monograph - carbaglu · 2018-01-03 · 5 back to table of contents _ 1.0 the urea cycle and urea...
TRANSCRIPT

®Carbaglucarglumic acid
Carbaglu® (carglumic acid)Monograph
PP-CBGL-US-0027

INDICATION AND IMPORTANT SAFETY INFORMATION Indications and Usage Carbaglu® (carglumic acid) is a Carbamoyl Phosphate Synthetase 1 (CPS 1) activator indicated as:
• Adjunctive therapy in pediatric and adult patients for the treatment of acute hyperammonemia due to the deficiency of the hepatic enzyme N-acetylglutamate synthase (NAGS). During acute hyperammonemic episodes concomitant administration of CARBAGLU with other ammonia lowering therapies such as alternate pathway medications, hemodialysis, and dietary protein restriction are recommended.
• Maintenance therapy in pediatric and adult patients for chronic hyperammonemia due to the deficiency of the hepatic enzyme N- acetylglutamate synthase (NAGS). During maintenance therapy, the concomitant use of other ammonia lowering therapies and protein restriction may be reduced or discontinued based on plasma ammonia levels.
Important Safety Information
HYPERAMMONEMIA :• Management of hyperammonemia due to N-acetylglutamate synthase (NAGS) deficiency and
CARBAGLU treatment should be initiated by a physician experienced in the treatment of metabolic disorders.
• Any episode of acute symptomatic hyperammonemia should be treated as a life-threatening emergency. Treatment of hyperammonemia may require dialysis, preferably hemodialysis, to remove a large burden of ammonia. Uncontrolled hyperammonemia can rapidly result in brain injury/damage or death, and prompt use of all therapies necessary to reduce plasma ammonia levels is essential.
• Ongoing monitoring of plasma ammonia levels, neurological status, laboratory tests and clinical responses in patients receiving CARBAGLU is crucial to assess patient response to treatment.
THERAPEUTIC MONITORING:• Plasma ammonia levels should be maintained within normal range for age via individual dose
adjustment.
NUTRITIONAL MANAGEMENT:• Since hyperammonemia is the result of protein catabolism, complete protein restriction is
recommended to be maintained for 24 to 48 hours and caloric supplementation should be maximized to reverse catabolism and nitrogen turnover.
Themostcommonadversereactionsin≥13%ofpatientsare:infections,vomiting,abdominalpain,pyrexia,tonsillitis, anemia, ear infection, diarrhea, nasopharyngitis, and headache.
ToreportSUSPECTEDADVERSEREACTIONS,contactRecordatiRareDiseasesInc.at1-888-575-8344,orFDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
No drug interaction studies have been performed with CARBAGLU. There is no human pregnancy data, but decreased survival and growth occurred in animal offspring.
Breast feeding by a mother taking CARBAGLU is not recommended.
USE IN OTHER FOODS AND LIQUIDS HAS NOT BEEN STUDIED CLINICALLY AND IS THEREFORE NOT RECOMMENDED.

Table of Contents
ABBREVIATIONS ........................................................................................................41.0 THE UREA CYCLE AND UREA CYCLE DISORDER ................................................52.0 NAGS DEFICIENCY ..............................................................................................6 2.1 HEREDITY ................................................................................................6 2.2 INCIDENCE ...............................................................................................6 2.3 PATHOPHYSIOLOGY ..................................................................................7 2.4 NEONATAL/EARLY ONSET FORMS .............................................................8 2.5 LATE ONSET FORMS ................................................................................9 2.6 DIAGNOSIS ...............................................................................................10 2.7 TREATMENT OF NAGS DEFICIENCY ...........................................................133.0 PRODUCT INFORMATION .....................................................................................15 3.1 GENERAL INFORMATION ...........................................................................15 3.2 COMPOSITION OF CARBAGLU TABLETS ....................................................15 3.3 ACTIVE PHARMACEUTICAL INGREDIENT: CARGLUMIC ACID .......................15 3.4 EXCIPIENTS ..............................................................................................16 3.5 DRUG PRODUCT .......................................................................................17 3.6 PACKAGING ..............................................................................................194.0 NONCLINICAL DATA .............................................................................................20 4.1 PHARMACOLOGY .....................................................................................20 4.2 PHARMACOKINETICS ................................................................................21 4.3 SAFETY PHARMACOLOGY .........................................................................22 4.4 TOXICOLOGY ............................................................................................225.0 CLINICAL DATA ....................................................................................................24 5.1 PHARMACODYNAMICS .............................................................................24 5.2 PHARMACOKINETICS ................................................................................25 5.3 EFFICACY .................................................................................................26 5.4 SAFETY ....................................................................................................30 5.5 CONCLUSION OF CLINICAL ASPECTS ........................................................31 5.6 RECOMMENDATIONS FOR CARBAGLU® (carglumic acid) USE ...................326.0 REFERENCES .......................................................................................................357.0 CARBAGLU PRESCRIBING INFORMATION ..........................................................41

4 Back to Table of Contents _
ABBREVIATIONS AE Adverse event
API Active pharmaceutical ingredient
ARG Arginase
ASL Argininosuccinate lyase
ASS Argininosuccinate synthetase
Cmax Maximum plasma concentration
CNS Central nervous system
CPS1 Carbamoyl phosphate synthetase 1
DNA Deoxyribonucleic acid
GLN Glutamine
GLU Glutamate
GMP Good Manufacturing Practices
GS Glutamine synthetase
HPA Hydantoin-5-propionic acid
HPLC High performance liquid chromatography
ICH International Conference on Harmonisation
INN International Non Proprietary Name
IR Infrared
LC/MS-MS Liquid chromatographic-tandem mass spectrometry
NAG N-acetylglutamate
NAGS N-acetylglutamate synthase
NOAEL No observable adverse effect level
NOEL No observable effect level
OTC Ornithine transcarbamylase
PK Pharmacokinetic
RH Relative humidity
SAE Serious adverse event
Tmax Time to reach maximum plasma concentration
UCD Urea cycle disorders
UV Ultraviolet
WHO World Health Organization

5 Back to Table of Contents _
1.0 THE UREA CYCLE AND UREA CYCLE DISORDER The urea cycle is the primary pathway for waste of nitrogenexcretioninhumans.Approximately80%of excreted nitrogen is in the form of urea, which is produced primarily in the liver (Burton 2000).
As presented in the diagram, the urea cycle is comprised of the following: • 5 catalytic enzymes - carbamoyl phosphate
synthetase 1 (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL) and arginase (ARG)
• A cofactor-producer enzyme: N-acetylglutamate synthase (NAGS)
• Two transporters - ornithine translocase (ORNT1) and citrin (Ah Mew et al 2015)
Although these enzymes are found in other tissues, the urea cycle occurs solely in the liver, and this organ consequently actsasthemainammoniadetoxificationcenter(Walser1983).
The NAGS, CPS1, and OTC enzymes are found within the mitochondria. The mitochondrial part of the urea cycle transforms an initial ammonia molecule into a molecule of citrulline. Citrulline then enters the cytosol where it binds a second ammonia molecule carried by aspartate before ultimate transformation to urea. This cytosolic part of the cycle is catalyzed by the ASS, ASL, and ARG enzymes. The final product of the urea cycle is de novo synthesis of arginine, which is a non-essential amino acid.
The activity of the urea cycle does not depend solely upon these 6 enzymes; it is also regulated by other enzyme systems, such as mitochondrial membrane transport systems, which allow aspartate-glutamate and citrulline-ornithine exchange (Summar 2001, Summar 2005a, Tuchman 2002).
Urea cycle disorders (UCD) are inherited metabolic disorders resulting from defects in the metabolism of the extra nitrogen from the breakdown of protein and other nitrogen-containing molecules.
Severe deficiency or total absence of activity of the cofactor-producing enzyme (NAGS) or any of the first four urea cycle enzymes (CPS1, OTC, ASS, ASL) results in the accumulation of ammonia and other precursor metabolites during the first few days of life (Ah Mew et al 2015). Ammonia is a toxic product of protein catabolism. UCD can present with extreme variability depending on residual urea cycle activity in the liver, and clinical presentation can be seen in all age groups (Summar 2001). The significant morbidity and mortality of these disorders arise from acute and chronic neurotoxicity associated with massive hyperammonemia (Bachmann 2005). The symptoms may range from life-threatening episodes of hyperammonemia in the newborn period (neonatal/early onset) to gastrointestinal disturbances, neurological symptoms such as recurrent headaches and drowsiness, and psychiatric disturbances including behavioral changes and hallucinations, from age one month into adulthood (late onset)(Kleppe2003,Summar2001).TheincidencefortheUnitedStatesispredictedtobe1UCDpatientforevery35,000birthspresentingabout113newpatientsperyearacrossallagegroups(Summar2013).
Enzymes of the Urea Cycle: NAGS: N-acetylglutamate synthaseCPS1: carbamoyl phosphate synthetase 1OTC: ornithine transcarbamylase ASS: argininosuccinate synthetase ASL: argininosuccinate lyase ARG: arginase
Acetyl CoA+
Glutamate
NAGS
CytosolM
itochondrium
CPS1
OTC
ASL
Fumarate
ASS
Urea
URINE
NH4+
Ornithine
Carbamoyl Phosphate
OrnithineAspartate
Citrulline
Arginine Argininosuccinic acid
Citrulline
ARG
N-acetylglutamate

6 Back to Table of Contents _
2.0 NAGS DEFICIENCY NAGS deficiency is the rarest of the UCDs (Caldovic 2010, Tuchman 2008). NAGS deficiency is an inherited, autosomalrecessivemetabolicdisorder(Caldovic2002,Caldovic2003,Caldovic2005).
NAGS is a mitochondrial liver enzyme that is essential for the urea cycle (Roth 2006). NAGS catalyzes the formation of N-acetylglutamate (NAG) from glutamate and acetyl-CoA, which then NAG acts as an allosteric activatorofCPS1,thefirstenzymeoftheureacycle(Cartegena2013).IfNAGSisdefective,NAGsynthesisis impaired and there is no activation of CPS1 to trigger the urea cycle. The activity of the urea cycle is regulated by the rate of synthesis of NAG.
The impairment of ammonia detoxification due to NAGS deficiency results in acute and chronic hyperammonemia, hyperglutaminemia and, eventually, hypocitrullinemia.
Hyperammonemia and hyperglutaminemia are particularly toxic to the central nervous system (CNS) (Albrecht 1998, Broere 2000).
NAGS deficiency represents a serious, life-threatening clinical condition (Caldovic 2002, Guffon 1995, Schubiger 1991). In published cases, NAGS deficiency has presented at ages ranging from the neonatal period to the fifth decade of life (Ah Mew 2011).
Patients with complete NAGS deficiency present with acute severe hyperammonemia within the first few days of life (Caldovic 2005, Nordenström 2007). The clinical course in the neonatal period may be lethal. Left untreated or insufficiently corrected, this condition leads to cerebral edema, coma, and eventually death. For those children who survive, psychomotor retardation is a frequent outcome (Schubiger 1991). Patients with partial NAGS deficiency (late onset) can present symptoms at almost any time of life due to any stressful event such as an infection, trauma, vaccination (Kingsley 2006), surgery, pregnancy, etc.
2.1 HEREDITYNAGS deficiency is transmitted as an autosomal recessive trait and consequently affects boys and girls to the same extent in both the neonatal onset and late onset forms. Heterozygous individuals are clinically asymptomatic (Caldovic 2005, Caldovic 2007).
The gene coding for NAGS has been identified and is located on chromosome 17 (Caldovic 2002, Elpeleg 2002).
2.2 INCIDENCENAGS deficiency is the rarest of the UCD (Caldovic 2010, Tuchman 2008).The true incidence is not known, althoughitissuspectedtobe<1in2millionbirths(Summar2013).TheincidencefortheUnitedStatesispredictedtobe1ureacycledisorderpatientforevery35,000birthspresentingabout113newpatientsperyearacrossallagegroups(Summar2013).

