ns w do alkoholi-2015 po 6
TRANSCRIPT

Chemia organicznadr hab. Danuta Kalemba, p. 205
tel. 42-6313428konsultacje wt. 13 15-14; śr. 1215-13
sem. II sem. III
Wykład: 20
Ćwiczenia: 16
Laboratorium: 20
Informacje dotycz ące przedmiotuTablica ogłosze ń chemii organicznej, II pi ętro, nowy gmach WBiNo ś
http://snack.p.lodz.pl/studia i studenci/ogłoszenia wykładowców/Kalemba
Rozwi ązywanie arkuszy – punkty do egzaminu
Kartkówki zapowiedziane z partii materiału – punkty do egzaminu
Egzamin pisemny w sesji letniej
Kilka bardzo wa Ŝnych rad• nie zamykaj szuflad z wiedz ą z chemii nieorganicznej i fizyki• uczestnicz w wykładach (korelacja z ćwiczeniami)
• bierz czynny udział w ćwiczeniach• zadawaj pytania • rozwi ązuj samodzielnie problemy• korzystaj z podr ęcznika i materiałów• korzystaj z konsultacji • wykorzystuj modele cz ąsteczkowe
Podręczniki• McMurry J., Chemia organiczna, tom I i II, PWN, 2000.• Mastalerz P., Podręcznik chemii organicznej, Wydawnictwo Chemiczne,
1998.• Morrison R.T., Boyd R.N., Chemia organiczna, tom I i II, PWN, 1997.• Hart H., Chemia organiczna. Krótki kurs. PZWL, 1999.
materiały do ćwiczeń
karta przedmiotuCel przedmiotu1. UmoŜliwienie zdobycia wiedzy dotyczącej budowy, właściwości fizycznych i przebiegureakcji chemicznych związków organicznych, w tym biocząsteczek.2. Wykształcenie umiejętności wykorzystania nabytej wiedzy do określania stereochemii i reaktywności poznanych klas związków organicznych.
Efekty kształceniaStudent:1. wyjaśnia podstawowe pojęcia i definicje2. rozpoznaje i rozróŜnia rodzaje izomerii i określa konfigurację związków organicznych3. klasyfikuje i nazywa związki organiczne4. przewiduje właściwości fizyczne i chemiczne związku na podstawie budowy cząsteczkowej5. przewiduje warunki i kierunki przemian związków ze wszystkich poznanych klas, w tymprostych cząsteczek biologicznych6. projektuje syntezy związków organicznych7. wykorzystując wiedzę na temat reaktywności klas związków organicznych zapisuje schematy reakcji dla konkretnych związków8. zapisuje i wyjaśnia mechanizmy podstawowych typów reakcji chemicznych9. znajduje, selekcjonuje i wykorzystuje wiedzę zawartą w podręcznikach i materiałachdydaktycznych do rozwiązywania problemów z zakresu chemii organicznej
Metody weryfikacji efektów kształcenia1-9: dwa kolokwia pisemne, aktywność w dyskusji na ćwiczeniach1-9: egzamin pisemny i ustny
Formy zaliczeniaPisemne kolokwia na ćwiczeniach - 20%Rozwiązane arkusze (zaliczenie po terminowym oddaniu)Egzamin pisemny i ustny - 70%.
Przeci ętne obci ąŜenie studenta prac ą własn ąUdział w konsultacjach 5Udział w pisemnych i/lub praktycznych formach weryfikacji 1Przygotowanie do ćwiczeń i kolokwiów 40 (2x15) – 16Wykonanie zadań domowych 30 Przygotowanie do egzaminu 60 (30+15) – 20
CHEMIA ORGANICZNA
= CHEMIA ZWIĄZKÓW WĘGLA
(minus H 2CO3 i pochodne)
•1770 Bergman : substancje organiczne (z organizmów Ŝywych - vis vitalis) i nieorganiczne
•1828 Wöhler: NH4CNO H2N-C-NH2
Omocznik
metan CH 4 DNA
1 atom C 10 10 atomów C
Dlaczego chemia organiczna?• związki organiczne>>> zwi ązki nieorganiczne
11 000 000 100 000
• rola w procesach Ŝyciowych
• właściwo ści zupełnie ró Ŝne
Dlaczego tak du Ŝo zwi ązków organicznych?
• atomy C ł ączą się w długie ła ńcuchy i pier ścienie
• atomy C przył ączają inne: H, O, N, Cl, Br, F, J, S, P
• kaŜde odmienne rozmieszczenie atomów to nowy
związek, np. C 15H24 – 500 związków poznane

