mathematical programming assisted drug design for nonclassical antifolates
TRANSCRIPT

Mathematical Programming Assisted Drug Design for NonclassicalAntifolates
Sanjeev Garg and Luke E. K. Achenie*
Department of Chemical Engineering, University of Connecticut, Storrs, Connecticut 06269
A concept from optimization theory, specifically, mathematical programming, isproposed for designing drugs with desired properties. The mathematical programmingformulation is solved to obtain the optimal descriptor values, which are employed inthe Cerius2 modeling environment to infer the optimal lead candidates, in the sensethat they exhibit both high selectivity and activity while ensuring low toxicity. It hasbeen observed that unique substituent groups and their molecular conformations areresponsible for attaining the goal of simultaneous high selectivity and activity. Bothlinear and nonlinear quantitative structure activity relationships (QSARs) have beendeveloped for use in the proposed approach. A comparative study of these models isdone, and it is shown that the QSARs are well represented by nonlinear models. Theproposed mathematical programming strategy has been demonstrated for a class ofnonclassical antifolates for Pneumocistis carinii and Toxoplasma gondii dihydofolatereductase. Some of the potential leads found in this study have biological propertiessimilar to those in the open literature. We believe the technique proposed is generaland can be applied to other structure based drug design.
1. Introduction
Pneumonia and toxoplasmosis are the major causes ofmorbidity and mortality in AIDS patients (Vita et al.,1987). Opportunistic pathogens, Pneumocystis carinii (pc)and Toxoplasma gondii (tg), respectively, cause thesediseases via the dihydrofolate reductase (DHFR) enzymein AIDS and other immunocompromised patients. Exist-ing therapies based on present drugs are either too toxicor not very selective between human DHFR and pcDHFR(or tgDHFR) (Walzer et al., 1988; Kovacs et al., 1988).Currently available antifolate therapies (namely, tri-methoprim and pyrimethamine) for pc and tg infections,are weak inhibitors of DHFR. On the other hand,trimetrexate and piritrexim, although 100-10,000 timesmore potent than trimethoprim and pyrimethamine, areunfortunately strong inhibitors of DHFR from mam-malian sources (Gangjee et al., 1993, 1995, 1996a;Rosowsky et al., 1994, 1995).
There has been a flurry of research activities reportedin the open literature (Chio et al., 1991; Piper et al., 1996;Gangjee et al., 1996b, 1997, 1998) focused on the designof drugs that are simultaneously active against pcDHFRand tgDHFR and relatively inactive (i.e., selective)against human DHFR. In these studies, typically theresearcher takes an antifolate backbone, changes someof the functional groups, synthesizes the molecule, andperforms bioassays to determine if it is a potential leadcandidate. This process is naturally very time-consumingand expensive. Compounding this problem is the fact thatthere are several hundred, even millions of possiblemolecules that can be screened through this approach.This is a Herculean task for any research group toaccomplish in a reasonable amount of time. Thus there
is a definite need for systematic, fast, and inexpensivetools for identifying a set of potential leads for subsequentdetailed experimental studies. Combinatorial chemistry(Fenniri, 2000) is one possible approach. We, however,propose a computer-aided molecular design approachbased on concepts in optimization theory, specifically,mathematical programming.
In mathematical programming, one formulates a per-formance objective to be optimized (for example, maxi-mization of selectivity), together with a set of constraintsto be satisfied (for example, activity should exceed a givenacceptable value). In addition, one needs to identify a setof variables (for example, a set of descriptors thatdescribe a molecule) to be systematically varied in orderto optimize the performance objective while satisfying theconstraints. This necessarily means that both the per-formance objective and the constraints should depend onthe given set of variables.
The mathematical programming approach has previ-ously been used successfully for product design. Forexample it has been used in refrigerant design (Duvediand Achenie, 1996; Churi and Achenie, 1996), polymerand polymer blend design (Vaidyanathan and El-Hal-wagi, 1994; Maranas, 1996), and for solvent design (Odeleand Macchietto, 1993; Pretel et al., 1994; Sinha et al.,1999). Thus, it seems reasonable to apply it to drugdesign.
2. Formulation
We propose a systematic mathematical programmingapproach to identify potential leads that are simulta-neously active against pcDHFR and tgDHFR and rela-tively inactive (i.e., selective) against human DHFR. Inthe suggested approach, we tackle two subproblems,namely, a forward problem and an inverse problem (seeFigure 1). In the forward problem we develop models to
* Phone: (860) 486-2756. Fax: (860) 486-2959. Email: [email protected].
