontuários revisados
Figura 9 – Representação do número de amostras utilizadas para cada técnica (RT-PCR e IHQ),
número de prontuários revisados e as respectivas instituições onde foram coletados.

25
Critérios de Inclusão (amostras provenientes do Hospital de Câncer de Barretos):
- Indivíduos maiores de 18 anos;
- Pacientes com as suas respectivas amostras de tecido congelado tumoral.
4.2 Linhagens celulares e cultura celular
Oito linhagens celulares de GBM imortalizadas foram utilizadas: SW1088, SW1783, U-
87 MG e A172, obtidas da ATCC (American Type Culture Collection), SNB-19 e GAMG foram
obtidas da DSMZ (German Collection of Microorganisms and Cell Cultures) e U251 e U373
foram fornecidas gentilmente pelo Professor Joseph Costello. Todas as linhagens celulares
foram mantidas em DMEM-10, a 37ºC e 5% CO2 85. Além disso, culturas celulares primárias
tumorais foram derivadas de biópsias cirúrgicas obtidas no Departamento de Neurocirurgia
do Hospital de Câncer de Barretos, São Paulo, Brasil. O estudo foi aprovado pelo Comitê de
Ética em Pesquisa local (nº 491/2011) e, os pacientes assinaram o termo de consentimento.
Cada amostra tumoral foi cortada em pequenos pedaços, removendo os vasos sanguíneos,
então ressuspendidos em tripsina (Gibco, Invitrogen) e incubados a 37ºC por 30 minutos.
Durante a incubação, a suspensão foi pipetada “up and down” várias vezes para a digestão
total dos tecidos 84.
A Autenticação das linhagens celulares foi realizada pelo Laboratório IdentiCell
Departamento de Medicina Molecular (MOMA) no Hospital Skejby - Universidade de Aarhus,
em Aarhus, Dinamarca) em Agosto de 2011. A genotipagem confirmou a identidade
completa de todas as linhagens, com exceção da U373, que se mostrou como um sub-clone
da linhagem celular U251 84. Também foi utilizada a linhagem celular de cérebro normal
denominada NHA (“normal human astrocytes” – astrócitos normais humanos).
4.3 Extração de RNA e RT-PCR (Reverse Transcription-Polymerase Chain Reaction)
Para avaliar a expressão das isoformas GNNK de KIT, nós utilizamos a extração de
RNA e RT-PCR. Todas as amostras congeladas foram coletadas no Hospital de Câncer de
Barretos (Brasil), o estudo contém 117 amostras tumorais congeladas de glioblastoma. Para

26
a purificação do RNA total desses tecidos congelados foi utilizado o RNeasy Mini Kit
(Qiagen). Recomenda-se a quantidade máxima de 30 mg de tecido congelado e o RNA foi
extraído de acordo com as instruções do fabricante. Para a extração dos tecidos parafinados
foi utilizado o PureLink FFPE RNA Isolation Kit – Ambion (Thermo Fisher Scientific) .Para as
linhagens celulares, a extração do RNA foi realizada com o TRIzol (Invitrogen), utilizando 1
mL do reagente para 5-10 x 106 células. Para determinar a integridade do RNA extraído foi
utilizado o Agilent 2100 bioanalyzer and RNA LabChip, como nosso objetivo era amplificar
um pequeno amplicon, utilizamos RIN>3.
Para a síntese do cDNA, nós utilizamos o kit Thermo Scientific Phusion RT-PCR de
acordo com as instruções do fabricante. O cDNA foi amplificado por PCR utilizando o método
padrão. Os primers foward e reverse utilizados para amplificar as isoformas GNNK de KIT
foram: 3’TGGGCAAGACTTCTGCCTAT e 5’CTCCTCAACAACCTTCCACTG, respectivamente. A
análise das isoformas GNNK de KIT foi observada em gel de agarose (4%).
4.4 Imunohistoquímica e Imunoflorescência para CD117
Cortes representativos de 4 µm de espessura foram submetidos à
imunohistoquímica. A expressão de KIT (CD117) foi avaliada por imunohistoquímica de
acordo com o sistema indireto do complexo estreptavidina-biotina peroxidase, utilizando o
anticorpo primário CD117 (diluição de 1:50; clone A4502, DAKO Corporation, Carpentaria,
CA, EUA), como descrito anteriormente 44, 46, 54. Resumidamente, lâminas desparafinizadas e
re-hidratadas foram submetidas a 10 minutos de incubação em peróxido de hidrogênio em
metanol (3%), a fim de inibir a peroxidase endógena. Não foi utilizada recuperação
antigênica. Após a incubação com o anticorpo primário, overnight a 4ᵒC, o anticorpo
secundário biotinilado polivalente foi aplicado durante 10 minutos seguido de incubação
com o complexo de estreptavidina-peroxidase. A reação imune foi visualizada por 3,3'-
Diamonobenzidine (DAB) como cromogênio (Ultravision Sistema de Detecção do Anti-
polivalente, HRP / DAB; Lab Vision, Fremont, CA, EUA). Controles positivos e negativos
apropriados foram incluídos em cada série. Os controles positivos incluíram um tumor
estromal gastrointestinal como previamente caracterizados superexpressão CD117. Os

27
controles negativos incluíram omissão do anticorpo primário e do controle negativo DAKO
(N1699, DAKO Corp., Carpentia, Califórnia, EUA). Foi realizada a contra-coloração das
lâminas com hematoxilina Gill-2 (25%). Em uma série de 21 casos, também foi realizada a
imuno-histoquímica com o mesmo método, mas utilizando o anticorpo primário p-KIT (Cell
Signalling Tyr 719) e o anticorpo primário SCF (diluição de 1:500; Santa Cruz Biotechnology).
Amostras de tumor coradas foram analisadas de acordo com um método semi
quantitativo previamente descrito e sem o conhecimento dos achados clínicos. Marcação de
CD117 na membrana celular com ou sem immunoreatividade citoplasmática em células
neoplásicas foi considerado específico. Também foi analisada a marcação do tecido
endotelial que parece específico em GBM 46, 59. A determinação da distribuição de
intensidade e extensão eram semi-quantitativamente pontuado como se segue: (-)
(negativo), (+) (≤ 5%), (++) (5-50%), e (+++) (> 50 %). As amostras com contagens de (-) e (+)
foram considerados negativos, e aqueles com as contagens (++) e (+++) foram considerados
positivos.
Para detectar a localização e a abundância relativa da proteína KIT, foi realizada a
Imunofluorescência (IF) nas células GAMG, de acordo com o método indireto, utilizando o
anticorpo primário contra CD117 (diluição 1:50; clone A4502, DAKO Corporation,
Carpentaria, CA, US). Aproximadamente 1x103 células foram plaqueadas em lamínulas,
esperou-se até atingir a confluência desejada, o meio de cultura foi aspirado, foram lavadas
três vezes em PBS 1x. As células foram fixadas com metanol gelado por 5 minutos a -20ºC,
lavadas três vezes com PBS 1x. As células foram incubadas com Block (Labvision) por 15
minutos a temperatura ambiente, depois incubadas com o anticorpo primário overnight a
4ºC, o anticorpo secundário (diluição 1:500; Goat anti-Rabbit IgG; TRITC – Invitrogen,
Thermo Fisher Scientific) foi aplicado por 1 hora a temperatura ambiente e as lamínulas
lavadas três vezes em PBS 1x. Na lâmina, foi colocada uma gota da solução de montagem
com DAPI e colocada a lamínula na lâmina.
4.5 Transfecção estável de linhagens celulares

28
Os construtos que codificam ambas isoformas GNNK de KIT, clonados no plasmídeo
pBluescript, foram fornecidos pela Dra Leonie Ashman 86. Então, cDNAs foram subclonadas
no plasmídeo pCDNA3. As linhagens celulares foram estavelmente transfectadas, utilizando
a seleção do antibiótico G418, com as isoformas GNNK+ e GNNK-. As mesmas linhagens
celulares foram transfectadas com o vetor vazio a fim de serem utilizadas como controle.
4.6 Ensaio de Viabilidade Celular
Para determinar a diferença de viabilidade celular entre as células transfectadas com
as isoformas e o vetor vazio, as células foram plaqueadas em placas de 96 poços em uma
densidade de 2x103 células por poço e foi permitida a aderência das células overnight em
DMEM 10% SFB. Ao final de 24h, a viabilidade celular foi quantificada utilizando Cell Titer96
Aqueous Cell Proliferation Assay (MTS) (Promega), sendo o tempo 0 (controle). A viabilidade
foi quantificada em tempos subsequentes (48h, 72h e 96h). Os resultados foram expressos
como a média ± DP das células viáveis relativas ao controle (tempo 0). Os gráficos foram
realizados utilizando o software GraphPad Prism. Os ensaios foram realizados em triplicata
por pelo menos três vezes (triplicata biológica e triplicata experimental).
4.7 Ensaio de Clonogenicidade
As células (1x103) foram plaqueadas em placas de 12 poços e incubadas por 24 horas
para sua aderência. O meio de cultura foi retirado e colocado novamente de acordo com as
condições: DMEM 0,5% SFB e DMEM 10% SFB por 10-15 dias. As colônias foram coradas com
Giemsa 20% em metanol por 45 minutos e contadas manualmente 87.
4.8 Ensaio de Migração (Wound healing)
As células foram plaqueadas em placas de 6 poços e cultivadas até cerca de 95% de
confluência. A monocamada de células foram lavadas com PBS, foi realizada uma “ranhura”

29
com uma ponteira de plástico de 200 µl e incubado com condições definidas (ou
concentrações fixas de inibidores). As “ranhuras” foram fotografadas em microscópio de
contraste de fase em 0, 24 e 48 horas. A distância de migração relativa foi calculada pela
seguinte fórmula: porcentagem do fechamento da ranhura (%) = 100 (A-B)/A, onde A é a
largura da ranhura antes da incubação e B é a largura da ranhura depois da incubação 85.
4.9 Ensaio de Invasão em Matrigel
A invasão de células foi medida utilizando os insertos BD BioCoat Matrigel (BD
Biosciences), como indicado nas instruções do fabricante. Em resumo, 2.5x104 células foram
plaqueadas nos insertos de 24 poços revestidos com matrigel com meio de cultura 0,5% de
SFB. DMEM 10% SFB foi utilizado como quimioatrativo. Foi permitida a invasão das células
por 48 horas. As células invasivas, ligadas à membrana do inserto, foram fixadas com
metanol e coradas com hematoxilina. As células invasivas foram fotografadas no
microscópio com aumento de 40x e contados todos os campos da membrana. Os resultados
foram expressos em relação ao controle (vetor vazio) como a porcentagem média ± DP da
invasão.
4.10 Western blot
Para avaliar o efeito das vias de sinalização intracelulares e o KIT, as células foram
cultivadas em placas de 6 poços, permitindo o crescimento de 85% de confluência e depois
foram colocadas em starvation (meio de cultura sem SFB) por 2 horas e em seguida
estimuladas com o ligante SCF. Nos tempos indicados, as células foram lavadas e raspadas
em PBS 1X gelado e lisadas em buffer contendo 50 mM Tris pH 7.6-8, 150 mM NaCl, 5 mM
EDTA, 1 mM Na3VO4, 10 mM NaF, 10 mM NaPyrophosphate, 1% NP-40 e 1/7 de coquetel de
inibidores de proteases (Roche). Para a quantificação da concentração das proteínas foi
utilizado o reagente Bradford (Sigma-Aldrich). Western blotting foi realizado utilizando o
método padrão 10% SDS-PAGE, carregado 20ug de proteína por poço. Todos os anticorpos
foram utilizados como recomendado pelo fabricante [KIT (A4502), p-KIT Tyr-719 (#3391S),

30
ERK (#4695), p-ERK (#4370), AKT (#4691), p-AKT (#4060) e β-actina (#4967), todos anticorpos
da Cell Signaling, exceto KIT, da Dako.
A detecção das proteínas através do Western blot foi realizada por
quimioluminescência (SuperSignal™ West Pico Chemiluminescent Substrate – Thermo Fisher
Scientific) no ChemiDoc MP System (Bio-Rad).
4.11 Ensaio in vivo em Membrana Corioalantóide (CAM) de embrião de galinha
Para avaliar a proliferação e angiogênese in vivo, foi utilizada a CAM, como já descrito
previamente 84, 85, 88. Ovos de galinha fertilizados foram incubados a 37ᵒC e 70% de umidade,
em que no terceiro dia de desenvolvimento, uma janela é aberta na casca dos ovos, sendo
coberta com fita adesiva e retornaram à incubação. No nono dia de desenvolvimento,
células tumorais, aproximadamente 3x106 células, foram ressuspendidas em 20 µl de
matrigel e colocadas/injetadas sobre a CAM. No 13º dia, os tumores formados foram
fotografados in ovo utilizando Stereomicroscópio (Olympus S2616). No 17ᵒ dia, os tumores
foram fotografados novamente in ovo. Os ovos de galinha foram sacrificados a -80ᵒC por 10
minutos, e a CAM de cada um foi retirada, fixada em paraformaldeído 4% e fotografada ex
ovo, além de ser incluída em parafina para análises histológicas posteriores O perímetro dos
tumores foram medidos utilizando o ImageJ nos dias 13 e 17. Os resultados são expressos
através da média da porcentagem do crescimento dos tumores de cada grupo, do dia 13
(considerado como 0%) até o dia 17, ± SD. Para o descarte, os ovos são autoclavados.
4.12 Análise Estatística
Os resultados foram estatisticamente relacionados com dados clínicos dos pacientes
utilizando o programa SPSS (versão 21.0, SPSS Inc). O teste χ2 foi aplicado para correlacionar
a expressão das isoformas de KIT e os parâmetros clínico-patológicos. Estimativa da
sobrevida global (morte por qualquer motivo) foi realizada utilizando o método de Kaplan-
Meier, e a diferença entre as curvas de sobrevida foram avaliadas com o teste log-rank.

