microbiological aspects of cleaning validation · microbiological aspects of cleaning validation ....
TRANSCRIPT

Dr. Tim Sandle
Pharmaceutical Microbiology Resources: www.pharmamicroresources.com
Microbiological Aspects of Cleaning Validation

Introduction
• Cleaning
• Cleaning validation & GMPs
• Microbiological concerns
• Microbiological risks
• Risk assessment
• Microbiological tests and acceptance criteria
• Why failures happen

What is cleaning?
• Cleaning is assessed based on the level of residues that remain, either those directly found on the equipment or those indirectly contained within the final rinse after water has passed through or over the equipment.
• Whether the residues remaining have been reduced to a satisfactory low level is based on predetermined acceptance criteria.
• An additional concern is with the microbial bioburden.

Cleaning validation
Cleaning validation - methodology applied to give the assurance that a cleaning process has removed residues and contaminants from a piece of equipment or machinery.
Residues:
• Microorganisms
• Active pharmaceutical ingredients
• Other process chemicals, such as buffers
• Cleaning agents themselves (detergents)
• Microbiological culture media
The ‘validation’ aspect refers to providing documented evidence that that the acceptance criteria have been met.

Relative risks – manual vs automated cleaning • Manual cleaning is inherently high-risk compared
with automated cleaning.
• There will be variations between each cleaner.
• Variations of time.
• Variations with quantities of cleaning chemicals used.
• Manual cleaning procedures may need to be monitored and maintained.

GMPs #1
• Code of Federal Regulations (CFR):
• PART 111--CURRENT GOOD MANUFACTURING PRACTICE INMANUFACTURING, PACKAGING, LABELING, OR HOLDING OPERATIONS FOR DIETARY SUPPLEMENTS
• § 111.27(d) You must maintain, clean, and sanitize, as necessary, all equipment, utensils, and any other contact surfaces used to manufacture, package, label, or hold components or dietary supplements.
• PART 211 -- CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS
• § 211.67 Equipment cleaning and maintenance
• PART 820—QUALITY SYSTEM REGULATION

GMPs #2
• FDA, Guide to Inspections Validation of Cleaning Processes. (Silver Spring, MD, 1993).
• Pharmaceutical Inspection Convention/Pharmaceutical Inspection Co-Operation Scheme (PIC/S). Validation Master Plan Installation And Operational Qualification Non-Sterile Process Validation Cleaning Validation. (Geneva, Switzerland, Sept., 2009).

Number of runs?
• No direct guidance relating to the number of runs.
• By convention, three runs are normally performed in order to demonstrate that the obtained results are not due to chance.

Microbial Aspects

Microbial aspects #1
• The chemical verification of cleaning validation is relatively well described. What is often less clear is the microbiological aspect.
• Divided into:
• The microorganisms themselves (a direct hazard)
• The presence of residues that potentially provide a microbial growth source (an indirect hazard).
• To evaluate these microbiological risks a sound microbiological sampling plan is required.

Microbial aspects #2

Microbial aspects #3
• The processes that form part of cleaning validation have an impact upon microorganisms remaining post-cleaning and on microbial survival.
• With cleanrooms, humidity control is recommended for areas where equipment is held and stored in order to reduce the level of moisture (given that moist environments support the survival and growth of many microorganisms and there is a particular association with Gram-negative bacteria and fungi).
• The microbiologist should input.

Cleaning chemicals and microbial survival #1
The types of cleaning agents selected will have different affects upon microorganisms depending on the nature of chemical and the species (and numbers) of microorganisms present on a given surface.
These cleaning agents, which are typically one caustic based agent (sodium hydroxide) and one acid based reagent (such as hydrochloric acid), should be included as part of the cleaning validation.
Many commercial cleaning agents are formulated cleaning (in addition to water) with a surfactant, an alkalinity source (such as sodium hydroxide), and a chelant.

