myopathies des ceintures - association.gens.free.frassociation.gens.free.fr/neurologia/emc...
TRANSCRIPT

Myopathies des ceintures [17-175-C-10] - Doi : 10.1016/S0246-0378(10)53745-X
G. Solé : Praticien hospitalier
Introduction Le terme de myopathie des ceintures est longtemps resté décrié, en raison de l'hétérogénéité des pathologies que l'on peut classer sous cette dénomination. Certains spécialistes ont voulu abandonner ce concept il y a quelques années. On lui préfère actuellement le terme plus restrictif de dystrophie des ceintures. Ces myopathies ont en commun la présence d'un déficit musculaire des ceintures associé à des lésions histologiques dites « dystrophiques », c'est-à-dire associant nécrose et régénération. On distingue les formes autosomiques dominantes, au nombre de sept actuellement, des formes récessives, au nombre de 15. Devant la découverte permanente de nouveaux gènes, il est fondamental de rappeler l'importance des aspects cliniques qui peuvent orienter les explorations anatomopathologiques et génétiques. Après un bref rappel historique, nous étudions d'abord les éléments communs à toutes les dystrophies des ceintures, puis les caractéristiques cliniques et paracliniques propres à chacune de ces pathologies. Dans un troisième temps, nous proposons une stratégie de diagnostic étiologique.
Historique
De la description clinique aux aspects biopsiques
Les premières descriptions de myopathie des ceintures remontent à la fin du XIXe siècle. Leyden fut le premier à décrire, en 1876, une forme de dystrophie musculaire plus bénigne que la myopathie de Duchenne [1]. Dans la première moitié du XXe siècle, plusieurs tentatives de classification se sont succédées et le concept de myopathie des ceintures a été fortement débattu [2]. Après la Seconde Guerre mondiale, le diagnostic des pathologies neuromusculaires connaît un nouvel essor grâce au développement des techniques d'électrophysiologie et surtout de microscopie optique puis électronique. C'est Stevenson qui introduisit, en 1953, le terme de dystrophie des ceintures autosomiques (autosomal limb-girdle muscular dystrophy [LGMD]) à partir d'une série de 51 familles irlandaises [3]. La classique série de Walton et Nattrass fut la première à individualiser clairement les myopathies des ceintures autosomiques récessives [4]. En raison de l'hétérogénéité clinique et des progrès des techniques histologiques, la nécessité de conserver ce terme de dystrophie des ceintures a été longuement discutée. Dans les années 1980, le terme plus général de « syndrome des ceintures » était alors communément utilisé [5].
Ère de la biologie moléculaire : définition et classification actuelle
Les progrès de la biologie moléculaire ont permis d'individualiser les différentes dystrophies des ceintures et de clarifier en partie leur hétérogénéité. Ces progrès moléculaires ont rendu nécessaire l'établissement d'une classification. En 1995, une définition de travail [6] et une classification des dystrophies des ceintures [7] ont été proposées. Les dystrophies des ceintures se définissent par un déficit moteur proximal, épargnant les muscles distaux, faciaux et oculomoteurs à la phase précoce de la pathologie et pour lequel la biopsie individualise des

lésions dystrophiques. La classification repose sur le mode de transmission : LGMD1 pour les formes autosomiques dominantes et LGMD2 pour les formes autosomiques récessives. Chaque pathologie est ensuite désignée par une lettre unique, attribuée principalement selon l'ordre chronologique des publications. Le Tableau 1 récapitule la classification actualisée en 2007 [8]. Les principales protéines impliquées dans les dystrophies musculaires des ceintures sont schématisées dans la Figure 1.
L'orientation diagnostique entre ces diverses formes se fait à partir d'un faisceau d'arguments : hérédité, présentation clinique, données anatomopathologiques, étude des protéines musculaires sur lame et en western blot.
Caractéristiques communes Toutes les dystrophies des ceintures présentent des aspects cliniques et paracliniques communs.
Aspects cliniques
L'âge de début est variable en fonction du type de dystrophie des ceintures. Les troubles débutent généralement à la ceinture pelvienne par une marche dandinante, éventuellement associée à des chutes. Les patients ont des difficultés à monter des escaliers et doivent s'aider de la rampe. Le relevé est myopathique avec un signe de Gowers. L'aggravation est presque toujours lentement progressive. La perte de la marche est inconstante. L'atteinte de la ceinture scapulaire se manifeste initialement par une difficulté à porter des charges lourdes. La musculature axiale peut être touchée dans certaines formes. Si cette atteinte débute avant la fin de la croissance, elle peut être responsable d'une scoliose. L'étude de la force musculaire analytique confirme le déficit prédominant aux ceintures et permet de rechercher une atteinte d'autres muscles (comme les muscles distaux) qui peut orienter vers une étiologie spécifique. On recherche aussi une pseudohypertrophie des mollets ou de la langue du type de celle observée dans les maladies de Duchenne et de Becker. Comme dans la majorité des atteintes musculaires primitives, on ne retrouve pas de déficit sensitif ni d'atteinte centrale, sauf exceptions. Il est fondamental de rechercher des signes associés comme un déficit distal, une atteinte de la musculature bulbaire, des rétractions ostéotendineuses ou des troubles cognitifs.
Aspects paracliniques
Trois examens principaux peuvent orienter le diagnostic. Le dosage des créatine-kinases (CK) est couramment réalisé. Leur élévation témoigne d'une lésion évolutive de la fibre musculaire. Elles sont fréquemment plus élevées en début de maladie. Le taux peut se normaliser par la suite en raison de la dégénérescence fibroadipeuse du tissu musculaire. L'imagerie musculaire est aussi souvent utile. Le scanner et l'imagerie par résonance magnétique (IRM) ont un apport généralement identique dans le cadre des dystrophies. On recherche la présence d'une amyotrophie et/ou d'une dégénérescence graisseuse et on analyse sa topographie. En scanner, le muscle normal présente une densité homogène, alors que le muscle pathologique apparaît hypodense et hétérogène (souvent marbré). Les aspects IRM sont globalement proches et dépendent des séquences utilisées. Le scanner est plus volontiers utilisé en raison de sa rapidité d'exécution et de sa meilleure disponibilité. L'IRM est en revanche bien plus utile s'il existe une hésitation entre myopathie héréditaire et myopathie inflammatoire. La topographie de l'atteinte peut orienter le diagnostic étiologique. L'électromyogramme (EMG) a une place

plus limitée. Il confirme généralement l'existence de signes myogènes, mais peut parfois être pris en défaut. Il est utile pour identifier certains diagnostics différentiels : dystrophie myotonique de type 2, atteinte neurogène, etc.
Aspects anatomopathologiques
La biopsie musculaire reste l'examen de choix afin de parvenir au diagnostic étiologique. Par opposition aux myopathies congénitales où il existe une anomalie du développement de la fibre musculaire, les dystrophies des ceintures se caractérisent par l'existence d'une nécrose des fibres matures. Afin de maintenir la fonction tissulaire, cette nécrose est suivie par une régénération. Ces deux phénomènes se succèdent en permanence et leur association sur la biopsie définit la lésion dystrophique [9].
Nécrose
Quand une lésion se produit sur la membrane de la fibre musculaire (sarcolemme), elle entraîne une entrée de calcium conduisant à une contraction anormalement soutenue des myofibrilles (unité contractile des fibres musculaires) puis à leur destruction. La région lésée est détruite par des protéases intrinsèques à la fibre musculaire. Les noyaux subissent alors une caryolyse complète. Sur le plan histologique, ces phénomènes se traduisent par la disparition des noyaux et une modification de la coloration de la fibre musculaire par l'hémalun-éosine : coloration pâle dans les régions nécrotiques de la fibre lésée (fibres dites « hyalines »), sombre dans les régions voisines où se situent les myofibrilles en hypercontraction [10]. La membrane lésée laisse échapper de la fibre une partie de ses composants et en particulier les CK.
Régénération
La régénération débute par l'activation et la multiplication des cellules satellites de la fibre musculaire. Ces cellules prolifèrent et forment des myoblastes qui fusionnent progressivement pour former un myotube [11]. Ces cellules sont caractérisées par des noyaux en position centrale et une basophilie liée à leur richesse en acide ribonucléique (ARN). Au fur et à mesure de l'enrichissement en myofibrilles, les noyaux sont repoussés en périphérie. Les myotubes prennent progressivement les caractéristiques de myofibres matures [10]. La persistance de noyaux centralisés est un bon marqueur de fibres régénérées.
Description habituelle
Dans les dystrophies des ceintures, la biopsie musculaire montre l'association caractéristique nécrose/régénération. Les fibres ont un diamètre variable et présentent des internalisations nucléaires. Des fibres lobulées peuvent être observées. Un infiltrat inflammatoire peut parfois être présent et faire poser par erreur le diagnostic de myosite. Au début de la maladie, la régénération compense la nécrose et le muscle garde une bonne trophicité. Le nombre de mitoses que peut subir une cellule satellite étant limité, ce phénomène s'épuise [11]. À un stade tardif, les fibres musculaires sont donc remplacées par de la fibrose et du tissu adipeux.
Dystrophies musculaires des ceintures autosomiques récessives