Szczególne miejsce atomu w ęgla
I II III IV V VI VII VIII
H He
Li Be B C N O F Ne
Na Mg Al Si P S Cl Ar
K Ca Br
J
1, 2 lub 3 éwalencyjne
oddaj ą é
5, 6 lub 7 éwalencyjnych
przyjmuj ą é4 é walencyjne
uwspólniaj ą é
chemia organiczna
biologia
Dlaczego chemia organiczna wa Ŝna?
biochemia
biotechnologia
ekologia
medycynachemia Ŝywno ści
• Ŝywno ść
• leki
• odzie Ŝ
• drewno, papier
• tworzywa sztuczne
• benzyna, oleje, gaz
• biopaliwa
• farby, barwniki
• ……
chemia nieorganiczna
fizykachemia fizyczna
Zadania chemii organicznej• poznanie struktury i wła ściwo ści nowych
związków
• synteza zwi ązków znanych z natury i nowych
Źródła zwi ązków organicznych
• węgiel
• ropa naftowa
• rośliny: zło Ŝone zwi ązki organiczne
Klasyfikacja zwi ązków organicznych
C, H
i inne pierwiastkiO, N, F, Cl, Br, I, S
C, H węglowodory
e
ALKANYNazewnictwo
C1 metan C2 etan C3 propan C4 butan C5 pentan ..........C10 dekan
C21 henejkozanC22 dokozanC23 trikozan
C30 triakontanC40 tetrakontanC50 pentakontanC100 hektan
Rdzenie liczebnikowe
½ hemi (semi)1 mono (uni)1½ (seskwi)2 di (bi)3 tri4 tetra5 penta6 heksa7 hepta8 okta9 nona10 deka
C11 undekanC12 dodekanC13 tridekan............C20 ejkozan
NazewnictwoIUPAC
(International Union of Pure and Applied Chemistry)
-L-
miejsce podstawnika miejsce grupy
funkcyjnej
L-
ilość atomów węgla w
szkielecie
RDZEŃ
(cyklo)alk
rodzaj podstawnika
PRZEDROSTEK
alkilo, alkenylo, alkinylo
hydroksy, formylo, okso
grupa funkcyjna
rodzina związków
PRZYROSTEK
-an, -en, -yn
-ol, -al, -onkwas -owy
aminaznaki interpunkcyjne-ł ączniki-1,2-przecinki-(kropki)-nawiasy
(cyklo)alkan
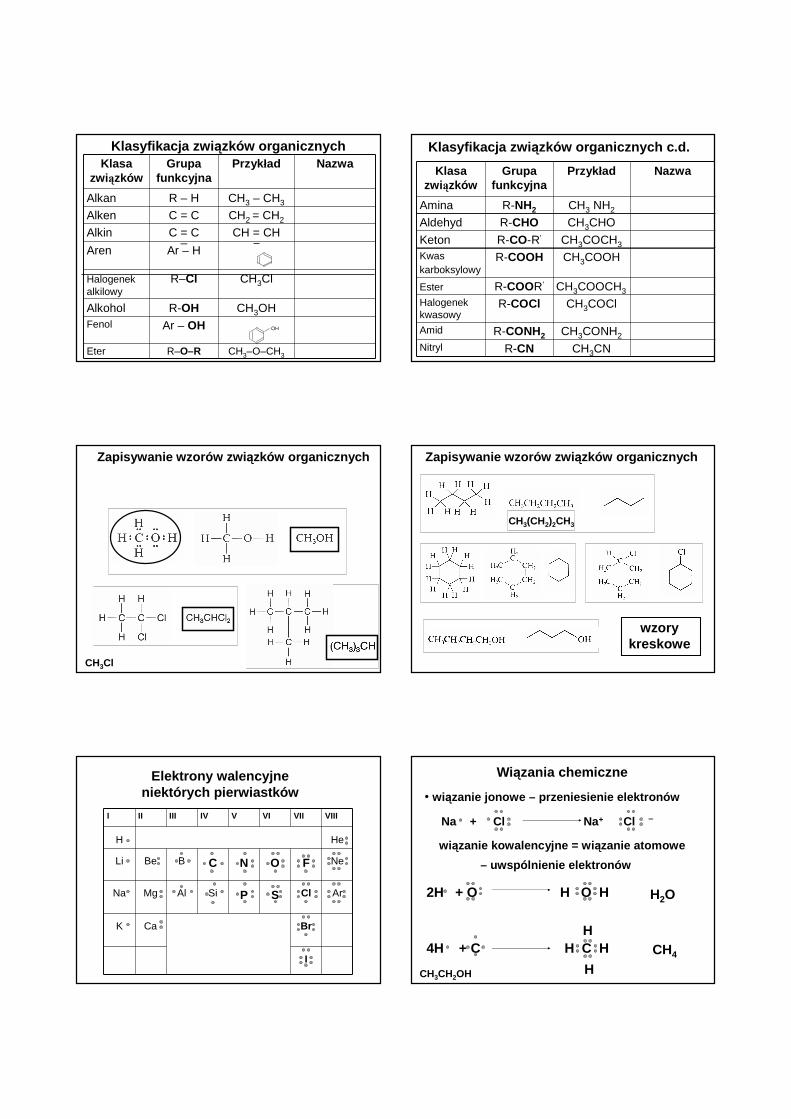
Klasyfikacja zwi ązków organicznychKlasa
związkówGrupa
funkcyjnaPrzyk ład Nazwa
Alkan R – H CH3 – CH3
Alken C = C CH2 = CH2
Alkin C = C CH = CH
Aren Ar – H
Halogenekalkilowy
R–Cl CH3Cl
Alkohol R-OH CH3OHFenol Ar – OH
Eter R–O–R CH3–O–CH3
OH
–
Klasyfikacja zwi ązków organicznych c.d.
Klasazwiązków
Grupa funkcyjna
Przyk ład Nazwa
Amina R-NH2 CH3 NH2
Aldehyd R-CHO CH3CHO
Keton R-CO-R’ CH3COCH3
Kwas karboksylowy
R-COOH CH3COOH
Ester R-COOR’ CH3COOCH3
Halogenek kwasowy
R-COCl CH3COCl
Amid R-CONH2 CH3CONH2
Nitryl R-CN CH3CN
Zapisywanie wzorów zwi ązków organicznych
CH3Cl
Zapisywanie wzorów zwi ązków organicznych
wzory kreskowe
CH3(CH2)2CH3
Elektrony walencyjne niektórych pierwiastków
I II III IV V VI VII VIII
H He
Li Be B C N O F Ne
Na Mg Al Si P S Cl Ar
K Ca Br
I
Wiązania chemiczne
• wiązanie jonowe – przeniesienie elektronów
Na + Cl Na+ Cl –
wiązanie kowalencyjne = wi ązanie atomowe
– uwspólnienie elektronów
CH4
H2O2H + O H O H
4H + C H C H
H
H
CH3CH2OH

Elektroujemno ść pierwiastkówzdolno ść do przyci ągania elektronów
I II III IV V VI VII
H2.2
Li1.0
Be1.6
B2.0
C2.5
N3.0
O3.4
F4.0
Na0.9
Mg1.3
Al.1.6
Si1.9
P2.2
S2.6
Cl3.2
K0.8
Ca1.0
Br3.0
I3.7
szereg elektroujemno ści wybranych pierwiastków F O N Cl Br I S C H4.0 2.1
Wiązanie kowalencyjne spolaryzowane
wiązania niepolarne
H H O O C C C Hwiązania polarne
H2O H– O–H NH3 H– N–H
H
δ-
δ+ δ+
δ+
δ-
δ+δ+
Wiązanie koordynacyjnewspólna para elektronów od jednego atomu
NH3 + H+ NH4+
H2O +H+ H3O+CH3CH2OH
kształt: azymutalna liczba kwantowa
l = 0 orbital s
l = 1 orbital p
Orbitale atomowe
1s 2s
p
orientacja: magnetyczna liczba kwantowa
px pzpy
Orbitale cz ąsteczkowe = molekularne
Wiązanie π
nakładanie równolegle p-p - wi ązanie π(nakładanie czołowe p-p - wi ązanie σ)nakładanie p(czołowo)-s - wi ązanie σ
orbitale atomowe
Wiązanie σ
orbitale cząsteczkowe
Hybrydyzacja atomu w ęgla
126C
1s 2s 2p
*C
sp 3 1é s + 3é p = 4é CH3 CH3
109°28’ C C 1.54 Å
C H 1.09 Å
H H 0.74 Å
sp3
Hybrydyzacja sp3
CH4 CH3CH3

120 ° C = C 1.34 Å
sp 2 1é s + 2é p = 3é sp 2 + 1é p
CH2 = CH2
Hybrydyzacja atomu w ęgla
126C
1s 2s 2p
*Csp2
Hybrydyzacja sp2
sp2
sp2
sp2
p
H2C CH CH3
*C
sp 1é s + 1é p = 2é sp + 2é p
sp
CH CH
180 ° C C 1.21 Å
Hybrydyzacja atomu w ęgla
126C
1s 2s 2p
Hybrydyzacja sp
spsp
pp
HC C CH3
*C
sp3 1é s + 3é p = 4é sp 3 109°28’ 1.54 Å
sp2 1é s + 2é p = 3é sp 2 + 1é p 120 ° 1.34 Å
sp 1é s + 1é p = 2é sp + 2é p 180 ° 1.21 Å
Hybrydyzacja atomu w ęgla
126C
1s 2s 2p
Alkany CnH2n+2
Węglowodory alifatyczne nasycone
prostoła ńcuchowe rozgał ęzione
C7H14
?