412 Biotechnol. Prog. 2001, 17, 412−418
10.1021/bp010034q CCC: $20.00 © 2001 American Chemical Society and American Institute of Chemical EngineersPublished on Web 05/10/2001

predict the selectivity and activity from molecular de-scriptors. In the “inverse” problem, we determine theoptimal values (based on selectivity and activity) of themolecular descriptors and infer an appropriate molecularstructure. We note that while the forward problem givesa unique solution (i.e., one-to-one relation), the inverseproblem is not unique. In other words, in the inverseproblem one can find more than one molecular structurewith given selectivity and activity values. The math-ematical programming framework allows us to identifya solution to the inverse problem in a systematic andefficient manner.
Forward Problem. In preparation for the forwardproblem we proceed as follows. Several antifolates eachwith different inhibitory activity characteristics, and witha general structure as shown, where W ) [N, CH], X )
[N, CH], Y ) [N, CH2], Z ) [N, CH2], R1 )[no substituent, H, Cl, CH3], R2 ) [H, CH3] and R3 )[H, CH3, CHO, CH2CCH, CH(CH3)2, CH2CH3], are fedinto the MSI Cerius2 modeling environment. The latterthen gives a unique set of descriptor values correspondingto each molecule. The descriptors available in the Cerius2
modeling environment are conformational, electronic,quantum mechanical (semiempirically calculated descrip-tors using MOPAC), topological (2-dimensional descrip-tors based on graph theory concepts), spatial, structuraland thermodynamic (see Table 1). We employ the GeneticFunction Approximation algorithm in Cerius2 to deter-mine the most important descriptors needed. Next wedevelop a quantitative structure activity relationship(QSAR) between the activities (from the open literature)of the antifolate molecules and the descriptor values.
QSAR models are also developed for selectivities of theantifolate molecules for pcDHFR (and tgDHFR) versusrlDHFR. The models considered are (1) linear regression,(2) genetic partial least-squares (G/PLS) (Dunn et al.,1996), and (3) artificial neural networks (ANN) (Schalkoff,1997). Note that models 2 and 3 are nonlinear. In Resultsand Discussion, we compare the various models.
G/PLS uses the conventional genetic programming(Goldberg, 1989) operators, namely, reproduction, cross-over, and mutation, to converge to better models. Ingenetic programming, a population of chromosomes(binary strings corresponding to unique real variablevalues) is selected in the mating pool (search space). Overthe generations (iterations) we converge to better chro-mosomes by reproduction (multiple copies of parentchromosomes are introduced in the mating pool) of goodchromosomes (those that have above average fitnessvalue based on the objective function) and by crossover(changing the chromosome string to have new descriptorvalues) of parent chromosomes to create new and hope-fully better children chromosomes. The G/PLS methodcombines the best features of Genetic Function Ap-proximation and Partial Least Squares (PLS). Eachgeneration has PLS applied to it instead of multiplelinear regression, and so each model can have more termsin it with minimal danger of overfitting.
A typical feed forward multilayer neural networkconsists of many layers of neural elements. Every neuralelement in a layer is interconnected with every elementin adjacent layers. The strength of every interconnectionis characterized by its weight. Information propagatesfrom the input layer (first layer) to the output layer (lastlayer). Each neural element weights the input it receivesfrom the elements in the previous layer using theappropriate interconnection weights. Subsequently, thesum of the weighted inputs is filtered through a sigmoidfunction to produce an output from the element. Theoutput from the output layer represents the final predic-
Figure 1. Schematic representation of molecular design process.
Biotechnol. Prog., 2001, Vol. 17, No. 3 413

tion of the neural network. The Stuttgart Neural Net-work Simulator (SNNS) (University of Stuttgart (SNNSUser Manual, Version 4.1, 1995)) is used in the presentstudy for generating ANN models.
Inverse Problem. The inverse problem is solved usingone of the QSAR models discussed earlier. We employ amathematical program to do model inversion. Thisresults in a set of optimal descriptor values for potentiallead candidates with both high selectivity and activityvalues. More formally we cast the inverse problem as
where d is the vector of descriptors. The Selectivity andActivity are models generated from the forward problem.Activity_low is the activity above which the drug has asignificant biological effect. The suggested formulationis done bearing in mind the fact that the selectivity of adrug is more critical than its activity since the dosageand/or its form can control the activity of any drug. Notethat the drug can be given at higher potency or frequency.On the other hand it can be given at lower potency orfrequency. The dosage form can be intravenous or oral.