31
Comparações únicas entre diferentes condições foram realizadas utilizando o teste t-
Student, e diferenças entre grupos foram realizadas utilizando ANOVA. Análises estatísticas
foram realizadas utilizando o Graph Pad Prism versão 5. O nível de significância adotado em
todas as análises foi p-valor<0,05.
4.13 Delineamento Experimental
4.13.1 Caracterizar a expressão de RNAm e proteína de KIT em glioblastoma
Para esta proposta, analisamos a predominância das isoformas GNNK de KIT em
linhagens celulares primárias e estabelecidas, em um grupo de amostras congeladas ou
parafinadas por conveniência (136 casos) de GBMs, que foram coletadas nos últimos anos
no Hospital de Câncer de Barretos e Hospital Pedro Hispano (Portugal), de acordo com o
Comitê de Ética e o consentimento do paciente. Além de analisar a expressão das isoformas,
analisamos também a expressão proteica de KIT (células neoplásicas e tecido endotelial) em
uma casuística de 92 pacientes, além de avaliar p-KIT e SCF em uma casuística de 21
pacientes, através de imunohistoquímica.
4.13.2 Impacto clínico das isoformas de KIT em pacientes com glioblastoma
Dados demográficos e informações clínicas relevantes, incluindo a idade, sexo, cor,
tabagista, etilista, índice de Karnofsky (KPS), lateralidade, radioterapia, quimioterapia e
recidiva foram coletados e associados à predominância das isoformas e expressão da
proteína, também foram associados à sobrevida global dos pacientes. Além disso, avaliamos
a predominância das isoformas e presença da proteína em relação à sobrevida global.
4.13.3 Análise do papel tumorigênico das isoformas de KIT em glioblastomas
Para determinar o papel tumorigênico das isoformas GNNK de KIT em GBM por meio
de estudos in vitro, utilizamos as linhagens celulares transfectadas com GNNK, que foram
estimuladas com 100 ng/ml de SCF humano e a viabilidade celular foi medida pelo ensaio de
MTS (Promega) 84, 85, 89. Também avaliamos as mudanças na migração e invasão celular nas

32
células transfectadas, pelo ensaio de wound healing e ensaio de invasão em matrigel (BD
Biosciences), respectivamente 84, 85, 89. Além disso, avaliamos a habilidade de uma única
célula formar uma colônia através do ensaio de clonogenicidade, como descrito 87. Para
todos os ensaios in vitro, os experimentos foram repetidos três vezes e as células
transfectadas com o vetor vazio foram utilizadas como controle.
4.13.4 Análise das funções bioquímicas das isoformas de KIT em linhagens celulares
de glioblastoma
Para avaliarmos as diferenças de sinalização das isoformas de KIT em linhagens
celulares de GBM, foram utilizadas as linhagens GAMG e U251. Para isto, linhagens celulares
de GBM transfectadas com as isoformas GNNK- e GNNK+ e vetor vazio (controle) foram
estimuladas com 100 ng/ml de SCF recombinante humano ao longo do tempo. Após cada
espaço de tempo (0, 5 min, 15 min, 30 min, 1 hora, 3 horas e 6 horas) os lisados foram
coletados para análise de western blot de KIT e vias de ativação intracelular. Para ativação
de KIT utilizamos um anticorpo contra o resíduo de fosfotirosina (Tyr719). Também
avaliamos as vias de sinalização intracelular MAPK e PI3K/AKT 84. A ativação e abundância da
proteína KIT também foram avaliadas por Imunofluorescência.
4.13.5 Ensaio in vivo em Membrana Corioalantóide (CAM) de embrião de galinha
Para avaliar a proliferação tumoral foram realizados 2 ensaios a fim de conseguirmos
ao menos 20 ovos por grupo (GNNK+, GNNK- e vetor vazio) para ambas as linhagens, GAMG
e U251. O ensaio tem a duração de 17 dias e, além de ser medido o perímetro do tumor
formado no 13º dia e 17º para a comparação do crescimento, também foi realizado a
imunohistoquímica desses tumores com a finalidade de observar a expressão de KIT.

33
RESULTADOS

34
5 RESULTADOS
5.1 Caracterizar a expressão de RNAm e proteína de KIT em glioblastoma
Para determinar o padrão de expressão das isoformas GNNK de KIT em GBM,
avaliamos 136 casos de GBM em tecidos congelados/parafinados (Figura 10). De um modo
geral verificou-se que RNAm de KIT está presente em 95% (129/136) dos tumores, sendo
apenas 5% (7/136) totalmente negativa para o RNAm de KIT. No que diz respeito as
isoformas GNNK, foram observados cinco padrões diferentes de expressão: tumores que
expressam 75% de GNNK+ e apenas 25% de GNNK-, correspondendo a 43% (58/136) dos
casos; tumores que expressam 75% de GNNK- e apenas 25% de GNNK+, correspondendo a
17% (23/136) dos casos; tumores que expressam 50% de cada uma das isoformas, o que
corresponde a 30% (41/136) dos casos; tumores que expressam apenas GNNK+,
correspondendo a 3% (4/136) dos casos e tumores que expressam apenas GNNK-,
correspondente a 2% (3/136) dos casos. Assim, em tecidos de GBM, RNAm de KIT é
altamente expresso, tendo a maioria das amostras de predominância da isoforma GNNK+.
0
10
20
30
40
50
% o
f cases
59/139
41/139
25/139
7/1394/139 3/139
GNNK+
GNNK-
Figura 10 - Análise da predominância das isoformas GNNK de KIT nos tecidos congelados e
parafinados de glioblastoma em cinco grupos: negativo para RNAm, 100% GNNK+, 100% GNNK-, 75%
GNNK+/25% GNNK-, 75% GNNK-/25% GNNK+ e 50% GNNK+/50% GNNK-. Foram avaliados 136 casos,
sendo 117 de tecidos congelados (Hospital de Câncer de Barretos - Brasil) e 19 de tecidos parafinados
(Hospital Pedro Hispano – Portugal).
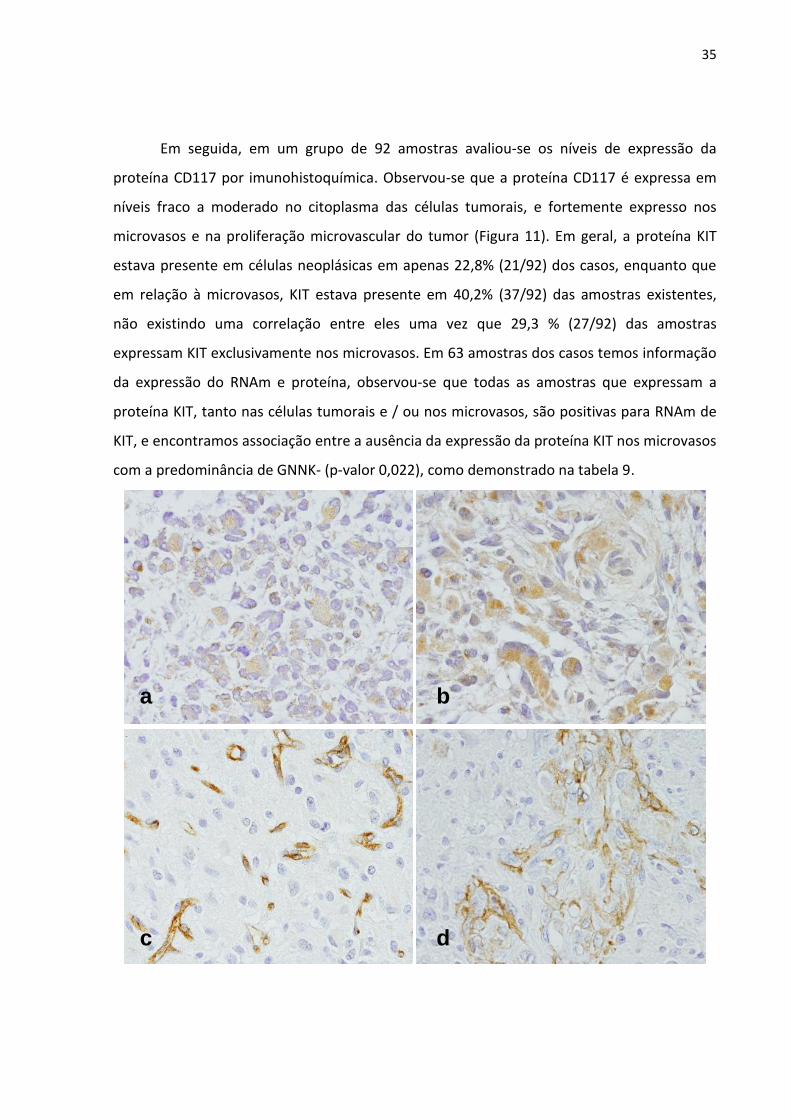
35
Em seguida, em um grupo de 92 amostras avaliou-se os níveis de expressão da
proteína CD117 por imunohistoquímica. Observou-se que a proteína CD117 é expressa em
níveis fraco a moderado no citoplasma das células tumorais, e fortemente expresso nos
microvasos e na proliferação microvascular do tumor (Figura 11). Em geral, a proteína KIT
estava presente em células neoplásicas em apenas 22,8% (21/92) dos casos, enquanto que
em relação à microvasos, KIT estava presente em 40,2% (37/92) das amostras existentes,
não existindo uma correlação entre eles uma vez que 29,3 % (27/92) das amostras
expressam KIT exclusivamente nos microvasos. Em 63 amostras dos casos temos informação
da expressão do RNAm e proteína, observou-se que todas as amostras que expressam a
proteína KIT, tanto nas células tumorais e / ou nos microvasos, são positivas para RNAm de
KIT, e encontramos associação entre a ausência da expressão da proteína KIT nos microvasos
com a predominância de GNNK- (p-valor 0,022), como demonstrado na tabela 9.
a
c
b
d

