mary v. relling and kathleen m....
TRANSCRIPT

93
C H A P T E R
4
PHARMACOGENETICS
Mary V. Relling and Kathleen M. Giacomini
Pharmacogenetics
is the study of the genetic basis for vari-ation in drug response. In this broadest sense, pharmacoge-netics encompasses pharmacogenomics, which employstools for surveying the entire genome to assess multigenicdeterminants of drug response. Until the technical advancesin genomics of the last few years, pharmacogenetics pro-ceeded using a forward genetic, phenotype-to-genotypeapproach. Drug response outliers were compared to indi-viduals with “normal” drug response to identify the phar-macologic basis of altered response. An inherited compo-nent to response was demonstrated using family studies orimputed through intra-
vs.
intersubject reproducibility stud-ies. With the explosion of technology in genomics, areverse genetic, genotype-to-phenotype approach is feasiblewhereby genomic polymorphisms can serve as the startingpoint to assess whether genomic variability translates intophenotypic variability.
Historical Context.
In the pre-genomics era, the frequency of geneticvariation was hypothesized to be relatively uncommon, and the dem-onstration of inherited drug-response traits applied to a relativelysmall number of drugs and pathways (Eichelbaum and Gross, 1990;Evans and Relling, 2004; Johnson and Lima, 2003). Historically,uncommon severe drug-induced phenotypes served as the triggers toinvestigate and document pharmacogenetic phenotypes. Prolongedneuromuscular blockade following normal doses of
succinylcholine
,neurotoxicity following
isoniazid
therapy (Hughes
et al.
, 1954), andmethemoglobinemia in glucose-6-phosphate dehydrogenase (G6PD)deficiency (Alving
et al.
, 1956) (
see
Chapter 39) were discovered tohave a genetic basis in the first half of the 20th century. In the 1970sand 1980s,
debrisoquine
hydroxylation and exaggerated hypotensiveeffects from that drug were related to an autosomal recessive inheriteddeficiency in the cytochrome P450 isoenzyme 2D6 (CYP2D6) (Evansand Relling, 2004). Since the elucidation of the molecular basis of thephenotypic polymorphism in CYP2D6 (Gonzalez
et al.
, 1988), themolecular bases of many other monogenic pharmacogenetic traitshave been identified (Meyer and Zanger, 1997).
Individuals differ from each other approximately every 300 to1000 nucleotides, with an estimated total of 3.2 million single
nucleotide polymorphisms (SNPs; single base pair substitutionsfound at frequencies
≥
1% in a population) in the genome (Sachidan-andam
et al.
, 2001; The International SNP Map Working Group,2001). Identifying which of these variants or combinations of vari-ants have functional consequence for drug effects is the task ofmodern pharmacogenetics.
Importance of Pharmacogenetics to Variability in Drug Response
Drug response is considered to be a gene-by-environmentphenotype. That is, an individual’s response to a drugdepends on the complex interplay between environmentalfactors and genetic factors (Figure 4–1). Variation in drugresponse therefore may be explained by variation in envi-ronmental and genetic factors, alone or in combination.What proportion of drug-response variability is likely tobe genetically determined? Classical family studies pro-vide some information (Weinshilboum and Wang, 2004).Because estimating the fraction of phenotypic variabilitythat is attributable to genetic factors in pharmacogeneticsusually requires administration of a drug to twins or triosof family members, data are somewhat limited. Twinstudies have shown that drug metabolism is highly herita-ble, with genetic factors accounting for most of the varia-tion in metabolic rates for many drugs (Vesell, 2000).Results from a twin study in which the half-life of
antipy-rine
was measured are typical (Figure 4–2). Antipyrine, apyrazolone analgesic, is eliminated exclusively by metab-olism and is a substrate for multiple CYPs. There is con-siderably greater concordance in the half-life of antipyrinebetween the monozygotic (identical) twin pairs in com-parison to the dizygotic (fraternal) twin pairs. Comparisonof intra-twin
vs.
inter-pair variability suggests thatapproximately 75% to 85% of the variability in pharma-cokinetic half-lives for drugs that are eliminated bymetabolism is heritable (Penno
et al.
, 1981). It has also
94
Section I / General Principles
been proposed that heritability can be estimated by com-paring intra-subject
vs.
inter-subject variability in drugresponse or disposition in unrelated individuals (Kalow
etal.
, 1998), with the assumption that high intra-subjectreproducibility translates into high heritability; thevalidity of this method across pharmacologic phenotypesremains to be established. In any case, such studies pro-vide only an estimate of the overall contribution of inheri-
tance to the phenotype; because multiple gene productscontribute to antipyrine disposition, most of which haveunelucidated mechanisms of genetic variability, the pre-dictability of antipyrine disposition based on knowngenetic variability is poor.
Another approach to estimating the degree of heritabil-ity of a pharmacogenetic phenotype uses
ex vivo
experi-ments with cell lines derived from related individuals.Inter-
vs.
intrafamily variability and relationships amongmembers of a kindred are used to estimate heritability.Using this approach with lymphoblastoid cells, cytotoxic-ity from chemotherapeutic agents was shown to be herita-ble, with about 20% to 70% of the variability in sensitivi-ty to
5-fluorouracil
and
docetaxel
estimated as inherited,depending upon dose (Watters
et al.
, 2004).For the “monogenic” phenotypic traits of G6PD deficien-
cy, CYP2D6 or thiopurine methyltransferase (TPMT)metabolism, it is possible to predict phenotype based ongenotype. Several genetic polymorphisms of drug metabo-lizing enzymes result in monogenic traits. Based on a retro-spective study, 49% of adverse drug reactions were associat-ed with drugs that are substrates for polymorphic drugmetabolizing enzymes, a proportion larger than estimated forall drugs (22%) or for top-selling drugs (7%) (Phillips
et al.
,2001). Prospective genotype determinations may result in theability to prevent adverse drug reactions (Meyer, 2000).
Defining multigenic contributors to drug response willbe much more challenging. For some multigenic pheno-types, such as response to antihypertensives, the largenumbers of candidate genes will necessitate a largepatient sample size to produce the statistical powerrequired to solve the “multigene” problem.
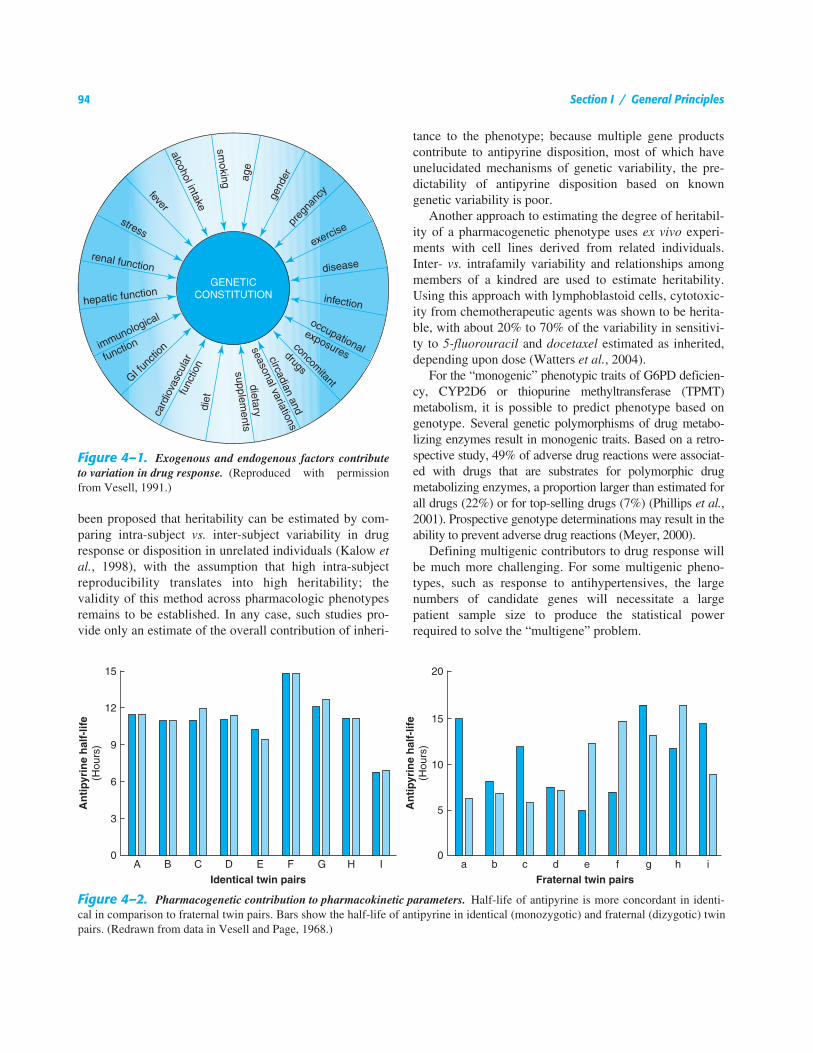
Figure 4–1.
Exogenous and endogenous factors contributeto variation in drug response.
(Reproduced with permissionfrom Vesell, 1991.)
Figure 4–2.
Pharmacogenetic contribution to pharmacokinetic parameters.
Half-life of antipyrine is more concordant in identi-cal in comparison to fraternal twin pairs. Bars show the half-life of antipyrine in identical (monozygotic) and fraternal (dizygotic) twinpairs. (Redrawn from data in Vesell and Page, 1968.)

Chapter 4 / Pharmacogenetics
95
GENOMIC BASIS OF PHARMACOGENETICS
Phenotype-Driven Terminology
Because initial discoveries in pharmacogenetics weredriven by variable phenotypes and defined by familyand twin studies, the classic genetic terms for monogen-ic traits apply to some pharmacogenetic polymorphisms.A trait (
e.g.
, CYP2D6 “poor metabolism”) is deemedautosomal recessive if the responsible gene is located onan autosome (
i.e.
, it is not sex-linked) and a distinct phe-notype is evident only with nonfunctional alleles onboth the maternal and paternal chromosomes. For manyof the earliest identified pharmacogenetic polymorphisms,phenotype did not differ enough between heterozygotesand homozygous “wild-type” individuals to distinguishthat heterozygotes exhibited an intermediate (or codom-inant) phenotype (
e.g.,
for CYP2D6-mediated debriso-quine metabolism). Other traits, such as TPMT, exhibitthree relatively distinct phenotypes, and thus weredeemed codominant even in the premolecular era. Withthe advances in molecular characterization of polymor-phisms and a genotype-to-phenotype approach, addi-tional polymorphic traits (
e.g.,
CYP2C19 metabolism ofdrugs such as
mephenytoin
and
omeprazole
) are nowrecognized to exhibit some degree of codominance.Some pharmacogenetic traits, such as the long QT syn-drome, segregate as dominant traits; the long QT syn-drome is associated with heterozygous loss-of-functionmutations of ion channels. A prolonged QT interval isseen on the electrocardiogram, either basally or in thepresence of certain drugs, and the individual is predis-posed to cardiac arrhythmias (
see
Chapter 34).In an era of detailed molecular characterization, two
major factors complicate the historical designation ofrecessive, codominant, and dominant traits. First, evenwithin a single gene, a vast array of polymorphisms (pro-moter, coding, noncoding, completely inactivating, ormodestly modifying) are possible, making the assignmentof “variant”
vs
. “wild-type” to an allele a designation thatdepends upon a complete survey of the gene’s polymor-phisms and is not necessarily easily assigned. Secondly,most traits (pharmacogenetic and otherwise) are multigen-ic, not monogenic. Thus, even if the designations of reces-sive, codominant, and dominant are informative for agiven gene, their utility in describing the genetic variabili-ty that underlies variability in drug response phenotype isdiminished, because most phenotypic variability is likelyto be multigenic.
Types of Genetic Variants
A
polymorphism
is a variation in the DNA sequence thatis present at an allele frequency of 1% or greater in a pop-ulation. Two major types of sequence variation have beenassociated with variation in human phenotype:
singlenucleotide polymorphisms
(SNPs) and
insertions/dele-tions
(indels) (Figure 4–3). In comparison to base pairsubstitutions, indels are much less frequent in the genomeand are of particularly low frequency in coding regions ofgenes (Cargill
et al.
, 1999; Stephens
et al.
, 2001). Singlebase pair substitutions that are present at frequencies of1% or greater in a population are termed single nucleotidepolymorphisms (SNPs) and are present in the humangenome at approximately 1 SNP every few hundred to athousand base pairs, depending on the gene region(Stephens
et al.
, 2001).SNPs in the coding region are termed
cSNPs
. cSNPs arefurther classified as nonsynonymous (or
missense
) if thebase pair change results in an amino acid substitution, orsynonymous (or
sense
) if the base pair substitution within acodon does not alter the encoded amino acid. Typically,substitutions of the third base pair, termed the
wobble posi-tion
, in a three base pair codon, such as the G to A substitu-tion in proline shown in Figure 4–3, do not alter the encod-ed amino acid. Base pair substitutions that lead to a stopcodon are termed
nonsense
mutations. In addition, about10% of SNPs can have more than two possible alleles (
e.g.,
a C can be replaced by either an A or G), so that the samepolymorphic site can be associated with amino acid substi-tutions in some alleles but not others.
Polymorphisms in noncoding regions of genes mayoccur in the 3' and 5' untranslated regions, in promoter orenhancer regions, in intronic regions, or in large regionsbetween genes, intergenic regions (Figure 4–4). Polymor-phisms in introns found near exon-intron boundaries areoften treated as a separate category from other intronicpolymorphisms since these may affect splicing, and there-by affect function. Noncoding SNPs in promoters orenhancers may alter
cis
- or
trans
-acting elements that reg-ulate gene transcription or transcript stability. NoncodingSNPs in introns or exons may create alternative exonsplicing sites, and the altered transcript may have fewer ormore exons, or shorter or larger exons, than the wild-typetranscript. Introduction or deletion of exonic sequence cancause a frame shift in the translated protein and therebychange protein structure or function, or result in an earlystop codon, which makes an unstable or nonfunctionalprotein. Because 95% of the genome is intergenic, mostpolymorphisms are unlikely to directly affect the encodedtranscript or protein. However, intergenic polymorphisms