We note that the mathematical programming formula-tion can handle objectives and constraints different fromthe suggested one above. The mathematical program issolved using successive quadratic programming (Biegleret al., 1997), a local optimization method. The optimaldescriptors values are then used to identify the appropri-ate substituents on the antifolate backbone. In otherwords, these descriptor values are used to infer theimportant structural features necessary to attain the
desired properties. These features are included in theCerius2 analog builder at the respective positions on thebackbone molecule. The Cerius2 analog builder generatesall the possible molecules. These are then used tocalculate the descriptors values, which are compared withthe values obtained in the inverse problem.
3. Results and Discussion
A set of 125 antifolate molecules was selected from theopen literature for the present study (Chio et al., 1991;Piper et al., 1996; Gangjee et al., 1996b, 1997, 1998).Each molecule has either a better activity or selectivitycompared to existing therapies. Unfortunately, none ofthem has both good activity and selectivity. The mol-ecules were partitioned into a training set of 95 and avalidation set of 30 using Ward’s hierarchic clusteringanalysis (HCA) (Willett, 1987). The goal of clusteranalysis is to partition the data set into classes orcategories consisting of antifolate candidates of compa-rable similarity. HCA clustering has two main steps: thefirst step creates a hierarchical structure of clustering,and the second step selects the clustering on the basis ofthe objective function value. In Ward’s method, theobjective function is defined as intracluster variancessummed over the clusters and is minimized for theselection process.
Forward Problem. The present mathematical pro-gramming framework tries to design for high selectivityand activity simultaneously. These molecules are repre-sented by various descriptors generated by semiempiricalmethods in the MSI Cerius2 modeling environment. Asubset of important descriptors for pcDHFR and tgDHFRactivity and selectivity were selected using G/PLS. Weobserve that the important descriptors for activity model-
Table 1. Descriptors Included in the Present Study
type descriptor details
conformational Energy the energy of the selected conformation
electronic Charge sum of partial chargesFcharge sum of formal chargesApol sum of atomic polarizabilitiesDipole dipole momentHOMO highest occupied molecular orbital energyLUMO lowest unoccupied molecular orbital energySr superdelocalizability
quantum mechanical LUMO_MOPAC lowest unoccupied molecular orbital energyDIPOL_MOPAC dipole momentHOMO_MOPAC highest occupied molecular orbital energyHf_MOPAC heat of formation
topological Wiener Index the sum of the chemical bonds existing betweenall pairs of heavy atoms in the molecule
Zagreb Index the sum of the square of vertex valenciesKier and Hall molecular
connectivity indexa series of numbers designated by “order” and
“subgraph type”
spatial RadofGyration radius of gyrationJurs descriptors Jurs charged partial surface area descriptorsShadow indices surface area projectionsArea molecular surface areaDensity densityPMI molecular volumeVm molecular volume
structural MW molecular weightRotlbonds number of rotatable bondsHbond acceptor number of hydrogen bond acceptorsHbond donor number of hydrogen bond donors
thermodynamic AlogP log of partition coefficientFh2o desolvation free energy for waterFoct desolvation free energy for octanolHf heat of formationMolref molar refractivity
Maximized
Selectivity (d)
Subject to Activity (d) g Activity_low
414 Biotechnol. Prog., 2001, Vol. 17, No. 3

ing are thermodynamic, electronic, and conformationaldescriptors, namely, Energy, S_aaCH, S_aaaC, HOMO,Shadow-XYfrac, S_aaN, and AlogP (see explanation inTable 1). The G/PLS in the MSI Cerius2 picks thedescriptors that a medicinal chemist is likely to pick.
Similarly, we observe that the important descriptorsfor selectivity modeling are spatial, electronic, andtopological descriptors, namely, S_aasC, S_aaaC,Dipole_Mag, HOMO_MOPAC, Jurs-PNSA-1, Dipole-X,Energy, S_ssCH2, Dipole-Y, HOMO, and LUMO (seeexplanation in Table 1). The existing libraries can be usedfor selecting the optimal descriptors in a more efficientway than genetic algorithms. Case specific details canalso be handled effectively with medicinal chemistryknowledge. Thus, it is to be emphasized that an efficientdrug design effort can be realized with the help ofmathematical programming.
It should be mentioned here that, for the two casespcDHFR and tgDHFR a few descriptors required formodeling are different. This can be attributed to thepossible differences in structure of the two enzymes, asthe structure of tgDHFR is yet to be determined. Thiscan also be verified with the docking experiments oftgDHFR with selective inhibitors.