36
Figura 11 - Análise da expressão da proteína KIT em cortes parafinados de glioblastoma por
Imunohistoquímica (IHC). a: GBM com baixa expressão nas células tumorais; b: GBM com expressão
moderada nas células tumorais; c: expressão de CD117 nos microvasos (tecido endotelial); d:
expressão de CD117 na proliferação microvascular (tecido endotelial) (Aumento 200x).
Além disso, avaliamos a expressão de isoformas KIT em 8 linhagens celulares de GBM
primárias e, mais uma vez, quase todas eles expressam predominantemente a isoforma
GNNK- (Figura 12). Esta mesma análise foi realizada em 5 destes casos, entretanto, na
amostra congelada do paciente e 3 deles apresentaram um padrão de expressão diferente,
onde a predominância da isoforma negativa não é observada. Curiosamente, em 3 das
linhagens celulares primárias, a proteina de KIT estava presente no western blot, enquanto
que, para os mesmos pacientes, utilizando as amostras de FFPE não fomos capazes de
detectar a proteína KIT por IHQ (Dados não mostrados).
HC
B7
HC
B2
HC
B1
8
Proteína Linhagem Celular
CD117
β-Actina
HC
B1
9
HC
B7
HC
B2
HC
B4
0
HC
B1
8
HC
B2
9
HC
B1
62
HC
B1
48
RNAm Linhagem Celular
KIT
hGUS
HC
B2
9
HC
B2
HC
B1
62
HC
B1
48
HC
B4
0
RNAm Pacientes
Figura 12 - Análise da expressão das isoformas GNNK de KIT em 8 linhagens celulares primárias de
glioblastoma e análise dos respectivos tecidos congelados dos pacientes. Análise da expressão
proteica de KIT das respectivas linhagens celulares.
Nesse conjunto, os dados sugerem que existe uma mudança da expressão das
isoformas de KIT, o que sempre favorece a predominância da negativa, em adaptação de
células tumorais para as condições de cultura e que culmina na expressão da proteína. Se KIT
é uma oncoproteina, a hipótese de que a mudança para a superexpressão da isoforma
GNNK- poderia ser um evento oncogênico em GBM.
Finalmente, nós nos perguntamos sobre o significado da expressão da proteína KIT
em tumores GBM, já que ele existe em uma freqüência muito menor. Assim, em uma coorte
de 21 pacientes em que avaliamos os níveis de expressão da proteína KIT, também
avaliamos a expressão do ligante de KIT, o SCF, e forma ativada de KIT, resíduo de
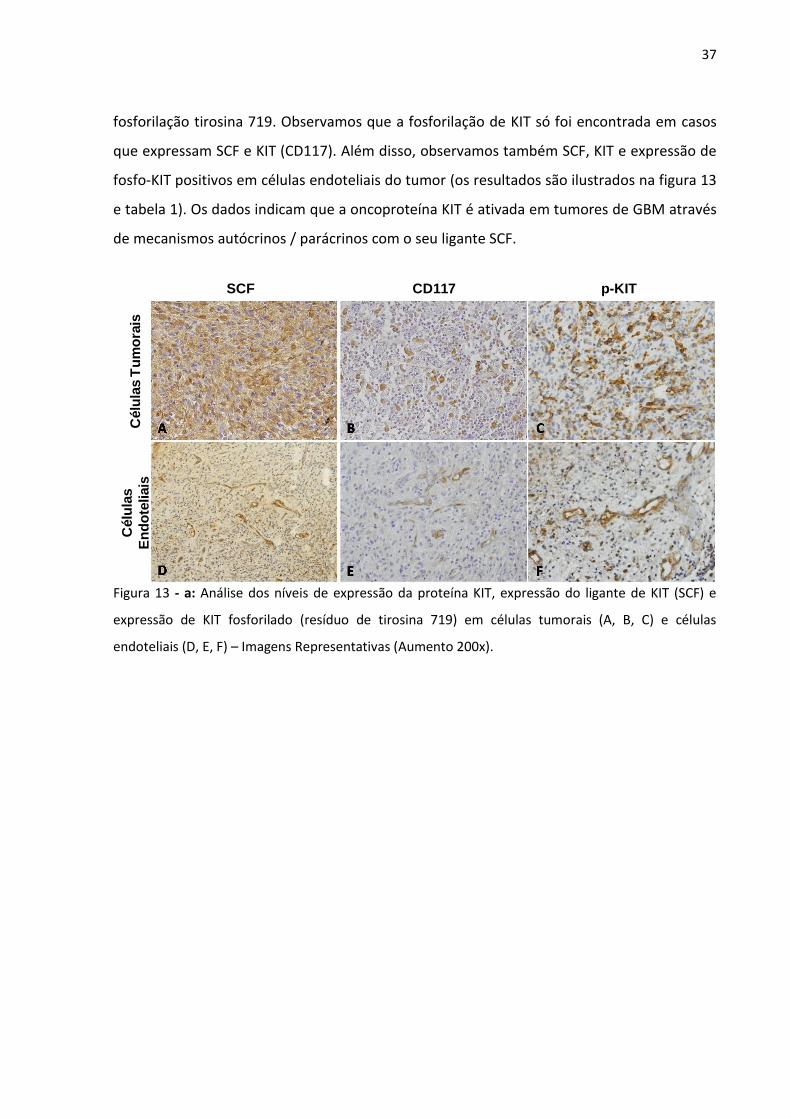
37
fosforilação tirosina 719. Observamos que a fosforilação de KIT só foi encontrada em casos
que expressam SCF e KIT (CD117). Além disso, observamos também SCF, KIT e expressão de
fosfo-KIT positivos em células endoteliais do tumor (os resultados são ilustrados na figura 13
e tabela 1). Os dados indicam que a oncoproteína KIT é ativada em tumores de GBM através
de mecanismos autócrinos / parácrinos com o seu ligante SCF.
SCF CD117 p-KIT
Célu
las T
um
ora
isC
élu
las
En
do
teliais
Figura 13 - a: Análise dos níveis de expressão da proteína KIT, expressão do ligante de KIT (SCF) e
expressão de KIT fosforilado (resíduo de tirosina 719) em células tumorais (A, B, C) e células
endoteliais (D, E, F) – Imagens Representativas (Aumento 200x).

38
Tabela 1 - Casuística dos 21 casos que foram analisados de acordo com a expressão da proteína KIT,
expressão do ligante de KIT (SCF) e expressão de KIT fosforilado (resíduo de tirosina 719) e suas
respectivas marcações.
Casos Células tumorais
Células endoteliais
SCF CD117 p-KIT SCF CD117 p-KIT
1 + - -
+ + +
2 + - n.d
+ - n.d
3 + - n.d
+ - n.d
4 - - -
+ + -
5 + + +
+ + +
6 + - -
+ - -
7 + + +
+ + +
8 + - n.d
+ - n.d
9 + - -
+ - -
10 + + +
+ + +
11 + - -
+ + -
12 + - -
- - -
13 + + +
+ - -
14 + - -
+ + -
15 - - -
+ - -
16 - - n.d
+ - n.d
17 - - n.d
+ - n.d
18 + - -
+ - -
19 + - n.d
+ - n.d
20 + - -
+ + +
21 + - n.d
+ - n.d
+: Expressão positiva; -: Expressão negativa; n.d.: não realizado
5.2 Impacto clínico das isoformas de KIT em pacientes com glioblastoma
No levantamento realizado dos casos de glioblastoma do Hospital de Câncer de
Barretos, revisamos os prontuários de 129 casos, entre o ano de 2002 e 2014; também
foram revisados 19 casos do Hospital Pedro Hispano (Braga, Portugal). Portanto, foram
revisados 148 prontuários, sendo esses pacientes com idades entre 18 e 79 anos, maioria do
sexo masculino (59,5%), maioria branca (62,2%) e quanto ao tabagismo, 63,5% não são
fumantes e 75% não são etilistas.

39
Em relação à história da doença, 58,1% dos pacientes apresentaram, no momento do
diagnóstico, o KPS (Índice de Karnofsky) ≥ 80, o que indica apenas alguns sinais e sintomas
da doença. Apenas 6 pacientes, apresentaram um tumor antes do tumor cerebral e em
outro local, dentre os locais: colo do útero, próstata, pele (melanoma) e tireoide. Dentre as
localizações mais frequentes estão os lobos temporal e parietal, seguidos dos lobos frontal e
occipital; acometendo as lateralidades com frequências bastante similares, lateralidade
direita (48,6%) e esquerda (48,6%), e ambas as lateralidades (1,4%). Cerca de 65,5% dos
pacientes realizaram apenas 1 cirurgia para ressecção do tumor. Em relação ao tratamento,
a maioria dos pacientes realizaram radioterapia (56,8%) e quanto à quimioterapia, apenas
37,2% foram submetidas, sendo o quimioterápico mais utilizado a temozolamida seguido de
carmustina. Dos 148 pacientes, 110 (74,4%) apresentaram óbito em decorrência do câncer,
18 (12,2%) houve a perda de seguimento e 20 (13,5%) estão vivos; 73% apresentaram
sobrevida global ≤ 15 meses (Tabela 2).
A partir da obtenção de todas as características e informações clínicas relevantes dos
pacientes, obtidas através de prontuários, realizamos as análises estatísticas utilizando o
teste de χ2 (Qui-quadrado), afim de verificar associações estatisticamente significativas
entre a expressão das isoformas e expressão da proteína com Idade, Sexo, Cor, Tabagismo,
Etilismo, KPS, Lateralidade, Radioterapia, Quimioterapia, Recidiva e Sobrevida.
Encontramos associação estatísticamente significativa entre a predominância da
isoforma GNNK- e a lateralidade dos tumores (p-valor 0,027), entre a expressão da proteína
KIT (tecido endotelial) e o KPS (p-valor 0,022) e entre a expressão da proteína KIT (tecido
endotelial) e a predominância da isoforma GNNK- (p-valor 0,022), listadas nas tabelas 4, 7 e
9, respectivamente. No restante das análises, não foram encontradas associações
estatisticamente significativas entre as isoformas e expressão da proteína com as variáveis
clínico patológicas e demográficas.

40
Tabela 2 – Características e histórico da doença de 148 pacientes com GBM do Hospital de
Câncer de Barretos e Hospital Pedro Hispano (Portugal).
Idade 18 - 79 Nº de cirurgias
KPS (diagnóstico) Uma 97 (65,5%)
< 80 43 (29,1%) Duas 25 (16,9%)
≥ 80 86 (58,1%) Três 4 (2,7%)
Ignorado 19 (12,8%) Quatro ou mais 3 (2,1%)
Tumor em outro local Ignorado 19 (12,8%)
Não 123 (83,1%) Radioterapia
Sim 6 (4,1%) Não 45 (30,4%)
Ignorado 19 (12,8%) Sim 84 (56,8%)
Glioblastoma Primário Ignorado 19 (12,8%)
Não 6 (4,1%) Quimioterapia
Sim 123 (83,1%) Não 74 (50%)
Ignorado 19 (12,8%) Sim 55 (37,2%)
Localização tumoral Ignorado 19 (12,8%)
Temporal 110 (40%) Status
Frontal 68 (24,7%) Óbito por câncer 110 (74,4%)
Parietal 65 (23,6%) Vivo (com ou sem doença) 20 (13,5%)
Occipital 25 (9,1%) Perda de seguimento 18 (12,2%)
Insular 7 (2,5%) Sobrevida global
Lateralidade ≤ 15 meses 108 (73%)
Direito 72 (48,6%) ˃ 15 meses 40 (27%)
Esquerdo 72 (48,6%)
Ambos 2 (1,4%)
Ignorado 2 (1,4%)
Indivíduos (N): 148
*Os 19 casos ignorados geralmente encontrados em algumas variáveis são referentes aos casos do Hospital Pedro Hispano (Portugal) que não conseguimos todas as informações clínicas.

41
Tabela 3 – Associação da expressão do RNA de KIT com as variáveis demográficas e clínico-
patológicas de pacientes com glioblastoma (n=136).
RNA negativo RNA positivo
Variável Categoria n n (%) n (%) p-valor
Idade ≤ 50 anos 45 2 (4,4%) 43 (95,6%)
˃ 50 anos 91 5 (5,5%) 86 (94,5%) 1,000
Sexo Feminino 56 2 (3,6%) 54 (96,4%)
Masculino 80 5 (6,3%) 75 (93,7%) 0,700
Cor Branco 85 1 (1,2%) 84 (98,8%)
Pardo 25 1 (4,0%) 24 (96,0%)
Negro 6 0 (0,0%) 6 (100,0%) 0,465
Tabagista Não 85 2 (2,4%) 83 (97,6%)
Sim 21 0 (0,0%) 21 (100,0%) 1,000
Etilista Não 101 2 (2,0%) 99 (98,0%)
Sim 6 0 (0,0%) 6 (100,0%) 1,000
KPS ˂ 80 38 2 (5,3%) 36 (94,7%)
≥ 80 81 5 (6,2%) 76 (93,8%) 1,000
Lateralidade Direito 67 5 (7,5%) 62 (92,5%)
Esquerdo 65 2 (3,1%) 63 (96,9%)
Ambos 2 0 (0,0%) 2 (100,0%) 0,498
Radioterapia Não 40 0 (0,0%) 40 (100,0%)
Sim 77 2 (2,6%) 75 (97,4%) 0,546
Quimioterapia Não 64 0 (0,0%) 64 (100,0%)
Sim 53 2 (3,8%) 51 (96,2%) 0,203
Recidiva Não 100 2 (2,0%) 98 (98,0%)
Sim 16 0 (0,0%) 16 (100,0%) 1,000
RNA
* Dados de alguns pacientes são ignorados.

42
Tabela 4 – Associação da predominância da isoforma GNNK- de KIT com as variáveis
demográficas e clínico-patológicas de pacientes com glioblastoma (n=136).
negativo positivo
Variável Categoria n n (%) n (%) p-valor
Idade ≤ 50 anos 45 37 (82,2%) 8 (17,8%)
˃ 50 anos 91 73 (80,2%) 18 (19,8%) 0,780
Sexo Feminino 56 47 (83,9%) 9 (16,1%)
Masculino 80 63 (78,7%) 17 (21,3%) 0,450
Cor Branco 85 66 (77,6%) 19 (22,4%)
Pardo 25 22 (88,0%) 3 (12,0%)
Negro 6 6 (100,0%) 0 (0,0%) 0,306
Tabagista Não 85 70 (82,4%) 15 (17,6%)
Sim 21 16 (76,2%) 5 (23,8%) 0,539
Etilista Não 101 81 (80,2%) 20 (19,8%)
Sim 6 6 (100,0%) 0 (0,0%) 0,591
KPS ˂ 80 38 33 (86,8%) 5 (13,2%)
≥ 80 81 63 (77,8%) 18 (22,2%) 0,243
Lateralidade Direito 67 57 (85,1%) 10 (14,9%)
Esquerdo 65 51 (78,5%) 14 (21,5%)
Ambos 2 0 (0,0%) 2 (100,0%) 0,027
Radioterapia Não 40 32 (80,0%) 8 (20,0%)
Sim 77 63 (81,8%) 14 (18,2%) 0,811
Quimioterapia Não 64 53 (82,8%) 11 (17,2%)
Sim 53 42 (79,3%) 11 (20,7%) 0,623
Recidiva Não 100 80 (80,0%) 20 (20,0%)
Sim 16 14 (87,5%) 2 (12,5%) 0,733
Predominância GNNK-
* Dados de alguns pacientes são ignorados.