7 Back to Table of Contents _
2.3 PATHOPHYSIOLOGYIn NAGS deficiency, the lack of adequate NAG results in failure to activate the enzyme CPS1. Signs and symptoms occur when ammonia fails to fix into carbamoyl phosphate effectively, thus disabling the urea cycle. This leads to accumulation of:
a) ammonia;b) glutamine (Gln) and eventually alanine;c) transamination products of pyruvate and glutamate (Glu).
The major cause of mortality and morbidity in many UCD, including NAGS deficiency, is hyperammonemia (Jouvet 2007). Most therapeutic interventions have focused on prevention and treatment of hyperammonemia. However, as a cohort of treated patients gets older, other complications have appeared even without a significant history of recurrent hyperammonemia (Bachmann 2005, Cederbaum 2001, Gomceli 2007, Nagata 1991, Smith 2005).
Most pathophysiologic studies to date have focused on the mechanisms of ammonia neurotoxicity (Brusilow1995,Gropman2004,Herndon2003).However,thespecificmechanismsarestillcontroversialand diverse. The brain, like the other extrahepatic organs, lacks a complete urea cycle. Hence, the brain relies on Gln synthesis for the removal of excess ammonia and the temporary storage of nitrogen. As this process is predominantly localized in the astrocytes, acute hyperammonemia causes increased intracellular production of Gln from Glu and ammonia via glutamine synthetase (GS), which leads to astrocyte swelling and dysfunction. Excess Gln is released into the extracellular space, and directly alters the local astrocyte-neuronal synaptic environment (Felipo 1994). A consequent imbalance of excitatory versus inhibitory neurotransmission occurs because of increased Glu production in conjunction with decreased synaptic uptake of Glu. Over time, chronic hyperammonemia causes changes reminiscent of Alzheimer type II astrocytosis (Robinson 1995).
Acute hyperammonemia also alters astrocyte protein expression (Jackson 1986), including changes in GS, glial fibrillary acidic protein, Glu transporter, nitric oxide synthase, and peripheral-type benzodiazepine receptors. All of these changes can result in alterations in the brain’s ability to remove additional ammonia, to regulate cerebral blood flow, and to maintain energy homeostasis and neurotransmission.

8 Back to Table of Contents _
2.4 NEONATAL/EARLY ONSET FORMSApproximately half of all reported cases of NAGS deficiency had onset of clinical symptoms in the neonatal period (Caldovic 2010). The clinical presentation of the disease is described below (Bachman 1988, Gessler 2010, Guffon 1995, Nordenström 2007, Roth 2006, Ah Mew 2011):
NAGSD Neonatal/Early Onset Clinical PresentationInfants delivered at term are usually relatively healthy and well
U
There is a symptom-free interval of several hours to several days prior to onset of the first symptoms; this period represents the time for ammonia to accumulate in the body.
Initial symptoms include:poor feedingvomitinggradual lethargymuscular hypotoniahypothermia polypnea resulting in respiratory alkalosis (This is highly characteristic of hyperammonemic encephalopathy).
U
Subsequently, there is a rapidly progressive neurological distress that is fatal if untreated; symptoms include:hypotonic comaconvulsionsapnea requiring assisted ventilationsigns of cerebral edema on brain imaging.

9 Back to Table of Contents _
2.5 LATE ONSET FORMSTheclinicalpictureanddevelopmentofthelateonsetformsvarywidely:theymayoccurinthefirstmonthsoflifeorinadulthood.Clinicalsymptomsresultprimarilyfromhyperammonemia,andthemostcommonclinicalfindingsinclude central nervous system symptoms, such as lethargy, irritability or somnolence. In general, patients with lateonsetformscanpresentwith(Bélanger-Quintana2003,Caldovic2005,Elpeleg1989,Cartagena2013):
Acute decompensation episodes are often triggered by an increase in protein intake following a change in dietary habit or have been precipitated by illness, pregnancy, or surgery (Elpeleg 1989, Kingsley 2006, Summar 2005a, Ah Mew 2011).
The prognosis for these forms varies according to age at onset, whether or not neurological impairment is present at the time of diagnosis, and whether any subsequent episodes of decompensation occur.
NAGSD Late Onset Clinical PresentationGastrointestinal presentations
(possibly accompanied by hepatic involvement):
chronic or paroxysmal vomiting
anorexia with refusal to eat high-protein foods, generally resulting
in delayed physical growth
abdominal pain
hepatomegaly
recurrent episodes of cytolysis
Neurological presentations with psychomotor retardation involving
either chronic encephalopathy with epileptic seizures or episodes
of headache or drowsiness and neurological accidents that may even comprise inaugural hyperammonemic
coma
Psychiatric presentations include behavioral changes,
hallucinations

10 Back to Table of Contents _
2.6 DIAGNOSISInordertodiagnoseNAGSdeficiency,thefirststepistosuspectureacycledisorders.
2.6.1 DIAGNOSIS OF UCD
TherearegeneralandspecificsignsandsymptomsofUCD,includingunexplainedcomaorencephalopathyassociatedwithcatabolicstress(newbornperiod,infectiousillness,andpostpartum),andnonspecificneurologicorgastrointestinalsymptomsespeciallyassociatedwithincreasedproteinintake(Kleppe2003).A wide spectrum of clinical presentations has been reported in the literature. The symptoms may range from life-threatening episodes of hyperammonemia in the newborn period (early onset) to gastrointestinal disturbances, neurological symptoms such as recurrent headaches and drowsiness, and psychiatric disturbances including behavioral changes and hallucinations, in adulthood (Summar 2001).
The diagnosis of a urea cycle disorder is based on clinical suspicion and biochemical and molecular genetic testing.Aplasmaammoniaconcentrationof150μmol/Lorhigherassociatedwithanormalaniongapandanormal plasma glucose concentration is an indication for the presence of a UCD. Plasma quantitative amino acidanalysisandmeasurementofurinaryoroticacidcandistinguishbetweenthespecificUCDs.Adefinitivediagnosis of a urea cycle defect depends on either molecular genetic testing or measurement of enzyme activity. Molecular genetic testing is possible for all urea cycle defects (Ah Mew et al 2015).
HYPERAMMONEMIA
Glutamine Low/Normal
Organic Acidemias
Glutamine Elevated
Citrulline Low Citrulline High
ASL deficiency
ASS deficiency
LP intolerance
Urinary Orotic Acid
Normal
Urinary Orotic Acid Elevated
OTC deficiency
OAT deficiencyCPS 1 deficiency
ARG1deficiency
HHH syndrome
NAGS: N-acetylglutamate synthase CPS 1: carbamoyl phosphate synthetase 1OTC: ornithine transcarbamylase OAT: ornithine aminotransferase ARG1: arginase HHH: hyperornithinaemia-hyperammonaemia-homocitrullinuria ASL: argininosuccinate lyase ASS: argininosuccinate synthase LP: lysinuric protein
Adapted from Häberle J, et al. (2012) Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis ;7:32.
NAGS deficiency
11 Back to Table of Contents _
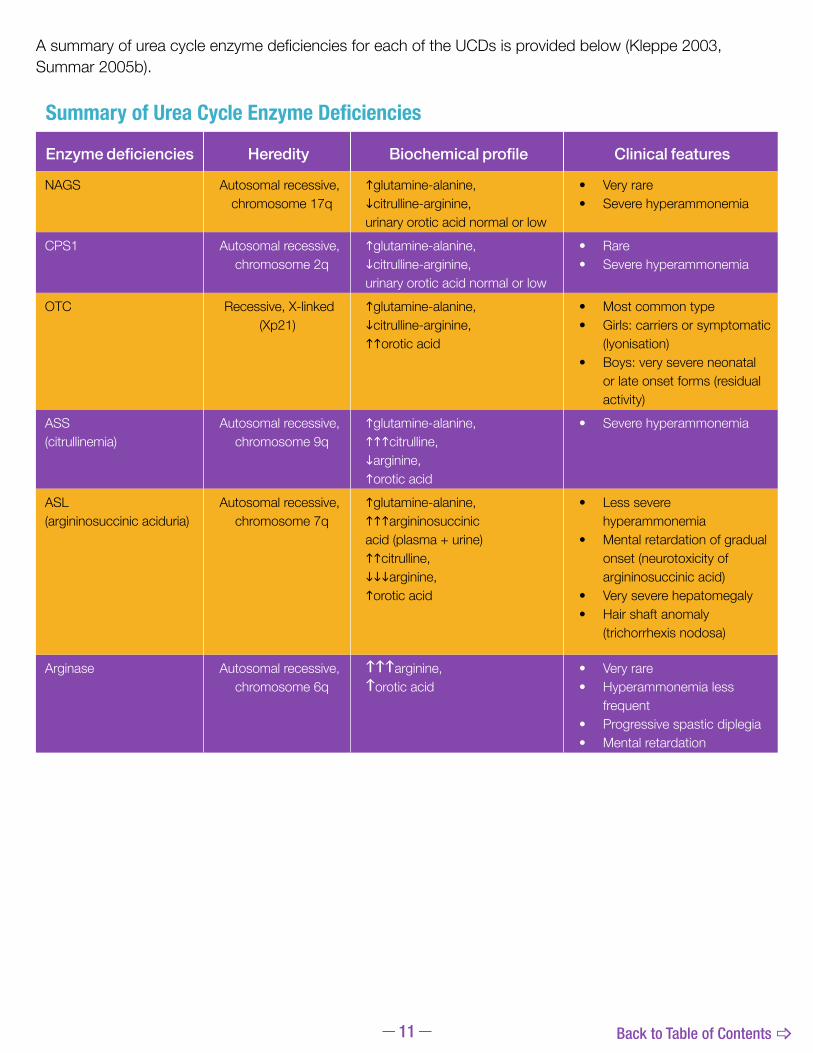
AsummaryofureacycleenzymedeficienciesforeachoftheUCDsisprovidedbelow(Kleppe2003,Summar 2005b).
Summary of Urea Cycle Enzyme Deficiencies
Enzyme deficiencies Heredity Biochemical profile Clinical features
NAGS Autosomal recessive,chromosome 17q
glutamine-alanine,citrulline-arginine, urinary orotic acid normal or low
• Very rare• Severe hyperammonemia
CPS1 Autosomal recessive,chromosome 2q
glutamine-alanine,citrulline-arginine, urinary orotic acid normal or low
• Rare• Severe hyperammonemia
OTC Recessive, X-linked(Xp21)
glutamine-alanine,citrulline-arginine,orotic acid
• Most common type• Girls: carriers or symptomatic
(lyonisation)• Boys: very severe neonatal
or late onset forms (residual activity)
ASS (citrullinemia)
Autosomal recessive,chromosome 9q
glutamine-alanine,citrulline, arginine,orotic acid
• Severe hyperammonemia
ASL (argininosuccinic aciduria)
Autosomal recessive,chromosome 7q
glutamine-alanine,argininosuccinicacid (plasma + urine)citrulline,arginine,orotic acid
• Less severe hyperammonemia
• Mental retardation of gradual onset (neurotoxicity of argininosuccinic acid)
• Very severe hepatomegaly• Hair shaft anomaly
(trichorrhexis nodosa)
Arginase Autosomal recessive,chromosome 6q
\\\arginine,\orotic acid
• Very rare • Hyperammonemia less
frequent• Progressive spastic diplegia• Mental retardation

12 Back to Table of Contents _
2.6.2 DIAGNOSIS OF NAGS DEFICIENCYDependingontheageatNAGSdeficiencyonset,someorallofthefollowingsymptomscanclinicallymanifestinpatients(Endo2004,Gomceli2007,Kleppe2003,Scaglia2004a):anorexia,irritability,heavyorrapid breathing, lethargy, vomiting, disorientation, somnolence, combativeness, and hypotonia. Coma and fatalcerebraledemawillbetheoutcomeifaneffectivetreatmentisnotimplementedrapidly.
DiagnosisofNAGSdeficiencyismadebasedonthefollowingassessments(Bachmann1982,Bachmann2003a,Bachmann2003b,Barsotti2001,Colombo1995,Huizenga1994,Saudubray2007,Steiner2001):
� Blood ammonia levels: theneonatalonsetformsofNAGSdeficiencyhaveextremelyhighbloodammonialevels.Affectednewbornsmayexperienceammonialevelsfrom>150µmol/Luptoorpossiblyexceeding1000µmol/L(normalconcentrationinnewborns:<100µmol/L).Inthelateonsetforms,although blood ammonia levels are always high during the acute phase, they may be normal at other times(thenormalrangevariesbetweenlaboratoriesbutisgenerally<50µmol/L).Thus,totalnitrogentesting must be performed carefully.
� Amino acids: plasma levels of Gln are elevated. Urinary amino acids are non-diagnostic in NAGS deficiencybutmaybeimportantinordertohelpruleouthyperammonemia-hyperornithinemia-homocitrullinuria or lysinuric protein intolerance.
� Urinary orotic acid: level is within normal range or low. � Urinary organic acids: arewithinreferencerangesinNAGSdeficiency.Itisessentialtoruleoutorganic
acid disorders, which can present with similar signs and symptoms including hyperammonemia. � Liver biopsy: might be helpful if performed under protein load procedure but the results are not always
conclusive; basal enzyme activity may not always be pathognomonic (Colombo 1982, Heckmann 2005, Jackson 1986). The invasive nature of a liver biopsy limits its current use.
� DNA testing: finalconfirmationcomesthroughmoleculardiagnosiswiththespecificDNAgenotype(mutation).Thishasbeenavailablesince2003.
Afterevaluationofsymptomsandbiochemicalparameters,theinitialdiagnosisofNAGSdeficiencycanbe supported by liver biopsy, but the results are not always conclusive. The DNA mutation analysis is now usedtoconfirmdiagnosis.Approximately20causativemutationshavebeenreportedworldwideforNAGSdeficiency(Caldovic2007,Haberle2003).
DetectionofamutationintheNAGSgene,mappedtochromosome17q21.31,confirmsthediagnosisofNAGSdeficiencyallowingspecifictherapy,detectionofcarriers,andprenataldiagnosis(Caldovic2002,Caldovic2003,Caldovic2005,Caldovic2007,Häberle2003,Häberle2004,Morizono2004).However,moleculargenetictestingresultsareusuallyavailablewithinfewweeks,thusgenetictestingisconsideredaconfirmatorydiagnostictool.TreatmentofacutehyperammonemiashouldnotbedelayedifNAGSdeficiencyissuspected.
UntilfinalconfirmationisavailablefromtheDNAtestingorhepaticenzymaticassay,adiagnosiscanusuallybenarroweddowntoeitherNAGSdeficiencyorCPS1deficiency.Bothofthesediseaseshavesimilarclinicalandbiochemicalfindingsincludinghyperammonemia,elevatedGlnlevels,nospecificincreaseofurea cycle intermediates, and normal orotic aciduria, with exclusion of organic aciduria (Bachmann 1982, Caldovic 2002, Caldovic 2010).