96
Section I / General Principles
may have biological consequences by affecting DNA ter-tiary structure, interaction with chromatin and topoi-somerases, or DNA replication. Thus, intergenic polymor-phisms cannot be assumed to be without pharmacogeneticimportance.
A remarkable degree of diversity in the types of inser-tions/deletions that are tolerated as germline polymor-phisms is evident. A common glutathione-S-transferaseM1 (
GSTM1
) polymorphism is caused by a 50-kilobase(kb) germline deletion, and the null allele has a populationfrequency of 0.3 to 0.5, depending on race/ethnicity. Bio-chemical studies indicate that livers from homozygousnull individuals have only ~50% of the glutathione conju-gating capacity of those with at least one copy of the
GSTM1
gene (Townsend and Tew, 2003a). The numberof TA repeats in the
UGT1A1
promoter affects the quanti-tative expression of this crucial glucuronosyl transferasein liver; although 4 to 9 TA repeats exist in germline-inherited alleles, 6 or 7 repeats constitute the most com-mon alleles (Monaghan
et al.
, 1996). Cystathionine
β
-synthase has a common 68 base pair insertion/deletionpolymorphism that has been linked to folate levels (Kraus
et al.
, 1998). Although in many of these cases the localsequence context of these insertions/deletions stronglysuggests mechanisms underlying the genomic alterations(
e.g.,
homologous recombination sites bracket the
GSTM1
deletion), high allele frequencies are maintained due toMendelian inheritance.
A
haplotype
, which is defined as a series of allelesfound at a linked locus on a chromosome, specifies theDNA sequence variation in a gene or a gene region onone chromosome. For example, consider two SNPs in
ABCB1
, which encodes for the multidrug resistance pro-tein, P-glycoprotein. One SNP is a T to A base pair sub-stitution at position 3421 and the other is a C to T changeat position 3435. Possible haplotypes would be T
3421
C
3435
,T
3421
T
3435
, A
3421
C
3435
, and A
3421
T
3435
. For any gene, indi-viduals will have two haplotypes, one maternal and onepaternal in origin, which may or may not be identical.Haplotypes are important because they are the functionalunit of the gene. That is, a haplotype represents the con-stellation of variants that occur together for the gene on
Figure 4–3.
Molecular mechanisms of genetic polymor-phisms.
The most common genetic variants are single nucleotidepolymorphism substitutions (SNPs).
Coding nonsynonymous
SNPsresult in a nucleotide substitution that changes the amino acidcodon (here proline to glutamine), which could change proteinstructure, stability, substrate affinities, or introduce a stop codon.
Coding synonymous
SNPs do not change the amino acid codon,but may have functional consequences (transcript stability, splic-ing). Noncoding SNPs may be in promoters, introns, or other regu-latory regions that may affect transcription factor binding, enhanc-ers, transcript stability, or splicing. The second major type ofpolymorphism is
indels (insertion/deletions)
. SNP indels can haveany of the same effects as SNP substitutions: short repeats in thepromoter (which can affect transcript amount), or larger insertions/deletions that add or subtract amino acids. Indels can also involvegene duplications, stably transmitted inherited germline gene repli-cation that causes increased protein expression and activity, or genedeletions that result in the complete lack of protein production. Allof these mechanisms have been implicated in common germlinepharmacogenetic polymorphisms. TPMT, thiopurine methyltrans-ferase; ABCB1, the multidrug resistance transporter (P-glycopro-tein); CYP, cytochrome P450; CBS, cystathionine
β
-synthase;UGT, UDP-glucuronyl transferase; GST, glutathione-S-transferase.
Figure 4–4.
Nomenclature of genomic regions.

Chapter 4 / Pharmacogenetics
97
each chromosome. In some cases, this constellation ofvariants, rather than the individual variant or allele, maybe functionally important. In others, however, a singlemutation may be functionally important regardless ofother linked variants within the haplotype(s).
Ethnic Diversity
Polymorphisms differ in their frequencies within humanpopulations (Burchard
et al.
, 2003; Rosenberg
et al.
,2002; Rosenberg
et al.
, 2003). Among coding regionSNPs, synonymous SNPs are present, on average, at high-er frequencies than nonsynonymous SNPs. Thus, for mostgenes, the nucleotide diversity, which reflects the numberof SNPs and the frequency of the SNPs, is greater for syn-onymous than for nonsynonymous SNPs. This factreflects selective pressures (termed
negative
or
purifyingselection
), which act to preserve the functional activity ofproteins, and therefore the amino acid sequence. Frequen-cies of polymorphisms in ethnically or racially diversehuman populations have been examined in whole genomescanning studies (Cargill
et al.
, 1999; Stephens
et al.
,2001). In these studies, polymorphisms have been classi-fied as either cosmopolitan or population (or race and eth-nic) specific. Cosmopolitan polymorphisms are thosepolymorphisms present in all ethnic groups, although fre-quencies may differ among ethnic groups. Cosmopolitanpolymorphisms are usually found at higher allele frequen-cies in comparison to population-specific polymorphisms.Likely to have arisen before migrations of humans fromAfrica, cosmopolitan polymorphisms are generally olderthan population-specific polymorphisms.
The presence of ethnic and race-specific polymor-phisms is consistent with geographical isolation of humanpopulations (Xie
et al.
, 2001). These polymorphismsprobably arose in isolated populations and then reached acertain frequency because they are advantageous (positiveselection) or more likely, neutral, conferring no advantageor disadvantage to a population. Large-scale sequencestudies in ethnically diverse populations in the UnitedStates demonstrate that African Americans have the high-est number of population-specific polymorphisms in com-parison to European Americans, Mexican Americans, andAsian Americans (Leabman
et al.
, 2003; Stephens
et al.
,2001). Africans are believed to be the oldest populationand therefore have both recently derived, population-spe-cific polymorphisms, and older polymorphisms thatoccurred before migrations out of Africa.
Consider the coding region variants of two mem-brane transporters identified in 247 ethnically diverseDNA samples (Figure 4–5). Shown are nonsynonymous
and synonymous SNPs; population-specific nonsynony-mous cSNPs are indicated in the figure. The multidrugresistance associated protein, MRP2, has a large num-ber of nonsynonymous cSNPs. There are fewer synony-mous variants than nonsynonymous variants, but theallele frequencies of the synonymous variants are great-er than those of the nonsynonymous variants (Leabman
et al.
, 2003). By comparison, DAT, the dopamine trans-porter, has a number of synonymous variants but nononsynonymous variants, suggesting that selective pres-sures have acted against substitutions that led to chang-es in amino acids.
In a survey of coding region haplotypes in 313 differ-ent genes in 80 ethnically diverse DNA samples, mostgenes were found to have between 2 and 53 haplotypes,with the average number of haplotypes in a gene being 14(Stephens
et al.
, 2001). Like SNPs, haplotypes may becosmopolitan or population specific and about 20% ofthe over 4000 identified haplotypes were cosmopolitan(Stephens
et al.
, 2001). Considering the frequencies of thehaplotypes, cosmopolitan haplotypes actually accountedfor over 80% of all haplotypes, whereas population-spe-cific haplotypes accounted for only 8%.
Polymorphism Selection
Genetic variation that results in penetrant and constitu-tively evident biological variation sometimes causes a“disease” phenotype. Cystic fibrosis, sickle-cell anemia,and Crigler-Najjar syndrome are examples of inheriteddiseases caused by single gene defects (Pani
et al.
, 2000).In the case of Crigler-Najjar syndrome, the same gene(
UGT1A1
) that is targeted by rare inactivating mutations(and associated with a serious disease) is also targeted bymodest polymorphisms (and associated with modesthyperbilirubinemia and altered drug clearance) (Mon-aghan
et al.
, 1996). Due to the disease, some evolutionaryselection against these single-gene polymorphisms ispresent. Polymorphisms in other genes have highly pene-trant effects in the drug-challenged but not in the constitu-tive state, which are the causes of monogenic pharmaco-genetic traits. There is unlikely to be any selectivepressure for or against these polymorphisms (Evans andRelling, 2004; Meyer, 2000; Weinshilboum, 2003). Thevast majority of genetic polymorphisms have a modestimpact on the affected genes, are part of a large array ofmultigenic factors that impact on drug effect, or affectgenes whose products play a minor role in drug action rel-ative to a large nongenetic effect. For example, phenobar-bital induction of metabolism may be such an overwhelm-ing “environmental” effect that polymorphisms in the

98
Section I / General Principles
affected transcription factors and drug-metabolizing geneshave modest effects in comparison.
PHARMACOGENETIC STUDY DESIGN CONSIDERATIONS
Pharmacogenetic Measures
What are pharmacogenetic traits and how are they mea-sured? A
pharmacogenetic trait
is any measurable or dis-cernible trait associated with a drug. Thus, enzyme activity,drug or metabolite levels in plasma or urine, blood pressureor lipid lowering produced by a drug, and drug-inducedgene expression patterns are examples of pharmacogenetictraits. Directly measuring a trait (
e.g.,
enzyme activity) has
the advantage that the net effect of the contributions of allgenes that influence the trait is reflected in the phenotypicmeasure. However, it has the disadvantage that it is alsoreflective of nongenetic influences (
e.g., diet, drug interac-tions, diurnal or hormonal fluctuation) and thus, may be“unstable.” For CYP2D6, if a patient is given an oral doseof dextromethorphan, and the urinary ratio of parent drug tometabolite is assessed, the phenotype is reflective of thegenotype for CYP2D6 (Meyer and Zanger, 1997). Howev-er, if dextromethorphan is given with quinidine, a potentinhibitor of CYP2D6, the phenotype may be consistent witha poor metabolizer genotype, even though the subject carrieswild-type CYP2D6 alleles. In this case, quinidine adminis-tration results in a drug-induced haplo-insufficiency, and theassignment of a CYP2D6 poor metabolizer phenotypewould not be accurate for that subject in the absence of qui-nidine. If a phenotypic measure, such as the erythromycin
Figure 4–5. Coding region polymorphisms in two membrane transporters. Shown are the dopamine transporter, DAT (encoded bySLCGA3) and multidrug resistance associated protein, MRP2 (encoded by ABCC2). Coding region variants were identified in 247 ethni-cally diverse DNA samples (100 African Americans, 100 European Americans, 30 Asians, 10 Mexicans, and 7 Pacific Islanders). Shownin light gray are synonymous variants, and in black, nonsynonymous variants. (Reproduced with permission from Shu et al., 2003.)