Dans la classification actuelle, les dystrophies des ceintures autosomiques récessives sont dénommées LGMD2. Il s'agit d'un groupe hétérogène pour lequel 15 loci sont actuellement décrits (dont seulement 14 sont présents dans l'actuelle classification de l'European Federation of Neurological Societies [EFNS]). Certaines formes sont fréquentes, alors que d'autres n'ont été décrites que sur une seule famille (Tableau 2).
Calpaïnopathie (LGMD2A)
Il s'agit de la première forme de dystrophie des ceintures autosomique récessive décrite. Pour de nombreux auteurs et en particulier pour Fardeau, à qui revient la description princeps [12], il s'agit de la forme la plus pure de dystrophie des ceintures. La calpaïne 3 (MIM 114240) appartient à une famille de protéases non lysosomales calcium-dépendantes. Son rôle ainsi que ses substrats restent encore mal connus. Elle intervient dans la dégradation des myofibrilles et des protéines du cytosquelette [13].
Épidémiologie
L'étude de la prévalence des différentes dystrophies des ceintures est difficile. L'existence d'isolats (île de la Réunion, Pays basque, etc.) peut fortement biaiser les estimations. Selon les études, les calpaïnopathies représentent entre 6 % et 22 % des dystrophies des ceintures [14, 15, 16, 17], dont elles sont généralement considérées comme la première étiologie.
Présentation clinique
La calpaïnopathie primaire a d'abord été décrite grâce aux études moléculaires menées sur une communauté réunionnaise [12]. La faiblesse musculaire débute entre la première et la troisième décennie, typiquement entre 10 et 15 ans [18, 19, 20]. Les premiers territoires touchés se situent au niveau de la ceinture pelvienne : grand fessier et adducteurs de cuisse. Il faut noter la préservation fréquente du quadriceps aux stades précoces. À la ceinture scapulaire, les muscles suivants sont atteints : grand dorsal, grand pectoral, grand dentelé, rhomboïde et biceps brachial (Figure 2A), l'atteinte du deltoïde, du triceps et des radiaux étant tardive. Il existe fréquemment un décollement des omoplates (Figure 2B). La dissociation entre l'amyotrophie du biceps et la conservation du triceps est typique (Figure 2A). L'aggravation est lentement progressive mais variable, y compris à l'intérieur d'une même famille [18]. Il peut apparaître des rétractions des tendons d'Achille généralement peu sévères. La perte de la marche se situe entre la troisième et la quatrième décennie. Il faut noter l'absence de pseudohypertrophie des mollets ou de macroglossie contrairement au phénotype Duchenne/Becker présent dans de nombreuses dystrophies des ceintures. Il peut exister une scoliose modérée et généralement non chirurgicale. La face est classiquement préservée. Il n'y a pas d'atteinte cardiaque ni de retard intellectuel. L'insuffisance respiratoire est peu fréquente. La durée de vie est proche de la normale [21].
Éléments paracliniques
Créatine-kinases et autres éléments biologiques
Au début de la maladie, les CK sont très élevées (jusqu'à 20 fois la normale) puis décroissent dans un second temps. Il peut exister une hyperéosinophilie sanguine.
Imagerie musculaire

L'imagerie musculaire est très intéressante au début de la maladie en raison de la sélectivité de l'atteinte. Il existe typiquement une atteinte précoce de la loge postérieure de cuisse avec préservation de la loge antérieure (Figure 3) [22]. Il faut noter que les muscles restant le plus longtemps épargnés sont : le vaste externe, le sartorius et le gracile [23]. Au niveau jambier, on peut observer une atteinte du soléaire et du jumeau interne alors que le jumeau externe est préservé [22]. À un stade plus évolué, l'atteinte du biceps brachial avec préservation du triceps est évocatrice (Figure 3).
Anatomopathologie
Avec les techniques habituelles, on observe des lésions typiques de dystrophie musculaire, avec une formule de nécrose/régénération. Bien que non spécifique, la présence de nombreuses fibres lobulées est évocatrice. On peut parfois observer un infiltrat inflammatoire à éosinophile pouvant faire errer le diagnostic [24]. Il est impossible d'utiliser des techniques d'immunomarquage sur lame pour étudier la calpaïne 3. Le western blot montre une diminution de la calpaïne 3 dans 80 % des cas [25]. Cette diminution peut toutefois être observée dans certaines autres dystrophies telles que les dysferlinopathies et titinopathies [26, 27]. On parle alors de déficit secondaire en calpaïne 3.
Génétique moléculaire
Le locus a été identifié en 1991 grâce aux études menées sur l'île de la Réunion par Fardeau [13, 28]. Les premières mutations du gène codant la calpaïne 3, CAPN3, ont été identifiées en 1995 [29]. D'autres isolats ont été décrits, en particulier dans le Pays basque [20]. La recherche directe de mutations est réalisée en première intention. Toutefois, 20 % à 25 % des patients ne présentent qu'une seule mutation [30]. Dans ce cas, une analyse de l'ARN messager peut être réalisée. Les mutations retrouvées sont extrêmement variées en dehors des mutations privées identifiées dans certains isolats : sud de l'île de la Réunion, Pays basque [12, 20], etc. Sur les larges séries, environ les deux tiers des patients mutés correspondent au phénotype décrit plus haut. Dix pour cent présentent un début précoce pouvant initialement faire évoquer une maladie de Duchenne, 3 % un déficit de début distal et 6 % une hypercréatine-kinasémie isolée [31].
À retenir LGMD2A : calpaïnopathie Origine géographique : ubiquitaire, île de la Réunion, Pays basque Clinique
• début : 2e décennie
• prédominance : loge postérieure de cuisse
• pas d'atteinte cardiaque
• peu d'atteinte respiratoire
• perte de la marche : 4e décennie

Paraclinique
• imagerie : atteinte loge postérieure de cuisse
• biopsie : infiltrat inflammatoire possible, immunomarquages normaux
• western blot : utile, mais parfois trompeur
• biologie moléculaire : indispensable mais difficile
Dysferlinopathie (LGMD2B)
La première description de dysferlinopathie a été faite sur une large famille palestinienne de dystrophie des ceintures. Il est maintenant bien établi qu'il existe une importante variabilité phénotypique dans cette pathologie [32]. Le déficit en dysferline peut aussi être responsable d'une présentation distale appelée myopathie de Miyoshi. La dysferline (MIM 603009) est une protéine de la membrane plasmique de 230 kDa. Son expression est ubiquitaire, mais plus forte dans le muscle squelettique, le muscle cardiaque et le rein. La dysferline joue un rôle dans la machinerie de réparation des membranes après lésion [33].
Épidémiologie
Les dysferlinopathies semblent un peu moins fréquentes que les calpaïnopathies. Dans les séries récentes, leur proportion évolue entre 1 % et 20 % des dystrophies des ceintures [14, 15, 16, 17].
Présentation clinique
Le tableau clinique est hétérogène [34, 35, 36]. Les premiers signes apparaissent généralement aux alentours de 20 ans, bien que des formes tardives ou au contraire très précoces aient été décrites [37]. L'atteinte débute aux membres inférieurs. Elle peut être proximale (phénotype LGMD) ou distale (phénotype Miyoshi). Dans les formes distales, les premiers muscles touchés se situent dans la loge postérieure de jambe : gastrocnémien et soléaire conduisant à une aréflexie achilléenne précoce [21]. Dans les formes de début proximal, le déficit débute par le grand fessier, le quadriceps et le psoas-iliaque. La ceinture scapulaire est habituellement atteinte dans un second temps. Même dans ces formes proximales, l'amyotrophie de la loge postérieure de jambe est très évocatrice de dysferlinopathie (Figure 4). Il existe une troisième présentation avec atteinte précoce du muscle tibial antérieur et préservation des muscles de la loge antéroexterne [34]. Dans les trois formes, il existe une importante variabilité interfamiliale et intrafamiliale de la topographie et de la chronologie de l'atteinte. Il faut noter qu'un aspect en « boule » du biceps est assez souvent retrouvé dans les dysferlinopathies. L'évolution est généralement lente. Parfois, il peut exister des aggravations plus aiguës, associées à la présence d'un infiltrat inflammatoire sur la biopsie. Ces formes « inflammatoires » ont souvent été traitées (sans succès) comme des polymyosites. Il n'y a pas d'atteinte axiale, faciale ou pharyngée avant un stade évolué. Il n'est pas décrit d'atteinte cardiaque. L'atteinte respiratoire est tardive.
Éléments paracliniques
Créatine-kinases