We use multiple linear regression to fit linear quan-titative structure activity relationships to pcDHFR andtgDHFR activity and pcDHFR and tgDHFR selectivity.Selectivity is defined as the ratio of rlDHFR to pcDHFRor tgDHFR. Selectivity values are obtained using therespective activity values and the activity values for ratliver (rl) DHFR, which is similar to human DHFR. Themodels are cross validated using the validation dataset. Linear models obtained in the present study aregiven as
The correlation coefficients values, r2, for these linearmodels are low (equal to 0.4951, 0.2791, 0.2425 and0.2515, respectively). The models are, therefore, deemedtoo inaccurate for the mathematical programming frame-work. The models have 6, 7, 3, and 1 outliers, respec-tively, for a 90% confidence interval.
We generate nonlinear QSAR models based on GeneticPartial Least Squares (G/PLS) to improve upon the linearmodels in Cerius2 modeling environment. These have theadvantage of having nonlinear interaction terms andhence better modeling capabilities. The G/PLS modelsobtained in the present study are
where < > represents a spline term.These G/PLS models show an improvement over the
linear models in terms of their correlation coefficientvalues. The r2 values for these models are 0.752, 0.632,0.762, and 0.647, respectively. The G/PLS models have5, 6, 6, and 2 outliers, respectively, for 90% confidenceinterval. We therefore observe that the QSAR models fornonclassical antifolates are nonlinear in nature andhence there is scope for further improvement.
Improved nonlinear ANN models are developed as aresult. A feed-forward neural network, with one hiddenlayer having a maximum of four neurons, appears to givethe smallest root-mean-square errors (for both the train-ing and validation sets), for modeling the pcDHFR andtgDHFR activity and selectivity. We initialize the net-work in SNNS using the randomized weights. On theother hand we use topological_order and scaled conjugategradient for update and learning, respectively.
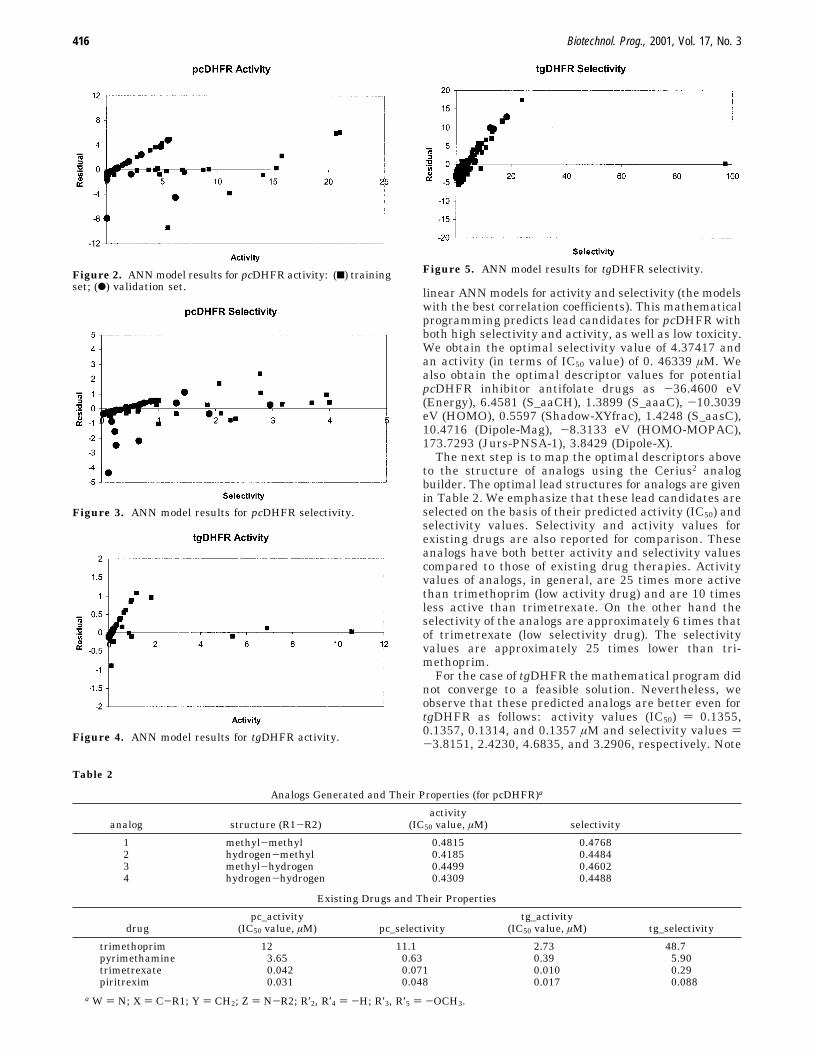
The ANN results showed remarkable improvementcompared to the linear and G/PLS models. Four ANNmodel results, in terms of residual plots, are depictedgraphically in Figures 2-5, and their correlation coef-ficient values in terms of r2 are equal to 0.9036, 0.7545,0.9756, and 0.9249, respectively. In Figure 2 for pcDHFRactivity, 2 and 1 outliers result from the training andvalidation sets, respectively. Note also that the residualsare well below 5% of the observed experimental values.Similarly, in Figure 3, for pcDHFR selectivity, 2 and 5outliers result from the training and validation sets,respectively. The residuals, in general, are below 15%of the experimental values. In Figures 4 and 5 (fortgDHFR), there are only a few outliers. We furtherobserve that for both activity and selectivity models, theresiduals are well below 15% of the observed experimen-tal values. In general, the residual plots seem to berandom, although there is some skewness at loweractivity and selectivity values. The departure fromrandomness in Figure 5 for tgDHFR selectivity may pointto a systematic error in the NN model. This will indicatenonoptimal selection of descriptors. A better understand-ing about these descriptors (for tgDHFR QSAR) can beobtained when the X-ray crystal structure of tgDHFRenzyme and its docking experiments with some inhibitorsbecome available.