Chapter 4 / Pharmacogenetics 99
breath test (for CYP3A), is not stable within a subject, this isan indication that the phenotype is highly influenced bynongenetic factors, and may indicate a multigenic or weaklypenetrant effect of a monogenic trait. Because most pharma-cogenetic traits are multigenic rather than monogenic (Fig-ure 4–6), considerable effort is being made to identify theimportant genes and their polymorphisms that influencevariability in drug response.
Most genotyping methods use germline DNA, that is,DNA extracted from any somatic, diploid cells, usuallywhite blood cells or buccal cells (due to their ready acces-sibility). DNA is extremely stable if appropriately extract-ed and stored, and unlike many laboratory tests, genotyp-ing need be performed only once, because DNA sequenceis generally invariant throughout an individual's lifetime.Although tremendous progress has been made in molecu-lar biological techniques to determine genotypes, relative-ly few are used routinely in patient care. Genotyping testsare directed at each specific known polymorphic siteusing a variety of strategies that generally depend at somelevel on the specific and avid annealing of at least one oli-gonucleotide to a region of DNA flanking or overlapping
the polymorphic site (Koch, 2004). Because genomicvariability is so common (with polymorphic sites everyfew hundred nucleotides), “cryptic” or unrecognized poly-morphisms may interfere with oligonucleotide annealing,thereby resulting in false positive or false negative geno-type assignments. Full integration of genotyping into ther-apeutics will require high standards for genotyping tech-nology, perhaps with more than one method required foreach polymorphic site.
One method to assess the reliability of genotypedeterminations in a group of individuals is to assesswhether the relative number of homozygotes to het-erozygotes is consistent with the overall allele frequencyat each polymorphic site. Hardy-Weinberg equilibriumis maintained when mating within a population is ran-dom and there is no natural selection effect on the vari-ant. Such assumptions are described mathematicallywhen the proportions of the population that are observedto be homozygous for the variant genotype (q2), homo-zygous for the wild-type genotype (p2), and heterozy-gous (2*p*q) are not significantly different from thatpredicted from the overall allele frequencies (p = fre-
Figure 4–6. Monogenic versus multigenic pharmacogenetic traits. Possible alleles for a monogenic trait (upper left), in which asingle gene has a low-activity (1a) and a high-activity (1b) allele. The population frequency distribution of a monogenic trait (bottomleft), here depicted as enzyme activity, may exhibit a trimodal frequency distribution with relatively distinct separation among lowactivity (homozygosity for 1a), intermediate activity (heterozygote for 1a and 1b), and high activity (homozygosity for 1b). This iscontrasted with multigenic traits (e.g., an activity influenced by up to four different genes, genes 2 through 5), each of which has 2, 3,or 4 alleles (a through d). The population histogram for activity is unimodal-skewed, with no distinct differences among the genotypicgroups. Multiple combinations of alleles coding for low activity and high activity at several of the genes can translate into low-, medi-um-, and high-activity phenotypes.

100 Section I / General Principles
quency of wild-type allele; q = frequency of variantallele) in the population. If proportions of the observedthree genotypes, which must add up to one, differ signif-icantly from those predicted, it may indicate that a geno-typing error may be present.
Because polymorphisms are so common, haplotype(the allelic structure that indicates whether polymor-phisms within a gene are on the same or different alleles)may also be important. Thus far, experimental methods tounambiguously confirm whether polymorphisms are alle-lic has proven to be feasible but technically challenging(McDonald et al., 2002). Most investigators use statisticalprobability to assign putative or inferred haplotypes; e.g.,because the two most common SNPs in TPMT (at 460 and719) often are allelic, a genotyping result showing het-erozygosity at both SNPs will have a >95% chance ofreflecting one allele wild-type and one allele variant atboth SNP positions (resulting in a “heterozygote” geno-type for TPMT). However, the remote prospect that eachof the two alleles carries a single SNP variant, therebyconferring a homozygous variant/deficient phenotype, is atheoretical possibility.
Candidate Gene versus Genome-Wide Approaches
Because pathways involved in drug response are oftenknown or at least partially known, pharmacogenetic stud-ies are highly amenable to candidate gene association
studies. After genes in drug response pathways are identi-fied, the next step in the design of a candidate gene asso-ciation pharmacogenetic study is to identify the geneticpolymorphisms that are likely to contribute to the thera-peutic and/or adverse responses to the drug. There areseveral databases that contain information on polymor-phisms and mutations in human genes (Table 4–1), whichallow the investigator to search by gene for polymor-phisms that have been reported. Some of the databases,such as the Pharmacogenetics and PharmacogenomicsKnowledge Base (PharmGKB), include phenotypic aswell as genotypic data.
Because it is currently not practical to analyze allpolymorphisms in a candidate gene association study, itis important to select polymorphisms that are likely to beassociated with the drug-response phenotype. For thispurpose, there are two categories of polymorphisms. Thefirst are polymorphisms that do not, in and of them-selves, cause altered function of the expressed protein(e.g., an enzyme that metabolizes the drug or the drugreceptor). Rather, these polymorphisms are linked to thevariant allele that produces the altered function. Thesepolymorphisms serve as biomarkers for drug-responsephenotype. However, their major shortcoming is thatunless they are in 100% linkage with the causative poly-morphism, they are not the best markers for the drug-response phenotype.
The second type of polymorphism is the causativepolymorphism, which directly precipitates the pheno-
Table 4–1Databases Containing Information on Human Genetic Variation
DATABASE NAME URL (Agency) DESCRIPTION OF CONTENTS
Pharmacogenetics and Pharmacogenomics Knowledge Base (PharmGKB)
www.pharmgkb.org(NIH Sponsored Research Network and Knowl-edge Database)
Genotype and phenotype data related to drug response
Single Nucleotide Polymorphism Database (dbSNP)
www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp(National Center for Biotechnology Information, NCBI)
SNP
Human Genome Variation Database (HGVbase)
hgvbase.cgb.ki.se/ Genotype/phenotype associa-tions
Human Gene Mutation Database (HGMD)
www.hgmd.org/ Mutations/SNPs in human genes
Online Mendelian Inher-itance in Man
www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM(NCBI)
Human genes and genetic disorders