13 Back to Table of Contents _
2.7 TREATMENT OF NAGS DEFICIENCYSincetheprognosisisstronglyinfluencedbycomaandpeakammonialevels,onceadiagnosisofaUCDismade,treatmentshouldbetailoredtothespecificureacycledisorder(Haberle,etal2012,Summar,Tuchman2001). Patients in hyperammonemic crisis should be transferred to a specialist center (Haberle, et al 2012). The primary goal is to rapidly return plasma ammonia concentration to normal physiologic levels, prevent recurrent hyperammonemia and to avoid the associated complications of the CNS (Valayannopoulos 2005). Early prompt diagnosis of hyperammonemia is crucial for a better outcome. The start of ammonia detoxificationandreversingcatabolismmustnotbedelayed(Haberle,etal2012).Long-termsecondarygoals include maintenance of optimal growth and neurological development by avoiding nutrient imbalance andinsufficiencyandaminoaciddeficiencies.Intheacutephaseofseverehyperammonemia,thebestwaytoreduceplasmaammoniaconcentrationquicklyisbydialysis.Othermethodsarehemofiltrationandhemodialysis and protein restriction diet (Ah Mew et al 2015).
Treatment of Acute Manifestations
• Pharmacological interventions should be performed to allow alternative pathway excretion of excessive nitrogen. These include nitrogen scavenger therapy (sodium phenylacetate and sodium benzoate).Deficientureacycleintermediatesneedtobereplaced,whichcanincludearginine(IVinfusion)and/orcitrulline(oralpreparation).InpatientswithNAGSdeficiency,replacementofn-acetylglutamate with the analog molecule carbamyl glutamate (or also known as Carbaglu® (carglumic acid)) can improve clinical symptoms. CARBAGLU should be added to the treatment regimen in a patient without a clear diagnosis at initial presentation [Dosing in adults and children is 100 mg/kg/day to 250 mg/kg/day divided into two to four doses] (Ah Mew et al 2015).
• Treat catabolic state with calories from glucose, fats, and essential amino acids. Complete restriction of protein should not exceed 12-24 hours because depletion of essential amino acids results in protein catabolism and nitrogen release. Enteral nutrition is preferred; however parenteral nutrition is an option. Placement of a nasogastric tube is needed for administration of essential amino acids, infant formula, and administration of cofactors like CARBAGLU. Other strategies to combat catabolism include low-dose continuous infusion of insulin with maintenance of glucose delivery (Ah Mew et al 2015 and Haberle, et al 2012).
• Reduce the risk for neurologic damage.Intravenousfluids(10%glucoseinfusion)shouldbeginwithappropriate electrolytes) for physiologic stabilization, cardiac pressors as necessary, and treatment of subclinical seizures (Ah Mew et al 2015 and Haberle, et al 2012).
Long-term Treatment of Manifestations. The goal of treatment is to achieve normal development and to preventhyperammonemiawhileprovidinggoodqualityoflifeandavoidingsideeffectsandcomplications.Thisis accomplished by decreasing nitrogen load with dietary restriction of protein, and using nitrogen scavengers (phenylbutarate and sodium benzoate) to provide alternative routes for nitrogen dispersal. Prompt replacement of citrulline or arginine may be necessary. CARBAGLU may be used to promote normal or near normal functionoftheCPS1enzymeinNAGSdeficiency(AhMewetal2015andHaberle,etal2012). Liver transplantation is the only curative treatment preventing further hyperammonemic crises, allowing normal diet, and stopping drug administration. It should be performed in a patient without severe neurological damage, while in a stable metabolic condition (Haberle, et al 2012).

14 Back to Table of Contents _
2.7.1 TREATMENT OF NAGS DEFICIENCY WITH CARBAGLU® (carglumic acid)MonotherapywithCARBAGLUisthetreatmentofchoiceforNAGSdeficiency(Haberle,etal2012).Itiscurrently the only available product indicated by the United States FDA as adjunctive therapy for the treatment ofacutehyperammonemiaduetoNAGSdeficiencyandasmaintenancetherapyforthetreatmentofchronichyperammonemiaduetoNAGSdeficiency.Givenorally,CARBAGLUactsasasubstituteforthemissingphysiologic activator and thereby stimulates the enzyme CPS1, triggering the urea cycle and normalizing blood ammonia concentration (Tuchman 2008). Any episode of acute symptomatic hyperammonemia should be treated as a life-threatening emergency. Uncontrolled hyperammonemias can rapidly result in brain injury, brain damage, and death. Prompt use of all therapies necessary to reduce plasma levels is essential.
CARBAGLU is a Carbamoyl Phosphate Synthetase 1 (CPS 1) activator indicated as adjunctive therapy for thetreatmentofacutehyperammonemiaduetothedeficiencyofthehepaticenzymeN-acetylglutamatesynthase (NAGS) and for maintenance therapy for the treatment of chronic hyperammonemia due to thedeficiencyofthehepaticenzymeN-acetylglutamatesynthase(NAGS).IntheacutestateofseverehyperammonemiaduetoNAGSdeficiency,treatmentforpediatricandadultpatientswithCARBAGLUis initiated at a dose of 100 mg/kg/day to 250 mg/kg/day. The total dose should be divided into 2 to 4 doses and rounded to the nearest 100 mg. Plasma ammonia levels are generally reduced within 24 hours, butitmaytake2-3daystorecovertowithinnormalranges.Duringacutehyperammonemicepisodes,concomitant administration of CARBAGLU with other ammonia lowering therapies, such as alternate pathway medications, hemodialysis, and dietary protein is recommended. The administration of ammonia scavengers,ofL-arginineorL-citrullineandinNAGSdeficiency,ofcarbamylglutamateishighlyvaluablefor treating acute hyperammonemic decompensation. Citrulline and/ or arginine administration aims at maximizing ammonia excretion through the urea cycle. (Haberle, et al 2012).
Once the ammonia levels have normalized, doses may be adjusted to establish a long-term treatment regimen that targets normal plasma ammonia levels for the age of the patient. Based on limited data in 22 patients receiving maintenance treatment with CARBAGLU in a retrospective case series, pediatric and adult maintenance doses were usually less than 100 mg/kg/day. During maintenance therapy the concomitant use of other ammonia lowering therapies and protein restriction may be reduced or discontinued based on plasma ammonia levels.
ManagementofhyperammonemiaduetoNAGSdeficiencyshouldbedoneincoordinationwithmedicalpersonnelexperienced in metabolic disorders. Ongoing monitoring of plasma ammonia levels, neurological status, laboratory tests and clinical responses in patients receiving treatment with CARBAGLU is crucial to assess patient response to treatment.
Contraindications: None
Warnings and Precautions Hyperammonemia: Monitor plasma ammonia levels during treatment.Prolonged exposure to elevated plasma ammonia levels can rapidly result in injury to the brain or death.Prompt use of all therapies necessary to reduce plasma ammonia levels is essential.
Therapeutic Monitoring:Plasma ammonia levels should be maintained within normal range for age via individual dose adjustment.
Nutritional Management: IntheinitialtreatmentofNAGSdeficiency,proteinrestrictionisrecommended.Whentheplasmaammonialevel is normalized, dietary protein intake can usually be reintroduced.
15 Back to Table of Contents _
3.0 PRODUCT INFORMATION
3.1 GENERAL INFORMATIONCarbaglu® (carglumic acid) was approved by the US Food and Drug Administration (FDA) in 2010 for the adjunctivetherapyforthetreatmentofacutehyperammonemiaduetoNAGSdeficiencyandforthemainte-nancetherapyforthetreatmentofchronichyperammonemiaduetoNAGSdeficiency.
The marketing authorization for CARBAGLU is held with Orphan Europe Sarl (Paris, France), part of the Recordati Group.
CARBAGLU is distributed in the United States by Recordati Rare Diseases, Inc. (Lebanon, New Jersey), part of the Recordati Group.
3.2 COMPOSITION OF CARBAGLU TABLETSEach CARBAGLU tablet contains 200 mg carglumic acid as active substance and is presented as a white elongated tablet (size 18.0 x 6.0 mm), scored on both sides and coded “C” on one side. It is available in 2 differentpacksizes:5-and60-tabletbottles(seealsoSection3.3).
3.3 ACTIVE PHARMACEUTICAL INGREDIENT: CARGLUMIC ACIDCARBAGLU was developed as a new chemical entity by Orphan Europe, part of the Recordati Group, and is distributed in the United States by Recordati Rare Diseases. Carglumic acid is the International Non Proprietary Name (INN) of the active pharmaceutical ingredient (API) which was proposed by Orphan Europe and agreed on by the World Health Organization (WHO Recommended INN List 46).
Name of ingredients Function Formula per tablet
Quantity per5-tablet container
Quantity per60-tablet container
Active substance:Carglumic acid active substance 200 mg 1000 mg 12000 mg
Excipients:Microcrystalline cellulose binder 270 mg 1350mg 16200 mgSodium lauryl sulfate wetting agent 0.5 mg 2.5 mg 30mgHypromellose binder 4 mg 20 mg 240 mgCroscarmellose sodium disintegrating agent 19 mg 90 mg 1140 mgSilica, colloidal anhydrous glidant 1.5 mg 7.5 mg 90 mgSodium stearyl fumarate lubricant 5 mg 25 mg 300mgPurifiedwater* wetting agent 30mg
Total 500 mg 2500 mg 30000 mg*substancedisappearingduringmanufacturing
16 Back to Table of Contents _
General Information
3.4 EXCIPIENTSThe excipients are microcrystalline cellulose, sodium lauryl sulfate, hypromellose, croscarmellose sodium, silica colloidal anhydrous and sodium stearyl fumarate. For safety reasons, no excipient from animal origin has been selected during the pharmaceutical development of the formulation.
All excipients are described in the European and US Pharmacopoeia and are tested for full compliance with theirofficialmonographsbeforethemanufactureofthefinishedproduct.
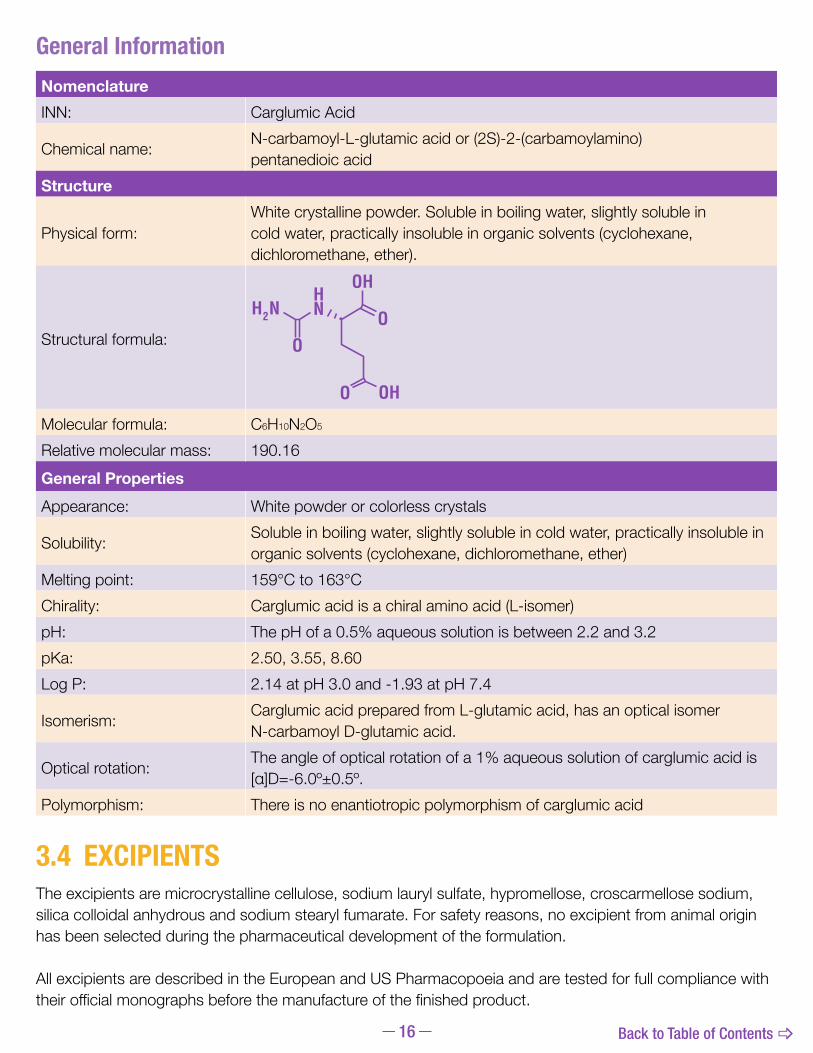
NomenclatureINN: Carglumic Acid
Chemical name: N-carbamoyl-L-glutamic acid or (2S)-2-(carbamoylamino) pentanedioic acid
Structure
Physical form:White crystalline powder. Soluble in boiling water, slightly soluble in cold water, practically insoluble in organic solvents (cyclohexane, dichloromethane, ether).
Structural formula:
Molecular formula: C6H10N2O5
Relative molecular mass: 190.16General PropertiesAppearance: White powder or colorless crystals
Solubility: Soluble in boiling water, slightly soluble in cold water, practically insoluble in organic solvents (cyclohexane, dichloromethane, ether)
Melting point: 159°Cto163°CChirality: Carglumic acid is a chiral amino acid (L-isomer)pH: ThepHofa0.5%aqueoussolutionisbetween2.2and3.2pKa: 2.50,3.55,8.60Log P: 2.14atpH3.0and-1.93atpH7.4
Isomerism: Carglumic acid prepared from L-glutamic acid, has an optical isomer N-carbamoyl D-glutamic acid.
Optical rotation: Theangleofopticalrotationofa1%aqueoussolutionofcarglumicacidis[α]D=-6.0º±0.5º.
Polymorphism: There is no enantiotropic polymorphism of carglumic acid