Izomeryzwiązki o takim samym wzorze sumarycznym,
ale ró Ŝnym uło Ŝeniu atomów w cz ąsteczce
Izomery strukturalne(konstytucyjne)
Izomeria szkieletowa
Izomeria poło Ŝenia
Izomeria grup
funkcyjnych
taka sama kolejno śćatomów, ale ró Ŝne uło Ŝenie w przestrzeni
Stereoizomery
Nazewnictwo alkan ówTworzenie nazwy alkanu: 1.Wybieramy najdłu Ŝszy łańcuch2.Numerujemy atomy w ęgla tak, aby lokanty okre ślające poło Ŝenia
podstawników były jak najni Ŝsze3.Podstawniki w formie przedrostków szeregujemy alf abetycznie4.Liczb ę takich samych podstawników okre ślamy przedrostkami:
di- (2); tri- (3); tetra- (4); penta- (5); heksa- (6);.. i.t.d.5.Na końcu umieszczamy nazw ę łańcucha w ęglowego
Rozgał ęzione podstawniki alkilowe nazywamy analogicznie jak alkany • numerację atomów węgla rozpoczynamy od atomu związanego
z głównym szkieletem,• nazwę podstawnika złoŜonego umieszczamy w nawiasie
Rozgał ęzione podstawniki alkilowe C3, C4, C5 maj ą nazwy zwyczajowe
izo, neo, sec-, tert-
Wyst ępowanie alkanów
woski ro ślinne, np. C 29 i C31 - wosk li ści
ropa naftowa
<20°C
C5-C6 20-60°C eter naftowyC6-C7 60-100°C lekka naftaC5-C10 40-205°C benzyna
175-325°C
>275°C
Właściwo ści fizyczne alkanów• niepolarne – nierozpuszczalne w wodzie• gęstość < 1g/ml
temp. wrzenia: pentan 36ºCizopentan 28 ºCneopentan 10 ºC
gazy ciecze
ciała stałe > C 17
Konformacje alkanów
rozmieszczenie atomów wynikaj ące ze swobodnego obrotu wokół wi ązania C–C
konformery – nietrwałeróŜnica energii – mała
projekcja Newmana
Projekcja Newmana etanu
konformacja naprzemianległa dalszyatom C
bli Ŝszyatom C
konformacja naprzeciwległa

Projekcja Newmana butanu
konformacja naprzemianległa
konformacja sko śna
konformacja naprzeciwległa
projekcja C2 – C3
H
Otrzymywanie alkanówMetody laboratoryjne
• uwodornienie alkenów
• z halogenków – poprzez reakcj ę Grignarda
• z halogenków –przez redukcj ę
2-bromobutanbromek sec-butylu
Właściwo ści chemiczne alkanówreakcje utleniania (spalania)CH4 + 2O2 CO2 + 2H2O + 890 kJ/molpłomie ń
spalanie ciepło spalania
6 CH4 + O2 2 CH CH + 2CO + 10H21500ºC
C4H4 + O2 CO2 + H2O + 2880 kJ/mol
CO + H2O trucizna
C + H2O sadza
HCHO + H2O smog
CH3COOH +H2O z olejów
płomie ń
Ciepło reakcji = zmiany energii reakcjirozrywanie wi ązań - potrzebna energia - reakcja endotermicznatworzenie wi ązań - wydziela si ę energia - reakcja egzotermiczna
Właściwo ści chemiczne alkanów
• reakcje halogenowania (światło hν lub temp.)
CH4 CH3Cl + HClCl2
Cl2 CH2Cl2 + HCl
Cl2 CHCl3 + HCl
Cl2 CCl4 + HCl
chlorek metylu
chlorek metylenu
chloroform
czterochlorek w ęgla
Mechanizm reakcji halogenowania alkanówpodstawienie wolnorodnikowe
reakcja ła ńcuchowa
nast ępnie reakcje (2), (3), (2), (3) itd., a Ŝ w końcu:
R.
(1) Cl2 2Cl
(2) Cl + CH4 HCl + CH3
(3) CH3 + Cl2 CH3Cl + Cl
ciepło lub
światło
etapy wzrostu łańcucha
etap inicjowania łańcucha
etapy zakończenia łańcucha
Cl + Cl Cl2
CH3 + CH3 CH3CH3
CH3 + Cl CH3Cl
ciepło lub
światło
R· - Rodnik alkilowy - płaski
hybrydyzacja sp 2 = 3 orbirale sp 2 + 1 orbital p
Rodnik stabilizowany przez hiperkoniugacj ę
Cl Cl Cl hν
homolityczne rozerwanie wi ązania
A B A + Bheterolityczne rozerwanie wi ązania
A B A + + B

Rzędowo ść atomów w ęgla
Rzędowo ść atomów wodoru = rz ędowo ści atomu C
H
R C H
H
R
R C H
H
R
R C R
R
R
R C H
R
= ilość grup R
1º 2º 3º 4º
1º
CH3
CH3 CH2 CH CH3
2º 3º(R2CH2) (R3CH)
Konkurencyjno ść reakcji
Łatwo ść odrywania si ę atomów wodoru =
Trwało ść i reaktywno ść wolnych rodników
2º
1º
3 ° >> 2° >> 1°Prawdopodobie ństwo podstawienia = f(ilo ści atomów H)
2° : 1° = 2 : 6 ale 2° = 1°
CH3CH2CH2 CH3CH2CH2Cl
CH3CH2CH3
CH3CHCH3 CH3CHCH3
Cl
Cl
Cl2
Cl2
oderwanieH 1°
oderwanieH 2°
Hiperkoniugacjaefekt stabilizuj ący poprzez delokalizacj ę elektronów
Wynik oddziaływania niesparowanego elektronu (w R •) lub orbitalu niewi ąŜącego π (wiązania C=C)
z zapełnionym orbitalem σ sąsiedniej grupy alkilowej
Konkurencyjno ść reakcji c.d.
CH3CH2CH3 CH3CH2CH2Cl + CH3CHCH3
Cl45% 55%
Cl2/hν
CH3CH2CH2CH3 CH3(CH2)3Cl + CH3CH2CHCH3
Cl28% 72%
Cl2/hν
CH3 CH3 CH3
CH3CHCH3 CH3CHCH2Cl + CH3CCH3
Cl64% 36%
Cl2/hν
3% 97%
2% 98%
ślady >99%
Br 2/hν
Reaktywno ść halogenów: F2 > Cl2 > Br 2 > (J2)Selektywno ść reakcji: Br 2 > Cl2 > F2
Cykloalkany CnH2n
Cyklopropan Cyklobutan Cyklopentan
napręŜenia a trwało ść
pierścienia
Cykloalkany CnH2n
Cykloheksan
trwała konformacja krzesłowa, bez
napręŜeń
sp3
109.5°
nietrwała konformacja łódkowa,
napręŜenia steryczne
Cykloalkany = węglowodory cykloalifatyczne
Właściwo ści fizyczne – jak alkany
Cykloalkany
Alkany