Inverse Problem. We achieve possible lead candi-dates using the above mathematical program with non-
pc_activity ) 117.025 + 0.020 × Energy + 0.088 ×S_aaCH - 14.289 × S_aaaC + 9.923 ×
HOMO + 0.999 × Shadow-XYfrac
pc_selectivity ) -9.497 + 0.234 ×S_aasC - 0.952 × S_aaaC - 0.028 ×
Dipole_Mag + 0.027 × Dipole-X - 1.313 ×HOMO_MOPAC - 0.001 × Jurs-PNSA-1
tg_activity ) -2.736 + 0.007 × Energy - 0.023 ×S_aaCH + 0.257 × S_aaN - 0.143 × AlogP
tg_selectivity ) 201.634 - 0.049 ×Energy + 11.472 × S_ssCH2 + 0.561 ×
Dipole-Y + 17.233 × HOMO - 17.118 × LUMO
tg_activity ) 0.129 + 0.003 ×<Energy - 139.676>2 + 60.872 ×
<4.734 - S_aaCH> + 0.153 ×<S_aaN - 12.603> + 5.910 × 107 ×
<0.132 - AlogP>
pc_activity ) 3.229 + 0.045 ×<Energy -19.602> + 88.469 ×<4.734 - S_aaCH> - 8.703 ×<S_aaaC - 0.822> + 9.732 ×
<HOMO + 10.072> + 389.776 ×<0.450 - Shadow-XYfrac>
pc_selectivity ) 0.323 + 0.416 ×<S_aasC - 3.617>2 + 1.174 ×<S_aasC - 4.831>2 - 0.067 ×
<Dipole_Mag - 5.303> + 2.036 ×<-8.337 - HOMO_MOPAC> + 0.020 ×
<148.674 - Jurs-PNSA-1>
tg_selectivity ) 4.645 + 0.495 ×<-39.952 - Energy> + 743.002 ×
<S_ssCH2 - 1.357>2 - 18321.790 ×<S_ssCH2 - 1.519>2 - 0.104 ×
{(Dipole-Y - 0.914)2} + 3779.5 ×<LUMO -2.096>2
Biotechnol. Prog., 2001, Vol. 17, No. 3 415
linear ANN models for activity and selectivity (the modelswith the best correlation coefficients). This mathematicalprogramming predicts lead candidates for pcDHFR withboth high selectivity and activity, as well as low toxicity.We obtain the optimal selectivity value of 4.37417 andan activity (in terms of IC50 value) of 0. 46339 µM. Wealso obtain the optimal descriptor values for potentialpcDHFR inhibitor antifolate drugs as -36.4600 eV(Energy), 6.4581 (S_aaCH), 1.3899 (S_aaaC), -10.3039eV (HOMO), 0.5597 (Shadow-XYfrac), 1.4248 (S_aasC),10.4716 (Dipole-Mag), -8.3133 eV (HOMO-MOPAC),173.7293 (Jurs-PNSA-1), 3.8429 (Dipole-X).
The next step is to map the optimal descriptors aboveto the structure of analogs using the Cerius2 analogbuilder. The optimal lead structures for analogs are givenin Table 2. We emphasize that these lead candidates areselected on the basis of their predicted activity (IC50) andselectivity values. Selectivity and activity values forexisting drugs are also reported for comparison. Theseanalogs have both better activity and selectivity valuescompared to those of existing drug therapies. Activityvalues of analogs, in general, are 25 times more activethan trimethoprim (low activity drug) and are 10 timesless active than trimetrexate. On the other hand theselectivity of the analogs are approximately 6 times thatof trimetrexate (low selectivity drug). The selectivityvalues are approximately 25 times lower than tri-methoprim.