Chapter 4 / Pharmacogenetics 101
type. For example, a causative SNP may change anamino acid residue at a site that is highly conservedthroughout evolution. This substitution may result in aprotein that is nonfunctional or has reduced function.Whenever possible, it is desirable to select polymor-phisms for pharmacogenetic studies that are likely to becausative (Tabor et al., 2002). If biological informationindicates that a particular polymorphism alters function,for example, in cellular assays of nonsynonymous vari-ants, this polymorphism is an excellent candidate to usein an association study.
A potential drawback of the candidate gene approachis that the wrong genes may be studied. Genome-wideapproaches, using gene expression arrays, genome-widescans, or proteomics, can complement the candidate geneapproach by providing a relatively unbiased survey of thegenome to identify previously unrecognized candidategenes. For example, RNA, DNA, or protein from patientswho have unacceptable toxicity from a drug can be com-pared with identical material from identically treatedpatients who did not have such toxicity. Patterns of geneexpression, clusters of polymorphisms or heterozygosity,or relative amounts of proteins can be ascertained usingcomputational tools, to identify genes, genomic regions,or proteins that can be further assessed for germline poly-morphisms differentiating the phenotype. Gene expres-sion and proteomic approaches have the advantage thatthe abundance of signal may itself directly reflect some ofthe relevant genetic variation; however, both types ofexpression are highly influenced by choice of tissue type,which may not be available from the relevant tissue; forexample, it may not be feasible to obtain biopsies of braintissue for studies on CNS toxicity. DNA has the advan-tage that it is readily available and independent of tissuetype, but the vast majority of genomic variation is not ingenes, and the large number of SNPs raises the danger oftype I error (finding differences that are false-positives).Nonetheless, technology is rapidly evolving for genome-wide surveys of RNA, DNA, and protein, and suchapproaches hold promise for future pharmacogenomicdiscoveries.
Functional Studies of Polymorphisms
For most polymorphisms, functional information is notavailable. Therefore, to select polymorphisms that arelikely to be causative, it is important to predict whether apolymorphism may result in a change in protein func-tion, stability, or subcellular localization. One way thatwe can gain an understanding of the functional effects ofvarious types of genomic variations is to survey the
mutations that have been associated with human Mende-lian disease. The greatest number of DNA variationsassociated with diseases or traits are missense and non-sense mutations, followed by deletions (Figure 4–7).Further studies have suggested that of amino acid replace-ments associated with human disease, there is a highrepresentation at residues that are most evolutionarilyconserved (Miller and Kumar, 2001; Ng and Henikoff,2003). These data have been supplemented by a largesurvey of genetic variation in membrane transportersimportant in drug response (Leabman et al., 2003). Thatsurvey shows that nonsynonymous SNPs that alter evo-lutionarily conserved amino acids are present at lowerallele frequencies on average than those that alter resi-dues that are not conserved across species. This suggeststhat SNPs that alter evolutionarily conserved residuesare most deleterious. The nature of chemical change ofan amino acid substitution determines the functionaleffect of an amino acid variant. More radical changes inamino acids are more likely to be associated with dis-ease than more conservative changes. For example, sub-stitution of a charged amino acid (Arg) for a nonpolar,uncharged amino acid (Cys) is more likely to affectfunction than substitution of residues that are morechemically similar (e.g., Arg to Lys).
Among the first pharmacogenetic examples to be dis-covered was glucose-6-phosphate dehydrogenase (G6PD)deficiency, an X-linked monogenic trait that results insevere hemolytic anemia in individuals after ingestion offava beans or various drugs, including many antimalarialagents (Alving et al., 1956). G6PD is normally present inred blood cells and helps to regulate levels of glutathione(GSH), an antioxidant. Antimalarials such as primaquine
Figure 4–7. DNA mutations associated with human diseases.Among the 27,027 DNA variations studied, 1,222 were associatedwith diseases. These 1,222 are sorted into functional groups in theproportions indicated by the pie chart. Missense and nonsensemutations account for the greatest fraction of DNA variationsassociated with Mendelian disease (Botstein and Risch, 2003).
102 Section I / General Principles
increase red blood cell fragility in individuals with G6PDdeficiency, leading to profound hemolytic anemia. Inter-estingly, the severity of the deficiency syndrome variesamong individuals and is related to the amino acid variantin G6PD. The severe form of G6PD deficiency is associ-ated with changes at residues that are highly conservedacross evolutionary history. Chemical change is also moreradical on average in mutations associated with severeG6PD deficiency in comparison to mutations associatedwith milder forms of the syndrome. Collectively, studiesof Mendelian traits and polymorphisms suggest that non-synonymous SNPs that alter residues that are highly con-served among species and those that result in more radicalchanges in the nature of the amino acid are likely to be thebest candidates for causing functional changes. The infor-
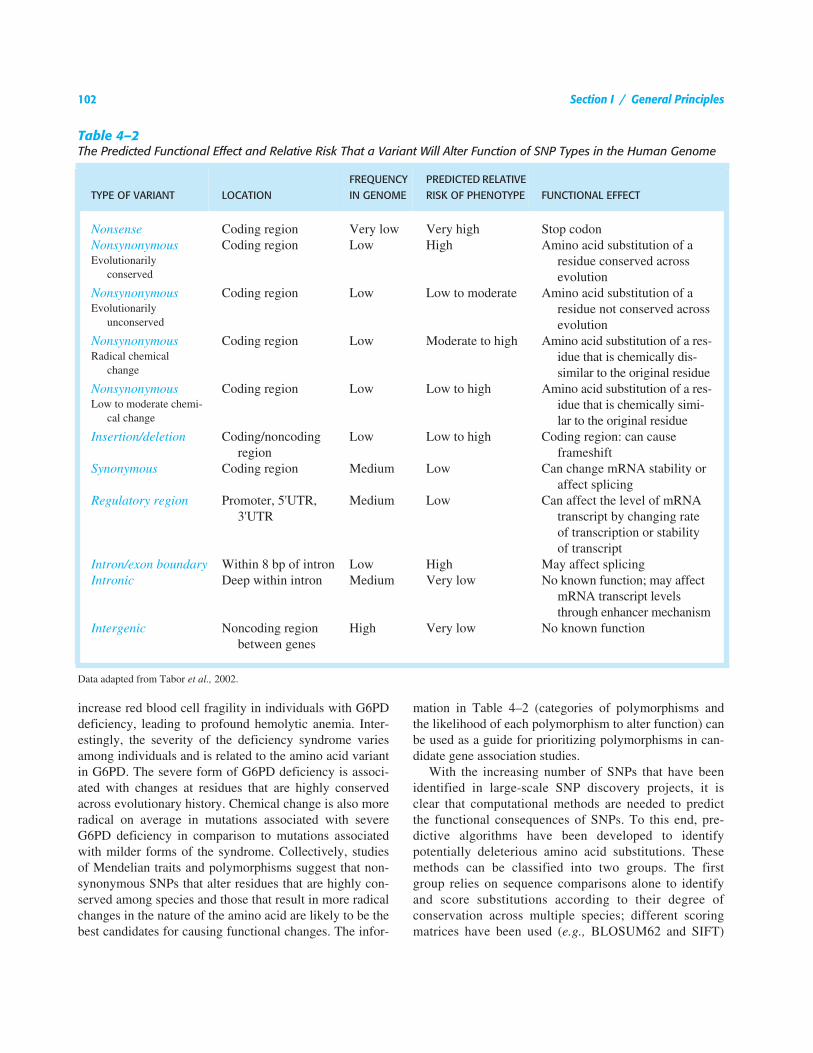
mation in Table 4–2 (categories of polymorphisms andthe likelihood of each polymorphism to alter function) canbe used as a guide for prioritizing polymorphisms in can-didate gene association studies.
With the increasing number of SNPs that have beenidentified in large-scale SNP discovery projects, it isclear that computational methods are needed to predictthe functional consequences of SNPs. To this end, pre-dictive algorithms have been developed to identifypotentially deleterious amino acid substitutions. Thesemethods can be classified into two groups. The firstgroup relies on sequence comparisons alone to identifyand score substitutions according to their degree ofconservation across multiple species; different scoringmatrices have been used (e.g., BLOSUM62 and SIFT)
Table 4–2The Predicted Functional Effect and Relative Risk That a Variant Will Alter Function of SNP Types in the Human Genome
TYPE OF VARIANT LOCATIONFREQUENCY IN GENOME
PREDICTED RELATIVE RISK OF PHENOTYPE FUNCTIONAL EFFECT
Nonsense Coding region Very low Very high Stop codonNonsynonymous Coding region Low High Amino acid substitution of a
residue conserved across evolution
Evolutionarily conserved
Nonsynonymous Coding region Low Low to moderate Amino acid substitution of a residue not conserved across evolution
Evolutionarily unconserved
Nonsynonymous Coding region Low Moderate to high Amino acid substitution of a res-idue that is chemically dis-similar to the original residue
Radical chemical change
Nonsynonymous Coding region Low Low to high Amino acid substitution of a res-idue that is chemically simi-lar to the original residue
Low to moderate chemi-cal change
Insertion/deletion Coding/noncoding region
Low Low to high Coding region: can cause frameshift
Synonymous Coding region Medium Low Can change mRNA stability or affect splicing
Regulatory region Promoter, 5'UTR, 3'UTR
Medium Low Can affect the level of mRNA transcript by changing rate of transcription or stability of transcript
Intron/exon boundary Within 8 bp of intron Low High May affect splicingIntronic Deep within intron Medium Very low No known function; may affect
mRNA transcript levels through enhancer mechanism
Intergenic Noncoding region between genes
High Very low No known function
Data adapted from Tabor et al., 2002.
Chapter 4 / Pharmacogenetics 103
(Henikoff and Henikoff, 1992; Ng and Henikoff, 2003).The second group of methods relies on mapping of SNPsonto protein structures, in addition to sequence compari-sons (Mirkovic et al., 2004). For example, rules havebeen developed that classify SNPs in terms of theirimpact on folding and stability of the native proteinstructure as well as shapes of its binding sites. Suchrules depend on the structural context in which SNPsoccur (e.g., buried in the core of the fold or exposed tothe solvent, in the binding site or not), and are inferredby machine learning methods from many functionallyannotated SNPs in test proteins.
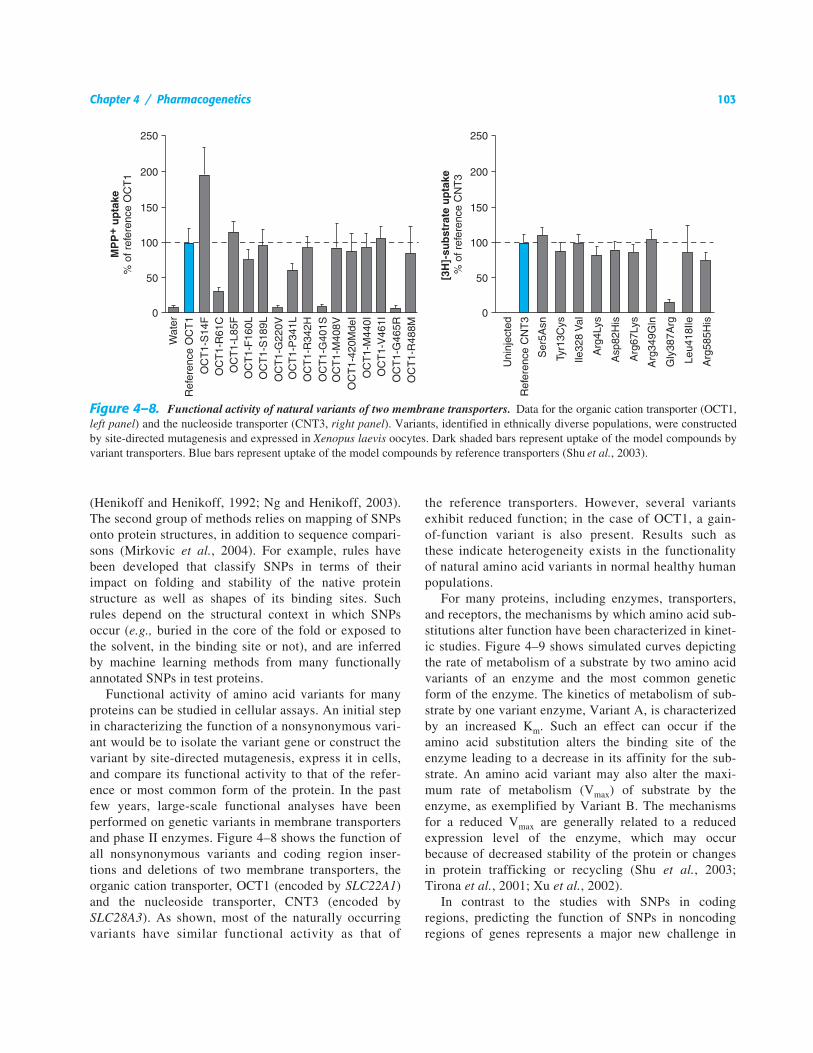
Functional activity of amino acid variants for manyproteins can be studied in cellular assays. An initial stepin characterizing the function of a nonsynonymous vari-ant would be to isolate the variant gene or construct thevariant by site-directed mutagenesis, express it in cells,and compare its functional activity to that of the refer-ence or most common form of the protein. In the pastfew years, large-scale functional analyses have beenperformed on genetic variants in membrane transportersand phase II enzymes. Figure 4–8 shows the function ofall nonsynonymous variants and coding region inser-tions and deletions of two membrane transporters, theorganic cation transporter, OCT1 (encoded by SLC22A1)and the nucleoside transporter, CNT3 (encoded bySLC28A3). As shown, most of the naturally occurringvariants have similar functional activity as that of
the reference transporters. However, several variantsexhibit reduced function; in the case of OCT1, a gain-of-function variant is also present. Results such asthese indicate heterogeneity exists in the functionalityof natural amino acid variants in normal healthy humanpopulations.
For many proteins, including enzymes, transporters,and receptors, the mechanisms by which amino acid sub-stitutions alter function have been characterized in kinet-ic studies. Figure 4–9 shows simulated curves depictingthe rate of metabolism of a substrate by two amino acidvariants of an enzyme and the most common geneticform of the enzyme. The kinetics of metabolism of sub-strate by one variant enzyme, Variant A, is characterizedby an increased Km. Such an effect can occur if theamino acid substitution alters the binding site of theenzyme leading to a decrease in its affinity for the sub-strate. An amino acid variant may also alter the maxi-mum rate of metabolism (Vmax) of substrate by theenzyme, as exemplified by Variant B. The mechanismsfor a reduced Vmax are generally related to a reducedexpression level of the enzyme, which may occurbecause of decreased stability of the protein or changesin protein trafficking or recycling (Shu et al., 2003;Tirona et al., 2001; Xu et al., 2002).
In contrast to the studies with SNPs in codingregions, predicting the function of SNPs in noncodingregions of genes represents a major new challenge in
Figure 4–8. Functional activity of natural variants of two membrane transporters. Data for the organic cation transporter (OCT1,left panel) and the nucleoside transporter (CNT3, right panel). Variants, identified in ethnically diverse populations, were constructedby site-directed mutagenesis and expressed in Xenopus laevis oocytes. Dark shaded bars represent uptake of the model compounds byvariant transporters. Blue bars represent uptake of the model compounds by reference transporters (Shu et al., 2003).