NazewnictwoTworzenie nazwy cykloalkanu: do nazwy alkanu o odpowiedniej ilości atomów w ęgla dodajemy przedrostek cyklo
alkilocykloalkanygdy liczba atomów w ęgla w pier ścieniu jest równa lub wi ększa ni Ŝ liczba atomów w ęgla w ła ńcuchu
cykloalkiloalkany
gdy liczba atomów w ęgla w pier ścieniu jest mniejsza ni Ŝ liczba atomów w ęgla w ła ńcuchu
Numeracj ę atomów w ęgla rozpoczyna si ę od miejsca przył ączenia podstawnika i dalej numeruje się tak, by lokanty były jak najni Ŝsze.
Podstawniki wymienia si ę alfabetycznie
Monopodstawiony cykloheksan konformacje
a (aksjalne) e (ekwatorialne)
Dipodstawiony cykloheksan: konformacje
e – a = a – e1,2-cis
e – e = a – a1,2-trans
Izomeryzwiązki o takim samym wzorze sumarycznym,
ale ró Ŝnym uło Ŝeniu atomów w cz ąsteczce
Izomery strukturalne(konstytucyjne)
Izomeria szkieletowa
Izomeria poło Ŝenia
Izomeria grup
funkcyjnych
taka sama kolejno śćatomów, ale ró Ŝne uło Ŝenie w przestrzeni
izomeria geometryczna
izomeria optyczna
Stereoizomery
cykloalkany i alkeny
cis, trans (Z), (E)
NazewnictwoStereoizomeria: izomeria geometryczna
alkeny i cykloalkany
cis - stereoizomer, w którym dwie grupy o większych masach znajdująsię po tej samej stronie wiązania podwójnego lub pierścienia
trans - stereoizomer, w którym dwie grupy o większych masachznajdują się po przeciwnych stronach wiązania podwójnego lub pierścienia
Z - stereoizomer, w którym dwie grupy „starsze ” w/g reguł Cahna, Ingolda, Preloga znajdują się po tej samej stronie wiązania podwójnego.
E - stereoizomer, w którym dwie grupy „starsze ” znajdują się po przeciwnych stronach wiązania podwójnego
Alkeny CnH2n Alkiny CnH2n-2
prostoła ńcuchowe rozgał ęzione
C7H14
ile?

NazewnictwoALKENY, ALKINY
wiązania podwójne: -en, -dien,- trien
wiązania potrójne: -yn lub -in (po spó łgłoskach g, k, l )
k na ko ńcu rdzenia nazwy wymienia si ę na c,
np. dekan ale decen, decyn .i decyl
gdy kilka wi ązań podwójnych i/lub potrójnych:
alkadien, alkatriyn
gdy wi ązania podwójne i potrójne:
najpierw wymieniamy -en, pó źniej -yn
hept-1-en-6-yn 24% 76%równowaga w obecno ści H+
Temp. wrz.
+4°C
Temp. wrz.
+1°C
but -2-en - izomery geometryczneStereo izomeria w alkenach
Właściwo ści fizyczne alkenów i alkinów• niepolarne – nierozpuszczalne w wodzie
• temperatura wrzenia i topnienia – podobne jak alkan ów
• gęstość (jak alkany) < 1g/ml
Otrzymywanie alkenów
z halogenków alkilowych
z alkoholi
z dihalogeno
alkanów
CH3CH2CH2CH3
butan
2-buten
H2 / katalizator
Właściwo ści chemiczne alkenówUwodornienie = redukcja = hydrogenacja
Otrzymywanie alkanów
Pt, Pd, Ni
Katalityczna addycja/ przył ączenie
Redukcja katalityczna alkenówWłaściwo ści chemiczne alkenów
Katalizator obni Ŝa energi ę aktywacji
Energia aktywacjiEakt
najmniejsza energia cz ąsteczek przy zderzeniu, potrzebna aby
reakcja mogła zaj ść
Szybko ść reakcji =częstotliwo ść zderzeń
x czynnik energetycznyx współczynnik steryczny
(właściwa orientacja)
Addycja elektrofilowa halogenowodorów
Reakcje alkenów
Reguła Markownikowa
H+ tam gdzie wi ęcej H
lub tak, Ŝe powstaje trwalszy karbokation
lub tak, Ŝe tworzy si ęhalogenek o wy Ŝszej
rzędowo ści
elektrofil: zawiera atom ubogi w elektrony, który mo Ŝe tworzy ć wiązanie przyjmuj ąc par ę elektronów od atomu w ęgla bogatego w elektrony
moŜe mieć ładunek dodatni

Mechanizm addycji elektrofilowej
karbokation –
produkt po średni
I etap: przył ączenie H + do C=C i utworzenie karbokationu
bromoetan = bromek etylu
II etap: przył ączenie Br - do karbokationu i utworzenie produktu
CH2=CH2 + HBr CH 3-CH2BrAddycja (przył ączenie) elektrofilowa
3° > 2° > 1° > kation metylowy
Trwało ść karbokationów – płaskie: sp 2
Stabilizacja karbokationu przez: • hiperkoniugacj ę
• efekt indukcyjny (spychanie elektronów wi ązań grup
alkilowych)
Addycja elektrofilowa wody do alkenów
etylen etanol
Otrzymywanie alkoholi
Addycja elektrofilowa i substytucja wolnorodnikowa halogenem
allilowy atom
wodoru
Utlenienie alkenów do dioli
Utlenienie alkenów z rozszczepieniem
Otrzymywanie kwasów karboksylowych
Ozonoliza alkenów (utlenienie)
Zn,
Otrzymywanie aldehydów i/lub ketonów
Alkadieny
Dieny sprz ęŜone
1.48Å
Addycja do dienu sprz ęŜonego