For the case of tgDHFR the mathematical program didnot converge to a feasible solution. Nevertheless, weobserve that these predicted analogs are better even fortgDHFR as follows: activity values (IC50) ) 0.1355,0.1357, 0.1314, and 0.1357 µM and selectivity values )-3.8151, 2.4230, 4.6835, and 3.2906, respectively. Note
Figure 2. ANN model results for pcDHFR activity: (9) trainingset; (b) validation set.
Figure 3. ANN model results for pcDHFR selectivity.
Figure 4. ANN model results for tgDHFR activity.
Table 2
Analogs Generated and Their Properties (for pcDHFR)a
analog structure (R1-R2)activity
(IC50 value, µM) selectivity
1 methyl-methyl 0.4815 0.47682 hydrogen-methyl 0.4185 0.44843 methyl-hydrogen 0.4499 0.46024 hydrogen-hydrogen 0.4309 0.4488
Existing Drugs and Their Properties
drugpc_activity
(IC50 value, µM) pc_selectivitytg_activity
(IC50 value, µM) tg_selectivity
trimethoprim 12 11.1 2.73 48.7pyrimethamine 3.65 0.63 0.39 5.90trimetrexate 0.042 0.071 0.010 0.29piritrexim 0.031 0.048 0.017 0.088
a W ) N; X ) C-R1; Y ) CH2; Z ) N-R2; R′2, R′4 ) -H; R′3, R′5 ) -OCH3.
Figure 5. ANN model results for tgDHFR selectivity.
416 Biotechnol. Prog., 2001, Vol. 17, No. 3

that a negative scaled selectivity value means that theselectivity is lower than the selectivity of the lowest drugin the training set. Analogs are, thus, approximately 3times more active than pyrimethamine but about 10times less active than piritrexim. In terms of selectivityvalues, they are approximately 30 times more selectivethan piritrixim and 2 times less selective than py-rimethamine. The tgDHFR structure is not known. Also,the docking experiments with inhibitors are not reported,and hence the descriptors selected for the modeling maynot be the optimal set. This can be the reason fornonconvergence of the mathematical program. Thus,better modeling strategies than QSAR, such as theComparative Molecular Field Analysis (CoMFA) need tobe applied for tgDHFR activity and selectivity modeling.
The present study thus elucidates the structuralfeatures required for the simultaneous high selectivityand activity values in the nonclassical antifolates. Weobserve that the W, X, Y, and Z positions are N, C-R,CH2, and N-R, respectively, with R ) [H, CH3] (seeantifolate structure in the section on formulation). Itappears that 3,5-OCH3 is critical for the simultaneoushigh selectivity and activity. It is interesting that two ofthe predicted optimal leads, namely, analogs 1 and 3 inTable 2, have already been synthesized and their bio-assay performed (Gangjee et al., 1995). Analog 1 hasreported values of 0.13 µM for activity and 1.3 forselectivity for pcDHFR, while analog 3 has reportedvalues of 0.029 µM for activity and 1.9 for selectivity.These values clearly show that these analogs have betteractivity compared to trimethoprim as well as betterselectivity compared to trimetrexate.
The various descriptor values for all four analogs aregiven in Table 3. We note that seven of these (Energy,S_aaCH, S_aaaC, HOMO, Shadow-XYfrac, HOMO-MOPAC, Jurs-PNSA-1) have values very close to theoptimal values. The remaining three (S_aasC, Dipole-Mag, Dipole-X) vary significantly from the optimalvalues. One possible explanation of this is that Cerius2
is not able to adequately capture some of the shape anddipole effects. This simply means that the modeling effortin the forward problem has to be improved.
4. Conclusions
This study reports the first effort, to the best of ourknowledge, of the application of mathematical program-ming in drug design. Various modeling techniques havebeen used to derive linear and nonlinear QSAR modelsfor the activity and selectivity values for a class ofnonclassical antifolates. A comparative study has beendone, and the best models, namely, ANN based nonlinearmodels, have been used for identifying potential leadsthrough the solution of the inverse problem. We empha-size that accurate modeling in the forward problem iscritical for the proposed approach because the QSAR
models are used in the inverse problem. Thus thesuggested lead candidates should be verified by actualsynthesis and bioassay. The results could be used, ifnecessary, for the refinement of the models. It is veryencouraging that two of the predicted optimal leads fromthe mathematical programming based approach, havealready been synthesized and their bioassay performed.