Acid and caustic?
There is a recurrent discussion about the need for both an acid and a caustic for cleaning validation.
Alkaline detergents are good for removing organic soils, i.e. oils, fats, proteins, starches, and carbohydrates. They hydrolyze peptide bonds and breaking down large, insoluble proteins into small, more easily soluble polypeptides.
Alkaline reagents are good for microbial kill.
Theoretically microbiological contamination, including viral residues, can be overcome by using sodium hydroxide in the cleaning process.
Sodium hydroxide dissolves proteinaceous soils and fatty oils by saphonification.
Sodium hydroxide effectively dissolves protein and organic matter.

Acid and caustic?
The use of an acid depends on which impurities are of a concern, and whether these will act as a barrier for the alkaline agent and protect microorganisms.
Acid detergents do not work well on heavy soils, oils, and glucans.
Phosphoric acid is useful in removing protein residues
Acid detergents are effective for the prevention or removal of water scale (calcium and magnesium carbonates), and aluminium oxide.
They are also good at bacteria kill, but they are rarely used without alkaline detergents.

Cleaning chemicals and microbial survival #2 • Cleaning agents should not be seen as disinfecting in
the sense that disinfection is defined as the known reduction of a population of microorganisms as demonstrated through controlled laboratory studies.
• Other aspects of the cleaning process can prove hostile to microbial survival:
• Temperature of the water used for cleaning (above 60oC)
• pH ranges below 4 and above 11.

Effect of water rinses #1 • Water is a key part of cleaning validation.
• Water rinses will remove cleaning chemicals and will siphon away any microorganisms in the planktonic state.
• Water is:
• Capable of wetting surface to penetrate the soil deposit
• Has the capacity to break the soil into fine particles
• Water holds small fine particles into suspension
• Water can prevent residues from redepositing onto a cleaned surface

Effect of water rinses #2
• Final water rinse is Water of Injections (this is for all types of pharmaceutical products).
• Water must meet the required microbiological specification e.g. with WFI – NMT 10 CFU/100mL for bioburden and NMT 0.25 EU/mL for bacterial endotoxin.
• Following rinsing equipment should not be left with residual water after cleaning.
• The last step of the cleaning procedure involve drying, perhaps with the addition of a solvent (such as 70% sterile isopropyl alcohol) or flushing with sterile compressed air.

Critical process and quality parameters
• Critical process parameters for cleaning validation are:
• Time (dirty and clean hold times and process run time).
• Activity (chemical exposure, number of water rinses etc).
• Chemical (concentration).
• Temperature (cleaning temperature).
• Critical quality attributes include:
• Water quality.
• Type of soil.
• Nature of the soil.
• Surface material and surface quality.

Risk assessment #1
• Need to assess worst-case factors:
• Stage of manufacture
• Type of soil
• Equipment design
• Ease of equipment drying
• Manual processing (as opposed to automated processing)
• Surface materials
• Equipment age
• Equipment damage
• Dirty storage areas
• Dirty hold time

Risk assessment #2
• The level of cleaning required relates to the stage in the process that the equipment is used for and the acceptable level of remaining microbial contamination (if any).
• With stages of manufacture, equipment used with products at early stage in the process chain generally requires lower levels of cleaning (compared with equipment used for product that is at an intermediate or final stage).
• Levels of cleaning need to greater if the cleaning is to be followed by sanitization or sterilization.
• The permitted microbial levels for equipment used in non-sterile processing require a separate risk assessment and limits setting compared with equipment used for sterile processing.