Znaczenie alkenów w syntezie organicznej
Dehydrohalogenacja dihalogenopochodnych
Otrzymywanie alkinów
Znalken
Reakcje alkinówAddycja wodoru
Addycja elektrofilowa do alkinówaddycja halogenów
addycja halogenowodorów
reakcja z bromem - reakcja charakterystyczna(odbarwienie roztworu bromu)
Odró Ŝnianie alkenów i alkinów od w ęglowodorów nasyconych
Addycja elektrofilowa wody do alkinów
synteza ketonów metylowych
Tworzenie acetylenków
Reakcje acetylenków
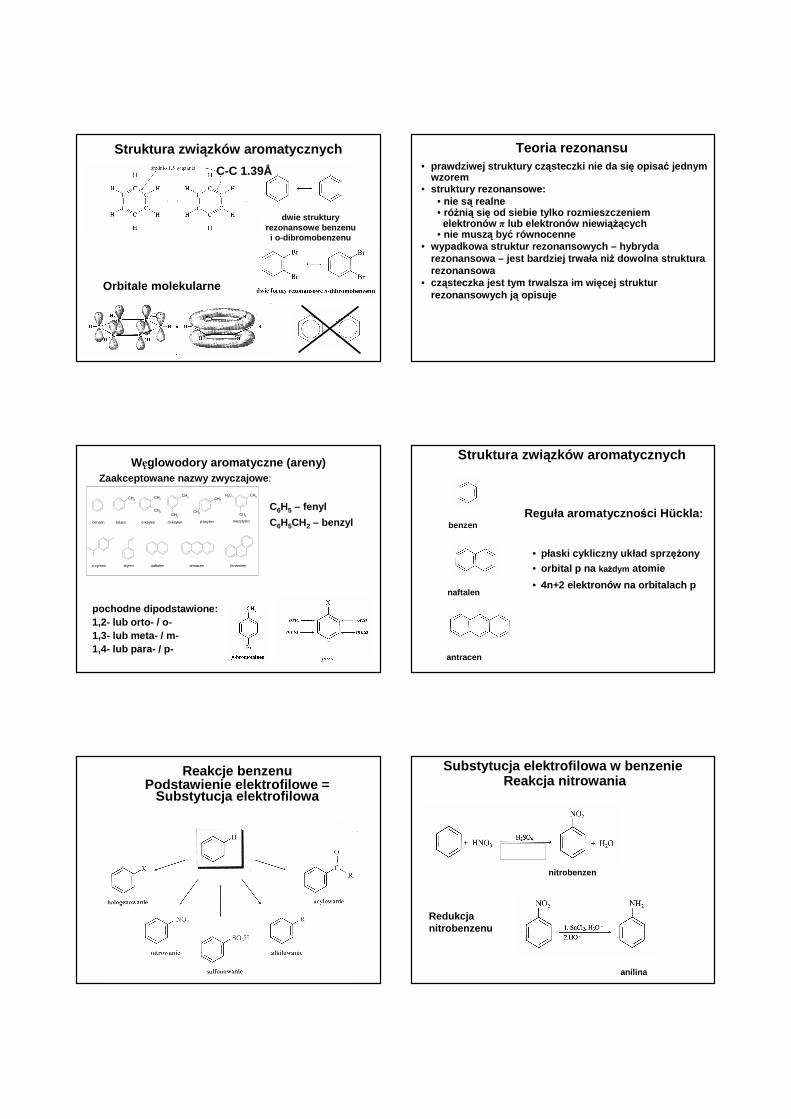
Struktura zwi ązków aromatycznych
dwie struktury rezonansowe benzenui o-dibromobenzenu
C-C 1.39Å
Orbitale molekularne
Teoria rezonansu• prawdziwej struktury cz ąsteczki nie da si ę opisa ć jednym
wzorem• struktury rezonansowe:
• nie s ą realne• róŜnią się od siebie tylko rozmieszczeniem
elektronów π lub elektronów niewi ąŜących• nie musz ą być równocenne
• wypadkowa struktur rezonansowych – hybryda rezonansowa – jest bardziej trwała ni Ŝ dowolna struktura rezonansowa
• cząsteczka jest tym trwalsza im wi ęcej struktur rezonansowych j ą opisuje
Węglowodory aromatyczne (areny)
CH3CH3
CH3
CH3
CH3
CH3
CH3
benzen toluen o-ksylen m-ksylen p-ksylen
p-cymen
CH3
CH3
CH3
mezytylen
styren naftalen antracen fenantren
Zaakceptowane nazwy zwyczajowe :
C6H5 – fenyl
C6H5CH2 – benzyl
pochodne dipodstawione: 1,2- lub orto- / o-1,3- lub meta- / m-1,4- lub para- / p-
Struktura zwi ązków aromatycznych
Reguła aromatyczno ści Hückla:
• płaski cykliczny układ sprz ęŜony• orbital p na kaŜdym atomie
• 4n+2 elektronów na orbitalach p
benzen
antracen
naftalen
Reakcje benzenuPodstawienie elektrofilowe =
Substytucja elektrofilowa
Substytucja elektrofilowa w benzenieReakcja nitrowania
nitrobenzen
Redukcja nitrobenzenu
anilina

Reakcja sulfonowania benzenu
kwas benzenosulfonowyfenol
Stapianie kwasu benzenosulfonowego z NaOH
Substytucja elektrofilowa halogenem = halogenowanie benzenu
Reakcja alkilowania benzenu halogenkami
toluen
karbokation
2º > 1º
Właściwo ści alkilobenzenów
toluen kwas benzoesowy
utlenianie ła ńcucha bocznego
o-ksylen kwas ftalowy
chlorowcowanie
chlorowcowanie w ła ńcuchu bocznymsubstytucja wolnorodnikowa
chlorowcowanie w pier ścieniusubstytucja elektrofilowa
Reakcja acylowania benzenu
acetylobenzen = fenylometyloketon
Reakcje alkilowania i acylowania
halogenkami z AlCl 3 =reakcje Friedla-
Craftsa
Właściwo ści acylobenzenówredukcja do n-alkilobenzenów
Mechanizm reakcji nitrowania
• atak elektrofila na benzen – przył ączenie elektrofilai utworzenie karbokationu – etap wolny
NO2+
(2)
• utworzenie elektrofila
=
struktury rezonansowe karbokationu hybryda rezonansowa
sp3
• oderwanie H +, przywrócenie układu aromatycznego – etap szybki
Substytucja elektrofilowa w benzenieOdczynniki elektrofilowe
kationalkilowy
kationacyliowy
struktury rezonansowe
FeBr3 + Br2

Substytucja elektrofilowa w monopodstawionym benzenie
Substytucja elektrofilowa w benzenie- szybko ść reakcji
Wpływ kieruj ący podstawnika
2
podstawniki aktywuj ące
kieruj ą w o- i p-
chlorowcedezaktywuj ą
kieruj ą w o- i p-
podstawniki dezaktywuj ące
kieruj ą w m-+
Izomeryzwiązki o takim samym wzorze sumarycznym,
ale ró Ŝnym uło Ŝeniu atomów w cz ąsteczce
Izomery strukturalne(konstytucyjne)
Izomeria szkieletowa
Izomeria poło Ŝenia
Izomeria grup funkcyjnych
taka sama kolejno ść atomów, ale róŜne uło Ŝenie w przestrzeni
izomeria geometryczna
izomeria optyczna
Stereoizomery
alkeny i cykloalkany
cis, trans (E), (Z)
R, S D, L
Konfiguracja: absolutna wzgl ędna
Izomeria optyczna
Warunek wystarczaj ący:
atom w ęgla z czterema ró Ŝnymi podstawnikami – sp 3
tzw. chiralny lub asymetryczny atom C
bromochlorojodometan
* *
Izomeria optyczna
kwas mlekowy
odbicia lustrzane =enancjomery
S R
**
Enancjomeryskr ęcają płaszczyzn ę światła spolaryzowanego o taki sam k ąt,
ale w przeciwnych kierunkach:w prawo – prawoskr ętne (+)
w lewo – lewoskr ętne (–)
wszystkie inne wła ściwo ści fizyczne oraz wszystkie wła ściwo ści chemiczne
(z wyj ątkiem reakcji ze zwi ązkami czynnymi optycznie) mają identyczne
Izomeria optyczna
Warunek konieczny: brak płaszczyzny symetrii
**