43
Tabela 5 – Associação da predominância da isoforma GNNK+ de KIT com as variáveis
demográficas e clínico-patológicas de pacientes com glioblastoma (n=136).
negativo positivo
Variável Categoria n(%) n (%) p-valor
Idade ≤ 50 anos 45 21 (46,7%) 24 (53,3%)
˃ 50 anos 91 53 (58,2%) 38 (41,8%) 0,202
Sexo Feminino 56 29 (51,8%) 27 (48,2%)
Masculino 80 45 (56,2%) 35 (43,8%) 0,607
Cor Branco 85 45 (52,9%) 40 (47,1%)
Pardo 25 12 (48,0%) 13 (52,0%)
Negro 6 4 (66,7%) 2 (33,3%) 0,793
Tabagista Não 85 46 (54,1%) 39 (45,9%)
Sim 21 10 (47,6%) 11 (52,4%) 0,593
Etilista Não 101 54 (53,5%) 47 (46,5%)
Sim 6 3 (50,0%) 3 (50,0%) 1,000
KPS ˂ 80 38 24 (63,2%) 14 (36,8%)
≥ 80 81 43 (53,1%) 38 (46,9%) 0,302
Lateralidade Direito 67 34 (50,7%) 33 (49,3%)
Esquerdo 65 37 (56,9%) 28 (43,1%)
Ambos 2 2 (100,0%) 0 (0,0%) 0,431
Radioterapia Não 40 20 (50,0%) 20 (50,0%)
Sim 77 42 (54,5%) 35 (45,5%) 0,640
Quimioterapia Não 64 31 (48,4%) 33 (51,6%)
Sim 53 31 (58,5%) 22 (41,5%) 0,278
Recidiva Não 100 54 (54,0%) 46 (46,0%)
Sim 16 7 (43,7%) 9 (56,3%) 0,446
Predominância GNNK+
* Dados de alguns pacientes são ignorados.

44
Tabela 6 – Associação da expressão da proteína KIT (células neoplásicas) com as variáveis
demográficas e clínico-patológicas de pacientes com glioblastoma (n=70).
positivo negativo
Variável Categoria n n (%) n (%) p-valor
Idade ≤ 50 anos 14 1 (7,1%) 13 (92,9%)
˃ 50 anos 56 15 (26,8%) 41 (73,2%) 0,164
Sexo Feminino 22 2 (9,1%) 20 (90,9%)
Masculino 48 14 (29,2%) 34 (70,8%) 0,074
Cor Branco 47 12 (25,5%) 35 (74,5%)
Pardo 17 4 (23,5%) 13 (76,5%)
Negro 5 0 (0,0%) 5 (100,0%) 0,715
Tabagista Não 48 10 (20,8%) 38 (79,2%)
Sim 14 5 (35,7%) 9 (64,3%) 0,296
Etilista Não 60 15 (25,0%) 45 (75,0%)
Sim 3 0 (0,0%) 3 (100,0%) 1,000
KPS ˂ 80 25 6 (24,0%) 19 (76,0%)
≥ 80 36 8 (22,2%) 28 (77,8%) 0,871
Lateralidade Direito 32 8 (25,0%) 24 (75,0%)
Esquerdo 34 8 (23,5%) 26 (76,5%)
Ambos 2 0 (0,0%) 2 (100,0%) 1,000
Radioterapia Não 26 8 (30,8%) 18 (69,2%)
Sim 44 8 (18,2%) 36 (81,8%) 0,226
Quimioterapia Não 46 10 (21,7%) 36 (78,3%)
Sim 24 6 (25,0%) 18 (75,0%) 0,758
Recidiva Não 64 16 (25,0%) 48 (75,0%)
Sim 5 0 (0,0%) 5 (100,0%) 0,583
Proteína - KIT (células neoplásicas)
* Dados de alguns pacientes são ignorados.

45
Tabela 7 – Associação da expressão da proteína KIT (tecido endotelial) com as variáveis
demográficas e clínico-patológicas de pacientes com glioblastoma (n=70).
positivo negativo
Variável Categoria n n (%) n (%) p-valor
Idade ≤ 50 anos 14 4 (28,6%) 10 (71,4%)
˃ 50 anos 56 25 (44,6%) 31 (55,4%) 0,275
Sexo Feminino 22 10 (45,5%) 12 (54,5%)
Masculino 48 19 (39,6%) 29 (60,4%) 0,643
Cor Branco 47 20 (42,6%) 27 (57,4%)
Pardo 17 7 (41,2%) 10 (58,8%)
Negro 5 2 (40,0%) 3 (60,0%) 1,000
Tabagista Não 48 20 (41,7%) 28 (58,3%)
Sim 14 6 (42,9%) 8 (57,1%) 0,937
Etilista Não 60 23 (38,3%) 37 (61,7%)
Sim 3 3 (100,0%) 0 (0,0%) 0,065
KPS ˂ 80 25 15 (60,0%) 10 (40,0%)
≥ 80 36 11 (30,6%) 25 (69,4%) 0,022
Lateralidade Direito 32 15 (46,9%) 17 (53,1%)
Esquerdo 34 12 (35,3%) 22 (64,7%)
Ambos 2 0 (0,0%) 2 (100,0%) 0,422
Radioterapia Não 26 13 (50,0%) 13 (50,0%)
Sim 44 16 (36,4%) 28 (63,6%) 0,263
Quimioterapia Não 46 20 (43,5%) 26 (56,5%)
Sim 24 9 (37,5%) 15 (62,5%) 0,630
Recidiva Não 64 26 (40,6%) 38 (59,4%)
Sim 5 2 (40,0%) 3 (60,0%) 1,000
Proteína - KIT (tecido endotelial)
* Dados de alguns pacientes são ignorados.
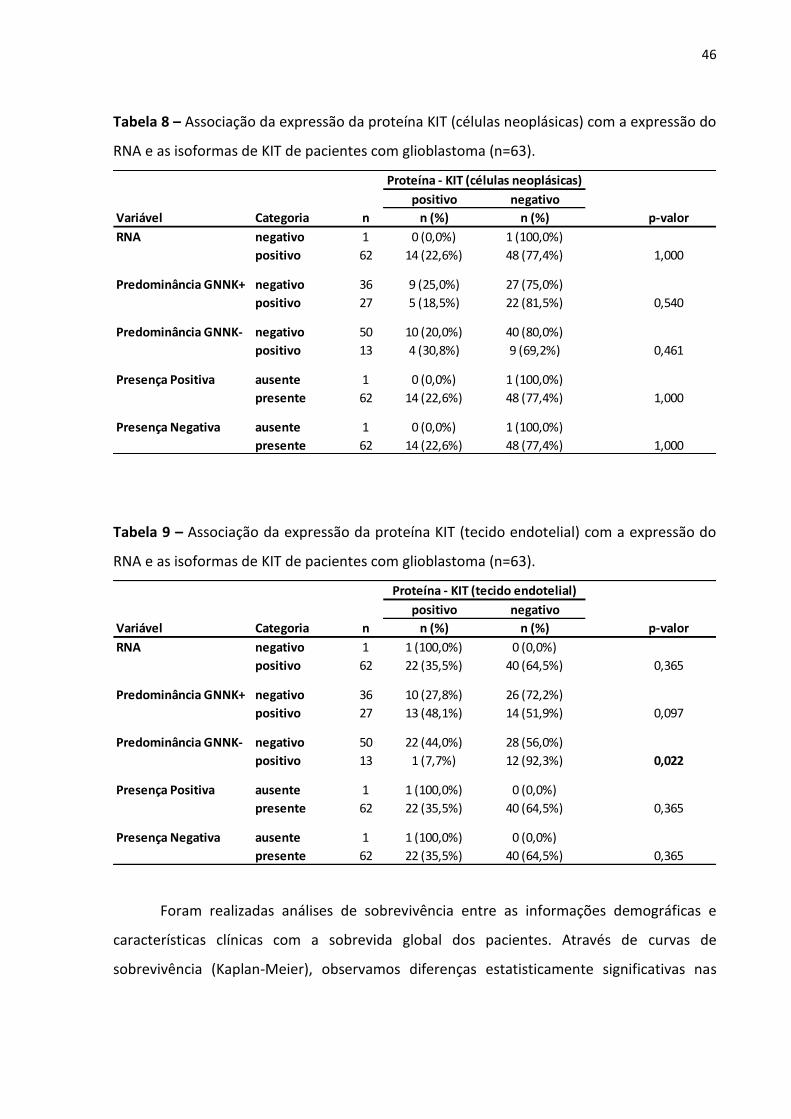
46
Tabela 8 – Associação da expressão da proteína KIT (células neoplásicas) com a expressão do
RNA e as isoformas de KIT de pacientes com glioblastoma (n=63).
positivo negativo
Variável Categoria n n (%) n (%) p-valor
RNA negativo 1 0 (0,0%) 1 (100,0%)
positivo 62 14 (22,6%) 48 (77,4%) 1,000
Predominância GNNK+ negativo 36 9 (25,0%) 27 (75,0%)
positivo 27 5 (18,5%) 22 (81,5%) 0,540
Predominância GNNK- negativo 50 10 (20,0%) 40 (80,0%)
positivo 13 4 (30,8%) 9 (69,2%) 0,461
Presença Positiva ausente 1 0 (0,0%) 1 (100,0%)
presente 62 14 (22,6%) 48 (77,4%) 1,000
Presença Negativa ausente 1 0 (0,0%) 1 (100,0%)
presente 62 14 (22,6%) 48 (77,4%) 1,000
Proteína - KIT (células neoplásicas)
Tabela 9 – Associação da expressão da proteína KIT (tecido endotelial) com a expressão do
RNA e as isoformas de KIT de pacientes com glioblastoma (n=63).
positivo negativo
Variável Categoria n n (%) n (%) p-valor
RNA negativo 1 1 (100,0%) 0 (0,0%)
positivo 62 22 (35,5%) 40 (64,5%) 0,365
Predominância GNNK+ negativo 36 10 (27,8%) 26 (72,2%)
positivo 27 13 (48,1%) 14 (51,9%) 0,097
Predominância GNNK- negativo 50 22 (44,0%) 28 (56,0%)
positivo 13 1 (7,7%) 12 (92,3%) 0,022
Presença Positiva ausente 1 1 (100,0%) 0 (0,0%)
presente 62 22 (35,5%) 40 (64,5%) 0,365
Presença Negativa ausente 1 1 (100,0%) 0 (0,0%)
presente 62 22 (35,5%) 40 (64,5%) 0,365
Proteína - KIT (tecido endotelial)
Foram realizadas análises de sobrevivência entre as informações demográficas e
características clínicas com a sobrevida global dos pacientes. Através de curvas de
sobrevivência (Kaplan-Meier), observamos diferenças estatisticamente significativas nas

47
variáveis idade, cor, KPS, radioterapia, quimioterapia e recidiva, como mostrado na tabela
10.
Tabela 10 – Associação entre os dados clínico-patológicos com a sobrevida de pacientes com
GBM.
Variável Categoria n p-valor
Idade ≤ 50 anos 34
˃ 50 anos 86 0,001
Sexo Feminino 48
Masculino 72 0,112
Cor Branco 69
Pardo 26
Negro 7 0,006
Tabagista Não 76
Sim 18 0,591
Etilista Não 87
Sim 8 0,060
KPS ˂ 80 40
≥ 80 65 0,042
Lateralidade Direito 57
Esquerdo 59
Ambos 2 0,271
Radioterapia Não 41
Sim 61 < 0,001
Quimioterapia Não 64
Sim 38 < 0,001
0.0 ± 0.0
4.1 ± 1.0
11.5 ± 1.1
4.6 ± 0.7
15.0 ± 1.3
9.0 ± 0.9
4.1 ± 1.3
7.6 ± 1.1
11.5 ± 1.2
9.3 ± 1.1
9.9 ± 1.2
8.6 ± 0.9
9.4 ± 1.0
8.0 ± 1.7
2.0 ± 0.8
8.8 ± 1.0
7.2 ± 1.4
Análises Univariadas
(meses ± DP)
14.1 ± 1.7
7.7 ± 0.8
11.1 ± 1.5
DP: Desvio-padrão; IC: 95% Intervalo de confiança
Além de análises de sobrevivência com as características demográficas e clínicas,
realizamos a mesma análise de sobrevivência entre a predominância das isoformas e a
sobrevida global. Não foram encontradas diferenças estatisticamente significantes entre as
curvas de sobrevivência, embora entre os casos que apresentam KIT positivo e KIT negativo
houve uma diferença importante, em que, os casos negativos pra KIT apresentam uma

48
melhor sobrevida. Além disso, há uma tendência interessante em que a predominância de
GNNK- parece aumentar a sobrevida dos pacientes (Tabela 11 e Figura 14).
Também tentamos associar a expressão da proteína KIT, tanto das células
neoplásicas quanto do tecido endotelial, com a sobrevida global, entretanto não
encontramos nenhuma diferença estatisticamente significativa (Tabela 11 e Figura 15).
Tabela 11 – Associação entre dados de expressão de KIT com a sobrevida de pacientes com
GBM.
Variável n p-valor
CD117 Tumor
14
49 0,517
CD117 Vasos
38
25 0,354
KIT RNAm
101
7 0,057
GNNK+ predominância
48
60 0,291
GNNK- predominância
24
84 0,933
10.8 ± 1.2
Positiva
Negativa
Positiva
Negativa 16.4 ± 2.3
Negativo
Positivo
Ausente
Presente
Positivo
Negativo
8.0 ± 1.0
7.0 ± 1.2
14.2 ± 3.9
8.3 ± 1.4
9.4 ± 0.9
8.8 ± 1.2
17.0 ± 2.7
Análises Univariadas
(meses ± DP)
6.1 ± 1.6
DP: Desvio-padrão; IC: 95% Intervalo de confiança

49
a.
b.

50
c.
Figura 14 – Curvas de sobrevivência de 136 pacientes, utilizando Kaplan Meier e teste de log-rank. a:
Curvas de sobrevivência com amostras negativas pra KIT, com predominância da isoforma GNNK+,
com predominância da isoforma GNNK- e expressão igual de ambas isoformas (p-valor 0,263); b:
Curvas de sobrevivência de amostras com KIT positivo e KIT negativo (p-valor 0,057); c: Curvas de
sobrevivência de amostras com a predominância de GNNK+ e amostras com predominância de
GNNK- (p-valor 0,291).
a.