104 Section I / General Principles
human genetics and pharmacogenetics. The principles ofevolutionary conservation that have been shown to beimportant in predicting the function of nonsynonymousvariants in the coding region need to be refined and test-ed as predictors of function of SNPs in noncodingregions. New methods in comparative genomics arebeing refined to identify conserved elements in noncod-ing regions of genes that may be functionally important(Bejerano et al., 2004; Boffelli et al., 2004; Brudno etal., 2003).
Pharmacogenetic Phenotypes
Candidate genes for therapeutic and adverse response canbe divided into three categories: pharmacokinetic, recep-tor/target, and disease-modifying.
Pharmacokinetics. Germline variability in genes thatencode determinants of the pharmacokinetics of a drug,in particular enzymes and transporters, affect drug con-centrations, and are therefore major determinants oftherapeutic and adverse drug response (Table 4–3;Nebert et al., 1996). Multiple enzymes and transportersmay be involved in the pharmacokinetics of a singledrug. Several polymorphisms in drug metabolizing enzymeswere discovered as monogenic phenotypic trait varia-tions, and thus may be referenced using their phenotypicdesignations (e.g., slow vs. fast acetylation, extensive vs.poor metabolizers of debrisoquine or sparteine) rather
than their genotypic designations that reference the genethat is the target of polymorphisms in each case (NAT2and CYP2D6 polymorphisms, respectively) (Grant et al.,1990). CYP2D6 is now known to catabolize the two ini-tial probe drugs (sparteine and debrisoquine), each ofwhich was associated with exaggerated responses in 5%to 10% of treated individuals. The exaggerated respons-es are an inherited trait (Eichelbaum et al., 1975; Mah-goub et al., 1977). At present, a very large number ofmedications (estimated at 15% to 25% of all medicinesin use) have been shown to be substrates for CYP2D6(Table 4–3). The molecular and phenotypic character-ization of multiple racial and ethnic groups has shownthat seven variant alleles account for well over 90% ofthe “poor metabolizer” low-activity alleles for this genein most racial groups; that the frequency of variantalleles varies with geographic origin; and that a smallpercentage of individuals carry stable duplications ofCYP2D6, with “ultra-rapid” metabolizers having up to13 copies of the active gene (Ingelman-Sundberg andEvans, 2001). Phenotypic consequences of the deficientCYP2D6 phenotype include increased risk of toxicity ofantidepressants or antipsychotics (catabolized by theenzyme), and lack of analgesic effects of codeine(anabolized by the enzyme); conversely, the ultra-rapidphenotype is associated with extremely rapid clearanceand thus inefficacy of antidepressants (Kirchheiner etal., 2001).
A promoter region variant in the enzyme UGT1A1,UGT1A1*28, which has an additional TA in comparisonto the more common form of the gene, has been associat-ed with a reduced transcription rate of UGT1A1 and lowerglucuronidation activity of the enzyme. This reducedactivity has been associated with higher levels of theactive metabolite of the cancer chemotherapeutic agentirinotecan (see Chapters 3 and 51). The metabolite, SN38,which is eliminated by glucuronidation, is associated withthe risk of toxicity (Iyer et al., 2002), which will be moresevere in individuals with genetically lower UGT1A1activity.
CYP2C19 codes for a cytochrome P450, historicallytermed mephenytoin hydroxylase, that displays penetrantpharmacogenetic variability, with just a few SNPsaccounting for the majority of the deficient, poor metabo-lizer phenotype (Mallal et al., 2002). The deficient pheno-type is much more common in Chinese and Japanesepopulations. Several proton pump inhibitors, includingomeprazole and lansoprazole, are inactivated by CYP2C19.Thus, the deficient patients have higher exposure to activeparent drug, a greater pharmacodynamic effect (highergastric pH), and a higher probability of ulcer cure than
Figure 4–9. Simulated concentration–dependence curvesshowing the rate of metabolism of a hypothetical substrate bythe common genetic form of an enzyme and two nonsynony-mous variants. Variant A exhibits an increased Km and likelyreflects a change in the substrate binding site of the protein bythe substituted amino acid. Variant B exhibits a change in themaximum rate of metabolism (Vmax) of the substrate. This maybe due to reduced expression level of the enzyme.
l

105
Table 4–3Examples of Genetic Polymorphisms Influencing Drug Response
GENE PRODUCT(GENE) DRUGS RESPONSES AFFECTED
Drug metabolizersCYP2C9 Tolbutamide, warfarin, phenytoin,
nonsteroidal antiinflammatoriesAnticoagulant effect of warfarin (Aithal et al.,
1999; Roden, 2003; Weinshilboum, 2003)CYP2C19 Mephenytoin, omeprazole, hexobar-
bital, mephobarbital, propranolol, proguanil, phenytoin
Peptic ulcer response to omeprazole (Kirchheiner et al., 2001)
CYP2D6 β blockers, antidepressants, anti-psychotics, codeine, debrisoquine, dextromethorphan, encainide, flecainide, fluoxetine, guanoxan, N-propylajmaline, perhexiline, phenacetin, phenformin, pro-pafenone, sparteine
Tardive dyskinesia from antipsychotics, narcotic side effects, codeine efficacy, imipramine dose requirement, β blocker effect (Kirchhei-ner et al., 2001; Weinshilboum, 2003)
CYP3A4/3A5/3A7 Macrolides, cyclosporine, tacrolimus, Ca2+ channel blockers, midazolam, terfenadine, lidocaine, dapsone, quinidine, triazolam, etoposide, teniposide, lovastatin, alfentanil, tamoxifen, steroids
Efficacy of immunosuppressive effects of tac-rolimus (Evans and Relling, 2004)
Dihydropyrimidine dehy-drogenase
Fluorouracil 5-Fluorouracil neurotoxicity (Chibana et al., 2002)
N-acetyltransferase (NAT2)
Isoniazid, hydralazine, sulfonamides, amonafide, procainamide, dapsone, caffeine
Hypersensitivity to sulfonamides, amonafide tox-icity, hydralazine-induced lupus, isoniazid neurotoxicity (Roden, 2003; Grant et al., 1990)
Glutathione transferases (GSTM1, GSTT1, GSTP1)
Several anticancer agents Decreased response in breast cancer, more toxic-ity and worse response in acute myelogenous leukemia (Townsend and Tew, 2003b)
Thiopurine methyltrans-ferase (TPMT)
Mercaptopurine, thioguanine, azathioprine
Thiopurine toxicity and efficacy, risk of second cancers (Relling and Dervieux, 2001; Wein-shilboum, 2003)
UDP-glucuronosyl-trans-ferase (UGT1A1)
Irinotecan, bilirubin Irinotecan toxicity (Iyer et al., 1998; Relling and Dervieux, 2001)
P-glycoprotein (ABCB1) Natural product anticancer drugs, HIV protease inhibitors, digoxin
Decreased CD4 response in HIV-infected patients, decreased digoxin AUC, drug resis-tance in epilepsy (Fellay et al., 2002; Quirk et al., 2004; Roden, 2003; Siddiqui et al., 2003)
UGT2B7 Morphine Morphine plasma levels (Sawyer et al., 2003)COMT Levodopa Enhanced drug effect (Weinshilboum, 2003)CYP2B6 Cyclophosphamide Ovarian failure (Takada et al., 2004)Targets and receptors
Angiotensin-converting enzyme (ACE)
ACE inhibitors (e.g., enalapril) Renoprotective effects, hypotension, left ventric-ular mass reduction, cough (van Essen et al., 1996; Roden, 2003)
(Continued)
106
Thymidylate synthase Methotrexate Leukemia response, colorectal cancer response (Krajinovic et al., 2002; Relling and Dervieux, 2001)
β2 Adrenergic receptor (ADBR2)
β2 Antagonists (e.g., albuterol, terbutaline)
Bronchodilation, susceptibility to agonist-induced desensitization, cardiovascular effects (e.g., increased heart rate, cardiac index, peripheral vasodilation) (Roden, 2003; Tan et al., 1997)
β1 Adrenergic receptor (ADBR1)
β1 Antagonists Response to β1 antagonists (Johnson and Lima, 2003)
5-Lipoxygenase (ALOX5)
Leukotriene receptor antagonists Asthma response (Drazen et al., 1999)
Dopamine receptors (D2, D3, D4)
Antipsychotics (e.g., haloperidol, clozapine, thioridazine, nemonapride)
Antipsychotic response (D2, D3, D4), antipsy-chotic-induced tardive dyskinesia (D3) and acute akathisia (D3), hyperprolactinemia in females (D2) (Arranz et al., 2000; Evans and McLeod, 2003)
Estrogen receptor α Estrogen hormone replacement therapy High-density lipoprotein cholesterol (Herrington et al., 2002)
Serotonin transporter (5-HTT)
Antidepressants (e.g., clomipramine, fluoxetine, paroxetine, fluvoxamine)
Clozapine effects, 5-HT neurotransmission, anti-depressant response (Arranz et al., 2000)
Serotonin receptor (5-HT2A)
Antipsychotics Clozapine antipsychotic response, tardive dyski-nesia, paroxetine antidepression response, drug discrimination (Arranz et al., 2000) (Murphy et al., 2003)
HMG-CoA reductase Pravastatin Reduction in serum cholesterolModifiers
Adducin Diuretics Myocardial infarction or strokes (Roden, 2003)Apolipoprotein E Statins (e.g., simvastatin), tacrine Lipid-lowering; clinical improvement in
Alzheimer’s (Evans and McLeod, 2003)Human leukocyte antigen Abacavir Hypersensitivity reactions (Mallal et al., 2002)Cholesteryl ester
transfer proteinStatins (e.g., pravastatin) Slowing atherosclerosis progression (Evans and
McLeod, 2003)Ion channels (HERG,
KvLQT1, Mink, MiRP1)
Erythromycin, cisapride, clarithromycin, quinidine
Increased risk of drug-induced torsades de pointes, increased QT interval (Roden, 2003; Roden, 2004)
Methylguanine-deoxy-ribonucleic acid methyltransferase
Carmustine Response of glioma to carmustine (Evans and McLeod, 2003)
Parkin Levodopa Parkinson’s disease response (Evans and McLe-od, 2003)
MTHFR Methotrexate Gastrointestinal toxicity (Ulrich et al., 2001)Prothrombin, factor V Oral contraceptives Venous thrombosis risk (Evans and McLeod, 2003)Stromelysin-1 Statins (e.g., pravastatin) Reduction in cardiovascular events and in repeat
angioplasty (Evans et al., 2003)Vitamin D receptor Estrogen Bone mineral density (Hustmyer et al., 1994)
Table 4–3Examples of Genetic Polymorphisms Influencing Drug Response (Continued)
GENE PRODUCT(GENE) DRUGS RESPONSES AFFECTED

Chapter 4 / Pharmacogenetics 107
heterozygotes or homozygous wild-type individuals (Fig-ure 4–10).
The anticoagulant warfarin is catabolized by CYP2C9.Inactivating polymorphisms in CYP2C9 are common(Goldstein, 2001), with 2% to 10% of most populationsbeing homozygous for low-activity variants, and are asso-ciated with lower warfarin clearance, a higher risk ofbleeding complications, and lower dose requirements(Aithal et al., 1999).
Thiopurine methyltransferase (TPMT) methylates thi-opurines such as mercaptopurine (an antileukemic drugthat is also the product of azathioprine metabolism).One in 300 individuals is homozygous deficient, 10%are heterozygotes, and about 90% are homozygous forthe wild-type alleles for TPMT (Weinshilboum andSladek, 1980). Three SNPs account for over 90% of theinactivating alleles (Yates et al., 1997). Because methy-lation of mercaptopurine competes with activation of thedrug to thioguanine nucleotides, the concentration of theactive (but also toxic) thioguanine metabolites isinversely related to TPMT activity and directly relatedto the probability of pharmacologic effects. Dose reduc-tions (from the “average” population dose) may berequired to avoid myelosuppression in 100% of homozy-gous deficient patients, 35% of heterozygotes, and only7% to 8% of those with homozygous wild-type activity(Relling et al., 1999). The rare homozygous deficientpatients can tolerate 10% or less of the mercaptopurinedoses tolerated by the homozygous wild-type patients,with heterozygotes often requiring an intermediate dose.Mercaptopurine has a narrow therapeutic range, and dos-ing by trial and error can place patients at higher risk of
toxicity; thus, prospective adjustment of thiopurine dosesbased on TPMT genotype has been suggested (Leskoand Woodcock, 2004). Life-threatening toxicity has alsobeen reported when azathioprine has been given topatients with nonmalignant conditions (such as Crohn’sdisease, arthritis, or for prevention of solid organ trans-plant rejection) (Evans and Johnson, 2001; Evans andRelling, 2004; Weinshilboum, 2003).
Pharmacogenetics and Drug Targets. Gene productsthat are direct targets for drugs have an important role inpharmacogenetics (Johnson and Lima, 2003). Whereashighly penetrant variants with profound functional con-sequences in some genes may cause disease phenotypesthat confer negative selective pressure, more subtle vari-ations in the same genes can be maintained in the popu-lation without causing disease, but nonetheless causingvariation in drug response. For example, complete inac-tivation via rare point mutations in methylenetetrahydro-folate reductase (MTHFR) causes severe mental retarda-tion, cardiovascular disease, and a shortened lifespan(Goyette et al., 1994). MTHFR reduces 5,10-CH2- to 5-CH3-tetrahydrofolate, and thereby interacts with folate-dependent one-carbon synthesis reactions, includinghomocysteine/methionine metabolism and pyrimidine/purine synthesis (see Chapter 51). This pathway is thetarget of several antifolate drugs (Figure 4–11). Whereasrare variants in MTHFR may result in early death, the677C→T SNP causes an amino acid substitution that ismaintained in the population at a high frequency (variantallele, q, frequency in most white populations = 0.4).This variant is associated with modestly lower MTHFR
Figure 4–10. Effect of CYP2C19 genotype on proton pump inhibitor (PPI) pharmacokinetics (AUC), gastric pH, and ulcer curerates. Depicted are the average variables for CYP2C19 homozygous extensive metabolizers (homEM), heterozygotes (hetEM), andpoor metabolizers (PM). (Reproduced with permission from Furuta et al., 2004.)