17 Back to Table of Contents _
3.5 DRUG PRODUCTThefinishedproductconsistsoftabletscontaining200mgofcarglumicacidwhichareeasilydispersibleinwater.The tablets are produced in strict compliance with the requirements of the Good Manufacturing Practices (GMP).
3.5.1 ROUTINE CONTROL TESTS OF THE DRUG PRODUCTInadditiontotheverificationofthegeneralcharacteristicsofthetablets(appearance,dimensions),thepharmaceuticaltests cover uniformity of mass and disintegration time, in compliance with the methods described in the European and US Pharmacopoeia. A limit of 1 minute for the disintegration time was set in the interest of providing good dispersion. Adissolutiontestwithalimitofminimum90%dissolutionwithin15minutesisalsoapartofthespecifications.Aproceduretotestthefinenessofthedispersion(untiltheendofshelflife)completesthepharmaceuticaltests.AsecondcarglumicacididentificationtestinthetabletsisperformedbyIRspectrophotometry.ThequalityspecificationsofCarbaglu® (carglumic acid) tablets at release are summarized below:
Specifications of CARBAGLU TabletsTests SpecificationsGeneral characteristics
Tablet appearanceWhite, bar-shaped tablets, scored on both sides and engraved on one side, size 18.0 x 6.0 mm
Pharmaceutical technical testsUniformity of dosage units L1=15.0;L2=25.0Disintegration time Complete disintegration within 1 minDissolution 90%within15minIdentification of carglumic acidHPLC retention time Identical to reference
IR spectrumIdentity of the tablet spectrum minus placebo spectrum compared to the reference spectrum
D-enantiomer of carglumic acid ≤0.1%Assay of carglumic acid 95.0%-105%oflabelclaim(190to210mg/tablet)Tests for impurities Hydantoin-5-propionic acid NMT0.08%Diaza-1,3-dione-2,4-carboxy-7-cycloheptane NMT0.08%Other impurities (individually) NMT0.10%Total of impurities NMT0.75%Water determination (moisture content) ≤5%Microbial limitsTotal viable aerobic countBacteria ≤1000CFU/gYeast and mold ≤100CFU/gTest for specified microorganismsEscherichia coli Absence in 1g of the productHPLC: high performance liquid chromatography; IR: infrared; NMT: no more than

18 Back to Table of Contents _
3.5.2 STABILITY STUDY RESULTS: DRUG PRODUCT Unopened Tablet ContainersIn addition to the routine tests performed for the release of the drug product, other tests were implemented during the stability studies in order to assess the pharmaceutical quality of the tablets: hardness, friability, watercontent(OrphanEurope,dataonfile).Breakabilitytestswerealsocarriedoutatthebeginningofthestudies and were repeated at the end of the studies.
Thestabilityresultssupportaprovisionalshelflifeof36monthsfortheunopenedcommerciallypackagedproductwhenitisstoredat2°Cto8°C(36°F–46°F,inarefrigerator).
Opened Tablet ContainersA complementary stability study was conducted on the drug product after opening the tablet container (Orphan Europe,dataonfile).Onetabletcontainerof3differentbatcheswasopenedforthefirsttimeafter12monthstorageat5°C.Onetabletcontainerwasplacedinaclimaticchamberat25°C+60%RHandtheotheroneinaclimaticchamberat30°C+60%RHfor1month.Thethirdtabletcontainerwasstoredduring1monthatroomtemperatureandwasopenedandclosed3timesperdaytomimictheconditionofuseoftheproduct.
The tests performed did not reveal any important degradation phenomenon and support a storage period of upto1monthafterfirstopeningofthetabletcontainer,atatemperaturebelow30°C(86°F).
19 Back to Table of Contents _
3.6 PACKAGINGCarbaglu® (carglumic acid) is available in 5 or 60 tablets in a polypropylene bottle with polyethylene cap and desiccant unit. The desiccant (white silica gel) is placed inside the stopper to prevent humidity problems due to the high amounts of disintegrating agent used in the formulation.

20 Back to Table of Contents _
4.0 NONCLINICAL DATA
4.1 PHARMACOLOGY
4.1.1 MECHANISM OF ACTIONThe mechanism of action of Carbaglu® (carglumic acid) is based on the structural similarity of carglumic acid with NAG and its ability to replace NAG as an activator of mitochondrial CPS1, thefirstenzymeoftheureacycle.
4.1.2 IN VIVO STUDIESEvidenceforpharmacologicalefficacyofcarglumicacidinthetreatment of hyperammonemia is based on the treatment of normal or partially hepatectomised rats receiving potentially lethal doses of ammonia, as reported in the following 2 publications:
• Kim et al. (1972) found that 1 mmol/kg of intraperitoneally administeredcarglumicacidprotected61%ofratsafterlethalintravenous dose of ammonium acetate (10.8 mmol/kg), and 4mmol/kgofcarglumicacidprotected85%oftheserats.
• Lee et al. (1998) performed a similar type of experiment in partially hepatectomized rats injected with ammonium acetate(3.4mmol/kg).Afteradministrationofcarglumicacid(1mmol/kg)therewasadecreaseinammonialevelsto278.09±60.02µmol/Lcomparedto415.72±166.38inthecontrolgroup(p<0.05).The correlation between blood ammonia level and behavioral abnormalities observed in hepatic encephalopathy was also investigated.
Concerningthebehavioralgradingscores,theresultsaftercarglumicacidalonewere1.09±0.30comparedto2.54±1.36inthecontrolgroup(p<0.01).Thustheprotectiveeffectonthebehavioralchangesignificantlycorrelatedwiththeireffectsonbloodammonialevel.
In both studies, rats received carglumic acid combined with arginine. Results showed that arginine increased theprotectiveeffectofcarglumicacidagainstammoniaintoxication.Itissuggestedthatthispotentializationby arginine might be due to an increase in the synthesis of NAG. In clinical use for the treatment of NAGS deficiency,thistheoreticalpotentializationwouldthereforebedependentonthelevelofresidualenzymeactivity.Asthebenefitofargininehasnotbeensofarconfirmedinclinicalpractice,theconcomitantuseofarginine cannot be systematically recommended but this option is left open to the clinician.
Another study found that the mitochondrial membrane is more permeable for carglumic acid than for NAG (Rubio1981).Thedifferentialpenetrationintomitochondriainfavorofcarglumicacidwasdemonstratedinmice models where 14C-labeled NAG could not be detected in liver mitochondria but 14C-labeled carglumic acid could be found.
Enzymes of the Urea Cycle: NAGS: N-acetylglutamate synthaseCPS1: carbamoyl phosphate synthetase 1OTC: ornithine transcarbamylase ASS: argininosuccinate synthetase ASL: argininosuccinate lyase ARG: arginase

21 Back to Table of Contents _
4.2 PHARMACOKINETICSThepharmacokinetic(PK)profileofcarglumicacidwasdeterminedbasedon2studiesusingradiolabelledcarglumic acid: 1 study was conducted in rats after single oral administration and 1 study was an in vitro studyusinghumanandrathepatocytes(OrphanEurope,dataonfile).
In addition, toxicokinetic data collected during 2-week and 6-month repeated dose toxicity studies in rats as well as data from an absorption, distribution, metabolism and elimination (ADME) study in dogs were available tocompletetheinformationonthePKofcarglumicacid(OrphanEurope,dataonfile).Inthesestudies,carglumic acid was measured in plasma and urine samples using a validated liquid chromatographic-tandem massspectrometry(LC/MS-MS)method,withahighlevelofsensitivityandspecificity.
4.2.1 ABSORPTIONTheabsorptionprofileinratsshowedagender-independentTmax of between 2 and 4 hours, without a dose proportional AUC or Cmax in the table below.
After repeated administration for 6 months, no accumulation phenomenon was shown in plasma.
4.2.2 METABOLISMThemetabolicprofilewasaddressedinvitro(onculturesofhumanandrathepatocytes)andinvivo(inratswith 14C carglumic acid) and did not reveal any metabolite formation. Nevertheless, in rats given a single oral dose of 500 mg/kg 14Ccarglumicacid,51%,37%and9%oftheradioactivityhasbeenshowntobeeliminated within 96 hours by urine, feces, and CO2, respectively. Therefore it is assumed that a low level of metabolization of carglumic acid should occur in vivo, leading to a total breakdown of the molecule into CO2 even if no intermediate such as 14C-glutamic acid or any other potential metabolite was detected under the experimental conditions.
Preliminary comparative analysis of the preclinical and clinical information on Carbaglu® (carglumic acid) metabolismcouldhaveledtotheconclusionthattheremaybesomepotentialdifferencesinthemetaboliteprofilesofcarglumicacidinratsandhumanswithregardstotheformationofHPA.Thismoleculeisacyclizationby-product of the drug substance, whose level in the drug product is strictly controlled in order to remain below orequalto0.1%m/m;itisalsoaknownmetaboliteofhistidinecatabolism,inparticularinratsandhumans,
Evolution of Cmax Versus the Administered Doses in Different Studies In Rats.
Daily doses (mg/kg) Time of sampling Cmax (µg/ml)
Male Female250 Day 12 of the 2-week
toxicity study 50.4 39.1
500 Day 1 of the 6-month toxicity study 76.7 68.7
1000 Day 1 of the 6-month toxicity study 85.9 95.5
500 End of the 6-month toxicity study 71.6 82.9

22 Back to Table of Contents _
and was investigated as a potential metabolite of carglumic acid. The assay of HPA by LC/MS-MS in plasma ofhealthyvolunteersshowedthatsomevolunteershadquantifiableamountsofHPAinafewsamplescollected after dosing with Carbaglu® (carglumic acid), which could be interpreted as evidence that HPA is a metabolite of carglumic acid, at least in small amounts in humans. There was no convincing evidence of metabolizationofcarglumicacidintoHPAasnoPKprofileofthispotentialmetabolitewasdetectedbutonlyisolatedfluctuationsofthiscompoundproducedendogenouslyviahistidinemetabolism.
An absorption, distribution, metabolism, and elimination study of carglumic acid after oral and intravenous administration was performed in dogs. A cross species comparative analysis of the preclinical and clinical information available was also done.
TheconclusiononCARBAGLUmetabolismisthatthePKpropertiesfitwellbetweenthe3species(rats,dogs, and humans).
4.2.3 ELIMINATIONInrats,carglumicacidiseliminatedmainlyinurine(50%)andinfeces(about40%)asunchangedcompound. Although no circulating metabolite was detected, a small percentage of the total radioactivity was found to be eliminated as expired CO2(about9%,72hourspost-dose).Theeliminationisbiphasicwithafirstrapideliminationphaseinwhichabout70%ofthedoseiseliminatedduringthefirst12hours.Thetotalexcretioninratsaccountsforabout97%oftheadministereddoseafter96hours.Toxicokineticdatashowed that T1/2isabout3to3.5hourswhatevertheadministereddoseinrats.
4.3 SAFETY PHARMACOLOGYThreesafetypharmacologystudiesconductedbyOrphanEuropeevaluatedtheeffectsofcarglumicacidon the behavior and body temperature in the rat (Irwin test), on respiratory parameters in the rat, and on the cardiovascularsysteminthedog(OrphanEurope,dataonfile).
Thesestudiesdidnotrevealanyrelevanteffectsfollowingsingledoseadministrationupto1000mg/kgofcarglumicacidinbothspecies.Inaddition,aninvitrostudyonisolatedcaninePurkinjefibres(OrphanEurope,dataonfile)confirmedtheabsenceofcardiovasculareffects,inparticularonactionpotentialduration of carglumic acid.
4.4 TOXICOLOGY4.4.1 GENERAL TOXICOLOGYBaseduponclinicalexperience,thetreatmentwithCARBAGLUmaybestartedasearlyastheveryfirstdayof life. Therefore toxicology studies were deliberately designed with consideration for dosing of infants and children.ThegeneraltoxicologicalprofileforCARBAGLUwasaddressedthroughsingledosetoxicitystudiesand repeated dose toxicity studies in rodents.
The repeated dose toxicity study program involved one study in newborn rats, administered from the 4th to the21stdayoflifeandone6-monthstudyinjuvenile/youngadultrats,withinterimevaluationat3monthsand including satellite groups of animals treated 4 weeks for micronucleus testing. Moreover, immunotoxicity endpoints and cell proliferation evaluation in selected organs were incorporated in the study.