51
b.
Figura 15 – Curvas de sobrevivência de 136 pacientes, utilizando Kaplan Meier e teste de log-rank. a:
Curvas de sobrevivência com amostras com expressão positiva e negativa da proteína KIT nas células
neoplásicas (p-valor 0,517); b: Curvas de sobrevivência de amostras com expressão positiva e
negativa da proteína KIT no tecido endotelial (p-valor 0,354).
5.3 Análise do papel tumorigênico das isoformas de KIT em glioblastomas
Para determinar o papel tumorigênico das isoformas GNNK de KIT, foram realizados
ensaios in vitro com a linhagem GAMG. Foi realizada a transfecção das células com os
respectivos plasmídeos (GNNK+, GNNK- e pCDNA3) e a seleção com 300ug/ml de G418, a
confirmação da transfecção pode ser vista na Figura 16.

52
GN
NK
+
GN
NK
-
pC
DN
A3
RN
Am
Prot
eína
hGUS
KIT
CD117
β-Actina
p-KIT (Y719)
Figura 16 - Confirmação da transfecção das isoformas GNNK+ e GNNK- de KIT na linhagem GAMG
através do RNAm, expressão da proteina KIT (CD117) e confirmação da ativação através da expressão
de KIT fosforilado (p-KIT 719). Observação: Devido à limitação da técnica na revelação do western
blot, não conseguimos demonstrar a expressão de KIT no vetor vazio (pCDNA3), já que nas linhagens
transfectadas, houve maior expressão da proteina KIT e a GAMG é uma linhagem que expressa a
proteína.
Os ensaios de viabilidade celular foram realizados com MTS (Promega), sendo
medida até 72 horas em intervalos de 24 horas, em que observou-se maior viabilidade em
GNNK- comparado à GNNK+ e ao veto vazio, não havendo diferenças com a estimulação de
SCF. Em relação ao ensaio de wound healing, monitoramos o fechamento da ranhura
durante 72 horas e também com a estimulação de SCF (100ng/ml), percebemos maior
potencial das células transfectadas em fechar a ranhura em comparação ao vetor vazio, sem
diferenças estatisticamente significativas com a estimulação de SCF. Para determinar a
capacidade de formação de colônias, utilizamos o ensaio de clonogenicidade, encontramos
maior formação de colônias nas células transfectadas com a isoforma GNNK- e, no ensaio de
invasão em matrigel (BD Biosciences) encontramos maior potencial de invasão também nas
células transfectadas com a isoforma GNNK-. Todas as diferenças foram estatisticamente
significativas.
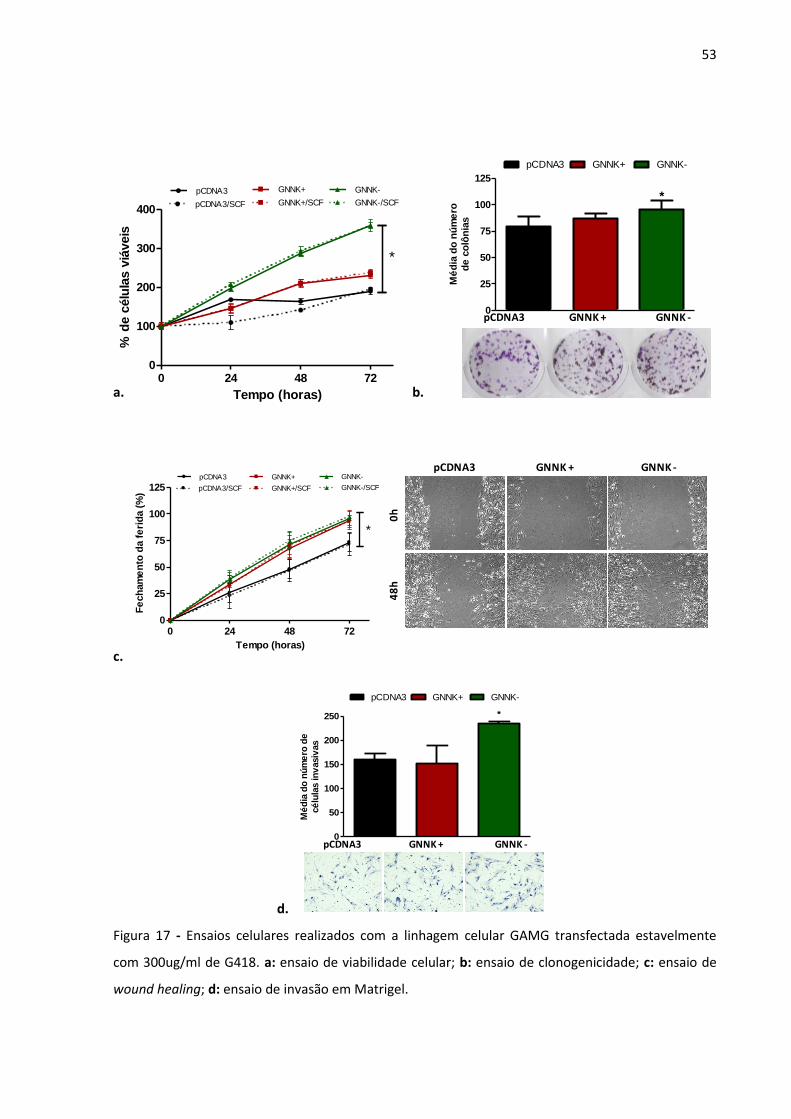
53
a. b.
c.
0 24 48 720
25
50
75
100
125pCDNA3 GNNK+ GNNK-
pCDNA3/SCF GNNK+/SCF GNNK-/SCF
*
Tempo (horas)
Fe
ch
am
en
to d
a f
eri
da (
%)
GNNK + GNNK -pCDNA3
0h
48
h
d.
0
50
100
150
200
250 *
pCDNA3 GNNK+ GNNK-
Mé
dia
do
nú
me
ro d
e
cé
lula
s in
vasiv
as
GNNK + GNNK -pCDNA3
Figura 17 - Ensaios celulares realizados com a linhagem celular GAMG transfectada estavelmente
com 300ug/ml de G418. a: ensaio de viabilidade celular; b: ensaio de clonogenicidade; c: ensaio de
wound healing; d: ensaio de invasão em Matrigel.
0 24 48 720
100
200
300
400
pCDNA3 GNNK+ GNNK-
pCDNA3/SCF GNNK+/SCF GNNK-/SCF
*
Tempo (horas)
% d
e c
élu
las v
iáveis
0
25
50
75
100
125
*
pCDNA3 GNNK+ GNNK-
Mé
dia
do
nú
me
ro
de
co
lôn
ias
Inva
sio
n
GNNK + GNNK -pCDNA3

54
E para a confirmação dos resultados, foram realizados os ensaios com a linhagem
celular U251, transfectada com as isoformas e o vetor vazio e, selecionada com 300ug/ml de
G418. Também encontramos maior viabilidade e maior potencial de migração nas células
transfectadas com a isoforma GNNK-. Em relação ao ensaio de clonogenicidade, foi
encontrado maior potencial de formação de colônias em GNNK+, entretanto com diferenças
não estatisticamente significativas.
a.
GN
NK
+
hGUS
GN
NK
-
pC
DN
A3
KIT
CD117
β-Actin
mR
NA
Pro
tein
b.
c. d.
Figura 18 - Ensaios celulares realizados com a linhagem celular U251 transfectada estavelmente com
300ug/ml de G418. a: Confirmação da transfecção das isoformas GNNK+ e GNNK- de KIT na linhagem
GAMG através do RNAm, expressão da proteina KIT (CD117); b: ensaio de viabilidade celular; c:
ensaio de clonogenicidade; d: ensaio de wound healing.
0 24 48 720
100
200
300 pCDNA3 GNNK+ GNNK-
*
Tempo (horas)
% d
as c
élu
las v
iáv
eis
0
50
100
150
200
*
pcDNA3 GNNK+ GNNK-
Mé
dia
do
nú
me
ro
de
co
lôn
ias
0 24 48 720
25
50
75
100
125 pCDNA3 GNNK+ GNNK-
*
Tempo (horas)
Fe
ch
am
en
to d
a f
eri
da (
%)
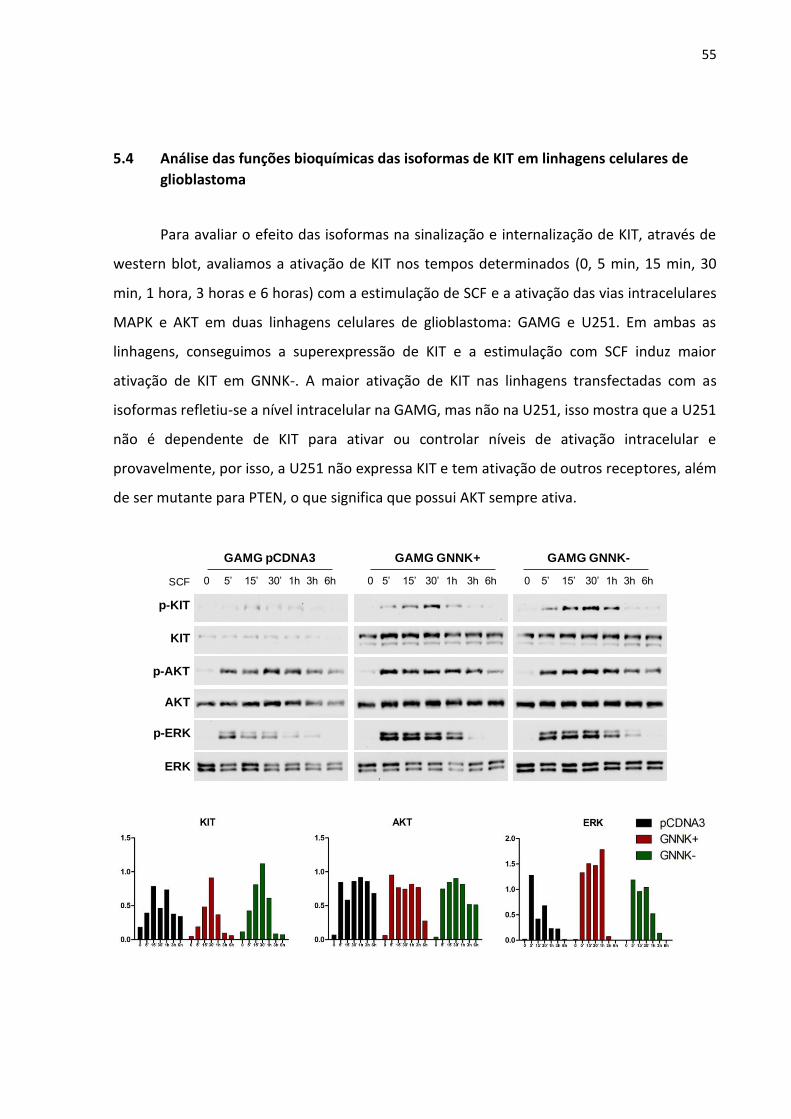
55
5.4 Análise das funções bioquímicas das isoformas de KIT em linhagens celulares de
glioblastoma
Para avaliar o efeito das isoformas na sinalização e internalização de KIT, através de
western blot, avaliamos a ativação de KIT nos tempos determinados (0, 5 min, 15 min, 30
min, 1 hora, 3 horas e 6 horas) com a estimulação de SCF e a ativação das vias intracelulares
MAPK e AKT em duas linhagens celulares de glioblastoma: GAMG e U251. Em ambas as
linhagens, conseguimos a superexpressão de KIT e a estimulação com SCF induz maior
ativação de KIT em GNNK-. A maior ativação de KIT nas linhagens transfectadas com as
isoformas refletiu-se a nível intracelular na GAMG, mas não na U251, isso mostra que a U251
não é dependente de KIT para ativar ou controlar níveis de ativação intracelular e
provavelmente, por isso, a U251 não expressa KIT e tem ativação de outros receptores, além
de ser mutante para PTEN, o que significa que possui AKT sempre ativa.
0 5’ 15’ 30’ 1h 3h 6h
GAMG pCDNA3 GAMG GNNK+ GAMG GNNK-
p-KIT
KIT
p-AKT
AKT
p-ERK
ERK
0 5’ 15’ 30’ 1h 3h 6h 0 5’ 15’ 30’ 1h 3h 6h SCF