Le taux de CK est très élevé : généralement de 30 à 200 fois la normale [32]. Il s'agit de la LGMD où les CK sont les plus élevées.
Imagerie
Dans la myopathie distale de Miyoshi, il existe une atteinte des adducteurs, du soléaire et des gastrocnémiens, avec une relative préservation du sartorius, du gracile et du compartiment antérieur de la jambe [38]. En revanche, il n'existe pas de travaux de grande ampleur étudiant l'imagerie musculaire dans les deux autres phénotypes.
Anatomopathologie
En dehors des lésions typiques de dystrophie musculaire, les immunomarquages sur lame montrent une absence ou une diminution de la fixation de l'anticorps antidysferline sur la membrane [39]. Ce déficit doit être confirmé en western blot soit sur le muscle, soit sur les leucocytes. Il existe couramment un infiltrat de cellules mononucléées dans l'endomysium et autour des vaisseaux, pouvant faire porter par erreur le diagnostic de myopathie inflammatoire [40]. Contrairement à la polymyosite, il n'existe pas d'invasion des fibres musculaires saines par les cellules inflammatoires, et le marquage human leukocyte antigen (HLA) de classe I est bien moins intense. La possibilité de dépôts amyloïdes sarcolemmaux et périmysiaux a été récemment rapportée [41].
Génétique moléculaire
Les premières mutations du gène DYSF codant la dysferline ont été identifiées en 1998 dans le phénotype Miyoshi [42]. Il existe une grande variété de mutations. Quatre-vingt pour cent des patients mutés présentent un phénotype distal postérieur de type Miyoshi, 8 % un phénotype proximal de type LGMD et 6 % une hyperCKémie isolée [21]. Le phénotype distal antérieur est rare.
À retenir LGMD2B : dysferlinopathie Origine géographique : ubiquitaire Clinique
• début : 3e décennie
• déficit distal des membres inférieurs fréquent
• pas d'atteinte cardiaque
• aggravation parfois rapide
• perte de la marche : 4e décennie
Paraclinique
• CK : très élevées

• biopsie : infiltrat inflammatoire possible, immunomarquage de la dysferline anormal
• western blot : dysferline absente ou diminuée
Sarcoglycanopathies (LGMD2C, 2D, 2E et 2F)
Les sarcoglycanopathies constituent un groupe d'affections proches des dystrophinopathies (myopathies de Duchenne et de Becker) sur le plan physiopathologique, clinique et paraclinique. Le principal élément les différenciant est la possibilité d'une atteinte des deux sexes dans les sarcoglycanopathies. Le premier sarcoglycane identifié est la forme ⍺, autrefois appelé adhaline ou dystrophin associated glycoprotein 50 (DAG50, en référence à son poids moléculaire). Le sarcoglycane γ a été isolé par la suite sous le nom de DAG35. Les autres sarcoglycanes ont été identifiés dans un second temps.
Les sarcoglycanopathies sont par définition des pathologies liées à des mutations dans un des quatre gènes codant respectivement le sarcoglycane :
• γ : SGCG pour la LGMD2C ;
• ⍺ : SGCA pour la LGMD2D ;
• β : SGCB pour la LGMD2E ;
• δ : SGCD pour la LGMD2F.
Physiopathologie
Les sous-unités ⍺ (MIM 600119), β (MIM 604286), γ (MIM 608896) et δ (MIM 601411) sont retrouvées dans la membrane de la fibre musculaire où elles s'assemblent pour former un complexe qui semble jouer un rôle important dans la stabilisation des protéines associées à la dystrophine [43].
Présentation clinique
Nous décrivons ici les différentes sarcoglycanopathies dans l'ordre de leur découverte qui correspond aussi à leur fréquence relative.
⍺-sarcoglycanopathie (forme caucasienne) : LGMD2D
Cette forme était auparavant dénommée adhalinopathie primaire. Dans la forme classique, les premiers symptômes sont proches de la myopathie de Duchenne, avec un début légèrement plus tardif (entre 15 et 20 ans) [44, 45]. La principale différence vient du fait que les filles sont autant touchées que les garçons. Il existe initialement des difficultés à la course puis à la montée des escaliers. La marche est dandinante et les mollets hypertrophiés. Il peut exister des rétractions, en particulier du tendon d'Achille. Aux membres supérieurs, l'atteinte des fixateurs de l'omoplate est précoce. Il existe une macroglossie, sans atteinte faciale ni des muscles de la déglutition. Il n'y a pas d'atteinte cognitive contrairement à la maladie de Duchenne [21]. Les complications cardiorespiratoires sont rares. Il existe une importante

variabilité clinique interfamiliale et intrafamiliale, le spectre s'étendant de formes paucisymptomatiques (pseudométaboliques) à des formes de type Duchenne (sans cardiopathie) en passant par des formes de type Becker [44].
γ-sarcoglycanopathie (forme maghrébine) : LGMD2C
Il s'agit de la forme décrite en Tunisie au début des années 1980 par Ben Hamida et al. [46]. Cette pathologie est mondialement distribuée, mais certains isolats ont été identifiés :
• au Maghreb (mutation c.521delT), où il s'agit de la plus fréquente des dystrophies des ceintures ;
• en Europe centrale dans la population tzigane (mutation p.C283Y) [47].
Le phénotype est souvent plus sévère (de type Duchenne) et moins variable que dans l'⍺-sarcoglycanopathie. Ici aussi, les filles sont autant touchées que les garçons, contrairement à la maladie de Duchenne. La pathologie débute par une atteinte proximale des membres inférieurs sans topographie spécifique, avec perte de la marche entre 12 et 15 ans, pseudohypertrophie des mollets et rétractions tendineuses fréquentes. Une scoliose est possible, de même qu'une atteinte cardiaque sous la forme d'une cardiomyopathie dilatée [48]. L'insuffisance respiratoire restrictive est fréquente, mais généralement modérée.
β-sarcoglycanopathie (forme nord-américaine) : LGMD2E
Cette forme a été décrite aux États-Unis dans la population amish, où il existe une mutation privée [49]. Le déficit débute après l'âge de 5 ans par une atteinte proximale pelvienne puis scapulaire d'évolution lente [45].
δ-sarcoglycanopathie (forme brésilienne) : LGMD2F
Il s'agit d'une forme très rare dont la présentation est sévère, de type Duchenne avec atteintes cardiaque et respiratoire [50].
Éléments paracliniques
Il n'existe pas de différence notable sur le plan paraclinique entre toutes les sarcoglycanopathies.
Créatine-kinases
Le taux de CK est élevé, en particulier au début de la maladie [45].
Imagerie
Les anomalies sont proches de celles observées dans les dystrophinopathies. Au niveau de la cuisse, on observe des anomalies aussi bien dans la loge antérieure que postérieure (contrairement à la LGMD2A). Les muscles sartorius et droit interne sont longtemps préservés [23].
Anatomopathologie