NotationAlogP thermodynamic descriptor, log of the
partition coefficientANN artificial neural networkantifolates compounds that interfere with the
utilization of folic acidd descriptor vectorDHFR dihyrofolate reductase enzymeDipole-Mag descriptor based on magnetic dipole
momentDipole-X descriptor based on dipole moment in
x-directionDipole-Y descriptor based on dipole moment in
y-directionEnergy conformational descriptorG/PLS genetic partial least squaresHCA hierarchical clustering analysisHOMO electronic descriptor, highest occu-
pied molecular orbital energyHOMO_MOPAC descriptor, HOMO based on MOPAC
calculationsJurs-PNSA-1 spatial descriptor, Jurs charged par-
tial surface areaLUMO electronic descriptor, lowest unoc-
cupied molecular orbital energypc Pneumocystis cariniiPLS partial least squaresQSAR quantitative structure activity rela-
tionshipr2 correlation coefficientrandomized_weights initiation function in ANNrl rat liverR1-R3 substituents groups on antifolatesScaled Conjugate
Gradientlearning function in ANN
S_aaaC descriptorS_aaCH descriptorS_aaN descriptorS_aasC descriptorShadow-XYfrac spatial descriptor, surface area pro-
jectionsS_ssCH2 descriptortg Toxoplasma gondiiTopological Order update function in ANNW functional group in antifolateX functional group in antifolateY functional group in antifolateZ functional group in antifolate
References and Notes
Biegler, L. T.; Grossmann, I. E.; Westerberg, A. W. SystematicMethods of Chemical Process Design; Prentice Hall Inc.: NewJersey, 1997.
Broughton, M. C.; Queener, S. F. Pneumocystis carinii dihydro-folate reductase used to screen potential antipneumocystisdrugs. Antimicrob. Agents Chemother. 1991, 35, 1348-1355.
Table 3. Descriptor Values for Analogs Generated(for pcDHFR)
descriptor analog 1 analog 2 analog 3 analog 4
Energy -36.9414 -48.0413 -28.1048 -44.8719S_aaCH 7.5229 9.3837 7.3605 9.1901S_aaaC 1.2317 1.1692 1.2139 1.1514HOMO -9.8493 -10.2549 -10.0768 -10.2627Shadow-XYfrac 0.6781 0.5231 0.4748 0.5076S_aasC 4.8740 3.8194 4.6868 3.6501Dipole-Mag 4.1671 4.1177 3.9786 3.6987HOMO_MOPAC -8.4703 -8.5284 -8.5584 -8.4792Jurs-PNSA-1 165.0326 170.5729 169.1242 176.0141Dipole-X 1.4128 1.8242 1.3793 1.7637
Biotechnol. Prog., 2001, Vol. 17, No. 3 417

Chio, L.-C; Queener, S. F. Identification of highly potent andselective inhibitors of Toxoplasma gondii dihydrofolate re-ductase. Antimicrob. Agents Chemother. 1991, 37, 1914-1923.
Churi, N.; Achenie, L. E. K. Novel mathematical programmingmodel for computer aided molecular design. Ind. Eng. Chem.Res. 1996, 35(10), 3788-3794.
De Vita, V. T., Jr.; Broder, S.; Fauci, A. S.; Kovacs, J. A.;Chabner, B. A. Ann. Intern. Med. 1987, 106, 568-581.
Dunn, W. J.; Rogers, D. Genetic partial least squares in QSAR.In Genetic Algorithms in Molecular Modeling; AcademicPress: London, 1996; pp 109-130.
Duvedi, A. P.; Achenie, L. E. K. Designing environmentally saferefrigerants using mathematical programming. Chem. Eng.Sci. 1996, 51, 3727-3739.
Fenniri, H. Combinatorial Chemistry: A Practical Approach.Oxford University Press: New York, 2000.
Gangjee, A.; Shi, J.; Queener, S. F.; Barrows, L. R.; Kisliuk, R.L. Synthesis of 5-methyl-5-deazononclassical antifolates asinhibitors of dihydrofolate reductases and as potential an-tineumocystis, antitoxoplasma, and antitumor agents. J. Med.Chem. 1993, 36, 3437-3443.
Gangjee, A., Vasudevan, A.; Queener, S. F.; Kisliuk, R. L.6-Substituted 2,4-diamino-5-methylpyrido[2,3-d]pyrimidinesas inhibitors of dihydrofolate reductases from Pneumocystiscarinii and Toxoplasma gondii and as antitumor agents. J.Med. Chem. 1995, 38, 1778-1785.
Gangjee, A.; Vasudevan, A.; Queener, S. F.; Kisliuk, R. L. 2,4-Diamino-5-deaza-6-substituted pyrido[2,3-d]pyrimidine an-tifolates as potent and selective nonclassical inhibitors ofdihydrofolate reductases. J. Med. Chem. 1996a, 39, 1438-1446.