56
Figura 19 - Efeito na ativação de KIT e vias intracelulares (MAPK e AKT) em células transfectadas com
as isoformas GNNK+ e GNNK- na linhagem celular GAMG. Os gráficos apresentam a quantificação das
proteínas, sendo (proteína fosforilada/proteína total).
U251 pCDNA3 U251 GNNK+ U251 GNNK-
0 5’ 15’ 30’ 1h 3h 6h
p-KIT
KIT
p-AKT
AKT
p-ERK
ERK
0 5’ 15’ 30’ 1h 3h 6h 0 5’ 15’ 30’ 1h 3h 6h
Figura 20 - Efeito na ativação de KIT e vias intracelulares em células transfectadas com as isoformas
GNNK+ e GNNK- na linhagem celular U251. O gráfico apresenta a quantificação das proteínas, sendo
(proteína fosforilada/proteína total).
Por imunoflorescência, avaliamos a internalização e abundância do receptor KIT, após
estimulação com SCF em 15 e 30 minutos. Entre os diferentes tempos de estimulação não
observamos grandes diferenças de marcação, entretanto, conseguimos observar uma
marcação mais forte, abundante e “granulada” intracelularmente, ao longo do tempo nas
células transfectadas, em especial em GNNK-, o que demonstra uma internalização mais
rápida do receptor nas células que superexpressam KIT em relação ao controle não
transfectado.
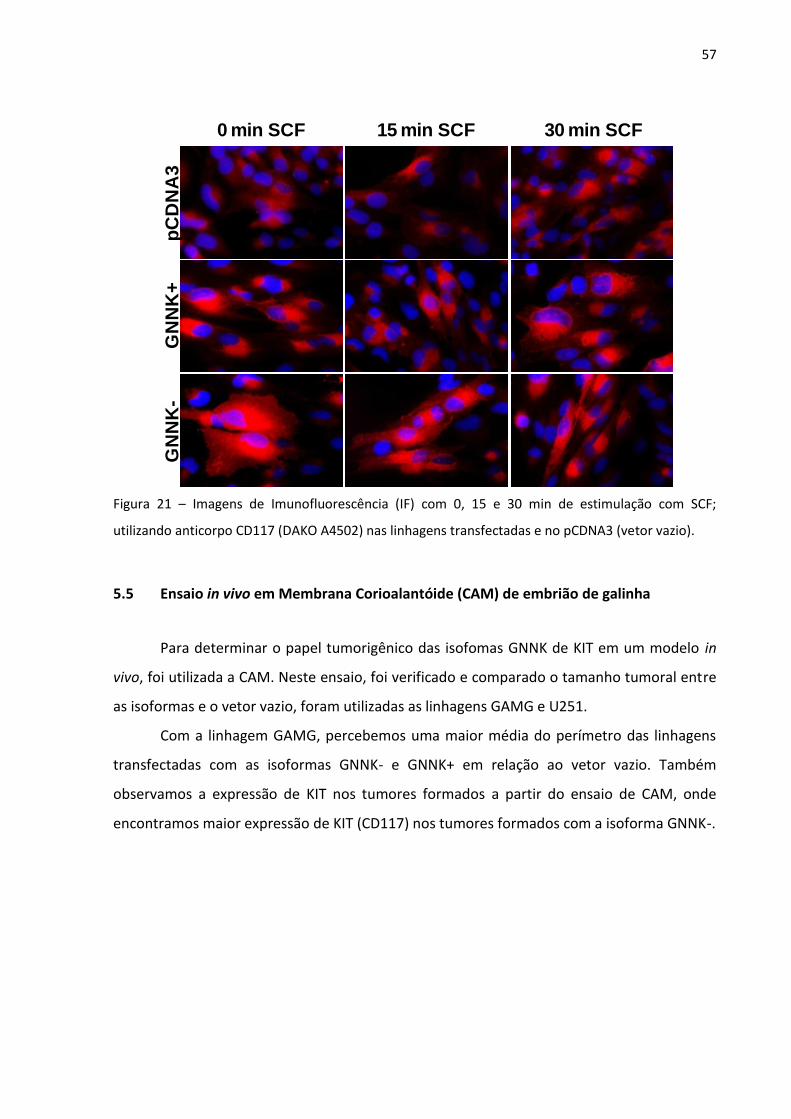
57
pC
DN
A3
GN
NK
+G
NN
K-
0 min SCF 15 min SCF 30 min SCF
Figura 21 – Imagens de Imunofluorescência (IF) com 0, 15 e 30 min de estimulação com SCF;
utilizando anticorpo CD117 (DAKO A4502) nas linhagens transfectadas e no pCDNA3 (vetor vazio).
5.5 Ensaio in vivo em Membrana Corioalantóide (CAM) de embrião de galinha
Para determinar o papel tumorigênico das isofomas GNNK de KIT em um modelo in
vivo, foi utilizada a CAM. Neste ensaio, foi verificado e comparado o tamanho tumoral entre
as isoformas e o vetor vazio, foram utilizadas as linhagens GAMG e U251.
Com a linhagem GAMG, percebemos uma maior média do perímetro das linhagens
transfectadas com as isoformas GNNK- e GNNK+ em relação ao vetor vazio. Também
observamos a expressão de KIT nos tumores formados a partir do ensaio de CAM, onde
encontramos maior expressão de KIT (CD117) nos tumores formados com a isoforma GNNK-.
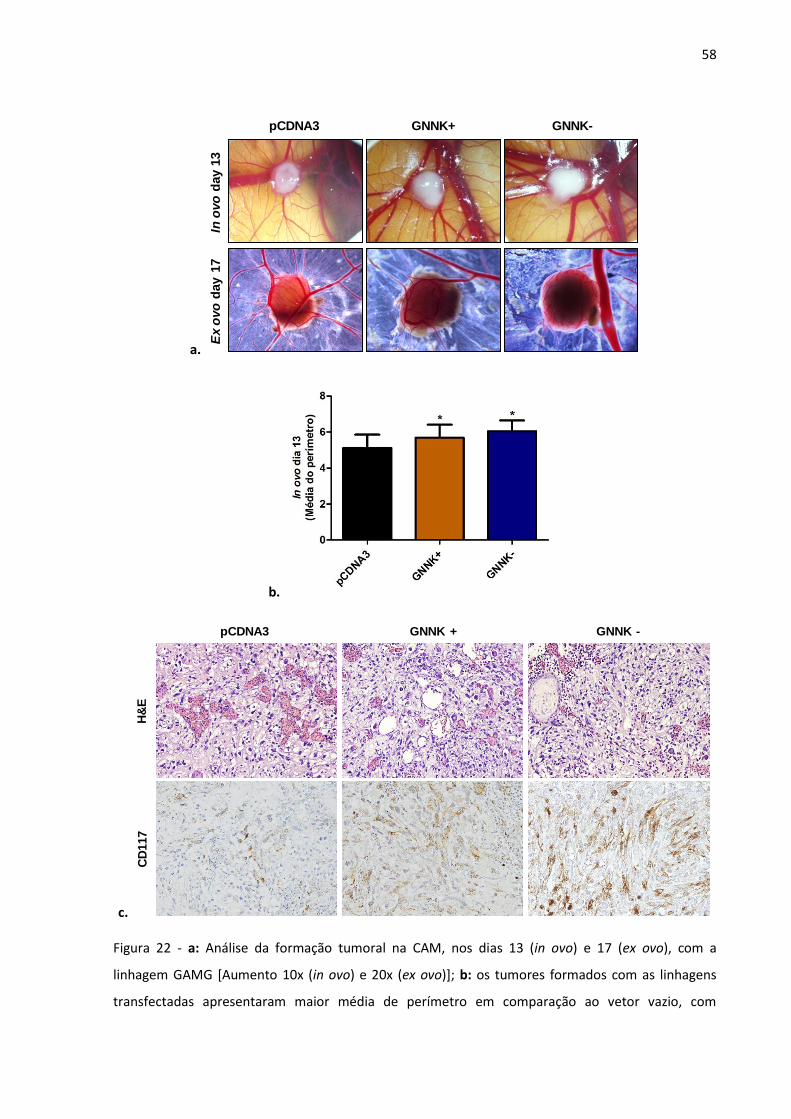
58
a.
pCDNA3 GNNK+ GNNK-
In o
vo
day 1
3E
x o
vo
day 1
7
b.
c.
GNNK + GNNK -pCDNA3
H&
EC
D117
Figura 22 - a: Análise da formação tumoral na CAM, nos dias 13 (in ovo) e 17 (ex ovo), com a
linhagem GAMG [Aumento 10x (in ovo) e 20x (ex ovo)]; b: os tumores formados com as linhagens
transfectadas apresentaram maior média de perímetro em comparação ao vetor vazio, com

59
resultados estatisticamente significativos (GNNK- p-valor <0,0001; GNNK+ p-valor 0,0154); c:
coloração de H&E e imunohistoquímica para CD117 (KIT) de cortes feitos a partir da CAM incluída em
parafina.
Nos ensaios com a linhagem U251, podemos confirmar os achados com a GAMG. Os
tumores formados com as isoformas GNNK- apresentaram maior média do perímetro em
relação ao vetor vazio (pCDNA3), sendo estatisticamente significativo.
a.
pCDNA3 GNNK+ GNNK-
In o
vo d
ay1
3Ex
ovo
day
17
b.
Figura 23 - a: Análise da formação tumoral na CAM, nos dias 13 (in ovo) e 17 (ex ovo), com a
linhagem U251 [Aumento 10x (in ovo) e 20x (ex ovo)]; b: os tumores formados com a linhagem
transfectada com a isoforma GNNK- apresentaram maior média de perímetro em comparação ao
vetor vazio, com resultados estatisticamente significativos (p-valor 0,0311).

60
DISCUSSÃO

61
6 DISCUSSÃO
O GBM é o tumor cerebral mais agressivo, sendo um dos maiores desafios no
tratamento do câncer. Embora com os avanços da medicina, o prognóstico para os pacientes
com GBM ainda é ruim devido à resistência terapêutica e a recorrência tumoral após a
remoção cirúrgica, diante disso, apresentam a sobrevida curta, cerca de 15 meses. Novas
vias moleculares importantes estão sendo associadas à biologia, patogênese e abordagem
terapêutica dos GBMs, entre elas os RTKs.
Estudos anteriores mostraram a presença frequente do RNAm total de KIT em tecidos
e linhagens celulares de GBM 90, no entanto não há relatos a respeito da expressão das
isoformas GNNK de KIT em GBMs. Nossos resultados nas linhagens celulares e tecidos
congelados/parafinados são os primeiros a abordar esta questão. Considerando que na
literatura ainda não existem dados sobre as isoformas GNNK de KIT em tecidos cerebrais
normais e tumorais e, baseado em resultados preliminares de linhagens celulares de GBM e
tecidos normais de cérebro (Martinho O, et al. dados pessoais), esperávamos observar a
frequente presença de ambas isoformas, com a predominância da isoforma GNNK- em
tumores primários. Entretanto, os resultados deste estudo apresentam a coexpressão de
ambas isoformas com a predominância da isoforma GNNK+ (45,3%) em tecidos de GBM,
diferente do encontrado em linhagens celulares de GBM. Esses resultados também são
completamente diferentes de estudos anteriores realizados com outros tipos tumorais,
como em leucemia mielóide aguda, mieloma múltiplo, TCGs testiculares e GISTs, em que
demonstram a coexpressão de ambas isoformas com a predominância da isoforma GNNK- 61,
65, 68, 69.
Além de caracterizar os tecidos de GBM quanto à expressão das isoformas, avaliamos
linhagens celulares primárias e seus respectivos tecidos tumorais congelados e, então,
encontramos uma mudança na expressão das isoformas do RNAm de KIT que ainda não foi
descrito em nenhum tipo tumoral. Verificamos a expressão proteica de KIT nas linhagens
primárias, enquanto que nos tecidos parafinados correspondentes, não encontramos a
expressão proteica, acredita-se que devido a utilização de técnicas distintas; western blot
para a linhagens e imunohistoquímica para os tecidos parafinados, é possível a diferença de
sensibilidade entre as técnicas. Esses dados sugerem que existe uma mudança de expressão

62
das isoformas de KIT, que favorece a predominância da isoforma GNNK- e a expressão da
proteína, na adaptação das células à condições de cultura celular.
Observamos também a expressão da proteina KIT em cortes histológicos de tecidos
parafinados através de imunohistoquímica, onde encontramos a expressão de KIT em níveis
fraco a moderado no citoplasma das células tumorais (22,8% dos casos) e forte no tecido
endotelial - microvasos e proliferação microvascular – (40,2% dos casos). As porcentagens de
expressão de KIT encontradas neste trabalho vão de encontro com os dados da literatura,
Gomes et al. encontraram expressão de KIT em 20,8% dos GBMs em células neoplásicas 46 e
Sihto et al. encontraram 59% de expressão nos GBMs em tecido endotelial, além disso, GBM
foi o único tipo tumoral entre 34 diferentes histologias tumorais a apresentar expressão de
KIT no tecido endotelial 59.
Além da análise da proteina KIT em 92 casos, em uma coorte de 21 pacientes, foi
avaliado também a expressão do ligante (SCF) e de KIT ativado (p-KIT Tyr 719), notamos que
nos casos onde há a expressão de p-KIT, há também a expressão de KIT total e do ligante
SCF, portanto, podemos sugerir a ativação de KIT através de mecanismos
autócrinos/parácrinos com o ligante SCF. Estes dados corroboram com os achados de
Stanulla e colaboradores (1995), que mostraram a coexpressão do receptor e do ligante na
maioria dos casos de KIT positivo, levando à ativação de KIT em linhagens celulares de
glioma 57. Demonstrada a ativação de KIT por mecanismos autócrinos, alguns trabalhos
evidenciaram uma maior indução da secreção de SCF em condições de hipóxia (característico
de glioblastomas que apresentam necrose) no microambiente tumoral, levando assim, ao
aumento dos níveis de HIF1α e VEGF e a ativação de KIT, promovendo angiogênese e
sobrevivência do tecido endotelial tumoral. Além disso, SCF atua como fator de
sobrevivência para células-tronco neurais 52, 59, 91, 92.
Quanto aos ensaios celulares, verificamos maior papel tumorigênico da linhagem
transfectada com a isoforma GNNK- em comparação à isoforma GNNK+ e pCDNA3 (vetor
vazio), principalmente em relação à viabilidade, potencial de migração e potencial invasivo,
que já foram descritos em outros trabalhos onde utilizaram fibloblastos de camundongo,
melanoma de camundongo (linhagens celulares NIH3T3 e B16F0, respectivamente) e
mastócitos 63, 67, 86, 93. Um estudo recente relatou um aumento na proliferação de astrócitos
de camundongos após a transfecção de cDNA de KIT humano, não especificando qual