Le diagnostic repose sur la normalité de l'immunomarquage de la dystrophine alors que le marquage membranaire des sarcoglycanes est très diminué. Cette étude ne permet pas de préciser avec certitude quel est le sarcoglycane en cause car ils sont généralement tous diminués. Le déficit doit ensuite être confirmé par western blot. Quand le marquage de plusieurs sarcoglycanes est altéré avec la même intensité, seule l'analyse génétique peut affirmer le diagnostic précis.
Génétique moléculaire
Le diagnostic de certitude repose sur l'étude des gènes SGCA, SGCG, SGCB, SGCD respectivement pour les déficits en ⍺-, γ-, β- et δ-sarcoglycane. Toutefois, l'étude génétique est longue et difficile.
À retenir Sarcoglycanopathies Origine géographique : ubiquitaire, Maghreb (LGMD2C), Gitans (LGMD2C), Amish (LGMD2E) Clinique
• pseudo-Duchenne ou pseudo-Becker
• sévérité variable
• atteinte cardiaque possible
Paraclinique
• immunomarquage et western blot anormaux
• biologie moléculaire : difficile
Téléthoninopathie : LGMD2G
Depuis les descriptions initiales de cette pathologie au Brésil [51], il n'existe pas d'autre description d'un déficit primaire en téléthonine. L'interaction entre la téléthonine (MIM 604488) et la titine semble jouer un rôle majeur au cours de la genèse des myofibrilles.
Il existe une importante variabilité inter- et intrafamiliale des cas décrits. Le déficit débute entre 9 et 15 ans par une faiblesse proximale touchant préférentiellement les loges antérieures de cuisse et de jambe. La perte de la marche est inconstante et généralement assez tardive alors qu'une atteinte cardiaque est fréquente.
Les CK sont élevées entre 3 et 30 fois la normale. L'étude du marquage de la téléthonine sur la biopsie musculaire oriente vers le diagnostic. La confirmation est assurée par l'étude du gène TCAP.
Déficit en TRIM32 : LGMD2H

La LGMD2H a été initialement décrite dans la population huttérite du Manitoba (Canada). Depuis la série initiale de Weiler et al. [52], les descriptions cliniques sont restées sommaires [53]. Le tableau débute avant 30 ans par un déficit pelvien et quadricipital peu évolutif parfois associé à des troubles de la conduction intracardiaque [53].
Le taux de CK est élevé jusqu'à 50 fois la limite supérieure de la normale [21]. La biopsie peut montrer, en microscopie électronique, de petites vacuoles à proximité des tubules T [21]. Le diagnostic repose sur l'étude du gène TRIM32.
⍺-dystroglycanopathies : LGMD2I, 2K, 2L, 2M
Il s'agit d'un ensemble de pathologies où il existe une anomalie de glycosylation de l'⍺-
dystroglycane (⍺DG). La forme la plus fréquente (LGMD2I) est due à un déficit en fukutin-related protein (FKRP). Lors de la maturation post-traductionnelle dans l'appareil de Golgi, les chaînes d'oses (monomères de glucides) sont liées aux protéines par des liaisons O-glycosidiques ou N-glycosidiques selon leur site d'ancrage. L'⍺DG (MIM 128239) est une glycoprotéine extracellulaire associée à la membrane et qui interagit avec plusieurs molécules de la matrice extracellulaire, en particulier avec la mérosine. Elle subit une O-glycosylation au cours de son processus de maturation (Figure 5). Un défaut de glycosylation altère les capacités de liaison de l'⍺DG à ses ligands.
Déficit en « fukutin-related protein » : LGMD2I
Cette pathologie a initialement été décrite dans une famille consanguine tunisienne [54]. Depuis, des cas ont été décrits dans le monde entier. La FKRP (MIM 606596) est une glycosyltransférase dont le déficit est responsable d'une glycosylation anormale de l'⍺DG.
Épidémiologie
La LGMD2I est une des causes les plus fréquentes de dystrophie des ceintures. Selon les séries, le déficit en FKRP rend compte de 2,6 % à 6,5 % des LGMD [14, 15, 16, 17].
Présentation clinique
Les troubles débutent à un âge variable : depuis la petite enfance jusqu'à la cinquième décennie. Il faut noter une importante variabilité clinique, aussi bien inter- qu'intrafamiliale [55].
Les formes de début précoce ont une présentation de type Duchenne, avec perte de la marche avant 10 ans [55, 56, 57]. Dans certains cas, il peut aussi exister un retard des acquisitions motrices et une hypotonie dans la première année de vie. La faiblesse musculaire touche préférentiellement les muscles proximaux des membres inférieurs ainsi que les muscles axiaux. Il peut exister une atteinte faciale modérée. Les patients présentent fréquemment une pseudohypertrophie des mollets, de la langue (Figure 6) et du brachioradial.
Le début peut être plus tardif, de type Becker. Il existe là aussi une hypertrophie des mollets et du brachioradial. Une cardiomyopathie dilatée est présente chez environ la moitié des patients

[57, 58]. Il peut aussi exister une insuffisance ventilatoire restrictive précoce [58]. Généralement, il n'y a pas d'atteinte cognitive contrairement à d'autres ⍺-dystroglycanopathies [57].
Éléments paracliniques
CK. Le taux est élevé entre 10 et 30 fois la normale [59].
Imagerie musculaire. Au niveau de la ceinture pelvienne, on observe une atteinte préférentielle des adducteurs et des muscles postérieurs de cuisse. Toutefois, le quadriceps est lui aussi touché et de façon nettement plus précoce que dans les calpaïnopathies [60]. Il existe par ailleurs une hypertrophie du sartorius et du gracile [23].
Anatomopathologie. En dehors des lésions dystrophiques habituelles, le marquage de l'⍺DG est diminué (mais cette analyse est parfois difficile). Cette anomalie est ensuite confirmée en western blot.
Génétique moléculaire. Le diagnostic de la LGMD2I repose sur l'analyse du gène FKRP qui ne comprend que quatre exons [56]. Des mutations faux-sens et non-sens ont été décrites. La mutation la plus fréquemment retrouvée (p.L276I) s'associe communément à un phénotype moins sévère. Des mutations de FKRP sont aussi responsables d'autres pathologies associant atteintes musculaire, cérébrale et oculaire comme le syndrome de Walker-Warburg [61].
À retenir LGMD2I : déficit en FKRP Origine géographique : ubiquitaire Clinique
• pseudo-Duchenne ou pseudo-Becker : hypertrophie des mollets, macroglossie
• atteinte cardiaque+++
• atteinte respiratoire
Paraclinique
• immunomarquage et western blot de l'⍺DG diminués (interprétation parfois difficile)
Déficit en POMT1 : LGMD2K
Cette dystrophie des ceintures est liée aux mutations du gène POMT1 [62]. Dincer et al., en 2003, ont rapporté les premiers cas sur des sujets turcs et britanniques [63]. Il existe un déficit proximal débutant avant 6 ans associé à une hypertrophie musculaire généralement modérée et à un retard mental (QI entre 50 et 76) ainsi qu'une microcéphalie sans autre anomalie à l'imagerie cérébrale. Un phénotype de début plus précoce et plus sévère est possible [64]. Aucune atteinte cardiaque n'est décrite. Sur la biopsie musculaire, on retrouve des