108 Section I / General Principles
activity (about 30% less than the 677C allele) and mod-est but significantly elevated plasma homocysteine con-centrations (about 25% higher) (Klerk et al., 2002). Thispolymorphism does not alter drug pharmacokinetics, butdoes appear to modulate pharmacodynamics by predis-posing to gastrointestinal toxicity to the antifolate drugmethotrexate in stem cell transplant recipients. Follow-ing prophylactic treatment with methotrexate for graft-versus-host disease, mucositis was three times morecommon among patients homozygous for the 677T allelethan those homozygous for the 677C allele (Ulrich et al.,2001).
Methotrexate is a substrate for transporters and anabo-lizing enzymes that affect its intracellular pharmacokine-tics and that are subject to common polymorphisms (Fig-ure 4–11). Several of the direct targets (dihydrofolatereductase, purine transformylases, and thymidylate syn-thase [TYMS]) are also subject to common polymor-phisms. A polymorphic indel in TYMS (two vs. threerepeats of a 28–base pair repeat in the enhancer) affectsthe amount of enzyme expression in both normal and
tumor cells. The polymorphism is quite common, withalleles equally split between the lower-expression two-repeat and the higher-expression three-repeat alleles. TheTYMS polymorphism can affect both toxicity and efficacyof anticancer agents (e.g., fluorouracil and methotrexate)that target TYMS (Krajinovic et al., 2002). Thus, thegenetic contribution to variability in the pharmacokineticsand pharmacodynamics of methotrexate cannot be under-stood without assessing genotypes at a number of differ-ent loci.
Many drug target polymorphisms have been shownto predict responsiveness to drugs (Table 4–3). Seroto-nin receptor polymorphisms predict not only theresponsiveness to antidepressants (Figure 4–12), butalso the overall risk of depression (Murphy et al.,2003). β adrenergic receptor polymorphisms have beenlinked to asthma responsiveness (degree of change in one-second forced expiratory volume after use of a β ago-nist) (Tan et al., 1997), renal function following angio-tensin-converting enzyme (ACE) inhibitors (van Essenet al., 1996), and heart rate following β-blockers (Tay-
Figure 4–11. Gene products involved in the pharmacogenetics of a single drug, methotrexate. Those involved directly in phar-macokinetics (transport and metabolism) of methotrexate are circled (e.g., SLC19A1, FPGS, MRPs, GGH); direct targets of the drug areoutlined in solid rectangles (e.g., DHFR, TYMS, GART, ATIC); and a gene product that may indirectly modulate the effect of methotrexate(e.g., MTHFR) is outlined with a dotted line. DHFR, dihydrofolate reductase; TYMS, thymidylate synthetase; GART, glycinamide ribo-nucleotide transformylase; ATIC, aminoimidazole carboxamide transformylase; MTHFR, methylenetetrahydrofolate reductase.

Chapter 4 / Pharmacogenetics 109
lor and Kennedy, 2001). Polymorphisms in 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductasehave been linked to the degree of lipid lowering follow-ing statins, which are HMG-CoA reductase inhibitors(see Chapter 35), and to the degree of positive effectson high-density lipoproteins among women on estrogen
replacement therapy (Herrington et al., 2002; Figure 4–13).Ion channel polymorphisms have been linked to a riskof cardiac arrhythmias in the presence and absence ofdrug triggers (Roden, 2004).
Polymorphism-Modifying Diseases and Drug Respons-es. Some genes may be involved in an underlying dis-ease being treated, but do not directly interact with thedrug. Modifier polymorphisms are important for the denovo risk of some events and for the risk of drug-induced events. The MTHFR polymorphism, for exam-ple, is linked to homocysteinemia, which in turn affectsthrombosis risk (den Heijer, 2003). The risk of a drug-induced thrombosis is dependent not only on the use ofprothrombotic drugs, but on environmental and geneticpredisposition to thrombosis, which may be affected bygermline polymorphisms in MTHFR, factor V, and pro-thrombin (Chanock, 2003). These polymorphisms do notdirectly act on the pharmacokinetics or pharmacodynam-ics of prothrombotic drugs, such as glucocorticoids,estrogens, and asparaginase, but may modify the risk ofthe phenotypic event (thrombosis) in the presence of thedrug.
Likewise, polymorphisms in ion channels (e.g., HERG,KvLQT1, Mink, and MiRP1) may affect the overall riskof cardiac dysrhythmias, which may be accentuated in thepresence of a drug that can prolong the QT interval insome circumstances (e.g., macrolide antibiotics, antihista-mines) (Roden, 2003). These modifier polymorphismsmay impact on the risk of “disease” phenotypes even inthe absence of drug challenges; in the presence of drug,the “disease” phenotype may be elicited.
Figure 4–12. Pharmacodynamics and pharmacogenetics.The proportion of patients requiring a dosage decrease for theantidepressant drug paroxetine was greater (p = 0.001) in theapproximately one-third of patients who have the C/C geno-type for the serotonin 2A receptor (5HT2A) compared to thetwo-thirds of patients who have either the T/C or T/T geno-type at position 102 (Murphy et al., 2003). The major reasonfor dosage decreases in paroxetine was the occurrence ofadverse drug effects. (Reproduced with permission from Greeret al., 2003.)
Figure 4–13. Effect of genotype on response to estrogen hormone replacement therapy. Depicted are pretreatment (base line)and posttreatment (follow-up) high-density lipoprotein (HDL) cholesterol levels in women of the C/C vs. C/T or T/T HMG-CoAreductase genotype. (Reproduced with permission from Herrington et al., 2002.)

110 Section I / General Principles
Cancer pharmacogenetics have an unusual aspect inthat tumors exhibit somatically-acquired mutations inaddition to the underlying germline variation of the host.Thus, the efficacy of some anticancer drugs depends onthe genetics of both the host and the tumor. For exam-ple, non-small-cell lung cancer is treated with an inhibi-tor of epidermal growth factor receptor (EGFR), gefi-tinib (see Chapter 51). Patients whose tumors haveactivating mutations in the tyrosine kinase domain ofEGFR appear to respond better to gefitinib than thosewithout the mutations (Lynch et al., 2004). Thus, thereceptor is altered, and at the same time, individualswith the activating mutations may be considered to havea distinct category of non-small-cell lung cancer. Asanother example, the TYMS enhancer repeat polymor-phism affects not only host toxicity, but also tumor sus-ceptibility to thymidylate synthase inhibitors (Evans andMcLeod, 2003; Villafranca et al., 2001; Relling andDervieux, 2001).
Pharmacogenetics and Drug Development
Pharmacogenetics will likely impact drug regulatory con-siderations in several ways (Evans and Relling, 2004;Lesko and Woodcock, 2004; Weinshilboum and Wang,2004). Genome-wide approaches hold promise for identi-fication of new drug targets and therefore new drugs. Inaddition, accounting for genetic/genomic interindividualvariability may lead to genotype-specific development ofnew drugs, and to genotype-specific dosing regimens.
Pharmacogenomics can identify new targets. Forexample, genome-wide assessments using microarraytechnology could identify genes whose expression differ-entiates inflammatory processes; a compound could beidentified that changes expression of that gene; and thenthat compound could serve as a starting point for anti-inflammatory drug development. Proof of principle hasbeen demonstrated for identification of antileukemicagents (Stegmaier et al., 2004) and antifungal drugs (Par-sons et al., 2004), among others.
Pharmacogenetics may identify subsets of patients whowill have a very high or a very low likelihood of respond-ing to an agent. This will permit testing of the drug in aselected population that is more likely to respond, mini-mizing the possibility of adverse events in patients whoderive no benefit, and more tightly defining the parame-ters of response in the subset more likely to benefit.Somatic mutations in the EGFR gene strongly identifypatients with lung cancer who are likely to respond to thetyrosine kinase inhibitor gefitinib (Lynch et al., 2004);germline variations in 5-lipoxygenase (ALOX5) determine
which asthma patients are likely to respond to ALOXinhibitors (Drazen et al., 1999); and vasodilation inresponse to β2 agonists has been linked to β2 adrenergicreceptor polymorphisms (Johnson and Lima, 2003).
A related role for pharmacogenomics in drug develop-ment is to identify which genetic subset of patients is athighest risk for a serious adverse drug effect, and to avoidtesting the drug in that subset of patients (Lesko andWoodcock, 2004). For example, the identification of HLAsubtypes associated with hypersensitivity to the HIV-1reverse transcriptase inhibitor abacavir (Mallal et al.,2002) could theoretically identify a subset of patients whoshould receive alternative therapy, and thereby minimizeor even abrogate hypersensitivity as an adverse effect ofthis agent. Children with acute myeloid leukemia who arehomozygous for germline deletions in GSH transferase(GSTT1) are almost three times as likely to die of toxicityas those patients who have at least one wild-type copy ofGSTT1 following intensively timed antileukemic therapybut not after “usual” doses of antileukemic therapy(Davies et al., 2001). These latter results suggest animportant principle: pharmacogenetic testing may help toidentify patients who require altered dosages of medica-tions, but will not necessarily preclude the use of theagents completely.
Pharmacogenetics in Clinical Practice
Despite considerable research activity, pharmacogeneticsis rarely utilized in clinical practice (Evans and Johnson,2001; Weinshilboum and Wang, 2004). There are threemajor types of evidence that should accumulate in orderto implicate a polymorphism in clinical care (Figure 4–14): screens of tissues from multiple humans linking thepolymorphism to a trait; complementary preclinical func-tional studies indicating that the polymorphism is plausi-bly linked with the phenotype; and multiple supportiveclinical phenotype/genotype studies. Because of the highprobability of type I error in genotype/phenotype associa-tion studies, replication of clinical findings will generallybe necessary. Although the impact of the polymorphismin TPMT on mercaptopurine dosing in childhood leuke-mia is a good example of a polymorphism for which allthree types of evidence are available, proactive individu-alized dosing of thiopurines based on genotype has notbeen widely incorporated into clinical practice (Lesko etal., 2004).
Most drug dosing takes place using a population“average” dose of drug. Adjusting dosages for variablessuch as renal or liver dysfunction is often accepted indrug dosing, even in cases in which the clinical outcome