23 Back to Table of Contents _
4.4.2 ACUTE TOXICITY STUDIESSingle dose toxicity studies were performed in the rat via oral and intravenous administration (Orphan Europe,dataonfile).
Singledosesofcarglumicacidupto2800mg/kgorallyand239mg/kgintravenouslyadministereddidnotinduce any mortality or abnormal clinical signs in adult rats. There were no variations of body weight and no abnormalities observed at necropsy.
4.4.3 REPEATED DOSE TOXICITY STUDIESTherepeateddosetoxicityprogramincludes2studiesintherat(OrphanEurope,dataonfile);asubacutetoxicitystudyperformedinnewbornratstoaddresstheintendeduseinthefirstpartoflife,andachronic6-month study in rats to address the planned long term exposure.
In the newborn rats receiving daily carglumic acid by oral gavage for 18 days as well as in the young rats receivingdailycarglumicacidfor26weeks,theNoObservableEffectLevel(NOEL)wasestablishedat500mg/kg/dayandtheNoObservedAdverseEffectLevel(NOAEL)wasestablishedat1000mg/kg/day.
A low level of metabolization was observed in vivo in rats. This level was too low to enable the detection of any metabolite explaining the metabolic pathway into expired CO2 but with reassuring bioassay results in human plasma demonstrating that HPA, the main impurity of Carbaglu® (carglumic acid), is unlikely to be one of its metabolites.
4.4.4 GENOTOXICITYA complete battery of genotoxicity studies was performed on carglumic acid and its 2 impurities hydantoin-5-propionicacid(HPA)anddiaza-1,3-dione-2,4-carboxy-7-cycloheptane(Diaza)athighdoses(OrphanEurope,dataonfile).
• Carglumic acid was tested for Ames test, bacterial reverse mutation test, in vitro chromosome aberration in human lymphocytes test, rat erythrocyte micronucleus test in bone marrow, without treatment and after 4 week treatment by oral route (gavage) in rats, up to 1000 mg/kg/day
• Diaza was tested for Ames test.• HPA was tested for Ames test, chromosome aberrations by in vitro human lymphocytes metaphase
analysis, in vivo study using the micronucleus test in mice up to 2000 mg/kg/day.
All those studies demonstrated that carglumic acid and its 2 impurities HPA and Diaza are devoid of genotoxicpotential(OrphanEurope,dataonfile).
4.4.5 CARCINOGENICITYThe carcinogenic potential of carglumic acid has not been evaluated. However, carglumic acid does not have structural analogy with known carcinogens. In addition, there were no immunotoxicity, genotoxic, or immunodepressivepropertiesbasedonabatteryofmutagenicitytests(OrphanEurope,dataonfile)andtests on rats treated with carglumic acid for 6 months. There was no cell proliferation or signs of hyperplasia inthetissuesandorgansofratsafterchronicadministration(OrphanEurope,dataonfile).

24 Back to Table of Contents _
4.4.6 REPRODUCTION TOXICITY STUDIESIn embryo-fetal developmental toxicity studies, pregnant rats and rabbits received oral carglumic acid during organogenesisatdosesupto1.3timesthemaximumrecommendedhumanstartingdosebasedonbodysurface area (mg/m2). Actual doses were 500 and 2000 mg/kg/day (rats) and 250 and 1000 mg/kg/day (rabbits).Thehighdosesresultedinmaternaltoxicityinbothratsandrabbits.Noeffectsonembryo-fetaldevelopment were observed in either species.
In a peri- and post-natal developmental study, female rats received oral carglumic acid from organogenesis throughday21post-partumatdosesupto1.3timesthemaximumrecommendedstartinghumandosebased on body surface area (mg/m2).Actualdoseswere500and2000mg/kg/day.Areductioninoffspringsurvivalwasseenatthehighdoseandareductioninoffspringgrowthwasseenatbothdoses.
Therewerenoeffectsonfertilityorreproductiveperformanceinfemaleratsatoraldosesupto2000mg/kg/day(1.3timesthemaximumrecommendedhumanstartingdosebasedonbodysurfacearea).Inaseparatestudy,matingandfertilitywereunaffectedinmaleratsatoraldosesupto1000mg/kg/day(0.6times the maximum recommended human starting dose based on body surface area).
5.0 CLINICAL DATA
5.1 PHARMACODYNAMICS
The mechanism of action of Carbaglu® (carglumic acid) is based on the structural similarity with NAG and its ability to replace the NAG function as activator of CPS1. CARBAGLU is able to restore the activity of the urea cycle, to normalizeplasmaammonialevels,whichareabnormallyelevatedinNAGSdeficiencypatients,andtofinallyrestoreureagenesis (O’Connor 1985).
Short term administration of CARBAGLU rapidly restores the ureagenesis capacity in patients with NAGS deficiencytonormal.(Caldovic2004,Tuchman2008).Thiseffectcorrelatedwith:
• Normalization of both plasma ammonia and glutamine levels• Markedly increased urea production
TheauthorsstudiedpatientswithNAGSdeficiencyandonesubjectheterozygousforthedisorderpriortoandafter3daysofCARBAGLUtreatment(2.2g/m2/day). One patient and her heterozygous mother were studied with the [15N] tracer and another patient with the [13C] tracer incorporated as the principal surrogate marker.Theresultsconfirmedrestorationtocontrollevelsofmarkedlydeficientureagenesisfollowingtheadministration of CARBAGLU, and suggested that the [13C] tracer method is reliable even if it does not contain ammonia and labels urea more slowly than [15N].
In conclusion, these results are consistent with a complete restoration of ureagenesis shortly after the initiation of treatment. The published experience (Caldovic 2004, Tuchman 2008) and the observations in NAGSdeficiencypatientstreatedwithCARBAGLUshowaquick,significantandsteadydecreaseinthekeybiologicalresponsemarkers,ammonemiaandglutaminemia,inresponsetothespecifictreatment.

25 Back to Table of Contents _
5.2 PHARMACOKINETICS5.2.1 PHARMACOKINETICS IN HEALTHY VOLUNTEERSA PK study was conducted in 12 healthy male adults after a single oral administration of 100 mg/kg in order todetermineplasmaconcentrationsandurineexcretionofcarglumicacid(OrphanEurope,dataonfile).Aspecificandhighlysensitiveassaymethodwasdevelopedandvalidatedforcarglumicaciddeterminationusing HPLC coupled with tandem mass spectrometry.
Theinter-subjectvariabilitywasmoderate(coefficientsofvariationaround30%formostparameters).Carglumic acid peaked in plasma after a few hours suggesting an active transport through the intestinal mucosa. The oral bioavailability of carglumic acid was low, probably due to weak intestinal absorption andnotbecauseofextensiveliveruptake.Anotheropenlabelstudy(OrphanEurope,dataonfile)with14C-labelled carglumic acid investigated the mass balance, PK and metabolism following a single oral administrationof70μCurie14C-labeledcarglumicacid(100mg/kg)in3healthymalesubjects.Themetaboliccapacityofcarglumicacidwasdemonstratedtobelimitedinatleast2outofthe3subjects(OrphanEurope,dataonfile).Theparentcompoundassayedwasshowntobeexcretedinlargeamountsinfeces(approximately60%inSubjects001and003butonly8.5%inSubject002),andtoalesserextentinurine(approximately9%).Theseresultsaccuratelymatchedtheradioactivityamountsexcretedinfeces(approximately72%inSubjects001and003butonly16.5%inSubject002)andperfectlymatchedtheradioactivityamountsexcretedinurine(approximately9%).
Theplasmaeliminationcurveofcarglumicacidwasbiphasicwitharapidphaseoverthefirst12hourspostdosing followed by a slow phase from 12 to 72 hours (no detectable concentration of the parent compound after this time point). The apparent terminal half-life was estimated to be around 21 hours. The plasma profileofcarglumicacidonlymatchedthefirstpeakofradioactivity.
5.2.2 PHARMACOKINETICS IN NAGS DEFICIENCY PATIENTSCarglumicacidplasmalevelsweremeasuredin11NAGSdeficiencypatients(OrphanEurope,dataonfile).Theageofthesepatientsrangedfrom1dayto13yearsandthedailydosegivenrangedfrom7.4to122.3mg/kg.Bloodwascollectedattroughand/oranticipatedpeak(around3hourspost-dose)times.
The range of plasma concentrations measured in these patients, including newborn infants, was consistent with the data obtained in healthy adults.
Pharmacokinetic Parameters for the Carbaglu® (carglumic acid) TabletPharmacokinetic parameter Median RangeTime to peak plasma concentration 3hours (2 – 4)Peak plasma concentration 2.6µg/mL (1.8–4.8)Apparent volume of distribution 2657 L (1616–5797)Terminal half-life 5.6 hours (4.3–9.5)Apparent total clearance 5.7 L/min (3.0–9.7)Renal clearance 290 mL/min (204 - 445)

26 Back to Table of Contents _
5.2.3 CONCLUSIONSThe results of the PK analyses found that:
• The apparent volume of distribution was 2657L (range:1616-5797). Protein binding has not been determined.• After single oral administration, Cmaxwasachievedin2-4hoursinbothhealthyandNAGSdeficiencypatients.• In humans, after a single oral dose of 100 mg/kg of body weight, carglumic acid was mainly eliminated
astheunchangedcompoundinfeces(around60%)andtoamuchlesserextentinurine(9%). 5.3 EFFICACY
5.3.1 RETROSPECTIVE STUDY DESCRIPTION (OrphanEurope,dataonfile)
Duetotherareandseriousnatureofthedisease,OrphanEuropeassessedtheefficacyofCarbaglu® (carglumic acid) based on data from uncontrolled, retrospective case series.
OrphanEuropeconductedaretrospectivestudyusingdatacollectedfrom23NAGSdeficiencypatients(14males and 9 females) who received treatment with CARBAGLU and who started treatment before December 2007(thecut-offdateforsafetydatacollectionwasDecember2008).
TheprimaryobjectivewastoreviewtheclinicalandbiologicalresponseofNAGSdeficiencypatientstoCARBAGLUwithinthefirst7daysoftreatment(shortterm)andatthelastreport(longterm).Fortheshortterm analysis, ammonemia was the primary biomarker supported by glutaminemia and citrullinemia results. Forthelongtermanalysisall3biomarkers,ammonemia,glutaminemiaandcitrullinemia,wereconsideredequally.
The secondary objectives were the evaluation of the clinical development, ie, neurological and psychomotor status, and anthropometric development (growth) parameters; the analysis of the implementation of a restrictive/protein-free diet and concomitant treatment; the analysis of the CARBAGLU doses prescribed as anindirectefficacyparameterandasadefinitionofthedoseresponsedetermination.
This report also included a short PK analysis and a short description of safety data.
Outofthe23patients,18(78.3%)wereunderlongtermcontinuoustreatmentwithCARBAGLUandprovidedatatosupporttheclaimfortheindicationofCARBAGLUinthemanagementofNAGSdeficiency(pg27).Intotal,5patientsdiscontinuedtreatment:3patientshadconfirmationofaheterozygoteNAGSgenemutationbyDNAtesting,and2patientsdied(seeSection5.3.5).The age at initiation of treatment was heterogeneous: 9 patients started at <1 month of age, 9 patients between2and11monthsofage,and5patientsbetween1and13yearsofage.
5.3.2 DIAGNOSISDiagnosisofNAGSdeficiencywasconfirmedbyDNAtestingin19ofthe23patients,allofwhichwereperformed in the same laboratory.