centralisations nucléaires particulièrement abondantes [62]. L'immunomarquage de l'⍺DG est diminué, anomalie confirmée en western blot.
Déficit en fukutine : LGMD2L
Dans un premier temps, le gène FKTN qui code la fukutine a été impliqué dans la dystrophie musculaire congénitale de Fukuyama, fréquente au Japon [65]. La fukutine (MIM 607440) est une protéine de 461 acides aminés, localisée dans l'appareil de Golgi et dans des granules sécrétoires. Une altération de sa fonction conduit à une anomalie de glycosylation de l'⍺DG.
Données cliniques
Godfrey et al. ont rapporté en 2006 trois enfants issus de deux familles différentes et présentant un tableau de dystrophie musculaire des ceintures récessive sans atteinte centrale [66]. Tous ont développé une hypotonie et un déficit moteur entre 4 et 10 mois. Lors d'épisodes fébriles, leur déficit s'est aggravé puis amélioré sous corticoïdes, faisant suspecter une origine inflammatoire. L'examen met en évidence une hypertrophie des membres inférieurs touchant plus particulièrement le jumeau externe au mollet. Dans cette description, les trois patients sont encore ambulatoires. Le développement intellectuel est normal. Il n'y a pas d'atteinte cardiaque décrite.
Éléments paracliniques
CK. Le taux est élevé et augmente au cours des épisodes fébriles.
Imagerie cérébrale. L'IRM cérébrale ne montre pas d'anomalie structurelle notable, contrairement à la dystrophie musculaire congénitale de Fukuyama. Dans cette dernière, on peut observer deux types de dysplasie corticale : une polymicrogyrie (cortex aminci et sillons peu profonds) ou une lissencéphalie de type II (cortex pachygyrique et surface lisse). Ces anomalies s'associent à une désorganisation caractéristique de la foliation cérébelleuse [67].
Anatomopathologie. Les techniques usuelles permettent de mettre en évidence des aspects de nécrose/régénération de type dystrophique. Il peut exister un infiltrat inflammatoire macrophagique et lymphocytaire T modéré [66]. Les immunomarquages sur lame montrent une réduction majeure de l'⍺DG, alors qu'il peut aussi exister une réduction modérée du
marquage de la laminine ⍺2, β1 et γ1.
À retenir LGMD2L : déficit en fukutine Origine géographique : Japon++, Europe Clinique
• déficit quadricipital
• hypertrophie des mollets

Paraclinique
• immunomarquage et western blot de l'⍺DG diminués
Déficit en POMT2 : LGMD2N
Il existe actuellement une seule description de dystrophie des ceintures liée à une mutation du gène POMT2 [68]. Le déficit débute vers l'âge de 5 ans par une atteinte proximale pelvienne et scapulaire modérée, sans anomalie cérébrale ou cardiaque. L'étude anatomopathologique retrouve des anomalies de type dystrophique associées à un infiltrat inflammatoire.
Déficit en POMGnT1
Cette dystrophie des ceintures est de description récente et n'a pas encore été incluse dans la classification de l'EFNS. Il existe une seule description de dystrophie des ceintures liée à des mutations du gène POMGnT1 [69]. À partir de 12 ans s'installe un déficit touchant les deux ceintures et la musculature axiale avec perte de la marche à 19 ans et hypertrophie des mollets. La biopsie musculaire montre des lésions dystrophiques sans autre élément d'orientation.
Titinopathie : LGMD2J
Cette forme de LGMD a été initialement décrite en Finlande par Udd et al. en 1992 dans une large famille consanguine où certains patients présentaient un phénotype récessif proximal de type LGMD et d'autres un phénotype distal dominant de type dystrophie musculaire tibiale (TMD) [70]. La confirmation moléculaire du lien entre les deux pathologies n'a eu lieu que 10 ans plus tard [27] : en cas de mutation hétérozygote du gène TTN, les individus présentent une atteinte distale (phénotype TMD), alors qu'en cas de mutation homozygote, ils sont porteurs d'une atteinte plus sévère et proximale (phénotype LGMD2J).
La titine (MIM 188840) est la plus grosse protéine connue chez l'homme (3 000 kDa) et l'une des plus abondantes du tissu musculaire. Elle s'étend sur toute la longueur du sarcomère, le long des filaments d'actine et de myosine. Elle assure le maintien de la stabilité du sarcomère en permettant une bonne répartition des forces de tension [71].
Présentation clinique
Udd et al. ont rapporté initialement une très large famille consanguine dans laquelle 12 membres présentaient une TMD et huit autres un tableau de dystrophie des ceintures [70]. Les formes de type dystrophie des ceintures sont nettement plus sévères que les formes distales. Leur début est plus précoce, entre la première et la troisième décennie. Tous les muscles proximaux sont déficitaires. La marche est perdue environ 20 ans après le début des symptômes. Certains patients ont développé ultérieurement un déficit distal. Il n'y a pas d'atteinte cardiaque décrite.
Éléments paracliniques
Le taux de CK est normal ou modérément élevé, jusqu'à cinq fois la normale. Sur la biopsie musculaire, les techniques usuelles mettent en évidence des lésions dystrophiques, mais il

n'existe pas de technique d'immunomarquage de cette protéine. En revanche, un déficit secondaire en calpaïne 3 est possible [27].
Génétique moléculaire
La mutation du gène TTN identifiée en Finlande à l'état hétérozygote pour les TMD et à l'état homozygote pour les LGMD2J altère 11 paires de bases situées dans région de la ligne M de la titine [72]. Une seconde mutation (p.L34315P) a ensuite été identifiée dans une famille française présentant un phénotype distal (TMD) [27].
À retenir LGMD2J : titinopathie Origine géographique : Finlande Clinique
• début : entre 2e et 3e décennie
• déficit distal des membres inférieurs possible
Paraclinique
• atteinte distale en imagerie
LGMD2M
Cette forme a été décrite en 2007 dans huit familles du Canada francophone [73]. Les études de liaison ont montré que le locus était situé sur le chromosome 11p mais le gène n'a pas été identifié et il n'y a pas eu de nouvelle description. Les patients présentent un tableau de déficit des ceintures de sévérité très variable avec amyotrophie quadricipitale asymétrique et débutant entre 11 et 50 ans. Le taux de CK est soit normal, soit élevé. La biopsie musculaire est aspécifique.
Dystrophies musculaires des ceintures autosomiques dominantes Dans la classification originelle de Walton et Nattrass, les formes autosomiques dominantes de dystrophies musculaires des ceintures n'existaient pas [4]. Toutefois, de nombreuses publications ont ultérieurement rapporté des cas dont le tableau clinique et anatomopathologique correspondait à une dystrophie des ceintures et dont la transmission n'était pas autosomique récessive, mais dominante. Dans la classification actuelle des dystrophies musculaires des ceintures, les formes autosomiques dominantes sont dénommées LGMD1 [9]. Actuellement, sept loci sont connus (Tableau 3).
Myotilinopathie : LGMD1A

Cette première forme de dystrophie des ceintures dominante a été décrite initialement en Virginie [74], les mutations du gène codant la myotiline (TTID) en 2000 [75]. La myotiline (MIM604103) est une protéine de 57 kDa associée aux filaments d'actine dans le disque Z par son extrémité N-terminale [76]. Elle joue un rôle de stabilisation et de fixation des filaments fins d'actine, permettant une bonne organisation du disque Z.
Présentation clinique et paraclinique
La description initiale a été réalisée dans une large famille de Virginie occidentale en 1988 [74]. Les troubles débutent dans la troisième décennie par un déficit proximal des membres inférieurs conduisant progressivement à une perte de la marche. L'atteinte des membres supérieurs est plus tardive. Dans quelques cas, le déficit est à prédominance distale et non proximale [77]. D'autres manifestations ont été décrites : aréflexie achilléenne et surtout dysarthrie avec voix nasonnée [75]. La pénétrance est incomplète et âge-dépendante. Une atteinte cardiaque est possible.
Le taux de CK peut être normal ou élevé. L'étude de la biopsie musculaire confirme le caractère dystrophique associé à des vacuoles bordées, alors que certains patients présentent un aspect de myopathie myofibrillaire.
Génétique moléculaire
La mutation p.C450T dans le gène TTID codant la myotiline a été identifiée en 2000 [75]. L'existence de mutations dans ce gène fut confirmée par la suite [77, 78]. Il faut noter le grand nombre de mutations de novo expliquant les nombreux cas sans histoire familiale. Le gène de la myotiline est aussi impliqué dans des myopathies à corps sphéroïdes et des myopathies myofibrillaires.
À retenir LGMD1A : myotilinopathie Origine géographique : ubiquitaire Clinique
• dysarthrie et dysphonie
• déficit distal possible
• atteinte cardiaque
Paraclinique
• biopsie : parfois aspect de myopathie myofibrillaire ou de vacuoles bordées
Laminopathie : LGMD1B
Cette dystrophie musculaire est apparentée à la myopathie d'Emery-Dreifuss (EDMD) dans sa forme autosomique dominante. Le même gène, LMNA codant la lamine A/C, a été impliqué