Gangjee, A.; Zhu, Y.; Queener, S. F.; Francom, P.; Broom, A. D.Nonclassical 2,4-diamino-8-deazafolate analogues as inhibi-tors of dihydrofolate reductases from rat liver, Pneumocystiscarinii, and Toxoplasma gondii. J. Med. Chem. 1996b, 39,1836-1845.
Gangjee, A.; Devraj, R.; Queener, S. F. Synthesis and dihydro-folate reductase inhibitory activities of 2,4-diamino-5-deazaand 2,4-diamino-5,10-dideaza lipophilic antifolates. J. Med.Chem. 1997, 40, 470-478.
Gangjee, A.; Vidwans, A. P.; Vasudevan, A.; Queener, S. F.;Kisliuk, R. L.; Cody, V.; Li, R.; Galitsky, N.; Luft, J. R.;Pangborn, W. Structure-based design and synthesis of lipo-philic 2,4-diamino-6-substituted quinazolines and their evalu-ation as inhibitors of dihydrofolate reductases and potentialantitumor agents. J. Med. Chem. 1998, 41, 3426-3434.
Goldberg, D. E. Genetics Algorithms in Search, Optimizationand Machine Learning; Addison-Wesley: Reading, MA, 1989.
Kovacs, J. A.; Allegra, C. A.; Swan, J. C.; Drake, J. C.; Parrillo,J. E.; Chabner, B. A.; Masur, H. Potent antipneumocystis andantitoxoplasma activities of piritrexim, a lipid-soluble anti-folate. Antimicrob. Agents Chemother. 1988, 32, 430-433.
Maranas, C. D. Optimal computer-aided molecular design: apolymer design case study. Ind. Eng. Chem.Res. 1996, 35,3403.
Odele, O.; Machietto, S. Computer aided molecular design: anovel method for optimal solvent selection. Fluid PhaseEquilib. 1993, 82, 47-54.
Piper, J. R.; Johnson, C. A.; Krauth, C. A.; Carter, R. L.; Hosmer,C. A.; Queener, S. F.; Borotz, S. E.; Pfefferkorn, E. R.Lipophilic antifolates as agents against opportunistic infec-tions. 1. Agents superior to trimetrexate and piritreximagainst Toxoplasma gondii and Pneumocystis carinii in invitro evaluations. J. Med. Chem. 1996, 39, 1271-1280.
Pretel, E. J.; Lopez, P. A.; Bottini, S. B.; Brignole, E. A.Computer-aided molecular design of solvents for separationprocesses. AIChE J. 1994, 40(8), 1349-1360.
Rosowsky, A.; Mota, C. E.; Wright, J. E.; Queener, S. F. 2,4-Diamino-5-chloroquinozoline analogues of trimetrexate andpiritrexim: Synthesis and antifolate activity. J. Med. Chem.1994, 37, 4522-4528.
Rosowsky, A.; Forsch, R. A.; Queener, S. F. 2,4-Diaminopyrido-[3,2-d]pyrimidine inhibitors of dihydrofolate reductase fromPneumocystis carinii and Toxoplasma gondii. J. Med. Chem.1995, 38, 2615-2620.
Schalkoff, R. J. Artificial Neural Networks; McGraw-Hill: NewYork, 1997.
Sinha, M.; Achenie, L. E. K.; Ostrovski, G. M. Environmentallybenign solvent design by global optimization. Comput. Chem.Eng. 1999, 23, 1381-1394.
Stuttgart Neural Network Simulator User manual, Version 4.1;Institute for Parallel and Distributed High PerformanceSystems: University of Stuttgart, Germany, 1995.
Vaidyanathan, R.; El-Halwagi, M. Computer aided design ofhigh performance polymers. J. Elastomers Plast. 1994, 26(3),277.
Venkatasubramanian, V.; Sundaram A.; Chan A.; Caruthers J.M. Computer-aided molecular design using neural networksand genetic algorithms. In Genetic Algorithms in MolecularModeling; Academic Press: London, 1996; pp 271-302.
Walzer, P. D.; Kim, C. K.; Foy, J. M.; Cushion, M. T. Inhibitorsof folic acid synthesis in the treatment of experimentalPneumocystiscariniipneumonia.Antimicrob.Agents.Chemther.1988, 32, 96.
Willett, P. Similarity and Clustering in Chemical InformationSystems; Research Studies Press Ltd.: England, 1987.
Accepted for publication April 2, 2001.
BP010034Q
418 Biotechnol. Prog., 2001, Vol. 17, No. 3