Izomeria optyczna
Czynno ść optyczna: zdolno ść skr ęcania płaszczyzny światła
płasko spolaryzowanego
pomiar skr ęcalno ści w polarymetrze α
skr ęcalno ść właściwawielko ść charakteryzuj ąca zwi ązki optycznie czynne
[α]D =α
l dα - odczyt ( °)l - długo ść rurki (dm)d - stęŜenie roztworu lub g ęstość cieczy (g/cm 3)
Izomeria optyczna
Konfiguracja: uło Ŝenie podstawników wokół
asymetrycznego atomu C
Konfiguracja absolutna: R i Swg reguł pierwsze ństwa podstawników
Konfiguracja wzgl ędna: D i Lw porównaniu z wzorcem: aldehyd glicerynowy
CH2OH
OHH
CHO
NazewnictwoStereoizomeria: izomeria geometryczna alkeny i cykl oalkany
Regu ły pierwsze ństwa grup w/g Cahna, Ingolda, Preloga
JeŜeli starszeństwa nie moŜna ustalić w/g tej reguły, porównuje się kolejne atomy w podstawnikach w/g tego samego kryterium
-CH2CH2F > -CH2CH3-OCH3 > -OH -CH2CH2Cl > -CH2CH2CH3
Atom o wyŜszej liczbie atomowej ma pierwszeństwo przed atomem o niŜszej liczbie atomowej
-OH > -CH3 -Br > -C6H5 -NH2 > -CH2CH2CH3-Br> -Cl
Atomy połączone wiązaniami wielokrotnymi rozpatruje się jako równowaŜne odpowiedniej liczbie atomów związanych wiązaniami pojedynczymi
C C
H H
H C N
N
N
C
CC CH C C
C
C
H
C
C
C NC C
C C
H
H H
C O
H
C O
O C
H
-COOH > -CHO > CHOH > CH3
Starszeństwo w szeregu utlenienia
Izomeria optyczna
Konfiguracja absolutna R / S
3
1
4
2
1
3 2
4
S
niezgodnie z ruchem wskazówek zegara
R
zgodnie z ruchem wskazówek zegara 1 > 2 > 3 > 4
HCH2CH3
ClCH3
CH2CH3
Cl CH3
H
11
22
33
R
zgodnie z ruchem wskazówek zegara
S
niezgodnie z ruchem wskazówek zegara
2-chlorobutan
C l
Starszeństwo podstawników: 1. chlor Cl
2. grupa etylowa CH2CH3
3. grupa metylowa CH3
4. wodór H
Izomeria optyczna
CHO
HO CH2OH
H
OH
CHOH
HOH2C
aldehyd glicerynowy
R (+) S (-)[α]D = 8.7° [α]D = - 8.7°
alanina
NH2
COOH
H3C
H HCOOH
NH2H3C
R (-) S (+)[α]D = - 8.5° [α]D = 8.5°
HO
HCOOH
CH3OH
COOH
H3C
H
R (-) (-3.8 °) S (+) (3.8 °)
kwas mlekowyrównomolowa mieszanina
enancjomerów
mieszanina racemiczna = racemat
optycznie nieczynna

Izomeria optyczna
konfiguracja wzgl ędnaodnoszona do aldehydu glicerynowego
projekcja Fischera = wzory Fischeracząsteczka rzutowana na płaszczyzn ę
Izomeria optyczna
zasady ustawiania cz ąsteczki z 1 chiralnym C do rzutowaniaw projekcji Fischera = wzory Fischera
• rysujemy krzy Ŝ: C* jest na przeci ęciu linii, podstawniki przy linii pionowej s ą pod, a przy poziomej nad płaszczyzn ą
• najdłu Ŝszy łańcuch w ęglowy ustawiamy pionowo• bardziej utleniony koniec ła ńcucha piszemy u góry• wpisujemy pozostałe podstawniki• jeśli grupa OH lub odpowiadaj ąca jej (np., X, NH 2) jest
po prawej stronie, to zwi ązek nale Ŝy do szeregu D,po lewej stronie – do szeregu L
kwas (R)-mlekowy kwas D-mlekowy
CHO
HO CH2OH
H
2
3 11
2
3SR
aldehyd glicerynowy
zgodnie z ruchem wskazówek zegara
niezgodnie z ruchem wskazówek zegara
OH
CHO
HOH2C
H
1. OH2. CHO3. CH2OH4. H
Izomeria optyczna
CHO
*CHOH
CH2OH
CHO
CH3
OHH
CH2OHCH2OH
OHH
CHO
wzór Fischera
CH2OH
HOH
CHO
Izomeria optyczna
dwa centra asymetriiwzory przestrzenne
2R, 3S 2S, 3R 2R, 3R 2S, 3S
Izomeria optyczna
projekcja Fischera = wzory Fischeradla zwi ązków z dwoma centrami asymetrii
• pionow ą prost ą przecinamy tyloma poziomymi, ile jest
chiralnych atomów w ęgla (C*)
• nadal aktualne s ą wszystkie poprzednie zało Ŝenia
• jeśli grupa OH lub odpowiadaj ąca jej (np., X, NH 2)
przy najni Ŝej poło Ŝonym C* jest po prawej stronie, to
związek nale Ŝy do szeregu D
Izomeria optyczna
dwa centra asymetriiwzory Fischera
enancjomery enancjomery
?diastereoizomery
H
H
CH2OH
OH
CHO
OH HOH
OH
CH2OH
H
CHO
H
OH
CH2OH
H
CHO
OH
A B C D
B+C, A+C, B+D, A+D
HOH
H
CH2OH
OH
CHO

Izomeria optyczna
dwa centra asymetrii i płaszczyzna symetrii
enancjomery enancjomeryH
HOH
OH
COOH
COOH
HOH
OHH
COOH
COOH
H
OHH
OH
COOH
COOH
HOH
HOH
COOH
COOH
2R, 3R 2S, 3S 2R, 3S 2S, 3R
płaszczyzna symetrii
=
kwas L-winowy kwas D-winowy kwas mezowinowy
forma mezo nieczynna optyczniezwiązek achiralny mimo C*
A B C
enancjomery
diastereoizomery: A+C, B+C
Izomeria optyczna
diastereoizomery• izomery optyczne nie b ędące odbiciami lustrzanymi • część atomów ma tak ą samą konfiguracj ę,
ale przynajmniej jeden inn ą
właściwo ści diastereoizomerów• mają róŜną skręcalno ść właściwą• mają róŜne wła ściwo ści fizyczne (np. temp. topnienia, rozpuszczalno ść)
• moŜna je rozdzieli ć
ilość izomerów optycznych zwi ązku o ncentrach chiralnych
2n
Przekształcanie wzorów tetraedrycznych we wzory Fischera
• ustawi ć cząsteczk ę tak, by wi ązania poziome były
skierowane przed, a pionowe za płaszczyzn ę• rozpłaszczy ć cząsteczk ę
Radawykona ć model tetraedrycznego (sp 3) atomu w ęgla
z plasteliny i zapałek i ... ćwiczy ć
D czy L ?
płaski karbokation sp 2atak
Przykład reakcji z utworzeniem C*addycja elektrofilowa do C=C
Halogenkichlorki, bromki, jodki , fluorki
Znaczenie: rozpuszczalnikianestetykiczynniki chłodz ącepestycydy
X = F, Cl, Br, Jwiązanie spolaryzowane:
związki polarne, ale nierozpuszczalne w wodzie
Halogenki alkilowewłaściwo ści fizyczne
• gęstość: monochlorki < 1 g/cm 3
bromki 1.2-14 g/cm 3
jodki 1.4-1.9 g/cm 3
di- i tripochodne – odpowiednio wy Ŝsze gęstości
• temperatura wrzenia: znacznie wy Ŝsza ni Ŝ odpowiednich w ęglowodorów
• chlorek propylu 47 °C• bromek propylu 71 °C• jodek propylu 102 °C