dans ces deux pathologies [79, 80]. La lamina nucléaire constitue un échafaudage protéique qui permet de maintenir l'intégrité structurelle de la membrane interne du noyau et des pores nucléaires. Les lamines A et C sont des filaments intermédiaires exprimés de façon ubiquitaire appartenant à cette lamina (MIM 150330). Elles sont toutes les deux codées par le même gène, LMNA, grâce à l'épissage alternatif du même ARN messager [81]. Elles jouent un rôle fondamental non seulement dans le maintien de la structure des noyaux, mais également dans un ensemble de régulations concernant l'enveloppe nucléaire, la structure de la chromatine, la division et la différenciation des cellules.
Épidémiologie
Il s'agit de la plus fréquente des dystrophies des ceintures autosomiques dominantes ; toutefois, elle reste plus rare que les calpaïnopathies.
Présentation clinique
Nous décrivons ici le phénotype LGMD et non les autres pathologies associées aux mutations de LMNA. Les symptômes débutent généralement dans la deuxième à troisième décennie, mais le spectre s'étend de 4 à 38 ans [82]. Le déficit moteur touche d'abord les membres inférieurs puis les membres supérieurs, et tout particulièrement le biceps brachial. La pénétrance de la pathologie est totale après 45 ans dans la description initiale faite aux Pays-Bas [82]. Des rétractions tendineuses modérées touchant les coudes et les tendons d'Achille peuvent apparaître tardivement dans l'histoire de la maladie. Quand elles sont présentes, elles restent au second plan contrairement au phénotype EDMD. De même, les muscles paravertébraux sont peu atteints contrairement au phénotype EDMD. Les laminopathies sont caractérisées par la présence d'une atteinte cardiaque extrêmement fréquente [83]. En général, cette atteinte débute avant la troisième décennie. Ces anomalies s'expriment sur deux versants :
• des troubles du rythme et de la conduction (bloc atrioventriculaire), parfois responsables de mort subite par tachycardie ventriculaire [83] ;
• une cardiomyopathie dilatée, moins fréquente et plus tardive.
Le risque de mort subite est majeur, y compris quand un pacemaker est en place. Les recommandations actuelles proposent plutôt la pose d'un défibrillateur implantable. La sévérité de ces troubles doit faire proposer un dépistage des apparentés asymptomatiques et un suivi cardiologique resserré de ces patients. L'atteinte cardiaque peut être isolée.
Éléments paracliniques
Créatine-kinases
Le taux de CK est normal à modérément élevé (jusqu'à 3 fois la normale).
Imagerie musculaire
Il existe une dégénérescence graisseuse des muscles fessiers, des vastes internes et externes, ainsi que des adducteurs [84]. En revanche, le droit fémoral, le sartorius et le gracile sont

épargnés. À l'étage jambier, le jumeau interne est le plus touché, alors que les loges antérieures ne sont pas atteintes.
Anatomopathologie
Les techniques usuelles mettent en évidence des anomalies dystrophiques peu abondantes [85]. Dans les fibres atrophiques, des vacuoles bordées peuvent être observées. L'immunomarquage de la lamine A/C n'est pas contributif.
Génétique moléculaire
Soixante-seize pour cent des mutations du gène LMNA apparaissent de novo [80]. En raison de la distribution ubiquitaire des lamines, de très nombreux tissus peuvent être touchés en cas de mutation de ce gène (ou d'un gène intervenant dans sa maturation) :
• muscle squelettique : LGMD1B (MIM 159001) étudiée ici, EDMD2 autosomique dominante (MIM 181350), myopathie congénitale avec syndrome de la colonne raide, dystrophie musculaire congénitale ;
• muscle cardiaque : troubles du rythme et de la conduction, cardiomyopathie dilatée de type 1A (MIM 115200) ;
• nerf périphérique : CMT2B1 autosomique récessif (MIM 605588), neuropathie axonale dominante avec dystrophie musculaire, atteinte cardiaque et leuconychies [86] ;
• tissu adipeux et troubles métaboliques : lipodystrophie familiale de type Dunnigan (MIM 151660), lipoatrophie associée à un diabète, une stéatose hépatique, une cardiomyopathie hypertrophique et des papules leucomélanodermiques (MIM 608056), probablement syndrome de résistance à l'insuline de type A ;
• peau : dermopathies restrictives ;
• os : dysplasie acromandibulaire de type A avec lipodystrophie (MIM 248370), dysplasie claviculaire de la progéria.
Une des formes les plus sévères de laminopathie est la progéria ou syndrome de Hutchinson-Gilford (MIM 176670). Cette longue liste illustre bien l'extrême variabilité phénotypique des laminopathies. Pour certaines de ces pathologies, il existe des points chauds de mutations sur le gène LMNA.
À retenir LGMD1B : laminopathie Origine géographique : ubiquitaire Clinique
• cardiopathie+++ (risque de mort subite)
• allélique avec la maladie d'Emery-Dreifuss autosomique dominante

• nombreux autres phénotypes
Cavéolinopathie : LGMD1C
Cette forme de dystrophie musculaire a été initialement identifiée chez des patients présentant un phénotype de dystrophie des ceintures modérée [87]. La cavéoline 3 (MIM 601253) est une protéine d'environ 20 kDa qui constitue un des composants principaux des caveolae, invaginations de la membrane plasmique de 50 à 100 nm impliquées dans la signalisation cellulaire [88].
Présentation clinique
Une faiblesse proximale apparaît vers 5 ans [87]. L'évolution est variable : certains individus présentent simplement un signe de Gowers à l'âge adulte, alors que d'autres ont un tableau bien plus sévère. Tous les patients présentent une pseudohypertrophie des mollets. Ce déficit peut s'associer à la rippling muscle disease. Dans cette pathologie, on retrouve une raideur musculaire, un myooedème à la percussion (Figure 7) et des contractions musculaires en « vague » (ou rippling) induites par l'activité ou la percussion.
Ces vagues sont électriquement silencieuses [89]. Dans ce cas, il existe fréquemment une hypertrophie musculaire. Ces deux phénotypes peuvent être associés chez un même patient. Enfin, certaines cavéolinopathies sont asymptomatiques, se traduisant uniquement par une hypercréatine-kinasémie isolée. Il n'y a pas de description d'atteinte cardiaque associée à la LGMD1C.
Éléments paracliniques
Le taux de CK est élevé de 3 à 25 fois la normale. L'étude anatomopathologique met en évidence des anomalies de type dystrophique sans caractéristique particulière. L'immunomarquage de la cavéoline 3 est réduit le long de la membrane plasmique. L'étude ultrastructurale met en évidence une diminution de la densité en caveolae.
Génétique moléculaire
Le gène CAV3 codant la cavéoline 3 est de petite taille. Peu de mutations ont été décrites dans le phénotype de dystrophie des ceintures. Dans de nombreux cas, les mutations surviennent de novo. Certaines mutations identifiées dans un premier temps se sont révélées non pathogènes [90].
À retenir LGMD1C : cavéolinopathie Origine géographique : ubiquitaire Clinique
• phénomène de rippling