Chapter 4 / Pharmacogenetics 111
of such adjustments has not been studied. Even thoughthere are many examples of significant effects of poly-morphisms on drug disposition (e.g., Table 4–3), there ismuch more hesitation from clinicians to adjust dosesbased on genetic testing than on indirect clinical mea-sures of renal and liver function. Whether this hesita-tion reflects resistance to abandon the “trial-and-error”approach that has defined most drug dosing, distrust ofthe genetic tests (which are constantly being refined),or unfamiliarity with the principles of genetics is notclear. Nonetheless, broad public initiatives, such as theNIH-funded Pharmacogenetics and Pharmacogenomics
Knowledge Base (www.pharmGKB.org), provide usefulresources to permit clinicians to access information onpharmacogenetics (see Table 4–1).
The fact that functionally important polymorphisms areso common means that complexity of dosing will be likelyto increase substantially in the postgenomic era. Even ifevery drug has only one important polymorphism to con-sider when dosing, the scale of complexity could be large.Many individuals take multiple drugs simultaneously fordifferent diseases, and many therapeutic regimens for a sin-gle disease consist of multiple agents. This situation trans-lates into a large number of possible drug-dose combina-
Figure 4–14. Three primary types of evidence in pharmacogenetics. Screens of human tissue (A) link phenotype (thiopurinemethyltransferase activity in erythrocytes) with genotype (germline TPMT genotype). The two alleles are separated by a slash (/); the*1 and *1S alleles are wild-type, and the *2, *3A, and *3C are nonfunctional alleles. Shaded areas indicate low and intermediate levelsof enzyme activity: those with the homozygous wild-type genotype have the highest activity, those heterozygous for at least one *1allele have intermediate activity, and those homozygous for two inactive alleles have low or undetectable TPMT activity (Yates et al.,1997). Directed preclinical functional studies (B) can provide biochemical data consistent with the in vitro screens of human tissue, andmay offer further confirmatory evidence. Here, the heterologous expression of the TPMT*1 wild-type and the TPMT*2 variant allelesindicate that the former produces a more stable protein, as assessed by Western blot (Tai et al., 1997). The third type of evidence comesfrom clinical phenotype/genotype association studies (C and D). The incidence of required dosage decrease for thiopurine in childrenwith leukemia (C) differs by TPMT genotype: 100%, 35%, and 7% of patients with homozygous variant, heterozygous, or homozygouswild-type, respectively, require a dosage decrease (Relling et al., 1999). When dosages of thiopurine are adjusted based on TPMT geno-type in the successor study (D), leukemic relapse is not compromised, as indicated by comparable relapse rates in children who werewild-type vs. heterozygous for TPMT. Taken together, these three data sets indicate that the polymorphism should be accounted for indosing of thiopurines. (Reproduced with permission from Relling et al., 1999.)

112 Section I / General Principles
tions. Much of the excitement regarding the promise ofhuman genomics has emphasized the hope of discoveringindividualized “magic bullets,” and ignored the reality ofthe added complexity of additional testing and need forinterpretation of results to capitalize on individualized dos-ing. This is illustrated in a potential pharmacogeneticexample in Figure 4–15. In this case, a traditional antican-cer treatment approach is replaced with one that incorpo-
rates pharmacogenetic information with the stage of thecancer determined by a variety of standardized pathologicalcriteria. Assuming just one important genetic polymor-phism for each of the three different anticancer drugs, 11individual drug regimens can easily be generated.
Nonetheless, the potential utility of pharmacogeneticsto optimize drug therapy is great. Once adequate geno-type/phenotype studies have been conducted, molecular
Figure 4–15. Potential impact of incorporation of pharmacogenetics into dosing of drugs for a relatively simple therapeuticregimen. The traditional approach to treatment for a disease (A), in this case a cancer, is based purely on stage of the cancer. Up tothree different drugs are used in combination, with intensity of dosing dependent on the stage of the cancer. With this strategy, somewith stage II disease are not receiving as much drug as they could tolerate; some patients with stage III or IV disease are undertreatedand some are overtreated. B illustrates a hypothetical patient population with eight different multilocus genotypes. It is assumed thateach of the three drugs is affected by just one genetic polymorphism (TYMS for methotrexate [MTX], MDR1 for paclitaxel, andGSTM1 for cyclophosphamide), and each polymorphism has just two important genotypes (one coding for low and one for high activi-ty). The possible multilocus genotypes are designated by the letters A to H, and the combinations of TYMS, MDR1 and GSTM1 geno-types giving rise to those multilocus genotypes are indicated in the table. If these three genotypes, along with stage of cancer, are usedto individualize dosages (C), so that those with low activity receive lower doses (LD) and those with higher activity receive higherdoses (HD) of the relevant drug, what began as a total of three drug regimens in the absence of pharmacogenetics becomes 11 regi-mens by using pharmacogenetics for dosage individualization.

Chapter 4 / Pharmacogenetics 113
diagnostic tests will be developed that detect >95% of theimportant genetic variants for the majority of polymor-phisms, and genetic tests have the advantage that theyneed only be conducted once during an individual’s life-time. With continued incorporation of pharmacogeneticsinto clinical trials, the important genes and polymor-phisms will be identified, and data will demonstratewhether dosage individualization can improve outcomesand decrease short- and long-term adverse effects. Signifi-cant covariates will be identified to allow refinement ofdosing in the context of drug interactions and diseaseinfluences. Although the challenges are substantial, account-ing for the genetic basis of variability in response to medi-cations is likely to become a fundamental component ofdiagnosing any illness and guiding the choice and dosageof medications.
BIBLIOGRAPHYAithal, G.P., Day, C.P., Kesteven, P.J., and Daly, A.K. Association of
polymorphisms in the cytochrome P450 CYP2C9 with warfarin doserequirement and risk of bleeding complications. Lancet, 1999,353:717–719.
Alving, A.S., Carson, P.E., Flanagan, C.L., and Ickes, C.E. Enzymaticdeficiency in primaquine-sensitive erythrocytes. Science, 1956,124:484–485.
Arranz, M.J., Munro, J., Birkett, J., et al. Pharmacogenetic prediction ofclozapine response [letter]. Lancet, 2000, 355:1615–1616.
Bejerano, G., Pheasant, M., Makunin, I., et al. Ultraconserved elementsin the human genome. Science, 2004, 304:1321–1325.
Boffelli, D., Nobrega, M.A., and Rubin, E.M. Comparative genomics atthe vertebrate extremes. Nat. Rev. Genet., 2004, 5:456–465.
Botstein, D., and Risch, N. Discovering genotypes underlying humanphenotypes: past successes for mendelian disease, future approachesfor complex disease. Nat. Genet., 2003, 33(suppl):228–237.
Brudno, M., Do, C.B., Cooper, G.M., et al. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genom-ic DNA. Genome Res., 2003, 13:721–731.
Burchard, E.G., Ziv, E., Coyle, N., et al. The importance of race and eth-nic background in biomedical research and clinical practice. N. Engl.J. Med., 2003, 348:1170–1175.
Cargill, M., Altshuler, D., Ireland, J., et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat.Genet., 1999, 22:231–238.
Chanock, S. Genetic variation and hematology: single-nucleotide poly-morphisms, haplotypes, and complex disease. Semin. Hematol., 2003,40:321–328.
Chibana, K., Ishii, Y., and Fukuda, T. Tailor-made medicine for bron-chial asthma. Nippon Rinsho, 2002, 60:189–196.
Davies, S.M., Robison, L.L., Buckley, J.D., et al. Glutathione S-trans-ferase polymorphisms and outcome of chemotherapy in childhoodacute myeloid leukemia. J. Clin. Oncol., 2001, 19:1279–1287.
Drazen, J.M., Yandava, C.N., Dube, L., et al. Pharmacogenetic associa-tion between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat. Genet., 1999, 22:168–170.
Eichelbaum, M., and Gross, A.S. The genetic polymorphism of debriso-quine/sparteine metabolism—clinical aspects. Pharmacol. Ther.,1990, 46:377–394.
Eichelbaum, M., Spannbrucker, N., and Dengler, H.J. Proceedings: N-oxidation of sparteine in man and its interindividual differences. Nau-nyn Schmiedebergs Arch. Pharmacol., 1975, 287(suppl):R94.
van Essen, G.G., Rensma, P.L., de Zeeuw, D., et al. Associationbetween angiotensin-converting-enzyme gene polymorphism and fail-ure of renoprotective therapy. Lancet, 1996, 347:94–95.
Evans, W.E., and Johnson, J.A. Pharmacogenomics: the inherited basisfor interindividual differences in drug response. Annu. Rev. GenomicsHum. Genet., 2001, 2:9–39.
Evans, W.E., and McLeod, H.L. Pharmacogenomics—drug disposition,drug targets, and side effects. N. Engl. J. Med., 2003, 348:538–549.
Evans, W.E., and Relling, M.V. Moving towards individualized medi-cine with pharmacogenomics. Nature, 2004, 429:464–468.
Fellay, J., Marzolini, C., Meaden, E.R., et al. Response to antiretroviraltreatment in HIV-1-infected individuals with allelic variants of themultidrug resistance transporter 1: a pharmacogenetics study. Lancet,2002, 359:30–36.
Furuta, T., Shirai, N., Sugimoto, M., et al. Pharmacogenomics of protonpump inhibitors. Pharmacogenomics, 2004, 5:181–202.
Goldstein, J.A. Clinical relevance of genetic polymorphisms in thehuman CYP2C subfamily. Br. J. Clin. Pharmacol., 2001, 52:349–355.
Gonzalez, F.J., Skoda, R.C., Kimura, S., et al. Characterization of thecommon genetic defect in humans deficient in debrisoquine metabo-lism. Nature, 1988, 331:442–446.
Goyette, P., Sumner, J.S., Milos, R., et al. Human methylenetetrahydro-folate reductase: isolation of cDNA, mapping and mutation identifica-tion. Nat. Genet., 1994, 7:195–200.
Grant, D.M., Morike, K., Eichelbaum, M., and Meyer, U.A. Acetylationpharmacogenetics. The slow acetylator phenotype is caused bydecreased or absent arylamine N-acetyltransferase in human liver. J.Clin. Invest., 1990, 85:968–972.
Greer, M., Murphy, J., et al. Pharmacogenomics of antidepressant medi-cation intolerance. Am. J. Psychiatry, 2003, 160:1830–1835.
den Heijer, M. Hyperhomocysteinaemia as a risk factor for venousthrombosis: an update of the current evidence. Clin. Chem. Lab.Med., 2003, 41:1404–1407.
Henikoff, S., and Henikoff, J.G. Amino acid substitution matrices fromprotein blocks. Proc. Natl. Acad. Sci. U.S.A., 1992, 89:10915–10919.
Herrington, D.M., Howard, T.D., Hawkins, G.A., et al. Estrogen-recep-tor polymorphisms and effects of estrogen replacement on high-den-sity lipoprotein cholesterol in women with coronary disease. N. Engl.J. Med., 2002, 346:967–974.
Hughes, H.B., Biehl, J.P., Jones, A.P., and Schmidt, L.H. Metabolism ofisoniazid in man as related to occurrence of peripheral neuritis. Am.Rev. Tuberc., 1954, 70:266–273.
Hustmyer, F.G., Peacock, M., Hui, S., et al. Bone mineral density inrelation to polymorphism at the vitamin D receptor gene locus. J.Clin. Invest., 1994, 94:2130–2134.
Ingelman-Sundberg, M., and Evans, W.E. Unravelling the functionalgenomics of the human CYP2D6 gene locus. Pharmacogenetics,2001, 7:553–554.
Iyer, L., Das, S., Janisch, L., et al. UGT1A1*28 polymorphism as adeterminant of irinotecan disposition and toxicity. Pharmacogenom-ics J., 2002, 2:43–47.
Iyer, L., King, C.D., Whitington, P.F., et al. Genetic predisposition tothe metabolism of irinotecan (CPT-11). Role of uridine diphosphateglucuronosyltransferase isoform 1A1 in the glucuronidation of its