Nazewnictwo halogenków alkilowych
halogenek alkilu = halogenek alkilowy = halogenoalk an
cis-4,5-dichloro-1-(2-chloroetenylo)-2,4-dimetylocykloheksen
Halogenki alkilowe – metody otrzymywaniaaddycja elektrofilowa do alkenów
addycja halogenu
dihalogenek alkilowy
addycja halogenowodoru
monohalogenek alkilowy
zgodnie z reguł ąMarkownikowa tworzy si ę
halogenek o wy Ŝszej rzędowo ści
HBr
Br
aleHBr
ROORBr
Halogenki alkilowe – metody otrzymywaniaaddycja wolnorodnikowa HBr do alkenów
niezgodna z reguł ą Markownikowa
Br.
atakuje pier ścien od góry lub od dołu, powstaje trwalszy karborodnik 3º, a nast ępnie halogenek o ni Ŝszej rz ędowo ści
HBrROOR
wolnorodnikowa substytucja w alkanach
Niedogodno ść reakcji: mieszanina produktów, wymagane wydzielanie
Br CH3
Halogenki alkilowe – metody otrzymywaniasubstytucja nukleofilowa w alkoholach
reakcja alkoholi z halogenowodorami
reakcja alkoholi z halogenkami tribromkiem/trichlorkiem fosforu lub chlorkiem tionylu (alkohole 1°i 2°)
Reakcje halogenków alkilowychprowadz ące do alkoholi
substytucja nukleofilowa
alkohole1°
alkohole3°
Substytucja nukleofilowa
odczynniki nukleofilowe: zasadowo ść a nukleofilowo ść
H2O lub OH – (mocna zasada, średni nukeofil)
X – (słaba zasada, mocny nukeofil)
NH3, RNH2, R2NH, R3N
CN –
HC C –
ROH lub CH 3O –
CH3COO –
nukleofil: zawiera atom bogaty w elektrony, który mo Ŝe tworzy ćwiązanie oddaj ąc par ę elektronów atomowi w ęgla ubogiemu w elektrony
moŜe mieć ładunek ujemny

Substytucja nukleofilowa inne wła ściwo ści halogenków alkilowych
synteza nitryli
synteza amin
synteza eterów
synteza alkinówalkilowanie
acetylenków
Reakcje halogenków alkilowychX = F, Cl, Br, J
atak nukleofila
Nu: lub Nu: -
Nu -nukleofil
wchodz ący
X - nukleofil odchodz ący
(opuszczaj ący)
substytucja nukleofilowa S N
eliminacja nukleofilowa E
dwie reakcje konkurencyjne, zachodz ące w tym samym środowisku, ale z ró Ŝną łatwo ścią i szybko ścią;
zwykle powstaj ą dwa produkty: substytucji i eliminacji
Substytucja i eliminacja nukleofilowa S N i E
Na proporcje produktów wpływa:
• budowa substratu: 1 ° - łatwiej S N, 3° - łatwiej E
• zasadowo ść i nukleofilowo ść odczynnika Nu, oraz jego st ęŜenie:
im wy Ŝsze te warto ści tym łatwiej E
• temperatura reakcji: im wy Ŝsza tym łatwiej E
• rozpuszczalnik: im bardziej polarny tym łatwiej E
Konkurencyjno ść reakcji S N i E
Substytucja i eliminacja nukleofilowa -przykłady reakcji halogenków
EtOH
Lub KOH,EtOH
Reguła Zajcewa(odwrotna do reguły Markownikowa)
w reakcji eliminacji oderwaniu ulega wodór od s ąsiedniego najmniej uwodornionego atomu w ęgla
CH3CH2CH=CH2
Substytucja i eliminacja nukleofilowa -konkurencyjno ść
SN E
Substytucja i eliminacja nukleofilowa -Konkurencyjno ść reakcji
Słaby Nu – przewaga S N

Mechanizm substytucji nukleofilowejatak Nu: na centrum
elektrofilowe od strony przeciwnej
• jeden etap• dwie cz ąsteczki
płaski stanprzej ściowy:
obydwie grupy poł ączone z C
odej ście grupy opuszczaj ącej
SN2
Substytucja nukleofilowa dwucz ąsteczkowa SN2
inwersja=odwróceniekonfiguracji R S
reakcja dwucz ąsteczkowa = reakcja drugiego rz ęduszybko ść reakcji zale Ŝy od st ęŜenia dwu reaguj ących substancji
szybko ść reakcji = k [RX] [Nu:]
halogenek 2 °
halogenek 1° łatwiej ni Ŝ 2°
metylowy >1 °>2°>3° (nie reaguj ą)
z produktu optycznie czynnego powstaje mieszanina e nancjomerów
reakcja z racemizacj ą
Mechanizm ??? – inny ni Ŝ SN2
Mechanizm substytucji nukleofilowej
halogenek 3 °brak miejsca na atak od strony
przeciwnej
3°C*
bromek tert-butylu tert-butanol
SN1
etap decyduj ącyo szybko ści
reakcji ?
ile cz ąsteczek bierze w nim
udział?
etap I wolny etap II szybki
jedna,tylko
substrat
ETAP I odszczepienie grupy
opuszczaj ącej:utworzenie karbokationu
ETAP II atak Nu:
wchodz ącegona karbokation:
przył ączenie
Substytucja nukleofilowa jednocz ąsteczkowa SN1
Substytucja nukleofilowa jednocz ąsteczkowa SN1
płaski karbokation
reakcja jednocz ąsteczkowa = reakcja pierwszego rz ęduszybko ść reakcji zale Ŝy od st ęŜenia jednego z substratów
szybko ść reakcji = k [RX]
reakcja dwuetapowao szybko ści reakcji decyduje etap najwolniejszy
racemizacja (C*)R R + SS S + R
3°>2°>1°
Reakcje eliminacji nukleofilowej –mechanizm analogiczny jak mechanizm S N
Reakcja E2
Reakcja E1
Substytucja i eliminacja nukleofilowa -przewidywanie mechanizmu i produktów
substrat 1 ° SN2 i E2 ( łatwiej S N)
substrat 3 ° SN1 i E1 ( łatwiej E)
substrat 2 ° mechanizm zale Ŝy od innych czynników:
zasadowo ść i nukleofilowo ść odczynnika Nu, st ęŜenie, temperatura