27 Back to Table of Contents _
5.3.3 CARBAGLU TREATMENTData available on dosing of CARBAGLU were assessed relative to short term and long term treatment. Analysisofshorttermtreatmentshowedthatdailydoseswerebetween100-250mg/kgduringthefirstfew days, administered 2 to 4 times a day. The dose was then slowly reduced over time depending upon the biological response. Extremely low doses were associated with failure to prevent metabolic decompensation.
Thestudyshowedatrendtowardmaintenanceoftheinitialdoseinthefirstfewdaysafterstartingtreatment. Those patients who started with a relatively high daily dose of CARBAGLU and responded positively to the treatment were slowly treated with relatively lower CARBAGLU doses.
Atthecut-offdatefortheefficacyanalyses(December31,2007)thedurationoftreatmentrangedfrom7.4monthsto 248.5 months (20.7 years). With the exception of 2 patients, the duration of exposure to CARBAGLU for the majorityofpatientsexceeded1year.MostpatientshadalongtermexposuretoCARBAGLUfor≥5years(14outof23patients).
Concomitanttreatmentswerespecificaminoacids,ammoniascavengersandhemodialysis.Hemodialysiswas administered only as a rescue therapy during a hyperammonemic crisis in 4 patients.
Diet analysis showed that the majority of patients quickly changed from a protein-restricted diet to a normal protein intake upon CARBAGLU initiation, with the exception of 1 patient who was kept on a mildly protein-restricted diet on a long term basis.
Confirmed NAGSdeficient patients
N=23Discontinued
N=2Reason for discontinuation:
Heterozygous for NAGS
DiscontinuedN=3
Reason for discontinuation:
Efficacy AnalysisN=21ª
Long term TreatmentN=18 b
- Death (N=2)- Heterozygous for NAGS (N=1)
Overall ExposureN=143
Long term TreatmentN=99
Short term TreatmentN=44
Non-NAGS DeficiencyN=76
Confirmed NAGS DeficiencyN=23
(Retrospective Study)
Study Population of the Retrospective Study
NAGS = N-acetylglutamate synthase; N = Number of patients.A. The development of ammonemia was assessed in 20 patients, since 1 patient
had no ammonia level reported prior to treatment with Carbaglu® (carglumic acid).B. One of these patients was previously diagnosed and treated as a patient with
CPS1 deficiency until a DNA confirmation of NAGS homozygous mutation.

28 Back to Table of Contents _
5.3.4 EFFICACY RESULTSPlasma ammonia concentrations, measured in 20 patients,showedasignificantandrapiddecreasein pathological plasma ammonia levels to normal ranges. Plasma ammonia reached normal levels withinthefirst24to48hoursafterinitiationofCARBAGLU treatment, with the exception of 1 patient whoseextremelyhighbaselineammonia(>1400μmol/L)onlyreachednormallevelsbyDay3.Atthelongtermtreatment evaluation, the ammonia levels were within the normal range in all patients receiving an adequate dose of CARBAGLU. Decompensations were only reported at the end of the data collection period in 2 patients due to inadequately low dosing.
Plasma Gln levels were elevated before CARBAGLU therapy in 16 patients, sometimes even despite sodium benzoate or phenylbutyrate treatment. They rapidlynormalizedwithinthefirst24to48hoursofCARBAGLU treatment and were maintained with continuous CARBAGLU treatment.
Plasmacitrullinelevelswerelowin13patientsbeforeinitiation of CARBAGLU treatment, as expected during hyperammonemic crisis, and normalized quicklyafterthefirstdoseofCARBAGLU.
Atbaseline,7of10patients(70%)presentedwithaffectedneurologicalstatus,only2ofwhomcontinuedtohaveanaffectedneurologicalstatusatthelongtermevaluation.Theother5patientswhopresentedwithaffectedneurologicaldevelopmentatbaselinehadnormalneurologicaldevelopmentatthelastfollow-up.Noneofthenon-affectedpatients(30%)atbaselinedevelopedaffectedneurologicalstatusatthelongtermevaluation.
Themean(95%confidenceinterval)changefrombaselineforheightinthe16patientswithdatawas+0.5,(-0.1,+1.2)andthemedianchangefrombaselineof+0.5.Themean(95%confidenceinterval)changefrombaselineforweightinthe16patientswithdatawas+1.9,(+0.4,+3.4)andthemedianchangefrombaselineof+0.9.These results show an improvement in height development and weight over time with CARBAGLU treatment.
Ammonia Response for 13 Evaluable NAGS Deficiency Patients at Baseline and After Treatment With Carbaglu® (carglumic acid)
Reduction in Glutamine Levels on Treament With Carbaglu Over Time
Mea
n GL
N (µ
mol
/L)
Days
Glutamine Response
0
200
Baseline Day 2 Day 4 Day 6 Long-term
400
600
800
1000
0
50
100
150
200
250
300
Mea
n NH
3 (µ
mol
/L)
NH3 Response
Baseline Day 2 Day 4 Day 6 Long-term
Days
Mea
n GL
N (µ
mol
/L)
Days
Glutamine Response
0
200
Baseline Day 2 Day 4 Day 6 Long-term
400
600
800
1000
0
50
100
150
200
250
300
Mea
n NH
3 (µ
mol
/L)
NH3 Response
Baseline Day 2 Day 4 Day 6 Long-term
Days

29 Back to Table of Contents _
5.3.5 PUBLISHED DATA• ThefirstpatientwithNAGSdeficiencydescribedintheliteratureandtreatedwithcarglumicacidstartedthe
treatmentin1980onhis10thdayoflife*(Bachmann1981,1982;Schubiger1991).Twomalesiblingshadpreviouslydiedwithinthefirst2weeksoflifeandhyperammonemiahadbeensuspectedinthesecondcase.In the neonatal period, the patient was adhered to regimen that involved restriction of protein intake, sodium benzoate infusions, and adequate caloric supply with glucose infusions. An accidental withdrawal of the benzoatesupplyledtohyperammonemiawithcoma,muscularhypotonia,andarespiratoryinsufficiencywhichnecessitatedprolongedartificialventilation.Carglumicacidwasthenstartedatadailydoseof100mg/kgallowing withdrawal of sodium benzoate and increase of alimentation. An initial increase to a daily dose of 750 mg/kg led to the clinical picture of a sympathomimetic-like reaction: tachycardia, profuse sweating, bronchial hypersecretion,increasedbodytemperatureandpersistentcrying.Attheageof13.5monthstheboywasdischarged fromthe hospital with the following therapy: enteral nutrition (1.5 g of protein/d), carglumic acid (180 mg/kg/d) and arginine. He was already developmentally delayed with pronounced muscular hypotonia. During his 9 years of life, he required 7 hospitalizations of short duration due to metabolic relapses usually caused by changes in nutrition or febrile infections. On several occasions increased transaminases were detected. The somatic development progressed along the 50th percentiles and the psychomotor development remained delayed. In 1990, the patient died at the age of 9.5 years after an episode of coma and generalized convulsions despite successfully lowering ammonia level by sodium benzoate infusion; the cause of death was not clearly elucidated and the relation to treatment was not assessable.
• Anotherpublication(Hinnie1997)reportedthecaseofa20-year-oldpatientwithNAGSdeficiencyfromthe United Kingdom (UK) who had been hospitalized on several occasions for hyperammonemia since the age of 1.5 years. During the last admission for severe hyperammonemia secondary to febrile illness, he developed pneumonia and generalized convulsions. Hemodialysis was performed for 5 weeks in order to control ammonia levels, during which time he developed 2 episodes of septicemia. Carglumic acid was introduced at a daily dose of 60 mg/kg following cessation of hemodialysis because ammonia levelsroseagainabove100µmol/L.Thepatienthassubsequentlymaintainedlevelsaround20-60µmol/Landhasbeencrisisfreefor2yearsoncarglumicacidandarginine.Unfortunately,hehasbeenleft with gross cerebral dysfunction and paraplegia with incontinence; prior to the last episode he had been fully ambulant with normal somatic development.
• OnecaseofneonatalNAGSdeficiencywasalsoreportedintheUK(Morris1998).At4monthsofage,the patient was started on a daily dose of carglumic acid (100 mg/kg) which allowed an increase in daily protein intake to 2.5 g/kg and a reduction in sodium benzoate dosage. At 20 months of age the patient had no neurological or developmental abnormalities and had experienced only one hyperammonemic episode precipitated by otitis media.
• AnothercaseofneonatalNAGSdeficiencywasreportedintheNetherlands.Thisneonatewasrescuedfrom (Huijmans 1998) was rescued from neonatal hyperammonemic coma by peritoneal dialysis, sodium benzoate and arginine infusions; he was then started on a daily dose of carglumic acid of 100 mg/kg at the 10th day of life. Blood ammonia completely normalized, even upon increasing the daily proteinintakeupto3g/kg.Hismentalandmotordevelopmentwasnormalattheageof1.3years.
• AnothercaseofneonatalNAGSdeficiencywasreportedinSweden(Nordenström2007).Atthe3rdday of life, the patient was rescued from hyperammonemic coma with restriction of oral food intake, infusions of glucose, sodium benzoate and arginine. At the 4th day of life, carglumic acid was given

30 Back to Table of Contents _
orallyatadailydoseof200mg/kg,dividedin3dosesandveno-venoushemodiafiltrationwasinitiated.Duetotechnicalproblem,thehemodiafiltrationwasdiscontinuedfor4h,resultinginincreasingammonialevels(from984to1187μmol/L).Onthe5thdayoflife,theammonialevelstartedtodecreaseandreachednormallevelsabout36hoursaftertheinitialdoseofcarglumicacid.Proteinintakewasgradually increased. Sodium benzoate and carglumic acid were gradually decreased and completely stoppedat13daysofage.Afterfeedingdifficultiesandelevatedammonialevels,sodiumbenzoateandcarglumic acid were reintroduced. Carglumic acid therapy was maintained at a daily dose of 50 mg/kg with normal plasma ammonia levels and a moderate protein-restricted diet was introduced after 5 months of age. At 2.5 years of age, the patient had normal psychomotor development.
• AnothercaseofneonatalNAGSdeficiencywasreportedinGermany(Gessler2010).Thepatientdevelopedslighthyperammonemiaonthe3rddayoflifeandwastreatedwithcarglumicacidbeforeconfirmationofNAGSdeficiency.(Oneyoungersiblinghadpreviouslydiedduetohyperammonemiain the neonatal period and another younger sibling who developed hyperammonemia on the 2nd day of life, was treated with arginine hydrochloride, sodium benzoate and protein restriction. After NAGS deficiencywassuspectedbyenzymeanalysisinthesecondsibling,sodiumbenzoatewasreplacedby carglumic acid. At 10 years of age, this sibling tested heterozygous for NAGS and carglumic acid treatment was stopped without deterioration of urea cycle function). After initiation of carglumic acid, the patient’s diet was supplemented with sodium benzoate and arginine. After 4 hospitalizations for hyperammonemiawithinthe3monthsofage,carglumicacidtreatmentwasstartedat3monthsofage at a daily dose of 250 mg/kg, divided in 4 doses. Sodium benzoate and protein restriction were discontinued, whereas arginine supplementation was maintained. At 14 months of age, the daily dose of carglumic acid was adjusted to about 200 mg/kg. The patient did not develop hyperammonemia in thefollowingyears.NAGSdeficiencywasconfirmedat8yearsofage.At13yearsofage,thepatienthad normal neurological development.
5.4 SAFETY5.4.1 CLINICAL SAFETY IN NAGS DEFICIENCY PATIENTSUptocut-offdateforthesafetyanalysis,December31,2008,18ofthe23patientssufferingfromNAGSdeficiencyintheretrospectivestudyhadexperiencedanadverseevent(AE).BecausetheseAEswerereported retrospectively, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Intotal,120AEswerereportedincluding37seriousadverseevents(SAEs)and83non-seriousAEs.ThereportedAEsweremainlyfrom3systemorganclasses:gastrointestinaldisorders(21%),infectionsandinfestations(19%),andnervoussystemdisorders(14%).Themostcommonadversereactionsin≥13%ofpatients treated with Carbaglu® (carglumic acid) are: infections, vomiting, abdominal pain, pyrexia, tonsilitis, anemia, ear infection, diarrhea, nasopharyngitis, and headache.
Two patients died due to an AE in the retrospective study. One patient died due to multiple organ failure. This patient showed poor compliance and, 1 month prior to the death, the daily dose of carglumic acid was reduced to 5 mg/kg, which was not in accordance with the recommended maintenance dose. Furthermore, a few days prior to the death, the patient experienced severe pneumopathy resulting in multiple organ failure. The second patient died due to a severe episode of hyperammonemia. The daily dose of carglumic