63
isoforma 57. Estudos funcionais em linhagem celular de fibroblastos de camundongos
(NIH3T3) demonstraram que essas células transfectadas com a isoforma GNNK- promove
maior formação de colônias independente de ancoragem e são tumorigênicas in vivo em
camundongos nude, enquanto células transfectadas com a isoforma GNNK+ não são 63.
Com base em estudos anteriores em outros tipos de células humanas e de
camundongos, foi encontrado nos resultados uma fosforilação, ubiquitinação e
internalização mais rápida de KIT em células GNNK- transfectadas 63, 64, 67. Para os nossos
achados, utilizamos as linhagens celulares de GBM, GAMG e U251; a GAMG é uma linhagem
celular que expressa o RNAm e a proteína KIT e a U251 é uma linhagem celular que expressa
apenas o RNAm de KIT. Em ambas linhagens transfectadas, observamos a superexpressão da
proteína KIT e maior ativação de p-KIT (com estimulação de SCF) nas células transfectadas
com a isoforma GNNK-. Também observamos, através da imunofluorescência, uma
marcação mais forte, abundante e granulada nas linhagens transfectadas, principalmente
em GNNK-, o que demonstra maior ativação de KIT e internalização mais rápida,
corroborando com estudos citados acima. De acordo com Phung et al. (2013), observaram
que a consecutiva eliminação de aminoácidos da sequência tetrapeptídica GNNK, aumenta
gradualmente a fosforilação, ubiquitinação, internalização e ativação downstream de MAPK,
além de aumentar a viabilidade celular, como já descrito em outros estudos. Esse trabalho
também mostrou a importância do domínio transmembrana responsável por controlar a
orientação dos domínios tirosina-quinase e, assim, sua atividade. Modificações no domínio
transmembrana, resulta na rotação dos domínios tirosina-quinase em torno do seu eixo,
levando a modificações quando o peptídeo é encurtado 67.
Dados na literatura mostram diferenças significantivas entre as vias de ativação
intracelular dependentes de isoformas transfectadas, um aumento da ativação de MAPK e
PI3K/AKT, entretanto segundo relatórios anteriores, o efeito das isoformas nas vias de
sinalização é dependente do tipo celular 63-66. Avaliamos as principais vias de sinalização,
MAPK e PI3K/AKT, na GAMG observamos um aumento da ativação de ERK nas linhagens
transfectadas em relação ao vetor vazio, o que não aconteceu em AKT; enquanto na U251, a
ativação de KIT não influenciou nas vias intracelulares nas linhagens transfectadas, isso se
deve a U251 ser mutante para PTEN, o que significa que possui AKT sempre ativa e ser

64
dependente de outros receptores, provavelmente de PDGFR. De acordo com um artigo do
grupo, foi observado que PDGFRA foi o único alvo do imatinib na linhagem celular U251 84.
Em relação aos ensaios in vivo, utilizando a CAM, encontramos um maior potencial
tumorigênico das células transfectadas com as isoformas GNNK, formando tumores com
maior perímetro em GNNK-, embora não existem relatos de estudos que tenham utilizado
essa metodologia para avaliação do papel tumorigênico das isoformas GNNK de KIT, de
acordo com Caruana e colaboradores (1999), fibroblastos transfectados com a isoforma
GNNK- levou a tumorigenicidade em camundongos, enquanto fibroblastos transfectados
com a isoforma GNNK+, não 63. Embora nossos resultados não são comparáveis com os
achados de Caruana et al. (1999), pois utilizamos linhagens celulares tumorais, portanto
formam tumores; se KIT é um oncogene, esperamos que ambas isoformas apresentem
potencial tumorigênico, é o que nossos resultados evidenciam. Por outro lado, a
predominância de GNNK+ que encontramos em grande parte dos tecidos tumorais, pode ter
um significado biológico/tumorigênico/terapêutico importante.
Diante de vários trabalhos interessados em elucidar o papel dos RTK em gliomas,
notamos que KIT está envolvido na patogênese molecular de alguns gliomas, seja por
amplificação gênica ou por mecanismos autócrinos/parácrinos. Dessa forma, o KIT poderia
ser um potencial alvo molecular na terapia antitumoral através dos inibidores de tirosina
quinase. O papel das isoformas de KIT em terapias anti-KIT é quase inexplorado. Em tumores
sólidos, a resposta do imatinib é altamente associada com a presença de mutações ativantes
no gene KIT como observado em GIST 48. Para os outros fármacos em estudo, não é claro
quais os biomarcadores de resposta, no entanto, eles mostram-se eficazes em casos
resistentes ao imatinib 48. Um estudo recente em células de mieloma tratadas com imatinib
mostrou uma inibição igual na ativação de KIT em ambas isoformas 65. As linhagens celulares
de GBM que foram utilizadas neste projeto não possuem mutações ativantes em KIT e o
Imatinib, em contraste com o sunitinib, como agente isolado, não apresentaria benefício
terapêutico 83, 84. Baseado nesses fatos espera-se que linhagens celulares de GBM tratadas
com imatinib não apresentem um efeito anti-tumorigênico acentuado, independentemente
da isoforma utilizada. Por outro lado, espera-se encontrar efeito inibitório com outros
fármacos anti-KIT. Com base na maior ativação da sinalização de KIT na isoforma GNNK-,
talvez essas células exibiriam uma resposta mais acentuada a alguns desses inibidores,

65
embora os resultados sejam inesperados. Atualmente, agentes farmacológicos que tem
como alvo as tirosina quinases, incluindo o KIT (por exemplo, imatinib, sunitinib, sorafenib e
dasatinib) já são utilizados na prática clínica para o tratamento de câncer. Alguns desses
medicamentos estão em fase de ensaios clínicos para glioblastomas 83. O papel das
isoformas de KIT em resposta às terapias alvo de KIT em GBMs ainda é desconhecido.
Com a finalidade de encontrar associações entre a presença das isoformas e a
expressão da proteína KIT com os dados clínico-patológicos: idade, sexo, raça, tabagismo,
etilismo, KPS, lateralidade, radioterapia, quimioterapia e recidiva realizamos o teste de χ2.
Encontramos apenas três associações com p-valor significativo. Entre elas: associação entre
a predominância da isoforma GNNK- com a lateralidade tumoral, em que ambos os lados
está associado à predominância da isoforma GNNK-; associação entre a expressão da
proteína KIT (tecido endotelial) com o KPS categorizado ˂ 80 e; associação entre a expressão
da proteína KIT (tecido endotelial) negativa com a predominância da isoforma GNNK-.
Com os mesmos dados clínico-patológicos que utilizamos para associar com as
isoformas e expressão da proteína KIT, também utilizamos para associar com a sobrevida
global dos pacientes. Não observamos resultados estatisticamente significativos entre a
sobrevida global associada aos dados de expressão de KIT, tanto RNAm quanto proteína.
Quanto à associação da sobrevida com os dados clínico-patológicos, observamos o aumento
da sobrevida em pacientes com idade menor ou igual a 50 anos, pacientes com KPS maior ou
igual a 80, pacientes que realizaram radioterapia, pacientes que realizaram quimioterapia e,
pacientes que apresentaram recidiva. Essas associações estão de acordo com dados já
publicados 25, 94, quanto à associação com o etilismo e tabagismo, dados anteriores também
não encontraram associações ou então observaram resultados inconclusivos 95.
Os fatores de risco ambientais para o desenvolvimento de GBMs ainda são
desconhecidos e apresentam alguns resultados inconclusivos. De acordo com vários estudos
citados por Ostrom e colaboradores (2014), fatores de risco tem sido estudados como
potenciais contribuidores para o risco de glioma, entre eles: condições alérgicas e doenças
atópicas estão relacionadas à diminuição do risco, enquanto exposição à radiação ionizante,
ao aumento do risco; quanto à radiação não-ionizante (celulares) e exposição ocupacional,
não foram obtidos resultados conclusivos 11. Sabemos que o tabagismo e o etilismo
aumentam o risco de vários tipos tumorais e certos componentes do cigarro e do álcool

66
podem penetrar na barreira hemato-encefálica, entretanto, segundo Braganza e
colaboradores (2014), não foi encontrada associação entre o tabagismo/etilismo e o
aumento do risco de glioma 95. Também não encontramos associações estatisticamente
significativas entre o tabagismo/etilismo com a expressão do RNAm, proteína de KIT e
sobrevida global.
Este é o primeiro trabalho a avaliar o impacto de cada isoforma em pacientes com
GBM, se as isoformas distintas de KIT levam a diferenças no comportamento bioquímico e
funcional de linhagens celulares de GBM, o impacto na sobrevida do paciente poderá ser
observado. Guerrini et al. relataram uma associação entre a diferença de expressão das
isoformas GNNK de KIT e a sobrevida global em pacientes com leucemia mieloide aguda, em
que pacientes que expressam somente a isoforma GNNK- apresentam maior sobrevida
global e sobrevida livre de progressão 70. Esses achados corroboram com os nossos
resultados, em que pacientes com a predominância da isoforma GNNK- apresentam a
tendência de maior sobrevida. Mais que isso, observamos uma tendência quanto à
expressão de RNAm de KIT, os pacientes que não expressam RNAm, apresentam maior
sobrevida (p-valor 0,057), embora os resultados não sejam estatisticamente significativos.
Esses dados parecem contraditórios em relação aos ensaios celulares, em que observamos,
nas linhagens transfectadas com a isoforma GNNK-, como sendo mais oncogênica, no
entanto, outros autores já demonstraram a internalização e degradação mediada por
ubiquitinação mais rápida em GNNK-, implicando em um “switch-off” mais rápido da
atividade de KIT. A expressão da proteína KIT também foi associada com a sobrevida global
dos pacientes, entretanto não encontramos diferenças estatisticamente significativas.

67
CONCLUSÕES

68
7 CONCLUSÕES
Diante dos resultados deste estudo, podemos concluir que a proteína não é expressa
na mesma proporção que o RNAm de KIT, este expresso em 95% das amostras, enquanto a
proteína nas células neoplásicas (22,8%) e no tecido endotelial (40,2%), além disso, as
isoformas de KIT são diferentemente expressas em tecidos normais e congelados versus
linhagens celulares de GBM, esses dados sugerem uma mudança da expressão das isoformas
de KIT em adaptação das células tumorais para as condições de cultura celular. KIT parece
ser ativado em GBM através de mecanismos autócrinos/parácrinos com o ligante SCF. Em
relação ao papel tumorigênico, a isoforma GNNK- está associada parece ser mais oncogênica
devido à maior viabilidade e fenótipo invasivo em linhagens celulares de GBM, e através de
ensaios in vivo, observamos maior formação tumoral com as células transfectadas com as
isoformas e aumento da expressão de KIT nos tumores em GNNK-. Observamos maior
ativação de KIT em GNNK- e sua influência na ativação de ERK nas linhagens transfectadas
(GAMG). Apesar de diferenças não significativas, pacientes que expressam o RNAm de KIT
apresentam pior prognóstico e pacientes com a predominância da isoforma GNNK-
apresentam uma tendência a maior sobrevida.

69
PERSPECTIVAS

70
8 PERSPECTIVAS
De acordo com os achados deste trabalho, notamos que KIT está envolvido na
patogênese molecular de alguns gliomas, seja por amplificação gênica, que já foi descrito em
outros trabalhos, ou por mecanismos autócrinos/parácrinos, além de observar diferenças no
papel funcional e tumorigênico das isoformas GNNK de KIT. Dessa forma, o KIT poderia ser
um potencial alvo molecular na terapia antitumoral dos GBMs através dos inibidores de
tirosina quinase, além do mais, o papel das isoformas de KIT em resposta às terapias alvo de
KIT em GBMs é desconhecido. Diante disso, pretendemos avaliar o papel dos inibidores de
KIT (Imatinib, Sorafenib, Sunitinib, Cediranib, Nilotinib e Dasatinib) nas células transfectadas
com as respectivas isoformas.

71
REFERÊNCIAS

72
REFERÊNCIAS
1. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9(4):138-41.
2. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology.
3. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27-36.
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74.
5. Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23(38):6445-70.
6. Martin-Moreno JM, Soerjomataram I, Magnusson G. Cancer causes and prevention: a condensed appraisal in Europe in 2008. Eur J Cancer. 2008;44(10):1390-403.
7. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492-507.
8. Buckner RL, Carroll DC. Self-projection and the brain. Trends Cogn Sci. 2007;11(2):49-57.
9. Chandana SR, Movva S, Arora M, Singh T. Primary brain tumors in adults. Am Fam Physician. 2008;77(10):1423-30.
10. Farrell CJ, Plotkin SR. Genetic causes of brain tumors: neurofibromatosis, tuberous sclerosis, von Hippel-Lindau, and other syndromes. Neurol Clin. 2007;25(4):925-46, viii.
11. Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, et al. The epidemiology of glioma in adults: a "state of the science" review. Neuro Oncol. 2014;16(7):896-913.
12. CBTRUS. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004-20062010.
13. Dubrow R, Darefsky AS. Demographic variation in incidence of adult glioma by subtype, United States, 1992-2007. BMC Cancer. 2011;11:325.
14. Estimativa 2014 - Incidência de Câncer no Brasil. In: INCA, editor.: Ministério da Saúde; 2014.