114 Section I / General Principles
active metabolite (SN-38) in human liver microsomes. J. Clin.Invest., 1998, 101:847–854.
Johnson, J.A., and Lima, J.J. Drug receptor/effector polymorphisms andpharmacogenetics: current status and challenges. Pharmacogenetics,2003, 13:525–534.
Kalow, W., Tang, B.K., and Endrenyi, L. Hypothesis: comparisons ofinter- and intra-individual variations can substitute for twin studies indrug research. Pharmacogenetics, 1998, 8:283–289.
Kirchheiner, J., Brosen, K., Dahl, M.L., et al. CYP2D6 and CYP2C19genotype-based dose recommendations for antidepressants: a firststep towards subpopulation-specific dosages. Acta. Psychiatr. Scand.,2001, 104:173–192.
Klerk, M., Verhoef, P., Clarke, R., et al. MTHFR 677C→T polymor-phism and risk of coronary heart disease: a meta-analysis. JAMA,2002, 288:2023–2031.
Koch, W.H. Technology platforms for pharmacogenomic diagnosticassays. Nat. Rev. Drug Discov., 2004, 3:749–761.
Krajinovic, M., Costea, I., and Chiasson, S. Polymorphism of thethymidylate synthase gene and outcome of acute lymphoblastic leu-kaemia. Lancet, 2002, 359:1033–1034.
Kraus, J.P., Oliveriusova, J., Sokolova, J., et al. The human cystathion-ine beta-synthase (CBS) gene: complete sequence, alternative splic-ing, and polymorphisms. Genomics, 1998, 52:312–324.
Leabman, M.K., Huang, C.C., DeYoung, J., et al. Natural variation inhuman membrane transporter genes reveals evolutionary and functionalconstraints. Proc. Natl. Acad. Sci. U.S.A., 2003, 100:5896–5901.
Lesko, L.J., and Woodcock, J. Opinion: Translation of pharmacogenom-ics and pharmacogenetics: a regulatory perspective. Nat. Rev. DrugDiscov., 2004, 3:763–769.
Lynch, T.J., Bell, D.W., Sordella, R., et al. Activating mutations in the epi-dermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med., 2004, 350:2129–2139.
McDonald, O.G., Krynetski, E.Y., and Evans, W.E. Molecular haplotyp-ing of genomic DNA for multiple single-nucleotide polymorphismslocated kilobases apart using long-range polymerase chain reactionand intramolecular ligation. Pharmacogenetics, 2002, 12:93–99.
Mahgoub, A., Idle, J.R., Dring, L.G., et al. Polymorphic hydroxylationof debrisoquine in man. Lancet, 1977, 2:584–586.
Mallal, S., Nolan, D., Witt, C., et al. Association between presenceof HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivityto HIV-1 reverse-transcriptase inhibitor abacavir. Lancet, 2002,359:727–732.
Meyer, U.A. Pharmacogenetics and adverse drug reactions. Lancet,2000, 356:1667–1671.
Meyer, U.A., and Zanger, U.M. Molecular mechanisms of genetic poly-morphisms of drug metabolism. Annu. Rev. Pharmacol. Toxicol.,1997, 37:269–296.
Miller, M.P., and Kumar, S. Understanding human disease mutationsthrough the use of interspecific genetic variation. Hum. Mol. Genet.,2001, 10:2319–2328.
Mirkovic, N., Marti-Renom, M.A., Weber, B.L., et al. Structure-basedassessment of missense mutations in human BRCA1: implications forbreast and ovarian cancer predisposition. Cancer Res., 2004,64:3790–3797.
Monaghan, G., Ryan, M., Seddon, R., et al. Genetic variation in biliru-bin UPD-glucuronosyltransferase gene promoter and Gilbert’s syn-drome. Lancet, 1996, 347:578–581.
Murphy, G.M. Jr., Kremer, C., Rodrigues, H.E., and Schatzberg, A.F.Pharmacogenetics of antidepressant medication intolerance. Am. J.Psychiatry, 2003, 160:1830–1835.
Nebert, D.W., McKinnon, R.A., and Puga, A. Human drug-metabolizingenzyme polymorphisms: effects on risk of toxicity and cancer. DNACell Biol., 1996, 15:273–280.
Ng, P.C., and Henikoff, S. SIFT: Predicting amino acid changes thataffect protein function. Nucleic Acids Res., 2003, 31:3812–3814.
Pani, M.A., Knapp, M., Donner, H., et al. Vitamin D receptor allelecombinations influence genetic susceptibility to type 1 diabetes inGermans. Diabetes, 2000, 49:504–507.
Parsons, A.B., Brost, R.L., Ding, H., et al. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cel-lular target pathways. Nat. Biotechnol., 2004, 22:62–69.
Penno, M.B., Dvorchik, B.H., and Vesell, E.S. Genetic variation in ratesof antipyrine metabolite formation: a study in uninduced twins. Proc.Natl. Acad. Sci. U.S.A., 1981, 78:5193–5196.
Phillips, K.A., Veenstra, D.L., Oren, E., et al. Potential role of pharma-cogenomics in reducing adverse drug reactions: a systematic review.JAMA, 2001, 286:2270–2279.
Quirk, E., McLeod, H., and Powderly, W. The pharmacogenetics of anti-retroviral therapy: a review of studies to date. Clin. Infect. Dis., 2004,39:98–106.
Relling, M.V., and Dervieux, T. Pharmacogenetics and cancer therapy.Nat. Rev. Cancer, 2001, 1:99–108.
Relling, M.V., Hancock, M.L., Rivera, G.K., et al. Mercaptopurine ther-apy intolerance and heterozygosity at the thiopurine S-methyltrans-ferase gene locus. J. Natl. Cancer Inst., 1999, 91:2001–2008.
Roden, D.M. Cardiovascular pharmacogenomics. Circulation, 2003,108:3071–3074.
Roden, D.M. Drug-induced prolongation of the QT interval. N. Engl. J.Med., 2004, 350:1013–1022.
Rosenberg, N.A., Li, L.M., Ward, R., and Pritchard, J.K. Informative-ness of genetic markers for inference of ancestry. Am. J. Hum. Genet.,2003, 73:1402–1422.
Rosenberg, N.A., Pritchard, J.K., Weber, J.L., et al. Genetic structure ofhuman populations. Science, 2002, 298:2381–2385.
Sachidanandam, R., Weissman, D., Schmidt, S.C., et al. A map ofhuman genome sequence variation containing 1.42 million singlenucleotide polymorphisms. Nature, 2001, 409:928–933.
Sawyer, M.B., Innocenti, F., Das, S., et al. A pharmacogenetic study ofuridine diphosphate-glucuronosyltransferase 2B7 in patients receivingmorphine. Clin. Pharmacol. Ther., 2003, 73:566–574.
Shu, Y., Leabman, M.K., Feng, B., et al. Evolutionary conservation pre-dicts function of variants of the human organic cation transporter,OCT 1. Proc. Natl. Acad. Sci. U.S.A., 2003, 100:5902–5907.
Siddiqui, A., Kerb, R., Weale, M.E., et al. Association of multidrugresistance in epilepsy with a polymorphism in the drug-transportergene ABCB1. N. Engl. J. Med., 2003, 348:1442–1448.
Stegmaier, K., Ross, K.N., Colavito, S.A., et al. Gene expression-basedhigh-throughput screening (GE-HTS) and application to leukemia dif-ferentiation. Nat. Genet., 2004, 36:257–263.
Stephens, J.C., Schneider, J.A., Tanguay, D.A., et al. Haplotype varia-tion and linkage disequilibrium in 313 human genes. Science, 2001,293:489–493.
Tabor, H.K., Risch, N.J., and Myers, R.M. Opinion: Candidate-geneapproaches for studying complex genetic traits: practical consider-ations. Nat. Rev. Genet., 2002, 3:391–397.
Tai, H.L., Krynetski, E.Y., Schuetz, E.G., et al. Enhanced proteolysis ofthiopurine S-methyltransferase (TPMT) encoded by mutant alleles inhumans (TPMT*3A, TPMT*2): mechanisms for the genetic polymor-phism of TPMT activity. Proc. Natl. Acad. Sci. U.S.A., 1997,94:6444–6449.

Chapter 4 / Pharmacogenetics 115
Takada, K., Arefayene, M., Desta, Z., et al. Cytochrome P450 pharma-cogenetics as a predictor of toxicity and clinical response to pulsecyclophosphamide in lupus nephritis. Arthritis Rheum., 2004,50:2202–2210.
Tan, S., Hall, I.P., Dewar, J., et al. Association between β2-adrenocep-tor polymorphism and susceptibility to bronchodilator desensitisa-tion in moderately severe stable asthmatics. Lancet, 1997, 350:995–999.
Taylor, D.R., and Kennedy, M.A. Genetic variation of the β2-adrenocep-tor: its functional and clinical importance in bronchial asthma. Am. J.Pharmacogenomics., 2001, 1:165–174.
The International SNP Map Working Group. A map of human genomesequence variation containing 1.4 million single nucleotide polymor-phisms. Nature, 2001.
Tirona, R.G., Leake, B.F., Merino, G., and Kim, R.B. Polymorphisms inOATP-C: identification of multiple allelic variants associated withaltered transport activity among European- and African-Americans. J.Biol. Chem., 2001, 276:35669–35675.
Townsend, D., and Tew, K. Cancer drugs, genetic variation and the glu-tathione-S-transferase gene family. Am. J. Pharmacogenomics,2003a, 3:157–172.
Townsend, D.M., and Tew, K.D. The role of glutathione-S-transferase inanti-cancer drug resistance. Oncogene, 2003b, 22:7369–7375.
Ulrich, C.M., Yasui, Y., Storb, R., et al. Pharmacogenetics of metho-trexate: toxicity among marrow transplantation patients varies withthe methylenetetrahydrofolate reductase C677T polymorphism.Blood, 2001, 98:231–234.
Vesell, E.S. Advances in pharmacogenetics and pharmacogenomics. J.Clin. Pharmacol., 2000, 40:930–938.
Vesell, E.S. Genetic and environmental factors causing variation in drugresponse. Mutat. Res., 1991, 247:241–257.
Vesell, E.S., and Page, J.G. Genetic control of drug levels in man: anti-pyrine. Science, 1968, 161:72–73.
Villafranca, E., Okruzhnov, Y., Dominguez, M.A., et al. Polymorphismsof the repeated sequences in the enhancer region of the thymidylatesynthase gene promoter may predict downstaging after preoperativechemoradiation in rectal cancer. J. Clin. Oncol., 2001, 19:1779–1786.
Watters, J.W., Kraja, A., Meucci, M.A., et al. Genome-wide discoveryof loci influencing chemotherapy cytotoxicity. Proc. Natl. Acad. Sci.U.S.A., 2004, 101:11809–11814.
Weinshilboum, R. Inheritance and drug response. N. Engl. J. Med.,2003, 348:529–537.
Weinshilboum, R.M., and Sladek, S.L. Mercaptopurine pharmacogenet-ics: monogenic inheritance of erythrocyte thiopurine methyltrans-ferase activity. Am. J. Hum. Genet., 1980, 32:651–662.
Weinshilboum, R., and Wang, L. Pharmacogenomics: bench to bedside.Nat. Rev. Drug Discov., 2004, 3:739–748.
Xie, H.G., Kim, R.B., Wood, A.J., and Stein, C.M. Molecular basis ofethnic differences in drug disposition and response. Annu. Rev. Phar-macol. Toxicol., 2001, 41:815–850.
Xu, Z.H., Freimuth, R.R., Eckloff, B., et al. Human 3'-phosphoadeno-sine 5'-phosphosulfate synthetase 2 (PAPSS2) pharmacogenetics:gene resequencing, genetic polymorphisms and functional character-ization of variant allozymes. Pharmacogenetics, 2002, 12:11–21.
Yates, C.R., Krynetski, E.Y., Loennechen, T., et al. Molecular diagnosisof thiopurine S-methyltransferase deficiency: genetic basis for azathi-oprine and mercaptopurine intolerance. Ann. Intern. Med., 1997,126:608–614.

116—blank