Risk assessment #3
• Hazards need to be identified and risk assessed, based on the severity of the hazard and the likelihood that the hazard will occur.
• Microorganisms represent a hazard:
• Microbial contamination on the equipment post-use (and pre-cleaning);
• The effects of hold time prior to cleaning (in relation to microbial proliferation and the release of endotoxin);
• Additional microbial challenges from the storage environment;
• The efficacy of the cleaning process to remove microorganisms and endotoxin;
• Storage of the equipment post-cleaning prior to use or a subsequent sanitization or sterilization step (in relation to recontamination from the environment).
• Plus the degree of severity should a level of microorganisms be present and the likelihood of microorganisms still being present after a cleaning or storage step. Likelihood is affected by the equipment design and easiness of cleaning.
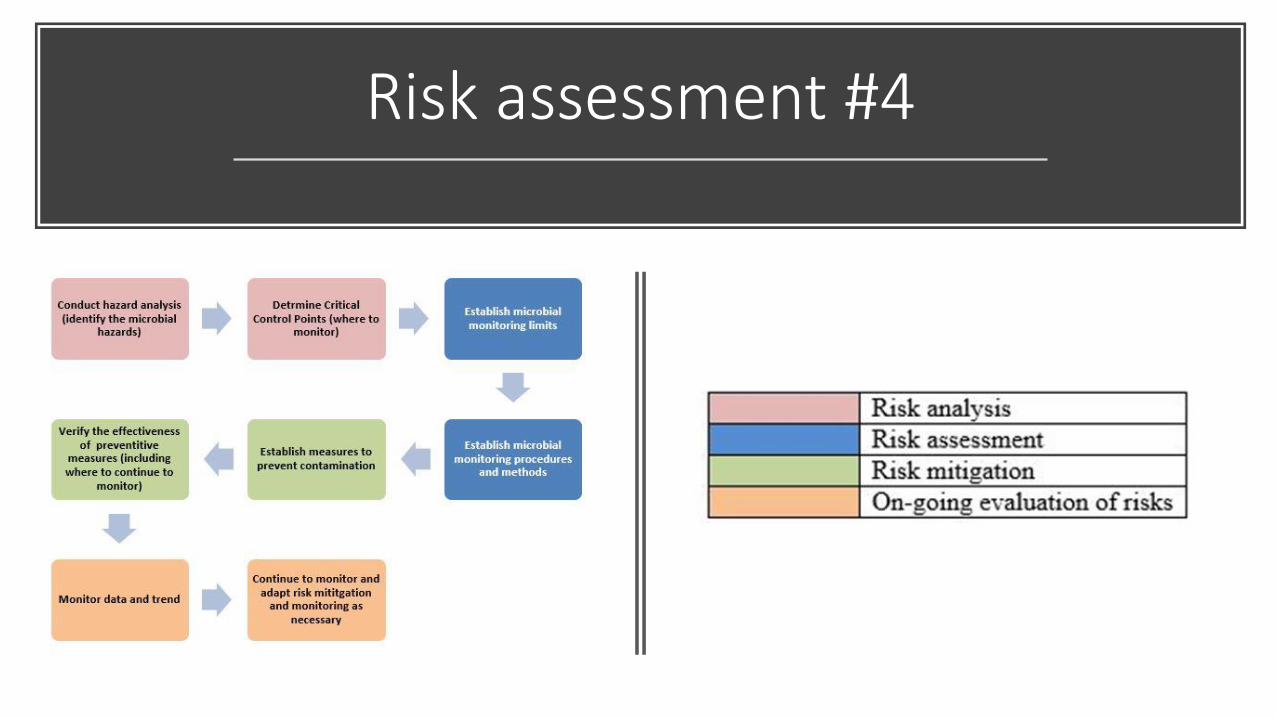
Risk assessment #4

Worst-case conditions for testing • Residues
• The ability of any residues to support and to promote microbial growth should be assessed.
• A viscous substance that does not readily support microbial growth may sometimes not be as great a risk as a growth-promoting substance like broth media residues.
• Time of testing
• The dirty-hold time is an important factor for consideration.
• Cleaning validation needs to be tested at the end of the hold time and the cleaning process.