31 Back to Table of Contents _
acid was very low at 6 mg/kg, which was not in accordance with the recommended maintenance dose. Neither of the fatal events was related to Carbaglu® (carglumic acid).
Inadditiontothe2above-mentionedNAGSdeficiencypatientswhodied,9otherpatientsexperiencedan SAE, none of which were related to CARBAGLU. These SAEs were mostly reported in 2 system organ classes: gastrointestinal disorders (10 SAEs, of which 6 were vomiting) and nervous system disorders (10 SAEs). The remaining SAEs were distributed in other system organ classes. None of the SAEs were considered as “related” to carglumic acid by the reporters.
Amongthe83non-seriousAEs,only1wasconsideredas“related”tocarglumicacidbyboththereporterand Orphan Europe. The patient experienced “poor acceptance” (due to product acidity) a few days after the initiation of treatment. Following temporary discontinuation, the AE abated. After reintroduction, nosimilareventoccurred.Inaddition,noneoftheNAGSdeficiencypatientspermanentlydiscontinuedCARBAGLU due to an AE related to the treatment.
5.5 CONCLUSION OF CLINICAL ASPECTSAlthoughsmall,thepatientpopulationwithconfirmedNAGSdeficiencythathasbeenstudiedincludesthemajorityoflivingpatientsknowntobeaffectedwithNAGSdeficiencyinEurope,andtherefore,itisrepresentative.
ThevalueoftheretrospectiveNAGSdeficiencydataisrepresentedbythelongterm(>187patient-years)follow-upoftheNAGSdeficiencypatientstreatedwithCARBAGLU.
These data demonstrated:
• Good metabolic control was achieved, normalizing ammonia and glutamine levels in the reported patients within a few hours after the initiation of CARBAGLU treatment.
• In spite of common childhood infectious episodes, vaccinations, and even surgical operations under generalanesthesia–alleventswhichmayeasilytriggerdecompensationepisodesinNAGSdeficiencyunderclassicaltherapy–anincreaseinplasmaammonialevelswasveryrareand,whenitoccurred,itwas only minimal.
• It is important to maintain adequate doses of CARBAGLU. Clinical signs of decompensation were only observed on 2 occasions in patients receiving inadequate (very low) dosing.
• A normal protein intake or moderate protein restriction was possible in all patients treated with CARBAGLU.• The need for arginine should be tested individually. Additional arginine supplementation was systematically
giveninafewpatientsbutoverallresultsinthesepatientswerenotdifferentfromthoseinpatientswhoreceived Carbaglu only.
• Noserioussafetyissuehasbeenidentified.ThesafetyprofilewiththerecommendeddosageregimenwashighlyacceptableinNAGSdeficiencypatients.Routinehematologyandbloodchemistrytestsdidnot show any abnormality that could be related to CARBAGLU therapy.
CarbaglutherapyoffersaspecificpharmacologicaltherapyforthetreatmentofhyperammonemiaduetoNAGSdeficiency:asadjunctivetherapyforacutehyperammonemiaandasmaintenancetherapyforchronichyperammonemia (see CARBAGLU Prescribing Information).

32 Back to Table of Contents _
5.6 RECOMMENDATIONS FOR CARBAGLU® (carglumic acid) USE5.6.1 INDICATIONS AND USAGECARBAGLU is a Carbamoyl Phosphate Synthetase 1 (CPS 1) activator indicated as:
• Adjunctive therapy in pediatric and adult patients for the treatment of acute hyperammonemia due to the deficiency of the hepatic enzyme N-acetylglutamate synthase (NAGS). During acute hyperammonemic episodes concomitant administration of CARBAGLU with other ammonia lowering therapies such as alternate pathway medications, hemodialysis, and dietary protein restriction are recommended.
• Maintenance therapy in pediatric and adult patients for chronic hyperammonemia due to the deficiency of the hepatic enzyme N- acetylglutamate synthase (NAGS). During maintenance therapy, the concomitant use of other ammonia lowering therapies and protein restriction may be reduced or discontinued based on plasma ammonia levels.
5.6.2 DOSAGECARBAGLU treatment should be initiated by a physician experienced in the treatment of metabolic disorders. Basedonclinicalexperience,thetreatmentmaybestartedasearlyasthefirstdayoflife.Therecommendedinitial dose in pediatric and adult patients for acute hyperammonemia is 100 mg/kg/day to 250 mg/kg/day. Dosing should be titrated based on individual patient plasma ammonia levels and clinical symptoms.
In the long term, pediatric and adult maintenance doses should be titrated to target normal plasma ammonia levels fortheage.Basedondatafromtheretrospectivestudy(seeSection5.3.1.2),dailymaintenancedoseswereusuallyless than 100 mg/kg.
Based on PK data and clinical experience, it is recommended to divide the total daily dose into 2 to 4 doses, rounded to the nearest 100 mg (ie, half a CARBAGLU tablet), to be given before meals or feedings. The breaking of the tablets in halves allows most of the required posology adjustments. There is no concern for thesafeuseofhalf-tabletsbecausethekeyelementforthetreatmentofNAGSdeficiencypatientsisthetotal daily dose of CARBAGLU.
5.6.3 PREPARATION AND ADMINISTRATIONPreparation for Oral Administration in Adults CARBAGLU tablets should not be swallowed whole or crushed. Disperse CARBAGLU tablets in water immediately before use. Each 200 mg tablet should be dispersed in a minimum of 2.5 mL of water and taken immediately. CARBAGLU tablets do not dissolve completely in water and undissolved particles of the tablet may remain in the mixing container. To ensure complete delivery of the dose, the mixing container should be rinsed with additional volumes of water and the contents swallowed immediately. USE IN OTHER FOODS AND LIQUIDS HAS NOT BEEN STUDIED CLINICALLY AND IS THEREFORE NOT RECOMMENDED.
Preparation for Nasogastric Tube Administration in Adults For patients who have a nasogastric tube in place, CARBAGLU should be administered as follows:
• Mix each 200 mg tablet in a minimum of 2.5 mL of water. Shake gently to allow for quick dispersal.• Administer the dispersion immediately through the nasogastric tube.• Flush with additional water to clear the nasogastric tube.

33 Back to Table of Contents _
Preparation for Oral Administration Using an Oral Syringe in Pediatrics For administration via oral syringe, Carbaglu® (carglumic acid) should be administered as follows:
• Mix each 200 mg tablet in 2.5 mL of water to yield a concentration of 80 mg/mL in a mixing container. Shake gently to allow for quick dispersal.
• Draw up the appropriate volume of dispersion in an oral syringe and administer immediately. Discard the unused portion.
• Refilltheoralsyringewithaminimumvolumeofwater(1-2mL)andadministerimmediately. Preparation for Nasogastric Tube Administration in Pediatrics For patients who have a nasogastric tube in place, CARBAGLU should be administered as follows:
• Mix each 200 mg tablet in 2.5 mL of water to yield a concentration of 80 mg/mL in a mixing container. Shake gently to allow for quick dispersal.
• Draw up the appropriate volume of dispersion and administer immediately through a nasogastric tube. Discard the unused portion.
• Flush with additional water to clear the nasogastric tube. USE IN OTHER FOODS AND LIQUIDS HAS NOT BEEN STUDIED CLINICALLY AND IS THEREFORE NOT RECOMMENDED.
CARBAGLU is a white and elongated tablet, scored and coded “C” on one side. Each tablet contains 200 mg of carglumic acid. CARBAGLU is available in 5 or 60 tablets in a polypropylene bottle with polyethylene capanddesiccantunit.NDC52276-312-05Bottlesof5tabletsNDC52276-312-60Bottlesof60tablets.
Storage Beforeopening,storerefrigeratedat2–8°C(36–46°F).Afterfirstopeningofthecontainer:
• Donotrefrigerate,donotstoreabove30°C(86°F).• Keep the container tightly closed in order to protect from moisture.• Write the date of opening on the tablet container.• Do not use after the expiration date stated on the tablet container.• Discardonemonthafterfirstopening.
5.6.4 IMPORTANT SAFETY INFORMATION Indications and Usage CARBAGLU is a Carbamoyl Phosphate Synthetase 1 (CPS 1) activator indicated as:
• Adjunctive therapy in pediatric and adult patients for the treatment of acute hyperammonemia due to the deficiency of the hepatic enzyme N-acetylglutamate synthase (NAGS). During acute hyperammonemic episodes concomitant administration of CARBAGLU with other ammonia lowering therapies such as alternate pathway medications, hemodialysis, and dietary protein restriction are recommended.
• Maintenance therapy in pediatric and adult patients for chronic hyperammonemia due to the deficiency of the hepatic enzyme N- acetylglutamate synthase (NAGS). During maintenance therapy, the concomitant use of other ammonia lowering therapies and protein restriction may be reduced or discontinued based on plasma ammonia levels.

34 Back to Table of Contents _
Important Safety Information
HYPERAMMONEMIA :• Management of hyperammonemia due to N-acetylglutamate synthase (NAGS) deficiency and
Carbaglu® (carglumic acid) treatment should be initiated by a physician experienced in the treatment of metabolic disorders.
• Any episode of acute symptomatic hyperammonemia should be treated as a life-threatening emergency. Treatment of hyperammonemia may require dialysis, preferably hemodialysis, to remove a large burden of ammonia. Uncontrolled hyperammonemia can rapidly result in brain injury/damage or death, and prompt use of all therapies necessary to reduce plasma ammonia levels is essential.
• Ongoing monitoring of plasma ammonia levels, neurological status, laboratory tests and clinical responses in patients receiving CARBAGLU is crucial to assess patient response to treatment.
THERAPEUTIC MONITORING:• Plasma ammonia levels should be maintained within normal range for age via individual dose adjustment.
NUTRITIONAL MANAGEMENT:• Since hyperammonemia is the result of protein catabolism, complete protein restriction is
recommended to be maintained for 24 to 48 hours and caloric supplementation should be maximized to reverse catabolism and nitrogen turnover.
Themostcommonadversereactionsin≥13%ofpatientsare:infections,vomiting,abdominalpain,pyrexia,tonsillitis, anemia, ear infection, diarrhea, nasopharyngitis, and headache.
ToreportSUSPECTEDADVERSEREACTIONS,contactRecordatiRareDiseasesInc.at1-888-575-8344,orFDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
No drug interactions have been identified. There is no human pregnancy data, but decreased survival and growth occurred in animal offspring.
Breast feeding by a mother taking CARBAGLU is not recommended.
USE IN OTHER FOODS AND LIQUIDS HAS NOT BEEN STUDIED CLINICALLY AND IS THEREFORE NOT RECOMMENDED.

35 Back to Table of Contents _
6.0 REFERENCESAhMewN,CaldovicL.(2011)N-acetylglutamatesynthasedeficiency:aninsightintothegenetics,epidemiology,pathophysiology,andtreatment.TheApplicationofClinicalGenetics4:128-135.
AhMewN,LanpherBC,GropmanA,etal.UreaCycleDisordersOverview.(2003)Apr29[Updated2015Apr9]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington,Seattle;1993-2015.Availablefrom:http://www.ncbi.nlm.nih.gov/books/NBK1217/
AlbrechtJ(1998)Rolesofneuroactiveaminoacidsinammonianeurotoxicity.JNeurosciRes.51:133-8.
BachmannC,KrahenbuhlS,ColomboJP(1981)N-acetylglutamatesynthetasedeficiency:adisorderofammoniadetoxication[letter]NEnglJMed304,9:543.
BachmannC,ColomboJPandJaggiK(1982)N-acetylglutamatesynthetase(NAGS)deficiency:diagnosis,clinicalobservationsandtreatment.AdvinExperMedBiol153:39-45.
Bachmann C, Brandis M, Weissebarth-Riedel E, Burghard R, Colombo JP (1988) N-acetylglutamate synthetasedeficiency,asecondpatientJInheritMetabDis11:191-193.
BachmannC(2003a)Inheritedhyperammonemias.In:BlauN,DuranM,BlaskovicsM,GibsonK,editors.Physician’s guide to the laboratory diagnosis of metabolic diseases. 2nd ed. Berlin, Heidelberg, New York: Springer p. 261-76.
BachmannC(2003b)Outcomeandsurvivalof88patientswithureacycledisorders:aretrospectiveevaluation. Eur J Pediatr. 162(6):410-6.
Bachmann C (2005) Long term outcome of urea cycle disorders. Acta Gastroenterol Belg. Oct-Dec; 68:466-8.
BarsottiRJ(2001)Measurementofammoniainblood.JPediatr.138:S11-S20.
Belanger-Quintana A (2002) Hyperammonemia as a cause of psychosis in an adolescent. Eur J Pediatr. 162(11):773-775.
BerryG,SteinerR(2001)LongtermmanagementofpatientswithUCDJPediatr138:S56-S61.
Broere D, van Gemert WG, Kneepkens CM, Neele DM, Manoliu RA, Rauwerda JA et al. (2000) A 6-year-oldboywithhyperammonaemia:partialN-acetylglutamatesynthasedeficiencyorportosystemicencephalopathy? Eur J Pediatr. 159(12):905-7.
Brown DD and Kies MW (1959) The mammalian metabolism of L-histidine. I. The enzymatic formation of L-hydantoin-5-propionicacid.JBiolChem234;12:3182-3187.
Brusilow SW (1995) Urea cycle disorders: clinical paradigm of hyperammonemic encephalopathy. Prog Liver Dis.13:293-309.