• hypertrophie musculaire
• formes asymptomatiques (hypercréatine-kinasémie)
Paraclinique
• immunomarquage et western blot : anomalie de la cavéoline (inconstant)
• biologie moléculaire parfois complexe à interpréter
LGMD1D
Il s'agit d'une forme rare de dystrophie des ceintures associant un déficit proximal progressif, une cardiomyopathie et des troubles rythmiques débutant chez l'adulte jeune [91]. Le gène n'est pas encore identifié.
LGMD1E
Dans cette forme de dystrophie décrite à une seule reprise, il existe un déficit proximal de début tardif associé à une dysphagie [92].
LGMD1F
Il s'agit à nouveau d'une forme rare d'âge de début variable et dont le déficit prédomine à la ceinture pelvienne avec parfois une atteinte distale associée [93].
LGMD1G
Dans cette forme rare, la ceinture pelvienne est nettement plus déficitaire que la ceinture scapulaire et tous les individus développent une limitation de flexion des doigts et des orteils [94].
Stratégie diagnostique Comme nous venons de le voir, il existe de multiples formes de dystrophies musculaires des ceintures. L'établissement d'une stratégie diagnostique univoque est complexe, en particulier en raison de la variabilité phénotypique présente dans chaque type de dystrophie des ceintures [9]. À tout moment, il faut savoir repartir à zéro pour tenter d'approcher le diagnostic.
Éléments d'orientation clinique
L'étude du mode de transmission est fondamentale. Dans les formes dominantes, la présence de certains signes chez le patient ou bien chez ses apparentés peut faire évoquer un diagnostic. Dans les myotilinopathies (LGMD1A) et laminopathies (LGMD1B), l'étude des protéines en cause n'est pas possible sur la biopsie. Ainsi, s'il existe une forte suspicion clinique, l'étude génétique peut être proposée en première intention (Figure 8). Concernant les formes récessives, il est le plus souvent nécessaire de recourir à la biopsie musculaire afin d'étudier les protéines musculaires par immunomarquage sur lame puis western blot. Il existe toutefois quelques exceptions où l'on peut se passer de la biopsie :

• populations avec effet fondateur fort : Maghreb et LGMD2C par exemple ;
• présence d'antécédents familiaux de myopathie distale (TMD) de transmission autosomique dominante orientant vers une mutation de TTN (LGMD2J).
Biopsie et étude des protéines musculaires
Dans la majorité des cas, les données cliniques sont insuffisantes pour mener directement à l'étude moléculaire. Dans ces cas, la biopsie musculaire s'avère indispensable.
Les techniques usuelles confirment la présence de lésions dystrophiques. Par ailleurs, elles peuvent montrer des signes d'orientation comme un infiltrat éosinophile dans les calpaïnopathies (LGMD2A). Toutefois, l'étude des protéines musculaires par immunomarquage sur lame puis par western blot reste indispensable dans la majorité des cas. Sur lame, des anticorps dirigés contre les protéines suivantes sont le plus souvent utilisés : dystrophine (DYS1, DYS2 et DYS3 afin d'éliminer une dystrophinopathie), dysferline, ⍺-, β-,
γ- et δ-sarcoglycanes, cavéoline, ⍺DG et éventuellement téléthonine. En western blot, les anticorps dirigés contre les protéines suivantes sont le plus souvent utilisés : dystrophine (DYS1 et DYS2), dysferline, ⍺- et γ-sarcoglycanes, calpaïne et éventuellement ⍺DG. En fonction des déficits retrouvés, une étude génétique adaptée peut être proposée (Figure 8).
Biopsie non contributive
Dans un nombre non négligeable de cas, ni les données cliniques initiales ni l'étude anatomopathologique ne permettent d'orienter le diagnostic. Il est alors nécessaire d'examiner à nouveau le patient à la recherche de signes évoquant un diagnostic différentiel.
S'il n'existe aucun élément d'orientation, il existe deux possibilités :
• le patient présente une dystrophie des ceintures due à une mutation dans un gène connu, mais dont l'étude protéique est difficile. C'est le cas par exemple dans les calpaïnopathies où le western blot est faussement négatif dans environ 20 % des cas [25]. Il existe aussi des descriptions de faux négatifs pour les ⍺-dystroglycanopathies liées à FKRP [15]. Ces éléments peuvent justifier l'étude systématique des gènes CAPN3 et FKRP. Dans le premier cas, il faut noter que cette analyse est longue et que les résultats peuvent être d'interprétation difficile ;
• le patient présente une dystrophie des ceintures dont le gène n'est pas encore connu ou non étudiable en pratique courante. Dans cette situation, il est nécessaire de mener une étude familiale la plus exhaustive possible afin de pouvoir mener des études moléculaires plus complexes.
Éléments de prise en charge

Que le diagnostic moléculaire soit posé ou non, la prise en charge pluridisciplinaire des patients atteints de dystrophie musculaire des ceintures est fondamentale. Compte tenu de l'absence de thérapeutique curative, les soins sont essentiellement symptomatiques et palliatifs. Le maillage du territoire français par les centres de références des maladies neuromusculaires devrait améliorer la prise en charge des patients tant sur le plan diagnostique que thérapeutique.
Bien que les phénomènes rétractiles soient généralement au second plan, ils peuvent parfois être une source de handicap. Ces rétractions sont d'autant plus fréquentes que la pathologie débute avant la phase de croissance. La prévention de ce type de complications repose sur la kinésithérapie éventuellement associée à un appareillage orthopédique, l'objectif étant de retarder la perte de la marche.
L'atteinte cardiaque est une autre complication à rechercher chez ces patients. Comme nous l'avons vu, la fréquence de cette atteinte est très variable selon le gène muté. Dans les laminopathies, le risque de mort subite est majeur. Cela impose donc une surveillance régulière par échocardiographie et enregistrement de l'électrocardiogramme (ECG) sur 24 heures (Holter-ECG). Dans certains cas, des investigations plus complexes peuvent s'imposer dans des centres spécialisés. La mise en place précoce de pacemakers, voire de défibrillateurs implantables est souvent justifiée. Au suivi cardiologique doit s'associer un suivi pneumologique à la recherche d'une insuffisance respiratoire restrictive par exploration fonctionnelle respiratoire et éventuellement un enregistrement polysomnographique.
Les thérapies géniques et cellulaires constituent des perspectives d'avenir intéressantes. Après plusieurs espoirs déçus, ce type de traitement s'imposera vraisemblablement dans les années à venir compte tenu de la recherche extrêmement active dans ce domaine.
Conclusion Comme nous venons de le voir, les dystrophies musculaires des ceintures constituent un groupe en partie hétérogène de pathologies. Les progrès de la biologie moléculaire ont permis une nette amélioration du rendement diagnostique au détriment de la simplicité. La présence d'une atteinte cardiaque possiblement grave (risque de mort subite et laminopathie) doit amener à pousser les investigations au plus loin afin de proposer des mesures de prévention (défibrillateur implantable, etc.) et un dépistage des apparentés. Hormis de rares cas, la biopsie musculaire avec études des protéines musculaires est indispensable au diagnostic.
Remerciements
je tiens à remercier le docteur Xavier Ferrer, le docteur Florian Perez et madame Evelyne Riblet pour leurs corrections et conseils toujours fructueux.
© 2010 Elsevier Masson SAS. Tous droits réservés.

Toute référence à cet article doit porter la mention : G. Solé. Myopathies des ceintures. EMC - Neurologie 2010:1-15 [Article 17-175-C-10].
Tableau 1 - Classification des dystrophies des ceintures selon l'EFNS (issue de [8]) Nom Localisation Gène Protéine Transmission autosomique dominante : LGMD1 LGMD1A 5q22-q34 TTID Myotiline LGMD1B 1q11-21 LMNA Lamine A/C LGMD1C 3p25 CAV3 Cavéoline 3 LGMD1D 6q23 LGMD1E 7q Transmission autosomique récessive : LGMD2 LGMD2A 15q15.1-q21.1 CAPN3 Calpaïne 3 LGMD2B 2p13 DYSF Dysferline LGMD2C 13q12 SGCG γ-sarcoglycane LGMD2D 17q12-q21.33 SGCA ⍺-sarcoglycane LGMD2E 4q12 SGCB β-sarcoglycane LGMD2F 5q33-q34 SGCD δ-sarcoglycane LGMD2G 17q11-q34.1 TCAP Téléthonine LGMD2H 9q31-q34.1 TRIM32 TRIM32 LGMD2I 19q13.3 FKRP Fukutin related protein LGMD2J 2q TTN Titine LGMD2K 9q34 POMT1 POMT1 LGMD2L 9q31 FKTN Fukutine LGMD2M 11p13 L GMD2N 19q13.3 POMT2 POMT2 Légende : Les LGMD1F, 1G et 2O sont absentes de cette classification.
Tableau 2 - Dystrophies musculaires des ceintures autosomiques récessives : formes « fréquentes » et formes exceptionnelles Formes les plus « fréquentes » Calpaïnopathie Dysferlinopathie Déficit en FKRP Sarcoglycanopathies ⍺ et γ
LGMD2A LGMD2B LGMD2I LGMD2D et 2C
Formes exceptionnelles Sarcoglycanopathies β et δ LGMD2E et 2F