Microbial tests
• Direct tests
• Surface sampling
• Contact plate
• Swab
• Indirect tests
• Final rinse tests
• Bioburden by membrane filtration
• Endotoxin testing
• Follow-up testing
• On-going in-process controls e.g. intermediate product bioburden testing
• Microorganism characterisation
• Hold time related testing
• Environmental monitoring

Locations for monitoring
• Locations need to be representative and of worst-case locations.
• Be aware of non-uniform contamination transfer.
• Equipment needs to be categorized between uniform contamination equipment and non-uniform contamination equipment.
• Define the most difficult-to-clean locations in equipment.
• Sites should be in a protocol.
• Assessment for location needs to be provided and justified.
• Sampling locations chosen arbitrarily or easiest-to-clean locations should be avoided.

Swabbing vs rinsing
• Rinse and swab measure two different things
• They will not necessarily correlate
• Area:
• Swabs focus on small area
• Rinses focus on larger area
• Representativeness:
• Swab measures worst case
• With appropriate locations
• Rinse measures average bioburden
• Failures:
• If a swab fails, a rinse may pass
• If a rinse fails, it is likely a swab will also fail

Limitations of microbial methods • Methods are limited:
• In terms of culture media
• Incubation time
• Incubation temperature
• Suitability of the medium
• In terms of the method, in general
• Swabs recovery 30-60%
• In terms of type of bacterial attachment to surfaces
• In terms of whether the organisms can be cultured
• Not capable of growing on the culture medium
• Too stressed to recover on the medium

Qualification of microbiological media
• Validate environmental monitoring media
• Confidence that residues of cleaning agents or disinfectants are not influencing the recovery of organisms on media used to perform routine environmental monitoring.
• Enables pharmaceutical and healthcare facilities to implement the use of adequate contact plates and swabs containing the appropriate neutralizers in the environmental monitoring process.

Microbial attachment to surfaces #1 • Bacteria attach to surfaces in different ways and according to different
circumstances.
• Bacterial adhesion is a consequence of the balance of attractive and repulsive physicochemical interactions between bacteria and surfaces.
• Bacteria prefer to grow on available surfaces (sessile form) rather than existing in planktonic form within an aqueous environment.
• The process of bacterial adhesion to a surface comprises of three stages:
• Transport.
• Initial adhesion (bioattachment).
• The cell may adhere to the surface either "reversibly" (that is, temporarily) or "irreversibly" (that is, permanently).
• Colonization.

Microbial attachment to surfaces #2
Irreversible attachment is harder to remove
• Irreversible bacterial adhesion occurs because of short-range molecular interactions like hydrogen, ionic, and covalent bonding, interactions involving extracellular structures, and secretions.
• Connected with biofilm formation.
Bioattachment occurs relatively slowly.
• Affected by the type of bacterium involved, the size of the bacterial population in the environment, and the duration of its growth phase.
Gram negative bacteria form biofilms more readily.
• This is because EPS and LPS are found in greater abundance.
• Important in relation to water, Gram negative bacteria constitute the majority of the bacterial populations found in aquatic environments.

Control is improtant
Microbial control is important, due to limitations of methods and surface attachment challenges.
This means:
• Observing time.
• Sticking with validated parameters
• Avoiding cross-contamination.

Microbial limits #1
•Acceptable levels need to be decided through risk assessment and it stands that the needs of non-sterile processing (where cleaned and sanitized equipment is acceptable) differs to sterile processing (where the equipment is subject to a sterilization step).
Regulatory agencies do not provide any direct
guidance about suitable microbiological test
limits.
•For bioburden: Not more than 10 CFU/100mL;
•For endotoxin: Not more than 0.25 EU/mL.
As a general indicator, for rinse water samples Water of Injection limits are often applied for the
final rinse water, namely:
•That is not more than 10 CFU per cm2 (for a contact plate) or per swab.
With surface sampling an equivalent value to
the bioburden test may be suitable