36 Back to Table of Contents _
BurtonB(2000)Ureacycledisorders.ClinLiverDis4(4):815-830.
Caldovic L, Morizono H, Panglao MG, Gallegos R, Yu X, Shi D, Malamy MH, Allewell N, Tuchman M (2002) Cloning and expression of the human N-acetylglutamate synthase gene. Biochem Biophysic Res Comm 299:581-586.
CaldovicL,MorizonoH,PanglaoMG,ChengSF,PackmanS,TuchmanM(2003)NullmutationsintheN-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Hum Genet.112:364-8.
CaldovicL,MorizonoH,DaikhinY,NissimI,McCarterRJ,YudkoffM,TuchmanM(2004)RestorationofureagenesisinN-acetylglutamatesynthasedeficiencybyN-carbamylglutamate.JPediatr.145(4):552-4.
Caldovic L, Morizono H, Panglao MG, Lopez GY, Shi D, Summar ML et al. (2005) Late onset N- acetylglutamatesynthasedeficiencycausedbyhypomorphicalleles.HumMutat.25:293-8.
Caldovic L, Morizono H, Tuchman M. (2007) Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 28:754-9.
CaldovicL,AhMewN,ShiD,MorizonoH,YudkoffM,TuchmanM(2010)N-acetylglutamatesynthase:structure,functionanddefectsMolGenetMetab100:S13-S19.
Carbaglu Prescribing Information (2015).
Cartegena.A,PrasadAN,RuparCA,StrongM,TuchmanM,AhMewN,andPrasadC(2013)RecurrentEncephalopathy:NAGS(N-AcetylglutamateSynthase)DeficiencyinAdultsCanJNeurolSci;40(1)3-9
Cederbaum JA, Lemons C, Rosen M, Ahrens M, Vonachen S, Cederbaum SD (2001) Psychosocial issues andcopingstrategiesinfamiliesaffectedbyureacycledisorders.JPediatr.138:S72-S80.
Colombo JP, Krahenbuhl S, Bachmann C and Aeberhard P (1982) N-acetylglutamate synthase: enzyme assayinhumanliver.JClinChemClinBiochem20:325-9.
ColomboJP(1995)N-acetylglutamatedeficiency:clinicalandbiochemicalfeatures.InternationalPediatrics10:109-113.
ElpelegOetal.(1990)Late-onsetformofpartialN-acetylglutamatesynthetasedeficiency.EurJPediatr149:634-636.
ElpelegO,ShaagA,Ben-ShalomE,SchmidT,BachmannC(2002)N-acetylglutamatesynthasedeficiencyand the treatment of hyperammonemic encephalopathy. Ann Neurol 52:845-849.
Endo F, Matsuura T, Yanagita K, Matsuda I (2004) Clinical manifestations of inborn errors of the urea cycle andrelatedmetabolicdisordersduringchildhood.JNutr.134(Suppl):S1605-S1609;discussion30S32S,67S-72S.

37 Back to Table of Contents _
Feillet F, Leonard JV (1998) Alternative pathway therapy for urea cycle disorders. J Inherit Metab Dis. 21(Suppl 1):101-11.
Felipo V, Kosenko E, Minana MD, Marcaida G, Grisolia S (1994) Molecular mechanism of acute ammonia toxicityandofitspreventionbyL-carnitine.AdvExpMedBiol.368:65-77.
Feoli-Fonseca JC, Lambert M, Oureshi IA, Mitchell AA, Melançon L, Dallaire L (1992) Chronic benzoate therapy in children with inborn errors of urea cycle synthesis. Clin Invest Med. 15:107A.
Gessler P, Buchal P, Schwenk H.U., Wermuth B (2010) Favourable long term outcome after immediate treatmentofneonatalhyperammonemiaduetoN-acetylglutamatesynthasedeficiency.EurJPediatr169(2):197-199.
GomceliYB,KutluG,CavdarL,SanivarF,InanLE(2007)Differentclinicalmanifestationsofhyperammonemicencephalopathy.EpilepsyBehav.10:583-7.
Gropman AL, Batshaw ML (2004) Cognitive outcome in urea cycle disorders. Mol Genet Metab. 81:S58-62.
GuffonN,Vianey-SabanC,BourgeoisJ,RabierD,ColomboJP,GuibaudP(1995)AnewneonatalcaseofN-acetylglutamatesynthasedefiencyJIMD18:61-65.
HäberleJ,SchmidtE,PauliS,KreuderJG,PleckoB,GallerA,etal.(2003)MutationanalysisinpatientswithN-acetylglutamatesynthasedeficiency.HumMutat.;21:593-7.
Häberle J, Koch HG (2004) Genetic approach to prenatal diagnosis in urea cycle defects. Prenat Diagn 24:378-83.
Heckmann M, Wermuth B, Häberle J, Koch HG, Gortner L, Kreuder JG (2005) Misleading diagnosis of partialN-acetylglutamatesynthasedeficiencybasedonenzymemeasurementcorrectedbymutationanalysis. Acta Paediatr 94:121-4.
HerndonE,JonesV,BennettM(2003)Lethargyandhyperammonemiainaneonate.LabMed34:779-81.
HinnieJ,ColomboJP,WermuthB,DryburghFJ(1997)N-acetylglutamatesynthetasedeficiencyrespondingtocarbamylglutamate.JInherMetabDis20:839-840.
HuijmansJGM,deKlerkJBC,JankieR,SlotemaR,WolffED,DuranM(1998)ThefateoforalcarbamylglutamateinN-acetylglutamatesynthetasedeficiency.JInherMetabDis21(Suppl2):28.
HuizengaJR,TangermanA,GipsCH(1994)Determinationofammoniainbiologicalfluids.AnnClinBiochem31(Pt6):529-43.
JacksonMJ,BeaudetAL,O’BrienWE(1986)Mammalianureacycleenzymes.AnnuRevGenet20:431-64.
Jouvet P, Touati G, Lesage F, Dupic L, Tucci M, Saudubray JM et al. (2007) Impact of inborn errors of metabolism on admission and mortality in a pediatric intensive care unit. Eur J Pediatr 166:461-5.

38 Back to Table of Contents _
Kim S, Paik WK, Cohen PP (1972) Ammonia intoxication in rats: protection by N-carbamyl-L-glutamate plus L-arginine.ProcNatAcadSciUSA69;12:3530-3533.
Kingsley JD, Varman M, Chatterjee A, Kingsley RA, Roth KS (2006) Immunizations for patients with metabolic disorders. Pediatrics 118:e460-70.
KleppeS,MianA,LeeB(2003)UreaCycleDisorders.CurrTreatOptionsNeurol5(4):309-19.
LeeBandGossJ(2001)Longtermcorrectionofureacycledisorders.JPediatr138:S62-S71.
Lee JH, Kim WH, Cho H, Kwak YS, Kim S and Paik WK (1998) N-carbamyl-L-glutamate plus L-arginine can protect ammonia intoxication in rats with reduced functional liver mass. Biochem Biophys Res Commun 248:391-394.
LeonardJV(2001)Thenutritionalmanagementofureacycledisorders.JPediatr138:S40-S5.
Leonard JV and McKiernan PJ (2004) The role of liver transplantation in urea cycle disorders. Mol Genet Metab 81:S74-S78.
Martinez-PardoM(2001)Orphandrugsandmetabolicdisorders.RevNeurol33:220-5.
MeyburgJ,HoersterF,LlindnerM,BodamerO,SchmidtJ,EngelmannG,OttM,HoffmanGF(2007)Livercelltransplantation(LCT)forcarbamoylphasphatesynthase1(CPS1)deficiencyandcitrullenaemia.JInheritMetabDis20:S83.
Morris AM, Richmond SWJ, Oddie SJ, Pourfarzam M, Worthington V, Leonard JV (1998) N-acetylglutamate synthetasedeficiency:favourableexperiencewithcarbamylglutamate.JInherMetabDis21:867-868.
Morizono H, Caldovic L, Shi D, Tuchman M (2004) Mammalian N-acetylglutamate synthase. Mol Genet Metab 81:S4-11.
Nagata N, Matsuda I, Matsuura T, Oyanagi K, Tada K, Narisawa K et al. (1991) Retrospective survey of urea cycle disorders: Part 2. Neurological outcome in forty-nine Japanese patients with urea cycle enzymopathies. Am J Med Genet 40:477-81.
Nordenström A, Halladin M, Hallberg B, Alm J (2007) A trial with N-carbamylglutamate may not detect all patientswithNAGSdeficiencyandneonatalonsetJIMDShortReport#50.
O’ConnorJE,JordaA,GrisoliaS(1985)Acuteandchroniceffectsofcarbamylglutamateonbloodureaandammonia.EurJPediatr143:196-7.
OrphanEurope,dataonfile(2009).
Robinson MB, Batshaw ML (1995) Neurotransmitter alterations in congenital hyperammonemia. Mental Retardation and Developmental Disabilities Research Reviews 1995 1:201-7.

39 Back to Table of Contents _
RothKS.N-acetylglutamatesynthetasedeficiency.Journal[serialontheInternet](2006)Availablefrom:http://www.emedicine.com/ped/topic10.htm.
RubioVandGrisoliaS(1981)HumancarbamylphosphatesynthetaseI.Enzyme26:233-239.
SaudubrayJM,RabierD(2007)Biomarkersidentifiedininbornerrorsforlysine,arginine,andornithine.JNutr.137(6Suppl2):1669S-72S.
Scaglia F, Brunetti-Pierri N, Kleppe S, Marini J, Carter S, Garlick P et al. (2004a) Clinical consequences ofureacycleenzymedeficienciesandpotentiallinkstoarginineandnitricoxidemetabolism.JNutr134:S2775-82;discussion96S-97S.
ScagliaF,CarterS,O’BrienWE,LeeB(2004b)Effectofalternativepathwaytherapyonbranchedchainamino acid metabolism in urea cycle disorder patients. Mol Genet Metab 81:S79-85. Schubiger G, Bachmann C, Barben P, Colombo JP, Tönz O, Schüpbach D (1991) N- acetylglutamate synthetasedeficiency:diagnosis,managementandfollow-upofararedisorderofammoniadetoxication.EurJPediatr150:353-356.
ShihVE(2007)Alternative-pathwaytherapyforhyperammonemia.NEnglJMed356:2321-2.
Smith W, Kishnani PS, Lee B, Singh RH, Rhead WJ, Sniderman King L et al. (2005) Urea cycle disorders: clinical presentation outside the newborn period. Crit Care Clin 21:S9-17.
SteinerRD,CederbaumSD(2001)Laboratoryevaluationofureacycledisorders.JPediatr138:S21-S9.
Summar M, Tuchman M (2001) Proceedings of a consensus conference for the management of patients withureacycledisorders.JPediatr138:S6-10.
Summar M (2005a) Unmasked adult-onset UCD in the critical care setting. Crit Care Clin 21: S1-S8.
SummarM(2005b)Ureacycledisordersoverview.GeneReviews.InitialpostingApril2003,lastupdateAugust 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1217/.
SummarMetal.(2013)Theincidenceofureacycledisorders.MolecularGeneticsandMetabolism110:179-180.
Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, Kirmse B; European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD). Electronic address: http://www.e-imd.org/en/index.phtml; Members of the Urea Cycle Disorders Consortium (UCDC). Electronic address: http://rarediseasesnetwork.epi.usf.edu/ucdc/.MolGenetMetab.2013Sep-Oct;110(1-2):179-80.doi:10.1016/j.ymgme.2013.07.008.Epub2013Jul18.
Tuchman M, Batshaw M (2002) Management of inherited disorders of ureagenesis. The Endocrinologist 12(2):99-109

40 Back to Table of Contents _
TuchmanM,CaldovicL,DaikhinY,HorynO,NissimI,NissimI,KorsonM,BurtonB,andYudkoffM(2008)N-carbamylglutamatemarkedlyenhancesureagenesisinN-acetylglutamatedeficiencyandpropionicacidemiaasmeasuredbyisotopicincorporationandbloodbiomarkers.PediatricResearch64:213-7.
Valayannopoulos V, Mention K, Guyot V, Rabier D, Lesage F, Dupic L, et al. (2005) Experience with carbamylglutamate treatment for neonatal hyperammonemia. J Inherit Metab Dis 28(Suppl 1).
WalserM(1983)Ureacycledisordersandanotherhereditaryhyperammo-nemicsyndromes.In:Themetabolic basis of inherited disease. Standbury JB, Wyngaarden JB, Fredrickson DS, Eds, McGraw Hill, NewYorkp.402-438

41 Back to Table of Contents _
7.0 CARBAGLU PRESCRIBING INFORMATION

42 Back to Table of Contents _

43 Back to Table of Contents _

44 Back to Table of Contents _