Téléthoninopathie Déficit en TRIM32 Titinopathie Déficit en POMT1 Déficit en fukutine Déficit en POMT2 Déficit en POMGnT1
LGMD2G LGMD2H LGMD2J LGMD2K LGMD2L LGMD2N LGMD2O (?) LGMD2M
Tableau 3 - Dystrophies musculaires des ceintures autosomiques dominantes : formes « fréquentes » et formes exceptionnelles Forme la plus « fréquente » Laminopathie LGM1B Formes rares Cavéolinopathie LGMD1C Myotilinopathie LGMD1A Formes exceptionnelles LGMD1D LGMD1E LGMD1F LGMD1G
Figure 1 :
Principales protéines impliquées dans les maladies musculaires. Elles peuvent se situer dans la membrane cellulaire (ou sarcolemme), le cytoplasme, le noyau ou au sein des myofibrilles. FKRP : fukutin-related protein. D'après Principales maladies neuromusculaires, Association française contre les myopathies (AFM).
Figure 2 :
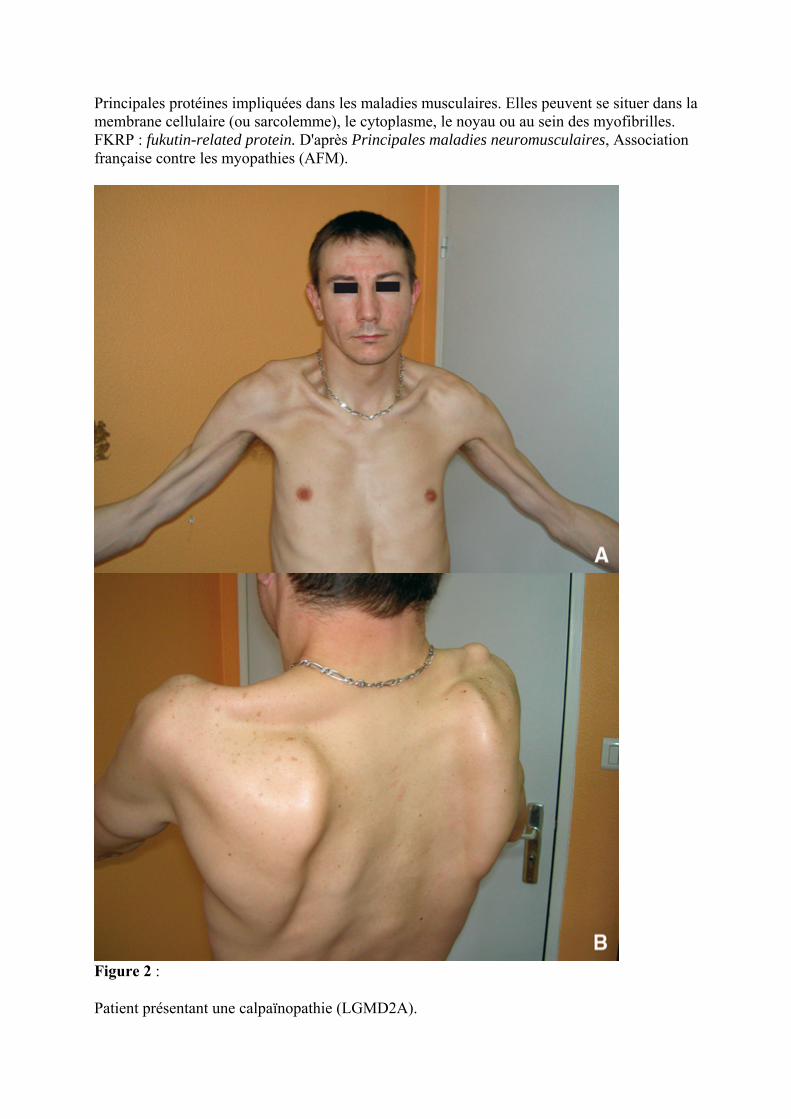
Patient présentant une calpaïnopathie (LGMD2A).
A. Limitation d'abduction des épaules. Noter la présence d'une amyotrophie bicipitale notable.
B. Décollement des omoplates chez le même patient.
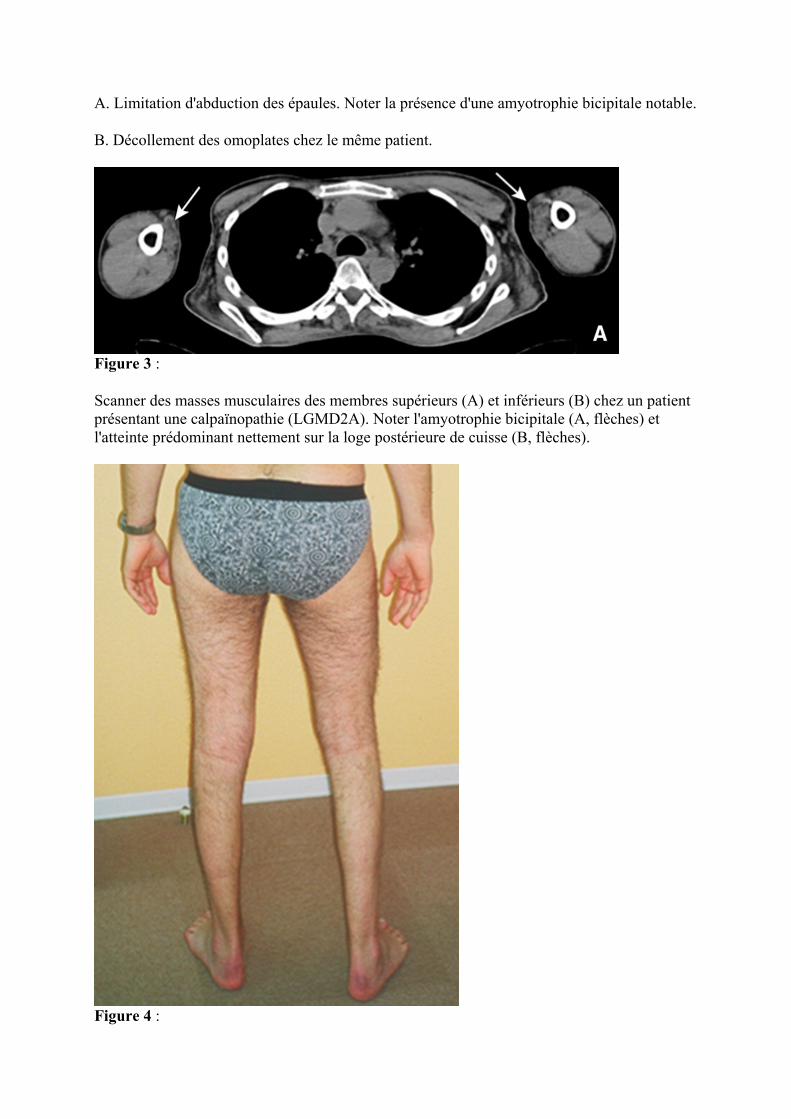
Figure 3 :
Scanner des masses musculaires des membres supérieurs (A) et inférieurs (B) chez un patient présentant une calpaïnopathie (LGMD2A). Noter l'amyotrophie bicipitale (A, flèches) et l'atteinte prédominant nettement sur la loge postérieure de cuisse (B, flèches).
Figure 4 :
Amyotrophie de la loge postérieure de jambe chez un patient présentant une dysferlinopathie (LGMD2B).
Figure 4 :
Amyotrophie de la loge postérieure de jambe chez un patient présentant une dysferlinopathie (LGMD2B).
Figure 6 :

Macroglossie chez une patiente porteuse d'une ⍺-dystroglycanopathie par mutation de FKRP (LGMD2I).
Figure 7 :
Zone de myooedème localisé (cercle) après percussion chez un patient présentant une muscle rippling disease.

Figure 8 :
Étude génétique. Les gènes à étudier sont encadrés en rouge. AD : autosomique dominant ; IM : immunomarquages ; WB : western blot ; FSH : myopathie facio-scapulo-humérale ; ⍺DG : ⍺-dystroglycane.