Dihalogenki alkilowesynteza alkenów synteza alkinów
synteza halogenków winylowych
Halogenki aromatyczne (arylowe)syntezasubstytucja elektrofilowa
właściwo ścisubstytucja nukleofilowa, ani S N1 ani SN2
Właściwo ści halogenków alkilowych i arylowychTworzenie zwi ązków Grignarda (magnezoorganicznych)
podatne na atak kwasów i elektrofili
Właściwo ści zwi ązków Grignarda
rozkład pod wpływem: wody, kwasów, alkoholi, amin
Halogenki R-X Alkohole R-OH
1°
2°
3°
Rzędowo ść alkoholi= rzędowo ści atomu w ęgla poł ączonego z OH
CH3OH ?
NazewnictwoStarszeństwo grup funkcyjnych wyst ępuj ących
w przyrostkach:R4N
+Cl > R-COOH > R-SO3H > R-COO-Na+
> R-COOR’ > R-COCl > R-CONH2 > R-CN
> R-CHO > R-CO-R’
> R-OH > Ar-OH > R-NH2
Tylko jedna „najstarsza” grupa funkcyjna mo Ŝe być w przyrostku !!! Jest to grupa decyduj ąca o klasyfikacji zwi ązku
alkanol, alkano diolalkohol alkilowy
nazwa systematycznanazwa zwyczajowa
Alkohole
podstawnikprzedrostek hydroksy
wiązanie spolaryzowane:związki polarne,
C1-C6 rozpuszczalne w wodzie
AlkoholeWłaściwo ści fizyczne
temperatura wrzenia
wiązanie wodorowe
Otrzymywanie alkoholiMetanol
Etanolfermentacja
metoda uniwersalna
elektrofilowe przył ączenie wody do etylenu
biopaliwo
trucizna!! LD50 10.6 g/kg

Otrzymywanie alkoholi z alkenów
reakcja hydroborowania
niezgodnie z reguł ąMarkownikowa:
alkohol o ni Ŝszej rz ędowo ści
H2O2,OH-
CH3CH2OH
reakcja addycji elektrofilowej wody
zgodnie z reguł ą Markownikowa: alkohol o wy Ŝszej rz ędowo ści
Otrzymywanie alkoholiz halogenków w reakcjach substytucji nukleofilowej
3°
1° i 2°
Otrzymywanie alkoholi redukcja zwi ązków karbonylowych
związek karbonylowy-C=O grupa karbonylowa
alkohol
[H]=reduktor
odczynniki redukuj ące: NaBH 4, LiAlH 4 (wodorki) lub H 2/ /kat.
redukcja aldehydów – 1 ° alkohole
redukcja ketonów – 2 ° alkohole
selektywna redukcja grupy karbonylowej
Otrzymywanie alkoholi
redukcja kwasów karboksylowych 1°alkohol
dwa alkohole: 1°(z cz ęści kwasowej) i inny redukcja estrów
Otrzymywanie alkoholi 1, 2 i 3°addycja zwi ązków Grignarda do zwi ązków
karbonylowych
Reakcje (wła ściwo ści chemiczne) alkoholi
Alkohole jako kwasy - rozerwanie wi ązania O–H
anionalkoksylowy
(CH3)3COH < CH3CH2OH < CH3OH < HOH < CF3OH
szeregi kwasowo ści
RH < NH3 < HC CH < CH3OH < HOH
rozerwanie wiązania
rozerwanie wiązania
Reakcje alkoholirozerwanie wi ązania O – H
Alkohole jako kwasy: alkoholany
reakcja z metalami (tylko litowce: K, Na)
reakcja z mocnymi zasadami
reakcje tworzenia estrów

reakcje utleniania alkoholi
Reakcje alkoholirozerwanie wi ązania O – H
1°alkohol – kwas karboksylowy [O]czynniki utleniaj ące
do kwasów: KMnO 4, H+
Na2Cr2O7 , H+
CrO3/H2SO4(odczynnik Jonesa)
1°alkohol – aldehyd reakcja wymaga specjalnych warunków (np. PCC)
2°alkohol – keton 3°alkohol – nie zachodzi
Reakcje alkoholirozerwanie wi ązania C – O
substytucja nukleofilowa halogenem
HCl, HBr
3° >2° >1°
SOCl2, PBr 3
protonowanie alkoholu: pierwszy etap reakcji elimina cji wody z alkoholi
Reakcje alkoholirozerwanie wi ązania C – O
Alkohole jako zasadyprotonowanie alkoholu: pierwszy etap reakcji S N1, SN2 i E
jonoksoniowy
3° SN1
1° S N2
mechanizm dehydratacji alkoholi E1 H2O + H+ H3O+
dehydratacja alkoholi - odszczepienie H 2O
kwas siarkowy
Reakcje alkoholirozerwanie wi ązania C – O
reaktywno ść alkoholi w reakcji eliminacji
3° >2° >1°
reguła Zajcewa
75%, 140°
60%, 100°
20%, 90°
Otrzymywanie dioli z alkenów reakcje utleniania
KMnO 4
reakcja rozszczepienia dioli wicynalnych
Reakcje dioli
KMnO4
H+
Diole Fenole
Właściwo ści fizyczne fenoli
• wysokowrz ące ciecze lub ciała stałe• trudno rozpuszczalne w wodzie
(poza fenolem 9g w 100g)
Fenole naturalne – z olejków eterycznych

Otrzymywanie fenoli
Otrzymywanie fenoli
Reakcje fenoli
utlenianie fenoli do chinonów
Reakcje fenoli
stabilizacja anionu fenolanowego przez rezonans
Kwasowo ść fenoli:nitrofenol > fenol > metylofenol > woda > alkohol
kwasowo ść fenoli
wzrostkwasowo ści
obni Ŝeniekwasowo ści
wpływ podstawnika na
kwasowo ść
Reakcje fenolisubstytucja elektrofilowa w fenolachgrupa OH bardzo silnie aktywuje pier ścień aromatyczny
nitrowanie bromowanie
alkilowanie
• CH3 słabo aktywuje o- i p-• OH silnie aktywuje o- i p-
EteryR – O – R (R – O – H, H – O – H)
Epoksydy = oksirany
Właściwo ści fizyczne eterów
• mało polarne• temperatura wrzenia:
heptan 98 °Ceter metylowo-pentylowy 100 °Cheksanol 157 °C
Otrzymywanie eterówsynteza z alkoholi
synteza Williamsona (reakcja S N halogenków 1 ° i 2° )
Reakcje eterów - bierne chemicznie
rozszczepienie
Tiole
Otrzymywanie tioli
z halogenków alkilowych - S N2
wodorosiarczek sodu
Właściwo ści tioli
disulfid
utlenianie