73
15. Yabroff KR, Harlan L, Zeruto C, Abrams J, Mann B. Patterns of care and survival for patients with glioblastoma multiforme diagnosed during 2006. Neuro Oncol. 2012;14(3):351-9.
16. Estimativa 2016 - Incidência de Câncer no Brasil. In: INCA, editor.: Ministério da Saúde; 2016.
17. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97-109.
18. Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011;12(9):495-508.
19. Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15(11):1311-33.
20. Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2(8):616-26.
21. Kleihues P, Ohgaki H. Phenotype vs genotype in the evolution of astrocytic brain tumors. Toxicol Pathol. 2000;28(1):164-70.
22. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014;23(10):1985-96.
23. Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95(2):190-8.
24. Bauchet L, Mathieu-Daude H, Fabbro-Peray P, Rigau V, Fabbro M, Chinot O, et al. Oncological patterns of care and outcome for 952 patients with newly diagnosed glioblastoma in 2004. Neuro Oncol. 2010;12(7):725-35.
25. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987-96.
26. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117-34.
27. Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61(2):203-12.

74
28. Zwick E, Bange J, Ullrich A. Receptor tyrosine kinases as targets for anticancer drugs. Trends Mol Med. 2002;8(1):17-23.
29. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177-82.
30. Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, Collins VP, et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9(8):2313-20.
31. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183-232.
32. Besmer P, Murphy JE, George PC, Qiu FH, Bergold PJ, Lederman L, et al. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320(6061):415-21.
33. Lev S, Blechman JM, Givol D, Yarden Y. Steel factor and c-kit protooncogene: genetic lessons in signal transduction. Crit Rev Oncog. 1994;5(2-3):141-68.
34. Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6(11):3341-51.
35. Qiu FH, Ray P, Brown K, Barker PE, Jhanwar S, Ruddle FH, et al. Primary structure of c-kit: relationship with the CSF-1/PDGF receptor kinase family--oncogenic activation of v-kit involves deletion of extracellular domain and C terminus. EMBO J. 1988;7(4):1003-11.
36. Blume-Jensen P, Ronnstrand L, Gout I, Waterfield MD, Heldin CH. Modulation of Kit/stem cell factor receptor-induced signaling by protein kinase C. J Biol Chem. 1994;269(34):21793-802.
37. Masson K, Heiss E, Band H, Ronnstrand L. Direct binding of Cbl to Tyr568 and Tyr936 of the stem cell factor receptor/c-Kit is required for ligand-induced ubiquitination, internalization and degradation. Biochem J. 2006;399(1):59-67.
38. Sun J, Pedersen M, Bengtsson S, Ronnstrand L. Grb2 mediates negative regulation of stem cell factor receptor/c-Kit signaling by recruitment of Cbl. Exp Cell Res. 2007;313(18):3935-42.
39. Zeng S, Xu Z, Lipkowitz S, Longley JB. Regulation of stem cell factor receptor signaling by Cbl family proteins (Cbl-b/c-Cbl). Blood. 2005;105(1):226-32.

75
40. Ropers HH, Craig IW. Report of the committee on the genetic constitution of chromosomes 12 and 13. Cytogenet Cell Genet. 1989;51(1-4):259-79.
41. Martin FH, Suggs SV, Langley KE, Lu HS, Ting J, Okino KH, et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63(1):203-11.
42. Ronnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004;61(19-20):2535-48.
43. Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23(1):16-43.
44. Martinho O, Goncalves A, Moreira MA, Ribeiro LF, Queiroz GS, Schmitt FC, et al. KIT activation in uterine cervix adenosquamous carcinomas by KIT/SCF autocrine/paracrine stimulation loops. Gynecol Oncol. 2008;111(2):350-5.
45. Martinho O, Gouveia A, Viana-Pereira M, Silva P, Pimenta A, Reis RM, et al. Low frequency of MAP kinase pathway alterations in KIT and PDGFRA wild-type GISTs. Histopathology. 2009;55(1):53-62.
46. Gomes AL, Reis-Filho JS, Lopes JM, Martinho O, Lambros MB, Martins A, et al. Molecular alterations of KIT oncogene in gliomas. Cell Oncol. 2007;29(5):399-408.
47. Ashman LK. The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol. 1999;31(10):1037-51.
48. Wiedmann MW, Caca K. Molecularly targeted therapy for gastrointestinal cancer. Curr Cancer Drug Targets. 2005;5(3):171-93.
49. Orr-Urtreger A, Avivi A, Zimmer Y, Givol D, Yarden Y, Lonai P. Developmental expression of c-kit, a proto-oncogene encoded by the W locus. Development. 1990;109(4):911-23.
50. Ida JA, Jr., Dubois-Dalcq M, McKinnon RD. Expression of the receptor tyrosine kinase c-kit in oligodendrocyte progenitor cells. J Neurosci Res. 1993;36(5):596-606.
51. Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest. 2004;113(9):1364-74.
52. Jin K, Mao XO, Sun Y, Xie L, Greenberg DA. Stem cell factor stimulates neurogenesis in vitro and in vivo. J Clin Invest. 2002;110(3):311-9.

76
53. Cetin N, Dienel G, Gokden M. CD117 expression in glial tumors. J Neurooncol. 2005;75(2):195-202.
54. Reis RM, Martins A, Ribeiro SA, Basto D, Longatto-Filho A, Schmitt FC, et al. Molecular characterization of PDGFR-alpha/PDGF-A and c-KIT/SCF in gliosarcomas. Cell Oncol. 2005;27(5-6):319-26.
55. Joensuu H, Puputti M, Sihto H, Tynninen O, Nupponen NN. Amplification of genes encoding KIT, PDGFRalpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol. 2005;207(2):224-31.
56. Holtkamp N, Ziegenhagen N, Malzer E, Hartmann C, Giese A, von Deimling A. Characterization of the amplicon on chromosomal segment 4q12 in glioblastoma multiforme. Neuro Oncol. 2007;9(3):291-7.
57. Stanulla M, Welte K, Hadam MR, Pietsch T. Coexpression of stem cell factor and its receptor c-Kit in human malignant glioma cell lines. Acta Neuropathol. 1995;89(2):158-65.
58. Blom T, Fox H, Angers-Loustau A, Peltonen K, Kerosuo L, Wartiovaara K, et al. KIT overexpression induces proliferation in astrocytes in an imatinib-responsive manner and associates with proliferation index in gliomas. Int J Cancer. 2008;123(4):793-800.
59. Sihto H, Tynninen O, Butzow R, Saarialho-Kere U, Joensuu H. Endothelial cell KIT expression in human tumours. J Pathol. 2007;211(4):481-8.
60. Reith AD, Ellis C, Lyman SD, Anderson DM, Williams DE, Bernstein A, et al. Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. EMBO J. 1991;10(9):2451-9.
61. Crosier PS, Ricciardi ST, Hall LR, Vitas MR, Clark SC, Crosier KE. Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood. 1993;82(4):1151-8.
62. Zhu WM, Dong WF, Minden M. Alternate splicing creates two forms of the human kit protein. Leuk Lymphoma. 1994;12(5-6):441-7.
63. Caruana G, Cambareri AC, Ashman LK. Isoforms of c-KIT differ in activation of signalling pathways and transformation of NIH3T3 fibroblasts. Oncogene. 1999;18(40):5573-81.
64. Young SM, Cambareri AC, Odell A, Geary SM, Ashman LK. Early myeloid cells expressing c-KIT isoforms differ in signal transduction, survival and chemotactic responses to Stem Cell Factor. Cell Signal. 2007;19(12):2572-81.

77
65. Montero JC, Lopez-Perez R, San Miguel JF, Pandiella A. Expression of c-Kit isoforms in multiple myeloma: differences in signaling and drug sensitivity. Haematologica. 2008;93(6):851-9.
66. Sun J, Pedersen M, Ronnstrand L. Gab2 is involved in differential phosphoinositide 3-kinase signaling by two splice forms of c-Kit. J Biol Chem. 2008;283(41):27444-51.
67. Phung B, Steingrimsson E, Ronnstrand L. Differential activity of c-KIT splice forms is controlled by extracellular peptide insert length. Cell Signal. 2013;25(11):2231-8.
68. Sakuma Y, Sakurai S, Oguni S, Hironaka M, Saito K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003;94(6):486-91.
69. Theou N, Tabone S, Saffroy R, Le Cesne A, Julie C, Cortez A, et al. High expression of both mutant and wild-type alleles of c-kit in gastrointestinal stromal tumors. Biochim Biophys Acta. 2004;1688(3):250-6.
70. Guerrini F, Galimberti S, Ciabatti E, Brizzi S, Testi R, Pollastrini A, et al. Molecular detection of GNNK- and GNNK+ c-kit isoforms: a new tool for risk stratification in adult acute myeloid leukaemia. Leukemia. 2007;21(9):2056-8.
71. Wang H, Xu T, Jiang Y, Xu H, Yan Y, Fu D, et al. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia. 2015;17(3):239-55.
72. Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-small-cell lung cancer. Clin Ther. 2005;27(10):1513-34.
73. Ribas A, Hersey P, Middleton MR, Gogas H, Flaherty KT, Sondak VK, et al. New challenges in endpoints for drug development in advanced melanoma. Clin Cancer Res. 2012;18(2):336-41.
74. Habeck M. FDA licences imatinib mesylate for CML. Lancet Oncol. 2002;3(1):6.
75. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061-8.
76. Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181(4):1126-41.
77. Reardon DA, Turner S, Peters KB, Desjardins A, Gururangan S, Sampson JH, et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Canc Netw. 2011;9(4):414-27.

78
78. Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14(11):1131-8.
79. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709-22.
80. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699-708.
81. Chamberlain MC. Emerging clinical principles on the use of bevacizumab for the treatment of malignant gliomas. Cancer. 2010;116(17):3988-99.
82. Brandsma D, van den Bent MJ. Molecular targeted therapies and chemotherapy in malignant gliomas. Curr Opin Oncol. 2007;19(6):598-605.
83. Ren H, Yang BF, Rainov NG. Receptor tyrosine kinases as therapeutic targets in malignant glioma. Rev Recent Clin Trials. 2007;2(2):87-101.
84. Martinho O, Silva-Oliveira R, Miranda-Goncalves V, Clara C, Almeida JR, Carvalho AL, et al. In Vitro and In Vivo Analysis of RTK Inhibitor Efficacy and Identification of Its Novel Targets in Glioblastomas. Transl Oncol. 2013;6(2):187-96.
85. Martinho O, Granja S, Jaraquemada T, Caeiro C, Miranda-Goncalves V, Honavar M, et al. Downregulation of RKIP is associated with poor outcome and malignant progression in gliomas. PLoS One. 2012;7(1):e30769.
86. Caruana G, Cambareri AC, Gonda TJ, Ashman LK. Transformation of NIH3T3 fibroblasts by the c-Kit receptor tyrosine kinase: effect of receptor density and ligand-requirement. Oncogene. 1998;16(2):179-90.
87. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315-9.
88. Moniz S, Martinho O, Pinto F, Sousa B, Loureiro C, Oliveira MJ, et al. Loss of WNK2 expression by promoter gene methylation occurs in adult gliomas and triggers Rac1-mediated tumour cell invasiveness. Hum Mol Genet. 2013;22(1):84-95.
89. Martinho O, Pinto F, Granja S, Miranda-Goncalves V, Moreira MA, Ribeiro LF, et al. RKIP inhibition in cervical cancer is associated with higher tumor aggressive behavior and resistance to cisplatin therapy. PLoS One. 2013;8(3):e59104.

79
90. Hamel W, Westphal M. The road less travelled: c-kit and stem cell factor. J Neurooncol. 1997;35(3):327-33.
91. Han ZB, Ren H, Zhao H, Chi Y, Chen K, Zhou B, et al. Hypoxia-inducible factor (HIF)-1 alpha directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF). Carcinogenesis. 2008;29(10):1853-61.
92. Jogi A, Ora I, Nilsson H, Lindeheim A, Makino Y, Poellinger L, et al. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci U S A. 2002;99(10):7021-6.
93. Chan EC, Bai Y, Bandara G, Simakova O, Brittain E, Scott L, et al. KIT GNNK splice variants: expression in systemic mastocytosis and influence on the activating potential of the D816V mutation in mast cells. Exp Hematol. 2013;41(10):870-81 e2.
94. Laws ER, Parney IF, Huang W, Anderson F, Morris AM, Asher A, et al. Survival following surgery and prognostic factors for recently diagnosed malignant glioma: data from the Glioma Outcomes Project. J Neurosurg. 2003;99(3):467-73.
95. Braganza MZ, Rajaraman P, Park Y, Inskip PD, Freedman ND, Hollenbeck AR, et al. Cigarette smoking, alcohol intake, and risk of glioma in the NIH-AARP Diet and Health Study. Br J Cancer. 2014;110(1):242-8.

80
ANEXOS

81
ANEXOS
ANEXO A

82

83

84

85
ANEXO B

86

87
ANEXO C

88

89

90

91

92

93

94

95
ANEXO D
Ficha de coleta utilizada para coletar dados clínico-patológicos de pacientes com
glioblastoma do Hospital de Câncer de Barretos – Fundação Pio XII, no período de 2002 –
2014.

96

97