Microbial limits #2
• For non-sterile equipment a formula devised by Docherty is also widely used.
• Based on the permitted microbial levels in the finished product and then working backwards to determine what might be permitted on the surface of an item of equipment (as colony forming units per square centimeter).
• Docherty's process became adjusted to a 'universal' figure of 25, e.g. 25 CFU per cm2.
Ref: Docherty, S. (1999) Establishing microbial cleaning limits for non-sterile manufacturing equipment, Pharmaceutical Engineering, 19 (3): 36-40

Documentation
• Types and number of samples e.g. rinses, swabs, contact plates.
• A sample diagram, showing sample locations.
• Reference to sampling SOPs.
• Verifying that the person who took the samples was appropriately trained.
• Check-list to record when samples are taken.
• Sample transfer conditions.
• A receipt section for the arrival of samples in the microbiology laboratory.
• Test details, including verification of testing.
• List of all consumables, culture media and lot numbers.
• A results section.
• Test limits.
• Note of any deviations from procedure.
• Laboratory management sign-off.

Cleaning validation challenges #1
• Reproducibility of manual cleaning of small parts and non-clean-in-place (CIP) systems.
• Effective design of fully automated CIP systems to avoid dead legs and lack of drainability.
• Build-up of physical protein residue on the walls of equipment cleaned by CIP.
• Failure of CIP systems over time due to blocked steam traps and poor maintenance practices.

Cleaning validation challenges #2
• Developing acceptance criteria and validation strategies.
• Achieving degradation or deactivation of proteins and their breakdown products
• Using inappropriate risk-assessment methodologies.
• Such as a weakness with a matrix approach.
• Addressing air-liquid interface issues.
• Ensuring suitable a high-level of cleanliness to prevent potential for microbial excursions and biofilms.
• Avoiding rouge formation on surface cleanliness.

Rouging
• Rouge formation is a steady chemical process that is underway in all metallic piping systems in contact with water and all stainless steels corrode over time.
• What varies is the pace of the reaction.
• Exacerbated by high temperature and particular metal compositions.
• The risk is with product contamination, through the presence of particulates, could occur.
• Plus an impact on water quality.
• Plus buildups of rouge byproducts can lead to blockages in filters.
• Localized corrosion (pitting) provides increased opportunities for microbial attachment.

Why failures happen
• Recovery of organisms in the rinse water may signal that either the chemical treatment was insufficient or that the number of rinses was inadequate.
• Resolved through altering the chemical treatment or by increasing the number of rinses.
• Insufficient chemical contact times
• Biofilms
• Poor equipment design
• Ball valves.
• Long piping with dead-legs.
• Sampling error
• Poor training

Equipment storage, post-cleaning • Control measures are required to prevent
recontamination.
• The main microbial risk arises from equipment either not being dry or becoming wet post-cleaning.
• A considerable risk arises from stagnant water remaining inside the equipment.
• Water remaining provides a growth source for many microorganisms in the vegetative state.

Microbial risks with wet equipment • Some Gram-negative growth examples, for doubling
times:
• Escherichia species: 19-20 minutes
• Klebsiella species: 38-40 minutes
• Acinetobacter species: 48 -55 minutes
• Pseudomonas species: 100 minutes
• Plus, they become harder to remove over time.

References
• Sandle, T. (2013). Bacterial Adhesion: an Introduction, Journal of Validation Technology, Vol. 19, Issue 2
• Sandle, T. (2015) The Rouging Effect in Pharmaceutical Water Systems: Causes and Strategies for Prevention, Journal of GXP Compliance, Vol. 19, Issue 1
• Sandle, T. (2017) Microbiological Aspects of Cleaning Validation, Journal of GxP Compliance, Vol. 21, Issue 5

Summary
Microbiological aspects of cleaning validation can
readily be captured within the broad cleaning validation
approach.
The variables that might lead to microorganisms surviving and the approaches need to
remove or inactivate microorganisms need to be
fully considered.
This needs to be captured in the cleaning validation
strategy.
